Бензотиофеновые соединения или их фармацевтически приемлемые соли - RU2158737C2
Код документа: RU2158737C2
Чертежи











Описание
Изобретение относится к области фармацевтической и органической химии, более конкретно к новым бензотиофеновым соединениям, которые можно использовать для лечения при различных медицинских показаниях, связанных с постменопаузальным синдромом, фиброидными заболеваниями матки, эндометриозом и пролиферацией клеток гладкой мускулатуры аорты. Далее настоящее изобретение относится к промежуточным соединениям, которые можно использовать для получения фармацевтически активных соединений настоящего изобретения и фармацевтических композиций.
"Постменопаузальный синдром" - это термин, который используют для описания различных патологических состояний, которые часто проявляются у женщин, которые вступают в, или у которых закончился период физиологических изменений, известных как менопауза. Хотя этот термин используют при различных патологиях, три основных эффекта постменопаузального синдрома, приводящих к наиболее длительным заболеваниям, включают: остеопороз, такие сердечно-сосудистые проявления, как гиперлипидемия, и эстроген-зависимые раковые заболевания, особенно рак груди и рак матки.
Термин остеопороз описывает группу заболеваний различной этиологии, но все они характеризуются потерей чистого веса костной массы на единицу объема. Следствием такой потери костной массы и возникающих переломов костей является нарушение функций скелета как структурной поддержки тела. Один из наиболее часто встречающихся типов остеопороза связан с менопаузой. Большинство женщин теряет от около 20% до 60% костной массы в трабекулярной части костей за 3/6 лет после прекращения месячных. Столь быстрая потеря обычно связана с возрастанием резорбции и образования костной ткани.
Однако резорбтивный цикл оказывается доминантным, и это приводит к чистой потере костной массы. Остеопороз является обычным и весьма серьезным заболеванием у женщин в постменопаузальном периоде.
Только в США насчитывается 25 миллионов женщин, подверженных этому заболеванию. Результатом остеопороза являются персональные повреждения и это приводит также к значительным экономическим потерям, связанным с хроническим характером заболевания и необходимостью интенсивной и длительной поддержки /госпитализации и домашний уход/ из-за последующих осложнений. Это особенно справедливо для пациентов более старшего возраста. Кроме того, хотя остеопороз обычно не рассматривают как угрожающее жизни состояние, от 20 до 30% смертельных исходов связано с переломами бедра у старых женщин. Значительный процент этой смертности может быть напрямую связан с постменопаузальным остеопорозом.
Наиболее повреждаемой тканью кости, на которую действует постменопаузальной остеопороз, является трабекулярная /губчатая/ ткань. Такая губчатая ткань, в основном, концентрируется вблизи концов кости /вблизи суставов/ и в отростках позвоночника. Губчатая ткань характеризуется небольшими остеоидными структурами, которые соединены друг с другом, а также с более твердой кортикальной /корковой/ тканью, которая составляет внешний слой и центральный стержень кости. Такая соединенная между собой губчатая сетка обеспечивает поддержку внешней корковой структуре и является критической для биохимической прочности всей структуры. При постменопаузальном остеопорозе происходит, главным образом, резорбция и потеря губчатой ткани, что приводит к переломам и разрушению кости. В свете потерь губчатой ткани у женщин в постменопаузальном периоде, вовсе не удивительно, что наиболее часто встречаются переломы, которые связаны с теми костями, которые в значительной степени зависят от поддержки губчатой ткани, то есть, позвоночник, шейка таких несущих вес костей, как бедро и предплечье. Действительно, переломы бедра, переломы collies и компрессионные переломы позвоночника являются признаками постменопаузального остеопороза.
К настоящему времени единственным общепринятым способом лечения постменопаузального остеопороза является эстрогензаменяющая терапия. Хотя такое лечение обычно бывает успешным, восприятие пациентом такого лечения обычно низко, так как лечение эстрогеном часто приводит к нежелательным побочным эффектам.
На протяжении пременопаузального периода большинство женщин менее подвержено сердечно-сосудистым заболеваниям, нежели наблюдается у мужчин того же возраста. Однако после менопаузы количество сердечно-сосудистых заболеваний у женщин медленно возрастает и приближается к количеству заболеваний, наблюдаемому у мужчин. Такую потерю защиты связывали с утратой эстрогена, и, в частности, с утратой эстрогеном способности регулировать уровни липидов в сыворотки. Природа способности эстрогена регулировать липиды в сыворотке не совсем понятна, но имеющиеся к настоящему времени доказательства указывают на то, что эстроген может увеличивать эффективность рецепторов липидов низкой плотности /ZDZ/ в печени, что приводит к удалению избыточного холестерина. Кроме того, эстроген, по-видимому, оказывает некоторое влияние на биосинтез холестерина, и обладает некоторыми другими благоприятными воздействиями на здоровье сердечно-сосудистой системы.
Имелось сообщение в литературе, что женщины в постменопаузальном периоде, которые получали эстрогенное лечение, демонстрировали возвращение концентраций липидов в сыворотке до значений, которые были у них в пременопаузальном периоде. Так, эфироген, по-видимому, является разумным лечением таких состояний. Однако побочные явления эстроген-заменяющей терапии неприемлемы для многих женщин, что ограничивает применение такого лечения. Идеальным лечением такого состояния должен быть агент, который смог бы регулировать уровень липидов в сыворотке как и эстроген, но который был бы лишен побочных эффектов и рисков, связанных с лечением эстрогеном.
Третья основная патология, связанная с постменопаузальным синдромом, представляет собой эстроген-зависимое раковое заболевание молочной железы и, в меньшей степени, эстроген-зависимые раковые заболевания других органов, в частности, матки. Хотя такие неоплазмы нередко связаны не только с женщинами в постменопаузальном периоде, они преобладают у населения преимущественно в пожилом, постменопаузальном возрасте. Существующая химиотерапия таких раковых заболеваний основана, в основном, на применении таких анти-эстрогеновых соединений, как, например, тамоксифен. Хотя такие смешанные агонист-антагонисты и оказывают благоприятное влияние при лечении этих раковых заболеваний и эстрогенные побочные эффекты переносимы в острых жизненных случаях, они вовсе не идеальны. Так например, эти агенты могут оказывать стимулирующее действие на некоторые популяции раковых клеток в матке за счет своих эстрогенных /агонист/ свойств, и поэтому могут оказаться контрапродуктивными в некоторых случаях. Более подходящим для лечения таких раковых заболеваний был бы агент, который был бы анти-эстрогенным соединением, обладающим незначительными свойствами агониста эстрогена /или вовсе не обладающим такими свойствами/ по отношению к репродуктивным тканям.
В ответ на существенную необходимость в новых фармацевтических агентах, которые были бы способны ослабить симптомы (наряду с другими) постменопаузального синдрома, в настоящем изобретении предложены бензотиофеновые соединения, их фармацевтические композиции, и способы использования таких соединений для лечения постменопаузального синдрома и других связанных с эстрогеном патологических состояний, которые были указаны ранее.
Фиброзы матки /фиброидные заболевания матки/ являются старой и постоянной клинической проблемой, которая известна под различными названиями, включая фиброидные заболевания матки, гипертрофию матки, лиемиому матки, миометриальную гипертрофию, фиброзы матки и фиброзный метрит. Важно, что фиброзы матки являются состоянием, при котором наблюдается неприемлемое образование фиброидной ткани на стенках матки.
Такое состояние является причиной дисменорреи и бесплодия у женщин. Точная причина такого состояния не совсем понятна, но существующие доказательства дают возможность предположить, что это неправильная реакция фиброидной ткани на эстроген. Такое состояние вызывают у кроликов за счет ежедневного введения эстрогенов в течение 3 месяцев. У морских свинок такое состояние можно вызвать при ежедневном введении эстрогена в течение 4 месяцев. Далее, у крыс эстроген также вызывает аналогичную гипертрофию.
Наиболее общий способ лечения фиброзов матки включает хирургическое вмешательство, но он дорогой и иногда вызывает такие осложнения, как образование абдоминальных спаек и инфекций. У некоторых пациентов начальная операция является лишь временным лечением, и фибромы вырастают снова. В этих случаях осуществляют гистерэктомию, которая эффективно прекращает образование фиброидов, но также и репродуктивную жизнь пациентки. Далее, можно вводить антогонисты гормона выделения гонадотропина, но их применение также лишь временно, так как они приводят к остепорозу. Таким образом, существует необходимость в новых способах лечения фиброзов матки и способы настоящего изобретения удовлетворяют этим требованиям.
Эндометриоз является состоянием серьезной дисменорреи, которое сопровождается сильной болью, кровотечением в эндометриальные массы или полость брюшины, и часто приводит к бесплодию. Причиной симптомов такого состояния, по-видимому, является эктопический эндометриальный рост, который неправильно реагирует на нормальный гормональный контроль, и расположены в неподходящих тканях. Из-за неподходящих положений для эндометриального роста, ткань, по-видимому начинает вырабатывать локальные подобные воспаления реакции, приводящие к инфильтрации макрогенов и каскаду событий, который приводит к началу болезненной реакции. Точная этиология такого заболевания не совсем понятна, и ее лечение гормонами неоднозначно, слабо мотивировано и отличается рядом нежелательных и возможно опасных побочных эффектов.
Одним из способов лечения этого заболевания является использование низких доз экстрогена для подавления эндометриального роста за счет негативного обратного действия на центральное выделение гонадотропина, и последующего выделения яичниками эстрогена; однако иногда необходимо непрерывно использовать эстроген для контроля за симптомами. Такое применение эстрогена часто приводит к нежелательным побочным эффектам, и даже к риску эндометриального ракового заболевания.
Другой способ лечения состоит в неправильном введении прогестинов, что вызывает аменоррею, и из-за подавления продуцирования эстрогена яичниками, может вызвать замедление эндометриального роста. Использование хронической прогестиновой терапии часто сопровождается нежелательными действиями прогестина на центральную нервную систему, и часто приводит к бесплодию за счет подавления функций яичников.
Третий способ лечения состоит во введении слабых андрогенов, которые эффективно контролируют эндометриоз; однако они вызывают серьезные эффекты маскулинизации. Некоторые способы лечения эндометриоза также приводят к осложнениям, которые сопровождаются средней степенью потери костной ткани при длительной терапии. Вот почему так необходимы новые способы лечения эндометриозов.
Настоящее изобретение относится к соединениям формулы I

где R1 представляет -H, -OH-, -O/C1-C4-алкил/, -OCOC6H5, -OCO/C1-C6-алкил/, или -OSO2/C2-C6 -алкил/;
R2 представляет -H, -OH, -O/C1-C4-алкил/, -OCOC6H5, -OCO/C1-C6-алкил/, -OSO2/C2 -C6алкил, или галоид;
R3 представляет 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино, диизопропиламино, или 1-гексаметиленимино;
n = 2 или 3;
Z представляет -O- или -S-;
или к их фармацевтически приемлемым солям.
Кроме того, в настоящем
изобретении предложены следующие промежуточные соединения, необходимые для получения фармацевтически активных соединений настоящего изобретения, некоторые из которых также являются фармацевтически
активными соединениями:

где R1a представляет -H или -OR7, где R7 представляет защищающую гидроксил группу;
R2a представляет -H, галоид, или -OR8, где R8 представляет защищающую гидроксил группу;
R3 представляет 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диизопропиламино или 1-гексаметиленимино;
R6 представляет -H или защищающую гидроксил группу, которую можно селективно удалять;
R9 представляет отщепляемую группу;
R11 либо отсутствует, либо представляет = 0;
n = 2 или 3; и
Z представляет -O или -S-;
или их фармацевтически приемлемые соли.
Предложен также способ получения соединений формулы:

где R1a представляет -H, или -OR7a, где R7a представляет -H или защищающую гидроксил группу;
R2a представляет -H, галоид, или -OR8a, где R8a представляет -H или защищающую гидроксил группу;
R3 представляет 1-пиперидинил, 1-пирролидино, метил-1-пирролидинил, диметил-1-пирролидино, 4-морфолино, диметиламино, диэтиламино, диизопропиламино, или 1-гексаметиленимино;
n представляет 2 или 3, и
Z представляет -O- или -S-;
или его фармацевтически приемлемых солей, отличающийся тем, что включает

где R1a и R2a имеют указанные ранее значения; и
R9 представляет отщепляемую группу;
b/ осуществление взаимодействия продукта со стадии a/, соединения формулы XIV:

с нуклеофильной группой формулы:

где R12 представляет -OH или -SH;
c/ восстановление продукта со стадии b/, соединения формулы XVI:
до получения соединения формулы:

d/ необязательное удаление R1a и/или R2a защищающих гидроксил групп, если они присутствуют, у продукта со стадии c/; и
e/ необязательное получение соли продукта со стадии c/ или d//.
Кроме того, настоящее изобретения относится к фармацевтическим композициям, содержащим соединение формулы I, необязательно содержащим эстроген или прогестин, и к использованию таких соединений отдельно, или в сочетании с эстрогеном или прогестином, для ослабления симптомов постменопаузального синдрома, особенно остеопороза, патологических состояний сердечно-сосудистой системы и эстроген-зависимых раковых заболеваний. В том смысле, как здесь использован, термин "эстроген" включает стероидные соединения эстрадиол, эстрон, коньюгированный эстроген Premarin® экуинэстроген 17 - этинилэстрадиол и т.п. В том смысле, как использован здесь, термин "прогестин" включает соединения, обладающие прогестероновой активностью, как например, прогестерон, норэтилнодрел, нонгестрел, мегестролацетат, норэтиндрон и т.п.
Соединения настоящего изобретения можно также использовать для подавления фиброидных заболеваний матки и эндометриоза у женщин, пролиферации клеток гладкой мускулатуры аорты, особенно повторного стеноза, у людей.
Один аспект настоящего изобретения включает соединение формулы I:

где R1 представляет -H, -OH, -O/C1-C4алкил/, -OCOC6H5, -OCO/C1-C6алкил/, или -OSO2/C4-C6алкил/;
R2 представляет -H, -OH, -O/C1-C4алкил, -OCOC6H5, OCO/C1-C6алкил/, -OSO2/C2-C6алкил/ или галоид;
R3 представляет 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино, диизопропиламино или 1-гексаметиленимино;
n = 2 или 3; и
Z представляет -O- или -S-;
или их фармацевтически приемлемые соли.
Общие термины, которые использованы при описании здесь соединений имеют свои обычные значения. Так например, "C1-C6алкил" относится к разветвленным или неразветвленным алифатическим цепям, содержащим от 1 до 6 атомов, включая такие фрагменты, как метил, этил, пропил, изопропил, бутил, н-бутил, пентил, изопентил, гексил, изогексил и т.п. Аналогично, термин "C1-C4алкокси" представляет C1 -C4алкильную группу, присоединенную через молекулу кислорода, и включает такие фрагменты, как, например, метокси, этокси, н-пропокси, изопропокси и т.п.
Исходные материалы
для одного из способов получения соединений формулы I настоящего изобретения, соединения формулы III, получают практически по способу описанному C. D. Johes в патенте США N 4418068 и 4133814, которые
включены сюда по ссылке. Соединения формулы III имеют следующее строение:

где R7 и R2a имеют указанные ранее значения.
R7 и R8 защищающие гидроксил группы представляют фрагменты, которые обычно отсутствуют в конечных, терапевтических активных соединениях формулы I, но которые намеренно вводят на некоторых стадиях синтеза для защиты группы, которая, в противном случае, может реагировать в химических реакциях, а затем удаляют на более поздних стадиях синтеза. Так как соединения, содержащие такие защитные группы, представляют ценность, главным образом, как химические промежуточные соединения /хотя некоторые производные также демонстрируют биологическую активность/, их конкретная структура не является критической. Целый ряд реакций для создания, удаления и, возможно, повторного введения таких защитных групп, описан в ряде стандартных работ, включая, например, Protective Groups in Organic Chemistry, Plenum Press (London and New Iork, 1973); Green, T.W., Protective Groups in Organic Synthesis, Wiley (New Iork, 1981); and The Peptides, Vol. I, Schrooder and Lubke, Academic Press, (London and New Iork, 1965).
Представители защищающих гидроксил групп включают, например, -C1-C4-алкил, -C1-C4-алкокси, -CO-/C1-C6алкил/, -SO2-/C4-C6алкил/, и -CO-Ar, где Ar представляет бензил или необязательно замещенный фенил. Термин "замещенный фенил" относится к фенильной группе, содержащей один или более из заместителей, выбранных из группы, состоящей из C1-C4алкила, C1-C4алкокси, гидрокси, нитро, галоида и три/хлор или фтор/метила. Термин "галоид" относится к брому, хлору, фтору и иоду.
Для соединений формулы III, предпочтительными R7 и R8/R2a/ заместителями являются метил, изопропил, бензил и метоксиметил. Соединения, в которых R7 и R8 каждый представляет метил, получают способом, описанным в вышеупомянутом патенте Джонса. Другой предпочтительной защищающей гидроксид группой является метоксиметил. Однако соединение формулы IV /представлена далее/ вначале получают с предпочтительными метильной или другими защищающими гидроксид группами. Затем эти защитные группы удаляют, получая фенольные фрагменты, которые затем снова защищают метоксиметильными защитными группами.
Соединения формулы II получают с R7 защищающими гидроксил группами, которые селективно удаляют, оставляя R8/R2a/ защищающую гидроксил группу как часть конечного продукта. То же самое справедливо, когда R8/R2a защищающую гидроксил группу селективно удаляют, оставляя R7 защищающую гидроксил защитную группу на месте. Так например, R7 представляет изопропил или бензил, а R8/R2a/ представляет метил. Изопропильный или бензильный фрагмент селективно удаляют стандартным способом, а R8 метильную защитную группу оставляют как часть конечного продукта.
Первые стадии способа настоящего изобретения получения некоторых соединений формулы I включают селективное введение отщепляемых групп в положение 3 соединений формулы III, осуществления реакции присоединения и продукта реакции с первой стадии с 4-/защищенная гидрокси/фенолом, и удаление фенольной защищающей гидроксил группы. Этот способ изображен на схеме I (см. в конце описания).
На первой стадии схемы I, соответствующую отщепляемую группу селективно вводят в положение 3 исходного соединения формулы III стандартным способом. Соответствующие R9 отщепляемые группы включают такие сульфонаты, как метансульфонат, 4-бромбензолсульфонат, толуолсульфонат, этансульфонат, изопропансульфонат, 4-метоксибензолсульфонат, 4-нитробензолсульфонат, 2-хлорбензолсульфонат, трифлат и т.п., такие галоиды, как бром, хлор и иод, и другие родственные отщепляемые группы. Однако для обеспечения нужного введения отщепляемой группы предпочтительны указанные галоиды, и наиболее предпочтителен бром.
Рассматриваемую реакцию ведут, используя стандартные способы. Так например, если используют предпочтительный галоидирующий агент, эквивалент такого галоидирующего агента, предпочтительно, брома подвергают взаимодействию с эквивалентном субстрата формулы III в присутствии такого подходящего растворителя, как, например, хлороформ или уксусная кислота. Реакцию ведут при температуре от около 40oC до около 80oC.
Продукт реакции с этой стадии, соединение формулы IV, затем подвергают взаимодействию с 4-/защищенная гидрокси/фенолом до получения соединений формулы IIа, в которых R6 представляет селективно удаляемую защищающую гидроксил группу. Обычно 4-гидрокси-защитный фрагмент фенола может быть любой известной защитной группой, которую можно селективно удалить, не удаляя при этом, например, R7, и, если они присутствуют, R8 фрагменты соединения формулы IIa. Предпочтительные R6 защитные группы включают метоксиметил, если R7 и/или R8 не являются метоксиметилом, и бензил. Из них наиболее предпочтителен бензил. Реагенты - 4-/защищенная гидрокси/фенолы коммерчески доступны, или их можно получить стандартными способами.
Эта реакция присоединения (сочетания) известна специалистам как реакция /Ullman/ Ульмана, и ее ведут стандартным способом [см. Advanced Organic Chemistry: Reactions, Mechanismg and Structure, Fouth Edition, 3-16, (J. march, ed. , John Wiley & Sons. Inc. 1992); Jones. C.D., J.Chem. Soc. Peck. Trans. I, 4:407 (1992)].
Обычно эквивалентные количества двух арильных субстратов в присутствии вплоть до эквимолярного количества катализатора - окиси меди /1/ и соответствующего растворителя, нагревают до кипения с обратным холодильником в инертной атмосфере. Предпочтительно подвергать взаимодействию эквивалент соединения формулы IV, в котором R9 представляет бром, с эквивалентным количеством 4-бензилоксифенола в присутствии эквивалента окиси меди /1/.
Подходящими для этой реакции растворителями являются растворители или смесь растворителей, которые остаются инертными на протяжении всей реакции. Обычно предпочтительными растворителями являются такие "затрудненные" основания, как, например, 2,4,6-коллидин.
Температура на этой стадии реакции должна быть достаточной для завершения этой реакции присоединения, и влияет на необходимое для этого время. Если реакционную смесь нагревают до температуры кипения с обратным холодильником, в инертной атмосфере, например, в атмосфере азота, время, необходимое для завершения реакции, обычно составляет от около 20 до около 60 часов.
После присоединения, в котором образуется соединение формулы IIa соединения формулы IIb получают, селективно удаляя R6 защищающую гидроксил группу соединения формулы IIa за счет хорошо известной реакции восстановления. При этом выбранный способ не должен влиять на R7 и, если присутствуют, R8 защищающие гидроксил группы.
Если R6 представляет предпочтительный бензильный фрагмент, а R7 и, если присутствует, R8 каждый являются метилом, эту стадию способа ведут за счет стандартного гидрогенолиза.
Обычно субстрат формулы IIa добавляют к подходящему растворителю или смеси растворителей, добавляя затем донор протонов для ускорения реакции и соответствующий катализатор гидрирования.
Подходящие катализаторы включают благородные металлы, и такие оксиды, как оксид палладия, патины и родия на таком носителе, как уголь или карбонат кальция. Из них предпочтителен палладий на угле, особенно 10%-ный палладий на угле.
Растворителями для этой реакции служат те растворители или смесь растворителей, которые остаются инертными на протяжении всей реакции. Обычно предпочтительны этилацетат и C1-C4-алифатические спирты, особенно этанол.
Для рассматриваемой реакции соляная кислота служит подходящим и предпочтительным донором протонов.
Если реакцию ведут при комнатной температуре и давлении от около 30 пси/2,11 кг/см2/ до около 50 пси /3,515 кг/см2/ она протекает очень быстро. За ходом реакции можно следить с помощью стандартных хроматографических методик, например с помощью тонкослойной хроматографии.
Соединения формулы IIa и IIb являются новыми соединениями, входят в круг соединений, охватываемых формулой II, и их можно использовать для получения фармацевтически активных соединений формулы I.
После получения соединения формулы IIb, его подвергают взаимодействию с соединением формулы V:
R3
-/CH2/n-Q (V)
где R3 и n имеют указанные ранее значения, а O представляет бром или, предпочтительно, хлор, до получения соединения формулы VI. Затем у
соединения формулы VI удаляют защиту до получения соединения формулы Ia. Эти стадии способа настоящего изобретения представлены на схеме II (см. в конце описания), где R3, R7,
R2a и n имеют указанные ранее значения, а R2 представляет -H, -OH или галоид.
На первой стадии способа, представленного на схеме II,. алкилирование ведут стандартными способами. Соединения формулы V коммерчески доступны или их получают хорошо известными специалистам способами. Предпочтительно использовать соль соляной кислоты соединения формулы V, особенно 2-хлорэтилпиперидингидрохлорид.
Обычно по крайней мере около 1 эквивалента субстрата формулы IIb подвергают взаимодействию с 2 эквивалентами соединения формулы V в присутствии, по крайней мере, около 4 эквивалентов карбоната щелочного металла, предпочтительно, карбоната цезия, в соответствующем растворителе.
Растворителями в этой реакции могут служить такие растворители или смеси растворителей, которые остаются инертными на протяжении всего хода реакции. Наиболее предпочтительным является N,N-диметилформамид, особенно в его безводной форме.
Температура на этой стадии должна быть достаточной для того, чтобы завершилась эта реакция алкилирования. Обычно комнатная температура бывает остаточной и предпочтительной.
Рассматриваемую реакцию ведут в инертной атмосфере, предпочтительно, в атмосфере азота.
В условиях предпочтительной реакции она завершается обычно примерно за 16-20 часов. За ходом реакции можно следить с помощью стандартных хроматографических методик.
В качестве альтернативы для получения соединений формулы VI соединение формулы IIb подвергают взаимодействию с
избытком алкилирующего агента формулы:
Q-/CH2/n -Q',
где Q и Q' одинаковы или различны и каждый представляет отщепляемую группу, в щелочном растворе. Подходящие
отцепляемые группы, перечисленные ранее, используют при получении соединений формулы IV.
Предпочтительный щелочной раствор для этой реакции алкилирования содержит карбонат кальция в инертном растворителе, например, таком как метилэтилкетон /МЕК/ или ДМФ. В таком растворе 4-гидроксигруппа бензоильного фрагмента соединения формулы IIb существует в виде феноксидного иона, который замещает одну из отщепляемых групп алкилирующего агента.
Эта реакция протекает лучше всего, если щелочной раствор, содержащий реагенты, доводят до температуры кипения с обратным холодильником, и оставляют до завершения. Если в качестве предпочтительного растворителя используют МЕК, время реакции составляет от около 6 до около 20 часов.
Продукт реакции с этой стадии подвергают затем реакции с 1-пиперидином, 1-пирролидином, метил-1-пирролидином, диметил-1-пирролидином, 4-морфолином, диметиламином, диэтиламином, диизопропиламином или 1-гексаметиленимином стандартным способом до получения соединений формулы VI.
Предпочтительно соль соляной кислоты пиперидина подвергать взаимодействию с алкилированным соединением формулы IIb в инертном растворителе, таком, как безводный ДМФ, и нагревать до температуры в интервале от около 60o до около 110oC. Если смесь нагревают до предпочтительной температуры около 90oC, реакция занимает всего от около 30 минут до около 1 часа. Однако изменения условия реакции влияют на промежуток времени, необходимый для завершения реакции. Естественно, за ходом реакции на этой стадии можно следить с помощью стандартных хроматографических методик.
Соединения формулы VI, в которых R7 и, если присутствует, R8 каждый представляет C1-C4 алкил, предпочтительно, метил, и в которых R2a представляет -H или галоид, являются новыми соединениями и фармацевтически активными для описанных здесь способов. Соответственно, такие соединения включены в определение соединений формулы I.
Некоторые предпочтительные соединения формулы I получают, отщепляя R7 и, если присутствует, R8 защищающие гидроксид группы соединений формулы VI, хорошо известными способами. Многочисленные реакции для введения и удаления таких защитных групп описаны во многих работах, включая, например, Protective Groups in Organic Chemistry, Plenum Press, (Lonlon and New York, 1973); Green. T.W. Protective Groups in Organic Synthesis, Wiley. (New York, 1981); and The Peptides, Vol.1, Schroodez and Lubke, Academic Press (London and New York, 1965).
Способы удаления предпочтительных R7 и/или R8 защищающих гидроксил групп, особенно метила и метоксиметила, указаны далее в примерах.
Соединения формулы Ia являются новыми; они фармацевтически активны в описанных здесь методиках, и охватываются определенной здесь формулой I.
Соединения формулы I, в которых R1 представляет -H получают по схеме III представленной далее. В этом способе,, 3-положение отщепляемую группу /R9/ вводят в 3-положение коммерчески доступного тианафталина /формулы VII/ до получения соединения формулы VIII, которое затем присоединяют к 4- /защищенная гидрокси/ фенолу, получая соединение формулы IX, где R6 представляет защищающую гидроксилгруппу, которую можно селективно удалить, а R9 представляет отщепляемую группу.
Соединение формулы VII коммерчески доступно, Получение соединений формул VIII и IX, включая определения для заместителей R6 и R9, а также предпочтительные реагенты и условия реакции, если нет других указаний, те же, что и указаны ранее для схемы I.
Затем соединения формулы IX арилируют в реакции присоединения Сузуки /см. Suzuki, A., Pure and Appl. Chem. 6/2/:212-222/ 1994//. Используя один из вариантов
присоединения Сузуки, соединение формулы IX селективно галоидируют в 2-положении, а затем присоединяют к соединению арилбороновой кислоты формулы XIa (способ A на схеме IV см. в конце описания), где
R2a, R2b, R3, R6 и n имеют указанные ранее значения, X представляет иод, бром или фтор, в порядке предпочтительности; и
X' представляет иод, бром
или фтор, в порядке предпочтительности, или трифлат.
Однако предпочтительно, арилбороновую кислоту формулы XIb получать из соединения формулы IX, а затем подвергать ее взаимодействию с галоидареном формулы XIb до получения новых промежуточных формулы IIc /cпособ B на схеме IV далее/. Такие новые промежуточные соединения можно использовать для получения фармацевтически активных соединений настоящего изобретения /формулы Ib/ в результате алкилирования и удаления защиты.
На первой стадии схемы IV иодируют или бромируют соединения формулы IX в положении 2 известными способами. Обычно соединение формулы IX подвергают взаимодействию с небольшим избытком н-бутиллития в гексане, в соответствующем растворителе и в инертной атмосфере, например, в атмосфере азота, а затем прикапывают небольшой избыток нужного галоидирующего агента в подходящем растворителе. Предпочтительно, чтобы галоидирующим агентом на этой стадии был иод, но можно также использовать и бром и N-бромсукцинимид.
Подходящие растворители включают инертные растворители или смеси растворителей, например, диэтиловый эфир и тетрагидрофуран /ТГФ/. Из них предпочтителен тетрагидрофуран, особенно безводный ТГФ.
Такую реакцию селективного галоидирования в 2-положение обычно ведут при температуре около -75oC до около 85oC.
В результате вышеуказанной реакции получают галоидарен формулы Xa, который затем соединяют с арилбороновой кислотой формулы XIa стандартным способом присоединения Сузуки, до получения соединений формулы IIc. Соединения формулы XIa, в которых R2a представляет -H, галоид или -OR8 /где R8 представляет защищающую гидроксил группу, определенную ранее/, получают из коммерчески доступных соединений известными специалистам способами /см. March J., and Suzuki, A, Supra).
В этой реакции присоединения небольшой избыток соединения формулы XIa подвергают взаимодействию с каждым эквивалентом соединения формулы Xa в присутствии палладиевого катализатора и соответствующего основания в таком инертном растворителе, как толуол.
Хотя в реакции присоединения Сузуки можно использовать различные палладиевые катализаторы, обычно выбирают специфический, для реакции катализатор. Так наиболее предпочтительно использовать в этой реакции тетракистрифенилфосфин.
Аналогично, в этой реакции присоединения можно использовать различные основания. Однако предпочтительно использовать карбонат щелочного металла, особенно, 1н карбонат натрия.
Температура, используемая на этой стадии должна быть достаточной для того, чтобы осуществить реакцию присоединения до конца. Обычно бывает достаточным и предпочтительным нагревать реакционную смесь до кипения с обратным холодильником в течение от около 2 до около 4 часов.
В способе B схемы IV соединение формулы Xb с арилбороновым заместителем в 2-положении получают хорошо известными способами. Обычно соединение формулы IX обрабатывают небольшим избытком н-бутиллития в гексане, в соответствующем растворителе и в такой инертной атмосфере, как азот, с последующим прикапыванием соответствующего триалкилбората.
Подходящие растворители включают инертный растворитель или смесь таких растворителей, как, например, диэтиловый эфир, диоксин и тетрагидрофуран /ТГФ/. Предпочтителен ТГФ, особенно безводный ТГФ.
Предпочтительным триалкилборатом, используемым в настоящем изобретении является триизопропилборат.
Продукт этой реакции, соединение формулы Xb, подвергают затем взаимодействию с арилгалидом или арилтрифлатом формулы XIb за счет стандартной реакции присоединения Сузуки, до получения соединений формулы IIc. Предпочтительными условиями реакции являются условия, указанные для взаимодействия соединений XIa и Xa на схеме IV, которые также приводят к получению соединений формулы IIc.
Превращение соединений формулы IIc в соединения формулы Ia осуществляют, как указано ранее для превращения соединений формулы IIa в соединения формулы Ia.
Соединения формулы IIc и IId являются новыми, и их можно использовать для получения фармацевтически активных соединений настоящего изобретения.
Соединения формулы XII и Ib также являются новыми и могут быть использованы в описанных здесь способах, и входят в определение формулы I.
Соединения формулы I, в которых или R1 или R2 представляет -H, а другой R1 или R2 заместитель представляет -OH, также получают из соединений формулы I, в которых как R1, так и R2 представляют -OH. Дигидрокси соединение формулы I превращают в смесь 6- и 4'-монотрифлатов, а трифлатный фрагмент восстанавливают водородом /см. J. M.et al., J.Org.Chem. 55:991 /1990//. Полученную смесь моногидроксипроизводных, либо в виде свободного основания, либо фармацевтически приемлемой соли, предпочтительно, в виде соли соляной кислоты, можно затем разделить за счет стандартных методик кристаллизации.
Вообще, дигидрокси соединение формулы I обрабатывают от около 4 до около 6 эквивалентами амино-основания, например, триэтиламина, в нереакционноспособном растворителе, с последующим добавлением 1 эквивалента трифторметансульфонового ангидрида. Получают статистическую смесь моно- и ди-трифлатов, и разделяют ее стандартными хроматографическими способами. Предпочтительным растворителем на этой стадии является безводный дихлорметан.
Если температуру реакции поддерживают в интервале от около 0oC до около 25oC, рассматриваемая реакция завершается за промежуток времени от около 1 до около 5 часов.
Выделенную смесь моно-трифлатных соединений затем гидрируют в нереакционноспособном растворителе, в присутствии от около 3 до около 6 эквивалентом амина-основания, предпочтительно, триэтиламина, и катализатора гидрирования, такого как палладий-на угле, который и предпочтителен. Предпочтительные растворители этой реакции включают этилацетат и этанол, или в другом варианте, их смесь. Если на этой стадии реакцию ведут при давлении 40 пси /9,84 кг/см2/ газообразного водорода, при температуре окружающей среды, время реакции составляет от около 2 часов до около 5 часов.
Полученная смесь моногидрокси производных формулы I обладает различными растворимостями в этилацетате, и 6-гидрокси-4'-водородные можно частично отделить от 6-водород-4'-гидроксипроизводных за счет селективной кристаллизации. Дальнейшего разделения, которое обеспечило бы получение чистых моногидроксисоединений формулы I, можно достичь, превращая обогащенные смеси в соли соляной кислоты с последующей кристаллизацией из смеси этилацетат/этанол.
Более прямым способом
получения соединений формулы I, в которых либо R1, либо R2 представляют -H, а другой R1 или R2 заместитель представляет -OH, а также альтернативным способом
получения соединений формулы I, в которых либо R1, либо R2 представляют -H, а другой R1 или R2 заместитель представляет -O-/C1-C4
алкил/, является способ, в котором используют соединение формулы:

где R3 и n имеют указанные ранее значения,
R1c представляет -OH или -O-/C1-C4-алкил/; и
R2c представляет -OH или -O-/C1-C4-алкил/;
при условии, что если R1c представляет -OH, R2c представляет -O-/C1-C4алкил/, если R1c представляет -O-/C1-C4-алкил/, тогда R2c представляет -OH.
В этом способе гидроксифрагмент такого соединения превращает в трифлатное производное за счет обработки трифторметансульфоновым ангидридом. Затем трифлатный фрагмент восстанавливают в стандартных условиях, предпочтительно, за счет каталитического гидрирования. Затем защищающий гидроксил фрагмент удаляют стандартным способом, таким, как те, которые описаны здесь, в результате чего получают соединение формулы I, в котором либо R1, либо R2 представляет -H, а другой R1 или R2 заместитель представляет -OH.
Другим вариантом, и предпочтительным, является способ получения соединений настоящего изобретения, представленный на схеме V, (см. в конце описания), где каждое из переменных имеет указанные ранее значения.
В этом способе атом серы соединения формулы IV окисляют до получения сульфоксида /формулы XIV/, который затем подвергают взаимодействию с нуклеофильной группой для введения атома кислорода или серы в качестве связующего соединения формулы I и формулы II. Затем сульфоксидный фрагмент соединений формулы XVI восстанавливают до получения некоторых соединений настоящего изобретения.
На первой стадии этого процесса соединение формулы IV селективно окисляют до сульфоксида. Для осуществления этой стадии процесса можно использовать ряд известных способов /см. Madesclaire. M., Tetrahedron, 42/20/; 5459-5495 /1986/; Trost B. M. et al. Tetrahedron Letters, 22/14/; 1287-1290/1981/; Drabowicz. J. et al., Synthetic Communications 11/12; 1025-1030 /1981/; Kramer. J. B. et al., 34th National Organic Symposium, Williamsburg. VA., June 11-15/ 1995/. Однако многие окисляющие агенты приводят лишь к малой конверсии в нужный продукт, наряду с избыточным окислением до сульфона. Однако в рассматриваемом новом способе происходит превращение соединения формулы IV в сульфоксид формулы XIV с высоким выходом при незначительном образовании сульфонов /или вовсе без образования сульфонов/. Этот новый способ включает осуществление взаимодействия соединения формулы IV с от около 1 до около 1,5 эквивалентами перекиси водорода в смеси с от около 20% до около 50% трифторуксусной кислоты в метиленхлориде. Реакцию ведут при температуре от около 10oC до около 50oC, и обычно требуется от около 1 до около 2 часов для завершения реакции. Далее, отщепляемую группу в 3-положении /R9/ заменяют нужным нуклеофильным производным формулы XV. Такие нуклеофильные производные получают стандартными способами.
На этой стадии процесса, кислотный протон нуклеофильной группы удаляют за счет обработки основанием, предпочтительно, небольшим избытком гидрида натрия или
трет-бутоксида калия, в полярном апротонном растворителе, предпочтительно, ДМФ или тетрагидрофуране. Можно использовать и другие основания, включая карбонат калия и карбонат цезия. Кроме того,
можно использовать и такие другие растворители, как диоксан или диметилсульфоксид. Реакцию депротонирования обычно ведут при температуре от около 0oC до около 30oC, и обычно для
завершения этой реакции необходимо около 30 минут. Затем соединение формулы XIV добавляют к раствору нуклеофила. Реакцию замещения ведут при температуре от около 0oC до около 50o
C, и обычно она занимает от около 1 до около 2 часов. Полученный продукт выделяют стандартными способами.
Если в качестве защищающей гидроксил группы используют бензильный фрагмент, гидрогенолиз сульфоксидного фрагмента обеспечит также удаление бензильной защитной группы, что исключит требование селективного удаления такой группы на более поздней стадии процесса.
На следующей стадии процесса настоящего изобретения новые сульфоксиды формул XVIa, b, c и d /общая формула XVI/ восстанавливают до бензотиофенового соединения формул IIg, Ic, IIe и Id, соответственно. До этого процесса восстановления соединения формулы IIg и IIe можно сначала алкилировать, как здесь указано. Восстановление сульфоксидных соединений можно осуществлять, используя один из многочисленных известных способов, включая, например, восстановление гидридом /литийалюминийгидрид/, каталитическое восстановление, трансфер-гидрогенолиз и триметилсилилиодид /ТМС-И/. При таком восстановлении выбор реагента зависит от совместимости других функциональных групп молекулы. Для соединений, описанных в настоящем изобретении наиболее предпочтительными реагентами являются литийалюминийгидрид /LiAlH4/ и трансфер - гидрогенолиз /палладиевая чернь/аммонийформат/. Для восстановления за счет LiAlH4 подходящими растворителями являются, например, диэтиловый эфир, диоксан и тетрагидрофуран /ТГФ/. Из них предпочтителен ТГФ, особенно безводный ТГФ. Для трансфер-гидрогенолиза наиболее предпочтительны спиртовые растворители, особенно этанол. Реакцию ведут при температуре от около 0oC до около 60oC, и для завершения реакции необходимо от около 0,5 часа до около 2 часов.
При желании можно удалить
защищающие гидроксил группы /группу/ у продуктов способа, представленного на схеме V, и у солей продукта на любой стадии способа. Соответственно, настоящее изобретение предлагает способ получения
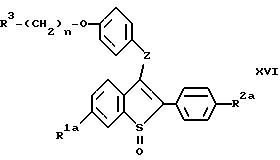
соединений формулы:
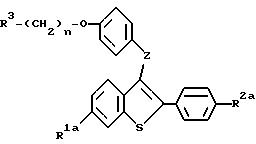
где R1a представляет -H или -OR7a, где R7a представляет -H или гидроксил защищающую группу;
R2a представляет -H, галоид, или -OR8a, где R8a представляет -H или гидроксил защищающую группу;
R3 представляет 1-пиперидинил, 1-пирролидино, метил-1-пирролидинил, диметил-1-пирролидино, 4-аморфолино, дидметиламино, диэтиламино, диизопропиламино, или 1-гексаметиленимино;
n=2 или 3; и
z представляет -O- или -S-;
или их фармацевтически приемлемых солей, включающий:
а/ окисление атома серы соединения формулы IV:

где R1a и R2a имеют указанные ранее значения; и
R9 представляет отщепляемую группу;
b/ осуществление взаимодействия продукта со стадии
a/, соединения формулы XIV:

с нуклеофильной группой формулы:

где R12 представляет -OH или -SH;
c/ восстановление продукта со стадии b/, соединения формулы XVI:

до получения соединения формулы:

d/ необязательно удаление R1a и/или R2a гидроксил защищающих групп /если они присутствуют/ продукта со стадии c/; и
e/ необязательное получение соли продукта со стадии c/ или d/.
Этот новый способ обеспечивает также получение новых соединений формул XIV и XVI a, b, c и d, каждое из которых представляет собой промежуточное соединение, пригодное для получения фармацевтически активных соединений настоящего изобретения.
Соединения формулы I, в которых Z представляет S, также получают используя способ схемы VI (см. в конце описания), где R1a представляет -H или -OR7, где R7 представляет гидроксил защищающую группу; R2a представляет -H, или -OR8, где R8 представляет защищающую гидроксил группу; R6 представляет защищающую гидроксил группу, которую можно селективно удалять; R9 представляет отщепляемую группу; и M представляет ион металла, в котором соединение IVa металлируют. Полученный продукт, соединение формулы XVII, подвергают взаимодействию с 4/ защищенный гидроксил/ фенилдисульфидом формулы XVII, и защитную фенольную группу соединения формулы IIe удаляют, получая соединения формулы IIf. Следует заметить, что если используют этот способ, R2 не может быть галоидом из-за химических ограничений.
На первых двух стадиях схемы VI соединение формулы IVa металлируют известными способами. Обычно и предпочтительно обрабатывать соединение формулы IVa небольшим избытком н-бутиллития в гексанах в подходящем растворителе, с последующим прикапыванием раствора дисульфидного соединения формулы XVIII в подходящем растворителе. Обе эти стадии реакции ведут в такой инертной атмосфере, как азот, причем подходящие растворители для обоих стадий включают один или более из таких инертных растворителей как диэтиловый эфир, диоксан и ТГФ. Из них наиболее предпочтителен ТГФ, и особенно его безводная форма. Стадии рассматриваемой схемы реакций ведут при температуре от около -78oC до около 85oC.
На первой стадии этой реакции получают металлированное соединение формулы XVII 4-/защищенный гидроксид/фенилдисульфид/ соединение формулы XVIII/, который подвергают взаимодействию с таким соединением формулы XVII, чтобы получить соединение формулы IIe, получают, защищая гидроксильную группу коммерческого 4-гидроксифенилсульфида подходящей защитной группой известными способами. Предпочтительной R6 защитной группой является метоксиметил, при условии, что R7 и R8, (если либо один, либо оба присутствуют), представляет защищающую гидроксил группу, отличную от метоксиметила. При этом обязательно R6 группа, защищающая гидроксил, представляет фрагмент, который отличается от R7 и R8 групп, защищающих гидроксил/, если они присутствуют/ так, чтобы группу R66 можно было бы селективно удалять стандартными способами до получения соединений формулы IIf.
Для того, чтобы удалить R6 защитную группу, соединение формулы IIe в протонном растворителе или смеси растворителей подвергают взаимодействию в кислотной среде, содержащей по крайней мере один эквивалент кислоты, предпочтительно, метансульфоновой кислоты, при нагревании от около 25oC до около 110oC. Обычно время реакции составляет от около 6 до около 24 часов, и за ходом реакции можно следить с помощью стандартных хроматографических методик.
подходящие для этой реакции растворители включают, например, воду и метанол.
Соединения формул IIa и IIf являются новыми, и их можно использовать для получения фармацевтически активных соединений формулы I, и они входят в определение формулы II.
Соединения формулы Id:

где R1b представляет -H или -OH;
R2b представляет -H или -OH;
R3 и n имеют указанные ранее значения,
получают, используя вышеуказанные процедуры, соответствующие стадиям процесса, представленным на схемах II и IV. Такие соединения формулы Id также являются новыми, и их можно использовать в способах настоящего изобретения, и они включены в вышеприведенное определение формулы I.
Соединения формулы I, в которых R1 и R2 представляют различные защищающие гидроксил группы, или каждый из которых R1 и R2 представляет группу, защищающую гидроксил, и другой представляет гидроксил, селективно получают, используя модифицированный 2-арилбензотиофеновый исходный материал указанной ранее формулы III, при условии, что защищающие гидроксил группы, обозначенные R7 и R8 отличаются достаточно для того, чтобы можно было удалить одну защитную группу, а другая при этом осталась бы нетронутой. Такие 2-арилбензотиофены получают хорошо известными способами.
Наиболее походящей для получения соединений формулы I, в которых R1 и R2 представляют различные защитные группы, является присоединение Сузуки, представленное ранее на схеме IV. Однако 6/защищенная гидрокси/бензотиофен-2-бороновую кислоту подвергают взаимодействию с соединением формулы XIb, в котором R2a представляет -R8, а R7 не одинаков с R8. Эта реакция позволяет получать соединения настоящего изобретения, в которых R7 и R8 представляют различные защищающие гидроксил группы, так что одну из защитных групп можно селективно удалить, а другая при этом остается как фрагмент конечного продукта. Предпочтительно, чтобы R7 защитную группу, особенно бензил или изопропил, удаляли до получения гидроксильного фрагмента, а R8 защитная группа, в частности, метил, оставалась.
Присоединение Сузуки осуществляют также, используя вышеуказанные процедуры, но заменяя соединение формулы X1b соединением формулы XIX:

где R8a представляет C1-C6-алкилсульфонат, предпочтительно, метансульфонат, или C4 -C6-арилсульфонат; а
R10 представляет отщепляемую группу, предпочтительно, бром или трифлат.
В этом способе 6-/защищенная гидрокси/бензотиофен-2-бороновую кислоту, как указано ранее, подвергают взаимодействию с соединением формулы XIX до получения соединения формулы XX, которое подвергают взаимодействию с трехбромистым бором в метиленхлориде до получения моногидрокси соединения, которое затем превращают, например, в бензильный фрагмент стандартными способами /формулы XXI/. 4'-сульфонатный сложный эфир затем селективно удаляют за счет основного гидролиза или, предпочтительно, за счет обработки LiAlH4, в подходящем апротонном растворителе, таком, как например, ТГФ. Эта реакция приводит к получению соединения формулы XXII, которое в конце, например, метилируют в положении 4 стандартными способами /формула IIIa/. Естественно, специалистам должно быть понятно, что для получения соединений формулы IIIa, в которых группы, защищающие гидроксил, отличаются от представленных на схеме VII (см. в конце описания), которые можно селективно удалять с получением моногидрокси соединений формулы I настоящего изобретения, можно использовать различные способы.
Затем соединения IIIa подвергают различным воздействиям описанным здесь для получения соединений формулы I и II настоящего изобретения.
Другие предпочтительные соединения формулы I получают, за счет замены гидроксильных групп в 6- и/или 4'-положениях /если присутствуют/ на фрагменте формулы -O-CO-/C1-C6алкил/, или -O-SO2-/C2-C6алкил/ хорошо известными способами. См., например патент США 438593.
Так например, если -O-CO/C1-C6алкильная/группа нужна, тогда моно- или гидрокси-соединение формулы I подвергают взаимодействию с таким агентом как ацилхлорид, бромид, цианид или азид, или с соответствующим ангидридом или смешанным ангидридом. Реакции обычно ведут в таком основном растворителе, как пиридин, хинолин или изохинолин, или в таком третичном амине, как триэтиламин, тибутиламин, метилпиперидин и т.п. Реакцию можно также вести в таком инертном растворителе, как этилацетат, диметилформамид, диметилсульфоксид, диоксан, диметоксиэтан, ацетонитрил, ацетон, метилэтилкетон и т.п. к которому был добавлен по крайней мере один эквивалент акцептора кислоты/исключения приведены далее/, например, третичный амин. При желании можно использовать такой катализатор ацилирования, как 4-диметиламинопиридин или 4-пирролидинопиридин. См., например, Haslam et al., Tetrahedzon 36:2409-2433 /1980/.
Рассматриваемые реакции ведут при умеренных температурах в интервале от около -25oC до около 100oC, часто в инертной атмосфере, такой, как газообразный азот. Однако обычно для того, чтобы реакция происходила, достаточно температуры окружающей среды.
Ацилирование гидроксильной группы в 6- и/или 4'-положении, можно также осуществить за счет реакций, катализируемых кислотой при взаимодействии с подходящей карбоновой кислотой в инертных органических растворителях. Используют такие кислотные катализаторы, как серная кислота, полифосфорная кислота, метансульфоновая кислота и т.п.
Вышеуказанные группы R1 и/или R2 соединений формулы I можно также получить за счет образования активного сложного эфира соответствующей кислоты, например, такого сложного эфира, получаемого из таких известных реагентов, как дициклогексилкарбодиимид, ацилимидазолы, нитрофенолы, пентахлорфенол, N-гидроксисукцинимид и 1-гидроксибензотриазол. См., например, Bull. Chem. Soc. Japan, 38:1979 /1965/ и Chem. Ber., 788 и 2024 /1970/.
Каждый из вышеуказанных способом, который приводит к получению -O-CO-/C1-C6алкильных/ фрагментов, можно осуществлять, как обсуждалось ранее, в растворителях. Естественно, те способы, в которых не получают кислотного продукта в ходе реакции, не требуют использования в реакционной смеси акцептора кислоты.
Если нужно получить соединение формулы I, в котором гидроксильную группу в 6- и/или 4'-положении соединения формулы I превращают в группу формулы -O-SO2/C2-C6-алкил/, моно- или дигидрокси соединение подвергают взаимодействию с, например, сульфоновым ангидридом или таким производным подходящей сульфоновой кислоты, как сульфонилхлорид, бромид, или сульфониламмониевой солью, как указано у Кинга и Монойра, Y. Amer. Chem. Soc., 97:2566-2567 /1975/.
Дигидрокси соединение можно также подвергать взаимодействию с подходящим сульфоновым ангидридом или смешаными сульфоновыми ангидридами. Такие реакции ведут в условиях, которые были указаны ранее при обсуждении реакций с галоидангидридами и т.п.
Хотя в способах настоящего изобретения можно использовать соединения формулы I в форме свободного основания, предпочтительно, получать и использовать форму фармацевтически приемлемой соли. Так, соединения, которые используют в способах настоящего изобретения, образуют фармацевтически приемлемые соли присоединения с широким кругом органических и неорганических кислот, и включают физиологически приемлемые соли, которые часто используют в фармакопии. Эти соли также составляют часть настоящего изобретения. Типичные неорганические кислоты, которые используют для получения таких солей, включают соляную, бромистоводородную, иодистоводородную, азотную, серную, фосфорную, гипофосфорную и т.п. Можно также использовать соли таких органических кислот, как алифатические моно- и дикарбоновые кислоты, фенилзамещенные алкановые кислоты, гидроксиалкановые и гидроксиалкандиовые кислоты, ароматические кислоты, алифатические и ароматические сульфоновые кислоты. Поэтому фармацевтически приемлемые соли включают ацетат, фенилацетат, трифторацетат, акрилакт, аскорбат, бензоат, хлорбензоат, динитробензоат, гидроксибензоат, метоксибензоат, метилбензоат, о-ацетоксибензоат, нафталин-2-бензоат, бромид, изобутират, фенилбутират, b-гидроксибутират, бутил-1,4-диоат, капрат, каприлат, хлорид, циннамат, цитрат, формат, фумарат, гликоллат, гептаноат, гиппурат, лактат, малат, малеет, гидроксималеат, малонат, манделат, мезилат, никотинат, изоникотинат, нитрат, оксалат, фталат, терефталат, фосфат, гидрофосфат, дигидрофосфат, метафосфат, пирофосфат, пропиолат, пропионат, фенилпропионат, салицилат, себакат, сукцинат, суберат, сульфат, пиросульфат, сульфит, бисульфит, сульфонат, бензолсульфонат, p-бромфенилсульфонат, хлорбензолсульфонат, этансульфонат, 2-гидроксиэтансульфонат, метансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, p-толуолсульфонат, ксилолсульфонат, тартрат и т.п. Предпочтительными солями являются гидрохлорид и оксалат.
Фармацевтически приемлемые соли присоединения кислот обычно получают за счет взаимодействия соединения формулы I с эквимолярным количеством или избытком кислоты. Реагенты обычно объединяют в таком общем растворителе, как диэтиловый эфир или этилацетат. Обычно соль выпадает из раствора за промежуток времени от коло 1 часа до 10 дней, и ее можно выделить фильтрованием или растворитель можно выпарить обычными способами.
Фармацевтически приемлемые соли обычно обладают повышенной растворимостью по сравнению с соединениями, из которых они получены, и поэтому более удобны для композиций в виде жидкостей или эмульсий.
Представители предпочтительных соединений настоящего изобретения включают следующие:
Группа I:
[6-метокси-2-/4-метоксифенил/-3-бром]бензо[b]тиофен-/S-оксид/;
[6-изопропокси-2-/4-метоксифенил/-3-бром]бензо[b]тиофен-/S -оксид/;
[6-метокси-2-/4-изопропоксифенил/-3-бром]бензо[b]тиофен-/S -оксид/;
[2-/4-метоксифенил/-3-бром]бензо[b]тиофен-/S/-оксид/;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4 метоксифенил/]бензо[b]тиофен-/S-оксид/;
[3-[4-[2-/1-пиперидинил/этокси]
фенокси] -2-4-метоксифенил/] - бензо[b] тиофен-/S-оксид/;
[6-бензилокси-2-/4-метоксифенил/-3-бром]бензо[b]тиофен-/S -оксид/;
[6-изопропокси-2-/4-метоксифенил/-3-бром]бензо[b]тиофен-/S -оксид/;
[6-метокси-2-/4-бензилоксифенил/-3-бром]бензо[b]тиофен-/S -оксид/;
[6-метокси-2-/4-изопропоксифенил/-3-бром]бензо[b]тиофен-/S -оксид/;
[6-бензилокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2- /4-метоксифенил/-3-бром]бензо[b]тиофен-/S-оксид/;
[6-изопропокси-3-[4-[4-/1-пиперидинил/этокси] фенокси] -2- /4-метоксифенил/]бензо[b]тиофен-/S-оксид/;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси]
-2- /4-бензилоксифенил/]бензо[b]тиофен-/S-оксид/;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2- [4-изопропоксифенил/]бензо[b]тиофен-/S -оксид/;
[6-метокси-2-/4-метоксифенил/-3-/4-метоксиметиленокси/тиофенокси] бензо[b]тиофен;
[6-метокси-2-/4-метоксифенил/-3-/4-гидрокси/тиофенокси] бензо[b]тиофен;
Группа II:
[3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4-гидроксифенил/] бензо[b] тиофен;
[3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4-гидроксифенил/] бензо[b] тиофенгидрохлорид;
[3-[4-[2-/1-пирролидинил/этокси] фенокси] -2-/4-гидроксифенил/] бензо[b] тиофен;
[3-[4-[2-/1-гексаметиленимино/этокси] фенокси]-2-/4-гидроксифенил/] бензо[b]тиофен;
[3-[4-[2-/1-N,
N-диэтиламино/этокси] фенокси] -2-/4-гидроксифенил/] бензо[b]тиофен;
[3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4-гидроксифенил/] бензо[b] тиофенгидрохлорид;
[3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/фенил/] бензо[b]тиофенгидрохлорид;
[3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4-фторфенил/] бензо[b]тиофен;
[6-метокси-2-/4-метоксифенил/-3-/4-бензилокси/фенокси]бензо[b]- тиофен;
[6-изопропокси-2-/4-метоксифенил/-3-/4-бензилокси/фенокси]бензо[b]- тиофен;
[6-метокси-2-/4-изопропоксифенил/-3-/4-бензилокси/фенокси]бензо[b]- тиофен;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-4-метоксифенил/] бензо[b]-тиофен;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4-метоксифенил/] бензо[b]-тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-пирролидинил/этокси] фенокси] 2-/4-метоксифенил/] бензо[b]-тиофен;
[6-метокси-3-[4-[2-/1-гексаметиленимино/этокси] фенокси] -2-/4- метоксифенил/]бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-N, N-диэтиламино/этокси]
фенокси]-2-/4- метоксифенил/]бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/морфолино/этокси]фенокси]-2-/4- метоксифенил/]бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[3-/пиперидино/пропокси] фенокси] -2-/4- метоксифенил/] бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[3-/1-N,
N-диэтиламино/пропокси]фенокси]-2-/4- метоксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4- гидроксифенил/] бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4- гидроксифенил/] бензо[b]тиофеноксалат;
[6-гидрокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4- гидроксифенил/]
бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-пирролидинил/этокси] фенокси] -2-/4- гидроксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-гексаметиленимино/этокси]фенокси]-2- /4-гидроксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-N, N-диэтиламино/этокси] фенокси]-2-/4- гидроксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/морфолино/этокси]фенокси]-2-/4- гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[3-/1-N,
N-диэтиламино/пропокси]фенокси]-2-/4- гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-N, N-диизопропиламино/-этокси]фенокси]-2-/4- гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[3-/пиперидино/пропокси] фенокси]-2-/4- гидроксифенил/] бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4- метоксифенил/]
бензо[b]тиофенгидрохлорид;
[6-бензилокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4- метоксифенил/]бензо[b]тиофен;
[6-бензилокси-3-[4-[2-/1-пирролидинил/этокси] фенокси]
-2-/4- метоксифенил/]бензо[b]тиофен;
[6-бензилокси-3-[4-[2-/1-гексаметилимино/этокси] фенокси]-2-/4- метоксифенил/]бензо[b]тиофен;
[6-бензилокси-3-[4-[2-/1-N,
N-диметиламино/этокси]фенокси]-2-/4- метоксифенил/]бензо[b]тиофен;
[6-бензилокси-3-[4-[2-/1-морфолино/этокси] фенокси]-2-/4- метоксифенил/] бензо[b]тиофен;
[6-изопропокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4- метоксифенил/]бензо[b]тиофен;
[6-изопропокси-3-[4-[3-пирролидинил/этокси]фенокси]-2-/4- метоксифенил/] бензо[b]тиофен;
[6-изопропокси-3-[4-[2-/1-гексаметилимино/этокси]фенокси]-2-/4- метоксифенил/]бензо[b]тиофен;
[6-изопропокси-3-[4-[2-/1-N, N-диметиламино/этокси]фенокси]-2-/4- метоксифенил/]бензо[b]тиофен;
[6-изопропокси-3-[4-[2-/1-морфолино/этокси]фенокси]-2-/4- метоксифенил/] бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-пиперидинил/этокси] фенокси]-2-/4- метоксифенил/] бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-пирролидинил/этокси]фенокси]-2-/4- метоксифенил/] бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-гексаметилимино/этокси ]фенокси]-2-/4- метоксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-N,N-диметиламино/этокси]фенокси]-2-/4- метоксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-морфолино/этокси]фенокси]-2-/4- метоксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-пиперидинил/этокси] фенокси]-2-/4- метоксифенил/] бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-пирролидинил/этокси]фенокси]-2-/4- метоксифенил/]
бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-гексаметилимино/этокси] фенокси]-2-/4- метоксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-N,
N-диметиламино/этокси]фенокси]-2-/4- метоксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-морфолино/этокси]фенокси]-2-/4- метоксифенил/]бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4- бензилоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-пирролидинил/этокси] фенокси] -2-/4- бензилоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-гексаметиленимино/этокси] фенокси]-2-/4- бензилоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-N,N-диметиламино/этокси]фенокси]-2-/4- бензилоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-морфолино/этокси] фенокси]-2-/4- бензилоксифенил/] бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4- изопропоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-пирролидинил/этокси] фенокси]-2-/4- изопропоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-гексаметиленимино/этокси]фенокси]-2-/4- изопропоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-N, N-диметиламино/этокси]фенокси]-2-/4- изопропоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-морфолино/этокси]фенокси]-2-/4- изопропоксифенил/] бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси]-2-/4- гидроксифенил/] бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-пирролидинил/этокси]фенокси]-2-/4- гидроксифенил/] бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-гексаметиленимино/этокси]фенокси]-2-/4- гидроксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-N, N-диметиламино/этокси] фенокси]-2-/4- гидроксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-морфолино/этокси] фенокси] -2-/4- гидроксифенил/] бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси]-2-/4- гидроксифенил/] бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-пирролидинил/этокси]фенокси]-2-/4- гидроксифенил/]
бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-гексаметиленимино/этокси] фенокси]-2-/4- гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-N, N-диметиламино/этокси]
фенокси]-2-/4- гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-морфолино/этокси] фенокси] -2-/4- гидроксифенил/] бензо[b]тиофенгидрохлорид;
[6-бензоилокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4- бензоилоксифенил/]бензо[b]тиофенгидрохлорид;
[6-этилсульфонилокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]
-2-/4-этилсульфонилоксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-4- этилсульфонилфенил/]бензо[b]тиофенгидрохлорид;
[6-этилсульфонилокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2- /4-гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси]
-2-/4- трифторметансульфонилоксифенил/]бензо[b]тиофен;
[3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4-бензоилоксифенил/] бензо[b] тиофенгидрохлорид;
[3-[4-[2-/1-пиперидинил/этокси]
фенокси] -2-/4- пивалоилоксифенил/]бензо[b]тиофенгидрохлорид;
[3-[4-[2-/1-пиперидинил/этокси] фенокси]-2-/4- бутилсульфонилоксифенил/] бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси]тиофенокси]-2-/4- метоксифенил] бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-пирролидинил/этокси] тиофенокси] -2-/4- метоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-гексаметиленимино/этокси]тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-N,N-диметиламино/этокси]тиофенокси]-2- /4-метоксифенил/][b]тиофен;
[6-метокси-3-[4-[2-/1-морфолино/этокси] тиофенокси]-2-/4- метоксифенил/] бензо[b]тиофен;
[6-бензилокси-3-[4-[2-/1-пиперидинил/этокси]тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофен;
[6-бензилокси-3-[4-[2-/1-пирролидинил/этокси] тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофен;
[6-бензилокси-3-[4-[2-/1-гексаметиленимино/этокси] тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофен;
[6-бензилокси-3-[4-[2-/1-N, N-диметиламино/этокси] тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофен;
[6-бензилокси-3-[4-[2-/1-морфолино/этокси] тиофенокси]
-2- /4-метоксифенил/]бензо[b]тиофен;
[6-изопропокси-3-[4-[2-/1-пиперидинил/этокси] тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофен;
[6-изопропокси-3-[4-[2-/1-пирролидинил/этокси]тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофен;
[6-изопропокси-3-[4-[2-/1-гексаметиленимино/этокси]тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофен;
[6-изопропокси-3-[4-[2-/1-N, N-диметиламино/этокси]тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофен;
[6-изопропокси-3-[4-[2-/1-морфолино/этокси]
тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-пиперидинил/этокси] тиофенокси] -2- /4-метоксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-пирролидинил/этокси] тиофенокси] -2- /4-метоксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-гексаметиленимино/этокси]тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-N, N-диметиламино/этокси]тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-морфолино/этокси]тиофенокси]-2- /4-метоксифенил/] бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-пиперидинил/этокси] тиофенокси] -2- /4-метоксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-пирролидинил/этокси]
тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-гексаметиленимино/этокси]тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-N, N-диметиламино/этокси]тиофенокси]-2- /4-метоксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-морфолино/этокси]тиофенокси]-2- /4-метоксифенил/]
бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси] тиофенокси] -2- /4-бензилоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-пирролидинил/этокси]
тиофенокси]-2- /4-бензилоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-гексаметиленимино/этокси] тиофенокси]-2- /4-бензилоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-N,
N-диметиламино/этокси] тиофенокси]-2- /4-бензилоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-морфолино/этокси] тиофенокси] -2- /4-бензилоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси]тиофенокси]-2- /4-изопропоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-пирролидинил/этокси] тиофенокси] -2- /4-изопропоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-гексаметиленимино/этокси]тиофенокси]-2- /4-изопропоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-N,
N-диметиламино/этокси]тиофенокси]-2- /4-изопропоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-морфолино/этокси] тиофенокси] -2-/4- изопропоксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси]тиофенокси]-2-/4- гидроксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-пирролидинил/этокси] тиофенокси]-2-/4- гидроксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-гексаметиленимино/этокси]тиофенокси]-2-/4- гидроксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-N, N-диметиламино/этокси]тиофенокси]-2-/4- гидроксифенил/]бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-морфолино/этокси]тиофенокси]-2-/4- гидроксифенил/] бензо[b]тиофен;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси]тиофенокси]-2-/4- гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-пирролидинил/этокси]
тиофенокси]-2-/4- гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-гексаметиленимино/этокси]тиофенокси]-2-/4- гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-N, N-диметиламино/этокси]тиофенокси]-2-/4- гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-морфолино/этокси]тиофенокси]-2-/4- гидроксифенил/]
бензо[b]тиофенгидрохлорид;
[6-метокси-3-[4-[2-/1-пиперидинил/этокси] тиофенокси]-2-/4- метоксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-пиперидинил/этокси]тиофенокси]-2-/4- гидроксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-пирролидинил/этокси] тиофенокси] -2-/4- гидроксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-гексаметиленимино/этокси] тиофенокси] -2-/4- гидроксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-N,
N-диметиламино/этокси]тиофенокси]-2- /4-гидроксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-морфолино/этокси] тиофенокси]-2- /4-гидроксифенил/]бензо[b]тиофен;
[6-гидрокси-3-[4-[2-/1-пиперидинил/этокси] тиофенокси]-2- /4-гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-пирролидинил/этокси]тиофенокси]-2- /4-гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-гексаметиленимино/этокси] тиофенокси]
-2- /4-гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-N,N-диметиламино/этокси]тиофенокси]-2- /4-гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-морфолино/этокси] тиофенокси] -2- /4-гидроксифенил/]бензо[b]тиофенгидрохлорид;
[6-гидрокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2- фенил]бензо[b]тиофенгидрохлорид;
Дальнейшие примеры представлены для иллюстрации настоящего изобретения, и никоим образом не ограничивают его объем.
Данные ЯМР, приводимые в этих примерах, получены на спектрометре GE 300 МГц, а в качестве растворителя использовали безводный d-6 ДМСО /если нет других указаний/.
Пример получения 1. [3-/4-Бензилокси/фенокси]бензо[b]тиофен

К раствору 3-бромбензо [b] тиофена /69,62 г, 0,325 моля/ в 55 мл безводного коллидина в атмосфере азота добавляют 4-бензилоксифенол /97,6 г, 0,488 моля/ и закись меди /23,3 г, 0,163 моля/. Полученную смесь нагревают до кипения с обратным холодильником в течение 24 часов. После охлаждения реакционную смесь разбавляют этилацетатом /200 мл/ и неочищенную смесь фильтруют через лепешку Целита Celite® Aldrich, Milwaukee WI/ для удаления неорганических солей. Полученный фильтрат промывают 1н. соляной кислотой /3х150 мл/. Органическую часть сушат над сульфатом натрия и концентрируют в вакууме до жидкости. Тионафталин удаляют перегонкой /10 мм рт.ст, 115 - 120oC/. Оставшуюся часть материала обрабатывают хроматографически/ двуокись кремния, гексаны:этилацетат 85:15/ до получения 12,2 г бензо[b]тиофена и 12,95 г /35% в расчете на выделенный исходный материал/ [3-/4-бензилокси/фенокси]бензо[b] тиофена в виде грязно-белого твердого вещества. Т. пл. 84-86oC.1H ЯМР (CDCl3/d: 7,91 - 7,83 /м, 2H/ 7,91 - 7,83 (м, 2H/, 7, 47 - 7,34 /м, 7H/, 7,04 /кв, JAB = 9,0 Гц, 4H/, 6,47 /c, 1H/, 5,07 /c, 2H/.
Элементный анализ для C21H16O2S:
Рассчитано: C 75,88 H
4,85
Найдено: C 75,75 H 5,00
Пример получения 2. [2-Иодо-3-/4-бензилокси/фенокси]бензо[b]тиофен

К раствору [3-/4-бензолокси/фенокси]бензо[b]тиофена /6,00 г, 18,1 ммоля/ в безводном тетрагидрофуране /100 мл/ в атмосфере азота при -78oC добавляют н-бутиллитий /12,4 мл, 19,9 ммоля, 1,6 М в гексанах/ по каплям через шприц. Раствор из бесцветного превращается в темно-оранжевый. После перемешивания в течение 20 минут при -78oC литиевые образцы обрабатывают I1 /5,03, 19,9 ммоля/, прикапывают через канюлю в виде раствора 50 мл тетрагидрофурана. После завершения добавления реакционная смесь становится светложелтой, и ее оставляют медленно нагреваться до комнатной температуры. Реакцию гасят, добавляя 0,1 и раствор сульфата натрия /200 мл/. Слои разделяют и водный экстрагируют этилацетатом /2х150 мл/. Органические слои объединяют, сушат /над сульфатом натрия/ и концентрируют в вакууме до получения масла, которое кристаллизуется при стоянии. После перекристаллизации из смеси гексанов/этилового эфира получают 7,10 г /86%/ [2-иодо-3-/4-бензилокси/фенокси] бензо[b] тиофена в виде белого кристаллического порошка. Т.плавления 87 - 92oC.
1H ЯМР /CDCl3/d: 7,72/д, J = 8,1 Гц, 1H/, 7,47 - 7,20 /м, 8H/, 6,89 /с, 4H/, 5,01 /с, 2H/.
Элементарный анализ для C21H15O2SI:
Рассчитано: C 55,03 H 3,30
Найдено: C
55,29 H 3,31
Пример получения 3. [2-/4-трет-Бутилоксифенил/-3-/4-бензилокси/фенокси] бензо[b]тиофен

К раствору [2-иодо-3-/4-бензилокси/фенокси]бензо[b]тиофена /4,50 г, 9,82 ммоля/ в 20 мл толуола добавляют 4-/трет-бутокси/-фенилбороновую кислоту /2,28 г, 11,75 ммоля/, а затем тетракисфенилфосфинпалладий /0,76 г, 0,66 ммоля/. К этому раствору добавляют 14,5 мл 2 н. раствора карбоната натрия. Полученную смесь нагревают до кипения с обратным холодильником в течение 3 часов. После охлаждения реакционную смесь разбавляют 150 мл этилацетата. Органическую часть промывают 0,1 г гидроксидом натрия /2х100 мл/, а затем сушат над сульфатом натрия. В результате концентрирования получают полутвердое вещество, которое растворяют в хлороформе и пропускают через слой двуокиси кремния. После концентрирования получают масло, которое после тщательного растирания с гексанами дает 4, 0 г /91%/ [2-/4-трет-бутилоксифенил/-3-/4-бензилокси/фенокси/бензо[b] тиофена в виде белого порошка. Т. плавления 105-108oC.
1H ЯМР /CDCl3/d: 7,77 /д,
J = 7,7 Гц, 1H/, 7,68 /д, J = 8,6 Гц, 2H/, 7,43 - 7,24 /м, 8H/, 6,98 /д, J = 8,6 Гц, 2H/, 6,89 /кв. JAB = 9,3 Гц, 4H/, 4,99 /с, 2H/, 1,36 /с, 9H/. Масс-спектр с полевой десорбцией: 480 ,
Элементный анализ для C31H28O3S:
Рассчитано: C 77,47 H 5,87
Найдено: C 77,35, H 5,99.
Пример получения 4. Аналогичным способом
получают [2-/4-метоксифенил/-3-/4-бензилокси/-фенокси] бензо[b] тиофен, используя 4-метоксифенилбороновую кислоту

Выход = 73%, Т.плавления 115 - 118oC.
1H ЯМР /CDCl3/d: 7,80 - 7,90 /м, 3H/, 7,33 - 7,53 /м, 8H/, 6,93 - 7,06 /м, 6H/, 5,00 /с, 2H/, 3,83 /с, 3H/. Масс-спектр с полевой дисорбцией 483.
Элементный анализ для C28H22O3S:
Рассчитано: C 76,69, H 5,06.
Найдено: C 76,52, H 5,09.
Пример получения 5. [2-/4-трет-Бутилоксифенил/-3-/4-гидрокси/фенокси] бензо[b]тиофен

К раствору [2-/4-трет-бутилоскифенил/-3-/4-бензилокси/фенокси]-бензо[b] тиофена /1,50 г, 3,37 ммоля/ в 30 мл абсолютного этанола, содержащего 1% концентрированной соляной кислоты, добавляют 0,50 г 10% палладия на угле. Полученную смесь гидрируют при 40 пси /2,81 кг/см2/ в течение 1 часа, после чего по данным тонкослойной хроматографии реакцию считают завершенной. Полученную смесь фильтруют через слой Целита и полученный фильтрат концентрируют в вакууме. Неочищенный продукт растворяют в минимальном количестве этилацетата и пропускают через короткую колонку с двуокисью кремния для удаления Целита, используя в качестве элюента этилацетат. В результате концентрирования получают белый твердый продукт, который тщательно растворяют со смесью гексаны/этиловый эфир. После фильтрования получают 868 мг /73%/ [2-/4-трет-бутилоксифенил/-3-/4-гидрокси/фенокси]-бензо[b]тиофена. Т. плавления 210-213oC.1 ЯМР /DMCO-d6/d: 9,13 /с, 1H/, 7,94 /д, J = 7,7 Гц, 1H/, 7,63 /д, J = 8,6 Гц, 2H/, 7,35 - 7,26 /м, 3H/, 7,01 - д, J = 8,6 Гц, 2H/, 6,70 /кв, JAB = 8,9 Гц, 4H/, 1,28 /с, 9H/.
Масс-спектр с полевой десорбцией: 390.
Элементный анализ для C24H22O3S:
Рассчитано: C 73,82, H 5,68
Найдено: C 73,98, H 5,84
Пример получения 6. Аналогичным способом получают [2-/4-метоксифенил/-3-/4-гидрокси/-фенокси]бензо[b]тиофен

Выход 80%. Т. плавления 120-125oC.
1H ЯМР /CDCl3/d: 7,80 - 7,90 /м, 3H/, 7,48 /м, 1H/, 7,30 - 7,48 /м, 2H/, 6,90 - 7,03 /м, 4H/, 6,76 - 6,86 /м, 2H/, 3,82 /с, 3H/.
Масс-спектр с полевой десорбцией: 348.
Элементный анализ для C21H16O3S:
Рассчитано: C 72,39 H 4,63
Найдено: C 72,68 H 4,82
Пример 1. [3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4-гидроксифенил/] бензо[b]тиофен

К раствору [2-/4-трет-бутилоксифенил/-3-/4-гидрокси/фенокси] бензо[b] тиофена /1,25 г, 3,20 ммолей/ в безводном N,N-диметилформамиде /10 мл/ при комнатной температуре добавляют карбонат цезия /5,70 г, 17,6 ммоля/. После двадцатиминутного перемешивания небольшими порциями добавляют 2-хлорэтилпиперидингдирохлорид /1,95 г, 10,56 ммоля/, полученную гетерогенную смесь интенсивно перемешивают в течение 24 часов. Затем реакционную смесь разбавляют 200 мл воды. Водную фазу экстрагируют этилацетатом трижды по 100 мл. Объединенные органические слои промывают водой /2х200 мл/. После сушки органического слоя над сульфатом натрия и концентрирования в вакууме получают масло. После хроматографической обработки /5-10% метанол/ хлороформ/ получают 1,47 г /91%/ [3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4-трет-бутилоксифенил/] бензо] [b]тиофена, который используют на следующей стадии без получения аналитических данных.
[3-[4-[1-/1-Пиперидинил/этокси] фенокси] -2-/4-трет-бутилкосифенил/]бензо[b] тиофен /1,37 г, 2,72 ммоля/ растворяют в трифторуксусной кислоте /10 мл при комнатной температуре.
После перемешивания в течение 15 минут, растворитель удаляют в вакууме. Остаток растворяют в этилацетате /20 мл/ и промывают насыщенным раствором бикарбоната натрия /3х10 мл/. Органический слой сушат над сульфатом натрия и концентрируют до тех пор, пока в растворе не образуется белый твердый осадок. Полученный продукт перекристаллизовывают из смеси этилацетат/этиловый эфир до получения 1,03 г /85% [3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4-гидроксифенил/]бензо[b] тиофена в виде бесцветных кристаллов. Т. плавления 169-172oC.
1H ЯМР /ДМСО-d6/d: 9,81 /c, 1H/, 7,93 /д, J = 7,7 Гц, 1H/, 7,54 /д, J = 8,5 Гц, 2H/, 7,36 - 7,26 /м, 3H/, 6.86 /с, 4H/, 6,78 /д, J = 8,6 Гц, 2H/, 4,10 /м, 2H/, 3,29 /м, 2H/, 2,95 - 2,75 /м, 4H/, 1,68 - 1,40 /м, 6H/.
Элементный анализ для C27H27NO3S • 0,55 CF3CO2
H:
Рассчитано: C 66,40 H 5,46 N 2,76
Найдено: C 65,99 H 5,49 N 2,61
Пример 2 [3-[[4-[2-/1-Пиперидинил/этокси]фенокси]-2-/4-гидроксифенил/] бензо[b] тиофен превращают в его
гидрохлорид с 90% выходом за счет обработки этиловый эфир • соляной кислотой в этилацетате

1H ЯМР /ДМСО-d6/d: 10,43 /м, 1H/, 9,89 /с, 1H/, 7,93 - 7,95 /м. 1H/, 7,60 - 7,64 /м, 2H/, 7,35 - 7,50 /м, 3H/, 6,83 - 7,03 /м, 6H/, 4,27 - 4,30 /м, 2H/, 3,40 - 3,60 /м, 4H/, 2,96 - 3,10 /м, 2H/, 1,70 - 1,95 /м, 5H/, 1,40 - 1,53 /м, 1H/. Масс-спектр с полевой десорбцией: 446.
Элементный анализ для C27H27NO3S • 1,0 HCl:
Рассчитано: C 67,28 H 5,86 N 2,91.
Найдено: C 67,07 H 5,66 N 2,96
Пример 3.
Аналогичным способом получены следующие соединения: [3-[4-[2-/1-пирролидинил/этокси]фенокси]-2-/4-гидроксифенл/]бензо[b] тиофен
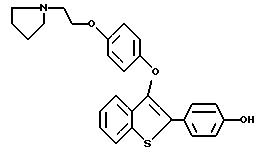
Т.плавления 150-155oC.1H ЯМР /ДМСО-d6/d: 9,79 /с, 1H/, 7,92 /д, J = 7,8 Гц, 1H/, 7,54 /д, J = 8,6 Гц, 2H/, 7,36 - 7,26 /м, 3H/, 6,84 /с, 4H/, 6,78 /д, J = 8,6 Гц, 2H/, 4,00 /шир. т., 2H/, 2,92 /м, 2H/, 2,85 /м, 4H/, 1,73 /м, 4H/. Элементный анализ для C26H25NO3S • 0,33 CF3 CO2H:
Рассчитано: C 68,25 H 5,44 H 2,99.
Найдено: C 68,29 H 5,46 N 3,19
Пример 4.
[3-[4-[2-/1-Гексаметиленимино/этокси]фенокси]-2-/4-гидроксифенил/]бензо[b] тиофен
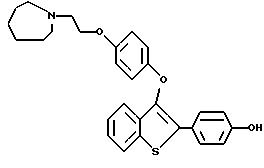
Т.плавления 189-191oC.1H ЯМР /ДМСО-d6/d: 7,91 /д, J = 7,6 Гц, 1H/, 7,52 /д, J = 8,5 Гц, 2H/, 7,34 - 7,25 /м, 3H/, 6,81 /с, 4H/, 6,75 /д, J = 8,6 Гц, 2H/, 3,89 /шир. т. , 2H/, 2, 75 /шир. т., 2H/, 2,68 /м, 4H/, 1,48 /м, 8H/. Элементный анализ для C28H29NO3S • 1,50 H2O:
Рассчитано: C 69,11 H 6,79 N 2,88
Найдено: C 69,25 H 6,79 N 2,58
Пример 5. [3-[4-[2-/1-N,N-Диметиламино/этокси]фенокси]-2- (4-гидроксифенил/]бензо[b]тиофен

Т. плавления 70oC.1H ЯМР /ДМСО-d6/d: 9,91 /шир. с. 1H/, 7,92 /д, J = 7,9 Гц, 1H/, 7,54 /д, J = 8,6 Гц, 2H/, 7,35 - 7,24 /м, 3H/, 6,82 /с, 4H/, 6,78 /д, J = 8,6 Гц, 2H/, 3,88 /шир. т., 2H/, 2,76 /шир., т., 2H/, 2,51 /м, 4H/, 0,91 /м, 6H/.
Масс-спектр с полевой десорбцией: 434.
Элементный
анализ для C26H27NO3S • 0,50 H2O:
Рассчитано: C 70,56, H 6,38 N 3,16.
Найдено: C 70,45 H 6,26 N 3,20.
Пример 6. [3-[4-[2-/1-Пиперидинил/этокси]фенокси]-2-/4-метоксифенил/] бензо[b] тиофенгидрохлорид

Т. плавления = 228-23-oC.1H ЯМР /ДМСО-d6/d: 7,96 /д, J = 7,5 Гц, 1H/, 7,66 /д, J = 8,8 Гц, 2H/, 7,35 - 7,50 /м, 3H/, 6,98 /д, J = 8,7 гц, 2H/, 6,86 - 6,90 /м, 4H/, 4,28 - 4,31 /м, 2H/, 3,74 /с, 3H/, 3,37 - 3,45 /м, 4H/, 2,92 - 2,96 /м, 2H/, 2,46 - 2,48 /м, 5H/, 1,74 /м, 1H/.
Масс-спектр с полевой десорбцией: 459. Элементный анализ для C28H29NO3S • 1,0 HCl:
Рассчитано: C 67,80 H 6,10 N 2,82
Найдено: 68,06 H 6,38 N 2,60
Альтернативный способ получения
[2-/4-трет-бутилоксифенил/-3-/4-бензилокси/фенокси]бензо[b]тиофена
Пример получения 7. [3-/4-Бензилокси/фенокси] [b] тиофен-2-бороновая кислота

К раствору [3-/4-бензилокси/фенилокси] бензо[b] тиофена /5,00 г, 15,1 ммоля/ в 20 мл безводного тетрагидрофурана при температуре -78oC в атмосфере азота добавляют по каплям через шприц -N-бутиллитий /9,90 мл, 15,8 ммоля, 1,6 М в гексанах/. После пятнадцатиминутного перемешивания через шприц добавляют В/О изо Pr/3 /3, 83 мл, 16,6 ммоля/, полученной смеси дают нагреться до 0oC. Реакцию гасят, разделяя реакционную смесь между этилацетатом и 1,0 и соляной кислотой /по 100 мл каждого/. Органический слой экстрагируют водой /1х100 мл/. Органический слой сушат над сульфатом натрия и концентрируют в вакууме до твердого вещества, которое тщательно растирают со смесью этиловый эфир/гексаны. После фильтрования получают 3,96 г /70%/ [3-/4-бензилокси/фенокси]бензо[b]тиофен-2-бороновой кислоты в виде белого твердого вещества. Т.плавления 115-121oC.1H ЯМР /ДМСО-d6 /d: 8,16 /д, J = 8,5 Гц, 1H/, 7,98 /д, J = 9,0 Гц, 1H/, 7,42-7,23 /м, 7H/, 6,90 /кв. JAB = 9,0 Гц, 4H/, 5,01 /с, 2H/. Элементный анализ для C21H17O4SB:
Рассчитано: C 67,04 H 4,55
Найдено: C 67,17 H 4,78
[3-/4-Бензилокси/фенокси] бензо[b] тиофен-2-бороновую кислоту подвергают взаимодействию с 4-/трет-бутокси/бромбензолом в условиях, описанных ранее для [2-иодо-3-/4-бензилокси/фенокси] бензо[b] тиофена и 4-/трет-бутокси/фенилбороновой кислоты, до получения [2-/4-трет-бутоксифенил/-3-/4-бензилокси/фенокси]бензо[b]тиофена с 81% выходом.
В следующих примерах использован этот способ:
Пример 7. [3-[4-[2-/1-Пиперидинил/этокси] -2-/фенил/] бензо[b] тиофенгидрохлорид

Т. плавления 223-226oC.1 ЯМР /ДМСО-d6/d: 7,99 /д, J = 8,2 Гц, 1H/, 7,71 /д, J = 7,3 Гц, 1H/, 7,44 - 7,30 /м, 7H/, 6,90 /с, 4H/, 4,27 /м, 2H/, 3,46 - 3,35 /м, 4H/, 2,97 - 2,88 /м, 2H/, 1,73 - 1,61 /м, 5H/, 1,34 /м, 1H/.
Элементный анализ для C27H27NO2S • 1,0 HCl:
Рассчитано: C 69,59 H 6,06 N 3,00
Найдено: C 69,88 H 6,11 N 3,19
Пример 8.
[3-[4-[2-/1-Пиперидинил/этокси]фенокси]-2-/4-фторфенил/]бензо[b]тиофен

Т.плавления 219-226oC.1H ЯМР /ДМСО-d6/d: 10,20 /шир, с. 1H/, 7,99 /д, J = 8,2 Гц, 1H/, 7,77 - 7,73 /м, 4H/, 7,42 - 7,25 /м, 5H/, 6,90 /с, 4H/, 4,27 /м, 2H/, 3,44 - 3,31 /м, 4H/, 2,96 - 2,89 /м, 2H/, 1,78 - 1,61 /м, 5H/, 1,34 /м, 1H/. Масс-спектр с полевой десорбцией: 447
Элементный анализ для C27H26NO2SF • 1,0 HCl:
Рассчитано: C 67,00 H 5,62 N 2,89
Найдено: C 67,26 H 5,67 N 3,03
Пример получения 8. [6-Метокси-2-/4-метоксифенил/-3-бром]бензо[b] тиофен

К раствору [6-метокси-2-/4-метоксифенил/] бензо[b]тиофена /27,0 г, 100 ммолей/ в 1,10 л хлороформа при 60oC добавляют бром /15,98 г, 100 ммолей/ по каплям в виде раствора в 200 мл хлороформа. После завершения добавления реакционную смесь охлаждают до комнатной температуры и растворитель удаляют в вакууме до получения 34,2 г /100%/ [6-метокси-2-/4-метоксифенил/-3-бром] бензо[b]тиофена в виде твердого вещества белого цвета. Т. плавления 83-85oC.
1H ЯМР /ДМСО-d6/d: 7,70 - 7, 62 /м, 4H/, 7,17 /дд, J = 8,6 Гц, 2,0 Гц, 1H/, 7,09 /д, J = 8,4 Гц, 2H/. Масс-спектр с полевой десорбцией: 349.
Элементный анализ для C16H13O2SBr;
Рассчитано: C 55,03 H 3,75
Найдено: C 54,79 H 3,76
Пример 9. [6-Метокси-2-/4-метоксифенил/-3-/4-бензилокси/фенокси]бензо[b] тиофен

К раствору [6-метокси-2-/4-метоксифенил/-3-бром]бензо[b]тиофена /34,00 г, 97,4 ммоля/ в 60 мл коллидина в атмосфере азота добавляют 4-бензилоксифенол /38,96 г, 194,8 ммоля/ и закись меди /14,5 г, 97,4 ммоля/. Полученную смесь нагревают до температуры кипения с обратным холодильником в течение 48 часов. После охлаждения до комнатной температуры смесь растворяют в ацетоне /200 мл/ и неорганическую твердую часть удаляют фильтрованием. Полученный фильтрат концентрируют в вакууме, а полученный остаток растворяют в 500 мл метиленхлорида. Метиленхлоридный раствор промывают 3 н. соляной кислотой /3х300 мл/, затем 1н. гидроксидом натрия /3х300 мл/. Органический слой сушат над сульфатом натрия и концентрируют в вакууме. Остаток помещают в 100 мл этилацетата, после чего образуется белое твердое вещество, которое собирают фильтрованием. Выделен [6-метокси-2-/4-метоксифенил/]бензо[b]тиофен /4,62 г, 17,11 ммоля/. Полученный фильтрат концентрируют в вакууме, затем пропускают через тонкий слой силикагеля/ элюент метиленхлорид/ для удаления основного материала. Полученный фильтрат концентрируют в вакууме, а остаток кристаллизуют из смеси гексаны/этилацетат до получения вначале 7,19 г [6-метокси-2-/4-метоксифенил/-3-/4-бензилокси/фенокси] бензо[b] тиофена в виде грязно-белого кристаллического твердого вещества. Маточный раствор концентрируют и обрабатывают хроматографически на силикагеле /гексаны/этилацетат 80:20/ до получения дополнительно 1,81 г продукта. Полный выход [6-метокси-2-/4-метоксифенил/-3-/4-бензилокси/фенокси]бензо[b]тиофена составляет 9,00 г /24% в расчете на выделенный исходный материал/. Основной экстракт подкисляют до pH 4 за счет 5 н. соляной кислоты и полученный осадок собирают фильтрованием и сушат до получения 13,3 г выделенного 4-бензилоксифенола. Т.плавления 100-103oC.2H ЯМР /CDCl3/: d: 7,60 /д, J = 08,0 Гц, 2H/, 7,39 - 7,24 /м, 7H/, 6,90 - 6,85 /м, 7H/, 4,98 /с, 2H/, 3,86 /с, 3H/, 3,81 /с, 3H/. Масс-спектр с полевой десорбцией: 468.
Элементный анализ для C29H24O4S:
Рассчитано: C 74,34 H 5,16.
Найдено: C 74,64 H 5,29,
Пример получения 9. [6-Метокси-2-/4-метоксифенил/-3-/4-гидрокси/фенокси]
бензо[b]тиофен

К раствору [6-метокси-2-/4-метоксифенил/-3-/4-бензилокси/фенокси] бензо[b] тиофена /1,50 г, 3,20 ммоля/ в 50 мл этилацетата и 10 мл 1% концентрированной соляной кислоты в этаноле добавляют 10% палладий на угле /300 мг/. Полученную смесь гидрируют при 40 пси /2,81 кг/см3/ в течение 20 минут, после чего по данным тонкослойной хроматографии реакции завершается. Полученную смесь пропускают через Целит для удаления катализатора, а полученый фильтрат концентрируют в вакууме до получения твердого вещества белого цвета. Неочищенный продукт пропускают через слой силикагеля /в качестве элюента используют хлороформ/. После концентрирования получают 1,10 г /91%/. [6-метокси-2-/4-метоксифенил/-3-/4-гидрокси/фенокси] бензо[b] тиофена в виде твердого вещества белого цвета. Т.плавления 123-126oC.1H ЯМР /ДМСО-d6/d: 9,10 /с, 1H/, 7, 59 /д, J = 8,8 Гц, 2H/, 7,52 /д, J = 2,1 Гц, 1H/, 7,14 /д, J = 8,8 Гц, 1H/, 6,95 /д, J = 8,8 Гц, 2H/, 6,89 /дд, J = 8,8, 2,1 Гц, 1H/, 6,72 /д, J = 9,0 Гц, 2H/, 6,63 /д, J = 9,0 Гц, 2H/, 3,78 /с, 3H/, 3,72 /с, 3H/. Масс-спектр с полевой десорбцией: 378. Элементный анализ для C22H18O4S:
Рассчитано: C 69,82 H 4,79
Найдено: C 70,06 H 4,98
Пример 10. [6-Метокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4-метоксифенил/]бензо [b]тиофен

К раствору [6-метокси-2-/4-метоксифенил/-3-/4-гидрокси/фенокси]бензо[b] тиофена /1,12 г, 2,97 ммоля/ в 7 мл безводного N,N-диметилформамида в атмосфере азота добавляют карбонат цезия /3,86 г, 11, 88 ммоля/. После перемешивания в течение 10 минут добавляют 2-хлорэтилпиперидингидрохлорид /1,10 г, 1,48 ммоля/. Полученную смесь перемешивают в течение 18 часов при комнатной температуре. Реакционную смесь разделяют между хлороформом и водой /по 100 мл каждого/. Водный слой экстрагируют хлороформом /3х50 мл/. Органические слои объединяют и промывают водой /2х100 мл/. После сушки органической части над сульфатом натрия и концентрирования получают масло, которое хроматографически обрабатывают на силикагеле /2% метанол/ /хлороформ/. Целевые фракции концентрируют до масла, которое растворяют в 10 мл этилацетата обрабатывают щавелевой кислотой /311 мг, 3,4 ммоля/. После перемешивания в течение 10 минут образуется белый осадок, который собирают фильтрованием и сушат до получения 1,17 г /70%/ всего [6-метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4-метоксифенил/] бензо[b]тиофена в виде оксалатной соли. Т.плавления 197-200oC /с разложением/.1H ЯМР /ДМСО-d6/d: 7,60 /д, J = 8,7 Гц, 2H/, 7,55 /д, J = 1,1 Гц, 1H/, 7,14 /д, J = 8,8 Гц, 1H/, 7,06 /д, J = 8,8 Гц, 2H/, 6,91 /дд, J = 8,8 Гц, 1H/, 6,87 /с, 4H/, 4,19 /шир.т., 2H/, 3,78 /с, 2H/, 3,72 /с, 3H/, 3,32 /шир. т., 2H/, 3,12 - 3,06 /м, 4H/, 1,69 - 1,47 /м, 4H/, 1,44 - 1,38 /м, 2H/. Масс-спектр с полевой десорбцией: 489.
Элементный анализ для C29H31NO4S • 0,88 HO2CCO2H:
Рассчитано: C 64,95 H 5,80 N 2,46
Найдено: C 64,92 H 5,77 N 2,54
Пример 11. Обработка свободного
основания этиловым эфиром соляной кислотой приводит к получению [6-метокси-3-[4-[2-/1-пиперидинил/этокси]-фенокси] -2-/4-метоксифенил/]бензо [b]тиофенгидрохлорида

Т.плавления 216 - 220oC.1H ЯМР (ДМСО-d6/d: 10,20 /шир.с., 1H/, 7,64 /д, J = 8,7 Гц, 2H/, 7, 59 /д, J = 1,5 Гц, 1H/, 7,18 /д, J = 9,0 Гц, 1H/, 7,00 /д, J = 8,7 Гц, 1H/, 6,96 /дд, J = 9,0, 1,5 Гц, 1H/, 6,92 /кв. JAB = 9,0 Гц, 4H/, 4,31 /м, 2H/, 3,83 /с, 3H/, 3,77 /с, 3H/, 3,43 /м, 4H/, 2,97 /м, 2H/, 1,7 /м, 5H/, 1,37 /м, 1H/. Масс-спектр с полевой десорбцией: 489. Элементный анализ для C29H31NO4S • 1,0 HCl
Рассчитано: C 66,21 H 6, 13 N 2,66
Найдено: C 66,46 H 6,16 N 2,74
Аналогичным способом получают соединения следующих примеров:
Пример 12. [6-Метокси-3-[4-[2-/1-пирролидинил/этокси]фенокси]-2-/1-метоксифенил/]бензо [b]тиофен

Т. плавления 95-98oC.1H ЯМР /ДМСО-d6/d: 7,64 /д, J = 9,0 Гц, 2H/, 7,58 /д, J = 2,0 Гц, 1H/, 7,18 /д, J = 9,0 Гц, 1H/, 7,00 /д, J = 9,0 Гц, 2H/, 6,94 /дд, J = 9,0, 2,0 Гц, 1H/, 6,86 /с, 4H/, 3,97 /т, J = 6,0 Гц, 2H/, 3,83 /с, 3H/, 3,76 /с, 3H/, 2,73 /т, J = 6,0 Гц, 2H/, 2,51 /т, 4H/, 1,66 /м, 4H/. Масс-спектр с полевой десорбцией: 477.
Элементный
анализ для C28H29NO4S:
Рассчитано: C 70,71 H 6,15 N 2,99.
Найдено: C 70,59 H 6,15 N 3,01.
Пример 13.
[6-Метокси-3-[4-[2-/1-гексаметиленимино/этокси]фенокси]-2-/4-метоксифенил/] бензо[b]тиофенгидрохлорид

Т. плавления 189 - 192oC.1H ЯМР /ДМСО-d6/d: 10,55 /шир. с., 1H/, 7,64 /д, J = 9,0 Гц, 2H/, 7,58 /д, J = 2,0 Гц, 1H/, 7,19 /д, J = 9,0 Гц, 1H/, 7,00 /д, J = 9,0 Гц, 2H/, 6,95 /дд, J = 9,0, 2,0 Гц, H/, 6,86 /с, 4H/, 3,94 /т, J = 6,0 Гц, 2H/, 3,83 /с, 3H/, 3,76 /с, 3H/, 2,80 /т, J = 6,0 Гц, 2H/, 2,66 /м, 4H/, 1,53 /м, 8H/. Элементный анализ для C30H33NO4S • 1,0 HCl:
Рассчитано: C 66,71 H 6,35 N 2,59
Найдено: C 66,43 H 6,46 N 2,84
Пример 14. [6-Метокси-3-[4-[2-/1-N, N-диэтиламино/этокси]фенокси]-2-/4-метоксифенил/] бензо[b]тиофенгидрохлорид

Т. плавления 196-198oC.1H ЯМР /ДМСО-d6/d: 10,48 /шир. с., 1H/, 7,64 /д, J = 9,0 Гц, 2H/, 7,59 /д, J = 2,0 Гц, 1H/, 7,19 /д, J = 9,0 Гц, 1H/, 7,00 /д, J = 9,0 Гц, 2H/, 6,97 /дд, J = 9,0, 2,0 Гц, 1H/, 6,87 /кв. JAB = 9,0 Гц, 4H/, 4,25 (м, 2H/, 3,83 /с, 3H/, 3,77 /с, 3H/, 3,09 /м, 4H/, 2,00 /м, 3H/, 1,88 /м, 3H/. Элементный анализ для C28H31NO4S • 1,5 HCl:
Рассчитано: C 63,18 H 6,15 N 2,63
Найдено: C 63,46 H 5,79 N 2,85
Пример 15. [6-Метокси-3[4-[2-/морфолино/этокси]фенокси] -2-/4-метоксифенил/]-бензо[b]тиофенгидрохлорид

T. плавления 208-211oC1H ЯМР /ДМСО-d6/ d: 10,6 /шир. с., 1H/, 7,63 /д, J = 9,0 Гц, 2H/, 7,60 /д, J = 2,0 Гц, 1H/, 7,20 /J = 9,0 Гц, 1H/ 7,00 /д, J = 9,00 Гц, 2H/, 6,97 /дд, J 9,0, 2,0 Гц, 1H/, 6,91 /кв., JAB=9,0 Гц, 4H/, 4,29 /м, 2H/, 4,08-3,91 /м, 4H/, 3,82 /с, 3H/, 3,77 /с, 3H/, 3,59-3,42 /м, 4H/, 3,21-3,10 /м, 2H/. Элементный анализ для C28H29NO5S • 1,0 HCl.
Рассчитано: C 63,09 H 5,73 N 2,65.
Найдено: C 63,39 H 5,80 N 2,40
Пример 16. [6-Метокси-3-[4-[3-пиперидино/протокси]
-фенокси]-2- /4-метоксифенил/]-бензо[b]тиофенгидрохлорид

T. плавления 195 - 200oC.1H ЯМР /ДМСО-d6/d: 9,90 шир.с., 1H/, 7,64 /д. J = 9,0 Гц, 2H/, 7,59 /д, J = 2,0 Гц, 1H/, 7,18 /д, J = 9,0 Гц, 1H/, 7,00 /д, J = 9,0 Гц, 2H/, 6,95 /дд. J = 9,0, 2,0 Гц, 1H/, 6,88 /с, 4H/, 3,97 /т, J = 6,0 Гц = 6,0 Гц, 2 H/, 3,83 /с, 3H/, 3,77 с, 3H/, 3,44 /м, 2H/, 3,15 /м, 2H/, 2,87 /м, 2H/, 2,12 /м, 2H/, 1,77 /м, 5H/, 1,39 /м, 1H/,
Элементный анализ для C30 H33NO4S • 1,15 HCl:
Рассчитано: C 66,01 H 6,40 N 2,73
Найдено: C 66,01 H 6,40 N 2,73.
Пример 17. [6-Метокси-3-[4-/1-N,
N-диэтиламино/пропокси]фенокси]- 2-/4-метоксифенил/]бензо[b]тиофенгидрохлорид

T. плавления 164 - 166oC.1H ЯМР /ДМСО-d6/ d: 9,77 /шир.с., 1H/, 7,64 /д, J = 9,0 Гц, 2H/, 7,59 /д, J = 2,0 Гц, 1H/, 7,18 /д, J = 9,0 Гц, 1H/, 7,00 /д, J = 9,0 Гц, 2H/, 6,95 /дд, J = 9,0, 2,0 Гц, 1H/, 6,89 /с, 4H/, 3,99 /т, J = 6,0 Гц, 2H/, 3,83 /с, 3H/, 3,77 /с, 3H/, 3,15 /м, 6H, 2,06 /м, 2H/, 1,20 /т, J = 7,0 Гц, 6H/. Элементный анализ для C29H33 NO4S • 1,0 HCl:
Рассчитано: C 65,96 H 6,49 N 2,65.
Найдено: C 66,25 H 6,64 N 2,84.
Пример 18 [6-Гидрокси-3-[4-[2-/1-пиперидинил/этокси] фенокси]
-2-/4- гидроксифенил/]бензо[b]тиофен

[6-Метокси-3[4-2-/1-пиперидинил/этокси] фенокси] -2-/4-метоксифенил/] бензо[b] тиофенгидрохлор /10,00 г, 19,05 ммоля/ растворяют в 500 мл безводного метиленхлорида и охлаждают до 8oC. К этому раствору добавляют трехбромистый бор /7,20 мл, 76,20 ммоля/. Полученную смесь перемешивают при 8oC в течение 2,5 часа. Реакцию гасят, выливая в перемешиваемый раствор насыщенного бикарбоната натрия /1 л/, охлажденный до 0oC. Выделяют метиленхлоридный слой, а оставшуюся твердую часть растворяют в смеси метанол/этилацетат. Водный слой затем экстрагируют смесью 5% метанол/этилацетат /3 х 500 мл/. Все органические экстракты /этилацетатные и метиленхлоридные/ объединяют и сушат над сульфатом натрия. После концентрирования в вакууме получают желто-коричневое тведое вещество, которое обрабатывают хроматографически /двуокись кремния, 1 - 7% метанол/хлороформ/ до получения 7,13 г /81%/ [6-гидрокси-3-[4- [2-[1-пиперидинл/этокси]фенокси]-2-/4-гидроксифенил/]бензо[b]тиофена в виде твердого вещества белого цвета. T. плавления в виде твердого вещества белого цвета. T. плавления 93oC.1H ЯМР /ДМСО-d6/ d: 9,73 /шир.с., 1H/, 7,45 /д, J = 8,5 Гц, 2H/, 7,21 /д, J = 1,8 Гц, 1H/, 7,04 /д, J 8,6Гц, 1H/, 6,84 /дд, J = 8,6, 1,8 Гц, 1H/ маскирован//, 6,81 /с, 4H/, 6,75 /д, J = 8,6 Гц, 2H/, 3,92 /т, J = 5,8 Гц, 2H/, 2,56 /т, J = 5,8 Гц, 2H/, 2,36 /м, 4H/, 1,43 /м, 4H/, 1,32 м, 2H/, Масс-спектр с полевой десорбцией: 462.
Элементный анализ C27H27NO4S: C 70,20 H 5,90 N 3.03/
Найдено: C 69,96 H 5,90 N 3,14
Пример 19.
[6-Гидрокси-3-[4-[2-/1-пиперидинил/этокси] фенокси]-2-/4-гидроксифенил[/ бензо[b] тиофен превращают в оксалатную соль с 80% выходом описанным ранее способом. Данные для
[6-гидрокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2/-4- гидроксифенил/]бензо[b]тиофеноксалата:

T. плавления 246 - 249oC /с разложение/.1H ЯМР /ДМСО-d6/ d: 7,45 /д, J = 8,6 Гц, 2H/, 7,22 /д, J = 1,8 Гц, 1H/, 7,05 /д, J = 8,6 Гц, 1H/, 6,87 /дд, J = 8,6 Гц, 1,8 Гц, 1H /маскирован//, 6,84 с., 4H/, 6,75 /д, J = 8.6 Гц, 2H/, 4,08 /шир. т., 2H/, 3,01 шир. т., 2H/, 2,79 /м, 4H/, 1,56 /м, 4H/, 1,40 /м, 2H/. Масс-спектр с полевой десорбцией: 462.
Элементный анализ для C27H27NO4S • 0,75 HO2CCO2H:
Рассчитано: C 64,63 H 5,42 N 2,64.
Найдено: C 64,61 H 5,55 N 2,62.
Пример 20. [6-Гидрокси-3-[4-[2-/1-пиперидинил/этокси] фенокси]-2-/4-гидроксифенил/] бензо[b] тиофен превращают в его гидрохлорид с 91% выходов, обрабатывая свободное
основание этилацетатом с этиловый эфир, соляной кислотой. Данные для [6-гидрокси-3-/4-[2-/1-пиперидинил/этокси]- фенокси]-2-/4-гидроксифенил/]бензо[b]тиофенгидрохлорида:

T. пл. 158 - 165oC.1H ЯМР /ДМСО-d6/ d: 9.97 /с, 1H/, 9,74 /c, 1H/, 7,40 /д, J = 8,6 гц, 2H/, 7,23 /д, J = 2,0 Гц, 1H/, 7,04 /д, J = 8,6 Гц, 1H/, 6,86 /кв. JAB = 9,3 Гц, 4H/, 6,76 /дд, J = 8,6, 2,0 Гц, 1H/, 6,74 /д, J = 8,6 Гц, 2H/, 4,26 /шир.т., 2H/, 3,37 /м, 4H/, 2,91 /м, 2H/, 1,72 /м, 5H/, 1,25 /м, 1H/. Масс-спектр с полевой десорбцией: 461.
Элементный анализ для C27H27NO4S • 1,0 HCl:
Рассчитано: C 65,11 H 5,67 N 2,81
Найдено: C 64,84 H 5,64 N 2,91.
Аналогичным образом получают соединения следующих примеров:
Пример 21.
[6-Гидрокси-3-[4-[2-/1-пирролидинил/этокси]фенокси]- 2-/4-гидроксифенил/]бензо[b]тиофен

T. плавления 99 - 113oC.1H ЯМР /DMCO-d6/ d: 9,75 /с, 1H/, 9,71 /с, 1H/, 7,50 /д, J = 9,0 Гц, 2H/, 7,25 /д, J = 2,0 Гц, 1H/, 7,09 /д, J = 9,0 Гц, 1H/, 6,85 /с, 1H/, 6,80 /дд, J = 9,0, 2,0 Гц, 1H/, 6,79 /д, J = 9,0 Гц, 2H/, 3,93 /м, 2H/, 2,73 /м, 2H/, 2,53 /м, 4H/, 0,96 /т, J = 7,0 Гц, 4H/.
Элементный анализ для C26H25NO4S • 0,5 H2O
Рассчитано: 68,40 H 5,74 N 3,07
Найдено: C 68,52 H 6,00 N 3,34
Пример 22. [6-Гидрокси-3-[4-[2-/1-гексаметиленимино/этокси]
фенокси]-2-/4-гидроксифенил/]бензо[b]тиофен

T. плавления 125 - 130oC.1 H ЯМР /DMCO-d6/ d: 9,75 /с, 1H/, 9,71 /с, 1H/, 7,50 /д, J = 9,0 Гц, 2H/, 7,26 /д, J = 2,0 Гц, 1H/, 7,09 /д, J = 9,0 Гц, 1H/, 6,85 /с, 3H/, 6,80 /дд, J = 9,0, 2,0 Гц, 1H/, 6,79 /д, J = 9,0 Гц/, 3,94 /т, J = 6,0 Гц, 2H/, 2,80 /т, J = 6,0 Гц, 2H/, 2,66 /м, 4H/, 1,53 /м, 8H/. Элементный анализ для C28H29NO4:
Рассчитано: C 70,71 H 6,15 N 2,94
Найдено: C 70,67 H 6,31 N 2,93
Пример 23. [6-Гидрокси-3-[4-[2-/1-N,N-диэтиламино/этокси]фенокси] -2-/4-гидроксифенил/]бензо[b]тиофен

T. плавления 137 - 141oC.1H ЯМР /ДМСО-d6/ d: 9,75 /с, 1H/, 9,71 /с, 1H/, 7,49 /д, J = 9,0 Гц, 1H/, 7,25 /д, J = 2,0 Гц, 1H/, 7,09 /д, J = 9,0 Гц, 1H/, 6,85 /с, 4H/, 6,80 /дд, J = 9,0, 2,0 Гц, 1H/, 6,79 /д, J = 9,0 Гц, 2H/, 3,95 /т. J = 6,0 Гц, 2H/, 2,74 /т, J = 6,0 Гц, 2H/, 2,51 /м, 4H/, 1,66 /м, 6H/. Элементный анализ для C26 H27NO4S:
Рассчитано: C 69,46 H 6,05 N 3,12
Найдено: C 69,76 H 5,85 N 3,40
Пример 24. [6-Гидрокси-3-[4-[2-/морфолино/этокси] фенокси] - 2-/4-гидроксифенил/]-бензо[b]тиофенгидрохлорид

T. плавления 157 - 162oC.1H ЯМР /ДМСО-d6/ d: 10,60 /шир. с., 1H/, 9,80 /с, 1H/, 9,75 /с, 1H/, 7,50 /д, J = 9,0 Гц, 2H/, 7,28 /д, J = 2,0 Гц, 1H/, 7,10 /д, J = 9, 0 Гц, 1H/, 6,92 /кв. JAB = 9,0 Гц, 4H/, 6,81 /дд, J = 9,0, 2,0 Гц, 1H/, 6,80 /д, J = 9,0 Гц, 2H/, 4,30 /м, 2H/, 3,95 /м, 2H/, 3,75 /м, 2H/, 3,51 /м, 4H/, 3,18 /м, 2H/. Элементный анализ для C26H25NO5S • HCl
Рассчитано: C 62,46 H 5,25 N 2,80
Найдено: C 69,69 H 5,43 N 2,92
Пример 25. [6-Гидрокси-3-[4-[3-/1-N, N-диэтиламино/пропокси] фенокси]-2-/4-гидроксифенил/]бензо[b]тиофенгидрохлорид

Т. плавления 185-191oC.1H ЯМР /ДМСО-d6/ d 9,94 /шир.с., 1H/, 9,81 /с, 1H/, 9,75 /с, 1H/, 7,50 /д, J = 9,0 Гц, 2H/, 7,27 /дд, J = 2,0 Гц, 1H/, 7,10 /д, J = 9,0 Гц, 1H/, 6,87 /с, 4H/, 6,80 /дд, J = 9,0 2,0 Гц, 1H/, 6,79 /д, J = 9,0 Гц, 2H/, 3,99 /т, J = 6,0 Гц, 2H/, 3,14 /м, 6H/, 2,08 /м, 2H/, 1,20 /т, J = 6,0 Гц, 6H/. Элементный анализ для C27H29NO4S • 1,3 HCl
Рассчитано: C 63,46 H 5,98 N 2,74
Найдено: C 63,23 H 6,03 N 3,14
Пример 26. [6-Гидрокси-3-[4-[2-/1-N,N-диэтиламино/этокси] фенокси]-2-/4-гидроксифенил/]бензо[b]тиофенгидрохлорид

Т. плавления 128-131oC.1H ЯМР /ДМСО-d6/d: 9,81 /шир. с., 1H/, 9,76 /с, 1H/, 9,02 /с, 1H/, 7,49 /д, J = 9,0 Гц, 2H/, 7,28 /м, 1H/, 7,09 /д, J = 9,0 Гц, 1H/, 6,90 /с, 4H/, 6,79 /м, 3H/, 4,19 /м, 2H/, 3, 68 /м, 2H/, 3,50 /м, 2H/, 1,31 /м, 12H/. Элементный анализ для C28H31NO4S • 1,33 HCl:
Рассчитано: C 63,94 H 6,19 N 2,66
Найдено: C 63,82 H 6, 53 N 2,61
Пример 27. [6-Гидрокси-3-[4-[3-/пиперидино/пропокси]фенокси -2-/4-гидроксифенил/]бензо[b]тиофенгидрохлорид

Т. плавления 258-262oC.1H ЯМР /ДМСО-d6/ d: 9,85 /шир. с., 1H/, 9,81 /с, 1H/, 9,75 /с, 1H/, 7,50 /д, J = 9,0 Гц, 2H/, 7,27 /д, J = 2,0 Гц, 1H/, 7,10 /д, J = 9,0 Гц, 1H/, 6,87/с, 4H/, 6,80 /дд, J = 9,0, 2,0 Гц 1H/, 6,79 /д, J = 9,0 Гц, 2H/, 3,97 /т, J = 6,0 Гц, 2H/, 3,44 /м, 2H/, 3,15 /м, 2H/, 2,88 /м, 2H/, 2,11 /м, 2H/, 1,73 /м, 5H/, 1,39 /м, 1H/.
Элементный анализ для C28H29NO4S • 0,75 HCl:
Рассчитано: C 66,87 H 5,96 N 2,78
Найдено: C 67,04 H 5,90
N 2,68
В другом варианте, как представлено на схеме III, соединение примера 19, получают, используя метоксиметильные /МОМ/ защитные группы вместо метокси. Способы в точности аналогичны
использованным ранее, за исключением того, что МОМ группы удаляют на конечной стадии кислотным гидролизом.
Пример получения 10.
[6-Метокси-2-/4-метоксиметилоксифенил/-3-/4-бензилокси/фенокси] бензо[b]тиофен

Т. плавления 94-96oC1H ЯМР /ДМСО-d6/ d: 7,65 /д, J = 2,0 Гц, 1H/, 7,64 /д, J = 8,6 Гц, 2H/, 7,43-7,32 /м, 5H/, 7,23 /д, J = 8,8 Гц, 1H/, 7,08 /д, J = 8,6 Гц, 2H/, 7,04 /дд, J = 8, 8, 2,0 Гц, 1H/, 6,92 /кв. JAB = 9,2 Гц, 4H/, 5,26 /с, 2H/, 5,21 /с, 2H/, 5,01 /с, 3H/, 3,40 /с, 3H//, 3,37 /с, 3H/. Масс-спектр с полевой десорбцией: 528
Пример получения 11. [6-Метокси-2-/4-метоксиметилоксифенил/-3-/4-гидрокси/фенокси] бензо[b]тиофен
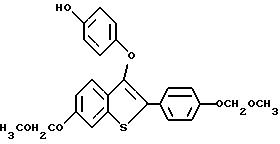
Т.плавления 90 - 91oC.1H ЯМР /ДМСО-d6/d: 9,15 /с, 1H/, 7,65 /д, J = 8,1 Гц, 2H/, 7,63 /д, J = 2,0 Гц, 1H/, 7,22 /д, J = 8,8 Гц, 1H/, 7,05 /дд, J = 8,8 Гц, 2,0 Гц, 1H/, 6,72 /кв. JAB = 9,1 Гц, 4H/, 5,26 /с, 2H/, 5,21 /с, 2H/, 3,40 /с, 3H/, 3,37 /с, 3H/. Масс-спектр с полевой десорбцией: 438.
Элементный анализ для C24H22O6S: C 65,74 H 5,06
Найдено: C 65,50 H 4,99
Пример 28. [6-Метокси-2-/4-метоксифенил/-3-бром]бензо[b]тиофен-/S оксид

К раствору [6-Метокси-2-/4-метоксифенил/-3-бром]бензо[b]тиофена /10,0 г, 28,6 ммоля/ в 50 мл безводного метиленхлорида добавляют 50 мл трифторуксусной кислоты. После перемешивания в течение 5 минут добавляют перекись водорода /4,0 мл, 28,6 ммоля, 30% водный раствор. Полученную смесь перемешивают при комнатной температуре в течение 2 часов. К темному раствору добавляют твердый бисульфит натрия /1,25 г/, а затем 15 мл воды. Полученную смесь интенсивно перемешивают в течение 15 минут, затем концентрируют в вакууме. Остаток разделяют между хлороформом и насыщенным раствором бикарбоната натрия /по 200 мл каждого/. Органический слой экстрагируют насыщенным раствором бикарбоната натрия. Затем органический слой сушат над сульфатом натрия и концентрируют в вакууме до получения твердого продукта, который тщательно растирают с этиловым эфиром/этилацетатом. После фильтрования получают 8,2 г /80%/ [6-метокси-2-/4-метоксифенил/-3-бром]бензо[b]тиофен-/S оксида/ в виде твердого вещества желтого цвета, которое можно перекристаллизовывать из этилацетата. Т. плавления 170 - 173oC.1H ЯМР /ДМСО-d6/d: 7,24 /д, J = 2,2 Гц, 1H/, 7,68 /д, J = 8,8 Гц, 2H/, 7,54 /д, J = 8,5 Гц, 1H/, 7,26 /дд, J = 8,5, 2,2 Гц, 1H/, 7,10 /д, J = 8,8 Гц, 2H/, 3,86 /с, 3H/, 3,80 /с, 3H/.
Элементный анализ для C16H13O3SBr:
Рассчитано: C 52,62 H 3,59
Найдено: C 52,40 H 3,55
Пример 29. Аналогичным способом
получают [2-/4-метоксифенил/-3-бром]бензо[b]тиофен-/S оксид/

Т.плавления 120-125o C.1H ЯМР /ДМСО-d6/d: 8,06 /д, J = 7,6 Гц, 1H/, 7,78 - 7,59 /м, 5H/, 7,13 /д, J = 8,7 Гц, 2H/, 3,81 /с, 3H/. Масс-спектр с полевой десорбцией: 335. Элементный анализ для C15H11O2SBr:
Рассчитано: C 53,75 H 3,31.
Найдено: C 53,71 H 3,46
Пример получения 12. Получение 4-[2-/1-пиперидинил/этокси]фенола

К раствору 4-бензилоксифенола /50,50 г, 0,25 моля/ в 350 мл безводного ДМФ добавляют 2-хлорэтилпиперидин /46,30 г, 0,25 моля/. После десятиминутного перемешивания добавляют карбонат калия /52,0 г 0,375 моля/ и карбонат цезия /85,0 0,25 моля/. Полученную гетерогенную смесь интенсивно перемешивают при комнатной температуре в течение 48 часов. Затем реакционную смесь выливают в 500 мл воды и экстрагируют метиленхлоридом. Затем органическую часть экстрагируют несколько раз 1н. гидроксидом натрия, и, наконец, промывают рассолом. Затем органический слой сушат над сульфатом натрия и концентрируют в вакууме до масла. В результате хроматографической обработки /SiO2 1: 1 гексаны/этилацетат/ получают 60,0 г /77%/ 4-/2-/1-пиперидинил/-этокси/феноксибензиловый эфир в виде бесцветного масла.
1H ЯМР /ДМСО-d6/d: 7,40 - 7,27 /м, 5H/, 6,
84 /кв. JAB = 11,5 Гц, 4H/, 4,98 /с, 2H/, 3,93 /т, J = 6,0 Гц, 2H/, 2,56 /т, J = 6,0 Гц, 2H/, 2,35 - 2,37 /м, 4H/, 1,48 - 1,32 /м, 6H/. Масс-спектр с полевой десорбцией: 311. Элементный
анализ для C20H25NO2:
Рассчитано: C 77,14 H 98,0 N 4,50.
Найдено: C 77,34 H 8,18 N 4,64
4-[2-/1-пиперидинил/этокси]
феноксибензиловый эфир /21,40 г, 68,81 моля/ растворяют в 200 мл 1:1 EtOH:EtOAc, содержащего 1% конц. HCl. Этот раствор переносят в склянку Парра и добавляют 3,4 г 5% палладия-на-угле. Полученную
смесь гидрируют при давлении 40 пси /2,81 кг/см2/, в течение 2 часов, а затем пропускают через слой Целита для удаления катализатора. Полученный фильтрат концентрируют в вакууме до
получения твердого вещества, которое затем суспендируют в этиловом эфире, и фильтруют до получения 12,10 г /83%/ 4-[2-/1-пиперидинил/этокси] фенола. Т.плавления 148 - 150oC.1H
ЯМР /ДМСО-d6/d: 8,40 /с, 1H/, 6,70 /кв. JAB = 11,5 Гц, 4H/, 3,93 /т, J = 6,0 Гц, 2H/, 2,59 /т, J = 6,0 Гц, 2H/, 2,42 - 2,38 /м, 4H/, 1,52 - 1,32 /м, 6H/. Масс-спектр с полевой
десорбцией: 221. Элементный анализ для C13H19NO2.
Рассчитано: C 70,56 H 8,09 N 4,50
Найдено: C 70,75 H 8,59 N 6,54.
Пример
30. [6-Метокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2- /4-метоксифенил/]-бензо[b]тиофен/S-оксид/

К раствору 4-[2-/1-пиперидинил/этокси]фенола /0,32 г, 1,43 ммоля/ в 5 мл безводного ДМФ при комнатной температуре добавляют гидрид натрия /0,57 г, 1,43 ммоля, 60% дисперсии в минеральном масле/. После перемешивания в течение 15 минут [6-метокси-2-/4-метоксифенил/-3-бром]бензо[b]тиофен/S-оксид/ /0,50 г, 1,37 ммоля/ добавляют небольшими порциями. После перемешивания в течение 1 часа о завершении реакции судят по данным ТСХ. Растворитель удаляют в вакууме, а остаток разделяют между водой и 10% этанол/этилацетатом. Органическую часть несколько раз промывают водой, а затем сушат над сульфатом натрия. После концентрирования в вакууме получают масло, которое тщательно растирают в этилацетатом/гексанами до получения 0,62 г /89%/ [6-метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2- /4-метоксифенил/]-бензо[b] тиофен/S-оксида/ в виде твердого вещества светло-желтого цвета. Т.плавления 97 - 100oC.1H ЯМР /ДМСО-d6/d: 7,68 /д, J = 2,1 Гц, 1H/, 7, 62 /д, J = 8,8 Гц, 2H/, 7,06 - 6,92 /м, 6H/, 6,85 /д, J = 8,8 Гц, 2H/, 3,94 /т, J = 6,0 Гц, 2H/, 3,81 /с, 3H/,
3,72 /с, 3H/, 2,56 /т, J = 6,0 Гц, 2H/, 2,39 - 2,32 /м, 4H/, 1,47 - 1,32 /м, 6H/. Элементный анализ для C29H31NO5S :
Рассчитано: C 68,89 H 6,18 N 2,77
Найдено: C 68,95 H 6,04 N 2,57
Пример 31. Аналогичным способом получают [3-[4-[2-/1-пиперидинил/этокси] фенокси]-2-/4-метоксифенил/] бензо[b]тиофен/S-оксид/

Масло.1H ЯМР /ДМСО-d6/d : 8,03 /м, 1H/, 7,65 /д, J = 8,7 Гц, 2H/, 7,53 - 7,50 /м, 2H/, 7,09 - 6,82 /с, 7H/, 3,94 /шир. т., J = 5,9 Гц, 2H/, 3,74 /с, 3H/, 2,56 /шир. т., J = 5,9 Гц, 2H/, 2,36 - 2,33 /м, 4H/, 1,56 - 1,31 /м, 6H/. Масс-спектр с полевой десорбцией: 475. Элементный анализ для C28H29NO4S : C 70,71 H 6,15 N 2,94. Найдено: C 70,44 H 6,43 N 3,20.
Пример 32. [6-Метокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2- /4-метоксифенил/]-бензо[b]тиофенгидрохлорид

К раствору 6-метокси-[3-[4-[2-/1-пиперидинил/этокси] фенокси]-2-/4-метоксифенил/] бензо[b]тиофен/S-оксида/ пример 30/ /3,00 г, 5,94 ммоля/ в 200 мл безводного ДГФ в атмосфере азота при 0oC добавляют литийалюминийгидрид /0,34 г 8,91 ммоля/ небольшими порциями. После десятиминутного перемешивания реакцию гасят, осторожно добавляя 5,0 мл 2,0 н. гидроксида натрия. Полученную смесь интенсивно перемешивают в течение 30 минут и добавляют дополнительно 2,0 н. гидроксид натрия для растворения солей. Затем полученную смесь распределяют между водой и 10% гидроксидом натрия. Слои разделяют и водный несколько раз экстрагируют 10% этанол/этилацетатом. Органический слой сушат над сульфатом натрия и концентрируют в вакууме до масла. Неочищенный продукт растворяют в 50 мл 1:1 этилацетат/этиловый эфир и обрабатывают избытком этилэфиргидрохлорида. Полученный осадок собирают и сушат до получения 2,98 /96%/ [6-метокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2- /4-метоксифенил/] -бензо[b]тиофенгидрохлорида в виде белого твердого вещества.
Соединение примера 6 получают из соединения примера 31 тем же способом.
Пример
получения 13. 6-Метоксибензо[b]тиофен-2-бороновая кислота

К раствору 6-метоксибензо[b] тиофена /18,13 г, 0,111 моля/ в 150 мл безводного тетрагидрофурана при -60oC добавляют н-бутиллитий /76,2 мл, 0,122 моля, 1,6 М раствор в гексанах/ по каплям через шприц. После перемешивания в течение 30 минут через шприц вводят триизопропилборат /28,2 мл, 0,122 моля/. Полученную смесь оставляют постепенно нагреваться до 0oC, а затем разделяют между 1 н. соляной кислотой и этилацетатом /300 мл каждого/. Органический слой сушат над сульфатом натрия. После концентрирования в вакууме получают твердый продукт белого цвета, который тщательно растирают с этиловым эфир/гексанами. После фильтрования получают 16,4 г /71%/ 6-метоксибензо[b] тиофен-2-бороновой кислоты в виде твердого вещества белого цвета. Т. плавления 200oC /с разложением/.
1H ЯМР /ДМСО-d6/d: 7,83 /c, 1H/, 7,78 /д, J = 8,6 Гц, 1H/, 7,51 /д, J = 2,0 Гц, 1H/, 6,97 /дд, J = 8,6, 2,0 Гц, 1H/, 3,82 /с, 3H/. Масс-спектр с полевой десорбцией: 208.
Пример получения 14. [6-Метокси-2-/4-метансульфонилоксифенил/]бензо[b] тиофен

К раствору 6-метоксибензо[b] тиофен-2-бороновой кислоты /3,00 г, 14,4 ммоля/ в 100 мл толуола добавляют 4-/матансульфонилокси/фенилбромид/ 3,98 г, 15,8 ммоля/, а затем 16 мл 2,0 н раствора карбоната натрия. После десятиминутного перемешивания добавляют тетракистрифенилфосфинпалладия /0,60 г, 0,52 моля/ и полученную смесь нагревают до кипения с обратным холодильником 5 часов. Затем реакционную смесь оставляют охлаждаться до комнатной температуры, после чего продукт осаждается из органической фазы. Водную фазу удаляют, а органический слой концентрируют в вакууме до получения твердого продукта. После тщательного растирания в этиловом эфире получают твердый продукт, который отфильтровывают и сушат в вакууме до получения 3,70 г /77%/ [6-метокси-2-/4-метансульфонилоксифенил/] бензо[b] тиофена в виде продукта желто-коричневого цвета. Т. плавления 197 -201oC.1H ЯМР /ДМСО-d6/d: 7,81-7,77 /м, 3H/, 7,71 /д, J = 8,8 Гц, 1H/, 7,54 /д, J = 2,3 Гц, 1H/, 7,40 /д, J = 8,7 Гц, 2H/, 6,98 /дд, J = 8,7, 1,5 Гц, 1H/, 3,80 /с, 3H/, 3,39 /с, 3H/. Масс-спектр с полевой десорбцией: 334. Элементный анализ для C16H14O4S2:
Рассчитано: C 57,46 H 4,21
Найдено: C 57,76 H 4,21.
Пример
получения 15. Аналогично примеру получения 14 получают [6-Метокси-2-/4-бензилоксифенил/]бензо[b]тиофен
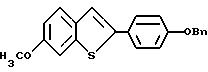
Выход 73%. Т. плавления 217 - 221oC.1H ЯМР /ДМСО-d/ d: 7,63 - 7,60 /м, 3H/, 7,59 - 7,26 /м, 7H/, 7,02 /д, J = 8,7 Гц, 2H/, 6,96 /дд, J = 8,8, 2,2 Гц, 1H/, 5,11 /с, 2H/, 3,88 /с, 3H/. Масс-спектр с полевой десорбцией: 346. Элементный анализ для C22H18O2S:
Рассчитано: C 76,27 H 5,24
Найдено: C 76,00 H 5,25.
Пример получения 16. [6-Гидрокси-2-/4-метансульфонилоксифенил/]бензо[b] тиофен

К раствору [6-метокси-2-/4-метансульфонилоксифенил/]бензо[b]тиофен /9,50 г, 28,40 ммоля/ в безводном метиленхлориде /200 мл/ при комнатной температуре в атмосфере азот добавляют трехбромистый бор /14,40 г, 5,36 мл, 56,8 ммоля/. Полученную смесь перемешивают при комнатной температуре в течение 3 часов. Реакцию гасят, медленно выливая реакционную смесь в избыток ледяной воды. После тридцатиминутного интенсивного перемешивания белый осадок собирают фильтрованием, несколько раз промывают водой, а затем сушат в вакууме до получения 8,92 /98%/ [6-гидрокси-2-/4-метансульфонилоксифенил/]бензо[b]тиофена в виде твердого вещества белого цвета. Т. плавления 239 - 243oC.
1H ЯМР /ДМСО-d6/d: 9,70 /с, 1H/, 7,76 /д, J = 8,7 Гц, 2H/, 7,72 /с, 1H/, 7,62 /д, J =
8,7, 1H/, 7,38 /д, J = 8,7 Гц, 2H/, 7,24 /д, J = 1,7 Гц, 1H/, 6,86 /дд, J = 8,7, 1,7 Гц, 1H/, 3,38 /с, 3H/. Масс-спектр с полевой десорбцией 320. Элементный анализ для C15H12
O4S2:
Рассчитано: C 56,23 H 3,77
Найдено: C 56,49 H 3,68
Пример получения 17. [6-Бензилокси-2-/4-метансульфонилоксифенил/]бензо[b]тиофен

К раствору [6-гидрокси-2-/4-метансульфонилоксифенил/] бензо[b] тиофена /3,20 г, 10,0 ммоля/ в 75 мл безводного ДМФ добавляют C2CO3 /5,75 г, 17,7 ммоля/, а затем бензилхлорид /1,72 мл, 11,0 ммоля/. Полученную смесь интенсивно перемешивают в течение 24 часов. Растворитель удаляют в вакууме, а твердый остаток суспендируют в 200 мл воды. Белый осадок собирают фильтрованием и несколько раз промывают водой. После сушки в вакууме неочищенный продукт суспендируют в смеси 1:1 гексаны:этиловый эфир. Твердую часть собирают до получения 3,72 г /91%/ [6-бензилокси-2-/4-метансульфонилоксифенил/] бензо[b] тиофена в виде твердого вещества белого цвета. Т. плавления 198 - 202oC.1H ЯМР /ДМСО-d6/d: 7,81 - 7,78 /м, 3H/, 7,72 /д, J = 8,7 Гц, 1H/, 7,64 /д, J = 2,2 Гц, 1H/, 7,47 - 7,30 /м, 7H/, 5,15 /с, 2H/, 3,39 /с, 3H/. Масс-спектр с полевой десорбцией: 410.
Пример получения 18. [6-Бензилокси-2-/4-гидроксифенил/]бензо[b]тиофен

К раствору [6-бензилокси-2-/4-метансульфонилоксифенил/]бензо[b]тиофена /12,50 г, 30,50 ммоля/ в 300 мл безводного ТГФ в атмосфере азота при комнатной температуре добавляют литийалюминийгидрид /2,32 г, 61,0 ммоля /небольшими порциями. Затем полученную смесь перемешивают при комнатной температуре в течение 3 часов а затем ее гасят, осторожно выливая в избыток холодной 1,0 и соляной кислоты. Водную фазу экстрагируют этилацетатом. Органическую часть несколько раз промывают водой, а затем сушат над сульфатом натрия и концентрируют в вакууме до получения твердого вещества. После хроматографической обработки /двуокись кремния, хлороформ/ получают 8,75 г /87%/ [6-бензилокси-2-/4-гидроксифенил/]бензо[b]тиофена в виде твердого вещества белого цвета. Т. плавления 212 - 216oC.1H ЯМР /ДМСО-d6/d: 9,70 /с, 1H/, 7,63 /д, J = 8,7 Гц, 1H/, 7,56 /д, J = 2,2 Гц, 1H/, 7,51 - 7,30 /м, 8H/, 7,00 /дд, J = 8,7, 2,2 Гц, 1H/, 6,80 /д, J = 8,6 Гц, 2H/, 5,13 /с, 2H/. Масс-спектр с полевой десорбцией 331. Элементный анализ для C21H16O2S:
Рассчитано: C 75,88 H 4,85
Найдено: C 75,64 H 4,85.
Пример получения 19. [6-бензилокси-2-/4-метоксифенил/]бензо[b]тиофен

К раствору [6-бензилокси-2-/4-гидроксифенил/] бензо[b]тиофена /8,50 г, 26,40 ммоля/ в 200 мл безводного ДМФ в атмосфере азота при комнатной температуре добавляют гидрид натрия /1,66 г, 41,5 ммоля/ небольшими порциями. После того, как прекращается выделение газа, прикапывают иодометан /3,25 мл, 52,18 ммоля/. Реакционную смесь перемешивают в течение 3 часов при комнатной температуре. Затем растворитель удаляют в вакууме, а остаток разделяют между водой и этилацетатом. Органическую фазу несколько раз промывают водой. Затем органическую часть сушат над сульфатом натрия и концентрируют в вакууме до получения 9,00 г /98%/ [6-бензилокси-2-/4-метоксифенил/]бензо[b]тиофена в виде твердого вещества белого цвета Т. плавления 180 - 185oC.1H ЯМР /ДМСО-d6/d: 7,67 - 7,58 /м, 5H/, 7,46 - 7,29 /м, 5H/, 7,02 /дд, J = 8,8, 2,2 Гц, 1H/, 6,98 /д, J = 8,7 Гц, 2H/, 5,13 /с, 2H/, 3,76 /с, 3H/. Масс-спектр с полевой десорбцией: 346. Элементный анализ для C22H18O2S:
Рассчитано: C 76,27 H 5,24.
Найдено: C 76,54 H 5,43.
Пример получения 20. [6-Бензилокси-2-/4-метоксифенил/]-3-бром]бензо[b] тиофен

[6-Бензилокси-2-/4-метоксифенил/] бензо[b] тиофен /10,0 г, 28,9 ммоля/ помещают в 200 мл хлороформа с 10,0 г твердого бикарбоната натрия при комнатной температуре. К этой суспензии добавляют бром /1,50 мл, 29,1 ммоля/ по каплям за 30 минут в виде раствора в 100 мл хлороформа. После завершения добавления, добавляют 200 мл воды и слои разделяются. Органическую фазу сушат над сульфатом натрия и концентрируют в вакууме до получения твердого вещества белого цвета. После кристаллизации из смеси метиленхлорид/метанол получают 10,50 г /84%/ [6-бензилокси-2-/4-метоксифенил/]-3-бром]бензо[b]тиофена в виде твердого вещества белого цвета. Т. плавления 146 - 150o C.1H ЯМР /ДМСО-d6/d: 7,70 /д, J = 2,2 Гц, 1H/, 7,65 - 7,60 /м, 3H/, 7,47 - 7,30 /м, 5H/, 7,19 /дд, J = 8,8, 2,2 Гц, 1H/, 7,06 /д, J = 8,7 Гц, 2H/, 5,17 /с, 2H/, 3,78 /с, 3H/. Масс-спектр с полевой десорбцией: 346.
Элементный анализ для C22H17O2SBr:
Рассчитано: C 62,13 H 4,03.
Найдено: C 61,87 H 4,
00
Пример получения 21. Аналогичным способом получают [6-метокси-2-/4-бенсизоксифенил/]-3-бром]бензо[b]тиофен

Выход 91%. Т. плавления 125 - 127oC.1H ЯМР /ДМСО-d6/d: 7,64 - 7,61 /м, 4H/, 7,46-7,31 /м, 5H/, 7,15-7,09 /м, 3H/, 5,15 /с, 2H/, 3,82 /с, 3H/. Масс-спектр с полевой десорбцией: 346.
Элементный анализ для C22H17O2SBr:
Рассчитано: C 62,13 H 4,03
Найдено: C
62,33 H 3,93.
Аналогично способу примера 28 получают соединения примеров 33 - 34.
Пример 33. [6-Бензилокси-2-/4-метоксифенил/]-3-бром]бензо[b]тиофен-/S-оксид/

Выделен в виде твердого вещества желтого цвета за счет кристаллизации из этилацетата. Т. плавления 202 - 205oC.1H ЯМР /ДМСО-d6/d: 7,80 /д, J = 2,2 Гц, 1H/, 7,68 /д, J = 8,7 Гц, 2H/, 7,55 /д, J = 8,4 Гц, 1H/, 7,47 - 7,32 /м, 6H/, 7,10 /д, J = 8,7 Гц, 2H/, 5,23 /с, 2H/, 3,80 /с, 3H/. Масс-спектр солевой десорбцией: 441. Элементный анализ для C22H17O3SBr,
Рассчитано: C 59,87 H 3,88.
Найдено: C 59,59 H 3, 78.
Пример 34. [6-Метокси-2-/4-бензилоксифенил/]-3-бром]бензо[b]тиофен-/S-оксид/

Выделен в виде твердого вещества желтого цвета за счет хроматографической обработки /двуокись кремния, CHCl3/. Т. плавления 119-123oC1H ЯМР /ДМСО-d6 /d: 7,73 /д, J= 2,2 Гц, 1H/, 7,68 /д, J=8,8 Гц, 2H/, 7,55 /д, J=8,5 Гц, 1H/, 7,46 - 7,31 /м, 5H/, 7,26 /дд, J = 8,5, 2,2 Гц, 1H/, 7,18 /д, J= 8,8 Гц, 2H/, 5,16 /с, 2H/, 3,86 /с, 3H/. Масс-спектр с полевой десорбцией: 441. Элементный анализ для C22H17O3SBr:
Рассчитано: C 59,87 H 3,88.
Найдено: C 60,13 H 4,10.
Аналогично способу примера 30 получают соединения примеров 35-36.
Пример 35. [6-бензилокси-3-[4-[2-/1-пиперидинил/этокси] фенокси]-2-/4- метоксифенил/]бензо[b]тиофен-/S-оксид/

Желтое масло.1H ЯМР /ДМСО-d6/d: 7,76 /д, J = 2,2 Гц, 1H/, 7,62 /д, J= 8,8 Гц, 2H/, 7,44-7,30 /м, 5H/, 7,12 /дд, J = 8,6, 2,2 Гц, 1H/, 7,03-6,93 /м, 5H/, 6,85 /д, J = 8,8 Гц, 2H/, 5,18 /с, 2H/, 3,94 /шир.т., J = 5,8 Гц, 2H/, 3,73 /с, 3H/, 2,56 /шир.т.J = 5,8 Гц, 2H/, 2,37 - 2,34 /м. 4H/, 2,37-2,34 /м, 4H/, 1,45 - 1,32 /м, 6H/. Масс-спектр с полевой десорбцией: 592. Элементарный анализ для C35H35NO5S:
Рассчитано: C 72,26 H 6,06 N 2,41.
Найдено: C 72,19 H 5,99 N 2,11
Пример 36. [6-Метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2- /4-бензилоксифенил/]бензо[b]тиофен-/S-оксид/

Твердое вещество желтого цвета. Т.плавления 89-93oC.1H ЯМР /ДМСО-d6d: 7,68/ д, J = 2,2 Гц, 1H/, 7,62 /д, J = 8,8 Гц, 2H/, 7,42 - 7,28 /м, 5H/, 7,08-6,92 /м, 6H/, 6,86 /д, J=8,8 Гц, 2H/, 5,09 /с, 2H/, 3,94 /шир.т, J = 5,8 Гц, 2H/, 3,81 /с, 3H/, 2,56 /шир., J = 5,8 Гц, 2H/, 2,37-2,34 /м, 4H/, 1,45 - 1,31 /м, 6H/. Масс-спектр с полевой десорбцией 592. Элементный анализ для C35H35NO5S. 0,25 EtOAc:
Рассчитано: C 71,62 H 6,18 N 2,32.
Найдено: C 71,32 H 5,96 N 2,71
аналогично способу примера 11 получают соединения примеров 37-38.
Пример 37. [6-Бензилокси-3-[4-[2-/1-пиперидинил/этокси] фенокси]
-2-/метоксифенил/] бензо[b]тиофен

Выделен с 95%-ным выходом, исходя из [6-бензилокси-2-/4-метоксифенил/-3-бром]бензо[b]тиофен-/S-оксида/. После хроматографической очистки /Двуокись кремния, 1-5% метанол/хлороформ/ получают грязно-белое твердое вещество. Т. плавления 105-108oC.1H ЯМР /ДМСО-d6/d: 7,62 /д, J = 2,2 Гц, 1H/, 7,59 /д, J = 8,8 Гц, 2H/, 7,45-7,30 /м, 5H/, 7,15 /дд, J = 8,6 Гц, 1H/, 7,00-6,94 /м, 3H/, 6,82 /с, 4H/, 5,13 /с, 2H/, 3,92 /шир.т.к, J = 5,8 Гц, 2H/, 3,72 /с, 3H/, 2,55 /шир.т. J = 5,8 Гц, 2H/, 2,37-2,34 /м, 4H/, 1,44 - 1,31 /м, 4H/. Масс-спектр с полевой десорбцией 565. Элементный анализ для C35 H35NO4S:
Раасчитано: C 74,31 H 6,24 N 2,48.
Найдено: C 74,35 H 6,07 N 2,76.
Пример 38.
[6-Метокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2- /4-бензилоксифенил/]бензо[b]тиофен

Выход 91%. Т. плавления 106-110oC.1H-ЯМР /ДМСО-d6/d: 7,59 /д, J = 8,8 Гц, 2H/, 7,54 /д, J = 2,2 Гц, 1H/, 7,42-7,28 /м, 5H/, 7,13 /д, J = 8,8 Гц, 1H/, 7,03 /д, J=8,8 Гц, 2H/, 6,82 /с, 4H/, 5,08 /с, 2H/, 3,92 /шир.т. J = 5,8 Гц, 2H/, 3,78 /c, 3H/, 2,55 /шир.т.J = 5,8 Гц, 2H/, 2,37 - 2,33 /м, 4H/, 1,44 - 1,31 /м, 4H/. Масс-спектр с полевой десорбцией 565.
Элементный анализ для C35H35N)4S:
Рассчитано: C 74,31 H 6,24 N 2,48
Найдено: C 74,25 H 6,17 N 2,73
Пример 39.
[6-Гидрокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2- /метоксифенил/]бензо[b]тиофен

К раствору [6-бензилокси-3-[4-[2-/1-пиперидинил/этокси] фенил]-2- /4-метоксифенил/] бензо[b] тиофена /8,50 г, 15,0 ммоля/ в 300 мл смеси 5:1 этанол/этилацетата добавляют палладиевую чернь /1,50 г/, формат аммония /3,50 г, 55,6 ммоля/ и 30 мл воды. Полученную смесь нагревают до кипения с обратным холодильником и за ходом реакции следят с помощью ТСХ. Примерно через 3 часа считают, что реакция завершилась и полученный раствор охлаждают до комнатной температуры. Реакционную смесь фильтруют через слой Целита для удаления катализатора, а полученный фильтрат концентрируют в вакууме до твердого вещества. Этот концентрат разделяют между насыщенным раствором бикарбоната натрия и 5% этанол/этилацетатом. Органическую фазу сушат над сульфатом натрия и концентрируют в вакууме. Неочищенный продукт обрабатывают хроматографически /двуокись кремния, 1-5% метанол/хлороформ/ до получения 6,50 г /91%/ [6-гидрокси-3-[4- [2-/1-пиперидинил/этокси]фенокси]-2-/4-метоксифенил/] бензо[b] - тиофена в виде пены, которая превращается в твердое соединение после тщательного растирания с гексанами. Т.плавления 174-176oC1H ЯМР /ДМСО-d6/d: 9,77 /с, 1H/, 7,56 /в, J = 8,8 Гц, 2H/, 7,23 /д, J = 2,0 Гц, 1H/, 7,07 /д, J = 8,6 Гц, 1H/, 6,93 /д, J = 8,8 Гц, 2H/, 6,81 /с, 4H/, 6,76 /дд, J = 8,7, 2,0 Гц, 1H/, 3,91 /шир.т. J = 5,9 Гц, 2H/, 3,71 /с, 3H/, 2,55 /шир. т. J = 5,9 Гц, 2H/, 2,38 - 2,33 /м, 4H/, 1,46 - 1,28 /м, 6H/. Масс-спектр с полевой десорбцией 475.
Элементный анализ:
Рассчитано для C28H29NO4S: C 70,71 H
6,15 N 2,94.
Найдено: C 70,46 H 5,93 N 2,71.
Пример 40. Аналогично способу примера 39 получают [6-метокси-3-[4-[2-/1-пиперидинл/этокси]фенокси]-2-/4-гидроксифенил/]
бензо[b]тиофен

Выход = 88%. Т. плавления 148-150oC.1H ЯМР /ДМСО-d6/d: 9,72 /с, 1H/, 7,51 /д, J = 2,0 Гц, 1H/, 7,48 /д, J = 8,6 Гц, 2H/, 7,11 /д, J = 8,8 Гц, 1H/, 6,88 /дд, J = 8,8, 2,2 Гц, 1H/, 6,81 /с, 4H/, 6,76 /д, J = 8,6 2H/, 3,91 /шир., т., J = 5,9 Гц, 2H/, 3,77 /с, 3H/, 2,55 /шир.т. J = 5,9 Гц, 2H/, 2,38 - 2,33 /м, 4H/, 1,46 - 1,28 /м, 6H/. Масс-спектр с полевой десорбцией: 475. Элементный анализ. Рассчитано для C28H29NO4S:
C 70,71 H 6,15 N 2,94
Найдено: C 71,00 H 6,17 N 2,94.
В другом варианте соединения примеров 39 и 40 можно получить непосредственно тем же способом трансфергидрогенолиза с 90% выходом из [6-метокси-3-[4-[2-/1-пиперидинл/этокси]фенокси]-2-2/4-бензилоксифенил/] бензо[b] тиофен-/S-оксида/ и [6-бензилокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4- метоксифенил/]бензо[b]-тиофен-/S-оксида/, соответственно.
Пример 41. [6-Гидрокси-3-[4-[2-/1-пиперидинл/этокси]фенокси]-2- /4-метоксифенил/] бензо[b] тиофен /пример 39/ превращают в гидрохлорид с 85%-ным выходом за счет обработки этиловый эфир/гидрохлоридом в этилацетате с последующей кристаллизацией из этанол/этилацетата.

Т. плавления 156-160oC1H ЯМР /ДМСО-d6/d: 10,28 /шир.с., 1H/, 9,85 /с, 1H/, 7,56 /д, J = 8,8 Гц, 2H/, 7,25 /д, J = 2,0 Гц, 1H/, 7,06 /д, J = 8,7 гц, 1H/, 6,93 /д, J = 8,8 Гц, 2H/, 6,87 /кв, JAB = 9,3 Гц, 4H/, 4,27 /шир., т., J = 5,9 Гц, 2H/, 3,71 /с. 3H/, 3,44 - 3,31 /м, 4H/, 2,98 - 2,88 /м, 2H/, 1,74 - 1,60 /м, 5H/, 1,36 - 1,29 /м, 1H/. Масс-спектр с полевой десорбцией: 475.
Элементный анализ для C28H29NO4S. 1,0 HCl
Рассчитано: C 65,68 H 5,90 N 2,73.
Найдено: C 65,98 H 6,11 N 2, 64.
Пример 42. Аналогично способу примера 41 получают [6-гидрокси -3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4- метоксифенил/] бензо[b]тиофенгидрохлорид

Т.плавления 215 - 217oC.1H ЯМР /ДМСО-d6/d: 10,28 /шир.с., 1H/, 9,80 /с, 1H/, 7,52 /д, J = 2,2 Гц, 1H/, 7,47 /д, J = 8,6 Гц, 2H/, 7,12 /д, J = 8,4 Гц, 1H/, 6,91 - 6,80 /м, 5H/, 6,78 /д, J = 8,6 Гц, 2H/, 4,27 /шир., т., J = 5,8 Гц, 2H/, 3,78 /с, 3H/, 3,43 - 3,34 /м, 4H/, 2, 97 - 2,91 /м, 2H/, 1,78 - 1,61 /м, 5H/, 1,36 - 1,29 /м, 1H, Масс-спектр с полевой десорбцией: 475.
Элементный анализ для Ct28H29NO4S•1,0 HCl
Рассчитано: C 65,68 H 5,90 N 2,73
Найдено: C 65,87 H 5,79 N 2,99.
Пример 43. [6-Бензилокси-3-[4-[2-/1-пиперидинил/этокси]
фенокси]-2-/4- бензоилоксифенил/]бензо[b]тиофенгидрохлорид

К раствору примера 20 /0,50 г, 1, 08 ммоля/ в 20 мл безводного тетрагидрофурана при 0oC добавляют триэтиламина /1,00 мл/. К этой смеси добавляют бензоилхлорид /0,28 мл, 2,35 ммоля/. После перемешивания в течение 2 часов при 0oC реакцию гасят, распределяя между этилацетатом/ насыщенным раствором бикарбоната натрия /по 100 мл каждого/. Слои разделяют и органику сушат над сульфатом натрия и концентрируют в вакууме до твердого вещества белого цвета. Неочищенный продукт растворяют в 10 мл этилацетата и обрабатывают этиловый эфир соляной кислотой. Образуется белый осадок, который собирают фильтрованием. После сушки получают 390 мг /50%/ [6-бензилокси-3-[4-[2-/1-пиперидинил/этокси] фенокси]-2-/4- бензилоксифенил/]бензо[b]-тиофенгидрохлорид в виде твердого вещества белого цвета. Т.плавления 200-204oC1H ЯМР /ДМСО -d6/d: 9,95 /шир.с, 1H/, 8,16 /м, 2H/, 8,12 /дд, J = 10,0, 2,0 Гц, 2H/, 7,87 /дд, J = 7,0, 2,0 Гц, 2H/, 7,78 /м, 2H/, 7,64 /м, 2H/, 7,42 /д, J = 7,0 Гц, 2H/, 7,34 /дд, J = 8,0, 2,0 Гц, 1H/, 7,00 /с, 4H/, 4,32 /м, 2H/, 3,45 /м, 4H/, 2,99 /м, 2H/, 1,75 /м, 5H/, 1,39 /м, 1H/.
Элементный анализ для C41H35NO6S • 1,5 HCl:
Рассчитано C 67,97 H 5,08 N 1,93.
Найдено: C 68,05 H 5,25 N 2,01
Таким же образом получают:
Пример 44.
[6-Этилсульфонилокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4-этилсульфонилоксифенил/]бензо[b]тиофенгидрохлорид
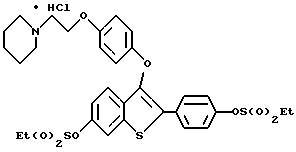
Выход = 72%. Т.плавления 110-115oC.1H ЯМР /ДМСО-d6/d: 10,15 /шир.с., 1H/, 8,15 /д, J = 2,0 Гц, 1H/, 7,85 /д, J = 7,0 Гц, 2H/, 7,43 /м, 3H/, 7,34 /дд, J = 9,0, 2,0 Гц, 1H/, 6,97 /м, 4H/, 4,31 /м, 2H/, 3,57 /м, 4H/, 3,44 /м, 4H/, 2,97 /м, 2H/, 1,76 /м, 5H/, 1,40 /м, 7H/. Элементный анализ для C31H35NO8S3 • 1,5 HCl
Рассчитано: C 54,57 H 5,32 N 2,05.
Найдено: C 54,36 H 5,37 N 2,05.
Аналогичным способом, используя
трифторметансульфоновый ангидрид получают:
Пример 45. [6-Метокси-3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4- трифторметансульфонилоксифенил/]бензо[b]тиофен

Выход = 81%. Масло.1H ЯМР /ДМСО-d6/d: 7,82 /д, J = 8,7 Гц, 2H/, 7,60 /д, J = 2,0 Гц, 1H/, 7,54 /д, J = 8,7 Гц, 2H/, 7,17 /д, J = 8,8 Гц, 1H/, 6,93 /дд, J = 8,8, 2,0 Гц, 1H/, 6,84 /с, 4H/, 3,92 /шир.т., J = 5,7 Гц, 2H/, 3,79 /с, 3H/, 2,56 /шир., т., J = 5,7 Гц, 2H/, 2,36 - 2,30 /м, 4H/, 1,44 - 1,31 /м, 6H/. Масс-спектр с полевой десорбцией. 607. Элементный анализ для C29H28NO6F3S2:
Рассчитано: C 57,32 H 4,64 N 2,30.
Найдено: C 57,16 H 4,52 N 2,01
Из примера 1 аналогичным способом получают:
Пример 46. [3-[4-[2-/1-Пиперидинил/этокси]фенокси]-2-/4-бензилоксифенил/]бензо [b]тиофенгидрохлорид

Выход 85%. Т. плавления 190-198oC.1H ЯМР /ДМСО-d6/d: 10,48 /шир.ст., 1H/, 8,00-8,10 /м, 2H/, 7,80 - 8,00 /м, 3H/, 7,60 - 7,53 /м, 4H/, 7,40 - 7,56 /м, 6H/, 6,93 /c, 2H/, 4,37 - 4,43 /м, 2H/, 3,0 - 3,05 /м, 2H/, 2,53 - 2,63 /м, 6H/, 1,75 - 1,95 /м, 3H/, 1,40 - 1,50 /м, 1H/. Масс-спектр с полевой десорбцией: 550.
Элементный анализ для C34H31NO4S • 1,0 HCl:
Рассчитано: C 74,29 H 5,68 N 2,55.
Найдено: C 74,52 H 5,80 N 2,59.
Пример 47. [3-[4-[2-/1-Пиперидинил/этокси]фенокси]-2-/4- пивалоилоксифенил/]бензо[b]тиофенгидрохлорид

Выход = 90%. Т.плавления 193-197oC.1H ЯМР /ДМСО-d6/d: 10,10 /шир.с., 1H/, 8,12 /д, J = 8,0 Гц, 1H/, 1H/, 7,85 /д, J = 8,6 Гц, 1H/, 7,40 - 7,53 /м, 3H/, 7,15 /д, J = 6,7 Гц, 2H/, 7,00 /с, 5H/, 4,33 - 4,40 /м. 2H/, 3,45 - 3,60 /м, 4H/, 3,00 - 3,10 /м, 2H/, 1,70 - 1,90 /м, 6H/, 1,40 /с, 9H/. Масс-спектр с полевой десорбцией: 529. Элементный анализ для C32H35NO4S • 1,0 HCl
Рассчитано: C 67,89 H 6,41 N 2,47.
Найдено: C 68,94
H 6,61 N 1,72
Пример 48. [3-[4-[2-/1-Пиперидинил/этокси]фенокси]-2-/4-бутилсульфонилоксифенил/] бензо[b]тиофенгидрохлорид

Выход = 85% твердого вещества белого цвета. Т.плавления 98 - 104oC.1H ЯМР /ДМСО-d6/d, 10,20 /шир., с., 1H/, 8,02 /д, J = 8,0 Гц, 1H/, 7,82 /д, J = 8,7 Гц, 2H/, 7,40 - 7,55 /м, 5H/, 7,00 /с. 4H/, 4,30 - 4,40 /м, 2H/, 3,46 - 3,66 /м, 6H/, 3,00 - 3,10 /м, 2H/, 1,70 - 1,95 /м, 6H/, 1,40 - 1,60 /м, 4H/, 0,87 /т, J = 7,3 Гц, 3H/. Масс-спектр с полевой десорбцией: 565.
Элементный анализ для C31H35NO5S2 • 1,0 HCl
Рассчитано: C 61,83 H 6,03 N 2,33.
Найдено: C 61,55 H 6,15 N 2,25.
Пример получения 21. [6-Гидрокси-3-[4-[2-/1-пиперидинил/этокси]тиофенокси]-2-/4-гидроксифенил/] бензо[b]тиофен

Получение 4-/метоксиметилокси/фенилдисульфида.
К раствору 4-гидроксифенилсульфида /650 мг, 2,60 ммоля/ в 10 мл безводного N,N-диметилформамида при 10oC добавляют гидрид натрия /230 мг, 5,75 ммоля/, 60% дисперсия в минеральном масле. После перемешивания в течение 15 минут добавляют через шприц хлорметилметиловый эфир /0,44 мл, 5,75 ммолей/. Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение 0,5 часа. Полученную смесь распределяют между рассолом и этилацетатом /по 20 мл каждого/. Выделенный водный слой экстрагируют этилацетатом /2 х 20 мл/. Органическую часть сушат над сульфатом натрия и концентрируют до желтого масла /993 мг, 100%/. Аналитический образец 4-/метоксиметилокси/фенилдисульфида получают в результате хроматографической обработки /двуокись кремния, 4% этилацетат/гексан/.1H ЯМР /ДМСО-d6/d: 7,40 /д, J = 6,9 Гц, 4H/, 7,00 /д, J = 6,9 Гц, 4H/, 5,15 /с, 4H/, 3,32 /с, 6H/. Масс-спектр с полевой десорбцией: 338. Элементный анализ для C16H18O4S2.
Рассчитано: C 56,78 H 5,36.
Найдено: C 57,08 H 5,44.
Пример 49. [6-Метокси-2-/4-метоксифенил/-3-/4-метоксиметиленокси/тиофенокси] бензо[b]тиофен

К раствору [6-метокси-2-/4-метоксифенил/-3-бром]бензо[b]тиофена /1,82 г, 5,2 ммоля/ в 10 мл безводного тетрагидрофурана в атмосфере азота при -60oC добавляют н-бутиллитий /3,15 мл, 5,0 ммоля, 1,6M раствор в гексанах/ по каплям через шприц. Полученную смесь нагревают до -20oC в течение 10 минут, затем снова охлаждают до -60oC. К литиевому образцу добавляют 4/метоксиметилокси/-фенилдисульфид /800 мг, 2,36 ммоля/, в 5 мл безводного тетрагидрофурана, и полученную смесь оставляют постепенно нагреваться до 0oC. После двадцатиминутного перемешивания реакцию гасят, распределяя между смесью рассола и этилацетата /по 50 мл каждого/. Слои разделяют и водную фазу экстрагируют этилацетатом /2 х 50 мл/. Органические слои объединяют, сушат над сульфатом натрия и концентрируют в вакууме до масла. В результате хроматографической обработки /двуокись кремния, 5% этилацетат /гексаны/ получают 287 мг /27%/ [6-метокси-2-/4-метоксифенил/-3-/4-метоксиметиленокси/тиофенокси] бензо[b]тиофена в виде бесцветного масла.1H ЯМР /ДМСО-d6/d: 7,59 /д, J = 8,4 Гц, 2H/, 7,58 /д, J = 2,0 Гц, 1H/, 7,52 /д, J = 8,8 Гц, 1H/, 7,03 - 6,85 /м, 7H/, 5,06 /с, 2H/, 3,79 /с, 3H/, 3,76 /с, 3H/. Масс-спектр с полевой десорбцией: 438. Элементный анализ для C24H22O4S2:
Рассчитано: C 65,73 H 5,06
Найдено: C 65,93 H 5,10
Пример 50. [6-Метокси-2-/4-метоксифенил/-3-/4-гидрокси/тиофенокси] бензо[b]тиофен

К раствору [6-метокси-2-/4-метоксифенил/-3-/4-метоксиметиленокси/тиофенокси] бензо[b]тиофена /233 мг, 0,53 ммоля/ в 10 мл смеси 1:1:2 метанол: вода: тетрагидрофуран добавляют метансульфоновую кислоту /0,2 мл, 2, 66 ммоля/. Полученную смесь нагревают до кипения с обратным холодильником в течение 5 часов. После охлаждения до комнатной температуры реакционную смесь разбавляют водой. Водную фазу экстрагируют дважды этилацетатом. Органический слой промывают несколько раз насыщенным раствором бикарбоната натрия. Органический слой сушат над сульфатом натрия и концентрируют в вакууме до получения 206 мг /99%/ [6-метокси-2-/4-метоксифенил/-3-/4-гидрокси/тиофенокси]бензо[b] тиофена в виде бесцветного масла.1H ЯМР /ДМСО-d6/d: 9,43 /с, 1H/, 7,63 /д, J = 8,4 Гц, 2H/, 7,61 /д, J = 2, 0 Гц, 1H/, 7,59 /д, J = 8,8 Гц, 1H/, 7,08 /д, J = 8,4 Гц, 2H/, 7,02 /дд, J = 8,8 Гц, 2,0 Гц, 1H/, 6,90 /д, J = 8,6 Гц, 2H/, 6,63 /д, J = 8,6 Гц, 2H/. Масс-спектр с полевой десорбцией: 395. Элементный анализ для C22H18O3S2: C 66,98 H 4,60 рассчитано;
Найдено: C 67,26 H 4,78.
Пример 51.
[6-Метокси-3-[4-[2-/1-пиперидинил/этокси]тиофенокси]-2-4-метоксифенил/] бензо[b]тиофен

К раствору [6-метокси-2-/4-метоксифенил/3-/4-гидрокси/тиофенокси]бензо [b] тиофена /242 мг, 0,61 ммоля/ в 8,0 мл безводного N, N-диметилформамида добавляют карбонат цезия /820 мг, 2,5 ммоля/ а затем 2-хлорэтилпиперидингидрохлорид /194 мг, 1,05 ммоля/. Полученную смесь перемешивают в течение 48 часов при комнатной температуре, а затем распределяют между рассолом и этилацетатом. Слои разделяют и водную фазу экстрагируют этилацетатом трижды. Органический слой сушат над сульфатом натрия и концентрируют в вакууме до масла. В результате хроматографической обработки /двуокись кремния, 0-2% метанол /хлороформ/ получают 244 мг /92% [6-метокси-3-[4-[2-/1-пиперидинил/этокси]тиофенокси]-2-/4-метоксифенил/] бензо[b]тиофена в виде масла янтарного цвета.
Пример 52. Образец [6-метокси-3-[4-[2-/1-пиперидинил/этокси]тиофенокси] -2-/4-метоксифенил/] бензо[b] тиофена превращают в гидрохлорид стандартным способом с 72% выходом.

Т. плавления 198-201o.1H ЯМР /ДМСО-d6/d: 7,63 /д, J = 8,6 Гц, 2H/, 7,62 /д, J = 2,0 Гц, 1H/, 7,58 /д, J = 8,2 Гц, 1H/, 7,07 /д, J=8,6 Гц, 2H/, 7,02/дд, J=8,2, 2,0 Гц, 1H/, 6,92 /кв. JAB=9,0 Гц, 4H/, 4,24 /шир. т., 2H/, 3,82 /с, 3H/, 3,80 /с, 3H/, 3,49-3,39 /м, 4H/, 2,93 /м, 2H/, 1,82-1,62 /м, 5H/, 1,38 /м, 1H/, Элементный анализ для C29H32NO3S2•1,0 HCl
Рассчитано: C 64,28 H 5,95 N 2,58
Найдено: C 64,09 H 6,08 N 2, 78
Пример 53. [6-Гидрокси-3-[4-[2-/1-пиперидинил/этокси]тиофенокси]-2-/4-гидроксифенил/] бензо[b]тиофен

К раствору
6-метокси-3-[4-[2-/1-пиперидинил/этокси]тиофенокси]-2-/4- метоксифенил/] бензо[b] тиофенгидрохлорида /160 мг, 0,29 ммоля/ в 15 мл безводного метиленхлорида при 0oC в атмосфере азота добавляют трехбромистый бор /0,15 мл/. Полученный темный раствор перемешивают в течение 1 часа при 0oC, а затем сразу выливают в перемешиваемый раствор этилацетата в насыщенном растворе бикарбоната натрия /по 50 мл/ каждого. Слои разделяют и водную фазу промывают этилацетатом /3 х 30 мл/. Органическую часть сушат над сульфатом натрия и концентрируют в вакууме до получения твердого вещества белого цвета. В результате хроматографической обработки /двуокись кремния 0-5% метанол/хлорофор/ получают 91 мг /60%/ [6-гидрокси-3-[4-[2-/1-пиперидинил/этокси] -2-/4-гидроксифенил/] бензо[b] тиофена в виде твердого вещества белого цвета. Т.плавления 123-127oC.1H ЯМР /ДМСО-d6 /d: 9,79 /с, 1H/, 9,71 /с, 1H/, 7,46 /д, J=8,4 Гц, 2H/, 7,42 /д, J=8,9 Гц, 1H/, 7,26 /д, J=2,0 Гц, 1H /, 6,91 /д, J=8,8 Гц, 2H/, 6,82-6,76 /м, 5H/, 3,91 /т, J=8,8 Гц, 2H/, 2,56 /т, J=5,8 Гц, 2H/, 2,40 /м, 4H/, 1,41-1,28 /м, 6H/. Масс-спектр с полевой десорбцией: 478. Элементный анализ для C27H27NO3S2:
Рассчитано: C 67,90 H 5,70 N 2,93
Найдено: C 68,14 H 5,84 N 2,65
Пример 54. [6-Гидрокси-3-[4-[2-1-пиперидинил/этокси]тиофенокси]-2-/4-гидроксифенил/] бензо[b]тиофенгидрохлорид

Т. плавления 180-190oC.1H ЯМР /ДМСО-d6/d: 9,86 /с, 1H/, 9,79 /с, 1H/, 7,46 /l, J=8,5 Гц, 2H/, 7,42 /д, J=8,7 Гц, 1H/, 7,29 /дБ J=2,0 Гц, 1H/, 6,96 /д, J= 8,7 Гц, 2H/, 6,86-6,81 /м, 5H/, 4,27 /м, 2H/, 3,41-3,37 /м, 4H/, 2,96-2,84 /м, 2H/, 1,77-1,60 /м, 5H/, 13,5-1,28/м,1H/. Масс-спектр с полевой десорбцией: 477. Элементный анализ для C27H27NO3S2•2,2 HCl
Рассчитано: C 58,13 H 5,28 N 2,51.
Найдено: C 58,11 H 5,10 N 2,61.
Тем же способом получают соединения:
Пример 55. [6-Метокси-3-[4-[2-/1-пирролидинил/этокси] тиофенокси]- 2-/4-метоксифенил/]бензо[b]тиофенгидрохлорид

Т. плавления 215-218oC.1 H ЯМР /ДМСО-d6/d 7,61-7,58 /м, 3H/, 7,52 /д, J= 8,8 Гц, 1H/, 7,04-6,95 /м, 5H/, 6,86 /д, J=8,8 Гц, 2H/, 4,22 /шир. т., 2H/, 3,79 /с, 3H/, 3,76 /с, 3H/, 3,47-3,42 /м, 4H/, 3,01 /м, 2H/, 1,94-1,80 /м, 4H/. Масс-спектр с полевой десорбцией: 491. Элементный анализ для C28H29NO3S2•1,0 HCl.
Рассчитано: C 63, 67 H 5,73 N 2,65.
Найдено: C 63,47 H 5,78 N 2,65.
Пример 56. [6-Гидрокси-3-[4-[2-/1-пирролидинил/этокси]тиофенокси]-2-/4- гидроксифенил/]бензо[b]тиофенгидрохлорид

Т. плавления 137-140oC /с разложением/.1H ЯМР /ДМСО-d6/d: 9,86/с, 1H/, 9,80 /с, 1H/, 7,46 /д, J=8,6 Гц, 2H/, 7,42 /д, J=8,7 Гц, 1H/, 7,29 /д, J=2,0 Гц, 1H/, 6,96 /д, J= 8,7 Гц, 2H/, 6,87-6,81 /м, 5H/, 4,21 /шир. т., 2H/, 3,53-3,41 /м, 4H/, 3,01 /м, 2H/, 1,95-1,82 /м, 4H/. Масс-спектр с полевой десорбцией: 464. Элементный анализ для C26H25NO3S2•1,0 HCl:
Рассчитано: C 62,45 H 5,24 N 2,80.
Найдено: C 62,36 H 5,37 N 2,61.
Пример
57
Из продукта примера 45 за счет гидрогенолиза трифлата, как указано далее, в примере 58, получают [6-метокси-3-[4-[2-/1-пиперидинил/-этокси]фенокси]-2-/фенил/]бензо [b]тиофенгидрохлорид

Т. плавления 187-195oC.1H ЯМР/ДМСО-d6/d: 7,66 /д, J =2,8 Гц, 2H/, 7,58 /д, J=2,0 Гц, 1H/, 7,39 /т, J=7,5 Гц, 2H/, 7,28 /м, 1H/, 7,17 /д, J=8,8 Гц, 1H/, 6,91 /дд, = 8,8 Гц, 2,0 Гц, 1H/, 6,89 /с, 4H/, 4,23 /шир. т., J=5,7 Гц, 2H/, 3,79/с, 3H/, 3,45-3,38/м, 4H/, 2, 98 /м, 2H/, 1,77-1,61 /м, 5H/, 1,31 /м, 1H/. Масс-спектр с полевой десорбцией: 460. Элементный анализ для C28H29NO3S•1,0 HCl:
Рассчитано: C 67,80 H 6, 10 N 2,84.
Найдено: C 67,62 H 5,89 N 2,67.
Пример 58
[6-гидрокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/фенил/] бензо[b] тиофенгидрохлорид

К раствору [6-гидрокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4-гидроксифенил /бензо[b] тиофенгидрохлорида (5,0 г, 10,0 ммоль) в 100 мл безводного метиленхлорида при 0oC в атмосфере N2 добавляют триэтиламин /8,38 мл, 60,0 ммоля/ а затем трифторметансульфоновый ангидрид /1,69 мл, 10,0 ммоля/. Полученную смесь оставляют постепенно нагреваться до комнатной температуры и перемешивают в течение 1,5 часа. Реакционную смесь гасят, выливая ее в 200 мл насыщенного раствора бикарбоната натрия. Затем водную фазу экстрагируют этилацетатом /3 х 100 мл/. Органический слой сушат над сульфатом натрия и концентрируют в вакууме до масла. В результате хроматографической обработки /0-3% метанол /хлороформ/ получают 2,82 г /39%/ [6-трифторметансульфонат-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4- трифторметансульфонатфенил/] бензо[b] тиофен, /1,82 г, 31%/ смеси 1:1 [6-трифторметансульфонат-3-[4-[2-/1-пиперидинил/этокси] -фенокси] -2-/4-фенил/] бензо[b] тиофена и [3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4-трифторметансульфонатфенил/] бензо [b] тиофена и 1,48 г /36%/ выделенного исходного материала в виде свободного основания.
К раствору смеси 1:1 трифлатных производных из последней реакции /0,50 г, 0,84 ммоля/ в 60 мл смеси этанол: этилацетат /5:1/ добавляют триэтиламин /2,0 мл/ и 5% палладий-на-угле /0,50 г/. Полученную смесь гидрируют при 40 пси /2,81 кг/см2/ в течение 2 часов. Затем полученную смесь фильтруют через Целлит /Celite®/ для удаления катализатора. Полученный фильтрат концентрируют до масла. Полученную смесь моногидрокси производных растворяют в этилацетате, из которого осаждается [3-[4-[2-/1-пиперидинил/этокси]фенокси]-2-/4-гидроксифенил/] бензо[b] тиофен. Полученный фильтрат состоит из смеси 4:1 моногидроксипроизводных, в которых [6-гидрокси-3-[4-[2-/1-пиперидинил/этокси] фенокси]-2-/фенил/[бензо[b]тиофен является основным компонентом. Полученный фильтрат концентрируют в вакууме, а полученную твердую часть растворяют в минимальном количестве этилацетата и обрабатывают этиловый эфир соляной кислотой. Полученную твердую часть перекристаллизовывают из этанола до получения 69 мг /18%/ изомерно чистого [6-гидрокси-3-[4-[2- /1-пиперединил/этокси]фенокси]-2-/фенил/]бензо[b]тиофенгидрохлорида. T. плавления 217 - 219oC.1H ЯМР /DMCO-d6/d: 9,87 /с, 1H/, 7,64 /д, J = 7,5 Гц, 2H/, 7,39-7,26 /м, 4H/, 7,10 /д, J = 8,6, 1H/, 6,89 /с, 4H/, 6,78 /дд, J = 8,6, 2,0 Гц, 1H/. 4,22 /шир.т., 2H/, 3,39-3,37 /м, 4H/, 2,97-2,90 /м, 2H/, 1,74-1,60 /м, 5H/, 1,39 /м, 1H/.
Масс-спектр с полевой десорбцией: 466.
Элементный анализ для C27H27NO3S • 1,0 HCl
Рассчитано: C 67,28 H 5,86 N 2,91.
Найдено: C 67,00 H 5,59 N 2,87.
В другом варианте соединения примера 58 получают за счет деметилирования продукта примера 57, как было указано в примере 18.
Пример 59.
[6-изопропокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4- метоксифенил/] бензо[b] тиофен-/S/-оксид/ получают как указано для [6-метокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4-метоксифенил/] бензо[b]тиофен-/S-оксида/. /пример 30/.

Желтое масло.1H ЯМР /ДМСО-d6/d: 7,69 /д, J = 2,0 1H/, 7,67 /д, J = 8,6 Гц, 2H/. 7,09-6,99 /м, 5H/, 6,96-6,87 /м, 3H/, 4,76 /септет, J = 6,0 Гц, 1H/, 3,99 /шир. т., J = 6,0 Гц, 2H/, 3,78 /с, 3H/, 2,61 /шир. т. J = 6,0 Гц, 2H/, 2,44-2,37 /м, 4H/, 1,53-1,43 /м, 4H/, 1,40-1,32 /м, 2H/, 1,29 /д, J = 6,0 Гц, 6H/. Масс-спектр полевой десорбции: 533. Элементный анализ для C31H35NO5S • 0,67 H2O:
Рассчитано: C 68,23 H 6,71 N 2,57.
Найдено: C 67,90 H 6,31 N 2,53.
[6-изопропокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4- метоксифенил/] бензо[b]тиофенгидрохлорид получают по способу, указанному для [6-метокси-3-[4-[2-/1-пиперидинил/этокси]
фенокси]-2-/4- метоксифенил/]бензо[b]тиофена /пример 32/

T. плавления 168 - 170oC1H ЯМР /ДМСО-d6/d: 10,37 /с, 1H/, 7,58 /д, J = 8,6 Гц, 2H/, 7,52 /д, J = 1,3 Гц, 1H/, 7,12 /д, J = 8,8 Гц, 1H/, 6,95 /д, J = 8,6 Гц, 2H/, 6,92-6,85 /м, 5H/, 4,64 /септет, J = 6,0 Гц, 1H/, 4,28 /шир. т. , J = 6,0 Гц, 2H/, 3,72 /с, 3H/, 3,44-3,37 /м, 4H/, 2,95-2,89 /м, 2H/, 1,73-1,60 /м, 5H/, 1,36-1,28 /м, 1H/, 1,25 /д, J = 6,0 Гц, 6H/.
Масс-спектр с полевой
десорбцией: 517. Элементный анализ для C31H35NO4S • HCl
Рассчитано: C 67,19 H 6,55 N 2,53.
Найдено: C 67,15 H 6,29 N 2,62.
[6-изопропокси-3-[4-[2-/1-пиперидинил/этокси] фенокси] -2-/4- метоксифенил/] бензо[b] тиофенгидрохлорид превращают в [6-гидрокси-3-[4-[2-/1-пиперидинил/этокси] фенокси/-2-/4-метоксифенил/] бензо[b]тиофен за счет обработки 2,0 эквивалентами BCl3 при температуре 0 - 10oC в безводном дихлорметане /в этих условиях метиловый эфир не расщепляется/.
Пример 60
[6-гидрокси-3-[4-[2-(1-пирролидинил)этокси] фенокси] -2-(4- метоксифенил)бензо[b]тиофен формулы:

Данные для (IA): C27H27NO4S • HCl
Элементный анализ:
Вычислено,%: C 65,12 H 5,67 N 2,81
Найдено,%: C 65,28 H 5,74 N 2,89
Масс-спектр (ПД+) (полевая десорбция+) : 462
Соединение формулы (IA) получают, исходя из [6-изопропокси-2-(4-метоксифенил)-3-бром] [b]тиофена, синтезированного согласно примеру получения 8 с использованием [6-изопропокси-2- (4-метоксифенил)]бензо[b]тиофена.
Затем согласно примеру 28 получают [6-изопропокси-2-(4- метоксифенил)-3-бром]бензо[b]тиофен-(S-оксид). Одновременно с этим, согласно методике примера получения 12, но используя 2-хлорэтилпирролидин, получают 4-(2-(1-пирролидинил)-этокси)фенил. Затем 4-(2-(1-пирролидинил)-этокси)фенил подвергают взаимодействию с [6-изопропокси-2-(4-метоксифенил)-3-бром]бензо[b] тиофен-(S-оксидом) в соответствии с методикой примера 30. Сульфоксидную группу затем восстанавливают согласно методике примера 32, получая хлористоводородную соль [6-изопропокси-3-[4-[2-(1-пирролидинил)-этокси]фенокси]-2-(4-метоксифенил) бензо[b]тиофена.
Наконец, изопропильную защитную группу удаляют согласно методике примера 59, получая хлористоводородную соль [6-гидрокси-3-[4-[2-(1- пирролидинил)этокси]фенокси]-2-(4-метоксифенил)-бензо[b]тиофена.
Тестовая процедура
Общая процедура подготовки
В примерах, которые
иллюстрируют способ, использована постменопаузальная модель, в которой определяют эффективность различных способов лечения в отношении циркулирующих липидов.
Семидесятипятидневных самок крыс штамма Spaque Dawley /весом от 200 до 225 г/ получают из Charles River Laborotories (portoge, MI). У животных либо удаляют оба яичника /O X/, либо проводят операцию по методу Шама в Чарльз Ривер Лабораториз, а затем поставляют спустя неделю. По прибытии их помещают в металлические подвесные клетки по 3-4 крысы в каждую клетку и дают пищу ad libitum /с содержанием кальция примерно 0, 5%/ и воду в течение одной недели. Температуру в комнате поддерживают при 22,2o ± 1,7oC при минимальной относительной влажности 40%. Фотопериод в комнате составляет 12 часов света и 12 часов темноты. Дозирующий режим. Отбор тканей.
После недельного периода акклиматизации /т.е. спустя две недели после овариэктомии/ начинают ежедневное введение тестового соединения. 17 α -этинилэстрадиол или тестовое соединение вводят орально /если нет других указаний/ в виде суспензии в 1% карбоксиметилцеллюлозе, или растворенное в 20% циклодекстрине. Животным вводят в дозу ежедневно в течение 4 дней. После завершения схемы дозировки животных взвешивают и анестезируют смесью кетамин : ксилазин /2:1 объем/объем/ и образцы крови отбирают, беря пункцию из сердца. Затем животных умерщвляют с помощью CO2, матку извлекают через разрез брюшины. Затем определяют вес влажной матки.
Анализ на холестерин
Образцам крови дают свернуться при комнатной температуре в течение 2 часов и сыворотку получают после центрифугирования в течение 10 минут при 3000 об/мин. Сыаороточный холестерин определяют с
помощью высокоэффективного анализа на холестерин Boehringer Mannheim Diagnostike. Холестерин окисляют до холест-4-ен-3-она и перекиси водорода. Затем перекись водорода реагируют с фенолом и
4-аминофеназолом в присутствии пероксидазы до получения p-хинониминового красителя, который определяют спектрофотометрически на длине волны 500 нм. Затем рассчитывают содержание холестерина по
стандартной кривой. Весь анализ проводится автоматически с использованием Biomek Automated Workstation/
Пероксидазный анализ содержание в матке зозинофилов /EPO/
Матки содержат при
4oC до момента проведения энзиматического анализа. Тогда матки гомогенизируют в 50 объемах мМ Трис буфера /pH 8,0/, содержащего 0,005% Тритон X-100. После добавления в Трис буфер 0,01%
перекиси водорода и 10 мМ O-фенилендиамина /конечные концентрации/ в течение одной минуты следят за возрастанием поглощения на 450 нм. Наличие зозинофилов в матках является показателем экстрогенной
активности соединения. Максимальную скорость с 15 секундным интервалом определяют относительно начального, линейного участка кривой реакции.
Источник соединения: 17 α - этинилэстрадиол получают от Сигмы Камикал Ко., Ст. Луис, МО.
Влияние соединений формулы I на сывороточный холестерин и определение агонист/не-агонистической активности
Данные, представленные далее в таблице 1, показыают результаты, полученные при сравнении крыс после овариэктомии, крыс, обработанных 17 α - этинилэстрадиолом /EE2; и орально
доступного из экстрогена/, и крыс, обработанных некоторыми соединениями настоящего изобретения. Хотя EE2 вызывает снижение сывороточного холестерина при оральном введении в дозах от 0,1
мг/кг/день, он также проявляет стимулирующее действие на матки, так что вес EE2 маток оказывается существенно выше, нежели вес маток у тестовых животных после овариэктомии. Такая реакция
матки на эстроген хорошо известна специалистам.
Соединение настоящего изобретения не только приводят к общему снижению сывороточного холестерина по сравнению с контрольными животными после овариэктомии, но вес матки лишь минимально возрастает либо слегка снижается для большинства тестированных соединений формулы I. По сравнению с известными специалистам эстрогенными соединениями, благоприятный эффект снижения сывороточного холестерина без нежелательного влияния на вес матки очень редок и желателен.
Как видно из приводимых далее результатов, эстрогенность можно также оценить, определяя обратную реакцию инфильтрации зозинофилов в матки. Соединения настоящего изобретения не вызывают какого-либо увеличения числа зозинофилов, наблюдаемых в строительном слое у овариэктомизированных крыс, хотя эстрадиол вызывает существенное ожидаемое возрастание инфильтрации зозинофилов.
Представленные далее в таблице 1 результаты отражают реакции 5-6 крыс в каждом эксперименте.
В дополнение к продемонстрированным преимуществам соединений настоящего изобретения, особенно при сравнении с эстрадиолом, приведенные выше результаты ясно показывают, что соединения формулы I не являются имитаторами эстрогена. Более того, ни в одном из случаев не наблюдалось нежелательных токсикологических действий /выражение/.
Тестовая
процедура для определения остеопороза
В соответствии с общей процедурой подготовки крыс обрабатывали ежедневно в течение 35 дней /по 6 крыс в группе/ и умертвили улучшением двуокисью углерода
на 36 день. Оказалось, что периода в 35 дней достаточно для того, чтобы обеспечить максимальное восстановление плотности костей, которое определяли как здесь указано. В момент умерщвления матку
удаляют, освобождают от наружной ткани и содержащуюся в ней жидкость убирают прежде чем определить влажный вес для подтверждения дефицита эстрогена, связанного с полной овариэктомией. Вес матки обычно
снижается и составляет около 75% как реакция на овириэктомию. Затем матку помещают в 10% нейтральный буферированный формалин для обеспечения последующего гистологического анализа.
Иссекают первую бедренную кость и проводят рентгеновское исследование и анализ по программе имидж-анализа /NIH имидж/ у дистального метафиза. Проксимальный аспект большой берцовой кости этих животных также сканируют с помощью количественной компьютерной томографии.
В соответствии с вышеописанной процедурой соединения настоящего изобретения и этинилэстрадиол /EE2/ в 20% гидроксипропил -β- циклодекстрине вводят орально тестовым животным. Данные относительно дистального метафиза большой берцовый кости представлены далее в таблицах 2 и 3, и представляют результаты обработки соединением формулы I по сравнению с интактными и овариэктомизированными тестовыми животными. Полученные результаты представлены как среднее ± средняя стандартная ошибка.
В заключении можно сказать, что овариэктомия текстовых животных вызывает существенное снижение плотности берцовой кости по сравнению с интактными животными, которым вводят только носитель. Оральное введение этинилэстрадиола /EE2/ предотвращает такую потерю, но при этом всегда присутствует риск стимуляции матки при такой обработке.
Соединения настоящего изобретения также предотвращают общую потерю костной ткани зависящим от дозы образом. Соответственно, соединения настоящего изобретения можно использовать для лечения постменопаузального синдрома, в частности остеопороза.
Анализ пролиферации MCF-7
Клетки аденокарциномы группы MCF-7 /ATCC НТВ 22/ поддерживают в МЕМ /минимальной поддерживающей среде, не
содержащей фенола, Сигма, Ст. Луис, МО/, дополненной 10% фетальной телячьей сывороткой /FBS/ /объем/объем/, L-глутамином /1 мМ/, пируватом натрия /1 мМ/, HEPES
//N-/2-гидроксиэтил/пиперазин-N'/2-этансульфоновая кислота/ 10 мМ/, незамещенными аминокислотами и бычьим инсулином /1 мкг/мл/ /поддерживающая среда/. За 10 дней до анализа клетки MCF-7 переводят на
поддерживающую среду, дополненную 10% декстраном, покрытым углем очищенной фетальной телячьей сывороткой /ДСС-FBS/ аналитическая среда/вместо 10% FBS, для того, чтобы исчерпать внутренние запасы
стероидов. Клетки МСР-7 удаляют из склянок, используя среду для диссоциации клеток /Ca++ /Mg++ свободная HBSS/ не содержащая фенола красного/, дополненную 10 мМ HEPES и 2 мМ
ЕДТА/. Клетки дважды промывают аналитической средой и доводят концентрацию до 8000 клеток/мл. Примерно 100 мл /8000 клеток/ добавляют в плоскодонные микрокультуральные ячейки /Костар 3596/ и
инкубируют при 37oC в 5% CO2 увлажненном инкубаторе в течение 48 часов для того, чтобы дать возможность клеткам прилипнуть и уравновеситься после переноса. Подготавливают
сериальные разбавления лекарств для ДМСО в качестве контроля разбавителя в аналитической среде и 50 мл переносят в три дублирующие микрокультуры, а затем 50 мл аналитической среды до конечного объема
200 мл. После дополнительных 48 часов при 37oC в 5% CO2 увлажненном инкубаторе, микрокультуры импульсно обрабатывают обработанным тритием тимидином /1 микрокюри/ячейку/ в течение
4 часов. Обработку культур завершают, замораживания при -70oC в течение 24 часов, а затем оттаивая и собирая микрокультуры с помощью полуавтоматического сборника клеток Скартона /Skarron/.
Образцы подсчитывают, используя жидкостной сцинтилляционный счетчик Wallac BetaPlace b. Результаты представлены в таблице 4 далее и демонстрируют ИК50 для некоторых соединений настоящего
изобретения.
Ингибирование опухоли молочной железы, вызванной ДМБА
Эстроген-зависимые опухоли молочной железы вызывают у самок крыс штамма Спраг-Доули, которых получают из
Харлан Индастриз, Индианаполис, Индиана. В возрасте около 55 дней крысам вводят орально и однократно 20 мг 7,12-диметилбенз/а/антрацена /ДМБА/. Спустя примерно 6 недель после введения ДМБА молочные
железы пальпируют с недельными интервалами на предмет появления опухолей. Как только появляется одна или более опухоль, наибольший и наименьший диаметры каждой из опухолей измеряют штангенциркулем,
измерения регистрируют и этих животных отбирают для экспериментов. Предпринята попытка равномерно распределить животных с различными размерами опухолей в обработанной и контрольной группах, с тем,
чтобы средние размеры опухолей были одинаково распределены между тестовыми группами. В каждом из экспериментов в контрольных группах и тестовых группах было по 5 - 9 животных.
Соединения формулы I вводят либо за счет внутрибрюшинных инъекций в 2% акации или орально. Те соединения, которые вводят орально, вводят либо растворенными, либо суспендированными в 0,2 мл кукурузного масла. При каждой обработке, включая контрольные обработки акацией и кукурузным маслом, препарат вводят один раз в день каждому тестируемому животному. После начальных измерений опухоли и отбора тестовых животных размеров, размеры опухолей определяют еженедельно вышеуказанным способом. Обработка и контроль за животными продолжаются в течение 3 - 5 недель, после чего определяют окончательную площадь опухолей. Определяют изменения средней площади опухолей для каждого из соединений и для контрольных животных.
Тест на фиброзы матки
Тест 1. Трем-двадцати женщинам с
фиброзами матки вводят соединение настоящего изобретения. Количество вводимого соединения составляет величину от 0,1 до 1000 кг/день, а период лечения составляет 3 месяца.
На протяжении времени приема и в течение вплоть до 3 месяцев после прекращения приема лекарства за женщинами ведется наблюдение относительно влияния на фиброзы матки.
Тест 2. Используют процедуру теста 1, но введение препарата продолжается в течение 6 месяцев.
Тест 3. Используют процедуру теста 1, но период введения препарата составляет 1 год.
Тест 4.
A. Фиброидные опухоли, вызванные у морских свинок
Для того, чтобы вызвать лейомиому у сексуально зрелых самок морских свинок, используют пролонгированную стимуляцию
эстрогеном. Животным вводят дозы эстрадиола 3-5 раз в неделю за счет инъекций в течение 2-4 месяцев или до появления опухоли. Обработка, которая состоит из соединения настоящего изобретения или
носителя, проводится ежедневно в течение 3-16 недель, а затем животных умертвляют, матки собирают и анализируют на предмет регрессии опухоли.
B. Имплантация фиброидной ткани матки
женщины голым мышам
Ткани из лейомиом женщины имплантируют в перитонеальную полость и/или миометриум матки сексуально зрелых, кастрированных самок голых мышей. Поставляют экзогенный эстроген
для того, чтобы вызвать рост эксплантированной ткани. В некоторых случаях собранные опухолевые клетки культивируют ин витро перед имплантацией. Обработку, состоящую из соединения настоящего
изобретения или носителя осуществляют за счет лаважа желудка ежедневно в течение 3-16 недель, имплантанты удаляют и определяют рост или регрессию. В момент умертвления матки отбирают и осматривают для
оценки состояния органа.
Тест 5.
A. Ткани фиброидных опухолей матки женщины собирают и сохраняют ин витро как первичные нетрансформированные культуры. Хирургические образцы пропускают через стерильное сито или сетку, или в другом варианте, отщипывают от окружающей ткани для получения одноклеточной суспензии. Клетки содержат в среде, содержащей 10% сыворотки и антибиотик. Определяют скорость роста в присутствии эстрогена или без него. Клетки анализируют по их способности продуцировать комплект компонента C3 и по их реакции на факторы роста и гормоны роста. Ин витро культуры оценивают по их пролиферативной реакции после обработки прогестинами, GnRH, соединением настоящего изобретения и носителем. Уровни стероидных гормональных рецепторов оценивают еженедельно для определения того, сохраняются ли важные клеточные характеристики ин витро. При этом используют ткани от 5 - 25 пациентов.
Активность в, по крайней мере, одном из вышеперечисленных тестов указывает на то, что соединения настоящего изобретения эффективны для лечения фиброза матки.
Тест на эндометриоз
В тестах 1 и 2 эффекты 14 и 21
дневного введения соединений настоящего изобретения на рост эксплантированной эндометриальной ткани можно исследовать.
Тест. 1.
В качестве тестовых животных используют от 12 до 30 взрослых самок крыс СД штамма. Их делят на три группы поровну. Контролируют эстральный цикл всех животных. В день проэструса каждую самку оперируют, у самок в каждой группе удаляют левый рог матки разделяют на мелкие квадраты и эти квадраты свободно пришивают в различные точки, соединение с мезентерическим потоком крови. Кроме того, у самок группы 2 удаляют яичники.
На следующий день после хирургического вмешательства животным группам 1 и 2 внутрибрюшинно вводят воду в течение 14 дней, тогда как животным группам 3 внутрибрюшинно вводят инъекции 1,0 мг соединения настоящего изобретения на 1 кг веса тела в течение такого же промежутка времени. После 14 дней обработки всех самок умерщвляют и эндометриальные эксплантаты, надпочечники, остатки матки и яичники, там, где возможно, удаляют и препарируют для гистологических исследований. Яичники и надпочечники взвешивают.
Тест. 2.
В качестве тестовых животных используют от 12 до 30 взрослых самок крыс штамма СД. Их делят на две равные группы. Контролируют эстральный цикл всех животных. В день проэструса всех самок оперируют. У самок всех групп удаляют левый рог матки, разрезают на мелкие квадраты и эти квадраты свободно пришивают в различные точки рядом с мезентерическим потоком крови.
Примерно через 50 дней после операции животным группы 1 вводят внутрибрюшинными инъекциями воду в течение 21 дня, тогда как животным группы 2 внутрибрюшинно вводят по 1,0 мг соединения настоящего изобретения на кг веса тела в течение такого же промежутка времени. После 21 дня обработки каждую из самок умерщвляют и эндометриальные эксплантаты и надпочечники удаляют и взвешивают. Эти эксплантаты измеряют, как показатели роста. Записывают циклы экструса.
Тест 3.
A. Хирургическое индуцирование эндометриоза
Аутотрансплантаты эндометриальной ткани используют для индуцирования эндометриоза у крыс
и/или кроликов. У самок животных в репродуктивном возрасте удаляют оба яичника и эстроген вводят экзогенно, обеспечивая тем самым специфический и постоянный уровень гормона. Аутологичную
эндометриальную ткань имплантируют в брюшину 5 - 150 животным и вводят эстроген для индуцирования роста эксплантированной ткани. Обработка, включающая соединение настоящего изобретения проводится за
счет лаважа желудка ежедневно в течение 3 - 16 недель, имплантаты удаляют и измеряют для определения роста или регрессии. В момент умерщвления отбирают интактный рог матки для оценки состояния
эндометрия.
B. Имплантация эндометриальной ткани женщины голым мышам
Ткань из язв эндометрия женщины имплантируют в брюшину сексуально зрелых кастрированных самок голых
мышей. Вводят экзогенный эстроген индуцирования роста эксплантированной ткани. В некоторых случаях собранные эндометриальные клетки культивируют ин витро перед имплантацией. Обработку, состоящую из
соединения настоящего изобретения вводят за счет лаважа желудка ежедневно в течение 3 - 16 недель, имплантаты удаляют и измеряют для определения роста или регрессии. В момент умерщвления отбирают
матку для определения состояния эндометрия.
Тест 4.
A. Собирают ткани эндометриальных язв женщины и сохраняют ин витро в качестве первичных нетрансформированных культур. Полученные хирургическим путем образцы продавливают через стерильную сетку или сито, или в другом варианте отщипывают от окружающей ткани для получения одноклеточной суспензии.
Клетки поддерживают в среде, содержащей 10% сыворотки и антибиотик. Скорости роста в присутствии эстрогена или без него определяют. Клетки оценивают по их способности продуцировать комплемент компонента C3 и по их реакции на факторы роста и гормоны роста. Культуры ин витро оценивают по их пролиферативной реакции после обработки протестинами, СпРН, соединением настоящего изобретения и носителем. Уровни стероидных гормональных рецепторов оценивают еженедельно для определения того, сохраняются ли важные клеточные характеристики ин витро. При этом используют ткани от 5 - 25 пациентов.
Активность в любом из вышеуказанных анализов указывает на то, что соединения настоящего изобретения можно использовать для лечения эндометриоза.
В настоящем изобретении предложен также способ облегчения постменопаузального синдрома у женщин, который включает вышеуказанный способ использования соединений формулы I и далее включает введение женщинам эффективного количества эстрогена или прогестина. Такие способы лечения особенно подходят для лечения остеопороза и снижения сывороточного холестерина, так как обеспечивает получение пациентом выгод от каждого фармацевтического агента, причем соединения настоящего изобретения будут ингибировать нежелательные побочные эффекты эстрогена и прогестина. Активность этих сочетаний лечения в любом из постменопаузальных тестов, указывает, что такие комбинации лечения можно использовать для облегчения симптомов постменопаузальных симптомов у женщин.
Коммерчески доступны различные формы эстрогена и прогестина. Агенты на базе эстрогена включают, например, этинилэстроген /0,01 - 0,03 мг/день/, местранол /0,05 - 0,15 мг/день/ и коньюгированные эстрогенные гормоны, такие, как премарин® /Wyest-Ayerst; 0,3-2,5 мг/день/. Агенты на базе прогестина включают, например, медроксипрогестерон, такой, как провера® /Апджон; 2, 5-10 мг/день/, норэтилнодрел /1,0 - 10,0 мг /день/ и норэтиндрон /0,5-2,0 мг/день/. Предпочтительным соединением на основе эстрогена является премарин, а норэтилнодрел и норэтиндрон являются предпочтительными агентами на основе прогестина.
Способ введения каждого из агентов на основе эстрогена или прогестина соответствует тем, которые известны специалистам. Для большинства способов настоящего изобретения соединения формулы I вводят непрерывно, от 1 до 3 раз в день. Однако циклическая терапия может быть особенно полезна при лечении эндометриоза или может быть активно использована во время болезненных приступов заболевания. В случае повторного стеноза лечение может быть ограничено короткими /1-6 месячными/ интервалами после таких медицинских процедур как ангиопластика.
В том смысле, как здесь использован, термин "эффективное количество" означает количество соединения настоящего изобретения, которое способно облегчить симптомы различных патологических состояний, здесь описанных. Конкретная доза соединения, вводимого по способу настоящего изобретения, должна, естественно, определяться конкретными обстоятельствами, сопутствующими заболеванию, включая, например, вводимое соединение, способ введения, состояние пациента и патологическое состояние, подлежащее лечению. Обычная дневная доза содержит нетоксичный уровень дозы от около 5 мг до около 600 мг/день соединения настоящего изобретения. Обычно предпочтительная дневная доза составляет от около 15 мг до около 80 мг/день.
Соединения настоящего изобретения можно вводить различными способами, включая оральный, ректальный, подкожный, внутривенный, внутримышечный, через кожу и через нос. Эти соединения предпочтительно формулируют перед введением, способ которого определяет врач. Так, другой аспект настоящего изобретения составляет фармацевтическая композиция, содержащая эффективное количество соединения формулы I, или его фармацевтически приемлемой соли, необязательно содержащая эффективное количество эстрогена или прогестина, и фармацевтически приемлемый носитель, разбавитель, или эксципиент.
Полное количество активных ингредиентов в таких композициях составляет от 0,1% до 99,9% по весу композиции. Под термином "фармацевтически приемлемый" подразумевают носитель, разбавитель, эксципиент и соль, которые совместимы с другими ингредиентами композиции, и не оказывают вредного воздействия на реципиента.
Фармацевтические композиции настоящего изобретения можно получить известными специалистам способами, используя хорошо известные и легко доступные ингредиенты. Так например, соединения формулы I, вместе с эстрогеном или прогестином, можно приготовить с обычными эксципиентами, разбавителями или носителями, в виде таблеток, капсул, суспензий, порошков и т.п. Примеры эксципиентов, разбавителей и носителей, которые пригодны для таких композиций, включают следующие: такие наполнители и увеличивающие объем агенты, как крахмал, сахара, маннит и производные кремния; такие связывающие агенты, как карбоксиметилцеллюлоза и другие производные целлюлозы, альгинаты, желатин и поливинилпирролидон; такие увлажняющие агенты, как глицерин; такие разрыхляющие агенты, как карбонат кальция и бикарбонат натрия; такие агенты для задержки растворения, как парафин; такие ускорители ресорбции, как соединения четвертичного аммония; такие поверхностно-активные агенты, как цетиловый спирт, моностеарат глицерина; такие адсорбирующие носители, как каолин и бентонит; и такие скользящие, как тальк, кальций и стеарат магния, и твердый полиэтиленгликоль.
Соединения можно также приготовить в виде элексиров или растворов для обычного перорального приема или в виде растворов для парэнтерального введения, например, внутримышечно, подкожно или внутривенно. В другом варианте эти соединения хорошо подходят для приготовления композиций в виде дозированных форм с замедленным выделением активного агента и т.п. Эти композиции могут быть так приготовлены, чтобы высвободить активный ингредиент только, или предпочтительно, в конкретном физиологическом окружении, возможно в течение некоторого промежутка времени. Покрытия, оболочки и защитные матрицы можно изготовить, например, из полимерных материалов или восков.
Соединения формулы I отдельно или в сочетании с фармацевтическим агентом настоящего изобретения обычно можно вводить в виде общепринятых композиций. Приводимые далее примеры таких композиций являются лишь иллюстративными и никоим образом не должны ограничивать объем настоящего изобретения.
Композиции
В приводимых далее примерах композиций "активный ингредиент" означает соединение формулы I или его
соль или сольват.
Форма 1: Желатиновые капсулы
Твердые желатиновые капсулы получают используя:
Ингредиент - Количество, мг/капсула
Активный ингредиент - 0,1
- 1000
Крахмал, /NF-Национальный справочник/ - 0 - 650
Пересыпающийся порошок крахмала - 0 - 650
Силиконовая жидкость, 350 сСт - 0 - 15
Указанную композицию можно
изменить в соответствии с разумными вариантами.
Форма 2: Таблетки
Композиции таблеток получают, используя следующие ингредиенты:
Ингредиент - Количество,
мг/таблетка
Активный ингредиент - 2,5 - 1000
Микрокристаллическая целлюлоза - 200 - 650
Двуокись кремния (белая сажа) - 10 - 650
Стеариновая кислота - 5 - 15
Все компоненты смешивают и прессуют в виде таблеток.
В другом варианте можно получить таблетки, каждая из которых содержит 2,5-1000,0 мг активного ингредиента, следующим
образом:
Форма 3: Таблетки.
Ингредиент - Количество, мг/таблетка
Активный ингредиент - 25 - 1000
Крахмал - 45
Микрокристаллическая целлюлоза
- 35
Поливинилпирролидон (10%-ный раствор в воде) - 4
Натрийкарбоксиметилцеллюлоза - 4,5
Стеарат магния - 0,5
Тальк - 1
Активный ингредиент, крахмал, и
целлюлозу пропускают через сито N 45 мешей США и тщательно перемешивают. С полученными порошками смешивают раствор поливинилпирролидона, все это пропускают через сито N 14 мешей США. Полученные
гранулы сушат при 50 - 60oC и пропускают через сито N 18 мешей США. Натрийкарбоксиметилцеллюлозу, стеарат магния и тальк, пропущенные предварительно через сито N 60 мешей США, добавляют к
гранулам, которые, после перемешивания составляющих, прессуют в таблетки.
Следующим образом получают суспензии, каждая из которых содержит 0,1-1000 кг медикамента на 5 мл дозу:
Форма 4: Суспензии.
Ингредиент - Количество, мг/5 мл:
Активный ингредиент - 0,1-1000
Натрийкарбоксиметилцеллюлоза - 50
Сироп - 1,25
Раствор
бензойной кислоты, мл - 0,10
Вкусовой агент - q.v.
Краситель - q.v.
Очищенная вода - До 5 мл
Медикамент пропускают через сито N 45 мешей США и
смешивают с натрийкарабоксиметилцеллюлозой и сиропом до получения однородной пасты. Добавляют при перемешивании раствор бензойной кислоты, вкусовой агент и краситель, разбавленные небольшим
количеством воды, после чего добавляют воду до нужного объема.
Аэрозольный раствор, содержащий следующие ингредиенты, получают следующим образом:
Форма 5: Аэрозоль.
Ингредиент - Количество, мас.%:
Активный ингредиент - 0,25
Этанол - 25,75
Пропеллант 22 /Хлордифторметан/ - 70,00
Активный ингредиент смешивают с
этанолом, полученную смесь добавляют к части пропелланта 22, охлажденного до 30oC, и переносят в заполняющее устройство. Затем нужное количество вводят в контейнеры из нержавеющей стали и
разбавляют остальным количеством пропелланта. Затем контейнер снабжают клапаном.
Следующим образом приготавливают суппозитории:
Форма 6: Суппозитории.
Ингредиент - Количество, мг/суппозиторий:
Активный ингредиент - 250
Глицериды насыщенных жирных кислот - 2000
Активный ингредиент пропускают через сито N 60 мешей США и
суспендируют в глицеридах насыщенных жирных кислот, которые предварительно расплавляют, используя минимальный необходимый нагрев. Затем полученную смесь выливают в формы для суппозиториев емкостью 2 г
и оставляют остывать.
Композиции для внутривенного введения получают следующим образом.
Форма 7: Раствор для внутривенного введения.
Ингредиент
- Количество
Активный ингредиент - 50 мг
Изотонический физиологический раствор - 1000 мл
Раствор вышеуказанных ингредиентов вводят пациенту внутривенно со скоростью около 1
мл в минуту.
Форма 8: Комбинированная капсула I.
Ингредиент - Количество, мг/капсула:
Активный ингредиент - 50
Премарин - 1
Авицел pH 101
- 50
Крахмал 1500 - 117,50
Силиконовое масло - 2
Твин 80 - 0,50
Cab-o-Sil - 0,25
Форма 9: Комбинированная капсула II.
Ингредиент
- Количество, мг/капсула:
Активный ингредиент - 50
Норэтилнодрел - 5
Авицел pH 101 - 82,50
Крахмал 1500 - 90
Силиконовое масло - 2
Твин 80 - 0,
50
Форма 10. Комбинированные таблетки.
Ингредиент - Количество, мг/капсула:
Активный ингредиент - 50
Премарин - 1
Кукурузный крахмал NF - 50
Повидон, K29-32 - 6
Авицел pH 101 - 41,50
Авицел pH 102 - 136,50
Кросповидон XZ10 - 2,50
Стеарат магния - 0,50
Cab-O-Sil - 0,50п
Реферат
Изобретение относится к бензотиофеновым соединениям формулы I, где R1 -Н, - ОН, -O(С1-С4алкил), - ОСОС6Н5-, ОСО(С1-С6алкил) или -OSO2(С2-С6алкил);
R2 - -Н, -ОН, -O(С1-С4алкил), ОСОС6Н5, ОСО(С1-С6алкил) , -OSO2(С2-С6алкил) или галоид; R3 - 1-пиперидинил,
1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино, диизопропиламино или 1-гексаметиленимино; n = 2 или 3; Z - -О- или -S-, или их фармацевтически
приемлемым солям. Технический результат - получение соединений, которые можно использовать для лечения при различных медицинских показаниях, связанных с постместруальным синдромом, фиброидными
заболеваниями матки, эндометризом и пролиферацией клеток гладкой мускулатуры аорты. 9 з.п. ф-лы, 3 табл.
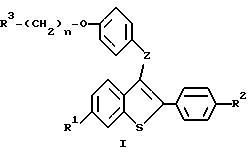
Формула

где R1 - -H, -OH, -O(C1 - C4алкил), -OCOC6H5, -OCO(C1 - C4алкил) или -OSO2(C2 - C6 алкил);
R2 - -H, -OH, -O(C1 - C4алкил), -OCOC6H5, -OCO(C1 - C6алкил), -OSO2(C2 - C6алкил) или галоид;
R3 - 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолино, диметиламино, диэтиламино, диизопропиламино или 1-гексаметиленимино;
n = 2 или 3;
Z - -O- или -S-;
или их фармацевтически приемлемые соли.

или его фармацевтически приемлемую соль.
Комментарии