Новые бициклические производные, способ их получения и фармацевтические композиции, содержащие их - RU2760554C1
Код документа: RU2760554C1
Описание
Настоящее изобретение относится к новым бициклическим производным, к способу их получения и к фармацевтическим композициям, содержащим их.
Соединения настоящего изобретения являются новыми и обладают очень ценными фармакологическими характеристиками в области апоптоза и онкологии.
Апоптоз, или запрограммированная гибель клеток, является физиологическим процессом, который имеет решающее значение для эмбрионального развития и поддержания тканевого гомеостаза.
Гибель клеток по типу апоптоза вовлекает морфологические изменения, такие как конденсация ядра, фрагментация ДНК, а также биохимический феномен, такой как активация каспаз, что вызывает повреждение ключевых структурных компонентов клетки, таким образом вызывая ее разборку и смерть. Регуляция процесса апоптоза является комплексной и задействует активацию или репрессию нескольких внутриклеточных путей передачи сигналов (Cory S. и др., Nature Review Cancer 2002, 2, 647-656).
Дерегулирование апоптоза вовлечено в определенные патологии. Повышенный апоптоз связан с нейродегенеративными заболеваниями, такими как болезнь Паркинсона, болезнь Альцгеймера и ишемия. Наоборот, недостаточности осуществления апоптоза играют важную роль в развитии злокачественных новообразований и их резистентности к химиотерапии, при аутоиммунных заболеваниях, воспалительных заболеваниях и вирусных инфекциях. Следовательно, отсутствие апоптоза является одним из характерных фенотипических признаков злокачественного новообразования (Hanahan D. и др., Cell 2000, 100, 57-70).
Антиапоптотические белки семейства Bcl-2 связаны с многочисленными патологиями. Задействование белков семейства Bcl-2 описано для многочисленных типов злокачественных новообразований, таких как рак ободочной кишки, рак молочной железы, мелкоклеточный рак легкого, немелкоклеточный рак легкого, рак мочевого пузыря, рак яичников, рак предстательной железы, хронический лимфоидный лейкоз, лимфома, миелома, острый миелоцитарный лейкоз, рак поджелудочной железы и т.д. Сверхэкспрессия антиапоптотических белков семейства Bcl-2 связана с онкогенезом, с устойчивостью к химиотерапии и с клиническим прогнозом пациентов, страдающих злокачественным новообразованием. В особенности, Mcl-1, член семейства антиапоптотических белков Bcl-2, сверхэкспрессируется при различных типах рака (Beroukhim R. и др., Nature 2010, 899-905). Таким образом, существует терапевтическая потребность в соединениях, которые ингибируют антиапоптотическую активность белков семейства Bcl-2.
Помимо того, что соединения настоящего изобретения являются новыми, они обладают проапоптотическими свойствами, что позволяет их применение при патологиях, в которые вовлечен дефект апоптоза, как, например, для лечения злокачественного новообразования и иммунопатологических и аутоиммунных заболеваний.
Более конкретно, настоящее изобретение относится к соединениям формулы (I):

где:

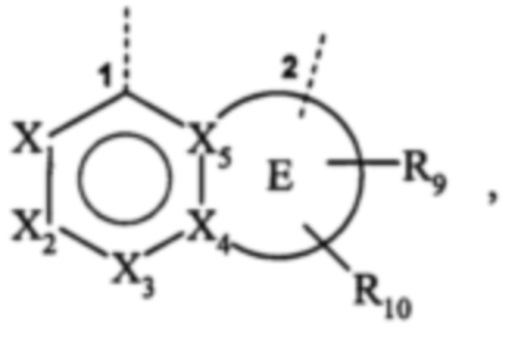
в которой 1 означает место присоединения к группе W и 2 означает место присоединения к фенильному кольцу, где:
- Е представляет собой фурильное, тиенильное или пирролильное кольцо,
- X1, Х3, Х4 и Х5 независимо друг от друга представляют собой атом углерода или атом азота,
- Х2 представляет собой группу C-R21 или атом азота, и
-

или заместители пары (R1, R2) вместе с атомами углерода, несущими их, образуют ароматическое или неароматическое кольцо, состоящее из 5-7 кольцевых членов, которое может содержать от 1 до 3 гетероатомов, выбранных из кислорода, серы и азота, при этом следует понимать, что полученное в результате кольцо может быть замещено 1-2 группами, выбранными из галогена, линейного или разветвленного (С1-С6)алкила, -алкил(С0-С6)-NR11R11', -NR13R13', -алкил(С0-С6)-Cy1 или оксо,
или заместители пары (R6, R7), когда они присоединены к двум расположенным рядом атомам углерода, вместе с атомами углерода, несущими их, образуют ароматическое или неароматическое кольцо, состоящее из 5-7 кольцевых членов, которое может содержать от 1 до 3 гетероатомов, выбранных из кислорода, серы и азота, при этом следует понимать, что полученное в результате кольцо может быть замещено группой, выбранной из линейной или разветвленной (С1-С6)алкильной группы, -NR13R13', -алкил(С0-С6)-Cy1 или оксо,
или заместители пары (R9, R10), когда они присоединены к двум расположенным рядом атомам углерода, вместе с атомами углерода, несущими их, образуют ароматическое или неароматическое кольцо, состоящее из 5-7 кольцевых членов, которое может содержать от 1 до 3 гетероатомов, выбранных из кислорода, серы и азота,
или заместители пары (R11, R11') вместе с атомом азота, несущим их, образуют ароматическое или неароматическое кольцо, состоящее из 5-7 кольцевых членов, которое может содержать в дополнение к атому азота от 1 до 3 гетероатомов, выбранных из кислорода, серы и азота, при этом следует понимать, что данный азот может быть замещен группой, представляющей собой атом водорода или линейную или разветвленную (С1-С6)алкильную группу,
или заместители пары (Rc, Rc') вместе с атомом азота, несущим их, образуют неароматическое кольцо, состоящее из 5-7 кольцевых членов, которое может содержать в дополнение к атому азота от 1 до 3 гетероатомов, выбранных из кислорода и азота, при этом следует понимать, что данный азот может быть замещен группой, представляющей собой линейную или разветвленную (С1-С6)алкильную группу,
при этом следует понимать, что:
- "арил" означает фенильную, нафтильную, бифенильную, инданильную или инденильную группу,
- "гетероарил" означает любую моно- или бициклическую группу, состоящую из 5-10 кольцевых членов, содержащую по меньшей мере один ароматический фрагмент и содержащую от 1 до 3 гетероатомов, выбранных из кислорода, серы и азота,
- "циклоалкил" означает любую моно- или бициклическую неароматическую карбоциклическую группу, содержащую от 3 до 10 кольцевых членов,
- "гетероциклоалкил" означает любую моно- или бициклическую неароматическую карбоциклическую группу, содержащую от 3 до 10 кольцевых членов и содержащую от 1 до 3 гетероатомов, выбранных из кислорода, серы и азота, которая может включать конденсированные, мостиковые или спиро-кольцевые системы,
причем арильные, гетероарильные, циклоалкильные и гетероциклоалкильные группы, определенные таким образом, и алкильные, алкенильные, алкинильные и алкокси группы могут быть замещены 1-4 группами, выбранными из необязательно замещенного линейного или разветвленного (С1-С6)алкила, необязательно замещенного линейного или разветвленного (С2-С6)алкенила, необязательно замещенного линейного или разветвленного (С2-С6)алкинила, необязательно замещенного линейного или разветвленного (С1-С6)алкокси, необязательно замещенного (С1-С6)алкил-S-, гидрокси, оксо (или N-оксида, в соответствующих случаях), нитро, циано, -С(O)-OR', -O-C(O)-R', -C(O)-NR'R'', -O-C(O)-NR'R'', -NR'R'', -(C=NR')-OR'', -O-P(O)(OR')2, -O-P(O)(O-M+)2, линейного или разветвленного (C1-С6)полигалогеналкила, трифторметокси, галогена или альдогексозы формулы:

где каждый R' является независимым;
при этом следует понимать, что R' и R'' независимо друг от друга представляют собой атом водорода или необязательно замещенную линейную или разветвленную (С1-С6)алкильную группу, и М+ представляет собой фармацевтически приемлемый одновалентный катион,
при условии, что


их энантиомерам, диастереоизомерам и атропоизомерам, и их солям присоединения с фармацевтически приемлемой кислотой или основанием.
Предпочтительно настоящее изобретение относится к соединениям формулы (I), где:
или заместители пары (R1, R2) вместе с атомами углерода, несущими их, образуют ароматическое кольцо, состоящее из 5-7 кольцевых членов, которое может содержать от 1 до 3 атомов азота,
или заместители пары (R9, R10), когда они присоединены к двум расположенным рядом атомам углерода, вместе с атомами углерода, несущими их, образуют неароматическое кольцо, состоящее из 5-7 кольцевых членов, которое может содержать от 1 до 3 гетероатомов, выбранных из кислорода, серы и азота,
или заместители пары (R11, R11') вместе с атомом азота, несущим их, образуют неароматическое кольцо, состоящее из 5-7 кольцевых членов, которое может содержать в дополнение к атому азота от 1 до 3 гетероатомов, выбранных из кислорода и азота, при этом следует понимать, что данный азот может быть замещен группой, представляющей собой линейную или разветвленную (С1-С6)алкильную группу,
причем арильные, гетероарильные, циклоалкильные и гетероциклоалкильные группы, определенные таким образом, и алкильные, алкенильные, алкинильные и алкоксигруппы могут быть замещены 1-4 группами, выбранными из необязательно замещенного линейного или разветвленного (С1-С6)алкила, необязательно замещенного линейного или разветвленного (С1-С6)алкокси, гидрокси, оксо (или N-оксида, в соответствующих случаях), -C(O)-OR', -C(O)-NR'R'', -O-C(O)-NR'R'', -NR'R'', -O-P(O)(OR')2, -O-P(O)(O-M+)2, линейного или разветвленного (C1-С6)полигалогеналкила, галогена или альдогексозы формулы:

где каждый R' является независимым;
при этом следует понимать, что R' и R'' независимо друг от друга представляют собой атом водорода или необязательно замещенную линейную или разветвленную (С1-С6)алкильную группу и М+ представляет собой фармацевтически приемлемый одновалентный катион.
Более конкретно, соединения формулы (I), которым отдают предпочтение, являются соединениями, где n означает целое число, равное 1.
В другом варианте осуществления изобретения предпочтительными являются соединения формулы (I-а):

где R1, R2, R3, R4, R5, R6, R7, R8, R9, R14, X1, X2, Х3 и W являются такими, как определено для формулы (I). Более конкретно, соединения формулы (I-а), которым отдают предпочтение, являются соединениями, где



Более конкретно, соединения формулы (I-а), которым отдают предпочтение, являются соединениями, где



Предпочтительно


В другом варианте осуществления изобретения предпочтительными являются соединения формулы (I-b):

где R1, R2, R3, R4, R5, R6, R7, R8, R9, R14, X1, X2, Х3 и W являются такими, как определено для формулы (I). Более конкретно, соединения формулы (I-b), которым отдают предпочтение, являются соединениями, где


Предпочтительно



В другом варианте осуществления изобретения предпочтительными являются соединения формулы (I-c):

где R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R14, X1, X2, Х3 и W являются такими, как определено для формулы (I). Более конкретно, соединения формулы (I-c), которым отдают предпочтение, являются соединениями, где



Более конкретно, соединения формулы (I-c), которым отдают предпочтение, являются соединениями, где


В другом варианте осуществления изобретения предпочтительными являются соединения формулы (I-d):

где R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R14, X1, X2, Х3 и W являются такими, как определено для формулы (I). Более конкретно, соединения формулы (I-d), которым отдают предпочтение, являются соединениями, где


В другом варианте осуществления изобретения предпочтительными являются соединения формулы (I-е):
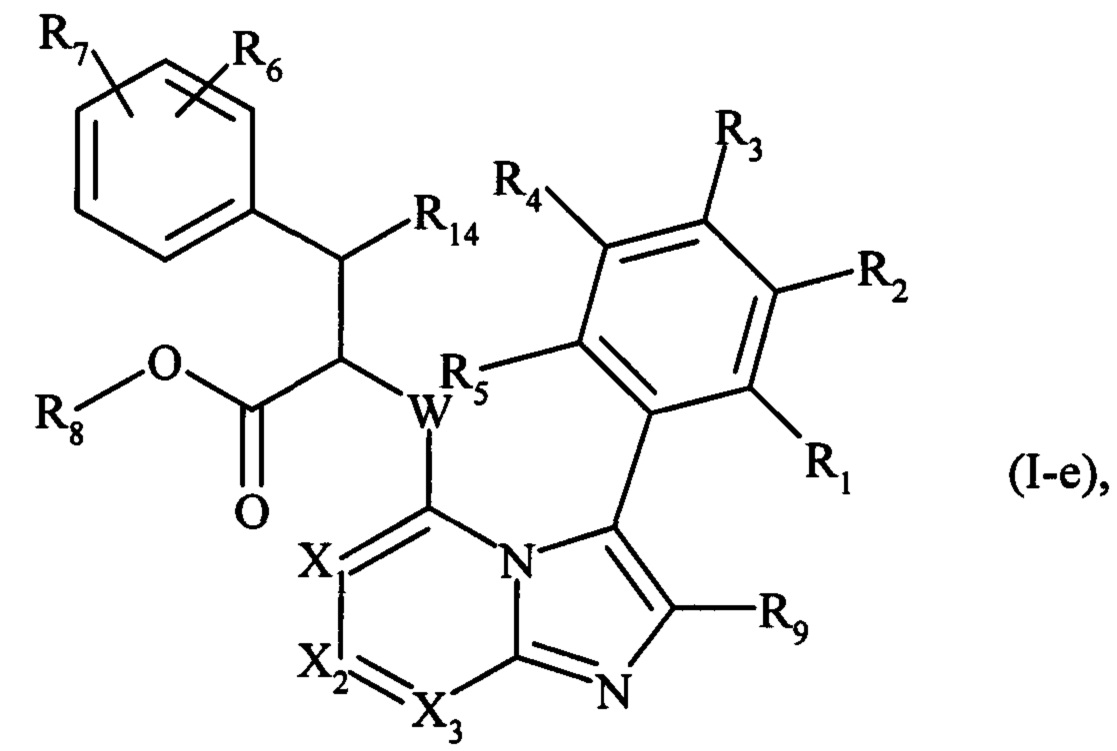
где R1, R2, R3, R4, R5, R6, R7, R8, R9, R14, X1, X2, X3 и W являются такими, как определено для формулы (I). Более конкретно, соединения формулы (I-е), которым отдают предпочтение, являются соединениями, где



Предпочтительно


Соединения формул (I-a), (I-b), (I-c) и (I-е) являются особенно предпочтительными. Соединения формул (I-a) и (I-b) являются еще более предпочтительными.
В другом варианте осуществления изобретения предпочтительными являются соединения формулы (I-f):

где Е, R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, R14, X1, X2, Х3, X4, X5 и W являются такими, как определено для формулы (I).
Атропоизомеры представляют собой стереоизомеры, возникающие из-за затрудненного вращения вокруг одинарной связи, где разности энергий вследствие пространственной деформации или других причин создают барьер для вращения, который является достаточно высоким для обеспечения выделения индивидуальных конформеров. Например, в случае соединений формулы (I-b) (то же самое можно продемонстрировать и для соединений формул (I-a), (I-c), (I-d) и (I-е)), атропоизомеры имеют следующее строение:

В случае соединений формул (I-а), (I-b), (I-c) и (I-d) предпочтительным атропоизомером является (Sa). В случае соединений формулы (I-е) предпочтительным атропоизомером является (Ra).
Предпочтительно по меньшей мере одна группа, выбранная из R2, R3, R4 и R5, не представляет собой атом водорода.
Предпочтительно R14 представляет собой атом водорода.
R21 представляет собой предпочтительно атом водорода, атом фтора, метильную группу или цианогруппу. Более предпочтительно R21 представляет собой атом водорода или атом фтора. Еще более предпочтительно R21 представляет собой атом водорода.
В предпочтительных соединениях изобретения R1 представляет собой линейную или разветвленную (С1-С6)алкильную группу или атом галогена. Более предпочтительно R1 представляет собой метильную группу, этильную группу, атом брома или атом хлора. Еще более предпочтительно R1 представляет собой метильную группу или этильную группу.
Предпочтительно R2 представляет собой атом галогена, гидроксигруппу, линейную или разветвленную (С1-С6)алкокси группу. Более предпочтительно R2 представляет собой метокси группу, гидроксигруппу, атом фтора, атом брома или атом хлора. Еще более предпочтительно R2 представляет собой атом хлора.
В некоторых предпочтительных вариантах осуществления изобретения когда заместители пары (R1, R2) вместе с атомами углерода, несущими их, образуют ароматическое кольцо,


R3 предпочтительно представляет собой атом водорода, гидроксигруппу, линейную или разветвленную (С1-С6)алкоксигруппу или -O-алкил(C1-C6)-NR11R11'. Предпочтительно R3 представляет собой -O-алкил(С1-С6)-NR11R11'.
R4 и R5 предпочтительно представляют собой атом водорода.
В предпочтительном варианте осуществления заместители пары (R1, R5) являются одинаковыми и заместители пары (R2, R4) являются одинаковыми. В предпочтительных соединениях изобретения заместители пары (R1, R5) являются одинаковыми и представляют собой (С1-С6)алкильную группу, предпочтительно метильную группу, в то время как заместители пары (R2, R4) являются одинаковыми и представляют собой атом галогена, предпочтительно атом хлора, или атом водорода.
В предпочтительных соединениях изобретения


где R11 и R11' являются такими, как определено для формулы (I).
В другом варианте осуществления изобретения Re представляет собой атом водорода, необязательно замещенную линейную или разветвленную (С1-С6)алкоксигруппу или группу -O-алкил(С1-С6)-R12. Предпочтительно R6 представляет собой 2,2,2-трифторэтокси группу, метоксигруппу или группу -O-алкил(C1-C6)-R12.
R7 предпочтительно представляет собой атом водорода.
В предпочтительных соединениях изобретения

где R12 является таким, как определено для формулы (I).
В другом варианте осуществления изобретения предпочтительными являются соединения формулы (I-g):
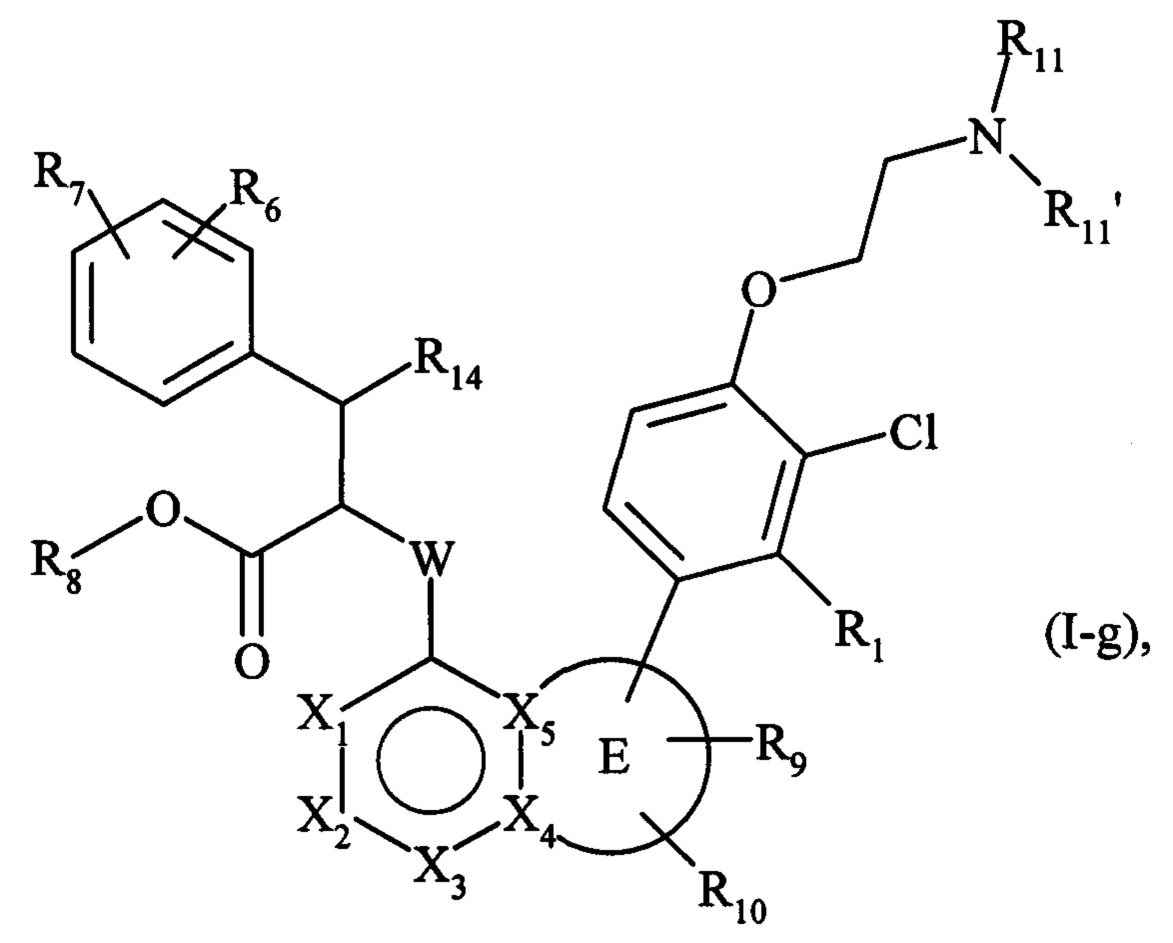
где R1, R6, R7, R8, R9, R10, R11, R11', R14, X1, X2, Х3, X4, X5, W и Е являются такими, как определено для формулы (I).
Предпочтительно R8 представляет собой атом водорода, группу -CHRaRb, необязательно замещенную линейную или разветвленную (С1-С8)алкильную группу или группу гетероарилалкил(С1-С6). Предпочтительно R8 представляет собой группу -CHRaRb, в которой Ra представляет собой атом водорода или метальную группу и Rb представляет собой -O-С(O)-O-(С1-С8)алкильную группу; -O-С(O)-O-циклоалкильную группу; группу -O-C(O)-NRcRc', в которой Rc и Rc' независимо друг от друга представляют собой атом водорода, линейную или разветвленную (С1-С8)алкильную группу, (С1-С6)алкокси(С1-С6)алкильную группу, (С1-С6)алкоксикарбонил(С1-С6)алкильную группу, или заместители пары (Rc, Rc') вместе с атомом азота, несущим их, образуют неароматическое кольцо, состоящее из 5-7 кольцевых членов, которое может содержать в дополнение к атому азота от 1 до 3 гетероатомов, выбранных из кислорода и азота; или группу -O-Р(O)(ОН)2. Предпочтительными группами R8 являются следующие: водород; метил; этил; (5-метил-2-оксо-1,3-диоксол-4-ил)метил; группа -CHRaRb, в которой Ra представляет собой метильную группу и Rb представляет собой группу -O-С(O)-O-СН2СН3 или группу -O-С(O)-N(СН3)2. Еще более предпочтительно R8 представляет собой водород.
В предпочтительных соединениях изобретения R9 представляет собой атом водорода, атом галогена, линейную или разветвленную (С1-С6)алкильную группу, линейную или разветвленную (С2-С6)алкенильную группу, линейную или разветвленную (С2-С6)алкинильную группу, арильную группу или гетероарильную группу. Более предпочтительно R9 представляет собой проп-1-ин-1-ильную группу, фенильную группу или фуран-2-ильную группу. В более предпочтительном варианте осуществления R9 представляет собой проп-1-ин-1-ильную группу, 4-фторфенильную группу или 5-фторфуран-2-ильную группу. Еще более предпочтительно R9 представляет собой 4-фторфенильную группу.
В предпочтительном варианте соединений формулы (I-c), предпочтительными R10 группами являются следующие: водород; метил; изопропил; 2,2,2-трифторэтил; бензил; 4-метоксибензил; фенэтил; 3-фенилпропил; циклопропилметил; циклопентилэтил; нафталин-1-илметил; 2-(нафталин-1-илокси)этил; бут-2-ин-1-ил; проп-2-ен-1-ил; бут-3-ен-1-ил. В другом варианте осуществления заместители пары (R9, R10), когда они присоединены к двум расположенным атомам, вместе с атомами углерода и азота, несущими их, образуют неароматическое кольцо, состоящее из 5-6 кольцевых членов.
В предпочтительном варианте соединений формулы (I-d) радикал R10 предпочтительно представляет собой атом водорода или атом галогена.
В предпочтительных соединениях изобретения R11 и R11' независимо друг от друга представляют собой линейную или разветвленную (С1-С6)алкильную группу, или заместители пары (R11, R11') вместе с атомом азота, несущим их, образуют неароматическое кольцо, состоящее из 5-7 кольцевых членов, которое может содержать в дополнение к атому азота от 1 до 3 гетероатомов, выбранных из кислорода, серы и азота, при этом следует понимать, что данный азот может быть замещен группой, представляющей собой атом водорода, линейную или разветвленную (С1-С6)алкильную группу. Более предпочтительно R11 и R11' представляют собой метильную группу, или заместители пары (R11, R11') вместе образуют 4-метилпиперазинильную группу или 4-этилпиперазинильную группу. В более предпочтительном варианте осуществления заместители пары (R11, R11') вместе образуют 4-метилпиперазинильную группу. В другом предпочтительном варианте осуществления R11 и R11' представляют собой метильную группу.
Предпочтительно R12 представляет собой -Cy5 или -Cy5-алкил(С0-С6)-Cy6. Предпочтительно R12 представляет собой -Cy5 или -Cy5-Cy6.
Cy5 предпочтительно представляет собой гетероарильную группу, в частности, пиримидинильную группу, пиразолильную группу, триазолильную группу, пиразинильную группу или пиридинильную группу. Более предпочтительно Cy5 представляет собой пиримидин-4-ильную группу, пиразол-5-ильную группу или пиразин-2-ильную группу. В предпочтительных соединениях изобретения Cy5 представляет собой пиримидин-4-ильную группу.
В другом варианте осуществления изобретения Cy5 представляет собой гетероарильную группу, которая замещена необязательно замещенной линейной или разветвленной (С1-С6)алкильной группой, необязательно замещенной линейной или разветвленной (С1-С6)алкокси группой, группой -NR'R'', или линейной или разветвленной (С1-С6)полигалогеналкильной группой, при этом следует понимать, что R' и R'' независимо друг от друга представляют собой атом водорода или необязательно замещенную линейную или разветвленную (С1-С6)алкильную группу.
Cy6 предпочтительно представляет собой фенильную группу.
Другими соединениями изобретения, которым отдают предпочтение, являются те, в которых,
R12 представляет собой

где p означает целое число, равное 0 или 1, и R16 представляет собой атом водорода, гидроксигруппу, необязательно замещенную линейную или разветвленную (С1-С6)алкильную группу, линейную или разветвленную (C1-С6)алкоксигруппу, группу -O-(CHR17-CHR18-O)q-R', группу -O-P(O)(OR')2, группу -O-P(O)(O-M+)2, группу -O-C(O)-NR19R20, ди(С1-С6)алкиламино(С1-С6)алкокси группу, атом галогена или альдогексозу формулы:

где каждый R' является независимым;
при этом следует понимать, что:

Альдогексоза в соответствии с изобретением предпочтительно представляет собой D-маннозу. Предпочтительно группа -(CH2)p-R16 расположена в орто-положении фенильной группы.
Из числа предпочтительных соединений изобретения могут быть упомянуты:
- (2R)-2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота;
- (2R)-2-{[5-{3-хлор-2-этил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота;
- N-[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}-D-фенилаланин;
- (2R)-2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1-бензотиофен-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота;
- (2R)-2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1-бензофуран-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота;
- (2R)-2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-фтор-2-(4-фторфенил)-1-бензофуран-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота;
- (2R)-2-{[3-{(3Sa)-3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1-метил-1H-индол-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота;
- (2R)-2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота;
- (2R)-2-[5-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-6-(4-фторфенил)-7-метилпирроло[2,3-d]пиримидин-4-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропановая кислота;
- 1-[(диметилкарбамоил)окси]этил(2R)-2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропаноат;
- 1-[(этоксикарбонил)окси]этил(2R)-2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропаноат;
- N-[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}-D-фенилаланин;
- N-[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[3,2-с]пиридин-4-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенилаланин;
- 2-{[(3Ra)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)имидазо[1,2-с]пиримидин-5-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота.
Изобретение также относится к способу получения соединений формулы (I), который отличается тем, что в качестве исходного вещества применяют соединение формулы (II-а):

где Z1 представляет собой бром или йод, Z2 представляет собой хлор, бром или гидрокси, и А является таким, как определено для формулы (I), в которой 1 означает место присоединения к группе Z2 и 2 означает место присоединения к группе Z1,
причем соединение формулы (II-а) подвергают сочетанию с соединением формулы (III):

где R6, R7, R14, W и n являются такими, как определено для формулы (I), и Alk представляет собой линейную или разветвленную (С1-С6)алкильную группу,
с получением соединения формулы (IV):

где R6, R7, R14, A, W и n являются такими, как определено для формулы (I), и Z1 и Alk являются такими, как определено выше,
соединение формулы (IV) далее подвергают сочетанию с соединением формулы (V):

где R1, R2, R3, R4 и R5 являются такими, как определено для формулы (I), и RB1 и RB2 представляют собой атом водорода, линейную или разветвленную (C1-С6)алкильную группу, или RB1 и RB2 вместе с атомами кислорода, несущими их, образуют необязательно метилированное кольцо,
с получением соединения формулы (VI):

где R1, R2, R3 R4, R5, R6, R7, R14, A, W и n являются такими, как определено для формулы (I), и Alk является таким, как определено выше,
Alk-O-C(O)- сложноэфирную функцию соединения формулы (VI) гидролизуют с получением карбоновой кислоты, которую необязательно можно подвергнуть реакции со спиртом формулы R8'-OH или хлорированным соединением формулы R8'-Cl, где R8' представляет собой линейную или разветвленную (С1-С8)алкильную группу, группу -CHRaRb, арильную группу, гетероарильную группу, группу арилалкил(С1-С6) или группу гетероарилалкил(С1-С6), Ra и Rb являются такими, как определено для формулы (I),
с получением соединения формулы (I), которое можно очистить в соответствии с обычными методиками разделения, которое превращают, при необходимости, в его соли присоединения с фармацевтически приемлемой кислотой или основанием и которое необязательно разделяют на его изомеры в соответствии с обычными методиками разделения,
при этом следует понимать, что в любое время, признанное подходящим в ходе описанного выше способа, некоторые группы (гидрокси, амино …) исходных реагентов или промежуточных соединений синтеза могут быть защищены, впоследствии лишены защиты и функционализированы, как того требует синтез.
В другом варианте осуществления изобретения соединения формулы (I) можно получить с использованием альтернативного способа, который отличается тем, что в качестве исходного вещества применяют соединение формулы (II-b):

где Z3 представляет собой йод, Z4 представляет собой хлор, гидрокси, и А является таким, как определено для формулы (I), в которой 1 означает место присоединения к группе Z4 и 2 означает место присоединения к группе Z3,
причем соединение формулы (II-b) подвергают сочетанию с соединением формулы (V):

где R1, R2, R3, R4 и R5 являются такими, как определено для формулы (I), и RB1 и RB2 представляют собой атом водорода, линейную или разветвленную (C1-С6)алкильную группу, или RB1 и RB2 вместе с атомами кислорода, несущими их, образуют необязательно метилированное кольцо,
с получением соединения формулы (VII):
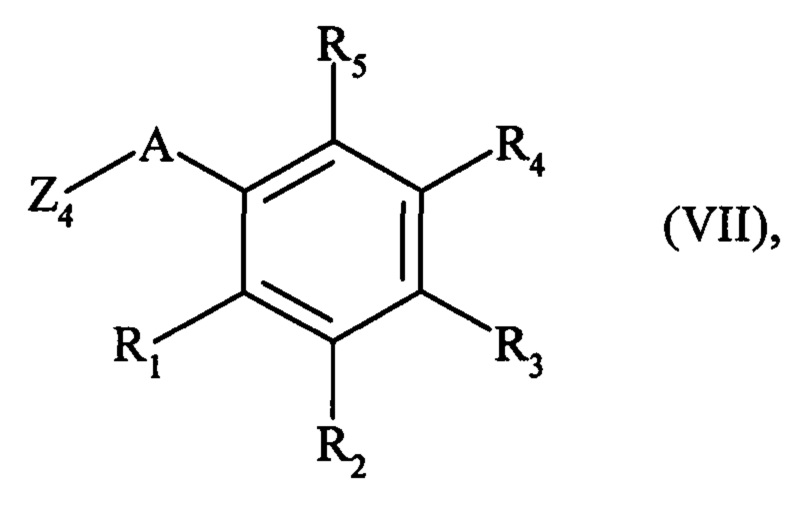
где R1, R2, R3 R4, R5 и А являются такими, как определено для формулы (I), и Z4 является таким, как определено выше,
соединение формулы (VII) далее подвергают сочетанию с соединением формулы (III):

где R6, R7, R14, W и n являются такими, как определено для формулы (I), и Alk представляет собой линейную или разветвленную (С1-С6)алкильную группу,
с получением соединения формулы (VI):

где R1, R2, R3 R4, R5, R6, R7, R14, А, W и n являются такими, как определено для формулы (I), и Alk является таким, как определено выше,
Alk-O-C(O)- сложноэфирную функцию соединения формулы (VI) гидролизуют с получением карбоновой кислоты, которую необязательно можно подвергнуть реакции со спиртом формулы R8'-OH или хлорированным соединением формулы R8'-Cl, где R8' представляет собой линейную или разветвленную (С1-С8)алкильную группу, группу -CHRaRb, арильную группу, гетероарильную группу, группу арилалкил(С1-С6) или группу гетероарилалкил(С1-С6), Ra и Rb являются такими, как определено для формулы (I),
с получением соединения формулы (I), которое можно очистить в соответствии с обычными методиками разделения, которое превращают, при необходимости, в его соли присоединения с фармацевтически приемлемой кислотой или основанием и которое необязательно разделяют на его изомеры в соответствии с обычными методиками разделения,
при этом следует понимать, что в любое время, признанное подходящим в ходе описанного выше способа, некоторые группы (гидрокси, амино …) исходных реагентов или промежуточных соединений синтеза могут быть защищены, впоследствии лишены защиты и функционализированы, как того требует синтез.
Соединения формул (II-а), (II-b), (III), (V), R8'-OH и R8'-Cl либо доступны для приобретения, либо могут быть получены специалистом в данной области техники с использованием обычных химических реакций, описанных в литературе.
Фармакологическое исследование соединений изобретения показало, что они обладают проапоптотическими свойствами. Способность реактивировать апоптотический процесс в раковых клетках представляет большой терапевтический интерес для лечения злокачественных новообразований и иммунопатологических и аутоиммунных заболеваний.
Более конкретно, соединения в соответствии с изобретением будут полезны для лечения хемо- или радиорезистентных злокачественных новообразований.
Из числа намеченных противоопухолевых терапий может быть упомянута, без какого-либо ограничения, терапия рака мочевого пузыря, головного мозга, молочной железы и матки, хронического лимфоидного лейкоза, рака ободочной кишки, пищевода и печени, лимфобластного лейкоза, острого миелоцитарного лейкоза, лимфом, меланом, злокачественных заболеваний крови, миелом, рака яичников, немелкоклеточного рака легкого, рака предстательной железы, рака поджелудочной железы и мелкоклеточного рака легкого.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим по меньшей мере одно соединение формулы (I) в комбинации с одним или несколькими фармацевтически приемлемыми наполнителями.
Из числа фармацевтических композиций в соответствии с изобретением могут быть упомянуты более конкретно те, которые подходят для перорального, парентерального, назального, чрес- или транскожного, ректального, перлингвального, офтальмологического или респираторного введения, в особенности таблетки или драже, сублингвальные таблетки, саше, пакеты, капсулы, глоссеты, пастилки, суппозитории, кремы, мази, гели для кожи и питьевые или инъекционные ампулы.
Дозировка варьируется в зависимости от пола, возраста и веса пациента, пути введения, природы терапевтического показания, или каких-либо сопутствующих лечений, и находится в диапазоне от 0.01 мг до 1 г в 24 часа за одно или несколько введений.
Кроме того, настоящее изобретение также относится к комбинации соединения формулы (I) с противоопухолевым средством, выбранным из генотоксичных средств, митотических ядов, антиметаболитов, ингибиторов протеасом, ингибиторов киназ и антител, а также к фармацевтическим композициям, содержащим такой тип комбинации и их применению для изготовления лекарственных средств для применения для лечения злокачественного новообразования.
Предпочтительно настоящее изобретение относится к комбинации соединения формулы (I) с ингибитором EGFR, а также к фармацевтическим композициям, содержащим такой тип комбинации.
В другом варианте осуществления настоящее изобретение относится к комбинации соединения формулы (I) с ингибитором mTOR/PI3K, а также к фармацевтическим композициям, содержащим такой тип комбинации.
В предпочтительном варианте осуществления настоящее изобретение относится к комбинации соединения формулы (I) с ингибитором MEK, а также к фармацевтическим композициям, содержащим такой тип комбинации.
Предпочтительно настоящее изобретение относится к комбинации соединения формулы (I) с ингибитором HER2, а также к фармацевтическим композициям, содержащим такой тип комбинации.
Предпочтительно настоящее изобретение относится к комбинации соединения формулы (I) с ингибитором RAF, а также к фармацевтическим композициям, содержащим такой тип комбинации.
В другом варианте осуществления настоящее изобретение относится к комбинации соединения формулы (I) с ингибитором EGFR/HER2, а также к фармацевтическим композициям, содержащим такой тип комбинации.
В предпочтительном варианте осуществления настоящее изобретение относится к комбинации соединения формулы (I) с таксаном, а также к фармацевтическим композициям, содержащим такой тип комбинации.
В другом варианте осуществления настоящее изобретение относится к комбинации соединения формулы (I) с ингибитором протеасом, иммуномодулятором или алкилирующим агентом, а также к фармацевтическим композициям, содержащим такой тип комбинации.
Комбинацию соединения формулы (I) с противоопухолевым средством можно вводить одновременно или последовательно. Предпочтительным путем введения является пероральный, причем соответствующие фармацевтические композиции могут обеспечивать незамедлительное или отсроченное высвобождение активных компонентов. Кроме того, соединения комбинации можно вводить в виде двух отдельных фармацевтических композиций, каждая из которых содержит один из активных компонентов, или в виде одной фармацевтической композиции, в которой активные компоненты находятся в смеси.
Для лечения злокачественного новообразования соединения изобретения также можно применять в комбинации с радиотерапией.
В заключение, соединения изобретения могут быть связаны с моноклональными антителами или их фрагментами или связаны с каркасными белками, которые могут относиться или не относиться к моноклональным антителам.
Фрагменты антител следует понимать как фрагменты Fv, scFv, Fab, F(ab')2, F(ab'), scFv-Fc типа или диатела, которые обычно имеют такую же специфичность связывания, что и антитело, из которого они происходят. В соответствии с настоящим изобретением, фрагменты антител изобретения могут быть получены исходя из антител с помощью методов, таких как переваривание ферментами, такими как пепсин или папаин, и/или посредством расщепления дисульфидных мостиков с помощью химического восстановления. Другим путем, фрагменты антител, включенных в настоящее изобретение, могут быть получены с использованием методик генетической рекомбинации, также хорошо известных специалисту в данной области техники, или даже посредством пептидного синтеза с помощью, например, автоматических пептидных синтезаторов, таких как те, которые поставляются компанией Applied Biosystems, и т.д.
Под каркасными белками, которые могут относиться или не относиться к моноклональным антителам, понимают белок, который содержит или не содержит укладку цепи иммуноглобулинов и который обеспечивает способность к связыванию, такую же, как и у моноклонального антитела. Специалисту в данной области техники известно, каким образом выбрать каркас белка. Более конкретно, известно, что должен быть выбран такой каркас, который будет демонстрировать несколько следующих отличительных признаков (Skerra A., J. Mol. Recogn. 2000, 13. 167-187): хорошая филогенетическая консервативность, прочная архитектура с хорошо известной трехмерной молекулярной организацией (как, например, на основании кристаллографии или ЯМР), небольшой размер, отсутствие или лишь низкая степень посттрансляционных модификаций, простота получения, экспрессии и очистки. Таким каркасным белком может быть, без ограничения перечисленным, структура, выбранная из группы, состоящей из фибронектина и предпочтительно десятого домена фибронектина типа III (FNfn10), липокалина, антикалина (Skerra A., J. Biotechnol. 2001, 74(4):257-75), белка Z, производного из домена В стафилококкового белка А, тиоредоксина А или любого белка с повторяющимся доменом, таким как "анкириновый повтор" (Kohl и др., PNAS 2003, 100(4), 1700-1705), "армадилло повтор", "богатый лейцином повтор" или "тетратрикопептидный повтор". Также можно упомянуть каркас, производный из токсинов (как, например, токсины скорпиона, насекомых, растений или моллюсков) или белковых ингибиторов нейрональной синтазы оксида азота (PIN).
Следующие Синтезы и Примеры иллюстрируют изобретение, но не ограничивают его каким-либо образом.
Общие Методики
Все реагенты, полученные из коммерческих источников, использовали без дополнительной очистки. Безводные растворители получали из коммерческих источников и использовали без дополнительной сушки.
Флэш-хроматографию выполняли на приборе ISCO CombiFlash Rf 200i с предварительно заправленными силикагелевыми картриджами (RediSep®Rf Gold High Performance).
Тонкослойную хроматографию проводили на 5×10 см пластинах, покрытых силикагелем Merck Type 60 F254.
Нагревание микроволновым излучением выполняли в приборе Anton Parr MonoWave или СЕМ Discover®.
Операции очистки с помощью препаративной ВЭЖХ выполняли на системе Armen Spot Liquid Chromatography с Gemini-NX® 10 мкм С18, 250 мм ×50 мм в.д. колонкой, работая при скорости потока 118 мл⋅мин-1 с УФ детектированием на диодной матрице (210-400 нм), используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов, если не указано иное.
Аналитическая ЖХ-МС: соединения настоящего изобретения характеризовали с помощью высокоэффективной жидкостной хроматографии - масс-спектроскопии (ВЭЖХ-МС) на приборе Agilent HP1200 с Agilent 6140 квадрупольным ЖХ/МС, работая с электрораспылительной ионизацией в режиме положительных или отрицательных ионов. Диапазон сканирования молекулярных масс составлял от 100 до 1350. Параллельное УФ-детектирование проводили на 210 нм и 254 нм. Образцы вводили в виде 1 мМ раствора в ACN, или в смеси ТГФ/Н2О (1:1) с помощью 5 мкл петлевого дозатора. ЖХМС анализы выполняли на двух приборах, один из которых работал с основными, а другой с кислыми элюентами.
Основная ЖХМС: Gemini-NX, 3 мкм, С18, 50 мм ×3.00 мм в.д. колонка при 23°С, при скорости потока 1 мл⋅мин-1, используя 5 мМ бикарбонат аммония (растворитель А) и ацетонитрил (растворитель В) с градиентом, начиная со 100%-ного растворителя А и завершая 100%-ным растворителем В в течение различных/определенных промежутков времени.
Кислая ЖХМС: ZORBAX Eclipse XDB-C18, 1.8 мкм, 50 мм ×4.6 мм в.д. колонка при 40°С, при скорости потока 1 мл⋅мин-1, используя 0.02% об./об. водную муравьиную кислоту (растворитель А) и 0.02% об./об. муравьиную кислоту в ацетонитриле (растворитель В) с градиентом, начиная со 100%-ного растворителя А и завершая 100%-ным растворителем В в течение различных/определенных промежутков времени.
1H-ЯМР исследования выполняли на спектрометре Bruker Avance III 500 МГц и спектрометре Bruker Avance III 400 МГц, используя ДМСО-d6 или CDCl3 в качестве растворителя.lH ЯМР данные представлены в виде дельта-значений, приведенных в миллионных долях (м.д.), используя остаточный пик растворителя (2.50 м.д. для ДМСО-d6 и 7.26 м.д. для CDCl3) в качестве внутреннего стандарта. Картины расщепления обозначены в виде: s (синглет), d (дублет), t (триплет), q (квартет), quint (квинтет), m (мультиплет), br s (широкий синглет), dd (дублет дублетов), td (триплет дублетов), dt (дублет дублетов), ddd (дублет дублета дублетов).
Газовую хроматографию, комбинированную с масс-спектрометрией низкого разрешения, выполняли на газовом хроматографе Agilent 6850 и масс-спектрометре Agilent 5975C, используя 15 м ×0.25 мм колонку с 0.25 мкм покрытием HP-5MS и гелием в качестве газа-носителя. Источник ионов: EI+, 70 эВ, 230°С, квадруполь: 150°С, интерфейс: 300°С.
Данные МСВР определяли на приборе Shimadzu IT-TOF, температура источника ионов 200°С, ESI +/-, напряжение ионизации: (+-)4.5 кВ. Разрешение по массам мин. 10000.
Элементарные анализы выполняли на элементном анализаторе Thermo Flash ЕА 1112.
Перечень сокращений
Общая Методика Ia
1 экв. соединения Синтеза 1а, 2 экв. подходящего производного сложного эфира молочной кислоты, 10 мл/ммольtBuOH и 5 экв. Cs2CO3 помещали в колбу и перемешивали при 55°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь концентрировали при пониженном давлении, нейтрализовали 1 М водным раствором HCl, разбавляли соляным раствором и экстрагировали с помощью EtOAc. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя, если не указано иное, гептан и EtOAc в качестве элюентов.
Общая Методика Ib
1 экв. соединения Синтеза 1а, 2 экв. подходящего аминокислотного производного, 10 мл/ммоль ДМСО и 3 экв. K2CO3 помещали в колбу и перемешивали при 45°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь нейтрализовали 1 М водным раствором HCl, разбавляли соляным раствором и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт, если не указано иное, очищали с помощью ЖХГВ хроматографии.
Общая Методика II
Стадия А
1 экв. подходящего производного сложного эфира 5-бромфуро[2,3-d]пиримидилмолочной кислоты, 1.25 экв. подходящего производного бороновой кислоты, 10 мол.% AtaPhos и 3 экв. Cs2CO3 растворяли в 1:1 смеси диоксана и воды (10 мл/ммоль производного сложного эфира 5-бромфуро[2,3-d]пиримидилмолочной кислоты) и перемешивали при 105°С в MW реакторе до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь нейтрализовали 1 М водным раствором HCl, разбавляли соляным раствором и экстрагировали с помощью ТГФ. Объединенные органические фазы сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с использованием препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов.
Стадия В
Полученное промежуточное соединение растворяли в 1:1 смеси диоксана и воды (25 мл/ммоль) и добавляли 10 экв. LiOH×Н2О. Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли соляным раствором, нейтрализовали 2 М водным раствором HCl, экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Диастереоизомеры очищали и разделяли с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов.
Общая Методика III
1 экв. подходящего 4-хлорпирроло[2,3-d]пиримидинового производного, 3 экв. подходящего аминокислотного производного, 10 мл/ммоль ДМСО и 4 экв. K2CO3 перемешивали при 150°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь подкисляли 1 М водным раствором HCl, осадок отфильтровывали и очищали с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов.
Общая Методика IVa
1 экв. подходящего 5-бромпирроло[2,3-d]пиримидинового производного, 3 экв. подходящего производного бороновой кислоты, 3 экв. ТВАОН, 0.2 экв. ацетата палладия, 0.4 экв. тетрафторбората трициклогексилфосфония и 3.5 мл/ммоль DME перемешивали в атмосфере N2 при 120°С в MW реакторе до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь фильтровали через целит и промывали МТВЕ и водой. Слои разделяли, водный слой промывали МТВЕ. Объединенные органические слои промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 40 мМ водный раствор NH4OAc (рН=4) и MeCN в качестве элюентов.
Общая Методика IVb
1 экв. подходящего 5-йодпирроло[2,3-d]пиримидинового производного, 3 экв. подходящего производного бороновой кислоты, 3 экв. ТВАОН, 0.2 экв. ацетата палладия, 0.4 экв. бутилди-1-адамантилфосфина и 7 мл/ммоль DME перемешивали в атмосфере N2 при нагревании в колбе с обратным холодильником до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь фильтровали через целит и концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии, используя ДХМ и МеОН в качестве элюентов.
Общая Методика V
1 экв. подходящего производного бензофуран-4-ола, 2.5 экв. подходящего производного сложного эфира молочной кислоты, 2.5 экв. DTAD и 2.5 экв. PPh3 растворяли в сухом толуоле (20 мл/ммоль) и перемешивали при 55°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь концентрировали и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов.
Общая Методика VI
1 экв. подходящего производного 3-бромбензофурана, 2 экв. подходящего производного бороновой кислоты, 2 экв. Cs2CO3, 10 мол. % Ataphos, 1.5 экв. тетрафторбората три-трет-бутилфосфония и ТГФ (10 мл/ммоль) и воду (4 мл/ммоль) перемешивали в атмосфере N2 при 110°С в MW реакторе до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь подкисляли 1 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенные органические слои промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов. Полученное промежуточное соединение растворяли в смеси диоксан : вода 1:1 (10 мл/ммоль), добавляли 10 экв. LiOH×H2O и смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли водой, подкисляли 1 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов.
Синтез 1а: 5-бром-4-хлор-6-(4-фторфенил)фуро[2,3-d]пиримидин
Стадия А: 2-(4-фторбензоил)пропандинитрил
81 мл 1 М раствора NaOEt в EtOH (81 ммоль) охлаждали до 0°С и добавляли 6.14 г малононитрила (93 ммоль). Смесь перемешивали при 0°С в течение 1 часа, затем добавляли 16.8 г 2-бром-1-(4-фторфенил)этанона (77.4 ммоль). Смесь перемешивали при 0°С в течение 1 часа, затем при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Летучие компоненты удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 2-(4-фторбензоил)пропандинитрила.lH ЯМР (400 МГц, CDCl3): 8.1 (m, 2Н), 7.24 (m, 2H), 4.41 (t, 1H), 3.75 (d, 2H)
Стадия В: 2-амино-5-(4-фторфенил)фуран-3-карбонитрил
6.56 г 2-(4-фторбензоил)пропандинитрила (28.5 ммоль) растворяли в 140 мл АсОН и добавляли 6 г Amberlite 15H+. Смесь перемешивали при 90°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь фильтровали, фильтрат концентрировали при пониженном давлении. Остаток перекристаллизовывали из ДХМ с получением 2-амино-5-(4-фторфенил)фуран-3-карбонитрила.1H ЯМР (400 МГц, ДМСО-d6): 7.69 (m, 2H), 7.24 (m, 2H), 6.96 (s, 1H)
Стадия С: 6-(4-фторфенил)-3Н-фуро[2,3-d]пиримидин-4-он
1290 мг 2-амино-5-(4-фторфенил)фуран-3-карбонитрила (6.38 ммоль) и 25.5 мл муравьиноуксусного ангидрида помещали в колбу и перемешивали при к.т. в течение 30 минут. Затем, летучие компоненты упаривали при пониженном давлении. Остаток растворяли в 51 мл АсОН и нагревали в MW реакторе при 160°С в течение 30 минут, затем при 180°С в течение 15 минут. Затем смесь охлаждали до к.т., и осадок отфильтровывали с получением 6-(4-фторфенил)-3H-фуро[2,3-d]пиримидин-4-она.1Н ЯМР (500 МГц, ДМСО-d6): 12.66 (br s, 1H), 8.15 (s, 1H), 7.99 (m, 2H), 7.47 (s, 1H), 7.33 (m, 2H)
Стадия D: 5-бром-6-(4-фторфенил)-3Н-фуро[2,3-d]пиримидин-4-он
1704 мг 6-(4-фторфенил)-3H-фуро[2,3-d]пиримидин-4-она (7.4 ммоль) растворяли в 74 мл АсОН, затем добавляли 1182 мг брома (7.4 ммоль). Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем фильтровали, фильтрат концентрировали при пониженном давлении. Остаток дигерировали 15 мл МеОН, отфильтровывали и сушили на воздухе с получением 5-бром-6-(4-фторфенил)-3H-фуро[2,3-d]пиримидин-4-она. МС: (М-H)+=309.0
Стадия Е: соединение Синтеза 1a
1680 мг 5-бром-6-(4-фторфенил)-3H-фуро[2,3-d]пиримидин-4-она (5.44 ммоль) растворяли в 12.7 мл POCl3 (136 ммоль) и добавляли 690 мкл DMA (5.44 ммоль). Смесь перемешивали при 110°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем охлаждали до 0°С и выливали в смесь льда и воды. Сырой продукт выделяли путем фильтрования и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением соединения Синтеза 1а.1H ЯМР (400 МГц, ДМСО-d6): 8.87 (s, 1Н), 8.16 (m, 2H), 7.47 (m, 2H)
Синтез 1b: 5-бром-4-хлор-6-этил-7H-пирроло[2,3-d]пиримидин
Стадия А: 6-амино-5-[(2-этил-1,3-диоксолан-2-ил)метил]пиримидин-4-ол
257 мг 6-амино-5-[(2-этил-1,3-диоксолан-2-ил)метил]-2-сульфанилпиримидин-4-ола (0.1 ммоль), 0.77 мл конц. водного раствора NH3, 768 мг Ni Ренея и 11 мл воды помещали в колбу в атмосфере N2 и нагревали в колбе с обратным холодильником до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Горячую реакционную смесь затем фильтровали через целит и промывали горячей водой. Фильтрат концентрировали при пониженном давлении. Сырой продукт (6-амино-5-[(2-этил-1,3-диоксолан-2-ил)метил]пиримидин-4-ол) использовали без дополнительной очистки.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 11.44 (br s, 1Н), 7.70 (s, 1Н), 6.07 (s, 2H), 3.89 (m, 4H), 2.62 (s, 2H), 1.53 (m, 2H), 0.81 (t, 3H)
МС (М+Н): 226.2
Стадия В: 6-этил-7H-пирроло[2,3-d]пиримидин-4-ол
4.193 г 6-амино-5-[(2-этил-1,3-диоксолан-2-ил)метил]пиримидин-4-ола (18.6 ммоль) растворяли в 280 мл 0.2 М водного раствора HCl. Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Осадок отфильтровывали, промывали водой и сушили с получением 6-этил-7H-пирроло[2,3-d]пиримидин-4-ола.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 11.67 (s, 1Н), 7.75 (s, 1Н), 6.12 (t, 1Н), 2.56 (m, 2H), 1.21 (t, 3H)
МС (М+Н): 164.2
Стадия С: 5-бром-6-этил-7Н-пирроло[2,3-d]пиримидин-4-ол
1.63 г 6-этил-7H-пирроло[2,3-d]пиримидин-4-ола (10 ммоль) растворяли в 20 мл ДМФА и охлаждали до 0°С. Добавляли 1 мл брома (20 ммоль) и смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли водой и водным раствором Na2S2O3 и экстрагировали с помощью ДХМ. Объединенные органические слои промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 5-бром-6-этил-7H-пирроло[2,3-d]пиримидин-4-ола.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 12.08 (s, 1Н), 11.83 (s, 1H), 7.80 (d, 1H), 2.60 (q, 2H), 1.16 (t, 3H)
MC (M+H): 243.8
Стадия D: соединение Синтеза 1b
1936 мг 5-бром-6-этил-7H-пирроло[2,3-d]пиримидин-4-ола (8 ммоль), 4.5 мл POCl3 и 969 мг N,N-диметиланилина (8 ммоль) помещали в колбу и перемешивали при 100°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем выливали в смесь льда и воды и экстрагировали с помощью ДХМ. Объединенные органические слои промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением соединения Синтеза 1b.
1Н ЯМР (400 МГц, CDCl3) δ: 9.79 (s, 1H), 8.59 (s, 1H), 2.91 (q, 2H), 1.37 (t, 3Н)
MC (M+H): 260.0
Синтез 1с: 3-бром-2-(4-фторфенил)бензофуран-4-ол
Стадия А: 2-(4-фторфенил)бензофуран-4-ол
2.37 г 2-бромрезорцина (12.5 ммоль) растворяли в 30 мл сухого ТГФ в атмосфере N2 и добавляли 4.17 мл TEA (30 ммоль) и 1.92 мл AcCl (27 ммоль) соответственно. После перемешивания смеси в течение 5 минут, добавляли 2.4 г 1-этинил-4-фторбензола (20 ммоль), 561 мг Pd(OAc)2 (2.5 ммоль), 1.45 г тетрафторбората три-трет-бутилфосфония (5 ммоль), 476 мг CuI (2.5 ммоль) и 10 мл сухого DIPA и смесь перемешивали при 80°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем добавляли 2 г LiOH×H2O и смесь перемешивали при 80°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем концентрировали при пониженном давлении и очищали с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов с получением 2-(4-фторфенил)бензофуран-4-ола.1H ЯМР (400 МГц, ДМСО-d6) δ: 10.00 (s, 1Н), 7.91 (m, 2H), 7.38 (s, 1H), 7.31 (t, 2H), 7.10 (t, 1H), 7.04 (d, 1H), 6.63 (dd, 1H)
Стадия В: [2-(4-фторфенил)бензофуран-4-ил]ацетат
456 мг 2-(4-фторфенил)бензофуран-4-ола (2 ммоль) растворяли в 10 мл сухого ТГФ, затем осторожно добавляли 156 мкл AcCl (2.2 ммоль) и затем 306 мкл TEA (2.2 ммоль). Смесь перемешивали в атмосфере N2 до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Растворитель затем удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением [2-(4-фторфенил) бензофуран-4-ил] ацетата.1H ЯМР (400 МГц, CDCl3) δ: 7.84 (m, 2H), 7.42 (d, 1H), 7.28 (t, 1H), 7.15 (t, 2H), 7.02 (d, 1H), 6.86 (s, 1H), 2.42 (s, 3Н)
Стадия С: [3-бром-2-(4-фторфенил)бензофуран-4-ил]ацетат
688 мг [2-(4-фторфенил)бензофуран-4-ил]ацетата (2.54 ммоль) и 589 мг NBS (3.31 ммоль) растворяли в 20 мл MeCN и перемешивали при 70°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Растворитель затем удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением [3-бром-2-(4-фторфенил)бензофуран-4-ил]ацетата.lH ЯМР (400 МГц, CDCl3) δ: 8.11 (m, 2H), 7.44 (dd, 1H), 7.34 (t, 1H), 7.19 (m, 2H), 7.00 (dd, 1H), 2.45 (s, 3Н)
Стадия D: соединение Синтеза 1с
175 мг [3-бром-2-(4-фторфенил)бензофуран-4-ил]ацетата (0.5 ммоль) и 150 мкл 1 М раствора NaOEt в EtOH и 5 мл EtOH перемешивали при к.т. в атмосфере N2 до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь разбавляли 50 мл конц. водного раствора NH4Cl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали с получением соединения Синтеза 1с.1Н ЯМР (400 МГц, ДМСО-d6) δ: 10.16 (br s, 1H), 8.08 (m, 2H), 7.38 (m, 2H), 7.17 (t, 1H), 7.08 (d, 1H), 6.70 (d, 1H)
Синтез 1d: 3-бром-6-фтор-2-(4-фторфенил)бензофуран-4-ол
Стадия А: 5-фтор-2-йодбензол-1,3-диол
3.81 г (29.7 ммоль) 5-фторбензол-1,3-диола растворяли в 600 мл воды и при 0°С добавляли 8.08 г (31.8 ммоль) йода и смесь перемешивали в течение 30 минут. Затем рН доводили до 3 с помощью раствора NaHCO3 и смесь перемешивали до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем рН доводили до 8 (с помощью раствора NaHCO3), добавляли 20 г Na2S2O3 и смесь экстрагировали с помощью EtOAc. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 5-фтор-2-йодбензол-1,3-диола.1Н ЯМР (400 МГц, ДМСО-d6): 10.54 (s, 2H), 6.19 (d, 2H)
Стадия В: (3-ацетокси-5-фтор-2-йодфенил)ацетат
4.78 г 3-бром-6-фтор-2-(4-фторфенил)бензофуран-4-ола (18.8 ммоль) растворяли в 150 мл ТГФ и добавляли 5.70 г TEA (56.5 ммоль), затем при к.т. по каплям добавляли 4.267 г Ас2О (41.4 ммоль). Смесь перемешивали до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением (3-ацетокси-5-фтор-2-йодфенил)ацетата.1H ЯМР (400 МГц, ДМСО-d6): 7.24 (d, 2H), 2.34 (s, 6H)
Стадия С: 6-фтор-2-(4-фторфенил)бензофуран-4-ол
5.9 г (3-ацетокси-5-фтор-2-йодфенил)ацетата (17.45 ммоль) растворяли в 70 мл сухого ТГФ и 70 мл сухого DIPA в атмосфере N2, затем добавляли 3.77 г 1-этинил-4-фторбензола (31.4 ммоль), 587 мг Pd(OAc)2 (2.62 ммоль), 1.52 г тетрафторбората три-трет-бутилфосфония (5.24 ммоль) и 500 мг CuI (2.62 ммоль) и смесь перемешивали при 60°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем добавляли 2.93 г LiOH×H2O и смесь перемешивали при 60°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем концентрировали при пониженном давлении и очищали с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов с получением 6-фтор-2-(4-фторфенил)бензофуран-4-ола.1Н ЯМР (400 МГц, ДМСО-d6): 10.60 (s, 1H), 7.89 (m, 2H), 7.38 (s, 1H), 7.32 (m, 2H), 6.99 (m, 1H), 6.48 (dd, 1H)
Стадия D: [6-фтор-2-(4-фторфенил)бензофуран-4-ил]ацетат
2.49 мг 6-фтор-2-(4-фторфенил)бензофуран-4-ола (10.1 ммоль) растворяли в 50 мл сухого ТГФ, затем осторожно добавляли 791 мкл AcCl (11.1 ммоль) и затем 1.55 мл TEA (11.1 ммоль). Смесь перемешивали в атмосфере N2 до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Растворитель затем удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением [6-фтор-2-(4-фторфенил)бензофуран-4-ил]ацетата.1H ЯМР (400 МГц, ДМСО-d6): 7.95 (m, 2H), 7.57 (m, 1H), 7.46 (s, 1H), 7.37 (m, 2H), 7.09 (dd, 1H), 2.40 (s, 3H)
Стадия Е: [3-бром-6-фтор-2-(4-фторфенил)бензофуран-4-ил]ацетат
2.96 г [6-фтор-2-(4-фторфенил)бензофуран-4-ил]ацетата (10.27 ммоль) и 2.28 г NBS (12.84 ммоль) растворяли в 120 мл MeCN и перемешивали при 60°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Растворитель затем удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением [3-бром-6-фтор-2-(4-фторфенил)бензофуран-4-ил]ацетата.1H ЯМР (400 МГц, ДМСО-d6): 8.07 (m, 2H), 7.69 (dd, 1H), 7.44 (m, 1H), 7.19 (m, 2H), 7.09 (dd, 1H), 2.41 (s, 3H)
Стадия F: соединение Синтеза 1d
3.35 г [3-бром-6-фтор-2-(4-фторфенил)бензофуран-4-ил]ацетата (9.12 ммоль) и 8.67 мл 1 М раствора NaOEt в EtOH и 90 мл EtOH перемешивали при к.т. в атмосфере N2 до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь разбавляли 50 мл водного конц. раствора NH4Cl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали с получением соединения Синтеза 1d.1H ЯМР (400 МГц, ДМСО-d6): 10.78 (s, 1H), 8.06 (m, 2H), 7.40 (m, 2H), 7.06 (dd, 1H), 6.54 (dd, 1H)
Синтез 2а: этил (2R)-2-ацетокси-3-(2-гидроксифенил)пропаноат
и
Синтез 2b: этил (2S)-2-ацетокси-3-(2-гидроксифенил)пропаноат
Стадия А: [2-(бромметил)фенил]ацетат
60.07 г 2-метилфенилацетата (400 ммоль) и 106.8 г NBS (600 ммоль) помещали в 1 л колбу. Добавляли 500 мл циклогексана и затем при интенсивном перемешивании добавляли 3.284 г AIBN (20 ммоль) в течение 30 минут. Смесь перемешивали при 80°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения, затем охлаждали до к.т. Осадок отфильтровывали и промывали циклогексаном. Маточный раствор концентрировали при пониженном давлении и сырой продукт использовали на Стадии В без дополнительной очистки.
Стадия В: соединения Синтезов 2а и 2b
23.10 г безводного LiCl (545 ммоль) и 65.36 г безводного ZnCl2 (479.6 ммоль) помещали в 2 л колбу, затем сушили при 160°С при давлении 0.1 мм Hg в течение 1 часа. После охлаждения до к.т., в атмосфере аргона добавляли 26.49 г магниевой стружки (1090 ммоль) и 1 л сухого, предварительного охлажденного (0°С) ТГФ. Полученную в результате смесь погружали в ледяную баню и затем перемешивали в течение 30 минут.100 г [2-(бромметил)фенил]ацетата (сырой продукт из Стадии А, ~436 ммоль) растворяли в 120 мл сухого ТГФ и добавляли к предварительно охлажденной неорганике в течение 15 минут. После добавления реагента полученную в результате смесь перемешивали в течение 45 минут, поддерживая температуру в диапазоне 0-5°С. Затем в течение 5 минут добавляли 64.82 мл этил 2-оксоацетата (654 ммоль, 50% в толуоле) и полученную в результате смесь перемешивали в течение еще 15 минут. Оставшуюся неорганику удаляли путем фильтрования, и фильтрат разбавляли 500 мл МеОН. Смесь перемешивали до тех пор, пока не завершилась внутримолекулярная миграция ацетильной группы от фенольного кислорода к алкильному кислороду. Затем добавляли 30 мл уксусной кислоты, летучие компоненты упаривали при пониженном давлении. К остатку добавляли 350 мл воды и смесь экстрагировали с помощью EtOAc. Объединенные органические слои промывали насыщенным водным раствором NaHCO3 и соляным раствором и затем сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Затем добавляли 100 мл гексана и смесь перемешивали в течение 30 минут при 0°С. Образовавшиеся белые кристаллы собирали с помощью фильтрования и промывали гексаном.1Н ЯМР (500 МГц, ДМСО-d6) δ: 9.53 (s, 1Н), 7.06 (t, 1H), 7.04 (d, 1H), 6.79 (d, 1H), 6.71 (t, 1H), 5.10 (dd, 1H), 4.05 (q, 2H), 3.06 (dd, 1H), 2.94 (dd, 1H), 2.00 (s, 3H), 1.09 (t, 3H)
Энантиомеры разделяли с помощью хиральной хроматографии. Колонка: OD; Элюенты: гептан/EtOH; энантиомер, элюирующийся раньше, собирали в качестве соединения Синтеза 2b с 99.8% ее и энантиомер, элюирующийся позже, собирали в качестве соединения Синтеза 2а с 99.9% ее.
Синтез 2с: этил (2R)-2-гидрокси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат
Стадия А: (2R)-2-гидрокси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропановая кислота
30.3 г соединения Синтеза 2а (120 ммоль), 38.9 г соединения Синтеза 5b (180 ммоль) и 47.2 г трифенилфосфина (180 ммоль) растворяли в 120 мл сухого толуола, затем добавляли 82 мл DEAD (180 ммоль, 40% в толуоле). Смесь перемешивали при 50°С в атмосфере азота до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Летучие компоненты упаривали при пониженном давлении. Затем добавляли 300 мл DEE, смесь подвергали действию ультразвука и фильтровали, промывали DEE. Фильтрат концентрировали при пониженном давлении. Остаток растворяли в 125 мл ТГФ, затем добавляли 24 г NaOH (0.6 моль), растворенного в 125 мл воды. Смесь перемешивали при 50°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Значение рН доводили до 5 с помощью конц. HCl и летучие компоненты удаляли при пониженном давлении. Добавляли 100 мл воды и 350 мл ДХМ, смесь перемешивали при 0°С и осадок отфильтровывали, промывали холодной водой и ДХМ и сушили при пониженном давлении с получением (2R)-2-гидрокси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропановой кислоты.1Н-ЯМР (400 МГц, ДМСО-d6) δ: 8.88 (d, 1H), 7.80 (d, 1H), 7.55 (dd, 1H), 7.49-7.44 (m, 1H), 7.26 (dd, 1H), 7.17-7.11 (m, 2H), 7.06 (t, 1H), 6.98 (d, 1H), 6.88 (t, 1H), 5.22 (s, 2H), 4.50 (d, 1H), 3.81 (dd, 1H), 3.77 (s, 3H), 3.73 (dd, 1H), 2.44 (dd, 1H)
Стадия В: соединение Синтеза 2с
51.7 г (2R)-2-гидрокси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропановой кислоты (136 ммоль) растворяли в 520 мл EtOH, затем добавляли 20 мл конц. H2SO4. Смесь перемешивали при 60°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли водой, нейтрализовали водным насыщенным раствором NaHCO3 и экстрагировали дихлорметаном. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя EtOAc и МеОН в качестве элюентов с получением соединения Синтеза 2с. МСВР: рассчитано для C23H24N2O5: 408.1685, найдено: 409.1757 (М+Н)
Синтез 2d; этил (2S)-2-гидрокси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат
Соединение Синтеза 2d получали путем, аналогичным получению соединения Синтеза 2с, но исходя из соединения Синтеза 2b взамен соединения Синтеза 2а.
Синтез 2е: этил (2R)-2-гидрокси-3-(2-метоксифенил)пропаноат
и
Синтез 2f: этил (2S)-2-гидрокси-3-(2-метоксифенил)пропаноат
Энантиомеры этил 2-гидрокси-3-(2-метоксифенил)пропаноата разделяли с помощью хиральной хроматографии; Колонка: AD, Элюент: 2-PrOH; энантиомер, элюирующийся раньше, собирали в качестве соединения Синтеза 2е с 99.8% ее. Энантиомер, элюирующийся позже, собирали в качестве соединения Синтеза 2f c 97.8% ее.
Синтез 2g: этил (2R)-2-гидрокси-3-[2-(пиразин-2-илметокси)фенил]пропаноат
Стадия А: этил (2R)-2-ацетокси-3-[2-(пиразин-2-илметокси)фенил]пропаноат
1 экв. соединения Синтеза 2а, 2 экв. пиразин-2-илметанола и 2 экв. трифенилфосфина растворяли в сухом толуоле (0.2 М для фенола), затем добавляли 2 экв. DTAD. Смесь перемешивали при 50°С в атмосфере азота. После достижения подходящей степени превращения летучие компоненты удаляли при пониженном давлении. Сырое промежуточное соединение очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением этил (2R)-2-ацетокси-3-[2-(пиразин-2-илметокси)фенил]пропаноата.
Стадия В: соединение Синтеза 2g
Этил (2R)-2-ацетокси-3-[2-(пиразин-2-илметокси)фенил]пропаноат растворяли в этаноле (0.5 М), затем добавляли 2 мол. % раствор NaOEt (1.0 М в этаноле). Полученную в результате смесь перемешивали при к.т. Если превращение не было завершено, добавляли дополнительное количество раствора NaOEt. Смесь концентрировали до половины ее объема, затем добавляли воду и соляной раствор, и смесь экстрагировали с помощью EtOAc. Объединенную органику сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя ДХМ и метанол в качестве элюентов с получением соединения Синтеза 2g.1Н ЯМР (400 МГц, ДМСО-d6) δ: 8.88 (s, 1Н), 8.64 (dd, 2H), 7.22-7.16 (m, 2H), 7.06 (d, 1H), 6.89 (t, 1H), 5.46 (d, 1H), 5.27 (dd, 2H), 4.29 (dq, 1H), 4.00 (q, 2H), 3.09 (dd, 1H), 2.79 (dd, 1H), 1.08 (t, 3H)
Синтез 2h: этил (2S)-2-гидрокси-3-[2-(2,2,2-трифторэтокси)фенил]пропаноат
Стадия А: этил (2S)-2-гидрокси-3-(2-гидроксифенил)пропаноат
13.633 г соединения Синтеза 2b (54 ммоль) растворяли в 200 мл сухого EtOH, затем добавляли 30 мл раствора NaOEt (1 М в EtOH) и смесь перемешивали при к.т. При необходимости, добавление раствора NaOEt повторяли до тех пор, пока не завершилось отщепление ацетильной группы. Смесь разбавляли 600 мл воды и смесь экстрагировали с помощью EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Полученный этил (2S)-2-гидрокси-3-(2-гидроксифенил)пропаноат использовали на следующей стадии без дополнительной очистки.
Стадия В: соединение Синтеза 2h
9.18 г этил (2S)-2-гидрокси-3-(2-гидроксифенил)пропаноата (43.7 ммоль) растворяли в 130 мл сухого ДМФА, затем добавляли 6.040 г Ka2CO3 (43.7 ммоль). После 5 минут перемешивания добавляли 7.7 мл 2,2,2-трифторэтилтрифторметансульфоната (48 ммоль) в течение 5 минут. Полученную в результате смесь перемешивали до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Реакционную смесь разбавляли соляным раствором, затем экстрагировали с помощью EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов.1Н ЯМР (500 МГц, ДМСО-d6) δ: 7.23 (t, 1Н), 7.18 (d, 1H), 7.06 (d, 1H), 6.95 (t, 1H), 5.50 (d, 1H), 4.75 (q, 2H), 4.22 (m, 1H), 4.02 (q, 2H), 3.00 (dd, 1H), 2.76 (dd, 1H), 1.09(t, 3H)
Синтез 2i: (2R)-2-амино-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропановая кислота
Стадия А: гидрохлорид этил (2R)-2-амино-3-(2-гидроксифенил)пропаноата
653 мг гидрохлорида (2R)-2-амино-3-(2-гидроксифенил)пропановой кислоты (3.0 ммоль) растворяли в 6 мл HCl (1.25 М в EtOH) и перемешивали при 60°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем реакционную смесь осторожно разбавляли 10% водным раствором NaHCO3 и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Продукт следует хранить в морозильной камере.
1Н ЯМР (500 МГц, ДМСО-d6) δ: 7.05-6.95 (m, 2H), 6.72 (dm, 1H), 6.69-6.63 (m, 1H), 4.02 (q, 2H), 3.65 (dd, 1H), 2.84 (dd, 1H), 2.78 (dd, 1H), 1.12 (t, 3H)
МСВР: рассчитано для С11Н15NО3: 209.1052; найдено: 210.1128 (М+Н)
Стадия В: этил (2R)-2-амино-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат
3.96 г гидрохлорида этил (2R)-2-амино-3-(2-гидроксифенил)пропаноата (18.9 ммоль) растворяли в 200 мл сухого толуола, затем добавляли 5.69 г PPh3 (21.7 ммоль) и 4.69 г соединения Синтеза 5b (21.7 ммоль), и смесь нагревали до 35°С, затем добавляли 5.0 г DTAD (21.7 ммоль) и смесь перемешивали при 45°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя EtOAc и МеОН в качестве элюентов.
1H ЯМР (500 МГц, ДМСО-d6) δ: 8.92 (d, 1H), 7.61 (d, 1H), 7.55 (dd, 1H), 7.46 (td, 1H), 7.20 (td, 1H), 7.17 (dd, 1H), 7.15 (dd, 1H), 7.06 (td, 1H), 7.04 (dd, 1H), 6.91 (td, 1H), 5.27/5.23 (d, 2H), 4.01 (q, 2H), 3.76 (s, 3H), 3.68 (dd, 1H), 3.08 (br, 2H), 3.03/2.83 (dd, 2H), 1.07 (t, 3H)
МСВР: рассчитано для C23H25N3O4: 407.1845; найдено: 408.1928 (М+Н)
Стадия С: соединение Синтеза 2i
3.20 г этил (2R)-2-амино-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноата (7.85 ммоль) растворяли в 10 мл ТГФ, затем добавляли 10 мл воды и 420 мг LiOH×H2O (10 ммоль) и смесь перемешивали при к.т. до тех пор, пока не завершится гидролиз. Затем смесь разбавляли водой и нейтрализовали 2 М водным раствором HCl. Образовавшийся осадок отфильтровывали, промывали водой и сушили с получением соединения Синтеза 2i.
1Н ЯМР (500 МГц, ДМСО-d6): 8.88 (d, 1Н), 7.82 (d, 1H), 7.54 (dd, 1H), 7.47 (m, 1H), 7.27 (dd, 1H), 7.23 (t, 1H), 7.16 (d, 1H), 7.06 (t, 1H), 7.05 (d, 1H), 6.93 (t, 1H), 5.26 (s, 2H), 3.76 (s, 3H), 3.59 (dd, 1H), 3.49/2.83 (dd, 2H)
MCBP: рассчитано для C21H21N3O4: 379.1532; найдено: 380.1610 (М+Н)
Синтез 3а: 2-хлор-3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенол
Стадия А: (4-бром-2-хлорфенокси)-триметилсилан
20.8 г 4-бром-2-хлорфенола (100 ммоль) растворяли в 150 мл сухого ТГФ, затем добавляли 24.2 г HMDS (150 ммоль). Реакционную смесь перемешивали при 85°С в атмосфере аргона в течение 1.5 часа, затем концентрировали при пониженном давлении. Полученный в результате сырой продукт использовали без дополнительной очистки.1H ЯМР (200 МГц, CDCl3): 7.49 (d, 1H), 7.23 (dd, 1H), 6.75 (d, 1H), 0.26 (s, 9H)
Стадия В: 4-бром-2-хлор-3-метилфенол
48 мл раствораnBuLi в гексанах (120 ммоль, 2.5 М в гексанах) при -78°С в атмосфере аргона по каплям добавляли к раствору 12.1 г сухого DIPA (120 ммоль) в 250 мл сухого ТГФ. Смесь перемешивали в течение 30 минут при такой же температуре, затем по каплям добавляли 28.0 г (4-бром-2-хлорфенокси)-триметилсилана (100 ммоль). Спустя 2.5 часа по каплям добавляли 21.3 г MeI (150 ммоль), затем охлаждающую баню удаляли и смесь перемешивали в течение ночи. Реакцию гасили 100 мл водного раствора NH3 и 200 мл насыщенного водного раствора NH4Cl и экстрагировали с помощью EtOAc. Органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Полученную в результате темную массу несколько раз нагревали в колбе с обратным холодильником с чистым гексаном (аликвоты 150-150 мл) и декантировали, оставляя черную смолу. Объединенные органические фазы концентрировали при пониженном давлении с получением 19.0 г сырого продукта, который использовали без дополнительной очистки.1Н ЯМР (200 МГц, CDCl3) δ: 7.32 (d, 1H), 6.76 (d, 1H), 5.62 (s, 1H), 2.49 (s, 3H)
Стадия С: (4-бром-2-хлор-3-метилфенокси)-триметилсилан
20.8 г HMDS (129 ммоль) добавляли к раствору 19.0 г 4-бром-2-хлор-3-метилфенола (86.0 ммоль) в 150 мл сухого ТГФ. Смесь перемешивали при 85°С под баллонным аргоном в течение 1.5 часа и затем концентрировали при пониженном давлении. Полученный продукт использовали без дополнительной очистки.1H ЯМР (200 МГц, CDCl3) δ: 7.30 (d, 1H), 6.63 (d, 1H), 2.50 (s, 3H), 0.28 (s, 9H)
Стадия D: соединение Синтеза 3а
Раствор 25.2 г (4-бром-2-хлор-3-метилфенокси)-триметилсилана (86.0 ммоль) в 250 мл сухого ТГФ охлаждали до -78°С под аргоном и затем добавляли по каплям 38 мл раствораnBuLi (94.6 ммоль, 2.5 М в гексанах). Спустя 5 минут по каплям добавляли 19.2 г 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолана (103 ммоль). Охлаждающую баню удаляли и смеси давали медленно нагреться до к.т. Затем смесь добавляли к 200 мл насыщенного водного раствора NH4Cl и экстрагировали с помощью EtOAc. Объединенные органические слои концентрировали при пониженном давлении и пропускали через набивку силикагеля, используя гексан и EtOAc в качестве элюентов. Сырой продукт перекристаллизовывали из смеси EtOAc и гексана с получением соединения Синтеза 3а.1Н ЯМР (500 МГц, ДМСО-d6) δ: 10.40 (s, 1H), 7.42 (d, 1H), 6.80 (d, 1H), 2.49 (s, 3H), 1.27 (s, 12H)
Синтез 3b: 1-[2-[2-хлор-3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]этил]-4-метилпиперазин
10.0 г соединения Синтеза 3а (37.2 ммоль,), 8.7 г 2-(4-метилпиперазин-1-ил)этанола (60.3 ммоль) и 15.8 г PPh3 (60.3 ммоль) растворяли в 100 мл сухого толуола и затем по каплям добавляли 27 мл DEAD (60.3 ммоль, 40% раствор в толуоле). Смесь перемешивали при 50°С в атмосфере аргона до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Летучие компоненты упаривали при пониженном давлении и добавляли 100 мл Et2O. Осажденные белые кристаллы отфильтровывали и промывали с помощью Et2O. Фильтрат концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя CHCl3 и МеОН в качестве элюентов. Полученное в результате светло-коричневое масло кристаллизовали из гексана с получением соединения Синтеза 3b в виде не совсем белого твердого вещества.1H ЯМР (500 МГц, ДМСО-d6) δ: 7.56 (d, 1Н), 6.99 (d, 1H), 4.15 (t, 2H), 2.72 (t, 2H), 2.51 (s, 3H), 2.50 (br s, 4H), 2.29 (br s, 4H), 2.13 (s, 3H), 1.29 (s, 12H)
Синтез 3с: 2-(3-хлор-2-метилфенил)-5,5-диметил-1,3,2-диоксаборинан
4.94 г (3-хлор-2-метилфенил)бороновой кислоты (29 ммоль) и 3.021 г неопентилгликоля (29 ммоль) перемешивали при к.т. в присутствии ионообменной смолы Amberlite 15H+ (высушенной толуолом) до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем фильтровали через целит и промывали 2-Ме-ТГФ. Фильтрат концентрировали при пониженном давлении с получением соединения Синтеза 3с.1Н ЯМР (400 МГц, CDCl3): 7.59 (dd, 1H), 7.38 (dd, 1H), 7.10 (t, 1H), 3.79 (s, 4H), 2.57 (s, 3H), 1.05 (s, 6H)
Синтез 4: этил (2R)-2-[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат
Используя Общую Методику Ia и соединение Синтеза 2с в качестве подходящего производного сложного эфира молочной кислоты, получали соединение Синтеза 4. МС: (М+Н)+=700.4
Синтез 5а: (E)-4-(диметиламино)-1,1-диметоксибут-3-ен-2-он
502.1 г 1,1-диметоксипропан-2-она (4.25 моль) и 506.4 г 1,1-диметокси-N,N-диметилметанамина (4.25 моль) смешивали в 2 л колбе и перемешивали при 105°С в течение 3 часов. Образовавшийся МеОН непрерывно удаляли с помощью дистилляции. Когда образование МеОН прекратилось (при температуре головки 65°С) реакционную смесь подвергали вакуумной дистилляции (медленно уменьшая давление до 30 мбар) для удаления побочных продуктов и непрореагировавших исходных веществ. Сырой продукт дистиллировали при 0.1 мбар. Фракции собирали при температуре головки в диапазоне 107-118°С (температура бани 160-165°С) с получением желтого масла.1Н ЯМР (500 МГц, ДМСО-d6) δ: 7.59 (d, 1H), 5.17 (d, 1H), 4.42 (s, 1H), 3.25 (s, 6H), 3.09 (s, 3H), 2.78 (s, 3H)
Синтез 5b: [2-(2-метоксифенил)пиримидин-4-ил]метанол
Стадия А: 4-(диметоксиметил)-2-(2-метоксифенил)пиримидин
К смеси 1.2 экв. соли уксусной кислоты и 2-метоксибензамидина и 1 экв. соединения Синтеза 5а в сухом метаноле (0.5 мл/ммоль) по частям добавляли 1.2 экв. NaOEt и смесь перемешивали при 75°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем реакционную смесь охлаждали и концентрировали при пониженном давлении. К остатку добавляли воду и соединения экстрагировали с помощью ДХМ. Объединенные органические слои сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 4-(диметоксиметил)-2-(2-метоксифенил)пиримидина.1Н ЯМР (400 МГц, ДМСО-d6) δ: 8.93 (d, 1H), 7.55-7.44 (m, 3Н), 7.16 (d, 1H), 7.06 (m, 1H), 5.31 (s, 1H), 3.76 (s, 3Н), 3.37 (s, 6H)
Стадия В: соединение Синтеза 5b
261 мг 4-(диметоксиметил)-2-(2-метоксифенил)пиримидина (1.0 ммоль) растворяли в 2 мл раствора HCl в диоксане (4 М раствор), затем добавляли 2 мл воды и эту смесь перемешивали при 50°С в течение 16 часов. Реакционную смесь охлаждали до 0°С, затем частями добавляли 320 мг NaOH (8.0 ммоль). Значение рН доводили до 8, используя 10% водный раствор K2CO3, затем добавляли 76 мг борогидрида натрия (2.0 ммоль) и смесь перемешивали в течение 30 минут при 0°С. Реакционную смесь разбавляли 5 мл воды и экстрагировали с помощью EtOAc. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением соединения Синтеза 5b.1Н ЯМР (400 МГц, ДМСО-d6) δ: 8.84 (d, 1H), 7.50-7.42 (m, 3Н), 7.14 (d, 1H), 7.03 (m, 1H), 5.66 (t, 1H), 4.58 (d, 2H), 3.75 (s, 3Н)
Синтез 6: (2R)-2-[(7-бензил-5-бром-6-этилпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановая кислота
Стадия А: 7-бензил-5-бром-4-хлор-6-этилпирроло[2,3-d]пиримидин
255 мг NaH (6.38 ммоль) и 50 мл сухого ТГФ загружали в 50 мл сосуд Шленка в атмосфере N2 и взвесь охлаждали до 0°С. Затем добавляли 1.792 г соединения Синтеза 1b (5.8 ммоль). После перемешивания смеси в течение 30 минут при 0°С, добавляли 773 мкл бензилбромида (6.38 ммоль) и смеси давали нагреться до к.т. и перемешивали до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем разбавляли насыщенным водным раствором NH4Cl и экстрагировали с помощью ДХМ. Объединенные органические слои промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 7-бензил-5-бром-4-хлор-6-этилпирроло[2,3-d]пиримидина.
1H ЯМР (400 МГц, CDCl3) δ: 8.60 (s, 1H), 7.33-7.26 (m, 3H), 7.06-7.04 (m, 2H), 5.54 (s, 2H), 2.79 (q, 2H), 1.07 (t, 3H)
MC(M+H): 351.8
Стадия В: соединение Синтеза 6
Используя Общую Методику III и 7-бензил-5-бром-4-хлор-6-этилпирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали соединение Синтеза 6. МС (М+Н): 279.2
Синтез 7а: N-[2-бензилокси-6-(2,2-дибромвинил)фенил]-3-хлор-2-метил-4-триизопропилсилилоксианилин
Стадия А: (4-бром-2-хлорфенокси)-триизопропилсилан
200 г 4-бром-2-хлорфенола (0.97 моль) и 126 мл TIPSCl (1.18 моль) растворяли в 1.6 л ДХМ. Добавляли 167 г имидазола (2.45 моль) и смесь перемешивали при к.т. в течение 2 часов. Затем летучие компоненты упаривали при пониженном давлении и остаток растворяли в 1.5 л EtOAc. Смесь промывали соляным раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Загрязнение триизопропилсилилгидроксид удаляли с помощью дистилляции (120°С при 0.01 мм Hg). Остаток фильтровали через короткую набивку силикагеля, используя гексан, и концентрировали при пониженном давлении. Продукт (бесцветное масло) использовали на следующей стадии без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ: 7.49 (d, 1H), 7.21 (dd, 1H), 6.78 (d, 1H), 1.31 (sept, 3H), 1.14 (d, 18H)
MC (EI, 70 эВ) m/z (% относительная интенсивность, [ион]): 63 (30), 79 (24), 93 (41), 170 (17), 235 (19), 251 (16), 265 (24), 293 (23), 319 (77), 321 (100), 323 (28), 362 (1, [М+])
Стадия В: (4-бром-2-хлор-3-метилфенокси)-триизопропилсилан
76.0 мл сухого DIPA (0.54 моль) растворяли в 1.2 л сухого ТГФ в атмосфере аргона и при -78°С по каплям добавляли 51.2 мл раствораnBuLi (0.512 моль, 10 М в гексанах). Смесь перемешивали в течение 45 минут при такой же температуре. Затем при -78°С по каплям добавляли 178 г (4-бром-2-хлорфенокси)-триизопропилсилана (0.488 моль) и белую суспензию перемешивали до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем при этой температуре добавляли 36.5 мл MeI (0.586 ммоль) и реакционную смесь перемешивали в течение ночи без дополнительного охлаждения. Летучие компоненты упаривали при пониженном давлении. Остаток растворяли в 1.5 л EtOAc, промывали соляным раствором. Органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт фильтровали через короткую набивку силикагеля, используя гексан в качестве элюента, и концентрировали при пониженном давлении с получением продукта в виде бледно-желтого масла.1H ЯМР (400 МГц, CDCl3) δ: 7.30 (d, 1H), 6.68 (d, 1H), 2.53 (s, 3H), 1.32 (sept, 3H), 1.14 (d, 18H)
Стадия С: N-бензил-3-хлор-2-метил-4-триизопропилсилилоксианилин
7.56 г (4-бром-2-хлор-3-метилфенокси)-триизопропилсилана (20 ммоль) и 4.29 г бензиламина (40 ммоль) растворяли в 16 мл сухого толуола, затем добавляли 450 мг Pd2dba3 (0.5 ммоль), 450 мг X-Phos (1 ммоль) и 9.77 г Cs2CO3 (30 ммоль) и смесь перемешивали при 100°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гексан и EtOAc в качестве элюентов с получением N-бензил-3-хлор-2-метил-4-триизопропилсилилоксианилина.
Стадия D: 3-хлор-2-метил-4-триизопропилсилилоксианилин
3.50 г N-бензил-3-хлор-2-метил-4-триизопропилсилилоксианилина (8.66 ммоль) растворяли в 100 мл МеОН и 20 мл EtOAc, затем добавляли 80 мг 10% Pd/C и смесь перемешивали в атмосфере Н2 при давлении 1 бар до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гексан и EtOAc в качестве элюентов с получением 3-хлор-2-метил-4-триизопропилсилилоксианилина.
1H ЯМР (400 МГц, ДМСО-d6) δ: 6.58 (d, 1Н), 6.50 (d, 1H), 4.68 (s, 2H), 2.11 (s, 3H), 1.24 (m, 3H), 1.06 (d, 18H)
MC: (M+H)+=314.2
Стадия Е: 3-бензилокси-2-бромбензалъдегид
4.554 г 2-бром-3-гидроксибензальдегида (22.65 ммоль), 4.262 г бензилбромида (24.92 ммоль) и 4.696 г K2CO3 (33.98 ммоль) растворяли в 20 мл ДМСО и перемешивали при 50°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем выливали в воду. Осадок отфильтровывали с получением 3-бензилокси-2-бромбензальдегида. МС (EI, 70 эВ) m/z (% относительная интенсивность, [ион]): 65 (10), 91 (100), 290 (5, [М+]), 292 (5, [М+])
Стадия F: 3-бензилокси-2-(3-хлор-2-метил-4-триизопропилсилилоксианилино)бензалъдегид
5.0 г 3-бензилокси-2-бромбензальдегида (17.17 ммоль), 5.391 г 3-хлор-2-метил-4-триизопропилсилилоксианилина (17.17 ммоль), 16.782 г Cs2CO3 (51.51 ммоль), 393 мг Pd2dba3 (0.43 ммоль) и 535 мг рац. BINAP (0.86 ммоль) смешивали в 85 мл толуола и перемешивали при 120°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Летучие компоненты удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 3-бензилокси-2-(3-хлор-2-метил-4-триизопропилсилилоксианилино)бензальдегида. МС: (М+Н)+=524.2
Стадия G: соединение Синтеза 7а
7.7 г 3-бензилокси-2-(3-хлор-2-метил-4-триизопропилсилилоксианилино)бензальдегида (14.69 ммоль) и 7.308 г четырехбромистого углерода (22.03 ммоль) растворяли в 160 мл ДХМ при 0°С, затем добавляли 11.56 г PPh3 (44.07 ммоль). Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем растворитель удаляли при пониженном давлении, остаток растворяли в Et2O. Затем добавляли гептан и образовавшийся осадок отфильтровывали, фильтрат концентрировали при пониженном давлении. Затем добавляли гептан и смесь перемешивали в течение 10 минут и снова фильтровали. Фильтрат концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением соединения Синтеза 7а.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 7.28-7.23 (m, 5H), 7.19 (s, 1H), 7.11 (dd, 2H), 7.05 (d, 1H), 6.60 (d, 1H), 6.41 (s, 1H), 6.22 (d, 1H), 5.08 (s, 2H), 2.30 (s, 3H), 1.25(m, 3H), 1.05(d, 18H)
MC: (M+H)+=680.0
Синтез 7b: этил (2R)-2-[1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)индол-7-ил]окси-3-(2-метоксифенил)пропаноат
Стадия А: [4-[7-бензилокси-2-(4-фторфенил)индол-1-ил]-2-хлор-3-метилфенокси]-триизопропилсилан
2720 мг соединения Синтеза 7а (4 ммоль), 1119 мг 4-фторфенилбороновой кислоты (8 ммоль), 4245 мг K3PO4 (20 ммоль), 90 мг Pd(OAc)2 (0.4 ммоль) и 328 мг SPhos (0.8 ммоль) смешивали в 60 мл сухого толуола в атмосфере N2 и перемешивали при 100°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем растворитель удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением [4-[7-бензилокси-2-(4-фторфенил)индол-1-ил]-2-хлор-3-метилфенокси]-триизопропилсилана.1Н ЯМР (400 МГц, CDCl3) δ: 7.33 (d, 2H), 7.29-t.22 (m, 2H), 7.18 (d, 1H), 7.16 (d, 1H), 7.10 (t, 2H), 6.94 (d, 1H), 6.92-6.84 (m, 4H), 6.73 (s, 1H), 6.61 (d, 1H), 4.94 (d, 1H), 4.89 (d, 1H), 1.97 (s, 3H), 1.31 (m, 3H), 1.13 (t, 18H)
Стадия В: 4-[7-бензилокси-2-(4-фторфенил)индол-1-ил]-2-хлор-3-метилфенол
2600 мг [4-[7-бензилокси-2-(4-фторфенил)индол-1-ил]-2-хлор-3-метилфенокси]-триизопропилсилана (2.96 ммоль), 2.96 мл раствора TBAF (2.96 ммоль, 1 М в ТГФ) и 50 мл ТГФ перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Растворитель затем удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 4-[7-бензилокси-2-(4-фторфенил)индол-1-ил]-2-хлор-3-метилфенола.
1H ЯМР (400 МГц, ДМСО-d6) δ: 10.27 (br s, 1H), 7.28-7.18 (m, 6H), 7.10 (t, 2H), 7.07-6.99 (m, 2H), 6.85-6.77 (m, 3H), 6.75 (s, 1H), 6.72 (d, 1H), 4.95 (d, 1H), 4.90 (d, 1H), 1.75(s, 3H)
MC: (M+H)+=458.0.
Стадия С: 7-бензилокси-1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)индол
1.2 г 4-[7-бензилокси-2-(4-фторфенил)индол-1-ил]-2-хлор-3-метилфенола (2.1 ммоль), 606 мг 1-(2-гидроксиэтил)-4-метилпиперазина (4.2 ммоль) и 2.1 г PPh3 (6.3 ммоль) растворяли в 50 мл сухого толуола в атмосфере N2 и смесь охлаждали до 0°С. Затем добавляли 1451 мг DTAD (6.3 ммоль) и смесь нагревали до 45°С и перемешивали до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Растворитель затем удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc и МеОН в качестве элюентов с получением 7-бензилокси-1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)индола. MC: (M+H)+=584.2
Стадия D: 1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)индол-7-ол
1280 мг 7-бензилокси-1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)индола (2.19 ммоль) растворяли в 100 мл EtOH, затем добавляли 100 мг 10% Pd/C. Смесь перемешивали в атмосфере Н2 при давлении 1 бар и к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь фильтровали через целит и фильтрат концентрировали с получением 1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)индол-7-ола.
1H ЯМР (400 МГц, ДМСО-d6) δ: 9.04 (br s, 1H), 7.25 (dd, 2H), 7.17-7.03 (m, 4H), 6.94 (d, 1H), 6.86 (t, 1H), 6.70 (s, 1H), 6.47 (d, 1H), 4.13 (m, 2H), 2.72 (t, 2H), 2.58-2.42 (br s, 4H), 2.40-2.17 (br s, 4H), 2.14 (s, 3H), 1.86 (s, 3H)
MC: (M+H)+=494.2
Стадия Е: соединение Синтеза 7b
494 мг 1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)индол-7-ола (1 ммоль), 449 мг соединения Синтеза 2f (2 ммоль) и 786 мг PPh3 (3 ммоль) растворяли в 10 мл сухого толуола в атмосфере N2 и смесь охлаждали до 0°С. Затем добавляли 691 мг DTAD (3 ммоль) и смесь нагревали до 45°С и перемешивали до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Растворитель затем удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc и МеОН в качестве элюентов с получением соединения Синтеза 7b в виде смеси диастереоизомеров.
1Н ЯМР (500 МГц, ДМСО-d6) δ: 7.43/6.98 (d, 1Н), 7.28 (m, 2H), 7.23/7.24 (d, 1H), 7.17/7.18 (t, 1Н), 7.14 (m, 2H), 7.12/6.88 (d, 1Н), 6.95/6.94 (t, 1Н), 6.91/6.91 (d, 1Н), 6.79/6.78 (s, 1Н), 6.73/6.75 (t, 1Н), 6.52/6.60 (d, 1Н), 6.46/6.40 (d, 1Н), 4.85/4.76 (dd, 1Н), 4.25-4.01 (m, 2H), 4.01-3.89 (m, 2H), 3.77/3.76 (s, 3Н), 2.70-2.60 (m, 3Н), 2.54-2.30 (m, 5H), 2.21 (br s, 4H), 2.13/2.09 (s, 3Н), 1.59/2.08 (s, 3Н), 0.99/0.98 (t, 3Н)
МС: (M+H)+=700.0
Пример 1: (2R)-2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Используя Общую Методику II и соединение Синтеза 4 в качестве подходящего 5-бромфуро[2,3-d]пиримидинового производного и соединение Синтеза 3b в качестве подходящего производного бороновой кислоты. Пример 1 получали в виде смеси диастереоизомеров. МСВР: рассчитано для C47H44ClFN6O7: 858.2944, найдено: 430.1547 и 430.1555 (М+2Н)
Пример 2: (2R)-2-{[5-{3-хлор-2-этил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Стадия А: 1-[2-(4-бром-2-хлорфенокси)этил]-4-метилпиперазин
10.373 г 4-бром-2-хлорфенола (50 ммоль), 14.442 г 2-(4-метилпиперазин-1-ил)этанола (100 ммоль) и 26.229 г PPh3 (100 ммоль) растворяли в 250 мл сухого толуола в атмосфере N2, затем добавляли 23.027 г DTAD (100 ммоль). Смесь перемешивали при 50°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Летучие компоненты упаривали при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя EtOAc и МеОН в качестве элюентов. МС (М+Н): 333.0
Стадия В: 1-[2-(4-бром-2-хлор-3-этилфенокси)этил]-4-метилпиперазин
2.0 г 1-[2-(4-бром-2-хлорфенокси)этил]-4-метилпиперазина (6 ммоль) растворяли в 50 мл сухого ТГФ в атмосфере N2 и охлаждали до -78°С. Добавляли 6 мл раствора LDA (12 ммоль в 2М ТГФ) и смесь перемешивали в течение 3 часов, затем добавляли 982 мг йодэтана (6.3 ммоль) и смеси давали нагреться до к.т. Ее гасили насыщенным водным раствором NH4Cl и экстрагировали с помощью EtOAc. Объединенный органический слой сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. МС (М+Н): 360.8
Стадия С: 1-[2-[2-хлор-3-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]этил]-4-метилпиперазин
2099 мг 1-[2-(4-бром-2-хлор-3-этилфенокси)этил]-4-метилпиперазина (5.8 ммоль) растворяли в 30 мл сухого ТГФ в атмосфере N2 и охлаждали до -78°С. По каплям добавляли 4.65 мл раствораnBuLi (11.61 ммоль в 2.5М ТГФ). Смесь перемешивали в течение 5 часов, затем добавляли 2.6 мл 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолана (12.77 ммоль) и смесь перемешивали в течение 30 минут. Затем смеси давали нагреться до к.т. и концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя EtOAc и МеОН в качестве элюентов. МС (М+Н): 409.2
Стадия D: Пример 2
Используя Общую Методику II и соединение Синтеза 4 в качестве подходящего 5-бромфуро[2,3-d]пиримидинового производного и 1-[2-[2-хлор-3-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]этил]-4-метилпиперазин в качестве подходящего производного бороновой кислоты, Пример 2 получали в виде смеси диастереоизомеров. МСВР: рассчитано для C48H46ClFN6O7: 872.3101, найдено: 437.1620 и 437.1620 (М+2Н)
Пример 3: (2R)-2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси}-3-(2-метоксифенил)пропановая кислота
Стадия А: этил (2R)-2-[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси-3-(2-метоксифенил)пропаноат
Используя Общую Методику Ia и соединение Синтеза 2е в качестве подходящего производного сложного эфира молочной кислоты, получали этил (2R)-2-[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси-3-(2-метоксифенил)пропаноат.1Н ЯМР (400 МГц, ДМСО-d6): 8.53 (s, 1H), 8.10 (m, 2Н), 7.47-7.36 (m, 3Н), 7.23 (m, 1H), 6.96 (m, 1H), 6.89 (t, 1H), 5.58 (m, 1H), 4.12 (q, 2Н), 3.79 (s, 3Н), 3.36 (m, 1H), 3.21 (m, 1H), 1.11 (t, 3Н)
Стадия В: Пример 3
Используя Общую Методику II и этил (2R)-2-[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси-3-(2-метоксифенил)пропаноат в качестве подходящего 5-бромфуро[2,3-d]пиримидинового производного и соединение Синтеза 3b в качестве подходящего производного бороновой кислоты, Пример 3 получали в виде смеси диастереоизомеров. МСВР: рассчитано для C36H36ClFN4O6: 674.2307, найдено: 675.2367 и 675.2364 (М+Н)
Пример 4: (2R)-2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси}-3-[2-(пиразин-2-илметокси)фенил]пропановая кислота
Стадия А: этил (2R)-2-[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси-3-[2-(пиразин-2-илметокси)фенил]пропаноат
Используя Общую Методику Ia и соединение Синтеза 2g в качестве подходящего производного сложного эфира молочной кислоты, получали этил (2R)-2-[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси-3-[2-(пиразин-2-илметокси)фенил]пропаноат. МС: (М+Н)+=595.0
Стадия В: Пример 4
Используя Общую Методику II и этил (2R)-2-[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси-3-[2-(пиразин-2-илметокси)фенил]пропаноат в качестве подходящего 5-бромфуро[2,3-d]пиримидинового производного и соединение Синтеза 3b в качестве подходящего производного бороновой кислоты, Пример 4 получали в виде смеси диастереоизомеров. МСВР: рассчитано для C40H38ClFN6O6: 752.2525, найдено: 753.2645 и 753.2606 (М+Н)
Пример 5: (2R)-2-{[6-(5-хлорфуран-2-ил)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}фуро[2,3-d]пиримидин-4-ил]окси}-3-[2-(пиразин-2-илметокси)фенил]пропановая кислота
Стадия А: 2-[2-(2-фурил)-2-оксоэтил]пропандинитрил
46.2 мл 1 М раствора NaOEt в EtOH (46.2 ммоль) и 400 мл EtOH охлаждали до 0°С и добавляли 3.2 г малононитрила (48.4 ммоль). Смесь перемешивали при 0°С в течение 1 часа, затем добавляли 8.35 г 2-бром-1-(2-фурил)этанона (44 ммоль). Смесь перемешивали при 0°С в течение 1 часа и затем при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Летучие компоненты удаляли при пониженном давлении, остаток дигерировали в Et2O, отфильтровывали, затем очищали с помощью флэш-хроматографии, используя ДХМ и EtOAc в качестве элюентов с получением 2-[2-(2-фурил)-2-оксоэтил]пропандинитрила. МС: (M+H)+=175.2
Стадия В: 2-амино-5-(2-фурил)фуран-3-карбонитрил
4.587 г 2-[2-(2-фурил)-2-оксоэтил]пропандинитрила (26.34 ммоль) растворяли в 150 мл EtOH и добавляли 4.6 г Amberlite 15H+. Смесь перемешивали при 90°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем фильтровали, промывали ДХМ и EtOAc. Фильтрат концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 2-амино-5-(2-фурил)фуран-3-карбонитрила. МС: (М+Н)+=175.4
Стадия С: 6-(2-фурил)-3Н-фуро[2,3-d]пиримидин-4-он
1310 мг 2-амино-5-(2-фурил)фуран-3-карбонитрила (7.52 ммоль) и 30 мл муравьиноуксусного ангидрида помещали в колбу и перемешивали при к.т. в течение 30 минут. Затем летучие компоненты упаривали при пониженном давлении и остаток растворяли в 60 мл АсОН и облучали при 180°С в течение 50 минут. Смесь охлаждали до к.т. и сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 6-(2-фурил)-3H-фуро[2,3-d]пиримидин-4-она.1Н ЯМР (400 МГц, ДМСО-d6): 12.68 (br s, 1Н), 8.14 (s, 1H), 7.84 (m, 1H), 7.08 (s, 1H), 6.94 (d, 1H), 6.67 (m, 1H)
Стадия D: 6-(5-хлор-2-фурил)-3Н-фуро[2,3-d]пиримидин-4-он
1.183 г 6-(2-фурил)-3H-фуро[2,3-d]пиримидин-4-она (5.85 ммоль) растворяли в 55 мл ТГФ и добавляли 860 мг NCS (6.44 ммоль). Смесь перемешивали при 40°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь охлаждали до 0°С, и осадок отфильтровывали и сушили с получением 6-(5-хлор-2-фурил)-3H-фуро[2,3-d]пиримидин-4-она. МС: (М+Н)+=237.0
Стадия Е: 5-бром-6-(5-хлор-2-фурил)-3Н-фуро[2,3-d]пиримидин-4-он)
1000 мг 6-(5-хлор-2-фурил)-3H-фуро[2,3-d]пиримидин-4-она (4.23 ммоль) растворяли в 40 мл АсОН, затем добавляли 776 мг брома (4.86 ммоль). Смесь перемешивали при 40°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем летучие компоненты удаляли при пониженном давлении. Остаток дигерировали ДХМ, затем отфильтровывали с получением 5-бром-6-(5-хлор-2-фурил)-3H-фуро[2,3-d]пиримидин-4-она. МС: (М-Н)+=314.8
Стадия F: 5-бром-4-хлор-6-(5-хлор-2-фурил)фуро[2,3-d]пиримидин 1110 мг 5-бром-6-(5-хлор-2-фурил)-3H-фуро[2,3-d]пиримидин-4-она (3.52 ммоль) растворяли в 8.21 мл POCl3 (88.1 ммоль), затем добавляли 447 мкл DMA (3.52 ммоль). Смесь перемешивали при 110°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем охлаждали до -78°С и добавляли лед. Смесь подвергали действию ультразвука и затем осадок отфильтровывали. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 5-бром-4-хлор-6-(5-хлор-2-фурил)фуро[2,3-d]пиримидина. МС: (М+Н)+=335.0
Стадия G: этил (2R)-2-[5-бром-6-(5-хлор-2-фурил)фуро[2,3-d]пиримидин-4-ил]окси-3-[2-(пиразин-2-илметокси)фенил]пропаноат
1 экв. 5-бром-4-хлор-6-(5-хлор-2-фурил)фуро[2,3-d]пиримидина, 2 экв. соединения Синтеза 2g, 10 мл/ммольtBuOH и 5 экв. Cs2CO3 помещали в колбу и перемешивали при 55°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем концентрировали при пониженном давлении, остаток разбавляли соляным раствором, нейтрализовали 1 М водным раствором HCl и экстрагировали с помощью EtOAc. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением этил (2R)-2-[5-бром-6-(5-хлор-2-фурил)фуро[2,3-d]пиримидин-4-ил]окси-3-[2-(пиразин-2-илметокси)фенил]пропаноата. МС: (М+Н)+=601.0
Стадия Н: Пример 5
Используя Общую Методику II и этил (2R)-2-[5-бром-6-(5-хлор-2-фурил)фуро[2,3-d]пиримидин-4-ил]окси-3-[2-(пиразин-2-илметокси)фенил]пропаноат в качестве подходящего 5-бромфуро[2,3-d]пиримидинового производного и соединение Синтеза 3b в качестве подходящего производного бороновой кислоты, Пример 5 получали в виде смеси диастереоизомеров. МСВР: рассчитано для C38H36Cl2N6O7: 758.2023, найдено: 759.2119 и 759.2156 (М+Н)
Пример 6: (2R)-3-{2-[(1-трет-бутил-1H-пиразол-5-ил)метокси]фенил}-2-{[5-{3-хлор-4-[2-(диметиламино)этокси]-2-метилфенил}-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси}пропановая кислота
Стадия А: 1-трет-бутил-5-(диметоксиметил)-1H-пиразол
1.2 экв. гидрохлорида трет-бутилгидразина и 1 экв. соединения Синтеза 5а растворяли в сухом метаноле (0.5 мл/ммоль), затем по частям добавляли 1.2 экв. NaOEt и смесь перемешивали при 75°С в течение 2 часов. Реакционную смесь охлаждали и концентрировали при пониженном давлении. Остаток разбавляли водой и смесь экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 1-трет-бутил-5-(диметоксиметил)-1H-пиразола.1H ЯМР (400 МГц, ДМСО-d6) δ: 7.34 (d, 1Н), 6.34 (d, 1H), 5.74 (s, 1H), 3.24 (s, 6H), 1.57 (s, 9H). Также был получен 1-трет-бутил-3-(диметоксиметил)-1H-пиразол.1Н ЯМР (400 МГц, ДМСО-d6) δ: 7.75 (d, 1H), 6.18 (d, 1H), 5.34 (s, 1H), 3.24 (s, 6H), 1.50 (s, 9H)
Стадия В: (1-трет-бутил-1Н-пиразол-5-ил)метанол
1 экв. 1-трет-бутил-5-(диметоксиметил)-1H-пиразола при 50°С перемешивали с 1 М водным раствором HCl (3 мл/ммоль) до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Реакционную смесь охлаждали до 0°С, затем частями добавляли 2.85 экв. твердого NaOH. Значение рН доводили до 8, используя 10% водный раствор K2CO3, затем частями добавляли 2 экв. борогидрида натрия, поддерживая температуру ниже 5°С, и перемешивали при 0°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь экстрагировали с помощью EtOAc, объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc с получением (1-трет-бутил-1H-пиразол-5-ил)метанола.1Н ЯМР (400 МГц, ДМСО-d6) δ: 7.27 (d, 1H), 6.19 (d, 1H), 5.31 (t, 1H), 4.61 (d, 2H), 1.56 (s, 9H)
Стадия С: (2R)-3-[2-[(2-трет-бутилпиразол-3-ил)метокси]фенил]-2-гидроксипропановая кислота
2.51 г соединения Синтеза 2а (9.96 ммоль), 2.0 г (1-трет-бутил-1Н-пиразол-5-ил)метанола (13 ммоль) и 3.39 г трифенилфосфина (13 ммоль) растворяли в 12 мл сухого толуола, затем добавляли 5.9 мл DEAD (13 ммоль). Смесь перемешивали при 50°С в атмосфере азота до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Летучие компоненты упаривали при пониженном давлении. Затем добавляли 30 мл Et2O, смесь подвергали действию ультразвука и фильтровали (для удаления PPh3 и PPh3O). Фильтрат концентрировали при пониженном давлении. Остаток растворяли в ТГФ и затем добавляли 2 г NaOH, растворенного в 8 мл воды. Смесь перемешивали при 50°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь подкисляли 2 М водным раствором HCl, и ТГФ удаляли при пониженном давлении. Остаток экстрагировали с помощью ДХМ, сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением (2R)-3-[2-[(2-трет-бутилпиразол-3-ил)метокси]фенил]-2-гидроксипропановой кислоты. МС (М+Н): 319.0
Стадия D: этил (2R)-3-[2-[(2-трет-бутилпиразол-3-ил)метокси]фенил]-2-гидроксипропаноат
7.2 г (2R)-3-[2-[(2-трет-бутилпиразол-3-ил)метокси]фенил]-2-гидроксипропановой кислоты растворяли в 75 мл EtOH и затем добавляли 2 мл конц. H2SO4. Смесь перемешивали при 60°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли водой, нейтрализовали насыщенным водным раствором NаНСО3 и экстрагировали дихлорметаном. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя EtOAc и МеОН в качестве элюентов с получением этил (2R)-3-[2-[(2-трет-бутилпиразол-3-ил)метокси]фенил]-2-гидроксипропаноата. МС (М+Н): 347.0
Стадия Е: этил (2R)-2-[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси-3-[2-[(2-трет-бутилпиразол-3-ил)метокси]фенил]пропаноат
Используя Общую Методику Ia и этил (2R)-3-[2-[(2-трет-бутилпиразол-3-ил)метокси]фенил]-2-гидроксипропаноат в качестве подходящего производного сложного эфира молочной кислоты, получали этил (2R)-2-[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси-3-[2-[(2-трет-бутилпиразол-3-ил)метокси]фенил]пропаноат. МС (М+Н): 636.6-638.6
Стадия F: 2-[2-хлор-3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]-N,N-диметилэтанамин
10.0 г соединения Синтеза 3а (37.2 ммоль), 5.366 г N,N-диметилэтаноламина (60.3 ммоль) и 15.8 г PPh3 (60.3 ммоль) растворяли в 100 мл сухого толуола и затем по каплям добавляли 27 мл DEAD (60.3 ммоль, 40% раствор в толуоле). Смесь перемешивали при 50°С в атмосфере аргона до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Летучие компоненты упаривали при пониженном давлении и добавляли 100 мл Et2O. Осажденные белые кристаллы отфильтровывали и промывали с помощью Et2O. Фильтрат концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя CHCl3 и МеОН в качестве элюентов. Полученное в результате светло-коричневое масло кристаллизовали из гексана с получением 2-[2-хлор-3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]-N,N-диметилэтан амина.
1Н ЯМР (200 МГц, CDCl3) δ: 7.63 (d, 1H), 6.75 (d, 1H), 4.15 (t, 2H), 2.81 (t, 2H), 2.60 (s, 3H), 2.38 (s, 6H), 1.33 (s, 12H)
MC(M+H): 340.1
Стадия G: Пример 6
Используя Общую Методику II и этил (2R)-2-[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]окси-3-[2-[(2-трет-бутилпиразол-3-ил)метокси]фенил]пропаноат в качестве подходящего 5-бромфуро[2,3-d]пиримидинового производного и 2-[2-хлор-3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]-N,N-диметилэтанамин в качестве подходящего производного бороновой кислоты, получали Пример 6. МСВР: рассчитано для C40H41ClFN5O6: 741.2729, найдено: 742.2813 и 742.2808 (М+Н) для двух диастереомеров.
Пример 7: N-[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]-2-метокси-D-фенилаланин
Стадия А: (2R)-2-[[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]амино]-3-(2-метоксифенил)пропановая кислота
Используя Общую Методику Ib и (2R)-2-амино-3-(2-метоксифенил)пропановую кислоту в качестве подходящего аминокислотного производного, получали (2R)-2-[[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]амино]-3-(2-метоксифенил)пропановую кислоту. МС: (М+Н)+=487.8
Стадия В: Пример 7
1 экв. (2R)-2-[[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]амино]-3-(2-метоксифенил)пропановой кислоты, 1.5 экв. соединения Синтеза 3b, 5 мол. % AtaPhos и 2 экв. Cs2CO3 перемешивали в 1:1 смеси ТГФ и воды (10 мл/ммоль 5-бромфуро[2,3-d]пиримидинового производного) и нагревали до 110°С в MW реакторе до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли соляным раствором, рН доводили до 4 с помощью 1 М водного раствора HCl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Полученную смесь диастереоизомеров очищали и разделяли с помощью ЖХГВ хроматографии. Пример 7 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C36H37ClFN5O5: 673.2467, найдено: 337.6286 (М+2Н)
Пример 8: N-[(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}-D-фенилаланин
и
Пример 9: N-[(5Ra)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}-D-фенилаланин
Стадия А: (2R)-2-[[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]амино]-3-(2-гидроксифенил)пропановая кислота
Используя Общую Методику Ib и D-(R)-2-амино-3-(2-гидроксифенил)-пропионовую кислоту в качестве подходящего аминокислотного производного, получали (2R)-2-[[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]амино]-3-(2-гидроксифенил)пропановую кислоту. МС: (М+Н)+=473.6
Стадия В: этил (2R)-2-[[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-5-ил]метокси]фенил]пропаноат
163 мг (2R)-2-[[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]амино]-3-(2-гидроксифенил)пропановой кислоты растворяли в 3 мл раствора HCl (1.25 М в EtOH) и перемешивали при 60°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь концентрировали при пониженном давлении и разбавляли водой. Осадок отфильтровывали и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением этил (2R)-2-[[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]амино]-3-(2-гидроксифенил)пропаноата. МС: (М+Н)+=501.6
Стадия С: этил (2R)-2-[[5-бром-6-(4-фторфенил)фуро[2,3-d}пиримидин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-5-ил]метокси]фенил]пропаноат
500 мг этил (2R)-2-[[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]амино]-3-(2-гидроксифенил)пропаноата (1 ммоль), 540 мг соединения Синтеза 5b (2.5 ммоль) и 656 мг PPh3 (2.5 ммоль) растворяли в 20 мл сухого толуола в атмосфере N2, затем добавляли 576 мг DTAD (2.5 ммоль). Смесь перемешивали при 60°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением этил (2R)-2-[[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-5-ил]метокси]фенил]пропаноата. МСВР (М+Н)+: 698.1402
Стадия D: Примеры 8 и 9
1 экв. этил (2R)-2-[[5-бром-6-(4-фторфенил)фуро[2,3-d]пиримидин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-5-ил]метокси]фенил]пропаноата, 1.5 экв. соединения Синтеза 3b, 5 мол. % AtaPhos и 2 экв. Cs2CO3 перемешивали в 1:1 смеси ТГФ и воды (10 мл/ммоль 5-бромфуро[2,3-d]пиримидинового производного) и нагревали до 70°С и перемешивали до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли соляным раствором, рН доводили до 4 с помощью 1 М водного раствора HCl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырое промежуточное соединение очищали с помощью флэш-хроматографии, используя ДХМ и МеОН в качестве элюентов. Затем его растворяли в смеси диоксан : вода 1:1 (20 мл/ммоль) и добавляли 10 экв. LiOH×H2O. Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли соляным раствором, нейтрализовали 2 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением смеси диастереоизомеров. Их разделяли и очищали с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов. Пример 8 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C47H45ClFN7O6: 857.3104, найдено: 429.6637 (М+2Н). Пример 9 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C47H45ClFN7O6: 857.3104, найдено: 429.6648 (М+2Н)
Пример 10: N-[7-метил-5-(нафталин-1-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
Стадия А: 4-хлор-5-йод-7-метилпирроло[2,3-d]пиримидин
В 50 мл сосуд Шленка в атмосфере N2 загружали 220 мг NaH (5.5 ммоль) и 40 мл сухого ТГФ, и взвесь охлаждали до 0°С. Затем добавляли 1471 мг 4-хлор-5-йод-7H-пирроло[2,3-d]пиримидина (5 ммоль). После 30 минут перемешивания добавляли 346 мкл MeI (5.5 ммоль) и смеси давали нагреться до к.т. и перемешивали до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем разбавляли насыщенным водным раствором NH4Cl и экстрагировали с помощью ДХМ. Объединенные органические слои промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 4-хлор-5-йод-7-метилпирроло[2,3-d]пиримидина.
1H ЯМР (400 МГц, ДМСО-d6) δ: 8.65 (s, 1Н), 7.98 (s, 1H), 3.83 (s, 3H)
MC: (M+H)+=294.0
Стадия В: 4-хлор-7-метил-5-(1-нафтил)пирроло[2,3-d]пиримидин
1 экв. 4-хлор-5-йод-7-метилпирроло[2,3-d]пиримидина, 1.1 экв. сложного неопентилгликолевого эфира 1-нафталинбороновой кислоты, 1.1 экв. карбоната серебра, 0.15 экв. Pd(PPh3)4 и 2-Ме-ТГФ (15 мл/ммоль 5-йодпирроло[2,3-d]пиримидинового производного) перемешивали в N2 атмосфере при 110°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь разбавляли соляным раствором, нейтрализовали 1 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 4-хлор-7-метил-5-(1-нафтил)пирроло[2,3-d]пиримидина. МС: (М+Н)+=294.2
Стадия С: Пример 10
Используя Общую Методику III и 4-хлор-7-метил-5-(1-нафтил)пирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали Пример 10. МСВР: рассчитано для C26H22N4O2: 422.1743, найдено: 423.1804 (М+Н)
Пример 11: N-[5-(нафталин-1-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
Стадия А: 7-(бензолсульфонил)-4-хлор-5-йодпирроло[2,3-d]пиримидин
В 50 мл сосуд Шленка в атмосфере N2 загружали 220 мг NaH (5.5 ммоль) и 40 мл сухого ТГФ, и взвесь охлаждали до 0°С. Затем добавляли 1471 мг 4-хлор-5-йод-7H-пирроло[2,3-d]пиримидина (5 ммоль). После 30 минут перемешивания добавляли 1.4 мл бензолсульфонилхлорида (5.25 ммоль) и смеси давали нагреться до к.т., и перемешивали до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем разбавляли насыщенным водным раствором NH4Cl и экстрагировали с помощью ДХМ. Объединенные органические слои промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Затем остаток дигерировали МТВЕ и затем отфильтровывали с получением 7-(бензолсульфонил)-4-хлор-5-йодпирроло[2,3-d]пиримидина.
1H ЯМР (400 МГц, CDCl3) δ: 8.75 (s, 1Н), 8.22 (m, 2H), 7.95 (s, 1H), 7.67 (m, 1H), 7.56 (m, 2H)
МС: (M+H)+=419.8
Стадия B: 7-(бензолсулъфонил)-4-хлор-5-(1-нафтил)пирроло[2,3-d]пиримидин
1 экв. 7-(бензолсульфонил)-4-хлор-5-йодпирроло[2,3-d]пиримидина, 1.1 экв. сложного неопентилгликолевого эфира 1-нафталинбороновой кислоты, 1.1 экв. карбоната серебра, 0.15 экв. Pd(PPh3)4 и 2-Ме-ТТФ (15 мл/ммоль 5-йодпирроло[2,3-d]пиримидинового производного) перемешивали в N2 атмосфере при 110°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь разбавляли соляным раствором, нейтрализовали 1 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 7-(бензолсульфонил)-4-хлор-5-(1-нафтил)пирроло[2,3-d]пиримидина.
1H ЯМР (400 МГц, CDCl3) δ: 8.82 (s, 1H), 8.31 (m, 2H), 7.94 (m, 2H), 7.84 (s, 1H), 7.71 (m, 1H), 7.60 (m, 2H), 7.56-7.48 (m, 3H), 7.48-7.38 (m, 2H)
MC: (M+H)+=420.0
Стадия С: Пример 11
Используя Общую Методику III и 7-(бензолсульфонил)-4-хлор-5-(1-нафтил)пирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали Пример 11. МСВР: рассчитано для C25H20N4O2: 408.1586, найдено: 409.1670 (М+Н)
Пример 12: N-[7-бензил-6-этил-5-(нафталин-1-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин, диастереоизомер 1
и
Пример 13: N-[7-бензил-6-этил-5-(нафталин-1-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин, диастереоизомер 2
Используя Общую Методику IVa и соединение Синтеза 6 в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и сложный неопентилгликолевый эфир 1-нафталинбороновой кислоты в качестве подходящего производного бороновой кислоты. Пример 12 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C34H30N4O2: 526.2369, найдено: 527.2431 (М+Н). Пример 13 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C34H30N4O2: 526.2369, найдено: 527.2423 (М+Н)
Пример 14: N-{6-этил-5-(нафталин-1-ил)-7-[2-(нафталин-1-илокси)этил]-7H-пирроло[2,3-d]пиримидин-4-ил}-D-фенилаланин, диастереоизомер 1
и
Пример 15: N-{6-этил-5-(нафталин-1-ил)-7-[2-(нафталин-1-илокси)этил]-7H-пирроло[2,3-d]пиримидин-4-ил}-D-фенилаланин, диастереоизомер 2
Стадия А: 5-бром-4-хлор-6-этил-7-[2-(1-нафтилокси)этил]пирроло[2,3-d]пиримидин
94 мг 2-(1-нафтилокси)этанола (0.5 ммоль), 131 мг PPh3 (0.5 ммоль) и 66 мг соединения Синтеза 1b (0.25 ммоль) растворяли в 2.5 мл сухого ТГФ в атмосфере N2 и охлаждали до 0°С. Затем по каплям добавляли 230 мкл DEAD (0.5 ммоль, 40% в толуоле). Смесь перемешивали при 40°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем летучие компоненты удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 5-бром-4-хлор-6-этил-7-[2-(1-нафтилокси)этил]пирроло[2,3-d]пиримидина.
1H ЯМР (400 МГц, ДМСО-d6) δ: 8.69 (s, 1Н), 7.80 (dd, 2H), 7.51-7.31 (m, 4H), 6.94 (d, 1Н), 4.90 (t, 2H), 4.52 (t, 2H), 3.08 (q, 2H), 1.26 (t, 3Н)
MC: (М+H)+=430.0
Стадия В: (2R)-2-[[5-бром-6-этил-7-[2-(1-нафтилокси)этил]пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановая кислота
Используя Общую Методику III и 5-бром-4-хлор-6-этил-7-[2-(1-нафтилокси)этил]пирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[[5-бром-6-этил-7-[2-(1-нафтилокси)этил]пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 12.96 (br s, 1Н), 8.24 (s, 1Н), 7.88 (d, 1Н), 7.82 (d, 1Н), 7.52-7.32 (m, 4H), 7.29-7.15 (m, 5H), 6.94 (d, 1Н), 6.38 (d, 1Н), 4.94 (q, 1Н), 4.72 (t, 2H), 4.45 (t, 2H), 3.28 (m, 1Н), 3.18 (dd, 1Н), 2.92 (q, 2H), 1.19 (t, 3Н)
MC: (M+H)+=559.2
Стадия С: Примеры 14 и 15
Используя Общую Методику IVa и (2R)-2-[[5-бром-6-этил-7-[2-(1-нафтилокси)этил]пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и сложный неопентилгликолевый эфир 1-нафталинбороновой кислоты в качестве подходящего производного бороновой кислоты. Пример 14 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C39H34N4O3: 606.2631, найдено: 607.2711 (М+Н). Пример 15 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C39H34N4O3: 606.2631, найдено: 607.2705 (М+Н)
Пример 16: N-[6-этил-5-(нафталин-1-ил)-7-(2-фенилэтил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин, диастереоизомер 1
и
Пример 17: N-[6-этил-5-(нафтталин-1-ил)-7-(2-фенилэтил)-7Н-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин, диастереоизомер 2
Стадия А: 5-бром-4-хлор-6-этил-7-фенэтилпирроло[2,3-d]пиримидин
3.1 мл 2-фенилэтанола (25.9 ммоль), 3.397 г PPh3 (12.95 ммоль) и 3.40 г соединения Синтеза 1b (12.95 ммоль) растворяли в 110 мл сухого ТГФ в атмосфере N2 и охлаждали до 0°С. Затем по каплям добавляли 11.87 мл DEAD (40% в толуоле). Смесь перемешивали при 40°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем летучие компоненты удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 5-бром-4-хлор-6-этил-7-фенэтилпирроло[2,3-d]пиримидина.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 8.61 (s, 1H), 7.32-7.16 (m, 3Н), 7.11 (m, 2H), 4.51 (t, 2H), 3.06 (t, 2H), 2.70 (q, 2H), 1.10 (t, 3Н)
MC: (М+H)+=364.0
Стадия В: (2R)-2-[(5-бром-6-этил-7-фенэтилпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановая кислота
Используя Общую Методику III и 5-бром-4-хлор-6-этил-7-фенэтилпирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[(5-бром-6-этил-7-фенэтилпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановую кислоту.
1H ЯМР (400 МГц, ДМСО-d6) δ: 12.80 (br s, 1H), 8.20 (s, 1H), 7.34-7.17 (m, 8H), 7.13 (m, 2H), 6.45 (d, 1H), 4.91 (q, 1H), 4.33 (t, 2H), 3.31 (dd, 1H), 3.18 (dd, 1H), 3.00 (t, 2H), 2.55 (q, 2H), 1.04 (t, 3Н)
MC: (M+H)+=493.2
Стадия С: Примеры 16 и 17
Используя Общую Методику IVa и (2R)-2-[(5-бром-6-этил-7-фенэтилпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановую кислоту в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и сложный неопентилгликолевый эфир 1-нафталинбороновой кислоты в качестве подходящего производного бороновой кислоты. Пример 16 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C35H32N4O2: 540.2525, найдено: 541.2592 (М+Н). Пример 17 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C35H32N4O2: 540.2525, найдено: 541.2619 (М+Н)
Пример 18: N-[6-этил-5-(нафталин-1-ил)-7-(3-фенилпропил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин, диастереоизомер 1
и
Пример 19: N-[6-этил-5-(нафталин-1-ил)-7-(3-фенилпропил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин, диастереоизомер 2
Стадия А: 5-бром-4-хлор-6-этил-7-(3-фенилпропил)пирроло[2,3-d]пиримидин
3.52 мл 3-фенилпропанола (25.9 ммоль), 3.397 г PPh3 (12.95 ммоль) и 3.4 г соединения Синтеза 1b (12.95 ммоль) растворяли в 110 мл сухого ТГФ в атмосфере N2 и охлаждали до 0°С. Затем по каплям добавляли 11.87 мл DEAD (40% в толуоле). Смесь перемешивали при 40°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем летучие компоненты удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 5-бром-4-хлор-6-этил-7-(3-фенилпропил)пирроло[2,3-d]пиримидина.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 8.60 (s, 1Н), 7.31-7.22 (m, 2H), 7.21-7.13 (m, 3Н), 4.32 (t, 2H), 2.85 (q, 2H), 2.65 (t, 2H), 2.05 (m, 2H), 1.16 (t, 3Н)
MC: (M+H)+=378.0
Стадия В: (2R)-2-[[5-бром-6-этил-7-(3-фенилпропил)пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановая кислота
Используя Общую Методику III и 5-бром-4-хлор-6-этил-7-(3-фенилпропил)пирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[[5-бром-6-этил-7-(3-фенилпропил)пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 12.95 (br s, 1H), 8.15 (s, 1H), 7.33-7.12 (m, 10H), 6.35 (d, 1H), 4.94 (q, 1H), 4.16 (t, 2H), 3.28 (dd, 1H), 3.16 (dd, 1H), 2.68 (q, 2H), 2.61 (t, 2H), 1.97 (m, 2H), 1.09 (t, 3H)
MC: (M+H)+=507.2
Стадия С: Примеры 18 и 19
Используя Общую Методику IVa и (2R)-2-[[5-бром-6-этил-7-(3-фенилпропил)пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и сложный неопентилгликолевый эфир 1-нафталинбороновой кислоты в качестве подходящего производного бороновой кислоты. Пример 18 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C36H34N4O2: 554.2682, найдено: 555.2742 (М+Н). Пример 19 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C36H34N4O2: 554.2682, найдено: 555.2756 (М+Н)
Пример 20: N-[(5Ra)-5-(3-хлор-2-метилфенил)-6-этил-7-метил-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
и
Пример 21: N-[(5Sa)-5-(3-хлор-2-метилфенил)-6-этил-7-метил-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
Стадия А: 5-бром-4-хлор-6-этил-7-метилпирроло[2,3-d]пиримидин
65 мг соединения Синтеза 1b (0.25 ммоль) растворяли в 1 мл сухого ТГФ, затем добавляли 20.3 мкл сухого МеОН (0.5 ммоль) и 0.5 мл раствора цианометилентрибутилфосфорана (0.5 ммоль, 1 М в толуоле). Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Летучие компоненты удаляли при пониженном давлении. Остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 5-бром-4-хлор-6-этил-7-метилпирроло[2,3-d]пиримидина.
Н ЯМР (400 МГц, CDCl3) δ: 8.56 (s, 1H), 3.84, (s, 3H), 2.91 (q, 2H), 1.26 (t, 3H)
MC: (M+H)+=274.0
Стадия В: (2R)-2-[[5-бром-6-этил-7-метилпирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановая кислота
Используя Общую Методику III и 5-бром-4-хлор-6-этил-7-метилпирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[[5-бром-6-этил-7-метилпирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту.
1Н ЯМР (500 МГц, ДМСО-d6) δ: 13.05 (br s, 1H), 8.17 (s, 1H), 7.32-7.25 (m, 2H), 7.25-7.18 (m, 3H), 6.32 (d, 1H), 4.97 (m, 1H), 3.68, (s, 3H), 3.29 (dd, 1H), 3.18 (dd, 1H),2.75(q, 2H),1.13(t, 3H)
MC: (M+H)+=403.0
Стадия С: Примеры 20 и 21
Используя Общую Методику IVa и (2R)-2-[[5-бром-6-этил-7-метилпирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и соединение Синтеза 3с в качестве подходящего производного бороновой кислоты, Пример 20 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C25H25ClN4O2: 448.1666, найдено: 449.1753 (М+Н). Пример 21 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C25H25ClN4O2: 448.1666, найдено: 449.1752 (М+Н)
Пример 22: N-[(5Ra)-5-(3-хлор-2-метилфенил)-7-(циклопропилметил)-6-этил-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
и
Пример 23: N-[(5Sa)-5-(3-хлор-2-метилфенил)-7-(циклопропилметил)-6-этил-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
Стадия А: 5-бром-4-хлор-7-(циклопропилметил)-6-этилпирроло[2,3-d]пиримидин
65 мг соединения Синтеза 1b (0.25 ммоль) растворяли в 1 мл сухого ТГФ, затем добавляли 40 мкл циклопропанметанола (0.5 ммоль) и 0.5 мл раствора цианометилентрибутилфосфорана (0.5 ммоль, 1 М в толуоле). Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Летучие компоненты удаляли при пониженном давлении. Остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 5-бром-4-хлор-7-(циклопропилметил)-6-этилпирроло[2,3-d]пиримидина.
1Н ЯМР (400 МГц, CDCl3) δ: 8.54 (s, 1H), 4.18 (d, 2H), 2.94 (q, 2H), 1.29 (t, 3H), 1.24-1.14 (m, 1H), 0.60-0.51 (m, 2H), 0.51-0.43 (m, 2H)
MC: (М+Н)+=314.0
Стадия В: (2R)-2-[[5-бром-7-(циклопропилметил)-6-этилпирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановая кислота
Используя Общую Методику III и 5-бром-4-хлор-7-(пиклопропилметил)-6-этилпирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[[5-бром-7-(циклопропилметил)-6-этилпирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту.
1Н ЯМР (500 МГц, ДМСО-d6) δ: 13.05 (br s, 1H), 8.15 (s, 1H), 7.32-7.26 (m, 2H), 7.26-7.20 (m, 3H), 6.34 (d, 1H), 4.94 (m, 1H), 4.05 (d, 2H) 3.29 (dd, 1H), 3.18 (dd, 1H), 2.78 (q, 2H), 1.28-1.20 (m, 1H), 1.16 (t, 3H), 0.47-0.42 (m, 2H), 0.42-0.37 (m, 2H)
MC: (M+H)+=443.0
Стадия С: Примеры 22 и 23
Используя Общую Методику IVa и (2R)-2-[[5-бром-7-(циклопропилметил)-6-этилпирроло [2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и соединение Синтеза 3с в качестве подходящего производного бороновой кислоты, Пример 22 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C28H29ClN4O2: 488.1979, найдено: 489.2064 (М+Н). Пример 23 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C28H29ClN4O2: 488.1979, найдено: 489.2048 (М+Н)
Пример 24: N-[(5Ra)-5-(3-хлор-2-метилфенил)-6-этил-7-(проп-2-ен-1-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
и
Пример 25: N-[(5Sa)-5-3-хлор-2-метилфенил)-6-этил-7-(проп-2-ен-1-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
Стадия А: 7-аллил-5-бром-4-хлор-6-этилпирроло[2,3-d]пиримидин
65 мг соединения Синтеза 1b (0.25 ммоль) растворяли в 1 мл сухого ТГФ, затем добавляли 34 мкл аллилового спирта (0.5 ммоль) и 0.5 мл раствора цианометилентрибутилфосфорана (0.5 ммоль, 1 М в толуоле). Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Летучие компоненты удаляли при пониженном давлении. Остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 7-аллил-5-бром-4-хлор-6-этилпирроло[2,3-d]пиримидина.
1Н ЯМР (400 МГц, CDCl3) δ: 8.57 (s, 1H), 6.02-5.90 (m, 1H), 5.25-5.16 (m, 1H), 5.00-4.85 (m, 3H), 2.87 (q, 2H), 1.26 (t, 3H)
MC: (M+H)+=300.0
Стадия В: (2R)-2-[[7-аллил-5-бром-6-этилпирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановая кислота
Используя Общую Методику III и 7-аллил-5-бром-4-хлор-6-этилпирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[[7-аллил-5-бром-6-этилпирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту.
1Н ЯМР (500 МГц, ДМСО-d6) δ: 13.06 (br s, 1H), 8.16 (s, 1H), 7.34-7.26 (m, 2H), 7.26-7.19 (m, 3H), 6.35 (d, 1H), 6.01-5.89 (m, 1H), 5.10 (dd, 1H), 5.01-4.93 (m, 1H), 4.87-4.73 (m, 3H), 3.29 (dd, 1H), 3.18 (dd, 1H), 2.70 (q, 2H), 1.12 (t, 3H)
MC: (M+H)+=429.0
Стадия С: Примеры 24 и 25
Используя Общую Методику IVa и (2R)-2-[[7-аллил-5-бром-6-этилпирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и соединение Синтеза 3с в качестве подходящего производного бороновой кислоты, Пример 24 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C27H27ClN4O2: 474.1823, найдено: 475.1908. Пример 25 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C27H27ClN4O2: 474.1823, найдено: 475.1909
Пример 26: N-[7-(бут-2-ин-1-ил)-(5Ra)-5-(3-хлор-2-метилфенил)-6-этил-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
и
Пример 27: N-[7-(бут-2-ин-1-ил)-(5Sa)-5-(3-хлор-2-метилфенил)-6-этил-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
Стадия А: 5-бром-7-бут-2-инил-4-хлор-6-этилпирроло[2,3-d]пиримидин
37 мкл 2-бутин-1-ола (0.5 ммоль), 131 мг PPh3 (0.5 ммоль) и 66 мг соединения Синтеза 1b (0.25 ммоль) растворяли в 2.5 мл сухого ТГФ в атмосфере N2 и охлаждали до 0°С. Затем по каплям добавляли 230 мкл DEAD (0.5 ммоль, 40% в толуоле). Смесь перемешивали при 40°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем летучие компоненты удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 5-бром-7-бут-2-инил-4-хлор-6-этилпирроло[2,3-d] пиримидина.
1Н ЯМР (400 МГц, CDCl3) δ: 8.59 (s, 1Н), 5.03 (q, 2H), 2.99 (q, 2H), 1.77 (t, 3H), 1.33(t, 3H)
MC: (M+H)+=312.0
Стадия В: (2R)-2-[(5-бром-7-бут-2-инил-6-этилпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановая кислота
Используя Общую Методику III и 5-бром-7-бут-2-инил-4-хлор-6-этилпирроло [2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[(5-бром-7-бут-2-инил-6-этилпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановую кислоту.
1Н ЯМР (500 МГц, ДМСО-d6) δ: 13.25 (br s, 1Н), 8.19 (s, 1Н), 7.30-7.24 (m, 2H), 7.24-7.16 (m, 3H), 6.45 (d, 1Н), 5.02-4.96 (m, 2H), 4.93 (q, 1Н), 3.30 (dd, 1Н), 3.19 (dd, 1Н), 2.80 (q, 2H), 1.74 (t, 3H), 1.19 (t, 3H)
MC: (M+H)+=441.0
Стадия С: Примеры 26 и 27
Используя Общую Методику IVa и (2R)-2-[(5-бром-7-бут-2-инил-6-этилпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановую кислоту в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и соединение Синтеза 3с в качестве подходящего производного бороновой кислоты. Пример 26 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C28H27ClN4O2: 486.1823, найдено: 487.1893 (М+Н). Пример 27 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C28H27ClN4O2: 486.1823, найдено: 487.1893 (М+Н)
Пример 28: N-[(5Ra)-5-(3-хлор-2-метилфенил)-6-этил-7-(2,2,2-трифторэтил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
и
Пример 29: N-[(5Sa)-5-(3-хлор-2-метилфенил)-6-этил-7-(2,2,2-трифторэтил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
Стадия А: 5-бром-4-хлор-6-этил-7-(2,2,2-трифторэтил)пирроло[2,3-d]пиримидин
72 мкл трифторэтанола (1 ммоль), 262 мг РРh3 (1 ммоль) и 130 мг соединения Синтеза 1b (0.5 ммоль) растворяли в 5 мл сухого ТГФ в атмосфере N2 и охлаждали до 0°С. Затем по каплям добавляли 460 мкл DEAD (0.5 ммоль, 40% в толуоле). Смесь перемешивали при 40°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем летучие компоненты удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 5-бром-4-хлор-6-этил-7-(2,2,2-трифторэтил)пирроло[2,3-d]пиримидина.
1Н ЯМР (400 МГц, CDCl3) δ: 8.62 (s, 1H), 4.90 (q, 2H), 2.94 (q, 2H), 1.28 (t, 3H)
MC: (M+H)+=342.0
Стадия В: (2R)-2-[[5-бром-6-этил-7-(2,2,2-трифторэтил)пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановая кислота
Используя Общую Методику III и 5-бром-4-хлор-6-этил-7-(2,2,2-трифторэтил)пирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[[5-бром-6-этил-7-(2,2,2-трифторэтил)пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту.
1H ЯМР (500 МГц, ДМСО-d6) δ: 13.11 (br s, 1H), 8.23 (s, 1H), 7.33-7.26 (m, 2H), 7.26-7.19 (m, 3H), 6.44 (d, 1H), 5.12 (q, 2H), 5.00-4.93 (m, 1H), 3.30 (dd, 1H), 3.20 (dd, 1H), 2.78 (q, 2H), 1.14 (t, 3H)
MC: (M+H)+=471.0
Стадия С: Примеры 28 и 29
Используя Общую Методику IVa и (2R)-2-[[5-бром-6-этил-7-(2,2,2-трифторэтил)пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и соединение Синтеза 3с в качестве подходящего производного бороновой кислоты, Пример 28 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C26H24ClF3N4O2: 516.1540, найдено: 517.1624 (М+Н). Пример 29 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C26H24ClF3N4O2: 516.1540, найдено: 517.1606 (М+Н)
Пример 30: N-[(5Ra)-5-(3-хлор-2-метилфенил)-7-(2-циклопентилэтил)-6-этил-7H-пирроло(2,3-d]пиримидин-4-ил]-D-фенилаланин
и
Пример 31: N-[(5Sa)-5-(3-хлор-2-метилфенил)-7-(2-циклопентилэтил)-6-этил-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
Стадия А: 5-бром-4-хлор-7-(2-циклопентилэтил)-6-этилпирроло[2,3-d]пиримидин
124 мкл 2-циклопентилэтанола (1 ммоль), 262 мг PPh3 (1 ммоль) и 130 мг соединения Синтеза 1b (0.5 ммоль) растворяли в 5 мл сухого ТГФ в атмосфере N2 и охлаждали до 0°С. Затем по каплям добавляли 460 мкл DEAD (0.5 ммоль, 40% в толуоле). Смесь перемешивали при 40°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем летучие компоненты удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 5-бром-4-хлор-7-(2-циклопентилэтил)-6-этилпирроло[2,3-d]пиримидина.
1Н ЯМР (400 МГц, CDCl3) δ: 8.55 (s, 1H), 4.31-4.20 (m, 2H), 2.89 (q, 2H), 1.91-1.72 (m, 5H), 1.69-1.57 (m, 2H), 1.57-1.46 (m, 2H), 1.28 (t, 3H), 1.23-1.05 (m, 2H)
MC: (M+H)+=356.0
Стадия В: (2R)-2-[[5-бром-7-(2-циклопентилэтил)-6-этилпирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановая кислота
Используя Общую Методику III и 5-бром-4-хлор-7-(2-циклопентилэтил)-6-этилпирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[[5-бром-7-(2-циклопентилэтил)-6-этилпирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту.
1Н ЯМР (500 МГц, ДМСО-d6) δ: 13.04 (br s, 1H), 8.17 (s, 1H), 7.32-7.26 (m, 2H), 7.25-7.19 (m, 3H), 6.32 (d, 1H), 5.00-4.92 (m, 1H), 4.17-4.09 (m, 2H), 3.29 (dd, 1H), 3.18 (dd, 1H), 2.74 (q, 2H), 1.79-1.70 (m, 3H), 1.70-1.62 (m, 2H), 1.60-1.50 (m, 2H), 1.50-1.42 (m, 2H), 1.15 (t, 3H), 1.12-1.01 (m, 2H)
MC: (M+H)+=485.2
Стадия С: Примеры 30 и 31
Используя Общую Методику IVa и (2R)-2-[[5-бром-7-(2-циклопентилэтил)-6-этилпирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и соединение Синтеза 3с в качестве подходящего производного бороновой кислоты. Пример 30 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C31H35ClN4O2: 530.2449, найдено: 531.2528 (М+Н). Пример 31 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C31H35ClN4O2: 530.2449, найдено: 531.2547 (М+Н)
Пример 32: N-[(5Ra)-5-(3-хлор-2-метилфенил)-6-этил-7-(нафталин-1-илметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
и
Пример 33: N-[(5Sa)-5-(3-хлор-2-метилфенил)-6-этил-7-(нафталин-1-илметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
Стадия А: 5-бром-4-хлор-6-этил-7-(1-нафтилметил)пирроло[2,3-d]пиримидин
158 мг 1-нафталинметанола (1 ммоль), 262 мг PPh3 (1 ммоль) и 130 мг соединения Синтеза 1b (0.5 ммоль) растворяли в 5 мл сухого ТГФ в атмосфере N2 и охлаждали до 0°С. Затем по каплям добавляли 460 мкл DEAD (0.5 ммоль, 40% в толуоле). Смесь перемешивали при 40°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем летучие компоненты удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 5-бром-4-хлор-6-этил-7-(1-нафтилметил)пирроло[2,3-d]пиримидина.
1Н ЯМР (400 МГц, CDCl3) δ: 8.58 (s, 1H), 8.09 (d, 1H), 7.95-7.89 (m, 1H), 7.79 (d, 1H), 7.66-7.54 (m, 2H), 7.25 (t, 1H), 6.45 (dd, 1H), 6.03 (s, 2H), 2.76 (q, 2H), 1.08(t, 3H)
MC: (M+H)+=400.0
Стадия В: (2R)-2-[[5-бром-6-этил-7-(1-нафтилметил)пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановая кислота
Используя Общую Методику III и 5-бром-4-хлор-6-этил-7-(1-нафтилметил)пирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[[5-бром-6-этил-7-(1-нафтилметил)пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту.
1H ЯМР (500 МГц, ДМСО-d6) δ: 13.14 (br s, 1H), 8.27 (d, 1H), 8.15 (s, 1H), 7.98 (d, 1H), 7.83 (d, 1H), 7.66-7.56 (m, 2H), 7.37-7.20 (m, 6H), 6.48 (d, 1H), 6.40 (d, 1H), 5.94 (s, 2H), 4.99 (q, 1H), 3.33 (dd, 1H), 3.22 (dd, 1H), 2.62 (q, 2H), 0.89 (t, 3H)
MC: (M+H)+=529.0
Стадия С: Примеры 32 и 33
Используя Общую Методику IVa и (2R)-2-[[5-бром-6-этил-7-(1-нафтилметил)пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и соединение Синтеза 3с в качестве подходящего производного бороновой кислоты. Пример 32 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C35H31ClN4O2: 574.2136, найдено: 575.2211 (М+Н). Пример 33 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C35H31ClN4O2: 574.2136, найдено: 575.2203 (М+Н)
Пример 34: N-[(5Ra)-5-(3-хлор-2-метилфенил)-6-этил-7-(4-метоксибензил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
и
Пример 35: N-[(5Sa)-5-(3-хлор-2-метилфенил)-6-этил-7-(4-метоксибензил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
Стадия А: 5-бром-4-хлор-6-этил-7-[(4-метоксифенил)метил]пирроло[2,3-d]пиримидин
138 мг 4-метоксибензилового спирта (1 ммоль), 262 мг PPh3 (1 ммоль) и 130 мг соединения Синтеза 1b (0.5 ммоль) растворяли в 5 мл сухого ТГФ в атмосфере N2 и охлаждали до 0°С. Затем по каплям добавляли 460 мкл DEAD (0.5 ммоль, 40% в толуоле). Смесь перемешивали при 40°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем летучие компоненты удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и ЕtOАс в качестве элюентов с получением 5-бром-4-хлор-6-этил-7-[(4-метоксифенил)метил]пирроло[2,3-d]пиримидина. МС: (М+Н)+=380.0
Стадия В: (2R)-2-[[5-бром-6-этил-7-[(4-метоксифенил)метил]пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановая кислота
Используя Общую Методику III и 5-бром-4-хлор-6-этил-7-[(4-метоксифенил)метил]пирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[[5-бром-6-этил-7-[(4-метоксифенил)метил]пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту.
1Н ЯМР (500 МГц, ДМСО-d6) δ: 13.07 (br s, 1H), 8.20 (s, 1Н), 7.33-7.17 (m, 5Н), 7.03 (d, 2Н), 6.85 (d, 2Н), 6.37 (d, 1Н), 5.37 (s, 2Н), 4.99 (q, 1Н), 3.69 (s, 3Н), 3.31 (dd, 1Н), 3.20 (dd, 1Н), 2.65 (q, 2Н), 0.91 (t, 3Н)
МС: (М+Н)+=508.8
Стадия С: Примеры 34 и 35
Используя Общую Методику IVa и (2R)-2-[[5-бром-6-этил-7-[(4-метоксифенил)метил]пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и соединение Синтеза 3с в качестве подходящего производного бороновой кислоты, Пример 34 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C32H31ClN4O3: 554.2085, найдено: 555.2176 (М+Н). Пример 35 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C32H31ClN4O3: 554.2085, найдено: 555.2140 (М+Н)
Пример 36: N-[7-бензил-(5Ra)-5-(3-хлор-2-метилфенил)-6-этил-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
и
Пример 37: N-[7-бензил-(5Sa)-5-(3-хлор-2-метилфенил)-6-этил-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
Используя Общую Методику IVa и соединение Синтеза 6 в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и соединение Синтеза 3с в качестве подходящего производного бороновой кислоты, Пример 36 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C31H29ClN4O2: 524.1979, найдено: 525.2048 (М+Н). Пример 37 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C31H29ClN4O2: 524.1979, найдено: 525.2064 (М+Н)
Пример 38: N-[(5Rа)-5-(3-хлор-2-метилфенил)-6-этил-7-(пропан-2-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
и
Пример 39: N-[(5Sa)-5-(3-хлор-2-метилфенил)-6-этил-7-(пропан-2-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-2)-фенилаланин
Стадия А: 5-бром-4-хлор-6-этил-7-изопропилпирроло[2,3-d]пиримидин
76 мкл 2-пропанола (1 ммоль), 262 мг РРh3 (1 ммоль) и 130 мг соединения Синтеза 1b (0.5 ммоль) растворяли в 5 мл сухого ТГФ в атмосфере N2 и охлаждали до 0°С. Затем по каплям добавляли 460 мкл DEAD (0.5 ммоль, 40% в толуоле). Смесь перемешивали при 40°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем летучие компоненты удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и ЕtOАс в качестве элюентов с получением 5-бром-4-хлор-6-этил-7-изопропилпирроло[2,3-d]пиримидина.
1Н ЯМР (400 МГц, CDCl3) δ: 8.53 (s, 1Н), 4.71 (sp, 1Н), 2.92 (q, 2Н), 1.72 (d, 6Н), 1.25 (t, 3Н)
МС: (М+Н)+=302.0
Стадия В: (2R)-2-[(5-бром-6-этил-7-изопропилпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановая кислота
Используя Общую Методику III и 5-бром-4-хлор-6-этил-7-изопропилпирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[(5-бром-6-этил-7-изопропилпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановую кислоту.
1Н ЯМР (500 МГц, ДМСО-d6) δ: 13.04 (br s, 1H), 8.14 (s, 1Н), 7.35-7.17 (m, 5Н), 6.33 (d, 1Н), 4.95 (q, 1H), 4.64 (sp, 1Н), 3.28 (dd, 1H), 3.17 (dd, 1Н), 2.76 (q, 2Н), 1.59 (d, 6Н), 1.11 (t, 3Н)
МС: (М+Н)+=431.2
Стадия С: Примеры 38 и 39
Используя Общую Методику IVa и (2R)-2-[(5-бром-6-этил-7-изопропилпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановую кислоту в качестве подходящего 5-бромпирроло[2,3-d]пиримидинового производного и соединение Синтеза 3с в качестве подходящего производного бороновой кислоты, Пример 38 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C27H29ClN4O2: 476.1979, найдено: 477.2057 (М+Н). Пример 39 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C27H29ClN4O2: 476.1979, найдено: 477.2063 (М+Н)
Пример 40: (2R)-2-[(7-бензил-(5Sa)-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-этил-7H-пирроло[2,3-d]пиримидин-4-ил)окси]-3-фенилпропановая кислота
Стадия А: 7-бензил-5-бром-4-хлор-6-этилпирроло[2,3-d]пиримидин
255 мг NaH (6.38 ммоль) и 50 мл сухого ТГФ загружали в 50 мл сосуд Шленка в атмосфере N2 и взвесь охлаждали до 0°С. Затем добавляли 1.792 г соединения Синтеза 1b (5.8 ммоль). После перемешивания смеси в течение 30 минут при 0°С, добавляли 773 мкл бензилбромида (6.38 ммоль) и смеси давали нагреться до к.т., и перемешивали до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем разбавляли насыщенным водным раствором NH4Cl и экстрагировали с помощью ДХМ. Объединенные органические слои промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 7-бензил-5-бром-4-хлор-6-этилпирроло[2,3-d]пиримидина.1Н ЯМР (400 МГц, CDCl3) δ: 8.58 (s, 1Н), 7.35-7.20 (m, 3Н), 7.10-6.96 (m, 2Н), 5.52 (s, 2Н), 2.78 (q, 2Н), 1.05 (t, 3Н)
Стадия В: метил (2R)-2-(7-бензил-5-бром-6-этилпирроло[2,3-d]пиримидин-4-ил)окси-3-фенилпропаноат
1.639 г 7-бензил-5-бром-4-хлор-6-этилпирроло[2,3-d]пиримидина (4.67 ммоль) растворяли в 47 мл сухого ДМСО, затем добавляли 2.948 г метил (2R)-2-гидрокси-3-фенилпропаноата (16.4 ммоль) и 7.234 г CS2CO3 (22.2 ммоль) и смесь перемешивали при 100°С в атмосфере N2 до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли водой и соляным раствором и экстрагировали с помощью ДХМ. Органический слой сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан иiРr2О в качестве элюентов с получением метил (2R)-2-(7-бензил-5-бром-6-этилпирроло[2,3-d]пиримидин-4-ил)окси-3-фенилпропаноата.
1Н ЯМР (400 МГц, CDCl3) δ: 8.29 (s, 1Н), 7.47 (d, 2Н), 7.36-7.19 (m, 6Н), 7.06-6.96 (m, 2Н), 5.60 (dd, 1Н), 5.47 (s, 2Н), 3.73 (s, 3Н), 3.41-3.28 (m, 2Н), 2.72 (q, 2Н), 1.03 (t, 3Н)
МС: (М+Н)+=494.2
Стадия С: метил (2R)-2-[7-бензил-(5Sa)-5-[3-хлор-2-метил-4-гидроксифенил]-6-этилпирроло[2,3-d]пиримидин-4-ил]окси-3-фенилпропаноат
Смесь 1.20 г метил (2R)-2-(7-бензил-5-бром-6-этилпирроло[2,3-d]пиримидин-4-ил)окси-3-фенилпропаноата (2.43 ммоль), 1.98 г соединения Синтеза 3а (7.21 ммоль), 110 мг Pd(OAc)2 (0.49 ммоль), 350 мг бутилдиадамантилфосфина (0.98 ммоль) и 7.35 мл 1 М водного раствора ТВАОН в 18 мл DME нагревали при MW облучении при температуре 100°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Реакционную смесь фильтровали через целит. К фильтрату добавляли воду, смесь подкисляли до рН=4 и экстрагировали с помощью МТВЕ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении.
Остаток нагревали в смеси 10 мл МеОН и 40 мкл конц. H2SO4 до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Летучие компоненты удаляли при пониженном давлении, остаток разбавляли водой, рН доводили до 5, и смесь экстрагировали с помощью ДХМ. Объединенные органические слои сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением метил (2R)-2-[7-бензил-(5Sа)-5-[3-хлор-2-метил-4-гидроксифенил]-6-этилпирроло[2,3-d]пиримидин-4-ил]окси-3-фенилпропаноата в виде диастереоизомера, элюирующегося позже.1Н ЯМР (500 МГц, ДMCO-d6) δ: 10.14 (s, 1H), 8.27 (s, 1H), 7.34-7.27 (m, 2H), 7.27-7.22 (m, 1H), 7.17-7.07 (m, 4H), 7.05 (d, 2H), 6.98 (dd, 1H), 6.64 (d, 2H), 5.60 (d, 1H), 5.51 (d, 1H), 5.43 (dd, 1H), 3.56 (s, 3H), 3.00 (dd, 1H), 2.85 (dd, 1H), 2.60-2.51 (m, 1H), 2.48-2.38 (m, 1H), 2.04 (s, 3H), 0.84 (t, 3H)
Стадия D: Пример 40
139 мг метил (2R)-2-[7-бензил-(5Sа)-5-[3-хлор-2-метил-4-гидроксифенил]-6-этилпирроло[2,3-d]пиримидин-4-ил]окси-3-фенилпропаноата (0.25 ммоль), 72 мг 1-(2-гидроксиэтил)-4-метилпиперазина (0.50 ммоль) и 166 мг связанного со смолой РРh3 (0.5 ммоль) растворяли в 3 мл сухого толуола в атмосфере N2, затем добавляли 115 мг DTAD (0.5 ммоль). Смесь перемешивали при 50°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем разбавляли ДХМ, фильтровали и фильтрат концентрированный при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан, ЕtOАс и МеОН в качестве элюентов. Полученное промежуточное соединение растворяли в 10 мл МеОН, затем добавляли 500 мг LiОН×Н2О и смесь перемешивали при 50°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь разбавляли соляным раствором, нейтрализовали 1 М водным раствором НСl и экстрагировали с помощью ДХМ. Органический слой сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 40 мМ водный раствор NH4OAC (рН=4, доводили с помощью АсОН) и MeCN в качестве элюентов с получением Примера 40. МСВР: рассчитано для C38H42ClN5O4: 667.2925, найдено: 668.2992 (М+Н)
Пример 41: N-[6-бром-7-(бут-3-ен-1-ил)-(5Rа)-5-(3-хлор-2-метилфенил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
и
Пример 42: N-[6-бром-7-(бут-3-ен-1-ил)-(5Sа)-5-(3-хлор-2-метилфенил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
Стадия А: 7-бут-3-енил-4-хлор-5-йодпирроло[2,3-d]пиримидин
5.0 г 4-хлор-5-йод-7H-пирроло[2,3-d]пиримидина (17 ммоль), 2.842 г К2СО3 (20.57 ммоль), 2.15 мл 4-бром-1-бутена (20.57 ммоль) и 26 мл сухого ДМФА перемешивали при к.т.в атмосфере N2 до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь выливали в воду и экстрагировали с помощью ЕtOАс. Объединенные органические слои промывали водой, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и ЕtOАс с получением 7-бут-3-енил-4-хлор-5-йодпирроло[2,3-d] пиримидина.
1Н ЯМР (400 МГц, CDCl3) δ: 8.62 (s, 1Н), 7.38 (s, 1Н), 5.82-5.69 (m, 1Н), 5.08 (s, 1Н), 5.04 (dd, 1Н), 4.33 (t, 2Н), 2.60 (q, 2Н)
МС: (М+Н)+=334.0
Стадия В: (2R)-2-[(7-бут-3-енил-5-йодпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановая кислота
Используя Общую Методику III и 7-бут-3-енил-4-хлор-5-йодпирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[(7-бут-3-енил-5-йодпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановую кислоту.1Н ЯМР (400 МГц, CDCl3) δ: 8.32 (s, 1Н), 7.38 (s, 1H), 7.35-7.28 (m, 3Н), 7.28-7.22 (m, 2Н), 7.02 (s, 1Н), 6.28 (d, 1Н), 5.80-5.67 (m, 1Н), 5.09-5.04 (m, 1Н), 5.04-5.00 (s, 1Н), 4.94-4.85 (m, 1Н), 4.22 (t, 2Н), 3.51 (dd, 1Н), 3.30 (dd, 1Н), 2.54 (q, 2Н)
Стадия С: (2R)-2-[[7-бут-3-енил-5-(3-хлор-2-метилфенил)пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановая кислота
Используя Общую Методику IVb и (2R)-2-[(7-бут-3-енил-5-йодпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановую кислоту в качестве подходящего 5-йодпирроло[2,3-d]пиримидинового производного и соединение Синтеза 3с в качестве подходящего производного бороновой кислоты, получали (2R)-2-[[7-бут-3-енил-5-(3-хлор-2-метилфенил)пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановую кислоту.1Н ЯМР (500 МГц, ДМСО-d6) δ: 12.86 (br s, 1Н), 8.24 (s, 1Н), 7.55-7.43 (m, 1Н), 7.33-6.95 (m, 6Н), 6.89-6.80 (m, 2Н), 5.84-5.40 (m, 1Н), 5.08-4.93 (m, 3Н), 4.84 (br s, 1Н), 4.37-4.15 (m, 2Н), 3.16 (d, 1Н), 2.85 (dd, 1H), 2.56 (q, 2H), 2.22-2.04 (s, 3H).
Стадия D: Примеры 41 и 42
512 мг (2R)-2-[[7-бут-3-енил-5-(3-хлор-2-метилфенил)пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановой кислоты (1 ммоль) растворяли в 4.5 мл сухого ДМФА и добавляли 187 мг NBS (1 ммоль). Смесь перемешивали при к.т.до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем выливали в воду и экстрагировали с помощью EtOAc. Объединенные органические слои промывали соляным раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 0.1% водный раствор ТФУ и MeCN в качестве элюентов с получением Примера 41 в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для С26H24BrСlN4O2: 538.0771, найдено: 541.0831 (М+Н). Пример 42 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C26H24BrClN4O2: 538.0771, найдено: 541.0835 (М+Н)
Пример 43: N-[6-бром-(5Ra)-5-(3-хлор-2-метилфенил)-7-(проп-2-ен-1-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
и
Пример 44: N-[6-бром-(5Sa)-5-(3-хлор-2-метилфенил)-7-(проп-2-ен-1-ил)-7H-пирроло[2,3-d]пиримидин-4-ил]-D-фенилаланин
Стадия А: 7-аллил-4-хлор-5-йодпирроло[2,3-d]пиримидин
176.5 мг 4-хлор-5-йод-7H-пирроло[2,3-d]пиримидина (0.6 ммоль), 100.7 мг К2СО3 (0.73 ммоль), 63 мкл аллилбромида (0.73 ммоль) и 1 мл сухого ДМФА перемешивали при к.т. в атмосфере N2 до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь выливали в воду и экстрагировали с помощью EtOAc. Объединенные органические слои промывали водой, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и ЕtOАс с получением 7-аллил-4-хлор-5-йодпирроло[2,3-d]пиримидина. МС: (М+Н)+=320.0
Стадия В: (2R)-2-[(7-аллил-5-йодпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановая кислота
Используя Общую Методику III и 7-аллил-4-хлор-5-йодпирроло[2,3-d]пиримидин в качестве подходящего 4-хлорпирроло[2,3-d]пиримидинового производного и D-фенилаланин в качестве подходящего аминокислотного производного, получали (2R)-2-[(7-аллил-5-йодпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановую кислоту.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 13.09 (br s, 1Н), 8.20 (s, 1Н), 7.43 (s, 1Н), 7.34-7.18 (m, 5Н), 6.52 (bd, 1Н), 6.05-5.90 (m, 1Н), 5.15 (dd, 1H), 5.07-4.94 (m, 2Н), 4.74 (d, 2Н), 3.38 (dd, 1Н), 3.15 (dd, 1Н)
МС: (М+Н)+=449.0
Стадия С: (2R)-2-[[7-аллил-5-(3-хлор-2-метилфенил)пирроло[2,3-й]пиримидин-4-ил]амино]-3-фенилпропановая кислота
Используя Общую Методику IVb и (2R)-2-[(7-аллил-5-йодпирроло[2,3-d]пиримидин-4-ил)амино]-3-фенилпропановую кислоту в качестве подходящего 5-йодпирроло[2,3-d]пиримидинового производного и соединение Синтеза 3с в качестве подходящего производного бороновой кислоты, получали (2R)-2-[[7-аллил-5-(3-хлор-2-метилфенил)пирроло[2,3-d)пиримидин-4-ил]амино]-3-фенилпропановую кислоту.
1H ЯМР (400 МГц, ДМСО-d6) δ: 12.89 (br s, 1Н), 8.23 (s, 1Н), 7.59-7.42 (br, 1Н), 7.31-7.10 (m, 6Н), 6.91-6.81 (br, 2Н), 6.12-5.98 (m, 1Н), 5.16 (dd, 1Н), 5.09-4.96 (m, 2Н), 4.90-4.76 (br, 3Н), 3.17 (dd, 1Н), 2.86 (dd, 1Н), 2.23-2.04 (br s, 3Н)
МС: (М+Н)+=447.0
Стадия D: Примеры 43 и 44
447 мг (2R)-2-[[7-аллил-5-(3-хлор-2-метилфенил)пирроло[2,3-d]пиримидин-4-ил]амино]-3-фенилпропановой кислоты (1 ммоль) растворяли в 4.5 мл сухого ДМФА и добавляли 187 мг NBS (1 ммоль). Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем выливали в воду и экстрагировали с помощью EtOAc. Объединенные органические слои промывали соляным раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 0.1% водный раствор ТФУ и MeCN в качестве элюентов с получением Примера 43 в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C25H22BrClN4O2: 524.0615, найдено: 525.0675 (М+Н). Пример 44 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C25H22BrClN4O2: 524.0615, найдено: 525.0674 (М+Н)
Пример 45: (2R)-2-[(7-бензил-5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-7H-пирроло[2,3-d]пиримидин-4-ил)окси]-3-фенилпропановая кислота
Стадия А: 7-бензил-4-хлор-5-йодпирропо[2,3-d]пиримидин
1.68 г 4-хлор-5-йод-7H-пирроло[2,3-d]пиримидина (6 ммоль), 1.24 мл бензилового спирта (12 ммоль), 3.144 г РРh3 (12 ммоль) и 60 мл сухого ТГФ охлаждали до 0°С в атмосфере N2, затем добавляли 5.5 мл раствора DEAD (12 ммоль, 40% в толуоле) и смесь перемешивали при 40°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь выливали в воду и экстрагировали с помощью Еt2О. Объединенные органические слои промывали водой, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и ЕtOАс с получением 7-бензил-4-хлор-5-йодпирроло[2,3-d]пиримидина.
1Н ЯМР (500 МГц, ДМСО-d6) δ: 8.67 (s, 1H), 8.12 (s, 1Н), 7.32 (t, 2Н), 7.28 (t, 1Н), 7.28 (d, 2Н), 5.47 (s, 2Н)
МС (М+Н): 369.9
Стадия В: метил (2R)-2-(7-бензил-5-йодпирроло[2,3-d]пиримидин-4-ил)окси-3-фенилпропаноат
1 экв. 7-бензил-4-хлор-5-йодпирроло[2,3-d]пиримидина, 3 экв. метил (2R)-2-гидрокси-3-фенилпропаноата, 3 экв. Cs2CO3 и сухой ДМСО (6 мл/ммоль) перемешивали при 100°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь подкисляли 1 М водным раствором НСl и экстрагировали с помощью ДХМ. Органический слой сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и ЕtOАс в качестве элюентов с получением метил (2R)-2-(7-бензил-5-йодпирроло[2,3-d]пиримидин-4-ил)окси-3-фенилпропаноата. МС (М+Н): 514.1
Стадия С: Пример 45
Используя Общую Методику IVb и метил (2R)-2-(7-бензил-5-йодпирроло[2,3-d]пиримидин-4-ил)окси-3-фенилпропаноат в качестве подходящего 5-йодпирроло[2,3-d]пиримидинового производного и соединение Синтеза 3b в качестве подходящего производного бороновой кислоты, получали метил (2R)-2-[7-бензил-5-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]пирроло[2,3-d]пиримидин-4-ил]окси-3-фенилпропаноат. Его растворяли в смеси диоксан : вода 1:1 (20 мл/ммоль) и добавляли 10 экв. LiОН×Н2О. Смесь перемешивали при к.т.до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли соляным раствором, нейтрализовали 2 М водным раствором НСl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 0.1% водный раствор ТФУ и MeCN в качестве элюентов с получением Примера 45. МСВР: рассчитано для C36H38ClN5O4: 639.2612, найдено: 640.2654 (М+Н)
Пример 46: N-[5-(3-хлор-2-метилфенил)-7,8-дигидро-6H-пиримидо[5,4-b]пирролизин-4-ил]-D-фенилаланин
210 мг 1:1 смеси Примеров 43 и 44 (смесь двух диастереоизомеров, 0.4 ммоль) растворяли в 3 мл МеОН и добавляли 70 мкл конц. H2SO4 (1.2 ммоль). Смесь перемешивали при к.т.до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь выливали в ледяную воду, нейтрализовали насыщенным водным раствором NaHCO3 и экстрагировали с помощью EtOAc. Объединенные органические фазы промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Затем остаток растворяли в сухом ТГФ (6 мл/ммоль) и охлаждали до 0°С. Добавляли раствор 5 экв. 9-борабицикло[3.3.1]нонана (0.5М в ТГФ) и смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем добавляли 20 экв. 2 М водного раствора NaOH и 20 мол. % PdCl2×dppf. Смесь перемешивали при 80°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь фильтровали через целит и промывали с помощью EtOAc. Слои фильтрата разделяли, водный слой подкисляли до рН 3 с помощью 2 М водного раствора НСl и затем экстрагировали с помощью EtOAc. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 40 мМ водный раствор NH4OAc (рН=4, доводили с помощью АсОН) и MeCN в качестве элюентов с получением Примера 46 в виде смеси диастереоизомеров. МСВР: рассчитано для C25H23ClN4O2: 446.1510, найдено: 447.159 и 447.1591 (М+Н)
Пример 47: N-[(5Ra)-5-(3-хлор-2-метилфенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4-ил]-D-фенилаланин
и
Пример 48: N-[(5Sа)-5-(3-хлор-2-метилфенил)-6,7,8,9-тетрагидропиримидо[5,4-b]индолизин-4-ил]-D-фенилаланин
1.29 г 1:1 смеси Примеров 41 и 42 (смесь двух диастереоизомеров, 2.3 ммоль) растворяли в 10 мл МеОН и добавляли 0.4 мл конц. H2SO4 (6.9 ммоль). Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь выливали в ледяную воду, нейтрализовали насыщенным водным раствором NаНСО3 и экстрагировали с помощью EtOAc. Объединенные органические фазы промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Затем остаток растворяли в сухом ТГФ (6 мл/ммоль) и охлаждали до 0°С. Добавляли раствор 5 экв. 9-борабицикло[3.3.1]нонана (0.5М в ТГФ) и смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем добавляли 20 экв. 2 М водного раствора NaOH и 20 мол. % PdCl2×dppf. Смесь перемешивали при 80°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь фильтровали через целит и промывали с помощью EtOAc. Слои фильтрата разделяли, водный слой подкисляли до рН 3 с помощью 2 М водного раствора НСl и затем экстрагировали с помощью EtOAc. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 0.1% водный раствор ТФУ и MeCN в качестве элюентов. Пример 47 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C26H25ClN4O2: 460.1666, найдено: 461.1747 (М+Н). Пример 48 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C26H25ClN4O2: 460.1666, найдено: 461.1752 (М+Н)
Пример 49: (2R)-2-{[(3Sa)-3-(3-хлор-4-гидрокси-2-метилфенил)-2-этил-1-бензотиофен-4-ил]окси}-3-фенилпропановая кислота
и
Пример 50: (2R)-2-{[(3Ra)-3-(3-хлор-4-гидрокси-2-метилфенил)-2-этил-1-бензотиофен-4-ил]окси}-3-фенилпропановая кислота
Стадия А: (2R)-2-(2-этилбензотиофен-4-ил)окси-3-фенилпропановая кислота
270 мг (2R)-2-гидрокси-3-фенилпропановой кислоты (1.63 ммоль), 40 мг CuI (0.21 ммоль) и 325 мг Cs2CO3 (1 ммоль) отмеривали в 7 мл пробирку, оснащенную навинчивающимся колпачком и резиновой прокладкой. Пробирку продували аргоном и с помощью шприца добавляли 5 мл сухого ДМФА и 288 мг 2-этил-4-йодбензо[b]тиофена (1 ммоль). Смесь перемешивали при 110°С в темноте в течение 20 часов. Все дальнейшие стадии осуществляли в темноте или при красном свете. Добавляли 10 мл воды и рН доводили до 3 с помощью 2 М водного раствора НСl. Затем смесь экстрагировали с помощью EtOAc. Объединенный органический слой сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали на пластинке для препаративной ТСХ (слой силикагеля, в качестве элюента использовали смесь толуол : АсОН 9:1) с получением (2R)-2-(2-этилбензотиофен-4-ил)окси-3-фенилпропановой кислоты.1Н ЯМР (500 МГц, ДМСО-d6) δ: 12.53 (br s, 1Н), 7.42-7.36 (m, 3Н), 7.30 (t, 2Н), 7.25-7.18 (m, 1Н), 7.13 (t, 1H), 7.07 (br, 1H), 6.65 (d, 1Н), 4.98 (dd, 1H), 3.29 (dd, 1Н), 3.22 (dd, 1Н), 2.89 (q, 2Н), 1.30 (t, 3Н)
Стадия В: метил (2R)-2-(2-этилбензотиофен-4-ил)окси-3-фенилпропаноат
1.434 г (2R)-2-(2-этилбензотиофен-4-ил)окси-3-фенилпропановой кислоты (4.39 ммоль) растворяли в 20 мл МеОН и добавляли 20 мкл конц. H2SO4. Смесь перемешивали при 80°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь концентрировали при пониженном давлении, затем разбавляли водой, нейтрализовали насыщенным водным раствором NаНСО3 и экстрагировали с помощью ДХМ. Объединенные органические фазы промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением метил (2R)-2-(2-этилбензотиофен-4-ил)окси-3-фенилпропаноата.1Н ЯМР (400 МГц, CDCl3) δ: 7.46-7.33 (m, 5Н), 7.33-7.26 (m, 1H), 7.16 (bd, 1Н), 7.13 (t, 1Н), 6.65 (d, 1Н), 4.99 (dd, 1H), 3.75 (s, 3Н), 3.46-3.32 (m, 2Н), 3.01-2.91 (m, 2Н), 1.42 (t, 3Н)
Стадия С: метил (2R)-2-(2-этил-3-йодбензотиофен-4-ил)окси-3-фенилпропаноат
1.278 г метил-(2R)-2-(2-этилбензотиофен-4-ил)окси-3-фенилпропаноата (3.75 ммоль), 2.284 г I2 (9 ммоль) и 2.5 г Ag2SO4 (8 ммоль) растворяли в 10 мл EtOH и перемешивали при к.т.до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем фильтровали, фильтрат концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 860 мг метил (2R)-2-(2-этил-3,7-дийодбензотиофен-4-ил)окси-3-фенилпропаноата, который растворяли в 20 мл ТГФ, добавляли 150 мг 10% Pd/C и смесь перемешивали при к.т. в атмосфере Н2 при давлении 4 бар до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь фильтровали через целит, фильтрат концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением метил (2R)-2-(2-этил-3-йодбензотиофен-4-ил)окси-3-фенилпропаноата.1Н ЯМP (500 МГц, ДМСО-d6) δ: 7.53 (d, 1Н), 7.49-7.41 (m, 2H), 7.34-7.27 (m, 2H), 7.26-7.18 (m, 2H), 6.77 (d, 1H), 5.33 (dd, 1H), 3.61 (s, 3H), 3.43 (dd, 1H), 3.32 (dd, 1H), 2.94-2.85 (m, 2H), 1.25 (t, 3H)
Стадия D: Примеры 49 и 50
320 мг метил (2R)-2-(2-этил-3-йодбензотиофен-4-ил)окси-3-фенилпропаноата (0.686 ммоль) и 368 мг соединения Синтеза 3а (1.37 ммоль) растворяли в 4 мл 2-Ме-ТГФ в атмосфере N2, затем добавляли 1.37 мл раствора ТВАОН (1.37 ммоль, 1 М в ТГФ) и 49 мг AtaPhos (0.069 ммоль) и смесь перемешивали при 90°С в закрытой пробирке до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли 30 мл ДХМ, промывали 10 мл 1 М водного раствора НСl. Органический слой концентрировали при пониженном давлении, затем растворяли в 5 мл МеОН. Добавляли 100 мг LiОН×Н2О и смесь перемешивали при 50°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли соляным раствором, нейтрализовали 1 М водным раствором НСl и экстрагировали с помощью ДХМ. Органический слой сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 0.1% водный раствор ТФУ и MeCN в качестве элюентов. Пример 49 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C26H23ClO4S: 466.1006, найдено: 465.0956 (М-Н). Пример 50 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C26H23ClO4S: 466.1006, найдено: 465.0971 (М-Н)
Пример 51: (2R)-2-[((3Sа)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-этил-1-бензотиофен-4-ил)окси]-3-фенилпропановая кислота
Стадия А: метил (2R)-2-[3-(3-хлор-4-гидрокси-2-метилфенил)-2-этилбензотиофен-4-ил]окси-3-фенилпропаноат
140 мг Примера 49 (0.3 ммоль) растворяли в 3 мл МеОН и добавляли 50 мкл конц. H2SO4. Смесь перемешивали при 80°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь концентрировали при пониженном давлении и остаток разбавляли водой, нейтрализовали насыщенным водным раствором NаНСО3 и экстрагировали с помощью ДХМ. Объединенные органические фазы промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением метил (2R)-2-[3-(3-хлор-4-гидрокси-2-метилфенил)-2-этилбензотиофен-4-ил]окси-3-фенилпропаноата.1H ЯМР (500 МГц, ДМСО-d6) δ: 10.02 (s, 1Н), 7.49 (d, 1Н), 7.23-7.12 (m, 4Н), 7.02 (d, 1Н), 6.92 (d, 1Н), 6.89-6.86 (m, 2Н), 6.62 (d, 1Н), 5.01 (dd, 1Н), 3.50 (s, 3Н), 2.72 (dd, 1Н), 2.60-2.51 (m, 2Н), 2.38 (dd, 1H), 1.96 (s, 3Н), 1.12 (t, 3Н)
Стадия В: Пример 51
63 мг метил (2R)-2-[3-(3-хлор-4-гидрокси-2-метилфенил)-2-этилбензотиофен-4-ил]окси-3-фенилпропаноата (0.13 ммоль), 23 мг 1-(2-гидроксиэтил)-4-метилпиперазина (0.156 ммоль) и 41 мг РРh3 (0.156 ммоль) растворяли в 2 мл сухого ТГФ в атмосфере N2, затем добавляли 36 мг DTAD (0.156 ммоль). Смесь перемешивали при 50°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан, EtOAc и МеОН в качестве элюентов. Полученное промежуточное соединение растворяли в 5 мл МеОН, затем добавляли 100 мг LiОН×Н2О и смесь перемешивали при 50°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли соляным раствором, нейтрализовали 1 М водным раствором НСl и экстрагировали с помощью ДХМ. Органический слой сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 40 мМ водный раствор NH4OAC (рН=4, доводили с помощью АсОН) и MeCN в качестве элюентов с получением Примера 51. МСВР: рассчитано для C33H37ClN2O4S: 592.2163, найдено: 593.2238 (М+Н)
Пример 52: (2R)-2-{[(3Ra)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1-бензотиофен-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
и
Пример 53: (2R)-2-{[(3Sa)-3-(3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1-бензотиофен-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Стадия А: (3-бромфенил) N,N-диэтилкарбамат
5.0 г 3-бромфенола (28.9 ммоль) и 4.31 г диэтилкарбамоилхлорида (31.8 ммоль) растворяли в 50 мл пиридина и перемешивали при 100°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением (3-бромфенил) N,N-диэтилкарбамата. МС (EI, 70 эВ) m/z (% относительная интенсивность, [ион]): 56 (9), 72 (42), 100 (100), 174 (4), 176 (4), 271 (4, [М+]), 273 (4, [М+])
Стадия В: (3-бром-2-йодфенил) N,N-диэтилкарбамат
2.72 г (3-бромфенил) N,N-диэтилкарбамата (10 ммоль) растворяли в 50 мл сухого ТГФ в атмосфере N2 и охлаждали до -78°С. Добавляли 6 мл раствора LDA (12 ммоль, 2М в ТГФ, гептане, этилбензоле) и смесь перемешивали при -78°С в течение 30 минут. Затем добавляли 3.18 г I2 (12.5 ммоль) и смесь перемешивали при -78°С в течение 30 минут, и затем ей давали нагреться до к.т. Затем смесь концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением (3-бром-2-йодфенил) N,N-диэтилкарбамата.1Н ЯМР (400 МГц, ДМСО-d6) δ: 7.60 (dd, 1Н), 7.35 (t, 1Н), 7.17 (dd, 1Н), 3.47 (q, 2Н), 3.31 (q, 2Н), 1.27 (t, 3Н), 1.14 (t, 3Н)
Стадия С: [3-бром-2-[2-(4-фторфенил)этинил]фенил] N,N-диэтилкарбамат
2.60 г (3-бром-2-йодфенил) N,N-диэтилкарбамата (6.53 ммоль), 863 мг 1-этинил-4-фторбензола (7.19 ммоль), 229 мг Pd(PPh3)2Cl2 (0.33 ммоль), 130 мг йодида меди(I) (0.65 ммоль) и 1.43 г диэтиламина (19.6 ммоль) растворяли в 25 мл сухого ДМФА и перемешивали при 50°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь разбавляли водой и экстрагировали с помощью EtOAc. Объединенные органические слои промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением [3-бром-2-[2-(4-фторфенил)этинил]фенил] N,N-диэтилкарбамата. МС (EI, 70 эВ) m/z (% относительная интенсивность, [ион]): 56 (2), 72 (35), 100 (100), 261 (2), 263 (2), 389 (2, [М+]), 391 (2, [М+])
Стадия D: [2-[2-(4-фторфенил)этинил]-3-метилсульфанилфенил] N,N-диэтилкарбамат
2.5 г [3-бром-2-[2-(4-фторфенил)этинил]фенил] N,N-диэтилкарбамата (6.56 ммоль) растворяли в 65 мл сухого ТГФ и охлаждали до -78°С, затем добавляли 4.3 мл раствораnBuLi (6.88 ммоль, 1.6 М в гексанах). Смесь перемешивали при -78°С в течение 30 минут. Затем добавляли 742 мг S2Ме2 (7.87 ммоль) и смесь перемешивали при -78°С в течение 30 минут, и затем ей давали нагреться до к.т. Смесь затем концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением [2-[2-(4-фторфенил)этинил]-3-метилсульфанилфенил] N,N-диэтилкарбамата. МС (EI, 70 эВ) m/z (% относительная интенсивность, [ион]): 56 (2), 72 (46), 100 (100), 342 (40), 357 (1, [М+])
Стадия Е: [2-(4-фторфенил)-3-йодбензотиофен-4-ил] N,N-диэтилкарбамат
1100 мг 2-[2-(4-фторфенил)этинил]-3-метилсульфанилфенил] N,N-диэтилкарбамата (3.08 ммоль) и 937 мг I2 (3.7 ммоль) растворяли в 20 мл ДХМ и перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем разбавляли 10% водным раствором Nа2S2О3 и экстрагировали с помощью ДХМ. Объединенные органические слои промывали соляным раствором с получением [2-(4-фторфенил)-3-йодбензотиофен-4-ил] N,N-диэтилкарбамата.
1Н ЯМР (400 МГц, CDCl3) δ: 7.74 (dd, 1Н), 7.56 (m, 2Н), 7.40 (t, 1Н), 7.18 (m, 2Н), 7.12 (dd, 1Н), 3.60 (q, 2Н), 3.46 (q, 2Н), 1.36 (t, 3Н), 1.26 (t, 3Н)
МС (EI, 70 эВ) m/z (% относительная интенсивность, [ион]): 72 (42), 100 (100), 170 (16), 342 (37), 369 (5), 469 (1, [М+])
Стадия F: [3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)бензотиофен-4-ил] N,N-диэтилкарбамат
1 экв. [2-(4-фторфенил)-3-йодбензотиофен-4-ил] N,N-диэтилкарбамата, 2 экв. соединения Синтеза 3b, 2 экв. CS2CO3, 0.1 экв. Ataphos и смесь ТГФ : вода 3:1 (10 мл/ммоль производного бензотиофена) перемешивали в N2 атмосфере при 70°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь разбавляли водой и экстрагировали с помощью ДХМ. Органическую фазу сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и ЕtOАс в качестве элюентов с получением [3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)бензотиофен-4-ил] N,N-диэтилкарбамата. МС: (М+Н)+=610.2
Стадия G: 3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)бензотиофен-4-ол
1.8 г [3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)бензотиофен-4-ил] N,N-диэтилкарбамата (3 ммоль) растворяли в 80 мл ЕtOН и добавляли 1.2 г NaOH (30 ммоль). Смесь перемешивали при 80°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя ДХМ и МеОН в качестве элюентов с получением 3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)бензотиофен-4-ола. МС: (М+Н)+=511.2
Стадия Н: Примеры 52 и 53
470 мг 3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)бензотиофен-4-ола (0.92 ммоль), 1.12 г соединения Синтеза 2d (2.76 ммоль) и 726 мг РРh3 (2.76 ммоль) растворяли в 10 мл сухого толуола, затем добавляли 635 мг DTAD (2.76 ммоль). Смесь перемешивали при 50°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и ЕtOАс в качестве элюентов. Образовавшееся промежуточное соединение растворяли в 10 мл смеси диоксан : вода 1:1, добавляли 400 мг LiОН×Н2О и смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь нейтрализовали с помощью 2 М водного раствора НСl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов. Пример 52 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C49H46ClFN4O6S: 872.2811, найдено: 437.1457 (М+2Н). Пример 53 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C49H46ClFN4O6S: 872.2811, найдено: 437.1491 (М+2Н)
Пример 54: 2-бензил-3-[3-(3-хлор-4-гидрокси-2-метилфенил)-2-этил-1-бензотиофен-4-ил]пропановая кислота
Стадия А: метил (Z)-2-бензил-3-(2-этилбензотиофен-4-ил)проп-2-еноат
576 мг 2-этил-4-йодбензо[b]тиофена (2 ммоль), 717 мг метил 2-бензилакрилата (4 ммоль), 556 мкл TEA (4 ммоль) и 24 мг PdCl2 (0.1 ммоль) растворяли в 10 мл ДМФА и перемешивали при 130°С в MW реакторе до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением метил (Z)-2-бензил-3-(2-этилбензотиофен-4-ил)проп-2-еноата.1Н ЯМР (400 МГц, CDCl3) соотношение диастереоизомеров 1.00 / 0.77 = главный / минорный, δ: 8.06-8.28 (s, 1Н), 7.68-7.76 (d, 1Н), 7.44-6.98 (m, 8Н), 4.25-3.93 (s, 2Н), 3.78-3.82 (s, 3Н), 2.97-2.99 (q, 2Н), 1.41-1.43 (t, 3Н)
Стадия В: метил 2-бензил-3-(2-этилбензотиофен-4-ил)пропаноат 432 мг метил (Z)-2-бензил-3-(2-этилбензотиофен-4-ил)проп-2-еноата (1.28 ммоль), 137 мг 10% Pd/C, 5 мл АсОН и 20 мл МеОН перемешивали в атмосфере Н2 при давлении 4 бар и к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь фильтровали через целит, фильтрат концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением метил 2-бензил-3-(2-этилбензотиофен-4-ил)пропаноата.1Н ЯМР (400 МГц, CDCl3) δ: 7.61 (d, 1Н), 7.38-7.05 (m, 7Н), 6.80 (s, 1Н), 3.50 (s, 3Н), 3.28-3.18 (m, 1Н), 3.11-3.00 (m, 3Н), 2.90 (q, 2Н), 2.86-2.77 (m, 1H), 1.35 (t, 3Н)
Стадия С: метил 2-бензил-3-(2-этил-3-йодбензотиофен-4-ил)пропаноат
346 мг метил 2-бензил-3-(2-этилбензотиофен-4-ил)пропаноата (1.02 ммоль), 305 мг I2 (1.2 ммоль) и 468 мг Ag2SO4 (1.5 ммоль) растворяли в 5 мл EtOH и перемешивали при к.т.до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь фильтровали, фильтрат концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением метил 2-бензил-3-(2-этил-3-йодбензотиофен-4-ил)пропаноата.1Н ЯМР (400 МГц, CDCl3) δ: 7.67 (dd, 1Н), 7.28-7.06 (m, 7Н), 4.29-4.17 (m, 1Н), 3.80-3.71 (m, 1Н), 3.32 (s, 3Н), 3.28-3.21 (m, 1Н), 3.08-3.00 (m, 2Н), 2.97 (q, 2Н), 1.35 (t, 3Н)
Стадия D: Пример 54
1 экв. метил 2-бензил-3-(2-этил-3-йодбензотиофен-4-ил)пропаноата, 2 экв. соединения Синтеза 3а, 2 экв. раствора ТВАОН (1M в воде), 0.1 экв. Ataphos и 2-Ме-ТГФ (5 мл/ммоль производного бензотиофена) перемешивали в атмосфере N2 при 100°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь разбавляли водой и экстрагировали с помощью ДХМ. Органический слой сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Образовавшееся промежуточное соединение растворяли в МеОН (5 мл/ммоль производного бензотиофена), добавляли 10 экв. LiОН×Н2О и смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь нейтрализовали с помощью 2 М водного раствора НСl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с использованием препаративной хроматографии с обращенной фазой, используя 0.1% водный раствор ТФУ и MeCN в качестве элюентов с получением Примера 54. МСВР: рассчитано для C27H25ClO3S: 464.1213, найдено: 463.1158 (М-Н)
Пример 55: (2R)-2-{[(1Rа)-1-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1H-индол-7-ил]окси}-3-(2-метоксифенил)пропановая кислота
и
Пример 56: (2R)-2-{[(1Sa)-1-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1H-индол-7-ил]окси}-3-(2-метоксифенил)пропановая кислота
600 мг соединения Синтеза 7b (0.86 ммоль) растворяли в 20 мл смеси диоксан : вода 1:1 и добавляли 600 мг LiОН×Н2О. Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли водой, подкисляли 1 М водным раствором НСl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении и очищали с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов. Пример 55 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C38H39ClFN3O5: 671.2562, найдено: 672.2618 (М+Н). Пример 56 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C38H39ClFN3O5: 671.2562, найдено: 672.2652 (М+Н)
Пример 57: (2R)-2-{[3-хлор-(1Sа)-1-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1H-индол-7-ил]окси}-3-(2-метоксифенил)пропановая кислота
240 мг соединения Синтеза 7b (0.34 ммоль) растворяли в 3 мл ДХМ и добавляли 46 мг NCS (0.34 ммоль). Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли водой и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Затем остаток растворяли в 5 мл смеси диоксан : вода 1:1 и добавляли 140 мг LiOH×H2O. Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли водой, подкисляли 1 М водным раствором НСl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов. Пример 57 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C38H38Cl2FN3O5: 705.2173, найдено: 706.2227 (М+Н)
Пример 58: (2R)-2-{[(1Ra)-1-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(фуран-2-ил)-1H-индол-7-ил]окси}-3-(2-метоксифенил)пропановая кислота
и
Пример 59: (2R)-2-{[(1Sа)-1-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(фуран-2-ил)-1H-индол-7-ил]окси}-3-(2-метоксифенил)пропановая кислота
и
Пример 60: (2R)-2-{[(1Sa)-1-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(5-фторфуран-2-ил)-1H-индол-7-ил]окси}-3-(2-метоксифенил)пропановая кислота
Стадия А: 4-[7-бензилокси-2-(5-фтор-2-фурил)индол-1-ил]-2-хлор-3-метилфенол
1360 мг соединения Синтеза 7а (2 ммоль), 848 мг 2-(5-фтор-2-фурил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (4 ммоль), 2123 мг K3РО4 (10 ммоль), 45 мг Pd(OAc)2 (0.2 ммоль) и 164 мг SPhos (0.4 ммоль) растворяли в 30 мл сухого толуола и перемешивали при 75°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Растворитель затем удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов. Затем добавляли 2 мл раствора TBAF (2 ммоль, 1 M в ТГФ) и 25 мл ТГФ и смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь концентрировали при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 4-[7-бензилокси-2-(5-фтор-2-фурил)индол-1-ил]-2-хлор-3-метилфенола. МС: (М+Н)+=448.0
Стадия В: 7-бензилокси-1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(5-фтор-2-фурил)индол
650 мг 4-[7-бензилокси-2-(5-фтор-2-фурил)индол-1-ил]-2-хлор-3-метилфенола (1.01 ммоль), 288 мг 1-(2-гидроксиэтил)-4-метилпиперазина (2 ммоль) и 786 мг PPh3 (3 ммоль) растворяли в 20 мл сухого толуола. Затем добавляли 690 мг DTAD (3 ммоль) и смесь перемешивали при 45°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя ДХМ и МеОН в качестве элюентов с получением 7-бензилокси-1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(5-фтор-2-фурил)индола. МС: (М+Н)+=574.2
Стадия С: смесь 1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(2-фурил)индол-7-ола и 1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(5-фтор-2-фурил)индол-7-ола
1300 мг 7-бензилокси-1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(5-фтор-2-фурил)индола (2.26 ммоль) растворяли в 100 мл МеОН и добавляли 100 мг 10% Pd/C. Смесь перемешивали в атмосфере Н2 при давлении 1 бар и к.т. в течение ночи. Смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением 7:3 смеси 1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(2-фурил)индол-7-ола (МС: (М+Н)+=466.2) и 1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(5-фтор-2-фурил)индол-7-ола (МС: (М+Н)+=484.2).
Стадия D: Примеры 58, 59 и 60
465 мг 7:3 смеси 1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(2-фурил)индол-7-ола и 1-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(5-фтор-2-фурил)индол-7-ола (1 ммоль), 449 мг этил (2S)-2-гидрокси-3-фенилпропаноата (2 ммоль) и 786 мг РРh3 (3 ммоль) растворяли в 10 мл сухого толуола. Затем добавляли 691 мг DTAD (3 ммоль) и смесь перемешивали при 45°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь концентрировали при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя ДХМ и МеОН в качестве элюентов. Затем продукт растворяли в 5 мл смеси диоксан : вода 1:1 и добавляли 140 мг LiОН×Н2О. Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли водой, подкисляли 1 М водным раствором НСl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов. Пример 58 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для С36Н38СlN3О6: 643.2449, найдено: 644.2512 (М+Н). Пример 59 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для С36Н38СlN3О6: 643.2449, найдено: 644.2521 (М+Н). Пример 60 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C36H37ClFN3O6: 661.2355, найдено: 662.2411 (М+Н)
Пример 61: (2R)-2-{[(3Ra)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1-бензофуран-4-ил]окси}-3-(2-метоксифенил)пропановая кислота
и
Пример 62: (2R)-2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1-бензофуран-4-ил]окси}-3-(2-метоксифенил)пропановая кислота
Стадия А: этил (2S)-3-(2-метоксифенил)-2-(n-толилсульфонилокси)пропаноат
3000 мг соединения Синтеза 2f (13.38 ммоль) растворяли в 10 мл пиридина и при 0°С добавляли 2933 мг TsCl (15.38 ммоль). Смесь перемешивали при к.т.до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли водой и экстрагировали с помощью EtOAc. Объединенные органические фазы промывали 1 М водным раствором лимонной кислоты, сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением этил (2S)-3-(2-метоксифенил)-2-(n-толилсульфонилокси)пропаноата. МС (EI, 70 эВ) m/z (% относительная интенсивность, [ион]): 65 (7), 77 (14), 91 (49), 123 (33), 133 (33), 165 (100), 207 (65), 307 (13), 512 (7, [М+])
Стадия В: этил (2R)-2-[3-бром-2-(4-фторфенил)бензофуран-4-ил]окси-3-(2-метоксифенил)пропаноат
1 экв. соединения Синтеза 1 с, 1.5 экв. этил (2S)-3-(2-метоксифенил)-2-(n-толилсульфонилокси)пропаноата, 2 экв. K2СО3 и ДМСО (10 мл/ммоль производного бензофурана) перемешивали при 60°С в атмосфере N2 до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли соляным раствором, нейтрализовали 1 М водным раствором НСl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением этил (2R)-2-[3-бром-2-(4-фторфенил)бензофуран-4-ил]окси-3-(2-метоксифенил)пропаноата. МС (EI, 70 эВ) m/z (% относительная интенсивность, [ион]): 91 (56), 133 (41), 165 (100), 207 (93), 281 (26), 305 (9), 512 (3, [М+]), 514 (3, [М+])
Стадия С: Примеры 61 и 62
Используя Общую Методику VI и этил (2R)-2-[3-бром-2-(4-фторфенил)бензофуран-4-ил]окси-3-(2-метоксифенил)пропаноат в качестве подходящего производного 3-бромбензофурана и соединение Синтеза 3b в качестве подходящего производного бороновой кислоты, Пример 61 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C38H38ClFN2O6: 672.2402, найдено: 673.2465 (М+Н). Пример 62 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C38H38ClFN2O6: 672.2402, найдено: 673.2486 (М+Н)
Пример 63: (2R)-2-{[(3Ra)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1-бензофуран-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
и
Пример 64: (2R)-2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1-бензофуран-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Стадия А: этил (2R)-2-[3-бром-2-(4-фторфенил)бензофуран-4-ил]окси-3-[2-[2-(2-метоксифенил)пиримидин-4-ил]оксифенил]пропаноат
Используя Общую Методику V и соединение Синтеза 1 с в качестве подходящего производного бензофуран-4-ола и соединение Синтеза 2d в качестве подходящего производного сложного эфира молочной кислоты, получали этил (2R)-2-[3-бром-2-(4-фторфенил)бензофуран-4-ил]окси-3-[2-[2-(2-метоксифенил)пиримидин-4-ил]оксифенил]пропаноат. МС: (М+Н)+=699.2
Стадия В: Примеры 63 и 64
Используя Общую Методику VI и этил (2R)-2-[3-бром-2-(4-фторфенил)бензофуран-4-ил]окси-3-[2-[2-(2-метоксифенил)пиримидин-4-ил]оксифенил]пропаноат в качестве подходящего 3-бромбензофуранового производного и соединение Синтеза 3b в качестве подходящего производного бороновой кислоты, Пример 63 получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C49H46ClFN4O7: 856.3039, найдено: 429.1582 (М+2Н). Пример 64 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C49H46ClFN4O7: 856.3039, найдено: 429.1604 (М+2Н)
Пример 65: (2R)-2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1-бензофуран-4-ил]окси}-3-[2-(2,2,2-трифторэтокси)фенил]пропановая кислота
Стадия А: этил (2R)-2-[3-бром-2-(4-фторфенил)бензофуран-4-ил]окси-3-[2-(2,2,2-трифторэтокси)фенил]пропаноат
Используя Общую Методику V и соединение Синтеза 1с в качестве подходящего производного бензофуран-4-ола и соединение Синтеза 2h в качестве подходящего производного сложного эфира молочной кислоты, получали этил (2R)-2-[3-бром-2-(4-фторфенил)бензофуран-4-ил]окси-3-[2-(2,2,2-трифторэтокси)фенил]пропаноат. МС: (M+Na)+=604.4
Стадия В: Пример 65
Используя Общую Методику VI и этил (2R)-2-[3-бром-2-(4-фторфенил)бензофуран-4-ил]окси-3-[2-(2,2,2-трифторэтокси)фенил]пропаноат в качестве подходящего производного 3-бромбензофурана и соединение Синтеза 3b в качестве подходящего производного бороновой кислоты, Пример 65 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C39H37ClF4N2O6: 740.2276, найдено: 741.2372 (М+Н)
Пример 66: (2R)-2-{[(3Sа)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-фтор-2-(4-фторфенил)-1-бензофуран-4-ил]окси}-3-[2-(2,2,2-трифторэтокси)фенил]пропановая кислота
Стадия А: этил (2R)-2-[3-бром-6-фтор-2-(4-фторфенил)бензофуран-4-ил]окси-3-[2-(2,2,2-трифторэтокси)фенил]пропаноат
Используя Общую Методику V и соединение Синтеза 1d в качестве подходящего производного бензофуран-4-ола и соединение Синтеза 2h в качестве подходящего производного сложного эфира молочной кислоты, получали этил (2R)-2-[3-бром-6-фтор-2-(4-фторфенил)бензофуран-4-ил]окси-3-[2-(2,2,2-трифторэтокси)фенил]пропаноат.1Н ЯМР (400 МГц, ДМСO-d6): 8.07 (m, 2Н), 7.43 (m, 3Н), 7.27 (m, 2Н), 7.11 (m, 1Н), 6.98 (m, 1Н), 6.55 (dd, 1Н), 5.23 (m, 1H), 4.82 (q, 2Н), 4.12 (q, 2Н), 3.37 (m, 1Н), 3.25 (m, 1Н), 1.10 (t, 3Н)
Стадия В: Пример 66
Используя Общую Методику VI и этил (2R)-2-[3-бром-6-фтор-2-(4-фторфенил)бензофуран-4-ил]окси-3-[2-(2,2,2-трифторэтокси)фенил]пропаноат в качестве подходящего 3-бромбензофуранового производного и соединение Синтеза 3b в качестве подходящего производного бороновой кислоты, Пример 66 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C39H36ClF5N2O6: 758.2182, найдено: 759.2244 (М+Н)
Пример 67: (2R)-2-{[(3Sа)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-фтор-2-(4-фторфенил)-1-бензофуран-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Стадия А: этил (2R)-2-[3-бром-6-фтор-2-(4-фторфенил)бензофуран-4-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат
Используя Общую Методику V и соединение Синтеза 1d в качестве подходящего производного бензофуран-4-ола и соединение Синтеза 2d в качестве подходящего производного сложного эфира молочной кислоты, получали этил (2R)-2-[3-бром-6-фтор-2-(4-фторфенил)бензофуран-4-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат.1Н ЯМР (400 МГц, ДМСО-d6): 8.86 (d, 1Н), 8.05 (m, 2Н), 7.61 (d, 1Н), 7.52 (dd, 1Н), 7.48-7.38 (m, 4Н), 7.25 (m, 1H), 7.21 (m, 1Н), 7.12 (m, 2Н), 7.03 (td, 1Н), 6.94 (td, 1Н), 6.67 (dd, 1Н), 5.40 (m, 1Н), 5.26 (s, 2Н), 4.15 (q, 2Н), 3.75 (s, 3Н), 3.56 (m, 1Н), 3.30 (m, 1Н), 1.12 (t, 3Н)
Стадия В: Пример 67
Используя Общую Методику VI и этил (2R)-2-[3-бром-6-фтор-2-(4-фторфенил)бензофуран-4-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат в качестве подходящего производного 3-бромбензофурана и соединение Синтеза 3b в качестве подходящего производного бороновой кислоты, Пример 67 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C49H45ClF2N4O7: 874.2945, найдено: 438.1543 (М+2Н)
Пример 68: (2R)-2-{[(3Rа)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1-метил-1H-индол-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
и
Пример 69: (2R)-2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)-1-метил-1H-индол-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Стадия А: 1-(бензолсулъфонил)-4-бензилоксииндол
7.0 г 4-бензилокси-1H-индола (31.35 ммоль) растворяли в 60 мл сухого ДМФА и при 0°С добавляли 1.317 г NaH (32.92 ммоль, 60% в минеральном масле). Смесь перемешивали в течение 1 часа, затем по каплям добавляли 6.09 г бензолсульфонилхлорида (34.48 ммоль) и смесь перемешивали при 0°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли водой и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и ЕtOАс в качестве элюентов с получением 1-(бензолсульфонил)-4-бензилоксииндола.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 7.97 (d, 2Н), 7.72 (d, 1Н), 7.69 (t, 1H), 7.59 (t, 2Н), 7.54 (d, 1Н), 7.47 (d, 2Н), 7.39 (t, 2Н), 7.33 (d, 1Н), 7.27 (t, 1H), 6.89 (d, 1Н), 6.85 (d, 1H), 5.20 (s, 2H)
MC (EI, 70 эВ) m/z (% относительная интенсивность, [ион]): 77 (32), 91 (100), 141 (18), 222 (6), 272 (11), 363 (10, [М+])
Стадия В: 1-(бензолсулъфонил)-4-бензилокси-2-йодиндол
5.08 г 1-(бензолсульфонил)-4-бензилоксииндола (13.98 ммоль) растворяли в 140 мл сухого ТГФ. При -78°С добавляли 8.54 мл раствора LDA (15.38 ммоль, 1.8М в смеси ТГФ-гептан-этилбензол) и реакционную смесь перемешивали в течение 1 часа. Затем добавляли 4.26 г йода (16.8 ммоль) и смесь перемешивали в течение 1 часа при -78°С. Смесь гасили насыщенным водным раствором NH4Cl и экстрагировали с помощью EtOAc. Объединенные органические фазы промывали водным раствором Nа2S2О3 и водой, затем сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 1-(бензолсульфонил)-4-бензилокси-2-йодиндола.1Н ЯМР (400 МГц, ДМСO-d6) δ: 7.86 (dd, 2Н), 7.75 (d, 1Н), 7.70 (d, 1Н), 7.61 (t, 2Н), 7.47 (dd, 2Н), 7.39 (t, 2Н), 7.33 (d, 1Н), 7.23 (t, 1Н), 7.18 (s, 1H), 6.90 (d, 1Н), 5.20 (s, 2Н)
Стадия С: 1-(бензолсульфонил)-4-бензилокси-2-(4-фторфенил)индол
5.8 г 1-(бензолсульфонил)-4-бензилокси-2-йодиндола (11.86 ммоль) и 3.16 г 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фторбензола (14.22 ммоль) растворяли в 75 мл ТГФ, затем добавляли 7.73 г CS2CO3 (23.72 ммоль), 420 мг Ataphos (0.59 ммоль) и 25 мл воды и смесь перемешивали при 70°С в атмосфере N2 до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 1-(бензолсульфонил)-4-бензилокси-2-(4-фторфенил)индола.1Н ЯМР (400 МГц, ДMCO-d6) δ: 7.79 (d, 1Н), 7.67 (m, 1Н), 7.60-7.48 (m, 6Н), 7.43-7.25 (m, 8Н), 7.00 (d, 1Н), 5.57 (s, 1H), 5.22 (s, 2Н)
Стадия D: 1-(бензолсульфонил)-4-бензилокси-2-(4-фторфенил)-3-йодиндол
4.92 г 1-(бензолсульфонил)-4-бензилокси-2-(4-фторфенил)индола (10.75 ммоль), 3.69 г Ag2SO4 (11.83 ммоль) и 3.0 г йода (11.83 ммоль) перемешивали в 100 мл EtOH при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 1-(бензолсульфонил)-4-бензилокси-2-(4-фторфенил)-3-йодиндола. МС: (М+Н)+=584.2
Стадия Е: 1-(бензолсулъфонил)-4-бензилокси-3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)индол
5.5 г 1-(бензолсульфонил)-4-бензилокси-2-(4-фторфенил)-3-йодиндола (9.42 ммоль), 4.46 г соединения Синтеза 3b (11.31 ммоль), 6.14 г Cs2CO3 (18.84 ммоль) и 354 мг Ataphos (0.5 ммоль) растворяли в 100 мл смеси ТГФ : вода 3:1 и перемешивали при 70°С в атмосфере N2 до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан, EtOAc и МеОН в качестве элюентов с получением 1-(бензолсульфонил)-4-бензилокси-3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)индола.
1H ЯМР (400 МГц, ДМСО-d6) δ: 7.85 (d, 1Н), 7.67 (t, 1Н), 7.61-6.90 (m, 2Н), 7.53-7.47 (m, 4Н), 7.4 (t, 1Н), 7.20-7.07 (m, 5Н), 6.96 (d, 1Н), 6.77 (d, 1H), 6.73 (d, 1Н), 6.66 (d, 2Н), 4.96 (d, 1Н), 4.86 (d, 1Н), 4.09 (m, 1Н), 4.00 (m, 1H), 3.34 (br s, 4Н), 2.75 (t, 2Н), 2.58 (br s, 4Н), 2.30 (s, 3Н), 1.81 (s, 3Н)
МС: (М+Н)+=724.2
Стадия F: 4-бензилокси-3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)-1Н-индол
6.5 г 1-(бензолсульфонил)-4-бензилокси-3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)индола (8.97 ммоль) растворяли в 100 мл ТГФ и 100 мл МеОН, затем добавляли 28.3 г Ва(ОН)2×8 Н2О (89.7 ммоль) и смесь перемешивали при 70°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем фильтровали, фильтрат концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя ДХМ и МеОН в качестве элюентов с получением 4-бензилокси-3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)-1H-индола. МС: (М+Н)+=584.2
Стадия G: 4-бензилокси-3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)-1-метилиндол
1.626 г 4-бензилокси-3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)-1H-индола (2.78 ммоль) растворяли в 25 мл сухого ДМФА и охлаждали до 0°С. Затем добавляли 123 мг NaH (3.06 ммоль, 60% в минеральном масле) и смесь перемешивали в течение 1 часа. Затем добавляли 395 мг метилйодида (2.78 ммоль) и смесь перемешивали в течение 1 часа. Смесь затем выливали в воду и экстрагировали с помощью ДХМ. Объединенные органические фазы промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя ДХМ и МеОН в качестве элюентов с получением 4-бензилокси-3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)-1-метилиндола.1Н ЯМР (400 МГц, ДМСО-d6) δ: 7.31 (dd, 2Н), 7.24-7.10 (m, 7Н), 6.97 (d, 1Н), 6.83-6.76 (m, 3Н), 6.68 (dd, 1Н), 5.01 (d, 1Н), 4.93 (d, 1Н), 4.14 (m, 1Н), 4.06 (m, 1Н), 3.63 (s, 3Н), 3.10-2.60 (br s, 8Н), 2.84 (br s, 2Н), 2.58 (s, 3Н), 2.04 (s, 3Н)
Стадия Н: 3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)-1-метилиндол-4-ол
1.6 г 4-бензилокси-3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)-1-метилиндола (2.68 ммоль) растворяли в 10 мл ДХМ и добавляли 1 экв. НВr (33% раствор в АсОН). Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем разбавляли 10% водным раствором K2СО3 и экстрагировали с помощью ДХМ. Объединенные органические фазы промывали соляным раствором, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя ДХМ и МеОН в качестве элюентов, затем с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов с получением 3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)-1-метилиндол-4-ола.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 9.02 (s, 1Н), 7.29-7.15 (m, 4H), 7.06-6.92 (m, 2H), 6.86 (d, 1H), 6.78 (d, 1H), 6.38 (dd, 1H), 4.07 (m, 2H), 3.58 (s, 3H), 2.70 (t, 2H), 2.58-2.40 (br s, 4H), 2.40-2.19 (br s, 4H), 2.19 (s, 3H), 2.09 (s, 3H)
MC: (M+H)+=508.2
Стадия I: этил (2S)-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]-2-(n-толилсулъфонилокси)пропаноат
3.668 г соединения Синтеза 2d (8.97 ммоль) растворяли в 12 мл пиридина и при 0°С добавляли 1.97 г TsCl (10.31 ммоль). Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли водой и экстрагировали с помощью EtOAc. Объединенные органические фазы промывали 1 М водным раствором лимонной кислоты, сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением этил (2S)-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]-2-(n-толилсульфонилокси)пропаноата.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 8.93 (d, 1Н), 7.58 (dd, 1Н), 7.52-7.43 (m, 2Н), 7.43-7.34 (m, 2Н), 7.26-7.15 (m, 4Н), 7.13-7.04 (m, 2Н), 6.93-6.83 (m, 2Н), 5.12 (d, 1Н), 5.03-4.92 (m, 2Н), 4.01 (q, 2Н), 3.79 (s, 3Н), 3.26 (dd, 1Н), 3.01 (dd, 1Н), 2.36 (s, 3Н), 1.12 (t, 3Н)
МС: (М+Н)+=563.2
Стадия J: Примеры 68 и 69
60 мг 3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)-1-метилиндол-4-ола (0.12 ммоль), 101 мг этил (2S)-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]-2-(n-толилсульфонилокси)пропаноата (0.18 ммоль) и 80 мг CS2CO3 (0.24 ммоль) растворяли в 2 мл сухого ДМФА и перемешивали при 50°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем добавляли 2 экв. LiОН×Н2О и смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь концентрировали и очищали с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов с получением Примера 68 в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C50H49ClFN5O6: 869.3355, найдено: 435.6743 (М+2Н). Пример 69 получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C50H49ClFN5O6: 869.3355, найдено: 435.6767 (М+2Н)
Пример 70: N-[3-(3-хлор-2-метилфенил)тиено[3,2-с]пиридин-4-ил]-D-фенилаланин
Стадия А: 4-бром-N-(диметоксиметил)тиофен-3-карбоксамид
5.01 г 4-бромтиофен-3-карбоновой кислоты (24.2 ммоль) растворяли в 25 мл изопропилацетата и добавляли 17.9 мл SOCl2 (242 ммоль) и смесь перемешивали при 50°С в течение 2 часов. Затем дистиллировали избыток SOCl2 и остаток растворяли в 25 мл изопропилацетата и охлаждали до 10°С. Добавляли 10.6 мл DIPEA (60.5 ммоль) и 4.0 мл диметилацеталя аминоацетальдегида (36.3 ммоль). Смеси давали нагреться до к.т. и перемешивали в атмосфере N2 в течение ночи. Смесь разбавляли 10% водным раствором Н3РО4 и экстрагировали изопропилацетатом. Объединенные органические фазы промывали 10% водным раствором KН2РО4 и соляным раствором, затем сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 4-бром-N-(диметоксиметил)тиофен-3-карбоксамида.
1Н ЯМР (400 МГц, ДМСО-d6) δ: 8.36 (t, 1Н), 7.93 (d, 1H), 7.72 (d, 1H), 4.48 (t, 1Н), 3.31-3.28 (m, 8Н)
МС (М+Н): 294.0
Стадия В: 3-бром-5H-тиено[3,2-с]пиридин-4-он
32 мг 4-бром-N-(диметоксиметил)тиофен-3-карбоксамида (0.102 ммоль) растворяли в 1 мл РРА и перемешивали при 100°С в атмосфере аргона до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Смесь затем выливали на лед, образовавшийся осадок отфильтровывали и промывали водой с получением 3-бром-5H-тиено[3,2-с]пиридин-4-она. МС (М+Н): 229.9
Стадия С: 3-бром-4-хлортиено[3,2-с]пиридин
1.06 г 3-бром-5H-тиено[3,2-с]пиридин-4-она (4.4 ммоль), 560 мкл N,N-диметиланилина (4.4 ммоль) и 8.37 мл РОСl3 (88 ммоль) перемешивали при 100°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Реакционную смесь затем выливали на лед и экстрагировали с помощью ДХМ. Объединенные органические фазы промывали насыщенным водным раствором NаНСО3 и соляным раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 3-бром-4-хлортиено[3,2-с]пиридина. МС (М+Н): 247.9
Стадия D: 3,4-дибромтиено[3,2-с]пиридин
735 мг 3-бром-4-хлортиено[3,2-с]пиридина (2.8 ммоль) и 2.288 г бромтриметилсилана (14.5 ммоль) растворяли в 15 мл пропионитрила и перемешивали при 100°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Реакционную смесь затем концентрировали при пониженном давлении и очищали с помощью препаративной хроматографии с обращенной фазой, используя 40 мМ водный раствор NH4OAC (рН=4, доводили с помощью АсОН) и MeCN в качестве элюентов с получением 3,4-дибромтиено[3,2-с]пиридина. МС (М+Н): 291.8
Стадия Е: (2R)-2-[(3-бромтиено[3,2-с]пиридин-4-ил)амино]-3-фенилпропановая кислота
340 мг 3,4-дибромтиено[3,2-с]пиридина (1.16 ммоль) и 718 мг D-фенилаланина (4.35 ммоль) растворяли в 7.5 мл сульфолана, затем добавляли 421 мг фторида калия (7.25 ммоль) и 2.23 г 4,7,13,16,21,24-гексаокса-1,10-диазабицикло[8.8.8]гексакозана (5.8 ммоль) и смесь перемешивали при 175°С в атмосфере аргона до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Реакционную смесь непосредственно инжектировали и очищали с помощью препаративной хроматографии с обращенной фазой, используя 40 мМ водный раствор NH4OAC (рН=4, доводили с помощью АсОН) и MeCN в качестве элюентов с получением (2R)-2-[(3-бромтиено[3,2-с]пиридин-4-ил)амино]-3-фенилпропановой кислоты.
Стадия F: Пример 70
189 мг (2R)-2-[(3-бромтиено[3,2-с]пиридин-4-ил)амино]-3-фенилпропановой кислоты (0.5 ммоль) и 341 мг (3-хлор-2-метилфенил)бороновой кислоты (2 ммоль) растворяли в 3.5 мл DME и затем добавляли 72 мг бутилди-1-адамантилфосфина (0.2 ммоль), 22 мг Pd(OAc)2 (0.1 ммоль) и 389 мг ТВАОН (1.5 ммоль) и смесь перемешивали при 100°С в атмосфере аргона до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь выливали в ледяную воду и экстрагировали с помощью МТВЕ. Водную фазу подкисляли до рН 2 и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью препаративной хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов с получением Примера 70. МСВР: рассчитано для C23H19ClN2O2S: 422.0856, найдено: 423.0937 и 423.0919 для двух диастереомеров (М+Н)
Пример 71: (2R)-2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Стадия А: 2-хлор-3-[2-(4-фторфенил)этинил]пиридин
В сухую колбу в 40 мл сухого TEA добавляли 3.85 г 3-бром-2-хлорпиридина (20 ммоль), 0.23 г CuI (1.2 ммоль) и 0.42 г PdCl2(PPh3)2 (0.6 ммоль). После перемешивания в течение 10 минут, добавляли 2.64 г 1-этинил-4-фторбензола (22 ммоль) и раствор нагревали до 100°С и перемешивали в течение ночи. Реакционную смесь охлаждали, разбавляли водой и затем экстрагировали с помощью EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 2-хлор-3-[2-(4-фторфенил)этинил]пиридина.1Н ЯМР (500 МГц, ДМСО-d6) δ 8.44 (dd, 1H), 8.14 (dd, 1Н), 7.68 (t, 2Н), 7.51 (dd, 1), 7.33 (t, 2Н)
Стадия В: 2-(4-фторфенил)тиено[2,3-b]пиридин
2.95 г 2-хлор-3-[2-(4-фторфенил)этинил]пиридина (12.7 ммоль) и 3.97 г Na2S (51 ммоль) помещали в 250 мл колбу. Добавляли 120 мл ДМФА и смесь перемешивали при 130°С в течение 2 часов. Затем реакционную смесь охлаждали, разбавляли водой и затем экстрагировали с помощью EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением 2-(4-фторфенил)тиено[2,3-b]пиридина. МС (М+Н): 230.2
Стадия С: N-оксид 2-(4-фторфенил)тиено[2,3-b]пиридина
1.94 г 2-(4-фторфенил)тиено[2,3-b]пиридина (8.4 ммоль) растворяли в ДХМ (50 мл) и охлаждали до 0°С. По частям добавляли 3.12 г МСРВА (12.6 ммоль) и перемешивали при к.т. в течение 6 часов. Затем смесь концентрировали при пониженном давлении и сырой продукт очищали с помощью флэш-хроматографии, используя ДХМ и метанол в качестве элюентов. МС (М+Н): 246.2
Стадия D: 4-хлор-2-(4-фторфенил)тиено[2,3-b]пиридин
1.56 г 2-(4-фторфенил)-7-оксидотиено[2,3-b]пиридин-7-ия (6.4 ммоль) растворяли в 50 мл СНСl3. Добавляли 15.7 мл РОСl3 (25.76 г, 168 ммоль) и реакционную смесь перемешивали при нагревании в колбе с обратным холодильником в течение 3 часов. Затем смесь охлаждали, добавляли лед и насыщенный водный раствор NaHCO3 и экстрагировали с помощью CHCl3. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя ДХМ и метанол в качестве элюентов с получением 4-хлор-2-(4-фторфенил)тиено[2,3-b]пиридина. МС (М+Н): 264.0
Стадия Е: 3-бром-4-хлор-2-(4-фторфенил)тиено[2,3-b]пиридин
1.15 г Вr2 (7.2 ммоль) по каплям добавляли к смеси 1.46 г 4-хлор-2-(4-фторфенил)тиено[2,3-b]пиридина (5.5 ммоль), 0.52 г K2НРО4 (3.0 ммоль), 0.46 г NaHCO3 (5.5 ммоль) и 1.12 г MgSO4 (9.2 ммоль) в 20 мл СНСl3. Смесь перемешивали в течение ночи при нагревании в колбе с обратным холодильником. Затем, реакционную смесь охлаждали и фильтровали. Фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя ДХМ и метанол в качестве элюентов с получением 3-бром-4-хлор-2-(4-фторфенил)тиено[2,3-b]пиридина.1Н ЯМР (500 МГц, ДМСО-d6) δ 8.59 (d, 1Н), 7.76 (m, 2Н), 7.71 (d, 1Н), 7.42 (m, 2Н)
Стадия F: 3-бром-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ол
Смесь 0.206 г 3-бром-4-хлор-2-(4-фторфенил)тиено[2,3-b]пиридина (0.6 ммоль), 0.492 г ацетата натрия (6 ммоль), 12 мл АсОН и 0.18 мл Н2О нагревали при 150°С с помощью MW облучения в течение 5 часов. Добавляли воду и продукт собирали с помощью фильтрования.1Н ЯМР (500 МГц, ДМСО-d6) δ 11.63 (br s, 1Н), 8.30 (br s, 1H), 7.72 (m, 2H), 7.38 (m, 2H), 6.87 (br s, 1H)
Стадия G: этил (2R)-2-[3-бром-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат
0.324 г 3-бром-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ола (1 ммоль), 0.613 г соединения Синтеза 2d (1.5 ммоль), 0.691 г DTAD (3 ммоль) и 0.787 г РРh3 (3 ммоль) растворяли в 10 мл сухого ТГФ в атмосфере N2 и смесь перемешивали при к.т.до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Растворитель затем удаляли при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением этил (2R)-2-[3-бром-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноата.1Н ЯМP (500 МГц, ДМСО-d6) δ 8.86 (d, 1Н), 8.33 (d, 1Н), 7.72 (m, 2Н), 7.61 (d, 1Н), 7.51 (dd, 1Н), 7.45 (td, 1Н), 7.44 (d, 1H), 7.39 (m, 2Н), 7.25 (td, 1Н), 7.14 (d, 1Н), 7.10 (d, 1H), 7.03 (td, 1H), 6.93 (t, 1Н), 6.88 (d, 1Н), 5.55 (dd, 1H), 5.30 (d, 1Н), 5.26 (d, 1H), 4.16 (m, 2Н), 3.75 (s, 3Н), 3.58 (dd, 1Н), 3.35 (dd, 1Н), 1.13 (t, 3Н)
Стадия Н: Пример 71
0.288 г (2R)-2-[3-бром-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноата (0.4 ммоль), 0.472 г соединения Синтеза 3b (1.2 ммоль), 0.028 г Ataphos (0.004 ммоль) и 0.392 г Cs2CO3 (1.2 ммоль) растворяли в смеси диоксана (4 мл) и воды (3 мл) и перемешивали в атмосфере N2 при 70°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли водой и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4 и концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя ДХМ и метанол в качестве элюентов. Полученное промежуточное соединение растворяли в смеси диоксана (7 мл) и воды (7 мл) и добавляли 0.168 г LiОН×Н2О (4 ммоль). Смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли соляным раствором, нейтрализовали 2 М водным раствором НСl и экстрагировали с помощью ДХМ. Объединенные органические фазы сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Диастереоизомеры очищали и разделяли с помощью препаративной хроматографии с обращенной фазой, используя 5 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов. Диастереомер, элюирующийся позже, собирали в качестве Примера 71. МСВР: рассчитано для C48H45ClFN5O6S: 873.2763; найдено: 437.6441 (М+2Н)
Пример 72: (2R)-2-[5-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-6-(4-фторфенил)-7-метилпирроло[2,3-d]пиримидин-4-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропановая кислота
Стадия А: этил 2-амино-5-(4-фторфенил)-1H-пиррол-3-карбоксилат
Раствор 3330 мг этил 3-амино-3-иминопропаноата (20 ммоль) и 4340 мг 2-бром-1-(4-фторфенил)этанона (20 ммоль) в 40 мл этанола перемешивали при к.т. в течение 30 минут, затем при 0°С добавляли 20 мл 1 М раствора NaOEt в этаноле (20 ммоль) и затем смесь перемешивали при 60°С в течение 90 минут. При комнатной температуре добавляли дополнительные 13 мл 1 М раствора NaOEt в этаноле (13 ммоль) и смесь перемешивали при 60°С в течение еще 1 часа. Реакционную смесь концентрировали при пониженном давлении, остаток разбавляли 40 мл воды и затем экстрагировали этилацетатом. Объединенную органическую фазу сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением этил 2-амино-5-(4-фторфенил)-1H-пиррол-3-карбоксилата.1Н ЯМР (400 МГц, ДМСО-d6) δ: 10.75 (br s, 1Н), 7.52 (m, 2H), 7.14 (m, 2H), 6.44 (d, 1H), 5.68 (br s, 2H), 4.14 (q, 2H), 1.25 (t, 3H)
Стадия В: 6-(4-фторфенил)-3,7-дигидропирроло[2,3-d]пиримидин-4-он
Раствор 6.83 г этил 2-амино-5-(4-фторфенил)-1H-пиррол-3-карбоксилата (27.5 ммоль) и 12 мл муравьиной кислоты в 50 мл формамида и 24 мл ДМФА перемешивали при 160°С в течение 16 часов в герметичном реакционном сосуде. Реакционную смесь охлаждали до комнатной температуры; добавляли 150 мл 2-пропанола. Осадок отфильтровывали, промывали гептаном, затем сушили при пониженном давлении с получением 6-(4-фторфенил)-3,7-дигидропирроло[2,3-d]пиримидин-4-она.1Н ЯМР (400 МГц, ДМСО-d6) δ: 12.36 (br s, 1Н), 11.88 (br s, 1Н), 7.88 (m, 3Н), 7.27 (t, 2Н), 6.93 (s, 1Н)
Стадия С: 4-хлор-6-(4-фторфенил)-7Н-пирроло[2,3-d]пиримидин
Раствор 4.50 г 6-(4-фторфенил)-3,7-дигидропирроло[2,3-d]пиримидин-4-она (19.6 ммоль) в 46 мл РОСl3 (491 ммоль) перемешивали при 90°С в течение 3 часов. Смесь концентрировали при пониженном давлении, остаток выливали на лед. Значение рН доводили до 7, используя твердый K2СО3, и затем смесь экстрагировали этилацетатом. Объединенную органическую фазу промывали соляным раствором, затем сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении с получением 4-хлор-6-(4-фторфенил)-7H-пирроло[2,3-d]пиримидина.1Н ЯМР (400 МГц, ДМСО-d6) δ: 13.04 (br s, 1Н), 8.60 (s, 1Н), 8.08 (m, 2Н), 7.37 (t, 2Н), 7.10 (d, 1Н)
Стадия D: 4-хлор-6-(4-фторфенил)-7-метилпирроло[2,3-d]пиримидин
К раствору 1.87 г 4-хлор-6-(4-фторфенил)-7H-пирроло[2,3-d]пиримидина (7.55 ммоль) в 38 мл ДМФА добавляли 1.286 г MeI (9.06 ммоль) и затем 1.15 г K2СО3 (8.30 ммоль), и смесь перемешивали при к.т.в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении. Остаток разбавляли соляным раствором, и смесь экстрагировали дихлорметаном. Объединенную органическую фазу сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении, затем остаток очищали с помощью флэш-хроматографии, используя гептан и этилацетат в качестве элюентов с получением 4-хлор-6-(4-фторфенил)-7-метилпирроло[2,3-d]пиримидина.1Н ЯМР (400 МГц, ДМСО-d6) δ: 8.69 (s, 1H), 7.79 (m, 2Н), 7.42 (m, 2Н), 6.80 (s, 1Н), 3.83 (s, 3Н)
Стадия Е: 5-бром-4-хлор-6-(4-фторфенил)-7-метилпирроло[2,3-d]пиримидин
К раствору 1.36 г 4-хлор-6-(4-фторфенил)-7-метилпирроло[2,3-d]пиримидина (5.20 ммоль) в 16 мл уксусной кислоты при 0°С по каплям добавляли 5.46 мл 1 М раствора Вr2 в уксусной кислоте (5.46 ммоль) и затем реакционную смесь перемешивали при к.т. в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении, затем остаток разбавляли насыщенным водным раствором NaHCO3 и экстрагировали этилацетатом. Объединенную органическую фазу сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и этилацетат в качестве элюентов с получением 5-бром-4-хлор-6-(4-фторфенил)-7-метилпирроло[2,3-d]пиримидина.1Н ЯМР (400 МГц, ДМСО-d6) δ: 8.73 (s, 1Н), 7.70 (m, 2Н), 7.47 (m, 2Н), 3.69 (s, 3Н)
Стадия F: этил (2R)-2-[5-бром-6-(4-фторфенил)-7-метилпирроло[2,3-d]пиримидин-4-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат
845 мг 5-бром-4-хлор-6-(4-фторфенил)-7-метилпирроло[2,3-d]пиримидина (2.48 ммоль) и 1.27 г соединения Синтеза 2с (3.11 ммоль) растворяли в 10 мл ДМФА, затем добавляли 2.43 г Cs2CO3 (7.44 ммоль) и смесь перемешивали при 60°С в течение 6 часов. Реакционную смесь концентрировали при пониженном давлении, остаток разбавляли соляным раствором и затем смесь экстрагировали этилацетатом. Объединенную органическую фазу сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении, затем остаток очищали с помощью флэш-хроматографии, используя гептан и этилацетат в качестве элюентов с получением этил (2R)-2-[5-бром-6-(4-фторфенил)-7-метилпирроло[2,3-d]пиримидин-4-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноата. МС (М+Н): 712.0
Стадия G: Пример 72
Используя Общую Методику II и этил (2R)-2-[5-бром-6-(4-фторфенил)-7-метилпирроло[2,3-d]пиримидин-4-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат взамен сложного эфира 5-бромфуро[2,3-d]пиримидилмолочной кислоты, и используя соединение Синтеза 3b в качестве подходящего производного бороновой кислоты Пример 72 получали в виде смеси диастереоизомеров. МСВР: рассчитано для C48H47ClFN7O6: 871.3260; найдено: 436.6703 и 436.6710 (М+2Н)
Пример 73: 2-{[3-{3,5-дихлор-2,6-диметил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Пример 74: 2-{[3-{2,6-диметил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Пример 75: 1-[(диметилкарбамоил)окси]этил (2R)-2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-6]пиридин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропаноат
591 мг гидрохлорида диметиламина (7.25 ммоль) и 1.20 мл пиридина (14.9 ммоль) растворяли в 18 мл сухого ДХМ в атмосфере азота, затем смесь охлаждали до -78°С и добавляли 990 мг 1-хлорэтилхлорформиата (6.9 ммоль). Реакционную смесь перемешивали при -78°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Холодную смесь фильтровали и фильтрат концентрировали при пониженном давлении (30 мбар), используя 30°С баню. Затем остаток растворяли в 2 мл сухого ДМФА в атмосфере азота, добавляли 60 мг Примера 71 (0.069 ммоль) и 223 мг Cs2CO3 (0.55 ммоль) и реакционную смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли соляным раствором, экстрагировали с помощью ЕtOАс. Объединенный органический слой сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью хроматографии с обращенной фазой, используя 5 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов с получением Примера 75 в виде смеси диастереоизомеров. МСВР: рассчитано для C53H54ClFN6O8S: 988.3397; найдено: 495.1782 и 495.1772 (М+2Н)
Пример 76: 1-[(этоксикарбонил)окси]этил (2R)-2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропаноат
668 мг ЕtOН (14.5 ммоль) и 1.26 г пиридина (15.6 ммоль) растворяли в 18 мл сухого ДХМ в атмосфере азота, затем смесь охлаждали до -78°С и добавляли 1.98 г 1-хлорэтил хлорформиата (13.8 ммоль). Реакционную смесь перемешивали при -78°С до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Холодную смесь фильтровали и фильтрат концентрировали при пониженном давлении (30 мбар), используя 30°С баню. Затем остаток растворяли в 2 мл сухого ДМФА в атмосфере азота, добавляли 60 мг Примера 71 (0.069 ммоль) и 223 мг Cs2CO3 (0.55 ммоль) и реакционную смесь перемешивали при к.т. до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь фильтровали и фильтрат очищали с помощью хроматографии с обращенной фазой, используя 5 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов с получением Примера 76 в виде смеси диастереоизомеров. МСВР: рассчитано для C53H53ClFN5O9S: 989.3237; найдено: 990.3342 и 990.3314 (М+Н).
Пример 77: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси}-3-гидрокси-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Пример 78: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси}-4-гидрокси-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)бутановая кислота
Пример 79: 2-O-[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]-3,4-дидеокси-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пентоновая кислота
Пример 80: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси}-3-[2-({2-[5-(гидроксиметил)пиридин-3-ил]пиримидин-4-ил}метокси)фенил]пропановая кислота
Пример 81: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси}-3-{2-[(2-{2-[(2-гидроксиэтокси)метил]фенил}пиримидин-4-ил)метокси]фенил}пропановая кислота
Пример 82: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси}-3-{2-[(2-{4-[2-(диметиламино)этокси]фенил}пиримидин-4-ил)метокси]фенил}пропановая кислота
Пример 83: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]окси}-3-[2-({2-[3-(фосфоноокси)фенил]пиримидин-4-ил}метокси)фенил]пропановая кислота
Пример 84: N-[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}-D-фенилаланин
Стадия А: этил (2R)-2-[[3-бром-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]-фенил]пропаноат
343 мг 3-бром-4-хлор-2-(4-фторфенил)тиено[2,3-b]пиридина (Стадия Е в Примере 71, 1.0 ммоль) и 455 мг соединения Синтеза 2i (1.20 ммоль) растворяли в 5 мл сухого ДМСО, затем добавляли 978 мг Cs2CO3 (3.0 ммоль) и смесь перемешивали в атмосфере азота при 100°C до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли соляным раствором, нейтрализовали 2 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Затем остаток растворяли в 1.5 мл 1.25 М раствора HCl в EtOH и смесь перемешивали при 60°C до тех пор, пока не завершится образование сложного эфира. Затем смесь осторожно нейтрализовали насыщенным водным раствором NaHCO3 и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов.
1H ЯМР (500 МГц, ДМСО-d6) δ: 8.77 (d, 1Н), 8.07 (d, 1Н), 7.6 (m, 2Н), 7.52-6.88 (m, 8Н), 7.37 (d, 1Н), 7.34 (m, 2Н), 7.05 (d, 1Н), 6.57 (d, 1Н), 5.23/5.19 (d+d, 2Н), 4.92 (m, 1Н), 4.12 (m, 2Н), 3.73 (s, 3Н), 3.44/3.25 (dd+dd, 2Н), 1.14 (t, 3Н)
МСВР: рассчитано для C36H30BrFN4O4S: 712.1155; найдено: 357.0649 (М+2Н)
Стадия В: этил (2R)-2-[[3-(3-хлор-4-гидрокси-2-метилфенил)-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат
178 мг этил (2R)-2-[[3-бром-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноата (0.249 ммоль) и 107 мг соединения Синтеза 3а (0.4 ммоль) растворяли в 1 мл 1,4-диоксана в атмосфере азота, затем добавляли 163 мг Cs2CO3 (0.50 ммоль), 0.5 мл воды и 28 мг AtaPhos (0.04 ммоль) и смесь перемешивали в микроволновом реакторе при 111°C в течение 15 минут. Затем смесь разбавляли соляным раствором, нейтрализовали 2 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением этил (2R)-2-[[3-(3-хлор-4-гидрокси-2-метилфенил)-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноата в виде смеси атропоизомеров. МСВР: рассчитано для C43H36ClFN4O5S: 774.2079; найдено: 388.1113 (М+2Н)
Стадия С: Пример 84
80 мг этил (2R)-2-[[3-(3-хлор-4-гидрокси-2-метилфенил)-2-(4-фторфенил)тиено[2,3-b]пиридин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноата (0.103 ммоль), 43 мг 2-(4-метилпиперазин-1-ил)этанола (0.30 ммоль) и 79 мг PPh3 (0.30 ммоль) растворяли в 1 мл сухого толуола, затем добавляли 69 мг DTAD (0.30 ммоль) и смесь перемешивали при 50°C в атмосфере азота до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь концентрировали при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя EtOAc и МеОН в качестве элюентов. Полученное сложноэфирное производное растворяли в 1 мл ТГФ, затем добавляли 80 мг LiOH×H2O и 1 мл воды и смесь перемешивали при к.т. до тех пор, пока не завершится гидролиз. Затем смесь разбавляли соляным раствором, нейтрализовали 2 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов с получением Примера 84 в виде 7:3 смеси диастереоизомеров. МСВР: рассчитано для C48H46ClN6O5S: 872.2923; найдено: 437.1540 и 437.1538 (М+2Н)
Пример 85: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[3,2-с]пиридин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Пример 86: N-[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[3,2-с]пиридин-4-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенилаланин
Стадия А: 3-бром-4-хлор-2-йодтиено[3,2-c]пиридин
4.97 г 3-бром-4-хлортиено[3,2-с]пиридина (20.0 ммоль) растворяли в 50 мл сухого ТГФ в атмосфере аргона и смесь охлаждали до -45°C. Затем по каплям добавляли 22 мл раствора Mg(TMP)Cl×LiCl (22 ммоль, 1M в ТГФ) и смесь перемешивали в течение 1 часа при -45°C, затем 1 час при 0°C, и затем ее снова охлаждали до -45°C. Затем по каплям добавляли 5.58 г йода (22 ммоль, растворенного в 20 мл сухого, холодного ТГФ) и смесь перемешивали при -45°C в течение 2 часов. Затем смеси давали нагреться до к.т. и концентрировали при пониженном давлении. Остаток выливали на 300 мл соляного раствора и экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали насыщенным водным раствором Na2S2O3, насыщенным водным раствором NH4Cl, затем водой и затем сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гексаны и EtOAc в качестве элюентов.
1Н ЯМР (500 МГц, ДМСО-d6) δ: 8.27 (d, 1Н), 8.17 (d, 1Н)
MCBP: рассчитано для C7H2BrClINS: 372.7824; найдено: 373.7916 (М+Н)
Стадия В: 3-бром-4-хлор-2-(4-фторфенил)тиено[3,2-c]пиридин
2.62 г 3-бром-4-хлор-2-йодтиено[3,2-с]пиридина (7.0 ммоль) и 2.33 г 2-(4-фторфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (10.5 ммоль) растворяли в 18 мл ТГФ в атмосфере аргона, затем добавляли 6.84 г Cs2CO3 (21 ммоль), 18 мл воды, 79 мг Pd(OAc)2 (0.35 ммоль) и 297 мгtBuXPhos (0.70 ммоль) и смесь перемешивали при 70°C до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем летучие компоненты упаривали при пониженном давлении. Остаток разбавляли водой и экстрагировали с помощью EtOAc. Объединенную органическую фазу промывали насыщенным водным раствором NH4Cl, затем соляным раствором и затем сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гексаны и EtOAc в качестве элюентов.
1Н ЯМР (500 МГц, ДМСО-d6) δ: 8.35 (d, 1H), 8.26 (d, 1Н), 7.74 (dd, 2Н), 7.42 (t, 2Н)
МСВР: рассчитано для C13H6BrClFNS: 340.9077; найдено: 341.9144 (М+Н)
Стадия С: этил (2R)-2-[[3-бром-2-(4-фторфенил)тиено[3,2-с]пиридин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат
343 мг 3-бром-4-хлор-2-(4-фторфенил)тиено[3,2-с]пиридина (1.0 ммоль) и 455 мг соединения Синтеза 2i (1.20 ммоль) растворяли в 5 мл сухого ДМСО, затем добавляли 978 мг Cs2CO3 (3.00 ммоль) и смесь перемешивали в атмосфере азота при 100°C до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь разбавляли соляным раствором, нейтрализовали 2 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Затем остаток растворяли в 1.5 мл 1.25 М раствора HCl в EtOH и смесь перемешивали при 60°C до тех пор, пока не завершится образование сложного эфира. Затем смесь осторожно нейтрализовали насыщенным водным раствором NaHCO3 и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов. МСВР: рассчитано для C36H30BrFN4O4S: 712.1155; найдено: 713.1209 (М+Н)
Стадия D: этил (2R)-2-[[3-(3-хлор-4-гидрокси-2-метилфенил)-2-(4-фторфенил)тиено[3,2-с]пиридин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат
31 мг этил (2R)-2-[[3-бром-2-(4-фторфенил)тиено[3,2-с]пиридин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноата (0.043 ммоль) и 24 мг соединения Синтеза 3а (0.09 ммоль) растворяли в 0.5 мл 1,4-диоксана в атмосфере азота, затем добавляли 33 мг Cs2CO3 (0.10 ммоль), 0.5 мл воды и 9.4 мг AtaPhos (0.013 ммоль) и смесь перемешивали в микроволновом реакторе при 111°C в течение 10 минут. Затем смесь разбавляли соляным раствором, нейтрализовали 2 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением этил (2R)-2-[[3-(3-хлор-4-гидрокси-2-метилфенил)-2-(4-фторфенил)тиено[3,2-с]пиридин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноата в виде смеси атропоизомеров. МСВР: рассчитано для C43H36ClFN4O5S: 774.2079; найдено: 775.2134 (М+Н)
Стадия Е: Пример 86
33 мг этил (2R)-2-[[3-(3-хлор-4-гидрокси-2-метилфенил)-2-(4-фторфенил)тиено[3,2-с]пиридин-4-ил]амино]-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноата (0.04 ммоль), 15 мг 2-(4-метилпиперазин-1-ил)этанола (0.10 ммоль) и 26 мг PPh3 (0.10 ммоль) растворяли в 1 мл сухого толуола, затем добавляли 23 мг DTAD (0.10 ммоль) и смесь перемешивали при 50°C в атмосфере азота до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь концентрировали при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя EtOAc и МеОН в качестве элюентов. Полученное сложноэфирное производное растворяли в 1 мл ТГФ, затем добавляли 80 мг LiOH×H2O и 1 мл воды и смесь перемешивали при к.т.до тех пор, пока не завершится гидролиз. Затем смесь разбавляли соляным раствором, нейтрализовали 2 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов с получением Примера 86 в виде 10:3 смеси диастереоизомеров. МСВР: рассчитано для C48H46ClFN6O5S: 872.2923; найдено: 437.1549 и 437.1532 (М+2Н)
Пример 87: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-с]пиридин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Пример 88: N-[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-с]пиридин-4-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенилаланин
Пример 89: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-d]пиридазин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Пример 90: N-[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)тиено[2,3-d]пиридазин-4-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенилаланин
Пример 91: 2-{[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-с]пиридазин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Пример 92: N-[5-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-6-(4-фторфенил)тиено[2,3-с]пиридазин-4-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенилаланин
Пример 93: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)фуро[2,3-b]пиридин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Пример 94: N-[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)фуро[2,3-b]пиридин-4-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенилаланин
Пример 95: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)фуро[3,2-с]пиридин-4-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Пример 96: N-[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)фуро[3,2-с]пиридин-4-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенилаланин
Пример 97а: 2-{[(3Sa)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)имидазо[1,2-с]пиримидин-5-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
и
Пример 97b: 2-{[(3Ra)-3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)имидазо[1,2-с]пиримидин-5-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Стадия А: 1-(2-бром-1,1-диметоксиэтил)-4-фторбензол
8.68 г 2-бром-1-(4-фторфенил)этанона (40.0 ммоль) растворяли в 80 мл МеОН, затем добавляли 8.75 мл СН(ОМе)3 (80.0 ммоль) и 380 мг TsOH×H2O (2.00 ммоль) и смесь перемешивали при нагревании в колбе с обратным холодильником до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь концентрировали при пониженном давлении и разбавляли с помощью Et2O. Смесь промывали 10% водным раствором K2CO3, сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении.1Н ЯМР (250 МГц, CDCl3) δ: 7.53-7.44 (m, 2Н), 7.11-7.01 (m, 2Н), 3.60 (s, 2Н), 3.22 (s, 6Н)
Стадия В: 5-хлор-2-(4-фторфенил)имидазо[1,2-с]пиримидин
В реакционный сосуд высокого давления, изготовленный из стали, загружали 648 мг 2-хлорпиримидин-4-амина (5.0 ммоль), 1.58 г 1-(2-бром-1,1-диметоксиэтил)-4-фторбензола (6.0 ммоль), 123 мг Sc(OTf)3 (0.25 ммоль) и 50 мл MeCN и смесь перемешивали при 120°C в течение 24 часов. Затем смесь разбавляли ДХМ и промывали насыщенным водным раствором NaHCO3. Водный слой экстрагировали с помощью ДХМ. Объединенный органический слой сушили над MgSO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гексаны и EtOAc в качестве элюентов. МСВР: рассчитано для C12H7ClFN3: 247.0312; найдено: 248.0397 (М+Н)
Стадия С: 3-бром-5-хлор-2-(4-фторфенил)имидазо[1,2-с]пиримидин
198 мг 5-хлор-2-(4-фторфенил)имидазо[1,2-с]пиримидина (0.80 ммоль) растворяли в 4.8 мл ДМФА, затем добавляли 142 мг NBS (0.80 ммоль) и смесь перемешивали при к.т. до тех пор, пока не израсходуется исходное вещество. Затем смесь выливали на насыщенный водный раствор NaHCO3 и образовавшийся осадок отфильтровывали и промывали водой. Сырой продукт очищали с помощью флэш-хроматографии, используя гексаны и EtOAc в качестве элюентов.
1H ЯМР (500 МГц, ДМСО-d6) δ: 8.03 (m, 2Н), 7.90 (d, 1Н), 7.73 (d, 1Н), 7.39 (m, 2Н)
МСВР: рассчитано для C12H6BrClFN3: 324.9418; найдено: 325.9496 (М+Н)
Стадия D: этил (2R)-2-[3-бром-2-(4-фторфенил)имидазо[1,2-с]пиримидин-5-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноат
102 мг 3-бром-5-хлор-2-(4-фторфенил)имидазо[1,2-с]пиримидина (0.312 ммоль) и 140 мг соединения Синтеза 2с (0.344 ммоль) растворяли в 3 мл сухого ДМСО в атмосфере азота, затем добавляли 305 мг CS2CO3 (0.936 ммоль) и смесь перемешивали при к.т.до тех пор, пока не наблюдали отсутствия дальнейшего желаемого превращения. Затем смесь разбавляли соляным раствором и водой, нейтрализовали 2 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов.
1Н ЯМР (500 МГц, ДМСО-d6) δ: 8.20 (d, 1Н), 8.03 (m, 2Н), 7.62 (d, 1Н), 7.56 (d, 1H), 7.50 (dd, 1Н), 7.49 (dd, 1H), 7.42 (ddd, 1Н), 7.35 (m, 2Н), 7.27 (ddd, 1H), 7.23 (d, 1Н), 7.13 (d, 1Н), 7.11 (d, 1Н), 7.01 (td, 1Н), 6.96 (td, 1Н), 5.80 (dd, 1H), 5.31/5.27 (d+d, 2Н), 4.18/4.15 (m+m, 2Н), 3.75 (s, 3Н), 3.62/3.36 (dd+dd, 2Н), 1.12 (t, 3Н)
МСВР: рассчитано для C35H29BrFN5O5: 697.1336; найдено: 698.1419 (М+Н)
Стадия Е: этил (2R)-2-[3-(3-хлор-4-гидрокси-2-метилфенил)-2-(4-фторфенил)имидазо[1,2-с]пиримидин-5-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси] фенил] пропаноат
150 мг этил (2R)-2-[3-бром-2-(4-фторфенил)имидазо[1,2-с]пиримидин-5-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноата (0.215 ммоль) и 80.8 мг соединения Синтеза 3а (0.301 ммоль) растворяли в 1 мл ТГФ в атмосфере азота, затем добавляли 140 мг Cs2CO3 (0.430 ммоль), 0.2 мл воды и 30.4 мг AtaPhos (0.043 ммоль) и смесь перемешивали в микроволновом реакторе при 100°C в течение 5 минут. Затем смесь разбавляли соляным раствором, нейтрализовали 2 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов с получением смеси диастереоизомеров. МСВР: рассчитано для C42H35ClFN5O6: 759.2260; найдено: 760.2370 и 760.2344 (М+Н)
Стадия F: Примеры 97а и 97b
11.4 мг этил (2R)-2-[3-(3-хлор-4-гидрокси-2-метилфенил)-2-(4-фторфенил)имидазо[1,2-с]пиримидин-5-ил]окси-3-[2-[[2-(2-метоксифенил)пиримидин-4-ил]метокси]фенил]пропаноата (0.015 ммоль), 7.2 мг 2-(4-метилпиперазин-1-ил)этанола (0.050 ммоль) и 13.1 мг PPh3 (0.050 ммоль) растворяли в 1 мл сухого толуола, затем добавляли 11.5 мг DTAD (0.050 ммоль) и смесь перемешивали при 50°C в атмосфере азота до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь концентрировали при пониженном давлении и остаток очищали с помощью флэш-хроматографии, используя EtOAc и МеОН в качестве элюентов. Полученное сложноэфирное производное растворяли в 1 мл ТГФ, затем добавляли 42 мг LiOH×Н2О и 1 мл воды и смесь перемешивали при к.т. до тех пор, пока не завершится гидролиз. Затем смесь разбавляли соляным раствором, нейтрализовали 2 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов. Пример 97а получали в виде диастереоизомера, элюирующегося раньше. МСВР: рассчитано для C47H45ClFN7O6: 857.3104; найдено: 429.6626 (М+2Н)
Пример 97b получали в виде диастереоизомера, элюирующегося позже. МСВР: рассчитано для C47H45ClFN7O6: 857.3104; найдено: 429.6638 (М+2Н)
Пример 98: N-[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)имидазо[1,2-с]пиримидин-5-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенилаланин
Пример 99: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)имидазо[1,2-а]пиразин-5-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Пример 100: N-[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)имидазо[1,2-а]пиразин-5-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенилаланин
Пример 101: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)имидазо[1,2-а]пиримидин-5-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Пример 102: N-[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)имидазо[1,2-а]пиримидин-5-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}-D-фенилаланин
Стадия А: 2-(4-фторфенил)-1H-имидазо[1,2-а]пиримидин-5-он
10.0 г 2-амино-1H-пиримидин-4-она (90.0 ммоль) и 9.77 г 2-бром-1-(4-фторфенил)этанона (45.0 ммоль) растворяли в 100 мл ДМФА и смесь перемешивали при 120°C до тех пор, пока не наблюдали отсутствия дальнейшего превращения. Затем смесь концентрировали при пониженном давлении и разбавляли EtOAc. Добавляли целит и летучие компоненты упаривали при пониженном давлении. Смесь очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов. Региоизомер, элюирующийся раньше, собирали в качестве 2-(4-фторфенил)-1H-имидазо[1,2-a]пиримидин-5-она.1H ЯМР (500 МГц, ДМСО-d6) δ: 12.98 (br s, 1Н), 8.14 (s, 1Н), 7.97 (m, 2Н), 7.90 (d, 1H), 7.27 (m, 2Н), 5.57 (d, 1Н)
Стадия В: 5-хлор-2-(4-фторфенил)имидазо[1,2-а]пиримидин
1.36 г 2-(4-фторфенил)-1H-имидазо[1,2-a]пиримидин-5-она (5.9 ммоль) и 16.6 мл POCl3 перемешивали при 93°C в течение 90 минут, затем смесь охлаждали до к.т. и концентрировали при пониженном давлении. Остаток выливали на ледяную воду. После того, как лед растаял, образовавшийся осадок отфильтровывали и промывали водой.1H ЯМР (500 МГц, ДМСО-d6) δ: 8.65 (s, 1Н), 8.55 (d, 1Н), 8.17 (m, 2Н), 7.45 (d, 1H), 7.33 (m, 2Н)
Стадия С: 3-бром-5-хлор-2-(4-фторфенил)имидазо[1,2-a]пиримидин
715 мг 5-хлор-2-(4-фторфенил)имидазо[1,2-a]пиримидина (2.89 ммоль) растворяли в 10 мл хлороформа, затем добавляли 570 мг NBS (3.20 ммоль) и смесь перемешивали при к.т.до тех пор, пока не израсходуется исходное вещество. Затем смесь концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии, используя гептан и EtOAc в качестве элюентов.
1H ЯМР (500 МГц, ДМСО-d6) δ: 8.52 (d, 1H), 8.10 (m, 2Н), 7.39 (m, 2Н), 7.38 (d, 1Н)
МСВР: рассчитано для C12H6BrClFN3: 324.9418; найдено: 325.9481 (М+Н)
Стадия D: 5-хлор-3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси] фенил]-2-(4-фторфенил)имидазо[1,2-a] пиримидин
620 мг 3-бром-5-хлор-2-(4-фторфенил)имидазо[1,2-a]пиримидина (1.93 ммоль) и 2.37 г соединения Синтеза 3b (6.0 ммоль) растворяли в 10 мл ТГФ в атмосфере азота, затем добавляли 1.30 г Cs2CO3 (4.00 ммоль), 3 мл воды и 273 мг AtaPhos (0.386 ммоль) и смесь перемешивали в микроволновом реакторе при 110°C в течение 10 минут. Затем смесь разбавляли соляным раствором и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью флэш-хроматографии, используя EtOAc и МеОН в качестве элюентов. МСНР: рассчитано для C26H26Cl2FN5O: 513.15; найдено: 514.1 (М+Н)
Стадия Е: Пример 102
341 мг 5-хлор-3-[3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил]-2-(4-фторфенил)имидазо[1,2-а]пиримидина (0.66 ммоль) и 300 мг соединения Синтеза 2i (0.76 ммоль) растворяли в 3 мл сухого ДМСО в атмосфере азота, затем добавляли 652 мг Cs2CO3 (2.0 ммоль) и смесь перемешивали в микроволновом реакторе при 160°C в течение 10 минут. Затем смесь разбавляли соляным раствором и водой, нейтрализовали 2 М водным раствором HCl и экстрагировали с помощью ДХМ. Объединенную органическую фазу сушили над Na2SO4, фильтровали и фильтрат концентрировали при пониженном давлении. Сырой продукт очищали с помощью хроматографии с обращенной фазой, используя 25 мМ водный раствор NH4HCO3 и MeCN в качестве элюентов с получением Примера 102 в виде смеси диастереоизомеров. МСВР: рассчитано для C47H46ClFN8O5: 856.3264; найдено: 429.1687 и 429.1705 (М+2Н)
Пример 103: 2-{[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)имидазо[1,2-а]пиридин-5-ил]окси}-3-(2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенил)пропановая кислота
Пример 104: N-[3-{3-хлор-2-метил-4-[2-(4-метилпиперазин-1-ил)этокси]фенил}-2-(4-фторфенил)имидазо[1,2-а]пиридин-5-ил]-2-{[2-(2-метоксифенил)пиримидин-4-ил]метокси}фенилаланин
ФАРМАКОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ
ПРИМЕР А: Исследование ингибирования Mcl-1 с использованием методики поляризации флуоресценции
Относительный потенциал связывания каждого соединения определяли с помощью поляризации флуоресценции (FP). Метод включает использование меченого флуоресцеином лиганда (флуоресцеин-βAla-Ahx-A-REIGAQLRRMADDLNAQY-OH; мол. масса 2.765), который связывается с Mcl-1 белком (при условии, что Mcl-1 соответствует UniProtKB® первичному номеру доступа: Q07820), что приводит к повышенный анизотропии, измеренной в единицах миллиполяризации (mP) с использованием ридера. Добавление соединения, которое конкурентно связывается с тем же самым сайтом в качестве лиганда, приводит к большей доли несвязанного лиганда в системе, определенной уменьшением единиц mP.
Метод 1: В ДМСО приготовляли 11-точечные серийные разведения каждого соединения и 2 мкл переносили в плоскодонный 384-луночный планшет с низкой связываемостью (конечная концентрация ДМСО 5%). Затем добавляли 38 мкл буфера (10 мМ 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота [HEPES], 150 мМ NaCl, 0.05% Tween 20, pH 7.4), содержащего меченый флуоресцеином лиганд (конечная концентрация 1 нМ) и Mcl-1 белок (конечная концентрация 5 нМ).
Аналитические планшеты инкубировали в течение ~ 2 часов при комнатной температуре перед тем, как измеряли FP на ридере Biomek Synergy2 (возбуждение 528 нм, испускание 640 нм, отсечение 510 нм) и рассчитывали единицы mP. Связывание возрастающих доз тестируемого соединения выражали в виде снижения mP в процентах по сравнению с окном, установленным между контролями 'только 5% ДМСО' и '100%-ное ингибирование'. Выполняли построение 11-точечных кривых зависимости доза-эффект с помощью программного обеспечения XL-Fit с использованием 4-параметрической логистической модели (сигмоидальная модель зависимости доза-эффект), и определяли ингибирующие концентрации, которые давали 50%-ное снижение mP (IC50). Полученные с использованием Метода 1 результаты представлены в Таблице 1 ниже; значения IC50 ингибирования Mcl-1, полученные с использованием Метода 1, подчеркиванием не выделены.
Метод 2: В ДМСО приготовляли 11-точечные серийные разведения каждого соединения и 2 мкл переносили в плоскодонный 384-луночный планшет с низкой связываемостью (конечная концентрация ДМСО 5%). Затем добавляли 38 мкл буфера (20 мМ Na2HPO4, 1 мМ EDTA, 50 мМ NaCl, pH 7.4), содержащего меченый флуоресцеином лиганд (конечная концентрация 10 нМ) и Mcl-1 белок (конечная концентрация 10 nM).
Аналитические планшеты инкубировали в течение ~ 2 часов при комнатной температуре перед тем, как измеряли FP на ридере Biomek Synergy2 (возбуждение 528 нм, испускание 640 нм, отсечение 510 нм) и рассчитывали единицы mP. Связывание возрастающих доз тестируемого соединения выражали в виде снижения mP в процентах по сравнению с окном, установленным между контролями 'только 5% ДМСО' и '100%-ное ингибирование' (50 мкМ немеченый лиганд). Выполняли построение 11-точечных кривых зависимости доза-эффект с помощью программного обеспечения XL-Fit с использованием 4-параметрической логистической модели (сигмоидальная модель зависимости доза-эффект), и определяли ингибирующие концентрации, которые давали 50%-ное снижение mP (IC50). Полученные с использованием Метода 2 результаты представлены в Таблице 1 ниже; значения IC50 ингибирования Mcl-1. полученные с использованием Метода 2, выделены подчеркиванием.
Результаты показывают, что соединения изобретения ингибируют взаимодействие между Mcl-1 белком и флуоресцентным пептидом, описанным выше.
ПРИМЕР В: In vitro цитотоксичность
Исследования цитотоксичности осуществляли на опухолевой линии Н929 множественной миеломы.
Клетки распределяли на микропланшетах и подвергали действию тестируемых соединений в течение 48 часов. Жизнеспособность клеток затем определяли количественно с помощью колориметрического анализа, микрокультурального исследования на основе тетразолия (Cancer Res., 1987, 47, 939-942).
Результаты выражены в значениях IC50 (концентрация соединения, которая ингибирует жизнеспособность клеток на 50%) и представлены в Таблице 1 ниже.
Результаты показывают, что соединения изобретения являются цитотоксическими.



НО: не определено
В случае частичных ингибиторов, для данной концентрации тестируемого соединения указано процентное значение ингибирования поляризации флуоресценции. Таким образом, 45.1% @10 мкМ означает, что 45.1%-ное ингибирование поляризации флуоресценции наблюдается при концентрации тестируемого соединения, равной 10 мкМ.
ПРИМЕР С: Количественное in vivo определение расщепленной формы PARP
Способность соединений изобретения индуцировать апоптоз оценивали путем измерения уровней расщепленной PARP на ксенотрансплантатной модели множественной миеломы с использованием клеток АМО-1.
1.107 клеток АМО-1 пересаживали подкожно иммуносупрессивным мышам (штамм SCID). Через 12-14 дней после пересадки, животных лечили внутривенным или пероральным путем различными соединениями. После лечения, опухолевые массы извлекали и лизировали, и расщепленную форму PARP в лизатах опухолей определяли количественно.
Количественную оценку проводили с использованием исследования "Meso Scale Discovery (MSD) ELISA platform", которое предназначено для специфического анализа на расщепленную форму PARP. Оценку выражали в виде фактора активации, который равняется соотношению между количеством расщепленной PARP у получающих лечение мышей и количеством расщепленной PARP у контрольных мышей.
Результаты (представленные в Таблице 2 ниже) показывают, что соединения изобретения способны in vivo индуцировать апоптоз в опухолевых клетках АМО-1.

ПРИМЕР D: Противоопухолевая активность in vivo
Противоопухолевую активность соединений изобретения оценивали на ксенотрансплантатной модели множественной миеломы с использованием клеток АМО-1.
1×107 клеток АМО-1 пересаживали подкожно иммуносупрессивным мышам (штамм SCID).
Через 6-8 дней после пересадки, когда опухолевая масса достигла приблизительно 150 мм3, мышей лечили различными соединениями согласно ежедневного плана (лечение в течение 5 дней). Опухолевую массу измеряли два раза в неделю с самого начала лечения.
Соединение изобретения обладает противоопухолевой активностью (регрессия опухоли) в модели АМО-1 множественной миеломы со значением ΔТ/С (квалификационный параметр активности продукта, который рассчитывают путем вычитания среднего объема опухоли в последний день лечения из среднего объема опухоли в первый день лечения и деления на объем опухоли контрольной группы, не получающей лечение, в последний день лечения животных, его получающих), равным -27%. Полученные результаты показывают, что соединения изобретения индуцируют значительную регрессию опухоли в период лечения.
ПРИМЕР Е: Фармацевтическая композиция: Таблетки
Реферат
Изобретение относится к бициклическим производным формулы (I), где R1, R2, R3, R4, R5, R6, R7, R8, R14, W, А и n являются такими, как определено в описании, к способу их получения и к фармацевтическим композициям, содержащим их. Соединения настоящего изобретения являются новыми и обладают очень ценными фармакологическими характеристиками в области апоптоза и онкологии. 13 н. и 27 з.п. ф-лы, 104 пр., 2 табл.

Формула




















































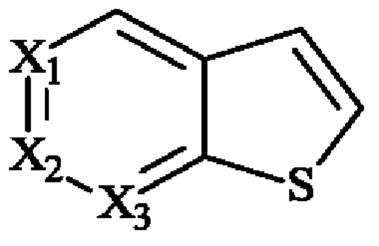



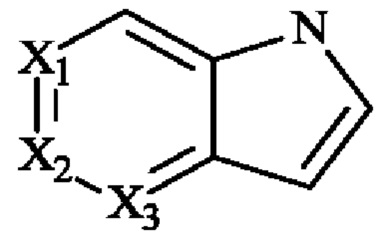


























Комментарии