Синтез карбамоилпиридоновых ингибиторов интегразы вич и промежуточных соединений - RU2527451C2
Код документа: RU2527451C2
Чертежи






Описание
Область изобретения
Настоящее изобретение включает модификации известных способов синтеза соединений, обладающих ингибиторной активностью в отношении интегразы ВИЧ.
Предшествующий уровень техники
В публикации международной заявки WO 2006/116764, опубликованной 2 ноября 2006 года, включенной посредством ссылки во всей своей полноте, описаны различные соединения и подробные схемы синтеза их получения. В частности, последовательность реакций показана на странице 79 указанной заявки, где 3-бензилокси-2-метил-1Н-пиридин-4-он формулы 3 бромируют до бромпиридина 4, который затем подвергают взаимодействию с метанолом и монооксидом углерода с получением метилового эфира никотиновой кислоты 5, который после нескольких стадий подвергают взаимодействию с бензиламином с образованием пиридина с амидсодержащей боковой цепью 10. Таким образом, амидная боковая цепь находится на месте перед образованием кольца Z1Z2 конечного продукта формулы (I) по реакции, показанной на странице 80, из соединения 16 до соединения 17-1.
Вторая последовательность реакций показана на странице 113 в WO 2006/116764, где пирролидиновому соединению 102 дают возможность конденсации до трициклического соединения 103, которое затем бромируют с получением соединения брома 104, которое затем подвергают взаимодействию с бензиламином с образованием трициклического соединения с амид-содержащей боковой цепью 105. Таким образом, бромирование происходит после образования кольца Z1Z2 конечного продукта формулы (I).
N-Метокси-N-метиламиды могут быть получены посредством Pd-катализируемого аминокарбонилирования арилбромидов, как описано J.R.Martinelli et al in Organic Letters, Vol.8, No.21, pages 4843-4846 (2006). Броманилины и броманизолы превращают в сложные эфиры, как описано J.Albaneze-Walker et al in Organic Letters, Vol.6, No.13, pages 2097-2100 (2004).
Краткое изложение сущности изобретения
Предложены способы, в которых использована ранняя стадия бромирования при получении соединений, полезных как обладающих ингибиторной активностью в отношении интегразы ВИЧ, как изложено в WO 2006/116764. Бромирование обеспечивает уходящую группу для присоединения амидной боковой цепи к пиридоновому кольцу.
Подробное описание изобретения
Предложен способ синтеза пиридонового соединения следующей формулы (АА), (ВВ) или (СС):


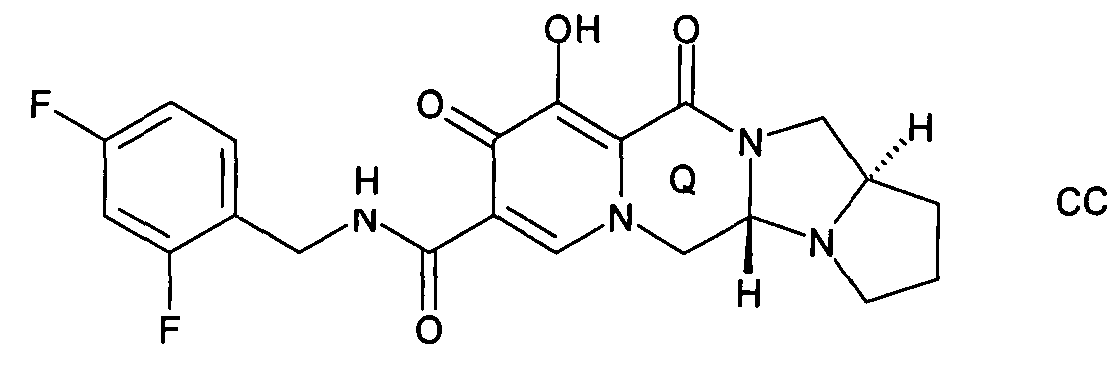
включающий стадии:
P-1) бромирования соединения следующей формулы (I-I) с получением соединения брома следующей формулы (II-II):

где R представляет собой -СНО, -СН(ОН)2, -CH(OH)(OR4), -CH(OH)-CH2OH или -CH(OR5)(OR6);
Р1 представляет собой Н или защитную группу гидроксила;
Р3 представляет собой Н или защитную группу карбоксила;
R4 представляет собой низший алкил;
R5 и R6 независимо представляют собой низший алкил или R5 и R6 могут представлять собой низший алкил и могут быть соединены с образованием 5-, 6- или 7-членного кольца,
и
Р-2) образования боковой цепи 2,4-ди-фторфенил-СН2-NН-С(0)- с использованием реагентов 2,4-ди-фторфенил-СН2-NН2 и монооксида углерода.
Термин «низший алкил», отдельно или в комбинации с любым другим термином, относится к насыщенному алифатическому углеводородному радикалу с прямой или разветвленной цепью, содержащему от 1 до 6 атомов углерода. Примеры алкильных радикалов включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изоамил, н-гексил и тому подобное, но не ограничиваются ими.
Термин «низший циклоалкил» относится к насыщенному или частично насыщенному карбоциклическому кольцу, состоящему из 3-6 атомов углерода в любой химически стабильной конфигурации. Примеры подходящих карбоциклических групп включают циклопропил, циклобутил, циклопентил, циклогексил и циклогексенил.
Термин «низший алкенил», отдельно или в комбинации с любым другим термином, относится к алкильной группе с прямой или разветвленной цепью с одной или двумя углерод-углеродными двойными связями. Примеры алкенильных радикалов включают этенил, пропенил, изопропенил, бутенил, изобутенил, пентенил, гексенил, гексадиенил и тому подобное, но не ограничиваются ими.
Термин «низший алкилен» относится к двухвалентному углеводородному радикалу с прямой или разветвленной цепью, предпочтительно имеющему от одного до шести атомов углерода, если не оговорено особо. Примеры «алкилена», как он использован здесь, включают метилен, этилен, пропилен, бутилен, изобутилен и тому подобное, но не ограничиваются ими.
Термин «низший алкенилен» относится к двухвалентному углеводородному радикалу с прямой или разветвленной цепью, имеющему одну или две углерод-углеродные двойные связи.
Термин «низший алкокси» относится к алкиловому эфирному радикалу, где термин «алкил» определен выше. Примеры подходящих алкиловых эфирных радикалов включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси и тому подобное, но не ограничиваются ими.
Термин «галоген» относится к фтору (F), хлору (Сl), брому (Вr) или иоду (I).
Термин «арил», отдельно или в комбинации с любым другим термином, относится к карбоциклической ароматической группировке (такой как фенил или нафтил), содержащей 6 атомов углерода, и более предпочтительно от 6 до 10 атомов углерода. Примеры арильных радикалов включают фенил, нафтил, инденил, азуленил, флуоренил, антраценил, фенантренил, тетрагидронафтил, инданил, фенантридинил и тому подобное, но не ограничиваются ими. Если не оговорено особо, термин «арил» также включает каждый возможный позиционный изомер ароматического углеводородного радикала, такой как 1-нафтил, 2-нафтил, 5-тетрагидронафтил, 6-тетрагидронафтил, 1-фенантридинил, 2-фенантридинил, 3-фенантридинил, 4-фенантридинил, 7-фенантридинил, 8-фенантридинил, 9-фенантридинил и 10-фенантридинил. Примеры арильных радикалов включают фенил, нафтил, инденил, азуленил, флуоренил, антраценил, фенантренил, тетрагидронафтил, инданил, фенантридинил и тому подобное, но не ограничиваются ими. Термин «аралкил» относится к алкильной группе, замещенной арилом. Примеры аралкильных групп включают бензил и фенетил, но не ограничиваются ими.
Термины «гетероциклическая группа» и «гетероцикл», как они использованы здесь, относятся к 3-7-членной моноциклической гетероциклической кольцевой или 8-11-членной бициклической гетероциклической кольцевой системе, любое кольцо которой является насыщенным, частично насыщенным или ненасыщенным и которая, в случае если она является моноциклической, может быть возможно конденсированной с бензольным кольцом. Каждый гетероцикл состоит из одного или более чем одного атома углерода и от одного до четырех гетероатомов, выбранных из группы, состоящей из N, О и S, и где гетероатомы азота и серы могут быть возможно окисленными, и атом азота может быть возможно четвертичным, включая любую бициклическую группу, в которой любое из определенных выше гетероциклических колец конденсировано с бензольным кольцом. Гетероциклическое кольцо может быть присоединено по любому атому углерода или гетероатому, при условии, что присоединение приводит к образованию стабильной структуры. Предпочтительные гетероциклы включают 5-7-членные моноциклические гетероциклы и 8-10-членные бициклические гетероциклы. Когда гетероциклическое кольцо имеет заместители, понятно, что заместители могут быть присоединены к любому атому в кольце, будь то гетероатом или атом углерода, при условии, что в результате получается стабильная химическая структура. «Гетероароматические группы» или «гетероарил» включены в гетероциклы, как они определены выше, и обычно относятся к гетероциклу, в котором кольцевая система представляет собой ароматический моноциклический или полициклический кольцевой радикал, содержащий от пяти до двадцати атомов углерода, предпочтительно от пяти до десяти атомов углерода, в котором один или более чем один кольцевой атом углерода, предпочтительно от одного до четырех, каждый замещен гетероатомом, таким как N, О, S и Р. Предпочтительные гетероарильные группы включают 5-6-членные моноциклические гетероарилы и 8-10-членные бициклические гетероарилы. Кроме того, в объем термина «гетероцикл», «гетероциклический» или «гетероциклил» включена группа, в которой неароматическое кольцо, содержащее гетероатом, конденсировано с одним или более чем одним ароматическим кольцом, например как в индолиниле, хроманиле, фенантридиниле или тетрагидрохинолиниле, где радикал или точка присоединения находится в неароматическом кольце, содержащем гетероатом. Если не оговорено особо, термин «гетероцикл», «гетероциклический» или «гетероциклил» также включает каждый возможный позиционный изомер гетероциклического радикала, например как 1-индолинил, 2-индолинил, 3-индолинил. Примеры гетероциклов включают имидазолил, имидазолиноил, имидазолидинил, хинолил, изохинолил, индолил, индазолил, индазолинолил, пергидропиридазил, пиридазил, пиридил, пирролил, пирролинил, пирролидинил, пиразолил, пиразинил, хиноксолил, пиперидинил, пиранил, пиразолинил, пиперазинил, пиримидинил, пиридазинил, морфолинил, тиаморфолинил, фурил, тиенил, триазолил, тиазолил, карболинил, тетразолил, тиазолидинил, бензофураноил, тиаморфолинилсульфон, оксазолил, оксадиазолил, бензоксазолил, оксопиперидинил, оксопирролидинил, оксоазепинил, азепинил, изоксозолил, изотиазолил, фуразанил, тетрагидропиранил, тетрагидрофуранил, тиазолил, тиадиазоил, диоксолил, диоксинил, оксатиолил, бензодиоксолил, дитиолил, тиофенил, тетрагидротиофенил, сульфоланил, диоксанил, диоксоланил, тетрагидрофуродигидрофуранил, тетрагидропиранодигидрофуранил, дигидропиранил, тетрагидрофурофуранил и тетрагидропиранофуранил.
Возможные заместители представляют собой гидрокси, галоген, амино и низший алкил.
Защитные группы могут быть выбраны из групп, известных специалистам в данной области техники, включая защитные группы, раскрытые в Greene, Theodora W.; Wuts, Peter G. M.. Protective Groups in Organic Synthesis. 2nd Ed. (1991), 473 pp. или Kocienski, Philip J. Protecting Groups. 3rd Ed. 2005, (2005), 679 pp.
Пиридоновое кольцо, изображенное в формуле (I-I) и (II-II), то есть к которому непосредственно присоединен -ОР1, превращается в формуле АА, ВВ и СС в кольцо, которое показано соседним с кольцом Q, а именно:
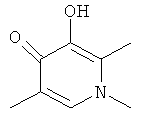
Таким образом, стадия (Р-2) может быть проведена до или после образования кольца Q, и такие стадии образования кольца Q описаны здесь и в WO 2006/1116764.
Согласно настоящему изобретению предложен способ, как описано выше, где указанную стадию (Р-2) проводят до образования кольца Q и где указанное пиридоновое соединение представляет собой соединение формулы АА, или формулы ВВ, или формулы СС.
Согласно настоящему изобретению предложен способ, как описано выше, где указанную стадию (Р-2) проводят после образования кольца Q и где указанное пиридоновое соединение представляет собой соединение формулы АА, или формулы ВВ, или формулы СС.
Согласно настоящему изобретению предложен способ, как описано выше, где указанное пиридоновое соединение представляет собой соединение формулы АА.
Согласно настоящему изобретению предложен способ, как описано выше, где указанное пиридоновое соединение представляет собой соединение формулы ВВ.
Согласно настоящему изобретению предложен способ, как описано выше, где указанное пиридоновое соединение представляет собой соединение формулы СС.
Кроме того, часть настоящего изобретения представляет собой новые промежуточные соединения, такие как соединения следующей ниже формулы (DD):

где Р1 является таким, как описано выше, в частности бензилом.
Предложен способ получения пиридонового соединения следующей формулы (АА), (ВВ) или (СС):



включающий стадии:
P-1) бромирования соединения следующей формулы (I-I) с получением соединения брома следующей формулы (II-II):

где R представляет собой -СНО, -СН(ОН)2, -CH(OH)(OR4), -CH(OH)-CH2OH или -CH(OR5)(OR6);
Р1 представляет собой Н или защитную группу гидроксила;
Р3 представляет собой Н или защитную группу карбоксила;
R4 представляет собой низший алкил;
R5 и R6 независимо представляют собой низший алкил или R5 и R6 могут представлять собой низший алкил и могут быть соединены с образованием 5-, 6- или 7-членного кольца,
и
Р-2) образования боковой цепи 2,4-ди-фторфенил-СН2-МН-С(0)- с использованием реагентов 2,4-ди-фторфенил-СН2-МН2 и монооксида углерода с получением соединения формулы Ill-Ill

P-3) конденсации и дебензилирования соединения формулы III-III с получением соединения формулы АА, ВВ или СС.
Согласно настоящему изобретению также предложены способы, как описано выше, где Р1 представляет собой бензил; Р3 представляет собой метил и R представляет собой -СНО, -CH(OH)(OR4), -CI(OR5)(OR5), где R4 и R5представляют собой низший алкил.
Кроме того, здесь описан способ получения соединения следующей формулы (I):
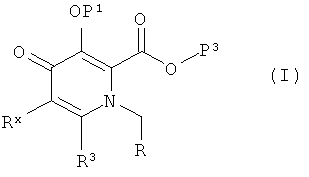
где R представляет собой -СНО, -СН(ОН)2 или -CH(OH)(OR4);
Р1 представляет собой Н или защитную группу гидроксила;
Р3 представляет собой Н или защитную группу карбоксила;
R3 представляет собой Н, галоген, гидрокси, возможно замещенный низший алкил, возможно замещенный циклоалкил, возможно замещенный низший алкенил, возможно замещенный низший алкокси, возможно замещенный низший алкенилокси, возможно замещенный арил, возможно замещенный арилокси, возможно замещенную гетероциклическую группу, возможно замещенный гетероциклокси и возможно замещенный амино;
R4 представляет собой низший алкил;
Rx представляет собой Н, галогено или R2-X-NR1-C(O)-;
R2 представляет собой возможно замещенный арил;
Х представляет собой простую связь, гетероатомную группу, выбранную из О, S, SO, SO2 и NH, или низший алкилен, или низший алкенилен, в каждый из которых может быть встроен гетероатом; и
R1 представляет собой Н или низший алкил, включающий стадии:
1) взаимодействия соединения формулы (II):

с амином формулы (III) или (IV):

где R5 и R6 независимо представляют собой низший алкил, или R5 и R6могут представлять собой алкил и могут быть соединены с образованием 5-, 6-или 7-членного кольца

с получением промежуточного соединения формулы (V) или (VI), соответственно:
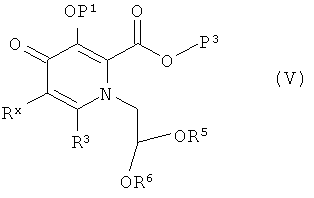

и
2) перефункционализации соединения формулы (V) или (VI) с получением соединения формулы (I).
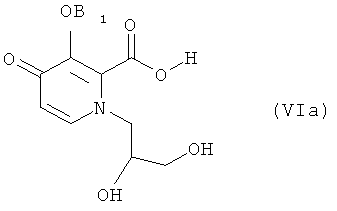
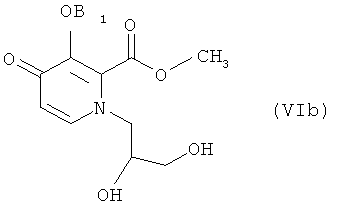
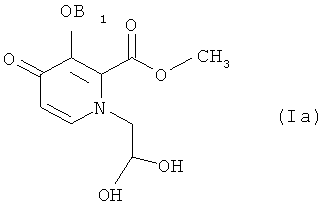
Конкретные соединения, используемые в этих способах, включают соединения следующих формул (IIа), (VIa), (VIb) и (Ia), использованные в разделе «Примеры» ниже:

Продукт (Iа) последовательности синтеза можно конденсировать с амином, например формулы H2NCH2CH2CH2OH, бромировать, если Rx представляет собой Н, карбонилировать и амидировать и, наконец, дебензилировать с получением соединения по WO 2006/116764, обозначенного (I-7) на странице 240, где (R)m представляет собой 4-F, и Ra представляет собой Н. Альтернативно, такое соединение может быть синтезировано, начиная с соединения формулы (I), где Rx представляет собой пара-F-фенил-СН2-NН-С(O)-, R3 представляет собой Н, Р1 представляет собой бензил (Вn) и Р3представляет собой защитную группу карбоксила.
Более конкретно, стадия (1) может быть проведена в протонном или апротонном растворителе, таком как ЕtOН, ТГФ (тетрагидрофуран) или ДМФА (диметилформамид), при температуре примерно 50-150ºС в течение примерно 1-10 часов.
Более конкретно, стадия (2) может быть проведена для исходного вещества диольного типа (VI) с использованием окислителя, такого как NalO4, RuO4 или Pb(OAc)4, в растворителе, таком как H2O, MeOH или СН3СN, при температуре окружающей среды в течение одного или более чем одного часа. Для исходного вещества ацетального типа, такого как (V), взаимодействие может быть проведено в кислоте, такой как НСl, СF3СООН или НСОН, возможно при нагревании.
Стадия (2) может также включать перефункционализацию по положению Rx, например Rx=H в Rx=Br возможно с последующей перефункционализацией в Rx=R2-X-NR1-C(O)-. Стадия (2) может также включать перефункционализацию Р3, например Р3=Н в Р3=Ме.
Более конкретно, стадия (Р-1) может быть проведена посредством обработки соединения формулы I-I источником брома, включая N-бромсукцинимид или бром, но не ограничиваясь ими, в растворителе, таком как N,N-диметилформамид, ТГФ или уксусная кислота и тому подобное. Это превращение может быть проведено, в частности, при температуре от -10ºС до 50ºС с получением соединения формулы II-II.
Более конкретно, стадия (Р-2) может быть проведена посредством обработки соединения формулы II-II 2,4-дифторфенил-СН2-NН2, монооксидом углерода, подходящим основанием и источником палладия(0) и возможно соответствующим лигандом в инертном растворителе, возможно при нагревании. Монооксид углерода может находиться при атмосферном давлении (14,7 фунт/кв.дюйм (101 кПа)) или при повышенном давлении, в частности в пределах вплоть до 60 фунт/кв.дюйм (413,7 кПа), но в некоторых случаях может потребоваться более высокое давление. Основания включают третичные амины, такие как диизопропилэтиламин и триэтиламин и тому подобное, но не ограничиваются ими. Неорганические основания, такие как ацетат калия и фосфат калия, также имеют важное значение. Подходящие источники Pd(0) включают тетракис(трифенилфосфин)палладий(0), но не ограничиваются им. В некоторых случаях Pd(II) предшественник может быть использован для получения Pd(0) in situ. Подходящие Pd(II) предшественники включают Pd(OAc)2, Pd(OCOCF3)2, но не ограничиваются ими, и лиганды включают Xantphos (9,9-диметил-4,6-бис(дифенилфосфино)ксантен), дифенилфосфиноферроцен (dppf), трифенилфосфин и тому подобное. Растворители включают N,N-диметилформамид, ТГФ, толуол, ДМСО (диметилсульфоксид) и тому подобное. Возможно используют нагревание смеси в пределах от температуры окружающей среды до 150ºС.
Согласно настоящему изобретению также предложены кристаллические формы соли соединения формулы АА (соединение 13, пример 1) и ее гидрата. Согласно настоящему изобретению предложены:
1) соль или гидрат соли соединения формулы АА:

2) кристаллическая форма натриевой соли или гидрата натриевой
соли соединения формулы АА:

3) кристаллическая форма соединения (2), имеющая одно или более чем одно физическое свойство, выбранное из группы, состоящей из (1) и (2):
1) имеющая характеристические дифракционные пики при углах 2-тета, равных 6,4º±0,2º; 9,2º±0,2º; 13,8º±0,2º; 19,2º±0,2º и 21,8º±0,2º, на картине дифракции рентгеновских лучей на порошке; и
2) имеющая характеристические инфракрасные спектры поглощения при 1641 см-1±2 см-1, 1536 см-1±2 см-1, 1503 см-1±2 см-1 и 1424 см-1±2 см-1;
4) кристаллическая форма соединения (2), имеющая характеристические дифракционные пики при углах 2-тета, равных 6,4º±0,2º; 9,2º±0,2º; 13,8º±0,2º; 19,2º±0,2º и 21,8º±0,2º, на картине дифракции рентгеновских лучей на порошке;
5) кристаллическая форма соединения (2), имеющая характеристические дифракционные пики при углах 2-тета, равных 6,4º±0,2º; 9,2º±0,2º; 13,8º±0,2º; 14,6º±0,2º; 15,2º±0,2º; 17,6º±0,2º; 19,2º±0,2º; 21,8º±0,2º; 24,1º±0,2º и 28,7º±0,2º, на картине дифракции рентгеновских лучей на порошке;
6) кристаллическая форма соединения (2), имеющая характеристические инфракрасные спектры поглощения при 1641 см-1±2 см-1, 1536 см-1±2 см-1, 1503 см-1±2 см-1 и 1424 см-1±2 см-1;
7) кристаллическая форма соединения (2), имеющая характеристические инфракрасные спектры поглощения при 1641 см-1±2 см-1, 1536 см-1±2 см-1, 1503 см-1±2 см-1, 1424 см-1±2 см-1, 1282 см-1±2 см-1, 1258 см-1±2 см-1, 1093 см-1±2 см-1 и 1069 см-1±2 см-1;
8) кристаллическая форма соединения (2), имеющая один или более чем один спектр, выбранный из группы, состоящей из (а)-(с):
a) картины дифракции рентгеновских лучей на порошке, по существу как показано на Фиг.1;
b) инфракрасных спектров поглощения, по существу как показано на Фиг.2; и
c)13С-ЯМР спектров в твердом состоянии, по существу как показано на Фиг.3.
9) кристаллическая форма соединения (2), имеющая одно или более чем одно физическое свойство, выбранное из группы, состоящей из (3) и (4):
3) имеющая характеристические дифракционные пики при углах 2-тета, равных 8,0º±0,2º; 9,3º±0,2º; 11,3º±0,2º; 16,0º±0,2º и 22,8º±0,2º, на картине дифракции рентгеновских лучей на порошке; и
4) имеющая характеристические инфракрасные спектры поглощения при 1637 см-1±2 см-1, 1536 см-1±2 см-1, 1501 см-1±2 см-1 и 1422 см-1±2 см-1;
10) кристаллическая форма соединения (2), имеющая характеристические дифракционные пики при углах 2-тета, равных 8,0º±0,2º; 9,3º±0,2º; 11,3º±0,2º; 16,0º±0,2º и 22,8º±0,2º, на картине дифракции рентгеновских лучей на порошке;
11) кристаллическая форма соединения (2), имеющая характеристические дифракционные пики при углах 2-тета, равных 8,0º±0,2º; 9,3º±0,2º; 11,3º±0,2º; 15,4º±0,2º; 16,0º±0,2º; 18,7º±0,2º; 19,1º±0,2º; 19,8º±0,2º; 22,8º±0,2º и 26,8º±0,2º, на картине дифракции рентгеновских лучей на порошке;
12) кристаллическая форма соединения (2), имеющая характеристические инфракрасные спектры поглощения при 1637 см-1±2 см-1, 1536 см-1±2 см-1, 1501 см-1±2 см-1 и 1422 см-1±2 см-1;
13) кристаллическая форма соединения (2), имеющая характеристические инфракрасные спектры поглощения при 1637 см-1±2 см-1, 1536 см-1±2 см-1, 1501 см-1±2 см-1, 1422 см-1±2 см-1, 1277 см-1±2 см-1, 1258 см-1±2 см-1, 1093 см-1±2 см-1 и 1069 см-1±2 см-1;
14) кристаллическая форма соединения (2), имеющая один или более чем один спектр, выбранный из группы, состоящей из (d) и (е):
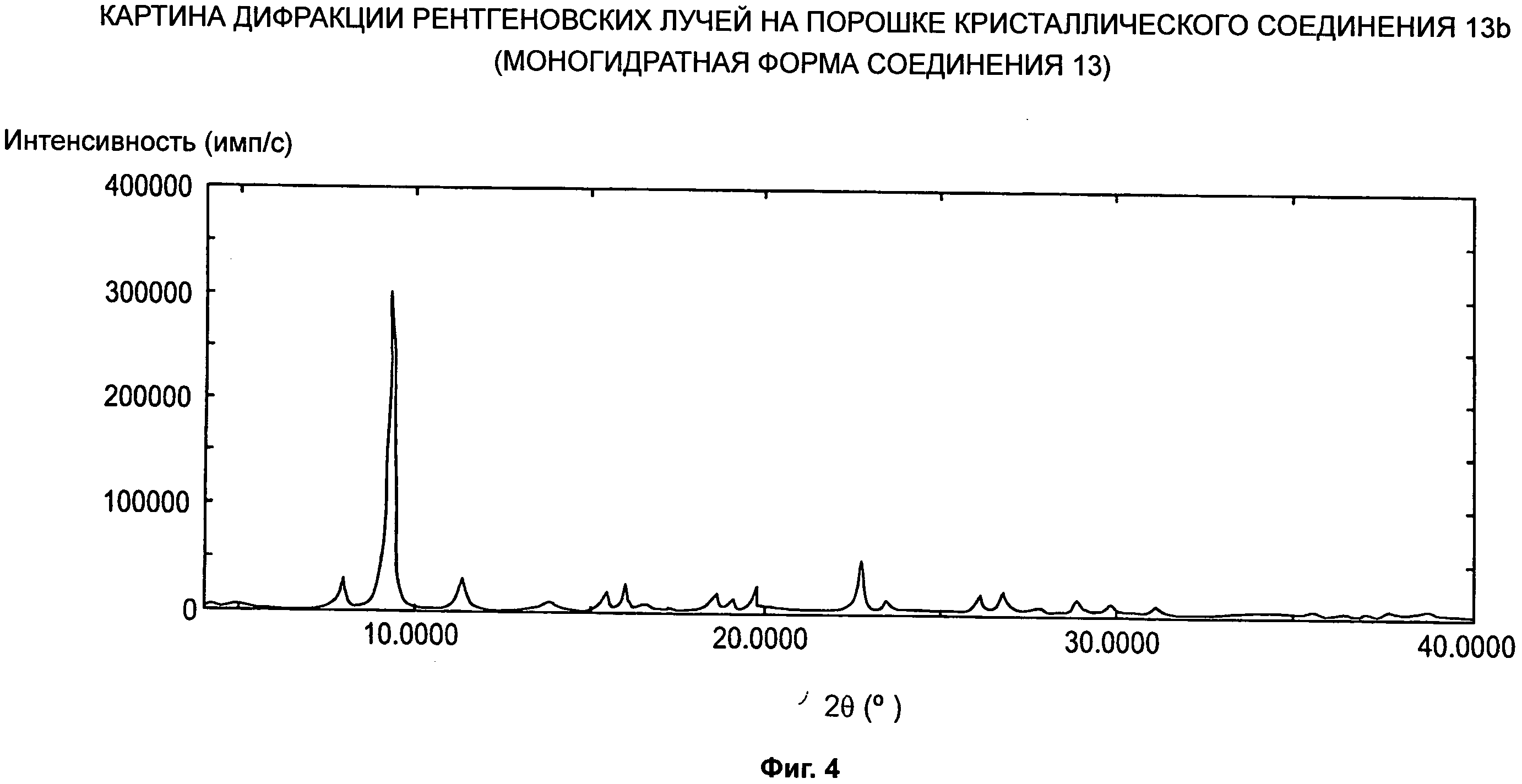
d) картины дифракции рентгеновских лучей на порошке, по существу как показано на Фиг.4; и
e) инфракрасных спектров поглощения, по существу как показано на Фиг.5;
15) фармацевтическая композиция, содержащая кристаллическую форму, как она определена в любом из (2)-(14);
16) способ получения кристаллических форм, как они определены в любом из (2)-(14).
Согласно настоящему изобретению предложены кристаллические формы соли соединения формулы АА, в частности натриевой соли.
Согласно настоящему изобретению предложены кристаллические формы гидрата соли соединения формулы АА, в частности натриевой соли.
Это изобретение также включает кристаллическую форму соединения формулы АА (соединение 12, пример 1). Подробности показаны в подпунктах с (17) по (22):
17) кристаллическая форма соединения формулы АА:

имеющая одно или более чем одно физическое свойство, выбранное из группы, состоящей из (5) и (б):
5) имеющая характеристические дифракционные пики при углах 2-тета, равных 5,4º±0,2º; 10,7º±0,2º; 12,3º±0,2º; 15,2º±0,2º и 16,4º±0,2º, на картине дифракции рентгеновских лучей на порошке; и
6) имеющая характеристические инфракрасные спектры поглощения при 1658 см-1±2 см-1, 1628 см-1±2 см-1, 1540 см-1±2 см-1 и 1498 см-1±2 см-1;
18) кристаллическая форма соединения формулы АА:

имеющая характеристические дифракционные пики при углах 2-тета, равных 5,4º±0,2º; 10,7º±0,2º; 12,3º±0,2º; 15,2º±0,2º и 16,4º±0,2º, на картине дифракции рентгеновских лучей на порошке;
19) кристаллическая форма соединения формулы АА:

имеющая характеристические дифракционные пики при углах 2-тета, равных 5,4º±0,2º; 10,7º±0,2º; 12,3º±0,2º; 14,3º±0,2º; 15,2º±0,2º; 16,4º±0,2º; 21,7º±0,2º; 24,9º±0,2º; 25,4º±0,2º и 27,9º±0,2º, на картине дифракции рентгеновских лучей на порошке;
20) кристаллическая форма соединения формулы АА:
имеющая характеристические инфракрасные спектры поглощения при 1658 см-1±2 см-1, 1628 см-1±2 см-1, 1540 см-1±2 см-1 и 1498 см-1±2 см-1;
21) кристаллическая форма соединения формулы АА:

имеющая характеристические инфракрасные спектры поглощения при 1658 см-1±2 см-1, 1628 см-1±2 см-1, 1540 см-1±2 см-1, 1498 см-1±2 см-1, 1355 см-1±2 см-1, 1264 см-1±2 см-1, 1238 см-1±2 см-1, 1080 см-1±2 см-1 и 1056 см-1±2 см-1;
22) кристаллическая форма соединения формулы AA:

имеющая один или более чем один спектр, выбранный из группы, состоящей из (О и (д):
f) картины дифракции рентгеновских лучей на порошке, по существу как показано на Фиг.6; и
g) инфракрасных спектров поглощения, по существу как показано на Фиг.7.
Кристаллы соединения 13 и 13b (моногидратная форма соединения 13) демонстрируют высокую растворимость в воде или физиологическом растворе, высокую биодоступность (БД), высокую максимальную концентрацию лекарственного средства (Сmах), короткое время достижения максимальной концентрации лекарственного средства (Тmах), высокую стабильность к действию тепла и света и/или легкость в обращении. Поэтому кристаллы соединения 13 и 13b подходят в качестве фармацевтических ингредиентов.
В следующих ниже примерах и в этом описании могут быть использованы следующие сокращения: Me (метил), Вn (бензил), водн. (водный), Et (этил), С (по Цельсию).
ПРИМЕРЫ
В следующие примеры, в которых показаны реакции бромирования и амидирования по настоящему изобретению, включены примеры С, пример 2 и пример СС.
Следующие примеры предназначены только для иллюстрации и не предназначены для ограничения объема изобретения.
Примеры 1 и 3
Исходным веществом в примере 1е и 3е является соединение формулы (На), которое также показано как соединение 5 ниже и соединение #101 на странице 113 в WO 2006/116764. Продукт, показанный ниже как соединение 8, является соединением формулы (I). Конечный продукт, показанный ниже как соединение 13, является соединением формулы (I-7) на странице 240 в WO 2006/116764, где (R)m представляет собой 2,4-ди-Р, и Ra представляет собой Н, при условии, однако, что в положении, обозначенном R16 в формуле (XXVI) на странице 65, находится альфа-метил.

Таким образом, в вышеприведенной последовательности по примеру 1 соединение 5 является идентичным соединению 101 на странице 113 в WO 2006/116764 и соединению формулы (IIа) способа по настоящему изобретению;
вышеприведенное соединение 6 является идентичным соединению формулы (VIa) способа по настоящему изобретению; вышеприведенное соединение 7 является идентичным соединению формулы (VIb) способа по настоящему изобретению; и соединение 8 является идентичным соединению формулы (1а) способа по настоящему изобретению. Стадия (1) способа по изобретению включает соединения с 5 по 6, приведенные выше, тогда как стадия (2) включает соединения с 6 по 8.
Пример 1а
К суспензии 2000 г соединения 1 (1,0 экв.) в 14,0 л MeCN добавляли 2848 г бензилбромида (1,05 экв.) и 2630 г К2СО3(1,2 экв.). Смесь перемешивали при 80ºС в течение 5 часов и охлаждали до 13ºС. Осадок отфильтровывали и промывали 5,0 л MeCN. Фильтрат концентрировали и к остатку добавляли 3,0 л ТГФ. Этот раствор ТГФ концентрировали с получением 3585 г неочищенного соединения 2 в виде масла. Соединение 2 использовали на следующей стадии без дополнительной очистки.
1H-ЯМР (300 МГц, CDCl3) δ 7.60 (d, J=5,7 Гц, 1Н), 7.4-7.3 (m, 5H), 6.37 (d, J=5,7 Гц, 1Н), 5.17 (s, 2H), 2.09 (s, 3H).
Пример 1b
К 904 г неочищенного соединения 2 добавляли 5,88 л ТГФ и этот раствор охлаждали до -60ºС. 5,00 л 1,0 М бис(триметилсилиламида) лития в ТГФ (1,25 экв.) по каплям добавляли в течение 2 часов к раствору соединения 2 при -60ºС. Затем добавляли раствор 509 г бензальдегида (1,2 экв.) в 800 мл ТГФ при -60ºС и эту реакционную смесь выдерживали при -60ºС в течение 1 часа. Раствор ТГФ выливали в смесь 1,21 л конц. HCl, 8,14 л ледяной воды и 4,52 л ЕЮАс при температуре ниже 2ºС. Органический слой промывали 2,71 л рассола (дважды) и водный слой экстрагировали 3,98 л ЕtOАс. Объединенные органические слои концентрировали. К смеси добавляли 1,63 л толуола и концентрировали (дважды) с получением толуольной суспензии соединения 3. Фильтрация, промывка 0,90 л холодного толуола и сушка дали 955 г соединения 3 (выход 74% от соединения 1) в виде кристаллического вещества.
1H-ЯМР (300 МГц, CDCl3) δ 7.62 (d, J=5,7 Гц, 1Н), 7.5-7.2 (m, 10Н), 6.38 (d, J=5,7 Гц, 1Н), 5.16 (d, J=11,4 Гц, 1Н), 5.09 (d, J=11,4 Гц, 1Н), 4.95 (dd, J=4,8, 9,0 Гц, 1Н), 3.01 (dd, J=9,0, 14,1 Гц, 1Н), 2.84 (dd, J=4,8, 14,1 Гц, 1Н).
Пример 1с
К раствору 882 г соединения 3 (1,0 экв.) в 8,82 л ТГФ добавляли 416 г Et3N (1,5 экв.) и 408 г метансульфонилхлорида (1,3 экв.) при температуре ниже 30ºС. После подтверждения того, что соединение 3 полностью израсходовано, к этой реакционной смеси при температуре ниже 30ºС добавляли 440 мл NMP и 1167 г DBU (1,8-диазабицикло[5.4.0]ундец-7-ен) (2,8 экв.) и реакционную смесь выдерживали в течение 30 минут. Смесь нейтрализовали 1,76 л 16% серной кислоты и органический слой промывали 1,76 л 2% водн. Na2SO3. После концентрирования органического слоя добавляли 4,41 л толуола и смесь концентрировали (три раза). После добавления 4,67 л гексана смесь охлаждали на ледяной бане. Фильтрация, промывка 1,77 л гексана и сушка дали 780 г соединения 4 (выход 94%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, CDCl3) δ 7.69 (d, J=5,7 Гц, 1Н), 7.50-7.25 (m, 10Н), 7.22 (d, J=16,2 Гц, 1Н), 7.03 (d, J=16,2 Гц, 1Н), 6.41 (d, J=5,7 Гц, 1Н), 5.27 (s, 2H).
Пример 1d
К смеси 822 г соединения 4 (1,0 экв.) и 11,2 г RuCl3·nH2O (0,02 экв.) в 2,47 л МеСN, 2,47 л ЕtOАс и 2,47 л H2O добавляли 2310 г NalO4 (4,0 экв.) при температуре ниже 25ºС. После выдерживания в течение 1 часа к этой смеси при температуре ниже 25ºС добавляли 733 г NaClO2 (3,0 экв.). После выдерживания в течение 1 часа осадок отфильтровывали и промывали 8,22 л ЕtOАс. К фильтрату добавляли 1,64 л 50% водн. Na2S2O3, 822 мл H2O и 630 мл конц. HCl. Водный слой экстрагировали 4,11 л ЕtOАс и органические слои объединяли и концентрировали. К остатку добавляли 4 л толуола и смесь концентрировали и охлаждали на водяной бане. Фильтрация, промывка 1 л толуола и сушка дали 372 г соединения 5 (выход 56%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, CDCl3) 5 7.78 (d, J=5,7 Гц, 1Н), 7.54-7.46 (m, 2H), 7.40-7.26 (m, 3H), 6.48 (d, J=5,7 Гц, 1Н), 5.6 (brs, 1Н), 5.31 (s, 2H).
Пример 1е
Смесь 509 г соединения 5 (1,0 экв.) и 407 г 3-амино-пропан-1,2-диола (2,5 экв.) в 1,53 л ЕtOН перемешивали при 65ºС в течение 1 часа и при 80ºС в течение 6 часов. После добавления 18,8 г 3-амино-пропан-1,2-диола (0,1 экв.) в 200 мл ЕtOН смесь перемешивали при 80ºС в течение 1 часа. После добавления 18,8 г 3-амино-пропан-1,2-диола (0,1 экв.) в 200 мл ЕtOН смесь перемешивали при 80ºС в течение 30 минут. После охлаждения и добавления 509 мл Н2O смесь концентрировали. К остатку добавляли 2,54 л H2O и 2,54 л AcOEt. После разделения водный слой промывали 1,02 л ЕtOАс. К водному слою добавляли 2,03 л 12% серной кислоты при температуре ниже 12ºС с получением кристаллического соединения 6. Фильтрация, промывка 1,53 л холодной Н2О и сушка дали 576 г соединения 6 (выход 83%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 7.67 (d, J=7,5 Гц, 1Н), 7.5-7.2 (m, 5H), 6.40 (d, J=7,5 Гц, 1Н), 5.07 (s, 2H), 4.2-4.0 (m, 1Н), 3.9-3.6 (m, 2H), 3.38 (dd, J=4,2, 10,8 Гц, 1Н), 3.27 (dd, J=6,0, 10,8 Гц, 1Н).
Пример 1f
К суспензии 576 г соединения 6 (1,0 экв.; содержит 5,8% Н2O) в 2,88 л NMP добавляли 431 г NаНСО3(3,0 экв.) и 160 мл метилиодида (1,5 экв.) и эту смесь перемешивали при комнатной температуре в течение 4 часов. После охлаждения до 5ºС к этой смеси при температуре ниже 10ºС добавляли 1,71 л 2 н. HCl и 1,15 л 20% водн. NaCl с получением кристаллического соединения 7. Фильтрация, промывка 1,73 л Н2O и сушка дали 507 г соединения 7 (выход 89%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 7.59 (d, J=7,5 Гц, 1Н), 7.40-7.28 (m, 5H), 6.28 (d, J=7,5 Гц, 1Н), 5.21 (d, J=5,4 Гц, 1Н), 5.12 (d, J=10,8 Гц, 1Н), 5.07 (d, J=10,8 Гц, 1Н), 4.83 (t, J=5,7 Гц, 1Н), 3.97 (dd, J=2,4, 14,1 Гц, 1Н), 3.79 (s, 3Н), 3.70 (dd, J=9,0, 14,4 Гц, 1Н), 3.65-3.50 (m, 1Н), 3.40-3.28 (m, 1Н), 3.26-3.14 (m, 1Н).
Пример 1q
К смеси 507 г соединения 7 (1,0 экв.) в 5,07 л MeCN, 5,07 л Н2O и 9,13 г АсОН (0,1 экв.) добавляли 390 г NalO4(1,2 экв.) и эту смесь перемешивали при комнатной температуре в течение 2 часов. После добавления 1,52 л 10% водн. Nа2S2O3 смесь концентрировали и охлаждали до 10ºС. Фильтрация, промывка Н2O и сушка дали 386 г соединения 8 (выход 80%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 7.62 (d, J=7,5 Гц, 1Н), 7.42-7.30 (m, 5H), 6.33 (d, J=6,0 Гц, 2H), 6.29 (d, J=7,5 Гц, 1Н), 5.08 (s, 2H), 4.95-4.85 (m, 1Н), 3.80 (s,3H),3.74(d,J=5,1 Гц, 2Н).
Пример 1h
После растворения смеси 378 г соединения 8 (1,0 экв.) в 3,78 л МеОН при нагревании этот раствор концентрировали. К остатку добавляли 1,51 л толуола и эту смесь концентрировали. К остатку добавляли 1,89 л толуола, 378 мл АсОН и 137 г (R)-3-амино-бутан-1-ола (1,3 экв.) и эту смесь нагревали до 90ºС, перемешивали при 90ºС в течение 2,5 часа и концентрировали. К остатку добавляли 1,89 л толуола и смесь концентрировали. Остаток экстрагировали 3,78 л и 1,89 л СНСl3 и промывали 2×1,89 л Н2O. Органические слои объединяли и концентрировали. К остатку добавляли 1,89 л ЕtOАс и эту смесь концентрировали. После добавления 1,89 л ЕtOАс, фильтрации, промывки 1,13 л ЕtOАс и сушки получили 335 г соединения 9 (выход 83%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, CDCl3) δ 7.70-7.58 (m, 2H), 7.40-7.24 (m, 3Н), 7.14 (d, J=7,5 Гц, 2H), 6.47 (d, J=7,5 Гц, 1Н), 5.35 (d, J=10,2 Гц, 1Н), 5.28 (d, J=10,2 Гц, 1Н), 5.12 (dd, J=3,9, 6,3 Гц, 1Н), 5.05-4.90 (m, 1Н), 4.07 (dd, J=3,9, 13,5 Гц, 1Н), 4.00-3.86 (m, 3Н), 2.23-2.06 (m, 1Н), 1.48 (ddd, J=2,4, 4,5, 13,8 Гц, 1Н), 1.30 (d, J=6,9 Гц, 3Н).
Пример 1i
К суспензии 332 г соединения 9 (1,0 экв.) в 1,66 л NMP добавляли 191 г NBS (N-бромсукцинимид) (1,1 экв.) и эту смесь перемешивали при комнатной температуре в течение 2 часов. После добавления 1,26 л Н2O смесь перемешивали в течение 30 минут. После добавления 5,38 л Н2O и выдерживания смеси при 10ºС в течение 30 минут и при 5ºС в течение 1 часа, фильтрации, промывки 1,33 л холодной Н2O и сушки получили 362 г соединения 10 (выход 89%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, CDCl3) δ 7.69-7.63 (m, 2H), 7.59 (s, 1Н), 7.38-7.24 (m, 3Н), 5.33 (d, J=10,2 Гц, 1Н), 5.25 (d, J=9,9 Гц, 1Н), 5.12 (dd, J=3,9, 5,7 Гц, 1Н), 5.05-4.90 (m, 1Н), 4.11 (dd, J=3,9, 13,2 Гц, 1Н), 4.02-3.88 (m, 3Н), 2.21-2.06 (m, 1Н), 1.49 (ddd, J=2,4, 4,5, 14,1 Гц, 1Н), 1.31 (d, J=6,9 Гц, 3Н).
Пример 1j
В атмосфере монооксида углерода смесь 33,5 г соединения 10 (1,0 экв.), 34,8 мл изо-Рr2NЕt (2,5 экв.), 14,3 мл 2,4-дифторбензиламина (1,5 экв.) и 4,62 г Pd(PPh3)4 (0,05 экв.) в 335 мл ДМСО перемешивали при 90ºС в течение 5,5 часов. После охлаждения осадок отфильтровывали и промывали 50 мл 2-пропанола. После добавления 502 мл Н2O и 670 мл AcOEt к фильтрату органический слой промывали 335 мл 0,5 н. водн. HCl и 335 мл Н2O и водный слой экстрагировали 335 мл AcOEt. Органические слои объединяли и концентрировали. К остатку добавляли 150 мл 2-пропанола и эту смесь концентрировали. После добавления 150 мл 2-пропанола, концентрирования, охлаждения до 20ºС и фильтрации получили неочищенное кристаллическое соединение 11. После растворения неочищенного кристаллического вещества в 380 мл ацетона с помощью нагревания осадок отфильтровывали и фильтрат концентрировали. После добавления 200 мл ЕtOН, концентрирования, добавления 150 мл ЕtOН, концентрирования, охлаждения и фильтрации получили неочищенное кристаллическое соединение 11. После растворения этого неочищенного кристаллического вещества в 450 мл ацетона с помощью нагревания раствор концентрировали. К остатку добавляли 150 мл 2-пропанола и смесь концентрировали (дважды). После охлаждения остатка, фильтрации, промывки 2-пропанолом и сушки получили 34,3 г соединения 11 (выход 84%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, CDCl3) δ 10.40 (t, J=6,0 Гц, 1Н), 8.35 (s, 1H), 7.66-7.58 (m, 2H), 7.42-7.24 (m, 5H), 6.78-6.74 (m, 2H), 5.30 (d, J=9,9 Гц, 1H), 5.26 (d, J=10,2 Гц, 1H), 5.15 (dd, J=3,9, 5,7 Гц, 1H), 5.05-4.90 (m, 1H), 4.64 (d, J=5,4 Гц, 2Н), 4.22 (dd, J=3,9, 13,5, 1H), 4.09 (dd, J=6,0, 13,2 Гц, 1H), 4.02-3.88 (m, 2H), 2.24-1.86 (m, 1H), 1.50 (ddd, J=2,4, 4,5, 14,1 Гц, 1H), 1.33 (d, J=7,2 Гц, 3Н).
Пример 1k
В атмосфере водорода смесь 28,0 г соединения 11 (1,0 экв.) и 5,6 г 10% Pd-C в 252 мл ТГФ и 28 мл МеОН перемешивали в течение 1 часа. После того как осадок (Pd-C) отфильтровали и промыли 45 мл ТГФ, добавляли 5,6 г 10% Pd-C и смесь перемешивали в течение 1,5 часа в атмосфере водорода. После того как Pd-C отфильтровали и промыли 150 мл смеси СНCl3/МеОН (9/1), фильтрат концентрировали. После растворения остатка в 1,38 л ЕtOН с помощью нагревания раствор постепенно охлаждали до комнатной температуры. После фильтрации фильтрат концентрировали и охлаждали. Фильтрация, промывка ЕЮН и сушка дали 21,2 г соединения 12 (выход 92%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 12.51 (s, 1H), 10.36 (t, J=5,7 Гц, 1 Н), 8.50 (s, 1H), 7.39 (td, J=8,7, 6,3 Гц, 1H), 7.24 (add, J=2,6, 9,5, 10,8 Гц, 1H), 7.12-7.00 (m, 1H), 5.44 (dd, J=3,9, 5,7 Гц, 1H), 4.90-4.70 (m, 1H), 4.65-4.50 (m, 1H), 4.54 (d, J=5,1 Гц, 2Н), 4.35 (dd, J=6,0, 13,8 Гц, 1H), 4.10-3.98 (m, 1H), 3.96-3.86 (m, 1H), 2.10-1.94 (m, 1H), 1.60-1.48 (m, 1H), 1.33 (d, J=6,9 Гц, 3Н).
Пример 1l
После растворения 18,0 г соединения 12 (1,0 экв.) в 54 мл ЕtOН с помощью нагревания с последующей фильтрацией к этому раствору при 80ºС добавляли 21,5 мл 2 н. водн. NaOH (1,0 экв.). Раствор постепенно охлаждали до комнатной температуры. Фильтрация, промывка 80 мл ЕtOН и сушка дали 18,8 г соединения 13 (выход 99%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 10.70 (t, J=6,0 Гц, 1H), 7.89 (s, 1H), 7.40-7.30 (m, 1H), 7.25-7.16 (m, 1H), 7.06-6.98 (m, 1H), 5.22-5.12 (m, 1H), 4.87-4.74 (m, 1H), 4.51 (d, J=5,4 Гц, 2Н), 4.35-4.25 (m, 1H), 4.16 (dd, J=1,8, 14,1 Гц, 1Н), 4.05-3.90 (m, 1H), 3.86-3.74 (m, 1H), 2.00-1.72 (m, 1H), 1.44-1.32 (m, 1H), 1.24 (d, J=6,9 Гц, 3Н).
Пример 1m
Пример 1m иллюстрирует способ получения кристаллического соединения 13b, которое является моногидратной формой соединения 13.
После растворения 30,0 г соединения 13 (1,0 экв.) в 600 мл раствора ТГФ-вода (8:2) при 30ºС к этому раствору добавляли 36,0 мл 2 н. водн. NaOH (1,0 экв.). Смесь перемешивали при комнатной температуре в течение 2 часов. Осадок отфильтровывали, промывали 150 мл раствора ТГФ-вода (8:2), 150 мл ТГФ. Сушка при 85ºС и установление требуемой влажности дали 30,4 г соединения 13b (моногидратная форма соединения 13, выход 93%) в виде кристаллического вещества.
Пример 3
Пример 3а
К суспензии 2000 г соединения 1 (1,0 экв.) в 14,0 л MeCN добавляли 2848 г бензилбромида (1,05 экв.) и 2630 г К2СО3(1,2 экв.). Смесь перемешивали при 80ºС в течение 5 часов и охлаждали до 13ºС. Осадок отфильтровывали и промывали 5,0 л MeCN. Фильтрат концентрировали и к остатку добавляли 3,0 л ТГФ. Этот раствор ТГФ концентрировали с получением 3585 г неочищенного соединения 2 в виде масла. Соединение 2 использовали на следующей стадии без дополнительной очистки.
1H-ЯМР (300 МГц, CDCl3) δ 7.60 (d, J=5,7 Гц, 1Н), 7.4-7.3 (m, 5H), 6.37 (d, J=5,7 Гц, 1Н), 5.17 (s, 2H), 2.09 (s, 3H).
Пример 3b
К 904 г неочищенного соединения 2 добавляли 5,88 л ТГФ и этот раствор охлаждали до -60ºС. 5,00 л 1,0 М бис(триметилсилиламида) лития в ТГФ (1,25 экв.) по каплям добавляли в течение 2 часов к раствору соединения 2 при -60ºС. Затем добавляли раствор 509 г бензальдегида (1,2 экв.) в 800 мл ТГФ при -60ºС и реакционную смесь выдерживали при -60ºС в течение 1 часа. Этот раствор ТГФ выливали в смесь 1,21 л конц. HCl, 8,14 л ледяной воды и 4,52 л ЕtOАс при температуре ниже 2ºС. Органический слой промывали 2,71 л рассола (дважды) и водный слой экстрагировали 3,98 л ЕtOАс. Объединенные органические слои концентрировали. К смеси добавляли 1,63 л толуола и концентрировали (дважды) с получением толуольной суспензии соединения 3. Фильтрация, промывка 0,90 л холодного толуола и сушка дали 955 г соединения 3 (выход 74% относительно соединения 1) в виде кристаллического вещества.
1H-ЯМР (300 МГц, CDCl3) δ 7.62 (d, J=5,7 Гц, 1Н), 7.5-7.2 (m, 10Н), 6.38 (d, J=5,7 Гц, 1Н), 5.16 (d, J=11,4 Гц, 1Н), 5.09 (d, J=11,4 Гц, 1Н), 4.95 (dd, J=4,8, 9,0 Гц, 1Н), 3.01 (dd, J=9,0, 14,1 Гц, 1Н), 2.84 (dd, J=4,8, 14,1 Гц, 1Н).
Пример 3с
К раствору 882 г соединения 3 (1,0 экв.) в 8,82 л ТГФ добавляли 416 г Еt3М (1,5 экв.) и 408 г метансульфонилхлорида (1,3 экв.) при температуре ниже 30ºС. После подтверждения того, что соединение 3 полностью израсходовано, к этой реакционной смеси при температуре ниже 30ºС добавляли 440 мл NMP и 1167 г DBU (2,8 экв.) и реакционную смесь выдерживали в течение 30 минут. Смесь нейтрализовали 1,76 л 16% серной кислоты и органический слой промывали 1,76 л 2% водн. Na2SO3. После концентрирования органического слоя добавляли 4,41 л толуола и смесь концентрировали (три раза). После добавления 4,67 л гексана смесь охлаждали на ледяной бане. Фильтрация, промывка 1,77 л гексана и сушка дали 780 г соединения 4 (выход 94%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, CDCl3) δ 7.69 (d, J=5,7 Гц, 1Н), 7.50-7.25 (m, 10Н), 7.22 (d, J=16,2 Гц, 1Н), 7.03 (d, J=16,2 Гц, 1Н), 6.41 (d, J=5,7 Гц, 1Н), 5.27 (s, 2H).
Пример 3d
К смеси 10,0 г соединения 4 и 13,6 мг RuCl3·nH2O в 95 мл MeCN и 10 мл воды добавляли смесь 155 мл воды, 7,2 г сероводородной кислоты и 15,5 г NalO4 в течение 2,5 часов при 20ºС. После выдерживания в течение 1 часа органический и водный слои разделяли и водный слой экстрагировали 30 мл этилацетата. Водный слой экстрагировали снова 30 мл этилацетата и органические слои объединяли. 6 мл 5% раствора NаНSO3 добавляли к объединенному органическому слою и слои разделяли. Органический слой доводили до pH 6,0 посредством добавления 4,0 г 2 М раствора NaOH и водный слой отделяли. После добавления 60 мл 5% раствора NаНСО3 и 257 мг TEMPO (2,2,6,6-тетраметил-1-пиперидинилокси) к этой реакционной смеси при 25ºС в течение 1 часа добавляли 25,9 г раствора NaClO и перемешивали в течение 30 минут, чтобы проконтролировать, что реакция закончилась. После разделения слоев добавляли 42,5 мл 5% раствора Na2SO3 и 30 мл AcOEt и разделяли. Водный слой экстрагировали 30 мл AcOEt и разделяли. К реакционной смеси при 20ºС в течение 1 часа добавляли 12% H2SO4 и смесь охлаждали до 5ºС. После перемешивания смеси в течение 30 минут смесь фильтровали, дважды промывали 30 мл воды и сушили с получением 5,7 г соединения 5 (выход 70%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, CDCl3) δ 7.78 (d, J=5,7 Гц, 1Н), 7.54-7.46 (m, 2H), 7.40-7.26 (m, 3H), 6.48 (d, J=5,7 Гц, 1Н), 5.6 (brs, 1Н), 5.31 (s, 2H).
Пример 3е
Смесь 509 г соединения 5 (1,0 экв.) и 407 г 3-амино-пропан-1,2-диола (2,5 экв.) в 1,53 л ЕtOН перемешивали при 65ºС в течение 1 часа и при 80ºС в течение 6 часов. После добавления 18,8 г 3-амино-пропан-1,2-диола (0,1 экв.) в 200 мл ЕtOН эту смесь перемешивали при 80ºС в течение 1 часа. После добавления 18,8 г 3-амино-пропан-1,2-диола (0,1 экв.) в 200 мл ЕtOН эту смесь перемешивали при 80ºС в течение 30 минут. После охлаждения и добавления 509 мл Н20 смесь концентрировали. К остатку добавляли 2,54 л H2O и 2,54 л AcOEt. После разделения водный слой промывали 1,02 л ЕtOАс. К водному слою добавляли 2,03 л 12% серной кислоты при температуре ниже 12ºС с получением кристаллического соединения 6. Фильтрация, промывка 1,53 л холодной Н20 и сушка дали 576 г соединения 6 (выход 83%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 7.67 (d, J=7,5 Гц, 1Н), 7.5-7.2 (m, 5H), 6.40 (d, J=7,5 Гц, 1Н), 5.07 (s, 2H), 4.2-4.0 (m, 1Н), 3.9-3.6 (m, 2H), 3.38 (dd, J=4,2, 10,8 Гц, 1Н), 3.27 (dd, J=6,0, 10,8 Гц, 1Н).
Пример 3f
К суспензии 576 г соединения 6 (1,0 экв.; содержит 5,8% Н2О) в 2,88 л NMP добавляли 431 г NаНСО3 (3,0 экв.) и 160 мл метилиодида (1,5 экв.), эту смесь перемешивали при комнатной температуре в течение 4 часов. После охлаждения до 5ºС к этой смеси при температуре ниже 10ºС добавляли 1,71 л 2 н. HCl и 1,15 л 20% водн. NaCl с получением кристаллического соединения 7. Фильтрация, промывка 1,73 л Н2O и сушка дали 507 г соединения 7 (выход 89%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 7.59 (d, J=7,5 Гц, 1Н), 7.40-7.28 (m, 5H), 6.28 (d, J=7,5 Гц, 1Н), 5.21 (d, J=5,4 Гц, 1Н), 5.12 (d, J=10,8 Гц, 1Н), 5.07 (d, J=10,8 Гц, 1Н), 4.83 (t, J=5,7 Гц, 1Н), 3.97 (dd, J=2,4, 14,1 Гц, 1Н), 3.79 (s, 3Н), 3.70 (dd, J=9,0, 14,4 Гц, 1Н), 3.65-3.50 (m, 1Н), 3.40-3.28 (m, 1Н), 3.26-3.14 (m, 1Н).
Пример 3g
К смеси 15,0 г соединения 7 (1,0 экв.) в 70,9 г MeCN добавляли смесь 60 мл Н2О, 2,6 г H2SO4 и 11,5 г NalO4 в диапазоне температур от 17ºС до 14ºС. После перемешивания реакционной смеси в течение 1 часа осадок отфильтровывали. Фильтрат добавляли в раствор 11,8 г натриевой соли аскорбиновой кислоты, 64 г воды и 60 мг H2SO4. После концентрирования смеси, охлаждения до 5ºС, фильтрации, промывки Н2O и сушки получили 12,9 г соединения 8 (выход 90%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 7.62 (d, J=7,5 Гц, 1Н), 7.42-7.30 (m, 5H), 6.33 (d, J=6,0 Гц, 2H), 6.29 (d, J=7,5 Гц, 1Н), 5.08 (s, 2H), 4.95-4.85 (m, 1Н), 3.80 (s, 3H), 3.74(d,J=5,1 Гц, 2Н).
Пример 3h
К смеси 10,0 г соединения 8 и 33,3 г диглима добавляли раствор 3,3 г (R)-3-амино-бутан-1-ола в 4,7 г диглима и 1,0 г уксусной кислоты при 60°С. После перемешивания реакционной смеси при 95ºС в течение 9 часов эту реакционную смесь охлаждали до -5ºС и фильтровали. Влажное кристаллическое вещество промывали и сушили с получением 8,3 г соединения 9 (78%).
Данные XRD (дифракции рентгеновских лучей):
1H-ЯМР (300 МГц, CDCl3) δ 7.70-7.58 (m, 2H), 7.40-7.24 (m, 3H), 7.14 (d, J=7,5 Гц, 2H), 6.47 (d, J=7,5 Гц, 1Н), 5.35 (d, J=10,2 Гц, 1Н), 5.28 (d, J=10,2 Гц, 1Н), 5.12 (dd, J=3,9, 6,3 Гц, 1Н), 5.05-4.90 (m, 1Н), 4.07 (dd, J=3,9, 13,5 Гц, 1Н), 4.00-3.86 (m, 3H), 2.23-2.06 (m, 1Н), 1.48 (ddd, J=2,4, 4,5, 13,8 Гц, 1Н), 1.30 (d, J=6,9 Гц, 3Н).
Пример 3i
К суспензии 5,7 г NBS в 26,5 г дихлорметана добавляли 10 г соединения 9 в 92,8 г дихлорметана при комнатной температуре. После перемешивания реакционной смеси в течение 6,5 часа, эту реакционную смесь добавляли к раствору 2,0 г Nа2SO3 и 40,3 г воды. Органический слой промывали разбавленным раствором NaOH и водой, дихлорметан концентрировали и заменяли на метанол. Смесь охлаждали до -5ºС и фильтровали и влажное кристаллическое вещество промывали и сушили с получением 10,3 г соединения 10 (84%).
1H-ЯМР (300 МГц, CDCl3) δ 7.69-7.63 (m, 2H), 7.59 (s, 1Н), 7.38-7.24 (m, 3Н), 5.33 (d, J=10,2 Гц, 1Н), 5.25 (d, J=9,9 Гц, 1Н), 5.12 (dd, J=3,9, 5,7 Гц, 1Н), 5.05-4.90 (m, 1Н), 4.11 (dd, J=3,9, 13,2 Гц, 1Н), 4.02-3.88 (m, 3H), 2.21-2.06 (m, 1Н), 1.49 (ddd, J=2,4, 4,5, 14,1 Гц, 1Н), 1.31 (d, J=6,9 Гц, 3Н).
Пример 3i
В атмосфере монооксида углерода смесь 25,0 г соединения 10, 11,6 г изо-Рr2NEt, 12,8 г 2,4-дифторбензиламина, 335 мг Pd(OAc)2 и 1,9 г 1,4-бис(дифенилфосфино)бутана в 188 мл DMA (диметилацетамид) перемешивали при 85ºС в течение 4 часов. После охлаждения реакционную смесь разделяли и 10/25 смеси использовали на следующей стадии. К этой реакционной смеси при 40ºС добавляли 6,6 г AcOEt, 29,9 г воды и 3 мг затравочных кристаллов. После перемешивания в течение 7 минут добавляли 29,9 г воды и охлаждали до комнатной температуры. Кристаллическое вещество отфильтровывали при комнатной температуре и промывали 47,2 г этанола с получением 10,1 г соединения 11 (выход 83%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, CDCl3) δ 10.40 (t, J=6,0 Гц, 1Н), 8.35 (s, 1H), 7.66-7.58 (m, 2H), 7.42-7.24 (m, 5H), 6.78-6.74 (m, 2H), 5.30 (d, J=9,9 Гц, 1H), 5.26 (d, J=10,2 Гц, 1H), 5.15 (dd, J=3,9, 5,7 Гц, 1H), 5.05-4.90 (m, 1H), 4.64 (d, J=5,4 Гц, 2Н), 4.22 (dd, J=3,9, 13,5, 1H), 4.09 (dd, J=6,0, 13,2 Гц, 1H), 4.02-3.88 (m, 2H), 2.24-1.86 (m, 1H), 1.50 (ddd, J=2,4, 4,5, 14,1 Гц, 1H), 1.33 (d, J=7,2 Гц, 3Н).
Пример 3k
В атмосфере водорода смесь 4,0 г соединения 11 и 0,8 г 5% Pd-C с влажностью 50% в 67,6 мл ТГФ и 1,6 мл H2O перемешивали в течение 1,5 часа при 50ºС. Затем к этой реакционной смеси добавляли смесь 80 мг NaHSO3 и 2,0 мл очищенной воды и реакционную смесь перемешивали в течение 1 часа, осадок отфильтровывали, промывали 20 мл ТГФ и фильтрат концентрировали до 11,97 г. После добавления 6,7 мл этанола и 33,6 мл очищенной воды в течение 1 часа реакционную смесь охлаждали до 25ºС. Фильтрация, промывка 26,9 мл ЕtOН и сушка дали 2,33 г соединения 12 (выход 82%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 12.51 (s, 1H), 10.36 (t, J=5,7 Гц, 1H), 8.50 (s, 1H), 7.39 (td, J=8,7, 6,3 Гц, 1H), 7.24 (ddd, J=2,6, 9,5, 10,8 Гц, 1H), 7.12-7.00 (m, 1H), 5.44 (dd, J=3,9, 5,7 Гц, 1H), 4.90-4.70 (m, 1H), 4.65-4.50 (m, 1H), 4.54 (d, J=5,1 Гц, 2H), 4.35 (dd, J=6,0, 13,8 Гц, 1H), 4.10-3.98 (m, 1H), 3.96-3.86 (m, 1H), 2.10-1.94 (m, 1H), 1.60-1.48 (m, 1H), 1.33 (d, J=6,9 Гц, 3Н).
Пример 3l
После растворения 18,0 г соединения 12 (1,0 экв.) в 54 мл ЕtOН с помощью нагревания с последующей фильтрацией к этому раствору при 80ºС добавляли 21,5 мл 2 н. водн. NaOH (1,0 экв.). Раствор постепенно охлаждали до комнатной температуры. Фильтрация, промывка 80 мл ЕtOН и сушка дали 18,8 г соединения 13 (выход 99%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 10.70 (t, J=6,0 Гц, 1Н), 7.89 (s, 1H), 7.40-7.30 (m, 1H), 7.25-7,16 (m, 1H), 7.06-6.98 (m, 1H), 5.22-5.12 (m, 1H), 4.87-4.74 (m, 1H), 4.51 (d, J=5,4 Гц, 2Н), 4.35-4.25 (m, 1H), 4.16 (dd, J=1,8, 14,1 Гц, 1H), 4.05-3.90 (m, 1H), 3.86-3.74 (m, 1H), 2.00-1.72 (m, 1H), 1.44-1.32 (m, 1H), 1.24 (d, J=6,9 Гц, 3Н).
Аппаратура и условия, использованные для составления графических материалов (Фиг.1-7), являются следующими.
Измерения картины дифракции рентгеновских лучей на порошке
Использованные условия измерения были такими же, как и общая метрология измерений по картине дифракции рентгеновских лучей на порошке, описанная в «Фармакопее Японии, издание пятнадцатое».
Измерительная аппаратура
Дифрактометр RINT TTR III
Методика
Режим сбора данных был следующим.
Метод измерения: метод параллельного пучка
Анод трубки: Сu
Излучение: Сu Кα
Ток генератора: 300 мА
Напряжение генератора: 50 кВ
Образец готовили на алюминиевой подложке.
Угол падения: 4º и 40º
Измерения при анализе методом инфракрасной спектроскопии
Режим сбора данных был следующим.
Измерительная аппаратура
Спектрофотометр FT/IR-4200 type А (от JASCO Corporation)
Методика
Метод измерения: метод НПО (нарушенного полного отражения)
Разрешение: 4 (см-1)
Детектор: DLATGS
Накопление: 32 сканирования
Измерение13С-ЯМР спектров в твердом состоянии
Спектр получали с использованием метода кросс-поляризации и вращения под магическим углом (CP/MAS). Режим сбора данных был следующим.
Измерительная аппаратура
Спектрометр: ЯМР-система Varian (частота 1Н: 599,8 МГц)
Методика
Проба: 3,2 мм Т3 НХ Probe
Ширина спектра: 43103,4 Гц
Время исследования: 0,04 с
Задержка повторного цикла: 10 с
Время контакта: 3 мс
Внешний стандарт: адамантан (углерод метина: 38,52 млн-1)
Температура: 10
Скорость MAS: 20 кГц.
Пример А
Исходным веществом в примере А является соединение 8, которое идентично соединению формулы (Ia). Таким образом, Пример А иллюстрирует способ получения промежуточного соединения для соединения формулы 17, приведенного ниже, которое является изомером соединения ZZ-2 на странице 237 в WO 2006/116764 Brian Johns et al.

Пример Аа
После растворения смеси 320 г соединения 8 (1,0 экв.) в 3,20 л МеОН с помощью нагревания раствор концентрировали. К остатку добавляли 1,66 л МеСN, 5,72 мл АсОН (0,1 экв.) и 82,6 г (5)-2-амино-пропан-1-ола (1,1 экв.) и эту смесь нагревали до 70ºС, перемешивали при 70ºС в течение 4 часов и концентрировали. К остатку добавляли 1,67 л 2-пропанола и эту смесь концентрировали (дважды). После охлаждения остатка, фильтрации, промывки 500 мл холодного 2-пропанола и сушки получили 167 г соединения 14 (выход 52%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, CDCl3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J=7,2, 1H), 5.46 (d, J=10,5 Гц, 1H), 5,23 (d, J=10,2 Гц, 1Н), 5.20 (dd, J=3,9, 9,6 Гц, 1H), 4.46-4.34 (m, 1H), 4.31 (dd, J=6,6, 8,7 Гц, 1H), 4.14 (dd, J=3,9, 12,3 Гц, 1H), 3.79 (dd, J=9,9, 12,3 Гц, 1H), 3.62 (dd, J=6,9, 8,7 Гц, 1H), 1.38 (d, J=6,3 Гц, 3Н).
Пример Ab
К суспензии 156 г соединения 14 (1,0 экв.) в 780 мл NMP добавляли 93,6 г NBS (1,1 экв.) и эту смесь перемешивали при комнатной температуре в течение 2,5 часов. Реакционную смесь добавляли в 3,12 л Н2O. Фильтрация, промывка 8,0 л Н2O и сушка дали 163 г соединения 15 (выход 84%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 8.37 (s, 1H), 7.55-7.50 (m, 2H), 7.42-7.25 (m, 3Н), 5.34 (dd, J=3,6, 9,9 Гц, 1H), 5.18 (d, J=10,8 Гц, 1H), 5.03 (d, J=10,5 Гц, 1H), 4.53 (dd, J=3,6, 12,0 Гц, 1H), 4.40-4.20 (m, 2H), 3.99 (dd, J=9,9, 11,7 Гц, 1H), 3.64 (dd, J=5,7, 8,1 Гц, 1H), 1.27 (d, J=6,3 Гц, 3Н).
Пример Ас
В атмосфере монооксида углерода смесь 163 г соединения 15 (1,0 экв.), 163 мл изо-Рr2Nеt (2,5 экв.), 68,4 мл 2,4-дифторбензиламина (1,5 экв.) и 22,5 г Pd(PPh3)4 (0,05 экв.) в 816 мл ДМСО перемешивали при 90ºС в течение 7 часов. После охлаждения, удаления осадка, промывки 50 мл ДМСО и добавления 11,3 г Pd(PPh3)4(0,025 экв.) реакционную смесь снова перемешивали при 90ºС в течение 2 часов в атмосфере монооксида углерода. После охлаждения, удаления осадка и добавления 2,0 л AcOEt и 2,0 л H2O органический слой промывали 1,0 л 1 н. водн. HCl и 1,0 л Н2О (дважды) и водный слой экстрагировали 1,0 л AcOEt. Органические слои объединяли и концентрировали. Колоночная хроматография остатка на силикагеле дала соединение 16 (выход 96%) в виде пены.
1H-ЯМР (300 МГц, CDCl3) δ 10.38 (t, J=6,3 Гц, 1Н), 8.39 (s, 1H), 7.75-7.25 (m, 7H), 6.90-6.70 (m, 2H), 5.43 (d, J=10,2 Гц, 1H), 5.24 (d, J=10,2 Гц, 1Н), 5.19 (dd, J=3,9, 9,9 Гц, 1Н), 4.63 (d, J=6,0 Гц, 2H), 4.50-4.25 (m, 3H), 3.86 (dd, J=9,9, 12,3 Гц, 1Н), 3.66 (dd, J=6,9, 8,4 Гц, 1Н), 1.39 (d, J=6,0 Гц, 3Н).
Пример Ad
В атмосфере водорода смесь 184 г соединения 16 (1,0 экв.) и 36,8 г 10% Pd-C в 3,31 л ТГФ и 0,37 л МеОН перемешивали в течение 3 часов. После фильтрации осадка (Pd-C), промывки смесью ТГФ/МеОН (9/1) и добавления 36,8 г 10% Pd-C эту смесь перемешивали в течение 20 минут в атмосфере водорода. После фильтрации осадка (Pd-C) и промывки смесью ТГФ/МеОН (9/1) фильтрат концентрировали. После добавления к остатку 200 мл AcOEt фильтрация дала неочищенное твердое соединение 17. Осадки объединяли и экстрагировали 4,0 л смеси СНCl3/МеОН (5/1). После концентрирования этого раствора СНСl3/МеОН и добавления к остатку 250 мл AcOEt фильтрация дала неочищенное твердое соединение 17. Неочищенные твердые вещества объединяли и растворяли в 8,2 л смеси MeCN/H2O (9/1) с помощью нагревания. После фильтрации фильтрат концентрировали. К остатку добавляли 1,5 л ЕtOН и смесь концентрировали (три раза). После охлаждения остатка, фильтрации и сушки получили 132 г соединения 17 (выход 88%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 11.47 (brs, 1Н), 10.31 (t, J=6,0 Гц, 1Н), 8.46 (s, 1Н), 7.40 (td, J=8,6, 6,9 Гц, 1Н), 7.24 (ddd, J=2,6, 9,4, 10,6, 1Н), 7.11-7.01 (m, 1Н), 5.39 (dd, J=4,1, 10,4 Гц, 1Н), 4.89 (dd, J=4,2, 12,3 Гц, 1Н), 4.55 (d, J=6,0 Гц, 2H), 4.40 (dd, J=6,8, 8,6 Гц, 1Н), 4.36-4.22 (m, 1Н), 4.00 (dd, J=10,2, 12,3 Гц, 1Н), 3.67 (dd, J=6,7, 8,6 Гц, 1Н), 1.34 (d, J=6,3 Гц, 3Н).
Пример Ае
После растворения 16,0 г соединения 17 (1,0 экв.) в 2,56 л ЕtOН и 0,64 л Н2О с помощью нагревания с последующей фильтрацией к этому раствору при 75ºС добавляли 39 мл 1 н. водн. NaOH (1,0 экв.). Раствор постепенно охлаждали до комнатной температуры. Фильтрация, промывка 80 мл ЕtOН и сушка дали 13,5 г соединения 18 (выход 80%) в виде кристаллического вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ 10.73 (t, J=6,0 Гц, 1Н), 7.89 (s, 1H), 7.40-7.30 (m, 1H), 7.25-7.16 (m, 1H), 7.07-6.98 (m, 1H), 5.21 (dd, J=3,8, 10,0 Гц, 1H), 4.58 (dd, J=3,8, 12,1 Гц, 1H), 4.51 (d, J=5,4 Гц, 2Н), 4.30-4.20 (m, 2H), 3.75 (dd, J=10,0, 12,1 Гц, 1H), 3.65-3.55 (m, 1H), 1.27 (d, J=6,1 Гц, 3Н).
Пример В
В этом примере использован способ включения кольцевого атома азота вместо атома кислорода в пироновое кольцо и образования эквивалента альдегида посредством катализируемого осмием окисления двойной связи. Таким образом, этот пример не является бромированием по изобретению.

Пример Ва
К бромбензольному (238 мл) раствору соединения А (23,8 г; 110 ммоль) добавляли диоксид селена (24,4 г; 220 ммоль). Эту реакционную смесь перемешивали в течение 13 часов при 140ºС с удалением воды с помощью насадки Дина-Старка. Нерастворимые частицы удаляли посредством фильтрации после охлаждения и растворитель выпаривали. К остатку добавляли толуол и осадки отфильтровывали. После концентрирования под вакуумом остаток очищали посредством колоночной хроматографии на силикагеле (гексан/этилацетат). Соединение В (16,5 г; 65%) получили в виде желтого масла.
1H-ЯМР (CDCl3) δ: 5.51 (2Н, s), 6.50 (1H, d, J=5,4 Гц), 7.36 (5H, s), 7.75 (1H, d, J=5,4 Гц), 9.88(1H, s).
Пример Bb
К охлажденному на льду водному (465 мл) раствору хлорита натрия (38,4 г; 424 ммоль) и сульфаминовой кислоты (54,9 г; 566 ммоль) добавляли ацетоновый (465 мл) раствор соединения В (46,5 г; 202 ммоль) и эту смесь перемешивали в течение 40 минут при комнатной температуре. После удаления ацетона под вакуумом осадки собирали посредством фильтрации и промывали холодной водой. Соединение С (42,8 г; 86%) получили в виде бесцветного кристаллического вещества.
1H-ЯМР (ДМСО-d6) δ: 5.12 (2Н, s), 6.54 (1H, d, J=5,6 Гц), 7.33-7.46 (5H, m), 8.20(1H, d, J=5,6 Гц).
Пример Be
Этанольный (17 мл) раствор аллиламина (13,2 г; 231 ммоль) добавляли к этанольной (69 мл) суспензии соединения С (17,2 г; 70 ммоль), затем смесь перемешивали в течение 4,5 часов при 50ºС и в течение 3 часов при 75ºС. К охлажденной реакционной смеси добавляли 2 н. соляную кислоту и лед и осадки собирали посредством фильтрации. Соединение D получили в виде бесцветного кристаллического вещества.
1H-ЯМР (CDCl3) δ: 4.37 (2Н, brs), 4.95 (2Н, s), 5.26-5.39 (2Н, m), 5.81-5.94 (1H, m), 6.32 (1H, dd, J=0,8, 7,2 Гц), 7.29-7.37 (3Н, m), 7.48-7.51 (2Н, m), 7.99 (1H, dd,J=0,8, 7,6 Гц), 8.11 (1H, brs).
Пример Bd
К ацетонитрильной (146 мл) суспензии соединения D (14,6 г; 51 ммоль) добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (15,5 г; 102 ммоль) и метилиодид (18,2 г; 128 ммоль) и эту смесь перемешивали в течение 15 часов при комнатной температуре. После выпаривания растворителя остаток очищали посредством колоночной хроматографии на силикагеле (хлороформ/метанол). Соединение Е (14,2 г; 93%) получили в виде бесцветного твердого вещества.
1H-ЯМР (CDCl3) δ: 3.75 (3Н, s), 4.40 (2H, d, J=5,7 Гц), 5.16-5.35 (2H, m), 5.29 (2H, s), 5.81-5.94 (1H, m), 6.62 (1H, d, J=7,5 Гц), 7.27-7.42 (6Н, m).
Пример Be
К диэтилэфирному (390 мл) раствору соединения Е (13,3 г; 44 ммоль) добавляли дигидрат осмата калия(VI) (1,62 г; 4,4 ммоль) и метапериодат натрия (28,1 г; 132 ммоль). Смесь перемешивали в течение 2,5 часов при комнатной температуре и осадки собирали посредством фильтрации. Собранное твердое вещество растворяли в смеси хлороформ-метанол и нерастворимые частицы отфильтровывали. Концентрирование под вакуумом дало неочищенный продукт соединения F (14,3 г).
1Н-ЯМР (ДМСО-d6) δ: 3.23 (3Н, s), 3.82 (3Н, s), 3.87 (2H, t, J=4,4 Гц), 4.62 (1H, dd, J=11,7, 4,8 Гц), 5.11 (2H, s), 6.31 (1H, d, J=7,5 Гц), 6.78 (1H, d, J=6,6 Гц), 7.33-7.40 (5H, m), 7.64 (1H, d, J=7,5 Гц).
Пример Bf
К хлороформному (108 мл) и метанольному (12 мл) раствору соединения F (11,7 г; неочищенный продукт) добавляли 3-аминопропанол (2,77 г; 36,9 ммоль) и уксусную кислоту (1,2 мл) и эту смесь перемешивали в течение 90 минут при 70ºС. После концентрирования под вакуумом остаток очищали посредством колоночной хроматографии на силикагеле (хлороформ/метанол). Соединение G (8,48 г; 72% за две стадии) получили в виде бесцветного кристаллического вещества.
1H-ЯМР (CDCl3) δ: 1.54-1.64 (1H, m), 1.85-2.01 (1H, m), 3.00 (1H, dt, J=3,6, 12,9 Гц), 3.74 (1H, dt, J=2,7, 12,3 Гц), 3.93 (1H, dd, J=5,1, 13,5 Гц), 4.07-4.21 (2H, m), 4.63-4.69 (1H, m), 4.94 (1H, t, J=4,8 Гц), 5.25 (2H, dd, J=10,2, 24,6 Гц), 6.56 (1H, d, J=7,5 Гц), 7.22-7.38 (5H, m), 7.63-7.66 (2H, m).
Пример Вg
К уксуснокислому (93 мл) раствору соединения G (6,1 г; 18,7 ммоль) по каплям добавляли уксуснокислый (31 мл) раствор брома (1,44 мл; 28,0 ммоль) в течение 15 минут. Смесь перемешивали в течение 3 часов при комнатной температуре. После добавления 5% водного гидросульфита натрия (8 мл) по каплям в течение 20 минут добавляли 2 н. гидроксид натрия (500 мл). Осадки собирали посредством фильтрации и промывали смесью дихлорметана и диизопропилового эфира. Соединение Н (6,02 г; 79%) получили в виде бесцветного кристаллического вещества.
1H-ЯМР (ДМСО-d6) δ: 1.55-1.74 (2Н, m), 3.12 (1Н, dt, J=3,0, 12,3 Гц), 3.84 (1Н, dt, J=2,7, 11,7 Гц), 4.00-4.05 (1Н, m), 4.20-4.26 (1Н, m), 4.40-4.46 (2Н, m), 5.03 (2Н, s), 5.15-5.17 (1Н, m), 7.31-7.40 (3H, m), 7.56-7.58 (2Н, m), 8.39 (1Н, s).
Пример Bh
К диметилсульфоксидному (1,42 мл) раствору соединения Н (71 мг; 0,175 ммоль) и тетракис(трифенилфосфин)палладия(0) (25 мг; 0,035 ммоль) добавляли 4-фторбензиламин (0,06 мл; 0,525 ммоль) и диизопропиламин (0,15 мл; 0,875 ммоль), затем эту смесь перемешивали в атмосфере монооксида углерода в течение 5 часов при 80ºС. После охлаждения добавляли насыщенный водный хлорид аммония и смесь экстрагировали этилацетатом. Экстракт промывали водой и сушили безводным сульфатом натрия. Растворитель удаляли под вакуумом и остаток очищали посредством колоночной хроматографии на силикагеле (этилацетат/метанол). Соединение I (74,5 мг; 89%) получили в виде бесцветного кристаллического вещества.
1H-ЯМР (ДМСО-d6) δ: 1.58-1.74 (2Н, m), 3.10-3.18 (1Н, m), 3.80-3.88 (1Н, m), 4.02-4.07 (1Н, m), 4.43-4.59 (5H, m), 5.05 (2Н, s), 5.20 (1Н, t, J=3,9 Гц), 7.13-7.19 (2Н, m), 7.32-7.40 (5H, m), 7.56-7.59 (2Н, m), 8.61 (1Н, s).
Пример С
Синтез метил-5-бром-1 -[2-гидрокси-2-(метилокси)этил]-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилата (в равновесии с соответствующим альдегидом).
Данный пример С демонстрирует перефункционализацию соединения 6, как показано выше в Примере 1 (соединение формулы (VI)), включая бромирование по положению Rx, с получением конечных продуктов 20 и 21 (соединение формулы (I)). Такие соединения с Вr по положению Rx могут быть подвергнуты взаимодействию, как в Примерах 1 и 2, с присоединением боковой цепи R2-X-NR1-C(O)-.

Пример Са
Метил-1-(2,3-дигидроксипропил)-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилат

В реактор загружали 1-(2,3-дигидроксипропил)-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоновую кислоту 6 (4,302 кг; 13,47 моль) с последующей загрузкой NаНСО3 (1,69 кг; 20,09 моль) и 242 г деионизированной воды. К этому добавляли 21,4 кг NMP, смесь перемешивали и температуру доводили до 28-35ºС. К этой реакционной смеси в течение 1-3 часов добавляли по каплям через капельную воронку диметилсульфат (2,34 кг; 18,30 моль), поддерживая температуру при 28-33ºС. Суспензию перемешивали при этой температуре в течение 14 часов. Когда процесс сочли завершенным, реакционную смесь охлаждали до 5ºС или ниже и смесь нейтрализовали до pH 6 посредством добавления HCI (561 мл конц. HCl в 2806 г деионизированной воды). В реакционный сосуд медленно загружали холодный 20% рассол, состоящий из 8,7 кг NaCl, 20 кг деионизированной воды и 14,8 кг льда при максимальной температуре 10ºС. Смесь перемешивали при 0-10ºС в течение 2,5 часов. Суспензию фильтровали под вакуумом и осадок на фильтре промывали 15 кг деионизированной воды два раза. Влажный твердый продукт сушили при 45-55ºС под вакуумом до достижения постоянной массы. Требуемый продукт, метил-1 -(2,3-дигидроксипропил)-4-оксо-3-[(фенилметил)-окси]-1,4-дигидро-2-пиридинкарбоксилат 7, получили в виде светло-желтого твердого вещества (3,77 кг; чистота 99,42% по ВЭЖХ; 84%).
1H-ЯМР (300 МГц, ДМСО-d6) δ 7.60 (d, J=7,5 Гц, 1Н), 7.36 (m, 5 H), 6.28 (d, J=7,5 Гц, 1Н), 5.23 (d, J=5,4 Гц, 1Н), 5.10 (Abq, J=10,8 Гц, 2H), 4.85 (m, 1Н), 3.98 (dd, J=14,3, 2,4 Гц, 1Н), 3.79 (s, 3H), 3.70 (dd, J=14,3, 9,0 Гц, 1Н), 3.58 (m, 1Н), 3.23 (m, 1Н).
Пример Cb
Метил-5-бром-1-(2,3-дигидроксипропил)-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилат

В реактор загружали метил-1-(2,3-дигидроксипропил)-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилат 7 (3,759 кг; 11,27 моль) и 188 л ДМФА. К этой перемешиваемой смеси при 18-20ºС добавляли N-бромсукцинимид (2,220 кг; 12,47 моль) в течение 20 минут через насыпную воронку. Полученную смесь перемешивали при комнатной температуре в течение 16 часов. На этот момент было установлено посредством ВЭЖХ наличие менее 1% исходного вещества. Половинную партию этой смеси обрабатывали посредством охлаждения до 10ºС и добавляли смесь лед/вода (12 кг льда в 35 кг деионизированной воды) и эту смесь перемешивали, затем фильтровали. Эту процедуру повторяли для второй половинной партии. Объединенный осадок на фильтре промывали 14 л воды и сушили в вакуумной печи с получением 4,033 кг метил-5-бром-1-(2,3-дигидроксипропил)-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилата 19 (91,6%) в виде желтоватого порошка с чистотой 99,2% по ВЭЖХ.
1H-ЯМР (300 МГц, Метанол-d4) δ 8.21 (s, 1H), 7.41-7.33 (m, 5H), 5.16 (s, 2H), 4.17 (dd, J=14,3, 2,4 Гц, 1H), 3.90 (dd, J=14,3, 9,0 Гц, 1H), 3.81 (s, 3H), 3.78 (m,1),3.52(dd,J=11,3,4,8rH, 1H), 3.41 (dd, J=11,3, 6,3 Гц, 1Н).
Пример Сс
Метил-5-бром-1-[2-гидрокси-2-(метилокси)этил]-4-оксо-3-[(фенилметил)-окси]-1,4-дигидро-2-пиридинкарбоксилат (в равновесии с соответствующим альдегидом)

В реактор загружали периодат натрия (1,67 кг; 7,8 моль) и 44 л деионизированной воды. К этой перемешиваемой смеси добавляли 8,5 кг льда. Ее перемешивали до тех пор, пока весь лед не расплавился и температура смеси не составила 1,4ºС. К этому добавляли метил 5-бром-1-(2,3-дигидроксипропил)-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридин-карбоксилат 19 (2,73 кг; 6,62 моль) через насыпную воронку. Смесь оставляли для нагревания до комнатной температуры и суспензию перемешивали в течение 16 часов. Образец контролировали посредством1H ЯМР, и было показано, что исходное вещество израсходовано. Эту смесь фильтровали и осадок на фильтре промывали 20 кг деионизированной воды. Эту процедуру повторяли до тех пор, пока не получали отрицательный результат по йодкрахмальной бумаге (4×20 л промывки). Твердые вещества сушили в вакуумной печи при 45-55ºС с получением метил-5-бром-1-(2,2-дигидрокси-этил)-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилата 20 (2,176 кг; 88%) в виде смеси с соответствующей альдегидной формой 21. Установили, что чистота составила 99,5% по ВЭЖХ.
1H-ЯМР (300 МГц, ацетон-d6) δ 8.12 (s, 1H), 7.49-7.30 (m, 5H), 5.56 (dd, J=6,0, 2,4 Гц, 1H), 5.23 (m, 1H), 5.20 (s, 2H), 3.97 (d, J=5,1 Гц, 2H), 3.87 (s, 3H).
Пример 2
Метил-5-({[(2,4-дифторфенил)метил]амино}карбонил)-1-[2-гидрокси-2-(метилокси)этил]-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилат (в равновесии с соответствующим альдегидом)
Этот пример демонстрирует взаимодействие соединения 5 формулы (II) с одним из соединений формулы (III) на стадии (1) и стадию перефункционализации (2) соединений формулы (V) (соединения 22, 23, 24 и 25) с получением соединений формулы (I).

Пример 2а
1-[2,2-Бис(метилокси)этил]-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоновая кислота
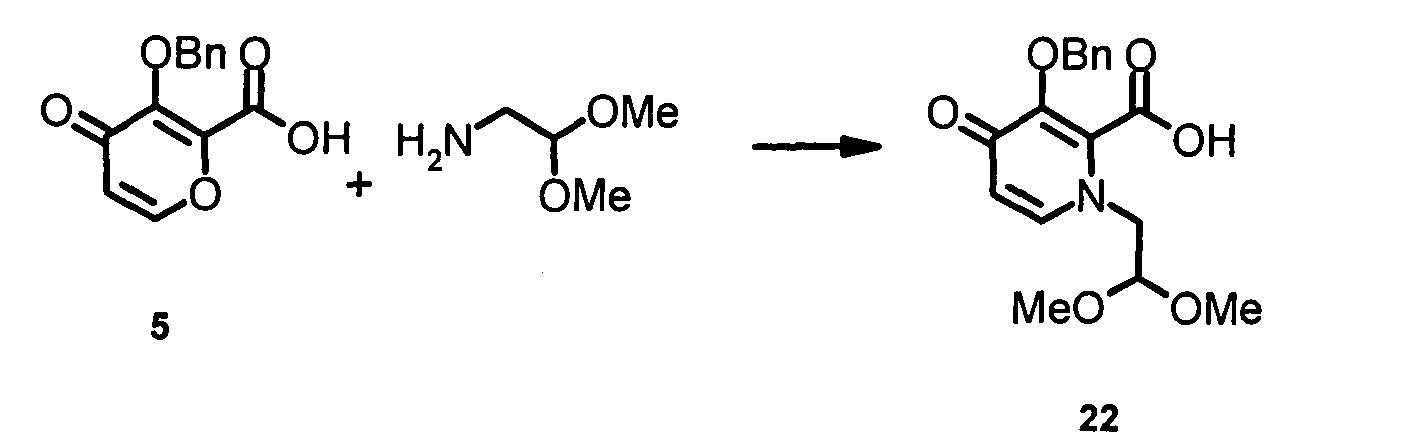
В колбу (1 л), загруженную 500 мл безводного этанола, добавляли 49,2 г (0,2 моль) 4-оксо-3-[(фенилметил)окси]-4Н-пиран-2-карбоновой кислоты 5.
Суспензию медленно нагревали до 55~60ºС, затем добавляли диметилацеталь 2-амино-ацетальдегида (84,1 г; 0,8 моль). Реакционную смесь доводили до 65ºС и дополнительно перемешивали в течение 18 часов. Растворитель удаляли при пониженном давлении с получением 1-[2,2-бис(метилокси)этил]-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридин-карбоновой кислоты 22 (неочищенная) в виде коричневого масла, которую использовали непосредственно на следующей стадии.
Пример 2b
Метил-1-[2,2-бис(метилокси)этил]-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилат

Неочищенную 1-[2,2-бис(метилокси)этил]-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоновую кислоту 22, полученную, как описано выше, растворяли в ДМФА (500 мл). К этому раствору добавляли NаНСО3 (50,5 г; 0,6 моль). Суспензию интенсивно перемешивали механической мешалкой при одновременном введении СН3I в ТВМЕ (тpeт-бутилметиловый эфир) (2,0 М; 300 мл) посредством капельной воронки в течение 30 минут. После добавления реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь затем разбавляли ЕtOАс (примерно 1,5 л) и промывали водой и рассолом. Органический слой сушили над безводным Na2SO4. Выпаривание растворителей дало метил-1-[2,2-бис(метилокси)этил]-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилат 23 в виде коричневого масла, который использовали непосредственно на следующей стадии.
Пример 2с
Метил-1-[2,2-бис(метилокси)этил]-5-бром-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилат

В колбу на 2 л, снабженную механической мешалкой, загружали метил-1-[2,2-бис(метилокси)этил]-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридин-карбоксилат 23, который получен выше, и 500 мл дихлорметана. В эту колбу порциями добавляли NBS (30 г; 0,17 моль). Реакционную смесь перемешивали при комнатной температуре до завершения взаимодействия (контролировали посредством ТСХ (тонкослойной хроматографии), примерно 6 часов). Эту смесь затем разбавляли дихлорметаном и промывали NaHCO3 (ss). Органическую фазу сушили над Na2SO4, затем растворители выпаривали. Неочищенный продукт очищали посредством колоночной хроматографии (силикагель, ЕtOН/ДХМ: 0-40%) с получением метил-1-[2,2-бис(метилокси)этил]-5-бром-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилата 24 в виде светло-коричневого твердого вещества (50 г; 60% за три стадии).
1H-ЯМР (400 МГц, хлороформ-d) δ млн-1 7.7 (s, 1H), 7.4 (m, 2H), 7.3 (d, J=7,9 Гц, 3Н), 5.3 (s, 2H), 4.4 (s, 1H), 3.8 (d, J=4,8 Гц, 2H), 3.8 (s, 3H), 3.4 (s, 6H). LC-MS (M+H+): рассчитано 426, обнаружено 426.
Пример 2d
Метил-1-[2,2-бис(метилокси)этил]-5-({[(2,4-дифторфенил)метил]амино}-карбонил)-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилат
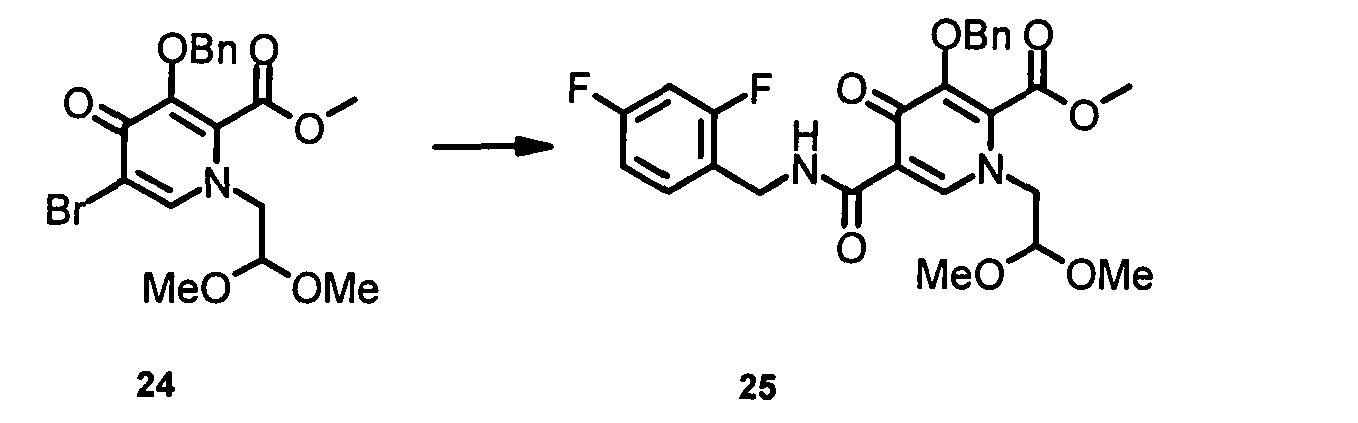
В сосуд под давлением загружали метил-1-[2,2-бис(метилокси)этил]-5-бром-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилат 24 (6,4 г; 15 ммоль), 2,4-дифторбензиламин (3,2 г; 22,5 ммоль), К3РО4 (9,45 г; 45 ммоль), Pd(OCOCF3)2 (398 мг; 1,2 ммоль), Xantphos (694 мг; 1,2 ммоль) и толуол (200 мл). Смесь продували СО (4x), затем нагревали до 100ºС в течение 22 часов в атмосфере СО (60 фунт/кв.дюйм (0,41 МПа)). После охлаждения до комнатной температуры твердые вещества отфильтровывали через целит и промывали ЕtOАс. Фильтрат концентрировали и остаток очищали посредством колоночной хроматографии (силикагель, ЕtOАс/гексан 0~80%) с получением метил-1-[2,2-бис(метилокси)этил]-5-({[(2,4-дифторфенил)метил]амино}карбонил)-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилата 25 в виде светло-коричневого масла (4,7 г; 61%).
1H ЯМР (400 МГц, хлороформ-d) δ млн-1 10.4 (m, 1H), 8.4 (s, 1H), 7.4 (m, 6H), 6.8 (d, J=9,3 Гц, 2H), 5.3 (s, 2H), 4.6 (d, J=5,7 Гц, 2H), 4.5 (s, 1H), 4.0 (d, J=4,8 Гц, 2H), 3.8 (s, 3Н), 3.4 (s, 6H). LC-MS (M+H+): рассчитано 517, обнаружено 517.
Пример 2е
Метил-5-({[(2,4-дифторфенил)метил]амино}карбонил)-1-[2-гидрокси-2-(метилокси)этил]-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилат (в равновесии с соответствующим альдегидом)
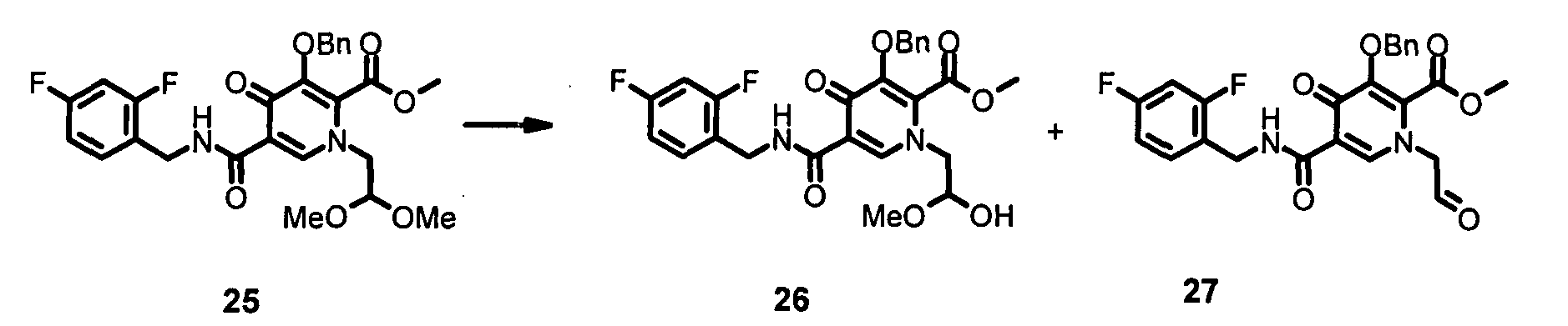
Метил-1 -[2,2-бис(метилокси)этил]-5-({[(2,4-дифторфенил)метил]амино}-карбонил)-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридинкарбоксилат 25 (11,6 г) обрабатывали 90% муравьиной кислотой (250 мл) при 40ºС в течение примерно 12 часов (контролировали посредством LC-MS (жидкостная хроматография-масс-спектрометрия)). После выпаривания растворителей при <40ºС остаток повторно растворяли в ЕtOАс (примерно 1 л) и полученный раствор промывали NаНСО3 и рассолом. Органическую фазу затем сушили над Na2SО4. После выпаривания растворителей указанные в заголовке соединения 26 и 27 получали в виде равновесной смеси приблизительно 80/20 (10,57 г).
1H-ЯМР (400 МГц, ДМСО-d6) δ млн-1 10.3 (m, 1H), 9.47 (s, альдегид-H. ~0.2Н), 8.4 (m, 1H), 7.3 (s, 6H), 7.2 (m, 1H), 7.0 (m, 1H), 6.3 (m, 2H), 5.1 (s, 3Н), 4.9 (m, 1H), 4.5 (m, 3Н), 3.9 (m, 2H), 3.8 (s, 3Н). LC-MS для 26 (М+Н+): рассчитано 503, обнаружено 503; для 27 (М+Н2О+Н+): рассчитано 489, обнаружено 489.
Пример СС
(4aS,13аR)-N-[(2,4-Дифторфенил)метил]-10-гидрокси-9,11 -диоксо-2,3,4а,5,9,11,13,13а-октагидро-1H-пиридо[1,2-а]пирроло[1',2':3,4]имидазо[1,2-d]пиразин-8-карбоксамид

Пример ССа: (4aS,13аR)-8-Бром-10-[(фенилметил)окси]-2,3,4а,5,13,13а-гексагидро-1 Н-пиридо[1,2-а]пирроло[1',2':3,4]имидазо[1,2-d]пиразин-9,11 -дион (DD)
В реактор загружали [(2Я)-2-пирролидинилметил]амин (0,75 кг) и добавляли 4,6 л ДМФА с последующим добавлением 0,45 кг ледяной уксусной кислоты. Затем добавляли ацетонитрил (41,4 л) и эту смесь перемешивали в течение 15 минут. К реакционной смеси добавляли метил-5-бром-1-(2,2-дигидроксиэтил)-4-оксо-3-[(фенилметил)окси]-1,4-дигидро-2-пиридин-карбоксилат (2,30 кг). После перемешивания в течение 20 минут при температуре окружающей среды смесь нагревали при 75-85ºС до тех пор, пока анализ ВЭЖХ не подтвердил израсходование бромидного исходного вещества (примерно 6 часов). По завершении смесь охлаждали до тех пор, пока дефлегмация не уменьшилась, затем загружали 6,9 л метанола и эту смесь нагревали с обратным холодильником в течение примерно 45 минут, затем охлаждали до 15ºС, фильтровали и сушили с получением (4аS,13аR)-8-бром-10-[(фенилметил)окси]-2,3,4а,5,13,13а-гексагидро-1H-пиридо[1,2-а]пирроло[1',2':3,4]имидазо[1,2-d]пиразин-9,11-диона (1,93 кг; 78%) в виде белого твердого вещества.
1H-ЯМР (300 МГц, ДМСО-d6) δ млн-1 8.65 (m, 1H), 7.54 (m, 2H), 7.33 (m, 3Н), 5.15 (d, 1H), 4.99 (d, 1H), 4.60 (m, 1H), 4.36 (m, 1H), 4.03 (m, 1H), 3.90 (m, 1H), 3.65 (m, 1H), 3.06-2.84 (m, 3Н), 1.92-1.60 (m, 4H).
Пример ССb: (4аS,13аR)-N-[(2,4-дифторфенил)метил]-9,11-диоксо-10-[(фенилметил)окси]-2,3,4а,5,9,11,13,13а-октагидро-1Н-пиридо[1,2-а]пирроло[1',2':3,4]имидазо[1,2-d]пиразин-8-карбоксамид
В реакционный сосуд загружали (4аS,13аR)-8-бром-10-[(фенилметил)окси]-2,3,4а,5,13,13а-гексагидро-1H-пиридо[1,2-а]пирроло[1',2':3,4]имидазо[1,2-d]пиразин-9,11-дион (1,4 кг), 2,4-дифторбензиламин (705 г), основание Хунига (1,4 л), dppf (60 г) и ДМСО (12 л). Смесь дегазировали азотом высокой степени чистоты 4 раза. К этой смеси добавляли трифторацетат палладия(II) (18 г) в ДМСО (2 л). Смесь снова дегазировали 3 раза азотом высокой степени чистоты и затем продували СО 3 раза и оставляли в атмосфере 45 фунт/кв.дюйм (0,31 МПа) СО. Эту смесь нагревали при 80ºС в атмосфере 45 фунт/кв.дюйм (0,31 МПа) СО до тех пор, пока ВЭЖХ-анализ не подтвердил, что взаимодействие завершено (24 часа). Смесь охлаждали до комнатной температуры и медленно переносили в ледяную суспензию хлорида аммония. Смесь фильтровали и промывали водой и изопропанолом. Остаток перекристаллизовывали из изопропанола с получением (4aS, 13аR)-N-[(2,4-дифторфенил)метил]-9,11-диоксо-10-[(фенилметил)окси]-2,3,4а,5,9,11,13,13а-октагидро-1Н-пиридо[1,2-а]пирроло[1',2':3,4]имидазо[1,2-d]пиразин-8-карбоксамида (952 г; 56%). Перекристаллизация маточного раствора от изопропанола дала вторую порцию кристаллов требуемого продукта в количестве 327 г (19%).
1H-ЯМР (300 МГц, ДМСО-d6) δ млн-1 10.44 (m, 1H), 8.55 (s, 1H), 7.56-7.07 (m, 8H), 5.18 (d, 1H), 5.03 (d, 1H), 4.62-4.54 (m, 4H), 4.06-3.60 (m, 3Н), 3.20-2.80 (m, 3H), 1.93-1.60(m, 4H).
Пример ССс: (4aS,13аR)-N-[(2,4-дифторфенил)метил]-10-гидрокси-9,11-диоксо-2,3,4а,5,9,11,13,13а-октагидро-1H-пиридо[1,2-а]пирроло[1',2':3,4]имидазо[1,2-d]пиразин-8-карбоксамид
В реакционный сосуд под давлением загружали (4aS,13aR)-N-[(2,4-дифторфенил)метил]-9,11-диоксо-10-[(фенилметил)окси]-2,3,4а,5,9,11,13,13а-октагидро-1H-пиридо[1,2-а]пирроло[1',2':3,4]имидазо[1,2-d]пиразин-8-карбоксамид (950 г), 192 г палладия на угле (влажность 50%), этанол (9,5 л) и концентрированный гидроксид аммония (124 мл). Смесь дегазировали азотом 3 раза и затем помещали в атмосферу 50 фунт/кв.дюйм (0,345 МПа) водорода до тех пор, пока взаимодействие не завершилось. Смесь снова дегазировали азотом и затем фильтровали через целит. Осадок на фильтре экстрагировали дефлегмирующим дихлорметаном и затем снова фильтровали. Объединенные фильтраты концентрировали до небольшого объема (4 л), подвергали азеотропной перегонке с этанолом (28,5 л) до конечного объема 9 л. Суспензию фильтровали, промывали этанолом и сушили с получением (4aS,13aR)-N-[(2,4-дифторфенил)метил]-10-гидрокси-9,11-диоксо-2,3,4а,5,9,11,13,13а-октагидро-1H-пиридо[1,2-а]пирроло[1',2':3,4]имидазо[1,2-d]пиразин-8-карбоксамида (616 г; 78,4%) в виде белого твердого вещества.
1H-ЯМР (300 МГц, ДМСО-d) δ млн-1 10.37 (m, 1H), 8.42 (s, 1H), 7.41-7.05 (m, 3Н), 4.72-4.53 (m, 4H), 4.05 (m, 1H), 3.86 (m, 1H), 3.70 (m, 1H), 3.16 (m, 1H), 2.88 (m, 2H), 1.92-1.60 (m, 4H).
Реферат
Изобретение относится к усовершенствованному способу получения пиридоновых соединений (АА),(ВВ) и (СС) соответствующих формул:. Указанные соединения обладают ингибирующим действием в отношении ВИЧ-интегразы. Способ заключается в проведении стадий: P-1) бромирования соединения формулы (I-I) с получением бром-соединения формулы (II-II):где значение R представляет собой -CHO, -CH(OH), -CH(OH)(OR), -CH(OH)-CHOH или -CH(OR)(OR); Pпредставляет собой бензил; Pпредставляет собой Н или защитную группу карбоксила; Rпредставляет собой низший алкил; Rи Rнезависимо представляют собой низший алкил или Rи Rмогут представлять собой алкил и быть соединены с образованием 5-, 6- или 7-членного кольца, P-2) образования боковой цепи 2,4-ди-фторфенил-CH-NH-C(O)- с использованием реагентов 2,4-ди-фторфенил-CH-NHи монооксида углерода, стадию образования кольца Q с помощью соответствующего амина, выбранного из 3-амино-бутан-1-ола, 2-амино-пропан-1-ола и 2-пирролидинилметиламина, и стадию дебензилирования с получением соединения формулы (AA), (BB) или (CC), где указанную стадию Р-2 проводят после образования кольца Q. Способ позволяет упростить получение целевых соединений за счет проведения региоселективного бромирования на первой стадии. 2 н. и 4 з.п. ф-лы, 3 пр., 7 ил.
Формула



включающий стадии:
- P-1) бромирования соединения следующей формулы (I-I) с получением соединения брома следующей формулы (II-II):

где R представляет собой -CHO, -CH(OH)2, -CH(OH)(OR4), -CH(OH)-CH2OH или -CH(OR5)(OR6);
P1 представляет собой бензил;
P3 представляет собой Н или защитную группу карбоксила;
R4 представляет собой низший алкил;
R5 и R6 независимо представляют собой низший алкил или R5 и R6 могут представлять собой алкил и быть соединены с образованием 5-, 6- или 7-членного кольца,
- P-2) образования боковой цепи 2,4-ди-фторфенил-CH2-NH-C(O)- с использованием реагентов 2,4-ди-фторфенил-CH2-NH2 и монооксида углерода,
- стадию образования кольца Q с помощью соответствующего амина, выбранного из 3-амино-бутан-1-ола, 2-амино-пропан-1-ола и 2-пирролидинилметиламина, и
- стадию дебензилирования с получением соединения формулы (AA), (BB) или (CC),
где указанную стадию Р-2 проводят после образования кольца Q.

где P1 представляет собой бензил.
Комментарии