Производные бензо(b)тиепин-1,1-диоксидов, способ их получения, лекарственное средство и способ его получения - RU2215001C2
Код документа: RU2215001C2
Чертежи
Описание
Изобретение относится к замещенным производным бензо(b)тиепин-1,1-диоксидов, их физиологически приемлемым солям и физиологически функциональным производным.
Описаны производные бензо(b)тиепин-1,1-диоксидов, так же как и их применение для лечения гиперлипидемии, а также атеросклероза и гиперхолистеринемии [заявка PCT/US 97/04076, публикация WO 97/33882].
Задачей данного изобретения являются другие соединения, оказывающие терапевтически применимое гиполипидемическое действие. В особенности задача состоит в том, чтобы найти новые соединения, которые по сравнению с описанными в уровне техники соединениями уже в низких дозах вызывают повышенное фекальное выделение желчных кислот. Особенно желательно было снижение эффективной дозы (ЭД200) по меньшей мере в 5 раз по сравнению с описанными в уровне техники соединениями.
Данное
изобретение, таким образом,
относится к соединениям формулы I,

где
R1 - метил, этил, пропил, бутил;
R2 - Н, ОН, NH2, NH-(С1-С6)-алкил);
R3 - остаток аминокислоты, остаток диаминокислоты, остаток триаминокислоты, остаток тетрааминокислоты, причем остаток аминокислоты, остаток диаминокислоты, остаток триаминокислоты или остаток тетрааминокислоты в случае необходимости одно- или многократно замещены аминозащитной группой.
R4 - метил, этил, пропил, бутил;
R5 - метил, этил, пропил, бутил;
Z - -(С= 0)n-С0-С16-алкил-, -(C=
O)n-С0-С16-алкил-NН-, -(С= O)n-С0-С16-алкил-О-, -(С=O)n-С1-С16-алкил-(C=O)m, ковалентная
связь;
n - 0 или 1;
m - 0 или 1;
а также их фармацевтически приемлемые соли и физиологически функциональные производные.
Предпочтительными являются соединения
формулы I, в которых один или более остатков имеют следующие значения:
R1 - этил, пропил, бутил;
R2 - Н, ОН, NH2, NН-(С1-С6
)-алкил;
R3 - остаток аминокислоты, остаток диаминокислоты, причем остаток аминокислоты или остаток диаминокислоты в случае необходимости
одно- или многократно замещены
аминозащитной группой;
R4 - метил, этил, пропил, бутил;
R5 - метил, этил, пропил, бутил;
Z - -(С= O)n-С0-С16-алкил-,
-(С= O)n-С0-С16-алкил-NН-, -(С= O)n-С0-С16-алкил-О-, -(С=O)n-С1-С16
-алкил-(С=O)m,
ковалентная связь;
n - 0 или 1;
m - 0 или 1;
а также их фармацевтически приемлемые соли.
Особенно предпочтительными являются
соединения формулы I, в которых
один или более остатков имеют следующие значения:
R1 - этил, бутил;
R2 - ОН;
R3 - остаток диаминокислоты,
причем остаток диаминокислоты в
случае необходимости одно- или многократно замещен аминозащитной группой;
R4 - метил;
R5 - метил;
Z - -(С=O)n-С1-С4
-алкил-, ковалентная связь;
а также их фармацевтически приемлемые соли.
Фармацевтически приемлемые соли вследствие их более высокой растворимости в воде по сравнению с исходными или основными соединениями особенно пригодны для медицинского применения. Эти соли должны содержать фармацевтически приемлемый анион или катион. Фармацевтически приемлемыми аддитивными солями кислот предложенных соединений являются соли неорганических кислот, таких как соляная кислота, бромистоводородная, фосфорная, метафосфорная, азотная, сульфоновая и серная кислоты, так же как и органических кислот, как, например, уксусная кислота, бензолсульфоновая, бензойная, лимонная, этансульфоновая, фумаровая, глюконовая, гликолевая, изотионовая, молочная, лактобионовая, малеиновая, яблочная, метансульфоновая, янтарная, пара-толуолсульфоновая, винная и трифторуксусная кислоты. Для медицинских целей особенно предпочтительными являются соли соляной кислоты. Фармацевтически приемлемыми основными солями являются соли аммония, соли щелочных металлов (такие как соли натрия и калия) и соли щелочноземельных металлов (такие как соли магния и кальция).
Соли с фармацевтически неприемлемым анионом равным образом охватываются данным изобретением в качестве необходимых промежуточных продуктов для получения или очистки фармацевтически приемлемых солей и/или для применения в нетерапевтических целях, например для применения in vitro.
Использованный здесь термин "физиологически функциональные производные" означает любое физиологически приемлемое производное предлагаемого соединения, например сложный эфир, который при введении млекопитающему, например человеку, способно образовывать (прямо или косвенно) такое соединение или его активный метаболит.
Дополнительным аспектом данного изобретения являются пролекарства предлагаемых соединений. Такие пролекарства могут быть метаболизированы in vivo до соединения согласно изобретению. Такие пролекарства сами могут быть или не быть активными.
Соединения согласно изобретению могут также находиться в различных полиморфных формах, например в виде аморфных и кристаллических полиморфных форм. Все полиморфные формы предлагаемых соединений относятся к данному изобретению и являются дополнительным аспектом данного изобретения.
Далее все указания на "соединение(ия) согласно формуле (I)" относятся к соединению(иям) формулы (I), как описано выше, а также к их солям, сольватам и физиологически функциональным производным.
Количество соединения согласно формуле (I), которое требуется для достижения желаемого биологического эффекта, зависит от ряда факторов, например от выбранного специфического соединения, предполагаемого способа введения и клинического состояния пациента. В основном дневные дозы находятся в диапазоне от 0,1 до 100 мг (обычно от 0,1 до 50 мг) в день на килограмм веса тела, например, 0, 1-10 мг/кг/день. Таблетки или капсулы могут содержать, например, от 0,01 до 100 мг, обычно от 0,02 до 50 мг. В случае фармацевтически приемлемых солей приведенные весовые значения относятся к весу освобожденного из соли иона бензо(b)тиепина. Для профилактики или терапии названных выше состояний соединения согласно формуле (I) могут применяться непосредственно в виде соединений, однако предпочтительно они представлены с подходящим носителем в виде фармацевтической композиции. Конечно, носитель должен быть пригодным, т.е. он является совместимым с другими ингредиентами композиции и безвреден для здоровья пациента. Носитель может быть твердым веществом, или жидкостью, или и тем и другим и предпочтительно приготовлен вместе с соединением в виде отдельной дозы, например в виде таблетки, которая может содержать от 0,05 до 95 вес.% активного вещества. Также могут иметь место другие фармацевтически активные вещества, включая другие соединения согласно формуле (I). Фармацевтические композиции согласно изобретению могут быть получены по одному из известных фармацевтических способов, которые в основном состоят в том, что ингредиенты смешивают с фармакологически приемлемыми носителями и/или вспомогательными веществами.
Фармацевтические композиции согласно изобретению являются пригодными для орального и перорального (например, сублингвального) введения, хотя наиболее подходящий способ введения в каждом отдельном случае зависит от вида и тяжести излечиваемого состояния и от вида применяемого соединения согласно формуле (I). Все препараты в виде драже и препараты пролонгированного действия в виде драже также относятся к объему данного изобретения. Предпочтительными являются формы, устойчивые к кислотам и желудочному соку. Подходящими устойчивыми к желудочному соку покрытиями являются ацетатфталат целлюлозы, поливинилацетатфталат, фталат гидроксипропилметилцеллюлозы и анионные полимеры метакриловой кислоты и метилового эфира метакриловой кислоты.
Фармацевтически пригодные соединения для орального введения могут быть представлены в виде отдельных единиц, как, например, капсул, оболочек облатки, таблеток для сосания или таблеток, каждая из которых содержит определенное количество соединения согласно формуле (I); в виде порошков или гранулятов; в виде растворов или суспензий в водной или неводной жидкости; или в виде эмульсии масло-в-воде или вода-в-масле. Такие композиции могут, как уже упоминалось, быть приготовлены любым подходящим фармацевтическим способом, который включает стадию, при которой контактируют активное вещество и носитель (который может состоять из одного или более дополнительных ингредиентов). Обычно композиции получают путем равномерного и гомогенного перемешивания активного вещества с жидким и/или тонкоизмельченным твердым носителем, после чего продукт в случае необходимости формуют. Так, например, может быть получена таблетка, в которой спрессован или отформован порошок или гранулят соединения в случае необходимости с одним или более дополнительными ингредиентами. Прессованные таблетки могут быть получены таблетированием соединения в свободнотекучей форме, как, например, порошок или гранулят, в случае необходимости смешанного со связующим, придающим скользкость средством, инертным разбавителем и/или одним (несколькими) поверхностно-активными/диспергирующими средствами в подходящем оборудовании. Оформленные таблетки могут быть получены путем формования порошкообразного, увлажненного жидким инертным разбавителем соединения на подходящем оборудовании.
Фармацевтическими композициями, пригодными для перорального (сублингвального) применения, являются таблетки для сосания, которые содержат соединение согласно формуле (I) с вкусовой добавкой, обычно сахарозой и гуммиарабиком или трагантом, и пастилки, которые содержат соединение в инертной основе, такой как желатина и глицерин или сахароза и гуммиарабик.
Предметом данного изобретения являются далее смеси изомеров формулы I, а также чистые стереоизомеры формулы I, a также смеси диастереомеров формулы I и чистые диастереомеры. Разделение смесей осуществляют хроматографическим способом.
Предпочтительными являются рацемические смеси, а также и энантиомерно
чистые соединения формулы I следующей структуры:


Под аминокислотами или остатками аминокислот понимают, например, стереоизомерные формы, т.е. D- или L-формы следующих соединений:
аланин; глицин; пролин;
цистеин; гистидин; глутамин;
аспарагиновая кислота; изолейцин; аргинин;
глутаминовая кислота; лизин; серин;
фенилаланин; лейцин; треонин;
триптофан; метионин; валин;
тирозин; аспарагин;
2-аминоадипиновая кислота; 2-аминоизомасляная кислота;
3-аминоадипиновая кислота; 3-аминоизомасляная кислота;
бета-аланин; 2-аминопимелиновая кислота;
2-аминомасляная кислота; 2,4-диаминомасляная кислота;
4-аминомасляная кислота; десмозин;
пиперидиновая кислота; 2, 4-диаминопимелиновая кислота;
6-аминокапроновая кислота; 2, 3-диаминопропионовая кислота;
2-аминогептановая кислота; N-этилглицин;
2-(2-тиенил)-глицин; 3-(2-тиенил)-аланин;
пеницилламин; саркозин;
N-этиласпарагин; N-метилизолейцин;
гидроксилизин; 6-N-метиллизин;
алло-гидроксилизин; М-метилвалин;
3-гидроксипролин; норвалин;
4-гидроксипролин; норлейцин;
изодесмозин; орнитин;
алло-изолейцин;
N-метилглицин.
Краткие обозначения аминокислот осуществляют общими обычными способами (Schroeder, Luebke, Nhe Peptides, том I, New York 1965, с. XXII-XXIII; Houben-Weyl, Methoden der Organischen Chemie, том XV/1 и 2, Штуттгарт 1974). Аминокислота pGlu означает пироглутамил, Nal - 3(2-нафтил)-аланин, Azagly-NH2 - соединение формулы NH2-NH-CONH2 и D-Asp-означает D-форму аспарагиновой кислоты. Пептиды по их химической природе являются амидами кислот и разлагаются при гидролизе аминокислоты.
Под остатками диаминокислот, триаминокислот, тетрааминокислот понимают пептиды, включающие от 2 до 4 названных аминокислот.
В качестве подходящих аминозащитных групп (Greene,
"Protective Groups in Organic Synthesis") в первую очередь использованы:
Arg(Tos), Arg(Mts), Arg(Mtr), Arg(PMV), Asp(OBzl),
Asp(OBut), Cys(4-MeBzl), Cys (Acm), Cys(SBut), Glu(OBzl), Glu(Obut),
His(Tos), His(Fmoc), His(Dnp), His(Trt), Lus(Cl-Z), Lys(Boc), Met(O), Ser(Bzl), Ser(But), Thr(Bzl), Thr(But), Nrp(Mts), Trp(CHO),
Tyr(Br-Z), Tyr(Bzl) или Туг(But).
В качестве аминозащитных групп предпочтительны отщепляемый при каталитическом гидрировании бензилоксикарбонильный-(Z)-остаток, отщепляемые слабыми кислотами 2-(3,5-диметилоксифенил)пропил(2)оксикарбонил(Ddz-) или тритилльный (Trt)-остатки, отщепляемые вторичными аминами 9-флуоренилметоксикарбонильный (Fmoc).
Данное изобретение
далее относится к способу получения производных бензо(b)тиепин-1,
1-диоксидов формулы I:

Способ получения соединений формулы I отличается тем, что амин формулы II, где R1, R2, R4 и R5 имеют приведенные в формуле I значения, подвергают взаимодействию с соединением формулы III, где R3 и Z имеют значения, приведенные в формуле I, с отщеплением воды, и образованием соединения формулы I и полученное соединение формулы I в случае необходимости переводят в физиологически приемлемую соль или физиологически функциональное производное. Если в качестве остатка R3 речь идет об остатке моноаминокислоты, этот остаток в случае необходимости может быть еще удлинен путем связывания с амином формулы II последовательно до остатка диаминокислоты, остатка триаминокислоты или остатка тетрааминокислоты.
Соединения формулы I и их фармацевтически приемлемые соли и физиологически функциональные производные представляют собой идеальное лекарственное средство для лечения нарушений липидного обмена, в частности гиперлипидемии. Соединения формулы I равным образом пригодны для влияния на уровень сывороточного холестерина, так же как и для профилактики и лечения атеросклеротических явлений. Эти соединения в случае необходимости могут также вводиться со статинами, как, например, симвастататин, флувастатин, правастатин, церивастатин, ловастатин или аторвастин. Следующие результаты иллюстрируют фармакологическую активность предлагаемых соединений.
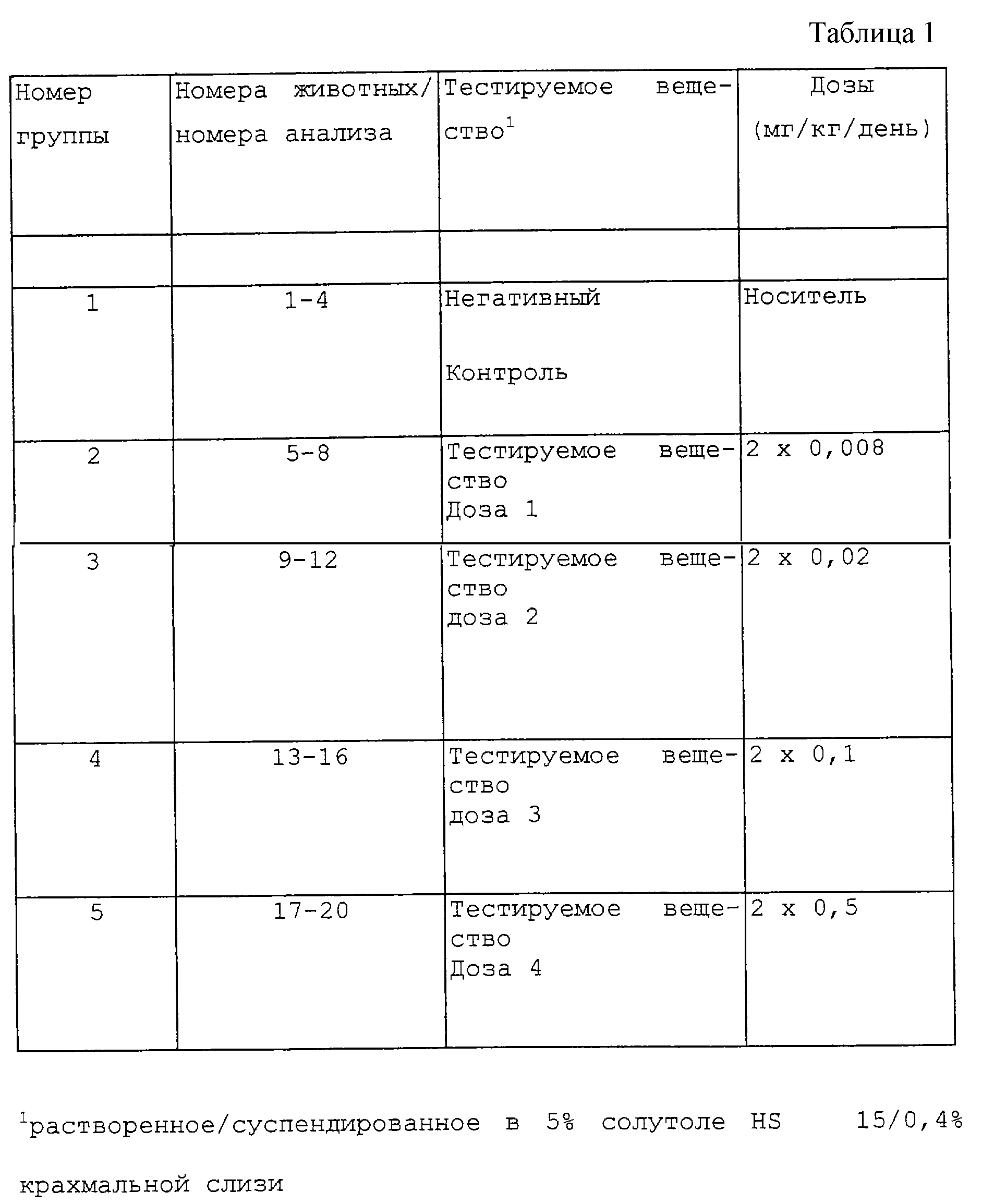
Биологическое исследование соединений согласно изобретению осуществляли путем выведения ЭД200. Было изучено действие предлагаемых соединений на транспорт желчных кислот в подвздошной кишке и фекальное выделение желчных кислот у крыс после орального введения дважды в день. Были опробованы смеси диастереомеров соединений.
Тестирование проводили следующим образом.
1) Состав тестируемых и сравнительных веществ.
Для приготовления водного раствора
использовали следующую рецептуру:
вещества растворяли в водном растворе, содержащем
адекватные объемы солутола (= гидроксистеарата полиэтиленгликоля 600; BASF, Ludwigshafen, Германия;
Chargennr. 1763) так, чтобы конечная концентрация солутола в водном растворе составляла 5%.
Растворы/суспензии вводили в дозе 5 мл/кг перорально.
2) Условия опыта
Самцов крыс
Wistar (Kastengrund, Hoechst AG, вес 250-350 г) в группах по 6 животных выдерживали в
течение 10 дней до начала тестирования (день 1) при перевернутом ритме дня/ночи (400-1600 в
темноте, 1600-400 при свете) и давали стандартную кормовую смесь
(Altromin, Lage, Германия). За три дня до начала тестирования (день 0) животных разделили на группы по 4
особи.
Разделение животных в тестируемых группах приведено в табл. 1.
3) Проведение эксперимента
После внутривенного или подкожного введения 5 микрокюри14С-таурохолата на крысу (день 0) давали носитель или тестируемое вещество в 700-800 и в 1500-1600 на следующий день (день 1) (лечение один день).
Исследование свежего кала для определения14С-таурохолата производили через 24
часа непосредственно после введения утренней дозы. Испражнения взвешивали, сохраняли при -18oС и
позднее суспендировали в 100 мл деминерализованной воды и гомогенизировали (Ultra Turrax,
Janke & Kunkel, IKA-Werk). Аликвотные части (0,5 г) взвешивали и сжигали на наконечнике для сжигания
(Combusto Cones, Canberra Packard) в аппарате для сжигания (Tri Carb(R) 307
combuster Canberra Packard GmbH, Франкфурт-на-Майне, Германия). Образующийся14CO2
абсорбировали на Саrbо-Sorb(R) (Canberra Packard). Последующее измерение
радиоактивности14C производили после добавления к пробе сцинтиллятора (Perma-Fluor complete
scintillation coctail 6013187, Packard) с помощью жидкостного сцинтилляционного счетчика (ЖСС).
Фекальное выделение14С-таурохолевой кислоты было рассчитано в виде кумулятивной и/или
процентной остаточной радиоактивности (смотри ниже).
4) Наблюдения и измерения
Фекальное выделение14С-ТХК было определено в сожженных аликвотных частях собранных
с интервалом 24 часа проб свежего кала, рассчитано как "кумулятивное процентное отношение" от введенной
активности и выражено как % остаточной активности (=оставшейся активности, то есть введенная
активность за вычетом уже выделенной активности). Для расчета кривой дозы-активность выделение14
С-таурохолевой кислоты выражено в виде процентной доли соответствующего значения для
контрольной группы (обработанной носителем). ЭД200, то есть доза, которая повышает фекальное выделение14С-таурохолевой кислоты на 200% по сравнению с контрольной группой, была
рассчитана путем интерполяции сигмовидной или линейной кривой доза-действие. Рассчитанная ЭД200
соответствует дозе, которая удваивает фекальное выделение желчных кислот.
5)
Результаты
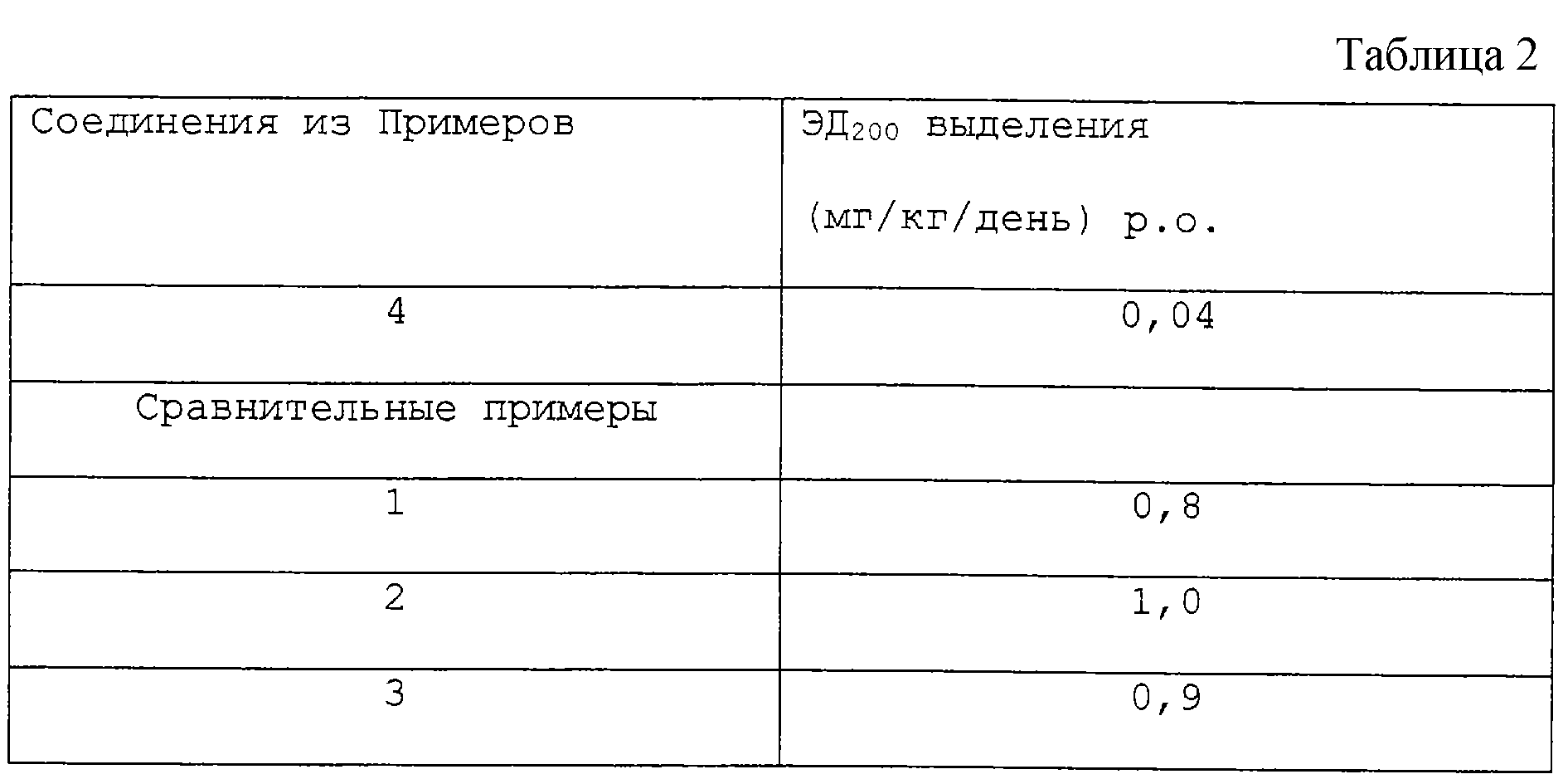
Табл. 2 демонстрирует измеренные значения ЭД200 выделения.
6) Обсуждение
Из данных измерений следует, что предлагаемые соединения формулы I по
сравнению с описанными в уровне техники соединениями обладают улучшенным действием в 20 раз.
Последующие примеры более подробно поясняют данное изобретение, не ограничивая его описанными в примерах продуктами и формами выполнения.
Пример 1


С46Н74 N6ОS (887,20). Масс-спектр (М+Н)+=887,5
Сравнительные примеры из PCT/US 97/04076;
Сравнительный пример 1


Сравнительный пример 2


Сравнительный пример 3


Примеры или сравнительные примеры получены следующим образом (показан синтез только

Схема 1

Синтез соединения 6 в виде смеси диастереомеров:
150 мг (0,35 ммоль) 1а/b и 245 мг (0,52 ммоль Fmoc-D-Lys (Вос)-ОН 5 (Fluka) в 6 мл ДМФА реагировали со 168 мг TOTU, 74 мг оксима и 0,5 мл NЭМ аналогично синтезу соединения 3. Выход 290 мг (94%) 6a/b в виде аморфного твердого вещества. ТСХ (этилацетат/н-гептан 2: 1) Rf= 0,6. C50H64N4C8S (881,15), Масс-спектр (М+Н)+=881,5.
Синтез соединения 7 в виде смеси
диастереомеров:
285 мг (0,32 ммоль) 6a/b растворяли в 5 мл ДМФА. После прибавления 0,6 мл диэтиламина
выдерживали 30 минут. Обработку осуществляли аналогично синтезу соединения 3. 173 мг
(81%) 7a/b в виде аморфного твердого вещества. ТСХ (метиленхлорид/метанол 15: 1). Rf=0,2, эдукт 6a/b
Rf=0,4. С35Н54N4096 (658,91)
Масс-спектр (М+Н)+=659,4.
Синтез соединения 8 в виде смеси диастереомеров:
168
мг (0,25 ммоль 7a/b подвергали взаимодействию аналогично синтезу соединений 6 и 7 и
получили 169 мг (75% на две стадии) 8а/b в виде аморфного твердого вещества. ТСХ (метиленхлорид/метанол 9: 1). Rf=0,3. C46H74N6O9S (887,20).
Масс-спектр (М+Н)+=887,5.
Реферат
Изобретение относится к замещенным производным бензо (b) тиепин-1,1-диоксидов и их аддитивным солям с кислотами формулы I, в которой R1 - метил, этил, пропил, бутил; R2 - Н, ОН; R3 - остаток аминокислоты, остаток диаминокислоты, причем остаток аминокислоты, остаток диаминокислоты в случае необходимости одно- или многократно замещены аминозащитной группой; R4 - метил, этил, пропил, бутил; R5 - метил, этил, пропил, бутил; Z - ковалентная связь. Описан также способ получения соединений, лекарственное средство на их основе и способ получения лекарственного средства. Соединения могут быть использованы в качестве гиполипидемических средств. 4 с. и 2 з.п.ф-лы, 2 табл.

Формула

где R1 - метил, этил, пропил, бутил;
R2 - Н, ОН;
R3 - остаток аминокислоты, остаток диаминокислоты, причем остаток аминокислоты, остаток диаминокислоты, в случае необходимости, одно- или многократно замещены аминозащитной группой;
R4 - метил, этил, пропил, бутил;
R5 - метил, этил, пропил, бутил;
Z - ковалентная связь,
а также их фармацевтически приемлемые соли.
R4 - метил, этил, пропил, бутил;
R5 - метил, этил, пропил, бутил;
Z - ковалентная связь,
а также их фармацевтически приемлемые соли.
R1 - этил, бутил;
R2 - ОН;
R3 - остаток диаминокислоты, причем остаток диаминокислоты, в случае необходимости, одно- или многократно замещен аминозащитной группой;
R4 - метил;
R5 - метил;
Z - ковалентная связь,
а также их фармацевтически приемлемые соли.

где R1, R2, R4 и R5 имеют приведенные в формуле I значения, подвергают взаимодействию с соединением формулы III
НО-Z-R3, III
где R3 и Z имеют значения, приведенные в формуле I,
с отщеплением воды и образованием соединения формулы I, полученное соединение формулы I в случае необходимости переводят в физиологически приемлемую соль.
Комментарии