Способ получения оптически активных производных амина - RU2385318C2
Код документа: RU2385318C2
Чертежи

Описание
Настоящее изобретение относится к способу получения оптически активной производного амина высокой чистоты, в котором предотвращается образование побочных продуктов
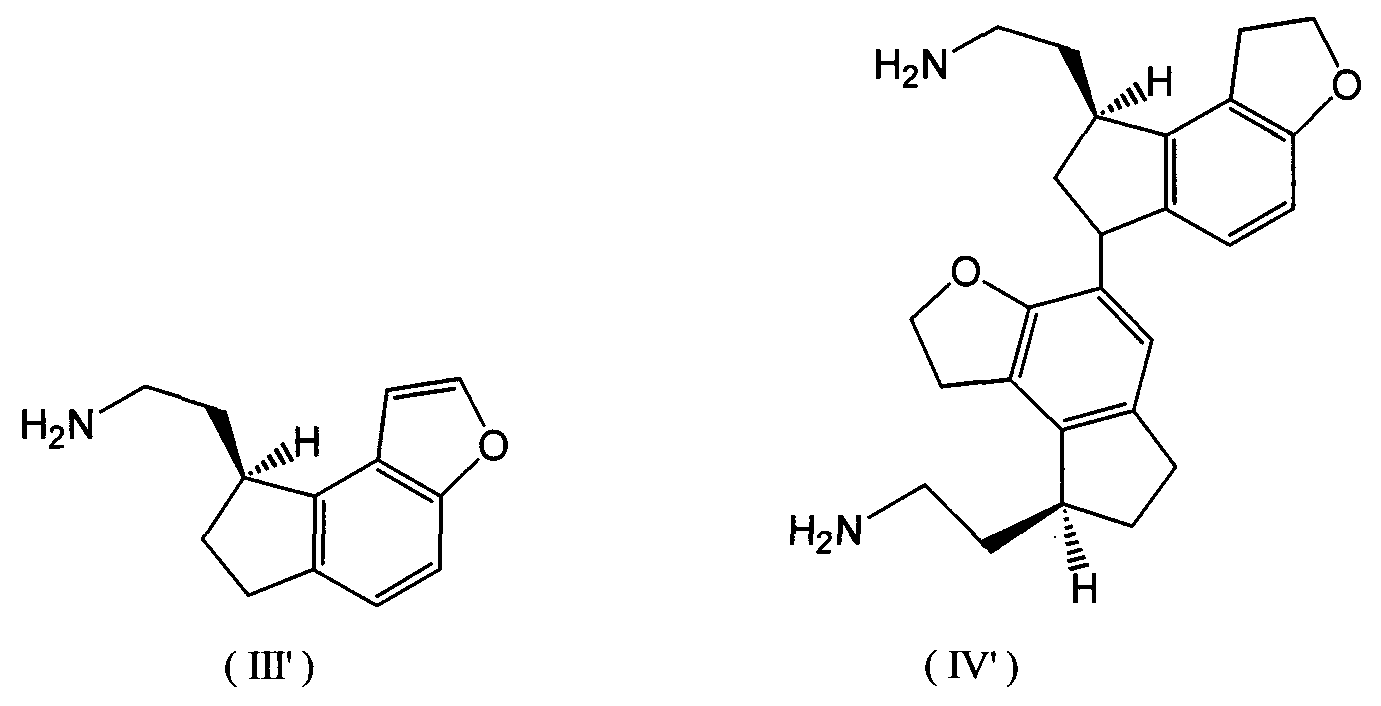
Хотя в патентных заявках JP-A 11-140073 и JP-A 2002-212063 и раскрыты способы получения гидрохлорида(S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламина путем ассиметричного восстановления исходного реагента, гидрохлорида (E)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-илиден)этиламина, оба способа не пригодны для промышленного способа получения, позволяющего предотвращать образование побочных продуктов и получать с высоким выходом кристаллы гидрохлорида (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламина высокой чистоты. В частности, проблемами являются контроль образования побочных продуктов, представленных следующими формулами (III') и (IV'):
С другой стороны, в статьях Liebigs. Ann. Chem., 10,945 (1989) и J. Chem. Soc. (A), 1324 (1968) описана димеризация соединения бензофурана в присутствии Pd-катализатора. Однако в данных случаях димеризация происходит в результате связывания двух ароматических колец, и структуры их димеров отличаются от тех димеров, которые образуются в результате реакции между ароматическим кольцом и бензильным фрагментом, то есть соединения, представленного в вышеприведенной формуле. Кроме того, в статье J. Chem. Soc. D, 736 (1970) раскрывается, что окисление бензильного фрагмента производного бензофурана происходит в присутствии Pd-катализатора, но образование димера в ней не описано.
Описание изобретения
Задачей настоящего изобретения является разработка промышленного способа получения оптически активной производной амина высокой чистоты и с высоким выходом, в котором контролируется образование побочных продуктов.
В результате интенсивных поисков решения вышеназванной проблемы авторы настоящего изобретения обнаружили, что на стадиях получения оптически активной производной амина можно контролировать образование побочных продуктов, представленных вышеприведенными формулами (III') и (IV'), путем регулирования pH и температуры реакционного раствора во время каталитического восстановления на Pd-C и pH и температуры раствора во время его последующей обработки, в результате чего и было создано настоящее изобретение.
А именно, настоящее изобретение предлагает:
(1) Способ получения (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламина или его соли, который включает стадию (i): стадию ассиметричного восстановления (E)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-илиден)этиламина или его соли на катализаторе и стадию (ii): стадию каталитического восстановления реакционного продукта, полученного на стадии (i), при температуре реакции от 40°C до 100°C и pH от 3 до 9 на катализаторе.
(2) Способ по вышеприведенному пункту (1), в котором температура реакции на стадии (ii) составляет от 50°С до 70°C.
(3) Способ по вышеприведенному пункту (1), в котором pH на стадии (ii) составляет от 5 до 7.
(4) Способ по вышеприведенному пункту (1), в котором катализатором на стадии (i) является Ru-BINAP катализатор (BINAP - это 2,2-бис-дифенилфосфино-1,1-бинафтил).
(5) Способ по вышеприведенному пункту (1), в котором катализатором на стадии (ii) является Pd-C катализатор.
(6) Способ получения кристаллов (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамида, включающий стадию (a): стадию пропионилирования аминогруппы (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламина или его соли, полученного в способе по вышеприведенному пункту (1), и стадию (b): стадию кристаллизации путем добавления водного растворителя к реакционному раствору, полученному на стадии (a).
(7) Кристаллы (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамида, в которых содержание каждого из соединений, представленных следующими формулами (I), (II), (III) и (IV), составляет 0,15% по массе или менее и суммарное содержание соединений, представленных следующими формулами (I)-(IV), составляет 0,2% по массе или менее

(8) Кристаллы (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамида, в которых содержание каждого из соединений, представленных следующими формулами (I), (III) и (IV), составляет 0,15% по массе или менее и содержание соединения, представленного следующей формулой (II), составляет от 0,02 до 0,15% по массе, и дополнительно суммарное содержание соединений, представленных следующими формулами (I)-(IV), составляет 0,2% по массе или менее
(9) Кристаллы по вышеприведенному пункту (7) или (8), в которых содержание соединения, представленного формулой (I), составляет 0,10% по массе или менее.
(10) Композиция, включающая (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамид и соединения, представленные следующими формулами (I), (II), (III) и (IV), в которой относительно 100 частей по массе (S)-N-[2-(i,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамида, содержание каждого из соединений, представленных следующими формулами (I), (II), (III) и (IV), составляет от 0 до 0,15 частей по массе и суммарное содержание соединений, представленных следующими формулами (I)-(IV), составляет от 0 до 0,2 частей по массе
(11) Композиция, включающая (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамид и соединения, представленные следующими формулами (I), (II), (III) и (IV), в которой относительно 100 частей по массе (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамида, содержание каждого из соединений, представленных следующими формулами (I), (III) и (IV), составляет от 0 до 0,15 частей по массе, и содержание соединения, представленного следующей формулой (II), составляет от 0,02 до 0,15 частей по массе, и дополнительно суммарное содержание соединений, представленных следующими формулами (I)-(IV), составляет от 0 до 0,2 частей по массе
(12) Композиция по вышеприведенным пунктам (10) или (11), в которой содержание соединения, представленного формулой (I), составляет от 0 до 0,10 частей по массе относительно 100 частей по массе (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил) этил]пропионамида.
(13) Кристаллы по вышеприведенному пункту (7) или композиция по вышеприведенному пункту (10), которую получают в промышленном масштабе.
(14) Способ получения 1,2,6,7-тетрагидро-8H-индена[5,4-b]фуран-8-она, включающий стадию восстановления 4,5-дибром-l,2,6,7-тетрагидро-8H-индено[5,4-b]фуран-8-она на Pd-C катализаторе при условии:
давление водорода (МПа)>-0,02 × суммарный объемный коэффициент массопередачи газ-жидкость (1/час)+0,43.
(15) Применение кристаллов по вышеприведенному пункту (7) для получения профилактического или терапевтического средства при нарушении сна.
(16) Композиция по вышеприведенному пункту (10), которая является профилактическим или терапевтическим средством при нарушении сна.
(17) Способ предотвращения или лечения нарушения сна, включающий введение кристаллов по вышеприведенному пункту (8) или композиции по вышеприведенному пункту (10).
Краткое описание чертежа
На чертеже приведена ВЭЖХ хроматограмма, показывающая результат анализа содержания соединений (I)-(IV) в кристаллах (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамида.
Предпочтительный вариант осуществления изобретения
Соли (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламина в настоящем изобретении включают соли неорганической кислоты, такой как хлористоводородная кислота, серная кислота и азотная кислота, соли органической кислоты, такой как муравьиная кислота, уксусная кислота, и трифторуксусная кислота, и другие подобные соли.
Кроме того, (E)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-илиден)этиламин, который используется в качестве исходного соединения для получения (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламина или его соли настоящего изобретения, может быть получен способом, описанным в патентной заявке JP-A 2002-212063, а именно способом каталитического восстановления 4,5-дибром-l,2,6,7-тетрагидро-8H-индено[5,4-b] фуран-8-она на катализаторе для каталитического восстановления, такого как Pd-C, затем взаимодействием полученного 1,2,6,7-тетрагидро-8H-индено[5,4-b]фуран-8-она с диэтилцианометилфосфонатом с последующим гидрированием на кобальтовом катализаторе, или другими аналогичными способами.
На стадии каталитического восстановления каталитическое восстановление 4,5-дибром-1,2,6,7-тетрагидро-8H-индено[5,4-b] фуран-8-она может быть проведено путем смешения 4,5-дибром-1,2,6,7-тетрагидро-8H-индено[5,4-b]фуран-8-она, органического растворителя и при необходимости основания, затем предпочтительно после продувки системы азотом, добавления катализатора восстановления с последующим созданием избыточного давления водорода и перемешиванием.
В данном случае, с точки зрения контроля образования побочного продукта (в частности, димера), предпочтительно, чтобы реакцию осуществляли при давлении водорода и суммарном объемном коэффициенте массопередачи газ-жидкость, удовлетворяющем неравенству:
давление водорода (МПа)>-0,02 × суммарный объемный коэффициент массопередачи газ-жидкость (1/час)+0,43.
При этом суммарный объемный коэффициент массопередачи газ-жидкость может быть определен методом с использованием Na2SO3, подробно описанным в приводимом далее примере 1.
Кроме того, давление водорода в настоящей реакции обычно составляет от 0,1 до 1 МПа, предпочтительно от 0,3 до 0,5 МПа.
В системе газ-жидкость скорость NA, с которой газ растворяется в растворе на единицу площади контакта, может быть выражена так же, как и в случае скорости растворения в системе твердое вещество-жидкость, скорости экстракции в системе жидкость-жидкость и явления переноса при теплопередаче конвекцией, и приводится в виде (коэффициент массопередачи) × (градиент концентраций).
NA=KL(C1-C) (1)
Здесь KL является коэффициентом массопередачи для жидкости, C1 является концентрацией, которая находится в равновесии с парциальным давлением газа в пузырьке газа, и С является концентрацией при насыщении в определенный момент времени, и (C1-C) является движущей силой абсорбции газа.
Кроме того, если A является площадью контакта газ-жидкость, и VL является объемом жидкости, и увеличение скорости VLdC/dt концентрации газа в растворе равно скорости растворения газа, то может быть получено следующее уравнение:
NAA=VLdC/dt (2)
В результате из формул(1) и (2) получают уравнение
dC/dt=KLA(C1-C/VL(3)
Более того, если A/VL обозначить через a - площадь поверхности раздела газ-жидкость на единицу площади, то получим уравнение
dC/dt=KLa(C1-C) (4)
Так как трудно определить непосредственно площадь поверхности раздела газ-жидкость а при операции перемешивания газ-жидкость, в качестве показателя для выражения способности газа абсорбироваться используют объемный коэффициент массопередачи для жидкости KLa, который является произведением а и коэффициента массопередачи для жидкости KL.
Кроме того, исходя из того факта, что при увеличении скорости перемешивания площадь поверхности раздела газ-жидкость становится больше, можно в целом сделать вывод, что KLa будет расти при увеличении скорости перемешивания.
Примеры органического растворителя, используемого в настоящей реакции, включают муравьиную кислоту, уксусную кислоту, метанол, этанол, N-метилпирролидон и другие подобные растворители, но особенно предпочтительным является метанол. Эти растворители могут использоваться по отдельности или в смеси 2 или более из них. Количество используемого растворителя составляет от 5 до 100 мл, предпочтительно от 15 до 25 мл, на 1 г исходного реагента.
Основания, используемые в настоящей реакции, включают безводный ацетат натрия, Et3N, пиридин, NaHCO3, Na2CO3 и другие подобные основания. В частности, предпочтительными являются безводный ацетат натрия и Et3N. Количество используемого основания обычно составляет от 2 до 3 молей.
Примеры катализатора каталитического восстановления, используемого в настоящей реакции, включают Pd-C, PtO2, Rh-Al2O3, (RhCl[Р(С6Н5)3]3) и другие подобные катализаторы. Количество используемого катализатора каталитического восстановления составляет от 1/10 моля до 5/1000 моля, предпочтительно от 1/100 моля до 3/100 моля, на 1 моль исходного реагента, используемого на стадии (i).
Реакционная температура настоящей реакции составляет обычно от 10°C до 100°C, предпочтительно от 30°C до 50°C, и время реакции составляет обычно от 1 до 50 часов, предпочтительно от 2 до 10 часов.
(E)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-илиден) этиламин, используемый в настоящем изобретении, может быть использован в форме свободного соединения или его соли. Примеры такой соли включают соль неорганической кислоты, такой как хлористоводородная кислота, серная кислота и азотная кислота, соль органической кислоты, такой как муравьиная кислота, уксусная кислота, и трифторуксусная кислота, и другие подобные кислоты.
Способ получения (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламина или его соли настоящего изобретения состоит из стадии (i) для ассиметричного восстановления исходного реагента (E)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-илиден)этиламина или его соли и стадии (ii) для превращения побочного продукта, присутствующего в реакционных продуктах, полученных на стадии (i), в целевое соединение путем каталитического восстановления.
Асимметрическое восстановление на стадии (i) проводят с использованием катализатора, и примеры такого катализатора для асимметрического восстановления включают оптически активный фосфиновый комплекс рутения (Ru-BINAP), оптически активный фосфиновый комплекс родия (Rh-BINAP), оптически активный фосфиновый комплекс иридия (Ir-BINAP) и другие подобные комплексы.
Примерами Ru-BINAP катализатора, в частности, являются Ru2Cl4[(R)-BINAP]2N(C2H5)3, {RuCl(бензол)[(R)-BINAP]}Cl, {RuCl(п-цимол)[(R)-BINAP]}Cl, {RuBr(п-цимол)[(R)-BINAP]}Br, {RuI(п-цимол)[(R)-BINAP]}I3, {RuI(п-цимол)[(R)-BINAP]}I и другие подобные соединения. Эти катализаторы могут быть получены известным способом, например способами, описанными в патентных заявках JP-B 07-57758, JP-A 11-140073 и других.
В качестве катализатора асимметрического восстановления предпочтительно использовать {RuCl(бензол)[(R)-BINAP]}Cl.
Когда в качестве исходного реагента в реакции асимметрического восстановления на стадии (i) используют соль (E)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-илиден)этил-амина, ее превращают в свободное соединение обработкой щелочью, растворяют в органическом растворителе, и в раствор добавляют катализатор асимметрического восстановления, и затем проводят реакцию асимметрического восстановления под давлением и в атмосфере водорода.
Примеры органического растворителя включают ароматические углеводороды (например, толуол, бензол, и другие), спирты (например, метанол, этанол и другие), алифатические сложные эфиры (этилацетат, н-пропилацетат, н-бутилацетат и другие), простые эфиры (например, изопропиловый эфир, диэтиловый эфир, тетрагидрофуран (THF) и другие), галогенированные углеводороды (например, дихлорметан, дихлорэтан и другие), амиды (например, N,N-диметилформамид и другие) и другие органические растворители. Эти растворители могут использоваться по отдельности или в смеси 2 или более из них, предпочтительными являются смешанный растворитель из толуола и метанола, смешанный растворитель из тетрагидрофурана и метанола, и другие подобные смеси. Количество используемого растворителя составляет от 1 до 1000 мл, предпочтительно от 2 до 20 мл, на 1 г исходного реагента.
Количество добавляемого катализатора асимметричного восстановления, используемого в настоящей реакции, составляет от 1/2 моля до 1/2000 моля, предпочтительно от 1/10 моля до 1/1000 моля, на 1 моль исходного реагента, и давление водорода составляет от 0,5 до 15 МПа, предпочтительно от 3 до 11 МПа.
Кроме того, температура реакции составляет от 0 до 150°C, предпочтительно от 10 до 80°C, и время реакции составляет от 0,5 до 200 часов, предпочтительно от 5 до 50 часов.
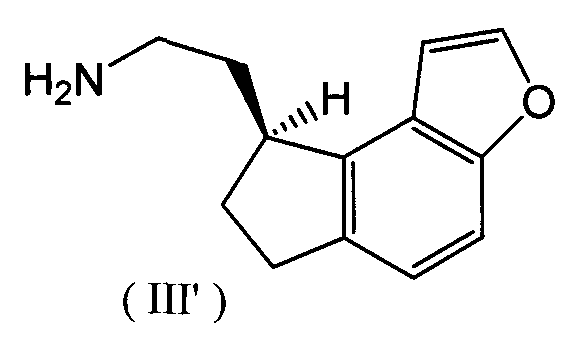
Реакцию каталитического восстановления упомянутой выше стадии (ii) проводят с использованием реакционного раствора, полученного на стадии (i). В реакционном растворе, полученном на стадии (i), соединение, представленное следующей формулой (III'), содержится в виде побочного продукта, и на стадии (ii) этот побочный продукт превращают путем каталитического восстановления в целевое соединение, то есть (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламин
Реакцию упомянутой выше стадии (ii) проводят следующим образом. А именно, к реакционному раствору, полученному на стадии (i), добавляют достаточное количество разбавленной хлористоводородной кислоты для растворения продукта реакции при температуре ниже 10°C, и реакционный продукт переводят в водный слой путем перемешивания или встряхивания, затем водный слой отделяют. Значение рН полученного водного слоя доводят до величины от 3 до 9, предпочтительно от 5 до 7, с помощью щелочи, такой как разбавленный водный раствор гидроксида натрия, и добавляют в него катализатор каталитического восстановления для восстановления под давлением и в атмосфере водорода.
Примеры катализатора каталитического восстановления, используемого в настоящей реакции, включают Pd-C, PtO2, Rh-Al2O3, (RhCl[Р(С6Н5)3]3) и другие подобные катализаторы. Количество используемого катализатора каталитического восстановления составляет от 1/2 моля до 1/2000 моля, предпочтительно от 1/10 моля до 1/500 моля, на 1 моль исходного реагента, используемого на стадии (i), и давление водорода составляет от 0,5 до 15 МПа, предпочтительно от 3 до 11 МПа.
Кроме того, температура реакции составляет от 40°C до 100°C, предпочтительно от 50°C до 70°C, и время реакции составляет от 0,5 до 200 часов, предпочтительно от 3 до 20 часов.
Реакционный раствор, полученный в результате реакции каталитического восстановления, фильтруют для удаления катализатора и обрабатывают с использованием известного, по существу, способа (например, концентрирования, кристаллизации, перекристаллизации, хроматографии и других способов) с получением (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламина.
Кроме того, образующийся (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламин может быть превращен в требуемую соль с помощью традиционного способа.
Когда реакцию и последующую обработку вышеупомянутой стадии (ii) проводят без корректировки pH, то есть в сильно кислой среде при pH ниже 1, образуются упомянутое выше производное бензофурана (III') в количестве от 5 до 10%, и димер дигидробензофурана (IV') в количестве 0,2%, которые являются побочными продуктами. В отличие от этого в результате осуществления настоящего изобретения образование этих побочных продуктов может быть снижено до величины менее 0,07% и менее 0,02% соответственно при величине pH от 3 до 9, предпочтительно pH от 5 до 7.
Способ получения кристалла (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамида настоящего изобретения описан ниже. Способ состоит из стадии (a) - пропионилирования аминогруппы (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламина, и стадии (b) - кристаллизации из реакционного раствора, полученного на стадии (a). А именно, на стадии (a) аминогруппа (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламина, полученного в вышеупомянутом способе, взаимодействует с пропионилирующим агентом с образованием пропионилата. Если исходный реагент, (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламин, используют в форме соли, его превращает в свободное соединение традиционным способом с последующим проведением реакции пропионилирования. Примеры пропионилирующего агента включают пропионилгалогениды, такие как пропионилхлорид и пропионилбромид. Количество используемого пропионилирующего агента составляет 1-2 моля на 1 моль (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламина.
Реакцию проводят в растворителе, и примеры растворителя включают простые эфиры, такие как тетрагидрофуран, диэтиловый эфир, диоксан и 1,2-диметоксиэтан, спирты, такие как метанол, этанол и пропанол, углеводороды, такие как бензол, толуол, циклогексан и гексан, амиды, такие как N,N-диметилформамид и N,N-диметилацетамид, галогенированные углеводороды, такие как дихлорметан, хлороформ, четыреххлористый углерод и 1,2-дихлорэтан, нитрилы, такие как ацетонитрил и пропиононитрил, сульфоксиды, такие как диметилсульфоксид, и другие подобные растворители, и смеси из них, в частности, предпочтительным является тетрагидрофуран. Время реакции обычно составляет от 5 минут до 48 часов, предпочтительно от 30 минут до 6 часов. Температура реакции обычно составляет от -20 до 200°C, предпочтительно от -10 до 50°C.
На стадии (b) кристаллы (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-илэтил]пропионамида кристаллизуют в результате добавления водного растворителя к реакционному раствору, полученному на стадии (a). Примеры водного растворителя включают водопроводную воду, чистую воду, очищенную воду и так далее. Количество добавляемого водного растворителя составляет по объему от 0,5 до 5 от объема реакционного раствора, полученного на стадии (a). Температура кристаллизации составляет обычно от -20 до 60°C, предпочтительно от -10 до 40°C.
Кристаллы (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамида получают с высоким выходом порядка 97%, отфильтровывая осажденные кристаллы. Кристаллы (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил] пропионамида высокой чистоты могут быть получены дополнительной перекристаллизацией полученных кристаллов из смеси этанол-вода (1:2).
Хотя кристаллы (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамида, полученные в способе настоящего изобретения, вероятно, содержат в качестве примесей соединения, представленные следующими формулами (I)-(IV), (в настоящем описании, далее, иногда называемые как соединение (I)-(IV) соответственно), содержание каждого из соединений (I)-(IV) составляет 0,15% по массе или менее, и, кроме того, суммарное содержание соединений (I)- (IV) составляет около 0,20% по массе или менее
Например, в кристаллах (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамида, полученных в способе настоящего изобретения, содержание каждого соединения (III) и (IV) составляет ниже предела обнаружения, то есть менее чем 0,02% по массе (ВЭЖХ), содержание соединения (I) составляет около 0,1% по массе или менее (предпочтительно 0,03% по массе или менее), и содержание соединения (II) составляет от 0,02 до 0,15% по массе, и, кроме того, суммарное содержание соединений (I)-(IV) составляет около 0,20% по массе или менее.
Как было описано выше, кристаллы (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамида высокой чистоты могут быть получены путем контролирования содержания примесей, в результате чего ожидаются улучшение кристалличности, сопровождаемое улучшением чистоты, улучшением стабильности и других подобных характеристик. Кроме того, когда (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамид применяют в качестве лекарственного препарата, уменьшение примесей имеет чрезвычайно важное значение с точки зрения гарантии качества для пациентов. Согласно настоящему изобретению такие кристаллы могут быть получены в промышленном масштабе. Кроме того, путем применения таких кристаллов может быть получена композиция настоящего изобретения в соответствии с известным способом.
(S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил) этил]пропионамид настоящего изобретения обладает физиологической активностью, такой как аффинность к рецептору мелатонина, кроме того, обладает низкой токсичностью и меньшими побочными эффектами. Поэтому он может применяться для предотвращения и лечения нарушения ритма сна-бодрствования, нарушения суточного ритма организма, нарушения биоритма в результате работы в три смены и так далее, сезонной депрессии, заболевания репродуктивной и нейроэндокринной систем, старческого слабоумия, болезни Альцгеймера, различных заболеваний, связанных со старением (например, профилактика старения и так далее), нарушения мозгового кровообращения (апоплексия мозга и так далее), травмы головы, травмы костного мозга, стресса, эпилепсии, спазма мышц, тревожного состояния, депрессии, болезни Паркинсона, гипертонии, глаукомы, рака, нарушения сна, диабета, мигрени и других подобных заболеваний, и, кроме того, он также эффективен для иммуномодуляции, нотропизма, стабилизации умственной деятельности и регулирования овуляция (например, контрацепции).
Соединения настоящего изобретения могут применяться в комбинации с антидепрессантами (например, имипрамином, кломипрамином, ноксиптилином, фенелзином, гидрохлоридом амитриптилина, гидрохлоридом нортриптилина, амоксапином, гидрохлоридом гимиансерина, гидрохлоридом мапротилина, сульпиридом, малеатом флувоксамина, гидрохлоридом тразодона, гидрохлоридом пароксетина, гидрохлоридом милнаципрана, флуоксетином, венлафаксином, митразапином, сертралином, циталопрамом, дулоксетином, ребоксетином, моклобемидом), транквилизаторами (например, диазепамом, оксазоламом, бромозепамом, алпразоламом, клоназепамом, буспироном, цитратом тандоспирона), нормотимическим средством (например, солью лития, вальпроевой кислотой, карбамазепином), средством против деменции (например, такрином, донепезилом, ривастигмином, галантамином, мемантином), нейролептиками (например, галоперидолом, оланзапином, рисперидоном, кветиапином, зипрасидоном, хлорпромазином, сульпиридом, арипипразолом), противоэпилептическим средством (например, фенобарбиталом, габапентином, тиагабином, прегабалином), средством для улучшения мозгового кровообращения, метаболическим стимулятором мозга и другими подобными средствами.
Примеры формы введения включают (1) введение одного препарата, полученного в результате разработки рецептуры соединения настоящего изобретения, и совместно используемого лекарственного препарата одновременно, (2) одновременное введение двух видов препаратов, полученных в результате разработки рецептуры соединения настоящего изобретения, и совместно используемого лекарственного препарата раздельно, через один и тот же путь введения, (3) последовательное и чередующееся введение двух видов препаратов, полученных в результате разработки рецептуры соединения настоящего изобретения, и совместно используемого лекарственного препарата раздельно, через один и тот же путь введения, (4) одновременное введение двух видов препаратов, полученных в результате разработки рецептуры соединения настоящего изобретения, и совместно используемого лекарственного препарата раздельно, через различные пути введения и (5) последовательное и чередующееся введение двух видов препаратов, полученных в результате разработки рецептуры соединения настоящего изобретения, и совместно используемого лекарственного препарата раздельно, через различные пути введения (например, введение в следующем порядке: соединение настоящего изобретения совместно используемый лекарственный препарат, или введение в обратном порядке). Доза совместно используемого лекарственного препарата может быть выбрана соответствующим образом на основе применяемой в клинике дозы. Кроме того, соотношение между соединением настоящего изобретения и совместно используемого лекарственного препарата может быть соответствующим образом выбрано в зависимости от субъекта введения, пути введения, заболевания субъекта, симптома, комбинации и других подобных параметров. Например, если субъектом введения является человек, то может быть использовано от 0,01 до 100 частей по массе совместно используемого лекарственного препарата на 1 часть по массе соединения настоящего изобретения.
(S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил) этил]пропионамид настоящего изобретения может быть использован в качестве исходного фармацевтического материала путем измельчения в струйной мельнице и так далее, и постоянство содержания соединения в препарате и другие подобные параметры могут быть гарантированы путем корректировки размера частиц (среднего размера) от 1 до 10 мкм.
Размер частиц может быть измерен следующим образом путем использования коммерчески доступной измерительной аппаратуры.
В 100 мл колбу Эрленмейера, снабженную пробкой, загружают 0,05 г образца и добавляют туда 50 мл дисперсионной среды. Смесь подвергают действию ультразвуковой волны в течение 5 минут при встряхивании и перемешивании с получением суспензии. К 40 мл дисперсионной среды добавляют около 100 мкл этой суспензии и тест проводят при следующих условиях.
Дисперсионная среда
0,1% раствор натрийлаурилсульфата, насыщенный (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамид-ом.
Аппаратура
Система HELOS system KF (Sympatec GmbH)
Сенсорный датчик HELOS sensor
Дисперсионная установка CUVETTE (смачиватель-диспергатор)
Стандартное программное обеспечение HELOS: WINDOX 3.2 (для Windows) или аналогичное
Условие измерения
Фокусное расстояние 100 мм
Скорость перемешивания 50%
Время забора образца 1 секунда
Время измерения 10 секунд
Соединение настоящего изобретения может быть безопасно введено перорально, или парентерально (например, местное, ректальное, внутривенное введение и так далее) само по себе, или путем получения из него фармацевтических препаратов, таких как таблетки (включая таблетки с сахарным покрытием, таблетки с пленочным покрытием), порошки, гранулы, капсулы, растворы, эмульсии, суспензии, инъекции, суппозитории, в форме с замедленным высвобождением и клейкие составы путем смешения с фармакологически приемлемым носителем традиционным способом (например, способом, описанным в Japanese Pharmacopoeia, и так далее). Содержание соединения в фармацевтической композиции составляет обычно от 0,01 до 100% по массе от суммарной массы композиции.
Настоящее изобретение будет далее объяснено в деталях посредством следующих справочных примеров и примеров, но эти примеры не ограничивают настоящее изобретение. Кроме того, каждое сокращение в справочных примерах и примерах имеет следующие значения.
DBF: 2,3-дигидробензофуран
FBA: 2,3- дигидробензофуран-5-карбальдегид
PPN: этил-(E)-3-(2,3-дигидробензофуран-5-ил)пропеноат
PPE: этил-3-(2,3-дигидробензофуран-5-ил)пропионат
DBA: 3-(6,7-дибром-2,3-дигидробензофуран-5-ил)пропионовая кислота
BIF: 4,5-дибром-l,2,6,7-тетрагидро-8H-индено[5,4-b]фуран-8-он
THI: 1,2,6,7-тетрагидро-8H-индено[5,4-b]фуран-8-он
ICN: (E)-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-илиден) ацетонитрил
EAI-HCl: гидрохлорид (E)-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-илиден)этиламина
(S)-AMI-HCl: гидрохлорид (S)-(1,6,7,8-тетрагидро-2H-индено [5,4-b]фуран-8-илиден)этиламина
Справочный пример 1
2,3-Дигидробензофуран-5-карбальдегид

2,3-Дигидробензофуран (100 г, 832 ммоль) и N,N-диметилформамид (134 г, 1830 ммоль) смешивали, нагревали и добавляли к ним оксихлорид фосфора (255 г, 1643 ммоль) при температуре от 70 до 80°C в течение 2 часов. Реакционную смесь нагревали при температуре от 80 до 90°C и перемешивали в течение 7,5 часов. Затем образующуюся смесь добавляли по каплям в воду (1000 г) при охлаждении и перемешивали при комнатной температуре в течение 5 часов. Образующуюся смесь экстрагировали толуолом, и экстракт промывали последовательно водой, насыщенным водным раствором бикарбоната натрия и водой, и органический слой концентрировали под вакуумом с получением толуольного раствора названного соединения (количество 340 г, кажущийся выход 100%).
Справочный пример 2
Этил-(E)-3-(2,3-дигидробензофуран-5-ил)пропеноат

К раствору (340 г) 2,3-дигидробензофуран-5-карбальдегида (832 ммоль) в толуоле, полученному на вышеприведенной стадии, добавляли по каплям триэтилфосфоноацетат (205 г, 916 ммоль) при охлаждении. Затем по каплям добавляли суспензию т-бутилата натрия (88,0 г, 1187 ммоль) в толуоле (530 г), и перемешивали в течение 1 часа, и далее добавляли по каплям уксусную кислоту (20 г) и воду (500 г). Реакционную смесь подогревали до комнатной температуры и слои разделяли. Органический слой промывали последовательно водой, насыщенным водным раствором бикарбоната натрия и водой и органический слой концентрировали до объема меньше 300 мл под вакуумом. Затем к остатку добавляли метанол (396 г) и остаток растворяли при нагревании. К раствору добавляли по каплям воду (500 г) при комнатной температуре и перемешивали до выпадения кристаллов, которые отфильтровывали и сушили при пониженном давлении с получением названного соединения (количество 161 г, выход 88,1%).
Справочный пример 3
Этил-3-(2,3-дигидробензофуран-5-ил)пропионат

Этил-(E)-3-(2,3-дигидробензофуран-5-ил)пропеноат (50,0 г, 227 ммоль) растворяли в уксусной кислоте (312 г) и реакционную систему заполняли азотом. Затем добавляли к раствору 5% Pd/C (4,96 г, в виде сухой массы) и подавали под давлением водород от 196 до 294 КПа. Реакцию проводили при 50°C в течение 1 часа при давлении от 196 до 294 КПа. Катализатор отфильтровывали и промывали уксусной кислотой (208 г) с получением раствора названного соединения в уксусной кислоте (количество 569 г, кажущийся выход 100%).
Справочный пример 4
3-(6,7-Дибром-2,3-дигидробензофуран-5-ил)пропионовая кислота
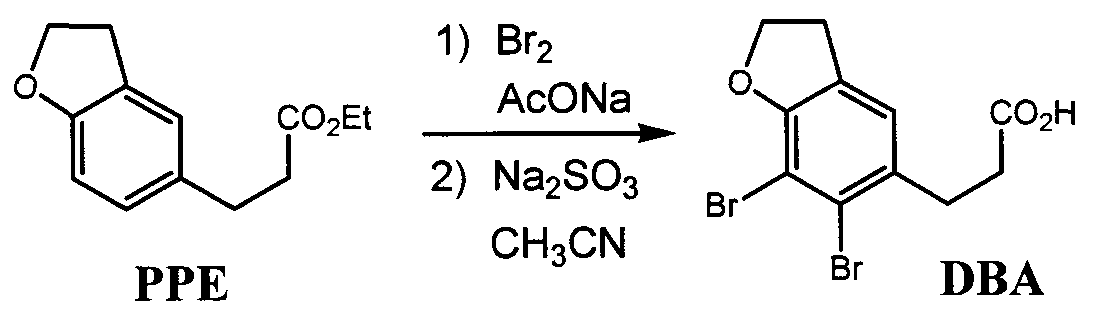
К раствору PPE в уксусной кислоте (569 г, 227 ммоль), полученному на вышеприведенной стадии, добавляли безводный ацетат натрия (18,6 г) и по каплям добавляли бром (222 г) при перемешивании и охлаждении в течение 2 часов. После взаимодействия в течение 4 часов при комнатной температуре добавляли по каплям реакционную смесь к охлажденному 15% водному раствору сульфита натрия (670 мл) и перемешивали в течение 30 минут. К реакционному раствору добавляли ацетонитрил (118 г) и проводили реакцию в течение 2 часов при нагревании с обратным холодильником, затем постепенно охлаждали и перемешивали в течение 1 часа до кристаллизации. Кристаллы отфильтровывали, промывали водой и сушили под вакуумом с получением названного соединения (количество 63,3 г, выход 73,2%).
Справочный пример 5
4,5-Дибром-1,2,6,7-тетрагидро-8H-индено[5,4-b]фуран-8-он

Смешивали 3-(6,7-дибром-2,3-дигидробензофуран-5-ил)пропионовую кислоту (40,0 г, 114 ммоль), o-дихлорбензол (182 г) и N,N-диметилформамид (0,1 г) и добавляли по каплям тионилхлорид (17,7 г, 149 ммоль) при температуре 42°C с последующим перемешиванием в течение от 30 до 40 минут с получением раствора кислого хлорида. Затем к раствору добавляли несколькими порциями безводный хлорид алюминия (17,5 г, 132 ммоль) при охлаждении льдом и перемешивали в течение 30 минут. Отдельно приготавливали метанол (475 г) и реакционный раствор добавляли по каплям в метанол для кристаллизации. К кристаллизационному раствору добавляли по каплям воду (76 г) при охлаждении и перемешивали в течение 30 минут. Кристаллы отфильтровывали и влажные кристаллы промывали последовательно метанолом, водой, насыщенным водным раствором бикарбоната натрия, водой и метанолом с последующей сушкой под вакуумом с получением 31,6 г названного соединения (выход 92,2%).
Пример 1
1,2,6,7-Тетрагидро-8H-индено[5,4-b]фуран-8-он

(1) Смешивали 4,5-дибром-1,2,6,7-тетрагидро-8H-индено[5,4-b]фуран-8-он (280 кг, 843 моль), безводный ацетат натрия (173 кг, 2109 моль), метанол (6384 л) и реакционную систему заполняли азотом. Затем к реакционной смеси добавляли 10% Pd/C (30,8 кг, в виде сухой массы), и подавали под давлением водород от 0,29 до 0,49 МПа, и проводили каталитическое восстановление при 40°C в течение 8 часов при перемешивании при такой скорости перемешивания, при которой суммарный объемный коэффициент массопередачи газ-жидкость KLa(1/час) составлял величину около 15. Катализатор отфильтровывали, и фильтрат концентрировали при пониженном давлении, и затем к остатку добавляли воду с последующим концентрированием при пониженном давлении для замещения растворителя, охлаждением и перемешиванием в течение 1 часа для вызревания. Кристаллизационный раствор фильтровали с получением влажных кристаллов названного соединения (количество 127 кг в виде сухой массы, выход 86,6%). Содержание димера во влажных кристаллах было меньше чем 0,1% по массе.
(2) Стадия очистки
Смешивали влажные кристаллы (127 кг в виде сухой массы), активированный уголь (6 кг, торговая марка Shirasagi A) и метанол (1723 л) и перемешивали в течение 1 часа с обратным холодильником и фильтровали. Фильтрат и промывки концентрировали при пониженном давлении, затем остаток кипятили в течение 1 часа с обратным холодильником и охлаждали. Добавляли воду (306 л) при охлаждении, выдерживали для вызревания в течение 1 часа и осадок отфильтровывали с последующей сушкой при пониженном давлении с получением названного соединения (количество 117 кг, выход 92,1%).
(3) Суммарный коэффициент массопередачи газ-жидкость
Здесь суммарный коэффициент массопередачи газ-жидкость определяли методом с Na2SO3.
1) Метод с Na2SO3 (метод с сульфитом натрия)
(a) Принцип метода
Сульфит натрия (Na2SO3) в водном растворе превращается в сульфат натрия (Na2SO4) в результате реакции с кислородом, который поступал в раствор из воздуха. Скорость реакции является достаточно быстрой по сравнению со скоростью абсорбции кислорода (абсорбция кислорода является лимитирующей стадией). Поэтому скорость абсорбции кислорода (NA) может быть определена в результате измерения изменения концентрации сульфита натрия.
Итак, суммарный объемный коэффициент массопередачи газ-жидкость KLa определяется следующим уравнением:
NA=KLa(C*-C)
Так как фактически в этой измерительной системе концентрация растворенного кислорода в водном растворе сульфита натрия может быть принята равной 0, то получаем следующее уравнение:
KLa=NA/C*
С другой стороны, растворимость кислорода в водном растворе может быть выражена следующим образом на основе закона Генри
C*=p/H
Из этих уравнений может быть вычислен KLa.
Кроме того, символы в вышеприведенном уравнении имеют следующие значения:
KLa: суммарный объемный коэффициент массопередачи газ-жидкость [1/час]
NA: скорость абсорбции кислорода [моль/л-час]
C: концентрация кислорода в жидкости [моль/л]
C*: растворимость кислорода при насыщении [моль/л]
p: парциальное давление кислорода в газовой фазе [Па]
H: константа Генри [Па-л/час]
(b) Метод измерения
(i) Чистую воду (475 мл, такой же объем жидкости, как при вводимой норме BIF 23,34 г) загружают в автоклав объемом 1 л (стеклянный реактор типа TEM-V-1000).
(ii) Добавляют Na2SO3 (9,5 г) и смешивают в течение 2 минут до растворения.
(iii) К водному раствору Na2SO3 добавляют приготовленный 0,1 моль/л раствор CuSO4 (4,75 мл) (концентрация CuSO4 после добавления=1x10-3 M) и реакционный раствор перемешивают медленно в течение 1 минуты (инициирование реакции).
(iv) Сразу же берут точно аликвоту 10 мл раствора и титруют согласно следующей методике (c) (объем, пошедший на титрование=T1 [мл])
(v) Реакционный раствор перемешивают при данной скорости вращения в течение данного времени Δθ (= 1,0 [час]). За это время определенное количество воздуха пропускают в верхнюю часть сосуда для предотвращения снижения парциального давления кислорода в газовой фазе в автоклаве (около 200 мл/л).
(vi) Берут точно аликвоту 10 мл раствора (v) и титруют согласно следующей методике (c) (объем, пошедший на титрование=T2 [мл])
(vii) По результату титрования рассчитывают скорость абсорбции кислорода NA согласно следующему уравнению. Здесь F обозначает коэффициент для 0,1N раствора йода.

(с) Метод титрования (метод титрования сульфита натрия)*1
(i) Заранее готовят колбу Эрленмейера емкостью 200 мл, содержащую чистую воду (100 мл), буфер уксусная кислота-ацетат натрия*2 (10 мл) и 0,1 N раствор йода (40 мл).
(ii) Осторожно добавляют в нее аликвоту раствора (10 мл).
(iii) По истечении 5 минут аликвоту раствора титруют 0,1N раствором тиосульфата натрия с использованием раствора крахмала*3 (0,5-1 мл) в качестве индикатора.
*1: Принцип титрования основан на том, что после окисления йодом сульфитного аниона, присутствующего в аликвоте раствора, оставшийся йод оттитровывают тиосульфатом натрия, и каждая стадия может быть представлена следующими уравнениями реакции:
окисление сульфитного аниона: Na2SO3+I2+Н2О → 2NaI+H2SO4 титрование йода: I2+2Na2S2O3 → 2NaI+Na2S4O6
*2: 75 г ацетата натрия (CH3COONa·3H20) растворяют в 500 мл водного раствора уксусной кислоты (CH3COOH:H20=1:2).
*3: 1,0 г крахмала протирают и смешивают с 10 мл воды, и полученную смесь вводят в 200 мл горячей воды. После кипячения до момента, когда раствор становится полупрозрачным, его охлаждают.
Справочный пример 6
(Е)-(1,6,7,8-Тетрагидро-2H-индено[5,4-b]фуран-8-илиден) ацетонитрил
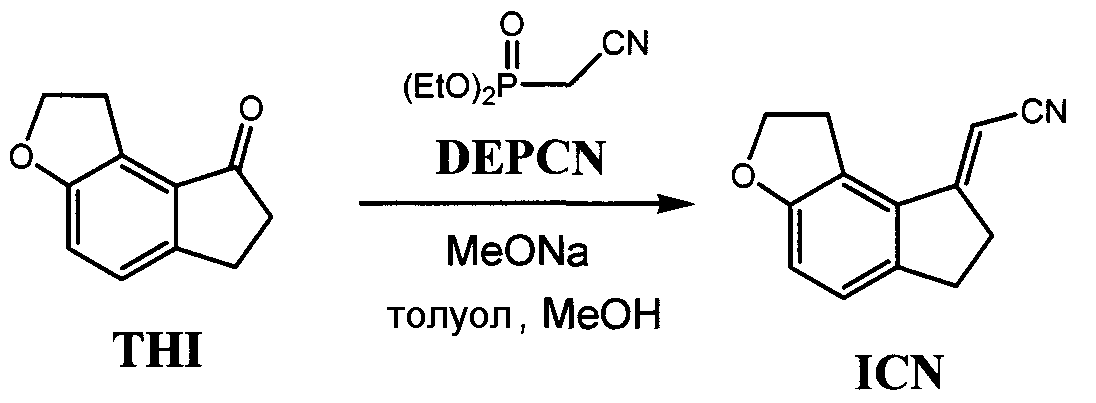
К раствору толуола (184 г), 1,2,6,7-тетрагидро-8H-индено[5,4-b]фуран-8-она (8,5 г, 48,9 ммоль) и диэтилцианометилфосфоната (10,4 г, 58,7 ммоль) добавляли по каплям 28% раствор метоксида натрия в метаноле (11,3 г) в течение 1 часа при охлаждении льдом и реакцию проводили в течение 4 часов. К реакционному раствору добавляли по каплям воду (85 г) и нагревали, затем разделяли слои. Органический слой промывали водой и фильтровали для удаления пылинок под давлением. Органический слой концентрировали при пониженном давлении, к остатку добавляли метанол и концентрировали при пониженном давлении для замещения растворителя. После перемешивания в течение 1 часа при нагревании с обратным холодильником раствор охлаждали и подвергали вызреванию в течение 1 часа. Кристаллизационный раствор отфильтровывали и кристаллы сушили при пониженном давлении с получением названного соединения (количество 8,1 г, выход 84,4%).
Справочный пример 7
Гидрохлорид (E)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-илиден)этиламина

К смешанной суспензии (E)-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-илиден)ацетонитрила (10,0 г, 50,7 ммоль) в толуоле (37,5 мл) и метаноле (12,5 мл) добавляли активированный кобальт (7,22 г) и 14,4% водный раствор гидроксида калия (1,4 г) и перемешивали в течение 6,5 часов при температуре от 34 до 50°C в атмосфере водорода (0,2 МПа). Реакционный раствор фильтровали и к фильтрату добавляли толуол (170 мл) и метанол (35 мл) для разделения слоев. К органическому слою добавляли 0,5N хлористоводородную кислоту (101 мл) и перемешивали в течение 30 минут при температуре от 25 до 30°C. Затем слои разделяли и добавляли активированный уголь (1 г) к водному слою с последующим перемешиванием. Активированный уголь удаляли фильтрацией с получением водного раствора названного соединения (246 г, вес чистого соединения 12,0 г, выход 99,6%).
Пример 2
Гидрохлорид (S)-2-(l,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламина

К водному раствору (E)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-илиден)этиламина (1979 кг, вес чистого соединения 122 кг, 513 моль) добавляли толуол (532 л) и 5% водный раствор гидроксида натрия (456 л) и перемешивали. Слои разделяли и к органическому слою добавляли метанол (155 кг) и [RuCl(бензол)(R)-BINAP]Cl (894 г) в атмосфере азота с последующим перемешиванием при 80°C в течение 15 часов в атмосфере водорода (4,9 МПа). Реакционный раствор охлаждали и добавляли воду (330 л) и концентрированную хлористоводородную кислоту (52,3 кг) при температуре ниже 30°C с последующим перемешиванием в течение 30 минут, затем слои разделяли. Водный слой промывали толуолом (195 л) и доводили pH до 6,0 путем добавления 5% водного раствора NaOH к водному слою (содержащему 5,0% соединения III'. Добавляли 5% Pd-C (50% влажности, 9,7 кг) и перемешивали при 60°C в течение 6 часов в атмосфере водорода (4,9 МПа). Реакционную смесь фильтровали, и pH фильтрата доводили до 6,0 с помощью 5% водного раствора NaOH или разбавленной хлористоводородной кислоты с последующим концентрированием при пониженном давлении. Остаток перекристаллизовывали из смешанного раствора н-бутанола и воды с получением названного соединения (88,6 кг, выход 73,0%, соединение (III') не обнаруживалось, соединение (IV') не обнаруживалось).
Кроме того, содержание соединения (III') и соединения (IV') (димера) в полученных кристаллов названного соединения определяли при помощи ВЭЖХ при следующих условиях.
детектор: УФ-денситометр (длина волны для измерения 220 нм)
колонка: Develosil UG-3, внутр. диаметр 4,6 мм x 75 мм
температура колонки: заданная температура около 25°C
подвижная фаза: смешанный раствор 0,1 моль/л дигидрофосфата калия (pH 3,0)/метанол (75:25)
Пример 3
(i) (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-
ил)этил]пропионамид

К смешанному раствору гидрохлорида (S)-2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этиламина (74 кг, 309 моль) в тетрагидрофуране (185 л) и водопроводной воде (259 л) добавляли 30% водный раствор гидроксида натрия (70 л) и пропионилхлорид (32,8 кг) и перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли водопроводную воду (592 л) и охлаждали. Осажденные кристаллы отфильтровывали и сушили при пониженном давлении с получением названного соединения (78,0 кг, выход 97,4%).
(ii) Стадия очистки
Кристаллы (77,3 кг, 298 моль), полученные в (i), растворяли в смешанном растворе (178 кг) этанола и очищенной воды (10:1) и добавляли активированный уголь (0,78 кг), затем перемешивали в течение 10 минут с последующей фильтрацией (промывали смешанным раствором (74 кг) этанола и очищенной воды (10:1)). К фильтрату добавляли воду (588 л) при нагревании, охлаждали, и осажденные кристаллы отфильтровывали, и сушили при пониженном давлении. Полученные кристаллы измельчали при помощи струйной мельницы с получением названного соединения (74,0 кг, выход 95,7%, соединение (I) 0,02%, соединение (II) 0,06%, соединение (III) и (IV) менее чем 0,02%, суммарный аналогичный материал 0,08%).
(iii) Условия анализа
Содержание соединений (I)-(IV) в кристаллах названного соединения, полученных в (ii), определяли с помощью ВЭЖХ при следующих условиях:
детектор: УФ-денситометр (длина волны для измерения 288 нм)
колонка: YMC-Pack ODS-AM AM-302,5 мкм, внутр. диаметр 4,6 мм×150 мм (фирмы YMC)
температура в колонке: заданная температура около 25°C
подвижная фаза:
градиент
Полученная ВЭЖХ хроматограмма приведена на чертеже. Как видно на чертеже 1, соединения (I) и (II) проявлялись с каждой стороны от основного пика (S)-N-[2-(1,6,7,8-тетрагидро-2H-индено[5,4-b]фуран-8-ил)этил]пропионамида, однако содержание каждого из соединений (III) и (IV) было ниже предела обнаружения, то есть менее чем 0,02%.
Промышленное применение
Согласно способу настоящего изобретения путем регулирования pH реакционного раствора на стадии каталитического восстановления и его последующей обработки могут быть получены с высоким выходом в промышленном масштабе оптически активные производные амина высокой чистоты, которые применяются в качестве лекарственного препарата и исходных фармацевтических материалов высокого качества.
Реферат
Изобретение относится к промышленному способу получения оптически активных производных амина высокой чистоты с высоким выходом при подавлении образования побочных продуктов, который включает асимметрическое восстановление (Е)-2-(1,6,7,8-тетрагидро-2Н-индено[5,4-b]фуран-8-илиден)этиленамина, каталитическое восстановление полученного продукта при температуре от 40 до 100°С и рН от 3 до 9, пропионилирование полученного (S)-2-(1,6,7,8-тетрагидро-2Н-индено[5,4-b]фуран-8-ил)этиламина, и затем кристаллизацию из реакционной смеси. 2 н. и 4 з.п. ф-лы., 1 табл., 1 ил.
Комментарии