Средство для ингибирования фермента тирозил-днк-фосфодиэстеразы 1 человека на основе производных пентафуранозилнуклеозидов - RU2748103C1
Код документа: RU2748103C1
Чертежи



Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к молекулярной биологии, медицинской химии и биохимии, конкретно к соединениям, представляющим собой производные пентафуранозилнуклеозидов (Фиг. 1.), обладающих биологической активностью в отношении фермента тирозил-ДНК-фосфодиэстеразы 1 человека (Tdp1), а именно проявлять ингибирующие свойства в отношении Tdp1.
Соединения, представленные в данном изобретении, могут использоваться для создания новых прототипов лекарственных средств, направленных на проведение комбинированной противоопухолевой терапии.
Уровень техники
Традиционная химиотерапия онкологических заболеваний направлена на повреждение ДНК злокачественных клеток, и одним из факторов, влияющих на ее результат, является эффективность систем репарации ДНК. Во многих раковых клетках отдельные ферменты репарации ДНК гиперэкспрессированы, что в сочетании с ускоренной клеточной пролиферацией приводит к резистентности таких опухолей к лекарственным препаратам. Развитие молекулярной биологии привело к принципиальному изменению подходов в создании новых противоопухолевых препаратов.
Нуклеозиды и их аналоги входят в состав биополимеров и участвуют в процессах хранения и реализации наследственной информации. В настоящее время известно около 100 лекарств на основе нуклеозидов, из них примерно четверть составляют противоопухолевые лекарства.
Важным стратегическим направлением лечения онкологических заболеваний является избирательное подавление активности ряда ферментов, вовлеченных в регуляцию жизнедеятельности клеток. В последнее время в качестве потенциальных лекарств рассматриваются соединения-ингибиторы репарации ДНК [Hosoya N., Miyagawa K., Targeting DNA damage response in cancer therapy. // Cancer Sci. 2014, 105, 370-388]. При лишении раковой клетки способности к восстановлению поврежденной ДНК, существенно возрастает эффективность традиционных методов лечения.
Поиск ингибиторов ключевых ферментов репарации ДНК относится к перспективным направлениям медицинской химии и является одним из путей создания эффективной терапии при лечении сердечно-сосудистых, нейродегенеративных и онкологических заболеваний [Laev S.S., Salakhutdinov N.F., Lavrik О.I., Tyrosyl-DNA phosphodiesterase inhibitors: Progress and potential // Bioorg. Med. Chem. 2016, 24, 5017-5027].
К числу перспективных мишеней относится фермент тирозил-ДНК-фосфодиэстераза 1 (Tdp1). Данный фермент является важной мишенью при проведении противоопухолевой терапии, основанной на применении ингибиторов топоизомеразы 1 (Top1) [Negrini S., Gorgoulis V.G., Halazonetis T.D., Genomic instability-an evolving hallmark of cancer. // Nat. Rev. Mol. Cell. Biol. 2010, 11, 220-228]. Tdp1 играет ключевую роль в удалении аддуктов Top1-ДНК, стабилизированных ингибиторами Top1, такими как камптотецин и его клиническими производными. Мутация в гене Tdp1 делает клетки гиперчувствительными к камптотецину - каноническому ингибитору Top1 [El-Khamisy S.F., Masutani М., Suzuki Н., Caldecott K.W., A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. // Nucleic Acids Res., 2003, 31, 5526-5533]. Было показано, что мыши, нокаутные по Tdp1, гиперчувствительны к камптотецину и его производным. И, наоборот, гиперэкспрессия Tdp1 приводит к защите клеток от повреждений ДНК, вызванных камптотецином [Nivens М.С., Pouliot J.J., Felder Т., Pena М.М.О., Spencer Н.Т., Galloway, А.Н., Engineered resistance to camptothecin and antifolates by retroviral coexpression of tyrosyl DNA phosphodiesterase-I and thymidylate synthase. // Cancer Chemother. Pharmacol., 2004, 53, 107-115; Alagoz M., Gilbert D.C., El-Khamisy S., Chalmers A.J., DNA Repair and Resistance to Topoisomerase I Inhibitors: Mechanisms, Biomarkers and Therapeutic Targets. // Curr. Med. Chem., 2012, 19, 3874-3885]. При подавлении экспрессии Tdp1 с помощью миноциклина усиливается антиметастатический эффект иринотекана и увеличивается продолжительность жизни экспериментальных животных, в то время как сам миноциклин обладает незначительной противоопухолевой активностью [Huang Н.-С., Liu J., Baglo Y., Rizvi I., Anbil S., Pigula M., Hasan Т., Mechanism-informed Repurposing of Minocycline Overcomes Resistance to Topoisomerase Inhibition for Peritoneal Carcinomatosis. // Mol. Cancer Ther., 2018, 17, 508-520]. Более того, в опухолях кишечника с гиперэкспрессией Tdp1 снижен ответ на терапию иринотеканом [Meisenberg С., Gilbert D.C., Chalmers A., Haley V., Gollins S., Ward S.E., El-Khamisy, S.F., Clinical and Cellular Roles for TDP1 and TOP1 in Modulating Colorectal Cancer Response to Irinotecan. // Mol. Cancer Ther. 2015, 14, 575-585]. Таким образом, подавление активности Tdp1 может значительно усилить терапевтический эффект ингибиторов Top1.
В литературе описан небольшой ряд ингибиторов Tdp1, эффективных при концентрациях в диапазоне значений IC50 0.15-100 мкМ. Данные ингибиторы представляют собой производные гликозидов: неомицина, нетилмицина, спектиномицина [Liao Z., Thibaut L., Jobson A., Pommier Y., Inhibition of Human Tyrosyl-DNA Phosphodiesterase by Aminoglycoside Antibiotics and Ribosome Inhibitors // Mol. Pharmacol. 2006, 70, 366-372]. Однако для проявления ингибиторных свойств необходимо использование данных соединений в достаточно высоких концентрациях, при этом селективность ингибирования фермента Tdp1 очень низкая. В том же диапазоне концентраций, что и гликозидные производные, ингибируют Tdp1 и диамидины [Huang S.N., Pommier Y., Marchand С., Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) inhibitors // Expert Opin. Ther. Pat. 2011, 9, 1285-1292]. Активно изучается способность ингибировать Tdp1 производными инденоизохинолина [Elsayed M.S.A., Su Y., Wang P., Sethi Т., Agama K., Ravji A., Redon C.E., Kiselev E., Horzmann K.A., Freeman J.L., Yves Pommier Y., Cushman M., Design and Synthesis of Chlorinated and Fluorinated 7 Azaindenoisoquinolines as Potent Cytotoxic Anticancer Agents That Inhibit Topoisomerase I // J. Med. Chem. 2017, 60, 5364-5376]. Исследована ингибиторная активность в отношении Tdp1 производных 2-циано-3-фенилпроп-2-ентиоамида и тиоксотиазолидинона. Было показано, что данные соединения ингибируют фермент со значениями IC50=0.9-100 мкМ [Sirivolu V.R., Vernekar S.K., Marchand С., Naumova А., Chergui A., Renaud A., Stephen A.G., Chen F., Sham Y.Y., Pommier Y., Wang Z., 5-Arylidenethioxothiazolidinones as inhibitors of tyrosyl-DNA phosphodiesterase I // J.Med.Chem. 2012, 55, 8671-8684]. Были обнаружены также и новые классы ингибиторов Tdp1, а именно аналоги бензопентатиепина, эффективных при концентрациях в диапазоне значений IC50 0.2-3.7 мкМ [Zakharenko A., Khomenko Т., Zhukova S., Koval О., Zakharova О., Anarbaev R., Lebedeva N., Korchagina D., Komarova N., Vasiliev V., Reynisson J., Volcho K., Salakhutdinov N., Lavrik O. // Bioorg. Med. Chem. 2015, 23, 2044-2052], производные кумаринов [Khomenko Т., Zakharenko A., Odarchenko Т., Arabshahi H.J., Sannikova V., Zakharova O., Korchagina D., Reynisson J., Volcho K., Salakhutdinov N., Lavrik O. // Bioorg. Med. Chem. 2016, 24, 5573-5581] и производные усниновой кислоты, эффективных при концентрациях в диапазоне значений IC50 0.2-1.9 мкМ - [Zakharenko A., Luzina О., Koval О., Nilov D., Gushchina I., Dyrkheeva N.,

Каталитическое действие Tdp1 заключается в гидролизе фосфотирозильной связи между 3'-концом ДНК и аминокислотным остатком тирозина Top1 в стабилизированном Top-1-ДНК-комплексе, при этом важную роль в процессе связывания Tdp1 с комплексом Тор-1-ДНК играют гидрофобные взаимодействия [Н.И. Речкунова, Н.А. Лебедева, О.И. Лаврик. Тирозил-ДНК-фосфодиэстераза 1 - новый участник репарации апуриновых/апиримидиновых сайтов в ДНК. Биоорганическая химия, 2015, 41, 531-538].
Существует два основных метода получения аналогов нуклеозидов, проявляющих ингибиторные свойства в отношении Tdp1.
Первый метод основан на модификации природных соединений [M.S. Drenichev, V.E. Oslovsky, S.N. Mikhailov. Cytokinin Nucleosides - Natural compounds with a unique spectrum of biological activities. Current Topics in Medicinal Chemistry, 16, 2562-2576, 2016].
При синтезе нуклеозидных аналогов вторым методом сначала получают производные гетероциклических оснований и моносахаридных производных, а затем проводят их конденсацию [С.Н. Михайлов, Ю.П. Лысов, Г.И. Яковлев. Применение функционально-компетентных аналогов нуклеозидов и нуклеотидов для изучения фермент-субстратных взаимодействий. Молекулярная биология, 33, 393-407, 1999]. В данном методе ключевой стадией является создание N-гликозидной связи. К настоящему времени разработаны удобные и эффективные методы получения рибонуклеозидов, исходя из триметилсилильных производных гетероциклических оснований и полностью ацилированной рибофуранозы в присутствии кислот Льюиса. [H.Vorbrüggen С. Ruh-Pohlenz. Handbook of nucleoside synthesis (Vol. 60). John Wiley & Sons, 2001]. Стереоселективность реакции конденсации гетероциклического основания и рибофуранозы определяется 2-О-ацильной группой, участвующей в образовании ацилоксониевого иона, и продуктами этих реакций являются природные β-нуклеозиды, в которых гетероциклическое основание находится в транс-положении по отношению к 2-О-ацильной группе. Когда 2-O-ацильная группа отсутствует, как в случае 2-дезоксирибозы, образуется смесь α,β-изомеров, что существенно затрудняет выделение и очистку искомых соединений. При гликозилировании производных D-арабинозы образуются α-нуклеозиды, в которых гетероциклическое основание находится в трансположении по отношению к 2-О-ацильной группе.
Раскрытие сущности изобретения
Изобретение решает задачу создания новых эффективных ингибиторов Tdp1, а также расширение ассортимента известных ингибиторов фермента Tdp1. Сущность изобретения заключается в повышении эффективности ингибирования фермента Tdp1 человека - важного фермента репарации ДНК. Так как С-концевой домен Tdp1 относится к семейству фосфолипаз [Н.И. Речкунова, Н.А. Лебедева, О.И. Лаврик. Биоорганическая химия, 2015, 41, 531-538], то для выполнения указанной задачи были получены аналоги нуклеозидов, содержащие ароматические и другие липофильные заместители которые способствуют лучшему связыванию с Tdp-1 за счет гидрофобных взаимодействий.
Наиболее близкими по структуре и свойствам к заявляемым соединениям являются аминогликозидный антибиотик неомицин В формулы A [Liao Z., Thibaut L., Jobson A., Pommier Y. // Mol. Pharmacol. 2006, 70, 366-372] и производные дисахаридных нуклеозидов общей формулы В (Фиг. 4) [Komarova А.О., Drenichev M.S., Dyrkheeva N.S., Kulikova I.V., Oslovsky V.E., Zakharova O.D., Zakharenko A.L., Mikhailov S.N., Lavrik O.I. Novel group of tyrosyl-DNA-phosphodiesterase 1 inhibitors based on disaccharide nucleosides as drug prototypes for anti-cancer therapy. Journal Of Enzyme Inhibition And Medicinal Chemistry, 2018, 33(1), 1415-1429, https://doi.org/10.1080/14756366.2018.1509210].
Недостатками неомицина В являются невысокая активность в отношении очищенного фермента Tdp1 (IC50 8 мМ) и существенная вероятность побочных эффектов, т.к. неомицин В ингибирует и другие ферменты семейства фосфолипаз, к которым принадлежит Tdp1 [Huang Y, Qureshi LA. Chen, H. // Mol. Cell Biochem. 1999, 197, 195-201].
Производные дисахаридных нуклеозидов ингибируют другой фермент системы репарации ДНК - поли(ADP-рибозо)полимеразу-1 (ПАРП-1), что может снизить селективность их действия в отношении Tdp1 в клетке [Efremova A.S., Zakharenko A.L., Shram S.I., Kulikova I.V., Drenichev M.S., Sukhanova M.V., Khodyreva S.N., Myasoedov N.F., Lavrik O.I., Mikhailov S.N. Disaccharide pyrimidine nucleosides and their derivatives: a novel group of cell-penetrating inhibitors of poly(ADP-ribose) polymerase-1. Nucleosides, Nucleotides and Nucleic Acids, 2013, 32, No 9, 510-528, DOI: 10.1080/15257770.2013.827793].
Настоящее изобретение позволяет повысить эффективность ингибирования фермента репарации Tdp1 путем применения новых производных нуклеозидов общей формулы (I), (II), (III), у которых выявлена высокая ингибиторная активность в отношении Tdp1 (IC50 0.13-49 мкМ), и расширить ассортимент известных ингибиторов Tdp1.
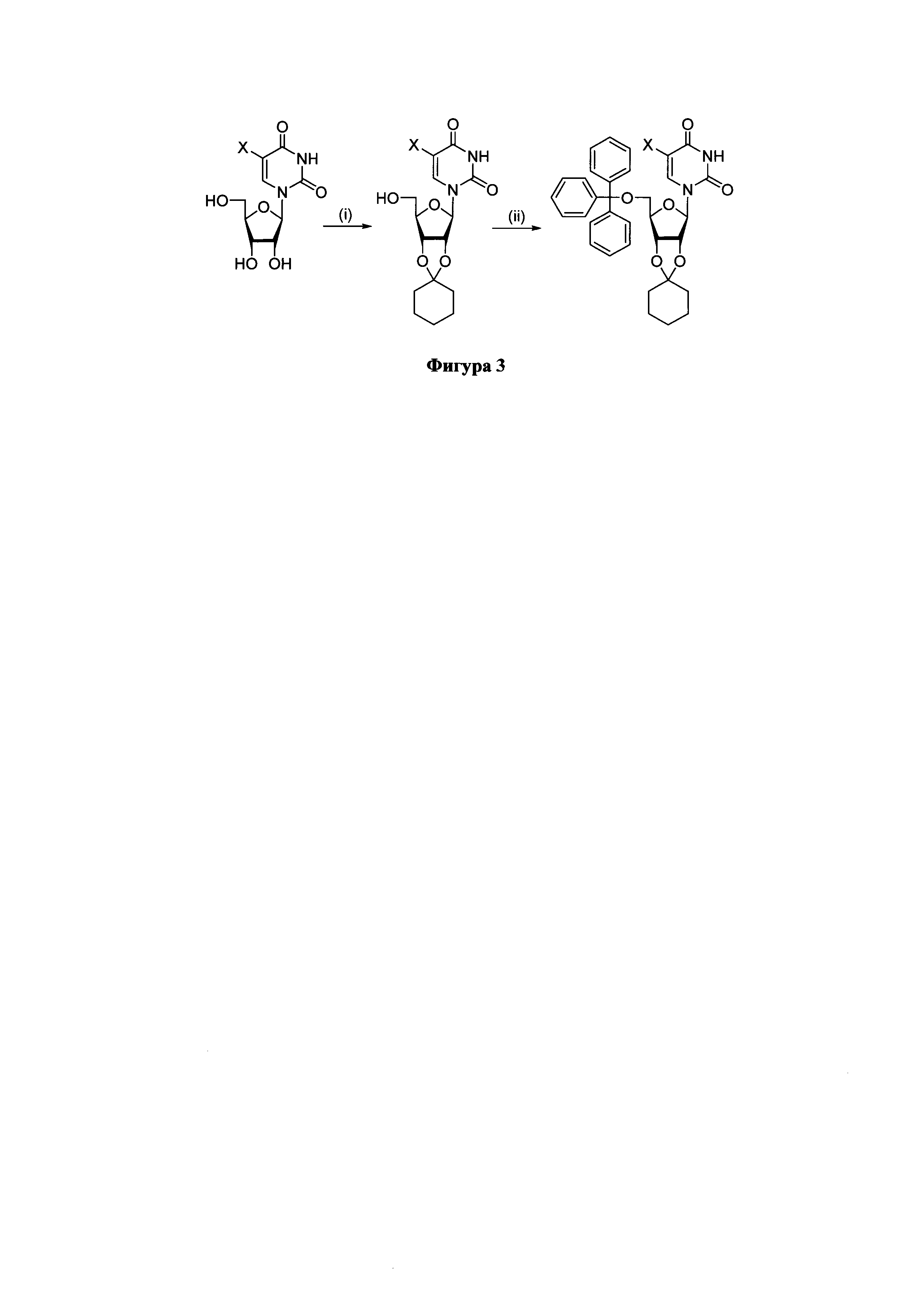
Для получения производных нуклеозидов, отвечающих формулам (I)-(II), применялись оба подхода (Фиг. 2). Для получения производных нуклеозидов, отвечающих формуле (III), применялся первый подход, заключающийся в последовательной защите 2',3'-цис-диольного фрагмента остатком циклогексанона и введении объемной трифенилметильной группы (Tr) в 5'-положение остатка рибофуранозы (Фиг. 3, Х=Н, Br, I).
Структуры полученных соединений подтверждены данными ЯМР-спектроскопии и масс-спектрометрии.
Для изучения ингибиторных свойств синтезированных соединений в отношении очищенного фермента Tdp1 использовалась тест-система на основе 16-мерного одноцепочечного олигонуклеотида, несущего флуорофор на 5'-конце и тушитель на 3'-конце. [Zakharenko et al. Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety. Bioorg. Med. Chem., 2015, 23, 2044-2052, doi: 10.1016/j.bmc.2015.03.020]. Находясь в пределах Ферстеровского радиуса, тушитель гасит флуоресценцию флуорофора. При инкубации такой ДНК с ферментом тушитель удаляется за счет активности Tdp1, что приводит к испусканию флуоресценции, регистрируемой в режиме реального времени. При использовании ингибиторов Tdp1 интенсивность испускаемой флуоресценции снижается.
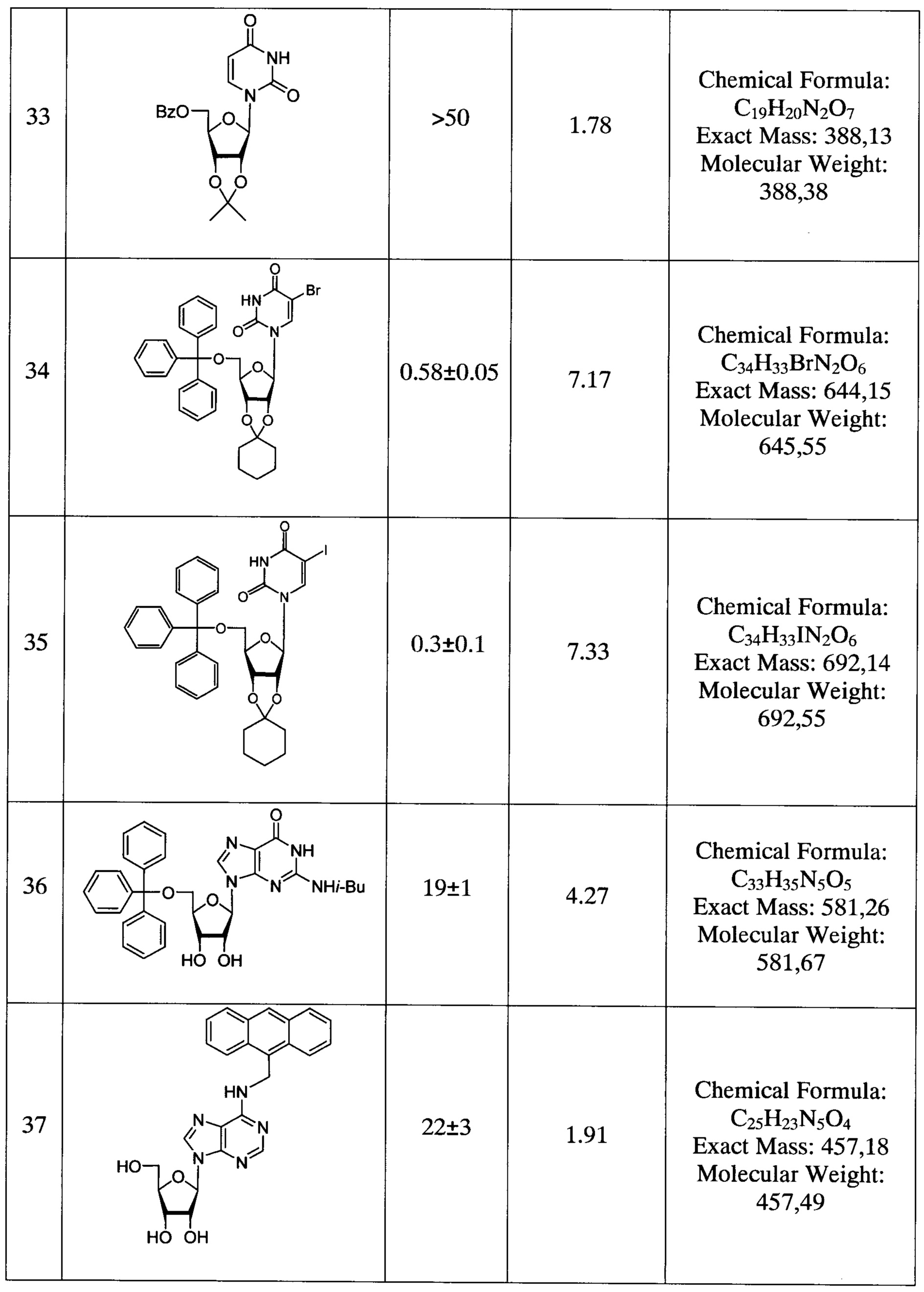
Было показано, что заявленные соединения проявляют высокую ингибиторную активность в отношении фермента Tdp1, с IC50 в субмикромолярном или нижнем микромолярном диапазонах (Табл. 1).
Согласно данным, представленным в Таблице 1, нуклеозидные производные, не содержащие липофильных групп, не ингибируют Tdp1, а для проявления ингибиторной активности в отношении Tdp1 в концентрациях 1-5 мкМ необходимо наличие остатка 2,3,5-три-O-бензоил-β-D-пентафуранозы (2,3,5-tri-O-Bz-β-D-Rib или др.). Введение меньшего числа бензоильных групп (Bz) в углеводный остаток нуклеозида приводит к снижению ингибиторного эффекта (Таблица 1, строки 2, 6, 7). Производные уридина, содержащие объемный заместитель (атом йода и т.п.) или гидрофобный заместитель в положении 5 пиримидина (Таблица 1, строки 13, 18), ингибируют Tdp1 в субмикромолярных концентрациях. Ингибиторной активностью в концентрациях 0.18-7 мкМ обладают пуриновые нуклеозиды (Таблица 1, строки 38-43). Замена О-бензоильных групп на О-изобутироильные группы приводит к исчезновению ингибиторной активности (Таблица 1, строка 44). Замена остатка 2,3,5-три-О-бензоил-β-D-рибофуранозы на остаток 2,3,5-три-O-бензоил-β-L-рибофуранозы также приводит к снижению ингибиторной активности у производных нуклеозидов (Таблица 1, строки 2 и 3, 15 и 16). Перспективным направлением химической модификации является введение одного или нескольких объемных заместителей в состав нуклеозидов (Таблица 1, одна тритильная группа и остаток циклогексана в составе соединений 30, 34-35, две тритильные группы в составе соединения 45), приводящее к ингибиторному эффекту в субмикромолярном диапазоне концентраций. Нуклеозиды, содержащие одну тритильную группу, проявляют меньшую ингибиторную активность (Таблица 1, строки 29, 36). Замена тритильной группы в составе структуры (III) на менее объемный остаток бензойной кислоты и замена 2',3'-O-циклогексилиденового фрагмента на менее объемный 2',3'-O-изопропилиденовый фрагмент снижают ингибиторную активность (Таблица 1, строки 32-33). Нуклеозиды, содержащие в составе только 2',3'-O-циклогексилиденовый фрагмент, не обладают заметной ингибиторной активностью в отношении Tdp1 (Таблица 1, строка 31). Производные пиримидиновых 5'-С-метилрибонуклеозидов ингибируют Tdp1 в концентрациях 3-6 мкМ, при этом величина ингибиторного эффекта мало зависит от 5-(R/S)-изомерии (Таблица 1, строки 19-20, 26-27). При этом замена пиримидинового остатка на пуриновый в 5'-С-метилнуклеозидах приводит к ингибированию Tdp1 в субмикромолярных концентрациях (Таблица 1, строка 39).
Согласно расчетам коэффициента распределения (Р) между фазами октанол-вода, проведенным в программе Instant J. Chem. (ChemAxon®), введение фенильных групп и/или остатков циклогексана в состав пуриновых и пиримидиновых нуклеозидов приводит к повышению значения logP, что может улучшить проникновение таких соединений через клеточную мембрану. Согласно данным, приведенным в таблице 1, ингибиторная активность модифицированных нуклеозидов в отношении Tdp1 усиливается с увеличением logP и количества фенильных групп.
Таким образом, в ряду производных нуклеозидов найдены новые эффективные ингибиторы важного фермента репарации ДНК Tdp1, сравнимые по величине ингибиторного эффекта с известными соединениями, описанными в литературных источниках.
Соединения с IC50 от 0.13 до 49 мкМ обладают незначительной цитотоксичностью на культурах клеток HeLa, А549 и здоровых фибробластов человека (СС50>50 мкМ) и после проведения углубленных фармакологических исследований, могут использоваться для дальнейшей разработки на их основе новых прототипов лекарственных средств, направленных на проведение комбинированной терапии опухолей.
Ниже приводятся конкретные примеры осуществления заявляемого технического решения, но которые не предназначены для ограничения объема притязаний.
Краткое описание фигур и таблиц
Фигура 1. Липофильные аналоги пентафуранозилнуклеозидов. На рисунке приведены общие формулы заявленных производных нуклеозидов, обладающих ингибиторной активностью в отношении фермента Tdp1 - фермента системы репарации ДНК.
Фигура 2. Синтез трибензоатов пентафуранозилнуклеозидов. Реагенты и условия. (i) BzCN/Et3N, диоксан, 20°С, 20-40 мин., 40-70%; (ii) пуриновое или пиримидиновое основание (В), BSA/DCE, 40 мин, 60°С, затем - TMSOTƒ 3 ч, 60°С, 49-60%.
Фигура 3. Синтез 5'-O-тритил-2',3'-O-циклогексилиденрибофуранозилнуклеозидов. Реагенты и условия. (i) С6Н10=O/(EtO)3CH, 3 ч, 20°С, 34-63%; (ii) TrCl/пиридин, 24 ч, 20°С. 49-79%.
Фигура 4. Ингибиторы Tdp1, содержащие несколько углеводных звеньев. На рисунке приведены структуры неомицина и дисахаридных нуклеозидов - известных ингибиторов Tdp1, содержащих углеводные остатки.
Таблица 1. Ингибирование Tdp1 производными нуклеозидов. В таблице приведены данные по ингибиторной активности (IC50) заявленных соединений в отношении Tdp1. Также в таблице приведены данные расчета коэффициента распределения Р между фазами октанол-вода для оценки проникновения полученных соединений через клеточную мембрану.
Осуществление изобретения
Структуры заявленных соединений подтверждали методами ЯМР-спектроскопии. ЯМР-спектры регистрировали на приборе Bruker АМХ 300 или Bruker АМХ 400 (Германия). Химические сдвиги (δ) в1H-ЯМР приведены в миллионных долях (м.д.) и измеряются относительно остаточного сигнала растворителя: CDCl3 - 7.26 м.д., DMSO-d6 - 2,50 м.д., D2O - 4,79 м.д. Величины констант спин - спинового взаимодействия (J) рассчитаны в герцах (Гц). При описании спектров1Н-ЯМР приняты следующие сокращения: s - синглет, br s - уширенный синглет, d - дублет, t - триплет, m - мультиплет. Химические сдвиги (δ) в13С-ЯМР приведены в миллионных долях (м.д.) и измеряются относительно остаточного сигнала растворителя: CDCl3 - 77.16 м.д., DMSO-d6 - 39,52 м.д. Для колоночной хроматографии использовали силикагель Kieselgel (0,040-0,063 мм, Merck). Очистку растворителей проводили по стандартным методикам. Углеводные компоненты для гликозилирования (Таблица 1, структуры 4-5, 21-23) получали по методике [Korbukh, I.A., Abramova, L.N., Preobrazhenskaya, M.N. (1978). Anomers of 1-O-acetyl-2,3,5-tri-O-benzoyl-D-arabinose. In: Townsend, L.B., Stuart Tipson, R. (Eds.), Nucleic Acid Chem. Improved and new synthetic procedures, methods and techniques. John Wiley and Sons], липофильные производные 5-C-метилрибонуклеозидов (Таблица 1, структуры 19-20, 26-28, 39) и производные 2-пиримидона и 4-пиримидона (Таблица 1, структуры 8-9) получали методами [Reist, E.J., Goodman, L, Baker, B.R. Potential anticancer agents. VIII. Synthesis of nucleosides derived from L-talofuranose. // J. Am. Chem. Soc. 1958, 80, 5775-5779; Vorbrüggen, H. Adventures in silicon-organic chemistry. // Acc. Chem. Res. 1995, 28, 509-520]. Синтез 2',3',5'-три-O-бензоилцитидина (25) проводили по методу [Prasad, А.K., Kumar, V., Malhotra, S., Ravikumar, V.T., Sanghvi, Y.S., Parmar, V.S. 'Green' methodology for efficient and selective benzoylation of nucleosides using benzoyl cyanide in an ionic liquid. // Bioorg. Med. Chem. 2005, 13, 4467-4472]. Значения коэффициента распределения вещества между фазами октанол-вода (logP) расчитывали с применением программы Instant J. Chem. (ChemAxon®).
Пример 1. Синтез 2',3',5'-три-O-бензоилуридина (2).
К раствору 1 г (4.7 ммоль, 1 eq) уридина в 40 мл сухого диоксана при перемешивании порциями добавляли 2.03 г (15.5 ммоль, 3.3 eq) бензоилцианида (BzCN), а затем приливали 2.2 мл (15.51 ммоль, 3.3 eq) триэтиламина (после добавления триэтиламина смесь приобретает зеленый оттенок). Смесь перемешивали 40 мин при комнатной температуре до гомогенности. Избыток BzCN нейтрализовали добавлением 15 мл МеОН, оставляли смесь на 30 мин при комнатной температуре, а затем упаривали в вакууме и переупаривали с CHCl3 (10 мл). Вещество перекристаллизовывали из метиленхлорида и сушили на вакуумном насосе 1 ч. Выход 1.84 г (70%) в виде белых пористых кристаллов, Тпл=153°С. Rf=0.45 (CH2Cl2/EtOH=99/1, v/v).1Н-ЯМР (400 МГц, DMSO-d6): 11.49 (d,4J=2.1, 1Н, NH3), 8.04 (dd, 2Н,3J=8.5,4J=1.4, o-Bz), 7.93-7.85 (m, 4Н, o-Bz), 7.83 (d, 1Н,3J6-5=8.1, H-6 Ura), 7.68 - 7.60 (m, 3H, p-Bz), 7.52 (dd, 2H,3J=8.5,3J=6.9, m-Bz), 7.49-7.41 (m, 4H, m-Bz), 6.16 (d, J1',2'=3.6, 1H, H-1'), 5.97-5.88 (m, 2H, H-2', H-3'), 5.67 (dd,2J5-6=8.1,4J=2.1, 1H, H-5 Ura), 4.74 (ddd, J4',3'=6.2, J4',5'a=3.6, J4',5'b=5.5, 1H, H-4'), 4.72 (dd, 1H, J5'a,4'=3.6, J5'a,5'b = - 11.8, Н-5'b), 4.64 (1H, J5'b,4'=5.5, J5'b,5'а = -11.8, Н-5'b).13С-ЯМР (100 МГц, DMSO-d6): 165.44 (С=O), 164.60 (С=O), 164.57 (С=O), 163.04 (С-4), 150.27 (С-2), 142.20 (С-6), 133.88, 133.78, 133.48, 129.27 (Bz), 129.18 (Bz), 128.69 (Bz), 128.53 (Bz), 128.43 (Bz), 102.24 (C-5), 89.55 (C-1'), 78.74 (C-4'), 73.16 (C-2'), 70.49 (C-3'), 63.62 (C-5').
Пример 2. Синтез 2',3',5'-три-O-бензоил-5-бромуридина (12).
Проводили аналогично примеру 1, исходя из 250 мг (0.77 ммоль) 5-бромуридина. Выход 198 мг (40%) в виде белого порошка, Тпл=195°С (разд.), Rf=0.96 (CH2Cl2/EtOH=99/1, v/v).1Н-ЯМР (400 МГц, CDCl3): 8.50 (br s, 1H, HN3), 8.13 (dd, 2H,3J=7.2,4J=1.4, o-Bz), 7.99 (dd, 2H,3J=7.2,4J=1.3, o-Bz), 7.94 (dd, 2H,3J=7.2,4J=1.3, o-Bz), 7.76 (s, 1H, H-6 5-BrUra), 7.66-7.55 (m, 3H, p-Bz), 7.53 (dd, 1H,3J=7.2,3J=6.4, m-Bz), 7.51 (dd, 1H,3J=7.2,3J=8.3, m-Bz), 7.42 (dd, 2H,3J=7.2,3J=8.3, m-Bz), 7.46-7.32 (m, 4H, m-Bz), 6.37 (d, 1H, J1',2'=6.0, H-1'), 5.89 (dd, 1H, J3',2'=6.0, J3',4'=3.5, H-3'), 5.73 (t, 1H, J2',1'=J2',3'=6.0, H-2'), 4.83 (dd, 1H, J5'a,5'b = - 13.6, J5'a,4'=3.9, H-5'a), 4.76-4.73 (m, 1H, H-4', перекрывается с сигналом Н-5'b), 4.72 (dd, 1Н, J5'b,5'a = - 13.6, J5'b,4'=3.4, Н-5'b).13C-ЯМР (100 МГц, DMSO-d6): 165.45 (С=O), 164.55 (С-4), 159.06 (С=O), 149.65 (С-2), 141.38 (С-6), 133.86 (Bz), 133.77 (Bz), 133.49 (Bz), 129.28 (Bz), 129.13 (Bz), 128.72 (Bz), 128.68 (Bz), 129.64 (Bz), 128.52 (Bz), 128.45 (Bz), 96.52 (C-5), 89.26 (С-1'), 78.98 (C-4'), 73.23 (C-2'), 70.25 (C-3'), 63.56 (C-5').
Пример 3. Синтез 2,'3',5'-три-О-бензоил-5-йодуридина (13).
Проводили аналогично примеру 1, исходя из 200 мг (0.54 ммоль) 5-иодуридина. Выход 117 мг (56%) в виде пены. Rf=0.96 (CH2Cl2/EtOH=99/1, v/v).1Н ЯМР (400 МГц, DMSO-d6): 11.86 (s, 1Н, HN3), 8.30 (s, 1H, Н-6 5-IUra), 8.02 (dd, 2Н,3J=8.4,4J=1.4, о-Bz), 7.91-7.85 (m, 4Н, o-Bz), 7.71 - 7.58 (m, 3Н, p-Bz), 7.51 (dd, 2H,3J=7.8,3J=6.9, m-Bz), 7.48-7.40 (m, 4H, m-Bz), 6.18 (d, 1H, J1',2'=3.5, H-1'), 5.97 - 5.90 (m, 2H, H-2', H-3'), 4.78 - 4.71 (m, 1H, H-4', перекрывается с сигналом H-5'a), 4.67 (dd, 1H, J5'a,5'b = - 12.0, J5'a,4'=3.8, H-5'а, перекрывается с сигналом Н-5'b), 4.65 (dd, 1Н, J5'b,5'a = - 12.0, J5'b,4'=5.6, H-5'b).13С ЯМР (100 МГц, CDCl3): 166.30 (C=O), 165.57 (C=O), 165.53 (C-4), 159.60 (C=O), 149.92 (C-2), 144.18 (C-6), 130.15 (Bz), 130.06 (Bz), 129.99 (Bz), 129.17 (Bz), 128.81 (Bz), 128.78 (Bz), 87.87 (С-1'), 81.27 (C-4'), 74.06 (C-2'), 71.62 (C-5, 5I-Ura) 69.92 (C-3'), 64.09 (C-5').
Пример 4. Синтез 2',3',5'-три-О-бензоил-5-метилуридина (15).
Проводили аналогично примеру 1, исходя из 200 мг (0.77 ммоль) 5-метилуридина (риботимидина). Выход 309 мг (70%) в виде пены. Rf=0.55 (CH2Cl2/EtOH=99/1, v/v).1Н-ЯМР (400 МГц, DMSO-d6): 11.46 (s,3NH), 8.03 (dd, 2H,3J=8.5,4J=1.4, o-Bz), 7.91 (dd, 2H,3J=8.5,4J=1.4, o-Bz), 7.87 (dd, 1H,3J=8.5,4J=1.4, o-Bz), 7.71-7.60 (m, 4H, p-Bz+H-6 Thy), 7.52 (dd, 2H,3J=7.6,3J=8.5, m-Bz), 7.49-7.40 (m, 4H, m-Bz), 6.20 (d, J1',2'=4.2, 1H, H-1'), 5.96-5.87 (m, 2H, H-2', H-3'), 4.78-4.73 (m, 1H, H-4', перекрывается с сигналом H-5'a), 4.73 (dd, 1H, J5'a,4'=3.5, J5'a,5'b = -12.5, H-5'a), 4.63 (1H, J5'b,4' = 5.8, J5'b,5'a = -12.5, Н-5'b), 1.68 (s, 3H, Me).13С-ЯМР (100 МГц, DMSO-d6): 165.44 (C=O), 164.60 (C=O), 164.57 (C=O), 163.70 (C-4), 150.80 (C-2), 136.00 (C-6), 133.88, 133.78, 133.48, 129.27 (Bz), 129.18 (Bz), 128.69 (Bz), 128.53 (Bz), 128.43 (Bz), 110.90 (C-5), 89.55 (С-1'), 78.74 (C-4'), 73.16 (C-2'), 70.49 (C-3'), 63.62 (C-5'), 12.40 (Me).
Пример 5. Синтез 2',3',5'-три-О-бензоил-5-хлоруридина (11).
К раствору 893 мг (1.6 ммоль, 1 eq) 2',3',5'-три-О-бензоилуридина в 15 мл сухого пиридина добавляли порциями 686 мг (5.1 ммоль, 3.2 eq) N-хлорсукциимида и оставляли перемешиваться на 15 мин при комнатной температуре до образования гомогенного раствора зеленоватого оттенка. Пиридин отгоняли в вакууме, остаток упаривали с этилацетатом (10 мл) и метиленхлоридом (10 мл). Вещество очищали колоночной хроматографией на силикагеле в системе CH2Cl2/EtOH=99/1 (v/v). Выход 909 мг (94%) в виде белых пористых кристаллов. Тпл=200°С (с разл.). Rf=0.58 (CH2Cl2/EtOH - 99/1, v/v).1H-ЯМР (400 МГц, DMSO-d6): 12.03 (s, 1H, HN3), 8.25 (s, 1H, H-6 5Cl-Ura), 8.02 (dd, 2H,3J=7.4,4J=1.3, o-Bz), 7.88 (dd,3J=7.2,4J=1.2, 4H, o-Bz), 7.72 - 7.58 (m, 3H, p-Bz), 7.57 - 7.36 (m, 6H, m-Bz), 6.20 (d, 1H, J1',2'=2.9, H-1'), 6.00 - 5.86 (m, 2H, H-2', H-3'), 4.82 - 4.74 (m, 1H, H-4', перекрывается с сигналом H-5'a), 4.69 (dd, 1H, J5'a,5'b = - 12.2, J5'a,4' = 3.4, H-5'а, перекрывается с сигналом Н-5'b), 4.67 (dd, 1Н, J5'b,5'а = - 12.2, J5'b,4' = 5.9, Н-5'b).13C ЯМР (100 МГц, DMSO-d6): 165.48 (C=O), 164.57 (C-4), 158.94 (C=O), 149.47 (C-2), 138.94 (C-6), 133.89 (Bz), 133.80 (Bz), 133.52 (Bz), 129.33 (Bz), 129.30 (Bz), 129.16 (Bz), 128.73 (Bz), 128.71 (Bz), 128.66 (Bz), 128.54 (Bz), 128.47 (Bz), 107.97 (C-5), 89.22 (C-1'), 79.03 (C-4'), 73.27 (C-2'), 70.26 (C-3'), 63.58 (C-5').
Пример 6. Синтез 1-(2,3,5-три-O-бензоил-β-L-рибофуранозил)урацила (3).
54 мг (0.48 ммоль, 2 eq) урацила и 120 мг (0.24 ммоль, 1 eq) 1-O-ацетил-2,3,5-три-O-бензоил-α,β-L-рибофуранозы сушили в вакуумном эксикаторе над P2O5 в течение 12 часов. Урацил растворяли в 5 мл в сухого 1,2-дихлорэтана, приливали 0.176 мл (0.72 ммоль, 3 eq) N,O-бис(триметилсилил)ацетамида (BSA) и нагревали с дефлегматором при 60°С 40 мин до образования гомогенного раствора. К полученному раствору добавляли 1-O-ацетил-2,3,5-три-O-бензоил-α,β-L-рибофуранозу и 0.65 мл (0.35 ммоль, 1.5 eq) триметилсилилтрифторметансульфоната (TMSOTƒ) и нагревали с дефлегматором при 60°С в течение 3 часов. Ход реакции контролировали по ТСХ в системе метиленхлорид/этанол - 99.4/0.6 (v/v). Реакционную смесь нейтрализовали 10% водным раствором бикарбоната натрия NaHCO3 (20 мл), продукт экстрагировали метиленхлоридом (2×20 мл). Органические слои отделяли, объединяли, промывали дистиллированной водой (2×20 мл), сушили над безводным сульфатом натрия (Na2SO4), фильтровали и упаривали в вакууме досуха. Остаток подвергали хроматографической очистке на колонке с силикагелем в системе метиленхлорид/этанол - 99.4/0.6 (v/v). Выход 66 мг (49%) в виде белой пены. Rf=0.52 (CH2Cl2/EtOH - 99.4/0.6, v/v). ЯМР-спектры идентичны 2',3',5'-три-O-бензоилуридину (2).
Пример 7. Синтез 1-(2,3,5-три-O-бензоил-β-L-рибофуранозил)тимина (16).
Проводили аналогично примеру 1, исходя из 75 мг (0.6 ммоль) тимина и 200 мг (0.4 ммоль) 1-O-ацетил-2,3,5-три-O-бензоил-α,β-L-рибофуранозы. Выход 114 мг (50%) в виде пены. Rf=0.12 (CH2Cl2/EtOH=99.4/0.6, v/v) ЯМР-спектры идентичны 2',3',5'-три-О-бензоил-5-метилуридину (15).
Пример 8. Синтез 5'-O-тритил-2',3'-O-циклогексилиденуридина (30).
500 мг (2.05 ммоль) уридина и 39 мг (0.205 ммоль) моногидрадата пара-толуолсульфокислоты суспендировали в 3.5 мл циклогексанона и перемешивали в течение 15 минут при комнатной температуре. К смеси приливали 0.682 мл (4.1 ммоль, 2eq.) этилортоформиата и оставляли перемешиваться 3 часа при комнатной температуре до образования раствора. К полученному раствору добавляли 217 мг (2.05 ммоль, 1 eq.) бикарбоната натрия, перемешивали в течение 15 минут и затем отфильтровывали. Осадок промывали ацетоном (2×25 мл). Фильтрат упаривали в вакууме при температуре бани 30°С, остаток переупаривали с толуолом (2×25 мл) до состояния зеленоватой маслянистой жидкости. Вещество подвергали хроматографической очистке на силикагеле в системе метиленхлорид/этанол в градиенте концентраций этанола от 0 до 2%. Фракции, содержащие вещество, собирали и упаривали в вакууме до состояния белой пены. Полученный 2',3'-O-циклогексалиденуридин (418 мг, 1.29 ммоль) [выход 63%, Rf=0.3 (CH2Cl2/EtOH - 97/3, v/v).1Н-ЯМР (400 МГц, DMSO-d6): 11.35 (d,4J=1.6, 1Н,3NH), 7.79 (d, 1H,3J=8.1, H-6 Ura), 5.84 (d, 1H, J1'2'=2.7, H-1'), 5.63 (dd, 1H,3J=8.0,4J=1.6, H-5 Ura), 5.05 (t, 1H, JOH,5',=5.1, 5'-OH), 4.89 (dd, 1H, J2'3'=6.3, J2'1'=2.7, H-2'), 4.74 (dd, 1H, J3'2'=6.3, J3'4'=3.6, H-3'), 4.06 (ddd, 1H, J4'3'=3.6, J4',5'a=3.4, J4',5'b=4.9, H-4'), 3.60 (ddd, 1H, J5'a,5'b= - 11.4, J5'a,4'=3.4, J5'a,OH = 5.1, H-5'a), 3.54 (ddd, 1H, J5'b,5'a = - 11.4, J5'b,4'=4.9, J5'b,OH=5.1, Н-5'b), 1.75-1.65 (m, 2H, С6Н10), 1.65-1.42 (m, 6H, С6Н10), 1.42-1.30 (m, 2H, С6Н10).13C ЯМР (100 МГц, DMSO-d6): 163.11 (C-4), 150.32 (C-2), 141.94 (C-6), 113.51 (C(CH2)5), 101.76 (C-5), 91.14 (С-1'), 86.60 (C-4'), 83.22 (C-2'), 80.12 (C-3'), 61.28 (C-5'), 36.59, 34.28, 24.42, 23.61, 23.20 (С6Н10)] растворяли в 20 мл сухого пиридина и добавляли 1.08 г (3.87 ммоль) тритилхлорида. Реакционную смесь оставляли при комнатной температуре на 18 ч. Избыток тритилхлорида нейтрализовали этанолом (10 мл). Реакционную смесь упаривали в вакууме. Остаток растворяли в этилацетате (40 мл), переносили в делительную воронку на 100 мл и промывали последовательно 10% водным раствором бикарбоната натрия NaHCO3 (30 мл) и дистиллированной водой (2×30 мл). Органический слой отделяли, сушили над безводным сульфатом натрия (Na2SO4), фильтровали и упаривали в вакууме досуха. Остаток подвергали хроматографической очистке на колонке с силикагелем в системе метиленхлорид/этанол - 99/1 (v/v). Фракции, содержащие вещество, собирали и упаривали в вакууме. Полученную пену сушили на вакуумном насосе 1 ч. Выход 504 мг (69%) в виде белой пены, Rf=0.75 (CH2Cl2/EtOH - 97/3, v/v).1Н ЯМР (400 МГц, DMSO-d6): 11.35 (d,3J=1.8, 1H,3HN), 7.72 (d,3J=8.1, 1H, H-6 Ura), 7.41 - 7.24 (m, 15H, Ph), 5.86 (d, J1'2'=1.8, H-1'), 5.50 (dd,4J=8.1,3J=2.1, 1H, H-5 Ura), 4.97 (dd, J2'3'=6.3, J2'1'=1.8, 1H, H-2'), 4.69 (dd, J3'2'=6.3, J3'4'=4.6, 1H, H-3'), 4.13 (ddd, 1H, J4',5'a=7.0, J4',5'b=3.4, J4'3'=4.6, H-4'), 3.34 (dd, 1H, J5'a,5'b = - 10.3, J5'a,4'=7.0, 1H, H-5'a) 3.11 (dd, 1H, J5'b,5'a = - 10.3, J5'b,4'=3.4, 1H, Н-5'b), 1.75-1.65 (m, 2H, С6Н10), 1.62-1.40 (m, 6H, С6Н10), 1.39-1.27 (m, 2H, С6Н10).13C ЯМР (100 МГц, DMSO-d6): 163.14 (C-4), 150.20 (C-2), 143.43 (Ph), 142.80 (C-6), 128.22 (Ph), 127.86 (Ph), 127.07 (Ph), 113.78 (C(CH2)5), 101.68 (C-5), 91.92 (С-1'), 86.22 (C-4'), 85.81 (Ph3C), 83.25 (C-2'), 80.15 (C-3'), 64.16 (C-5'), 36.53, 34.30, 24.40, 23.60, 23.18 (С6Н10).
Пример 9. Синтез 5'-O-тритил-2',3'-O-циклогексилиден-5-бромуридина (34).
Проводили аналогично примеру 7, исходя из 250 мг (0.62 ммоль) 2',3'-O-циклогексилиден-5-бромуридина. Выход 316 мг (79%) в виде пены. Rf=0.59 (CH2Cl2/EtOH - 99/1, v/v).1Н-ЯМР (400 МГц, DMSO-d6): 11.87 (s, 1Н,3HN), 8.22 (s, 1H, H-6 Ura), 7.41 - 7.24 (m, 15H, Ph), 5.83 (d, J1'2'=2.0, 1H, H-1'), 5.01 (dd, J2'3'=6.4, J2'1'=2.0, 1H, H-2'), 4.65 (dd, J3'2'=6.4, J3'4'=4.5, H-3'), 4.13 (ddd, 1H, J4',5'a=7.2, J4',5'b=3.3, J4'3'=4.5, H-4'), 3.34 (dd, 1H, J5'a,5'b = -10.4, J5'a,4'=7.2, 1H, H-5'a), 3.12 (dd, 1H, J5'b,5'а = -10.4, J5'b,4'=3.3, H-5'b), 1.75-1.62 (m, 2H, С6Н10), 1.62-1.40 (m, 6H, С6Н10), 1.40-1.26 (m, 2H, С6Н10).13С-ЯМР (100 МГц, DMSO-d6): 159.17 (C-4), 149.57 (C-2), 143.47 (Ph), 142.14 (C-6), 128.19 (Ph), 127.86 (Ph), 127.05 (Ph), 113.82 (C(CH2)5), 96.07 (C-5), 92.43 (C-1'), 86.23 (C-4'), 85.91 (Ph3C), 83.12 (C-2'), 80.15 (C-3'), 64.15 (C-5'), 36.48, 34.29, 24.39, 23.59, 23.17 (С6Н10).
Пример 10. Синтез 5'-O-тритил-2',3'-O-циклогексилиден-5-йодуридина (35).
Проводили аналогично примеру 7, исходя из 300 мг (0.67 ммоль) 2',3'-O-циклогексилиден-5-йодуридина. Выход 225 мг (48%) в виде белой пены. Rf=0.83 (CH2Cl2/EtOH - 99/1, v/v).1Н-ЯМР (400 МГц, DMSO-d6): 11.73 (s, 1Н,3HN), 8.21 (s, 1H, H-6 Ura), 7.42 - 7.21 (m, 15H, Tr), 5.82 (d, 1H, J1'2'=1.9, H-1'), 5.00 (dd, 1H, J2'3'=6.4, J2'1'=2.0, H-2'), 4.62 (dd, 1H, J3'2'=6.4, J3'4'=4.5, H-3'), 4.11 (ddd, 1H, J4',5'a=7.2, J4',5'b=3.3, J4'3'=4.5, H-4'), 3.26 (dd, 1H, J5'a,5'b = - 10.5, J5'a,4'=7.3, 1H, H-5'a), 3.11 (dd, 1H, J5'b,5'a = - 10.5, J5'b,4'=3.3, Н-5'b).13С-ЯМР (100 МГц, DMSO-d6): 160.58 (C-4), 149.94 (C-2), 146.92, 143.49, 128.19, 127.86, 127.03, 113.80 (C(CH2)5), 92.45 (С-1'), 86.18 (C-4'), 85.94 (Ph3C), 83.05 (C-2'), 80.22 (C-3'), 69.76 (C-5, 5-IUra), 64.20 (C-5'), 36.46, 34.28, 24.40, 23.59, 23.17 (С6Н10).
Пример 11. Синтез 6-хлор-9-(2',3',5'-три-O-бензоил-β-D-рибофуранозил)пурина (40).
К суспензии инозина (952 мг, 3.549 ммоль) и BzCN (2286 мг, 17.432 ммоль) в сухом ацетонитриле (7 мл) добавляли Et3N (2.425 мл, 17.446 ммоль) и перемешивали при комнатной температуре. Реакцию контролировали с помощью ТСХ (CH2Cl2:EtOH-25:1). Через 24 часа к реакционной смеси добавляли МеОН (3 мл) и оставляли перемешиваться при комнатной температуре на 10 минут, после чего упаривали в вакууме. Остаток растворяли в CH2Cl2 (35 мл), и промывали водой (4×30 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и упаривали в вакууме. Остаток хроматографировали на колонке с силикагелем. Колонку промывали системой CH2Cl2:EtOH-99:1 (v/v), CH2Cl2:EtOH-95:5 (v/v), продукт элюировали системой CH2Cl2:EtOH-90:10 (v/v). Фракции, содержащие продукт, объединяли, упаривали и сушили в вакуумном эксикаторе над P2O5. Выход 2',3',5'-три-О-бензоилинозина 1460 мг, 71% (белая пена). Rƒ=1.17 (CH2Cl2-EtOH, 25:1). Полученный 2',3',5'-три-O-бензоилинозин 43 (1161 мг, 1.999 ммоль) растворяли в сухом дихлорэтане (5 мл), затем добавляли сухой N,N-диметилформамид (0.68 мл, 8.833 ммоль) и хлорокись фосфора POCl3 (1.7 мл, 18.59 ммоль) и оставляли перемешиваться при 60°С. Реакцию контролировали с помощью ТСХ (CH2Cl2:EtOH-20:1, v/v). Через 10 часов реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (50 мл), и промывали 10% водным раствором бикарбоната натрия (4×30 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и упаривали в вакууме. Остаток хроматографировали на колонке с силикагелем. Колонку промывали системой CH2Cl2:EtOH-99:1 (v/v), продукт элюировали системой CH2Cl2:EtOH-97:3 (v/v). Фракции, содержащие продукт, объединяли, упаривали и сушили в вакуумном эксикаторе над P2O5. Выход 1048 мг, 87% (белая пена). Rƒ=0.48 (CH2Cl2-EtOH, 95:5, v/v).1Н-ЯМР (400 МГц, CDCl3): δ=8.61 (s, 1H, Н-2), 8.27 (s, 1Н, Н-8), 8.07 (dd, 2Н, Jh1-h2=Jh5-h4=8.3, JH1-H3=JH5-H3=1-3, H1-Bz, H5-Bz), 8.02 (dd, 2Н, JH1-Н2=JH5-Н4=8.3, JH1-H3=JH5-H3=1-3, H1-Bz, H5-Bz), 7.92 (dd, 2H, JH1-H2=JH5-H4=8.3, Jh1-H3=JH5-H3=1.3, H1-Bz, H5-Bz), 7.65-7.35 (m, 9H, Bz), 6.45 (d, 1H, J1'2'=5.1, H-1'), 6.41 (dd, 1H, J2'1'=5.1, J2'3'=5.6, H-2'), 6.25 (dd, 1H, J3'2'=5.6, J3'4'=4.8, H-3'), 4.93 (dd, 1H, J5'a,5'b = -12.2, J5'a4'=3.3, H-5'a), 4.85 (ddd, 1H, J4'3'=4.8, J4'5'a=3.3, J4'5'b=4.2, H-4'), 4.70 (dd, 1H, J5'b5'a = -12.2, J5'b4'=4.2, Н-5'b).
Пример 12. Синтез 2,6-дихлор-9-(2',3',5'-три-O-бензоил-β-D-рибофуранозил)пурина (42).
К раствору 2,6-дихлорпурина (617 мг, 3.265 ммоль) [Steklov, М.Y., Tararov, V.I., Romanov, G.A., Mikhailov, S.N. (2011) Facile synthesis of 8-azido-6-benzylaminopurine. Nucleosides Nucleotides Nucleic Acids. 30, 503-511] и 1-О-ацетил-2,3,5-три-O-бензоил-α/β-D-рибофуранозы (1976 мг, 3.917 ммоль) в сухом ацетонитриле (25 мл) добавляли BSA (1.6 мл, 6.54 ммоль) и нагревали с обратным водяным холодильником при 70°С в течение 10 минут, после чего к смеси добавляли 2М раствор TMSOTf в сухом DCE (2.5 мл, 13.78 ммоль), и продолжали нагревать при 70°С.Реакцию контролировали с помощью ТСХ (CH2Cl2:EtOH-25:1). Через 2 часа реакционную смесь охлаждали до комнатной температуры, разбавляли CH2Cl2 (40 мл), и промывали последовательно 10% водным раствором NaHCO3 (2×20 мл) и водой (2×20 мл). Органический слой отделяли, сушили над безводным Na2SO4, фильтровали и упаривали в вакууме. Остаток хроматографировали на колонке с силикагелем. Колонку промывали системой CH2Cl2:EtOH-99.5:0.5 (v/v), CH2Cl2:EtOH-99:1 (v/v), продукт элюировали системой CH2Cl2:EtOH-98:2 (v/v). Фракции, содержащие продукт, объединяли, упаривали и сушили в вакуумном эксикаторе над P2O5. Выход 1236 мг (60%) в виде белой пены. Rƒ=0.59 (CH2Cl2-EtOH, 25:1, v/v).1Н-ЯМР (400 МГц, CDCl3): δ=8.28 (s, 1Н, H-8), 8.06 (dd, 2H, JH1-H2=JH5-H4=8.3, JH1-h3=Jh5-h3=1.3, H1-Bz, H5-Bz), 8.03 (dd, 2H, Jh1-h2=Jh5-h4=8.3, JH1-H3=JH5-H3=1-3, H1-Bz, H5-Bz), 7.94 (dd, 2H, JH1-H2=JH5-H4=8.3, JH1-H3=JH5-H3=1.3, H1-Bz, H5-Bz), 7.63-7.35 (m, 9H, Bz), 6.48 (d, 1H, J1'2'=5.4, H-1'), 6.18 (dd, 1H, J2'1'=5.4, J2'3'=5.9, H-2'), 6.13 (dd, 1H, J3'2'=5.9, J3'4'=4.2, H-3'), 4.92 (dd, 1H, J5'a5'b =-12.1, J5'a4'=3.3, H-5'a), 4.86 (ddd, 1H, J4'3'=4.2, J4'5'a=3.3, J4'5'b=4.1, H-4'), 4.73 (dd, 1H, J5'b5'a = -12.1, J5'b4'=4.1, H-5'b).
Пример 13. Синтез 2-фтор-6-хлор-9-(2',3',5'-три-O-бензоил-β-D-рибофуранозил)пурина (41).
Проводили аналогично примеру 12, исходя из 2-фтор-6-хлорпурина (929 мг, 5.384 ммоль) [Gray NS, Kwon S, Schultz PG. Combinatorial synthesis of 2,9-disubstituted purines. Tetrahedron Lett 1997; 38: 1161-1164] и 1-O-ацетил-2,3,5-три-O-бензоил-α/β-D-рибофуранозы (3259 мг, 6.460 ммоль). Выход 1236 мг (60%) в виде белой пены. Rƒ=0.59 (CH2Cl2-EtOH, 25:1).1Н-ЯМР (400 МГц, CDCl3): δ=8.26 (s, 1Н, H-8), 8.08 (dd, 2H, JH1-H2=JH5-H4=8.4, JH1-H3=JH5-H3=1.3, H1-Bz, H5-Bz), 8.03 (dd, 2H, Jh1-h2=Hh5-H4=8.4, JH1-H3=JH5-H3=1.3, H1-Bz, H5-Bz), 7.92 (dd, 2H, Jh1-h2=Jh5-h4=8.4, JH1-H3=JH5-H3=1.3, H1-Bz, H5-Bz), 7.63-7.35 (m, 9H, Bz), 6.43 (d, 1H, J1'2'=5.5, H-1'), 6.22 (dd, 1H, J2'1'=5.5, J2'3'=5.8, H-2'), 6.13 (dd, 1H, J3'2'=5.8, J3'4'=4.6, H-3'), 4.91 (dd, 1H, J5'a5'b = -12.2, J5'a4'=3.3, H-5'a), 4.85 (ddd, 1H, J4'3'=4.6, J4'5'a=3.3, J4'5'b=4.0, H-4'), 4.73 (dd, 1H, J5'b5'a = -12.2, J5'b4'=4.0, Н-5'b).
Пример 14. Влияние производных нуклеозидов на активность фермента Tdp1.
Рекомбинантную тирозил-ДНК-фосфодиэстеразу 1 человека (КФ 3.1.4.) экспрессировали в системе Escherichia coli (плазмида рЕТ 16В-Tdp1) и выделяли как описано [Interthal Н, Pouliot JJ, Champoux JJ. // Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 12009-12014].
В качестве тест-системы для определения ингибирующих свойств предлагаемых соединений использовали реакцию удаления тушителя флуоресценции Black Hole Quencher 1 (BHQ1) с 3'-конца олигонуклеотида, катализируемую Tdp1. На 5'-конце олигонуклеотида находится (5,6)-FAM - флуорофор, интенсивность флуоресценции которого возрастает при удалении тушителя. Для измерения флуоресценции использовали флуориметр POLARstar OPTIMA производства BMG LABTECH.
Реакционные смеси объемом 200 мкл содержали буфер (50 мМ Tris-HCl, рН 8.0; 50 мМ NaCl; 7 мМ меркаптоэтанол), олигонуклеотид (50 нМ) и различные концентрации ингибиторов. Реакция запускалась добавлением Tdp1 до конечной концентрации 1.3 нМ. Измерения проводили в линейном диапазоне зависимости скорости реакции от времени (до 8 минут) через каждые 55 секунд. Влияние предлагаемых соединений оценивали по величине IC50 (концентрация ингибитора, при которой активность фермента снижена наполовину). Обсчет значений IC50 проводили с помощью программы MARS Data Analisys 2.0 (BMG LABTECH).
Величины IC50 для изученных соединений приведены в Таблице 1.








Реферат
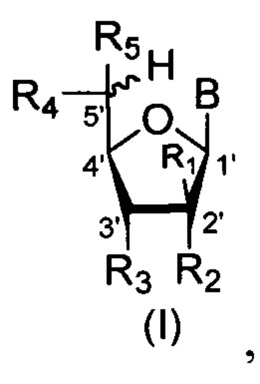
Изобретение относится к области молекулярной биологии, медицинской химии, биохимии, а именно к применению производных пентафуранозилнуклеозидов общей формулы (I-II) вкачестве ингибиторов тирозил-ДНК-фосфодиэстеразы 1 человека:где в формуле (I) В = урацил-1-ил, или 5-метилурацил-1-ил (тимин-1-ил), или 5-(2-метоксикарбонилэтен-1-ил)-6-метилурацил-3-ил, или 6-метилурацил-1-ил, или 4-O-метилурацил-1-ил, или 5-фторурацил-1-ил, или 5-хлорурацил-1-ил, или 5-бромурацил-1-ил, или 5-йодурацил-1-ил, или 4-пиримидон-1-ил, или 2-пиримидон-1-ил, или цитозин-1 -ил, или 6-хлорпурин-9-ил, или 2-фтор-6-хлорпурин-9-ил, или 2,6-дихлорпурин-9-ил, или гипоксантин-9-ил, или N6-бензоиладенин-9-ил, или N6,N6-дибензоиладенин-9-ил, R1=R5=H, R2=R3=R4=OBz (Bz-остаток бензойной кислотыили В = 5-йодурацил-1-ил, R1=R3=R4=OBz, R2=R5=H; или 1'-O-α- или β-аномеры или их смеси, где В=ОАс (Ac-остаток уксусной кислотыR1=R5=H, R2=R3=R4=OBz; или 1'-O-α- или β-аномеры или их смеси, где В=ОМе, R1=R3=R4=OBz, R2=R5=H; или 5'-(R или S)-изомеры или их смеси, где В = урацил-3-ил, или цитозин-1-ил, или N6-бензоиладенин-9-ил, R1=H, R2=R3=R4=OBz, R5=CH3; или В=N6-трифенилметиладенин-9-ил или N2-изобутироилгуанин-9-ил, R1=R5=H, R2=R3=OH, R4=O-трифенилметилили 1'-О-α- или β-аномеры 5'-(R или S)-изомеров или их смеси, где В = ОАс или ОМе, R1=H, R2=R3=R4=OBz, R5=CH3; или В = урацил-1-ил, или 5-бромурацил-1-ил, или 5-йодурацил-1-ил, R1=R5=H, R2, R3=2',3'-O-циклогексилиденR4=О-трифенилметилтакже их аналоги L-ряда общей формулы (II):где В = урацил-1-ил или 5-метилурацил-1-ил (тимин-1-ил); или 1'-О-α- или β-аномеры или их смеси, где В = ОАс. Технический результат: предложено новое применение производных пентафуранозилнуклеозидов общей формулы (I-II) в качестве ингибиторов тирозил-ДНК-фосфодиэстеразы, которые могут быть использованы для проведения комбинированной противоопухолевой терапии. 4 ил., 1 табл., 20 пр.
Формула






Комментарии