Соли изофосфорамидного иприта и его аналогов - RU2527531C2
Код документа: RU2527531C2
Чертежи























Описание
Предпосылки создания изобретения
Аутопсии солдат, погибших во время Первой мировой войны, показали, что сернистый иприт оказывает диспропорциональный эффект на быстро делящиеся клетки, на основании чего было сделано предположение, что соединения сернистого иприта могут оказывать противоопухолевый эффект. В действительности, ранее некоторые исследователи пытались лечить раковые заболевания путем непосредственного введения сернистого иприта в опухолевую ткань. Эти исследования ограничивались чрезвычайно высокой токсичностью соединений сернистого иприта, поэтому были предложены аналоги азотистого иприта, такие как мехлорэтамин, которые являются менее токсичными.


Поскольку большинство аналогов мехлорэтамина обладают недостаточной селективностью действия, были разработаны пролекраства, такие, как соединения фосфорамида, которые могут активироваться за счет высокой концентрации фосфорамидаз, присутствующих в неопластических клетках. Наиболее эффективными считаются два фосфорамидных алкилирующих агента, циклофосфамид (СРА, от англ. Cyclophosphamide) и его изомерное соединение Ифосфамид (Ifos).


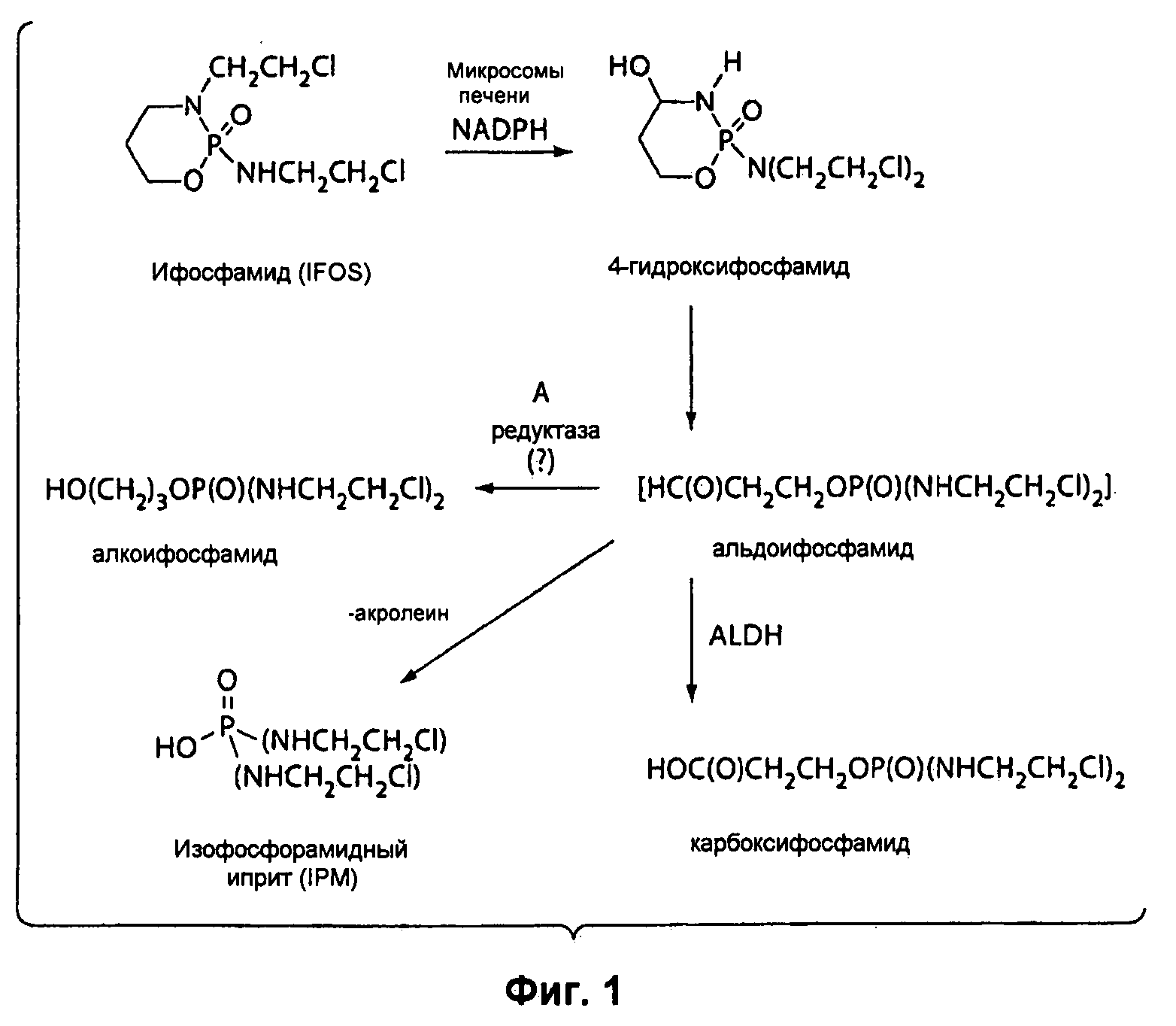
Согласно Рис.1, изофосфорамидный иприт (IPM, от англ. Isophosphoramide Mustard) является обычным метаболитом СРА и Ifos. Считается, что IPM обладает лишь частью той противоопухолевой активности, которой обладают СРА и Ifos. Попытки использовать чистый IPM в качестве противоопухолевого агента оказались неудачными, частично по причине нестабильности соединения. IPM был синтезирован, была проведена предварительная оценка его биологических свойств, но, к сожалению, IPM является слишком нестабильным соединением, чтобы можно было его выделить и использовать для лечения человека.
Краткое описание изобретения
В настоящем изобретении раскрываются кристаллические соединения, имеющие формулу (I)

где А+ представляет собой алифатическое аммониевое соединение; а Х и Y независимо друг от друга являются уходящими группами.
Согласно определенным вариантам своего осуществления, изобретение относится к фармацевтическим композициям, включающим соединение по формуле (I) и фармацевтически приемлемый растворитель или наполнитель. Также в изобретении описываются способы получения таких соединений и композиций.
Кроме того, в настоящем изобретении описывается лиофилизат и способ получения лиофилизата, включающего изофосфорамидный иприт (IPM) и/или аналог IPM, один или более эквивалент основы, и эксципиент. Согласно определенным вариантам осуществления изобретения, способ включает обеспечение контакта кристаллической соли IPM или его аналога и алифатического амина, такого, как трис(гидроксиметил)аминометан (Tris), или смеси IPM и его аналогов и одного или более алифатического амина (предпочтительно, в соотношении, приблизительно, 1:1 IPM или его аналога к амину или аминам) с водой с последующей лиофилизацией полученной смеси. Согласно определенным вариантам осуществления изобретения, исходная смесь и конечный лиофилизат содержать эксципиент, такой как маннитол, безводная лактоза, сахароза, D(+)-трегалоза, декстран 40 или повидон (PVP К24), предпочтительно маннитол.
Такие композиции включают лиофилизаты, предпочтительно, содержащие эксципиент, такой как маннитол, безводная лактоза, сахароза, D(+)-трегалоза, декстран 40 или повидон (PVP K24), предпочтительно маннитол, и соединение по формуле

где А+ представляет собой аммониевое соединение, выбранное из группы протонированных (сопряженных с кислотой) или четвертичных форм алифатических аминов и ароматических аминов, включая основные аминокислоты, гетероциклические амины, замещенные и незамещенные пиридины, гуанидины и амидины; а Х и Y независимо друг от друга являются уходящими группами.
Также в настоящем изобретении раскрываются фармацевтические композиции, адаптированные для перорального введения, включающие фармацевтически приемлемый растворитель или эксципиент и соединение, заявленное в соответствии с настоящим изобретением.
Согласно определенным вариантам своего осуществления, изобретение относится к способам лечения гиперпролиферативных заболеваний, например, при помощи соединений или композиций, заявленных в соответствии с изобретением. Согласно определенным вариантам своего осуществления, изобретение относится к лечению гиперпролиферативных заболеваний, к которым относятся лейкозы, включая острые лейкозы (такие, как острый лимфоцитарный лейкоз, острый миелоцитарный лейкоз, острый миелогенный лейкоз и миелобластный, промиелоцитарный, миеломоноцитарный, моноцитарный лейкозы и эритролейкоз), хронические лейкозы (такие, как хронический миелоцитарный (гранулоцитарный) лейкоз, хронический миелогенный лейкоз и хронический лимфоцитарный лейкоз), истинная полицитемия, лимфома, болезнь Ходжкина, неходжкинская лимфома (медленно растущие формы и высокодифференцированные формы), множественная миелома, макроглобулинемия Вальденстрема, болезнь тяжелых цепей, миелодиспластический синдром, волосяноклеточный лейкоз и миелодисплазия.
Дополнительными примерами заболеваний, для лечения которых могут использоваться соединения и композиции, заявленные в соответствии с настоящим изобретением, являются твердые опухоли, такие как саркомы и карциномы, включая, без ограничений указанными, фибросаркому, миксосаркому, липосаркому, хондросаркому, остеогенную саркому, циклофосфамид (СРА)-устойчивую саркому, и другие саркомы, синовиому, мезотелиому, опухоль Эвинга, лейомиосаркому, рабдомиосаркому, карциному толстой кишки, лимфонеоплазии, рак поджелудочной железы, рак груди, рак легких, рак яичников, рак предстательной железы, гепатоцеллюлярную карциному, плоскоклеточную карциному, базальноклеточную карциному, аденокарциному, карциному потовых желез, карциному сальных желез, папиллярную карциному, папиллярные аденокарциномы, медуллярную карциному, бронхогенную карциному, почечноклеточную карциному, гепатому, карциному желчных протоков, хориокарциному, опухоль Вильмса, цервикальный рак, опухоли яичек, карциному желчного пузыря и опухоли ЦНС (такие как глиома, астроцитома, медуллобластома, краниофарингиома, эпендимома, пинеалома, гемангиобластома, невринома слухового нерва, олигодендроглиома, менингиома, меланома, нейробластома и ретинобластома).
Краткое описание Рисунков
На Рисунке 1 представлена схема, иллюстрирующая метаболизм ифосфамида, включая образование акролеина и изофосфорамидного иприта.
На Рисунке 2 представлены результаты рентгеновской порошковой дифрактометрии IPM•Tris.
На Рисунке 3 представлена дифференциальная сканирующая калориметрия (DSC, от англ. Differential Scanning Calorimetry) IPM•Tris.
На Рисунке 4 представлен термогравиметрический анализ (TGA, от англ. Thermogravimetric Analysis) IPM•Tris.
На Рисунке 5 представлены результаты сканирующей электронной микроскопии (SEM, от англ. Scanning Electron Microscope) кристаллической соли IPM•Tris.
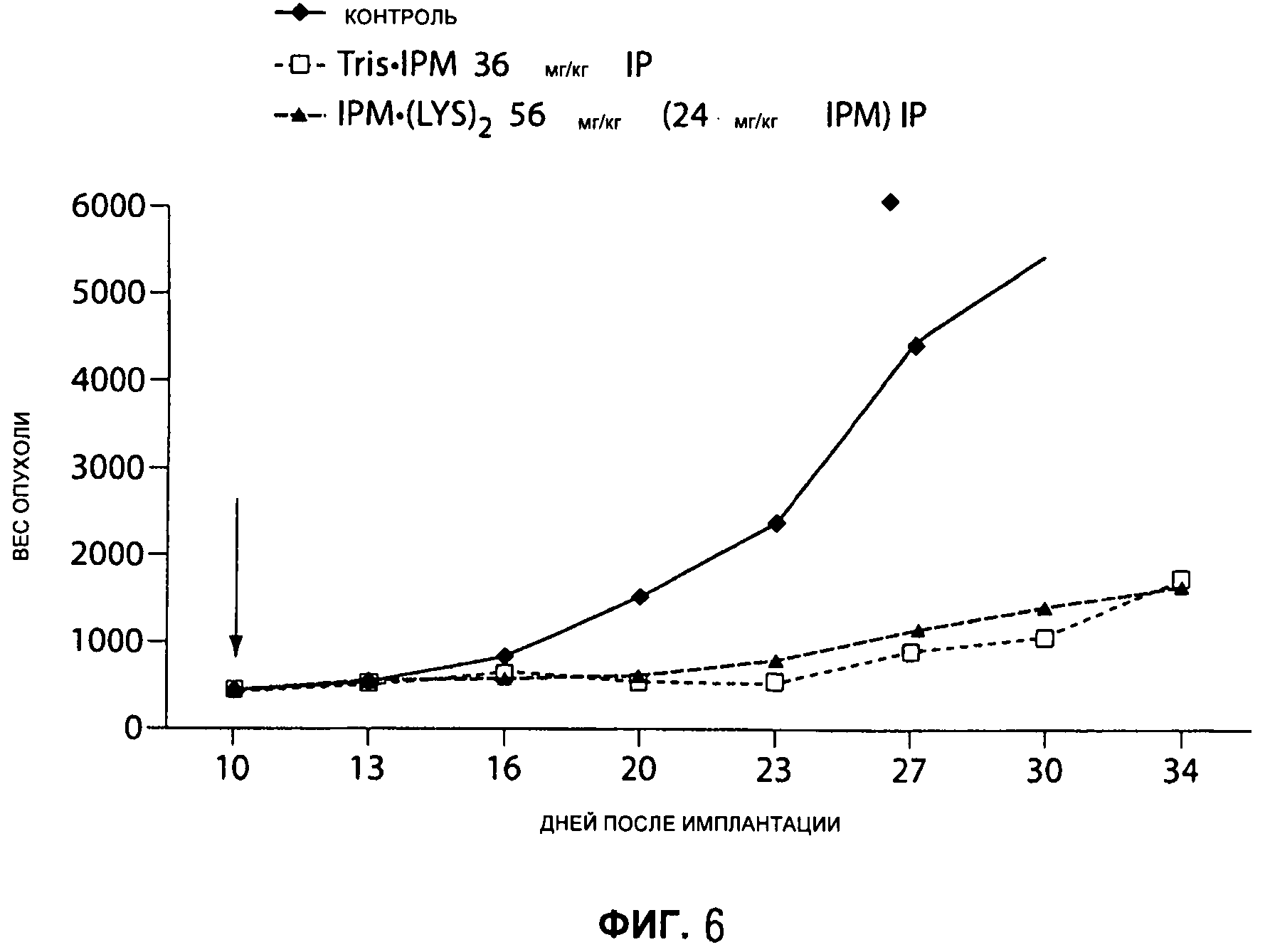
На Рисунке 6 представлен сравнительный анализ действия IPM•(LYS)2 и IPM•Tris на ксенотрансплантат рака груди человека МХ-1.
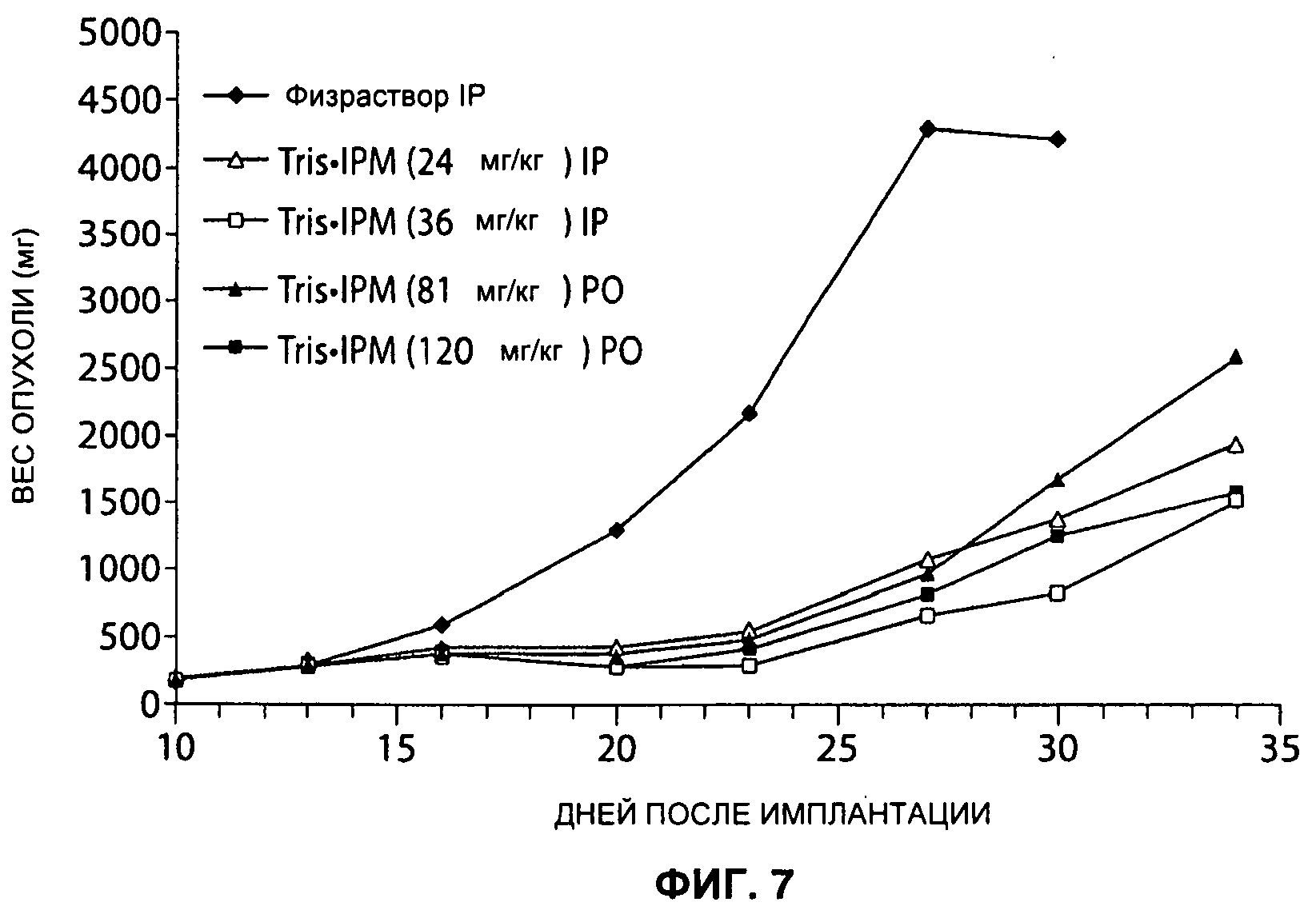
На Рисунке 7 представлен сравнительный анализ противоопухолевой активности IP и IPM•Tris при пероральном введении на МХ-1 ксенотрансплантат рака груди человека.
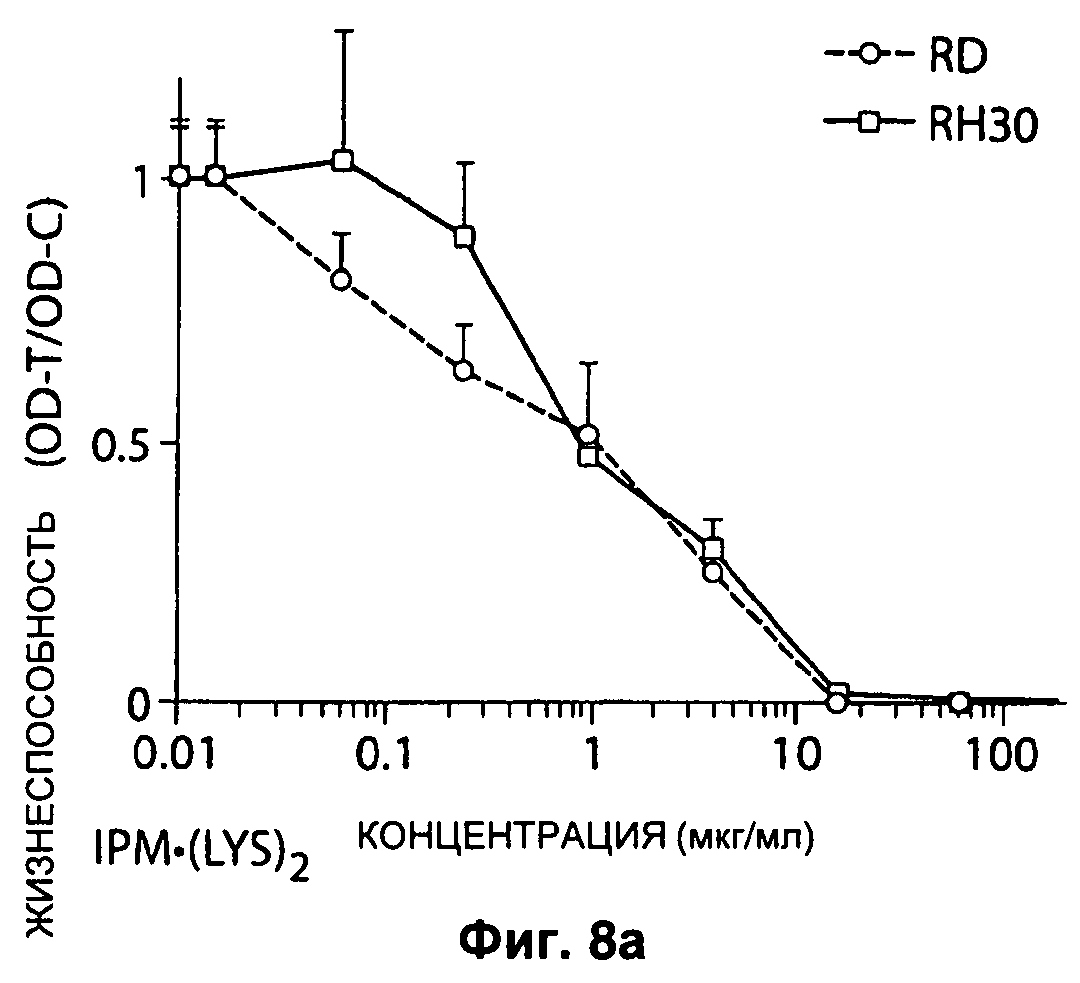
На Рисунке 8а представлена вариабельность маркеров RD и RH30 клеток рабдомиосаркомы при обработке их IPM•(LYS)2 в различных концентрациях.
На Рисунке 8b представлена вариабельность маркеров SKES1 и SKPNDW клеток саркомы Эвинга при обработке их IPM•(LYS)2 в различных концентрациях.
На Рисунке 8 с представлена вариабельность маркеров OS230, OS229, OS222 и SaOS клеток остеосаркомы при обработке их IPM•(LYS)2 в различных концентрациях.
На Рисунке 8d представлена вариабельность маркеров SY01 и HSSYII клеток синовиальной саркомы при обработке их IPM•(LYS)2 в различных концентрациях.
На Рисунке 9а представлена вариабельность маркеров RH30 клеток рабдомиосаркомы при обработке их IPM•(LYS)2 в различных концентрациях как один раз в день, так и три раза в день.
На Рисунке 9b представлена вариабельность маркеров OS229 клеток остеосаркомы при обработке их IPM•(LYS)2 в различных концентрациях как один раз в день, так и три раза в день.
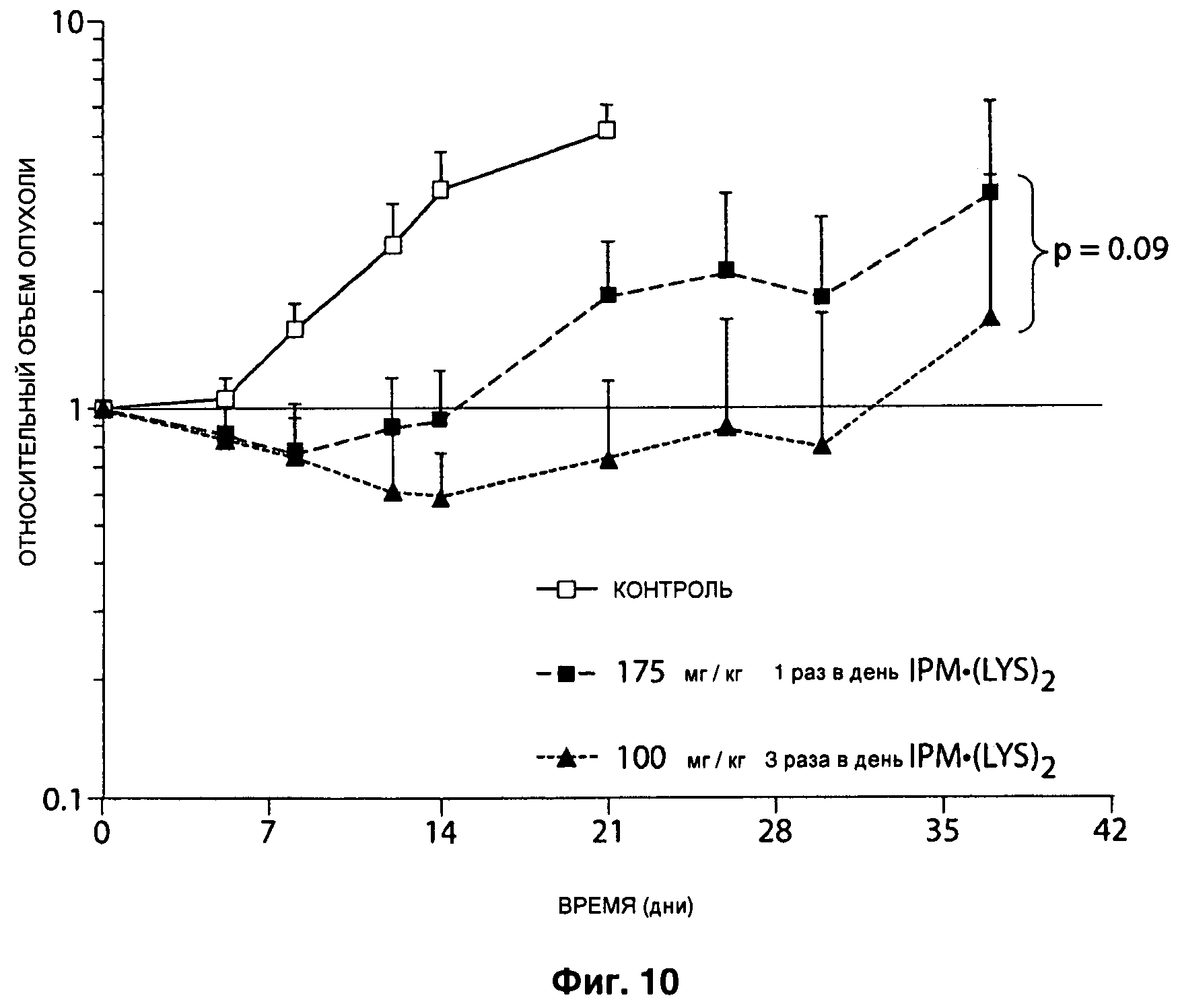
На Рисунке 10 показано, что введение трех максимальных толерантных доз (MTD, от англ. Maximum Tolerated Dose) IPM•(LYS)2 в клетки циклофосфамид-устойчивой остеосаркомы с маркером OS31, имплантированные мышам женского пола с тяжелым комбинированным иммунодефицитом (scid*/* мыши) СВ17, приводит к существенному замедлению опухолевого роста, в каждом из трех режимов дозирования (контрольном, 175 мг/кг ежедневно 1 раз в день, и 100 мг/кг ежедневно 3 раза в день).
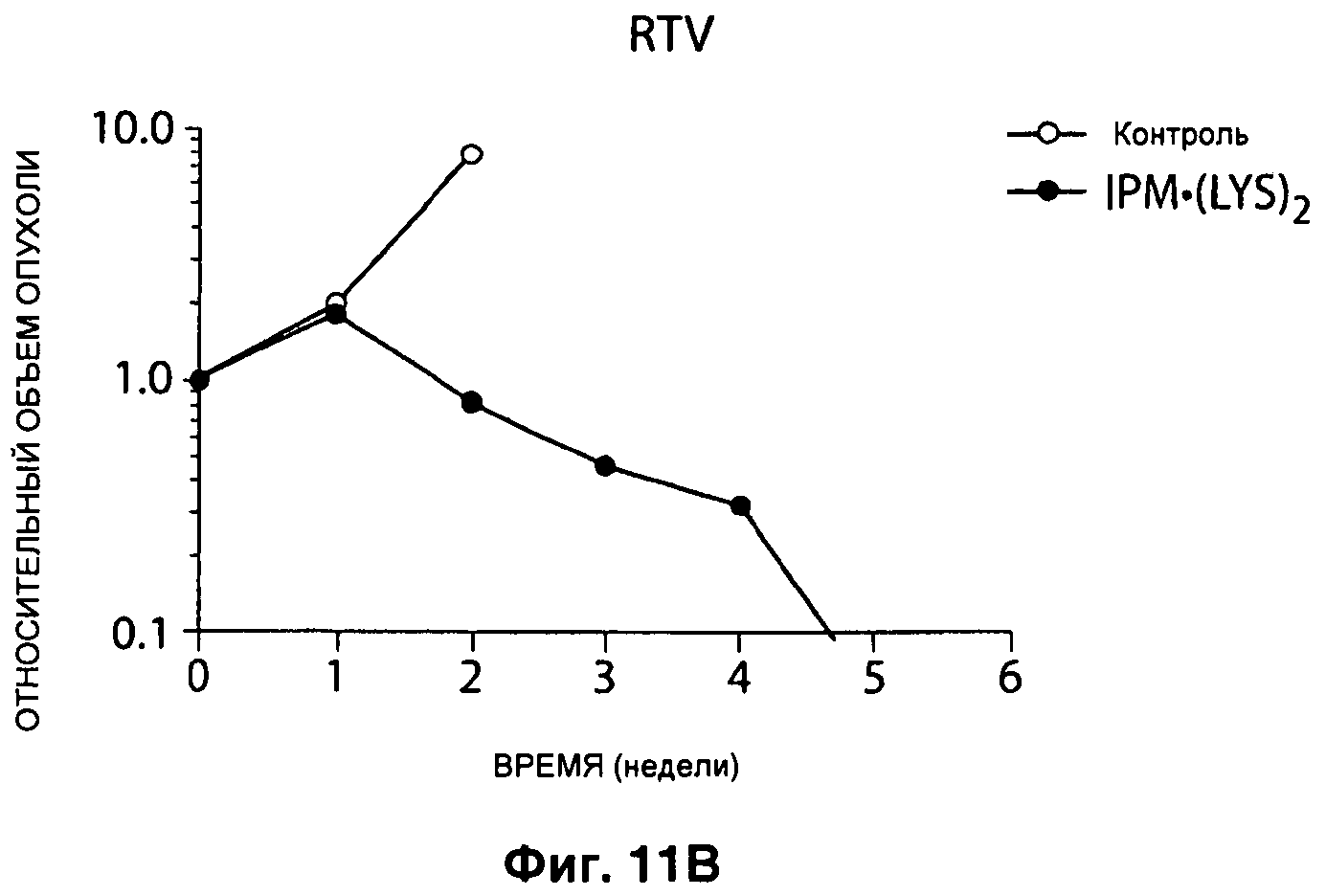
На Рисунке 11 а показана устойчивость клеток остеосаркомы с маркером OS31, имплантированных scid*/* мышам женского пола линии СВ17, к циклофосфамиду, выраженная через относительный объем опухоли, при этом лечение включает введение IPM•(LYS)2 и плацебо.
На Рисунке 11b показана устойчивость клеток остеосаркомы с маркером OS33, имплантированных scid*/* мышам женского пола линии СВ17, к циклофосфамиду, выраженная через относительный объем опухоли, при этом лечение включает введение IPM•(LYS)2 и плацебо.
На Рисунке 12 представлена активность IPM•(LYS)2, выраженная через относительный объем опухоли, при введении его в клетки остеосаркомы с маркером OS31, имплантированные scid*/* мышам женского пола линии СВ17, в дозе 100 мг/кг ежедневно 3 раза в день при сравнении с плацебо.
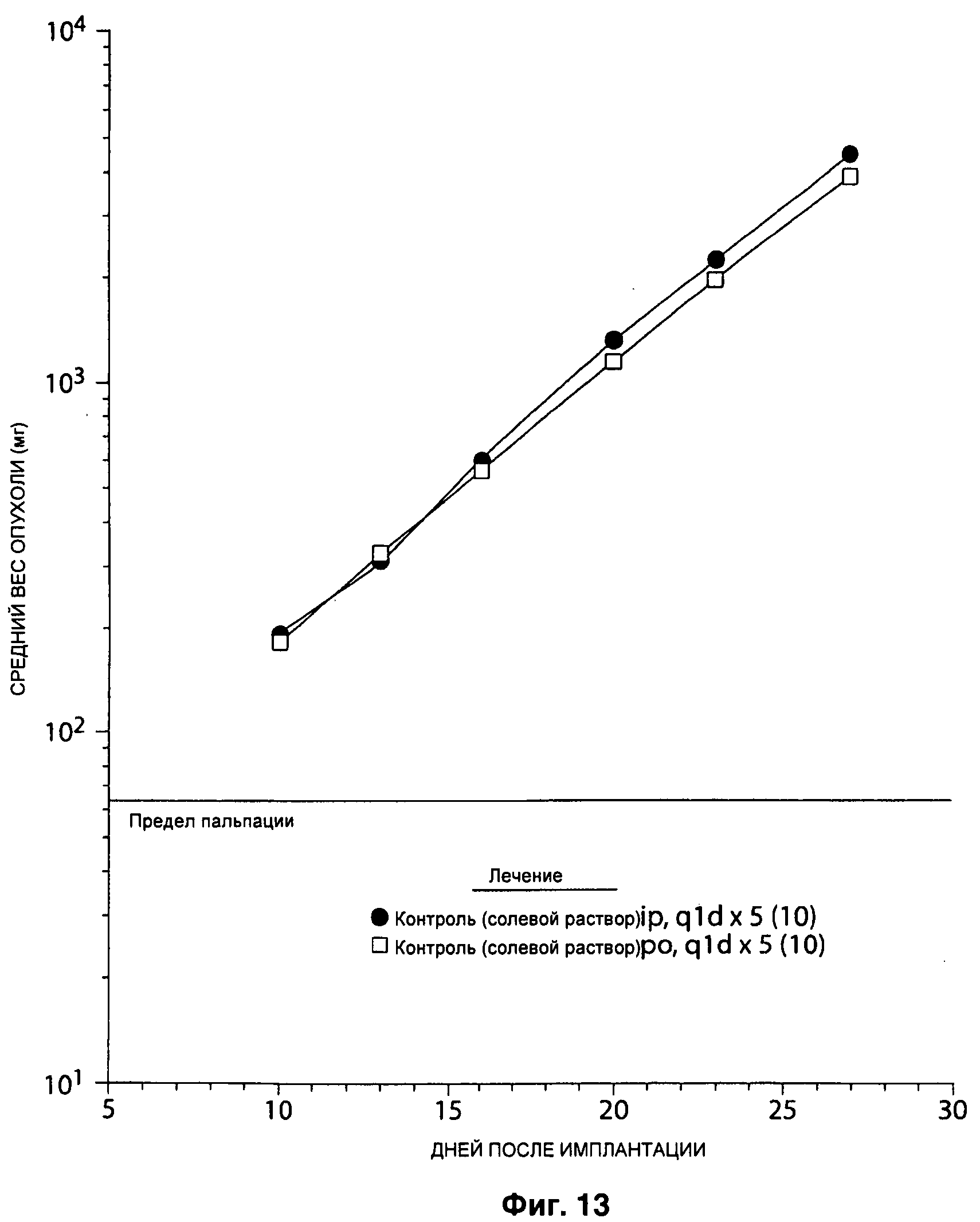
На Рисунке 13 показан ответ опухоли молочной железы SC MX-1 на интраперитонеальное и пероральное введение солевого раствора ежедневно 1 раз в день в течение 5 дней (режим q1d×5).
На Рисунке 14 показан ответ опухоли молочной железы SC MX-1 на интраперитонеальное введение IPM•Tris, IPM и IPM•(LYS)2 в режиме q1d×5.
На Рисунке 15 показан ответ опухоли молочной железы SC MX-1 на пероральное введение IPM•Tris, IPM и IPM•(LYS)2 в режиме q1d×5.
На Рисунке 16 показан ответ опухоли молочной железы SC MX-1 на интраперитонеальное и пероральное введение солевого раствора в режиме q1d×5.
На Рисунке 17 показан ответ опухоли молочной железы SC MX-1 на интраперитонеальное введение IPM•Tris, IPM и IPM•(LYS)2 в режиме q1d×5.
На Рисунке 18 показан ответ опухоли молочной железы SC МХ-1 на пероральное введение IPM•Tris, IPM и IPM•(LYS)2 в режиме q1d×5.
На Рисунке 19 показан ответ опухолей молочной железы МХ-1 на лечение IPM•Tris в комбинации с доксорубицином в дозе 12 мг/кг в день IPM'Tris, которую вводят однократно в течение 5 дней (q1d×5) и 8 мг/мл доксорубицина, которую вводят 4 раза в день в течение 3 дней (q4d×3).
На Рисунке 20 представлен эффект IPM•Tris на выживаемость, в комбинации с доксорубицином, в дозе 12 мг/кг в день IPM•Tris Q1D×5 и 8 мг/кг доксорубицина Q4D×3.
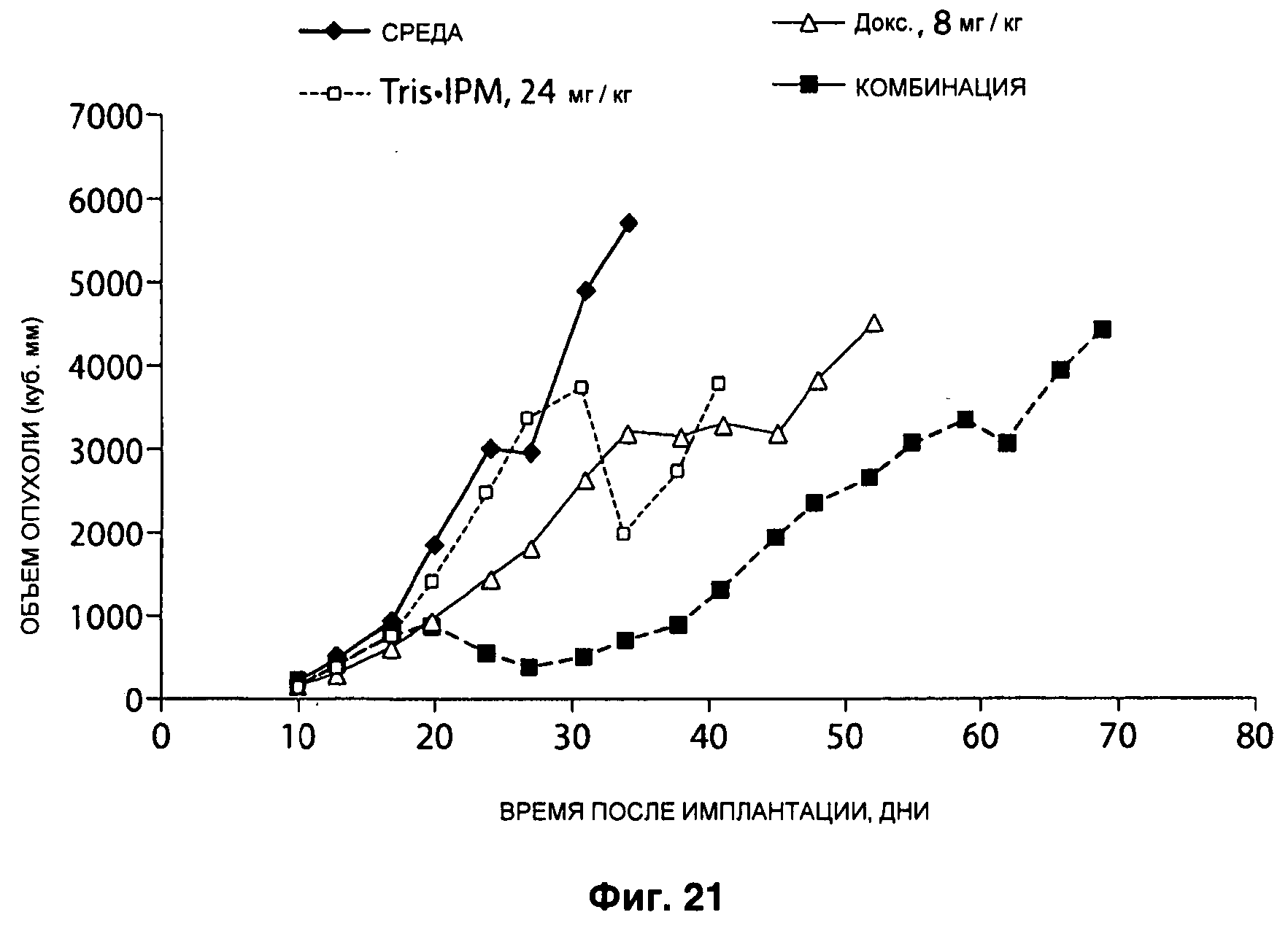
На Рисунке 21 показан ответ опухолей молочной железы МХ-1 на лечение IPM•Tris в комбинации с доксорубицином в дозе 24 мг/кг в день IPM•Tris Q1D×5 и 8 мг/кг в день доксорубицина Q4D×3.
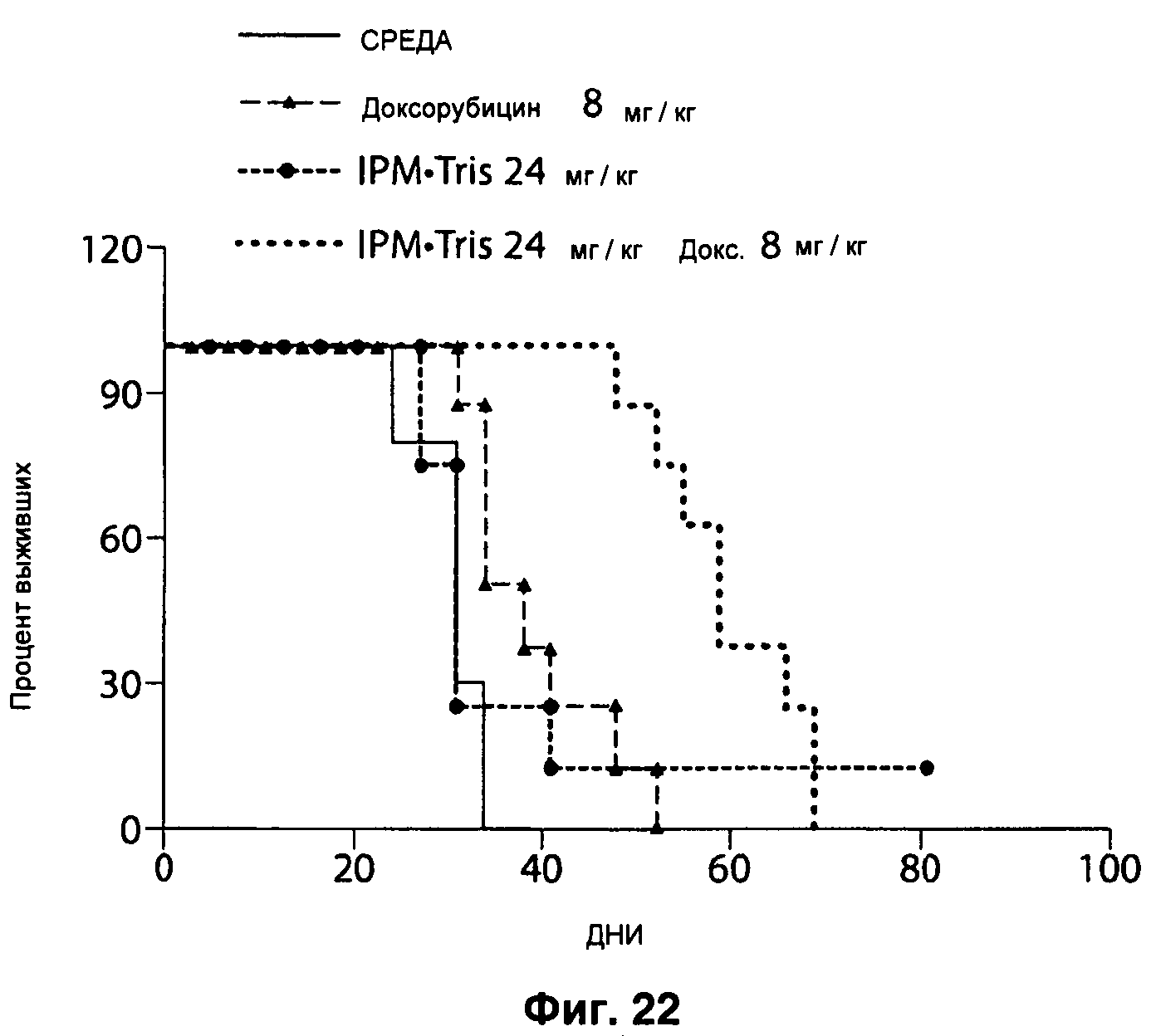
На Рисунке 22 показан эффект IPM•Tris на выживаемость, в комбинации с доксорубицином, в дозе 24 мг/кг в день IPM•Tris Q1D×5 и 8 мг/кг доксорубицина Q4D×3.
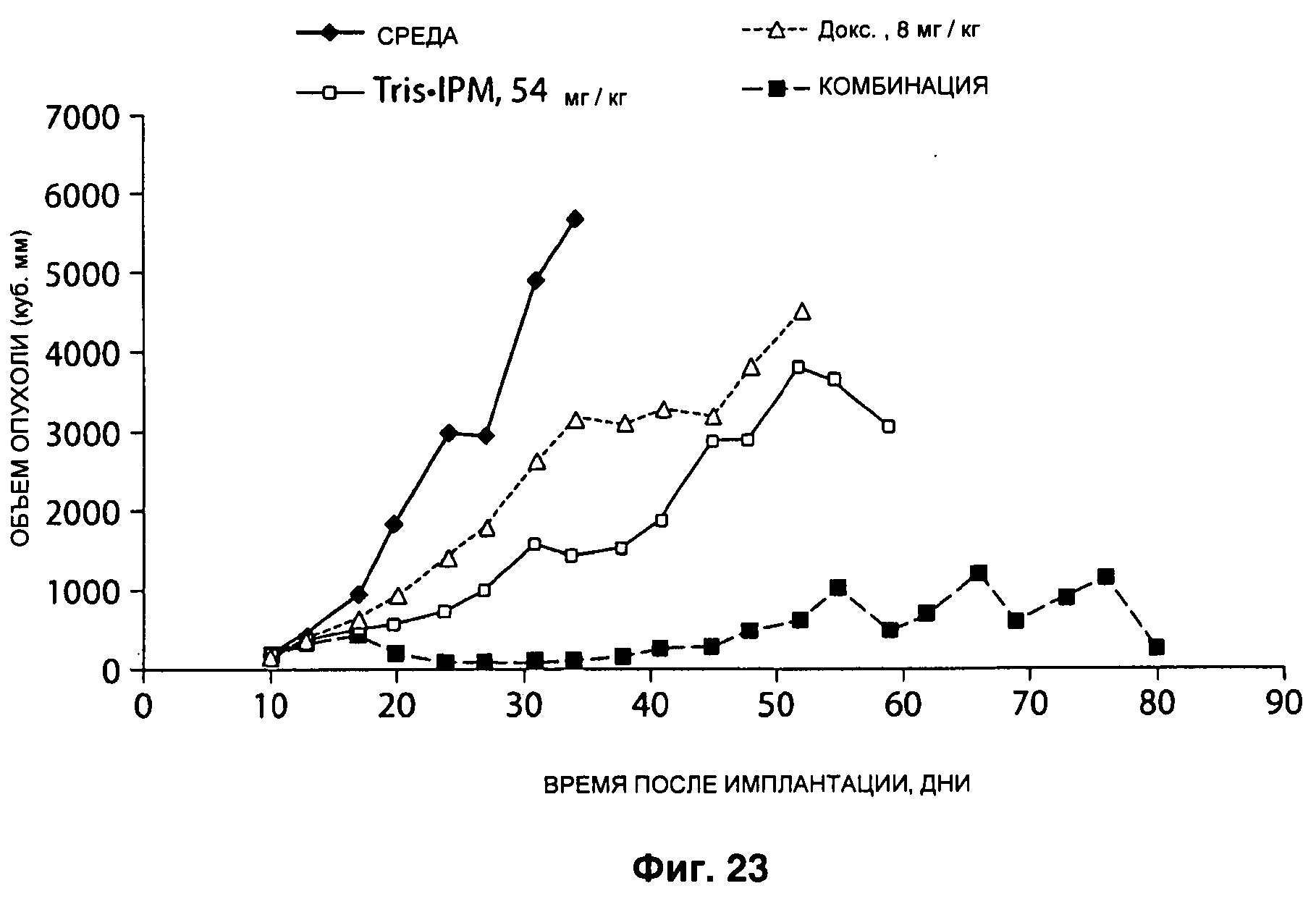
На Рисунке 23 показан ответ опухолей молочной железы МХ-1 на лечение IPM•Tris в комбинации с доксорубицином в дозе 54 мг/кг в день IPM•Tris Q1D×5 и 8 мг/кг в день доксорубицина Q4D×3.
На Рисунке 24 эффект IPM•Tris на выживаемость, в комбинации с доксорубицином, в дозе 54 мг/кг в день IPM•Tris Q1D×5 и 8 мг/кг доксорубицина Q4D×3.
На Рисунке 25 показана токсичность IPM•Tris/доксорубицина при таком комбинированном введении.
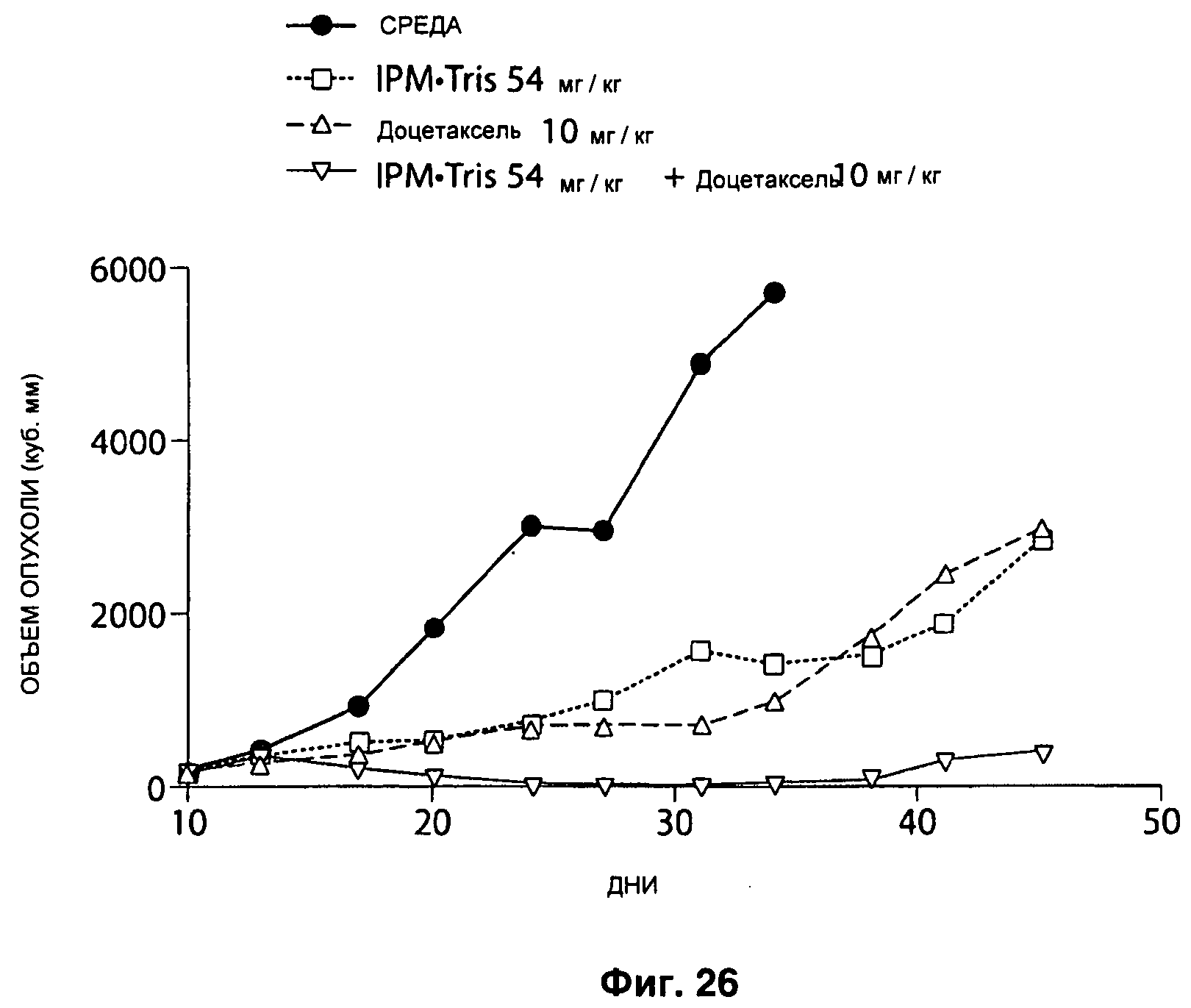
На Рисунке 26 ответ опухолей молочной железы МХ-1 на лечение IPM•Tris в комбинации с доцетакселем в дозе 54 мг/кг в день IPM•Tris Q1D×5 интраперитонеально и 10 мг/кг в день доцетакселя Q6D×3 внутривенно.
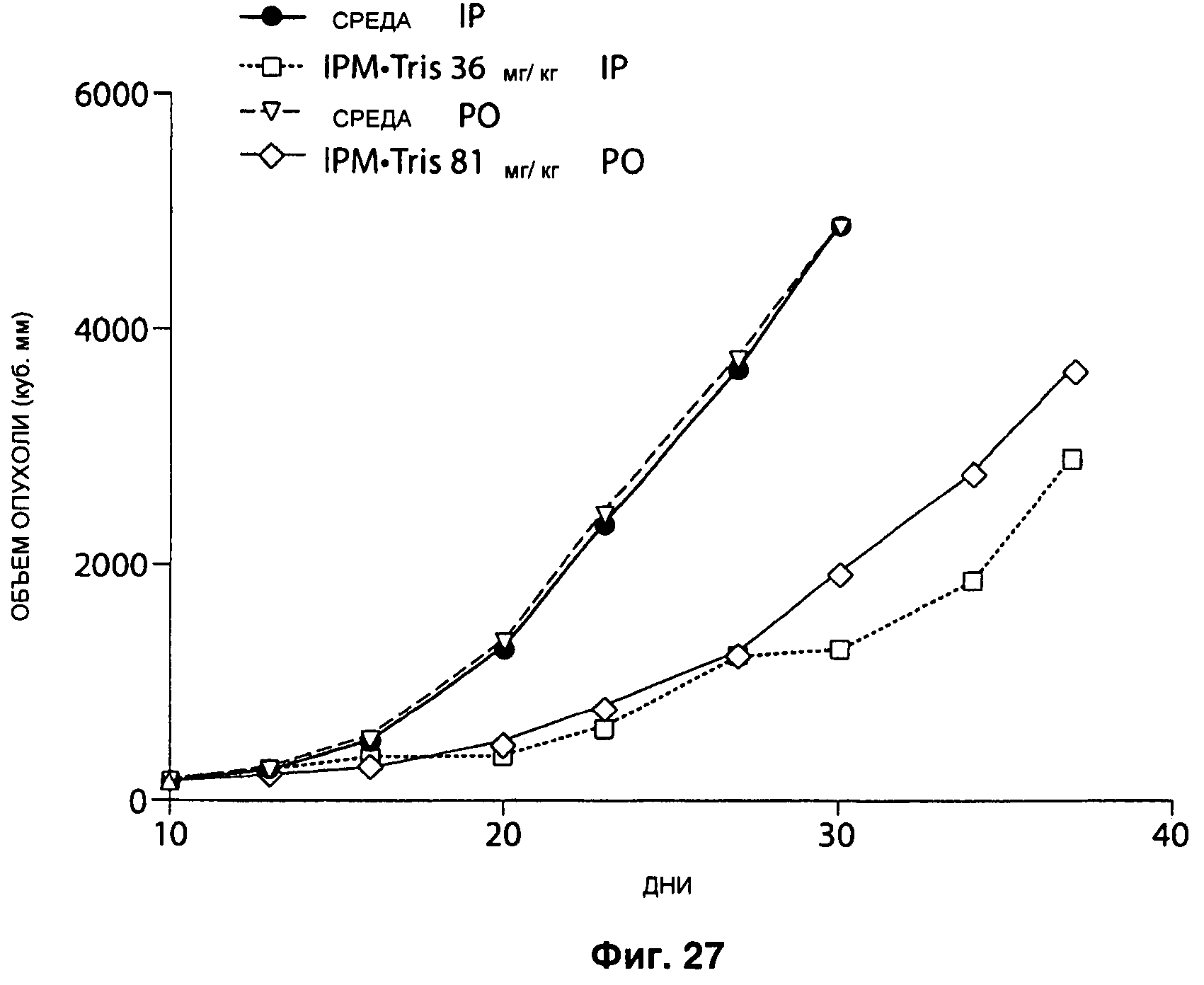
На Рисунке 27 ответ опухолей молочной железы МХ-1 на лечение IPM•Tris в дозе 36 мг/кг в день интраперитонеально или в дозе 81 мг/кг в день перорально.
На Рисунке 29 представлена фармакокинетика IPM•Tris при пероральном и внутривенном введении крысам женского пола Спраг-Доули.
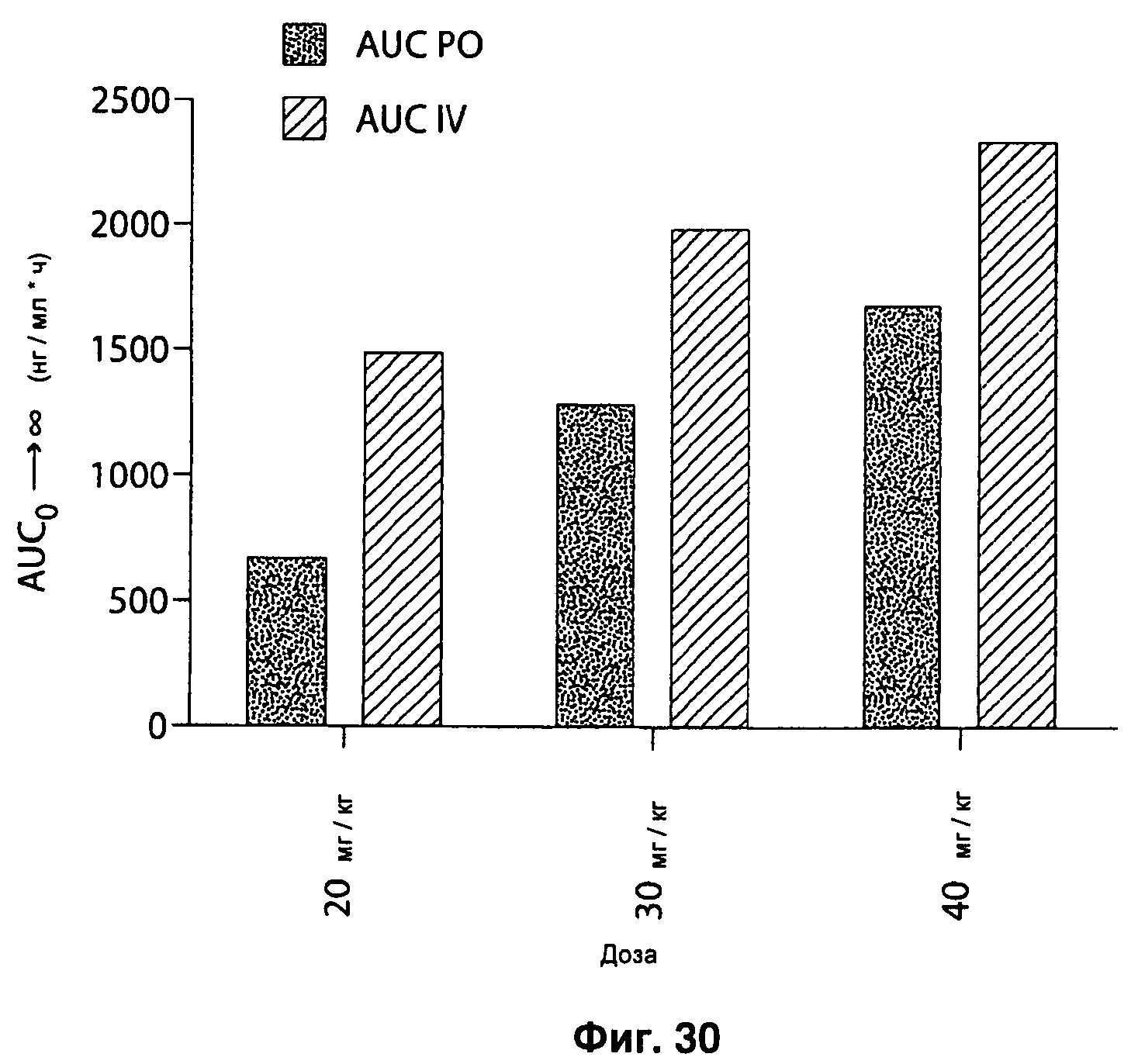
На Рисунке 30 представлена площадь под фармакокинетической кривой (AUC) с увеличением доз IPM•Tris при пероральном и внутривенном введении.
На Рисунке 31 представлена Cmax с увеличением доз IPM•Tris при пероральном и внутривенном введении.
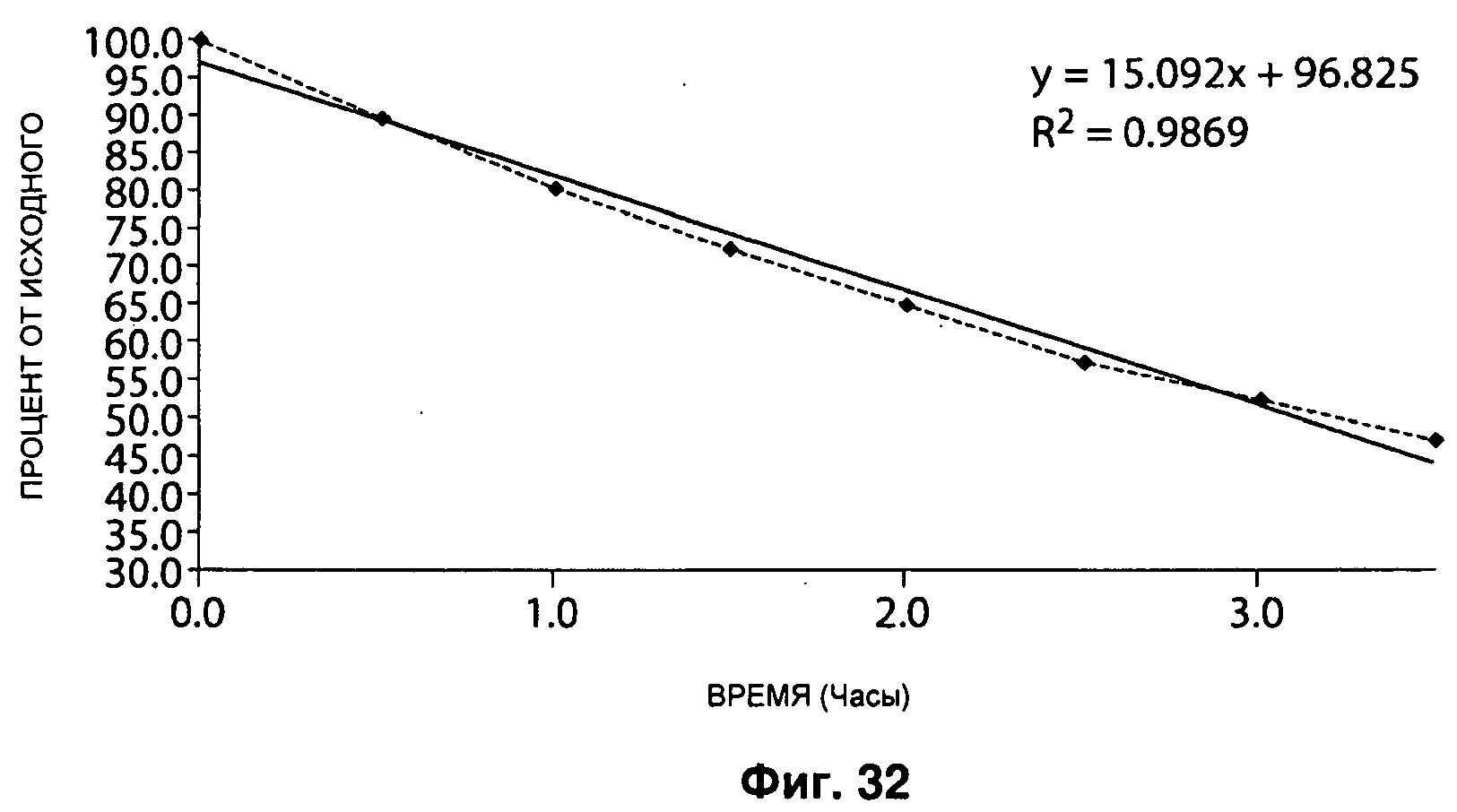
На Рисунке 32 представлена стабильность IPM в буферном растворе при pH 7.0 и 25°С.
Подробное описание изобретения
I. Соли IPM и его аналогов
Заявленные композиции включают кристаллические соли IPM или его аналогов. Согласно определенным вариантам осуществления изобретения, заявленные соли включают один или более катион. Согласно определенным вариантам осуществления изобретения, катионами могут быть сопряженная с аминным основанием кислота или четвертичный аммониевый катион.
Согласно определенным вариантам осуществления изобретения, заявленные кристаллические соединения включают соли IPM или его аналогов. Такие соединения включают кристаллические соединения, имеющие формулу (I):
где А+ представляет собой аммониевое соединение, выбранное из группы протонированных (сопряженных с кислотой) или четвертичных форм алифатических аминов и ароматических аминов; а Х и Y независимо друг от друга являются уходящими группами. Согласно определенным вариантам осуществления изобретения, Х и Y независимо друг от друга представляют собой галоген. Предпочтительно, Х и Y являются одинаковыми соединениями. Согласно таким вариантам осуществления изобретения, Х и Y оба представляют собой Cl.
Отдельными примерами подходящих сопряженных с аминными основаниями кислот являются, без ограничений указанными, кислоты сопряженные с моно-, бис- или трис-(2-гидроксиэтил)амином, 2-гидрокси-терт-бутиламином, N,N-диметил-N-(2-гидроксиэтил)амином, и трис(гидроксиметил)аминометаном (Tris).
Согласно определенным вариантам своего осуществления, изобретение относится к соединениям, включающим кристаллические соли IPM или его аналогов, при этом IPM или его аналог и противоион, предпочтительно, Tris, находятся в соотношении от 2:1 до 1:2, предпочтительно, 1:1. Согласно определенным вариантам осуществления изобретения, кристаллическая композиция включает более чем одну полиморфную форму кристаллов, например, две, три, четыре или даже пять полиморфных кристаллических форм. Согласно еще одному альтернативному варианту осуществления изобретения, кристаллическая композиция включает единичную полиморфную кристаллическую форму. Согласно определенному варианту осуществления изобретения, такие соли являются более стабильными, чем IPM и его аналоги в форме свободных кислот.
Согласно определенным вариантам осуществления изобретения, соединение представляет собой кристаллическую соль с соотношением IPM:Tris, составляющим 1:1.
Согласно еще одному варианту осуществления изобретения, температура плавления кристаллического твердого вещества составляет, приблизительно, от 100 до 110°С, приблизительно, от 102 до 108°С, приблизительно, от 103 до 106°С, или даже от 105 до 106°С.
Согласно определенным вариантам осуществления изобретения, соединение, например, кристаллическая соль с соотношением IPM:Tris 1:1, является, по крайней мере, приблизительно, на 80% химически чистой, по крайней мере, на 85% химически чистой, по крайней мере, на 90% химически чистой, по крайней мере, на 95% химически чистой, по крайней мере, на 98% химически чистой, или даже по крайней мере на 99% химически чистой. Согласно данным вариантам осуществления изобретения, содержание примесей в изолированном веществе не превышает 1% по весу. Согласно определенным вариантам осуществления изобретения, степень химической чистоты измеряют относительно всех других компонентов композиции, в то время как согласно другим вариантам осуществления изобретения (например, если соединение является частью фармацевтической композиции или лиофилизированной смеси), степень химической чистоты может измеряться относительно продуктов распада соединения (например, фосфорсодержащих продуктов распада соединения), или побочных продуктов, образующихся в процессе получения соединения (например, фосфорсодержащих продуктов распада соединения), тем самым исключаются другие соединения, целенаправленно добавляемые в композицию.
Согласно определенным вариантам осуществления изобретения, заявленные соединения представляют собой соли IPM или его аналогов, при этом период полураспада такой соли при комнатной температуре (например, около 23°С) в присутствии воды, больше, чем период полураспада IPM (например, в форме свободной кислоты) в присутствии воды при тех же условиях. Согласно данному определенному варианту осуществления изобретения, соль IPM имеет период полураспада в присутствии воды, равный периоду полураспада самого IPM или превышающий его, по крайней мере, в два раза в присутствии воды, более предпочтительно превышающий его, по крайней мере, в пять раз.
Согласно определенным вариантам осуществления изобретения, заявленные соединения представляют собой соли IPM или его аналогов, при этом указанные соли являются стабильными при комнатной температуре в присутствии воды, по крайней мере, в течение одного дня, двух дней, трех дней, четырех дней, пяти дней, шести дней, или даже в течение недели.
Используемый здесь термин "стабильный" означает, что химическая чистота соли IPM или его аналога по прошествии периода времени (например, одного месяца, двух месяцев, трех месяцев, шести месяцев, года и т.д.) составляет, по крайней мере, 90%, по крайней мере, 95%, по крайней мере, 97%, или даже, по крайней мере, 99% от исходной величины, которая может быть определена, например, при помощи HPLC с использованием испарительного детектора светорассеяния (ELSD, от англ. Evaporate Light Scattering Detection). Такое исследование может осуществляться, например, с использованием колонки С18 и изократической системы с мобильной фазой, включающей 0,005 М гептафтормасляной кислоты и 0,1% трифторуксусной кислоты в воде.
Согласно определенным вариантам своего осуществления, настоящее изобретение относится к лиофилизатам, включающим соединение, имеющее формулу:
где А+ представляет собой аммониевое соединение, выбранное из группы протонированных (сопряженных с кислотой) или четвертичных форм алифатических аминов и ароматических аминов, включая основные аминокислоты, гетероциклические амины, замещенные и незамещенные пиридины, гуанидины и амидины; а Х и Y независимо друг от друга являются уходящими группами.
Примерами подходящих аминных оснований (и их соответствующих аммониевых ионов), которые могут использоваться в заявленных соединениях, являются, без ограничений указанными, пиридин, N,N-диметиламинопиридин, диазабициклононан, диазабициклоундецен, N-метил-N-этиламин, диэтиламин, триэтиламин, диизопропилэтиламин, моно-, бис- или трис-(2-гидроксиэтил)амин, 2-гидрокси-терт-бутиламин, трис-(гидроксиметил)аминометан, N,N-диметил-N-(2-гидроксиэтил)амин, три-(2-гидроксиэтил)амин и N-метил-D-глюкамин.
Согласно определенным вариантам своего осуществления, изобретение относится к лиофилизатам, включающим соединение, имеющее формулу:

При этом согласно формуле, В может представлять собой, для каждого n, независимо выбранную основную молекулу. Согласно одному варианту воплощения формулы, соединение В может быть выбрано из группы основных аминокислот, ароматических аминов, замещенных и незамещенных пиридинов, циклических и ациклических гуанидинов, а также циклических и ациклических амидинов. Обычно, n представляет собой число от 1 до 3, таким образом, что формула может включать различные основные группы. Специалисту в данной области очевидно, что представленная структура изофосфорамидного иприта включает кислый протон, и, соответственно, существует преимущественно в форме его сопряженного основания при физиологическом показателе pH и в присутствии основания, такого, как соединение В. Аналогично, В, представляя собой основную группу, существует преимущественно в форме сопряженной кислоты при физиологическом показателе pH и в присутствии изофосфорамидного иприта и аналогов изофосфорамидного иприта. Возможные варианты рассматриваемых соединений представлены в Таблице 1.
II. Композиции и Методы
Согласно определенным вариантам своего осуществления, настоящее изобретение относится к фармацевтической композиции, включающей соль IPM или его аналога и фармацевтически приемлемый растворитель или эксципиент. Согласно данным вариантам осуществления изобретения, концентрация соли IPM или его аналога в растворе составляет, приблизительно, от 3 мг/мл до, приблизительно, 30 мг/мл, или даже больше. Такие солевые растворы можно приготовить, например, путем растворения кристаллического соединения, имеющего, формулу (I), или лиофилизата IPM или его аналога, заявленного в соответствии с настоящим изобретением, в солевом растворе, например, с одновременным помешиванием при комнатной температуре. Согласно данному варианту осуществления изобретения, солевой раствор приготавливают таким образом, что концентрация хлорида натрия составляет, приблизительно, 0,5%, 0,9%, 2,5%, 2,7%, 3,0%, 4,0%, или даже 5,0%.
Согласно определенным вариантам осуществления изобретения, из кристаллического соединения, имеющего формулу (I) или лиофилизата IPM или его аналога, заявленных в соответствии с настоящим изобретением, может быть приготовлен водный раствор. Такой водный раствор (например, в воде или изотоническом солевом растворе) остается стабильным при комнатной температуре в течение, по крайней мере, 60 минут, 80 минут, 100 минут, 120 минут, 140 минут, или даже, в течение, приблизительно, 160 минут при комнатной температуре.
Согласно определенным вариантам осуществления изобретения, кристаллическое соединение, имеющее формулу (I), такое как соль IPM и Tris, имеет растворимость в воде, по крайней мере, приблизительно, 30 мг/мл, 40 мг/мл, 50 мг/мл, 60 мг/мл, 70 мг/мл или даже 80 мг/мл. Согласно определенным вариантам осуществления изобретения, кристаллическое соединение, имеющее формулу (I), такое, как соль IPM или Tris, имеет растворимость в воде, по крайней мере, приблизительно, 200 мг/мл, по крайней мере, 500 мг/мл, по крайней мере, 800 мг/мл, по крайней мере, 1000 мг/мл, по крайней мере, 1200 мг/мл или даже, по крайней мере, 1400 мг/мл. Согласно определенным вариантам осуществления изобретения, показатель pH раствора кристаллического соединения в воде, составляет, приблизительно, от 4,5 до, приблизительно, 10, например, приблизительно, от 5,0 до 8,5, предпочтительно, приблизительно от 5,0 до, приблизительно, 7.0. Согласно определенным вариантам осуществления изобретения, показатель pH такого раствора равен, приблизительно, 5,0.
Согласно определенным вариантам своего осуществления, изобретение относится к набору, включающему кристаллическое соединение, имеющее формулу I, и солевой раствор.
Лиофилизаты, описанные в настоящем изобретении, включают IPM или его аналоги, которые составляют рецептуру с одним или более эквивалентом основания. Поскольку IPM и его аналоги являются кислото-неустойчивыми и кислыми соединениями, заявленные лиофилизаты являются значительно более стабильными и имеют ряд других преимуществ. Преимущества заявленных композиций, касающиеся их синтеза, стабильности и биологической доступности, будут очевидны специалистам после рассмотрения настоящей заявки. Дополнительными преимуществами IPM и IPM аналогов, которые составляют рецептуру вместе с одним или более эквивалентом основания, являются повышенная растворимость в воде или биологических жидкостях организма.
Согласно определенным вариантам осуществления изобретения, заявленные лиофилизаты, представляют собой соли изофосфорамидного иприта или аналогов изофосфорамидного иприта, включая один или более катион. Согласно определенным вариантам осуществления изобретения, катионом может быть сопряженная с аминным основанием кислота, или четвертичный аммониевый катион. Подходящими противоионами для изофосфорамидного иприта или его аналогов являются сопряженные с основанием кислоты (в данном описании используемые термины, которые относятся к аминам, должны интерпретироваться как их сопряженные кислоты, если в тексте дополнительно не оговаривается, что речь идет об изолированных аминах), включая основные аминокислоты, гетероциклические амины, ароматические амины, пиридины, гуанидины и амидины. Из алифатических аминов наиболее предпочтительными для использования в заявленных соединениях являются ациклические алифатические амины, и также циклические и ациклические ди- и триалкиламины. Кроме того, подходящими противоионами, которые могут использоваться в заявленных соединениях, являются четвертичные аммониевые противоионы. Согласно определенным вариантам осуществления изобретения, такой лиофилизат может дополнительно содержать наполнитель. Подходящими наполнителями являются, без ограничений указанными, маннитол, безводная лактоза, сахароза, D(+)-трегалоза, декстран 40 и повидон (PVP K24).
Согласно определенным вариантам осуществления изобретения, заявленные соединения и композиции, такие как лиофилизаты, являются стабильными при комнатной температуре в течение, по крайней мере, двух недель, по крайней мере, трех недель, по крайней мере, месяца, по крайней мере, в течение двух месяцев, по крайней мере, в течение трех месяцев, или даже, по крайней мере, в течение шести месяцев. Согласно определенным вариантам осуществления изобретения, соли являются стабильными при низких температурах (например, 0°С, 2°С, 4°С, 6°С и т.д.) в течение, по крайней мере, двух недель, по крайней мере, трех недель, по крайней мере, месяца, по крайней мере, в течение двух месяцев, по крайней мере, в течение трех месяцев, или даже, по крайней мере, в течение шести месяцев. Согласно данным вариантам осуществления изобретения, заявленные соединения и композиции, такие как лиофилизаты, являются стабильными в течение, по крайней мере, месяца, по крайней мере, двух месяцев, по крайней мере, четырех месяцев, или даже в течение, по крайней мере, шест и месяцев при низкой температуре (например, от 0°С до 20°С, от 0°С до 10°С, или даже от 2°С до 8°С). Согласно данным определенным вариантам осуществления изобретения, лиофилизат содержит соль IPM или его аналога. Согласно определенным вариантам осуществления изобретения, лиофилизат содержит IPM•Tris или IPM(LYS)2, предпочтительно, IPM•Tris, и, согласно наиболее предпочтительным вариантам осуществления изобретения, такие композиции дополнительно содержат наполнитель, такой как маннитол.
Согласно дополнительному варианту осуществления изобретения, описанные выше соли могут включать второй амин или аммониевую группу. Согласно одному варианту осуществления изобретения, заявленные лиофилизаты содержат более чем один эквивалент амина на каждый эквивалент изофосфорамидного иприта или аналога изофосфорамидного иприта. Такие варианты включают соединения, в которых соотношение амина к изофосфорамидному иприту или его аналогу не является целым числом. Согласно определенным вариантам осуществления изобретения, соотношение амина к изофосфорамидному иприту или аналогу изофосфорамидного иприта составляет два к одному или три к одному. В действительных вариантах осуществления изобретения, получают соли, содержащие два эквивалента азотистого основания на эквивалент изофосфорамидного иприта. Согласно определенным вариантам осуществления изобретения, основание амина, которое используется для получения солей изофосфорамидного иприта или аналога изофосфорамидного иприта, содержит более чем одну азотистую группу; такие основания могут обозначаться термином "многоосновные". Более конкретно, определенными примерами многоосновных оснований, которые могут использоваться в соответствии с настоящим изобретением, являются основания с двумя аминными группами; их обозначают термином "двухосновные". Например, одной из подходящих двухосновных молекул является N,N-диметиламинопиридин, который включает две группы аминного основания.
Определенные заявленные лиофилизаты, включающие изофосфорамидный иприт или его аналоги, содержат две уходящие группы. Без ограничения одной определенной теорией, считается, что две уходящие группы in vivo замещаются биомолекулами нуклеофила, такими, как нуклеиновые кислоты и белки, что обеспечивает перекрестную связь с биомолекулами. Термин "уходящая группа" относится к группе, которая может быть замещена нуклеофилом. Относительно заявленных соединений, термин "уходящая группа" относится к группе, которая может быть замещена с образованием промежуточного соединения азиридиния, или может быть непосредственно замещена биомолекулой нуклеофила, такой, как нуклеиновая кислота, с образованием, например, 7-алкилированного гуанидинового соединения. Примерами походящих уходящих групп являются галогены и сульфонаты (-SO2R). Согласно одному варианту воплощения заявленных солей аналогов изофосфорамида, соединение представляет собой соединение "смешанных" уходящих групп, включая две различные уходящие группы, например, галоген и сульфонат, или два различных галогена, например, бром и хлор. В патенте США номер 6,197,760 Struck раскрывается способ получения таких соединений с разными уходящими группами.
Согласно определенным вариантам осуществления изобретения, лиофилизаты заявленных солей имеют лучшую стабильность при восстановлении в растворе по сравнению с лиофилизатами чистого изофосфорамидного иприта. Согласно данным вариантам осуществления изобретения, лиофилизат, приготовленный из заявленных солей IPM или его аналогов и эксципиента, такой, как лиофилизат IPM или его аналога и Tris, дополнительно содержащий эксципиент, например, наполнитель, такой, как маннитол, и восстановленный в солевом растворе (предпочтительно, 5% растворе хлорида натрия) сохраняет >90% активности в течение, по крайней мере, 30 минут, 60 минут, 90 минут, 120 минут, 140 минут, или даже в течение, приблизительно, 160 минут.
Согласно данному варианту осуществления изобретения, при растворении соли IPM или его аналога, например, IPM•Tris, или лиофилизата из соли IPM или его аналога, например, IPM•Tris, и дополнительного эксципиента, например, наполнителя, такого как маннитол, в солевом растворе сохраняется, по крайней мере, 96%, по крайней мере 97%, по крайней мере, 98%, или даже, по крайней мере, 99% химической чистоты соединения в течение, по крайней мере, 30 минут, 60 минут, 90 минут, 3 часов или даже 4,5 часов и более при комнатной температуре. Согласно определенным вариантам осуществления изобретения, такие восстановленные растворы являются более стабильными, чем восстановленные растворы IPM•(LYS)2 при одинаковых условиях. Согласно данным вариантам осуществления изобретения, восстановленный IPM•(LYS)2распадается, по крайней мере, в 1,25 раз быстрее, по крайней мере, в 1,5 раза быстрее, по крайней мере, в два раза быстрее, или даже, по крайней мере, в три или четыре раза быстрее, чем соль IPM или его аналога.
Согласно определенным вариантам осуществления изобретения, когда лиофилизат включает соль IPM или его аналога и эксципиент, например, если лиофилизат включает соль IPM или его аналога, Tris, и маннитол, такая смесь имеет растворимость в воде, по крайней мере, приблизительно, 30 мг/мл, 40 мг/мл, 50 мг/мл, 60 мг/мл, 70 мг/мл или даже 80 мг/мл.
Согласно определенным вариантам осуществления изобретения, лиофилизаты заявленных солей IPM или его аналогов являются более стабильными, чем лиофилизаты чистого изофосфорамидного иприта, то есть в форме свободной кислоты. Согласно предпочтительным вариантам осуществления изобретения, лиофилизат заявленных солей имеет более длительный срок хранения, чем лиофилизаты чистого изофосфорамидного иприта, предпочтительно, по крайней мере, в два раза более длительный срок хранения, еще более предпочтительно, по крайней мере, в пять раз более длительный срок хранения. Согласно определенным вариантам осуществления изобретения, лиофилизат состоит из Tris соли IPM, которая может быть как в кристаллической, так и не в кристаллической форме перед растворением.
Как описано выше, согласно определенным вариантам осуществления изобретения, такие лиофилизаты дополнительно содержат эксципиент, например, наполнитель, предпочтительно маннитол. Согласно определенным вариантам осуществления изобретения, лиофилизат содержит наполняющий агент, выбранный из группы, включающей маннитол, безводную лактозу, сахарозу, D(+)-трегалозу, декстран 40 и повидон (PVP K24), предпочтительно, маннитол. Согласно определенным вариантам осуществления изобретения, добавление такого эксципиента может улучшать стабильность лиофилизата, по сравнению с лиофилизатом, не содержащим такого наполнителя. Согласно данному варианту осуществления изобретения, такой лиофилизат является стабильным при температуре, приблизительно, -70°С, приблизительно, -20°С, или даже 5°С, например, в течение одного месяца, двух месяцев, трех месяцев, шести месяцев, девяти месяцев, одного года или даже в течение двух лет и более.
Согласно определенным вариантам осуществления изобретения, если лиофилизат содержит наполнитель, такой, как маннитол, его количество составляет, приблизительно, от 1% до 10%, или, приблизительно, от 1% до 5% (вес/объем). Перед процессом лиофилизации или после восстановления, такие композиции могут содержать, приблизительно, от 15 мг/мл до, приблизительно, 25 мг/мл IPM, и/или амина, такого как Tris в концентрации, приблизительно, от 0,5 до 1,5 М, предпочтительно, приблизительно эквимолярно количеству IPM. Согласно определенным вариантам осуществления изобретения, при получении раствора перед лиофилизацией, вместо последовательного добавления IPM и амина, такого как Tris, как отдельных компонентов, их добавляют вместе в форме кристаллической соли IPM•Tris, как раскрывается в заявке.
Согласно определенным вариантам своего осуществления, изобретение относится к наборам, содержащим заявленный лиофилизат и солевой раствор.
Соединения, заявленные в соответствии с настоящим изобретением, могут вводиться перорально, местно, чрескожно, парентерально, посредством ингаляции или при помощи спрея; при этом они могут вводиться посредством лекарственных форм, содержащих подходящие нетоксичные фармацевтически приемлемые эксципиенты, адъюванты и носители.
Согласно определенным вариантам осуществления изобретения, предпочтительным является парентеральное введение заявленных солей IPM или его аналогов посредством инъекций. Согласно определенным вариантам осуществления изобретения, предпочтительным является пероральное введение заявленных солей IPM или его аналогов. Согласно определенным вариантам осуществления изобретения, при пероральном введении солей IPM и его аналогов наблюдаемые фармакокинетические параметры аналогичны таковым при внутривенном введении. Заявленные агенты могут входить в состав лекарственных форм, содержащих однократную дозу препарата, или несколько доз, в зависимости от конкретного заболевания, состояния пациента, токсичности соединения и других факторов, которые определяет специалист.
Терапевтически эффективное количество соединения или соединений, которые вводятся пациенту, может различаться в зависимости от желаемых эффектов и факторов, указанных выше.
Фармацевтические композиции, предназначенные для введения пациенту, могут содержать носители, уплотнители, растворители, консерванты, буферные агенты, поверхностно-активные вещества и другие агенты в дополнение к основным активным молекулам. Фармацевтические композиции также могут включать один или более дополнительный активный ингредиент, такой, как противомикробный агент, противовоспалительный агент, анестетик и подобные ингредиенты. Фармацевтические композиции могут содержать дополнительные компоненты, такие, как носители. Фармацевтически приемлемые носители, подходящие для таких композиций, обычно широко применяются. В издании Remington's Pharmaceutical Science, by E.W.Martin Publishing Co., Easton, PA, 21stEdition (2006), описываются соединения и композиции, подходящие для фармацевтической доставки заявленных соединений.
В целом, природа носителя зависит от того, какой способ введения композиции будет использоваться. Например, композиции для парентерального введения обычно содержат подходящие для инъекционного введения жидкости, к которым относятся фармацевтически и физиологически приемлемые жидкости, такие, как вода, физиологический раствор, сбалансированные солевые растворы, водная декстроза, глицерол или подобные носители. Для твердых композиций (например, порошков, пилюль, таблеток, или капсул) традиционными нетоксичными твердыми носителями являются, например, фармацевтические пригодные маннитол, лактоза, крахмал или стеарат магния. Помимо биологически нейтральных носителей, фармацевтические композиции, предназначенные для введения пациенту, могут содержать небольшие количества нетоксичных вспомогательных компонентов, таких как увлажняющие или эмульгирующие агенты, консерванты, буферные агенты и подобные вещества, например ацетат натрия или сорбитанмонолаурат.
Согласно определенным вариантам осуществления изобретения, заявленное соединение входит в состав лекарственной формы для перорального введения, такой, как пилюли, таблетки или капсулы. Согласно определенным вариантам осуществления изобретения, пероральной лекарственной формой является капсула.
Согласно определенным вариантам осуществления изобретения, такая пероральная лекарственная форма содержит, по крайней мере, один эксципиент, глидант, любрикант и/или дезинтегрант. Согласно данным вариантам осуществления изобретения, такими подходящими эксципиентами, глидантами, растворителями или любрикантами, и/или дезинтегрантами являются, без ограничений указанными, тальк, коллоидальная двуокись кремния, крахмал, силикат кальция, карбонат магния, оксид магния, лаурилсульфат магния, лаурилсульфат натрия, лактоза, микрокристаллическая целлюлоза, гидроксипропилметилцеллюлоза, декстроза, глюкоза, сахароза, крахмал, производные крахмала, карбонат кальция, двухосновный фосфат кальция, карбонат магния, стеарат магния, стеарат кальция, стеарилфумарат натрия, полиэтиленгликоль 4000, полиэтиленгликоль 6000, бензоат натрия, слабоминерализованное масло, гидрогенированные овощные масла, стеариновая кислота, глицерилбегенат, нерастворимые ионообменные смолы, натриевая соль гликолята крахмала, натриевая карбоксиметилцеллюлоза (кроскармеллоза натрия), камеди (например, агар, гуар, ксантан), альгиновая кислота, альгинат натрия и кроспивидон.
Согласно данным вариантам осуществления изобретения, пероральные лекарственные формы содержат заявленное соединение и по крайней мере, один эксципиент, глидант, растворитель, любрикант и/или дезинтегрант; предпочтительно, по крайней мере, один эксципиент, глидант, растворитель, любрикант и/или дезинтегрант, подходящий для составления единой рецептуры с гигроскопичным активным агентом. Согласно данным вариантам осуществления изобретения, пероральные лекарственные формы содержат, по крайней мере, один эксципиент, глидант, растворитель, любрикант и/или дезинтегрант, выбранный из группы, включающей микрокристаллическую целлюлозу, лактозу, натриевую карбоксиметиллцелюлозу, стеарат магния, двухосновный фосфат кальция, натриевую соль гликолята крахмала, гидроксипропилметилцеллюлозу и маннитол.
Согласно определенным вариантам осуществления изобретения, заявленное соединение составляет рецептуру для введения человеку. Согласно данному аспекту изобретения, фармацевтическая композиция содержит, приблизительно, от 0,1 мг/мл до, приблизительно, 250 мг/мл, например, приблизительно, от 20 до 100 мг/мл соединения соли IPM или его аналога.
Согласно одному аспекту, заявленные лекарственные формы содержат единичную дозу препарата. Например, такие лекарственные формы могут содержать, приблизительно, от 1 мг до, приблизительно, 100 мг, или от 100 мг до, приблизительно, 1500 мг, более конкретно, приблизительно, от 5 мг до, приблизительно, 200 мг, или от 200 мг, до приблизительно, 1500 мг заявленной соли IPM или его аналога на одну лекарственную форму. Согласно определенным вариантам осуществления изобретения, лекарственная форма может содержать, приблизительно, 15 мг, приблизительно, 30 мг, приблизительно, 45 мг, приблизительно, 60 мг, приблизительно, 75 мг, или даже, приблизительно, 77 мг заявленной соли IPM или его аналога.
Согласно определенным вариантам осуществления изобретения, предполагается, что заявленные соединения вводятся посредством инъецируемого и/или имплантируемого лекарственного депо, например, содержащего мультивезикулярные липосомы, такие как DepoFoam (SkyePharma, Inc, San Diego, Ca) (см, например, Chamberlain et al. Arch. Neuro. 1993, 50, 261-264; Katri et al. J.Pharm. Sci. 1998, 87, 1341-1346; Ye et al., J.Control Release 2000, 64, 155-166; and Howell, Cancer J. 2001, 7, 219-227).
Настоящее изобретение относится к способам лечения патологических состояний, характеризующихся аномальной или патологической пролиферативной активностью или неоплазией, заключающимся во введении одного или более заявленного соединения или композиции субъекту.
К состояниям, для лечения которых может использоваться заявленный способ, относятся такие состояния, которые характеризуются аномальным клеточным ростом и/или дифференцировкой клеток, такие как рак и другие неопластические состояния. Типичные примеры пролиферативных заболеваний, для лечения которых могут использоваться заявленные соединения и композиции, представлены ниже.
Примерами гематологических заболеваний, для лечения которых могут применяться заявленные соединения и композиции, являются лейкозы, включая острые лейкозы (такие как острый лимфоцитарный лейкоз, острый миелоцитарный лейкоз, острый миелогенный лейкоз и миелобластный, промиелоцитарный, миеломоноцитарный, моноцитарный лейкозы и эритролейкоз), хронические лейкозы (такие, как хронический миелоцитарный (гранулоцитарный) лейкоз, хронический миелогенный лейкоз и хронический лимфоцитарный лейкоз), истинная полицитемия, лимфома, болезнь Ходжкина, неходжкинская лимфома (медленно растущие формы и высокодифференцированные формы), множественная миелома, макроглобулинемия Вальденстрема, болезнь тяжелых цепей, миелодиспластический синдром, волосяноклеточный лейкоз и миелодисплазия.
Дополнительными примерами патологических состояний, для лечения которых могут применяться заявленные соединения и композиции, являются твердые опухоли, такие как саркомы и карциномы, включая фибросаркому, миксосаркому, липосаркому, хондросаркому, остеогенную саркому, циклофосфамид-устойчивую саркому, и другие саркомы, синовиому, мезотелиому, опухоль Эвинга, лейомиосаркому, рабдомиосаркому, карциному толстой кишки, лимфонеоплазии, рак поджелудочной железы, рак груди, рак легких, рак яичников, рак предстательной железы, гепатоцеллюлярную карциному, плоскоклеточную карциному, базальноклеточную карциному, аденокарциному, карциному потовых желез, карциному сальных желез, папиллярную карциному, папиллярные аденокарциномы, медуллярную карциному, бронхогенную карциному, почечноклеточную карциному, гепатому, карциному желчных протоков, хориокарциному, опухоль Вильмса, цервикальный рак, опухоли яичек, карциному желчного пузыря и опухоли ЦНС (такие как глиома, астроцитома, медуллобластома, краниофарингиома, эпендимома, пинеалома, гемангиобластома, невринома слухового нерва, олигодендроглиома, менингиома, меланома, нейробластома и ретинобластома).
Согласно определенным вариантам осуществления изобретения, заявленные соединения превосходят чистые СРА и Ifos по противоопухолевой активности в отношении СРА и/или Ifos-устойчивых опухолей. Таким образом, согласно одному аспекту своего осуществления, заявленный способ включает лечение субъекта, страдающего от СРА и/или Ifos-устойчивого неопластического состояния, заявленной солью IPM или его аналога.
Согласно некоторым вариантам осуществления изобретения, заявленные соединения обладают меньшей токсичностью по сравнению с СРА и/или Ifos. Например, введение высоких доз СРА и/или Ifos может приводить к токсическому действию на почки, мочевой пузырь и/или центральную нервную систему в результате накопления определенных метаболитов, таких, как хлорацетальдегид и акролеин. Согласно некоторым вариантам осуществления изобретения, при применении заявленных соединений снижается или прекращается образование указанных веществ или других токсичных метаболитов, при этом сохраняется эффективность заявленных соединений. Заявленные в настоящем изобретении соединения, таким образом, обладают способностью оказывать терапевтическое действие и при этом уменьшать нежелательные побочные эффекты, такие как гибель здоровых клеток почек, мочевого пузыря и центральной нервной системы, которые могут быть обусловлены воздействием метаболитов СРА и/или Ifos. Соответственно, заявленные в настоящем изобретении соединения являются полезной альтернативой СРА и/или Ifos.
Например, заявленные соединения являются полезными при подготовке пациентов к трансплантации клеток крови или костного мозга. СРА и Ifos обычно применяют при трансплантации клеток крови и костного мозга, а заявленные соединения являются хорошей альтернативой указанным веществам, например, благодаря своей низкой токсичности и/или повышенной активности. Кроме того, заявленные соединения могут также применяться в случае трансплантации клеток крови и костного мозга тогда, когда применение СРА и Ifos является неправильным, например, в случае, когда высокие дозы СРА и Ifos становятся слишком токсичными. Заявленные соединения могут вводиться за минуты, дни, недели или месяцы перед трансплантацией, в частности, за несколько дней или месяцев перед трансплантацией. Кроме того, заявленные соединения могут вводиться в составе лекарственных форм, содержащих единичные, множественные и/или повторяющиеся дозы препаратов, и/или в комбинации с другими агентами в препаратах клеток крови или трансплантате костного мозга.
Согласно определенным вариантам осуществления изобретения, заявленные соединения являются полезными в случае так называемых тренировочных режимов при трансплантации костного мозга или клеток крови, например, в качестве заменителей для СРА и Ifos. Кроме того, заявленные соединения могут вводиться без дополнительных защитных мер, таких, как введение препарата Месны и/или внутривенная гидратация, которые всегда применяются при введении СРА и Ifos.
Согласно другому варианту осуществления изобретения, заявленные соединения могут использоваться в комбинации с СРА и/или Ifos, например, при подготовке пациентов к трансплантации клеток крови и костного мозга, при применении тренировочных режимов при трансплантации клеток крови и костного мозга. Композиции, содержащие одно или более заявленное соединение в комбинации с СРА и/или Ifos, имеют дополнительные преимущества, такие, как пониженная токсичность и/или повышенная активность, по сравнению с чистыми СРА и/или Ifos.
Согласно определенным вариантам осуществления изобретения, субъекту вводится приблизительно, от 0,2 мг/кг в день до, приблизительно, 20 мг/кг в день заявленной соли IPM или его аналога. Например, субъекту может вводиться, приблизительно, от 0,5 мг/кг в день до, приблизительно, 10 мг/кг в день, например, приблизительно, от 1 мг/кг в день до, приблизительно, 7,5 мг/кг в день заявленного соединения.
Согласно определенным вариантам осуществления изобретения, IPM, его аналог или соль вводят субъекту в дозе (например, ежедневной дозе), превышающей 1,0 г, превышающей, приблизительно, 1,5 г, превышающей, приблизительно, 2,0 г, или даже превышающей, приблизительно, 2,5 г. Согласно определенным вариантам осуществления изобретения, солью IPM является IPM•Tris, которую вводят в дозе вплоть до 2,0 г, 2,5 г или даже 3,0 г.
Согласно определенным вариантам осуществления изобретения, IPM или его аналог вводят субъекту в форме соли, таким образом, что доза (например, ежедневная доза) IPM или его аналога (то есть, если учитывать только IPM анион в составе соли и не учитывать вес противоиона или других компонентов композиции) составляет, приблизительно, более 0,4 г, приблизительно, более 0,6 г, приблизительно, более 0,8 г, или даже, приблизительно, более 1,0 г. Согласно определенным вариантам осуществления изобретения, IPM вводят в составе заявленной композиции в дозе, которая составляет, приблизительно, более 0,4 г, приблизительно, более 0,6 г, приблизительно, более 0,8 г или даже, приблизительно, более 1,0 г, например, вплоть до 2,0 г, 2,5 г или даже 3,0 г.
Согласно определенным вариантам осуществления изобретения, курс соли IPM или его аналога может включать общее количество IPM или его аналога (то есть, если учитывать только IPM анион в составе соли и не учитывать вес противоиона или других компонентов композиции), которое составляет, приблизительно, более 0,8 г, приблизительно, более 1,0 г, приблизительно, более 1,5, или даже, приблизительно, более 2,0 г.
Согласно определенным вариантам осуществления изобретения, доза соли IPM или его аналога может вводиться один раз в неделю, три раза в неделю, пять раз в неделю, один раз в день или даже два раза в день, предпочтительно, один раз в день. Согласно данным вариантам осуществления изобретения, курс введения соли IPM или его аналога включает введение двух или более последовательных доз. Согласно определенным вариантам осуществления изобретения, курс лечения включает введение дозы соли IPM или его аналога один раз в день в течение двух, трех, четырех или даже пяти дней, предпочтительно, трех дней. Такие дозы могут вводиться последовательно, т.е каждый день или непоследовательно.
Согласно определенным вариантам осуществления изобретения, единичная доза (например, ежедневная доза) может входить в состав более чем одной лекарственной формы, то есть единичная доза может содержаться в двух и более капсулах, таблетках или пилюлях. Согласно определенным вариантам осуществления изобретения, ежедневная доза в составе нескольких лекарственных форм может вводиться однократно, или путем последовательного приема нескольких лекарственных форм с определенными интервалами в течение одного дня.
Согласно другому варианту осуществления заявленного способа, субъект получает дозу (например, ежедневную дозу), которая составляет, приблизительно, от 1 до, приблизительно, 1500 мг/м2, например, приблизительно, от 1 до 700 мг/м2, приблизительно, от 5 до 1000 мг/м2, приблизительно, от 5 до 700 мг/м2, приблизительно, от 5 до 500 мг/м2, приблизительно, от 600 до 1200 мг/м2, приблизительно, от 100 до 1500 мг/м2, приблизительно, от 30 до 600 мг/м2, приблизительно, от 10 до 600 мг/м2, или, приблизительно, от 10 до 100 мг/м2 соли IPM или его аналога, заявленного в соответствии с настоящим изобретением. Например, доза составляет, приблизительно, 10 мг/м2, приблизительно, 12, или даже 14 мг/м2.
Согласно определенным вариантам осуществления способа лечения гиперпролиферативных заболеваний, заявленного в настоящем изобретении, заявленное соединение вводят субъекту в режиме, включающем ежедневный прием нескольких доз. Согласно данным вариантам осуществления изобретения, соединение вводят, по крайней мере, два дня подряд в течение пяти различных дней. Согласно одному варианту такого режима дозирования, соединение вводят субъекту последовательно, например, в течение двух-пяти последовательных дней. Альтернативно, соединение вводят субъекту непоследовательно, например, через день.
Согласно определенным вариантам осуществления заявленного способа, субъекту дополнительно с заявленными соединениями и композициями вводят один или более дополнительный лекарственный агент. Например, к дополнительным лекарственным агентам, которые могут вводиться совместно с заявленными соединениями, относятся агенты, связывающие микротрубочки, интерколяторы ДНК или перекрестно-связывающие ДНК соединения, ингибиторы синтеза ДНК, ингибиторы транскрипции ДНК и/или РНК, антитела, ферменты, ингибиторы ферментов, генные регуляторы и/или ингибиторы ангиогенеза.
К агентам, связывающим микротрубочки, относятся агенты, которые взаимодействуют с тубулином и таким образом стабилизируют или дестабилизируют формирование микротрубочек, что приводит к нарушению деления клеток. Примерами агентов, связывающих микротрубочки, которые могут использоваться в комбинации с IPM или его аналогом, или его солью, являются, без ограничений указанными, паклитаксель, доцетаксель, винбластин, виндезин, винорелбин (навелбин), эпотилоны, колхицин, доластатин 15, нокодазол, подофиллотоксин и ризоцин. Также могут применяться аналоги и производные указанных соединений, которые хорошо известны специалистам. Например, подходящие эпотилоны и аналоги эпотилона, которые могут вводиться совместно с заявленными соединениями, описаны в заявке на международный патент WO No. 2004/018478, которая включена в настоящее описание в качестве ссылки. В настоящее время считается, что таксоиды, такие, как паклитаксель и доцетаксель, являются особенно полезными лекарственными агентами для введения совместно с заявленными соединениями. Примеры дополнительных подходящих таксоидов, включая аналоги паклитакселя, раскрываются в патентах США номер 6,610,860 Holton, 5,530,020 Gurram et al., и 5,912,264 Wittman et al., которые включены в настоящее описание в качестве ссылок.
Подходящими регуляторами транскрипции ДНК и/или РНК являются, без ограничений указанными, актиномицин D, даунорубицин, доксорубицин, а также аналоги и производные указанных соединений, которые подходят для совместного введения с заявленными соединениями.
К интеркаляторам и агентам, перекрестно связывающим ДНК, которые могут использоваться совместно с заявленными соединениями, являются, без ограничений указанными, цисплатин, карбоплатин, оксалиплатин, митомицины, такие, как митомицин С, блеомицин, хлорамбуцил, циклофосфамид и их аналоги и производные.
К ингибиторам синтеза ДНК, которые являются подходящими лекарственными агентами, относятся, без ограничений указанными, метотрексат, 5-фтор-5'-деоксиуридин, 5-фторурацил и их аналоги.
Примерами подходящих ингибиторов ферментов, которые могут использоваться в комбинации с заявленными соединениями, относятся, без ограничений указанными, камптотецин, этопозид, форместан, трихостатин и их аналоги и производные.
Подходящими лекарственными агентами, которые могут использоваться совместно с заявленными соединениями и влияют на регуляцию генов, являются агенты, которые приводят к увеличению или уменьшению экспрессии одного или более генов, к ним относятся, без ограничений указанными, ралоксифен, 5-азацитидин, 5-аза-2'-деоксицитидин, тамоксифен, 4-гидрокситамоксифен, мифепристон и их производные и аналоги.
Ингибиторы ангиогенеза известны из области техники, примерами подходящих ингибиторов ангиогенеза являются, без ограничений указанными, ангиостатин К1-3, стауроспорин, генистеин, фумагилин, медроксипрогестерон, сурамин, интерферон-альфа, ингибиторы металлопротеиназ, фактор тромбоцитов 4, соматостатин, тромбоспондин, эндостатин, талидомид, а также их производные и аналоги.
Другие лекарственные средства, в особенности противоопухолевые агенты, которые не попадают или попадают под одну или несколько приведенных выше классификаций, также подходят для введения в комбинации с заявленными соединениями. Например, к таким агентам относятся адриамицин, апигенин, рапамицин, зебуларин, циметидин, а также их производные и аналоги.
Согласно определенным вариантам осуществления изобретения, соль IPM или его аналога вводят в комбинации с регулятором транскрипции ДНК и/или РНК, таким как доксорубицин. Согласно альтернативным вариантам осуществления изобретения, соль IPM или его аналога вводят в комбинации с агентом, связывающим микротрубочки, таким как доцетаксель или паклитаксель.
Согласно определенным вариантам осуществления изобретения, описанные комбинации могут быть синергичными по свой природе, что означает, что достигаемый терапевтический эффект от введения комбинации соли IPM или его аналога и другого лекарственного агента (или агентов) превышает сумму индивидуальных эффектов двух и более агентов, если их вводят отдельно в тех же количествах.
Согласно определенным вариантам осуществления изобретения, такой синергизм действия позволяет вводить субтерапевтические дозы соли IPM или его аналога. Согласно определенным вариантам осуществления изобретения, введение субтерапевтической дозы позволяет уменьшить или избежать нежелательных побочных эффектов, ассоциированных с введением высоких дозировок соли IPM или его аналога, например, способы, заявленные в соответствии с настоящим изобретением. имеют преимущества перед существующими схемами комбинированной противоопухолевой терапии, поскольку позволяют достичь более выраженного эффекта при использовании низких дозировок.
Согласно определенным вариантам осуществления изобретения, эффективность дополнительного агента улучшается, когда он вводится в комбинации с солью IPM или его аналога. Согласно данным вариантам осуществления изобретения, дополнительный лекарственный агент является химиотерапевтическим агентом, включая, без ограничений указанными, агенты, связывающие микротрубочки, интеркаляторы или кросс-линкеры ДНК, ингибиторы синтеза ДНК, ингибиторы транскрипции ДНК и/или РНК, антитела, ферменты, ингибиторы ферментов, генные регуляторы, и/или ингибиторы ангиогенеза. Согласно данным вариантам осуществления изобретения, эффективность агента, связывающего микротрубочки, такого как доцетаксель или паклитаксель, улучшается при введении в комбинации с солью IPM или его аналога. Согласно альтернативным вариантам осуществления изобретения, эффективность ингибитора транскрипции ДНК и/или РНК, такого как доксорубицин, улучшается при введении в комбинации с солью IPM или его аналога. Согласно определенным вариантам осуществления изобретения, солью IPM или его аналога является IPM•Tris.
Используемый здесь термин "субтерапевтическая доза" относится к дозе, которая может стабилизировать или уменьшать объем опухоли, но при этом не считается эффективной терапевтической дозой, или даже к дозе, которая сама по себе не оказывает какого-либо измеримого терапевтического эффекта.
Согласно определенным вариантам осуществления изобретения, соль IPM или его аналога, вводится в дозе, приблизительно, от 100 до, приблизительно, 500 мг, приблизительно, от 150 мг до, приблизительно, 400 мг, или даже приблизительно, от 175 мг до, приблизительно, 300 мг. Согласно определенным вариантам осуществления изобретения, соль IPM или его аналога вводится в дозе, приблизительно, 150 мг, приблизительно, 175 мг, приблизительно, 185 мг, приблизительно, 190 мг, приблизительно, 200 мг, приблизительно, 225 мг, приблизительно, 250 мг, приблизительно, 275 мг, приблизительно, 285 мг, приблизительно, 290 мг, или даже приблизительно, 300 мг. Согласно данным вариантам осуществления изобретения, соль IPM или его аналога в таких дозах вводится перорально.
Согласно определенным вариантам осуществления изобретения, соль IPM или его аналога вводится в комбинации с доксорубицином в дозе, приблизительно, от 100 мг до, приблизительно, 200 мг, приблизительно, 110 мг до, приблизительно, 180 мг, или даже, приблизительно, от 115 мг до, приблизительно, 150 мг. Согласно определенным вариантам осуществления изобретения, соль IPM или его аналога вводится в комбинации с доксорубицином в дозе, приблизительно, 100 мг, приблизительно, 110 мг, приблизительно, 115 мг, приблизительно, 125 мг, приблизительно, 135 мг, приблизительно, 140 мг, приблизительно, 145 мг, приблизительно, 155 мг, приблизительно, 165 мг, приблизительно, 175 мг, приблизительно, 185 мг, или даже приблизительно 200 мг.
Согласно определенным вариантам осуществления изобретения, соль IPM или его аналога вводится в комбинации с доцетакселем в дозе, приблизительно, от 50 мг до, приблизительно, 200 мг, приблизительно, от 75 мг до, приблизительно, 195 мг, или даже, приблизительно, от 80 мг до, приблизительно, 190 мг. Согласно определенным вариантам осуществления изобретения, соль IPM или его аналога вводится в комбинации с доцетакселем в дозе, приблизительно, 50 мг, приблизительно, 70 мг, приблизительно, 80 мг, приблизительно, 90 мг, приблизительно, 95 мг, приблизительно, 100 мг, приблизительно, 110 мг, приблизительно, 120 мг, приблизительно, 130 мг, приблизительно, 140 мг, приблизительно, 150 мг, приблизительно 160 мг, приблизительно, 170 мг, приблизительно, 180 мг, приблизительно, 190 мг или даже, приблизительно, 200 мг.
III. Определения
Следующие объяснения терминов и примеры приведены здесь для лучшего описания заявленных соединений, композиций и способов и предназначены для того, чтобы помочь специалисту в данной области практически осуществить изобретение. Также должно быть понятно, что используемая в описании терминология относится к определенным вариантам осуществления изобретения и примерам и не ограничивается этими спецификациями.
Интервалы и пределы в настоящем описании выражаются от одной "приблизительной" величины и/или до другой "приблизительной" величины. В том случае, когда представлен такой интервал, другой вариант осуществления изобретения включает интервал от одной определенной величины и/или до другой определенной величины. Аналогично, когда величины выражены через приближение, с использованием антецедента "приблизительно", должно быть понятно, что определенные величины относятся к другому варианту осуществления изобретения. Также должно быть понятно, что конечные значения каждого интервала являются существенными по отношению к другому конечному значению, и не зависят от другого конечного значения.
Термин "ациклический алифатический амин" относится к алифатическому амину, как описано выше, при этом, по крайней мере, одна из алифатических групп является ацикличной.
Используемый здесь термин "алифатический амин" относится к соединению, имеющему формулу NR1R2R3, при этом, по крайней мере, один R1-3является алифатической группой.
Термин "ингибитор ангиогенеза", используемый в настоящем описании, относится к молекуле, включая, без ограничений указанными, биологические молекулы, такие как пептиды, белки, ферменты, полисахариды, олигонуклеотиды, ДНК, РНК, рекомбинантные векторы, и малые молекулы, которые ингибируют рост кровеносных сосудов. Ангиогенез вовлечен в некоторые патологические процессы, такие, которые участвуют в патогенезе таких заболеваний, как диабетическая ретинопатия, хронические воспалительные заболевания, ревматоидный артрит, дерматит, псориаз, язвы желудка и большинство типов твердых опухолей человека.
Термин "гетероциклический амин" относится к соединению, имеющему формулу NR1R2R3, при этом, по крайней мере, один R1-3, является гетероцикличной группой или R1, R2 и/или R3 вместе посредством их общего атома азота образуют кольцо.
Термин "уходящая группа" относится к группе, которая может быть замещена нуклеофилом. Относительно заявленных соединений, термин "уходящая группа" относится к группе, которая может быть замещена с образованием промежуточного соединения азиридиния, или может быть непосредственно замещена биомолекулой нуклеофила, такой как нуклеиновая кислота, с образованием, например, 7-алкилированного гуанидинового соединения. Примерами походящих уходящих групп являются галогены и сульфонаты (-SO2R). Согласно одному варианту воплощения заявленных солей аналогов изофосфорамида, соединение представляет собой соединение "смешанных" уходящих групп, включая две различные уходящие группы, например, галоген и сульфонат, или два различных галогена, например, бром и хлор. В патенте США номер 6,197,760 Struck раскрывается способ получения таких соединений с разными уходящими группами.
Термин "неоплазия" обозначает процесс аномального или неконтролируемого клеточного роста. Неоплазия является одним из примеров пролиферативных заболеваний. Продуктом неоплазии является неоплазм (опухоль), которая представляет собой аномальный рост ткани, который является следствием избыточного деления клеток. Опухоль, которая не обладает способностью к метастазированию, обозначается термином "доброкачественная". Опухоль, которая прорастает в соседние ткани и/или обладает способностью к метастазированию обозначается как "злокачественная".
Термин "необязательный" или "необязательно" означает, что описанное далее явление или условие может происходить, но не является необходимым, при этом описание включает частные случаи, когда указанное явление или условие происходит, и когда оно не происходит.
Используемый здесь термин "стабильный" означает, что соединение распадается не более чем на 5%, предпочтительно, не более чем на 2% или даже не более чем на 1% в течение пяти дней, или в течение обозначенного периода времени. Такой распад может контролироваться при помощи1H ЯМР, HPLC или других подходящих способов.
Используемый здесь термин "лечение", как хорошо понятно из области техники, означает подход для достижения благоприятных или желаемых результатов, включая клинические результаты. К благоприятным или желаемым клиническим результатам относятся, без ограничений указанными, уменьшение или исчезновение одного или более симптомов или патологических состояний, уменьшение продолжительности болезни, стабилизация (то есть, отсутствие ухудшений) состояния больного, предотвращение распространения заболевания, отсрочка или замедление прогрессирования заболевания, улучшение или временное облегчение состояния больного, и ремиссия (как частичная, так и полная), включая как регистрируемые, так и нерегистрируемые результаты. Термин "лечение" также может означать пролонгирование продолжительности жизни по сравнению с ожидаемой продолжительностью при отсутствии лечения.
IV. Примеры
Данное изобретение также раскрывается ниже с помощью примеров, которые не ограничивают объем изобретения.
Пример 1
В реактор загружают сначала Tris (103,3 мг) и MeCN (3 мл), затем IPM (200,5 мг) в MeCN (3 мл). Реакционную смесь перемешивают в течение ночи. Затем собирают твердые частицы при помощи фильтрации, и отжатый осадок отмывают MeCN. Отжатый осадок высушивают под действием вакуума до постоянного веса с получением конечного продукта (296 мг). Конечный продукт подвергают рентгеновской порошковой дифрактометрии для подтверждения кристаллической структуры (Рисунок 2). Кристаллическую структуру также подтверждают при помощи DSC, при которой острый пик появляется при 105,77 (Рисунок 3). Кроме того, отмечается потеря веса на 0,7692% соли IPM•Tris приблизительно при 125° при TGA (Рисунок 4). Наконец, SEM показала, что IPM•Tris имеет пластинчатую кристаллическую структуру.
Стабильность кристаллической соли IPM•Tris контролировали при помощи1H ЯМР, было показано, что соль остается стабильной при комнатной температуре вплоть до шести дней. Стабильность кристаллической структуры контролировали при помощи DSC, которая показала, что кристаллы IPM•Tris не абсорбируют воду и не изменяют своей структуры в течение десяти дней при комнатной температуре.
Пример 2
В реактор загружают Tris (8,563 г) и DMF (40 мл), после чего нагревают до образования чистого раствора. После того, как раствор охладится до комнатной температуры, добавляют IPM. Смесь перемешивают при комнатной температуре до образования чистого раствора. Затем к раствору добавляют ацетонитрил (40 мл) и небольшое количество ядер кристаллизации, после чего медленно добавляют МТВЕ (240 мл) с образованием жидкой массы. Полученную массу перемешивают в течение еще одного часа, после чего преципитат собирают путем фильтрации, а отжатый осадок отмывают МТВЕ (80 мл). Отжатый осадок высушивают под действием вакуума до постоянного веса при комнатной температуре с получением конечного продукта (23,2 г).
Пример 3
Фрагменты опухоли молочной железы МХ-1 человека весом 30-40 мг после in vivo пассажа имплантируют подкожно в жировое тело молочных желез nu/nu мышам и позволяют им вырасти до размера 75-200 мг перед началом лечения. Лечение начинают на 10 день после имплантации опухоли; лекарственный препарат вводят интраперитонеально один раз в день ежедневно в течение 5 дней. IMP•(LYS)2 (43% IPM и 57% LYS) и IPM•Tris демонстрируют одинаковую активность в отношении МХ-1 опухолей, когда дозы стандартизированы по отношению к IPM (см. Рисунок 6).
Пример 4
Фрагменты опухоли молочной железы МХ-1 человека весом 30-40 мг после in vivo пассажа имплантируют подкожно в жировое тело молочных желез nu/nu мышам и позволяют им вырасти до размера 75-200 мг перед началом лечения. Лечение начинают на 10 день после имплантации опухоли; лекарственный препарат вводят интраперитонеально или перорально один раз в день ежедневно в течение 5 дней. IPM•Tris, который может быть синтезирован как описано в Примере 1, в максимально толерантных дозах для каждого введения, был одинаково активен в отношении МХ-1 опухолей как при пероральном, так и при системном введении (см. Рисунок 7).
Пример 5
Исследование пролиферации клеток
Ингибирование роста определяют микрокультуральным тетразолиевым методом. Кратко, клетки помещают в 96-луночные планшеты для микротитрования с плоским дном в количестве 500 клеток на лунку в 100 мкл среды. Планшеты инкубируют в течение ночи, после чего добавляют 100 мкл среды, содержащей IPM•(LYS)2 до достижения специфической конечной концентрации и конечного объема 200 мкл на лунку. Представленные данные отражают среднее значение жизнеспособности и величину ошибки (стандартное отклонение для каждого эксперимента, осуществленного трижды). Через 120 часов измеряют относительную метаболическую активность обработанных и необработанных клеток, регистрируя митохондриальную конверсию 3-[4,5-диметилтиазон-2-ил]-2,5-дифенилтетразолиум бромида (МТТ, Sigma, St. Louis, МО) в формазин. По окончании лекарственной обработки, в каждую лунку добавляют 250 мкг МТТ и инкубируют при 37°С, 5% СО2 в течение 6 часов. Кристаллы формазина растворяют в DMSO и измеряют оптическую плотность при 595 нм на спектрофотометре VERSAmax (Molecular Devices, Sunnyvale, CA). Жизнеспособность определяют как поглощение при 595 нм в обработанных образцах, деленное на поглощение при 570 нм в контрольных образцах. IC50определяют как концентрацию, при которой жизнеспособность обработанных клеток составляет 50% от величины контрольных клеток (обработанные клетки/контрольные клетки = 0,5).
Мышиные ксенотрансплантантные модели
Мышам женского пола с тяжелым комбинированным иммунодефицитом СВ17 (scid*/* мыши) (Taconic Farms, Germantown, NY) подкожно в область паха имплантируют опухоль. Опухолевая клеточная линия OS31 получена из госпиталя St. Judes по исследованию рака у детей (St. Judes Children's Cancer Research Hospital) и была описана выше (20). Перед трансплантацией OS1 опухоли мышей анестезируют 4% изофлураном. Затем в паховой области производят небольшой разрез и подкожно имплантируют фрагмент опухоли размером 4 мм на 4 мм.
Scid */* мышам женского пола СВ17 (Taconic Farms, Germantown, NY) в хвостовую вену вводят IPM•(LYS)2 ежедневно в течение 1 или 3 дней, начиная с 1 дня, лечение включает 2 цикла с интервалом 21 день. В каждую группу, получающую лечение, включают пять мышей, не имеющих опухоли. Мышам вводят IPM•(LYS)2 в дозах 75 мг/кг в день, 100 мг/кг в день, 150 мг/кг в день или 200 мг/кг в день. Токсический эффект определяют как потерю массы тела более чем или на 20% от исходной массы тела животного на момент выборки, или гибель животного. MTD определяют как наибольшую дозу, при которой не наблюдается токсического эффекта.
Когда опухоли достигают размера приблизительно 0,20-0,7 см в диаметре, мышей, имеющих опухоли, случайным образом разделяют на группы по 5-8 мышей: 1 лечебная группа и 1 контрольная группа. Мыши из лечебной группы получают интраперитонеальную инъекцию тазидотина в дозе 90 мг/кг в день ежедневно в течение 5 дней, начиная на 1 и 21 день. Допуская сферическую форму опухоли, ее объем рассчитывают по формуле: мм3=*/6(D)d2, где D - максимальный диаметр, a d - диаметр, перпендикулярный D. Объем опухоли выражают в относительных опухолевых единицах (RTV, от англ. Relative Tumor Volume), при этом объем опухоли в любой заданной временной точке делится на исходный объем опухоли. RTV мышам в лечебной и контрольной группах измеряют минимум один раз в неделю.
Оценка ответа опухоли на лечение и статистический анализ у мышей
Согласно критериям, предложенным Houghton et al., прогрессирующее заболевание определяется при регрессии исходного объема опухоли менее, чем на 50% в течение всей продолжительности исследования (RTV>0,5), и при увеличении объема опухоли на конец исследования более чем на 25% (RTV>1,25). Стабильная регрессия опухолевого заболевания определяется, когда объем опухоли в течение всего исследования не превышает 50% от исходного объема опухоли (RTV>0,5) и на момент окончания исследования объем опухоли не увеличивается более чем на 25% (RTV<1,25). Частичный ответ определяется как более чем 50% регрессия объема опухоли (RTV<0,5) но с измеряемой массой опухоли более чем 0,10 см3. Потеря измеримой массы опухоли (<0,10 см3) в любой временной точке в течение исследования (6 недель) определяется как полный ответ (CR, от англ. Complete Response). Устойчивый CR определяется как потеря измеримой массы опухоли (<0,10 см3) в любой временной точке после начала лечения без возобновления роста в течение 6-недельного периода исследования. Мыши, умершие до 9 недели исследования или до того, как объем опухоли увеличится в 4 раза по сравнению с исходным, исключаются.
Статистический анализ основан на безсобытийной выживаемости (EFS, от англ. Free-Event Survival). Событием считается относительный объем опухоли 4х (то есть, объем в четыре раза превышающий исходный) или гибель. EFS определяется как период времени от начала исследования до события. Для тех опухолей, которые, не достигнут события в течение 6 недель, то есть к моменту окончания исследования, время EFS исключается из этого периода. Для сравнения распределения безсобытийной выживаемости между контрольными и лечебными группами используется точный логранговый критерий. Кроме того, сравнивают RTVs на 22 день в контрольной и лечебной группах с использованием критерия Уилкоксона-Манна-Уитни. Он позволяет проводить сравнение опухолевого объема после одного цикла лечения тазидотином и на момент события или рядом с ним для мышей из контрольной группы.
Биологические данные
Используемые опухолевые клеточные линии включают клетки рабдомиоскаркомы линий RD и RH30 (American Type Culture Collection, Manassas, VA), клеточную линию остеосаркомы Saos-2, клетки саркомы Эвинга линий SKPNDW и SKES1, и клетки синовиальной саркомы линий HSSYII и SYOI. Клетки выращивают в монослое при 37°С, 5% CO2 в среде MEM (Saos-2, SYO-1, HSSY-II), DME (SK-PN-DW, RD) или RPMI (RH30), обогащенной 10% фетальной бычьей сывороткой (Invitrogen, Carlsbad, CA), 0,5% раствором пенициллин/стрептомицин (Invitrogen, Carlsbad, CA) и 1% глутамином (Invitrogen, Carlsbad, CA). Результаты представлены ниже (Рисунки 8 и 9).
Устойчивость опухолей к циклофосфамиду (СРА) и ифосфамиду (IFOS) является основным препятствием в лечении рака. У мышей с ксенотрансплантатами клеток СРА-устойчивой остеосаркомы человека отмечается более чем 5-кратное уменьшение роста саркомы при лечении IPM•(LYS)2; лечение СРА не имело эффекта. (Рисунки 10-12).
Клинические исследования на людях
В исследованиях безопасности препарата и исследованиях с целью определения оптимальной дозы I фазы использовался IPM•(LYS)2, который вводили ежедневно в течение трех последовательных дней каждые четыре недели (1 цикл). Результаты показали очевидную клиническую активность в отношении саркомы (2/11 субъекта, включая, по крайней мере, одного, у кого лечение IFOS было неэффективным) и мезотелиомы (1 субъект с продолжительной стабилизацией процесса). Максимальная толерантная доза (MTD) IPM•(LYS)2 при таком режиме составляла 400 мг/м2 в день. Был отмечен незначительный токсический эффект в отношении костного мозга, случаев геморрагического цистита (токсическое действие на мочевой пузырь) и токсического действия на ЦНС не зарегистрировано. Дозолимитирующая токсичность определялась дисбалансом электролитов. Такая MTD сопоставима с дозами для IFOS, более 25 г/м2, указанная доза обеспечивает уровень препарата в сыворотке в 25 раз больший, чем при введении доз, убивающих 50% клеток саркомы человека.
Пример 6
Материалы и Методы
Животные: Пяти-шестинедельных мышей мужского пола CD2F1 получают в Фредерикском Научно-исследовательском центре по изучению рака (Frederick, MD).
Опухолевая модель: Мышам интраперитонеально при помощи иглы 23 калибра имплантируют один миллион клеток лейкемии мышей Р388. Опухолевую клеточную линию Р388 поддерживают при помощи in vivo пассажа. День имплантации опухоли обозначают как 0 день, лечение начинают на 1 день после имплантации. Для исследования отбиралось достаточное количество животных, таким образом, чтобы разброс их массы тела был как можно более узким.
Технология приготовления лекарственного средства: IPM•(LYS)2, полученный во флаконах с предварительно отвешенной дозой в 100 мг, разводят солевым раствором на первый день лечения (день 1) в концентрации 70 мг/мл. Затем часть полученного раствора дополнительно разводят до более низких концентраций 46,65, 23,35, 14, 9,35, 4,65 и 2,8 мг/мл. В последующие дни лечения изготавливают раствор IPM•(LYS)2 с концентрацией 14 мг/мл и часть этого раствора дополнительно разводят до более низких концентраций. Все инъекции осуществляют с учетом точно измеренной массы тела, вводимый объем составляет 0,2 мл на 10 г массы тела.
Лечение: Исследование включает восемь лечебных групп по восемь мышей в каждой группе и две контрольные группы, получающие плацебо, по десять животных в группе, итого 84 животных на первый день лечения. На 1 день однократно вводят (q1d×1) в дозировках 1400, 933 и 467 мг/кг перорально и в дозировке 280 мг/кг интраперитонеально. Также IPM•(LYS)2вводят ежедневно в течение пяти последовательных дней (q1d×5) в дозировках 280, 187 и 93 мг/кг перорально и в дозировке 56 мг/кг интраперитонеально. Одна контрольная группа получала солевой раствор перорально на 1 день. Вторая контрольная группа получала солевой раствор перорально в режиме q1d×5.
Продолжительность исследования: Исследование продолжалось 61 день после трансплантации опухоли. Каждое животное в состоянии агонии подвергали эвтаназии до окончания исследования.
Анализируемые параметры: количество случаев неспецифической гибели, средний день смерти, и увеличение продолжительности жизни на основании среднего дня смерти и выраженное в процентах (%ILS, от англ. Increase of Lifespan); среднее время выживания и %ILS, рассчитанное на основе среднего времени выживания.
Статистический анализ: Индивидуальное время выживания животных используют в качестве конечной точки для построения таблиц продолжительности жизни (стратифицированный анализ Каплана-Мейра с последующим логранговым тестом Мантель-Хэнзеля) для статистического сравнения данных выживаемости среди групп. Анализ при помощи таблиц продолжительности жизни позволяет сравнить данные выживаемости между группами с учетом животных, которые не достигли конечной точки по причине их исключения.
Результаты
Средний день смерти животных в обеих контрольных группах, получавших плацебо, 11 день, смерть наступала между 10 и 14 днем. Асцит был выявлен у всех животных.
IPM•(LYS)2 при однократном пероральном введении на 1 день оказывал токсическое действие на мышей в дозировках 1400 и 933 мг/кг. В группе, получавшей препарат в дозе 1400 мг/кг, одно животное погибло и четыре животных были умерщвлены по причине агонии на 5 день, два животных были умерщвлены на 6 день, последнее животное погибло на 10 день. В группе, получавшей лечение препаратом в дозировке 933 мг/мг, шесть животных были умерщвлены на 5 день по причине агонии, оставшиеся два животных в группе погибли на 9 день. Аутопсия показала отсутствие опухоли у животных в обеих группах. Перед гибелью или эвтаназией животных была отмечена существенная потеря массы тела на 24% и 22% у животных, получавших IPM•(LYS)2 в дозировках 1400 и 933 мг/кг, соответственно. IPM•(LYS)2IPM•(LYS)2, вводимый однократно перорально в дозе 467 мг/кг переносился лучше; однако, двое животных были подвергнуты эвтаназии на 8 день по причине агонального состояния. Максимальная потеря массы тела в этой группе составила 8%. Оставшиеся шесть животных в группе погибли между 11 и 15 днем, при аутопсии был обнаружен асцит. ILS для этой лечебной группы составила 9%, при этом расчет основан на среднем дне смерти или среднем времени выживания. Статистическое сравнение данных выживаемости для данной группы и контрольной плацебо-группы (группа 1, получавшая q1d×1) не выявило существенных различий.
IPM•(LYS)2 при однократном интраперитонеальном введении в дозе 280 мг/кг хорошо переносился с минимальной потерей массы тела (4%, 1 г) и без летальных исходов. Средний день смерти в данной группе - 34,5 с ILS 214%. Среднее время выживания составило 38,0 дней с ILS 245%. Кроме того, двое животных остались живы на момент окончания исследования, то есть на 61 день. Аутопсия показала отсутствие опухоли. Статистический анализ данных выживаемости для данной группы и контрольной группы, получавшей плацебо (группа 1, q1d×1) показал существенное различие.
IPM•(LYS)2 при введении в дозировках 280, 187 и 93 мг/кг в режиме q1d×5 переносился хорошо, без обусловленной лечением гибели животных. Минимальная потеря средней массы тела (4%, 1 г) наблюдалась в группе, получавшей IPM•(LYS)2 в дозе 280 мг/кг. В двух других группах, получавших меньшие дозы препарата, потери массы тела отмечено не было. Средний день смерти и среднее время выживания составили 17,0, 17,0 и 15,0 день с показателем ILS 55%, 55% и 36% для дозировок 280, 187 и 93 мг/кг, соответственно. Статистическое сравнение данных выживаемости для каждой из указанных трех групп с данными выживаемости в контрольной группе, получавшей плацебо (группа 6, получавшая q1d×5), показало существенное увеличение продолжительности жизни в каждой лечебной группе.
IPM•(LYS)2 при введении в дозе 56 мг/кг в режиме q1d×5 оказался достаточно эффективным в отношении лейкоза Р388/0, средний день смерти - 28,5. Первая смерть животного произошла на 24 день, а последняя - на 33 день, четверо из восьми животных в группе дожили до окончания исследования, то есть до 61 дня. Показатель ILS, рассчитанный на основании среднего дня смерти составил 159%. Среднее время выживания для этой лечебной группы составило >47, 0 дней с рассчитанным показателем ILS 327%. Этот лечебный режим хорошо переносился, без обусловленной лечением гибели животных и со средней потерей массы тела 4% (1 г). При сравнении данных выживаемости в этой группе с данными в контрольной группе (группа 6, q1d×5) было выявлено существенное различие.
Краткие данные статистического анализа
Выводы
IPM•(LYS)2 оказывал токсическое действие на мышей при однократном пероральном введении в дозировках 1400 и 933 мг/кг. Однократное пероральное введение препарата в дозировке 467 мг/кг хорошо переносилось животными, но незначительно увеличивало продолжительность жизни; увеличение не было статистически значимым. IPM•(LYS)2 при однократном интраперитонеальном введении в дозе 280 мг/кг оказался относительно эффективным и приводил к существенному увеличению продолжительности жизни: 2 случая выживания до 61 дня исследования.
IPM•(LYS)2 при пероральном введении в режиме 1qd×5 в дозировках 280, 187 и 93 переносился значительно лучше и был более эффективным в отношении лейкоза Р388/0, чем при однократном введении в более высоких дозах. Лечение всеми тремя дозами препарата перорально в режиме q1d×5 приводило к существенному увеличению продолжительности жизни. Введение препарата интраперитонеально в режиме q1d×5 в дозе 56 мг/кг способствовало существенному увеличению продолжительности жизни, четверо из восьми животных дожили до окончания исследования, то есть до 61 дня.
Пример 7
Материалы и Методы
Животные: Пятинедельных бестимусных мышей женского пола NCr-nu/nu получают в лаборатории Taconic Farms (Germantown, NY).
Опухолевая модель: Фрагменты опухоли груди человека МХ-1 весом 30-40 мг, поддерживаемые in vivo пассажем, имплантируют мышам подкожно в жировое тело молочной железы при помощи троакарной иглы 12 калибра и оставляют расти. День имплантации опухоли обозначают как 0 день. До начала лечения опухолям позволяют вырасти до размера 138-245 мг (138-245 мм3). Для исследования отбирают достаточное количество животных, таким образом, чтобы разброс размера опухоли на момент начала лечения (10 день после имплантации опухоли) был как можно меньше. Выбранных животных с нужным размером опухоли распределяют в различные лечебные группы, таким образом, чтобы средний размер опухоли на первый день лечения у животных как можно меньше различался (172-197 мг).
Технология приготовления лекарственного средства: IPM•Tris (205 мг IPM на флакон), который может быть получен, как описано в Примере 1, и IPM разводят солевым раствором на первый день лечения (день 10) в концентрации 6,0 мг/мл и затем вновь разводят солевым раствором до более низких концентраций 4,05, 1,8 и 1,2 мг/мл. IPM•(LYS)2 (100 мг IPM•Лизин на флакон) разводят солевым раствором на первый день лечения в концентрации 14 мг/мл и затем вновь разводят солевым раствором до более низких концентраций 9,35, 4,2 и 2, 8 мг/мл. Каждую концентрацию затем разделяют на аликвоты для ежедневного использования, замораживают, хранят при -20°С и размораживают при необходимости. Все инъекции осуществляют с учетом точно измеренной массы тела, вводимый объем составляет 0,2 мл на 10 г массы тела.
Лечение: Исследование включает двенадцать лечебных групп по восемь мышей в каждой группе и две контрольные группы, получавшие плацебо, по десять мышей в каждой группе, итого 116 мышей на первый день исследования. Все лекарственные агенты (и плацебо) вводят ежедневно в течение пяти последовательных дней (q1d×5). Оба препарата, IPM•Tris и IPM, вводят интраперитонеально в дозировках 36 и 24 мг/кг и перорально в дозировках 120 и 81 мг/кг. IPM•(LYS)2 вводят интраперитонеально в дозировках 84 и 56 мг/кг и перорально в дозировках 280 и 187 мг/кг. Контрольные группы получают плацебо (солевой раствор), как перорально, так и интраперитонеально.
Измерение массы тела и размера опухоли: Размер подкожных опухолей и вес животных измеряют два раза в неделю, начиная с первого дня лечения. Объем опухоли определяют при помощи медицинского циркуля (мм), расчет производят по формуле для эллипсоидной сферы:
L×W2/2=мм2
где L и W обозначают наибольший и наименьший перпендикулярный размер, полученный при каждом измерении. Эта формула также используется для расчета веса опухоли, с допущением единиц плотности (1 мм3=1 мг).
Продолжительность исследования: Исследование продолжается в течение 50 дней после имплантации опухоли. Каждое животное в состоянии агонии и животное, у которого опухоль достигала размера 4,000 мг или начинала изъязвляться, подвергали эвтаназии до окончания исследования.
Анализируемые параметры: количество случаев неспецифической гибели, количество случаев частичной или полной регрессии опухоли, количество выживших без опухоли, кроме того, определяют индивидуальное время, за которое происходило удвоение объема опухоли, для каждого животного. Среднее время, за которое происходило удвоение объема опухоли, в лечебной группе (Т) и контрольной группе (С) использовали для расчета общей задержки роста средней опухоли (Т-С).
Статистический анализ: Индивидуальное время, за которое происходит удвоение объема опухоли, для каждого животного используют в качестве конечной точки для t-критерия Стьюдента /критерия суммы рангов Манна-Уитни или анализе таблиц продолжительности жизни для статистического сравнения данных роста между группами. Анализ таблиц продолжительности жизни (стратифицированный анализ Каплана-Мейра с последующим логранговым тестом Мантель-Хэнзеля) позволяет провести сравнение данных роста между группами с учетом животных, которые не достигли конечной точки по причине их исключения.
Результаты
Опухоли у животных из обеих контрольных групп, получавших плацебо, хорошо выросли у всех 10 животных. В среднем удвоение исходного объема опухоли происходило к 7,4 и 7,2 дню, соответственно, в группах, получавших препарат интраперитонеально и перорально. В указанных двух группах не было отмечено уменьшения средней массы тела во время исследования.
Интраперитонеальное введение IPM•Tris в дозировках 36 и 24 мг/кг приводило к задержке роста опухоли (Т-С) на 10,2 и 7,7 день, соответственно. При введении высокой дозы был отмечен один случай выживания животного с отсутствием опухоли. Обе дозировки переносились с максимальными потерями средней массы тела на 10% (2 г) и 0% для дозировок 36 мг/кг и 24 мг/кг, соответственно. Интраперитонеальное введение IPM в дозировках 36 и 24 мг/кг приводило к задержке роста опухоли (Т-С) на 5,2 и 2,6 дни, соответственно. Обе дозировки переносились с максимальными потерями средней массы тела на 0% и 5% (1 г) для дозировок 36 мг/кг и 24 мг/кг, соответственно. Время удвоения объема опухоли при введении IPM было статистически меньше, чем время при введении соответствующей дозы IPM•Tris (р=0,000 для дозы 36 мг/кг; р=0,021 для дозы 24 мг/кг). Интраперитонеальное введение IPM•(LYS)2 в дозировках 84 и 56 мг/кг, что соответствует IPM в дозах 36 и 24 мг/кг, приводило к задержке роста опухоли на 24,3 и 9,0 дни, соответственно. Более высокая доза IPM•(LYS)2оказалась токсичной - две мыши погибли на 20 день и две мыши были подвергнуты эвтаназии по причине агонии и чрезмерной потери массы тела. Более низкая доза IPM•(LYS)2 переносилась с максимальной потерей массы тела на 15% (3 г). Переносимая доза IPM•(LYS)2 сравнима по своей активности с таковой для IPM•Tris, составляющей 24 мг/кг (р=0,766) и обладает большей активностью, чем IPM в дозе 24 мг/кг (р=0,0047).
Пероральное введение IPM•Tris в дозировках 120 и 81 мг/кг приводило к задержке роста опухоли на 8,7 и 9,0 день, соответственно. Более высокая доза IPM•Tris вызывала максимальную потерю массы тела на 15% (3 г), одно животное пришлось умертвить, поскольку масса тела животного была менее 14 г. Низкая доза переносилась с максимальной потерей средней массы тела на 10% (2 г). Пероральное введение IPM в дозировках 120 и 81 мг/кг приводило к задержке роста опухоли на 4,6 и 4,0 дни, соответственно. Обе дозы переносились с максимальной потерей средней массы тела на 5% (1 г). Время удвоения объема опухоли статистически не отличалось от времени при введении соответствующей дозы IPM•Tris (р=0,1174 для дозы 120 мг/кг; р=0,1152 для дозы 81 мг/кг). Пероральное введение IPM•(LYS)2 в дозировках 280 и 187 мг/кг, что соответствует IPM в дозах 120 и 81 мг/кг, приводило к задержке роста опухоли на 5,0 и 3,5 дни, соответственно. Обе дозировки переносились с максимальными потерями средней массы тела на 5% (1 г) и 0%. Более высокая доза IPM•(LYS)2 сравнима по активности с соответствующей дозой IPM•Tris и IPM, составляющей 120 мг/кг (р=0,1000 и р=0,9143, соответственно). Более низкая доза IPM•(LYS)2 показала меньшую активность, по сравнению с соответствующей дозой IPM•Tris, составляющей 81 мг/кг (р=0,0290) и сопоставимую активность с соответствующей дозой IPM, составляющей 81 мг/кг (р=0,3073).
Краткие данные статистического анализа
Ответ ксенотрансплантантов рака груди человека МХ-1 на лечение (1) плацебо - интраперитонеально и перорально, (2) интраперитонеально IPM•Tris, IPM и IPM•(LYS)2, и (3) перорально IPM•Tris, IPM и IPM•(LYS)2 представлен на Рисунках 13, 14 и 15, соответственно.
Выводы
При интраперитональном введении эквивалентных IPM доз противоопухолевая активность IPM•Tris была выше, чем у IPM (в обеих дозировках) и сопоставима с активностью IPM•(LYS)2 (в обеих дозировках). При пероральном введении эквивалентных IPM доз, противоопухолевая активность IPM•Tris была сопоставима с активностью IPM (в обеих дозировках), сопоставима с IPM•(LYS)2 при его введении в более высоких дозах, и выше, чем активность IPM•(LYS)2 при использовании более низких дозировок.
Пример 8
Материалы и Методы
Животные: Пятинедельных бестимусных мышей женского пола NCr-nu/nu получают в лаборатории Harlan (Prattville, AL).
Опухолевая модель: Фрагменты опухоли груди человека МХ-1 весом 30-40 мг, поддерживаемые in vivo пассажем, имплантируют мышам подкожно в жировое тело молочной железы при помощи троакарной иглы 12 калибра и оставляют расти. День имплантации опухоли обозначают как 0 день. До начала лечения опухолям позволяют вырасти до размера 113-245 мг (138-245 мм3). Для исследования отбирают достаточное количество животных, таким образом, чтобы разброс размера опухоли на момент начала лечения (6 день после имплантации опухоли) был как можно меньше. Выбранных животных с нужным размером опухоли распределяют в различные лечебные группы, таким образом, чтобы средний размер опухоли на первый день лечения у животных как можно меньше различался (144-162 мг).
Технология приготовления лекарственного средства: IPM•Tris (205 мг IPM на флакон, Cardinal Health), который может быть получен, как описано в Примере 1, и IPM разводят солевым раствором в каждый день лечения в концентрации 13,5 мг/мл и затем вновь разводят солевым раствором до более низких концентраций 9, 6, 4,05, 2,7, 1,8 и 1,2 мг/мл. IPM (Eagle-Picher Pharmaceutical Service) разводят солевым раствором в каждый день лечения в концентрации 6,0 мг/мл и затем вновь разводят солевым раствором до более низких концентраций 4,05, 1,8 и 1,2 мг/мл. IPM•(LYS)2 (100 мг IPM•Лизин на флакон, University of Iowa) разводят солевым раствором в каждый день лечения до концентрации 14 мг/мл и затем вновь разводят солевым раствором в каждый до более низких концентраций 9,35, 4,2 и 2, 8 мг/мл. Все полученные растворы после приготовления рецептур хранят на льду и вводят в течение 30 минут после приготовления. Все инъекции осуществляют с учетом точно измеренной массы тела, вводимый объем составляет 0,2 мл на 10 г массы тела.
Лечение: Исследование включает шестнадцать лечебных групп по восемь мышей в каждой группе и две контрольные группы, получавшие плацебо, по десять мышей в каждой группе, итого 148 мышей на первый день исследования. Все лекарственные агенты (и плацебо) вводят ежедневно в течение пяти последовательных дней (q1d×5). IPM•Tris вводят интраперитонеально в дозировках 81, 54, 36 и 24 мг/кг и перорально в дозировках 270, 180,120 и 81 мг/кг. IPM вводят интраперитонеально в дозировках 36 и 24 мг/кг и перорально в дозировках 120 и 81 мг/кг. IPM•(LYS)2 вводят интраперитонеально в дозировках 84 и 56 мг/кг и перорально в дозировках 280 и 187 мг/кг. Контрольные группы получают плацебо (солевой раствор), как перорально (группа 10), так и интраперитонеально (группа 1).
Измерение массы тела и размера опухоли: Размер подкожных опухолей и вес животных измеряют два раза в неделю, начиная с первого дня лечения. Объем опухоли определяют при помощи медицинского циркуля (мм), расчет производят по формуле для эллипсоидной сферы:
L×W2/2=мм2
где L и W обозначают наибольший и наименьший перпендикулярный размер, полученный при каждом измерении. Эта формула также используется для расчета веса опухоли, учитывая единицы плотности (1 мм3=1 мг).
Продолжительность исследования: Исследование продолжается в течение 52 дней после имплантации опухоли. Каждое животное в состоянии агонии и животное, у которого опухоль достигала размера 4,000 мг или начинала изъязвляться, умерщвляли до окончания исследования.
Анализируемые параметры: количество случаев неспецифической гибели, количество случаев частичной или полной регрессии опухоли, количество выживших без опухоли, кроме того, определяют индивидуальное время, за которое происходило удвоение объема опухоли, для каждого животного. Среднее время, за которое происходило удвоение объема опухоли, в лечебной группе (Т) и контрольной группе (С) использовали для расчета общей задержки роста средней опухоли (Т-С).
Статистический анализ: Индивидуальное время, за которое происходит удвоение объема опухоли, для каждого животного используют в качестве конечной точки для t-критерия Стьюдента /критерия суммы рангов Манна-Уитни для статистического сравнения данных роста между группами.
Результаты: Опухоли у животных из обеих контрольных групп, получавших плацебо, хорошо выросли у всех 10 животных. В среднем удвоение исходного объема опухоли происходило к 9,2 и 8,9 дню, соответственно, в группах, получавших плацебо интраперитонеально и перорально. В указанных двух группах не было отмечено уменьшения средней массы тела во время исследования.
Интраперитонеальное введение IPM•Tris в дозе 81 мг/кг оказывало токсическое действие на мышей, что привело к гибели трех животных, максимальной потере средней массы тела на 20% (4,5 г); одно животное было умерщвлено по причине терминального состояния. Более низкие дозировки 54, 36 и 24 мг/кг переносились с максимальной потерей средней массы тела на 5% (1,2 г), 6% (1,3 г) и 1% (0,2 г), соответственно. Дозы 54, 35 и 24 мг/кг приводили к задержке роста опухоли (Т-С) на 4,4, 3,3 и 0,9 дни, соответственно. Интраперитонеальное введение IPM в дозировках 36 и 24 мг/кг приводило к задержке роста опухоли на 1,1 и 0,3 дни, соответственно. Обе дозировки переносились с максимальными потерями средней массы тела на 8% (1,8 г) и 2% (0,4 г) для дозировок 36 мг/кг и 24 мг/кг, соответственно. Время удвоения объема опухоли при введении IPM было статистически меньше, чем время при введении наибольших переносимых доз IPM•Tris, но статистически одинаковым при введении соответствующих доз IPM•Tris (р=0,0148 для дозы 54 мг/кг; р=0,1879 для дозы 36 мг/кг). Интраперитонеальное введение IPM•(LYS)2 в дозировках 84 и 56 мг/кг, что соответствует IPM в дозах 36 и 24 мг/кг, приводило к задержке роста опухоли на 0,5 и 0,3 дни, соответственно. Обе дозы переносились с максимальной потерей средней массы тела на 1% (0,3 г) и 0% для доз 36 и 24 мг/кг, соответственно. Время удвоения объема опухоли при введении IPM•(LYS)2 было статистически меньше, чем время при введении наибольших переносимых доз IPM•Tris, но статистически одинаковым при введении соответствующих доз IPM•Tris (р=0,0104 для дозы 54 мг/кг; р=0,1578 для дозы 36 мг/кг).
Пероральное введние введение IPM•Tris в дозировках 270, 180 и 120 мг/кг оказывал токсическое действие на мышей, семь животных пришлось умертвить из-за терминального состояния, семь животных погибли при введении дозы 180 мг/кг, трое - при введении дозы 120 мг/кг, соответственно. Дозы 180 и 120 мг/кг приводили к максимальной потере средней массы тела на 23% (5,3 г) и 20% (4,7 г), соответственно. Более низкая доза - 81 мг/кг переносилась с максимальной потерей массы тела на 10% (2,2 г). Доза 81 мг/кг вызывала задержку роста опухоли на 2,6 день. Пероральное введение IPM в дозе 120 мг/кг оказывало токсическое действие на мышей, привело к гибели двух животных и максимальной потере средней массы тела на 18% (3,8 г). Более низка доза 82 мг/кг переносилась с максимальной потерей средней массы тела на 9% (2 г) и приводила к задержке роста опухоли на 2,3 день. Время удвоения объема опухоли статистически не отличалось от времени при введении соответствующей дозы IPM•Tris (p=0,2932 для дозы 81 мг/кг). Пероральное введение IPM•(LYS)2 в дозировках 280 и 187 мг/кг, что соответствует IPM в дозах 120 и 81 мг/кг, приводило к задержке роста опухоли на 3,9 и 4,7 дни, соответственно. Обе дозировки переносились с максимальными потерями средней массы тела на 14% (3 г) и 7% (1,6 г). IPM•(LYS)2 в более низкой дозе обладал такой же активностью, что и IPM•Tris в соответствующей дозе 81 мг/кг (p=0,8785).
Краткие данные статистического анализа
Ответ ксенотрансплантантов рака груди человека МХ-1 на лечение (1) плацебо - интраперитонеально и перорально, (2) интраперитонеально IPM•Tris, IPM и IPM•(LYS)2, и (3) перорально IPM•Tris, IPM и IPM•(LYS)2 представлен на Рисунках 16, 17 и 18, соответственно.
Выводы
При интраперитонеальном введении эквивалентных доз IPM и IPM•Tris противоопухолевая активность IPM•Tris была сопоставима с активностью IPM и IPM•(LYS)2 (в более высоких дозах). Более низкая доза оказалась неэффективной в отношении МХ-1 опухоли в данном исследовании. Наибольшая толерантная доза IPM•Tris была более эффективной, чем наибольшая тестируемая доза как IPM и IPM•(LYS)2. При пероральном введении эквивалентных доз, противоопухолевая активность IPM•Tris была сопоставима с активностью как IPM, так и IPM•(LYS)2, но в более низких дозах. IPM•Tris и IPM были токсичны для мышей в дозе 120 мг/кг.
В предыдущем исследовании МХ-1 опухолей (Пример 7) при интраперитонеальном введении эквивалентных доз, противоопухолевая активность IPM•Tris была выше, чем активность IPM (в обеих дозировках) и сопоставима с противоопухолевой активностью IPM•(LYS)2 (в обеих дозировках). При пероральном введении эквивалентных доз, противоопухолевая активность IPM•Tris была сопоставима с активностью IPM (в обеих дозировках), сопоставима с активностью IPM•(LYS)2 в более высокой дозе, и превышала активность IPM•(LYS)2 в низких дозировках.
При сравнении результатов двух исследований, активность всех трех агентов при интраперитонеальном введении была ниже в последнем исследовании (например, для IPM•Tris в дозе 36 мг/кг - величина Т-С составила 10,2 дней vs. 3,3 дня; для IPM в дозе 36 мг/кг - величина Т-С составила 5,2 дня vs. 1,1 день; и для IPM•(LYS)2 в дозе 56 мг/кг - величина Т-С составила 9,9 дней vs. 0,3 дня). Активность IPM•Tris при пероральном введении также была ниже (сопоставимые величины для IPM и IPM•(LYS)2) в этом исследовании (например, для IPM•Tris в дозе 81 мг/кг - величина Т-С составила 9,0 дней vs. 2,6 дня; для IPM в дозе 81 мг/кг - величина Т-С составила 4,0 дня vs. 2,3 дня; и для IPM•(LYS)2 в дозе 280 мг/кг - величина Т-С составила 5,0 дней vs. 3,9 дней). Причины такой пониженной активности не являются очевидными, наблюдаются ее вариации от исследования к исследованию в различных биологических системах. Если рассматривать исключительно размеры опухоли в каждом исследовании, то в обоих исследованиях у животных из контрольных групп опухоли росли с соизмеримыми скоростями. Средний вес опухоли на первый день лечения был незначительно больше в Примере 7 (интервал 172=197 мг), чем в данном примере (интервал 144-162 мг); однако, это незначительное различие на должно оказывать существенного влияния на противоопухолевую активность препарата.
Пример 9 Материалы и Методы
Три группы по восемь мышей в каждой получали IPM•Tris, который может быть получен, как описано в Примере 1; три группы по восемь животных получали доксорубицин, одна группа из десяти мышей получала плацебо, а восемнадцать групп по восемь мышей в каждой получали комбинацию IPM•Tris /доксорубицин. Опухолевые фрагменты МХ-1 (30-40 мг, после in vivo пассажа) имплантировали подкожно в жировое тело молочных желез бестимусным мышам женского пола. IPM•Tris (или его плацебо) вводили интраперитонеально ежедневно в течение пяти последовательных дней (q1d×5) в трех дозах (12, 24 и 54 мг/кг), при этом рецептура IPM•Tris включает 258,9 мг IPM (MW 221,02), 141,9 мг Tris (MW 121,14, молярное соотношение IPM:Tris 1:1) и 3% маннитол. Доксорубицин (или его плацебо) вводили внутривенно каждый четвертый день посредством трех инъекций (q4d×3) в дозе 8 мг/кг. Лекарственные растворы приготавливали в день введения, растворы IPM•Tris хранили на льду после приготовления.
Лечение начинали, когда опухоли достигали размера приблизительно 175 мг (интервал от 100 мг до 250 мг). Каждую опухоль измеряли циркулем в двух направлениях, после чего рассчитывали ее массу при помощи формулы для удлиненного эллипсоида (а×b2/2), где а - самый длинный размер, a b - самый короткий размер, учитывая единицы плотности (1 мм3=1 мг). Измерения осуществляют дважды в неделю, а противоопухолевую активность оценивают, сравнивая задержку роста опухоли в лечебных группах и контрольных группах, получавших плацебо, оценивая частичную и полную опухолевую регрессию и количество выживших животных, не имеющих опухоли. Надо отметить, что в группе, получавшей комбинацию IPM•Tris (в дозе 54 мг/кг) и доксорубицин (в дозе 8 мг/кг), двое животных умерли на ранней стадии исследования в результате токсического действия препаратов. Результаты представлены на Рисунках 19-25.
Комбинированное введение IPM•Tris и доксорубицина оказывало выраженное противоопухолевое действие, ингибирование роста опухоли в результате активности данной комбинации существенно превышало показатели, которые наблюдается при введении каждого из агентов по отдельности, кроме того, данная комбинация значительно увеличивала продолжительность жизни животных, по сравнению с каждым агентом в отдельности. Фактически, данная комбинация обладает синергичным действием, которое по своей эффективности превышает эффективность индивидуальных препаратов при введении их в тех же дозах, но по отдельности; даже в том случае, если доза IPM•Tris при его индивидуальном введении слишком мала для достижения какого-либо улучшения и сопоставима с действием плацебо в контрольной группе. Несмотря на то, что животные, получавшие комбинированное лечение, теряли в весе во время лечения (средний вес животных уменьшился на 20%, на конец исследования, 22 день), они быстро восстанавливали его после завершения введения препарата (полное восстановление отмечалось к 38 дню), что позволяет предположить обратимость токсического действия препаратов. Полученные данные также позволяют предположить, что комбинированная терапия IPM•Tris и доксорубицином может быть полезной для лечения любого типа рака, который отвечает на лечение IPM•Tris или доксорубицином при их индивидуальном применении или в комбинации, включая, без ограничений указанными, рак груди, рак яичников и саркому.
Пример 10
Фрагменты (30-40 мг каждый) МХ-1 рака груди человека после in vivo пассажа подкожно имплантируют голым мышам в жировое тело молочных желез, позволяют им вырасти до размера 75-198 мг, после чего начинают лечение. IPM•Tris (54 мг/кг), который может быть получен как описано в Примере 1, и доцетаксель (10 мг/кг) вводят в режиме q1d5×5 интраперитонеально и q6d×3 внутривенно, соответственно, начиная с десятого дня после имплантации опухоли. Комбинация указанных двух агентов обладает усиленным противоопухолевым действием, по сравнению с индивидуальным эффектом каждого из агентов при их отдельном введении.
Пример 11
Фрагменты (30-40 мг каждый) МХ-1 рака груди человека после in vivo пассажа подкожно имплантируют голым мышам в жировое тело молочных желез, позволяют им вырасти до размера 75-198 мг, после чего начинают лечение. Было показано, что IPM•Tris (36 мг/кг), который может быть получен как описано в Примере 1, при интраперитонеальном введении подавляет опухолевый рост приблизительно с той же активностью, что IPM•Tris (81 мг/кг) при пероральном введении, как показано на Рисунке 27. Кроме того, пероральное или системное введение IPM•Tris приводит к одинаковому увеличению продолжительности жизни мышей, имеющих ксенотрансплантат МХ-1. Среднее время выживания для животных из группы, получавшей препарат интраперитонеально, составило 39 дней, среднее время выживания для животных из группы, получавшей препарат перорально, составило 37,5 дней, а среднее время выживания для животных из контрольной группы составило 30 дней, как показано на Рисунке 28. Эквивалентная противоопухолевая активность указанных пероральных и интраперитонеальных доз находится в ожидаемых пределах для фармакокинетики IPM•Tris при пероральном и системном введении.
Пример 12
Крысам Спраг-Доули вводят IPM•Tris, который может быть получен, как описано в Примере 1, один раз в день через зонд (перорально) или путем болюсного внутривенного введения. IPM•Tris вводят в дозах 20,30 или 40 мг/кг; образцы крови для анализа фармакокинетики забирают перед введением препарата и через 0,5, 1, 2, 4, 6, 8, 12 и 24 часа после введения, из ретроорбитального синуса. У трех животных из группы берут образцы в каждую временную точку. Фармакокинетика перед введением дозы и через 0.5, 1, 2 и 4 часа представлены на Рисунке 29, при этом Tmax составляет, приблизительно, 0,5 ч. При оценке конечного t1/2 он составил от 0,25 до 0, 64 ч. Для каждой дозы использовался показатель AUC, чтобы оценить абсолютную биоэквивалентность перорально вводимого IPM•Tris через соотношение (AUC после пероральной дозы)/(AUC после внутривенной дозы). Показатели AUC для каждой дозы показаны на Рисунке 30, а показатели Cmax для каждой дозы показаны на Рисунке 31. Оба показателя, AUC и Cmax, линейно увеличивались с увеличением дозы перорально- или внугривенно вводимого IPM•Tris.
Биодоступность IPM•Tris в дозах 20, 30 и 40 мг/кг при пероральном введении составляла 48%, 65% и 73%, соответственно. Общая средняя биодоступность составляет 62% у особей женского пола. Аналогичная фармакокинетика наблюдается у крыс; однако, средняя биодоступность у особей мужского пола составила 41%.
Пример 13
Стабильность IPM•Tris /маннитол и IPM•(LYS)2 в растворе
Стабильность при восстановлении в растворе композиции IPM•Tris оценивают в 5% растворе хлорида натрия для инъекций. Было показано, что концентрация IPM сохраняет >90% активности в течение 2,0 часов.
Композицию IPM•(LYS)2 оценивают в 0,9% растворе хлорида натрия для инъекций. Было показано, что концентрация IPM сохраняет >90% активности в течение 1,0 часов.
Стабильность IPM•Tris /маннитол в растворе
В Таблице 1 представлены данные стабильности при восстановлении в растворе для IPM•Tris /маннитола в присутствии 25 мл 5% раствора хлорида натрия для инъекций. Аликвоты собирают в заданное время ~1 ч, 1,5 ч, 2,5 ч, 3,5 ч, 4,0 ч и 5,0 ч. Анализируют активность IPM в образцах.
Стабильность IPM•(LYS)2 в растворе
В Таблице 2 представлены данные стабильности при восстановлении в растворе для IPM•(LYS)2 в присутствии 25 мл 0,9% раствора хлорида натрия для инъекций. Аликвоты собирают в заданное время ~ 1 ч, 1,5 ч, 2,5 ч, 3,5 ч, 4,0 ч и 5,0 ч. Анализируют активность IPM•(LYS)2 в образцах.
Выводы
Композиция IPM/трометамин/маннитол сохраняет 90% активности в течение 2,0 часов при растворении в 5% растворе хлорида натрия. Композиция IPM•(LYS)2 сохраняет 90% активность в течение 1,0 часа в присутствии 0,9% раствора хлорида натрия. Разница в стабильности при восстановлении в растворе (>90% активности) между двумя композициями составляет 1,0 час, то есть отличается в два раза. Увеличение времени стабильности может делать процесс приготовления и введения препарата более удобным для врачей.
Пример 14
Стабильность IPM в растворе
Стабильность при восстановлении в растворе композиции IPM оценивают при pH 7 при температуре приблизительно 25°С в течение 3,5 часов, как показано ниже в Таблице 3 и представлено на Рисунке 32.
Стабильность композиции IPM•Tris/маннитол в растворе
В Таблице 4 представлены данные стабильности при восстановлении в растворе для композиции IPM•Tris/маннитол в присутствии 25 мл 5% раствора хлорида натрия для инъекций. Аликвоты собирают в заданное время: 1,5 ч, 3,0 ч, и 4,5 ч.
Стабильность композиции IPM•(LYS)2 в растворе
В Таблице 5 представлены данные стабильности при восстановлении в растворе для композиции IPM•(LYS)2 в присутствии 25 мл 0,9% раствора хлорида натрия для инъекций. Аликвоты собирают в заданное время: 1,5 ч, 3,0 ч, и 4,5 ч.
Пример 15
Стабильность твердой композиции IPM•Tris
Стабильность твердого лиофилизата IPM•Tris/маннитол
Стабильность твердого лиофилизата IPM•(LYS)2
Пример 16
Эквиваленты
Специалист в данной области техники способен понять и осуществить без дополнительных экспериментов, выходящих за рамки рутинных процедур, многочисленные эквиваленты описанных здесь изобретений, относящихся к веществам и способам. Такие эквиваленты входят в объем изобретения и охвачены формулой изобретения.
Все приведенные ниже источники литературы и публикации включены в настоящее описание в качестве ссылок.
Реферат
Настоящее изобретение относится к эффективному для лечения гиперпролиферативных заболеваний кристаллическому соединению, имеющему формулу (I)где Aпредставляет собой сопряженную кислоту трис(гидроксиметил)аминометана (Tris); Х и Y независимо друг от друга представляют собой хлор, температура плавления которого составляет 103,37°С-105,77°С, и спектр рентгеновской порошковой дифракции представлен на Фигуре 2, а также его применению для лечения различных онкологических заболеваний. Предложено новое кристаллическое соединение, обладающее повышенной стабильностью, эффективное для лечения гиперпролиферативных заболеваний. 7 н. и 8 з.п. ф-лы, 32 ил., 2 табл., 16 пр.
Формула

где A+ представляет собой сопряженную кислоту трис(гидроксиметил)аминометана (Tris);
Х и Y независимо друг от друга представляют собой хлор;
температура плавления кристаллического соединения составляет от приблизительно 103,37°С до приблизительно 105,77°С; и
кристаллическое соединение имеет спектр рентгеновской порошковой дифракции, представленный на Фигуре 2.
Комментарии