Способ получения n-[(3-аминооксетан-3-ил)метил]-2-(1,1-диоксо-3,5-дигидро-1,4-бензотиазепин-4ил)-6-метил-хиназолин-4-амина - RU2664643C2
Код документа: RU2664643C2
Описание
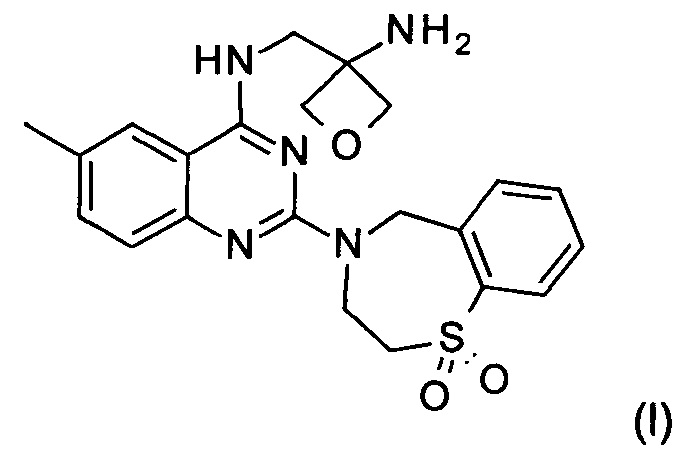
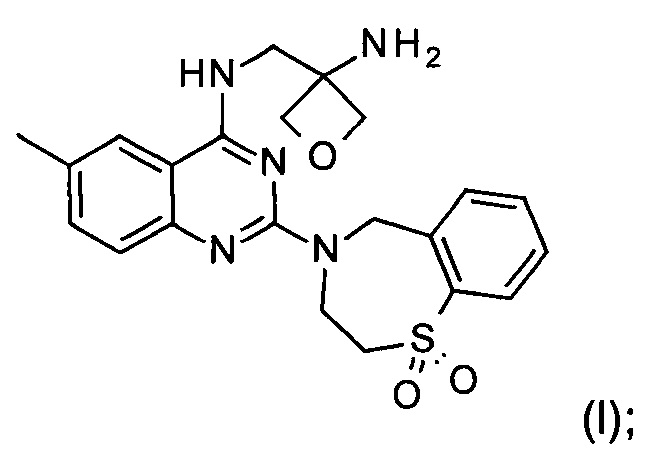
Настоящее изобретение относится к способу получения соединения формулы (I):

и его фармацевтически приемлемых солей присоединения кислоты, которые полезны для профилактики и лечения инфекции, вызванной респираторно-синцитиальным вирусом (RSV) у млекопитающего или человека.
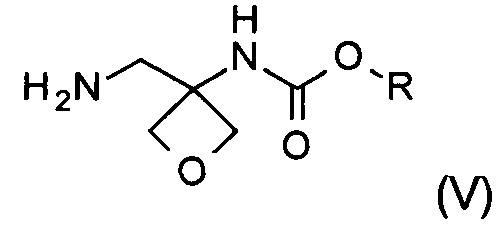
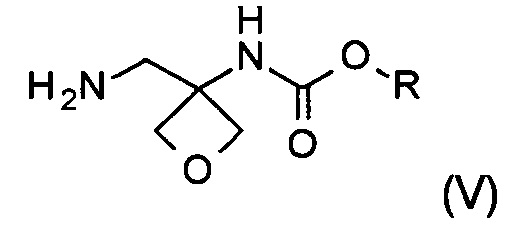
Другой аспект настоящего изобретения относится к новому способу получения соединения формулы (V):

где R представляет собой C1-6алкил, C1-6алкоксифенил-CxH2x- или фенил-СхН2х-. Соединение формулы (V) представляет собой важное промежуточное соединение при синтезе и производстве фармацевтически активного соединения формулы (I), описанного в патентной заявке WO 2013020993 А1.
Предшествующий уровень техники
В патентной заявке WO 2013020993 А1 раскрыты способы синтеза для получения соединения формулы (I).
Однако, согласно способу синтеза по заявке WO 2013020993 А1, снятие защиты с одного из промежуточных соединений для синтеза соединения формулы (I), 3-(аминометил)-N,N-дибензил-оксетан-3-амина, путем гидрогенирования при помощи палладия на углероде будет приводить к выходу остатка тяжелого металла, который не приемлем для химической технологии и производства в крупном масштабе. Кроме того, другое промежуточное соединение для синтеза соединения формулы (I), трет-бутил-[(3-аминооксетан-3-ил)метил]карбамат, является нестабильным в качестве первичного амина.
В данном изобретении разработан простой и эффективный способ синтеза соединений формулы (I). Данный способ синтеза может быть применен в промышленном масштабе и позволяет получать продукт с хорошим выходом, желаемой чистотой и стабильностью без применения тяжелого металла в качестве катализатора.
Подробное описание изобретения
Определения
В контексте данного документа термин "C1-6алкил" означает алкильную группу с линейной или разветвленной цепью, содержащей от 1 до 6, в частности от 1 до 5 атомов углерода, например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, 3-метилбутил, 1,1-диметилпропил, n-гексил, 2-этилбутил и т.п. Конкретные "C1-6алкильные" группы представляют собой трет-бутил и 1,1-диметилпропил.
Термин "СхН2х" означает насыщенную алкильную группу с линейной или разветвленной цепью, содержащей 1-6, в частности 1-4, атомов углерода.
Термин "C1-6алкокси" означает группу C1-6алкил-О-, где "C1-6алкил" представляет собой группу, определенную выше, например, метокси, этокси, пропокси, изо-пропокси, н-бутокси, изо-бутокси, 2-бутокси, трет-бутокси и т.п. Конкретные группы "C1-6алкокси" представляют собой метокси и этокси, в частности метокси.
Термин "C1-6алкоксифенил" означает фенил, замещенный группой C1-6алкокси, определение которой дано выше, в орто-, мета- или пара-положениях. Конкретная "C1-6алкоксифенильная" группа представляет собой 4-метоксифенил.
Термин "амино" относится к первичному (-NH2), вторичному (-NH-) или третичному амино

Термин "гидрокси" относится к группе -ОН.
Термин "НА" относится к органическим и неорганическим кислотам, таким как соляная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота, уксусная кислота, L-винная кислота, лимонная кислота, L-молочная кислота, малеиновая кислота, фумаровая кислота, янтарная кислота, метансульфоновая кислота, бензолсульфоновая кислота, бензойная кислота, п-толуолсульфоновая кислота, щавелевая кислота, п-нитробензойная кислота, салициловая кислота и 4-хлорбензойная кислота и т.п.
Термин "соль присоединения кислоты" относится к обычным солям присоединения кислот, которые получены из приемлемых нетоксичных органических или неорганических кислот. Соли присоединения кислот включают, например, соли, полученные из органических и неорганических кислот, таких как соляная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота, уксусная кислота, L-винная кислота, лимонная кислота, L-молочная кислота, малеиновая кислота, фумаровая кислота, янтарная кислота, метансульфоновая кислота, бензолсульфоновая кислота, бензольная кислота, п-толуолсульфоновая кислота, щавелевая кислота, п-нитробензойная кислота, салициловая кислота и 4-хлорбензойная кислота и т.п.


Проблемы способа по WO 2013020993 А1 решены согласно настоящему изобретению посредством способа получения соединений формулы (I), показанного на схеме 1:
Схема 1
где R представляет собой C1-6алкил, C1-6алкоксифенил-CxH2x- или фенил-СхН2х-.
Изобретение относится к способу получения соединения формулы (I):

и его фармацевтически приемлемой соли присоединения кислоты, включающему следующие стадии:
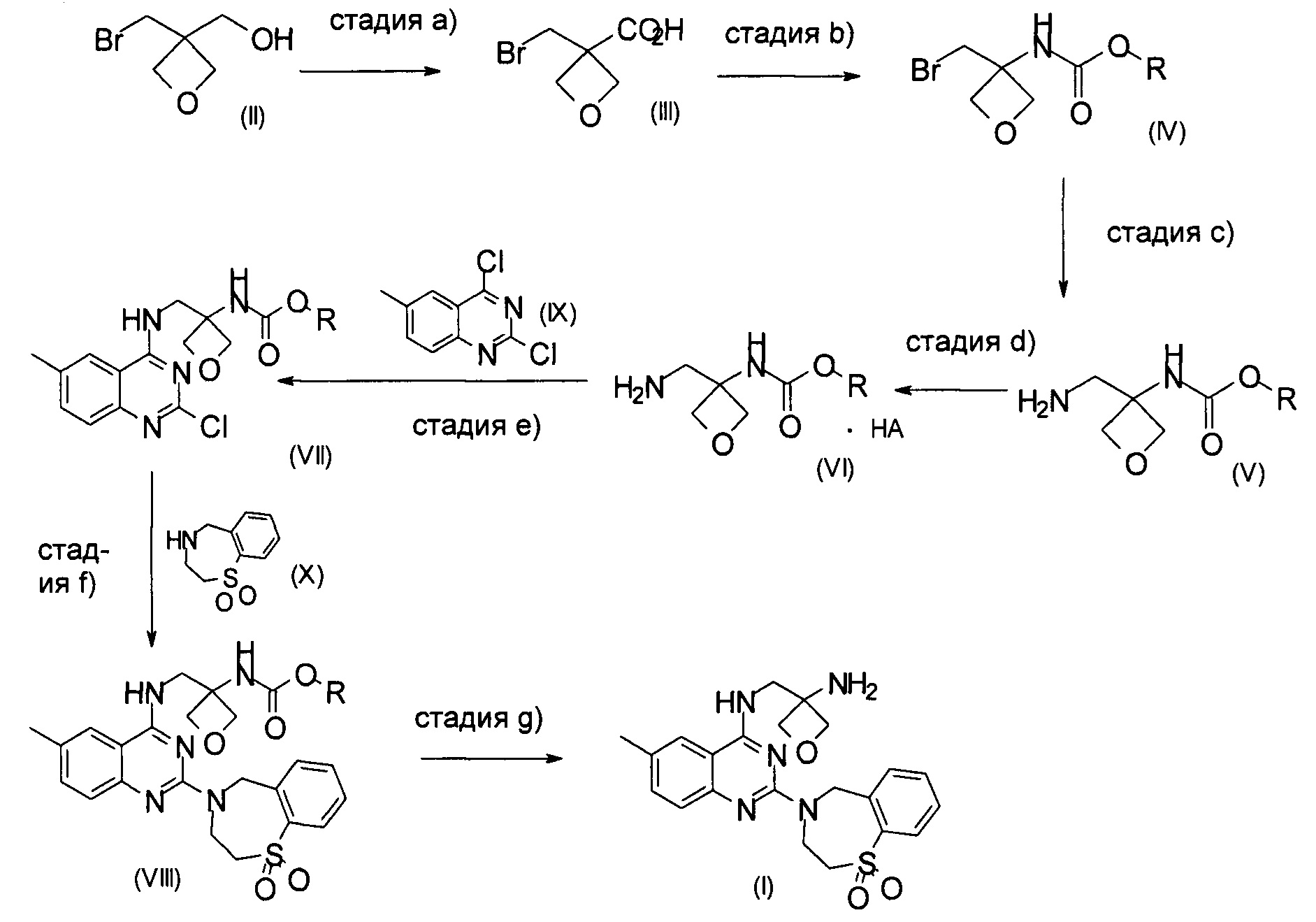
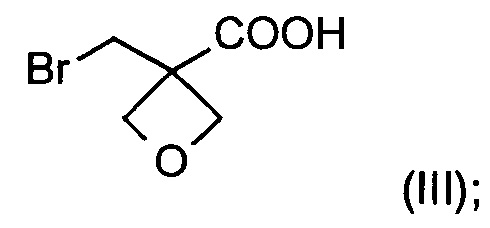
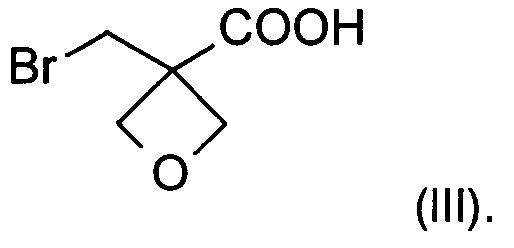
а) окисление [3-(бромметил)оксетан-3-ил]метанола формулы (II) с образованием соединения формулы (III)
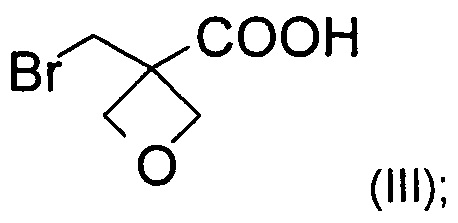
b) превращение карбоксильной группы соединения формулы (III) в карбамат с образованием соединения формулы (IV)
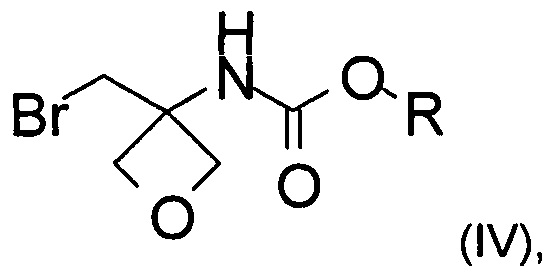
где R представляет собой C1-6алкил, C1-6алкоксифенил-СхН2х- или фенил-СхН2х-;
с) аминирование соединения формулы (IV) с образованием соединения формулы (V)
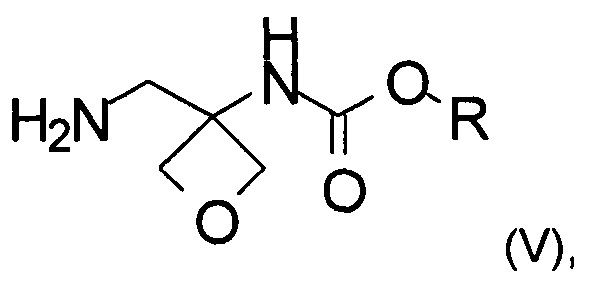
где R является таким, как определено выше;
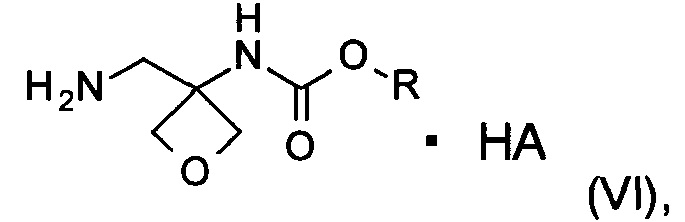
d) образование соли соединения формулы (V) при помощи кислоты с образованием соединения формулы (VI)
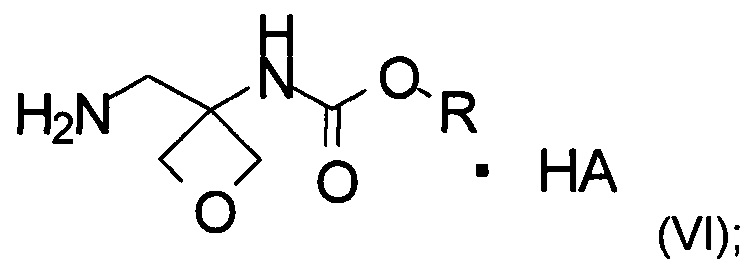
где R является таким, как определено выше;
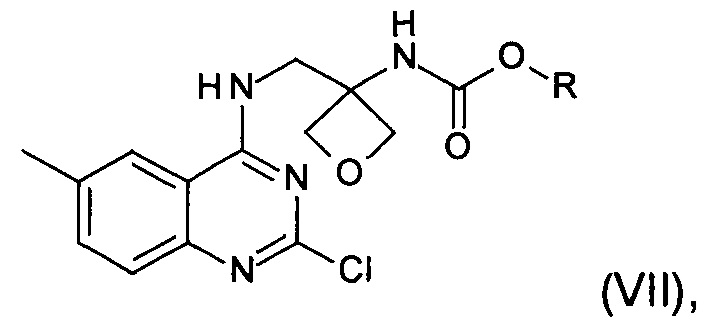
е) реакция замещения соединения формулы (VI) соединением формулы (IX) с образованием соединения формулы (VII)

где R является таким, как определено выше;
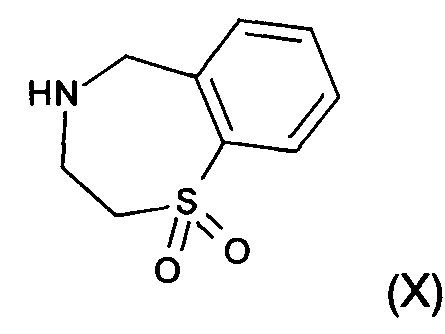
f) реакция замещения соединения формулы (VII) соединением формулы (X) с образованием соединения формулы (VIII)
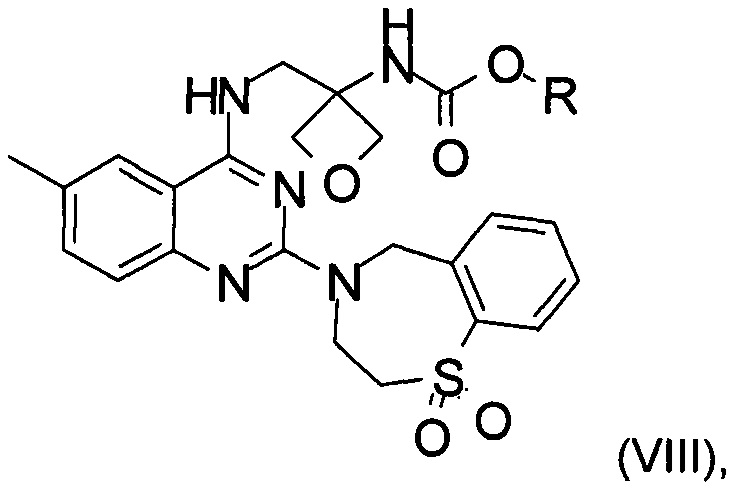
где R является таким, как определено выше;
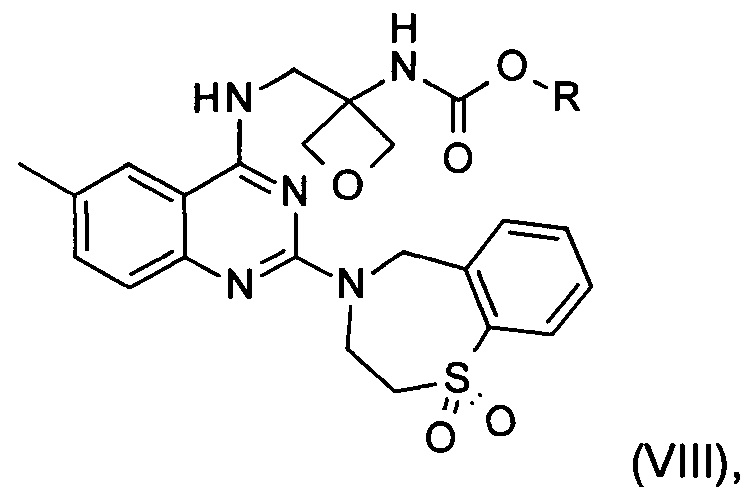
g) снятие защиты с соединения формулы (VIII) с образованием соединения формулы (I)

и при необходимости образование фармацевтически приемлемой соли присоединения кислоты.
Стадии (а), (b) и (с) способа синтеза приводят к соединению формулы (V), которое является новым и представляет собой другой важный аспект настоящего изобретения.
Подробное описание стадий способа по настоящему изобретению заключается в следующем.
На стадии а) получают карбоновую кислоту формулы (III) путем окисления [3-(бромметил)оксетан-3-ил]метанола формулы (II).
Данную реакцию проводят с окислителем при температуре реакции в диапазоне от 0°С до 100°С, в частности от 15°С до 25°С. Порядок добавления реагентов может быть таким, как удобно.
Реакция может быть проведена в различных растворителях, в частности, реакционный растворитель представляет собой воду, ацетонитрил, дихлорметан, этилацетат или изопропилацетат; или в сорастворителе, который представляет собой смесь двух или более видов растворителей, выбранных из воды, ацетонитрила, дихлорметана, этилацетата и изопропилацетата. В частности, растворитель представляет собой сорастворитель: вода с ацетонитрилом.
Окислитель, применяемый в данной реакции, представляет собой гипохлорит натрия, перманганат калия, 2,2,6,6-тетраметилпиперидиноокси или хлорхромат пиридиния; или соокислитель, который представляет собой смесь двух или более видов окислителей, выбранных из гипохлорита натрия, перманганата калия, 2,2,6,6-тетраметилпиперидиноокси и хлорхромата пиридиния. Конкретный окислитель представляет собой соокислитель: 2,2,6,6-тетраметилпиперидиноокси с гипохлоритом натрия. Как правило, реакция окисления завершается спустя 1-24 часа, в частности 4-6 часов.
На стадии b) происходит превращение карбоновой кислоты формулы (III) в карбамат формулы (IV) через перегруппировку Курциуса.
Реакцию проводят с участием азидного реагента и основания в органическом растворителе и с последующим добавлением спирта при температуре в диапазоне от 0°С до 100°С, в частности при 80°С.
На данной стадии соединение формулы (III) смешивают с азидным реагентом, в частности дифенилфосфорилазидом, и основанием в органическом растворителе с образованием активного промежуточного соединения 3-(бромметил)-3-изоцианато-оксетана, который в дальнейшем может быть превращен в карбаматы формулы (IV) при добавлении различных спиртов.
Основание, применяемое в данной реакции, представляет собой триэтиламин, диизопропилэтиламин или 4-метилморфолин, в частности 4-метилморфолин.
Реакция может быть проведена во многих органических растворителях. В частности, растворитель, применяемый на стадии b), представляет собой ацетонитрил, толуол, хлорбензол, дихлорметан. В частности, растворитель представляет собой толуол.
Температура реакции находится в диапазоне от 0°С до 100°С, в частности 80°С.
Обычно спирт, применяемый на стадии b), представляет собой трет-бутанол, 2-метил-2-бутанол, бензиловый спирт или 4-метоксифенилметанол, в частности 4-метоксифенилметанол.
На стадии с) протекает аминирование соединения формулы (IV) с образованием аминосоединения формулы (V).
Реакцию проводят с участием аминирующего агента при температуре реакции в диапазоне от 0°С до 60°С, в частности, в диапазоне от 25°С до 30°С.
С целью образования первичного амина соединение формулы (IV) и аминирующий агент, в частности жидкий аммиак, загружают в автоклав для получения соединения формулы (V).
Температура реакции, как правило, находится в диапазоне от 0°С до 60°С, в частности, в диапазоне от 25°С до 30°С.
Реакция обычно завершается спустя 1-24 часа, в частности через 8 часов.
На стадии d) происходит образование соли соединения формулы (V) при помощи кислоты с образованием соединения формулы (VI).
Кислота, применяемая в данной реакции, включает различные органические и неорганические кислоты, такие как соляная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота, уксусная кислота, L-винная кислота, лимонная кислота, L-молочная кислота, малеиновая кислота, фумаровая кислота, янтарная кислота, метансульфоновая кислота, бензолсульфоновая кислота, бензойная кислота, п-толуолсульфоновая кислота, щавелевая кислота, п-нитробензойная кислота, салициловая кислота, 4-хлорбензойная кислота и т.п., в частности 4-хлорбензойная кислота.
На стадии е) протекает реакция замещения соединения формулы (VI) соединением формулы (IX) с образованием соединения формулы (VII).
Реакция может быть проведена в органическом растворителе. В частности, реакцию проводят в тетрагидрофуране, 2-метилтетрагидрофуране или ацетонитриле, в частности в тетрагидрофуране.
Конкретный диапазон температуры реакции составляет от 10°С до 30°С.
На стадии f) протекает реакция замещения соединения формулы (VII) соединением формулы (X) с образованием соединения формулы (VIII). Данную реакцию проводят в органическом растворителе с кислотным катализатором при температуре в диапазоне от 0°С до 100°С, в частности, от 60°С до 80°С.
Реакцию проводят в органическом растворителе. В частности, реакцию проводят в тетрагидрофуране, 2-метилтетрагидрофуране, ацетонитриле, толуоле, метаноле, этаноле или изопропаноле, в частности в этаноле.
Кислотный катализатор, применяемый в реакции, представляет собой соляную кислоту, бромистоводородную кислоту, серную кислоту, фосфорную кислоту, метансульфоновую кислоту или хлорид аммония, в частности хлорид аммония.
На стадии g) проводят снятие защиты с соединения формулы (VIII) с образованием соединения формулы (I). Реакцию проводят в органическом растворителе с кислотой при температуре в диапазоне от 0°С до 100°С, в частности, от 10°С до 40°С.
Органический растворитель, применяемый в реакции, представляет собой дихлорметан, этилацетат, изопропилацетат, тетрагидрофуран или диоксан, в частности дихлорметан.
Кислота, применяемая в данной реакции, представляет собой соляную кислоту, бромистоводородную кислоту, серную кислоту, фосфорную кислоту, метансульфоновую кислоту или трифторуксусную кислоту, в частности трифторуксусную кислоту.
Далее изобретение проиллюстрировано следующими примерами, которые не должны толковаться в качестве ограничивающих изобретение в объеме определенных процедур, описанных в данном документе.
Изобретение также относится к соединению формулы (V):

где R представляет собой C1-6алкил, C1-6алкоксифенил-СхН2х- или фенил-СхН2х-.
Изобретение также относится к соединению формулы (III):
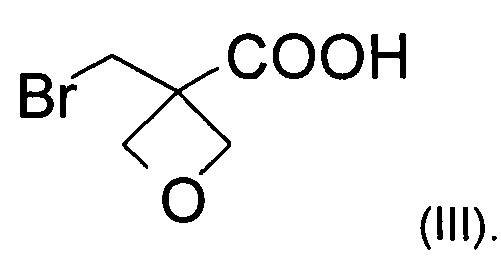
Примеры
Пример 1
Получение 3-(бромметил)оксетан-3-карбоновой кислоты:

В 100 мл колбу загружали [3-(бромметил)оксетан-3-ил]метанол (17,3 г, 9,6 ммоль), затем 25 мл воды и 5,3 мл ацетонитрила и TEMPO (153 мг, 0,96 ммоль). Смесь охлаждали до 10°С. 15,3 г гипохлорида натрия (14%) добавляли в течение 10 мин при внутренней температуре, поддерживаемой между 15°С и 20°С. Реакционную смесь перемешивали при комнатной температуре до тех пор, пока по данным ВЭЖХ не был израсходован [3-(бромметил)оксетан-3-ил]метанол. рН полученной смеси доводили до 8-9 и экстрагировали дважды 20 мл EtOAc. рН водного слоя доводили до 1-2 5Н водным раствором H2SO4 и экстрагировали дихлорметаном. После удаления дихлорметана получали 3-(бромметил)оксетан-3-карбоновую кислоту. MS набл. (ESI+) [(М+Н)+] 194, 1Н ЯМР (400 МГц, метанол-d4) d ppm, 9,40-9,90 (s, 1 Н), 5,00-5,02 (d, J=6,8 Гц, 2 H), 4,56-4,57 (d, J=6,8 Гц, 2 H), 3,97 (s, 2 Н).
Пример 2
Получение (4-метоксифенил)метил-N-[3-(бромметил)оксетан-3-ил]карбамата
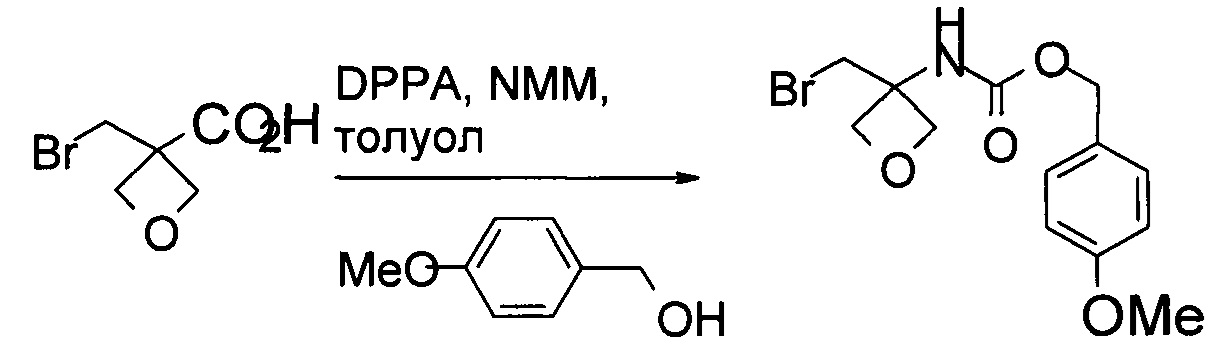
В реактор 1 загружали 3-(бромметил)оксетан-3-карбоновую кислоту (1,2 кг, 6,15 моль), затем 6,4 кг толуола. Реакционную смесь охлаждали до 5°С. Затем к этой реакционной смеси медленно добавляли NMM (0,72 кг, 7,12 моль). После добавления раствор перемешивали 10 минут при комнатной температуре.
В реактор 2 загружали дифенилфосфорилазид (1,76 кг, 6,39 моль), затем 3,2 кг толуола. Смесь нагревали до 80°С. Раствор из реактора 1 по каплям добавляли в реактор 2. После добавления реакционную смесь перемешивали в течение 30 мин при 80°С. К реакционной смеси затем медленно добавляли раствор 4-метоксифенилметанола (0,82 кг, 5,94 моль) в 1,58 кг толуола. После добавления реакционную смесь выдерживали в течение 75 мин при 80°С. Протекание реакции контролировали посредством ВЭЖХ. После завершения реакции смесь охлаждали до комнатной температуры и последовательно промывали 6,0 кг воды, 6,24 кг 4% водного раствора карбоната натрия и 3,0 кг воды. Органическую фазу концентрировали до сухости при пониженном давлении, и осадок перекристаллизовывали в системе н-гептан/этанол. Суспензию разделяли при помощи центрифугирования и влажный осадок промывали 1 кг н-гептана. Влажный осадок высушивали в вакуумной печи в течение 24 часов до получения 1,46 кг указанного в заголовке соединения, выход 72%. MS набл. (ESI+) [(М+Н)+] 330. 1Н ЯМР (400 МГц, CD3Cl-d3) d ppm 7,30-7,33 (d, J=8,4 Гц, 2H), 6,91-6,93 (d, J=8,4 Гц, 2H), 5,33 (s, 1H), 5,06 (s, 2H), 4,70-4,72 (d, J=6,4 Гц, 2H), 4,51-4,53 (d, J=6,4 Гц, 2H), 4,00 (s, 2H), 3,84 (s, 3H).
Пример 3
Получение трет-бутил-N-[3-(бромметил)оксетан-3-ил]карбамата
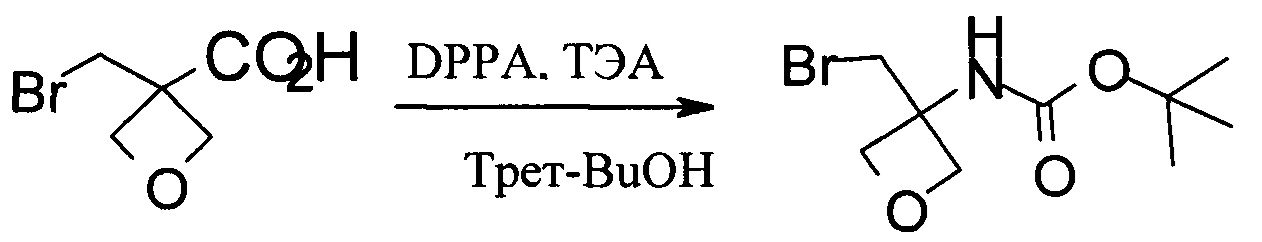
В 50 мл колбу загружали 3-(бромметил)оксетан-3-карбоновую кислоту (2,0 г, 10,3 ммоль), затем 20 мл безводного трет-бутанола. Реакционную смесь охлаждали до 5°С. Затем добавляли ТЭА (1,1 г, 11,3 ммоль). После этого добавления прибавляли порциями DPPA (3,1 г, 10,8 ммоль). Смесь нагревали с обратным холодильником в течение ночи. Затем органическую фазу концентрировали до сухости при пониженном давлении, и осадок растворяли в 30 мл EtOAc. Органическую фазу промывали 10 мл раствора Na2CO3 и 10 мл солевого раствора. После удаления растворителей получали трет-бутил-N[3-(бромметил)оксетан-3-ил]карбамат.
Пример 4
Получение бензил-N-[3-(бромметил)оксетан-3-ил]карбамата
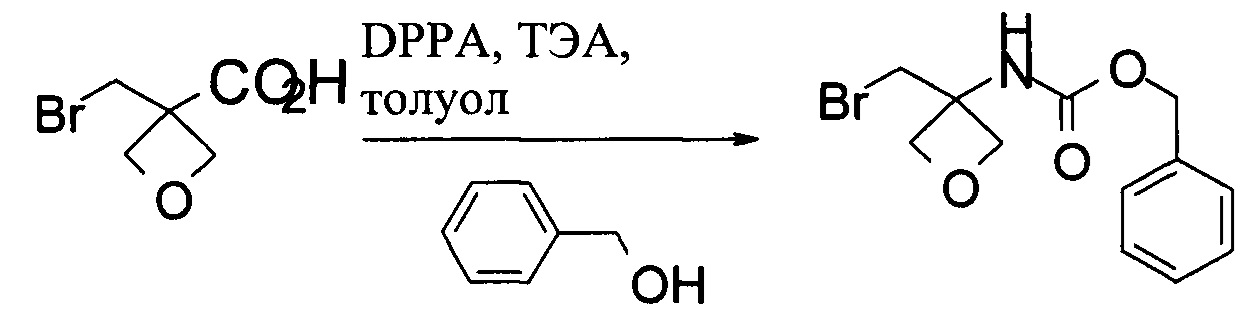
В 100 мл колбу загружали 3-(бромметил)оксетан-3-карбоновую кислоту (5,0 г, 25,6 ммоль), затем 50 мл безводного толуола. Затем добавляли ТЭА (2,87 г, 28,2 ммоль). После добавления ТЭА прибавляли порциями DPPA (7,64 г, 26,9 ммоль). Смесь нагревали при 65-70°С в течение 1 часа. Затем к реакционной смеси добавляли бензиловый спирт (4,2 г, 38,4 ммоль) и нагревали при 80°С в течение 2 часов. Полученную смесь охлаждали до комнатной температуры и разводили 50 мл EtOAc. Реакционную смесь промывали 30 мл воды, 30 мл 10% водного раствора Na2CO3 и 30 мл солевого раствора. Органическую фазу концентрировали для удаления большинства растворителей при пониженном давлении и добавляли 15 мл гептана. Суспензию перемешивали при комнатной температуре в течение 2 часов и отделяли посредством фильтрации. Влажный осадок высушивали в вакуумной печи до получения 4,95 г бензил-N-[3-(бромметил)оксетан-3-ил]карбамата.
Пример 5
Получение 1,1-диметилпропил-N-[3-(бромметил)оксетан-3-ил]карбамата
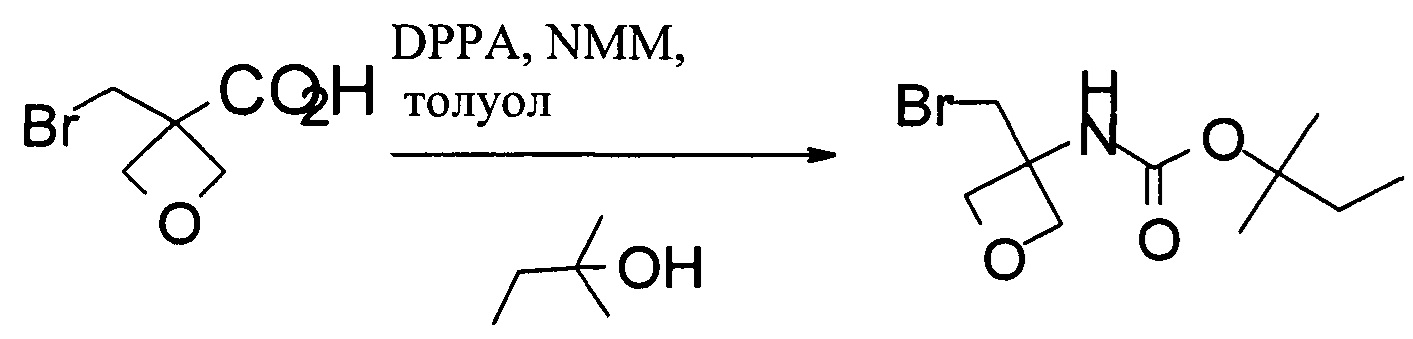
В 250 мл колбу загружали 3-(бромметил)оксетан-3-карбоновую кислоту (20,4 г, 100 ммоль), затем 120 мл безводного толуола. Добавляли NMM (12,1 г, 120 ммоль) в течение 30 мин. После добавления в течение 30 мин добавляли DPPA (30,3 г, 110 ммоль) в 80 мл толуола. Смесь нагревали при 80-85°С в течение 40 мин. Затем к реакционной смеси добавляли трет-амиловый спирт (44 г, 150 ммоль) и нагревали при 80°С в течение 3 ч. Полученную смесь охлаждали до комнатной температуры и промывали 100 мл воды, 30 мл 10% водного раствора Na2CO3 и 30 мл солевого раствора. Органическую фазу концентрировали в вакууме, и неочищенный продукт очищали при помощи флэш-хроматографии. Продукт разжижали в гептане. После фильтрования и высушивания получали 12,3 г 1,1-диметилпропил-N-[3-(бромметил)оксетан-3-ил]карбамата.
Пример 6
Получение (4-метоксифенил)метил-N-[3-(аминометил)оксетан-3-ил]карбамата

В 10 л автоклав загружали (4-метоксифенил)метил-N-[3-(бромметил)оксетан-3-ил]карбамат (1,1 кг, 3,33 моль) и 5,5 л жидкого аммиака. Реакционную смесь перемешивали при 25-30°С в течение 8 часов. Затем аммиак аккуратно высвобождали. К осадку добавляли 5,5 л 2-метилтетрагидрофурана. Смесь переносили в делительную воронку. К смеси затем добавляли 1,1 л 3 Н раствора NaOH. Водную фазу экстрагировали 4,4 л 2-метилтетрагидрофурана. Комбинированную органическую фазу дважды промывали 1,1 л насыщенного водного раствора NaCl. После разделения фаз органическую фазу концентрировали в вакууме до приблизительно 1 л. Неочищенный осадок использовали непосредственно без дополнительной обработки.
Пример 7
Получение бензил-N-[3-(аминометил)оксетан-3-ил]карбамата
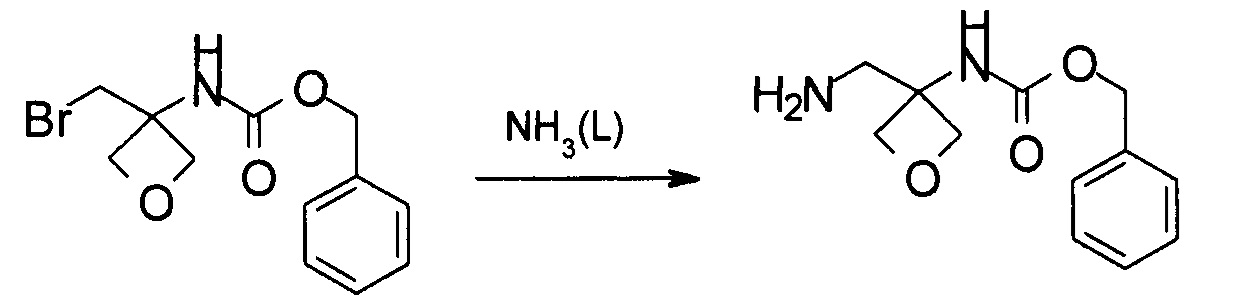
Указанное в заголовке соединение получают аналогично примеру 6, используя бензил-N-[3-(бромметил)оксетан-3-ил]карбамат, который получали в примере 4, вместо (4-метоксифенил)метил-N-[3-(бромметил)оксетан-3-ил]карбамата.
Пример 8
Получение 1,1-диметилпропил-N-[3-(аминометил)оксетан-3-ил]карбамата
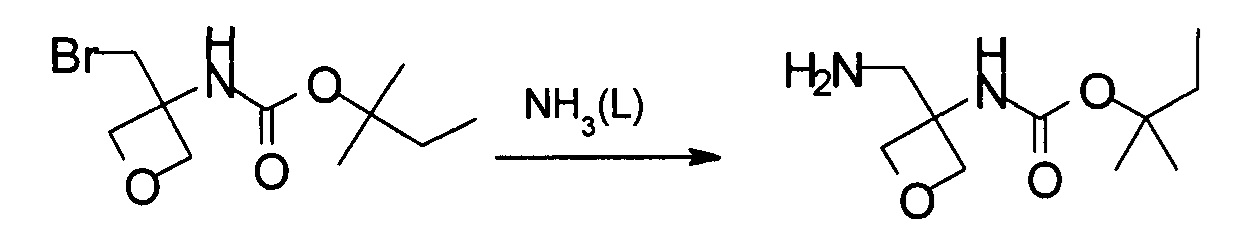
Указанное в заголовке соединение получают аналогично примеру 6, используя 1,1-диметилпропил-N-[3-(бромметил)оксетан-3-ил]карбамат, который получали в примере 5, вместо (4-метоксифенил)метил-N-[3-(бромметил)оксетан-3-ил]карбамата.
Пример 9
Получение трет-бутил-N-[3-(аминометил)оксетан-3-ил]карбамата

Указанное в заголовке соединение получают аналогично примеру 6, используя трет-бутил-N-[3-(бромметил)оксетан-3-ил]карбамат, который получали в примере 3, вместо (4-метоксифенил)метил-N-[3-(бромметил)оксетан-3-ил]карбамата.
Пример 10
Получение соли присоединения 4-хлорбензойной кислоты к (4-метоксифенил)метил-N-[3-(аминометил)оксетан-3-ил]карбамату
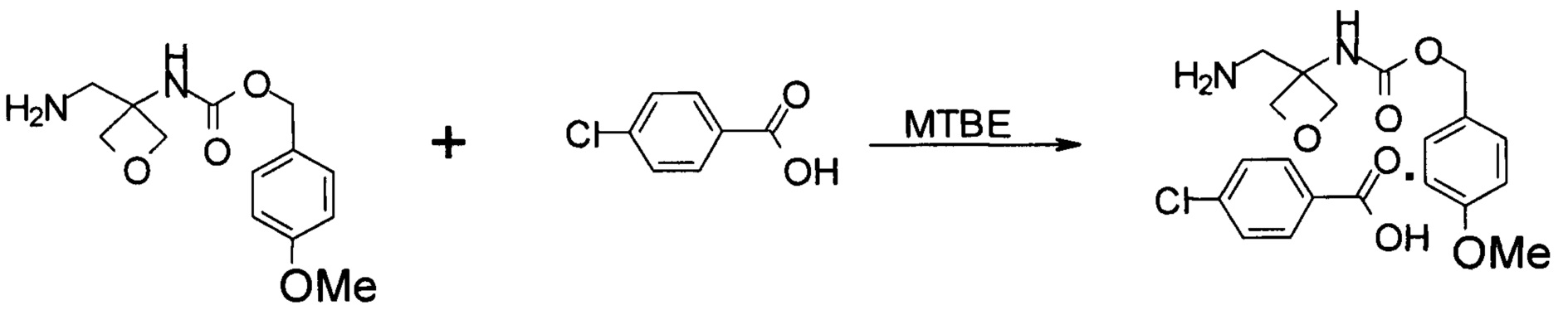
К осадку, полученному в примере 6, затем добавляли 4-хлорбензойную кислоту (420 г, 2,68 моль) и 2 л МТВЕ. Смесь перемешивали при 15-25°С в течение 14 часов. Применяли вакуумную фильтрацию для получения твердого вещества и влажный осадок промывали 1 л МТВЕ. Влажный осадок высушивали в вакуумной печи в течение 24 часов, получая 0,81 кг желаемой соли с выходом 57,5%. MS набл. (ESI+) [(М+Н)+] 423. 1Н ЯМР (400 МГц, ДМСО-d6) d ppm 8,32 (s, 1Н), 7,88-7,91 (d, J=8,4 Гц, 2H), 7,42-7,45 (d, J=8,4 Гц, 2H), 7,29-7,31 (d, J=8,4 Гц, 2H), 6,90-6,92 (d, J=8,4 Гц, 2H), 4,94 (s, 2 Н), 4,54-4,56 (d, J=6,4 Гц, 2H), 4,44-4,45 (d, J=6,4 Гц, 2H), 3,75 (s, 3Н), 3,19 (s, 2Н).
Пример 11
Получение (4-метоксифенил)метил-N-[3-[[(2-хлор-6-метил-хиназолин-4-ил)амино]метил]оксетан-3-ил]карбамата
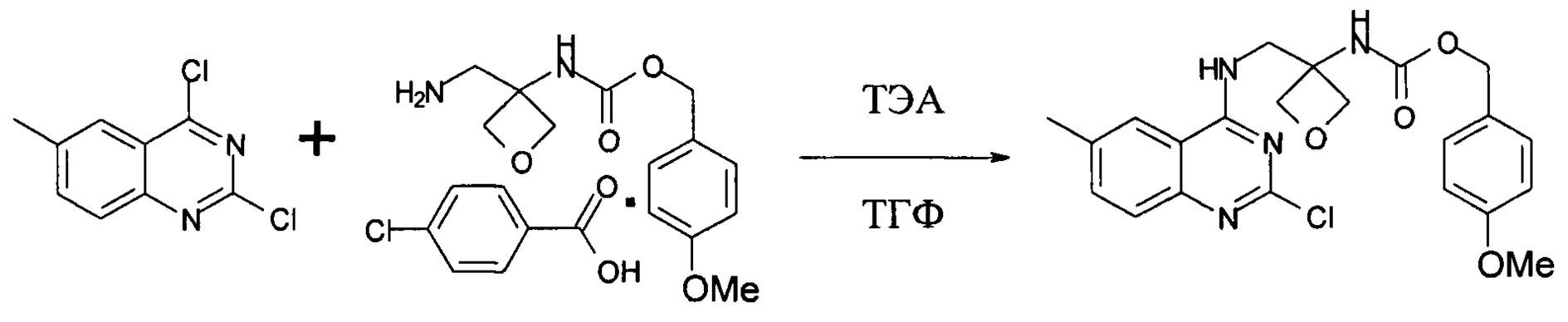
В 250 л реактор, покрытый с внутренней стороны эмалью, загружали соль присоединения 4-хлорбензойной кислоты к (4-метоксифенил)метил-N-[3-(аминометил)оксетан-3-ил]карбамату (8,1 кг, 19,2 моль) и 61,6 кг тетрагидрофурана. Затем к раствору добавляли ТЭА (5,9 кг, 58,3 моль). Затем смесь охлаждали до 10-15°С. Затем к смеси добавляли 2,4-дихлор-6-метил-хиназолин (3,99 кг, 18,7 моль), поддерживая температуру реакции в диапазоне 10-30°С. Затем реакционную смесь перемешивали при 22-27°С в течение 20 часов. Ход реакции контролировали посредством ВЭЖХ. После завершения реакции реакционную смесь концентрировали в вакууме при температуре ниже 40°С до 24,3-32,4 л в течение 3,5 часов, поддерживая температуру бани в диапазоне 15-25°С. Затем к осадку добавляли 80,2 кг воды в течение 100 мин. Смесь перемешивали при 15-25°С в течение 3,5 часов. Суспензию разделяли центрифугированием и промывали 48 кг воды четырьмя порциями в течение 50 мин до получения 23,6 кг влажного (4-метоксифенил)метил-N-[3-[[(2-хлор-6-метил-хиназолин-4-ил)амино]метил]оксетан-3-ил]карбамата.
В 250 л реактор, покрытый с внутренней стороны эмалью, загружали 23,6 кг влажного (4-метоксифенил)метил-N-[3-[[(2-хлор-6-метил-хиназолин-4-ил)амино]метил]оксетан-3-ил]карбамата, 24,0 кг МТВЕ и 7,0 кг этилацетата. Смесь перемешивали при 15-25°С в течение 2,5 часов. Суспензию разделяли при помощи центрифугирования и промывали 6,0 кг МТВЕ. Влажный осадок высушивали в вакуумной печи при 38-42°С с выпуском азота в течение 3 часов и затем при 40-52°С в течение 17 часов до получения 7,9 кг указанного в заголовке соединения с выходом 92%. MS набл. (ESI+) [(М+Н)+] 443. 1Н-ЯМР (400 Гц, ДМСО-d6) d ppm 8,67-8,69 (t, J=5,6 Гц, 1H), 8,09 (s, 1H), 7,85 (s, 1H), 7,64-7,67 (m, 1H), 7,53-7,55 (m, 1H), 7,26-7,29 (d, J=8,4 Гц, 2H), 6,88-6,90 (d, J=8,4 Гц, 2H), 4,96 (s, 2H), 4,62-4,64 (d, J=6,4 Гц, 2H), 4,51-4,53 (d, J=6,4 Гц, 2H), 4,07-4,09 (d, J=6,4 Гц, 2H), 3,74 (s, 3H), 2,47(s, 3H).
Пример 12
Получение (4-метоксифенил)метил-N-[3-[[[2-(1,1-диоксо-3,5-дигидро-1,4-бензотиазепин-4-ил)-6-метил-хиназолин-4-ил]амино]метил]оксетан-3-ил]карбамата.
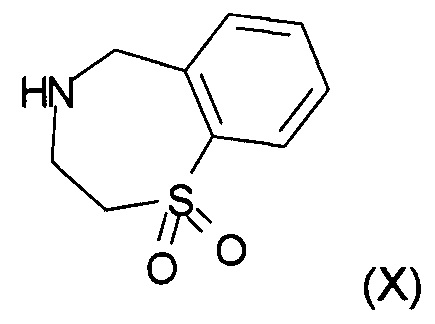
Получение промежуточного соединения формулы (X): 2,3,4,5-тетрагидро-1,4-бензотиазепин-1,1-диоксида:
Стадия 1: Получение 2-фенилсульфанилэтанамина:

В реактор загружали 56,1 кг воды, затем NaOH (7,0 кг, 175 моль). Механически перемешивали до растворения всего NaOH с образованием раствора. Охлаждали раствор до 25°С и к раствору добавляли тиофеноксид натрия (50,7 кг, водный раствор) и 2-хлорэтиламида гидрохлорид (17,7 кг, 153 моль). Смесь перемешивали при 25°С в течение 15 часов. Ход реакции контролировали посредством ВЭЖХ. После завершения реакции смесь дважды экстрагировали 61,1 кг EtOAc. Объединенную органическую фазу концентрировали до приблизительно 92 л и применяли на следующей стадии без дополнительной очистки.
Стадия 2: Получение N-(2-фенилсульфанилэтил)ацетамида:

Осадок с последней стадии нагревали до 45°С и к раствору медленно добавляли АсОН (14,0 кг, 233 моль), поддерживая температуру реакции ниже 60°С. Ход реакции контролировали посредством ВЭЖХ. После завершения реакции раствор охлаждали до 45°С и концентрировали в вакууме для удаления 55 л EtOAc. Затем смесь охлаждали до температуры ниже 25°С и к раствору медленно прибавляли 62,0 кг н-гептана. После добавления суспензию охлаждали до 0°С и выдерживали в течение 1 часа. Твердое вещество собирали при помощи центрифугирования.
Влажный осадок высушивали в вакуумной печи в течение 22 часов до получения 22,2 кг N-(2-фенилсульфанилэтил)ацетамида с выходом 74%. MS набл. (ESI+) [(М+Н)+] 196. 1Н-ЯМР (400 Гц, ДМСО-d6) d ppm 8,07 (s, 1Н), 7,18-7,40 (m, 5Н), 3,21-3,26 (m, 2Н), 2,99-3,03 (m, 2Н), 1,80 (s, 3Н).
Стадия 3: Получение 1-(3,5-дигидро-2Н-1,4-бензотиазепин-4-ил)этанона:
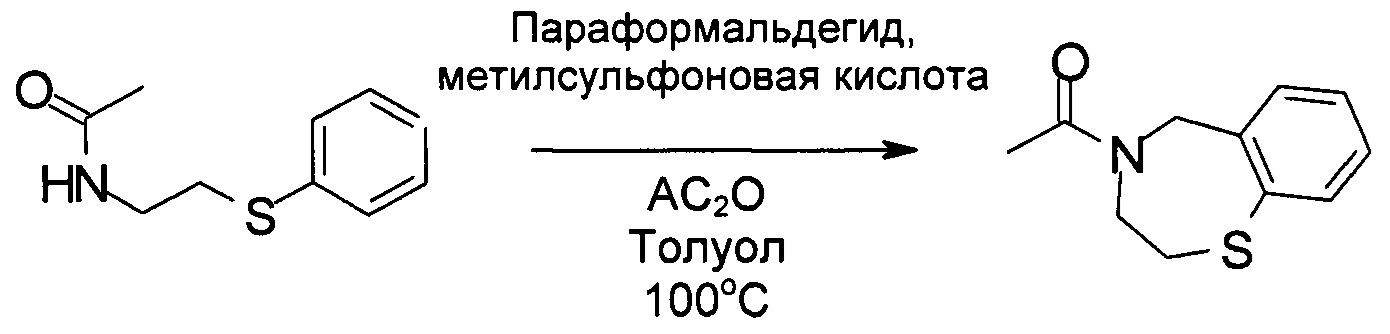
В реактор загружали N-(2-фенилсульфанилэтил)ацетамид (22,2 кг, 114 моль) и 124,7 кг толуола. Затем к раствору добавляли параформальдегид (2,1 кг, 70 моль), метилсульфоновую кислоту (10,9 кг, 113 моль) и Ac2O (14,0 кг, 137 моль). Реакционную смесь нагревали до 75-80°С и затем в реактор порциями загружали параформальдегид (4,9 кг, 163 моль), поддерживая температуру реакции ниже 80°С. После добавления реакционную смесь нагревали до 100-105°С и выдерживали в течение 1 часа. Ход реакции контролировали посредством ВЭЖХ. После завершения реакции реакционную смесь охлаждали до 30°С и в реактор добавляли 71,1 кг воды. Разделяли фазы и органический раствор промывали 63,1 кг насыщенного водного раствора NaHCO3, а затем 63,1 кг солевого раствора. Затем органическую фазу концентрировали под вакуумом для удаления всего органического растворителя и осадок использовали непосредственно на следующей стадии без дальнейшей очистки.
Стадия 4: получение 1-(1,1-диоксидо-2,3-дигидро-1,4-бензотиазепин-4(5Н)-ил)этанона:

К остатку, полученному на последней стадии, добавляли 112,8 кг муравьиной кислоты и 12,8 кг воды. Смесь охлаждали до 0°С. К реакционной смеси медленно добавляли 80,4 кг H2O2 (35%), поддерживая температуру реакции ниже 10°С. После добавления реакционную смесь перемешивали в течение 1 часа при 10°С. Затем реакционную смесь нагревали до 25°С и перемешивали в течение 3 часов. Ход реакции контролировали посредством ВЭЖХ. После завершения реакции к реакционной смеси добавляли 177,3 кг воды и 235,1 кг ДХМ. Фазы разделяли и водный слой снова экстрагировали 165,9 кг ДХМ. Объединенную органическую фазу промывали 112,7 кг насыщенного водного раствора Na2SO3, 112,1 кг насыщенного водного раствора Na2CO3 и 103,0 кг насыщенного водного раствора NaCl. Затем органическую фазу концентрировали под вакуумом для удаления органического растворителя. Затем осадок диспергировали в 54,3 кг EtOH и перемешивали в течение 1 часа при 55-65°С. Суспензию разделяли при помощи центрифугирования и влажный осадок высушивали в вакуумной печи в течение 12 часов до получения 17,4 кг 1-(1,1-диоксидо-2,3-дигидро-1,4-бензотиазепин-4(5Н)-ил)этанона с выходом 64%. MS набл. (ESI+) [(М+Н)+] 240. 1Н-ЯМР (400 Гц, ДМСО-d6) d ppm 7,92-8,00 (m, 1Н), 7,55-7,74 (m, 3Н), 4,60-4,88 (m, 2Н), 4,05 (brs, 2Н), 3,48-3,70 (m, 2Н), 3,53 (d, J=8.0 Гц, 3Н).
Стадия 5: Получение 2,3,4,5-тетрагидро-1,4-бензотиазепин-1,1-диоксида:

В реактор загружали 1-(1,1-диоксидо-2,3-дигидро-1,4-бензотиазепин-4(5Н)-ил)этанона (16,6 кг, 69,4 моль), 55,2 кг EtOH и 55,8 кг водного раствора NaOH (11,1 кг NaOH в 44,7 кг H2O). Реакционную смесь нагревали до 74-79°С и выдерживали в течение 24 часов при данной температуре. Ход реакции контролировали посредством ВЭЖХ. После завершения реакции смесь охлаждали до 50-55°С и органический растворитель удаляли при пониженном давлении. Затем к реактору добавляли 104,1 кг воды, смесь охлаждали до 0-7°С и выдерживали в течение 1 часа. Суспензию разделяли при помощи центрифугирования и влажный осадок дважды промывали 44,7 кг воды. Влажный осадок высушивали в вакуумной печи в течение 24 часов до получения 9,9 кг 2,3,4,5-тетрагидро-1,4-бензотиазепин-1,1-диоксида с выходом 72,3%. MS набл. (ESI+) [(М+Н)+] 198. 1Н-ЯМР (400 Гц, ДМСО-d6) d ppm 7,89 (dd, J=1,2, 7,6 Гц, 1H), 7,56 (t, J=7,6 Гц, 1H), 7,47 (t, J=7,6 Гц, 1H), 7,42 (d, J=7,6 Гц, 1H), 4,04 (s, 2Н), 3,30-3,32 (m, 2Н), 3,25-3,30 (m, 2Н), 2,64 (s, 1Н).
Получение (4-метоксифенил)метил-N-[3-[[[2-(1,1-диоксо-3,5-дигидро-1,4-бензотиазепин-4-ил)-6-метил-хиназолин-4-ил]амино]метил]оксетан-3-ил]карбамата:
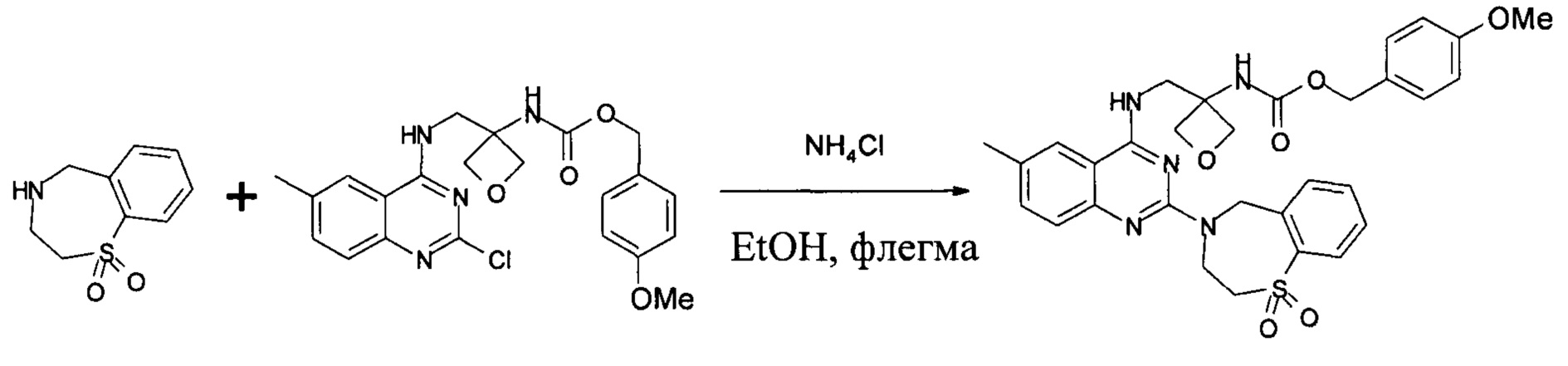
В 250 л реактор, покрытый с внутренней стороны эмалью, загружали 63 кг EtOH, а затем (4-метоксифенил)метил-N-[3-[[(2-хлор-6-метио-хиназолин-4-ил)амино]метил]оксетан-3-ил]карбамат (7,8 кг, 17,6 моль), 2,3,4,5-тетрагидро-1,4-бензотиазепин-1,1-диоксид (3,89 кг, 19,7 моль) и хлорид аммония (49 г, 0,92 моль). Реакционную смесь перемешивали при 68-72°С в течение 20 часов. Ход реакции контролировали посредством ВЭЖХ. После завершения реакции реакционную смесь медленно охлаждали до 20-25°С. Твердые вещества собирали вакуумной фильтрацией и промывали 15,6 кг EtOH двумя порциями. Влажный осадок высушивали в вакуумной печи с выпуском азота при 38-42°С в течение приблизительно 4 часов, и затем нагревали до 50-55°С в течение 30 часов до получения 11,0 кг указанного в заголовке соединения с выходом 88%. MS набл. (ESI+) [(М+Н)+] 604. 1Н-ЯМР (400 Гц, ДМСО) d ppm 9,57(s, 1Н), 8,14 (s,1H), 7,93-7,95 (d, 1H, J=8), 7,68 (m, 3H), 7,57-7,58 (m, 1H), 7,22-7,23 (d, 2H, J=4), 7,68-7,69 (d,2H, J=4), 4,98-5,17(m, 2H), 4,26-4,68 (m, 5H), 3,74-4,1 (m, 3H), 3,4-3,46 (t, 1H, J=8), 2,51 (s, 3H), 2,39 (s, 3H).
Пример 13
Получение N-[(3-аминооксетан-3-ил)метил]-2-(1,1-диоксо-3,5-дигидро-1,4-бензотиазепин-4-ил)-6-метил-хиназолин-4-амина.

В 250 л реактор, покрытый с внутренней стороны эмалью, загружали N-[3-[[[2-(1,1-диоксо-3,5-дигидро-1,4-бензотиазепин-4-ил)-6-метил-хиназолин-4-ил]амино]метил]оксетан-3-ил]карбамат (10,8 кг, 24,6 моль) и 120 кг дихлорметана. Затем к смеси порциями добавляли 16,0 кг 1Н раствора NaOH. После разделения фаз водную фазу экстрагировали 14,0 кг дихлорметана. Объединенную органическую фазу промывали 25 кг 20% водного раствора NaCl, затем переносили в 100 л реактор, покрытый с внутренней стороны эмалью, и концентрировали до 30-35 л при температуре ниже 35°С в вакууме для получения раствора 1.
В другой 250 л реактор, покрытый с внутренней стороны эмалью, загружали 26,0 кг дихлорметана и 16,0 кг трифторуксусной кислоты. Смесь охлаждали до 15-20°С и к раствору порциями добавляли указанный раствор 1. Смесь перемешивали в течение 30 минут при 15-25°С и затем охлаждали до 0-10°С. К смеси добавляли 39,8 кг ДМФ и затем раствор концентрировали до 62-65 л при 15-30°С в вакууме в течение 16,5 часов для получения раствора 2.
В 300 л реактор, покрытый с внутренней стороны эмалью, загружали 128,3 кг 1,5 Н раствора NaOH и охлаждали до 5-7°С. Затем в реактор добавляли 3,0 кг диметилформамида, а затем раствор 2. Суспензию перемешивали при 7-11°С в течение 30 минут. Твердое вещество собирали вакуумной фильтрацией и промывали 101 кг воды. Затем в 250 л реактор, покрытый с внутренней стороны эмалью, загружали влажный осадок, а затем 54,0 кг EtOH. Смесь нагревали до 74-78°С и перемешивали в течение 4,5 часов. Затем смесь охлаждали до 20-25°С. Твердое вещество собирали вакуумной фильтрацией и влажный осадок промывали 15,0 кг EtOH. Влажный осадок высушивали в вакуумной печи при 48-52°С с выпуском азота в течение 20 часов до получения 5,82 кг указанного в заголовке соединения с выходом 85%. MS набл. (ESI+) [(М+Н)+] 440. 1Н-ЯМР (400 Гц, METHANOL-D4) d ppm 7,98 (d, J=7,6 Гц, 1H), 7,86 (d, J=7,6 Гц, 1H), 7,73 (s, 1Н), 7,60 (t, J=7,6 Гц, 1H), 7,32-7,47 (m, 3Н), 5,53 (s, 2Н), 4,58 (brs, 2Н), 3,84 (s, 2Н), 3,53 (t, J=4,8 Гц, 2H), 2,41 (2, 3Н), 2,21 (m, 2Н), 1,97-2,04 (m, 2Н), 1,82-1,91 (m, 2Н).
Реферат
Изобретение относится к новому способу получения соединения формулы (I). Технический результат: разработан новый способ получения соединения формулы (I), которое полезно для профилактики и лечения инфекции, вызванной респираторно-синцитиальным вирусом (RSV) у млекопитающего или человека. Способ позволяет получать продукт с хорошим выходом и чистотой. 4 н. и 17 з.п. ф-лы, 1 табл., 13 пр.
Формула








Комментарии