Способ и композиция для лечения ракового заболевания, тозилат и фармацевтически приемлемые соли n-(4-хлор-3-(трифторметил)фенил)-n'-(4-(2-(n-метилкарбамоил)-4-пиридилокси)фенил)мочевины - RU2316326C2
Код документа: RU2316326C2
Чертежи



Описание
Область техники, к которой относится изобретение
Данное изобретение относится к производным арилмочевины в комбинации с цитотоксическими или цитостатическими агентами и к их использованию для лечения опосредованных киназой raf заболеваний, таких как раковые опухолевые заболевания.
Уровень техники
Онкоген p21ras представляет собой главный агент, участвующий в возникновении и развитии солидных форм рака у человека, и является мутантным в 30% всех случаев рака у человека (см. статьи Bolton и соавт., Ann. Rep. Med. Chem., 29, 165-74, (1994); Bos, Cancer Res., 40, 4682-9, (1989)). В своей нормальной немутантной форме белок ras является ключевым элементом каскада сигнальной трансдукции, направляемого рецепторами фактора роста почти во всех тканях (см. статью Avruch и соавт., Trends Biochem. Sci., 19, 279-83, (1994)). С биохимической точки зрения онкоген ras является белком типа ГТФазы, связывающим гуаниновые нуклеотиды, который существует в активированной ГТФ-связанной форме и неактивной ГДФ-связанной форме. Эндогенная ГТФазная активность этого белка строго саморегулируется, а также контролируется другими регуляторными белками. Мутанты обладают пониженной эндогенной ГТФазной активностью. Вследствие этого белок доставляет конститутивные сигналы роста действующим после него эффекторам, таким как киназа raf. Это приводит к раковому росту клеток, которые несут данные мутации (Magnuson и соавт., Semin. Cancer Biol., 5, 247-53, (1994)). Было показано, что ингибирование эффекта активного газ путем подавления сигнального пути киназы raf посредством введения дезактивирующих антител против киназы raf или коэкспрессии доминантно отрицательной киназы raf или доминантно отрицательного МЕК, субстрата киназы raf, приводит к реверсии трансформированных клеток к фенотипу нормального роста (см. статьи Daum и соавт., Trends Biochem., Sci., 19, 474-80, (1994); Friedman и соавт., J. Biol. Chem., 269, 30105-8, (1994); Kocj и соавт., Nature, 349, 426-28, (1991)). В данных источниках дополнительно показано, что ингибирование экспрессии raf антисмысловой РНК блокирует пролиферацию клеток в мембранно-ассоциированных онкогенах. Аналогично, ингибирование киназы raf (антисмысловыми олигодезоксинуклеотидами) коррелировало in vitro и in vivo с ингибированием роста множества типов опухолей человека (Monia и соавт., Nat. Med., 2, 668-75, (1996)). Следовательно, соединения, которые могут действовать в качестве ингибиторов киназы raf, представляют важную группу химиотерапевтических агентов для использования при лечении ряда онкологических заболеваний различного типа.
Раскрытие изобретения
Основной задачей настоящего изобретения является разработка цитотоксических и/или цитостатических агентов в комбинации с производными арилмочевины - ингибиторами киназы raf, что обеспечит: (1) улучшенную эффективность для снижения роста опухоли или даже устранение опухоли по сравнению с каждым агентом, введенным в отдельности, (2) введение меньших количеств химиотерапевтических агентов, (3) химиотерапевтическое лечение, которое характеризуется достаточно высокой переносимостьюу пациентов и снижением отрицательных фармакологических побочных действий, наблюдаемых при введении одного химиотерапевтического агента или при использовании некоторых видов комбинированной терапии, (4) расширение спектра поддающихся лечению различных типов рака млекопитающих, прежде всего, человека, (5) более высокий уровень ответной реакции у пациентов, проходящих лечение, (6) увеличение продолжительности жизни пациентов, проходящих лечение, по сравнению с пациентами, проходящими курс лечения стандартной химиотерапией, (7) увеличение продолжительности опухолевой прогрессии и/или (8) по крайней мере сопоставимые эффективность и переносимость по сравнению с агентами, введенными в отдельности и по сравнению с известными случаями, в которых комбинация других агентов приводит к антагонистическому действию.
Краткое описание чертежей
На фиг.1 представлена чувствительность устойчивых подкожных ксенотрансплантатов опухоли DLD-1 ободочной кишки человека к соединению А и камптозару, введенным каждое в отдельности и в комбинации.
На фиг.2 представлена чувствительность устойчивых подкожных ксенотрансплантатов опухоли MiaPaCa-2 поджелудочной железы человека к соединению А и гемзару, введенным каждое в отдельности и в комбинации.
На фиг.3 представлена чувствительность устойчивых подкожных ксенотрансплантатов NCl-H460 опухоли NSCLC человека к соединению А и навельбину, введенным каждое в отдельности и в комбинации.
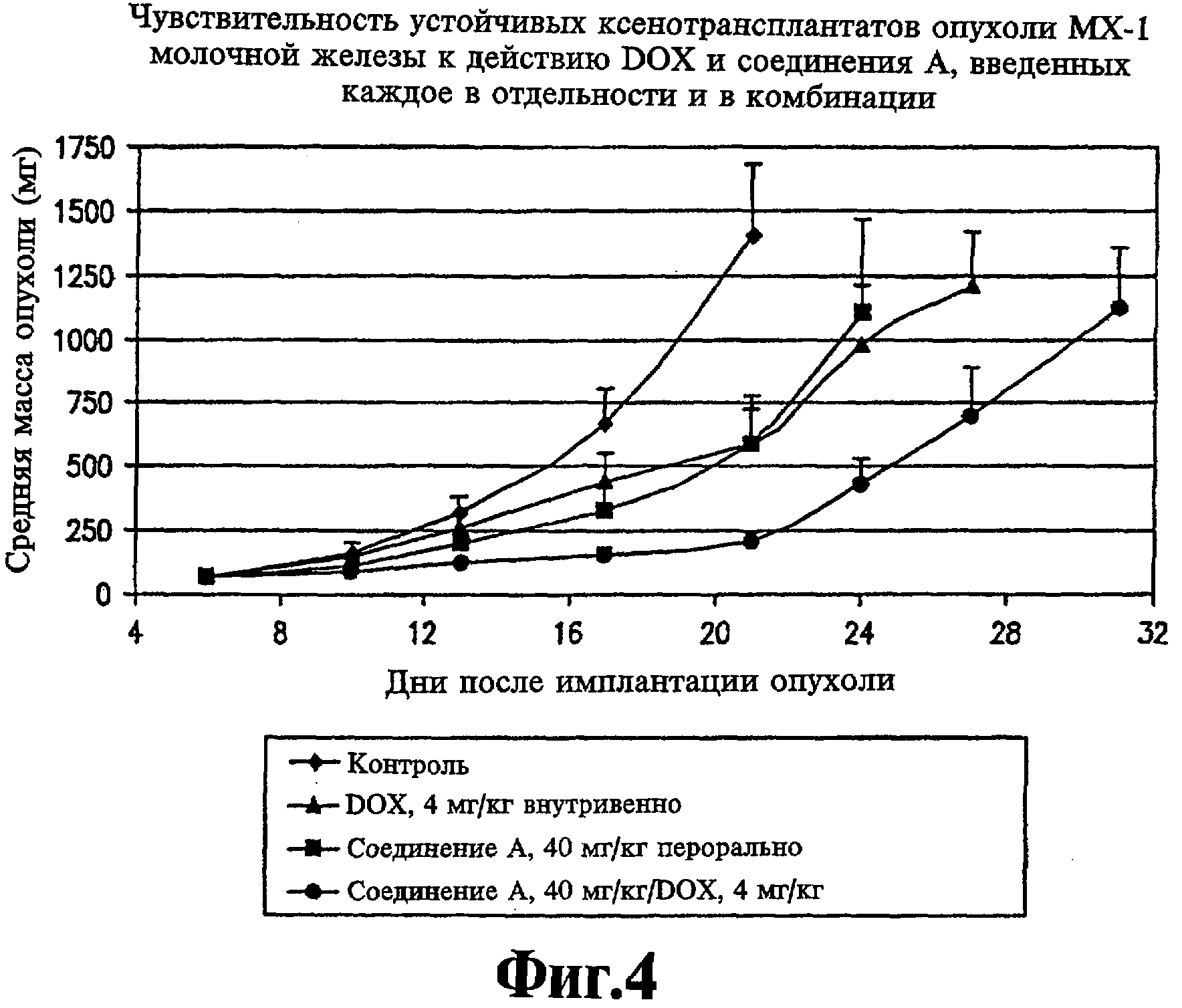
На фиг.4 представлена чувствительность устойчивых ксенотрансплантатов опухоли МХ-1 молочной железы человека к соединению А и DOX (доксорубицину), введенным каждое в отдельности и в комбинации.
На фиг.5 представлена чувствительность устойчивых ксенотрансплантатов опухоли А-549 немелкоклеточного рака легких к соединению А и гефинитибу, введенным каждое в отдельности и в комбинации.
Осуществление изобретения
Настоящее изобретение относится к комбинации, включающей производное арилмочевины в смеси по крайней мере с одним другим химиотерапевтическим (а) цитотоксическим агентом, или (б) цитостатическим агентом, или с фармацевтически приемлемыми солями любого компонента.
Другой аспект настоящего изобретения относится к комбинации цитотоксического или цитостатического агента и (1) замещенного мостикового производного арилмочевины или (2) замещенного мостикового производного арилмочевины, содержащего по крайней мере один фрагмент мостиковой арилмочевины с заместителем (заместителями) в другом (удаленном) кольце или (3) замещенного γ-карбоксамидом мостикового производного арилмочевины, или (4) соединения, или фармацевтически приемлемой соли соединения формулы I

В формуле I D представляет собой -NH-C(O)-NH-, A - замещенная структура, содержащая до 40 атомов углерода формулы -L-(M-L1 )q, где L представляет собой 5- или 6-членную циклическую структуру, связанную непосредственно с D, L1 содержит замещенную циклическую структуру, имеющую по меньшей мере 5 членов, М является мостиковой группой, содержащей по меньшей мере один атом, q - целое число от 1 до 3, и каждая циклическая структура в L и L1 содержит 0-4 члена группы, состоящей из азота, кислорода и серы, а В представляет собой замещенный или незамещенный арил или гетероарил, до трициклического арила или гетероарила, содержащего до 30 атомов углерода с по меньшей мере одной 6-членной циклической структурой, связанной непосредственно с D, содержащей 0-4 члена группы, состоящей из азота, кислорода и серы,
причем L1 является замещенным по меньшей мере одним заместителем, выбранным из группы, состоящей из -SO2Rx, -С(O)Rxи -C(NRy) Rz,
Ry - водород или структура на основе углерода, содержащая до 24 атомов углерода, необязательно содержащая гетероатомы, выбранные из N, S и О, и необязательно галогензамещенная вплоть до образования пергалоидного соединения,
Rz - водород или структура на основе углерода, содержащая до 30 атомов углерода, необязательно содержащая гетероатомы, выбранные из N, S и О, и необязательно замещенная галогеном, гидроксигруппой и заместителями на основе углерода, содержащими до 24 атомов углерода, которые необязательно содержат гетероатомы, выбранные из N, S и О, и необязательно являются замещенными галогеном;
Rx означает Rz или NRaRb, где Ra и Rb
а) независимо представлены водородом, структурой на основе углерода, содержащей до 30 атомов углерода, необязательно содержащей гетероатомы, выбранные из N, S и О, и необязательно замещенной галогеном, гидроксигруппой и заместителями на основе углерода, содержащими до 24 атомов углерода, которые необязательно содержат гетероатомы, выбранные из N, S и О, и необязательно замещены галогеном, или
-OSi(Rf)3, где Rf - водород или структура на основе углерода, содержащая до 24 атомов углерода, необязательно содержащая гетероатомы, выбранные из N, S и О, и необязательно замещенная галогеном, гидроксигруппой и заместителями на основе углерода, содержащим до 24 атомов углерода, которые необязательно содержат гетероатомы, выбранные из N, S и О, и необязательно являются замещенными галогеном, или
b) Ra и Rb вместе образуют 5-7-членную гетероциклическую структуру из 1-3 гетероатомов, выбранных из N, S и О, или замещенную 5-7-членную гетероциклическую структуру из 1-3 гетероатомов, выбранных из N, S и О, замещенную галогеном, гидроксигруппой или заместителями на основе углерода, содержащими до 24 атомов углерода, которые необязательно содержат гетероатомы, выбранные из N, S и О, необязательно замещенными галогеном, или
c) один из Ra или Rb, представляет собой -С(O)-, C1-C5 двухвалентную алкиленовую группу или замещенную C1-C5 двухвалентную алкиленовую группу, связанную со структурой L с образованием циклической структуры из по меньшей мере 5 членов, в которой заместители замещенной С1-С5двухвалентной алкиленовой группы выбраны из группы, состоящей из галогена, гидроксигруппы и заместителей на основе углерода, содержащих до 24 атомов углерода, которые необязательно содержат гетероатомы, выбранные из N, S и О, и являются необязательно замещенными галогеном;
где В является замещенным, L является замещенным или L1 является дополнительно замещенным, при этом заместители выбраны из группы, состоящей из галогена до образования пергалоидного соединения, и Wn, где n=0-3;
причем каждый W независимо выбран из группы, состоящей из -CN, -CO2R7, -C(O)NR7R7, -C(O)-R7, -NO2, -OR7, -SR7, -NR7 R7, -NR7C(O)OR7, -NR7C(O)R7, -Q-Ar и структур на основе углерода, содержащих до 24 атомов углерода, необязательно содержащих гетероатомы, выбранные из N, S и О, и необязательно замещенные одним или более заместителей, независимо выбранных из группы, состоящей из -CN, -CO2R7, -C(O)R7, -C(O)NR7 R7, -OR7, -SR7, -NR7R7, -NO2, -NR7C(O)R7, -NR7C(O)OR7 и галогена вплоть до образования пергалоидного соединения при том, что каждый из R7независимо выбран из Н или структуры на основе углерода, содержащей до 24 атомов углерода, необязательно содержащей гетероатомы, выбранные из N, S и О, и необязательно замещенной галогеном,
где Q представляет собой -О-, -S-, -N(R7)-, -(CH2)m-, -С(O)-, -СН(ОН)-, -(СН2)m О-, -(CH2)mS-, -(CH2)mN(R7)-, -O(CH2)m- CHXa-, -CXa2-, -S-(CH2)m- и -N(R7)(CH2)m-, где m=1-3, а Хa представляет собой галоген, и
Ar представляет собой 5- или 6-членную ароматическую структуру, содержащую 0-2 члена, выбранных из группы, состоящей из азота, кислорода и серы, которая является необязательно замещенной галогеном до образования пергалоидного соединения, и необязательно замещенной Zn1, где n1=0-3 и каждый Z независимо выбран из группы, состоящей из -CN, -CO2R7, -C(O)R7, -C(O)NR7R7, -NO2, -OR7 , -SR7, -NR7R7, -NR7C(O)OR7, -NR7C(O)R7 и структурой на основе углерода, содержащей до 24 атомов углерода, необязательно содержащей гетероатомы, выбранные из N, S и О, и необязательно замещенной одним или более заместителями, выбранными из группы, состоящей из -CN, -CO2R7, -COR7, -C(O)NR7R7, -OR7, -SR7, -NO2, -NR7R7, -NR7C(O)OR7 и -NR7C(O)OR7 при R7 таком, как определено выше.
В формуле I подходящие группы гетероарила включают, без ограничения перечисленным, ароматические циклы из 5-12 атомов углерода или циклические системы, содержащие 1-3 цикла, по меньшей мере один из которых является ароматическим, в котором один или более, например 1-4, атомов углерода в одном или более циклов возможно замещены атомами кислорода, азота или серы. Каждый цикл, как правило, содержит 3-7 атомов, например, В может быть 2- или 3-фурилом, 2- или 3-тиенилом, 2- или 4-триазинилом, 1-, 2- или 3-пирролилом, 1-, 2-, 4- или 5-имидазолилом, 1-, 3-, 4- или 5-пиразолилом, 2-, 4- или 5-оксазолилом, 3-, 4- или 5-изоксазолилом, 2-, 4- или 5-тиазолилом, 3-, 4- или 5-изотиазолилом, 2-, 3-или 4-пиридилом, 2-, 4-, 5- или 6-пиримидинилом, 1,2,3-триазол-1-, -4- или -5-илом, 1,2,4-триазол-1-, -3- или -5-илом, 1- или 5-тетразолилом, 1,2,3-оксадиазол-4- или -5-илом, 1,2,4-оксадиазол-3- или -5-илом, 1,3,4-тиадиазол-2- или -5-илом, 1,2,4-оксадиазол-3- или -5-илом, 1,3,4-тиадиазол-2- или -5-илом, 1,3,4-тиадиазол-3- или -5-илом, 1,2,3-тиадиазол-4- или -5-илом, 2-, 3-, 4-, 5- или 6-2Н-тиопиранилом, 2-, 3-или 4-4Н-тиопиранилом, 3- или 4-пиридазинилом, пиразинилом, 2-, 3-, 4-, 5-, 6- или 7-бензофурилом, 2-, 3-, 4-, 5-, 6- или 7-бензотиенилом, 1-, 2-, 3-, 4-, 5-, 6- или 7-индолилом, 1-, 2-, 4- или 5-бензимидазолилом, 1-, 3-, 4-, 5-, 6-или 7-бензопиразолилом, 2-, 4-, 5-, 6- или 7-бензоксазолилом, 3-, 4-, 5- 6- или 7-бензизоксазолилом, 1-, 3-, 4-, 5-, 6- или 7-бензотиазолилом, 2-, 4-, 5-, 6- или 7-бензизотиазолилом, 2-, 4-, 5-, 6- или 7-бенз-1,3-оксадиазолилом, 2-, 3-, 4-, 5-, 6-, 7- или 8-хинолинилом, 1-, 3-, 4-, 5-, 6-, 7-, 8- изохинолинилом, 1-, 2-, 3-, 4- или 9-карбазолилом, 1-, 2-, 3-, 4-, 5-, 6-, 7-, 8- или 9-акридинилом или 2-, 4-, 5-, 6-, 7- или 8-хиназолинилом или дополнительно необязательно замещенными фенилом, 2- или 3-тиенилом, 1,3,4-тиадиазолилом, 3-пиррилом, 3-пиразолилом, 2-тиазолилом или 5-тиазолилом и т.п. Например, В может быть 4-метил-фенилом, 6-метил-2-тиенилом, 4-метил-2-тиенилом, 1-метил-3-пиррилом, 1-метил-3-пиразолилом, 5-метил-2-тиазолилом или 5-метил-1,2, 4-тиадиазол-2-илом.
Подходящие алкильные группы или алкильные фрагменты групп, например алкоксигруппа и т.п., во всех случаях включают метил, этил, пропил, бутил и т.п., в том числе все изомеры с прямой цепью и разветвленные изомеры, такие как изопропил, изобутил, втор-бутил, трет-бутил и т.п.
Подходящие арильные группы, которые не содержат гетероатомы, включают, например, фенил и 1- и 2-нафтил.
Термин "циклоалкил", как используют в данном контексте, относится к циклическим структурам, содержащим или не содержащим алкильные заместители, так, например, "C4 циклоалкил" включает метил-замещенные группы циклопропила, а также группы циклобутила. Термин "циклоалкил", как используют в данном контексте, включает также насыщенные гетероциклические группы.
Подходящие галогеновые группы включают F, Cl, Br и/или I от одной до образования пергалоидного соединения (т.е. когда все атомы Н в группе замещены атомами галогена), что является возможным, когда алкильная группа замещена галогеном, смешанное замещение типами атомов галогенов также является возможным в данной структуре.
Изобретение относится также к соединениям формулы I, как таковым.
Изобретение относится также к фармацевтическому препарату, включающему (I) (а) производное арилмочевины, например соединения А (определенного ниже) и (б) по крайней мере один другой цитотоксический или цитостатический агент в количестве, которое обеспечивает совместную эффективность для лечения онкологического заболевания, причем любой компонент (а) или (б) может также присутствовать в форме фармацевтически приемлемой соли, если в соединении содержится по крайней мере одна солеобразующая группа, (2) с одним или более фармацевтически приемлемым носителем.
Изобретение относится также к способу лечения онкологического заболевания, которое можно лечить введением производного арилмочевины, мишенью которого является киназа raf, и по крайней мере одного другого химиотерапевтического агента, который является цитотоксическим или цитостатическим агентом. Производное арилмочевины и цитотоксический или цитостатический агент можно вводить млекопитающему в количестве, обеспечивающем совместное терапевтически эффективное действие на пролиферативные заболевания, включая, без ограничения перечисленным, онкологические заболевания ободочной кишки, желудка, легких, поджелудочной железы, яичников, предстательной железы, лейкоз, меланому, печеночно-клеточный рак, рак почек, головы и шеи, глиому и рак молочной железы. Таким образом, производное арилмочевины эффективно при лечении опосредованных киназой raf онкологических заболеваний. Кроме того, эти соединения эффективны также при лечении онкологических заболеваний, неопосредованных киназой raf.
В предпочтительном варианте воплощения настоящего изобретения цитотоксический или цитостатический агент по настоящему изобретению включает, без ограничения перечисленным, иринотекан, винорельбин, гемцитабин, гефинитиб, паклитаксель, таксотер, доксорубицин, цисплатин, карбоплатин, BCNU (кармустин), CCNU (ломустин), DTIC (дакарбазин), мелфалан, циклофосфамид, ара А, ара С, этопозид, винкристин, винбластин, актиномицин D, 5-фторурацил, метотрексат, герцептин и митомицин С.
В предпочтительном варианте настоящее изобретение относится к способу лечения онкологического заболевания у млекопитающих, прежде всего, человека, причем способ включает введение производного арилмочевины в комбинации с цитотоксическим или цитостатическим химиотерапевтическим агентом, включающим, без ограничения перечисленным, ингибиторы ДНК топоизомеразы I и II, интеркаляторы ДНК, алкилирующие агенты, деструкторы микротрубочек, агонисты или антагонисты рецепторов гормонов и ростовых факторов, другие ингибиторы киназы и антиметаболиты.
В более предпочтительном варианте настоящее изобретение относится к способу лечения онологического заболевания у млекопитающих, прежде всего, человека, причем способ влючает введение производного арилмочевины в комбинации с иринотеканом.
В другом предпочтительном варианте настоящее изобретение относится к способу лечения онкологического заболевания у млекопитающих, прежде всего, человека, причем способ влючает введение производного арилмочевины в комбинации с паклитакселем.
В еще одном предпочтительном варианте настоящее изобретение относится к способу лечения онкологического заболевания у млекопитающих, прежде всего, человека, причем способ включает введение производного арилмочевины в комбинации с винорельбином.
В другом предпочтительном варианте настоящее изобретение относится к способу лечения онкологического заболевания у млекопитающих, прежде всего, человека, причем способ влючает введение производного арилмочевины в комбинации с гефинитибом.
В еще одном предпочтительном варианте настоящее изобретение относится к способу лечения онкологического заболевания у млекопитающих, прежде всего, человека, причем способ влючает введение производного арилмочевины в комбинации с доксорубицином.
В другом предпочтительном варианте настоящее изобретение относится к способу лечения онкологического заболевания у млекопитающих, прежде всего, человека, причем способ влючает введение производного арилмочевины в комбинации с гемцитабином.
В еще одном варианте способы по настоящему изобретению можно использовать для лечения ряда онкологических заболеваний человека, включая, без ограничения перечисленным, онкологические заболевания поджелудочной железы, легких, ободочной кишки, яичников, предстательной железы, лейкоз, меланому, печеночно-клеточный рак, рак почек, головы и шеи, глиому и карциномы молочной железы.
В другом варианте воплощения настоящего изобретения описан способ для введения химиотерапевтических агентов, включающих производные арилмочевины и цитотоксические и цитостатические агенты, пациенту с использованием перорального приема или внутривенной инъекции или инфузии.
В еще одном варианте воплощения композицию, включающую производные арилмочевины и цитотоксический или цитостатический агент, можно вводить пациенту в форме таблетки, жидкости, геля для местного применения, ингалятора или в форме композиции с замедленным высвобождением.
В одном варианте воплощения изобретения производное арилмочевины можно вводить одновременно с цитотоксическим или цитостатическим агентом онкологическому пациенту в виде одного состава или в более типичном случае в виде отдельных составов и часто с использованием различных способов введения. Вводить составы можно также последовательно в любом порядке.
В предпочтительном варианте воплощения производное арилмочевины можно вводить в сочетании с цитотоксическим или цитостатическим агентом, причем производное арилмочевины можно вводить пациенту один или более раз в сутки в течение периода вплоть до 28 календарных дней, с одновременным или дробным введением цитотоксического или цитостатического агента в течение одного и того же общего периода лечения.
В другом предпочтительном варианте воплощения изобретения производные арилмочевины можно вводить пациенту в виде пероральной, внутривенной, внутримышечной, подкожной или парентеральной лекарственной формы в диапазоне от приблизительно 0,1 до приблизительно 300 мг/кг общей массы тела.
В еще одном предпочтительном варианте цитотоксический или цитостатический агент можно вводить пациенту в виде внутривенной, внутримышечной, подкожной или парентеральной лекарственной формы в диапазоне от приблизительно 0,1 до приблизительно 300 мг/кг общей массы тела.
В предпочтительном варианте воплощения изобретения производным арилмочевины является тозилат N-(4-хлор-3-(трифторметил)фенил)-N'-(4-(2-(N-метилкарбамоил)-4-пиридилокси)фенил)мочевины. Масштабируемый синтез производного арилмочевины описан в статье Organic Process Research and Development (2002), т.6, No6, стр.777-781 и в находящейся одновременно на рассмотрении заявке №09/948915, поданной 10 сентября 2001 г, включенных в данное описание в качестве ссылок.
Кроме того, настоящее изобретение относится к способу ингибирования пролиферации опухолевых клеток, включающему контактирование опухолевых клеток с фармацевтическим препаратом или продуктом по настоящему изобретению, прежде всего, к способу лечения пролиферативного заболевания, причем способ включает контактирование субъекта, клеток, тканей или жидкости организма упомянутого субъекта с предположительным диагнозом опухолевого заболевания с фармацевтической композицией или продуктом по настоящему изобретению.
Настоящее изобретение относится также к композициям, содержащим производное арилмочевины и другой цитотоксический или цитостатический агенты в количествах по настоящему изобретению. Кроме того, настоящее изобретение относится к наборам, включающим отдельные дозы двух упомянутых химиотерапевтических агентов в отдельных емкостях. Комбинации по настоящему изобретению могут также образоваться in vivo, например, в организме пациента.
Термин "цитотоксический" относится к агенту, который можно вводить, чтобы вызвать гибель или удаление опухолевых клеток. Термин "цитостатический" относится к агенту, который можно вводить для подавления пролиферации опухоли, а не для индукции цитотоксического снижения числа клеток, при этом происходит удаление опухолевых клеток из общей жизнеспособной популяции клеток пациента. Описанные выше химиотерапевтические агенты, например иринотекан, винорельбин, гемцитабин, доксорубицин и паклитаксель, относятся к цитотоксическим агентам. Гефинитиб относится к цитостатическим агентам. Такие цитотоксические и цитостатические агенты нашли широкое применение при использовании их в качестве химиотерапевтических средств для лечения различных типов онкологических заболеваний и являются широко известными средствами.
Иринотекан (СРТ) выпускается с торговым названием камптозар фирмой Pharmacia & Upjohn Co., Kalamazoo, MI. Иринотеканом называется камптотецин или ингибитор топоизомеразы I. Не углубляясь в теоретические подробности, можно предположить, что при блокировании указанного фермента в клетках происходит нарушение процессов клеточной репликации и таким образом контролируется рост опухоли. Предполагают, что цитотоксический эффект происходит за счет повреждения двойной спирали ДНК, которое индуцируется в процессе синтеза ДНК при взаимодействии репликационных ферментов с третичным комплексом, включающим топоизомеразу I, ДНК и иринотекан или SN-38 (его активный метаболит). Предполагают, что превращение иринотекана в SN-38 происходит в печени. Обычно иринотекан вводят инъекцией или внутривенной инфузией.
Винорельбин (тартрат винорельбина) характеризуется молекулярной формулой С45Н54N4O8 ·2С4Н6О6, молекулярной массой 1079,12, и выпускается с торговым названием навельбин фирмой Glaxo SmithKline, Research Triangle Park. Винорельбином является полусинтетический алкалоид из барвинка розового, обладающий противоопухолевой активностью. Химическое название - [соль R-(R,R)-2,3-дигидроксибутандиовой кислоты] и 3', 4'-дидегидро-4'-дезокси-С-норвинкалейкобластина (1:2). Не углубляясь в теоретические подробности, можно предположить, что противоопухолевая активность винорельбина проявляется в основном за счет ингибирования митоза на метафазной стадии благодаря его взаимодействию с тубулином. Винорельбин влияет 1) на процесс метаболизма аминокислот, цАМФ и глутатиона, 2) на зависимую от кальмодулина АТФазную активность, связанную с транспортом Са++, 3) на процесс дыхания клеток и 4) на процесс биосинтеза нуклеиновых кислот и липидов. Винорельбин обычно вводят внутривенной инъекцией (в/в) или другим пригодным способом инфузии. Винорельбин обычно готовят в нормальном солевом растворе, D5W или в других совместимых растворах.
Гемцитабин выпускается с торговым названием гемзар фирмы Eli Lilly & Co., Indianapolis, IN. Гемзар является антиметаболитом, родственным цитарабину. Гемзар назначают пациентам, прошедшим предварительный курс лечения 5-фторурацилом. Гемзар является пиримидиновым аналогом, который проявляет активность в широком диапазоне против солидных опухолей, включая, без ограничения перечисленным, карциномы молочной железы, яичников, поджелудочной железы и легких. Предполагают, что указанный агент включается в ДНК быстрорастущих раковых клеток, влияя на репликацию. Гемзар является нуклеиновым аналогом, который нарушает синтез ДНК в S-фазе клеточного цикла и блокирует развитие клеток на границе фаз G 1/S. Предполагают, что метаболизм гидрохлорида гемцитабина происходит при участии нуклеозидкиназ с образованием активных дифосфатной и трифосфатной форм, которые ингибируют рибонуклеотидредуктазу и которые конкурируют с ЦТФ за включение в ДНК, соответственно. Гемзар обычно вводят внутривенной инъекцией (в/в) или другим пригодным путем инфузии.
Гефинитиб выпускается с торговым названием иресса ZD 1839, фирмой Astra-Zeneca. Ирессой вляется 4-анилинохиназолин, причем предполагают, что этот агент ингибирует киназную активность регулятора эпидермального фактора роста (EGFR). Исследования механизма действия показали, что иресса является АТФ-конкурентным ингибитором EGFR и блокирует автофосфорилирование рецептора, стимулированного при связывании с EGF или αTGF. Показано, что иресса характеризуется пероральной биодоступностью и доклинической эффективностью против опухолевых моделей, в которых экспрессируется EGFR и один из его лигандов - αTGF. Показано также, что иресса ингибирует in vitro пролиферацию клеточных линий, которые характеризуются сверхэкспрессией EGFR или Her2. В клинических испытаниях ирессу вводят перорально по непрерывной суточной схеме в дозе вплоть до 800 мг/сут.
Доксорубицин (DOX) выпускается с торговым названием адриамицин фирмой Adria. Предполагают, что DOX - антрациклин, участвует в процессе интеркаляции ДНК и взаимодействует с ДНК топоизомеразой II, индуцируя разрывы в двойной спирали ДНК. DOX проявляет широкий спектр противоопухолевой активности. В клинике DOX вводят внутривенно по дробной схеме введения. В основном DOX выводится с помощью желчи, при этом не наблюдается энтерогепатической циркуляции. Ограничивающая дозу острая токсичность DOX связана с миелосупрессией. Другие известные, но обычно не ограничивающие дозу типы токсичности относятся к повреждению/изъязвлению желудочно-кишечного тракта, алопеции и местному повреждению/изъязвлению тканей в местах инъекции по причине проникновения лекарственного средства из сосудов в ткани.
Паклитаксель выпускается под торговым названием таксол фирмой Bristol-Myers Squibb Со. Паклитаксель - 4,10-диацетат2-бензоат-13-сложного эфира (5β,20-эпокси-1,2α,4,7β,10β,13β -гексагидрокси-такс-11-ен-9-она и (2R,3S)-N-бензоил-3-фенилизосерина, характеризуется молекулярной формулой С47Н53NO14 и молекулярной массой 853,9. Агент проявляет сильные липофильные свойства в воде. Паклитаксель является противомикротрубочковым агентом, который ускоряет сборку микротрубочек из димеров тубулина и стабилизирует микротрубочки, предотвращая деполимеризацию. Не вдаваясь в теоретические подробности, можно предположить, что такая стабилизация происходит благодаря ингибированию нормальной динамической реорганизации сети микротрубочек, которая является важной для обеспечения интерфазных и митозных функций живых клеток. Предполагают также, что паклитаксель индуцирует образование аномальной сети или пучков микротрубочек в процессе клеточного цикла и многочисленные звездообразные пучки микротрубочек в процессе митоза. Паклитаксель вводят внутривенной инъекцией или другим пригодным путем инфузии.
Указанные и другие цитотоксические/цитостатические агенты можно вводить в виде стандартных составов с использованием стандартных схем лечения, которые известны для применения каждого агента в отдельности.
Производное арилмочевины может ингибировать фермент - киназу raf. Кроме того, такие соединения могут ингибировать передачу сигнала с помощью рецепторов ростовых факторов. Такие соединения ранее описаны в заявке на выдачу патента, регистрационный номер 09/425228, поданной 26 октября 1999 г., которая полностью включена в данное описание в качестве ссылки.
Производные арилмочевины можно вводить пероральным, дермальным, парентеральным способами, инъекцией, ингаляцией или в виде спрея, сублингвальным, ректальным или вагинальным способами в виде стандартных составов для введения лекарственных форм. Термин "введение инъекцией" включает внутривенную, внутрисуставную, внутримышечную, подкожную и парентеральную инъекцию, а также использование методов инфузии. Дермальное введение может включать местное нанесение или чрескожное введение. Одно или более соединений могут присутствовать в смеси с одним или более нетоксичным, фармацевтически приемлемым носителем и при необходимости других активных ингредиентов.
Композиции, предназначенные для перорального применения, могут быть приготовлены в соответствии с любым подходящим способом, известным в области техники, которым получают фармацевтически композиции. Данные композиции могут содержать один или более агентов, выбранных из группы, состоящей из разбавителей, подсластителей, вкусовых добавок, окрашивающих агентов и консервирующих агентов с целью получения приятных на вкус препаратов. Таблетки содержат активный ингредиент в смеси с нетоксическими фармацевтически приемлемыми наполнителями, которые являются походящими для производства таблеток. Данные наполнители могут быть представлены, например, инертными разбавителями, такими как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; агентами, вызывающими грануляцию или дезинтеграцию, например, кукурузным крахмалом или альгиновой кислотой, и связующими агентами, например стеаратом магния, стеариновой кислотой или тальком. Таблетки могут быть непокрытыми или они могут быть покрыты с помощью известных технологий с целью задержки дезинтеграции и всасывания в желудочно-кишечном тракте и таким образом обеспечения задержанного действия в течения более длительного периода. Например, может быть использован такой задерживающий время материал, как глицерилмоностеарат или глицерилдистеарат. Данные соединения могут быть также приготовлены в твердой форме с быстрым освобождением.
Препараты для перорального применения могут быть также представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым наполнителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водой или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом.
Водные суспензии содержат активные материалы в смеси с наполнителями, пригодными для приготовления водных суспензий. Данные наполнители представляют собой суспендирующие агенты, например натриевую соль карбоксиметилцеллюлозы, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия, поливинилпирролидон, камедь трагаканта и камедь акации; диспергирующие или увлажняющие компоненты могут быть представлены природным фосфатидом, например лецитином, или продуктами конденсации алкиленоксида с жирными кислотами, например полиоксиэтилен стеаратом, или продуктами конденсации этиленоксида с алифатическими спиртами с длинной цепью, например гептадекаэтилен оксицетанолом, или продуктами конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот, и гекситола, такими как полиоксиэтиленсорбитол моноолеат, или продуктами конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот, ингидридами гекситола, например полиэтиленсорбитан моноолеатом. Водные суспензии могут также содержать один или более консервирующих агентов, например этил n-пропил р-гидроксибензоат, один или более окрашивающих агентов, одну или более вкусовых добавок и один или более подсластителей, таких как сахароза или сахарин.
В диспергирующихся порошках и гранулах, подходящих для приготовления водной суспензии путем добавления воды, активный ингредиент представлен в смеси с диспергирующими или увлажняющими агентами и суспендирующими агентами, примеры которых уже приведены выше. Могут также присутствовать дополнительные наполнители, например подсластители, вкусовые добавки и окрашивающие агенты.
Соединения могут также быть в форме неводных жидких препаратов, например масляных суспензий, которые можно приготовить суспендированием активных ингредиентов в растительном масле, например арахисовом масле, оливковом масле, кунжутном масле или арахисовом?? масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загущающий компонент, например пчелиный воск, твердый парафин или цетиловый спирт. Подсластители, такие как приведенные выше, и вкусовые добавки могут быть добавлены для получения приятных на вкус пероральных препаратов. Данные композиции могут быть законсервированы добавлением антиоксиданта, такого как аскорбиновая кислота.
Фармацевтические композиции, соответствующие изобретению, могут быть также в форме эмульсий масло в воде. Масляная фаза может быть представлена растительным маслом, например оливковым маслом или арахисовым маслом, или минеральным маслом, например жидким парафином, или их смесями. Подходящие эмульгирующие агенты могут быть представлены природными камедями, например камедью акации или камедью трагаканта, природными фосфатидами, например соевым лецитином, и сложными эфирами или частичными сложными эфирами, полученными из жирных кислот и ангидридом гекситола, например сорбитан моноолеатом, и продуктами конденсации данных частичных сложных эфиров, например полиоксиэтиленсорбитан моноолеатом. Эмульсии могут также содержать подсластители и вкусовые добавки.
Сиропы и эликсиры могут быть приготовлены с подсластителями, например глицерином, пропиленгликолем, сорбитом или сахарозой. Данные препараты могут также содержать деэмульгатор, консервант и вкусовую добавку и окрашивающий агент.
Соединения могут также применяться в форме суппозиториев для ректального введения лекарственного вещества. Данные композиции можно приготовить путем смешивания лекарственного вещества с подходящим нераздражающим наполнителем, который является твердым при обычных температурах, но жидким при ректальной температуре и будет вследствие этого плавиться в прямой кишке с освобождением лекарственного вещества. Данные материалы включают масло какао и полиэтиленгликоли.
Соединения по настоящему изобретению можно также вводить чрескожным способом с использованием способов, известных в данной области техники (см., например, статью Chien "Transdermal Controlled Systemic medications", Marcel Dekker, Inc., 1987 и заявку Lipp et al. WO 94/04157 от 3 марта 1994 г.). Например, раствор или суспензия производного арилмочевины в подходящем летучем растворителе, по выбору содержащем агенты для ускорения проникновения, можно смешивать с другими добавками, известными в данной области техники, такими как матричные материалы и бактериоциды. После стерилизации полученную смесь можно перерабатывать в лекарственные формы с использованием известных способов. Кроме того, после обработки эмульгирующими агентами и водой раствор или суспензию производного арилмочевины можно перерабатывать в лосьон или мазь.
Подходящие растворители для получения чрескожных систем доставки известны в данной области техники и включают диметилсульфоксид, низшие спирты, такие как этанол или изопропиловый спирт, низшие кетоны, такие как ацетон, сложные эфиры низших карбоновых кислот, такие как этилацетат, полярные простые эфиры, такие как тетрагидрофуран, низшие углеводороды, такие как гексан, циклогексан или бензол, или галогенированные углеводороды, такие как дихлорметан, хлороформ, трихлортрифторэтан или трихлорфторметан. Подходящие растворители могут также включать смеси одного или более материалов, выбранных из ряда: низшие спирты, низшие кетоны, сложные эфиры низших карбоновых кислот, полярные простые эфиры, низшие углеводороды, галогенированные углеводороды.
Подходящие материалы, ускоряющие проникновение в чрескожных системах доставки, известны в данной области техники и включают, например, одноатомные или многоатомные спирты, такие как этанол, полиэтиленгликоль или бензиловый спирт, насыщенные или ненасыщенные С8-С18жирные спирты, такие как лауриловый спирт или цетиловый спирт, насыщенные или ненасыщенные С8-С18жирные кислоты, такие как стеариновая кислота, насыщенные или ненасыщенные жирные сложные эфиры, содержащие вплоть до 24 атомов углерода, такие как метиловые, этиловые, пропиловые, изопропиловые, н-бутиловые, втор-бутиловые, изобутиловые, трет-бутиловые или моноглицериновые эфиры уксусной кислоты, капроновой кислоты, лауриновой кислоты, миристиновой кислоты, стеариновой кислоты или пальмитиновой кислоты, или диэфиры насыщенных или ненасыщенных дикарбоновых кислот, содержащих вплоть до 24 атомов углерода, такие как диизопропиладипат, диизобутиладипат, диизопропилсебацинат, диизопропилмалеат или диизопропилфумарат. Дополнительные материалы, ускоряющие проникновение, включают фосфатидилпроизводные, такие как лецитин или цефалин, тепены, амиды, кетоны, мочевины и их производные, и простые эфиры, такие как диметилизосорбид и моноэтиловый эфир диэтиленгликоля.
Подходящие составы, ускоряющие проникновение, могут также включать смеси одного или более материалов, выбранные из ряда: одноатомные или многоатомные спирты, насыщенные или ненасыщенные С8 -С18жирные спирты, насыщенные или ненасыщенные С8-С18жирные кислоты, насыщенные или ненасыщенные жирные сложные эфиры, содержащие вплоть до 24 атомов углерода, диэфиры насыщенных или ненасыщенных дикарбоновых кислот, содержащие вплоть до 24 атомов углерода, фосфатидилпроизводные, терпены, амиды, кетоны, мочевины и их производные, а также простые эфиры.
Подходящие связующие материалы для чрескожных систем доставки известны в данной области техники и включают полиакрилаты, силиконы, полиуретаны, блокполимеры, сополимеры стирола и бутадиена, а также природные и синтетические каучуки. В качестве матричных компонентов можно также использовать простые эфиры целлюлозы, производные полиэтиленов и силикатов. Для повышения вязкости матрицы можно включать другие добавки, такие как вязкие смолы или масла.
Настоящее изобретение включает также наборы для лечения онкологических заболеваний млекопитающих. Такие наборы можно использовать для лечения пациента, страдающего от стимулированного киназой raf онкологического заболевания, а также для лечения онкологических заболеваний, не стимулированных киназой raf. Набор может включать единый фармацевтический состав, содержащий производное арилмочевины и цитотоксический или цитостатический агент. В другом варианте набор может содержать производное арилмочевины и цитотоксический или цитостатический агент в отдельных составах. Набор может также содержать инструкции по введению соединений онкологическому пациенту, нуждающемуся в лечении. Набор можно также использовать для лечения онкологических заболеваний различного типа, которые включают, без ограничения перечисленным, рак ободочной кишки, предстательной железы, лейкоз, меланому, печеночно-клеточный рак, рак почек, головы и шеи, глиому, рак легких, поджелудочной железы, яичников и молочной железы.
Для специалиста в данной области техники представляется очевидным, что конкретный способ введения зависит от ряда факторов, которые обычно учитывают при введении терапевтических средств. Однако следует также понимать, что конкретный уровень дозы для каждого пациента зависит от множества факторов, включающих активность используемого конкретного соединения, возраст пациента, массу тела пациента, общее состояние здоровья пациента, пол пациента, диета пациента, время введения, путь введения, степень выведения лекарственного средства, комбинация лекарственных средств и тяжесть патологического состояния, которое подвергается лечению. Кроме того, для специалиста в данной области техники представляется очевидным, что оптимальный курс лечения, то есть режим лечения и суточное число доз производного арилмочевины или его фармацевтически приемлемой соли, введенных в течение определенного количества дней, определяется врачом с использованием стандартных методов анализа.
Применение комбинации производных арилмочевины и цитотоксических или цитостатических агентов является более эффективным по сравнению с известным действием каждого противоопухолевого агента в отдельности. Например, при комбинационной терапии с использованием производных арилмочевины и цитотоксических агентов иринотекана, гемцитабина, винорельбина или паклитакселя достигается по крайней мере аддитивная противоопухолевая эффективность по сравнению с введением либо производного арилмочевины, либо цитотоксических агентов в отдельности. В основном применение цитотоксических и/или цитостатических агентов в комбинации с ингибиторами киназы raf - производными арилмочевины, обеспечивает: (1) улучшенную эффективность при снижении роста опухоли или даже устранение опухоли по сравнению с другим химиотерапевтическим агентом, введенным в отдельности, (2) введение меньших количеств химиотерапевтических агентов, (3) химиотерапевтическое лечение, которое характеризуется достаточно высокой переносимостью у пациентов и снижением отрицательных фармакологических побочных действий, наблюдаемых при введении более высоких доз одного химиотерапевтического агента или при использовании других видов комбинационной терапии, (4) расширение спектра различных поддающихся терапии типов рака, для лечения млекопитающих, прежде всего, человека, (5) более высокий уровень ответной реакции пациентов, подвергающихся лечению, (6) увеличение продолжительности жизни у пациентов, подвергающихся лечению, по сравнению с пациентами, проходящими курс лечения стандартной химиотерапией, (7) увеличение продолжительности опухолевой прогрессии и/или (8) по крайней мере сопоставимые эффективность и переносимость по сравнению с агентами, введенными в отдельности и по сравнению с известными случаями, в которых комбинация других агентов приводит к антагонистическому действию.
Производное арилмочевины можно вводить пациенту в дозе в диапазоне от приблизительно 0,1 до приблизительно 300 мг/кг общей массы тела. Суточная доза для перорального введения составляет предпочтительно от 0,1 до 300 мг/кг общей массы тела. Суточная доза для введения инъекцией, которая включает внутривенную, внутримышечную, подкожную и парентеральную инъекцию, а также способы инфузии, составляет предпочтительно от 0,1 до 300 мг/кг общей массы тела. Суточная вагинальная доза предпочтительно составляет от 0,1 до 300 мг/кг общей массы тела. Суточная местная доза предпочтительно составляет от приблизительно 0,1 до приблизительно 300 мг/кг общей массы тела, причем дозу вводят от 1 до 4 раз в сутки. При чрескожном введении требуется поддерживать концентрацию, которая должна обеспечиваться суточной дозой от 1 до 300 мг/кг. Для всех перечисленных выше способов введения предпочтительная доза составляет от 0,1 до 300 мг/кг. Суточная доза при введении ингаляцией предпочтительно составляет от 0,1 до 300 мг/кг общей массы тела.
Цитотоксический или цитостатический агент можно вводить пациенту в дозе в диапазоне от приблизительно 0,1 до приблизительно 300 мг/кг общей массы тела. Агенты можно также вводить с использованием стандартных способов, используемых в химиотерапии онкологических заболеваний.
Как в случае производного арилмочевины, так и в случае цитотоксического или цитостатического агента введенная доза может быть изменена в зависимости от любых более эффективных или неожиданных результатов, которые могут наблюдаться в описанных в данном изобретении условиях.
Производные арилмочевины можно вводить пероральным, местным, парентеральным, ректальным способами, ингаляцией или инъекцией. Введение инъекцией включает внутривенный, внутримышечный, подкожный и парентеральный способы, а также способы инфузии. Производное арилмочевины может присутствовать в смеси с одним или более нетоксичных фармацевтически приемлемых носителей и при необходимости других активных компонентов. Предпочтительным способом введения производного арилмочевины является пероральное введение.
Цитотоксический или цитостатический агент можно вводить пероральным, местным, парентеральным, ректальным способами, ингаляцией или инъекцией. Введение инъекцией включает внутривенный, внутримышечный, подкожный и парентеральный способы, а также способы инфузии. Агенты можно вводить любым стандартным способом, известным для введения таких соединений. Предпочтительным способом введения цитотоксического/цитостатического агентов по настоящему изобретению обычно является тот же способ инъекции, который известен для введения агента в отдельности. Любой цитотоксический или цитостатический агент можно вводить в комбинации в производным арилмочевины любым из упомянутых способов ведения.
Для введения производных арилмочевины и цитотоксического/ цитостатического агента любым из описанных выше способов введения производное арилмочевины можно вводить одновременно с цитотоксическим или цитостатическим агентом. При этом пациенту вводят единый состав, содержащий производное арилмочевины и цитотоксический/цитостатический агент, или вводят пациенту производное арилмочевины и цитотоксический/ цитостатический агенты в отдельных составах в один и тот же момент времени.
В другом варианте производное арилмочевины можно вводить последовательно с цитотоксическим/цитостатическим агентом. Производное арилмочевины можно вводить до введения цитотоксического/ цитостатического агента. Например, производное арилмочевины можно вводить один или более раз в сутки в течение периода вплоть до 28 календарных дней с последующим введением цитотоксического/ цитостатического агента. Кроме того, цитотоксический/цитостатический агент можно вводить первым с последующим введением производного арилмочевины. Выбор порядка введения производного арилмочевины относительно цитотоксического/цитостатического агента может зависеть от свойств различных агентов. Цитотоксический или цитостатический агент можно также вводить с использованием любой схемы лечения, обычно используемой для таких агентов.
В других схемах лечения производное арилмочевины и цитотоксический/цитостатический агент можно вводить один или более раз в сутки в день введения.
Любые из способов и схем лечения могут быть изменены в зависимости от более эффективных или неожиданных результатов, которые можно наблюдать в определенных в данном изобретении условиях.
Предполагается, что специалист в данной области техники может использовать данное изобретение на практике в полном объеме с использованием представленного выше описания. Следовательно, следующие предпочтительные варианты воплощения изобретения приведены только для его иллюстрации и ни в коем случае не ограничивают объема изобретения.
Во всех фрагментах описания и следующих примерах, если не указано иное, все величины температуры приведены в градусах Цельсия, а весовые части и проценты в мас.%.
В экспериментах, описанных ниже в примерах, использовали производное арилмочевины (соединение А): тозилат N-(4-хлор-3-(трифторметил)фенил)-N'-(4-(2-(N-метилкарбамоил)-4-пиридилокси)-фенил)мочевины.
Примеры
Животные
Во всех испытаниях in vivo использовали самок мышей Ncr nu/nu (фирмы Taconic Farms, Germantown, Нью-Йорк), включая модели опухолей DLD-1 и NCl-H460. Для испытаний используют самок мышей СВ-17 SCID (фирмы Taconic Farms, Germantown, Нью-Йорк), включая модель опухоли Mia-РаСа-2. Мышей выращивают и выдерживают в виварии фирмы Bayer Co., отделение сравнительной медицины, West-Haven, CT, согласно инструкциям IACUC фирмы Bayer и рекомендациям государственных и федеральных служб по обращению и уходу персонала за лабораторными животными. Мыши получали корм и воду по желанию.
Соединения
Во всех экспериментах используют соединение А (партия 9910071). Соединение А представляет сухой порошок, цвет изменяется от белого до цвета слоновой кости или светло-желтого. Соединение А хранят в темноте вплоть до использования.
Камптозар (партия 09FDY и партия 27FMR) получают на фирме Pharmacia-Upjohn в виде раствора с концентрацией 20 мг/мл. Раствор хранят при комнатной температуре, как указано в инструкции по применению.
Гемзар (гидрохлорид гемцитабина) получают на фирме Eli Lilly and Co. в виде сухого порошка, который хранят при комнатной температуре, как указано в инструкции по применению.
Навельбин (тартрат винорельбина) получают на фирме Glaxo Wellcome в виде раствора с концентрацией 10 мг/мл. Раствор хранят при 4°С, как указано в инструкции по применению.
DOX (гидрохлорид доксорубицина) получают на фирме Bedford Laboratories (партия 110033) в виде лиофилизированного порошка красного/оранжевого цвета, который хранят при 4°С и защищают от света.
Гефинитиб (ZD1839) (4-(3-хлор-4-фторанилино)-7-метокси-6-(3-морфолинопропокси)хиназолин был синтезирован на фирме Albany Medical Research (Syracuse, Нью-Йорк). ZD1839 хранят в темноте при комнатной температуре вплоть до использования.
Наполнители
Кремофор EL/этанол (50:50) (Sigma Cremophor EL, номер по каталогу С-5135, 500 г, 95%-ный этиловый спирт) получают в виде исходного раствора, флакон с которым заворачивают в алюминиевую фольгу и хранят при комнатной температуре. Готовят 4-кратный концентрированный раствор соединения А (4х) (самая высокая доза) в указанном растворе кремофора EL в этаноле (50:50). Свежий раствор 4х готовят каждые 3 дня. Конечные растворы доз готовят в день использования разбавлением до концентрации 1х дистиллированной водой, не содержащей эндотоксины (фирмы Gibco, номер по каталогу 15230-147), и перед введением дозы перемешивают на мешалке типа Вортекс. Низкие дозы получают разбавлением до концентрации 1х раствором кремофор EL/этанол/вода (12,5:12, 5:75). В качестве носителя для камптозара и гемзара используют 0,9% солевой раствор, а в качестве носителя для навельбина используют D5W. Все носители и растворы соединений хранят при комнатной температуре и флаконы с растворами заворачивают в фольгу.
Линии опухолевых клеток
Карциному LDL-1 ободочной кишки человека и карциному MiaPaCa-2 поджелудочной железы человека получают в американском банке культуральных тканей American Type Tissue Culture Collection Repository. Опухоль МХ-1 молочной железы человека получают в банке опухолей NCl. Опухоли культивируют путем серийных пассажей in vivo подкожных фрагментов (3×3 мм), трансплантированных в бок мыши с использованием калибровочного троакара 12. Новую серию пассажей проводят каждые три или четыре недели.
Линии NCI-H460 и А549 немелкоклеточной карциномы легких человека получают в американском банке культур тканей American Type Tissue Culture Collection Repository. Клетки NCI-H460 культивируют и проводят пассажи in vitro с использованием среды DMEM (фирмы Gibco, номер по каталогу 11995-065:500 мл), дополненной следующими компонентами: 10% инактивированная нагреванием фетальная сыворотка крупного рогатого скота (фирмы JRH Biosciences, номер по каталогу 12106-500М), 2 мМ L-глютамин (фирмы Gibco, номер по каталогу 25030-81), 10 мМ буферный раствор HEPES (фирмы Gibco, номер по каталогу 15630-080) и пенициллин-стрептомицин (фирмы Gibco, номер по каталогу 15140-122:5 мл/50 мл DMEM). Клетки А549 культивируют и проводят пассажи с использованием среды RPMI (фирмы Gibco, номер по каталогу 11875-085:1000 мл), дополненной 10% инактивированной нагреванием фетальной сывороткой крупного рогатого скота (фирмы JRH Biosciences, номер по каталогу 12106-500М). Все клетки культивируют при 37°С и 5% CO2 в клеточном инкубаторе "Fisher Scientific 610 CO2 incubator".
Эксперименты с опухолевыми ксенотрансплантатами
Самкам мышей трансплантируют подкожно опухолевые фрагменты DLD-1, МХ-1 или Mia-PaCa-2 из пассажей in vivo. Эксперименты с клетками NCI-Н460 и А549 проводят после сбора клеток из культуры in vitro добавлением смеси трипсина-ЭДТА (фирмы Gibco, номер по каталогу 25200-056) в течение 2 мин с последующим центрифугированием клеток и суспендированием полученного осадка в HBSS (фирмы Gibco, номер по каталогу 14025-092) до конечной концентрации клеток 3-5×107 живых клеток/мл. В правый бок каждой мыши вводят подкожной инъекцией по 0,1 мл клеточной суспензии. Все эксперименты по лечению начинают после того, как все экспериментальные мыши имели устойчивые опухоли размером в диапазоне от 100 до 150 мг. Затем один раз в сутки регистрируют общее состояние здоровья и смертность мышей. Регистрируют размеры опухоли и массу тела два раза в неделю, начиная с первого дня лечения. Животных забивают по рекомендациям фирмы Bayer IACUC. Случаи лечения, при которых наблюдалось более 20% летальности и/или более 20% потери массы тела, регистрируют как "токсичные".
Массу опухоли рассчитывают по уравнению (I×w2)/2, где I и w означают большой и малый размеры, определенные при каждом измерении. Время завершения каждого эксперимента определяют как среднее время, в течение которого опухоли в контрольной группе достигали указанного размера, которое незначительно больше, чем продолжительность эксперимента. Противоопухолевую эффективность определяют с помощью следующих параметров: полная регрессия (ПР), то есть случаи, когда размеры опухоли составляют величины ниже предела измерения как по длине (3 мм), так и по ширине; частичная регрессия (ЧР), то есть случаи, когда размеры опухоли снижаются на более 50%, но на менее 100% от исходного размера; и процент подавления роста опухоли (% ПРО). Величину ПРО рассчитывают по уравнению [(Т-С)/С]×100, где Т и С означают периоды времени, в течение которых средние опухоли в леченой (Т) и контрольной (С) группах соответственно достигают размеров, необходимых для оценки в данном эксперименте.
Результаты
Комбинация соединения А и цитотоксических/цитостатических агентов
Наиболее интенсивная комбинированная химиотерапия, предписываемая для клинического использования соединения А для лечения онкологических заболеваний, включает ежедневное введение соединения А, которое вводили одновременно с дробным введением цитотоксических/цитостатических агентов, таких, например, как камптозар, гемзар, навельбин или DOX, в соответствии с современной клинической практикой для каждого из указанных агентов. Для исследования возможных взаимодействий таких агентов авторами предложена данная модель схем введения агентов в доклинических испытаниях, которая заключается в наложении схем введения индивидуальных агентов (qd×9 для соединения А и q4d×3 для камптозара, гемзара, навельбина или DOX) с обоими типами терапии в каждом эксперименте, начиная указанные схемы в один и тот же день. Другая схема комбинированной химиотерапии заключается в ежедневном введении соединения А одновременно с непрерывным введением цитостатических агентов, таких как иресса. Для исследования возможных взаимодействий таких агентов авторами предложена модель схем введения агентов в доклинических испытаниях, которая заключается в наложении схем введения индивидуальных агентов (qd×9 или 10 как для соединения А, так и для ирессы). Такие схемы названы "параллельная терапия". В каждый эксперимент по лечению включены контрольная группа из 10-20 животных, которые не проходят курс лечения, и группы, проходящие курс лечения, содержащие по 10 мышей в каждой группе.
Пример 1
В первом эксперименте камптозар вводят внутрибрюшинно в дозе 40 мг/кг. Соединение А вводят перорально по схеме qd×9 в дозе 80 мг/кг. Все курсы лечения начинают в день 7 после трансплантации опухоли, когда у всех животных образуются небольшие по размеру, но устойчивые ксенотрансплантаты опухоли DLD-1 ободочной кишки человека со средним размером 108 мг. Контрольные опухоли прогрессируют у всех животных, причем среднее время удвоения размеров опухоли составляет 4,4 сут. Время завершения эксперимента для расчета параметров снижения роста опухоли определяют как время, в течение которого увеличение роста опухоли составляет три удвоения массы. Среднее время, в течение которого опухоли в контрольной группе достигают указанного размера, составляет 10,4 сут.
При введении камптозара в отдельности наблюдается удовлетворительная переносимость при минимальной потере массы тела и отсутствии летальности. При уровне дозы 40 мг/кг ПРО составляет 71% и не наблюдается полной или частичной регрессии опухоли.
При введении соединения А в отдельности в дозе 80 мг/кг также наблюдается удовлетворительная переносимость при отсутствии значительной потери массы тела и летальности. ПРО для соединения А составляет 100%.
При введении комбинации камптозара с соединением А не наблюдается увеличение степени потери массы тела и отсутствует летальность. Противоопухолевая активность при параллельной терапии составляет по крайней мере аддитивную величину, то есть ПРО равно 229%, что связано с 3 ЧР.
Пример 2
Во втором эксперименте исследовали действие гемзара, введенного внутрибрюшинно в дозе 120 мг/кг по схеме q4d×3, и действие соединения А, введенного перорально по схеме qd×9 в дозе 40 мг/кг. Все эксперименты по лечению начинали в день 7 после трансплантации опухоли, когда у всех животных образуются небольшие по размеру, но устойчивые ксенотрансплантаты опухоли MiaPaCa поджелудочной железы человека со средним размером 108 мг. Контрольные опухоли прогрессируют у всех животных, причем среднее время удвоения размеров опухоли составляет 4,1 сут. Время завершения эксперимента для расчета параметров снижения роста опухоли определяют как время, в течение которого увеличение роста опухоли составляет два удвоения массы. Среднее время, в течение которого опухоли в контрольной группе достигают указанного размера, составляет 5,8 сут.
При введении гемзара в отдельности наблюдается удовлетворительная переносимость при отсутствии потери массы тела и летальности. При данном уровне дозы ПРО составляет 154% и не наблюдается полной или частичной регрессии опухоли. При введении соединения А в отдельности в дозе 80 мг/кг также наблюдается удовлетворительная переносимость при отсутствии значительной потери массы тела и летальности. ПРО для соединения А составляет 112%. При введении комбинации гемзара с соединением А не наблюдается увеличение степени потери массы тела и отсутствует летальность. Противоопухолевая активность при параллельной терапии в дозе гемзара 120 мг/кг и соединения А 40 мг/кг составляет по крайней мере аддитивную величину, то есть ПРО равно 222%, что связано с 2 ЧР.
Пример 3
В третьем эксперименте исследовали действие комбинации соединения А, введенного перорально по схеме qd×9 в дозе 40 мг/кг, и действие навельбина, введенного внутривенно в дозе 6,7 мг/кг по схеме q4d×3. Все эксперименты по лечению начинали в день 6 после трансплантации опухоли, когда у всех животных образуются небольшие по размеру, но устойчивые ксенотрансплантаты опухоли NCl-H460 немелкоклеточного рака легких человека со средним размером 100 мг. Контрольные опухоли прогрессируют у всех животных, причем среднее время удвоения размеров опухоли составляет 3,1 сут. Время завершения эксперимента для расчета параметров снижения роста опухоли определяют как время, в течение которого увеличение роста опухоли составляет три удвоения массы. Среднее время, в течение которого опухоли в контрольной группе, не подвергавшейся лечению, достигают указанного размера, составляет 7,4 сут. При введении навельбина в дозе 6,7 мг/кг (максимальная переносимая доза) наблюдается потеря массы тела в среднем на 19% в течение периода лечения данным агентом в отдельности. При этом ПРО составляет 32%. Соединение А также проявляет удовлетворительную переносимость при отсутствии значительной потери массы тела, ПРО составляет 104%. При введении указанной комбинации наблюдается удовлетворительная переносимость, отсутствие летальности и средняя потеря массы тела 14% (меньше величины, наблюдаемой при введении навельбина в отдельности). Противоопухолевая активность при введении указанной комбинации также составляет аддитивную величину, при этом ПРО равно 133%.
Пример 4
В четвертом эксперименте исследовали действие комбинации соединения А, введенного перорально по схеме qd×9 в дозе 40 мг/кг, и DOX, введенного внутривенно в дозе 4 мг/кг по схеме q4d×3. Все эксперименты по лечению начинали в день 6 после трансплантации опухоли, когда у всех животных образуются небольшие по размеру, но устойчивые опухоли со средним размером 66 мг. Контрольные опухоли прогрессируют у всех животных, причем среднее время удвоения размеров опухоли составляет 3,7 сут. Время завершения эксперимента для расчета параметров снижения роста опухоли определяют как время, в течение которого увеличение роста опухоли составляет четыре удвоения массы. Среднее время, в течение которого опухоли в контрольной группе, не подвергавшейся лечению, достигают указанного размера, составляет 14,5 сут. При введении DOX в дозе 4 мг/кг наблюдается удовлетворительная переносимость при потере массы тела в среднем на 5% в течение периода лечения данным агентом в отдельности. При этом ПРО составляет 43%. Соединение А также проявляет удовлетворительную переносимость при отсутствии значительной потери массы тела, ПРО составляет 46%. При введении указанной комбинации наблюдается удовлетворительная переносимость, отсутствие летальности и средняя потеря массы тела 12%. Противоопухолевая активность при введении указанной комбинации также составляет аддитивную величину, при этом ПРО равно 133%.
Пример 5
В пятом эксперименте исследовали действие комбинации соединения А, введенного перорально по схеме qd×9 в дозе 80 мг/кг, и гефинитиба (ирессы), введенного перорально в дозе 150 мг/кг по схеме qd×9. Все эксперименты по лечению начинали в день 15 после трансплантации опухоли, когда у всех животных образуются небольшие по размеру, но устойчивые ксенотрансплантаты опухоли А549 немелкоклеточного рака легких человека со средним размером 110 мг. Контрольные опухоли прогрессируют у всех животных, причем среднее время удвоения размеров опухоли составляет 10,5 сут. Время завершения эксперимента для расчета параметров снижения роста опухоли определяют как время, в течение которого увеличение роста опухоли составляет одно удвоение массы. При введении ирессы в дозе 150 мг/кг наблюдается удовлетворительная переносимость, отсутствие потери массы тела и летальности в течение периода лечения данным агентом в отдельности. При этом ПРО составляет 110%, что связано с 1 ЧР. Соединение А, введенное в отдельности, также проявляет удовлетворительную переносимость при отсутствии значительной потери массы тела или летальности, ПРО составляет 218%, что связано с 1 ПР и 2 ЧР. При введении указанной комбинации наблюдается один случай неспецифичного летального исхода в группе из 10 мышей и средняя потеря массы тела 10%. Противоопухолевая активность при введении указанной комбинации также составляет аддитивную величину, при этом ПРО равно 314%, что связано с 6 ПР и 3 ЧР.
Представленные выше примеры можно воспроизвести с аналогичными результатами при замене описанных в общем виде или конкретных реагентов и/или рабочих условий по настоящему изобретению на реагенты и условия, представленные в описанных выше примерах.
При ознакомлении с представленным ниже описанием изобретения специалист в данной области техники может провести различные изменения и модификации изобретения с целью его использования в различных областях применения и условиях, не выходя за пределы объема и сущности настоящего изобретения.
Реферат
Настоящее изобретение относится к производным арилмочевины, предназначенным для использования при лечении опосредованных киназой raf заболеваний, таких как раковые заболевания. Композиция для лечения ракового заболевания содержит терапевтически эффективное количество ингибитора киназы raf, которым является фармацевтически приемлемая соль N-(4-хлор-3-(трифторметил)фенил-N'-(4-(2-(N-метилкарбамоил)-4-пиридилокси)фенил)мочевины, в частности тозилат N-(4-хлор-3-(трифторметил)фенил-N'-(4-(2-(N-метилкарбамоил)-4-пиридилокси)фенил)мочевины. Изобретение обеспечивает расширение спектра поддающихся лечению различных типов рака млекопитающих, более высокий уровень ответной реакции у пациентов, проходящих лечение, введение меньших количеств химиотерапевтических агентов. 4 н. и 7 з.п. ф-лы, 5 ил.

Комментарии