Новые производные peg - RU2697551C2
Код документа: RU2697551C2
Чертежи

Описание
Область техники
[0001] Настоящее изобретение относится к новому производному PEG, полезному к качестве терапевтического средства против злокачественных опухолей, и содержащему его фармацевтическому продукту.
Уровень техники
[0002] Производные цитозина, такие как цитарабин, 1-(2'-циано-2'-дезокси-β-D-арабинофуранозил)цитозин, гемцитабин, децитабин, 5-азацитидин, RX-3117 (Rexahn) и SGI-110 (Astex), обладают эффектами ингибирования рака ДНК полимераз или регулирования цикла раковых клеток (G2/M блокировка) и вызывания дифференцировки лейкозных клеток. Поэтому, производные цитозина полезны в качестве терапевтических средств против злокачественных опухолей, таких как острый миелобластный лейкоз, острый лимфоцитарный лейкоз, злокачественная лимфома, множественная миелома, рак поджелудочной железы, рак легких и рак молочной железы (патентная литература 1, непатентные литературы 1-3). Лечение злокачественных опухолей при использовании указанных производных цитозина включает введение внутривенным вливанием, которое обычно поддерживается в течение нескольких часов до нескольких недель (непатентная литература 1).
[0003] Кроме того, митомицин C представляет собой противораковое средство, используемое при лечении хронического лимфоцитарного лейкоза, хронического миелолейкоза, рака желудка, колоректального рака, рака легких, рака поджелудочной железы, рака печени, рака шейки матки, рака матки, опухоли головы и шеи, и опухоли мочевого пузыря. Но митомицин C при этом обычно вводится внутривенно ежедневно. Гефитиниб и эрлотиниб являются молекулярно нацеленными противоопухолевыми средствами, которые селективно ингибируют тирозинкиназу рецептора эпителиального фактора роста (EGFR) и используются, например, против мелкоклеточного рака легких, рака поджелудочной железы, глиобластомы мозга и плоскоклеточной карциномы головы и шеи. Кроме того, лапатиниб и сунитиниб тоже являются ингибиторами тирозинкиназы и используются, например, против рака молочной железы. Однако у таких молекулярно нацеленных лекарственных средств в свою очередь существуют проблемы с неблагоприятными последствиями, такими как острое повреждение легких и интерстициальный пневмонит.
[0004] Паклитаксел и доцетаксел представляют собой противораковые средства, используемые при лечении, например, рака легкого, рака яичников, рака молочной железы, рака головы и шеи, и прогрессирующей саркомы Капоши. Однако основанные на таксане противораковые средства обладают побочными эффектами, такими как миелосуппрессия, такая как лейкопения и периферические нервные расстройства, и низкой растворимостью в воде. Кремофор предназначен для использования в качестве способствующего растворению средства, но поскольку кремофор вызывает тяжелые аллергические симптомы, необходима предварительная обработка антагонистами гистамина H1/H2, которая требует сложных манипуляций при введении в клинических условиях. Кроме того, имеются случаи, при которых используется человеческий сывороточный альбумин в качестве способствующего растворению средства; при этом имеются опасения рисков, таких как отсутствие человеческого сывороточного альбумина и вирусная инфекция, такая как СПИД.
[0005] Общепринятые противораковые средства, сформированные из соединений с низкой молекулярной массой, клинически применялись при внутривенном введении или пероральном введении; однако, в сложившихся обстоятельствах, они все еще являются малодоступными и только очень небольшая часть введенного средства достигает опухоли. При этом данные противораковые средства распределяются системно, что приводит к системной токсичности. Поскольку дозирование определяется балансом между эффектом и токсичностью, возникает системная токсичность. Таким образом, в большинстве случаев, достаточное количество противоракового средства, требуемое для проявления эффективности лекарственного средства, не обеспечивается.
В последние годы было разработано несколько систем доставки лекарственных средств, с целью преодоления проблем, описанных выше. Например, иллюстративными примерами систем доставки лекарственных средств (здесь и далее обозначенных как DDS) включают способы физического встраивания противоракового средства с низкой молекулярной массой в фосфолипидные липосомы, полимерные мицеллы или водорастворимый полимер, все из которых формируются из компонентов биосовместимого материала, или формирования химических ковалентных связей между двумя.
[0006] Что касается получения липосом для внутривенного введения, осуществляется контроль размера частиц, который должен составлять от 200 до 300 нм, для того, чтобы липосомы могли проникать через капилляры кровеносных сосудов без каких-либо проблем и могли проникать через новые кровеносные сосуды вблизи опухоли и, в дополнение к этому, после того как поверхность мембран частиц липосом покрывается полиэтиленгликолем (здесь и далее обозначенный как PEG), имеющим молекулярную массу около 2000, как правило, прекращается поглощение липосом фагоцитами in vivo.
При получении полимерных мицелл, размер частиц контролируется в пределах 50 нм, и поверхность мембран частиц покрывают PEG, при этом можно избежать поглощения мицелл фагоцитами in vivo, и сообщалось, что мицеллы полимера могут легко проникнуть через новые кровеносные сосуды вблизи опухоли.
Однако, в сложившихся обстоятельствах, вышеуказанные препараты, содержащие мелкие частицы нано размера, имеют относительно короткий период полураспада в крови, нацеливание на опухоли также является неудовлетворительным, и истинная цель по существу не достигнута.
[0007] Тем временем, недавно были предприняты попытки клинического применения производных, в которых противораковое средство химически ковалентно связано с синтетическим PEG с высокой молекулярной массой, обладающим высокой биосовместимостью и высокой растворимостью в воде, в частности PEG, имеющим четыре цепи (молекулярная масса 40000), который с трудом образует раствор высокой вязкости.
Перечень ссылок
Непатентная литература
[0008] Непатентная литература 1: J. Med. Chem., Vol. 34, 2917-2919 (1991)
Непатентная литература 2: J. Med. Chem., Vol. 36, 4183-4189 (1993)
Непатентная литература 3: package insert for CYLOCIDE™ injections
Сущность изобретения
Техническая проблема
[0009] Однако, поскольку внимание в избытке уделялось контролируемому высвобождению данных производных в кровь, химическое связывание между PEG и противораковым средством ограничивалось сложноэфирным связыванием или карбаматным связыванием, оба из которых относительно легко расщепляются лиазами, такими как эстераза или карбоксилаза, в крови. Таким образом, долговременная стабильность в крови по существу не достигается, эффективность нацеливания на опухоль ткани не является удовлетворительной и первоначальная цель DDS не достигается.
Недавно было сообщено, что PEG с четырьмя разветвленными цепями и SN-38, который является активным ингредиентом противоракового средства (CPT-11), ковалентно связывается карбаматным связыванием, которое с трудом расщепляется в кислоте или щелочи; в то же время, в определенной степени просто преодолевает недостаточное карбаматное связывание. Кроме того, условия для химической реакции, используемые в синтезирующем карбаматную связь органе, являются суровыми (сильнощелочные условия), и данные условия не подходят для внутримолекулярных сложноэфирных связей или для связывания противоракового средства, которое чувствительно к щелочам.
Поэтому, задачей настоящего изобретения является обеспечение нового терапевтического средства против злокачественной опухоли, которое сократит негативные побочные эффекты, такие как тяжелая желудочно-кишечная токсичность или костная токсичность, будет иметь устойчивый противоопухолевый эффект и позволит улучшить средства для введения и частоту введения
Решение задачи
[0010] Таким образом, авторы настоящего изобретения уделили внимание устойчивости производного PEG в крови, высоким свойствам кластеризации опухолевых тканей и усилению долговременного устойчивого воздействия противоопухолевого средства, при этом авторы провели исследования с учетом свойств контролируемого высвобождения в крови, связанного с лиазой в крови, на модификации SN-38, который является активным веществом CPT-11, путем сложноэфирного связывания или карбаматного связывания между гидроксильной группой SN-38 и полиэтиленгликолем. При этом было обнаружено, что противоопухолевое активное вещество высвобождалось в кровь быстрее, чем ожидалось для человека, требовалось большое количество производного полиэтиленгликоля для введения, и не было достигнуто усиления достаточного долговременного эффекта и усиления безопасности.
Таким образом, авторы провели дополнительные исследования, и в результате, авторы обнаружили, что соединение формулы (1), в котором полиэтиленгликоль с четырьмя разветвленными цепями, имеющими метилкарбоксильную группу, введенную в каждую из концевых цепей, является амидно связанным с первичной или вторичной аминогруппой противоракового средства, такого как производное цитозина, митомицина C или паклитаксела, или с первичной или вторичной аминогруппой молекулярно нацеленного лекарственного средства против рака, такого как гефитиниб, эрлотиниб, лапатиниб или сунитиниб, прямо или через спейсер, основанный на аминокислоте, такой как β-аланин, обладает отличным долговременным противоопухолевым эффектом и высокой безопасностью, и данное соединение обеспечивает улучшенный терапевтический эффект в отношении злокачественных опухолей в малых дозах и небольшом количестве раз введения, по сравнению с общепринятыми противоопухолевыми средствами. Таким образом, авторы завершили настоящее изобретение.
[0011] Иными словами, настоящее изобретение представлено следующими пунктами [1]-[11].
[0012] [1] Соединение формулы (1) или его соль:
[0013]
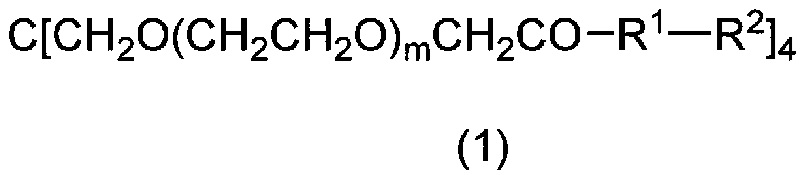
[0014] где R1 представляет собой простую связь, -N(R3)(CH2)n1CO- или -N(R4)(CH2)n2N(R5)CO(CH2)n3CO-, где R3 представляет собой атом водорода или алкильную группу; R4 и R5, которые являются одинаковыми или отличаются друг от друга, каждый представляет собой атом водорода или алкильную группу, или R4 и R5 связаны вместе и представляют собой алкиленовую группу, имеющую 1-4 атома углерода; и n1, n2 и n3, которые являются одинаковыми или отличаются друг от друга, каждый равен целому числу от 1 до 3;
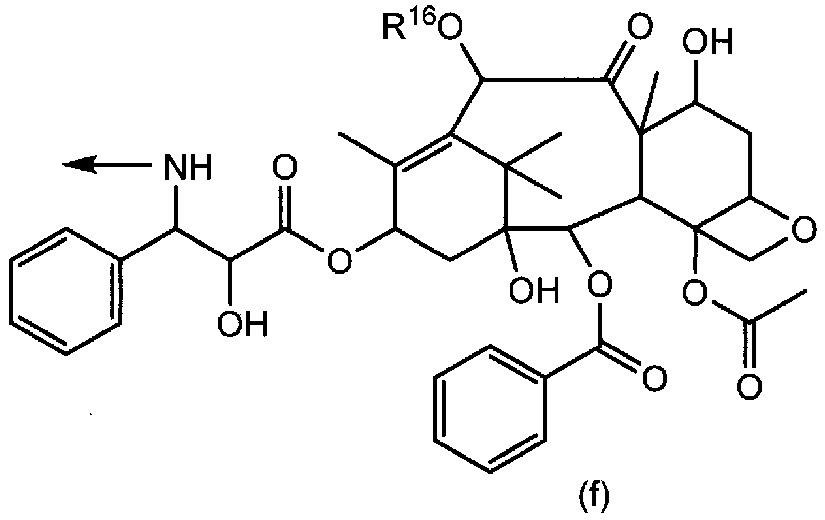
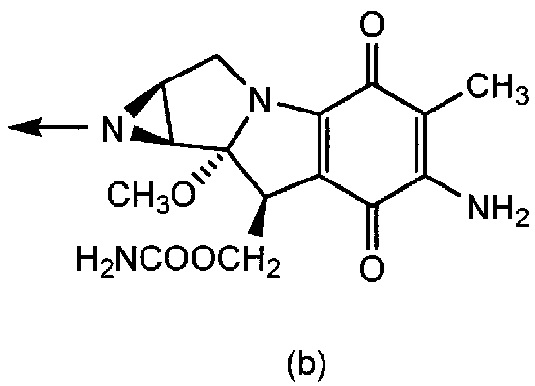
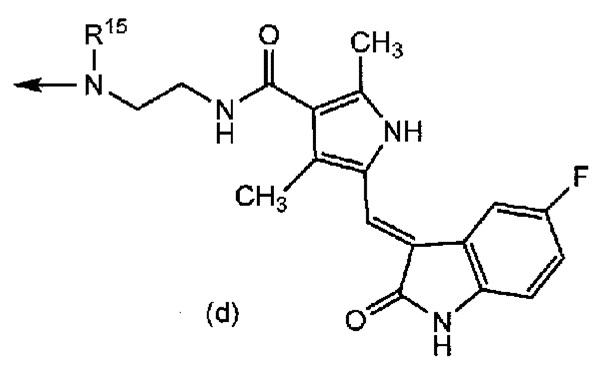
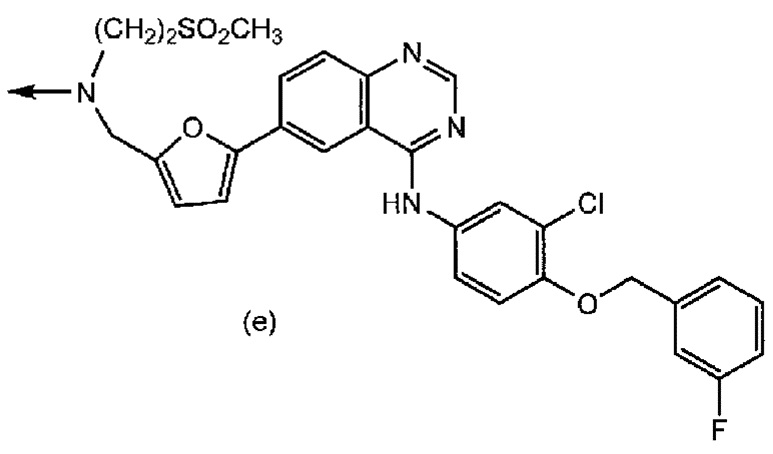
R2 представляет собой группу формул (a), (b), (c), (d), (e) или (f):
[0015]
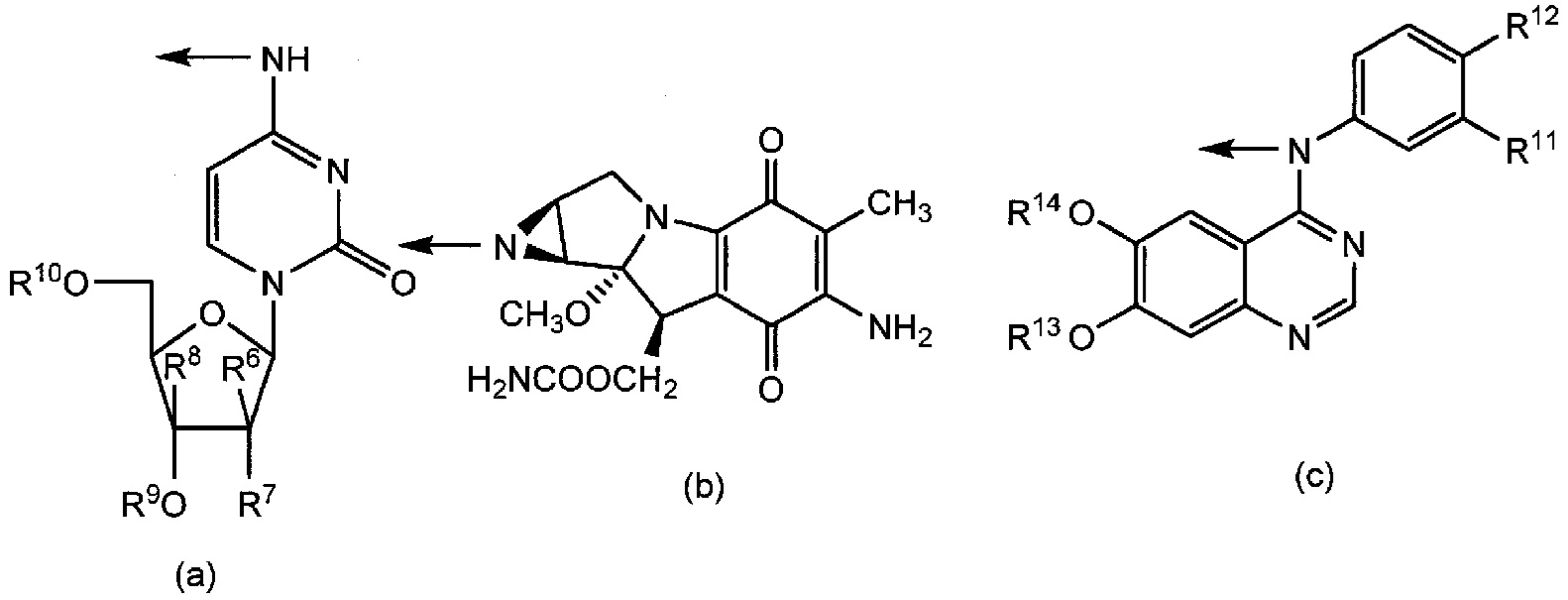

[0016] где R6 представляет собой гидроксильную группу, цианогруппу или атом галогена; R7 представляет собой атом водорода или атом галогена; R8 представляет собой атом водорода или этинильную группу; R9 и R10, которые являются одинаковыми или отличаются друг от друга, каждый представляет собой атом водорода или триалкилсилильную группу, или R9 и R10 связаны вместе и представляют собой тетраалкилсилоксисилильную группу; R11 представляет собой атом галогена или этинильную группу; R12 представляет собой атом водорода или атом галогена; R13 представляет собой алкильную группу или алкоксиалкильную группу; R14 представляет собой алкоксиалкильную группу или морфолиноалкильную группу; R15 представляет собой алкильную группу; и R16 представляет собой атом водорода или алканоильную группу;
m равен числу от 10 до 1000; и
стрелка обозначает место связывания.
[2] Соединение по пункту [1] или его соль, где R2 представляет собой группу формулы (a) или (b).
[3] Соединение по пункту [1] или [2] или его соль, где R2 представляет собой группу формулы (a).
[4] Соединение по любому из пунктов [1]-[3] или его соль, где R2 представляет собой группу формулы (a); R6 представляет собой гидроксильную группу, цианогруппу или атом галогена; R7 представляет собой атом водорода или атом галогена; R8 представляет собой атом водорода или этинильную группу; и R9 и R10 каждый представляет собой атом водорода.
[5] Соединение по любому из пунктов [1]-[4] или его соль, где R1 представляет собой простую связь, -NH(CH2)n1CO-, -NH(CH2)n2NHCO(CH2)n3CO- или следующую формулу:
[0017]

[0018] где n1, n2 и n3 имеют такие же значения, как определено выше, соответственно.
[6] Лекарственное средство, содержащее соединение, представленное в любом из пунктов [1]-[5], или его соль.
[7] Лекарственное средство по пункту [6], где указанное лекарственное средство представляет собой терапевтическое средство против злокачественной опухоли.
[8] Фармацевтическая композиция, содержащая соединение по любому из пунктов [1]-[5] или его соль и фармацевтически приемлемую соль.
[9] Применение соединения по любому из пунктов [1]-[5] или его соли для изготовления терапевтического средства против злокачественной опухоли.
[10] Соединение по любому из пунктов [1]-[5] или его соль, для применения для лечения злокачественной опухоли.
[11] Способ лечения злокачественной опухоли, включающий введение эффективного количества соединения по любому из пунктов [1]-[5] или его соли.
Преимущественные эффекты изобретения
[0019] Поскольку соединение (1) по настоящему изобретению обладает отличным противоопухолевым действием в отношении злокачественных опухолей и обладает улучшенным эффектом продолжительности действия, соединение (1) проявляет отличное противоопухолевое действие в отношении злокачественных опухолей в малой дозе (в виде преобразования в активное вещество оригинального противоопухолевого средства), при небольшом числе раз введения и низкой частоте введения по сравнению с обычными противоопухолевыми средствами, а также сокращает неблагоприятные побочные эффекты. Поэтому, когда при лечении злокачественной опухоли применяют соединение (1) по настоящему изобретению, снижается нагрузка как на пациентов, так и на лечащих врачей, и достигаются отличные противоопухолевые эффекты в отношении злокачественных опухолей.
Краткое описание фигур
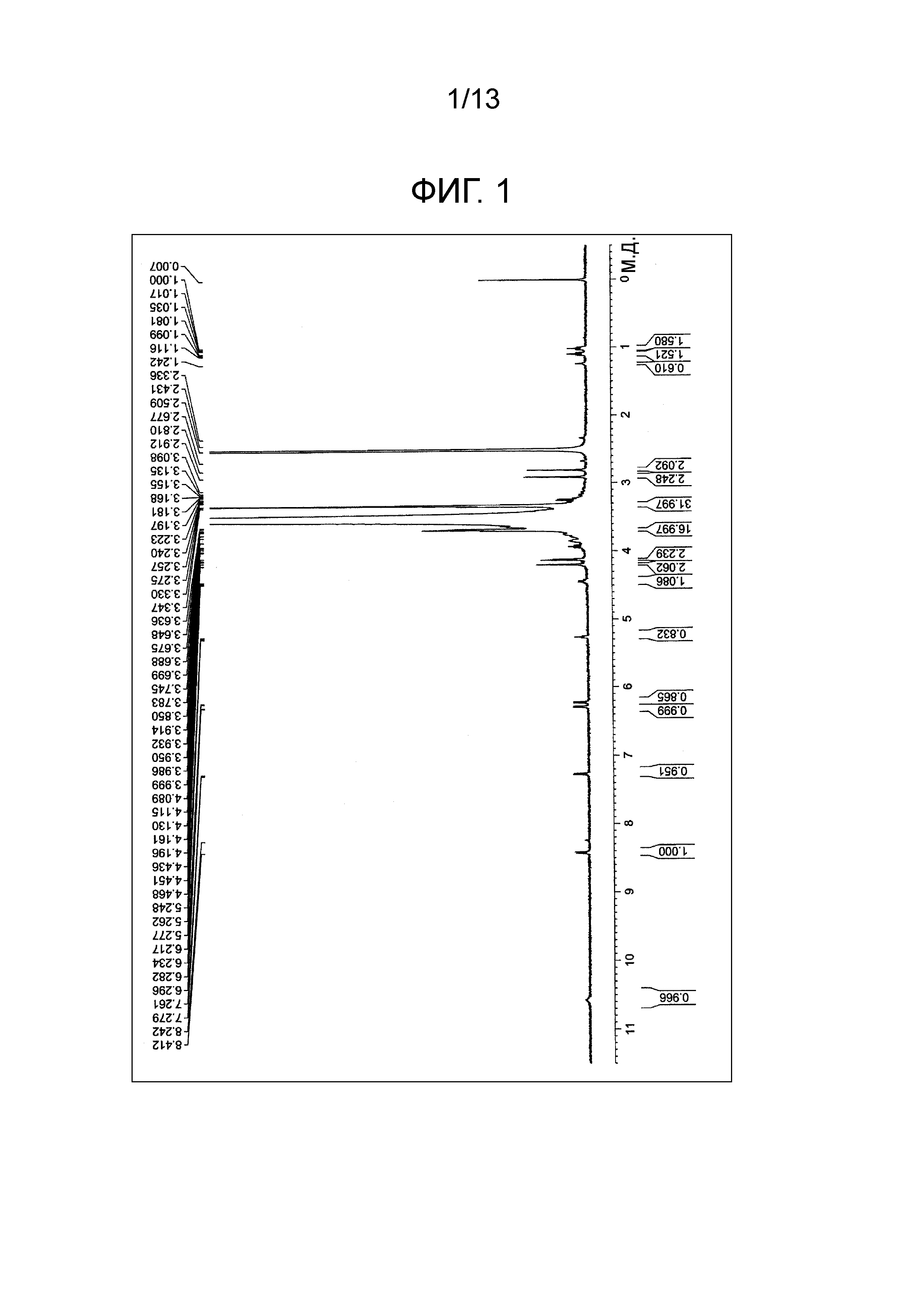
[0020] На фиг.1 проиллюстрирована диаграмма ЯМР спектра соединения (1a).
На фиг.2 проиллюстрирована диаграмма ЯМР спектра соединения (1b).
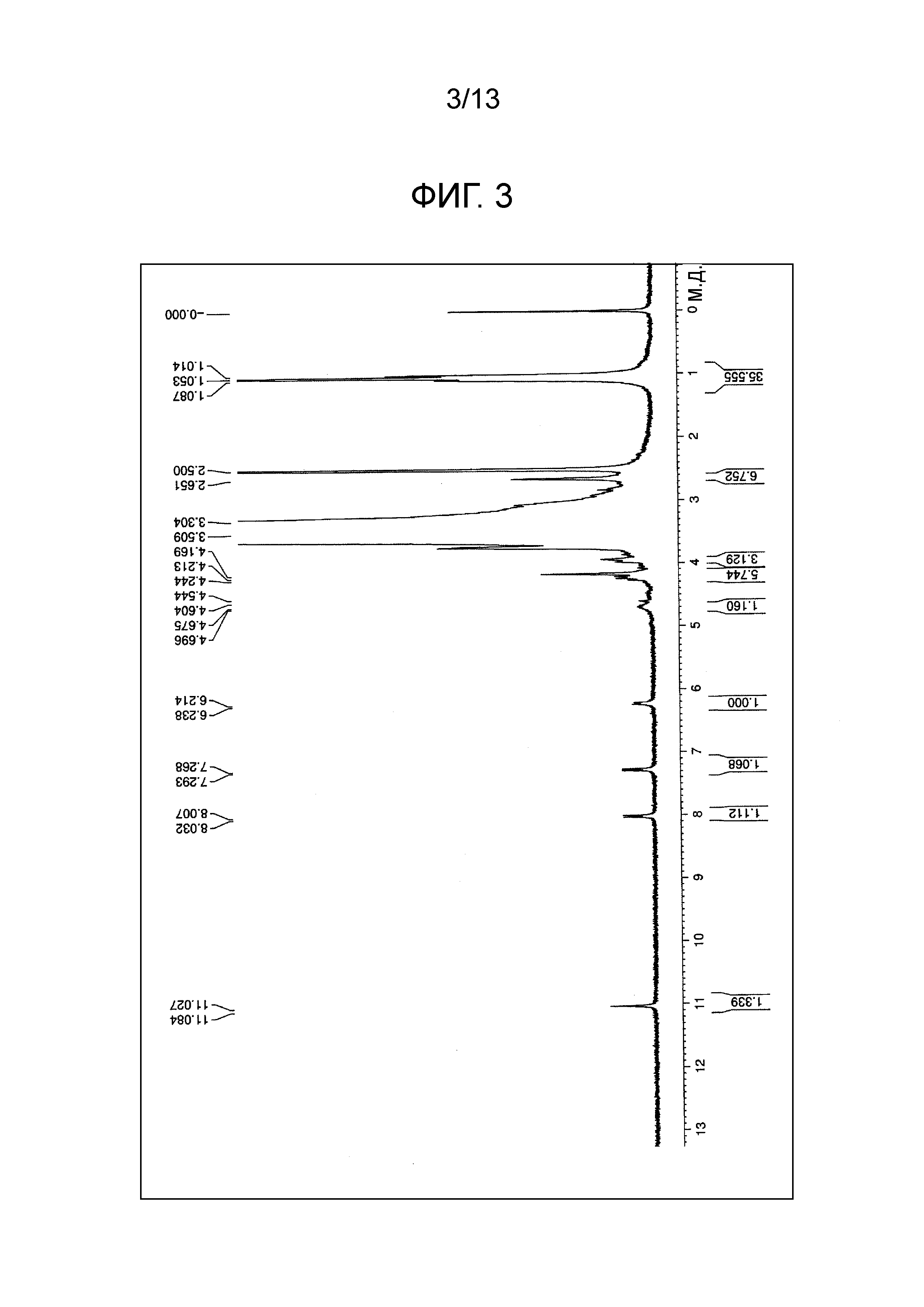
На фиг.3 проиллюстрирована диаграмма ЯМР спектра соединения (1c).
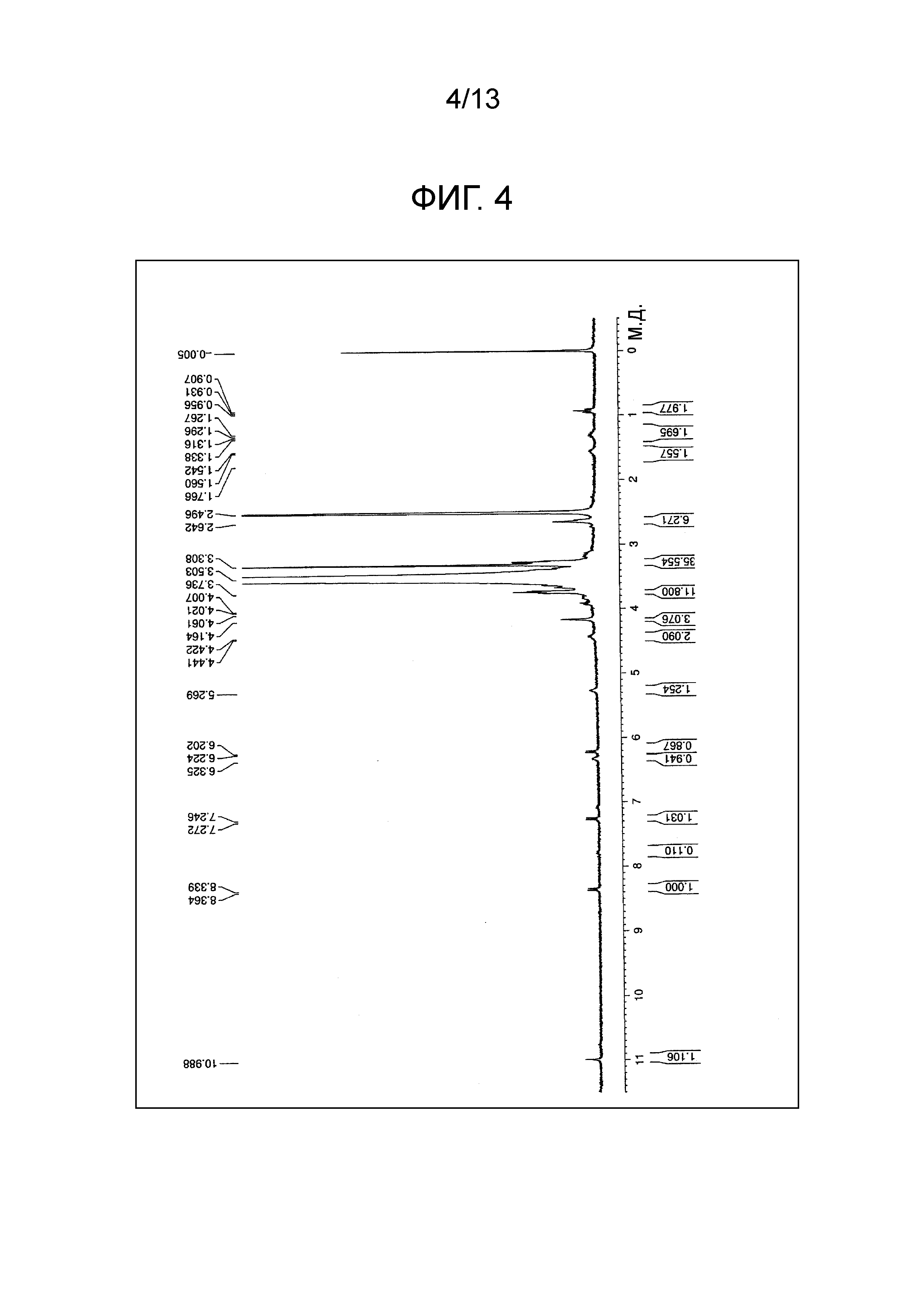
На фиг.4 проиллюстрирована диаграмма ЯМР спектра соединения (1d).
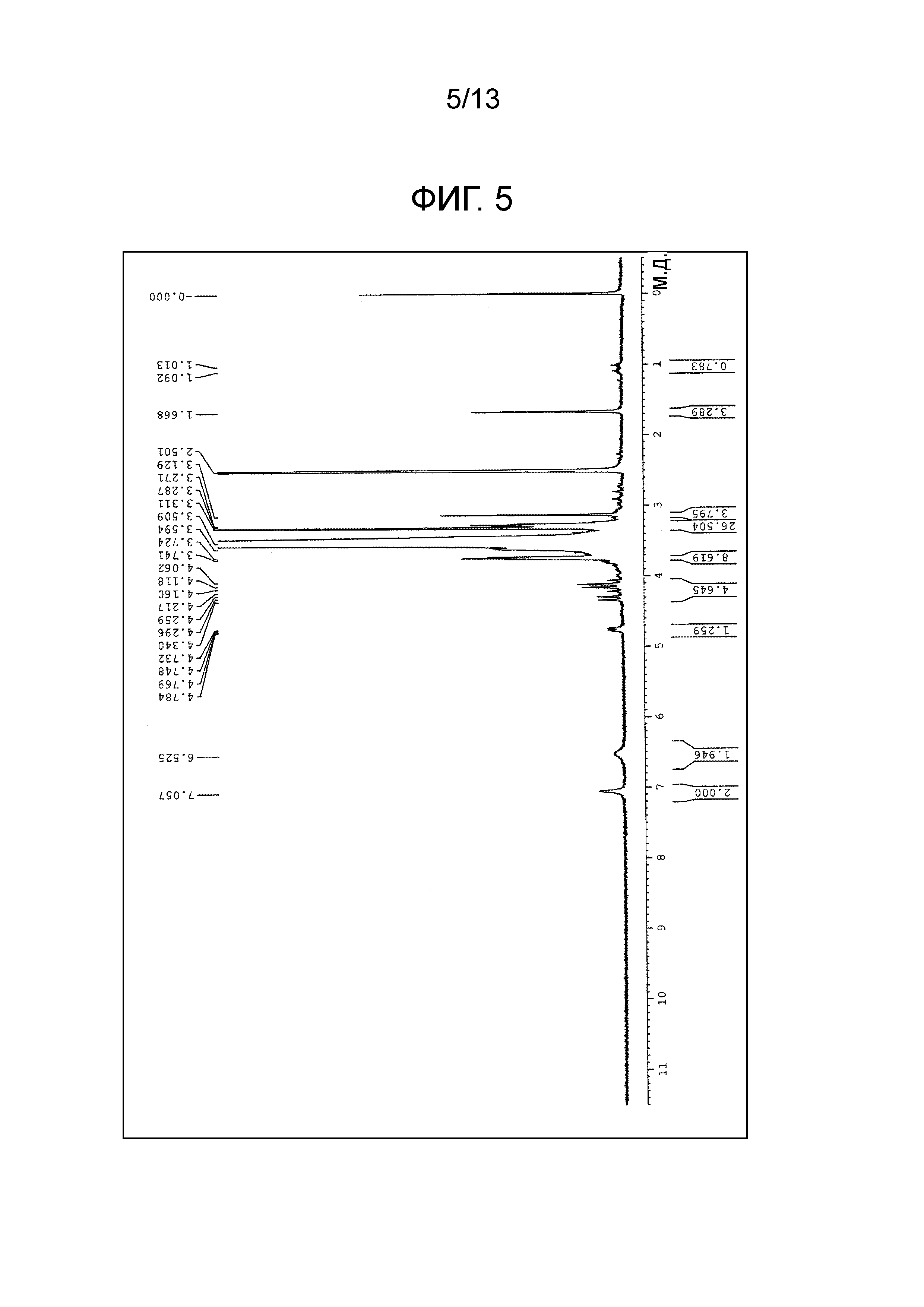
На фиг.5 проиллюстрирована диаграмма ЯМР спектра соединения (1e).
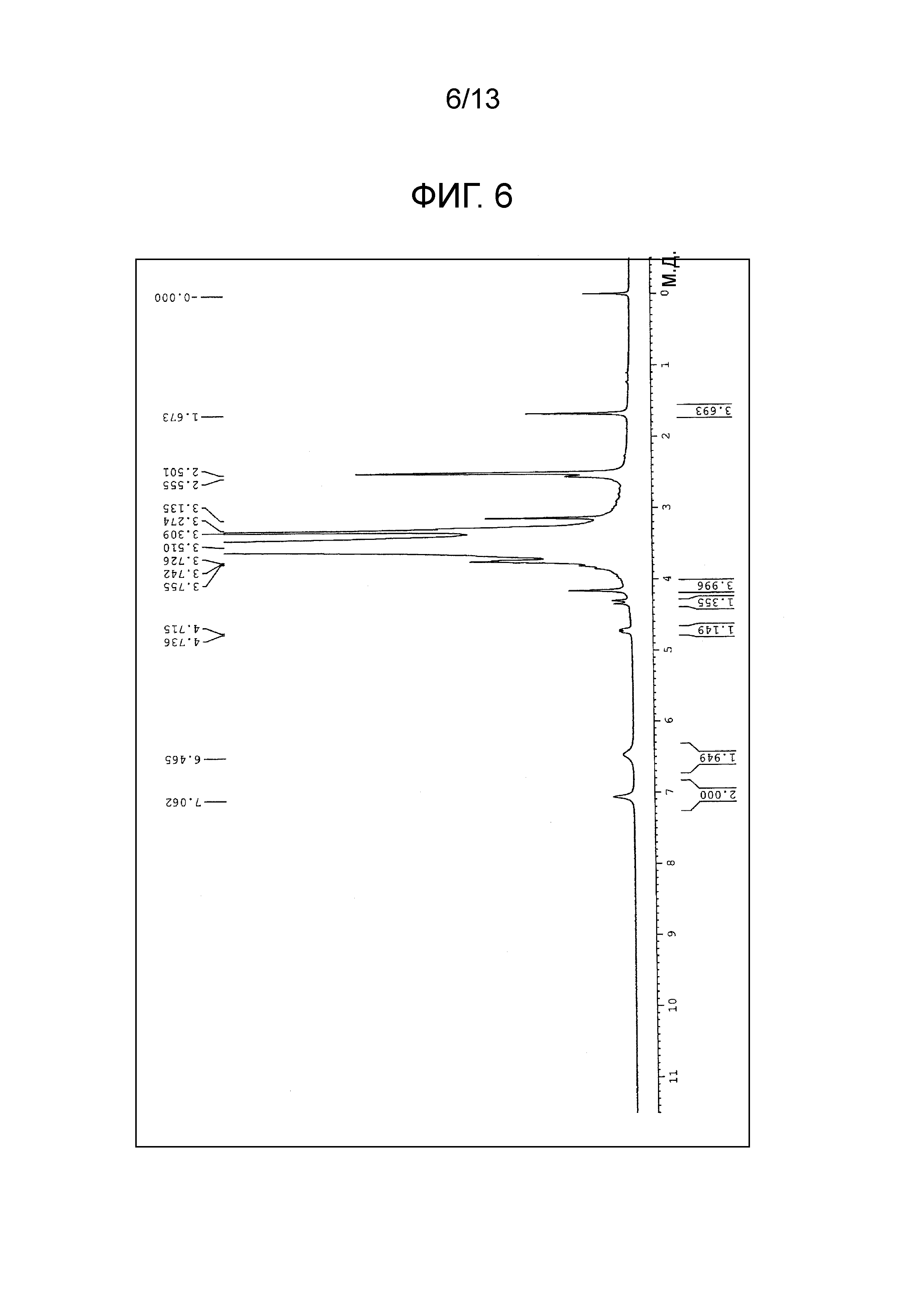
На фиг.6 проиллюстрирована диаграмма ЯМР спектра соединения (1f).
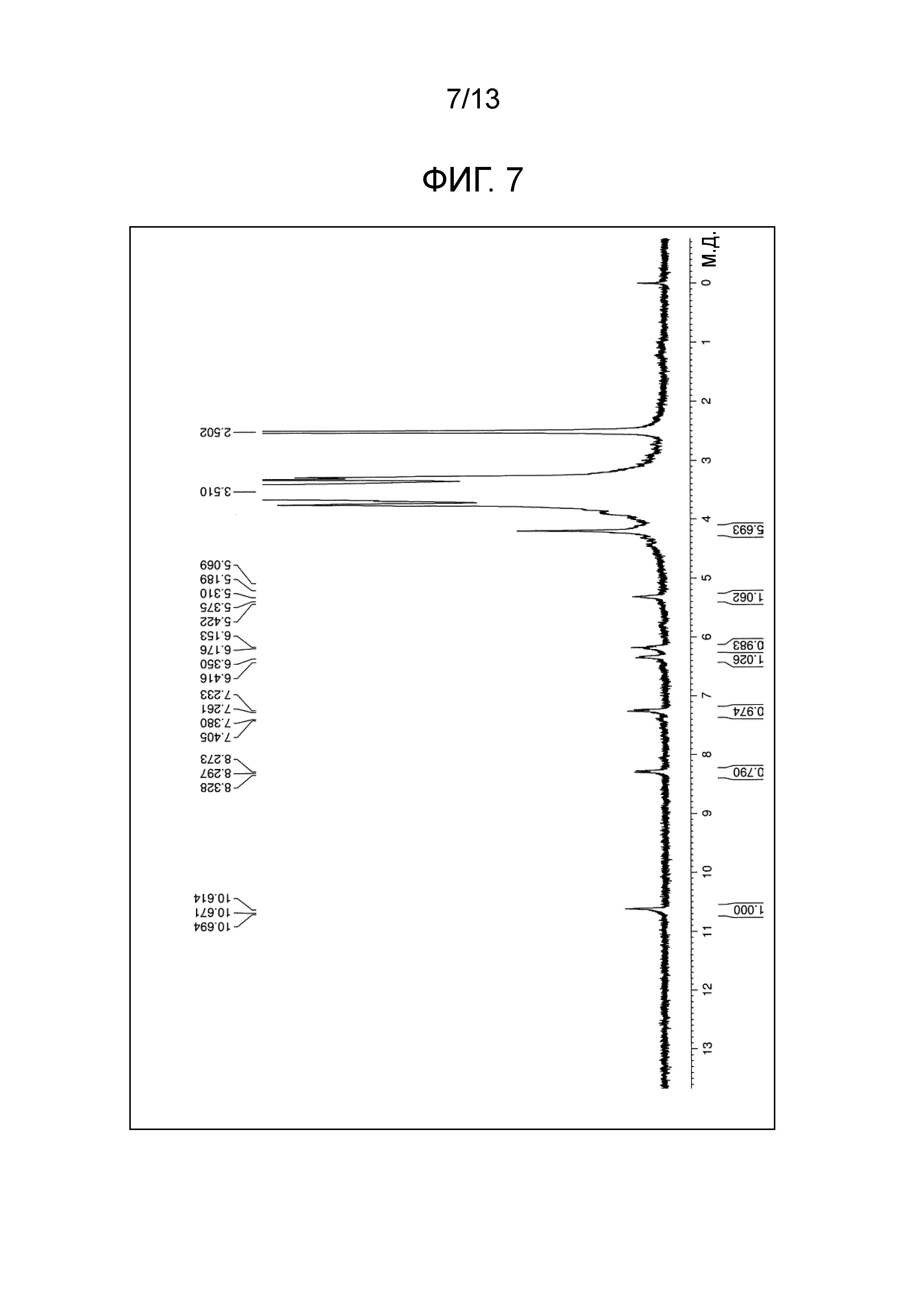
На фиг.7 проиллюстрирована диаграмма ЯМР спектра соединения (1g).
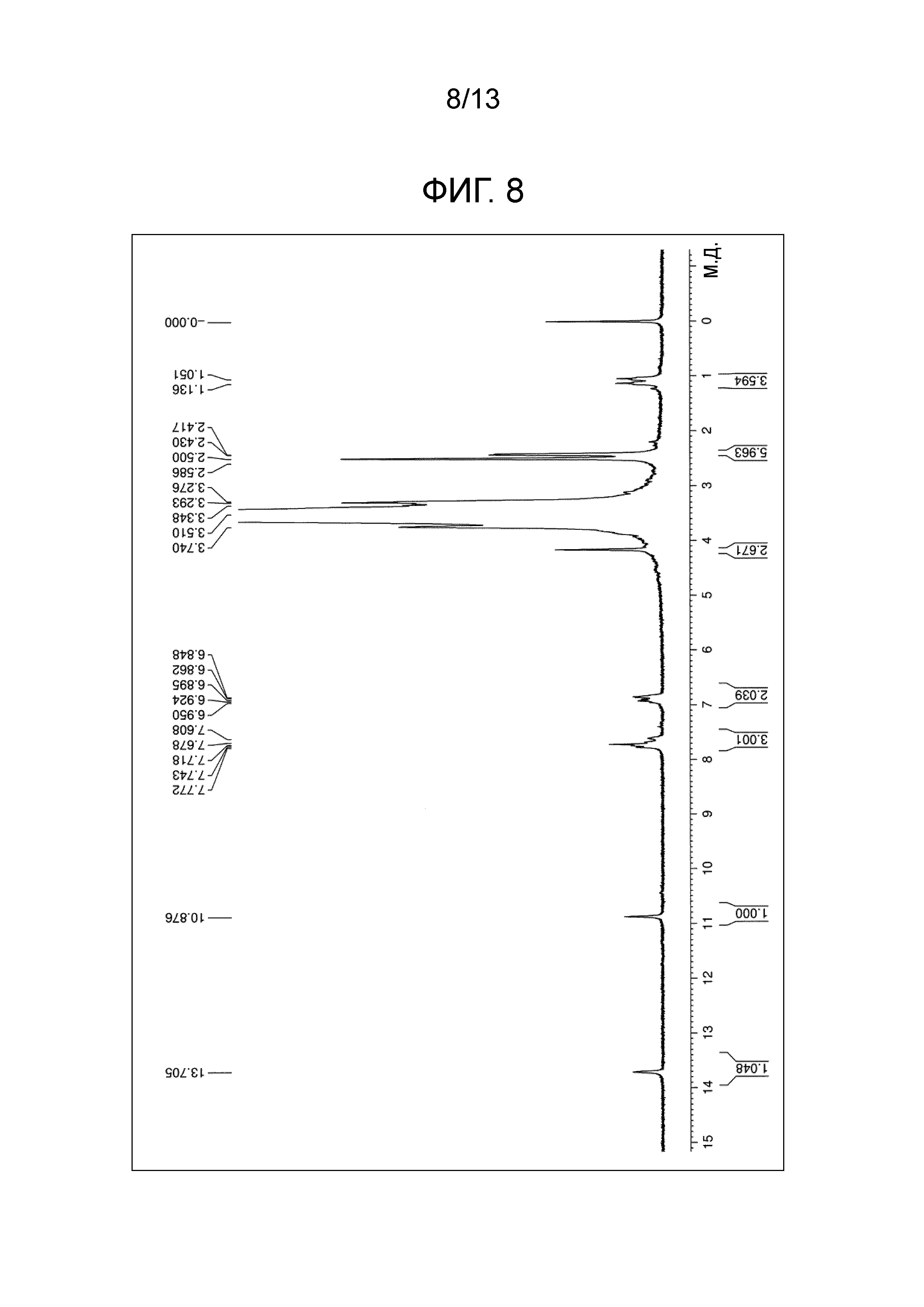
На фиг.8 проиллюстрирована диаграмма ЯМР спектра соединения (1h).
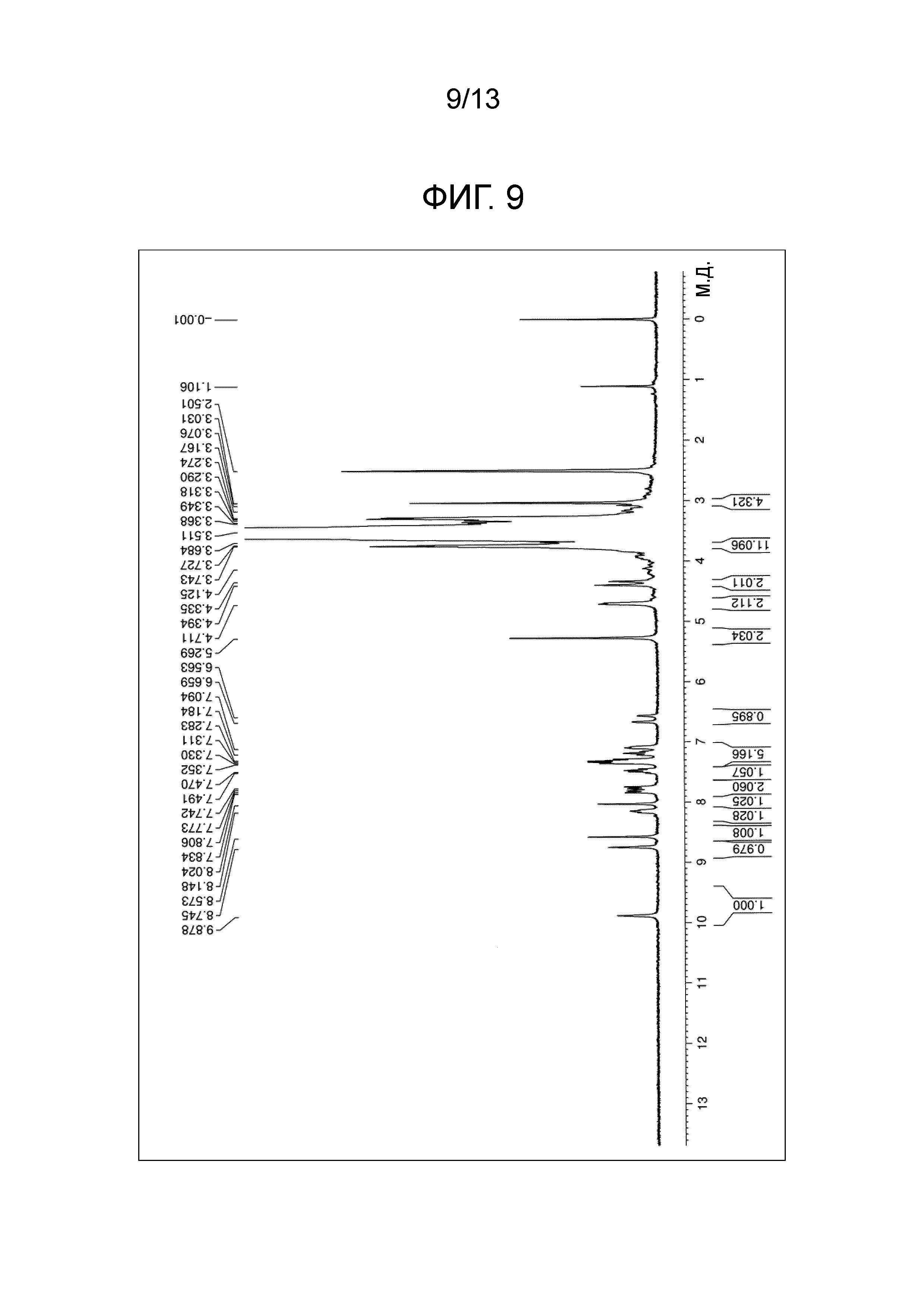
На фиг.9 проиллюстрирована диаграмма ЯМР спектра соединения (1i).
На фиг.10 проиллюстрирована диаграмма ЯМР спектра соединения (1j).
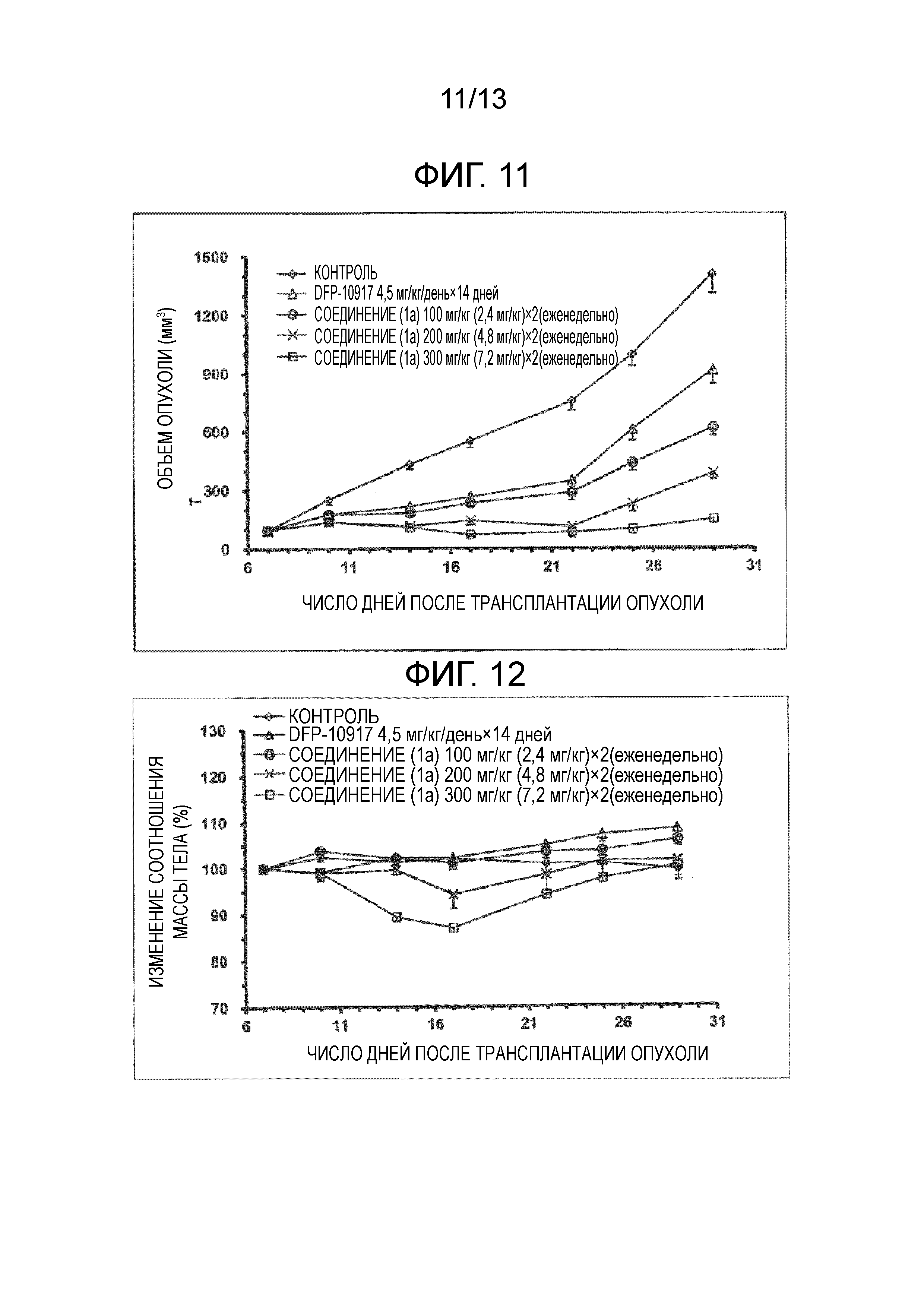
На фиг.11 проиллюстрированы изменения объема опухоли после трансплантации опухоли. Значение в скобках для дозы представляет собой дозу в пересчете на DFP-10917.
На фиг.12 показано отношение изменения массы тела (%), после трансплантации опухоли. Значение в скобках для дозы представляет собой дозу в пересчете на DFP-10917.
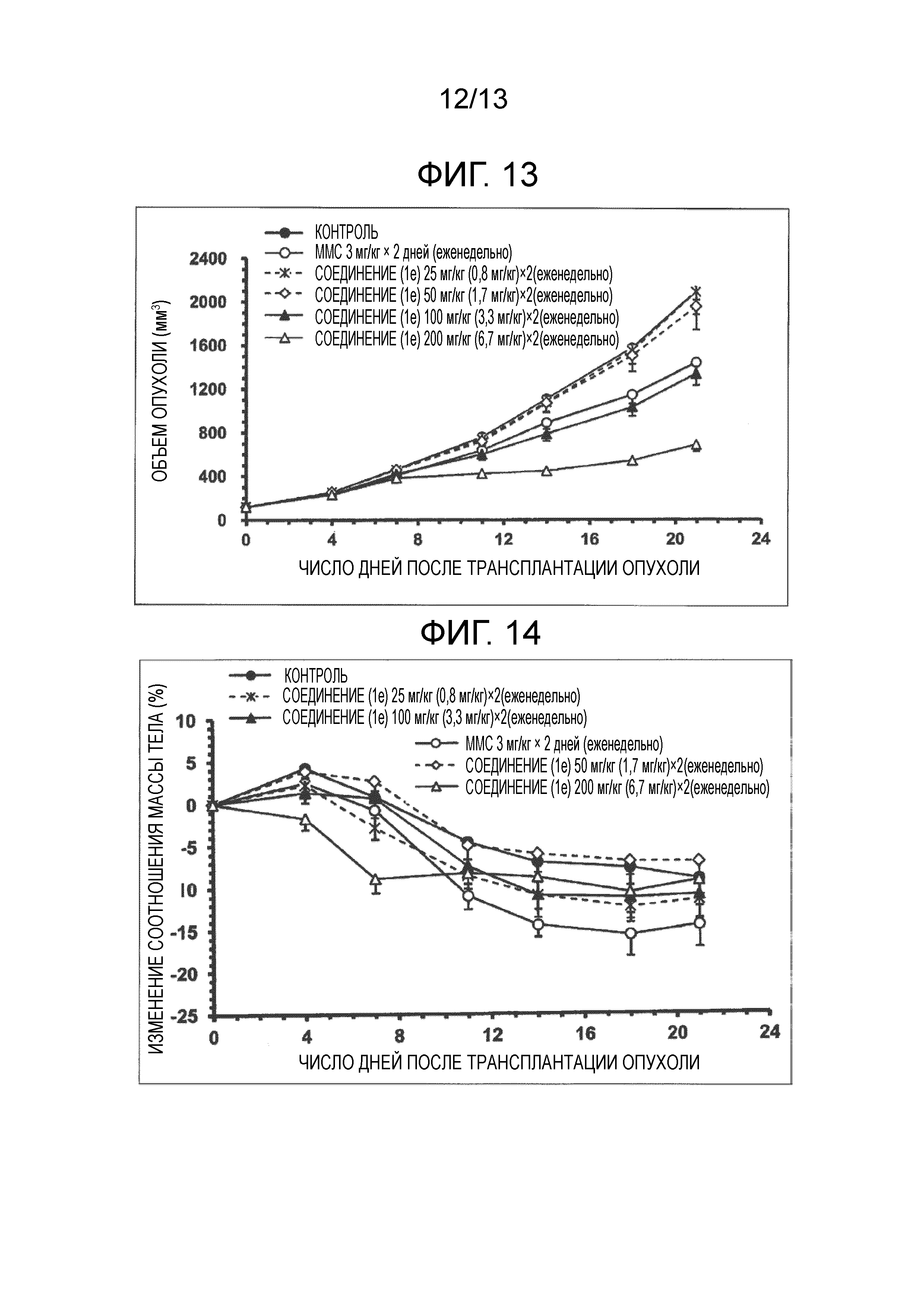
На фиг.13 проиллюстрированы изменения объема опухоли после трансплантации опухоли. Значение в скобках для дозы представляет собой дозу в пересчете на MMC.
На фиг.14 показано отношение изменения массы тела (%), после трансплантации опухоли. Значение в скобках для дозы представляет собой дозу в пересчете на MMC.
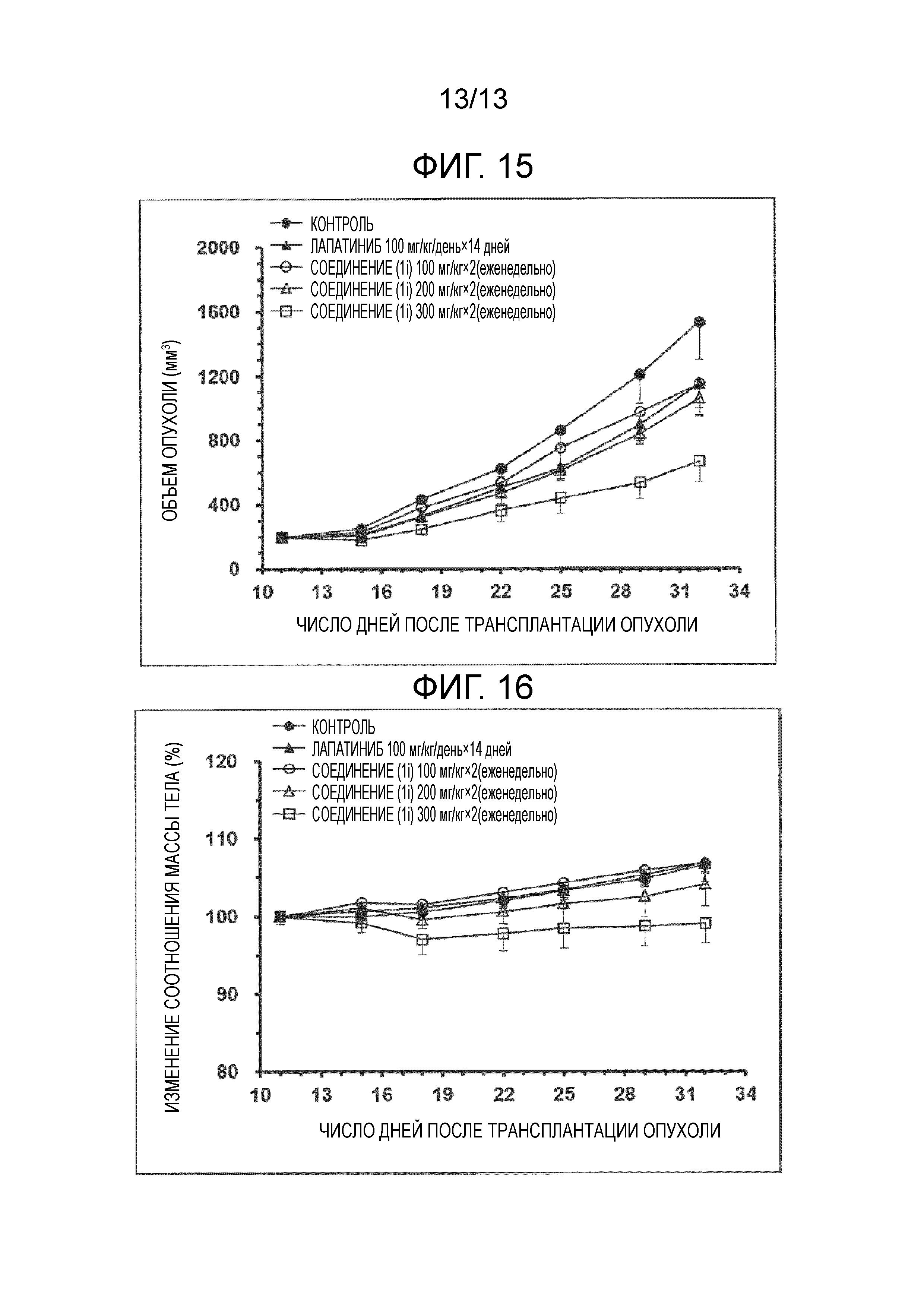
На фиг.15 представлены изменения объема опухоли после трансплантации опухоли.
На фиг.16 представлены изменения массы тела (в %) после трансплантации опухоли.
Описание вариантов осуществления
[0021] В формуле (1) R1 представляет собой простую связь, -N(R3)(CH2)n1CO- или -N(R4)(CH2)n2N(R5)CO(CH2)n3CO-, где R3 представляет собой атом водорода или алкильную группу; R4 и R5, которые могут быть одинаковыми или отличаться друг от друга, каждый представляет собой атом водорода или алкильную группу, или R4 и R5 связаны вместе и представляют собой алкиленовую группу, имеющую 1-4 атома углерода; и n1, n2 и n3, которые могут быть одинаковыми или отличаться друг от друга, каждый равен целому числу от 1 до 3.
[0022] Группа R3, R4 или R5 предпочтительно представляет собой атом водорода и, когда данная группа представляет собой алкильную группу, данная алкильная группа может представлять собой линейную или разветвленную алкильную группу, имеющую 1-6 атомов углерода. Прежде всего, линейную или разветвленную алкильную группу, имеющую 1-4 атома углерода, являющейся предпочтительной, например, метильную группу, этильную группу и изопропильную группу, которые являются более предпочтительными.
Алкиленовая группа, имеющая 1-4 атома углерода, которая совместно образована R4 и R5, может представлять собой метиленовую группу, этиленовую группу, триметиленовую группу или тетраметиленовую группу, и этиленовая группа является более предпочтительной.
[0023] R3 более предпочтительно представляет собой атом водорода. Что касается R4 и R5, более предпочтительным является, когда оба представляют собой атом водорода, или R4 и R5 образуют C1-3алкиленовую группу.
[0024] n1, n2 и n3, каждый равен целому числу 1, 2 или 3, и среди них 2 является более предпочтительным.
[0025] Более предпочтительные примеры R1 включают простую связь, -NH(CH2)n1CO-, -NH(CH2)n2NHCO(CH2)n3CO- и
[0026]
[0027] Еще более предпочтительные конкретные примеры R1 включают простую связь, -NHCH2CH2CO- и
[0028]

[0029]
[0030] R2 представляет собой группу формул (a), (b), (c), (d), (e) или (f):
[0031]
[0032] где R6 представляет собой гидроксильную группу, цианогруппу или атом галогена; R7 представляет собой атом водорода или атом галогена; R8 представляет собой атом водорода или этинильную группу; R9 и R10, которые являются одинаковыми или отличаются друг от друга, каждый представляет собой атом водорода или триалкилсилильную группу, или R9 и R10 связаны вместе и представляют собой тетраалкилсилоксисилильную группу; R11 представляет собой атом галогена или этинильную группу; R12 представляет собой атом водорода или атом галогена; R13 представляет собой алкильную группу или алкоксиалкильную группу; R14 представляет собой алкоксиалкильную группу или морфолиноалкильную группу; R15 представляет собой алкильную группу; R16 представляет собой атом водорода или алканоильную группу; и стрелка обозначает место связывания.
[0033] R6 представляет собой гидроксильную группу, цианогруппу или атом галогена; однако, цианогруппа является особо предпочтительной. R7 представляет собой атом водорода или атом галогена. В данном случае, атомом галогена предпочтительно является атом фтора.
[0034] R8 представляет собой атом водорода или этинильную группу.
[0035] Примеры триалкилсилильной группы R9 или R10 включают триметилсилильную группу, триэтилсилильную группу, изоизопропилсилильную группу, диметилбутилсилильную группу и диметилпентилсилильную группу. Тетраалкилсилоксисилильная группа совместно образованная R9 и R10, предпочтительно представляет собой группу следующей формулы (g):
[0036]
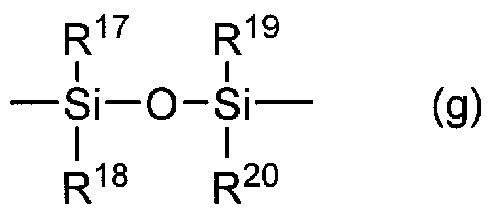
[0037] где R17, R18, R19 и R20, каждый представляет собой алкильную группу, например, метильную группу, этильную группу, изопропильную группу, бутильную группу, и пентильная группа является предпочтительной.
[0038] R11 представляет собой атом галогена или этинильную группу. R12 представляет собой атом водорода или атом галогена.
[0039] R13 представляет собой алкильную группу или алкоксиалкильную группу. В данном случае, алкильная группа предпочтительно представляет собой алкильную группу, имеющую 1-6 атомов углерода, и более предпочтительно метильную группу. Что касается алкоксиалкильной группы, C1-6алкокси-C1-6алкильная группа является предпочтительной; например, метоксиэтильная группа, метоксипропильная группа и этоксиэтильная группа являются более предпочтительными; и метоксиэтильная группа является особо предпочтительной.
[0040] R14 представляет собой алкоксиалкильную группу или морфолиноалкильную группу. В данном случае, алкоксиалкильная группа предпочтительно представляет собой C1-6алкокси-C1-6алкильную группу; более предпочтительно метоксиэтильную группу, метоксипропильную группу или этоксиэтильную группу; и особо предпочтительна метоксиэтильная группа. Морфолиноалкильная группа предпочтительно представляет собой морфолино-C1-4алкильную группу, и более предпочтительно морфолинопропильную группу.
[0041] R15 предпочтительно представляет собой C1-6алкильную группу; и более предпочтительно метильную группу, этильную группу, пропильную группу или бутильную группу.
[0042] R16 предпочтительно представляет собой атом водорода или алканоильную группу, имеющую 2-6 атомов углерода; и более предпочтительно атом водорода или ацетильную группу.
[0043] Группа формул (a), (b), (c), (d), (e) или (f) представляет собой группу, полученную из противоракового средства. Формула (a) представляет собой группу, полученную из основанного на арабинофуранозилцитозине противоракового средства. Формула (b) представляет собой группу, полученную из митомицина C. Формула (c) представляет собой группу, полученную из ингибитора тирозинкиназы EGFR, такого как гефинитиб или эрлотиниб. Формула (d) представляет собой группу, полученную из ингибитора тирозинкиназы PDGFR, такого как сунитиниб. Формула (e) представляет собой группу, полученную из ингибитора тирозинкиназы EGFR, такого как лапатиниб. Формула (f) представляет собой группу, полученную из основанного на таксане противоракового средства, такого как паклитаксел или доцетаксел.
[0044] Предпочтительные примеры структуры формулы (a) включают β-D-арабинофуранозилцитозин, 2'-циано-2'-дезокси-β-D-арабинофуранозилцитозин, 2'-дезокси-2',2'-дифтор-β-D-арабинофуранозилцитозин и 3'-этинил-β-D-арабинофуранозилцитозин.
[0045] Конкретные примеры группы формулы (c) включают группу формулы (c1) и группу формулы (c2):
[0046]
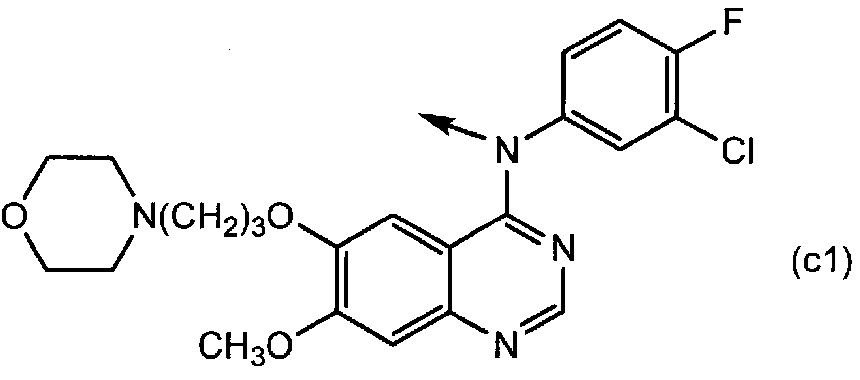

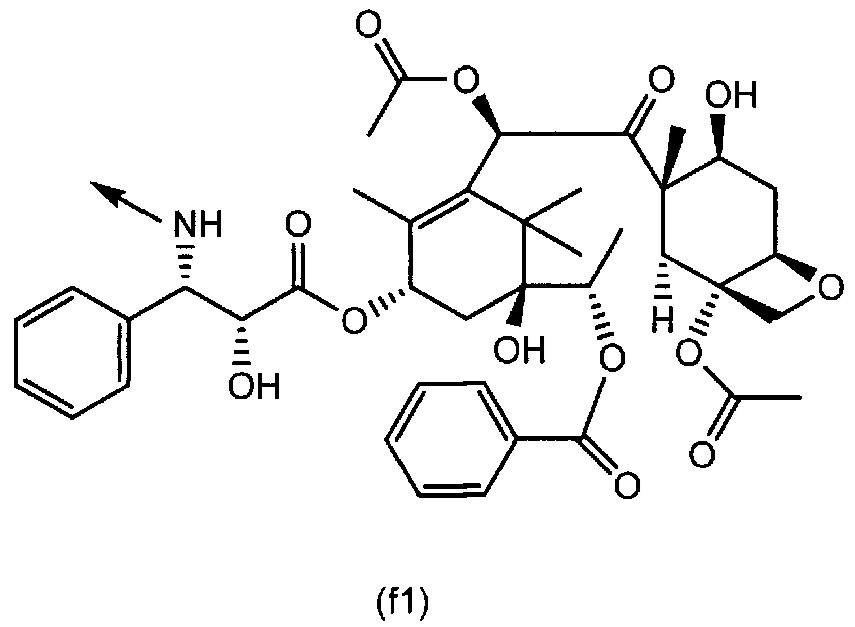
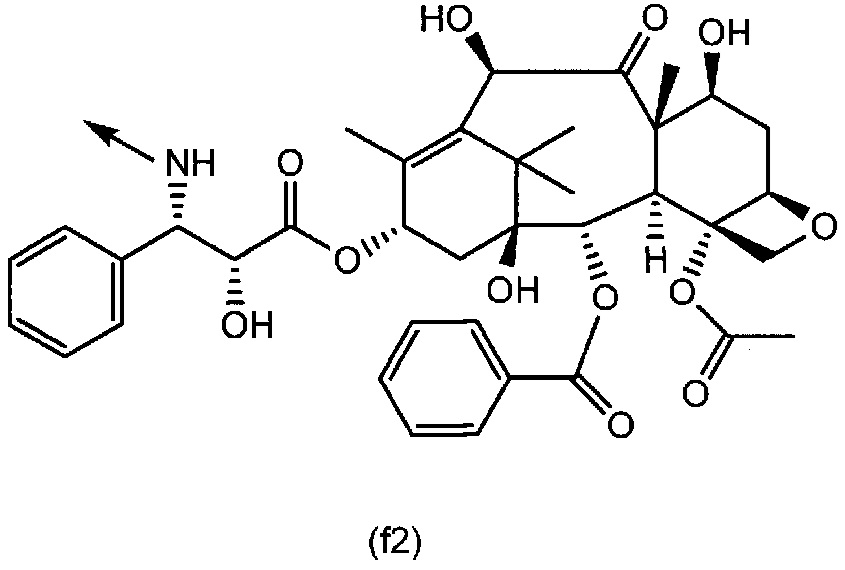
[0047] Конкретные примеры группы формулы (f) включают группу формулы (f1) и группу формулы (f2):
[0048]
[0049] m равен числу от 10 до 1000. Более предпочтительным значением m является 100-500, и еще более предпочтительным значением m является 200-300. m является числом, производным от полиэтиленгликолевой группы, и обычно является средним значением.
[0050] Соли соединения (1) по настоящему изобретению не являются особенно ограниченными до тех пор, пока они являются фармацевтически приемлемыми солями, и их примеры включают соли неорганических кислот, такие как гидрохлорид, сульфат и нитрат; и соли органических кислот, такие как ацетат, цитрат, тартрат, оксалат и малат. Поскольку соединение (1) по настоящему изобретению или его соль имеют асимметричный атом углерода, существуют их пространственные изомеры, и включают их оптически активные вещества, энантиомеры и их смеси.
[0051] Соединение (1) по настоящему изобретению или его соль могут быть получены, например, по следующей реакционной схеме:
[0052]
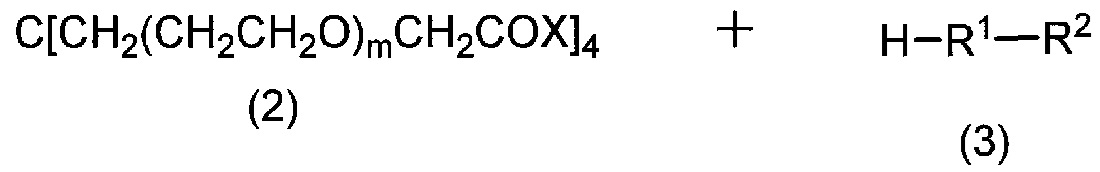
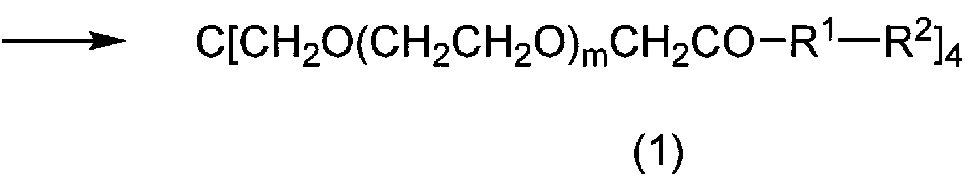
[0053] где X представляет собой гидроксильную группу, атом галогена или активный сложноэфирный фрагмент карбоксильной группы; и R1, R2 и m имеют такие же значения, как указано выше, соответственно.
[0054] То есть, соединение (1) по настоящему изобретению или его соль могут быть получены путем связывания карбоксильной группы производного тетракарбоновой кислоты формулы (2) с аминогруппой соединения формулы (3).
[0055] Производное тетракарбоновой кислоты (2) получают, например, путем взаимодействия пентаэритритола с этиленоксидом, с последующим карбоксиметилированием реакционного продукта и последующим галогенированием или активируемой этерификацией карбоксильной группы. Примеры атома галогена включают атом хлора и атом брома. Примеры активированного сложного эфира включают сукцинимид и смешанный ангидрид кислоты.
[0056] Среди производных соединения (3), соединение, в котором R1 представляет собой -N(R3)(CH2)n1CO- или -N(R4)(CH2)n2N(R5)CO(CH2)n3CO-, получают, например, путем взаимодействия соединения (3), в котором R1 представляет собой простую связь, с HN(R3)(CH2)n1COY или HN(R4)(CH2)n2N(R5)CO(CH2)n3COY. В данном случае, R3, R4, R5, n1, n2 и n3 имеют такие же значения, как указано выше, соответственно; и Y представляет собой гидроксильную группу, атом галогена или активированный сложноэфирный остаток.
Данное взаимодействие представляет собой реакцию амидного сочетания карбоновой кислоты и может быть осуществлено при использовании конденсирующего агента, такого как HBTU или DCC, в присутствии основания.
[0057] Взаимодействие между производным тетракарбоновой кислоты (2) и соединением (3) представляет собой реакцию амидного сочетания карбоновой кислоты и может быть осуществлено в общеизвестных условиях реакции амидирования. Например, реакция может быть осуществлена в условиях от 0°C до 150°C в присутствии амина, такого как триэтиламин или N,N-диметиланилин.
[0058] После завершения реакции, целевое вещество может быть очищено и выделено путем промывки, перекристаллизации и различными хроматографическими средствами.
[0059] Соединение (1) по настоящему изобретению или его соль обладают отличной противоопухолевой активностью в отношении злокачественной опухоли и сниженными неблагоприятными побочными эффектами, такими как снижение массы тела, и отличная противоопухолевая активность в отношении злокачественной опухоли достигается без необходимости постоянной инфузии, которая длится несколько часов или дольше. Поэтому, соединение (1) или его соль полезны в качестве отличного терапевтического средства против злокачественной опухоли со снижением обремененности для пациентов и лечащих врачей.
[0060] Соединение (1) по настоящему изобретению или его соль является таким, в котором в качестве связи между PEG и противораковым средством существует только амидная связь, и, таким образом, соединение или соль может избежать быстрого разложения лиазой в крови, такой как эстераза или карбоксилаза. В отличие от липосом или полимерных мицелл микрокарпускулярные препараты, которые могут стать объектом нападения фагоцитов, соединение (1) или соль, само по себе имеющее очень малый молекулярный размер, не подходит для того, чтобы быть атакованным фагоцитами вследствие характеристик PEG, и очень стабильно в крови. Кроме того, поскольку скорость почечной экскреции очень низка из-за большой молекулярной массы, направленное воздействие на опухоль очень эффективно. Это приводит к тому, что дозировка может быть существенно снижена по сравнению со случаями обычных низкомолекулярных противораковых средств, которые были ведены способами, такими как внутривенное введение или пероральное введение. Кроме того, в значительной степени можно уменьшить не только число введений, но также можно сократить продолжительность введения противоракового средства до времени около 30 минут, которое традиционно опиралось на длительное время постоянной инфузии.
[0061] Примеры злокачественной опухоли, которая становится мишенью описываемого соединения (1) по настоящему изобретению или его соли, включают рак головы и шеи, рак пищевода, рак желудка, рак толстой кишки, рак прямой кишки, рак печени, рак желчного пузыря и желчных протоков, рак поджелудочной железы, рак легких, рак молочной железы, рак яичников, рак шейки матки, рак матки, рак почек, рак мочевого пузыря, рак предстательной железы, опухоль яичка, саркому мягких тканей и костей, лейкемию, злокачественную лимфому, множественную миелому, рак кожи, опухоль головного мозга и мезотелиальную опухоль.
[0062] По поводу применения соединения (1) по настоящему изобретению или его соли, может быть получена фармацевтическая композиция различных форм путем смешивания данного соединения или его соли с фармацевтически приемлемым носителем, в случае необходимости. Примеры форм фармацевтической композиции включают пероральную форму, инъецируемый препарат, суппозиторий, пластырь и мазь; однако, желательно получение данного соединения или его соли в форме инъецируемого препарата.
[0063] Что касается фармацевтически приемлемого носителя, могут быть использованы различные органические или неорганические материалы носителей, которые обычно используются при получении композиций, и носитель включают в качестве эксципиента, связующего, дезинтегранта, лубриканта или красителя для твердых лекарственных средств; и в качестве растворителя, средства, способствующего растворению, суспендирующего агента, изотонического агента, буфера или смягчающего агента для жидких лекарственных средств. Кроме того, также могут быть использованы лекарственные добавки, такие как антисептик, антиоксидант, краситель, подсластитель и стабилизатор, в случае необходимости.
При получении твердого перорального лекарственного средства к соединению (1) по настоящему изобретению добавляют, например, эксципиент или наполнитель, связующее, дезинтегрант, лубрикант, краситель и вкусовой агент/корригент, и затем, например, таблетки, покрытые таблетки, гранулированный препарат, порошок и капсулы могут быть получены общеизвестными способами.
При получении инъецируемого препарата к соединению (1) по настоящему изобретению добавляют, например, поддерживающий pH агент, буферирующий агент, стабилизатор, изотонический агент и местный анестетик, и подкожно инъецируемый препарат, внутримышечно инъецируемый препарат и внутривенно инъецируемый препарат могут быть получены общеизвестными способами.
[0064] Для применения указанного лекарственного препарата по настоящему изобретению при лечении злокачественной опухоли в кровеносной системе, предпочтительно, чтобы указанный лекарственный препарат вводился средствами внутривенного введения в течение одного часа или средствами внутривенной капельной инфузии в течение нескольких часов после разведения лекарственного препарата физиологическим раствором или инфузионным раствором глюкозы.
Примеры
[0065] Здесь и далее, настоящее изобретение будет конкретно описано при помощи примеров и примеров тестирования; однако, они предназначены только для описания в иллюстративных целях и не предназначены для ограничения объема настоящего изобретения.
[0066] Пример 1
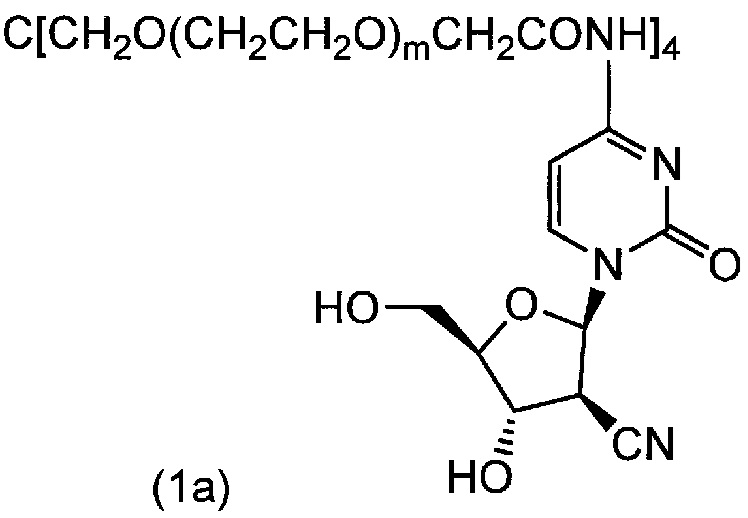
В атмосфере азота 5,0 моль гидрохлорида 1-(2'-циано-2'-дезокси-β-D-арабинофуранозил)цитозина, 10,0 моль триэтиламина и 8 моль диметилформамида добавляли в реакционный сосуд, и туда добавляли 1,0 моль тетра(сукцинимидилкарбоксиметилполиэтиленгликоль) пентаэритритола. Полученную смесь нагревали до 100°C и подвергали взаимодействию при перемешивании в течение 3 часов. Реакционную смесь охлаждали до 20°C-25°C, и затем реакционную смесь вносили в 100 мл метил-трет-бутилового эфира и перемешивали в течение одного часа. Исходные вещества отделяли фильтрованием. Остаток повторно подвергали данной операции, добавляя этанол при температуре от 50°C до 60°C, перемешивали, затем охлаждали до 20±5°C, перемешивали в течение 16 часов и промывали метил-трет-бутиловым эфиром, и полученный этанольный раствор охлаждали. Таким образом, соединение (1a) (m=230 в среднем) получали в виде белого порошка (выход 88%). Точка плавления: 54°C. Диаграмма ЯМР спектра соединения (1a) представлена на фиг.1.
[0067]
[0068] Пример 2
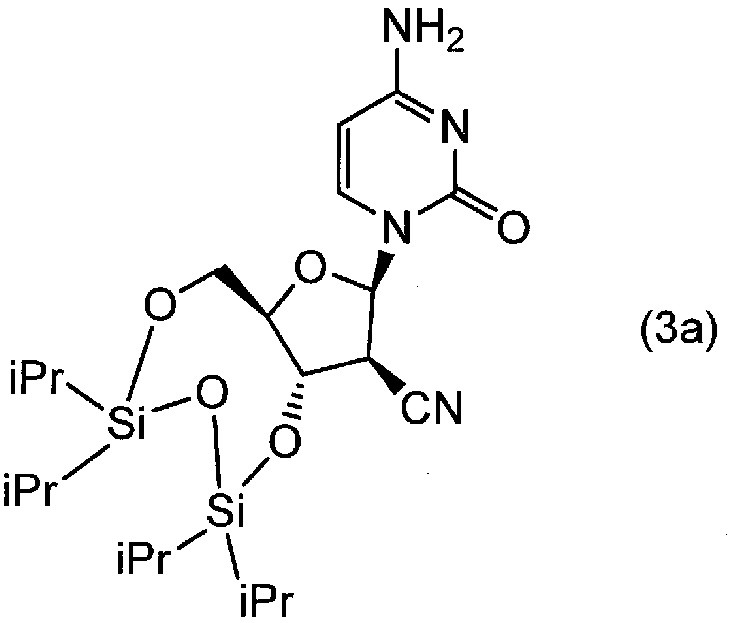
(1) 1 моль гидрохлорида 1-(2'-циано-2'-дезокси-β-D-арабинофуранозил)цитозина и 100 мл пиридина вводили в реакционный сосуд в атмосфере азота, и туда по каплям добавляли 1,2 моль тетраизопропилдисилоксандихлорида при температуре от 20°C до 25°C. Полученную смесь нагревали до 45±5°C и перемешивали в течение 2 часов. Туда добавляли гексан при температуре от 20°C до 25°C, и продукт выделяли в виде твердого вещества. Продукт вносили в воду при температуре от 20°C до 25°C, и полученную смесь перемешивали в течение 1 часа и 30 минут. После фильтрования полученной смеси, остаток промывали водой и гексаном, и сушили до твердого вещества упариванием. Таким образом, получали соединение (3a).
[0069]
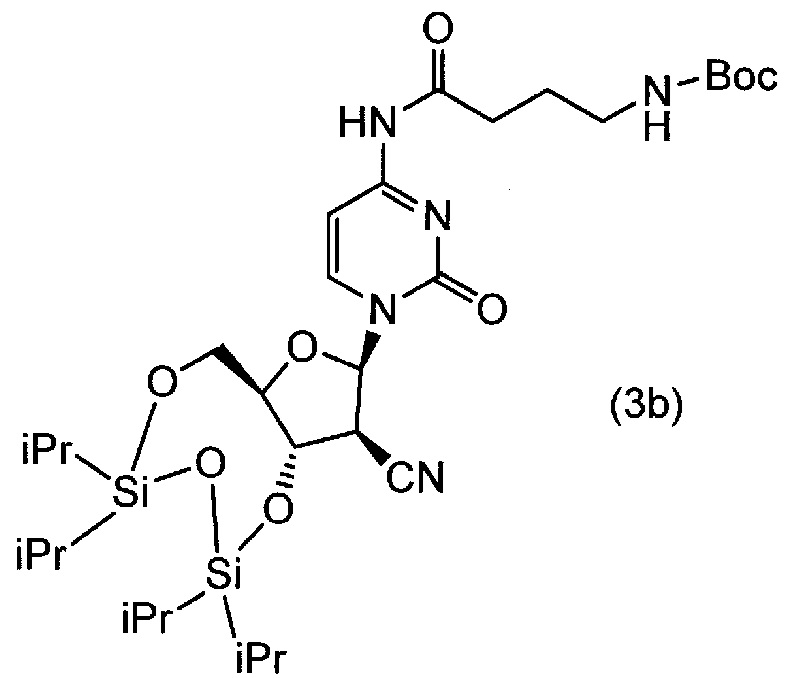
[0070] (2) 1,2 моль N-трет-бутоксикарбонилаланин, 1,5 моль HBTU, 2,0 моль триэтиламина и 300 мл дихлорметана вводили в реакционный сосуд в атмосфере азота, и полученную смесь перемешивали в течение одного часа при температуре от 20°C до 25°C. Туда добавляли 1 моль соединения (3a) при температуре от 20°C до 25°C, и полученную смесь нагревали до 35±5°C и перемешивали в течение 16 часов. Туда добавляли насыщенный водный раствор NaHCO3, и органическую фазу отделяли. Полученную органическую фазу промывали, соответственно, насыщенным водным раствором NaHCO3, насыщенным NH4Cl и физиологическим раствором, и полученную органическую фазу сушили над безводным Na2SO4. После фильтрования, фильтрат концентрировали при пониженном давлении и подвергали колоночной хроматографии на силикагеле (при элюировании этилацетатом). Таким образом, получали соединение (3b) (выход 77%).
[0071]
[0072] (3) 1 моль соединения (3b), 2 моль уксусной кислоты и 200 мл тетрагидрофурана вводили в реакционный сосуд в атмосфере азота, полученную смесь охлаждали до 0±5°C, и затем туда добавляли 1,5 моль тетра-н-бутиламмонийфторида (TBAF) при 0±5°C. Полученной смеси давали возможность взаимодействовать в течение одного часа, и затем отгоняли растворитель.
Реакционную смесь, описанную выше, и 300 мл этилацетата вводили в реакционный сосуд в атмосфере азота, и туда добавляли 2M смесь HCl/этилацетат при 25±5°C. Исходные вещества быстро растворялись в растворе концентрированная HCl/этилацетат, и спустя 5 минут, требуемый продукт начинал отделяться в виде твердой фракции. Полученную смесь перемешивали в течение одного часа, фильтровали и затем промывали этилацетатом. Таким образом, получали соединение (3c).
[0073]
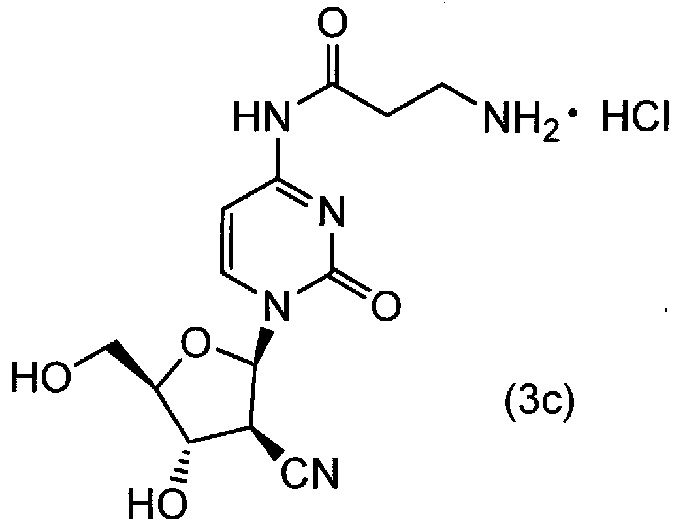
[0074] (4) соединение (3c) подвергали взаимодействию с простым тетра(сукцинимидилкарбоксиметилполиэтиленгликоль) пентаэритритоловым эфиром по методике, аналогично примеру 1, и целевое соединение (1b) (m=230 в среднем) получали в виде белого порошка (выход 83%). Диаграмма ЯМР спектра соединения (1b) представлена на фиг.2.
[0075]
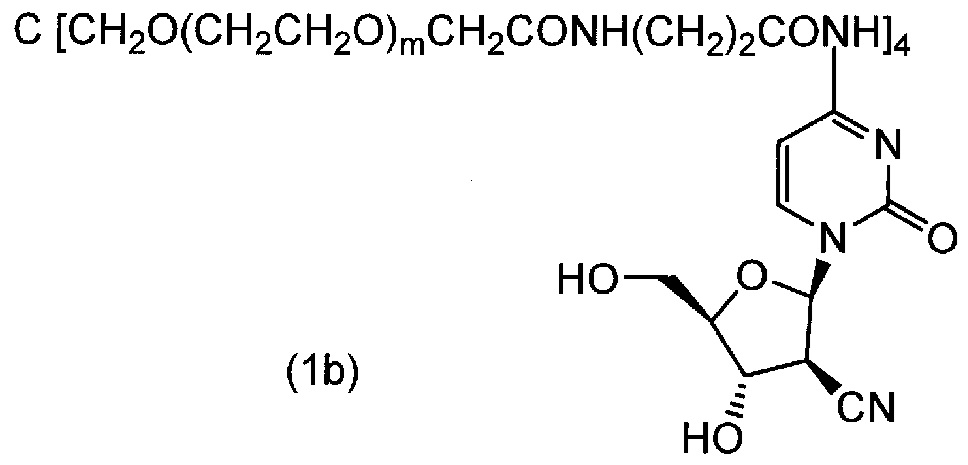
[0076] Пример 3
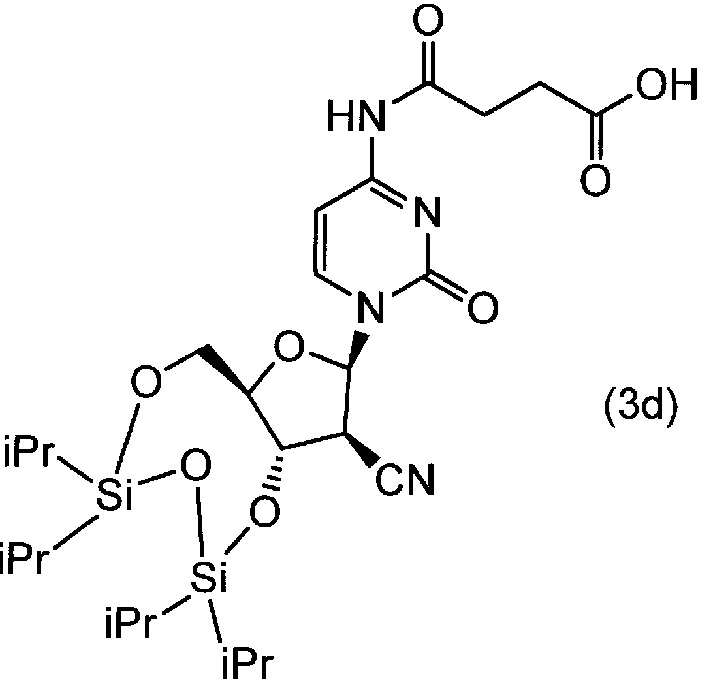
(1) 1 моль соединения (3a), полученного в примере 2(1), 4 моль янтарного ангидрида и 50 мл пиридина вводили в реакционный сосуд в атмосфере азота, и полученную смесь нагревали до 40±5°C и перемешивали в течение 2 часов. Реакционную смесь добавляли в смесь воды и этилацетата, pH полученной смеси доводили до 5 при помощи 1M хлористоводородной кислоты, и полученную смесь перемешивали в течение 30 минут при 30±5°C. Органическую фазу отделяли, водную фазу три раза экстрагировали этилацетатом, и полученные таким образом экстрагированные органические фазы объединяли и промывали физиологическим раствором. Полученную органическую фазу сушили над безводным Na2SO4 и фильтровали, и затем фильтрат концентрировали при пониженном давлении и затем сушили. Таким образом, получали соединение (3d).
[0077]
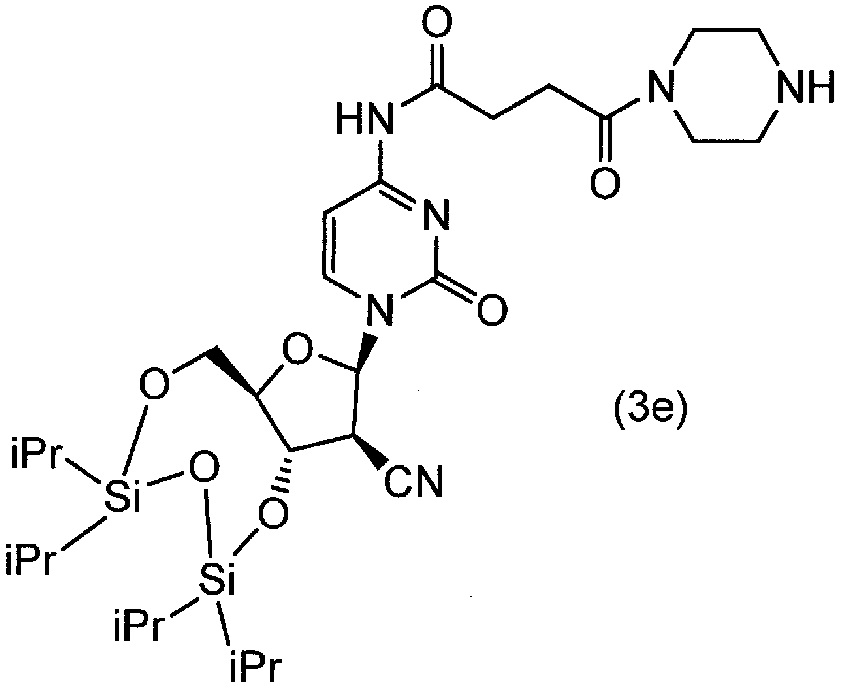
[0078] (2) 1 моль соединения (3d), 2 моль HBTU, 4 моль триэтиламина и 100 мл диметилформамида вводили в реакционный сосуд в атмосфере азота, и полученную смесь перемешивали в течение 5 минут при 30±5°C. Туда добавляли 4 моль пиперидина, и полученную смесь перемешивали в течение одного часа при 30±5°C. Туда добавляли этилацетат и воду, органическую фазу отделяли, и полученную органическую фазу промывали два раза физиологическим раствором и сушили над безводным Na2SO4. После фильтрования, фильтрат концентрировали при пониженном давлении, подвергали колоночной хроматографии на силикагеле при элюировании смесью дихлорметан/MeOH. Элюированную фракцию сушили до твердого вещества при пониженном давлении и получали соединение (3e).
[0079]
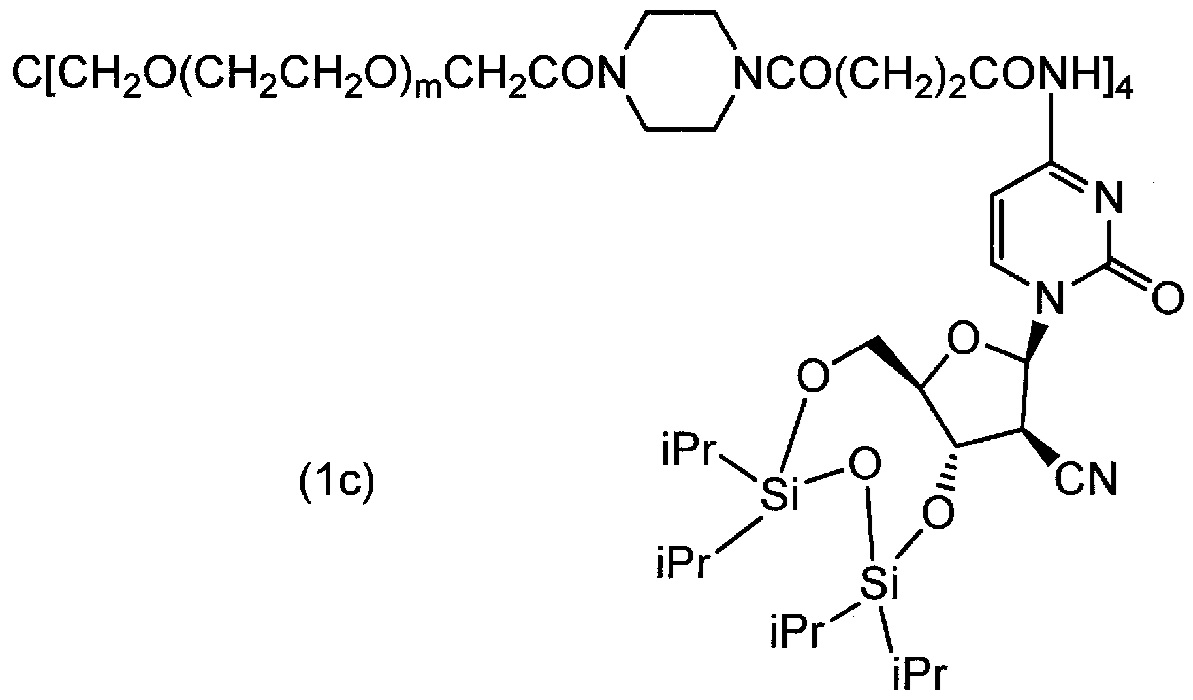
[0080] (3) соединение (3e) подвергали взаимодействию с простым тетра(сукцинимидилкарбоксиметилполиэтиленгликоль) пентаэритритоловым эфиром по методике, аналогично примеру 1, и целевое соединение (1c) (m=230 в среднем) получали в виде белого порошка (выход 94%). Диаграмма ЯМР спектра соединения (1c) представлена на фиг.3.
[0081]
[0082] Пример 4
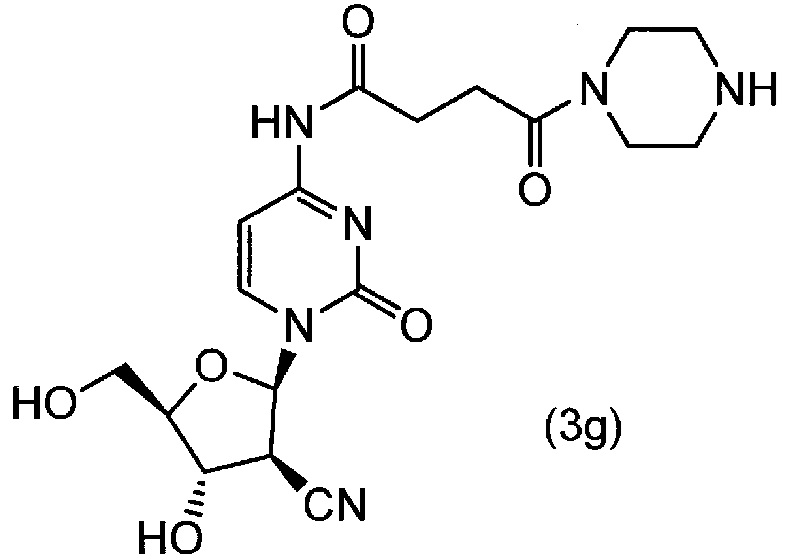
(1) 1 моль соединения (3e) примера 3(2), 2,3 моль уксусной кислоты и 50 мл диметилформамида вводили в реакционный сосуд в атмосфере азота, и полученную смесь охлаждали до 0±5°C. Туда добавляли 1,6 моль тетра-н-бутиламмонийфторида (TBAF) при 0±5°C, и полученную смесь перемешивали в течение одного часа. Таким образом, получали соединение (3g).
[0083]
[0084] (2) Соединение (3g) подвергали взаимодействию с простым тетра(сукцинимидилкарбоксиметилполиэтиленгликоль) пентаэритритоловым эфиром по методике, аналогично примеру 1, и получали соединение (1d) (m=230 в среднем). Диаграмма ЯМР спектра соединения (1d) представлена на фиг.4.
[0085]

[0086] Пример 5
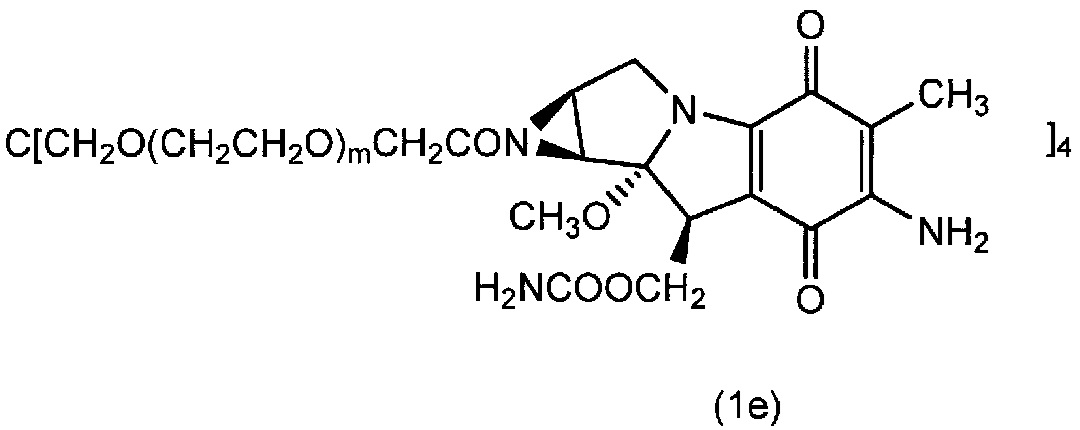
1,0 моль тетра(карбоксиметилполиэтиленгликоль) пентаэритритола, 4,8 моль митомицина C, 5,0 моль HBTU, 8 моль триэтиламина и 300 мл диметилформамида добавляли в реакционный сосуд в атмосфере азота, и полученной смеси давали возможность взаимодействовать в течение 3 часов при 40±5°C. Полученную смесь охлаждали до 20°C-25°C и затем обрабатывали по методике, аналогично примеру 1. Таким образом, получали соединение (1e) в виде белого порошка (m=230 в среднем) (выход 91.4%). Диаграмма ЯМР спектра соединения (1e) представлена на фиг.5.
[0087]
[0088] Пример 6
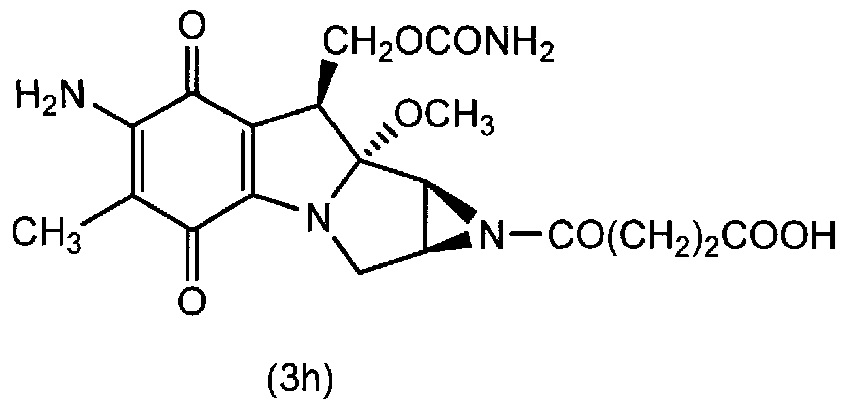
(1) Соединение (3h) получали по методике, аналогично примеру 3(1), используя митомицин C вместо соединения (3a).
[0089]
[0090] (2) Соединение (3i) получали по методике, аналогично примеру 4(1), используя соединение (3h) вместо соединения (3e).
[0091]
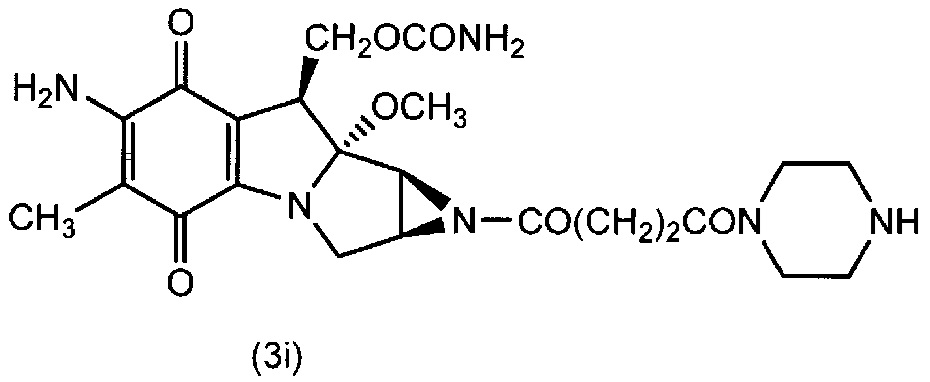
[0092] (3) Соединение (1f) получали (m=230 в среднем) по методике, аналогично примеру 4(2), используя соединение (3i) вместо соединения (3g). Диаграмма ЯМР спектра соединения (1f) представлена на фиг.6.
[0093]

[0094] Пример 7
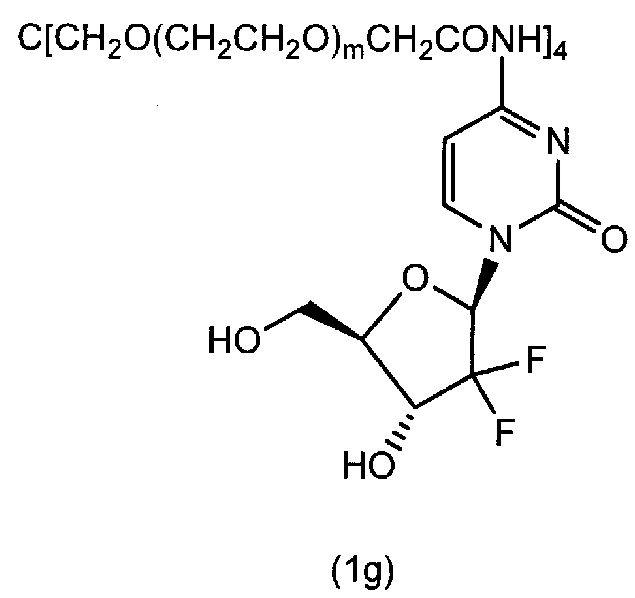
Соединение (1g) (m=230 в среднем) получали в виде белого порошка (выход 82.3%) по методике, аналогично примеру 1, используя гемцитабин и тетра(сукцинимидилкарбоксиметилполиэтиленгликоль)пентаэритритол. Точка плавления 57°C. Диаграмма1H-ЯМР спектра представлена на фиг.7.
[0095]
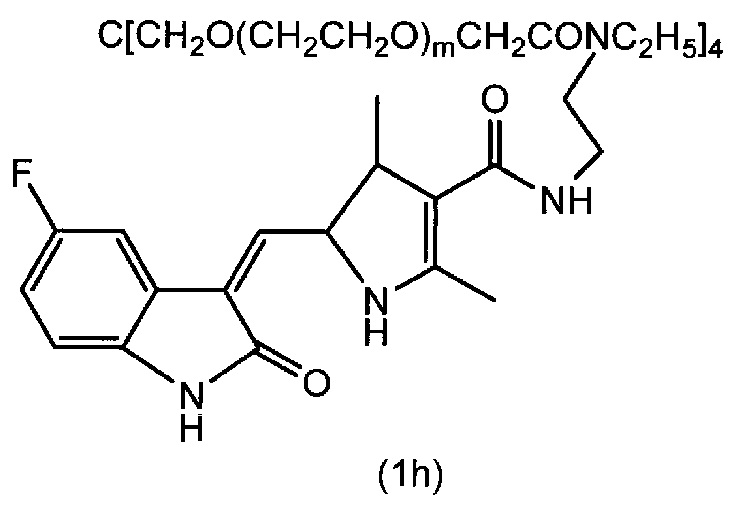
[0096] Пример 8
Соединение (1h) (m=230 в среднем) получали в виде желтого порошка (выход 87%) по методике, аналогично примеру 1, используя деэтилированную форму сунитиниба и тетра(сукцинимидилкарбоксиметилполиэтиленгликоль)пентаэритритол. Точка плавления 55°C. Диаграмма1H-ЯМР спектра представлена на фиг.8.
[0097]
[0098] Пример 9
Соединение (1i) получали в виде белого порошка (выход 87,8%) по методике, аналогично примеру 4, используя лапатиниб, тетра(карбоксиметилполиэтиленгликоль)пентаэритритол и HBTU. Точка плавления 56°C. Диаграмма1H-ЯМР спектра представлена на фиг.9.
[0099]

[0100] Пример 10
(1) Паклитаксел подвергали взаимодействию с трифторуксусной кислотой и таким образом отделяли трет-бутоксикарбонильную группу паклитаксела.
(2) Соединение (1j) получали (выход 84,2%) по методике, аналогично примеру 1, используя трет-бутоксикарбонил-отсоединенную форму паклитаксела и тетра(сукцинимидилкарбоксиметилполиэтиленгликоль)пентаэритритол. Диаграмма1H-ЯМР спектра представлена на фиг.10.
[0101]

[0102] Пример тестирования 1
5×106 клеток рака поджелудочной железы трансплантировали в правую часть живота голых мышей BALB/c, и спустя 7 дней, во временной точке, на которой средний размер опухоли достигал 100 мм3, начинали введение лекарственного средства. Вплоть до 29 дней от времени введения лекарственного средства, измеряли массу тела мышей и объем опухоли. Результаты представлены на фиг.11, фиг.12 и в таблице 1. Непрерывную подкожную инфузию гидрохлорида 1-(2'-циано-2'-дезокси-β-D-арабинофуранозил)цитозина (DFP-10917) при дозе 4,5 мг/кг/день постоянно проводили в течение двух недель с помощью микронасоса, встроенного в тело мыши. В отличие от этого, соединение (1a) внутривенно вводили раз в неделю при дозе 100 мг/кг, 200 мг/кг или 300 мг/кг. Дозирование соединением (1a) проводили введением раз в неделю дозы 2,4 мг/кг, 4,8 мг/кг или 7,2 мг/кг, в пересчете на DFP-10197. В качестве контроля вводили буферный раствор (pH=5,0) ацетата натрия.
[0103] Исходя из фиг.11 и фиг.12, было подтверждено, что соединение (1a) оказывает отличный терапевтический эффект на опухоль при внутривенном введении раз в неделю, практически без снижения массы тела.
[0104]
[0105] Пример тестирования 2
Противоопухолевую активность митомицина C и соединения (1e) исследовали по методике, аналогично примеру тестирования 1. Митомицин C (MMC) внутривенно вводили раз в неделю при дозе 3 мг/кг/день. В отличие от этого, соединение (1e) внутривенно вводили раз в неделю при дозе 25 мг/кг, 50 мг/кг, 100 мг/кг или 200 мг/кг. Дозирование соединением (1e) проводили введением раз в неделю дозы 0,8 мг/кг, 1,7 мг/кг, 3,3 мг/кг или 6,7 мг/кг, в пересчете на MMC.
[0106] Исходя из фиг.13 и фиг.14, было подтверждено, что соединение (1e) оказывает отличный терапевтический эффект на опухоль при внутривенном введении раз в неделю, практически без снижения массы тела.
[0107] Пример тестирования 3
5×106 клеток рака легких человека A549 трансплантировали в правую часть живота голых мышей BALB/c, и спустя 14 дней, во временной точке, на которой средний размер опухоли достигал 127 мм3, начинали введение лекарственного средства. Соединение (1a) внутривенно вводили раз в неделю при дозе 200 мг/кг (количество в пересчете на DFP-10917 составляло 4,8 мг/кг), и пеметрексед, который является стандартным препаратом для рака легких, внутрибрюшинно вводили раз в неделю при дозе 300 мг/кг. В качестве контроля, внутривенно вводили физиологический раствор раз в неделю. Обследование проводили в течение двух недель (в общей сложности, два раза).
В результате, соединение (1a), которое вводили при дозе 4,8 мг/кг в пересчете на DFP-10917, оказывает терапевтический эффект на опухоль, эквивалентный эффекту у группы, которой вводили 300 мг/кг пеметрекседа.
[0108]
[0109] Пример тестирования 4
1×107 клеток рака молочной железы человека BT474 трансплантировали в правую часть живота голых мышей BALB/c, и лекарственные средства вводили по методике, аналогично примеру тестирования 3. DFP-10917 и соединение (1a) вводили внутривенно раз в неделю, в общей сложности, два раза. В качестве контроля вводили физиологический раствор раз в неделю.
[0110] Исходя из результатов, показанных в таблице 3, соединение (1a) оказывает противоопухолевый эффект эквивалентный эффекту гемцитабина, несмотря на то, что соединение (1a) вводили при дозе 17,6/200, в пересчете на DFP-10917, по отношению к дозе самого DFP-10917.
[0111]
[0112] Пример тестирования 5
5×106 клеток рака поджелудочной железы человека Panc-1 трансплантировали в правую часть живота голых мышей BALB/c, и лекарственные средства вводили по методике, аналогично примеру тестирования 3. Соединение (1g) внутривенно вводили раз в неделю, в общей сложности, два раза или дважды в неделю. Гемцитабин вводили таким же образом по 100 мг/кг четырьмя разделенными порциями около 3 дней, и внутривенное введение повторяли, в общей сложности, четыре раза. В качестве контроля вводили физиологический раствор раз в неделю.
[0113] Исходя из результатов, представленных в таблице 4, соединение (1g) оказывает противоопухолевый эффект, который составил около 1/2 эффекта гемцитабина, несмотря на то, что соединение (1g) вводили при дозе от 1/35 до 1/40, в пересчете на гемцитабин, по отношению к дозе самого гемцитабина.
[0114]
[0115] Пример тестирования 5
Клетки клеточного рака легких человека A549 трансплантировали в правую часть живота голых мышей BALB/c, и спустя 11 дней, во временной точке, на которой средний размер опухоли достигал 200 мм3, начинали введение лекарственного средства. График дозирования представлен в таблице 5 и таблице 6.
[0116] Результаты предоставлены в таблице 5, таблице 6, на фиг.15 и фиг.16.
[0117]
[0118]
[0119] Исходя из таблицы 5, соединение (1h) оказывает противоопухолевый эффект, эквивалентный эффекту деэтилированной формы сунитиниба, при дозе от 4/50 до 8/50 в пересчете на сунитиниб, по отношению к дозе самого сунитиниба. Исходя из таблицы 6 и фиг.15, соединение (1i) оказывает противоопухолевый эффект превышающий эффект лапатиниба при дозе от 11,6/100 до 17,4/100, в пересчете на лапатиниб, по отношению к дозе самого лапатиниба.
Кроме того, следует признать, что соединение (1h) и соединение (1i) не вызывают какого-либо снижения массы тела при дозах, описанных выше, и было подтверждено, что эти соединения являются также высоко безопасными (фиг.16).
[0120] Пример тестирования 6
1×107 клетки рака молочной железы человека BT474 трансплантировали в правую часть живота голых мышей BALB/c, и когда объем опухоли увеличился до 100-150 мм3, вводили лекарственные средства. Введение реализовывали раз в неделю в течение двух недель.
[0121] Исходя из результатов, показанных в таблице 7, соединение (1j) показывает соотношение ингибирования роста опухоли 36%-37,5% и оказывает эффект, эквивалентный или превышающий эффект паклитакселя в качестве контроля, который давал соотношение ингибирования роста опухоли 29,8%. Таким образом, было подтверждено, что соединение (1j) является соединением, при помощи которого можно добиться превосходного баланса между эффективностью и безопасностью, и что соединение (1j), само по себе, обладает высокой растворимостью в воде, а также, что соединение может являться новым терапевтическим средством против злокачественной опухоли, которое может значительно улучшить средства для введения в клинических условиях, по сравнению с паклитакселом, который умеренно растворим в воде.
[0122]
Реферат
Настоящее изобретение относится к соединению формулы (1) или его фармацевтически приемлемой солигде Rпредставляет собой простую связь, -N(R)(CH)CO- или -N(R)(CH)N(R)CO(CH)CO- (где Rпредставляет собой атом водорода или алкильную группу, имеющую 1-6 атомов углерода; Rи Rсвязаны вместе и представляют собой алкиленовую группу, имеющую 1-4 атома углерода; и n1, n2 и n3 являются одинаковыми или отличаются друг от друга, каждый равен целому числу от 1 до 3); Rпредставляет собой (2'-циано-2'-дезокси-β-D-арабинофуранозил)цитозин или группу формул (b), (d), (e) или (f),,,,где Rпредставляет собой алкильную группу, имеющую 1-6 атомов углерода; и Rпредставляет собой атом водорода или алканоильную группу, имеющую 2-6 атомов углерода; m равен числу от 10 до 1000; и стрелка обозначает место связывания. Также раскрыты варианты применения указанного выше соединения, лекарственное средство и фармацевтическая композиция для лечения злокачественной опухоли, его содержащие, и способ лечения злокачественной опухоли с его использованием. Технический результат - разработка нового терапевтического средства против злокачественной опухоли, которое является весьма безопасным, позволяет сократить негативные побочные эффекты, такие как тяжелая желудочно-кишечная токсичность или костная токсичность, имеет устойчивый противоопухолевый эффект и позволяет улучшить средства для введения и частоту введения. 6 н. и 2 з.п. ф-лы, 16 ил., 7 табл., 10 пр.
Формула
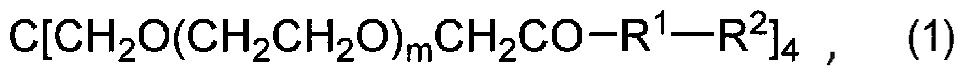

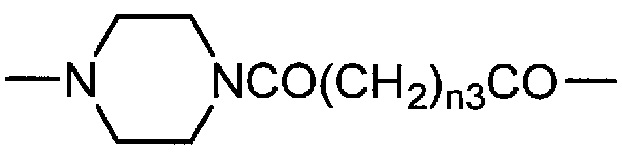
Документы, цитированные в отчёте о поиске
Твердые формы анти-egfr антител
Комментарии