Ингибиторы моноаминоксидазы (мао-в) - RU2392267C2
Код документа: RU2392267C2
Описание
Данное изобретение относится к бензилоксипроизводным общей формулы

где
R1 и R2 независимо друг от друга представляют собой Н или C1-C6-алкил;
R3 и R4 независимо друг от друга представляют собой Н или C1-C6-алкил;
R5 представляет собой галоген, CN, (С1-С6)-алкил или (С1-С6)-алкокси;
n, m или о представляют собой 0, 1 или 2;
а также к их фармацевтически приемлемым солям.
Данное изобретение включает как индивидуальные изомеры соединений формулы I, так и их рацемические и нерацемические смеси.
Соединения формулы I и их фармацевтически приемлемые соли, как индивидуальные изомеры соединений формулы I, так и их рацемические и нерацемические смеси (в дальнейшем: Фармацевтическое Соединение) обладают фармакологической активностью и пригодны для использования в качестве фармацевтических средств. В частности, Фармацевтические Соединения ингибируют активность моноамин оксидазы В.
Моноамин оксидаза (МАО) является флавин-содержащим ферментом, отвечающим за окислительное дезаминирование эндогенных моноаминных нейротрансмиттеров, таких как дофамин, серотонин, адреналин или норадреналин, и следовых аминов, например фенилэтиламина, а также ряда аминных ксенобиотиков. Данный фермент существует в двух формах, МАО-А и МОА-В, кодируемых разными генами (A.W.Bach et al., Proc. Natl. Acad. Sci. USA 1988, 85, 4934-4938) и отличающихся тканевым распределением, строением и субстратной специфичностью. МАО-А обладает более высоким сродством к серотонину, октопамину, адреналину и норадреналину; тогда как фенилэтиламин и тирамин являются природными субстратами для МАО-В. Считается, что дофамин окисляется обеими изоформами. МАО-В широко распределен в некоторых органах, включая мозг (A.M.Cesura and A.Pletscher, Prog. Drug Research 1992, 38, 171-297). Полагают, что с возрастом МАО-В-активность мозга увеличивается. Это увеличение было объяснено глиозом, связанным со старением (C.J.Fowler et al., J.Neural.Transm.1980, 49, 1-20). Кроме того, активность МАО-В значительно выше в мозге пациентов с болезнью Альцгеймера (P.Dostert et al., Biochem. Pharmacol. 1989, 38, 555-561) и, как было найдено, высоко выражена в астроцитах вокруг старческих бляшек (Saura et.al., Neoroscience 1994, 70, 755-774). В связи с этим, так как окислительное дезаминирование первичных моноаминов под действием МАО приводит к NH3, альдегидам и H2O2, веществам с установленной или потенциальной токсичностью, предполагается, что существует разумное объяснение использованию селективных МАО-В-ингибиторов для лечения слабоумия и болезни Паркинсона. Ингибирование МАО-В вызывает снижение ферментной инактивации дофамина и, таким образом, пролонгирование работоспособности нейротрансмиттера в дофаминергических нейронах. Также дегенеративные процессы, связанные с возрастом и болезнями Альцгеймера и Паркинсона, можно объяснить окислительным стрессом из-за увеличенной активности МАО и, как следствие, увеличенного образования H2O2 под действием МАО-В. Поэтому МАО-В-ингибиторы могут влиять как на уменьшение образования радикалов кислорода, так и повышение уровней моноаминов в мозге.
Учитывая вышеупомянутое влияние МАО-В на неврологические нарушения, существует значительный интерес получить эффективные и селективные ингибиторы, которые позволили бы контролировать эту ферментативную активность. Например, фармакология некоторых известных МАО-В обсуждается D.Bentue-Ferrer et.al. в CNS Drugs 1996, 6, 217-236. Главным ограничением активности необратимого и неселективного МАО-ингибитора является необходимость соблюдения диетических ограничений из-за риска проявления гипертонического криза при употреблении тирамина с пищей, а также возможность взаимодействий с другими лекарственными препаратами (D.M.Gardner et al., J.Clin. Psychiatry 1996, 57, 99-104), а к обратимым и селективным МАО-ингибиторам, в особенности к МАО-В, эти неблагоприятные факты относятся в меньшей степени. Таким образом, существует потребность в МАО-В-ингибиторах с высокой селективностью и без неблагоприятных побочных эффектов, обычных для необратимых МАО-ингибиторов с низкой селективностью к ферменту.
Фармацевтические Соединения соответственно полезны как селективные ингибиторы моноамин оксидазы В, например, при лечении или предотвращении болезней и состояний, при которых играет роль или включается активность моноамин оксидазы В. В особенности подобные состояния включают острые и/или хронические неврологические нарушения.
Острые и/или хронические неврологические нарушения включают психоз, шизофрению, болезнь Альцгеймера, когнитивные расстройства и нарушения памяти, подобные умеренному когнитивному расстройству, возрастному когнитивному спаду, мультиинфарктной деменции, болезни Паркинсона, нарушению памяти, связанному с депрессией или тревогой, синдрому Дауна, инсульту, травматическому повреждению мозга и синдрому дефицита внимания. Другие состояния, которые можно лечить, представляют собой ограниченную функцию мозга, вызванную операциями шунтирования или трансплантациями, недостаточное мозговое кровоснабжение, травмы спинного мозга, травмы головы, гипоксию, вызванную беременностью, остановку сердца и гипогликемию. Также состояниями, которые можно лечить, являются острая и хроническая боль, хорея Хантингтона, боковой амиотрофический склероз (ALS), слабоумие, вызванное ВИЧ, травмы глаз, ретинопатия, идиопатический паркинсонизм или лекарственный паркинсонизм, а также состояния, которые ведут к глутамат-недостаточным функциям, таким, например, как спазмы мышц, конвульсии, мигрени, недержание мочи, никотиновая зависимость, психотические приступы, опиатная зависимость, тревога, рвота, дискенезия и депрессия.
В одном воплощении острым и/или хроническим неврологическим нарушением является болезнь Альцгеймера. В другом воплощении острое и/или хроническое неврологическое нарушение представляет собой умеренное когнитивное расстройство или старческое слабоумие.
Таким образом, задача настоящего изобретения заключается в создании соединений, которые должны обладать обозначенными выше полезными свойствами. Было найдено, что соединения формулы I настоящего изобретения и их фармацевтически приемлемые соли показывают потенциал, чтобы считаться высокоселективными МАО-В-ингибиторами. Кроме того, предметами настоящего изобретения также являются лекарства, основанные на соединении формулы I в соответствии с данным изобретением, способ производства соединений формулы I и их фармацевтически приемлемых солей, а также применение соединений формулы I для лечения или предотвращения болезней, опосредованных ингибиторами моноамин оксидазы В, и соответственно применение для изготовления соответствующих лекарств.
Следующие определения общих терминов, используемых в настоящей заявке, применяются независимо от того, появляются ли эти термины отдельно или в комбинации. Следует отметить, что используемые в описании и приложенной формуле изобретения формы единственного числа включают и формы множественного числа, за исключением тех случаев, когда из контекста ясно не следует противоположное.
Термин «С1-С6-алкил» («низший алкил»), используемый в настоящей заявке, обозначает неразветвленные или разветвленные насыщенные углеводородные остатки с 1-6 атомами углерода, предпочтительно с 1-4 атомами углерода, такие как метил, этил, н-пропил, изо-пропил, н-бутил, втор-бутил, трет-бутил и т.п.
Термин «галоген» обозначает фтор, хлор, бром и иод.
«Алкокси» или «(С1-С6)-алкокси» означает остаток -O-R, где R представляет собой остаток низшего алкила, как определено здесь. Примеры алкоксирадикалов включают, но не ограничиваются ими, метокси, этокси, изопропокси и т.п.
Термин «фармацевтически приемлемые соли» соединения означает соли, которые являются фармацевтически приемлемыми: которые в целом являются безопасными, нетоксичными и не являются нежелательными ни биологически, ни как-либо иначе не подходящими и которые обладают требуемой фармакологической активностью исходного соединения. Данные соли получают из неорганической или органической кислоты или основания.
Такие соли включают:
(1) кислотно-аддитивные соли, образованные неорганическими кислотами, такими как соляная кислота, бромисто-водородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п.; или образованные органическими кислотами, например уксусной кислотой, бензолсульфоновой кислотой, бензойной кислотой, камфорсульфоновой кислотой, лимонной кислотой, этансульфоновой кислотой, фумаровой кислотой, глюкогептиловой кислотой, глюконовой кислотой, глутаминовой кислотой, гликолевой кислотой, гидроксинафтойной кислотой, 2-гидроксиэтансульфоновой кислотой, молочной кислотой, малеиновой кислотой, яблочной кислотой, миндальной кислотой, метансульфоновой кислотой, муконовой кислотой, 2-нафталенсульфоновой кислотой, пропионовой кислотой, салициловой кислотой, янтарной кислотой, дибензоил-L-винной кислотой, винной кислотой, п-толуолсульфоновой кислотой, триметилуксусной кислотой, 2,2,2-трифторуксусной кислотой и т.п.; или
(2) соли, образованные либо при замещении кислотного протона в исходном соединении ионом металла, например ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; или при координации с органическим или неорганическим основанием. Приемлемые органические основания включают диэтаноламин, этаноламин, N-метилглюкамин, триэтаноламин, трометамин и т.п. Приемлемые неорганические основания включают гидроксид алюминия, гидроксид кальция, гидроксид калия, карбонат натрия и гидроксид натрия.
Следует понимать, что все ссылки на фармацевтически приемлемые соли включают формы включения растворителя (сольваты) или кристаллические формы (полиморфы) той же кислотно-аддитивной соли.
«Фармацевтически приемлемый», например фармацевтически приемлемый носитель, эксципиент и т.д., означает фармакологически приемлемый и по существу нетоксичный для субъекта, которому вводится конкретное соединение.
«Терапевтически эффективное количество» означает количество, которое эффективно, для того чтобы предотвратить, облегчить или улучшить симптомы болезни или продлить жизнь объекта, проходящего курс лечения.
Кроме того, как используется здесь, термин «млекопитающее, которое нуждается в лечении острого и/или хронического неврологического нарушения» означает млекопитающее, например человека, которое страдает или подвергается риску страдать острым и/или хроническим неврологическим нарушением.
Используемые термины «лечить», «лечимый», «лечение» и т.п., применимые к острому и/или хроническому неврологическому нарушению, относятся как к способам, которые замедляют, улучшают, ослабляют или устраняют подобное нарушение или любые симптомы, связанные с данным нарушением, беспокоящие субъекта, так и к способам, которые предотвращают данное нарушение или любые симптомы его проявления.
Среди соединений настоящего изобретения предпочтительными являются некоторые соединения формулы I или их фармацевтически приемлемые соли.
Предпочтительными соединениями формулы I являются те, в которых о представляет собой 1, a m представляет собой 0, например следующие соединения:
2-(4-бензилокси-фенокси)-N-метил-ацетамид,
2-[4-(4-циано-бензилокси)-фенокси]-N-метил-ацетамид,
2-[4-(4-хлор-бензилокси)-фенокси]-N-метил-ацетамид,
2-[4-(2-фтор-бензилокси)-фенокси]-N-метил-ацетамид,
2-[4-(3-фтор-бензилокси)-фенокси]-N-метил-ацетамид,
2-[4-(4-фтор-бензилокси)-фенокси]-N-метил-ацетамид,
(RS)-2-[4-(3-фтор-бензилокси)-фенокси]-N-метил-пропионамид,
(RS)-2-[4-(3-хлор-бензилокси)-фенокси]-N-метил-пропионамид,
(S или R)-2-[4-(3-хлор-бензилокси)-фенокси]-N-метил-пропионамид,
(RS)-2-[4-(4-циано-бензилокси)-фенокси]-N-метил-пропионамид,
(RS)-2-[4-(3-фтор-бензилокси)-фенокси]-N-метил-бутирамид,
(RS)-2-[4-(3-хлор-бензилокси)-фенокси]-N-метил-бутирамид или
(RS)-2-[4-(4-циано-бензилокси)-фенокси]-N-метил-бутирамид.
Также предпочтительными являются соединения формулы I, в которых о представляет собой 2 и m представляет собой 0, например 3-[4-(3-фтор-бензилокси)-фенокси]-пропионамид.
Другой предпочтительной группой соединений формулы I являются те, где о представляет собой 0, а m представляет собой 2, например 2-[4-(3-фтор-бензилокси)-фенил]-этиловый эфир карбаминовой кислоты.
Также предпочтительными являются соединения формулы I, где о представляет собой 0 и m представляет собой 1, например 4-(3-фтор-бензилокси)-бензиловый эфир метилкарбаминовой кислоты.
Кроме того, предпочтительной группой соединений формулы I являются те, где о представляет собой 1 и m представляет собой 1, например 2-[4-(3-фтор-бензилокси)-бензилокси]-ацетамид.
Данные соединения общей формулы I и их фармацевтически приемлемые соли можно получить известными из предшествующего уровня техники методами, например, описанными ниже способами, при которых
а) соединение формулы

подвергают взаимодействию с соединением формулы

где Y является уходящей группой, с получением соединения формулы

где заместители такие, как описано выше, или
б) соединение формулы

где R6 представляет собой С1-С6-алкил, подвергают взаимодействию с амином формулы

с получением соединения формулы

где заместители являются такими, как описано выше, или
в) соединение формулы
подвергают взаимодействию с KOCN или с R3-N=C=O (VII) с получением соединения формулы

где заместители являются такими, как описано выше, или
г) соединение формулы

подвергают взаимодействию с соединением формулы HNR3R4 (V) с получением соединения формулы

где заместители являются такими, как описано выше, и
при желании переводят полученные соединения в фармацевтически приемлемые кислотно-аддитивные соли.
Согласно настоящему изобретению на схемах 1-3 показана возможность получения соединений общей формулы I.
Схема 1
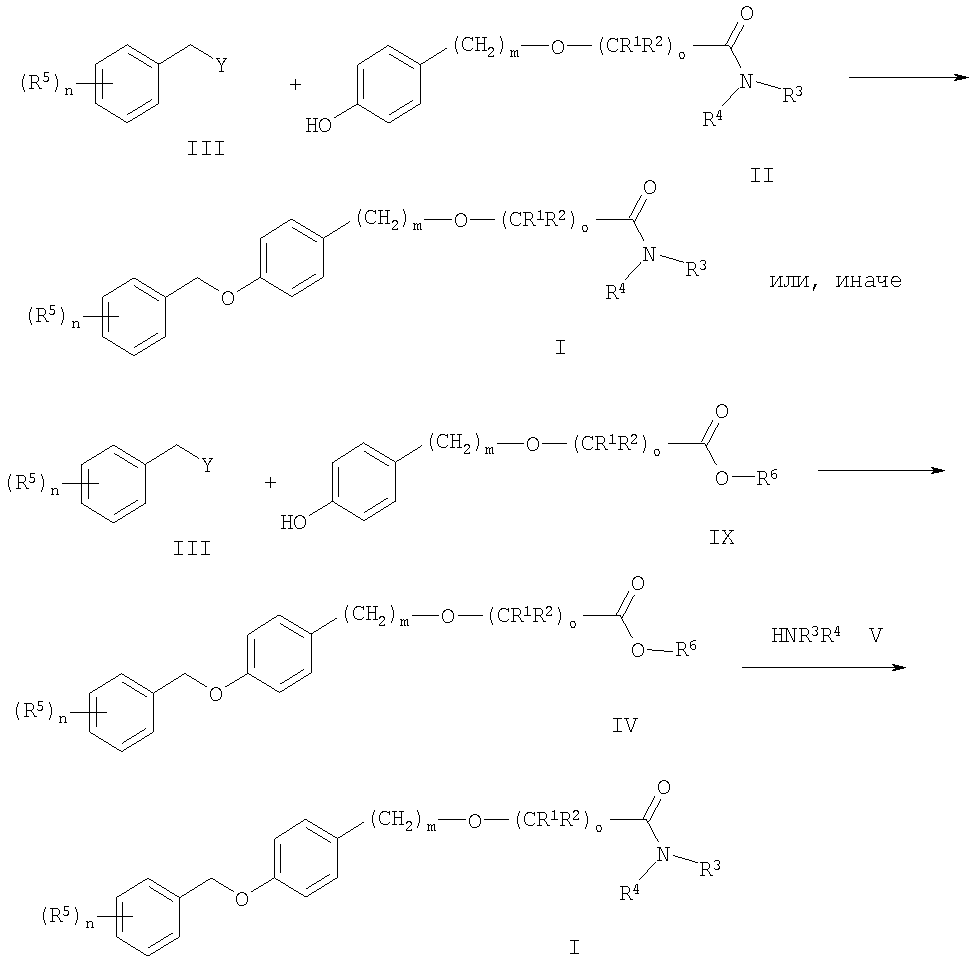
Заместители, n, m и о имеют вышеописанные значения, и Y является уходящей группой.
Соединения общей формулы I можно получить синтезом эфиров по Вильямсону, начиная с соответствующих паразамещенных фенолов формулы II по реакции с бензильными галогенидами, тозилатами, мезилатами или трифлатами формулы III. В качестве оснований, например, можно использовать алкоголяты или карбонаты (карбонат натрия, калия или цезия). Предпочтительными растворителями являются низшие спирты, ацетонитрил или низшие кетоны при температуре от 20°С до температуры дефлегмации. Другой способ представляет собой взаимодействие бензиловых спиртов формулы III с соответствующими фенолами формулы II по реакции сочетания Мицунобу. Обычно реакцию проводят в инертных растворителях, например в диэтиловом эфире или тетрагидрофуране, используя диалкилазодикарбоксилаты, в присутствии фосфинов (например, трибутил- или трифенилфосфина). В случае, когда R7 представляет собой NR3R4, в ходе вышеупомянутых реакций алкилирования непосредственно получают требуемые соединения формулы I.
Эфир формулы IV можно превратить в требуемый конечный продукт общей формулы I, используя стандартные процедуры: аминолизом с HNR3R4 в растворителях подобных метанолу, тетрагидрофурану и т.п., или омылением до соответствующей кислоты (например, с LiOH или КОН в метаноле), активированием кислоты посредством кислотного хлорида (тионилхлорида или оксалилхлорида) или активированием с N,N'-дициклогексилкарбодиимидом (ДЦК), N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлоридом (ЭДК) и т.п. и взаимодействием с амином HNR3R4.
Соединения общей формулы II и IX можно получить, используя соединение формулы IV или I с необязательно замещенным бензильным остатком, который может функционировать как переходная группа и отщепляться в ходе гидрогенолиза. Полученные в результате фенолы формулы II и IX затем можно вновь алкилировать другой бензильной группой при обозначенных выше условиях. Как известно квалифицированному специалисту в данной области, этот процесс возможен только в том случае, когда другие заместители и функциональные группы стабильны при вышеупомянутых условиях реакции гидрогенолиза и алкилирования.
На схеме 2 показан способ получения промежуточных соединений для соединений формулы I. Аналогично процедурам, описанным ранее, моно-алкилирование фенола Х с бензильными галогенидами, тозилатами, мезилатами или трифлатами формулы III по реакции синтеза эфиров Вильямсона приводит к гидроксильным производным формулы XI. Другой способ представляет собой взаимодействие бензиловых спиртов формулы III с соответствующими фенолами формулы Х по реакции сочетания Мицунобу. Обычно реакцию проводят в инертных растворителях, например в диэтиловом эфире или тетрагидрофуране, используя диалкилазодикарбоксилаты, в присутствии фосфинов (например трибутил- или трифенилфосфина). Соединения формулы XI можно далее алкилировать под действием сложноэфирных производных формулы XII с получением соединений формулы IV. Данную реакцию алкилирования также можно проводить при уже описанных ранее условиях, в основном, хорошо известных квалифицированному специалисту в данной области.
Соединения формулы XIV можно получить присоединением по Михаэлю гидроксильных производных, соответственно их солей, формулы XI к акрилатам формулы XIII в растворителях, инертных при этих реакционных условиях, предпочтительно в неразбавленных акрилатах при температуре от комнатной до температуры дефлегмации. Основаниями для получения алкоголятов могут быть, например, натрий или гидрид натрия. Для того чтобы получить соединения формулы I, обозначенные выше промежуточные соединения формулы IV и XIV обрабатывают аминами формулы V согласно описанным выше процедурам.
Схема 2
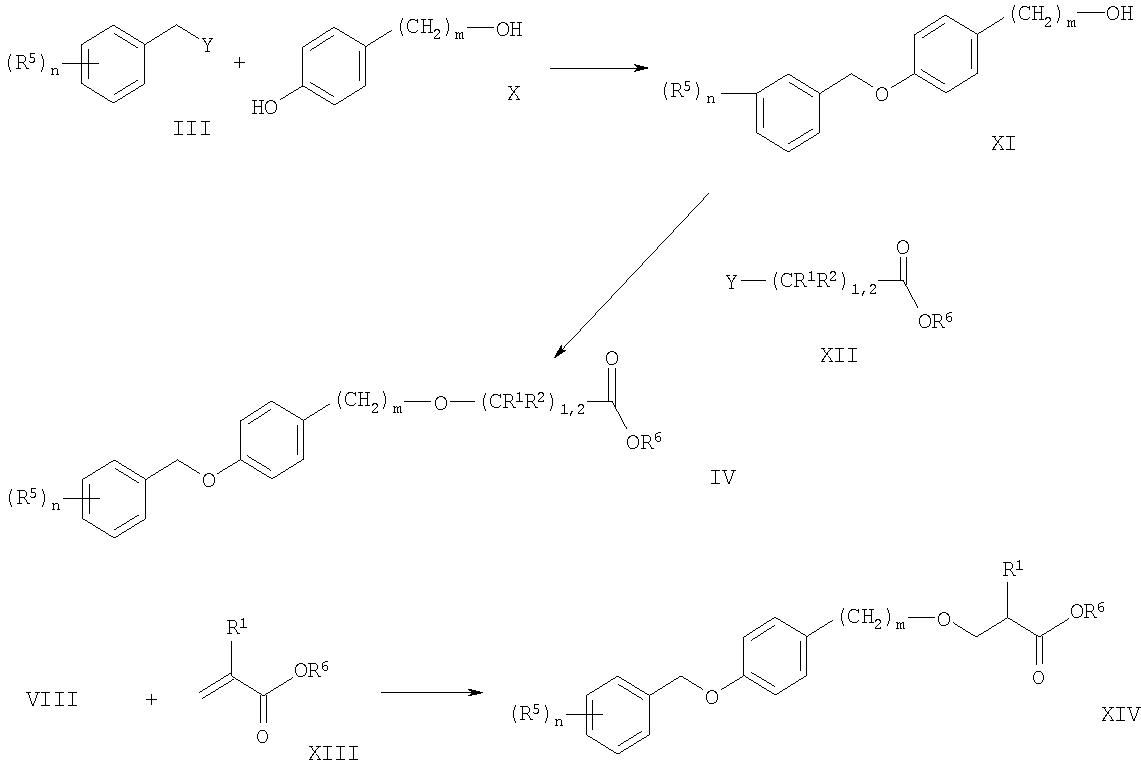
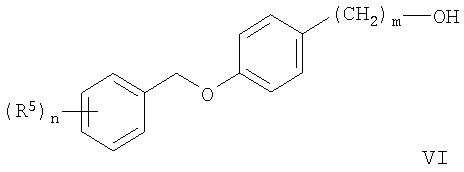
Заместители, n и m имеют вышеописанные значения. На схеме 3 показано получение соединений формулы Ia и Ib, где n=0. Реакция гидроксильных производных формулы VI с цианатом калия или алкилизоцианатами в герметичных сосудах с использованием растворителей подобных дихлорметану или толуолу при температуре от комнатной до 100°С приводит к образованию карбаматов формулы Ia.
Карбонаты формулы VIII образуются в ходе реакции гидроксильных производных формулы VI с фенил хлорформиатами, предпочтительно замещенными фенил хлорформиатами, например 4-нитрофенил хлорформиатами. Соединения формулы Ib получают при взаимодействии этих карбонатов с аминами общей формулы V предпочтительно в герметично закрытых пробирках и в растворителях, инертных при данных реакционных условиях, например тетрагидрофуране или диоксане при температуре от 0 до 60°С.
Схема 3

Заместители, n и m имеют вышеописанные значения. Также соединения общей формулы I могут существовать в оптически чистой форме. Согласно известным способам разделение на антиподы можно осуществить либо на ранней стадии синтеза, начинающегося с соединений формулы XII, в ходе солеобразования с оптически активным амином, таким как, например, (+)- или (-)-1-фенилэтиламин или (+)- или (-)-1-нафтилэтиламин, и разделения диастереомерных солей фракционной кристаллизацией или дериватизацией с хиральным вспомогательным веществом, таким как, например, (+)- или (-)-2-бутанол, (+)- или (-)-1-фенилэтанол, (+)- или (-)-ментол и разделением диастереомерных продуктов хроматофафически и/или кристаллизацией и последующим разрывом связи с хиральным вспомогательным веществом; либо на самой последней стадии при разделении энантиомеров формулы I хроматографически на хиральной фазе. Более того, соединения формулы I также можно получить из энантиочистых промежуточных соединений, полученных биотрансформацией, например при гидролизе сложных эфиров формулы IV, IX, XII или XIV под действием ферментов, таких как гидролазы или липазы. Для определения абсолютной конфигурации полученных производных чистые диастереомерные соли или производные можно проанализировать традиционными спектроскопическими методами, наиболее подходящим из которых является рентгеновская спектроскопия на монокристаллах.
Фармацевтически приемлемые соли соединений формулы I можно легко получить согласно известным методам, принимая во внимание природу превращаемого в соль соединения. Для образования фармацевтически приемлемых солей основных соединений формулы I подходящими неорганическими или органическими кислотами являются такие, например, как соляная кислота, бромисто-водородная кислота, серная кислота, азотная кислота, фосфорная кислота или лимонная кислота, муравьиная кислота, фумаровая кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метансульфоновая кислотой, п-толуолсульфоновая кислота и т.п. Соединения, содержащие щелочные или щелочноземельные металлы, например, натрий, калий, кальций, магний и т.п., основные амины или основные аминокислоты подходят для образования фармацевтически приемлемых солей кислотных соединений.
Как уже было сказано, соединения формулы 1 и их фармацевтически приемлемые соли представляют собой ингибиторы моноамин оксидазы В и могут быть использованы для лечения или предотвращения заболеваний, при которых могут быть полезны МАО-В-ингибиторы. Эти заболевания включают острые и хронические неврологические нарушения, когнитивные расстройства и нарушения памяти. Излечиваемыми неврологическими нарушениями являются, например, травматические или хронические дегенеративные процессы нервной системы, такие как болезнь Альцгеймера, другие виды слабоумия, минимальное когнитивное ухудшение или болезнь Паркинсона. Другие показания включают психиатрические расстройства, такие как депрессия, тревога, приступ паники, социальная фобия, шизофрения, расстройства питания и метаболические расстройства, например ожирение, а также предотвращение и лечение абстинентных синдромов, вызванных употреблением алкоголя, никотина и других препаратов, вызывающих привыкание. Другими излечиваемыми показаниями могут быть синдром дефицита значимости (G.M.Sullivan, международная заявка № WO 01/34172 А2), периферическая невропатия, вызванная химиотерапией рака (G.Bobotas, международная заявка №WO 97/33572 А1), или лечение рассеянного склероза (R.Y.Harris, международная заявка № WO 96/40095 А1) и другие нейровоспалительные заболевания.
Используя следующий метод, проверяли фармакологическую активность соединений.
Фармакологическую активность Фармацевтических Соединений можно показать, например, следующим образом:
кДНК, кодирующей МАО-А и МАО-В человека, временно трансфицировали в EBNA клетки, используя описанную Schlaeger и Christensen процедуру [Cytotechnology 15:1-13 (1998)]. После трансфекции клетки гомогенизировали в гомогенизаторе Polytron в 20 мМ Tris HCl буфере, рН 8,0, содержащем 0,5 мМ EGTA и 0,5 мМ фенилметансульфонилфторида. Клеточные мембраны получали центрифугированием при 45000 × g и после двукратного промывания с 20 мМ Tris HCl буфером, рН 8,0, содержащим 0,5 мМ EGTA, мембраны ресуспендировали в вышеописанном буфере и аликвоты хранили при -80°С до использования.
Ферментативную активность МАО-А и МАО-В проверяли на 96-луночном планшете, используя подходящий спектрофотометрический анализ, согласно адаптированному методу, описанному Zhou и Panchuk-Voloshina [Analitical Biochemistry 253:169-174 (1997)]. Кратко, аликвоты мембран инкубировали в 0,1 М калий-фосфатном буфере при рН 7,4 в течение 30 минут при 37°С без или с различными концентрациями соединений. После этого начинали ферментативную реакцию, добавляя МАО субстрат тирамин вместе с 1 Ед/мл пероксидазы хрена (Roche Biochemical) и 80 мкМ N-ацетил-3,7-дигидроксифеноксазином (Amplex Red, Molecular Probes). Затем образцы инкубировали в течение 30 минут при 37°С в конечном объеме 200 мкл и определяли оптическую плотность на длине волны 570 нм, используя считывающее устройство для планшетов SpectraMax (Molecular Devices). Фоновую (неспецифическую) оптическую плотность для МАО-А определяли в присутствии 10 мкМ клоргилина, а для МАО-В - в присутствии 10 мкМ L-депренила.
Значения ИК50 определяли по кривым ингибирования, полученным при использовании девяти дублированных концентраций ингибитора, подбором данных по четырехпараметрическому логистическому уравнению, используя компьютерную программу.
Соединения настоящего изобретения представляют собой специфические МАО-В-ингибиторы. Значения ИК50 соединений формулы I, измеренные в ходе ранее описанного анализа, находятся в пределах 1 мкМ или менее и идеально 0,1 мкМ или менее. В таблице ниже показаны характерные значения ИК50 соединений формулы I в одной из их энатиомерных форм:
Фармацевтические Соединения можно использовать в качестве лекарств, например, в форме фармацевтических препаратов. Фармацевтические препараты можно вводить перорально, например, в форме таблеток, таблеток, покрытых оболочкой, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Однако эффективным также может быть ректальное введение, например в форме суппозиториев, или парентеральное, например в форме растворов для инъекций.
Фармацевтические Соединения можно переработать с фармацевтически инертными, неорганическими или органическими носителями для получения фармацевтических препаратов. Лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли и т.п. можно использовать, например, в качестве таких носителей для таблеток, таблеток, покрытых оболочкой, драже и твердых желатиновых капсул. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и т.п.; однако в зависимости от природы активного вещества для мягких желатиновых капсул носители обычно не требуются. Подходящими носителями для получения растворов и сиропов являются, например, вода, полиолы, сахароза, инвертный сахар, глюкоза и т.п. Адъюванты, такие как спирты, полиолы, глицерин, растительные масла и т.п., могут быть использованы для водных инъекционных растворов водорастворимых солей соединений формулы I, но, как правило, они не являются необходимыми. Подходящими носителями для суппозиториев являются, например, природные или отвержденные масла, воски, жиры, полужидкие или жидкие полиолы и т.п.
Кроме того, фармацевтические препараты могут содержать консерванты, солюбилизаторы, стабилизаторы, смачивающие вещества, эмульгаторы, подсластители, красители, ароматизаторы, соли для регулирования осмотического давления, буферы, маскирующие агенты или антиоксиданты. Также они могут содержать другие терапевтически ценные вещества.
Дозировка может варьироваться в широких пределах и, конечно, должна быть подобрана по индивидуальным потребностям в каждом конкретном случае. В общем, эффективная дозировка для перорального или парентерального введения находится в интервале 0,01-20 мг/кг/день, для всех описанных показаний дозировка 0,1-10 мг/кг/день является предпочтительной. Суточная дозировка для взрослого человека весом 70 кг соответственно находится в интервале 0,7-1400 мг в день, предпочтительно в интервале 7-700 мг в день.
Следующие примеры иллюстрируют изобретение. Они не ограничивают объем изобретения, а только поясняют его.
Пример 1
2-[4-(4-Фтор-бензилокси)-фенокси]-ацетамид
а) 4-(4-Фтор-бензилокси)-фенол
К раствору 10 г гидрохинона в 90 мл ацетонитрила добавляли 8,58 г 4-фторбензилбромида и 15,7 г карбоната калия. Смесь нагревали до 90°С и перемешивали в течение 18 часов. Для завершения процесса реакционную смесь охлаждали до комнатной температуры и добавляли холодную воду. Получившееся твердое вещество фильтровали, дважды промывали холодной водой и высушивали. Для очистки и отделения от диэфира неочищенный продукт хроматографировали на силикагеле, используя в качестве элюента смесь гептана и этилацетата в соотношении 4:1. 4-(4-Фтор-бензилокси)-фенол был получен в виде белого твердого вещества массой 4,05 г (27% от теоретического); MS: m/e=218 (М)+.
б) Этиловый эфир [4-(4-фтор-бензилокси)-фенокси]-уксусной кислоты
К раствору 3 г 4-(4-фтор-бензилокси)-фенола в 40 мл 2-бутанона добавляли 2,5 г карбоната калия и 1,6 мл этил-бромацетата. Смесь перемешивали в течение 3 часов при 80°С, затем для полного завершения реакции в течение 24 часов постепенно добавляли четырьмя порциями 1,7 г карбоната калия и 1,9 мл этил-бромацетата. Для завершения процесса реакционную смесь охлаждали до комнатной температуры, добавляли воду и экстрагировали с этилацетатом. Органический слой отделяли, высушивали над сульфатом магния и выпаривали при пониженном давлении. Этиловый эфир [4-(4-фтор-бензилокси)-фенокси]-уксусной кислоты получали в виде коричневатых кристаллов массой 2,95 г (71% от теоретического), которые были достаточно чистыми для того, чтобы использовать их в следующей стадии без дополнительной очистки; MS: m/e=304 (М)+.
в) 2-[4-(4-Фтор-бензилокси)-фенокси]-ацетамид
К раствору 500 мг этилового эфира [4-(4-фтор-бензилокси)-фенокси]-уксусной кислоты в 10 мл тетрагидрофурана добавляли 3,3 мл раствора гидроксида натрия (1 н.). Смесь нагревали до 50°С в течение 2 часов, затем охлаждали до комнатной температуры и добавляли 3,3 мл соляной кислоты (1 н.). Пока тетрагидрофуран выпаривался при пониженном давлении, кислота выпадала в осадок, после чего ее собирали на воронке для фильтрования. После промывания водой и сушки при пониженном давлении получали 376 мг [4-(4-фтор-бензилокси)-фенокси]-уксусной кислоты, которую сразу использовали для дальнейшего превращения.
Неочищенную кислоту растворяли в 5 мл N,N-диметилформамида, добавляли 293 мг 1,1'-карбонил-диимидазола и полученный в результате раствор нагревали до 50°С в течение 1 часа. Затем смесь охлаждали до комнатной температуры, добавляли 0,16 мл раствора гидроксида аммония (25%) и продолжали перемешивать при комнатной температуре в течение 18 часов. Для завершения процесса к реакционной смеси добавляли воду, чтобы осадить продукт, который собирали на воронке для фильтрования и промывали водой. 2-[4-(4-Фтор-бензилокси)-фенокси]-ацетамид массой 337 мг (74% от теоретического) получали в виде твердого вещества белого цвета после кристаллизации из воды; MS: m/e=276 (М+Н)+.
Пример 2
2-(4-Бензилокси-фенокси)-N-метил-ацетамид
а) 4-Бензилокси-фенол
Аналогично процедуре, описанной в примере 1(а), проводили алкилирование гидрохинона с бензилбромидом с получением 4-бензилокси-фенола в виде бесцветного твердого вещества.
б) Метиловый эфир (4-бензилокси-фенокси)-уксусной кислоты
Аналогично процедуре, описанной в примере 1(б), проводили алкилирование 4-бензилокси-фенола с метил-бромацетатом, используя в качестве основания карбонат цезия, с получением метилового эфира (4-бензилокси-фенокси)-уксусной кислоты в виде кристаллов белого цвета; MS: m/e=272 (М)+.
в) 2-(4-Бензилокси-Фенокси)-N-метил-ацетамид
Аналогично процедуре, описанной в примере 1(в), проводили реакцию (4-бензилокси-фенокси)-уксусной кислоты с метиламином, используя в качестве конденсирующего реагента 1,1'-карбонил-диимидазол. 2-(4-Бензилокси-фенокси)-N-метил-ацетамид был получен в виде белых кристаллов; MS: m/e=272 (М+Н)+.
Пример 3
2-[4-(4-Циано-бензилокси)-фенокси]-N-метил-ацетамид
а) 2-(4-Гидрокси-фенокси)-N-метил-ацетамид
Раствор 4,7 г 2-(4-бензилокси-фенокси)-N-метил-ацетамида [пример 2(в)] в 150 мл тетрагидрофурана гидрировали при атмосферном давлении и комнатной температуре, используя в качестве катализатора 470 мг 10% Pd/C. Для завершения процесса реакционную смесь фильтровали через фильтр Dicalite и полученный в результате раствор выпаривали при пониженном давлении. Остаток в эфире растирали в порошок и твердое вещество собирали на воронке для фильтрования. После сушки получали 2,95 г (93% от теоретического) 2-(4-гидрокси-фенокси)-N-метил-ацетамида в виде кристаллов белого цвета; MS: m/e=181 (М)+.
б) 2-[4-(4-Циано-бензилокси)-фенокси]-N-метил-ацетамид
К раствору 500 мг 2-(4-гидрокси-фенокси)-N-метил-ацетамида в 25 мл 2-бутанона добавляли 762 мг карбоната калия и 595 мг 4-бромметил-бензонитрила. Реакционную смесь перемешивали при комнатной температуре в течение 60 часов. Для завершения процесса к реакционной смеси добавляли воду и затем экстрагировали этилацетатом. Водный слой вновь экстрагировали с этилацетатом, объединенные органические слои высушивали над сульфатом магния и выпаривали при пониженном давлении. Остаток растирали в эфире и полученные кристаллы собирали на воронке для фильтрования. После сушки получали 744 мг (95% от теоретического) 2-[4-(4-циано-бензилокси)-фенокси]-N-метил-ацетамида в виде белых кристаллов; MS: m/e=297 (М+Н)+.
Пример 4
2-[4-(4-Хлор-бензилокси)-фенокси]-N-метил-ацетамид
Аналогично процедуре, описанной в примере 3(6), проводили алкилирование 2-(4-гидрокси-фенокси)-N-метил-ацетамида [пример 3(а)] с 1-бромметил-4-хлорбензолом в 2-бутаноне, используя в качестве основания карбонат калия, с получением 2-[4-(4-хлор-бензилокси)-фенокси]-N-метил-ацетамида в виде кристаллов белого цвета; MS: m/e=306 (М+Н)+.
Пример 5
2-[4-(2-Фтор-бензилокси)-фенокси]-N-метил-ацетамид
Аналогично процедуре, описанной в примере 3(б), проводили алкилирование 2-(4-гидрокси-фенокси)-N-метил-ацетамида [пример 3(а)] с 1-бромметил-2-фторбензолом в 2-бутаноне, используя в качестве основания карбонат калия, с получением 2-[4-(2-фтор-бензилокси)-фенокси]-N-метил-ацетамида в виде кристаллов белого цвета; MS: m/e=290 (М+Н)+.
Пример 6
2-[4-(3-Фтор-бензилокси)-фенокси]-N-метил-ацетамид
Аналогично процедуре, описанной в примере 3(б), проводили алкилирование 2-(4-гидрокси-фенокси)-N-метил-ацетамида [пример 3(а)] с 1-бромметил-3-фторбензолом в 2-бутаноне, используя в качестве основания карбонат калия, с получением 2-[4-(3-фтор-бензилокси)-фенокси]-N-метил-ацетамида в виде кристаллов белого цвета; MS: m/e=290 (М+Н)+.
Пример 7
2-[4-(4-Фтор-бензилокси)-фенокси]-N-метил-ацетамид
Смесь 500 мг этилового эфира [4-(4-фтор-бензилокси)-фенокси]-уксусной кислоты [пример 1(б)] и 2 мл метиламина (приблизительно 8 М в этаноле) перемешивали в течение 18 часов при 80°С. Для завершения процесса раствор охлаждали до комнатной температуры и добавляли воду. Чистый продукт осаждали и собирали на воронке для фильтрования. После сушки получали 398 мг (86% от теоретического) 2-[4-(4-фтор-бензилокси)-фенокси]-N-метил-ацетамида в виде белых кристаллов; MS: m/e=290 (М+Н)+.
Пример 8
2-[4-(4-Фтор-бензилокси)-фенокси]-N,N-диметил-ацетамид
Аналогично процедуре, описанной в примере 7, проводили аминолиз этилового эфира [4-(4-фтор-бензилокси)-фенокси]-уксусной кислоты с диметиламином с получением 2-[4-(4-фтор-бензилокси)-фенокси]-N,N-диметил-ацетамида в виде кристаллов белого цвета; MS: m/e=304 (М+Н)+.
Пример 9
(RS)-2-(4-Бензилокси-фенокси)-N-метил-пропионамид
а) Метиловый эфир (RS)-2-(4-Бензилокси-фенокси)-пропионовой кислоты
Аналогично процедуре, описанной в примере 1(б), проводили алкилирование 4-бензилокси-фенола [пример 2(а)] с метил-(RS)-2-бромпропионатом в ацетоне, используя в качестве основания карбонат цезия, с получением метилового эфира (RS)-2-(4-бензилокси-фенокси)-пропионовой кислоты в виде светло-желтого масла; MS: m/e=304 (M+NH4)+.
б) (RS)-2-(4-Бензилокси-Фенокси)-N-метил-пропионамид
Аналогично процедуре, описанной в примере 7, проводили аминолиз метилового эфира (RS)-2-(4-бензилокси-фенокси)-пропионовой кислоты с метиламином с получением (RS)-2-(4-бензилокси-фенокси)-N-метил-пропионамида в виде твердого вещества белого цвета; MS: m/e=286 (М+Н)+.
Пример 10
(RS-2-[4-(2-Фтор-бензилокси)-фенокси]-N-метил-пропионамид
а) (RS)-2-(4-Гидрокси-Фенокси)-N-метил-пропионамид
Аналогично процедуре, описанной в примере 3(а), проводили гидрогенолиз (RS)-2-(4-бензилокси-фенокси)-N-метил-пропионамида с получением (RS)-2-(4-гидрокси-фенокси)-N-метил-пропионамида в виде кристаллов белого цвета; MS: m/e=194 (М-Н)-.
б) (RS)-2-[4-(2-Фтор-бензилокси)-фенокси]-N-метил-пропионамид
Аналогично процедуре, описанной в примере 3(б), проводили алкилирование (RS)-2-(4-гидрокси-фенокси)-N-метил-пропионамида с 2-фторбензилбромидом в 2-бутаноне, используя в качестве основания карбонат калия, с получением (RS)-2-[4-(2-фтор-бензилокси)-фенокси]-N-метил-пропионамида в виде кристаллов белого цвета; MS: m/e=304 (М+Н)+.
Пример 11
(RS)-2-[4-(3-Фтор-бензилокси)-фенокси]-N-метил-пропионамид
Аналогично процедуре, описанной в примере 3(б), проводили алкилирование (RS)-2-(4-гидрокси-фенокси)-N-метил-пропионамида с 3-фторбензилбромидом в 2-бутаноне, используя в качестве основания карбонат калия, с получением (RS)-2-[4-(3-фтор-бензилокси)-фенокси]-N-метил-пропионамида в виде твердого вещества белого цвета; MS: m/e=304 (М+Н)+.
Пример 12
(RS)-2-[4-(3-Хлор-бензилокси)-фенокси]-N-метил-пропионамид
Аналогично процедуре, описанной в примере 3(б), проводили алкилирование (RS)-2-(4-гидрокси-фенокси)-N-метил-пропионамида с 3-хлорбензилбромидом в 2-бутаноне, используя в качестве основания карбонат калия, с получением (RS)-2-[4-(3-хлор-бензилокси)-фенокси]-N-метил-пропионамида в виде твердого вещества белого цвета; MS: m/e=320 (М+Н)+.
Пример 13
(R или S)-2-[4-(3-Хлор-бензилокси)-фенокси]-N-метил-пропионамид и (S или R)-2-[4-(3-хлор-бензилокси)-фенокси]-N-метил-пропионамид
Разделение 300 мг двух изомеров (RS)-2-[4-(3-хлор-бензилокси)-фенокси]-N-метил-пропионамида (пример 12) осуществляли на препаративной хиральной ВЭЖХ-колонке (CHIRALPAK® AD, давление: 20 бар, скорость потока: 35 мл/мин), используя в качестве элюента смесь н-гептана и этанола в соотношении 85:15. Первым элюировали 122 мг (41% от теоретического) (R или S)-2-[4-(3-хлор-бензилокси)-фенокси]-N-метил-пропионамида, затем 860 мг (39% от теоретического) (S или R)-2-[4-(3-хлор-бензилокси)-фенокси]-N-метил-пропионамида, оба изомера в виде твердого вещества белого цвета.
Пример 14
(RS)-2-[4-(4-Циано-бензилокси)-фенокси]-N-метил-пропионамид
Аналогично процедуре, описанной в примере 3(б), проводили алкилирование (RS)-2-(4-гидрокси-фенокси)-N-метил-пропионамида с 4-бромметил-бензонитрилом в 2-бутаноне, используя в качестве основания карбонат калия, с получением (RS)-2-[4-(4-циано-бензилокси)-фенокси]-N-метил-пропионамида в виде твердого вещества белого цвета; MS: m/e=311 (М+Н)+.
Пример 15
(RS)-2-(4-Бензилокси-фенокси)-N-метил-бутирамид
а) Этиловый эфир (RS)-2-(4-бензилокси-фенокси)-масляной кислоты
Аналогично процедуре, описанной в примере 1(б), проводили алкилирование 4-бензилокси-фенола [пример 2(а)] с этил-(RS)-2-бромбутиратом в ацетоне, используя в качестве основания карбонат цезия, с получением этилового эфира (RS)-2-(4-бензилокси-фенокси)-масляной кислоты в виде коричневого масла; MS: m/e=314 (М)+.
б) (RS)-2-(4-Бензилокси-фенокси)-N-метил-бутирамид
Аналогично процедуре, описанной в примере 7, проводили аминолиз этилового эфира (RS)-2-(4-бензилокси-фенокси)-масляной кислоты с метиламином с получением (RS)-2-(4-бензилокси-фенокси)-N-метил-бутирамида в виде твердого вещества белого цвета; MS: m/e=300 (М+Н)+.
Пример 16
(RS)-2-[4-(2-Фтор-бензилокси)-фенокси]-N-метил-бутирамид
а) (RS)-2-(4-Гидрокси-фенокси)-N-метил-бутирамид
Аналогично процедуре, описанной в примере 3(а), проводили гидрогенолиз (RS)-2-(4-бензилокси-фенокси)-N-метил-бутирамида с получением (RS)-2-(4-гидрокси-фенокси)-N-метил-бутирамида в виде кристаллов белого цвета; MS: m/e=210 (М+Н)+.
б) (RS)-2-[4-(2-Фтор-бензилокси)-фенокси]-N-метил-бутирамид
Аналогично процедуре, описанной в примере 3(б), проводили алкилирование (RS)-2-(4-гидрокси-фенокси)-N-метил-бутирамида с 2-фторбензилбромидом в 2-бутаноне, используя в качестве основания карбонат калия, с получением (RS)-2-[4-(2-фтор-бензилокси)-фенокси]-N-метил-бутирамида в виде твердого вещества белого цвета; MS: m/e=318 (М+Н)+.
Пример 17
(RS)-2-[4-(3-Фтор-бензилокси)-фенокси]-N-метил-бутирамид
Аналогично процедуре, описанной в примере 3(б), проводили алкилирование (RS)-2-(4-гидрокси-фенокси)-N-метил-бутирамида с 3-фторбензилбромидом в 2-бутаноне, используя в качестве основания карбонат калия, с получением (RS)-2-[4-(3-фтор-бензилокси)-фенокси]-N-метил-бутирамида в виде твердого вещества белого цвета; MS: m/e=318 (М+Н)+.
Пример 18
(RS)-2-[4-(3-Хлор-бензилокси)-фенокси]-N-метил-бутирамид
Аналогично процедуре, описанной в примере 3(б), проводили алкилирование (RS)-2-(4-гидрокси-фенокси)-N-метил-бутирамида с 3-хлорбензилбромидом в 2-бутаноне, используя в качестве основания карбонат калия, с получением (RS)-2-[4-(3-хлор-бензилокси)-фенокси]-N-метил-бутирамида в виде твердого вещества белого цвета; MS: m/e=334 (М+Н)+.
Пример 19
(RS)-2-[4-(4-Циано-бензилокси)-фенокси]-N-метил-бутирамид
Аналогично процедуре, описанной в примере 3(б), проводили алкилирование (RS)-2-(4-гидрокси-фенокси)-N-метил-бутирамида с 4-бромметил-бензонитрилом в 2-бутаноне, используя в качестве основания карбонат калия, с получением (RS)-2-[4-(4-циано-бензилокси)-фенокси]-N-метил-бутирамида в виде твердого вещества белого цвета; MS: m/e=325 (М+Н)+.
Пример 20
2-(4-Бензилокси-фенокси)-2,N-диметил-пропионамид
Аналогично процедуре, описанной в примере 7, проводили аминолиз этилового эфира 2-(4-бензилокси-фенокси)-2-метил-пропионовой кислоты с метиламином с получением 2-(4-бензилокси-фенокси)-2,N-диметил-пропионамида в виде твердого вещества белого цвета; MS: m/e=300 (М+Н)+.
Пример 21
2-[4-(3-Фтор-бензилокси)-фенокси]-2,N-диметил-пропионамид
а) 2-(4-Гидрокси-фенокси)-2,N-диметил-пропионамид
Аналогично процедуре, описанной в примере 3(а), проводили гидрогенолиз 2-(4-бензилокси-фенокси)-2,N-диметил-пропионамида [пример 20] с получением 2-(4-гидрокси-фенокси)-2,N-диметил-пропионамида в виде твердого вещества белого цвета; MS: m/e=209 (M)+.
б) 2-[4-(3-Фтор-бензилокси)-Фенокси1-2,N-диметил-пропионамид
Аналогично процедуре, описанной в примере 3(б), проводили алкилирование 2-(4-гидрокси-фенокси)-2,N-диметил-пропионамида с 3-фторбензилбромидом в 2-бутаноне, используя в качестве основания карбонат калия, с получением 2-[4-(3-фтор-бензилокси)-фенокси]-2,N-диметил-пропионамида в виде коричневого масла; MS: m/e=318 (М+Н)+.
Пример 22
2-[4-(3-Хлор-бензилокси)-фенокси]-2,N-диметил-пропионамид
Аналогично процедуре, описанной в примере 3(б), проводили алкилирование 2-(4-гидрокси-фенокси)-2,N-диметил-пропионамида с 3-хлорбензилбромидом в 2-бутаноне, используя в качестве основания карбонат калия, с получением 2-[4-(3-хлор-бензилокси)-фенокси]-2,N-диметил-пропионамида в виде коричневого масла; MS: m/e=334 (М+Н)+.
Пример 23
3-[4-(3-Фтор-бензилокси)-фенокси]-пропионамид
а) Метиловый эфир 3-[4-(3-фтор-бензилокси)-фенокси]-пропионовой кислоты
К раствору 1,5 г 4-(3-фтор-бензилокси)-фенола (полученного по процедуре, схожей с описанной в примере 1(а) для 4-(4-фтор-бензилокси)-фенола) и 1,4 мг гидрохинона в 5 мл метилакрилата добавляли натрий и нагревали с обратным холодильником в течение 7,5 часов. Затем смесь охлаждали до комнатной температуры и нейтрализовали уксусной кислотой. После выпаривания при пониженном давлении остаток растворяли в эфире и этилацетате и полученный в результате раствор экстрагировали три раза водой. Органический слой высушивали над сульфатом магния и затем выпаривали при пониженном давлении. Неочищенный продукт перекристаллизовывали в небольшом объеме метанола. Метиловый эфир 3-[4-(3-фтор-бензилокси)-фенокси]-пропионовой кислоты получали в виде кристаллов белого цвета массой 1,1 г (54% от теоретического); MS: m/e=304 (М+Н)+.
б) 3-[4-(3-Фтор-бензилокси)-фенокси]-пропионовая кислота
Раствор 500 мг метилового эфира 3-[4-(3-фтор-бензилокси)-фенокси]-пропионовой кислоты массой в смеси 5 мл тетрагидрофурана и 25 мл 19% соляной кислоты нагревали в течение 7 часов при 70°С. Для завершения процесса выпаривали тетрагидрофуран при пониженном давлении и водный слой экстрагировали три раза 40 мл этилацетата. Объединенные органические слои высушивали над сульфатом магния и выпаривали при пониженном давлении. Неочищенный продукт растирали в эфире и твердый продукт собирали на воронке для фильтрования. После сушки получали 3-[4-(3-фтор-бензилокси)-фенокси]-пропионовой кислоты массой 114 мг (30% от теоретического) в виде белых кристаллов.
в) 3-[4-(3-Фтор-бензилокси)-фенокси]-пропионамид
Раствор 70 мг 3-[4-(3-фтор-бензилокси)-фенокси]-пропионовой кислоты в 4 мл дихлорметана (плюс 1 капля N,N-диметилформамида) охлаждали до 0°С и добавляли 0,03 мл оксалилхлорида. В течение 1,5 часов осуществляли перемешивание, после чего большую часть дихлорметана выпаривали. Готовили раствор 0,5 мл водного гидроксида аммония (25%) в 1 мл тетрагидрофурана и охлаждали до 0°С. Раствор кислотного хлорида добавляли к вышеупомянутому раствору и смесь оставляли нагреваться до комнатной температуры при продолжении перемешивания в течение выходных дней. Для завершения процесса реакционную смесь выпаривали при пониженном давлении и остаток растирали в эфире с получением 3-[4-(3-фтор-бензилокси)-фенокси]-пропионамид массой 42 мг (60% от теоретического) в виде белого твердого вещества; MS: m/e=307 (M+NH4)+.
Пример 24
2-[4-(3-Фтор-бензилокси)-фенил]-этиловый эфир карбаминовой кислоты
а) 2-[4-(3-Фтор-бензилокси)-фенил]-этанол
К смеси 5,0 г 2-(4-гидроксифенил)-этанола и 5,0 г карбоната калия в 100 мл ацетонитрила добавляли по каплям в атмосфере аргона при 0°С 4,5 мл 3-фторбензилбромида. После завершения добавления смесь продолжали перемешивать в течение 15 минут при 0°С, затем реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 18 часов. Для завершения процесса растворитель выпаривали при пониженном давлении, затем остаток растворяли в этилацетате и раствор промывали водой. Органический слой высушивали над сульфатом магния и выпаривали при пониженном давлении. Для очистки полученный неочищенный продукт (10,2 г желтого масла) хроматографировали на силикагеле, используя в качестве элюента смесь дихлорметана и метанола в соотношении 95:5. После перекристаллизации из смеси эфира и циклогексана получали 4,1 г (46% от теоретического) 2-[4-(3-фтор-бензилокси)-фенил]-этанола в виде твердого вещества белого цвета; MS: m/e=247 (М+Н)+.
б) 2-[4-(3-Фтор-бензилокси)-фенил]-этиловый эфир карбаминовой кислоты
К суспензии 1,0 г 2-[4-(3-фтор-бензилокси)-фенил]-этанола и 0,66 г изоцианата калия в 2 мл бензола по каплям при перемешивании в атмосфере аргона при комнатной температуре добавляли 0,62 мл трифторуксусной кислоты. После перемешивания при комнатной температуре в течение 18 часов реакционную смесь разбавляли водой, после чего экстрагировали три раза дихлорметаном. Объединенные органические слои высушивали над карбонатом калия и выпаривали. Получали 2-[4-(3-фтор-бензилокси)-фенил]-этиловый эфир карбаминовой кислоты массой 0,52 г (44% от теоретического) в виде твердого вещества белого цвета; MS: m/e=290 (М+Н)+.
Пример 25
4-(3-Фтор-бензилокси)-бензиловый эфир карбаминовой кислоты
а) 4-(3-Фтор-бензилокси)-бензальдегид
К смеси 4-гидрокси-бензальдегида массой 6,0 г и карбоната калия массой 13,58 г в 60 мл N,N-диметилформамида по каплям при комнатной температуре добавляли раствор 3-фторбензилбромида массой 11,14 г в 30 мл N,N-диметилформамида. Через 3 часа реакционную смесь разбавляли водой и экстрагировали эфиром. Органический слой промывали водой, высушивали над сульфатом магния и выпаривали до получения масла (9,85 г), которое кристаллизовали; MS: m/e=230 (М)+. Неочищенный продукт использовали на следующей стадии без дополнительной очистки.
б) [4-(3-Фтор-бензилокси)-фенил]-метанол
Раствор 3,79 г 4-(3-фтор-бензилокси)-бензальдегида в 10 мл тетрагидрофурана по каплям при комнатной температуре добавляли к суспензии алюмогидрида лития массой 1,25 г в 40 мл тетрагидрофурана. Смесь оставляли на 2 часа при комнатной температуре, затем последовательно при охлаждении по каплям добавляли 1,25 мл воды, потом 3,75 мл раствора гидроксида натрия (1 н.) и снова 1,25 мл воды. Смесь фильтровали через фильтр Dicalit и тетрагидрофуран удаляли из раствора при пониженном давлении. Остаточную фазу экстрагировали этилацетатом, объединенные органические слои высушивали над сульфатом магния и выпаривали при пониженном давлении. Остаток кристаллизовали из смеси эфира и н-гексана, получая 2,26 г (59% от теоретического) [4-(3-фтор-бензилокси)-фенил]-метанола в виде твердого вещества белого цвета; MS: m/e=232 (M)+.
в) 4-(3-Фтор-бензилокси)-бензиловый эфир карбаминовой кислоты
Аналогично процедуре, описанной в примере 24(б), проводили реакцию [4-(3-фтор-бензилокси)-фенил]-метанола с изоцианатом калия и трифторуксусной кислотой в бензоле с получением 4-(3-фтор-бензилокси)-бензилового эфира карбаминовой кислоты в виде твердого вещества белого цвета; MS: m/e=293 (M+NH4)+.
Пример 26
4-(3-Фтор-бензилокси)-бензиловый эфир метилкарбаминовой кислоты
Раствор 1,0 г [4-(3-фтор-бензилокси)-фенил]-метанола [пример 25б)], 0,03 мл триэтиламина и 0,245 г метилизоцианата в 40 мл дихлорметана нагревали в течение 3 дней при 40°С в герметично закрытой стеклянной пробирке. В этом случае, как показал ЯМР, степень превращения достигала 50%. Для завершения процесса реакционную смесь охлаждали и выпаривали. Остаток растворяли в 2 мл пиридина и 1 мл ангидрида уксусной кислоты, раствор перемешивали при комнатной температуре в течение 3 часов. Затем смесь выпаривали при пониженном давлении. Для очистки полученный неочищенный продукт хроматографировали на силикагеле, используя в качестве элюента смесь гептана и этилацетата в соотношении 2:1. 4-(3-Фтор-бензилокси)-бензиловый эфир метилкарбаминовой кислоты получали массой 580 мг (47% от теоретического) в виде твердого вещества белого цвета; MS: m/e=289 (М)+.
Пример 27
2-[4-(3-Фтор-бензилокси)-бензилокси]-ацетамид
К раствору 500 мг [4-(3-фтор-бензилокси)-фенил]-метанола [пример 25(б)] в 10 мл тетрагидрофурана при комнатной температуре добавляли 57 мг гидрида натрия (55% дисперсия в масле) и перемешивали в течение 1 часа. Затем добавляли 183 мг хлорацетамида и смесь нагревали с обратным холодильником в течение 48 часов. Для завершения процесса к охлажденной смеси добавляли воду и экстрагировали этилацетатом. Органический слой высушивали над сульфатом магния и выпаривали при пониженном давлении. Для очистки полученный неочищенный продукт хроматографировали на силикагеле, используя в качестве элюента градиент дихлорметан - смесь дихлорметана и метанола в соотношении 4:1. После кристаллизации из эфира получали 2-[4-(3-фтор-бензилокси)-бензилокси]-ацетамид массой 29 мг (5% от теоретического) в виде твердого вещества белого цвета; MS: m/e=307 (М+NH4)+.
Пример А
Таблетки следующего состава готовят традиционным способом:
Пример Б
Таблетки следующего состава готовят традиционным способом:
Пример В
Готовят капсулы следующего состава:
Активный ингредиент с подходящим размером частиц, кристаллическую лактозу и микрокристаллическую целлюлозу смешивают друг с другом до получения однородной смеси, просеивают и затем смешивают с тальком и стеаратом магния. Конечной смесью заполняют твердые желатиновые капсулы подходящего размера.
Пример Г
Раствор для инъекции может быть следующего состава и приготовлен традиционным способом:
Реферат
Настоящее изобретение относится к новым соединениям общей формулы (I) ! ! где R1, R2 независимо друг от друга представляют собой Н или C1-С6-алкил; R3, R4 независимо друг от друга представляют собой Н или C1-С6-алкил; R5 представляет собой галоген, CN; n, m или о представляют собой 0, 1 или 2; а также к их фармацевтически приемлемым солям. Данные соединения обладают свойствами ингибитора моноаминооксидазы В и могут использоваться для получения фармацевтических препаратов с соответствующей активностью. 3 н. и 11 з.п. ф-лы.
Формула
где R1, R2 независимо друг от друга представляют собой Н или C1-С6-алкил;
R3, R4 независимо друг от друга представляют собой Н или C1-С6-алкил;
R5 представляет собой галоген, CN;
n, m или о представляют собой 0,1 или 2;
а также их фармацевтически приемлемые соли,
за исключением
4-(бензилокси)-фенил-N-бутилкарбамата,
2-[4-(фенилметокси)фенокси]ацетамида,
4-(фенилметокси)фенилового эфира диметилкарбаминовой кислоты,
2-[4-(3-хлорбензокси)бензокси]ацетамида,
4-бензилокси-бензилового эфира диметилкарбаминовой кислоты,
4-бензилокси-фенилового эфира метилкарбаминовой кислоты,
2-[4-[(4-хлорфенил)метокси]фенокси]-2-метил-пропионамида и
2-[4-[(4-хлорфенил)метокси]фенокси]-2-метил-]-N-(1-метилэтил)-пропионамида,
а также при условии, что исключены соединения общей формулы (I), где один из R1 или R2 представляет собой C1-С6-алкил, а другой представляет собой Н;
R3 и R4 представляют собой Н;
n, m представляют собой 0; и
о представляет собой 1.
2-[4-(фенилметокси)фенокси]ацетамида,
2-[4-[(4-хлорфенил)метокси]фенокси]-2-метил-пропионамида и
2-[4-[(4-хлорфенил)метокси]фенокси]-2-метил-N-(1-метилэтил)пропионамида,
а также при условии, что исключены соединения общей формулы (I), где один из R1 или R2 представляет собой C1-С6-алкил, а другой представляет собой Н;
R3 и R4 представляют собой Н;
n, m представляют собой 0; и
о представляет собой 1.
2-(4-бензилокси-фенокси)-N-метил-ацетамид,
2-[4-(4-циано-бензилокси)-фенокси]-N-метил-ацетамид,
2-[4-(4-хлор-бензилокси)-фенокси]-N-метил-ацетамид,
2-[4-(2-фтор-бензилокси)-фенокси]-N-метил-ацетамид,
2-[4-(3-фтор-бензилокси)-фенокси]-N-метил-ацетамид,
2-[4-(4-фтор-бензилокси)-фенокси]-N-метил-ацетамид,
(RS)-2-[4-(3-фтор-бензилокси)-фенокси]-N-метил-пропионамид,
(RS)-2-[4-(3-хлор-бензилокси)-фенокси]-N-метил-пропионамид,
(S или R)-2-[4-(3-хлор-бензилокси)-фенокси]-N-метил-пропионамид,
(RS)-2-[4-(4-циано-бензилокси)-фенокси]-N-метил-пропионамид,
(RS)-2-[4-(3-фтор-бензилокси)-фенокси]-N-метил-бутирамид,
(RS)-2-[4-(3-хлор-бензилокси)-фенокси]-N-метил-бутирамид, или
(RS)-2-[4-(4-циано-бензилокси)-фенокси]-N-метил-бутирамид.
где R1, R2 независимо друг от друга представляют собой Н или C1-С6-алкил;
R3, R4 независимо друг от друга представляют собой Н или C1-С6-алкил;
R5 представляет собой галоген, CN;
n, m или о представляют собой 0, 1 или 2;
или их фармацевтически приемлемой соли,
и фармацевтически приемлемые эксципиенты, за исключением следующих соединений:
4-(бензилокси)-фенил-N-бутилкарбамата,
2-[4-(фенилметокси)фенокси]ацетамида,
4-(фенилметокси)фенилового эфира диметилкарбаминовой кислоты,
2-[4-(3-хлорбензокси)бензокси]ацетамида,
4-бензилокси-бензилового эфира диметилкарбаминовой кислоты,
4-бензилокси-фенилового эфира метилкарбаминовой кислоты,
2-[4-[(4-хлорфенил)метокси]фенокси]-2-метил-пропионамида и
2-[4-[(4-хлорфенил)метокси]фенокси]-2-метил-N-(1-метилэтил)-пропионамида,
а также при условии, что исключены соединения общей формулы (I), где один из R1 или R2 представляет собой C1-С6-алкил, а другой представляет собой Н;
R3 и R4 представляют собой Н;
n, m представляют собой 0; и
о представляет собой 1.
где R1, R2 независимо друг от друга представляют собой Н или C1-С6-алкил;
R3, R4 независимо друг от друга представляют собой Н или C1-С6-алкил;
R5 представляет собой галоген, CN;
n, m или о представляют собой 0, 1 или 2;
за исключением
4-(бензилокси)-фенил-N-бутилкарбамата,
2-[4-(фенилметокси)фенокси]ацетамида,
4-(фенилметокси)фенилового эфира диметилкарбаминовой кислоты,
2-[4-(3-хлорбензокси)бензокси]ацетамида,
4-бензилокси-бензилового эфира диметилкарбаминовой кислоты,
4-бензилокси-фенилового эфира метилкарбаминовой кислоты,
2-[4-[(4-хлорфенил)метокси]фенокси]-2-метил-пропионамида и
2-[4-[(4-хлорфенил)метокси]фенокси]-2-метил-N-(1-метилэтил)-пропионамида,
а также при условии, что исключены соединения общей формулы (I), где один из R1 или R2 представляет собой C1-С6-алкил, а другой представляет собой Н;
R3 и R4 представляют собой Н;
n, m представляют собой 0; и
о представляет собой 1;
а также его фармацевтически приемлемых солей для изготовления фармацевтического препарата, обладающего свойством ингибитора моноаминооксидазы В.
Комментарии