Кристаллы лобаплатина, способы получения и применения в фармацевтике - RU2648990C1
Код документа: RU2648990C1
Чертежи






Описание
Область техники
Настоящее изобретение относится к области фармацевтических препаратов, в частности, к новым кристаллам лобаплатина, способам их получения и к их применениям в фармацевтике, которые относятся к области медикаментов.
Предпосылки создания изобретения
Лобаплатин (D19466) представляет собой противоопухолевое средство на основе платины третьего поколения, после цисплатина и карбоплатина, с химическим названием цис-[транс-1,2-циклобутан бис(метиламин)-N, N']-[(2S)-лактат-O1,O2]-платина(II), молекулярной формулой C9H18N2O3Pt, молекулярным весом 397,34 и химической структурной формулой, представленной в виде формулы (1) следующим образом:

Лобаплатин относится к алкилирующему средству (обобщенно) с функцией алканизации. Более того, он обладает высокой противоопухолевой активностью, например, он проявляет сильное ингибирующее действие на опухоль AH135 in vitro, меланому B16, опухоль толстой кишки 115, лейкемию мыши P338 in vivo и т. д. Лобаплатин обладает характеристиками сильной противораковой активности, низкой токсичности, без кумулятивной токсичности и нефротоксичности, кроме того, низкой токсичности в отношении костного мозга и без тромбоцитопении. Лобаплатин для инъекций, имеющийся в продаже, применяют в основном для лечения рака молочной железы, мелкоклеточного рака легких и хронического миелоидного лейкоза.
Первый рецепт данного лекарственного средства принадлежит ASTA Pharmaceutical Co., Ltd. немецкой компании (ASTA Medica AG), при этом впервые способ получения лобаплатина был описан в основном патенте EP 0324154. Способ получения тригидрата лобаплатина был раскрыт в последующем патенте EP 0611303, согласно которому продукт получают путем перекристаллизации безводного лобаплатина с образованием кристаллического продукта, содержащего три молекулы воды. В патенте EP 0611303 указано, что лобаплатин, полученный с помощью способа получения (из примера 1a), характеризуется свойством расплывания и, таким образом, лобаплатин является липким, и из него сложно изготавливать препараты.
Краткое описание изобретения
Техническая задача, решаемая настоящим изобретением, заключается в том, что существующий в настоящее время безводный лобаплатин имеет ряд недостатков, касающихся расплывания, больших трудностей при его переработке в препараты и недостаточной стабильности. В настоящем изобретении предусматривают новые кристаллы лобаплатина, являющиеся наиболее идеальными для обеспечения высокой биодоступности, хорошей стабильности, высокой растворимости, хорошей текучести, они с трудом впитывают влагу, поэтому не являются липкими, а также характеризуются хорошим выходом и чистотой.
Специалист в данной области техники знает, что одно и то же лекарственное средство, имеющее разные кристаллические формы, может иметь различия в биодоступности, а также в стабильности, текучести, сжимаемости и т. д. Такие физико-химические свойства могут оказывать определенное влияние на применение лекарственных средств. Полиморфизм лекарственных средств стал необходимой и важной частью процесса изучения лекарственных средств, контроля качества и процесса тестирования для фармацевтического производства. Изучение полиморфизма лекарственного средства целесообразно в отношении биологической активности, выбираемой среди новых лекарственных соединений, целесообразно в отношении улучшения биодоступности и усиления клинического лечебного действия, целесообразно в отношении выбора и разработки способов введения лекарственного средства и определения многопрофильных параметров для фармацевтических препаратов чтобы, таким образом, улучшить качество продукта.
Благодаря постоянным исследованиям и улучшениям изобретены новые кристаллические формы лобаплатина, а также способы их получения и их применения в фармацевтике.
Более конкретно, то для того, чтобы решить технические проблемы, упомянутые выше, настоящее изобретение предусматривает следующие несколько видов технических решений для новых кристаллов лобаплатина.
(1) Дигидрат лобаплатина
Во-первых, настоящее изобретение предусматривает дигидрат лобаплатина (также называемый кристаллом A лобаплатина), способы его получения и его применения в фармацевтике. Подробные описания приведены ниже.
Настоящее изобретение предусматривает кристалл соединения лобаплатина, характеризующийся двумя молекулами кристаллической воды, находящимися в структуре кристалла.
Предпочтительно, кристаллическая форма указанного соединения лобаплатина по настоящему изобретению представляет собой кристаллическую форму A, и при этом на рентгенограмме PXRD присутствуют дифракционные пики при значениях 2θ приблизительно 11,04, 12,32, 12,61, 13,85, 15,14, 15,55, 16,68, 17,67, 17,86, 19,03, 20,06, 21,00, 22,68, 22,92, 23,76, 25,39, 25,58, 26,37, 26,77, 27,00, 27,71, 28,13, 29,71, 31,42, 31,94, 32,89, 34,29, 34,60, 36,10, 36,93, 37,66, 40,78, 43,41, при этом граница погрешности значений 2θ составляет 0,2.
Предпочтительно, температура плавления Tт.п. указанного кристалла соединения лобаплатина составляет 220±5°С.
Предпочтительно, кристаллическая форма указанного кристалла соединения лобаплатина является кристаллической формой A, которая относится к орторомбической системе с пространственной группой P212121, параметрами элементарной ячейки a = 10,601 (2) Å, b = 14,020 (3), c = 9,759 (2) Å, α = β = γ = 90,0°, объемом элементарной ячейки V = 1450,5 (5) Å3 и числом ассиметричных единиц Z = 4 в элементарной ячейке.
В то же время настоящее изобретение дополнительно предусматривает способ получения вышеупомянутого соединения лобаплатина, отличающийся тем, что включает следующие стадии:
добавление тригидрата лобаплатина к растворителю для кристаллизации из суспензии с образованием смеси во взвешенном состоянии, перемешивание смеси, осаждение кристалла, удаление растворителя, а затем высушивание с получением кристалла.
Предпочтительно, в вышеупомянутом способе после удаления растворителя кристалл промывают этиловым эфиром перед высушиванием, где указанное высушивание представляет собой вакуумное высушивание.
Предпочтительно, в вышеупомянутом способе отношение массы указанного тригидрата лобаплатина к объему указанного кристаллизующего растворителя составляет 1(г):15-30(мл).
Предпочтительно, в вышеупомянутом способе указанный кристаллизующий растворитель выбирают из метил-трет-бутилового эфира, толуола, этилового эфира, бутилацетата, 1,4-диоксана или н-гептана.
Предпочтительно, в вышеприведенном способе указанное взвешенное состояние получают при комнатной температуре, предпочтительно, указанное взвешенное состояние получают в течение 45-50 ч.
В то же время настоящее изобретение предусматривает фармацевтическую композицию, отличающуюся тем, что вышеупомянутое соединение лобаплатина применяют в качестве активного компонента.
Предпочтительно, количество кристалла лобаплатина в минимальной единице указанной фармацевтической композиции составляет 5 мг, 10 мг или 50 мг.
Предпочтительно, указанная фармацевтическая композиция находится в любой клинически приемлемой фармацевтической лекарственной форме.
Предпочтительно, указанная лекарственная форма представляет собой лиофилизированный препарат для инъекций.
В то же время настоящее изобретение дополнительно предусматривает применение вышеупомянутого кристалла соединения лобаплатина или вышеупомянутой фармацевтической композиции для получения противораковых лекарственных средств.
В то же время настоящее изобретение дополнительно предусматривает применение вышеупомянутого кристалла соединения лобаплатина или вышеупомянутой фармацевтической композиции для лечения рака, главным образом для лечения одного из рака молочной железы, мелкоклеточного рака легких или хронического миелоидного лейкоза.
Настоящее изобретение дополнительно предусматривает применение вышеупомянутой кристаллической формы лобаплатина для получения фармацевтических композиций и фармацевтических препаратов.
(2) Кристалл B лобаплатина
Во-вторых, настоящее изобретение предусматривает кристалл B лобаплатина, способы его получения и его применения в фармацевтике. Подробные описания приведены ниже.
Настоящее изобретение предусматривает кристалл соединения лобаплатина, отличающийся тем, что кристаллическая форма соединения лобаплатина является кристаллической формой B, и при этом на рентгенограмме PXRD присутствуют дифракционные пики при значениях 2θ приблизительно 8,25, 9,77, 11,70, 13,13, 15,28, 16,48, 17,22, 17,74, 19,01, 19,56, 22,28, 23,72, 24,04, 24,30, 25,62, 26,20, 28,57, 30,22, 30,61, при этом граница погрешности значений 2θ составляет 0,2.
Предпочтительно, температура плавления Tт.п. указанного кристалла соединения лобаплатина согласно настоящему изобретению составляет 230±5°С.
Указанную температуру плавления измеряют посредством DSC со скоростью нагревания равной 10°С/мин и оценивают по максимальному пику.
В то же время настоящее изобретение дополнительно предусматривает способ получения кристалла лобаплатина с кристаллической формой B, включающий следующие стадии:
добавление безводного метанола или безводного этанола к тригидрату лобаплатина с получением смеси, перемешивание смеси при комнатной температуре до растворения твердых веществ, удаление нерастворимых веществ из смеси, медленное выпаривание смеси, осаждение кристалла, выделение кристалла, а затем высушивание кристалла с получением белого порошка, который представляет собой кристалл B лобаплатина.
Предпочтительно, в вышеописанном способе получения отношение массы указанного тригидрата лобаплатина к объему указанного безводного метанола составляет 1(г):40-50(мл); отношение массы указанного тригидрата лобаплатина к объему указанного безводного этанола составляет 1(г):80-90(мл).
В то же время настоящее изобретение дополнительно предусматривает способ получения кристалла лобаплатина с кристаллической формой B, включающий следующую стадию b):
добавление безводного метанола к дигидрату лобаплатина с получением смеси, перемешивание смеси при комнатной температуре до растворения твердых веществ, удаление нерастворимых веществ из смеси, медленное выпаривание смеси, осаждение кристалла, выделение кристалла и высушивание кристалла с получением белого порошка, который представляет собой кристалл B лобаплатина.
В то же время настоящее изобретение дополнительно предусматривает способ получения кристалла лобаплатина с кристаллической формой B, который включает следующую стадию b):
добавление органического растворителя к дигидрату лобаплатина с образованием смеси во взвешенном состоянии, перемешивание смеси при комнатной температуре, осаждение кристалла, выделение кристалла и высушивание кристалла с получением белого порошка, который представляет собой кристалл B лобаплатина.
Предпочтительно, в вышеупомянутом способе получения способ получения указанного дигидрата лобаплатина включает следующую стадию a):
добавление растворителя для кристаллизации из суспензии к тригидрату лобаплатина с образованием смеси во взвешенном состоянии, перемешивание смеси, осаждение кристалла, промывание этиловым эфиром после удаления растворителя, а затем вакуумное высушивание кристалла с получением кристалла дигидрата лобаплатина.
Предпочтительно, в вышеупомянутом способе отношение массы указанного тригидрата лобаплатина к объему кристаллизующего растворителя на стадии a) составляет 1(г):15-30(мл).
Предпочтительно, в вышеупомянутом способе указанный растворитель для кристаллизации на стадии a) выбирают из метил-трет-бутилового эфира, толуола, этилового эфира, бутилацетата, 1,4-диоксана или н-гептана.
Предпочтительно, в вышеупомянутом способе после выделения кристалла кристалл промывают этиловым эфиром перед высушиванием кристалла, где на стадии b) указанное высушивание представляет собой вакуумное высушивание.
Предпочтительно, в вышеупомянутом способе указанное взвешенное состояние на стадии b) получают при комнатной температуре, преимущественно, указанное взвешенное состояние получают в течение 45-50 ч.
Предпочтительно, в вышеупомянутом способе отношение массы дигидрата лобаплатина к объему безводного метанола на указанной стадии b) составляет 1(г):40-50(мл).
Предпочтительно, в вышеупомянутом способе органический растворитель на указанной стадии b) выбирают из н-гексана, ацетона, этилацетата, нитрометана, ацетонитрила, тетрагидрофурана, 2-бутанона или дихлорметана, и при этом отношение массы дигидрата лобаплатина к объему органического растворителя составляет 1(г):15-30(мл).
В то же время настоящее изобретение предусматривает фармацевтическую композицию, отличающуюся тем, что вышеупомянутый кристалл B лобаплатина применяют в качестве активного компонента.
Предпочтительно, количество кристалла лобаплатина в минимальной единице указанной фармацевтической композиции составляет 5 мг, 10 мг или 50 мг.
Предпочтительно, форма указанной фармацевтической композиции представляет собой любую клинически приемлемую фармацевтическую лекарственную форму.
Предпочтительно, указанная лекарственная форма представляет собой лиофилизированный препарат для инъекций.
В то же время настоящее изобретение дополнительно предусматривает применение вышеупомянутого кристалла лобаплатина или вышеупомянутой фармацевтической композиции для получения противораковых лекарственных средств.
В то же время настоящее изобретение дополнительно предусматривает применение вышеупомянутого кристалла соединения лобаплатина или вышеупомянутой фармацевтической композиции для лечения рака, в частности, для лечения одного из рака молочной железы, мелкоклеточного рака легких или хронического миелоидного лейкоза.
(3) Кристалл F лобаплатина
В-третьих, настоящее изобретение предусматривает кристалл F лобаплатина, способы его получения и его применения в фармацевтике. Подробные описания приведены ниже.
Настоящее изобретение предусматривает кристалл соединения лобаплатина, отличающийся тем, что кристаллическая форма соединения является кристаллом F, и при этом на рентгенограмме PXRD присутствуют дифракционные пики при значениях 2θ приблизительно 8,21, 11,60, 12,99, 15,24, 16,44, 17,11, 17,55, 18,42, 19,01, 19,20, 19,42, 21,81, 22,17, 22,42, 23,33, 23,85, 24,18, 24,40, 24,77, 25,46, 25,98, 26,13, 27,89, 28,42, 29,03, 30,32, 31,17, 31,94, 33,30, 36,20, 37,62, 39,66, при этом граница погрешности значений 2θ составляет 0,2.
Температура плавления Tт.п. указанного кристалла соединения лобаплатина составляет 229±5°С.
Более того, указанную температуру плавления измеряют посредством DSC со скоростью нагревания, равной 10°С/мин, и оценивают по максимальному пику.
В то же время настоящее изобретение дополнительно предусматривает способ получения указанного кристалла F лобаплатина, который отличается тем, что включает следующую стадию b):
добавление метанола или этанола к дигидрату лобаплатина с получением смеси, перемешивание смеси при комнатной температуре до растворения твердых веществ, фильтрование смеси с удалением нерастворимых веществ, медленное добавление органического растворителя к смеси, осаждение кристалла, выделение кристалла, а затем высушивание кристалла с получением белого порошка, который представляет собой кристалл F лобаплатина.
Предпочтительно, способ получения указанного дигидрата лобаплатина включает следующую стадию a):
добавление растворителя для кристаллизации из суспензии к тригидрату лобаплатина с образованием смеси во взвешенном состоянии, перемешивание смеси, осаждение кристалла из смеси, промывание кристалла этиловым эфиром после удаления растворителя, а затем вакуумное высушивание кристалла с получением кристалла дигидрата лобаплатина.
Предпочтительно, отношение массы указанного тригидрата лобаплатина к объему растворителя для кристаллизации на стадии a) составляет 1(г):15-30(мл).
Предпочтительно, указанный растворитель для кристаллизации на стадии a) выбирают из метил-трет-бутилового эфира, толуола, этилового эфира, бутилацетата, 1,4-диоксана или н-гептана.
Предпочтительно, на стадии b) после выделения кристалла кристалл промывают этиловым эфиром перед высушиванием кристалла, где указанное высушивание представляет собой вакуумное обезвоживание.
Предпочтительно, указанный органический растворитель на указанной стадии b) выбирают из диметилового эфира этиленгликоля, н-гексана, этилацетата, ацетона, нитрометана, ацетонитрила, тетрагидрофурана или метиленхлорида.
Предпочтительно, отношение массы указанного дигидрата лобаплатина к объему органического растворителя на указанной стадии b) составляет 1(г):120-200(мл).
Предпочтительно, отношение массы указанного дигидрата лобаплатина к объему метанола составляет 1(г):40-50(мл); отношение массы указанного дигидрата лобаплатина к объему этанола составляет 1(г):80-90(мл).
В то же время настоящее изобретение предусматривает фармацевтическую композицию, отличающуюся тем, что вышеупомянутый кристалл F лобаплатина применяют в качестве активного компонента.
Предпочтительно, количество кристалла лобаплатина в минимальной единице указанной фармацевтической композиции составляет 5 мг, 10 мг или 50 мг.
Предпочтительно, форма указанной фармацевтической композиции представляет собой любую клинически приемлемую фармацевтическую лекарственную форму.
Предпочтительно, указанная лекарственная форма представляет собой лиофилизированный препарат для инъекций.
В то же время настоящее изобретение дополнительно предусматривает применение вышеупомянутого кристалла лобаплатина или вышеупомянутой фармацевтической композиции для получения противораковых лекарственных средств.
В то же время настоящее изобретение дополнительно предусматривает применение вышеупомянутого кристалла лобаплатина или вышеупомянутой фармацевтической композиции для лечения рака, в частности, для лечения одного из рака молочной железы, мелкоклеточного рака легких или хронического миелоидного лейкоза.
Исходный материал тригидрата лобаплатина, применяемый в настоящем изобретении, получают согласно способу из примеров в патенте EP 0611303.
Вышеупомянутый кристалл лобаплатина применяют в качестве активного компонента в указанной фармацевтической композиции, а количество кристалла лобаплатина в минимальной единице указанной фармацевтической композиции составляет 5 мг, 10 мг или 50 мг. Новый кристалл лобаплатина может быть получен в виде фармацевтической композиции с одним или несколькими фармацевтически приемлемыми носителями или наполнителями. Кроме того, указанная фармацевтическая композиция может быть получена в любой из фармацевтически приемлемых лекарственных форм, клинически подходящих для материала, включая лекарственные формы, вводимые не через желудочно-кишечный тракт, такие как инъекционный состав, состав для трансдермального введения, респираторные лекарственные формы, составы для мукозального введения через полость и другие части тела и т. д., преимущественно лиофилизированный порошок для инъекций.
Фармацевтические носители или наполнители могут быть выбраны из одного или нескольких из воды для инъекций, маннита, лактозы, полиэтиленгликолей, полисорбата 80, пропиленгликоля, винной кислоты, лимонной кислоты, аскорбиновой кислоты, эдетата динатрия, натрия кальция эдетата, бисульфита натрия, глюкозы, хлорида натрия, соевого масла, соевого лецитина, фосфолипидов яичных желтков, дистеарила фосфатидилэтаноламина, декстрана, глицина, глицерина.
Способ получения вышеупомянутых композиций и составов, как правило, известен специалисту в данной области техники. Как активная форма соединений лобаплатина согласно настоящему изобретению, так и составы лобаплатина, имеющиеся в продаже, являются лобаплатином, а точнее безводным лобаплатином, так что соединения лобаплатина согласно настоящему изобретению применимы для лечения всех заболеваний, при которых используют имеющиеся в продаже продукты на основе лобаплатина.
Лобаплатин, а именно цис-платина-[транс-1,2-циклобутан-бис-(метиламин)-N, N']-(2S)-лактат-O1, O2]-платина(II), относится к алкилирующему средству и цитотоксическому лекарственному средству, а также известен как биоалкилирующее средство. Карбоний ионы или другие соединения, имеющие активные электрофильные группы, могут быть образованы in vivo из лобаплатина и могут быть ковалентно связаны с группами (такими как амино, меркапто, гидроксильная, карбоксильная группа, группа фосфорной кислоты и т. д.), имеющими высокое содержание электронов, в биологических макромолекулах (ДНК, РНК, ферментах) клеток. Из-за утраты активности или разрыва молекул ДНК вследствие этого, может произойти гибель опухолевых клеток. Вот почему лобаплатин обладает сильной противоопухолевой активностью. Фармакокинетические исследования показывают, что после внутривенной инъекции лобаплатина весь лобаплатин и свободный лобаплатин, что образуется, оказывают противоопухолевое воздействие в сыворотках, другими словами, лобаплатин играет эффективную роль в этом в форме безводного лобаплатина, независимо от состояния материалов.
Вышеупомянутые три вида новых кристаллических форм лобаплатина по настоящему изобретению разработаны с устранением недостатков аморфного лобаплатина, а именно легкого расплывания, из-за чего он прилипает, недостаточной стабильности, трудностей хранения и т. д. Новые кристаллические формы лобаплатина обладают такими характеристиками, как высокая биодоступность, хорошая стабильность, они с трудом расплываются и т. д. Более того, было неожиданно обнаружено, что новые формы лобаплатина имеют преимущества перед тригидратом лобаплатина, такие как высокая растворимость, высокие выход и чистота, а также лучшая стабильность по сравнению с тригидратом лобаплатина. Следовательно, создание новых кристаллических форм целесообразно в отношении выбора и разработки способов введения лекарственного средства и определения технологических параметров для фармацевтических препаратов, чтобы, таким образом, улучшить качество производства лекарственных средств. Новые соединения лобаплатина по настоящему изобретению являются весьма стабильными при нормальных температурах, они с трудом расплываются и поэтому не являются липкими, они имеют хорошую подвижность, причем по сравнению с аморфным лобаплатином они обладают преимуществом, заключающимся в удобстве использования при хранении, транспортировке, а также получении и обработке.
Описание графических материалов
Фиг. 1: рентгеновская дифракционная рентгенограмма дигидрата лобаплатина.
Фиг. 2: проекционный рисунок молекулярной стереохимической структуры дигидрата лобаплатина.
Фиг. 3: дифференциально-термический анализ (термограмма DSC) дигидрата лобаплатина.
Фиг. 4: термогравиметрический анализ (термограмма TGA) дигидрата лобаплатина.
Фиг. 5: рентгеновская дифракционная рентгенограмма кристалла B лобаплатина.
Фиг. 6: дифференциально-термический анализ (термограмма DSC) кристалла B лобаплатина.
Фиг.7: термогравиметрический анализ (термограмма TGA) кристалла B лобаплатина.
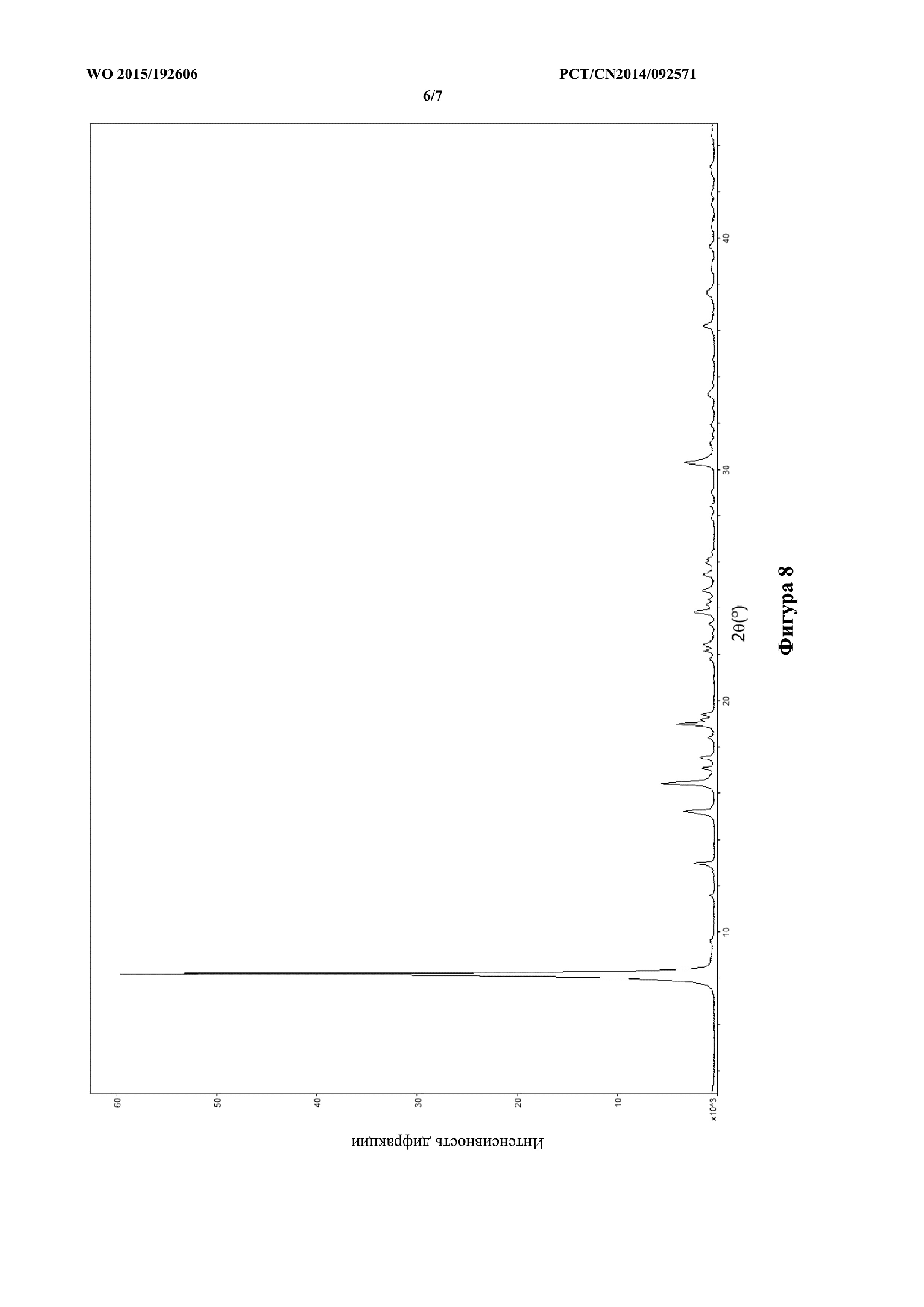
Фиг. 8: рентгеновская дифракционная рентгенограмма кристалла F лобаплатина.
Фиг. 9: дифференциально-термический анализ (термограмма DSC) кристалла F лобаплатина.
Фиг.10: термогравиметрический анализ (термограмма TGA) кристалла F лобаплатина.
Варианты осуществления изобретения
(1) Первый вариант осуществления настоящего изобретения
Настоящее изобретение предусматривает дигидрат лобаплатина, характеризующийся такой морфологией кристалла, при которой он хорошо растворим, имеет высокий выход и превосходную стабильность. Конкретные описания представлены ниже.
Настоящее изобретение предусматривает дигидрат лобаплатина c кристаллической формой A и его идентифицирующие данные рентгеновской дифракции (PXRD) являются следующими.
Кристаллическую форму A дигидрата лобаплатина анализируют с помощью рентгеновского дифрактометра типа Bruker D8 advance XRD, который производит компания Bruker. Условия измерений являются следующими: CuKa (40 кВ, 40 мА), скорость сканирования 2°/мин (значения 2θ), диапазон сканирования 3°-45° (значения 2θ). Кристаллическая форма A дигидрата лобаплатина характеризуется абсорбционными пиками со следующими характеристиками, представленными в таблице 1-a, и ее дифракционная рентгенограмма представлена на фиг. 1.
Таблица 1-a. Результат измерения с помощью рентгеновской дифракции (PXRD) кристаллической формы А дигидрата лобаплатина
Соединение лобаплатина с кристаллической формой A является бесцветным и прозрачным столбчатым кристаллом, как определено путем проведения экспериментов рентгеноструктурного анализа. Соединение лобаплатина с кристаллической формой A относится к орторомбической сингонии с пространственной группой P212121, параметрами элементарной ячейки a = 10,601 (2), b = 14,020 (3), c = 9,759 (2) Å, α = β = γ = 90,0°, объемом элементарной ячейки V = 1450,5 (5) Å3 и числом асимметричных единиц Z = 4 в элементарной ячейке.
Данные интенсивности дифракции получают с помощью дифрактометра Bruker SMART APEX-II с такими параметрами: излучение CuKα, графитовый монохроматор, диаметр отдельного сосуда ф = 0,50 мм, расстояние между кристаллом и CCD детектором d = 60,3 мм, напряжение на лампе 40 кВ, ток электронной лампы 30 мА, режим сканирования φ/ω скан. Число всех полученных точек дифракции равняется 5844, из которых число независимых точек дифракции равняется 2376, и число наблюдаемых точек равняется 2376 (| F | 2≥2σ| F | 2).
Структуру кристалла анализируют посредством прямого способа (Shelxs97), при этом могут быть получены все положения 17 атомов, не являющихся водородом. Структурные параметры являются уточненными, а атомные частицы определяются посредством метода наименьших квадратов. Получают положения атомов водорода посредством способа геометрического расчета и разностного метода Фурье. И наконец, фактор надежности R1 = 0,0569, wR2 = 0,1491 (w = 1 / σ | F | 2), S = 1,077. В конце определяли стехиометрическую формулу C9H18N2O3Pt • 2H2O, при этом рассчитанный молекулярный вес составлял 433,36 и рассчитанная плотность кристалла составляла 1,975 г/см3. Полученный новый кристалл лобаплатина является дигидратом лобаплатина, определенным посредством структурного анализа, со структурой молекулы, представленной следующей формулой (2):

Проекционный рисунок стереохимической структуры молекулы дигидрата лобаплатина с кристаллической формой A представлен на фигуре 2.
Дифференциально-термический анализ (DSC-TGA) дигидрата лобаплатина с кристаллической формой A осуществляют с помощью дифференциальных термоанализаторов моделей NETZSCH DSC 204 F1, NETZSCH TG 209 F1, которые производит компания NETZSCH. Термограмма DSC представлена на фигуре 3, а термограмма TGA представлена на фигуре 4. Результаты показывают, что измеренная посредством DSC и оцененная посредством максимального пика температура плавления Tт.п. составляла 220 ± 5°С, при скорости нагревания 10°С/мин. В частности, на термограмме DSC присутствует широкий эндотермический пик около 117°С. Данный пик может быть вызван потерей двух молекул кристаллической воды, судя по совмещению с данными относительно монокристалла и данными TGA. На термограмме DSC присутствует экзотермический пик при 220 ± 5°С. Этот пик является пиком плавления и разложения, судя по совмещению данных TGA и данных относительно температуры плавления, записанных в европейском патенте EP 0611303. На термограмме TGA присутствует 9,49% потеря веса перед 150°С, которая означает, что потеря веса вызвана потерей двух молекул кристаллической воды.
В то же время настоящее изобретение предусматривает способ получения новой кристаллической формы A лобаплатина, которую просто получить, с ней легко работать, и она подходит для расширенного производства, включающий следующие стадии:
взвешивание и помещение тригидрата лобаплатина в емкость, добавление органического растворителя в емкость с образованием смеси во взвешенном состоянии, перемешивание смеси при комнатной температуре в течение 45-50 ч., фильтрование смеси, промывание продукта, полученного путем фильтрования, этиловым эфиром, а затем вакуумное высушивание с получением белого порошка, который представляет собой дигидрат лобаплатина. Органический растворитель выбирают из метил-трет-бутилового эфира, толуола, этилового эфира, бутилацетата, 1,4-диоксана или н-гептана. Отношение массы тригидрата лобаплатина к объему органического растворителя составляет 1(г):15-30(мл).
(2) Второй вариант осуществления настоящего изобретения
Настоящее изобретение предусматривает новый кристалл лобаплатина, названный кристаллической формой B, который хорошо растворим и имеет превосходную стабильность.
С одной стороны, настоящее изобретение предусматривает кристалл B лобаплатина, который характеризуется морфологией кристалла B. Точные описания полученной кристаллической формы B лобаплатина согласно настоящему изобретению приведены ниже.
Кристаллическую форму B лобаплатина анализируют с помощью рентгеновского дифрактометра модели Bruker D8 advance XRD, который производит компания Bruker. Условия измерений являются следующими: CuKa (40 кВ, 40 мА), скорость сканирования 2°/мин (значения 2θ), диапазон сканирования 3°-45° (значения 2θ). Кристаллическая форма B лобаплатина характеризуется абсорбционными пиками со следующими характеристиками, представленными в таблице 1-b, и ее дифракционная рентгенограмма представлена на фиг. 5.
Таблица 1-b. Результаты измерений кристаллической формы B лобаплатина посредством рентгеновской дифракции (PXRD)
Дифференциально-термический анализ (DSC-TGA) новой кристаллической формы B лобаплатина осуществляют с помощью дифференциальных термоанализаторов моделей NETZSCH DSC 204 F1, NETZSCH TG 209 F1, которые производит компания NETZSCH. Термограмма DSC представлена на фигуре 6, а термограмма TGA представлена на фигуре 7. Результаты показывают, что измеренная посредством DSC и оцененная посредством максимального пика температура плавления Tт.п. составляла 230 ± 5°С, при скорости нагревания 10°С/мин. В частности, на термограмме DSC присутствует экзотермический пик при 230 ± 5°С, который является пиком плавления и разложения, судя по совмещению данных TGA и данных относительно температуры плавления, записанных в европейском патенте EP 0611303. На термограмме TGA отсутствует потеря веса перед 150°С, это означает, что кристаллическая форма B лобаплатина не является сольватом.
В то же время настоящее изобретение предусматривает способ получения новой кристаллической формы B лобаплатина, которую просто получить, с ней легко работать, и она подходит для расширенного производства.
В предпочтительном варианте осуществления настоящего изобретения способы получения указанной новой кристаллической формы B лобаплатина согласно настоящему изобретению включают следующие стадии:
a) получение дигидрата лобаплатина: взвешивание и помещение тригидрата лобаплатина в емкость, добавление 15-30 мл органического растворителя из расчета на 1 г тригидрата лобаплатина в емкость с образованием смеси во взвешенном состоянии, перемешивание смеси при комнатной температуре в течение 45-50 ч, фильтрование смеси, промывание продукта, полученного путем фильтрования, этиловым эфиром, а затем вакуумное высушивание с получением белого порошка, который представляет собой дигидрат лобаплатина;
где органический растворитель выбирают из метил-трет-бутилового эфира, толуола, этилового эфира, бутилацетат, 1,4-диоксана или н-гептана;
b) получение целевого кристалла: взвешивание полученного на стадии a дигидрата лобаплатина, помещение взвешенного дигидрата лобаплатина в емкость, добавление безводного метанола в емкость, перемешивание при комнатной температуре до растворения твердых веществ, фильтрование для удаления нерастворимых веществ, медленное выпаривание на проветриваемой кухне и выделение кристалла после осаждения кристалла, промывание кристалла 2-3 раза этиловым эфиром, а затем вакуумное высушивание кристалла с получением белого порошка, который представляет собой новую кристаллическую форму B лобаплатина.
Отношение массы дигидрата лобаплатина к объему безводного метанола на указанной стадии b составляет 1(г):40-50(мл).
В другом предпочтительном варианте осуществления способы получения указанной новой кристаллической формы B лобаплатина предпочтительно согласно настоящему изобретению также могут быть такими:
a) получение дигидрата лобаплатина: взвешивание и помещение тригидрата лобаплатина в емкость, добавление 15-30 мл органического растворителя в пересчете на 1 г тригидрата лобаплатина в емкость с образованием смеси во взвешенном состоянии, перемешивание смеси при комнатной температуре в течение 45-50 ч., фильтрование смеси, промывание продукта, полученного путем фильтрования, этиловым эфиром, а затем вакуумное высушивание с получением белого порошка, который представляет собой дигидрат лобаплатина;
где органический растворитель выбирают из метил-трет-бутилового эфира, толуола, этилового эфира, бутилацетат, 1,4-диоксана или н-гептана;
b) получение целевого кристалла: взвешивание полученного на стадии a дигидрата лобаплатина, помещение взвешенного дигидрата лобаплатина в емкость, добавление органического растворителя в емкость с образованием смеси во взвешенном состоянии, перемешивание при комнатной температуре в течение 45-50 часов, осаждение кристалла, фильтрование и выделение кристалла, промывание кристалла 2-3 раза этиловым эфиром, а затем вакуумное высушивание с получением белого порошка, который представляет собой новую кристаллическую форму B лобаплатина.
Указанный органический растворитель на стадии b выбирают из н-гексана, ацетона, этилацетата, нитрометана, ацетонитрила, тетрагидрофурана, 2-бутанона или дихлорметана. Отношение массы дигидрата лобаплатина к объему органического растворителя составляет 1(г):15-30(мл).
В еще одном предпочтительном варианте осуществления способы получения указанной новой кристаллической формы B лобаплатина, предпочтительно согласно настоящему изобретению, могут также быть такими: добавление безводного метанола или безводного этанола к тригидрату лобаплатина, перемешивание при комнатной температуре до растворения твердых веществ, удаление нерастворимых веществ, медленное выпаривание, выделение кристалла после осаждения кристалла, высушивание с получением белого порошка, который представляет собой кристаллическую форму B лобаплатина. Отношение массы тригидрата лобаплатина к объему безводного этанола составляет 1(г):80-90(мл).
(3) Третий вариант осуществления настоящего изобретения
Настоящее изобретение предусматривает новый кристалл лобаплатина, названный кристаллической формой F, который хорошо растворим и имеет превосходную стабильность.
С одной стороны, настоящее изобретение предусматривает кристалл F лобаплатина, который характеризуется морфологией кристаллической формы F. Далее точные описания полученной кристаллической формы F лобаплатина согласно настоящему изобретению приведены ниже.
Кристаллическую форму F лобаплатина анализируют с помощью рентгеновского дифрактометра модели Bruker D8 advance XRD, который производит компания Bruker. Условия измерений являются следующими: CuKa (40 кВ, 40 мА), скорость сканирования 2°/мин (значения 2θ), диапазон сканирования 3°-45° (значения 2θ). Кристаллическая форма F лобаплатина характеризуется абсорбционными пиками со следующими характеристиками, представленными в таблице 1-c, и ее дифракционная рентгенограмма представлена на фиг. 8.
Таблица 1-c. Результаты измерений кристаллической формы F лобаплатина посредством рентгеновской дифракции (PXRD)
Дифференциально-термический анализ (DSC-TGA) новой кристаллической формы F лобаплатина осуществляют с помощью дифференциальных термоанализаторов моделей NETZSCH DSC 204 F1, NETZSCH TG 209 F1, которые производит компания NETZSCH. Термограмма DSC представлена на фигуре 9, а термограмма TGA представлена на фигуре 10. Результаты показывают, что измеренная посредством DSC и оцененная посредством максимального пика температура плавления Tт.п. составляет 229 ± 5°С, при скорости нагревания 10°С/мин. В частности, на термограмме DSC присутствует экзотермический пик при 229 ± 5°С, который является пиком плавления и разложения, судя по совмещению данных TGA и данных относительно температуры плавления, записанных в европейском патенте EP 0611303. На термограмме TGA присутствует 1,97% потеря веса перед 150°С, и при совмещении с данными DSC можно сделать вывод о том, что потеря веса происходит из-за оставшегося растворителя.
В то же время настоящее изобретение предусматривает способ получения новой кристаллической формы F лобаплатина, которую просто получить, с ней легко работать, и она подходит для расширенного производства.
В предпочтительном варианте осуществления настоящего изобретения способы получения новой кристаллической формы F лобаплатина согласно настоящему изобретению включают следующие стадии:
a) получение дигидрата лобаплатина: взвешивание и помещение тригидрата лобаплатина в емкость, добавление 15-30 мл органического растворителя в пересчете на 1 г тригидрата лобаплатина в емкость с образованием смеси во взвешенном состоянии, перемешивание смеси при комнатной температуре в течение 45-50 ч., фильтрование смеси, промывание продукта, полученного путем фильтрования, этиловым эфиром, а затем вакуумное высушивание с получением белого порошка, который представляет собой дигидрат лобаплатина;
где органический растворитель выбирают из метил-трет-бутилового эфира, толуола, этилового эфира, бутилацетат, 1,4-диоксана или н-гептана;
b) получение целевого кристалла: взвешивание полученного на стадии a дигидрата лобаплатина, помещение взвешенного дигидрата лобаплатина в емкость, добавление метанола или этанола в емкость с образованием смеси, перемешивание смеси при комнатной температуре до растворения твердых веществ, фильтрование для удаления нерастворимых веществ, медленное добавление органического растворителя к отфильтрованной жидкости, фильтрование и выделение кристалла после осаждения кристалла, промывание кристалла этиловым эфиром, а затем вакуумное высушивание с получением белого порошка, который представляет собой новую кристаллическую форму F лобаплатина.
Указанный органический растворитель на стадии b выбирают из диметилового эфира этиленгликоля, н-гексана, этилацетата, ацетона, нитротолуола, ацетонитрила, тетрагидрофурана, дихлорметана. Отношение массы дигидрата лобаплатина к объему органического растворителя составляет 1(г):120-200(мл).
Отношение массы дигидрата лобаплатина к объему метанола на указанной стадии b составляет 1(г):40-50(мл), а отношение массы дигидрата лобаплатина к объему этанола на указанной стадии b составляет 1(г):80-90(мл).
Примеры
Способы получения кристалла лобаплатина в формах A, B и F согласно настоящему изобретению, а также способы скрининга и отбора разных новых кристаллических форм, идентификации и определения их свойств описаны в нижеследующих примерах, в частности, в настоящем изобретении.
Пример 1. Скрининг и анализы разных кристаллических форм
1.1. Скрининги посредством способа кристаллизации выпариванием при комнатной температуре
Во флакон объемом 10 мл отбирали 20 мг образца тригидрата лобаплатина и во флакон добавляли 3 мл безводного этанола или безводного метанола с получением смеси. После полного растворения тригидрата лобаплатина смесь медленно выпаривали при 25°С и получали сухие твердые вещества. Полученные твердые вещества анализировали с помощью PXRD. Полученные результаты представлены в таблице 2.
Таблица 2. Результаты эксперимента по кристаллизация выпариванием при комнатной температуре
Результаты показали, что при сравнении кристаллическая форма, полученная с помощью безводного метанола, была такой же как и кристаллическая форма, полученная с помощью безводного этанола, которую временно назвали кристаллической формой B.
1.2. Скрининги посредством способа кристаллизации из суспензии
Во флакон объемом 10 мл отбирали 20 мг образца тригидрата лобаплатина и во флакон добавляли 4 мл органического растворителя, упомянутого в таблице 3, с получением суспензии. Полученную суспензию подвергали вибрации в течение 5 ч. При 25°С, а затем удаляли растворитель с получением твердых веществ. После высушивания полученные твердые вещества анализировали с помощью PXRD. Результаты представлены в таблице 3.
Таблица 3. Результаты эксперимента по кристаллизации из суспензии

Результаты показали, что посредством кристаллизации из суспензии было получено 9 видов кристаллических форм, которые временно назвали кристаллическими формами A, B, C, D, E, G, H, I, L.
1.3. Скрининги посредством способа кристаллизации растворением и осаждением
Раствор готовили посредством добавления 20 мг образца тригидрата лобаплатина к 3 мл безводного метанола или безводного этанола. Нижеприведенные органические растворители постепенно добавляли до осаждения твердых веществ, а затем удаляли надосадочную жидкость с получением твердых веществ. После высушивания твердые вещества анализировали посредством PXRD. Полученные результаты представлены в таблице 4.
Таблица 4. Результаты эксперимента по кристаллизации с помощью растворителя

Результаты показали, что посредством кристаллизации с помощью растворителя было получено 7 видов кристаллических форм, которые временно назвали кристаллическими формами F, J, K, M, N, O, P.
1.4. Определение характеристик разных кристаллических форм
Помимо измерения PXRD в отношении образцов кристаллических форм A-P также может быть выполнено определение характеристик с помощью DCS и TGA. Названия, модели и производители каждого из применяемых инструментов представлены в таблице 5.
Таблица 5. Модели и производители каждого из инструментов, применяемых для определения характеристик кристаллических форм

Результаты измерений были следующими.
Кристаллическая форма A характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 11,04, 12,32, 12,61, 13,85, 15,14, 15,55, 16,68, 17,67, 17,86, 19,03, 20,06, 21,00,2 2,68, 22,92, 23,76, 25,39, 25,58, 26,37, 26,77, 27,00, 27,71, 28,13, 29,71, 31,42, 31,94, 32,89, 34,29, 34,60, 36,10, 36,93, 37,66, 40,78, 43,41, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма A характеризовалась тем, что на термограмме DSC присутствовал экзотермический пик около 220 ± 5°C.
Кристаллическая форма B характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 8,25, 9,77, 11,70, 13,13, 15,28, 16,48, 17,22, 17,74, 19,01, 19,56, 22,28, 23,72, 24,04, 24,30, 25,62, 26,20, 28,57, 30,22, 30,61, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма B характеризовалась тем, что на термограмме DSC присутствовал экзотермический пик около 230 ± 5°C.
Кристаллическая форма C характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 6,79, 8,07, 12,24, 12,61, 13,50, 16,50, 17,83, 18,32, 18,79, 20,09, 21,64, 22,27, 23,19, 24,73, 27,34, 28,35, 29,12, 31,92, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма C характеризовалась тем, что на термограмме DSC присутствовал экзотермический пик около 228 ± 5°C.
Кристаллическая форма D характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 6,76, 11,07, 12,35, 12,65, 13,88, 15,18, 15,56, 16,68, 17,70, 17,90, 20,08, 21,02, 22,70, 22,92, 25,41, 25,64, 26,41, 26,79, 27,02, 28,15, 31,44, 31,96, 32,96, 34,34, 34,62, 36,93, 40,82, 43,46, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма D характеризовалась тем, что на термограмме DSC присутствовал экзотермический пик около 218 ± 5°C.
Кристаллическая форма E характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 6,61, 8,09, 12,38, 13,03, 15,40, 16,66, 17,47, 19,07, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма E характеризовалась тем, что на термограмме DSC присутствовал экзотермический пик около 214 ± 5°C.
Кристаллическая форма F характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 8,21, 11,60, 12,99, 15,24, 16,44, 17,11, 17,55, 18,42, 19,01, 19,20, 19,42, 21,81, 22,17, 22,42, 23,33, 23,85, 24,18, 24,40, 24,77, 25,46, 25,98, 26,13, 27,89, 28,42, 29,03, 30,32, 31,17, 31,94, 33,30, 36,20, 37,62, 39,66, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма F характеризовалась тем, что на термограмме DSC присутствовал экзотермический пик около 229 ± 5°C.
Кристаллическая форма G характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 8,62, 10,82, 11,03, 12,26, 12,59, 13,82, 15,12, 15,57, 16,59, 17,43, 17,65, 18,48, 19,46, 20,11, 20,37, 21,01, 22,66, 22,86, 24,60, 25,40, 26,33, 26,77, 27,00, 28,11, 29,79, 31,42, 31,94, 32,87, 34,25, 34,58, 36,06, 40,76, 42,75, 43,39, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма H характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 8,35, 8,53, 8,68, 12,97, 15,24, 17,41, 18,40, 19,13, 19,48, 20,37, 24,68, 25,41, 30,33, 31,66, 36,34, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма I характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 6,75, 8,39, 11,07, 11,59, 12,32, 12,63, 12,99, 15,20, 16,80, 17,07, 17,57,19,14,19,46, 21,00, 22,13, 22,84, 23,29, 23,77, 24,22, 25,82, 26,76, 28,38, 30,34, 30,83, 31,90, 33,63, 36,32, 38,47, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма J характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 5,94, 8,35, 9,87, 13,05, 15,28, 16,66, 19,15, 22,22, 22,68, 25,09, 30,71, 33,56, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма K характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 8,29, 11,02, 12,31, 12,61, 13,84, 15,14, 15,53, 16,70, 17,66, 19,05, 20,06, 20,98, 22,68, 22,90, 25,60, 26,37, 26,77, 26,98, 27,68, 28,23, 29,75, 31,40, 31,88, 32,90, 33,81, 34,29, 34,60, 36,10, 36,84, 37,64, 39,93, 40,76, 41,51, 42,36, 42,70, 43,39, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма L характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 6,71, 7,91, 10,75, 11,84, 14,06, 14,29, 15,85, 16,78, 17,29, 19,76, 20,20, 20,63, 21,08, 21,58, 21,89, 22,17, 23,87, 25,09, 26,83, 27,02, 28,73, 29,18, 29,92, 30,56, 31,61, 33,95, 40,33, 41,33, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма M характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 8,05, 13,03, 15,20, 16,19, 17,47, 18,77, 19,32, 24,06, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма N характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 7,94, 12,67, 14,83, 16,32, 17,16, 18,71, 21,83, 22,44, 24,10, 24,89, 27,97, 30,02, 30,48, при этом граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма O характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 6,75, 8,15, 16,29, 18,95, 22,23, 24,52, 29,93, где граница погрешности значений 2θ составляла 0,2.
Кристаллическая форма P характеризовалась тем, что на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 6,61, 8,17, 13,34, 16,52, 20,10, 24,97, 27,02, 33,99, 41,06, при этом граница погрешности значений 2θ составляла 0,2.
1.5. Повторные эксперименты и эксперименты по увеличению в масштабе
На 100 мг образцов с кристаллическими формами A-P проводили расширенные и повторные испытания для проверки воспроизводимости кристаллических форм согласно вышеупомянутым «1.2 Скринингам посредством способа кристаллизации из суспензии». Результаты представлены в таблице 6 ниже.
Таблица 6. Результаты повторных экспериментов и экспериментов по увеличению в масштабе

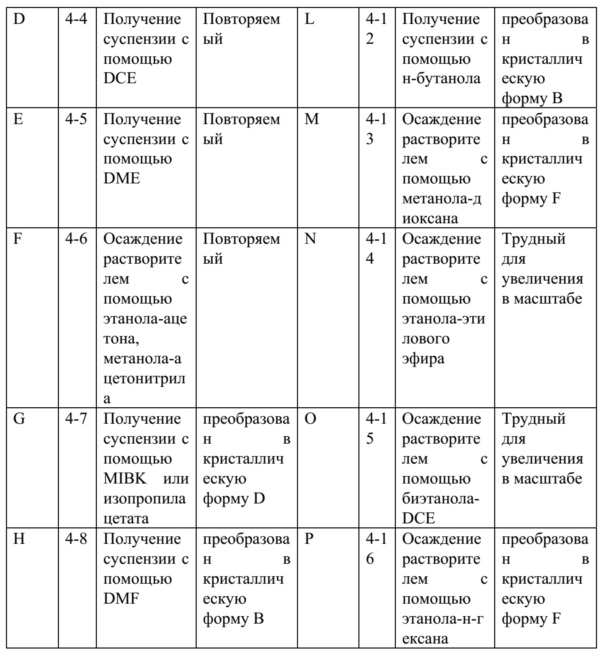
Результаты показали, что кристаллические формы A-F были стабильными. Тогда как некоторые из кристаллических форм G-P имели трудности с укрупнением и некоторые имели проявление феномена кристаллической трансформации, поэтому были непригодны для дальнейшего исследования.
Таким образом, проводили скрининг исходных материалов с помощью кристаллизации посредством способов выпаривания при нормальных температурах, кристаллизации из суспензии и кристаллизации растворением и осаждением и т. п. Рентгенограммы анализировали после применения PXRD для определения характеристик, при этом предварительно было определено, что у лобаплатина может существовать 16 видов кристаллических форм A-P. После процедуры проверки путем повторных экспериментов и экспериментов по увеличению в масштабе определили, что кристаллические формы A-F являлись сравнительно стабильными с лучшей воспроизводимостью. Тогда как с некоторыми из других кристаллических форм было трудно осуществлять увеличенное в масштабе производство с более низким выходом, о некоторых сделали заключение, что это нестабильные кристаллические формы, так как наблюдали явление кристаллической трансформации. В связи с этим далее для проведения всестороннего исследования выбрали кристаллические формы A-F.
Пример 2. Получение нового кристалла лобаплатина, названного кристаллической формой A, а также свойства продуктов из него и сравнительные анализы
2.1. Получение дигидрата лобаплатина, названного кристаллической формой A
Пример получения 1
Взвешивали 1 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 15 мл толуола с получением суспензии. Суспензию перемешивали при комнатной температуре в течение 48 ч. До осаждения кристаллов. Кристаллы отделяли путем фильтрации и промывали 2-3 раза этиловым эфиром. Наконец, после вакуумного высушивания получали 0,85 г белого порошка, который представлял собой дигидрат лобаплатина.
Пример получения 2
Взвешивали 1 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 15 мл этилового эфира с получением суспензии. Суспензию перемешивали при комнатной температуре в течение 45 ч. До осаждения кристаллов. Кристаллы отделяли путем фильтрации и промывали 2-3 раза этиловым эфиром. Наконец, после вакуумного высушивания получали 0,88 г белого порошка, который представлял собой дигидрат лобаплатина.
Пример получения 3
Взвешивали 1 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 20 мл бутилацетата с получением суспензии. Суспензию перемешивали при комнатной температуре в течение 50 ч. До осаждения кристаллов. Кристаллы отделяли путем фильтрации и промывали 2-3 раза этиловым эфиром. Наконец, после вакуумного высушивания получали 0,83 г белого порошка, который представлял собой дигидрат лобаплатина.
Пример получения 4
Взвешивали 1 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 25 мл 1,4-диоксана с получением суспензии. Суспензию перемешивали при комнатной температуре в течение 48 ч до осаждения кристаллов. Кристаллы отделяли путем фильтрации и промывали 2-3 раза этиловым эфиром. Наконец, после вакуумного высушивания получали 0,90 г белого порошка, который представлял собой дигидрат лобаплатина.
Пример получения 5
Взвешивали 1 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 30 мл н-гептана с получением суспензии. Суспензию перемешивали при комнатной температуре в течение 46 ч. До осаждения кристаллов. Кристаллы отделяли путем фильтрации и промывали 2-3 раза этиловым эфиром. Наконец, после вакуумного высушивания получали 0,87 г белого порошка, который представлял собой дигидрат лобаплатина.
Пример получения 6
Взвешивали 1 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 15 мл метил-трет-бутилового эфира с получением суспензии. Суспензию перемешивали при комнатной температуре в течение 48 ч до осаждения кристаллов. Кристаллы отделяли путем фильтрации и промывали 2-3 раза этиловым эфиром. Наконец, после вакуумного обезвоживания получали 0,92 г белого порошка, который представлял собой дигидрат лобаплатина.
Образцы, полученные согласно вышеупомянутым примерам получения, анализировали с помощью XRD дифракции посредством способа 1.4 в вышеизложенном примере 1. Определили, что все шесть образцов имели одинаковую кристаллическую форму, и характеристические пики были следующими: на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 11,04, 12,32, 12,61, 13,85, 15,14, 15,55, 16,68, 17,67, 17,86, 19,03, 20,06, 21,00, 22,68, 22,92, 23,76, 25,39, 25,58, 26,37, 26,77, 27,00, 27,71, 28,13, 29,71, 31,42, 31,94, 32,89, 34,29, 34,60, 36,10, 36,93, 37,66, 40,78, 43,41, при этом граница погрешности значений 2θ составляла 0,2.
Результаты элементных анализов: C9H18N2O3Pt*2H2O M = 433,36
Вычисленное значение (%): C 24,95 H 5,11 N 6,46 Pt 45,01
Измеренное значение (%): C 24,94 H 5,08 N 6,41 Pt 45,07
Такую кристаллическую форму назвали кристаллической формой A.
2.2. Определение свойств продуктов на основе кристаллов лобаплатина с кристаллической формой A и их сравнительные анализы
1. Тестируемые образцы
Дигидрат Лобаплатина под номером 1-6 согласно настоящему изобретению: дигидрат лобаплатина с кристаллической формой A получали с помощью способа из примера получения 1-6 соответственно.
Сравнительный образец 1: лобаплатин, получаемый с помощью способа из примера 1a, записанного в патенте EP 0324154, и конкретный способ получения указаны ниже:
3,8 г (0,01 моль) цис-[транс-1,2-бутил-бис(метиламин)-N,N']-дихлор-платины(II) суспендировали в 20 мл воды с получением смеси, и смесь нагревали до 40°С. К смеси добавляли 3,39 г (0,02 моль) нитрата серебра и смесь перемешивали в течение 1,5 часа. Смесь оставляли охлаждаться в холодильнике для осаждения хлорида серебра. Осажденный хлорид серебра затем фильтровали и промывали 10 мл воды. Фильтрат пропускали через колонку, содержащую 100 мл основного ионообменника, и промывали 150 мл воды. Затем фильтрат вливали по каплям в 4,5 г (0,01 моль, 20% водный раствор) L-молочной кислоты. После перемешивания при комнатной температуре в течение 3 дней реакционную смесь концентрировали, остаток растворяли в метаноле и перемешивали до его исчезновения с дополнительным активированным углем. Затем активированный уголь фильтровали и к фильтрату добавляли этиловый эфир. Твердое вещество, полученное путем быстрого концентрирования, представляло собой аморфный лобаплатин.
Сравнительный образец 2: тригидрат лобаплатина, получаемый согласно способу, записанному в примере патента EP 0611303, и конкретный способ получения указаны ниже:
3,8 г (0,01 моль) цис-[транс-1,2-бутил-бис(метиламин)-N,N']-дихлор-платину (II) суспендировали в 20 мл воды с получением смеси и смесь нагревали до 40°С. К смеси добавляли 3,39 г (0,02 моль) нитрата серебра и смесь перемешивали в течение 1,5 часа. Смесь оставляли охлаждаться в холодильнике для осаждения хлорида серебра. Осадок хлорида серебра затем фильтровали и промывали 10 мл воды. Фильтрат пропускали через колонку, содержащую 100 мл основного ионообменника, и промывали 150 мл воды. Затем фильтрат вливали по каплям в 4,5 г (0,01 моль, 20% водный раствор) L-молочной кислоты. После перемешивания при комнатной температуре в течение 3 дней реакционную смесь концентрировали до приблизительно 20 мл и оставляли охлаждаться в холодильнике на ночь для осаждения кристалла. Затем кристалл фильтровали, а фильтрат концентрировали и оставляли охлаждаться в холодильнике на ночь вновь для осаждения кристалла. Кристалл фильтровали и собирали фильтрат. Кристаллы объединяли и перекристаллизовывали из 20 мл воды/ацетона (1/1, об./об.), а кристалл, полученный вследствие этого, представлял собой тригидрат лобаплатина.
2. Определение морфологии
Сравнительный образец 1: полученный лобаплатин был аморфным;
Сравнительный образец 2: на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 6,71, 8,35, 12,89, 15,14, 16,74, 17,45, 19,01, 19,40, 22,07, 22,76, 23,16, 24,30, 25,21, 25,74, 27,08, 30,26, 30,79, при этом граница погрешности значений 2θ составляла меньше 0,2. Температура плавления данного образца, составляющая 210°С (разложение), была описана в патенте EP 0611303.
Образцы 1-6 с кристаллической формой A согласно настоящему изобретению: на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 11.04, 12.32, 12.61, 13.85, 15.14, 15.55, 16.68, 17.67, 17.86, 19.03, 20.06, 21.00, 22.68, 22.92, 23.76, 25.39, 25.58, 26.37, 26.77, 27.00, 27.71, 28.13, 29.71, 31.42, 31.94, 32.89, 34.29, 34.60, 36.10, 36.93, 37.66, 40.78, 43.41, при этом граница погрешности значений 2θ составляла меньше 0,2. На термограмме DSC присутствовал широкий эндотермический пик около 117°С, и этот пик мог быть вызван потерей двух молекул кристаллической воды, судя по совмещению с данными относительно монокристалла и данными TGA. Также присутствовал экзотермический пик при 220 ± 5°С, который являлся пиком плавления и разложения, судя по совмещению с данными TGA и данными относительно температуры плавления, записанными в европейском патенте EP 0611303. На термограмме TGA присутствовала 9,49% потеря веса перед 150°С, которая вызвана потерей двух молекул кристаллической воды. Все данные показали, что образцы 1-6 имели одинаковую кристаллическую форму, как и дигидрат лобаплатина.
3. Исследование растворимости
Готовили контрольные растворы тригидрата лобаплатина с концентрациями 60 мкг/мл, 80 мкг/мл, 200 мкг/мл, 400 мкг/мл и 800 мкг/мл, чтобы сделать стандартную кривую посредством способа HPLC. Полученная стандартная кривая представляла собой Y = 4,8641X + 20,5794, R = 0,9998. Готовили насыщенные водные растворы (суспензии) образца 6 дигидрата лобаплатина и сравнительного образца 2 соответственно. Растворы подвергали вибрации в течение 6 ч. в шейкере при 25°С с последующей фильтрацией и разбавляли до соответствующих кратностей для анализа с помощью HPLC. Результаты растворимости представлены в таблице 7 ниже.
Таблица 7. Результаты исследования растворимости
Результаты показали, что растворимость дигидрата лобаплатина, полученного согласно настоящему изобретению, была лучше, чем у тригидрата лобаплатина.
4. Исследование качества новых кристаллических форм лобаплатина методом сопоставления
Взвешивали по 20 мг образцов 1-6 дигидрата лобаплатина и сравнительных образцов 1-2 соответственно. Значения содержания влаги, примесей, значения содержания активных ингредиентов и выходы продуктов рассматривали в качестве показателей для исследования качества и выходов продуктов. Результаты представлены в таблице 8 ниже.
Таблица 8. Исследование качества дигидрата лобаплатина с кристаллической формой A методом сопоставления

Вышеуказанные результаты показали, что по сравнению с безводным лобаплатином и тригидратом лобаплатина новый кристалл лобаплатина A, полученный по настоящему изобретению, обладает характеристиками высоких значений содержания, низких значений содержания примесей и хороших выходов.
Примечание 1. Способ определения значений содержания: измеряли с помощью высокоэффективной жидкостной хроматографии; условия хроматографирования являлись следующими: силикагель с привитым к нему октадецилсиланом в качестве наполнителя, отношение раствора дигидрофосфата калия к ацетонитрилу 92:8 в качестве подвижной фазы, длина волны детектирования 210 нм, температура колонки 40°С, число теоретических тарелок, рассчитанное по пикам лобаплатина, должно быть не меньше 1000 и степень разделения пиков лобаплатина и пиков примесей должна отвечать требованиям. Получение раствора продукта для контроля: точно взвешивали 10 мг продукта на основе тригидрата лобаплатина для контроля и фиксировали в 50 мл мерной колбе, разбавляли водой до метки и хорошо встряхивали. Получение тестируемых растворов: точно взвешивали 20 мг образца в указанном порядке и фиксировали в 100 мл мерной колбе, разбавляли водой до метки и хорошо встряхивали. Измерения и результаты: отмеряли ровно по 10 мкл растворов продукта для контроля и растворов образца соответственно, вводили в жидкостной хроматограф и регистрировали хроматограммы. Вычисленные с учетом площадей пиков по методу внешнего стандарта значения содержания вычисляли относительно безводного лобаплатина со стандартным диапазоном 97,0%-102%.
Примечание 2. Способ проверки на присутствие примесей: лобаплатин, 1,2-диаминометилциклобутан(CBMA), молочную кислоту и другие известные и неизвестные примеси определяли с помощью TLC. Проявитель: этанол: хлороформ: 25% водный раствор аммиака: вода = 53: 39: 15: 1,5 (объемное отношение), пластины для TLC: силикагель 60 F25410 × 10 пластин для TLC. Пластины помещали в пары йода для окрашивания с 0,3% реагентом нингидрином и хромогенным реагентом нитрозо-диметиланилином после проявления. Проверяли на присутствие CBMA и неизвестных примесей.
Примечание 3. Способ определения значений содержания влаги: применяли способ Карла Фишера. Теоретическое содержание влаги для дигидрата лобаплатина составляло 8,77%, а теоретическое содержание влаги для тригидрата лобаплатина составляло 11,96%.
5. Исследование стабильности
Образец 6, полученный в примерах по настоящему изобретению, и полученный сравнительный образец 2 оставляли соответственно при таких условиях: температура печи 60°С, относительная влажность приблизительно 95% и емкость для тестирования на светостабильность при освещении приблизительно 4500 люкс (обычный термостат с функцией освещения). Через 5 дней и 10 дней образцы вынимали для проведения тестов PXRD и анализов HPLC, чтобы исследовать стабильность образцов при условиях высокой температуры, высокой влажности и освещения. Результаты представлены в таблице 9.
Таблица 9. Результаты оценки стабильности

Вышеуказанные результаты эксперимента показали, что лобаплатин согласно настоящему изобретению характеризовался новой кристаллической формой, которая имела более высокую растворимость, чем растворимость тригидрата лобаплатина, при этом выходы и чистота были идеальными. Новая кристаллическая форма лобаплатина имела хорошую стабильность и не проявляла феномена кристаллической трансформации согласно результатам исследования при высокой температуре, высокой влажности и освещении. Значения содержания активных ингредиентов были выше, чем у тригидрата лобаплатина, и не имели никаких видимых изменений согласно результатам HPLC, означая, что новая кристаллическая форма лобаплатина согласно настоящему изобретению была более стабильной, чем кристаллическая форма тригидрата лобаплатина, она с трудом расплывалась, поэтому не являлась липкой, и имела хорошую текучесть.
Пример 3. Получение нового кристалла лобаплатина, названного кристаллической формой B, а также свойства продуктов из него и сравнительные анализы
3.1. Получение нового кристалла лобаплатина, названного кристаллической формой B
Пример получения 1
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 30 мл толуола с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,73 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 40 мл безводного метанола с получением смеси. Смесь перемешивали при комнатной температуре до растворения твердых веществ. Смесь фильтровали для удаления нерастворимых веществ, медленно выпаривали в вытяжном шкафу до осаждения кристалла, а затем кристалл фильтровали и выделяли. Затем кристалл промывали 2-3 раза этиловым эфиром и после вакуумного высушивания получали 0,74 г белого порошка, который представлял собой новую кристаллическую форму B лобаплатина.
Пример получения 2
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 15 мл метил-трет-бутилового эфира с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания 1,84 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 50 мл безводного метанола с получением смеси. Смесь перемешивали при комнатной температуре до растворения твердых веществ. Смесь фильтровали для удаления нерастворимых веществ, медленно выпаривали в вытяжном шкафу до осаждения кристалла, а затем кристалл фильтровали и выделяли. Затем кристалл промывали 2-3 раза этиловым эфиром и после вакуумного высушивания получали 0,76 г белого порошка, который представлял собой новую кристаллическую форму B лобаплатина.
Пример получения 3
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 20 мл бутилацетата с получением суспензии. Суспензию перемешивали в течение 50 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,68 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 20 мл н-гексана с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали 2-3 раза этиловым эфиром и после вакуумного высушивания получали 0,75 г белого порошка, который представлял собой новую кристаллическую форму B лобаплатина.
Пример получения 4
a) Получение дигидрата лобаплатина: Взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 25 мл 1,4-диоксана с получением суспензии. Суспензию перемешивали в течение 45 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,76 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 15 мл ацетона с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали 2-3 раза этиловым эфиром и после вакуумного высушивания получали 0,78 г белого порошка, который представлял собой новую кристаллическую форму B лобаплатина.
Пример получения 5
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 30 мл н-гептана с получением суспензии. Суспензию перемешивали в течение 50 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,75 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 18 мл этилацетата с получением суспензии. Суспензию перемешивали в течение 50 ч. при комнатной температуре для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали 2-3 раза этиловым эфиром и после вакуумного высушивания получали 0,75 г белого порошка, который представлял собой новую кристаллическую форму B лобаплатина.
Пример получения 6
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 15 мл этилового эфира с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,78 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 25 мл нитрометана с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали 2-3 раза этиловым эфиром и после вакуумного высушивания получали 0,77 г белого порошка, который представлял собой новую кристаллическую форму B лобаплатина.
Пример получения 7
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 18 мл метил-трет-бутилового эфира с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,82 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 30 мл тетрагидрофурана с получением суспензии. Суспензию перемешивали в течение 46 ч. при комнатной температуре для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали 2-3 раза этиловым эфиром и после вакуумного высушивания получали 0,79 г белого порошка, который представлял собой новую кристаллическую форму B лобаплатина.
Пример получения 8
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 25 мл метил-трет-бутилового эфира с получением суспензии. Суспензию перемешивали в течение 46 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,83 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 15 мл дихлорметана с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали 2-3 раза этиловым эфиром и после вакуумного высушивания получали 0,76 г белого порошка, который представлял собой новую кристаллическую форму B лобаплатина.
Пример получения 9
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 30 мл метил-трет-бутилового эфира с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,85 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 25 мл ацетонитрила с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали 2-3 раза этиловым эфиром и после вакуумного высушивания получали 0,78 г белого порошка, который представлял собой новую кристаллическую форму B лобаплатина.
Пример получения 10
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 15 мл метил-трет-бутилового эфира с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,84 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 20 мл 2-бутанонас получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали 2-3 раза этиловым эфиром и после вакуумного высушивания получали 0,73 г белого порошка, который представлял собой новую кристаллическую форму B лобаплатина.
Пример получения 11
Взвешивали 1 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 40 мл безводного метанола с получением смеси. Смесь перемешивали при комнатной температуре до растворения твердых веществ. Смесь фильтровали для удаления нерастворимых веществ, а затем смесь медленно выпаривали для осаждения кристалла. Кристалл фильтровали и выделяли. После высушивания получали 0,68 г белого порошка, который представлял собой кристаллическую форму B лобаплатина.
Пример получения 12
Взвешивали 1 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 85 мл безводного этанола с получением смеси. Смесь перемешивали при комнатной температуре до растворения твердых веществ. Смесь фильтровали для удаления нерастворимых веществ, а затем смесь медленно выпаривали для осаждения кристалла. Кристалл фильтровали и выделяли. После высушивания получали 0,70 г белого порошка, который представлял собой кристаллическую форму B лобаплатина.
Образцы, полученные согласно стадии a) в вышеуказанных примерах получения 1-10, анализировали с помощью XRD дифракции посредством способа 1.4 в вышеизложенном примере 1. Определили, что все 10 образцов имели одинаковую кристаллическую форму, которая представляла собой кристаллическую форму A. И подробные определенные данные были такими же, как в предыдущей первой части варианта осуществления, являющегося «первым вариантом осуществления настоящего изобретения», и как в предыдущем примере 2.
В то же время образцы, полученные посредством стадии b) в вышеуказанных примерах получения 1-10 и примерах 11-12, анализировали с помощью XRD дифракции посредством способа 1.4 в вышеизложенном примере 1. Определили, что все 12 образцов имели одинаковую кристаллическую форму, а характеристические пики были следующими: на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 8,25, 9,77, 11,70, 13,13, 15,28, 16,48, 17,22, 17,74, 19,01, 19,56, 22,28, 23,72, 24,04, 24,30, 25,62, 26,20, 28,57, 30,22, 30,61, при этом граница погрешности значений 2θ составляла 0,2. Такую кристаллическую форму назвали кристаллической формой B.
3.2. Определение свойств продуктов с кристаллической формой B и их сравнительные анализы
1. Тестируемые образцы
Образцы 1-12: соединения лобаплатина с кристаллической формой B получали по способу из примеров получения 1-12 в примере 3 настоящего изобретения соответственно.
Сравнительный образец 1: был таким же, как предыдущий образец из сравнительного образца 1 в части 2.2 примера 2. Лобаплатин, полученный по способу из примера 1a в патенте EP 0324154, и конкретные способы получения были такими же, как в предыдущем сравнительном образце 1 в части 2.2 примера 2.
Сравнительный образец 2: был таким же, как предыдущий образец из сравнительного образца 2 в части 2.2 примера 2. Тригидрат лобаплатина получали по способу из примера в патенте EP 0611303, и конкретные способы получения были такими же, как в случае предыдущего сравнительного образца 2 в части 2.2 примера 2.
2. Определение морфологии
Результаты определения были такими же, как у сравнительного образца 1 и сравнительного образца 2 в части 2.2 примера 2, в частности, результаты указаны ниже:
Сравнительный образец 1: полученный лобаплатин был аморфным;
Сравнительный образец 2: на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 6,71, 8,35, 12,89, 15,14, 16,74, 17,45, 19,01, 19,40, 22,07, 22,76, 23,16, 24,30, 25,21, 25,74, 27,08, 30,26, 30,79, при этом граница погрешности значений 2θ составляла меньше 0,2. Температура плавления данного образца, составляющая 210°С (разложение), была описана в патенте EP 0611303.
Образцы 1-12 согласно настоящему изобретению: на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 8,25, 9,77, 11,70, 13,13, 15,28, 16,48, 17,22, 17,74, 19,01, 19,56, 22,28, 23,72, 24,04, 24,30, 25,62, 26,20, 28,57, 30,22, 30,61, при этом граница погрешности значений 2θ составляла меньше 0,2. Также присутствовал экзотермический пик при 230 ± 5°С, который являлся пиком плавления и разложения, судя по совмещению с данными TGA и данными относительно температуры плавления, записанными в европейском патенте EP 0611303. Все данные показали, что образцы 1-12 имели одинаковую кристаллическую форму, которой являлась новая кристаллическая форма B лобаплатина.
3. Исследование растворимости
Готовили контрольные растворы тригидрата лобаплатина с концентрациями 60 мкг/мл, 80 мкг/мл, 200 мкг/мл, 400 мкг/мл и 800 мкг/мл, чтобы сделать стандартную кривую посредством способа HPLC. Полученная стандартная кривая представляла собой Y = 4,8641X + 20,5794, R = 0,9998. Готовили насыщенные водные растворы (суспензии) образца 1 с новой кристаллической формой B лобаплатина и сравнительного образца 2 соответственно. Растворы подвергали вибрации в шейкере при 25°С в течение 6 ч. с последующей фильтрацией и разбавляли до соответствующих кратностей для анализа с помощью HPLC. Результаты растворимости представлены в таблице 10 ниже.
Таблица 10. Результаты исследования растворимости
Результаты показали, что растворимость соединения лобаплатина, полученного согласно настоящему изобретению, была лучше, чем у тригидрата лобаплатина.
4. Исследование качества новых кристаллических форм лобаплатина методом сопоставления
Взвешивали по 50 мг образцов 1-12 соединений лобаплатина и сравнительных образцов 1-2 соответственно. Значения содержания влаги, примесей, значения содержания активных ингредиентов и выходы продуктов рассматривали в качестве показателей для исследования качества и выходов продуктов. Теоретическое содержание влаги для тригидрата лобаплатина составляло 11,96%. Результаты представлены в таблице 11 ниже.
Таблица 11. Исследование качества новой кристаллической формы B лобаплатина методом сопоставления

Вышеуказанные результаты показали, что по сравнению с безводным лобаплатином и тригидратом лобаплатина новая кристаллическая форма B лобаплатина, полученная по настоящему изобретению, обладает характеристиками высоких значений содержания, низких значений содержания примесей и хороших выходов.
Примечание 1. Способ определения значений содержания: был таким же, как в предыдущей части 2.2 примера 2.
Примечание 2. Способ проверки на присутствие примесей: был таким же, как в предыдущей части 2.2 примера 2.
Примечание 3. Способ определения значений содержания влаги: был таким же, как в предыдущей части 2.2 примера 2.
5. Исследование стабильности
Образец 1, полученный в примере 3 настоящего изобретения, и полученный сравнительный образец 2 оставляли соответственно при таких условиях: температура печи 60°С, относительная влажность приблизительно 95% и емкость для тестирования на светостабильность при освещении приблизительно 4500 люкс (обычный термостат с функцией освещения). Через 5 дней и 10 дней образцы вынимали для проведения тестов PXRD и анализов HPLC, чтобы исследовать стабильность образцов при условиях высокой температуры, высокой влажности и освещения. Результаты представлены в таблице 12.
Таблица 12. Результаты оценки стабильности

Вышеуказанные результаты эксперимента показали, что новая кристаллическая форма B лобаплатина согласно настоящему изобретению имела более высокую растворимость, чем растворимость тригидрата лобаплатина, при этом выходы и чистота были идеальными. Новая кристаллическая форма B лобаплатина имела хорошую стабильность и не проявляла феномена кристаллической трансформации согласно результатам исследования при высокой температуре, высокой влажности и освещении. Значения содержания активных ингредиентов были выше, чем у тригидрата лобаплатина, и не имели никаких видимых изменений согласно результатам HPLC, означая, что новая кристаллическая форма лобаплатина согласно настоящему изобретению была более стабильной, чем кристаллическая форма тригидрата лобаплатина, она с трудом расплывалась, поэтому не являлась липкой, и имела хорошую текучесть.
Пример 4. Получение нового кристалла лобаплатина, названного кристаллической формой A, а также свойства продуктов из него и сравнительные анализы
4.1. Получение нового кристалла лобаплатина, названного кристаллической формой F
Пример получения 1
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 30 мл толуола с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,73 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 40 мл метанола с получением смеси. Смесь перемешивали при комнатной температуре до растворения твердых веществ. Смесь фильтровали для удаления нерастворимых веществ, а затем к смеси медленно добавляли 120 мл диметилового эфира этиленгликоля для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали этиловым эфиром и после вакуумного высушивания получали 0,71 г белого порошка, который представлял собой новую кристаллическую форму F лобаплатина.
Пример получения 2
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 15 мл метил-трет-бутилового эфира с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,84 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 50 мл метанола с получением смеси. Смесь перемешивали при комнатной температуре до растворения твердых веществ. Смесь фильтровали для удаления нерастворимых веществ, а затем к смеси медленно добавляли 150 мл н-гексана для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали этиловым эфиром и после вакуумного высушивания получали 0,68 г белого порошка, который представлял собой новую кристаллическую форму F лобаплатина.
Пример получения 3
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 20 мл бутилацетата с получением суспензии. Суспензию перемешивали в течение 50 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,68 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 80 мл этанола с получением смеси. Смесь перемешивали при комнатной температуре до растворения твердых веществ. Смесь фильтровали для удаления нерастворимых веществ, а затем к смеси медленно добавляли 200 мл этилацетата для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали этиловым эфиром и после вакуумного высушивания получали 0,70 г белого порошка, который представлял собой новую кристаллическую форму F лобаплатина.
Пример получения 4
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 25 мл 1,4-диоксана с получением суспензии. Суспензию перемешивали в течение 45 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,76 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 90 мл этанола с получением смеси. Смесь перемешивали при комнатной температуре до растворения твердых веществ. Смесь фильтровали для удаления нерастворимых веществ, а затем к смеси медленно добавляли 180 мл ацетона для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали этиловым эфиром и после вакуумного высушивания получали 0,72 г белого порошка, который представлял собой новую кристаллическую форму F лобаплатина.
Пример получения 5
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 30 мл н-гептана с получением суспензии. Суспензию перемешивали в течение 50 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,75 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 45 мл метанола с получением смеси. Смесь перемешивали при комнатной температуре до растворения твердых веществ. Смесь фильтровали для удаления нерастворимых веществ, а затем к смеси медленно добавляли 160 мл нитрометана для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали этиловым эфиром и после вакуумного высушивания получали 0,69 г белого порошка, который представлял собой новую кристаллическую форму F лобаплатина.
Пример получения 6
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 15 мл этилового эфира с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,78 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 40 мл метанола с получением смеси. Смесь перемешивали при комнатной температуре до растворения твердых веществ. Смесь фильтровали для удаления нерастворимых веществ, а затем к смеси медленно добавляли 150 мл ацетонитрила для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали этиловым эфиром и после вакуумного высушивания получали 0,73 г белого порошка, который представлял собой новую кристаллическую форму F лобаплатина.
Пример получения 7
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 15 мл метил-трет-бутилового эфира с получением суспензии. Суспензию перемешивали в течение 48 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,84 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 85 мл этанола с получением смеси. Смесь перемешивали при комнатной температуре до растворения твердых веществ. Смесь фильтровали для удаления нерастворимых веществ, а затем к смеси медленно добавляли 180 мл тетрагидрофурана для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали этиловым эфиром и после вакуумного высушивания получали 0,67 г белого порошка, который представлял собой новую кристаллическую форму F лобаплатина.
Пример получения 8
a) Получение дигидрата лобаплатина: взвешивали 2 г тригидрата лобаплатина и помещали в емкость, а также в емкость добавляли 20 мл метил-трет-бутилового эфира с получением суспензии. Суспензию перемешивали в течение 46 ч. при комнатной температуре, фильтровали и продукт, полученный путем фильтрования, промывали этиловым эфиром, а затем после вакуумного высушивания получали 1,80 г белого порошка, который представлял собой дигидрат лобаплатина.
b) Получение целевого кристалла: взвешивали 1 г дигидрата лобаплатина из стадии a и помещали в емкость, а также в емкость добавляли 40 мл метанола с получением смеси. Смесь перемешивали при комнатной температуре до растворения твердых веществ. Смесь фильтровали для удаления нерастворимых веществ, а затем к смеси медленно добавляли 150 мл дихлорметана для осаждения кристалла. Кристалл фильтровали и выделяли. Затем кристалл промывали этиловым эфиром и после вакуумного высушивания получали 0,66 г белого порошка, который представлял собой новую кристаллическую форму F лобаплатина.
Образцы, полученные согласно стадии a) в вышеуказанных примерах получения 1-8, анализировали с помощью XRD дифракции посредством способа 1.4 в вышеизложенном примере 1. Определили, что все 8 образцов имели одинаковую кристаллическую форму, которая представляла собой кристаллическую форму F. И подробные определенные данные были такими же, как и в предыдущей первой части варианта осуществления, являющегося «первым вариантом осуществления настоящего изобретения», и как в предыдущем примере 2.
В то же время образцы, полученные в вышеуказанных примерах получения 1-8 посредством способа 1.4 из примера 1, анализировали с помощью XRD дифракции. Определили, что все 8 образцов имели одинаковую кристаллическую форму, а характеристические пики были следующими: на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 8,21, 11,60, 12,99, 15,24, 16,44, 17,11, 17,55, 18,42, 19,01, 19,20, 19,42, 21,81, 22,17, 22,42, 23,33, 23,85, 24,18, 24,40, 24,77, 25,46, 25,98, 26,13, 27,89, 28,42, 29,03, 30,32, 31,17, 31,94, 33,30, 36,20, 37,62, 39,66, при этом граница погрешности значений 2θ составляла 0,2. Такую кристаллическую форму назвали кристаллической формой F.
4.2. Определение свойств продуктов с кристаллической формой F и их сравнительные анализы
1. Тестируемые образцы
Образцы 1-8: получали по способу из примеров получения 1-8 в настоящем изобретении соответственно.
Сравнительный образец 1: был таким же, как предыдущий образец из сравнительного образца 1 в части 2.2 примера 2. Лобаплатин, полученный согласно способу из примера 1a в патенте EP 0324154, и конкретные способы получения были такими же, как у предыдущего сравнительного образца 1 в части 2.2 примера 2.
Сравнительный образец 2: был таким же, как предыдущий образец из сравнительных образцов 2 в части 2.2 примера 2. Лобаплатин получали согласно способу из примера в патенте EP 0611303, и конкретные способы получения были такими же, как в случае предыдущего сравнительного образца 2 в части 2.2 примера 2.
2. Определение морфологии
Результаты определения были такими же, как у сравнительного образца 1 и сравнительного образца 2 в части 2.2 примера 2, в частности, результаты указаны ниже.
Сравнительный образец 1: полученный лобаплатин был аморфным;
Сравнительный образец 2: на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 6,71, 8,35, 12,89, 15,14, 16,74, 17,45, 19,01, 19,40, 22,07, 22,76, 23,16, 24,30, 25,21, 25,74, 27,08, 30,26, 30,79, при этом граница погрешности значений 2θ составляла меньше 0,2. Температура плавления данного образца, составляющая 210°С (разложение), была описана в патенте EP 0611303.
Образцы 1-8 согласно настоящему изобретению: на рентгенограмме PXRD присутствовали дифракционные пики при значениях 2θ приблизительно 8,21, 11,60, 12,99, 15,24, 16,44, 17,11, 17,55, 18,42, 19,01, 19,20, 19,42, 21,81, 22,17, 22,42, 23,33, 23,85, 24,18, 24,40, 24,77, 25,46, 25,98, 26,13, 27,89, 28,42, 29,03, 30,32, 31,17, 31,94, 33,30, 36,20, 37,62, 39,66, при этом граница погрешности значений 2θ составляла меньше 0,2. На термограмме DSC присутствовал экзотермический пик около 229°С, который являлся пиком плавления и разложения, судя по совмещению с данными TGA и данными относительно температуры плавления, записанными в европейском патенте EP 0611303. Все данные показали, что образцы 1-8 имели одинаковую кристаллическую форму, которой являлась новая кристаллическая форма F лобаплатина.
3. Исследование растворимости
Готовили контрольные растворы тригидрата лобаплатина с концентрациями 60 мкг/мл, 80 мкг/мл, 200 мкг/мл, 400 мкг/мл и 800 мкг/мл, чтобы сделать стандартную кривую посредством способа HPLC. Полученная стандартная кривая представляла собой Y = 4,8641X + 20,5794, R = 0,9998. Готовили насыщенные водные растворы (суспензии) образца 6 нового кристалла лобаплатина и сравнительного образца 2 соответственно. Растворы подвергали вибрации в шейкере при 25°С в течение 6 ч. с последующей фильтрацией и разбавляли до соответствующих кратностей для анализа с помощью HPLC. Результаты растворимости представлены в таблице 13 ниже.
Таблица 13. Результаты растворимости
Результаты показали, что растворимость соединения лобаплатина, полученного согласно настоящему изобретению, была лучше, чем у тригидрата лобаплатина.
4. Исследование качества новых кристаллических форм лобаплатина методом сопоставления
Взвешивали по 20 мг образцов 1-8 дигидрата лобаплатина и сравнительного образца 1-2 соответственно. Значения содержания влаги, примесей, значения содержания активных ингредиентов и выходы продуктов рассматривали в качестве показателей для исследования качества и выходов продуктов. Результаты представлены в таблице 14 ниже.
Таблица 14. Исследование качества нового кристалла лобаплатина методом сопоставления

Вышеуказанные результаты показали, что по сравнению с безводным лобаплатином и тригидратом лобаплатина новая кристаллическая форма F лобаплатина, полученная по настоящему изобретению, характеризуется высокими значениями содержания, низкими значениями содержания примесей и хорошими выходами.
Примечание 1. Способ определения значений содержания: был таким же, как в предыдущей части 2.2 примера 2.
Примечание 2. Способ проверки на присутствие примесей: был таким же, как в предыдущей части 2.2 примера 2.
Примечание 3. Способ определения значений содержания влаги: был таким же, как в предыдущей части 2.2 примера 2.
5. Исследование стабильности
Образец 6, полученный в примере по настоящему изобретению, и полученный сравнительный образец 2 оставляли соответственно при таких условиях: температура печи 60°С, относительная влажность приблизительно 95% и емкость для тестирования на светостабильность при освещении приблизительно 4500 люкс (обычный термостат с функцией освещения). Через 5 дней и 10 дней образцы вынимали для проведения тестов PXRD и анализов HPLC, чтобы исследовать стабильность образцов при условиях высокой температуры, высокой влажности и освещения. Результаты представлены в таблице 15.
Таблица 15. Результаты оценки стабильности

Вышеуказанные результаты эксперимента показали, что новая кристаллическая форма F лобаплатина согласно настоящему изобретению имела более высокую растворимость, чем растворимость тригидрата лобаплатина, при этом выходы и чистота были идеальными. Новая кристаллическая форма лобаплатина F имела хорошую стабильность и не проявляла феномена кристаллической трансформации согласно результатам исследования при высокой температуре, высокой влажности и освещении. Значения содержания активных ингредиентов были выше, чем у тригидрата лобаплатина, и не имели никаких видимых изменений согласно результатам HPLC, означая, что новый кристалл лобаплатина был стабильным.
Реферат
Настоящее изобретение относится к новому кристаллу соединения лобаплатина (кристаллическая форма А). Кристалл характеризуется двумя молекулами кристаллической воды, находящимися в структуре кристалла. Также предложены способ получения кристалла А, новые кристаллы лобаплатина, представляющие собой кристаллические формы B и F, способы их получения, фармацевтическая композиция и применения новых кристаллов для получения противораковых лекарственных средств. По сравнению с существующим лобаплатином и тригидратом лобаплатина кристаллы A, B и F лобаплатина обладают лучшей стабильностью и растворимостью и могут быть более эффективно использованы при лечении форм рака, таких как рак молочной железы, мелкоклеточный рак легких или хронический миелоидный лейкоз. 13 н. и 28 з.п. ф-лы, 10 ил., 15 табл., 26 пр.
Комментарии