Применение аналогов ацилфульвена и фармацевтическая композиция на их основе - RU2145849C1
Код документа: RU2145849C1
Чертежи














Описание
Эта работа финансируется частично грантом для исследовательских работ CA-37641 от Национальных Институтов Здравоохранения. Правительство США имеет права на это изобретение.
Предпосылки изобретения
Многочисленные химиотерапевтические агенты обладают лечебной способностью в случаях некоторых гематологических злокачественных заболеваний и прогрессирующих быстро
пролиферирующих твердых опухолях. Лечебная химиотерапия стала более успешной при обнаружении новых не перекрестно резистентных агентов и более эффективном использовании существующих агентов.
Воздействия, которые увеличивают эффективность общепринятых средств, включают в себя более эффективные курсы введения многочисленных лекарственных средств, максимальное снижение токсичности
лекарственных средств и расширенное применение вспомогательных лекарственных средств, хирургическое вмешательство и лучевую терапию.
Несмотря на последние достижения, больные со многими видами злокачественных заболеваний остаются под значительным риском рецидивов и смертельного исхода. После рецидива у некоторых больных может начаться ремиссия уже при начальном курсе лечения. Однако часто необходимы более высокие дозы начального химиотерапевтического средства или применение дополнительных средств, что свидетельствует о развитии по меньшей мере частичной устойчивости к лекарствам. Недавнее исследование показывает, что устойчивость может развиться одновременно к нескольким средствам, в том числе к таким, которые больной не получал. Развитие опухолей, устойчивых к многочисленным лекарственным средствам (mdr), может зависеть от массы опухоли и является основной причиной безуспешного лечения. Для преодоления этой устойчивости к лекарственным средствам применяют химиотерапию высокими дозами с облучением и аллогенной или аутогенной пересадкой костного мозга или без них. Химиотерапию высокими дозами можно проводить исходными лекарственными средствами или в измененном виде с применением дополнительных средств. Возможность такого подхода была продемонстрирована на гематопоэтических и твердых опухолях. Разработка новых лекарственных средств, не перекрестно резистентных с фенотипами mdr, необходима для повышения лечебных возможностей существующих схем лечения и для облегчения лечебных воздействий на ранее прошедших лечение больных.
Недавно противоопухолевая активность in vitro нового класса природных продуктов, названных иллудинами, была исследована Kelner, M. et. al., Cancer Res. 47: 3186 (1987), включенной здесь в качестве ссылки. Иллудин S и иллудин M являются другими известными типами существующих иллудинов. Иллудины имеют химическую структуру, полностью отличающуюся от других химиотерапевтических агентов. Соединения иллудина были ранее очищены и представлены для их оценки в Отделение лечения рака Национального института рака (NC IDCT) согласно программе скрининга лекарственных средств in vivo, но они имели низкий терапевтический индекс в других системах экспериментальных опухолей в соответствии с исследованиями NCI. Очень высокая токсичность иллудинов препятствовала их применению в лечении опухолей человека.
Таким образом, существует потребность в химиотерапевтичеких средствах, токсичных в отношении опухолей и особенно в отношении mdr опухолей и имеющих приемлемый терапевтический индекс, чтобы быть эффективными при лечении in vivo. Данное изобретение удовлетворяет эту потребность и обеспечивает относительные преимущества.
Сущность изобретения
Способ
ингибирования роста опухолевых клеток у субъекта заключается в воздействии на опухоль терапевтического количества аналога иллудина S или иллудина M, имеющего структуру

или

где аналог способен ингибировать рост опухолевых клеток без чрезмерной токсичности для субъекта. В формулах
R1 обозначает алкил или водород;
R2 обозначает алкил; и
R3 обозначает спирт или эфир.
Краткое описание чертежей
Фиг. 1 показывает чувствительность клеток карциномы молочной железы и клеток миелоидного лейкоза по
сравнению с другими опухолями к иллудину S.
Фиг. 2 показывает активные сайты иллудина S.
Фиг. 3 показывает ЯМР-спектр, демонстрирующий присутствие промежуточного продукта короткого действия в кислоте. Сигнал A исходит от водорода на двойной связи в 5-членном кольце (иллудин M). Сигнал B исходит от атома водорода на короткоживущем промежуточном продукте и является результатом раскрытия циклопропанового кольца (но перед тем, как реагирует двойная связь). Сигналы, указанные как C, исходят от продукта, образующегося при реакции двойной связи. Со временем сигнальные пики от иллудина M будут исчезать и пики по положению C будут становиться преобладающими сигналами. Сигнал B будет исчезать одновременно с сигналом A, подтверждая, что это короткоживующий промежуточный продукт, образующийся из иллудина M.
Фиг. 4 показывает действие иллудина S на рост опухоли Molt-4 в бестимусных (лишенных зобной железы) мышах (Balb/c).
Фиг. 5 показывает действие дегидроиллудина M на рост опухоли.
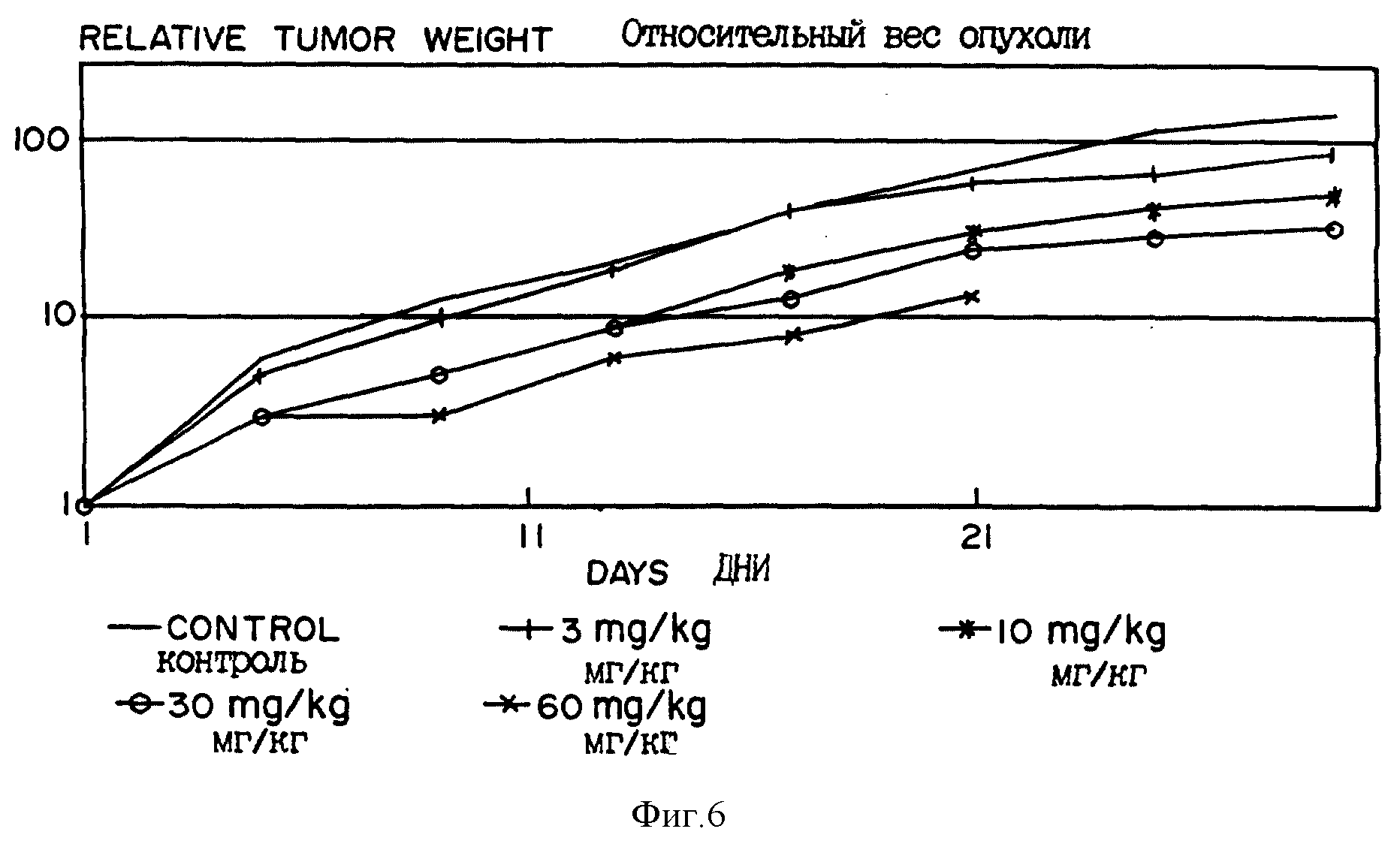
Фиг. 6 показывает ответную реакцию ксенотрансплантата HL 60/MR1 на дегидроиллудин M.
Фиг. 7 показывает поглощение иллудина S с помощью относительно чувствительных HL 60 клеток и устойчивых B-клеток.
Фиг. 8 показывает, что быстрое внутриклеточное накопление иллудина S HL 60 клетками насыщается при высоких концентрациях.
Фиг. 9 показывает анализ начального поглощения иллудина S HL 60 клетками при различных концентрациях, согласующийся с константами насыщения Михаэлиса-Ментона.
Фиг. 10 показывает действие 6-гидроксиметилацилфульвена и других обычных противораковых агентов на легочную аденокарциному MV 522 человека.
Фиг. 11 показывает действие различных доз 6-гидроксиметилацилфульвена на легочную аденокарциному MV 522 человека.
Подробное описание изобретения
Способ ингибирования роста опухолевых
клеток у субъекта заключается в воздействии на опухоль терапевтического количества аналога иллудина S или иллудина M, имеющего структуру

где аналог способен ингибировать рост опухолевых клеток без чрезмерной токсичности для индивидуума и где
R1 обозначает алкил или водород; R2 обозначает алкил и R3 обозначает спирт или эфир.
Аналог может быть любым соединением, имеющим указанную структуру. Примерами эффективных
аналогов являются

Способ ингибирования роста опухолевых клеток заключается также в воздействии на опухоль терапевтического количества аналога иллудина S или иллудина M, имеющего структуру
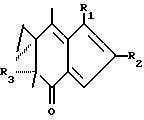
где аналог способен ингибировать рост опухолевых клеток без чрезмерной токсичности для индивидуума и где
R1 обозначает алкил, алкоксил или водород; R2 обозначает алкил и R3 обозначает спирт или эфир.
Аналог может быть любым соединением, имеющим указанную структуру. Примерами эффективных аналогов являются


Аналог может также представлять собой аналог ацилфульвена, имеющий структуру

где R представляет собой
CH2OH,



где R1 - алкил, арил, NH2, NHR или NR2.
Под "ингибированием" подразумевают либо уменьшение скорости роста опухолевых клеток по сравнению со скоростью, которая наблюдалась бы без обработки, либо уменьшение клеточной массы опухоли в размере. Ингибирование также включает полную регрессию (обратное развитие) опухоли. Таким образом, аналоги могут быть либо цитостатическими, либо цитотоксическими для опухолевых клеток.
Субъектом может быть любое животное, имеющее опухоль. Аналоги эффективны на опухолях человека in vivo, а также на линиях опухолевых клеток человека in vitro.
На опухоль можно воздействовать аналогом любым эффективным способом, многие из которых хорошо известны в данной области. Путь введения в субъект может включать внутривенный, пероральный, внутрибрюшинный, а также метод пероральной и назальной ингаляции. Предпочтительный путь введения зависит от субъекта и от типа опухоли.
Заявители неожиданно обнаружили, что могут быть получены аналоги иллудина S и M, которые менее токсичны, чем иллудин S и M, но являются более эффективными химиотерапевтическими средствами in vivo. Как отмечалось выше, иллудин S и M имеют низкий терапевтический индекс вследствие чрезвычайной токсичности и, следовательно, не могут быть применимы терапевтически для человека. Заявители обнаружили, что различные модификации иллудина S и M ингибируют нуклеофилы при реакциях с соединением. Это приводит к менее легкому раскрытию циклопропанового кольца и уменьшает токсичность соединения in vivo, что дает высокий терапевтический индекс. Заявители также обнаружили, что аналог ацилфульвена, 6-гидроксиметилацилфульвен, значительно более эффективен и менее токсичен, чем известные ранее аналоги иллудина S и M.
Различные перечисленные R группы определяются областью, не влияющей на жизненно важные функциональные группы иллудинов или их аналогов и, следовательно, могут быть разнообразными заместителями. Таким образом, заявители предлагают широкий состав различных R групп, например алкил, который включает в себя любую функциональную группу, присоединенную к алкильной группе (т. е. алкилфульвен или сама функциональная группа). R группы включают нитро, галоген, алкилгалогенид, спирт, пероксид, эндопероксид, первичный, вторичный или третичный амин, простой эфир, сложный эфир, сульфгидрил, тиоалкил, тиоэфир, дитиоэфир, дисульфид, карбоновую кислоту, енамин, имин или оксим.
Терапевтически эффективное количество аналога варьирует в зависимости от субъекта. Однако было обнаружено, что можно вводить относительно высокие дозы благодаря пониженной токсичности по сравнению с иллудином S и M. Терапевтическое количество между 30 и 112000 мкг на 1 кг веса тела особенно эффективно для внутривенного введения, тогда как 300 - 112000 мкг на 1 кг веса тела эффективно при внутрибрюшинном введении. Как будет понятно специалисту в этой области, количество может варьировать в зависимости от способа введения. Кроме того, количество может варьировать, если аналог соединен с токсином.
Аналоги могут быть присоединены к реагенту с образованием комплекса, который связан с опухолеспецифическим антигеном. Такие способы хорошо известны в этой области и в них может применяться линкер, который служит для соединения реагента с аналогом. Такое присоединение может включать любую химическую связь, например, ковалентную связь. Таким реагентом может быть любой реагент, специфически связывающийся с опухолеспецифическим антигеном на опухолевой клетке или в зоне опухолевой клетки. Обычно таким реагентом является антитело, поликлональное или моноклональное. Эти комплексы затем могут применяться в терапии. Способы данного изобретения могут практиковаться на любых опухолевых клетках, но особенно эффективны против опухолевых клеток миелоида (спинного или костного мозга), эпидермоида, T-клеточной лейкемии и карциномы легких, яичников и молочной железы.
Предложено также соединение, имеющее
структуру

где R обозначает метил, R1, R2 и R3 обозначают метил или алкил.
Предложено также соединение, имеющее структуру

где R обозначает H или метил; и R1, R2 и R3 обозначают метил или алкил.
Пример I.
Синтез дегидроиллудина M.
Смесь иллудина M (200 мг) и дихромата пиридиния (1 г) в сухом дихлорметане (60 мл) перемешивают при комнатной температуре в колбе, снабженной резиновой перегородкой (мембраной) для поддержания атмосферы аргона. Спустя 20 часов реакционную смесь разбавляют диэтиловым эфиром (20 мл) и фильтруют через короткую колонку силикагеля. Затем колонку элюируют диэтиловым эфиром и объединенный фильтрат концентрируют, получая остаток, который хроматографируют на силикагеле со смесью гексан-этилацетат (10:1) в качестве элюента.
Целевое соединение получают в ранних фракциях при хроматографии. Выход составляет 140 мг белых кристаллов, плавящихся при 64-65oC. Данные ЯМР-спектра согласуются с данным соединением.
Пример II.
Синтез фульвена.
Иллудин S (50 мг) растворяют в воде (2 мл) и к раствору добавляют 3 н. соляную кислоту (2 мл). Полученный раствор вскоре становится мутным (в пределах 30 минут) и образуется желтый осадок. Смесь помещают в холодильник на ночь, затем ее экстрагируют хлороформом (10 мл). Желтый раствор в хлороформе сушат (MgSO4) и растворитель удаляют при пониженном давлении, оставляя оранжево-желтую смолу. Этот материал хроматографируют на силикагеле с применением смеси гексан:этилацетат (6:1) в качестве элюента, получая фульвен (20 мг) и бисфульвен (10 мг). Данные ЯМР-спектра согласуются с данным соединением.
Альтернативно общий синтез фульвена может быть осуществлен следующим способом:

Взаимодействие известного 1,1-диацетилциклопропана с дианионовым производным циклопентадиена дает диол, который при мягкой кислотной обработке дает диолкетон. Селективное удаление третичной гидроксильной группы дает целевой фульвен.
Пример III.
Исследования in vitro.
Для оценки цитотоксического действия различные концентрации иллудинов добавляют к культурам клеток и выдерживают клетки в течение 48 часов, затем определяют рост клеток/жизнеспособность с помощью исключения трипана синего. В качестве альтернативы исследованию с помощью 48-часового непрерывного экспонирования клетки высевают в жидкую культуру на 96-луночные планшеты, обрабатывают различными концентрациями иллудинов в течение 2 часов, проводят импульсное мечение [3H]-тимидином в течение 1-2 часов и харвестируют клетки на стеклянных фильтрах. Фильтровальную бумагу добавляют во флаконы, содержащие сцинтилляционную жидкость, и оставшуюся на них радиоактивность определяют в бета-счетчике (сцинтилляционном).
При скрининге чувствительности других линий клеток твердых опухолей к иллудину S было замечено, что линия клеток молочной железы, MCF-7, была заметно чувствительной (фиг. 1). Было обнаружено также, что другая линия клеток молочной железы, поддерживаемая в нашей лаборатории, MDA-231, также заметно чувствительна к иллудину S (фиг. 1).
Исследования с дегидроиллудином M показали, что этот аналог также обнаруживает избирательную токсичность в отношении клеток миелоидной лейкемии и линий клеток карциномы молочной железы MCF-7 и MDA-231 (таблица 1).
Поскольку прежние исследования показали, что CEM mdr варианты не были устойчивы к иллудину S, несколько других типов mdr клеток исследовали на чувствительность к иллудину S и дегидроиллудину M. Эти дочерние mdr клеточные линии показали увеличение в 200 - 800 раз устойчивости к многочисленным общепринятым химиотерапевтическим средствам, однако обнаружили минимальную устойчивость (или не обнаружили устойчивости) к иллудину S или дегидроиллудину M (таблица 2). Таким образом, mdr клетки, связанные или не связанные с белком gp 170, были все еще чувствительны к токсичности иллудина. Эти исследования показывают, что новая иллудиновая структура сообщает относительно неперекрестную устойчивость резинстентным ко множеству лекарственных средств линиям кроветворных клеток. Производное иллудинов, дегидроиллудин M, несколько менее токсично, чем родительское соединение иллудина, но результаты (таблица 2) свидетельствуют о том, что в различных mdr клеточных линиях нет перекрестной устойчивости к этому соединению.
Исследовали действие иллудина S и дегидроиллудина M на клеточные линии L12100, мышиные линии клеток костного мозга GFU-gm и C1498 (клеточная линия AML). Иллудин S был наиболее сильным средством, когда-либо испытанным в этом тесте, и обнаружил наибольшее дифференциальное действие, когда-либо замеченное, между размерами зон линий L1210, AML клеток лейкоза и CFU-gm (таблица 3). Производное дегидроиллудин M, менее токсичное, было явно более избирательным в отношении линии AML. Оно ингибировало образование колоний AML при концентрациях, когда оно уже не действовало на клетки CFU-gm (таблица 4).
Пример IV.
Структурно-функциональные исследования.
Структурно-функциональные исследования осуществляют путем синтеза производных иллудина и испытания их токсичности in vitro для клеток лейкемии HL 60 (таблица 5). Это исследование определило три критических сайта для токсичности иллудина. Они включают в себя циклопропановое кольцо (сайт A), сайт ненасыщенной связи альфа/бета (сайт B) и кетоногруппу (сайт C) (фиг. 2). Изменение в любом из этих сайтов приводит к уменьшению токсичности до 4 log. В противоположность этому некольцевая первичная гидроксильная группа не участвует в токсичности (фиг. 2, сайт D). Различные большие химические группы могут быть присоединены к этому сайту без изменения токсичности. Многие производные с заметным уменьшением в токсичности (по сравнению с иллудином S или M) все еще являются более сильными, чем обычные химиотерапевтические средства, такие как BCNU или цис-платина (таблица 5).
Пример V.
Структурно-функциональные исследования: химические.
Иллудин M легко превращается в стабильные ароматические соединения (при обработке разбавленной HCl), которые более чем в 1000 раз менее токсичны в исследованиях, проводимых на культуре клеток. Образование связи хлор-углерод, раскрытие циклопропанового кольца и выделение третичного гидроксила (в виде воды) являются синхронными. Полученный промежуточный продукт можно обнаружить ЯМР-спектроскопией реакционной смеси (фиг. 3). Однако этот промежуточный продукт является очень реакционноспособным и быстро превращается в фенол в результате разрушения вторым нуклеофилом, т.е. водой. Поэтому при кислотных условиях иллудин M является явно бифункциональным.
Описанные выше исследования показывают, что токсичность иллудинов связана с легкостью удаления третичного гидроксила и раскрытия цилкопропанового кольца. Было обнаружено, что токсичность иллудина зависит от объединенных эффектов циклопропановой группы (сайт A, фиг. 2), двух двойных связей (конъюгированный диен) (сайт B) и кетона (сайт C). Было высказано предположение, что окисление вторичной гидроксильной группы в пятичленном кольце до кетона может изменить силу или избирательность молекулы путем дальнейшей делокализации электрона внутри молекулы. Новая кетонная группа действует как "электронный сток", так что электроны циклопропановых C-C связей делокализуются по направлению к кетону скорее, чем к атому углерода, несущему третичный гидроксил. Это означает, что начальный карбокатион, образующий разрывы связи углерод-кислород (кислород третичного гидроксила) не так стабилен, как в случае иллудина M. Поэтому разрыв связи углерод-кислород менее выгоден и реакционная способность уменьшается. Это производное кетона, названное дегидроиллудин M, было синтезировано и проявило меньшую токсичность для клеток HL-60 in vitro, чем иллудин S или M (таблица 4). Как обсуждалось выше, токсичность дегидроиллудина M, по-видимому, относительно избирательна для миелоидных клеток и клеток карциномы молочной железы in vitro (фиг. 1 и таблица 1).
В соответствии с высказанной выше гипотезой находятся результаты кинетики реакции иллудина M и дегидроиллудина M с разбавленной HCl. В разбавленной HCl иллудин M подвергается псевдореакции первого порядка (k = 4,7 • 10-3 мин-1, t 1/2 = 148 минут). Дегидроиллудин M также показал кинетику первого порядка, но эта реакция была значительно более медленной (k = 2 • 10-4 мин-1, t 1/2 = 2765 минут). В реакции с дегидроиллудином M ЯМР-спектроскопией не смогли обнаружить промежуточного продукта. Предположительно он образуется слишком медленно и является слишком короткоживущим, чтобы его можно было бы детектировать. Более низкая реакционная способность, обнаруженная дегидроиллудином M, предполагает, что он более избирателен в его реакции с нуклеофилом и, следовательно, имеет более низкую токсичность по сравнению с иллудином M.
Также изучают взаимодействие иллудинов с встречающимся в природе нуклеофилом, глутатионом. При широком диапазоне pH, от 3 до 9, глутатион самопроизвольно реагирует с иллудином M, иллудином S или дегидроиллудином M, образуя продукты, аналогичные продуктам реакции иллудина M и HCl. Скорость реакции оптимизируют при pH 6,1 - 7,0, что свидетельствует о том, что реакция происходит внутри клеток.
Затем исследуют токсичность иллудинов по отношению к клеточной линии карциномы молочной железы MCF7-wt и ее mdr устойчивой дочерней линии MCF/Adr • gp 170 - отрицательная дочерняя клеточная линия устойчива к лекарственным средствам на основе 50-кратного увеличения активности глутатионтрансферазы, что приводит к 200-800-кратному уменьшению в чувствительности к общепринятым химиотерапевтическим средствам. Эта линия также обнаруживает 4,1-кратное уменьшение в содержании глутатиона. Эта дочерняя линия обнаруживает 4,2-кратное уменьшение в чувствительности к иллудину S (IC50 родительской линии 0,88 нмоль/1; дочерней линии 3,70 нмоль/1) по сравнению с 200-800-кратным уменьшением с другими агентами. Кинетические исследования способности иллудинов ингибировать глутатионтрансферазу показывают, что нет прямого ингибирования активности этого фермента. Эти результаты показывают, что в то время как токсичность иллудина обратно коррелирована с содержанием внутриклеточного глутатиона, она не коррелирует с активностью глутатионтрансферазы.
Пример VI.
Исследования на животных.
При помощи способов, представленных Leonard. J.E. et al., Cancer Res. 47: 2899-02 (1987) и Dillman, R.O. et al., Cancer Res. 45: 5632: 36 (1985), включенных здесь в виде ссылок, ксенотрансплантаты Molt-4 (T-клеточная лейкемия человека) были внедрены в 4-недельных бестимусных Balb/C nu/nu мышей. После 3-недельных доз общего облучения тела (600 cGy) мышам инъецируют в бок клетки Molt-4 вместе с облученными (6000 cGy) питающими клетками HT-1080. Двое животных получают только облученные питающие клетки HT-1080, чтобы убедиться, что эти клетки не индуцируют образование опухолей. Животных наблюдали в отношении развития Molt-4 опухолей и, когда опухоли становятся прощупываемыми (приблизительно 4 х 4 мм при 5-7 днях), мышей разделяют случайным образом на группы из 5 мышей, как описано ранее. Контрольные мыши получают внутрибрюшинно солевой раствор, а обрабатываемые мыши получают либо 300 мкг/кг иллудина S, либо 30 или 300 мкг/кг дегидроиллудина M внутрибрюшинно (IP) два раза в неделю. У мышей, получающих иллудин S, наблюдается задержка в росте опухолей (фиг. 4).
В противоположность этому на "голых мышах", получивших дегироиллудин M при низкой дозе 30 мкг/кг (это соединение впоследствии было нетоксичным для мышей при 60000 мкг/кг IP дважды в неделю), три из пяти опухолей показывают полную рергессию (обратное развитие), но две опухоли не дают ответной реакции (фиг. 5). Эти две, по-видимому, устойчивые опухоли извлекают и исследуют in vitro на устойчивость к иллудину S и дегидроиллудину M. Двое из исследуемых животных наблюдались в течение более 12 недель без регрессии опухолей.
Эти эксперименты повторяют с применением разных источников бестимусных голых мышей. В этих исследованиях наблюдают незначительное действие иллудинов на рост опухолей. Причина такой вариабельности в ответе на ксенотрансплантаты Molt-4 возможно связана с низкими дозами дегидроиллуина M, изменениями в метаболизме глутатиона у разных животных или с распределением лекарственного средства.
Затем эффективность дегидроиллудина M подвергают скринингу, на сингенной модели с применением мышиных клеток SL-2. SL-2 лейкемия - лимфомные клетки инъецируют подкожно и они метастазируют к лимфатическим узлам, селезенке и легким. Эффективность лекарственных средств на этой модели определяют по увеличению продолжительности жизни (ILS). Клетки SL-2 вводят при концентрации 2,5 млн. клеток на животное и обработку откладывают на 7 дней до тех пор, пока опухоль не становится прощупываемой. Это относительно строгий тест на образовавшихся опухолях и он отличается от обычных скринингов лекарственных средств, в которых обычно используют только 0,5 млн. клеток и начинают обработку лекарственными средствами при 3 днях. Дегидроиллудин M обнаружил небольшое действие при 30 мг/кг IP два раза в неделю, ILS 5%, и 60 мг/кг IP два раза в неделю, ILS 18%. При введении IV (внутривенно) при 0,03 мг/кг два раза в неделю ILS увеличивалось до 38%. Это предполагает, что лекарственное средство метаболизировалось печенью и, вероятно, более эффективно при введении внутривенно (IV).
В ходе этих экспериментов in vivo стало ясно на основании экспериментов in vitro, что тканеспецифичность иллудинов зависит от присутствия активного зависимого от энергии насоса. При исследовании клеток SL-2 и Molt-4 было обнаружено, что у них отсутствует механизм поглощения. Поэтому исследования направили на модели ксенотрансплантатов, ипользующие клетки миелоидного происхождения (опухолей спинного или костного мозга).
Человеческие клетки HL-60, способные расти в качестве ксенотрансплататов в голых мышах без облучения животных, были получены от Др. Theodore Brightman (NCI). Эти клетки, обозначаемые как HL-60 MRI клетки, обнаружили наличие зависимого от энергии насоса поглощения, что не было неожиданным, поскольку их родительские клетки обладают таким насосом. Дегидроиллудин M индуцировал зависимым от дозы образом ингибирование опухолей при введении IP (внутрибрюшинно) два раза в неделю (фиг. 6). MTD IP доза для дегидроиллудина M достигает в этих исследованиях при двух дозах в неделю при внутрибрюшинном введении. Подобная регрессия опухолей наблюдается и при внутривенном введении дегидроиллудина M.
Одновременно снова исследуют действие in vivo дегидроиллудина M. Сначала исследуют действие этого соединения на L1210 клетки. Доза 2,5 мг/кг IP, даваемая ежедневно в течение 5 дней, приводит только к ILS 9%. Затем дегидроиллудин M вводят при помощи 24-часового вливания (5,0 мг/кг); ILS было 11%. После того как стало известно о присутствии зависимого от энергии поглощения в миелоцитах человека, дегидроиллудин M подвергают скринингу на эффективность in vivo на модели сингенных мышей AML с применением C1498 клеток, давая однократную ударную дозу иллудина S, 2,5 кг/кг IP, приводящую к ILS 35%. Второе испытание с применением той же самой дозы, вводимой IP один раз в день в течение 5 дней, приводит к ILS 44%. Поскольку эти животные могут переносить 60 мг/кг IP или 1 мг/кг IV (в хвостовую вену) при режиме дважды в неделю в течение 4 недель без обнаружения потери веса или снижения поглощения пищи/воды, можно, кроме того, оптимизировать как схему доз, так и схему обработки.
Пример VII.
Эксперимент с HL 60/MRI мышами с ацилфульвеном и дегидроиллудином M.
30 мышей инъецируют подкожно над плечом 500000 HL 60/MRI клетками (опухолевые клетки миелоидного лейкоза человека). Обработку начинают на 11-ый день, лучше, чем сразу. Эта задержка в начальной обработке является самым строгим тестом для определения, эффективно ли испытуемое соединение. В результате задержания обработки, опухолевые клетки становятся прочно внедренными.
Мышей делят на 6 групп по 5 мышей. Одна группа является контролем и эти животные получают плацебо, раствор, применяемый для разведения агента. Другие группы получают следующие соединения и дозы: соединение дегидроиллудина M при 1,0 мг/кг, дегидроиллудин M при 3,0 мг/кг, ацилфульвен при 0,3 мг/кг, ацилфульвен при 1,0 мг/кг, ацилфульвен при 3,0 мг/кг. Все животные получают плацебо или лекарственные средства путем внутривенной инъекции в хвостовую вену. Плацебо или лекарственные средства вводят дважды в неделю.
Результаты суммированы в таблице 6. Как дегидроиллудин M, так и соединение ацилфульвена являются эффективными при ингибировании роста опухолей и обнаруживают зависимое от дозы ингибирование (чем больше вводят лекарственного средства, тем меньше растут опухоли). Животные, получающие наивысшее количество любого лекарственного средства, демонстрируют доказательства какого-либо неблагоприятного действия, такого как уменьшение приема пищи или воды или статистически значимое снижение веса тела. Эти результаты показывают, что можно вводить более высокие дозы любого из этих лекарственных средств, а также что лекарственное средство можно было бы вводить при более эффективной схеме доз для ежедневного введения.
Пример VIII.
Общие способы скрининга in vitro и исследования поглощения соединений клетками.
Согласно предположению по предшествующим примерам и с нашим сосредоточением на механизмах действия иллудинов и тканевой специфичности другие линии клеток миелоидной лейкемии могут быть скринированы на быстрое поглощение иллудина (KGI, KLI a, HEL K562, OCI-MI, AML-193).
Способы для скрининга in vitro соединений иллудина описаны детально в предшествующих примерах. Цитотоксичность новых аналогов для клеточных линий сначала оценивают в 5 log-диапазоне с применением тестов роста или образования колоний на полутвердой среде и определения ингибирования включения тимидина. Ингибирование включения тимидина используют, поскольку более ранние исследования показывают, что включение тимидина преимущественно ингибируется иллудинами и тесно коррелирует со смертью клеток. Аналоги подвергают скринингу в отношении нормальных предшественников клеток костного мозга и многочисленных клеточных линий, включающих в себя различные лейкозы, B- и T-клеток, и твердые опухоли (меланому, опухоль яичников).
Тестирование in vitro дегидроиллудина M на различных клеточных линиях, в том числе MDR линиях, можно также проводить на ленточных линиях, дефектных в отношении репарации ДНК, и на клетках-предшественниках нормального костного мозга. Могут быть получены другие аналоги. Поскольку эти аналоги будут иметь изменения в известных активных сайтах, можно ожидать, что будут приводить к сходному ингибированию опухолей. Скрининг для этих аналогов может включать в себя применение различных mdr клеток (чтобы убедиться, что не происходит перекрестная устойчивость) и клеточных линий, дефектных в отношении репарации ДНК.
Тестирование in vitro может также исследовать чувствительность других клеточных линий молочной железы, чтобы определить, не имеют ли они также преимущественной чувствительности к иллудину S, дегидроиллудину M и аналогу фульвена.
Пример IX.
Оценка поглощения иллудинов в опухолевые клетки.
В то время как человеческие миелоидные опухолевые клетки чувствительны к иллудинам, их нормальные предшественники, гранулоциты/макрофаги образующие единицы, относительно устойчивы к иллудинам (1,5 - 2,0 log), что свидетельствует об отсутствии транспортной системы в некоторых нормальных клетках костного мозга, что обеспечивает терапевтический предел безопасности.
Удельное поглощение иллудина S исследуют с применением относительно чувствительных HL 60 клеток и устойчивых B-клеток. При 37oC HL 60 миелоидные лейкозные клетки показывают быстрое поглощение иллудина S, тогда как относительно нечувствительные B-клетки 8392 обнаруживают сравнительно небольшое включение лекарственного средства (фиг. 7). Внутриклеточное накопление иллудинов в этой B-клеточной линии было медленным и линейным в течение 7 часов (r - 0,984), когда была достигнута внутриклеточная концентрация, приближающаяся к концентрации в инкубационной смеси. HL 60 клетки, напротив, быстро накапливают токсин и внутриклеточное накопление достигает плато в пределах одного часа. HL 60 клетки, выдерживаемые с 10 нМ иллудином S, концентрируют токсин в 19 раз, тогда как B-клетки не концентрируют активно токсин. Быстрое внутриклеточное накопление иллудина S HL 60 клетками насыщается при высоких концентрациях (фиг. 8). В противоположность этому накопление иллудина S в B-клетках 8392 остается зависимым от концентрации. Анализ начального поглощения иллудина S HL 60 клетками при различных концентрациях показывает, что приток иллудина S соответствует кинетике насыщения Михаэлиса-Ментона (фиг. 9). Vмакс для HL 60 клеток была 27 пмоль/мин/мг белка и Km была 4,2 мкМ. Это указывает на то, что HL 60 клетки имеют очень высокую транспортную способность для иллудинов, т.к. Vmax для иллудинов в 5 раз больше Vmax для фолата, требующегося для клеток витамина.
Холод (4oC), 1% азид и блокаторы метаболизма 2-дезоксиглюкоза и антимицин A блокируют поглощение иллудина S в HL 60 клетки, но оказывают незначительное действие на нечувствительные B-клетки 8392 (таблица 7). Эти исследования свидетельствуют о том, что иллудин S транспортируется в клетки HL 60 и концентрируется в них при помощи зависимой от энергии транспортной системы, тогда как транспорт в нечувствительные B-клетки происходит только в результате диффузии, т.е. пассивного или не требующего энергии транспорта. Клетки MCF-7 опухоли молочной железы также показывают ингибирование поглощения холодом. Обнаружение зависимого от энергии транспортного механизма объясняет, почему клетки миелоида и опухоли молочной железы так чувствительны к иллудинам при коротких периодах экспозиции, а B-клетки нечувствительны.
Пример X.
Синтез и структура соединений 2,5,6,7-тетраметил-1-инденона и дегидроптерозина.
Сначала 2,4,5,6-тетраметил-1, 3-инданон синтезируют путем приготовления раствора 1,2,3-триметилбензола и метилмалонилхлорида в сероуглероде и добавления по каплям трихлорида алюминия в течение двух часов. Смесь кипятят с обратным холодильником в течение еще 2 часов, добавляют измельченный лед и экстрагируют трижды хлороформом. Объединенный экстракт промывают раствором соли, сушат и растворитель удаляют, получая остаток, который очищают хроматографией с применением 1% этилацетата в бензоле. Удаление растворителя и очистка сублимированием дает целевой продукт.
2,5,6,7-тетраметил-1-инденон получают
восстановлением 2,4,5,6-тетраметилинданона цинковой пылью при 50oC. Продукт очищают хроматографией с 1% этилацетатом в бензоле, получая два изомера. Основной изомер обрабатывают 10%
гидроксидом калия, затем очищают сублимацией. Соединение имеет структуру

Синтез дегидроптерозина O: 3-ацетокси-6 (β-метокси) этил-2,5,7-триметил-1-инданон растворяют в тетрагидрофуране и 10% гидроксиде калия и кипятят с обратным холодильником в течение 2 часов. Затем раствор экстрагируют трижды эфиром и объединенные экстракты хроматографируют с 2% этилацетатом в бензоле, получая соединение дегидроптерозин O. Соединение имеет структуру

R = H дегидроптерозин B,
R = CH3 дегидроптерозин O.
Оба соединения токсичны для клеток in vitro и обладают противогрибковыми свойствами.
Пример XI.
Синтез аналогов ацилфульвена.
Многие аналоги ацилфульвена, обладающие ключевыми структурными особенностями, необходимыми для противоопухолевой активности, могут быть получены исходя из иллудина S или общим синтезом из простых предшественников. Иллудин S получают ферментацией Omphalotus illudeus. Путем растворения этого соединения в воде и добавления разбавленной H2SO4 его превращают в ацилфульвен (R1 = H, R2 = H, R3 = CH3) с выходом 55%. Большое число аналогов можно получить из ацилфульвена модификацией заместителя R2, например при R2, обозначающем гидроксиметил, бром, иод, хлор, фтор, нитрогруппу, п-гидроксибензил, п-метоксибензил, R2 может также быть полициклическими или гетероциклическими ароматическими группами.

R1 группа может быть ацилом или алкилом. Аналоги с различными R1 группами (и также R2 группами) могут быть получены посредством общего синтеза, как описано.


Альдольная конденсация 1,1-диацетилциклопропана с дианионом, полученным из соответствующим образом замещенного циклопентадиена, дает промежуточный продукт, легко превращаемый в ацилфульвен. Может быть получено широкое разнообразие соединений т.к. R2 и R3 могут быть алкилом, арилом или замещенным алкилом или арилом.
Синтез ацилфульвена. Иллудин S (2 г) (9,2 ммоль) растворяют в 700 мл воды с последующим добавлением 4 М H2SO4 (236 мл). Раствор перемешивают в течение ночи и экстрагируют этилацетатом. Органическую фазу промывают насыщенным NaHCO3, водой и раствором соли, сушат над MgSO4 и концентрируют. Хроматография на диоксиде кремния с гексаном и этилацетатом дает 0,82 г ацилфульвена (50%).
1H ЯМР δ: 0,73 - 1,50 (м, 4H), 1,38 (с, 3H), 2,00 (с, 3H), 2,15 (с, 3H), 3, 93 (с, 1H), 6,43 (с, 1H), 7,16 (с, 1H).
MS: m/z 216 (M+), 202 (M+ -CH2), 188 (M+ -CH2CH2), 173 (M+ -2CH2CH3), 170 (M+ -2CH2-H2O).
УФ 325 нм (8,3 • 103), 235 нм (16,6 • 103).
Соединение имеет структуру

Синтез 6-гидроксиметилфульвена. Ацилфульвен (550 мг, 2,5 ммоль) растворяют в 41 мл TNF и добавляют 30% формальдегидводного раствора (40 мл). Затем добавляют 4 н. раствор H2SO4 (26,4 мл) для доведения конечной концентрации H2SO4 до 1 н.
Раствор перемешивают в течение ночи и экстрагируют этилацетатом. Органическую фазу промывают насыщенным раствором NaHCO3, водой и раствором соли и сушат над MgSO4.
Хроматография на силикагале с гексаном и этилацетатом дает 400 мг гидроксиметилфульвена (64%).
1H ЯМР δ: 0,72 - 1,48 (м, 4H), 1,38 (с, 3H), 2,15 (с, 3H), 2,19 (с, 3H), 3,90 (с, 1H), 4,66 (д, J=2,1 Гц, 2H), 7,10 (с, 1H).
MS: m/z 246 (M+), 228 (M+ -H2O), 218 (M+ -CH2CH2), 186 (M+ -CH3-CH2-CH2-OH), 185 (M+ -H2O-CH2 -CH2-CH3).
УФ 233 нм: (1,0•104), 325 нм: (7,7•103).
Соединение имеет структуру
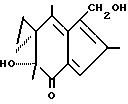
Иодофульвен. К раствору ацилфульвена (60 мг, 0,28 ммоль) в 15 мл CH2Cl2 добавляют трифторацетат серебра (63 мг, 0,29 ммоль). Раствор иода (70,5 мг, 0,28 ммоль) в 8 мл CH2Cl2 добавляют по каплям при 0oC. Смесь перемешивают при этой температуре в течение 3 часов, затем фильтруют через целит и элюируют эфиром. Концентрирование фильтрата дает иодофульвен в виде смолы (73 мг, 77%).
1 H-ЯМР

MS: m/z 342 (M+), 314 (M+ -CH2CH2), 299 (M+ -CH2CH2-CH3), 296 (M+ -CH2-CH2-H2O), 215 (M+ -I), 187 (M+ -I-CH2CH2), 127 (I+).
Соединение имеет структуру

Бромфульвен. Ацилфульвен (60 мг, 0,28 ммоль) растворяют в 9 мл ацетонитрила при 0oC. Добавляли N-бромсукцинимид (50 мг, 0,28 ммоль) и смесь перемешивают при той же температуре в течение 3,5 часов. Для остановки реакции используют воду, а для экстракции продукта используют эфир. Эфирный слой промывают водой и раствором соли и сушат над MgSO4. Хроматография дает бромфульвен в виде оранжевых кристаллов (77 мг, 94%; их перекристаллизовывают из смеси этилацетат-гексан, т.пл. 92-94oC).
1H-ЯМР δ: 0,75 - 1,55 (м, 4H), 1,40 (с, 3H), 2,12 (с, 3H), 2,33 (с, 3H), 3,89 (с, 1H), 7,15 (с, 1H).
MS: m/z 295 (M+), 293 (M+ -2), 267, 265 (M+ -CH2CH2), 252, 250 (M+ -CH2CH2-CH3), 249, 247 (M+ -CH2CH2-H2O), 215 (фульвен-1).
Соединение имеет структуру

n-Гидроксибензилфульвен. Фенол (40 мг, 0,4 ммоль) добавляют к раствору гидроксиметилфульвена (70 мг, 0,28 ммоль) в сухом CH2Cl2 (25 мл). Смесь охлаждают до -78oC и добавляют по каплям эфират трифторида бора (0,3 мл, 2,7 ммоль). Реакционную смесь перемешивают при той же температуре в течение 1 часа и добавляют воду для остановки реакции. Органический слой промывают H2O, NaHCO3 и раствором соли и сушат над MgSO4. Хроматография на силикагеле со смесью гексан-этилацетат дает 90 мг (98%) красных кристаллов (т.пл. 143 - 144oC).
1H-ЯМР δ: 0,59 - 1,43 (м, 4H), 1,36 (с, 3H), 1,76 (с, 3H), 2,07 (с, 3H), 3,95 (с, 1H), 3,97 (д, J=7,2 Гц, 2H), 4,83 (с, 1H), 6,74 (д, J=8,4 Гц, 2H), 6,91 (д, J=8,4 Гц, 2H), 7,22 (с, 1H).
MS: m/z 322 (M+), 294 (M+ -2CH2), 279 (M+ -2CH2-CH3), 251 (M+ -2CH2 -CH3-CO), 215, 107.
УФ 288 нм (1,2•104, с точками перегиба при 243 и 262 нм), 325 (7,1•103), 410 нм (2,6•103).
Соединение имеет структуру

n-Метоксибензилфульвен. Анизол (0,04 мл, 0,37 ммоль) добавляют к раствору гидроксиметилфульвена (10 мг, 0,04 ммоль) в сухом CH2Cl2 (5 мл). Смесь охлаждают до -78oC и добавляют по каплям эфират трихлорида бора (0,04 мл, 0, 36 ммоль). Реакцию перемешивают при той же температуре в течение 1 часа и добавляют воду для остановки реакции.
Органический слой промывают H2O, насыщенным NaHCO3 и солевым раствором и сушат над MgSO4. Концентрирование раствора дает остаток, который сушат в вакууме, получая продукт с количественным выходом (14 мг).
1H-ЯМР δ: 0,59 - 1,40 (м, 4H), 1,36 (с, 3H), 1,76 (с, 3H), 2,07 (с, 3H), 3,78 (с, 3H), 3,95 (с, 1H), 3,99 (д, J=16,12 Гц, 2H), 6,81 (д, J=8,8 Гц, 2H), 6,96 (д, J=8,3 Гц, 2H), 7,22 (с, 1H).
MS: m/z=336 (M+), 308 (M+ -CH2CH2), 215, 121 ((CH3-Ph-OCH3)+).
УФ 410 нм (2,7•103), 325 нм (7,0•103), 267 нм (1,0•104), 245 нм (перегиб), 226 нм (1,9•104), 203 нм (1,4•104).
Соединение имеет структуру

Пример XII.
Исследование аналогов ацилфульвена на культуре клеток in vitro.
Исследование in vitro с применением опытов на клеточных культурах показывает, что аналоги 6-гидроксиметилацилфульвен, бромацилфульвен и иодацилфульвен заметно токсичны для опухолевых клеток-мишеней HL 60 и MV 522 как при 2, так и при 48 часах экспозиции (таблица 8 и 9). Отношение относительной токсичности (2-часовая токсичность /48-часовая токсичность) предполагает, что эти аналоги более эффективны in vitro, чем родительское соединение иллудин S или аналоги, описанные в исходной патентной заявке.
Пример XIII.
Исследования in vitro.
На основании описанных результатов скрининга in vitro один из аналогов, 6-гидроксиметилацилфульвен, был выбран для исследований in vitro для определения, действительно ли этот аналог более сильнодействующий, чем ацилфульвен. Ксенотрансплантатом были опять клетки карциномы легких человека MV 522, т. к. они представляют собой модель, не реагирующую на общепринятые противораковые агенты, и которая путем образования метастаз (не ограничиваются местной опухолевой инвазией). В качестве фармацевтических контролей были выбраны обычные противораковые средства цис-платина, таксол, митомицин C, адриамицин, а также иллудин S. Была включена в опыт также контрольная группа, которая получала только растворитель, используемый для растворения лекарственных средств, смесь 40% диметилсульфоксида и обычного солевого раствора (40% DMSO/NS). Аналог 6-гидроксиметилацилфульвен действительно индуцировал регрессию опухоли в животных. Истинная регрессия (обратное развитие) опухолей под влиянием противоракового средства никогда не отмечается на этой модели. Общепринятые противораковые средства и иллудин S не ингибируют рост опухолей (фиг. 10). Животные получают только 9 доз 6-гидроксиметилацилфульвена. Не были обнаружены токсические побочные действия в этих животных, такие как уменьшение активности, прироста веса, приема пищи или приема воды. Не было значительного увеличения в продолжительности жизни животных, обработанных цисплатиной, таксолом, митомицином C и адриамицином, по сравнению с животными, обработанными DMSO/NS (контроль). Иллудин действительно вызывает преждевременную смерть (вследствие токсичности этого лекарственного средства) (таблица 10). Животные, получавшие 6-гидроксиметилацилфульвен, живут значительно дольше, чем контрольные животные (обработанные DMSO/NS), животные, обработанные цис-платиной, таксолом, митомицином C и адриамицином (p<0,001 для всех групп) (таблица 10).
Этот эксперимент повторяют с различными дозами аналога 6-гидроксиметилацилфульвена для определения, существует ли зависимость ответной реакции от дозы. В качестве фармацевтических контролей опять включают таксол и низкую дозу иллудина S (таблица 11). Также опять включают контрольную группу, получающую только фармацевтический растворитель (40% DMSO/NS). Регрессию опухолей опять отмечают при 10 мг/кг, а ингибирование роста опухоли отмечают при 1 и 5 мг/кг. Таксол и низкая доза иллудина S опять не ингибируют рост опухолей (фиг. 11). В этом втором эксперименте также не было заметного увеличения в продолжительности жизни животных, обработанных таксолом или иллудином S, по сравнению с контролями (40% DMSO/NS). Животные, получавшие 10 мг/кг 6-гидркосиметилацилфульвена и 5 мг/кг 6-гидроксиметилацилфульвена, живут значительно дольше, чем контрольные животные (обработанные DMSO/NS) или обработанные таксолом или иллудином S животные. Величина вероятности (или значимости) для обработанных 10 мг/кг 6-гидроксиметилацилфульвена по сравнению с контрольными или обработанными таксолом и иллудином S животными была менее 0,001 в каждом случае (p < 0,001). Величина вероятности для обработанных 5 мг/кг 6-гидроксиметилацилфульвена животных по сравнению с контрольными и обработанными таксолом и иллудином S животными была менее 0,001 в каждом случае (p < 0,001). Обработанные 1 мг/кг 6-гидроксиметилацилфульвена животные также жили значительно дольше, чем контрольная группа (p < 0,01).
Эксперимент повторяют третий раз (таблица 12). Количество вводимого внутрибрюшинно (вб) таксола увеличивают для демонстрации максимальной дозы и добавляют подкожное введенную дозу, чтобы показать, что этот путь введения может быть более эффективным. Испытывают опять также адриамицин и митомицин C. В этот эксперимент включают два новых производных ацилфульфена (иодацилфульвен и п-гидроксибензилфульвен). Дикетоновый аналог включают для демонстрации заметного улучшения 6-гидроксиметилацилфульвена. 6-Гидроксиметилацилфульвен заметно увеличивает продолжительность жизни по сравнению с контролями и обработанными другими агентами животными. Величина вероятности (или значимости) для 6-гидроксиметилацилфульвена по сравнению с контрольной группой, обработанной митомицином C группой, обработанной адриамицином группой и обеими обработанными таксолом группами менее 0,0005 (p < 0,0005). Это чрезвычайно значительный эффект. Важно отметить, что на основании длительности жизни двух животных можно думать, что, возможно, они могли бы быть излеченными. Хотя дикетон также заметно увеличивает продолжительность жизни по сравнению с контролями (p < 0,002) и обработанными другими лекарственными средствами животными, он не так эффективен, как 6-гидроксиметилацилфульвен. Следует заметить, что продолжительность жизни обработанных высокой дозой таксола животных (6 мг/кг IP) не только уменьшается по сравнению с обработанными 4 мг/кг таксола животными, но она была теперь меньше, чем продолжительность жизни необработанных или контрольных животных. Другие новые аналоги, бромацилфульвен и п-гидроксибензилфульвен также эффективны в этой модели.
Хотя изобретение описано со ссылкой на предпочтительный в настоящее время вариант, следует учесть, что могут быть сделаны различные модификации без отхода от сущности и характера данного изобретения. В соответствии с этим изобретение ограничивается только следующей далее формулой изобретения.
Ниже представлены фармацевтические композиции лекарственных форм, содержащие соединение формулы I, которые предназначены для терапевтического и профилактического использования при лечении людей.
(i) Таблетки 1 - мг/таблетка
Соединение формулы I - 100,0
Лактоза - 77,5
Повидон - 15,0
Натрий кроскармелоза - 12,0
Микрокристаллическая целлюлоза - 92,5
Стеарат магния - 3,0
Всего: - 300,0
(ii) Таблетки 2 - мг/таблетка
Соединение формулы I - 20,0
Микрокристаллическая целлюлоза - 410,0
Крахмал - 50,0
Натрий крахмал
гликолят - 15,0
Стеарат магния - 5,0
Всего: - 500,0
(iii) Капсулы - мг/капсула
Соединение формулы I - 10,0
Коллоидная двуокись кремния - 1,5
Лактоза - 465,1
Предварительно желатинизированный крахмал - 120,0
Стеарат магния - 3,0
Всего: - 600,0
(iv) Раствор для инъекций 1 (мг/мл) - мг/мл
Соединение формулы I (в форме свободной кислоты) - 1,0
Двуосновный фосфат натрия - 12,0
Одноосновный фосфат натрия - 0,7
Хлорид натрия - 4,5
1 н. раствор гидроксида
натрия (доведение pH до 7,0 - 7,5) - Сколько понадобится
Вода для инъекций - Сколько понадобится до 1 мл
(v) Раствор для инъекций 2 (10 мг/мл) - мг/мл
Соединение формулы I
(в форме свободной кислоты) - 10,0
Одноосновный фосфат натрия - 0,3
Двуосновный фосфат натрия - 1,1
Полиэтиленгликоль 400 - 200,0
0,1 н. раствор гидроксида натрия
(доведение pH до 7,0 - 7,5) - Сколько понадобится
Вода для инъекций - Сколько понадобится до 1 мл
(vi) Аэрозоли - мг/флакон
Соединение формулы I - 20,0
Олеиновая
кислота - 10,0
Трихлормонофторметан - 5000,0
Дихлордифторметан - 10000,0
Дихлортетрафторэтан - 5000,0
Приведенные выше композиции можно получать с помощью хорошо
известных в фармацевтике способов.
Реферат
Предложено новое средство и фармацевтическая композиция на его основе для ингибирования роста опухолевых клеток у субъекта. В качестве такого средства предложены новые аналоги ацилфульвена, имеющего структуру (I), где R обозначает (a), а R1 обозначает алкил, арил, NH2, NHR или NR2. Средство и фармацевтическая композиция на его основе способны ингибировать рост опухолевых клеток без чрезмерной токсичности для субъекта. 2 с. и 8 з.п. ф-лы, 12 табл., 11 ил.

CH2OH,





Формула

в которой R выбран из группы, состоящей из CH2OH,



где R1 представляет собой алкил, арил, NH2, NH (алкил) или N (алкил)2.

в которой R выбран из группы, состоящей из
CH2OH,


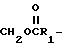
где R1 представляет собой алкил, арил, NH2, NH (алкил) или N (алкил)2 в сочетании с фармацевтически приемлемым носителем.
Комментарии