Производные 4- или 5-аминосалициловой кислоты - RU2414476C2
Код документа: RU2414476C2
Чертежи













Описание
Настоящее изобретение относится к соединениям, пригодным для лечения кишечных заболеваний, таких как воспалительное заболевание кишечника (IBD) и спастический колит (IBS), и химиопрофилактики рака ободочной кишки. В частности, были созданы производные 4- и 5-аминосалициловой кислоты, содержащие высвобождающую сероводород часть молекулы, связанную при помощи азогруппы, сложноэфирной, ангидридной, сложнотиоэфирной или амидной связи с молекулой 5- или 4-аминосалициловой кислоты.
Воспалительное заболевание кишечника (IBD) является общим названием заболеваний, вызывающих воспаление тонкой кишки и ободочной кишки. Неспецифический язвенный колит является наиболее распространенным воспалительным заболеванием кишечника, которое поражает разные отделы желудочно-кишечного (GI) тракта, в частности нижний отдел желудочно-кишечного тракта и более конкретно ободочную и/или прямую кишку. Вторым воспалительным заболеванием кишечника является болезнь Крона, которая поражает главным образом тонкую кишку (подвздошную кишку) и толстую кишку (ободочную кишку).
Неспецифический язвенный колит трудно диагностировать, так как симптомы данного заболевания подобны другим заболеваниям кишечника и болезни Крона. Болезнь Крона отличается от неспецифического язвенного колита тем, что вызывает более глубокое воспаление стенки тонкой кишки. Кроме того, болезнь Крона обычно возникает в тонкой кишке, хотя указанное заболевание может также поражать ротовую полость, пищевод, желудок, двенадцатиперстную кишку, толстую кишку, аппендикс и анус.
Неспецифический язвенный колит поражает людей любого возраста, но чаще всего возникает в возрасте 15-30 лет или менее часто в возрасте 50-70 лет. Данное заболевание иногда возникает у детей и подростков. Неспецифический язвенный колит в равной степени поражает мужчин и женщин и, по-видимому, является наследственным заболеванием в некоторых семьях.
Кроме того, важно отметить, что примерно у 5 процентов людей неспецифический язвенный колит является причиной возникновения рака ободочной кишки. Риск возникновения рака повышается в зависимости от продолжительности и степени поражения ободочной кишки. Например, если поражены только нижний отдел ободочной кишки и прямая кишка, риск возникновения рака не превышает среднего показателя. Однако при поражении всей ободочной кишки риск возникновения рака может в 32 раза превышать средний показатель. Таким образом, вполне возможно, что лекарственные средства, пригодные для лечения IBD, могут быть также использованы для профилактики рака ободочной кишки.
Патогенез IBD, по-видимому, определяется многофакторными взаимодействиями генетических факторов, иммунологических факторов и факторов окружающей среды. Недавно полученные данные позволяют предположить, что патологическая активация иммунной системы слизистой оболочки под воздействием антигенов является главным фактором патогенеза IBD.
Вслед за представлением антигена при возникновении воспалительного процесса сразу же происходит образование цитокинов, мелких пептидных молекул гликопротеина, которые направляют сигналы в разные популяции клеток, определяющие направление последующих иммунной и воспалительной реакций. Провоспалительные цитокины включают интерлейкин (IL)-1, IL-6, IL-8 и альфа-фактор некроза опухолей (TNF-α). Макрофаги являются основным источником цитокинов, при этом эпителиальные клетки также способны продуцировать ряд указанных пептидных факторов.
Хелперные Т-клетки (Th) являются еще одним важным источником цитокинов. Th1-клетки, ассоциированные с клеточноопосредованной иммунной реакцией, продуцируют IL-2, гамма-интерферон (IFN-γ) и TNF-α. Ключевой фактор транскрипции, участвующий в регулировании воспаления, NFkB, который специфически определяет патогенез IBD, регулирует количество цитокинов, продуцируемых Th1-клетками (см. публикацию Neurath et al. (1996) Nature Med. 2: 998-1004). Th2-клетки усиливают синтез антител В-клетками и продуцируют IL-4, IL-5, IL-6 и IL-10.
Считается, что хемокины также способствуют патогенезу колита. Хемокины являются провоспалительными белками, которые участвуют в иммунных и воспалительных реакциях в результате хемоаттракции и активации лейкоцитов. Например, RANTES является С-С хемокином, который стимулирует рекрутинг и активацию клеток воспалительного инфильтрата, таких как моноциты, лимфоциты, мастоциты и эозинофилы. Недавно было установлено, что число RANTES увеличивается во время хронической фазы колита (см. публикацию Ajuebor et al. (2001) J Immunol. 166: 552-558).
Лечение неспецифического язвенного колита зависит от тяжести заболевания. Большинство людей проходит курс лекарственного лечения. В тяжелых случаях субъекту может понадобиться хирургическое вмешательство для удаления пораженной ободочной кишки.
Спастический колит (IBS) является распространенным, но плохо исследованным заболеванием, вызывающим разные кишечные симптомы, которые включают боль в брюшной полости, диарею и/или констипацию, вздутие, газообразование и спазмы. Так как указанные симптомы могут быть вызваны целым рядом разных заболеваний кишечника, IBS обычно диагностируют только после исключения более серьезного заболевания. Существует все больше данных, указывающих на роль воспаления в патогенезе IBS.
Проводимое лечение направлено на то, чтобы вызвать и сохранить ремиссию и улучшить качество жизни людей, страдающих IBD/IBS. В настоящее время существует несколько типов лекарственных средств, предназначенных для достижения указанной цели.
Аминосалицилаты, которые являются лекарственными средствами, содержащими 5-аминосалициловую кислоту (5-ASA; мезаламин) или 4-аминосалициловую кислоту (4-ASA), помогают лечить воспаление. Однако мезаламин и 4-ASA могут всасываться при прохождении по желудочно-кишечному тракту, что отрицательно влияет на количество мезаламина, достигающего нижнего отдела желудочно-кишечного тракта, в частности ободочной и прямой кишки. Поэтому были созданы разные препараты на основе мезаламина с целью защиты мезаламина при прохождении препарата по кишечнику и верхнему отделу желудочно-кишечного тракта.
Кроме того, было создано несколько пролекарств мезаламина, которые обеспечивают доставку мезаламина в ободочную кишку. Указанные пролекарства обычно в меньшей степени всасываются в кишечнике и верхнем отделе желудочно-кишечного тракта, благодаря чему они легче достигают ободочной кишки.
Сульфасалазин представляет собой комбинацию сульфапиридина и 5-ASA и используется для достижения и сохранения ремиссии. Сульфасалазин метаболизируется в организме с образованием 5-ASA и сульфапиридина. Сульфапиридиновый компонент доставляет противовоспалительную 5-ASA в тонкую кишку.
Однако сульфапиридин может вызывать побочные эффекты, такие как тошнота, рвота, жжение в сердце, диарея и головная боль. Указанные вредные побочные эффекты обычно приписывают активности сульфапиридина в желудочно-кишечном тракте, а также всасыванию организмом.
Другие лекарственные средства на основе 5-ASA, такие как олсалазин, ипсалазид и балсалазид, которые имеют разные носители, вызывают меньше побочных эффектов и могут быть использованы людьми, плохо переносящими сульфасалазин. В отличие от сульфасалазина распад указанных соединений 5-ASA в кишечнике может не вызывать образования нежелательных продуктов обмена веществ.
Как правило, соединения 5-ASA вводят перорально, с помощью клизмы или в суппозитории в зависимости от места воспаления в ободочной кишке. Большинство людей, страдающих легкой или умеренной формой неспецифического язвенного колита, сначала подвергают лечению указанной группой лекарственных средств. Однако такое лечение нельзя считать оптимальным главным образом из-за плохой эффективности данного лекарственного средства, которое, кроме того, характеризуется плохой совместимостью с организмом субъекта.
Другими применяемыми лекарственными средствами являются кортикостероиды, такие как преднизон, гидрокортизон, будезонид и т.д., и иммуномодуляторы, такие как азатиоприн и 6-меркаптопурин (6-МР). Указанные лекарственные средства могут вызывать побочные эффекты, такие как гипертензия, повышенный риск возникновения инфекций и т.д.
Сульфасалазин, олсалазид и балсалазид являются производными мезаламина, в которых носитель, не являющийся мезаламином, связан с мезаламином при помощи диазосвязи. Указанные пролекарства плохо всасываются в кишечнике и верхнем отделе желудочно-кишечного тракта и поэтому могут достигать ободочной кишки, где они расщепляются азоредуктазами микрофлоры кишечника с высвобождением мезаламина и носителя непосредственно в ободочную кишку.
Другие производные мезаламина включают носитель, присоединенный к мезаламину при помощи карбоксильных и гидроксильных функциональных групп молекулы. Наряду с вышеуказанными лекарственными средствами в научной литературе были описаны препараты на основе сложных эфиров или амидов аминокислот, таких как L-серин и L-глицин, или с добавлением других биологических соединений, таких как таурин. Активность указанных пролекарств основана на действии карбоксипептидаз и аминопептидаз А, вызывающих высвобождение мезаламина (R. Pellicciari et al. (1993) Journal of Medicinal Chemistry, 36, pg. 4201-7).
Большинство ранее известных носителей, присоединенных к мезаламину, являются инертными. Таким образом, к 5-ASA или 4-ASA желательно присоединить носители, которые также являются биологически активными и пригодными для лечения IBD/IBS.
Часть молекулы, способная высвобождать сероводород (H2S) в ткань, обычно связана при помощи азогруппы, сложноэфирной, ангидридной, сложнотиоэфирной или амидной связи с молекулой 4- или 5-аминосалициловой кислоты (4- или 5-ASA) с образованием производного 4- или 5-ASA по настоящему изобретению. Благодаря присоединению H2S-высвобождающей части молекулы к 4- или 5-ASA при помощи ковалентной связи производные по настоящему изобретению могут действовать в качестве пролекарств, которые плохо всасываются в кишечнике и верхнем отделе желудочно-кишечного тракта и, таким образом, могут легко достигать ободочной кишки.
В научной литературе хорошо описаны противовоспалительные свойства 4- или 5-ASA и их применение для лечения неспецифического язвенного колита. 4- или 5-ASA уменьшает воспаление в кишечнике, диарею (частый стул), ректальное кровотечение и боль в желудке. Недавно было установлено, что H2S действует в качестве нейромодулятора и оказывает противовоспалительное действие. Кроме того, установлено, что H2S модулирует ноцицепцию при расширении прямой и ободочной кишки (см. публикацию Distrutti et al. (2005) Evidence That Hydrogen Sulfide Exerts Antinociceptive Effects in the Gastrointestinal Tract by Activating KATP Channels. J. Pharm. and Exp. Ther. 316:325-335, которая включена в настоящее описание изобретения в качестве ссылки). И наконец, как известно, H2S является релаксантом гладких мышц в тканях кишечника (см. публикацию Teague, B. et al. (2002) The Smooth Muscle Relaxant effect of Hydrogen Sulfide In Vitro: Evidence for a Physiological Role to Control Intestinal Contractility. Br. J. Pharmacol. 137: 139-145, которая включена в настоящее описание изобретения в качестве ссылки).
Весьма удивительным является то, что H2S-высвобождающая часть, связанная ковалентной связью с 4- или 5-ASA, характеризуется лучшей способностью высвобождения H2S по сравнению с отдельно используемой частью H2S. Из вышеизложенного следует, что H2S может высвобождаться как в период времени, когда H2S-высвобождающая часть связана ковалентной связью с 4- или 5-ASA, так и после отщепления H2S-высвобождающей части от 4- или 5-ASA в результате гидролиза или расщепления разными ферментами, присутствующими в желудочно-кишечном тракте, с высвобождением двух активных ингредиентов, а именно 4- или 5-аминосалициловой кислоты и H2S-высвобождащей части, которые оказывают дальнейшее действие.
Производные по настоящему изобретению оказывают более эффективное действие по сравнению с отдельно используемой 4- или 5-ASA, отдельно используемой H2S-высвобождающей частью и смесью 4- или 5-ASA и H2S-высвобождающей части в отношении уменьшения воспаления, диареи и скрытой крови в кале у субъектов, страдающих колитом, и ослабления висцеральной боли, обусловленной расширением прямой и ободочной кишки. Кроме того, производные по настоящему изобретению уменьшают также уровни мРНК циклооксигеназы (СОХ)-1, СОХ-2, конститутивной эндотелиальной синтазы оксида азота (eNOS) и индуцибельной NOS (iNOS), которые, как считается, являются ферментами, опосредующими воспаление.
Таким образом, одним объектом настоящего изобретения являются производные по настоящему изобретению, предназначенные для лечения воспалительного заболевания желудочно-кишечного (GI) тракта, такого как воспалительное заболевание кишечника (IBD) и спастический колит (IBS). Не ограничивая себя какой-либо теорией, можно отметить, что сероводород, высвобождаемый из части молекулы, высвобождающей сероводород, оказывает противовоспалительное действие в результате ингибирования NFkB, фактора транскрипции, регулирующего экспрессию нескольких провоспалительных генов. Кроме того, считается, что антиноцицептивное действие H2S может затрагивать АТР-чувствительные К+ (КАТР) каналы.
Другим объектом настоящего изобретения являются производные 4- или 5-ASA по настоящему изобретению, которые эффективно снижают жизнеспособность раковых клеток НТ-29 в ободочной кишке человека и, таким образом, позволяют предотвращать и/или лечить рак ободочной кишки.
Соединения по настоящему изобретению имеют нижеследующую общую формулу,

в которой:
А означает
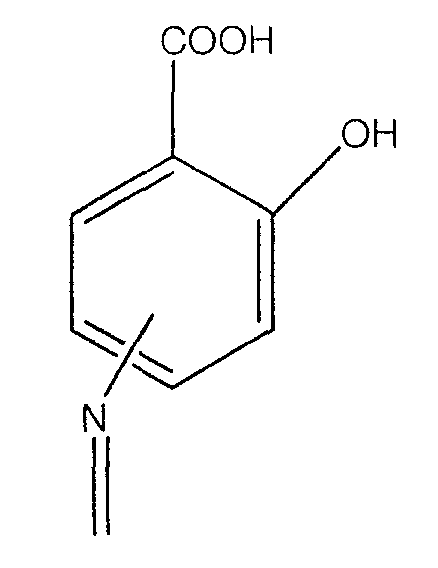
где -N= находится в положении 4 или 5,

где -NH находится в положении 4 или 5,

где -NH2 находится в положении 4 или 5, или

где -NH2 находится в положении 4 или 5;
L означает О, О-С=О, S, N или ковалентную связь и образует сложноэфирную связь, ангидридную связь, сложнотиоэфирную связь, амидную связь или связующую азогруппу; и
R означает высвобождающую сероводород часть молекулы, которая высвобождает H2S в ткань. Очевидно, что в настоящем изобретении может быть использована любая нетоксичная, эффективно высвобождающая сероводород часть молекулы, которая высвобождает H2S в присутствии ткани.
В предпочтительном варианте осуществления изобретения R выбирают из группы, включающей:
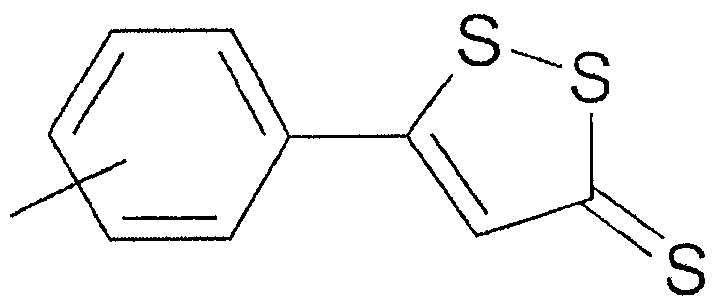





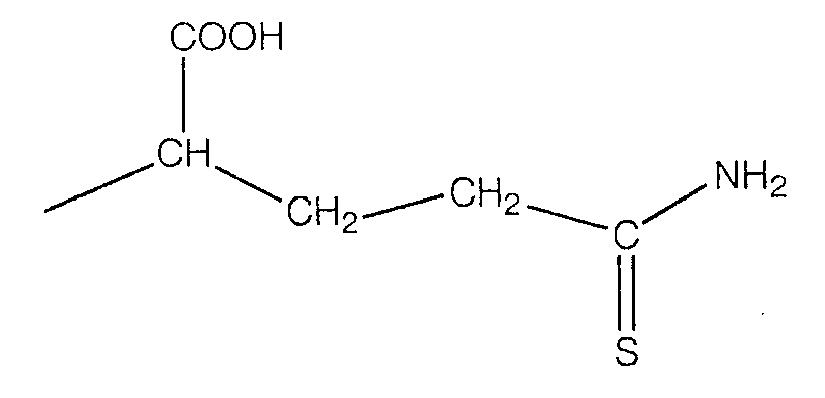

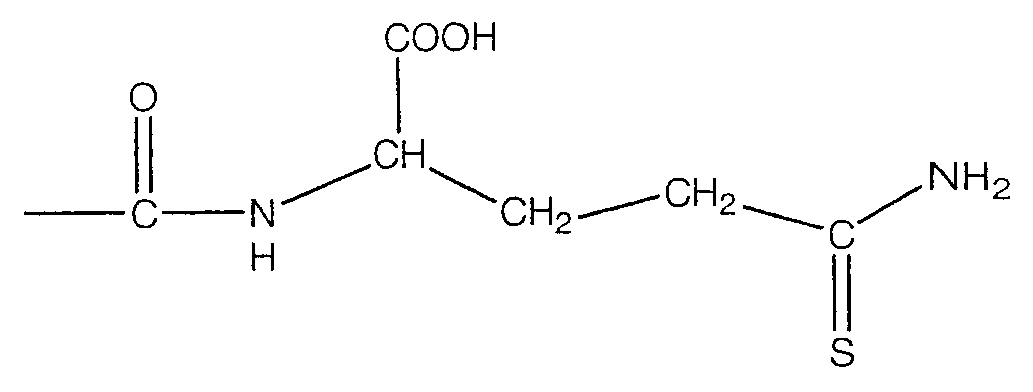
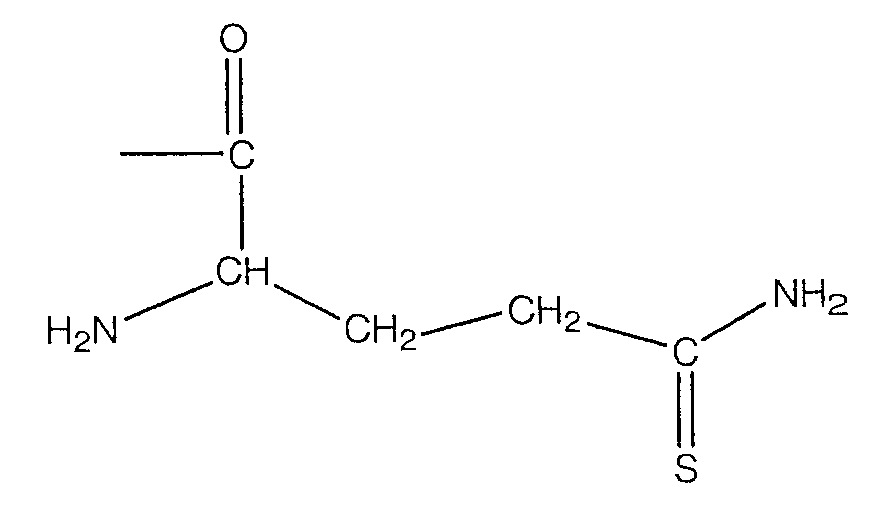

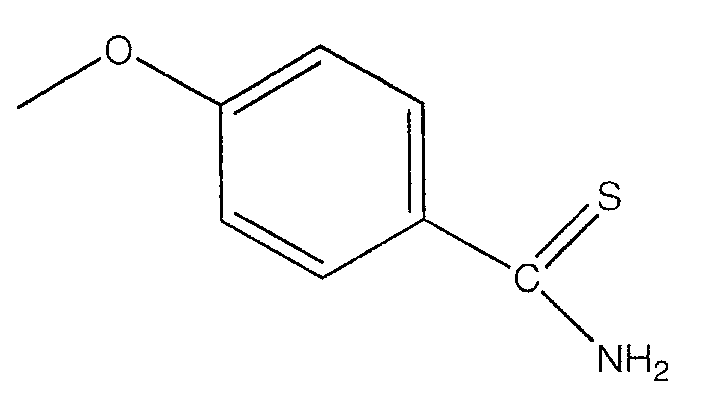
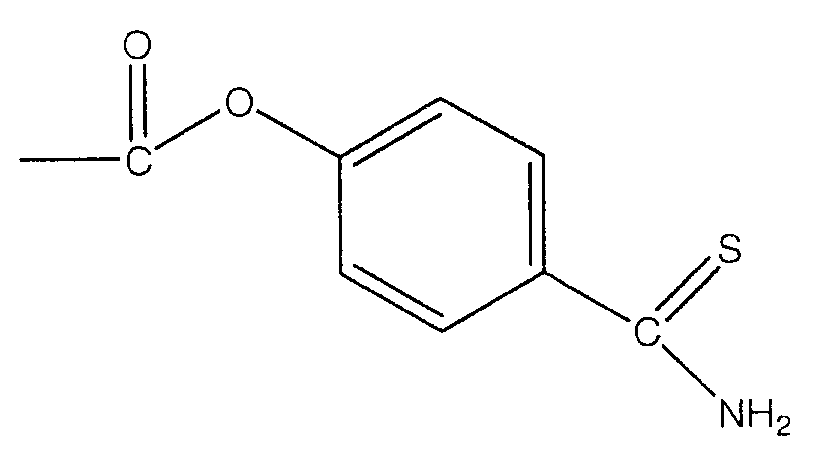
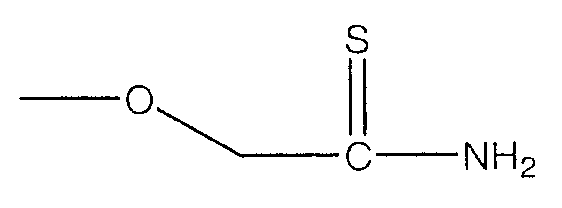

и

Все вышеуказанные части молекулы высвобождают H2S в биологические ткани; однако механизм действия большинства H2S-высвобождающих частей отличается от N-ацетилцистеина. Хорошо известно, что N-ацетилцистеин превращается в цистеин в разных тканях и в результате метаболизма цистеина in vivo образуется H2S. H2S продуцируют главным образом два типа пиридоксиль-5'-фосфат-зависимых ферментов, опосредующих метаболизм L-цистеина, а именно цистатион-γ-лиаза и цистатион-β-синтаза (см. публикацию Fujii et al. (2005) Hydrogen Sulfide as a Endogenous Modulator of Biliary Bicarbonate Excretion in the Rat Liver. Antioxid. Redox. Signal. 7:788-794, которая включена в настоящее описание изобретения в качестве ссылки).
В объем настоящего изобретения входят также фармацевтически приемлемые соли, такие как, например, соли щелочных металлов и щелочноземельных металлов, нетоксичных аминов и аминокислот. Предпочтительными солями являются соли аргинина и агматина. В объем настоящего изобретения входят также фармацевтически приемлемые кислотно-аддитивные соли.
Другим объектом настоящего изобретения является фармацевтическая композиция, содержащая соединения по настоящему изобретению и фармацевтически приемлемый наполнитель или носитель, которая предназначена, в частности, для лечения воспалительного заболевания желудочно-кишечного тракта.
Другие варианты осуществления настоящего изобретения относятся к способам лечения воспалительного заболевания желудочно-кишечного тракта, такого как воспалительное заболевание кишечника (IBD) и спастический колит (IBS), у субъекта, нуждающегося в таком лечении, которые включают введение указанному субъекту эффективного количества производных 4- или 5-ASA и их солей. Кроме того, настоящее изобретение относится к способам лечения или профилактики рака ободочной кишки у нуждающегося субъекта, которые включают введение указанному субъекту эффективного количества производных 4- или 5-ASA и их солей.
Другой вариант осуществления настоящего изобретения относится к применению производных 4- или 5-ASA и их солей по настоящему изобретению для приготовления лекарственного средства, предназначенного для лечения воспалительного заболевания желудочно-кишечного тракта. Настоящее изобретение относится также к применению производных 4- или 5-ASA и их солей для лечения воспалительного заболевания желудочно-кишечного тракта.
Предпочтительные соединения имеют нижеследующие формулы:
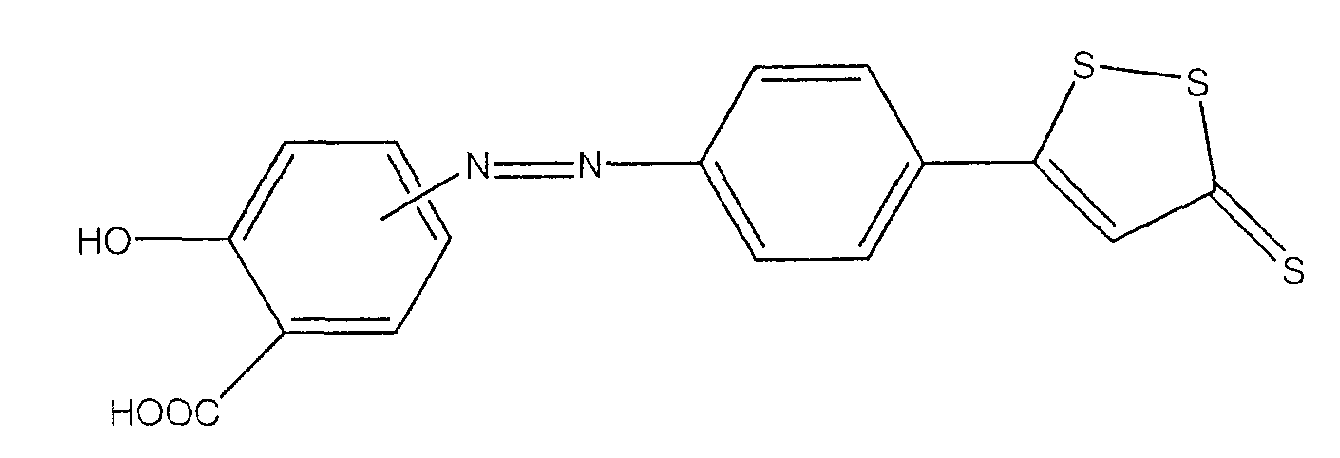
2-гидрокси-4- или 5-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилазо]-бензойная кислота (II),
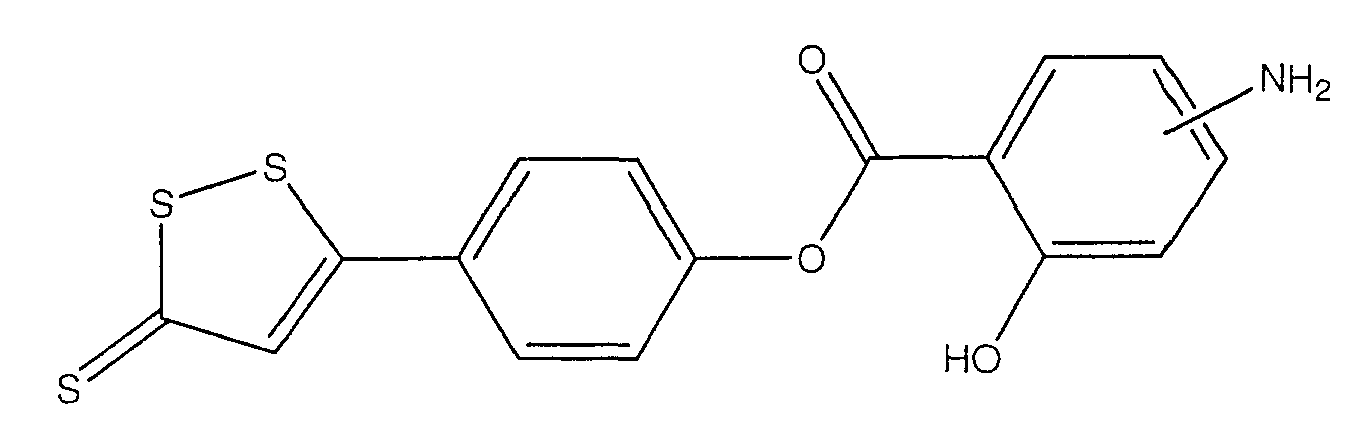
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фениловый эфир 4- или 5-амино-2-гидроксибензойной кислоты (III),
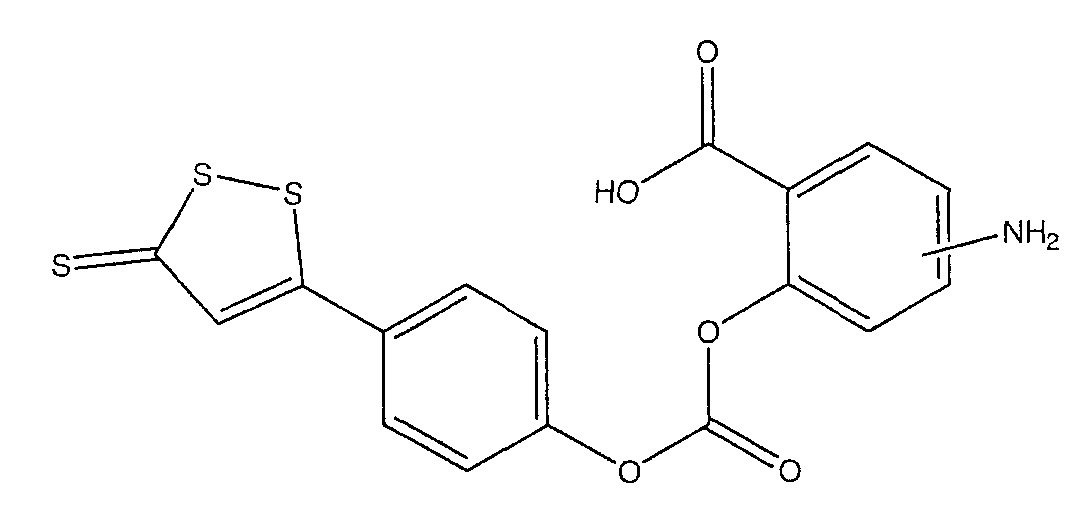
4- или 5-амино-2-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбонилокси]бензойная кислота (IV),
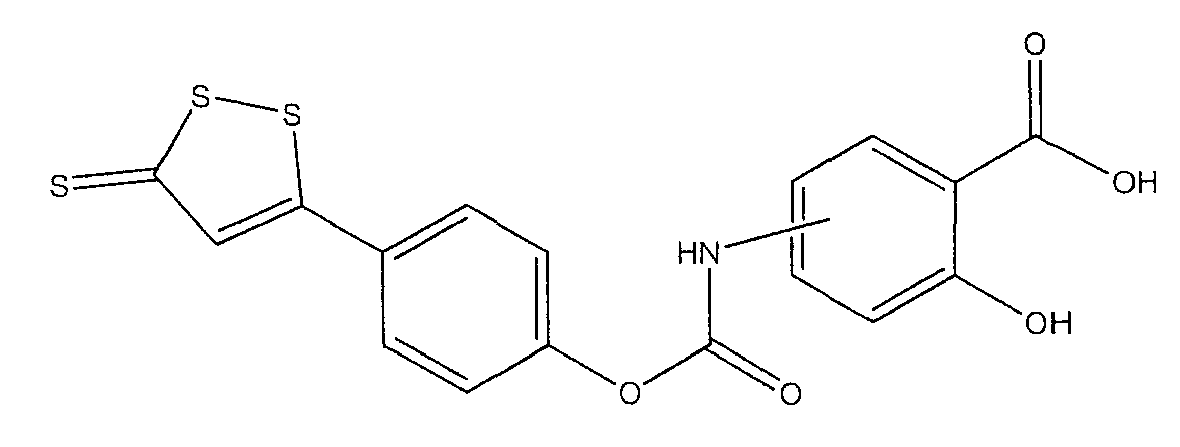
2-гидрокси-4- или 5-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбониламино]бензойная кислота (V),
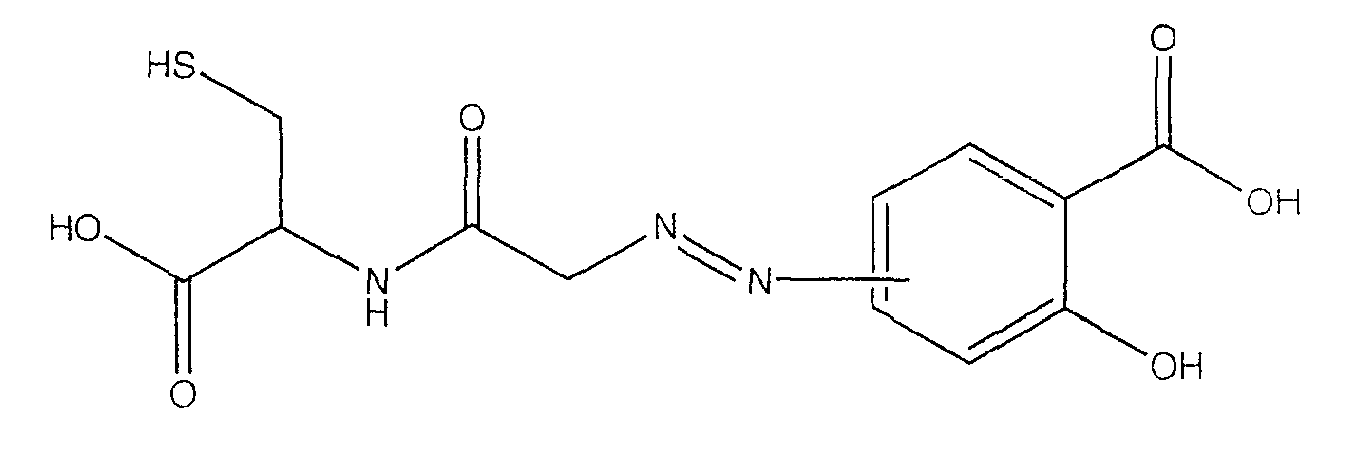
4- или 5-{[(1-карбокси-2-меркаптоэтилкарбамоил}метил]азогидроксибензойная кислота (VI),

(1-карбокси-2-меркаптоэтилкарбамоил)метиловый эфир 4- или 5-амино-2-гидроксибензойной кислоты (VII),
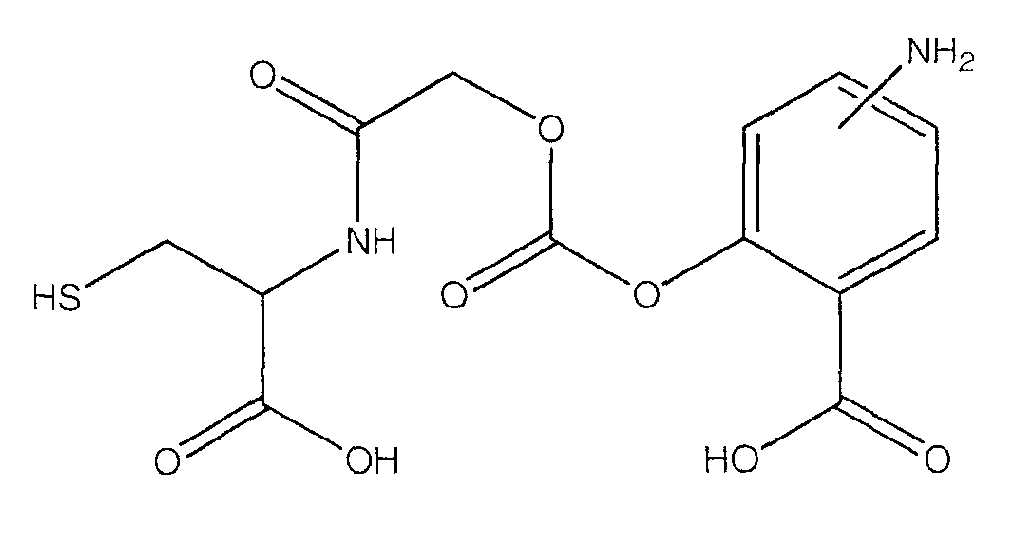
4- или 5-амино-2-[(1-карбокси-2-меркаптоэтилкарбамоил)метоксикарбонилокси]бензойная кислота (VIII),
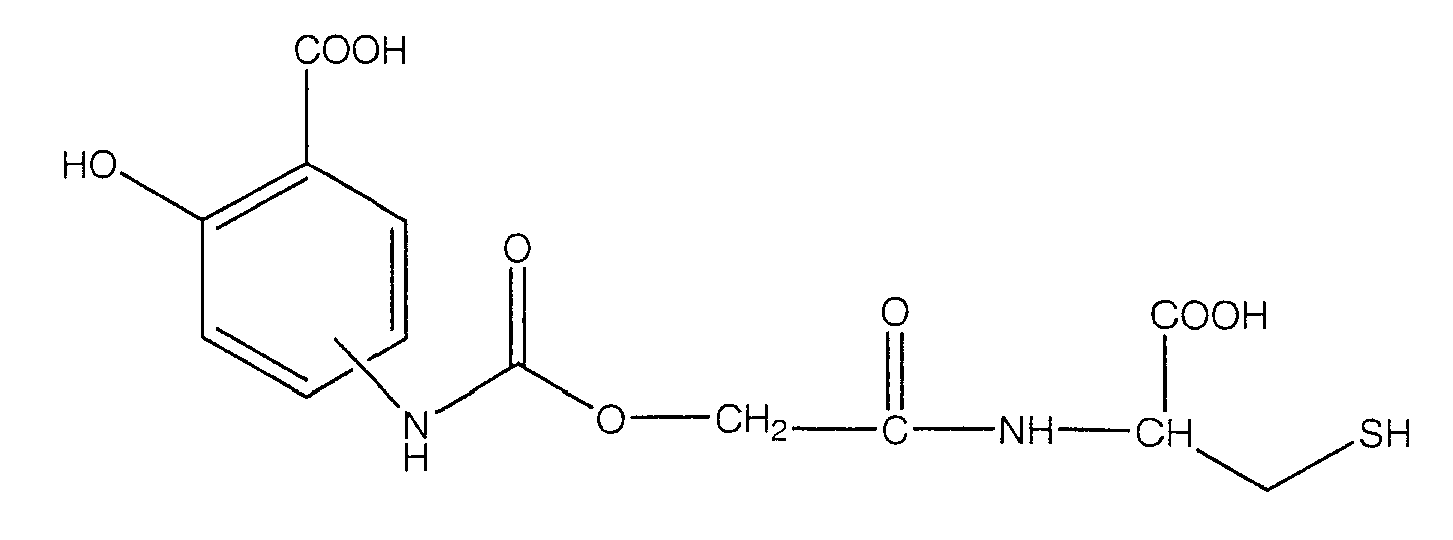
4- или 5-[(1-карбокси-2-меркаптоэтилкарбамоил)метоксикарбониламино]-2-гидроксибензойная кислота (IX),
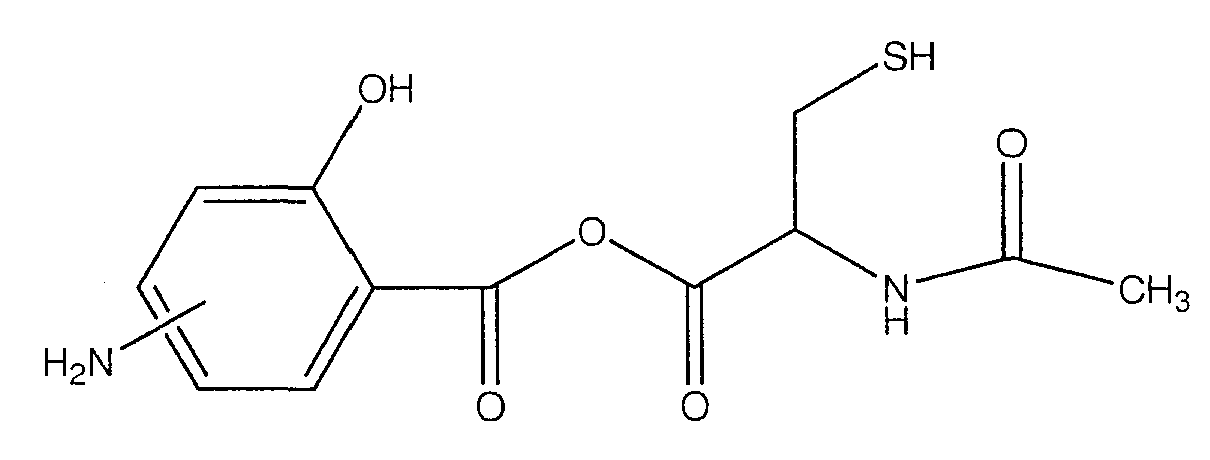
ангидрид 4- или 5-амино-2-гидроксибензойной кислоты с N-ацетилцистеином (Х),
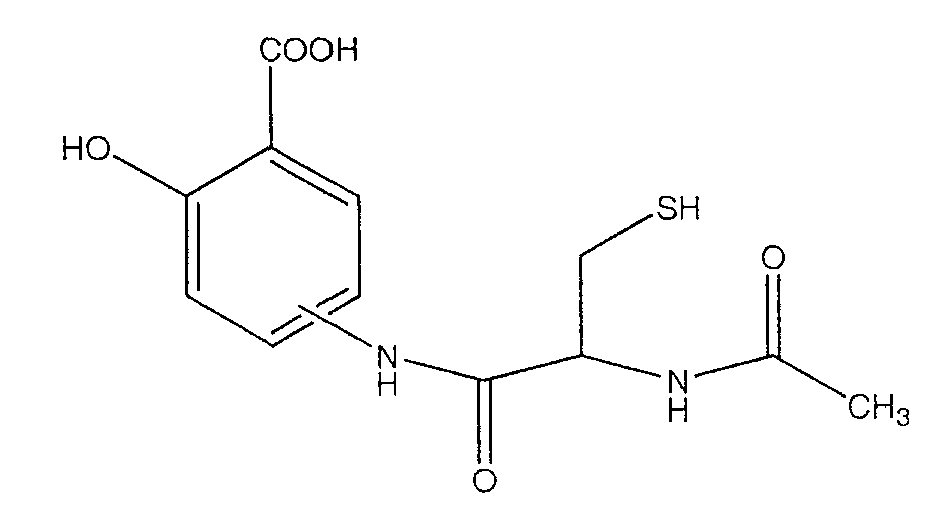
4- или 5-(2-ацетиламино-3-меркаптопропиониламино)-2-гидроксибензойная кислота (XI),
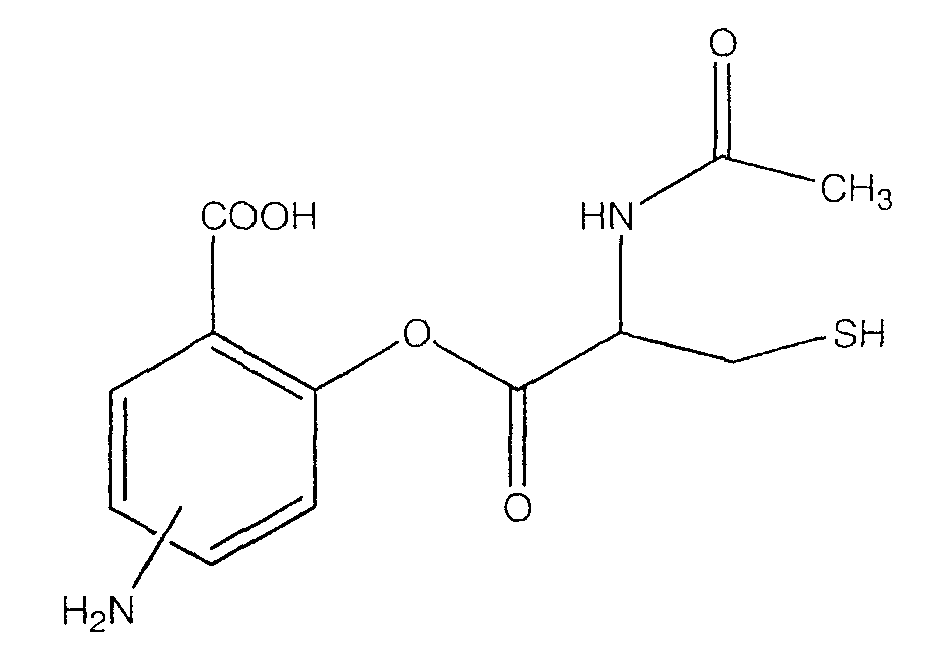
2-(2-ацетиламино-3-меркаптопропионилокси)-4- или 5-аминобензойная кислота (XII),

2-гидрокси-4- или 5-({4-[4-(4-метоксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксиметил}азо)бензойная кислота (XIII),

4- или 5-амино-2-{4-[4-(4-метоксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксиметоксикарбонилокси}бензойная кислота (XIV),
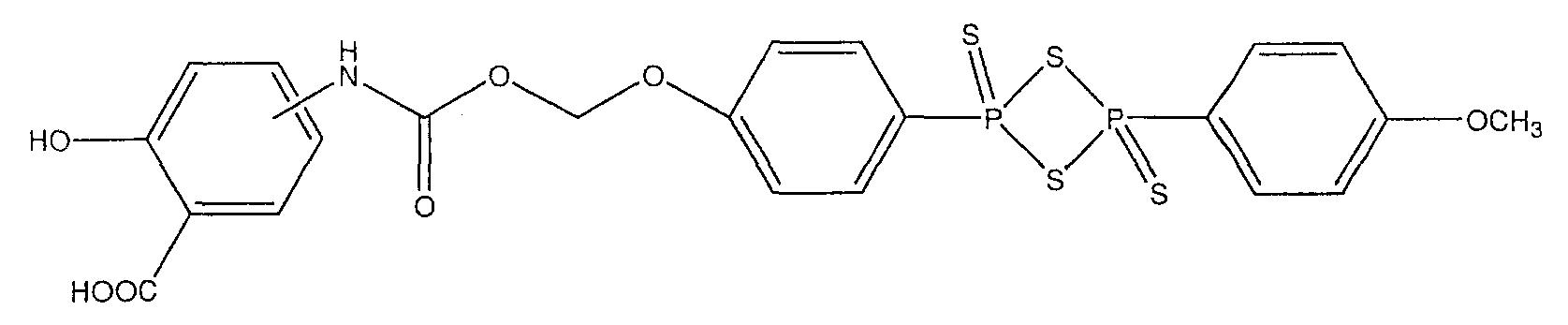
2-гидрокси-4- или 5-{4-[4-(4-метоксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксиметоксикарбониламино}бензойная кислота (XV),

4-[4-(4-метоксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксиметиловый эфир 4- или 5-амино-2-гидроксибензойной кислоты (XVI),
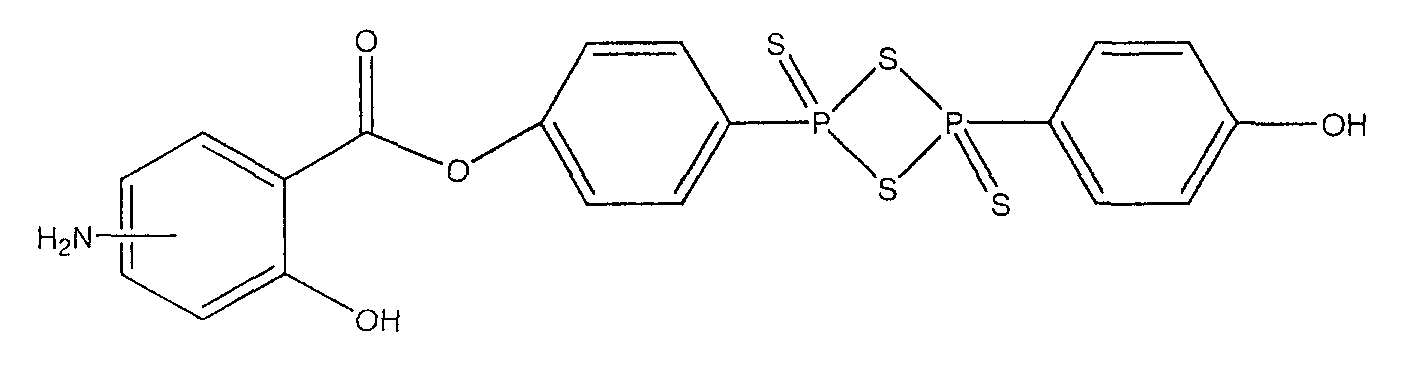
4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]фениловый эфир 4- или 5-амино-2-гидроксибензойной кислоты (XVII),
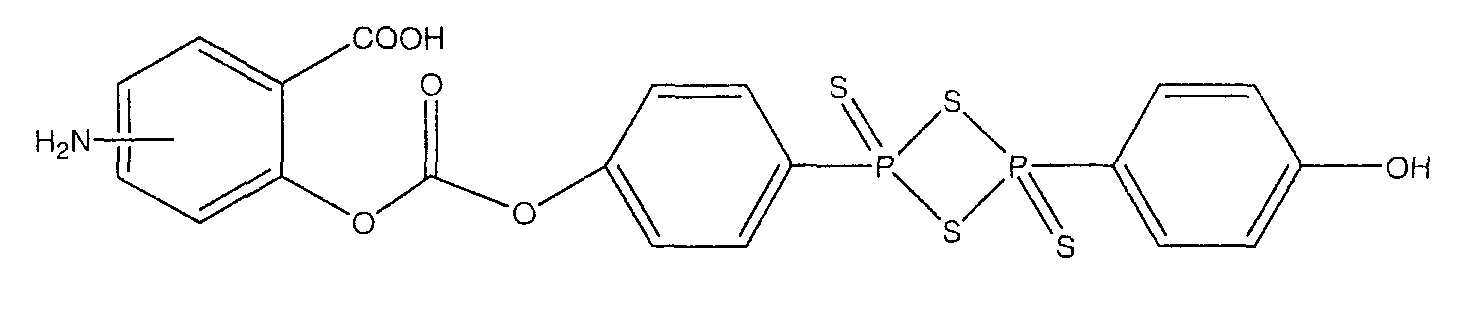
4- или 5-амино-2-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбонилокси}бензойная кислота (XVIII),

2-гидрокси-4- или 5-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбониламино}бензойная кислота (XIX),
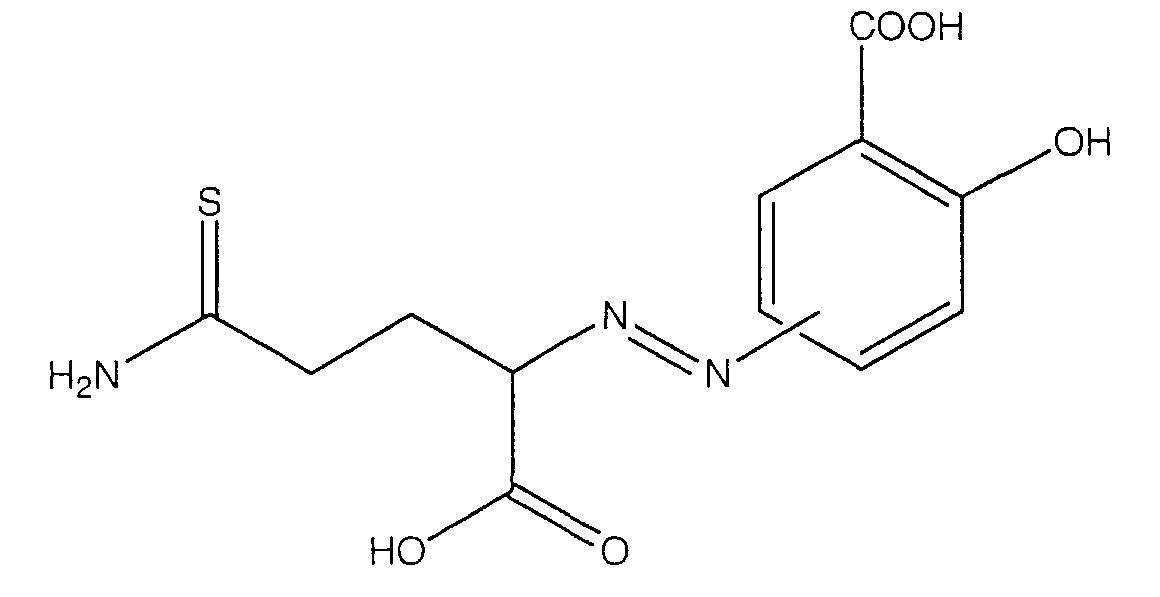
4- или 5-(1-карбокси-3-тиокарбамоилпропилазо)-2-гидроксибензойная кислота (ХХ),
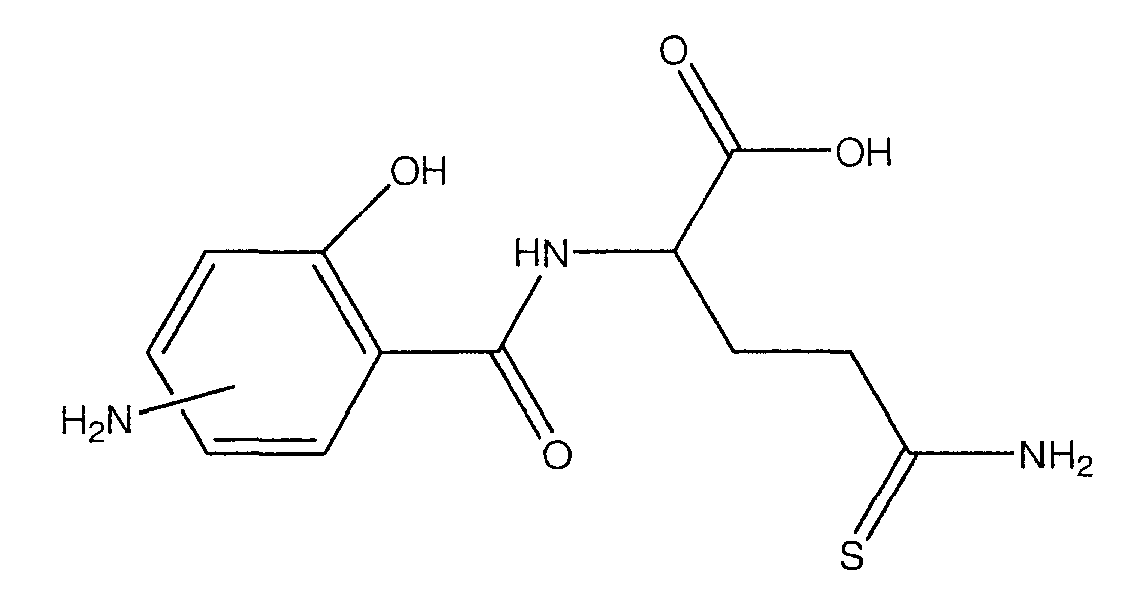
2-(4- или 5-амино-2-гидроксибензоиламино)-4-тиокарбамоилмасляная кислота (XXI),
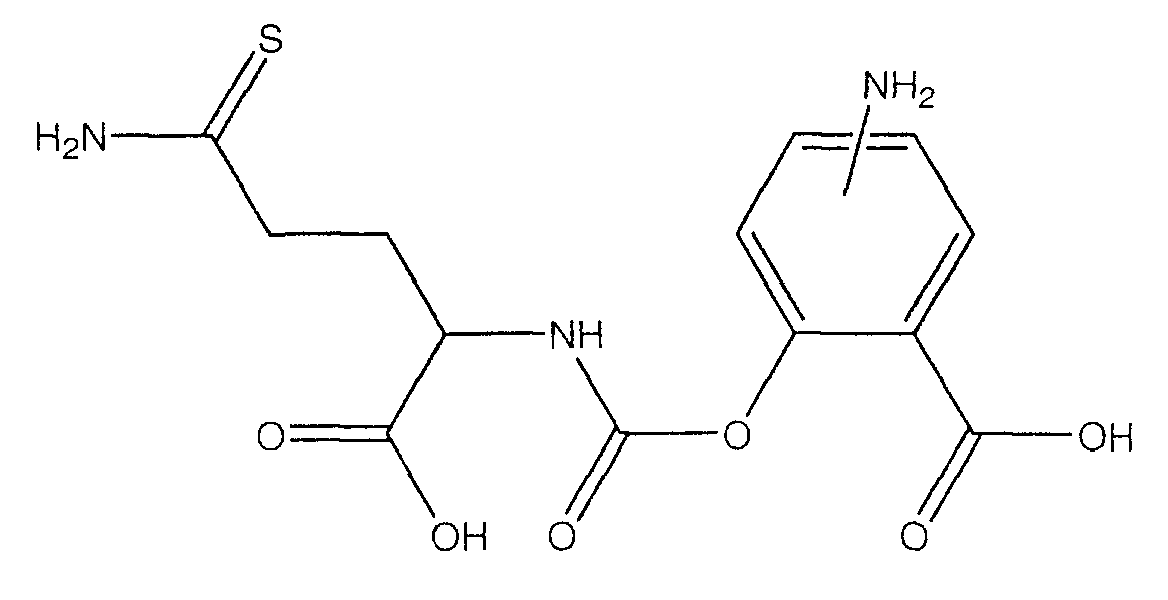
4- или 5-амино-2-(1-карбокси-3-тиокарбамоилпропилкарбамоилокси)бензойная кислота (XXII),
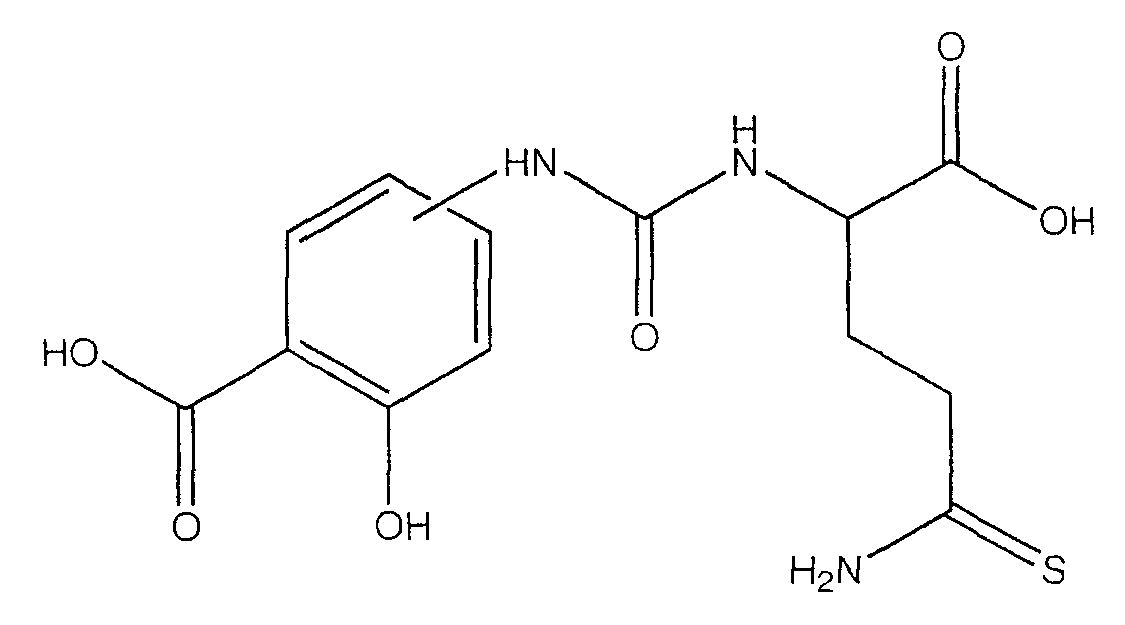
2-гидрокси-4- или 5-[3-(1-гидроксиметил-3-тиокарбамоилпропил)-уреидо]бензойная кислота (XXIII),

4- или 5-амино-2-(2-амино-4-тиокарбамоилбутирилокси)бензойная кислота (XXIV),

4- или 5-(2-амино-4-тиокарбамоилбутириламино)-2-гидроксибензойная кислота (XXV),
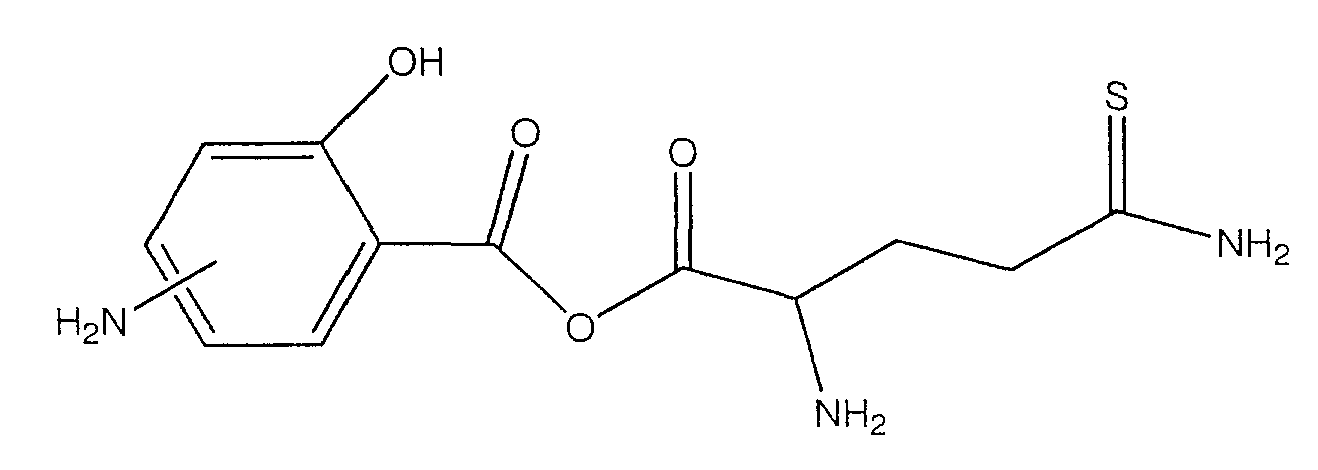
ангидрид 4- или 5-амино-2-гидроксибензойной кислоты с 2-амино-4-тиокарбамоилмасляной кислотой (XXVI),

4-тиокарбамоилфенил-4- или 5-амино-2-гидроксибензоат (XXVII),

4- или 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойная кислота (XXVIII),

2-гидрокси-4- или 5-(4-тиокарбамоилфеноксикарбониламино)бензойная кислота (XXIX),

тиокарбамоилметиловый эфир 4- или 5-амино-2-гидроксибензойной кислоты (ХХХ),
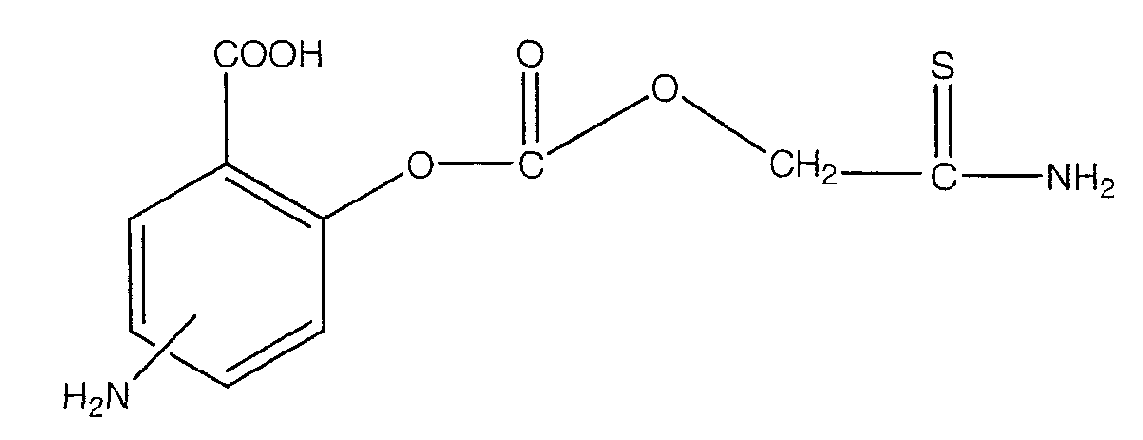
4- или 5-амино-2-тиокарбамоилметоксикарбонилоксибензойная кислота (XXXI),

2-гидрокси-4- или 5-тиокарбамоилметоксикарбониламинобензойная кислота (XXXII),
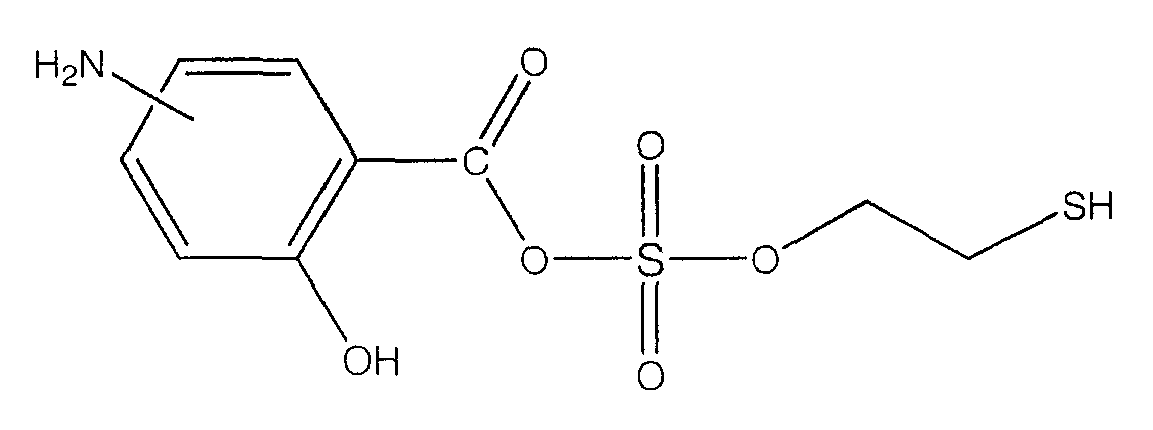
ангидрид 4- или 5-амино-2-гидроксибензойной кислоты с моно(2-меркаптоэтиловым) эфиром серной кислоты (XXXIII),

4- или 5-амино-2-(2-меркаптоэтоксисульфонилокси)бензойная кислота (XXXIV) и
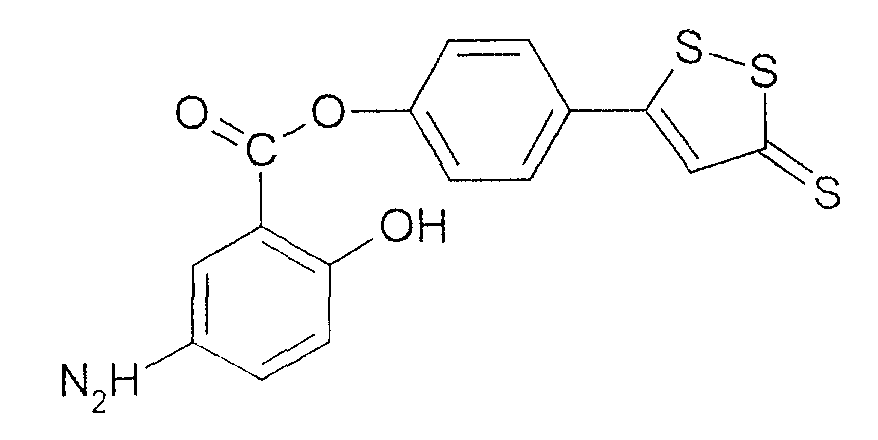
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фениловый эфир 5-амино-2-гидроксибензойной кислоты (XXXV).
Наиболее предпочтительными соединениями являются нижеследующие соединения:

4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фениловый эфир 5-амино-2-гидроксибензойной кислоты (XXXV),
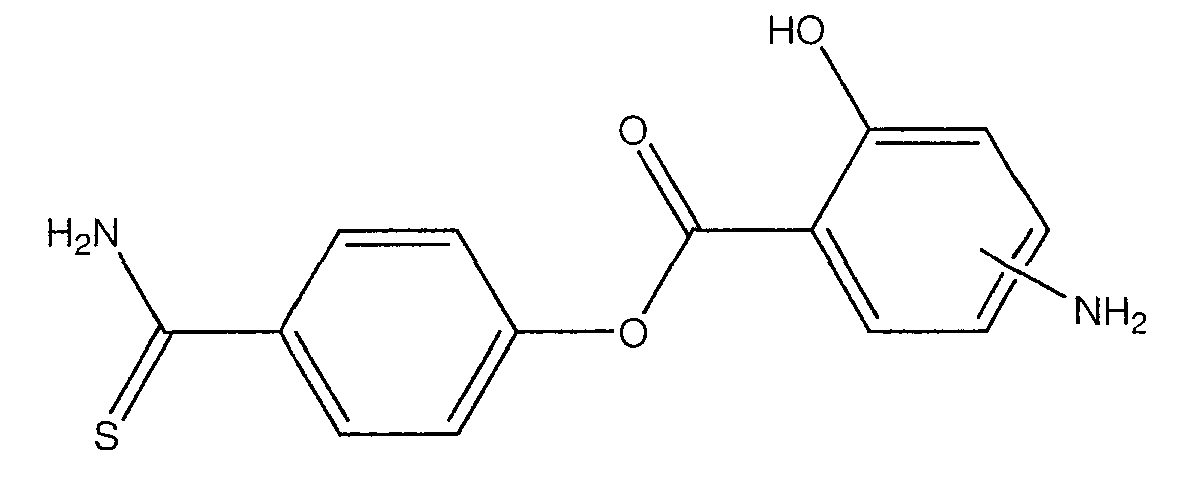
4-тиокарбамоилфенил-4- или 5-амино-2-гидроксибензоат (XXVII) и
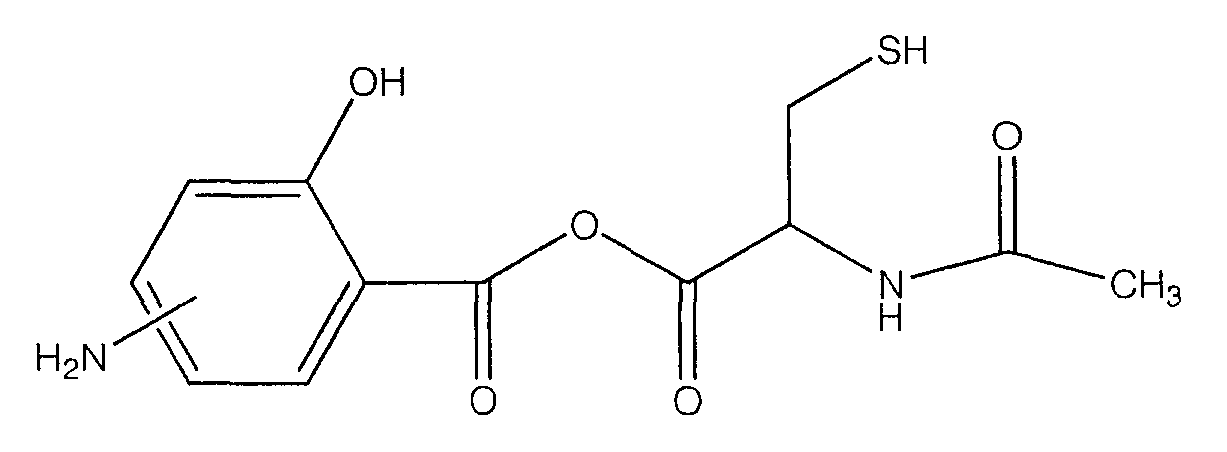
ангидрид 4- или 5-амино-2-гидроксибензойной кислоты с N-ацетилцистеином (Х).
Краткое описание чертежей
На фиг.1 показана оценка интенсивности заболевания у мышей, страдающих TNBS-индуцированным колитом, после лечения возрастающими дозами мезаламина и соединения XXXV по настоящему изобретению.
На фиг.2 показана активность миелопероксидазы (МРО) у мышей, страдающих TNBS-индуцированным колитом, после лечения возрастающими дозами мезаламина и соединения XXXV по настоящему изобретению.
На фиг.3 показана оценка интенсивности заболевания у мышей, страдающих TNBS-индуцированным колитом, после лечения соединением XXXV, отдельно используемым мезаламином, отдельно используемым 5-п-гидроксифенил-1,2-дитион-3-тионом (ADT-OH) и смесью мезаламина и ADT-OH.
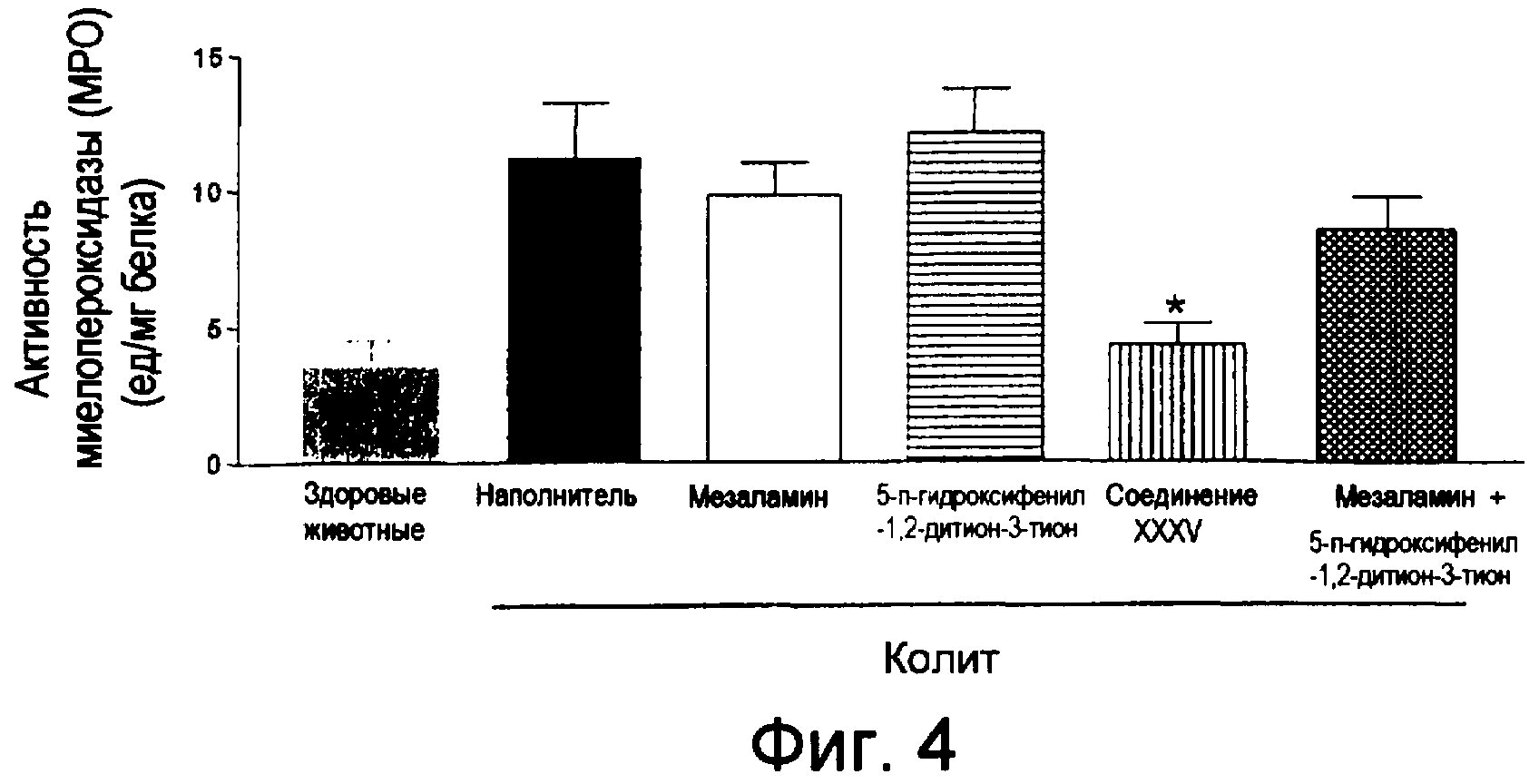
На фиг.4 показана активность миелопероксидазы (МРО) у мышей, страдающих TNBS-индуцированным колитом, после лечения соединением XXXV, отдельно используемым мезаламином, отдельно используемым ADT-OH и смесью мезаламина и ADT-OH.
На фиг.5 показана оценка интенсивности заболевания у мышей, страдающих TNBS-индуцированным колитом, после лечения соединением XXVII, отдельно используемым мезаламином, отдельно используемым 4-гидрокситиобензамидом (4-НТВ) и смесью мезаламина и 4-НТВ.
На фиг.6 показана активность миелопероксидазы (МРО) у мышей, страдающих TNBS-индуцированным колитом, после лечения соединением XXVII, отдельно используемым мезаламином, отдельно используемым 4-НТВ и смесью мезаламина и 4-НТВ.
На фиг.7 показана активность миелопероксидазы (МРО) у мышей, страдающих TNBS-индуцированным колитом, после лечения 50 мг/кг мезаламина и эквимолярной дозой соединения XXXV по настоящему изобретению.
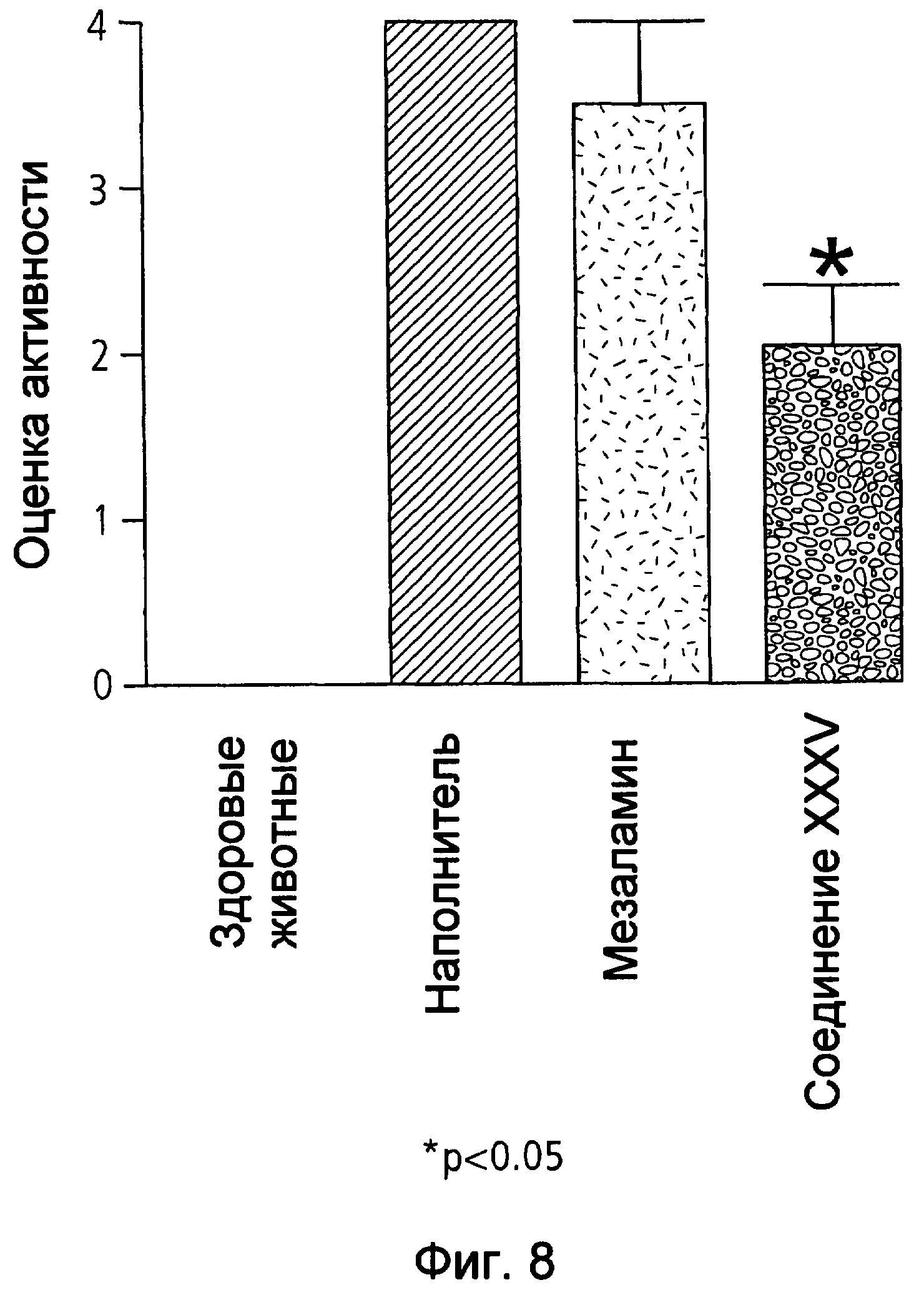
На фиг.8 показана оценка интенсивности заболевания у мышей, страдающих TNBS-индуцированным колитом, после лечения наполнителем (1% СМС), 50 мг/кг мезаламина и эквимолярной дозой соединения XXXV по настоящему изобретению.
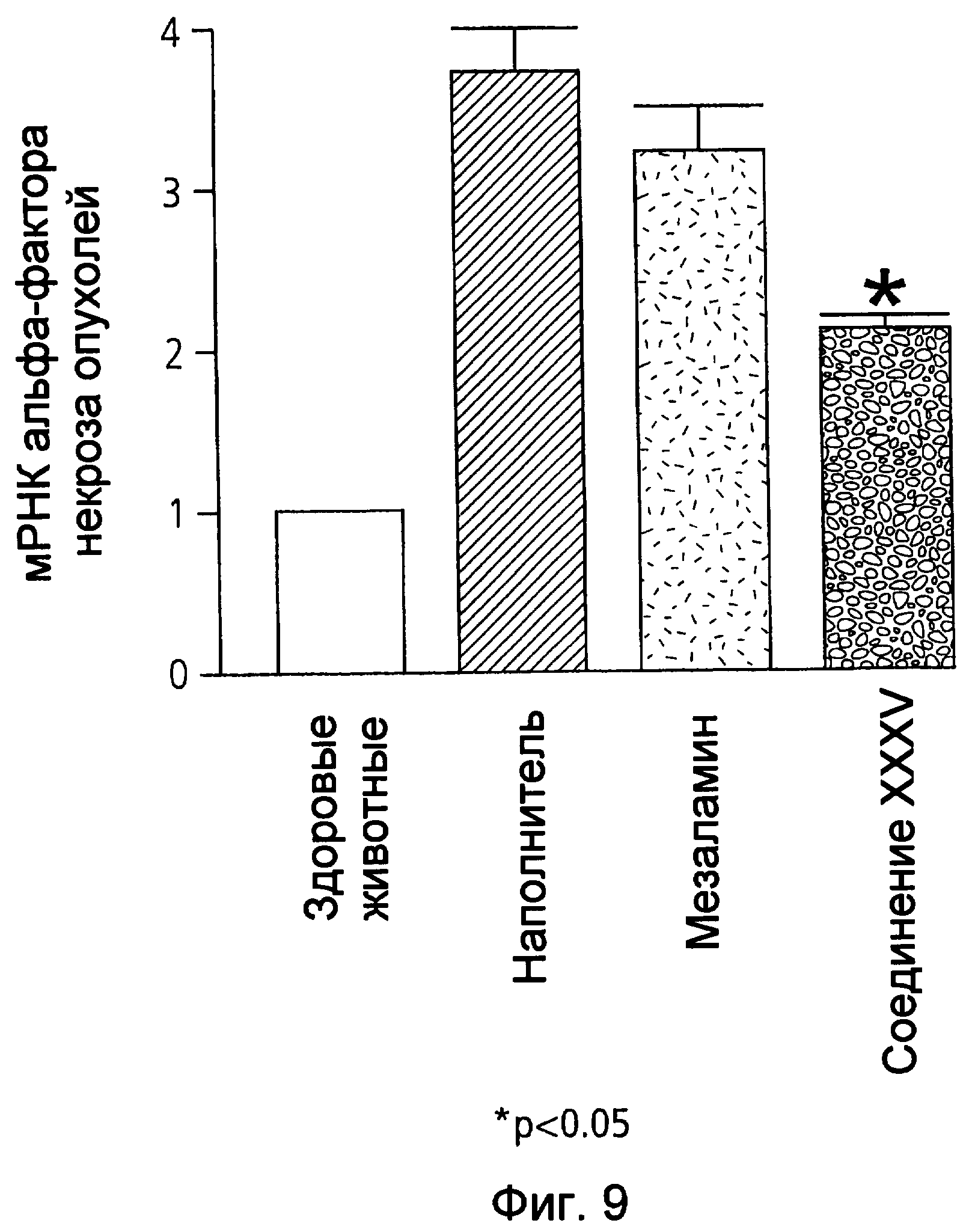
На фиг.9 показана экспрессия мРНК фактора некроза опухолей (TNF-α) в ободочной кишке у мышей, страдающих TNBS-индуцированным колитом, после лечения наполнителем (1% СМС), 50 мг/кг мезаламина и эквимолярной дозой соединения XXXV по настоящему изобретению.
На фиг.10 показана экспрессия мРНК гамма-интерферона (IFN-γ) у мышей, страдающих TNBS-индуцированным колитом, после лечения наполнителем (1% СМС), 50 мг/кг мезаламина и эквимолярной дозой соединения XXXV по настоящему изобретению.
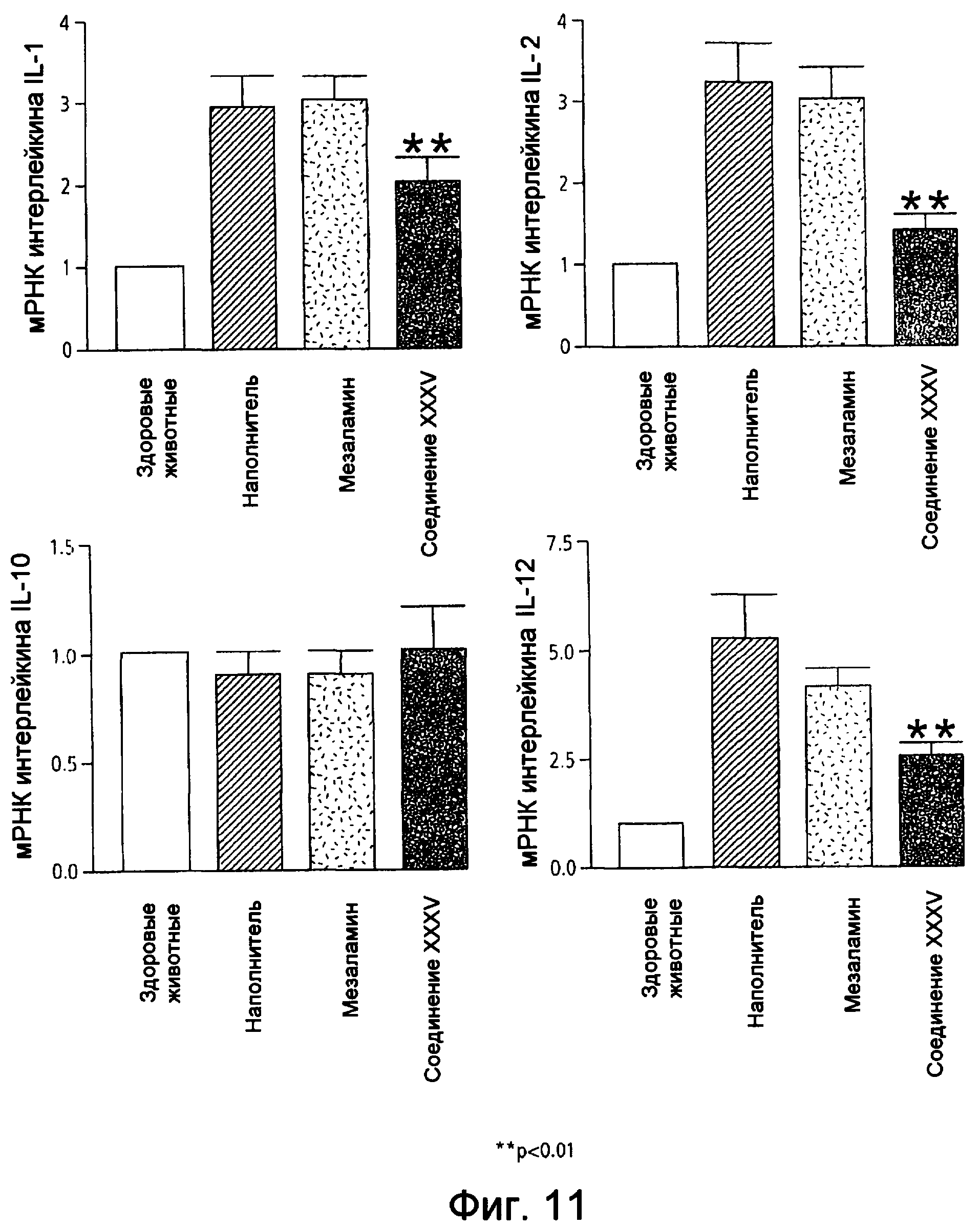
На фиг.11 показана экспрессия мРНК интерлейкина (IL), а именно IL-1, -2, -10 и -12, у мышей, страдающих TNBS-индуцированным колитом, после лечения наполнителем (1% СМС), 50 мг/кг мезаламина и эквимолярной дозой соединения XXXV по настоящему изобретению.
На фиг.12 показаны уровни мРНК RANTES в ободочной кишке у мышей, страдающих TNBS-индуцированным колитом, после лечения наполнителем (1% СМС), 50 мг/кг мезаламина и эквимолярной дозой соединения XXXV по настоящему изобретению.
На фиг.13 показана экспрессия мРНК СОХ-1 и СОХ-2 в ободочной кишке у мышей, страдающих TNBS-индуцированным колитом, после лечения наполнителем (1% СМС), 50 мг/кг мезаламина и эквимолярной дозой соединения XXXV по настоящему изобретению.
На фиг.14 показана экспрессия мРНК eNOS и iNOS в ободочной кишке у мышей, страдающих TNBS-индуцированным колитом, после лечения наполнителем (1% СМС), 50 мг/кг мезаламина и эквимолярной дозой соединения XXXV по настоящему изобретению.
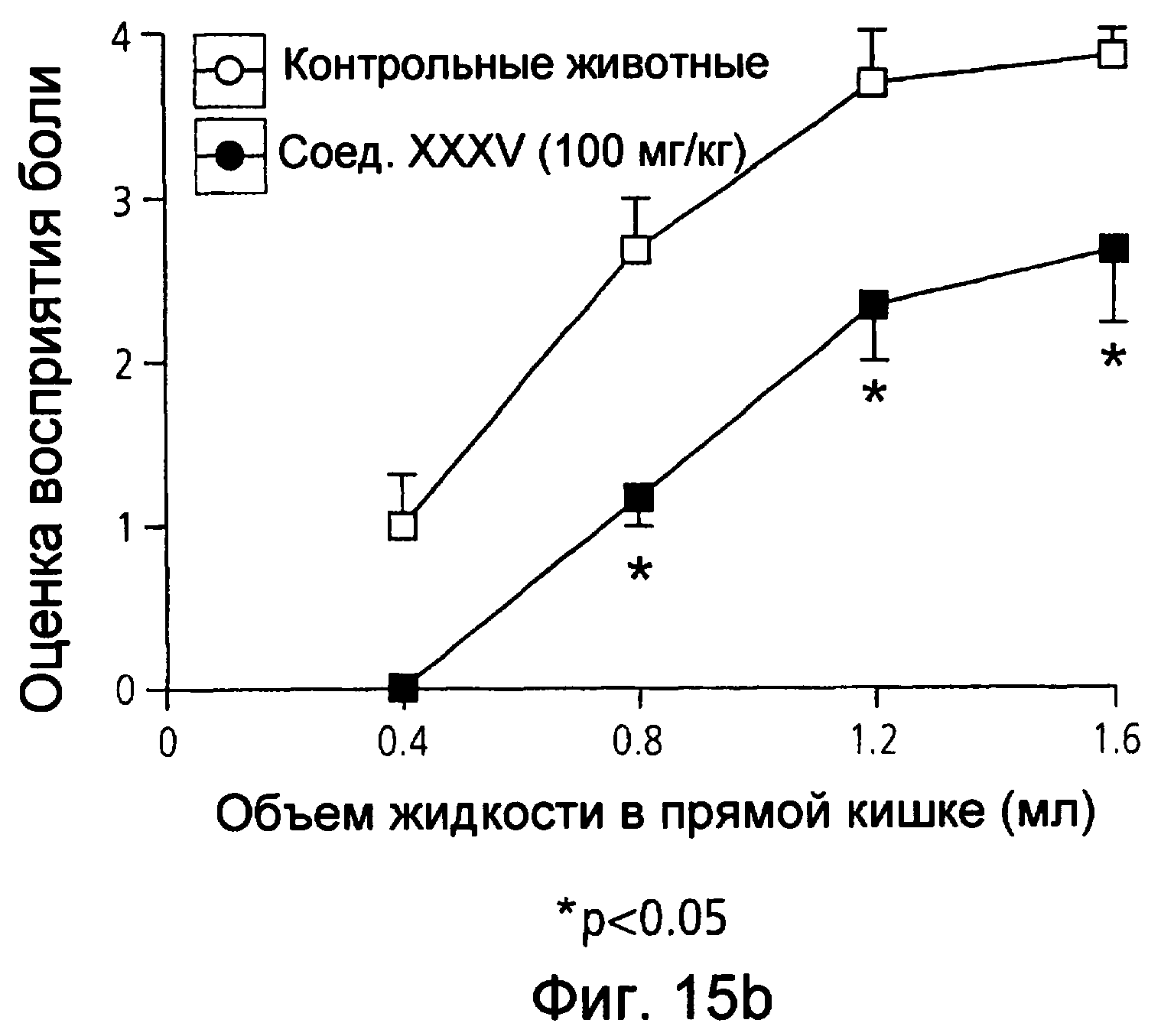
На фиг.15(а) и (b) показана оценка восприятия висцеральной боли крысами в животной модели при использовании соответственно мезаламина и соединения XXXV по настоящему изобретению.
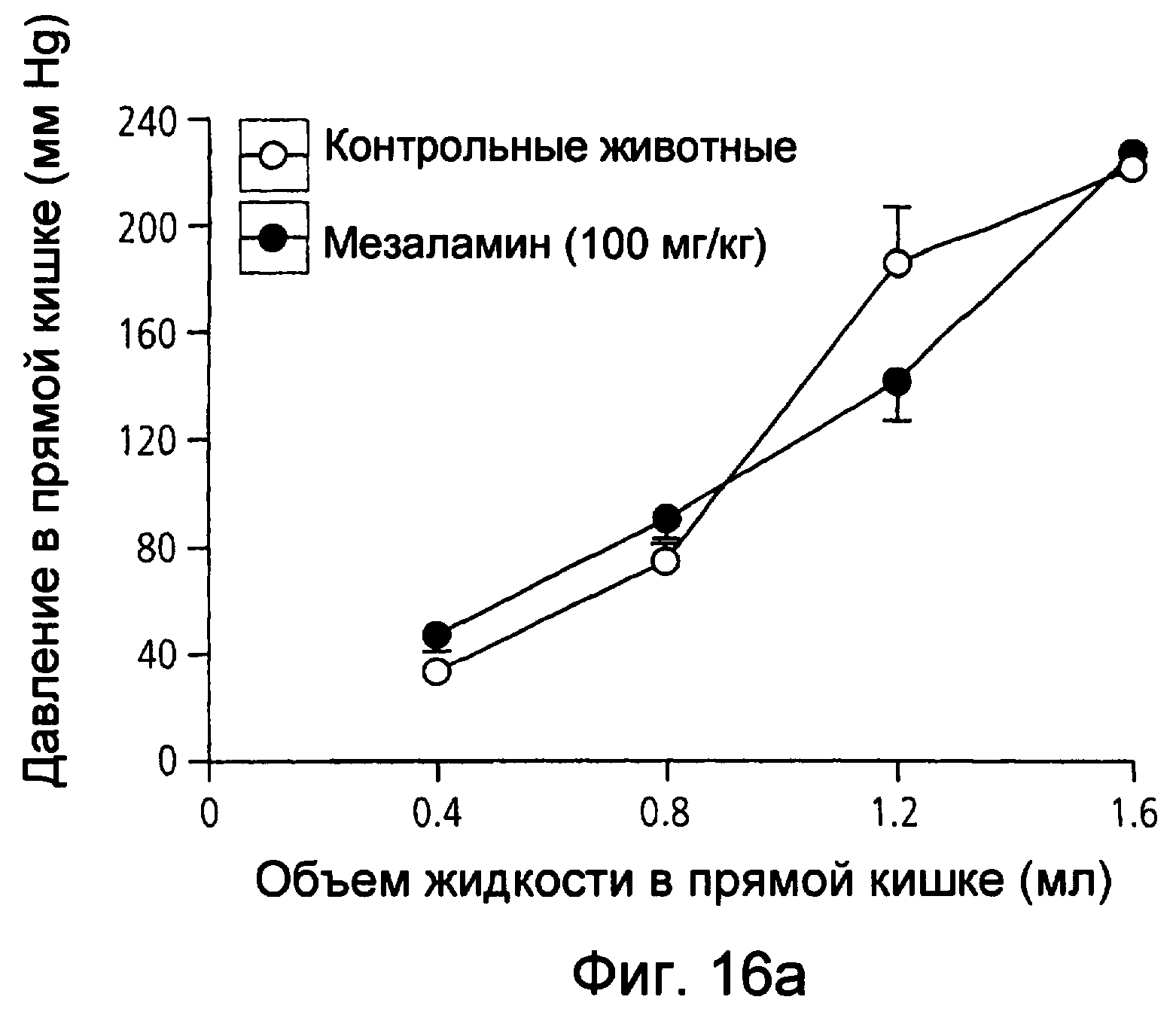
На фиг.16(а) и (b) показано давление в прямой кишке в животной модели восприятия висцеральной боли крысами при использовании соответственно мезаламина и соединения XXXV по настоящему изобретению.
На фиг.17 показаны оценки восприятия боли при использовании мезаламина, соединения XXXV и соединения XXVII с глибенкламидом или без указанного лекарственного средства.
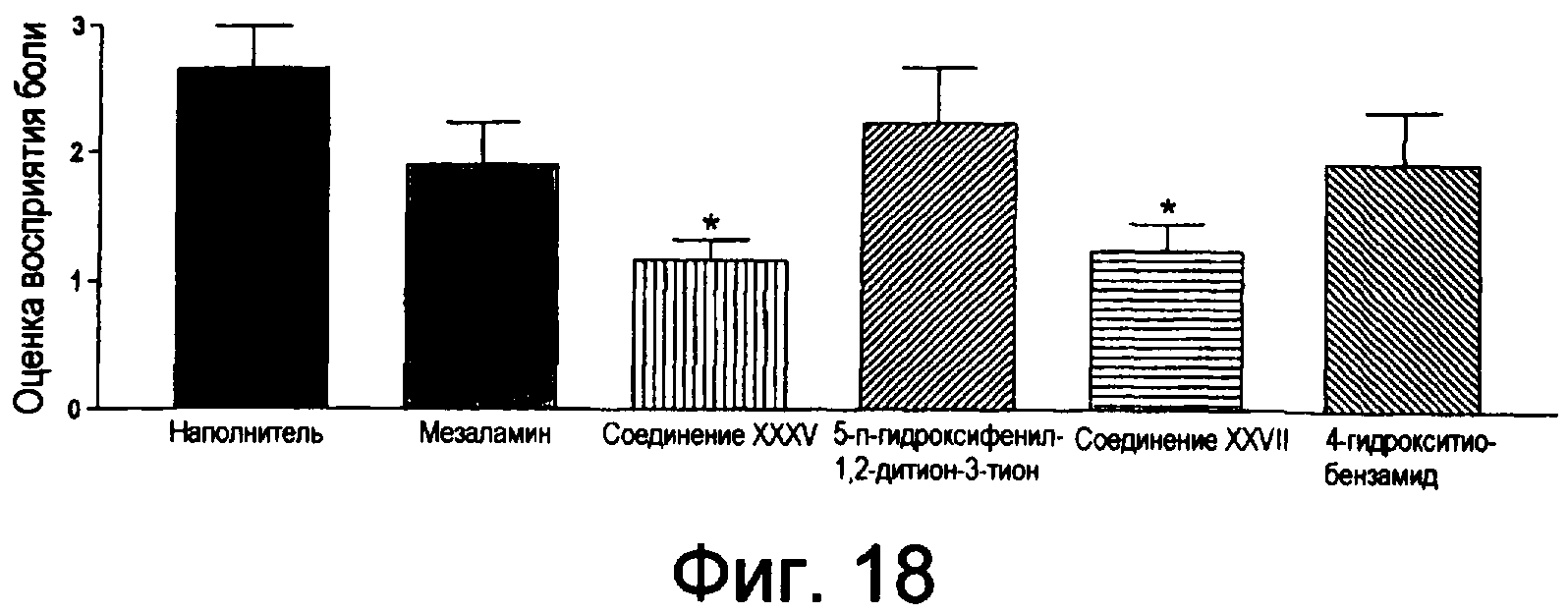
На фиг.18 показаны оценки восприятия боли при использовании соединения XXXV, соединения XXVII, мезаламина, ADT-OH и 4-НВТ.
На фиг.19 показана адгезия лейкоцитов в ответ на внутрижелудочное введение аспирина.
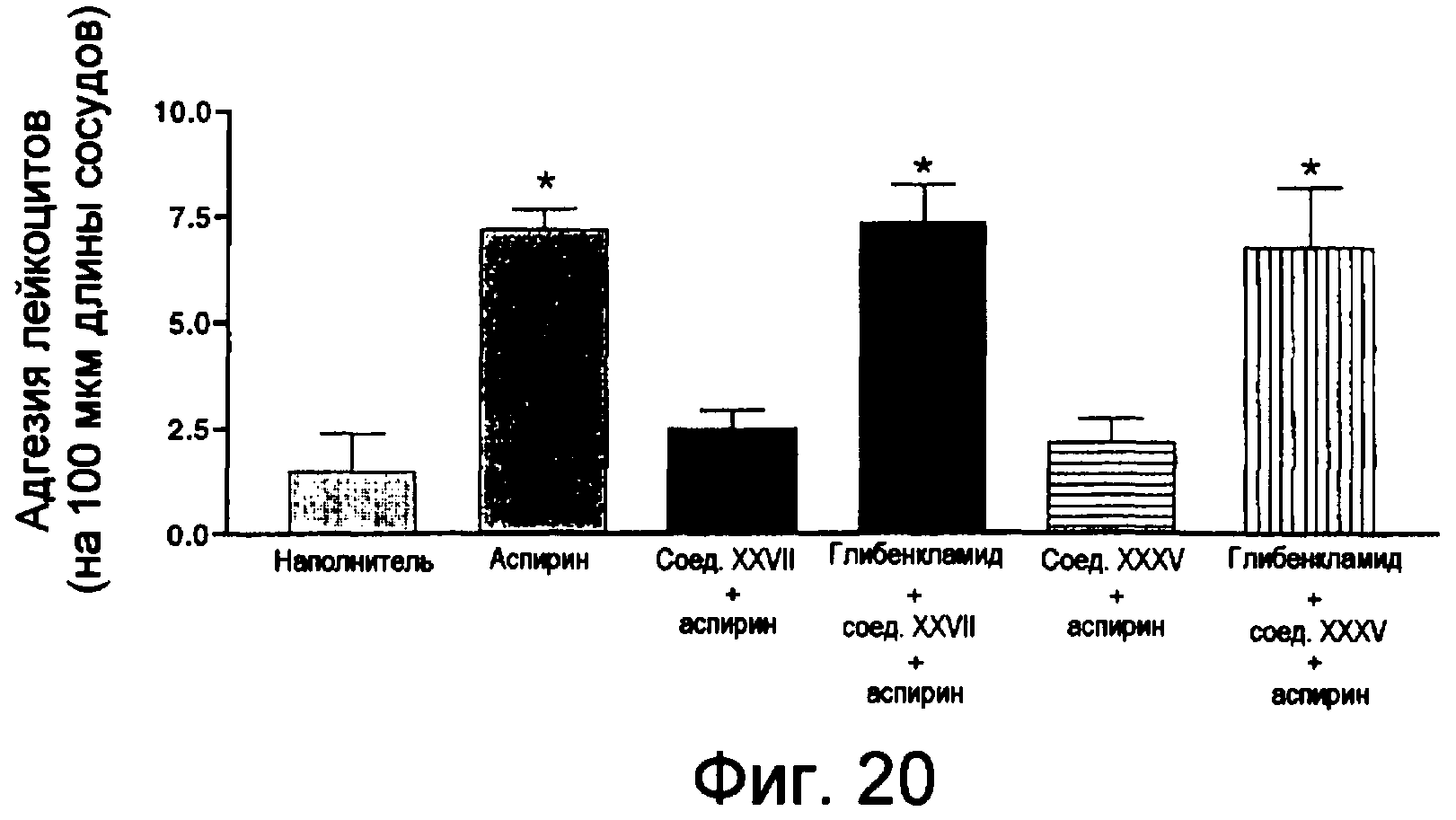
На фиг.20 изображена столбчатая диаграмма адгезии лейкоцитов в конце эксперимента (по истечении 60-65 минут).
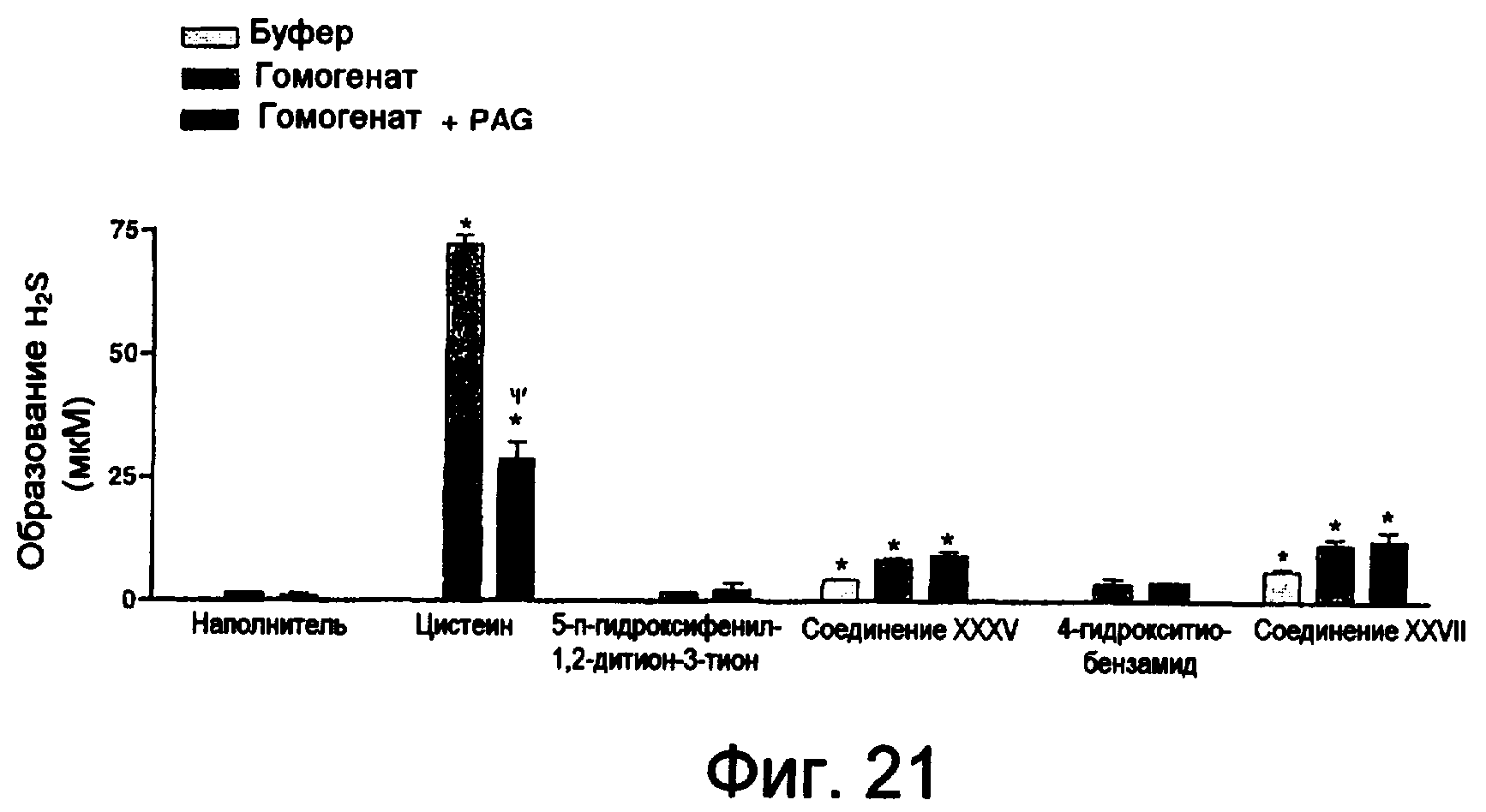
На фиг.21 изображена столбчатая диаграмма, показывающая образование H2S цистеином, ADT-OH, соединением XXXV, 4-НТВ и соединением XXVII.
На фиг.22 изображена кривая изменения реакции в зависимости от концентрации, показывающая вазорелаксирующее действие H2S-высвобождающих частей молекулы по настоящему изобретению.
Подробное описание предпочтительного варианта осуществления изобретения
Настоящее изобретение далее будет описано со ссылкой на предпочтительные варианты осуществления изобретения. Однако следует отметить, что указанные варианты осуществления изобретения приведены только в целях иллюстрации изобретения и не ограничивают объем изобретения, представленного в формуле изобретения.
Соединения по настоящему изобретению содержат две активные части: 4- или 5-ASA и часть, высвобождающую сероводород, которые связаны друг с другом при помощи азогруппы, сложноэфирной, ангидридной, сложнотиоэфирной или амидной связи. Наличие ферментов азоредуктазы делает возможным высвобождение 4- или 5-ASA из пролекарств, связанных азогруппой, обеспечивая таким образом направленную доставку в ободочную кишку и уменьшение системного всасывания. Аналогичным образом, присутствие карбоксипептидаз и аминопептидаз А также делает возможным высвобождение 4- или 5-ASA из пролекарств, связанных соответственно сложноэфирной и амидной связью. Эстеразы и тиоэстеразы также расщепляют соответственно сложноэфирные и сложнотиоэфирные связи. И наконец, липазы расщепляют ангидридные связи. Соединения по настоящему изобретению могут быть получены при использовании известных исходных веществ и реагентов.
Соединения по настоящему изобретению могут быть использованы для профилактики или лечения разных заболеваний, в частности воспалительных заболеваний желудочно-кишечного тракта, которые включают, не ограничиваясь ими, воспалительные заболевания ротовой полости, такие как воспаление слизистой оболочки, инфекционные заболевания (например, вирусные, бактериальные и грибковые заболевания) и болезнь Крона; воспалительные заболевания пищевода, такие как эзофагит, поражения, вызванные химическими веществами (например, поражение, возникающее при проглатывании щелока), гастроэзофагеальная рефлюксная болезнь, рефлюкс желчной кислоты, язва пищевода Баррета, болезнь Крона и сужение пищевода; воспалительные заболевания, такие как гастрит (например, Helicobacter pyroli, пептический эзофагит и атрофический гастрит), глютеновая болезнь, пептическая язва, предопухолевые состояния желудка, неязвенная диспепсия и болезнь Крона; воспалительные заболевания желудка, такие как болезнь Крона, чрезмерное развитие микрофлоры, пептическая язва и фиссуры тонкой кишки; воспалительные заболевания ободочной кишки, такие как болезнь Крона, неспецифический язвенный колит, спастический колит, инфекционный колит (например, псевдомембранозный колит, в частности колит, вызванный бактериями рода Clostridium difficile, энтерит, вызванный бактериями рода Salmonella, инфекционные заболевания, вызванные шигеллами, йерсиниоз, криптоспиридиоз, микроспиридиоз и вирусные инфекции), лучевой колит, колит у хозяина с ослабленным иммунитетом (например, тифлит), предопухолевые состояния ободочной кишки (например, дисплазия, воспалительные заболевания кишечника и полипозный колит), проктит, воспаление, обусловленное геморроем, прокталгия и трещины прямой кишки; заболевания печени, желчного пузыря и/или желчных протоков, такие как холангит, склерозирующий холангит, первичный цирроз желчных протоков и холецистит; и кишечный абсцесс.
В зависимости от конкретного состояния или заболевания, подлежащего лечению, соединения по настоящему изобретению можно вводить субъектам в любых терапевтически приемлемых, эффективных и безопасных дозах, которые могут быть легко определены специалистом в данной области. Указанные соединения наиболее желательно вводят в количестве от около 1 до около 2000 мг/сутки в виде однократной дозы или разделенных доз, хотя доза может быть изменена в зависимости от массы тела и состояния здоровья подлежащего лечению субъекта и конкретного способа введения. Однако наиболее желательная доза находится в пределах от около 0,1 до около 100 мг/кг, предпочтительно от около 5 до 90 мг/кг и более предпочтительно от около 5 до 50 мг/кг. Тем не менее доза может быть изменена в зависимости от массы тела и состояния здоровья подлежащего лечению субъекта, индивидуальной реакции на указанное лекарственное средство, а также от типа выбранного фармацевтического препарата и времени введения. В некоторых случаях более приемлемой может быть доза ниже нижнего предела вышеуказанного диапазона, в то время как в других случаях могут быть использованы более высокие дозы без возникновения вредных побочных эффектов при условии, что такие большие дозы сначала делят на несколько меньших доз, предназначенных для введения в течение дня.
Соединения по настоящему изобретению можно вводить в виде любого фармацевтического препарата, тип которого зависит от способа введения. Указанные фармацевтические композиции могут быть получены стандартными методами с использованием совместимых фармацевтически приемлемых наполнителей или носителей. Примеры таких композиций включают капсулы, таблетки, чрескожные пластыри, лепешки, пастилки, аэрозоли, сиропы, порошки, гранулы, гели, эликсиры, суппозитории и тому подобные, растворы, приготовленные по индивидуальному рецепту, инъецируемые препараты, ректальные, назальные, глазные, вагинальные и другие препараты. Предпочтительным способом введения является пероральное и ректальное введение.
Для перорального введения можно использовать таблетки, содержащие разные наполнители, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, дикальцийфосфат и глицин, наряду с разными дезинтеграторами, такими как крахмал (предпочтительно кукурузный крахмал, картофельный крахмал или крахмал из кассавы), альгиновая кислота и определенные комплексные силикаты, и гранулирующими связывающими веществами, такими как поливинилпирролидон, сахароза, желатин и аравийская камедь. Кроме того, в процессе таблетирования могут быть использованы смазывающие вещества, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции подобного типа могут быть также использованы для наполнения желатиновых капсул; предпочтительные вещества, используемые для указанной цели, включают также лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли. При получении водных суспензий и/или эликсиров для перорального введения активный ингредиент может быть объединен с подсластителями или ароматизаторами, красителями и при желании с эмульгирующими и/или суспендирующими агентами вместе с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин и разные комбинации указанных веществ.
Можно приготовить лекарственную форму с немедленным действием, регулируемым действием, замедленным действием, пролонгированным действием или целенаправленным пролонгированным действием. Определения вышеуказанных терминов известны специалистам в данной области. Кроме того, на профиль действия лекарственной формы может влиять состав полимерной смеси, состав матрицы с покрытием, состав частиц, состав частиц с покрытием, состав ионообменной смолы, осмотические характеристики композиции или биологически разлагающаяся полимерная композиция. Не ограничивая себя какой-либо теорией, можно отметить, что на действие лекарственного препарата может влиять благоприятная диффузия, растворимость, эрозия, ионный обмен, осмос или комбинация вышеуказанных факторов.
Для парентерального введения можно использовать раствор активного соединения в кунжутном или арахисовом масле либо в водном растворе пропиленгликоля. Водные растворы при необходимости должны быть должным образом забуферены (рН предпочтительно выше 8), при этом водный разбавитель прежде всего должен быть изотоническим. Для внутривенных инъекций могут быть использованы водные растворы. Все указанные растворы получают в стерильных условиях, создаваемых стандартными фармацевтическими методами, хорошо известными специалистам в данной области.
Ниже приведены не ограничивающие настоящее изобретение примеры, которые должны позволить специалисту в данной области реализовать и использовать данное изобретение.
Получение соединений
Пример 1
Синтез 2-гидрокси-5-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилазо]бензойной кислоты (4) [соединение формулы II]

Синтез трет-бутилового эфира (4-пропенилфенил)карбаминовой кислоты (2)
К раствору 4-пропенилфениламина (1) (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли триэтиламин (15,0 ммоль) и ди-трет-бутилдикарбонат (15,0 ммоль), перемешивая смесь при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом трет-бутиловый эфир (4-пропенилфенил)карбаминовой кислоты (2) (выход 90%).
Синтез 5-(4-аминофенил)-[1,2]дитиол-3-тиона (3)
Трет-бутиловый эфир (4-пропенилфенил)карбаминовой кислоты (2, 4,5 ммоль) и серу (31,5 ммоль) нагревали в диметилформамиде (500 мл) в течение 8 часов, растворитель удаляли и остаток почти полностью растворяли в толуоле. Раствор в толуоле экстрагировали 2Н водным раствором гидроксида натрия с образованием осадка оранжевого твердого вещества. Продукт растворяли в кипящей воде, обрабатывали 4Н раствором хлористоводородной кислоты в течение 30 минут при комнатной температуре и добавляли 4Н раствор NaOH, получая при этом требуемый продукт (3) (выход 55%).
Синтез 2-гидрокси-5-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилазо]бензойной кислоты (4)
5-(4-Аминофенил)-[1,2]дитиол-3-тион (3, 0,56 ммоль) растворяли в смеси 5 мл концентрированной HCl и 2,5 мл воды и диазотировали раствором нитрита натрия (0,56 ммоль). Тем временем в воде растворяли салициловую кислоту (0,56 ммоль), гидроксид калия (1,12 ммоль) и карбонат натрия. Суспензию диазосоединения добавляли порциями к щелочному раствору салициловой кислоты и в течение всей реакции щелочность поддерживали на достаточно высоком уровне, добавляя дополнительные количества раствора гидроксида калия. Через 2 дня реакционную смесь нагревали в течение 30 минут при 50°С. Азосоединение (4) осаждали при помощи HCl и отфильтровывали (выход 85%), получая при этом соединение формулы II, 2-гидрокси-5-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилазо]бензойную кислоту.
Пример 2
Синтез 2-гидрокси-4-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилазо]бензойной кислоты (2) [соединение формулы II]

Синтез 2-гидрокси-4-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилазо]бензойной кислоты (2)
4-Амино-2-гидроксибензойную кислоту (1, 1 ммоль) растворяли в смеси 10 мл концентрированной HCl и 5 мл воды и диазотировали раствором нитрита натрия (1 ммоль). Суспензию диазосоединения добавляли порциями к раствору 5-фенил-[1,2]дитиол-3-тиона (1 ммоль) в диметилформамиде. Через 2 дня реакционную смесь нагревали в течение 30 минут при 50°С. Смесь охлаждали, азосоединение (2) осаждали при помощи HCl и отфильтровывали (выход 65%), получая при этом соединение формулы II, 2-гидрокси-5-[4-(5-тиоксо-5Н-[1,2]-дитиол-3-ил)фенилазо]бензойную кислоту.
Пример 3
Общий способ синтеза 4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 4- или 5-амино-2-гидроксибензойной кислоты (4) [соединение формулы XXXV]

Синтез 5-п-гидроксифенил-1,2-дитион-3-тиона (ADT-OH)
Анетол (1) (32,5 г, 0,21 моль) и серу (45 г, 1,40 моль) нагревали в диметилформамиде (250 мл) в течение 8 часов, растворитель удаляли, и остаток почти полностью растворяли в толуоле. Раствор в толуоле экстрагировали 2Н водным раствором гидроксида натрия с образованием осадка оранжевого твердого вещества (8,5 г), температура плавления выше 300°С. Продукт растворяли в кипящей воде и добавляли хлористоводородную кислоту, получая при этом оранжевый осадок (2) (выход 50%), температура плавления 188-189°С.1H ЯМР (ДМСО) δ 6,86 (д, 2H), 7,68 (с, 1H), 7,75 (д, 2H), 10,51 (с, -OH); MS (ESI), m/z 225 (M-).

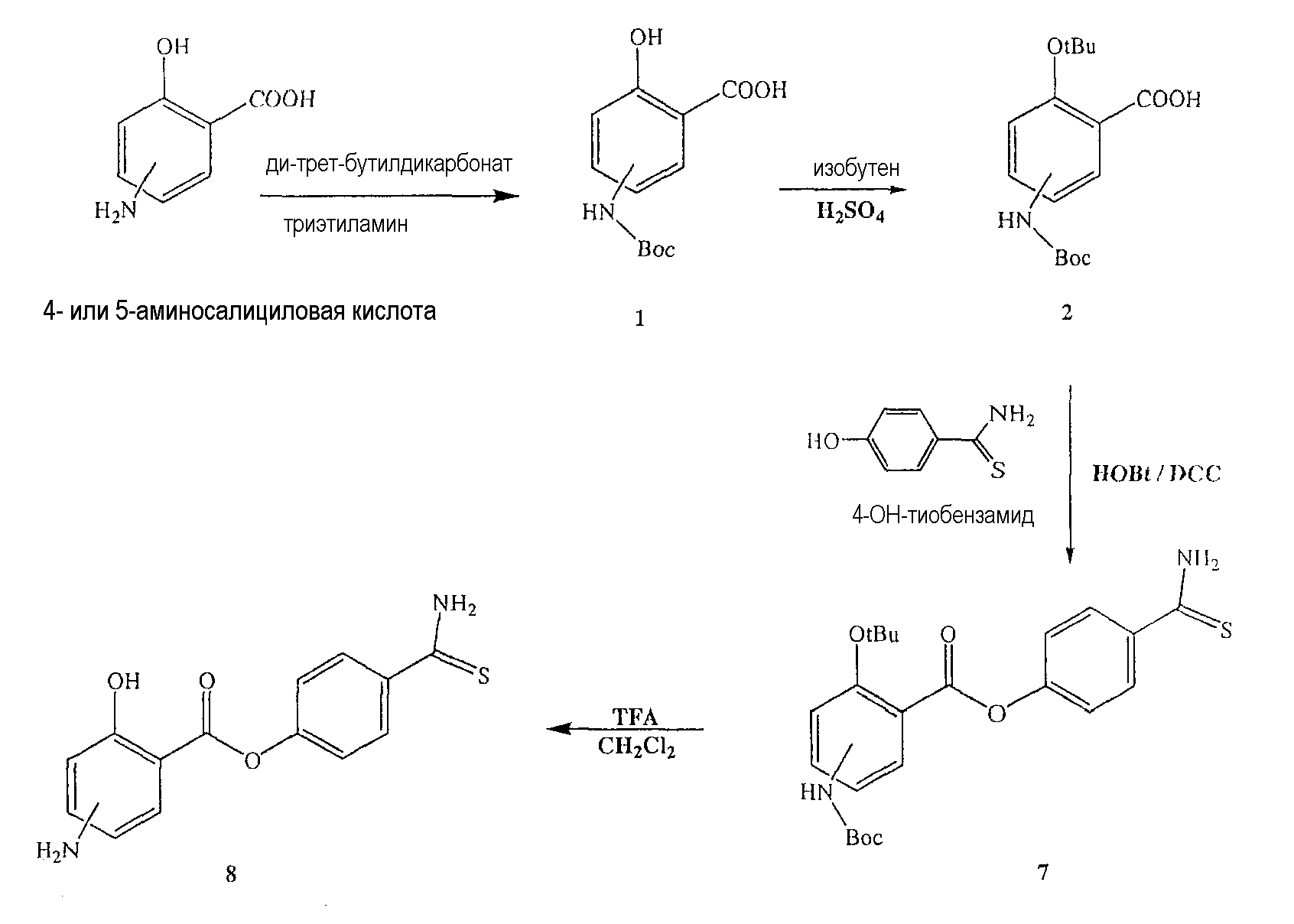
Синтез 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойной кислоты (1)
К раствору 4- или 5-аминосалициловой кислоты (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли триэтиламин (15,0 ммоль) и ди-трет-бутилдикарбонат (15,0 ммоль), перемешивая смесь при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойную кислоту (1) (выход 80%).
Синтез 4- или 5-трет-бутоксикарбониламино-2-трет-бутоксибензойной кислоты (2)
Соединение (1) (12,0 ммоль), концентрированную H2SO4 (6,0 ммоль) и DCM (100 мл) перемешивали в атмосфере газообразного изобутилена под давлением 0,35 атм (5 фунтов/кв.дюйм) в течение 6 часов при комнатной температуре. Раствор промывали холодным 10% NaHCO3 (2×100 мл) и насыщенным раствором соли (100 мл), сушили (Na2SO4) и упаривали. Остаток растворяли в MeOH/CCl4 (1:1) (400 мл), промывали водой (300 мл) и экстрагировали МеОН/водой (1:1) (2×200 мл). Экстракт сушили (Na2SO4) и упаривали с образованием белого твердого вещества (2), которое перекристаллизовывали DCM/гексаном (выход 83%).
Синтез 4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 4- или 5-амино-2-гидроксибензойной кислоты (4)
К раствору 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойной кислоты (2) (3,0 ммоль) в 50 мл диметилформамида добавляли гидроксибензотриазол (3,3 ммоль) и DCC (3,3 ммоль), перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли 5-п-гидроксифенил-1,2-дитион-3-тион (ADT-OH) (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный промежуточный продукт (3) обрабатывали раствором 40% TFA (трифторуксусной кислотой) в CH2Cl2. Через 2 часа растворитель удаляли с образованием соединения (3) в виде неочищенного остатка. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом 4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фениловый эфир 4- или 5-амино-2-гидроксибензойной кислоты (4) [соединение формулы XXXV] (выход 40%).
Соединение: 4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фениловый эфир 5-амино-2-гидроксибензойной кислоты (4):1H ЯМР (ДМСО) δ 7,07 (д, 2H), 7,38 (д, 2H), 7,46 (д, 2H), 7,79 (с, 1H), 7,85 (с, 1H), 8,01 (д, 2H), 10,35 (с, -OH); MS (ESI), m/z 362 (M+).
Пример 4
Общий способ синтеза 2-(трет-бутоксикарбонил)-4- или 5-аминофенилгидрокарбоната (5) и 3-(трет-бутоксикарбонил)-4- или 5-гидроксифенилкарбаминовой кислоты (6)

Синтез 4- или 5-амино-2-этоксикарбонилоксибензойной кислоты (1) и 4- или 5-этоксикарбониламино-2-гидроксибензойной кислоты (2)
4- или 5-Аминосалициловую кислоту (3,0 ммоль) растворяли в 40 мл хлороформа в круглодонной колбе, оснащенной сушильной трубкой. Постепенно добавляли этилхлорформиат (3,0 ммоль) и нагревали раствор с обратным холодильником в течение 2 часов. Хлороформ выпаривали в вакууме, и остаток поглощали простым эфиром. Эфирную фазу обесцвечивали углем, фильтровали и удаляли растворитель в вакууме. Полученный остаток растворяли в этаноле, и продукт осаждали н-гексаном в виде неочищенного маслянистого полутвердого вещества. Неочищенный продукт очищали флэш-хроматографией на силикагеле, производя элюирование диэтиловым эфиром/гексаном (7:3, об/об), в результате чего были получены указанные в заголовке соединения: 4- или 5-амино-2-этоксикарбонилоксибензойная кислота (1: выход 58%) и 4- или 5-этоксикарбониламино-2-гидроксибензойная кислота (2: выход 34%).
Синтез трет-бутилового эфира 4- или 5-амино-2-этоксикарбонилоксибензойной кислоты (3)
К раствору соединения (1) (3,0 ммоль) в 50 мл диметилформамида добавляли гидроксибензотриазол (3,3 ммоль) и DCC (3,3 ммоль), перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли трет-бутанол (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9,5/0,5), получая при этом трет-бутиловый эфир 4- или 5-амино-2-этоксикарбонилоксибензойной кислоты (3) (выход 55%).
Синтез трет-бутилового эфира 4- или 5-этоксикарбониламино-2-гидроксибензойной кислоты (4)
Соединение (4) было получено способом, описанным для получения соединения (3). Выход 74%.
Синтез 2-(трет-бутоксикарбонил)-4- или 5-аминофенилгидрокарбоната (5)
К раствору соединения (3) (3,5 г, 0,011 моль) в этаноле (80 мл) добавляли 1Н раствор NaOH (40 мл). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Затем раствор делали нейтральным, добавляя 1Н раствор HCl. Этанол удаляли и остаток экстрагировали этилацетатом (3×150 мл); органические слои промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель, получая при этом 2-(трет-бутоксикарбонил)-4- или 5-аминофенилгидрокарбонат (5) (3 г, 0,010 моль, выход 89%) в виде белого твердого вещества.
Синтез 3-(трет-бутоксикарбонил)-4- или 5-гидроксифенилкарбаминовой кислоты (6)
Соединение (6) было получено способом, описанным для получения соединения (5). Выход 91%.
Пример 5
Общий способ синтеза 4- или 5-амино-2-(1-карбокси-3-тиокарбамоилпропилкарбамоилокси)бензойной кислоты (11) [соединение формулы XXII] и 4- или 5-[3-(1-карбокси-3-тиокарбамоилпропил)уреидо]-2-гидроксибензойной кислоты (12)

Синтез 5-тио-L-глутамин-OtBu (2)
L-Глутамин-OtBu.HCl (1) (1,2 ммоль, 0,3 г) и реагент Лавессона (0,75 ммоль, 0,3 г) добавляли к бензолу (20 мл) и смесь нагревали с обратным холодильником в течение 15 минут. Затем реакционную смесь охлаждали и упаривали в вакууме. Неочищенный продукт очищали хроматографией на 100 г силикагеля и элюировали смесями этилацетата и н-гексана. Было получено 0,2 г (выход 76%) продукта (2) в виде белого твердого вещества.1H ЯМР (CDCl3) δ 1,4 (с, 9H), 1,8-2,8 (м, 5H), 4,0-4,8 (м, 3H); MS (ESI), m/z 219 (M+).

Синтез 2-(трет-бутоксикарбонил)-4- или 5-трет-бутоксикарбониламинофенилгидрокарбоната (7)
К раствору соединения (5) (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли триэтиламин (15,0 ммоль) и ди-трет-бутил-дикарбонат (15,0 ммоль), перемешивая смесь при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом 2-(трет-бутоксикарбонил)-4- или 5-трет-бутоксикарбониламинофенилгидрокарбонат (7) (выход 80%).
Синтез 3-(трет-бутоксикарбонил)-4- или 5-гидроксифенилкарбаминовой кислоты (8)
Соединение (6) (12,0 ммоль), концентрированную H2SO4 (6,0 ммоль) и DCM (100 мл) перемешивали в атмосфере газообразного изобутилена под давлением 0,35 атм (5 фунтов/кв.дюйм) в течение 6 часов при комнатной температуре. Раствор промывали холодным 10% NaHCO3 (2×100 мл) и насыщенным раствором соли (100 мл), сушили (Na2SO4) и упаривали. Остаток растворяли в MeOH/CCl4 (1:1) (400 мл), промывали водой (300 мл) и экстрагировали MeOH/водой (1:1) (2×200 мл). Экстракт сушили (Na2SO4) и упаривали с образованием белого твердого вещества (8), которое перекристаллизовывали DCM/гексаном (выход 83%).
Синтез 4- или 5-амино-2-(1-карбокси-3-тиокарбамоилпропилкарбамоилокси)бензойной кислоты (11)
К раствору соединения (7) (3,0 ммоль) в 50 мл диметилформамида добавляли гидроксибензотриазол (3,3 ммоль) и DCC (3,3 ммоль), перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли трет-бутиловый эфир 2-амино-4-тиокарбамоилмасляной кислоты (3,0 ммоль) и триэтиламин (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный промежуточный продукт (9) обрабатывали раствором 40% TFA в CH2Cl2. Через 2 часа растворитель удаляли с образованием соединения (11) в виде неочищенного остатка. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом 4- или 5-амино-2-(1-карбокси-3-тиокарбамоилпропилкарбамоилокси)-бензойную кислоту (11) (выход 45%) [соединение формулы XXII].
Синтез 4- или 5-[3-(1-карбокси-3-тиокарбамоилпропил)уреидо]-2-гидроксибензойной кислоты (12)
Соединение (12) было получено способом, описанным для получения соединения (11). Выход 38%.
Пример 6
Общий способ синтеза 4- или 5-амино-2-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбонилокси]бензойной кислоты (15) [соединение формулы IV] и 2-гидрокси-4- или 5-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбониламино]бензойной кислоты (16) [соединение формулы V]

Синтез 5-п-гидроксифенил-1,2-дитион-3-тиона (ADT-OH)
Анетол (1) (32,5 г, 0,21 моль) и серу (45 г, 1,40 моль) нагревали в диметилформамиде (250 мл) в течение 8 часов; растворитель удаляли и остаток почти полностью растворяли в толуоле. Раствор в толуоле экстрагировали 2Н водным раствором гидроксида натрия с образованием осадка оранжевого твердого вещества (8,5 г), температура плавления выше 300°С. Продукт растворяли в кипящей воде и добавляли хлористоводородную кислоту, получая при этом оранжевый осадок (2) (выход 50%), температура плавления 188-189°С.1H ЯМР (ДМСО) δ 6,86 (д, 2H), 7,68 (с, 1H), 7,75 (д, 2H), 10,51 (с, -OH); MS (ESI), m/z 225 (M-).

Синтез 2-(трет-бутоксикарбонил)-4- или 5-трет-бутоксикарбониламинофенилгидрокарбоната (7)
К раствору соединения (5) (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли триэтиламин (15,0 ммоль) и ди-трет-бутил-дикарбонат (15,0 ммоль), перемешивая при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом 2-(трет-бутоксикарбонил)-4- или 5-трет-бутоксикарбониламинофенилгидрокарбонат (7) (выход 80%).
Синтез 3-(трет-бутоксикарбонил)-4- или 5-гидроксифенилкарбаминовой кислоты (8)
Соединение (6) (12,0 ммоль), концентрированную H2SO4 (6,0 ммоль) и DCM (100 мл) перемешивали в атмосфере газообразного изобутена под давлением 0,35 атм (5 фунтов/кв.дюйм) в течение 6 часов при комнатной температуре. Раствор промывали холодным 10% NaHCO3 (2×100 мл) и насыщенным раствором соли (100 мл), сушили (Na2SO4) и упаривали. Остаток растворяли в MeOH/CCl4 (1:1) (400 мл), промывали водой (300 мл) и экстрагировали MeOH/водой (1:1) (2×200 мл). Экстракт сушили (Na2SO4) и упаривали с образованием белого твердого вещества (8), которое перекристаллизовывали DCM/гексаном (выход 83%).
Синтез 4- или 5-амино-2-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбонилокси]бензойной кислоты (15)
К раствору соединения (7) (3,0 ммоль) в 50 мл диметилформамида добавляли гидроксибензотриазол (3,3 ммоль) и DCC (3,3 ммоль), перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли 5-п-гидроксифенил-1,2-дитион-3-тион (ADT-OH) (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный промежуточный продукт (13) обрабатывали раствором 40% TFA в CH2Cl2. Через 2 часа растворитель удаляли с образованием соединения (15) в виде неочищенного остатка. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом 4- или 5-амино-2-[4-(5-тиоксо-5Н-[1,2]-дитиол-3-ил)феноксикарбонилокси]бензойную кислоту (15) (выход 45%), соединение формулы IV.
Синтез 2-гидрокси-4- или 5-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбониламино]бензойной кислоты (16)
Соединение (16), соединение формулы V, было получено способом, описанным для получения соединения (15). Выход 38%.
Пример 7
Общий способ синтеза 4- или 5-амино-2-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбонилокси}бензойной кислоты (19) [соединение формулы XIV] и 2-гидрокси-4- или 5-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]-дитиадифосфетан-2-ил]феноксикарбониламино}бензойной кислоты (20) [соединение формулы XIII]

Синтез ангидрида (п-гидроксифенил)дитиофосфоновой кислоты
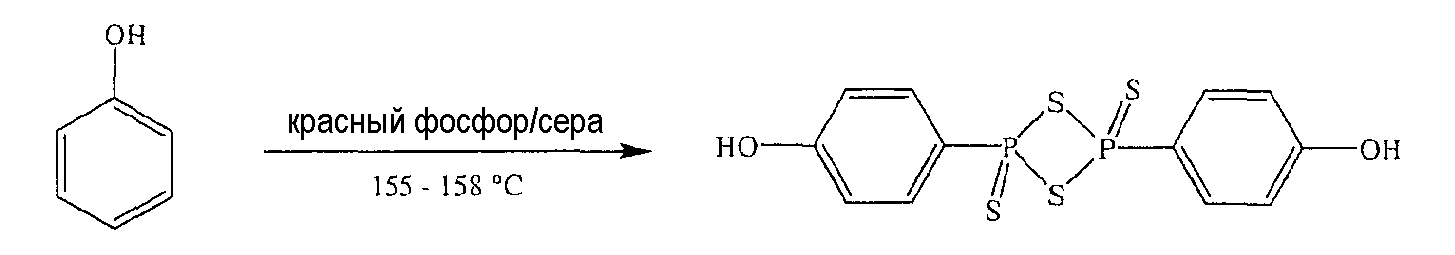
Красный фосфор (4 г, 0,129 моль), серу (4 г, 0,125 моль) и PhOH (4 г, 0,042 моль) нагревали в течение 5,5 часа при 155-158°С; реакционную смесь охлаждали при комнатной температуре и осадок собирали (5,5 г, выход 34%), температура плавления 224-226°С. Результаты анализов ЯМР и масс-спектроскопии (MS) соответствовали структуре ангидрида п-гидроксифенилдитиофосфоновой кислоты.
Синтез 2-(трет-бутоксикарбонил)-4- или 5-трет-бутоксикарбониламинофенилгидрокарбоната (7)
К раствору соединения (5) (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли триэтиламин (15,0 ммоль) и ди-трет-бутил-дикарбонат (15,0 ммоль), перемешивая смесь при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом 2-(трет-бутоксикарбонил)-4- или 5-трет-бутоксикарбониламинофенилгидрокарбонат (7) (выход 80%).
Синтез 3-(трет-бутоксикарбонил)-4- или 5-гидроксифенилкарбаминовой кислоты (8)
Соединение (6) (12,0 ммоль), концентрированную H2SO4 (6,0 ммоль) и DCM (100 мл) перемешивали в атмосфере газообразного изобутена под давлением 0,35 атм (5 фунтов/кв.дюйм) в течение 6 часов при комнатной температуре. Раствор промывали холодным 10% NaHCO3 (2×100 мл) и насыщенным раствором соли (100 мл), сушили (Na2SO4) и упаривали. Остаток растворяли в MeOH/CCl4 (1:1) (400 мл), промывали водой (300 мл) и экстрагировали MeOH/водой (1:1) (2×200 мл). Экстракт сушили (Na2SO4) и упаривали с образованием белого твердого вещества (8), которое перекристаллизовывали DCM/гексаном (выход 83%).
Синтез 4- или 5-амино-2-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбонилокси}бензойной кислоты (19)
К раствору соединения (7) (3,0 ммоль) в 50 мл диметилформамида добавляли гидроксибензотриазол (3,3 ммоль) и DCC (3,3 ммоль, перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли ангидрид п-гидроксифенилдитиофосфоновой кислоты (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный промежуточный продукт (17) обрабатывали раствором 40% TFA в CH2Cl2. Через 2 часа растворитель удаляли с образованием соединения (19) в виде неочищенного остатка. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом 4- или 5-амино-2-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбонилокси}бензойную кислоту (19) (выход 65%), соединение формулы XIV.
Синтез 2-гидрокси-4- или 5-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбониламино}бензойной кислоты (20)
Соединение (20), соединение формулы XIII, было получено способом, описанным для получения соединения (19). Выход 48%.
Пример 8
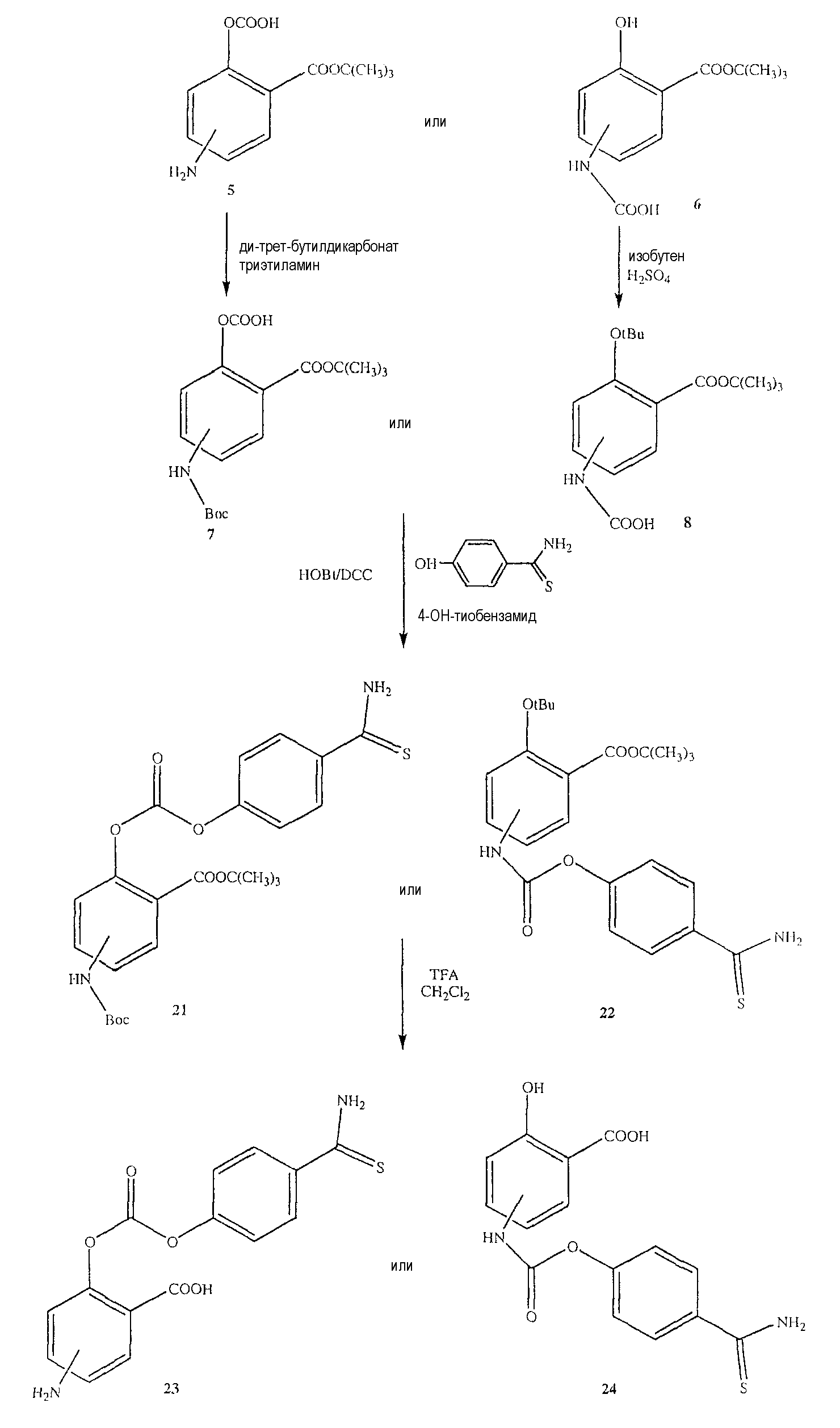
Общий способ синтеза 4- или 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты (23) [соединение формулы XXVIII] и 2-гидрокси-4- или 5-(4-тиокарбамоилфеноксикарбониламин)бензойной кислоты (24) [соединение формулы XXIX]
Синтез 2-(трет-бутоксикарбонил)-4- или 5-трет-бутоксикарбониламинофенилгидрокарбонат (7)
К раствору соединения (5) (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли триэтиламин (15,0 ммоль) и ди-трет-бутил-дикарбонат (15,0 ммоль), перемешивая смесь при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом 2-(трет-бутоксикарбонил)-4- или 5-трет-бутоксикарбониламинофенилгидрокарбонат (7) (выход 80%).
Синтез 3-(трет-бутоксикарбонил)-4- или 5-гидроксифенилкарбаминовой кислоты (8)
Соединение (6) (12,0 ммоль), концентрированную H2SO4 (6,0 ммоль) и DCM (100 мл) перемешивали в атмосфере газообразного изобутена под давлением 0,35 атм (5 фунтов/кв.дюйм) в течение 6 часов при комнатной температуре. Раствор промывали холодным 10% NaHCO3 (2×100 мл) и насыщенным раствором соли (100 мл), сушили (Na2SO4) и упаривали. Остаток растворяли в MeOH/CCl4 (1:1) (400 мл), промывали водой (300 мл) и экстрагировали MeOH/водой (1:1) (2×200 мл). Экстракт сушили (Na2SO4) и упаривали с образованием белого твердого вещества (8), которое перекристаллизовывали DCM/гексаном (выход 83%).
Синтез 4- или 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)-бензойной кислоты (23)
К раствору соединения (7) (3,0 ммоль) в 50 мл диметилформамида добавляли гидроксибензотриазол (3,3 ммоль) и DCC (3,3 ммоль), перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли 4-гидрокситиобензамид (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный промежуточный продукт (21) обрабатывали раствором 40% TFA в CH2Cl2. Через 2 часа растворитель удаляли с образованием соединения (23) в виде неочищенного остатка. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом 4- или 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойную кислоту (23) (выход 71%), соединение формулы XXVII.
Синтез 2-гидрокси-4- или 5-(4-тиокарбамоилфеноксикарбониламино)бензойной кислоты (24)
Соединение (24), соединение формулы XXIX, было получено способом, описанным для получения соединения (23). Выход 68%.
Пример 9
Общий способ синтеза 2-(4- или 5-амино-2-гидроксибензоиламино)-4-тиокарбамоилмасляной кислоты (6)[соединение формулы XXI]

Синтез 5-тио-L-глутамин-OtBu (2)
L-Глутамин-OtBu.HCl (1) (1,2 ммоль, 0,3 г) и реагент Лавессона (0,75 ммоль, 0,3 г) добавляли к бензолу (20 мл) и смесь нагревали с обратным холодильником в течение 15 минут. Реакционную смесь охлаждали и упаривали в вакууме. Неочищенный продукт очищали хроматографией на 100 г силикагаля и элюировали смесями этилацетата и н-гексана. Было получено 0,2 г (выход 76%) продукта (2) в виде белого твердого вещества.1H ЯМР (CDCl3) δ 1,4 (с, 9H), 1,8-2,8 (м, 5H), 4,0-4,8 (м, 3H); MS (ESI), m/z 219 (M+).
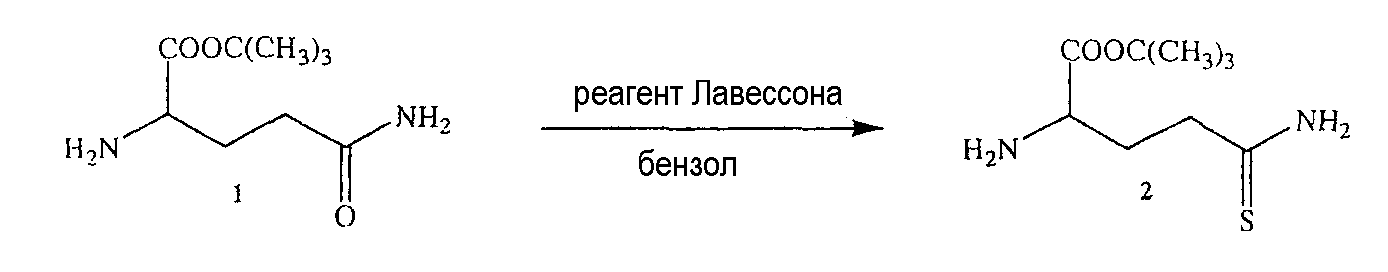
Синтез 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойной кислоты (1)
К раствору 4- или 5-аминосалициловой кислоты (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли триэтиламин (15,0 ммоль) и ди-трет-бутилдикарбонат (15,0 ммоль), перемешивая смесь при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойную кислоту (1) (выход 80%).
Синтез 4- или 5-трет-бутоксикарбониламино-2-трет-бутоксибензойной кислоты (2)
Соединение (1) (12,0 ммоль), концентрированную H2SO4 (6,0 ммоль) и DCM (100 мл) перемешивали в атмосфере газообразного изобутилена под давлением 0,35 атм (5 фунтов/кв.дюйм) в течение 6 часов при комнатной температуре. Раствор промывали холодным 10% NaHCO3 (2×100 мл) и насыщенным раствором соли (100 мл), сушили (Na2SO4) и упаривали. Остаток растворяли в MeOH/CCl4 (1:1) (400 мл), промывали водой (300 мл) и экстрагировали MeOH/водой (1:1) (2×200 мл). Экстракт сушили (Na2SO4) и упаривали с образованием белого твердого вещества (2), которое перекристаллизовывали DCM/гексаном (выход 83%).
Синтез 2-(4- или 5-амино-2-гидроксибензоиламино)-4-тиокарбамоилмасляной кислоты (6)
К раствору 4- или 5-трет-бутоксикарбониламино-2-трет-бутоксибензойной кислоты (2) (3,0 ммоль) в 50 мл диметилформамида добавляли гидроксибензотриазол (3,3 ммоль) и DCC (3,3 ммоль), перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли трет-бутиловый эфир 2-амино-4-тиокарбамоилмасляной кислоты (3,0 ммоль) и триэтиламин (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органические слои промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный промежуточный продукт (5) обрабатывали раствором TFA (40%) в CH2Cl2. Через 2 часа растворитель удаляли с образованием соединения (6) в виде неочищенного остатка. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом 2-(4- или 5-амино-2-гидроксибензоиламино)-4-тиокарбамоил-масляную кислоту (6), соединение формулы XXI, (выход 80%). MS (ESI), m/z 298 (M+).
Пример 10
Общий способ синтеза 4-тиокарбамоилфенилового эфира 4- или 5-амино-2-гидроксибензойной кислоты (8) [соединение формулы XXVII]
Синтез 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойной кислоты (1)
К раствору 4- или 5-аминосалициловой кислоты (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли триэтиламин (15,0 ммоль) и ди-трет-бутилдикарбонат (15,0 ммоль), перемешивая смесь при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойную кислоту (1) (выход 80%).
Синтез 4- или 5-трет-бутоксикарбониламино-2-трет-бутоксибензойной кислоты (2)
Соединение (1) (12,0 ммоль), концентрированную H2SO4 (6,0 ммоль) и DCM (100 мл) перемешивали в атмосфере газообразного изобутилена под давлением 0,35 атм (5 фунтов/кв.дюйм) в течение 6 часов при комнатной температуре. Раствор промывали холодным 10% NaHCO3 (2×100 мл) и насыщенным раствором соли (100 мл), сушили (Na2SO4) и упаривали. Остаток растворяли в MeOH/CCl4 (1:1) (400 мл), промывали водой (300 мл) и экстрагировали MeOH/водой (1:1) (2×200 мл). Экстракт сушили (Na2SO4) и упаривали с образованием белого твердого вещества (2), которое перекристаллизовывали DCM/гексаном (выход 83%).
Синтез 4-тиокарбамоилфенилового эфира 4- или 5-амино-2-гидроксибензойной кислоты (8)
К раствору 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойной кислоты (2) (3,0 ммоль) в 50 мл диметилформамида добавляли гидроксибензотриазол (3,3 ммоль) и DCC (3,3 ммоль), перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли 4-гидрокситиобензамид (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный промежуточный продукт (7) обрабатывали раствором 40% TFA в CH2Cl2. Через 2 часа растворитель удаляли с образованием соединения (8) в виде неочищенного остатка. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом 4-тиокарбамоилфениловый эфир 4- или 5-амино-2-гидроксибензойной кислоты (8), соединение формулы XXVII, (выход 48%).
Пример 11
Общий способ синтеза 4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]фенилового эфира 4- или 5-амино-2-гидроксибензойной кислоты (10) [соединение формулы XVII]
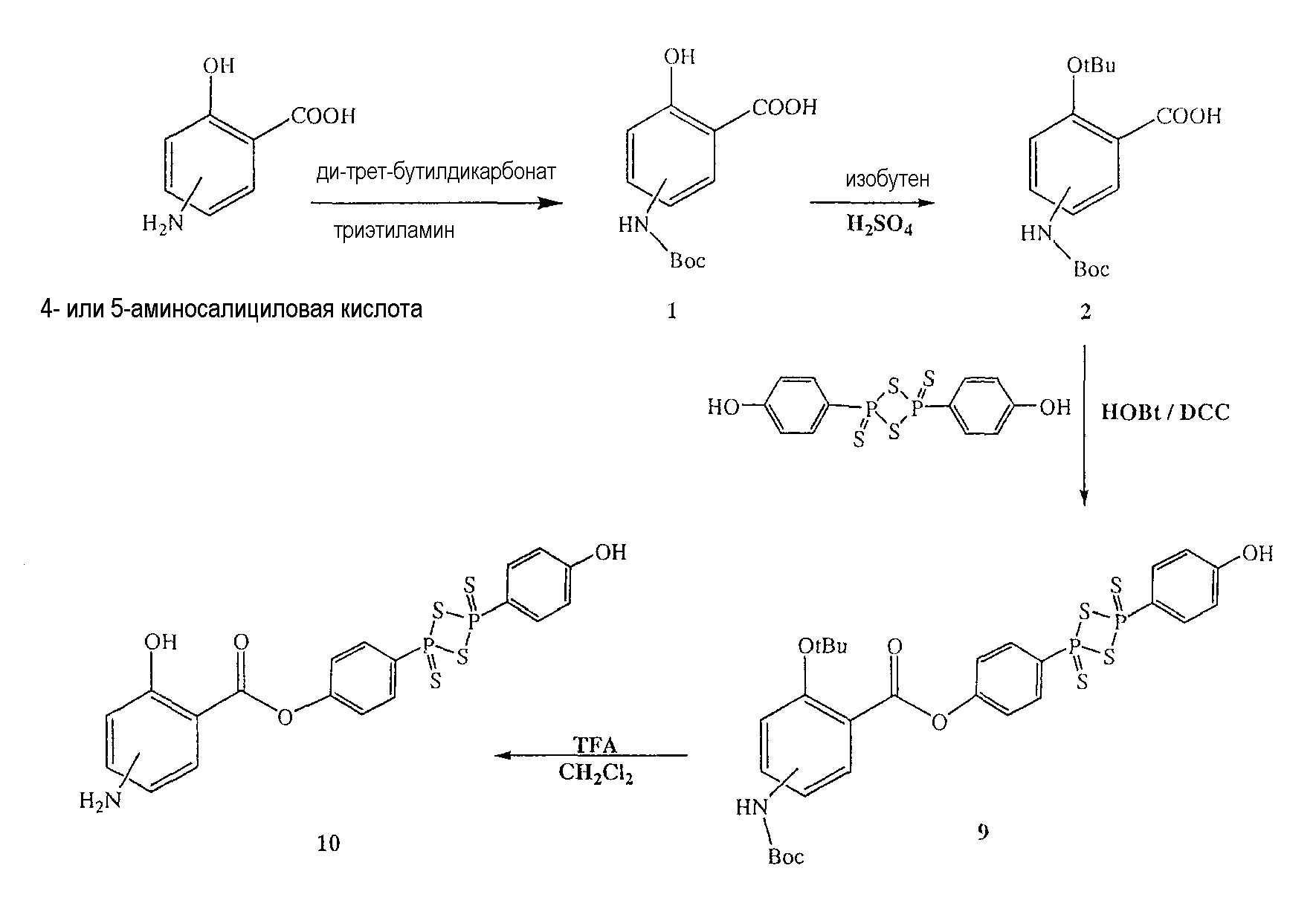
Синтез ангидрида (п-гидроксифенил)дитиофосфоновой кислоты
Красный фосфор (4 г, 0,129 моль), серу (4 г, 0,125 моль) и PhOH (4 г, 0,042 моль) нагревали в течение 5,5 часа при 155-158°С; реакционную смесь охлаждали при комнатной температуре и осадок собирали (5,5 г, выход 34%), температура плавления 224-226°С. Результаты анализов ЯМР и масс-спектроскопии (MS) соответствовали структуре ангидрида (п-гидроксифенил)дитиофосфоновой кислоты.

Синтез 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойной кислоты (1)
К раствору 4- или 5-аминосалициловой кислоты (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли триэтиламин (15,0 ммоль) и ди-трет-бутилдикарбонат (15,0 ммоль), перемешивая смесь при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойную кислоту (1) (выход 80%).
Синтез 4- или 5-трет-бутоксикарбониламино-2-трет-бутоксибензойной кислоты (2)
Соединение (1) (12,0 ммоль), концентрированную H2SO4 (6,0 ммоль) и DCM (100 мл) перемешивали в атмосфере газообразного изобутилена под давлением 0,35 атм (5 фунтов/кв.дюйм) в течение 6 часов при комнатной температуре. Раствор промывали холодным 10% NaHCO3 (2×100 мл) и насыщенным раствором соли (100 мл), сушили (Na2SO4) и упаривали. Остаток растворяли в MeOH/CCl4 (1:1) (400 мл), промывали водой (300 мл) и экстрагировали MeOH/водой (1:1) (2×200 мл). Экстракт сушили (Na2SO4) и упаривали с образованием белого твердого вещества (2), которое перекристаллизовывали DCM/гексаном (выход 83%).
Синтез 4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]-дитиадифосфетан-2-ил]фенилового эфира 4- или 5-амино-2-гидроксибензойной кислоты (10)
К раствору 4- или 5-трет-бутоксикарбониламино-2-трет-бутоксибензойной кислоты (2) (3,0 ммоль) в 50 мл диметилформамида добавляли гидроксибензотриазол (3,3 ммоль) и DCC (3,3 ммоль), перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли ангидрид п-гидроксифенилдитиофосфоновой кислоты (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный промежуточный продукт (9) обрабатывали раствором TFA (40%) в CH2Cl2. Через 2 часа растворитель удаляли с образованием соединения (10) в виде неочищенного остатка. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом 4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]фениловый эфир 4- или 5-амино-2-гидроксибензойной кислоты (10), соединение формулы XVII, (выход 73%).
Синтез меркаптоэтансульфоната 4- или 5-амино-2-гидроксибензойной кислоты (2)

Смесь 2-меркаптоэтилового эфира сернистой кислоты (0,1 моль) в 100 мл этилацетата добавляли к раствору 4- или 5-аминосалициловой кислоты (1) (0,1 моль в 100 мл этилацетата) в течение 30-45 минут при 20-25°С в инертной атмосфере. Затем смесь перемешивали при 0-5°С в течение 1 часа и фильтровали, получая при этом меркаптоэтансульфонат 4- или 5-амино-2-гидроксибензойной кислоты (2) (выход 98%).
Пример 12
Синтез 4- или 5-амино-2-(2-ацетиламино-3-меркаптопропионилокси)-бензойной кислоты (3) [соединение формулы XII]
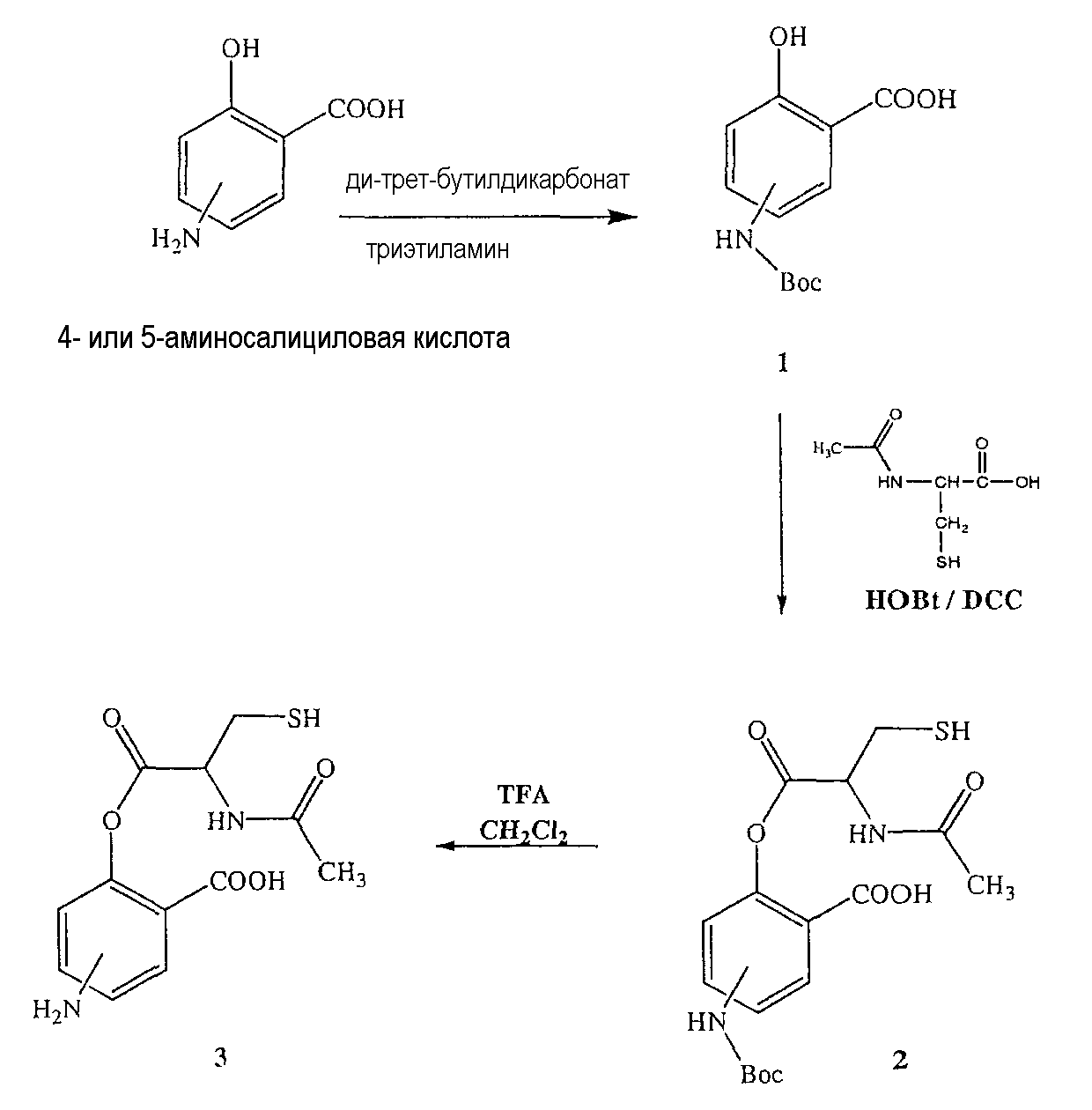
Синтез 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойной кислоты (1)
К раствору 4- или 5-аминосалициловой кислоты (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли триэтиламин (15,0 ммоль) и ди-трет-бутилдикарбонат (15,0 ммоль), перемешивая смесь при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойную кислоту (1) (выход 80%).
Синтез 4- или 5-амино-2-(2-ацетиламино-3-меркаптопропионилокси)бензойной кислоты (3)
К раствору 2-ацетиламино-3-меркаптопропионовой кислоты (3,0 ммоль) в 50 мл диметилформамида добавляли гидроксибензотриазол (3,3 ммоль) и DCC (3,3 ммоль), перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойную кислоту (2) (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный промежуточный продукт (2) обрабатывали раствором TFA (40%) в CH2Cl2. Через 2 часа растворитель удаляли с образованием соединения (3) в виде неочищенного остатка. Остаток вводили в открытую коолонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом 4- или 5-амино-2-(2-ацетиламино-3-меркаптопропионилокси)бензойную кислоту (3), соединение формулы XII, (выход 52%).
Пример 13
Синтез ангидрида 4- или 5-амино-2-гидроксибензойной кислоты с 2-ацетиламино-3-меркаптопропионовой кислотой (4) [соединение формулы Х]

Синтез 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойной кислоты (1)
К раствору 5-аминосалициловой кислоты (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли триэтиламин (15,0 ммоль) и ди-трет-бутилдикарбонат (15,0 ммоль), перемешивая смесь при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойную кислоту (1) (выход 80%).
Синтез 4- или 5-трет-бутоксикарбониламино-2-трет-бутоксибензойной кислоты (2)
Соединение (1) (12,0 ммоль), концентрированную H2SO4 (6,0 ммоль) и DCM (100 мл) перемешивали в атмосфере газообразного изобутилена под давлением 0,35 атм (5 фунтов/кв.дюйм) в течение 6 часов при комнатной температуре. Раствор промывали холодным 10% NaHCO3 (2×100 мл) и насыщенным раствором соли (100 мл), сушили (Na2SO4) и упаривали. Остаток растворяли в MeOH/CCl4 (1:1) (400 мл), промывали водой (300 мл) и экстрагировали MeOH/водой (1:1) (2×200 мл). Экстракт сушили (Na2SO4) и упаривали с образованием белого твердого вещества (2), которое перекристаллизовывали DCM/гексаном (выход 83%).
Синтез ангидрида 4- или 5-амино-2-гидроксибензойной кислоты с 2-ацетиламино-3-меркаптопропионовой кислотой (4)
К раствору 4- или 5-трет-бутоксикарбониламино-2-трет-бутоксибензойной кислоты (2) (3,0 ммоль) в 50 мл диметилформамида добавляли DCC (3,3 ммоль), перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли 2-ацетиламино-3-меркаптопропионовую кислоту (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органические слои промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный промежуточный продукт (3) обрабатывали раствором TFA (40%) в CH2Cl2. Через 2 часа растворитель удаляли с образованием соединения (4) в виде неочищенного остатка. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом ангидрид 4- или 5-амино-2-гидроксибензойной кислоты с 2-ацетиламино-3-меркаптопропионовой кислотой (4), соединение формулы Х, (выход 68%).
Пример 14
Синтез 4- или 5-(2-ацетиламино-3-меркаптопропиониламино)-2-гидроксибензойной кислоты (5) [соединение формулы XI]

Синтез 4- или 5-(9Н-флуорен-9-илметоксикарбониламино)-2-гидроксибензойной кислоты (1)
К раствору 4- или 5-аминосалициловой кислоты (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли Na2CO3 (10%) (15 мл) и Fmoc-OSu (15,0 ммоль), перемешивая смесь при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойную кислоту (1) (выход 90%).
Синтез трет-бутилового эфира 4- или 5-амино-2-трет-бутоксибензойной кислоты (3)
Соединение (1) (12,0 ммоль), концентрированную H2SO4 (6,0 ммоль) и DCM (100 мл) перемешивали в атмосфере газообразного изобутилена под давлением 0,49 атм (7 фунтов/кв.дюйм) в течение 24 часов при комнатной температуре. Раствор промывали холодным 10% NaHCO3 (2×100 мл) и насыщенным раствором соли (100 мл), сушили (Na2SO4) и упаривали. Остаток растворяли в MeOH/CCl4 (1:1) (400 мл), промывали водой (300 мл) и экстрагировали MeOH/водой (1:1) (2×200 мл). Экстракт сушили (Na2SO4) и упаривали с образованием белого твердого вещества (2). Неочищенный промежуточный продукт (2) обрабатывали раствором диэтиламина (33%) в THF (тетрагидрофуране). Через 2 часа растворитель удаляли с образованием соединения (3) в виде неочищенного остатка. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом трет-бутиловый эфир 4- или 5-амино-2-трет-бутоксибензойной кислоты (3) (выход 67%).
Синтез 4- или 5-(2-ацетиламино-3-меркаптопропиониламино)-2-гидроксибензойной кислоты (5)
К раствору 2-ацетиламино-3-меркаптопропионовой кислоты (3,0 ммоль) в 50 мл диметилформамида добавляли гидроксибензотриазол (3,3 ммоль) и DCC (3,3 ммоль), перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли трет-бутиловый эфир 4- или 5-амино-2-трет-бутоксибензойной кислоты (3) (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный промежуточный продукт (4) обрабатывали раствором TFA (40%) в CH2Cl2. Через 2 часа растворитель удаляли с образованием соединения (5) в виде неочищенного остатка. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом 4- или 5-(2-ацетиламино-3-меркаптопропиониламино)-2-гидроксибензойную кислоту (5), соединение формулы XI, (выход 78%).
Пример 15
Синтез 4- или 5-амино-2-(2-меркаптоэтоксисульфонилокси)бензойной кислоты (3) [соединение формулы XXXIV]
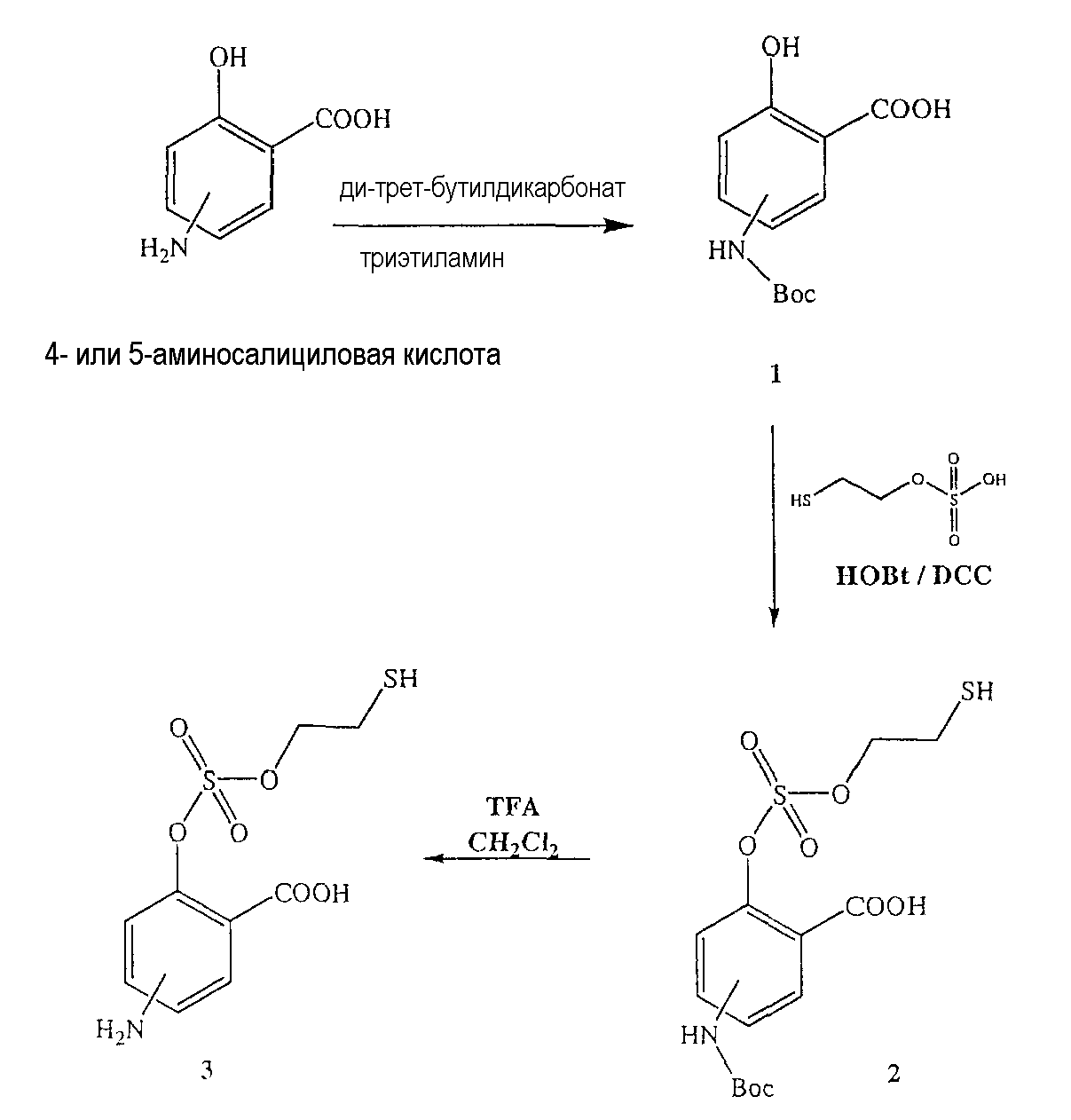
Синтез 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойной кислоты (1)
К раствору 4- или 5-аминосалициловой кислоты (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли триэтиламин (15,0 ммоль) и ди-трет-бутилдикарбонат (15,0 ммоль), перемешивая смесь при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойную кислоту (1) (выход 80%).
Синтез 4- или 5-амино-2-(2-меркаптоэтоксисульфонилокси)бензойной кислоты (3)
К раствору моно(2-меркаптоэтилового) эфира серной кислоты в 50 мл диметилформамида добавляли гидроксибензотриазол (3,3 ммоль) и DCC (3,3 ммоль), перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойную кислоту (2) (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органический слой промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный промежуточный продукт (2) обрабатывали раствором TFA (40%) в CH2Cl2. Через 2 часа растворитель удаляли с образованием соединения (3) в виде неочищенного остатка. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом 4- или 5-амино-2-(2-меркаптоэтоксисульфонилокси)бензойную кислоту (3), соединение формулы XXXIV, (выход 57%).
Пример 16
Синтез ангидрида 4- или 5-амино-2-гидроксибензойной кислоты с моно(2-меркаптоэтиловым) эфиром серной кислоты (4) [соединение формулы XXXIII]
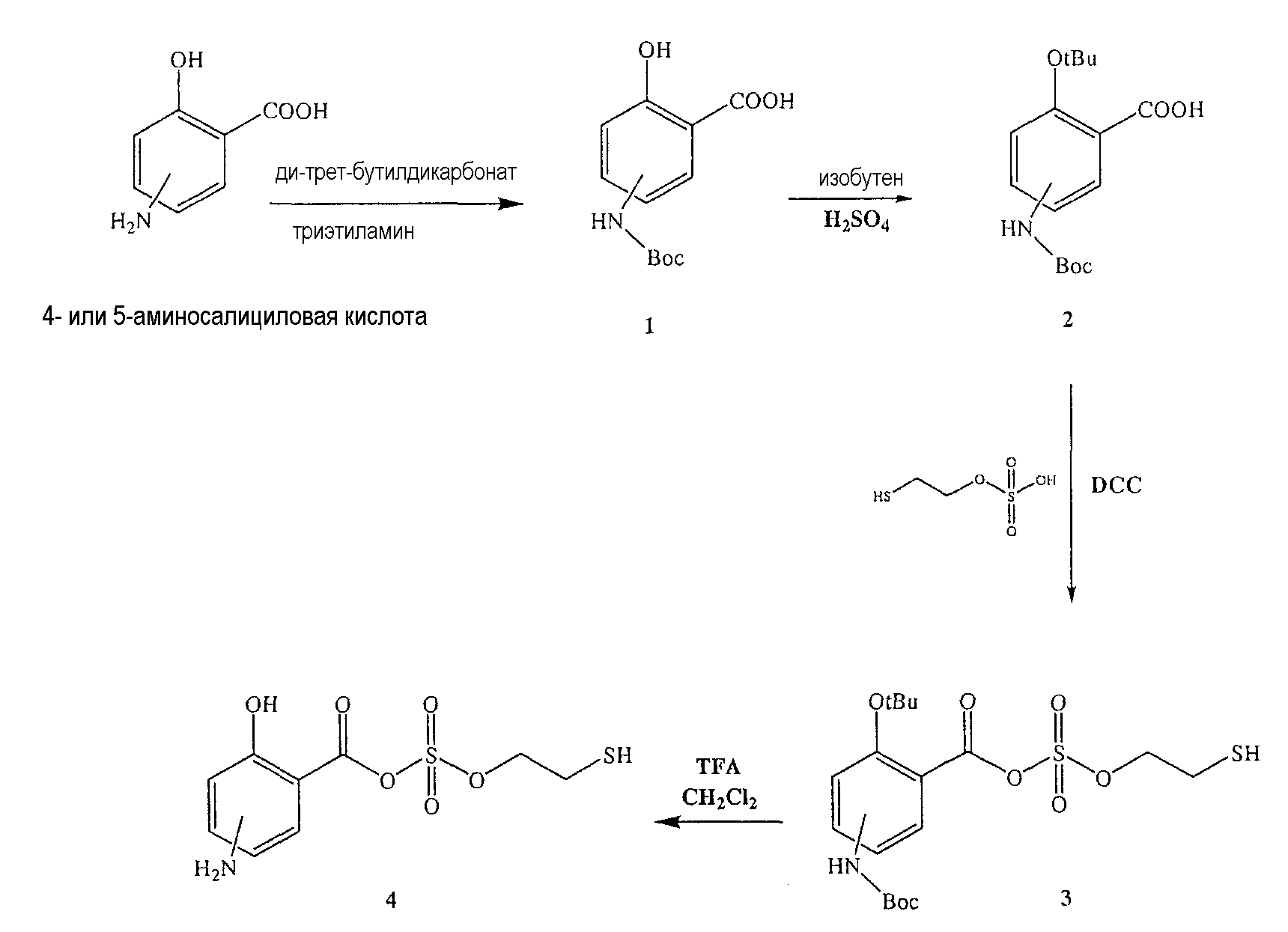
Синтез 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойной кислоты (1)
К раствору 5-аминосалициловой кислоты (10,0 ммоль) в 25 мл диоксана и 12,5 мл воды добавляли триэтиламин (15,0 ммоль) и ди-трет-бутилдикарбонат (15,0 ммоль), перемешивая смесь при 0°С в течение 1/2 часа. Реакционную смесь перемешивали механической мешалкой в течение 24 часов при комнатной температуре. Растворитель выпаривали и к остатку по каплям добавляли 3М раствор HCl (15 мл). Осадок фильтровали, промывали водой и сушили. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (9/1), получая при этом 4- или 5-трет-бутоксикарбониламино-2-гидроксибензойную кислоту (1) (выход 80%).
Синтез 4- или 5-трет-бутоксикарбониламино-2-трет-бутоксибензойной кислоты (2)
Соединение (1) (12,0 ммоль), концентрированную H2SO4 (6,0 ммоль) и DCM (100 мл) перемешивали в атмосфере газообразного изобутилена под давлением 0,35 атм (5 фунтов/кв.дюйм) в течение 6 часов при комнатной температуре. Раствор промывали холодным 10% NaHCO3 (2×100 мл) и насыщенным раствором соли (100 мл), сушили (Na2SO4) и упаривали. Остаток растворяли в MeOH/CCl4 (1:1) (400 мл), промывали водой (300 мл) и экстрагировали MeOH/водой (1:1) (2×200 мл). Экстракт сушили (Na2SO4) и упаривали с образованием белого твердого вещества (2), которое перекристаллизовывали DCM/гексаном (выход 83%).
Синтез ангидрида 4- или 5-амино-2-гидроксибензойной кислоты с моно(2-меркаптоэтиловым) эфиром серной кислоты (4)
К раствору 4- или 5-трет-бутоксикарбониламино-2-трет-бутоксибензойной кислоты (2) (3,0 ммоль) в 50 мл диметилформамида добавляли DCC (3,3 ммоль), перемешивая смесь при 0°С в течение 1 часа. К реакционной смеси добавляли моно(2-меркаптоэтиловый) эфир серной кислоты (3,0 ммоль) и перемешивали механической мешалкой в течение 3 часов при 0°С и в течение 72 часов при комнатной температуре. Смесь фильтровали и фильтрат упаривали при пониженном давлении, чтобы удалить растворитель. Полученный таким образом маслянистый остаток растворяли в этилацетате; органические слои промывали насыщенным раствором соли, сушили над безводным MgSO4, фильтровали и выпаривали растворитель. Неочищенный промежуточный продукт (3) обрабатывали раствором TFA (40%) в CH2Cl2. Через 2 часа растворитель удаляли с образованием соединения (4) в виде неочищенного остатка. Остаток вводили в открытую колонку с силикагелем и элюировали CH2Cl2/MeOH (8/2), получая при этом ангидрид 4- или 5-амино-2-гидроксибензойной кислоты с моно(2-меркаптоэтиловым) эфиром серной кислоты (4), соединение формулы XXXIII (выход 68%).
Исследование соединений
Пример 17
4-(5-Тиоксо-5Н-[1,2]дитиол-3-ил)фениловый эфир 5-амино-2-гидроксибензойной кислоты [соединение, определяемое в настоящем описании изобретения как соединение XXXV]
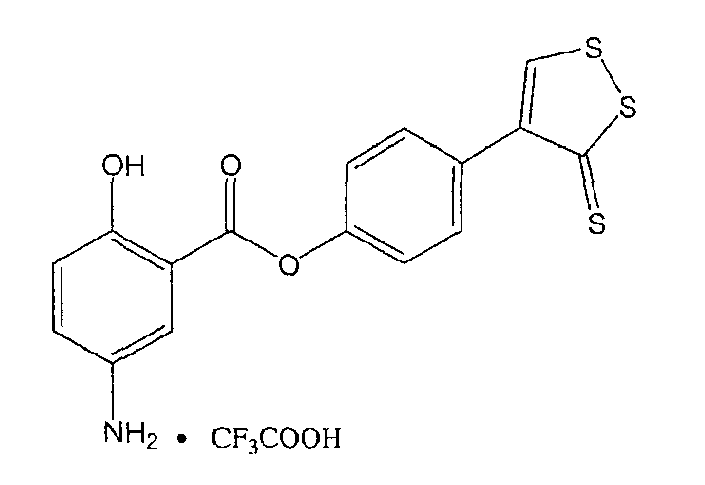
Тонкослойную хроматографию выполняли на пластинках с силикагелем 50 Macherey-Nagel™, используя флуоресцентный индикатор, и визуализировали пластинки ультрафиолетовым светом (254 нм). Для хроматографии на колонках использовали Kieselgel™ 60. Все синтетические реагенты были приобретены в химической компании Aldrich-Sigma и использованы без очистки. Растворители характеризовались аналитической или более высокой чистотой и были использованы в том виде, как они были поставлены. Растворы сушили над Na2SO4 и для удаления растворителей в вакууме использовали роторный испаритель R-114 Buchi™. Достоверность структур проверяли спектроскопическими методами при помощи1Н-ЯМР и13С-ЯМР. Спектры были получены в приборе Varian Mercury Plus 400. Спектры регистрировали в ДМСО. Для обозначения максимальных характеристик были использованы следующие аббревиатуры: с (синглет), д (дублет). Химические сдвиги определяли относительно Me4Si, используемого в качестве внутреннего эталона. Масс-спектры синтезированных продуктов были получены при помощи масс-спектрометра API 2000 Applayed Biosystem™. Температуру плавления определяли в устройстве для измерения температуры в горячем состоянии Kofler™ и не корректировали.
1H ЯМР (ДМСО): δ 7,07 (д, 2H), 7,38 (д, 2H), 7,46 (д, 2H), 7,79 (с, 1H), 7,85 (с, 1H), 8,01 (д, 2H), 10,35 (с, -OH);
13C ЯМР (ДМСО): δ 114,6, 119,6, 123,9, 127,7, 128,7, 129,4, 129,8, 136,1, 153,8, 158,8, 165,4, 173,2, 189,7, 216,2;
MS (EI), m/e 362 (M+);
температура плавления: 93-95°С.
Пример 18
4-Тиокарбамоилфениловый эфир 5-амино-2-гидроксибензойной кислоты [соединение, определяемое в настоящем описании изобретения как соединение XXVII]

Тонкослойную хроматографию выполняли на пластинках с силикагелем 50 Macherey-Nagel, используя флуоресцентный индикатор, и визуализировали пластинки ультрафиолетовым светом (254 нм). Для хроматографии на колонках использовали Kieselgel 60. Все синтетические реагенты были приобретены в химической компании Aldrich-Sigma и использованы без очистки. Растворители характеризовались аналитической или более высокой чистотой и были использованы в том виде, как они были поставлены. Растворы сушили над Na2SO4 и для удаления растворителей в вакууме использовали роторный испаритель R-114 Buchi. Достоверность структур проверяли спектроскопическими методами при помощи1Н-ЯМР и13С-ЯМР. Спектры были получены в приборе Varian Mercury Plus 400. Спектры регистрировали в ДМСО. Для обозначения максимальных характеристик были использованы следующие аббревиатуры: с (синглет), д (дублет). Химические сдвиги определяли относительно Me4Si, используемого в качестве внутреннего эталона. Масс-спектры синтезированных продуктов были получены при помощи масс-спектрометра API 2000 Applayed Biosystem. Температуру плавления определяли в устройстве для измерения температуры в горячем состоянии Kofler и не корректировали.
1H ЯМР (ДМСО): δ 7,03 (д, 1H), 7,31 (д, 2H), 7,32 (с, 1H), 7,71 (д, 1H), 7,97 (д, 1H), 9,55 (с, NH2), 9,91 (с, NH2), 10,25 (с, -OH);
13C ЯМР (ДМСО): δ 114,4, 119,5, 122,1, 122,7, 129,2, 129,5, 138,1, 152,1, 157,7, 165,9, 189,7, 199,7;
MS (EI), m/e 289 (M+);
температура плавления: 193-195°С.
Испытание соединений
Пример 19
Исследование диапазона дозирования гидрохлорида 4-(тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 2-гидрокси-5-аминобензойной кислоты (соединение XXXV) в модели TNBS-индуцированного колита у мышей
В нижеследующем примере использована стандартная экспериментальная животная модель колита, индуцированного у мышей путем введения в ободочную кишку 2,4,6-тринитробензолсульфоновой кислоты (TNBS). Подробное описание указанной модели приведено в публикации Santucci et al. (2003) Gastroenterology 124:1381-94, которая включена в настоящее описание изобретения в качестве ссылки. Мышам Balb/c в возрасте 6-8 недель вводили в ободочную кишку TNBS в дозе 1,5 мг в 0,1 мл 30% этанола. Мышей произвольно распределяли в разные экспериментальные группы (n=6 в каждой группе). Через один час после введения TNBS и затем через каждые 12 часов в течение 5 дней мышам перорально вводили наполнитель (1% карбоксиметилцеллюлоза (СМС)), 5-ASA (мезаламин) (25, 50 или 75 мг/кг) или эквимолярные дозы гидрохлорида 4-(тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 2-гидрокси-5-аминобензойной кислоты (соединение XXXV) (130 мг/кг) или 66% (100 мг/кг), 50% (66 мг/кг) и 25% (33 мг/кг) указанной дозы. В последний день исследования у мышей определяли (слепым методом) наличие диареи и скрытой крови в фекалиях и измеряли массу тела. На основании полученных данных высчитывали “оценку интенсивности заболевания” (по шкале от 0 до 4 в соответствии с описанием, приведенным в вышеуказанной публикации). Затем мышей умерщвляли и иссекали ободочную кишку для измерения активности миелопероксидазы (МРО) в качестве маркера инфильтрации гранулоцитов. Все результаты сравнивали с результатами, полученными у здоровых мышей.
Данные оценки интенсивности заболевания и активности МРО показаны соответственно на фиг.1 и фиг.2. На фиг.1 показано, что соединение XXXV превосходит мезаламин в отношении уменьшения оценки интенсивности заболевания при введении в эквимолярных дозах, равных 50 мг/кг и 75 мг/кг. Кроме того, как показано на фиг.2, активность МРО значительно сократилась (почти наполовину) при введении самых высоких испытанных доз.
Пример 20
Сравнение индекса интенсивности заболевания и активности МРО соединения XXXV с отдельно используемым 5-ASA (мезаламин), отдельно используемым ADT-OH и смесью мезаламина и ADT-OH
На фиг.3 и 4 показаны соответственно значения индекса интенсивности заболевания и активности МРО при использовании экспериментальной животной модели колита, аналогичной описанной выше, в которой соединение XXXV (130 мг/кг) сравнивали с эквимолярными дозами двух его компонентов: мезаламина (50 мг/кг) и 5-п-гидроксифенил-1,2-дитион-3-тиона (ADT-OH) (80 мг/кг) и смеси мезаламина (50 мг/кг) и ADT-OH (80 мг/кг). Значение *р<0,05 по сравнению с группой, которой вводили носитель. Каждая группа состояла по меньшей мере из 5 крыс.
На фиг.3 показано, что соединение XXXV почти в два раза эффективнее ослабляет симптомы заболевания по сравнению с отдельно используемым мезаламином, отдельно используемым ADT-OH или смесью мезаламина и ADT-OH. Кроме того, на фиг.4 показано, что соединение XXXV в значительной степени уменьшает воспаление, о чем свидетельствует сокращение инфильтрации гранулоцитов (пониженная активность МРО).
Пример 21
Сравнение индекса интенсивности заболевания и активности МРО 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты (соединение XXVII)
На фиг.6 и 7 показаны соответственно значения индекса интенсивности заболевания и активности МРО при использовании экспериментальной животной модели колита, аналогичной описанной выше, в которой соединение XXVII (100 мг/кг) сравнивали с эквимолярными дозами двух его компонентов: мезаламина (50 мг/кг) и 4-гидрокситиобензамида (4-НТВ) (50 мг/кг), отдельно используемого мезаламина (50 мг/кг) и отдельно используемого 4-НТВ (50 мг/кг). Значение *р<0,05 по сравнению с группой, которой вводили носитель. Каждая группа состояла по меньшей мере из 5 крыс.
На фиг.5 показано, что соединение XXVII почти в три раза эффективнее ослабляет симптомы заболевания по сравнению с отдельно используемым мезаламином, отдельно используемым 4-НТВ или смесью мезаламина и 4-НТВ. Кроме того, на фиг.6 показано, что соединение XXVII в значительной степени уменьшает воспаление, о чем свидетельствует сокращение инфильтрации гранулоцитов (пониженная активность МРО).
Пример 22
Действие мезаламина и соединения XXXV при лечении TNBS-индуцированного колита у мышей
Была использована животная модель, аналогичная описанной выше. В данном примере действие мезаламина (50 мг/кг) сравнивали с действием эквимолярных доз соединения XXXV. Помимо измерения тяжести колита путем определения оценки интенсивности заболевания и активности МРО были исследованы ткани для определения числа генов воспалительных цитокинов и других медиаторов.
В частности, в соответствии с описанием, приведенным в публикации Wallace et al. (1999) Gastroenterology 117:557-566, которая включена в настоящее описание изобретения в качестве ссылки, у мышей измеряли экспрессию мРНК альфа-фактора некроза опухолей (TNF-α), гамма-интерферона (IFN-γ), интерлейкина (IL)-1, IL-2, IL-10, IL-12 p40 в ободочной кишке, RANTES, циклооксигеназы (СОХ)-1, СОХ-2, конститутивной эндотелиальной синтазы оксида азота (eNOS) и индуцибельной NOS (iNOS).
Для обнаружения и количественного определения мРНК определенного цитокина/хемокина/фермента использовали полимеразную реакцию синтеза цепи с обратной транскрипцией (RT-PCR). Глицеральдегид-3-фосфат-дегидрогеназу (GAPDH) использовали в качестве “обязательного гена” для экспрессии мРНК (то есть в качестве внутреннего эталона). Для каждого образца определяли отношение амплификации гена-мишени к амплификации GAPDH (экспрессию каждого гена измеряли методом денситометрии на гелях). Затем относительную амплификацию (экспрессию) гена-мишени в тканях экспериментальных групп сравнивали с экспрессией в тканях здоровых контрольных животных. Таким образом, данные, показанные на фиг.7-14, представляют относительную экспрессию гена-мишени (нормализованную в отношении экспрессии GAPDH) в виде отношения к экспрессии у здоровых контрольных животных.
Со ссылкой на фиг.7-14 можно отметить, что соединение XXXV превосходило мезаламин по всем показателям. Особенно интересным является то, что соединение XXXV подавляло экспрессию мРНК нескольких провоспалительных цитокинов и хемокинов, опосредующих патогенез воспалительного заболевания кишечника. Однако соединение XXXV не подавляло экспрессию мРНК IL-10, который является противовоспалительным цитокином.
Кроме того, соединение XXXV подавляло мРНК СОХ-1 и СОХ-2. СОХ-1 и СОХ-2 участвуют в синтезе простагландинов, которые играют важную роль в воспалении. Соединение XXXV подавляло также мРНК eNOS и iNOS. Как eNOS, так и iNOS опосредуют заболевания желудочно-кишечного тракта.
Пример 23
Сравнение действия соединения XXXV с мезаламином в отношении ингибирования жизнеспособности раковых клеток ободочной кишки человека НТ-29 in vitro
Клетки НТ-29 выращивали в культуре стандартными методами. Указанные клетки подвергали воздействию наполнителя (ДМСО), мезаламина или соединения XXXV. Были испытаны концентрации в диапазоне от 0,1 до 10 мкМ, причем каждую концентрацию испытывали в 6 лунках. Жизнеспособность клеток измеряли в конце 72-часового воздействия испытуемых лекарственных средств при помощи анализа МТТ [бромид 3-(4,5-диметилтиаксол-2-ил)-2,5-дифенилтетразолия], описанного в публикации Carmichael et al. (1978) Cancer Res. 47, 936-942, которая включена в настоящее описание изобретения в качестве ссылки. Степени жизнеспособности клеток высчитывали в виде процентного значения относительно клеток, обработанных наполнителем (ДМСО); полученные результаты приведены в таблице.
Пример 24
Сравнение действия соединения XXXV с мезаламином в модели восприятия висцеральной боли у крыс
В нижеследующем примере была использована модель восприятия висцеральной боли у крыс, которая является предклинической моделью спастического колита. Крыс (самцы, Wistar, 200-250 г, предоставлены компанией Charles River, Monza, Italy) содержали в пластиковых клетках в контролируемых условиях с 12-часовым циклом чередования света и темноты при включении освещения в 7,00 часов. Крысы имели свободный доступ к водопроводной воде и стандартному лабораторному корму. Перед началом экспериментов крыс индивидуально тренировали, помещая их на 2-3 часа в день в клетку из плексигласа в течение 2-3 дней. Такая тренировка позволяла крысам привыкнуть к среде обитания с ограничением подвижности. Крыс не кормили в течение 12 часов до регистрации расширения ободочной и прямой кишок (CRD). Эксперименты выполняли слепым методом с использованием бодрствующих крыс, при этом исследователь не был осведомлен о типе лекарственного средства, вводимого каждому животному.
В день испытания крыс подвергали воздействию седативного средства путем ингаляции простого эфира, вводили в прямую кишку латексный баллон длиной 2 см на расстояние 2 см от анального отверствия и фиксировали у основания хвоста. Баллон был соединен двухцилиндровой канюлей с датчиком давления для постоянного контроля за давлением в прямой кишке при помощи компьютера (PowerLab PC, A.D. Instruments, Milford, MA, USA) и со шприцем для увеличения/уменьшения объема баллона. Крыс помещали в клетку небольшого размера (20×8×8 см), расположенную на приподнятой платформе из плексигласа (Plexiglas™), и предоставляли им возможность проснуться и адаптироваться в течение 1 часа. После восстановления от воздействия седативного средства животных подвергали процедуре CRD и исследовали поведенческие реакции. В ночь перед выполнением экспериментов баллоны раздували и оставляли на ночь, в результате чего латекс растягивался и баллоны становились эластичными.
Процедуру CDR продолжительностью 20 секунд выполняли через каждые 5 минут с увеличением количества вводимой воды на 0,4 мл, начиная с 0,4 мл и кончая 1,6 мл. Для точного измерения параметров ободочной кишки и восприятия боли расширение повторяли дважды для каждого вводимого количества воды, и усредняли данные, полученные при каждом анализе. Каждого животного подвергали двум сериям CRD. Через двадцать минут после первой серии CRD (0,4 мл - 1,6 мл воды) внутрибрюшинно (i.p.) вводили лекарственные средства и выполняли вторую серию CRD. Затем оценивали и сравнивали поведенческие реакции во время первой и второй серий CRD.
Поведенческую реакцию на CRD оценивали путем измерения абдоминального сгибательного рефлекса (AWR) методом полуколичественной оценки (1). AWR является непроизвольным двигательным рефлексом, подобным висцеромоторному рефлексу, но большим преимуществом AWR в отличие от висцеромоторного рефлекса является то, что в данном случае не требуется хирургического вскрытия брюшной полости для имплантации регистрирующих электродов и проводов в мышцы стенки брюшной полости, что может вызывать дополнительное повышение чувствительности (см. публикацию Ness, T.J. and Gebhart, G.F. (1990) Pain 41:167-234, которая включена в настоящее описание изобретения в качестве ссылки).
Измерение AWR включало визуальное наблюдение за реакцией животного на возрастающее расширение ободочной и прямой кишки (CRD) действующим вслепую исследователем и присвоение оценки AWR по шкале поведенческой реакции, ранее описанной в публикации Al-Chaer, E.D. et al. (2000) Gastroenterology 19: 1276-85, включенной в настоящее описание изобретения в качестве ссылки, в которой оценка 0 соответствует отсутствию поведенческой реакции на CRD, оценка 1 соответствует кратковременному движению головы в начале стимуляции с последующей неподвижностью, оценка 2 соответствует слабому сокращению мышц брюшной полости, при этом крыса не отрывает живот от платформы, оценка 3 соответствует сильному сокращению мышц брюшной полости с отрыванием живота от платформы, и оценка 4 соответствует очень сильному сокращению мышц брюшной полости, о чем свидетельствует выгибание туловища, поднятие живота, таза и мошонки.
Действие мезаламина и соединения XXXV на расширение ободочной кишки и восприятие боли определяли, используя в общей сложности 8 голодающих крыс. Для исследования воздействия, оказываемого введением мезаламина и соединения XXXV на устранение боли, вызываемой CRD, после первой серии CRD 4 крысам внутрибрюшинно (i.p.) вводили мезаламин в дозе 100 мг/кг или соединение XXXV в дозе 100 мг/кг, после чего выполняли вторую серию CRD. Результаты указанных экспериментов показаны на фиг.15(а) и (b).
Для определения воздействия мезаламина или соединения XXXV на гладкие мышцы ободочной кишки расширение ободочной и прямой кишки во время CRD определяли на основании объема и давления, создаваемого в ободочной и прямой кишке, и выражали в мл/мм Hg. Результаты показаны на фиг.16(а) и (b).
Все данные выражены в виде среднего значения ± стандартная ошибка среднего (SEM) при величине выборки, равной 4 крысам в группе; статистическое сравнение парных данных выполняли в соответствии с ранговым критерием со знаком Вилкоксона. Ассоциированная вероятность (значение р) менее 5% считалась значимой.
На фиг.15(а) и (b) показано, что соединение XXXV является более эффективным, чем мезаламин (и наполнитель) в отношении ослабления висцеральной боли, вызываемой расширением ободочной и прямой кишок. Кроме того, соединение XXXV успешно уменьшает давление в прямой кишке, как показано на фиг.16(b).
Таким образом, соединение XXXV, которое, как было установлено, оказывает также эффективное противовоспалительное действие, пригодно для лечения разных воспалительных заболеваний пищеварительного тракта, а также функциональных желудочно-кишечных заболеваний, таких как спастический колит, диспепсия и т.д., которые характеризуются повышенной висцеральной ноцицепцией (при наличии или отсутствии воспаления).
Пример 25
Оценки восприятия боли при введении соединения XXXV и соединения XXVII с глибенкламидом или без указанного средств
Вышеописанную модель восприятия висцеральной боли крысами использовали для сравнения оценок восприятия боли при введении соединения XXXV и соединения XXVII вместе с глибенкламидом или без указанного средства, которое является ингибитором АТР-чувствительных К+ (КАТР) каналов.
На фиг.17 показана оценка восприятия боли, возникающей в результате введения в ободочную и прямую кишки 0,8 мл воды, в группах крыс (по меньшей мере 5 животных в группе), которым вводили наполнитель, мезаламин (100 мг/кг), соединение XXXV (100 мг/кг) или соединение XXVII (100 мг/кг). Соединения XXXV и XXVII значительно ослабляли восприятие боли (*р<0,05 по сравнению с группой, которой вводили наполнитель), в то время как мезаламин не оказывал значительного действия. Подобное ослабление восприятия боли соединением XXXV и соединением XXVII блокировалось в результате предварительного введения глибенкламида (внутрибрюшинное введение 10 мг/кг за 30 минут до исследования), при этом предварительное введение глибенкламида не влияло на восприятие боли в группах животных, которым вводили наполнитель или мезаламин, из чего следует, что антиноцицептивное действие соединений XXXV и XXVII может быть опосредовано АТР-чувствительнымии К+ (КАТР) каналами.
Пример 26
Сравнение оценок восприятия боли для соединения XXXV и соединения XXVII с мезаламином
На фиг.19 показаны результаты эксперимента с использованием вышеописанной модели восприятия боли. Действие мезаламина (50 мг/кг) сравнивали с действием эквимолярных доз соединения XXXV (130 мг/кг), ADT-OH (80 мг/кг), соединения XXVII (100 мг/кг) и 4-НТВ (50 мг/кг). Только соединение XXXV и соединение XXVII значительно ослабляли восприятие боли (*р<0,05) по сравнению с группой животных, которым вводили наполнитель.
Пример 27
Воздействие соединений XXXV и XXVII на адгезию лейкоцитов к эндотелию сосудов in vivo
Адгезию лейкоцитов исследовали при помощи прижизненной микроскопии, подробно описанной в публикации Wallace et al., (1993) Am. J. Physiol. 265: 993-998, которая включена в настоящее описание изобретения в качестве ссылки. Крыс анестезировали пентобарбиталом натрия (внутрибрюшинное введение 60 мг/кг), и делали разрезы с прижиганием в области живота. Для облегчения дыхания выполняли трахеотомию. Крыс клали на спину и через разрез на животе временно выводили на поверхность тела сегмент брыжейки. Брыжейку осторожно помещали на оптически прозрачный смотровой столик, обеспечивающий трансиллюминацию сегмента ткани размером 2 см2. Всю обнаженную ткань покрывали марлей, пропитанной физиологическим раствором, для минимизации обезвоживания. Температуру смотрового столика поддерживали, равной 37°С, и брыжейку обливали подогретым физиологическим раствором с бикарбонатным буфером (рН 7,4). Для наблюдения за микроциркуляцией в брыжейке использовали прижизненный микроскоп (Nikon L25/0,35) и окуляр с 10-кратным увеличением. Для исследования были выбраны посткапиллярные венулы с диаметром от 20 до 40 мкм. Видеокамера, смонтированная на микроскопе (цифровая камера 5000 Panasonic™), проецировала изображения на монитор, с которого изображения записывали при помощи видеокассетного магнитофона для последующего анализа с воспроизведением. Изображения микроциркуляции в брыжейке регистрировали в течение 5 минут до введения аспирина (исходный период), во время введения аспирина (0-5 минут) и через каждые 15 минут в течение 60 минут. Производили количественное определение адгезии лейкоцитов слепым методом на основании видеозаписей изображений сосудов, произведенных в течение 5-минутных периодов, в виде числа лейкоцитов, остававшихся неподвижными на стенке сосуда в течение 30 или более секунд (на 100 мкм длины венулы). Группам крыс (по меньшей мере 5 животных в группе) предварительно вводили соединение XXXV (130 мг/кг), соединение XXVII (100 мг/кг), мезаламин (50 мг/кг) или наполнитель за 60 минут до введения аспирина (или наполнителя). Указанные лекарственные средства вводили внутрижелудочно. В некоторых эспериментах крысам вводили глибенкламид (внутрибрюшинно 10 мг/кг) или наполнитель за 30 минут до введения вышеуказанных соединений.
На фиг.19 показана адгезия лейкоцитов в ответ на внутрижелудочное введение аспирина и действие указанных соединений. Аспирин значительно увеличивал адгезию лейкоцитов по сравнению с исходным периодом (*р<0,05 по сравнению с наполнителем). Предварительное введение соединения XXXV, но не мезаламина, предотвращало вызываемое аспирином увеличение адгезии лейкоцитов. Отдельно вводимый глибенкламид не влиял на адгезию лейкоцитов и на величину вызванной аспирином адгезии лейкоцитов. Глибенкламид также не оказывал действия в группе животных, которым вводили мезаламин вместе с аспирином. Однако глибенкламид блокировал ингибирующее действие соединения XXXV на вызванную аспирином адгезию лейкоцитов.
На фиг.20 показана адгезия лейкоцитов в последний период времени выполнения эксперимента (60-65 минут). Приведенный график иллюстрирует способность соединения XXXV и соединения XXVII подавлять вызванную аспирином адгезию лейкоцитов и возможность блокирования указанного ингибирующего воздействия на адгезию лейкоцитов путем предварительного введения глибенкламида.
Пример 28
Образование H2S 4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фениловым эфиром 5-амино-2-гидроксибензойной кислоты (соединение XXXV) и 4- или 5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислотой (определяемой как соединение XXVII)
Два соединения, соединение XXXV и соединение XXVII, были испытаны в отношении образования H2S в трех разных условиях. Кроме того, были измерены концентрации H2S, образованные в течение 1 часа 1 мМ концентрациями L-цистеина, H2S-высвобождающей части соединения XXXV, ADT-OH (5-(4-аминофенил)-[1,2]дитиол-3-тион), H2S-высвобождающей части соединения XXVII, 4-НВТ (4-гидрокситиобензамид). Высвобождение H2S исследовали в трех условиях: (i) при нахождении соединения в буфере, (ii) при нахождении соединения в гомогенате печени и (iii) при нахождении соединения в гомогенате печени вместе с ингибитором цистатионин-γ-лиазы (PAG = DL-пропаргилглицин; 2 мМ). Результаты показаны на фиг.13. Значение *р<0,05 сравнивали с высвобождением в группе животных, которым вводили наполнитель. Значение **р>0,05 сравнивали с показателем, полученным в соответствующей группе животных, которым вводили гомогенат. Способность ферментов продуцировать H2S определяли, используя реактор, описанный в публикации Khan et al., (1980) Microchem. J. 25: 388-395, которая включена в настоящее описание изобретения в качестве ссылки. В реактор вводили два мл анализируемой реакционной смеси. Указанная смесь содержала 1 мМ L-цистеина (или соединения), 2 мМ пиридоксаль-5'-фосфата, 100 мМ калий-фосфатного буфера (рН=7,4). Через смесь пропускали постоянный поток азота, используя газоподающий капилляр. Реакции инициировали путем переноса пробирок с ледяной бани на водяную баню с температурой 37°С. С потоком азота подавали сернистую кислоту во второй реактор, содержащий 4 мл буферного раствора сульфидного антиоксиданта (SAOB), состоящего из 2М КОН, 1М салициловой кислоты и 0,22М аскорбиновой кислоты при рН 12,8 [5]. После инкубации при 37°С в течение 90 минут к смеси добавляли 1 мл 10% раствора трихлоруксусной кислоты для прекращения реакции. Остаток H2S в смеси отводили с потоком азота в течение последующих 60 минут инкубации при 37°С. Концентрацию сульфида в растворе SAOB измеряли при помощи электрода, чувствительного к сульфиду (электрод S2-/Ag+ модели 9616, Orion Research, Beverly, MA, USA). Для исследований, в которых испытуемые соединения инкубировали в гомогенате печени, 100-150 мг выделенной печени крыс гомогенизировали в 1 мл охлаждаемого льдом экстрактора белка Т-PER. Гомогенаты добавляли к реакционной смеси в 10% концентрации (масс/об). 2 мМ DL-пропаргилглицина инкубировали с гомогенатами печени в течение 5 минут при 37°С до взаимодействия с ферментом. См. публикацию Khan, S.U., Morris, G.F. and Hidiroglou, M. (1980) Rapid estimation of sulfide in rumen and blood with sulfide-specific ion electrode. Microchem. J. 25: 388-395, которая включена в настоящее описание изобретения в качестве ссылки.
Результаты, показанные на фиг.21, позволяют предположить, что производные 4- или 5-ASA по настоящему изобретению и, в частности, соединения XXXV и XXVII обладают нижеследующими отличительными признаками:
1. Указанные производные спонтанно высвобождают H2S (в буфере), который желателен для местного действия в кишечнике. Отдельно используемые H2S-высвобождающие части, ADT-OH, 4-НТВ и L-цистеин не высвобождают значительного количества H2S при инкубации в одном буфере;
2. Высвобождение H2S увеличивается в присутствии ткани;
3. Высвобождение H2S из производных 4- или 5-ASA (отличных от ангидрида 4- или 5-амино-2-гидроксибензойной кислоты с N-ацетилцистеином (формула Х)) происходит независимо от активности двух основных ферментов, необходимых для эндогенного синтеза H2S (цистатионин-β-синтаза и цистатионин-γ-лиаза). Подобный результат продемонстрирован отсутствием воздействия ингибитора указанных ферментов (PAG; DL-пропаргилглицин) на образование H2S соединением XXXV и соединением XXVII. В отличие от этого высвобождение H2S L-цистеином было значительно ингибировано при использовании PAG;
4. Концентрации H2S, продуцированного соединением XXXV и соединением XXVII, находятся в пределах 10-20 мкМ при использовании 1 мМ соединения. Концентрации мезаламина в ободочной кишке могут быть равны 5 мМ после введения субъектам обычных доз данного лекарственного средства (Dif. Dis. Sci. 1989; 34:573-578). Эндогенные концентрации H2S могут достигать 160 мкМ (Antioxid. Redox. Signal. 2003; 5, 493-501). Соединения XXXV и XXVII высвобождают H2S в концентрациях, находящихся в пределах физиологического диапазона, что минимизирует вероятность H2S-обусловленной токсичности. Однако следует отметить, что когда H2S-высвобождающей частью (соединения формулы Х) является н-ацетилцистеин, необходимо использовать меньшую дозу вследствие большего высвобождения H2S цистеином.
Пример 29
Вазорелаксирующее действие H2S-высвобождающих частей молекулы
Нижеследующие эксперименты были выполнены главным образом в соответствии с описанием, приведенным в публикации Bucci, M. et al. (2004) Diabetic mouse angiopathy is linked to progressive sympathetic receptor deletion coupled to an enhanced caveolin-1 expression. Arterioscler. Thromb. Vasc. Biol. 24: 721-726, которая включена в настоящее описание изобретения в качестве ссылки. Мышей CD-1 умерщвляли, быстро иссекали грудную аорту и очищали ее от жира и соединительной ткани. Вырезали кольца длиной 1,5-2 мм и помещали в ванну для выделенных органов (Fort 10 World Precision Instruments, USA), наполненную насыщенным газом раствором Кребса (95% О2 + 5% СО2) при 37°С. Изменения изометрического натяжения регистрировали при помощи системы сбора данных PowerLab™ (Ugo Basile, Italy). Раствор Кребса имел следующий состав (моль/л): NaCl 0,118, KCl 0,0047, MgCl2 0,0012, KH2PO4 0,0012, CaCl2 0,0025, NaHCO3 0,025 и глюкоза 0,010. Кольца сначала растягивали до достижения натяжения в покое, равного 1,5 г, оставляли для уравновешивания по меньшей мере на 40 минут, в течение указанного времени натяжение при необходимости доводили до 1,5 г и периодически меняли раствор в ванне. В предварительном исследовании было установлено, что натяжение в покое, равное 1,5 г, обеспечивает оптимальное натяжение для стимуляции сокращающими агентами.
В каждом эксперименте кольца стандартизировали, используя 1 мкмоль/л L-фенилэфрина (РЕ), до достижения воспроизводимых реакций. Для оценки вазорелаксирующего действия испытуемых соединений были построены кривые кумулятивных данных изменения реакции в зависимости от концентрации (10 нМ-3 мМ) с использованием колец, предварительно подвергнутых сжатию под действием L-фенилэфрина (1 мкМ), для нижеследующих соединений:
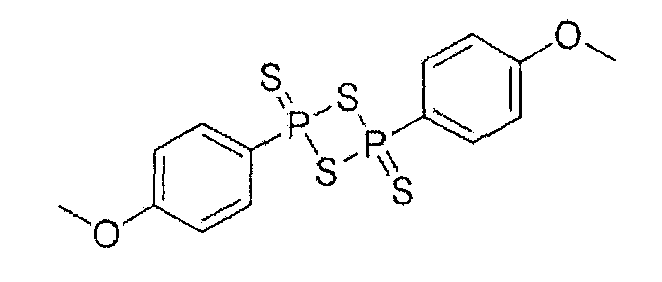
2,4-бис(4-метоксифенил)-1,3,2,4-дитиадифосфетан-2,4-дисульфида (реагент Лавессона), тиоацетамида, NaHS, 4-НТВ и Na2S. Наполнитель является буфером, а не соединением. Кривые для испытуемых соединений были построены в присутствии эндотелия. Для оценки целостности эндотелия была построена кривая кумулятивных данных изменения реакции в зависимости от концентрации для Ach (10 нМ-30 мкМ) с использованием колец, предварительно подвергнутых сжатию под воздействием L-фенилэфрина (РЕ).
Полученные данные показаны на фиг.22 и выражены в виде среднего значения ± стандартная ошибка среднего (SEM). Уровень статистической значимости был определен при помощи двумерного дисперсионного анализа (ANOVA) с последующим применением критерия Бонферрони для множественного сравнения с использованием программного обеспечения GraphPad™.
На фиг.22 показано, что все H2S-высвобождающие части по настоящему изобретению, а именно реагент Лавессона, 4-НТВ и тиоацетамид, оказывают значительное вазорелаксирующее действие, зависящее от концентрации, по сравнению с наполнителем. Кроме того, все кривые % релаксации были сравнимы с кривыми, полученными при использовании NaHS и Na2S.
Реферат
Настоящее изобретение относится к новым производным 4- или 5-аминосалициловой кислоты формулы (I) и к фармацевтической композиции на их основе, предназначенных для лечения воспалительных заболеваний кишечника, спастического колита, для профилактики/лечения рака ободочной кишки: ! ! А означает ! ! где -N=, -NH, -NH2 и -NH2, соответственно, находятся в положении 4 или 5; ! L означает О, O-С=O, S, N или ковалентную связь; и ! R - часть молекулы, которая высвобождает сероводород. Технический результат - получение новых биологически активных субстанций. 6 н. и 15 з.п. ф-лы, 23 ил.
Формула

в которой А означает
где -N = находится в положении 4 или 5,

где -NH находится в положении 4 или 5,

где -NH2 находится в положении 4 или 5, или

где -NH2 находится в положении 4 или 5;
L означает О, O-С=O, S, N или ковалентную связь; и
R означает высвобождающую сероводород часть молекулы, которая
высвобождает сероводород в ткань, выбранную из группы, состоящей из:
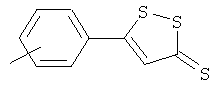

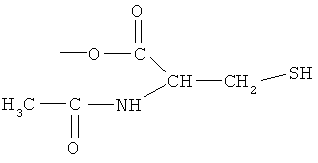



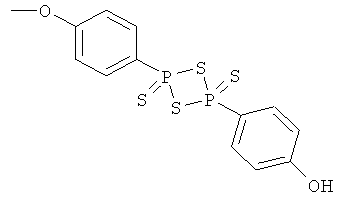


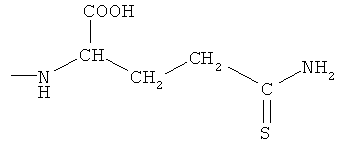


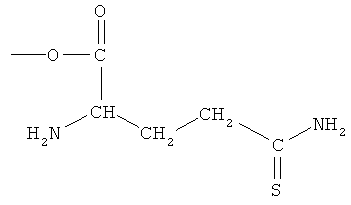
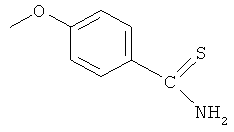
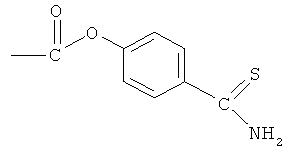
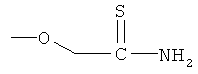

и
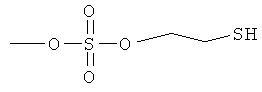
или его соли.
2-гидрокси-4-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилазо]бензойной кислоты;
2-гидрокси-5-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилазо]бензойной кислоты;
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 4-амино-2-гидроксибензойной кислоты;
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 5-амино-2-гидроксибензойной кислоты;
4-амино-2-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбонилокси]бензойной кислоты;
5-амино-2-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбонилокси]бензойной кислоты;
2-гидрокси-4-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбониламино]бензойной кислоты;
2-гидрокси-5-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбониламино]бензойной кислоты;
4-{[(1-карбокси-2-меркаптоэтилкарбамоил)метил]азо}-2-гидроксибензойной кислоты;
5-{[(1-карбокси-2-меркаптоэтилкарбамоил)метил]азо}-2-гидроксибензойной кислоты;
(1-карбокси-2-меркаптоэтилкарбамоил)метилового эфира 4-амино-2-гидроксибензойной кислоты;
(1-карбокси-2-меркаптоэтилкарбамоил)метилового эфира 5-амино-2-гидроксибензойной кислоты;
4-амино-2-[(1-карбокси-2-меркаптоэтилкарбамоил)-метоксикарбонилокси] бензойной кислоты;
5-амино-2-[(1-карбокси-2-меркаптоэтилкарбамоил)-метоксикарбонилокси] бензойной кислоты;
4-{[1-карбокси-2-меркаптоэтилкарбамоил)метоксикарбониламино]-2-гидроксибензойной кислоты;
5-{[1-карбокси-2-меркаптоэтилкарбамоил)метоксикарбониламино]-2-гидроксибензойной кислоты;
ангидрида 4-амино-2-гидроксибензойной кислоты с N-ацетилцистеином;
ангидрида 5-амино-2-гидроксибензойной кислоты с N-ацетилцистеином;
4-(2-ацетиламино-3-меркаптопропиониламино)-2-гидроксибензойной кислоты;
5-(2-ацетиламино-3-меркаптопропиониламино)-2-гидроксибензойной кислоты;
2-(2-ацетиламино-3-меркаптопропионилокси)-4-аминобензойной кислоты;
2-(2-ацетиламино-3-меркаптопропионилокси)-5-аминобензойной кислоты;
2-гидрокси-4-({4-[4-(4-метоксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксиметил}азо)бензойной кислоты;
2-гидрокси-5-({4-[4-(4-метоксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил] феноксиметил}азо)бензойной кислоты;
4-амино-2-{4-[4-(4-метоксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксиметоксикарбонилокси}бензойной кислоты;
5-амино-2-{4-[4-(4-метоксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксиметоксикарбонилокси}бензойной кислоты;
2-гидрокси-4-{4-[4-(4-метоксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксиметоксикарбониламино}бензойной кислоты;
2-гидрокси-5-{4-[4-(4-метоксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксиметоксикарбониламино}бензойной кислоты;
4-[4-(4-метоксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]-дитиадифосфетан-2-ил]феноксиметилового эфира 4-амино-2-гидроксибензойной кислоты;
4-[4-(4-метоксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]-дитиадифосфетан-2-ил]феноксиметилового эфира 5-амино-2-гидроксибензойной кислоты;
4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]-дитиадифосфетан-2-ил]фенилового эфира 4-амино-2-гидроксибензойной кислоты;
4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]-дитиадифосфетан-2-ил]фенилового эфира 5-амино-2-гидроксибензойной кислоты;
4-амино-2-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбонилокси}бензойной кислоты;
5-амино-2-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбонилокси}бензойной кислоты;
2-гидрокси-4-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил}феноксикарбониламино}бензойной кислоты;
2-гидрокси-5-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил}феноксикарбониламино}бензойной кислоты;
4-(1-карбокси-3-тиокарбамоилпропилазо)-2-гидроксибензойной кислоты;
5-(1-карбокси-3-тиокарбамоилпропилазо)-2-гидроксибензойной кислоты;
2-(4-амино-2-гидроксибензоиламино)-4-тиокарбамоилмасляной кислоты;
2-(5-амино-2-гидроксибензоиламино)-4-тиокарбамоилмасляной кислоты;
4-амино-2-(1-карбокси-3-тиокарбамоилпропилкарбамоилокси)-бензойной кислоты;
5-амино-2-(1-карбокси-3-тиокарбамоилпропилкарбамоилокси)-бензойной кислоты;
2-гидрокси-4-[3-(1-карбокси-3-тиокарбамоилпропил)уреидо]бензойной кислоты;
2-гидрокси-5-[3-(1-карбокси-3-тиокарбамоилпропил)уреидо]бензойной кислоты;
4-амино-2-(2-амино-4-тиокарбамоилбутирилокси)бензойной кислоты;
5-амино-2-(2-амино-4-тиокарбамоилбутирилокси)бензойной кислоты;
4-(2-амино-4-тиокарбамоилбутириламино)-2-гидроксибензойной кислоты;
5-(2-амино-4-тиокарбамоилбутириламино)-2-гидроксибензойной кислоты;
ангидрида 4-амино-2-гидроксибензойной кислоты с 2-амино-4-тиокарбамоилмасляной кислотой;
ангидрида 5-амино-2-гидроксибензойной кислоты с 2-амино-4-тиокарбамоилмасляной кислотой;
4-тиокарбамоилфенил-4-амино-2-гидроксибензоата;
4-тиокарбамоилфенил-5-амино-2-гидроксибензоата;
4-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты;
5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты;
2-гидрокси-4-(4-тиокарбамоилфеноксикарбониламино)бензойной кислоты;
2-гидрокси-5-(4-тиокарбамоилфеноксикарбониламино)бензойной кислоты;
тиокарбамоилметилового эфира 4-амино-2-гидроксибензойной кислоты;
тиокарбамоилметилового эфира 5-амино-2-гидроксибензойной кислоты;
4-амино-2-тиокарбамоилметоксикарбонилоксибензойной кислоты;
5-амино-2-тиокарбамоилметоксикарбонилоксибензойной кислоты;
2-гидрокси-4-тиокарбамоилметоксикарбониламинобензойной кислоты;
2-гидрокси-5-тиокарбамоилметоксикарбониламинобензойной кислоты;
ангидрида 4-амино-2-гидроксибензойной кислоты с моно(2-меркаптоэтиловым) эфиром серной кислоты;
ангидрида 5-амино-2-гидроксибензойной кислоты с моно(2-меркаптоэтиловым) эфиром серной кислоты;
4-амино-2-(2-меркаптоэтоксисульфонилокси)бензойной кислоты;
5-амино-2-(2-меркаптоэтоксисульфонилокси)бензойной кислоты;
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 4-амино-2-гидроксибензойной кислоты;
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 5-амино-2-гидроксибензойной кислоты;
или его соль.
2-гидрокси-4-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилазо]бензойной кислоты;
2-гидрокси-5-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилазо]бензойной кислоты;
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 4-амино-2-гидроксибензойной кислоты;
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 5-амино-2-гидроксибензойной кислоты;
4-амино-2-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбонилокси]бензойной кислоты;
5-амино-2-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбонилокси]бензойной кислоты;
2-гидрокси-4-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбониламино]бензойной кислоты;
2-гидрокси-5-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбониламино]бензойной кислоты;
ангидрида 4-амино-2-гидроксибензойной кислоты с N-ацетилцистеином;
ангидрида 5-амино-2-гидроксибензойной кислоты с N-ацетилцистеином;
4-(2-ацетиламино-3-меркаптопропиониламино)-2-гидроксибензойной кислоты;
5-(2-ацетиламино-3-меркаптопропиониламино)-2-гидроксибензойной кислоты;
2-(2-ацетиламино-3-меркаптопропионилокси)-4-аминобензойной кислоты;
2-(2-ацетиламино-3-меркаптопропионилокси)-5-аминобензойной кислоты;
4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]-дитиадифосфетан-2-ил]фенилового эфира 4-амино-2-гидроксибензойной кислоты;
4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]-дитиадифосфетан-2-ил]фенилового эфира 5-амино-2-гидроксибензойной кислоты;
4-амино-2-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбонилокси}бензойной кислоты;
5-амино-2-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбонилокси}бензойной кислоты;
2-гидрокси-4-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбониламино}бензойной кислоты;
2-гидрокси-5-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбониламино}бензойной кислоты;
2-(4-амино-2-гидроксибензоиламино)-4-тиокарбамоилмасляной кислоты;
2-(5-амино-2-гидроксибензоиламино)-4-тиокарбамоилмасляной кислоты;
4-амино-2-(1-карбокси-3-тиокарбамоилпропилкарбамоилокси)бензойной кислоты;
5-амино-2-(1-карбокси-3-тиокарбамоилпропилкарбамоилокси)бензойной кислоты;
4-тиокарбамоилфенил-4-амино-2-гидроксибензоата;
4-тиокарбамоилфенил-5-амино-2-гидроксибензоата;
4-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты;
5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты;
2-гидрокси-4-(4-тиокарбамоилфеноксикарбониламино)бензойной кислоты;
2-гидрокси-5-(4-тиокарбамоилфеноксикарбониламино)бензойной кислоты;
ангидрида 4-амино-2-гидроксибензойной кислоты с моно(2-меркаптоэтиловым)эфиром серной кислоты;
ангидрида 5-амино-2-гидроксибензойной кислоты с моно(2-меркаптоэтиловым) эфиром серной кислоты;
4-амино-2-(2-меркаптоэтоксисульфонилокси)бензойной кислоты;
5-амино-2-(2-меркаптоэтоксисульфонилокси)бензойной кислоты;
или его соль.
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 4-амино-2-гидроксибензойной кислоты, и
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 5-амино-2-гидроксибензойной кислоты, или его соль.
ангидрида 4-амино-2-гидроксибензойной кислоты с N-ацетилцистеином,
ангидрида 5-амино-2-гидроксибензойной кислоты с N-ацетилцистеином, или его соль.
4-тиокарбамоилфенил-4-амино-2-гидроксибензоата,
4-тиокарбамоилфенил-5-амино-2-гидроксибензоата; или его соль.
2-гидрокси-4-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилазо]бензойной кислоты;
2-гидрокси-5-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилазо]бензойной кислоты;
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 4-амино-2-гидроксибензойной кислоты;
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фенилового эфира 5-амино-2-гидроксибензойной кислоты;
4-амино-2-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбонилокси]бензойной кислоты;
5-амино-2-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбонилокси]бензойной кислоты;
2-гидрокси-4-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбониламино]бензойной кислоты;
2-гидрокси-5-[4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)феноксикарбониламино]бензойной кислоты;
ангидрида 4-амино-2-гидроксибензойной кислоты с N-ацетилцистеином;
ангидрида 5-амино-2-гидроксибензойной кислоты с N-ацетилцистеином;
4-(2-ацетиламино-3-меркаптопропиониламино)-2-гидроксибензойной кислоты;
5-(2-ацетиламино-3-меркаптопропиониламино)-2-гидроксибензойной кислоты;
2-(2-ацетиламино-3-меркаптопропионилокси)-4-аминобензойной кислоты;
2-(2-ацетиламино-3-меркаптопропионилокси)-5-аминобензойной кислоты;
4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]фенилового эфира 4-амино-2-гидроксибензойной кислоты;
4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[l,3,2,4]дитиадифосфетан-2-ил]фенилового эфира 5-амино-2-гидроксибензойной кислоты;
4-амино-2-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбонилокси}бензойной кислоты;
5-амино-2-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбонилокси}бензойной кислоты;
2-гидрокси-4-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил]феноксикарбониламино}бензойной кислоты;
2-гидрокси-5-{4-[4-(4-гидроксифенил)-2,4-дитиоксо-2λ5,4λ5-[1,3,2,4]дитиадифосфетан-2-ил] феноксикарбониламино}бензойной кислоты;
2-(4-амино-2-гидроксибензоиламино)-4-тиокарбамоилмасляной кислоты;
2-(5-амино-2-гидроксибензоиламино)-4-тиокарбамоилмасляной кислоты;
4-амино-2-(1-карбокси-3-тиокарбамоилпропилкарбамоилокси)бензойной кислоты;
5-амино-2-(1-карбокси-3-тиокарбамоилпропилкарбамоилокси)бензойной кислоты;
4-тиокарбамоилфенил-4-амино-2-гидроксибензоата;
4-тиокарбамоилфенил-5-амино-2-гидроксибензоата;
4-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты;
5-амино-2-(4-тиокарбамоилфеноксикарбонилокси)бензойной кислоты;
2-гидрокси-4-(4-тиокарбамоилфеноксикарбониламино)бензойной кислоты;
2-гидрокси-5-(4-тиокарбамоилфеноксикарбониламино)бензойной кислоты;
ангидрида 4-амино-2-гидроксибензойной кислоты с моно(2-меркаптоэтиловым) эфиром серной кислоты;
ангидрида 5-амино-2-гидроксибензойной кислоты с моно(2-меркаптоэтиловым) эфиром серной кислоты;
4-амино-2-(2-меркаптоэтоксисульфонилокси)бензойной кислоты;
5-амино-2-(2-меркаптоэтоксисульфонилокси)бензойной кислоты, или его фармацевтически приемлемой соли.
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)-фениловый эфир 4-амино-2-гидроксибензойной кислоты;
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)-фениловый эфир 5-амино-2-гидроксибензойной кислоты;
ангидрид 4-амино-2-гидроксибензойной кислоты с N-ацетилцистеином;
ангидрид 5-амино-2-гидроксибензойной кислоты с N-ацетилцистеином;
4-тиокарбамоилфенил-4-амино-2-гидроксибензоат;
4-тиокарбамоилфенил-5-амино-2-гидроксибензоат,
или его фармацевтически приемлемую соль.
4-(5-тиоксо-5Н-[1,2]дитиол-3-ил)фениловым эфиром 5-амино-2-гидроксибензойной кислоты.
Комментарии