Способ лечения недержания мочи с помощью (s)-оксибутинина и (s)-дезэтилоксибутинина - RU2181589C2
Код документа: RU2181589C2
Чертежи









Описание
Область техники, к которой относится изобретение
Изобретение
относится к способу лечения недержания мочи с помощью оптически чистого (S)-оксибутинина и (S)-дезэтилоксибутинина (S-DEO), к фармацевтическим составам, включающим оптически чистый (S)-оксибутинин или
S-DEO, а также к процессу получения одиночных энантиомеров DEO.
Предпосылки создания изобретения
Рацемический оксибутинин используется терапевтически при лечении
гипермотильности кишечника и при лечении недержания мочи вследствие нестабильности мышцы, выталкивающей мочу. Рацемический оксибутинин оказывает прямое антиспазматическое воздействие на гладкие мышцы
и подавляет действие ацетилхолина на гладкие мышцы. Он обладает лишь одной пятой антихолинергической активности атропина на мышцу, выталкивающую мочу у кроликов, но по антиспазматической активности
превышает последний в четыре-десять раз. Он отличается крайней избирательностью в отношении мускариновых рецепторов в присутствии холиномиметических рецепторов и, как следствие, не оказывает
блокирующего воздействия на скелетные нервно-мышечные синапсы либо вегетативные ганглии.
Рацемический оксибутинин расслабляет гладкие мышцы мочевого пузыря и у пациентов, находящихся в состоянии, характеризующемся непроизвольными сокращениями мочевого пузыря. Цистометрические исследования показали, что рацемический оксибутинин повышает емкость пузыря, уменьшает частоту непроизвольных сокращений мышцы, выталкивающей мочу, и задерживает первоначальное желание к опорожнению пузыря. Следовательно, он пригоден для лечения и предотвращения как недержания мочи, так и частого произвольного мочеиспускания. Эффективность рацемического оксибутинина в отношении мочевого пузыря приписывают сочетанию антимускаринового, прямого спазматического и местного анестезирующего воздействия на гладкую мышцу, выталкивающую мочу. По причине антимускариновой активности рацемического лекарственного средства очень частыми побочными эффектами являются ксеростомия (сухость во рту) и мидриаз (расширение зрачков) с вовлечением мускариновых холинергических рецепторов. По крайней мере один ученый указывал на "неизбежные симптомы мидриаза, ксеростомии, тахикардии и т. д.", которые сопровождают введение рацемического оксибутинина (Лиш и др., Lish (Arch. Int. Pharmacodyn. 156, 467-488 (1965), 481). Частым следствием высокой частоты антихолинергических побочных эффектов (от 40 до 80%) является снижение дозировки либо прерывание курса лечения.
Результаты фармакологических исследований отдельных энантиомеров позволяет предположить, что эффективным энантиомером является R энантиомер. Норона-Блоб (Noronha-Blob) и др. (J. Pharmacol. Exp. Ther. 256, 562-567 (1991)) пришли к выводу, что холинергический антагонизм рацемического оксибутинина (измеряемый in vitro по его сродству к подтипам рецепторов M1, M2 и М3 и in vivo по различным физиологическим реакциям) может приписываться, главным образом, активности R энантиомера. Они установили, что по всем реакциям упорядоченная активность рацемического оксибутинина и его энантиомеров является одинаковой, а именно, активность (R)-оксибутинина превосходит либо равняется активности рацемического оксибутинина, которая намного превосходит активность (S)-оксибутинина, причем активность (S)-оксибутинина ниже активности (R)-оксибутинина на 1-2 порядка.
Краткое
изложение сущности изобретения
Неожиданно установили, что по существу оптически чистый S энантиомер оксибутинина и его дезэтилового метаболита обладает великолепными лечебными свойствами при
лечении недержания мочи.
Оптически чистый (S)-оксибутинин (S-OXY) и (S)-дезэтилоксибутинин (S-DEO) обеспечивают подобное лечение с одновременным существенным снижением отрицательных эффектов, которые возникают, главным образом, вследствие антихолинергической активности и которые связаны с введением рацемического оксибутинина. К их числу относятся, но ими не ограничиваются ксеростомия, мидриаз, сонливость, тошнота, запоры, учащенное сердцебиение и тахикардия. Особую терапевтическую ценность имеет сокращение побочных эффектов, оказываемых рацемическим оксибутинином в отношении сердечно-сосудистой системы, в частности учащенного сердцебиения и тахикардии вследствие введения (S)-оксибутинина или (S-DEO).
Активными компонентами этих составов и способов
являются оптические изомеры оксибутинина и дезэтилоксибутинина. Получение рацемического оксибутинина раскрыто в описании изобретения к патенту Великобритании 940540. В химическом отношении активными
соединениями являются: (1) S энантиомер 4-(диэтиламино)-2-бутинил α-циклогексил-α-гидроксибензолацетата, известный также как 4-(диэтиламино)-2-бутинилфенилциклогексилгликолят, именуемый
в последующем оксибутинином и (2) S энантиомер 4-(этиламино)-2-бутинил α-циклогексил-α-гидроксибензолацетат, именуемый в последующем дезэтилоксибутинином. Американский Совет по
стандартизации присвоил хлористоводородной соли рацемического оксибутинина родовое наименование оксибутинина хлорид; он продается под торговым названием Diropan®. Изомер оксибутинина,
имеющий S абсолютную стереохимию (Регистрационный номер 119618-22-3), является правовращающим и представлен формулой I:

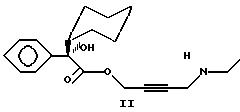
S энантиомер дезэтилоксибутинина представлен Формулой II:
Синтезирование (S)-оксибутинина описано (Качур (Kachur) и др. , J. Pharmacol. Exp. Ther. , 247, 867-872 (1988)), однако сам (S)-оксибутинин в настоящее время коммерчески недоступен. Все клинические результаты, о которых сообщалось, были получены с рацемической смесью несмотря на то, что фармакология отдельных энантиомеров была описана на морских свинках и крысах (см. Качур и др., J. Pharmacol. Exp. Ther., 247, 867-872 (1988) и Норона-Блоб и др. ,), J. Pharmacol. Exp. Ther., 256, 562-567 (1991)). (S)-дезэтилоксибутинин ранее не описывался; синтез этого соединения осуществляется в соответствии со способом, описание которого приведено далее.
Одним из своих аспектов изобретение относится к способу лечения недержания мочи с предотвращением сопутствующей вероятности возникновения отрицательных эффектов, который включает введение человеку, нуждающемуся в подобном лечении, терапевтически эффективного количества (S)-оксибутинина, (S)-дезэтилоксибутинина или фармацевтически приемлемой соли любого из них, по существу свободных от соответствующего R энантиомера. Термин "по существу, свободных от соответствующего R энантиомера", используемый в данном описании, означает, что составы содержат, как минимум, 90 мас.% (S)-оксибутинина или (S)-дезэтилоксибутинина и 10 мас.% или менее (R)-оксибутинина или (R)-дезэтилоксибутинина.
В более предпочтительном варианте осуществления составы содержат, как минимум, 99 мас.% S энантиомера и 1% или менее R энантиомера. По существу, оптически чистый (S)-оксибутинин или (S)-дезэтилоксибутинин могут вводиться парентарально, ректально, внутрипузырно, чрескожно, перорально либо аэрозольным способом, причем предпочтение отдается пероральному и чрескожному пути введения, в дозе приблизительно от 1 до 100 мг в день.
В другом аспекте изобретение относится к фармацевтической стандартной дозированной форме в виде таблетки либо капсулы, включающей терапевтически эффективное количество (S)-оксибутинина, (S)-дезэтилоксибутинина либо фармацевтически приемлемой соли любого из них, по существу свободных от соответствующего R стереоизомера, и фармацевтически приемлемый носитель. Таблетка либо капсула в предпочтительном варианте содержит от 0,5 до 25 мг (S)-оксибутинина или (S)-дезэтилоксибутинина и готовится традиционными способами, общеизвестными в данной отрасли. Изобретение относится также к дозированной форме в виде чрескожного средства. Чрескожное введение улучшается посредством включения в чрескожное средство доставки усилителя проникновения, например, как описано в заявке РСТ WO 92/20377.
Последующий аспект изобретения относится к процессу получения дезэтилоксибутинина, в предпочтительном варианте одиночного энантиомера DEO, в более предпочтительном варианте S-DEO, который включает этапы, во-первых, реагирования метил-α-циклогексил-α-гидроксибензолацетата с 4-[N-этил-(4-метоксифенил)метиламино] -2-бутин-1-олом в присутствии безводного основания для получения 4-[N-этил-(4-метоксифенил)метиламино]-2-бутинил α-циклогексил-α-гидроксибензолацетата; затем последовательно с карбонхлоридатом и метанолом для получения 4-(этиламино)-2-бутинил α-циклогексил-α-гидроксибензолацетата (дезэтилоксибутинина). Процесс может дополнительно включать этап реагирования 4-этил-4-метоксибензолметанамина с 2-пропин-1-олом и формальдегидом либо эквивалентом формальдегида в присутствии соли меди (I) для получения 4-[N-этил-(4-метоксифенил)метиламино]-2-бутин-1-ола, необходимого для первого этапа.
Подробное описание изобретения
S энантиомеры оксибутинина и DEO могут быть получены повторным растворением промежуточного продукта манделовой кислоты с последующей этерификацией.
Этерификация может осуществляться, как описано Качуром (упоминавшаяся работа) для OXY либо посредством усовершенствованного способа, изображенного на представленной далее схеме А для S-DEO.
Графическое изображение рацемических, амбискалемических и скалемических или энантиомерно чистых соединений, используемое в данном описании, заимствовано из работы Мера (Maehr) J. Chem. Ed., 62, 114-120 (1985). Так, сплошные и ломаные клинья (наподобие показанных в формуле I), используются для обозначения абсолютной конфигурации хирального элемента; очертания клина и штрих-пунктирная либо ломанная линии (наподобие показанных в формуле III) обозначают энантиометрически чистые соединения неопределенной абсолютной конфигурации.
Общий процесс для получения DEO включает:
(a) реагирование N-этил-4-метоксибензолметананамина с 2-пропин-1-олом и параформальдегидом в инертном растворителе в присутствии хлорида меди для получения
4-[N-этил-(4-метоксифенил)метиламино]-2-бутин-1-ола (V);
(b) реагирование одиночного энантиомера метил α-циклогексил-α-гидроксифенолацетата (IV) с
4-[N-этил-(4-метоксифенил)метиламино] -2-бутин-1-олом (V) в присутствии каталитического количества метоксида натрия в толуоле для получения одиночного энантиомера 4-[N-этил-(4-метоксифенил)метиламино]
-2-бутинил α-циклогексил-α-гидроксибензолацетата (VI);
(c) реагирование 4-[N-этил-(4-метоксифенил)метиламино] -2-бутинил α-циклогексил-α-гидроксибензолацетата (VI)
последовательно с α-хлорэтилкарбонхлоридатом в дихлорэтане, затем с метанолом, для образования одиночного энантиомера DEO (VII).
Процесс, очевидно, пригоден для получения рацемического DEO из рацемического метил α-циклогексил-α-гидроксибензолацетата. Параформальдегид используется в качестве удобного источника формальдегида, но он может быть замещен любым источником формальдегида, что хорошо известно в данной отрасли техники. Подобным же образом α-хлорэтилкарбонхлоридат используется для деалкилирования, однако могут быть использованы и другие карбонохлоридаты (например, винил).
В альтернативном варианте S энантиомеры OXY и DEO могут быть получены повторным растворением рацемического оксибутинина либо DEO с помощью традиционных приемов, например, фракционной кристаллизации диастереомерных солей хиральными кислотами. Могут также использоваться и другие стандартные способы повторного растворения, известного специалистам в данной области, включая, но не ограничиваясь, простую кристаллизацию и хроматографирование на хиральном субстрате.
Величина профилактической либо терапевтической дозы (S)-оксибутинина или S-DEO при остром либо хроническом заболевании будет изменяться в зависимости от тяжести и природы состояния, подлежащего лечению, и пути введения. Доза и, возможно, частота введения дозы будет также изменяться в зависимости от возраста, массы тела и реакции отдельного пациента. В целом общая суточная доза (S)-оксибутинина или S-DEO для описанных здесь состояний составляет приблизительно от 1 до 100 мг в виде разовой дозы либо небольшой дозы, повторяемой через определенные интервалы времени, причем предпочтение отдается небольшой дозе, повторяемой через определенные интервалы времени. При лечении пациента курс начинается с более низкой дозы, возможно, приблизительно от 0,25 до 25 мг с возрастанием приблизительно до 100 мг в зависимости от общей реакции пациента. Рекомендуется также, чтобы пациенты старше 65 лет и пациенты с нарушением функции печени либо почек первоначально получали более низкие дозы, которые должны подбираться исходя из индивидуальной реакции(-ий) и уровня(-ей) в крови. В некоторых случаях может возникнуть необходимость использования дозировок, выходящих за пределы указанных диапазонов, что вполне понятно для специалистов в данной области. Далее следует заметить, что практикующий либо лечащий врач знает, как и когда прервать, изменить либо прекратить проведение курса лечения в зависимости от индивидуальной реакции пациента. Термины "терапевтически эффективное количество" и "количество, достаточное для лечения недержания, но недостаточное для причинения отрицательных эффектов" входят в объем вышеописанных дозировочных количеств и схемы применения лекарственного средства.
Для обеспечения пациента эффективным количеством (S)-оксибутинина или S-DEO может использоваться любой приемлемый путь введения. Например, могут быть использованы пероральная, ректальная, парентальная (подкожная, внутримышечная, внутривенная), чрескожная, аэрозольная и т.п. формы введения. В дополнение к этому лекарственное средство может вводиться непосредственно в мочевой пузырь через уретру, как описано для рацемического оксибутинина Массадом (Massad) и др. (J. Urol. 148, 595-597 (1992)). К числу дозированных лекарственных форм относятся таблетки, пастилки, дисперсии, суспензии, растворы, капсулы, чрескожные системы доставки и т.п.
Фармацевтические составы настоящего изобретения включают (S)-оксибутинин либо S-DEO в качестве активного ингредиента либо его фармацевтически приемлемую соль и, кроме того, могут также включать фармацевтически приемлемый носитель либо, факультативно, другие терапевтические ингредиенты.
Термины "фармацевтически приемлемые соли" либо "его фармацевтически приемлемую соль" относятся к солям, полученным из фармацевтически приемлемых нетоксичных кислот. К числу фармацевтически приемлемых кислых солей для соединений настоящего изобретения относятся уксусная, бензолсульфоновая (безилатная), бензойная, камфорсульфоновая, лимонная, этансульфоновая, фумаровая, глюконовая, глутаминовая, бромистоводородная, хлористоводородная, изэтионовая, молочная, малеиновая, яблочная, миндальная, метансульфоновая, слизевая, азотная, памовая, пантотеновая, фосфорная, янтарная, серная, винная, р-толуолсульфоновая и т.п. Особо пригодной была хлористоводородная соль, которая и использовалась в описываемых далее исследованиях.
К числу составов настоящего изобретения относятся суспензии, растворы, эликсиры либо твердые дозированные лекарственные формы. В случае пероральных твердых лекарственных форм (таких как порошки, капсулы и таблетки) пригодны такие носители, как крахмалы, сахара и микрокристаллическая целлюлоза, разбавители, гранулирующие средства, смазывающие вещества, вяжущие вещества, вещества, способствующие распаду, и т. п. Твердым пероральным препаратам отдается предпочтение по сравнению с жидкими пероральными препаратами.
Благодаря простоте введения таблетки и капсулы представляют одну из более предпочтительных пероральных дозированных лекарственных форм, причем в этом случае используются твердые фармацевтические носители. В случае необходимости таблетки могут покрываться с помощью стандартных водных или неводных способов.
В дополнение к обычным вышеупомянутым дозированным лекарственным формам соединения настоящего изобретения могут также вводиться средствами контролируемого выделения либо такими средствами доставки, которые описаны в патентах США 3845770, 3916899, 3536800, 3598123 и 4008719, а также в заявке РСТ WO 92/20377, описание которых включено в настоящее описание в качестве справочного материала.
Фармацевтические составы настоящего изобретения, пригодные для перорального введения, могут быть представлены в виде дискретных стандартных дозированных лекарственных форм, например капсул, крахмальных облаток или таблеток, каждая из которых включает предпочтительное количество активного ингредиента в виде порошка или гранул либо в виде раствора или суспензии в водной жидкости, неводной жидкости, эмульсии типа "масло-в-воде" либо жидкой эмульсии типа "вода-в-масле". Подобные составы могут готовиться любым из фармакологических способов, однако все способы включают этап соединения активного ингредиента с носителем, который представляет собой один или более необходимых ингредиентов. В целом составы готовят однородным и тщательным смешиванием ингредиента с жидкими носителями, либо тонко измельченными твердыми носителями, либо с тем и другим и затем в случае необходимости приданием продукту необходимой формы в соответствии с тем, что известно для рацемической смеси.
Например, таблетка может быть изготовлена факультативно прессованием либо формованием с одним либо более дополнительными ингредиентами. Прессованные таблетки могут готовиться прессованием в подходящей машине активного ингредиента в свободно-текучей форме, например порошке, либо в форме гранул, факультативно смешанных с вяжущим средством, смазывающим средством, инертным разбавителем, поверхностно-активным веществом либо веществом, способствующим распаду. Формованные таблетки могут изготовляться формованием, в подходящей машине, смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Все вышеупомянутые способы хорошо известны специалистам в области фармакологам. В состав каждой таблетки может входить приблизительно от 0,5 до 25 мг активного ингредиента.
Примеры
Пример 1. Пероральная стандартная лекарственная форма (см.
табл. 1а).
(S)-оксибутинин или дезэтилоксибутинин смешивают с лактозой или целлюлозой до образования гомогенной смеси. Добавляют красочный лак и продолжают смешивание. Добавляют стеарат кальция и образовавшуюся смесь прессуют в таблетки с помощью полого вогнутого пуансона 9/32 дюйма (7 мм). Таблетки с иным содержанием действующего вещества могут быть приготовлены посредством изменения соотношения активного ингредиента и наполнителей либо конечной массы таблетки.
При проведении описанных далее исследований была установлена вызывающая удивление пригодность S энантиомера как OXY, так и DEO.
Энантиомеры оксибутинина
Связывание (R)- и (S)-оксибутинина с подтипами мускариновых рецепторов человека M1, M2, M3 и М4
Материалы и методы
Эксперименты проводили на мембранах, приготовленных из клеток SF9, инфицированных бакуловирусами для экспрессирования рекомбинантных
подтипов мускариновых рецепторов человека M1, М2, М3 и М4.
Пробы на связывание приведены в табл. 1.
После инкубирования пробы подвергали ускоренному фильтрованию под вакуумом через стекловолоконные фильтры GF/B (компания Whatman) и промывали ледяным буфером, используя бранделовский харвестер клеток. Связанную радиоактивность определяли жидкостным сцинтилляционным счетчиком (LS 6000, компания Beckman), используя жидкую сцинтилляционную смесь (формула 99, компания DuPont NEN).
Схема
проведения экспериментов
Соединения испытывали на каждом рецепторе в 10 концентрациях с повтором для получения конкурентных кривых. В каждом эксперименте эталонное соединение для исследуемого
рецептора одновременно испытывали в 8 концентрациях с повтором для получения конкурентной кривой для оценки этого эксперимента.
Анализ и выражение результатов
Специфическое
связывание радиолиганда на каждом рецепторе определяли как разницу между полным связыванием и неспецифическим связыванием, определявшимся в присутствии избыточного количества немеченого лиганда.
Значения IC50 (концентрации, необходимые для ингибирования 50% специфического связывания) определяли нелинейным регрессивным анализом конкурентных кривых. Эти параметры были получены
подбором эмпирической кривой с помощью программы SigmaplotTM. IC50 для R- и S-OXY приведены в табл. 2.
Эти результаты свидетельствуют о том, что S-OXY обладает меньшим сродством к подтипам мускариновых рецепторов по сравнению с R-OXY.
Связывание (R)-оксибутинина и (S)-оксибутинина с кальциевыми каналами
Материалы и методы
Пробы на связывание ставили с использованием методов, приведенных в табл. 3.
Условия экспериментов приведены в табл. 4.
После инкубирования пробы подвергали ускоренному фильтрованию под вакуумом через стекловолоконные фильтры GF/B (компания Whatman) и промывали ледяным буфером, используя бранделовский харвестер клеток. Связанную радиоактивность определяли жидкостным сцинтилляционным счетчиком (LS 6000, компания Beckman), используя жидкую сцинтилляционную смесь (формула 99, компания DuPont NEN).
Схемы проведения экспериментов
Соединения
испытывали на каждом рецепторе в концентрациях 10-5 М с повтором. В каждом эксперименте эталонное соединение для исследуемого рецептора одновременно испытывали в 8 концентрациях с повтором
для получения конкурентной кривой для оценки этого эксперимента.
Анализ и выражение результатов
Специфическое связывание радиолиганда на каждом рецепторе определяли как
разницу между полным связыванием и неспецифическим связыванием, определявшимся в присутствии избыточного количества немеченого лиганда. Средние значения, выраженные в виде процента ингибирования
специфического связывания, представлены в табл. 5. IС50 (концентрации, необходимые для ингибирования 50% специфического связывания) определяли нелинейным регрессивным анализом
соответствующих конкурентных кривых. Эти параметры были получены подбором эмпирической кривой с помощью программы SigmaplotTM.
Эти результаты свидетельствуют о том, что S-OXY обладает активностью блокирования проникновения кальция, подобной R-OXY.
Энантиомеры дезэтилоксибутинина
Основным метаболитом рацемического оксибутинина является RS
дезэтилоксибутинин (DEO). R и S энантиомеры DEO не описывались и антиспазматическая активность и активность блокирования проникновения кальция отдельных энантиомеров, R- и S- DEO была до проведения
наших исследований неизвестной. Мы синтезировали эти энантиомеры и изучили их антимускариновое, спазмолитическое действие и активность блокирования проникновения кальция на моделях связывания
рецептора и функции мочевого пузыря. Мы установили, что каждый энантиомер метаболита сохраняет относительный фармакологический профиль "родительского" оксибутинового энантиомера.
Связывание на подтипах мускаринового рецептора
Ингибирование (%) специфического связывания радиолигандов, вызванное тремя концентрациями каждого соединения (R-, S- и RS-DEO) испытывали на
клонированных подтипах мускариновых рецепторов человека (М1-М4), как описывалось ранее для энантиомеров оксибутинина. В табл. 6 и 7 приведено ингибирование (%) на каждом подтипе. Дополнительно к этому
определяли значения IC50 для подтипов рецепторов человека Ml и М2; они представлены в табл 6.
Эти результаты свидетельствуют о том, что S-DEO обладает меньшим сродством к подтипам мускариновых рецепторов, чем R- либо рацемический DEO.
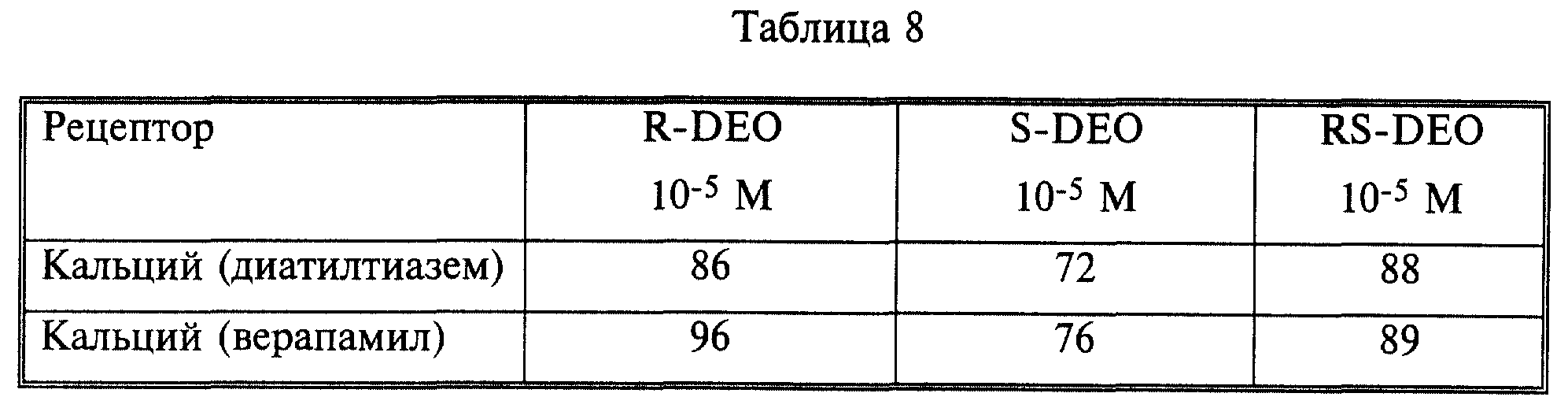
Связывание на кальциевых каналах
Ингибирование (%) специфического связывания радиолигандов, вызванное каждым
соединением (R-, S- и RS-DEO), испытывали на участках дилтиазема и верапамила кальциевого канала типа L. Результаты представлены в табл. 8.
Приведенные результаты свидетельствуют о том, что S-DEO обладает активностью блокирования проникновения кальция, подобной активности R- и рацемического DEO.
Определение функциональных характеристик
антимускариновой/антиспазматической активности
Влияние R-, S- и RS-оксибутинина (OXY), а также R-, S- и RS-DEO изучали на модели функционирования мочевого пузыря in vitro. Как описывается
далее, изолированные полоски гладких мышц мочевого пузыря морских свинок помещали в тканевую ванночку и вызывали их сокращение либо с помощью мускаринового агониста карбахола, либо посредством
повышенных концентраций экзогенного калия.
Материалы и методы
Полоски мочевого пузыря. Эксперименты проводили с использованием методов, подобных описанным Качуром и др. 1988,
а также Норона-Блобом и Качуром, 1991. Полоски ткани (длиной приблизительно 10 мм и шириной 1,5 мм) извлекали из тела мочевого пузыря морских свинок-самцов (линия Hartley, масса 400-600 г) (Elm Hill
Breeding Laboratories, Chelmsford, MA). Ткани суспендировали в оксигенированном буфере следующего состава, мМ: NaCl 133; КС1 4,7; CaCl2 2,5; MgSО4 0,6; NaH2PO4 1,3; NaHCO3 16,3 и глюкоза 7,7, при температуре 37,5oС. Сокращения регистрировали с помощью изометрических датчиков (Модель FT-10) и чернильных полиграфов (Модель 7)
(Astro-Med, Inc., Grass Instrument Div. , West Warwick, RI). На всех тканях постоянно поддерживалось остаточное напряжение 0,5 г.
В каждом эксперименте из одного мочевого пузыря вырезали до семи полосок. Они суспендировались в отдельных тканевых камерах и уравновешивались раствором тканевой ванночки в течение часа перед продолжением эксперимента.
Карбохол-индуцированные сокращения. В одной серии экспериментов внимание сосредоточено на антихолинергическом действии оксибутинина. В этих экспериментах для оценки жизнеспособности каждой ткани и для использования в качестве эталонной рамки сокращения каждой полоски ткани вначале регистрировали в ответ на воздействие тканевой среды, в которой NaC1 был заменен КСl, до получения концентрации КСl в среде на уровне 137,7 мМ. После этого следовал возврат к стандартной среде, затем воздействие постепенно возрастающих концентраций карбахола с раздельным воздействием каждой концентрации только после регистрации максимальной реакции. После этого, оставив одну полоску необработанной и/либо одну полоску, подверженную воздействию 17 мМ этанола, в качестве контрольной ткани(-ей), все остальные полоски подвергали в течение одного часа воздействию одной концентрации антагониста. Этаноловые контроли использовали в том случае, когда по причине плохой растворимости необходимо было готовить маточные растворы экспериментальных веществ в этаноле, в результате чего тканевые ванночки испытывали воздействие эффективной концентрации 17 мМ этанола. В конечном счете повторно была произведена запись реакций на возрастающие концентрации карбахола после воздействия 137,7 мМ КСl.
Калий-индуцированные сокращения. Внимание во второй серии экспериментов фокусировалось на спазмолитическом действии исследуемых веществ. Сокращения регистрировались в ответ на последовательно возрастающую концентрацию калия в среде.
Анализ данных. Для определения, снижали ли антагонисты максимальную реакцию на воздействие агонистов, максимальное напряжение, развиваемое каждой полоской во время второй серии определений, выражали в виде процента от максимального напряжения, развивавшегося во время первого определения воздействия концентраций. После этого полученные данные для каждого антагониста анализировали на различие, связанное с обработкой, посредством дисперсионного анализа. Поскольку на каждой полоске мочевого пузыря испытывали лишь одну концентрацию, для оценки рА2 и наклона линии регрессии Шильца использовали модифицированный вариант методики Арунлакшана и Шильда (1959). Во-первых, для каждой полоски на основании второго набора данных по возрастанию концентрации определяли концентрации агониста, вызывающие полумаксимальную реакцию (ЕС50). ЕС определяли посредством подгонки кривых линейной регрессии к логарифму концентрации лекарственного средства и реакциям, группирующим полумаксимальный уровень реакций. Для каждой полоски, обработанной лекарственным средством, вычисляли "коэффициент концентрации" (CR) как отношение ЕС обработанной ткани к ЕС необработанной ткани. Для каждого эксперимента, в котором две или более полоски подвергались воздействию одного и того же химиката, но в различных концентрациях, вычерчивали график зависимости между логарифмом этого соотношения минус единица (т.е. log(CR-l)) и логарифмом концентрации антагониста, воздействию которого подвергали полоску, для получения "графиков Шилда". Для определения рА2 и наклона регрессионной кривой использовали регрессионный анализ, соотносящий log(CR-l) с логарифмом концентрации антагониста. И, наконец, эксперименты группировались по химическим веществам и вычисляли среднее ± среднеквадратичную погрешность рА2 и наклона. С помощью стандартных способов для наклона определяли 95% доверительные уровни (CL) исходя из его среднеквадратичной погрешности. Для экспериментов, в которых воздействию данного химического вещества подвергали лишь одну полоску, pKD вычисляли как (концентрация антагониста)/(СR-1), после чего отрицательный логарифм KD объединяли со значениями рА2 для получения расширенного набора значений рА2.
Результаты
Воздействие рацемического оксибутинина и DEO, а также их соответствующих энантиомеров на карбохол-индуцированное сокращение обобщено в приведенной далее табл. 9. Представленные
данные являются обобщением анализа Шилда, дающим значения рА2 (среднее ± среднеквадратичная погрешность) и наклона (среднее ± среднеквадратичная погрешность).
Эти результаты свидетельствуют, что как S-OXY, так и S-DEO являются менее эффективными антагонистами мускариновых рецепторов мочевого пузыря по сравнению с R- и рацемическим OXY, а также R- и рацемическим DEO.
Влияние рацемического оксибутинина и его энантиомеров на калий-индуцированное сокращение обобщено в приведенной далее табл. 10. (Представленные значения являются величиной сокращения, вызываемого 137,7 мМ К+ после 60 мин воздействия соединения, разделенной на величину сокращения, индуцированного перед воздействием лекарственного средства).
Эти результаты свидетельствуют о том, что оксибутинин и его энантиомеры, а также дезэтилоксибутинин и его энантиомеры являются равно эффективными спазмолитиками гладких мышц мочевого пузыря.
Выводы
Общеизвестно, что нормальное опорожнение мочевого пузыря опосредуется холинергическими механизмами, в то время как нестабильность мочевого пузыря, наблюдаемая у пациентов, страдающих
недержанием мочи, по-видимому, связывается с нехолинергическими сокращениями мочевого пузыря. Андерсон (Andersson) и др. (Neurourol Urodun 5, 579-586 (1986)) показали на животных, что устойчивая к
атропину мышца, выталкивающая мочу, обладает высокой чувствительностью к антагонистам кальция.
Исследование рецептор-связующего аффинитета R- и (S)-оксибутинина к рецепторным участкам для блокаторов кальциевых каналов дилтиазема и верапамила, описанное выше, позволяет сделать вывод о том, что (S)-оксибутинин и (S)-дезэтилоксибутинин оказывают терапевтическое воздействие на непроизвольное мочеиспускание, в то время как (в отличие от R-изомеров и рацематов) они оказывают весьма незначительное воздействие на механизм нормального опорожнения мочевого пузыря. Оба соединения демонстрируют также значительное ослабленное антихолинергическое побочное воздействие по сравнению с соответствующим R-изомером и рацематом. Особого внимания заслуживает отсутствие побочного воздействия на сердечно-сосудистую систему, что является следствием антихолинергического действия рацемического оксибутинина. Мы пришли к заключению, что (S)-оксибутинин и (S)-дезэтилоксибутинин являются эффективными медикаментозными средствами для лечения недержания мочи у людей со значительно сокращенным побочным воздействием по сравнению с рацематами или чистыми R-энантиомерами.
Метил (R)-α-циклогексил-α-гидроксибензолацетат (IV)
К смеси (R)-α-циклогексил-α-гидроксибензолуксусной кислоты (III) (12,2 г, 52,1 ммоль) и К2
СО3 (10,8 г, 78,2 ммоль) в 100 мл ацетона прикапывали при 0oС (ледяная баня) метила иодид (МеI) (13,0 мл, 208 ммоль). После добавления (приблизительно в течение 1 часа) МеI
реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь фильтровали через слой целита и дважды промывали ацетоном. Фильтрат концентрировали до получения суспензии белого цвета,
которую разбавляли водой и экстрагировали гептаном. Объединенные экстракты промывали водой, рассолом, сушили и концентрировали до получения продукта (R)-IV (11,9 г, выход 92%) в виде твердого вещества
белого цвета.
Метил (S)-α-циклогексил-α-гидроксибензолацетат (IV).
Выполнив вышеприведенную процедуру, из (S)-III (10,6 г, 45,3 ммоль) получили (S)-IV (11, 2 г, выход 100%) в виде твердого вещества белого цвета.
4-[N-этил-(4-метоксифенил)метиламино]-2-бутинил(R)-α-циклогексил-α-гидроксибензолацетат (VI).
К раствору (R)-IV (11,9 г, 47,7 ммоль) и 4-[N-этил-(4-метоксифенил)метиламино] -2-бутин-1-ола (V) (9,30 г, 39,9 ммоль) в 120 мл толуола добавляли NaOMe (0,222 г, 4,11 ммоль). Реакционную смесь перемешивали с нагревом в колбе с обратным холодильником в течение 5 часов и с помощью аппарата Дина-Старка отбирали в целом 6 мл растворителя. Реакционную смесь охлаждали до комнатной температуры, разводили этилацетатом, промывали водой, рассолом, сушили и концентрировали. Остаток хроматографировали на силикагеле (элюирование 1, 2,5 и затем 5% МеОН и СН2Cl2) до получения продукта (R)-IV (14,1 г, выход 79%) в виде масла.
4-[N-этил-(4-метоксифенил)метиламино]-2-бутинил(S)-α-циклогексил-α-гидроксибензолацетат (VI).
Выполнив вышеприведенную процедуру, из (S)-IV (4,24 г, выход 58%) и V (4,24 г, 18,2 ммоль) получили (S)-VI (4,24 г, выход 58%) в виде масла.
Рацемический 4-[N-этил-(4-метоксифенил)метиламино]-2-бутинил(S)-α-циклогексил-α-гидроксибензолацетат.
Выполнив вышеприведенную процедуру, из рацемического IV (2,98 г, 12,0 ммоль) и V (2,48 г, 10,6 ммоль) получили рацемический предшественник DEO (2,05 г, выход 43%) в виде масла.
4-(этиламино)-2-бутинил(R)-α-циклогексил-α-гидроксибензолацетата хлористоводородная соль (VII)-HCl.
Раствор (R)-VI (14,0 г, 31,2 ммоль) и α-хлорэтилкарбонхлоридата (4,0 мл, 37,4 ммоль) в 1,2-дихлорэтане перемешивали с нагревом в колбе с обратным холодильником в течение 1 часа. После охлаждения реакционную смесь концентрировали и к остатку добавляли 200 мл МеОН. Реакционную смесь перемешивали с нагревом в колбе с обратным холодильником в течение 20 мин и охлаждали до комнатной температуры. Смесь концентрировали и остаток хроматографировали на силикагеле (элюирование 1% и затем 50% МеОН в СН2Сl2, затем смешивали с эфиром до получения продукта (R)-(VII)-HC1 (8,93 г, выход 87%) в виде твердого вещества коричнево-желтого цвета. Твердое вещество коричнево-желтого цвета подвергали дальнейшей очистке посредством перекристаллизации из EtOH/Et2O и последующей обработки 10% водным раствором К2СО3 и EtOAc, активировали углем и раствором 1N HC1 в эфире до получения (R)-DEO-HC1 (6,44 г) в виде твердого вещества не совсем белого цвета.
4-(этиламино)-2-бутинил(S)-α-циклогексил α-гидроксибензолацетата хлористо-водородная соль (VII)-HCl.
Выполнив вышеприведенную процедуру, из (S)-VI (11,4 г, 25,4 ммоль) получили (S)-DEO-HCl (5,27 г, выход 57%) в виде твердого вещества не совсем белого цвета.
Рацемическая 4-(этиламино)-2-бутинил-α-циклогексил-α-гидроксибензолацетата хлористоводородная соль.
Выполнив вышеприведенную процедуру, из (±) предшественника (2, 28 г, 5,08 ммоль) получили (±)-DEO-HC1 (0,63 г) в виде твердого вещества не совсем белого цвета.
S-оксибутинин может быть приготовлен таким же образом посредством замены 4-(диэтиламино)-2-бутил-1-ола на защищенный промежуточный V.
4-[N-этил-(4-метоксифенил)метиламино] -2-бутин-1-ол (V), используемый в качестве промежуточного вещества, синтезируется
следующим образом:
N-этил-4-метоксибензолметанамин:
К смеси анисальдегида (15,6 г, 115 ммоль) и этиламина (2,0 М в THF, 87 мл, 174 ммоль) в 1,2-дихлорэтане (450 мл) добавляли ледяную
уксусную кислоту (10,0 мл, 174 ммоль) в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, после чего охлаждали до 0oС с помощью ледяной бани.
Порциями добавляли NaBH(OAc) (36,9 г, 174 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали и остаток разбавляли основным раствором (10 г NaOH в
100 мл воды) для придания раствору незначительной основности. Этот водный слой экстрагировали эфиром. Объединенные экстракты промывали водой, рассолом, сушили и концентрировали. Остаток
хроматографировали на силикагеле (элюирование 5% МеОН в CH2Cl2, затем 50% МеОН в СН2Cl2, содержащем 4% EtN) до получения продукта (11,2 г, выход 59%) в виде
масла.
4-[N-этил-(4-метоксифенил)метиламино]-2-бутин-1-ол (V):
Смесь N-этил-4-метоксибензолметанамина (13,3 г, 80,6 ммоль), параформальдегида (3,63 г), пропаргилового спирта
(6,33 г, 113 ммоль) и CuCl (0,311 г) в 350 мл 1,4-диоксана перемешивали при нагревании в колбе с обратным холодильником в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры и
концентрировали. Остаток разводили 200 мл 50% NH4OH и экстрагировали EtOAc. Объединенные экстракты промывали водой, рассолом, сушили и концентрировали. Остаток хроматографировали на
силикагеле (элюирование 2,5% МеОН в СН2Cl2, затем 5% МеОН в CH2Cl2) до получения продукта V (15,1 г, выход 81%) в виде масла.
Реферат
Изобретение относится к медицине, в частности к урологии, и касается лечения недержания мочи. Для этого предлагается использовать введение терапевтически эффективного количества (S)-оксибутинина, (S)-дезэтилоксибутинина или его фармацевтически приемлемой соли, по существу свободного от соответствующего R энантиомера. Раскрываются также фармацевтические составы в виде таблеток и чрескожных средств, включающих (S)-оксибутинин или (S)-дезэтилоксибутинин и приемлемый носитель, а также синтез дезэтилоксибутинина. Способ предотвращает отрицательные эффекты терапии, сопутствующие введению рацемического оксибутинина. 8 с. и 23 з.п. ф-лы, 11 табл., 1 ил.
Формула
31.01.1995 - для пунктов, содержащих признаки, относящиеся к (S)-оксибутинину;
07.06.1995 - для пунктов, содержащих признаки, относящиеся к (S)-дезэтилоксибутинину.
Комментарии