Ингибиторы бета-лактамаз - RU2445314C1
Код документа: RU2445314C1
Чертежи


Описание
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет предварительной заявки на патент США № 61/011533 (поданной 18 января 2008 г.), содержание которой включено в настоящее описание путем ссылки во всей ее полноте.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым ингибиторам бета-лактамаз и их применению против устойчивости бактерий к действию антибиотиков. Более конкретно, изобретение относится к композициям и способам для преодоления устойчивости бактерий к действию антибиотиков.
УРОВЕНЬ ТЕХНИКИ
Устойчивость бактерий к действию антибиотиков стала одной из наиболее серьезных проблем современного здравоохранения. В публикации Cohen, Science 1992, 257: 1051-1055 сообщается, что инфекции, вызванные устойчивыми к действию медицинских препаратов бактериями, часто приводят к более длительному пребыванию в больнице, более высокой смертности и повышенным затратам на лечение. В публикации Neu, Science 1992, 257: 1064-1073 сообщается, что необходимость в новых антибиотиках будет постоянно возрастать в силу того, что бактерии обладают удивительной способностью вырабатывать устойчивость к действию новых лекарственных средств, быстро превращая их в неэффективные. В публикации Anderson, Nature America 1999, 5; 147-149 отмечается, что процесс распространения явления устойчивости к действию антибиотиков носит пандемический характер, и утверждается, что решение растущей серьезной проблемы здравоохранения потребует междисциплинарного подхода.
Данный кризис вызвал проведение различных исследований для выяснения механизмов, лежащих в основе устойчивости бактерий. Так, в публикации Coulton et al., Progress in Medicinal Chemistry 1994, 31: 297-349 заявляется, что широкое применение пенициллинов и цефалоспоринов привело к возникновению β-лактамаз, семейству бактериальных ферментов, которые катализируют гидролиз β-лактамового кольца, которое присутствует в большинстве применяемых в настоящее время антибиотиков. Совсем недавно в публикации Dudley, Pharmacotherapy 1995, 15: 9S-14S было сообщено, что устойчивость, опосредованная β-лактамазами, является главным аспектом причины развития устойчивости бактерий к действию антибиотиков. В настоящее время доступными полусинтетическими или природными ингибиторами β-лактамаз являются клавулановая кислота, которая представляет собой метаболит грибков Streptomyces clavuligerus, и два полусинтетических антибиотика, сульбактам и тазобактам. В патентных документах US 5698577, US 5510343, US 6472406 и публикациях Hubschwerlen et al., J. Med. Chem. 1998, 41: 3961 и Livermore et al., J. Med. Chem. 1997, 40: 335-343 описаны некоторые синтетические ингибиторы β-лактамаз.
Другими источниками информации, относящимися к данному вопросу, являются следующие.
В патентном документе US 2003/0199541 A1 раскрыты несколько азабициклических соединений, включая конкретные 7-оксо-6-диазабицикло[3.2.1]октан-2-карбоксамиды, и их применение в качестве противобактериальных средств.
В патентном документе US 2004/0157826 A1 описаны несколько гетеробициклических соединений, включая конкретные производные диазепинкарбоксамида и диазепинкарбоксилата, и их применение в качестве противобактериальных средств и ингибиторов β-лактамаз.
В патентном документе WO 2008/039420 A2 раскрыты конкретные 7-оксо-2,6-диазабицикло[3.2.0]гептан-6-сульфоокси-2-карбоксамиды и их применение в качестве ингибиторов бета-лактамаз.
В публикации Poole, Cell Mol. Life Set 2004, 61: 2200-2223 приводится обзор устойчивости бактериальных патогенов к β-лактамовым антибиотикам и подходы для преодоления этой устойчивости.
Доступные в настоящее время ингибиторы β-лактамаз неспособны противодействовать постоянно увеличивающемуся разнообразию β-лактамаз. Поэтому существует необходимость в новых ингибиторах β-лактамаз.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к определенным соединениям диазабициклического карбоксамида и карбоксилата, которые являются ингибиторами бета-лактамаз. Соединения и их фармацевтически приемлемые соли применяют в комбинации с бета-лактамовыми антибиотиками при лечении бактериальных инфекций, в частности бактериальных инфекций, устойчивых к действию антибиотиков. Более конкретно, настоящее изобретение включает соединения формулы I:

и их фармацевтически приемлемые соли, где:
связь, обозначенная как "a", является простой связью или двойной связью;
когда связь a является простой связью, X является:
(1) CH2,
(2) CH2CH2,
(3) CH2CH2CH2,
(4) CH-CH,
(5) CH2-CH-CH, или
(6) CH-CH-CH2;
когда связь a является двойной связью, X является:
(1) CH,
(2) CH-CH2, или
(3) CH-CH=CH;
R1 является:
(1) C(O)N(R3)R4,
(2) C(O)OR3, или
(3) C(O)OR5;
R2 является SO3M, OSO3M, SO2NH2, PO3M, OPO3M, CH2CO2M, CF2CO2M, или CF3;
M является H или фармацевтически приемлемым катионом;
R3 является:
(1) C1-8алкилом, замещенным суммарно от 1 до 4 заместителями, выбранными из группы, состоящей из (i) от нуля до 2 N(RA)RB, (ii) от нуля до 2 RC, и (iii) от нуля до 1 из AryA, HetA, или HetB,
(2) CycA,
(3) HetA,
(4) AryA,
(5) HetB, или
(6) AryB;
R4 является H или C1-8алкилом, необязательно замещенным с помощью N(RA)RB;
или, в качестве варианта, когда R1 является C(O)N(R3)R4, R3 и R4 вместе с атомом N, к которому они оба присоединены, образуют насыщенное моноциклическое кольцо с числом членов от 4 до 9, содержащее 1 гетероатом в дополнение к азоту, присоединенному к R3 и R4, выбранному из N, O, и S, где S необязательно окислена до S(O) или S(O)2; где моноциклическое кольцо необязательно конденсировано, соединено мостиком или спиро с насыщенным гетероциклическим кольцом с числом членов от 4 до 7, содержащим от 1 до 3 гетероатомов, независимо выбранных из N, O и S, где S необязательно окислена до S(O) или S(O)2, с образованием бициклической кольцевой системы, где образованная таким образом моноциклическая кольцевая или бициклическая кольцевая система необязательно замещена с помощью 1 или 2 заместителей, каждый из которых независимо является: (1) C1-6алкилом, (2) C1-6фторалкилом, (3) (CH2)1-2G, где G является OH, O-C1-6алкилом, O-C1-6фторалкилом, N(RA)RB, C(O)N(RA)RB, C(O)RA, CO2RA, или SO2RA, (4) O-C1-6алкилом, (5) O-C1-6фторалкилом, (6) OH, (7) оксо, (8) галогеном, (9) N(RA)RB, (10) C(O)N(RA)RB, (11) C(O)RA, (12) C(O)-C1-6фторалкилом, (13) C(O)ORA, или (14) S(O)2RA;
R5 является C1-8алкилом, замещенным 1 или 2 заместителями, каждый из которых независимо является N(RA)C(O)-AryA;
CycA является C4-9циклоалкилом, который необязательно замещен суммарно с помощью от 1 до 4 заместителей, выбранных из от нуля до 2 (CH2)nN(RA)RB и от нуля до 2 (CH2)nRC;
HetA является насыщенным или мононенасыщенным гетероциклическим кольцом с числом членов от 4 до 9, содержащим от 1 до 3 гетероатомов, независимо выбранных из N, O и S, где любая S в кольце необязательно окислена до S(O) или S(O)2 и либо 1, либо 2 атома углерода в кольце необязательно окислены до C(O); где кольцо необязательно конденсировано с C3-7циклоалкилом; и где необязательно конденсированное, насыщенное или мононенасыщенное кольцо необязательно замещено суммарно с помощью от 1 до 4 заместителей, выбранных из от нуля до 2 (CH2)nN(RA)RB и от нуля до 2 (CH2)nRC;
AryA является фенилом, который необязательно замещен суммарно с помощью от 1 до 4 заместителей, выбранных из от нуля до 2 (CH2)nN(RA)RB и от нуля до 2 (CH2)nRC;
HetB является гетероароматическим кольцом с 5 или 6 членами, содержащим от 1 до 4 гетероатомов, выбранных из от 1 до 3 атомов N, из нуля или 1 атома О, и из нуля или 1 атома S; где гетероароматическое кольцо необязательно конденсировано с насыщенным гетероциклическим кольцом с числом членов от 5 до 7, содержащим 1 или 2 гетероатома, независимо выбранных из N, O и S, где любая S в кольце необязательно окислена до S(O) или S(O)2 и либо 1, либо 2 атома углерода в неконденсированном кольце необязательно окислены до C(O); и где необязательно конденсированное гетероароматическое кольцо необязательно замещено суммарно с помощью от 1 до 4 заместителей, выбранных из от нуля до 2 (CH2)nN(RA)RB и от нуля до 2 (CH2)nRC;
AryB является бициклической кольцевой системой, в которой фенил конденсирован с насыщенным гетероциклическим кольцом с числом членов от 5 до 7, содержащим от 1 до 3 гетероатомов, независимо выбранных из N, O и S, где любая S в кольце необязательно окислена до S(O) или S(O)2, и где бициклическая кольцевая система необязательно замещена суммарно с помощью от 1 до 4 заместителей, выбранных из от нуля до 2 (CH2)nN(RA)RB и от нуля до 2 (CH2)nRC;
каждое n независимо является целым числом, которое равняется 0, 1, 2, или 3;
каждый RA является независимо H или C1-8алкилом;
каждый RB является независимо H или C1-8алкилом;
каждый RC является независимо C1-6алкилом, OH, O-C1-8алкилом, OC(O)-C1-8алкилом, C(=NH)NH2, NH-C(=NH)NH2, галогеном, CN, C(O)RA, C(O)ORA, C(O)N(RA)RB, SO2RA, SO2N(RA)RB, пиридилом, пирролидинилом, пиперидинилом, пиперазинилом, морфолинилом, или тиоморфолинилом;
и при условии, что:
(A) когда R1 является C(O)OR3 и R3 является AryA, тогда AryA не является (i) незамещенным фенилом, (ii) фенилом, замещенным с помощью NH2, (iii) фенилом, замещенным с помощью OH, (iii) фенилом, замещенным с помощью O-C1-6алкилом, (iv) фенилом, замещенным одним или более галогенами, или (v) фенилом, замещенным с помощью C1-6алкилом;
(B) когда R1 является C(O)OR3 и R3 является C1-6алкилом, замещенным с помощью HetB , тогда HetB не является пиридилом;
(C) когда R1 является C(O)OR3 и R3 является CH2-AryA или CH2CH2-AryA, тогда AryA не является (i) незамещенным фенилом, (ii) фенилом, замещенным с помощью NH2, OH, O-C1-6алкила, или C1-6алкила, или (iii) фенилом, замещенным одним или более галогенами;
(D) когда R1 является C(O)N(R3)R4, R3 является AryA, CH2-AryA или CH2CH2-AryA, и R4 является H или C1-6алкилом, тогда AryA не является незамещенным фенилом, фенилом, замещенным с помощью N(CH3)2, или фенилом, замещенным с помощью C(O)NH2;
(E) когда R1 является C(O)N(R3)R4, R3 является C1-6алкилом, замещенным с помощью HetB, и R4 является H или C1-6алкилом, тогда HetB не является пиридилом; и
(F) когда R1 является C(O)OR3 и R3 является C1-6алкилом, замещенным с помощью RC, тогда RC не является C(O)NH2.
Соединения формулы I ингибируют β-лактамазы и синергитически усиливают противобактериальное действие β-лактамовых антибиотиков (например, имипенема, цефтазидима и пиперациллина) против микроорганизмов, обычно устойчивых к действию β-лактамовых антибиотиков в результате присутствия β-лактамаз. Соединения настоящего изобретения эффективны против β-лактамаз класса A и класса C, и их комбинация с бета-лактамовыми антибиотиками, такими как имипенем, цефтазидим или пиперациллин, может обеспечить эффективное лечение бактериальных инфекций, вызванных β-лактамазами класса A и класса C, продуцируемыми микроорганизмами. Соответственно, настоящее изобретение включает комбинации соединения формулы I с β-лактамовым антибиотиком, подходящим для использования против β-лактамазы класса C, продуцируемой такими бактериями, как Pseudomonas spp., и против β-лактамазы класса A, продуцируемой такими бактериями, как Klebsiella spp. Изобретение также включает композиции, содержащие соединение формулы I или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель. Кроме того, изобретение включает способы лечения бактериальных инфекций и ингибирования бактериального роста путем применения соединения формулы I или его соли, или комбинации, или композиции, содержащей соединение или его соль.
Варианты осуществления, подварианты осуществления, аспекты и характерные черты настоящего изобретения далее либо будут описаны, либо становятся очевидными из последующего описания, примеров и прилагаемых пунктов формулы изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг.1 приведена порошковая рентгенограмма для кристаллического моногидрата, описанного в примере 1D.
На фиг.2 приведена кривая дифференциальной сканирующей калориметрии (DSC) для кристаллического моногидрата, описанного в примере 1D.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Как уже было отмечено выше, настоящее изобретение включает соединения формулы I, где соединения являются ингибиторами бета-лактамаз, подходящими для применения в комбинации с бета-лактамовыми антибиотиками для лечения бактериальных инфекций.
Термин "ингибитор β-лактамазы" относится к соединению, которое способно ингибировать активность β-лактамазы. Ингибирование активности β-лактамазы означает ингибирование активности β-лактамазы класса A, C, или D. В случае противомикробных применений предпочтительно, чтобы концентрация, при которой достигается 50% ингибирование, составляла примерно 100 микрограмм/мл или ниже, или примерно 50 микрограмм/мл или ниже, или примерно 25 микрограмм/мл. Термины β-лактамазы "класса A", "класса C", и "класса D" являются хорошо известными терминами для специалистов в этой области, и они описаны в монографии Waley, The Chemistry of β-лактамаза. Page Ed., Chapman & Hall, London, (1992) 198-228.
Термин "β-лактамаза" обозначает белок, способный инактивировать β-лактамовый антибиотик. β-Лактамаза может являться ферментом, который катализирует гидролиз β-лактамового кольца β-лактомового антибиотика. Особый интерес здесь представляют микробные β-лактамазы. β-Лактамаза может являться, например, сериновой β-лактамазой. Представляющий интерес β-лактамазы являются лактамазами, которые описаны, например, в монографии Waley, The Chemistry of β-лактамаза, Page Ed., Chapman & Hall, London, (1992) 198-228. Представляющий здесь особый интерес β-лактамазы включают β-лактамазу класса C из Pseudomonas aeruginosa или Enterobacter cloacae P99 (далее обозначаемую как P99 β-лактамаза) и бета-лактамазу класса A из Klebsiella spp.
Термин "антибиотик" относится к соединению или композиции, которые снижают жизнеспособность микроорганизма, или которые ингибируют рост или пролиферацию микроорганизма. Фраза "ингибирует рост или пролиферацию" означает увеличение времени генерации (то есть времени, требующегося для деления бактериальной клетки или для удвоения популяции), по меньшей мере, примерно в 2 раза. Предпочтительными антибиотиками являются антибиотики, которые могут увеличивать время генерации, по меньшей мере, примерно в 10 раз или более (например, по меньшей мере, примерно в 100 раз или даже до бесконечности, как в случае смерти всех клеток). При использовании в этом описании дополнительно предполагается, что антибиотик включает противомикробное, бактериостатическое, или бактерицидное средство. Примеры антибиотиков, подходящих для применения с точки зрения настоящего изобретения, включают пенициллины, цефалоспорины и карбапенемы.
Термин "β-лактамовый антибиотик" относится к соединению с антибиотическими свойствами, которое содержит β-лактамовую функциональность. Неограничивающие примеры β-лактамовых антибиотиков, подходящих с точки зрения изобретения, включают пенициллины, цефалоспорины, пенемы, карбапенемы, и монобактамы.
Первым вариантом осуществления настоящего изобретения (иначе обозначаемый здесь как "вариант осуществления Е1") является соединение формулы I (иначе обозначаемое здесь как "соединение I"), определяемое выше (то есть определяемое выше в разделе "Сущность изобретения"), или его фармацевтически приемлемая соль; и при условии, что:
(A) когда R1 является C(O)OR3, тогда R3 не является AryA;
(B) когда R1 является C(O)OR3, тогда R3 не является C1-8алкилом, замещенным с помощью HetB;
(C) когда R1 является C(O)OR3, тогда R3 не является C1-8алкилом, замещенным с помощью AryA;
(D) когда R1 является C(O)N(R3)R4; R3 является AryA или C1-8алкилом, замещенным с помощью AryA, и R4 является H или C1-8алкилом, тогда AryA не является незамещенным фенилом, фенилом, замещенным с помощью 1 или 2 N(RA)RB, или фенилом, замещенным с помощью 1 или 2 C(O)N(RA)RB;
(E) когда R1 является C(O)N(R3)R4 и R4 является H или C1-8алкилом, тогда R3 не является C1-8алкилом, замещенным с помощью HetB; и
(F) когда R1 является C(O)OR3 и R3 является C1-8алкилом, замещенным с помощью RC, тогда RC не является C(O)N(RA)RB.
Вторым вариантом осуществления настоящего изобретения (вариант осуществления Е2) является определяемое выше соединение формулы I, или его фармацевтически приемлемая соль; и при условии, что:
(A) когда R1 является C(O)OR3, тогда R3 не является AryA;
(B) когда R1 является C(O)OR3, тогда R3 не является C1-8алкилом, замещенным с помощью HetB;
(C) когда R1 является C(O)OR3, тогда R3 не является C1-8алкилом, замещенным с помощью AryA;
(D) когда R1 является C(O)N(R3)R4, тогда R3 не является AryA или C1-8алкилом, замещенным с помощью AryA;
(E) когда R1 является C(O)N(R3)R4, тогда R3 не является C1-8алкилом, замещенным с помощью HetB; и
(F) когда R1 является C(O)OR3, тогда R3 не является C1-8алкилом, замещенным с помощью RC.
Третьим вариантом осуществления настоящего изобретения (вариант осуществления Е3) является соединение формулы I, или его фармацевтически приемлемая соль; где:
R1 является:
(1) C(O)N(R3)R4, или
(2) C(O)OR3;
R3 является:
(1) C1-8алкилом, замещенным суммарно от 1 до 4 заместителями, выбранными из группы, состоящей из (i) от нуля до 2 N(RA)RB, (ii) от нуля до 2 RC, и (iii) от нуля до 1 из AryA, HetA, или HetB,
(2) CycA,
(3) HetA,
(4) AryA, или
(5) HetB;
R4 является H или C1-8алкилом, необязательно замещенным с помощью N(RA)RB;
HetA является насыщенным гетероциклическим кольцом с числом членов от 4 до 9, содержащим от 1 до 3 гетероатомов, независимо выбранных из N, O и S, где насыщенное гетероциклическое кольцо является необязательно суммарно замещенным с помощью от 1 до 4 заместителей, выбранных из от нуля до 2 (CH2)nN(RA)RB и от нуля до 2 (CH2)nRC;
каждый RC является независимо C1-6алкилом, OH, O-C1-8алкилом, C(=NH)NH2, NH-C(=NH)NH2, галогеном, CN, пиридилом, пирролидинилом, или пиперидинилом; и
все другие переменные определены выше; и при условии, что:
(A) когда R1 является C(O)OR3 и R3 является AryA, тогда AryA не является (i) незамещенным фенилом, (ii) фенилом, замещенным с помощью NH2, (iii) фенилом, замещенным с помощью OH, (iii) фенилом, замещенным с помощью O-C1-6алкила, (iv) фенилом, замещенным одним или более галогенами; или (v) фенилом, замещенным с помощью C1-6алкила;
(B) когда R1 является C(O)OR3 и R3 является C1-6алкилом, замещенным с помощью HetB, тогда HetB не является пиридилом;
(C) когда R1 является C(O)OR3 и R3 является CH2-AryA или CH2CH2-AryA, тогда AryA не является (i) незамещенным фенилом, (ii) фенилом, замещенным с помощью NH2, OH, O-C1-6алкила, или C1-6алкила, или (iii) фенилом, замещенным одним или более галогенами;
(D) когда R1 является C(O)N(R3)R4, R3 является AryA, CH2-AryA или CH2CH2-AryA, и R4 является H или C1-6алкилом, тогда AryA не является ни незамещенным фенилом, ни фенилом, замещенным с помощью N(CH3)2; и
(E) когда R1 является C(O)N(R3)R4, R3 является C1-6алкилом, замещенным с помощью HetB, и R4 является H или C1-6алкилом, тогда HetB не является пиридилом.
Четвертым вариантом осуществления настоящего изобретения (вариант осуществления Е4) является соединение формулы I, определенное в варианте осуществления Е3, или его фармацевтически приемлемая соль; и при условии, что:
(A) когда R1 является C(O)OR3, тогда R3 не является AryA;
(B) когда R1 является C(O)OR3, тогда R3 не является C1-6алкилом, замещенным с помощью HetB;
(C) когда R1 является C(O)OR3, тогда R3 не является C1-6алкилом, замещенным с помощью AryA;
(D) когда R1 является C(O)N(R3)R4, тогда R3 не является AryA или C1-6алкилом, замещенным с помощью AryA; и
(E) когда R1 является C(O)N(R3)R4, тогда R3 не является C1-6алкилом, замещенным с помощью HetB.
Пятым вариантом осуществления настоящего изобретения (вариант осуществления Е5) является соединение формулы I, определенное в варианте осуществления Е3, или его фармацевтически приемлемая соль; и при условии, что:
(A) когда R1 является C(O)OR3, тогда R3 не является AryA;
(B) когда R1 является C(O)OR3, тогда R3 не является C1-6алкилом, замещенным с помощью HetB;
(C) когда R1 является C(O)OR3, тогда R3 не является C1-6алкилом, замещенным с помощью AryA;
(D) когда R1 является C(O)N(R3)R4, тогда R3 не является AryA или C1-6алкилом, замещенным с помощью AryA; и
(E) когда R1 является C(O)N(R3)R4, тогда R3 не является C1-6алкилом, замещенным с помощью HetB.
Шестым вариантом осуществления настоящего изобретения (вариант осуществления Е6) является соединение формулы I, или его фармацевтически приемлемая соль, где связь "a" является простой связью; X является -CH2- или
-CH2CH2-; и все другие переменные определены выше или определены в любом из вышеизложенных вариантов осуществления.
Седьмым вариантом осуществления настоящего изобретения (вариант осуществления Е7) является соединение формулы I, или его фармацевтически приемлемая соль, где связь "a" является простой связью; X является -CH2-, и все другие переменные определены выше или определены в любом из вышеизложенных вариантов осуществления.
Восьмым вариантом осуществления настоящего изобретения (вариант осуществления Е8) является соединение формулы I, или его фармацевтически приемлемая соль, где связь "a" является простой связью; X является -CH2CH2-; и все другие переменные определены выше или определены в любом из вышеизложенных вариантов осуществления.
Девятым вариантом осуществления настоящего изобретения (вариант осуществления Е9) является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; и все другие переменные определены выше или определены в любом одном из предшествующих вариантов осуществления. В аспекте этого варианта осуществления R1 является C(O)NH(R4).
Десятым вариантом осуществления настоящего изобретения (вариант осуществления Е10) является соединение формулы I, или его фармацевтически приемлемая соль, где R2 является OSO3M; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Одиннадцатым вариантом осуществления настоящего изобретения (вариант осуществления Е11) является соединение формулы I, или его фармацевтически приемлемая соль, где R2 является OSO3H; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Двенадцатым вариантом осуществления настоящего изобретения (вариант осуществления Е12) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является: (1) C1-4алкилом, замещенным суммарно от 1 до 4 заместителями, выбранными из группы, состоящей из (i) от нуля до 2 N(RA)RB, (ii) от нуля до 2 RC, и (iii) от нуля до 1 из AryA, HetA, или HetB, (2) CycA, (3) HetA, (4) AryA, или (5) HetB; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Тринадцатым вариантом осуществления настоящего изобретения (вариант осуществления Е13) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является (CH2)2-3N(RA)RB, (CH2)1-3-AryA, (CH2)1-3-HetA, (CH2)1-3-HetB, CycA, HetA, AryA, или HetB; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Четырнадцатым вариантом осуществления настоящего изобретения (вариант осуществления Е14) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetA, CH2-HetA, CH2CH2-HetA, CH(CH3)-HetA, или CH(CH2OH)-HetA; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е14 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 определен выше в варианте осуществления Е14; и все другие переменные определены выше в варианте осуществления Е14.
Пятнадцатым вариантом осуществления настоящего изобретения (вариант осуществления Е15) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetA, CH2-HetA, или CH2CH2-HetA; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е15 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 определен выше в варианте осуществления Е15; и все другие переменные определены выше в варианте осуществления Е15.
Шестнадцатым вариантом осуществления настоящего изобретения (вариант осуществления Е16) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetA или CH2-HetA; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е16 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 определен выше в варианте осуществления Е16; и все другие переменные определены выше в варианте осуществления Е16.
Семнадцатым вариантом осуществления настоящего изобретения (вариант осуществления Е17) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetA; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е17 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 определен выше в варианте осуществления Е17; и все другие переменные определены выше в варианте осуществления Е17.
Восемнадцатым вариантом осуществления настоящего изобретения (вариант осуществления Е18) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetA; HetA является насыщенным гетероциклом, выбранным из группы, состоящей из пирролидинила, пиперидинила, азепанила, и азоканила; где насыщенный гетероцикл необязательно замещен с помощью N(RA)RB и необязательно замещен с помощью 1 или 2 (CH2)nRC; каждый RC является независимо C1-6алкилом, OH, O-C1-8алкилом, C(=NH)NH2, NH-C(=NH)NH2, галогеном, CN, пиридилом, пирролидинилом, или пиперидинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Девятнадцатым вариантом осуществления настоящего изобретения (вариант осуществления Е19) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetA; HetA является:

где символ звездочки обозначает место присоединения HetA к остальной части соединения; T является H или RC; RC является C1-6алкилом, OH, O-C1-8алкилом, C(=NH)NH2, NH-C(=NH)NH2, галогеном, CN, пиридилом, пирролидинилом, или пиперидинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Двадцатым вариантом осуществления настоящего изобретения (вариант осуществления Е20) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetA; HetA является:


где символ звездочки обозначает место присоединения HetA к остальной части соединения; T является H или RC; RC является С1-6алкилом, OH, O-C1-8алкилом, C(=NH)NH2, NH-C(=NH)NH2, галогеном, CN, пиридилом, пирролидинилом, или пиперидинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. В аспекте этого варианта осуществления T является H.
Двадцать первым вариантом осуществления настоящего изобретения (вариант осуществления Е21) является соединение формулы I, или его фармацевтически приемлемая соль, где HetA является необязательно конденсированным, насыщенным гетероциклическим кольцом, выбранным из группы, состоящей из азетидинила, пирролидинила, оксопирролидинила (например, 2-оксо-пирролидинила), пиперидинила, пиперазинила, тетрагидропиранила, тетрагидротиопиранила, морфолинила, 1,1-диоксидотетрагидро-тиопиранила, азепанила, оксазепанила, азоканила, и азабицикло[3.1.0]циклогексила, где гетероцикл необязательно замещен с помощью 1 или 2 (CH2)nN(RA)RB и необязательно замещен помощью 1 или 2 (CH2)nRC, и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е21 является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetA, и HetA и все другие переменные определены выше в варианте осуществления Е21. Вторым подвариантом осуществления варианта осуществления Е21 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 является HetA; и HetA и все другие переменные определены выше в варианте осуществления Е21. В аспекте этого варианта осуществления и его подвариантах осуществления HetA необязательно монозамещен с помощью (CH2)nN(RA)RB и необязательно замещен с помощью 1 или 2 (CH2)nRC.
Двадцать вторым вариантом осуществления настоящего изобретения (вариант осуществления Е22) является соединение формулы I, или его фармацевтически приемлемая соль, где HetA является насыщенным гетероциклическим кольцом, выбранным из группы, состоящей из пирролидинила, пиперидинила, азепанила, и азоканила; где гетероцикл необязательно замещен с помощью 1 или 2 (CH2)nN(RA)RB и необязательно замещен с помощью 1 или 2 (CH2)nRC; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е22 является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetA; и HetA и все другие переменные определены выше в варианте осуществления Е22. Вторым подвариантом осуществления варианта осуществления Е22 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 является HetA; и HetA и все другие переменные определены выше в варианте осуществления Е22. В аспекте этого варианта осуществления и его подвариантах осуществления HetA необязательно монозамещен с помощью (CH2)nN(RA)RB и необязательно замещен с помощью 1 или 2 (CH2)nRC.
Двадцать третьим вариантом осуществления настоящего изобретения (вариант осуществления Е23) является соединение формулы I, или его фармацевтически приемлемая соль, где HetA определен или в варианте осуществления Е21, или в варианте осуществления E22; каждый RC является независимо OH, O-C1-4алкилом, C(=NH)NH2, NH-C(=NH)NH2, Cl, Br, F, или CN; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. В первом подварианте осуществления R3 является HetA; и HetA определен выше в варианте осуществления Е21. Во втором подварианте осуществления R1 является C(O)N(R3)R4; R3 является HetA; и HetA определен выше в варианте осуществления Е22. В аспекте этого варианта осуществления и его подвариантах осуществления HetA необязательно монозамещен с помощью (CH2)nN(RA)RB и необязательно замещен с помощью 1 или 2 (CH2)nRC.
Двадцать четвертым вариантом осуществления настоящего изобретения (вариант осуществления Е24) является соединение формулы I, или его фармацевтически приемлемая соль, где HetA является гетероциклическим кольцом, определенным или в варианте осуществления Е21 или в варианте осуществления E22; гетероциклическое кольцо в HetA необязательно замещено с помощью галогена, C1-3алкила, O-C1-3алкила, NH2, N(H)-C1-3алкила, N(-C1-3алкила)2, CH2NH2, CH2N(H)-C1-3алкила, CH2N(-C1-3алкила)2, или пиперидинила; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. В первом подварианте осуществления R3 является HetA; и HetA определен выше в варианте осуществления Е24. Во втором подварианте осуществления, R1 является C(O)N(R3)R4; R3 является HetA; и HetA определен выше в варианте осуществления Е24.
Двадцать пятым вариантом осуществления настоящего изобретения (вариант осуществления Е25) является соединение формулы I, или его фармацевтически приемлемая соль, где HetA является гетероциклическим кольцом, определенным или в варианте осуществления Е21 или в варианте осуществления E22; гетероциклическое кольцо в HetA необязательно замещено с помощью F, CH3, OCH3, NH2, N(H)CH3, N(CH3)2, CH2NH2, CH2N(H)CH3, CH2N(CH3)2, или пиперидинила; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. В первом подварианте осуществления R3 является HetA; и HetA определен выше в варианте осуществления Е25. Во втором подварианте осуществления R1 является C(O)N(R3)R4; R3 является HetA; и HetA определен выше в варианте осуществления Е25.
Двадцать шестым вариантом осуществления настоящего изобретения (вариант осуществления Е26) является соединение формулы I, или его фармацевтически приемлемая соль, где HetA является гетероциклическим кольцом, выбранным из группы, состоящей из азетидинила, пирролидинила, пиразолидинила, пиперидинила, пиперазинила, азепанила, оксазепанила, оксазолидинила, изоксазолидинила, морфолинила, и тетрагидропиранила, где гетероциклическое кольцо необязательно замещено с помощью 1 или 2 заместителей, каждый из которых является независимо C1-3алкилом, CH2NH2, CH2N(H)-C1-3алкилом, CH2N(-C1-3алкил)2, O-C1-3алкилом, Cl, Br, F, NH2, N(H)-C1-3алкилом, N(-C1-3алкил)2, C(O)NH2, C(O)N(H)-C1-3алкилом, C(O)N(-C1-3алкил)2, C(O)-C1-3алкилом, C(O)O-C1-3алкилом, OC(O)-C1-3алкилом, S(O)2-C1-3алкилом, S(O)2NH2, S(O)2N(H)-C1-3алкилом, или S(O)2N(-C1-3алкил)2; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е26 является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetA, CH2-HetA, CH2CH2-HetA; и HetA и все другие переменные определены выше в варианте осуществления Е26. Вторым подвариантом осуществления варианта осуществления Е26 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)OR3; R3 является HetA, CH2-HetA, CH2CH2-HetA; и HetA и все другие переменные определены выше в варианте осуществления Е26. В аспекте этого варианта осуществления и его подвариантах осуществления R2 является OSO3H.
Двадцать седьмым вариантом осуществления настоящего изобретения (вариант осуществления Е27) является соединение формулы I, или его фармацевтически приемлемая соль, где HetA является гетероциклическим кольцом выбранными из группы, состоящей из азетидинила, пирролидинила, пиразолидинила, пиперидинила, пиперазинила, азепанила, оксазепанила, оксазолидинила, изоксазолидинила, морфолинила, и тетрагидропиранила, где гетероциклическое кольцо необязательно замещено с помощью 1 или 2 заместителей, каждый из которых является независимо CH3, CH2NH2, CH2N(H)CH3, CH2N(CH3)2, OCH3, Cl, Br, F, NH2, N(H)CH3, N(CH3)2, C(O)NH2, C(O)N(H)CH3, C(O)N(CH3)2, C(O)CH3, C(O)OCH3, OC(O)CH3, S(O)2CH3, S(O)2NH2, S(O)2N(H)CH3, или S(O)2N(CH3)2; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е27 является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetA, CH2-HetA, CH2CH2-HetA; и HetA и все другие переменные определены выше в варианте осуществления Е27. Вторым подвариантом осуществления варианта осуществления Е27 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)OR3; R3 является HetA, CH2-HetA, CH2CH2-HetA; и HetA и все другие переменные определены выше в варианте осуществления Е27. В аспекте этого варианта осуществления и его подвариантах осуществления R2 является OSO3H.
Двадцать восьмым вариантом осуществления настоящего изобретения (вариант осуществления Е28) является соединение формулы I, или его фармацевтически приемлемая соль, где AryA является фенилом, который необязательно замещен с помощью 1 или 2 заместителей, каждый из которых является независимо C1-3алкилом, CH2NH2, CH2N(H)-C1-3алкилом, CH2N(-C1-3алкил)2, O-C1-3алкилом, Cl, Br, F, NH2, N(H)-C1-3алкилом, N(-C1-3алкил)2, C(O)NH2, C(O)N(H)-C1-3алкилом, C(O)N(-C1-3алкил)2, C(O)-C1-3алкилом, C(O)O-C1-3алкилом, OC(O)-C1-3алкилом, S(O)2-C1-3алкилом, S(O)2NH2, S(O)2N(H)-C1-3алкилом, S(O)2N(-C1-3алкил)2, пирролидинилом, пиперидинилом, морфолинилом, CH2-пирролидинилом, CH2-пиперидинилом, или CH2-морфолинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е28 является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является AryA; и AryA и все другие переменные определены выше в варианте осуществления Е28. Вторым подвариантом осуществления варианта осуществления Е28 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)OR3; R3 является AryA; и AryA и все другие переменные определены выше в варианте осуществления Е28. В аспекте этого варианта осуществления и его подвариантах осуществления R2 является OSO3H.
Двадцать девятым вариантом осуществления настоящего изобретения (вариант осуществления Е29) является соединение формулы I, или его фармацевтически приемлемая соль, где AryA является фенилом, который необязательно замещен с помощью 1 или 2 заместителей, каждый из которых является независимо CH3, CH2NH2, CH2N(H)CH3, CH2N(CH3)2, OCH3, Cl, Br, F, NH2, N(H)CH3, N(CH3)2, C(O)NH2, C(O)N(H)CH3, C(O)N(CH3)2, C(O)CH3, C(O)OCH3, OC(O)CH3, S(O)2CH3, S(O)2NH2, S(O)2N(H)CH3, или S(O)2N(CH3)2, пирролидинилом, пиперидинилом, морфолинилом, CH2-пирролидинилом, CH2-пиперидинилом, или CH2-морфолинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е29 является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является AryA; и AryA и все другие переменные определены выше в варианте осуществления Е29. Вторым подвариантом осуществления варианта осуществления Е29 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)OR3; R3 является AryA; и AryA и все другие переменные определены выше в варианте осуществления Е29. В аспекте этого варианта осуществления и его подвариантах осуществления R2 является OSO3H.
Тридцатым вариантом осуществления настоящего изобретения (вариант осуществления Е30) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetB; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е30 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 определен выше в варианте осуществления Е30; и все другие переменные определены выше в варианте осуществления Е30.
Тридцать первым вариантом осуществления настоящего изобретения (вариант осуществления Е31) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetB; HetB является гетероароматическим кольцом, выбранным из группы, состоящей из пирролила, пиразолила, имидазолила, пиридила, и пиримидинила, где гетероароматическое кольцо необязательно монозамещено с помощью (CH2)nN(RA)RB и необязательно замещено с помощью 1 или 2 (CH2)nRC групп; каждый RC является независимо C1-6алкилом, OH, O-C1-8алкилом, C(=NH)NH2, NH-C(=NH)NH2, галогеном, CN, пиридилом, пирролидинилом, или пиперидинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Тридцать вторым вариантом осуществления настоящего изобретения (вариант осуществления Е32) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetB; HetB является пиридилом, который необязательно монозамещен с помощью N(RA)RB и необязательно замещен с помощью 1 или 2 RC групп; каждый RC является независимо C1-6алкилом, OH, O-C1-8алкилом, C(=NH)NH2, NH-C(=NH)NH2, галогеном, CN, пиридилом, пирролидинилом, или пиперидинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Тридцать третьим вариантом осуществления настоящего изобретения (вариант осуществления Е33) является соединение формулы или его фармацевтически приемлемая соль, где R3 является HetB; HetB является

Тридцать четвертым вариантом осуществления настоящего изобретения (вариант осуществления Е34) является соединение формулы I, или его фармацевтически приемлемая соль, где HetB является гетероароматическим соединением, выбранным из группы, состоящей из пирролила, пиразолила, имидазолила, пиридила, пиримидинила, тиазолила, пиперидотиазолила, пирролидотиазолила, пиперидопиридила, и пирролидопиридила, где гетероароматическое кольцо необязательно замещено с помощью 1 или 2 (CH2)nN(RA)RB и необязательно замещено с помощью 1 или 2 (CH2)nRC групп; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е34 является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является HetB; и HetB и все другие переменные определены выше в варианте осуществления Е34. Вторым подвариантом осуществления варианта осуществления Е34 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 является HetB; и HetB и все другие переменные определены выше в варианте осуществления Е34. В аспекте этого варианта осуществления и его подвариантах осуществления гетероароматическое кольцо в HetB необязательно монозамещено с помощью (CH2)nN(RA)RB и необязательно замещенно с помощью 1 или 2 (CH2)nRC. В другом аспекте этого варианта осуществления и его подвариантах осуществления гетероароматическое кольцо в HetB необязательно монозамещено с помощью NH2, N(H)-C1-3алкила, N(-C1-3алкил)2, CH2NH2, CH2N(H)-C1-3алкила, или CH2N(-C1-3алкил)2; и необязательно замещено с помощью 1 или 2 заместителей, каждый из которых является независимо C1-3алкилом, пирролидинилом, пиперидинилом, пиперазинилом, морфолинилом, или тиоморфолинилом. В еще одном аспекте этого варианта осуществления и его подвариантах осуществления гетероароматическое кольцо в HetB необязательно монозамещено с помощью NH2, N(H)CH3, N(CH3)2, CH2NH2, CH2N(H)CH3, или CH2N(CH3)2; и необязательно замещено с помощью 1 или 2 заместителей, каждый из которых является независимо CH3, пирролидинилом, пиперидинилом, пиперазинилом, морфолинилом, или тиоморфолинилом. В еще одном аспекте этого варианта осуществления и его подвариантах осуществления R2 является OSO3H.
Тридцать пятым вариантом осуществления настоящего изобретения (вариант осуществления Е35) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является AryA; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е35 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 является AryA; и AryA и все другие переменные определены выше в варианте осуществления Е35. В аспекте этого варианта осуществления и его подвариантах осуществления R2 является OSO3H.
Тридцать шестым вариантом осуществления настоящего изобретения (вариант осуществления Е36) является соединение формулы I, или его фармацевтически приемлемая соль, где AryA является фенилом, который необязательно замещен с помощью 1 или 2 заместителей, каждый из которых является независимо C1-3алкилом, CH2NH2, CH2N(H)-C1-3алкилом, CH2N(-C1-3алкил)2, O-C1-3алкилом, Cl, Br, F, NH2, N(H)-C1-3алкилом, N(-C1-3алкил)2, C(O)NH2, C(O)N(H)-C1-3алкилом, C(O)N(-C1-3алкил)2, C(O)-C1-3алкилом, C(O)O-C1-3алкилом, OC(O)-C1-3алкилом, S(O)2-C1-3алкилом, S(O)2NH2, S(O)2N(H)-C1-3алкилом, S(O)2N(-C1-3алкил)2, пирролидинилом, пиперидинилом, морфолинилом, CH2-пирролидинилом, CH2-пиперидинилом или CH2-морфолинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е36 является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является AryA; и AryA и все другие переменные определены выше в варианте осуществления Е36. Вторым подвариантом осуществления варианта осуществления Е36 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 является AryA; и AryA и все другие переменные определены выше в варианте осуществления Е36. В аспекте этого варианта осуществления и его подвариантах осуществления R2 является OSO3H.
Тридцать седьмым вариантом осуществления настоящего изобретения (вариант осуществления Е37) является соединение формулы I, или его фармацевтически приемлемая соль, где AryA является фенилом, который необязательно замещен с помощью 1 или 2 заместителей, каждый из которых является независимо CH3, CH2NH2, CH2N(H)CH3, CH2N(CH3)2, OCH3, Cl, Br, F, NH2, N(H)CH3, N(CH3)2, C(O)NH2, C(O)N(H)CH3, C(O)N(CH3)2, C(O)CH3, C(O)OCH3, OC(O)CH3, S(O)2CH3, S(O)2NH2, S(O)2N(H)CH3, или S(O)2N(CH3)2, пирролидинилом, пиперидинилом, морфолинилом, CH2-пирролидинилом, CH2-пиперидинилом, или CH2-морфолинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е37 является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является AryA; и AryA и все другие переменные определены выше в варианте осуществления Е37. Вторым подвариантом осуществления варианта осуществления Е37 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 является AryA; и AryA и все другие переменные определены выше в варианте осуществления Е37. В аспекте этого варианта осуществления и его подвариантах осуществления R2 является OSO3H.
Тридцать восьмым вариантом осуществления настоящего изобретения (вариант осуществления Е38) является соединение формулы I, или его фармацевтически приемлемая соль, где AryA является фенилом, который необязательно замещен с помощью 1 или 2 заместителей, каждый из которых является независимо CH3, CH2NH2, CH2N(H)CH3, CH2N(CH3)2, OCH3, Cl, Br, F, NH2, N(H)CH3, N(CH3)2, C(O)NH2, C(O)N(H)CH3, C(O)N(CH3)2, C(O)CH3, C(O)OCH3, OC(O)CH3, S(O)2CH3, S(O)2NH2, S(O)2N(H)CH3, S(O)2N(CH3)2, пирролидинилом, пиперидинилом, морфолинилом, CH2-пирролидинилом, CH2-пиперидинилом, или CH2-морфолинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е38 является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является AryA; и AryA и все другие переменные определены выше в варианте осуществления Е38. Вторым подвариантом осуществления варианта осуществления Е38 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 является AryA; и AryA и все другие переменные определены выше в варианте осуществления Е37. В аспекте этого варианта осуществления и его подвариантах осуществления R2 является OSO3H.
Тридцать девятым вариантом осуществления настоящего изобретения (вариант осуществления Е39) является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 и R4 вместе с атомом N, к которому они оба присоединены, образуют гетероцикл, выбранный из группы, состоящей из:


где кольцо необязательно замещено с помощью 1 или 2 заместителей, каждый из которых является независимо C1-3алкилом, CF3, CH2OH, CH2O-C1-3алкилом, CH2OCF3, CH2NH2, CH2N(H)-C1-3алкилом, CH2N(-C1-3алкил)2, O-C1-3алкилом, OCF3, оксо, Cl, Br, F, NH2, N(H)-C1-3алкилом, N(-C1-3алкил)2, C(O)NH2, C(O)N(H)-C1-3алкилом, C(O)N(-C1-3алкил)2, C(O)-C1-3алкилом, C(O)O-C1-3алкилом, или S(O)2-C1-3алкилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. В аспекте этого варианта осуществления R2 является OSO3H.
Сороковым вариантом осуществления настоящего изобретения (вариант осуществления Е40) является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 и R4 вместе с атомом N, к которому они оба присоединены, образуют гетероцикл, выбранный из группы, состоящей из:


где кольцо необязательно замещено с помощью 1 или 2 заместителей, каждый из которых является независимо CH3, CF3, CH2OH, CH2OCH3, CH2OCF3, CH2NH2, CH2N(H)CH3, CH2N(CH3)2, OCH3, OCF3, оксо, Cl, Br, F, NH2, N(H)CH3, N(CH3)2, C(O)NH2, C(O)N(H)CH3, C(O)N(CH3)2, C(O)CH3, C(O)OCH3, или S(O)2CH3; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. В аспекте этого варианта осуществления R2 является OSO3H.
Сорок первым вариантом осуществления настоящего изобретения (вариант осуществления Е41) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является AryB; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е41 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 является AryB; и AryB и все другие переменные определены выше в варианте осуществления Е41. В аспекте этого варианта осуществления и его подварианте осуществления R2 является OSO3H.
Сорок вторым вариантом осуществления настоящего изобретения (вариант осуществления Е42) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является AryB; AryB является бициклическим кольцом, выбранным из группы, состоящей из 1,2,3,4-тетрагидрохинолинила, 1,2,3,4-тетрагидроизохинолинила, 2,3-дигидро-1H-изоиндолила и 2,3-дигидро-1H-индолила, где бициклическое кольцо необязательно замещено с помощью 1 или 2 заместителями, каждый из которых является независимо C1-3алкилом, CH2NH2, CH2N(H)-C1-3алкилом, CH2N(-C1-3алкил)2, O-C1-3алкилом, Cl, Br, F, NH2, N(H)-C1-3алкилом, N(-C1-3алкил)2, C(O)NH2, C(O)N(H)-C1-3алкилом, C(O)N(-C1-3алкил)2 , C(O)-C1-3алкилом, C(O)O-C1-3алкилом, OC(O)-C1-3алкилом, S(O)2-C1-3алкилом, S(O)2NH2, S(O)2N(H)-C1-3алкилом, S(O)2N(-C1-3алкил)2, пирролидинилом, пиперидинилом, морфолинилом, CH2-пирролидинилом, CH2-пиперидинилом, или CH2-морфолинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е42 является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является AryB; и AryB и все другие переменные определены выше в варианте осуществления Е42. Вторым подвариантом осуществления варианта осуществления Е42 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 является AryB; и AryB и все другие переменные определены выше в варианте осуществления Е42. В аспекте этого варианта осуществления и его подвариантах осуществления R2 является OSO3H.
Сорок третьим вариантом осуществления настоящего изобретения (вариант осуществления Е43) является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является AryB; AryB является бициклическим кольцом, выбранным из группы, состоящей из 1,2,3,4-тетрагидрохинолинила, 1,2,3,4-тетрагидроизохинолинила, 2,3-дигидро-1H-изоиндолила и 2,3-дигидро-1H-индолила, где бициклическое кольцо необязательно замещено с помощью 1 или 2 заместителями, каждый из которых является независимо CH3, CH2NH2, CH2N(H)CH3, CH2N(CH3)2, OCH3, Cl, Br, F, NH2, N(H)CH3, N(CH3)2, C(O)NH2, C(O)N(H)CH3, C(O)N(CH3)2, C(O)CH3, C(O)OCH3, OC(O)CH3, S(O)2CH3, S(O)2NH2, S(O)2N(H)CH3, или S(O)2N(CH3)2, пирролидинилом, пиперидинилом, морфолинилом, CH2-пирролидинилом, CH2-пиперидинилом, или CH2-морфолинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. Первым подвариантом осуществления варианта осуществления Е43 является соединение формулы I, или его фармацевтически приемлемая соль, где R3 является AryB, и AryB и все другие переменные определены выше в варианте осуществления Е43. Вторым подвариантом осуществления варианта осуществления Е43 является соединение формулы I, или его фармацевтически приемлемая соль, где R1 является C(O)N(R3)R4; R3 является AryB, и AryB и все другие переменные определены выше в варианте осуществления Е43. В аспекте этого варианта осуществления и его подвариантах осуществления R2 является OSO3H.
Сорок четвертым вариантом осуществления настоящего изобретения (вариант осуществления Е44) является соединение формулы I, или его фармацевтически приемлемая соль, где R4 является H или C1-4алкилом, необязательно замещенным с помощью N(RA)RB; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. В аспекте этого варианта осуществления R4 является H или C1-4алкилом.
Сорок пятым вариантом осуществления настоящего изобретения (вариант осуществления Е45) является соединение формулы I, или его фармацевтически приемлемая соль, где R4 является H, C1-3алкилом, или (CH2)2-3N(RA)RB; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления. В аспекте этого варианта осуществления R4 является H или C1-3алкилом.
Сорок шестым вариантом осуществления настоящего изобретения (вариант осуществления Е46) является соединение формулы I, или его фармацевтически приемлемая соль, где R4 является H или метилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Сорок седьмым вариантом осуществления настоящего изобретения (вариант осуществления Е47) является соединение формулы I, или его фармацевтически приемлемая соль, где R4 является H; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Сорок восьмым вариантом осуществления настоящего изобретения (вариант осуществления Е48) является соединение формулы I, или его фармацевтически приемлемая соль, где каждый RA является независимо H или C1-4алкилом; каждый RB является независимо H или C1-4алкилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Сорок девятым вариантом осуществления настоящего изобретения (вариант осуществления Е49) является соединение формулы I, или его фармацевтически приемлемая соль, где каждый RA является независимо H или C1-3алкилом; каждый RB является независимо H или C1-3алкилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Пятидесятым вариантом осуществления настоящего изобретения (вариант осуществления Е50) является соединение формулы I, или его фармацевтически приемлемая соль, где каждый RA является независимо H или CH3; каждый RB является независимо H или CH3; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Пятьдесят первым вариантом осуществления настоящего изобретения (вариант осуществления Е51) является соединение формулы I, или его фармацевтически приемлемая соль, где каждый RC является независимо C1-4алкилом, OH, O-C1-4алкилом, C(=NH)NH2, NH-C(=NH)NH2, галогеном, CN, пиридилом, пирролидинилом, или пиперидинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Пятьдесят вторым вариантом осуществления настоящего изобретения (вариант осуществления Е52) является соединение формулы I, или его фармацевтически приемлемая соль, где каждый RC является независимо OH, O-C1-4алкилом, C(=NH)NH2, NH-C(=NH)NH2, Cl, Br, F, или CN; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Пятьдесят третьим вариантом осуществления настоящего изобретения (вариант осуществления Е53) является соединение формулы I, или его фармацевтически приемлемая соль, где каждый RC является независимо C1-3алкилом, O-C1-3алкилом, Cl, Br, F, C(O)NH2, C(O)N(H)-C1-3алкилом, C(O)N(-C1-3алкил)2, C(O)-C1-3алкилом, C(O)O-C1-3алкилом, OC(O)-C1-3алкилом, S(O)2-C1-3алкилом, S(O)2NH2, S(O)2N(H)-C1-3алкилом, S(O)2N(-C1-3алкил)2, пирролидинилом, пиперидинилом, морфолинилом, CH2-пирролидинилом, CH2-пиперидинилом, или CH2-морфолинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Пятьдесят четвертым вариантом осуществления настоящего изобретения (вариант осуществления Е54) является соединение формулы I, или его фармацевтически приемлемая соль, где каждый RC является независимо CH3, OCH3, Cl, Br, F, C(O)NH2, C(O)N(H)CH3, C(O)N(CH3)2, C(O)CH3, C(O)OCH3, OC(O)CH3, S(O)2CH3, S(O)2NH2, S(O)2N(H)CH3, или S(O)2N(CH3)2, пирролидинилом, пиперидинилом, морфолинилом, CH2-пирролидинилом, CH2-пиперидинилом, или CH2-морфолинилом; и все другие переменные определены выше или определены в любом из предшествующих вариантов осуществления.
Если однозначно не заявлено иначе, или же это ясно из контекста, то условия A-F, установленные для определения соединения I в разделе "Сущность изобретения", справедливы здесь для предшествующих и последующих вариантов осуществления. Из контекста ясно, например, что когда любой один из вариантов осуществления E17-E120 включают в приведенное ранее определение соединения I, ни одно из условий не применяется. Кроме того, в тех случаях, когда любой вариант осуществления ссылается на вариант осуществления Е1 или вариант осуществления Е2, и включает вариант осуществления Е1 или вариант осуществления Е2, он включает установленные в них условия A-F в тех случаях, когда применяется любой из них. Кроме того, следует иметь в виду, что определения переменных в условиях могут быть специально предназначены для отражения определений переменных в вариантах осуществления, которые в них включаются. Например, когда вариант осуществления Е9 (то есть R1 является C(O)N(R3)R4) включают в вариант осуществления Е1, то условие может быть сформулировано следующим образом (где условия A, B, C и F не применяются) - и при условии, что:
(D) когда R3 является AryA или C1-6алкилом, замещенным с помощью AryA, и R4 является H или C1-6алкилом, тогда AryA не является ни незамещенным фенилом, ни фенилом, замещенным с помощью 1 или 2 N(RA)RB; и
(E) когда R4 является H или C1-6алкилом, тогда R3 не является C1-6алкилом, замещенным с помощью HetB.
В качестве еще одного примера, когда соединение определенно во втором подварианте осуществления варианта осуществления Е36 (то есть R1 является C(O)N(R3)R4; R3 является AryA; и AryA и все другие переменные определены выше в варианте осуществления Е36), то тогда следует иметь в виду, что применяется следующее условие: и при условии, что AryA не является незамещенным фенилом, фенилом, замещенным с помощью N(CH3)2, или фенилом, замещенным с помощью C(O)NH2. Кроме того, следует иметь в виду, что необязательные условия, установленные в вариантах осуществления E1 и E2, могут, будучи соответствующим образом приспособленные, быть применены в качестве варианта. Условием на основе варианта осуществления Е1, например, является: и при условии, что AryA не является незамещенный фенилом, фенилом, замещенным с помощью 1 или 2 NH2, N(H)-C1-3алкила и N(-C1-3алкил)2, или фенилом, замещенным с помощью 1 или 2 C(O)NH2, C(O)N(H)-C1-3алкила и C(O)N(-C1-3алкил)2.
Первый класс соединений настоящего изобретения (в качестве варианта называемый здесь как "класс C1") включает соединения формулы I и их фармацевтически приемлемые соли, где R1 является C(O)N(R3)R4; R3 является HetA; и все другие переменные определены выше. В аспекте этого класса R4 является H.
Первый подкласс первого класса (в качестве варианта называемый здесь как "подкласс C1-S1") включает соединения формулы I и их фармацевтически приемлемые соли, где R1 является C(O)N(R3)R4; R3 является HetA; и HetA является насыщенным гетероциклическим кольцом, выбранным из группы, состоящей из пирролидинила, пиперидинила, азепанила, и азоканила, где насыщенный гетероцикл необязательно монозамещен с помощью N(RA)RB и необязательно замещен с помощью 1 или 2 (CH2)nRC; и все другие переменные определены выше. В аспекте этого подкласса R4 является H.
Второй подкласс первого класса (подкласс C1-S2) включает соединения формулы I и их фармацевтически приемлемые соли, где все переменные точно определены в подклассе C1-S1, за исключением того, что каждый RC является независимо OH, O-C1-4алкилом, C(=NH)NH2, NH-C(=NH)NH2, Cl, Br, F, или CN. В аспекте этого подкласса R4 является H.
Третий подкласс первого класса (подкласс C1-S3) включает соединения формулы I, выбранные из группы, состоящей из:


и их фармацевтически приемлемые соли; где T является H или (CH2)2-3RC; и RC и R2 каждый независимо определены выше или определены в любом из предшествующих вариантов осуществления. В одном аспекте этого подкласса T является H. В другом аспекте этого подкласса R2 является OSO3H или SO3H. В еще одном аспекте этого подкласса T является H и R2 является OSO3H или SO3H. В еще одном аспекте этого подкласса T является H и R2 является OSO3H. В еще одном аспекте этого подкласса каждый RC является независимо C1-6алкилом, OH, O-C1-8алкилом, C(=NH)NH2, NH-C(=NH)NH2, галогеном, CN, пиридилом, пирролидинилом, или пиперидинилом. В характерной черте этого аспекта R2 является OSO3H.
Второй класс соединений настоящего изобретения (класс C2) включает соединения формулы I и их фармацевтически приемлемые соли, где R1 является C(O)N(R3)R4; R3 является HetB; и все другие переменные определены выше. В аспекте этого класса R4 является H.
Первый подкласс второго класса (подкласс C2-S1) включает соединения формулы I и их фармацевтически приемлемые соли, где R1 является C(O)N(R3)R4; R3 является HetB; и HetB является гетероароматическим кольцом, выбранным из группы, состоящей из пирролила, пиразолила, имидазолила, пиридила, и пиримидинила, где гетероароматическое кольцо необязательно монозамещено с помощью (CH2)nN(RA)RB и необязательно замещено с помощью 1 или 2 (CH2)nRC групп; и все другие переменные определены выше. В аспекте этого подкласса R4 является H.
Второй подкласс второго класса (подкласс C2-S2) включает соединения формулы I и их фармацевтически приемлемые соли, где все переменные точно определены в подклассе C2-S1, за исключением того, что каждый RC является независимо OH, O-C1-4алкилом, C(=NH)NH2, NH-C(=NH)NH2, Cl, Br, F, или CN. В аспекте этого подкласса R4 является H.
Третий подкласс второго класса (подкласс C2-S3) включает соединения формулы I и их фармацевтически приемлемые соли, где R1 является C(O)N(R3)R4; R3 является HetB; и HetB является пиридилом, который необязательно монозамещен с помощью N(RA)RB и необязательно замещен с помощью 1 или 2 RC групп. В аспекте этого подкласса R4 является H.
Четвертый подкласс второго класса (подкласс C2-S4) включает соединения формулы I и их фармацевтически приемлемые соли, где все переменные точно определены в подклассе C2-S3, за исключением того, что каждый RC является независимо OH, O-C1-4алкилом, C(=NH)NH2, NH-C(=NH)NH2, Cl, Br, F, или CN. В аспекте этого подкласса R4 является H. В еще одном аспекте этого подкласса каждый RC является независимо C1-6алкилом, OH, O-C1-8алкилом, C(=NH)NH2, NH-C(=NH)NH2, галогеном, CN, пиридилом, пирролидинилом, или пиперидинилом.
Пятый подкласс второго класса (подкласс C2-S5) включает соединения формулы I, выбранные из группы, состоящей из:


и их фармацевтически приемлемые соли, где RC является C1-6алкилом, OH, O-C1-8алкилом, C(=NH)NH2, NH-C(=NH)NH2, галогеном, CN, пиридилом, пирролидинилом, или пиперидинилом; и R2, RA и RB каждый независимо определены выше. В аспекте этого подкласса N(RA)RB является NH(C1-4алкил) или N(C1-4алкил)2 и RC является O-C1-4алкилом. В другом аспекте этого подкласса R2 является OSO3H или SO3H. В еще одном аспекте этого подкласса R2 является OSO3H. В еще одном аспекте этого подкласса N(RA)RB является NH(C1-4алкил) или N(C1-4алкил)2; RC является O-C1-4алкилом; и R2 является OSO3H или SO3H. Другие аспекты этого подкласса включают соединения формулы B1a, B1b, и B1c и их фармацевтически приемлемые соли, где RC, R2, RA и RB каждый независимо определены в любом из предшествующих вариантов осуществления; то есть каждая уникальная комбинация этих переменных составляет отличающийся аспект.
Третий класс соединений настоящего изобретения (класс C3) включает соединения формулы I, выбранные из группы, состоящей из:



где T является H, C1-3алкилом, пирролидин-3-илом, пиперидин-4-илом, (CH2)2-3-O-C1-3алкилом, (CH2)2-3OH, (CH2)2-3F, (CH2)2-3-пиперидинилом, (CH2)2-3-пирролидинилом; и T' является H, Cl, Br, F, C1-3алкилом, O-C1-3алкилом, OH, NH2, N(H)-C1-3алкилом, или N(-C1-3алкил)2; и R2 определен выше.
Первый подкласс третьего класса (подкласс C3-S1) включает соединения формулы (A1)-(A20) и их фармацевтически приемлемые соли; где R2 является OSO3H; и все другие переменные определены выше в классе C1.
Второй подкласс третьего класса (подкласс C3-S2) включает соединения формулы (A1)-(A20) и их фармацевтически приемлемые соли; где T является H, CH3, пирролидин-3-илом, пиперидин-4-илом, (CH2)2-3OCH3, (CH2)2-3OH, (CH2)2-3F, (CH2)2-3-пиперидинилом, (CH2)2-3-пирролидинилом; T' является H, F, O-C1-3алкилом, OH, NH2, N(H)CH3, N(CH3)2; и R2 определен выше. В аспекте этого подкласса R2 является OSO3H.
Третий подкласс третьего класса (подкласс C3-S3) включает соединения формулы (A1)-(A20) и их фармацевтически приемлемые соли; где T является H; T' является H, F, OCH3, или OH; и R2 является OSO3H.
Четвертый класс соединений настоящего изобретения (класс C4) включает соединения формулы I, выбранные из группы, состоящей из:


где V, V', V'', Y, Y' и Z каждые независимо выбраны из группы, состоящей из H, CH3, пирролидинила, пиперидинила, пиперазинила, морфолинила, тиоморфолинила, CH2-пирролидинила, CH2-пиперидинила, CH2-пиперазинила, CH2-морфолинила, CH2-тиоморфолинила, NH2, N(H)CH3, N(CH3)2, CH2NH2, CH2N(H)CH3 и CH2N(CH3)2; при условии, что:
(i) по меньшей мере, один из V, V' и V'' является H; и
(ii) по меньшей мере, один из Y и Y' является H.
Первый подкласс четвертого класса (подкласс C4-S1) включает соединения формулы (B1)-(B9) и их фармацевтически приемлемые соли; где, по меньшей мере, два из V, V' и V'' являются H; и R2 является OSO3H.
Другим вариантом осуществления настоящего изобретения является соединение, выбранное из группы, состоящей из названных соединений примеров 1-117 (или, в качестве варианта, соединений 1-117) и их фармацевтически приемлемых солей.
Другим вариантом осуществления настоящего изобретения является соединение, выбранное из группы, состоящей из названных соединений примеров 1-13 (то есть соединения 1-13) и их фармацевтически приемлемых солей.
Другим вариантом осуществления настоящего изобретения является соединение, выбранное из группы, состоящей из соединений 1, 2, 4 и 6-9 и их фармацевтически приемлемых солей.
Другим вариантом осуществления настоящего изобретения является соединение, выбранное из группы, состоящей из:
(2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[4-(аминометил)фенил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-[(3R)пирролидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-6-(сульфоокси)-N-(1,2,3,4-тетрагидроизохинолин-6-ил)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-(5-пиперидин-4-илпиридин-2-ил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
пиперидин-4-илметил(2S,5R)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксилат; и их фармацевтически приемлемые соли.
Еще одним вариантом осуществления настоящего изобретения является (2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (то есть соединение примера 1, или, в качестве варианта, соединение 1) или его фармацевтически приемлемая соль.
Еще одним вариантом осуществления настоящего изобретения является (2S,5R)-N-[4-(аминометил)фенил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (то есть соединение примера 9, или, в качестве варианта, соединение 9) или его фармацевтически приемлемая соль.
Еще одним вариантом осуществления настоящего изобретения является (2S,5R)-7-оксо-N-[(3R)пирролидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (то есть соединение примера 14, или соединение 14) или его фармацевтически приемлемая соль.
Другим вариантом осуществления настоящего изобретения является соединение 1 в форме кристаллического моногидрата. Кристаллический моногидрат характеризуется порошковой рентгенограммой, приведенной на фиг.1, и кривой дифференциальной сканирующей калориметрии (DSC), приведенной на фиг.2. Кристаллический моногидрат может быть получен, как описано в части A в примере 1D. В одном варианте осуществления кристаллический моногидрат характеризуется порошковой рентгенограммой, полученной при помощи медного Kα излучения (то есть источником излучения является комбинация Cu Kα1 и Kα2 излучения), которая включает 2Θ значения (то есть отражения при 2Θ значения) в градусах около 15,6, 17,4 и 20,4. В этом варианте осуществления и любых аналогичных вариантах осуществления, которые следуют далее, термин "около" подразумевает изменение каждого из 2Θ значений. В другом варианте осуществления кристаллический моногидрат характеризуется порошковой рентгенограммой, полученной при помощи медного Kα излучения, которая включает 2Θ значения в градусах около 15,6, 17,4, 20,4, 24,0, 26,3 и 29,3. В еще одном варианте осуществления кристаллический моногидрат характеризуется порошковой рентгенограммой, полученной при помощи медного Kα излучения, которая включает 2Θ значения в градусах около 13,5, 15,5, 15,6, 17,4, 18,7, 19,7, 20,4, 21,7, 22,6, 24,0, 24,3, 25,9, 26,3, 26,6, 27,0, 27,5, 29,3, 30,0, 31,3, 32,4, 32,9, 33,1, 34,0, 34,7, 35,5 и 38,9.
В еще одном варианте осуществления кристаллический моногидрат соединения 1 характеризуется диаграммой распределения интенсивности (PDF trace), полученной из порошковой рентгенограммы на фиг.1. Диаграмма распределения интенсивности дает отпечаток межатомных расстояний, которые определяют кристаллический моногидрат. Диаграмма распределения интенсивности может быть получена так, как описано в патентном документе WO 2005/082050. В одном аспекте этого варианта осуществления кристаллический моногидрат характеризуется частями диаграммы распределения интенсивности, соответствующими 2Θ значениям в градусах около 15,6, 17,4 и 20,4 на порошковой рентгенограмме. В другом аспекте этого варианта осуществления кристаллический моногидрат характеризуется частями диаграммы распределения интенсивности, соответствующими 2Θ значениям в градусах около 15,6, 17,4, 20,4, 24,0, 26,3 и 29,3 на порошковой рентгенограмме. В еще одном аспекте этого варианта осуществления кристаллический моногидрат характеризуется частями диаграммы распределения интенсивности, соответствующими 2Θ значениям в градусах около 13,5, 15,5, 15,6, 17,4, 18,7, 19,7, 20,4, 21,7, 22,6, 24,0, 24,3, 25,9, 26,3, 26,6, 27,0, 27,5, 29,3, 30,0, 31,3, 32,4, 32,9, 33,1, 34,0, 34,7, 35,5 и 38,9 на порошковой рентгенограмме.
Термин "около или примерно" при изменении количества (например, кг, л, или эквивалентов) вещества или композиции или величины физического свойства, или величины параметра, характеризующего стадию процесса (например, температура, при которой проводят стадию процесса), или других подобных параметров, относится к отклонению численной величины, которое может происходить, например, при обычных методиках измерения, манипулирования и отбора проб, используемых при получении, исследовании и/или применении вещества или композиции; вследствие случайной ошибки в этих методиках; вследствие различий в производстве, источнике, или чистоте ингредиентов, используемых для получения или применения композиций, или в осуществлении методик; и других подобных причин. В конкретном случае 2Θ значений в градусах на описанной здесь порошковой рентгенограмме термин "около" обычно означает величину ±0,1.
Другим вариантом осуществления настоящего изобретения является соединение формулы I, или его фармацевтически приемлемая соль, определенные выше или определенные в любом из вышеизложенных вариантов осуществления, подвариантах осуществления, аспектах, классах или подклассах, где соединение или его соль находится в практически чистой форме. Используемый здесь термин "практически чистая" означает, соответственно, что, по меньшей мере, около 60 мас.%, обычно, по меньшей мере, около 70 мас.%, предпочтительно, по меньшей мере, около 80 мас.%, более предпочтительно, по меньшей мере, около 90 мас.% (например, примерно от 90 мас.% до 99 мас.%), еще более предпочтительно, по меньшей мере, около 95 мас.% (например, примерно от 95 мас.% до 99 мас.%, или примерно от 98 мас.% до 100 мас.%), и наиболее предпочтительно, по меньшей мере, около 99 мас.% (например, 100 мас.%) продукта, содержащего соединение формулы I или его соли (например, продукт, выделенный из реакционной смеси, дающий соединение или соль), составляют соединение или соль. Степень чистоты соединений или солей может быть определена с помощью стандартного метода анализа, такого как тонкослойная хроматография, гель-электрофорез, высокоэффективная жидкостная хроматография, и/или масс-спектрометрия. Если используют более чем один метод анализа, и методы обеспечивают в условиях эксперимента значительные различия в степени определяемой чистоты, то тогда метод, обеспечивающий самую высокую степень чистоты, имеет приоритетное значение. Соединение или соль 100% чистоты является соединением или солью, которые не содержат поддающиеся обнаружению примеси, определяемые стандартным методом анализа. Что касается соединения изобретения, которое имеет один или более центров асимметрии и может существовать в виде смесей стереоизомеров, то практически чистое соединение может являться или практически чистой смесью стереоизомеров, или практически чистым индивидуальным диастереомером или энантиомером.
Другие варианты осуществления настоящего изобретения включают следующие:
(a) Фармацевтическая композиция, включающая эффективное количество определенного выше соединения формулы I, или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
(b) Фармацевтическая композиция по пункту (a), дополнительно включающая эффективное количество β-лактамового антибиотика.
(c) Фармацевтическая композиция по пункту (b), где бета-лактамовый антибиотик выбирают из группы, состоящей из имипенема, эртапенема, меропенема, дорипенема, биапенема, панипенема, тикарциллина, ампициллина, амоксициллина, карбенициллина, пиперациллина, азлоциллина, мезлоциллина, тикарциллина, цефоперазона, цефотаксима, цефтриаксона, и цефтазидима.
(d) Фармацевтическая композиция по пункту (b), где β-лактамовым антибиотиком является имипенем.
(e) Фармацевтическая композиция по пункту (b), где β-лактамовым антибиотиком является цефтазидим.
(f) Фармацевтическая композиция по пункту (b), где β-лактамовым антибиотиком является пиперациллин.
(g) Фармацевтическая композиция по пункту (a), дополнительно включающая эффективные количества β-лактамового антибиотика и ингибитора DHP.
(h) Фармацевтическая композиция по пункту (g), где бета-лактамовым антибиотиком является имипенем, и ингибитором DHP является циластатин или его фармацевтически приемлемая соль.
(i) Комбинация эффективных количеств определенного выше соединения формулы или его фармацевтически приемлемой соли и β-лактамового антибиотика.
(j) Комбинация по пункту (i), где бета-лактамовый антибиотик выбирают из группы, состоящей из имипенема, эртапенема, меропенема, дорипенема, биапенема, панипенема, тикарциллина, ампициллина, амоксициллина, карбенициллина, пиперациллина, азлоциллина, мезлоциллина, тикарциллина, цефоперазона, цефотаксима, цефтриаксона, и цефтазидима.
(k) Комбинация по пункту (i), где β-лактамовым антибиотиком является имипенем.
(l) Комбинация по пункту (i), где β-лактамовым антибиотиком является цефтазидим.
(m) Комбинация по пункту (i), где β-лактамовым антибиотиком является пиперациллин.
(n) Комбинация эффективных количеств определенного выше соединения формулы I или его фармацевтически приемлемой соли, β-лактамового антибиотика и ингибитора DHP.
(o) Комбинация по пункту (n), где бета-лактамовым антибиотиком является имипенем, и ингибитором DHP является циластатин или его фармацевтически приемлемая соль.
(p) Способ лечения бактериальной инфекции, который включает введение субъекту при необходимости такого лечения терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли необязательно в комбинации с эффектиным количеством бета-лактамового антибиотика.
(q) Способ лечения бактериальной инфекции, который включает введение субъекту при необходимости такого лечения терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли в комбинации с эффективными количествами бета-лактамового антибиотика и ингибитора DHP.
(r) Способ лечения бактериальной инфекции, который включает введение субъекту при необходимости такого лечения терапевтически эффективного количества композиции по пунктам (a), (b), (c), (d), (e), (f), (g) и (h).
(s) Способ лечения бактериальной инфекции, который включает введение субъекту при необходимости такого лечения терапевтически эффективного количества комбинации по пунктам (i), (j), (k), (l), (m), (n) и (o).
(t) Способ лечения бактериальной инфекции по пунктам (p), (q), (r), или (s), где бактериальная инфекция обусловлена Pseudomonas spp. или Klebsiella spp.
Настоящее изобретение также включает соединение формулы I или его фармацевтически приемлемую соль, (i) для применения в лекарственном средстве, (ii) для применения в качестве лекарственного средства для лечения бактериальной инфекции, или (iii) для применения при получении (или производстве) лекарственного средства для лечения бактериальной инфекции. В этих применениях соединения настоящего изобретения могут необязательно быть использованы в комбинации с одним или более β-лактамовыми антибиотиками и/или одним или более ингибиторами DHP.
Дополнительные варианты осуществления изобретения включают фармацевтические композиции, комбинации и способы по приведенным выше пунктам (a)-(t) и применения, заявленные в предыдущем параграфе, где используемым здесь соединением настоящего изобретения является соединение одного из описанных выше вариантов осуществления, подвариантов осуществления, классов или подклассов. В этих вариантах осуществления соединение может необязательно быть использовано в форме фармацевтически приемлемой соли.
Дополнительные варианты осуществления настоящего изобретения включают каждые из фармацевтических композиций, комбинаций, способов и применений, заявленных в предыдущих параграфах, где используемое здесь соединение настоящего изобретения или его соль являются практически чистыми. Что касается фармацевтической композиции, включающей соединение формулы I или его соль и фармацевтически приемлемый носитель и необязательно одно или более вспомогательных веществ, то следует иметь в виду, что термин "практически чистое" относится непосредственно к соединению формулы I или его соли; то есть чистоте активного ингредиента в композиции.
Используемый здесь термин "алкил" относится к одновалентному линейному или разветвленному, насыщенному алифатическому углеводородному радикалу, имеющему количество углеродных атомов в указанном интервале. Так, например, "C1-8алкил" (или "C1-C8алкил") относится к любому из октильных, гептильных, гексильных и пентильных алкильных изомеров, так же как и н-, изо-, втор- и трет-бутилу, н- и изопропилу, этилу и метилу. В качестве другого примера, "C1-6алкил" (или "C1-C6алкил") относится к любому из гексильных и пентильных алкильных изомеров, так же как и н-, изо-, втор- и трет-бутилу, н- и изопропилу, этилу и метилу. В качестве еще одного примера, "C1-4алкил" относится к н-, изо-, втор- и трет-бутилу, н- и изопропилу, этилу и метилу.
Термин "галогеналкил" относится к определенной выше алкильной группе, в которой один или более атомов водорода были замещены галогеном (то есть F, Cl, Br и/или I). Так, например, "C1-6галогеналкил" (или "C1-C6галогеналкил") относится к определенной выше C1-C6 линейной или разветвленной алкильной группе с одним или более галогенными заместителями. Термин "фторалкил" имеет аналогичное значение, за исключением того, что галогенные заместители ограничиваются фтором. Подходящие фторалкилы включают ряд (CH2)0-4CF3 (то есть трифторметил, 2,2,2-трифторэтил, 3,3,3-трифтор-н-пропил, и так далее).
Термин "галоген" (или "гало") относится к фтору, хлору, брому и иоду (в качестве варианта, обозначается как фтор, хлор, бром, и иод).
Термин "циклоалкил" относится к любому одновалентному моноциклическому кольцу алкана, имеющему количество углеродных атомов в указанном интервале. Так, например, "C4-9циклоалкил" (или "C4-C9циклоалкил") относится к циклобутилу, циклопентилу, циклогексилу, циклогептилу, циклооктилу, и циклононилу, и "C4-7циклоалкил" относится к циклобутилу, циклопентилу, циклогексилу и циклогептилу.
Термин "C(O)" относится к карбонилу, термины "S(O)2" и "SO2" каждый относятся к сульфонилу. Термин "S(O)" относится к сульфинилу.
Символ "*" в конце связи относится к месту присоединения функциональной группы или другого химического фрагмента к остальной молекуле, частью которой он является.
Если однозначно не накладываются ограничения, то термин "замещенный" относится к одному и нескольким замещениям при помощи названного заместителя, при условии что такое одно и несколько замещений (включая несколько замещений в одном и том же положении) химически возможны. Если однозначно не заявлено иначе, то замещение при помощи названного заместителя дозволено на любом атоме в кольце (например, циклоалкиле, фениле, гетероароматическом кольце, или насыщенном гетероциклическом кольце), при условии что такое замещение в кольце химически возможно и приводит к стабильному соединению. Однако следует иметь в виду, что может быть оговорена степень замещения. Например, выражение "необязательно суммарно замещен с помощью от 1 до 4 заместителей, выбранных из от нуля до 2 N(RA)RB и от нуля до 2 RC" означает, что может необязательно присутствовать в совокупности 4 заместителя максимально с 2 заместителями N(RA)RB и максимально с 2 RC группами. В качестве другого примера, выражение "AryA не является ни незамещенным фенилом, ни фенилом, замещенным с помощью 1 или 2 N(RA)RB" означает, что AryA не является фенилом, фенилом, монозамещенным с помощью N(RA)RB, или фенилом, дизамещенным с помощью N(RA)RB, или же AryA определен где-нибудь в другом месте.
Определяемый здесь HetA является насыщенным или мононенасыщенным гетероциклическим кольцом с числом членов от 4 до 9, содержащим от 1 до 3 гетероатомов, независимо выбранных из N, O и S, где кольцо необязательно конденсировано с C3-7циклоалкилом. Насыщенные гетероциклические кольца, подходящие для использования в качестве HetA, включают, например, азетидинил, пиперидинил, морфолинил, тиоморфолинил, тиазолидинил, изотиазолидинил, оксазолидинил, изоксазолидинил, пирролидинил, имидазолидинил, пиперазинил, тетрагидрофуранил, тетрагидротиенил, пиразолидинил, гексагидропиримидинил, тиазинанил, миазепанил, азепанил, диазепанил, азоканил (=октагидроазоцинил), азонанил (=октагидро-1H-азонинил), тетрагидропиранил, тетрагидротиопиранил, и диоксанил. Подходящие мононенасыщенные гетероциклические кольца включают, например, кольца, соответствующие насыщенным кольцам, представленным в предыдущем предложении, за исключением того, что они содержат двойную связь (например, двойную связь углерод-углерод). Насыщенные гетероциклические кольца, конденсированные с циклоалкилом и подходящие для использования в качестве HetA, включают, например,

Определяемый здесь HetB является необязательно замещенным гетероароматическим кольцом, содержащим от 1 до 4 гетероатомов, выбранных из от 1 до 3 атомов N, нуля или 1 атома O, и нуля или 1 атома S, где гетероароматическое кольцо необязательно конденсировано с насыщенным гетероциклическим кольцом с числом членов от 5 до 7, содержащим 1 или 2 гетероатома, независимо выбранных из N, O и S, где любая S в кольце необязательно окислена до S(O) или S(O)2 и либо 1, либо 2 атома углерода в неконденсированном кольце необязательно окислены до C(O). Гетероароматические кольца, подходящие для использования в качестве HetB, включают, например, пиридил (также называемый как пиридинул), пирролил, пиразинил, пиримидинил, пиридазинил, триазинил, имидазолил, пиразолил, триазолил, оксазолил, изооксазолил, оксадиазолил, оксатриазолил, тиазолил, изотиазолил, и тиадиазолил. Конденсированные кольца, подходящие для использования в качестве HetB, включают, например,


Если однозначно не заявлено иначе в конкретном контексте, то любые из описанных здесь различных циклических колец и кольцевых систем могут быть присоединены к остальной части соединения по любому атому в кольце (то есть любому углеродному атому или любому гетероатому), при условии что это приводит к образованию стабильного соединения.
Если однозначно не заявлено иначе, то все приводимые здесь интервалы являются включающими в себя интервалами. Например, гетероароматическое кольцо, описываемое как содержащее от "1 до 4 гетероатомов", означает, что кольцо может содержать 1, 2, 3 или 4 гетероатома. Кроме того, следует иметь в виду, что любой приводимый здесь интервал включает внутри себя область всех подинтервалов внутри этого интервала. Так, например, предполагается, что гетероциклическое кольцо, описываемое как содержащее от "1 до 4 гетероатомов", включает в качестве его аспектов гетероциклические кольца, содержащие от 2 до 4 гетероатомов, 3 или 4 гетероатома, от 1 до 3 гетероатомов, 2 или 3 гетероатома, 1 или 2 гетероатома, 1 гетероатом, 2 гетероатома, 3 гетероатома, и 4 гетероатома.
Когда любая переменная (например, RA или RB) появляется более чем один раз в любой составляющей, или в формуле I, или в любой другой формуле, изображающей и описывающей соединения настоящего изобретения, ее определение для каждого случая появления не зависит от ее определения для каждого другого случая появления. Кроме того, комбинации заместителей и/или переменных являются дозволенными только в том случае, когда такие комбинации приводят к образованию стабильных соединений.
"Стабильным" соединением является соединение, которое может быть получено и выделено, и чья структура и свойства остаются или могут в результате принятых мер оставаться практически неизменными в течение периода времени, достаточного для того, чтобы иметь возможность использовать соединение для описанных здесь целей (например, введение субъекту в терапевтических целях). Соединения настоящего изобретения ограничиваются стабильными соединениями, описываемыми формулой I.
Соединения настоящего изобретения имеют, по меньшей мере, два центра асимметрии и могут иметь один или более дополнительных центров в результате присутствия определенных заместителей и/или заместительных групп. Соответственно, соединения изобретения могут существовать в виде смесей стереоизомеров, или в виде индивидуальных диастереомеров, или энантиомеров. Все изомерные формы этих соединений, или индивидуально, или в смесях, входят в объем настоящего изобретения.
Термин "соединение" относится к свободной форме соединения и любому его гидрату или сольвату, при условии, что они являются стабильными. Гидратом является соединение, образовавшее комплекс с водой, и сольватом является соединение, образовавшее комплекс с органическим растворителем.
Как было указано выше, соединения настоящего изобретения могут быть использованы в форме фармацевтически приемлемых солей. Термин "фармацевтически приемлемая соль" относится к соли, которая обладает эффективностью исходного соединения и которая не является биологически или иным образом нежелательной (например, не является ни токсичной, ни иным образом опасной для реципиента). Подходящей фармацевтически приемлемой солью является соль, образованная путем обработки соединения изобретения (например, соединения формулы I) с помощью одномолярного эквивалента слабого основания (например, карбонатом натрия, бикарбонатом натрия, бикарбонатом калия, или ацетатом натрия). В этом случае M является катионом, таким как Na+, в случае обработки с помощью натриевого основания.
Когда M является H (например, R2 является OSO3H), и соединение изобретения содержит внутреннее основание, которое способно к протонированию (например, R1 содержит основный азот), следует иметь в виду, что соединение может существовать в форме, в которой внутреннее основание полностью протонировано с помощью M=H, так что R2 обладает отрицательным зарядом (например, R2=OSO3-) и внутреннее основание имеет положительный заряд, или частично протонировано, так что R2 обладает частичным отрицательным зарядом, или не является протонированным. Аналогично, когда M является H и соединение изобретения содержит два или более внутренних основания, которые способны к протонированию (например, R1 содержит два или более основных азота), следует иметь в виду, что соединение может существовать в форме, в которой одно или другое из внутренних оснований полностью протонировано с помощью M=H, или что два или более из внутренних оснований каждые достаточно протонированы, так что R2 обладает отрицательным зарядом, или что одно или более из оснований частично протонированы, так что R2 обладает частичным отрицательным зарядом, или что ни одно из оснований не протонировано. Настоящее изобретение включает все такие формы соединения. Хотя эти соединения могут находиться в форме внутренней соли (то есть цвиттер-иона), их рассматривают в качестве соединений изобретения, а не в качестве их фармацевтически приемлемых солей.
С другой стороны, для соединения изобретения, которое содержит внутреннее основание (например, R1 содержит основный азот), фармацевтически приемлемой солью является соль, образованная путем обработки соединения соответствующим количеством кислоты (например, хлористоводородной кислоты, трифторуксусной кислоты, метансульфоновой кислоты, или другой подобной кислоты), так что внутреннее основание протонируется кислотой с положительным зарядом протонированного основания, компенсированным отрицательным противоионом (например, хлоридом, трифторидом, метансульфонатом, или другим подобным противоионом). Для соединений изобретения, содержащих два внутренних основания (например, R1 содержит два основных азота), другой фармацевтически приемлемой солью является соль, образованная в результате обработки соединения с помощью соответствующего количества кислоты, так что одно из внутренних оснований протонируется группой сульфоновой кислоты, присутствующей в молекуле (то есть R2 имеет отрицательный заряд), и другое протонируется кислотой с положительным зарядом протонированного основания, компенсированным подходящим отрицательным противоионом. Еще одна фармацевтически приемлемая соль для соединений изобретения, содержащих два внутренних основания, может быть получена путем обработки соединения с помощью соответствующей кислоты (например, серной кислоты, HCl, метансульфоновой кислоты, или TFA), так что группа сульфоновой кислоты, присутствующая в молекуле, остается протонированной (то есть M = H) и внутренние основания являются протонированными и к тому же связанными с подходящим отрицательным противоионом (например, сульфанатом). Как является очевидным из вышеизложенного, определенная природа и тип фармацевтически приемлемой соли, которые могут быть получены, будут зависеть от природы конкретного соединения, подвергаемого обработке (например, присутствия или отсутствия основных азотов в R1), и используемых условий обработки; например, они будут зависеть от выбора и количества кислоты или основания, с помощью которого обрабатывается соединение, pH обрабатывающей среды, количества и выбора буфера (если его используют), и других подобных факторов. Следует иметь в виду, что настоящее изобретение включает в себя все типы и формы фармацевтически приемлемых солей соединений настоящего изобретения.
Как уже было заявлено выше, настоящее изобретение включает фармацевтические композиции, содержащие соединение формулы I настоящего изобретения, необязательно один или более других активных компонентов (например, β-лактамовый антибиотик), и фармацевтически приемлемый носитель. Характеристики носителя будут зависеть от способа введения. Под понятием "фармацевтически приемлемый" подразумевается то, что ингредиенты фармацевтической композиции должны быть совместимы друг с другом, не препятствуют эффективности активного ингредиента (ингредиентов), и не являются опасными (например, токсичными) для реципиента. Таким образом, композиции согласно изобретению могут, помимо ингибитора, содержать разбавители, наполнители, соли, буферы, стабилизаторы, солюбилизаторы, и другие хорошо известные в технике материалы.
Кроме того, как уже было заявлено выше, настоящее изобретение включает способ лечения бактериальной инфекции, который включает введение субъекту при необходимости такого лечения терапевтически эффективного количества соединения формулы I, или его фармацевтически приемлемой соли, необязательно в комбинации с бета-лактамовым антибиотиком и/или ингибитором DHP. Используемый здесь термин "субъект" (или, в качестве варианта, "пациент") относится к животному, предпочтительно, млекопитающему, наиболее предпочтительно человеку, который являлся объектом лечения, исследования или эксперимента. Термин "введение" и его варианты (например, "введение" соединения) по отношению к соединению формулы I означает обеспечение человека при необходимости его лечения соединением или его фармацевтически приемлемой солью. Когда обеспечивают соединением или его солью в комбинации с одним или более другими активными веществами (например, антибиотиком карбапенемом или ингибитором DHP, или и тем и другим), следует иметь в виду, что "введение" и его варианты каждый включают обеспечением соединением или его солью и другими веществами одновременно или в разное время. Когда вещества комбинации вводят одновременно, они могут быть введены вместе в одной композиции или они могут быть введены раздельно. Следует иметь в виду, что "комбинацией" активных веществ может являться одна композиция, содержащая все активные вещества, или множество композиций, каждая из которых содержит одно или более активных веществ. В случае двух активных веществ комбинацией может являться или одна композиция, включающая оба вещества, или две отдельных композиции, каждая из которых включает одно из веществ; в случае трех активных веществ комбинацией может являться или одна композиция, включающая все три вещества, три отдельных композиции, каждая из которых включает одно из веществ, или две композиции, одна из которых включает два вещества и другая включает третье вещество; и так далее.
Композиции и комбинации настоящего изобретения вводят соответствующим образом в эффективных количествах. Используемый здесь термин "эффективное количество" означает то количество активного соединения или фармацевтического средства, вызывающего биологическую или лечебную ответную реакцию в ткани, системе, животном или человеке, которое определяется исследователем, ветеринаром, врачом или другим клиницистом. В одном варианте осуществления эффективным количеством является "терапевтически эффективное количество" для облегчения симптомов заболевания или состояния, подвергаемых лечению (например, лечение состояний, связанных с бактериальной инфекцией и/или устойчивостью бактерий к действию лекарственного средства). В другом варианте осуществления эффективным количеством является "профилактически эффективное количество" для профилактики симптомов заболевания или состояния, которые хотят предотвратить. Термин здесь также включает количество активного соединения, достаточное для ингибирования β-лактамазы, и в силу чего вызывается искомая ответная реакция (то есть "ингибирующее эффективное количество"). Когда активное соединение (то есть активный ингредиент) вводят в виде соли, ссылки на количество активного ингредиента относятся к форме свободной кислоты или форме свободного основания соединения.
Введение композиции настоящего изобретения осуществляется соответствующим образом парентерально, перорально, сублингвально, трансдермально, местно, интраназально, интратрахеально, или интраректально, где композицию соответствующим образом готовят для введения путем выбранного способа при помощи хорошо известных в технике методов приготовления, включая, например, методы приготовления и введения лекарственных форм, описанных в главах 39, 41, 42, 44 и 45 монографии Remington - The Science и Practice of Pharmacy. 21st edition, 2006. В одном варианте осуществления соединения изобретения вводят внутривенно в больничных условиях. В другом варианте осуществления введением является пероральное введение в форме таблетки или капсулы или другой подобной форме. Когда осуществляют системное введение, терапевтическую композицию вводят соответствующим образом при дозе, достаточной для достижения концентрации ингибитора в крови, по меньшей мере, около 1 микрограмм/мл, предпочтительно около 10 микрограмм/мл, и более предпочтительно около 25 микрограмм/мл. Для локализованного введения эффективными могут являться значительно более низкие концентрации, чем эти, и допустимыми могут являться значительно более высокие концентрации.
Внутривенное введение соединения изобретения может быть осуществлено путем растворения порошкообразной формы соединения с помощью приемлемого растворителя. Подходящие растворители включают, например, физиологические растворы (например, 0,9% раствор хлорида натрия для инъекций) и стерильную воду (например, стерильную воду для инъекций, бактериостатическую воду для инъекций с метилпарабеном и пролпилпарабеном, или бактериостатическую воду для инъекций с 0,9% бензилового спирта). Порошкообразная форма соединения может быть получена путем облучение соединения гамма-лучами или путем лиофилизации раствора соединения, после чего порошок можно хранить (например, в герметичной ампуле) при комнатной температуре или ниже, до тех пор пока его не подвергают растворению. Концентрация соединения в получаемом растворе может находиться, например, в интервале примерно от 0,1 мг/мл до 20 мг/мл.
Настоящее изобретение также включает способ ингибирования бактериального роста, который включает введение в бактериальную клеточную культуру, или в бактериально инфицированную клеточную культуру, ткань, или организм, ингибирующего эффективного количества соединения формулы I. Дополнительные варианты осуществления изобретения включают только что описанный способ ингибирования бактериального роста, где используемым в нем соединением настоящего изобретения является соединение одного из описанных выше вариантов осуществления, подвариантов осуществления или классов. В этих вариантах осуществления соединение может необязательно быть использовано в форме фармацевтически приемлемой соли. Способ может включать введение соединения формулы I в экспериментальную клеточную культуру in vitro для предотвращения роста устойчивых к действию β-лактама бактерий. Способ может, в качестве варианта, включать введение соединения формулы I животному, включая человека, для предотвращения роста устойчивых к действию β-лактама бактерий in vivo. В этих случаях соединение формулы I обычно вводят совместно с β-лактамовым антибиотиком.
Соединения изобретения могут применяться для лечения, профилактики или ингибирования бактериального роста или инфекций, вызванных бактериями, которые устойчивы к действию β-лактамовых антибиотиков. Более конкретно, бактериями могут являться β-лактамазовые положительные штаммы, которые обладают высокой устойчивостью к действию β-лактамовых антибиотиков. Термины "малоустойчивые" и "высокоустойчивые" широко используются обычными специалистами в этой области (смотрите, например, публикации Payne et al., Antimicrobial Agents и Chemotherapy 38:767-772 (1994); Hanaki et al., Antimicrobial Agents и Chemotherapy 30:11.20-11.26 (1995)). Для целей этого изобретения штаммами бактерий, которые обладают высокой устойчивостью к имипенему, являются штаммы, против которых минимальная концентрация ингибитора (MIC) для имипенема составляет >16 мкг/мл, и штаммами бактерий, которые имеют малую устойчивость к действию имипенема, являются штаммы, против которых минимальная концентрация ингибитора (MIC) для имипенема составляет >4 мкг/мл.
Соединения изобретения могут быть использованы в комбинации с антибиотическими средствами для лечения инфекций, вызванных штаммами, продуцирующими β-лактамазу класса C, помимо тех инфекций, которые относят к антибактериальному спектру антибиотического средства. Примерами бактерий, продуцирующих β-лактамазу класса C, являются Pseudomonas aeruginosa, Enter obacler cloacae, Klebsiella pneumoniae, Escherichia coli и Acinetobacter baumannii.
Обычно полезно использовать соединение формулы I в смеси или в сочетании с карбапенемом, пенициллином, цефалоспорином, или другим β-лактамовым антибиотиком, или его пролекарством. Обычно полезно использовать соединение формулы I в комбинации с одним или более β-лактамовыми антибиотиками в связи со свойствами соединений ингибировать β-лактамазy класса C. Как уже отмечалось, соединение формулы I и β-лактамовый антибиотик могут быть введены раздельно (одновременно или в различное время) или в форме одной композиции, содержащей оба активных ингредиентов.
Карбапенемы, пенициллины, цефалоспорины и другие β-лактамовые антибиотики, подходящие для использования в настоящем изобретении, включают как те, по поводу которых известно, что они проявляют нестабильность или же являются чувствительными к действию β-лактамаз класса C, так и те, по поводу которых известно, что они обладают некоторой степенью устойчивости к действию β-лактамазы класса C.
Когда соединения формулы I объединяют с антибиотиком карбапенемом, к ним также может быть добавлен ингибитор дегидропептидазы (DHP). Многие карбапенемы чувствительны к воздействию ренального фермента, известного как DHP. Это воздействие или распад может снижать эффективность карбапенемного антибактериального средства. Ингибиторы DHP и их использование с карбапенемами раскрыты, например, в патентных документах US 4539208, US 4616038, US 4880793 и US 5071843. Предпочтительным ингибитором DHP является 7-(L-2-амино-2-карбоксиэтилтио)-2-(2,2-диметилциклопропанкарбоксамид)-2-гептеновая кислота или ее фармацевтически приемлемая соль.
Карбапенемы, подходящие для совместного введения с соединениями настоящего изобретения, включают имипенем, меропенем, биапенем, (4R,5S,6S)-3-[3S,5S)-5-(3-карбоксифенилкарбамоил)пирролидин-3-илтио]-6-(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло-[3.2.0]гепт-2-ен-2-карбоновую кислоту, хлорид (1S,5R,6S)-2-(4-(2-(((карбамоилметил)-1,4-диазонийбицикло-[2.2.2]-окт-1-ил)-этил-(1,8-нафтосультам)метил)-6-[1(R)-гидроксиэтил]-1-метилкарбапен-2-ем-3-карбоксилата хлорида*хлорида38?*, BMS181139 ([4R-[4альфа,5бета,6бета(R*)]]-4-[2-[(аминоиминометил)амино]-этил]-3-[(2-цианоэтил)тио]-6-(1-гидроксиэтил)-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновую кислоту), BO2727 (моногидрохлорид [4R-3[3S*,5S*(R*)],4альфа,5бета,6бета(R*)]]-6-(1-гидроксиэтил)-3-[[5-[1-гидрокси-3-(метиламино)пропил]-3-пирролидинил]тио]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты), E1010 (гидрохлорид (1R,5S,6S)-6-[1(R)-гидроксиметил]-2-[2(S)-[1(R)-гидрокси-1-[пирролидин-3(R)-ил]-метил]пирролидин-4(S)-илсульфанил]-1-метил-1-карба-2-пенем-3-карбоновой кислоты) и S4661 ((1R,5S,6S)-2-[(3S,5S)-5-(сульфамоиламинометил)пирролидин-3-ил]тио-6-[(1R)-1-гидроксиэтил]-l-метилкарбапен-2-ем-3-карбоновую кислоту), хлорид (1S,5R,6S)-1-метил-2-{7-[4-(аминокарбонилметил)-1,4-диазонийбицикло(2.2.2)-октан-1-ил]-метилфлуорен-9-он-3-ил}-6-(1R-гидроксиэтил)карбапен-2-ем-3-карбоксилата.
Пенициллины, подходящие для совместного введения с соединениями настоящего изобретения, включают бензилпенициллин, феноксиметилпенициллин, карбенициллин, азидоциллин, пропициллин, ампициллин, амоксициллин, эпициллин, тикарциллин, циклациллин, пирбенициллин, азлоциллин, мезлоциллин, сульбенициллин, пиперациллин, и другие известные пенициллины. Пенициллины могут быть использованы в форме их пролекарств; например, в виде in vivo гидролизующихся эфиров, например ацетоксиметиловых, пивалоилоксиметиловых, α-этоксикарбонилоксиэтиловых и фталидиловых эфиров ампициллина, бензилпенициллина и амоксициллина; в виде альдегидных или кетонных аддуктов пенициллинов, содержащих 6-α-аминоацетамидную боковую цепь (например, гетациллин, метампициллин и аналогичные производные амоксициллина); и в виде эфиров карбенициллина и тикарциллина, например фениловых и инданиловых α-эфиров.
Цефалоспорины, подходящие для совместного введения с соединениями настоящего изобретения, включают цефатризин, цефалоридин, цефалотин, цефозолин, цефалексин, цефацетрил, цефапирин, цефамандола нафат, цефрадин, 4-гидроксицефалексин, цефалоглицин, цефоперазон, цефсулодин, цефтазидим, цефуроксим, цефметазол, цефотаксим, цефтриаксон, и другие известные цефалоспорины, каждый из которых может быть использован в форме его пролекарства.
β-Лактамовые антибиотики помимо пенициллинов и цефалоспоринов, которые могут быть совместно введены с соединениями настоящего изобретения, включают азтреонам, латамоксеф (торговая марка моксалактам), и другие известные β-лактамовые антибиотики, такие как карбапенемы типа имипенема, меропенема или (4R,5S,6S)-3-[(3S,5S)-5-(3-карбоксифенилкарбамоил)пирролидин-3-илтио]-6-(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты, каждый из которых может быть использован в форме его пролекарства.
В одном варианте осуществления антибиотик, совместно вводимый с соединением настоящего изобретения, выбирают из группы, состоящей из имипенема, меропенема и (4R,5S,6S)-3-[(3S,5 S)-5-(3-карбоксифенилкарбамоил)пирролидин-3-илтио]-6-(1R)-1-гидроксиэтил]-4-метил-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбоновой кислоты.
В другом варианте осуществления антибиотик, совместно вводимый с соединением настоящего изобретения, выбирают из группы пенициллинов, состоящей из ампициллина, амоксициллина, карбенициллина, пиперациллина, азлоциллина, мезлоциллина, и тикарциллина. Такие пенициллины могут необязательно быть использованы в форме их фармацевтически приемлемых солей, например их натриевых солей. Ампициллин или амоксициллин могут, в качестве варианта, быть использованы в виде тонкодисперсных частиц в форме цвиттер-ионов (обычно в виде тригидрата ампициллина или тригидрата амоксициллина) для использования в суспензиях, вводимых в форме инъекций или инфузий. В аспекте этого варианта осуществления пенициллином, совместно вводимым с соединением настоящего изобретения, является амоксициллин, необязательно в форме его натриевой соли или тригидрата.
В другом варианте осуществления антибиотик, совместно вводимый с соединением настоящего изобретения, выбирают из группы цефалоспоринов, состоящей из цефотаксима, цефтриаксона и цефтазидима, каждый из которых необязательно используют в форме их фармацевтически приемлемых солей, например их натриевых солей.
При совместном введении с β-лактамовым антибиотиком комбинация соединения изобретения и антибиотика может давать синергетический эффект. Термины "синергетический эффект" и "синергизм" указывают на то, что достигаемый эффект в случае совместного введения двух или более лекарственных средств представляет собой большую величину, чем можно было бы ожидать, исходя из эффекта, достигаемого при индивидуальном введении соединений. Не углубляясь в теорию этого вопроса, тем не менее, считается, что соединения настоящего изобретения являются ингибитором β-лактамаз, который препятствует разложению β-лактамовых антибиотиков, тем самым повышая их эффективность и обеспечивая синергетический эффект.
Используемые здесь сокращения включают следующие: acac = ацетилацетонат; AIBN = 2,2-азобисизобутиронитрил; BLI = ингибитор бета-лактамазы; Bn = бензил; BOC (или Boc) = третбутилоксикарбонил; BOC-ON = 2-(третбутоксикарбонилокси-амино)-2-фенилацетонитрил; BOC-OSN = N-третбутоксикарбонилокси)-сукцинимид; BOP = бензотриазол-1-илокси)трис(диметиламино)-фосфоний гексафторфосфат; BSA = бычий сывороточный альбумин; CBZ (или Cbz) = карбобензокси (в качестве варианта, бензилоксикарбонил); COD = циклооктадиенеил; DBU = 1,8-диазабицикло-[5.4.0]ундец-7-ен; DCC = дициклогексилкарбодиимид; DCE = 1,2-дихлорэтан; DCM = дихлорметан; DIPEA = диизопропилэтиламин (или основание Хюнига); DMAC = N,N-диметилацетамид; DMAP = 4-диметиламинопиридин N,N-диметиламинопиридин; DME = 1,2-диметоксиэтан; DMF = N,N-диметилформамид; ДМСО = диметилсульфоксид; EDC = 1-этил-3-(3-диметиламинопропил)карбодиимид; DSC = дифференциальная сканирующая калориметрия; Et = этил; EtOAc = этилацетат; HMDS = гексаметилдисилазид; HOBT = 1-гидроксибензотриазол; HOPO = 2-гидроксипиридин-N-оксид; HPLC = высокоэффективная жидкостная хроматография; IPA = изопропиловый спирт; IPAc = изопропилацетат; i-Pr = изопропил; LC/MS = жидкостная хроматография/масс-спектрометрия; Me = метил; MHBII = среда Мюллера-Хинтона типа II; MIC = минимальная ингибирующая концентирация; MSA = метансульфоновая кислота; NMP = N-метилпирролидинон; PG = защитная группа; Ph = фенил; TEA = триэтиламин; TFA = трифторуксусная кислота; TFE = 2,2,2,-трифторэтанол; THF = тетрагидрофуран; TLC = тонкослойная хроматография; TSB = триптиказо-соевый бульон; TsOH = пара-толуолсульфоновая кислота; XRPD = порошковая рентгенограмма.
Соединения настоящего изобретения могут быть легко получены в соответствии со следующими далее схемами реакций и примерами или их модификациями, используя легкодоступные исходные материалы, реагенты и традиционные методики синтеза, включающие, например, методики, описанные в патентном документе US 7112592. В этих реакциях можно также использовать варианты, которые сами по себе известны обычным специалистам в этой области, и не упоминается более подробно. Кроме того, другие методы получения соединений изобретения будут легко понятны для обычного специалиста в этой области с помощью следующих далее схем реакций и примеров. Если не указано иначе, то все переменные определены выше.
Карбоксамидные соединения настоящего изобретения, в которых a является простой связью и X является (CH2)1-3, могут быть получены, как изображено на схеме 1:


Бициклическое промежуточное соединение A может быть получено, как описано в патентном документе US 7112592, или при помощи его обычных модификаций. Боковая цепь может быть присоединена путем реакции кислоты A с амином B (где при необходимости в амин вводят защитную группу) при известных в технике стандартных условиях реакции образования амида. Например, раствор кислоты A и амина B (1-2 молярных эквивалентов) в растворителе (например, галогеналкане, таком как сухой дихлорметан или хлороформ) может перемешиваться при комнатной температуре при последовательном добавлении триэтиламина (1-2 эквивалента), HOBT (1-2 эквивалента), и EDC (1-2 эквивалента) при комнатной температуре в атмосфере азота. Полученную реакционную смесь можно затем перемешивать при комнатной температуре до тех пор, пока полностью не завершится реакция (например, в течение примерно от 8 до 24 часов), и затем реакционная смесь может быть сконцентрирована под вакуумом, и остаток, очищенный при помощи колонной хроматографии на силикагеле или с помощью HPLC, дает амид C. Снятие защитной группы, представляющей бензиловый эфир, с получением промежуточного соединения гидроксилактама D, может быть осуществлено путем гидрирования или, в некоторых случаях, путем гидролиза, катализируемого кислотой. Например, к раствору бензилового эфира в подходящем растворителе (например, спирте, таком как метанол или этанол, алкилацетате, таком как EtOAc, или эфире, таком как THF) может быть добавлен палладий на угле (0,05-0,5 экв.), и полученную смесь перемешивают в атмосфере водорода (1-3 атмосферы) до тех пор, пока реакция не завершится (например, в течение примерно от 1 до 24 часов), что определяется с помощью соответствующих методов текущего контроля, таких как TLC или HPLC. После завершения реакции гидроксилактам D может быть выделен с помощью традиционных методов. Например, реакционная смесь может быть отфильтрована, и фильтрат сконцентрирован с получением неочищенного гидроксилактама D, который во многих случаях может быть использован непосредственно на следующей стадии без дополнительной очистки. Если дополнительная очистка необходима или желательна, неочищенный гидроксилактам D может быть очищен с помощью колонной хроматографии на силикагеле или HPLC с получением чистого гидроксилактама D. Сульфатизация промежуточного соединения D с получением сульфата E может быть осуществлена при помощи сульфатизирующего реагента в соответствующем растворителе. Так, к раствору гидроксилактама D в апротонном растворителе (например, третичном амиде, таком как пиридин, DMF, или DMAC) при комнатной температуре может быть добавлен комплекс триоксида серы с пиридином (2-10 эквивалентов). Полученная смесь может быть перемешана при комнатной температуре до тех пор, пока не завершится реакция (например, в течение примерно от 4 до 24 часов), завершение которой контролируется с помощью HPLC или LC/MS. Для доведения реакции до конца при необходимости может быть добавлено дополнительное количество комплекса триоксида серы с пиридином. Очищенный продукт реакции может быть получен при помощи традиционных методов, таких как фильтрование реакционной смеси, концентрирование фильтрата под вакуумом, суспендирование концентрата в насыщенном водном растворе дигидрофосфата калия, промывка водного раствора с помощью подходящего органического растворителя (например, EtOAc), добавление к водному слою избытка кислого сульфата тетрабутиламмония, экстракция смеси органическим растворителем (например, EtOAc 4X), объединение органических слоев, сушка объединенных органических слоев над сульфатом натрия, и концентрирование под вакуумом с получением тетрабутиламмониевой соли промежуточного соединения E. В случаях, когда в боковой цепи отсутствует защитная группа, продуктом реакции сульфатизации является соединение формулы I настоящего изобретения. Когда защитная группа вводится в боковую амидную цепь (например, бензиламин или бензиловый эфир, BOC амин, или CBZ амин), группа может быть удалена при помощи известного в технике метода с получением соединения формулы I. Более конкретно, в соединениях формулы I, содержащих аминогруппу в боковой цепи (например, R3 является аминоалкилом), аминогруппу обычно защищают для исключения протекания нежелательных побочных реакций в процессе синтеза. Защита может быть соответствующим образом осуществлена путем использования BOC, CBZ, или аналогичной защитной группы.
Карбоксилатные соединения настоящего изобретения, в которых a является простой связью и X является (CH2)1-3, могут быть получены, как изображено на схеме 2:

На схеме 2 эфир G получают путем взаимодействия промежуточного соединения бициклической производной мочевины A со спиртом с боковой цепью F (где при необходимости в спирт вводят защитную группу) в присутствии этерифицирующего реагента (например, 1-2 эквивалентов DCC или EDC) в присутствии катализатора (например, 0,05-0,25 эквивалентов DMAP) в апротонном растворителе (например, эфире, таком как диэтиловый эфир или THF, или галогеналкане, таком как дихлорметан) при температуре в интервале примерно от 0°C до 35°C до тех пор, пока не завершится реакция (например, в течение примерно 1-24 часов), завершение которой контролируется с помощью TLC или HPLC. Промежуточное соединение G может затем превращено в соединение настоящего изобретения путем последовательных синтезов (дебензилирование, сульфатация, и снятие защиты в боковой цепи (если это необходимо)), аналогичных синтезам, приведенным на схеме 1 для синтеза амидных аналогов.
Карбоксамидные соединения настоящего изобретения, в которых a является простой связью и X является CH=CH (смотрите соединение T ниже), и соединения, в которых a является двойной связью и X является CH2 (смотрите соединение W ниже), могут быть получены, как изображено на схеме 3:


Как показано на схеме 3, защищенная аминокислота J может быть подвергнута реакции сочетания с амином B (1-2 молярных эквивалентов) в растворителе (например, галогеналкане, таком как сухой дихлорметан или хлороформ) при перемешивании при комнатной температуре при последовательном добавлении триэтиламина (1-2 эквивалентов), HOBT (1-2 эквивалентов), и EDC (1-2 эквивалентов) в атмосфере азота. Полученная реакционная смесь может затем быть перемешена при комнатной температуре до тех пор, пока не завершится реакция (например, в течение примерно от 8 до 24 часов), и промежуточный амид может быть извлечен с помощью известных методов (например, концентрирования реакционной смеси под вакуумом, очистки остатка с помощью колонной хроматографии на силикагеле или HPLC). Затем защитная BOC группа может быть удалена при помощи хорошо известных в технике методов с получением амина, который может быть подвергнут восстановительному амминированию с альдегидом L в результате реакции с восстановителем, таким как цианоборгидрид натрия (1-3 молярных эквивалентов) или другим подобным восстановителем, при температуре от 0°C до комнатной температуры в спиртовом растворителе, таком как метанол, этанол, или другие подобные спирты. Реакционная смесь может перемешиваться до тех пор, пока не завершится реакция (например, в течение примерно от 1 до 24 часов), затем с помощью известных методов извлекают амин K (например, концентрируют реакционную смесь под вакуумом и очищают остаток с помощью хроматографии). Затем амин K может быть ацилирован с помощью соединения M в присутствии сильного основания, такого как DBU (~1 эквивалент) или другого подобного основания, в ароматическом углеводородном растворителе, таком как бензол, толуол, или другой подобный растворитель, с получением промежуточного соединения мочевины, которое извлекают при помощи известных методов (например, промывкой реакционной смеси водным раствором кислоты, концентрированием промытой смеси под вакуумом, и очисткой остатка с помощью хроматографии). Затем ацетатная защитная группа может быть удалена при помощи хорошо известных методов, и полученная первичная гидроксильная группа может быть активирована путем реакции с от ~1 до 1,5 эквивалентов тозилхлорида, трифлатного ангидрида, или других подобных соединений в ненуклеофильном растворителе (например, дихлорметане, эфире, бензоле, и так далее) при низкой температуре (например, от 0°C до 25°C). Затем активированная гидроксильная группа может быть обработана с помощью ненуклеофильного основания, такого как DBU, третбутилат калия, третбутиллитий, или другое подобное основание, при низкой температуре (например, от 0°C до 25°C) с получением циклизированного промежуточного соединения Q, которое может быть извлечено и очищено с помощью стандартных методов выделения. Для циклизации диолефинового промежуточного соединения Q могут быть использованы хорошо известные специалистам в этой области методы метатезиса олефинов. Так, например, соединение Q может быть обработано каталитическим количеством (от 0,05 до 0,25 эквивалентов) катализатора Граббса для метатезиса олефинов в подходящем растворителе (например, бензоле, толуоле, тетрагидрофуране, или другом подобном растворителе) при температуре около 25°C с получением циклогексанового продукта, после чего защитная группа PG на гидроксилактаме может быть удалена при помощи хорошо известных методов, и полученный гидроксилактам сульфатируют с получением сульфата. Например, раствор гидроксилактама в апротонном растворителе (например, третичном амиде, таком как пиридин, DMF, или DMAC) может быть обработан комплексом триоксида серы с пиридином (2-10 эквивалентов) примерно при 25°C с получением требуемого продукта, который может быть извлечен и очищен с помощью стандартных методов с получением соединения T. В случаях, когда в боковой цепи защитная группа отсутствует, продуктом реакции сульфатизации является соединение формулы I настоящего изобретения. Когда защитную группу вводят в боковую амидную цепь, (например, бензиламин или эфир, BOC амин, или CBZ амин), группа может быть удалена при помощи известного в технике метода с получением соединения формулы I. Бета-гамма олефин в соединении T может быть изомеризован в сопряженное соединение с амидкарбонилом боковой цепи путем обработки соединения T с помощью ненуклеофильного основания (например, третбутилата калия, гидрида натрия, или другого подобного основания) в ненуклеофильном растворителе (например, третбутаноле, тетрагидрофуране, эфире, или другом подобном растворителе) при температуре от 0°C до 25°C. В качестве варианта, олефин может быть изомеризован в сопряженное соединение в кислотных условиях с использованием такой кислоты, как трифторметансульфоновая кислота или другая подобная кислота, или кислотной ионообменной смолы в ненуклеофильном растворителе (например, третбутаноле, тетрагидрофуране, эфире, или другом подобном растворителе) при температуре от 0°C до 25°C. Полученный олефиновый изомер W может быть извлечен и выделен с помощью стандартных методов выделения.
Вообще говоря, когда химическую группу в соединении называют здесь "защищенной" или говорится о введении "защитной группы", это означает, что химическую группу используют в модифицированной форме для предотвращения нежелательных побочных реакций в защищенном положении. Защитные группы, подходящие для использования при получении соединений настоящего изобретения, и методы введения и удаления таких защитных групп являются хорошо известными в технике и включают группы и методы, описанные в монографиях Protective Groups in Organic Chemistry, ed. J.F. W. McOmie, Plenum Press, 1973 и в T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis. John Wiley & Sons, 3rd edition, 1999, и 2nd edition, 1991, содержания которых приводится здесь путем ссылки на них.
Настоящее изобретение включает также способ (в качестве варианта, называемый способом P) получения соединения формулы P-II:

который включает:
(A) контактирование илида кетосульфоксония формулы P-I:

с иридиевым, родиевым, или рутениевым катализатором с получением соединения P-II;
где:
pG является защитной группой для амина, выбранной из группы, состоящей из карбаматов и бензиламинов;
RU является CH3 или фенилом;
RV является CH3 или фенилом;
R4 является H или C1-4алкилом;
T' является H, Cl, Br, F, C1-3алкилом, O-C1-3алкилом, OH, NH2, N(H)-C1-3алкилом, или N(-C1-3алкил)2;
p является нулем, 1 или 2; q является нулем, 1 , или 2; и p + q = нулю, 1, 2, или 3.
Соединение P-II является промежуточным соединением, используемым в синтезе конкретных соединений настоящего соединения. Защитная группа для амины PG может являться карбаматом (то есть защитной группой формулы

Другие варианты осуществления соединения P-II включают следующие: (1) RU и RV являются оба CH3 или оба фенилом; (2a) T' является H или F; (2b) T' является H; (3a) R4 является H или CH3; (3b) R4 является H. Один или более из этих вариантов осуществления (1)-(3) могут быть объединены друг с другом и/или с вариантами осуществления, описанными выше для PG, где каждая такая комбинация является отдельным вариантом осуществления соединения P-II.
Стадия A включает внутримолекулярное введение NH при помощи илида кетосульфоксония с образованием циклического продукта. Химические свойства илида, используемого на стадии A, обеспечивают преимущество безопасности в сравнении с альтернативными методами, в которых используется диазометан (взрывоопасен), для образования диазокетона, который затем может быть использован при циклизации. Стадия A может также обеспечивать высокий выход. Например, выход на стадии A при использовании каталитического количества [Ir(COD)Cl]2 может составлять 85% или выше.
Стадию A проводят в органическом растворителе. Подходящие растворители включают толуол, дихлорметан, DCE, DMF, THF, хлорбензол, 1,2-дихлорбензол, циклопентилметиловый эфир, ацетонитрил, IPAc, нитрометан, трифторметилбензол, метилэтилкетон, DME, и 2-MeTHF. Предпочтительным растворителем является толуол.
Циклизацию на стадии A проводят в присутствии Ir, Rh, или Ru катализатора. Подходящие катализаторы включают [Ir(COD)Cl]2, RuCl2(PPh3)3, Ru(ДМСО)4Cl2, [RuCl2(цимол)]2, [RuI2(цимол)]2, (циклопентадиенил)Ru(PPh3)2, (инден)RuCl(PPh3)2, Rh2(OAc)4, Rh2(TFA)4, (COD)2IrBF4, IrCI(CO)(PPh3)2, IrCl(CO)3, Ir(COD)(acac), Ir(CO)2(acac), (метилциклопентадиенил)(COD)Ir, или ((циклогексил)3P)3(COD)Ir(пиридин). Класс подходящих катализаторов состоит из [Ir(COD)Cl]2, RuCl2(PPh3)3, Ru(ДМСО)4Cl2, [RuCl2(цимол)]2, [RuI2(цимол)]2, (циклопентадиенил)Ru(PPh3)2, (инден)RuCl(PPh3)2, Rh2(OAc)4, Rh2(TFA)4. предпочтительным катализатором является [Ir(COD)Cl]2. Катализатор обычно используют в количестве примерно от 0,25 до 5 мольных процентов от количества соединения P-I, и более типично используют в количестве примерно от 0,5 до 2 мольных процентов.
Реакция на стадии A может быть соответствующим образом проведена при температуре в интервале примерно от 50°C до 130°C, и ее обычно проводят при температуре в интервале примерно от 70°C до 110°C.
Вариант осуществления способа P включает только что описанную выше стадию A, где PG является Cbz, для получения соединения P-IIa (= соединению P-II, в котором PG замещена с помощью Cbz), и дополнительно включает:
(B) обработку соединения P-IIa с помощью восстановителя с получением соединения формулы P-III:

(C) контактирование соединения P-III с сульфонилгалогенидом формулы R^-SO2W в присутствии основания третичного амина с получением соединения формулы P-IV:

где W является галогеном; и R^ является (1) фенилом, необязательно замещенным с помощью от 1 до 3 заместителей, каждый из которых является независимо C1-4алкилом, C1-4галогеналкилом, O-C1-4алкилом, O-C1-4галогеналкилом, Cl, Br, F, или NO2, (2) C1-4алкилом; или (3) C1-4галогеналкилом.
Стадию B проводят в органическом растворителе. Подходящие растворители включают толуол, дихлорметан, THF, изопропиловый спирт, и ацетонитрил. Предпочтительными растворителями являются толуол и THF.
Подходящие восстановители на стадии B включают LiBH4, NaBH4, KBH4, (Me4N)BH4, LiAlH(O-t-Bu)3, LiBH(OEt)3, и Al(O-i-Pr)3/IPA. Класс подходящих восстановителей состоит из LiBH4, NaBH4, и KBH4. Предпочтительные восстановители включают LiBH4 и NaBH4. Восстановитель обычно используют в количестве примерно от 1 до 2 эквивалентов на эквивалент соединения P-IIa, и более типично используют в количестве примерно от 1 до 1,3 эквивалентов.
Реакция на стадии B может быть проведена соответствующим образом при температуре в интервале примерно от -20°C до 40°C, и ее обычно проводят при температуре в интервале примерно от -15°C до 0°C.
Стадию C проводят в органическом растворителе. Подходящие растворители включают дихлорметан, THF, этилацетат, и MTBE. Предпочтительным растворителем является дихлорметан.
Примеры сульфонилгалогенидов, подходящих для использования на стадии C, включают метансульфонилхлорид, хлорметансульфонил-хлорид, дихлорметансульфонилхлорид, бензолсульфонилхлорид, п-трифторметилбензолсульфонилхлорид, п-толуолсульфонилхлорид, п-бромбензолсульфонилхлорид, п-фторбензолсульфонилхлорид, и п-метоксибензолсульфонилхлорид. Класс подходящих сульфонилгалогенидов состоит из хлорметансульфонилхлорида, п-трифторметилбензолсульфонилхлорида и п-бромбензолсульфонил-хлорида. Предпочтительным сульфонилгалогенидом является п-трифторметилбензолсульфонилхлорид. Сульфонилгалогенид обычно используют в количестве примерно от 1 до 2 эквивалентов на эквивалент соединения P-III, и более типично используют в количестве примерно от 1 до 1,5 эквивалентов (например, около 1,3 эквивалентов).
Подходящим третичным амином на стадии C является три-C1-4алкиламин. Класс подходящих аминов состоит из TEA, DIPEA, и диэтилизопропиламина. DIPEA является предпочтительным основанием. Основание обычно используют в количестве примерно от 1 до 3 эквивалентов на эквивалент соединения P-III, и более типично используют в количестве примерно от 1,1 до 2 эквивалентов (например, около 1,8 эквивалентов).
Реакция на стадии C может быть проведена соответствующим образом при температуре в интервале примерно от 0°C до 40°C, и ее обычно проводят при температуре в интервале примерно от 10°C до 25°C.
Другой вариант осуществления способа P включает только что описанные выше стадии A, B, и C, где PG является Cbz, для получения соединения P-IV, и дополнительно включает:
(D) контактирование соединения P-IV с N-Boc-O-бензилгидроксиамином в присутствии основания с получением соединения формулы P-V:

(E) обработку соединения P-V кислотой с получением соединения формулы P-VI:

Стадию D проводят в органическом растворителе. Подходящие растворители включают DMAC, DMF, NMP, THF и DME. Предпочтительным растворителем является NMP.
Подходящие основания на стадии D включают Li третбутилат, Na третбутилат, K третбутилат, карбонат цезия, KHMDS, и NaHMDS. Класс подходящих оснований состоит из Li третбутилата, Na третбутилата и K третбутилат. Предпочтительным основанием является K третбутилат. Основание обычно используют в количестве примерно от 1 до 2 эквивалентов на эквивалент соединения P-IV, и более типично используют в количестве примерно от 1 до 1,5 эквивалентов (например, около 1,3 эквивалентов).
N-Boc-O-бензилгидроксиламин обычно используют на стадии D в количестве примерно от 1 до 2 эквивалентов на эквивалент соединения P-IV, и более типично используют в количестве примерно от 1 до 1,5 эквивалентов (например, около 1,3 эквивалентов).
Реакция на стадии D может быть проведена соответствующим образом при температуре в интервале примерно от 30°C до 60°C и ее обычно проводят при температуре в интервале примерно от 35°C до 45°C.
Стадию E проводят в органическом растворителе. Подходящие растворители включают DCM и ацетонитрил.
Подходящие кислоты на стадии E включают сульфоновые кислоты. Подходящие кислоты на стадии E включают метансульфоновую кислоту, трифторметансульфоновую кислоту, хлорметансульфоновую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, п-бромбензолсульфоновую кислоту, п-метоксибензолсульфоновую кислоту, и п-трифторметилбензол-сульфоновую кислоту. Класс подходящих кислот состоит из п-толуолсульфоновой кислоты и метансульфоновой кислоты. Предпочтительной кислотой является метансульфоновая кислота. Кислоту обычно используют в количестве примерно от 1 до 6 эквивалентов на эквивалент соединения P-V, и более типично используют в количестве примерно от 3 до 5 эквивалентов.
Реакция на стадии E может быть проведена соответствующим образом при температуре в интервале примерно от 25°C до 60°C, и ее обычно проводят при температуре в интервале примерно от 30°C до 40°C.
Другой вариант осуществления способа Р включает только что описанные выше стадии A, B, C, D и E, где PG является Cbz, для получения соединения P-VI, и дополнительно включает:
(F) контактирование соединения P-VI с фосгеном, дифосгеном или трифосгеном в присутствии основания третичного амина, и затем добавление водного раствора кислоты с получением соединения формулы P-VII:

(G) контактирование соединения P-VII с источником водорода в присутствии катализатора гидрогенолиза и в присутствии Boc-образующего реагента с получением соединения формулы P-VIII:

Стадию F проводят в органическом растворителе. Подходящие растворители включают DCM и ацетонитрил. Предпочтительным растворителем является DCM.
Подходящие кислоты на стадии F включают хлористоводородную кислоту, серную кислоту, трифторуксусную кислоту, и фосфорную кислоту. Предпочтительной кислотой является фосфорная кислота. Кислоту обычно используют в количестве примерно от 1 до 6 эквивалентов на эквивалент соединения P-VI, и более типично используют в количестве примерно от 3 до 5 эквивалентов (например, около 3,2 эквивалентов).
Подходящим третичным амином на стадии F является три-C1-4алкиламин. Класс подходящих аминов состоит из TEA, DIPEA, и диэтилизопропиламина. DIPEA является предпочтительным основанием. Основание обычно используют в количестве примерно от 1 до 6 эквивалентов на эквивалент соединения P-VI, и более типично используют в количестве примерно от 3 до 5 эквивалентов (например, около 3,2 эквивалентов).
Трифосген, дифосген, или фосген обычно используют на стадии F в количестве примерно от 0,5 до 1 эквивалента на эквивалент соединения P-VI, и более типично используют в количестве примерно от 0,7 до 1 эквивалента (например, около 0,8 эквивалента). Трифосген является более предпочтительным по сравнению с дифосгеном и фосгеном.
Контактирование соединения P-VI с трифосгеном, дифосгеном, или фосгеном на стадии F может быть проведено соответствующим образом при температуре в интервале примерно от -15°C до 0°C, и его обычно проводят при температуре в интервале примерно от 35°C до 45°C. Последующее добавление и реакция с кислотой может быть проведена соответствующим образом при температуре в интервале примерно от 0°C до 25°C.
Стадию G проводят в органическом растворителе. Подходящие растворители включают этилацетат, DMAC, третбутанол, и THF. Предпочтительным растворителем является THF.
Подходящие Boc-образующие реагенты на стадии G включают дитретбутилкарбонат, третбутилхлорформиат, BOC-ON и BOC-OSN. Предпочтительным реагентом является дитретбутилкарбонат. Реагент обычно используют в количестве примерно от 0,9 до 3 эквивалентов на эквивалент соединения P-VII, и более типично используют в количестве примерно от 0,9 до 1,5 эквивалентов (например, примерно от 0,95 до 1,1 эквивалента).
Источником водорода на стадии G является обычно газообразный водород, необязательно в смеси с газом-носителем, который является химически инертным при условиях реакции, используемых на стадии G (например, азот или благородный газ, такой как гелий или аргон). Давление не является определяющим аспектом на стадии G, хотя целесообразным является использование атмосферного давления и давления выше атмосферного. Давление обычно составляет, по меньшей мере, около 2 psig (около 115 кПа). Источником водорода, в качестве варианта, может являться молекула - переносчик водорода, такая как формиат аммония, циклогексен, или циклогексадиен.
Расход водорода не является определяющим параметром способа, хотя обычно используют, по меньшей мере, стехиометрическое количество газообразного водорода или другого источника водорода.
Катализатор гидрогенолиза включает нанесенный на носитель или без носителя металл Группы 8 или нанесенное на носитель или без носителя соединение, соль или комплекс металла Группы 8. Катализатор, который обычно используют на стадии G, является нанесенным на носитель или ненанесенным металлическим Pd или нанесенным на носитель или без носителя соединением, солью или комплексом Pd. Подходящие носители катализатора включают уголь, оксид кремния, оксид алюминия, карбид кремния, фторид алюминия, и фторид кальция. Класс подходящих катализаторов состоит из Pd черни (то есть, тонкодисперсных частиц металлического палладия), Pd(OH)2, и Pd/C (то есть палладия на носителе из угля). Pd/C является предпочтительным катализатором гидрогенолиза. Катализатор обычно используют в количестве примерно от 5 до 20 мас.% относительно количества соединения VI, и более типично используют в количестве примерно от 5 до 15 мас.% (например, около 10 мас.%).
Реакция на стадии G может быть проведена соответствующим образом при температуре в интервале примерно от 10°C до 50°C, и ее обычно проводят при температуре в интервале примерно от 15°C до 30°C.
Подвариант осуществления способа P включает только что описанную стадию A, где соединением формулы P-II является соединение p-2:

где стадия A включает:
(A) контактирование илида кетосульфоксония p-1:

с катализатором, выбранным из группы, состоящей из димера хлорида циклооктадиениридия, RuCl2(PPh3), Ru(ДМСО)4Cl2, и Rh2(TFA)4, с получением соединения p-2.
Другой подвариант осуществления способа P включает только что описанную в предыдущем подварианте осуществления стадию A для получения соединения p-2, и дополнительно включает:
(B) обработку соединения p-2 с помощью восстановителя, выбранного из группы, состоящей из Li боргидрида, Na боргидрида и K боргидрида, с получением соединения p-3:

(C) контактирование соединения p-3 с сульфонилгалогенидом формулы R^-SO2W в присутствии основания три-C1-4алкиламина с получением соединения формулы p-4:

где
W является хлором; и
R^ является метилом, хлорметилом, фенилом, 4-бромфенилом, 4-трифторметилфенилом, или 4-метилфенилом.
Другой подвариант осуществления способа P включает только что описанные в предыдущем подварианте осуществления стадии A, B и C для получения соединения p-4, и дополнительно включает:
(D) контактирование соединения p-4 с N-Boc-O-бензилгидроксиламином в присутствии основания, выбранного из группы, состоящей из Li третбутилата, Na третбутилата, K третбутилата и K амилата с получением соединения p-5:

(E) обработку соединения p-5 с помощью кислоты, выбранной из группы, состоящей из метансульфоновой кислоты, хлорметансульфоновой кислоты, п-толуолсульфоновой кислоты и бензолсульфоновой кислоты, с получением соединения формулы p-6:

Другой подвариант осуществления способа P включает только что описанные в предыдущем подварианте осуществления стадии A, B, C, D и E для получения соединения p-6, и дополнительно включает:
(F) контактирование соединения p-6 с трифосгеном в присутствии основания три-C1-4алкиламина, и затем добавление водного раствора фосфорной кислоты с получением соединения p-7:

(G) контактирование соединения p-7 с водородом в присутствии Pd катализатора и Boc-образующего реагента, выбранного из группы, состоящей из дитретбутилкарбоната и BOC-ON, с получением соединения p-8:

Растворители, реагенты, катализаторы, реакционные количества, реакционные температуры, и так далее, описанные выше для стадий A-F в способе P, приводящие к получению соединения P-VIII, и в его вариантах осуществления, применимы для стадий A-F, описанных в предыдущих подвариантах осуществления, приводящих к получению соединения p-8, за исключением тех случаев, когда в подвариантах осуществления на одну или более из этих переменных накладываются четко сформулированные ограничения. Например, подвариант осуществления способа P, описывающий получение соединения p-2 из соединения p-1, ограничивает катализатор, используемый на стадии A, до конкретной группы Ir, Ru и Rh катализаторов. Соответственно, предлагаемое для способа P приведенное выше расширенное раскрытие подходящих катализаторов не применимо к этому подварианту осуществления.
Следует иметь в виду, что предполагается, что растворители, реагенты, катализаторы, реакционные количества, реакционные температуры, и так далее, описанные выше применительно к способу P и его вариантам осуществления и подвариантам осуществления, являются только иллюстрациями, не ограничивающими рамки способа. Например, органическим растворителем, используемым на любой из стадий A-G, может являться любое органическое вещество, которое при условиях реакции, используемых на данной стадии, находится в жидком состоянии, является химически инертным, и может растворять, суспендировать, и/или диспергировать реагирующие вещества и любые реагенты, для того чтобы привести в соприкосновение реагирующие вещества и реагенты и обеспечить протекание реакции. Аналогичные соображения применимы и к выбору оснований, катализаторов, и других реагентов, используемых на стадиях способа. Кроме того, каждая из стадий может быть проведена при любой температуре, при которой может заметно протекать реакция, с помощью которой получают требуемый продукт. Реагирующие вещества, катализаторы и реагенты на данной стадии могут быть использованы в любых количествах, которые приводят к образованию, по меньшей мере, некоторого количества требуемого продукта. Само собой разумеется, что целью на каждой стадии обычно является достижение высокой степени конверсии (например, по меньшей мере, около 60% и, предпочтительно, более высокой) исходных материалов в сочетании с высоким выходом (например, по меньшей мере, около 50% и, предпочтительно, более высоким) требуемых продуктов, и предпочтительным является выбор растворителей, реагентов, катализаторов, реакционных количеств, температур, и так далее, при котором могут быть обеспечены относительно высокие степени конверсии и выходы продукта, и более предпочтительным выбором являются условия, при которых могут быть достигнуты оптимальные степени конверсии и выходы. Конкретные растворители, реагенты, катализаторы, реакционные количества, реакционные температуры, и так далее, описанные выше применительно к способу P и его вариантам осуществления и подвариантам осуществления, могут обеспечить высокие оптимальные степени конверсии и выходы.
Настоящее изобретение также включает соединение, выбранное из группы, состоящей из:




где:
PG является защитной группой для амина, выбранной из группы, состоящей из карбаматов и бензиламинов;
RU является CH3 или фенилом;
RV является CH3 или фенилом;
R4 является H или C1-4алкилом;
T' является H, Cl, Br, F, C1-3алкилом, O-C1-3алкилом, OH, NH2, N(H)-C1-3алкилом, или N(-C1-3алкил)2;
p является нулем, 1 или 2; q является нулем, 1, или 2; p+q = нулю, 1, 2, или 3; и
R^ является:
(1) фенилом, необязательно замещенным с помощью от 1 до 3 заместителей, каждый из которых является независимо C1-4алкилом, C1-4галогеналкилом, OC1-4алкилом, O-C1-4галогеналкилом, Cl, Br, F, или NO2;
(2) C1-4алкилом; или
(3) C1-4галогеналкилом.
Настоящее изобретение также включает соединение, выбранное из группы, состоящей из:
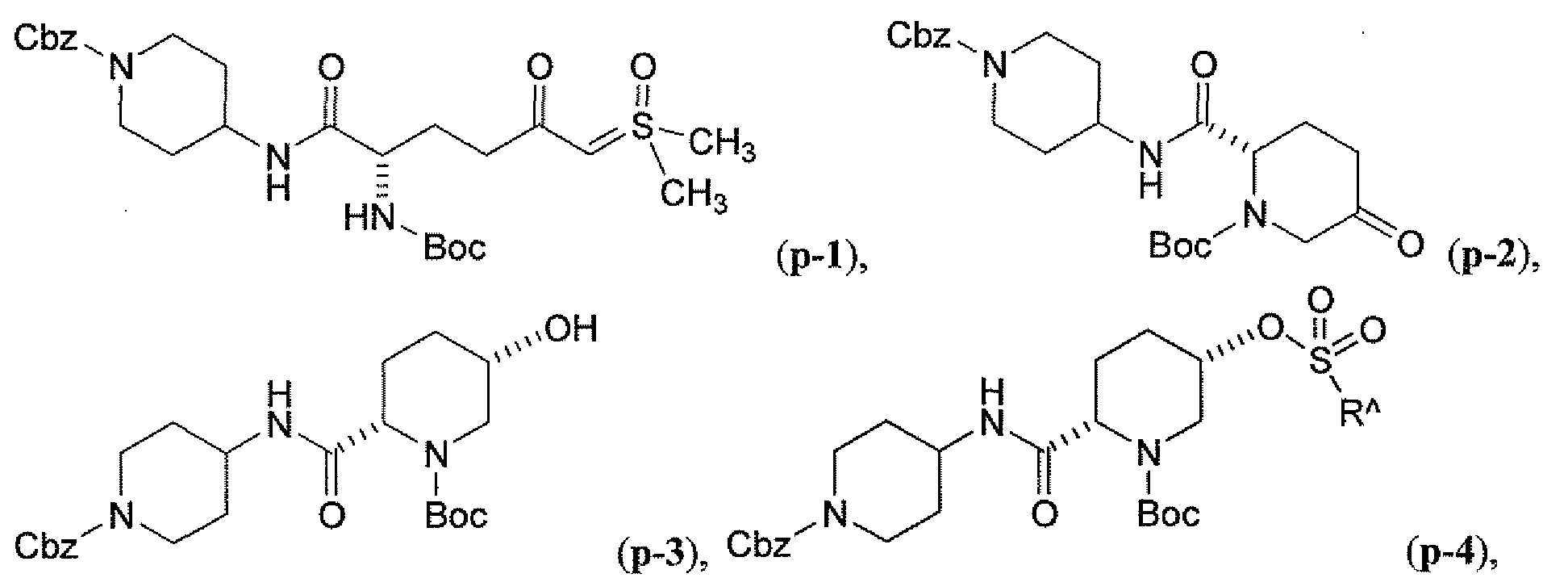

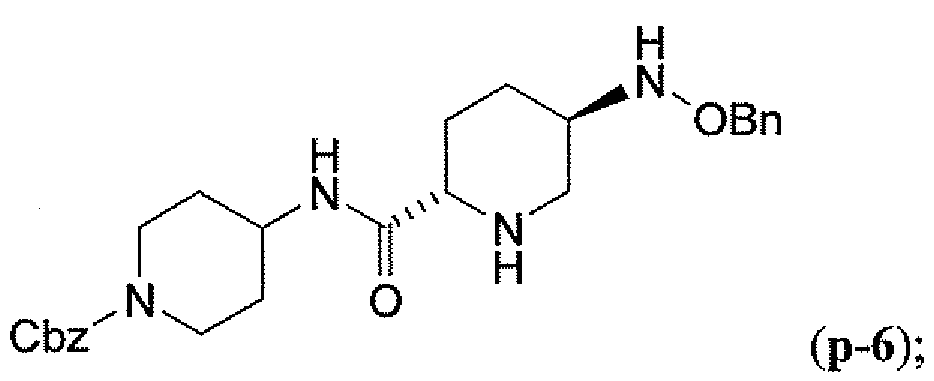
где R^ является метилом, хлорметилом, фенилом, 4-бромфенилом, 4-трифторметилфенилом, или 4-метилфенилом.
Следующие примеры служат только для иллюстрации изобретения и его осуществления. Примеры не следует рассматривать в качестве ограничений объема или сущности изобретения.
ПРЕПАРАТИВНЫЙ ПРИМЕР 1
(4R,6S)-3-(Бензилокси)-2-оксо-1,3-диазабицикло[2.2.1]-гептан-6-карбоновая кислота



Стадия 1: 2-Аллил 1-третбутил(2S,4S)-4-гидроксипирролидин-1,2-ди карбоксилат
Дитретбутилдикарбонат (0,532 мл, 2,291 ммоль) добавляли к раствору цис-4-гидрокси-L-пролина (265 мг, 2,02 ммоль) в DMF (5 мл) и водного раствора гидроксида натрия (2 мл, 2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли аллилбромид (0,18 мл, 2,08 ммоль), и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли с помощью этилацетата и промывали водным разбавленным раствором HCl, водой, насыщенным раствором бикарбоната натрия и рассолом, сушили над сульфатом магния, фильтровали и концентрировали под вакуумом с получением названного соединения в виде прозрачного масла.
Стадия 2: Гидрохлоридная соль аллил (4S)-4-гидрокси-L-пролината
Добавляли хлористоводородную кислоту (4,2 M раствор в диоксане, 5 мл, 21 ммоль) к раствору 2-аллил 1-третбутил (2S,4S)-4-гидроксипирролидин-1,2-дикарбоксилата (1,05 г, 4,87 ммоль) в дихлорметане (20 мл). Полученную смесь перемешивали при комнатной температуре в течение 4 часов, затем концентрировали под вакуумом с получением названного соединения.
Стадия 3: Аллил (4S)-4-гидрокси-1-(трифторацетил)-L-пролинат
Добавляли THF (18 мл) к гидрохлоридной соли аллил (4S)-4-гидрокси-L-пролината (1,011 г, 4,87 ммоль). Полученную суспензию охлаждали до 0°C, и добавляли триэтиламин (3,0 мл, 21,5 ммоль) и затем трифторуксусный ангидрид (2 мл, 14,2 ммоль). Полученную смесь перемешивали при 0°C в течение 30 минут, затем добавляли воду. Полученный раствор перемешивали при комнатной температуре в течение 30 минут, затем разбавляли с помощью этилацетата и промывали 1н раствором HCl, водой, разбавленным раствором бикарбоната натрия, сушили над сульфатом магния, фильтровали и концентрировали под вакуумом. Для того чтобы гидролизовать какой-либо трифторацетатный эфир, который мог образоваться в качестве побочного продукта, остаток помещали в тетрагидрофуран (11,5 мл) и воду (11,5 мл). Полученный мутный раствор перемешивали при комнатной температуре в течение 7 часов. Полученный прозрачный раствор разбавляли с помощью этилацетата и промывали водным 5% раствором бикарбоната натрия и рассолом, затем сушили над сульфатом магния, фильтровали, концентрировали под вакуумом с получением неочищенного продукта. Неочищенный продукт очищали с помощью хроматографии на силикагеле с получением названного соединения в виде бледно-коричневого масла.
Стадия 4: Аллил (4R)-4-[(бензилокси)амино]-1-(трифторацетил)-L-пролинат
Раствор аллил (4S)-4-гидрокси-1-(трифторацетил)-L-пролината (844 мг, 3,16 ммоль) в ацетонитриле (16 мл) охлаждали до -10°C и добавляли 2,6-лутидин (0,62 мл, 5,32 ммоль), затем трифторметансульфоновый ангидрид (0,85 мл, 5,15 ммоль). После окончания добавления температуру повышали до 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа, затем добавляли O-бензилгидроксиламин (1 мл, 8,67 ммоль) и 2,6-лутидин (0,62 мл, 5,32 ммоль). Реакционную смесь подогревали до комнатной температуры в течение ночи. Затем реакционную смесь разбавляли с помощью этилацетата и промывали водным 5% раствором бикарбоната натрия и рассолом, сушили над сульфатом магния, фильтровали и концентрировали под вакуумом. Коричневый масляный остаток (2,57 г) подвергали хроматографии на силикагеле, элюировали сначала смесью 95:5 дихлорметан:этилацетат и в конце 80:20 дихлорметан:этилацетат с получением названного соединения в виде бледно-желтого твердого вещества.
Стадия 5: Аллил (4R)-4-[(бензилокси)амино]-L-пролинат
Раствор аллил (4R)-4-[(бензилокси)амино]-1-(трифторацетил)-L-пролината (1,19 г, 3,20 ммоль) в метаноле (9,5 мл) медленно добавляли к раствору боргидрида натрия (312 мг, 8,25 ммоль) в метаноле (9,5 мл) при -10°C. Реакционную смесь медленно подогревали до 0°C, затем перемешивали при 0°C в течение 3 часов. Добавляли дополнительное количество боргидрида натрия (0,29 г, 7,67 ммоль) при 0°C, и реакционную смесь перемешивали при 0°C в течение трех часов, затем добавляли силикагель (для предварительной абсорбции неочищенного продукта для хроматографии), и растворитель удаляли под вакуумом. Остаток подвергали хроматографии на силикагеле, элюировали смесью 15:9:1 дихлорметан:этилацетат:метанол с получением названного соединения в виде бесцветного масла.
Стадия 6. 2-Аллил 1-третбутил (2S,4R)-4-[(бензилокси)амино]пирролидин-1,2-дикарбоксилат
Раствор аллил (4R)-4-[(бензилокси)амино]-L-пролината (1,2 г, 4,34 ммоль) в дихлорметане (29 мл) добавляли к дитретбутилдикарбонату (1,1 мл, 4,34 ммоль), и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали под вакуумом, и остаток подвергали хроматографии на силикагеле, элюировали гексаном, затем смесью 4:1 гексан:этилацетат с получением названного соединения в виде прозрачной смолы.
Стадия 7: 2-Аллил 1-третбутил (2S,4R)-4-{(бензилокси)-[(трихлорметокси)карбонил]амино}пирролидин-1,2-дикарбоксилат
Дифосген (0,1 мл, 0,804 ммоль) медленно добавляли к раствору 2-аллил 1-третбутил (2S,4R)-4-[(бензилокси)амино]пирролидин-1,2-дикарбоксилата (261 мг, 0,745 ммоль) и триэтиламина (0,13 мл, 0,933 ммоль) в дихлорметане (4 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 4 часов, затем выдерживали при комнатной температуре в течение ночи. Реакционную смесь подвергали хроматографии на силикагеле, элюировали сначала гексаном, затем смесью 4:1 гексан:этилацетат с получением названного соединения в виде прозрачной смолы.
Стадия 8: Аллил (4R,6S)-3-(бензилокси)-2-оксо-1,3-диазабицикло[2.2.1]гептан-6-карбоксилат
Хлористоводородную кислоту (4,2 M раствор в диоксане, 16 мл, 70,4 ммоль) добавляли к 2-аллил 1-третбутил (2S,4R)-4-{(бензилокси)[(трихлорметокси)карбонил]амино}пирролидин-1,2-дикарбоксилату (80 мг, 1,49 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи, затем растворитель удаляли под вакуумом. Добавляли к остатку дихлорметан (82 мл) и затем триэтиламин (0,62 мл, 4,45 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи, затем растворитель удаляли под вакуумом. Остаток подвергали хроматографии на силикагеле (хроматографическая система ISCO) с использованием градиента от гексана в течение 2 минут до смеси 7:3 гексан:этилацетат в течение 6 минут, с выдержкой в течение 3 минут и с переходом к 100% EtOAc в течение 8 минут с получением названного соединения в виде прозрачного масла.
Стадия 9: (4R,6S)-3-(Бензилокси)-2-оксо-1,3-диазабицикло-[2.2.1]гептан-6-карбоновая кислота
2-Этилгексаноат натрия (0,5 M в этилацетате, 2,5 мл, 1,25 ммоль) добавляли к раствору аллил (4R,6S)-3-(бензилокси)-2-оксо-1,3-диазабицикло[2.2.1]гептан-6-карбоксилата (459 мг, 1,52 ммоль), комплекса 1,1'-бис(дифенилфосфино)ферроценпалладий(II)-дихлорида с дихлорметаном (116 мг, 0,14 ммоль) в тетрагидрофуране (7,6 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов (образовывался осадок). Добавляли ацетон (37 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 часов, и смесь центрифугировали. Собирали твердое вещество, промывали его ацетоном и эфиром и сушили под вакуумом с получением названного соединения в виде коричневого твердого вещества. LC-MS (положительная ионизация) m/e 363 (M+H)
ПРИМЕР 1
(2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
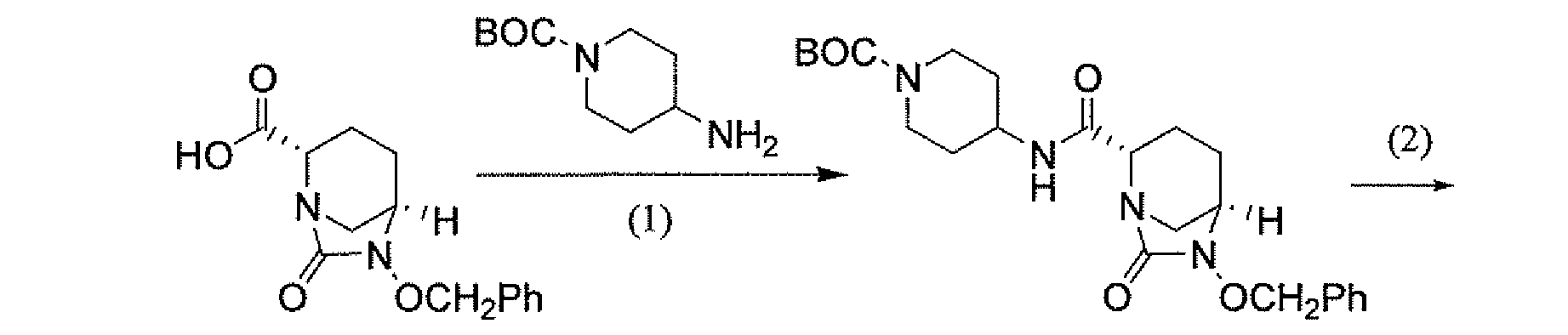

Стадия 1: третбутил 4-({[(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пиперидин-1-карбоксилат
К раствору (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (0,141 г, 0,509 ммоль) (примечание: это промежуточное соединение раскрыто в патентном документе US 7112592 в примере 32b) в сухом дихлорметане (3 мл) последовательно добавляли 4-амино-1-BOC-пиперидин (0,1532 г, 0,765 ммоль), триэтиламин (0,16 мл, 1,148 ммоль), HOBT (0,1145 г, 0,748 ммоль), и EDC (0,1455 г, 0,759 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали под вакуумом, и остаток очищали с помощью HPLC (колонка 30×100 мм Waters Sunfire; 5 микрон; 35 мл/минута; 210 нм; от 15% до 100% CH3CN + 0,05% TFA/вода + 0,05% TFA в течение 15 минут; требуемый продукт элюируют 50% CH3CN + 0,05% TFA/вода + 0,05% TFA) с получением названного соединения.
Стадия 2: третбутил 4-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пиперидин-1-карбоксилат
Палладий на угле (30,5 мг; 10% Pd/C) добавляли к раствору третбутил 4-({[(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло-[3.2.1]окт-2-ил]карбонил}амино)пиперидин-1-карбоксилата (151 мг, 0,33 ммоль) в метаноле (3 мл), и полученную смесь перемешивали в атмосфере водорода (баллон) в течение 3 часов. Анализ с помощью TLC показывал, что реакция прошла до конца. Реакционную смесь фильтровали через микрофильтр, и фильтрат концентрировали под вакуумом с получением названного соединения в виде желтого масла.
Стадия 3: N,N,N-Трибутилбутан-1-аминий[({(2S,5R)-7-оксо-2-[(пиперидин-4-иламино)карбонил]-1,6-диазабицикло[3.2.1]окт-6-ил}окси)сульфонил]оксиданид
К раствору третбутил 4-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пиперидин-1-карбоксилата (36 мг, 0,098 ммоль) в пиридине (0,5 мл) добавляли комплекс триоксида серы с пиридином (70 мг, 0,440 ммоль). Смесь перемешивали при комнатной температуре в атмосфере азота в течение ночи. Анализ с помощью LC/MS показывал, что реакция прошла не полностью. Реакционную смесь фильтровали, и твердое вещество промывали сухим пиридином и дихлорметаном. Фильтрат собирали и концентрировали под вакуумом. Остаток повторно растворяли в сухом пиридине (0,75 мл), и добавляли комплекс триоксида серы с пиридином (31 мг) и затем активированные 4A молекулярные сита. Реакционную смесь перемешивали в течение 4 часов, но изменений, контролируемых с помощью LC/MS, практически не наблюдалось. Реакционную смесь фильтровали, и сита промывали дихлорметаном. Фильтрат концентрировали под вакуумом и суспендировали в насыщенном водном растворе дигидрофосфата калия. Полученную смесь промывали этилацетатом. Собирали водный слой и добавляли гидросульфат тетрабутиламмония (0,034 мг, 0,098 ммоль). Смесь перемешивали в течение 10 минут, затем экстрагировали с помощью EtOAc (4X). Органические слои объединяли, сушили над сульфатом натрия, и концентрировали под вакуумом с получением названного соединения в виде желтого масла.
Стадия 4: (2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору N,N,N-трибутилбутан-1-аминий[({(2S,5R)-7-оксо-2-[(пиперидин-4-иламино)карбонил]-1,6-диазабицикло[3.2.1]-окт-6-ил}окси)сульфонил]оксиданида (22,4 мг, 0,050 ммоль) в безводном дихлорметане (2 мл) при 0°C в атмосфере азота добавляли по каплям трифторуксусную кислоту (0,1 мл, 1,298 ммоль). Реакционную смесь перемешивали в течение 1 часа, затем концентрировали под вакуумом. К остатку добавляли эфир, и полученный белый осадок собирали центрифугированием. Твердое вещество промывали эфиром (2X) с получением названного соединения, загрязненного гидросульфатом тетрабутиламмония и пиридином. Твердое вещество растирали с ацетонитрилом (2X), и собирали белое твердое вещество центрифугированием с получением названного соединения в виде белого твердого вещества. LC-MS (отрицательная ионизация) m/e 347 (M-H); LC-MS (положительная ионизация) m/e 349 (M+H), 381 (M+Na);1H ЯМР (600 МГц, D2O; без отнесения) (δ, м.д.) 4,19 (1H, ушир.д, J=2,5 Гц), 3,98-4,06 (2H, м), 3,47 (2H, ушир.д, J=13 Гц), 3,31 (1H, ушир.д, J=12 Гц), 3,12 (2H, ушир.дд, J=13,3 Гц), 3,06 (1H, д, J=12 Гц), 2,04-2,21 (м, 4H), 1,87-1,95 (1H, м), 1,72-1,83 (м, 3H).
ПРИМЕР 1A
(2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид

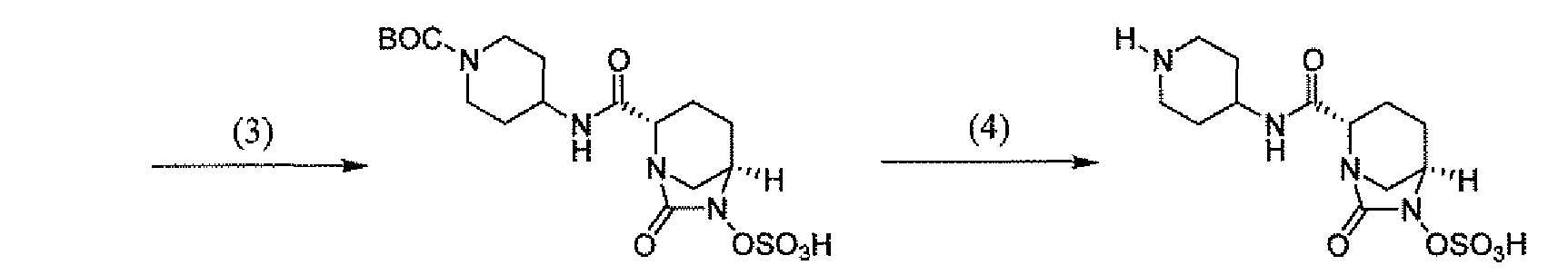
Стадия 1: третбутил 4-({[(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пиперидин-1-карбоксилат
К раствору (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (1,484 г, 5,37 ммоль) в сухом дихлорметане (60 мл) последовательно добавляли триэтиламин (1,88 мл, 13,49 ммоль), иодид 2-хлор-1-метилпиридиния (1,60 г, 6,26 ммоль), и 4-амино-1-BOC-пиперидин (1,30 г, 6,49 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь затем нагревали до 50°C в течение 1 часа. Реакционную смесь концентрировали под вакуумом и очищали с помощью хроматографии на силикагеле на Isco Combiflash (40 г силикагеля, 40 мл/минута, 254 нм, от 15% до 100% EtOAc/гексан до 14 объемов колонки, затем 100% EtOAc для 4 объемов колонки; названное соединение элюировали при 65% этилацетат/гексан) с получением названного соединения в виде бледно-оранжевого твердого вещества.
Стадия 2: третбутил 4-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пиперидин-1-карбоксилат
Палладий на угле (394 мг; 10% Pd/C) добавляли к раствору продукта со стадии 1 (1,81 г, 3,95 ммоль) в метаноле (50,6 мл), и полученную смесь перемешивали в атмосфере водорода (баллон) в течение ночи. Анализ с помощью LC/MS указывал на то, что реакция прошла неполностью. К реакционной смеси добавляли уксусную кислоту (6 капель) и дополнительное количество катализатора (159 мг 10% Pd/C), и полученную смесь перемешивали в атмосфере водорода (баллон) в течение еще 90 минут. К реакционной смеси добавляли дополнительное количество катализатора (0,2085 г 10% Pd/C) и продолжали перемешивание в атмосфере водорода в течение еще 2,5 часов, после чего с помощью анализа LC-MS делали вывод о том, что реакция прошла полностью. Реакционную смесь фильтровали через слой целлита, и собранное твердое вещество хорошо промывали с помощью MeOH. Фильтрат концентрировали под вакуумом с получением названного соединения в виде бесцветного масла, которое использовали без очистки на следующей стадии.
Стадия 3 : третбутил-4-({[(2S,5R)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пиперидин-1-карбоксилат
К раствору продукта со стадии 2 (1,455 г, 3,95 ммоль; теоретический выход стадии 2) в сухом пиридине (30 мл) добавляли комплекс триоксида серы с пиридином (3,2 г, 20,11 ммоль) при комнатной температуре в атмосфере азота. Полученную густую смесь перемешивали в течение недели. Реакционную смесь фильтровали, и белое нерастворимое твердое вещество хорошо промывали дихлорметаном. Фильтрат концентрировали под вакуумом. Остаток затем отгоняли в виде азеотропа с толуолом для удаления избытка пиридина с получением названного соединения, которое затем использовали без очистки на следующей стадии.
Стадия 4: (2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К смеси продукта со стадии 3 (1,772 г, 3,95 ммоль; теоретический выход стадии 3) в сухом дихлорметане (30 мл) при 0°C в атмосфере азота медленно добавляли трифторуксусную кислоту (6,1 мл, 79 ммоль). Реакционная смесь мгновенно превращалась в раствор. Через 1 час к реакционной смеси добавляли дополнительное количество трифторуксусной кислоты (8 мл). Реакционную смесь перемешивали при 0°C до тех пор, пока с помощью анализа LC-MS можно было сделать вывод о завершении реакции, затем концентрировали под вакуумом. Остаток растирали с эфиром (3X) для удаления избытка TFA и органических примесей. Полученное белое нерастворимое твердое вещество собирали с помощью центрифугирования, сушили под вакуумом, затем очищали с помощью препаративной HPLC (колонка 250×21,2 мм Phenomenex Synergi Polar-RP 80A; 10 микрон; 35 мл/минута; 210 нм; от 0% до 30% метанол/вода в течение 15 минут; названное соединение элюировали при 10% метанол/вода). Фракции, содержащие названное соединение, объединяли и лиофилизировали в течение ночи с получением названного соединения в виде белого твердого вещества. LC-MS (режим отрицательной ионизации) m/e 347 (M-H).
ПРИМЕР 1C
(2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
Стадия 1: Бензил 4-[(третбутоксикарбонил)амино]пиперидин-1-карбоксилат

4-(N-BOC-амино)пиперидин (17 кг, 84,88 моль) растворяли в DCM (90 кг), добавляли триэтиламин (10,14 кг, 100,16 моль), и полученный раствор охлаждали до 0-5°C. Добавляли в течение 45 минут бензилхлорформиат (16,51 кг, 96,76 моль), поддерживая при этом температуру <25°C, после чего раствор подвергали старению в течение 30 минут при 20°C. Затем в течение 10 минут добавляли 2M HCl (61 кг, 118,13 моль), поддерживая при этом температуру < 25°C. Смесь перемешивали в течение 10 минут, и затем перемешивание прекращали и давали возможность фазам расслоиться. Затем фазы друг от друга разделяли, и органическую фазу подвергали перегонки под вакуумом до объема 35 л. Затем добавляли изопропилацетат (89 кг), и смесь концентрировали с помощью вакуумной перегонки при температуре ниже, чем 35°C, до объема ~50 л для кристаллизации названного продукта. Затем в течение 10 минут добавляли гептан (47 кг), и полученную суспензию охлаждали и подвергали старению при 20°C в течение 20 минут, после чего подвергнутую старению суспензию фильтровали, промывали гептаном (17 кг), и сушили на фильтре в токе N2 с получением названного продукта в виде белого твердого вещества (24,7 кг, 87 %).1H ЯМР (CDCl3) 7,33 (5H, м), 5,13 (2H, с), 4,47 (1H, м), 4,11 (2H, м), 3,61 (1H, м), 2,93 (2H, м), 1,94 (2H, м), 1,45 (9H, с) и 1,30 (2H, м).
Стадия 2: Бензил 4-аминопиперидин-1-карбоксилат
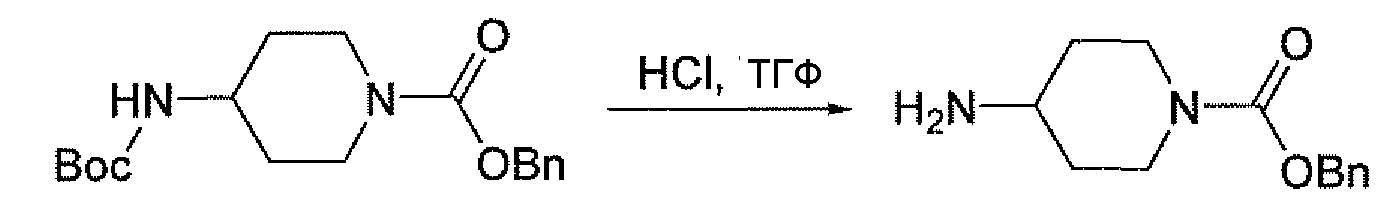
4-(N-BOC амино)-CBz пиперидин (24,4 кг, 73,42 моль), THF (65 кг) и 5 M HCl (23,0 кг, 110,13 моль) смешивали и нагревали до 30-35°C в течение ~2 часов, и затем при 55°C в течение ночи. После охлаждения реакционной смеси до 10°C добавляли дихлорметан (97 кг) и 10 M NaOH (7,97 кг, 145,12 моль), поддерживая при этом температуру <25°C. Фазы разделяли, и органическую фазу промывали 25 мас.% раствором NaCl (27,5 кг). Промытую органическую фазу подвергали отгонке при атмосферном давлении до объема 70 л. Затем добавляли дихлорметан (162 кг), и смесь концентрировали с помощью отгонки до объема 120 л с получением названного продукта в виде раствора в DCM (17,2 кг, 100%.).1H ЯМР (CDCl3) 7,33 (5H, м), 5,14 (2H, с), 4,14 (2H, ушир.с), 2,87 (3H, м), 1,83 (2H, м), 1,66 (3H, м) и 1,28 (2H, м).
Стадия 3: Бензил 4-{[1-(третбутоксикарбонил)-5-оксо-L-пропил]амино}пиперидин-1-карбоксилат

2-Гидроксипиридин-N-оксид (811 г, 7,3 моль), L-пиро-глутаминовую кислоту (9,43 кг, 73 моль), бензил 4-аминопиперидин-1-карбоксилат (17,1 кг в дихлорметане, объем 120 л, 73 моль) и дихлорметан (80 кг) смешивали вместе и подвергали старению при 20°C в течение 10 минут с образованием густой суспензии. К суспензии добавляли порциями гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (16,8 кг, 87,6 моль), поддерживая при этом температуру <30°C. Затем суспензию подвергали старению при 25°C в течение 30 минут, после чего добавляли 1 M хлористоводородную кислоту (94 кг, 85,5 моль). Фазы оставляли расслаиваться в течение ночи и затем разделяли, и затем органическую фазу промывали 2 M карбонатом натрия (109 кг) и затем растворитель заменяли на ацетонитрил до конечного объема 50 л. Добавляли толуол (88,2 кг) и смесь охлаждали до 0°C. К смеси добавляли дитретбутилдикарбонат (18,32 кг, 83,95 моль) и 4-диметиламинопиридин (223 г, 1,83 моль), и раствор нагревали до 25°C и подвергали старению в течение ночи. Смесь затем концентрировали с помощью отгонки до объема 80 л. Добавляли дополнительное количество толуола (88,2 кг), и смесь дополнительно концентрировали до 50 л. Добавляли изопропилацетат (30 кг), и полученную суспензию подвергали старению в течение 10 минут. Затем к суспензии добавляли по каплям гептан (70 кг) в течение 30 минут, и суспензию подвергали старению в течение 30 минут, затем фильтровали, промывали смесью изопропилацетат/гептан (22,5 кг/17,4 кг), и затем сушили под вакуумом при 55°C с получением названного продукта в виде белого твердого вещества (27,5 кг, 95,5 мас.%, 82%).1H ЯМР (CDCl3) 7,33 (5H, м), 6,19 (1H, м), 5,13 (2H, с), 4,48 (1H, дд), 4,15 (2H, м), 3,97 (1H, м), 2,95 (2H, м), 2,73 (1H, д, тр), 4,65 (1H, м), 2,61 (1H, м), 2,18 (2H, м), 1,45 (9H, с) и 1,30 (2H, м).
Стадия 4: Бензил 4-({N-(третбутоксикарбонил)-6-[диметил-(оксидо)-λ4-сульфанилиден]-5-оксо-L-норлейцил}амино)пиперидин-1-карбоксилат

Третбутилат калия (9,58 кг, 85,38 моль) добавляли тремя порциями к раствору иодида триметилсульфоксония (18,79 кг, 85,38 моль) в DMF (115 кг) при 15-25°C. Суспензию подвергали старению при 20-25°C в течение одного часа, затем добавляли четырьмя порциями исходный реагент бензил 4-{[1-(третбутоксикарбонил)-5-оксо-L-пролил]амино}пиперидин-1-карбоксилат (27,17 кг, 60,99 моль) в течение 30 минут, затем подвергали старению в течение 30 минут при 20°C. Добавляли воду (54 кг) и материал затравки (10 г) (примечание: кристаллизация может проходить без использования затравки, но предпочтительно использовать затравку, так как она обычно обеспечивает получение продукта с более постоянным качеством и более высокий выход), и суспензию подвергали старению при 20°C в течение 30 минут. Добавляли в течение 1 часа 10% раствор NaCl (543 кг), поддерживая при этом температуру ниже 25°C. Суспензию затем охлаждали до 3°C в течение 1 часа и подвергали старению в течение ночи при 3°C, после чего суспензию фильтровали, промывали три раза водой (136 л, 82 л, 82 л), и сушили под вакуумом при 55°C, с получением названного продукта в виде желтого твердого вещества (32,8 кг, 83%).1H ЯМР (CDCl3) 7,49 (1H, ушир.с), 7,33 (5H, м), 5,83 (1H, ушир.с), 5,13 (2H, с), 4,48 (1H, с), 4,08 (3H, м), 3,96 (1H, м), 3,45 (3H, с), 3,41 (3H, с), 3,03 (2H, м), 2,41 (1H, м), 2,24 (1H, м), 1,94 (4H, м), 1,68 (5H, с) и 1,44 (12H, с).
Стадия 5: Третбутил (2S)-2-[({1-[(бензилокси)карбонил]
пиперидин-4-ил}амино)карбонил]-5-оксопиперидин-1-карбоксилат

Из раствора димера хлорида циклооктадиениридия (336,3 г, 0,502 моль) в толуоле (318 кг) удаляли кислород путем дегазирования с помощью 3 циклов вакуумной дегазации с последующим барботированием азота через объем раствора в течение 30 минут, после чего раствор нагревали до 105°C. Из раствора исходного реагента илида (27,0 кг, 50,22 моль) в DMF (128 кг) при 25°C удаляли кислород путем дегазирования с помощью 3 циклов вакуумной дегазации с последующим барботированием азота через объем раствора в течение 30 минут. Дегазированный раствор затем добавляли к горячему раствору катализатора в течение 30 минут, поддерживая при этом температуру реакционной смеси выше 102°C. Реакционную смесь подвергали старению при 105°C в течение 40 минут, и затем охлаждали до 20°C. Органическую реакционную смесь промывали дважды 5 мас.% раствором хлорида лития (81 л × 2), затем водой (81 л). Органическую и водную фазы разделяли, и затем удаляли из органической фазы толуол путем отгонки под вакуумом до объема 130 л, и отогнанную фазу хранили охлажденной, перед тем как ее непосредственно использовать на следующей стадии.
Стадия 6: третбутил (2S,5S)-2-[({1-[(бензилокси)карбонил]
пиперидин-4-ил}амино)карбонил]-5-гидроксипиперидин-1-карбоксилат

Раствор боргидрида лития (22,2 кг 4,1 M раствора в THF, 101,9 моль) разбавляли с помощью THF (290 кг), после чего добавляли метанол (3,26 кг) при 20°C, и подвергали старению в течение 30 минут, перед тем как охладить до -4°C. К подвергнутому старению раствору боргидрида добавляли раствор кетона в толуоле (46,8 кг в ~4 мл/г растворе в толуоле, 101,9 моль), поддерживая при этом температуру реакционной смеси <0°C. Реакцию останавливали с помощью раствора уксусной кислоты (30,6 кг, 509,5 моль, растворенной в 183 кг метанола), поддерживая при этом температуру <20°C. После остановки реакции реакционную смесь затем подвергали старению при 20°C в течение 1 часа, после чего ее концентрировали под вакуумом до объема 184 л. Добавляли метанол (203 кг), и смесь отгоняли под вакуумом до объема 184 л. Добавляли изопропанол (294 кг), и смесь отгоняли под вакуумом до объема 184 л, поддерживая при этом внутреннюю температуру ~30°C. Добавляли затравку (5 г) (примечание: кристаллизация может проходить без использования затравки, но предпочтительно использовать затравку, так как она обычно обеспечивает получение продукта с более постоянным качеством и более высокий выход), и смесь подвергали старению в течение 1 часа для образования затравочного слоя. Затем добавляли воду (560 кг) в течение ~60 минут, затем добавляли изопропанол (111 кг). Суспензию фильтровали, промывали три раза с помощью MTBE (30 кг, 35 кг, 5 кг), и затем сушили под вакуумом при 55°C с получением названного продукта (26,74 кг, 57% выход по илиду).1H ЯМР (CDCl3) 7,33 (5H, м), 6,17 (1H, ушир.с), 5,13 (2H, с), 4,61 (1H, м), 4,11 (3H, м), 3,94 (1H, м), 3,64 (1H, м), 2,98 (2H, м), 2,59 (1H, дд), 2,33 (1H, м), 1,94 (4H, м), 1,71 (1H, м), 1,63 (2H, м), 1,48 (9H, с) и 1,35 (2H, м).
Стадия 7: Третбутил (2S,5S)-2-[({1-[(бензилокси)карбонил]пиперидин-4-ил}амино)карбонил]-5-({[4-(трифторметил)фенил]сульфонил}окси)пиперидин-1-карбоксилат

Исходный реагент спирт (26,6 кг, 57,7 моль) растворяли в дихлорметане (120 кг) и пропускали через картридж с активированным углем. К раствору спирта добавляли N,N-диметиламинопиридин (1,06 кг, 8,66 моль) и TEA (11,1 кг, 109,63 моль), затем добавляли 4-трифторметилбензолсульфонилхлорид (18,0 кг, 73,6 моль) в виде раствора в дихлорметане (30 кг) в течение 20 минут при температуре <25°C. Смесь затем подвергали старению в течение 3 часов, после чего добавляли воду (110 кг), поддерживая при этом температуру <25°C. Фазы разделяли, и органическую фазу промывали дважды водой (80 кг × 2), затем водным раствором HCl (15 л концентрированной 37 мас.% HCl в воде (80 кг)). Органический слой разбавляли с помощью дихлорметана (75 кг) и подвергали отгонке под вакуумом до 72 л. Затем добавляли MTBE (157 кг) и смесь отгоняли под вакуумом до 170 л для кристаллизации продукта. Суспензию подвергали старению в течение 1 часа и затем добавляли гептан (58 кг) в течение ~20 минут, и суспензию подвергали старению при 20°C в течение 18 часов. Подвергнутую старению суспензию затем фильтровали, промывали гептаном (20 кг) и MTBE (40 кг), и сушили на фильтре в токе азота в стечение 24 часов с получением названного продукта (38,3 кг, 98%).1H ЯМР (CDCl3) 8,20 (2H, д), 7,85 (2H, д), 7,33 (5H, м), 6,09 (1H, ушир.с), 5,13 (2H, с), 4,59 (1H, м), 4,46 (1H, м), 4,10 (3H, м), 3,91 (1H, м), 2,96 (2H, м), 2,75 (1H, м), 2,33 (1H, м), 1,58 (1H, м), 1,48 (9H, с) и 1,35 (2H, м).
Стадия 8: Бензил 4-[({(2S,5R)-5-[(бензилокси)амино]
пиперидин-2-ил}карбонил)амино]пиперидин-1-карбоксилат

N-Boc-O-бензилгидроксиламин (8,65 г, 38,7 ммоль, в виде раствора в объеме 38 мл DMAC) добавляли к раствору третбутилата калия (4,35 г, 38,7 ммоль) в DMAC (80 мл), поддерживая при этом температуру между 18°C и 25°C. Раствор подвергали старению в течение 30 минут, после чего он превращался в суспензию. Добавляли к суспензии исходный реагент сульфонат (20 г, 29,9 ммоль), растворенный в DMAC (40 мл), в течение 15 минут при 20°C, и полученную смесь нагревали до 40°C в течение 3,5 часов и затем выдерживали при 20°C в течение ночи. К смеси добавляли воду (350 мл), поддерживая при этом температуру <30°C. Затем добавляли DCM (350 мл), и разделяли фазы. Органическую фазу промывали три раза водой (350 мл × 3). Затем промытую органическую фазу подвергали отгонке при атмосферном давлении до объема 90 мл, после чего добавляли метансульфоновую кислоту (10 мл), и раствор нагревали до 35-40°C в течение 8 часов. Затем раствор охлаждали до 20°C и добавляли 2н NaOH (200 мл), затем добавляли DCM (90 мл). Фазы разделяли, и органическую фазу промывали водой (90 мл), и затем растворитель заменяли при атмосферном давлении на ацетонитрил объемом 50 мл. Для кристаллизации продукта добавляли p-толуолсульфоновую кислоту (4 г, 1 эквивалент в расчете на продукт) в виде раствора в ацетонитриле (40 мл) при 40°C. Затем добавляли MTBE (45 мл), и охлаждали суспензию до 20°C, подвергали ее старению при 20°C в течение 1 часа, и затем фильтровали с получением названного продукта в виде монотозилатной кристаллической соли (9,8 г, 53%).1H ЯМР (CDCl3) 7,75 (1H, ушир.с), 7,59 (2H, д), 7,36 (5H, м), 7,20 (3H, м), 7,14 (2H, д), 6,98 (2H, д), 5,30 (1H, м), 5,10 (2H, м), 4,37 (2H, с), 3,88 (3H, м), 3,61 (2H, м), 3,23 (2H, м), 2,22 (3H, с), 1,65 (1H, м), 1,26 (5H, м) и 1,21 (3H, м).
Стадия 9: Бензил 4-({[(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пиперидин-1-карбоксилат

Бензил 4-[({(2S,5R)-5-[(бензилокси)амино]пиперидин-2-ил}карбонил)амино]пиперидин-1-карбоксилат в форме тозилатной соли (8,1 кг, 12,68 моль) суспендировали в дихлорметане (108 кг), после чего добавляли 5 мас.% NaHCO3 (42 кг, 25,36 моль), и полученную двухфазную смесь интенсивно перемешивали в течение 30 минут. Фазы разделяли, и органическую фазу промывали водой (40,5 кг). Затем органическую фазу подвергали отгонке при атмосферном давлении до объема ~20 л, затем добавляли DCM (108 кг). Добавляли затем DIPEA (5,25 кг, 40,58 моль), и смесь охлаждали до 0~5°C. Добавляли четырьмя порциями трифосген (3,01 кг, 10,14 моль), поддерживая при этом температуру <10°C. Через 30 минут добавляли разбавленный раствор фосфорной кислоты (4,97 кг 85 мас.% фосфорная кислота в 32 кг воды), и смесь подвергали старению при 20°C в течение ночи. Фазы разделяли, и органическую фазу промывали с помощью 5 мас.% NaHCO3 (26 кг) и воды (25 кг). Органическую фазу затем подвергали отгонке при атмосферном давлении до объема 30 л. Затем добавляли этанол (77 кг) и вводили затравку (10 г) (примечание: кристаллизация может проходить без использования затравки, но предпочтительно использовать затравку, так как она обычно обеспечивает получение продукта с более постоянным качеством и более высокий выход). Суспензию подвергали отгонке под вакуумом до объема 33 л, затем по каплям добавляли гептан (55 кг). Затем суспензию охлаждали до 0°C, фильтровали, промывали смесью 3:1 гептан:этанол (30 л), и сушили на фильтре в токе азота с получением названного продукта (5,90 кг, 94%).1H ЯМР (CDCl3) 7,35 (10 H, м), 6,57 (1H, д), 5,14 (2H, c), 5,07 (1H, д), 4,92 (1H, д), 4,13 (2H, м), 3,95 (1H, м), 3,89 (1H, д), 3,31 (1H, c), 2,99 (3H, м), 2,65 (1H, д), 2,38 (1H, м), 1,94 (4H, м), 1,62 (2H, м) и 1,34 (2H, м).
Стадия 10: Третбутил 4-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пиперидин-1-карбоксилат

Бензил 4-({[(2R,5S)-6-(бензилокси)-7-оксо-1,6-диазабицикло-[3.2.1]окт-2-ил]карбонил}амино)пиперидин-1-карбоксилат исходный реагент (1,9 кг и 97 мас.%) и Boc2O (0,776 кг) загружали в стеклянную колбу, и растворяли твердые вещества в THF (15 л). Раствор затем загружали в реактор для гидрирования вместе с Pd(OH)2 (184,3 г) и еще одной порцией THF (10,8 л). Реакцию проводили при парциальном давлении H2 45 psig, 23°C в течение 5 часов. После завершения реакции, которое определяли с помощью HPLC анализа, раствор фильтровали через материал solka flok для удаления катализатора, и осадок на фильтре промывали с помощью THF. Затем в фильтрате и промывках заменяли растворитель на EtOAc путем отгонки под вакуумом до объема 10 л. Использовали приблизительно 30 л EtOAc для замены растворителя, и с помощью1Н ЯМР было определено, что содержание THF после отгонки при постоянном объеме (10 л при максимальной температуре 20°C) составляло ~4 мол.% THF:EtOAc. Полученную суспензию в EtOAc подвергали старению при комнатной температуре в течение 1 часа, после чего добавляли гексаны (4 л) в течение 1 часа при комнатной температуре. Суспензию подвергали старению в течение еще 1 часа, после чего определяли концентрацию в надосадочной жидкости (задано: ~6 мг/г). Твердое вещество затем фильтровали и промывали раствором 60% EtOAc/гексаны (3×3 л) и сушили под вакуумом и в токе N2 при комнатной температуре с получением названного продукта (80% выход при выделении продукта).1H ЯМР (400 МГц, CDCl3): 8,60 (ушир.c, 1H), 6,67 (д, J=8,2 Гц, 1H), 4,12-4,00 (м, 2H), 4,00-3,91 (м, 1H), 3,89 (д, J=7,8 Гц, 1H), 3,81-3,76 (м, 1H), 3,19 (дт, J=11,2, 2,9 Гц, 1H), 2,90 (т, J=11,9 Гц, 2H) 2,82 (д, J=11,3 Гц, 1H), 2,45 (дд, J=15,0, 6,7 Гц, 1H), 2,21-2,11 (м, 1H), 2,02-1,85 (м, 3H), 1,80-1,69 (м, 1H), 1,48 (c, 9H), 1,44-1,30 (м, 2H)
Стадия 11: Сульфатная соль тетрабутиламмония

Третбутил 4-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло-[3.2.1]окт-2-ил]карбонил}амино)пиперидин-1-карбоксилат (3,0 кг), THF (30 л), 2-пиколин (1,61 л) и комплекс пиридин-SO3 (4,54 кг) загружали в колбу в атмосфере азота. Экзотермический эффект не наблюдался. Гетерогенную смесь подвергали перемешиванию в течение ночи (~15 часов). Затем добавляли DCM (8 л), и смесь концентрировали путем отгонки под вакуумом, удаляя ~30 л THF/DCM. Добавляли дополнительное количество DCM (28 л), затем добавляли воду (20 л). Колбу помещали на ледяную баню и добавляли K2HPO4 (2,20 кг) в течение 4 минут, затем споласкивали водой (1 л). Добавляли затем BU4NHSO4 (2,90 кг) в течение 10 минут с последующим добавлением воды (4 л). Двухфазную смесь перемешивали в течение 30 минут, после чего нижний органический слой переносили в экстрактор емкостью 100 л через проходной фильтр. Оставшийся в колбе водный слой промывали дополнительным количеством DCM (2×4 л), и его затем также переносили в экстрактор. Было также перенесено небольшое количество водного слоя (~2 л), и два слоя разделяли. Органический слой возвращали в экстрактор и промывали водой (1×6 л); величина pH составляла 4,5. Органический слой отделяли и загружали в новую колбу через проходной фильтр. Смесь подвергали операции по замене растворителя на 2,2,2-трифторэтанол путем отгонки под вакуумом (конечный объем 34 л) и использовали как есть на следующей стадии. Содержание воды, определенное титрованием по методу Карла Фишера, составляло 1900 ч/млн.
При проведении эксперимента в меньшем масштабе с использованием этой же методики в результате испарения получали твердое вещество, для которого снимали1H ЯМР спектры.1H ЯМР (400 МГц, CDCl3): 6,65 (д, J=8,4 Гц, 1H), 4,37-4,32 (м, 1H), 4,18-4,00 (м, 2H), 4,00-3,89 (м, 1H), 3,87 (д, J=7,7 Гц, 1H), 3,36-3,27 (м, 9H), 2,95-2,79 (м, 2H), 2,75 (д, J=11,4 Гц, 1H), 2,42 (дд, J=15,0, 6,9 Гц, 1H) 2,24-2,11 (м, 2H), 1,96-1,81 (м, 3H), 1,74-1,60 (м, 8H), 1,47 (с, 9H), 1,46 (м, 8H), 1,39 (м, 2H), 1,01 (т, J=7,3, 12H)
Стадия 12: (2S,5R)-7-Оксо-N-пиперидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид

Раствор соли Bu4N+-OSO3 в TFE (34 л) использовали в том же виде, в котором он был получен на предыдущей стадии с предполагаемым выходом 100%. Реакционную смесь охлаждали на ледяной бане, и добавляли HBF4-Et2O (1,57 л) с помощью капельной воронки в течение 11 минут при температуре от 18°C до 22°C. Полученную белую суспензию подвергали перемешиванию в течение ночи (12 часов). Удаляли TFE (~15 л) путем отгонки под вакуумом. Затем добавляли DCM (15 л). В экстрактор емкостью 100 л загружали апирогенную воду (35 л) и NaHCO3 (274 г), и раствор охлаждали до 13°C. В экстрактор при помощи вакуума переносили реакционную смесь с температурой 11-13°C. Реакционную колбу споласкивали дополнительным количеством DCM (5 л), и суспензию также переносили в экстрактор. Реакционную смесь нагревали до 18,5°C и добавляли апирогенную воду (12 л) для растворения всех твердых веществ. Конечная величина pH составляла 4,5. Органический слой отделяли, и водный слой промывали с помощью DCM (2×16 л). Анализ водного слоя показал присутствие в нем 2,38 кг вещества (83,8%).
Водный слой загружали в чистую колбу. Раствор концентрировали путем отгонки под вакуумом и затем путем азеотропной перегонки с IPA. В этот момент1H ЯМР анализ отношения IPA:H2O указывал на присутствие 13,4 л воды и 24,6 л IPA. Добавляли IPA (22 л). Белое кристаллическое белое вещество отфильтровывали, промывали смесью 7:1 IPA:апирогенная вода (16 л) и сушили под вакуумом и в токе азота при комнатной температуре с получением названного продукта в форме кристаллического канального гидрата, 1,5 мас.% воды. (Выход = 1,715 кг, 57,4% суммарно по стадиям 11 и 12.)1H ЯМР (400 МГц, ДМСО-d6): 8,3 (ушир.с, 2H), 8,21 (д, J=7,8 Гц, 1H), 4,01 (с, 1H), 3,97-3,85 (м, 1H), 3,75 (д, J=6,5 Гц, 1H) 3,28 (дд, J=12,9, 2,5 Гц, 2H) 3,05-2,93 (м, 4H), 2,08-1,97 (м, 1H), 1,95-1,79 (м, 3H), 1,75-1,59 (м, 4H).
ПРИМЕР 1D
Кристаллический моногидрат (2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида
Раздел A: Получение
Аморфный (2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (1 г) и деионизованную воду (5 мл) добавляли в стеклянную колбу, и полученную суспензию перемешивали при комнатной температуре до тех пор, пока порошковая рентгенограмма (смотрите часть B) не указывала на то, что полностью произошло превращение в другую форму. Затем кристаллическое вещество собирали путем гравитационного фильтрования и сушили при комнатной температуре.
В качестве варианта, кристаллическая суспензия может быть выделена путем испарительного удаления, которое, в силу того что кристаллический гидрат растворим в воде (около 55 г/мл при комнатной температуре), может приводить к более высокому выходу.
Сушка может вызывать дегидратацию кристаллов, и, поэтому, не должны использоваться обычные методы сушки с использованием вакуума и/или высоких температур. Путем контролирования относительной влажности среды, при которой проводится сушка, можно минимизировать или исключить дегидратацию. Например, для исключения дегидратации, кристаллы могут быть подвергнуты сушке с использованием тока азота при контролировании содержание влаги (например, в интервале значений относительной влажности примерно от 40% до 70%).
Кристаллический гидрат также может быть получен путем суспендирования в смеси изопропилового спирта и воды и использования любого из упомянутых выше методов выделения. Подходящее отношение изопропилового спирта к воде составляет около 7:1 по объему.
Раздел В: Определение характеристик
Порошковую рентгенограмму кристаллического моногидрата, полученного в соответствии с методом, описанном в разделе A, снимали на порошковом рентгеновском дифрактометре Philips Panalytical X'Pert Pro с пультом P W3040/60 при непрерывным сканировании от 4 до 40 градусов 2Θ. В качестве источника использовали медное излучение K-альфа 1 (Kaα1) и K-альфа 2 (Kα2). Эксперимент проводили с образцом при комнатной температуре и при контакте с окружающей средой. Порошковая рентгенограмма показана на фиг.1. Порошковая рентгенограмма включают следующие значения 2Θ и соответствующие d-постоянные:
Кристаллический моногидрат, полученный в соответствии с методом, описанным в разделе A, исследовали с помощью дифференциального сканирующего калориметра TA Instruments DSC Q 1000 (DSC) при скорости нагрева 10°C/минута от 25°C до 350°C в открытом алюминиевом тигле в атмосфере азота. Кривая DSC (см. фиг.2) характеризовалась эндотермом вследствие потери воды в начале появления эндотерма при температуре 22,5°C и изменением энтальпии, равным 186 джоуль/грамм. Разложение наблюдалось при температуре выше 270°C.
Термогравиметрический анализ (TGA) кристаллического моногидрата, полученного в соответствии с методом, описанным в разделе A, проводили на приборе TA Instruments TGA Q 500 в атмосфере азота при скорости нагрева 10°C/минута от 25°C до 300°C. Термогравиметрический анализ показал потерю массы на 4,9 мас.% вплоть до 100°C с последующим разложением выше 270°C. Потеря массы на 4,9 мас.% соответствует потере 1 моля воды на моль соединения, что соответствует моногидрату.
ПРИМЕР 2
(2S,5R)-N-[(4S)-Азепан-4-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид


Стадия 1: Третбутил (4S)-4-({[(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)азепан-1-карбоксилат
К раствору (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (51,7 мг, 0,187 ммоль) в сухом дихлорметане (2 мл) последовательно добавляли раствор третбутил (4S)-4-аминоазепан-1-карбоксилата (69 мг, 0,275 ммоль), триэтиламин (0,090 мл, 0,646 ммоль), HOBT (42,5 мг, 0,278 ммоль), и EDC (54,7 мг, 0,285 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Концентрировали реакционную смесь под вакуумом, и остаток очищали с помощью HPLC на колонке 30×100 мм Waters Sunfire, элюировали с помощью смеси от 15% до 100% CH3CN+0,05% TFA/вода + 0,05% TFA в течение 15 минут с получением после лиофилизации названного соединения в виде белого твердого вещества.
Стадия 2: Третбутил (4S)-4-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)азепан-1-карбоксилат
Палладий на угле (11,8 мг; 10% Pd/C) добавляли к раствору третбутил (4S)-4-({[(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)азепан-1-карбоксилата (49,7 мг, 0,33 ммоль) в метаноле (1,5 мл), и полученную смесь перемешивали в атмосфере водорода (баллон) в течение 3 часов. Анализ с помощью TLC показал, что реакция прошла полностью. Реакционную смесь фильтровали через микрофильтр, и фильтрат концентрировали под вакуумом с получением загрязненного примесями названного соединения в виде белой пены (44,8 мг), которое использовали без очистки на следующей стадии.
Стадия 3: N,N,N-Трибутилбутан-1-аминий{[((2S,5R)-2-{[(4S)-азепан-4-иламино]карбонил}-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил)окси]сульфонил}оксиданид
К раствору загрязненного примесями третбутил (4S)-4-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)азепан-1-карбоксилата (40,2 мг, 0,105 ммоль) в пиридине (1 мл) добавляли комплекс триоксида серы с пиридином (42,1 мг, 0,265 ммоль). Смесь перемешивали при комнатной температуре в атмосфере азота в течение ночи. Анализ с помощью LC/MS показал, что реакция прошла не до конца. Добавляли дополнительное количество пиридина, и затем дополнительное количество комплекса триоксида серы с пиридином (40 мг). Полученную смесь перемешивали при комнатной температуре в течение пяти часов. Реакционную смесь фильтровали, и твердое вещество промывали дихлорметаном. Фильтрат концентрировали под вакуумом и суспендировали в насыщенном водном растворе дигидрофосфата калия. Полученную смесь экстрагировали этилацетатом. Водный слой собирали и добавляли гидросульфат тетрабутиламмония (0,036 мг, 0,105 ммоль). Смесь перемешивали в течение 10 минут, затем экстрагировали с помощью EtOAc (4X). Органические слои объединяли, сушили над сульфатом натрия, и концентрировали под вакуумом с получением названного соединения в виде бесцветного масла.
Стадия 4: (2S,5R)-N-[(4S)-Азепан-4-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору N,N,N-трибутилбутан-1-аминий{[((2S,5R)-2-{[(4S)-азепан-4-иламино]карбонил}-7-оксо-1,6-диазабицикло-[3.2.1]окт-6-ил)окси]сульфонил}оксиданида (30,9 мг, 0,067 ммоль) в безводном дихлорметане (7 мл) при 0°C в атмосфере азота добавляли по каплям трифторуксусную кислоту (0,5 мл, 6,5 ммоль). Реакционную смесь перемешивали в течение двух часов, затем концентрировали под вакуумом. К остатку добавляли эфир, и полученный белый осадок собирали путем центрифугирования. Осадок очищали с помощью HPLC на колонке Phenomenex Synergy Polar-RP 80A и лиофилизировали с получением названного соединения в виде белого твердого вещества. LC-MS (отрицательная ионизация) m/e 361 (M-H); LC-MS (положительная ионизация) m/e 385 (M+Na);1H ЯМР (600 МГц, D2O; без отнесения) (δ, м.д.) 4,17 (1H, ушир.д, J=3 Гц), 3,96-4,03 (2H, м), 3,27-3,38 (3H, м), 3,15-3,22 (2H, м), 3,02 (1H, д, J=12 Гц), 1,62-2,18 (м, 12H).
ПРИМЕР 3
(2S,5R)-N-[(4R)-Азепан-4-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид


Названное соединение можно получить путем замены в методике синтеза примера 2 третбутил (4R)-4-аминоазепан-1-карбоксилат на третбутил (4S)-4-аминоазепан-1-карбоксилат.
ПРИМЕР 4
(2S,5R)-7-Оксо-N-[(3R)пирролидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]-октан-2-карбоксамид


Стадия 1: Третбутил (3R)-3-({[(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пирролидин-1-карбоксилат
К раствору (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (53 мг, 0,192 ммоль) в сухом дихлорметане (2 мл) добавляли последовательно раствор третбутил (3R)-3-аминопирролидин-1-карбоксилата (55 мг, 0,288 ммоль), триэтиламин (0,061 мл, 0,441 ммоль), HOBT (44,1 мг, 0,288 ммоль), и EDC (55,2 мг, 0,288 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение шести часов. Реакционную смесь концентрировали под вакуумом, и остаток очищали с помощью HPLC на колонке 30×100 мм Waters Sunfire, элюируя с помощью смеси от 15% до 100% CH3CN + 0,05% TFA/вода + 0,05% TFA в течение 15 минут с получением названного соединения.
Стадия 2: Третбутил (3R)-3-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пирролидин-1-карбоксилат
Палладий на угле (9,18 мг; 10% Pd/C) добавляли к раствору третбутил (3R)-3-({[(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пирролидин-1-карбоксилат (38 мг, 0,085 ммоль) в метаноле (2 мл), и полученную смесь перемешивали в атмосфере водорода (баллон) в течение 3 часов. Анализ с помощью TLC показал, что реакция прошла полностью. Реакционную смесь фильтровали через микрофильтр, и фильтрат концентрировали под вакуумом с получением загрязненного примесями названного соединения в виде масла.
Стадия 3: N,N-Дибутилбутан-1-аминий({[(2S,5R)-2-({[(3R)-1-(третбутоксикарбонил)пирролидин-3-ил]амино}карбонил)-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]окси}сульфонил)оксиданид
К раствору третбутил (3R)-3-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пирролидин-1-карбоксилата (30 мг, 0,085 ммоль) в пиридине (1 мл) добавляли комплекс триоксида серы с пиридином (53,9 мг, 0,339 ммоль) и 4A молекуклярные сита. Смесь перемешивали при комнатной температуре в атмосфере азота в течение ночи. Анализ с помощью LC/MS показал, что реакция прошла не полностью. Реакционную смесь фильтровали, и фильтрат концентрировали под вакуумом. Остаток подвергали хроматографии на HPLC для извлечения непрореагировавшего исходного реагента, который повторно подвергался взаимодействию при условиях реакции. Объединенный продукт суспендировали в насыщенном водном растворе дигидрофосфата калия. Полученную смесь промывали этилацетатом. Водный слой собирали и добавляли гидросульфат тетрабутиламмония. Смесь перемешивали в течение 10 минут, затем экстрагировали с помощью EtOAc (4X). Органические слои объединяли, сушили над сульфатом натрия, и концентрировали под вакуумом с получением названного соединения в виде бесцветного масла.
Стадия 4: (2S,5R)-7-Оксо-н-[(3R)пирролидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору N,N-дибутилбутан-1-аминий({[(2S,5R)-2-({[(3R)-1-(третбутоксикарбонил)пирролидин-3-ил]амино}карбонил)-7-оксо-1,6-диазабицикло[3.2.1]окт-6-ил]окси}сульфонил)оксиданида (2 мг, 0,046 ммоль) в безводном дихлорметане (0,5 мл) при 0°C в атмосфере азота добавляли по каплям трифторуксусную кислоту (0,525 мл, 0,046 ммоль). Реакционную смесь перемешивали в течение двух часов, затем концентрировали под вакуумом. К остатку добавляли эфир, и полученный белый осадок собирали с помощью центрифугирования. Очищали при помощи HPLC на колонке Phenomenex Synergy Polar-RP 80A и лиофилизировали с получением названного соединения в виде белого твердого вещества. LC-MS (отрицательная ионизация) m/e 333 (M-H); LC-MS (положительная ионизация) m/e 336 (M+H);1H ЯМР (600 МГц, Д2O; без отнесения) (δ, м.д.) 4,50-4,54 (1H, м), 4,20 (1H, дд, J=3,6 Гц), 4,03 (1H, ушир.д, J=7 Гц), 3,54 (1H, дд, J=7,13 Гц), 3,40-3,48 (1H, м), 3,30-3,35 (2H, м), 3,24 (1H, дд, J=5,13 Гц)} 3,07 (1H, д, J=12 Гц), 2,31-2,37 (1H, м), 2,15-2,20 (1H, м), 2,00-2,10 (2H, м), 1,88-1,98 (1H, м), 1,76-1,84 (1H, м).
ПРИМЕР 5
(2S,5R)-N-Азокан-5-ил-7-оксо-6-(сульфоокси)-1,6-диазабицикло-[3.2.1]октан-2-карбоксамид


Названное соединение можно получить путем замены в методике синтеза примера 1 третбутил 5-аминоазокан-1-карбоксилата на 4-амино-1-BOC-пиперидин.
ПРИМЕР 6
(2S,5R)-7-Оксо-N-пиридин-4-ил-6-(сульфоокси)-1,6-диазабицикло-[3.2.1]октан-2-карбоксамид
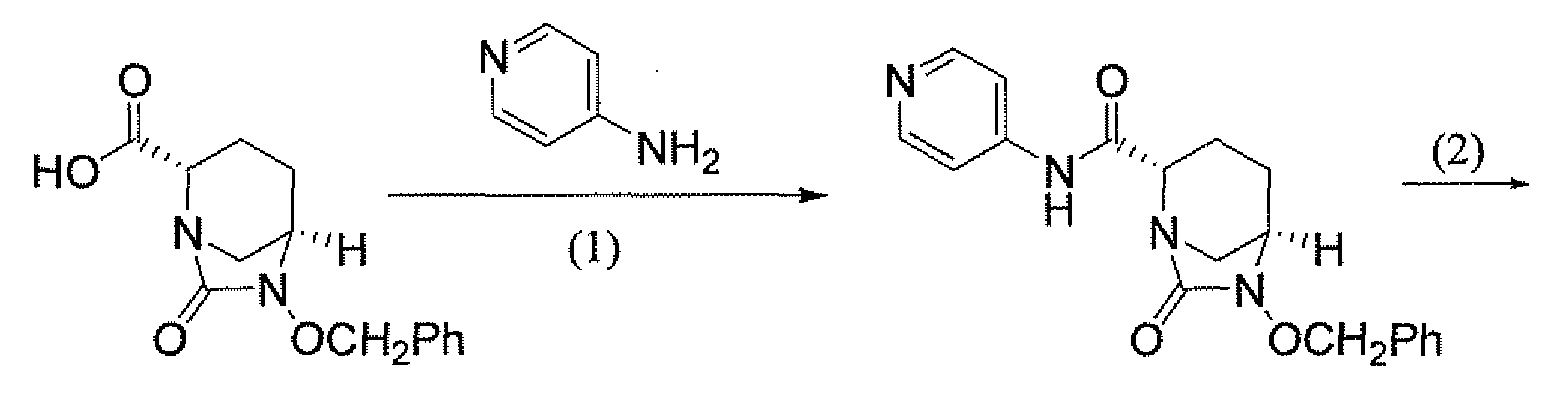

Стадия 1: (2S,5R)-6-(Бензилокси)-7-оксо-N-пиридин-4-ил-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (51,5 мг, 0,186 ммоль) в сухом дихлорметане (5 мл) последовательно добавляли триэтиламин (0,065 мл, 0,466 ммоль), иодид 2-хлор-1-метилпиридиния (63,4 мг, 0,248 ммоль), и 4-аминопиридин (19,2 мг, 0,204 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь затем нагревали до 50°C в течение 1,5 часов. Анализ с помощью LC/MS показал, что реакция прошла полностью. Реакционную смесь концентрировали и очищали с помощью HPLC на колонке 30×100 мм Waters Sunfire с получением после лиофилизации названного соединения в виде оранжевого твердого вещества.
Стадия 2: (2S,5R)-6-Гидрокси-7-оксо-N-пиридин-4-ил-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
Палладий на угле (13,2 мг; 10% Pd/C) добавляли к раствору (2S,5R)-6-(бензилокси)-7-оксо-N-пиридин-4-ил-1,6-диазабицикло-[3.2.1]октан-2-карбоксамида (52,7 мг, 0,15 ммоль; объединенный продукт двух экспериментов) в метаноле (1,5 мл), и полученную смесь перемешивали в атмосфере водорода (баллон) в течение 5 часов. Анализ с помощью TLC и HPLC показал присутствие небольшого количества непрореагировавшего исходного реагента. Добавляли дополнительное количество катализатора (5,6 мг), и полученную смесь перемешивали в атмосфере водорода (баллон) в течение еще 1 часа. Реакционную смесь фильтровали через микрофильтр, и фильтрат концентрировали под вакуумом с получением названного соединения в виде бесцветного масла.
Стадия 3: (2S,5R)-7-Оксо-N-пиридин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору (2S,5R)-6-гидрокси-7-оксо-N-пиридин-4-ил-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (9,2 мг, 0,035 ммоль) в сухом пиридине (0,5 мл) добавляли осушенные 4A молекулярные сита и комплекс триоксида серы с пиридином (22 мг, 0,138 ммоль) при комнатной температуре в атмосфере азота. Смесь перемешивали в течение четырех часов. Реакционную смесь фильтровали, и твердое вещество промывали дихлорметаном, ацетонитрилом, и метанолом. Фильтрат концентрировали под вакуумом, и остаток растирали с этилацетатом. Остаток сушили под вакуумом, растворяли в насыщенном водном растворе дигидрофосфата натрия и очищали с помощью HPLC на колонке Phenomenex Synergi Polar-RP 80A с получением после лиофилизации названного соединения в виде белого твердого вещества. LC-MS (отрицательная ионизация) m/e 341 (M-H); LC-MS (положительная ионизация) m/e 343 (M+H);1H ЯМР (600 МГц, D2O; неотнесенный) (δ, м.д.) 8,57 (2H, ушир.с), 8,15 (2H, ушир.с), 4,27 (1H, ушир.д, J=7 Гц), 4,20 (1H, ушир.с), 3,33 (1H, д, J=12 Гц), 3,10 (1H, д, J=12 Гц), 2,28-2,32 (1H, м), 2,08-2,11 (1H, м), 1,93-1,98 (1H, м), 1,83-1,88 (1H, м).
ПРИМЕР 7
(2S,5R)-N-(2-Метоксипиридин-4-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
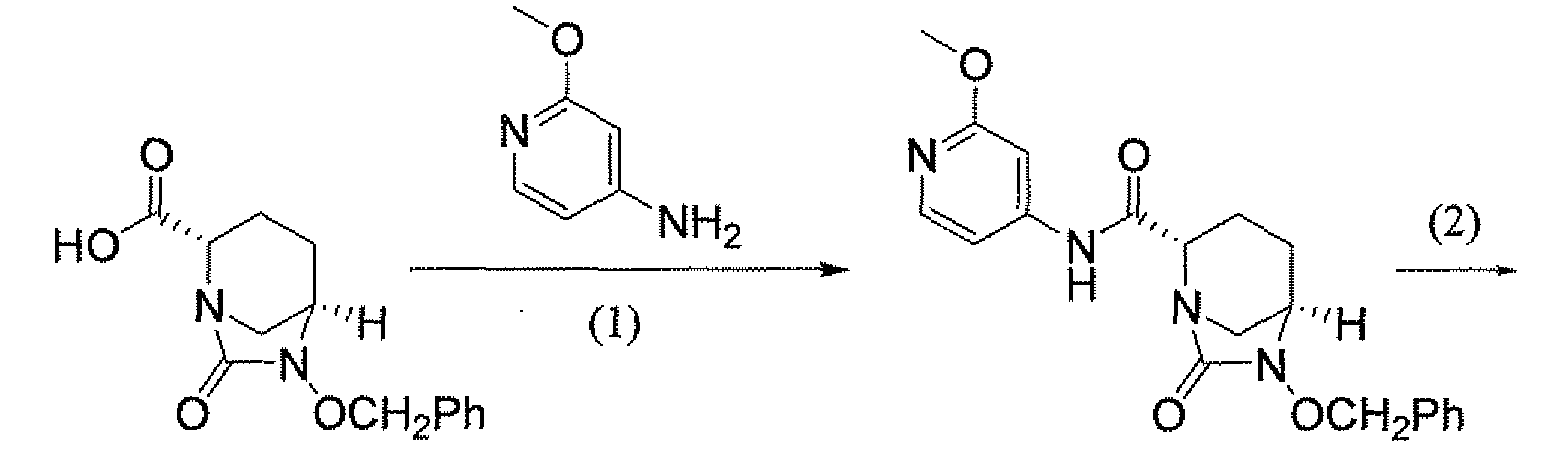

Стадия 1: (2S,5R)-6-(Бензилокси)-N-(2-метоксипиридин-4-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (38,9 мг, 0,141 ммоль) в сухом дихлорметане (2 мл) последовательно добавляли триэтиламин (0,049 мл, 0,352 ммоль), иодид 2-хлор-1-метилпиридиния (53,3 мг, 0,209 ммоль), и 2-метокси-4-аминопиридин (20,2 мг, 0,163 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь затем нагревали до 50°C в течение 1,5 часов. Анализ с помощью LC/MS показал, что реакция прошла полностью. Реакционную смесь концентрировали и очищали с помощью HPLC на колонке 30×100 мм Waters Sunfire с получением после лиофилизации названного соединения в виде оранжевого твердого вещества.
Стадия 2: (2S,5R)-6-Гидрокси-N-(2-метоксипиридин-4-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
Палладий на угле (13,4 мг; 10% Pd/C) добавляли к раствору (2S,5R)-6-(бензилокси)-N-(2-метоксипиридин-4-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (43,6 мг, 0,114 ммоль) в метаноле (1 мл), и полученную смесь перемешивали в атмосфере водорода (баллон) в течение ночи. Анализ с помощью HPLC показал, что реакция прошла полностью. Реакционную смесь фильтровали через микрофильтр, и фильтрат концентрировали под вакуумом с получением названного соединения в виде загрязненного примесями бесцветного масла.
Стадия 3: (2S,5R)-N-(2-Метоксипиридин-4-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору (2S,5R)-6-гидрокси-N-(2-метоксипиридин-4-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (33 мг, 0,114 ммоль) в сухом пиридине (1 мл) добавляли комплекс триоксида серы с пиридином (111 мг, 0,696 ммоль) при комнатной температуре в атмосфере азота. Смесь перемешивали в течение четырех часов. Реакционную смесь фильтровали, и твердое вещество промывали дихлорметаном, ацетонитрилом, и метанолом. Фильтрат концентрировали под вакуумом, и остаток растирали с этилацетатом, затем сушили под вакуумом, растворяли в насыщенном растворе дигидрофосфата натрия и очищали с помощью HPLC на колонке Phenomenex Synergi Polar-RP 80A с получением белового твердого вещества, которое дополнительно очищали на колонке Waters Sunfire с получением белого твердого вещества, которое дополнительно очищали с помощью HPLC на колонке Phenomenex Synergi Polar-RP 80A с получением после лиофилизации названного соединения в виде белого твердого вещества. LC-MS (отрицательная ионизация) m/e 371 (M-H); LC-MS (положительная ионизация) m/e 373 (M+H);1H ЯМР (600 МГц, D2O; без отнесения) (δ, м.д.) 8,10 (1H, ушир.д, J=6 Гц), 7,59 (1H, с), 7,41 (1H, д, J=6 Гц), 4,26 (1H, ушир.д, J=7 Гц), 4,23 (1H, ушир.с), 4,07 (3H, с), 3,36 (1H, д, J=12 Гц), 3,12 (1H, д, J=12 Гц), 2,29-2,33 (1H, м), 2,10-2,14 (1H, м), 1,93-1,99 (1H, м), 1,84-1,90 (1H, м).
ПРИМЕР 8
(2S,5R)-N-[2-(Диметиламино)пиридин-4-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид


Стадия 1: (2S,5R)-6-(Бензилокси)-N-[2-(диметиламино)пиридин-4-ил]-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (37,3 мг, 0,135 ммоль) в сухом дихлорметане (2,5 мл) последовательно добавляли триэтиламин (0,047 мл, 0,338 мммоль), иодид 2-хлор-1-метилпиридиния (38,3 мг, 0,15 ммоль), и 2-диметиламино-4-аминопиридин (21,7 мг, 0,158 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь затем нагревали до 50°C в течение 1,5 часов. Анализ с помощью LC/MS показал, что реакция прошла полностью. Реакционную смесь концентрировали и очищали с помощью HPLC на колонке 30×100 мм Waters Sunfire с получением после лиофилизации названного соединения в виде кремового твердого вещества.
Стадия 2: (2S,5R)-N-[2-(Диметиламино)пиридин-4-ил]-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
Палладий на угле (10,4 мг; 10% Pd/C) добавляли к раствору (2S,5R)-6-(бензилокси)-N-[2-(диметиламино)пиридин-4-ил]-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (47,8 мг, 0,121 ммоль) в метаноле (2 мл), и полученную смесь перемешивали в атмосфере водорода (баллон) в течение ночи. Анализ с помощью HPLC показал, что реакция прошла полностью. Реакционную смесь фильтровали через микрофильтр, и фильтрат концентрировали под вакуумом с получением названного соединения в виде бесцветного масла, которое использовали на следующей стадии без дополнительной очистки.
Стадия 3: (2S,5R)-N-[2-(Диметиламино)пиридин-4-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору (2S,5R)-N-[2-(диметиламино)пиридин-4-ил]-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (37 мг, 0,121 ммоль) в сухом пиридине (1,5 мл) добавляли комплекс триоксида серы с пиридином (92 мг, 0,578 ммоль) при комнатной температуре в атмосфере азота. Смесь перемешивали в течение семи часов. Анализ аликвоты с помощью ЯМР показал, что реакция прошла неполностью. Добавляли дополнительное количество пиридина (2 мл) и комплекс триоксида серы с пиридином (60 мг), и полученную смесь перемешивали при комнатной температуре в атмосфере азота. Реакционную смесь концентрировали под вакуумом, и остаток очищали с помощью HPLC на колонке Phenomenex Synergi Polar-RP 80A с получением загрязненного примесями продукта, который дополнительно очищали на колонке Waters Sunfire с получением после лиофилизации названного соединения в виде белого твердого вещества. LC-MS (отрицательная ионизация) m/e 384 (M-H); LC-MS (положительная ионизация) m/e 386 (M+H);1H ЯМР (600 МГц, D2O; unreferenced) (δ, м.д.) 7,77 (1H, ушир.д, J=6 Гц), 7,41 (1H, с), 6,94 (1H, д, J=6 Гц), 4,23 (2H, ушир.с), 3,36 (1H, д, J=12 Гц), 3,18 (6H, с), 3,11 (1H, д, J=12 Гц), 2,29-2,32 (1H, м), 2,10-2,14 (1H, м), 1,93-1,99 (1H, м), 1,84-1,90 (1H, м).
ПРИМЕР 9
(2S,5R)-N-[4-(Аминометил)фенил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид


Стадия 1: Третбутил [4-({[(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)бензил]карбамат
К раствору (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (29,9 мг, 0,108 ммоль) в сухом дихлорметане (3 мл) последовательно добавляли триэтиламин (0,038 мл, 0,271 ммоль), иодид 2-хлор-1-метилпиридиния (41,0 мг, 0,160 ммоль), и 4-(N-BOC-аминометил)анилин (30,6 мг, 0,138 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь затем нагревали до 60°C в течение 2 часов. Анализ с помощью LC/MS показал отсутствие непрореагировавшего исходного материала. Реакционную смесь концентрировали и очищали с помощью HPLC на колонке Waters Sunfire с получением названного соединения.
Стадия 2: Третбутил [4-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил)карбонил}амино)бензил] карбамат
Палладий на угле (7,8 мг; 10% Pd/C) добавляли к раствору продукта со стадии 1 (35 мг, 0,073 ммоль) в метаноле (2 мл), и полученную смесь перемешивали в атмосфере водорода (баллон) в течение ночи. Анализ с помощью LC-MS показал, что реакция прошла полностью. Реакционную смесь фильтровали через микрофильтр, и фильтрат концентрировали под вакуумом, который отгонялся из азеотропа с толуолом, с получением названного соединения в виде белого твердого вещества.
Стадия 3: Третбутил [4-({[(2S,5R)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)бензил]карбамат
К раствору продукта со стадии 2 (28,9 мг, 0,074 ммоль) в сухом пиридине (1 мл) добавляли комплекс триоксида серы с пиридином (60,3 мг, 0,379 ммоль) при комнатной температуре в атмосфере азота. Смесь перемешивали при комнатной температуре в течение ночи. Анализ с помощью LC-MS показал, что реакция прошла примерно на 50%. В реакционную смесь добавляли дополнительное количество комплекса триоксида серы с пиридином (64,4 мг) и продолжали перемешивание при комнатной температуре. Через 7 часов анализ с помощью LC-MS показал, что в реакционной смеси присутствует преимущественно продукт. Реакционную смесь фильтровали, и нерастворимое твердое вещество хорошо промывали дихлорметаном. Фильтрат концентрировали под вакуумом, и остаток подвергали отгонке в виде азеотропа с толуолом для удаления избытка пиридина. Полученное таким образом неочищенное названное соединение использовали без дополнительной очистки в следующей реакции.
Стадия 4: (2S,5R)-N-[4-(аминометил)фенил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К смеси продукта со стадии 3 (34,8 мг, 0,074 ммоль) в сухом дихлорметане (3 мл) при 0°C в атмосфере азота добавляли трифторуксусную кислоту (1,0 мл, 13 моль). При добавлении трифторуксусной кислоты все твердое вещество мгновенно растворялось, раствор перемешивали в течение 1,5 часов, в результате чего анализ с помощью LC-MS показал, что реакция прошла полностью. Реакционную смесь концентрировали под вакуумом, и остаток растирали с эфиром для удаления избытка трифторуксусной кислоты и органических примесей. Полученное клейкое твердое вещество сушили под вакуумом и хранили в морозильнике в течение ночи. Неочищенный продукт очищали с помощью HPLC на колонке Phenomenex Synergi Polar-RP 80A с получением после лиофилизации загрязненного примесями названного соединения в виде белого твердого вещества. Твердое вещество растирали с ацетонитрилом (3X) с получением чистого названного соединения в виде белого твердого вещества. LC-MS (отрицательная ионизация) m/e 369 (M-H); LC-MS (положительная ионизация) m/e 354 (M+H-NH3);1H ЯМР (600 МГц, D2O; без отнесения) (δ, м.д.) 7,53 (2H, д, J=8,5 Гц), 7,46 (2H, д, J=8,5 Гц), 4,23 (2H, ушир.с), 4,11 (2H, с), 3,39 (1H, д, J=12 Гц), 3,18 (1H, д, J=12 Гц), 2,27-2,31 (1H, м), 2,09-2,14 (1H, м), 1,90-2,00 (1H, м), 1,813-1,89 (1H, м).
ПРИМЕР 10
(2S,5R)-7-оксо-2-[(пиперидин-4-иламино)карбонил]-1,6-диазабицикло[3.2.1 ]октан-6-сульфоновая кислота

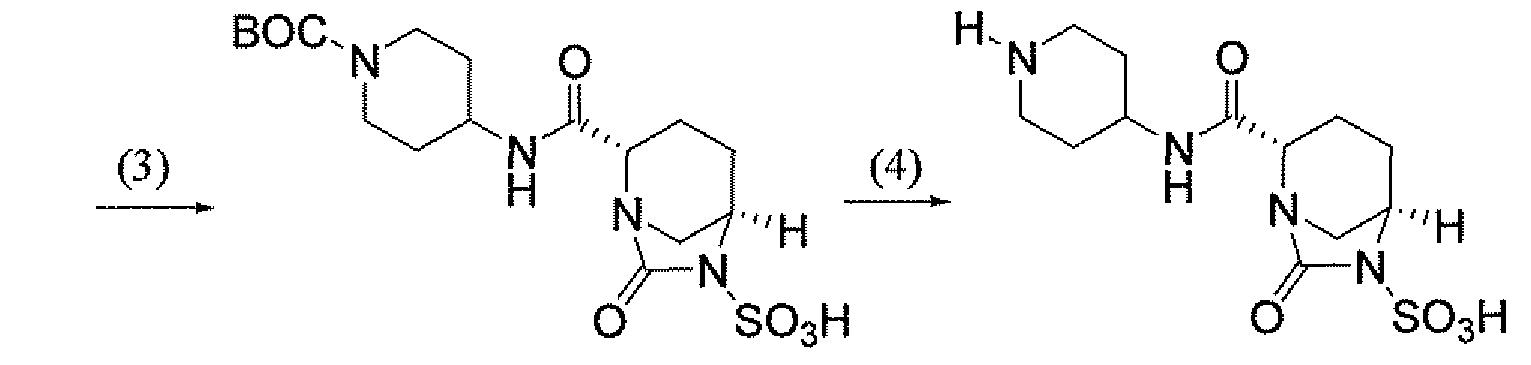
Стадия 1: Третбутил 4-[({(2S,5R)-7-оксо-6-[(феноксикарбонотиоил)окси]-1,6-диазабицикло[3.2.1]окт-2-ил}карбонил)амино]пиперидин-1-карбоксилат
Раствор фенилхлортионокарбоната (1,25 экв.) в дихлорметане добавляли к раствору пиридина (1,25 экв.), 4-диметиламинопиридина (0,1 экв.) и третбутил 4-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)
пиперидин-1-карбоксилата (см. пример 1, стадия 2) в дихлорметане. Полученную смесь перемешивали при комнатной температуре в течение ночи, затем охлаждали на ледяной бане, и реакцию останавливали путем добавления воды. Слои разделяли, и водный слой экстрагировали дихлорметаном. Объединенные органические слои сушили над сульфатом натрия, фильтровали, и концентрировали под вакуумом. Остаток очищали с помощью хроматографии на силикагеле с получением названного соединения.
Стадия 2: Третбутил 4-({[(2S,5R)-7-оксо-1,6-диазабицикло-[3.2.1]окт-2-ил]карбонил}амино)пиперидин-1-карбоксилат
AIBN (0,1 экв.) добавляли к раствору третбутил 4-[({(2S,5R)-7-оксо-6-[(феноксикарбонотиоил)окси]-1,6-диазабицикло[3.2.1]окт-2-ил}карбонил)амино]пиперидин-1-карбоксилата в сухом бензоле, и полученную смесь нагревали до кипения с обратным холодильником. Добавляли раствор гидрида трибутилолова (1,25 экв.) в бензоле в течение одного часа, и полученную смесь кипятили с обратным холодильником в течение еще 3 часов. Реакционную смесь концентрировали под вакуумом, и остаток очищали с помощью хроматографии на силикагеле с получением названного соединения.
Стадия 3: (2S,5R)-2-({[1-(Третбутоксикарбонил)пиперидин-4-ил]амино}карбонил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-сульфоновая кислота
Продукт стадии 2 подвергали сульфатации в соответствии с методикой стадии 3 примера 1 с получением названного соединения.
Стадия 4: (2S,5R)-7-оксо-2-[(пиперидин-4-иламино)карбонил]-1,6-диазабицикло[3.2.1]октан-6-сульфоновая кислота
Снимали защиту с (2S,5R)-2-({[1-(третбутоксикарбонил)пиперидин-4-ил]амино}карбонил)-7-оксо-1,6-диазабицикло[3.2.1]-октан-6-сульфоновой кислоты в соответствии с методикой стадии 4 примера 1 с получением названного соединения.
ПРИМЕР 11
(4R,6S)-2-Оксо-N-пиперидин-4-ил-3-(сульфоокси)-1,3-диазабицикло[2.2.1]гептан-6-карбоксамид

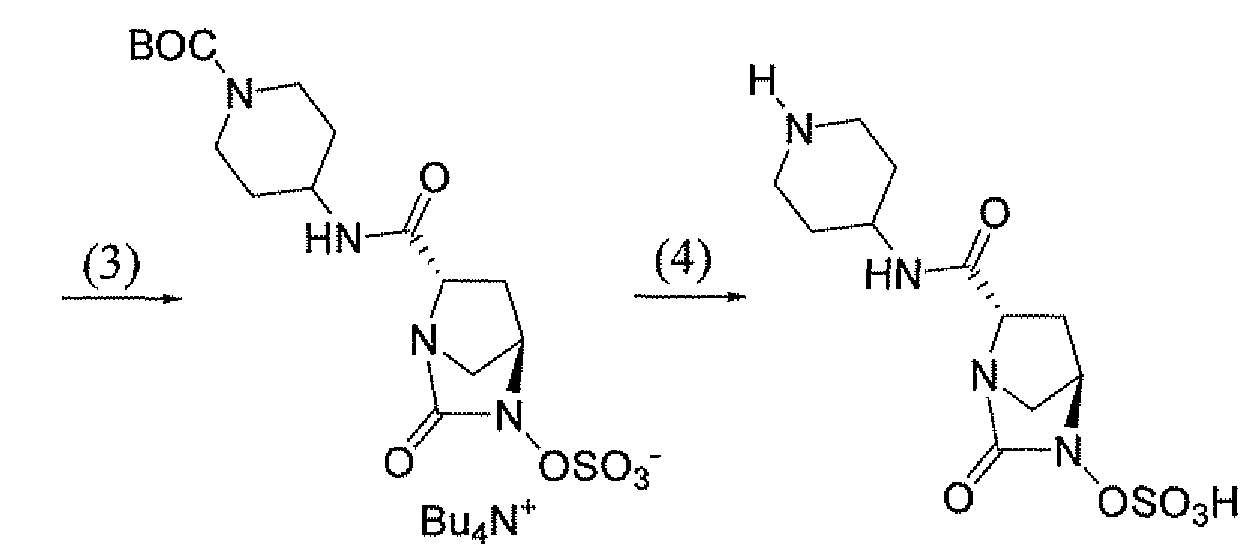
Названное соединение можно получить путем замены в методике примера 1 (4R,6S)-3-(бензилокси)-2-оксо-1,3-диазабицикло[2.2.1]-гептан-6-карбоновой кислоты на (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновую кислоту.
ПРИМЕР 12
(4R,6S)-2-Оксо-N-[(4S)-азепан-4-ил]-3-(сульфоокси)-1,3-диазабицикло[2.2.1 ]гептан-6-карбоксамид


Названное соединение можно получить путем замены в методике примера 2 (4R,6S)-3-(бензилокси)-2-оксо-1,3-диазабицикло-[2.2.1]гептан-6-карбоновой кислоты на (2S,5R)-6-(фенилметокси)- 7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновую кислоту.
ПРИМЕР 13
(4R,6S)-2-Оксо-N-пиридин-4-ил-3-(сульфоокси)-1,3-диазабицикло[2.2.1]гептан-6-карбоксамид


Названное соединение можно получить путем замены в методике примера 6 (4R,6S)-3-(бензилокси)-2-оксо-1,3-диазабицикло-[2.2.1]гептан-6-карбоновой кислоты на (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновую кислоту.
ПРИМЕР 14
(2S,5R)-7-Оксо-N-[(3S)пирролидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид

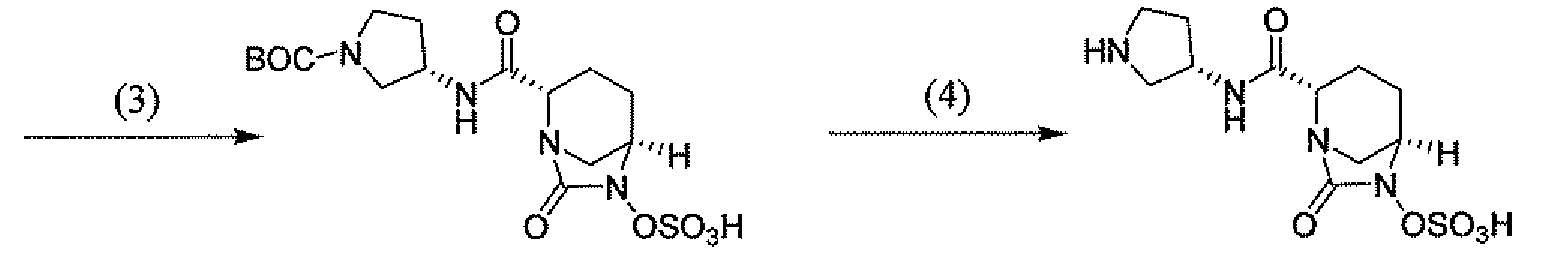
Стадия 1: Третбутил (3S)-3-({[(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пирролидин-1-карбоксилат
К раствору (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (1 г, 3,62 ммоль) в сухом дихлорметане (30 мл) последовательно добавляли диметиламинопиридин (884 мг, 7,24 ммоль), EDC (1,388 г, 7,24 ммоль), и третбутил (3S)-3-аминопирролидин-1-карбоксилат (742 мг, 3,98 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение недели. Реакционную смесь затем концентрировали под вакуумом, и остаток очищали с помощью HPLC на колонке 30 × 100 мм Waters Sunfire, элюировали с помощью смеси 15% до 100% CH3CN + 0,05% TFA/вода + 0,05% TFA в течение 15 минут с получением названного соединения в виде белого твердого вещества.
Стадия 2: Третбутил (3S)-3-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пирролидин-1-карбоксилат
Палладий на угле (335 мг; 10% Pd/C) добавляли к раствору продукта со стадии 1 (1,4 г, 3,15 ммоль) в метаноле (30 мл), и полученную смесь перемешивали в атмосфере водорода (баллон) в течение 1 часа. Анализ с помощью LC-MS показал, что реакция прошла полностью. Реакционную смесь фильтровали, и фильтрат концентрировали под вакуумом с получением названного соединения в виде масла, которое использовали без очистки на следующей стадии.
Стадия 3: Третбутил 3-({[(2S,5R)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пирролидин-1-карбоксилат
К раствору продукта со стадии 2 (1,11 г, 3,15 ммоль, теоретический выход стадии 2) в пиридине (10 мл) добавляли комплекс триоксида серы с пиридином (2,51 г, 15,75 ммоль). Смесь перемешивали при комнатной температуре в атмосфере азота в течение ночи. Добавляли дихлорметан, и смесь фильтровали. Собранное твердое вещество промывали дихлорметаном (4X), и объединенные фильтраты концентрировали под вакуумом. Остаток использовали без очистки на следующей стадии.
Стадия 4: (2S,5R)-7-оксо-N-[(3S)пирролидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору продукта со стадии 3 (1,37 г, 3,15 ммоль, теоретический выход стадии 3) в безводном дихлорметане (5 мл) при 0°C в атмосфере азота добавляли по каплям TFA (2 мл, 26 ммоль). Реакционную смесь перемешивали в течение двух часов, затем концентрировали под вакуумом. К остатку добавляли эфир, и полученный белый осадок собирали с помощью центрифугирования (растирание с эфиром повторяли еще два раза). Полученное твердое вещество очищали с помощью HPLC на колонке Phenomenex Synergy Polar-RP 80A, элюировали смесью метанол/вода и лиофилизировали с получением названного соединения в виде белого твердого вещества. LC-MS (режим отрицательной ионизации) m/e 333 (M-H). LC-MS (положительная ионизация) m/e 335 (M+H), 357 (M+Na);1H ЯМР (600 МГц, D2O; без отнесения) (δ, м.д.) 4,51 (1H, м), 4,16 (1H, ушир.д, J=2,6 Гц), 3,99 (1H, д, J=7 Гц), 3,54 (1H, дд, J=7,13 Гц), 3,40-3,50 (1H, м), 3,30-3,40 (1H, м), 3,20-3,30 (2H, м), 3,02 (1H, д, J=12 Гц), 2,30-2,40 (1H, м), 2,10-2,20 (1H, м), 2,00-2,10 (2H, м), 1,83-1,93 (1H, м), 1,72-1,80 (1H, м).
ПРИМЕР 15
(2S,5R)-N-[(3R,4S)-3-Фторпиперидин-4-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид


Стадия 1: Третбутил-(3R,4S)-4-({[(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)-3-фторпиперидин-1-карбоксилат
К раствору (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (2,108 г, 7,63 ммоль) в безводном диметилформамиде (15 мл) добавляли BOP (4,05 г, 9,15 ммоль), и полученную смесь перемешивали при комнатной температуре в атмосфере азота в течение 5 минут. Затем добавляли диизопропилэтиламин (2,66 мл, 15,26 ммоль), и затем раствор третбутил (3R,4S)-4-амино-3-фторпиперидин-1-карбоксилата (1,665 г, 7,63 ммоль) в 20 мл дихлорметана. Полученный раствор перемешивали при комнатной температуре в атмосфере азота в течение 2 часов, затем концентрировали под вакуумом, и остаток распределяли между этилацетатом и водой. Водный слой промывали дважды этилацетатом. Органический слой промывали рассолом, сушили над сульфатом натрия, и концентрировали под вакуумом. Остаток очищали с помощью флэш-хроматографии на силикагеле (установка Isco Combiflash - 120 г силикагеля, 80 мл/мин, 254 нм, от 0% до 100% EtOAc/гексан на 6 объемах колонки, затем 100% EtOAc на 9 объемах колонки; названное соединение элюировали с помощью 100% EtOAc). Собирали фракции, содержащие чистое названное соединение, и концентрировали их под вакуумом с получением коричневого твердого вещества. Фракции, содержащие загрязненный примесями продукт, также собирали и повторно подвергали очистке с помощью HPLC (колонка 30×100 мм Sunfire, 5 микрон, 35 мл/мин, от 10% до 100% CH3CN + 0,1% TFA/вода + 0,1% TFA в течение 15 минут; названное соединение элюировали при 70% CH3CN + 0,1% TFA). Фракции, содержащие чистый продукт, объединяли и концентрировали под вакуумом. Полученный водный остаток затем экстрагировали этилацетатом. Органический слой собирали и сушили над сульфатом магния. Концентрирование под вакуумом давало белое твердое вещество, которое объединяли с материалом, выделенное с помощью хроматографии на силикагеле, с получением названного соединения.
Стадия 2: Третбутил-(3R,4S)-4-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)-3-фторпиперидин-1-карбоксилат
К раствору продукта со стадии 1 (3,0175 г, 6,33 ммоль) в метаноле (80 мл) и этилацетате (20 мл) добавляли 10% палладий на угле (0,73 г, 6,86 ммоль), и реакционную смесь перемешивали в атмосфере водорода (баллон) в течение ночи. Анализ с помощью LC/MS показал, что реакция прошла полностью. Реакционную смесь фильтровали через микрофильтр, и собранное твердое вещество хорошо промывали метанолом. Фильтрат концентрировали под вакуумом и подвергали отгонке в виде азеотропа с толуолом с получением названного соединения в виде желтой пены, которое использовали непосредственно на следующей стадии без очистки.
Стадия 3: Третбутил (3R,4S)-4-({[(2S,5R)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)-3-фторпиперидин-1-карбоксилат
К раствору продукта со стадии 2 (2,59 г, 6,7 ммоль, теоретический выход стадии 2) в пиридине (30 мл) добавляли комплекс триоксида серы с пиридином (5,40 г, 34 ммоль). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 3 часов, затем добавляли дополнительное количество комплекса триоксида серы с пиридином (5,40 г, 34 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли дихлорметан, и смесь фильтровали. Собранное твердое вещество тщательно промывали дихлорметаном, и объединенные фильтраты концентрировали под вакуумом с получением неочищенного названного соединения. Остаток использовали без очистки на следующей стадии.
Стадия 4: (2S,5R)-N-[(3R,4S)-3-фторпиперидин-4-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору продукта со стадии 3 (3,13 г, 6,7 ммоль, теоретический выход стадии 3) в безводном дихлорметане (50 мл) при 0°C в атмосфере азота добавляли по каплям трифторуксусную кислоту (10 мл, 130 ммоль). Реакционную смесь подогревали до комнатной температуры, затем перемешивали в течение двух часов. Добавляли дополнительное количество трифторуксусной кислоты (6 мл, 78 моль), и реакционную смесь перемешивали при комнатной температуре еще в течение 3 часов, затем концентрировали под вакуумом. К остатку добавляли эфир, и полученный белый осадок собирали с помощью центрифугирования (растирание с эфиром повторяли еще два раза). Полученное твердое вещество очищали с помощью HPLC на колонке Phenomenex Synergy Polar-RP 80A, элюировали смесью метанол/вода и лиофилизировали с получением названного соединения в виде кремового твердого вещества, которое по данным ЯМР содержало ~6% пиридина. Этот загрязненный примесями продукт растирали и дважды диспергировали с помощью ультразвука с ацетонитрилом (твердое вещество выделяли центрифугированием) с получением названного соединения в виде белого твердого вещества. LC-MS (режим отрицательной ионизации) m/e 365 (M-H).
ПРИМЕР 16
(2S,5R)-7-Оксо-6-(сульфоокси)-N-(1,2,3,4-тетрагидроизохинолин-6-ил)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид


Стадия 1: Третбутил-6-({[(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)-3,4-дигидроизохинолин-2(1H)-карбоксилат
К раствору (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (53,3 мг, 0,193 ммоль) в сухом дихлорметане (2 мл) последовательно добавляли триэтиламин (0,067 мл, 0,482 ммоль), иодид 2-хлор-1-метилпиридиния (58,7 мг, 0,230 ммоль), и 6-амино-2-N-BOC-1,2,3,4-тетрагидроизохинолин (54,8 мг, 0,221 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь затем нагревали до 50°C в течение 45 минут, затем реакционный продукт концентрировали под вакуумом. Попытки растворить реакционный продукт в элюэнте (2:1:1 CH3CN/ДМСО/вода) для HPLC были неудачными, поэтому его распределяли между водным слоем и дихлорметаном. Органический слой собирали, сушили над сульфатом натрия, концентрировали под вакуумом и откладывали для отдельной очистки. Водный слой также собирали и очищали с помощью HPLC (колонка 30×100 мм Waters Sunfire; 5 микрон; 35 мл/мин; 210 нм; от 15% до 100% CH3CN + 0,05% TFA/вода + 0,05% TFA в течение 15 минут; названное соединение элюировали при 80% CH3CN + 0,05% TFA/вода + 0,05% TFA). Фракции, содержащие названное соединение, лиофилизировали в течение ночи с получением названного соединения в виде белого клейкого твердого вещества. Органический слой с операции распределения неочищенного продукта очищали с помощью препаративной TLC (пластинка 1000 микрон силикагель при элюировании смесью 50% этилацетат/гексан) с получением названного соединения. Обе партии названного соединения объединяли и использовали без дополнительной очистки на следующей стадии.
Стадия 2: Третбутил-6-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)-3,4-дигидроизохинолин-2(1H)-карбоксилат
К раствору продукта со стадии (79,6 мг, 0,157 ммоль) в метаноле (4 мл) и этилацетате (2 мл) добавляли 10% палладий на угле (18 мг), и реакционную смесь перемешивали в атмосфере водорода (баллон) в течение ночи. Анализ с помощью LC/MS показал, что реакция прошла неполностью, поэтому добавляли дополнительное количество 10% палладия на угле (10 мг), и реакционную смесь перемешивали в атмосфере водорода (баллон) в течение еще 6 часов. Реакционную смесь фильтровали через микрофильтр, и собранное твердое вещество хорошо промывали метанолом. Фильтрат концентрировали под вакуумом и отгоняли в виде азеотропа с толуолом с получением названного соединения в виде светло-коричневого масла, которое использовали непосредственно на следующей стадии без очистки.
Стадия 3: Третбутил-6-({[(2S,5R)-7-оксо-6-сульфоокси-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)-3,4-дигидроизохинолин-2(1H)-карбоксилат
К раствору продукта со стадии 2 (64,6 мг, 0,155 ммоль) в пиридине (1,5 мл) добавляли комплекс триоксида серы с пиридином (129,5 мг, 0,814 ммоль). Смесь перемешивали при комнатной температуре в атмосфере азота в течение недели. Затем добавляли дихлорметан, и смесь фильтровали. Собранное твердое вещество тщательно промывали дихлорметаном, и объединенные фильтраты концентрировали под вакуумом с получением неочищенного названного соединения. Остаток использовали без очистки на следующей стадии.
Стадия 4: (2S,5R)-7-оксо-6-(сульфоокси)-N-(1,2,3,4-тетра-гидроизохинолин-6-ил)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору продукта со стадии 3 (77 мг, 0,155 ммоль, теоретический выход стадии 3) в безводном дихлорметане (3 мл) при 0°C в атмосфере азота добавляли по каплям трифторуксусную кислоту (1 мл, 13 ммоль). Реакционную смесь перемешивали в течение одного часа, затем концентрировали под вакуумом. К остатку добавляли эфир, и полученный белый осадок собирали с помощью центрифугирования (растирание с эфиром повторяли еще два раза). Полученное твердое вещество очищали с помощью HPLC на колонке Phenomenex Synergy Polar-RP 80A, элюировали смесью метанол/вода и лиофилизировали с получением названного соединения в виде белого твердого вещества. LC-MS (режим отрицательной ионизации) m/e 395 (M-H).
ПРИМЕР 17
(2S,5R)-7-Оксо-N-(5-пиперидин-4-илпиридин-2-ил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид



Стадия 1: (2S,5R)-6-(бензилокси)-N-(5-бромпиридин-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (0,400 г, 1,448 ммоль) в сухом дихлорметане (17,66 мл) последовательно добавляли триэтиламин (0,504 мл, 3,62 ммоль), иодид 2-хлор-1-метилпиридиния (0,433 г, 1,694 ммоль), и 2-амино-5-бромпиридин (0,311 г, 1,795 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь нагревали до 50°C в течение 1 часа, затем очищали на установке HPLC, связанной с масс-спектрометром, (колонка 30×100 мм Waters Sunfire; 5 микрон; 50 мл/мин; ацетонитрил/вода с 0,1% TFA в течение 15 минут). Фракции, содержащие названное соединение, концентрировали под вакуумом, затем лиофилизировали в течение ночи с получением названного соединения в виде желтого твердого вещества.
Стадия 2: 6-({[(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)-3',6'-дигидро-3,4'-би-пиридин-1'(2'Η)-карбоксилат
Третбутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2H)-карбоксилат (215 мг, 0,696 ммоль) добавляли к продукту со стадии 1 (150 мг, 0,348 ммоль) в реакционную колбу, затем добавляли дихлорид бис(трифенилфосфин)-палладия(II) (24 мг, 0,035 ммоль), затем 1 M водный раствор карбоната натрия (0,869 мл, 0,869 ммоль) и ацетонитрил (0,899 мл). Реакционную смесь дегазировали, затем помещали на короткое время на предварительно нагретую до 70°C масляную баню, затем охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали под вакуумом, и остаток очищали с помощью колонной хроматографии на силикагеле, элюировали смесью этилацетат/гексан (0-50% для 1500 мл, затем 50-100% для 750 мл) с получением названного соединения в виде желтого масла.
Стадия 3: 4-[6-({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пиридин-3-ил]пиперидин-1-карбоксилат
К смеси продукта со стадии 2 (50 мг, 0,094 ммоль) в этилацетате (6 мл) добавляли 10% палладий на угле (9,97 мг). Реакционную смесь перемешивали в атмосфере водорода (баллон) в течение ночи, затем фильтровали. Фильтрат концентрировали под вакуумом с получением названного соединения в виде бесцветного масла, которое использовали без очистки на следующей стадии.
Стадия 4: 4-[6-({[(2S,5R)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)пиридин-3-ил]пиперидин-1-карбоксилат
К раствору продукта со стадии 3 (30 мг, 0,067 ммоль) в сухом пиридине (1,5 мл) добавляли комплекс пиридина с триоксидом серы (53,6 мг, 0,337 ммоль) при комнатной температуре, в темноте, в атмосфере азота. Реакционную смесь перемешивали в течение недели, затем фильтровали (собранное твердое вещество хорошо промывали дихлорметаном). Фильтрат концентрировали под вакуумом с получением названного соединения в виде бесцветного масла, которое использовали без очистки на следующей стадии.
Стадия 5: (2S,5R)-7-оксо-N-(5-пиперидин-4-илпиридин-2-ил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
К раствору продукта со стадии 4 (35 мг, 0,067 ммоль; теоретический выход стадии 4) в сухом дихлорметане (3 мл) добавляли трифторуксусную кислоту (0,00513 мл, 0,067 ммоль) при 0°C в атмосфере азота. Реакционную смесь перемешивали в течение 30 минут, затем концентрировали под вакуумом. Остаток растирали с эфиром для удаления избытка трифторуксусной кислоты и растворимых в органике примесей. Полученное твердое вещество сушили, растворяли в воде, и очищали с помощью препаративной HPLC на колонке Phenomenex Synergy Polar-RP 80A, элюировали смесью метанол/вода и лиофилизировали с получением названного соединения. LC-MS (режим отрицательной ионизации) m/e 424 (M-H).
ПРИМЕР 18
Пиперидин-4-илметил(2S,5R)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксилат



Стадия 1: [1-(третбутоксикарбонил)пиперидин-4-ил]метил-(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат
Гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (109 мг, 0,57 ммоль) и 4-диметиламинопиридин (69,6 мг, 0,57 ммоль) добавляли последовательно к раствору (2S,5R)-6-(фенилметокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (105 мг, 0,38 ммоль) в сухом дихлорметане при комнатной температуре. Затем добавляли третбутил 4-(гидроксиметил)пиперидин-1-карбоксилат (123 мг, 0,57 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи, и затем концентрировали под вакуумом. Остаток очищали с помощью препаративной HPLC с получением названного соединения.
Стадия 2: [1-(третбутоксикарбонил)пиперидин-4-ил]метил-(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат
К смеси продукта со стадии 1 (100 мг, 0,211 ммоль) в метаноле добавляли 10% палладий на угле (6,74 мг). Реакционную смесь перемешивали в атмосфере водорода (баллон) в течение ночи, затем фильтровали. Фильтрат концентрировали под вакуумом с получением названного соединения, которое использовали без очистки на следующей стадии.
Стадия 3: [1-(третбутоксикарбонил)пиперидин-4-ил]метил-(2S,5R)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксилат
К раствору продукта со стадии 2 (50 мг, 0,13 ммоль) в сухом пиридине (1 мл) добавляли комплекс пиридина с триоксидом серы (104 мг, 0,652 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь перемешивали в течение ночи, затем фильтровали (собранное твердое вещество хорошо промывали дихлорметаном). Фильтрат концентрировали под вакуумом с получением названного соединения, которое использовали без очистки на следующей стадии.
Стадия 4: Пиперидин-4-илметил(2S,5R)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксилат
TFA добавляли к продукту со стадии 3 в 0°C в атмосфере азота. Реакционную смесь перемешивали в течение 1 часа, затем концентрировали под вакуумом. Остаток растирали с эфиром для удаления избытка трифторуксусной кислоты и растворимых в органике примесей. Полученное твердое вещество сушили, растворяли в воде, и очищали с помощью препаративной HPLC на колонке Phenomenex Synergy Polar-RP 80A, элюировали смесью метанол/вода и лиофилизировали с получением названного соединения. LC-MS (режим отрицательной ионизации) m/e 362 (M-H).
ПРИМЕРЫ 19-56
Методику, описанную в примере 1A, использовали для получения следующих соединений, где указанный в качестве исходного реагента на стадии 1 амин заменяли на 4-амино-1-BOC-пиперидин.










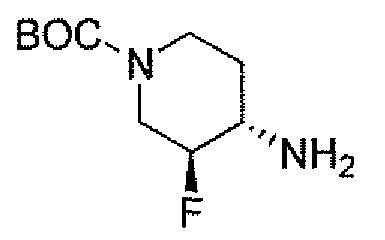












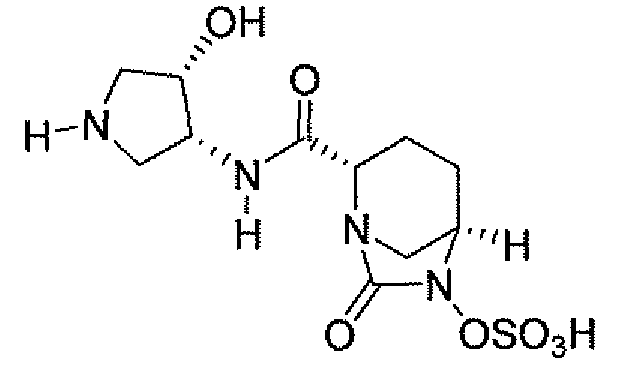

























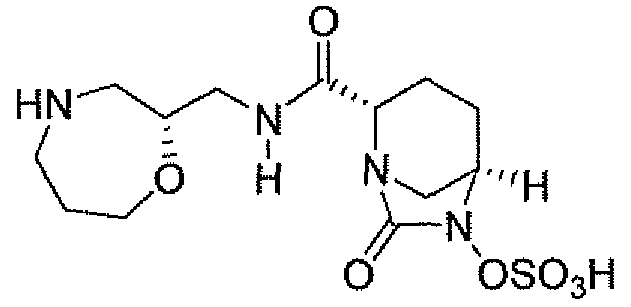


























ПРИМЕРЫ 57-90
Методика, описанная в примере 6, была использована для получения соединений примеров 57-77 и 80-90, где указанный в качестве исходного материала амин был заменен на стадии 1 на 4-аминопиридин. Методика, описанная в примере 17, была использована для получения соединений примеров 78 и 79, где указанный в качестве исходного материала пиридин был заменен на стадии 1 на 2-амино-5-бромпиридин.







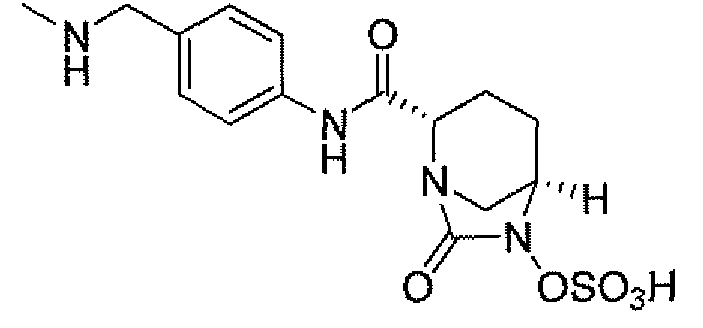









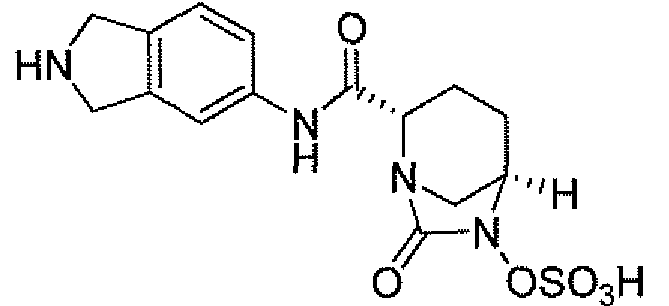


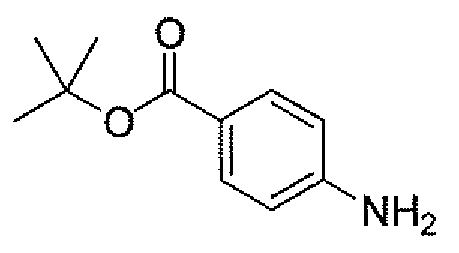

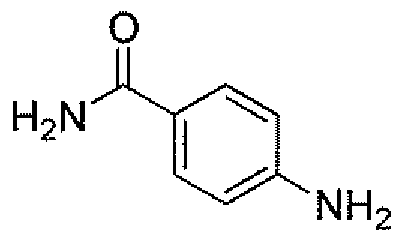

















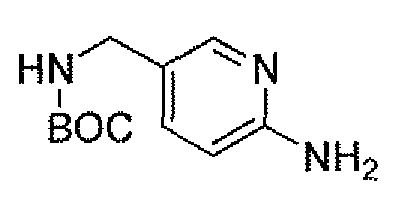














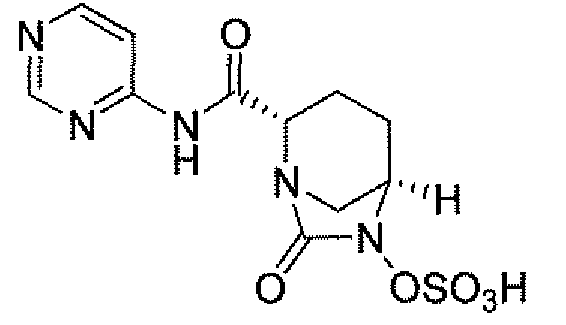





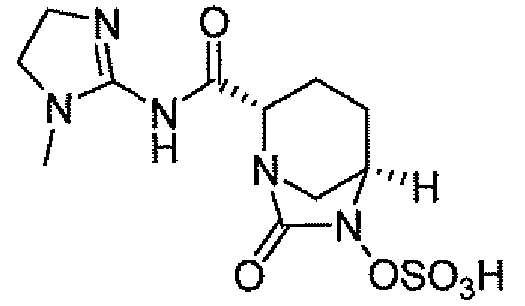






ПРИМЕРЫ 91-117
Методика, описанная в примере 18, была использована для получения следующих соединений, где указанный в качестве исходного материала спирт был заменен на стадии 1 на третбутил 4-(гидрокси-метил)пиперидин-1-карбоксилат.
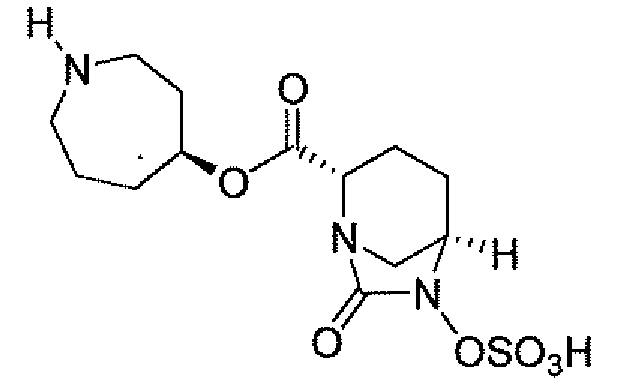

























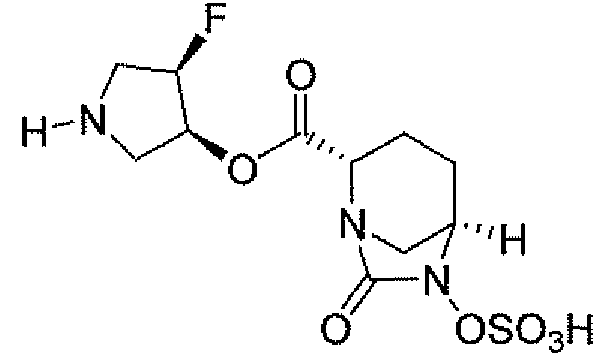



















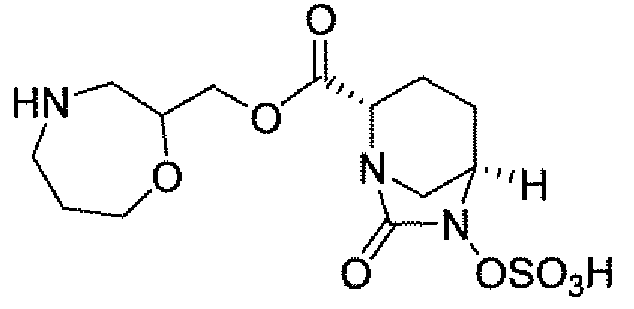




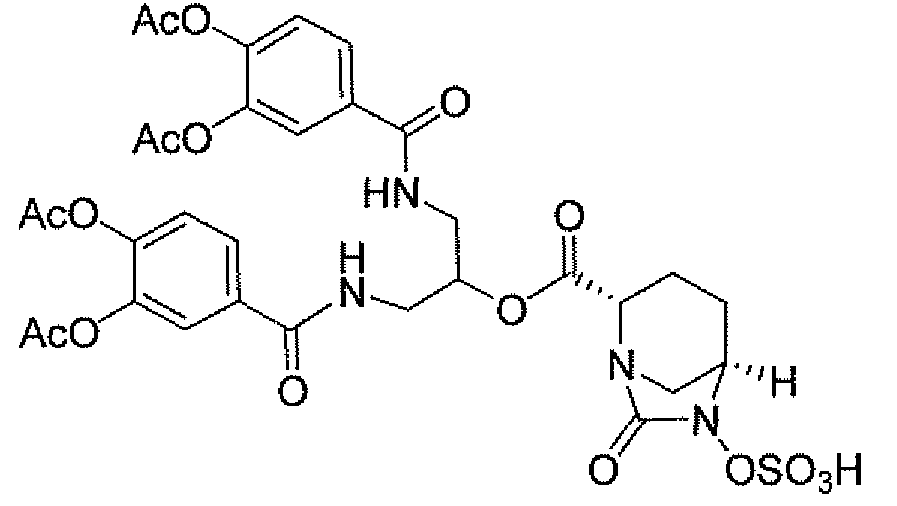
ПРИМЕР 118
Ферментная активность: определение IC50
Активность фермента класса C определяли в присутствии испытываемого ингибитора с помощью спектрофотометрического исследования по отношению к выпускаемому промышленностью субстрату, нитроцефину. Фермент AmpC (P. aeruginosa.) и субстрат растворяли в 100 мМ KH2PO4 буфера (pH 7). Буфер также содержит 0,005% BSA. При исследовании испытываемый ингибитор растворяли в ДМСО и разбавляли 1:20, в результате чего конечная концентрация находилась в интервале от 50 мкМ до 0,0002 мкМ. В 96-луночном микропланшете испытуемый ингибитор инкубировали с ферментом бета-лактамазы в течение 40 минут при температуре окружающей среды, добавляли раствор субстрата, и продолжали инкубирование в течение еще 40 минут. Спектрофотометрическую реакцию останавливали путем добавления 2,5 н уксусной кислоты, и измеряли поглощение при длине волны 492 нм. Величину IC50 определяли из полулогарифмических графических зависимостей ингибирования фермента от концентрации ингибитора, при этом кривую получали с помощью 4-параметрической аппроксимации.
Активность фермента класса А определяли с помощью такого же протокола испытания, который был описан выше для фермента класса C, за исключением того, что вместо фермента AmpC использовали фермент KPC-2 (K. pneumoniae).
Характерные соединения настоящего изобретения проявляют в этом исследовании ингибирующую активность по отношению к β-лактамазам класса C и класса A. Например, в этом исследовании подвергали испытаниям соединения примеров 1, 2, 4 и 6-9, и было обнаружено, что они имеют следующие значения IC50, приведенные в таблице 2. В таблице 3 приведены данные исследования для других примеров соединений.
Протокол исследования явления синергизма
В исследовании определяли концентрацию ингибитора β-лактамаз, необходимую для снижения MIC для β-лактамового антибиотика на половину, на четверть, на одну восьмую, на одну шестнадцатую и на одну тридцать вторую по отношению к штаммам бактерий, которые обычно устойчивы к действию исследуемого антибиотика. Это исследование проводят путем титрования BLI при последовательном разведении по горизонтали титрационного микропланшета, и, в то же самое время, титруя антибиотик при последовательном разведении вниз по вертикали титрационного микропланшета, и затем инокулируя планшет при помощи исследуемого бактериального штамма и давая возможность бактериям размножаться в течение ночи. Каждая лунка, расположенная в шахматном порядке на этом планшете, содержит различную комбинацию концентраций ингибитора и антибиотика, давая возможность провести полное определение любого явления синергизма между ними.
Комбинации бактериальный штамм/антибиотик:
CL 5701 (Pseudomonas aeruginosa; Pa AmpC)/имипенем
MB 2646 (Enterobacter cloacae; P99)/цефтазидим
CL 5513 {Klebsiella pneumoniae; SHV-5)/цефтазидим
CL 6188 {Acinetobacter baumanii; Oxa40)/имипенем
CL 6569 (Klebsiella pneumoniae; KPC-2)/имипенем
CL 5761 {Klebsiella pneumoniae; KPC-3)/имипенем
CLB 21648 {Acinetobacter baumanii; Ab AmpC)/имипенем
Обычный метод "шахматной доски":
1. Все лунки в рядах B-H микропланшетов MIC 2000 заполняют 100 мкл MHBII + 1% ДМСО.
2. Все лунки в ряду A микропланшетов MIC 2000 заполняют 100 мкл 2X MHBII + 2% ДМСО.
3. 100 мкл 4X требуемой конечной концентрации антибиотика добавляют в лунку A1 планшетов MIC 2000.
4. 100 мкл 2X требуемой конечной концентрации антибиотика добавляют в лунки A2-A12 планшетов MIC 2000.
5. 100 мкл последовательно разводят от ряда A до ряда G каждого планшета MIC 2000.
6. 100 мкл удаляют из каждой лунки в ряду G каждого планшета MIC 2000.
7. 100 мкл 2X требуемой конечной концентрации антибиотика (в MHBII + 1% ДМСО) добавляют во все лунки в колонку 1 титрационных микропланшетов.
8. 100 мкл последовательно разводят от колонки 1 до колонки 11 каждого планшета MIC 2000.
9. 100 мкл удаляют из каждой лунки в колонке 11 каждого планшета MIC 2000.
10. Планшеты затем инокулируют с помощью инокулятора MIC 2000 путем выращивания испытуемого штамма (в TSB) в течение ночи.
11. Планшеты выдерживают при 37°C примерно в течение 20 часов и оценивают рост визуально.
Среды (все стерилизуют в автоклаве перед любым добавлением ДМСО):
Триптиказо-соевый бульон (BBL™) приготавливали так, как предписано на бутылке.
Синергизм может быть выражен как отношение минимальной ингибирующей концентрации (MIC) испытываемого антибиотика в отсутствие ингибитора β-лактамазы к MIC этого же испытываемого антибиотика в присутствии ингибитора β-лактамазы. Отношение, равное единице (1), указывает, что ингибитор β-лактамазы не воздействует на эффективность антибиотика. Отношение, большее чем единица (1), указывает, что ингибитор β-лактамазы вызывает синергетический эффект при совместном введении с антибиотическим средством. Предпочтительные ингибиторы β-лактамаз настоящего изобретения характеризуются синергизмом с отношением, по меньшей мере, около 2, более предпочтительно, чтобы соединения характеризовались синергизмом с отношением, по меньшей мере, около 4, еще более предпочтительно по меньшей мере, около 8, и наиболее предпочтительно, по меньшей мере, около 16. В качестве варианта, синергетический эффект может быть выражен в виде коэффициента, опять же, использующего концентрацию BLI для низшего MIC для антибиотика. Таким образом, если MIC для антибиотика составляет 20 мкг/мл и 1,5 мкМ концентрация BLI снижает MIC до 5 мкг/мл, синергетический эффект является четырехкратным или проявляется "4X синергизм" при 1,5 мкМ BLI.
Характерные соединения настоящего изобретения проявляют синергетический эффект. Например, было обнаружено, что соединения примеров 1, 2, 4 и 6-9 имели 2X синергизм при концентрациях в интервале от 100 мкМ или меньше. Синергетические концентрации для примеров 1, 2, 4 и 6-9 против штамма CL5701 P. aeruginosa и штамма CL6569 Klebsiella pneumoniae приведены в таблице 2.






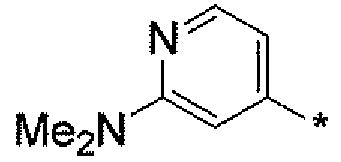

1. Это концентрации для 2X, 4X и 8X с имипенемом против штамма CL5701 P. aeruginosa. Например, концентрация 6,25 мкМ соединения примера 1 снижает MIC для имипенема против штамма CL5701 P. aeruginosa в 8 раз (8X синергизм).
2. Это концентрации для 16X, 32X и 64X с имипенемом против штамма CL6569 K. pneumoniae. Например, концентрация 12,5 мкМ соединение примера 1 снижает MIC для имипенема против штамма CL6569 K. pneumoniae в 64 раза (64X синергизм).
Следует отметить, что данные, приведенные в таблице 3, получены таким же способом и с использованием таких же ферментов, что и в таблице 2. Примеры 2, 6, 7 и 8 в таблице 3 дублируют данные, приведенные для этих примеров в таблице 2, за исключением того, что значения IC50 для AmpC в таблице 3 для примеров 7 и 8 составляют 11 нМ и 16 нМ против 1,1 нМ и 1,6 нМ, соответственно, для соответствующих примеров в таблице 2, и значение IC50 для KPC-2 в таблице 3 для примера 8 составляет 16 нМ против 1,6 нМ в таблице 2.
Кроме того, следует отметить, что соединение примера 1 в таблице 2 и соединение примера 1A в таблице 3 являются одним и тем же соединением. Значения IC50для KPG-2 для примеров 1 в таблице 2 и 1A в таблице 3 составляют 208 нМ и 210 нМ, соответственно. Эти два значения могли быть получены в разных экспериментах или они могут представлять данные одного и того же эксперимента.
Несмотря на то что приведенное выше описание раскрывает основные идеи настоящего изобретения с помощью примеров, приводимых с целью иллюстрации, тем не менее, осуществление изобретения охватывает все обычные изменения, усовершенствования и/или модификации, которые подпадают под объем следующих пунктов формулы изобретения. Содержание всех упоминаемых здесь публикаций, патентов и патентных заявок приводится здесь путем ссылки на них, где в случае любых противоречий настоящее описание будет иметь преимущественную силу.
Реферат
В настоящей заявке описаны замещенные бициклические бета-лактамы формулы I: ! !которые являются ингибиторами β-лактамаз класса А и класса С, где X, R1 и R2 определены в тексте заявки, а также способ их получения. Соединения формулы I и их фармацевтически приемлемые соли применяют для получения фармацевтической композиции и в производстве лекарственного средства. Заявленные соединения применяются для лечения бактериальных инфекций необязательно в комбинации с β-лактамовым антибиотиком. В частности соединения могут быть использованы с такими β-лактамовыми антибиотиками, как, например, имипенемом, пиперациллином, или цефтазидимом, против микроорганизмов, устойчивых к действию β-лактамовых антибиотиков вследствие присутствия β-лактамаз. 6 н. и 22 з.п. ф-лы, 118 пр., 3 табл., 3 сх., 2 ил.
Формула

или его фармацевтически приемлемая соль,
где Х является:
(1) CH2 или
(2) CH2CH2;
R1 является C(O)N(R3)R4,
R2 является OSO3M;
М является Н или фармацевтически приемлемым катионом;
R3 является:
(1) С1-8алкилом, замещенным с помощью HetA,
(2) HetA,
(3) AryA,
(4) HetB или
(5) AryB;
R4 является Н или С1-8алкилом;
или в качестве варианта R3 и R4 вместе с атомом N, к которому они оба присоединены, образуют насыщенное моноциклическое кольцо с числом членов от 4 до 6, содержащее 1 гетероатом в дополнение к азоту, присоединенному к R3 и R4, представляющий собой N; где моноциклическое кольцо необязательно конденсировано или спиро с насыщенным гетероциклическим кольцом с числом членов от 5 до 6, содержащим 1 гетероатом, представляющий собой N, с образованием бициклической кольцевой системы, где образованная таким образом моноциклическая кольцевая или бициклическая кольцевая система необязательно замещена с помощью 1 заместителя, который является (3) (CH2)1-2G, где G является N(RA)RB, или (2) N(RA)RB;
HetA является насыщенным или мононенасыщенным гетероциклическим кольцом с числом членов от 4 до 9, содержащим от 1 до 2 гетероатомов, независимо выбранных из N, О и S, где любая S в кольце необязательно окислена до S(O)2, и 1 атом углерода в кольце необязательно окислен до С(O); где кольцо необязательно конденсировано с C3-7циклоалкилом и где необязательно конденсированное, насыщенное или мононенасыщенное кольцо необязательно замещено суммарно с помощью 1 заместителя, выбранного из (CH2)nN(RA)RB и (CH2)nRC;
AryA является фенилом, который необязательно замещен суммарно с помощью 1 заместителя, выбранного из (CH2)nN(RA)RB и (CH2)nRC,
HetB является гетероароматическим кольцом с 5 или 6 членами, содержащим 1 гетероатом, выбранным из от 1 до 2 атомов N и из 1 атома S; где гетероароматическое кольцо необязательно конденсировано с насыщенным гетероциклическим кольцом с числом членов от 5 до 7, содержащим 1 гетероатом, представляющий собой N, где необязательно конденсированное гетероароматическое кольцо необязательно замещено суммарно с помощью от 1 до 2 заместителями, выбранными из (CH2)nN(RA)RB и (СН2)nRC;
AryB является бициклической кольцевой системой, в которой фенил конденсирован с насыщенным гетероциклическим кольцом с числом членов от 5 до 6, содержащим 1 гетероатом, представляющий собой N;
каждое n независимо является целым числом, которое равняется 0 или 1;
каждый RA является независимо Н или С1-8алкилом;
каждый RB является независимо Н или С1-8алкилом;
каждый RC является независимо C1-6алкилом, ОН, О-С1-8алкилом, галогеном, C(O)ORA, C(O)N(RA)RB, SO2N(RA)RB, пиридилом, пирролидинилом, пиперидинилом, пиперазинилом или морфолинилом;
и при условии, что:
(D) когда R является AryA или С1-8алкилом, замещенным с помощью AryA, тогда AryA не является незамещенным фенилом, фенилом, замещенным с помощью N(RA)R8, или фенилом, замещенным с помощью C(O)N(RA)RB.


















где T является H, C1-3алкилом, пирролидин-3-илом или пиперидин-4-илом и Т' является H, Cl, Br, F, C1-3алкилом, О-С1-3алкилом, ОН, NH2, N(H)-C1-3алкилом, или N(-С1-3алкил)2.









где V, V', V”, Y, Y' и Z каждый независимо выбран из группы, состоящей из H, CH3, пирролидинила, пиперидинила, пиперазинила, морфолинила, CH2-пирролидинила, CH2-пиперидинила, CH2-пиперазинила, CH2-морфолинила, NH2, N(Н)СН3, N(CH3)2, CH2NH2, CH2N(H)CH3 и CH2N(CH3)2, при условии, что:
(i) по меньшей мере, один из Y и Y' является H.
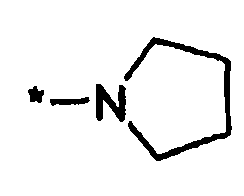

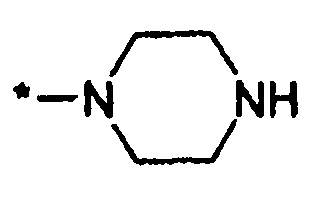


где кольцо необязательно замещено с помощью CH2NH2, CH2N(H)-C1-3алкилом, CH2N(-С1-3алкил)2, NH2, N(H)-C1-3алкилом или N(-С1-3алкил)2.
(2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[(4S)-азепан-4-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[(4R)-азепан-4-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-[(3R)пирролидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-[(3S)пирролидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-азокан-5-ил-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-пиридин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(2-метоксипиридин-4-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамцд;
(2S,5R)-N-[2-(диметиламино)пиридин-4-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[4-(аминометил)фенил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-2-[(пиперидин-4-иламино)карбонил]-1,6-диазабицикло[3.2.1]октан-6-сульфоновая кислота;
(4R,6S)-2-оксо-N-пиперидин-4-ил-3-(сульфоокси)-1,3-диазабицикло[2.2.1]гептан-6-карбоксамид;
(4R,6S)-2-оксо-N-[(4S)-азепан-4-ил]-3-(сульфоокси)-1,3-диазабицикло[2.2.1]гептан-6-карбоксамид;
(4R,6S)-2-оксо-N-пиридин-4-ил-3-(сульфоокси)-1,3-диазабицикло[2.2.1]гептан-6-карбоксамид; и их фармацевтически приемлемые соли.
(2S,5R)-7-оксо-N-[(3R)пирролидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[(3R,4S)-3-фторпиперидин-4-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-6-(сульфоокси)-N-(1,2,3,4-тетрагидроизохинолин-6-ил)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-(5-пиперидин-4-илпиридин-2-ил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
диастереомер 1 (2S,5R)-7-оксо-N-[(3)пиперидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
диастереомер 2 (2S,5R)-7-оксо-N-[(3)пиперидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
(2S,5R)-7-оксо-N-азетидин-3-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-[(3R)пирролидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-[(4R)-азепан-4-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-[1-метилпиперидин-4-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-[(3S,4S)-3-фторпиперидин-4-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид или его 3R,4R диастереомер или их смесь;
(2S,5R)-7-оксо-N-[(3S,4R)-3-фторпиперидин-4-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-[(3S,4R)-3-метоксипиперидин-4-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(1,1-диоксидотетрагидро-2Н-тиопиран-4-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[(3R,4R)-4-аминопирролидин-3-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[(3R,4R)-4-гидроксипирролидин-3-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[(3R,4S)-4-гидроксипирролидин-3-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[(3R,4S)-4-фторпирролидин-3-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[(3S,4R)-4-фторпирролидин-3-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-[(3S)-1-пиперидин-4-ил-2-оксопирролидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-[(3R)-1-пиперидин-4-ил-2-оксопирролидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[(3S,4R)-3-фторазепан-4-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[(3R,4S)-3-фторазепан-4-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[(3-фторазетидин-3-ил)метил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-(пирролидин-2-илметил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R-)-7-оксо-N-(пиперидин-2-илметил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-(пиперидин-4-илметил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[(2S)-1,4-оксазепан-2-илметил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[(2R)-1,4-оксазепан-2-илметил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-(2-пиперидин-4-илэтил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-(2-пиперидин-1-илэтил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-(2-пиперазин-1-илэтил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-3-азабицикло[3,1.0]гекс-6-ил-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-метил-7-оксо-N-пиперидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-2-{[2-(аминометил)пиперидин-1-ил]карбонил}-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-7-он;
(2S,5R)-2-[(4-аминопиперидин-1-ил)карбонил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-7-он;
(2S,5R)-2-(пиперазин-1-илкарбонил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-7-он;
(2S,5R)-2-(2,7-диазаспиро[3.5]нон-2-илкарбонил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-7-он;
(2S,5R)-2-(гексагидропирроло[3,4-с]пиррол-2(1H)-илкарбонил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-7-он;
(2S,5R)-2-{[(3R)-3-аминопирролидин-1-ил]карбонил}-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-7-он;
(2S,5R)-2-{[(3S)-3-аминопирролидин-1-ил]карбонил}-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-7-он;
(2S,5R)-2-{[3-(диметиламино)пирролидин-1-ил]-карбонил}-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-7-он;
(2S,5R)-N-[4-(аминометил)фенил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[3-(аминометил)фенил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[2-(аминометил)фенил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-{4-[(метиламино)метил]фенил}-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-{3-[(метиламино)метил]фенил}-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-{4-[(диметиламино)метил]фенил}-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-{4-[(пирролидинил)метил]фенил}-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-6-(сульфоокси)-N-(1,2,3,4-тетрагидроизохинолин-6-ил)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(2,3-дигидро-1H-изоиндол-5-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(2,3-дигидро-1H-индол-5-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
4-({[(2S,5R)-7-оксо-6-(сульфоокси)-1,6-диазабицикло-[3.2.1]окт-2-ил]карбонил}амино)бензойная кислота;
(2S,5R)-N-[4-(аминокарбонил)фенил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[4-(аминосульфонил)фенил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбокрамид;
(2S,5R)-N-[3-(аминокарбонил)фенил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-пиридин-3-ил-6-(сульфоокси)-1,6-диазабицикло-[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-пиридин-2-ил-6-(сульфоокси)-1,6-диазабицикло-[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(2,6-дипирролидин-1-илпиридин-4-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(6-аминопиридин-2-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[4-(диметиламино)пиридин-2-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[4-(аминометил)пиридин-2-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[5-(аминометил)пиридин-2-ил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(4-пиперидин-4-илпиридин-2-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(6-пиперидин-4-илпиридин-2-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(5-пиперазин-1-илпиридин-2-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(5-морфолин-4-илпиридин-2-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(5-пирролидин-1 -ил-пиридин-2-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-пиразин-2-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1] октан-2-карбоксамид;
(2S,5R)-7-оксо-N-пиримидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-(2-пиперизин-1-илпиримидин-4-ил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(1-метил-1H-имидазол-2-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(1-метил-4,5-дигидро-1H-имидазол-2-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-6-(сульфоокси)-N-1,3-тиазол-2-ил-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-6-(сульфоокси)-N-(4,5,6,7-тетрагидро[1,3]-тиазоло[5,4-с]пиридин-2-ил)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-(6,7-дигидро-5H-пирроло[3,4-b]пиридин-2-ил)-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид; и
их фармацевтически приемлемые соли.
(2S,5R)-7-оксо-N-пиперидин-4-ил-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-N-[4-(аминометил)фенил]-7-оксо-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-[(3R)пирролидин-3-ил]-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-6-(сульфоокси)-N-(1,2,3,4-тетрагидроизохинолин-6-ил)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
(2S,5R)-7-оксо-N-(5-пиперидин-4-илпиридин-2-ил)-6-(сульфоокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид; и
их фармацевтически приемлемые соли.

который включает:
(А) контактирование илида кетосульфоксония формулы P-I

с иридиевым, родиевым или рутениевым катализатором с получением соединения P-II,
где PG является Cbz;
Соединение P-II является соединением P-IIa

RU является CH3 или фенилом;
RV является CH3 или фенилом;
R4 является Н или С1-4алкилом;
Т' является H, Cl, Br, F, С1-3алкилом, O-C1-3алкилом, OH, NH2, N(H)-C1-3алкилом, или N(-С1-3алкил)2;
р является нулем, 1 или 2; q является нулем, 1 или 2 и p+q = нулю, 1, 2, или 3;
(В) обработку соединения P-IIa восстановителем с получением соединения формулы P-III

и (С) контактирование соединения P-III с сульфонилгалогенидом формулы R^-SO2W в присутствии основания третичного амина с получением соединения формулы P-IV

где W является галогеном и
R^ является;
(1) фенилом, необязательно замещенным с помощью от 1 до 3 заместителей, каждый из которых является независимо С1-4алкилом, С1-4галогеналкилом, О-С1-4алкилом, O-C1-4галогеналкилом, Cl, Br, F, или NO2;
(2) С1-4алкилом или
(3) С1-4галогеналкилом;
(D) контактирование соединения P-IV с N-Boc-O-бензилгидроксиламином в присутствии основания с получением соединения формулы P-V

(Е) обработку соединения P-V кислотой с получением соединения формулы P-VI

(F) контактирование соединения P-VI с фосгеном, дифосгеном или трифосгеном в присутствии третичного амина и затем добавление водного раствора кислоты с получением соединения формулы P-VII

и (G) контактирование соединения P-VII с источником водорода в присутствии катализатора гидрогенолиза и в присутствии Boc-образующего реагента с получением соединения формулы P-VIII


который включает:
(А) контактирование илида кетосульфоксония р-1:

с катализатором, выбранным из группы, состоящей из димера хлорида циклооктадиен иридия, RuCl2(PPhs), Ru(DMSO)4Cl2 и Rh2(TFA)4, с получением р-2;
(В) обработку соединения р-2 восстановителем, выбранным из группы, состоящей из Li боргидрида, Na боргидрида и K боргидрида, с получением соединения р-3

и (С) контактирование соединения р-3 с сульфонилгалогенидом формулы R^-SO2W в присутствии основания три-С1-4алкиламина с получением соединения формулы р-4

где W является хлором и
R^ является метилом, хлорметилом, фенилом, 4-бромфенилом, 4-трифторметилфенилом или 4-метилфенилом;
(D) контактирование соединения р-4 с N-Boc-O-бензилгидроксиламином в присутствии основания, выбранного из группы, состоящей из Li трет-бутилата, Na трет-бутилата, K трет-бутилата и K амилата, с получением соединения р-5

и (Е) обработку соединения р-5 кислотой, выбранной из группы, состоящей из метансульфоновой кислоты, хлорметансульфоновой кислоты, п-толуолсульфоновой кислоты и бензолсульфрновой кислоты, с получением соединения формулы р-6

(F) контактирование соединения р-6 с трифосгеном в присутствии основания три-С1-4алкиламина и затем добавление водного раствора фосфорной кислоты с получением соединения р-7

и (G) контактирование соединения р-7 с водородом в присутствии Pd-катализатора и Boc-образующего реагента, выбранного из группы, состоящей из дитрет-бутилкарбоната и Boc-ON, с получением соединения р-8







где PG является Cbz;
RU является CH3 или фенилом;
RV является CH3 или фенилом;
R4 является H или С1-4алкилом;
Т' является H, Cl, Br, F, C1-3алкилом, O-С1-3алкилом, OH, NH2, N(H)-C1-3алкилом, или N(-С1-3алкил)2;
р является нулем, 1 или 2; q является нулем, 1 или 2 и p+q = нулю, 1, 2, или 3;
R^ является:
(1) фенилом, необязательно замещенным с помощью от 1 до 3 заместителей, каждый из которых является независимо С1-4алкилом, С1-4галогеналкилом, О-С1-4алкилом, O-C1-4галогеналкилом, Cl, Br, F, или NO2;
(2) С1-4алкилом или
(3) С1-4галогеналкилом.






где R^ является метилом, хлорметилом, фенилом, 4-бромфенилом, 4-трифторметилфенилом, или 4-метилфенилом.
Комментарии