Замещенные 3-фенилпропионовые кислоты и их применение - RU2553263C2
Код документа: RU2553263C2
Описание
Данная заявка касается новых производных 3-фенилпропионовой кислоты, способа их получения, их применения для лечения и/или профилактики заболеваний, а также их применения для изготовления лекарственных средств для лечения и/или профилактики заболеваний, в частности, для лечения и/или профилактики сердечно-сосудистых заболеваний.
Одной из важнейших клеточных систем переноса в клетках млекопитающих является циклический гуанозинмонофосфат (цГМФ). Вместе с монооксидом азота (NO), который высвобождается из эндотелия и переносит гормональные и механические сигналы, он образует систему NO/цГМФ. Гуанилатциклазы катализируют биосинтез цГМФ из гуанозинтрифосфата (ГТФ). Известные до сих пор представители этого семейства могут подразделяться как по структурным признакам, так и по типу лигандов на две группы: гуанилатциклазы, имеющие форму частиц, способные стимулироваться натрийуретическими пептидами, и растворимые гуанилатциклазы, стимулируемые NO. Растворимые гуанилатциклазы состоят из двух субъединиц и по всей вероятности содержат один гем на каждый гетеродимер, который является частью регуляторного центра. Последний имеет центральное значение для механизма активации. NO может связываться с атомом железа гема и таким образом заметно повышать активность фермента. Составы, не содержащие гем, напротив, не могут стимулироваться под действием NO. Монооксид углерода (СО) также в состоянии прикрепляться к центральному атому железа гема, причем это стимулирование под действием СО заметно меньше, чем стимулирование с помощью NO.
Благодаря образованию цГМФ и вытекающему из этого регулированию фосфодиэтераз, ионных каналов и протеинкиназ играет гуанилатциклаза решающую роль при различных физиологических процессах, в частности, при релаксации и пролиферации гладких мышечных клеток, агрегации и адгезии тромбоцитов и нейрональной передаче сигнала, а также при заболеваниях, которые основываются на нарушении указанных выше процессов. При патофизиологических условиях система NO/цГМФ может подавляться, что может приводить, например, к повышенному кровяному давлению, активации тромбоцитов, увеличенной клеточной пролиферации, эндотелиальной дисфункции, атеросклерозу, стенокардии, сердечной недостаточности, тромбозам, инсульту и инфаркту миокарда.
Возможность лечения заболеваний такого типа, нацеленная на воздействие на сигнальный путь цГМФ в организме, независящая от NO, представляет собой перспективный подход благодаря высокой эффективности, которую следует ожидать, и незначительным побочным эффектам.
Для терапевтической стимуляции растворимой гуанилатциклазы до сегодняшнего дня применялись исключительно такие соединения как органические нитраты, действие которых основывается на NO. Этот NO образуется в результате биоконверсии и активирует растворимую гуанилатциклазу путем прикрепления к центральному атому железа гема. Помимо побочных эффектов, к решающим недостаткам этого способа лечения относится развитие невосприимчивости [O.V.Evgenov с соавт., Nature Rev. Drug Disc. 5 (2006), 755].
В последние годы были идентифицированы вещества, которые стимулируют растворимую гуанилатциклазу напрямую, то есть без предварительного высвобождения NO. С использованием индазольного производного YC-1 впервые был описан NO-независимый, однако гемзависимый стимулятор растворимой гуанилатциклазы (рГЦ) [Evgenov с соавт., там же]. Исходя из YC-1, были обнаружены другие вещества, которые имеют более высокую способность, чем YC-1, и не обладают существенным подавлением фософодиэстераз (ФДЭ). Это привело к идентифицированию пиразо-лопиридиновых производных BAY 41-2272, BAY 41-8543 и BAY 63-2521. Эти соединения совместно с приведенными в недавних публикациях, структурно отличными веществами CMF-1571 и А-350619 образуют новый класс стимуляторов рГЦ [Evgenov с соавт., там же]. Общей характерной особенностью этого класса веществ является NO-независимое и селективное активирование гемсодержащей рГЦ. Кроме того, эти стимуляторы рГЦ в сочетании с N0 демонстрируют синергетический эффект в отношении активации рГЦ, который основывается на стабилизации комплекса нитрозилгем. Точный центр связывания этих стимуляторов рГЦ с самой рГЦ до сегодняшнего дня является предметом дискуссий. Если из растворимой гуанилатциклазы удаляют группу гема, этот фермент все еще проявляет детектируемую каталитическую базальную активность, то есть по-прежнему образуется цГМФ. Сохраняющаяся каталитическая базальная активность фермента, не содержащего гем, не может быть простимулирована ни одним из указанных выше стимуляторов [Evgenov с соавт., там же].
Помимо этого, с помощью BAY 58-2667 в качестве прототипа этого класса были идентифицированы NO- и гемнезависимые активаторы рГЦ. Общими характерными признаками этих веществ является то, что в комбинации с NO они оказывают только дополнительный эффект на активацию фермента, и что эта активация окисленного или не содержащего гем фермента заметно сильнее в сравнении с ферментом, содержащим гем [Evgenov с соавт., там же; J.P.Stasch с соавт., Br.J.Pharmacol. 136 (2002), 773; J.P.Stasch с соавт., J.Clin. Invest. 116 (2006), 2552]. Спектроскопические исследования дают возможность увидеть, что BAY 58-2667 вытесняет окисленную группу гема, которая в результате ослабления связывания железа с гистидином лишь слабо соединена с рГЦ. Также было показано, что характеристичный мотив связывания рГЦ-гем Tyr-x-Ser-x-Arg является совершенно необходимым как для взаимодействия отрицательно заряженных пропионовых кислот группы гема, так и для действия BAY 58-2667. В свете вышеизложенного принимают, что центр связывания BAY 58-2667 с рГЦ является идентичным центру связывания группы гема [J.P.Stasch с соавт., J.Clin. Invest. 116 (2006), 2552].
Так, соединения, описанные в настоящем изобретении, так же в состоянии активировать форму растворимой гуанилатциклазы, не содержащую гем. Это также подтверждается тем, что эти активаторы нового типа, с одной стороны, не проявляют синергетического эффекта совместно с N0 для фермента, содержащего гем, а, с другой стороны, их действие не может блокироваться с помощью гемзависимого ингибитора растворимой гуанилатциклазы 1H-1,2,4-оксадиазоло[4,3-а]хиноксалин-1-она (ODQ), а даже усиливается этим веществом [сравните с O.V. Evgenov с соавт., Nature Rev. Drug Disc. 5 (2006), 755; J.P.Stasch с соавт., J.Clin. Invest. 116 (2006), 2552].
Следовательно, задачей данного изобретения было предоставление новых соединений, которые действуют описанным выше образом как активаторы растворимой гуанилатциклазы, и в качестве таковых могут использоваться, в частности, для лечения и профилактики сердечно-сосудистых заболеваний.
В международной заявке WO 00/64888-A1, европейских патентах ЕР 1216980-А1, ЕР 1375472-А1, ЕР 1452521-А1, патентах США US 2005/0187266-А1 и US 2005/0234066-A1 описываются различные производные арилалканкарбоновых кислот в качестве агонистов РАПП (рецепторов, активируемых пролифератором пероксисом) для лечения диабета, дислипидемии, артериосклероза, общего ожирения и других заболеваний. В европейских патентах ЕР 1312601-А1 и ЕР 1431267-А1 показываются замещенные арилалканкарбоновые кислоты в качестве антагонистов рецепторов простагландина PGE2 для лечения, например, болевых состояний, урологических заболеваний, болезни Альцгеймера и рака. Кроме того, в международной заявке WO 2005/086661-А2 арилалканкарбоновые кислоты заявляются в качестве модуляторов GPR40 для лечения диабета и дислипидемии, а в международных заявках WO 2004/099170-А2, W02006/050097-A1, а также WO 2006/055625-A2 описываются фенилзамещенные карбоновые кислоты в качестве ингибиторов протеинтирозин-фосфатазы-1 В (ПТФ-1 В) для лечения диабета, рака и нейродегенеративных заболеваний. Кроме того, из международных заявок WO 96/12473-A1 и WO 96/30036-A1 известны отдельные фенилацетамидозамещенные фенилалканкарбоновые кислоты, которые в форме нековалентных смесей улучшают доставку пептидных биологически активных веществ внутри тела. Недавно в международной заявке WO 2009/127338-А1 были предложены производные карбоновых кислот с оксогетероциклическими заместителями, которые действуют как активаторы растворимой гуанилатциклазы.
Предметом данного изобретения являются соединения общей формулы (I)
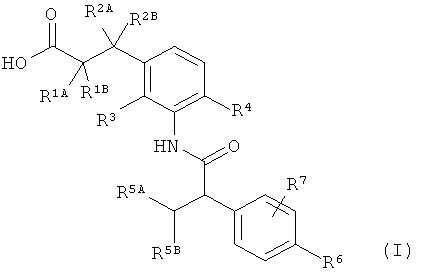
в которой
R1A представляет собой водород, фтор, метил, трифторметил, этил, 1,1-дифторэтил, 2,2,2-трифторэтил, н-пропил, циклопропил или циклобутил,
R1B является водородом или метилом,
R2A представляет собой водород, метил, трифторметил, этил, 1,1-дифторэтил, 2,2,2-трифторэтил или н-пропил,
R2B является водородом или метилом, или
R1A и R2A связаны друг с другом и вместе с атомами углерода, с которыми они соединены, образуют циклопропильное кольцо формулы

или
R2A и R2B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют циклическую группу формулы



n обозначает число 1, 2 или 3,
R3 является водородом, фтором, метилом или трифторметилом,
R4 представляет собой водород, фтор, хлор, цианогруппу, метил, трифторметил или этил,
R5A представляет собой метил, трифторметил или этил,
R5B является трифторметилом,
или
R5A и R5B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют дифторзамещенное циклоалкильное кольцо формулы
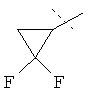

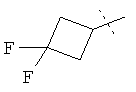

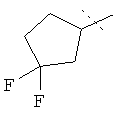
R6 представляет собой водород, фтор, хлор, бром, цианогруппу, алкил с 1-4 атомами углерода, алкенил с 2-4 атомами углерода, циклопропил или циклобутил, причем
алкил с 1-4 атомами углерода и алкенил с 2-4 атомами углерода могут содержать до трех атомов фтора в качестве заместителей,
а
циклопропил и циклобутил могут содержать до двух атомов фтора в качестве заместителей,
и
R7 представляет собой водород, фтор, хлор, цианогруппу, метил, трифторметил, этил, метокси- или трифторметоксигруппу,
а также их соли, сольваты и сольваты солей.
Соединения согласно изобретению представляют собой соединения формулы (I) и их соли, сольваты и сольваты этих солей, охватываемые формулой (I) соединения с формулами, указанными ниже, и их соли, сольваты и сольваты солей, а также соединения, охватываемые формулой (I), приведенные далее в качестве примеров исполнения изобретения и их соли, сольваты и сольваты солей, если в случае охватываемых формулой (I), приведенных далее соединений речь уже не идет о солях, сольватах и сольватах солей.
Соединения согласно изобретению в зависимости от их структуры могут существовать в различных стереоизомерных формах, то есть в форме конфигурационных изомеров или при необходимости также в виде конформационных изомеров (энантиомеров и/или диастереомеров, включая таковые в случае атропных изомеров). Поэтому настоящее изобретение включает энантиомеры и диастереомеры и их соответствующие смеси. Из таких смесей энантиомеров и/или диастереомеров индивидуальные стереоизомерные компоненты могут выделяться известным способом; предпочтительно для этого применяются хроматографические способы, особенно ВЭЖХ (высокоэффективная жидкостная хроматография) на ахиральной или соответственно хиральной фазах.
Поскольку соединения согласно изобретению могут существовать в таутомерных формах, данное изобретение включает все без исключения таутомерные формы.
В качестве солей в рамках настоящего изобретения предпочтительны физиологически приемлемые соли соединений согласно изобретению. Также включены соли, которые сами не являются подходящими для фармацевтического применения, однако могут применяться, например, для выделения или очистки соединений согласно изобретению.
Физиологически приемлемые соли соединений согласно изобретению прежде всего включают соли обычных оснований, такие как приводимые в качестве примерных и предпочтительных соли щелочных металлов (например, натриевые и калиевые соли), соли щелочноземельных металлов (например, кальциевые и магниевые соли) и аммониевые соли, являющиеся производными аммиака или органических аминов с числом атомов углерода от 1 до 16, такие как приводимые в качестве примерных и предпочтительных этиламин, диэтиламин, триэтиламин, этилдиизопропиламин, моноэтаноламин, диэтаноламин, триэтаноламин, дициклогексиламин, диметиламиноэтанол, прокаин, дибензиламин, N-метилморфолин, N-метилпиперидин, аргинин, лизин и этилендиамин.
Сольватами в рамках изобретения обозначаются такие формы соединений согласно изобретению, которые в твердом или жидком состоянии в результате координации с молекулами растворителя образуют комплекс. Гидраты являются частной формой сольватов, в случае которых эта координация осуществляется с водой. В качестве сольватов в рамках настоящего изобретения предпочтительны гидраты.
Кроме этого, данное изобретение также включает пролекарства из соединений согласно изобретению. Термин «пролекарство» при этом обозначает соединения, которые сами могут быть биологически активными или неактивными, однако в процессе их периода нахождения в организме превращаются в соединения согласно изобретению (например, в результате метаболизма или гидролитически).
Особенно данное изобретение включает способные к гидролизу сложноэфирные производные карбоновых кислот формулы (I) согласно изобретению. По этим понимают сложные эфиры, которые в физиологических средах, в условиях биологических испытаний, описанных в дальнейшем, и прежде всего, в испытаниях на живом объекте (in vivo) могут ферментативным или химическим путем гидролизоваться до свободных карбоновых кислот в качестве преимущественно биологически активных соединений. В качестве таких сложных эфиров предпочитают сложные алкиловые эфиры с 1-4 атомами углерода в алкиле, в которых алкильная группа может быть линейной или разветвленной. Особенно предпочтительными являются сложные метиловые, этиловые или mpem-бутиловые эфиры.
В рамках данного изобретения заместители, если не указано иное, имеют следующие значения:
алкил с 1-4 атомами углерода в рамках изобретения представляет собой линейный или разветвленный алкильный остаток с числом атомов углерода от 1 до 4. В качестве примерных и предпочтительных следует назвать: метил, этил, н-пропил, изопропил, н-бутил, изобутил, вторбутил и трет-бутил.
Алкенил с 2-4 атомами углерода и алкенил с 2-3 атомами углерода в рамках изобретения представляют собой неразветвленный или разветвленный алкенильный остаток с одной двойной связью и числом атомов углерода от 2 до 4 или соответственно 2 или 3. Предпочтительным является неразветвленный или разветвленный алкенильный остаток с 2 или 3 атомами углерода. В качестве примерных и предпочтительных следует назвать: винил, аллил, н-проп-1-ен-1-ил, изопропенил, н-бут-1-ен-1-ил, н-бут-2-ен-2-ил, н-бут-3-ен-2-ил, 2-метилпроп-1-ен-1-ил и 2-метилпроп-2-ен-1-ил.
В рамках данного изобретения справедливо то, что для всех остатков, которые встречаются многократно, их значения не зависят друг от друга. Если остатки в соединениях согласно изобретению являются замещенными, то эти остатки могут, если не указано иное, содержать один или несколько заместителей. Предпочтительным является замещение одним или двумя, или тремя одинаковыми или разными заместителями. Особенно предпочтительным является замещение одним или двумя одинаковыми или разными заместителями.
В одной определенной форме исполнения данное изобретение включает соединения формулы (I), в которой
R1A представляет собой водород, фтор, метил, трифторметил, этил, 1,1-дифторэтил, 2,2,2-трифторэтил или н-пропил,
R1B является водородом или метилом,
R2A представляет собой водород, метил, трифторметил, этил, 1,1-дифторэтил, 2,2,2-трифторэтил или н-пропил,
R2B является водородом или метилом, или
R1A и R2A связаны друг с другом и вместе с атомами углерода, с которыми они соединены, образуют циклопропильное кольцо формулы
R2A и R2B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют циклическую группу формулы
n обозначает число 1, 2 или 3,
R3 является водородом, фтором, метилом или трифторметилом,
R4 представляет собой водород, фтор, хлор, цианогруппу, метил, трифторметил или этил,
R5A представляет собой метил, трифторметил или этил,
R5B является трифторметилом,
или
R5A и R5B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют дифторзамещенное циклоалкильное кольцо формулы
R6 представляет собой водород, фтор, хлор, бром, цианогруппу, алкил с 1-4 атомами углерода или алкенил с 2-4 атомами углерода, причем алкил с 1-4 атомами углерода и алкенил с 2-4 атомами углерода, со своей стороны, могут содержать до трех атомов фтора в качестве заместителей,
и
R7 представляет собой водород, фтор, хлор или метил,
а также их соли, сольваты и сольваты солей.
Предпочтительными в рамках данного изобретения являются соединения формулы (I), в которых
R1A представляет собой водород, метил, трифторметил, этил, н-пропил, циклопропил или циклобутил,
R1B является водородом или метилом,
R2A представляет собой водород, метил, трифторметил, этил или н-пропил,
R2B является водородом или метилом, или
R2A и R2B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют циклическую группу формулы
n обозначает число 1 или 2,
R3 является водородом, фтором или метилом,
R4 представляет собой водород, фтор, хлор, цианогруппу, метил или трифторметил,
R5A представляет собой метил или этил,
R5B является трифторметилом,
или
R5A и R5B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют дифторзамещенное циклоалкильное кольцо формулы
R6 представляет собой фтор, хлор, алкил с 1-4 атомами углерода, алкенил с 2-3 атомами углерода, циклопропил или циклобутил, причем
алкил с 1-4 атомами углерода и алкенил с 2-3 атомами углерода могут содержать до трех атомов фтора в качестве заместителей,
а
циклопропил и циклобутил могут содержать до двух атомов фтора в качестве заместителей,
и
R7 представляет собой водород, фтор, хлор, метил или метоксигруппу,
а также их соли, сольваты и сольваты солей.
Еще один предпочтительный вариант исполнения данного изобретения включает соединения формулы (I), в которой
R1A представляет собой водород, метил, трифторметил, этил или н-пропил,
R1B является водородом или метилом,
R2A представляет собой водород, метил, трифторметил или этил,
R2B является водородом или метилом, или
R2A и R2B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют циклическую группу формулы
n обозначает число 1 или 2,
R3 является водородом или фтором,
R4 представляет собой водород, фтор, хлор, цианогруппу, метил или трифторметил,
R5A представляет собой метил или этил,
R5B является трифторметилом,
или
R5A и R5B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют дифторзамещенное циклоалкильное кольцо формулы
R6 представляет собой фтор, хлор, алкил с 1-4 атомами углерода или алкенил с 2-3 атомами углерода, причем алкил с 1-4 атомами углерода и алкенил с 2-3 атомами углерода, со своей стороны, могут содержать до трех атомов фтора в качестве заместителей,
и
R7 представляет собой водород, фтор или хлор,
а также их соли, сольваты и сольваты этих солей.
Особенно предпочтительными в рамках данного изобретения являются соединения формулы (I), в которой
R1A представляет собой водород, метил или этил,
R1B является водородом,
R2A представляет собой водород, метил, трифторметил, этил или н-пропил,
R2A является водородом или метилом,
или
R2A и R2B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют циклическую группу формулы
n обозначает число 1 или 2,
R3 является водородом,
R4 представляет собой фтор, хлор или метил,
R5A представляет собой метил,
R5B является трифторметилом, или
R5A и R5B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют дифторзамещенное циклопентильное кольцо формулы
R6 представляет собой фтор, хлор, метил, трифторметил, этил, 1,1-дифторэтил, 2,2,2-трифторэтил, изопропил, трет-бутил, 1,1,1-трифтор-2-метилпропан-2-ил, винил, 1-фторвинил, циклопропил, 2,2-дифторциклопропил, циклобутил или 3,3-дифторциклобутил,
и
R7 представляет собой водород, фтор, хлор или метил, а также их соли, сольваты и сольваты солей.
Другой особенно предпочтительный вариант исполнения данного изобретения включает соединения формулы (I), в которой
R1A представляет собой водород, метил или этил,
R1B является водородом,
R2A представляет собой водород или метил,
R2B является водородом,
или
R2A и R2B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют циклическую группу формулы
n обозначает число 1 или 2,
R3 является водородом,
R4 представляет собой фтор, хлор или метил,
R5A представляет собой метил,
R5B является трифторметилом, или
R5A и R5B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют дифторзамещенное циклопентильное кольцо формулы
R6 представляет собой атом хлора, метил, трифторметил, этил, 1,1-дифторэтил, 2,2,2-трифторэтил, изопропил, трет-бутил, 1,1,1-трифтор-2-метилпропан-2-ил, винил или 1-фторвинил,
и
R7 представляет собой водород или фтор,
а также их соли, сольваты и сольваты солей.
Особая форма исполнения данного изобретения включает соединения формулы (I), в которой
R1A представляет собой водород, метил или этил,
а
R1A, R2A и R2B соответственно являются атомами водорода,
а также их соли, сольваты и сольваты этих солей.
Другая особая форма исполнения данного изобретения включает соединения формулы (I), в которой
R2A представляет собой метил, трифторметил, этил или н-пропил,
а
R1A, R1B и R2B соответственно являются атомами водорода,
а также их соли, сольваты и сольваты этих солей.
Другая особая форма исполнения данного изобретения включает соединения формулы (I), в которой
R1A и R1B соответственно являются атомами водорода,
а
R2A и R2B соответственно представляют собой метил,
а также их соли, сольваты и сольваты этих солей.
Другая особая форма исполнения данного изобретения включает соединения формулы (I), в которой
R1A и R1B соответственно являются атомами водорода, а
R2A и R2B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют циклопропильное или циклобутильное кольцо формулы
а также их соли, сольваты и сольваты этих солей.
Другая особая форма исполнения данного изобретения включает соединения формулы (I), в которой
R3 является водородом,
и
R4 представляет собой фтор или хлор,
а также их соли, сольваты и сольваты солей.
Другая особая форма исполнения данного изобретения включает соединения формулы (I), в которой
R5A является метилом,
и
R5B является трифторметилом,
а также их соли, сольваты и сольваты солей.
Еще один отдельный вариант исполнения данного изобретения включает соединения формулы (I), в которой
R5A и R5B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют дифторзамещенное циклопентильное кольцо формулы
а также их соли, сольваты и сольваты солей.
Другая особая форма исполнения данного изобретения включает соединения формулы (1-А)
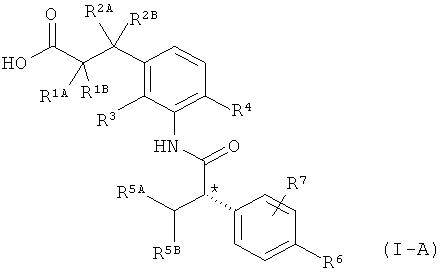
в которой атом углерода, обозначенный *, содержит фенилацетамидную группу представленной S-конфигурации,
а
остатки R1A, R1B, R2A, R2B, R3, R4, R5A, R5B, R6 и R7 соответственно имеют значения, приведенные выше,
а также их соли, сольваты и сольваты солей.
Определения остатков, приведенные в отдельности в соответствующих комбинациях или предпочтительных комбинациях остатков, независимо от этих соответствующих указанных комбинаций остатков также произвольно заменяются определениями остатков других комбинаций.
Наиболее предпочтительными являются комбинации двух или более из указанных выше предпочтительных областей.
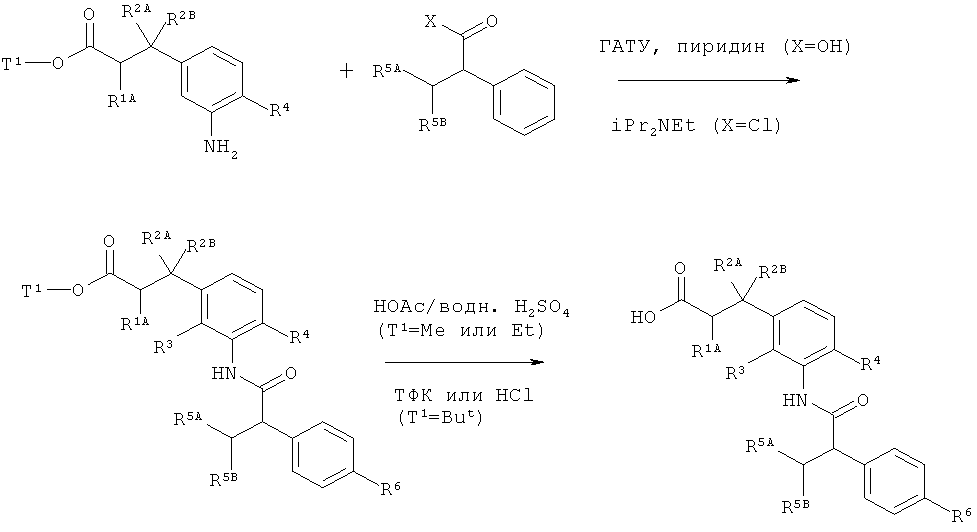
Другим объектом изобретения является способ получения соединений формулы (I) согласно изобретению, отличающийся тем, что карбоновую кислоту формулы (II)

в которой R5A, R5B, R6 и R7 имеют указанные выше значения,
в инертном растворителе, с помощью конденсирующего агента или через промежуточную стадию соответствующего хлорангидрида карбоновой кислоты, в присутствии основания подвергают сочетанию с амином формулы (III)

в которой R1A, R1B, R2A, R2B, R3 и R4 имеют указанные выше значения,
а
Т1 представляет собой алкил с 1-4 атомами углерода или бензил,
с образованием амида карбоновой кислоты формулы (IV)
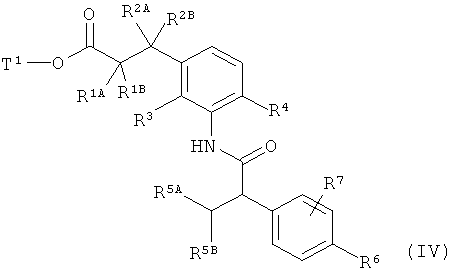
в которой R1A, R1B, R2A, R2B, R3, R4, R5A, R5B, R6, R7 и Т1 имеют значения, указанные выше,
а затем отщепляют остаток сложного эфира Т1 при помощи основного или кислотного сольволиза или, в случае если Т1 представляет собой бензил, также с помощью гидрогенолиза, с получением карбоновой кислоты формулы (I),
и при необходимости соединения формулы (I) с помощью методов известных специалисту разделяют на их энантиомеры и/или диастереомеры и/или превращают в их сольваты, соли и/или сольваты солей с помощью соответствующих растворителей (I) и/или оснований (ii).
Инертными растворителями для стадии процесса (II)+(III)→(IV) [сочетание с образованием амида] являются, например, простые эфиры, такие как простой диэтиловый эфир, простой метил-трет-бутиловый эфир, тетрагидрофуран, 1,4-диоксан, простой гликольдиметиловый эфир или простой диэтиленгликольдиметиловый эфир, углеводороды, такие как бензол, толуол, ксилол, гексан, циклогексан или нефтяные фракции, галогенуглеводороды, такие как дихлорметан, трихлорметан, тетрахлорметан, 1,2-дихлорэтан, трихлорэтилен или хлорбензол, или другие растворители, такие как ацетон, ацетонитрил, этилацетат, пиридин, диметилсульфоксид (ДМСО), N,N-диметилформамид (ДМФА), N,N'-диметилпропиленмочевина (ДМПМ) или N-метилпирролидон (NMP). Так же возможно использовать смеси указанных растворителей. Предпочтительно применяются дихлорметан, тетрагидрофуран, диметилформамид или смеси этих растворителей.
В качестве конденсирующих агентов для этой реакции сочетания подходят, например, карбодиимиды, такие как N,N'-диэтил-, N,N'-дипропил-, N,N'-диизопропил-, N,N'-дициклогексилкарбодиимид (ДЦК) или гидрохлорид N-(3-диметиламиноизопропил)-N'-этилкарбодиимида (ЭДК), производные фосгена, такие как N,N'-карбонилдиимидазол (КДИ), 1,2-оксазолиевые соединения, такие как 2-этил-5-фенил-1,2-оксазолий-3-сульфат или перхлорат 2-трет-бутил-5-метилизоксазолия, ациламиносоединения, такие как 2-этокси-1-этокси-1,2-дигидрохинолин или изобутилхлорформиат, 1-хлор-2-метил-1-диметиламино-1-пропен, ангидрид пропанфосфоновой кислоты, сложный диэтиловый эфир цианофосфоновой кислоты, бис(2-оксо-3-оксазолидинил)фосфорилхлорид, гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония, гексафторфосфат бензотриазол-1-илокситрис(пирролидино)фосфония (Ру-ВОР), тетрафторборат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (ТБТУ), гексафторфосфат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (ГБТУ), тетрафторборат 2-(2-оксо-1-(2H)-пиридил)-1,1,3,3-тетраметилурония (ТПТУ), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (ГАТУ) или тетрафторборат O-(1H-6-хлорбензотриазол-1-ил)-1,1,3,3-тетраметилурония (TCTU), при необходимости в сочетании с другими вспомогательными веществами, такими как 1-гидроксибензотриазол (HOBt) или N-гидроксисукцинимид (HOSu), а также в качестве оснований карбонаты щелочных металлов, например, карбонаты натрия или калия, или органические основания, такие как триэтиламин, N-метилморфолин, N-метипиперидин, N,N-диизопропилэтиламин, пиридин или 4-N,N-диметиламинопиридин. Предпочтительно используются гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (ГАТУ) или тетрафторборат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (ТБТУ), соответственно в комбинации с пиридином или N,N-диизопропилэтиламином, или гидрохлорид N-(3-диметиламиноизопропил)-N'-этилкарбодиимида (ЭДК) в сочетании с 1-гидроксибензотриазолом (HOBt) и триэтиламином, или 1-хлор-2-метил-1-диметиламино-1-пропен совместно с пиридином.
Реакция (II)+(III)→(IV), как правило, проводится в интервале температур от 0°С до +60°С, предпочтительно от +10°С до +40°С.
При использовании одного из соответствующих соединению (II) хлорангидридов карбоновых кислот это сочетание с аминным компонентом (III) проводится в присутствии обычного органического вспомогательного основания, такого как триэтиламин, N-метилморфолин, N-метилпиперидин, N,N-диизопропилэтиламин, пиридин, 4-N,N-диметиламинопиридин, 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ) или 1,5-диазабицикло[4.3.0]нон-5-ен (ДБН). Предпочтительно применяется триэтиламин или N,N-диизопропилэтиламин.
Взаимодействие амина (III) с хлорангидридом карбоновой кислоты, как правило, осуществляется в интервале температур от -20°С до +60°С, предпочтительно в интервале от -10°С до +30°С.
Само получение хлорангидрида карбоновой кислоты осуществляется обычным способом путем обработки карбоновой кислоты (II) тионилхлоридом или оксалилхлоридом.
Отщепление сложноэфирной группы Т1 на стадии процесса (IV)→(I) проводится по обычным способам, путем того, что сложный эфир в инертном растворителе обрабатывают кислотами или основаниями, причем в случае последнего варианта, образующаяся сначала соль, переводится в свободную карбоновую кислоту в результате обработки кислотой. В случае сложного mpem-бутилового эфира это расщепление сложного эфира предпочтительно осуществляется с помощью кислот. Сложные бензиловые эфиры предпочтительно расщепляются путем гидрогенолиза (гидрирования) в присутствии подходящего катализатора, такого как, например, палладий на активированном угле.
В качестве инертного растворителя для этой реакции подходят вода или обычные для расщепления сложных эфиров органические растворители. К таким предпочтительно относятся спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол или mpem-бутанол, или простые эфиры, такие как простой диэтиловый эфир, тетрагидрофуран, диоксан или простой гликольдиметиловый эфир, или другие растворители, такие как ацетон, дихлорметан, диметилформамид или диметилсульфоксид. Также возможно использовать смеси указанных растворителей. В случае гидролиза сложного эфира с применением основания предпочтительно используются смеси воды с диоксаном, тетрагидрофураном, метанолом и/или этанолом. В случае взаимодействия с трифторуксусной кислотой предпочтительно применяется дихлорметан, а в случае взаимодействия с хлороводородом предпочтительно тетрагидрофуран, простой диэтиловый эфир, диоксан или вода.
В качестве оснований подходят обычные неорганические основания. К таким относятся, прежде всего, гидроксиды щелочных или щелочноземельных металлов, такие как, например, гидроксиды лития, натрия, калия или бария, или карбонаты щелочных или щелочноземельных металлов, такие как карбонаты натрия, калия или кальция. Предпочтительными являются гидроксиды лития, натрия или калия.
В качестве кислот для расщепления сложных эфиров, как правило, подходящими являются серная кислота, хлороводород/соляная кислота, бромоводород/бромоводородная кислота, фосфорная кислота, уксусная кислота, трифторуксусная кислота, толуолсульфокислота, метансульфокислота или трифторметансульфокислота, или их смеси при необходимости с добавлением воды. Предпочтительными являются хлороводород или трифторуксусная кислота в случае сложных mpem-бутиловых эфиров и соляная кислота в случае сложных метиловых эфиров.
Это расщепление сложного эфира, как правило, осуществляется в интервале температур от -20°С до +100°С, предпочтительно от 0°С до +60°С.
Интермедиаты формулы (II) могут получаться, например, с помощью того, что сложный эфир карбоновой кислоты формулы (V)
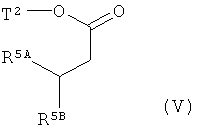
в которой R5A и R5B имеют значения, указанные выше,
а
Т2 представляет собой алкил с 1-4 атомами углерода или бензил,
сначала депротонируют в инертном растворителе с помощью основания, затем в присутствии подходящего палладиевого катализатора арилируют с помощью фенилбромида формулы (VI)

в которой R6 и R7 имеют значения, указанные выше, с получением соединения формулы (VII)
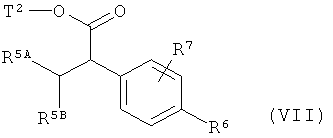
в которой R5A, R5B, R6, R7 и Т2 имеют значения, указанные выше,
а после этого отщепляют сложноэфирный остаток Т2 путем основного или кислотного сольволиза, или в случае если Т2 представляет собой бензил, также с помощью гидрогенолиза, с получением карбоновой кислоты (II).
Реакция арилирования на стадии процесса (V)+(VI)→(VII) предпочтительно проводится в толуоле или смесях толуол/тетрагидрофуран в интервале температур от +20°С до +100°С. В качестве основания для депротонирования сложного эфира (V) при этом предпочтительно используется бис(триметилсилил)амид лития. Подходящими палладиевыми катализаторами являются, например, ацетат палладия (II) или трис(дибензилиденацетон)дипалладий, соответственно в сочетании с богатыми электронами, пространственно требовательными фосфиновыми лигандами, такими как 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил или 2-ди-трет-бутилфосфино-2'-(N,N-диметиламино)бифенил [сравн. например, с W.A.Moradi, S.L.Buchwald, J.Am. Chem. Soc. 123. 7996-8002 (2001)].
Отщепление сложноэфирной группы Т2 на стадии процесса (VII)→(II) осуществляется таким же образом, как описано ранее для сложноэфирного остатка Т1.
Интермедиаты формулы (II-A)
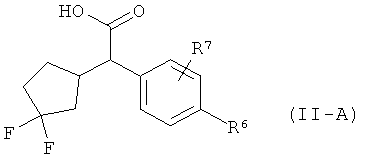
в которой R6 и R7 имеют значения, указанные выше,
в качестве альтернативы также могут получаться в результате того, что сложный эфир фенилуксусной кислоты формулы (VIII)

в которой R6, R7 и Т2 имеют значения, указанные выше,
сначала с помощью вызываемого под действием основания присоединения 2-циклопентен-1-она переводят в соединение формулы (IX)

в которой R6, R7 и Т2 имеют значения, указанные выше, затем это соединение фторируют с помощью 1,1'-[(трифтор-λ4-сульфанил)имино]бис(2-метоксиэтана) в условиях катализа трифторидом бора с получением соединения формулы (VII-A)
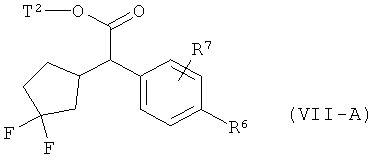
в которой R6, R7 и Т2 имеют значения, указанные выше,
а затем, в свою очередь, отщепляют сложноэфирную группу Т2 с получением карбоновой кислоты (II-A).
На стадии процесса (VIII)→(IX) для депротонирования сложного эфира (VIII) предпочтительно применяется амидное основание, такое как диизопропиламид лития или бис(триметилсилил)амид лития. Для деоксифторирования в превращении (IX)→(VII-A) вместо указанного выше 1,1'-[(трифтор-λ4-сульфанил)имино]бис(2-метоксиэтана) (деоксофтор реагент) при необходимости также могут применяться другие известные фторирующие реагенты, такие как трифторид диэтиламиносеры (DAST) или трифторид морфолиносеры (Morpho-DAST), [для последовательности реакций (VIII)→(IX)→(VII-A) сравн., например, с Т.Mase с соавт., J.Org. Chem. 66 (20), 6775-6786 (2001)].
Интермедиаты формулы (III) могут получаться, например, в результате того, что или
[А] сложный эфир фосфоноуксусной кислоты формулы (X)

в которой R1A и Т1 имеют значения, указанные выше,
и
R8 представляет собой алкил с 1-4 атомами углерода,
в инертном растворителе подвергают превращению в реакции олефинирования, вызываемой действием основания, с 3-нитробензоильным соединением формулы (XI)
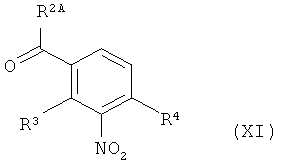
в которой R2A, R3 и R4 имеют значения, указанные выше, с образованием соединения формулы (XII)

в которой R1A, R2A, R3, R4 и Т1 имеют значения, указанные выше,
а это соединение потом в присутствии подходящего палладиевого или платинового катализатора гидрируют с получением сложного эфира 3-(3-аминофенил)пропионовой кислоты формулы (III-A)
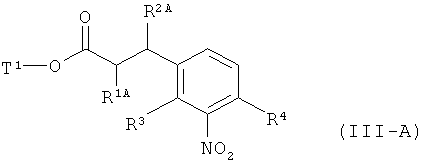
в которой R1A, R2B, R3, R4 и Т1 имеют значения, указанные выше,
или
[В] сложный эфир акриловой кислоты формулы (XIII)
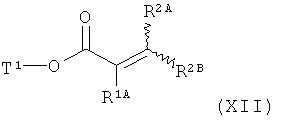
в которой R1A, R2A, R2B и Т1 имеют значения, указанные выше, в инертном растворителе подвергают взаимодействию либо в условиях (i) катализа родием(1) с фенилборной кислотой формулы (XIV)

в которой R3 и R4 имеют значения, указанные выше,
PG в качестве инертной защитной группы на амине является бензилом или п-метоксибензилом,
либо в условиях (ii) катализа медью (I) с фенилмагниевым реагентом формулы (XV)
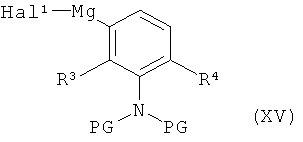
в которой R3, R4 и PG имеют значения, указанные выше,
а
Hal1 представляет собой хлор или бром,
с получением соединения формулы (XVI)

в которой R1A, R2A, R2B, R3, R4, PG и Т1 имеют значения, указанные выше,
а затем удаляют защитные группы на амине PG в соответствии с обычными методами путем гидрогенолиза или окислительным способом с получением сложного эфира 3-(3-аминофенил)пропионовой кислоты формулы (III-B)
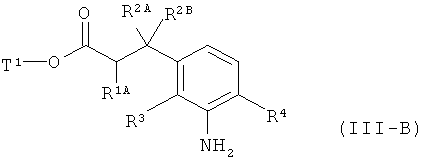
в которой R1A, R2A, R2B, R3, R4 и Т1 имеют значения, указанные выше,
или
[С] сложный эфир акриловой кислоты формулы (XVII)
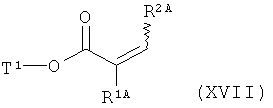
в которой R1A, R2A и Т1 имеют значения, указанные выше,
в инертном растворителе в условиях палладиевого катализа вводят в реакцию сочетания с 3-амино- или 3-нитрофенилбромидом формулы (XVIII)

в которой R3 и R4 имеют значения, указанные выше,
и
R9 является амино- или нитрогруппой,
с получением соединения формулы (XIX)

в которой R1A, R2A, R3, R4, R9 и Т1 имеют значения, указанные выше,
а это соединение в присутствии подходящего палладиевого или платинового катализатора гидрируют с получением сложного эфира 3-(3-аминофенил)пропионовой кислоты формулы (III-C)

в которой R1A, R2A, R3, R4 и Т1 имеют значения, указанные выше,
или
[D] сложный эфир формулы (XX)

в которой R1A, R1B и Т1 имеют значения, указанные выше,
в инертном растворителе после а-депротонирования алкилируют 3-бромбензилгалогенидом формулы (XXI)
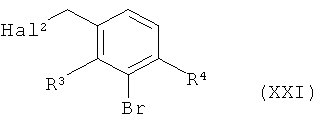
в которой R3 и R4 имеют значения, указанные выше,
и
Hal2 представляет собой хлор, бром или йод,
с получением соединения формулы (XXII)

в которой R1A, R1B, R3, R4 и Т1 имеют значения, указанные выше,
затем вводят во взаимодействие с бензиламином в присутствии основания и палладиевого катализатора с получением соединения формулы (XXIII)
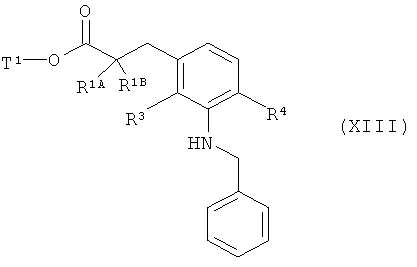
в которой R1A, R1B, R3, R4 и Т1 имеют значения, указанные выше,
а потом N-бензильную группу удаляют путем гидрогенолиза с получением сложного эфира 3-(3-аминофенил)пропионовой кислоты формулы (III-D)

в которой R1A, R1B, R3, R4 и Т1 имеют значения, указанные выше.
Для депротонирования сложного фосфонового эфира (X) в реакции олефинирования (X)+(XI)→(XII) особенно подходящими являются ненуклеофильные сильные основания, такие как, например, гидрид натрия или калия, бис(триметилсилил)амиды лития, натрия или калия или диизопропиламид лития; предпочтительно применяется гидрид натрия.
Гидрирование на стадии процесса (XII)→(III-А) или соответственно (XIX)→(III-C), как правило, проводится в атмосфере водорода при постоянном нормальном давлении. При этом в качестве катализатора предпочтительно используется палладий на активированном угле (в качестве материала-носителя). Удаление защитной группы (или групп) на амине в превращениях (XVI)→(III-В) и (XXIII)→(III-D) обычно осуществляется путем гидрогенолиза по аналогичной методике, в случае, если PG в (XVI) является п-метоксибензилом, в качестве альтернативы это может происходить также путем окисления, например, с помощью 2,3-дихлор-5,6-дициано-1,4-бензохинона (ДДХ) или церий (IV) аммонийнитрата.
В качестве палладиевого катализатора для взаимодействия (XVII)+(XVIII)→(XIX) [реакция Хека] предпочтительно используется ацетат палладия(II) в сочетании с фосфиновым лигандом, таким как, например, трифенил- или три-2-толилфосфин [для реакции (XIII)+(XIV)→(XVI) сравн., например, с N.Miyaura с соавт., Organometallics 16, 4229 (1997), а также Т.Hayashi, Synlett, Special Issue 2001, 879-887; для взаимодействия (XIII)+(XV)→(XVI) сравн., например, с Р.Knochel с соавт., Tetrahedron 56, 2727-2731 (2000), Angew. Chem. 120. 6907-6911 (2008)].
Для α-депротонирования сложного эфира (XX) в реакции алкилирования (XX)+(XXI)→(XXII) точно так же особенно подходящими являются ненуклеофильные сильные основания, такие как, например, гидрид натрия или калия, бис(триметилсилил)амиды лития, натрия или калия или диизопропиламид лития; предпочтительно для этого применяется диизопропиламид лития.
Для реакции (XXII)+бензиламин→(XXIII) [сочетание Бухвальда-Хартвига] в качестве палладиевого катализатора предпочтительно применяется трис(дибензилиденацетон)дипалладий (0) в сочетании с (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафтилом в качестве фосфинового лиганда и тpeт-бутилатом натрия или калия в качестве основания [сравн., например, с J.P.WoIfe und S.L.Buchwald, Organic Syntheses, Coll. Vol.10, 423 (2004), Vol.78, 23 (2002)].
Описанные выше стадии процесса могут проводиться при нормальном, при повышенном или при пониженном давлении (например, в области от 0,5 до 5 бар); как правило, в каждом из случаев работают при нормальном давлении.
Разделение соединений согласно изобретению на соответствующие энантиомеры и/или диастереомеры при необходимости, в зависимости от целесообразности, может также осуществляться уже на стадии соединений (II), (III), (IV), (VII), (XVI), (XXII) или (XXIII), которые потом далее соответственно подвергаются превращениям в описанных ранее последовательностях процессов в изолированной форме. Такое разделение стереоизомеров может проводиться согласно обычным, известным специалисту методам. Предпочтительно применяются хроматографические способы на ахиральных или хиральных разделяющих фазах; в случае карбоновых кислот в качестве промежуточных или конечных продуктов как альтернатива может также осуществляться разделение через диастереомерные соли.
Соединения формул (V), (VI), (VIII), (X), (XI), (XIII), (XIV), (XV), (XVII), (XVIII), (XX) и (XXI) являются или коммерчески доступными, или описаны как таковые в литературе, или они могут получаться по аналогии с методами, опубликованными в литературе, по способам, очевидным для специалиста. Многочисленные подробные методики, а также литературные данные для получения исходных веществ также находятся в экспериментальной части в разделе, касающемся получения исходных соединений и интермедиатов.
Получение соединений согласно изобретению в качестве примеров может быть наглядно представлено с помощью следующих схем реакций:
Схема 1
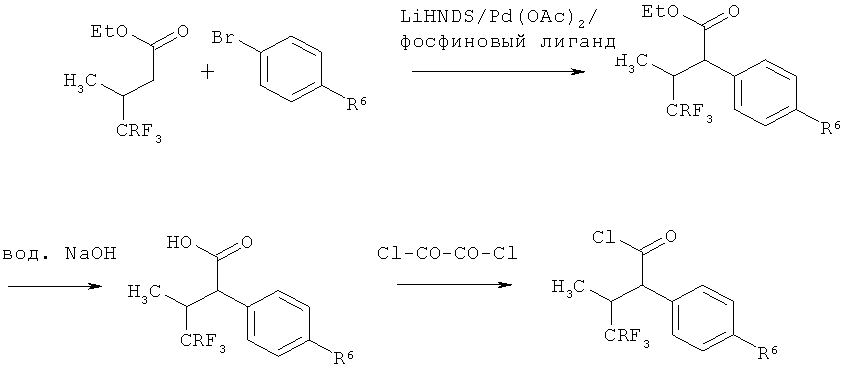
Схема 2
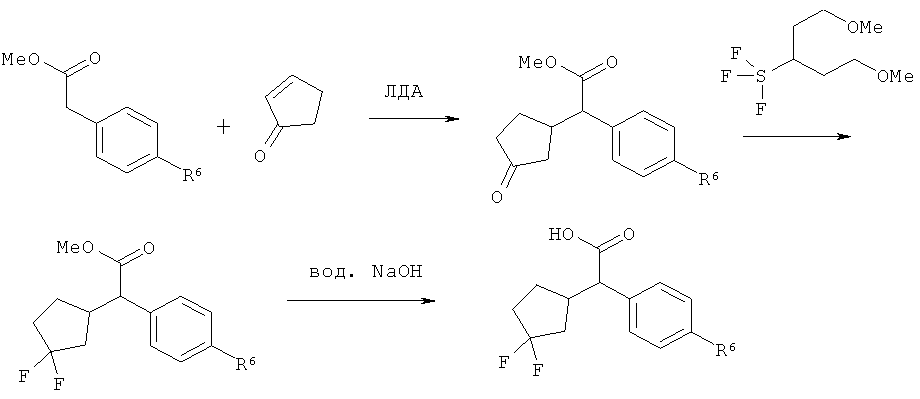
Схема 3
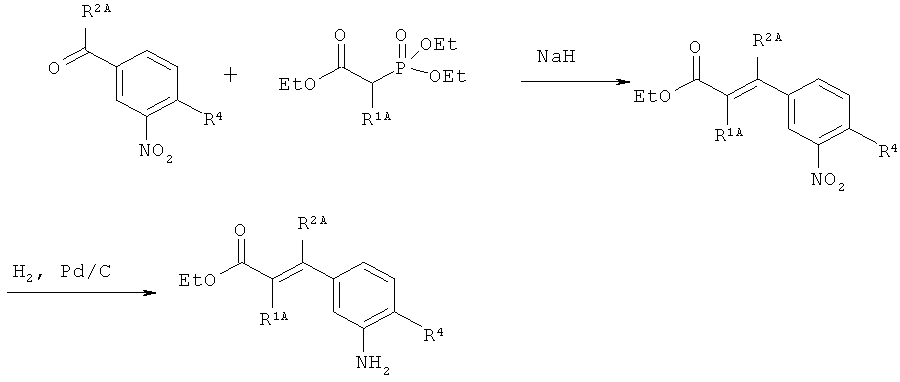
Схема 4

Схема 5

[PMB = п-метоксибензил; A=CH2 или O; R=метил или бензил].
Схема 6

[Bn = бензил].
Схема 7
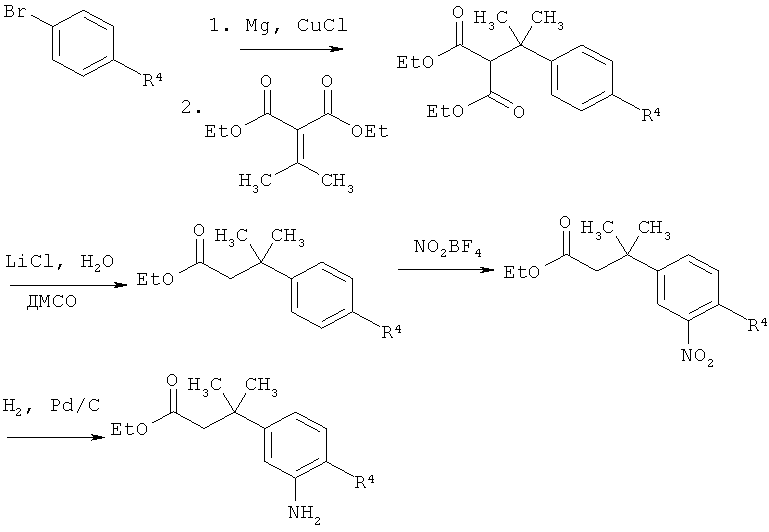
Схема 8
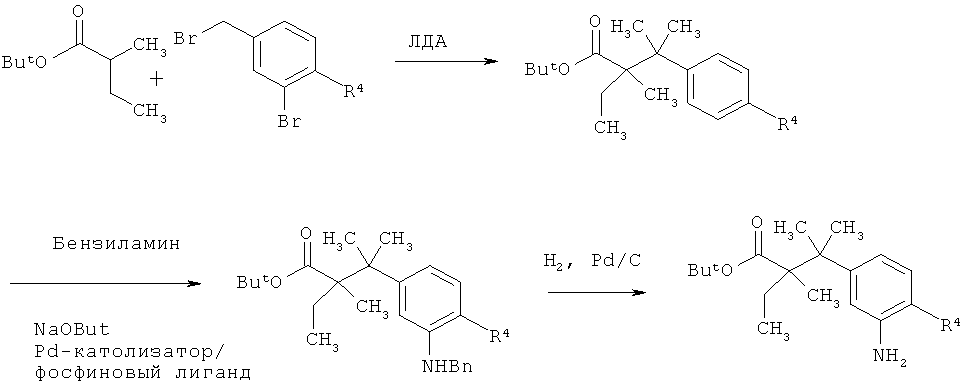
Схема 9
Соединения согласно изобретению обладают ценными фармакологическими свойствами и могут применяться для профилактики и лечения заболеваний у людей и животных.
Соединения согласно изобретению представляют собой мощные активаторы растворимой гуанилатциклазы. Они приводят к расслаблению сосудов, подавлению агрегации тромбоцитов и к снижению кровяного давления, а также к увеличению коронарного кровотока. Этим эффектам способствует непосредственная гемнезависимая активация растворимой гуанилатциклазы и рост внутриклеточного содержания циклического гуанозинмонофосфата (цГМФ).
Кроме того, соединения согласно изобретению обладают хорошими фармакокинетическими характеристиками, особенно в отношении их биодоступности и их периода полувыведения в организме.
Таким образом, соединения согласно изобретению могут использоваться в лекарственных средствах для лечения и/или профилактики сердечно-сосудистых заболеваний, таких как, например, повышенное кровяное давление (гипертония) и сердечная недостаточность, стабильная и нестабильная стенокардия, легочная артериальная гипертензия (ЛАГ) и другие формы легочной гипертензии (ЛГ), почечная гипертензия, заболевания периферических и сердечных сосудов, а также аритмий, для лечения тромбоэмболических заболеваний и ишемий, таких как инфаркт миокарда, инсульт, транзисторные и ишемические приступы, а также нарушения периферического кровообращения, для предотвращения рестенозов, таких как после тромболитической терапии, чрескожной транслюминальной ангиопластики (ЧТА), чрескожной транслюминальной коронарной ангиопластики (ЧТКА) и шунтирования, для лечения артериосклероза, для стимуляции заживления ран, а также для лечения остеопороза, глаукомы и гастропареза.
Согласно данному изобретению понятие сердечная недостаточность включает как острые, так и хронические формы проявления сердечной недостаточности, также как и специфические или родственные нозологические формы от этого, такие как острая декомпенсированная сердечная недостаточность, недостаточность правых отделов сердца, недостаточность левых отделов сердца, глобальная сердечная недостаточность, ишемическая кардиомиопатия, дилатационная кардиомиопатия, гипертрофическая кардиомиопатия, идиопатическая кардиомиопатия, врожденный порок сердца, порок клапанов сердца, сердечная недостаточность при пороках клапанов сердца, стеноз митрального клапана, недостаточность митрального клапана, стеноз аортального клапана, недостаточность клапана аорты, трикуспидальный стеноз, недостаточность трехстворчатого клапана, стеноз клапана легочной артерии, недостаточность клапана легочной артерии, комбинированные пороки клапанов сердца, воспаление сердечной мышцы (миокардит), хронический миокардит, острый миокардит, вирусный миокардит, сердечная недостаточность, вызванная диабетом, токсическая алкогольная кардиомиопатия, кардиальные болезни накопления, а также диастолическая и систолическая сердечная недостаточность.
Кроме того, соединения согласно изобретению могут применяться для лечения и/или профилактики первичного и вторичного феномена Рейно, нарушений микроциркуляции, перемежающейся хромоты, шума в ушах, периферических и автономных нейропатий, диабетических микроангиопатий, диабетической ретинопатии, диабетических язв на конечностях, CREST-синдрома, эритематоза, оникомикоза, а также ревматических заболеваний.
Соединения согласно изобретению, помимо этого, могут находить применение для предотвращения ишемических и/или реперфузионных повреждений органов или тканей, а также в качестве добавляемых веществ для перфузионных или консервирующих растворов для органов, частей органов, тканей или частей тканей человеческого или животного происхождения, особенно при хирургических вмешательствах или в области медицины трансплантаций.
Кроме того, соединения согласно изобретению подходят для лечения и/или профилактики заболеваний почек, особенно при отказе почек и почечной недостаточности. Согласно данному изобретению понятия почечной недостаточности и отказа почек включают как острые, так и хронические формы их проявления, также как и лежащие в их основе или родственные этим заболеваниям почек, такие как почечная гипоперфузия, интрадиалитическая гипотензия, обструктивная уропатия, гломерулопатии, гломерулонефрит, острый гломерулонефрит, гломерулосклероз, тубулоинтерсциальные заболевания, нефропатические заболевания, такие как первичные и врожденные заболевания почек, воспаления почек, иммунологические заболевания почек, такие как отторжение почечного трансплантата, и заболевания почек, вызванные иммунным комплексом, нефропатия, вызванная токсическими веществами, нефропатия, вызванная рентгеноконтрастным веществом, диабетическая и недиабетическая нефропатия, пиелонефрит, кисты почек, нефросклероз, гипертензивный нефросклероз и нефротический синдром, которые в плане диагностики могут характеризоваться, например, через анормально пониженное выведение креатинина и/или воды, анормально повышенными концентрациями в крови мочевины, азота, калия и/или креатинина, измененной активностью почечных ферментов, таких как, например, глютамилсинтетазы, измененными осмотической концентрацией мочи или количеством мочи, повышенной микроальбуминурией, макроальбуминурией, повреждениями гломерул и артериол, тубулярной дилатацией, гиперфосфатемией и/или потребностью в диализе. Данное изобретение также включает применение соединений согласно изобретению для лечения и/или профилактики вторичных проявлений почечной недостаточности, таких как, например, гипертония, отек легких, сердечная недостаточность, уремия, анемия, электролитные нарушения (к примеру, гиперкалиемия, гипонатриемия) и нарушения в метаболизме костей и углеводов.
Кроме того, соединения согласно изобретению подходят для лечения и/или профилактики заболеваний мочеполовой системы, таких как, например, гиперактивность мочевого пузыря, нарушения мочеиспускания, симптомы со стороны нижних мочевыводящих путей (LUTS), недержание, доброкачественная гиперплазия предстательной железы (ДГПЖ), эректильная дисфункция и женская половая дисфункция.
Помимо этого, соединения согласно изобретению могут использоваться для лечения астматических заболеваний, хронической обструктивной болезни легких (ХОБЛ) и респираторного дистресс-синдрома.
Соединения, описанные в данном изобретении, также представляют собой активные вещества для лечения заболеваний в центральной нервной системе, которые характеризуются нарушениями системы NO/цГМФ. Особенно они являются подходящими для улучшения восприятия, способности к концентрации, способности к обучению или функций памяти после когнитивных нарушений, как те, что встречаются, в частности, при обстоятельствах/заболеваниях/синдромах, таких как «умеренные когнитивные нарушения», связанные с возрастом задержки процесса обучения и нарушения памяти, связанная с возрастом потеря памяти, сосудистая деменция, черепно-мозговая травма, кровоизлияние в мозг, приобретенное слабоумие, которое встречается после инсультов («постинсультная деменция»), посттравматическая черепно-мозговая травма, общие нарушения концентрации, нарушения концентрации у детей с проблемами обучения и запоминания, болезнь Альцгеймера, болезнь диффузных телец Леви, приобретенное слабоумие с дегенерацией лобных долей головного мозга, включая синдром Пика, болезнь Паркинсона, прогрессирующий ядерный паралич, приобретенное слабоумие с кортикобазальной дегенерацией, боковой амиотрофический склероз (БАС), болезнь Хантингтона, множественный склероз, таламическая дегенерация, болезнь Крейтцфельда-Якоба, слабоумие, вызванное ВИЧ, шизофрения с деменцией или психоз Корсакова. Они также подходят для лечения заболеваний центральной нервной системы, таких как тревожные, напряженные и депрессивные состояния, сексуальных расстройств и нарушений сна, обусловленных состоянием центральной нервной системы, а также для регулирования патологических нарушений приема пищи, стимулянтов и наркотических средств.
Кроме того, соединения согласно изобретению подходят также для регулирования мозгового кровообращения и представляют собой эффективные средства для борьбы с мигренями. Также они подходят для профилактики и борьбы с последствиями случаев церебральных инфарктов (апоплексических ударов), таких как кровоизлияния в мозг, церебральная ишемия и черепно-мозговая травма. Также соединения согласно изобретению могут использоваться для подавления болевых состояний.
Наряду с этим, соединения согласно изобретению обладают противовоспалительным действием и, таким образом, могут использоваться в качестве противовоспалительных средств для лечения и/или профилактики заражения крови, множественных повреждений органов, воспалительных заболеваний почек, хронических воспалений кишечника, таких как язвенный колит (Colitis ulcerosa) и болезнь Крона (Morbus Crohn), панкреатита, перитонита, ревматоидных заболеваний, воспалительных заболеваний кожи и воспалительных глазных заболеваний.
Благодаря своему профилю действия соединения согласно изобретению особенно подходят для лечения и/или профилактики сердечно-сосудистых заболеваний, таких как сердечная недостаточность, стенокардия, гипертензия, легочная гипертензия, ишемия, заболевания сосудов, нарушения микроциркуляции, тромбоэмболические заболевания и артериосклероз.
Другим объектом данного изобретения является применение соединений согласно изобретению для лечения и/или профилактики заболеваний, особенно заболеваний, указанных выше.
Другим объектом данного изобретения является применение соединений согласно изобретению для изготовления лекарственного средства для лечения и/или профилактики заболеваний, особенно заболеваний, указанных выше.
Другим объектом данного изобретения является применение соединений согласно изобретению в способе лечения и/или профилактики заболеваний, особенно заболеваний, указанных выше.
Другим объектом данного изобретения является способ лечения и/или профилактики заболеваний, особенно заболеваний, указанных выше, с применением эффективного количества по меньшей мере одного из соединений согласно изобретению.
Соединения согласно изобретению могут использоваться самостоятельно или в случае необходимости в комбинации с другими активными веществами. Другим объектом данного изобретения являются лекарственные средства, содержащие по меньшей мере одно из соединений согласно изобретению и одно или несколько других активных веществ, особенно для лечения и/или профилактики указанных выше заболеваний. Как подходящие комбинации активных веществ в качестве примерных и предпочтительных следует привести:
- органические нитраты и доноры NO, такие как, например, нитропруссид натрия, нитроглицерин, мононитрат изосорбида, динитрат изосорбида, молсидомин или SIN-1, а также ингаляционный NO;
- соединения, которые ингибируют распад циклического гуанозинмонофосфата (цГМФ), такие как, например, ингибиторы фосфодиэстераз (ФДЭ) 1, 2 и/или 5, особенно ингибиторы ФДЭ 5, такие как сильденафил, варденафил и тадалафил;
- NO-независимые, однако гемзависимые стимуляторы гуанилатциклазы, такие как соединения, описанные, в частности, в международных заявках WO 00/06568, WO 00/06569, WO 02/42301 и WO 03/095451;
- средства с антитромботическим действием, например, и предпочтительно из группы средств, подавляющих агрегацию тромбоцитов, антикоагулянтов или профибринолитических веществ;
- активные вещества, снижающие кровяное давление, например, и предпочтительно из группы антагонистов кальция, антагонистов ангиотензина АН, ингибиторов ангиотензинпревращающего фермента (АСЕ), антагонистов эндотелина, ингибиторов ренина, блокаторов альфа-рецепторов, блокаторов бета-рецепторов, антагонистов минералокортикоидных рецепторов, а также диуретиков; и/или
- активные вещества, изменяющие жировой обмен, например, и предпочтительно из группы агонистов рецепторов тиреоидных гормонов, ингибиторов синтеза холестерина, таких как приводимые в качестве примерных и предпочтительных ингибиторы HMG-CoA-редуктазы или синтеза сквалена, ингибиторы АСАТ, ингибиторы белка переноса холестеринового эфира (СЕТР), ингибиторы микросомального белка, переносящего триглицериды (МТР), агонисты альфа-рецепторов, активируемых пролифератором пероксисом (РАПП), гамма-РАПП- и/или дельта-РАПП, средства, подавляющие абсорбцию холестерина, ингибиторы липазы, полимерные адсорберы желчной кислоты, средства, подавляющие реабсорбцию желчной кислоты и антагонисты липопротеина (а).
Под средствами, обладающими антитромботическим действием, предпочтительно понимают соединения из группы средств, подавляющих агрегацию тромбоцитов, антикоагулянтов или профибринолитических веществ.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации со средством, подавляющим агрегацию тромбоцитов, таким как, приводимые в качестве примерных и предпочтительных, аспирин, клопидогрель, тиклопидин или дипиридамол.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с ингибитором тромбина, таким как, приводимые в качестве примерных и предпочтительных, ксимелагатран, мелагатран, бивалирудин или клексан.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с антагонистом GPIIb/IIIa, таким как, приводимые в качестве примерных и предпочтительных, тирофибан или абциксимаб.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с ингибитором фактора Xa, таким как, приводимые в качестве примерных и предпочтительных, ривароксабан, апиксабан, фидексабан, разаксабан, фондапаринукс, идрапаринукс, DU-176b, PMD-3112, YM-150, KFA-1982, EMD-503982, МСМ-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 или SSR-128428.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с гепарином или производным низкомолекулярного гепарина (НМГ).
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с антагонистом витамина К, таким как приводимый в качестве примерного и предпочтительного кумарин.
Под средствами, снижающими кровяное давление, предпочтительно понимают соединения из группы антагонистов кальция, антагонистов ангиотензина АН, ингибиторов ангиотензинпревращающего фермента (АСЕ), антагонистов эндотелина, ингибиторов ренина, блокаторов альфа-рецепторов, блокаторов бета-рецепторов, антагонистов минералокортикоидных рецепторов, а также диуретиков.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с антагонистом кальция, таким как приводимые в качестве примерных и предпочтительных, нифедипин, амлодипин, верапамил или дитиазем.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с блокатором альфа-1-рецепторов, таким как приводимый в качестве примерного и предпочтительного, празозин.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с блокатором бета-рецепторов, такими как приводимые в качестве примерных и предпочтительных, пропранолол, атенолол, тимолол, пиндолол, алпренолол, окспренолол, пенбутолол, бупранолол, метипранолол, надолол, мепиндолол, каразолол, соталол, метопролол, бетаксолол, селипролол, бисопролол, картеолол, эсмолол, лабеталол, карведилол, адапролол, ландиолол, небиволол, эпанолол или бусиндолол.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с антагонистом ангиотензина АН, таким как приводимые в качестве примерных и предпочтительных, лозартан, кандесартан, вальсартан, телмисартан или эмбурсатан.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с ингибитором ангиотензинпревращающего фермента (АСЕ), таким как приводимые в качестве примерных и предпочтительных эналаприл, каптоприл, лизиноприл, рамиприл, делаприл, фозиноприл, квиноприл, периндоприл или трандоприл.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с антагонистом эндотелина, таким как приводимые в качестве примерных и предпочтительных босентан, дарусентан, амбрисентан или ситаксентан.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с ингибитором ренина, таким как приводимые в качестве примерных и предпочтительных алискирен, SPP-600 или SPP-800.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с антагонистом минерал кортикоидных рецепторов, таким как приводимые в качестве примерных и предпочтительных спиронолактон или эплеренон.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с диуретиком, таким как приводимый в качестве примерного и предпочтительного фуросемид.
Под средствами, изменяющими жировой обмен, предпочтительно понимают соединения из группы ингибиторов белков переноса холестеринового эфира (СЕТР), агонистов рецепторов тиреоидных гормонов, ингибиторов синтеза холестерина, таких как ингибиторы гидроксиметилглутарил-кофермент A-редуктазы (HMG-CoA) или синтеза сквалена, ингибиторы АСАТ, ингибиторы микросомального белка, переносящего триглицериды (МТР), агонистов альфа-, гамма- и/или дельта-РАПП-рецепторов, средств, подавляющих абсорбцию холестерина, полимерных адсорберов желчной кислоты, средств, подавляющих реабсорбцию желчной кислоты, ингибиторов липазы, а также антагонистов липопротеина (а).
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с ингибитором белков переноса холестеринового эфира (СЕТР), таким как приводимые в качестве примерных и предпочтительных торсетрапиб (СР-529 414), JJT-705 или вакцина CETP-vaccine (Avant).
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с агонистом рецепторов тиреоидных гормонов, таким как приводимые в качестве примерных и предпочтительных D-тироксин, 3,5,3'-трийодотиронин (Т3), CGS 23425 или акситиром (CGS 26214).
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с ингибитором гидроксиметилглутарил-кофермент А-редуктазы (HMG-CoA) из класса статинов, таким как приводимые в качестве примерных и предпочтительных ловастатин, симвастатин, правастатин, флувастатин, аторвастатин, розувастатин или питавастатин.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с ингибитором синтеза сквалена, таким как приводимые в качестве примерных и предпочтительных BMS-188494 или TAK-475.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с ингибитором АСАТ, таким как приводимые в качестве примерных и предпочтительных авасимиб, мелинамид, пактимиб, эфлусимиб или SMP-797.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с ингибитором микросомального белка, переносящего триглицериды (МТР), таким как приводимые в качестве примерных и предпочтительных имплитапид, BMS-201038, R-103757 или JTT-130.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с агонистом гамма-рецептора, активируемого пролифератором пероксисом (РАПП), таким как приводимые в качестве примерных и предпочтительных пиоглитазон или розиглитазон.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с агонистом дельта-РАПП, таким как приводимые в качестве примерных и предпочтительных GW 501516 или BAY 68-5042.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации со средством, подавляющим абсорбцию холестерина, таким как приводимые в качестве примерных и предпочтительных эзетимиб, тиквесид или памаквесид.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с ингибитором липазы, таким как приводимый в качестве примерного и предпочтительного орлистат.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с полимерным адсорбером желчной кислоты, таким как приводимые в качестве примерных и предпочтительных холестирамин, колестипол, колесольвам, колестагель или колестимид.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации со средством, подавляющим реабсорбцию желчной кислоты, таким как приводимые в качестве примерных и предпочтительных ингибиторы ASBT (=1 ВАТ), как например, AZD-7806, S-8921, AK-105, BARI-1741, SC-435 или SC-635.
В случае одной предпочтительной формы исполнения изобретения соединения согласно изобретению вводят в комбинации с антагонистом липопротеина (а), таким как приводимые в качестве примерных и предпочтительных гемкабен кальций (С1-1027) или никотиновая кислота.
Другим предметом данного изобретения являются лекарственные средства, которые содержат по меньшей мере одно соединение согласно изобретению, обычно вместе с одним или несколькими инертными, нетоксичными, фармацевтически пригодными вспомогательными веществами, а также их применение для целей, указанных выше.
Соединения согласно изобретению могут действовать системно или местно. Для этой цели они могут применяться подходящим способом, таким как, например, орально, парентерально, пульмонально, назально, сублингвально, лингвально, буккально, ректально, на коже, трансдермально, конъюктивально, внутрь уха или в виде имплантатов или соответственно стентов.
Для этих способов применения соединения согласно изобретению могут выпускаться в подходящих формах применения.
Для орального применения согласно вопросам уровня техники подходят действующие формы применения, выделяющие соединения согласно изобретению быстро и/или видоизмененно, которые содержат эти соединения согласно изобретению в кристаллической и/или аморфизированной и/или растворенной форме, такие как, например, таблетки (без покрытия или с покрытием, например, оболочками, устойчивыми к действию желудочного сока, или растворяющимися с задержкой, или нерастворимыми, которые контролируют высвобождение соединения согласно изобретению), таблетки, быстро распадающиеся в ротовой полости, или пленочные покрытия/облатки, пленочные покрытия/лиофилизаты, капсулы (например, твердые или мягкие желатиновые капсулы), драже, грануляты, пеллеты, порошки, эмульсии, суспензии, аэрозоли или растворы.
Парентеральное применение может происходить в обход стадии ресорбции (например, внутривенно, внутриартериально, внутрикардиально, внутрипозвоночно или эндолюмбально) или с включением ресорбции (например, внутримышечно, подкожно, внутрикожно, через кожу или внутрь брюшины). Для парентерального использования в качестве форм применения, среди прочего, подходят готовые формы для инъекций и внутривенного введения в форме растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Для прочих способов применения подходят, например, лекарственные формы для ингаляций (среди прочего, порошковые ингаляторы, аэрозоли), капли, растворы или спреи для носа, таблетки, предназначенные для применения лингвально, подъязычно или буккально, пленки/облатки или капсулы, суппозитории, ушные или глазные препараты, вагинальные капсулы, водные суспензии (лосьоны, болтушки), липофильные суспензии, мази, кремы, трансдермальные терапевтические системы (например, пластыри), молочко, пасты, пены, присыпки, имплантаты или стенты.
Предпочтительными являются оральное и парентеральное применения, особенно оральное и внутривенное применение.
Соединения согласно изобретению могут переводиться в указанные формы применения. Это может делаться по известным способам путем смешивания с инертными, нетоксичными, фармацевтически пригодными вспомогательными веществами. К этим вспомогательным веществам причисляют, среди прочего, вещества-носители, например, микрокристаллическую целлюлозу, лактозу, маннитол), растворители (например, жидкие полиэтиленгликоли), эмульгаторы и диспергирующие или смачивающие средства (например, додецилсульфат натрия, полиоксисорбитанолеат), связующие средства (например, поливинилпирролидон), синтетические и природные полимеры (например, альбумин), стабилизаторы (например, антиоксиданты, такие как, к примеру, аскорбиновая кислота), красители (например, неорганические пигменты, такие как, к примеру, оксид железа) и модификаторы вкуса и запаха.
Как правило, оказалось предпочтительным при парентеральном применении для достижения эффективных результатов вводить количества приблизительно от 0,001 до 1 мг/кг, предпочтительно примерно от 0,01 до 0,5 мг/кг массы тела. При оральном применении дозировка составляет примерно от 0,01 до 100 мг/кг, предпочтительно примерно от 0,01 до 20 мг/кг и наиболее предпочтительно от 0,1 до 10 мг/кг массы тела.
Несмотря на это, если нужно, может быть необходимым отклоняться от указанных количеств, а именно, в зависимости от массы тела, способа применения, индивидуальной переносимости по отношению к активному веществу, типа готового препарата и момента времени или соответственно промежутка, в который осуществляется применение. Так, в некоторых случаях может быть достаточно обходиться меньшими чем указанные выше минимальные количества, в то время как в других случаях приведенные верхние границы должны быть превышены. В случае применения более значительных количеств может быть рекомендовано распределять эти количества на несколько разовых доз в течение дня.
Следующие примеры исполнения более подробно поясняют изобретение. Изобретение не ограничивается исключительно этими примерами.
Процентные величины в следующих тестах и примерах, если не указано иное, представляют собой массовые проценты; части являются массовыми частями. Соотношения растворителей, соотношения разбавителей и величины концентраций растворов жидкость/жидкость соответственно относятся к объему.
А. Примеры Сокращения и аббревиатуры:
Методы ГХ-МС и ЖХ-МС:
Метод 1 (ГХ-МС):
Прибор: Micromass GCT, GC 6890; колонка: Restek RTX-35, 15 м×200 мкм×0,33 мкм; постоянный ток гелия: 0,88 мл/мин; нагревательная камера: 70°С; испаритель: 250°С; градиент: 70°С, 30°С/мин→310°C (3 мин выдержки).
Метод 2 (ЖХ-МС):
Тип прибора для МС: Micromass ZQ; тип прибора для ВЭЖХ: HP 1100 Series; UV DAD; колонка: Phenomenex Gemini 3 мкм 30 мм×3,00 мм; элюент А: 1 л воды + 0,5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 90% А → 2,5 мин 30% А → 3,0 мин 5% А → 4,5 мин 5% А; расход жидкости: 0,0 мин 1 мл/мин → 2,5 мин/3,0 мин/4,5 мин 2 мл/мин; нагревательная камера: 50°С; УФ-детектирование: 210 нм.
Метод 3 (ЖХ-МС):
Тип прибора для МС: Micromass ZQ; тип прибора для ВЭЖХ: Waters Alliance 2795; колонка: Phenomenex Synergi 2,5 мкм MAX-RP 100A Mercury 20 мм×4 мм; элюент А: 1 л воды + 0,5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 90% А → 0,1 мин 90% А → 3,0 мин 5% А → 4,0 мин 5% А → 4,01 мин 90% А; расход жидкости: 2 мл/мин; нагревательная камера: 50°С; УФ-детектирование: 210 нм.
Метод 4 (ЖХ-МС):
Прибор: Micromass Quattro Premier, оснащенный Waters UPLC Acquity; колонка: Thermo Hypersil GOLD 1,9 мкм 50 мм×1 мм; элюент А: 1 л воды + 0,5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 90% А → 0,1 мин 90% А → 1,5 мин 10% А → 2,2 мин 10% А; расход жидкости: 0,33 мл/мин; нагревательная камера: 50°С; УФ-детектирование: 210 нм.
Метод 5 (ЖХ-МС):
Тип прибора для МС: Waters Micromass Quattro Micro; тип прибора для ВЭЖХ: Agilent 1100 Serie; колонка: Thermo Hypersil GOLD 3 мкм 20 мм×4 мм; элюент А: 1 л воды + 0,5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 100% А → 3,0 мин 10% А → 4,0 мин 10% А → 4,01 мин 100% А (расход 2,5 мл/мин) → 5,00 мин 100% А; нагревательная камера: 50°С; расход жидкости: 2 мл/мин; УФ-детектирование: 210 нм.
Метод 6 (ЖХ-МС):
Прибор: Waters Acquity SQD UPLC System; колонка: Waters Acquity UPLC HSS T3 1,8 мкм, 50 мм×1 мм; элюент А: 1 л воды + 0,25 мл 99%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила+0,25 мл 99%-ной муравьиной кислоты; градиент: 0,0 мин 90% А → 1,2 мин 5% А → 2,0 мин 5% А; расход жидкости: 0,40 мл/мин; нагревательная камера: 50°С; УФ-детектирование: 210-400 нм.
Метод 7 (ЖХ-МС):
Тип прибора для МС: Waters ZQ; тип прибора для ВЭЖХ: Agilent 1100 Series; UV DAD; колонка: Thermo Hypersil GOLD 3 мкм 20 мм×4 мм; элюент А: 1 л воды + 0,5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 100% А → 3,0 мин 10% А → 4,0 мин 10% А → 4,1 мин 100% А (расход 2,5 мл/мин); нагревательная камера: 55°С; расход жидкости: 2 мл/мин; УФ-детектирование: 210 нм.
Метод 8 (ГХ-МС):
Прибор: Micromass GCT, GC 6890; колонка: Restek RTX-35, 15 м ×200 мкм×0,33 мкм; постоянный ток гелия: 0,88 мл/мин; нагревательная камера: 70°С; испаритель: 250°С; градиент: 70°С, 30°С/мин → 310°С (12 мин выдержки).
Метод 9 (ЖХ-МС):
Прибор: Micromass Quattro Premier, оснащенный Waters UPLC Acquity; колонка: Thermo Hypersil GOLD 1,9 мкм 50 мм×1 мм; элюент А: 1 л воды + 0,5 мл 50%-ной муравьиной кислоты, элюент В: 1 л ацетонитрила + 0,5 мл 50%-ной муравьиной кислоты; градиент: 0,0 мин 90% А → 0,3 мин 90% А → 3,0 мин 10% А → 4,8 мин 10% А; расход жидкости: 0,33 мл/мин; нагревательная камера: 50°С; УФ-детектирование: 210 нм.
Метод 10 (ГХ-МС):
Прибор: Thermo DFS, Trace GC Ultra; колонка: Restek RTX-35, 15 м×200 мкм×0,33 мкм; постоянный ток гелия: 1,20 мл/мин; нагревательная камера: 60°С; испаритель: 220°С; градиент: 60°С, 30°С/мин → 300°С (3,33 мин выдержки).
Исходные соединения и интермедиаты:
Пример 1А
Трет-бутил-3-(3-амино-2-метилфенил)пропаноат

В атмосфере аргона прикалывали 201 мл (1,39 моль) трет-бутилпроп-2-еноата к раствору 100 г (463 ммоль) 1-бром-2-метил-3-нитробензола, 322 мл (2,31 моль) триэтиламина, 28,18 г (92,58 ммоль) три-2-толилфосфина и 10,39 г (46,29 ммоль) ацетата палладия(II) в 2 литрах ДМФА, а смесь после этого перемешивали 36 ч при 125°С. После охлаждения до комнатной температуры эту реакционную смесь перемешивали с насыщенным водным раствором хлорида аммония и органическую фазу отделяли. Водную фазу трижды экстрагировали трет-бутилметиловым эфиром, а объединенные органические фазы промывали насыщенным раствором хлорида натрия и сушили над сульфатом натрия. После фильтрации растворитель удаляли в вакууме досуха. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле (растворитель петролейный эфир/этилацетат 9:1). Было получено 89 г (338 ммоль, 73% от теор.) промежуточного продукта трет-бутил-(2Е)-3-(2-метил-3-нитрофенил)проп-2-еноата в виде бесцветного твердого вещества. 88 г (334 ммоль) этого твердого вещества растворили в 2 литрах этанола, при комнатной температуре смешали с 7 г палладия на угле (10%) и гидрировали 18 ч при нормальном давлении. По достижении полного превращения реакционный раствор фильтровали через кизельгур, а полученный фильтрат упаривали в вакууме. Было получено 61,3 г (260,5 ммоль, 78% от теор.) целевого соединения в виде бесцветного твердого вещества.
ЖХ-МС (Метод 2): Rt=1,84 мин; m/z=236 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6, δ /м.д.): 6,77 (1H, т), 6,47 (1H, д), 6,36 (1H, д), 4,72 (2H, с), 2,14 (2Н, т), 2,36 (2Н, т), 1,95 (3H, с), 1,39 (9Н, с).
Пример 2А
Этил-3-(3-амино-2-метилфенил)пропаноат
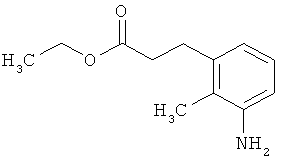
В атмосфере аргона прикалывали 10,844 г (108 ммоль) этилпроп-2-еноата к раствору 7,8 г (36,1 ммоль) 1-бром-2-метил-3-нитробензола, 20 мл (180,5 ммоль) триэтиламина, 2,197 г (7,22 ммоль) три-2-толилфосфина и 810 мг (3,6 ммоль) ацетата палладия(II) в 200 мл ДМФА, а смесь после этого перемешивали 36 ч при 125°С. После охлаждения до комнатной температуры эту реакционную смесь перемешивали с насыщенным водным раствором хлорида аммония и органическую фазу отделяли. Водную фазу трижды экстрагировали трет-бутилметиловым эфиром, а объединенные органические фазы промывали насыщенным раствором хлорида натрия и сушили над сульфатом натрия. После фильтрации растворитель удаляли в вакууме досуха. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле (растворитель петролейный эфир/этилацетат 3:1). Было получено 6,6 г (27,2 ммоль, содержание 97%, 75% от теор.) промежуточного продукта этил-(2Е)-3-(2-метил-3-нитрофенил)проп-2-еноата в виде бесцветного твердого вещества. 6,6 г (27,2 ммоль, содержание 97%) этого твердого вещества растворили в 200 мл этанола, при комнатной температуре смешали с 500 мг палладия на угле (10%) и в течение ночи гидрировали при нормальном давлении. По достижении полного превращения реакционный раствор фильтровали через кизельгур, а полученный фильтрат упаривали в вакууме. Было получено 5,47 г (26,38 ммоль, содержание 97%, 97% от теор.) целевого соединения в виде бесцветного твердого вещества.
ЖХ-МС (Метод 3): Rt=1,07 мин; m/z=208 (M+H)+.
Пример 3А
Трет-бутил-(2Е)-3-(4-фтор-3-нитрофенил)акрилат

В атмосфере аргона 0,65 г (16,3 ммоль) гидрида натрия (в виде 60%-ной суспензии в минеральном масле) помещали в 25 мл ТГФ и охлаждали до 0°С. После этого медленно прикалывали 4,29 г (17 ммоль) сложного трет-бутилового эфира диэтилфосфоноуксусной кислоты. Спустя 30 мин добавляли 2,5 г (14,8 ммоль) 4-фтор-3-нитробензальдегида. Реакционную смесь перемешивали 3 ч при Ткомн., потом выливали в 100 мл воды и трижды экстрагировали соответственно порциями по 100 мл этилацетата. Объединенные органические фазы сушили над сульфатом магния и упаривали. Остаток очищали с помощью флэш-хроматографии (силикагель, растворитель циклогексан/этилацетат 50:1). Было получено 3,37 г (85% от теор.) целевого соединения.
ГХ-МС (Метод 1): Rt=6,45 мин; m/z=211 (M-tBu)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,49 (с, 9Н), 6,69 (д, 1Н), 7,59-7,76 (м, 2Н), 8,19 (ддд, 1Н), 8,50 (дд, 1Н).
Пример 4А
Сложный трет-бутиловый эфир 3-(3-амино-4-фторфенил)пропановой кислоты

535 мг (2,00 ммоль) сложного mpem-бутилового эфира (2Е)-3-(4-фтор-3-нитрофенил)проп-2-еновой кислоты растворяли в 1 мл этанола и 1 мл ТГФ и смешивали с 21,3 мг палладия на угле (10%). При Ткомн. в течение ночи гидрировали в атмосфере водорода при нормальном давлении. Затем эту реакционную смесь фильтровали с отсасыванием через кизельгур, остаток дополнительно промывали ТГФ, а фильтрат упаривали. Было получено 479 мг (100% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,06 мин; m/z=184 (M-C6H8)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ=6,84 (дд, 1Н), 6,58 (дд, 1Н), 6,36-6,29 (м, 1Н), 5,00 (с, 2Н), 2,64 (т, 2Н), 2,42 (т, 2Н), 1,36 (с, 9Н).
Пример 5А
трет-бутил-(2Е)-3-(4-хлор-3-нитрофенил)проп-2-еноат

В атмосфере аргона 1,19 г (29,64 ммоль, 60%-ного) гидрида натрия суспендировали в 25 мл толуола и 25 мл ТГФ и охлаждали до 0°С. После этого медленно прикалывали 7,28 мл (30,99 ммоль) трет-бутил(диэтоксифосфорил)ацетата и эту смесь перемешивали 30 мин при 0°С. Затем к реакционной смеси добавляли 5 г (26,94 ммоль) 4-хлор-3-нитробензальдегида и загруженную реакционную смесь нагревали до комнатной температуры. Эту смесь перемешивали 2 ч при комнатной температуре, а затем добавляли 50 мл воды. После отделения органической фазы водную фазу еще трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом натрия. После фильтрации растворитель удаляли в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (растворитель циклогексан/этилацетат 9:1). Было получено 6,77 г (23,86 ммоль, 77% от теор.) целевого соединения.
МС (DCI): m/z=301 (M+NH4)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ=8,46 (д, 1Н), 8,07 (дд, 1Н), 7,71 (д, 1Н), 7,51 (д, 1Н), 6,75 (д,1Н), 1,49 (с, 9Н).
Пример 6А
Трет-бутил-3-(3-амино-4-хлорфенил)пропаноат

К раствору 6,74 г (23,76 ммоль) трет-бутил-(2Е)-3-(4-хлор-3-нитрофенил)проп-2-еноата в 200 мл этанола и 20 мл ТГФ при комнатной температуре добавляли 500 мг палладия на угле (10%) и 12 ч гидрировали при нормальном давлении. После полного прохождения реакции (ТСХ-контроль; элюент: циклогексан/этилацетат 1:1) реакционную смесь фильтровали через кизельгур, а фильтрат упаривали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 4:1→2:1). Было получено 1,40 г (5,47 ммоль, 23% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,14 мин; m/z=256 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ=7,08 (д, 1Н), 6,62 (с, 1Н), 6,39 (дд, 1Н), 5,22 (с, 2Н), 2,66 (т, 2Н), 2,45 (т, 2Н), 1,37 (с, 9Н).
Пример 7А
Метил-3-(3-амино-4-хлорфенил)пропаноат

К раствору 1,0 г (3,91 ммоль) трет-бутил-3-(3-амино-4-хлорфенил)пропаноата в 20 мл метанола при кипячении с обратным холодильником прикалывали 0,86 мл (11,7 ммоль) тионилхлорида. Смесь перемешивали 1,5 ч при кипении с обратным холодильником, а потом после охлаждения разбавляли дихлорметаном. Этот раствор выливали в воду, а после разделения фаз органическую фазу промывали насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушили над сульфатом натрия и упаривали в вакууме. Остаток сушили в высоком вакууме. Было получено 745 мг (90,3% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=0,91 мин; m/z=213/215 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=2,52-2,60 (м, 2Н), 2,62-2,77 (м, 2Н), 5,22 (с, 2Н), 6,39 (дд, 1Н), 6,62 (д, 1Н), 7,06 (д, 1Н).
Пример 8А
Метил-{1-[3-(дибензиламино)-4-фторфенил]циклопропил}ацетат

Получение раствора А: 688 мг (16,2 ммоль) хлорида лития в атмосфере аргона растворяли в 50 мл ТГФ, а затем смешивали с 789 мг (32,5 ммоль) магниевой стружки и 23 мкл (0,023 ммоль) 1 М раствора диизобутилалю-минийгидрида в ТГФ. Реакционный раствор перемешивали 10 мин при комнатной температуре, а потом охлаждали до -10°С. Далее добавляли 5 г (13,5 ммоль) N,N-дибензил-5-бром-2-фторанилина (регистрационный номер CAS 869529-97-5) и этот раствор дополнительно перемешивали прим. 1 ч при -10°С.
Получение раствора В: 110 мг (2,6 ммоль) хлорида лития и 128 мг (1,3 ммоль) хлорида меди(1) в атмосфере аргона при комнатной температуре суспендировали в 10 мл ТГФ, а затем смешивали с 1,65 мл (12,98 ммоль) хлор(триметил)силана, а также 1,46 г (12,98 ммоль) метилхлорпропилиденацетата (регистрационный номер CAS 110793-87-8). Этот раствор затем перемешивали еще 1 ч при Ткомн.
Полученный выше раствор А охлаждали до -40°C. Затем медленно прикалывали раствор В. Объединенные теперь растворы медленно нагревали до -20°С и дополнительно перемешивали 1 ч при этой температуре. Потом к реакционной смеси добавляли 50 мл охлажденного льдом, полунасыщенного раствора хлорида аммония. После разделения фаз водную фазу еще трижды экстрагировали этилацетатом, объединенные органические фазы сушили над сульфатом магния и упаривали досуха. Полученный сырой продукт очищали хроматографически на силикагеле (элюент: циклогексан/этилацетат 10:1). Было выделено 2,1 г (5,2 ммоль, 39% от теор.) целевого соединения.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 7,33-7,25 (8Н, м), 7,25-7,18 (2Н, м), 7,02-6,94 (1Н, м), 6,78-6,69 (2Н, м), 4,27 (4Н, с), 3,43 (ЗН, с), 2,48 (2Н, с), 0,78-0,73 (2Н, м), 0,63-0,58 (2Н, м).
ЖХ-МС (Метод 5): Rt=2,99 мин; m/z=404 (М+Н)+.
Пример 9А
Метил-[1-(3-амино-4-фторфенил)циклопропил]ацетат

К раствору 2,1 г (5,2 ммоль) метил-{1-[3-(дибензиламино)-4-фторфенил]циклопропил}ацетата в 100 мл этанола при комнатной температуре добавляли 200 мг палладия на угле (10%) и гидрировали 12 ч при нормальном давлении. После полного прохождения реакции (ТСХ-контроль; элюент: циклогексан/этилацетат 1:1) реакционную смесь фильтровали через кизельгур, а фильтрат упаривали в вакууме. Сырой продукт очищали с помощью хроматографии через силикагель (элюент: циклогексан/этилацетат 10:1). Было получено 647 мг (2,9 ммоль, 56% от теор.) целевого соединения.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 6,88-6,78 (1Н, м), 6,70-6,62 (1Н, м), 6,44-6,35 (1Н, м), 4,98 (2Н, уш. с), 3,51 (3H, с), 2,55 (2Н, с), 0,84-0,79 (2Н, м), 0,78-0,73 (2Н, м).
ГХ-МС (Метод 1): Rt=5,67 мин; m/z=224 (М+Н)+.
Пример 10А
5-Бром-2-хлор-N,N-бис(4-метоксибензил)анилин

В атмосфере аргона 5,07 г (126,93 ммоль, 60%-ного) гидрида натрия суспендировали в 150 мл ТГФ и охлаждали до 0°С. После этого медленно прикалывали 10,70 г (51,81 ммоль) 5-бром-2-хоранилина, растворенного в 10 мл ТГФ, и эту смесь перемешивали 30 мин при 0°С. Затем к реакционной смеси добавляли 25 г (124,34 ммоль) 4-метоксибензилхлорида и потом реакционную массу нагревали до комнатной температуры. Эту смесь перемешивали 2 ч при Ткомн., а затем медленно выливали в 150 мл ледяной воды. После отделения органической фазы водную фазу еще трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом натрия. После фильтрации растворитель удаляли в вакууме. Сырой продукт очищали с помощью хроматографии [колонка: Kromasil Si 6012, 350 мм×30 мм; элюент А: изогексан, элюент В: этилацетат; градиент: 0 мин 98% А → 4,65 мин 98% А → 13 мин 87% А → 13,01 мин 98% А → 13,28 мин 98% А; расход: 70 мл/мин; температура: 20°С; УФ-детектирование: 265 нм]. Было получено 12,37 г (27,69 ммоль, 57% от теор.) целевого соединения.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 7,37 (1Н, д), 7,26-7,19 (5Н, м), 7,19-7,14 (1Н, м), 6,86 (4Н, д), 4,11 (4Н, с), 3,71 (6Н, с).
ЖХ-МС (Метод 4): Rt=1,68 мин; m/z=446 (М)+.
Пример 11А
{3-[Бис(4-метоксибензил)амино]-4-хлорфенил}борная кислота
В атмосфере аргона при -78°С к раствору 5,2 г (11,64 ммоль) 5-бром-2-хлор-N,N-бис(4-метоксибензил)анилина в 100 мл смеси ТГФ/диэтиловый эфир (1:1) медленно прикалывали 6,1 мл (15,25 ммоль) 2,5 М раствора н-бутиллития в гексане. После того как реакционный раствор дополнительно перемешивали 60 мин при -78°С, медленно добавили 4,3 мл (18,62 ммоль) триизопропилбората. Этот реакционный раствор затем дополнительно перемешивали еще 15 мин при -78°С, потом медленно нагревали до комнатной температуры и перемешивали еще 3 ч при этой температуре. После этого добавили 150 мл ледяной воды. После отделения органической фазы водную фазу еще трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом натрия. После фильтрации растворитель удаляли в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: сначала циклогексан/этилацетат 10:1→9:1→4:1, затем дихлорметан/метанол 95:5). Таким образом было получено 2,54 г (6,17 ммоль, 53% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,20 мин; m/z=412 (М+Н)+.
Пример 12А
Бензилоксетан-3-илиденацетат

В атмосфере аргона при 0°С растворяли 3,0 г (41,63 ммоль) оксетан-3-она (регистрационный номер CAS 6704-31-0) в 50 мл дихлорметана, а затем смешивали с 18,8 г (45,79 ммоль) бензил(трифенил-λ5-фосфанилиден)ацетата. После этого реакционную смесь медленно нагревали до комнатной температуры и дополнительно перемешивали еще 15 мин. Реакционный раствор потом упаривали досуха. Остаток переводили в 25 мл диэтилового эфира и перемешивали, а эту смесь выдерживали 12 ч при 4°С. Выпавший трифенилфосфиноксид отфильтровывали, а фильтрат упаривали досуха. Полученный сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат4:1→1:1). Было получено 4,2 г (20,57 ммоль, 49% от теор.) целевого соединения.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 7,42-7,30 (5Н, м), 5,85-5,80 (1Н, м), 5,39-5,34 (2Н, м), 5,27-5,22 (2Н, м), 5,13 (2Н, с).
МС (DCI): m/z=205 (М+Н)+.
Пример 13А
Бензил-(3-{3-[бис(4-метоксибензил)амино]-4-хлорфенил}оксетан-3-ил)ацетат
В атмосфере аргона при комнатной температуре к раствору 45 мг (0,09 ммоль) димера хлорида (1 г,52)-циклоокта-1,5-диенродия(1) в 25 мл диоксана последовательно добавляли 1,6 мл (2,37 ммоль) 1,5 М водного раствора гидроксида калия, 272 мг (1,82 ммоль) бензилоксетан-3-илиденацетата, а также 750 мг (1,82 ммоль) {3-[бис(4-метоксибензил)амино]-4-хлорфенил}борной кислоты. Затем реакционный раствор перемешивали 4 ч при комнатной температуре. После прошедшего взаимодействия этот раствор был упарен досуха, а остаток переводили в 25 мл воды и 25 мл этилацетата. После разделения фаз водную фазу еще трижды экстрагировали этилацетатом, объединенные органические фазы сушили над сульфатом магния и упаривали досуха. Полученный сырой продукт очищали с помощью хроматографии на силикагеле (элюент:циклогексан/этилацетат 4:1→1:1). Было выделено 669 мг (1,17 ммоль, 64% от теор.) целевого соединения.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 7,34-7,26 (4Н, м), 7,18-7,13 (4Н, м), 7,12-7,07 (2Н, м), 6,86-6,76 (6Н, м), 4,90 (2Н, с), 4,72 (2Н, д), 4,57 (2Н, д), 4,01 (6Н, с), 3,08 (2Н, с).
ЖХ-МС (Метод 6): Rt=1,45 мин; m/z=572 (М)+.
Аналогично примеру синтеза 13А было получено следующее соединение:

Пример 15А
Бензил-[3-(3-амино-4-хлорфенил)оксетан-3-ил]ацетат
К раствору 660 мг (1,15 ммоль) бензил-(3-{3-[бис(4-метоксибензил)амино]-4-хлорфенил}оксетан-3-ил)ацетата в 30 мл дихлорметана и 6 мл воды при комнатной температуре добавляли 576 мг (2,54 ммоль) 2,3-дихлор-5,6-дициано-1,4-бензохинона (ДДХ) и перемешивали 2 ч. После полного прохождения реакции (ТСХ-контроль; элюент: циклогексан/этилацетат 2:1) к реакционному раствору добавляли 25 мл насыщенного раствора гидрокарбоната натрия. После разделения фаз водную фазу еще трижды экстрагировали дихлорметаном, объединенные органические фазы сушили над сульфатом магния и упаривали досуха. Полученный сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 5:1). Было выделено 360 мг (0,98 ммоль, содержание 90%, 85% от теор.) целевого соединения.
ЖХ-МС (Метод 4): Rt=1,18 мин; m/z=332 (М+Н)+.
Аналогично примеру синтеза 15А было получено следующее соединение:

Пример 17А
3-Бром-2-фторанилин

2,0 г (9,09 ммоль) 3-бром-2-фторнитробензола растворяли в 10 мл диоксана и при Ткомн. смешивали с 8,62 г (45,45 ммоль) хлорида олова(И). После добавления нескольких капель 1 н. соляной кислоты смесь нагревали 2 ч при 70°C. После охлаждения реакционную смесь упаривали в вакууме, а остаток переводили в этилацетат. Раствор последовательно промывали: дважды 1 н. водным раствором едкого натра, водой и насыщенным раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 10:1). Было получено 997 мг (57,7% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=0,88 мин; m/z=189/191 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=5,43 (с, 2Н), 6,66-6,85 (м, 3Н).
Пример 18А
Трет-бутил-(2Е)-3-(3-амино-2-фторфенил)акрилат
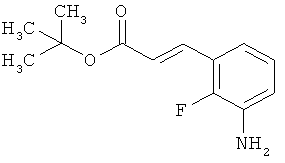
Раствор 1,41 г (7,42 ммоль) 3-бром-2-фторанилина и 2,85 г (22,3 ммоль) сложного трет-бутилового эфира акриловой кислоты в 8 мл ДМФА смешивали с 5,2 мл (37,1 ммоль) триэтиламина. Из колбы трижды откачивали воздух и заполняли ее аргоном, прежде чем добавили 451 мг (1,48 ммоль) три-2-толилфосфина и 166,6 мг (0,74 ммоль) ацетата палладия (II). Реакционный сосуд снова дважды вакуумировали и заполняли аргоном, а потом смесь нагревали примерно до 140°С. Спустя 2 ч интенсивного перемешивания реакционную смесь охладили и вылили в насыщенный раствор гидрокарбоната натрия. Эту смесь трижды экстрагировали этилацетатом, а объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 10:1). Было получено 1660 мг целевого продукта (94,3% от теор.).
ЖХ-МС (Метод 6): Rt=1,12 мин; m/z=279 (M+H+CH3CN)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,48 (с, 9Н), 5,27 (с, 2Н), 6,45 (д, 1Н), 6,73-7,02 (м, 3Н), 7,59 (д, 1Н).
Пример 19А
Трет-бутил-3-(3-амино-2-фторфенил)пропаноат

К раствору 1660 мг (7,0 ммоль) трет-бутил-(2Е)-3-(3-амино-2-фторфенил)акрилата в смеси из 5 мл этанола и 3 мл ТГФ добавляли палладий на угле (10%) и в течение ночи интенсивно перемешивали в атмосфере водорода при нормальном давлении. Затем реакционную смесь фильтровали через кизельгур, а остаток на фильтре многократно промывали смесью этанол/ТГФ. Объединенный фильтрат упаривали в вакууме, а остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 20:1→10:1). Было получено 1350 мг целевого продукта (80,6% от теор.).
ЖХ-МС (Метод 6): Rt=1,07 мин; m/z=225.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,36 (с, 9Н), 2,45 (т, 2Н), 2,74 (т, 2Н), 5,00 (с, 2Н), 6,24-6,46 (м, 1Н), 6,51-6,66 (м, 1Н), 6,66-6,82 (м, 1Н).
Пример 20А
Сложный этиловый эфир (Е/Z)-3-(4-фтор-3-нитрофенил)-2-метилпроп-2-еновой кислоты

3,17 г гидрида натрия (60%-ная суспензия в минеральном масле, 79,36 ммоль) суспендировали в 90 мл смеси ТГФ/ДМФА (2:1). Эту смесь охлаждали до 0°С и по каплям прибавляли раствор 19,76 г (82,96 ммоль) сложного триэтилового эфира 2-фосфонопропионовой кислоты в 60 мл смеси ТГФ/ДМФА (2:1). Спустя 30 мин при 0°С к этому прикалывали раствор 12,2 г (72,14 ммоль) 4-фтор-3-нитробензальдегида в 60 мл смеси ТГФ/ДМФА (2:1). После окончания прибавления реакционную смесь медленно нагревали до Ткомн. и перемешивали 2 ч при этой температуре. После этого реакционную смесь выливали в воду. Трижды экстрагировали этилацетатом, а объединенные органические фазы упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 20:1). Было получено 15,2 г (83,2% от теор.) целевого продукта в виде смеси изомеров E/Z (E/Z 91:9).
ЖХ-МС (Метод 6): Z-изомер: Rt=1,11 мин; m/z=254 (М+Н)+; Е-изомер: Rt=1,14 мин; m/z=254 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): Е-изомер: δ [м.д.]=1,28 (т, 3Н), 4,22 (кв, 2Н), 7,59-7,73 (м, 2Н), 7,92 (ддд, 1Н), 8,24 (дд, 1Н).
Пример 21А
Сложный этиловый эфир (+/-)-3-(3-амино-4-фторфенил)-2-метилпропановой кислоты

15,2 г (60,02 ммоль) сложного этилового эфира (Е/Z)-3-(4-фтор-3-нитрофенил)-2-метилпроп-2-еновой кислоты (E/Z 91:9) в смеси из 100 мл этанола и 100 мл ТГФ смешивали с палладием на угле (10%) и в течение ночи интенсивно перемешивали в атмосфере водорода при нормальном давлении. Реакционную смесь затем фильтровали через целит, остаток промывали смесью этанол/дихлорметан, а объединенный фильтрат упаривали в вакууме. Продукт сушили в высоком вакууме. Было получено 13,34 г целевого продукта (98,7% от теор.).
ЖХ-МС (Метод 6): Rt=0,98 мин; m/z=226 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,04 (д, 3Н), 1,12 (т, 3Н), 2,46-2,50 (м, 1Н), 2,55-2,66 (м, 1Н), 2,66-2,78 (м, 1Н), 4,01 (кв, 2Н), 5,00 (с, 2Н), 6,18-6,35 (м, 1Н), 6,55 (дд, 1Н), 6,84 (дд, 1Н).
Полученный выше рацемат был разделен на энантиомеры с помощью ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×20 мм; объем впрыска: 0,15 мл; температура: 30°С; элюент: 90% изогексана/10% этанола; расход: 15 мл/мин; детектирование: 220 нм]. Исходя из 7,25 г рацемата было получено 3,43 г энантиомера 1 (Пример 22А) и 3,35 г энантиомера 2 (Пример 23А):
Пример 22А
Сложный этиловый эфир (+)-(2S)-3-(3-амино-4-фторфенил)-2-метилпропановой кислоты

Выход: 3,43 г
ЖХ-МС (Метод 6): Rt=0,97 мин; m/z=226 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,04 (д, 3Н), 1,12 (т, 3Н), 2,46-2,50 (м, 1Н), 2,55-2,66 (м, 1Н), 2,66-2,78 (м, 1Н), 4,01 (кв, 2Н), 5,00 (с, 2Н), 6,18-6,35 (м, 1Н), 6,55 (дд, 1Н), 6,84 (дд, 1Н).
[α]D20=+18.3°, C=0,465, хлороформ.
Пример 23А
Сложный этиловый эфир (-)-(2R)-3-(3-амино-4-фторфенил)-2-метилпропановой кислоты

Выход: 3,35 г
ЖХ-МС (Метод 6): Rt=0,97 мин; m/z=226 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,04 (д, 3Н), 1,12 (т, 3Н), 2,46-2,50 (м, 1Н), 2,55-2,66 (м, 1Н), 2,68-2,79 (м, 1Н), 4,01 (кв, 2Н), 5,00 (уш. с, 2Н), 6,30 (дд, 1Н), 6,55 (дд, 1Н), 6,84 (дд, 1Н).
[α]D20=-31.4°, c=0,520, хлороформ.
Пример 24А
Сложный этиловый эфир (Е/Z)-3-(4-хлор-3-нитрофенил)-2-метилпроп-2-еновой кислоты
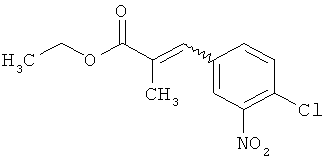
4,74 г гидрида натрия (60%-ная суспензия в минеральном масле, 118,56 ммоль) суспендировали в 93 мл смеси ТГФ/ДМФА (1:1). Эту смесь охлаждали до 0°С и по каплям прибавляли 26,6 мл (123,92 ммоль) сложного триэтилового эфира 2-фосфонопропионовой кислоты. Спустя 30 мин при 0°С к этому прикалывали 20,0 г (107,78 ммоль) 4-хлор-3-нитробензальдегида. После окончания прибавления реакционную смесь медленно нагревали до Ткомн. и перемешивали еще 3 ч при этой температуре. После этого реакционную смесь выливали в воду. Трижды экстрагировали этилацетатом, а объединенные органические фазы упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 70:1→50:1). Было получено 26,7 г (91,9% от теор.) целевого продукта в виде смеси изомеров E/Z (E/Z 91:9).
ЖХ-МС (Метод 4): Z-изомер: Rt=1,32 мин; m/z=255; Е - изомер: Rt=1,36 мин; m/z=270 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): Е - изомер: δ [м.д.]=1,28 (т, 3Н), 2,06 (д, 3Н), 4,22 (кв, 2Н), 7,56-7,67 (м, 1Н), 7,75-7,87 (м, 2Н), 8,17 (д, 1Н).
Пример 25А
Сложный этиловый эфир (+/-)-3-(3-амино-4-хлорфенил)-2-метилпропановой кислоты

10,0 г (37,08 ммоль) сложного этилового эфира (Е/Z)-3-(4-хлор-3-нитрофенил)-2-метилпроп-2-еновой кислоты (E/Z 91:9) растворяли в 25 мл этилацетата и 25 мл уксусной кислоты и добавляли палладий на угле (10%). Реакционную смесь в общей сложности 6 ч интенсивно перемешивали в атмосфере водорода при нормальном давлении, причем спустя 2 ч к ней еще раз были добавлены 25 мл уксусной кислоты и дополнительные порции 10% палладия на угле. Затем фильтровали через целит, а остаток промывали смесью этанол/дихлорметан. Объединенный фильтрат промывали насыщенным раствором гидрокарбоната натрия, сушили над сульфатом натрия и упаривали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 30:1→10:1). Было получено 4,01 г целевого продукта (44,7% от теор.).
ЖХ-МС (Метод 6): Rt=1,06 мин; m/z=242 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-ае): 5 [м.д.]=1,05 (д, 3Н), 1,12 (т, 3Н), 2,47-2,50 (м, 1Н), 2,56-2,67 (м, 1Н), 2,67-2,78 (м, 1Н), 4,02 (кв, 2Н), 5,23 (с, 2Н), 6,35 (дд, 1Н), 6,58 (д,1Н), 7,05 (д, 1Н).
Полученный выше рацемат был разделен на энантиомеры с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak OJ-H, 5 мкм, 250 мм×20 мм; объем впрыска: 0,15 мл; температура: 35°С; элюент: 50% изогексана/50% изопропанола; расход: 15 мл/мин; детектирование: 220 нм]. Исходя из 10,3 г рацемата, было получено 4,0 г энантиомера 1 (Пример 26А} и 3,7 г энантиомера 2 (Пример 27А):
Пример 26А
Сложный этиловый эфир (-)-(2R)-3-(3-амино-4-хлорфенил)-2-метилпропановой кислоты

Выход: 4,0 г
ЖХ-МС (Метод 7): Rt=2,27 мин; m/z=196/198.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,05 (д, 3Н), 1,12 (т, 3Н), 2,47-2,50 (м, 1Н), 2,54-2,66 (м, 2Н), 2,68-2,80 (м, 1Н), 4,02 (кв, 2Н), 5,23 (с, 2Н), 6,35 (дд, 1Н), 6,58 (д,1Н), 7,05 (д, 1Н).
[α]D20=-35,8°, c=0,560, хлороформ.
Пример 27А
Сложный этиловый эфир (+)-(2S)-3-(3-амино-4-хлорфенил)-2-метилпропановой кислоты
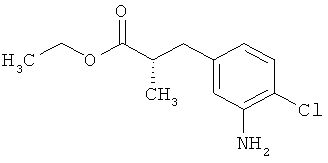
Выход: 3,7 г
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,05 (д, 3Н), 1,12 (т, 3Н), 2,47-2,50 (м, 1Н), 2,56-2,67 (м, 1Н), 2,67-2,81 (м, 1Н), 4,02 (кв, 2Н), 5,23 (уш. с, 2Н), 6,35 (дд, 1Н), 6,58 (д, 1Н), 7,05 (д, 1Н).
[α]D20=+35,1°, с=0,525, хлороформ.
Пример 28А
Этил-(2Е/Z)-2-(4-хлор-3-нитробензилиден)бутаноат

1,19 г гидрида натрия (60%-ная суспензия в минеральном масле, 29,64 ммоль) суспендировали в 50 мл смеси ТГФ/ДМФА (1:1). Эту смесь охлаждали до 0°С и по каплям прибавляли 7,3 мл (30,99 ммоль) сложного триэтилового эфира 2-фосфономасляной кислоты. Спустя 30 мин при -10°С к этому порциями добавляли 5,0 г (26,94 ммоль) 4-хлор-3-нитробензальдегида. После окончания прибавления реакционную смесь перемешивали 5 ч при 0°С, а потом медленно в течение ночи нагревали до Ткомн. После этого реакционную смесь выливали в воду. Трижды экстрагировали этилацетатом, а объединенные органические фазы упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 6:1). Было получено 7,05 г (92,1% от теор.) целевого продукта в виде смеси изомеров E/Z.
ЖХ-МС (Метод 6): Rt=1,24 мин и 1,26 мин; ионизация отсутствует.
Пример 29А
(+/-)-Этил-2-(3-амино-4-хлорбензил)бутаноат

7,05 г (24,84 ммоль) этил-(2Е/Z)-2-(4-хлор-3-нитробензилиден)бутаноата растворяли в 35 мл этилацетата и 35 мл уксусной кислоты и добавляли палладий на угле (10%). Реакционную смесь в общей сложности 6 ч интенсивно перемешивали в атмосфере водорода при нормальном давлении, причем спустя 4 ч к ней добавляли дополнительные порции 10% палладия на угле. Затем фильтровали через целит, а остаток промывали смесью этилацетат/ТГФ. Объединенный фильтрат промывали насыщенным раствором гидрокарбоната натрия, сушили над сульфатом натрия и упаривали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 30:1→10:1). Было получено 4,12 г целевого продукта (64,9% от теор.).
ЖХ-МС (Метод 6): Rt=1,14 мин; m/z=210.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,84 (т, 3Н), 1,10 (т, 3Н), 1,42-1,59 (м, 2Н), 2,40-2,80 (м, 4Н), 4,01 (кв, 2Н), 5,23 (с, 2Н), 6,34 (дд, 1Н), 6,58 (д, 1Н),7,05(д,1Н).
Полученный выше рацемат был разделен на энантиомеры с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak OJ-H, 5 мкм, 250 мм х 20 мм; объем впрыска: 0,43 мл; температура: 30°С; элюент:этанол; расход: 15 мл/мин; детектирование: 220 нм]. Исходя из 3,22 г рацемата, было получено 1,22 г энантиомера 1 (Пример 30А) и 1,27 г энан-тиомера 2 (Пример 31А):
Пример 30А
(-)-Этил-(2R)-2-(3-амино-4-хлорбензил)бутаноат
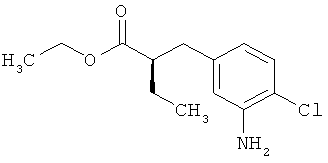
Выход: 1,22 г ЖХ-МС (Метод 6): Rt=1,14 мин; m/z=210.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,84 (т, 3Н), 1,10 (т, 3Н), 1,42-1,56 (м, 2Н), 2,39-2,48 (м, 1Н), 2,56-2,73 (м, 3Н), 4,01 (кв, 2Н), 5,11-5,27 (м, 2Н), 6,34 (дд, 1Н), 6,58 (д, 1Н), 7,05 (д, 1Н).
[α]D20=-28,1°, c=0,510, хлороформ.
Пример 31А
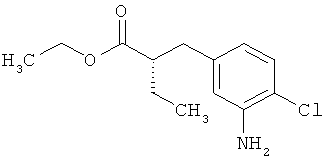
Выход: 1,27 г ЖХ-МС (Метод 6): Rt=1,15 мин; m/z=210.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,84 (т, 3Н), 1,10 (т, 3Н), 1,46-1,55 (м, 2Н), 2,42-2,49 (м, 1Н), 2,54-2,69 (м, 3Н), 4,01 (кв, 2Н), 5,22 (с, 2Н), 6,34 (дд, 1Н), 6,57 (д,1Н), 7,05 (д,1Н).
[α]D20=+34,1°, c=0,550, хлороформ.
Пример 32А
Трет-бутил-(2Е/Z)-3-(4-хлор-3-нитрофенил)бут-2-еноат

2,87 г гидрида натрия (60%-ная суспензия в минеральном масле, 71,65 ммоль) суспендировали в 80 мл ТГФ. Эту смесь охлаждали до 0°С и по каплям прибавляли 17,6 мл (74,9 ммоль) трет-бутил(диэтоксифосфорил)ацетата. Спустя 30 мин при 0°С к этому добавляли 13,0 г (65,1 ммоль) 4-хлор-3-нитроацетофенона. После окончания прибавления реакционную смесь медленно нагревали до Ткомн. и перемешивали еще 1,5 ч при Ткомн, прежде чем потом выливали эту смесь в воду. Трижды экстрагировали этилацетатом, а объединенные органические фазы упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 20:1→10:1). Было получено 17,03 г (87,8% от теор.) целевого продукта в виде смеси изомеров E/Z (E/Z приблизительно 1:1).
ЖХ-МС (Метод 5): изомер 1: Rt=2,61 мин; m/z=255; изомер 2: Rt=2,77 мин; m/z=224.
Пример 33А
Сложный трет-бутиловый эфир (+/-)-3-(3-амино-4-хлорфенил)бутановой кислоты
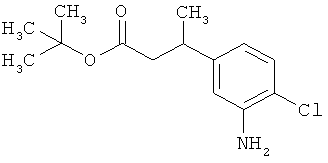
11,5 г (38,62 ммоль) трет-бутил-(2Е/Z)-3-(4-хлор-3-нитрофенил)бут-2-еноата (E/Z примерно 1:1) растворяли в 60 мл этилацетата и 60 мл уксусной кислоты и добавляли палладий на угле (10%). Реакционную смесь 6 ч интенсивно перемешивали при нормальном давлении в атмосфере водорода. Затем фильтровали через целит, а остаток промывали этилацетатом. Объединенный фильтрат промывали насыщенным раствором гидрокарбоната натрия, сушили над сульфатом натрия и упаривали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 30:1). Было получено 3,90 г (37,4% от теор.) целевого продукта.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,14 (д, 3Н), 1,31 (с, 9Н), 2,38 (дд, 2Н), 2,95 (кв, 1Н), 5,21 (уш. с, 2Н), 6,42 (дд, 1Н), 6,65 (д, 1Н), 7,06 (д, 1Н).
Полученный выше рацемат был разделен на энантиомеры с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×20 мм; объем впрыска: 0,15 мл; температура: 30°С; элюент: 90% изогексана/10% этанола; расход: 15 мл/мин; детектирование: 220 нм]. Исходя из 5,0 г рацемата, было получено 2,1 г энантиомера 1 (Пример 34А) и 1,8 г энантиомера 2 (Пример 35А):
Пример 34А
Сложный трет-бутиловый эфир (+)-(33)-3-(3-амино-4-хлорфенил)бутановой кислоты
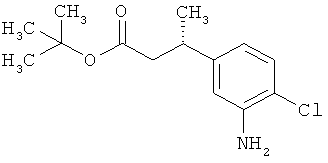
ЖХ-МС (Метод 4): Rt=1,34 мин; m/z=270 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1.14 (д, 3Н), 1,31 (с, 9Н), 2,19-2,45 (м, 2Н), 2,95 (кв, 1Н), 5,20 (с, 2Н), 6,42 (дд, 1Н), 6,65 (д, 1Н), 7,06 (д, 1Н).
[α]D20=+20,9°, с=0,670, хлороформ.
Пример 35А
Сложный трет-бутиловый эфир (-)-(3R)-3-(3-амино-4-хлорфенил)бутановой кислоты

ЖХ-МС (Метод 4): Rt=1,34 мин; m/z=214 (M+H-C4H8)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,14 (д, 3Н), 1,31 (с, 9Н), 2,38 (дд, 2Н), 2,95 (кв, 1Н), 5,20 (уш. с, 2Н), 6,42 (дд, 1Н), 6,65 (д, 1Н), 7,06 (д, 1Н).
[α]D20=-24,1°, с=0,570, хлороформ.
Пример 36А
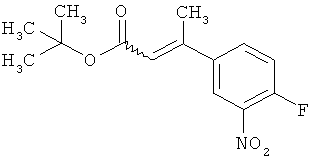
4,81 г гидрида натрия (60%-ная суспензия в минеральном масле, 120,13 ммоль) суспендировали в смеси из 120 мл ТГФ и 120 мл ДМФА. Эту смесь охлаждали до 0°С и по каплям прибавляли 29,5 мл (125,59 ммоль) трет-бутил(диэтоксифосфорил)ацетата. Спустя 30 мин при 0°С к этому добавляли 20,0 г (109,21 ммоль) 4-фтор-3-нитроацетофенона. После окончания прибавления реакционную смесь медленно нагревали до Ткомн. и перемешивали еще 3,5 ч при Ткомн, прежде чем потом выливали эту смесь в воду. Трижды экстрагировали этилацетатом, а объединенные органические фазы упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 50:1). Было получено 7,24 г (23,6% от теор.) целевого продукта в виде смеси изомеров E/Z (E/Z приблизительно 1,2:1).
ЖХ-МС (Метод 4): изомер 1: Rt=1,34 мин; m/z=208; изомер 2: Rt=1,42 мин; m/z=208.
Пример 37А
Сложный трет-бутиловый эфир (+/-)-3-(3-амино-4-фторфенил)бутановой кислоты

7,24 г (25,74 ммоль) трет-бутил-(2Е/Z)-3-(4-фтор-3-нитрофенил)бут-2-еноата (E/Z примерно 1,2:1) растворяли в 200 мл этанола и добавляли палладий на угле (10%). Реакционную смесь в течение ночи интенсивно перемешивали в атмосфере водорода при нормальном давлении. Затем фильтровали через целит, а остаток дважды промывали этилацетатом. Объединенные фильтраты упаривали в вакууме, а остаток сушили в высоком вакууме. Было получено 6,02 г целевого продукта (92,4% от теор.).
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,14 (д, 3Н), 1,31 (с, 9Н), 2,37 (дд, 2Н), 2,95 (кв, 1Н), 4,98 (с, 2Н), 6,36 (ддд, 1Н), 6,62 (дд, 1Н), 6,85 (дд, 1Н).
Полученный выше рацемат был разделен на энантиомеры с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak OJ-H, 5 мкм, 250 мм×20 мм; объем впрыска: 0,15 мл; температура: 35°С; элюент: 65% изогексана/35% этанола; расход: 15 мл/мин; детектирование: 220 нм]. Исходя из 6,0 г рацемата, было получено 2,44 г энантиомера 1 (Пример 38А) и 1,92 г энантиомера 2 (Пример 39А):
Пример 38А
Сложный трет-бутиловый эфир (+)-(3S)-3-(3-амино-4-фторфенил)бутановой кислоты
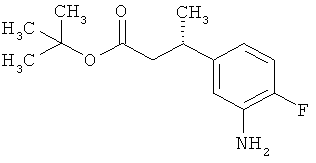
ЖХ-МС (Метод 6): Rt=1,11 мин; m/z=254 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,14 (д, 3Н), 1,31 (с, 9Н), 2,18-2,46 (м, 2Н), 2,95 (кв, 1Н), 4,99 (уш. с, 2Н), 6,36 (ддд, 1Н), 6,61 (дд, 1Н), 6,85 (дд, 1Н).
[α]D20=+22,5°, с=0,570, хлороформ.
Пример 39А
Сложный трет-бутиловый эфир (-)-(3R)-3-(3-амино-4-фторфенил)бутановой кислоты
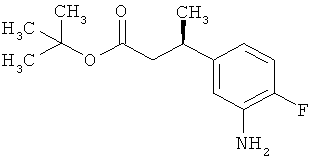
ЖХ-МС (Метод 6): Rt=1,11 мин; m/z=254 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,14 (д, 3Н), 1,31 (с, 9Н), 2,26-2,45 (м, 2Н), 2,95 (кв, 1Н), 4,99 (уш. с, 2Н), 6,36 (ддд, 1Н), 6,62 (дд, 1Н), 6,85 (дд, 1Н).
[α]D20=-23,2°, с=0,510, хлороформ.
Пример 40А
Этил-3-(3-бром-4-фторфенил)акрилат

К раствору 6,5 г (31,7 ммоль) 3-бром-4-фторбензилового спирта и 13,25 г (38 ммоль) этоксикарбонилметиленфосфорана в 390 мл толуола добавили 9,65 г (111 ммоль) двуокиси марганца. Реакционную смесь нагрели до кипения с обратным холодильником, спустя 1 ч добавили дополнительные 9,65 г двуокиси марганца и в течение ночи дополнительно кипятили с обратным холодильником. После охлаждения эту смесь фильтровали через целит, а фильтрат упаривали. Остаток очищали с помощью флэш-хроматографии на силикагеле (элюент: циклогексан/этилацетат 5:1). Было получено 7,05 г (81% от теор.) целевого продукта в форме смеси E/Z-изомеров.
ЖХ-МС (Метод 4): Rt=1,33 мин и 1,35 мин; m/z=273/275 (М+Н)+.
Пример 41А
Рац-этил-2-(3-бром-4-фторфенил)-транс-циклопропанкарбоксилат

В атмосфере аргона помещали 381 мг (9,52 ммоль) гидрида натрия (60%-ная суспензия в парафиновом масле) в 20 мл ДМСО и при Ткомн. за один прием добавляли 2,1 г (9,52 ммоль) йодида триметилсульфоксония. После окончания газовыделения медленно прикалывали 2,0 г (7,3 ммоль) этил-3-(3-бром-4-фторфенил)акрилата, растворенного в 10 мл ДМСО. Эту реакционную смесь в течение ночи нагревали при 50°С, потом охлаждали до Ткомн. и без дополнительной обработки очищали с помощью флэш-хроматографии на силикагеле (элюент:изогексан/этилацетат 100:1). Было получено 907 мг (43% от теор.) целевого продукта.
ЖХ-МС (Метод 6): Rt=1,20 мин; m/z=289 (М+Н)+.
ГХ-МС (Метод 1): Rt=5,85 мин; m/z=287/289 (М+Н)+.
1H-ЯМР (500 МГц, CDCl3): δ [м.д.]=1,20-1,33 (м, 4Н), 1,56-1,63 (м, 1Н), 1,80-1,88 (м, 1Н), 2,43-2,52 (м, 1Н), 4,17 (кв 2Н), 7,00-7,06 (м, 2Н), 7,28 (д, 1Н).
Пример 42А
Рац-этил-2-[3-(бензиламино)-4-фторфенил]-транс-циклопропан-карбоксилат
В атмосфере аргона суспендировали 361,5 мг (3,8 ммоль) mpem-бутилата натрия в 12,9 мл толуола и последовательно добавляли 900 мг (3,1 ммоль) (+/-)-транс-этил-2-(3-бром-4-фторфенил)циклопропанкарбоксилата, 403 мг (3,8 ммоль) бензиламина, 28,7 мг (0,03 ммоль) трис(дибензилиденацетон)дипалладия и 19,5 мг (0,03 ммоль) рац.-2,2'-бис(дифенилфосфино)-1,1'-бинафтила. Эту смесь в течение 4 ч нагревали при 110°C. Затем реакционную смесь охлаждали до Ткомн, добавляли 100 мл этилацетата и 50 мл насыщенного раствора хлорида аммония и фильтровали через целит.Органическую фазу отделяли, промывали соответственно порциями по 50 мл насыщенного раствора хлорида аммония и насыщенного раствора хлорида натрия, сушили над сульфатом магния и упаривали. Сырой продукт очищали с помощью препаративной ВЭЖХ. Было получено 262 мг целевого соединения с чистотой 66% (18% от теор.).
ЖХ-МС (Метод 6): Rt=1,28 мин; m/z=314 (M+H)+.
Пример 43А
Рац-этил-2-[3-амино-4-фторфенил]-транс-циклопропанкарбоксилат
262 мг (чистота 66%, 0,55 ммоль) (+/-)-этил-2-[3-(бензиламино)-4-фторфенил]-транс-циклопропанкарбоксилата растворяли в 5 мл смеси этанол/ТГФ (1:1), смешивали с 26 мг палладия на угле (10%) и гидрирова-ли в течение 24 ч при Ткомн при давлении водорода 1 бар. Затем реакционную смесь фильтровали через целит, остаток дополнительно промывали этанолом, а фильтрат упаривали. Полученный таким образом сырой продукт очищали при помощи препаративной ВЭЖХ. Было получено 87 мг (69% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=0,96 мин; m/z=224 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-D6): δ [м.д.]=1,17-1,23 (м, 3Н), 1,23-1,28 (м, 1Н), 1,39 (дт, 1Н), 1,67-1,81 (м, 1Н), 2,21-2,31 (м, 1Н), 4,09 (кв, 2Н), 6,31 (ддд, 1Н), 6,55 (дд,1Н), 6,86 (дд, 1Н).
Пример 44А
3-Амино-4-фторацетофенон
К раствору 3 г (16,4 ммоль) 4-фтор-3-нитроацетофенона в 7,8 мл 12 Н соляной кислоты при 0°C в течение 15 мин прикалывали раствор 11,1 г (89 ммоль) дигидрата хлорида олова в 12 мл воды. Затем реакционную смесь в течение 15 мин нагревали до кипения с обратным холодильником, а потом дополнительно перемешивали в течение ночи при Ткомн. После этого реакционную смесь выливали на лед, с помощью 50%-ного водного раствора гидроксида натрия доводили до pH 12 и экстрагировали этилацетатом. Органическую фазу промывали насыщенным раствором поваренной соли, сушили над сульфатом магния и упаривали. Было получено 2,47 г (90% чистоты, 87% от теор.) целевого соединения.
ЖХ-МС (Метод 5): Rt=1,32 мин; m/z=154 (М+Н)+.
Пример 45А и Пример 46A
Этил-(2Е)-3-(3-амино-4-фтофенил)-2-метилбут-2-еноат
и
этил-(2Z)-3-(3-амино-4-фтофенил)-2-метилбут-2-еноат
К суспензии 1,29 г гидрида натрия (60%-ная суспензия в парафиновом масле; 32,3 ммоль) в 24,7 мл ТГФ при 0°С медленно прикалывали 6,92 мл (7,68 г; 32,3 ммоль) сложного триэтилового эфира 2-фосфонопропионовой кислоты. Реакционную смесь перемешивали 30 мин, а затем добавляли 2,47 г (90% чистоты, 14,5 ммоль) 3-амино-4-фторацетофенона. Эту реакционную смесь сначала перемешивали 1 ч при Ткомн., затем 2 ч при кипении с обратным холодильником, потом снова охлаждали до Ткомн. и дополнительно перемешивали в течение ночи. Потом эту реакционную массу выливали в воду и трижды экстрагировали соответственно порциями по 100 мл этилацетата. Объединенные органические фазы сушили над сульфатом магния, упаривали, а остаток очищали с помощью флэш-хроматографии на силикагеле (элюент: толуол/этилацетат 5:1). В виде отдельных изомеров было получено 612 мг (15% от теор.) 2Е-изомера (Пример 45А) и 529 мг (13% от теор.) 27-изомера (Пример 46А).
2Е-изомер (Пример 45А):
ЖХ-МС (Метод 6): Rt=1,05 мин; m/z=238 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,22-1,29 (м, 3Н), 1,69 (д, 3Н), 2,11 (д, 3Н), 4,17 (д, 2Н), 6,30 (ддд, 1Н), 6,56 (дд, 1Н), 6,97 (дд, 1Н).
2 г-изомер (Пример 46А):
ЖХ-МС (Метод 6): Rt=0,99 мин; m/z=238 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,85 (т, 3Н), 1,86-1,92 (м, 3Н), 1,94-2,01 (м, 3Н), 3,82 (кв, 2Н), 6,24 (ддд, 1Н), 6,51 (дд, 1Н), 6,87 (дд, 1Н).
Пример 47А

К раствору 48 мг (0,2 ммоль) этил-(2Е)-3-(3-амино-4-фторфенил)-2-метилбут-2-еноата в 5 мл метанола добавляли 4,8 мг палладия на угле (10%). Реакционную массу гидрировали в течение ночи при давлении водорода 1 бар. Затем фильтровали через целит, а фильтрат упаривали. Было получено 35,8 мг (74% от теор.) целевого соединения, которое содержало приблизительно 20% эритроизомера.
ЖХ-МС (Метод 6): Rt=1,02 мин; m/z=240 (M+H)+.
Пример ЛВА
Рац-эртпро-этил-3-(3-амино-4-фторфенил)-2-метилбутаноат
К раствору 30 мг (0,13 ммоль) этил-(2Z)-3-(3-амино-4-фторфенил)-2-метилбут-2-еноата в 3,1 мл метанола добавляли 3 мг палладия на угле (10%). Реакционную массу гидрировали в течение ночи при давлении водорода 1 бар. Затем фильтровали через целит, а фильтрат упаривали. Было получено 22,5 мг (74% от теор.) целевого соединения, которое содержало приблизительно 5% mpeo-изомера.
ЖХ-МС (Метод 6): Rt=1,04 мин; m/z=240 (M+H)+.
Пример 49А
Трет-бутил-(2Е)-3-(4-фтор-3-нитрофенил)акрилат

В атмосфере аргона помещали 0,65 г гидрида натрия (60%-ная суспензия в парафиновом масле; 16,3 ммоль) в 25 мл ТГФ и охлаждали до 0°С. Затем медленно прикалывали 4,29 г (17 ммоль) сложного mpem-бутилового эфира диэтилфосфоноуксусной кислоты. Спустя 30 мин перемешивания добавляли 2,5 г (14,8 ммоль) 4-фтор-3-нитробензальдегида. Реакционную смесь перемешивали 3 ч при Ткомн., затем выливали в 100 мл воды и трижды экстрагировали соответственно порциями по 100 мл этилацетата. Объединенные органические фазы сушили над сульфатом магния и упаривали. Остаток очищали с помощью флэш-хроматографии на силикагеле (элюент: циклогексан/этилацетат 50:1). Было получено 3,37 г (85% от теор.) целевого продукта.
ГХ-МС (Метод 1): Rt=6,45 мин; m/z=211 (M-tBu)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,49 (с, 9Н), 6,69 (д, 1Н), 7,59-7,76 (м,2Н), 8,19 (ддд, 1Н), 8,50 (дд, 1Н).
Пример 50А
7рет-бутил-(2Е)-3-(4-циано-3-нитрофенил)акрилат

К раствору 500 мг (1,87 ммоль) трет-бутил-(2Е)-3-(4-фтор-3-нитрофенил)акрилата в 5,4 мл ДМФА добавляли 134 мг (2,06 ммоль) цианида калия. После перемешивания в течение ночи при Ткомн. реакционную смесь непосредственно очищали при помощи флэш-хроматографии (элюент: смесь циклогексан/этилацетат). Было получено 57 мг (11% от теор.) целевого соединения.
ЖХ-МС (Метод 5): Rt=2,40 мин; m/z=292 (M+NH4)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,39-1,58 (м, 9Н), 6,91 (д, 1Н), 7,73 (д, 1Н), 8,19 (д, 1Н), 8,26-8,38 (м, 1Н), 8,72 (д, 1Н).
Пример 51А
трет-бутил-3-(3-амино-4-цианофенил)пропаноат

К раствору 48,9 мг (0,18 ммоль) трет-бутил-(2Е)-3-(4-циано-3-нитрофенил)акрилата в 4,4 мл этанола добавляли 4,9 мг палладия на угле (10%). Реакционную массу гидрировали при Ткомн. в течение ночи при давлении водорода 1 бар. Затем фильтровали через целит, а фильтрат упаривали. Было получено 43,5 мг (99% от теор.) целевого соединения с чистотой 85%.
ЖХ-МС (Метод 6): Rt=1,06 мин; m/z=247 (М+Н)+.
Пример 52А
Этил-(3R)-4,4,4-трифтор-3-метилбутаноат

К 287 г (1,65 моль) (3R)-4,4,4-трифтор-3-метилбутановой кислоты [A.Gerlach и U.Schuiz, Speciality Chemicals Magazine 24 (4), 37-38 (2004); CAS Acc.-Nr. 142: 179196] в 580 мл этанола при комнатной температуре медленно добавляли 133 мл (1,82 моль) тионилхлорида. Затем реакционный раствор нагревали до 80°С и 2 ч перемешивали при этой температуре. После этого охлаждали до комнатной температуры, медленно добавляли 250 мл воды и трижды экстрагировали порциями по 150 мл метил-трет-бутилового эфира. Объединенные органические фазы сушили над сульфатом натрия. После фильтрации растворитель был удален в вакууме при 30°С и давлении 300 мбар. Затем сырой продукт перегоняли при 100 мбар и температуре в головной части колонки 65°С. Было выделено 225,8 г (113 моль, 74% от теор.) целевого соединения в виде бесцветной жидкости.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 4,10 (2Н, кв), 2,88-2,72 (1Н, м), 2,66-2,57 (1Н, м), 2,46-2,36 (1Н, м), 1,19 (3Н, т), 1,11 (3Н, д).
ГХ-МС (Метод 1): Rt=1,19 мин; m/z=184 (М)+.
[α]D20=+16,1°, с=0,41, метанол.
Пример 53А
Этил-4,4,4-трифтор-3-метил-2-(4-метилфенил)бутаноат (смесь диастереомеров)
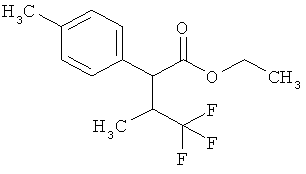
В атмосфере аргона 196,9 мг (0,88 ммоль) ацетата палладия(II) и 724,8 мг (1,84 ммоль) 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенила помещали в 50 мл безводного толуола. К этому реакционному раствору затем медленно добавляли 43,8 мл (43,8 ммоль) 1 М раствора гексаметилдисилазида лития в ТГФ и 10 мин перемешивали при Ткомн.. После этого реакционный раствор охлаждали до -10°С, медленно добавляли 7 г (38,0 ммоль) (+/-)-этил-4,4,4-трифтор-3-метилбутаноата и дополнительно перемешивали 10 мин при -10°С. Затем прикалывали 5 г (29,2 ммоль) 4-бромтолуола, растворенного в 50 мл толуола, и этот реакционный раствор нагревали сначала до Ткомн., а потом до 80°С. Смесь перемешивали 2 ч при этой температуре, потом охлаждали до Ткомн. и дополнительно перемешивали в течение ночи. После прохождения реакции (ТСХ-контроль; элюент: циклогексан/дихлорметан 2:1) реакционную смесь фильтровали через кизельгур, остаток многократно промывали этилацетатом и дихлорметаном, а объединенный фильтрат упаривали в вакууме. Полученный сырой продукт очищали с помощью хроматографии на силикагеле (элюент: петролейный эфир/дихлорметан 4:1→3:1). Было выделено 3,91 г (14,3 ммоль, 48,8% от теор.) целевого соединения в виде бесцветной жидкости.
1H-ЯМР (400 МГц, ДМСО-D6, δ/м.д.): 7,26 (2Н, д), 7,20-7,12 (2Н, м), 4,17-3,95 (2Н, м), 3,74 (0.25Н, д), 3,66 (0.75Н, д), 3,35-3,07 (1Н, м), 2,29 (2.25Н, с), 2,28 (0.75Н, с), 1,17 (0,75Н, д), 1,11 (3Н, т), 0,76 (2.25Н, д).
ГХ-МС (Метод 1): Rt=4,20 мин; m/z=275 (М+Н)+ (диастереомер 1); Rt=4,23 мин; m/z=275 (M+H)+ (диастереомер 2).
Пример 54А
Этил-(3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноат
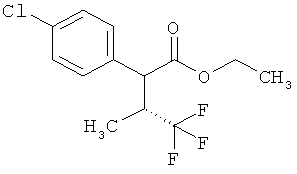
Приготовление раствора А: 163,9 мл 1 М раствора гексаметилдисилазида лития в толуоле в атмосфере аргона охлаждали до температуры от -10°С до -20°С (охлаждение с помощью бани ацетон/сухой лед) и медленно добавляли 20 г (108,6 ммоль) этил-(3R)-4,4,4-трифтор-3-метилбутаноата, растворенного в 150 мл толуола, причем обращали внимание на то, чтобы температура не превышала -10°С. Этот раствор затем дополнительно перемешивали 10 мин при температуре не более -10°С.
Приготовление раствора В: 27,03 г (141,2 ммоль) 1-бром-4-хлорбензола в атмосфере аргона при Т«омн. растворяли в 100 мл толуола и добавляли 731 мг (3,26 ммоль) ацетата палладия (II) и 2,693 г (6,84 ммоль) 2'-(дициклогексилфосфанил)-N,N-диметилбифенил-2-амина. Этот раствор дополнительно перемешивали 10 мин при Ткомн.
Сначала удаляли охлаждающую баню от раствора А. Затем к еще холодному раствору А медленно прикалывали раствор В. Объединенные теперь растворы медленно нагревали до Ткомн. и перемешивали 1 ч при этой температуре. Затем реакционный раствор нагревали до 80°С (внутренняя температура) и перемешивали при этой температуре 3 ч. После этого реакционный раствор медленно охлаждали до Ткомн. и дополнительно перемешивали еще 12 ч. Потом реакционную смесь фильтровали через кизельгур, остаток многократно дополнительно промывали толуолом, а объединенный фильтрат упаривали в вакууме. Полученный сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/дихлорметан 4:1). Было выделено 27,4 г (92,98 ммоль, 86% от теор.) целевого соединения в виде желтого масла с соотношением диастереомеров 3:1.
ГХ-МС (Метод 1): Rt=4,45 мин; m/z=294 (М)+ (диастереомер 1); Rt=4,48 мин; m/z=294 (M)+ (диастереомер 2).
Аналогично примерам синтеза 53А и 54А были получены следующие соединения:
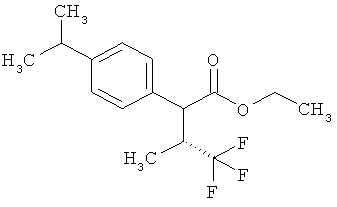
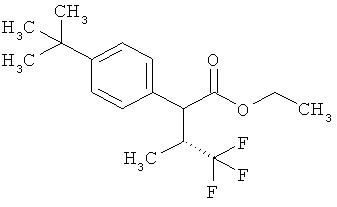
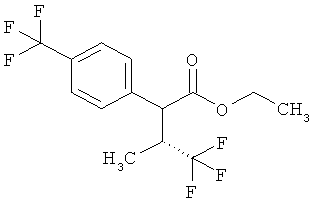

Пример 59А
Этил-2-[4-(бромметил)фенил]-4,4,4-трифтор-3-метилбутаноат

2,25 г (8,2 ммоль) этил-4,4,4-трифтор-3-метил-2-(4-метилфенил)бутаноата, 1,53 г (8,6 ммоль) N-бромсукцинимида и 67 мг (0,41 ммоль) 2,2'-азобис-2-метилпропаннитрила в 36 мл трихлорметана перемешивали в течение ночи при кипячении с обратным холодильником. После полностью завершенного превращения отфильтровывали сукцинимид, остаток на фильтре дополнительно промывали дихлорметаном, а фильтрат упаривали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 40:1). Было выделено 2,667 г (7,5 ммоль, 92% от теор.) желтого масла.
ГХ-МС (Метод 1): Rt=5,72 мин; m/z=373 (М-Br)+ (диастереомер 1); Rt=5,74 мин; m/z=373 (M-Br)+ (диастереомер 2).
Пример 60А
Этил-4,4,4-трифтор-3-метил-2-[4-(2,2,2-трифторэтил)фенил]бутаноат

К 3,77 г (10,67 ммоль) этил-2-[4-(бромметил)фенил]-4,4,4-трифтор-3-метилбутаноата в 40 мл 1-метилпирролидин-2-она добавляли 529 мг (2,78 ммоль) йодида меди(1) и 4 г (20,82 ммоль) метил-2,2-дифтор-2-(фторсульфонил)ацетата и в течение ночи перемешивали при 80°С. После прошедшего взаимодействия реакционный раствор медленно выливали в 100 мл воды со льдом. Полученную смесь затем трижды экстрагировали диэтиловым эфиром. Объединенные органические фазы сушили над сульфатом магния. После фильтрации растворитель удаляли в вакууме. Полученный сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/дихлорметан 4:1). Было выделено 1,48 г (4,32 ммоль, 41% от теор.) целевого соединения в виде желтоватого масла.
ГХ-МС (Метод 1): Rt=4,06 мин; m/z=342 (M)+ (диастереомер 1); Rt=4,09 мин; m/z=342 (М)+ (диастереомер 2).
МС (DCI): m/z=360 (M+NH4)+.
Пример 61А
1-Бром-4-(2-бром-1-фторэтил)бензол

5,0 г (27,31 ммоль) 4-бромстирола растворяли в 40 мл дихлорметана, охлаждали до 0°С и смешивали с 13,21 г (81,94 ммоль) тригидрофторида триэтиламина. После этого в три приема добавляли 5,83 г (32,78 ммоль) N-бромсукцинимида. Смесь перемешивали в течение ночи при Ткомн. После разбавления дихлорметаном реакционную смесь выливали в ледяную воду. Органическую фазу последовательно промывали 1 н. соляной кислотой, водой и насыщенным раствором гидрокарбоната натрия, сушили над сульфатом магния и упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: пентан). Было выделено 4,14 г (53,8% от теор.) целевого соединения.
ГХ-МС (Метод 1): Rt=4,94 мин; m/z=277/281/283 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-D6): δ [м.д.]=3,75-4,04 (м, 2Н), 5,84 (дт, 1Н), 7,31-7,51 (м, 2Н), 7,55-7,78 (м, 2Н).
Пример 62А
1-Бром-4-(1-фторвинил)бензол

К охлажденному до 0°С раствору 1,0 г (3,55 ммоль) 1-бром-4-(2-бром-1-фторэтил)бензола в 10 мл пентана в несколько порций добавляли 796 мг (7,09 ммоль) трет-бутилата калия. Полученную в результате суспензию перемешивали 30 мин при 0°С, а потом 1 ч при Ткомн. Отфильтровывали твердое вещество, а фильтрат промывали насыщенным раствором хлорида аммония, сушили над сульфатом магния и осторожно упаривали в вакууме. Было получено 0,61 г (85,6% от теор.) целевого соединения.
ГХ-МС (Метод 1): Rt=3,14 мин; m/z=200/202 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=5,10 (дд, 1Н), 5,47 (дд, 1Н), 7,48-7,61 (м, 2Н), 7,62-7,72 (м, 2Н).
Пример 63А
Сложный этиловый эфир (3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутановой кислоты (смесь диастереомеров)

24,4 мл (24,4 ммоль) 1 М раствора гексаметилдисилазида лития в толуоле охлаждали до -10°С и по каплям добавляли раствор 3,0 г (16,29 ммоль) этил-(3R)-4,4,4-трифтор-3-метилбутаноата в 15 мл абс. толуола. Эту смесь дополнительно перемешивали 10 мин. Затем при -10°С прикалывали предварительно приготовленный раствор 3,92 г (21,18 ммоль) 1-бром-4-этилбензола, 110 мг (0,49 ммоль) ацетата палладия (II) и 404 мг (1,03 ммоль) 2'-дициклогексилфосфино-2-(N,N-диметиламино)бифенила в 20 мл абс. толуола. Полученную в результате реакционную смесь перемешивали сначала 1 ч при Ткомн., потом 3 ч при 80°С. После этого смесь упаривали в вакууме, а остаток переводили в этилацетат и выливали в воду. Водную фазу повторно экстрагировали этилацетатом, а объединенные органические фазы промывали насыщенным раствором хлорида аммония и насыщенным раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Из этого остатка после хроматографии на силикагеле (элюент: сначала циклогексан, потом градиент циклогексан/этилацетат 200:1→50:1) получили 3,051 г целевого соединения (64,9% от теор., соотношение диастереомеров приблизительно 3:1).
ЖХ-МС (Метод 4): Rt=1,52 мин; m/z=289 (M+H)+ (побочный диастереомер); Rt=1,54 мин; m/z=289 (М+Н)+ (основной диастереомер).
1H-ЯМР (400 МГц, ДМСО-d6): основной диастереомер: δ [м.д.]=0,76 (д, 3Н), 1,13 (т, 3Н), 1,17 (т, 3Н), 2,55-2,63 (м, 2Н), 3,21-3,31 (м, 1Н), 3,67 (д, 1Н), 3,95-4,16 (м, 2Н), 7,15-7,23 (м, 2Н), 7,25-7,31 (м, 2Н).
Аналогичным образом, исходя из этил-(3R)-4,4,4-трифтор-3-метилбутаноата и соответствующего фенилбромида, были получены оба следующих соединения:
Пример 64А
Сложный этиловый эфир (3R)-4,4,4-трифтор-3-метил-2-(4-винилфенил)бутановой кислоты (смесь диастереомеров)
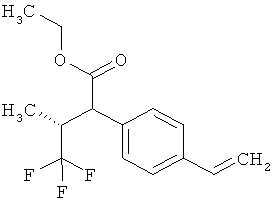
ГХ-МС (Метод 1): Rt=4,64 мин и 4,66 мин; m/z=286 (М)+.
1H-ЯМР (400 МГц, ДМСО-d6): основной диастереомер: δ [м.д.]=0,79 (д, 3Н), 1,12 (т, 3Н), 3,22-3,32 (м, 1Н), 3,73 (д, 1Н), 3,99-4,17 (м, 2Н), 5,28 (д, 1Н), 5,84 (д, 1Н), 6,72 (дд, 1Н), 7,34-7,40 (м, 2Н), 7,45-7,51 (м, 2Н).
Пример 65А
Сложный этиловый эфир (3R)-4,4,4-трифтор-2-[4-(1-фторвинил)фенил]-3-метилбутановой кислоты

ГХ-МС (Метод 1): Rt=4,60 мин и 4,63 мин; m/z=304 (М)+ ЖХ-МС (Метод 6); Rt=1,29 мин и 1,30 мин; m/z=279.
1H-ЯМР (400 МГц, ДМСО-d6): основной диастереомер: δ [м.д.]=0,79 (д, 3Н), 1,12 (т, 3Н), 3,34-3,38 (м, 1Н), 3,81 (д, 1Н), 3,99-4,17 (м, 2Н), 4,97 (дд, 1Н), 5,42 (дд, 1Н), 7,46-7,49 (м, 2Н), 7,63 (д, 2Н).
Пример 66A
Метил-(4-хлорфенил)-(3-оксоциклопентил)ацетат

В атмосфере аргона 14,8 мл (105,6 ммоль) диизопропиламина помещали в 150 мл ТГФ, охлаждали до -30°С и медленно добавляли 42,3 мл (105,75 ммоль) 2,5 М раствора н-бутиллития в гексане. Затем реакционный раствор нагревали до -20°С, медленно добавляли 15 г (81,25 ммоль) метил-(4-хлорфенил)ацетата, растворенного в 90 мл ТГФ, и 2 ч дополнительно перемешивали при этой температуре. Потом реакционный раствор охлаждали до -78°С и медленно добавляли 7,2 мл (86,1 ммоль) 2-циклопентен-1-она, растворенного в 60 мл ТГФ. После оконченного прибавления раствор дополнительно перемешивали 1 ч при этой температуре. После ТСХ-контроля (элюент: циклогексан/этилацетат 9:1) к реакционной массе добавляли насыщенный раствор хлорида аммония и переводили в этилацетат. Водную фазу дважды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом магния. После фильтрации растворитель удаляли в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 4:1). Было выделено 15,65 г (58,67 ммоль, 72% от теор.) целевого соединения в виде желтоватого масла.
ГХ-МС (Метод 1): Rt=7,02 мин; m/z=266 (M)+ (диастереомер 1); Rt=7,04 мин; m/z=266 (М)+ (диастереомер 2).
MC (DCI): m/z=284 (M+NH4)+.
Пример 67А
Метил-(4-хлорфенил)-(3,3-дифторциклопентил)ацетат
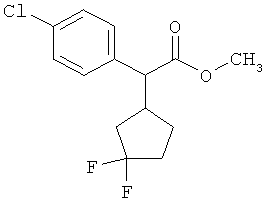
В атмосфере аргона загружали 82,5 мл (82,14 ммоль) 50%-ного раствора 1,1'-[(трифтор-λ4-сульфанил)имино]бис(2-метоксиэтана) (деоксофтор реагента) в ТГФ, разбавленного 200 мл толуола, охлаждали до 5°С и медленно прибавляли 744 мкл (5,87 ммоль) 1 М раствора комплекса трифторида бора с диэтиловым эфиром. Эту смесь дополнительно перемешивали 2 ч при 5°С. Затем к реакционному раствору медленно прибавляли 15,65 г (58,67 ммоль) метил-(4-хлорфенил)-(3-оксоциклопентил)ацетата, растворенного в 200 мл толуола, потом нагревали до 55°С и 60 ч дополнительно перемешивали при этой температуре. После этого реакционную смесь выливали в охлажденную до 0°С смесь, состоящую из 100 мл толуола и 100 мл 2 М водного раствора гидроксида натрия. Органическую фазу отделяли, а водную фазу еще трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом натрия. После фильтрации растворитель удаляли в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 7:1). Было выделено 13,24 г (45,86 ммоль, 78% от теор.) целевого соединения в виде бесцветного масла.
МС (DCI): m/z=306 (M+NH4)+.
ГХ-МС (Метод 1): Rt=5,83 мин; m/z=288 (M)+ (диастереомер 1); Rt=5,86 мин; m/z=288 (М)+ (диастереомер 2).
Пример 68А
(+/-)-Этил-(2,2-дифторциклопентил)ацетат
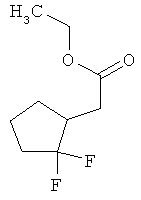
К раствору 52,8 мл (399,5 ммоль) трифторида диэтиламиносеры (DAST) в 150 мл абс. дихлорметана при Ткомн. прикалывали 17,0 г (99,88 ммоль) (+/-)-этил-2-оксоциклопентилацетата. Эту смесь кипятили в течение ночи с обратным холодильником. После охлаждения к этому были добавлены дополнительные 13,2 мл (99,88 ммоль) трифторида диэтиламиносеры (DAST), и смесь снова перемешивалась 36 ч при кипячении с обратным холодильником. После охлаждения разбавляли дихлорметаном, осторожно добавляли насыщенный раствор гидрокарбоната натрия и потом интенсивно перемешивали. Органическую фазу последовательно промывали насыщенным раствором гидрокарбоната натрия, дважды 1 н. соляной кислотой и насыщенным раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Из темно-коричневого остатка продукт выделяли с помощью колоночной хроматографии на силикагеле (элюент: пентан/дихлорметан 10:1→1:1). Было получено 7,52 г (39% от теор.) целевого соединения.
ГХ-МС (Метод 1): Rt=2,88 мин; m/z=172.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,18 (т, 3Н), 1,33-1,48 (м, 1Н), 1,61-1,77 (м, 2Н), 1,92-2,20 (м, 3Н), 2,24-2,38 (м, 1Н), 2,43-2,60 (м, 2Н), 4,07 (кв, 2Н).
Пример 69А
Сложный этиловый эфир (4-хлорфенил)-(2,2-дифторциклопентил)уксусной кислоты (смесь диастереомеров)

22,6 мл (22,6 ммоль) 1 М раствора гексаметилдисилазида лития в толуоле охлаждали до -20°С и по каплям добавляли раствор 2,90 г (15,09 ммоль) (+/-)-этил-(2,2-дифторциклопентил)ацетата в 20 мл абс. толуола. Эту смесь перемешивали 10 мин при -20°С. После удаления охлаждения прикалывали предварительно приготовленный раствор 3,75 г (19,61 ммоль) 4-бромхлорбензола, 110 мг (0,49 ммоль) ацетата палладия (II) и 374 мг (0,95 ммоль) 2'-дициклогексилфосфино-2-(N,N-диметиламино)бифенила в 20 мл абс. толуола. Полученную в результате реакционную смесь перемешивали сначала 1 ч при Ткомн., а потом 2 ч при 90°С. После охлаждения реакционную смесь выливали в воду. Водную фазу трижды экстрагировали этилацетатом, а объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме. Из этого остатка после хроматографии на силикагеле (элюент: циклогексан/этилацетат 50:1) получили 2,70 г целевого соединения (59,1% от теор., соотношение диастереомеров приблизительно 1:4,3).
ГХ-МС (Метод 1): Rt=6,09 мин и 6,20 мин.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,01-1,27 (м, 3Н), 1,37-1,50 (м, 1Н), 1,51-1,75 (м, 3Н), 1,94-2,23 (м, 3Н), 2,84-3,07 (м, 1Н), 3,55-3,79 (м, 1Н), 3,93-4,20 (м, 2Н), 7,29-7,53 (м, 4Н).
Пример 70А
(+)-(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановая кислота

5,086 г (17,26 ммоль) этил-(3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноата растворяли в 68 мл диоксана и добавляли 34 мл 1 н. водного раствора гидроксида натрия. Эту реакционную массу перемешивали 2 ч при 50°С. Потом реакционную смесь подкисляли с помощью 1 н. соляной кислоты до pH 1 и несколько раз экстрагировали дихлорметаном. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над сульфатом натрия и упаривали в вакууме. Было получено 3,9 г (14,63 ммоль, 85% от теор., 83% de) целевого соединения.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 12,95-12,73 (1Н, уш.с), 7,49-7,34 (4Н, м), 3,68 (1Н, д), 3,31-3,18 (1Н, м), 1,20 (0,25Н, д), 0,78 (2,75Н, д).
ГХ-МС (Метод 1): Rt=4,85 мин; m/z=266 (М)+.
[α]D20=+57,2°, c=0,41, метанол.
Аналогично примеру синтеза 70А были получены соединения, приведенные в следующей таблице:


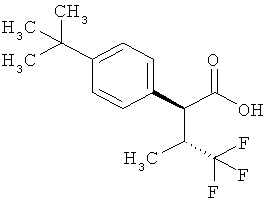
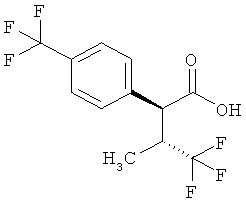


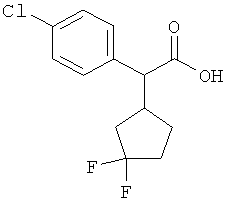
Пример 78А
(3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутановая кислота (смесь диастереомеров)

3,0 г сложного этилового эфира (3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутановой кислоты (примерно 88% чистоты, приблизительно 9,16 ммоль; смесь диастереомеров) растворяли в смеси, соответственно по 12,4 мл каждого, из метанола, ТГФ и воды и порциями добавляли 5,49 г (137,35 ммоль) гидроксида натрия. Реакционную смесь перемешивали 9 ч при 40°С. После охлаждения летучие растворители в значительной мере были удалены в вакууме, а остаток разбавили водой. Подкисляли с помощью соляной кислоты, а водную фазу трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом натрия, упаривали в вакууме, а остаток сушили в высоком вакууме. Было получено 2,61 г целевого соединения в виде сырого продукта, который далее не подвергался очистке (соотношение диастереомеров примерно 9:1).
ЖХ-МС (Метод 6): Rt=1,08 мин; m/z=259 (М-Н)- (побочный диастереомер); Rt=1,11 мин; m/z=259 (М-Н)- (основной диастереомер).
1H-ЯМР (400 МГц, ДМСО-d6): основной диастереомер: δ [м.д.]=0,76 (д, 3Н), 1,17 (т, 3Н), 2,54-2,66 (м, 4Н), 3,10-3,29 (м, 1Н), 3,56 (д, 1Н), 7,14-7,22 (м, 2Н), 7,22-7,32 (м, 2Н), 12,58 (уш. с, 1Н).
Аналогичным образом (температура реакции: от Ткомн. до 40°С; время реакции: 9-12 ч) из соответствующих сложных эфиров были получены оба следующих производных карбоновой кислоты:
Пример 79А
(3R)-4,4,4-трифтор-3-метил-2-(4-винилфенил)бутановая кислота (смесь диастереомеров)

Соотношение диастереомеров приблизительно 10:1.
ЖХ-МС (Метод 6): Rt=1,04 мин; m/z=257 (М-Н)- (побочный диастереомер); Rt=1,06 мин; m/z=257 (М-Н)- (основной диастереомер).
1Н-ЯМР (400 МГц, ДМСО-d6): основной диастереомер: δ [м.д.]=0,78 (д, 3Н), 3,18-3,31 (м, 1Н), 3,62 (д, 1Н), 5,28 (д, 1Н), 5,84 (д, 1Н), 6,73 (дд, 1Н), 7,31-7,39 (м, 2Н), 7,40-7,54 (м, 2Н), 12,74 (уш.с, 1Н).
Пример 80А
(3R)-4,4,4-трифтор-2-[4-(1-фторвинил)фенил]-3-метилбутановая кислота (смесь диастереомеров)

Соотношение диастереомеров приблизительно 9:1.
ГХ-МС (Метод 1): Rt=4,97 мин; m/z=276.
1H-ЯМР (400 МГц, ДМСО-d6): основной диастереомер: δ [м.д.]=0,78 (д, 3Н), 3,16-3,29 (м, 1Н), 3,70 (д, 1Н), 4,96 (дд, 1Н), 5,34 (д, 1Н), 5,47 (д, 1Н), 7,39-7,51 (м, 2Н), 7,58-7,69 (м, 2Н), 12,83 (уш.с, 1Н).
Пример 81А
(4-Хлорфенил)-(2,2-дифторциклопентил)уксусная кислота (смесь диастереомеров)

2,70 г (8,92 ммоль) сложного этилового эфира (4-хлорфенил)-(2,2-дифторциклопентил)уксусной кислоты (смеси изомеров) растворяли в 10 мл метанола, 10 мл ТГФ и 5 мл воды и при Ткомн. добавляли 7,13 г (89,18 ммоль) 50%-ного водного раствора гидроксида натрия. Эту реакционную смесь перемешивали в течение ночи при Ткомн. Потом разбавляли водой и с помощью соляной кислоты устанавливали кислый рН. Водную фазу трижды экстрагировали этилацетатом, а объединенные органические фазы сушили над сульфатом натрия, упаривали в вакууме и остаток сушили в высоком вакууме. Получили 2,39 г целевого соединения (97,6% от теор., соотношение диастереомеров примерно 1:1).
ЖХ-МС (Метод 6): Rt=1,05 мин и 1,07 мин; m/z=273 (М-Н)-.
Пример 82А
(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноилхлорид

19,5 г (73,13 ммоль) (2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты растворяли в 860 мл дихлорметана и смешивали с 0,5 мл ДМФА. Затем при температуре от -5°С до -10°С (охлаждающая баня лед/ацетон) медленно прикалывали 73 мл (146,26 ммоль) 2 М раствора оксалилхлорида в дихлорметане и смесь дополнительно перемешивали 1 ч при этой температуре. После полного прохождения реакции реакционный раствор упаривали в вакууме, а полученный остаток извлекали в 200 мл дихлорметана и затем снова упаривали досуха. Было получено 20.1 г (70,5 ммоль, 96% от теор.) целевого соединения в виде бесцветного масла. Полученный таким образом продукт был использован в последующих реакциях без дополнительной очистки и без дополнительной спектроскопической характеризации.
Аналогичным образом были получены соединения, приведенные в следующей таблице:
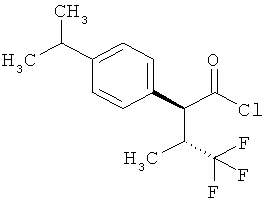
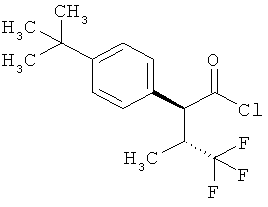

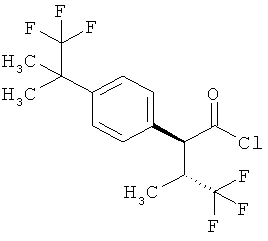

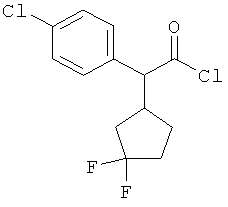
Пример 89А
Трет-бутил-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)пропаноат

18 г (70,38 ммоль) (2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноилхлорида растворяли в 500 мл ТГФ, добавляли 18,4 мл (105,57 ммоль) N,N-диизопропилэтиламина и охлаждали до -10°С. Затем медленно добавляли 20,07 г (70,38 ммоль) трет-бутил-3-(3-амино-4-хлорфенил)пропаноата, растворенного в 500 мл ТГФ, причем при этом обращали внимание, чтобы во время добавления температура не превышала 0°С. После этого смесь дополнительно перемешивали еще 1 ч. Потом к реакционному раствору добавляли воду и этилацетат, органическую фазу отделяли, а водную фазу еще трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом натрия и упаривали в роторном испарителе. Остаток очищали при помощи хроматографии на силикагеле (элюент: циклогексан/этилацетат 20:1). Получили 30,13 г (59,74 ммоль, 85% от теор.) целевого соединения.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 9,82 (1Н, с), 7,50-7,42 (4Н, м), 7,39-7,32 (2Н, м), 7,07-7,01 (1Н, м), 4,12 (1Н, д), 3,42-3,29 (1Н, м), 2,75 (2Н, т), 2,46 (2Н, т), 1,31 (9Н, с), 0,80 (3Н, д).
ЖХ-МС (Метод 7): Rt=3,03 мин; m/z=502/504 (M-H)+.
Аналогичным образом были получены соединения, приведенные в следующей таблице:
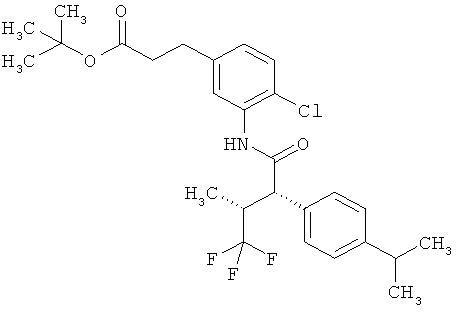



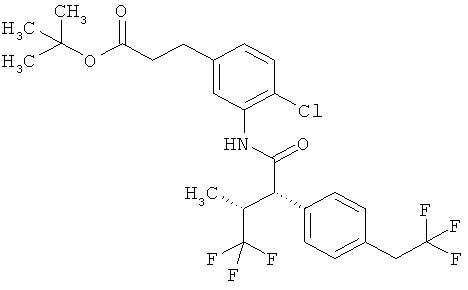





Пример 100А
Метил-[1-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]-амино}-4-фторфенил)циклопропил]ацетат

Раствор 70 мг (0,31 ммоль) (2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты, 84 мг (0,31 ммоль) метил-[1-(3-амино-4-фторфенил)циклопропил]ацетата, 179 мг (0,47 ммоль) гексафторфосфата 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (ГАТУ) и 0,6 мл пиридина в 2,4 мл ДМФА перемешивали в течение ночи при комнатной температуре. После окончания реакции реакционную массу без дополнительной обработки непосредственно разделяли при помощи препаративной ВЭЖХ. Было получено 106 мг (0,22 ммоль, 72% от теор.) целевого соединения в виде бесцветного масла.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 10,02 (1Н, с), 7,71 (1Н, дд), 7,52-7,38 (4Н, м), 7,15-7,06 (1Н, м), 7,05-6,98 (1Н, м), 4,11 (1Н, д), 3,48 (3Н, с), 3,42-3,25 (1Н, м), 2,57 (2Н, с), 0,90-0,84 (2Н, м), 0,81-0,74 (5Н, м).
ЖХ-МС (Метод 6): Rt=1,33 мин; m/z=472 (М+Н)+.
Аналогичным образом было получено следующее соединение:

Общая методика 1: Амидное сочетание производных 4,4,4-трифтор-3-метил-2-фенилбутановой кислоты с анилинами с применением ГАТУ
К раствору соответствующего производного 4,4,4-трифтор-3-метил-2-фенилбутановой кислоты (примерно от 0,8 до 1,5 экв., от 0,15 до 1,5 моль/л) и анилина (примерно от 0,8 до 1,5 экв., от 0,15 до 1,5 моль/л) в смеси из ДМФА и пиридина (соотношение в смеси приблизительно от 3:1 до 1,5:1) при Ткомн. добавляют ГАТУ (от 1,0 до 2,0 экв.). В качестве альтернативы вместо пиридина также может применяться N,N-диизопропилэтиламин (от 2,0 до 5,0 экв.). Полученная в результате смесь перемешивается от 4 ч до 48 ч при температуре от Ткомн. до 60°С. При необходимости спустя 24 ч добавляется дополнительная часть анилина или карбоновой кислоты и ГАТУ. По окончании реакции сырой продукт после удаления растворителя в вакууме может очищаться при помощи препаративной ОФ ВЭЖХ (элюент: градиент ацетонитрил/вода) или в качестве альтернативы после водной обработки реакционной смеси при помощи хроматографии на силикагеле (элюент: смеси циклогексан/этилацетат или дихлорметан/метанол).
Следующие примеры были получены в соответствии с общей методикой 1:
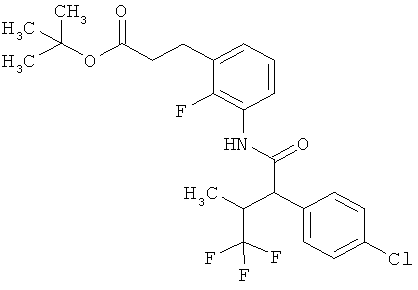

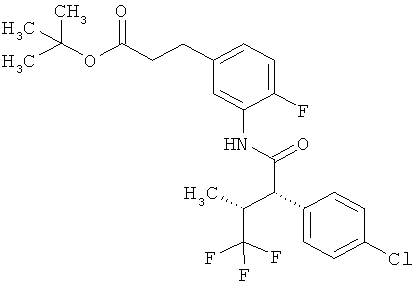
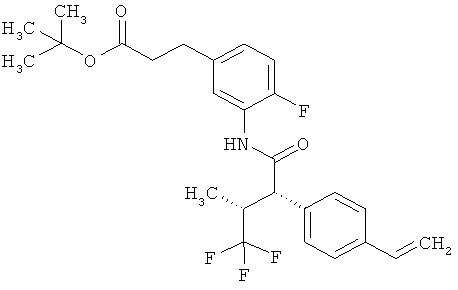


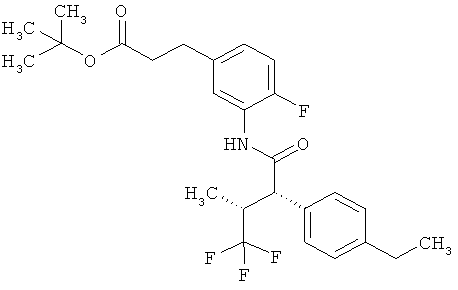
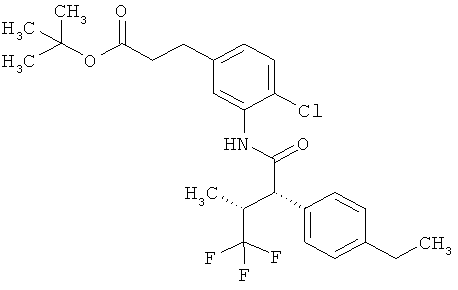

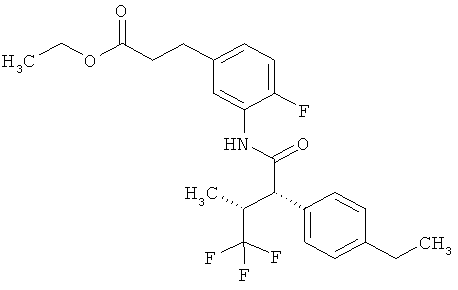


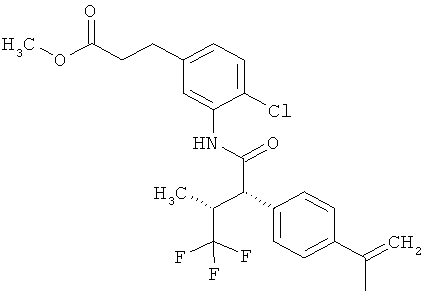
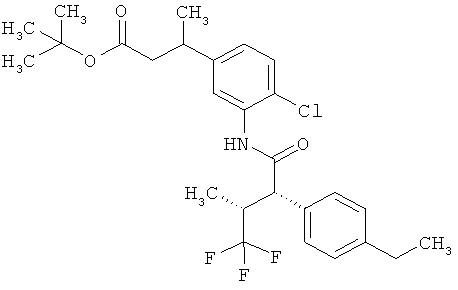
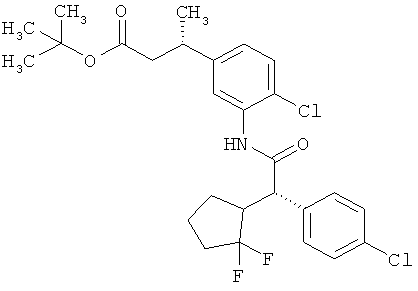
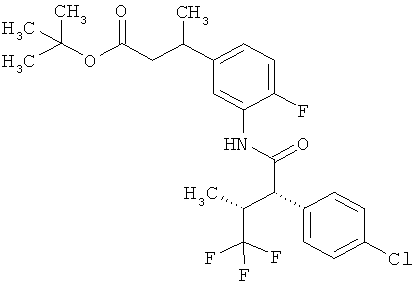



Пример123А
Этил-(2R)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-2-метилпропаноат
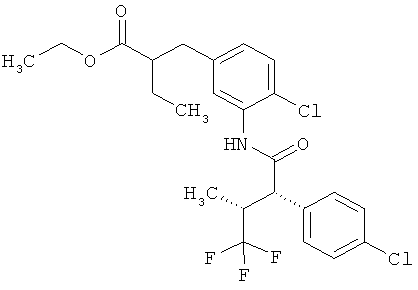
500 мг (2,07 ммоль) сложного этилового эфира (-)-(2R)-3-(3-амино-4-хлорфенил)-2-метилпропановой кислоты и 607 мг (2,28 ммоль) (2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты растворяли в смеси из 2,0 мл ДМФА и 1,0 мл пиридина и при Ткомн. смешивали с 1022 мг (2,69 ммоль) ГАТУ. Реакционную смесь перемешивали в течение ночи при Ткомн. После этого разбавляли этилацетатом, а этот раствор последовательно промывали 1 н. соляной кислотой, водой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия. Органическую фазу сушили над сульфатом магния и упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 40:1). Таким образом было получено 998 мг (98,4% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,41 мин; m/z=490/492 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 0,99-1,09 (м, 6Н), 1,54-1,74 (м, 1Н), 2,59-2,73 (м, 2Н), 2,74-2,88 (м, 1Н), 3,97 (кв, 2Н), 4,12 (д, 1Н), 6,99 (дд, 1Н), 7,25-7,37 (м, 2Н), 7,40-7,55 (м, 4Н), 9,81 (с, 1Н).
Пример 124А
(+)-Этил-(2S)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-2-метилпропаноат

Метод А:
1,50 г (6,21 ммоль) сложного этилового эфира (+)-(2S)-3-(3-амино-4-хлорфенил)-2-метилпропановой кислоты и 1,82 г (6,83 ммоль) (3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты (в виде смеси диастереомеров приблизительно 9:1) растворяли в смеси из 6,3 мл ДМФА и 3,2 мл пиридина и при Ткомн. смешивали с 2,83 г (7,45 ммоль) ГАТУ. Реакционную смесь перемешивали в течение ночи при Ткомн. После этого разбавляли этилацетатом, а этот раствор последовательно промывали насыщенным раствором хлорида аммония и насыщенным раствором хлорида натрия. Органическую фазу сушили над сульфатом магния и упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 50:1→40:1). Полученную при этом смешанную фракцию (которая содержала побочный диастереомер) разделяли при помощи повторной хроматографии на силикагеле (элюент: циклогексан/этилацетат 40:1). В общей сложности получили 2,46 г (80,8% от теор.) целевого соединения.
Метод В:
К 7,60 г (28,50 ммоль) (3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты (в виде смеси диастереомеров приблизительно 9:1) добавляли 30 мл дихлорметана и 1 каплю ДМФА. К раствору, охлажденному до -10°С, по каплям добавляли 4,48 мл (51,3 ммоль) оксалилхлорида, так чтобы температура не превышала -5°С. Реакционную смесь перемешивали 1 ч при температуре от -5°С до 0°С, а затем примерно 30 мин без охлаждения при поднятии температуры до Ткомн., прежде чем упаривали в вакууме. Остаток переводили в дихлорметан, а этот раствор снова упаривали в вакууме. После повторного проведения этого процесса полученный хлорангидрид непродолжительное время сушили в высоком вакууме и непосредственно вводили в дальнейшее превращение без дополнительной очистки.
К раствору 6,05 г (25,03 ммоль) сложного этилового эфира (+)-(2S)-3-(3-амино-4-хлорфенил)-2-метилпропановой кислоты в 25 мл абс. ТГФ добавляли 6,1 мл (35,04 ммоль) N,N-диизопропилэтиламина. Полученный в результате раствор охлаждали до -10°С и по каплям прибавляли раствор полученного выше хлорангидрида (7,85 г, 27,5 ммоль) примерно в 10 мл абс. ТГФ, причем температуру поддерживали ниже 0°С. Затем реакционную смесь перемешивали 1 ч при температуре от -10°С до 0°С, а потом добавляли этилацетат и три капли воды. Спустя 10 мин смесь, дополнительно разбавленную этилацетатом, последовательно промывали 1 н. соляной кислотой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Сырой продукт в течение 4 ч перемешивали в 50 мл диизопропилового эфира. После фильтрации полученное твердое вещество еще раз перемешивали с 40 мл диизопропилового эфира. Это полученное твердое вещество тщательно высушивали в высоком вакууме. Таким образом было получено 8,32 г целевого соединения. Полученные выше фильтраты объединяли и упаривали в вакууме. Из остатка при помощи хроматографии на силикагеле (элюент: циклогексан/этилацетат 50:1) были выделены дополнительные 1,75 г продукта. В общей сложности таким образом было получено 10,07 г (82,1% от теор.) целевого соединения.
ЖХ-МС (Метод 7): Rt=2,95 мин; m/z=490 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 1,00-1,10 (м, 6Н), 2,58-2,72 (м, 2Н), 2,72-2,83 (м, 1Н), 3,34-3,44 (м, 1Н), 3,96 (кв, 2Н), 4,12 (д, 1Н), 6,99 (дд, 1Н), 7,27-7,38 (м, 2Н), 7,42-7,51 (м, 4Н), 9,82 (с, 1Н).
[α]D20=+94°, с=0,58, хлороформ. Пример 125А
Сложный этиловый эфир (+)-(2S)-2-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}бензил)бутановой кислоты
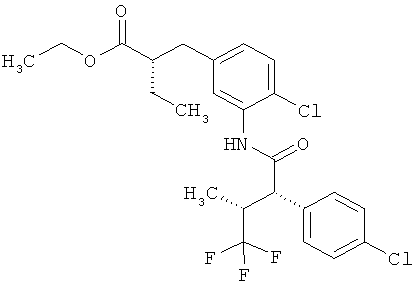
К 3,3 г (12,38 ммоль) (3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты (в виде смеси диастереомеров приблизительно 9:1) добавляли 13 мл дихлорметана и 1 каплю ДМФА. К раствору, охлажденному до -10°С, по каплям добавляли 1,94 мл (22,28 ммоль) оксалилхлорида, так чтобы температура не превышала -5°С. Затем реакционную смесь перемешивали 1 ч при температуре от -5°С до 0°С, прежде чем упаривали в вакууме. Остаток переводили в дихлорметан, и этот раствор снова упаривали в вакууме. После повторного проведения этого процесса полученный хлорангидрид непродолжительное время сушили в высоком вакууме и непосредственно вводили в дальнейшее превращение без дополнительной очистки.
К раствору 1,22 г (4,77 ммоль) (+)-этил-(2S)-2-(3-амино-4-хлорбензил)бутаноата в 4,8 мл абс. ТГФ добавляли 1,2 мл (6,68 ммоль) N,N-диизопропилэтиламина. Полученный в результате раствор охлаждали до -10°С и по каплям прибавляли раствор полученного выше хлорангидрида (1,5 г, 5,25 ммоль) в 2 мл абс. ТГФ, причем температуру поддерживали ниже 0°С. Затем реакционную смесь перемешивали 1 ч при температуре от -10°С до 0°С, а потом добавляли этилацетат и три капли воды. Спустя 10 мин смесь, дополнительно разбавленную этилацетатом, последовательно промывали 1 н. соляной кислотой, насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Желаемый продукт выделяли при помощи хроматографирования остатка на силикагеле (элюент: циклогексан/этилацетат 40:1). Было получено 2,13 г (88,5% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,46 мин; m/z=504 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,76-0,88 (м, 6Н), 1,02 (т, 3Н), 1,45-1,56 (м, 2Н), 2,47 (д, 1Н), 2,72 (д, 2Н), 3,34-3,43 (м, 1Н), 3,93 (кв.д, 2Н), 4,12 (д, 1Н), 6,98 (дд, 1Н), 7,29-7,38 (м, 2Н), 7,40-7,52 (м, 4Н), 9,81 (с, 1Н).
[α]D20=+62,6°, с=0,515, хлороформ.
В соответствии с аналогичной методикой было получено следующее соединение:
Пример 126А
Сложный этиловый эфир (+)-(2R)-2-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}бензил)бутановой кислоты
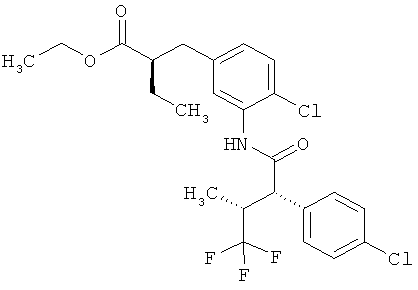
ЖХ-МС (Метод 6): Rt=1,46 мин; m/z=504/506 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 0,84 (т, 3Н), 1,04 (т, 3Н), 1,46-1,56 (м, 2Н), 2,45-2,49 (м, 1Н), 2,70-2,74 (м, 2Н), 3,34-3,42 (м, 1Н), 3,95 (кв, 2Н), 4,12 (д, 1Н), 6,98 (дд, 1Н), 7,30-7,37 (м, 2Н), 7,42-7,50 (м, 4Н), 9,81 (с, 1Н).
[α]D20=+52,3°, с=0,485, хлороформ.
Пример 127А
Этил-2-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-л7ранс-циклопропанкарбоксилат
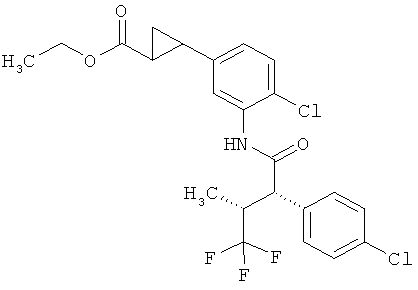
К раствору 90 мг (0,34 ммоль) (2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты в 1,5 мл смеси 4:1 из ДМФА и пиридина добавляли 167 мг (0,44 ммоль) ГАТУ. Спустя 30 мин перемешивания при Ткомн., к этому добавляли 83 мг (0,37 ммоль) рац.-этил-2-[3-амино-4-фторфенил]-транс-циклопропанкарбоксилата. Реакционную смесь перемешивали в течение ночи при Ткомн., потом разбавляли этилацетатом (приблизительно 50 мл) и промывали насыщенным раствором поваренной соли. Органическую фазу сушили над сульфатом магния и упаривали, а остаток очищали с помощью препаративной ВЭЖХ. Было получено 123 мг (77% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,35 мин; m/z=472 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d5): δ [м.д.]=0,79 (д, 3Н), 1,19 (т, 3Н), 1,25-1,34 (м, 1Н), 1,42 (дт, 1Н), 1,73-1,91 (м, 1Н), 2,31-2,45 (м, 1Н), 3,96-4,20 (м, 3Н), 6,83-7,00 (м, 1Н), 7,13 (дд, 1Н), 7,45 (с, 4Н), 7,57-7,68 (м, 1Н), 10,06 (с, 1Н).
Пример 128А
Этил-трео-3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-2-метилбутаноат

К раствору 35,5 мг (0,13 ммоль) (2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты в 0,98 мл ДМФА добавляли 0,15 мкл (0,19 ммоль) пиридина и 75,8 мг (0,2 ммоль) ГАТУ. Реакционную смесь 30 мин перемешивали при Ткомн., а затем добавляли 35 мг (0,15 ммоль) рац.-трео-этил-3-[3-амино-4-фторфенил]-2-метилбутаноата. После перемешивания в течение ночи реакционную смесь непосредственно очищали с помощью препаративной ВЭЖХ. Было получено 34 мг (52% от теор.) целевого соединения.
ЖХ-МС (Метод 5): Rt=2,89 мин; m/z=488 (М+Н)+.
Пример 129А
Этил-эритро-3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-2-метилбутаноат

К раствору 22,3 мг (84 мкмоль) (2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты в 0,62 мл ДМФА добавляли 9,5 мкл (117 мкмоль) пиридина и 47,7 мг (125 мкмоль) ГАТУ. Реакционную смесь 30 мин перемешивали при Ткомн., а затем добавляли 22 мг (92 мкмоль) рац.-эритро-этил-3-[3-амино-4-фторфенил]-2-метилбутаноата. После перемешивания в течение ночи реакционную смесь непосредственно очищали с помощью препаративной ВЭЖХ. Было получено 10,7 мг (24% от теор.) целевого соединения.
ЖХ-МС (Метод 4): Rt=1,57 мин; m/z=488 (М+Н)+.
Пример 130А
трет-бутил-3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-цианофенил)пропаноат

К раствору 41,3 мг (0,16 ммоль) (2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты в 1,16 мл дихлорметана при 0°С добавляли 155 мкл (0,31 ммоль) 2 М раствора оксалилхлорида в дихлорметане и каплю ДМФА. Спустя 1 ч перемешивания при 0°С реакционную массу упаривали, оставшийся осадок растворяли в 1 мл ТГФ, добавляли 32 мкл (0,19 ммоль) N,N-диизопропилэтиламина, охлаждали до 0°С и прибавляли раствор 42 мг (0,17 ммоль) трет-бутил-3-(3-амино-4-цианофенил)пропаноата в 2 мл ТГФ. Реакционную смесь перемешивали в течение ночи при Ткомн.. Затем эту смесь выливали в 10 мл воды и экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом магния и упаривали. Остаток очищали с помощью препаративной ВЭЖХ. Было получено 16,5 мг (22% от теор.) целевого соединения.
ЖХ-МС (Метод 7): Rt=2,86 мин; m/z=439 (M-tBu)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,81 (д, 3Н), 1,27-1,35 (м, 9Н), 2,83 (т, 2Н), 4,01 (д, 1Н), 7,21 (дд, 1Н), 7,30 (с, 1Н), 7,40-7,52 (м, 4Н), 7,69 (д, 1Н), 10,47 (с, 1Н).
Пример 131А
Метил-(2Е)-3-(2-метил-3-нитрофенил)акрилат

В атмосфере аргона прикалывали 119,5 г (1,39 моль) сложного метилового эфира акриловой кислоты к смеси из 100 г (0,463 моль) 2-бром-6-нитротолуола, 323 мл (2,31 моль) триэтиламина, 28,2 г (92,6 ммоль) три-2-толилфосфина и 10,4 г (46,3 ммоль) ацетата палладия (II) в 2,0 литрах ДМФА и затем эту смесь перемешивали 36 ч при 125°С. После охлаждения до комнатной температуры реакционную смесь перемешивали с 4 литрами насыщенного водного раствора хлорида аммония и трижды экстрагировали в общей сложности 5 литрами диэтилового эфира. Объединенные органические фазы промывали водой и насыщенным раствором хлорида натрия и сушили над сульфатом натрия. После фильтрации растворитель удаляли в вакууме досуха. Полученный остаток очищали с помощью флэш-хроматографии на силикагеле (элюент: петролейный эфир/этилацетат 6:1). Продукт перемешивали с гептаном, а выпавшее твердое вещество фильтровали с отсасыванием и сушили в высоком вакууме. Получали 48,7 г целевого соединения (46,6% от теор.).
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=2,41 (с, 3Н), 3,76 (с, 3Н), 6,63 (д, 1Н), 7,48 (т, 1Н), 7,84-7,95 (м, 2Н), 8,00 (д, 1Н).
Пример 132А
Метил-3-(3-амино-2-метилфенил)пропаноат

48,7 г (220,1 ммоль) метил-(2Е)-3-(2-метил-3-нитрофенил)акрилата растворяли в 2,2 литра метанола, а раствор гидрировали в установке для гидрирования непрерывного действия («H-Cube midi» фирмы Thales Nano, Будапешт) при расходе 6-10 мл/мин и при температуре реакции 35-40°С, а также при максимальном давлении водорода. После оконченного взаимодействия раствор, содержащий продукт, упаривали в вакууме. Было получено 40,0 г целевого продукта в виде твердого вещества (92,1% от теор.).
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,97 (с, 3Н), 2,45 (т, 2Н), 2,78 (т, 2Н), 3,58 (с, 3Н), 4,75 (с, 2Н), 6,37 (д, 1Н), 6,50 (д, 1Н), 6,79 (т, 1Н).
Пример 133А
Метил-3-(3-амино-4-хлор-2-метилфенил)пропаноат

К раствору 2,0 г (10,3 ммоль) метил-3-(3-амино-2-метилфенил)пропаноата в 10 мл ацетонитрила при Ткомн. прибавляли 1,38 г (10,3 ммоль) N-хлорсукцинимида. Реакционную смесь перемешивали 30 мин, а потом разбавляли этилацетатом. Эту смесь последовательно промывали нас. раствором гидрокарбоната натрия и нас. раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Из сырого продукта после хроматографии на силикагеле (элюент: циклогексан/этилацетат 10:1) выделили 279 мг целевого продукта (11,8% от теор.).
ЖХ-МС (Метод 4): Rt=1,11 мин; m/z=228 (M+H)+.
1H-ЯМР (500 МГц, ДМСО-d6): δ [м.д.]=2,06 (с, 3Н), 2,46 (т, 2Н), 2,78 (т, 2Н), 3,58 (с, 3Н), 4,94 (с, 2Н), 6,42 (д, 1Н), 6,98 (д, 1Н).
Пример134А
3-Бром-6-хлор-2-метиланилин
К раствору 12,0 г (64,5 ммоль) 3-бром-2-метиланилина в 150 мл ацетонитрила при Ткомн. прибавляли 8,61 г (64,5 ммоль) N-хлорсукцинимида. Реакционную смесь перемешивали 7 ч при 60°С, а после охлаждения упаривали в вакууме. Остаток переводили в дихлорметан, а эту смесь последовательно промывали нас. раствором гидрокарбоната натрия и нас. раствором хлорида натрия, сушили над сульфатом натрия и упаривали в вакууме. Из сырого продукта после хроматографии на силикагеле (элюент: циклогексан/этилацетат от 40:1 до 20:1) выделили 3,78 г целевого продукта (26,6% от теор.).
ГХ-МС (Метод 1): Rt=5,07 мин; m/z=218/220 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=2,24 (с, 3Н), 5,39 (с, 2Н), 6,80 (д, 1Н), 7,03 (д, 1Н).
Пример 135А
трет-бутил-(2Е)-3-(3-амино-4-хлор-2-метилфенил)-2-метилакрилат
и
трет-бутил-2-(3-амино-4-хлор-2-метилбензил)акрилат
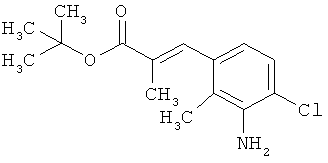
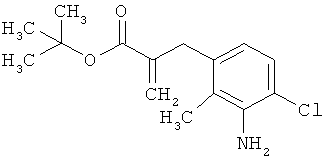
1,50 г (6,80 ммоль) 3-бром-6-хлор-2-метиланилина, 2,90 г (20,4 ммоль) трет-бутилметакрилата и 4,74 мл (34,0 ммоль) триэтиламина растворяли в 10,0 мл ДМФА. Реакционный раствор трижды вакуумировали и каждый раз продували аргоном. После добавления 152,7 мг (0,68 ммоль) ацетата палладия(II) и 414,1 мг (1,36 ммоль) три-2-толилфосфина снова дважды вакуумировали и соответственно продували аргоном. Потом реакционную смесь перемешивали 2 ч при 150°С. После охлаждения смесь отфильтровывали через целит, а остаток дополнительно промывали ДМФА. Фильтрат упаривали в вакууме и остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 100:1). Получили 1,59 г смеси обоих указанных соединений (соотношение приблизительно 2:1, 83% от теор.).
ЖХ-МС (Метод 4): Rt=1,45 мин; m/z=226 (M-C4H8) и Rt=1,49 мин; m/z=282 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): трет-бутил-(2Е)-3-(3-амино-4-хлор-2-метилфенил)-2-метилакрилат: δ [м.д.]=1,49 (с, 9Н), 1,75 (д, 3Н), 2,02 (с, 3Н), 5,12 (с, 2Н), 6,44 (д, 1Н), 7,11 (д, 1Н), 7,51 (с, 1Н).
1H-ЯМР (400 МГц, ДМСО-d6): трет-бутил-2-(3-амино-4-хлор-2-метилбензил)акрилат: δ [м.д.]=1,42 (с, 9Н), 1,98 (с, 3Н), 3,45 (с, 2Н), 4,97 (с, 2Н), 5,15 (д, 1Н), 6,01 (д, 1Н), 6,38 (д, 1Н), 7,02 (д, 1Н).
Пример 136А
(+/-)-трет-бутил-3-(3-амино-4-хлор-2-метилфенил)-2-метилпропаноат
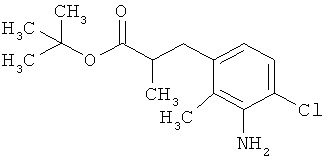
К 354 мг (14,6 ммоль) магниевой стружки и нескольким кристалликам йода добавляли раствор 1,58 г (5,61 ммоль) смеси трет-бутил-(2Е)-3-(3-амино-4-хлор-2-метилфенил)-2-метилакрилата и трет-бутил-2-(3-амино-4-хлор-2-метилбензил)акрилата (Пример 135А) в 5,0 мл метанола. Эту смесь перемешивали при Ткомн. (сначала при охлаждении) в течение ночи. Затем при охлаждении льдом добавляли 50 мл 1 н. соляной кислоты. Потом в результате добавления 10%-ного водного раствора гидроксида натрия величину pH смеси устанавливали примерно равной 10. Смесь трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме. Из сырого продукта после хроматографии на силикагеле (элюент: циклогексан/этилацетат от 50:1 до 40:1) выделили 962 мг целевого продукта (60,5% от теор.).
ЖХ-МС (Метод 9): Rt=2,30 мин; m/z=284 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,02 (д, 3Н), 1,32 (с, 9Н), 2,06 (с, 3Н), 2,46 (дд, 1Н), 2,80 (дд, 1Н), 4,94 (уш. с, 2Н), 6,38 (д, 1Н), 6,97 (д, 1Н).
Полученный выше рацемат был разделен на энантиомеры с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak OJ-H, 5 мкм, 250 мм×20 мм; расход: 20 мл/мин; детектирование: 220 нм, объем впрыска: 0,28 мл; температура: 22°С; элюент: 93% изогексана/7% изопропанола]. Исходя из 962 мг рацемата, было получено 434 мг энантиомера 1 (Пример 137А) и 325 мг энантиомера 2 (Пример 138А):
Пример 137А
(-)-Трет-бутил-(2R)-3-(3-амино-4-хлор-2-метилфенил)-2-метилпропаноат
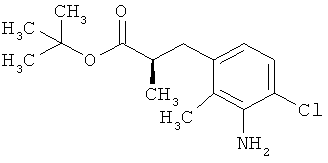
Выход:434 мг
ЖХ-МС (Метод 4): Rt=1,44 мин; m/z=284 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,03 (д, 3Н), 1,32 (с, 9Н), 2,06 (с, 3Н), 2,46 (дд, 1Н), 2,80 (дд, 1Н), 4,93 (с, 2Н), 6,38 (д, 1Н), 6,97 (д, 1Н).
[α]D20=-37,3°, с=0,455, хлороформ.
Пример138А
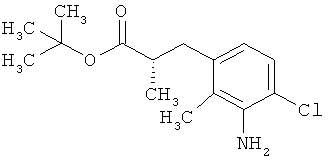
Выход: 325 мг
ЖХ-МС (Метод 4): Rt=1,44 мин; m/z=284 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,03 (д, 3Н), 1,32 (с, 9Н), 2,06 (с, 3Н), 2,46 (дд, 1Н), 2,80 (дд, 1Н), 4,93 (с, 2Н), 6,38 (д, 1Н), 6,97 (д, 1Н).
[α]D20=+35,0°, с=0,455, хлороформ.
Пример 139А
Этил-4,4,4-трифтор-3-(4-фтор-3-нитрофенил)бут-2-еноат

К охлажденной на ледяной бане суспензии 1,86 г (60%-ная суспензия в минеральном масле, 46,4 ммоль) гидрида натрия в смеси из 70 мл ТГФ и 20 мл ДМФА медленно прикалывали 10,9 г (48,5 ммоль) сложного этилового эфира диэтилфосфоноуксусной кислоты. После окончания прибавления смесь перемешивали еще 30 мин при 0°С, прежде чем добавляли 10,0 г (42,2 ммоль) 2,2,2-трифтор-1-(4-фтор-3-нитрофенил)этанона. Эту реакционную смесь перемешивали в течение ночи при Ткомн., а потом выливали в воду. Трижды экстрагировали этилацетатом, а объединенные органические фазы упаривали в вакууме. Из сырого продукта после хроматографии на силикагеле (элюент: циклогексан/этилацетат от 20:1 до 10:1) выделили 9,23 г целевого продукта (71,2% от теор.).
ГХ-МС (Метод 1): Rt=4,51 мин; m/z=262 (M-C2H5O)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,04 (т, 3Н), 4,03 (кв, 2Н), 6,96 (д, 1Н), 7,67-7,76 (м, 1Н), 7,78-7,86 (м, 1Н), 8,16 (дд, 1Н).
Пример 140А
(+/-)-Этил-3-(3-амино-4-фторфенил)-4,4,4-трифторбутаноат

5,0 г (16,3 ммоль) этил-4,4,4-трифтор-3-(4-фтор-3-нитрофенил)бут-2-еноата растворяли в 133 мл этанола и в атмосфере аргона добавляли 866 мг палладия на угле (10%). Реакционную смесь интенсивно перемешивали в течение ночи при Ткомн. в атмосфере водорода (при нормальном давлении). После этого отфильтровывали через целит, а остаток дополнительно промывали этилацетатом. Фильтрат упаривали в вакууме, а полученный продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 50:1). Получили 3,91 г целевого продукта (85,9% от теор.).
ЖХ-МС (Метод 6): Rt=0,97 мин; m/z=280 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,08 (т, 3Н), 2,85 (дд, 1Н), 2,99 (дд, 1Н), 3,81-3,92 (м, 1Н), 3,94-4,07 (м, 2Н), 5,21 (с, 2Н), 6,40-6,58 (м, 1Н), 6,77 (дд, 1Н), 6,96 (дд, 1Н).
Пример 141А
Трет-бутил-2-метилбутаноат
15,0 г (124,4 ммоль) хлорангидрида 2-метилмасляной кислоты растворяли в 150 мл абс. ТГФ, охлаждали до 0°С и по каплям прибавляли 114 мл (114 ммоль) 1 М раствора трет-бутилата калия в ТГФ. После окончания прибавления перемешивали 1 ч при 0°С, потом 5 ч при Ткомн., прежде чем в вакууме удаляли примерно половину растворителя. После добавления диэтилового эфира при интенсивном перемешивании по каплям добавляли нас. раствор гидрокарбоната натрия. После разделения фаз водную фазу экстрагировали диэтиловым эфиром и объединенные органические фазы промывали нас. раствором карбоната натрия, сушили над сульфатом натрия и упаривали в вакууме. Сырой продукт очищали с помощью перегонки в вакууме (19 мм рт.ст., 40-45°С). В общей сложности получили 6,35 г целевого продукта (32,3% от теор.).
ГХ-МС (Метод 1): Rt=1,53 мин; m/z=85.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,84 (м, 3Н), 1,01 (д, 3Н), 1,33-1,41 (м, 1Н), 1,39 (с, 9Н), 1,48-1,55 (м, 1Н), 2,13-2,26 (м, 1Н).
Пример 142А
2-Бром-4-(бромметил)-1-хлорбензол

Стадия 1:
199,0 г (0,845 моль) 3-бром-4-хлорбензойной кислоты растворяли в 2,5 литрах ТГФ, охлаждали до -10°С и при этой температуре добавляли 1,69 литра (1,69 моль) 1 М раствора борана в ТГФ. Реакционную смесь в течение ночи нагревали до Ткомн., прежде чем добавили насыщенный водный раствор хлорида аммония. После добавления воды дважды экстрагировали этилацетатом, объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме. Было получено в виде сырого продукта 206 г (3-бром-4-хлорфенил)метанола, который использовался на следующей стадии без дополнительной очистки.
Стадия 2:
260 г (примерно 1,05 моль) сырого (3-бром-4-хлорфенил)метанола растворяли в 2,86 литра дихлорметана, охлаждали до -5°С и медленно прибавляли 127,1 г (44,6 мл, 460 ммоль) трибромида фосфора. По окончании прибавления перемешивали еще 1 ч при -5°С, прежде чем разбавляли дихлорметаном и водой. Органическую фазу отделяли, сушили над сульфатом магния и упаривали в вакууме. Получили в виде сырого продукта 280,5 г (примерно 84% от теор.) 2-бром-4-(бромметил)-1-хлорбензола.
ГХ-МС (Метод 1): Rt=5,36 мин; m/z=281/283/285 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=4,71 (с, 2Н), 7,49 (дд, 1Н), 7,63 (д, 1Н), 7,89 (д, 1Н).
Пример 143А
(+/-)-Трет-бутил-2-(3-бром-4-хлорбензил)-2-метилбутаноат

5,8 мл (41,6 ммоль) диизопропиламина в атмосфере аргона растворяли в 50 мл сухого ТГФ и охлаждали до -30°С. Прикалывали 16,6 мл (41,6 ммоль) раствора н-бутиллития (2,5 М раствор в гексане), а полученную в результате смесь нагревали до 0°С, прежде чем охлаждали до -70°С. Добавили раствор 5,06 г (32,0 ммоль) трет-бутил-2-метилбутаноата в 20 мл ТГФ, причем температуру реакции поддерживали ниже -60°С. После 4 ч перемешивания при -60°С добавляли раствор 10,0 г (35,2 ммоль) 2-бром-4-(бромметил)-1-хлорбензола в 30 мл ТГФ, причем температуру снова поддерживали ниже -60°С. Потом реакционную смесь в течение ночи медленно нагревали до Ткомн., до того, как добавляли насыщенный водный раствор хлорида аммония и этилацетат. После разделения фаз водную фазу дважды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 100:1). Получили 7,62 г (65,9% от теор.) целевого соединения.
ГХ-МС (Метод 1): Rt=6,52 мин; m/z=306 (M-C4H7)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,83 (т, 3Н), 0,93 (с, 3Н), 1,32-1,45 (м, 10Н), 1,60-1,73 (м, 1Н), 2,62 (д, 1Н), 2,91 (д, 1Н), 7,18 (дд, 1Н), 7,47-7,56 (м, 2Н).
Пример 144А
(+/-)-Грет-бутил-2-[3-(бензиламино)-4-хлорбензил]-2-метилбутаноат
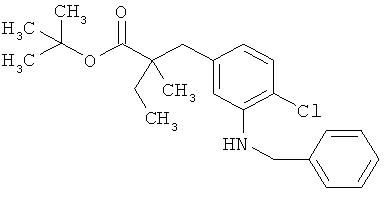
В атмосфере аргона в высушенную колбу отвешивали 1,59 г (16,6 ммоль) трет-бутилата натрия и добавляли 34,6 мл абс. толуола. К этому друг за другом добавляли 5,0 г (13,8 ммоль) (+/-)-трет-бутил-2-(3-бром-4-хлорбензил)-2-метилбутаноата, 1,8 мл (16,6 ммоль) бензиламина, 633 мг (0,69 ммоль) трис(дибензилиденацетон)дипалладия и 344 мг (0,55 ммоль) (+/-)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила. Потом реакционную смесь перемешивали в течение 2,0 ч при 110°С. После охлаждения к этой реакционной смеси добавили насыщенный водный раствор хлорида аммония и этилацетат и отфильтровали с отсасыванием через кизельгур. После разделения фаз органическую фазу промывали нас. раствором хлорида аммония и нас. раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 80:1). Получили 4,44 г целевого соединения в еще немного загрязненном виде (примерно 83% от теор.).
ЖХ-МС (Метод 6): Rt=1,57 мин; m/z=388 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,70 (т, 3Н), 1,13-1,22 (м, 1Н), 1,35 (с, 9Н), 1,39 (с, 3Н), 1,39-1,50 (м, 1Н), 2,42 (д, 1Н), 2,66 (д, 1Н), 4,26-4,46 (м, 2Н), 6,00 (т, 1Н), 6,26-6,35 (м, 1Н), 7,11 (д, 1Н), 7,16-7,23 (м, 1Н), 7,28-7,34 (м,4Н), 7,45-7,55 (м, 1Н).
Пример 145А
(+/-)-трет-бутил-2-(3-амино-4-хлорбензил)-2-метилбутаноат
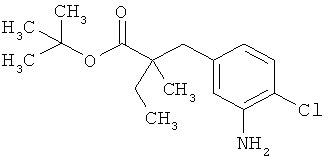
2,20 г (примерно 5,67 ммоль) (+/-)-трет-бутил-2-[3-(бензиламино)-4-хлорбензил]-2-метилбутаноата растворяли в 130 мл этилацетата и добавляли 100 мг палладия на угле (10%). Реакционную смесь перемешивали в течение ночи при Ткомн. в атмосфере водорода при нормальном давлении. Потом эту реакционную смесь отфильтровывали с отсасыванием через кизельгур, остаток тщательно промывали этилацетатом, а объединенный фильтрат упаривали. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат от 50:1 до 30:1). Получили 924 мг (54,7% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,34 мин; m/z=298 (М+Н)+.
Полученный выше рацемат был разделен на энантиомеры с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak OJ-H, 5 мкм, 250 мм×20 мм; расход: 20 мл/мин; детектирование: 220 нм, объем впрыска: 0,30 мл; температура: 35°С; элюент: 70% изогексана/30% этанола]. Исходя из 924 мг рацемата, было получено 405 мг энантиомера 1 (Пример 146А) и 403 мг энантиомера 2 (Пример 147А):
Пример 146А
(-)-7рет-бутил-2-(3-амино-4-хлорбензил)-2-метилбутаноат (энантиомер 1)

Выход: 405 мг
ЖХ-МС (Метод 6): Rt=1,32 мин; m/z=298 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,81 (т, 3Н), 0,93 (с, 3Н), 1,28-1,37 (м, 1Н), 1,38 (с, 9Н), 1,59-1,71 (м, 1Н), 2,45 (д, 1Н), 2,74 (д, 1Н), 5,14-5,22 (м, 2Н), 6,31 (дд, 1Н), 6,57 (д, 1Н), 7,04 (д, 1Н).
[α]D20=-11,8°, с=0,50, хлороформ.
Пример 147А
(+)-трет-бутил-2-(3-амино-4-хлорбензил)-2-метилбутаноат (энантиомер 2)
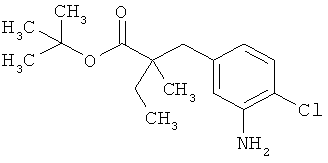
Выход:403 мг ЖХ-МС (Метод 6): Rt=1,32 мин; m/z=298 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,75-0,85 (м, 3Н), 0,93 (с, 3Н), 1,30-1,37 (м, 1Н), 1,39 (с, 9Н), 1,58-1,70 (м, 1Н), 2,45 (д, 1Н), 2,74 (д, 1Н), 5,09-5,23 (м, 2Н), 6,31 (дд, 1Н), 6,57 (д, 1Н), 7,04 (д, 1Н).
[α]D20=+12,0°, с=0,420, хлороформ.
Пример 148А
2,2,2-Трифтор-1-(3-нитрофенил)этанон

20,0 г (114,9 ммоль) 2,2,2-трифторацетофенона помещали в 80 мл конц. серной кислоты и охлаждали до -10°С. К этой смеси прикалывали предварительно приготовленный при -10°С раствор 4,8 мл (114,8 ммоль) азотной кислоты в 20 мл конц. серной кислоты, так чтобы температура реакции не превышала -5°С. По окончании прибавления реакционную смесь перемешивали 1 ч при температуре между -10°С и 0°С, а потом осторожно выливали в ледяную воду. При помощи добавления 50%-ного водного раствора гидроксида натрия устанавливали показатель pH смеси примерно на 9-10. Трижды экстрагировали этилацетатом, объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: сначала циклогексан/дихлорметан от 2:1 до 1:1, под конец чистый дихлорметан). Было получено 19,2 г целевого продукта (76,2% от теор.).
ЖХ-МС (Метод 6): Rt=0,81 мин; m/z=236.
ГХ-МС (Метод 1): Rt=3,19 мин; m/z=150 (M-CF3)+.
Пример 149А
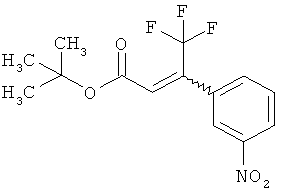
К охлажденной до 0°С суспензии 4,41 г (60%-ная суспензия в минеральном масле, 110,4 ммоль) гидрида натрия в смеси из 37,2 мл ТГФ и 37,2 мл ДМФА прикапывали 25,9 мл (110,4 ммоль) трет-бутил(диэтоксифосфорил)ацетата. Спустя 30 мин к этому добавляли 18,6 г (84,9 ммоль) 2,2,2-трифтор-1-(3-нитрофенил)этанона, охлаждающую баню убирали, а реакционную смесь перемешивали 2 ч при Ткомн.. Потом эту реакционную смесь выливали в воду и после насыщения хлоридом натрия трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат от 100:1 до 20:1). Получили 18,0 г целевого продукта в виде смеси E/Z-изомеров (66,8% от теор.).
ЖХ-МС (Метод 6): Rt=1,25 мин; ионизация отсутствует.
МС (DCI): m/z=335 (M+H2O)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,17/1,50 (2 с, совм, 9Н), 6,93/7,14 (2д, совм., 1Н), 7,74-7,94 (м, 2Н), 8,16/8,23 (2с, совм., 1Н), 8,30-8,42 (м, 1Н).
Пример 150А
(+/-)-Трет-бутил-3-(3-аминофенил)-4,4,4-трифторбутаноат
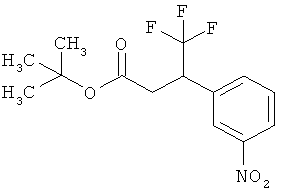
18,0 г (56,7 ммоль) трет-бутил-(2Е/Z)-4,4,4-трифтор-3-(3-нитрофенил)бут-2-еноата растворяли в 100 мл этанола и удаляли кислород с помощью аргона. После добавления 1,21 г палладия на угле (10%) эту смесь интенсивно перемешивали при Ткомн. в течение ночи в атмосфере водорода при нормальном давлении. Потом эту реакционную смесь отфильтровывали через целит, остаток тщательно промывали этанолом, фильтрат упаривали в вакууме, а получившиеся продукт в течение ночи сушили в высоком вакууме. Получили 13,7 г целевого продукта (83,7% от теор.).
ЖХ-МС (Метод 6): Rt=1,02 мин; m/z=290 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,27 (с, 9Н), 2,70 (дд, 1Н), 2,89 (дд, 1Н), 3,62-3,79 (м, 1Н), 5,11-5,17 (м, 2Н), 6,43-6,56 (м, 3Н), 6,99 (т, 1Н).
Пример 151А
(+/-)-трет-бутил-3-(3-амино-4-хлорфенил)-4,4,4-трифторбутаноат

13,6 г (47,0 ммоль) (+/-)-трет-бутил-3-(3-аминофенил)-4,4,4-трифторбутаноата помещали в 100 мл ацетонитрила и при Ткомн. прибавляли 6,28 г (47,0 ммоль) N-хлорсукцинимида. Реакционную смесь сначала перемешивали 12 ч при 60°С, а потом оставляли стоять 3 дня при Ткомн. После упаривания в вакууме остаток очищали при помощи хроматографии на силикагеле (элюент: циклогексан/этилацетат 100:1) и выделяли желаемый целевой продукт. Получили 4,49 г целевого соединения (29,5% от теор.).
ЖХ-МС (Метод 6): Rt=1,17 мин; m/z=324 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,27 (с, 9Н), 2,72 (дд, 1Н), 2,91 (дд, 1Н), 3,74-3,86 (м, 1Н), 5,43 (с, 2Н), 6,55 (дд, 1Н), 6,79 (д, 1Н), 7,17 (д, 1Н).
Полученный выше рацемат был разделен на энантиомеры с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak OJ-H, 5 мкм, 250 мм×20 мм; расход: 15 мл/мин; детектирование: 220 нм, объем впрыска: 0,20 мл; температура: 35°С; элюент: 70% изогексана/30% изо-пропанола]. Исходя из 4,49 г рацемата, было получено 2,02 г энантиомера 1 {Пример 152А) и 2,04 г энантиомера 2 (Пример 153А):
Пример 152А
(-)-Трет-бутил-(3R)-3-(3-амино-4-хлорфенил)-4,4,4-трифторбутаноат

Выход: 2,02 г
ЖХ-МС (Метод 6): Rt=1,17 мин; m/z=324 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,27 (с, 9Н), 2,72 (дд, 1Н), 2,91 (дд, 1Н), 3,75-3,85 (м, 1Н), 5,40-5,46 (м, 2Н), 6,55 (дд, 1Н), 6,79 (д, 1Н), 7,17 (д, 1Н).
[α]D20=-69,4°, с=0,520, хлороформ.
Пример 153А
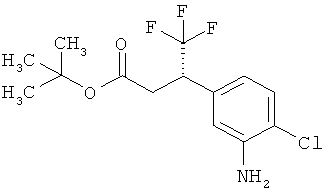
Выход: 2,04 г
ЖХ-МС (Метод 6): Rt=1,17 мин; m/z=324 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,27 (с, 9Н), 2,71 (дд, 1Н), 2,91 (дд, 1Н), 3,74-3,86 (м, 1Н), 5,38-5,46 (м, 2Н), 6,55 (дд, 1Н), 6,73-6,80 (м, 1Н), 7,17 (д, 1Н).
[α]D20=+66,3°, с=0,495, хлороформ.
Пример 154А
4,0 г (35,0 ммоль) циклобутилуксусной кислоты растворяли в 20 мл дихлорметана, добавляли каплю ДМФА и после охлаждения до 0°С по каплям прибавляли 4,0 мл (45,6 ммоль) дихлорангидрида щавелевой кислоты. Реакционную смесь перемешивали 2 ч при температуре между 0°С и 10°С, а потом упаривали в вакууме на холоду. Остаток извлекали в абс.дихлорметан и повторно на холоду упаривали в вакууме. Этот процесс повторяли еще один раз, прежде чем полученный хлорангидрид непродолжительное время, в течение 5 мин, сушили в высоком вакууме. Потом осадок переводили в 20 мл абс. ТГФ, охлаждали до 0°С и по каплям добавляли 28 мл (28 ммоль) 1 М раствора трет-бутилата калия в ТГФ. По окончании прибавления охлаждение убирали и дополнительно перемешивали 1 ч при Ткомн., прежде чем выливали в воду. Трижды экстрагировали дихлор-метаном, а объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме. Было получено 3,88 г сырого целевого продукта (примерно 65% от теор.).
ГХ-МС (Метод 1): Rt=2,29 мин; m/z=97 (M-C3H5O2)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,38 (с, 9Н), 1,60-1,89 (м, 5Н), 1,95-2,11(м,2Н), 2,28 (д,2Н).
Пример 155А
Трет-бутилциклопропилацетат
10,0 г (99,9 ммоль) циклопропилуксусной кислоты растворяли в 50 мл дихлорметана, добавляли каплю ДМФА и после охлаждения до 0°С по каплям прибавляли 9,6 мл (109,9 ммоль) дихлорангидрида щавелевой кислоты. Реакционную смесь перемешивали 2 ч при температуре между 0°С и 10°С, а потом упаривали в вакууме на холоду. Остаток непродолжительное время (примерно 5 мин) сушили в высоком вакууме, затем переводили в 20 мл абс. ТГФ, охлаждали до 0°С и по каплям добавляли 89,9 мл (89,9 ммоль) 1 М раствора трет-бутилата калия в ТГФ. По окончании прибавления охлаждение убирали и дополнительно перемешивали 2 ч при Ткомн., прежде чем в значительной мере удаляли в вакууме ТГФ (давление до 150 мм рт.ст., водяная баня примерно 30°С). К остатку добавляли диэтиловый эфир и 0,5 Н водный раствор едкого натра. После разделения фаз органическую фазу сушили над сульфатом магния, упаривали в вакууме, а остаток непродолжительное время сушили в высоком вакууме. Было получено 8,38 г сырого целевого продукта (примерно 53% от теор.).
ГХ-МС (Метод 1): Rt=1,80 мин; m/z=100 (M-C4H8)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,05-0,14 (м, 2Н), 0,38-0,51 (м, 2Н), 0,81-0,99 (м, 1Н), 1,40 (с, 9Н), 2,10 (д, 2Н).
Пример 156А
(+/-)-Трет-бутил-3-(3-бром-4-хлорфенил)-2-циклобутилпропаноат

2.9 мл (20,8 ммоль) диизопропиламина в атмосфере аргона растворяли в 30 мл сухого ТГФ и охлаждали до -20°С. Прикалывали 8,3 мл (20,8 ммоль) раствора н-бутиллития (2,5 М раствор в гексане), а полученную в результате смесь перемешивали 30 мин при -20°С, прежде чем охлаждали до -78°С. При этой температуре добавили раствор 2,60 г (примерно 15,3 ммоль, неочищенного) трет-бутилциклобутилацетата в 10 мл ТГФ. После 4 ч перемешивания при -78°С добавляли раствор 3,95 г (13,9 ммоль) 2-бром-4-(бромметил)-1-хлорбензола в 10 мл ТГФ. Эту реакционную смесь в течение ночи медленно нагревали до Ткомн., до того как добавили насыщенный водный раствор хлорида аммония. Трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме. Остаточное твердое вещество размешивали в 30 мл смеси циклогексан/дихлорметан (1:1) и отфильтровывали. Это твердое вещество повторно размешивали в 10 мл смеси циклогексан/дихлорметан (1:1) и опять отфильтровывали, этот процесс повторяли еще один раз. Все фильтраты собирали, объединяли и упаривали в вакууме. Затем этот осадок дополнительно очищали с помощью хроматографии на силикагеле (элюент: от чистого циклогексана до циклогексан/дихлорметан от 20:1 до 10:1). Таким образом получили 2,24 г целевого соединения (43,2% от теор.).
ГХ-МС (Метод 1): Rt=6,92 мин; m/z=318 (М-С4Н7)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,27 (с, 9Н), 1,71-1,84 (м, 4Н), 1,90-1,94 (м, 1Н), 1,96-2,04 (м, 1Н), 2,33-2,44 (м, 1Н), 2,53-2,60 (м, 1Н), 2,61-2,71 (м, 1Н), 7,22 (дд, 1Н), 7,52 (д, 1Н), 7,57 (д, 1Н).
Аналогичным образом был получен следующий пример:
Пример 157А
Трет-бутил-3-(3-бром-4-хлорфенил)-2-циклопропилпропаноат
Исходя из трет-бутилциклопропилацетата и 2-бром-4-(бромметил)-1-хлорбензола, получили 3,13 г целевого соединения (45% от теор.).
ГХ-МС (Метод 1): Rt=6,41 мин; m/z=301/304 (М-C4H8)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,22 (тт, 2Н), 0,40-0,50 (м, 2Н), 0,82-0,93 (м, 1Н), 1,28 (с, 9Н), 1,82-1,88 (м, 1Н), 2,81-2,89 (м, 2Н), 7,24 (дд, 1Н), 7,52 (д, 1Н), 7,60 (д, 1Н).
Пример158А
(+/-)-Трет-бутил-3-[3-(бензиламино)-4-хлорфенил]-2-циклобутилпропаноат
В сухую колбу в атмосфере аргона отвешивали 848,6 мг (8,83 ммоль) трет-бутилата натрия, 337 мг (0,39 ммоль) трис(дибензилиденацетон)дипалладия и 183 мг (0,29 ммоль) рац.-2,2'-бис(дифенилфосфино)-1,1'-бинафтила, выдерживали 10 мин в высоком вакууме, а потом подавали аргон. К этому друг за другом добавляли 5 мл абс.толуола, 0,96 мл (8,83 ммоль) бензиламина и раствор 2,75 г (7,36 ммоль) (+/-)-трет-бутил-3-(3-бром-4-хлорфенил)-2-циклобутилпропаноата в 5 мл абс. толуола. Реакционную смесь вакуумировали еще трижды и соответственно каждый раз снова подавали аргон, а потом 3 ч перемешивали при 110°С. После охлаждения реакционную смесь выливали в насыщенный водный раствор хлорида аммония. Трижды экстрагировали этилацетатом. Органические фазы объединяли, сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/дихлорметан от 4:1 до 2:1, потом чистый дихлорметан). Получили 1,95 г целевого соединения (65,1% от теор.).
ЖХ-МС (Метод 4): Rt=1,90 мин; m/z=400 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,26 (с, 9Н), 1,54-1,76 (м, 4Н), 1,78-1,86 (м, 2Н), 2,19-2,38 (м, 2Н), 2,38-2,45 (м, 2Н), 4,33-4,44 (м, 2Н), 5,95 (т, 1Н), 6,25-6,40 (м, 2Н), 7,11 (д, 1Н), 7,23 (тд, 1Н), 7,27-7,37 (м, 4Н).
Аналогичным образом был получен следующий пример:
Пример159А
Трет-бутил-3-[3-(бензиламино)-4-хлорфенил]-2-циклопропилпропаноат
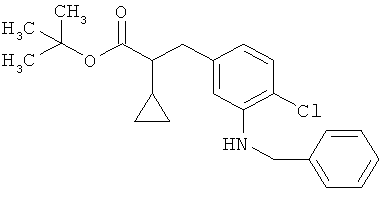
Исходя из трет-бутил-3-(3-бром-4-хлорфенил)-2-циклопропилпропаноата и бензиламина получили 2,51 г целевого соединения (74,7% от теор.).
ЖХ-МС (Метод 6): Rt=1,55 мин; m/z=386 (M+H)+.
Пример 160А

1,85 г (4,63 ммоль) (+/-)-трет-бутил-3-[3-(бензиламино)-4-хлорфенил]-2-циклобутилпропаноата растворяли в 10 мл этилацетата, удаляли кислород с помощью аргона и добавляли 98 мг (0,093 ммоль) палладия на угле (10%). Реакционную смесь перемешивали 4 ч при Ткомн. в атмосфере водорода при нормальном давлении, а потом оставляли стоять на выходные. Смесь отфильтровывали через целит, остаток дополнительно промывали этилацетатом, фильтрат упаривали в вакууме, а получившийся из фильтрата остаток сушили в высоком вакууме. Этот остаток (смесь состава примерно 1:1 из исходных веществ и целевого продукта) повторно растворяли примерно в 10 мл этилацетата, удаляли кислород с помощью аргона и снова добавляли 98 мг (0,093 ммоль) палладия на угле (10%). Эту реакционную смесь повторно перемешивали 4 ч при Ткомн. в атмосфере водорода при нормальном давлении. Потом фильтровали через целит, дополнительно промывали этилацетатом, фильтрат упаривали в вакууме, а остаток сушили в высоком вакууме. После хроматографической очистки на силикагеле (элюент: циклогексан/этилацетат 20:1) получили 1,12 г целевого продукта (78,2% от теор.).
ЖХ-МС (Метод 6): Rt=1,34 мин; m/z=310 (М+Н)+ 254 (M-C4H7)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,29 (с, 9Н), 1,68-1,86 (м, 4Н), 1,87-1,95 (м, 1Н), 1,96-2,07 (м, 1Н), 2,32-2,48 (м, 4Н), 5,21 (с, 2Н), 6,33 (дд, 1Н), 6,57 (д,1Н), 7,04 (д, 1Н).
Полученный выше рацемат был разделен на энантиомеры с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×20 мм; расход: 20 мл/мин; детектирование: 230 нм, объем впрыска: 0,80 мл; температура: 25°С; элюент: 95% изогексана/5% этанола]. Исходя из 790 мг рацемата, было получено 318 мг энантиомера 1 (Пример 161А) и 339 мг энантиомера 2 (Пример 162А):
Пример 161А
Трет-бутил-3-(3-амино-4-хлорфенил)-2-циклобутилпропаноат (энантиомер 1)
Выход: 318 мг
ЖХ-МС (Метод 4): Rt=1,70 мин; m/z=254 (M-C4H7)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,29 (с, 9Н), 1,68-1,85 (м, 4Н), 1,87-1,94 (м, 1Н), 1,96-2,06 (м, 1Н), 2,31-2,48 (м, 4Н), 5,21 (с, 2Н), 6,33 (дд, 1Н), 6,57 (д, 1Н), 7,04 (д, 1Н).
Пример 162А
Трет-бутил-3-(3-амино-4-хлорфенил)-2-циклобутилпропаноат (энантиомер 2)
Выход:339 мг
ЖХ-МС (Метод 4): Rt=1,71 мин; m/z=254 (M-C4H7)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,29 (с, 9Н), 1,67-1,84 (м, 4Н), 1,87-1,94 (м, 1Н), 1,95-2,05 (м, 1Н), 2,32-2,48 (м, 4Н), 5,22 (с, 2Н), 6,33 (дд, 1Н), 6,57 (д, 1Н), 7,04 (д, 1Н).
Пример 163А
(+/-)Трет-бутил-3-(3-амино-4-хлорфенил)-2-циклопропилпропаноат
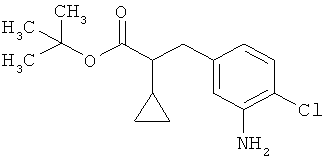
2,50 г (4,63 ммоль) (+/-)-трет-бутил-3-[3-(бензиламино)-4-хлорфенил]-2-циклопропилпропаноата растворяли в 160 мл этилацетата, удаляли кислород с помощью аргона и добавляли 150 мг палладия на угле (10%). Реакционную смесь перемешивали 8 ч при Ткомн. в атмосфере водорода при нормальном давлении. Затем эту смесь фильтровали через целит, дополнительно промывали этилацетатом, фильтрат упаривали в вакууме, а остаток сушили в высоком вакууме. Этот остаток очищали с помощью хроматографии на силикагеле (элюент: от чистого циклогексана до циклогексан/этилацетат 20:1). Получили 1,41 г целевого продукта (73,6% от теор.).
ЖХ-МС (Метод 6): Rt=1,28 мин; m/z=240 (M-C4H7)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,10-0,18 (м, 1Н), 0,19-0,26 (м, 1Н), 0,37-0,52 (м, 2Н), 0,79-0,92 (м, 1Н), 1,30 (с, 9Н), 1,73 (тд, 1Н), 2,65-2,74 (м, 2Н), 5,10-5,25 (м, 2Н), 6,35 (дд, 1Н), 6,59 (д, 1Н), 6,99-7,06 (м, 1Н).
Пример 164А
Метил-3-(3-амино-4-хлорфенил)гекс-2-еноат
и
метил-3-(3-амино-4-хлорфенил)гекс-3-еноат
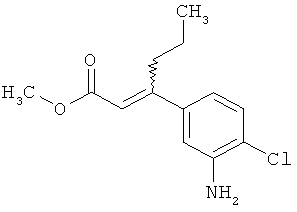

К смеси 10,0 г (48,4 ммоль) 5-бром-2-хлоранилина и 8,69 г (67,8 ммоль) метил-(2Е)-гекс-2-еноата в 100 мл ДМФА добавляли 33,8 мл (242,2 ммоль) триэтиламина. Смесь трижды вакуумировали и каждый раз снова подавали аргон. После добавления 1,09 г (4,84 ммоль) ацетата палладия (II) и 2,95 г (9,69 ммоль) три-2-толилфосфина снова дважды вакуумировали и соответственно заполняли аргоном, до того, как потом эту реакционную смесь перемешивали 4 ч при 150°С. После охлаждения эту смесь выливали на воду, насыщали хлоридом натрия и трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме, под конец в высоком вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 50:1). Получили 7,70 г смеси обоих целевых соединений (62,7% от теор., соотношение примерно 1,5:1 в пользу α,β-ненасыщенного изомера).
ЖХ-МС (Метод 6): метил-3-(3-амино-4-хлорфенил)гекс-2-еноат: Rt=1,04 мин, m/z=254 (М+Н)+; метил-3-(3-амино-4-хлорфенил)гекс-3-еноат: Rt=1,12 мин, m/z=254(M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): метил-3-(3-амино-4-хлорфенил)гекс-2-еноат: δ [м.д.]=0,85 (т, 3Н), 1,29-1,41 (м, 2Н), 2,92-3,00 (м, 2Н), 3,46 (с, 3Н), 5,45 (с, 2Н), 5,98 (с, 1Н), 6,69 (дд, 1Н), 6,94 (д, 1Н), 7,20 (д, 1Н).
1H-ЯМР (400 МГц, ДМСО-d6): метил-3-(3-амино-4-хлорфенил)гекс-3-еноат: δ [м.д.]=1,00 (т, 3Н), 2,15 (квинт, 2Н), 3,56 (с, 3Н), 3,66 (с, 2Н), 5,26-5,31 (м, 2Н), 5,84 (т, 1Н), 6,54 (дд, 1Н), 6,79 (д, 1Н), 7,09 (д, 1Н).
Пример 165А
(+/-)-Метил-3-(3-амино-4-хлорфенил)гексаноат
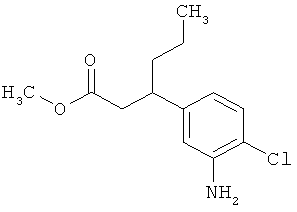
7,70 г (30,3 ммоль) смеси из метил-3-(3-амино-4-хлорфенил)гекс-2-еноата и метил-3-(3-амино-4-хлорфенил)гекс-3-еноата (примерно 1,5:1, Пример 164А) растворяли в 45 мл этилацетата, добавляли 646 мг (0,303 ммоль) палладия на угле (5%) и перемешивали при Ткомн. в атмосфере водорода при нормальном давлении. Спустя 10 ч реакционную смесь отфильтровывали через целит, дополнительно промывали этилацетатом, а фильтрат упаривали. Остаток переводили примерно в 50 мл этилацетата, снова добавляли приблизительно 650 мг палладия на угле (5%) и перемешивали при Ткомн. в атмосфере водорода при нормальном давлении. Спустя дополнительные 36 ч реакционную смесь снова отфильтровывали через целит, дополнительно промывали этилацетатом, а фильтрат упаривали. Остаток переводили в 800 мл этилацетата, снова добавляли приблизительно 650 мг палладия на угле (5%) и перемешивали 24 ч при Ткомн. в атмосфере водорода при нормальном давлении. Реакционную смесь еще раз отфильтровывали через целит, дополнительно промывали этилацетатом, а фильтрат упаривали. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат от 50:1 до 10:1). В общей сложности получили 4,79 г смеси из целевого продукта и исходных веществ. Эту смесь растворяли в 180 мл этилацетата, снова добавляли 603 мг (0,566 ммоль) палладия на угле (10%) и перемешивали в течение ночи при Ткомн. в атмосфере водорода при нормальном давлении. Реакционную смесь отфильтровывали через целит, дополнительно промывали этилацетатом, фильтрат упаривали, а остаток сушили в высоком вакууме. Так было получено 4,45 г (примерно 57% от теор.) целевого продукта.
ЖХ-МС (Метод 4): Rt=1,50 мин; m/z=256 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (т, 3Н), 1,03-1,15 (м, 2Н), 1,39-1,56 (м, 2Н), 2,46 (дд, 1Н), 2,59 (дд, 1Н), 2,78-2,89 (м, 1Н), 3,50 (с, 3Н), 5,22 (уш. с, 2Н), 6,39 (дд, 1Н), 6,61 (д, 1Н), 7,06 (д, 1Н).
Пример 166А и Пример 167А
Метил-3-(3-амино-4-хлорфенил)пент-2-еноат
и
метил-3-(3-амино-4-хлорфенил)пент-3-еноат
К смеси 5,0 г (24,2 ммоль) 5-бром-2-хлоранилина и 5,53 г (48,4 ммоль) ме-тил-2-пентеноата в 50 мл ДМФА добавляли 16,9 мл (121 ммоль) триэтиламина. Смесь трижды вакуумировали и каждый раз снова подавали аргон. После добавления 544 мг (2,42 ммоль) ацетата палладия(II) и 1,47 г (4,84 ммоль) три-2-толилфосфина снова дважды вакуумировали и соответственно заполняли аргоном, а потом реакционную смесь перемешивали 6 ч при 150°С. После охлаждения эту смесь выдерживали в течение ночи при Ткомн. и после этого выливали в воду. Трижды экстрагировали этилацетатом. Органические фазы объединяли, сушили над сульфатом магния, упаривали в вакууме, а остаток сушили в высоком вакууме. Из этого остатка с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат от 50:1 до 10:1) были выделены оба изомерных целевых продукта в разделенном виде. Получили 0,85 г метил-3-(3-амино-4-хлорфенил)пент-2-еноата (14,6% от теор.), а также 3,05 г метил-3-(3-амино-4-хлорфенил)пент-3-еноата (52,5% от теор.).
Метил-3-(3-амино-4-хлорфенил)пент-2-еноат (Пример 166A):
ЖХ-МС (Метод 6): Rt=1,09 мин; m/z=240 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,97 (т, 3Н), 2,98 (кв, 2Н), 3,66 (с, 3Н), 5,45 (с, 2Н), 5,96 (с, 1Н), 6,70 (дд, 1Н), 6,95 (д, 1Н), 7,21 (д, 1Н).
Метил-3-(3-амино-4-хлорфенил)пент-3-еноат (Пример 167А):
ЖХ-МС (Метод 6): Rt=1,00 мин; m/z=240 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,75 (д, 3Н), 3,47 (с, 2Н), 3,56 (с, 3Н), 5,28 (с, 2Н), 5,94 (кв, 1Н), 6,54 (дд, 1Н), 6,77 (д, 1Н), 7,09 (д, 1Н).
Пример 168А
(+/-)-Метил-3-(3-амино-4-хлорфенил)пентаноат

3,05 г (12,7 ммоль) метил-3-(3-амино-4-хлорфенил)пент-3-еноата и 0,85 г (3,55 ммоль) метил-3-(3-амино-4-хлорфенил)пент-2-еноата вместе растворяли в 500 мл этилацетата, добавляли 346 мг (0,325 ммоль) палладия на угле (10%) и перемешивали в течение ночи при Ткомн. в атмосфере водорода при нормальном давлении. После этого реакционную смесь отфильтровывали через целит, дополнительно промывали этилацетатом и фильтрат упаривали. После высушивания остатка в высоком вакууме получили 3,73 г целевого продукта (94,8% от теор.).
ГХ-МС (Метод 1): Rt=6,07 мин; m/z=242 (М)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,71 (т, 3Н), 1,42-1,49 (м, 1Н), 1,55-1,61 (м, 1Н), 2,42-2,48 (м, 1Н), 2,60 (дд, 1Н), 2,68-2,78 (м, 1Н), 3,50 (с, 3Н), 5,22 (с, 2Н), 6,39 (дд, 1Н), 6,61 (д, 1Н), 7,05-7,08 (м, 1Н).
Пример 169А
Сложный этиловый эфир (3R)-2-(4-хлор-2-фторфенил)-4,4,4-трифтор-3-метилбутановой кислоты (смесь диастереомеров)

81,5 мл (81,5 ммоль) 1 М раствора гексаметилдисилазида лития в толуоле охлаждали до -20°С и по каплям добавляли раствор 10,0 г (50,3 ммоль) этил-(3R)-4,4,4-трифтор-3-метилбутаноата в 50 мл абс. толуола. Эту смесь дополнительно перемешивали 10 мин. Затем при -20°С прикалывали предварительно приготовленный раствор 14,8 г (70,6 ммоль) 1-бром-4-хлор-2-фторбензола, 366 мг (1,63 ммоль) ацетата палладия (II) и 1,35 г (3,42 ммоль) 2-дициклогексилфосфино-2'-(N,N-Диметиламино)бифенила в 50 мл абс. толуола. По окончании прибавления охлаждение убирали, а полученную в результате реакционную смесь перемешивали сначала 1 ч при Ткомн., потом в течение ночи при 80°С. После охлаждения смесь фильтровали через целит, несколько раз дополнительно промывали толуолом, а полученный фильтрат упаривали в вакууме. Из этого остатка после хроматографии на силикагеле (элюент: циклогексан/этилацетат 100:1→100:4) получили 4,26 г целевого соединения (25,1% от теор.).
230 ГХ-МС (Метод 1): Rt=4.21 мин; m/z=312 (M)+.
Аналогичным образом был получен следующий пример:
Пример 170А
Сложный этиловый эфир (3R)-2-(4-хлор-3-фторфенил)-4,4,4-трифтор-3-метилбутановой кислоты (смесь диастереомеров)

Исходя из 2,0 г этил-(3R)-4,4,4-трифтор-3-метилбутаноата и 2,96 г 1-бром-4-хлор-3-фторбензола, получили 2,47 г целевого соединения.
ГХ-МС (Метод 1): Rt=4,33 мин+4,36 мин; оба m/z=312 (М)+.
1H-ЯМР (400 МГц, ДМСО-d6): основной диастереомер δ [м.д.]=0,80 (д, 3Н), 1,08-1,19 (м, 3Н), 3,34-3,41 (м, 1Н), 3,88 (д, 1Н), 4,01-4,18 (м, 2Н), 7,28-7,34 (м, 1Н), 7,51-7,64 (м, 2Н).
Пример 171А
Этил-(3R)-2-(4-хлор-2-метилфенил)-4,4,4-трифтор-3-метилбутаноат (смесь диастереомеров)

22,5 мл (22,5 ммоль) 1 М раствора гексаметилдисилазида лития в толуоле охлаждали до -20°С и по каплям добавляли раствор 2,76 г (50,3 ммоль) этил-(3R)-4,4,4-трифтор-3-метилбутаноата в 15 мл абс. толуола. Эту смесь дополнительно перемешивали 10 мин. Затем при -20°С прикалывали предварительно приготовленный раствор 4,0 г (19,5 ммоль) 2-бром-5-хлортолуола, 101 мг (0,45 ммоль) ацетата палладия(II) и 371 мг (0,94 ммоль) 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенила в 15 мл абс. толуола. По окончании прибавления охлаждение убирали, а полученную в результате реакционную смесь перемешивали сначала 1 ч при Ткомн., потом в течение ночи при 100°С. После охлаждения смесь фильтровали через целит, несколько раз дополнительно промывали толуолом, а полученный фильтрат упаривали в вакууме. Получили 3,10 г целевого соединения в виде неочищенного продукта, который непосредственно подвергался дальнейшим превращениям.
ГХ-МС (Метод 1): Rt=4,72 мин; m/z=308 (M)+.
Пример 172А
Этил-(3R)-2-(4-хлор-3-метилфенил)-4,4,4-трифтор-3-метилбутаноат (смесь диастереомеров)

29,2 мл (29,2 ммоль) 1 М раствора гексаметилдисилазида лития в толуоле охлаждали до -10°С и по каплям добавляли раствор 4,30 г (23,4 ммоль) этил-(3R)-4,4,4-трифтор-3-метилбутаноата в 26 мл абс. толуола. Эту смесь дополнительно перемешивали 10 мин. Затем при -10°С прикалывали предварительно приготовленный раствор 5,0 г (19,5 ммоль, 80%-ного) 5-бром-2-хлортолуола, 131 мг (0,58 ммоль) ацетата палладия(II) и 483 мг (1,23 ммоль) 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенила в 26 мл абс. толуола. Полученную в результате реакционную смесь перемешивали сначала 1 ч при Ткомн., потом 4 ч при 80°С. После охлаждения смесь разбавляли этилацетатом, дважды промывали водным раствором гидрокарбоната натрия и один раз нас. раствором хлорида натрия, сушили над сульфатом натрия и упаривали в вакууме. Получили 7,80 г целевого соединения в виде неочищенного продукта, который непосредственно подвергался дальнейшим превращениям.
ЖХ-МС (Метод 4): Rt=1,55 мин; m/z=309 (М+Н)+.
Аналогичным образом был получен следующий пример:
Пример 173А
Этил-(3R)-2-(3,4-дихлорфенил)-4,4,4-трифтор-3-метилбутаноат (смесь диастереомеров)
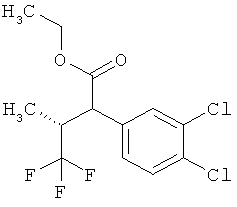
Исходя из 3,91 г этил-(3R)-4,4,4-трифтор-3-метилбутаноата и 5,0 г 4-бром-1,2-дихлорбензола, получили 7,54 г целевого соединения в виде неочищенного продукта.
ЖХ-МС (Метод 4): Rt=1,54 мин; m/z=329 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): оба диастереомера δ [м.д.]=0,80/1,17 (2д, совм., 3Н), 1,10-1,15 (м, 3Н), 3,30-3,41 (м, 1Н), 3,89/3,94 (2д, совм., 1Н), 4,01-4,18 (м, 2Н), 7,38-7,48 (м, прим. 1Н), 7,59-7,68 (м, прим. 1Н), 7,74/7,75 (2д, совм., 1Н).
Пример 174А
(3R)-2-(4-хлор-2-фторфенил)-4,4,4-трифтор-3-метилбутановая кислота (смесь диастереомеров)
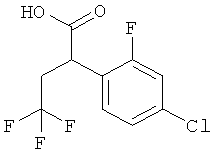
4,26 г (13,6 ммоль) сложного этилового эфира (3R)-2-(4-хлор-2-фторфенил)-4,4,4-трифтор-3-метилбутановой кислоты (смесь диастереомеров) растворяли в смеси из 22 мл метанола, 22 мл ТГФ и 11 мл воды и при 0°С добавляли 10,9 г 50%-ного водного раствора гидроксида натрия. Реакционную смесь перемешивали в течение ночи при Ткомн.. Затем в вакууме удаляли значительную часть органических растворителей. Оставшаяся смесь была разбавлена водой и экстрагирована диэтиловым эфиром. После разделения фаз органическую фазу отбрасывали, а водную фазу подкисляли полуконцентрированной соляной кислотой (pH приблизительно 2) и несколько раз экстрагировали этилацетатом. Объединенные фазы этилацетата сушили над сульфатом натрия и упаривали в вакууме. Получили 3,38 г (76,7% от теор.) целевого продукта в виде смеси диастереомеров, который мог использоваться в последующих реакциях без дополнительной очистки.
ЖХ-МС (Метод 4): Rt=1,25 мин; m/z=283 (M-H)-.
1H-ЯМР (400 МГц, ДМСО-d6): основной диастереомер δ [м.д.]=0,87 (д, 3Н), 3,27-3,37 (м, 1Н), 4,02 (д, 1Н), 7,35 (дд, 1Н), 7,45-7,52 (м, 2Н), 13,02 (уш. с, 1Н).
Аналогичным образом были получены обе следующие карбоновые кислоты:
Пример 175А
(3R)-2-(4-хлор-3-фторфенил)-4,4,4-трифтор-3-метилбутановая кислота (смесь диастереомеров)

Соотношение диастереомеров примерно 1:1.
ГХ-МС (Метод 1): Rt=4,79 мин; m/z=284 (М)+.
1H-ЯМР (400 МГц, ДМСО-d6): оба диастереомера δ [м.д.]=0,80/1,19 (2д, совм., 3Н), 3,18-3,29 (м, 1Н), 3,74/3,77 (2дд, совм., 1Н), 7,28 (д, 1Н), 7,43-7,65 (м, 2Н), 12,91/13,24 (2 уш. с, совм., 1Н).
Пример 176А
(3R)-2-(4-хлор-3-метилфенил)-4,4,4-трифтор-3-метилбутановая кислота (смесь диастереомеров)
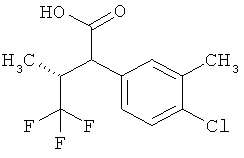
Соотношение диастереомеров примерно 5:1.
1H-ЯМР (400 МГц, ДМСО-d6): оба диастереомера δ [м.д.]=0,78/1,11 (2д, совм., 3Н), 2,31/2,32 (2с, совм., 3Н), 3,24-3,30 (м, 1Н), 3,61/3,64 (2д, совм., 1Н), 7,20-7,50 (м, 5Н), 12,80 (уш. с, 1Н).
Пример 177А
(3R)-2-(4-хлор-2-метилфенил)-4,4,4-трифтор-3-метилбутановая кислота (смесь диастереомеров)
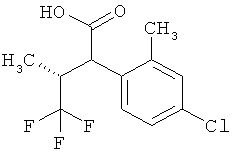
3,10 г (неочищенного, примерно 10,04 ммоль) этил-(3R)-2-(4-хлор-2-метилфенил)-4,4,4-трифтор-3-метилбутаноата (смесь диастереомеров) растворяли в смеси из 10 мл метанола, 10 мл ТГФ и 5 мл воды и при 0°С добавляли 8,03 г 50%-ного водного раствора гидроксида натрия. Реакционную смесь перемешивали в течение ночи при Ткомн.. После этого подкисляли 1 н. соляной кислотой (pH приблизительно 2) и трижды экстрагировали этилацетатом. Объединенные органические фазы промывали нас. раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат от 50:1 до 4:1). Получили 1,46 г (51,8% от теор.) целевого продукта в виде смеси диастереомеров (приблизительно 5:1).
ГХ-МС (Метод 1): Rt=5,14 мин; m/z=280 (М)+.
1H-ЯМР (400 МГц, ДМСО-d6): оба диастереомера δ [м.д.]=0,76/1,11 (2д, совм., 3Н), 2,34/2,36 (2с, совм., 3Н), 3,33-3,38 (м, прим. 1Н, перекрывается), 3,81/3,88 (2д, совм., 1Н), 7,27-7,41 (м, 3Н), 12,81 (уш. с, 1Н).
Пример 178А
(3R)-2-(3,4-дихлорфенил)-4,4,4-трифтор-3-метилбутановая кислота (смесь диастереомеров)
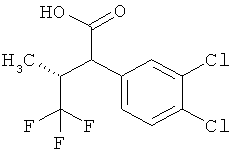
3,77 г (неочищенного, приблизительно 11,5 ммоль) этил-(3R)-2-(3,4-дихлорфенил)-4,4,4-трифтор-3-метилбутаноата (смесь диастереомеров) растворяли в смеси из 14 мл метанола, 14 мл ТГФ и 5 мл воды и при 0°С добавляли 9,16 г 50%-ного водного раствора гидроксида натрия. Реакционную смесь перемешивали примерно 6 ч при 40°С. Затем подкисляли 1 н. соляной кислотой (pH приблизительно 2) и трижды экстрагировали этилацетатом. Объединенные органические фазы промывали нас. раствором хлорида натрия, сушили над сульфатом натрия и упаривали в вакууме. Получили 3,94 г целевого соединения в виде сырого продукта, который мог использоваться в последующих реакциях без дополнительной очистки.
1H-ЯМР (400 МГц, ДМСО-d6): оба диастереомера δ [м.д.]=0,80/1,19 (2д, совм., 3Н), 3,21-3,30 (м, 1Н), 3,69-3,82 (м, 1Н), 7,42 (дд, 1Н), 7,63-7,67 (м, 1Н), 7,70-7,73 (м, 1Н), 12,97 (уш. с, 1Н).
Пример 179А
(3R)-2-(4-хлор-2-фторфенил)-4,4,4-трифтор-3-метилбутаноилхлорид (смесь диастереомеров)

660 мг (2,32 ммоль) (3R)-2-(4-хлор-2-фторфенил)-4,4,4-трифтор-3-метилбутановой кислоты (смесь диастереомеров) растворяли в 2 мл дихлорметана. После добавления одной маленькой капли ДМФА реакционный раствор охлаждали до температуры от -5°С до 0°С и по каплям добавляли 0,4 мл (4,64 ммоль) оксалилхлорида. Охлаждение убирали, а реакционную смесь перемешивали 1 ч при Ткомн. до окончания выделения газа. После этого реакционную массу упаривали в вакууме. Остаток дважды извлекали абс. дихлорметаном, каждый раз снова упаривали в вакууме и в заключение остаток сушили в высоком вакууме. Получили 640 мг целевого продукта, который непосредственно подвергался дальнейшим превращениям без дополнительной очистки.
Следующие примеры были получены в соответствии с Общей методикой 1 (Амидное сочетание производных 4,4,4-трифтор-3-метил-2-фенилбутановой кислоты с анилинами с применением ГАТУ в ДМФА с использованием пиридина или N,N-диизопропилэтиламина в качестве основания):
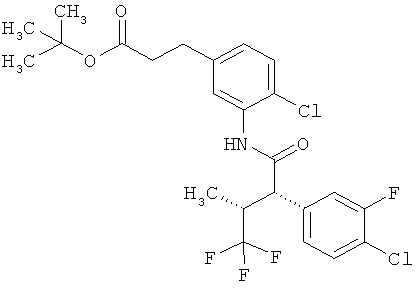


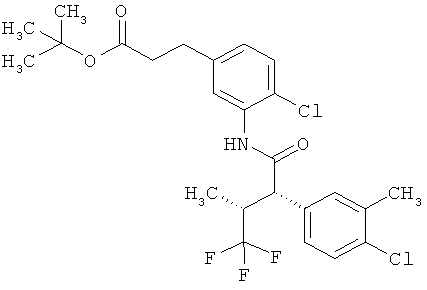
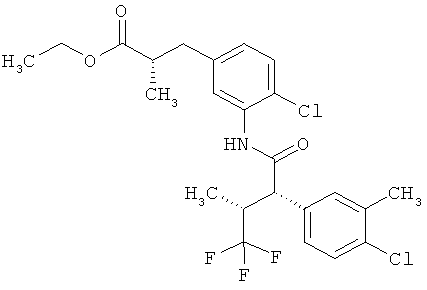





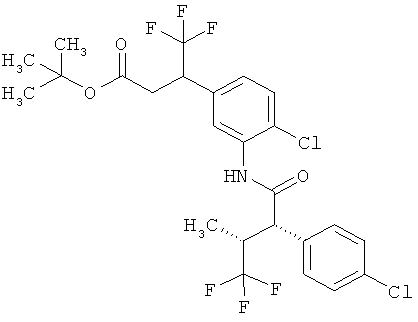


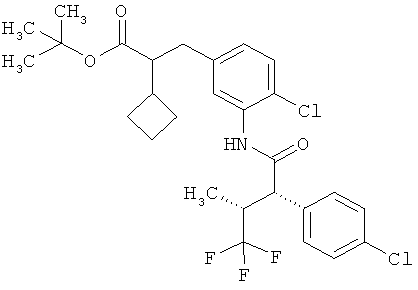

Пример 196А
(+)-Этил-(2S)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлор-2-фторфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-2-метилпропаноат

280,7 мг (1,16 ммоль) сложного этилового эфира (+)-(2S)-3-(3-амино-4-хлорфенил)-2-метилпропановой кислоты растворяли в 1,5 мл абс. ТГФ, добавляли 0,26 мл (1,48 ммоль) N,N-диизопропилэтиламина и после охлаждения до -10°С по каплям прибавляли раствор 320 мг (неочищенного, приблизительно 1,06 ммоль) приготовленного in situ (3R)-2-(4-хлор-2-фторфенил)-4,4,4-трифтор-3-метилбутаноилхлорида в 0,5 мл абс. ТГФ. Реакционную смесь по окончании прибавления в течение 30 мин перемешивали при температуре между -10°С и 0°С. Потом после добавления нескольких капель воды разбавляли дихлорметаном. Эту смесь промывали 1 н. соляной кислотой и нас. раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали сначала при помощи препаративной ОФ ВЭЖХ (элюент: метанол/вода), потом при помощи хроматографии на силикагеле (элюент: циклогексан/этилацетат 40:1). Получили 144 мг целевого соединения (26,9% от теор.).
ЖХ-МС (Метод 6): Rt=1,42 мин; m/z=508 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,86 (д, 3Н), 1,02-1,12 (м, 6Н), 2,63-2,72 (м, 2Н), 2,76-2,86 (м, 1Н), 3,34-3,44 (м, 1Н), 3,93-4,02 (м, 2Н), 4,36 (д, 1Н), 7,03 (дд, 1Н), 7,25-7,29 (м, 1Н), 7,32-7,38 (м, 2Н), 7,51 (дд, 1Н), 7,61 (т, 1Н), 10,02 (с, 1Н).
[α]D20=+90°, с=0,30, хлороформ.
Пример 197А
Метил-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-2-метилфенил)пропаноат

265 мг (1,16 ммоль) метил-3-(3-амино-4-хлор-2-метилфенил)пропаноата растворяли в 1,5 мл абс. ТГФ, добавляли 0,28 мл (1,63 ммоль) N,N-диизопропилэтиламина и после охлаждения до -10°С по каплям прибавляли раствор 398 мг (неочищенного, приблизительно 1,40 ммоль) приготовленного in situ (2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноилхлорида в 0,5 мл абс. ТГФ. Реакционную смесь по окончании прибавления в течение 1 ч нагревали от -10°С до Ткомн., а потом разбавляли этилацетатом. Эту смесь промывали 1 н. соляной кислотой и нас. раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали при помощи препаративной ОФ ВЭЖХ (элюент: метанол/вода). Получили 485 мг целевого соединения (87,5% от теор.).
ЖХ-МС (Метод 6): Rt=1,25 мин; m/z=476 (М+Н)+.
Пример 198А
(+)-Трет-бутил-(2R)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-2-метилфенил)-2-метилпропаноат
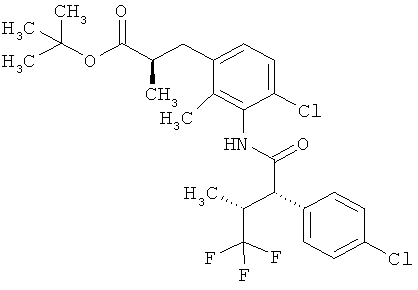
200 мг (0,705 ммоль) (-)-трет-бутил-(2R)-3-(3-амино-4-хлор-2-метилфенил)-2-метилпропаноата растворяли в 1 мл абс. ТГФ, добавляли 0,17 мл (0,987 ммоль) N,N-диизопропилэтиламина и после охлаждения до -10°С по каплям прибавляли раствор 241 мг (неочищенного, приблизительно 0,846 ммоль) приготовленного in situ (2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноилхлорида в 0,2 мл абс. ТГФ. Реакционную смесь по окончании прибавления в течение 2 ч нагревали от -10°С до Ткомн., а потом выливали в воду. Водную фазу трижды экстрагировали этилацетатом и объединенные органические фазы промывали 1 н. соляной кислотой и нас. раствором хлорида натрия, сушили над сульфатом магния, упаривали в вакууме, а остаток сушили в высоком вакууме. Получили 282 мг целевого соединения (75,2% от теор.).
ЖХ-МС (Метод 6): Rt=1,45 мин; m/z=530 (М-Н)-.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 1,03 (уш. с, 3Н), 1,30 (с, 9Н), 1,50 (уш. с, 1Н), 2,15 (уш. с, 1Н), 2,42 (уш. с, 1Н), 2,69-2,92 (м, 1Н), 3,34-3,45 (м, 1Н), 3,94 (д, 1Н), 7,03 (д, 1Н), 7,23 (уш. с, 1Н), 7,45 (с, 4Н), 9,83/9,91 (2 уш. с, совм., 1Н) [сигналы значительно уширены вследствие наличия ротамеров}.
[α]D20=+68,9°, с=0,50, хлороформ.
Аналогичным образом был получен следующий пример:
Пример 199А
(+)-Трет-бутил-(2S)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-2-метилфенил)-2-метилпропаноат

Исходя из 200 мг (+)-трет-бутил-(2S)-3-(3-амино-4-хлор-2-метилфенил)-2-метилпропаноата и 241 мг свежеприготовленного (2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноилхлорида, получили 287 мг целевого продукта (75,2% от теор.).
ЖХ-МС (Метод 6): Rt=1,51 мин; m/z=530 (М-Н)-.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 1,04 (уш. с, 3Н), 1,29 (с, 9Н), 1,51 (уш. с, 1Н), 2,15 (уш. с, 1Н), 2,56-2,68 (м, 1Н), 2,79 (уш. с, 1Н), 3,34-3,45 (м, 1Н), 3,94 (уш. д, 1Н), 7,03 (д, 1Н), 7,15 (уш. с, 1Н), 7,23 (уш. с, 1 Н), 7,45 (с, 4Н), 9,87 (уш. с, 1Н) [сигналы значительно уширены вследствие наличия ротамеров].
[α]D20=+116,1°, с=0,520, хлороформ.
Пример 200А
(+)-Трет-бутил-2-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}бензил)-2-метилбутаноат (диастереомер А)

225 мг (0,756 ммоль) (-)-трет-бутил-2-(3-амино-4-хлорбензил)-2-метилбутаноата (энантиомер 1) и 231 мг (0,907 ммоль) (+)-(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты растворяли в 0,9 мл пиридина и 2,7 мл ДМФА и при Ткомн. добавляли 345 мг (0,907 ммоль) ГА-ТУ. Реакционную смесь в течение ночи перемешивали при 45°С, до того как добавили дополнительные 0,5 экв. (+)-(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты и 0,6 экв. ГАТУ. Реакционную смесь еще раз перемешивали 3 ч при 45°С, а потом после охлаждения разбавляли этилацетатом. Эту смесь промывали 1 н. соляной кислотой и нас. раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали при помощи препаративной ОФ ВЭЖХ (элюент: ацетонитрил/вода) и последующей хроматографии на силикагеле (элюент: циклогексан/этилацетат 40:1). Получили 177 мг целевого продукта (35,6% от теор.).
ЖХ-МС (Метод 4): Rt=1,97 мин; m/z=544 (М-Н)-.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,76-0,83 (м, 6Н), 0,91 (с, 3Н), 1,31 (с, 9Н), 1,33-1,40 (м, 1Н), 1,57-1,67 (м, 1Н), 2,57 (д, 1Н), 2,85 (д, 1Н), 3,35-3,43 (м, 1Н), 4,07-4,13 (м, 1Н), 6,95 (дд, 1Н), 7,30-7,36 (м, 2Н), 7,41-7,48 (м, 4Н),9,82(с, 1Н).
[α]D20=+63,2°, с=0,365, хлороформ.
Пример 201А
Метил-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)гексаноат (смесь диастереомеров)
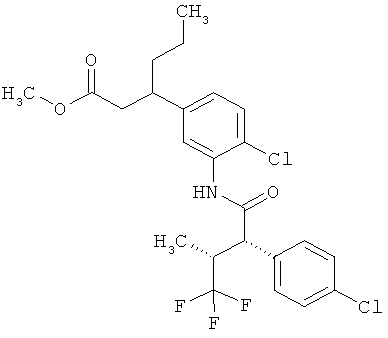
1,45 г (5,67 ммоль) (+/-)-метил-3-(3-амино-4-хлорфенил)гексаноата и 1,81 г (6,80 ммоль) (+)-(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты растворяли в 5,0 мл пиридина и 10,0 мл ДМФА и при Ткомн. добавляли 2,80 г (7,37 ммоль) ГАТУ. Реакционную смесь в течение ночи перемешивали при Ткомн., а потом разбавляли этилацетатом. Эту смесь промывали 1 н. соляной кислотой и нас. раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали при помощи хроматографии на силикагеле (элюент: сначала циклогексан, потом циклогексан/этилацетат 50:1). В двух фракциях получили в общей сложности 2,02 г целевого продукта (70,6% от теор.).
ЖХ-МС (Метод 6): Rt=1,43 мин; m/z=504 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): оба диастереомера δ [м.д.]=0,74-0,85 (м, 6Н), 0,98-1,16 (м, 2Н), 1,42-1,61 (м, 2Н), 2,49 (дд, прибл, 1Н, перекрываются), 2,64 (дд, 1Н), 2,84-3,02 (м, 1Н), 3,37-3,42 (м, 1Н), 3,47/3,48 (2 с, совм., 3Н), 4,12 (д, 1Н), 7,05 (дд, 1Н), 7,31-7,38 (м, 2Н), 7,41-7,55 (м, 4Н), 9,83 (с, 1Н).
Пример 202А
Метил-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)пентаноат (смесь диастереомеров)

500 мг (2,07 ммоль) (+/-)-метил-3-(3-амино-4-хлорфенил)пентаноата и 668,9 мг (2,48 ммоль) (+)-(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты растворяли в 1,7 мл пиридина и 3,3 мл ДМФА и при Ткомн. добавляли 1,02 г (2,69 ммоль) ГАТУ. Реакционную смесь в течение ночи перемешивали при Ткомн., а потом разбавляли этилацетатом. Эту смесь промывали 1 н. соляной кислотой и нас. раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали при помощи хроматографии на силикагеле (элюент: сначала циклогексан, потом циклогексан/этилацетат 50:1). Получили 675 мг целевого продукта (66,6% от теор.).
257 ЖХ-МС (Метод 6): Rt=1,39 мин; m/z=490 (M-H)-.
1H-ЯМР (400 МГц, ДМСО-d6): оба диастереомера δ [м.д.]=0,65-0,74 (м, 3Н), 0,80 (д, 3Н), 1,43-1,67 (м, 2Н), 2,49 (дд, прибл., 1Н, перекрываются), 2,65 (дд, 1Н), 2,80-2,92 (м, 1Н), 3,35-3,43 (м, 1Н), 3,47/3,48 (2с, совм., 3Н), 4,13 (д, 1Н), 7,05 (дд, 1Н), 7,36 (дд, 2Н), 7,43-7,51 (м, 4Н), 9,84 (с, 1Н).
Пример 203А
(+)-Трет-бутил-(3S)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-4,4,4-трифторбутаноат
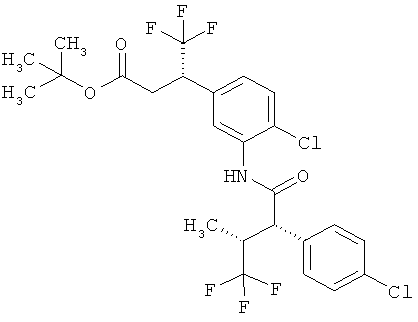
2,0 г (6,18 ммоль) (+)-трет-бутил-(3S)-3-(3-амино-4-хлорфенил)-4,4,4-трифторбутаноата и 1,98 г (7,41 ммоль) (+)-(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты растворяли в 5,0 мл пиридина и 10,0 мл ДМФА и при Ткомн. добавляли 3,05 г (8,03 ммоль) ГАТУ. Реакционную смесь в течение ночи перемешивали при Ткомн., до того как добавили дополнительные 1,98 г (7,41 ммоль) (+)-(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты и 3,05 г (8,03 ммоль) ГАТУ. Реакционную смесь еще раз перемешивали в течение 8 ч при 40°С, а потом после охлаждения разбавляли этилацетатом. Эту смесь промывали 1 н. соляной кислотой и нас. раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали при помощи хроматографии на силикагеле (элюент: сначала циклогексан, потом циклогексан/этилацетат 50:1). Полученный таким образом продукт (2,7 г) еще раз дополнительно очищался с помощью повторной хроматографии на силикагеле (элюент: циклогексан/этилацетат 100:1). Получили 1,80 г целевого продукта (50,9% от теор.).
ЖХ-МС (Метод 4): Rt=1,74 мин; m/z=570 (М-Н)-.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 1,21 (с, 9Н), 2,77 (дд, 1Н), 2,94 (дд, 1Н), 3,36-3,46 (м, 1Н), 3,99-4,09 (м, 1Н), 4,15 (д, 1Н), 7,17-7,29 (м, 1Н), 7,42-7,52 (м, 5Н), 7,59-7,65 (м, 1Н), 9,94 (с, 1Н).
[α]D20=+84,0°, с=0,48, хлороформ. Аналогичным образом был получен следующий пример:
Пример 204А
(+)-Трет-бутил-(3R)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-4,4,4-трифторбутаноат

Исходя из 1,0 г (3,09 ммоль) (-)-трет-бутил-(3R)-3-(3-амино-4-хлорфенил)-4,4,4-трифторбутаноата и 988 мг (3,71 ммоль) (+)-(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты, было получено 1,2 г (68% от теор.) целевого продукта.
ЖХ-МС (Метод 4): Rt=1,75 мин; m/z=570 (М-Н)-.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 1,21 (с, 9Н), 2,78 (дд, 1Н), 2,93 (дд, 1Н), 3,35-3,47 (м, 1Н), 3,99-4,10 (м, 1Н), 4,15 (д, 1Н), 7,19-7,28 (м, 1Н), 7,40-7,52 (м, 5Н), 7,60-7,66 (м, 1Н), 9,93 (с, 1Н).
[α]D20=+42,7°, с=0,48, хлороформ.
Пример 205А

В атмосфере аргона прикалывали 201 мл (1,39 моль) трет-бутилпроп-2-еноата к раствору 100 г (463 ммоль) 1-бром-2-метил-3-нитробензола, 322 мл (2,31 моль) триэтиламина, 28,18 г (92,58 ммоль) три-2-толилфосфина и 10,39 г (46,29 ммоль) ацетата палладия (II) в 2 литрах ДМФА и после этого смесь перемешивали 36 ч при 125°С. После охлаждения до комнатной температуры эту реакционную смесь перемешивали с насыщенным водным раствором хлорида аммония и отделяли органическую фазу. Водную фазу трижды экстрагировали трет-бутилметиловым эфиром, а объединенные органические фазы промывали насыщенным раствором хлорида натрия и сушили над сульфатом натрия. После фильтрации растворитель досуха удаляли в вакууме. Полученный остаток очищали при помощи флэш-хроматографии на силикагеле (элюент: петролейный эфир/этилацетат 9:1). Получили 89 г (338 ммоль, 73% от теор.) промежуточного продукта - трет-бутил-(2Е)-3-(2-метил-3-нитрофенил)проп-2-еноата в виде бесцветного твердого вещества.
88 г (334 ммоль) этого твердого вещества растворяли в 2 литрах этанола, при комнатной температуре добавляли 7 г палладия на угле (10%) и гидрировали 18 ч при нормальном давлении. После полного прохождения реакции фильтровали через кизельгур, а полученный фильтрат упаривали в вакууме. Получили 61,3 г (260,5 ммоль, 78% от теор.) целевого соединения в виде бесцветного твердого вещества.
ЖХ-МС (Метод 2): Rt=1,84 мин; m/z=236 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 6,77 (1Н, т), 6,47 (1Н, д), 6,36 (1Н, д), 4,72 (2Н, с), 2,14 (2Н, т), 2,36 (2Н, т), 1,95 (3Н, с), 1,39 (9Н, с).
Пример 206А
Трет-бутил-3-(3-бром-4-хлорфенил)-2,2-диметилпропаноат

4,0 мл (28,8 ммоль) диизопропиламина в атмосфере аргона растворяли в 50 мл сухого ТГФ и охлаждали до -30°С. Прикалывали 11,5 мл (28,8 ммоль) раствора н-бутиллития (2,5 М раствор в гексане). Полученную в результате смесь нагревали до 0°С, а потом охлаждали до -70°С. Затем добавили раствор 2,77 г (19,2 ммоль) трет-бутил-2-метилбутаноата в 20 мл ТГФ, причем температуру реакции поддерживали ниже -60°С. После 4 ч перемешивания при -60°С добавляли раствор 6,0 г (21,1 ммоль) 2-бром-4-(бромметил)-1-хлорбензола в 30 мл ТГФ, причем температуру реакции снова поддерживали ниже -60°С. Реакционную смесь в течение ночи перемешивали при медленном нагревании до Ткомн., а потом добавили насыщенный водный раствор хлорида аммония и этилацетат. После разделения фаз водную фазу дважды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 10:1→4:1). Получили 5,6 г (84% от теор.) целевого соединения.
ГХ-МС (Метод 1): Rt=6,16 мин; m/z=290/292 (M-C4H8)+.
Пример 207А
Трет-бутил-3-[3-(бензиламино)-4-хлорфенил]-2,2-диметилпропаноат
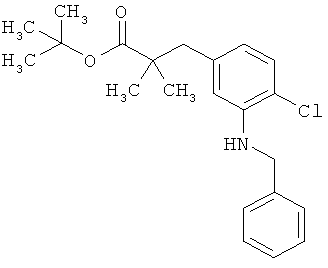
В атмосфере аргона в сухую колбу отвешивали 1,73 г (17,95 ммоль) трет-бутилата натрия и смешивали с 40 мл абс. толуола. К этому последовательно прибавляли 5,2 г (14,96 ммоль) трет-бутил-3-(3-бром-4-хлорфенил)-2,2-диметилпропаноата, 1,96 мл (17,95 ммоль) бензиламина, 685 мг (0,75 ммоль) трис(дибензилиденацетон)дипалладия, а также 373 мг (0,60 ммоль) (+/-)-2,2'-бис(дифенилфосфино)-1,1'-бинафтила. Реакционную смесь 2 ч перемешивали при 110°С, затем охлаждали до Ткомн. и дополнительно перемешивали при этой температуре в течение ночи. Потом к реакционной смеси прибавляли насыщенный водный раствор хлорида аммония и этилацетат и фильтровали через кизельгур с отсасыванием. После разделения фаз органическую фазу промывали насыщенным раствором хлорида аммония и насыщенным раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали с помощью препаративной ВЭЖХ (элюент: ацетонитрил/вода). Получили 2,78 г целевого соединения (50% от теор.).
ЖХ-МС (Метод 6): Rt=1,53 мин; m/z=374/376 (М+Н)+.
Пример 208А
Трет-бутил-3-(3-амино-4-хлорфенил)-2,2-диметилпропаноат
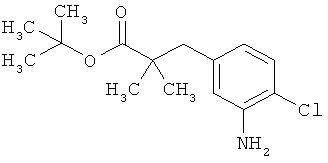
2,7 г (примерно 7,22 ммоль) трет-бутил-3-[3-(бензиламино)-4-хлорфенил]-2,2-диметилпропаноата растворяли в 150 мл этилацетата и добавляли 100 мг палладия на угле (10%). Реакционную смесь перемешивали при Ткомн. в течение ночи в атмосфере водорода при нормальном давлении. Потом эту смесь фильтровали через кизельгур с отсасыванием, остаток тщательно промывали этилацетатом, а объединенный фильтрат упаривали. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 10:1→7:1). Получили 1,49 г (72,7% от теор.) целевого соединения.
ЖХ-МС (Метод 4): Rt=1,46 мин; m/z=284/286 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 7,05 (1Н, д), 6,57 (1Н, д), 6,32 (1Н, дд), 5,20 (2Н, с), 2,60 (2Н, с), 1,38 (9Н, с), 1,05 (6Н, с).
Пример 209А
N,N-дибензил-5-бром-2-хлоранилин

В атмосфере аргона 9,69 г (242,16 ммоль, 60%-ной суспензии в минеральном масле) гидрида натрия суспендировали в 100 мл ТГФ и охлаждали до 0°С. После этого медленно прикалывали 20,0 г (96,86 ммоль) 5-бром-2-хлоранилина, растворенного в 50 мл ТГФ, и смесь перемешивали 30 мин при 0°С. После этого к реакционной смеси медленно прибавляли 39,76 г (232,47 ммоль) бензилбромида, растворенного в 150 мл ТГФ, и потом реакционную смесь нагревали до комнатной температуры. Эту смесь перемешивали в течение ночи при Ткомн. и после этого осторожно выливали в 150 мл ледяной воды. После отделения органической фазы водную фазу еще трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом натрия. После фильтрации растворитель удалялся в вакууме. К полученному сырому продукту добавляли изопропанол, а образовавшиеся кристаллы отфильтровывали с отсасыванием и сушили в высоком вакууме при 40°С. Получили 14 г целевого соединения. Фильтрат упаривали, а полученный остаток очищали при помощи хроматографии на силикагеле (элюент: циклогексан/этилацетат 20:1). Таким образом получили дополнительные 7,57 г целевого соединения (общий выход: 21,57 г, 58% от теор.).
ЖХ-МС (Метод 6): Rt=1,53 мин; m/z=386/388 (М+Н)+.
Аналогично примеру 209А было получено следующее соединение:

Пример 211А
[4-Хлор-3-(дибензиламино)фенил]борная кислота

В атмосфере аргона при -78°С к раствору 15 г (38,79 ммоль) N,N-дибензил-5-бром-2-хлоранилина в 350 мл смеси ТГФ/диэтиловый эфир (1:1) медленно прикалывали 20,2 мл (50,42 ммоль) 2,5 М раствора н-бутиллития в гексане. После того как реакционный раствор дополнительно перемешивали 60 мин при -78°С, к нему медленно прибавляли 14,3 мл (62,1 ммоль) триизопропилбората. Реакционный раствор затем еще перемешивали дополнительно 15 мин при -78°С, потом медленно нагревали до комнатной температуры и в течение ночи перемешивали еще при этой температуре. После этого добавляли 150 мл ледяной воды. После отделения органической фазы водную фазу еще трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом натрия. После фильтрации растворитель удаляли в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 10:1→4:1). Получили 9 г (66% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,21 мин; m/z=352 (М+Н)+.
Пример 212А
Трет-бутилциклобутилиденацетат
В атмосфере аргона при комнатной температуре растворяли 3,0 г (42,8 ммоль) циклобутанона в 160 мл дихлорметана, а затем прибавляли 20,95 г (55,64 ммоль) трет-бутил(трифенил-λ5-фосфанилиден)ацетата и 0,68 г (5,56 ммоль) бензойной кислоты. Реакционную смесь перемешивали в течение ночи при комнатной температуре, а потом упаривали досуха. Остаток перемешивали с 25 мл диэтилового эфира и эту смесь выдерживали 12 ч при 4°С. Выпавший трифенилфосфиноксид отфильтровывали и фильтрат упаривали досуха. Полученный сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 20:1). Получили 9,3 г (99% от теор.) целевого соединения.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): δ,47-5,41 (1Н, м), 3,05-2,95 (2Н, м), 2,82-2,74 (2Н, м), 2,06-1,95 (2Н, м), 1,50 (9Н, с).
ГХ-МС (Метод 1): Rt=3,01 мин; m/z=112 (М-C4H8)+.
Пример 213А
Трет-бутилциклопропилиденацетат
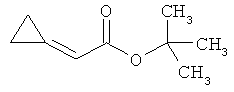
К раствору 9,65 г (55,34 ммоль) [(1-этоксициклопропил)окси](триметил)силана, 25 г (66,41 ммоль) трет-бутил(трифенил-λ5-фосфанилиден)ацетата и 8,11 г (66,41 ммоль) бензойной кислоты в 240 мл ТГФ при комнатной температуре прикалывали 55 мл (55 ммоль) 1 М раствора тетра-н-бутиламмонийфторида в ТГФ. Спустя 1 ч перемешивания реакционную смесь нагревали до 80°С и дополнительно перемешивали 2 ч при этой температуре. Затем с помощью роторного испарителя отгоняли растворитель (200 мбар, температура бани 40°С). Полученный остаток переводили в диэтиловый эфир, охлаждали до 4°С и оставляли стоять 1 ч при этой температуре. Выпавший осадок (трифенилфосфиноксид) отфильтровывали. После этого с помощью роторного испарителя из фильтрата удаляли растворитель. Полученный сырой продукт очищали хроматографически на силикагеле (элюент: циклогексан/этилацетат 20:1). Получили 3,58 г (42% от теор.) целевого соединения.
ГХ-МС (Метод 1): Rt=2,45 мин; m/z=98 (M-C4H8)+.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 1,18-1,26 (м, 2Н), 1,34-1,41 (м, 3Н), 1,44 (с, 9Н), 6,06-6,13 (м, 1Н).
Пример 214А
Этил-(3,3-ди метоксициклобутилиден)ацетат

Раствор 3,93 г (44,59 ммоль) 1,1-диметоксиэтена и 5 г (44,59 ммоль) этил-бута-2,3-диеноата в 50 мл толуола нагревали до кипения с обратным холодильником и перемешивали 24 ч. После охлаждения до комнатной температуры из реакционной смеси удаляли растворитель, а полученный сырой продукт очищали хроматографически на силикагеле (элюент: циклогексан/этилацетат 20:1). Получили 1,9 г (21% от теор.) целевого соединения в виде бесцветной жидкости, которая использовалась в последующих реакциях без дополнительной характеризации.
Пример 215А
Трет-бутил-{1-[4-хлор-3-(дибензиламино)фенил]циклопропил}ацетат

Получение раствора А: 300 мг (0,69 ммоль) N,N-дибензил-2-хлор-5-йоданилина в атмосфере аргона растворяли в 3 мл ТГФ и охлаждали до -78°С. Затем медленно прикалывали 0,4 мл (0,80 ммоль) 2 М раствора изопропилмагнийхлорида в ТГФ. После этого реакционный раствор медленно нагревали до -40°С и 30 мин дополнительно перемешивали при этой температуре.
Получение раствора В: 6 мг (0,14 ммоль) хлорида лития и 13 мг (0,07 ммоль) хлорида меди(1) в атмосфере аргона при комнатной температуре суспендировали в 12 мл ТГФ, а затем добавляли 84 мкл (0,66 ммоль) хлор(триметил)силана, а также 102 мг (0,66 ммоль) трет-бутилциклопропилиденацетата. После этого раствор еще 1 ч дополнительно перемешивали при Ткомн..
Раствор В охлаждали до -40°С и медленно прикалывали к раствору А. Затем полученную реакционную смесь еще 1 ч дополнительно перемешивали при -40°С. Потом к реакционной смеси добавляли 20 мл охлажденного льдом, полунасыщенного водного раствора хлорида аммония. После разделения фаз водную фазу еще трижды экстрагировали этилацетатом, а объединенные органические фазы сушили над сульфатом магния, фильтровали и упаривали досуха. Полученный сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 20:1). Получили 135 мг (42% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,73 мин; m/z=462/464 (М+Н)+.
Аналогично Примеру 13А были получены следующие соединения:


Пример 218А
Этил-{1-[4-хлор-3-(дибензиламино)фенил]-3-оксоциклобутил}ацетат

770 мг (1,52 ммоль) этил-{1-[4-хлор-3-(дибензиламино)фенил]-3,3-диметоксициклобутил}ацетата растворяли в 10 мл ТГФ, смешивали с 2 мл 1 М соляной кислоты и 1 ч перемешивали при 50°С. Затем реакционный раствор разбавляли 10 мл воды и 10 мл этилацетата. После разделения фаз органическую фазу сушили над сульфатом магния, фильтровали и упаривали досуха с помощью роторного испарителя. Получили 607 мг целевого соединения (87% от теор.).
ЖХ-МС (Метод 6): Rt=1,44 мин; m/z=462/464 (М+Н)+.
Пример 219А
Этил-{1-[4-хлор-3-(дибензиламино)фенил]-3,3-дифторциклобутил}ацетат
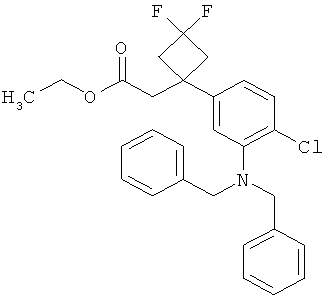
В атмосфере аргона к 2 мл дихлорметана добавляли 0,3 мл (2,27 ммоль) [этил(трифтор-λ4-сульфанил)амино]этана. Реакционный раствор охлаждали до 0°С и медленно прибавляли 175 мг (0,38 ммоль) этил-{1-[4-хлор-3-(дибензиламино)фенил]-3-оксоциклобутил}ацетата в 3 мл дихлорметана. Затем раствор медленно нагревался до комнатной температуры и в течение ночи дополнительно перемешивался при этой температуре. После этого реакционную смесь выливали в 50 мл ледяной воды, а органическую фазу отделяли. Водную фазу еще трижды экстрагировали дихлорметаном. Объединенные органические фазы сушили над сульфатом магния. После фильтрации растворитель удаляли в вакууме, а полученный сырой продукт очищали с помощью препаративной ВЭЖХ (элюент: метанол/вода 8:2). Получили 59 мг целевого соединения (32% от теор.).
ЖХ-МС (Метод 6): Rt=1,53 мин; m/z=484/486 (М+Н)+.
Пример 220А

135 мг (0,29 ммоль) трет-бутил-{1-[4-хлор-3-(дибензиламино)фенил]-циклопропил}ацетата растворяли в 10 мл этилацетата, добавляли 15 мг палладия на угле (10%) и 2 ч перемешивали при Ткомн. в атмосфере водорода при нормальном давлении. После этого реакционную смесь отфильтровывали через целит, остаток дополнительно промывали этилацетатом, а фильтрат упаривали. Получили 73 мг целевого соединения (89% от теор.).
ЖХ-МС (Метод 6): Rt=1,15 мин; m/z=282/284 (М+Н)+.
Аналогично примеру 220А были получены следующие соединения:

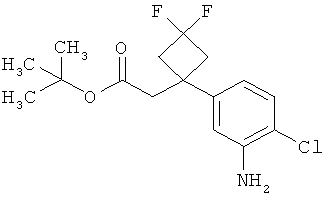
Пример 223А
Трет-бутил-(2Е)-3-(3-амино-4-цианофенил)-2-метилакрилат
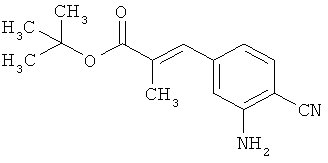
В атмосфере аргона смесь из 2,0 г (10,15 ммоль) 2-амино-4-бромбензонитрила, 2,165 г (2,5 мл, 15,23 ммоль) трет-бутил-2-метилакрилата, 93 мг (0,10 ммоль) трис(дибензилиденацетон)дипалладия, 41 мг (0,20 ммоль) три-трет-бутилфосфина и 2,4 мл (11,17 ммоль) N,N-дициклогексилметиламина в 20 мл диоксана нагревали до 120°С и в течение ночи перемешивали при этой температуре. По результатам контроля реакции (ТСХ, элюент: циклогексан/этилацетат 9:1) еще раз были добавлены 10 мг трис(дибензилиденацетон)дипалладия, 10 мг три-трет-бутилфосфина и 500 мкл трет-бутил-2-метилакрилата и смесь перемешивали дополнительные 4 ч при 120°С. Затем реакционную смесь фильтровали через целит, а фильтрат упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 9:1→4:1). Получили 1,375 г целевого соединения (52% от теор.).
ЖХ-МС (Метод 4): Rt=1,33 мин; m/z=259 (М+Н)+.
Пример 224А
Трет-бутил-3-(3-амино-4-цианофенил)-2-метилпропаноат
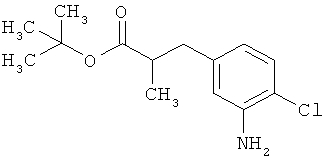
1370 мг (5.3 ммоль) трет-бутил-(2Е)-3-(3-амино-4-цианофенил)-2-метилакрилата растворяли в 30 мл этилацетата, прибавляли 282 мг палладия на угле (10%) и при Ткомн. перемешивали в течение 3 дней в атмосфере водорода при нормальном давлении. Затем реакционную смесь отфильтровывали через целит, остаток на фильтре дополнительно промывали этилацетатом, а объединенный фильтрат упаривали. Сырой продукт очищали с помощью препаративной ВЭЖХ (элюент: ацетонитрил/вода). Получили 870 мг целевого соединения (63% от теор.).
ЖХ-МС (Метод 6): Rt=1,04 мин; m/z=261 (М+Н)+.
Аналогично примеру 54А было получено следующее соединение:

Пример 226А
Этил-(3R)-2-[4-(2,2-дихлор-3-оксоциклобутил)фенил]-4,4,4-трифтор-3-метилбутаноат

3,83 г (13,38 ммоль) этил-(3R)-4,4,4-трифтор-3-метил-2-(4-винилфенил)-бутаноата растворяли в 50 мл диэтилового эфира и друг за другом прибавляли 2,67 г (20,74 ммоль) цинк-медной пары и 6,5 мл 1,2-диметоксиэтана. Затем к полученной суспензии медленно прикалывали 4 мл (36,1 ммоль) трихлорацетилхлорида. Потом реакционный раствор нагревали при кипении с обратным холодильником и перемешивали в течение ночи. После добавления дихлорметана реакционную смесь последовательно промывали водой и насыщенным раствором хлорида натрия. Органическую фазу сушили над сульфатом магния, фильтровали и упаривали в вакууме. Полученный сырой продукт очищали хроматографически на силикагеле (элюент: циклогексан/этилацетат 4:1). Получили 4,57 г (86% от теор.) целевого соединения в виде желтоватого масла, которое использовалось в дальнейших реакциях без дополнительной характеризации.
Пример 227А
Этил-(3R)-4,4,4-трифтор-3-метил-2-[4-(3-оксоциклобутил)фенил]бутаноат

К 4,57 г (11,51 ммоль) этил-(3R)-2-[4-(2,2-дихлор-3-оксоциклобутил)фенил]-4,4,4-трифтор-3-метилбутаноата и 3,76 г (57,5 ммоль) цинковой пыли в 100 мл ТГФ добавляли 100 мл насыщенного водного раствора хлорида аммония, а затем перемешивали 5 ч при 75°С. После охлаждения до комнатной температуры и добавления дихлорметана реакционную смесь промывали водой. После разделения фаз водную фазу дополнительно трижды экстрагировали дихлорметаном. Затем объединенные органические фазы сушили над сульфатом магния, фильтровали и упаривали в вакууме. Получили 1,21 г целевого соединения (32% от теор.).
ГХ-МС (Метод 1): Rt=6,52 мин, m/z=286 (M-C2H2O)+ (диастереомер 1); Rt=6,55 мин, m/z=286 (M-C2H2O)+ (диастереомер 2).
МС (DCI): m/z=346 (M+NH4)+.
Пример 228А
Этил-(3R)-2-[4-(3,3-дифторциклобутил)фенил]-4,4,4-трифтор-3-метилбутаноат

В атмосфере аргона загружали 7,3 мл (5,16 ммоль) 50%-ного раствора 1,1'-[(трифтор-λ4-сульфанил)имино]бис(2-метоксиэтана) (деоксо-фторреагента) в ТГФ, разбавленного 20 мл толуола, охлаждали до 5°С и медленно прибавляли 47 мкл (0,37 ммоль) 1 М раствора комплекса трифторида бора с диэтиловым эфиром. Эту смесь дополнительно перемешивали 2 ч при 5°С. Затем к реакционному раствору медленно прибавляли 1,21 г (3,69 ммоль) этил-(3R)-4,4,4-трифтор-3-метил-2-[4-(3-оксоциклобутил)фенил]бутаноата, растворенного в 20 мл толуола, потом нагревали до 55°С и перемешивали дополнительно 48 ч при этой температуре. После этого реакционную смесь выливали в охлажденную до 0°С смесь, состоящую из 20 мл толуола и 20 мл 2 М водного раствора гидроксида натрия. Органическую фазу отделяли, а водную фазу еще трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом натрия. После фильтрации растворитель удаляли в вакууме. Сырой продукт очищали хроматографически на силикагеле (элюент: циклогексан/этилацетат 10:1). Выделили 558 мг (43% от теор.) целевого соединения в виде желтоватой жидкости.
ГХ-МС (Метод 1): Rt=5,40 мин, m/z=350 (M)+ (диастереомер 1); Rt=5,44 мин, m/z=350 (M)+ (диастереомер 2).
MC (DCI): m/z=368 (М+NH4)+.
Пример 229А
Этил-(3R)-2-[4-(2,2-дифторциклопропил)фенил]-4,4,4-трифтор-3-метилбутаноат

1,58 г (5,52 ммоль) этил-(3R)-4,4,4-трифтор-3-метил-2-(4-винилфенил)бутаноата, 23 мг (0,55 ммоль) фторида натрия и 24 мг (0,11 ммоль) 2,6-дитрел7-бутил-4-метилфенола нагревали до 110°С и перемешивали 5 мин. Затем медленно прикалывали 1,9 мл (9,38 ммоль) триметилсилилдифтор(фторсульфонил)ацетата и эту смесь дополнительно перемешивали 60 мин при 110°С (осторожно: выделение газа спустя примерно 30 мин). После охлаждения до комнатной температуры и добавления этилацетата и насыщенного водного раствора гидрокарбоната натрия органическую фазу отделяли, сушили над сульфатом магния, фильтровали и упаривали досуха. Сырой продукт очищали хроматографически на силикагеле (элюент: циклогексан/дихлорметан 4:1). Получили 1,5 г целевого соединения (81% от теор.).
ГХ-МС (Метод 1): Rt=4,99 мин, m/z=336 (М)+ (диастереомер 1); Rt=5,01 мин, m/z=336 (М)+ (диастереомер 2).
MC (DCI): m/z=354 (M+NH4)+.
Аналогично примеру 70А были получены соединения, приведенные в следующей таблице:
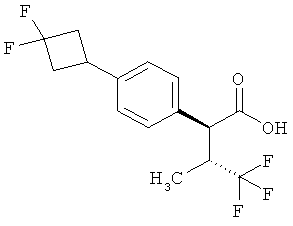
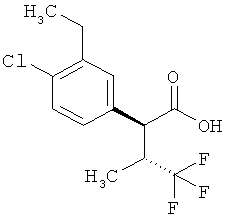

Аналогично примеру 82А были получены соединения, приведенные в следующей таблице:


Пример 235А
Трет-бутил-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-2,2-диметилпропаноат

400 мг (1,50 ммоль) (2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты растворяли в 24 мл дихлорметана, добавляли 320 мг (2,40 ммоль) 1-хлор-N,N,2-триметилпроп-1-ен-1-амина и 30 мин перемешивали при комнатной температуре. Затем к этому добавляли 364 мкл (4,5 ммоль) пиридина, а также 510 мг (1,80 ммоль) трет-бутил-3-(3-амино-4-хлорфенил)-2,2-диметилпропаноата и смесь дополнительно перемешивали 2 ч при комнатной температуре. После этого реакционную смесь упаривали в вакууме, а полученный сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 20:1). Получили 462 мг целевого соединения (58% от теор.).
ЖХ-МС (Метод 6): Rt=1,53 мин; m/z=530/532 (М-Н)-.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 1,02 (с, 3Н), 1,05 (с, 3Н), 1,26 (с, 9Н), 2,65-2,78 (м, 2Н), 3,27-3,44 (м, 1Н, частично перекрывается сигналом H2O), 4,10 (д, 1Н), 6,96 (дд, 1Н), 7,31 (д, 1Н), 7,35 (д, 1Н), 7,41-7,51 (м, 4Н), 9,83 (с, 1Н).
Аналогичным образом были получены соединения, приведенные в следующей таблице:





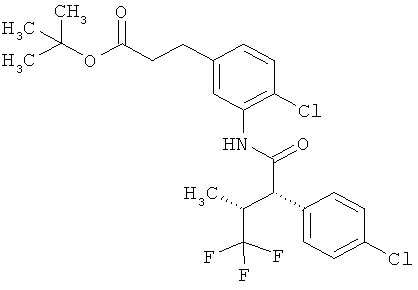


Аналогично примеру 89А были получены соединения, приведенные в следующей таблице:

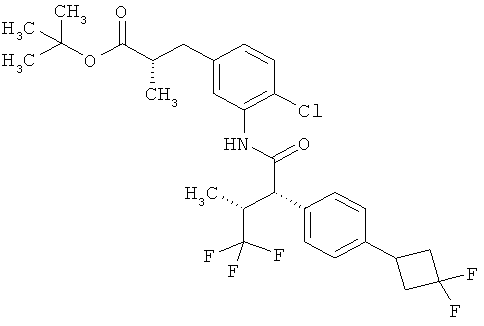
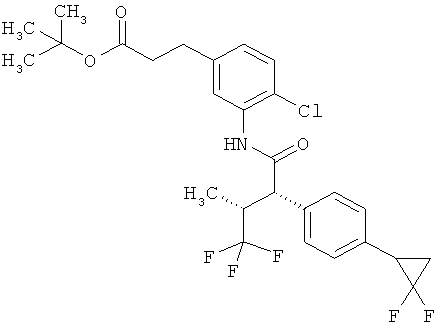
Пример 247А
Трет-бутил-3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-2-метилфенил)пропаноат

Смесь 100 мг (0,38 ммоль) (2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутановой кислоты, 88 мг (0,38 ммоль) трет-бутил-3-(3-амино-2-метилфенил)пропаноата, 213 мг (0,56 ммоль) гексафторфосфата 2-(1Н-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (ГАТУ) и 1 мл пиридина в 4 мл ДМФА перемешивали в течение ночи при комнатной температуре. По окончании взаимодействия эта реакционная смесь непосредственно разделялась на свои компоненты без дополнительной обработки с помощью препаративной ВЭЖХ (элюент: ацетонитрил/вода). Получили 151 мг (83% от теор.) целевого соединения в виде бесцветного масла.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 9,68 (1Н, с), 7,46 (4Н, с), 7,09-6,93 (3Н, м), 3,94 (1Н, д), 3,43-3,28 (1Н, м, частично перекрывается сигналом H2O), 2,78 (2Н, т), 2,41 (2Н, т), 1,91 (ЗН, с), 1,35 (9Н, с), 0,80 (3Н, д).
ЖХ-МС (Метод 4): Rt=1,57 мин; m/z=482 (М-Н)-.
Аналогичным образом было получено следующее соединение:
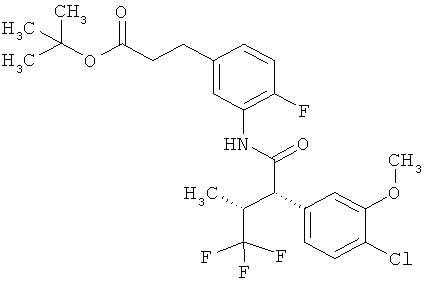
Пример 249А
Диэтил-[2-(4-хлорфенил)пропан-2-ил]малонат

В атмосфере аргона к 254 мг (10,45 ммоль) магниевой стружки в 5 мл диэтилового эфира медленно прибавляли 1 г (5,23 ммоль) 1-бром-4-хлорбензола в 2,5 мл диэтилового эфира. После начала реакции к реакционной смеси добавляли дополнительные 1 г (5,23 ммоль) 1-бром-4-хлорбензола в 2,5 мл диэтилового эфира. Эту реакционную смесь дополнительно перемешивали 30 мин при комнатной температуре, добавляли 103 мг (1,05 ммоль) хлорида меди(1), а потом охлаждали до -10°С. Затем к этому медленно прикалывали 2,09 г (10,45 ммоль) диэтилпропан-2-илиденмалоната. Далее реакционную смесь нагревали до кипения с обратным холодильником и дополнительно перемешивали 3 ч при этой температуре. После этого очень медленно добавляли 20 мл ледяной 1 М соляной кислоты. После разделения фаз водную фазу еще трижды экстрагировали диэтиловым эфиром. Объединенные органические фазы сушили над сульфатом магния и потом упаривали досуха. Сырой продукт очищали с помощью препаративной ВЭЖХ (элюент: метанол/вода 70:30). Таким образом получили 800 мг целевого соединения (25% от теор.).
МС (DCI): m/z=330 (M+NH4)+.
ГХ-МС (Метод 1): Rt=6.19 мин; m/z=312 (M)+.
Пример 250А
Этил-3-(4-хлорфенил)-3-метилбутаноат
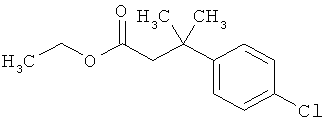
Раствор 796 мг (2,55 ммоль) диэтил-[2-(4-хлорфенил)пропан-2-ил]малоната, 216 мг (5,10 ммоль) хлорида лития и 46 мкл (2,55 ммоль) воды в 5 мл ДМСО нагревали до кипения с обратным холодильником и перемешивали 4 ч при этой температуре. После охлаждения до комнатной температуры к реакционной смеси добавляли 20 мл диэтилового эфира и 20 мл воды. После разделения фаз органическую фазу еще трижды промывали водой, и сушили эту органическую фазу над сульфатом магния, а затем упаривали досуха. Сырой продукт очищали с помощью препаративной ВЭЖХ (элюент: метанол/вода 70:30). Получили 276 мг целевого соединения (45% от теор.).
МС (DCI): m/z=258 (M+NH4)+.
ГХ-МС (Метод 1): Rt=4.99 мин; m/z=240/242 (M)+.
Пример 251А
Этил-3-(4-хлор-3-нитрофенил)-3-метилбутаноат

276 мг (1,45 ммоль) этил-3-(4-хлорфенил)-3-метилбутаноата растворяли в 10 мл дихлорметана и охлаждали до 0°С. Затем порциями добавляли 278 мг (1,38 ммоль) тетрафторбората нитрония и эту смесь перемешивали 4 ч при температуре между 0°С и 10°С. После этого добавляли 10 мл воды, а также 10 мл дихлорметана и разделяли фазы. Органическую фазу сушили над сульфатом магния и упаривали досуха. Остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/ этилацетат 10:1). Получили 223 мг целевого соединения (68% от теор.).
МС (DCI): m/z=303 (M+NH4)+.
ГХ-МС (Метод 1): Rt=6,39 мин; m/z=285 (M)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,00 (т, 3Н), 1,38 (с, 6Н), 2,74 (с, 2Н), 3,89 (кв, 2Н), 7,66-7,76 (м, 2Н), 8,03 (д, 1Н).
Пример 252А
Этил-3-(3-амино-4-хлорфенил)-3-метилбутаноат
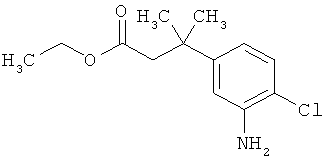
К раствору 213 мг (0,75 ммоль) этил-3-(4-хлор-3-нитрофенил)-3-метилбутаноата в 10 мл этилацетата прибавляли 40 мг палладия на угле (10%). Реакционную смесь гидрировали в течение ночи при Ткомн. с давлением водорода 1 бар. Затем фильтровали через целит, а фильтрат упаривали. Получили 166 мг (87% от теор.) целевого соединения в виде желтоватого масла.
ЖХ-МС (Метод 6): Rt=1,05 мин; m/z=256/258 (М+Н)+.
Аналогично примеру 235А было получено следующее соединение:

Примеры исполнения:
Общая методика 2: Расщепление сложных трет-бутиловых эфиров до соответствующих карбоновых кислот с помощью трифторуксусной кислоты
К раствору соответствующего сложного трет-бутилового эфира в дихлорметане (концентрация примерно от 0,1 до 2,0 моль/л; дополнительно при желании с каплей воды) при температуре от 0°С до Ткомн. по каплям добавляется трифторуксусная кислота (ТФК), пока не достигается соотношение дихлорметан/ТФК примерно от 2:1 до 1:2 (об/об). Смесь перемешивается 1-24 ч при Ткомн., при необходимости эта смесь нагревается до 40°С, пока не достигается полное превращение. После этого реакционная смесь упаривается в вакууме. Сырой продукт может очищаться с помощью хроматографии на силикагеле (элюирование смесями дихлорметан/этилацетат или циклогексан/этилацетат, при необходимости с добавлением незначительных количеств уксусной кислоты, или смесями дихлорметан/метанол), при помощи кристаллизации из ацетонитрила или смесей вода/ацетонитрил, или путем препаративной ОФ ВЭЖХ (элюент: градиент ацетонитрил/вода).
Следующие примеры были получены в соответствии с общей методикой 2:
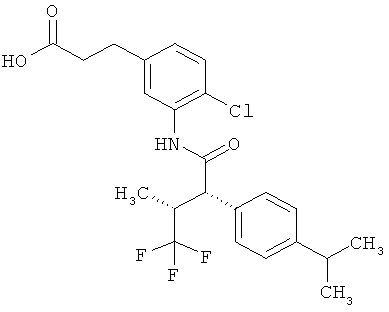

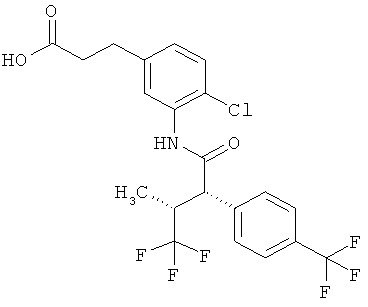

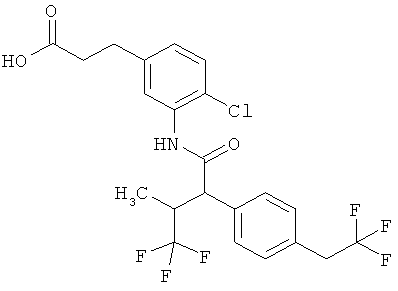
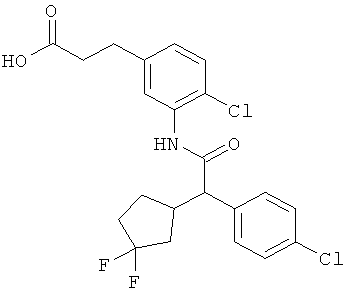
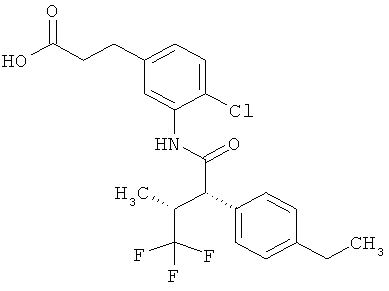




Пример 12
(+)-3-(4-Фтор-3-{[(28,3/?)-4,4,4-трифтор-3-метил-2-(4-винилфенил)-бутаноил]амино}фенил)пропановая кислота

283 мг (0,590 ммоль) сложного трет-бутилового эфира (+)-3-(4-фтор-3-{[(2S,3R)-4,4,4-трифтор-3-метил-2-(4-винилфенил)бутаноил]амино}фенил)-пропановой кислоты растворяли в 5,9 мл 4Н раствора хлороводорода в диоксане и 24 ч перемешивали при Ткомн.. Затем в вакууме удаляли летучие компоненты. Остаток очищали с помощью проводимой дважды препаративной ОФ ВЭЖХ (элюент: градиент ацетонитрил/вода). Получили 48 мг (19,2% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,14 мин; m/z=424 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 2,63-2,82 (м, 4Н), 3,58-3,66 (м, 1Н), 4,08 (д, 1Н), 5,27 (д, 1Н), 5,83 (д, 1Н), 6,72 (дд, 1Н), 6,89-7,03 (м, 1Н), 7,12 (дд, 1Н), 7,41 (д, 2Н), 7,47 (д, 2Н), 7,65 (дд, 1Н), 10,00 (с, 1Н), 12,12 (уш. с, 1Н).
[α]D20=+149,5°, с=0,310, хлороформ.
Пример 13
(+)-3-(4-Хлор-3-{[(2S,3R)-4,4,4-трифтор-3-метил-2-(4-винилфенил)-бутаноил]амино}фенил)пропановая кислота

249,0 мг (0,502 ммоль) сложного трет-бутилового эфира (+)-3-(4-хлор-3-{[(2S,3R)-4,4,4-трифтор-3-метил-2-(4-винилфенил)бутаноил]амино}фенил)-пропановой кислоты растворяли в 3,8 мл 4Н раствора хлороводорода в диоксане и 24 ч перемешивали при Ткомн.. Затем реакционную массу замораживали (-78°С), а потом подвергали лиофильной сушке в высоком вакууме. Остаток очищали с помощью препаративной ОФ ВЭЖХ (элюент: градиент ацетонитрил/вода). Получили 167,4 мг (75,8% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,16 мин; m/z=440 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 2,48 (т, 2Н), 2,76 (т, 2Н), 3,35-3,42 (м, 1Н), 4,09 (д, 1Н), 5,27 (д, 1Н), 5,84 (д, 1Н), 6,73 (дд, 1Н), 7,04 (дд, 1Н), 7,34 (д, 1Н), 7,39-7,52 (м, 5Н), 9,79 (с, 1Н), 12,14 (уш. с, 1Н).
[α]D20=+88,8°, с=0,325, хлороформ.
Пример 14
(+)-3-[4-Хлор-3-({(2S,3R)-4,4,4-трифтор-2-[4-(1-фторвинил)фенил]-3-метилбутаноил}амино)фенил]пропановая кислота

К охлажденному до 0°С раствору 212 мг (0,449 ммоль) сложного метилового эфира (+)-3-[4-хлор-3-({(2S,3R)-4,4,4-трифтор-2-[4-(1-фторвинил)фенил]-3-метилбутаноил}амино)фенил]пропановой кислоты в смеси из соответственно по 1,0 мл каждого, метанола, ТГФ и воды добавляли 16,1 мг (0,674 ммоль) гидроксида лития. Эту смесь затем нагревали до Ткомн. и 3 ч перемешивали при Ткомн., потом разбавляли водой и с помощью 1 н. соляной кислоты устанавливали кислую среду (pH примерно 2). Трижды экстрагировали этилацетатом. Органические фазы объединяли и упаривали в вакууме. Сырой продукт сначала предварительно очищали с помощью ОФ ВЭЖХ (элюент: градиент ацетонитрил/вода). Образовавшийся в ходе основного гидролиза 2R-диастереомер затем отделялся с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel ChiralpakAD-H, 5 мкм, 250 мм×20 мм; объем впрыска: 0,25 мл; температура: 35°С; элюент: 90% изогексана/10% этанола, расход: 15 мл/мин; детектирование: 220 нм]. Таким образом было получено 74,0 мг (36,0% от теор.) целевого соединения.
ЖХ-МС (Метод 4): Rt=1,33 мин; m/z=458 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,81 (д, 3Н), 2,47 (т, 2Н), 2,76 (т, 2Н), 3,35-3,43 (м, 1Н), 4,15 (д, 1Н), 4,95 (дд, 1Н), 5,39 (дд, 1Н), 7,04 (дд, 1Н), 7,26-7,44 (м, 2Н), 7,44-7,59 (м, 2Н), 7,59-7,68 (м, 2Н),9,82 (с, 1Н), 12,11 (уш. с,1Н).
[α]D20=+69,2°, с=0,405, хлороформ.
Пример 15
(+)-3-(4-Хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)пропановая кислота
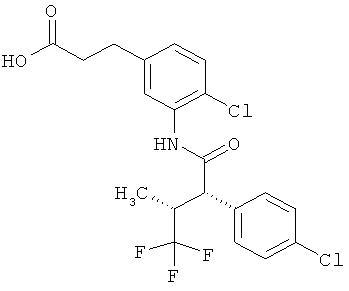
30,13 г (59,74 ммоль) трет-бутил-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)пропаноата растворяли в 1000 мл дихлорметана и при Ткомн. прибавляли 92 мл трифторуксусной кислоты. Реакционную смесь перемешивали 3,5 ч при Ткомн.. Потом добавляли дихлорметан и воду. Органическую фазу отделяли, сушили с сульфатом магния и упаривали в вакууме. Остаток тщательно сушили в высоком вакууме. Получили 26,31 г (98,3% от теор.) целевого соединения.
ЖХ-МС (Метод 7): Rt=2,51 мин; m/z=446/448 (М-Н)-.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 12,14 (1Н, с), 9,83 (1Н, с), 7,50-7,43 (4Н, м), 7,39 (1Н, д), 7,35 (1Н, д), 7,05 (1Н, дд), 4,12 (1Н, д), 3,43-3,28 (1Н, м), 2,76 (2Н, т), 2,48 (2Н, т), 0,80 (ЗН, д).
[α]D20=+100,1°, с=0,42, метанол.
Пример 16
(+)-(2R)-3-(4-хлор-3-[[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-2-метилпропановая кислота
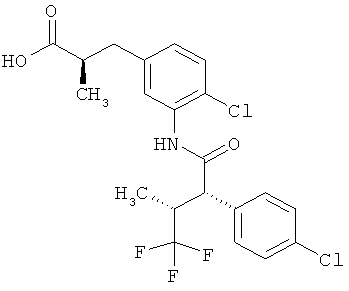
990 мг (2,02 ммоль) этил-(2R)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-2-метилпропаноата растворяли в 5,7 мл уксусной кислоты и добавляли 2,7 мл концентрированной соляной кислоты. Смесь перемешивали 1 ч при 100°С. После охлаждения реакционную массу упаривали в вакууме. Остаток переводили в этилацетат и несколько раз промывали водой с добавлением нескольких капель насыщенного раствора гидрокарбоната натрия. Органическую фазу сушили над сульфатом магния и упаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (элюент: сначала дихлорметан, потом дихлорметан/этилацетат 10:1). Получили 652 мг (69,9% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,21 мин; m/z=462 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 1,02 (д, 3Н), 2,55-2,62 (м, 2Н), 2,78-2,88 (м, 1Н), 3,35-3,43 (м, 1Н), 4,12 (д, 1Н), 7,01 (дд, 1Н), 7,32-7,39 (м, 2Н), 7,42-7,50 (м, 4Н), 9,83 (с, 1Н), 12,16 (уш. с, 1Н).
[α]D20=+60,56°, с=0,530, хлороформ.
Пример 17
(+)-(2S)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-2-метилпропановая кислота
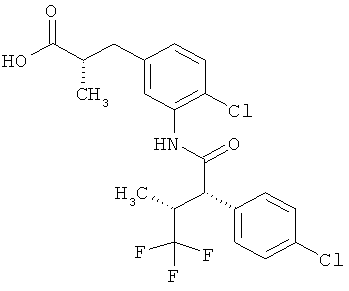
Метод А:
Смесь из 2,45 г (5,0 ммоль) этил-(2S)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-2-метилпропаноата, 6,0 мл уксусной кислоты и 20 мл 20%-ной водной серной кислоты перемешивали 7 ч при кипении с обратным холодильником. После охлаждения реакционную смесь выливали в воду. Водную фазу трижды экстрагировали этилацетатом, и объединенные органические фазы упаривали в вакууме. Остаток снова переводили в этилацетат и несколько раз промывали водой с добавлением нескольких капель насыщенного раствора гидрокарбоната натрия. Органическую фазу сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 10:1→4:1). Получили 1,88 г (81,4% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,22 мин; m/z=462 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 1,02 (д, 3Н), 2,55-2,61 (м, 2Н), 2,78-2,88 (м, 1Н), 3,35-3,43 (м, 1Н), 4,12 (д, 1Н), 7,01 (дд, 1Н), 7,32-7,39 (м, 2Н), 7,43-7,50 (м, 4Н), 9,83 (с, 1Н), 12,16 (уш. с, 1Н).
[α]D20=+101,2°, с=0,590, хлороформ.
Метод В:
Смесь из 12,99 г (26,49 ммоль) (+)-этил-(2S)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-2-метилпропаноата, 60 мл уксусной кислоты и 60 мл 30%-ной водной серной кислоты перемешивали 3 ч при кипении с обратным холодильником (температура бани 140°С). После охлаждения реакционную смесь выливали в воду. Водную фазу трижды экстрагировали этилацетатом, а объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над сульфатом натрия и упаривали в вакууме. Остаток в течение ночи сушили в высоком вакууме. Полученный таким образом сырой продукт перемешивали с 90 мл диизопропилового эфира сначала 1 ч при 50°С, а потом 4 ч при Ткомн.. После фильтрации твердое вещество сушили в высоком вакууме. Получили 7,84 г (64% от теор.) целевого соединения (фракция 1). Из фильтрата после упаривания и повторной обработки 30 мл диизопропилового эфира выделяли дополнительную порцию. После сушки в высоком вакууме получили 1,65 г (13,5% от теор.) слегка загрязненного целевого соединения (фракция 2).
ЖХ-МС (Метод 4): Rt=1,39 мин; m/z=461/463 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 1,02 (д, 3Н), 2,55-2,61 (м, 2Н), 2,77-2,88 (м, 1Н), 3,34-3,43 (м, 1Н), 4,12 (д, 1Н), 7,01 (дд, 1Н), 7,31-7,39 (м, 2Н), 7,41-7,51 (м, 4Н), 9,83 (с, 1Н), 12,15 (с, 1Н).
[α]D20=+127,6°, с=0,575, хлороформ. Пример 18
(+)-[1-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)циклопропил]уксусная кислота

106 мг (0,23 ммоль) метил-[1-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)циклопропил]ацетата растворяли в 4 мл ледяной уксусной кислоты и 2 мл концентрированной соляной кислоты и перемешивали 1 ч при 100°С. Затем реакционную смесь разбавляли 10 мл воды, а потом этот водный раствор трижды экстрагировали соответственно порциями по 10 мл этилацетата. Объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме. Полученный сырой продукт очищали с помощью препаративной ОФ ВЭЖХ (элюент: градиент ацетонитрил/вода). Получили 64 мг целевого соединения (0,14 ммоль, 89% от теор.).
ЖХ-МС (Метод 4): Rt=1,34 мин; m/z=458 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 10,03 (1Н, с), 7,69-7,59 (1Н, м), 7,52-7,34 (4Н, м), 7,11-7,00 (2Н, м), 4,11 (1Н, д), 3,43-3,27 (1Н, м), 2,36 (2Н, с), 0,88-0,82 (2Н, м), 0,78 (3Н, д), 0,72-0,64 (2Н, м).
[α]D20=+108,7°, с=0,36, метанол.
Общая методика 3: Расщепление сложных этиловых или метиловых эфиров до соответствующих карбоновых кислот с помощью смеси соляной кислоты или серной кислоты с уксусной кислотой
Раствор соответствующего сложного этилового или метилового эфира в смеси из уксусной кислоты и концентрированной соляной кислоты или из уксусной кислоты и 10%-ной или полуконцентрированной серной кислоты перемешивается при температуре от 80°С до 130°С (при необходимости при кипении с обратным холодильником) в течение времени от 30 мин до 12 ч. После охлаждения эту реакционную смесь или непосредственно упаривают в вакууме или выливают в воду, водную фазу экстрагируют этилацетатом или дихлорметаном, а объединенные органические фазы упаривают в вакууме. Сырой продукт может очищаться с помощью хроматографии на силикагеле (элюирование смесями дихлорметан/этилацетат или циклогексан/этилацетат, при необходимости с добавлением незначительных количеств уксусной кислоты, или смесями дихлорметан/метанол), с помощью кристаллизации из ацетонитрила или смесей вода/ацетонитрил или с помощью препаративной ОФ ВЭЖХ (элюент: градиент ацетонитрил/вода).
Следующие примеры были получены в соответствии с общей методикой 3:
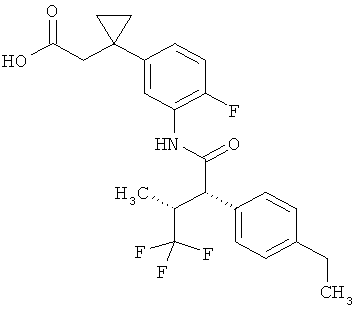


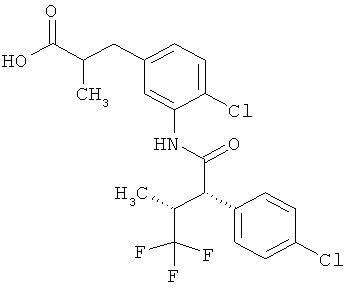
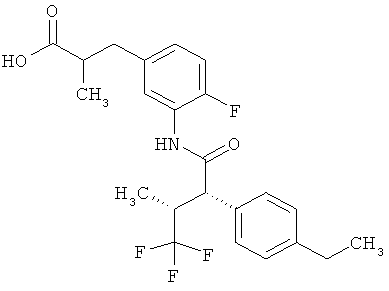



Пример 27
(+)-[3-(4-Хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)оксетан-3-ил]уксусная кислота

Раствор 120 мг (0,21 ммоль) бензил[3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)оксетан-3-ил]ацетата в 15 мл этилацетата смешивали с 25 мг палладия на угле (10%). В атмосфере водорода гидрировали при нормальном давлении в течение 2 ч. После этого реакционную смесь фильтровали через тонсил, остаток на фильтре дополнительно промывали этилацетатом, а объединенный фильтрат упаривали на роторном испарителе. Получили 98 мг (0,2 ммоль, 97% от теор.) целевого соединения.
ЖХ-МС (Метод 4): Rt=1,28 мин; m/z=488/490 (M-H)-.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 12,76-11,52 (1Н, уш. с), 9,88 (1Н, с), 7,52 (1Н, д), 7,50-7,39 (5Н, м), 7,10 (1Н, дд), 4,79-4,71 (2Н, м), 4,71-4,64 (2Н, м), 4,14 (1Н, д), 3,42-3,28 (1Н, м), 3,03 (2Н, с), 0,80 (3Н, д).
[α]D20=+88,4°, с=0,355, метанол.
Пример 28
[1-(4-Хлор-3-{[(3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)циклобутил]уксусная кислота
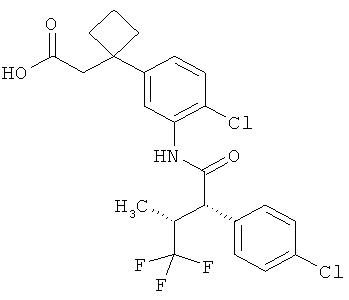
38 мг (0,08 ммоль) метил-[1-(4-хлор-3-{[(3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)циклобутил]ацетата растворяли в 9,5 мл диоксана и прибавляли 0,15 мл 1 н. водного раствора гидроксида натрия. Реакционную массу перемешивали в течение ночи при 80°С. Потом эту реакционную смесь подкисляли 1 н. соляной кислотой до pH 1 и несколько раз экстрагировали этилацетатом. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над сульфатом натрия и упаривали в вакууме. Сырой продукт очищали с помощью препаративной ВЭЖХ. Получили 22 мг (0,05 ммоль, 60% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,28 мин; m/z=488/490 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 11,88 (1Н, уш. с), 9,95 (0,5Н, с), 9,81 (0,5Н, с), 7,54-7,31 (6Н, м), 7,06-6,96 (1Н, м), 4,14 (1Н, д), 3,43-3,27 (0,5Н, м), 3,27-3,14 (0.5Н, м), 2,70 (1Н, с), 2,69 (1Н, с), 2,34-2,17 (4Н, м), 2,10-1,95 (1Н, м), 1,81-1,66 (1Н, м), 1,25 (1,5Н, д), 0,80 (1,5Н, д).
Пример 29
(+)-(2R)-2-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}бензил)бутановая кислота

К 1,96 г (3,89 ммоль) сложного этилового эфира (+)-(2R)-2-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}бензил)-бутановой кислоты прибавляли 15,2 мл уксусной кислоты и 7,6 мл концентрированной соляной кислоты. Реакционную смесь перемешивали 5 ч при кипении с обратным холодильником (температура бани 140°С). После охлаждения добавляли воду. Несколько раз экстрагировали дихлорметаном, а объединенные органические фазы промывали насыщенным раствором хлорида натрия, сушили над сульфатом натрия и упаривали в вакууме.
После хроматографирования остатка на силикагеле (элюент: циклогексан/этилацетат 10:1→2:1) получили 1,46 г (78,6% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,25 мин; m/z=476 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 0,82-0,89 (м, 3Н), 1,42-1,54 (м, 2Н), 2,41 (т, 1Н), 2,64 (дд, 1Н), 2,75 (дд, 1Н), 4,12 (д, 1Н), 7,00 (дд, 1Н), 7,31-7,39 (м, 1Н), 7,42-7,50 (м, 3Н), 9,82 (с, 1Н), 12,16 (уш. с, 1Н).
[α]D20=+92,7°, с=0,380, метанол. По аналогичной методике было получено следующее соединение:
Пример 30
(+)-(2S)-2-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}бензил)бутановая кислота
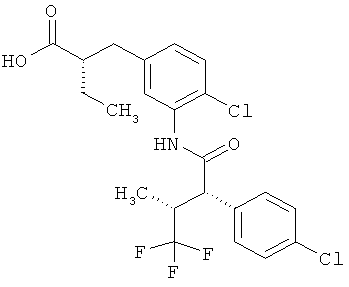
325 ЖХ-МС (Метод 5): Rt=2,66 мин; m/z=476 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 0,85 (т, 3Н), 1,43-1,52 (м, 2Н), 2,26-2,47 (м, 1Н), 2,59-2,69 (м, 1Н), 2,70-2,82 (м, 1Н), 3,34-3,44 (м, 1Н), 4,12 (д, 1Н), 7,00 (дд, 1Н), 7,30-7,39 (м, 2Н), 7,40-7,52 (м, 4Н), 9,82 (с, 1Н), 12,13 (уш. с, 1Н).
[α]D20=+143,1°, с=0,380, хлороформ.
Пример 31 и Пример 32
3-[4-Хлор-3-({4,4,4-трифтор-3-метил-2-[4-(2,2,2-трифторэтил)фенил]-бутаноил}амино)фенил]пропановая кислота (энантиомеры 1 и 2)

120 мг (0,24 ммоль) рацемической 3-[4-хлор-3-({4,4,4-трифтор-3-метил-2-[4-(2,2,2-трифторэтил)фенил]бутаноил}амино)фенил]пропановой кислоты (пример 5) разделяли на энантиомеры с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×20 мм; элюент: изогексан/этанол 85:15 (об/об); расход: 15 мл/мин; УФ-детектирование: 220 нм; температура: 35°С].
Пример 31
(+)-3-[4-Хлор-3-({(2S,3R)-4,4,4-трифтор-3-метил-2-[4-(2,2,2-трифторэтил)-фенил]бутаноил}амино)фенил]пропановая кислота (энантиомер 1)
Выход: 48 мг
Rt=5,75 мин; химическая чистота > 99%; > 99% ее
[колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×4,6 мм; элюент: изогексан/этанол +0,2% ТФК +1% воды) 85:15 (об/об); расход: 1 мл/мин; температура: 35°С; УФ-детектирование: 220 нм].
[α]D20=+91,8°, с=0,405, метанол.
ЖХ-МС (Метод 4): Rt=1,33 мин; m/z=496 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6, δ/м.д.): 12,24-12,02 (1Н, уш. с), 9,80 (1Н, с), 7,46 (2Н, д), 7,43-7,39 (1Н, м), 7,35 (3Н, т), 7,04 (1Н, дд), 4,11 (1Н, д), 3,64 (2Н, кв), 3,44-3,27 (1Н, м), 2,76 (2Н, т), 2,48 (2Н, т), 0,79 (3Н, д).
Пример 32
(-)-3-[4-Хлор-3-({(2R,3S)-4,4,4-трифтор-3-метил-2-[4-(2,2,2-трифторэтил)-фенил]бутаноил}амино)фенил]пропановая кислота {энантиомер 2)

Выход:52 мг
Rt=6,85 мин; химическая чистота > 97,4%; > 99% ее
[колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×4,6 мм; элюент: изогексан/(этанол + 0,2% ТФК + 1% воды) 85:15 (об/об); расход: 1 мл/мин; температура: 35°С; УФ-детектирование: 220 нм].
[α]D20=-94,3°, с=0,40, метанол.
ЖХ-МС (Метод 4): Rt=1,33 мин; m/z=496 (M+H)+.
Примеры 33-36
3-(4-Хлор-3-{[(4-хлорфенил)-(3,3-дифторциклопентил)ацетил]амино}-фенил)пропановая кислота (изомеры 1-4)
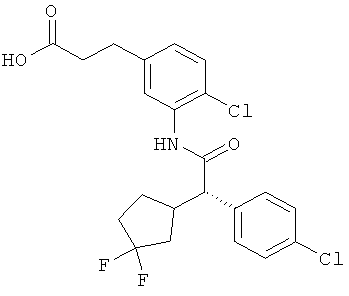
44 мг (0,096 ммоль) смеси диастереомеров 3-(4-хлор-3-{[(4-хлорфенил)-(3,3-дифторциклопентил)ацетил]амино}фенил)пропановой кислоты (Пример 6) дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralcel OJ-H, 5 мкм, 250 мм×20 мм; элюент: изогексан/этанол 70:30 (об/об); расход: 15 мл/мин; УФ-детектирование: 220 нм; температура: 35°С].
При этом были получены четыре различные фракции, которые состояли соответственно из смеси двух изомеров. Эти фракции были разделены с помощью еще одной препаративной ВЭЖХ на хиральной фазе на отдельные изомеры [Фракции 1 и 2: колонка: Daicel Chiralpak AS-H, 5 мкм, 250 мм×20 мм; элюент: изогексан/изопропанол 75:25 (об/об); расход: 15 мл/мин; УФ-детектирование: 220 нм; температура: 35°С. Фракции 3 и 4: колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×20 мм; элюент: изогексан/этанол 80:20 (об/об); расход: 15 мл/мин; УФ-детектирование: 220 нм; температура: 35°С]:
Пример 33 (изомер 1):
Выход:8 мг
Rt=6,49 мин; химическая чистота > 99%
[колонка: Daicel Chiralpak AS-H, 5 мкм, 250 мм×4,6 мм; элюент: изогексан/изопропанол 75:25 (об/об); расход: 1 мл/мин; УФ-детектирование: 220 нм; температура: 35°С].
Пример 34 (изомер 2):
Выход: 11 мг
Rt=9,08 мин; химическая чистота > 98,5%
[колонка: Daicel Chiralpak AS-H, 5 мкм, 250 мм×4,6 мм; элюент: изогексан/изопропанол 75:25 (об/об); расход: 1 мл/мин; УФ-детектирование: 220 нм; температура: 35°С].
Пример 35 (изомер 3):
Выход: 12 мг Rt=7,19 мин; химическая чистота > 99%
[колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×4,6 мм; элюент: изогексан/этанол 80:20 (об/об); расход: 1 мл/мин; УФ-детектирование: 220 нм; температура: 30°С].
Пример 36 (изомер 4):
Выход: 9 мг
Rt=8,58 мин; химическая чистота > 97,5%
[колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×4,6 мм; элюент: изогексан/этанол 80:20 (об/об); расход: 1 мл/мин; УФ-детектирование: 220 нм; температура: 30°С].
Пример 37
3-(3-{[(3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-2-метилпропановая кислота (смесь диастереомеров)

300 мг (0,633 ммоль) этил-3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-2-метилпропаноата (смесь диастереомеров) растворяли в смеси из соответственно каждого по 1,0 мл метанола, ТГФ и воды и при 0°С прибавляли 265,5 мг (6,33 ммоль) гидроксида лития. Смесь перемешивали сначала 1 ч при 0°С, а затем 1 ч при Ткомн.. После этого раствор разбавляли водой и устанавливали кислую среду с помощью 1 н. соляной кислоты (pH примерно 2). Водную фазу трижды экстрагировали диэтиловым эфиром и один раз этилацетатом. Объединенные органические фазы сушили над сульфатом натрия и упаривали в вакууме. Было получено 294 мг (99,7% от теор.) целевого соединения в виде смеси четырех диастереомеров.
ЖХ-МС (Метод 6): Rt=1,18 мин; m/z=446 (М+Н)+.
Пример 38 и Пример 39
3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-2-метилпропановая кислота (диастереомеры 1 и 2)

Полученную выше смесь диастереомерных 3-(3-{[(3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-2-метилпропановых кислот (Пример 37) дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AS-H, 5 мкм, 250 мм×20 мм; объем впрыска: 0,25 мл; температура: 40°С; элюент: 90% изогексана/10% (этанол + 0,2% ТФК + 1% воды); расход: 15 мл/мин; детектирование: 220 нм]. Исходя из 260 мг смеси диастереомеров, помимо двух других изомеров было получено 52 мг изомера 1 (Пример 38) и 54 мг изомера 2 (Пример 39):
Пример 38 (диастереомер 1Y
(+)-(2S)-3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]-амино}-4-фторфенил)-2-метилпропановая кислота

Изомер 1 с помощью препаративной ОФ ВЭЖХ (элюент: ацетонитрил/вода) еще раз очищали дополнительно. Получили 32 мг.
ЖХ-МС (Метод 6): Rt=1,18 мин; m/z=446 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 1,01 (д, 3Н), 2,51-2,58 (м, 2Н), 2,76-2,86 (м, 1Н), 3,35 (дд, 1Н), 4,11 (д, 1Н), 6,87-7,00 (м, 1Н), 7,12 (дд, 1Н), 7,41-7,49 (м, 4Н), 7,63 (дд, 1Н), 10,04 (с, 1Н), 12,11 (уш. с, 1Н).
[α]D20=+150,4°, с=0,50, хлороформ.
Пример 39 (диастереомер 2):
(+)-(2R)-3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-2-метилпропановая кислота
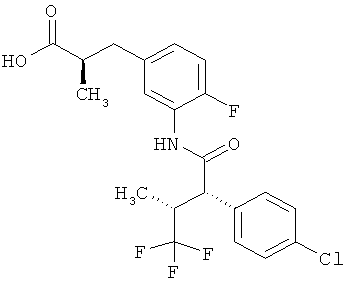
Изомер 2 еще раз дополнительно очищали с помощью препаративной ОФ ВЭЖХ (элюент: ацетонитрил/вода). Получили 21 мг.
ЖХ-МС (Метод 6): Rt=1,18 мин; m/z=446 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 1,02 (д, 3Н), 2,53-2,58 (м, 2Н), 2,77-2,87 (м, 1Н), 3,30-3,41 (м, 1Н), 4,11 (д, 1Н), 6,89-7,00 (м, 1Н), 7,12 (дд, 1Н), 7,41-7,48 (м, 4Н), 7,63 (дд, 1Н), 10,04 (с, 1Н), 12,12 (уш. с, 1Н).
[α]D20=+131,6°, с=0,530, хлороформ.
Пример 40 и Пример 41
3-(4-Хлор-3-{[(2S,3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутаноил]-амино}фенил)-2-метилпропановая кислота (диастереомеры 1 и 2)

Полученную выше смесь диастереомерных 3-(4-хлор-3-{[(2S,3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-2-метилпропановых кислот (Пример 24) дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: хиральная фаза силикагеля на основе селектора поли(N-метакрилоил-1--изолейцин-3-пентиламида), 430 мм×40 мм; объем впрыска: 2,0 мл; температура: 24°С; элюент: 40% изогексана/60% этилацетата; расход: 80 мл/мин; детектирование: 265 нм]. Исходя из 514 мг смеси диастереомеров, получили 178 мг диастереомера 1 (Пример 40) и 218 мг диастереомера 2 (Пример 41):
Пример 40 (диастереомер 1):
(+)-(2R)-3-(4-хлор-3-{[(2S,3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-2-метилпропановая кислота

ЖХ-МС (Метод 6): Rt=1,25 мин; m/z=456 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 0,98-1,05 (м, 3Н), 1,17 (т, 3Н), 2,55- 2,63 (м, 4Н), 2,78-2,88 (м, 1Н), 3,28-3,37 (м, 1Н), 4,06 (д, 1Н), 6,99 (дд, 1Н), 7,20 (д, 2Н), 7,34 (дд, 3Н), 7,41 (д, 1Н), 9,73 (с, 1Н), 12,15 (с, 1Н).
[α]D20=+52°, с=0,500, хлороформ.
Пример 41 (диастереомер 2):
(+)-(2S)-3-(4-хлор-3-{[(2S,3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-2-метилпропановая кислота
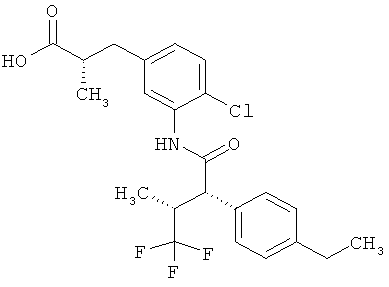
ЖХ-МС (Метод 6): Rt=1,27 мин; m/z=456 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,78 (д, 3Н), 1,02 (д, 3Н), 1,17 (т, 3Н), 2,54-2,64 (м, 4Н), 2,77-2,87 (м, 1Н), 3,28-3,37 (м, 1Н), 4,06 (д, 1Н), 6,99 (дд, 1Н), 7,21 (д, 2Н), 7,34 (дд, 3Н), 7,41 (д, 1Н), 9,74 (с, 1Н), 12,16 (уш.с, 1Н).
[α]D20=+75,0°, с=0,640, хлороформ.
Пример 42 и Пример 43
3-(3-{[(2S,3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-2-метилпропановая кислота (диастереомеры 1 и 2)

Полученную выше смесь диастереомерных 3-(3-{[(2S,3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-2-метилпропановых кислот (Пример 23) дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AS-H, 5 мкм, 250 мм×20 мм; объем впрыска: 0,30 мл; температура: 30°С; элюент: 92% изогексана/8% (этанол + 0,2% ТФК + 1% воды); расход: 15 мл/мин; детектирование: 220 нм]. Исходя из 509 мг смеси диастереомеров, получили 209 мг диастереомера 1 (Пример 42) и 220 мг диастереомера 2 (Пример 43):
Пример 42 (диастереомер 1):
(+)-(2S)-3-(3-{[(2S,3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-2-метилпропановая кислота

ЖХ-МС (Метод 6): Rt=1,22 мин; m/z=440 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,77 (д, 3Н), 1,01 (д, 3Н), 1,17 (т, 3Н), 2,55-2,63 (м, 4Н), 2,76-2,86 (м, 1Н), 3,25-3,39 (м, 1Н), 4,05 (д, 1Н), 6,88-6,98 (м, 1Н), 7,11 (дд, 1Н), 7,17-7,24 (м, 2Н), 7,29-7,38 (м, 2Н), 7,66 (дд, 1Н), 9,97 (с, 1Н), 12,13 (уш.с, 1Н).
[α]D20=+162,1°, с=0,500, хлороформ.
Пример 43 (диастереомер 2):
(+)-(2R)-3-(3-{[(2S,3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-2-метилпропановая кислота
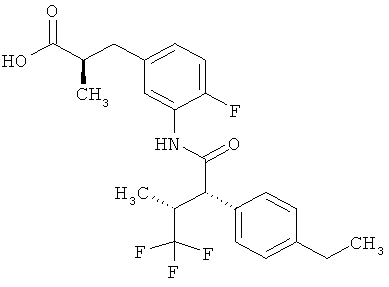
ЖХ-МС (Метод 6): Rt=1,22 мин; m/z=440 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,77 (д, 3Н), 1,01 (д, 3Н), 1,17 (т, 3Н), 2,53-2,64 (м, 4Н), 2,76-2,87 (м, 1Н), 3,26-3,38 (м, 1Н), 4,04 (д, 1Н), 6,87-6,97 (м, 1Н), 7,11 (дд, 1Н), 7,17-7,23 (м, 2Н), 7,28-7,38 (м, 2Н), 7,65 (дд, 1Н), 9,97 (с, 1Н), 12,12(уш.с, 1Н).
[α]D20=+94,0°, с=0,620, хлороформ.
Пример 44 и Пример 45
3-(4-Хлор-3-{[(2S,3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)бутановая кислота (диастереомеры 1 и 2)

Полученную выше смесь диастереомерных 3-(4-хлор-3-{[(2S,3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)бутановых кислот (Пример 7) дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AS-H, 5 мкм, 250 мм×20 мм; объем впрыска: 0,20 мл; температура: 30°С; элюент: 90% изогексана/10% (изопропанол + 0,2% ТФК + 1% воды); расход: 15 мл/мин; детектирование: 220 нм]. Исходя из 210 мг смеси диастереомеров, получили 110 мг диастереомера 1 (Пример 44) и 99 мг диастереомера 2 (Пример 45):
Пример 44 (диастереомер 1):
(+)-(3S)-3-(4-хлор-3-{[(2S,3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)бутановая кислота

ЖХ-МС (Метод 6): Rt=1,26 мин; m/z=456 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 1,13-1,21 (м, 6Н), 2,45 (д, 2Н), 2,59 (кв, 2Н), 3,08 (кв, 1Н), 3,27-3,38 (м, 1Н), 4,07 (д, 1Н), 7,06 (дд, 1Н), 7,21 (д, 2Н), 7,35 (дд, 3Н), 7,46 (д, 1Н), 9,72 (с, 1Н), 12,05 (уш.с, 1Н).
[α]D20=+86,8°, с=0,440, хлороформ. Пример 45 (диастереомер 2):
(+)-(3R)-3-(4-хлор-3-{[(2S,3R)-2-(4-этилфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)бутановая кислота

ЖХ-МС (Метод 6): Rt=1,26 мин; m/z=456 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 1,11-1,22 (м, 6Н), 2,45 (д, 2Н), 2,55-2,63 (м, 2Н), 3,08 (кв, 1Н), 3,28-3,38 (м, 1Н), 4,08 (д, 1Н), 7,06 (дд, 1Н), 7,20 (д, 2Н), 7,35 (дд, 3Н), 7,47 (д, 1Н), 9,72 (с, 1Н), 12,05 (уш.с, 1Н).
[α]D20=+68,0°, с=0,415, хлороформ. Пример 46 и Пример 47
3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-хлорфенил)бутановая кислота (диастереомеры 1 и 2)
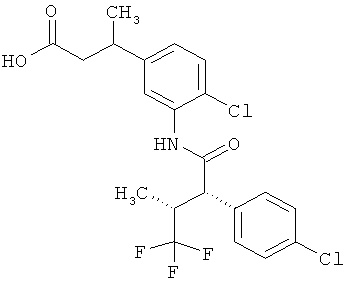
Полученную выше смесь диастереомерных 3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-хлорфенил)бутановых кислот (Пример 8) дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AS-H, 5 мкм, 250 мм×20 мм; объем впрыска: 0,30 мл; температура: 30°С; элюент: 90% изогексана/10% изопропанола; расход: 15 мл/мин; детектирование: 220 нм]. Исходя из 250 мг смеси диастереомеров, получили 116 мг диастереомера 1 (Пример 46) и 113 мг диастереомера 2 (Пример 47):
Пример 46 (диастереомер 1):
(+)-(3S)-3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-хлорфенил)бутановая кислота
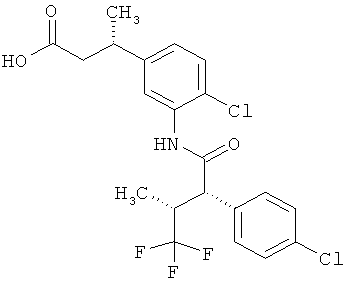
ЖХ-МС (Метод 4): Rt=1,36 мин; m/z=462 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 1,16 (д, 3Н), 2,45 (д, 2Н), 3,03-3,14 (м, 1Н), 3,33-3,42 (м, 1Н), 4,13 (д, 1Н), 7,08 (дд, 1Н), 7,36 (д, 1Н), 7,41 (д, 1Н), 7,43-7,51 (м, 4Н), 9,81 (с, 1Н), 12,05 (с, 1Н).
[α]D20=+88,6°, с=0,435, хлороформ.
Пример 47 (диастереомер 2):
(+)-(3R)-3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-хлорфенил)бутановая кислота
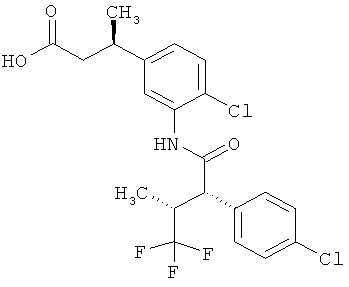
ЖХ-МС (Метод 4): Rt=1,36 мин; m/z=462 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 1,16 (д, 3Н), 2,45 (д, 2Н), 3,09 (кв, 1Н), 3,33-3,42 (м, 1Н), 4,13 (д, 1Н), 7,08 (дд, 1Н), 7,35 (д, 1Н), 7,42 (д, 1Н), 7,43-7,51 (м, 4Н), 9,81 (с, 1Н), 12,05 (с, 1Н).
[α]D20=+57,9°, с=0,365, хлороформ.
Пример 48 и Пример 49
3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)бутановая кислота (диастереомеры 1 и 2)
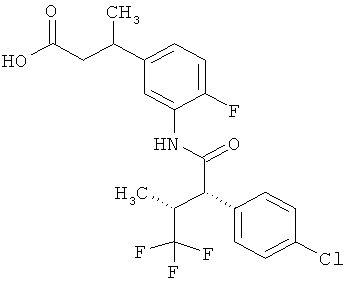
Полученную выше смесь диастереомерных 3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)бутановых кислот (Пример 9) дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AS-H, 5 мкм, 250 мм×20 мм; объем впрыска: 0,25 мл; температура: 30°С; элюент: 85% изогексана/15% изопропанола; расход: 15 мл/мин; детектирование: 220 нм]. Исходя из 295 мг смеси диастереомеров, получили 121 мг диастереомера 1 (Пример 48) и 111 мг диастереомера 2 (Пример 49):
Пример 48 (диастереомер 1):
(+)-(38)-3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)бутановая кислота
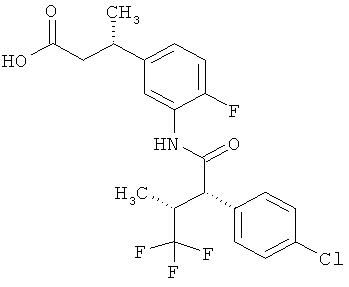
ЖХ-МС (Метод 6): Rt=1,14 мин; m/z=446 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 1,16 (д, 3Н), 2,44 (д, 2Н), 3,02-3,12 (м, 1Н), 3,33-3,42 (м, 1Н), 4,12 (д, 1Н), 7,00-7,04 (м, 1Н), 7,13 (дд, 1Н), 7,43-7,48 (м, 4Н), 7,68 (дд, 1Н), 10,04 (с, 1Н), 12,03 (с, 1Н).
[α]D20=+142,0°, с=0,350, хлороформ.
Пример 49 (диастереомер 2):
(+)-(3R)-3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)бутановая кислота
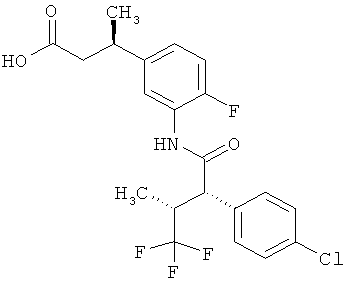
ЖХ-МС (Метод 6): Rt=1,14 мин; m/z=446 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 1,15 (д, 3Н), 2,44 (д, 2Н), 3,08 (кв, 1Н), 3,30-3,42 (м, 1Н), 4,12 (д, 1Н), 6,94-7,06 (м, 1Н), 7,13 (дд, 1Н), 7,40-7,50 (м, 4Н), 7,68 (дд, 1Н), 10,04 (с, 1Н), 12,04 (уш. с, 1Н).
[α]D20=+139,8°, с=0,405, хлороформ.
Общая методика 4: Кислотный гидролиз сложных этиловых эфиров
Соответствующий сложный этиловый эфир растворяется в смеси состава 7:2 из уксусной кислоты и концентрированной соляной кислоты (примерно 10 мл/ммоль субстрата) и нагревается вплоть до полного превращения при 100°С (как правило, в промежутке между 1 ч и 8 ч). Затем реакционная масса охлаждается, выливается в воду и несколько раз экстрагируется дихлорметаном. Объединенные органические фазы трижды промывают насыщенным раствором поваренной соли, сушат над сульфатом магния и упаривают. В случае необходимости этот остаток очищают при помощи флэш-хроматографии или препаративной ВЭЖХ.
В соответствии с общей методикой 4 были получены следующие примеры соединений:
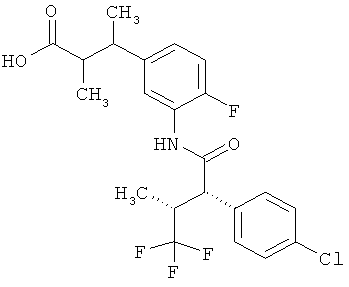


Пример 53 и Пример 54
2-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-транс-циклопропанкарбоновая кислота (диастереомеры 1 и 2)

71 мг смеси диастереомеров 2-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-транс-циклопропанкарбоновой кислоты (Пример 52) растворяли в 2 мл этанола и 2 мл изогексана и дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×200 мм; объем впрыска: 0,25 мл; температура: 30°С; элюент: 15% изопропанола/85% изогексана; расход: 15 мл/мин; детектирование: 220 нм]. Было получено 36 мг диастереомера 1 (Пример 53) и 37 мг диастереомера 2 [Пример 54):
Пример 53 (диастереомер 1):
ЖХ-МС (Метод 6): Rt=1,15 мин; m/z=444 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 1,24 (ддд, 1Н), 1,38 (дт, 1Н), 1,64-1,80 (м, 1Н), 2,35 (ддд, 1Н), 4,12 (д, 1Н), 6,85-7,01 (м, 1Н), 7,13 (дд, 1Н), 7,37-7,56 (м, 4Н), 7,62 (дд, 1Н), 10,06 (с, 1Н).
[α]D20=+291,4°, с=0,48, хлороформ.
Пример 54 (диастереомер 2):
ЖХ-МС (Метод 6): Rt=1,15 мин; m/z=444 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 1,24 (ддд, 1Н), 1,38 (дт, 1Н), 1,64-1,76 (м, 1Н), 2,29-2,40 (м, 2Н), 4,12 (д, 1Н), 6,92 (ддд, 1Н), 7,13 (дд, 1Н), 7,39-7,52 (м, 4Н), 7,62 (дд, 1Н), 10,06 (с, 1Н).
[α]D20=+44,3°, с=0,40, хлороформ.
Пример 55
3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-цианофенил)пропановая кислота

16,5 мг (33 мкмоль) трет-бутил-3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-цианофенил)пропаноата растворяли в 1,1 мл дихлорметана и добавляли 275 мкл трифторуксусной кислоты. Эту реакционную смесь перемешивали 1,5 ч при Ткомн., затем разбавляли 20 мл дихлорметана и упаривали в вакууме. Остаток в течение ночи сушили в высоком вакууме. Получили 14,8 мг (97% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,10 мин; m/z=439 (M+NH4)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,81 (д, 3Н), 2,80-2,94 (м, 2Н), 4,01 (д, 1Н), 7,22 (дд, 1Н), 7,32 (с, 1Н), 7,40-7,52 (м, 4Н), 7,69 (д, 1Н), 10,48 (с, 1Н).
Пример 56
(+/-)-3-(3-{[2-(4-Хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-2-фторфенил)пропановая кислота (диастереомер 1)

270 мг (0,553 ммоль) сложного mpem-бутилового эфира (+/-)-3-(3-{[2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-2-фторфенил)-пропановой кислоты (диастереомер 1, Пример 102А) растворяли в 0,2 мл дихлорметана и при Ткомн. добавляли 0,85 мл трифторуксусной кислоты. Реакционную смесь перемешивали 4 ч при Ткомн., а потом упаривали в вакууме. Остаток очищали с помощью препаративной ОФ ВЭЖХ (смесь ацетонитрил/вода). Получили 188 мг (78,7% от теор.) целевого соединения.
ЖХ-МС (Метод 6): Rt=1,16 мин; m/z=432 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 2,48-2,53 (м, 2Н), 2,82 (т, 2Н), 3,35-3,48 (м, 1Н), 4,13 (д, 1Н), 6,88-7,13 (м, 2Н), 7,37-7,51 (м, 4Н), 7,54-7,76 (м, 1Н), 10,04 (с, 1Н), 12,19 (уш. с, 1Н).
Аналогичным образом было получено следующее соединение:
Пример 57
(+/-)-3-(3-{[2-(4-Хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-2-фторфенил)пропановая кислота {диастереомер 2)

ЖХ-МС (Метод 6): Rt=1,15 мин; m/z=432 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,22 (д, 3Н), 2,52-2,56 (м, 2Н), 2,83 (т, 2Н), 3,22 (дд, 1Н), 4,15 (д, 1Н), 6,98-7,10 (м, 2Н), 7,36-7,43 (м, 2Н), 7,45-7,53 (м, 2Н), 7,62 (тд, 1Н), 10,13 (с, 1Н), 12,19 (с, 1Н).
Следующие примеры были получены в соответствии с Общей методикой 2 (расщепление сложных трет-бутиловых эфиров до соответствующих карбоновых кислот с помощью трифторуксусной кислоты):



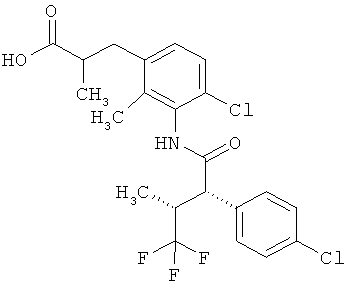

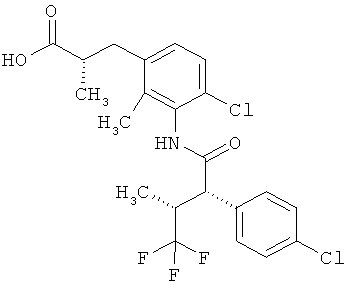





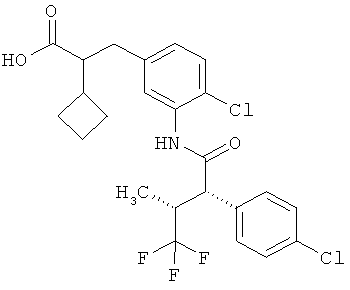

Пример 71
(+)-2-(4-Хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}бензил)-2-метилбутановая кислота {диастереомер А)

302 мг (0,553 ммоль) (+)-трет-бутил-2-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}бензил)-2-метилбутаноата {диастереомера А) растворяли в 2,3 мл дихлорметана и при Ткомн. добавляли 2 мл ТФК. Спустя 30 мин реакционную смесь упаривали в вакууме, а остаток сушили в высоком вакууме. Потом этот остаток очищали с помощью препаративной ОФ ВЭЖХ (элюент: ацетонитрил/вода). Получили 110,8 мг целевого продукта (40,9% от теор.).
ЖХ-МС (Метод 4): Rt=1,50 мин; m/z=490 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,71-0,86 (м, 6Н), 0,92 (с, 3Н), 1,30-1,43 (м, 1Н), 1,54-1,69 (м, 1Н), 2,60 (д, 1Н), 2,86 (д, 1Н), 3,34-3,45 (м, 1Н), 4,11 (д, 1Н), 6,86-7,00 (м, 1Н), 7,25-7,36 (м, 2Н), 7,39-7,52 (м, 4Н), 9,83 (с, 1Н), 12,28 (с, 1Н).
[α]D20=+74,0°, с=0,280, хлороформ.
Следующие примеры были получены в соответствии с Общей методикой 3 (расщепление сложных метиловых или этиловых эфиров до соответствующих карбоновых кислот в смесях из соляной кислоты или серной кислоты с уксусной кислотой):





Пример 79 и Пример 80
(+)-3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-4,4,4-трифторбутановая кислота (диастереомеры 1 и 2)
Полученную выше смесь диастереомерных 3-(3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}-4-фторфенил)-4,4,4-трифторбутановых кислот (Пример 78) дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×20 мм; расход: 15 мл/мин; детектирование: 230 нм; объем впрыска: 0,80 мл; температура: 45°С; элюент: 92% изогексана/8% изопропанола]. Исходя из 1,95 г смеси диастереомеров, получили 556 мг диастереомера 1 (Пример 79) и 730 мг диастереомера 2 (Пример 80):
Пример 79 (диастереомер 1):
Диастереомер 1 был еще раз дополнительно очищен с помощью препаративной ОФ ВЭЖХ (элюент: метанол/вода). Получили 418 мг.
ЖХ-МС (Метод 4): Rt=1,49 мин; m/z=500 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 2,82 (дд, 1Н), 2,94 (дд, 1Н), 3,37-3,44 (м, 1Н), 4,02 (тд, 1Н), 4,13 (д, 1Н), 7,17-7,30 (м, 2Н), 7,40-7,50 (м, 4Н), 7,87 (д, 1Н), 10,18 (с, 1Н), 12,53 (уш. с, 1Н).
[α]D20=+130°, с=0,29, хлороформ.
Пример 80 (диастереомер 2):
Диастереомер 2 был еще раз дополнительно очищен с помощью препаративной ОФ ВЭЖХ (элюент: метанол/вода). Получили 352 мг.
ЖХ-МС (Метод 6): Rt=1,18 мин; m/z=500 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,79 (д, 3Н), 2,82 (дд, 1Н), 2,94 (дд, 1Н), 3,94-4,08 (м, 1Н), 4,13 (д, 1Н), 7,17-7,33 (м, 2Н), 7,40-7,52 (м, 4Н), 7,88 (д, 1Н), 10,18 (с, 1Н).
[α]D20=+104°, с=0,260, хлороформ.
Пример 81
3-(4-Хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)гексановая кислота (смесь диастереомеров)
1,50 г (2,97 ммоль) метил-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)гексаноата (смесь диастереомеров) растворяли в 5 мл уксусной кислоты, добавляли 5 мл 30%-ной серной кислоты и нагревали до кипения с обратным холодильником (температура бани примерно 140°С). Спустя 1,5 ч к этому добавляли дополнительные 2,5 мл уксусной кислоты, а реакционную смесь перемешивали еще 2,5 ч при кипении с обратным холодильником. После охлаждения эту смесь оставляли стоять в течение ночи при Ткомн., потом выливали в воду и трижды экстрагировали диэтиловым эфиром. Объединенные органические фазы промывали 5%-ным раствором гидрокарбоната натрия и нас. раствором хлорида натрия, сушили над сульфатом магния, упаривали в вакууме, а остаток сушили в высоком вакууме. Было получено 1,44 г целевого продукта (98,8% от теор.).
ЖХ-МС (Метод 6): Rt=1,29 мин; m/z=490 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): оба диастереомера δ [м.д.]=0,74-0,83 (м, 6Н), 0,99-1,14 (м, 2Н), 1,37-1,60 (м, 2Н), 2,39 (дд, 1Н), 2,85-2,99 (м, 1Н), 3,36-3,44 (м, 1Н), 4,13 (д, 1Н), 6,97-7,10 (м, 1Н), 7,32-7,40 (м, 2Н), 7,42-7,53 (м, 4Н), 9,83 (с, 1Н), 12,02 (уш. с, 1Н).
Пример 82 и Пример 83
(+)-3-(4-Хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)гексановая кислота {диастереомеры 1 и 2)

Полученную выше смесь диастереомерных 3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)гексановых кислот (Пример 81) дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×20 мм; расход: 20 мл/мин; детектирование: 230 нм; объем впрыска: 0,60 мл; температура: 25°С; элюент: 95% изогексана/5% изопропанола]. Исходя из 59,2 мг смеси диастереомеров, получили 19 мг диастереомера 1 (Пример 82) и 17 мг диастереомера 2 (Пример 83):
Пример 82 (диастереомер 1):
(+)-(3S)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)гексановая кислота
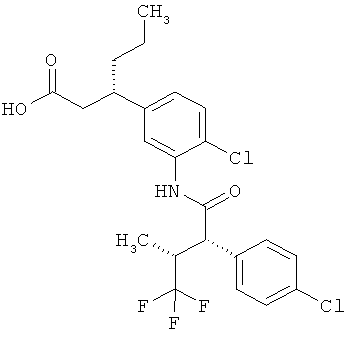
ЖХ-МС (Метод 6): Rt=1,27 мин; m/z=490 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,72-0,86 (м, 6Н), 0,98-1,19 (м, 2Н), 1,37-1,61 (м, 2Н), 2,34-2,44 (м, 1Н), 2,88-2,97 (м, 1Н), 3,34-3,43 (м, 1Н), 4,13 (д, 1Н), 7,05 (дд, 1Н), 7,26-7,40 (м, 2Н), 7,41-7,63 (м, 4Н), 9,82 (с, 1Н), 12,02 (с, 1Н).
[α]D20=+52°, с=0,30, хлороформ.
Согласно такой же препаративной методике ВЭЖХ было разделено также большее количество (1,40 г) диастереомерных 3-(4-xnop-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)гексановых кислот (Пример 81). Полученный диастереомер 1 в этом случае был еще раз дополнительно очищен с помощью ОФ ВЭЖХ [колонка: Sunfire 250 мм×20 мм; элюент: 80% ацетонитрила/5% водн. ТФК (1%-ной)/15% воды]. Таким образом было получено 337 мг чистого диастереомера 1.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,74-0,86 (м, 6Н), 0,97-1,16 (м, 2Н), 1,40-1,60 (м, 2Н), 2,35-2,44 (м, 1Н), 2,89-2,97 (м, 1Н), 3,35-3,43 (м, 1Н), 4,13 (д, 1Н), 7,05 (дд, 1Н), 7,30-7,40 (м, 2Н), 7,41-7,54 (м, 4Н), 9,83 (с, 1Н), 12,02 (уш. с, 1Н).
[α]D20=+86°, с=0,480, хлороформ.
Пример 83 (диастереомер 2):
(+)-(3R)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)гексановая кислота
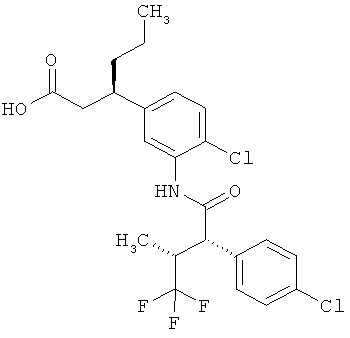
ЖХ-МС (Метод 6); Rt=1,27 мин; m/z=490 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,77-0,83 (м, 6Н), 1,00-1,12 (м, 2Н), 1,41-1,60 (м, 2Н), 2,35-2,44 (м, 1Н), 2,88-2,98 (м, 1Н), 3,35-3,45 (м, 1Н), 4,13 (д, 1Н), 7,05 (дд, 1Н), 7,32-7,40 (м, 2Н), 7,43-7,64 (м, 4Н), 9,82 (с, 1Н), 12,04 (уш. с, 1Н).
[α]D20=+22,1°, с=0,40, хлороформ.
Пример 84 и Пример 85
(+)-3-(4-Хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-4,4,4-трифторбутановая кислота (диастереомеры 1 и 2)
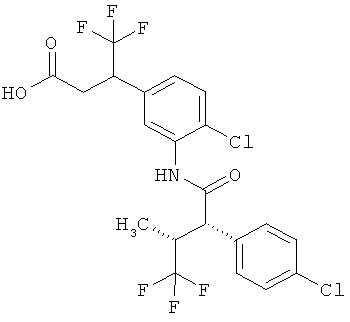
Полученную выше смесь диастереомерных 3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-4,4,4-трифторбутановых кислот (Пример 66) дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×20 мм; расход: 15 мл/мин; детектирование: 220 нм; объем впрыска: 0,25 мл; температура: 30°С; элюент: 93% изогексана/7% изопропанола]. Исходя из 150 мг смеси диастереомеров, получили 70 мг диастереомера 1 (Пример 84) и 79 мг диастереомера 2 (Пример 85):
Пример 84 (диастереомер 1):
(+)-(3S)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-4,4,4-трифторбутановая кислота
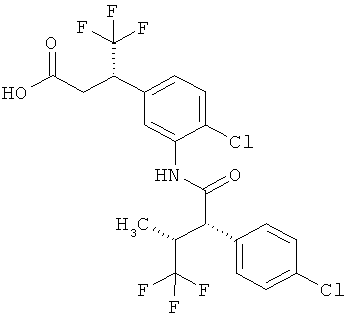
ЖХ-МС (Метод 6): Rt=1,18 мин; m/z=516 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 2,84 (дд, 1Н), 2,95 (дд, 1Н), 3,36-3,43 (м, 1Н), 4,06 (тд, 1Н), 4,14 (д, 1Н), 7,26 (дд, 1Н), 7,40-7,52 (м, 5Н), 7,60 (д, 1Н), 9,95 (с, 1Н), 12,54 (уш. с, 1Н).
[α]D20=+78°, с=0,52, хлороформ.
В качестве альтернативы (+)-(3S)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-4,4,4-трифторбутановая кислота также могла получаться следующим способом:
1,76 г (3,08 ммоль) (+)-трет-бутил-(3S)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-4,4,4-трифторбутаноата (Пример 203А) растворяли в 4,9 мл дихлорметана и при Ткомн. добавляли 4,7 мл ТФК. Реакционную смесь перемешивали 2 ч при Ткомн., а потом упаривали в вакууме. Остаток переводили в этилацетат и промывали нас. раствором гидрокарбоната натрия и нас. раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали с помощью препаративной ОФ ВЭЖХ (элюент: метанол/вода). Получили 1,30 г целевого продукта (81,9% от теор.).
ЖХ-МС (Метод 4): Rt=1,46 мин; m/z=515 (М)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 2,83 (дд, 1Н), 2,95 (дд, 1Н), 3,37-3,45 (м, 1Н), 4,06 (тд, 1Н), 4,14 (д, 1Н), 7,26 (дд, 1Н), 7,43-7,52 (м, 5Н), 7,60 (д, 1Н), 9,95 (с, 1Н), 12,56 (уш. с, 1Н).
[α]D20=+79,9°, с=0,475, хлороформ.
Пример 85 (диастереомер 2):
(+)-(3R)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-4,4,4-трифторбутановая кислота
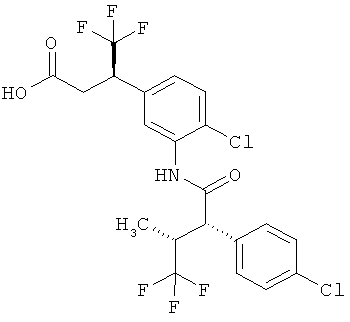
ЖХ-МС (Метод 6): Rt=1,19 мин; m/z=516 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 2,84 (дд, 1Н), 2,95 (дд, 1Н), 3,28-3,44 (м, 1Н), 3,95-4,11 (м, 1Н), 4,15 (д, 1Н), 7,22-7,30 (м, 1Н), 7,41-7,53 (м, 5Н), 7,57-7,70 (м, 1Н), 9,95 (с, 1Н), 12,55 (уш. с, 1Н).
[α]D20=+40,2°, с=0,52, хлороформ.
В качестве альтернативы (+)-(3R)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-4,4,4-трифторбутановая кислота также могла получаться следующим способом:
1,17 г (2,04 ммоль) (+)-трет-бутил-(3R)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)-4,4,4-трифторбутаноата (Пример 204А) растворяли в 4,9 мл дихлорметана и при Ткомн. добавляли 3,2 мл ТФК. Реакционную смесь перемешивали 2 ч при Ткомн., а потом упаривали в вакууме. Остаток переводили в этилацетат и промывали нас. раствором гидрокарбоната натрия и нас. раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. Сырой продукт очищали с помощью препаративной ОФ ВЭЖХ (элюент: метанол/вода). Получили 0,76 г целевого продукта (72% от теор.).
ЖХ-МС (Метод 6): Rt=1,19 мин; m/z=516 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,80 (д, 3Н), 2,83 (дд, 1Н), 2,94 (дд, 1Н), 3,37-3,47 (м, 1Н), 3,93-4,10 (м, 1Н), 4,15 (д, 1Н), 7,26 (дд, 1Н), 7,43-7,52 (м, 5Н), 7,58-7,66 (м, 1Н), 9,95 (с, 1Н), 12,57 (уш. с, 1Н).
[α]D20=+44,8°, с=0,47, хлороформ. Пример 86
3-(4-Хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)пентановая кислота (смесь диастереомеров)

650 мг (1,33 ммоль) метил-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метил-бутаноил]амино}фенил)пентаноат (смесь диастереомеров) растворяли в 2 мл уксусной кислоты, добавляли 1 мл 30%-ной серной кислоты и нагревали до кипения с обратным холодильником (температура бани примерно 140°С). Спустя 1,5 ч реакционную смесь охлаждали и выливали в воду. Трижды экстрагировали этилацетатом. Объединенные органические фазы промывали нас. раствором гидрокарбоната натрия и нас. раствором хлорида натрия, сушили над сульфатом магния и упаривали в вакууме. После сушки в высоком вакууме остаток очищали с помощью хроматографии на силикагеле (элюент: циклогексан/этилацетат 50:1). Получили 600 мг целевого продукта (95% от теор.).
ЖХ-МС (Метод 6): Rt=1,24 мин; m/z=476 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): оба диастереомера δ [м.д.]=0,69 (тд, 3Н), 0,80 (д, 3Н), 1,40-1,52 (м, 1Н), 1,55-1,68 (м, 1Н), 2,40 (дд, 1Н), 2,55-2,59 (м, 1Н), 2,78-2,89 (м, 1Н), 3,36-3,43 (м, 1Н), 4,13 (д, 1Н), 7,04 (дд, 1Н), 7,31-7,41 (м, 2Н), 7,42-7,57 (м, 4Н), 9,84 (с, 1Н), 12,04 (уш. с, 1Н).
Пример 87 и Пример 88
(+)-3-(4-Хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)пентановая кислота (диастереомеры 1 и 2)
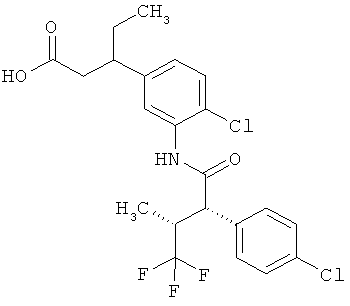
Полученную выше смесь диастереомерных 3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)пентановых кислот (Пример 86) дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×20 мм; расход: 18 мл/мин; детектирование: 230 нм; объем впрыска: 0,25 мл; температура: 25°С; элюент: 95% изогексана/5% изопропанола]. Исходя из 545 мг смеси диастереомеров, получили 140 мг диастереомера 1 (Пример 87) и 156 мг диастереомера 2 (Пример 88):
Пример 87 (диастереомер 1):
(+)-(3S)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)пентановая кислота

ЖХ-МС (Метод 4): Rt=1,47 мин; m/z=476 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,69 (т, 3Н), 0,80 (д, 3Н), 1,37-1,51 (м, 1Н), 1,54-1,68 (м, 1Н), 2,35-2,44 (м, 1Н), 2,55-2,59 (м, 1Н), 2,80-2,87 (м, 1Н), 3,36-3,40 (м, 1Н), 4,13 (д, 1Н), 7,04 (дд, 1Н), 7,32-7,40 (м, 2Н), 7,42-7,50 (м, 4Н), 9,83 (с, 1Н), 12,03 (уш. с, 1Н).
[α]D20=+87,0, с=0,47, хлороформ.
Пример 88 (диастереомер 2):
(+)-(3R)-3-(4-хлор-3-{[(2S,3R)-2-(4-хлорфенил)-4,4,4-трифтор-3-метилбутаноил]амино}фенил)пентановая кислота

ЖХ-МС (Метод 4): Rt=1,47 мин; m/z=476 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=0,69 (т, 3Н), 0,80 (д, 3Н), 1,39-1,50 (м, 1Н), 1,56-1,65 (м, 1Н), 2,35-2,45 (м, 1Н), 2,52-2,58 (м, 1Н), 2,80-2,87 (м, 1Н), 3,35-3,41 (м, 1Н), 4,13 (д, 1Н), 7,04 (дд, 1Н), 7,31-7,41 (м, 2Н), 7,42-7,52 (м, 4Н), 9,83 (с, 1Н), 12,04 (уш. с, 1Н).
[α]D20=+71,4, с=0,48, хлороформ.
Следующие примеры были получены в соответствии с общей методикой 2 (расщепление сложных трет-бутиловых эфиров до соответствующих карбоновых кислот с помощью трифторуксусной кислоты):
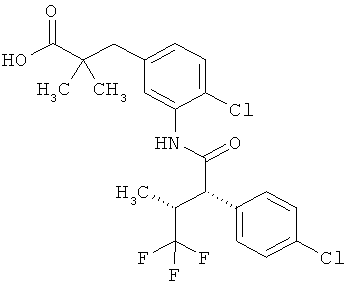
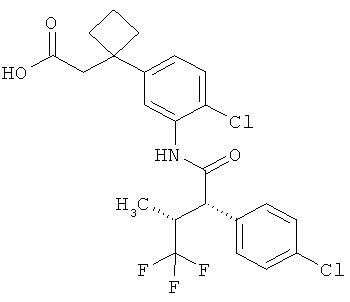

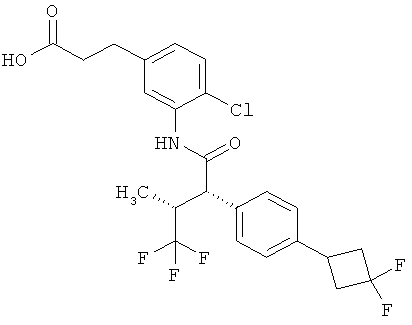
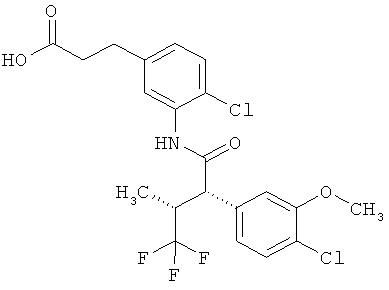
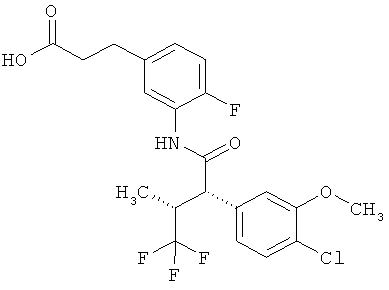



Следующие примеры были получены в соответствии с общей методикой 3 (расщепление сложных метиловых или этиловых эфиров до соответствующих карбоновых кислот в смесях из соляной кислоты или серной кислоты с уксусной кислотой):


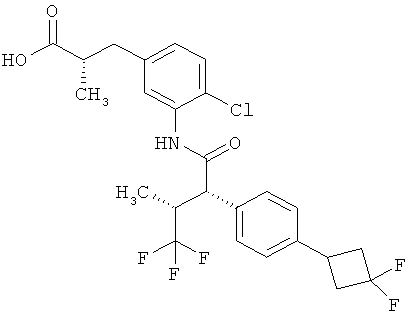


Пример 103 и Пример 104
(2S)-3-[4-хлор-3-({4,4,4-трифтор-3-метил-2-[4-(2,2,2-трифторэтил)фенил]бутаноил}амино)фенил]-2-метилпропановая кислота (диастереомеры 1 и 2)

74 мг (0,15 ммоль) смеси изомеров (2S)-3-[4-хлор-3-({4,4,4-трифтор-3-метил-2-[4-(2,2,2-трифторэтил)фенил]бутаноил}амино)фенил]-2-метилпропановой кислоты (Пример 99) дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralpak AY-H, 5 мкм, 250 мм×20 мм; элюент: изогексан/этанол 1:1 (об/об); расход: 15 мл/мин; УФ-детектирование: 220 нм; температура: 45°С]:
Пример 103 (диастереомер 1):
Выход:39 мг
Rt=3,72 мин; химическая чистота > 98%; > 99% de
[колонка: Daicel Chiralpak AY-H, 5 мкм, 250 мм×4 мм; элюент: изогексан/(изопропанол + 0,2% ТФК + 1% воды) 70:30 (об/об); расход: 1 мл/мин; температура: 45°С; УФ-детектирование: 220 нм].
ЖХ-МС (Метод 6): Rt=1,16 мин; m/z=508/510 (М-Н)-.
Пример 104 (диастереомер 2):
Выход:39 мг Rt=6,09 мин; химическая чистота > 98%; > 99% de
[колонка: Daicel Chiralpak AY-H, 5 мкм, 250 мм×4 мм; элюент: изогексан/(изопропанол + 0,2% ТФК + 1% воды) 70:30 (об/об); расход: 1 мл/мин; температура: 45°С; УФ-детектирование: 220 нм].
ЖХ-МС (Метод 6): Rt=1,16 мин; m/z=508/510 (М-Н)-.
Примеры 105-108
(2S)-3-(4-хлор-3-{[(4-хлорфенил)-(3,3-дифторциклопентил)ацетил]амино}-фенил)-2-метилпропановая кислота (изомеры 1-4)
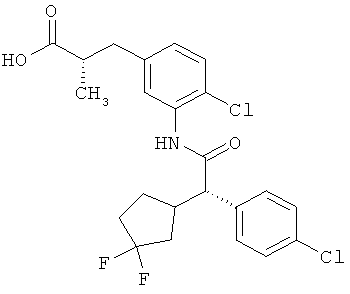
205 мг (0,44 ммоль) смеси диастереомеров (2S)-3-(4-хлор-3-{[(4-хлорфенил)-(3,3-дифторциклопентил)ацетил]амино}фенил)-2-метилпропановой кислоты (Пример 20) дополнительно разделяли с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralcel OJ-H, 5 мкм, 250 мм×20 мм; элюент: изогексан/этанол 70:30 (об/об); расход: 25 мл/мин; УФ-детектирование: 230 нм; температура: 25°С]. При этом получали две различные фракции, которые соответственно состояли из смеси двух изомеров. Обе эти фракции были разделены на отдельные изомеры с помощью еще одной препаративной ВЭЖХ на хиральной фазе [Фракция 1: колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×20 мм; элюент: изогексан/изопропанол 80:20 (об/об); расход: 20 мл/мин; УФ-детектирование: 230 нм; температура: 25°С. Фракция 2: колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×20 мм; элюент: изогексан/этанол 90:10 (об/об); расход: 20 мл/мин; УФ-детектирование: 230 нм; температура: 25°С]:
Пример 105 (изомер 1):
Выход:30 мг Rt=15,70 мин; химическая чистота > 89,8%
[колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×4 мм; элюент: изогексан/изопропанол 70:30 (об/об); расход: 1 мл/мин; УФ-детектирование: 230 нм; температура: 25°С].
ЖХ-МС (Метод 6): Rt=1,22 мин; m/z=470/472 (М+Н)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,02 (д, 3Н), 1,21-1,35 (м, 1Н), 1,45-1,58 (м, 1Н), 1,84-2,20 (м, 3Н), 2,28-2,43 (м, 1Н), 2,53-2,62 (м, 2Н, частично перекрывается сигналом ДМСО), 2,76-2,90 (м, 2Н), 3,75 (д, 1Н), 7,03 (дд, 1Н), 7,32-7,49 (м, 6Н), 9,74 (с, 1Н), 12,04-12,35 (уш. с, 1Н).
Пример 106 (изомер 2):
Выход: 35 мг Rt=20,07 мин; химическая чистота > 98,9%
[колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×4 мм; элюент: изогексан/изопропанол 70:30 (об/об); расход: 1 мл/мин; УФ-детектирование: 230 нм; температура: 25°С].
ЖХ-МС (Метод 5): Rt=2,58 мин; m/z=470/472 (M+H)+.
1H-ЯМР (400 МГц, ДМСО-d6): δ [м.д.]=1,02 (д, 3Н), 1,52-1,69 (м, 2Н), 1,81-1,96 (м, 1Н), 1,98-2,29 (м, 3Н), 2,52-2,62 (м, 2Н, частично перекрывается сигналом ДМСО), 2,78-2,92 (м, 2Н), 3,78 (д, 1Н), 7,03 (дд, 1Н), 7,33-7,48 (м, 6Н), 9,78 (с, 1Н), 12,04-12,26 (уш. с, 1Н).
Пример 107 (изомер 3):
Выход: 37 мг
Rt=14,17 мин; химическая чистота > 95,7%
[колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм×4 мм; элюент: изогексан/этанол 90:10 (об/об); расход: 1 мл/мин; УФ-детектирование: 230 нм; температура: 25°С].
ЖХ-МС (Метод 5): Rt=2,57 мин; m/z=470/472 (М+Н)+.
1H-ЯМР: смотрите Пример 106 (изомер 2).
Пример 108 (изомер 4Y
Выход:29 мг
Rt=17,77 мин; химическая чистота > 99,5%
[колонка: Daicel Chiralpak AD-H, 5 мкм, 250 мм х 4 мм; элюент: изогексан/этанол 90:10 (об/об); расход: 1 мл/мин; УФ-детектирование: 230 нм; температура: 25°С].
ЖХ-МС (Метод 5): Rt=2,58 мин; m/z=470/472 (М+Н)+.
1H-ЯМР: смотрите пример 105 (изомер 1). Примеры 109-112
(2R)-3-(4-хлор-3-{[(4-хлорфенил)-(3,3-дифторциклопентил)ацетил]амино}-фенил)-2-метилпропановая кислота (изомеры 1-4)
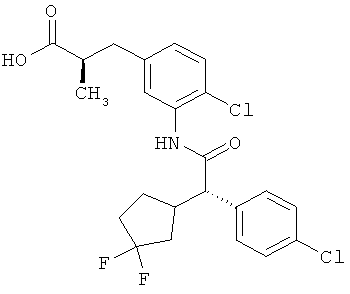
160 мг (0,32 ммоль) смеси диастереомеров (2R)-3-(4-хлор-3-{[(4-хлорфенил)-(3,3-дифторциклопентил)ацетил]амино}фенил)-2-метилпропановой кислоты (Пример 21) разделяли на 4 изомера с помощью препаративной ВЭЖХ на хиральной фазе [колонка: Daicel Chiralcel OJ-H, 5 мкм, 250 мм×20 мм; элюент: изогексан/этанол/метанол 90:5:5 (об/об); расход: 20 мл/мин; УФ-детектирование: 230 нм; температура: 25°С]:
Пример 109 (изомер 1):
Выход: 16,7 мг Rt=10,49 мин; химическая чистота > 92,8%
[колонка: Daicel Chiralpak OJ-H, 5 мкм, 250 мм×4 мм; элюент: изогексан/этанол/метанол 90:5:5 (об/об); расход: 1 мл/мин; УФ-детектирование: 230 нм; температура: 25°С].
ЖХ-МС (Метод 4): Rt=1,39 мин; m/z=470/472 (M+H)+.
Пример 110 (изомер 2):
Выход: 24,4 мг Rt=12,26 мин; химическая чистота > 94,7%
[колонка: Daicel Chiralpak OJ-H, 5 мкм, 250 мм×4 мм; элюент: изогексан/этанол/метанол 90:5:5 (об/об); расход: 1 мл/мин; УФ-детектирование: 230 нм; температура: 25°С].
ЖХ-МС (Метод 4): Rt=1,39 мин; m/z=470/472 (M+H)+.
398 Пример 111 (изомер 3):
Выход: 22 мг Rt=18,89 мин; химическая чистота > 97,8%
[колонка: Daicel Chiralpak OJ-H, 5 мкм, 250 мм×4 мм; элюент: изогексан/этанол/метанол 90:5:5 (об/об); расход: 1 мл/мин; УФ-детектирование: 230 нм; температура: 25°С].
ЖХ-МС (Метод 4): Rt=1,39 мин; m/z=470/472 (М+Н)+.
Пример 112 (изомер 4):
Выход: 25 мг Rt=28,37 мин; химическая чистота > 97,8%
[колонка: Daicel Chiralpak OJ-H, 5 мкм, 250 мм×4 мм; элюент: изогексан/этанол/метанол 90:5:5 (об/об); расход: 1 мл/мин; УФ-детектирование: 230 нм; температура: 25°С].
ЖХ-МС (Метод 4): Rt=1,39 мин; m/z=470/472 (М+Н)+
Следующий пример был получен в соответствии с общей методикой 3:

В. Оценка фармакологической эффективности
Фармакологическая эффективность соединений согласно изобретению может быть показана в следующих биологических испытаниях:
В-1. Стимуляция рекомбинантной растворимой гуанилатциклазы (рГШ in vitro (вне организма)
Исследования стимуляции рекомбинантной растворимой гуанилатциклазы (рГЦ) с помощью соединений согласно изобретению в присутствии или в отсутствие нитропруссида натрия, а также в присутствии или в отсутствие гемзависимого ингибитора рГЦ 1H-1,2,4-оксадиазоло[4,3-а]хиноксалин-1-она (ODQ) проводятся в соответствии с методом, подробно описанным в следующей литературной ссылке: М.Hoenicka, E.M.Becker, H.Apeler, Т.Sirichoke, H.Schroeder, R.Gerzer und J.-P.Stasch, «Purified soluble guanylyl cyclase expressed in a baculovirus/Sf9 system: Stimulation by YC-1, nitric oxide, and carbon oxide», J. Mol. Med. 77 (1999), 14-23. He содержащая гем гуанилатциклаза получается в результате добавления твин 20 к буферу для образца (0,5% в конечной концентрации).
Активация рГЦ с помощью исследуемого вещества приводится как x-кратная стимуляция базальной активности. Этот результат для примера 15 показан в таблице 1А, а для примера 17 в таблице 1В:
Из таблиц 1А и 1В очевидно, что достигается стимуляция как гемсодержащего, так и не содержащего гем фермента. Кроме того, комбинация примера 15 или соответственно примера 17 с 2-(N,N-диэтиламино)диазенолат-2-оксидом (DEA/NO), донором N0, не показывает синергетического эффекта, то есть эффективность DEA/NO не потенциируется, как этого можно было бы ожидать в случае активатора рГЦ, действующего по гемзависимому механизму. Помимо этого, эффективность активатора рГЦ согласно изобретению не блокируется 1Н-1,2,4-оксадиазоло[4,3-а]хиноксалин-1-оном (ODQ), гемзависимым ингибитором растворимой гуанилатциклазы, а даже усиливается. Следовательно, результаты в таблицах 1А и 1В подтверждают механизм действия соединений согласно изобретению в качестве активаторов растворимой гуанилатциклазы.
В-2. Эффективность на репортерной клеточной линии рекомбинантной гуанилатциклазы
Клеточная эффективность соединений согласно изобретению определяется на репортерной клеточной линии рекомбинантной гуанилатциклазы, как описано в публикации F. Wunderc соавт., Anal. Biochem. 339.104-112 (2005).
Репрезентативные результаты для соединений согласно изобретению приведены в таблице 2:
В-3. Стимуляция активности фермента рГЦ
Растворимая гуанилатциклаза (рГЦ) при стимуляции превращает ГТФ в цГМФ и пирофосфат (PPi). PPi обнаруживается с помощью описанного ниже теста. Сигнал, возникающий в тесте, усиливается по мере прогрессирующего взаимодействия и служит в качестве критерия для активности рГЦ-фермента при указанной стимуляции.
Для проведения этого теста 29 мкл раствора фермента [0-10 нМ растворимой гуанилатциклазы (полученной согласно Honicka с соавт., J. Mol. M(3d. 77, 14-23 (1999)) в 50 мМ ТЭА, 2 мМ MgCl2, 0,1% БСА (фракция V), 0,005% Brij®, pH 7,5] помещают в микропланшет и к нему добавляют 1 мкл вещества, которое нужно исследовать (в виде последовательно разбавленного раствора в ДМСО). Загруженную массу в течение 10 мин инкубируют при комнатной температуре. Затем добавляют 20 мкл диагностической смеси [1,2 нМ люциферазы светлячка (Photinus pyralis-Luciferase, фирмы Promega), 29 мкМ дегидролюциферина (приготовленного согласно Bitler & McElroy, Arch. Biochem. Biophys. 72, 358 (1957)), 122 мкМ люцифе-рина (фирмы Promega), 153 мкМ АТФ (фирмы Sigma) и 0,4 мМ ДТТ (фирмы Sigma) в 50 mM ТЭА, 2 мМ MgCb, 0,1% БСА (фракция V), 0,005% Brij®, phi 7,5]. Ферментативная реакция запускается в результате добавления 20 мкл раствора субстрата [1,25 мМ гуанозин-5'-трифосфата (фирмы Sigma) в 5С мМ ТЭА, 2 мМ MgCl2, 0,1% БСА (фракция V), 0,005% Brij®, pH 7,5] и непрерывно измеряется люминометрически. Степень стимуляции под действием подлежащего исследованию вещества может определяться относительно сигнала нестимулированной реакции.
В эезультате добавления 25 мкМ 1Н-1,2,4-оксадиазоло[4,3-а]хиноксалин-1-она (ODQ) к ферментному раствору и последующей 30-минутной инкубации исследуется активация не содержащей гем гуанилатциклазы и сравнивается со стимуляцией нативного фермента.
Репрезентативные результаты, касающиеся соединений согласно изобретению, приведены в таблице 3:
В-{4. Радиотелеметрическое измерение кровяного давления и частоты сердечных сокращений у бодрствующих крыс со спонтанной гипертензией (СГ-крыс)
Для описанных далее измерений у бодрствующих СГ-крыс используется имеющаяся в продаже телеметрическая система фирмы Data Sciences International DSI, США.
Эта система состоит из 3 основных компонентов: (1) имплантируемые передатчики, (2) приемные датчики, которые с помощью концентратора соединены с (3) компьютером сбора данных. Телеметрическое оборудование делает возможным непрерывную регистрацию кровяного давления и частоты сердечных сокращений у бодрствующих животных в их привычной среде обитания.
Исследования проводятся на взрослых женских особях крыс со спонтанной гипертензией (СГ-крысах) с массой тела > 200 г. Подопытные животные после имплантации передатчика содержатся по отдельности в клетках из макролона типа 3. Они имеют свободный доступ к стандартному корму и воде. Ритм день/ночь в экспериментальной лаборатории изменяется с помощью освещения помещения в 6:00 часов утра и в 19:00 часов вечера.
Использованные телеметрические передатчики (ТАМ РА-С40, DSI) имплантируются подопытным животным хирургическим путем в асептических условиях по меньшей мере за 14 дней до первого испытания. Животные, инструментально подготовленные таким образом, могут использоваться снова после заживания ранки и зарастания имплантата.
Для имплантации животных подвергают наркозу на пустой желудок с помощью пентобарбитала (нембутал, фирмы Sanofi, 50 мг/кг интраперитонеально) и выбривают и дезинфицируют обширную область на брюхе. После вскрытия брюшной полости вдоль белой линии живота наполненный жидкостью измерительный катетер системы вводится выше разветвления в краниальном направлении в нисходящую аорту и фиксируется с помощью клея для тканей (VetBonD™, 3М). Корпус передатчика фиксируется интраперитонеально на мускулатуре стенки брюшной полости, а разрез закрывается послойно. Послеоперационно для профилактики инфекции вводится антибиотик (Tardomyocel COMP, Bayer AG, 1 мл/кг п/к).
Процесс испытаний:
Вещества, подлежащие испытаниям, в каждом случае вводятся группе животных (n=6) орально с помощью желудочного зонда. В соответствии с дозой применения, составляющей 5 мл/кг массы тела, исследуемые вещества растворяются в подходящих смесях растворителей или суспендируются в 0,5%-ной тилозе. Группа животных, подвергшихся введению растворителя, используется в качестве контрольной.
Измерительное телеметрическое оборудование конфигурируется для 24 животных. Каждый эксперимент регистрируется под одним номером эксперимента.
За живущей в установке инструментированной крысой закреплена соответственно собственная приемная антенна (1010 Receiver, фирмы DSI). Имплантированные передатчики могут активироваться извне с помощью встроенного электромагнитного реле и в процессе испытаний включаются на передачу. Излучаемые сигналы с помощью компьютера для сбора данных (Dataquest™ A.R.T. для операционной системы Windows, DSI) могут регистрироваться в режиме реального времени и соответственно обрабатываться. Хранение данных осуществляется соответственно в открытой для этой цели директории, которая имеет номер эксперимента.
В стандартном процессе на протяжении каждого 10-ти секундного периода измеряются: (1) систолическое кровяное давление (СКД), (2) диастолическое кровяное давление (ДКД), (3) среднее артериальное давление (САД) и (4) частота сердечных сокращений (ЧСС).
Регистрация измеряемых величин повторяется под управлением компьютера с 5-ти минутными интервалами. Исходные данные, собранные в виде абсолютных величин, корректируются на диаграмме с измеренным на текущий момент барометрическим давлением и накапливаются в отдельных файлах. Другие технические подробности приведены в документации фирмы-изготовителя (DSI).
Введение испытуемых соединений осуществляется в день эксперимента в 9:00 часов. По окончании приема в течение 24 часов измеряются описанные выше параметры. После окончания эксперимента собранные отдельные данные сортируются с помощью аналитического программного обеспечения (Dataquest™ A.R.T. Analysis). В качестве исходной величины принимается момент времени за 2 часа до приема вещества, так что выделенный массив данных охватывает промежуток времени от 7:00 часов в день эксперимента до 9:00 часов следующего дня.
Эти данные через предварительно устанавливаемое время усредняются путем определения средней величины (15-минутная средняя величина, 30-ти минутная средняя величина) и переносятся на информационный носитель в виде текстовых данных. Предварительно отсортированные таким образом и сжатые измеренные величины переносятся в приложение Excel и представляются в виде таблицы.
С. Примеры исполнения для фармацевтических композиций
Соединения согласно изобретению могут переводиться в фармацевтические композиции следующим образом:
Таблетки:
Состав:
100 мг соединения согласно изобретению, 50 мг лактозы (моногидрата), 50 мг кукурузного крахмала (натурального), 10 мг поливинилпирролидона (ПВП 25) (фирмы BASF, Людвигсхафен, Германия) и 2 мг стеарата магния.
Масса таблетки 212 мг. Диаметр 8 мм, радиус кривизны 12 мм.
Изготовление:
Смесь из соединения согласно изобретению, лактозы и крахмала гранулируется с 5%-ным раствором (масс./масс.) ПВП в воде. Этот гранулят после сушки 5 минут перемешивается со стеаратом магния. Эта смесь прессуется с помощью обычного таблеточного пресса (размер таблеток смотрите выше). В качестве ориентировочной величины для прессования применяется прессующее усилие 15 кН.
Суспензия для орального применения:
Состав:
1000 мг соединения согласно изобретению, 1000 мг этанола (96%), 400 мг Rhodigel® (ксантановая камедь фирмы FMC, Пенсильвания, США) и 99 г воды.
Однократной дозе в 100 мг соединения согласно изобретению соответствуют 10 мл суспензии для орального применения.
Изготовление:
Компонент Rhodigel суспендируют в этаноле, соединение согласно изобретению прибавляют к этой суспензии. При перемешивании осуществляют добавление воды. До завершения набухания Rhodigel перемешивают примерно в течение 6 ч.
Раствор для орального применения:
Состав:
500 мг соединения согласно изобретению, 2,5 г полисорбата и 97 г полиэтиленгликоля 400. Однократной дозе в 100 мг соединения согласно изобретению соответствуют 20 г раствора для орального применения.
Изготовление:
Соединение согласно изобретению при перемешивании суспендируют в смеси из полиэтиленгликоля и полисорбата. Процесс перемешивания продолжают до полного растворения соединения согласно изобретению.
Раствор для внутривенного применения (в/в):
Соединение согласно изобретению с концентрацией ниже растворимости при насыщении растворяется в физиологически допустимом растворителе (например, изотоническом растворе поваренной соли, 5%-ном растворе глюкозы и/или 30%-ном растворе ПЭГ 400). Этот раствор фильтруется в стерильных условиях и заливается в стерильные и апирогенные емкости для инъекций.
Реферат
Изобретение относится к производным 3-фенилпропионовой кислоты формулы (I), где Rпредставляет собой водород, метил, этил, циклопропил или циклобутил, Rявляется водородом или метилом, Rпредставляет собой водород, метил, трифторметил, этил или н-пропил, Rявляется водородом или метилом или Rи Rсвязаны друг с другом и вместе с атомами углерода, с которыми они соединены, образуют циклопропильное кольцо формулыв которой Rи Rимеют значения, указанные выше, или Rи Rсвязаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют циклическую группу формулыилив которой n обозначает число 1 или 2, Rявляется водородом, фтором или метилом, Rпредставляет собой водород, фтор, хлор или цианогруппу, Rпредставляет собой метил, Rявляется трифторметилом или Rи Rсвязаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют дифторзамещенное циклоалкильное кольцо формулыRпредставляет собой хлор, алкил с 1-4 атомами углерода, алкенил с 2-4 атомами углерода, циклопропил или циклобутил, причем алкил с 1-4 атомами углерода и алкенил с 2-4 атомами углерода могут содержать до трех атомов фтора, циклопропил и циклобутил до двух атомов фтора в качестве заместителей, и Rпредставляет собой водород, фтор, хлор, метил или метоксигруппу. Также изобретение относится к лекарственному средству, содержащему указанные соединения и способу получения соединений формулы (I). Технический результат - соединения формулы (I) активируют форму растворимой гуанилатциклазы, не содержащей гем, и предназначены для применения в способе лечения и/или профилактики сердечной недостаточности, стенокардии, гипертензии, легочной гипертензии, ише
Формула
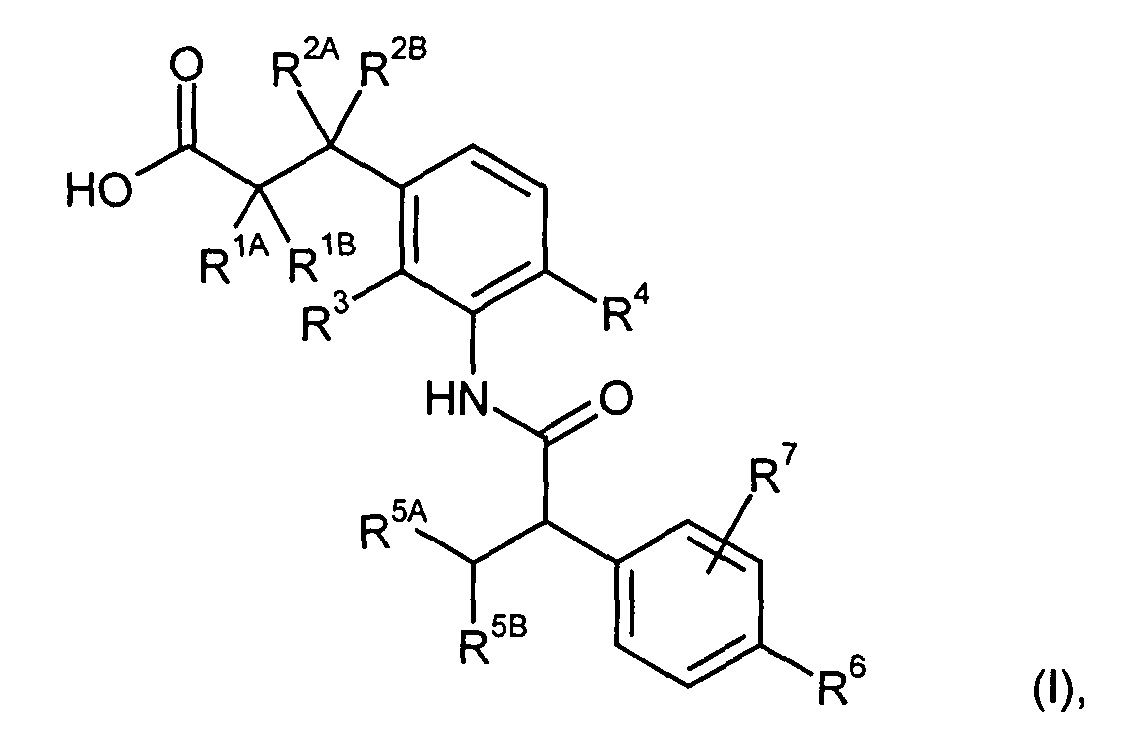
в которой
R1A представляет собой водород, метил, этил, циклопропил или циклобутил,
R1B является водородом или метилом,
R2A представляет собой водород, метил, трифторметил, этил или н-пропил,
R2B является водородом или метилом или
R1A и R2A связаны друг с другом и вместе с атомами углерода, с которыми они соединены, образуют циклопропильное кольцо формулы
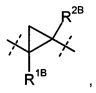
в которой
R1B и R2B имеют значения, указанные выше, или
R2A и R2B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют циклическую группу формулы


в которой
n обозначает число 1 или 2,
R3 является водородом, фтором или метилом,
R4 представляет собой водород, фтор, хлор или цианогруппу,
R5A представляет собой метил,
R5B является трифторметилом или
R5A и R5B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют дифторзамещенное циклоалкильное кольцо формулы
R6 представляет собой хлор, алкил с 1-4 атомами углерода, алкенил с 2-4 атомами углерода, циклопропил или циклобутил, причем алкил с 1-4 атомами углерода и алкенил с 2-4 атомами углерода могут содержать до трех атомов фтора, а циклопропил и циклобутил до двух атомов фтора в качестве заместителей, и
R7 представляет собой водород, фтор, хлор, метил или метоксигруппу, а также его физиологически приемлемые соли.
R1A представляет собой водород, метил, этил, циклопропил или циклобутил,
R1B является водородом или метилом,
R2A представляет собой водород, метил, трифторметил, этил или н-пропил,
R2B является водородом или метилом или
R2A и R2B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют циклическую группу формулы


в которой
n обозначает число 1 или 2,
R3 является водородом, фтором или метилом,
R4 представляет собой водород, фтор, хлор или цианогруппу,
R5A представляет собой метил,
R5B является трифторметилом или
R5A и R5B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют дифторзамещенное циклоалкильное кольцо формулы

R6 представляет собой хлор, алкил с 1-4 атомами углерода, алкенил с 2-3 атомами углерода, циклопропил или циклобутил, причем алкил с 1-4 атомами углерода и алкенил с 2-3 атомами углерода могут содержать до трех атомов фтора, а циклопропил и циклобутил до двух атомов фтора в качестве заместителей, и
R7 представляет собой водород, фтор, хлор, метил или метоксигруппу, а также его физиологически приемлемые соли.
R1A представляет собой водород, метил или этил,
R1B является водородом,
R2A представляет собой водород, метил, трифторметил, этил или н-пропил,
R2B является водородом или метилом или
R2A и R2B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют циклическую группу формулы

в которой
n обозначает число 1 или 2,
R3 является водородом,
R4 представляет собой фтор или хлор,
R5A представляет собой метил,
R5B является трифторметилом или
R5A и R5B связаны друг с другом и вместе с атомом углерода, с которым они соединены, образуют дифторзамещенное циклопентильное кольцо формулы
R6 представляет собой хлор, метил, трифторметил, этил, 1,1-дифторэтил, 2,2,2-трифторэтил, изопропил, трет-бутил, 1,1,1-трифтор-2-метилпропан-2-ил, винил, 1-фторвинил, циклопропил, 2,2-дифторциклопропил, циклобутил или 3,3-дифторциклобутил и
R7 представляет собой водород, фтор, хлор или метил,
а также его физиологически приемлемые соли.

в которой
R5A, R5B, R6 и R7 имеют значения, указанные в пп.1-3,
в инертном растворителе, с помощью конденсирующего агента или через промежуточную стадию соответствующего хлорангидрида карбоновой кислоты, в присутствии основания подвергают сочетанию с амином формулы (III)

в которой
R1A, R1B, R2A, R2B, R3 и R4 имеют значения, указанные в пп.1-3, а
Т1 представляет собой алкил с 1-4 атомами углерода или бензил,
с образованием амида карбоновой кислоты формулы (IV)
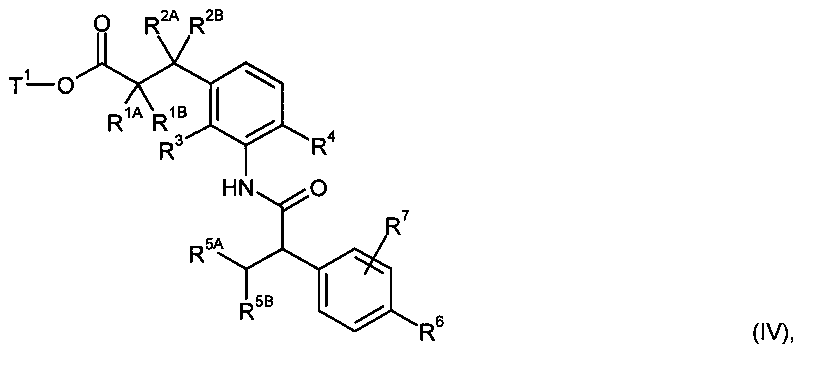
в которой R1A, R1B, R2A, R2B, R3, R4, R5A, R5B, R6, R7 и T1 имеют значения, указанные выше,
а затем отщепляют остаток сложного эфира Т1 при помощи основного или кислотного сольволиза или в случае, если Т1 представляет собой бензил, также с помощью гидрогенолиза, с получением карбоновой кислоты формулы (I), и при необходимости соединения формулы (I) с помощью методов, известных специалисту, разделяют на их энантиомеры и/или диастереомеры и/или превращают в их физиологически приемлемые соли с помощью соответствующих (i) растворителей и/или (ii) оснований.
Комментарии