Аналоги циклопамина - RU2486194C2
Код документа: RU2486194C2
Описание
В данной заявке заявлен приоритет согласно заявкам USSN 60/878018, поданной 28 декабря 2006 и USSN 60/941596, поданной 1 июня 2007, обе из которых включены сюда посредством ссылок полностью.
УРОВЕНЬ ТЕХНИКИ
Данное изобретение в общем относится к аналогам циклопамина и их фармацевтическим композициям, и способам получения таких аналогов и композиций. Эти соединения и композиции применяются для лечения расстройств, медиированных «ежовым» сигнальным путем, таких как рак.
Было показано, что ингибирование «ежового» пути при определенных раковых заболеваниях дает ингибирование роста опухоли. Например было показано, что анти-«ежовые» антитела антагонизируют функцию «ежового» пути и ингибируют рост опухолей. Также было показано, что низкомолекулярное ингибирование активности «ежового» пути вызывает смерть клеток во множестве типов рака.
Исследования в этой области преимущественно сфокусированы на выяснение биологии «ежового» пути и открытии новых ингибиторов «ежового» пути. Хотя ингибиторы «ежового» пути были идентифицированы, до сих пор существует необходимость в идентификации более мощных ингибиторов «ежового» пути.
В публикации PCT WO 2006/026430, опубликованной 9 марта 2006 и принадлежащей тому же правообладателю, что и данная заявка, описано множество аналогов циклопамина, где особое внимание уделяется тем, в которых имеется ненасыщенность в кольце А или В. В данном изобретении неожиданно мощные аналоги содержат полностью насыщенные кольца А и В.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
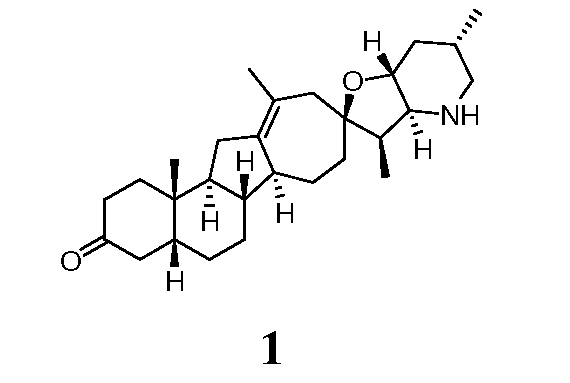
Данное изобретение относится к аналогам стероидных алкалоидов, таких как циклопамин, фармацевтическим композициям, содержащим эти соединения, и способам получения этих соединений. В одном варианте, данное изобретение относится к соединению, представленному следующей структурой:

или его фармацевтически приемлемой соли;
где R1 является H, алкилом, -OR, амино, сульфонамидо, сульфамидо, -OC(O)R5, -N(R5)C(O)R5 или сахаром;
R2 является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, нитрилом или гетероциклоалкилом;
или R1 и R2 вместе образуют =O, =S, =N(OR), =N(R), =N(NR2), =C(R)2;
R3 является H, алкилом, алкенилом или алкинилом;
R4 является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом, гетероаралкилом, галоалкилом, -OR5, -C(O)R5, -CO2R5, -SO2R5, -C(O)N(R5)(R5), -[C(R)2]q-R5, -[(W)-N(R)C(O)]qR5, -[(W)-C(O)]qR5, -[(W)-C(O)O]qR5, -[(W)-OC(0)]qR5, -[(W)-SO2]qR5, -[(W)-N(R5)SO2]qR5, -[(W)-C(O)N(R5)]qR5, -[(W)-O]qR5, -[(W)-N(R)]qR5, -W-NR53+X- или -[(W)-S]qR5;
где каждый W независимо является дирадикалом;
каждый q независимо равен 1, 2, 3, 4, 5 или 6;
X- является галогенидом;
каждый R5 независимо является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом, гетероаралкилом или -[C(R)2]P-R6; где p равно 0-6; или два любых R5 могут быть взяты вместе с образованием 4-8-членного необязательно замещенного кольца, которое содержит 0-3 гетероатома, выбранных из N, O, S и P;
каждый R6 независимо является гидроксилом, -N(R)COR, -N(R)C(O)OR, -N(R)SO2(R), -C(O)N(R)2, -OC(O)N(R)(R), -SO2N(R)(R), -N(R)(R), -COOR, -C(O)N(OH)(R), -OS(O)2OR, -S(O)2OR, -OP(O)(OR)(OR), -NP(O)(OR)(OR) или -P(O)(OR)(OR); и
каждый R независимо является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом или аралкилом;
при условии, что если R2, R3 и R4 являются H; R1 не является гидроксилом или сахаром; далее
при условии, что если R4 является гидроксилом, то R1 не является сахаром или гидроксилом, и R1 и R2 не являются C=O.
В определенных вариантах, R1 является H, гидроксилом, алкоксилом, арилокси или амино. В других вариантах, R1 и R2 взятые вместе с атомом углерода, к которому они присоединены, образуют =O, =N(OR) или =S. В других вариантах R3 является H и/или R4 является H, алкилом, гидроксилом, аралкилом, -[C(R)2]q-R5, -[(W)-N(R)C(O)]qR5, -[(W)-N(R)SO2]qR5, -[(W)-C(O)N(R)]qR5, -[(W)-O]qR5, -[(W)-C(O)]qR5 или -[(W)-C(O)O]qR5. В других вариантах, R1 является H или -OR, R2 является H или алкилом, и R4 является H. В других, R2 является H или алкилом, R3 является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом или аралкилом; и/или R4 является H, алкилом, аралкилом, -[(W)-N(R)C(O)]qR5, -[(W)-N(R)SO2]qR5, -[(W)-C(O)N(R)]qR5, -[(W)-O]qR5, -[(W)-C(O)]qR5, или -[(W)-C(O)O]qR5. В других вариантах R1 является сульфонамидо.
В другом варианте данное изобретение относится к соединениям, выбранным из группы, включающей:




или их фармацевтически приемлемым солям.
В определенных вариантах указанные выше соединения выделены.
В другом варианте данное изобретение относится к выделенным соединениям, выбранным из группы, включающей:

или их фармацевтически приемлемым солям.
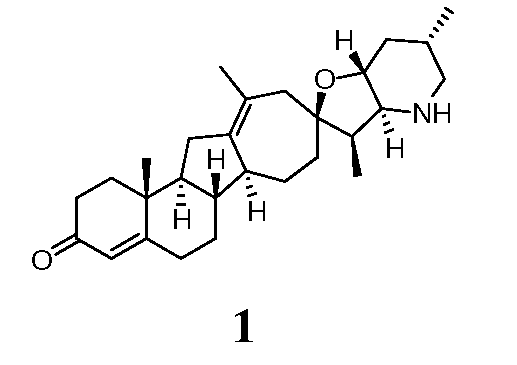
В другом варианте данное изобретение относится к соединению, представленному следующей формулой:
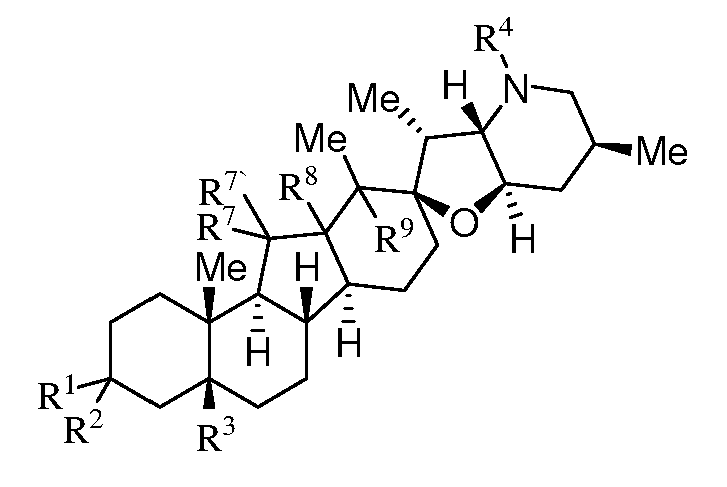
или его фармацевтически приемлемой соли;
где R1 является H, алкилом, -OR, амино, сульфонамидо, сульфамидо, -OC(O)R5, -N(R5)C(O)R5 или сахаром;
R2 является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, нитрилом или гетероциклоалкилом;
или R1 и R2 вместе образуют =O, =S, =N(OR), =N(R), =N(NR2), =C(R)2;
R3 является H, алкилом, алкенилом или алкинилом;
R4 является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом, гетероаралкилом, галоалкилом, -OR5, -C(O)R5, -CO2R5, -SO2R5, -C(O)N(R5)(R5), -[C(R)2]q-R5, -[(W)-N(R)C(O)]qR5, -[(W)-C(O)]qR5, -[(W)-C(O)O]qR5, -[(W)-OC(O)]qR5, -[(W)-SO2]qR5, -[(W)-N(R5)SO2]qR5, -[(W)-C(O)N(R5)]qR5, -[(W)-O]qR5, -[(W)-N(R)]qR5, -W-NR53+X-, или -[(W)-S]qR5;
где каждый W независимо является дирадикалом;
каждый q независимо равен 1, 2, 3, 4, 5 или 6;
X- является галогенидом;
каждый R независимо является алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом, гетероаралкилом или -[C(R)2]P-R6; где p равно 0-6; или любые два R5 могут быть взяты вместе с образованием 4-8-членного необязательно замещенного кольца, которое содержит 0-3 гетероатома, выбранных из N, O, S и P;
каждый R6 независимо является гидроксилом, -N(R)COR, -N(R)C(O)OR, -N(R)SO2(R), -C(O)N(R)2, -OC(O)N(R)(R), -SO2N(R)(R), -N(R)(R), -COOR, -C(O)N(OH)(R), -OS(O)2OR, -S(O)2OR, -OP(O)(OR)(OR), -NP(O)(OR)(OR) или -P(O)(OR)(OR);
каждый R независимо является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом или аралкилом;
каждый R7 и R7' является H; или R7 и R7' вместе образуют =O;
R8 и R9 являются H; или
R8 и R9 вместе образуют связь; и
при условии, что если R3, R4, R8, R9 являются H и R7 и R7' вместе образуют =O; R1 не может быть гидроксилом и R2 не может быть H;
при условии, что если R3, R4, R8, R9 являются H и R7 и R7' вместе образуют =O; R1 не может быть ацетатом и R2 не может быть H;
при условии, что если R3, R4, R8, R9 являются H и R7 и R7' являются H; R1 и R2 вместе не могут быть =O; и
при условии, что если R3, R4, R8, R9 являются H и R7 и R7' являются H; R1 и R2 не могут быть H.
В некоторых вариантах соединение является эпимерно чистым и/или выделенным. В других вариантах R4 является алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом, гетероаралкилом, галоалкилом, -OR5, -[C(R)2]q-R5, -[(W)-N(R)C(O)]qR5, -[(W)-C(O)]qR5, -[(W)-C(O)O]qR5, -[(W)-OC(O)]qR5, -[(W)-SO2]qR5, -[(W)-N(R5)SO2]qR5, -[(W)-C(O)N(R5)]qR5, -[(W)-O]qR5, -[(W)-N(R)]qR5, или -[(W)-S]qR5. Каждый из R7 и R7' может быть H. Кроме того, R1 и R2 вместе образуют =O.
В другом варианте данное изобретение относится к соединениям, выбранным из группы, включающей:

или их фармацевтически приемлемым солям.
В определенных вариантах указанные выше соединения являются эпимерно чистыми и/или выделенными.
В другом варианте данное изобретение относится к эпимерно чистому соединению, выбранному из группы, включающей:

или их фармацевтически приемлемым солям.
Другой аспект данного изобретения относится к фармацевтической композиции, включающей любое из указанных выше соединений и фармацевтически приемлемый эксципиент.
В одном варианте данное изобретение относится к способу получения циклопропильных производных циклопамина и родственных аналогов, имеющих формулу 136:

где
Y является CR7R8;
R1 является H, алкилом, амино, гидроксилом, карбоксилом, карбамоилом, алкокси, гидроксилом, сахаром или защищенной гидроксильной группой;
R2 является H, алкилом, алкенилом, алкинилом, нитрилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом, или гетероаралкилом; или R1 и R2 вместе образуют =O, =S, =N(OR9), =N(R9), =C(R9)2 или =N(N(R9)2);
каждый из R3, R4 и R5 независимо является Н, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом или гетероаралкилом; или R3 и R4 или R4 и R5 вместе образуют связь;
R6 является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом, гетероаралкилом, галоалкилом, -OR9, -C(O)R9, -CO2R9, -SO2R9, -C(O)N(R9)(R9), -[C(R9)2]qR9, -[(W)-N(R9)C(O)]qR9, -[(W)-C(O)]qR9, -[(W)-C(O)O]qR9, -[(W)-OC(O)]qR9, -[(W)-SO2]qR9, -[(W)-N(R9)SO2]qR9, -[(W)-C(O)N(R9)]qR9, -[(W)-O]qR9, -[(W)-N(R9)]qR9, -[(W)-S]qR9, или защитной группой азота; где каждый W независимо является a дирадикалом; каждый q независимо равен 1, 2, 3, 4, 5 или 6;
каждый из R7 и R8 независимо является H, алкилом, алкенилом, арилом, нитрилом, амидо, галогенидом или сложным эфиром; и
каждый R9 независимо является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом или гетероаралкилом.
Способ включает стадии взаимодействия соединения формулы 136a с циклопропанирующим агентом на основе фосфата галоалкилцинка с получением соединения формулы 136:

где
R1, R2, R3, R4, R5, R6 такие, как определены для соединения 136.
В другом варианте данное изобретение относится к способам получения соединения формулы 137:

где
Y является CR7R8;
R1 является H, алкилом, амино, гидроксилом, карбоксилом, карбамоилом, алкокси, гидроксилом, сахаром или защищенной гидроксильной группой;
R2 является H, алкилом, алкенилом, алкинилом, нитрилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом или гетероаралкилом; или R1 и R2 вместе образуют =O, =S, =N(OR9), =N(R9), =C(R9)2 или =N(N(R9)2);
каждый из R3, R4 и R5 независимо является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом или гетероаралкилом; или R3 и R4 или R4 и R5 вместе образуют связь;
R6 является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом, гетероаралкилом, галоалкилом, -OR9, -C(O)R9, -CO2R9, -SO2R9, -C(O)N(R9)(R9), -[C(R9)2]qR9, -[(W)-N(R9)C(O)]qR9, -[(W)-C(O)]qR9, -[(W)-C(O)O]qR9, -[(W)-OC(O)]qR9, -[(W)-SO2]qR9, -[(W)-N(R9)SO2]qR9, -[(W)-C(O)N(R9)]qR9, -[(W)-O]qR9, -[(W)-N(R9)]qR9, -[(W)-S]qR9 или защитной группой азота;
где каждый W независимо является дирадикалом;
каждый q независимо является 1, 2, 3, 4, 5 или 6;
каждый R7 и R8 независимо является H, алкилом, алкенилом, арилом, нитрилом, амидо, галогенидом или сложным эфиром; и
каждый R9 независимо является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом или гетероаралкилом. Способ включает стадии: взаимодействия соединения формулы 137a с циклопропанирующим агентом на основе фосфата галоалкилцинка;

где
R1, R2, R3, R4, R5, R6 такие, как определены для соединения 137; с получением соединения формулы 137b

где
R1, R2, R3, R4, R5, R6 и Y такие, как определены для соединения 137; с последующим взаимодействием соединения формулы 137b с кислотой с получением соединения формулы 137.
В определенных вариантах R7 и R8 оба могут быть H; в других вариантах R1 может быть защищенным гидроксилом; и/или R6 является защитной группой азота.
В определенных вариантах циклопропанирующий агент на основе фосфата галоалкилцинка получают объединением фосфорной кислоты формулы 141a, диалкилцинка и дигалоалкилана формулы 141b:

где каждый из X и X' независимо является хлоридом, бромидом или йодидом;
каждый из R7 и R8 независимо является H, алкилом, галогенидом, амидо, нитро или сложным эфиром;
каждый из R10 и R11 независимо является алкилом, алкенилом, аралкилом, арилом, гетероарилом, гетероаралкилом; или R10 и R11 вместе имеют формулу 141c, 141d или 141e;

где m независимо равно для каждого случая 0, 1, 2, 3 или 4; n независимо равно для каждого случая 0, 1 или 2; и каждый из R12, R13, R14, R15, R16, R17 и R18 независимо является алкилом, арилом, аралкилом или галогенидом.
В другом варианте данное изобретение относится к способу получения соединения формулы 142:

Способ включает стадии взаимодействия соединения формулы 142a с циклопропанирующим агентом с получением соединения формулы 142b; и


объединения соединения формулы 142b с кислотой с получением соединения формулы 142;
где
Y является CR7R8; R1 является защищенной гидроксильной группой;
R2 является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом или гетероаралкилом; каждый из R3, R4 и R5 независимо является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом или гетероаралкилом; или
R3 и R4 или R4 и R5 вместе образуют связь; R6 является защитной группой азота;
каждый из R7 и R8 независимо является H, алкилом, алкенилом, арилом, нитрилом, амидо, галогенидом или сложным эфиром; и
каждый R9 независимо является H, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом или гетероаралкилом.
В определенных вариантах R7 и R8 оба могут быть H; в других вариантах защищенной гидроксильной группой может быть сложный эфир или карбонат; защитой азота может быть карбамат или амид; R7 и R8 оба могут быть H и защита азота может быть карбаматом или амидом; R2 и R3 могут быть H и R4 и R5 взятые вместе могут образовывать связь; и/или циклопропанирующий агент получают из дигалоалкана и металлических соединений (например, диалкилцинка или пары цинк-медь).
В определенных вариантах циклопропанирующий агент получают из соединений дигалоалкана и соединений диалкилцинка, и соединений фосфорной кислоты или ее соли. Соединения фосфорной кислоты имеют структуру формулы 151:

или ее соль;
где
каждый из R10 и R11 независимо является алкилом, алкенилом, аралкилом, арилом, гетероарилом, гетероаралкилом; или R10 и R11 вместе имеют формулу 151a, 151b или 151c;
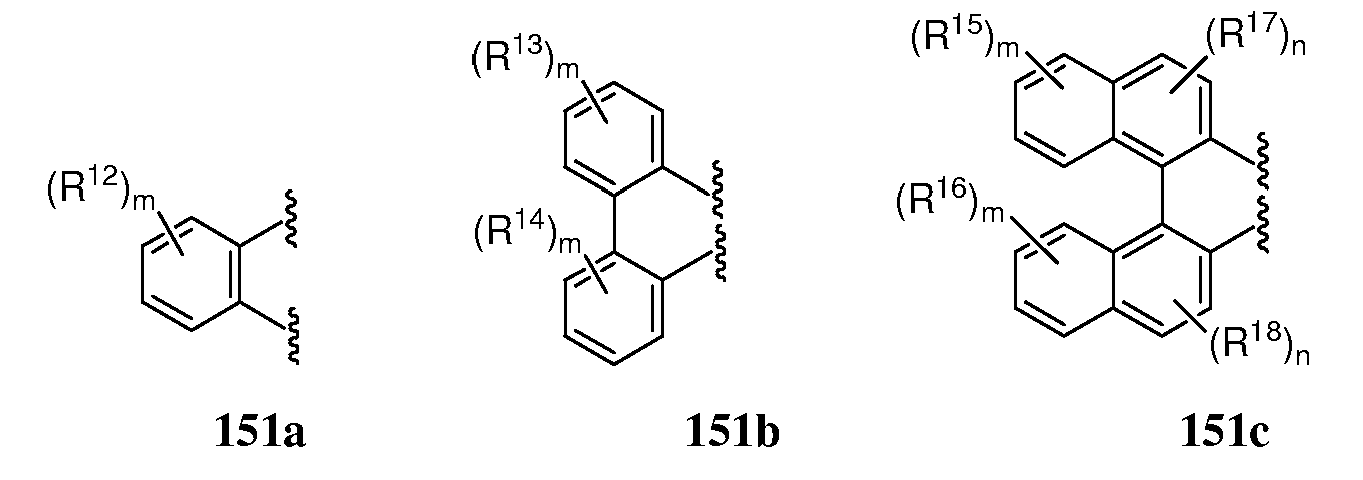
где
m независимо для каждого случая равно 0, 1, 2, 3 или 4; n независимо для каждого случая равно 0, 1 или 2; каждый из R12, R13, R14, R15, R16, R17 и R18 независимо является алкилом, арилом, аралкилом или галогенидом.
В определенных вариантах кислотой является кислота Бренстеда (например, уксусная кислота, трифторметансульфоновая кислота, фосфорная кислота, метансульфоновая кислота или HCl). В других вариантах кислотой является кислота Льюиса (например, BF3, хлорид цинка, метансульфонат цинка или соль цинка).
Данное изобретение также относится к способу получения соединения формулы 156:

Способ включает стадии:
взаимодействия соединения формулы 156a с циклопропанирующим агентом с получением соединения формулы 156b; и

объединения соединения формулы 156b с кислотой с получением соединения формулы 156;
где R1 является защитной группой кислорода, выбранной из группы, включающей формиат, ацетат, хлорацетат, дихлорацетат, трихлорацетат, пивалоат, бензоат, алкилкарбонат, алкенилкарбонат, арилкарбонаты, аралкилкарбонат, 2,2,2-трихлорэтилкарбонат, алкоксиметиловый эфир, аралкоксиметиловый эфир, алкилтиометиловый эфир, аралкилтиоэфир, арилтиоэфир, триалкилсилиловый эфир, алкиларилсилиловый эфир, бензиловый эфир, арилметиловый эфир, аллиловый эфир; и R2 является защитной группой азота, выбранной из группы, включающей формил, хлорацетил, трихлорацетил, трифторацетил, фенилацетил, бензоилы, алкилкарбаматы, аралкилкарбаматы, арилкарбаматы, аллил, аралкил, триарилметил, алкоксиметил, аралкоксиметил, N-2-цианоэтил, диарилфосфинамиды, диалкилфосфинамидаты, диарилфосфинамидаты и триалкилсилил.
В определенных вариантах циклопропанирующий агент получают объединением фосфорной кислоты формулы 58a, диалкилцинка и дигалоалкилана формулы 158b:
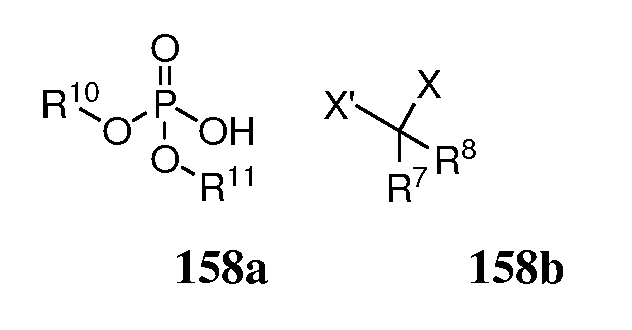
где
каждый из X и X' независимо является хлоридом, бромидом или йодидом; каждый из R7 и R8 независимо является H, алкилом, галогенидом, амидо или сложным эфиром; каждый R10 и R11 независимо является алкилом, алкенилом, аралкилом, арилом, гетероарилом, гетероаралкилом; или R10 и R11 взятые вместе имеют формулу 158c, 158d или 158e;

где
m независимо для каждого случая равно 0, 1, 2, 3 или 4; n независимо для каждого случая равно 0, 1 или 2; каждый из R12, R13, R14, R15, R16, R17 и R18 независимо является алкилом, арилом, аралкилом или галогенидом.
Защитная группа кислорода может быть, в некоторых вариантах, выбрана из алкилкарбонатов, аралкилкарбонатов (например, бензилкарбоната), бензоатов, пивалоата или формиата. Защитная группа азота может быть выбрана из бензоила, трихлорацетила, трифторацетила, формила, алкилкарбаматов, аралкилкарбаматов (например, бензилкарбамата), арилкарбаматов, диарилфосфинамидов, диалкилфосфинамидатов или диарилфосфинамидатов.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Определения терминов, применяемых в данном описании, включают принятые в данной области техники определения, известные для каждого термина в химической и фармацевтической областях. Где это приемлемо, представлены примеры. Определения применяются к терминам так, как они применяются во всем описании, если не указано иначе в конкретных случаях, либо отдельно, либо как часть большей группы.
В данном описании определение для каждого выражения, например, алкил, m, n, и т.д., если оно возникает более одного раза в структуре, является независимым от его определений в любой части той же структуры.
Термин "ациламино" относится к группе, которая может быть представлена общей формулой:

где R50 и R54 независимо является водородом, алкилом, алкенилом или -(CH2)m-R61,
где R61 является арилом, циклоалкилом, циклоалкенилом, гетероциклом или полициклом; и m равно нулю или целому числу в интервале от 1 до 8.
Термины "алкенил" и "алкинил" относятся к ненасыщенным алифатическим группам, аналогичным по длине и возможному замещению описанным ниже алкилам, но которые содержат, по крайней мере, одну двойную или тройную связь, соответственно.
Термины "алкоксил" или "алкокси" относятся к алкильной группе, такой как определена выше, имеющей присоединенный к ней кислородный радикал. Типовые алкоксильные группы включают метокси, этокси, пропилокси, трет-бутилокси и подобные.
Термин "алкил" относится к радикалу насыщенных алифатических групп, включающих прямые алкильные группы, разветвленные алкильные группы, циклоалкильные (алициклические) группы, замещенные алкилом циклоалкильные группы и замещенные циклоалкилом алкильные группы. В определенных вариантах прямые или разветвленные алкилы имеют 30 или менее атомов углерода в основной цепи (например, C1-C30 для прямой цепи, C3-C30 для разветвленной цепи), 20 или менее. Также, определенные циклоалкилы имеют 3-10 атомов углерода в кольцевой структуре, и другие имеют 5, 6 или 7 атомов углерода в кольцевой структуре.
Термин "алкилтио" относится к алкильной группе, такой как определена выше, имеющей присоединенную к ней серу. В определенных вариантах "алкилтио" группа представлена одним из -S-алкила, -S-алкенила, -S-алкинила и -S-(CH2)m-R61, где m и R61 такие, как определены выше. Типовые алкилтиогруппы включают метилтио, этилтио и подобные.
Термин "амидо" известен в данной области техники как амино-замещенный карбонил, и включает группу, которая может быть представлена общей формулой:

где R50 и R51 каждый независимо являются водородом, алкилом, алкенилом, -(CH2)m-R61, R61 является арилом, циклоалкилом, циклоалкенилом, гетероциклом или полициклом; и m равно нулю или целому числу в интервале от 1 до 8, или R50 и R51, взятые вместе с атомом N, к которому они присоединены, образуют гетероцикл, имеющий от 4 до 8 атомов в кольцевой структуре. Определенные варианты амида в соответствии с данным изобретением не включают имиды, которые могут быть нестабильными.
Термины "амин" и "амино" известны в данной области техники и оба относятся к незамещенным и замещенным аминам, например, группе, которая может быть представлена общими формулами:

где R50 и R51 (и необязательно R52) каждый независимо является водородом, алкилом, алкенилом или -(CH2)m-R61, где R61 такой, как определен выше. Таким образом, термин "алкиламин" включает аминовую группу, такую как определена выше, имеющую присоединенный к ней замещенный или незамещенный алкил, т.е., по крайней мере, один из R50 и R51 является алкильной группой.
Термин "аралкил", в данном описании, относится к алкильной группе, замещенной арильной группой (например, ароматической или гетероароматической группой).
Термин "арил" в данном описании включает 5-, 6- и 7-членные ароматические группы с одним кольцом, которые могут включать от нуля до четырех гетероатомов, например, бензол, антрацен, нафталин, пирен, пиррол, фуран, тиофен, имидазол, оксазол, тиазол, триазол, пиразол, пиридин, пиразин, пиридазин и пиримидин и подобные. Эти арильные группы, имеющие гетероатомы в кольцевой структуре, также могут быть обозначены как "арилгетероциклы" или "гетероароматические соединения." Ароматическое кольцо может быть замещено в одном или более положениях кольца такими заместителями, как описаны выше, например, галогеном, азидом, алкилом, аралкилом, алкенилом, алкинилом, циклоалкилом, гидроксилом, алкоксилом, амино, нитро, сульфгидрилом, имино, амидо, фосфонатом, фосфинатом, карбонилом, карбоксилом, силилом, простым эфиром, алкилтио, сульфонилом, сульфонамидо, кетоном, альдегидом, сложным эфиром, гетероциклилом, ароматическими или гетероароматическими группами, -CF3, -CN или подобными. Термин "арил" также включает полициклические кольцевые системы, имеющие два или более циклических кольца, где два или более атомов углерода являются общими для двух соседних колец (кольца являются «конденсированными кольцами»), где, по крайней мере, одно из колец является ароматическим, например, другие циклические кольца могут быть циклоалкилами, циклоалкенилами, циклоалкинилами, арилами и/или гетероциклилами.
Термин «кислота Бренстеда» относится к любому веществу, которое может действовать как донор иона водорода (протона).
Термин "карбоксил" включает сложные эфиры, тиокарбоксил, альдегиды, кетоны и подобные и, таким образом, включает такие группы, которые могут быть представлены общими формулами:
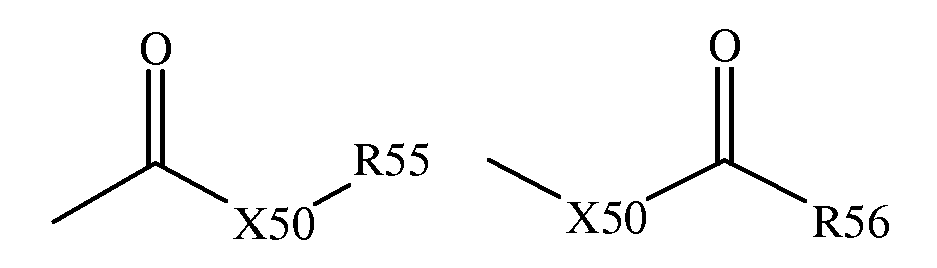
где X50 является связью или является кислородом или серой, и каждый из R55 и R56 независимо является водородом, алкилом, алкенилом, -(CH2)m-R61, где m и R61 такие, как определены выше.
Термин «дирадикал» относится к любому ряду двухвалентных групп, выбранных из алкила, алкенила, алкинила, арила, циклоалкила, гетероциклоалкила, аралкила, гетероарила и гетероаралкила. Например,





Термин "галоалкил", в данном описании, относится к алкильной группе, в которой любой из от 1 до всех атомов водорода замещен галогенидом. "Пергалоалкил" является соединением, в котором все атомы водорода замещены галогенидом.
Термин "гетероатом" в данном описании означает атом любого элемента, отличного от углерода или водорода. Примеры гетероатомов включают бор, азот, кислород, фосфор, серу и селен.
Термины "гетероциклил" или "гетероциклическая группа" относится к 3-10-членным кольцевым структурам, в некоторых случаях, 3-7-членным кольцам, где кольцевые структуры включают от одного до четырех гетероатомов. Гетероциклы также могут быть полициклами. Гетероциклильные группы включают, например, тиофен, тиантрен, фуран, пиран, изобензофуран, хромен, ксантен, феноксантин, пиррол, имидазол, пиразол, изотиазол, изоксазол, пиридин, пиразин, пиримидин, пиридазин, индолизин, изоиндол, индол, индазол, пурин, хинолизин, изохинолин, хинолин, фталазин, нафтиридин, хиноксалин, хиназолин, циннолин, птеридин, карбазол, карбонил, фенантридин, акридин, пиримидин, фенантролин, феназин, фенарсазин, фенотиазин, фуразан, феноксазин, пирролидин, оксолан, тиолан, оксазол, пиперидин, пиперазин, морфолин, лактоны, лактамы, такие как азетидиноны и пирролидиноны, сультамы, сультоны и подобные. Гетероциклическое кольцо может быть замещено в одном или более положениях такими заместителями, как описаны выше, например, галогеном, алкилом, аралкилом, алкенилом, алкинилом, циклоалкилом, гидроксилом, амино, нитро, сульфгидрилом, имино, амидо, фосфонатом, фосфинатом, карбонилом, карбоксилом, силилом, простым эфиром, алкилтио, сульфонилом, кетоном, альдегидом, сложным эфиром, гетероциклилом, ароматической или гетероароматической группой, -CF3, -CN или подобными.
Термин "выделенный" в сочетании с соединением в соответствии с данным изобретением означает то, что соединение не находится в клетке или организме, и соединение отделено от некоторых или всех компонентов, которые обычно сопровождают его в природе.
Термин «кислота Льюиса» относится к любому веществу, которое может действовать в качестве акцептора электронной пары.
Если количество атомов углерода не обозначено, «низший алкил» означает алкильную группу, такую как определена выше, но имеющую от одного до десяти атомов углерода, в некоторых вариантах, от одного до шести атомов углерода, в основной цепи. Также «низший алкенил» и «низший алкинил» имеет подобные длины цепей. Определенные алкильные группы включают низшие алкилы. В некоторых вариантах заместитель, обозначенный здесь как алкил, является низшим алкилом.
В данном описании термин "нитро" означает -NO2; термин "галоген" означает -F, -Cl, -Br или -I; термин "сульфгидрил" означает -SH; термин "гидроксил" означает -OH; и термин "сульфонил" означает -SO2-.
Термин "оксо" относится к карбонильному кислороду (=O).
Термины "полициклил" или "полициклическая группа" относятся к двум или более кольцам (например, циклоалкилам, циклоалкенилам, циклоалкинилам, арилам и/или гетероциклилам), в которых два или более атомов углерода являются общими для двух соседних колец, например, кольца являются "конденсированными кольцами". Кольца, которые соединены через не соседние атомы, называются "мостиковыми" кольцами. Каждое из колец полицикла может быть замещено такими заместителями, как описаны выше, например, галогеном, алкилом, аралкилом, алкенилом, алкинилом, циклоалкилом, гидроксилом, амино, нитро, сульфгидрилом, имино, амидо, фосфонатом, фосфинатом, карбонилом, карбоксилом, силилом, простым эфиром, алкилтио, сульфонилом, кетоном, альдегидом, сложным эфиром, гетероциклилом, ароматической или гетероароматической группой, -CF3, -CN или подобными.
Термин "эпимерно чистое" в сочетании с соединением в соответствии с данным изобретением означает, что соединение по существу не содержит стереоизомеры соединения, где конфигурация стереогенного центра, к которому присоединен R3, инвертирована. Например, эпимерно чистое соединение представлено следующей формулой:

где R1, R2, R3, R4, R7, R7', R8 и R9 такие, как определены выше, и по существу не содержит соединений, представленных следующей формулой:

где R1, R2, R3, R4, R7, R7', R8 и R9 такие, как определены выше. Эпимерно чистые соединения содержат менее 20% массовых, менее около 15% массовых, менее около 10% массовых, менее около 5% массовых или менее около 3% массовых стереоизомерных соединений, где конфигурация стереогенного центра, который связан с R3, инвертирована относительно соединения.
Фраза «защитная группа» в данном описании означает временные заместители, которые защищают потенциально реакционноспособную функциональную группу от нежелательных химических превращений. Примеры таких защитных групп включают сложные эфиры карбоновых кислот, силиловые эфиры спиртов, и ацетали и кетали альдегидов и кетонов, соответственно. Химия защитных групп широко представлена (Greene, T. W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 2nd ed.; Wiley: New York, 1991). В некоторых случаях защищенная функциональная группа и защитная группа вместе обозначаются как одна группа. Например, фрагмент, показанный ниже, иногда обозначается как бензилкарбонат; т.е. защищенный (подчеркнутый) атом O составляет часть карбоната.
Также, показанный ниже фрагмент, в котором защищенный N составляет часть карбамата, обозначен как бензилкарбамат.

Термин «сахар» в данном описании относится к природному или синтетическому моносахариду, дисахариду или олигосахариду, содержащему одно или более пиранозных или фуранозных кольца. Сахар может быть ковалентно связан со стероидным алкалоидом в соответствии с данным изобретением через простую эфирную связь или через алкильную связь. В определенных вариантах сахаридная группа может быть ковалентно связана со стероидным алкалоидом в соответствии с данным изобретением в аномерном центре сахаридного кольца. Сахара могут включать, но не ограничены ими, рибозу, арабинозу, ксилозу, ликсозу, аллозу, альтрозу, глюкозу, маннозу, гулозу, идозу, галактозу, талозу, глюкозу и трегалозу.
Термин "сульфонамидо" или "сульфонамид" в данном описании включает группу, имеющую любую из следующих формул:

где R50 и R56 такие, как определены выше.
Термины "трифлил", "тозил", "мезил" и "нонафлил" относятся к трифторметансульфонилу, п-толуолсульфонилу, метансульфонилу и нонафторбутансульфонилу, соответственно. Термины "трифлат", "тозилат", "мезилат" и "нонафлат" относятся к функциональным группам сложного эфира трифторметансульфоната, сложного эфира п-толуолсульфоната, сложного эфира метансульфоната и сложного эфира нонафторбутансульфоната, и молекулам, которые содержат группы, соответственно.
Термин "тиоксо" относится к карбонильной сере (=S).
Должно быть понятно, что "замещение" или "замещен" включают безоговорочное условие, что такое замещение соответствует разрешенной валентности замещенного атома и заместителя, и что замещение дает стабильное соединение, например, которое не может спонтанно измениться вследствие, например, перегруппировки, циклизации, отщепления и т.д.
Определенные соединения в соответствии с данным изобретением могут существовать в конкретных геометрических или стереоизомерных формах. Данное изобретение охватывает все такие соединения, включая цис- и транс-изомеры, R- и S-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры, их рацемические смеси и их другие смеси, которые попадают в объем данного изобретения. Дополнительные асимметричные атомы углерода могут присутствовать в заместителе, таком как алкильная группа. Все такие изомеры, а также их смеси, включены в данное изобретение.
Как указано выше, определенные варианты данного изобретения могут содержать оснóвную функциональную группу, такую как амино или алкиламино, и поэтому способны к образованию фармацевтически приемлемых солей с фармацевтически приемлемыми кислотами. Термин "фармацевтически приемлемые соли" в этом смысле относится к относительно не токсичным, неорганическим и органическим кислотно-аддитивным солям соединений в соответствии с данным изобретением. Эти соли могут быть получены in situ в носителе для введения или в процессе получения дозированной формы, или отдельным взаимодействием очищенного соединения в соответствии с данным изобретением в форме свободного основания с подходящей органической или неорганической кислотой, и выделением полученной соли во время последующей очистки. Типовые соли включают гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, валерат, олеат, пальмитат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат и лаурилсульфонат и подобные. (См., например, Berge et al. (1977) "Pharmaceutical Salts", J. Pharm. Sci. 66:1-19)
Фармацевтически приемлемые соли соединений в соответствии с данным изобретением включают обычные нетоксичные соли или четвертичные аммониевые соли соединений, например, из нетоксичных органических или неорганических кислот. Например, такие обычные нетоксичные соли включают такие, которые получены из неорганических кислот, таких как хлористоводородная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная и подобные; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, пальмитиновая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изотионовая и подобные.
В других случаях, соединения в соответствии с данным изобретением могут содержать одну или более кислотных функциональных групп и, поэтому, способны образовывать фармацевтически приемлемые соли с фармацевтически приемлемыми основаниями. Термин "фармацевтически приемлемые соли" в этих случаях относится к относительно не токсичным неорганическим и органическим основно-аддитивным солям соединений в соответствии с данным изобретением. Эти соли также могут быть получены in situ в носителе для введения или в процессе получения дозированной формы, или отдельным взаимодействием очищенного соединения в форме свободной кислоты с подходящим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла, с аммиаком, или с фармацевтически приемлемым органическим первичным, вторичным или третичным амином. Типовые щелочные или щелочноземельные соли включают соли лития, натрия, калия, кальция, магния и алюминия, и подобные. Типовые органические амины, применяемые для получения основно-аддитивных солей, включают этиламин, диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и подобные. (См., например, Berge et al, выше)
Синтез стероидных алкалоидных соединений
Стероидные алкалоидные производные с увеличенным кольцом, описанные выше, могут быть получены непосредственно из природных стероидных алкалоидов или их синтетических аналогов. В определенных случаях исходными стероидными алкалоидами могут быть циклопамин или иервин. Эти стероидные алкалоиды могут быть закуплены из коммерческих источников или экстрагированы из Veratrum Californicum. Коротко, способ в соответствии с данным изобретением включает стадии циклопропанирования подходящих исходных стероидных алкалоидных производных с последующей перегруппировкой с расширением кольца циклопропильных производных. В некоторых случаях может быть желательно подходящим образом защищать или другим образом трансформировать реакционноспособные функциональные группы, присутствующие в молекуле до циклопропанирования. Например, спирт, присутствующий на R1, и вторичный азот, присутствующий на конденсированном фурано-пиперидиновом кольце, могут быть замещены до циклопропанирования. В определенных вариантах могут быть предпочтительны защитные группы, которые эффективно добавляют и удаляют из алкалоида, в процессе синтеза дают промежуточные соединения с улучшенными технологическими свойствами, и которые позволяют эффективную очистку полученных синтетических производных.
Примеры защитных групп кислорода включают, но не ограничены ими, формиат, ацетат, хлорацетат, дихлорацетат, трихлорацетат, пивалоат, бензоаты, алкилкарбонат, алкенилкарбонат, арилкарбонаты, аралкилкарбонат (например, бензилкарбонат), 2,2,2-трихлорэтилкарбонат, алкоксиметиловый эфир, аралкоксиметиловый эфир, алкилтиометиловый эфир, аралкилтиоэфир, арилтиоэфир, триалкилсилиловый эфир, алкиларилсилиловый эфир, бензиловый эфир, арилметиловый эфир и аллиловый эфир.
Примеры защитных групп азота включают, но не ограничены ими, формил, хлорацетил, трихлорацетил, трифторацетил, фенилацетил, бензоилы, бензамиды, алкилкарбаматы, аралкилкарбаматы (например, бензилкарбаматы), арилкарбаматы, аллил, аралкил, алкоксиметил, аралкоксиметил, N-2-цианоэтил, диарилфосфинамиды, диалкилфосфинамидаты, диарилфосфинамидаты и триалкилсилил.
Дополнительные защитные группы, которые могут применяться в способах в соответствии с данным изобретением, описаны у Green T. W.; Wuts P. G. Protective Groups in Organic Synthesis 3rd Edition, John Wiley & Sons, Inc. 1999.
Множество циклопропанирующих агентов могут применяться для циклопропанирования стероидного алкалоида. 1,1-галоалкилметаллические комплексы и реакционноспособные соединения, обозначенные как карбеноиды, обычно применяют для циклопропанирования олефинов. Эти реагенты обычно получают с применением дийодалкана или диазоалкана и металлических или металлорганических соединений, таких как Et2Zn, iBu3Al, самарий, медь, родий или палладий. В определенных вариантах Et2Zn и дийодметан применяют для получения 1,1-галоалкилметаллических соединений.
Реакционноспособность и простота применения 1,1-галоалкилцинковых комплексов могут быть модифицированы добавлением определенных реагентов, таких как кислоты. Полагают, что добавление кислоты к 1,1-галоалкилцинковым соединениям дает смешанную соль алкилцинка. В описанных ниже примерах биарилфосфорную кислоту объединяют с дийодметаном и диэтилцинком с получением предполагаемого циклопропанирующего агента на основе фосфата галоалкилцинка. Множество фосфорных кислот может применяться для получения предполагаемых фосфатов галоалкилцинка.
Также могут применяться другие известные методы циклопропанирования, такие, в которых применяются серные илиды для взаимодействия с олефином, конъюгированным с карбонилом, для присоединения CH2 или CH-алкила или CH-арила, и катализированное металлом разложение соединений диазоалкила и α-диазокарбонила, таких как диазометан и этилдиазоацетат: эти методы легко дают циклопропаны, имеющие алкильный, арильный, алкоксикарбонильный (-COOR) или ацильный заместители. Дополнительные циклопропанирующие агенты описаны в Masalov et al., Organic Letters (2004) Vol. 6, pp. 2365-8 и Hansen et al., Chem. Comm. (2006) 4838-40.
Циклопропильное кольцо может быть замещено или не замещено. Если циклопропильное кольцо замещено, группы, присоединенные к метилену циклопропана, будут установлены в кольцо D после перегруппировки и расширения кольца.
Реакции циклопропанирования могут проводиться в апротонном растворителе. Подходящие растворители включают простые эфиры, такие как диэтиловый эфир, 1,2-диметоксиэтан, диглим, трет-бутилметиловый эфир, тетрагидрофуран и подобные; галогенированные растворители, такие как хлороформ, дихлорметан, дихлорэтан и подобные; алифатические или ароматические растворители, такие как бензол, ксилол, толуол, гексан, пентан и подобные; сложные эфиры и кетоны, такие как этилацетат, ацетон и 2-бутанон; полярные апротонные растворители, такие как ацетонитрил, диметилсульфоксид, диметилформамид и подобные; или сочетания двух или более растворителей. В определенных вариантах дихлорметан является растворителем, применяемым для циклопропанирования с применением диалкилцинка и дийодметана.
В описанных ниже примерах раствор, содержащий циклопропанирующий агент, получают сначала добавлением раствора фосфорной кислоты к раствору диэтилцинка, с последующим добавлением дийодметана к реакционному раствору. Циклопропанирующий субстрат затем добавляют к этому раствору. Альтернативно, циклопропанирующий агент может быть получен в присутствии циклопропанирующего субстрата изменением порядка добавления реагентов. В определенных вариантах, реакцию циклопропанирования проводят сначала добавлением фосфорной кислоты к раствору диалкилцинка, с последующим добавлением циклопропанирующего субстрата, и, наконец, добавляют дигалоалкан. С применением этого метода циклопропанирующий агент получают в контролируемых условиях, и затем его сразу же подвергают взаимодействию с циклопропанирующим субстратом. Описанные здесь методы циклопропанирования также могут применяться для циклопропанирования других полициклических соединений, например, имеющих стероидные основные цепи.
После синтеза циклопропанированной стероидной алкалоидной основной цепи, соединение может быть превращено с применением множества реакций функционализации, известных в данной области техники. Типовые примеры включают реакции сочетания палладия с алкенилгалогенидами или арилгалогенидами, окисления, восстановления, реакции с нуклеофилами, реакции с электрофилами, перициклические реакции, радикальные реакции, введение защитных групп, удаление защитных групп и подобные.
В присутствии кислот Льюиса или Бренстеда, циклопропильные аналоги претерпевают перегруппировку и расширение кольца с получением стероидных алкалоидных аналогов, в которых кольцо D расширено на один атом углерода.
Циклопропанирование и расширение кольца могут проходить в две стадии в одном реакторе, или в две стадии в двух реакторах. Если циклопропанирование и расширение кольца проводят в одном реакторе, кислоту, применяемую для инициации перегруппировки с расширением кольца, добавляют после завершения реакции циклопропанирования. В определенных условиях соли цинка, образующиеся в процессе циклопропанирования стероидного алкалоида, сами могут действовать как кислоты Льюиса для катализа перегруппировки с расширением кольца. Реакционноспособность солей цинка, образованных после циклопропанирования, может быть модифицирована добавлением кислот для получения более активных кислот Льюиса.
Как описано ниже в примерах, метансульфоновую кислоту добавляют в реактор циклопропанирования после завершения циклопропанирования. Дополнительные примеры подходящих кислот включают, но не ограничены ими, соли цинка, соединения бора, соли магния, соли титана, соли индия, соли алюминия, соли олова, соли лантана, трифторметансульфоновую кислоту, диарилоксифосфорные кислоты, уксусную кислоту и HCl. В определенных вариантах данного изобретения применяемой кислотой Льюиса является соль цинка или BF3.
Эти аналоги с расширенным кольцом могут быть далее функционализированы с применением множества реакций функционализации, известных в данной области техники. Типовые примеры включают реакции сочетания палладия с алкенилгалогенидами или арилгалогенидами, окисления, восстановления, реакции с нуклеофилами, реакции с электрофилами, перициклические реакции, радикальные реакции, добавления защитных групп, удаление защитных групп и подобные.
Применение соединений
«Ежовый» сигнальный путь является существенным на многих стадиях развития, особенно при формировании лево-правой симметрии. Потеря или снижение «ежового» сигнального пути приводит к множественным нарушениям развития и патологиям, одним из наиболее существенных из которых является циклопия.
Было показано, что множество опухолей и пролиферативных состояний зависят от «ежового» пути. На рост таких клеток и выживание влияет лечение соединениями в соответствии с данным изобретением. Недавно было описано, что активирование мутаций «ежового» пути возникает в спорадичной карциноме базальных эпидермоцитов (Xie et al. (1998) Nature 391: 90-2) и примитивных нейроэктодермальных опухолях центральной нервной системы (Reifenberger et al. (1998) Cancer Res 58: 1798-803). Неконтролируемая активация «ежового» пути также была показана во множестве типов рака, таких как раки ЖК пути, включая рак поджелудочной железы, рак пищевода, рак желудка (Berman et al. (2003) Nature 425: 846-51, Thayer et al. (2003) Nature 425: 851-56) рак легких (Watkins et al. (2003) Nature 422: 313-317), рак простаты (Karhadkar et al (2004) Nature 431: 707-12, Sheng et al. (2004) Molecular Cancer 3: 29-42, Fan et al. (2004) Endocrinology 145: 3961-70), рак грудной железы (Kubo et al. (2004) Cancer Research 64: 6071-74, Lewis et al. (2004) Journal of Mammary Gland Biology и Neoplasia 2: 165-181) и печеночно-клеточный рак (Sicklick et al. (2005) ASCO conference, Mohini et al. (2005) AACR conference).
Например, было показано, что низкомолекулярное ингибирование «ежового» пути ингибирует рост карциномы базальных клеток (Williams, et al., 2003 PNAS 100: 4616-21), медуллобластомы (Berman et al., 2002 Science 297: 1559-61), рака поджелудочной железы (Berman et al., 2003 Nature 425: 846-51), раков желудочно-кишечного тракта (Berman et al., 2003 Nature 425: 846-51, опубликованная заявка РСТ WO 05/013800), рака пищевода (Berman et al., 2003 Nature 425: 846-51), рака легких (Watkins et al., 2003. Nature 422: 313-7) и рака простаты (Karhadkar et al., 2004. Nature 431: 707-12).
В дополнение, было показано, что множество типов рака включают неконтролируемую активацию «ежового» пути, например, рак молочной железы (Kubo et al., 2004. Cancer Research 64: 6071-4), печеночно-клеточный рак (Patil et al., 2005. 96th Annual AACR conference, abstract #2942 Sicklick et al., 2005. ASCO annual meeting, abstract #9610), гематологические злокачественные образования (Watkins and Matsui, неопубликованные результаты), базальная карцинома (Bale & Yu, 2001. Human Molec. Genet. 10:757-762 Xie et al., 1998 Nature 391: 90-92), медуллобластома (Pietsch et al., 1997. Cancer Res. 57: 2085-88) и рак желудка (Ma et al., 2005 Carcinogenesis May 19, 2005 (Epub)). Как показано в примерах, описанные здесь соединения модулируют «ежовый» путь, и показано, что выбранные соединения ингибируют рост опухоли. Поэтому полагают, что эти соединения могут быть полезны для лечения множества состояний, такие как различные виды рака.
Фармацевтические композиции
В другом варианте, в данном изобретении представлены фармацевтически приемлемые композиции, которые содержат терапевтически эффективное количество одного или более описанных здесь соединений, вместе с одним или более фармацевтически приемлемыми носителями (добавками) и/или разбавителями. Фармацевтические композиции в соответствии с данным изобретением могут быть составлены для введения в твердой или жидкой форме, включая те, которые адаптированы для следующего: (1) оральное введение, например, пропитки (водные или неводные растворы или суспензии), таблетки, например, предназначенные для буккальной, подъязычной и системной абсорбции, капсулы, болюсы, порошки, гранулы, пасты для нанесения на язык; (2) парентеральное введение, например, подкожной, внутримышечной, внутривенной или эпидуральной инъекцией, в виде, например, стерильного раствора или суспензии, или композиции замедленного высвобождения; (3) местное введение, например, в виде крема, мази и пластыря с контролируемым высвобождением, или спрея, наносимого на кожу; (4) вагинальное или ректальное, например, в виде пессариев, крема или пены; (5) подъязычное введение; (6) введение в глаз; (7) чрезкожное введение; (8) введение в легкие, или (9) введение в нос.
Примеры подходящих водных и неводных носителей, которые могут применяться в фармацевтических композициях в соответствии с данным изобретением, включают воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и подобные) и их подходящие смеси, растительные масла, такие как оливковое масло, и органические сложные эфиры для инъекций, такие как этилолеат. Подходящая текучесть может поддерживаться, например, с помощью покрытий, таких как лецитин, сохранением требуемого размера частиц для дисперсий, и применением поверхностно-активных веществ.
Эти композиции также могут содержать адъюванты, такие как консерванты, смачивающие агенты, эмульгирующие агенты, диспергирующие агенты, лубриканты и/или антиоксиданты. Профилактика воздействия микроорганизмов на соединения в соответствии с данным изобретением может обеспечиваться введением различных антибактериальных и противогрибковых агентов, например, парабена, хлорбутанола, фенола, сорбиновой кислоты и подобных. Также может быть желательно включать изотонические агенты, такие как сахара, хлорид натрия и подобные, в композиции. Кроме того, пролонгированная абсорбция фармацевтической формы для инъекций может быть достигнута добавлением агентов, которые задерживают абсорбцию, таких как моностеарат алюминия и желатин.
Методы получения таких композиций включают стадию смешивания соединения в соответствии с данным изобретением и носителем и, необязательно, одним или более необязательными ингредиентами. В общем, композиции получают однородным и тщательным смешиванием соединения в соответствии с данным изобретением с жидкими носителями или тонкоизмельченными твердыми носителями, или обоими, и затем, при необходимости, формованием продукта.
Если соединения в соответствии с данным изобретением вводят в виде фармацевтического средства, человеку и животным, они могут даваться как таковые или в виде фармацевтической композиции, содержащей, например, от около 0,1 до 99%, или от около 10 до 50%, или от около 10 до 40%, или от около 10 до 30, или от около 10 до 20%, или от около 10 до 15% активного ингредиента в сочетании с фармацевтически приемлемым носителем.
Действительные уровни дозирования активных ингредиентов в фармацевтических композициях в соответствии с данным изобретением могут варьироваться таким образом, чтобы получить количество активного ингредиента, которое эффективно для достижения желаемой терапевтической реакции конкретного пациента, композиции и способа введения, при этом не токсичное для пациента.
Выбранные уровни дозирования зависят от множества факторов, включая активность конкретного применяемого соединения в соответствии с данным изобретением, или его сложного эфира, соли или амида, способа введения, времени введения, скорости выведения или метаболизма конкретного вводимого соединения, скорости и продолжительности абсорбции, длительности лечения, других лекарственных соединений и/или продуктов, применяемых в сочетании с конкретным применяемым соединением, возрастом, полом, массой тела, состоянием, общим состоянием здоровья и анамнезом пациента, подвергаемого лечению, и подобных факторов, известных специалистам в данной области техники.
В общем, подходящая суточная доза соединения в соответствии с данным изобретением равна тому количеству соединения, которое составляет минимальную дозу, эффективную для получения терапевтического эффекта. Такая эффективная доза обычно зависит от описанных выше факторов. В общем, пероральные, внутривенные и подкожные дозы соединений в соответствии с данным изобретением для пациента, при применении для указанного действия, варьируются от около 0,0001 до около 100 мг, или от около 0,001 до около 100 мг, или от около 0,01 до около 100 мг, или от около 0,1 до около 100 мг, или от около 1 до около 50 мг на килограмм массы тела в сутки.
Пациентом, получающим такое лечение, является любое нуждающееся в нем животное, включая приматов в частности, человека, и других млекопитающих, таких как лошади, крупный рогатый скот, свиньи и овцы; и птица, и домашние животные в целом.
ПРИМЕРЫ
Выше представлено общее описание данного изобретения, но оно будет более понятно с помощью представленных ниже примеров, которые включены только для целей иллюстрации определенных аспектов и вариантов данного изобретения, и не ограничивают данное изобретение.
Пример 1
Стадия А

Перекристаллизованный циклопамин 2 (14,1 г, 34,0 ммоль, 1 экв.) растворяют в безводном ДХМ (70 мл) и безводном MeOH (29 мл). Прозрачный раствор охлаждают и добавляют триэтиламин (10,4 г, 102,7 ммоль, 3 экв.), затем бензилхлорформиат (6,20 г, 36,3 ммоль, 1,1 экв.). После завершения добавления раствор перемешивают на ледяной бане в течение 30 мин. Три порции бензилхлорформиата (3×0,35 г, 3,46 ммоль, 0,03 экв.) добавляют в течение более 3 ч. Реакционную смесь медленно гасят водой (71 мл), сохраняя температуру ниже 20°С. Смесь перемешивают в течение 15 мин, затем слои формируются и разделяются. Органический слой сушат над сульфатом натрия и фильтруют. Объединенный фильтрат буферируют безводным пиридином (30 мл), концентрируют и растворитель меняют на дополнительный безводный пиридин (43 мл) и концентрируют.
Раствор соединения в пиридине (43 мл) далее разбавляют дополнительным безводным пиридином (85 мл). В реакционную смесь медленно добавляют хлорид триметилацетила (8,3 г, 68,7 ммоль, 2 экв.), и реакционную смесь нагревают до 45°С. Реакционную смесь перемешивают при температуре 45°С в течение 30 мин. Реакционную смесь охлаждают и гасят добавлением безводного MeOH (4,5 мл). Резко охлажденную смесь перемешивают при кт в течение 40 мин и затем разбавляют толуолом (97 мл) и затем обрабатывают водой (35 мл) и 10% масс. водным раствором карбоната натрия (100 мл). После энергичного перемешивания слои разделяют, и органический слой дважды промывают водой (2×100 мл), сушат над сульфатом натрия и фильтруют. Фильтровальную лепешку промывают толуолом (49 мл) и выбрасывают. Объединенные фильтраты концентрируют, и растворитель меняют концентрацией на толуол (145 мл) и затем концентрируют досуха. Продукт перекристаллизовывают из толуола и гептана. Кристаллический продукт выделяют фильтрованием с отсасыванием, промывают холодным гептаном и сушат до постоянной массы с получением 15,1 г желаемого продукта.
Стадия В

Бис(2,6-диметилфенил)фосфат (10,65 г, 34,8 ммоль, 3,1 экв.) сушат концентрированием из безводного ДХМ (42 мл) и хранят в атмосфере азота. Затем фосфат повторно растворяют в безводном ДХМ (110 мл). В отдельной колбе получают раствор чистого диэтилцинка (4,17 г, 33,8 ммоль, 3,0 экв.) в безводном ДХМ (35 мл), который охлаждают до -25°С. Раствор фосфата медленно переносят в реактор, содержащий раствор диэтилцинка в течение более 1 ч, сохраняя температуру при или ниже -10°С. Прозрачный раствор фосфата этилцинка нагревают до 0°С и перемешивают в течение 15 мин. Дийодметан (9,25 г, 34,5 ммоль, 3,0 экв.) медленно добавляют в раствор фосфата этилцинка, сохраняя температуру реакции между 0 и 5°С. После завершения добавления раствор карбеноида цинка перемешивают в течение еще 20 мин.
В отдельной колбе соединение 3 (7,20 г, 11,4 ммоль, 1 экв.) растворяют в безводном ДХМ (36 мл) и переносят в реакционную колбу. После завершения добавления ледяную баню удаляют, и реакционную смесь нагревают до кт. Через 6 ч содержимое колбы охлаждают до -53°С. Добавляют раствор метансульфоновой кислоты (3,38 г, 35,2 ммоль, 3,1 экв.) в безводном ДХМ (3 мл), сохраняя температуру реакции ниже -45°С. Через 10 мин к реакционной смеси добавляют морфолин (20 г, 230 ммоль, 20 экв.), сохраняя температуру ниже -40°С. Реакционную смесь нагревают до кт в течение ночи. Соли морфолина удаляют фильтрацией, и фильтровальную лепешку промывают ДХМ (22 мл). Объединенные фильтраты промывают 2N водной хлористоводородной кислотой (2×140 мл), 5% водным бикарбонатом натрия (140 мл), 5% водным бикарбонатом натрия (70 мл) и 5% водным бисульфитом натрия (70 мл) и насыщенным раствором соли (144 мл). Органический слой сушат над сульфатом магния и фильтруют. Не доводя до сухого состояния, раствор ДХМ концентрируют, и растворитель меняют на метанол (280 мл). Суспензию охлаждают на ледяной бане и перемешивают в течение 40 минут. Твердые вещества выделяют фильтрацией, дважды промывают холодным метанолом (2×25 мл) и сушат до постоянной массы с получением 5,94 г желаемого продукта.
Стадия С

В круглодонной колбе соединение 4 (11,67 г, 18,1 ммоль, 1 экв.) и 20% гидроксид палладия на влажном углероде (2,40 г, 1,71 ммоль, 0,09 экв.) помещают в атмосферу азота и разбавляют EtOAc (115 мл) и толуолом (60 мл). Раствор дегазируют азотом (3X) с применением циклов вакуумирования/продувки, и процесс повторяют с водородом. Суспензию энергично перемешивают при кт в течение 1,5 ч. Атмосферу водорода меняют на азот. К реакционной смеси добавляют этилендиамин (0,57 г, 9,5 ммоль, 0,52 экв.) и полученную смесь перемешивают в течение 20 мин. Раствор фильтруют в азоте, и фильтрат промывают 2% (масс./масс.) водным раствором этилендиамина (125 мл), затем водой (130 мл) и затем сушат над сульфатом натрия. Сушащий агент удаляют фильтрацией, и фильтрат концентрируют досуха в вакууме. Оставшиеся твердые вещества выгоняют толуолом (2×55 мл) на роторном испарителе, и полученный продукт используют без дальнейшей очистки на следующей стадии.
Продукт с предыдущей стадии растворяют в безводном ДХМ (26 мл). Полученный прозрачный раствор добавляют к 1 M раствору DIBAL в ДХМ (65 мл, 65 ммоль, 3,6 экв.), сохраняя температуру реакции от -10 до -25°С. Через 30 мин реакцию гасят ацетоном (13 мл), сохраняя температуру реакции при или ниже 0°С. После перемешивания гашенной реакционной смеси в течение 17 мин, ее порциями добавляют в колбу, содержащую холодный перемешанный раствор 20% (масс./масс.) водного виннокислого калия-натрия (200 мл). Полученную гелеобразную суспензию перемешивают при кт в течение 15 ч. После перемешивания чистые слои разделяют, и водный слой снова экстрагируют ДХМ (30 мл). Объединенные органические слои промывают водой (60 мл) и сушат над сульфатом натрия. Осушающий агент удаляют фильтрацией и выбрасывают. Фильтрат концентрируют в вакууме, и растворитель меняют на толуол (225 мл добавленные частями). Полученный раствор далее концентрируют до суспензии (50 мл) и разбавляют гептаном (115 мл). Полученную смесь нагревают до тех пор, пока она не станет гомогенной (92°С). Раствор медленно охлаждают в течение более 12 ч до 15°С, и затем в течение еще 16 ч. Кристаллический продукт выделяют фильтрацией с отсасыванием, промывают гептаном (2×75 мл) и сушат до постоянной массы с получением 7,70 г желаемого продукта.
В круглодонную колбу последовательно добавляют гомо-аллильный спирт (7,50 г, 17,6 ммоль, 1 экв.), три-трет-бутоксид алюминия (6,10 г, 24,8 ммоль, 1,4 экв.), безводный толуол (115 мл) и 2-бутанон (90 г, 1,24 моль, 7 экв.). Суспензию нагревают в атмосфере азота до 75°С в течение 16 ч. Температуру реакции затем понижают до 49°С. К перемешиваемой суспензии добавляют водный 20% (масс./масс.) раствор виннокислого калия-натрия (226 г). Суспензию перемешивают при кт в течение 3,5 ч. Слои разделяют. Органический слой промывают водный 20% виннокислым калием-натрием (2×250 мл) и водой (225 мл), затем сушат над сульфатом натрия и фильтруют. Остаток промывают толуолом (30 мл) и выбрасывают. Объединенные органические слои концентрируют досуха. Остаточные реакционные растворители удаляют из продукта концентрированием из 2-пропанола (250 мл добавляют порциями) до конечной массы раствора 44 г. Растворитель меняют из 2-пропанола на гептан (275 мл добавляют порциями) до конечной массы раствора 41 г, полностью осаждая желаемый продукт. Суспензию разбавляют дополнительным гептаном (40 мл), перемешивают при кт в течение 1 ч и фильтруют. Продукт промывают н-гептаном (17 мл) и сушат с получением 5,4 г желаемого продукта.
Стадия D

В круглодонную колбу загружают исходный материал (110 мг, 0,26 ммоль, 1 экв.) и 10% палладий на угле (106 мг). Твердые вещества суспендируют в пиридине (4 мл). Суспензию помещают в атмосферу водорода (1 атм.), и смесь перемешивают в течение ночи при кт. Реакционную смесь фильтруют через Celite®, и фильтрат концентрируют в вакууме. Неочищенный продукт очищают с применением флэш-хроматографии на силикагеле (MeOH/ДХМ 5:95) с получением 93 мг желаемого соединения. ([M+H] = 426,6 m/z).
Пример 2
Стадия А

Циклопамин 2 (5,02 г, 12,2 ммоль, 1,0 экв.) растворяют в безводном пиридине (25 мл). Добавляют ДМАП (300 мг, 2,44 ммоль, 0,2 экв.) и триэтиламин (5,5 мл, 39,1 ммоль, 3,2 экв.), затем BtO-Cbz (10,5 г, 39,1 ммоль, 3,2 экв.), и нагревают при 40°С в течение 2 ч. Смесь охлаждают до кт, обрабатывают 30 мл воды, нагревают с получением гомогенного раствора и охлаждают до комнатной температуры. Образовавшийся белый осадок собирают фильтрацией, фильтровальную лепешку промывают порциями воды (3×50 мл) и сушат на воздухе с получением 9,53 г неочищенного продукта, который кристаллизуют из толуола/гептана (1:9, 70 мл) с получением 6,75 г желаемого продукта.
Стадия В

К раствору диэтилцинка (572 мг, 482 мкл, 4,63 ммоль, 3,00 экв.) в 5,0 мл ДХМ при -20°С добавляют раствор бис-(2,6-диметилфенил)фосфорной кислоты (1,42 г, 4,63 ммоль, 3,00 экв.) в ДХМ (15 мл), сохраняя температуру реакции ниже -8°С. Раствор выстаивают в течение 15 мин при 0°С, добавляют чистый дийодметан (1,24 г, 374 мкл, 3,00 экв.), выстаивают в течение 15 мин при 0°С, затем добавляют раствор (БиCBzциклопамин, 1,05 г, 1,54 ммоль, 1,0 экв.) в ДХМ (10 мл). Охлаждающую баню заменяют на водяную баню при кт, и выдерживают при кт в течение 4.5 ч. Смесь охлаждают до -76°С на бане сухой лед-ацетон, и по каплям обрабатывают раствором метансульфоновой кислоты в ДХМ (0,6 мл 50% об./об. раствор 4,63 ммоль, 3,0 экв.), сохраняя температуру реакции ниже -74°С. Смесь выстаивают в течение 15-20 мин и по каплям гасят морфолином (2,69 г, 2,70 мл, 20 экв.), сохраняя температуру реакции ниже -65°С. Охлаждающую баню удаляют, реакционную смесь перемешивают в течение 16-18 ч, белый осадок отфильтровывают, и фильтрат последовательно промывают 2,0 M HCl (2×20 мл), насыщенным бикарбонатом натрия (2×20 мл), водой (2×20 мл) и насыщенным раствором соли (20 мл). Сушат над сульфатом магния, концентрируют в вакууме досуха, и неочищенный остаток очищают флэш-хроматографией на силикагеле (гексан/EtOAc 17:3→4:1) с получением 924 мг (1,33 ммоль, 86%) желаемого продукта.
Стадия С

К раствору соединения 7 (4,05 г, 5,83 ммоль, 1 экв.) в растворе EtOAc:толуол (2:1, 60 мл) добавляют 20% гидроксид палладия на угле (823 мг, 0,583 ммоль, 0,1 экв.). Колбу вакуумируют и заполняют водородом три раза. Смесь перемешивают в атмосфере водорода в течение 1 ч. Добавляют чистый этилендиамин (0,38 мл), перемешивают в течение 1 ч, и катализатор отфильтровывают. Фильтровальную лепешку дважды промывают EtOAc:толуолом (2:1, 12 мл). Объединенные фильтраты промывают 2% водным раствором этилендиамина (3×20 мл), сушат над сульфатом натрия и концентрируют в вакууме с получением 2,46 г в виде белого кристаллического твердого вещества.
Стадия D

В круглодонную колбу последовательно загружают гомо-аллильный спирт 8 (7,50 г, 17,6 ммоль, 1 экв.), три-трет-бутоксид алюминия (6,10 г, 24,8 ммоль, 1,4 экв.), безводный толуол (115 мл) и 2-бутанон (90 г, 1,24 моль, 7 экв.). Суспензию нагревают в атмосфере азота до 75°С в течение 16 ч. Затем температуру реакции понижают до 49°С. Водный 20% (масс./масс.) раствор тартрата натрия (226 г) добавляют к перемешиваемой суспензии. Суспензию перемешивают при кт в течение 3,5 ч. Слои разделяют. Органический слой промывают водным 20% виннокислым калием-натрием (2×250 мл) и водой (225 мл), затем сушат над сульфатом натрия и фильтруют. Остаток промывают толуолом (30 мл) и выбрасывают. Объединенные органические слои концентрируют досуха. Остаточные реакционные растворители удаляют из продукта концентрированием из 2-пропанола (250 мл добавляют порциями) до конечной массы раствора 44 г. Растворитель меняют с 2-пропанола на н-гептан (275 мл добавляют порциями) до конечной массы раствора 41 г, полностью осаждая желаемый продукт. Суспензию разбавляют дополнительным н-гептаном (40 мл), перемешивают при кт в течение 1 ч и фильтруют. Продукт промывают н-гептаном (17 мл) и сушат с получением 5,4 г желаемого продукта.
Стадия Е

В круглодонную колбу загружают исходный материал (110 мг, 0,26 ммоль, 1 экв.) и 10% палладий на угле (106 мг). Твердые вещества суспендируют в пиридине (4 мл). Суспензию помещают в атмосферу водорода (1 атм.), и смесь перемешивают в течение ночи при кт. Реакционную смесь фильтруют через Celite®, и фильтрат концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией на силикагеле (MeOH/ДХМ 5:95) с получением 93 мг желаемого соединения. ([M+H] = 426,6 m/z).
Пример 3

В герметично закрытую пробирку загружают кетон 6 (85 мг, 0,199 ммоль, 1 экв.) и добавляют триэтиленгликоль (2 мл), затем моногидрат гидразина (500 мг, 10 ммоль, 50 экв.) и карбонат калия (138 мг, 1 ммоль, 5 экв.). Пробирку герметично закрывают, и реакционную смесь нагревают при 150°С в течение 16 ч. Реакционную смесь охлаждают до кт и добавляют воду. Остаток экстрагируют хлороформом (3X). Объединенные органические слои промывают водой, сушат над Na2SO4 и концентрируют досуха. Бесцветное масло очищают флэш-хроматографией на силикагеле (ДХМ/MeOH 96:4). Очищенные фракции объединяют и концентрируют досуха. Полученное масло растворяют в МТБЭ и промывают водой (2X), 2N NaOH и затем насыщенным раствором соли. Объединенные органические слои сушат над Na2SO4, фильтруют и выпаривают с получением 64 мг желаемого продукта в виде белой пены. ([M+H] = 412,7 m/z).
Пример 4

В герметично закрытую пробирку загружают соединение 5 (223 мг, 0,52 ммоль, 1 экв.) и ДМФ (1 мл). Добавляют 2-бромпропан (1,3 г, 10,5 ммоль, 20 экв.) и Na2CO3 (73 мг, 0,68 ммоль, 1,3 экв.), и пробирку герметично закрывают и нагревают до 50°С. Смесь перемешивают в течение 16 ч, к этому моменту наблюдается уже практически 70% превращение. Добавляют еще (0,26 г, 2,12 ммоль, 4 экв.). Реакционную смесь перемешивают в течение 2 ч и добавляют еще 2-бромпропан (0,13 г, 1,1 ммоль, 2 экв.). Реакционную смесь перемешивают еще в течение 1 часа. Реакционную смесь охлаждают до кт и добавляют воду. Остаток экстрагируют МТБЭ (3X). Органические слои объединяют, промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют досуха. Белую пену очищают флэш-хроматографией на силикагеле (ДХМ/MeOH 99:1) с получением 206 мг N-изопропилового производного в виде белой пены.
N-изопропиловое производное (205 мг, 0,44 ммоль, 1 экв.) растворяют в 4-метоксипиридине (1,5 мл). Колбу помещают в инертную атмосферу и добавляют Pd/C 10% (влажный, Aldrich Degussa тип E101, 40 мг). Колбу герметично закрывают и три раза продувают азотом, и оставляют на 16 ч в 1 атм. водорода. К реакционной смеси добавляют Celite®. Смесь фильтруют через небольшой слой Celite® и промывают EtOAc. Органический слой промывают 1N HCl водн. (2x), затем водой. Органический слой сушат над Na2SO4, фильтруют через хлопок и выпаривают с получением 34 мг неочищенного продукта. Водный слой нейтрализуют 2N KOH и экстрагируют ДХМ (3X). Объединенные органические слои промывают водой, сушат над Na2SO4, фильтруют через хлопок и объединяют с первыми 34 мг неочищенного продукта. Неочищенный продукт очищают флэш-хроматографией на силикагеле гексан/EtOAc (6:4) с получением 80 мг желаемого продукта. ([M+H] = 468,7 m/z).
Пример 5
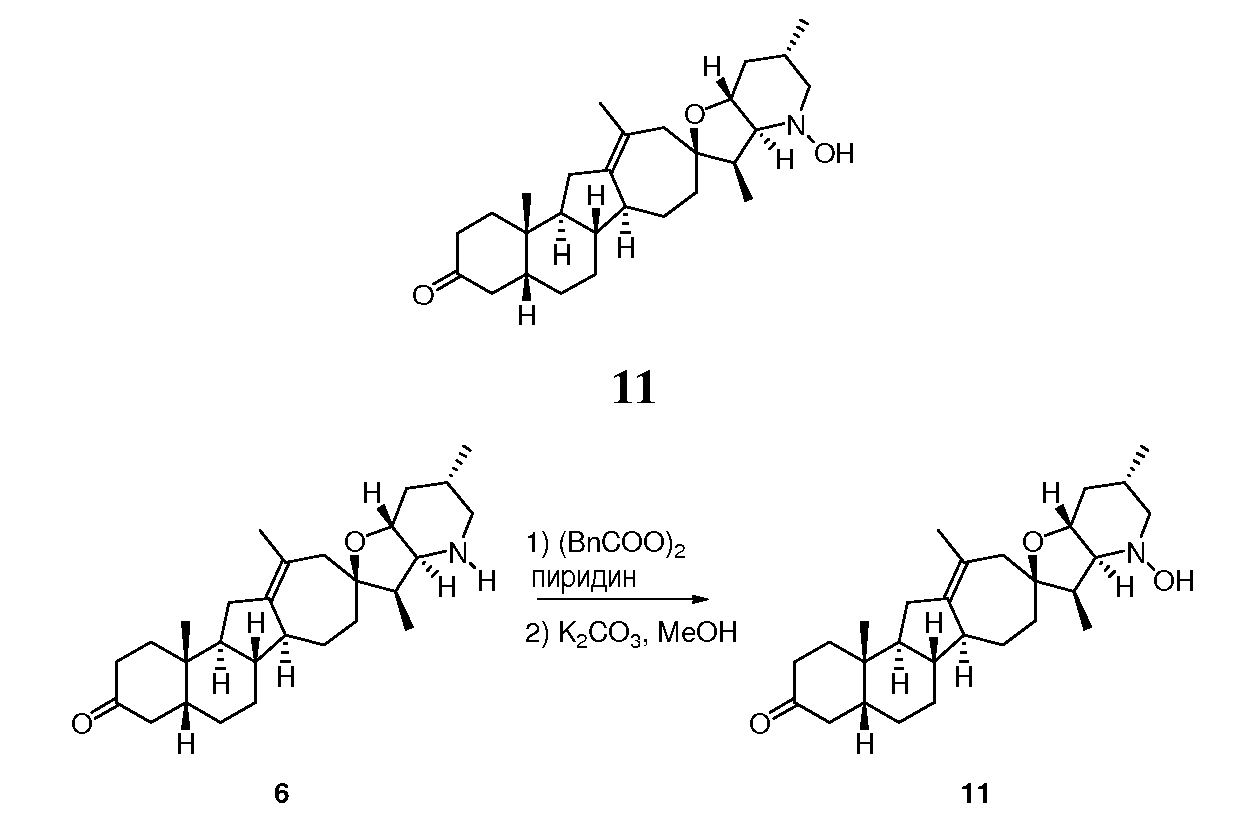
В круглодонной колбе соединение 6 (88 мг, 0,21 ммоль, 1 экв.) растворяют в безводном ТГФ (1 мл). Смесь охлаждают до 0°С. Последовательно добавляют пиридин (84 мкл, 1 ммоль, 5 экв.) и перекись бензоила (150 мг, 0,62 ммоль, 3 экв.). Гомогенную смесь постепенно нагревают до кт в течение более 2 ч и перемешивают в течение ночи при кт. Реакцию гасят добавлением насыщенного NaHCO3. Остаток экстрагируют МТБЭ. Объединенные органические слои промывают водой, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Неочищенный остаток очищают флэш-хроматографией на силикагеле (гексан/EtOAc (9:1 до 4:1)) с получением N-O производного продукта (60 мг, 0,11 ммоль) в виде белой пены. Эту пену растворяют в 2 мл MeOH, затем в 2N водном KOH (0,4 мл). Реакционную смесь перемешивают в течение 1 ч. Большую часть MeOH выпаривают в потоке азота и добавляют 1N HCl (500 мкл). Продукт экстрагируют ДХМ (3X). Объединенные органические слои промывают водой, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Неочищенный остаток очищают флэш-хроматографией на силикагеле (гексан/EtOAc (от 88:12→1:1)) с получением 11 мг желаемого продукта. ([M+H] = 442,5 m/z).
Пример 6

Стадия А

В круглодонной колбе соединение 6 (89 мг, 0,209 ммоль, 1 экв.) и N-(бензилоксикарбонил)-аминоацетальдегид (148 мг, 0,85 ммоль, 4 экв.) растворяют в ДХМ (2 мл). Добавляют триацетоксиборгидрид натрия (177 мг, 0,85 ммоль, 4 экв.), и реакционную смесь перемешивают в течение 3 ч при кт. Смесь выливают в насыщенный водный раствор ΝaHCO3, и остаток экстрагируют ДХМ (3x). Объединенные органические слои промывают водой, сушат над Na2SO4, фильтруют через хлопок и выпаривают с получением пенистого твердого вещества (247 мг). Неочищенный продукт растворяют в EtOAc (2 мл) и обрабатывают 4M HCl (156 мкл). Через 30 мин медленно формируется белый осадок. Полученную суспензию перемешивают в течение 15 мин. Фильтрация дает 120 мг белого твердого вещества. Продукт растворяют в EtOAc и обрабатывают насыщенным водным раствором NaHCO3. Органический слой собирают, и водный слой экстрагируют EtOAc (2X). Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4. Фильтрация и выпаривание дают желаемое промежуточное соединение. Этот продукт используют на следующей стадии без очистки.
Стадия В

Весь продукт со стадии A растворяют в EtOAc (3 мл) и обрабатывают Pd/C 10% (30 мг, влажный, Alrich Degussa тип E101). Колбу герметично закрывают и три раза продувают водородом и оставляют на ночь в 1 атм. водорода. Через 16 ч смесь фильтруют через небольшой слой Celite® и промывают EtOAc с получением 52 мг амина в виде белой пены.
Стадия С

В круглодонную колбу, содержащую амин 14 (52 мг, 0,11 ммоль, 1 экв.) загружают 1H-тетразол-5-уксусную кислоту (21 мг, 0,166 ммоль, 1,5 экв.), ДХМ (2 мл), EDCl (42 мг, 0,22 ммоль, 2 экв.) и N,N-диизопропилэтиламин (57 мг, 0,44 ммоль, 4 экв.). Полученный желтый раствор перемешивают при кт в течение 4 ч. Реакцию гасят добавлением насыщенного водного раствора NaHCO3, и остаток экстрагируют ДХМ (3X). Объединенные органические слои сушат над Na2SO4, фильтруют через хлопок и выпаривают с получением 62 мг беловатого твердого вещества. Этот продукт очищают флэш-хроматографией на силикагеле (MeOH/ДХМ 5:95→10:90) с получением 31 мг желаемого продукта. ([M+H] = 579,7 m/z).
Пример 7


В круглодонную колбу загружают исходный материал (47 мг, 0,110 ммоль, 1 экв.) и карбонат калия (150 мг, 1,09 ммоль, 10 экв.). Твердые вещества суспендируют в 2 мл ДХМ. Добавляют йодметан (14 мкл, 0,22 ммоль, 2 экв.), и смесь перемешивают в течение 2 при кт. ТСХ (ДХМ/MeOH 95:5) показывает >90% завершение. К реакционной смеси добавляют йодметан (14 мкл, 0,22 ммоль, 2 экв.), затем ее перемешивают в течение 2 ч. К реакционной смеси добавляют воду. Фазы разделяют, и органическую фазу сушат и концентрируют досуха. Остаток очищают флэш-хроматографией на силикагеле (ДХМ/MeOH 100:0→98:2) с получением 34 мг желаемого продукта ([M+H] = 440,5 m/z).
Пример 8

В круглодонную колбу загружают исходный материал (59 мг, 0,126 ммоль, 1 экв.) и карбонат калия (350 мг, 2,5 ммоль, 20 экв.). Твердые вещества суспендируют в 3 мл ДХМ. Добавляют йодметан (80 мкл, 1,29 ммоль, 10 экв.) и смесь перемешивают в течение ночи при кт. К реакционной смеси добавляют воду. Органическую фазу отделяют, и водный слой повторно экстрагируют ДХМ. Объединенные органические слои сушат и концентрируют досуха. Остаток очищают флэш-хроматографией на силикагеле. ДХМ/MeOH (95:5→90:10) с получением 52 мг желаемого продукта. ([M+H] = 639,5 m/z).
Пример 9

Стадия А

В круглодонной колбе соединение 5 (50 мг, 0,12 ммоль, 1 экв.) и N-(трет-бутоксикарбонил)-аминоацетальдегид (6 мг, 0,38 ммоль, 3,1 экв.) растворяют в ДХМ (2 мл). Добавляют триацетоксиборгидрид натрия (8 мг, 0,38 ммоль, 3,1 экв.), и реакционную смесь перемешивают в течение 2 ч при кт. Смесь выливают в насыщенный водный раствор ΝaHCO3, и остаток экстрагируют ДХМ (3x). Объединенные органические слои промывают водой, сушат над Na2SO4, фильтруют через хлопок и выпаривают с получением пенистого твердого вещества (95 мг). Неочищенный продукт очищают флэш-хроматографией на силикагеле (EtOAc/Гексан 1:1) с получением 55 мг соединения 18.
Стадия B
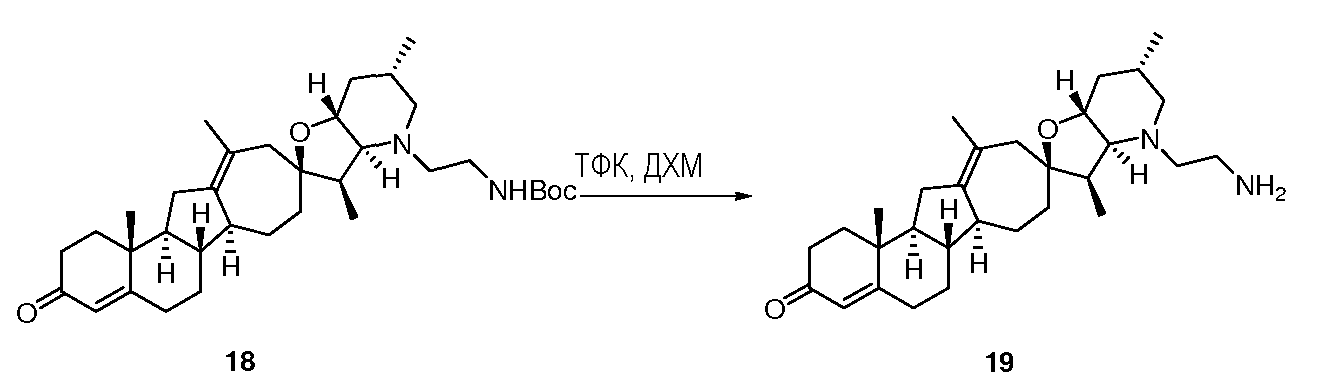
В круглодонную колбу загружают исходный материал 18 (800 мг, 1,4 ммоль, 1 экв.). Твердое вещество растворяют в растворе ДХМ и ТФК (10 мл, 1:1). Раствор перемешивают в течение 45 мин при кт. Реакционную смесь разделяют между раствором 10% карбоната натрия и ДХМ. Органические слои отделяют и промывают 10% карбонатом натрия. Органическую фазу концентрируют досуха. Остаток используют без дальнейшей очистки на следующей стадии.
Стадия С

В круглодонную колбу загружают исходный материал (300 мг, 0,64 ммоль, 1 экв.), растворенный в ТГФ/АЦН (1:1, 4 мл). В реакционную смесь загружают 37% формальдегид в воде (240 мкл, 3,22 ммоль, 5 экв.) и цианоборгидрид натрия (64 мг, 1 ммоль, 1,6 экв.). Смесь перемешивают в течение 30 мин при кт. Затем реакционную смесь разделяют между раствором насыщенного водного раствора бикарбоната натрия и ДХМ. Органический слой отделяют, сушат и концентрируют досуха. Неочищенный продукт очищают флэш-хроматографией на силикагеле (MeOH/ДХМ 5:95→10:90) с получением желаемого материала.
Стадия D

В круглодонную колбу загружают исходный материал 20 (30 мг, 0,06 ммоль, 1 экв.) и 10% палладий на угле (30 мг). Твердые вещества суспендируют в пиридине (2 мл). Суспензию помещают в атмосферу водорода, и смесь перемешивают в течение ночи при кт. Реакционную смесь фильтруют на Celite®, и фильтрат концентрируют досуха. Неочищенный продукт очищают флэш-хроматографией на силикагеле (MeOH/ДХМ 5:95→10:90) с получением желаемого продукта. ([M+H] = 497,7 m/z).
Пример 10


В круглодонную колбу загружают исходный материал (85 мг, 0,20 ммоль, 1 экв.), растворенный в ДХМ (4 мл). В реакционную смесь добавляют N-(2-оксоэтил)ацетамид (80 мг, 0,70 ммоль, 3,5 экв.) и триацетоксиборгидрид натрия (170 мг, 0,80, 4 экв.). Смесь перемешивают в течение 1 часа при кт. Реакцию разделяют между раствором насыщенного водного раствора бикарбоната натрия и ДХМ. Органический слой отделяют, сушат и концентрируют досуха. Неочищенный продукт очищают флэш-хроматографией на силикагеле (MeOH/ДХМ 5:95) с получением желаемого продукта. ([M+H] = 511,7 m/z).
Пример 11

Соединение 22 синтезируют по методике, описанной в примере 9, используя N-метил-N-(2-оксоэтил)ацетамид вместо N-(2-оксоэтил)ацетамида. ([M+H] = 525,7 m/z).
Пример 12

Соединение 23 синтезируют по методике, описанной в примере 10, используя N-(2-оксоэтил)-3-фенилпропанамид вместо N-(2-оксоэтил)ацетамида. ([M+H] = 601,8 m/z).
Пример 13

Соединение 24 синтезируют по методике, описанной в примере 10, используя N-метил-N-(2-оксоэтил)-3-фенилпропанамид вместо N-(2-оксоэтил)ацетамида. ([M+H] 615,9 m/z)
Пример 14

Стадия А

В круглодонную колбу загружают соединение 6 (4,23 г, 9,94 ммоль, 1 экв.) и ТГФ (60 мл). Добавляют триэтиламин (6,92 мл, 49,7 ммоль, 5,0 экв.) и бензилхлорформиат (1,54 мл, 10,93 ммоль, 1,1 экв.), и смесь перемешивают в течение 1 часа при кт. Реакционную смесь разделяют между насыщенным водным бикарбонатом (100 мл) и EtOAc (100 мл). Фазы разделяют, и органические фазы сушат (Νa2SO4) и концентрируют досуха. Неочищенный продукт очищают флэш-хроматографией на силикагеле (EtOAc/Гексан 2:98→14:86) с получением 3,75 г продукта.
Стадия B
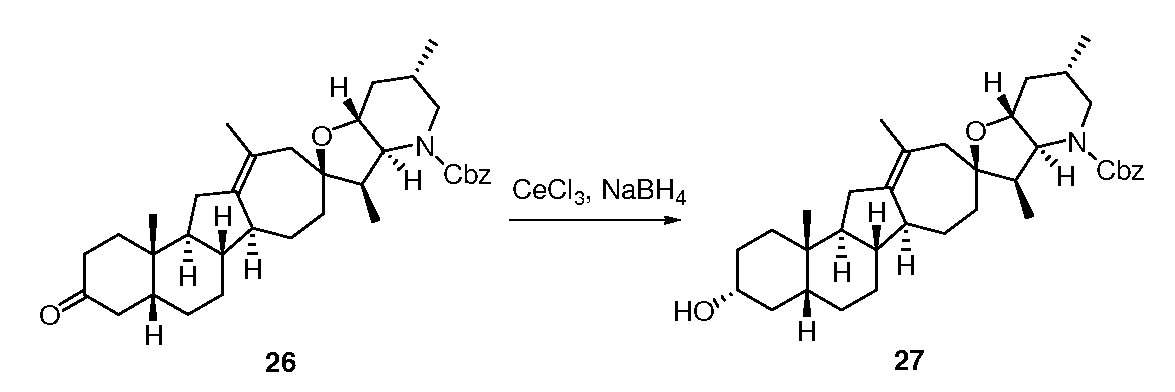
Раствор в MeOH (10 мл) гептагидрата трихлорида церия (260 мг, 0,69 ммоль, 1,3 экв.) при 0°С обрабатывают боргидридом натрия (24 мг, 0,65 ммоль, 1,2 экв.), перемешивают в течение 15 мин и затем охлаждают до -78°С. Добавляют раствор в ТГФ (10 мл) кетона 26 (300 мг, 0,54 ммоль, 1 экв.), и смесь перемешивают в течение 1 ч и затем нагревают до кт. Добавляют воду (50 мл) и EtOAc (50 мл), перемешивают, и слои разделяют. Органический слой собирают, промывают насыщенным раствором соли (30 мл), сушат над сульфатом натрия и концентрируют до белого остатка. Неочищенный продукт очищают флэш-хроматографией на силикагеле (простой эфир/гексан 2:3→1:1) с получением 235 мг 3-бета спирта 27.
Стадия С

Соединение 27 (235 мг, 0,42 ммоль, 1 экв.) растворяют в EtOAc (7 мл) в колбе с мешалкой и резиновой прокладкой. Раствор барботируют азотом и добавляют Pd/C 10% (влажный, Aldrich Degussa тип E101, 50 мг). Эту смесь барботируют азотом и затем водородом и перемешивают при кт в течение 3 ч. Смесь затем барботируют азотом, фильтруют через 0,45 мкм полиэтиленовую мембрану и концентрируют до прозрачного масла. Масло очищают флэш-хроматографией на силикагеле (NH4OH(водн.)/MeOH/ДХМ 0,5:2:97,5→0,5:6:93,5) с получением 130 мг соединения 25 в виде белого порошка. ([M+H] = 427,4 m/z)
Пример 15

Стадия А

Раствор в ТГФ (10 мл) кетона 26 (300 мг, 0,54 ммоль, 1 экв.) при -78°С обрабатывают K-Selectride® (три-втор-бутилборгидрид калия) (0,58 мл, 0,58 ммоль, 1,1 экв.) и перемешивают в течение 60 мин. Добавляют метанол (1 мл), и раствор нагревают до кт. Добавляют воду (50 мл) и EtOAc (50 мл), перемешивают и слои разделяют. Органический слой промывают насыщенным раствором соли (30 мл), сушат над сульфатом натрия и концентрируют до белого остатка. Неочищенный продукт очищают флэш-хроматографией на силикагеле (простой эфир/гексан 2:3→1:14) с получением 170 мг чистого 3-альфа спирта 29.
Стадия B

Соединение 29 (170 мг, 0,30 ммоль, 1 экв.) растворяют в EtOAc (5 мл) в колбе с мешалкой и резиновой прокладкой. Раствор барботируют азотом, и добавляют Pd/C 10% (влажный, Aldrich Degussa тип E101, 35 мг). Эту смесь барботируют азотом и затем водородом и перемешивают при кт в течение 3 ч. Смесь затем барботируют азотом, фильтруют через 0,45 мкм полиэтиленовую мембрану и концентрируют до прозрачного масла. Масло очищают флэш-хроматографией на силикагеле (NH4OH(водн.)/MeOH/ДХМ 0,5:2:97,5→0,5:6:93,5) с получением 76 мг соединения 28 в виде белого порошка ([M+H] = 427,4 m/z).
Пример 16

Стадия А

Соединение 27 (100 мг, 0,18 ммоль, 1 экв.) с хлоридом бензилтриэтиламмония (8 мг, 0,36 ммоль, 0,2 экв.) растворяют в ДХМ (5 мл) и энергично перемешивают с диметилсульфатом (130 мкл, 1,43 ммоль, 8 экв.) и 50% водным гидроксидом калия (0,5 мл) при кт в течение 18 ч. Смесь разделяют между водой (30 мл) и EtOAc (30 мл), и органический слой промывают насыщенным раствором соли, сушат над сульфатом натрия и концентрируют до прозрачного масла. Неочищенный простой эфир очищают флэш-хроматографией на силикагеле (простой эфир/гексан 3:7→9:113) с получением 75 мг метилового эфира в виде прозрачного масла.
Стадия B

Соединение 31 (66 мг, 0,115 ммоль, 1 экв.) растворяют в EtOAc (5 мл) в колбе с мешалкой и резиновой прокладкой. Раствор барботируют азотом и добавляют Pd/C 10% (влажный, Aldrich Degussa тип E101, 20 мг). Эту смесь барботируют азотом и затем водородом и перемешивают при кт в течение 3 ч. Затем смесь барботируют азотом, фильтруют через 0,45 мкм полиэтиленовую мембрану и концентрируют до прозрачного масла. Масло очищают флэш-хроматографией на силикагеле (NH4OH(водн.)/MeOH/ДХМ 0,5:2:97,5→0,5:6:93,5) с получением 22 мг соединения 30 в виде белого порошка ([M+H] = 441,4 m/z).
Пример 17
Стадия А

Соединение 27 (100 мг, 0,18 ммоль, 1 экв.) растворяют в ДХМ (5 мл), и добавляют 4-диметиламинопиридин (4 мг, 0,35 ммоль, 0,2 экв.), N,N-диизопропилэтиламин (0,15 мл, 0,9 ммоль, 5 экв.) и уксусный ангидрид (0,070 мл, 0,72 ммоль, 4 экв.). После перемешивания в течение 12 ч при кт раствор разделяют между EtOAc (30 мл) и 5% водным бикарбонатом натрия (15 мл). Органический слой промывают насыщенным раствором соли, сушат над сульфатом натрия и концентрируют до прозрачного масла. Неочищенный сложный эфир очищают хроматографией на силикагеле (простой эфир/гексан 3:7→9:113) с получением 100 мг сложного эфира в виде прозрачного масла.
Стадия B
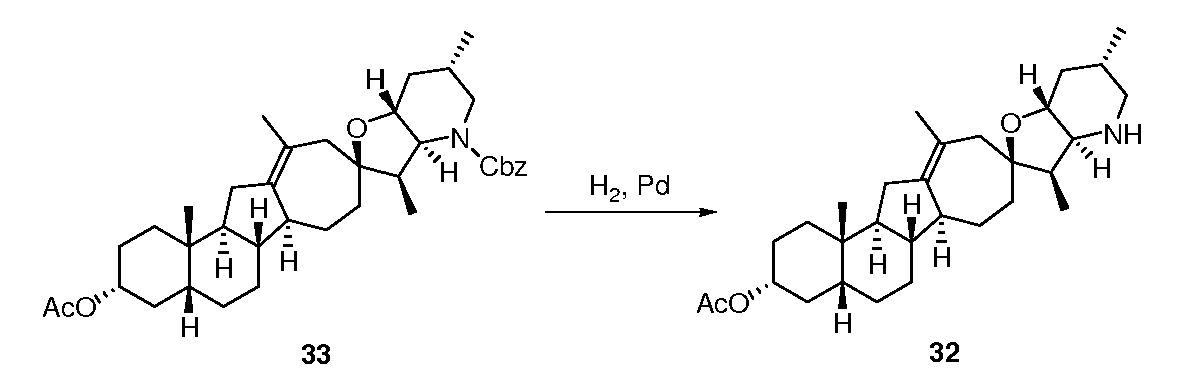
Соединение 33 (100 мг, 0,18 ммоль, 1 экв.) растворяют в EtOAc (5 мл) в колбе с мешалкой и резиновой прокладкой. Раствор барботируют азотом и добавляют Pd/C 10% (влажный, Aldrich Degussa тип E101, 20 мг). Эту смесь барботируют азотом и затем водородом и перемешивают при кт в течение 3 ч. Затем смесь барботируют азотом, фильтруют через 0,45 мкм полиэтиленовую мембрану и концентрируют до прозрачного масла. Масло очищают флэш-хроматографией на силикагеле (NH4OH(водн.)/MeOH/ДХМ 0,5:2:97,5→0,5:6:93,5) с получением 45 мг соединения 32 в виде белого порошка ([M+H] = 469,4 m/z).
Пример 18

Соединение 34 синтезируют по методике, описанной в примере 16, с применением соединения 29 вместо соединения 27. ([M+H] = 441,4 m/z).
Пример 19

Соединение 35 синтезируют по методике, описанной в примере 17, с применением соединения 29 вместо соединения 27. МС ([M+H] = 469,4 m/z)
Пример 20

Стадия А

Раствор в этаноле (5 мл) соединения 26 (185 мг, 0,3 ммоль, 1 экв.) обрабатывают гидрохлоридом гидроксиламина (140 мг, 2 ммоль, 6 экв.), ацетатом натрия (160 мг, 2 ммоль, 6 экв.) и водой (0,5 мл), и смесь перемешивают при кт в течение 1 ч. Смесь разделяют между EtOAc и водой (50 мл каждого). Органический слой промывают насыщенным раствором соли (30 мл), сушат над сульфатом натрия и концентрируют до белого остатка. Неочищенный продукт очищают хроматографией на силикагеле (простой эфир/гексан 2:3 → 1:1) с получением 193 мг оксима 37.
Стадия B

Соединение 37 (65 мг, 0,113 ммоль) растворяют в EtOAc (7 мл) в колбе с мешалкой и резиновой прокладкой. Раствор барботируют азотом и добавляют Pd/C 10% (влажный, Aldrich Degussa тип E101, 20 мг). Эту смесь барботируют азотом и затем водородом и перемешивают при кт в течение 3 ч. Затем смесь барботируют азотом, фильтруют через 0,45 мкм полиэтиленовую мембрану и концентрируют до прозрачного масла. Масло очищают флэш-хроматографией на силикагеле (NH4OH(водн.)/MeOH/ДХМ 0,5:2:97,5→0,5:6:93,5) с получением 15 мг соединения 36 в виде белого порошка, смесь цис и транс изомеров оксима ([M+H] = 440,3 m/z).
Пример 21

Стадия А

Соединение 27 (42 мг, 0,075 ммоль, 1 экв.) растворяют в ДХМ (5 мл), и добавляют 4-диметиламинопиридин (2 мг, 0,02 ммоль, 0,2 экв.), N-Cbz глицин (23 мг, 0,110 ммоль, 1,5 экв.) и диизопропилкарбодиимид (0,023 мл, 0,150 ммоль, 2 экв.). После перемешивания в течение 12 ч при кт раствор разделяют между EtOAc (30 мл) и 5% водным бикарбонатом натрия (15 мл). Органический слой промывают насыщенным раствором соли, сушат над сульфатом натрия и концентрируют до прозрачного масла. Неочищенный сложный эфир очищают флэш-хроматографией на силикагеле (простой эфир/гексан 2:3 → 1:1) с получением 35 мг сложного эфира в виде прозрачного масла.
Стадия B

Соединение 39 (235 мг, 0,42 ммоль, 1 экв.) растворяют в EtOAc (7 мл) в колбе с мешалкой и резиновой прокладкой. Раствор барботируют азотом и добавляют Pd/C 10% (влажный, Aldrich Degussa тип E101, 20 мг). Эту смесь барботируют азотом и затем водородом и перемешивают при кт в течение 3 ч. Затем смесь барботируют азотом, фильтруют через 0,45 мкм полиэтиленовую мембрану и концентрируют до прозрачного масла. Масло очищают флэш-хроматографией на силикагеле (NH4OH(водн.)/MeOH/ДХМ 0,5:2:97,5→0,5:6:93,5) с получением 17 мг желаемого продукта в виде белого порошка ([M+H] = 452,4 m/z).
Пример 22

Соединение 40 синтезируют по методике, описанной в примере 21, используя соединение 29 вместо соединения 27. ([M+H] = 452,4 m/z)
Пример 23
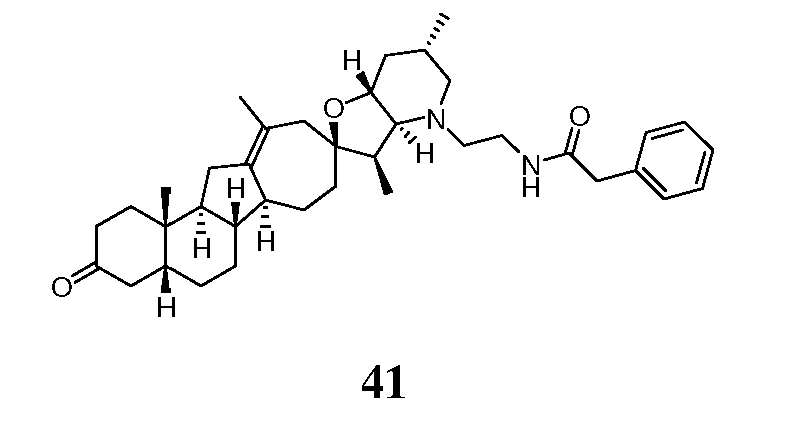
Соединение 41 синтезируют по методике, описанной в примере 10, используя N-(2-оксоэтил)-2-фенилацетамид вместо N-(2-оксоэтил)ацетамида. ([M+H] = 587,7 m/z).
Пример 24

Стадия А

В круглодонную колбу загружают спирт 29 (7,60 г, 13,53 ммоль, 1 экв.) и растворяют в ДХМ (115 мл). В реакционную смесь добавляют триэтиламин (8,21 г, 81 ммоль, 6,0 экв.). Смесь охлаждают до 0°С, и в нее добавляют метансульфонилхлорид (1,86 г, 16,2 ммоль, 1,2 экв.). Через 30 мин реакционную смесь разделяют между насыщенным водным раствором бикарбоната натрия и EtOAc. Органический слой отделяют, сушат над сульфатом натрия и концентрируют досуха. Остаток очищают флэш-хроматографией на силикагеле (EtOAc/гексан 10 → 25%) с получением желаемого продукта мезилата.
В круглодонную колбу загружают мезилат (9,1 г, 14,22 ммоль, 1 экв.) и растворяют в 50 мл DMPU. В реакционную смесь добавляют азид натрия (4,62 г, 71,1 ммоль, 5,0 экв.) и нагревают до 60°С. Смесь перемешивают в течение 17 ч. Затем реакционную смесь охлаждают до кт и добавляют воду. Смесь перемешивают в течение 30 мин. Смесь фильтруют в вакууме, промывают водой и сушат на воздухе, и применяют непосредственно без очистки на следующей стадии.
Стадия B

В круглодонную колбу загружают азид 43 (8,35 г, 14,23 ммоль, 1 экв.) и добавляют ТГФ (120 мл). В реакционную смесь затем добавляют трифенилфосфин (11,2 г, 42,7 ммоль, 3,0 экв.). Смесь нагревают до 50°С и перемешивают в течение 20 ч. Затем реакционную смесь охлаждают до кт, и растворитель удаляют в вакууме. Остаток очищают флэш-хроматографией на силикагеле (MeOH/ДХМ 10% → 20%) с получением амина.
В круглодонную колбу загружают амин (5,10 г, 9,09 ммоль, 1 экв.) и растворяют в ДХМ (60 мл). В реакционную смесь добавляют N,N-диизопропилэтиламин (5,88 г, 45,5 ммоль, 5,0 экв.). Смесь охлаждают до 0°С и добавляют метансульфонилхлорид (2,08 г, 18,2 ммоль, 2,0 экв.). Через 30 минут реакционную смесь разделяют между насыщенным водным раствором бикарбоната натрия и EtOAc. Органический слой собирают, сушат над сульфатом натрия и концентрируют досуха. остаток очищают флэш-хроматографией на силикагеле (EtOAc/гексан 10 → 30%) с получением Cbz-защищенного метансульфонамида.
Стадия С

В круглодонную колбу загружают Cbz-защищенный метансульфонамид (5,37 г, 8,41 ммоль, 1 экв.) и 10% палладий на угле (1,0 г). Твердые вещества суспендируют в 2-пропаноле (50 мл). Суспензию помещают в атмосферу водорода, и смесь перемешивают в течение 4 ч при 25°С. Затем реакционную смесь фильтруют на Celite®, и фильтрат концентрируют досуха. Затем остаток очищают флэш-хроматографией на силикагеле (ДХМ/MeOH 0 → 5%) с получением желаемого продукта. [M+H] = 505,6 m/z.
Альтернативный синтез соединения 42

Перекристаллизованный циклопамин (2,07 г) загружают в реактор подходящего размера и помещают в инертную атмосферу. Последовательно добавляют EtOAc (7,6 г), триэтиламин (1,53 г) и ДМАП (307 мг). Суспензию нагревают до 40°С. Добавляют Cbz-OBt тремя порциями в течение более 90 минут, сохраняя внутреннюю температуру ниже 45°С. Реакционную смесь перемешивают при 40°С в течение 90 минут. Температуру сохраняют пока к реакционной смеси медленно добавляют метанол (26,4 г). Полученную суспензию охлаждают до комнатной температуры и перемешивают в течение, по крайней мере, 15 часов. Неочищенный продукт собирают фильтрацией и промывают метанолом (5 г). Белое твердое вещество сушат в вакууме до постоянной массы и перекристаллизовывают из гептана (30,3 г) и толуола (3,2 г) с получением соединения 24a (3,0 г).

Твердый бис(2,6-диметилфенил)гидрофосфат и 24a заранее сушат и помещают в атмосферу азота. Чистый диэтилцинк (722 мг) загружают в реактор подходящего размера, содержащий ДХМ (9,0 г). Растворы в ДХМ фосфата (1,83 г в 17,9 г) и IPI-332690 (1,34 г в 3,6 г) последовательно добавляют при температуре 25°С или ниже. Добавляют дийодметан (1.58 г), и реакционную смесь перемешивают при 28°С в течение 4-6 часов. Реакционную смесь охлаждают до -45°С, и добавляют раствор метансульфоновой кислоты в ДХМ (566 мг в 1,5 г). Через 15 минут добавляют морфолин (1,711 г), и смесь нагревают до комнатной температуры в течение ночи. Органический слой дважды промывают 2N HCl (2×13,6 г), затем, последовательно, 4,8% масс. карбоната натрия (водн.), 4,8% масс. сульфита натрия (водн.) и 4,8% масс. насыщенного раствора соли (13,6 г каждого). Органический слой сушат, фильтруют, концентрируют до 4 г и разбавляют изопропанолом (4 г). Продукт кристаллизуют из раствора медленным добавлением метанола (9,3 г). Фильтрация с метанольной промывкой (2,6 г) и сушка дают 1,09 г соединения 24b (79% выделено).

Pd/C катализатор Johnson Matthey A-305038-5 (890 мг) загружают в реактор подходящего размера, затем добавляют соединение 24b (2,24 г). Реактор продувают N2 и последовательно добавляют толуол (21,8 г) и 2-пропанол (6,7 г). Систему дегазируют и помещают в атмосферу азота, и процесс повторяют с водородом. Систему энергично перемешивают в одной атмосфере в течение 4-5 часов. Реакцию отслеживают либо ТСХ, либо ВЭЖХ. Если она не завершена, реакционную смесь продувают инертным газом, добавляют еще катализатор (145 мг) и атмосферу водорода возвращают еще на час. Добавляют этилендиамин (12,9 мг), и смесь перемешивают в течение 15 минут. Катализатор удаляют фильтрацией с промывкой толуол:ИПА (3:1). Фильтрат и промывки концентрируют, и растворитель меняют на толуол. Продукт кристаллизуют из толуола (19,0 г) и гептана (18,0 г) с получением соединения 24c в виде белого кристаллического твердого вещества (1,34 г, выход 98%).
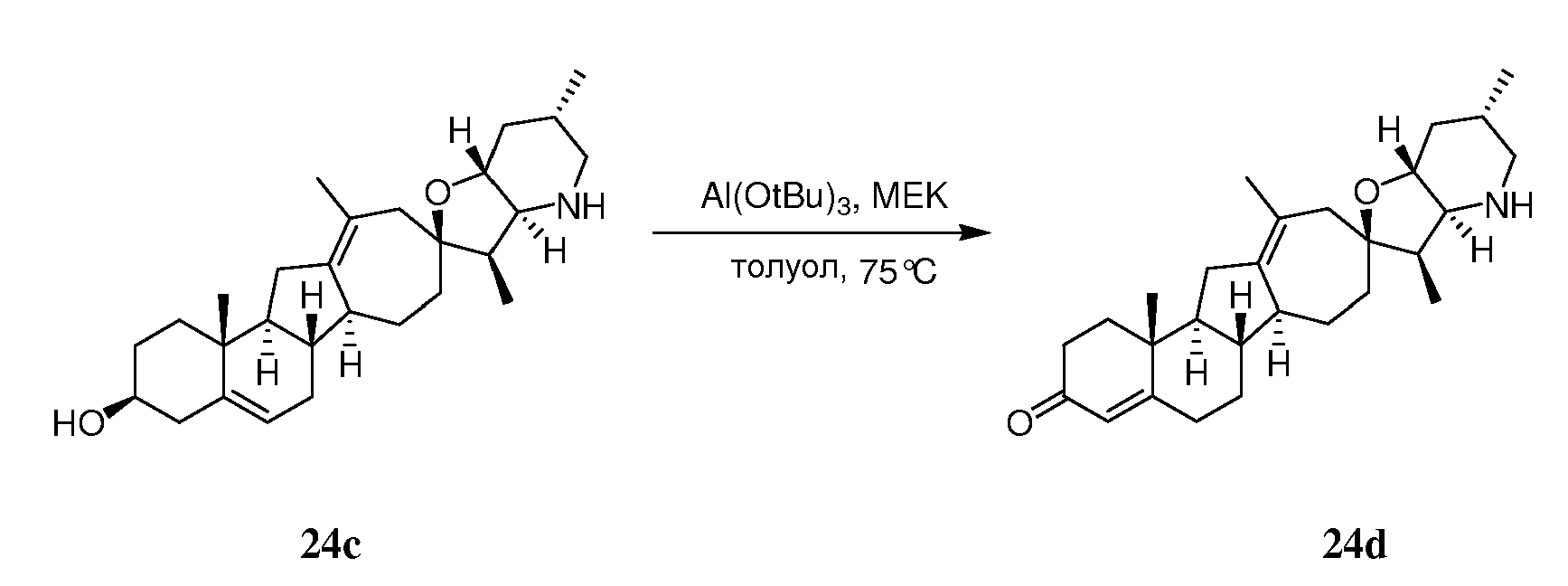
Соединение 24c (644 мг) загружают в реактор подходящего размера, затем добавляют трет-бутоксид алюминия (525 мг), толуол (8,34 г, 15 объем.) и 2-бутанон (7,83 г, 15 объем.). Содержимое колбы дегазируют с применением циклов вакуумирования/продувки азотом для удаления кислорода, и реакционную смесь нагревают при 75°С при энергичном перемешивании в течение 16-18 часов. Реакцию гасят добавлением водного виннокислого калия-натрия (2,6 г в 10,3 г воды), и смесь энергично перемешивают в течение одного часа при 45°С. Водный и органический слои разделяют. Водный слой повторно экстрагируют смесью толуола (2,9 г) и EtOAc (2,9 г). Органические слои объединяют и промывают свежим раствором виннокислого калия-натрия (2,6 г в 10,3 г воды), и затем водой (12,9 г). Полученный органический слой сушат над сульфатом натрия (1,97 г), фильтруют и концентрируют в вакууме. Продукт кристаллизуют через добавление и изменение концентрации растворителя, сначала на ИПА (6,5 г) и затем на гептан (7,7 г). Густую суспензию в гептане (~2,7 г) перемешивают в течение ночи и твердые вещества собирают фильтрацией. Вакуумная сушка дает соединение 24d (550 мг) с 85% выходом.

Енон 24d (459 мг) и 5% палладий на угле Johnson-Matthey (A503023-5, 101 мг) загружают в многогорлый реактор подходящего размера. Реактор продувают азотом и добавляют 3-пиколин (2,2 г) в качестве растворителя. Начинают перемешивание, и реактор сначала дегазируют с применением азота, и затем перемешивают в водороде при атмосферном давлении в течение 8 часов. В конце реакции катализатор удаляют фильтрацией через среду 0,2 микрона, промывая АЦН (1,4 мл). Фильтрат и промывку объединяют в чистом реакторе, оборудованном механической мешалкой, внутренним датчиком температуры, в атмосфере азота. В реактор добавляют раствор лимонной кислоты (3,7 г) в воде (9,2 мл) при 30°С или ниже, и IPI-335589 медленно кристаллизуют из раствора в виде цитрата при 20, а затем 0°С. Кристаллический продукт выделяют фильтрацией с отсасыванием, и промывают водой (3,7 мл). После сушки цитрат, 24e, выделяют в виде гидрата (3-5% масс воды) с 89,5% выходом (622 мг) при соотношении β:α около 90:1.

Соединение 24e (1,50 г) загружают в реактор подходящего размера вместе с 2-метилтетрагидрофураном (7,7 г) и 1M карбонатом натрия (9,0 мл). Раствор бензилхлорформиата (454 мг) в 2-метилтетрагидрофуране (300 мг) добавляют через добавляющую воронку, и реакционную смесь выстаивают при температуре окружающей среды в течение 1-2 часов. После завершения реакции перемешивание останавливают, слои разделяют, и органический слой дважды промывают водой (2×6 г). Органический слой сушат над сульфатом натрия (3 г), фильтруют и концентрируют. Остаточную воду далее снижают концентрацией из свежего 2-метилтетрагидрофурана (6,5 г), и продукт переносят в виде раствора в безводном 2-метилтетрагидрофуране на следующую стадию.

Коммерческий 1M K-Selectride в ТГФ (1,20 г) загружают в сухой реактор в атмосфере азота, разбавляют безводным 2-метилтетрагидрофураном (2,10 г) и охлаждают до -65°С. Раствор соединения 24f (0,41 г) в 2-метилтетрагидрофуране (1,5 г) затем медленно добавляют в реактор, контролируя внутреннюю температуру -65±5°C. Реакционную смесь перемешивают в течение 2 часов и нагревают до -20°С в течение приблизительно 1 часа, и перемешивают еще час. Реакцию отслеживают ВЭЖХ, и реакции, которые не завершились, стимулируют к завершению дополнительным K-selectride. Реакцию гасят при низкой температуре MeOH (0,33 г), затем 3M NaOH (2,4 г) при -20°С, и 15% перекисью водорода в воде (1,04 г) при 5°С или ниже, затем перемешивают в течение ночи при температуре окружающей среды. Слои разделяют, и органический слой промывают последовательно 1M водным NaOH (2 мл), 0,5 M водным Na2SO3 (2 мл) и водой (2 мл), доводя до рН 3 добавлением HCl. Органический слой сушат над сульфатом натрия (0,82 г), фильтруют и концентрируют. Продукт 24g (0,457 г) повторно концентрируют из ДХМ (0,9 г) и применяют на следующей стадии.

Соединение 24g (1,36 г) загружают с безводным ДХМ (18,1 г) в реактор подходящего размера, помещают в инертную атмосферу и охлаждают до -20°С. Добавляют триэтиламин (0,61 мг) с последующим медленным добавлением метансульфонилхлорида (373 мг) в безводном ДХМ (300 мг). Реакционную смесь перемешивают в течение 1 часа при -20°С. Реакцию отслеживают ВЭЖХ. Незавершенные реакции стимулируют дополнительным метансульфонилхлоридом. После завершения реакцию гасят водой (13,6 г) и нагревают. Слои разделяют, и органический слой промывают 2,5% масс. бикарбоната натрия (13,8 г) и затем водой (10,9 г). Органический слой сушат над сульфатом натрия (4 г), фильтруют и концентрируют. Меняют растворитель продукта добавлением и концентрацией до трет-бутилметилового эфира (10,9 мл) и затем 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидинона (DMPU, 4,7 мл). Раствор DMPU используют в следующей реакции.
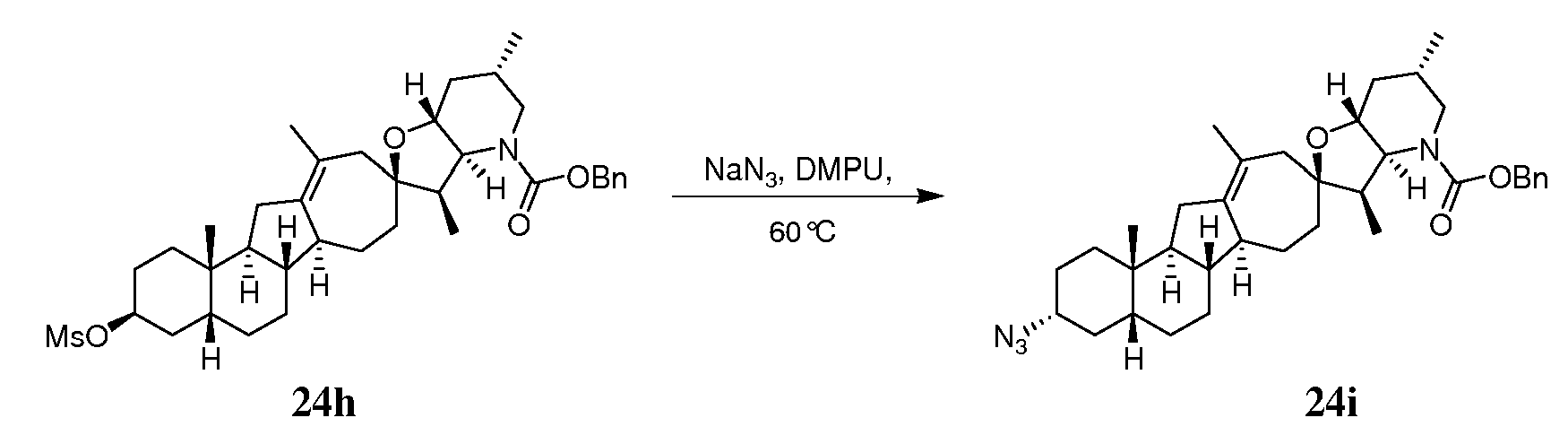
Азид натрия (0,74 г) загружают в реактор подходящего размера. В реактор добавляют раствор соединения 24h (1,46 г) в DMPU (5,9 г), промывая дополнительным DMPU (1,9 г). Суспензию нагревают до 60°С в течение 15 часов, сохраняя азотную чистку в течение всей реакции. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют МТБЭ (11,7 г). Органический раствор промывают 3 раза 2% насыщенным раствором соли (3×8 г), сушат над сульфатом натрия (4,4 г), фильтруют и концентрируют. Продукт концентрируют из ТГФ (6,4 г) и применяют сразу в следующей реакции.

Неочищенное соединение 24i (1,34 г) растворяют и переносят в реактор подходящего размера с ТГФ (12,6 г). Добавляют трифенилфосфин (0,70 г) и воду (0,44 г), и реакционную смесь нагревают до 55°С в течение 15-24 часов. После завершения реакцию охлаждают до температуры окружающей среды, сушат над сульфатом магния (1,4 г), фильтруют и концентрируют. Твердые вещества растворяют и концентрируют из трех порций ДХМ (3×9 г) и очищают хроматографией на силикагеле с применением ДХМ/MeOH/Et3N градиентов для удаления примесей на основе реагентов. Объединенные фракции концентрируют досуха, растворяют в ДХМ (6,8 г) и снова концентрируют досуха с получением аморфного твердого вещества (1,12 г), которое применяют в следующей реакции.

Соединение 24j (1,09 г) растворяют и переносят в реактор подходящего размера с безводным ДХМ (15,8 г) и помещают в атмосферу азота. Раствор охлаждают до 0°С. Последовательно добавляют N,N-диизопропилэтиламин (357 мг) и чистый метансульфонилхлорид (0,165 мл), сохраняя температуру ниже 5°С. Реакцию отслеживают ВЭЖХ. Незавершенные реакции стимулируют к завершению дополнительным метансульфонилхлоридом. Реакцию гасят 0,4 M водным бикарбонатом натрия (11,4 г) и нагревают до температуры окружающей среды. Слои разделяют, и водную фазу снова экстрагируют ДХМ (5,8 г). Объединенные органические слои сушат над сульфатом магния (0,55 г), фильтруют и концентрируют. Продукт 24k растворяют и отгоняют из 2-пропанола (4,0 г) для удаления остаточного ДХМ, и применяют сразу в следующей реакции.

Aldrich Degussa тип E101 NE/W 10% Pd/C (249 мг) загружают в реактор подходящего размера и помещают в атмосферу азота. Раствор в 2-пропаноле (9,8 г) соединения 24k (1,24 г) добавляют в реактор. Систему дегазируют и помещают в атмосферу азота, и процесс повторяют с водородом. Реакционную смесь перемешивают при 1 атм. водорода при температуре окружающей среды в течение 8 часов. Инертную атмосферу возвращают в реактор, и к реакционной смеси добавляют вторую партию катализатора (125 мг), суспендированного в 2-пропаноле (0,5 г). Реакционную смесь дегазируют и помещают в атмосферу азота, и процесс повторяют с водородом. Реакционную смесь перемешивают при 1 атм. водорода в течение еще 15 часов при температуре окружающей среды. Реакцию отслеживают ВЭЖХ. Незавершенные реакции обрабатывают дополнительным катализатором и водородом. После завершения реакционную смесь фильтруют, обрабатывают активированным углем (200 мг) и снова фильтруют. Раствор, высушенный частичной концентрацией, переносят в реактор и разбавляют безводным 2-пропанолом до 0,09 M по отношению к теоретическому выходу. 1,25 M HCl раствор в 2-пропаноле (1,64 г) добавляют в течение 20 минут. Гидрохлорид медленно кристаллизуют при осторожном перемешивании, и выделяют фильтрацией. Кристаллы промывают 2-пропанолом (2,5 г) и сушат в вакууме с получением соединения 42 (916 мг, выход 80%) в виде 1:1 ИПА сольвата.
Пример 25

Стадия А

В круглодонную колбу загружают амин 42 (1,1 г, 2,1 ммоль, 1 экв.), сухой тетрагидрофуран (10 мл) и пиридин (880 мкл, 10,9 ммоль, 5 экв.). Охлажденную (0°С) смесь обрабатывают бензоилпероксидом (1,6 г, 6,5 ммоль, 3 экв.). Смесь перемешивают в течение 2 часов при 0°С, затем в течение ночи при 25°С. Реакционную смесь разбавляют МТБЭ и промывают смесью насыщенного водного NaHCO3 с 1N NaOH до разделения слоев. Органический слой собирают, и водный слой повторно экстрагируют один раз МТБЭ. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют досуха. Неочищенное масло растворяют в 5 мл CH2Cl2, загружают в колонку с SiO2 (40 г) и элюируют из гексана/EtOAc (10% до 50%) с получением бензоильного производного 48 (380 мг) ([M+H] = 625,4 m/z).
Стадия В

В круглодонную колбу загружают соединение 48 (374 мг, 0,6 ммоль, 1 экв.) и MeOH (5 мл). Раствор обрабатывают при 25°C в присутствии 2N KOH (0,3 мл, 0,6 ммоль, 1 экв.). Смесь перемешивают в течение 3 ч. Растворитель удаляют в вакууме. К остатку добавляют МТБЭ, затем нейтрализуют 1N HCl. Слои разделяют, и водный слой экстрагируют двумя порциями CH2Cl2. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют досуха. Неочищенный продукт (380 мг) растворяют в CH2Cl2, загружают в колонку SiO2 (12 г) и элюируют гексаном/EtOAc (0% до 100%) с получением гидроксиламина 47. Продукт лиофилизируют из t-BuOH/7% H2O с получением 213 мг соединения 47 в виде белого порошка ([M+H] = 521,4 m/z).
Пример 26

Стадия А

В высушенную струйной воздушной сушилкой колбу загружают сухой CH2Cl2 (5 мл) и бензиловый спирт (785 мкл, 7,58 ммоль, 1,3 экв.). Охлажденный (0°С) раствор обрабатывают изоцианатом хлорсульфонила (506 мкл, 5,83 ммоль, 1 экв.). Затем добавляют ДМАП (1,4 г, 11,6 ммоль, 2 экв.), и смесь перемешивают в течение 1 ч при 25°C. После завершения растворения ДМАП реакционная смесь на короткое время остается прозрачной. Затем формируется белый хлопьевидный осадок. Смесь разбавляют CH2Cl2 (30 мл) и промывают тремя порциями (30 мл каждая) воды. Органический слой сушат над Na2SO4, фильтруют и выпаривают досуха. Желаемое белое твердое вещество 51 используют на следующей стадии без очистки.
Стадия В

В круглодонную колбу загружают соединение 52 (30 мг, 0,053 ммоль, 1 экв.) и соединение 51 (18 мг, 0,053 ммоль, 1 экв.). Оба реагента растворяют в CH2Cl2 (2 мл), и раствор перемешивают при 25°C. Неочищенный продукт загружают в колонку SiO2 (4 г) и элюируют гексаном/EtOAc (0% до 50%) с получением 16 мг сульфамоилированного производного 53 ([M+Na] = 796,4 m/z).
Стадия С

В круглодонную колбу загружают соединение 53 (16 мг, 0,021 ммоль, 1 экв.) и 11 мг 10% Pd/C (влажный, Aldrich Degussa тип E101). Материал суспендируют в 2-пропаноле (3 мл). Колбу герметично закрывают и три раза продувают водородом и оставляют на ночь в 1 атм. водорода. Суспензию фильтруют через 0,2 микрона Acrodisc, промывают 2-пропанолом, и растворитель удаляют в вакууме. Остаток очищают на колонке SiO2 (1 г), элюируя CH2Cl2/MeOH (5% до 10%). Основной продукт лиофилизируют из t-BuOH/7% H2O с получением 9 мг сульфамида 50 ([M+H] = 506,4 m/z).
Пример 27

Стадия А

В круглодонную колбу загружают циклопамин 4-ен-3-он (3,5 г, 8,5 ммоль, 1 экв.) и пиридин (70 мл). В реактор загружают Pd/C (10% Pd, 500 мг). Реакционную смесь помещают в 1 атмосферу водорода. Через 3,5 ч ЖХМС показывает полное потребление исходного материала. Катализатор отфильтровывают на фильтре Acrodisk 0,2 микрона и промывают толуолом. Растворитель удаляют азеотропной перегонкой с толуолом (2×10 мл). Желаемый продукт 56, 3,5 г ([M+H] = 412,5 m/z) применяют как есть на следующей стадии.
Стадия В

В круглодонную колбу загружают соединение 56 (1,2 г, 2,8 ммоль, 1 экв.), CH2Cl2 (10 мл) и триэтиламин (1,9 мл, 14,2 ммоль, 5 экв.). Охлажденный (0°С) раствор обрабатывают CBz-Cl (440 мкл, 2,8 ммоль, 1 экв.). Через 1 час ЖХМС показывает полное потребление исходного материала. Смесь разбавляют водой. Слои разделяют, и органический слой дважды промывают водой. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют досуха. Продукт очищают хроматографией на колонке (SiO2, 40 г), элюируя гексаном/EtOAc (0 до 20%) с получением соединения 57 (891 мг) ([M+Na] = 468,4 m/z).
Стадия С

В круглодонной колбе кетон 57 два раза подвергают азеотропной перегонке с CH2Cl2 и сушат в вакууме в течение 1 ч. В атмосфере азота кетон 2 (693 мг, 1,27 ммоль, 1 экв.) растворяют в безводном ТГФ (20 мл) и раствор охлаждают до -78°С. По каплям добавляют 1M раствор K-selectride в ТГФ (1,9 мл, 1,9 ммоль, 1,5 экв.). Через 1 час реакция завершается по ТСХ. Реакцию гасят добавлением 2,6 мл 5N NaOH с последующим медленным добавлением 2,6 мл 30% масс. H2O2. Полученную смесь перемешивают в течение ночи. Смесь разделяют между водой и EtOAc. Водный слой снова экстрагируют EtOAc. Объединенные органические слои промывают сначала водой (буферированной небольшим количеством хлорида аммония), затем насыщенным раствором соли. Органический слой сушат, фильтруют и концентрируют с получением неочищенной пены (840 мг). Неочищенный продукт растворяют в CH2Cl2, загружают в колонку SiO2 (40 г) и элюируют гексаном/EtOAc (0 до 50%) с получением соединения 58 (565 мг).
Стадия D
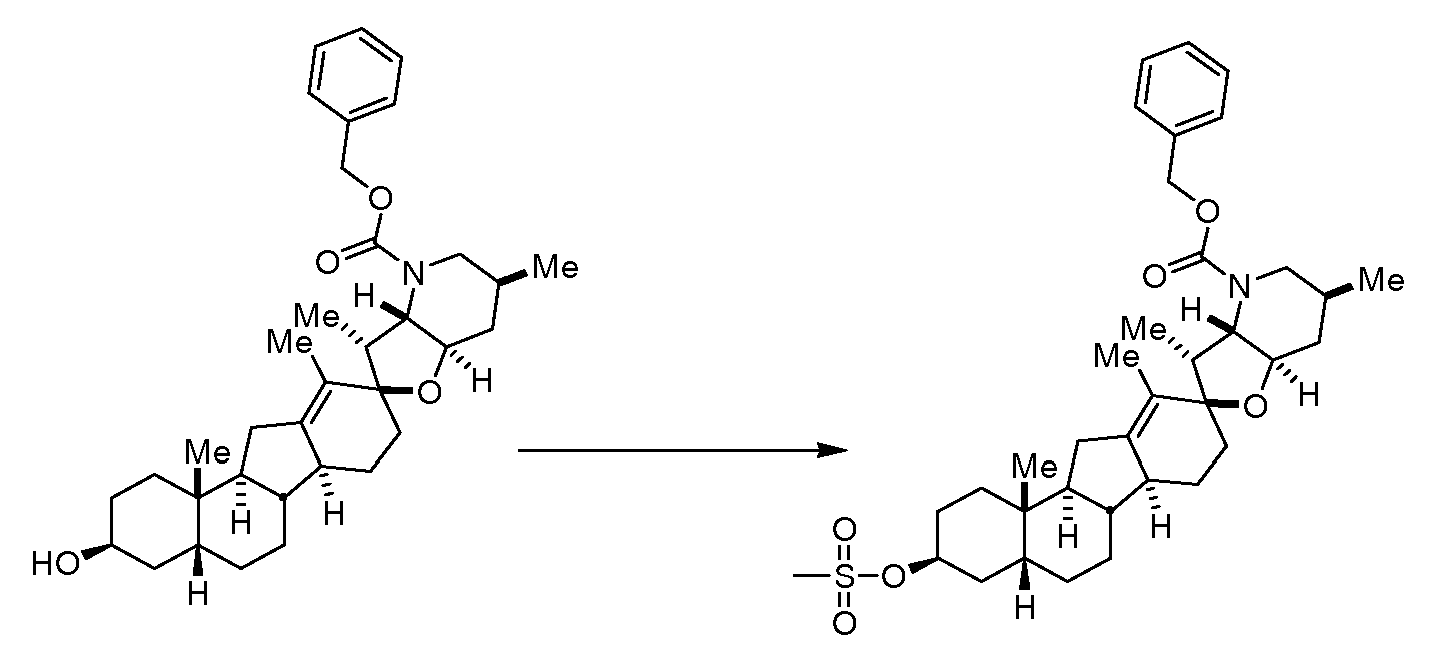

В круглодонной колбе в атмосфере азота спирт 58 (530 мг, 0,98 ммоль, 1 экв.) растворяют в 5 мл безводного CH2Cl2 и триэтиламине (800 мкл, 5,81 ммоль, 6 экв.). Реакционную смесь охлаждают до 0°С и по каплям добавляют Ms-Cl (112 мкл, 1,45 ммоль, 1,5 экв.). Смесь перемешивают при 0°С в течение 30 мин. ТСХ (гексан:EtOAC, 7:3) показывает ~70% превращение. В реактор добавляют 70 мкл триэтиламина (70 мкл, 0,5 экв.) и Ms-Cl (10 мкл, 0,1 экв.). Через 90 мин добавляют раствор насыщенного бикарбоната, и остаток экстрагируют CH2Cl2. Органический слой промывают водой, сушат и концентрируют до беловатой пены. Продукт растворяют в CH2Cl2 и очищают SiO2 (40 г), элюируя гексаном/EtOAc (0% до 50%) с получением соединения 59 (430 мг).
Стадия Е

В круглодонной колбе мезилат 59 (420 мг, 0,67 ммоль, 1 экв.) растворяют в 2 мл DMPU. Раствор обрабатывают азидом натрия (218 мг, 3,4 ммоль, 5 экв.) при 60°С в течение 5 ч. Смесь охлаждают до 25°C, затем выливают в ледяную воду для получения белого твердого вещества. Соединение экстрагируют МТБЭ (3 раза). Объединенные органические слои промывают водой (2X), затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют до белой пены (342 мг). Желаемый продукт 60 применяют как есть на следующей стадии.
Стадия F

В круглодонной колбе, оборудованной конденсатором, азид 60 (336 мг, 0,58 ммоль, 1 экв.) растворяют в 7 мл ТГФ и 140 мкл воды и обрабатывают трифенилфосфином (462 мг, 1,76 ммоль, 3 экв.). Смесь нагревают до 70°С в течение ночи. ТСХ (гексан/EtOAc, 7:3) подтверждает завершение реакции. Реакционную смесь концентрируют досуха. Неочищенный продукт растворяют в CH2Cl2, загружают в 12 г SiO2 и элюируют CH2Cl2/MeOH (0 до 20%) с получением амина 61 (254 мг).
Стадия G
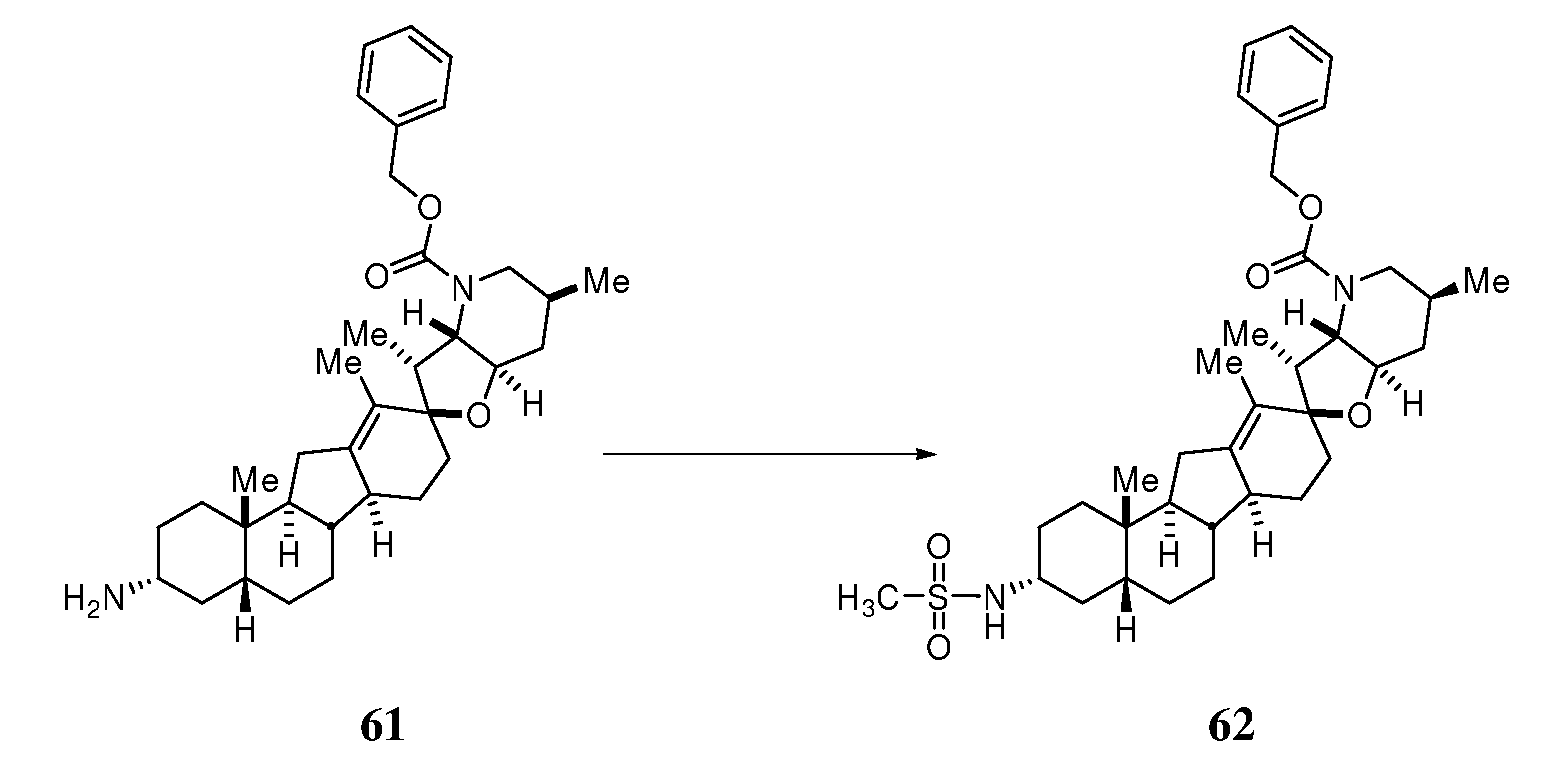
В круглодонной колбе в азоте амин 61 (248 мг, 0,45 ммоль, 1 экв.) растворяют в 7 мл безводного CH2Cl2 и N,N-диизопропилэтиламине (237 мкл, 0,91 ммоль, 2 экв.). Реакционную смесь охлаждают до 0°С и по каплям добавляют Ms-Cl (70 мкл, 1,45 ммоль, 1,5 экв.). Смесь перемешивают при 0°С в течение 2 ч. ТСХ (гексан/EtOAc, 7:3) показывает незначительное количество амина. В смесь добавляют 10 мкл Ms-Cl (0,2 экв.), и нагревают до 25°C в течение 1 ч. Реакционную смесь разбавляют CH2Cl2, затем насыщенным раствором NaHCO3. Слои разделяют. Водный слой экстрагируют одной порцией CH2Cl2. Объединенные органические слои промывают водой, сушат над Na2SO4, фильтруют и концентрируют досуха. Неочищенный продукт (326 мг) загружают в колонку SiO2 (12 г) и элюируют гексаном/EtOAc (0 до 50%) с получением сульфонамида 62 (256 мг).
Стадия Н

В круглодонную колбу загружают сульфонамид 62 (250 мг, 0,4 ммоль, 1 экв.) и 50 мг 10% Pd/C (влажный, Aldrich Degussa тип E101 лот 08331KC). Материал суспендируют в EtOAc (5 мл). Колбу герметично закрывают и три раза продувают водородом и перемешивают в 1 атм. водорода. Через 3 ч наблюдается некоторое превращение, но большая часть исходного материала остается. Суспензию фильтруют через 0,2 микрона Acrodisc, промывают 2-пропанолом. Раствор фильтрата повторно помещают в условия реакции добавлением 54 мг катализатора. Реакция завершается через 3 ч. Суспензию фильтруют через 0,2 микрона Acrodisc, промывают 2-пропанолом, и растворитель концентрируют досуха. Неочищенный продукт (200 мг) загружают в колонку SiO2 (12 г), и соединение элюируют с применением градиента CH2Cl2/MeOH (0 до 10%) с получением свободного амина. Продукт лиофилизируют из t-BuOH/7% H2O с получением 175 мг соединения 55 в виде белого порошка ([M+H] = 491,3 m/z).
Пример 28: Ингибирование «ежового» пути в культуре клеток
Губительное действие на специфичные к «ежовому» пути раковые клетки может быть подтверждено в данном анализе. Клетки C3H10T1/2 дифференцируют в остеобласты при контакте с обработанным ультразвуком «ежовым» пептидом (Shh-N). При дифференциации эти остеобласты выделяют высокие уровни щелочной фосфатазы (ЩФ), которые могут быть измерены в ферментном анализе (Nakamura et al., 1997 BBRC 237: 465). Соединения, которые блокируют дифференциацию C3H10T1/2 в остеобласты (Shh-зависимое событие), поэтому, могут быть идентифицированы снижением образования ЩФ (van der Horst et al., 2003 Bone 33: 899). Подробный анализ представлен ниже. Приблизительные результаты (EC50 для ингибирования) анализа дифференциации показаны ниже в таблице 1.
Протокол анализа
Культура клеток
Фибробласты мезодермы эмбрионов мыши C3H10T1/2 (получены из ATCC) культивируют в среде Basal MEM Media (Gibco/Invitrogen) с добавлением 10% чистого инактивированного ФРФБ (Hyclone), 50 единиц/мл пенициллина и 50 мкг/мл стрептомицина (Gibco/Invitrogen) при 37°С с 5% CO2 в атмосфере воздуха.
Анализ щелочной фосфатазы
Клетки C3H10T1/2 помещают в 96-луночные планшеты с плотностью 8×103 клеток/лунку. Клетки выращивают до конфлюэнтности (72 ч). После обработки «ежовым» ультразвуком (250 нг/мл) и/или соединением клетки лизируют в 110 мкл лизисного буфера (50 мМ Tris pH 7,4, 0,1% TritonX100), планшеты обрабатывают ультразвуком, и лизаты центрифугируют через 0,2 мкм PVDF планшеты (Corning). 40 мкл лизатов анализируют на активность ЩФ в растворе щелочного буфера (Sigma), содержащем 1 мг/мл п-нитрофенилфосфата. После инкубирования в течение 30 мин при 37°С планшеты считывают на планшетном ридере Envision при 405 нм. Общий белок количественно оценивают с помощью набора для анализа белка BCA от Pierce согласно инструкции производителя. Активность ЩФ нормализуют к общему белку. Примечание: "A" показывает, что IC50 менее 20 нМ, "B" показывает, что IC50 составляет 20-100 нМ, "C" показывает, что IC50 > 100 нМ.
Пример 29: модель рака поджелудочной железы
Активность соединения 42 тестируют на модели рака поджелудочной железы человека: клетки BXPC-3 имплантируют подкожно в подвздошную область правой лапы мыши. На 42 день после имплантации опухоли мышей произвольно делят на две группы, получающие либо носитель (30% HPBCD), либо соединение 42. Соединение 42 дают в дозе 40 мг/кг/сутки. После введения 25 суточных доз соединение 42 статистически снижает рост объема опухоли на 40% по сравнению с носителем (p=0,0309). В конце исследования опухоли собирают через 4 часа после последней дозы для оценки целевой реакции q-RT-PCR анализом генов HH пути. Анализ человеческой Gli-1 показал отсутствие модулирования. Анализ уровней Gli-1 в мРНК мышей показал устойчивую понижающую регуляцию в группе, обработанной соединениями, по сравнению с группой, обработанной носителем.
Пример 30: модель медуллобластомы
Активность соединения 42 также оценивают на модели медуллобластомы у трансгенных мышей. У мышей, которые является гетерозиготными для мутации с потерей функции в супрессорах опухоли Patched1 (Ptch1) и Hypermethylated in Cancer (Hic1) развивают спонтанную медуллобластому. Так же как и человеческая медуллобластома, эти опухоли демонстрируют полное гиперметилирование промотора оставшейся аллели Hic1, а также потерю экспрессии дикой аллели Ptch1. При помещении в виде подкожных аллотрансплантатов, эти опухоли агрессивно растут и являются зависимыми от «ежового» пути. Эту модель применяют для оценки эффективности и перорально вводимого соединения, и для сопоставления активности с облучением в плазме и опухолях. Пероральное введение (ПВ) одной дозы соединения 42 приводит к зависимой от дозы понижающей регуляции HH пути в подкожно имплантированных опухолях, что измеряется пониженной экспрессией Gli-1 мРНК через 8 часов после введения дозы.
Суточное (QD) введение соединения ПО приводит к зависимому от дозы ингибированию роста опухоли, где более явная регрессия опухоли наблюдается при наиболее высоких дозах. Приблизительная эффективная пероральная суточная доза для 50% ингибирования роста опухоли (ED50) составляет 4 мг/кг. Когда животных обрабатывают QD в течение 21 дня, наблюдается долговременное выживание после отмены лечения (>60 дней), при незначительном или отсутствии повторного роста опухоли.
Пример 31: модель рака легких
Для тестирования активности соединения 42 на модель опухоли SCLC человека клетки LX22 подкожно имплантируют в подвздошную область правой лапы. LX22 в первую очередь являются моделью ксенотрансплантата SCLC, полученной у не подвергнутых химиотерапии пациентов, который поддерживается передачей от мыши к мыши. Эта опухоль реагирует на химиотерапию этопозидом/карбоплатином тем, что сильно совпадает с клинической ситуацией. LX22 регрессируют во время химиотерапии, проходят период ремиссии и затем начинают восстанавливаться. В модели LX22 тестируется активность агента, содержащего только соединение, и его способность модулировать химиорезистентное восстановление. На 32 день после имплантации опухоли мышей произвольно делят на три группы, получающие носитель (30% HBPCD), соединение или химиотерапевтическую комбинацию этопозида и карбоплатина (E/P). Соединение 42 вводят в дозе 40 мг/кг/сутки, и после 16 последовательных доз не остается измеряемых различий между группами, получающими лечение и носитель. Этопозид вводят в.в. в дозе 12 мг/кг в дни 34, 35, 36 и 48, а карбоплатин вводят в.в. в дозе 60 мг/кг в дни 34, 41 и 48 после имплантации опухоли. На 50 день мышей, обработанных E/P, далее произвольно делят на группы, получающие либо носитель (30% HPBCD), либо соединение после лечения. Соединение вводят в виде пероральной мультидозы MTD 40 мг/кг/сутки, и после 35 последовательных доз наблюдается значительное отставание в восстановлении опухоли в леченной группе, по сравнению с группой, получающей носитель (p=0,0101).
Ссылки
Все патенты США и опубликованные заявки на патент США, указанные здесь, включены сюда посредством ссылок.
Эквиваленты
Специалисты в данной области техники поймут, или смогут удостовериться с помощью обычных экспериментов, что существует множество эквивалентов описанных здесь конкретных примеров данного изобретения. Такие эквиваленты включены в объем формулы изобретения.
Реферат
В изобретении представлены новые производные циклопамина, имеющие следующую формулу:где R- Н, алкил, -OR, амино, сульфонамидо, сульфамидо, OC(O)R, -N(R)C(O)R; R- Н, гетероциклоалкил; или Rи Rвместе образуют =O, =S, =N(OR), N(NR) или -C(R); R- Н; R- Н, алкил, OR, -C(O)R, [(W)-C(O)O]R, -W-NR Xи др., где W - двухвалентный алкильный радикал, X - галогенид, R- Н, алкил, алкенил, алкинил, арил, циклоалкил и др. Соединения обладают ингибирующим действием на рост опухолей. 6 н. и 32 з.п. ф-лы, 27 пр., 1 табл.
Формула

или его фармацевтически приемлемая соль;
где R1 является Н, алкилом, -OR, амино, сульфонамидо, сульфамидо, -OC(O)R5, -N(R5)С(О)R5;
R2 является Н или гетероциклоалкилом;
или R1 и R2 вместе образуют =O, =S, =N(OR), =N(R), =N(NR2) или =C(R)2;
R3 является Н;
R4 является Н, алкилом, -OR5, -C(O)R5, -CO2R5, -SO2R5, C(O)N(R5)(R5), -[C(R)2]q-R5, -[(W)-N(R)C(O)]qR5, -[(W)-C(O)]qR5, -[(W)-C(O)O]qR5, -[(W)-OC(O)]qR5, -[(W)-SO2]qR5, -[(W)-C(O)N(R5)]qR5, -[(W)-O]qR5, -[(W)-N(R)]qR5 или -W-NR53+X-;
где каждый W независимо является двухвалентным алкильным радикалом;
каждый q независимо равен для каждого случая 1, 2, 3, 4, 5 или 6;
каждый R независимо представляет собой Н, алкил или арил;
X- является галогенидом;
каждый R5 независимо является Н, алкилом, алкенилом, алкинилом, арилом, циклоалкилом, гетероциклоалкилом, аралкилом, гетероарилом или гетероаралкилом;
при условии, что если R2, R3 и R4 являются Н; R1 не является гидроксилом;
далее
при условии, что если R4 является гидроксилом, то R1 не является гидроксилом; далее
при условии, что если R4 является гидроксилом, то R1 и R2 вместе не являются С=O;
и где
каждый случай алкила относится к C1-С30 алкилу,
каждый случай алкенила относится к С2-С30 алкенилу,
каждый случай алкинила относится к С2-С30 алкинилу,
каждый случай циклоалкила относится к С3-С10 циклоалкилу,
каждый случай арила относится к 6-10-членному арилу;
каждый случай гетероциклоалкила относится к 3-7-членному гетероциклоалкилу, который включает 1-4 гетероатома, независимо выбранных из азота, кислорода и серы, и
каждый случай гетероарила относится к 5-6-членному гетероарилу, который включает 1-4 гетероатома, независимо выбранных из азота, кислорода и серы.
где R1 представляет собой Н, -OR, сульфонамидо или -OC(O)R5;
R2 является H;
или R1 и R2 вместе образуют =O или=N(OR);
R3 является H;
R4 является Н, С1-С6, алкилом, -OR5, -[(W)-N(R)C(O)]qR5, -[(W)-N(R)]qR5 или -W-NR53+X-;
где каждый W независимо является двухвалентным C1-С6алкильным радикалом;
q равен 1;
каждый R независимо является Н или C1-С6алкилом;
X- является галогенидом;
каждый R5 независимо является Н, C1-С6алкилом, фенилС1-С6алкилом или гетероарC1-С6алкилом, где гетероарильная часть представляет собой тетразолил.
где R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют =O, =N(OR) или =S.
где R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют =O или =N(OR).
где R1 является Н, -OR, сульфонамидо или -OC(O)R5;
R2 является Н;
или R1 и R2 вместе образуют =O или =N(OR);
R3 является Н;
R4 является Н, C1-С6алкил, гидрокси или -[(W)-N(R)C(O)]qR5;
и каждый R5 независимо является C1-С6алкилом или фенилС1-С6алкилом.
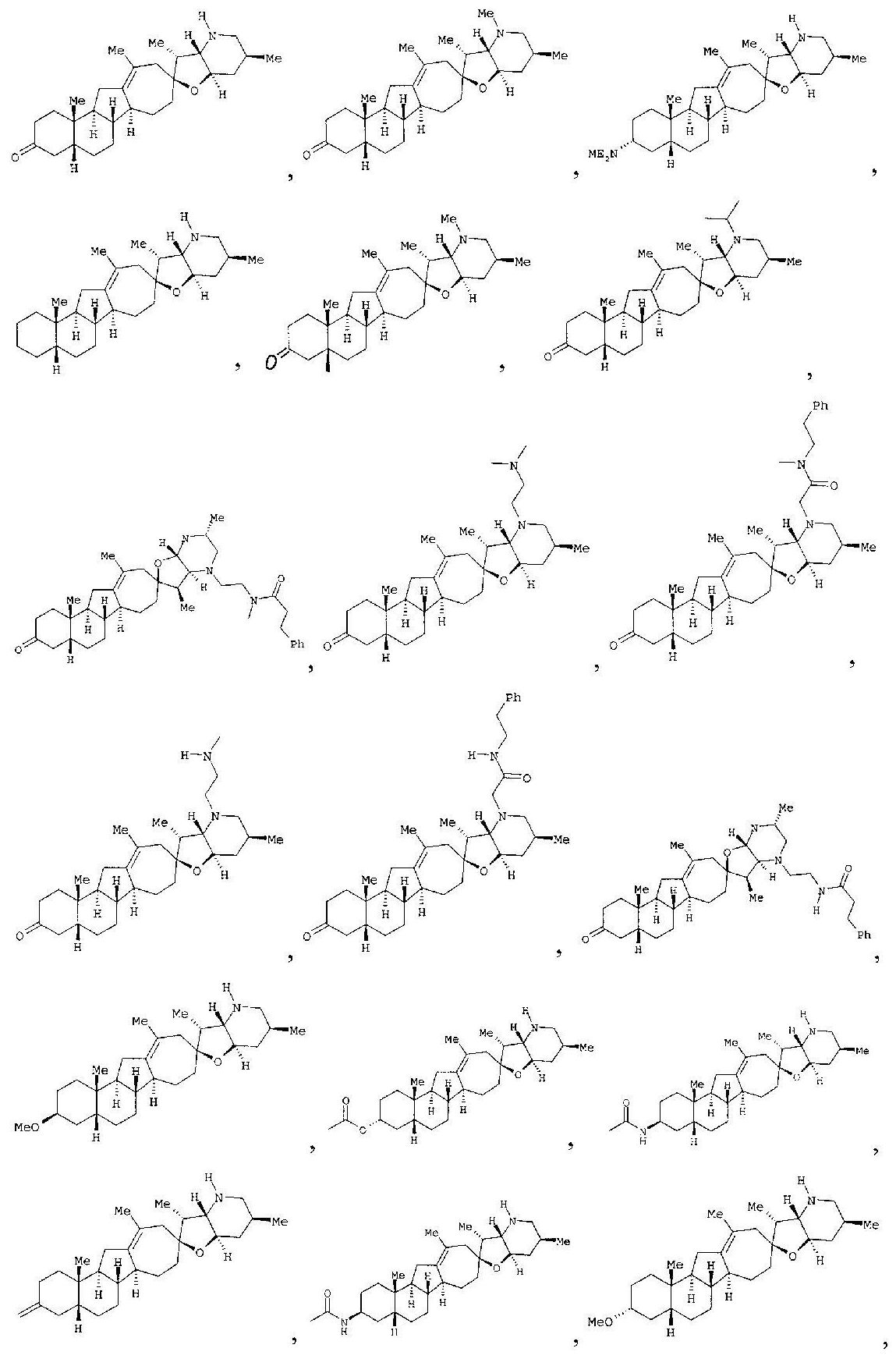










или его фармацевтически приемлемая соль.

или его фармацевтически приемлемую соль, и, по меньшей мере, один фармацевтически приемлемый эксципиент.
Комментарии