Ациламино-замещенные производные конденсированных циклопентанкарбоновых кислот и их применение в качестве фармацевтических средств - RU2529484C2
Код документа: RU2529484C2
Описание
Настоящее изобретение относится к соединениям формулы I,

где A, Y, Z, R3 по R6, R20 по R22 и R50 имеют значения, указанные ниже, и являются ценными фармацевтически активными соединениями. А именно, они являются ингибиторами рецептора-2 генов эндотелиальной дифференцировки (Edg-2, EDG2) который активируется лизофосфатидиловой кислотой (LPA), и также называется рецептором LPA1, и применяются для лечения таких заболеваний, как, атеросклероз, инфаркт миокарда и сердечная недостаточность. Изобретение также относится к способам получения соединений формулы I, их применению и содержащим их фармацевтическим композициям.
LPA представляет собой группу эндогенных лизофосфолипидных производных, включающую, например, 1-олеоил-sn-глицерол-3-фосфат. LPA активирует сопряженные с G-белком рецепторы (GPCR) из семейства рецепторов генов эндотелиальной дифференцировки, которые относятся к лизофосфолипидным рецепторам. Сигналы LPA вызывают набор различных плейотропных биологических откликов во множестве различных видов клеток, которые вмешиваются в такие процессы, как пролиферация клеток, рост клеток, клеточная гипертрофия, трансдифференцировка, клеточная ретракция, клеточная контрактура, миграция клеток, выживание клеток или воспаление. Семейство Edg рецепторов, первоначально идентифицируемое как семейство сиротских GPCR, в настоящее время включает восемь различных членов, которым недавно дали наименование в соответствии с соответствующим им лигандом как рецепторы LPA или рецепторы S1P (рецепторы сфингозин-1-фосфата). В соответствии с номенклатурой Международного союза фундаментальной и клинической фармакологии (IUPHAR) рецепторы LPA Edg-2, Edg-4 и Edg-7 также называются рецепторами LPA1, LPA2 и LPA3 (ср. I. Ishii et al., Annu. Rev. Biochem. 73 (2004), 321-354).
LPA образуется в основном во внеклеточных областях различными путями, главным образом, за счет фактора подвижности раковых клеток аутотаксина, который, как недавно было установлено, идентичен лизофосфолипазе D. LPA может также получаться по альтернативным механизмам, включающим гидролиз фосфолипазы (PLA1 и PLA2), или другим механизмам, например синтеза «de novo» фосфолипидов. Несмотря на то, что LPA, в отличие от других фосфолипидов, хорошо растворима в воде, в плазме она переносится различными связывающими белками, такими как альбумином и гельсолином, которые проявляют высокую аффинность к LPA, и из которых она может высвобождаться. В патофизиологических условиях уровни LPA могут повышаться до нежелательных количеств, а потому наращивать опосредованную LPA передачу сигнала и вызывать разрушительные процессы, например аномальную пролиферацию клеток. Блокирование сигналов LPA, например ингибиторами Edg-2, позволяет предотвратить такие процессы.
Например, повышенное выделение LPA наблюдалось в процессе активации тромбоцитов и свертывания крови и в областях воспаления (T. Sano et al., J. Biol. Chem. 277 (2002), 21197-21206). После острого инфаркта миокарда (AMI) у человека уровни LPA в сыворотке значительно увеличиваются до почти 6-кратного повышения концентраций, и, считалось, что LPA задействована в патофизиологических процессах в сердечно-сосудистой системе, связанных с AMI (X. Chen et al., Scand. J. Clin. Lab. Invest. 63 (2003), 497-503). Дальнейшие исследования подтвердили значимость LPA и ее рецептора Edg-2 для патофизиологических процессов после инфаркта миокарда, например ремоделирования сердца, и для профилактики сердечной гипертрофии и сердечной недостаточности (J. Chen et al., J. Cell. Biochem. 103 (2008), 1718-1731). Было показано, что LPA получается в ходе мягкого окисления частиц липопротеина низкой плотности (LDL) и накапливается в липидном ядре атеросклеротических бляшек человека (W. Siess et al., Proc. Natl. Acad. Sci. 96 (1999), 6931-6936). Более того, LPA была отнесена к важным биологически активным компонентом moxLDL (слабоокисленному липопротеину низкой плотности), вызывающим активацию тромбоцитов, и было показано, что воздействие LPA, moxLDL или экстрактов липидного ядра из атеросклеротических бляшек человека на активацию тромбоцитов можно блокировать ингибитором рецепторов Edg-2/Edg-7 диоктаноилглицеролпирофосфатом DGPP(8:0), указывая на причинную роль опосредованного LPA сигнала рецепторов Edg в агрегировании тромбоцитов и применимости таких LPA ингибиторов рецепторов для лечения сердечно-сосудистых заболеваний (E. Rother et al., Circulation 108 (2003), 741-747).
Дополнительные результаты подчеркивают разрушительную роль LPA в процессе инициирования и развития сердечно-сосудистых заболеваний, например атеросклероза, ремоделирования левого желудочка и сердечной недостаточности. LPA вызывает чувствительные к коклюшному токсину, опосредованные NFκB (ядерный фактор каппа B) провоспалительные отклики эндотелиальных клеток, в том числе повышение уровня хемокинов, например моноцитарного хемоаттрактантного белка-1 (MCP-1) и интерлейкина-8 (IL8) (A. Palmetshofer et al., Thromb. Haemost. 82 (1999), 1532-1537), а также воздействие молекул адгезии эндотелиальных клеток, например E-селектина или молекулы-1 межклеточной адгезии (ICAM-1) (H. Lee et al., Am. J. Physiol. 287 (2004), C1657-C1666). Прямые свидетельства участия рецепторов Edg-2 получены в ходе недавних исследований, которые показывают, что LPA индуцирует окислительный стресс в клетках гладкой мускулатуры сосудов и эндотелиальных клетках, который сглаживался фармакологическим ингибированием DGPP(8:0) или THG1603, специфическим антагонистом рецептора Edg-2 (U. Kaneyuki et al., Vascular Pharmacology 46 (2007), 286-292; S. Brault et al., Am. J. Physiol. Regul. Integr. Comp. Physiol. 292 (2007), R1174-R1183). В клетках гладкой мускулатуры сосудов LPA приводит к чувствительному к коклюшному токсину выбросу Ca2+ из внутреннего депо, к активации митоген-активируемой протеинкиназы с молекулярным весом 42 кДа (p42MAPK) и к пролиферации клеток (S. Seewald et al., Atherosclerosis 130 (1997), 121-131). Показано, что внутрисосудистая инъекция LPA индуцирует образование неоинтимы in vivo (K. Yoshida et al., Circulation 108 (2003), 1746-1752). На отдельных взрослых кардиомиоцитах LPA приводит к клеточной гипертрофии и к активации различных киназ, которые, как известно, играют важную роль в гипертрофическом отклике (Y.-J. Xu et al., Biochemical Pharmacology 59 (2000), 1163-1171). Исследования на неонатальных миоцитах подтвердили роль LPA в индуцировании гипертрофии и вскрыли значимость зависимого от rho-киназы канала (R. Hilal-Dandan et al., J. Mol. Cell. Cardiol. 36 (2004), 481-493). Значимость rho-киназы подчеркивает участие рецепторов Edg-2, которые, в отличие от рецепторов Edg-7, связаны с белками Gα12/13. Более того, LPA сглаживает силу сокращения препаратов миокарда желудочков и предсердий человека и ухудшает изопреналин-индуцированное фракционное сокращение изолированных желудочковых миоцитов взрослых крыс. Указанные эффекты исчезали после предварительного инкубирования с коклюшным токсином, указывая на значимость GPCR-опосредованных и Gαi/0-опосредованных каналов (B. Cremers et al., J. Mol. Cell. Cardiol. 35 (2003), 71-80). Было также обнаружено, что LPA приводит более активной генерации матрицы и пролиферации сердечных фибробластов (J. Chen et al., FEBS Letters 580 (2006), 4737-4745).
Важность воздействия на сигналы Edg-2 рецептора и опосредованные LPA эффекты при множестве заболеваний получила подтверждение в рамках фармакологических подходов с использованием специализированных соединений или мышей с нокаутом рецептора Edg-2 или посредством экспериментального подавления рецепторов Edg-2. Например, роль LPA-активированных рецепторов Edg при почечной недостаточности была продемонстрирована для различных видов ингибиторов рецепторов Edg-2/Edg-7. В одном из подходов было показано, что индуцированный LPA пролиферативный отклик мезангиальных клеток можно ингибировать соединением DGPP(8:0) (Y. Xing et al., Am. J. Physiol. Cell Physiol. 287 (2004), F1250-F1257). В другом подходе с использованием ингибитора рецепторов Edg-2/Edg-7 VPC12249 на модели реперфузии почечной ишемии мышей in vivo было продемонстрирована двойная роль LPA как ренопротектора. Несмотря на продемонстрированную пользу сигнала рецепторов Edg-4, сигналы рецепторов Edg-2 и Edg-7 усугубляли почечное поражение, скорее всего, из-за более глубокой инфильтрации лейкоцитов в ткань почек, а потому должны блокироваться для лечения или профилактики острой почечной недостаточности, вызванной ишемией/реперфузией (M. D. Okusa et al., Am. J. Physiol. Renal Physiol. 285 (2003), F565-F574). Критическая роль рецепторов Edg-2 в развитии тубулоинтерстициального фиброза была подтверждена на модели односторонней закупорки мочеточника (J. P. Pradere et al., J. Am. Soc. Nephrol. 18 (2007), 3110-3118). В данной модели почечная недостаточность купировалась у мышей с нокаутом рецептора Edg-2 или за счет фармакологического лечения ингибитором рецептора Edg-2/Edg-7 Ki16425. Воздействие рецепторной системы LPA/Edg-2 при легочном фиброзе и прохождении жидкости через сосуды недавно было подтверждено выводами о том, что биологически активное содержание LPA увеличивалось в бронхоальвеолярной жидкости людей, страдающих идиопатическим легочным фиброзом. Мыши с нокаутом Edg-2 рецептора были защищены от индуцированного блеомицином поражения легких и прохождения жидкости через сосуды по сравнению с нативными мышами из одного помета (A. M. Tager et al., Nat. Med. 14 (2008), 45-54).
Прямое участие рецепторов Edg-2 недавно было доказано в случая прогрессии костных метастазов in vivo. Прогрессия снижалась при фармакологическом лечении ингибитором рецептора Edg-2/Edg-7 Ki16425, а также на тот же порядок величины после специфического подавления рецепторов Edg-2 (A. Boucharaba et al., Proc. Natl. Acad. Sci. 103 (2006), 9643-9648). Значимость рецепторов Edg-2 также была показана in vitro в отношении пролиферации клеток рака простаты и метастатического потенциала клеток карциномы толстой кишки человека (R. Guo et al., Endocrinology 147 (2006), 4883-4892; D. Shida et al., Cancer Res. 63 (2003), 1706-1711).
Роль опосредованного LPA сигнала рецепторов Edg-2 была также продемонстрирована в модели нейропатической боли in vivo. Интратекальная инъекция LPA моделировала поведенческие, морфологические и биохимические изменения, подобные тем, которые наблюдались после поражения периферического нерва. Не предусматривающая дублирования функция рецепторов Edg-2 была продемонстрирована для мышей без рецептора Edg-2, у которых не проявлялись признаки нейропатической боли после поражения нерва. Поэтому сигнал рецептора Edg-2 рассматривается как важный элемент возникновения нейропатической боли (M. Inoue et al., Nat. Med. 10 (2004), 712-718). Поэтому очевидно, что ингибирование рецептора Edg-2 и воздействия LPA за счет использования подходящих ингибиторов является привлекательным подходом для лечения различных заболеваний.
Уже были описаны некоторые соединения, которые проявляют ингибирующее действие Edg-2. Например, в качестве соединений, структурно связанных с LPA, можно назвать приведенные выше соединения DGPP(8:0) или VPC12249. В WO 02/29001 и WO 2005/115150 описаны аминосоединения, содержащие фосфатную группу, фосфонатную группу или гидроксильную группу, которые проявляются активность в качестве агонистов или антагонистов рецепторов LPA. Азольные соединения антагонисты рецепторов LPA, для которых характерна карбаматная группа в 4-положении азольного кольца, описаны в EP 1258484. Использование азольных соединений, других гетероциклов и других соединений для модулирования рецепторов Edg-2, Edg-3, Edg-4 и Edg-7 описано в WO 03/062392. Соединения, проявляющие активность антагониста рецептора LPA, особенно Edg-2, в состав которых входят β-аланиновые группы, содержащие бифенил-2-карбонильную группу при аминогруппе или же спиртовую группу и по меньшей мере три циклических группы, описаны в EP 1533294 и EP 1695955, соответственно. Вместе с тем по-прежнему существует потребность в других ингибиторах Edg-2, которые проявляют благоприятный набор характеристик и могут использоваться при лечении заболеваний, например подобных указанным выше, а также других заболеваний, при которых определенную роль играет сигнал LPA и рецепторы Edg-2. Настоящее изобретение восполняет такую потребность за счет ациламинозамещенных конденсированных производных циклопентанкарбоновой кислоты приведенной ниже формулы I.
Некоторые ациламинозамещенные конденсированные производные циклопентанкарбоновой кислоты, которые структурно отличаются от соединений настоящего изобретения, уже были описаны ранее, например соединение 2-бензоиламиноиндан-2-карбоновой кислоты в работе R. Lohmar et al., Chem. Ber. 113 (1980), 3706-3715. 2-Ациламиноиндан-2-карбоновые кислоты, которые характеризуются арильным или гетероарильным заместителем при бензольном кольце индана и которые контролируют функционирование рецептора GPR34 и тем самым ингибируют выброс гистамина, описаны в WO 2006/088246 (EP 1849465), в их числе и соединения формулы I, в которых конденсированное циклопентановое кольцо, приведенное в формуле I наряду с кольцом А, представляет собой индановое кольцо, содержащее 4-хлорфенильный заместитель в 5-положении, группы R3 по R6 и R20 являются атомами водорода, группа R50 является гидроксилом или этоксигруппой, а циклический остаток, содержащий группы Y, Z, R21 и R22, представляет собой 4-(2-метил-1H-бензоимидазол-1-илметил)-фенил, каковой остаток также может называться 4-(2-метилбензоимидазол-1-илметил)-фенилом. Соединения формулы I, в которых конденсированное циклопентановое кольцо, описанное в формуле I наряду с кольцом А, представляет собой незамещенное индановое кольцо, группы R3 по R6 и R20 являются атомами водорода, группой R50 является гидроксил, а циклический остаток, содержащий группы Y, Z, R21 и R22, представляет собой 6,2',4'-трихлорбифенил-3-ил, 6-хлор-[1,1',4',1'']терфенил-3-ил или 4-хлор-3-(2-фенилэтинил)-фенил, были описаны в WO 2006/044975, которое относится к противоопухолевым средствам.
Предметом настоящего изобретения является соединение формулы I в любой из его стереоизомерных форм или сочетании стереоизомерных форм в любом соотношении или его физиологически приемлемая соль или физиологически приемлемый сольват любого из этих соединений,

где
кольцо A является циклоалкановым кольцом с числом членов от 3-х до 7-и, бензольным кольцом или моноциклическим 5-членным или 6-членным ароматическим гетероциклическим кольцом, содержащим 1 или 2 одинаковых или разных гетерочлена, выбранных из группы, содержащей N, N(R0), O и S, причем циклоалкановое кольцо может необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей фтор и (C1-C4)-алкил, а бензольное и гетероциклическое кольца могут необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген, R1, HO-, R1-O-, R1-C(O)-O-, R1-S(O)2-O-, R1-S(O)m-, H2N-, R1-NH-, R1-N(R1)-, R1-C(O)-NH-, R1-C(O)-N(R71)-, R1-S(O)2-NH-, R1-S(O)2-N(R71)-, R1-C(O)-, HO-C(O)-, R1-O-C(O)-, H2N-C(O)-, R1-NH-C(O)-, R1-N(R1)-C(O)-, H2N-S(O)2-, R1-NH-S(O)2-, R1-N(R1)-S(O)2-, NC-, O2N-, фенил и Het;
Y выбирают из группы, содержащей N(R10), S, O, C(R12)=C(R13), N=C(R14) и C(R15)=N;
Z выбирают из группы, содержащей N и C(R16);
R0 выбирают из группы, содержащей водород и R2;
R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, независимо от каждой другой группы R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, выбирают из группы, содержащей (C1-C6)-алкил, (C2-C6)-алкенил, (C2-C6)-алкинил, (C3-C7)-циклоалкил и (C3-C7)-циклоалкил-(C1-C4)-алкил-, причем все они могут необязательно иметь один или более одинаковых или разных заместителей R70;
R3 и R5 выбирают, независимо друг от друга, из группы, содержащей водород, (C1-C4)-алкил, фенил-(C1-C4)-алкил-, фенил и гидрокси;
R4 и R6 выбирают, независимо друг от друга, из группы, содержащей водород и (C1-C4)-алкил;
R10 выбирают из группы, содержащей водород и R11;
R12, R13, R14, R15 и R16 выбирают, независимо друг от друга, из группы, содержащей водород, галоген, (C1-C4)-алкил, HO-(C1-C4)-алкил-, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, H2N-, (C1-C4)-алкил-NH-, (C1-C4)-алкил-N((C1-C4)-алкил)-, (C1-C4)-алкил-C(O)-, NC- и O2N-;
R20 выбирают из группы, содержащей водород и (C1-C4)-алкил;
Одной из групп R21 и R22 является группа формулы II

а другую из групп R21 и R22 выбирают из группы, содержащей водород, галоген, R30, HO-, R30-O-, R30-C(O)-O-, R30-S(O)2-O-, R30-S(O)m-, H2N-, R30-NH-, R30-N(R30)-, R30-C(O)-NH-, R30-C(O)-N(R71)-, R30-S(O)2-NH-, R30-S(O)2-N(R71)-, R30-C(O)-, HO-C(O)-, R30-O-C(O)-, H2N-C(O)-, R30-NH-C(O)-, R30-N(R30)-C(O)-, H2N-S(O)2-, R30-NH-S(O)2-, R30-N(R30)-S(O)2-, NC-, O2N- и Het1;
R23является прямой связью или цепью, содержащей от 1 до 5 членов цепи, из которых 0, 1 и или 2 члена цепи являются одинаковыми или разными гетерочленами цепи, выбранными из группы, содержащей N(R25), O, S, S(O) и S(O)2, но два гетерочлена цепи могут находиться в соседних позициях, только при условии, что один из них выбирают из группы, содержащей S(O) и S(O)2, а другой выбирают из группы, содержащей N(R25), O и S, тогда как другие члены цепи являются одинаковыми или разными группами C(R26)(R26), где две соседние группы C(R26)(R26) могут соединяться друг с другом двойной или тройной связью;
R24 выбирают из группы, содержащей водород, R31, HO-, R31-O-, R31-C(O)-O-, R31-S(O)m-, H2N-, R31-NH-, R31-N(R31)-, R31-C(O)-NH-, R31-C(O)-N(R71)-, R31-S(O)2-NH-, R31-S(O)2-N(R71)-, R31-C(O)-, HO-C(O)-, R31-O-C(O)-, H2N-C(O)-, R31-NH-C(O)-, R31-N(R31)-C(O)-, H2N-S(O)2-, R31-NH-S(O)2-, R31-N(R31)-S(O)2-, NC- и моноциклическое, бициклическое или трициклическое кольцо с числом членов от 3-х до 10-и, которое является насыщенным или ненасыщенным и содержит 0, 1, 2 или 3 одинаковых или разных гетерочлена кольца, выбранных из группы, содержащей N, N(R32), O, S, S(O) и S(O)2, причем кольцо может необязательно иметь на атомах углерода кольца один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген, R33, HO-, R33-O-, R33-C(O)-O-, R33-S(O)2-O-, R33-S(O)m-, H2N-, R33-NH-, R33-N(R33)-, R33-C(O)-NH-, R33-C(O)-N(R71)-, R33-S(O)2-NH-, R33-S(O)2-N(R71)-, H2N-S(O)2-NH-, R33-NH-S(O)2-NH-, R33-N(R33)-S(O)2-NH-, H2N-S(O)2-N(R71)-, R33-NH-S(O)2-N(R71)-, R33-N(R33)-S(O)2-N(R71)-, R33-C(O)-, HO-C(O)-, R33-O-C(O)-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)-, H2N-S(O)2-, R33-NH-S(O)2-, R33-N(R33)-S(O)2-, NC-, O2N-, оксо, фенил и Het;
при условии, что общее число атомов C, N, O и S, присутствующих в двух группах R23 и R24, составляет не менее 5;
R25 выбирают из группы, содержащей водород и (C1-C4)-алкил;
R26, независимо от каждой другой группы R26, выбирают из группы, содержащей водород, фтор, (C1-C4)-алкил и HO-, или две группы R26, связанные с одним атомом углерода, представляют собой оксо, или две группы R26 или одна группа R25 и одна группа R26, вместе с входящими в них членами цепи, образуют моноциклическое кольцо с числом членов от 3-х до 7-и, которое является насыщенным и содержит 0, 1 или 2 одинаковых или разных гетерочлена кольца, выбранных из группы, содержащей N, N(R34), O, S, S(O) и S(O)2, причем кольцо может необязательно иметь на атомах углерода кольца один или более одинаковых или разных заместителей, выбранных из группы, содержащей фтор и (C1-C4)-алкил;
R31 выбирают из группы, содержащей (C1-C6)-алкил, (C2-C6)-алкенил и (C2-C6)-алкинил, причем все они могут необязательно иметь один или более одинаковых или разных заместителей R70;
R32 и R34 выбирают, независимо друг от друга, из группы, содержащей водород, R35, R35-S(O)2-, R35-C(O)-, R35-O-C(O)-, фенил и Het;
R50 выбирают из группы, содержащей R51-O- и R52-N(R53)-;
R51 выбирают из группы, содержащей водород и R54;
R52 выбирают из группы, содержащей водород, R55, NC- и R56-S(O)2-;
R53 выбирают из группы, содержащей водород и R57;
R56 выбирают из группы, содержащей R58 и фенил;
R60, независимо от каждой другой группы R60, выбирают из группы, содержащей водород и (C1-C4)-алкил;
R70 выбирают из группы, содержащей HO-, R71-O-, R71-C(O)-O-, R71-S(O)m-, H2N-, R71-NH-, R71-N(R71)-, R71-C(O)-NH-, R71-C(O)-N(R71)-, R71-S(O)2-NH-, R71-S(O)2-N(R71)-, HO-C(O)-, R71-O-C(O)-, H2N-C(O)-, R71-NH-C(O)-, R71-N(R17)-C(O)-, H2N-S(O)2-, R71-NH-S(O)2-, R71-N(R71)-S(O)2-, NC-, оксо, фенил и Het2;
R71, независимо от каждой другой группы R71, выбирают из группы, содержащей (C1-C4)-алкил, (C3-C4)-циклоалкил и (C3-C4)-циклоалкил-(C1-C2)-алкил-;
Het, независимо от каждой другой группы Het, является моноциклическим гетероциклическим кольцом с числом членов от 4-х до 7-и, которое содержит 1, 2 или 3 одинаковых или разных гетерочлена кольца, выбранных из группы, содержащей N, N(R60), O, S, S(O) и S(O)2, причем кольцо является насыщенным или ненасыщенным и может необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген, (C1-C4)-алкил и R70;
Het1 является моноциклическим гетероциклическим кольцом с числом членов от 4-х до 7-и, которое содержит 1 или 2 одинаковых или разных гетерочлена, выбранных из группы, содержащей N, N(R60), O, S, S(O) и S(O)2, причем кольцо является насыщенным и может необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей фтор и (C1-C4)-алкил;
Het2 является моноциклическим гетероциклическим кольцом с числом членов 5 или 6, которое содержит 1, 2 или 3 одинаковых или разных гетерочлена, выбранных из группы, содержащей N, N(R60), O и S, причем кольцо является ароматическим и может необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген, (C1-C4)-алкил, (C1-C4)-алкил-O- и NC-;
m, независимо от каждого другого числа m, является целым числом, выбранным из группы, содержащей 0, 1 и 2;
фенил, независимо от каждой другой группы фенила, может необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген, (C1-C4)-алкил, (C1-C4)-алкил-O- и NC-, если не указано иное;
циклоалкил, независимо от каждой другой группы циклоалкила и независимо от других заместителей циклоалкила, может необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей фтор и (C1-C4)-алкил;
алкил, алкенил, алкинил, независимо от каждой другой группы алкила, алкенила, алкинила и независимо от других алкильных заместителей, алкенила, алкинила, может необязательно иметь один или более фтор-заместителей;
при условии что соединение формулы I не является 2-[(6,2',4'-трихлорбифенил-3-карбонил)амино]индан-2-карбоновой кислотой, 2-[6-хлор-[1,1',4',1'']терфенил-3-карбонил)амино]индан-2-карбоновой кислотой, 2-(4-хлор-3-фенилэтинилбензоиламино)-индан-2-карбоновой кислотой, 5-(4-хлорфенил)-2-[4-(2-метил-1H-бензоимидазол-1-илметил)-бензоиламино]-индан-2-карбоновой кислотой или этиловым эфиром 5-(4-хлорфенил)-2-[4-(2-метил-1H-бензоимидазол-1-илметил)-бензоиламино]-индан-2-карбоновой кислоты.
Если структурные элементы, например группы, заместители или числа, могут встречаться несколько раз в соединениях формулы I, все они не зависят друг от друга и могут в каждом случае иметь любое указанное значение, и они в каждом случае могут быть одинаковыми или отличаться от любого такого другого элемента. В диалкиламиногруппе, например, алкильные группы могут быть одинаковыми или разными.
Алкильные группы, т.е. остатки насыщенной углеводородной группы, могут иметь вид линейной (прямой) или разветвленной цепи. Сказанное также верно в случае, когда эти группы замещаются или входят в состав другой группы, например алкил-O-группы (группа алкилокси, группа алкокси) или HO-замещенной алкильной группы (гидроксиалкильная группа). В зависимости от соответствующего определения, число атомов углерода в алкильной группе может быть 1, 2, 3, 4, 5 или 6, либо 1, 2, 3 или 4, либо 1, 2 или 3, либо 1 или 2, либо 1. Примерами алкила являются метил, этил, пропил, в том числе н-пропил и изопропил, бутил, в том числе н-бутил, втор-бутил, изобутил и трет-бутил, пентил, в том числе н-пентил, 1-метилбутил, изопентил, неопентил и трет-пентил, и гексил, в том числе н-гексил, 3,3-диметилбутил и изогексил. Примерами алкил-O-групп являются метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси, н-пентокси. Примерами алкил-S(O)m- являются метилсульфанил-(CH3-S-), метансульфенил-(CH3-S(O)-), метансульфонил-(CH3-S(O)2-), этилсульфанил-(CH3-CH2-S-), этансульфинил-(CH3-CH2-S(O)-), этансульфонил-(CH3-CH2-S(O)2-), 1-метилэтилсульфанил-((CH3)2CH-S-), 1-метилэтансульфинил-((CH3)2CH-S(O)-), 1-метилэтансульфонил-((CH3)2CH-S(O)2-). В одном осуществлении настоящего изобретения в качестве числа m выбирает 0 или 2, причем все числа m независимы друг от друга и могут быть одинаковыми или разными. В другом осуществлении число m во всех случаях, независимо от его значения в других случаях, равно 0. В другом осуществлении число m во всех случаях, независимо от его значения в других случаях, равно 2.
Замещенная алкильная группа может быть замещена в любой позиции, при условии, что соответствующее соединение в достаточной степени устойчиво и пригодно в качестве фармацевтически активного соединения. Условием, согласно которому конкретная группа и соединение формулы I должны быть в достаточной степени устойчивы и пригодны в качестве фармацевтически активного соединения, в целом распространяется применительно к определению всех групп соединений формулы I. Алкильная группа, которая может необязательно иметь один или более фтор-заместителей, может быть незамещена, т.е. не нести фтор-заместителей, или быть замещена, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или 11-ю фтор-заместителями или 1, 2, 3, 4 или 5-ю фтор-заместителями, которые могут находиться в любых позициях. Например, во фтор-замещенной алкильной группе одна или несколько метиловых групп могут содержать три фтор-заместителя на каждую группу и присутствовать в виде трифторметиловых групп, и (или) одна или несколько метиленовых групп (CH2) могут содержать два фтор-заместителя на каждую группу и присутствовать в виде дифторметиленовых групп. Объяснения в отношении замещения группы фтором также применимы в случае, когда группа дополнительно содержит другие заместители и(или) входит в состав другой группы, как, например, алкил-O-группы. Примерами фтор-замещенной алкильной группы являются трифторметил, 2-фторэтил, 1-фторэтил, 1,1-дифторэтил, 2,2,2-трифторэтил, пентафторэтил, 3,3,3-трифторпропил, 2,2,3,3,3-пентафторпропил, 4,4,4-трифторбутил и гептафторизопропил. Примерами фтор-замещенных алкил-O-групп являются трифторметокси, 2,2,2-трифторэтокси, пентафторэтокси и 3,3,3-трифторпропокси. Примерами фтор-замещенных алкил-S(O)m-групп являются трифторметилсульфанил-(CF3-S-), трифторметансульфинил-(CF3-S(O)-) и трифторметансульфонил-(CF3-S(O)2-).
Вышеприведенные объяснения в отношении алкильных групп применимы соответствующим образом к остаткам ненасыщенной углеводородной группы, т.е. к алкенильным группам, которые в одном из осуществлений настоящего изобретения содержат одну двойную связь, и алкинильным группам, которые в одном из осуществлений настоящего изобретения содержат одну тройную связь. Так, например, алкенильные группы и алкинильные группы также могут быть линейными или разветвленными, а замещенные алкенильные и алкинильные группы могут быть замещены в любой позиции, при условии, что полученное соединение в достаточной степени устойчиво и пригодно в качестве фармацевтически активного соединения. Двойные связи и тройные связи могут присутствовать в любой позиции. Число атомов углерода в алкенильной или алкинильной группе может быть 2, 3, 4, 5 или 6, например, 2, 3, 4 или 5. Примерами алкенила и алкинила являются этенил (винил), проп-1-энил, проп-2-энил (аллил), бут-2-энил, 2-метилпроп-2-энил, 3-метилбут-2-энил, гекс-3-энил, гекс-4-энил, 4-метилгекс-4-энил, проп-1-инил, проп-2-инил (пропаргил), бут-2-инил, бут-3-инил, 4-метилпент-2-инил, гекс-4-инил и гекс-5-инил. В одном осуществлении настоящего изобретения алкенильная или алкинильная группа содержит не менее трех атомов углерода и связана с остальной молекулой при помощи атома углерода, который не входит в двойную связь или тройную связь.
Вышеприведенные объяснения в отношении алкильных групп применимы соответствующим образом к алкандиильным группам (двухвалентным алкильным группам), включая цепи одной или более групп C(R26)(R26), которые как таковые, а также цепи этих групп являются алкандиильными группами, в случае если для R26 выбирают водород и (C1-C4)-алкил, или замещенные алкандиильные группы, в случае если R26 любой группы имеет значение, отличающееся от водорода и (C1-C4)-алкила. Аналогичным образом алкильная часть замещенной алкильной группы также может рассматриваться как алкандиильная группа. Таким образом, алкандиильные группы могут быть линейными или разветвленными, связи с соседними группами могут находиться в любой позиции и могут начинаться с одного или с разных атомов углерода, а также они могут быть замещены фтором. Примерами алкандиильных групп являются -CH2-, -CH2-CH2-, -CH2-CH2-CH2-, -CH2-CH2-CH2-CH2-, -CH2-CH2-CH2-CH2-CH2-, -CH(CH3)-, -C(CH3)2-, -CH(CH3)-CH2-, -CH2-CH(CH3)-, -C(CH3)2-CH2-, -CH2-C(CH3)2-. Примерами фтор-замещенных алкандиильных групп, которые могут содержать 1, 2, 3, 4, 5 или 6 фтор-заместителей являются, например, -CHF-, -CF2-, -CF2-CH2-, -CH2-CF2-, -CF2-CF2-, -CF(CH3)-, -C(CF3)2-, -C(CH3)2-CF2-, -CF2-C(CH3)2-. Кроме того, вышеприведенные объяснения применимы соответствующим образом к двухвалентным остаткам ненасыщенной углеводородной группы, т.е. ненасыщенным алкандиильным группам, таким как алкенедиильные группы и алкинедиильные группы, которые могут встречаться в группе R23, в случае если две смежные группы C(R26)(R26) соединены между собой двойной связью или тройной связью, и которые в одном из осуществлений настоящего изобретения содержат одну двойную связь или одну тройную связь, соответственно, которая может присутствовать в любой позиции, и такие группы могут быть необязательно замещены фтор-заместителями. Примерами таких ненасыщенных двухвалентных групп являются -CH=CH-, -CH2-CH=CH-, -CH=CH-CH2-, -CH2-CH=CH-CH2-, -C≡C-, -CH2-C≡C-, -C≡C-CH2-, -C(CH3)2-C≡C-, -C≡C-C(CH3)2-, -CH2-C≡C-CH2-.
Число атомов углерода в кольце в (C3-C7)-циклоалкильной группе может быть 3, 4, 5, 6 или 7. Примерами циклоалкила являются циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Циклоалкильные группы, которые могут необязательно иметь один или более (C1-C4)-алкил-заместителей, могут быть незамещенными, т.е. не нести алкил-заместителей, или быть замещенными, например, 1, 2, 3 или 4-мя одинаковыми или разными (C1-C4)-алкил-заместителями, например метиловыми группами, которые могут находиться в любых позициях. Примерами таких алкил-замещенных циклоалкильных групп являются 1-метилциклопропил, 2,2-диметилциклопропил, 1-метилциклопентил, 2,3-диметилциклопентил, 1-метилциклогексил, 4-метилциклогексил, 4-изопропилциклогексил, 4-трет-бутилциклогексил и 3,3,5,5-тетраметилциклогексил. Циклоалкильные группы, которые могут необязательно иметь один или более фторосодержащих заместителей, могут быть незамещены, т.е. не нести фторосодержащих заместителей, или быть замещены, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или 11-ю фтор-заместителями или 1, 2, 3, 4, 5, или 6-ю фтор-заместителями. Фтор-заместители могут находиться в любых позициях в циклоалкильной группе, а также могут находиться в алкильном заместителе циклоалкильной группы. Примерами таких фтор-замещенных циклоалкильных групп являются 1-фторциклопропил, 2,2-дифторциклопропил, 3,3-дифторциклобутил, 1-фторциклогексил, 4,4-дифторциклогексил и 3,3,4,4,5,5-гексафторциклогексил. Циклоалкильные группы также могут одновременно быть замещенны фтором и алкилом. Примерами группы (C3-C7)-циклоалкил-(C1-C4)-алкил- являются циклопропилметил-, циклобутилметил-, циклопентилметил-, циклогексилметил-, циклогептилметил-, 1-циклопропилэтил-, 2-циклопропилэтил-, 1-циклобутилэтил-, 2-циклобутилэтил-, 1-циклопентилэтил-, 2-циклопентилэтил-, 1-циклогексилэтил-, 2-циклогексилэтил-, 1-циклогептилэтил-, 2-циклогептилэтил-. Объяснения в отношении циклоалкильных групп применимы соответствующим образом к ненасыщенным циклоалкильным группам, таким как циклоалкенильные группы, которые могут встречаться в группе R24 и которые в одном осуществлении настоящего изобретения содержат одну двойную связь, которая может присутствовать в любой позиции, и двухвалентные циклоалкильные группы (циклоалкандиильные группы) которые могут встречаться в случае, когда две группы R26вместе с входящими в их состав членами цепи образуют кольцо. Аналогичным образом циклоалкильная часть замещенной циклоалкильной группы также может рассматриваться как циклоалкандиильная группа. Так, например, связи, посредством которых циклоалкандиильная группа, такая, как кольцо, образованное двумя группами R26вместе с входящими в их состав членами цепи, соединяется с соседними группами, могут находиться в любых позициях и могут начинаться с одного или с разных атомов углерода в кольце.
В замещенных фенильных группах, включая фенильные группы, которые представляют моноциклическое, бициклическое или трициклическое кольцо с числом членов от 3-х до 10-и, представляющее R24, заместители могут находиться в любых позициях. В монозамещенных фенильных группах заместитель может находиться в позиции 2, позиции 3 или позиции 4. В дизамещенных фенильных группах заместители могут находиться в позиции 2,3, позиции 2,4, позиции 2,5, позиции 2,6, позиции 3,4 или позиции 3,5. В тризамещенных фенильных группах заместители могут находиться в позиции 2,3,4, позиции 2,3,5, позиции 2,3,6, позиции 2,4,5, позиции 2,4,6 или позиции 3,4,5. Если фенильная группа имеет четыре заместителя, некоторыми из которых могут быть атомы фтора, заместители, например, могут находиться в позиции 2,3,4,5, позиции 2,3,4,6 или позиции 2,3,5,6. Если полизамещенная фенильная группа или любая другая полизамещенная группа, такая как гетероарильная группа, имеет разные заместители, каждый заместитель может находиться в любой подходящей позиции, и настоящее изобретение содержит все позиционные изомеры. Число заместителей в замещенной фенильной группе может быть 1, 2, 3, 4 или 5. В одном осуществлении настоящего изобретения замещенная фенильная группа, а также еще одна замещенная группа, такая как гетероарильная группа, имеет 1, 2 или 3, например 1 или 2, одинаковых или разных заместителей.
В гетероциклических группах, в том числе в группах Het, Het1 и Het2 и гетероциклических кольцах, которые могут присутствовать в структурных элементах соединений формулы I, таких как кольцо А или кольцо с числом членов от 3-х до 10-и, представляющее R24, или кольцо, образуемое группой R25 и группой R26, вместе с входящими в них членами цепи, например гетерочленами кольца, указанными в соответствующем определении, могут присутствовать в любом сочетании и находиться в любых подходящих позициях кольца, при условии, что полученные группа и соединение формулы I являются в достаточной степени устойчивыми и пригодными в качестве фармацевтически активного соединения. В одном осуществлении настоящего изобретения два атома кислорода в любом гетероциклическом кольце в соединениях формулы I не могут находиться в соседних позициях в кольце. В другом осуществлении два гетерочлена кольца из группы, содержащей атомы O, S и N, имеющие атом водорода или заместитель, не могут находиться в соседних позициях в кольце. Примерами таких групп являются гетерочлены кольца O, S и N(R32) или O, S и N(R34) или O, S и N(R60). В другом осуществлении настоящего изобретения два гетерочлена кольца из группы, содержащей S(O) и S(O)2, не могут находиться в соседних позициях в кольце. В ароматическом гетероциклическом кольце выбор членов гетероциклического кольца и их позиций ограничивается требованием, чтобы кольцо было ароматическим, т.е. содержало циклическую систему из шести делокализованных пи-электронов. Остаток моноциклического 5-членного или 6-членного ароматического гетероциклического кольца, который может присутствовать в группах Het, Het2, и кольца с числом членов от 3-х до 10-и, представляющего, например, R24, также может быть обозначен как моноциклическая 5-членная или 6-членная гетероарильная группа. Атом азота в кольце в такой гетероарильной группе, содержащей группу R32 или R60 соответственно, представляет собой атом азота в 5-членном кольце, например пиррол, пиразол, имидазол или триазол, к которому присоединен экзоциклический атом или группа, например атом водорода, и может присутствовать только один раз в 5-членном ароматическом кольце, аналогично членам гетероциклического кольца O и S. Примерами колец, из которых может быть получена такая гетероарильная группа, являются пиррол, фуран, тиофен, имидазол, пиразол, триазол, включая [1,2,3]триазол и [1,2,4]триазол, оксазол ([1,3]оксазол), изоксазол ([1,2]оксазол), тиазол ([1,3]тиазол), изотиазол ([1,2]тиазол), оксадиазолы, включая [1,2,4]оксадиазол, [1,3,4]оксадиазол и [1,2,5]оксадиазол, тиадиазолы, включая [1,3,4]тиадиазол, пиридин, пиридазин, пиримидин, пиразин, триазины, включая [1,2,3]триазин, [1,2,4]триазин и [1,3,5]триазин. Приведенные объяснения в отношении моноциклических 5-членных или 6-членных гетероарильных групп применимы соответствующим образом к моноциклическому 5-членному или 6-членному ароматическому гетероциклическому кольцу, представляющему кольцо А в формуле I, в котором атом азота в кольце, содержащий группу R0, также может присутствовать только один раз в 5-членном кольце, таком как пиррол, пиразол или имидазол. Точно так же, члены гетероциклического кольца O и S могут присутствовать только один раз в кольце А. В одном осуществлении настоящего изобретения моноциклическая 5-членная или 6-членная гетероарильная группа содержит 1 или 2 одинаковых или разных гетерочленов кольца, в другом осуществлении настоящего изобретения такая гетероарильная группа содержит один гетерочлен кольца, который определяются, как указано, и в еще одном осуществлении настоящего изобретения, такой гетероарил выбирают из группы, содержащей тиофенил, тиазолил и пиридинил. Моноциклическая 5-членная или 6-членная гетероарильная группа может присоединяться посредством любого атома углерода в кольце или, в случае 5-членного кольца, содержащего гетерочлен кольца N(R32) или N(R60), посредством атома азота в кольце, причем в последнем случае связь, посредством которой гетероарильная группа соединяется с остальной молекулой, заменяет группу R32 или R60. В одном осуществлении настоящего изобретения моноциклическая 5-членная или 6-членная гетероарильная группа присоединяется посредством атома азота в кольце. Например, тиофенильная группа (тиенильная группа) может представлять собой тиофен-2-ил (2-тиенил) или тиофен-3-ил (3-тиенил), фуранил может представлять собой фуран-2-ил или фуран-3-ил, пиридинил (пиридил) может представлять собой пиридин-2-ил, пиридин-3-ил или пиридин-4-ил, пиразолил может представлять собой 1H-пиразол-3-ил, 1H-пиразол-4-ил или 2H-пиразол-3-ил, имидазолил может представлять собой 1H-имидазол-1-ил, 1H-имидазол-2-ил, 1H-имидазол-4-ил или 3H-имидазолил-4-ил, тиазолил может представлять собой тиазол-2-ил, тиазол-4-ил или тиазол-5-ил.
В замещенных моноциклических 5-членных или 6-членных гетероарильных группах заместители могут находиться в любых позициях, например в тиофен-2-ильной группе или фуран-2-ильной группе в позиции 3 и(или) в позиции 4 и(или) в позиции 5, в тиофен-3-ильной группе или фуран-3-ильной группе в позиции 2 и(или) в позиции 4 и(или) в позиции 5, в пиридин-2-ильной группе в позиции 3 и(или) в позиции 4 и(или) в позиции 5 и(или) в позиции 6, в пиридин-3-ильной группе в позиции 2 и(или) в позиции 4 и(или) в позиции 5 и(или) в позиции 6, в пиридин-4-ильной группе в позиции 2 и(или) в позиции 3 и(или) в позиции 5 и(или) в позиции 6. В одном осуществлении настоящего изобретения замещенная моноциклическая 5-членная или 6-членная гетероарильная группа имеет 1, 2 или 3, например 1 или 2, одинаковых или разных заместителя. Как правило, наряду с возможностью содержания заместителей, указанных в определении группы, подходящие атомы азота в кольце в моноциклической 5-членной или 6-членной гетероарильной группе, а также в других гетероциклических группах, например в моноциклическом, бициклическом или трициклическом кольце с числом членов от 3-х до 10-и, представляющем R24, или в ароматическом кольце А, или ароматическом кольце, содержащем группы Y и Z, которые представлены в формуле I, например атом азота в пиридинильной группе или атом азота в [1,2,5]оксадиазолильной группе, могут также включать оксидо-заместитель -O‾ и присутствовать в виде N-оксида.
Вышеприведенные объяснения в отношении моноциклических 5-членных или 6-членных ароматических гетероциклических групп применимы соответствующим образом к бициклическим, ароматическим, гетероциклическим группам, рассматриваемым ниже, которые могут присутствовать в кольце с числом членов от 3 до 10, представляющем R24, и которые также могут быть обозначены как бициклическая гетероарильная группа.
Кроме моноциклических 5-членных или 6-членных ароматических гетероциклических групп группа Het влючает моноциклические частично ненасыщенные, т.е. неароматические гетероциклические группы с числом членов от 4 до 7 и насыщенные гетероциклические группы с числом членов от 4 до 7. Насыщенные гетероциклические группы с числом членов от 4 до 7 также входят в группу Het1. Таким образом, кольца групп Het и Het1 могут иметь 4 члена, 5 членов, 6 членов или 7 членов, например 5 членов или 6 членов. В одном осуществлении настоящего изобретения частично ненасыщенная группа Het имеет в кольце одну или две двойные связи, в другом осуществлении - одну двойную связь, которые могут присутствовать в любой позиции. В одном осуществлении настоящего изобретения 4-членная группа Het является ненасыщенной. В одном осуществлении настоящего изобретения группа Het представляет собой ненасыщенную группу с числом членов от 4 до 7 или 5-членную или 6-членную ароматическую группу, в другом осуществлении группа Het представляет собой насыщенную группу с числом членов от 4 до 7 и в другом осуществлении группа Het представляет собой 5-членную или 6-членную ароматическую группу. Группы Het и Het1 могут присоединяться посредством любого атома углерода в кольце или атома азота в кольце. Примерами групп Het и Het1 являются азетидинил, включая азетидин-1-ил, оксетанил, включая оксетан-3-ил, тетрагидрофуранил, включая тетрагидрофуран-2-ил и тетрагидрофуран-3-ил, тетрагидротиофенил, включая тетрагидротиофен-2-ил и тетрагидротиофен-3-ил, 1-оксо-тетрагидротиофенил, включая 1-оксо-тетрагидротиофен-2-ил и 1-оксо-тетрагидротиофен-3-ил, 1,1-диоксо-тетрагидротиофенил, включая 1,1-диоксо-тетрагидротиофен-2-ил и 1,1-диоксо-тетрагидротиофен-3-ил, пирролидинил, включая пирролидин-1-ил, пирролидин-2-ил и пирролидин-3-ил, тетрагидропиранил, включая тетрагидропиран-2-ил, тетрагидропиран-3-ил и тетрагидропиран-4-ил, тетрагидротиопиранил, включая тетрагидротиопиран-2-ил, тетрагидротиопиран-3-ил и тетрагидротиопиран-4-ил, пиперидинил, включая пиперидин-1-ил, пиперидин-2-ил, пиперидин-3-ил и пиперидин-4-ил, 1,2,3,4-тетрагидропиридинил, включая 1,2,3,4-тетрагидропиридин-1-ил, 1,2,3,6-тетрагидропиридинил, включая 1,2,3,6-тетрагидропиридин-1-ил, оксепанил, включая оксепан-2-ил, оксепан-3-ил и оксепан-4-ил, азепанил, включая азепан-1-ил, азепан-2-ил, азепан-3-ил и азепан-4-ил, 1,3-диоксоланил, включая 1,3-диоксолан-2-ил и 1,3-диоксолан-4-ил, имидазолидинил, включая имидазолидин-1-ил, имидазолидин-2-ил и имидазолидин-4-ил, [1,3]оксазолидинил, включая [1,3]оксазолидин-2-ил, [1,3]оксазолидин-3-ил, [1,3]оксазолидин-4-ил и [1,3]оксазолидин-5-ил, [1,3]тиазолидинил, включая [1,3]тиазолидин-2-ил, [1,3]тиазолидин-3-ил, [1,3]тиазолидин-4-ил и [1,3]тиазолидин-5-ил, [1,3]диоксанил, включая [1,3]диоксан-2-ил, [1,3]диоксан-4-ил и [1,3]диоксан-5-ил, [1,4]диоксанил, включая [1,4]диоксан-2-ил, пиперазинил, включая пиперазин-1-ил и пиперазин-2-ил, морфолинил, включая морфолин-2-ил, морфолин-3-ил и морфолин-4-ил, тиоморфолинил, включая тиоморфолин-2-ил, тиоморфолин-3-ил и тиоморфолин-4-ил, 1-оксо-тиоморфолинил, включая 1-оксо-тиоморфолин-2-ил, 1-оксо-тиоморфолин-3-ил и 1-оксо-тиоморфолин-4-ил, 1,1-диоксо-тиоморфолинил, включая 1,1-диоксо-тиоморфолин-2-ил, 1,1-диоксо-тиоморфолин-3-ил и 1,1-диоксо-тиоморфолин-4-ил, [1,3]диазепанил, [1,4]диазепанил, [1,4]оксазепанил или [1,4]тиазепанил. Кроме оксо-групп в членах кольца S(O) и S(O)2 и алкильных групп, представляющих R60, группы Het и Het1 могут необязательно иметь в качестве заместителей на атомах углерода в кольце один или более, например 1, 2, 3, 4 или 5, либо 1, 2, 3 или 4, либо 1, 2 или 3 одинаковых или разных заместителей, как указано, которые могут находиться в любых позициях.
Моноциклическое, бициклическое или трициклическое кольцо с числом членов от 3-х до 10-и, которое может быть насыщенным или ненасыщенным и содержит 0, 1, 2 или 3 одинаковых или разных гетерочленов, выбранных из группы, содержащей N, N(R32), O, S, S(O) и S(O)2, причем кольцо может представлять R24, может содержать 3, 4, 5, 6, 7, 8, 9 или 10 членов кольца. В одном из осуществлений настоящего изобретения бициклическое или трициклическое кольцо конденсировано или связано мостиковыми связями. Ненасыщенное кольцо может быть частично ненасыщенным и содержать, например, одну или две двойные связи в кольце, или в случае моноциклического или бициклического кольца, быть ароматическим в одном или обоих кольцах, а число двойных связей внутри ненасыщенного кольца может составлять одну, две, три, четыре или пять двойных связей. В бициклическом кольце два отдельных кольца могут, независимо друг от друга, быть насыщенными или частично ненасыщенными, или ароматическими, а в трициклическом кольце отдельные кольца, независимо друг от друга, могут быть, в частности, насыщенными или частично ненасыщенными. В одном осуществлении настоящего изобретения 3-членная или 4-членная группа является насыщенной. В 3-членных по 10-членных структурах моноциклическое, бициклическое или трициклическое кольцо может представлять собой карбоциклическое кольцо, то есть содержать 0 (ноль) гетерочленов кольца, или гетероциклическое кольцо, в котором, как отмечалось выше, могут присутствовать гетерочлены. В бициклическом гетероциклическом кольце одно или оба отдельных кольца могут содержать гетерочлены, а в трициклическом кольце одно или несколько отдельных колец могут содержать гетерочлены. В случае когда в качестве гетерочленов в бициклическом или трициклическом кольце выступают атомы азота, они также могут находиться в конденсированных положениях или положениях, соединенных мостиковыми связями. Свободная связь, посредством которой кольцо связывается с группой R23, может находиться при любом подходящем углеродном атоме кольца или азотном атоме кольца. В одном из осуществлений изобретения свободная связь находится при кольцевом атоме углерода. Кроме оксо-групп в членах кольца S(O) и S(O)2 и заместителей R32 при кольцевых атомах азота, кольца от 3-членной до 10-членной структуры могут необязательно иметь в качестве заместителей при атомах углерода в кольце один или более, например, 1, 2, 3, 4 или 5, либо 1, 2, 3 или 4, либо 1, 2 или 3 одинаковых или разных заместителей, как указано, которые могут находиться в любых позициях.
Кольца от 3-членной до 10-членной структуры, моноциклические, бициклические или трициклические, содержат (C3-C7)-циклоалкильные группы, фенильные группы, моноциклические, 5-членные или 6-членные - ароматические гетероциклические группы и моноциклические, от 4-членных до 7-членных, - частично ненасыщенные и насыщенные группы, согласно определению групп Het, Het1 и Het2. Все эти группы, поэтому являются примерами указанных колец от 3-членных до 10-членных, и все пояснения, приведенные выше в отношении таких групп, соответственно, касаются указанных колец от 3-членного до 10-членного, если в определении указанных колец от 3-членного до 10-членного не указано иначе. Поэтому, например, заместители в таких группах, такие как в фенильной группе, которая относится к указанным кольцам от 3-членного до 10-членного, или в моноциклической 5-членной или 6-членной ароматической гетероциклической группе, представляющей группу Het или Het2, которая относится к указанным кольцам от 3-членного до 10-членного, могут соответствовать приведенным в определении R24. В качестве дополнительных примеров циклических групп, которые составлены указанными кольцами от 3-членного до 10-членного, можно привести (C5-C7)-циклоалкенильные группы, нафтильные группы и гидрогенизированные нафтильные группы, инденильные группы и гидрогенизированные инденильные группы, бициклические гетероциклические группы, а также бициклоалкильные, бициклоалкенильные и трициклоалкильные группы и их гетероаналоги.
Число атомов углерода в кольце (C5-C7)-циклоалкенильной группы, представляющей R24, может составлять 5, 6 или 7. Примерами циклоалкенильных групп являются циклопентенильные группы, включающие циклопент-1-енил, циклопент-2-енил и циклопент-3-енил, циклогексильные группы, включающие циклогекс-1-енил, циклогекс-2-енил и циклогекс-3-енил, а также циклогептильные группы, включающие циклгепт-1-енил, циклогепт-2-енил, циклогепт-3-нил и циклогепт-4-енил. Циклоалкенильные группы, представляющие R24, могут быть незамещенными или замещенными, как это отмечается в случае колец от 3-членного до 10-членного, которые представляют R24, например, с одним или несколькими, или 1, 2, 3 или 4, или 1, 2 или 3 одинаковым или разными (C1-C4)-алкильными заместителями, например метильными группами, которые могут вводиться в любые положения. Примерами таких алкилзамещенных циклоалкенильных групп являются 1-метилциклопент-2-енил, 1-метилциклопент-3-енил, 2,3-диметилциклогекс-2-енил и 3,4-диметилциклогекс-3-енил. Циклоалкенильные группы также могут необязательно иметь один или более фтор-заместителей, т.е. они могут быть незамещенными и не нести фтор-заместителей, или замещенными, например, 1, 2, 3, 4, 5, 6 или 7, или 1, 2, 3, 4 или 5, или 1, 2, 3 или 4 фтор-заместителями. Циклоалкенильные группы также могут быть одновременно замещены фтором и алкилом. Атомы фтора могут находиться в любых позициях в циклоалкенильной группе, а также могут находиться в алкильном заместителе циклоалкенильной группы. Примерами таких фторзамещенных циклоалкильных групп являются 1-фторциклогекс-2-енил, 1-фторциклогекс-3-енил, 4,4-дифторциклогекс-2-енил.
Нафталинильными (нафтильными) группами, представляющими R24, могут быть нафталин-1-ил (1-нафтил) и нафталин-2-ил (2-нафтил), и они могут быть замещены одним или несколькими, например, 1, 2, 3, 4 или 5, или 1, 2 или 3, например 1 или 2, одинаковыми или различными заместителями, как это отмечалось выше. Заместители в замещенной нафтильной группе могут находиться в любом положении, например во 2-положении, 3-положении, 4-положении, 5-положении, 6-положении, 7-положении или 8-положении в случае монозамещенной нафталин-1-ильной группы и в 1-положении, 3-положении, 4-положении, 5-положении, 6-положении, 7-положении или 8-положении в случае монозамещенной нафталин-2-ильной группы. Аналогичным образом, в нафтильной группе, которая содержит два или более заместителей, такие заместители могут располагаться в кольце, с которым связана остальная часть молекулы, и (или) в другом кольце. Примерами гидрогенизированных нафтильных групп, представляющих R24, являются дигидронафталинильная, в том числе 1,4-дигидронафталинильная, тетрагидронафталинильная, в том числе 1,2,3,4-тетрагидронафталинильная и 5,6,7,8-тетрагидронафталинильная, октагидронафталинильная, в том числе 1,2,3,4,5,6,7,8-октагидронафталинильная, а также декагидронафталинильная. Гидрогенизированные нафталинильные группы могут быть связаны с остальной частью молекулы через любой углеродный атом кольца в насыщенном или частично ненасыщенном или ароматическом кольце и могут быть необязательно замещены одним или несколькими, например 1, 2, 3, 4 или 5, или 1, 2 или 3, например 1 или 2, одинаковыми или различными заместителями, как это отмечалось выше, которые могут находиться в любых положениях.
Инденильными группами, представляющими R24, могут быть 1H-инден-1-ил, 1H-инден-2-ил, 1H-инден-3-ил, 1H-инден-4-ил, 1H-инден-5-ил, 1H-инден-6-ил или 1H-инден-7-ил, например, и они могут быть необязательно замещены одним или несколькими, например 1, 2, 3, 4 или 5, или 1, 2 или 3, например 1 или 2, одинаковыми или различными заместителями, как это отмечалось выше, которые могут находиться в любых положениях. Примеры гидрогенизированных инденильных групп, представляющих R24, включают инданильные (2,3-дигидро-1Н-инденильные) и октагидро-1Н-инденильные, которые могут быть связаны с остальной частью молекулы через любой углеродный атом кольца в насыщенном или частично ненасыщенном или ароматическом кольце, например через 1-положение, 2-положение, 4-положение или 5-положение в случае инданильной группы, и могут быть необязательно замещены одним или несколькими, например 1, 2, 3, 4 или 5, или 1, 2 или 3, например, 1 или 2 одинаковыми или различными заместителями, как это отмечалось выше, которые могут находиться в любых положениях.
В одном из осуществлений настоящего изобретения бициклические гетероциклические группы, представляющие R24, являются конденсированными бициклическими группами, в которых два кольца имеют общую связь, и могут быть насыщенными, частично ненасыщенными или ароматическими, как это отмечалось выше в отношении колец от 3-членного до 10-членного, представляющих R24 в общем случае. Их кольца могут содержать 1,2, 3, 4 или 5 двойных связей. Оба кольца могут быть насыщенными, или же одно из колец может быть насыщенным или частично ненасыщенным, а другое кольцо частично ненасыщенным или ароматическим, или же оба кольца могут быть ароматическими, то есть содержать циклическую систему из шести делокализованных пи-электронов. В одном из осуществлений настоящего изобретения оба кольца являются ароматическими или одно из колец является ароматическим, а другое кольцо - частично ненасыщенным и содержит по меньшей мере одну двойную связь из-за конденсации с ароматическим кольцом. В одном из осуществлений настоящего изобретения бициклическая гетероциклическая группа образована 8-, 9- или 10-членными кольцами и двумя 5-членными кольцами или двумя 6-членными кольцами, или 6-членным кольцом, конденсированным с 5-членным кольцом или 7-членным кольцом, конденсированным с 5-членным кольцом, в другом осуществлении - 9- или 10-членным кольцом и двумя 6-членными конденсированными кольцами или 6-членным кольцом, конденсированным с 5-членным кольцом. Гетерочлены кольца могут присутствовать в обоих кольцах бициклической гетероциклической группы или только в одном из колец, а второе кольцо может не содержать гетерочленов. В обоих кольцах также могут присутствовать кольцевые атомы азота. Атом азота в кольце не только является гетероатомом в других кольцах, от 3-членного до 10-членного, представляющих R24, например насыщенных кольцах, атом азота кольца, содержащий группу R32, но также может быть кольцевым атомом азота конденсированного 5-членного кольца в ароматической бициклической гетероциклической группе, например в конденсированном пирроле, пиразоле, имидазоле или триазоле, с которым связан экзоциклический атом или группа. К примерам колец, производным которых может быть конденсированная бициклическая гетероциклическая группа, относятся индол, изоиндол, бензо[b]тиофен, бензофуран, бензо[1,3]диоксол ([1,3]бензодиоксол, 1,2-метилендиоксибензол), бензо[1,3]оксазол, бензо[1,3]тиазол, бензоимидазол, хроман, изохроман, бензо[1,4]диоксан ([1,4]бензодиоксан, 1,2-этилендиоксибензол), хинолин, изохинолин, циннолин, хиназолин, хиноксалин, фталазин, пирролоазепины, имидазоазепины, тиенотиофены, тиенопирролы, тиенопиридины, нафтиридины и соответствующие кольца, в которых одна или несколько или же все двойные связи гидрогенизированы, то есть заменены одинарными связями, такие как, например, 2,3-дигидро-1H-индол, 2,3-дигидро-1H-изоиндол, 2,3-дигидробензофуран, 1,2,3,4-тетрагидрохинолин, 5,6,7,8-тетрагидрохинолин, декагидрохинолин, 1,2,3,4-тетрагидроизохинолин, 5,6,7,8-тетрагидроизохинолин, декагидроизохинолин. Бициклическая гетероциклическая группа может присоединяться посредством любого атома углерода в кольце или атома азота в кольце. В одном из осуществлений изобретения бициклическая гетероароматическая группа связывается через кольцевой атом углерода. Например, в качестве индолильной группы могут выступать индол-1-ил, индол-2-ил, индол-3-ил, индол-4-ил, индол-5-ил, индол-6- или индол-7-ил, бензоимидазольной группой могут быть 1H-бензоимидазол-1-ил, 1H-бензоимидазол-2-ил, 1H-бензоимидазол-4-ил, 1H-бензоимидазол-5-ил, 1H-бензоимидазол-6-ил или 1H-бензоимидазол-7-ил, бензо[1,4]диоксанильной группой могут быть бензо[1,4]диоксан-2-ил, бензо[1,4]диоксан-5-ил или бензо[1,4]диоксан-6-ил, хинолинильной группой (хинолильной группой) могут быть хинолин-2-ил, хинолин-3-ил, хинолин-4-ил, хинолин-5-ил, хинолин-6-ил, хинолин-7-ил или хинолин-8-ил, изохинолинильной группой могут быть изохинолин-1-ил, изохинолин-3-ил, изохинолин-4-ил, изохинолин-5-ил, изохинолин-6-ил, изохинолин-7-ил или изохинолин-8-ил. В замещенной бициклической гетероароматической группе заместители могут находиться в любых положениях, например в индол-2-ильной группе в положении 1 и (или) в положении 3 и (или) в положении 4 и (или) в положении 5 и (или) в положении 6 и (или) в положении 7, в индол-5-ильной группе в положении 1 и (или) в положении 2, и (или) в положении 3, и (или) в положении 4, и (или) в положении 6, и (или) в положении 7, в 1Н-бензоимидазол-2-ильной группе в положении 1 и (или) в положении 4 и (или) в положении 5 и (или) в положении 6 и (или) в положении 7. Как правило, кроме перечисленных выше ингредиентов, бициклическая гетероциклическая группа может также содержать соответствующие кольцевые атомы азота в ароматическом кольце, например атома азота в хинолинильной группе или в изохинолинильной группе, оксидный заместитель -O‾ и присутствовать в форме N-оксида.
В одном из осуществлений настоящего изобретения бициклоалкильные, бициклоалкенильные и трициклоалкильные группы, представляющие R24, являются соединенными мостиковыми связями бициклические и трициклические группы, содержащие от 6 до 10 членов, в другом осуществлении от 7 до 10 членов, которые могут содержать атомы углерода только в составе кольца, то есть они могут быть производными карбоциклических бициклоалканов, бициклоалкенов и трициклоалканов, или которые, как показано выше, также могут содержать гетерочлены, то есть они могут быть производными соответствующих гетероаналогов аза-, оксо- и тиобициклоалканов, -бициклоалкенов и -трициклоалканов. Если их кольца содержат гетерочлены, то в одном из осуществлений они содержат один или два гетерочлена, в другом осуществлении - один гетерочлен, например гетерочлены, выбранные из ряда, включающего N, N(R28) и O. Гетерочлены могут присутствовать в любом желаемом положении в бициклической или трициклической системе, в том числе в положениях в мостиковых связях, а в случае атомов азота - в положениях на концах мостиковой связи. Бициклоалкенилы и их гетероциклические аналоги могут содержать одну или несколько двойных связей в кольцах. В одном из осуществлений настоящего изобретения они содержат одну или две двойные связи в кольце, в другом осуществлении - одну двойную связь. Бициклоалкильные, бициклоалкенильные и трициклоалкильные группы могут присоединяться к остальной части молекулы посредством атома углерода в кольце или атома азота в кольце. Свободная связь может находиться в любом стереохимическом положении, например в экзо-положении или в эндо-положении. Бициклоалкильная, бициклоалкенильная и трициклоалкильная группы и их гетероциклические аналоги могут быть необязательно замещены, как показано выше, например заместителями, выбранными из ряда, включающего (C1-C4)-алкил, (C2-C5)-алкенил, HO-, HO-CH2- (гидроксиметил-) и оксо- в любых положениях. К примерам бициклоалкильных, бициклоалкенильных и трициклоалкильных групп, а также их гетероциклических аналогов относятся норборнил (бицикло[2.2.1]гептил), бицикло[3.1.1]гептил, бицикло[3.1.1]гепт-2-енил, бицикло[2.2.2]октил, бицикло[2.2.2]окт-2-енил, бицикло[3.2.1]октил, 7-азабицикло[2.2.1]гептил, 1-азабицикло[2.2.2]октил, бицикло[2.2.2.]окт-2-енил, трицикло[4.4.0.03,8]децил), адамантил (трицикло[3.3.1.13,7]децил), норадамантил (трицикло[3.3.1.03,7]нонил), трицикло[2.2.1.02,6]гептил.
В качестве галогена выступает фтор, хлор, бром или иод. В одном из осуществлений настоящего изобретения галогеном является фтор, хлор или бром, в другом осуществлении - фтор или хлор.
Оксо-группа, то есть атом кислорода с двойной связью, которая связана с атомом углерода, заменяет два атома водорода при атоме углерода в исходной системе. Например, если CH2-группа заменяется оксо-группой, она превращается в карбонильную группу (C(O), C=O). Оксо-группа не может выступать в качестве заместителя при атоме углерода в ароматическом кольце, например фенильной группе.
Настоящее изобретение включает все стереоизомерные формы соединений формулы I, например все энантиомеры и диастереомеры, в том числе цис/транс-изомеры. Аналогичным образом, изобретение включает смеси двух или более стереоизомерных форм, например смеси энантиомеров и (или) диастереомеров, в том числе цис/транс-изомеров во всех соотношениях. Асимметрические центры, присутствующие в соединениях формулы I, например в незамещенных или замещенных алкильных группах, могут независимым друг от друга образом принимать S-конфигурацию или R-конфигурацию. Изобретение касается энантиомеров, как лево-, так и правовращающих антиподов, в энантиомерно чистой форме и в значительной мере в энантиомерно чистой форме, а также в форме рацематов и в форме смесей двух энантиомеров во всех соотношениях. Аналогичным образом, изобретение относится к диастереомерам в форме чистых и в значительной мере чистых диастереомеров, а также в форме смесей двух и более диастереомером во всех соотношениях. Изобретение также включает все цис/транс-изомеры соединений формулы I в чистой форме и в значительной мере в чистой форме, а также в форме смесей цис-изомеров и транс-изомеров во всех соотношениях. Цис/транс-изомерия может наблюдаться, например, в замещенных кольцах и при двойной связи. Индивидуальные стереоизомеры, при необходимости, могут получаться разделением смеси с использованием традиционных методов, например хроматографией или кристаллизацией, или за счет использования стереохимически однородных исходных соединений при синтезе, или в ходе стереоселективных реакций. В одном из вариантов перед разделением стереоизомеров можно провести их модификацию. Разделение смеси стереоизомеров может проводиться на стадии соединения формулы I или на стадии промежуточного соединения в ходе синтеза. Изобретение также включает все таутомерные формы соединений формулы I.
Физиологически приемлемые соли, в том числе фармацевтически используемые соли соединений формулы I, как правило, включают нетоксичный солевой компонент. Они могут содержать неорганические или органические солевые компоненты. Такие соли могут получаться, например, из соединений формулы I, которые содержат кислотную группу, например группу карбоновой кислоты (гидроксикарбонильная группа, HO-C(O)-), а также нетоксичные неорганические или органические основания. К подходящим основаниям, например, относятся соединения щелочных металлов или соединения щелочноземельных металлов, например гидроксид натрия, гидроксид калия, карбонат натрия или гидрокарбонат натрия, или аммиак, органические аминосоединения и гидроксиды четвертичного аммония. Реакции соединений формулы I с основаниями для получения солей, как правило, проводятся стандартными методами в растворителе или разбавителе. Примерами солей кислотных групп являются натриевые, калиевые, магниевые или кальциевые соли или аммониевые соли, которые могут также нести одну или несколько органических групп при атоме азота. Соединения формулы I, которые содержат основную, то есть протонируемую группу, например аминогруппу или основный гетероцикл, могут присутствовать в форме своих солей присоединения кислоты, образованных физиологически приемлемыми кислотами, например как соли хлористоводородной кислоты, бромистоводородной кислоты, фосфорной кислоты, серной кислоты, уксусной кислоты, бензойной кислоты, метансульфоновой кислоты, п-толуилсульфоновой кислоты, которые, как правило, могут получаться из соединений формулы I посредством реакции с кислотой в растворителе или разбавителе в соответствии со стандартными методами. Если соединения формулы I одновременно содержат кислую и основную группу в молекуле, изобретение также включает внутренние соли (бетаины, цвиттерионы) в дополнение к солевым формам, указанным выше. Настоящее изобретение также включает все соли соединений формулы I, которые из-за низкой физиологической переносимости не могут непосредственно применяться в качестве фармацевтических средств, но пригодны как интермедиаты химических реакций или для приготовления физиологически приемлемых солей, например посредством анионного обмена или катионного обмена. Настоящее изобретение также включает все сольваты соединений формулы I и их соли, в том числе физиологически приемлемые сольваты, например гидраты, то есть аддукты с водой, а также аддукты со спиртами, например (C1-C4)-алканолами, а также активные метаболиты соединений формулы I и пролекарства соединений формулы I, то есть соединений, которые in vitro необязательно проявляют фармакологическую активность, но которые in vivo превращаются в фармакологически активные соединения формулы I, например соединения, которые посредством метаболического гидролиза превращаются в соединение формулы I, например соединения, в которых присутствует карбоксильная группа в этерифицированной форме или форме амида.
Как отмечалось выше, гетерочлены в кольце А, включающем два атома углерода, которые также являются частью конденсированного 5-членного кольца, описанного в формуле I и содержащего группы R3 по R6, могут присутствовать в любых сочетаниях и могут находиться в любом подходящем положении. Например, в случае если А представлено пиридиновым или тиофеновым кольцом, атом азота или атом серы кольца может находиться в положении рядом с указанным 5-членным кольцом, или же в положении, которое удалено от указанного 5-членного кольца. В случае если кольцом А является 6-членное гетероциклическое кольцо, в которое входят два гетероатома кольца N, например оба гетероатома кольца могут находиться в двух положениях, которые примыкают к указанному 5-членному кольцу, и 6-членным кольцом будет пиразиновый цикл, или же один из них может присутствовать в положении, примыкающем к указанному 5-членному кольцу, а второй находиться в удаленном положении, и 6-членным кольцом будет пиримидиновый или пиридазиновый цикл, или же оба гетероатома могут находиться в удаленных положениях, и 6-членным кольцом будет пиридазиновый цикл. В одном из осуществлений изобретения гетероатомы в гетероциклическом кольце, представляющем кольцо А, выбирают из N и S, в другом осуществлении оба они N. В одном из осуществлений изобретения циклоалкановое кольцо, представляющее А, является 5-членным, 6-членным или 7-членным, в другом осуществлении 5-членным или 6-членным, в еще одном осуществлении 6-членным, а циклоалкановое кольцо, таким образом, представляет собой циклопентан, циклогексан или циклогептан, которые, как отмечалось выше, все могут быть замещены. В одном из осуществлений настоящего изобретения кольцо А представляет собой циклогексановое кольцо, бензольное кольцо, пиридиновое кольцо, пиразиновое кольцо или моноциклическое 5-членное ароматическое гетероциклическое кольцо, содержащее 1 или 2 одинаковых или разных гетерочленов, выбранных из ряда, включающего N, N(R1), O и S, например 1 гетерочлен выбирают из ряда, включающего N(R1), O и S, например тиофеновое кольцо, причем указанное кольцо может содержать указанные выше заместители. В другом осуществлении настоящего изобретения кольцо А представляет собой бензольное кольцо, пиридиновое кольцо, пиразиновое кольцо или моноциклическое 5-членное ароматическое гетероциклическое кольцо, содержащее 1 или 2 одинаковых или разных гетерочлена, выбранных из ряда, включающего N, N(R1), O и S, например 1 гетерочлен выбирают из ряда, включающего N(R1), O и S, например тиофеновое кольцо, причем указанное кольцо может содержать указанные выше заместители. В другом осуществлении настоящего изобретения кольцо А представляет собой бензольное кольцо или моноциклическое 5-членное ароматическое гетероциклическое кольцо, содержащее 1 или 2 одинаковых или разных гетерочлена, выбранных из ряда, включающего N, N(R1), O и S, например 1 гетерочлен выбирает из ряда, включающего N(R1), O и S, например тиофеновое кольцо, причем указанное кольцо может содержать указанные выше заместители. В другом осуществлении кольцо А представляет собой бензольное кольцо, пиразиновое кольцо или тиофеновое кольцо, в еще одном осуществлении - бензольное кольцо или тиофеновое кольцо, каковые кольца могут содержать все указанные выше заместители. В другом осуществлении кольцо А представляет собой бензольное кольцо, которое может необязательно содержать указанные выше заместители. В еще одном осуществлении настоящего изобретения кольцо А представляет собой циклоалкановое кольцо, которое может содержать указанные выше заместители.
Число заместителей, которые могут присутствовать в кольце А, зависит от размера и типа кольца А и числа гетерочленов кольца. В одном осуществлении настоящего изобретения число заместителей может быть 1, 2, 3 или 4, в другом осуществлении - 1, 2 или 3, в другом осуществлении - 1 или 2 и еще в одном осуществлении - 1. Например, в случае бензольного кольца, представляющего А, которое может иметь или не иметь заместители, число заместителей может быть 1, 2, 3 или 4, либо 1, 2 или 3, либо 1 или 2, например 1. В случае пиридинового кольца число заместителей может быть 1, 2 или 3, либо 1 или 2, например 1, в случае пиразинового кольца число заместителей может быть 1 или 2, например 1, в случае тиофенового кольца число заместителей может быть 1 или 2, например 1, в случае тиазолового кольца число заместителей может быть 1. В одном осуществлении настоящего изобретения циклоалкановое кольцо, представляющее А, не имеет никаких заместителей. В другом осуществлении настоящего изобретения кольцо А не имеет никаких заместителей, и атомы углерода в кольце содержат, таким образом, атомы водорода. Заместители в кольце А могут присутствовать в любой подходящей позиции. В одном осуществлении настоящего изобретения в соединениях формулы I, в которых кольцом А может быть замещенное бензольное кольцо, заместители, которые могут присутствовать в позициях 5 и 6 инданового кольца, составляющего указанное бензольное кольцо, представляющее А, выбирают из группы, содержащей галоген, R1, HO-, R1-O-, R1-C(O)-O-, R1-S(O)2-O-, R1-S(O)m-, H2N-, R1-NH-, R1-N(R1)-, R1-C(O)-NH-, R1-C(O)-N(R71)-, R1-S(O)2-NH-, R1-S(O)2-N(R71)-, R1-C(O)-, HO-C(O)-, R1-O-C(O)-, H2N-C(O)-, R1-NH-C(O)-, R1-N(R1)-C(O)-, H2N-S(O)2-, R1-NH-S(O)2-, R1-N(R1)-S(O)2-, NC- и O2N-. В другом осуществлении настоящего изобретения в соединениях формулы I, в которых кольцом А может быть необязательно замещенное бензольное кольцо, заместители, которые могут присутствовать в кольце А, выбирают из группы, содержащей галоген, R1, HO-, R1-O-, R1-C(O)-O-, R1-S(O)2-O-, R1-S(O)m-, H2N-, R1-NH-, R1-N(R1)-, R1-C(O)-NH-, R1-C(O)-N(R71)-, R1-S(O)2-NH-, R1-S(O)2-N(R71)-, R1-C(O)-, HO-C(O)-, R1-O-C(O)-, H2N-C(O)-, R1-NH-C(O)-, R1-N(R1)-C(O)-, H2N-S(O)2-, R1-NH-S(O)2-, R1-N(R1)-S(O)2-, NC- и O2N-. В другом осуществлении настоящего изобретения заместители в бензольном кольце или гетероциклическом кольце, представляющем А, выбирают из группы, содержащей галоген, R1, HO-, R1-O-, R1-C(O)-O-, R1-S(O)2-O-, R1-S(O)m-, H2N-, R1-NH-, R1-N(R1)-, R1-C(O)-NH-, R1-C(O)-N(R71)-, R1-S(O)2-NH-, R1-S(O)2-N(R71)-, R1-C(O)-, HO-C(O)-, R1-O-C(O)-, H2N-C(O)-, R1-NH-C(O)-, R1-N(R1)-C(O)-, H2N-S(O)2-, R1-NH-S(O)2-, R1-N(R1)-S(O)2-, NC- и O2N-, в другом осуществлении - из группы, содержащей галоген, R1, HO-, R1-O-, R1-C(O)-O-, R1-S(O)m-, H2N-, R1-NH-, R1-N(R1)-, R1-C(O)-NH-, R1-C(O)-N(R71)-, R1-S(O)2-NH-, R1-S(O)2-N(R71)-, NC- и O2N-, в другом осуществлении - из группы, содержащей галоген, R1, R1-O-, R1-S(O)m-, NC- и O2N-, например из группы, содержащей галоген, (C1-C4)-алкил, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, NC- и O2N-, в другом осуществлении - из группы, содержащей галоген, R1, R1-O-, R1-S(O)m- и NC-, например из группы, содержащей галоген, (C1-C4)-алкил, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m- и NC-, в другом осуществлении - из группы, содержащей галоген, R1, R1-O- и NC-, например, из группы, содержащей галоген, (C1-C4)-алкил, (C1-C4)-алкил-O- и NC-, в другом осуществлении - из группы, содержащей галоген, R1 и R1-O-, например из группы, содержащей галоген, (C1-C4)-алкил и (C1-C4)-алкил-O-. В одном осуществлении настоящего изобретения заместители в бензольном кольце или гетероциклическом кольце, представляющем А, выбирают из группы, содержащей галоген и (C1-C4)-алкил. В одном осуществлении настоящего изобретения число нитро-заместителей (O2N-) в кольце А не превышает двух, в другом осуществлении - не превышает одного. В одном осуществлении настоящего изобретения число нитрогрупп в соединении формулы I не превышает двух.
В случае если кольцо А является бензольным кольцом, соединения формулы I также могут быть представлены формулой Ia,

где Y, Z, R3 пo R6, R20 пo R22 и R50 определяют как в соединениях формулы I, R7 определяется как заместители, которые могут необязательно присутствовать в бензольном кольце, представляющем кольцо А в соединениях формулы I, т.е. R7 выбирают из группы, содержащей галоген, R1, HO-, R1-O-, R1-C(O)-O-, R1-S(O)2-O-, R1-S(O)m-, H2N-, R1-NH-, R1-N(R1)-, R1-C(O)-NH-, R1-C(O)-N(R71)-, R1-S(O)2-NH-, R1-S(O)2-N(R71)-, R1-C(O)-, HO-C(O)-, R1-O-C(O)-, H2N-C(O)-, R1-NH-C(O)-, R1-N(R1)-C(O)-, H2N-S(O)2-, R1-NH-S(O)2-, R1-N(R1)-S(O)2-, NC-, O2N-, фенил и Het, или из любой другой группы заместителей, указанной здесь, например из группы, содержащей галоген, (C1-C4)-алкил, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m- и NC-, или из группы, содержащей галоген и (C1-C4)-алкил, и число r имеет значение 0, 1, 2, 3 или 4, либо 0, 1, 2 или 3, либо 0, 1 или 2, либо 0 или 1. В одном осуществлении настоящего изобретения число r в соединениях формулы Ia имеет значение 0, т.е. бензольное кольцо, представленное в формуле Ia, не имеет заместителя R7. Заместители R7 могут находиться при любом из четырех атомов углерода бензольного кольца, приведенного в формуле Ia, которые не является частью конденсированного 5-членного кольца, содержащего группы R3 по R6. Все другие такие атомы углерода в бензольном кольце, которые не содержат заместителя R7, содержат атомы водорода. Иными словами, в случае если число r имеет значение 0, например, бензольное кольцо содержит четыре атома водорода.
Аналогичным образом, в случае если кольцо А является пиридиновым кольцом, пиридазиновым кольцом, тиофеновым кольцом или циклогексановым кольцом, соединения формулы I могут быть представлены формулами Ib-1, Ib-2, Ic, Id-1, Id-2 и Ie,

где Y, Z, R3 пo R6, R20 пo R22 и R50 определяют как в соединениях формулы I, R7 определяют как заместители, которые могут присутствовать в кольце А в соединениях формулы I, т.е. в случае соединений формул Ib-1, Ib-2, Ic, Id-1 и Id-2, R7 выбирают из группы, содержащей галоген, R1, HO-, R1-O-, R1-C(O)-O-, R1-S(O)2-O-, R1-S(O)m-, H2N-, R1-NH-, R1-N(R1)-, R1-C(O)-NH-, R1-C(O)-N(R71)-, R1-S(O)2-NH-, R1-S(O)2-N(R71)-, R1-C(O)-, HO-C(O)-, R1-O-C(O)-, H2N-C(O)-, R1-NH-C(O)-, R1-N(R1)-C(O)-, H2N-S(O)2-, R1-NH-S(O)2-, R1-N(R1)-S(O)2-, NC-, O2N-, фенил и Het, или из любой другой группы заместителей, указанной здесь, например из группы, содержащей галоген, (C1-C4)-алкил, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m- и NC-, и в случае соединений формулы Ie R7 выбирают из группы, содержащей фтор и (C1-C4)-алкил, и число r имеет значение 0, 1, 2 или 3, либо 0, 1 или 2, либо 0 или 1, в случае соединений формул Ib-1 и Ib-2, и имеет значение 0, 1 или 2, либо 0 или 1 в случае соединений формул Ic, Id-1 и Id-2, и имеет значение 0, 1, 2, 3, 4, 5, 6, 7 или 8, либо 0, 1, 2, 3 или 4, либо 0, 1 или 2, например в случае соединений формулы Ie. В одном осуществлении настоящего изобретения число r в соединениях формул Ib-1, Ib-2, Ic, Id-1, Id-2 и Ie имеет значение 0, т.е. пиридиновое кольцо, пиридазиновое кольцо, тиофеновое кольцо и циклогексановое кольцо, представленные в формулах, не содержат заместителя R7. Заместители R7 могут находиться при любом из атомов углерода кольца, в частности, при атомах углерода кольца, которые не является частью конденсированного 5-членного кольца, содержащего группы R3 по R6. В тех положениях при атомах углерода кольца, где заместитель R7 отсутствует, находятся атомы водорода.
В группе C(R12)=C(R13), представляющей двухвалентную группу Y, атом углерода, несущий группу R13, связан с кольцевым атомом углерода, несущим группу R21, и атом углерода, несущий группу R12, связан с кольцевым атомом углерода, несущим группу C(O)-N(R20). В группе N=C(R14) атом углерода, несущий группу R14, связан с кольцевым атомом углерода, несущим группу R21, и атом азота связан с кольцевым атомом углерода, несущим группу C(O)-N(R20). В группе C(R15)=N атом азота связан с кольцевым атомом углерода, несущим группу R21, и атом углерода, несущий группу R15, связан с кольцевым атомом углерода, несущим группу C(O)-N(R20). В одном осуществлении настоящего изобретения Y выбирают из ряда, включающего S, C(R12)=C(R13), N=C(R14) и C(R15)=N, в другом осуществлении - из ряда, включающего S, C(R12)=C(R13) и C(R15)=N. В одном из осуществлений настоящего изобретения Y выбирают из ряда, включающего S и C(R12)=C(R13), в другом осуществлении - из ряда, включающего C(R12)=C(R13) и C(R15)=N. В еще одном осуществлении изобретения Y представляет собой C(R12)=C(R13). В другом осуществлении настоящего изобретения Y представляет собой C(R15)=N.
В одном осуществлении настоящего изобретения трехвалентная группа Z является C(R16). В другом осуществлении Z представляет собой C(R16), а Y выбирают из ряда, включающего S, C(R12)=C(R13) и C(R15)=N. В другом осуществлении Z представляет собой C(R16), а Y выбирают из ряда, включающего S и C(R12)=C(R13). В другом осуществлении Z представляет собой C(R16), а Y выбирают из группы, содержащей C(R15)=N и C(R12)=C(R13). В указанном последнем осуществлении ароматическое кольцо в соединениях формулы I, содержащих группы кольца Y и Z, представляет собой пиридиновое кольцо или бензольное кольцо, соответственно, а соединения формулы I представляют собой соединения формулы If или формулы Ig.

где A, R3 по R6, R12, R13, R15, R16, R20 по R22 и R50 определяются как в соединениях формулы I или имеют свои другие указанные значения. В одном осуществлении настоящего изобретения группой Z является C(R16) и группой Y является S. В другом осуществлении настоящего изобретения группой Z является C(R16) и группой Y является C(R15)=N. В другом осуществлении настоящего изобретения группой Z является C(R16) и группой Y является C(R12)=C(R13), т.е. в настоящем осуществлении соединениями формулы I являются соединениями формулы Ig. В другом осуществлении настоящего изобретения в соединениях формулы Ia группой Z является C(R16) и группой Y является C(R12)=C(R13), т.е. соединениями настоящего осуществления являются соединения формулы Ih,

где R3 по R6, R12, R13, R16, R20 по R22 и R50 определяются как в соединениях формулы I или имеют свои другие указанные значения. R7 и r в соединениях формулы Ih определяются так же, как в соединениях формулы Ia, и так же, как в соединениях формулы Ia, заместители R7 могут присутствовать на любом из четырех атомов углерода конденсированного бензольного кольца, описанного формулой Ih, которые не являются частью конденсированного пятичленного кольца, содержащего с R3 по R6группы, а все прочие подобные атомы углерода бензольного кольца, которые не имееют заместителя R7, содержат атомы водорода. Все объяснения в отношении групп и все определения и осуществления, указанные выше или ниже в отношении соединений формулы I, применимы соответствующим образом к соединениям со всеми формулами, которые представляют подгруппы соединений формулы I, включая соединения формул с Ia по Ih.
В одном осуществлении настоящего изобретения R0 выбирают из группы, содержащей галоген и (C1-C4)-алкил, в другом осуществлении - из группы, содержащей водород и метил. В одном осуществлении настоящего изобретения R0 является водородом. В другом осуществлении настоящего изобретения R0 является (C1-C4)-алкилом, например метилом.
В одном осуществлении настоящего изобретения R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, независимо от каждой другой группы R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, выбирают из группы, содержащей (C1-C6)-алкил, (C2-C4)-алкенил, (C2-C4)-алкинил, (C3-C7)-циклоалкил и (C3-C7)-циклоалкил-(C1-C2)-алкил-, в другом осуществлении - из группы, содержащей (C1-C4)-алкил, (C2-C4)-алкенил, (C2-C4)-алкинил, (C3-C7)-циклоалкил и (C3-C7)-циклоалкил-(C1-C2)-алкил-, в другом осуществлении - из группы, содержащей (C1-C6)-алкил, (C3-C7)-циклоалкил и (C3-C7)-циклоалкил-(C1-C2)-алкил-, в другом осуществлении - из группы, содержащей (C1-C4)-алкил, (C3-C7)-циклоалкил и (C3-C7)-циклоалкил-(C1-C2)-алкил-, в другом осуществлении - из группы, содержащей (C1-C6)-алкил, (C3-C7)-циклоалкил и (C3-C7)-циклоалкил-CH2-алкил-, в другом осуществлении - из группы, содержащей (C1-C6)-алкил и (C3-C7)-циклоалкил, в другом осуществлении - из группы, содержащей (C1-C4)-алкил и (C3-C7)-циклоалкил, которые могут необязательно иметь один или более одинаковых или разных заместителей R70, причем в этих группах помимо заместителей R70 могут необязательно присутствовать один или более фтор-заместителей, а в циклоалкильных группах - могут присутствовать один или более заместителей (C1-C4)-алкил, применительно к алкилу, алкенилу, алкинилу и циклоалкильным группам в целом. В одном осуществлении настоящего изобретения R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, независимо от каждой другой группы R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, выбирают из группы, содержащей (C1-C6)-алкил, в другом осуществлении - из группы, содержащей (C1-C4)-алкил, которые могут необязательно иметь один или более одинаковых или разных заместителей R70. В одном осуществлении настоящего изобретения (C3-C7)-циклоалкильные группы, встречающиеся в R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, представляют собой, независимо от каждой другой группы R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, (C3-C6)-циклоалкил, в другом осуществлении - (C3-C4)-циклоалкил, например циклопропил, в другом осуществлении - (C5-C6)-циклоалкил, например циклогексил. В одном осуществлении настоящего изобретения число заместителей R70 в любой из групп R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, независимо от каждой другой группы R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, равно 0, 1, 2, 3 или 4, в другом осуществлении - 0, 1, 2 или 3, в другом осуществлении - 0, 1 или 2, в другом осуществлении - 0 или 1. В одном осуществлении настоящего изобретения любая из групп R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, независимо от каждой другой группы R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, не содержит заместитель R70, но лишь может необязательно иметь один или более фтор-заместителей, а в случае циклоалкильных групп - один или более заместителей вида (C1-C4)-алкил. В другом осуществлении настоящего изобретения любая из групп R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, независимо от каждой другой группы R1, R2, R11, R30, R33, R35, R54, R55, R57 и R58, не содержит ни заместитель R70, ни фтор-заместители, ни - в случае циклоалкильных групп - заместители вида (C1-C4)-алкил.
В одном осуществлении настоящего изобретения группа фенил-(C1-C4)-алкила-, представляющая R3 или R5, является бензильной группой, в которой фенильная группа может необязательно иметь заместители, как указано в отношении фенильных групп в целом. В одном осуществлении настоящего изобретения одну из групп R3 и R5 выбирают из группы, содержащей водород, (C1-C4)-алкил, фенил-(C1-C4)-алкил-, фенил и гидрокси, а другую из групп R3 и R5 выбирают из группы, содержащей водород, (C1-C4)-алкил, фенил-(C1-C4)-алкил- и фенил. В одном осуществлении настоящего изобретения группы R3 и R5 выбирают, независимо друг от друга, из группы, содержащей водород, (C1-C4)-алкил, фенил-(C1-C4)-алкил- и фенил. В другом осуществлении R3 и R5 выбирают, независимо друг от друга, из группы, содержащей водород и (C1-C4)-алкил, в другом осуществлении - из группы, содержащей водород и метил. В другом осуществлении R3 и R5 представляют собой водород.
В одном осуществлении настоящего изобретения R4 и R6 выбирают, независимо друг от друга, из группы, содержащей водород и метил. В другом осуществлении R4 и R6 представляют собой водород.
В одном осуществлении настоящего изобретения R3 и R4 идентичны и их выбирают из группы, содержащей водород и метил, в другом осуществлении R3 и R4 представляют собой водород. В другом осуществлении R5 и R6 идентичны и их выбирают из группы, содержащей водород и метил, в другом осуществлении R5 и R6 представляют собой водород. В другом осуществлении R3, R4, R5 и R6 идентичны и их выбирают из группы, содержащей водород и метил. В другом осуществлении R3, R4, R5 и R6 представляют собой водород.
В одном осуществлении настоящего изобретения R10 выбирают из группы, содержащей водород и метил. В другом осуществлении R10представляет собой водород. В другом осуществлении настоящего изобретения R10является (C1-C4)-алкилом, например метилом.
В одном осуществлении настоящего изобретения R12, R13, R14, R15 и R16 выбирают, независимо друг от друга, из группы, содержащей водород, галоген, (C1-C4)-алкил, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, H2N-, (C1-C4)-алкил-NH-, (C1-C4)-алкил-N((C1-C4)-алкил)-, NC- и O2N-, в другом осуществлении - из группы, содержащей водород, галоген, (C1-C4)-алкил, (C1-C4)-алкил-O-, NC- и O2N-, в другом осуществлении - из группы, содержащей водород, галоген, (C1-C4)-алкил, (C1-C4)-алкил-O- и O2N-, в другом осуществлении - из группы, содержащей водород, галоген, (C1-C4)-алкил, (C1-C4)-алкил-O- и NC, в другом осуществлении - из группы, содержащей водород, галоген, (C1-C4)-алкил и (C1-C4)-алкил-O-, в другом осуществлении - из группы, содержащей водород, галоген и (C1-C4)-алкил. В одном осуществлении настоящего изобретения R12 и R13 выбирают, независимо друг от друга, из группы, содержащей водород, галоген, (C1-C4)-алкил, (C1-C4)-алкил-O- и NC-, в другом осуществлении - из группы, содержащей водород, галоген, (C1-C4)-алкил и NC-, в другом осуществлении - из группы, содержащей водород, галоген и NC-, в другом осуществлении - из группы, содержащей водород и галоген, в другом осуществлении - из группы, содержащей водород, хлор и фтор, в другом осуществлении - из группы, содержащей водород и фтор. В одном осуществлении настоящего изобретения R12 является водородом и R13 - фтором, или R12 является фтором, а R13 - водородом. В другом осуществлении R12 и R13 представляют собой водород. В одном осуществлении настоящего изобретения R14 и R15 выбирают, независимо друг от друга, из группы, содержащей водород, галоген, (C1-C4)-алкил и (C1-C4)-алкил-O-, в другом осуществлении - из группы, содержащей водород, галоген и (C1-C4)-алкил, в другом осуществлении - из группы, содержащей водород и галоген, в другом осуществлении - из группы, содержащей водород, хлор и фтор. В другом осуществлении R14 и R15 представляют собой водород. В одном осуществлении настоящего изобретения R16 выбирают из группы, содержащей водород, галоген, (C1-C4)-алкил и (C1-C4)-алкил-O-, в другом осуществлении - из группы, содержащей водород, галоген и (C1-C4)-алкил, в другом осуществлении - из группы, содержащей водород и галоген, в другом осуществлении - из группы, содержащей водород, хлор и фтор. В другом осуществлении настоящего изобретения R16 является водородом.
В одном осуществлении настоящего изобретения R20 выбирают из группы, содержащей водород и метил. В другом осуществлении R20 представляет собой водород. В другом осуществлении R20 является (C1-C4)-алкилом, например метилом.
В одном осуществлении настоящего изобретения группа R21 является группой формулы II, т.е. формулы R24-R23-, которая соединяется с остальной молекулой посредством группы R23, как обозначено в отношении данной группы и в целом конечной черточкой, означающей свободную связь, и группу R22 выбирают из группы, содержащей водород, галоген, R30, HO-, R30-O-, R30-C(O)-O-, R30-S(O)2-O-, R30-S(O)m-, H2N-, R30-NH-, R30-N(R30)-, R30-C(O)-NH-, R30-C(O)-N(R71)-, R30-S(O)2-NH-, R30-S(O)2-N(R71)-, R30-C(O)-, HO-C(O)-, R30-O-C(O)-, H2N-C(O)-, R30-NH-C(O)-, R30-N(R30)-C(O)-, H2N-S(O)2-, R30-NH-S(O)2-, R30-N(R30)-S(O)2-, NC-, O2N- и Het1. В другом осуществлении настоящего изобретения группа R22 является группой формулы II, и группу R21 выбирают из группы, содержащей водород, галоген, R30, HO-, R30-O-, R30-C(O)-O-, R30-S(O)2-O-, R30-S(O)m-, H2N-, R30-NH-, R30-N(R30)-, R30-C(O)-NH-, R30-C(O)-N(R71)-, R30-S(O)2-NH-, R30-S(O)2-N(R71)-, R30-C(O)-, HO-C(O)-, R30-O-C(O)-, H2N-C(O)-, R30-NH-C(O)-, R30-N(R30)-C(O)-, H2N-S(O)2-, R30-NH-S(O)2-, R30-N(R30)-S(O)2-, NC-, O2N- и Het1.
В одном осуществлении настоящего изобретения одну из групп R21и R22, которая не является группой формулы II, выбирают из группы, содержащей водород, галоген, R30, R30-O-, R30-C(O)-O-, R30-S(O)m-, H2N-, R30-NH-, R30-N(R30)-, R30-C(O)-NH-, R30-C(O)- и NC-, в другом осуществлении - из группы, содержащей водород, галоген, (C1-C4)-алкил, HO-(C1-C4)-алкил-, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, H2N-, (C1-C4)-алкил-NH-, ди((C1-C4)-алкил)N-, (C1-C4)-алкил-C(O)- и NC-, в другом осуществлении - из группы, содержащей водород, галоген, (C1-C4)-алкил, HO-(C1-C4)-алкил-, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, (C1-C4)-алкил-C(O)- и NC-, в другом осуществлении - из группы, содержащей галоген, (C1-C4)-алкил, HO-(C1-C4)-алкил-, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, H2N-, (C1-C4)-алкил-NH-, ди((C1-C4)-алкил)N-, (C1-C4)-алкил-C(O)- и NC-, в другом осуществлении - из группы, содержащей (C1-C4)-алкил, HO-(C1-C4)-алкил-, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, H2N-, (C1-C4)-алкил-NH-, ди((C1-C4)-алкил)N-, (C1-C4)-алкил-C(O)- и NC-, в другом осуществлении - из группы, содержащей (C1-C4)-алкил, HO-(C1-C4)-алкил-, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, (C1-C4)-алкил-NH-, ди((C1-C4)-алкил)N- и (C1-C4)-алкил-C(O)-. В одном осуществлении настоящего изобретения одну из групп R21и R22, которая не является группой формулы II, выбирают из группы, содержащей (C1-C4)-алкил, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, (C1-C4)-алкил-NH- и ди((C1-C4)-алкил)N-, в другом осуществлении - из группы, содержащей (C1-C4)-алкил, (C1-C4)-алкил-O- и (C1-C4)-алкил-S(O)m-, в другом осуществлении - из группы, содержащей (C1-C4)-алкил-O- и (C1-C4)-алкил-S(O)m-. В другом осуществлении ту группу R21 и R22, которая не является группой формулы II, выбирают из группы, в которую входят (C1-C4)-алкил, HO-(C1-C4)-алкил-, (C1-C4)-алкил-O- и (C1-C4)-алкил-C(O)-, в другом осуществлении - из группы, содержащей (C1-C4)-алкил, HO-(C1-C4)-алкил- и (C1-C4)-алкил-O-, в другом осуществлении - из группы, содержащей (C1-C4)-алкил и (C1-C4)-алкил-O-. В другом осуществлении одна из групп R21и R22, которая не является группой формулы II, представляет собой (C1-C4)-алкил-O-, например метокси или этокси.
В одном осуществлении настоящего изобретения, в случае если группа R21 является группой формулы II, группу R22 выбирают из группы, содержащей (C1-C4)-алкил и (C1-C4)-алкил-O-, и в другом осуществлении - (C1-C4)-алкил-O-, а в случае если группа R22 является группой формулы II, группу R21 выбирают из группы, содержащей водород, галоген, R30, HO-, R30-O-, R30-C(O)-O-, R30-S(O)2-O-, R30-S(O)m-, H2N-, R30-NH-, R30-N(R30)-, R30-C(O)-NH-, R30-C(O)-N(R71)-, R30-S(O)2-NH-, R30-S(O)2-N(R71)-, R30-C(O)-, HO-C(O)-, R30-O-C(O)-, H2N-C(O)-, R30-NH-C(O)-, R30-N(R30)-C(O)-, H2N-S(O)2-, R30-NH-S(O)2-, R30-N(R30)-S(O)2-, NC-, O2N- и Het1, или согласно определению в любом осуществлении или ином определении R21, приведенном в настоящем документе.
Число членов в цепи, представляющей R23, может быть 1, 2, 3, 4 или 5. В одном осуществлении настоящего изобретения двухвалентная группа R23 представляет собой прямую связь, т.е. группа R24 присоединена непосредственно к кольцу, содержащему группы Y и Z, как показано в формуле I. В другом осуществлении R23 представляет собой прямую связь или цепь, содержащую 1, 2, 3 или 4 члена цепи. В другом осуществлении R23 представляет собой прямую связь или цепь, содержащую 2, 3 или 4 члена цепи, в другом осуществлении - прямую связь или цепь, содержащую 2 или 3 члена цепи, в другом осуществлении - прямую связь или цепь, содержащую 3 члена цепи, причем в этих осуществлениях члены цепи определяются согласно определению выше или ниже. В другом осуществлении R23 представляет собой цепь, содержащую 1, 2, 3, 4 или 5 членов цепи, в другом осуществлении - цепь, содержащую 1, 2, 3 или 4 члена цепи, в другом осуществлении - цепь, содержащую 2, 3 или 4 члена цепи, в другом осуществлении - цепь, содержащую 2 или 3 члена цепи, в другом осуществлении - цепь, содержащую 3 члена цепи, причем в этих осуществлениях члены цепи определяются согласно определению выше или ниже. В одном осуществлении настоящего изобретения 0 или 1 член цепи, представляющей R23, является гетерочленом цепи, а в другом осуществлении один из членов цепи, представляющей R23, является гетерочленом цепи, причем в этих осуществлениях гетерочлены цепи определяются согласно определению выше или ниже. В другом осуществлении настоящего изобретения ни один из членов цепи, представляющей R23, не является гетерочленом цепи. В одном осуществлении настоящего изобретения гетерочлены цепи, представляющей R23, выбирают из группы, содержащей N(R25), O, S и S(O)2. В другом осуществлении настоящего изобретения гетерочлены цепи, представляющей R23, выбирают из группы, содержащей N(R25), O и S, в другом осуществлении - из группы, содержащей N(R25) и O, в другом осуществлении - из группы, содержащей O и S, в другом осуществлении - из группы, содержащей N(R25), O и S(O)2, в другом осуществлении - из группы, содержащей N(R25) и S(O)2, в другом осуществлении - из группы, содержащей O и S(O)2. В другом осуществлении настоящего изобретения гетерочлены, которые могут присутствовать в цепи, представляющей R23, представляют собой О (кислород), в другом осуществлении гетерочлены которые могут присутствовать в цепи, представляющей R23, представляют собой N(R25). В другом осуществлении настоящего изобретения 0 или 1 гетерочлен присутствует в цепи, представляющей R23, представляя собой О (кислород), и в другом осуществлении один гетерочлен, который присутствует в цепи, представляет собой O. В другом осуществлении настоящего изобретения 0 или 1 гетерочлен присутствует в цепи, представляющей R23, представляя собой N(R25), и в другом осуществлении один гетерочлен, который присутствует в цепи, представляет собой N(R25).
Гетерочлены цепи, представляющей R23, могут присутствовать в любых позициях цепи, при условии, что полученная группа соответствует параметрам, указанным выше в отношении R23и соединений настоящего изобретения в целом. В случае если две смежные группы C(R26)(R26) в цепи, представляющей R23, соединены между собой двойной связью или тройной связью, в одном осуществлении настоящего изобретения гетерочлены цепи не присутствуют в позициях, смежных с такой двойной или тройной связью. Гетерочлены цепи могут присутствовать в одном конце или в обоих концах цепи и, таким образом, могут соединяться прямой связью с группой R24 и(или) с кольцом, содержащим группы Y и Z, как показано в формуле I, и(или) внутри цепи. В случае если один или два гетерочлена присутствуют в цепи, представляющей R23, в одном осуществлении настоящего изобретения по меньшей мере один из терминальных членов цепи является гетерочленом, и в другом осуществлении терминальный член цепи, присоединенный к группе R24, является гетерочленом, и в другом осуществлении терминальный член цепи, присоединенный к кольцу, содержащему группы Y и Z, является гетерочленом. В одном осуществлении настоящего изобретения один из членов цепи, представляющей R23, является гетерочленом, и данный гетерочлен является терминальным членом цепи, присоединенным к группе R24. В другом осуществлении один из членов цепи, представляющей R23, является гетерочленом, и данный гетерочлен является терминальным членом цепи, присоединенным к кольцу, содержащему группы Y и Z, как показано в формуле I.
В случае если две смежные группы C(R26)(R26) в цепи, представляющей R23, соединены между собой двойной связью или тройной связью, такая цепь содержит ненасыщенную двухвалентную группу -C(R26)=C(R26)-, в которой R26 определяют согласно описанию выше и в одном осуществлении настоящего изобретения выбирают из группы, содержащей водород и (C1-C4)-алкил, или ненасыщенную группу формулы -C≡C-. Члены цепи, не соединенные между собой двойной связью или тройной связью, соединяются между собой одинарной связью. В случае если две смежные группы C(R26)(R26) соединены двойной связью, одна из групп R26 в каждой из смежных групп C(R26)(R26) может рассматриваться как свободная связь, причем две свободные связи образуют вторую связь между соответствующими атомами углерода. В случае если две смежные группы C(R26)(R26) соединены тройной связью, обе группы R26 в каждой из смежных групп C(R26)(R26) могут рассматриваться как свободная связь, причем обе пары свободных связей образуют вторую и третью связь между соответствующими атомами углерода. В одном осуществлении настоящего изобретения указанная ненасыщенная группа присутствует не более одного раза в цепи, представляющей R23. Указанная ненасыщенная группа может присутствовать в любой позиции цепи, представляющей R23, и встречается в любом конце цепи и, таким образом, может соединяться прямой связью с группой R24 и(или) с кольцом, содержащим группы Y и Z, как показано в формуле I, либо быть внутри цепи. В одном осуществлении настоящего изобретения указанная ненасыщенная группа не прилегает к гетерочлену. В одном осуществлении настоящего изобретения цепь, представляющая R23, не содержит двойную или тройную связь. В другом осуществлении две смежные группы C(R26)(R26) могут быть соединены между собой двойной связью. В другом осуществлении две смежные группы C(R26)(R26) могут быть соединены между собой тройной связью. В другом осуществлении две смежные группы C(R26)(R26) соединены между собой тройной связью, т.е. в данном осуществлении цепь, представляющая R23, содержит тройную связь. В другом осуществлении группа R23 представляет собой группу формулы -C≡C-.
В одном осуществлении настоящего изобретения R23 выбирают из прямой связи и из одной или более цепей, присутствующих в следующих примерах групп формулы II, которые присоединены к кольцу, содержащему группы Y и Z, как показано в формуле I, свободной связью, представленной терминальной чертой, и из каковых групп формулы II получают в свою очередь группы R23 путем удаления группы R24:
причем в этих группах формулы II группы R24, R25 и R26 определяются согласно определению выше или ниже.
В одном осуществлении настоящего изобретения R24 выбирают из группы, содержащей R31, R31-O-, R31-S(O)m-, H2N-, R31-NH-, R31-N(R31)-, R31-C(O)-NH-, R31-C(O)-N(R71)-, HO-C(O)-, R31-O-C(O)-, H2N-C(O)-, R31-NH-C(O)-, R31-N(R31)-C(O)-, NC- и моноциклическое, бициклическое или трициклическое кольцо с числом членов от 3-х до 10-и, в другом осуществлении - из группы, содержащей R31, R31-O-, R31-S(O)m-, NC- и моноциклическое, бициклическое или трициклическое кольцо с числом членов от 3-х до 10-и, в другом осуществлении - из группы, содержащей R31, R31-O- и моноциклическое, бициклическое или трициклическое кольцо с числом членов от 3-х до 10-и, в другом осуществлении - из группы, содержащей (C1-C6)-алкил, (C1-C6)-алкил-O- и моноциклическое, бициклическое или трициклическое кольцо с числом членов от 3-х до 10-и, причем во всех этих осуществлениях моноциклическое, бициклическое или трициклическое кольцо с числом членов от 3-х до 10-и определяют согласно определению выше или ниже, может быть насыщенным или ненасыщенным и содержит 0, 1, 2 или 3 одинаковых или разных гетерочлена кольца, выбранных из группы, содержащей N, N(R32), O, S, S(O) и S(O)2 и может необязательно иметь на атомах углерода кольца один или более одинаковых разных заместителей, выбранных из группы, содержащей галоген, R33, HO-, R33-O-, R33-C(O)-O-, R33-S(O)2-O-, R33-S(O)m-, H2N-, R33-NH-, R33-N(R33)-, R33-C(O)-NH-, R33-C(O)-N(R71)-, R33-S(O)2-NH-, R33-S(O)2-N(R71)-, H2N-S(O)2-NH-, R33-NH-S(O)2-NH-, R33-N(R33)-S(O)2-NH-, H2N-S(O)2-N(R71)-, R33-NH-S(O)2-N(R71)-, R33-N(R33)-S(O)2-N(R71)-, R33-C(O)-, HO-C(O)-, R33-O-C(O)-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)-, H2N-S(O)2-, R33-NH-S(O)2-, R33-N(R33)-S(O)2-, NC-, O2N-, оксо, фенил и Het, или имеет другие указанные здесь значения. В другом осуществлении настоящего изобретения R24представляет собой моноциклическое, бициклическое или трициклическое кольцо с числом членов от 3-х до 10-и, определяемое согласно определению выше или ниже, которое может быть насыщенным или ненасыщенным и содержит 0, 1, 2 или 3 одинаковых или разных гетерочлена кольца, выбранных из группы, содержащей N, N(R32), O, S, S(O) и S(O)2, причем кольцо может необязательно иметь на атомах углерода кольца один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген, R33, HO-, R33-O-, R33-C(O)-O-, R33-S(O)2-O-, R33-S(O)m-, H2N-, R33-NH-, R33-N(R33)-, R33-C(O)-NH-, R33-C(O)-N(R71)-, R33-S(O)2-NH-, R33-S(O)2-N(R71)-, H2N-S(O)2-NH-, R33-NH-S(O)2-NH-, R33-N(R33)-S(O)2-NH-, H2N-S(O)2-N(R71)-, R33-NH-S(O)2-N(R71)-, R33-N(R33)-S(O)2-N(R71)-, R33-C(O)-, HO-C(O)-, R33-O-C(O)-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)-, H2N-S(O)2-, R33-NH-S(O)2-, R33-N(R33)-S(O)2-, NC-, O2N-, оксо, фенил и Het, или имеет другие указанные здесь значения.
В одном осуществлении настоящего изобретения моноциклическое, бициклическое или трициклическое кольцо с числом членов от 3-х до 10-и, представляющее R24, является моноциклическим или бициклическим кольцом, и в другом осуществлении оно является моноциклическим кольцом, причем все кольца могут быть необязательно замещены, как указано выше или ниже. В одном осуществлении настоящего изобретения моноциклическое, кольцо, представляющее R24, имеет от 3 до 7 членов, в другом осуществлении - 3 члена, 5 членов или 7 членов, в другом осуществлении - 3 члена, 5 членов или 6 членов, в другом осуществлении - 5 членов или 6 членов, в другом осуществлении - 6 членов, причем все кольца могут быть необязательно замещены, как указано выше или ниже. В одном осуществлении настоящего изобретения бициклическое или трициклическое кольцо, представляющее R24, имеет от 7 до 10 членов, причем все кольца могут быть необязательно замещены, как указано выше или ниже. В одном осуществлении настоящего изобретения кольцо, представляющее R24, является насыщенным кольцом или ненасыщенным кольцом, включая частично ненасыщенное, т.е. неароматическое кольцо, которое содержит 0, 1, 2 или 3, например 0, 1 или 2 двойных связей в кольце, или ароматическое кольцо, причем все кольца могут быть необязательно замещены, как указано выше или ниже. В другом осуществлении кольцо, представляющее R24, является насыщенным кольцом или частично ненасыщенным кольцом, которое содержит 0, 1, 2 или 3, например 0, 1 или 2 двойных связей в кольце, причем все кольца могут быть необязательно замещены, как указано выше или ниже. В другом осуществлении настоящего изобретения кольцо, представляющее R24, является ароматическим кольцом, в другом осуществлении ароматическое кольцо выбирают из бензола, ароматических 5-членных и 6-членных моноциклических гетероциклов, нафталина и ароматических 9-членных и 10-членных бициклических гетероциклов, в другом осуществлении ароматическое кольцо выбирают из бензола, ароматических 5-членных и 6-членных моноциклических гетероциклов, в другом осуществлении ароматическое кольцо выбирают из бензола и тиофена, причем все кольца могут быть необязательно замещены, как указано выше или ниже. В другом осуществлении кольцо, представляющее R24, является бензольным кольцом, которое может быть необязательно замещено, как указано выше или ниже, т.е. заместителями, указанными выше или ниже в отношении кольца с числом членов от 3 до 10, представляющего R24. В плане остатков в данном последнем осуществлении R24 является фенильной группой, которая может быть необязательно замещена, как указано выше или ниже, т.е. заместителями, указанными выше или ниже в отношении кольца с числом членов от 3 до 10, представляющего R24.
В одном осуществлении настоящего изобретения число гетерочленов, которые могут присутствовать в кольце с числом членов от 3 до 10, представляющем R24, составляет 0, 1 или 2, в другом осуществлении настоящего изобретения число гетерочленов равно 0 или 1, а в другом осуществлении настоящего изобретения число гетерочленов равно 0 (нулю), т.е. в данном последнем осуществлении кольцо с числом членов от 3 до 10, представляющее R24, является карбоциклическим кольцом, причем все кольца могут быть необязательно замещены, как указано выше или ниже. В одном осуществлении настоящего изобретения гетерочлены, которые могут присутствовать в кольце с числом членов от 3 до 10, представляющем R24, выбирают из N, N(R32), O, S и S(O)2, в другом осуществлении - из N, N(R32), O и S, в другом осуществлении - из N, O и S, в другом осуществлении - из N(R32), O и S, в другом осуществлении - из N и S.
В одном осуществлении настоящего изобретения число заместителей, которые могут присутствовать на атомах углерода в кольце с числом членов от 3 до 10, представляющем R24, составляет 1, 2, 3, 4 или 5, в другом осуществлении число заместителей, которые могут присутствовать в атомах углерода, составляет 1, 2, 3 или 4, в другом осуществлении число заместителей, которые могут присутствовать в атомах углерода, составляет 1, 2 или 3, в другом осуществлении число заместителей, которые могут необязательно присутствовать в атомах углерода, составляет 1 или 2.
В одном осуществлении настоящего изобретения заместители, которые могут необязательно присутствовать на атомах углерода в кольце с числом членов от 3 до 10, представляющем R24, включая бензольное кольцо или фенильную группу, соответственно, представляющих R24, выбирают из группы, содержащей галоген, R33, HO-, R33-O-, R33-S(O)m-, H2N-, R33-NH-, R33-N(R33)-, R33-C(O)-NH-, R33-C(O)-N(R71)-, R33-S(O)2-NH-, R33-S(O)2-N(R71)-, H2N-S(O)2-NH-, R33-NH-S(O)2-NH-, R33-N(R33)-S(O)2-NH-, H2N-S(O)2-N(R71)-, R33-NH-S(O)2-N(R71)-, R33-N(R33)-S(O)2-N(R71)-, HO-C(O)-, R33-O-C(O)-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)-, NC-, оксо, фенил и Het, в другом осуществлении - из группы, содержащей галоген, R33, HO-, R33-O-, R33-S(O)m-, H2N-, R33-NH-, R33-N(R33)-, R33-C(O)-NH-, R33-C(O)-N(R71)-, R33-S(O)2-NH-, R33-S(O)2-N(R71)-, H2N-S(O)2-NH-, R33-NH-S(O)2-NH-, R33-N(R33)-S(O)2-NH-, H2N-S(O)2-N(R71)-, R33-NH-S(O)2-N(R71)-, R33-N(R33)-S(O)2-N(R71)-, HO-C(O)-, R33-O-C(O)-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)- и NC-, в другом осуществлении - из группы, содержащей галоген, R33, HO-, R33-O-, R33-S(O)m-, H2N-, R33-NH-, R33-N(R33)-, R33-C(O)-NH-, R33-S(O)2-NH-, H2N-S(O)2-NH-, R33-NH-S(O)2-NH-, R33-N(R33)-S(O)2-NH-, HO-C(O)-, R33-O-C(O)-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)- и NC-, в другом осуществлении - из группы, содержащей галоген, R33, HO-, R33-O-, R33-S(O)m-, H2N-, R33-NH-, R33-N(R33)-, R33-C(O)-NH-, R33-S(O)2-NH-, H2N-S(O)2-NH-, R33-NH-S(O)2-NH-, R33-N(R33)-S(O)2-NH-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)- и NC-, в другом осуществлении - из группы, содержащей галоген, R33, HO-, R33-O-, R33-S(O)m-, H2N-, R33-NH-, R33-N(R33)-, R33-C(O)-NH-, R33-S(O)2-NH-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)- и NC-, в другом осуществлении - из группы, содержащей галоген, R33, HO-, R33-O-, R33-S(O)m-, H2N-, R33-NH-, R33-N(R33)-, R33-C(O)-NH-, R33-C(O)-N(R71)-, R33-S(O)2-NH-, R33-S(O)2-N(R71)-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)- и NC-, в другом осуществлении - из группы, содержащей галоген, R33, HO-, R33-O-, R33-S(O)m-, R33-C(O)-NH-, R33-C(O)-N(R71)-, R33-S(O)2-NH-, R33-S(O)2-N(R71)-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)- и NC-, в другом осуществлении - из группы, содержащей галоген, R33, HO-, R33-O-, R33-S(O)m-, R33-C(O)-NH-, R33-S(O)2-NH-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)- и NC-, в другом осуществлении - из группы, содержащей галоген, R33, HO-, R33-O-, R33-C(O)-NH-, R33-S(O)2-NH-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)- и NC-, в другом осуществлении - из группы, содержащей галоген, R33, R33-O-, R33-C(O)-NH-, R33-S(O)2-NH-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)- и NC-, в другом осуществлении - из группы, содержащей галоген, R33, R33-O- и NC-, в другом осуществлении - из группы, содержащей галоген, R33 и R33-O-, в другом осуществлении - из группы, содержащей галоген и R33, причем во всех этих осуществлениях R33 и R71 определяются согласно определению выше или ниже, и R33 может необязательно иметь один или более одинаковых или разных заместителей R70. В одном осуществлении настоящего изобретения группы R33в этих заместителях в кольце, представляющем R23, выбирают, независимо друг от друга, из группы, содержащей (C1-C6)-алкил, (C3-C7)-циклоалкил и (C3-C7)-циклоалкил-(C1-C4)-алкил-, в другом осуществлении - из группы, содержащей (C1-C6)-алкил, (C3-C6)-циклоалкил и (C3-C6)-циклоалкил-(C1-C2)-алкил-, в другом осуществлении - из группы, содержащей (C1-C6)-алкил, (C3-C6)-циклоалкил и (C3-C6)-циклоалкил-CH2-, в другом осуществлении - из группы, содержащей (C1-C6)-алкил, циклопропил и циклопропил-CH2-, например из группы, содержащей (C1-C6)-алкил, в другом осуществлении - из группы, содержащей (C1-C4)-алкил, циклопропил и циклопропил-CH2-, например из группы, содержащей (C1-C4)-алкил. В одном осуществлении настоящего изобретения число заместителей R70, которые могут необязательно присутствовать в этих группах R33 помимо каких-либо фтор-заместителей, а в случае циклоалкильных групп - каких-либо заместителей (C1-C4)-алкил, равно, независимо друг от друга, 0, 1, 2 или 3, в другом осуществлении - 0, 1 или 2, в другом осуществлении - 0 или 1, в другом осуществлении - 0. В одном осуществлении настоящего изобретения заместители R70 в этих группах R33 выбирают, независимо друг от друга, из группы, содержащей HO-, R71-O-, R71-C(O)-O-, H2N-, R71-NH-, R71-N(R71)-, R71-C(O)-NH-, R71-C(O)-N(R71)-, R71-S(O)2-NH- и R71-S(O)2-N(R71)-, в другом осуществлении - из группы, содержащей HO-, R71-C(O)-O-, H2N-, R71-C(O)-NH- и R71-S(O)2-NH-, в другом осуществлении - из группы, содержащей HO-, R71-C(O)-O- и R71-C(O)-NH-, в другом осуществлении - из группы, содержащей HO- и R71-C(O)-NH-, в другом осуществлении - из группы, содержащей HO- и R71-O-, и еще в одном осуществлении настоящего изобретения заместители R70 в этих группах R33 представляют собой HO-. В одном осуществлении настоящего изобретения группы R71, присутствующие в этих группах R33, выбирают, независимо друг от друга, из группы, содержащей (C1-C4)-алкил, циклопропил и циклопропил-, в другом осуществлении - из группы, содержащей (C1-C4)-алкил и циклопропил, в другом осуществлении - из группы, содержащей (C1-C4)-алкил. В одном осуществлении настоящего изобретения R24 представляет собой бензольное кольцо или тиофеновое кольцо, например бензольное кольцо, или в случае соотвествующих остатков R24 представляет собой фенильную группу или тиофенильную (тиенильную) группу, например фенильную группу, причем все группы могут быть необязательно замещены, как указано выше.
Примерами конкретных остатков бензольного и тиофенового колец, т.е. конкретных фенильных и тиофенильных групп, представляющих R24, из каковых одного или нескольких примеров выбирают группу R24в одном осуществлении настоящего изобретения, являются фенил, 2-фторфенил, 3-фторфенил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил, 3-бромфенил, 2,3-дихлорфенил, 3,4-дихлорфенил, 2,5-дифторфенил, 2,5-дихлорфенил, 2-хлор-6-фторфенил, 3,4,5-трифторфенил, 3-метилфенил (м-толил), 3-этилфенил, 3-изопропилфенил, 3-циклопропилфенил, 3-трет-бутил-5-метилфенил, 3-трифторметилфенил, 3-(2-гидроксиэтил)-фенил, 3-(2-гидрокси-2-метилпропил)-фенил, 3-(2-ацетиламиноэтил)-фенил, 2-фтор-5-метилфенил, 3-хлор-2-метилфенил, 5-хлор-2-метилфенил, 5-хлор-2-фтор-3-метилфенил, 2-фтор-3-трифторметилфенил, 2-фтор-5-трифторметилфенил, 4-фтор-3-трифторметилфенил, 5-фтор-3-трифторметилфенил, 3-хлор-4-трифторметилфенил, 5-хлор-2-трифторметилфенил, 5-хлор-3-трифторметилфенил, 3-этоксифенил, 2-пропоксифенил, 3-изопропоксифенил, 3-трифторметоксифенил, 3-(2,2,2-трифторэтокси)-фенил, 5-хлор-2-метоксифенил, 3-хлор-4-метоксифенил, 5-фтор-3-изопропоксифенил, 2-фтор-3-трифторметоксифенил, 4-метокси-3,5-диметилфенил, 3-метокси-5-трифторметилфенил, 3-метилсульфанилфенил, 3-этилсульфанилфенил, 3-трифторметилсульфанилфенил, 3-этансульфонилфенил, 3-ацетиламинофенил, 3-метансульфониламинофенил, 3-диметиламиносульфониламинофенил, 3-цианофенил, 2-тиенил, 3-тиенил, 4-метил-2-тиенил, 5-метил-3-тиенил.
В одном осуществлении настоящего изобретения общее число атомов C, N, O и S, присутствующее в двух группах R23 и R24, т.е. в замещающей группе R24-R23- в кольце, содержащем группы Y и Z, которые представлены в формуле I, равно по меньшей мере 6, в другом осуществлении - не менее 7, в другом осуществлении - не менее 8, в другом осуществлении - не менее 9.
В одном осуществлении настоящего изобретения R25 выбирают из группы, содержащей водород и метил, в другом осуществлении R25 представляет собой водород. В другом осуществлении настоящего изобретения R25является (C1-C4)-алкилом, например метилом.
В одном осуществлении настоящего изобретения R26, независимо от каждой другой группы R26, выбирают из группы, содержащей водород, фтор, метил и HO-, в другом осуществлении - из группы, содержащей водород, фтор и (C1-C4)-алкил, в другом осуществлении - из группы, содержащей водород, фтор и метил, в другом осуществлении - из группы, содержащей водород и фтор, в другом осуществлении - из группы, содержащей водород и метил, и еще в одном осуществлении R26 представляет собой водород, или во всех этих осуществлениях две группы R26, связанные с одним атомом углерода, представляют собой оксо, или две группы R26 или одна группа R25 и одна группа R26, вместе с входящими в них членами цепи, образуют моноциклическое кольцо с числом членов от 3 до 7, которое является насыщенным и содержит 0, 1 или 2 одинаковых или разных гетерочленов, выбранных из группы, содержащей N, N(R34), O, S, S(O) и S(O)2, причем кольцо может необязательно иметь на атомах углерода кольца один или более одинаковых или разных заместителей, выбранных из группы, содержащей фтор и (C1-C4)-алкил. В другом осуществлении настоящего изобретения R26, независимо от каждой другой группы R26, выбирают из группы, содержащей водород, фтор, метил и HO-, в другом осуществлении - из группы, содержащей водород, фтор и (C1-C4)-алкил, в другом осуществлении - из группы, содержащей водород, фтор и метил, в другом осуществлении - из группы, содержащей водород и фтор, в другом осуществлении - из группы, содержащей водород и метил, и еще в одном осуществлении R26- водород, или во всех этих осуществлениях две группы R26 или одна группа R25 и одна группа R26, вместе с входящими в них членами цепи, образуют моноциклическое кольцо с числом членов от 3 до 7, которое является насыщенным и содержит 0, 1 или 2 одинаковых или разных гетерочленов, выбранных из группы, содержащей N, N(R34), O, S, S(O) и S(O)2, причем кольцо может необязательно иметь на атомах углерода кольца один или более одинаковых или разных заместителей, выбранных из группы, содержащей фтор и (C1-C4)-алкил. В другом осуществлении настоящего изобретения R26, независимо от каждой другой группы R26, выбирают из группы, содержащей водород, фтор, метил и HO-, в другом осуществлении - из группы, содержащей водород, фтор и (C1-C4)-алкил, в другом осуществлении - из группы, содержащей водород, фтор и метил, в другом осуществлении - из группы, содержащей водород и фтор, в другом осуществлении - из группы, содержащей водород и метил, и еще в одном осуществлении все группы R26представляют собой водород.
В одном осуществлении настоящего изобретения число групп R26 в цепи, представляющей R23, которые являются HO-, равно 0, 1 или 2, в другом осуществлении равно 0 или 1, в другом осуществлении равно 0, в другом осуществлении равно 1. В одном осуществлении настоящего изобретения группа HO-, представляющая R26, не присутствует на атоме углерода, прилегающем к гетерочлену в цепи, представляющей R23. В другом осуществлении группа HO-, представляющая R26, не присоединяется к атому углерода, который присоединен к соседней группе C(R26)(R26) двойной связью. В одном осуществлении настоящего изобретения число групп R26 в цепи, представляющей R23, которые являются (C1-C4)-алкилом, таким как метил, равно 0, 1 или 2, в другом осуществлении равно 0 или 1, в другом осуществлении равно 0, в другом осуществлении равно 1, в другом осуществлении равно 2. В одном осуществлении настоящего изобретения число групп R26 в цепи, представляющей R23, которые являются фтором, равно 0, 1, 2, 3 или 4, в другом осуществлении 0, 1, 2 или 3, в другом осуществлении равно 0, 1 или 2, в другом осуществлении равно 0 или 1, в другом осуществлении равно 0, в другом осуществлении равно 1, в другом осуществлении равно 2. В одном осуществлении настоящего изобретения число оксо-заместителей в цепи, представляющей R23, которые образуются путем присоединения двух групп R26 к одному атому углерода, равно 0, 1 или 2, в другом осуществлении равно 0 или 1, в другом осуществлении равно 0, в другом осуществлении равно 1. В одном осуществлении настоящего изобретения R26, оксо-заместитель в цепи, представляющей R23, не присутствует на атоме углерода, прилегающем к гетерочлену, выбранному из группы, содержащей S(O) и S(O)2, в другом осуществлении - из группы, содержащей S, S(O) и S(O)2, в другом осуществлении - из группы, содержащей O, S, S(O) и S(O)2.
В одном осуществлении настоящего изобретения число колец, образуемых двумя группами R26или одной группой R25и одной группой R26, вместе с входящими в их состав членами цепи, равно 0, 1 или 2, в другом осуществлении равно 0 или 1, в другом осуществлении равно 1, в другом осуществлении равно 0. В одном осуществлении настоящего изобретения кольцо, образуемое двумя группами R26или одной группой R25и одной группой R26, вместе с входящими в них членами цепи, представляет собой кольцо с 3, 4, 5 или 6 членами, в другом осуществлении - кольцо с 3, 5 или 6 членами, в другом осуществлении - кольцо с 3 членами, в другом осуществлении - кольцо с 5 или 6 членами. В одном осуществлении настоящего изобретения две группы R26, вместе с входящими в них членами цепи, могут образовать кольцо, что невозможно в случае одной группы R25и одной группы R26. В одном осуществлении настоящего изобретения число членов цепи, входящих в кольцо, образуемое двумя группами R26или одной группой R25и одной группой R26, равно 1, 2, 3 или 4, в другом осуществлении равно 1, 2 или 3, в другом осуществлении равно 1 или 2, в другом осуществлении равно 1. Если такое кольцо содержит только один член цепи, две группы R26, образующие кольцо, присоединяются к одному и тому же атому углерода в цепи и указанный один член цепи является атомом углерода, несущим две группы R26. Примерами колец, образуемых двумя группами R26, присоединенными к одному и тому же атому углерода и одному члену цепи, являются циклоалкановые кольца, например циклопропан, циклобутан, циклопентан или циклогексан, и гетероциклические кольца, например тетрагидротиофен, тетрагидротиопиран, оксетан, тетрагидрофуран, тетрагидропиран, азетидин, пирролидин или пиперидин, например циклопропан, которые содержат любые соседние члены цепи, представляющей R23 и(или) группу R24 и(или) кольцо, содержащее группы Y и Z, которые представлены в формуле I, на том же атоме углерода в кольце, причем такие кольца могут быть необязательно замещены, как было указано. В случае если кольцо, образуемое двумя группами R26или одной группой R25и одной группой R26, вместе с входящими в них членами цепи, состоит из двух членов цепи, две группы R26, образующие кольцо, присоединяются к двум соседним атомам углерода в цепи или одна группа R26 присоединяется к атому углерода, примыкающему к группе N (R25), соответственно. Примерами колец, образуемых в таком случае, также являются циклоалкановые кольца, например циклопропан, циклобутан, циклопентан или циклогексан, и гетероциклические кольца, например тетрагидротиофен, тетрагидротиопиран, оксетан, тетрагидрофуран, тетрагидропиран, азетидин, пирролидин или пиперидин, например циклопропан, которые содержат любые соседние члены цепи, представляющей R23 и(или) группу R24 и(или) кольцо, содержащее группы Y и Z, которые представлены в формуле I, на двух соседних атомах углерода в кольце или на атоме азота в кольце и соседнем атоме углерода в кольце, причем такие кольца могут быть необязательно замещены, как было указано.
В случае если кольцо, образуемое двумя группами R26или одной группой R25и одной группой R26, вместе с входящими в них членами цепи, состоит из более чем одного члена цепи, помимо как минимум одной группы C(R26)(R26), входящие в них члены цепи могут также быть гетерочленами, включая группу N(R25), которые в таком случае являются гетерочленом образованного кольца. В одном осуществлении настоящего изобретения общее число гетерочленов в таком кольце равно 0, 1 или 2, в другом осуществлении равно 0 или 1, в другом осуществлении равно 0, в другом осуществлении равно 1. В одном осуществлении настоящего изобретения гетерочлены в таком кольце выбирают из группы, содержащей N, N(R34), O и S, в другом осуществлении - из группы, содержащей N, N(R34) и O, в другом осуществлении - из группы, содержащей N и N(R34), в другом осуществлении - из группы, содержащей N(R34) и O, в другом осуществлении - из группы, содержащей N(R34), в другом осуществлении гетероатомом в таком кольце являтся N, и еще в одном осуществлении гетероатомом в таком кольце являтся O, причем гетероатом N в кольце, образуемом двумя группами R26 или одной группой R25 и одной группой R26, вместе с входящими в него членами цепи, является атомом азота гетерочлена N(R25).
В одном осуществлении настоящего изобретения число заместителей, которые могут необязательно присутствовать в кольце, образуемом двумя группами R26 или одной группой R25 и одной группой R26, вместе с входящими в него членами цепи, равно 0, 1, 2, 3 или 4, в другом осуществлении равно 0, 1, 2 или 3, в другом осуществлении равно 0, 1 или 2, в другом осуществлении равно 0 или 1, в другом осуществлении равно 0. В одном осуществлении настоящего изобретения (С1-С4)-алкил-заместители, которые присутствуют в кольце, образуемом двумя группами R26 или одной группой R25 и одной группой R26, вместе с входящими в него членами цепи, являются метилом. В одном осуществлении настоящего изобретения заместители, которые присутствуют в кольце, образуемом двумя группами R26 или одной группой R25 и одной группой R26, вместе с входящими в него членами цепи, являются фтором, в другом осуществлении они являются одинаковыми или разными (С1-С4)-алкильными группами, например метилом.
Примеры конкретных групп R23, включая входящие в них конкретные группы R26, приведены в следующих примерах групп формулы II, которые присоединены к кольцу, содержащему группы Y и Z, как показано в формуле I, свободной связью, представленной терминальной чертой или терминальной линией в структурной формуле, и из каковых групп формулы II получают в свою очередь группы R23 путем удаления группы R24, причем в эти группы формулы II группа R24 определяется согласно определению выше или ниже:














В одном осуществлении настоящего изобретения R23 выбирают из прямой связи и из одной или более цепей R23 в предыдущих примерах групп формулы II и, аналогичным образом, группу формулы II выбирают из группы R24 и из одного или более предыдущих примеров групп формулы II.
В одном осуществлении настоящего изобретения число заместителей R70, которые могут необязательно присутствовать в группе R31, равно 0, 1, 2 или 3, в другом осуществлении равно 0, 1 или 2, в другом осуществлении равно 0 или 1, в другом осуществлении равно 0. В одном осуществлении настоящего изобретения R31выбирают из группы, содержащей (C1-C6)-алкил, в другом осуществлении - из группы, содержащей (C1-C4)-алкил, которые могут быть необязательно замещены одним или более одинаковыми или разными заместителями R70.
В одном осуществлении настоящего изобретения R32 и R34 выбирают, независимо друг от друга, из группы, содержащей водород, R35, R35-C(O)-, R35-O-C(O)-, фенил и Het, в другом осуществлении - из группы, содержащей водород, R35, R35-C(O)-, R35-O-C(O)-, фенил и Het2, в другом осуществлении - из группы, содержащей водород, R35, R35-C(O)-, R35-O-C(O)- и фенил, в другом осуществлении - из группы, содержащей водород, R35, R35-C(O)- и R35-O-C(O)-, в другом осуществлении - из группы, содержащей водород, R35 и R35-C(O)-, в другом осуществлении - из группы, содержащей водород, R35, фенил и Het, в другом осуществлении - из группы, содержащей водород, R35, фенил и Het2, в другом осуществлении - из группы, содержащей водород, R35и фенил, в другом осуществлении - из группы, содержащей водород и R35, причем в этих осуществлениях группу Het или Het2, встречающуюся в R32 и R34 в одном осуществлении настоящего изобретения выбирают из пиридинила и тиофенила. В одном осуществлении настоящего изобретения группы R35, встречающиеся в R32 и R34, выбирают, независимо друг от друга, из (C1-C6)-алкила, (C3-C7)-циклоалкила и (C3-C7)-циклоалкил-(C1-C4)-алкила-, в другом осуществлении из (C1-C6)-алкила, (C3-C7)-циклоалкила и (C3-C7)-циклоалкил-(C1-C2)-алкила-, в другом осуществлении из (C1-C6)-алкила, (C3-C7)-циклоалкила и (C3-C7)-циклоалкил-CH2-, в другом осуществлении из (C1-C6)-алкила и (C3-C7)-циклоалкила, в другом осуществлении из (C1-C6)-алкила, в другом осуществлении из (C1-C4)-алкила, которые могут необязательно иметь один или более одинаковых или разных заместителей R70, причем в этих группах помимо заместителей R70 могут необязательно присутствовать один или более фтор-заместителей, а в циклоалкильных группах - могут необязательно присутствовать один или более (C1-C4)-алкил-заместителей, применительно к алкильным и циклоалкильным группам в целом.
В одном осуществлении настоящего изобретения число заместителей R70, которые могут необязательно присутствовать в группе R35 и встречаются в R32 и R34 помимо каких-либо фтор-заместителей, а в случае циклоалкильной группы - алкильных заместителей, равно, независимо друг от друга, 0, 1, 2, 3 или 4, в другом осуществлении - 0, 1, 2 или 3, в другом осуществлении - 0, 1 или 2, в другом осуществлении - 0 или 1, в другом осуществлении - 0. В одном осуществлении настоящего изобретения заместители R70, которые могут необязательно присутствовать в группе R35 и встречаются в R32 и R34, выбирают, независимо друг от друга, из группы, содержащей HO-, R71-O-, NC-, фенил и Het2, в другом осуществлении - из группы, содержащей фенил и Het2, в другом осуществлении - из группы, содержащей фенил, причем фенил и Het2 определяются и могут быть необязательно замещены согласно указанию.
В одном осуществлении настоящего изобретения R50выбирают из R51-O- и R52-NH-, в другом осуществлении - из R51-O- и H2N-. В другом осуществлении R50 представляет собой R51-O-.
В одном осуществлении настоящего изобретения R51 является водородом. В другом осуществлении настоящего изобретения R51представляет собой R54.
В одном осуществлении настоящего изобретения R52 выбирают из группы, содержащей водород, R55 и R56-S(O)2-, в другом осуществлении - из группы, содержащей водород, (C1-C4)-алкил, который может быть необязательно замещен одним или более одинаковыми или разными заместителями R70, и R56-S(O)2-, в другом осуществлении - из группы, содержащей водород, незамещенный (C1-C4)-алкил и R56-S(O)2-, в другом осуществлении - из группы, содержащей водород, незамещенный метил и R56-S(O)2-, в другом осуществлении - из группы, содержащей водород и (C1-C4)-алкил, который может быть необязательно замещен одним или более одинаковыми или разными заместителями R70, в другом осуществлении - из группы, содержащей водород и незамещенный (C1-C4)-алкил, в другом осуществлении - из группы, содержащей водород и незамещенный метил. В другом осуществлении настоящего изобретения R52 является водородом.
В одном осуществлении настоящего изобретения R53 выбирают из группы, содержащей водород и (C1-C4)-алкил, который может быть необязательно замещен одним или более одинаковыми или разными заместителями R70, в другом осуществлении - из группы, содержащей водород и незамещенный (C1-C4)-алкил, в другом осуществлении - из группы, содержащей водород и незамещенный метил. В другом осуществлении настоящего изобретения R53 является водородом.
В одном осуществлении настоящего изобретения R54 выбирают из (C1-C6)-алкила, (C3-C7)-циклоалкила и (C3-C7)-циклоалкил-(C1-C4)-алкила-, в другом осуществлении из (C1-C6)-алкила, (C3-C7)-циклоалкила и (C3-C7)-циклоалкил-(C1-C2)-алкила-, в другом осуществлении из (C1-C6)-алкила, (C3-C7)-циклоалкила и (C3-C7)-циклоалкил-CH2-, в другом осуществлении из (C1-C6)-алкила и (C3-C7)-циклоалкила, в другом осуществлении из (C1-C6)-алкила, в другом осуществлении из (C1-C4)-алкила, в другом осуществлении из (C1-C3)-алкила, которые могут необязательно иметь один или более одинаковых или разных заместителей R70, причем в этих группах помимо заместителей R70 могут необязательно присутствовать один или более фтор-заместителей, а в циклоалкильных группах - могут необязательно присутствовать один или более (C1-C4)-алкил-заместителей, применительно к алкильным и циклоалкильным группам в целом. В одном осуществлении настоящего изобретения число заместителей R70, которые могут необязательно присутствовать в группе R54 помимо каких-либо фтор-заместителей, а в случае циклоалкильной группы - каких-либо алкильных заместителей, равно 0, 1 или 2, в другом осуществлении - 0 или 1, в другом осуществлении - 1, в другом осуществлении - 0. В другом осуществлении настоящего изобретения группа R54 не замещается ни R70, ни фтор-заместителями, ни алкильными заместителями - в случае циклоалкильной группы, - и, таким образом, R54 в данном осуществлении выбирают, например, из группы, содержащей (C1-C6)-алкил, (C2-C6)-алкенил, (C2-C6)-алкинил, (C3-C7)-циклоалкил и (C3-C7)-циклоалкил-(C1-C4)-алкил-, или из группы, содержащей (C1-C6)-алкил, (C3-C7)-циклоалкил и (C3-C7)-циклоалкил-CH2-, или из группы, содержащей (C1-C6)-алкил, или из группы, содержащей (C1-C4)-алкил, или из группы, содержащей (C1-C3)-алкил, причем все перечисленные соединения являются незамещенными. В одном осуществлении настоящего изобретения заместители R70, которые могут необязательно присутствовать в группе R54, выбирают, независимо друг от друга, из группы, содержащей HO-, R71-O-, R71-C(O)-O-, HO-C(O)- и R71-O-C(O)-, в другом осуществлении - из группы, содержащей HO-, R71-O- и R71-C(O)-O-, в другом осуществлении - из группы, содержащей HO- и R71-C(O)-O-.
В одном осуществлении настоящего изобретения R56 выбирают из группы, содержащей фенил, который может быть необязательно замещен согласно определению выше или ниже, и незамещенный (C1-C4)-алкил, в другом осуществлении - из группы, содержащей фенил, который может быть необязательно замещен согласно определению выше или ниже, и незамещенный метил, в другом осуществлении - из незамещенного (C1-C4)-алкила, в другом осуществлении - из незамещенного (C1-C3)-алкила. В другом осуществлении R56 является незамещенным метилом, в другом осуществлении - фенилом, который может быть необязательно замещен согласно определению.
В одном осуществлении настоящего изобретения R60 выбирают из группы, содержащей водород и метил. В другом осуществлении R60 представляет собой водород. В другом осуществлении R60 является (C1-C4)-алкилом, например метилом.
В одном осуществлении настоящего изобретения группу R70 во всех случаях выбирают, независимо от групп R70в других случаях, из группы, содержащей HO-, R71-O-, R71-C(O)-O-, R71-S(O)m-, H2N-, R71-NH-, R71-N(R71)-, R71-C(O)-NH-, R71-C(O)-N(R71)-, R71-S(O)2-NH-, R71-S(O)2-N(R71)-, HO-C(O)-, R71-O-C(O)-, H2N-C(O)-, R71-NH-C(O)-, R71-N(R17)-C(O)-, NC-, оксо, фенил и Het2, в другом осуществлении - из группы, содержащей HO-, R71-O-, R71-C(O)-O-, R71-S(O)m-, H2N-, R71-NH-, R71-N(R71)-, R71-C(O)-NH-, R71-S(O)2-NH-, HO-C(O)-, R71-O-C(O)-, H2N-C(O)-, R71-NH-C(O)-, R71-N(R17)-C(O)-, NC-, оксо, фенил и Het2, в другом осуществлении - из группы, содержащей HO-, R71-O-, R71-C(O)-O-, R71-S(O)m-, HO-C(O)-, R71-O-C(O)-, H2N-C(O)-, R71-NH-C(O)-, R71-N(R17)-C(O)-, NC-, оксо, фенил и Het2, в другом осуществлении - из группы, содержащей HO-, R71-O-, R71-C(O)-O-, R71-S(O)m-, H2N-, R71-NH-, R71-N(R71)-, R71-C(O)-NH-, R71-S(O)2-NH-, NC-, оксо, фенил и Het2, в другом осуществлении - из группы, содержащей HO-, R71-O-, R71-C(O)-O-, R71-S(O)m-, NC-, оксо, фенил и Het2, в другом осуществлении - из группы, содержащей HO-, R71-O-, R71-S(O)m-, NC-, оксо, фенил и Het2, в другом осуществлении - из группы, содержащей HO-, R71-O-, R71-C(O)-O-, R71-S(O)m-, NC-, фенил и Het2, в другом осуществлении - из группы, содержащей HO-, R71-O-, NC-, фенил и Het2, в другом осуществлении - из группы, содержащей HO-, R71-O-, фенил и Het2, в другом осуществлении - из группы, содержащей HO-, R71-O- и фенил, в другом осуществлении - из группы, содержащей HO- и R71-O-, в другом осуществлении - из группы, содержащей HO- и R71-C(O)-O-, в другом осуществлении - из группы, содержащей фенил и Het2, в другом осуществлении - из группы, содержащей фенил, в другом осуществлении - из группы, содержащей HO-C(O)-, R71-O-C(O)-, H2N-C(O)-, R71-NH-C(O)-, R71-N(R17)-C(O)-, в другом осуществлении - из группы, содержащей HO-C(O)- и R71-O-C(O)-, и в другом осуществлении R70 является HO-, причем R71, фенил и Het2 определяются и могут быть необязательно замещены согласно определению выше или ниже. В последнем из перечисленных осуществлений, в котором R70 является HO-, (C1-C6)-алкильная группа, например, которая может быть необязательно замещена указанной R70, может, среди прочего, быть такой группой, как (C1-C6)-алкил, HO-(C1-C6)-алкил-, т.е. гидрокси-(C1-C6)-алкил-, (HO)2(C2-C6)-алкил-, т.е. дигидрокси-(C2-C6)-алкил-, и (C1-C4)-алкильной группой, которая может быть необязательно замещена R70 и может, среди прочего, быть такой группой, как (C1-C4)-алкил, HO-(C1-C4)-алкил-, т.е. гидрокси-(C1-C4)-алкил-, (HO)2(C2-C4)-алкил-, т.е. дигидрокси-(C2-C4)-алкил-, причем алкильные группы могут быть необязательно замещены одним или более фтор-заместителями. В одном осуществлении настоящего изобретения атом углерода не присоединен к более чем одной группе HO-.
В одном осуществлении настоящего изобретения R71 выбирают из (C1-C4)-алкила, циклопропила и циклопропил-CH2-, в другом осуществлении - из (C1-C4)-алкила и циклопропила, в другом осуществлении - из (C1-C4)-алкила, в другом осуществлении - из (C1-C3)-алкила, если не указано иное.
Предметом изобретения являются все соединения формулы I, где один или несколько структурных элементов, таких как группы, заместители и числа, определяются как в любом из указанных осуществлений или определений элементов или имеют одно или несколько конкретных значений, приведенных здесь в качестве примеров элементов, причем все комбинации одного или нескольких указанных осуществлений и (или) определений и (или) конкретных значений элементов являются предметом настоящего изобретения. Также в отношении всех таких соединений формулы I все их стереоизомерные формы и сочетания стереоизомерных форм в любом соотношении и их физиологически приемлемые соли и физиологически приемлемые сольваты любого из них являются предметом настоящего изобретения.
Аналогичным образом, также в отношении всех конкретных соединений, раскрываемых в настоящем изобретении, как, например, образцы соединений, которые представляют осуществления изобретения, в которых различные группы и числа в общем определении соединений формулы I имеют конкретные значения, присутствующие в соответствующем конкретном соединении, подразумевается, что все их стереоизомерные формы и сочетания стереоизомерных форм в любом соотношении и их физиологически приемлемые соли и физиологически приемлемые сольваты любого из них являются предметом настоящего изобретения. Предметом изобретения также являются все конкретные соединения, раскрываемые в настоящем изобретении, независимо от того, представлены ли они как свободное соединение и (или) конкретная соль, как в виде свободного соединения, так и в виде всех его физиологически приемлемых солей, и если раскрывается конкретная соль, то дополнительно в виде этой конкретной соли и физиологически приемлемых сольватов любой из них. Например, в случае конкретного соединения 2-{2-хлор-5-[2-(3-хлорфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновой кислоты, которая представлена в виде свободного соединения, предметом изобретения являются 2-{2-хлор-5-[2-(3-хлорфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота и ее физиологически приемлемые соли и физиологически приемлемые сольваты любой из них.
Таким образом, предметом настоящего изобретения также является соединение формулы I, которое выбирает из любых конкретных соединений формулы I, которые раскрываются в настоящем изобретении, или любое из конкретных соединений формулы I, которые раскрываются в настоящем изобретении, независимо от того, представлены ли они как свободное соединение и (или) конкретная соль, например соединение формулы I, которое выбирается из
2-[4-метилсульфанил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-ацетил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-этил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-этокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-{4-метокси-3-[2-(3-трифторметилсульфанилфенил)-этокси]-бензоиламино}-индан-2-карбоновой кислоты,
2-[4-метокси-3-(1-м-толилциклопропилметокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-{3-[2-(3-цианофенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновой кислоты,
5-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-5,6-дигидро-4H-циклопента[c]тиофен-5-карбоновой кислоты,
5-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-5,6-дигидро-4H-циклопента[b]тиофен-5-карбоновой кислоты,
2-{[5-ацетил-4-(2-м-толилэтокси)-тиофен-2-карбонил]-амино}-индан-2-карбоновой кислоты,
2-[3-фтор-4-метокси-5-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-метокси-3-(3-м-толилпропил)-бензоиламино]-индан-2-карбоновой кислоты,
5-фтор-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-5,6-диметилиндан-2-карбоновой кислоты,
2-[4-циано-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-{3-[2-(3-хлорфенил)-этокси]-4-метилбензоиламино}-индан-2-карбоновой кислоты,
2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[3-(2-м-толилэтокси)-4-трифторметилбензоиламино]-индан-2-карбоновой кислоты,
2-{3-[2-(2-фтор-5-метилфенил)-этокси]-4-трифторметилбензоиламино}-индан-2-карбоновой кислоты,
2-(3-{2-[3-(2-гидроксиэтил)-фенил]-этокси}-4-метоксибензоиламино}-индан-2-карбоновой кислоты,
2-{[6-метокси-5-(2-м-толилэтокси)-пиридин-3-карбонил]-амино}-индан-2-карбоновой кислоты,
2-[(3'-метансульфониламино-6-метоксибифенил-3-карбонил)-амино]-индан-2-карбоновой кислоты,
2-[(3'-диметиламиносульфониламино-6-метоксибифенил-3-карбонил)-амино]-индан-2-карбоновой кислоты,
2-[(6-метокси-3'-трифторметоксибифенил-3-карбонил)-амино]-индан-2-карбоновой кислоты,
2-[(3'-цианометил-6-метоксибифенил-3-карбонил)-амино]-индан-2-карбоновой кислоты,
2-[(3'-изопропил-6-метоксибифенил-3-карбонил)-амино]-индан-2-карбоновой кислоты,
2-[(3'-хлор-6-метокси-2'-метилбифенил-3-карбонил)-амино]-индан-2-карбоновой кислоты,
2-{[5-(3-хлорфенил)-6-метоксипиридин-3-карбонил]-амино}-индан-2-карбоновой кислоты, и
2-[3-(2,2-дифтор-2-фенилэтокси]-4-метоксибензоиламино}-индан-2-карбоновой кислоты,
или которое является любым из этих соединений или их физиологически приемлемой солью или физиологически приемлемым сольватом любого из них, где соединение формулы I является предметом настоящего изобретения в любой из его стереоизомерных форм или сочетании стереоизомерных форм в любом соотношении, где это применимо.
В качестве примера соединений настоящего изобретения, которые в отношении любых структурных элементов определяются как в указанных осуществлениях настоящего изобретения или в определениях таких элементов, могут быть приведены соединения формулы I, в которых
кольцо A является циклогексановым кольцом, бензольным кольцом, пиридиновым кольцом, пиридазиновым кольцом или тиофеновым кольцом, причем циклогексановое кольцо может необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей фтор и (C1-C4)-алкил, а бензольное, пиридиновое, пиридазиновое и тиофеновое кольца могут необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген, R1, HO-, R1-O-, R1-C(O)-O-, R1-S(O)2-O-, R1-S(O)m-, H2N-, R1-NH-, R1-N(R1)-, R1-C(O)-NH-, R1-C(O)-N(R71)-, R1-S(O)2-NH-, R1-S(O)2-N(R71)-, R1-C(O)-, HO-C(O)-, R1-O-C(O)-, H2N-C(O)-, R1-NH-C(O)-, R1-N(R1)-C(O)-, H2N-S(O)2-, R1-NH-S(O)2-, R1-N(R1)-S(O)2-, NC- и O2N-;
Y выбирают из группы, содержащей S, C(R12)=C(R13) и C(R15)=N;
Z является C(R16);
и все другие числа и группы определены как в общем определении соединений формулы I или в любых указанных осуществлениях настоящего изобретения или в определениях структурных элементов, в любой из их стереоизомерных форм или сочетании стереоизомерных форм в любом соотношении, и их физиологически приемлемых солях и физиологически приемлемых сольватах любой из них.
В качестве дополнительного подобного примера могут быть приведены соединения формулы I, в которых кольцо A является бензольным кольцом, пиридиновым кольцом, пиразиновым кольцом или тиофеновым кольцом, которые могут необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген, (C1-C4)-алкил и (C1-C4)-алкил-O-;
Y выбирают из группы, содержащей S, C(R12)=C(R13) и C(R15)=N;
Z является C(R16);
R3 и R5 выбирают, независимо друг от друга, из группы, содержащей водород и (C1-C4)-алкил;
R4 и R6 - водород;
R12, R13, R15 и R16 выбирают, независимо друг от друга, из группы, содержащей водород, галоген, (C1-C4)-алкил, (C1-C4)-алкил-O- и NC-;
R20 - водород;
и все другие числа и группы определены как в общем определении соединений формулы I или в любых указанных осуществлениях настоящего изобретения или в определениях структурных элементов, в любой из их стереоизомерных форм или сочетании стереоизомерных форм в любом соотношении, и их физиологически приемлемых солях и физиологически приемлемых сольватах любой из них.
В качестве дополнительного подобного примера могут быть приведены соединения формулы I, в которых R21 выбирают из группы, содержащей водород, галоген, (C1-C4)-алкил, HO-(C1-C4)-алкил-, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, H2N-, (C1-C4)-алкил-NH-, ди((C1-C4)-алкил)N-,(C1-C4)-алкил-C(O)- и NC-;
R22 является группой формулы II;

R23 представляет собой прямую связь или цепь, содержащую 2, 3 или 4 члена цепи, из которых 0 или 1 член цепи являются гетерочленами, выбранных из группы, содержащей N(R25), O, S, S(O) и S(O)2, и другие члены цепи являются одинаковыми или разными группами C(R26)(R26), где две соседние группы C(R26)(R26) могут соединяться друг с другом двойной или тройной связью;
и все другие числа и группы определены как в общем определении соединений формулы I или в любых указанных осуществлениях настоящего изобретения или в определениях структурных элементов, в любой из их стереоизомерных форм или сочетании стереоизомерных форм в любом соотношении, и их физиологически приемлемых солях и физиологически приемлемых сольватах любой из них.
В качестве дополнительного подобного примера могут быть приведены соединения формулы I, в которых R24 - моноциклическое кольцо с числом членов от 3 до 7 или бициклическое кольцо с числом членов от 7 до 10, которое является насыщенным или ненасыщенным и содержит 0, 1 или 2 одинаковых или разных гетерочлена, выбранных из группы, содержащей N, N(R32), O, S, S(O) и S(O)2, причем кольца могут необязательно иметь на атомах углерода кольца один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген, R33, HO-, R33-O-, R33-S(O)m-, H2N-, R33-NH-, R33-N(R33)-, R33-C(O)-NH-, R33-C(O)-N(R71)-, R33-S(O)2-NH-, R33-S(O)2-N(R71)-, H2N-S(O)2-NH-, R33-NH-S(O)2-NH-, R33-N(R33)-S(O)2-NH-, H2N-S(O)2-N(R71)-, R33-NH-S(O)2-N(R71)-, R33-N(R33)-S(O)2-N(R71)-, HO-C(O)-, R33-O-C(O)-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)-, NC-, оксо, фенил и Het;
R32 выбирают из группы, содержащей водород, R35, R35-C(O)-, R35-O-C(O)- и фенил;
и все другие числа и группы определены как в общем определении соединений формулы I или в любых указанных осуществлениях настоящего изобретения или в определениях структурных элементов, в любой из их стереоизомерных форм или сочетании стереоизомерных форм в любом соотношении, и их физиологически приемлемых солях и физиологически приемлемых сольватах любой из них.
В качестве дополнительного подобного примера могут быть приведены соединения формулы I, в которых кольцо A является бензольным кольцом, пиридиновым кольцом, пиразиновым кольцом или тиофеновым кольцом, которые могут необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген, (C1-C4)-алкил и (C1-C4)-алкил-O-;
Y выбирают из группы, содержащей S, C(R12)=C(R13) и C(R15)=N;
Z является C(R16);
R3 и R5 выбирают, независимо друг от друга, из группы, содержащей водород и (C1-C4)-алкил;
R4 и R6 - водород;
R12, R13, R15 и R16 выбирают, независимо друг от друга, из группы, содержащей водород, галоген, (C1-C4)-алкил, (C1-C4)-алкил-O- и NC-;
R20 - водород;
R21 выбирают из группы, содержащей водород, галоген, (C1-C4)-алкил, HO-(C1-C4)-алкил-, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, H2N-, (C1-C4)-алкил-NH-, ди((C1-C4)-алкил)N-,(C1-C4)-алкил-C(O)- и NC-;
R22 является группой формулы II;
R23 представляет собой прямую связь или цепь, содержащую 2, 3 или 4 члена цепи, из которых 0 или 1 член цепи являются гетерочленами, выбранных из группы, содержащей N(R25), O, S, S(O) и S(O)2, и другие члены цепи являются одинаковыми или разными группами C(R26)(R26), где две соседние группы C(R26)(R26) могут соединяться друг с другом двойной или тройной связью;
R24 - моноциклическое кольцо с числом членов от 3 до 7 или бициклическое кольцо с числом членов от 7 до 10, которое является насыщенным или ненасыщенным и содержит 0, 1 или 2 одинаковых или разных гетерочлена, выбранных из группы, содержащей N, N(R32), O, S, S(O) и S(O)2, причем кольца могут необязательно иметь на атомах углерода кольца один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген, R33, HO-, R33-O-, R33-S(O)m-, H2N-, R33-NH-, R33-N(R33)-, R33-C(O)-NH-, R33-C(O)-N(R71)-, R33-S(O)2-NH-, R33-S(O)2-N(R71)-, HO-C(O)-, R33-O-C(O)-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)-, NC-, оксо, фенил и Het;
R32 выбирают из группы, содержащей водород, R35, R35-C(O)-, R35-O-C(O)- и фенил;
и все другие числа и группы определены как в общем определении соединений формулы I или в любых указанных осуществлениях настоящего изобретения или в определениях структурных элементов, в любой из их стереоизомерных форм или сочетании стереоизомерных форм в любом соотношении, и их физиологически приемлемых солях и физиологически приемлемых сольватах любой из них.
В качестве дополнительного подобного примера могут быть приведены соединения формулы I, в которых кольцо A является бензольным кольцом, которое может необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген, (C1-C4)-алкил и (C1-C4)-алкил-O-;
Y является C(R12)=C(R13);
Z является C(R16);
R3, R4, R5 и R6 - водород;
R12, R13 и R16 выбирают, независимо друг от друга, из группы, содержащей водород, галоген, (C1-C4)-алкил, (C1-C4)-алкил-O- и NC-;
R20 - водород;
R21 выбирают из группы, содержащей водород, галоген, (C1-C4)-алкил, HO-(C1-C4)-алкил-, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, (C1-C4)-алкил-C(O)- и NC-;
R22 является группой формулы II;
R23 представляет собой прямую связь или цепь, содержащую 2, 3 или 4 члена цепи, из которых 0 или 1 член цепи являются гетерочленами, выбранных из группы, содержащей N(R25), O, S, S(O) и S(O)2, и другие члены цепи являются одинаковыми или разными группами C(R26)(R26);
R24 является бензольным кольцом, которое может необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген, R33, HO-, R33-O-, R33-S(O)m-, H2N-, R33-NH-, R33-N(R33)-, R33-C(O)-NH-, R33-S(O)2-NH-, HO-C(O)-, R33-O-C(O)-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-C(O)- и NC-;
при условии, что общее число атомов C, N, O и S, присутствующих в двух группах R23 и R24, составляет не менее 5;
R25 выбирают из группы, содержащей водород и (C1-C4)-алкил;
R26, независимо от каждой другой группы R26, выбирают из группы, содержащей водород, фтор, (C1-C4)-алкил и HO-, или две группы R26, присоединенные к одному и тому же атому углерода в цепи, вместе с атомом углерода, к которому они присоединены, образуют циклопропановое кольцо;
R33 выбирают, независимо от каждой другой группы R33, из группы, содержащей (C1-C4)-алкил, (C3-C7)-циклоалкил и (C3-C7)-циклоалкил-(C1-C2)-алкил-, причем все они могут необязательно иметь один или более одинаковых или разных заместителей R70;
R50 выбирают из группы, содержащей R51-O- и R52-N(R53)-;
R51 выбирают из группы, содержащей водород и (C1-C4)-алкил;
R52 выбирают из группы, содержащей водород и (C1-C4)-алкил;
R53 выбирают из группы, содержащей водород и (C1-C4)-алкил;
R70 выбирают из группы, содержащей HO- и R71-O-;
R71 являют (C1-C4)-алкил;
m, независимо от каждого другого числа m, является целым числом, выбранным из группы, содержащей 0 и 2;
циклоалкил, независимо от каждой другой группы циклоалкила и независимо от других заместителей на циклоалкиле, может необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей фтор и (C1-C4)-алкил;
алкил, независимо от каждой другой группы алкила и независимо от других заместителей на алкиле, может необязательно иметь один или более фтор-заместителей в любой из их стереоизомерных форм или сочетании стереоизомерных форм в любом соотношении, и их физиологически приемлемых солях и физиологически приемлемых сольватах любой из них.
Еще одним предметом настоящего изобретения являются способы для получения соединений формулы I, которые изложены ниже и при помощи которых могут быть получены описываемые соединения. Например, соединения формулы I могут быть получены путем взаимодействия соединения формулы III с соединением формулы IV с образованием амидной связи.

Кольцо A и группы Y, Z, R3 по R6, R20 по R22 и R50 в соединениях формул III и IV определены как в соединениях формулы I, а также могут присутствовать функциональные группы - в защищенной форме или в форме группы-предшественника, которая впоследствии преобразуется в конечную группу. Группа G в соединениях формулы IV может быть HO-(гидрокси), т.е. соединение формулы IV может, таким образом, быть карбоновой кислотой, или другой группой, которая может быть замещена группой N (R20) в соединении формулы III в реакции замещения, например группой арилокси, такой как необязательно замещенная группа фенокси или алкилокси, такой как группа (C1-C4)-алкил-O-, например группа (C1-C3)-алкил-O-, такой как метокси или этокси, или галоген, например хлор или бром, и соединение формулы IV может, таким образом, быть реакционно-способным эфиром, таким как ариловый эфир или алкиловый эфир, например метиловый эфир или этиловый эфир, или галоидангидридом, например хлорангидридом или бромангидридом, соответствующей карбоновой кислоты. Также можно использовать соединение формулы III и (или) соединение формулы IV, и полученные соединения формулы I, в виде соли, например соли присоединения кислоты, такой как гидрогалид, например гидрохлорид, соединения формулы III, и (или) соль щелочных металлов, например соль натрия, соединения формулы IV, в которой G является HO-. Аналогичным образом, во всех других реакциях получения соединений формулы I, включая получение исходных соединений, также могут быть использованы соединения и (или) продукты получаться в виде соли.
При использовании соединения формулы IV, в котором G является HO-, группа карбоновой кислоты HO-C(O)-, как правило, активируется in situ при помощи стандартного связывающего амида или преобразуется в производное реактивной карбоновой кислоты, которое может быть получено in situ или выделено. Например, соединение формулы IV, в котором G является HO-, может быть преобразовано в галоидангидрид, например соединение формулы IV, в которой G является Cl или Br, путем обработки тионилхлоридом, пентахлоридом фосфора, трибромидом фосфора или оксалилхлоридом, или путем обработки алкилхлорформиатом, таким как хлорформиат этила или хлорформиат изобутила, для получения смешанного ангидрида. В качестве стандартных связующих реагентов могут использоваться пропанфосфоновый ангидрид, N,N'-карбонилдиазолы, такие как N,N'-карбонилдиимидазол (CDI), карбодиимиды, такие как 1,3-диизопропилкарбодиимид (DIC), 1,3-дициклогексилкарбодиимид (DCC) или гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC), карбодиимиды вместе с такими добавками, как 1-гидроксибензотриазол (HOBT) или 1-гидрокси-7-азабензотриазол (HOAT), связующие реагенты на основе урония, такие как O-(7-азабензотриазол -1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HBTU) или O-(циано(этоксикарбонил)метиленамино)-N,N,N',N'-тетраметилуроний тетрафторбората (TOTU), и связующие реагенты на основе фосфония, такие как (бензотриазол-1-илокси)трис(диметиламино)фосфоний гексафторфосфата (BOP), (бензотриазол-1-илокси)трипирролидинфосфоний гексафторфосфата (PyBOP) или бромтрипирролидинфосфоний гексафторфосфата (PyBroP).
Условия проведения реакции для получения соединения формулы I из соединений формул III и IV зависят от особенностей каждого конкретного случая, например группы G или применяемого агента реакции сочетания, и хорошо известны квалифицированному специалисту, имеющему общее представление о данной области. Например, в случае если соединение формулы IV, в котором G представляет собой алкил-O-, например метокси или этокси, реагирует с соединением формулы III, реакция, как правило, проводится в инертном растворителе, например углеводороде или хлорированном углеводороде, например, бензоле, толуоле, ксилоле, хлорбензоле, дихлорметане, хлороформе или дихлорэтане, в эфире, например тетрагидрофуране (THF), диоксане, дибутиловом эфире, диизопропиловом эфире или диметоксиэтане (DME), или в смеси растворителей при повышенных температурах, например при температурах от примерно 40°C до примерно 140°C, в частности при температурах от примерно 50°C до примерно 120°C, например примерно при температуре кипения растворителя. В случае если соединение формулы IV, в котором G представляет собой галоген, например хлор или бром, вступает в реакцию с соединением формулы III, реакция, как правило, также проводится в инертном растворителе, например углеводороде или хлорированном углеводороде, или эфире, подобно приведенному выше случаю, сложном эфире, например этилацетате или бутилацетате, нитриле, например ацетонитриле, или в воде, или в смеси растворителей, в том числе смеси воды с органическим растворителем, которые смешивается или не смешивается с водой, при температурах от примерно -10°C до примерно 100°C, в частности, при температурах от примерно 0°C до примерно 80°C, например примерно при комнатной температуре. Предпочтительно проводить реакцию соединения формулы IV, в котором G представляет собой галоген, с соединением формулы III в присутствии основания, например третичного амина, например триэтиламина, этилдиизопропиламина, N-метилморфолина или пиридина, или неорганического основания, например гидроксида, карбоната, гидрокарбоната щелочного металла, например гидроксида натрия, гидроксида калия, карбоната натрия или гидрокарбоната натрия.
В случае если соединение формулы IV, в котором G - HO-, реагирует с соединением формулы III, а карбоксильная группа активируется посредством амидного агента сочетания, например карбодиимидом или TOTU, реакция обычно проводится в безводной среде в инертном апротонном растворителе, например эфире, например THF, диоксане или DME, амиде, например N,N-диметилформамиде (DMF) или N-метилпирролидоне (NMP), при температурах от примерно -10°C до примерно 40°C, в частности при температурах от примерно 0°C до примерно 30°C в присутствии основания, например третичного амина, например триэтиламина, этилдиизопропиламина или N-метилморфолина. В случае если соединение формулы III используется в форме соли присоединения кислоты в реакции с соединением формулы IV, как правило, добавляются достаточные количества основания, чтобы выделить свободное соединение формулы III.
Как отмечалось выше, в процессе образования амидной связи между соединениями формул III и IV функциональные группы в соединениях формул III и IV могут присутствовать в защищенной форме или в форме группы-предшественника. В зависимости от особенностей конкретной ситуации, возможно, будет необходимо или целесообразно, во избежание нежелательного течения реакции или побочных реакций, временно блокировать любые функциональные группы защитными группы, а позже удалить их, или же оставить функциональные группы в форме групп-предшественников, которые впоследствии трансформируются в желаемую конечную группу. Это соответствующим образом относится ко всем реакциям в процессе синтеза соединений формулы I, в том числе описанного ниже синтеза промежуточных соединений, а также синтеза исходных соединений и структурных элементов. Соответствующие стратегии синтеза широко используются в области техники. Подробную информацию о защитных группах и их введении и удалении можно найти, например, в P. G. M. Wuts и T. W. Greene, Greene's Protective Groups in Organic Synthesis, 4. ed. (2007), John Wiley & Sons. К примерам защитных групп, которые могут быть здесь указанны, относятся бензильные защитные группы, которые могут существовать в форме бензиловых эфиров гидроксигрупп, а также бензиловых эфиров карбоновых кислот, из которых бензильную группу можно удалить каталитическим гидрированием в присутствии палладиевого катализатора, трет-бутильные защитные группы, которые могут существовать в форме трет-бутиловых эфиров карбоновых кислот, из которых трет-бутильную группу можно удалить обработкой трифторуксусной кислотой, ацильные защитные группы, которые могут использоваться для защиты гидроксильных групп и аминогрупп в форме эфиров и амидов и которые могут расщепляться посредством кислотного или щелочного гидролиза, а также алкилоксикарбонильные защитные группы, которые могут существовать в форме трет-бутоксикарбонильных производных аминогрупп и которые могут расщепляться обработкой трифторуксусной кислотой. Нежелательных реакций групп, например, карбоксильных групп, присутствующих в соединении формулы III, в случае если R50 представляет собой HO-, можно также избежать, если использовать их в реакции соединений формул III и IV в форме других эфиров, например в форме алкиловых эфиров, например метилового или этилового эфира, которые могут расщепляться посредством гидролиза, например в присутствии гидроксида щелочного металла, например гидроксида натрия или гидроксида лития. К примерам групп-предшественников, которые могут быть здесь указанны, относятся нитрогруппы, которые могут быть трансформированы в аминогруппы в процессе каталитического гидрирования или восстановлением дитионитом натрия, например, а также цианогруппы (NC-, N≡C-), которые могут быть преобразованы в карбоксамидные группы и карбоксильные кислые группы в процессе гидролиза. Другим примером группы-предшественника является оксогруппа, которая представляет группы R3 и R4 вместе, или две группы R5 и R6 вместе, и которая может изначально присутствовать в процессе синтеза соединений формулы I, в которых R3 или R5 представляют собой гидроксил. В одном из подходов к синтезу таких соединений формулы I соединение формулы III, в котором группы R3 и R4вместе являются оксо-группами или же группы R5 и R6 вместе являются оксо-группами, может быть получено из соответствующего соединения, которое содержит атом брома вместо группы R20-NH-, по реакции с азидом натрия с последующим взаимодействием с трибутилоловогидридом, как это описано в L. Benati et al., J. Org. Chem. 64 (1999), 7836-7841, при этом полученное аминосоединение вступает в реакцию с соединением формулы IV, оксо-группа восстанавливается, например комплексным гидридом, например боргидридом натрия, или реагирует с металлоорганическим соединением, например реактивом Гриньяра, и наконец, удаляются все защитные группы. Если в соединениях формул III и IV присутствуют какие-либо защитные группы или группы-предшественники, и непосредственный продукт реакции соединений формул III и IV не является конечным указанным в заголовке соединением, то удаление защитной группы или трансформация в указанное в заголовке соединение также, как правило, может проводиться in situ.
Соединения формулы III имеются в продаже или же могут быть получены в соответствии или по аналогии с методами, описанными в литературе, например посредством диалкилирования производного аминоуксусной кислоты формулы VI соединением формулы V, как это описано, например, в работе Kotha et al., J. Org. Chem. 65 (2000), 1359-1365.

Кольцо A и группы R3 по R6 в соединении формулы V определены как в соединениях формулы I, а также могут присутствовать функциональные группы - в защищенной форме или в форме группы-предшественника, которая впоследствии преобразуется в конечную группу. Группы L1 в алкилирующем соединении формулы V являются уходящими группами, такими как галоген, например хлор или бром, или группами сульфонилокси, например метансульфонилокси или трифторметансульфонилокси. Группа PG1 в соединении формулы VI является защитной группой карбоксильной группы аминоуксусной кислоты и может быть такой группой, как (C1-C4)-алкил, например метил, этил или трет-бутил, или бензил. Группа PG2 в соединении формулы VI является двухвалентной защитной группой аминогруппы аминоуксусной кислоты и может быть атомом углерода, и группа -N=PG2, таким образом, может быть изоциановой группой -N=C, или атомом углерода, содержащем две фенильные группы, и группа -N=PG2, таким образом, может быть бензгидрилиденаминовой группой, например -N=C(фенил)2. Реакция алкилирования соединения формулы VI с соединением формулы V проводится в присутствии основания, например алкоксида щелочного металла, например трет-бутоксида калия, или же гидрида щелочного металла, например гидрида натрия, или же карбоната щелочного металла, например карбоната калия, с добавлением катализатора межфазового переноса, например, гидросульфат тетрабутиламмония, в условиях межфазового переноса твердое-жидкость, в инертном растворителе, например подобном амиду DMF или NMP, или подобном нитрилу ацетонитриле, при температурах от примерно -40°C до примерно 80°C, в зависимости от особенностей каждого конкретного случая. После алкилирования защитная группа PG2 отщепляется, например обработкой хлористоводородной кислотой в этаноле в случае изоцианогруппы или водной хлористоводородной кислотой в случае бензгидрилиденаминогруппы, с возможным сопутствующим отщеплением защитной группы PG1, с тем чтобы получить соединение формулы III, в которой R50 является (C1-C4)-алкил-O-, например метокси, этокси или трет-бутокси, или бензилокси, или HO-, а R20 является водородом. Соединения формулы III, в которых R20 не является водородом, могут получаться из соединений, в которых R20 является водородом, посредством алкилирования или ацилирования и последующего восставновления полученного амида до амина. При желании соединения формулы III, в которых R50 является HO-, могут быть получены посредством кислого или основного гидролиза из соединений, в которых, например, R50 является (C1-C4)-алкил-O-, или гидрированием из соединений, в которых R50 представляет собой бензилокси.
Исходные соединения формулы V могут быть получены из соответствующих дигидроксисоединений, которые содержат гидроксигруппы вместо групп L1, обработкой галогенирующим агентом, например тионилхлоридом или трибромидом фосфора, или же сульфонилирующим агентом, например метансульфонилхлоридом или трифторметансульфоновым ангидридом, или из соответствующих углеводородов, которые содержат атомы водорода вместо групп L1, посредством бензильного бромирования, например при действии N-бромсукцинимида. Указанные дигидроксисоединения могут быть получены из соответствующих дикарбоновых кислот, которые содержат карбоксильные группы HO-C(O) вместо групп L1-C(R3)(R4)- и L1-C(R5)(R6)-, или этерифицированные карбоксильные группы, посредством восстановления, например, литийалюминийгидридом, в случае если все группы R3 по R6 являются атомами водорода, или в реакции с металлорганическим соединением, например реактивом Гриньяра, или литийорганическим соединением, например метиллитием, а также, в одном из вариантов, посредством восстановления, если группы R3 по R6 не являются атомами водорода. Соединения формулы III, в которых A является циклоалкановым кольцом, могут также получаться гидрированием в присутствии катализатора гидрирования из группы переходных металлов, например платинового катализатора, например из соответствующих соединений, в которых кольцо А является ненасыщенным циклом, в частности в случае соединений формулы III, в которых A - циклогексановое кольцо, которые могут получаться из соответствующих соединений, в которых А - бензольное кольцо. В другом подходе соединения формулы III могут получаться из соответствующих кетонов, то есть соединений формулы III, в которых две группы R20-NH- и R50-C(O)- заменены на оксогруппу, в соответствии с классическими методами синтеза аминокислот, например реакция Штрекера или реакция Бухерера-Бергса. Все перечисленные реакции являются стандартными реакциями, которые хорошо известны специалистам в области.
Соединения формулы IV также имеются в продаже или же могут быть получены в соответствии или по аналогии с методами, описанными в литературе. Традиционно, в методах синтеза для получения соединений формулы IV получают соединения, в которых группа G в соединениях формулы IV является группой, подобной (C1-C4)-алкил-O-, а группа G-C(O)-, таким образом представляет собой (C1-C4)-алкил эфирную группу, или группа G-C(O)- представляет собой любую другую эфирную группу, например бензиловый эфир фенил-CH2-O-C(O)-, а группа G, таким образом, является бензилоксигруппой. Соединения формулы IV, в которых G представляет собой HO-, могут быть получены из таких соединений формулы IV посредством кислого или основного гидролиза алкиловых эфиров, или гидрированием бензиловых эфиров в стандартных условиях. Соединения формулы IV, в которых G представляет собой HO-, могут затем трансформироваться в соединения формулы IV, в которых G представляет собой галоген, как это уже разъснялось выше, каковые указанные последними соединения могут трансформироваться в соединения, в которых G является арилокси, например посредством реакции с гидроксиаренами, например фенолом. Ниже приводятся примеры различных методов синтеза для получения соединений формулы IV, в которых группа R23 в группе R24-R23-, то есть в группе формулы II, которая представляет одну из групп R21 и R22, имеет различные значения.
В методе получения соединений формулы IV, в которых группа R23 представляет собой член терминальной цепи, связанный с кольцом, содержащим группы Y и Z, является гетерочленом, соединение формулы VII реагирует с соединением формулы VIII, с получением соединения формулы IVa.

В соединениях формул IVa, VII и VIII группы Y, Z и R24 определяются как в соединениях формулы I. Группу R80 выбирают из группы, содержащей водород, галоген, R30, HO-, R30-O-, R30-C(O)-O-, R30-S(O)2-O-, R30-S(O)m-, H2N-, R30-NH-, R30-N(R30)-, R30-C(O)-NH-, R30-C(O)-N(R71)-, R30-S(O)2-NH-, R30-S(O)2-N(R71)-, R30-C(O)-, HO-C(O)-, R30-O-C(O)-, H2N-C(O)-, R30-NH-C(O)-, R30-N(R30)-C(O)-, H2N-S(O)2-, R30-NH-S(O)2-, R30-N(R30)-S(O)2-, NC-, O2N- и Het1, т.е. имеет значение той группы R21или R22 в соединениях формулы I, которая не является группой формулы II. Также функциональные группы в соединениях формул IVa, VII и VIII могут присутствовать в защищенной форме или в форме группы-предшественника, которая впоследствии преобразуется в конечную группу. Группа G1-C(O)- является эфирной группой, и группа G1 является группой, такой как (C1-C4)-алкил-О- или бензилокси. Группа X является гетерочленом, как указано в определении R23, то есть группой, выбранной из группы, содержащей N(R25), O, S, S(O) и S(O)2, в частности, из группы, содержащей N(R25), О и S. Группы R23a и X вместе представляют группу R23, как указано выше, в которой терминальный член цепи, являющийся гетерочленом, связан с кольцом, содержащим группы Y и Z. Таким образом, R23 представляет собой прямую связь или цепь, содержащую от 1 до 4 членов цепи, из которых 0 или 1 член цепи являются гетерочленами, выбранными из группы, содержащей N(R25), O, S, S(O) и S(O)2, при условии, что терминальный член цепи, прилегающий к группе L2, может быть только гетерочленом, что приводит к образованию соединения формулы IVa, в котором одну из групп Х и указанный терминальный член цепи выбирают из группы, содержащей S(O) и S(O)2, а другую группу выбирают из группы, содержащей N(R25), O и S, тогда как другие члены цепи являются одинаковыми или разными группами C(R26)(R26), где две соседние группы C(R26)(R26) могут соединяться друг с другом двойной или тройной связью. В соответствии с обозначениями связей, соединяющих группы R80 и XH в соединениях формулы VII, а также группы R80 и X-R23a-R24 в соединениях формулы IVa, не связанные с конкретным углеродным атомом кольца, каждая из указанных двух групп может располагаться в любом из двух положений участка C=C кольца, содержащего группы Y и Z, что показано в формуле. То есть R80 может находиться при кольцевом углероде, который соседствует к группой Y и второй группе при кольцевом атоме углерода, который соседствует с группой Z, а также R80 может быть при кольцевом атоме углерода, который соседствует с группой Z и второй группе при кольцевом атоме углерода, который соседствует с группой Y. Это относится ко всем определяемым ниже соединениям, содержащим группу R80 и вторую группу, в котором связи соединяющие группу с кольцом, содержащим группы Y и Z, не являются связями с конкретными атомами углерода кольца. Группа L2 в соединениях формулы VIII является уходящей группой, которая может быть заменена группой X, такой как галоген, например хлор или бром, группой сульфонилокси, такой как, например, метансульфонилокси, трифторметансульфонилокси или толуол-4-сульфонилокси, или гидрокси.
Реакция соединения формулы VII с соединением формулы VIII представляет собой реакцию нуклеофильного замещения и может проводиться в стандартных условиях для таких реакций, которые хорошо известны специалистам в данной области. Реакция, как правило, проводится в инертном растворителе, например углеводороде или хлорированном углеводороде, например бензоле, толуоле, ксилоле, хлорбензоле, дихлорметане, хлороформе или дихлорэтане, в эфире, например тетрагидрофуране (THF), диоксане, дибутиловом эфире, диизопропиловом эфире или DME, в спирте, например метаноле, этаноле или изопропаноле, в кетоне, например ацетоне или бутан-2-оне, в сложном эфире, например этилацетате или бутилацетате, нитриле, например ацетонитриле, амиде, например DMF или NMP, в сульфоксиде, например DMSO, или в сульфоне, например сульфолане, или в смеси растворителей при температурах от примерно -10°C до примерно 120°C, в частности при температурах от примерно 0°C до примерно 100°C в зависимости от особенностей конкретного случая. Во многих случаях для усиления нуклеофильности соединения формулы VII и (или) связывания кислоты, которая высвобождается во время реакции, предпочтительно добавлять основание, например третичный амин, например триэтиламин, этилдиизопропиламин или N-метилморфолин, или неорганическое основание, например гидрид, гидроксид, карбонат или гидрокарбонат щелочного металла, например гидрид натрия, гидроксид натрия, гидроксид калия, карбонат натрия, карбонат калия, карбонат цезия или гидрокарбонат натрия, или же алкоксид или амид, например метоксид натрия, этоксид натрия, метоксид калия, трет-бутоксид калия, амид натрия или диизопропиламид лития. Соединение формулы VII может также обрабатываться основанием и преобразовываться в соль на отдельной стадии. Соединения формулы VIII, в которых группа L2 представляет собой гидрокси, могут с готовностью реагировать с соединениями формулы VII в условиях реакции Мицунобу в присутствии азодикарбоксилата, например диэтилазодикарбоксилата или диизопропилазодикарбоксилата, и фосфина, например трифенилфосфина или трибутилфосфина, в инертном апротонном растворителе, например эфире, таком как THF или диоксан (cf. O. Mitsunobu, Synthesis (1981), 1-28).
В рамках другого метода можно получить соединения формулы IVa в реакции соединения формулы IX с соединением формулы X.

В соединениях формул IX и X группы Y, Z и R24 определяются как в соединениях формулы I. Группа R80 определяется как в соединениях формул IVa и VII, т. е. имеет значение той группы R21или R22 в соединениях формулы I, которая не является группой формулы II. Также функциональные группы в соединениях формул IX и X могут присутствовать в защищенной форме или в форме группы-предшественника, которая впоследствии преобразуется в конечную группу. Группа G1-C(O)- является эфирной группой, и группа G1 - группой, такой как (C1-C4)-алкил-О- или бензилокси. Группа X является гетерочленом, как указано в определении R23, то есть группой, выбранной из группы, содержащей N(R25), O, S, S(O) и S(O)2, в частности, из группы, содержащей N(R25), О и S. В соединении формулы X группы R23a и X вместе представляют группу R23, как указано выше, в которой терминальный член цепи, являющийся гетерочленом, связан с кольцом, содержащим группы Y и Z, в полученных соединениях формулы IVa. Таким образом, R23a представляет собой прямую связь или цепь, содержащую от 1 до 4 членов цепи, из которых 0 или 1 член цепи являются гетерочленом, выбранными из группы, содержащей N(R25), O, S, S(O) и S(O)2, при условии, что терминальный член цепи, прилегающий к группе X, может быть гетерочленом, только если одну из групп Х и указанный терминальный член цепи выбирают из группы, содержащей S(O) и S(O)2, а другую группу выбирают из группы, содержащей N(R25), O и S, тогда как другие члены цепи являются одинаковыми или разными группами C(R26)(R26), где две соседние группы C(R26)(R26) могут соединяться друг с другом двойной или тройной связью. Группа L3 в соединениях формулы IX является уходящей группой, которая может быть заменена группой X, такой как галоген, например фтор, хлор, бром или йод, или группой сульфонилокси, такой как метансульфонилокси или трифторметансульфонилокси. Реакция соединения формулы IX с соединением формулы X формально является реакцией нуклеофильного замещения в кольце, содержащем группы Y и Z, которые могут, в особенности, осуществляться в случае соединений формулы IX, которые восприимчивы к такой реакции из-за присутствия электрон-выводящих заместителей или гетероатомов кольца. Реакция может проводиться в стандартных условиях для таких реакций, которые хорошо известны специалистам в данной области. Пояснения к условиям проведения реакции, например растворителям или основаниям, которые добавляются для более эффективного проведения реакции, приведенные выше в отношении реакции соединения формулы VII с соединением формулы VIII, соответствующим образом касаются и реакции соединения формулы IX с соединением формулы X.
Пояснения по реакции соединения формулы VII с соединением формулы VIII также соответствующим образом относятся к реакциям получения соединений формулы I, в которых гетероатом цепи в группе R23 не находится в терминальном положении цепи, которое соседствует с кольцом, включающим группы Y и Z, но отделено от указанного кольца одной или несколькими группами C(R26)(R26), каковые реакции относятся к такому же типу, что и реакции, перечисленные выше. В качестве примера можно привести получение соединения формулы IVb из соединения формулы XI и соединения формулы XII.
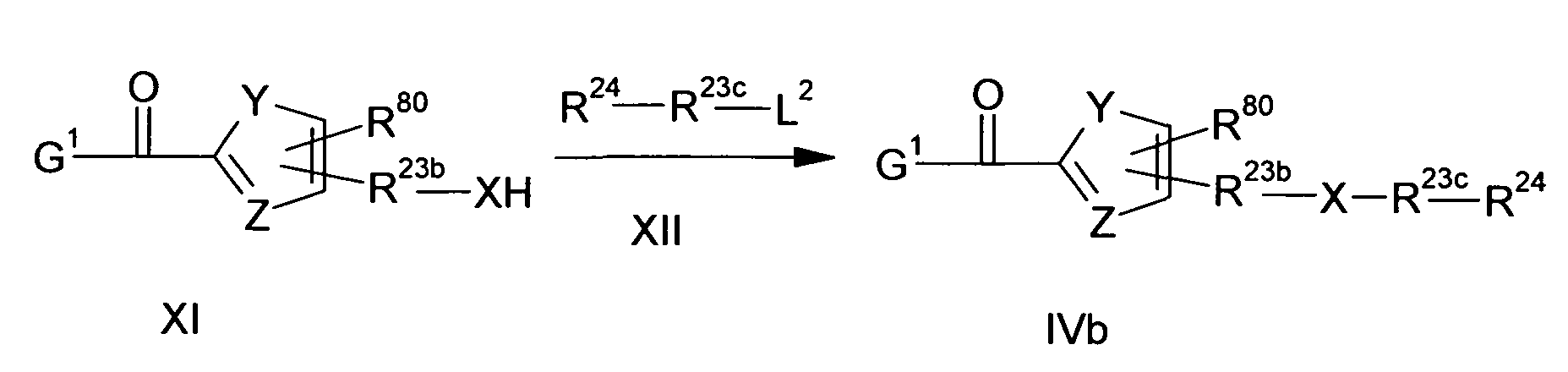
В соединениях формул IVb, XI и XII группы Y, Z и R24 определяются как в соединениях формулы I. Группа R80 определяется как в соединениях формул IVa и VII, т. е. имеет значение той группы R21или R22 в соединениях формулы I, которая не является группой формулы II. Также функциональные группы в соединениях формул IX и X могут присутствовать в защищенной форме или в форме группы-предшественника, которая впоследствии преобразуется в конечную группу. Также функциональные группы в соединениях формул IVb, XI и XII могут присутствовать в защищенной форме или в форме группы-предшественника, которая впоследствии преобразуется в конечную группу. Группа G1-C(O)- является эфирной группой, и группа G1 является группой, такой как (C1-C4)-алкил-О- или бензилокси. Группа X является гетерогруппой, как указано в определении R23, то есть группой, выбранной из группы, содержащей N(R25), O, S, S(O) и S(O)2, в частности из группы, содержащей N(R25), О и S. Группы R23b, R23cи X в соединениях формулы IVb вместе представляют группу R23, как указано выше, в которой X является указанной гетерогруппой цепи. В случае если R23 содержит только одну гетерогруппу, группа R23b в соединениях формул IVb и XI является цепью, содержащей от 1 до 4 одинаковых или разных групп C(R26)(R26), и группа R23c в соединениях формул IVb и XII является прямым соединением или цепью, содержащей от 1 до 3 одинаковых или разных групп C(R26)(R26), при условии, что общее число групп C(R26)(R26) не превышает 4, причем две соседние группы C(R26)(R26) могут соединяться друг с другом двойной или тройной связью. В группе R23c в соединениях формул IVb и XII вместо одной из групп может присутствовать дополнительная гетерогруппа, выбранная из группы, содержащей N(R25), O, S, S(O) и S(O)2, может присутствовать вместо одной из групп C(R26)(R26), при условии, что такая дополнительная гетерогруппа может присутствовать только в терминальной позиции, примыкающей к группе L2, если одну из групп X и указанный член цепи в терминальной позиции выбирают из группы, содержащей S(O) и S(O)2, а другую группу выбирают из группы, содержащей N(R25), О и S. Уходящая группа L2 в соединениях формулы XII определяется как в соединениях формулы VIII. Соответственно, как указано выше в отношении синтеза соединений формулы IVa, которые могут быть получены путем взаимодействия соединения формулы VII с соединением формулы VIII, а также путем взаимодействия соединения формулы IX с соединением формулы X, соединения формулы IVb также могут быть получены путем взаимодействия соединения, которое определяется как соединение формулы XI, но содержит группу L2 вместо группы XH, с соединением, которое определяется как соединение формулы XII, но содержит группу XH вместо группы L2.
В способе получения соединений формулы IV, в котором группа R23 является цепью, которая не содержит какой-либо гетерогруппы, карбонильное соединение формулы XIII конденсируется с соединением формулы XIV для получения олефина формулы IVc, который может впоследствии гидрироваться для получения соединения формулы IVd, соответственно, или вступает в реакцию с металлоорганическим соединением формулы XV для получения спирта формулы IVe, который также может впоследствии гидрироваться для получения соединения формулы IVf.

В соединениях формул IVc по IVf, XIII, XIV и XV группы Y, Z и R24 определяются, как в соединениях формулы I. Группа R80 определяется как в соединениях формул IVa и VII, т. е. имеет значение той группы R21или R22 в соединениях формулы I, которая не является группой формулы II. Также функциональные группы в соединениях формул IVc по IVf, XIII, XIV и XV могут присутствовать в защищенной форме или в форме группы-предшественника, которая впоследствии преобразуется в конечную группу. Группа G1-C(O)- является эфирной группой, и группа G1является группой, такой как (C1-C4)-алкил-О-или бензилокси. Группы Ra и Rb выбирают, независимо друг от друга, из водорода и (C1-C4)-алкила. Группа R23d представляет собой прямую связь или цепь, содержащую от 1 до 3 одинаковых или разных групп C(R26)(R26), группа R23e представляет собой прямую связь или цепь, содержащую от 1 до 4 одинаковых или разных групп C(R26)(R26). Группа L4 в соединениях формулы XIV является группой, допускающей формирование двойной связи между атомом углерода, к которому присоединена группа L4, и атомом углерода альдегидной группы или кетоновой группы, к которому присоединена группа Ra в соединении формулы XIII в реакции конденсации. Примерами подходящих реакций конденсации являются реакция Виттига и реакция Виттига-Хорнера, а примерами подходящих групп L4, таким образом, являются трехзамещенные фосфонильные группы, например трифенилфосфонильная группа, с анионом, например анионом галогена в качестве противоиона, и ди((C1-C4)-алкил)фосфонильные группы, например диэтилфосфонильная группа. Группа L5 в соединениях формулы XV является металлом, например литий или магний галогенсодержащей группой, например, MgCl, MgBr или MgI, и соединение формулы XV, поэтому является литийорганическим соединением или реактивом Гриньяра. Реакция Виттига и реакция Виттига-Хорнера, а также присоединение металлорганического соединения формулы XV к соединению формулы XIII могут проводиться в стандартных условиях в инертном растворителе, например углеводороде, например бензоле или толуоле, или в эфире, например диэтиловом эфире, THF, диоксане или DME. Реакция Виттига и реакция Виттига-Хорнера проводятся в присутствии основания, например гидрида, например гидрида натрия, или амида, например амида натрия или диизопропиламида лития, или алкоксида, например трет-бутоксида калия. В зависимости от конкретного случая вместо использования фосфониевой соли и ее депротонирования, в реакции можно напрямую использовать стабильные фосфорные илиды. Гидрирование двойной связи в соединениях формулы IVc для получения соединений формулы IVd, или бензильной гидроксигруппы в соединениях формулы IVe для получения соединений формулы IVf может проводиться в присутствии катализатора гидрирования, например палладия на активированном угле.
В зависимости от особенностей каждого конкретного случая для приготовления соединений формулы IV могут использоваться различные другие реакции. В качестве примера получения соединений, в которых группа R23 представляет собой цепочку, включающую три группы C(R26)(R26) и без гетероатомных элементов, можно привести альдольную реакцию соединения формулы XIIIa, которое представляет собой соединение формулы XIII, в котором группа Ra представляет собой метил, с альдегидом формулы XVI, которая приводит к соединению формулы IVg или формулы IVm, которые могут восстанавливаться до соединения, например формулы IVh, IVk или IVn.

В соединениях формул IVg по IVn и XIIIa группы Y и Z определяются как в соединениях формулы I. Группа R24a в соединениях формул IVg по IVn и XVI является группой R31 или кольцом с числом членов от 3 до 10, поскольку она может представлять группу R24 в соединениях формулы I, которая связана через атом углерода кольца, в частности ароматического кольца, как, например, необязательно замещенная фенильная, нафтильная или гетероарильная группа. Группа R80 определяется как в соединениях формул IVa и VII, т. е. имеет значение той группы R21или R22 в соединениях формулы I, которая не является группой формулы II. Также функциональные группы в соединениях формул IVg по IVn, XIIIa и XVI могут присутствовать в защищенной форме или в форме группы-предшественника, которая впоследствии преобразуется в конечную группу. Группа G1-C(O)- является эфирной группой, и группа G1является группой, такой как (C1-C4)-алкил-О-или бензилокси.
Реакция соединения формулы XIIIa с соединением формулы XIV для получения продукта альдольного присоединения формулы IVm или продукта конденсации формулы IVg может проводиться в стандартных условиях для альдольной реакции в присутствии основания, например гидроксида щелочного металла, например гидроксида натрия или гидроксида калия, или алкоксида, например метоксида натрия или этоксида натрия, или амида, например диизопропиламида лития, в растворителе, например в спирте, например, метаноле или этаноле, или в эфире, например диэтиловом эфире, THF или диоксане. При более низких температурах, например при температурах от примерно -80°C до примерно -30°C, может получаться соединение формулы IVm, а при более высоких температурах, например при температурах от примерно 10°C до примерно 60°C, или при обработке соединения формулы IVm кислотой, может получаться соединение формулы IVg. Кетоновая функция в соединениях формулы IVg и IVm может быть восстановлена до спиртовой функции, например комплексным гидридом, например боргидридом, например боргидридом лития или боргидридом натрия, с образованием соединений формулы IVh или IVn, соответственно, которые могут быть преобразованы в соединение формулы IVk посредством дегидрирования в присутствии кислоты и (или) гидрированием в присутствии катализатора, такого как, например палладия на активированном угле.
В качестве дополнительного примера реакций, которые могут использоваться для получения соединений формулы IV, можно привести катализируемые переходными металлами реакции сочетания C-C, с помощью которых можно получать соединения, где группа R23 представляет собой простую связь или включает цепочку из двух групп C(R26)(R26), связанных друг с другом тройной связью, то есть группу формулы -C≡C-, в положении, соседствующем с кольцом, содержащим группы Y и Z. Такие соединения могут быть получены из соединения формулы IX и борной кислоты, например необязательно замещенной фенилборной кислоты, циклоалкилборной кислоты или гетероарилборной кислоты, или этина, например замещенного фенилэтина. В качестве катализатора таких реакций может использоваться соединение палладия, например бис(трифенилфосфин)палладий(II) хлорид или тетракис(трифенилфосфин)палладий(0) и соединения меди, например иодид меди (I). Дополнительная информация о таких реакциях приводится, например, в N. Miyaura et al., Chem. Rev. 95 (1995), 2457-2483; и R. Chinchilla et al., Chem. Rev. 107 (2007), 874-922.
Порядок введения групп в процессе синтеза соединения формулы I может также отличаться от приведенного выше. Например, вместо получения вначале соединения формулы IVa из соединения формулы VII и соединения формулы VIII или из соединения формулы IX и соединения формулы X с последующим взаимодействием соединения формулы IVa с соединением формулы III с образованием соединения формулы I, соединение формулы III может также взаимодействовать с соединением формулы VII или соединением формулы IX, и полученное соединение формулы XVII или XVIII вступает в реакцию с соединением формулы VIII или X, соответственно, чтобы получить соединение формулы Ik.

В соединениях формул Ik, XVII и XVIII кольцо А и группы Y, Z, R3 по R6, R20, R24 и R50 определены как в соединениях формулы I. Группы X, R23a и R80 определены как в соединениях формулы IVa. Таким образом, R80 имеет значение той группы R21 или R22 в соединениях формулы I, которая не является группой формулы II. Группа X является гетерогруппой, как указано в определении R23, то есть группой, выбранной из группы, содержащей N(R25), O, S, S(O) и S(O)2, в частности, из группы, содержащей N(R25), О и S. Группы R23a и X вместе представляют группу R23, как указано выше, в которой терминальный член цепи, являющийся гетерочленом, связан с кольцом, содержащим группы Y и Z. Таким образом, R23 представляет собой прямую связь или цепь, содержащую от 1 до 4 членов цепи, из которых 0 или 1 член цепи являются гетерочленами, выбранными из группы, содержащей N(R25), O, S, S(O) и S(O)2, при условии, что терминальный член цепи, прилегающий к группе X, может быть только гетерочленом, если одну из групп Х и указанный терминальный член цепи выбирают из группы, содержащей S(O) и S(O)2, а другую группу выбирают из группы, содержащей N(R25), O и S, тогда как другие члены цепи являются одинаковыми или разными группами C(R26)(R26), где две соседние группы C(R26)(R26) могут соединяться друг с другом двойной или тройной связью. Также функциональные группы в соединениях формул Ik, XVII и XVIII могут присутствовать в защищенной форме или в форме группы-предшественника, которая впоследствии преобразуется в конечную группу. Как указано выше, а также применимо ко всем соединениям, которые содержат группу R80 и другую группу, которые связаны с кольцом, содержащим группы Y и Z, связями, не направленными на конкретный атом углерода кольца, группы R80 и X в соединениях формулы XVII, группы R80 и L3 в соединениях формулы XVIII и группы R80 и X-R23a-R24 в соединениях формулы Ik могут быть расположены в каждой из двух позиций связи С=С в кольце, содержащем группы Y и Z. Приведенные выше пояснения в отношении реакции соединения формулы III с соединением формулы IV, реакции соединения формулы VII с соединением формулы VIII, а также реакции соединения формулы IX с соединением формулы X относятся, соответственно, и к реакции соединения формулы III с соединением формулы VII или соединения формулы IX, реакции соединения формулы XVII с соединением формулы VIII, а также реакции соединения формулы XVIII с соединением формулы X. Порядок введения групп в ходе синтеза соединения формулы I можно также менять в других реакциях. Например, соединение формулы XVIII может использоваться в катализируемой переходными металлами реакции сочетания C-C, как это упоминалось выше, или же соединение формулы XIIIa может взаимодействовать с соединением формулы III, и полученное соединение модифицируется при группе CH3-C(O)- с образованием соединения формулы I.
Исходные соединения и структурные элементы для синтеза соединения формулы I имеются в продаже или могут быть получены в соответствии с методами, описанными в литературе, или же по аналогии с такими методами. В качестве примера можно привести получение соединений формулы VIII, в которой R24 является необязательно замещенной фенильной или нафтильной группой, R23a является необязательно алкилзамещенной группой CH2CH2, а L2 является гидроксигруппой, при котором используется реакция сочетания арилгалогенидов с енолятами эфиров, описанная в работе M. Jørgensen et al., J. Am. Chem. Soc. 124 (2002), 12557-12565. В указанной реакции возможно алкилзамещенный эфир уксусной кислоты, например трет-бутиловый эфир уксусной кислоты или метиловый эфир изомасляной кислоты, депротонируется основанием, например дициклогексиламидом лития, и реагирует с необязательно замещенным арилбромидом в присутствии палладиевого соединения, например бис(дибензилиденацетон)палладия или трис(дибензилиденацетон)дипалладия, и три(трет-бутил)фосфином, чтобы получить 2-(необязательно замещенный арил) эфир уксусной кислоты, который может необязательно иметь алкильный заместитель в положении 2 группы уксусной кислоты. Восстановление сложноэфирной функции в стандартных условиях, например алюмогидридом лития, затем позволяет получить 2-(необязательно замещенный арил)этанол, который может необязательно иметь алкильный заместитель в положении 2.
Для получения других соединений формулы I могут проводиться различные преобразования функциональных групп в соединениях формулы I, или в интермедиатах, или в исходных соединениях в стандартных условиях в процессе синтеза соединений формулы I. Например, гидроксильная группа может этерифицироваться с образованием эфира карбоновой кислоты или эфира сульфоновой кислоты, или же с образованием простого эфира. Этерификация гидроксильных групп может успешно проводиться посредством алкилирования при воздействии соответствующего галоидного соединения, например бромида или иодида, в присутствии основания, например карбоната щелочного металла, например карбоната калия или карбоната цезия, в инертном растворителе, например амиде, таком как DMF или NMP, или кетоне, например ацетоне или бутан-2-оне, или с соответствующим спиртом в условиях реакции Мицунобу, которая упоминалась выше. Гидроксигруппа может быть превращена в галоидную при обработке галогенирующим агентом. Атом галогена может заменяться различными группами в реакции замещения, которая также может представлять собой реакцию, катализируемую переходным металлом. Нитрогруппа может восстанавливаться до аминогруппы, например посредством каталитического гидрирования. Аминогруппа может модифицироваться в стандартных условиях алкилирования, например посредством реакции с галоидным соединением или в ходе восстановительного аминирования карбонильного соединения, или в стандартных условиях ацилирования или сульфонирования, например в реакции с активированной карбоновой кислотой или производным карбоновой кислоты, таким как хлорангидрид или ангидрид или хлорангидрид сульфоновой кислоты. Сложноэфирная группа карбоновой кислоты может подвергаться гидролизу в кислых или основных условиях с образованием карбоновой кислоты. Карбоксильная группа может активироваться или переводиться в реакционно-способное производное, как показано выше, в отношении соединений формулы IX, и вступать в реакцию со спиртом или амином, или аммиаком с образованием эфира или амида. Первичный амид может дегидрироваться с образованием нитрила. Атом серы в алкил-S-группе или в гетероциклическом кольце, или же атом серы, присутствующий в цепочке, которая образует группу R23, может окисляться перекисью, например перекисью водорода или надкислотой, с образованием сульфоксидной группы S(O) или сульфоновой группы S(O)2. Карбоксильная группа, сложноэфирная группа карбоновой кислоты и кетогруппа могут восстанавливаться до спирта, например комплексным гидридом, например алюмогидридом лития, боргидридом лития или боргидридом натрия. Все реакции получения соединений формулы I известны по существу и могут быть проведены в соответствии со способом, который известен специалистам в области, согласно или аналогично методам, которые описаны в стандартной литературе, например в Houben-Weyl, Methods of Organic Chemistry, Thieme; или Organic Reactions, John Wiley & Sons; или R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2. ed. (1999), John Wiley & Sons, и в цитируемых там источниках. Более того, кроме химических методик в растворах, соединения формулы I могут также получаться в ходе твердофазного химического синтеза.
Еще одним предметом настоящего изобретения являются новые исходные соединения и промежуточные соединения, получаемые при синтезе соединений формулы I, в том числе соединений формул III, IV, IV, IVb, IVc, IVd, IVe, IVf, IVg, IVh, IVk, IVm, IVn, V, VI, VII, VIII, IX, X, XI, XII, XIII, XIV, XV, XVI, XVII и XVIII, в которых кольцо A и группы G, G1, L1, L2, L3, PG1, PG2, X, Y, Z, R3 по R6, R20 по R23, R23a, R23b, R23c, R24, R24a, R50, R80, Ra и Rb согласно определению выше, в любой из их стереоизомерных форм или сочетании стереоизомерных форм в любом соотношении, и их соли и сольваты любой из них, а также их использование в качестве синтетических промежуточных или исходных соединений. Все общие объяснения, спецификации осуществлений и определения чисел и групп, приведенные выше в отношении соединений формулы I, применимы соответствующим образом к указанным промежуточным и исходным соединениям. Предметом настоящего изобретения являются, в частности, новые конкретные исходные и промежуточные соединения, раскрываемые в настоящем изобретении. Независимо от того, представлены ли они как свободное соединение и (или) конкретная соль, они являются предметом настоящего изобретения как в виде свободных соединений, так и в виде их солей, и если раскрывается конкретная соль, то дополнительно в виде этой конкретной соли.
Соединения формулы I ингибируют рецептор Edg-2 (рецептор LPA1), что может быть показано в ходе фармакологических испытаний, описанных ниже, а также в других тестах, которые известны специалистам в области. Поэтому соединения формулы I и их физиологически приемлемые соли и сольваты являются важными фармацевтически активными соединениями. Соединения формулы I и их физиологически приемлемые соли и сольваты могут использоваться для лечения сердечно-сосудистых заболеваний, например сердечной недостаточности, в том числе систолической сердечной недостаточности, диастолической сердечной недостаточности, диабетической сердечной недостаточности и сердечной недостаточности с сохраняющейся фракцией выброса, кардиомиопатии, инфаркта миокарда, ремоделирования миокарда, в том числе ремоделирования сердца после инфаркта или операции на сердце, ремоделирования сосудов, в том числе ригидности сосудов, гипертензии, в том числе легочной гипертензии, портальной гипертензии и систолической гипертензии, атеросклероза, окклюзионного периферического поражения артерий (PAOD), рестеноза, тромбоза или нарушений проницаемости сосудов, в качестве кардиопротектора, например кардиопротектора после инфаркта миокарда или после операции на сердце, в качестве ренопротектора или для лечения воспалений или воспалительных заболеваний, например ревматоидного артрита, остеоартрита, заболеваний почек, например медуллярного некроза почки или почечной недостаточности, в том числе почечной недостаточности после ишемии/реперфузии, легочных заболеваний, например, хронического обструктивного заболевания легких (ХОЗЛ), астмы или синдрома острой дыхательной недостаточности (СОДН), иммунологических заболеваний, аллергических заболеваний, роста опухоли, метастазирования, метаболических заболеваний, фиброзных заболеваний, например легочного фиброза, в том числе идиопатического фиброза легких, кардиофиброза, сосудистого фиброза, периваскулярного фиброза, почечного фиброза, в том числе тубулоинтерстициального почечного фиброза, фиброза печени, фиброзных состояний кожи, в том числе образования келоида, коллагеноза, склеродермы, прогрессирующего системного склероза и нефрогенной фиброзирующей дермопатии, или других типов фиброза, в том числе контрактуры Дюпуитрена, псориаза, боли, например нейропатической боли, диабетической боли или воспалительной боли, прурита, ретинального повреждения при ишемии/реперфузии, дегенерации желтого пятна, психиатрических заболеваний, нейродегенеративных заболеваний, нарушений черепно-мозгового нерва, расстройств периферической нервной системы, эндокринных расстройств, например гипертиреоза, избыточного рубцевания или нарушений заживления ран. Под лечением заболеваний следует понимать термин, означающий терапию существующих патологических изменений или нарушений в организме или существующих симптомов в целях их облегчения, ослабления или устранения, а также профилактику или предупреждение патологических изменений или нарушений в организме или симптомов у людей или животных, которые им подвержены и нуждаются в такой профилактике или предупреждении, в целях предупреждения или подавления их возникновения или ослабления в случае их проявлений. Например, у пациентов, которые по причине своей истории болезни подвержены риску инфаркта миокарда, за счет такого профилактического или предупредительного медицинского лечения можно предотвратить наступление или повторное наступление инфаркта миокарда или снизить его остроту и последствия, или же у пациентов, которые чувствительны к нарушениям заживления ран, такое профилактическое или предупредительное медицинское лечение может благоприятно отразиться на заживлении ран после хирургического вмешательства. Лечение заболеваний может проводиться как в острых, так и в хронических случаях. Эффективность соединений формулы I может быть продемонстрирована в фармакологических тестах, описанных ниже, а также в других тестах, которые известны специалистам в области.
Соединения формулы I и их физиологически приемлемые соли и сольваты, поэтому могут применяться на животных, в частности, млекопитающих и в особенности, на человеке, как фармацевтические или лекарственные средства сами по себе, в смесях друг с другом или в форме фармацевтических композиций. Предметом настоящего изобретения также являются соединения формулы I и их физиологически приемлемые соли и сольваты для применения в качестве лекарственных средств, а также фармацевтических композиций и лекарственных средств, которые включают эффективную дозу по меньшей мере одного соединения формулы I и (или) его физиологически приемлемой соли и (или) его сольвата в качестве активного компонента, и фармацевтически приемлемый носитель, то есть один или несколько фармацевтически безвредных или безопасных носителей и (или) формообразующих веществ, а также, возможно, одно или несколько других фармацевтически активных соединений. Предметом настоящего изобретения также являются соединения формулы I и их физиологически приемлемые соли и сольваты, используемые в лечении заболеваний, указанных выше или ниже, в том числе лечение любого из названных заболеваний, например сердечной недостаточности или фиброзных заболеваний, таких как легочный фиброз, кардиофиброз, сосудистый фиброз, периваскулярный фиброз, почечный фиброз, фиброз печени или фиброзные заболевания кожи, применение соединений формулы I и их физиологически приемлемых солей и сольватов для получения лекарственного средства для лечения заболеваний, указанных выше или ниже, в том числе лечение любого из названных заболеваний, например сердечной недостаточности или фиброзных заболеваний, таких как легочный фиброз, кардиофиброз, сосудистый фиброз, периваскулярный фиброз, почечный фиброз, фиброз печени или фиброзные заболевания кожи, причем лечение заболеваний заключается в их лечении и профилактике, как указано выше, а также в их применение для получения лекарственного средства для ингибирования рецептора Edg-2 (рецептора LPA1). Предметом настоящего изобретения также являются способы лечения заболеваний, указанных выше или ниже, в том числе лечение любого из названных заболеваний, например сердечной недостаточности или фиброзных заболеваний, таких как легочный фиброз, кардиофиброз, сосудистый фиброз, периваскулярный фиброз, почечный фиброз, фиброз печени или фиброзные заболевания кожи, которые включают введение эффективного количества по меньшей мере одного соединения формулы I и (или) его физиологически приемлемой соли и (или) его физиологически приемлемого сольвата человеку или животному, нуждающемуся в этом. Соединения формулы I и содержащие их фармацевтические композиции и лекарственные средства могут вводиться энтерально, например путем перорального, сублингвального или ректального введения, парентерально, например путем внутривенной, внутримышечной, подкожной или внутрибрюшинной инъекции или инфузии, или иным способом, таким как местное, чрескожное, трансдермальное, внутрисуставное, интраназальное или внутриглазное применение.
Соединения формулы I и их физиологически приемлемые соли и сольваты также могут использоваться в сочетании с другими фармацевтически активными соединениями, причем в таком сочетании использование соединений формулы I и (или) их физиологически приемлемых солей и (или) сольватов и одного или более других фармацевтически активных соединений может иметь место в одной и той же фармацевтической композиции или в двух или более фармацевтических композициях для раздельного, одновременного или последовательного введения. Примерами таких других фармацевтически активных соединений являются ингибиторы ангиотензин-преобразующего фермента (ACE), рамиприл, антагонисты 1 подтипа рецептора ангиотензина II (AT1), ирбесартан, антиаритмические средства, дронедарон, активаторы альфа-рецепторов, активируемых пролифераторами пероксисом (PPAR-α), активаторы гамма-рецепторов, активируемых пролифераторами пероксисом (PPAR-γ), пиоглитазон, розиглитазон, простаноиды, антагонисты рецепторов эндотелина, бозентан, ингибиторы эластазы, антагонисты кальция, бета-блокаторы, диуретики, антагонисты рецепторов альдостерона, эплеренон, ингибиторы ренина, ингибиторы rho киназы, активаторы растворимой гуанилатциклазы (sGC), сенсибилизаторы sGC, ингибиторы фосфодиэстеразы (PDE), ингибитора фосфодиэстеразы 5 типа (PDE5), доноры NO, препараты дигиталиса, ингибиторы ангиотензин-преобразующего фермента/нейтральной эндопептидазы (ACE/NEP), статины, ингибиторы обратного захвата желчной кислоты, антагонисты рецепторов тромбоцитарного фактора роста (PDGF), антагонисты вазопрессина, акваретики, ингибиторы систем ионного обмена натрий-протоны подтипа 1 (NHE1), антагонисты фактора II/фактора IIa, антагонисты фактора IX/фактора IXa, антагонисты фактора X/фактора Xa, антагонисты фактора XIII/фактора XIIIa, антикоагулянты, антитромботические средства, ингибиторы агрегации тромбоцитов, профибринолитики, ингибиторы тромбин-активируемого фибринолиза (TAFI), ингибитор активатора плазминогена-1 (PAI 1), кумарины, гепарины, антагонисты тромбоксана, антагонисты серотонина, ингибиторы циклооксигеназы, ацетилсалициловая кислота, терапевтические антитела, антагонисты гликопротеина IIb/IIIa (GPIIb/IIIa), в том числе абциксимаб, ингибиторы химазы, цитостатики, таксаны, паклитаксел, доцетаксел, ингибиторы ароматазы, антагонисты рецепторов эстрогена, модуляторы селективных рецепторов эстрогена (SERM), ингибиторы тирозинкиназы, иматиниб, ингибиторы рецепторов тирозинкиназы, ингибиторы RAF киназы, ингибиторы p38 митоген-активируемой протеинкиназы (p38 MAPK), пирфендион, ингибиторы мультикиназ, а также сорафениб. Предметом настоящего изобретения также является указанное сочетание использования одного или более соединений формулы I, раскрываемых в настоящем изобретении, и их физиологически приемлемых солей и сольватов с одним или более, например с одним или двумя, из указанных других фармацевтически активных соединений.
Фармацевтические композиции и лекарственные средства настоящего изобретения обычно содержат от примерно 0,5 до примерно 90 процентов по весу соединений формулы I и (или) их физиологически приемлемых солей и (или) сольватов, с количеством активного ингредиента формулы I и (или) его физиологически приемлемой соли и (или) сольвата, которое обычно составляет от примерно 0,2 мг до примерно 1000 мг, в частности от примерно 0,2 мг до примерно 500 мг, например от примерно 1 мг до примерно 300 мг, в однократной дозе. В зависимости от вида фармацевтической композиции и других особенностей конкретного случая количества могут отличаться от указанных. Фармацевтические композиции и лекарственные средства могут получаться известным для данной области способом. С этой целью соединения формулы I и (или) их физиологически приемлемые соли и (или) сольваты смешиваются с одним или несколькими твердыми или жидкими носителями и (или) формообразующими веществами, при желании, также в сочетании с одним или несколькими другими фармацевтически активными соединениями, например перечисленными выше, и переводятся в удобную форму для дозировки и введения, которая затем может применяться для лечения людей или в ветеринарной медицине.
В качестве носителей, которые также могут рассматриваться как разбавители или объемообразующие и формообразующие средства, могут применяться подходящие органические и неорганические соединения, которые не реагируют нежелательным образом с соединениями формулы I. В качестве примеров видов эксципиетов или добавок, которые могут содержаться в фармацевтических композициях и лекарственных средствах, можно лубриканты средства, консерванты, загустители, стабилизаторы, дезинтегрирующие вещества, увлажняющие вещества, вещества, обеспечивающие эффект депо, эмульгаторы, соли, например для воздействия на осмотическое давление, буферные вещества, красители, отдушки и ароматические вещества. Примерами носителей и эксципиентов являются вода, растительные масла, воски, спирты, например этанол, изопропанол, 1,2-пропандиол, бензиловые спирты, глицерин, полиолы, полиэтиленгликоли или полипропиленгликоли, триацетат глицерина, поливинилпирролидон, желатин, целлюлоза, углеводы, например лактоза или крахмал, например кукурузный крахмал, хлорид натрия, стеариновая кислота и ее соли, например стеарат магния, тальк, ланолин, вазелиновое масло, или их смеси, например физиологический раствор, или смеси воды с одним или несколькими органическими растворителями, например смеси воды со спиртами. Для перорального и ректального использования могут использоваться такие формы, как, например, таблетки, таблетки с пленочным покрытием, таблетки с покрытием сахарами, гранулы, капсулы из жесткого и мягкого желатина, суппозитории, растворы, в том числе масляные, спиртовые или водные растворы, сиропы, соки или капли, а также суспензии или эмульсии. При парентеральном введении, например посредством инъекции или вливания, могут использоваться такие фармацевтические формы, как, например, водные растворы. Для местного применения могут использоваться такие фармацевтические формы, как мази, кремы, пасты, лосьоны, гели, спреи, пены, аэрозоли, растворы или порошки. Другими подходящими фармацевтическими формами, например, являются импланты и пластыри, а также формы, приспособленные для ингаляции. Соединения формулы I и их физиологически приемлемые соли могут также лиофилизоваться, а полученные лиофилизаты использоваться, например, для приготовления инъекционных составов. В частности, для местного применения также пригодны липосомальные составы. Фармацевтические композиции и лекарственные средства могут также содержать один или несколько других активных ингредиентов и (или), например, один или несколько витаминов.
Как и во всех случаях, дозировка соединений формулы I зависит от обстоятельств в каждом конкретном случае и корректируется врачом в соответствии со стандартными правилами и процедурами. Например, она зависит от вводимого соединения формулы I и его активности и продолжительности действия, от характера и остроты индивидуального синдрома, от пола, возраста и веса и индивидуального отклика человека или животного, получающего лечение, от того, проводится ли лечение в остром, хроническом или профилактическом вариантах, или от того, вводятся ли другие фармацевтически активные соединения в дополнение к соединению формулы I. Как правило, в случае введения взрослому весом примерно 75 кг будет достаточно дозы от примерно 0,1 мг до примерно 100 мг на кг в день, в частности от примерно 1 мг до примерно 10 мг на кг в день (в каждом случае в мг на кг веса тела). Дневная доза может вводиться в форме разовой дозы или разделяться на несколько отдельных доз, например, две, три или четыре отдельные дозы. Введение может также осуществляться непрерывно, например посредством непрерывной инъекции или вливания. В зависимости от индивидуального поведения в каждом конкретном случае, возможно, придется отклониться от предписанных дозировок в сторону увеличения или уменьшения.
В дополнение к применению в качестве фармацевтически активного соединения при лечении человека и в ветеринарной медицине соединения формулы I могут также использоваться в качестве дополнительного средства для биохимических исследований или в качестве научного инструмента, или в целях диагностики, например, для in-vitro диагностики биологических образцов, если предполагается провести ингибирование рецептора Edg-2. Соединения формулы I и их соли могут также использоваться как интермедиаты для получения других фармацевтически активных веществ.
Ниже приведены примеры для иллюстрации настоящего изобретения.
Сокращения
Реакции, как правило, проводятся в токе аргона. Если соединения примеров, содержащие основную группу, очищаются препаративной высокоэффективной жидкостной хроматографией (ВЭЖХ) на обращеннофазовой колонке (ОФ), и, как обычно, элюент представляет собой градиентную смесь воды и ацетонитрила, содержащую трифторуксусную кислоту, они частично получаются в форме солей, полученных при присоединении трифторуксусной кислоты, в зависимости от тонкостей методов обработки, например условий упаривания или лиофилизации. В названиях соединений примеров и их структурных формулах такое содержание трифторуксусной кислоты не оговаривается.
Полученные соединения, как правило, характеризовали на основе спектроскопических и хроматографических данных, в частности масс-спектрам (МС) и временам удерживания ВЭЖХ (Rt; в мин), которые регистрировали посредством комбинированного анализа ВЭЖХ/МС (ЖХ/МС), и (или) по спектрам ядерного магнитного резонанса (ЯМР). Если не указано иначе, спектры1H-ЯМР регистрировали на 500 МГц в D6-DMSO в качестве растворителя при 298 K. При описании ЯМР спектров приводят химический сдвиг δ (в м.д.), число атомов водорода (H) и мультиплетность (с: синглет, д: дублет, дд: дублет дублетов, т: триплет, дт: дублет триплетов, к: квартет, м: мультиплет, ш: уширение) для пиков, определяемых по распечатке. При описании МС, как правило, приводят массовое число (m/z) пика молекулярного иона [M], например [M+], или родственного иона, например иона [M+1], например [(M+1)+], то есть протонированного молекулярного иона [(M+H)+], сокращаемого как [MH+], или иона [M-1], например [(M-1)‾], то есть депротонированного молекулярного иона [(M-H)‾], который образуется в зависимости от используемого метода ионизации. В общем случае методом ионизации была ионизация электрораспылением (ЭСИ) или химическая ионизация при атмосферном давлении (APCI). Ниже приводятся особенности применения методов ЖХ/МС.
Метод LC1
Колонка: YMC J'sphere H80, 20×2,1 мм, 4 мкм; 30°C; поток: 1,0 мл/мин; элюент A: ACN; элюент B: вода + 0,05% ТФУ; градиент: от 4% A + 96% B до 95% A + 5% B в течение 2,4 мин, затем до 4% A + 96% B в течение 0,05 мин, затем 4% A + 96% B в течение 0,05 мин; метод ионизации МС: ЭСИ+
Метод LC2
Колонка: YMC J'sphere H80, 20×2,1 мм, 4 мкм; 30°C; поток: 1,0 мл/мин; элюент A: ACN; элюент B: вода + 0,05% ТФУ; градиент: от 4% A + 96% B до 95% A + 5% B в течение 2,4 мин, затем до 4% A + 96% B в течение 0,05 мин, затем 4% A + 96% B в течение 0,05 мин.; метод ионизации МС: ЭСИ+
Метод LC3
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; поток: 1,3 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% ТФУ; градиент: от 5% A + 95% B до 95% A + 5% B в течение 2,5 мин, затем 95% A + 5% B в течение 0,5 мин, затем до 5% A + 95% B в течение 0,2 мин; метод ионизации МС: ЭСИ+
Метод LC4
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; поток: 1,0 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% ТФУ; градиент: от 5% A + 95% B до 95% A + 5% B в течение 3,4 мин, затем 95% A + 5% B в течение 1,0 мин, затем до 5% A + 95% B в течение 0,2 мин; затем 5% A + 95% B в течение 0,5 мин; метод ионизации МС: ЭСИ+
Метод LC5
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; поток: 1,3 мл/мин; элюент A: ACN + 0,08% муравьиной кислоты; элюент B: вода + 0,1% муравьиной кислоты; градиент: от 5% A + 95% B до 95% A + 5% B в течение 2,5 мин, затем 95% A + 5% B в течение 0,5 мин.; метод ионизации МС: ЭСИ+
Метод LC6
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; поток: 1,3 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: от 5% A + 95% B до 95% A + 5% B в течение 2,5 мин, затем до 5% A + 95% B в течение 0,5 мин; метод ионизации МС: ЭСИ+
Метод LC7
Колонка: Thermo Javelin C18, 40×2,1 мм, 5 мкм; поток: 1,0 мл/мин; элюент A: ACN + 0,1% TFA; элюент B: вода + 0,1% TFA; градиент: от 2% A + 98% B до 80% A + 20% B в течение 7,0 мин, затем до 100% A + 0% B в течение 0,2 мин, затем 100% A + 0% B в течение 1,0 мин, затем до 2% A + 98% B в течение 0,3 мин, затем 2% A + 98% B в течение 0,5 мин; метод ионизации МС: ЭСИ+
Метод LC8
Колонка: Thermo Javelin C18, 40×2,1 мм, 5 мкм; поток: 1,0 мл/мин; элюент A: ACN + 0,1% TFA; элюент B: вода + 0,1% TFA; градиент: от 2% A + 98% B до 80% A + 20% B в течение 5,0 мин, затем до 100% A + 0% B в течение 0,2 мин, затем 100% A + 0% B в течение 1,0 мин, затем до 2% A + 98% B в течение 0,3 мин, затем 2% A + 98% B в течение 0,5 мин; метод ионизации МС: ЭСИ+
Метод LC9
Колонка: HP Waters Atlantis dC18, 50×2,1 мм, 5 мкм; поток: 0,6 мл/мин; элюент A: ACN + 0,1% TFA; элюент B: вода + 0,1% TFA; градиент: от 2% A + 98% B до 80% A + 20% B в течение 5,0 мин, затем до 100% A + 0% B в течение 0,2 мин, затем 100% A + 0% B в течение 1,0 мин, затем до 2% A + 98% B в течение 0,3 мин, затем 2% A + 98% B в течение 0,5 мин; метод ионизации МС: ЭСИ+
Метод LC10
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; поток: 1,3 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: 5% A + 95% B в течение 0,5 мин, затем до 95% A + 5% B в течение 3,0 мин, затем до 5% A + 95% B в течение 0,5 мин; метод ионизации МС: ЭСИ+
Метод LC11
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; поток: 1,3 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: от 5% A + 95% B до 95% A + 5% B в течение 2,5 мин, затем 95% A + 5% B в течение 0,5 мин; метод ионизации МС: ЭСИ+
Метод LC12
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; поток: 1 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: 2% A + 98% B в течение 1 мин, затем до 95% A + 5% B в течение 4 мин, затем 95% A + 5% B в течение 1,25 мин; метод ионизации МС: ЭСИ+
Метод LC13
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; поток: 1,3 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: 5% A + 95% B в течение 0,5 мин, затем до 95% A + 5% B в течение 3 мин, затем 95% A + 5% B в течение 0,5 мин; метод ионизации МС: ЭСИ+
Метод LC14
Колонка: Waters XBridge C18, 50×4,6 мм, 2,5 мкм; поток: 1,3 мл/мин, 50°C; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: 5% A + 95% B в течение 0,3 мин, затем до 95% A + 5% B в течение 3,2 мин, затем 95% A + 5% B в течение 0,5 мин; метод ионизации МС: ЭСИ+
Метод LC15
Колонка: Waters XBridge C18, 50×4,6 мм, 2,5 мкм; поток: 1,3 мл/мин, 50°C; элюент A: ACN + 0,1% муравьиной кислоты; элюент B: вода + 0,1% муравьиной кислоты; градиент: от 3% A + 97% B до 60% A + 40% B в течение 3,5 мин, затем до 98% A + 2% B в течение 0,5 мин, затем 98% A + 2% B в течение 1 мин., затем до 3% A + 97% B в течение 0,2 мин, затем 3% A + 97% B в течение 1,3 мин; метод ионизации МС: APCI+
Метод LC16
Колонка: Waters XBridge C18, 50×4,6 мм, 2,5 мкм; поток: 1,3 мл/мин, элюент A: ACN + 0,08% муравьиной кислоты; элюент B: вода + 0,1% муравьиной кислоты; градиент: от 3% A + 97% B до 60% A + 40% B в течение 3,5 мин, затем до 98% A + 2% B в течение 0,5 мин, затем 98% A + 2% B в течение 1 мин, затем до 3% A + 97% B в течение 0,2 мин, затем 3% A + 97% B в течение 1,3 мин; метод ионизации МС: ЭСИ‾
Метод LC17
Колонка: Waters XBridge C18, 50×4,6 мм, 2,5 мкм; поток: 1,3 мл/мин, элюент A: ACN + 0,08% муравьиной кислоты; элюент B: вода + 0,1% муравьиной кислоты; градиент: от 3% A + 97% B до 60% A + 40% B в течение 3,5 мин, затем до 98% A + 2% B в течение 0,5 мин, затем 98% A + 2% B в течение 1 мин, затем до 3% A + 97% B в течение 0,2 мин, затем 3% A + 97% B в течение 1,3 мин; метод ионизации МС: ЭСИ+
Метод LC18
Колонка: Waters XBridge C18, 50×4,6 мм, 2,5 мкм; поток: 1,7 мл/мин, 50°C, элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: 5% A + 95% B в течение 0,2 мин, затем до 95% A + 5% B в течение 2,2 мин, затем 95% A + 5% B в течение 1,1 мин, затем до 5% A + 95% B в течение 0,1 мин, затем 5% A + 95% B в течение 0,9 мин; метод ионизации МС: ЭСИ+
Метод LC19
Колонка: Waters XBridge C18, 50×4,6 мм, 2,5 мкм; поток: 1,3 мл/мин, 50°C, элюент A: ACN + 0,1% муравьиной кислоты; элюент B: вода + 0,1% муравьиной кислоты; градиент: от 3% A + 97% B до 60% A + 40% B в течение 3,5 мин, затем до 98% A + 2% B в течение 0,5 мин, затем 98% A + 2% B в течение 1 мин, затем до 3% A + 97% B в течение 0,2 мин, затем 3% A + 97% B в течение 1,3 мин; метод ионизации МС: ЭСИ+
Метод LC20
Колонка: Waters XBridge C18, 50×4,6 мм, 2,5 мкм; поток: 1,3 мл/мин, элюент A: ACN + 0,08% муравьиной кислоты; элюент B: вода + 0,1% муравьиной кислоты; градиент: от 3% A + 97% B до 98% A + 2% B в течение 18 мин, затем 98% A + 2% B в течение 1 мин, затем до 3% A + 97 % B в течение 0,5 мин, затем 3% A + 97% B в течение 0,5 мин; метод ионизации МС: ЭСИ+
Метод LC21
Колонка: Waters XBridge C18, 50×4,6 мм, 2,5 мкм; поток: 1,3 мл/мин, 50°C, элюент A: ACN + 0,1% муравьиной кислоты; элюент B: вода + 0,1% муравьиной кислоты; градиент: от 3% A + 97% B до 60% A + 40% B в течение 3,5 мин, затем до 98% A + 2% B в течение 0,5 мин, затем 98% A + 2% B в течение 1 мин., затем до 3% A + 97% B в течение 0,2 мин, затем 3% A + 97% B в течение 1,3 мин; метод ионизации МС: ЭСИ‾
Метод LC22
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; поток: 1 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: от 5% A + 95% B до 95% A + 5% B в течение 3,7 мин; метод ионизации МС: ЭСИ+
Пример 1
Метиловый эфир 2-[4-бром-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты

Стадия 1: Метиловый эфир 4-бром-3-(2-м-толилэтокси)-бензойной кислоты
Метиловый эфир 4-бром-3-гидроксибензойной кислоты (1,00 г, 4,32 ммоль) и трифенилфосфин (1,36 г, 5,19 ммоль) растворили в ТГФ. После добавления 2-(3-метилфенил)-этанола (2-м-толилэтанола) (0,707 г, 5,19 ммоль) смесь охлаждали в ледяной бане, с последующим постепенным добавлением ДИАД (1,05 г, 5,19 ммоль) в процессе перемешивания. После удаления ледяной бани смесь непрерывно перемешивали в течение ночи при комнатной температуре. Летучие фракции удаляли под вакуумом и остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 1,55 г указанного в заголовке соединения.
Стадия 2: 4-Бром-3-(2-м-толилэтокси)-бензойная кислота
Соединение стадии 1 (0,50 г, 1,43 ммоль) растворяли в диоксане (5 мл), добавляли гидроксид лития (7,1 мл 1М водного раствора (т.е. 1 моль/л)) и оставляли реакцию на ночь. Смесь разделяли в 2н. хлористоводородной кислоте и EA, водную фазу экстрагировали EA и органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния с получением 0,414 г указанного в заголовке соединения.
1H ЯМР: δ = 13,2 (уш.с, 1H); 7,7 (д, 1H); 7,52 (д, 1H); 7,42 (дд, 1H); 7,22-7,11 (м, 3H); 7,02 (д, 1H); 4,30 (т, 2H); 3,03 (т, 2H); 2,29 (с, 3H)
Стадия 3: Метиловый эфир 2-[4-бром-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты
Соединение стадии 2 (0,410 г, 1,22 ммоль) растворяли в ДМФ (5 мл), добавляли EDIA (0,790 г, 6,12 ммоль), HOBT (33 мг, 0,244 ммоль) и гидрохлорид метилового эфира 2-амино-индан-2-карбоновой кислоты (0,246 г, 1,47 ммоль), смесь охлаждали в ледяной бане и добавляли EDC (352 мг, 1,84 ммоль). Смесь перемешивали в течение ночи. Летучие фракции удаляли под вакуумом, смесь разделяли в 2н. хлористоводородной кислоте и EA, органическую фракцию высушивали над сульфатом магния и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 0,56 г указанного в заголовке соединения.
ЖХ/МС (метод LC1): Rt = 1,98 мин.; м/z = 508,1/510,1 [MH+]
Пример 2
2-[4-Бром-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Соединение примера 1 (60 мг, 0,118 ммоль) растворяли в диоксане (1,5 мл), добавляли гидроксид лития (0,59 мл 1М водного раствора) и оставляли реакцию в течение 20 мин при темп. 60°C. Смесь разделяли в 2н хлористоводородной кислоте и EA, водную фазу экстрагировали EA и органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния под вакуумом. Остаток перемешивали в течение ночи со смесью диэтилового эфира и HEP, фильтровали и твердый продукт сушили под вакуумом с получением 43 мг указанного в заголовке соединения.
1H-ЯМР: δ = 12,45 (уш.с, 1H); 8,87 (с, 1H); 7,64 (д, 1H); 7,46 (д, 1H); 7,37 (дд, 1H); 7,25-7,11 (м, 7H); 7,02 (д, 1H); 4,27 (т, 2H); 3,60 (д, 2H); 3,37 (д, 2H); 3,02 (т, 2H); 2,28 (с, 3H)
Пример 3
Метиловый эфир 2-[4-метилсульфанил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты

Стадия 1: Метиловый эфир 4-нитро-3-(2-м-толилэтокси)-бензойной кислоты
Метиловый эфир 3-гидрокси-4-нитробензойной кислоты (1,00 г, 5,07 ммоль) и 2-(3-метилфенил)-этанол (0,829 г, 6,09 ммоль) вступали в реакцию по аналогии со стадией 1 примера 1 с получением 1,39 г указанного в заголовке соединения.
1H-ЯМР: δ = 7,96 (д, 1H); 7,75 (с, 1H); 7,63 (д, 1H); 7,18 (т, 1H); 7,11 (с, 1H); 7,08 (д, 1H); 7,03 (д, 1H); 4,41 (т, 2H); 3,89 (с, 3H); 3,01 (т, 2H); 2,28 (с, 3H)
Стадия 2: Метиловый эфир 4-метилсульфанил-3-(2-м-толилэтокси)-бензойной кислоты
Соединение стадии 1 (900 мг, 2,85 ммоль) растворяли в 1,3-диметил-2-имидазолидиноне (6 мл) с добавлением метанэтиолата натрия (0,23 г, 3,29 ммоль). Реакционную смесь оставляли при комнатной температуре в течение 60 ч, после чего разделяли в насыщенном растворе хлорида натрия и EA и водную фазу экстрагировали EA. Органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 600 мг указанного в заголовке соединения.
1H-ЯМР: δ = 7,56 (д, 1H); 7,39 (с, 1H); 7,23 (д, 1H); 7,21-7,12 (м, 3H); 7,02 (д, 1H); 4,25 (т, 2H); 3,82 (с, 3H); 3,01 (т, 2H); 2,41 (с, 3H); 2,29 (с, 3H)
Стадия 3: 4-метилсульфанил-3-(2-м-толилэтокси)-бензойная кислота
Соединение стадии 2 (450 мг, 1,42 ммоль) гидролизировали по аналогии с примером 2 с получением 395 мг указанного в заголовке соединения.
1H-ЯМР: δ = 12,8 (уш.с, 1H); 7,55 (д, 1H); 7,38 (с, 1H); 7,23-7,10 (м, 4H); 7,02 (д, 1H); 4,25 (т, 2H); 3,82 (с, 3H); 3,00 (т, 2H); 2,41 (с, 3H); 2,29 (с, 3H)
Стадия 4: Метиловый эфир 2-[4-метилсульфанил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты
Соединение стадии 3 (395 мг, 1,31 ммоль) растворяли в ДХМ (5 мл). ДМФ (29 мг, 0,39 ммоль) и оксалилхлорид (3,9 мл 2 М раствора в ДХМ) добавляли при комнатной температуре. Смесь перемешивали в течение 60 мин., выпаривали до сухого состояния под вакуумом, растворяли в диоксане и повторно выпаривали. Остаток растворяли в ДХМ (2 мл) и раствор постепенно добавляли в процессе перемешивания к охлажденной на льду смеси гидрохлорида метилового эфира 2-аминоиндан-2-карбоновой кислоты (5,42 г, 23,8 ммоль), EA и избыточного насыщенного водного раствора гидрокарбоната натрия. Через 2 ч органическую фазу отгоняли, промывали насыщенным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Получили 582 мг указанного в заголовке соединения.
1H-ЯМР: δ = 8,86 (с, 1H); 7,5 (дд, 1H); 7,36 (д, 1H); 7,25-7,11 (м, 8H); 7,02 (д, 1H); 4,23 (т, 2H); 3,61 (д, 2H); 3,60 (с, 3H); 3,37 (д, 2H); 3,01 (т, 2H); 2,39 (с, 3H); 2,28 (с, 3H)
Пример 4
2-[4-Метилсульфанил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Соединение примера 3 (580 мг, 1,22 ммоль) гидролизировали по аналогии с примером 2 с получением 480 мг указанного в заголовке соединения.
1H-ЯМР: δ = 12,4 (уш.с, 1H); 8,73 (с, 1H); 7,50 (дд, 1H); 7,35 (д, 1H); 7,24-7,11 (м, 8H); 7,02 (д, 1H); 4,24 (т, 2H); 3,59 (д, 2H); 3,37 (д, 2H); 3,01 (т, 2H); 2,39 (с, 3H); 2,28 (с, 3H)
Пример 5
2-[4-Метансульфинил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Соединение примера 4 (100 мг, 0,216 ммоль) растворяли в уксусной кислоте (7,5 мл), добавляли перекись водорода (74 мг 30-процентного раствора в воде, 0,65 ммоль), и реакционную смесь оставляли при комнатной температуре в течение 9 ч. Смесь разделяли в EA и 1-процентном водном растворе сульфита натрия, водную фазу экстрагировали EA и органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток перемешивали с диэтиловым эфиром, фильтровали и сушили под вакуумом с получением 79 мг указанного в заголовке соединения.
ЖХ/МС (метод LC1): Rt = 1,48 мин; м/z = 478,2 [MH+]
Пример 6
2-[4-Метансульфонил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Соединение примера 4 (140 мг, 0,30 ммоль) растворяли в уксусной кислоте (7,5 мл), добавляли перекись водорода (103 мг 30-процентного раствора в воде, 0,91 ммоль), и реакционную смесь оставляли при темп. 70°C в течение 8 ч. Смесь разделяли в EA и 1-процентном водном растворе сульфита натрия, водную фазу экстрагировали EA и органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток перемешивали с диэтиловым эфиром, фильтровали и сушили под вакуумом с получением 143 мг указанного в заголовке соединения.
ЖХ/МС (метод LC1): Rt = 1,55 мин; м/z = 494,0 [MH+]
Пример 7
Метиловый эфир 2-[4-ацетил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты

4-Ацетил-3-гидроксибензойная кислота (M. E. Zwaagstra et al., J. Med. Chem. 40 (1997), 1075-1089) вступила в реакцию с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 3 примера 1. Из полученного метилового эфира 2-[4-ацетил-3-гидроксибензоиламино]-индан-2-карбоновой кислоты получили указанное в заголовке соединение в результате реакции с 2-(3-метилфенил)-этанолом по аналогии со стадией 1 примера 1.
ЖХ/МС (метод LC1): Rt = 1,83 мин; м/z = 472,2 [MH+]
Пример 8
2-[4-Ацетил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Из соединения примера 7 получили указанное в заголовке соединение путем гидролиза с гидроксидом лития по аналогии с примером 2.
ЖХ/МС (метод LC1): Rt = 1,67 мин; м/z = 458,0 [MH+]
Пример 9
2-[4-(1-Гидроксиэтил)-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Соединение примера 7 (300 мг, 0,636 ммоль) растворяли в 3 мл этанола, смесь охлаждали в ледяной бане и добавляли борогидрид натрия (24,1 мг, 0,636 ммоль). Реакционную смесь доводили до комнатной температуры и продолжали перемешивать в течение 4 ч. Смесь разделяли в EA и насыщенном водном растворе гидрокарбоната натрия, водную фазу экстрагировали EA и органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением смеси метилового эфира 2-[4-(1-гидроксиэтил)-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты и этилового эфира 2-[4-(1-гидроксиэтил)-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты.
ЖХ/МС (метод LC1): Rt = 1,70 мин; м/z = 474,2 [MH+] (метиловый эфир 2-[4-(1-гидроксиэтил)-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты)
ЖХ/МС (метод LC1): Rt = 1,76 мин; м/z = 488,2 [MH+] (этиловый эфир 2-[4-(1-гидроксиэтил)-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты)
Из смеси метилового эфира и этилового эфира получили указанное в заголовке соединение путем гидролиза с гидроксидом лития по аналогии с примером 2.
ЖХ/МС (метод LC1): Rt = 1,54 мин.; м/z = 460,2 [MH+]
Пример 10
2-[4-Этил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Соединение примера 9 (80 мг, 0,174 ммоль) растворяли в метаноле и гидрировали в реакторе гидрирования H-cube™ (ThalesNano, Будапешт, Венгрия) под давлением водорода 100 бар в присутствии 10% палладия на активированном угле. Реакционную смесь выпаривали до сухого состояния, и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN).
1H-ЯМР: δ = 12,3 (уш.с, 1H); 8,72 (с, 1H); 7,40-7,33 (м, 2H); 7,25-7,12 (м, 7H); 7,10 (д, 1H); 7,02 (д, 1H); 4,20 (т, 2H); 3,59 (д, 2H); 3,37 (д, 2H); 3,02 (т, 2H); 2,56-2,51 (м, 2H); 2,27 (с, 3H); 1,02 (т, 3H)
Пример 11
2-[4-(1-Фторэтил)-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Смесь метилового эфира 2-[4-(1-гидроксиэтил)-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты и этилового эфира 2-[4-(1-гидроксиэтил)-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты, полученную в примере 9 (50 мг) растворяли в ДХМ и добавляли двумя порциями трифторид диэтиламиносеры (33 мг, 0,204 ммоль) (вторая порция через 3,5 ч). После полного превращения, зарегистрированного с помощью ВЭЖХ, смесь разделяли в EA и насыщенном водном растворе гидрокарбоната натрия, и водную фазу экстрагировали EA. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток перемешивали со смесью HEP/диэтилового эфира, фильтровали и сушили под вакуумом. Полученный эфир гидролизировали по аналогии с примером 2 с получением 12 мг указанного в заголовке соединения.
1H-ЯМР: δ = 12,4 (уш.с, 1H); 8,82 (с, 1H); 7,49 (д, 1H); 7,41 (с, 1H); 7,37 (д, 1H); 7,25-7,12 (м, 6H); 7,10 (д, 1H); 7,03 (д, 1H); 5,83/5,74 (дкв, 1H); 4,26 (т, 2H); 3,58 (д, 2H); 3,39 (д, 2H); 3,02 (д, 2H); 2,27 (с, 3H); 1,40/1,35 (дд, 3H)
Пример 12
2-[4-Этокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: Этиловый эфир 4-этокси-3-гидроксибензойной кислоты
Этиловый эфир 3,4-дигидроксибензойной кислоты (3,00 г, 16,0 ммоль) суспендировали в ДМФ (10 мл), добавляли карбонат калия (2,21 г, 16,0 ммоль), смесь перемешивали в течение 5 мин при комнатной температуре, а затем добавляли йодоэтан (2,49 г, 16,0 ммоль). Смесь перемешивали в течение ночи, повторно добавляли карбонат калия и йодоэтан, и смесь повторно перемешивали в течение ночи. Смесь разделяли в 2н. хлористоводородной кислоте и EA, водную фазу экстрагировали EA, и объединенные органические экстракты промывали насыщенным раствором гидрокарбоната натрия и насыщенным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток очищали с помощью хроматографии на силикагеле (градиент HEP/EA).
ЖХ/МС (метод LC1): Rt = 1,28 мин; м/z = 211,1 [MH+]
Стадия 2: 2-[4-Этокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота
Из соединения стадии 1 получали указанное в заголовке соединение в результате реакции с 2-(3-метилфенил)-этанолом по аналогии со стадией 1 примера 1, гидролиза эфирной группы по аналогии с примером 2, реакции полученной карбоновой кислоты с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 1 примера 15 и гидролиза эфирной группы по аналогии с примером 2.
1H-ЯМР: δ = 12,4 (уш.с, 1H); 8,64 (с, 1H); 7,46 (д, 1H); 7,41 (с, 1H); 7,22-7,10 (м, 7H); 7,02 (д, 1H); 6,99 (д, 1H); 4,16 (т, 2H); 4,04 (кв., 2H); 3,53 (д, 2H); 3,3-3,4 (2H); 2,98 (т, 2H); 2,28 (с, 3H); 1,32 (т, 3H)
Пример 13
Метиловый эфир 2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты

Стадия 1: 4-Метокси-3-(2-м-толилэтокси)-бензойная кислота
Из метилового эфира 3-гидрокси-4-метоксибензойной кислоты указанное в заголовке соединение получили в результате реакции с 2-(3-метилфенил)-этанолом по аналогии со стадией 1 примера 1 и гидролиза эфирной группы по аналогии с примером 2.
1H-ЯМР: δ = 12,65 (уш.с, 1H); 7,56 (дд, 1H); 7,44 (д, 1H); 7,19 (т, 1H); 7,17-7,15 (м, 1H); 7,12 (д, 1H); 7,06-7,02 (м, 2H); 4,19 (т, 2H); 3,83 (с, 3H); 3,01 (т, 2H); 2,29 (с, 3H)
Стадия 2: Метиловый эфир 2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты
Соединение стадии 1 (1,98 г, 6,91 ммоль) растворяли в тионилхлориде (10 мл) и перемешивали в течение 20 мин при 60°C. Раствор выпаривали под вакуумом до сухого состояния и остаток выпаривали дважды с диоксаном под вакуумом. Остаток растворяли в небольшом количестве ДХМ и добавляли к хорошо перемешанной смеси гидрохлорида метилового эфира 2-амино-индан-2-карбоновой кислоты (1,50 г, 6,58 ммоль) в EA и избыточного насыщенного водного раствора гидрокарбоната натрия. Смесь перемешивали в течение 30 мин при комнатной температуре. Фазы разделяли, водную фазу экстрагировали EA и объединенные органические фазы промывали солевым раствором, сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток перемешивали с диэтиловым эфиром в течение ночи, фильтровали и сушили под вакуумом с получением 1,92 мг указанного в заголовке соединения.
1H-ЯМР: δ = 8,78 (с, 1H); 7,50 (дд, 1H); 7,43 (д, 1H); 7,24-7,14 (м, 6H); 7,11 (д, 1H); 7,03 (д, 1H); 7,01 (д, 1H); 4,17 (т, 2H); 3,80 (с, 3H); 3,59 (д, 2H); 3,59 (с, 3H); 3,36 (д, 2H); 3,00 (т, 2H); 2,28 (с, 3H)
Пример 14
2-[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Соединение примера 13 (1,92 мг, 4,18 ммоль) растворяли в диоксане (40 мл), добавляли гидроксид лития (10 мл 1М водного раствора) и смесь перемешивали в течение 30 мин при темп. 60°C. Смесь разделяли в 2н. хлористоводородной кислоте и EA, водную фазу экстрагировали EA, и объединенные органические экстракты промывали солевым раствором, сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток перемешивали в течение ночи в EA, фильтровали и кристаллы сушили под вакуумом с получением 1,35 г указанного в заголовке соединения.
1H-ЯМР: δ = 12,36 (уш.с, 1H); 8,63 (с, 1H); 7,50 (дд, 1H); 7,43 (д, 1H); 7,23-7,13 (м, 6H); 7,11 (д, 1H); 7,03 (д, 1H); 7,00 (д, 1H); 4,17 (т, 2H); 3,79 (с, 3H); 3,58 (д, 2H); 3,37 (д, 2H); 3,00 (т, 2H); 2,28 (с, 3H)
Пример 15
Метиловый эфир 2-{4-метокси-3-[2-(3-метилсульфанилфенил)-этокси]-бензоиламино}-индан-2-карбоновой кислоты

Стадия 1: Метиловый эфир 2-(3-ацетокси-4-метоксибензоиламино)-индан-2-карбоновой кислоты
3-Ацетокси-4-метоксибензойную кислоту (5,00 г, 23,8 ммоль) растворяли в ДХМ (50 мл). ДМФ (167 мг, 2,38 ммоль) и оксалилхлорид (35,6 мл 2 М раствора в ДХМ) добавляли при комнатной температуре. Смесь перемешивали в течение 20 мин, выпаривали до сухого состояния под вакуумом, остаток повторно растворяли в ДХМ и повторно выпаривали. Остаток растворяли в ДХМ и постепенно добавляли к перемешанной смеси гидрохлорида метилового эфира 2-аминоиндан-2-карбоновой кислоты (5,42 г, 23,8 ммоль), EA и избыточного насыщенного водного раствора гидрокарбоната натрия. Через 90 мин органическую фазу отгоняли и промывали насыщенным раствором гидрокарбоната натрия, 2 М хлористоводородной кислоты и насыщенным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния с получением 8,95 г указанного в заголовке соединения.
1H-ЯМР: δ = 8,88 (с, 1H); 7,80 (дд, 1H); 7,62 (д, 1H); 7,28-7,13 (м, 5H); 3,81 (с, 3H); 3,60 (с, 3H); 3,58 (д, 2H); 3,37 (д, 2H); 2,27 (с, 3H)
Стадия 2: Метиловый эфир 2-(3-гидрокси-4-метоксибензоиламино)-индан-2-карбоновой кислоты
Соединение стадии 1 (3,44 г, 8,97 ммоль) растворяли в метаноле (50 мл), добавляли карбонат калия (248 мг, 1,79 ммоль) и смесь перемешивали в течение 2 ч при комнатной температуре. Смесь выпаривали до сухого состояния, остаток разделяли в EA и 1н. хлористоводородной кислоте и водную фазу экстрагировали EA. Объединенные органические экстракты сушили над сульфатом магния, фильтровали и выпаривали до сухого состояния с получением 2,80 г указанного в заголовке соединения.
1H-ЯМР: δ = 9,14 (с, 1H); 8,71 (с, 1H); 7,32 (дд, 1H); 7,29 (д, 1H); 7,24-7,20 (м, 2H); 7,19-7,13 (м, 2H); 6,93 (д, 2H); 3,80 (с, 3H); 3,60 (с, 3H); 3,56 (д, 2H); 3,38 (д, 2H)
Стадия 3: Метиловый эфир 2-{4-метокси-3-[2-(3-метилсульфанилфенил)-этокси]-бензоиламино}-индан-2-карбоновой кислоты
Соединение стадии 2 (0,380 г, 1,11 ммоль) и трифенилфосфин (0,461 г, 1,67 ммоль) растворяли в ТГФ. Добавляли 2-(3-метилсульфанилфенил)-этанол (0,281 г, 1,67 ммоль) и ДИАД (0,359 г, 1,67 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Летучие фракции удаляли под вакуумом, и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением 0,217 г указанного в заголовке соединения.
1H-ЯМР: δ = 8,78 (с, 1H); 7,50 (д, 1H); 7,41 (с, 1H); 7,28-7,20 (м, 4H); 7,20-7,13 (м, 2H); 7,13-7,08 (м, 2H); 7,00 (д, 1H); 4,18 (т, 2H); 3,79 (с, 3H); 3,62-3,55 (м, 5H); 3,38 (д, 2H); 3,01 (т, 2H); 2,45 (с, 3H)
Пример 16
2-{4-Метокси-3-[2-(3-метилсульфанилфенил)-этокси]-бензоиламино}-индан-2-карбоновая кислота

Соединение примера 15 (195 мг, 0,397 ммоль) растворяли в диоксане (2 мл), добавляли гидроксид лития (1,99 мл 1М водного раствора, 1,99 ммоль) и перемешивали смесь при темп. 60°C в течение 1 ч. Смесь разделяли в 2н. хлористоводородной кислоте и EA, водную фазу экстрагировали EA и объединенные органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния с получением 180 мг указанного в заголовке соединения.
1H-ЯМР: δ = 12,37 (уш.с, 1H); 8,65 (с, 1H); 7,47 (д, 1H); 7,41 (с, 1H); 7,29-7,18 (м, 4H); 7,18-7,08 (м, 4H); 7,01 (д, 1H); 4,17 (т, 2H); 3,79 (с, 3H); 3,54 (д, 2H); 3,37 (д, 2H); 3,01 (т, 2H); 2,45 (с, 3H)
Пример 17
2-{3-[2-(3-Метансульфинилфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Соединение примера 16 (35 мг, 0,078 ммоль) растворяли в уксусной кислоте (2,5 мл), добавляли перекись водорода (43 мг 30-процентного раствора в воде, 0,38 ммоль), и смесь перемешивали при комнатной температуре в течение 2 ч. Смесь разделяли в EA и 1-процентном водном растворе сульфита натрия, водную фазу экстрагировали EA, и объединенные органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния с получением 36 мг указанного в заголовке соединения.
1H-ЯМР: δ = 12,3 (уш.с, 1H); 8,62 (с, 1H); 7,65 (с, 1H); 7,56-7,47 (м, 4H); 7,43 (д, 1H); 7,23-7,19 (м, 2H); 7,17-7,12 (м, 2H); 7,00 (д, 1H); 4,24 (т, 2H); 3,80 (с, 3H); 3,57 (д, 2H); 3,38 (д, 2H); 3,14 (т, 2H); 2,72 (с, 3H)
Пример 18
2-{3-[2-(3-Метансульфонилфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Указанное в заголовке соединение синтезировали по аналогии с примером 17, с тем отличием, что температура реакции была 70°C. Выход: 36 мг.
1H-ЯМР: δ = 12,35 (уш.с, 1H); 8,64 (с, 1H); 7,92 (с, 1H); 7,78 (д, 1H); 7,70 (д, 1H); 7,58 (дд, 1H); 7,49 (дд, 1H); 7,42 (д, 1H); 7,22-7,19 (м, 2H); 7,18-7,12 (м, 2H); 7,00 (д, 1H); 4,24 (т, 2H); 3,79 (с, 3H); 3,56 (д, 2H); 3,37 (д, 2H); 3,19 (с, 3H); 3,18 (т, 2H)
По аналогии с вышеприведенными примерами подготовили образцы соединений формулы Im, перечисленные в таблице 1. В формулах групп R90 в таблице 1 линия, перечеркнутая знаком










































































Пример 92 (исходное соединение)
(1-м-Толилциклопропил)-метанол

м-Толилацетонитрил (1,00 г, 7,62 ммоль) и 1,2-дибромэтан (1,86 г, 9,91 ммоль) растворяли в ДМФ (5 мл). Смесь охлаждали в ледяной бане и медленно добавляли при перемешивании трет-бутоксид калия (855 мг, 19,1 ммоль). После перемешивания в течение 30 мин смесь разделяли в EA и воде. Органический слой промывали водой, сушили над хлоридом натрия, сливали и выпаривали до сухого состояния. После очистки остатка хроматографией на силикагеле (градиент HEP/EA) получали смесь примерно 1:1 смесь 1-м-толилциклопропанкарбонитрила и исходного соединения м-толилацетонитрила.
Полученную смесь нитрилов (500 мг) растворяли в этаноле (2 мл) и 50-процентном водном растворе гидроксида калия (2 мл). Смесь в укупоренном сосуде нагревали в микроволновом реакторе при 140°С в течение 4 ч. Затем смесь разделяли в EA и 2н. хлористоводородной кислоте, водную фазу экстрагировали EA и объединенные органические экстракты сушили над хлоридом натрия, сливали и выпаривали до сухого состояния с получением смеси амида 1-м-толилциклопропанкарбоновой кислоты и м-толилацетамида.
Полученную смесь амидов (600 мг) растворяли в уксусной кислоте (8,5 мл) и уксусном ангидриде (14,5 мл), смесь охлаждали в ледяной бане, добавляли нитрит натрия (1,97 г, 28,5 ммоль) и смесь перемешивали в течение 2 ч при комнатной температуре. Добавляли воду (15 мл) и смесь нагревали до 60°С в течение 30 мин. После выпаривания до сухого состояния смесь разделяли в EA и 2н. хлористоводородной кислоте, водную фазу экстрагировали EA и объединенные органические экстракты сушили над сульфатом натрия, сливали и выпаривали до сухого состояния с получением смеси приблизительно 1:1 1-м-толилциклопропанкарбоновой кислоты и м-толилуксусной кислоты.
Полученную смесь кислот (430 мг) растворяли в диметоксиэтане (8 мл) и добавляли при перемешивании NMM (272 мг, 2,68 ммоль) и хлорформиат изобутила (367 мг, 2,68 ммоль). Через несколько минут смесь фильтровали и в прозрачный фильтрат добавляли борогидрид натрия (369 мг, 9,76 ммоль). После осторожного добавления воды (4 мл; при бурном образовании водорода) продолжали перемешивать в течение нескольких минут до прекращения реакции, смесь разделяли в EA и 2н. хлористоводородной кислоте, водную фазу экстрагировали EA и объединенные органические экстракты сушили над сульфатом натрия, сливали и выпаривали до сухого состояния. Данный остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением 133 мг указанного в заголовке соединения.
1H-ЯМР: δ = 7,18-7,06 (м, 3H); 6,98 (д, 1H); 4,59 (т, 1H); 3,51 (д, 2H); 0,83-0,78 (м, 2H); 0,70-0,67 (м, 2H)
Пример 93 (исходное соединение)
2-(2-Фтор-5-метилфенил)-этанол

По аналогии с методом, описанным в M. Jørgensen et al., J. Am. Chem. Soc. 124 (2002), 12557-12565, в первой колбе дициклогексиламин (3,06 г, 16,9 ммоль) растворяли в толуоле и охлаждали в ледяной бане. Добавляли н-бутиллитий (6,14 мл, 2,5 М раствор в гексане). Через 5 мин медленно добавляли трет-бутилацетат (1,78 г, 15,3 ммоль). Во вторую колбу поместили тетрафторборат три-(трет-бутил)фосфоний (83 мг, 0,30 ммоль) и трис(дибензилиденацетон)дипалладий(0) (146 мг, 0,153 ммоль) и тщательно продували аргоном. Добавляли толуол (100 мл), затем 3-бром-4-фтортолуол (2,90 г, 15,3 ммоль) и содержимое первой колбы. После перемешивания в течение ночи образовавшуюся суспензию фильтровали через тонкий слой силикагеля, который повторно промывали диэтиловым эфиром. Фильтрат выпаривали под вакуумом и остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 2,81 г трет-бутилового эфира (2-фтор-5-метилфенил)-уксусной кислоты.
1H-ЯМР: δ = 7,12-7,07 (м, 2H); 7,03 (т, 1H); 3,54 (с, 2H); 2,25 (с, 3H); 1,39 (с, 9H)
В колбу поместили алюмогидрид лития (0,846 г, 22,3 ммоль) и продували аргоном. Добавляли ТГФ (8 мл) и полученный трет-бутиловый эфир (2-фтор-5-метилфенил)-уксусной кислоты медленно добавляли при перемешивании. Реакция началась непосредственно после этого. Через 2 мин добавили диэтиловый эфир (30 мл) и EA (2,5 мл). Затем осторожно и постепенно добавляли воду при перемешивании до образования сероватого осадка. Раствор слили и осадок промыли EA. Объединенные растворы сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 0,55 г указанного в заголовке соединения.
1H-ЯМР: δ = 7,10 (д, 1H); 7,05-6,96 (м, 2H); 4,70 (т, 1H); 3,56 (дт, 2H); 2,71 (т, 2H); 2,25 (с, 3H)
Пример 94
2-{3-[2-(3-Цианофенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 3-[2-(3-бромфенил)-этокси]-4-метоксибензойной кислоты
Метил-3-гидрокси-4-метоксибензоат (500 мг, 2,75 ммоль) и трифенилфосфин (1,08 г, 4,12 ммоль) растворяли в ТГФ (13 мл), раствор охлаждали в ледяной бане и последовательно добавляли 2-(3-бромфенил)-этанол (662 мг, 3,29 ммоль) и ДИАД (886 мг, 4,12 ммоль). Перемешивание продолжали в течение 3 ч при комнатной температуре. Реакционную смесь выпаривали до сухого состояния, и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением 900 мг указанного в заголовке соединения.
1H-ЯМР: δ = 7,62-7,58 (м, 2H); 7,45 (д, 1H); 7,41 (д, 1H); 7,35 (д, 1H); 7,28 (дд, 1H); 7,08 (д, 1H); 4,22 (т, 2H); 3,83 (с, 3H); 3,80 (с, 3H); 3,01 (т, 2H)
Стадия 2: Метиловый эфир 3-[2-(3-цианофенил)-этокси]-4-метоксибензойной кислоты
В колбу поместили цианид цинка (129 мг, 1,10 ммоль) и тетракис(трифенилфосфин)палладий(0) (63 мг, 0,0547 ммоль). В атмосфере аргона к смеси добавляли раствор стадии 1 (400 мг, 1,10 ммоль) в ДМФ (1,9 мл). После перемешивания при 150°С в течение 1 ч и охлаждения смесь разбавляли трет-бутилметиловым эфиром и фильтровали через целит. Фильтрат промывали водой, сушили над сульфатом магния, фильтровали и выпаривали до сухого состояния с получением 275 мг указанного в заголовке соединения.
1H-ЯМР: δ = 7,85 (с, 1H); 7,70 (дд, 1H); 7,58 (дд, 1H); 7,51 (дд, 1H); 7,44 (д, 1H); 7,07 (д, 1H); 4,26 (т, 2H); 3,82 (с, 3H); 3,80 (с, 3H); 3,11 (т, 2H)
Стадия 3: 3-[2-(3-Цианофенил)-этокси]-4-метоксибензойная кислота
Соединение стадии 2 (274 мг, 0,883 ммоль) растворяли в диоксане (4,5 мл), добавляли гидроксид лития (4,42 мл 1М водного раствора, 4,42 ммоль) и перемешивали смесь при темп. 60°C в течение 30 мин. Смесь разделяли в 2н. хлористоводородной кислоте и EA, водную фазу экстрагировали EA и объединенные органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением 160 мг указанного в заголовке соединения.
1H-ЯМР: δ = 12,65 (уш.с, 1H); 7,83 (с, 1H); 7,73-7,68 (м, 2H); 7,60-7,50 (м, 2H); 7,45 (д, 1H); 7,04 (д, 1H); 4,25 (т, 2H); 3,81 (с, 3H); 3,11 (т, 2H)
Стадия 4: Метиловый эфир 2-{3-[2-(3-цианофенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновой кислоты
Из соединения стадии 3 и гидрохлорида метилового эфира 2-аминоиндан-2-карбоновой кислоты получали указанное в заголовке соединение с выходом 79%, по аналогии со стадией 1 примера 15.
ЖХ/МС (метод LC2): Rt = 1,63 мин; м/z = 471,1 [MH+]
Стадия 5: 2-{3-[2-(3-цианофенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота
Из соединения стадии 4 получали указанное в заголовке соединение с выходом 37% путем гидролиза с гидроксидом лития по аналогии со стадией 3, с тем отличием, что реакция проводилась при комнатной температуре.
1H-ЯМР: δ = 12,35 (уш.с, 1H); 8,61 (с, 1H); 7,84 (с, 1H); 7,72-7,68 (2d, 2H); 7,54-7,49 (м, 2H); 7,43 (д, 1H); 7,24-7,20 (м, 2H); 7,18-7,13 (м, 2H); 7,00 (д, 1H); 4,22 (т, 2H); 3,79 (с, 3H); 3,58 (д, 2H); 3,37 (д, 2H); 3,11 (т, 2H)
Пример 95
2-{3-[2-(3-Карбамоилфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Соединение примера 94 вступало в реакцию с гидроксидом лития при температуре 60°С в течение 50 мин, по аналогии со стадией 3 примера 94. Полученную смесь указанного в заголовке соединения и 2-{3-[2-(3-карбоксифенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновой кислоты (пример 96) разделяли препаративной ОФ ВЭЖХ (градиент вода/ACN).
1H-ЯМР: δ = 12,3 (с, 1H); 8,61 (с, 1H); 7,91 (с, 1H); 7,83 (с, 1H); 7,72 (д, 1H); 7,53-7,47 (м, 2H); 7,45 (д, 1H); 7,37 (т, 1H); 7,31 (с, 1H); 7,23-7,19 (м, 2H); 7,17-7,12 (м, 2H); 7,00 (д, 1H); 4,22 (т, 2H); 3,78 (с, 3H); 3,59 (д, 2H); 3,38 (д, 2H); 3,10 (т, 2H)
Пример 96
2-{3-[2-(3-Карбоксифенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Указанное в заголовке соединение получили в соответствии с методами, описанными в примере 95.
1H-ЯМР: δ = 13,0-12,2 (уш., 2H); 8,62 (с, 1H); 7,94 (с, 1H); 7,80 (д, 1H); 7,58 (д, 1H); 7,50 (дд, 1H); 7,47-7,42 (м, 2H); 7,22-7,19 (м, 2H); 7,17-7,12 (м, 2H); 7,00 (д, 1H); 4,22 (т, 2H); 3,78 (с, 3H); 3,59 (д, 2H); 3,37 (д, 2H); 3,12 (т, 2H)
Пример 97
Этиловый эфир 5-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-5,6-дигидро-4H-циклопента[c]тиофен-5-карбоновой кислоты

Этиловый эфир (бензгидрилиденамино)-уксусной кислоты (0,113 г, 0,414 ммоль) растворяли в ДМФ (3 мл) и охлаждали в ледяной бане. Добавляли трет-бутоксид калия (94,8 мг, 0,828 ммоль) и смесь перемешивали в течение 10 мин. Смесь охлаждали до -30°С и добавляли одной порцией 3,4-бис-хлорметилтиофен (50 мг, 0,276 ммоль). Затем смесь снова помещали в ледяную баню и реакцию проводили в течение 20 мин. Смесь подкисляли 2н. хлористоводородной кислоты, перемешивали в течение 10 мин и разделяли в воде и диэтиловом эфире. Водную фазу нейтрализовали насыщенным раствором гидрокарбоната натрия и экстрагировали EA. Объединенные экстракты EA промывали насыщенным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток растворяли в диэтиловом эфире, фильтровали, выпаривали до сухого состояния, подкисляли хлористым водородом в метаноле и выпаривали до сухого состояния. Остаток перемешивали со смесью диэтилового эфира/HEP и твердое вещество отфильтровывали и сушили под вакуумом для получения неочищенного гидрохлорида этилового эфира 5-амино-5,6-дигидро-4Н-циклопента[с]тиофен-5-карбоновой кислоты. Часть неочищенного соединения (27 мг, 0,109 ммоль) вступала в реакцию без дополнительной очистки с 4-метокси-3-(2-м-толилэтокси)-бензойной кислотой по аналогии со стадией 1 примера 15. После очистки препаративной ОФ ВЭЖХ (градиент вода/ACN) получили 23 мг указанного в заголовке соединения.
1H-ЯМР: δ = 8,78 (с, 1H); 7,50 (дд, 1H); 7,41 (д, 1H); 7,22-7,14 (м, 2H); 7,11 (д, 1H); 7,08-6,99 (м, 4H); 4,18 (т, 2H); 4,05 (кв., 2H); 3,80 (с, 3H); 3,33 (д, 2H); 3,13 (д, 2H); 3,00 (т, 2H); 2,28 (с, 3H); 1,09 (т, 3H)
Пример 98
5-[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-5,6-дигидро-4H-циклопента[c]тиофен-5-карбоновая кислота

Соединение примера 97 (21 мг, 0,0438 ммоль) гидролизировали по аналогии со стадией 3 примера 94. После выпаривания до сухого состояния остаток перемешивали с диэтиловым эфиром, фильтровали и сушили под вакуумом с получением 16 мг указанного в заголовке соединения.
1H-ЯМР: δ = 8,69 (с, 1H); 7,48 (д, 1H); 7,41 (с, 1H); 7,21-7,09 (м, 3H); 7,05-6,98 (м, 4H); 4,19 (т, 2H); 3,80 (с, 3H); 3,30 (д, 2H); 3,11 (д, 2H); 3,00 (т, 2H); 2,29 (с, 3H)
Пример 99
5-[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-5,6-дигидро-4H-циклопента[b]тиофен-5-карбоновая кислота
Начиная с 2,3-бис-хлорметилтиофена, указанное в заголовке соединение получали по аналогии с примерами 97 и 98.
ЖХ/МС (метод LC1): Rt = 1,60 мин.; м/z = 452,0 [MH+]
Пример 100
2-Хлор-5-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-5,6-дигидро-4H-циклопента[b]тиофен-5-карбоновая кислота

Стадия 1: 5-хлор-2,3-бис-хлорметилтиофен
2,3-бис-хлорметилтиофен (500 мг, 2,76 ммоль) растворяли в уксусной кислоте (10 мл). Добавляли сульфурилхлорид (372 мг, 2,76 ммоль) и смесь перемешивали в течение 1 ч при комнатной температуре. Смесь разделяли в EA, воде и избытке гидрокарбоната натрия в твердом состоянии и водную фазу экстрагировали EA. Объединенные органические экстракты сушили над сульфатом магния, фильтровали и выпаривали до сухого состояния с получением 240 мг указанного в заголовке соединения.
1H-ЯМР: δ = 7,13 (с, 1H); 5,16 (с, 2H); 4,76 (с, 2H)
Стадия 2: 2-Хлор-5-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-5,6-дигидро-4H-циклопента[b]тиофен-5-карбоновая кислота
Из соединения стадии 1 получали указанное в заголовке соединение в результате реакции с этиловым эфиром (бензгидрилиденамино)-уксусной кислоты по аналогии со стадией 1 примера 97, реакции с 4-метокси-3-(2-м-толилэтокси)-бензойной кислотой по аналогии со стадией 3 примера 1 и гидролиза эфира по аналогии с примером 2.
ЖХ/МС (метод LC1): Rt = 1,74 мин.; м/z = 486,0/488,0 [MH+]
Пример 101
6-[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-6,7-дигидро-5H-[1]пириндин-6-карбоновая кислота

Стадия 1: Этиловый эфир 6-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-6,7-дигидро-5H-[1]пириндин-6-карбоновой кислоты
Метиловый эфир изоцианоуксусной кислоты (112 мг, 1,13 ммоль) и 2,3-бис-хлорметилпиридин (200 мг, 1,14 ммоль) растворяли в ДМФ. Добавляли трет-бутоксид калия (0,255 г, 2,27 ммоль) и реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Смесь разделяли в EA и насыщенном водном растворе гидрокарбоната натрия, водную фазу экстрагировали EA и органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (градиент HEP/EA) для получения метилового эфира 6-изоциано-6,7-дигидро-5Н-[1]пириндин-6-карбоновой кислоты. Это соединение добавляли к раствору тионилхлорида (147 мг, 1,23 ммоль) в этаноле (1 мл) и нагревали с обратным холодильником в течение ночи. Смесь выпаривали до сухого состояния и остаток перемешивали с HEP и сушили под вакуумом. Полученный продукт вступал в реакцию с 4-метокси-3-(2-м-толилэтокси)-бензойной кислотой по аналогии со стадией 1 примера 15 и указанное в заголовке соединение очищали препаративной ОФ ВЭЖХ (градиент вода/ACN).
ЖХ/МС (метод LC1): Rt = 1,22 мин; м/z = 475,2 [MH+]
Стадия 2: 6-[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-6,7-дигидро-5H-[1]пириндин-6-карбоновая кислота
Соединение стадии 1 гидролизировали с гидроксидом лития по аналогии со стадией 3 примера 94 с получением 5 мг указанного в заголовке соединения.
ЖХ/МС (метод LC1): Rt = 1,11 мин; м/z = 447,1 [MH+]
Пример 102
2-{[5-Ацетил-4-(2-м-толилэтокси)-тиофен-2-карбонил]-амино}-индан-2-карбоновая кислота

Указанное в заголовке соединение синтезировали в результате реакции метилового эфира 5-ацетил-4-гидрокситиофен-2-карбоновой кислоты с 2-(3-метилфенил)-этанолом по аналогии со стадией 1 примера 1, последующего гидролиза эфира по аналогии с примером 2, реакции с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 1 примера 15 и гидролиза эфира по аналогии с примером 2.
ЖХ/МС (метод LC1): Rt = 1,66 мин; м/z = 464,0 [MH+]
Пример 103
2-{5-[2-(3-Хлорфенил)-этокси]-4-метокси-2-нитробензоиламино}-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 5-[2-(3-хлорфенил)-этокси]-4-метокси-2-нитробензойной кислоты
Метил-3-гидрокси-4-метоксибензоат и 2-(3-хлорфенил)-этанол вступали в реакцию по аналогии со стадией 1 примера 1. Полученный метиловый эфир 3-[2-(3-хлорфенил)-этокси]-4-метоксибензойной кислоты (0,750 г, 2,34 ммоль) медленно добавляли к охлажденной в ледяной бане 100% азотной кислоте (10 мл). Ледяную баню удаляли, и перемешивание продолжали в течение ночи. Смесь осторожно переносили в перемешиваемую смесь EA, воды и избытка гидрокарбоната натрия и экстрагировали EA. Объединенные органические экстракты промывали насыщенным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Твердый остаток экстрагировали диэтиловым эфиром и эфирный раствор выпаривали. Остаток перемешивали с HEP и твердое вещество отфильтровывали и сушили под вакуумом с получением 0,680 г указанного в заголовке соединения.
1H-ЯМР: δ = 7,63 (с, 1H); 7,45 (с, 1H); 7,37 (с, 1H); 7,37-7,27 (м, 3H); 4,36 (т, 2H); 3,90 (с, 3H); 3,81 (с, 3H); 3,10 (т, 2H)
Стадия 2: 2-{5-[2-(3-Хлорфенил)-этокси]-4-метокси-2-нитробензоиламино}-индан-2-карбоновая кислота
Из соединения стадии 1 получали указанное в заголовке соединение в результате гидролиза эфирной группы по аналогии с примером 2, реакции полученной карбоновой кислоты с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 1 примера 15 и гидролиза эфирной группы по аналогии с примером 2.
1H-ЯМР: δ = 12,5 (с, 1H); 9,08 (с, 1H); 7,59 (с, 1H); 7,45 (с, 1H); 7,38-7,28 (м, 3H); 7,25-7,20 (м, 2H); 7,18-7,12 (м, 2H); 6,97 (с, 1H); 4,31 (т, 2H); 3,87 (с, 3H); 3,56 (д, 2H); 3,3 (д, 2H); 3,10 (т, 2H)
Пример 104
2-{2-Бром-5-[2-(3-хлорфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 2-бром-5-[2-(3-хлорфенил)-этокси]-4-метоксибензойной кислоты
Метил-3-гидрокси-4-метоксибензоат и 2-(3-хлорфенил)-этанол вступали в реакцию по аналогии со стадией 1 примера 1. Полученный метиловый эфир 3-[2-(3-хлорфенил)-этокси]-4-метоксибензойной кислоты (300 мг, 0,935 ммоль) и ацетат натрия (230 мг, 2,81 ммоль) растворяли в уксусной кислоте (10 мл), добавляли бром (224 мг, 1,40 ммоль) и смесь перемешивали при 95 °C с контролем реакции каждый час. Когда реакция прекратилась, дополнительно добавляли бром. Через 5 ч реакция была завершена. Летучие фракции выпаривали под вакуумом, остаток разделяли в EA и насыщенном водном растворе гидрокарбоната натрия и водную фазу экстрагировали EA. Объединенные органические экстракты промывали насыщенным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Данный остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 180 мг указанного в заголовке соединения.
1H-ЯМР: δ = 7,45-7,42 (м, 1H); 7,38 (с, 1H); 7,36-7,26 (м, 3H); 7,25 (с, 1H); 4,21 (т, 2H); 3,85 (с, 3H); 3,80 (с, 3H); 3,04 (т, 2H)
Стадия 2: 2-{2-бром-5-[2-(3-хлорфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота
Из соединения стадии 1 получали указанное в заголовке соединение в результате гидролиза эфирной группы по аналогии с примером 2, реакции полученной карбоновой кислоты с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 1 примера 15 и гидролиза эфирной группы по аналогии с примером 2.
ЖХ/МС (метод LC1): Rt = 1,64 мин; м/z = 544,0/546,0 [MH+]
Пример 105
2-{2-Хлор-5-[2-(3-хлорфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 2-хлор-5-[2-(3-хлорфенил)-этокси]-4-метоксибензойной кислоты
Метил-3-гидрокси-4-метоксибензоат и 2-(3-хлорфенил)-этанол вступали в реакцию по аналогии со стадией 1 примера 1. Полученный метиловый эфир 3-[2-(3-хлорфенил)-этокси]-4-метоксибензойной кислоты (300 мг, 0,935 ммоль), N-хлорсукцинимид (381 мг, 2,81 ммоль) и тетрахлорид циркония (129 мг, 0,57 ммоль) суспендировали в ДХМ (4 мл) и смесь перемешивали с обратным холодильником в течение 5 ч. до полного использования исходного материала. Смесь разделяли в EA и насыщенном водном растворе гидрокарбоната натрия, водную фазу экстрагировали EA и объединенные органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 118 мг указанного в заголовке соединения.
ЖХ/МС (метод LC1): Rt = 1,82 мин; м/z = 355,0/357,0 [MH+]
Стадия 2: 2-{2-Хлор-5-[2-(3-хлорфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота
Из соединения стадии 1 получали указанное в заголовке соединение в результате гидролиза эфирной группы по аналогии с примером 2, реакции полученной карбоновой кислоты с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 1 примера 15 и гидролиза эфирной группы по аналогии с примером 2.
ЖХ/МС (метод LC1): Rt = 1,64 мин; м/z = 500,1/502,1 [MH+]
Пример 106
2-[3-Фтор-4-метокси-5-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: 3-Ацетокси-5-амино-4-метокси-бензойная кислота
3-ацетокси-4-метокси-5-нитробензойную кислоту (4,50 г, 17,6 ммоль) (R. T. Borchardt et al., J. Med. Chem. 25 (1982), 312-323; F. Tiemann et al., Ber. dt. Chem. Ges. 9 (1876), 937) растворяли в этаноле (180 мл) и 0,5 М хлористом водороде в метаноле (4 мл) и гидрировали в реакторе гидрирования H-cube™ под давлением водорода 100 бар при темп. 40°С в присутствии 10% палладия на активированном угле. Смесь выпаривали до сухого состояния с получением 4,1 г указанного в заголовке соединения.
ЖХ/МС (метод LC1): Rt = 0,75 мин; м/z = 226,0 [MH+]
Стадия 2: 3-Фтор-5-гидрокси-4-метоксибензойная кислота
Соединение стадии 1 (2,0 г, 8,88 ммоль) растворяли в водном растворе тетрафторборной кислоты (48%, 4,5 мл), добавляли нитрит натрия (612 мг, 8,88 ммоль) при 0°С и смесь перемешивали при комнатной температуре в течение 60 мин. Летучие фракции выпаривали, добавляли толуол в маслянистый остаток и смесь нагревали при 100°С в течение 4 ч. Смесь разделяли в EA и 2н хлористоводородной кислоте, водную фазу экстрагировали EA и объединенные органические экстракты сушили над хлоридом натрия, сливали и выпаривали до сухого состояния. Остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением 170 мг указанного в заголовке соединения.
1H-ЯМР: δ = 12,9 (уш.с, 1H); 10,1 (с, 1H); 7,29 (д, 1H); 7,18 (дд, 1H); 3,85 (с, 3H)
Стадия 3: 3-Ацетокси-5-фтор-4-метоксибензойная кислота
Соединение стадии 2 (169 мг, 913 ммоль) суспендировали в уксусном ангидриде (1,75 мл) и нагревали при температуре 100°С в течение 3 ч. Раствор охлаждали, добавляли воду (2 мл) и перемешивали смесь при 60°С в течение 1 ч. После охлаждения образовались кристаллы, которые отфильтровывали и высушивали под вакуумом с получением 120 мг указанного в заголовке соединения.
1H-ЯМР: δ = 13 (уш.с, 1H); 7,67 (дд, 1H); 7,55 (д, 1H); 3,92 (с, 3H); 2,32 (с, 3H)
Стадия 4: Метиловый эфир 2-(3-фтор-5-гидрокси-4-метоксибензоиламино)-индан-2-карбоновой кислоты
Соединение стадии 3 (120 мг, 0,526 ммоль) вступало в реакцию с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 1 примера 15. Полученный продукт растворяли в метаноле (0,77 мл), добавляли карбонат калия (2 мг) и смесь перемешивали при комнатной температуре в течение 30 мин. Растворитель выпаривали, остаток разделяли в EA и насыщенном растворе хлорида натрия и водную фазу экстрагировали EA. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния с получением 60 мг указанного в заголовке соединения.
ЖХ/МС (метод LC1): Rt = 1,35 мин; м/z = 360,0 [MH+]
Стадия 5: 2-[3-Фтор-4-метокси-5-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота
Из соединения стадии 4 получали указанное в заголовке соединение в результате реакции с 2-(3-метилфенил)-этанолом по аналогии со стадией 1 примера 1 и гидролиза по аналогии с примером 2.
1H-ЯМР: δ = 12,4 (уш.с, 1H); 8,76 (с, 1H); 7,92-7,84 (м, 2H); 7,25-7,09 (м, 7H); 7,03 (д, 1H); 4,27 (т, 2H); 3,73 (с, 3H); 3,60 (д, 2H); 3,37 (д, 2H); 3,05 (т, 2H); 2,28 (с, 3H)
Пример 107
2-[4-Метокси-3-нитро-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали из 3-ацетокси-4-метокси-5-нитробензойной кислоты в результате реакции с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты, гидролиза группы ацетокси по аналогии со стадией 4 примера 106 и реакции полученного продукта с 2-(3-метилфенил)-этанолом при последующем гидролизе эфира по аналогии со стадией 5 примера 106.
ЖХ/МС (метод LC1): Rt = 1,77 мин; м/z = 491,0 [MH+]
Пример 108
2-{[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-метиламино}-индан-2-карбоновая кислота

Стадия 1: 1,3-диметилспиро(имидазолидин-5,2'-индан)-2,4-дион
Спиро[имидазолидин-4,2'-индан]-2,5-дион(2-инданон гидантоин) (200 мг, 0,989 ммоль) и трет-бутоксид калия (255 мг, 2,28 ммоль) суспендировали в ДМФ (2 мл) и перемешивали в течение 20 мин. при комнатной температуре. После добавления йодометана (323 мг, 2,28 ммоль) смесь перемешивали в течение ночи. Повторно добавляли трет-бутоксид калия, перемешивали в течение 20 мин, добавляли йодометан и перемешивали в течение ночи при комнатной температуре. Реакционную смесь разделяли в EA и 2н. хлористоводородной кислоте, водную фазу экстрагировали EA и объединенные органические экстракты сушили над хлоридом натрия, сливали и выпаривали до сухого состояния. После очистки остатка хроматографией на силикагеле получали смесь монометилированного и диметилированного продукта. Данную смесь растворяли в 3:1 смеси 0,3н раствора гидроксида калия и диоксана и перемешивали в течение ночи при комнатной температуре. Смесь разделяли в EA и воде и водную фазу экстрагировали EA. Объединенные органические экстракты сушили над хлоридом натрия, сливали и выпаривали до сухого состояния с получением 90 мг указанного в заголовке соединения.
ЖХ/МС (метод LC1): Rt = 1,10 мин; м/z = 231,1 [MH+]
Стадия 2: 2-{[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-метиламино}-индан-2-карбоновая кислота
Соединение стадии 1 (90 мг, 0,391 ммоль) растворяли в смеси метанола и 50-процентного раствора гидроксида натрия и перемешивали в микроволновом реакторе при темп. 140°С в течение 3 ч до завершения гидролиза. Смесь выпаривали до сухого состояния и остаток суспендировали в смеси воды (6 мл) и диоксана (3 мл) и охлаждали в ледяной бане. Избыток 4-метокси-3-(2-м-толилэтокси)-бензоилхлорида, который приготовили непосредственно перед применением путем растворения соответствующей бензойной кислоты в тионилхлориде, перемешивания смеси при 60°С в течение 20 мин, выпаривания до сухого состояния и растворения остатка в диоксане, постепенно добавляли в смесь в процессе перемешивания. Реакционную смесь перемешивали в течение 1 ч в ледяной бане. Затем смесь разделяли в 2н. хлористоводородной кислоте и EA, водную фазу экстрагировали EA и объединенные органические экстракты сушили над хлоридом натрия, сливали и выпаривали до сухого состояния под вакуумом. Остаток очищали хроматографией на силикагеле (градиент ДХМ/метанол/ гидроксид аммония).
1H-ЯМР: δ = 12,3 (уш.с, 1H); 7,25-7,08 (м, 7H); 7,05-6,97 (м, 4H); 4,15 (т, 2H); 3,79 (с, 3H); 3,63 (д, 2H); 3,40 (д, 2H); 3,00 (т, 2H); 2,98 (с, 3H); 2,27 (с, 3H)
Пример 109
2-(3-Бензолсульфонилокси-4-метоксибензоиламино)-индан-2-карбоновая кислота

Соединение стадии 2 примера 15 (200 мг, 0,586 ммоль) растворяли в ACN (3 мл), добавляли карбонат калия (243 мг, 1,7 ммоль) и бензолсульфонилхлорид (155 мг, 0,88 ммоль) и смесь перемешивали в течение 30 мин. Смесь разделяли в EA и насыщенном растворе хлорида натрия, водную фазу экстрагировали EA и объединенные органические экстракты сушили над сульфатом магния, фильтровали и выпаривали до сухого состояния. Остаток растворяли в диоксане (0,8 мл), добавляли гидроксид лития (0,8 мл 1н. водного раствора) и смесь перемешивали при комнатной температуре в течение 2,5 ч. Смесь разделяли в 2н хлористоводородной кислоте и EA, водную фазу экстрагировали EA и объединенные органические экстракты сушили над сульфатом магния, фильтровали и выпаривали до сухого состояния. Остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением 115 мг указанного в заголовке соединения.
1H-ЯМР: δ = 12,4 (с, 1H); 8,77 (с, 1H); 7,87-7,77 (м, 4H); 7,72 (д, 1H); 7,67-7,61 (м, 2H); 7,76-7,71 (м, 2H); 7,21-7,16 (м, 2H); 7,10 (д, 1H); 3,57 (д, 2H); 3,48 (с, 3H); 3,38 (д, 2H)
По аналогии с примером 109 получали образцы соединений формулы In, перечисленные в таблице 2. Соединения могут быть названы как 2-[3-(R91-сульфонилокси)-4-метоксибензоиламино]-индан-2-карбоновая кислота, например, как 2-[3-толуол-3-сульфонилокси)-4-метоксибензоиламино]-индан-2-карбоновая кислота в случае примера 111.

Пример 113
2-[4-метокси-3-(2-м-толилоксиацетил)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 3-(2-бромацетил)-4-метоксибензойной кислоты
Метиловый эфир 3-ацетил-4-метоксибензойной кислоты (T. Nagano et al., J. Am. Chem. Soc. 75 (1953), 6237-6238) (1,25 г) растворяли в смеси уксусной кислоты (7 мл) и бромистоводородной кислоты (3 мл), раствор охлаждали в ледяной бане и добавляли бром (0,961 г). Смесь медленно нагревали до комнатной температуры и проводили реакцию в течение 2 ч. Затем смесь выпаривали до сухого состояния под вакуумом, остаток разделяли в EA и насыщенном водном растворе гидрокарбоната натрия, водную фазу экстрагировали EA и объединенные органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. После перемешивания со смесью EA и HEP часть указанного в заголовке соединения кристаллизовалась и ее отфильтровывали. Фильтрат выпаривали до сухого состояния и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Всего получили 1,18 г указанного в заголовке соединения.
ЖХ/МС (метод LC2): Rt = 1,40 мин; м/z = 287,0/289,0 [MH+]
Стадия 2: Метиловый эфир 4-метокси-3-(2-м-толилоксиацетил)-бензойной кислоты
Соединение стадии 1 (1,18 г, 4,12 ммоль) и карбонат калия (1,72 г, 12,4 ммоль) суспендировали в ДМФ (10 мл), добавляли м-крезол (450 мг, 4,12 ммоль) и смесь перемешивали при комнатной температуре в течение 2 ч. Летучие фракции выпаривали под вакуумом, остаток разделяли в EA и воде и водную фазу экстрагировали EA. Объединенные органические экстракты промывали раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением 0,49 г указанного в заголовке соединения.
1H-ЯМР: δ = 8,28 (д, 1H); 8,18 (дд, 1H); 7,37 (д, 1H); 7,13 (дд, 1H); 6,74 (д, 1H); 6,72 (с, 1H); 6,67 (д, 1H); 5,30 (с, 2H); 4,03 (с, 3H); 3,85 (с, 3H); 2,26 (с, 3H)
Стадия 3: 2-[4-метокси-3-(2-м-толилоксиацетил)-бензоиламино]-индан-2-карбоновая кислота
Из соединения стадии 2 получали указанное в заголовке соединение в результате гидролиза эфирной группы по аналогии с примером 2, реакции полученной карбоновой кислоты с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 1 примера 15 и гидролиза эфирной группы по аналогии с примером 2.
1H-ЯМР: δ = 12,4 (с, 1H); 8,88 (с, 1H); 8,22 (д, 1H); 8,10 (дд, 1H); 7,29 (д, 1H); 7,23-7,19 (м, 2H); 7,18-7,10 (м, 3H); 6,73 (д, 1H); 6,70 (с, 1H); 6,65 (д, 1H); 5,28 (с, 2H); 3,98 (с, 3H); 3,57 (д, 2H); 3,40 (д, 2H); 2,24 (с, 3H)
Пример 114
2-[3-(1-гидрокси-2-м-толилоксиэтил)-4-метоксибензоиламино]-индан-2-карбоновая кислота

Соединение примера 113 (113 мг, 0,246 ммоль) растворяли в смеси метанола (2 мл) и этанола (2 мл). При охлаждении в ледяной бане к перемешиваемому раствору добавляли боргидрид натрия (28 мг, 0,738 ммоль) и смесь перемешивали на ледяной бане в течение 2 ч. Летучие фракции выпаривали, остаток разделяли в диэтиловом эфире и разбавленной хлористоводородной кислотой, водную фазу экстрагировали диэтиловым эфиром, объединенные органические экстракты фильтровали через тонкий слой силикагеля, сушили сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток перемешивали со смесью EA и HEP и фильтровали с получением 112 мг указанного в заголовке соединения.
ЖХ/МС (метод LC2): Rt = 1,51 мин.; м/z = 462,1 [MH+]
Пример 115
2-[4-метокси-3-(2-м-толилоксиэтил)-бензоиламино]-индан-2-карбоновая кислота

Соединение примера 114 (20 мг, 0,043 ммоль) растворяли в этаноле (2 мл), добавляли 0,5 М раствор хлористого водорода в метаноле (0,2 мл) и смесь гидрировали в течение ночи в присутствии палладия на угле (10%) при комнатной температуре под давлением водорода 5 бар (полная конверсия исходного соединения). После фильтрации через тонкий слой силикагеля и выпаривания остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN).
ЖХ/МС (метод LC2): Rt = 1,69 мин; м/z = 446,0 [MH+]
Пример 116
2-[4-метокси-3-(3-м-толилпропил)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 3-(1,3-дигидрокси-3-м-толилпропил)-бензойной кислоты
Метиловый эфир 3-ацетил-4-метоксибензойной кислоты (150 мг, 0,720 ммоль) растворяли в ТГФ (3 мл), охлаждали до -78 °C, и свежеприготовленный раствор диизопропиламида лития (полученного путем добавления н-бутиллития в н-гексане (0,317 мл, 2,5 М раствор) к диизопропиламину (80,1 мг, 0,792 ммоль) в ТГФ (3 мл) при 0°С и в процессе перемешивания в течение 10 мин.) постепенно добавляли при перемешивании. Через 10 мин добавляли 3-метилбензалдегид (86,5 мг, 0,720 ммоль) при -78 °C. Через 30 мин при темп. -78°С добавляли 2н. хлористоводородную кислоту и EA и после удаления охлаждающей бани смесь доводили до комнатной температуры. Фазы разделяли, водную фазу экстрагировали EA три раза, объединенные органические экстракты сушили над хлоридом натрия, сливали и выпаривали до сухого состояния. Остаток растворяли в метаноле (5 мл), добавляли боргидрид натрия (28,7 мг, 0,761 ммоль) и смесь перемешивали при комнатной температуре в течение 30 мин. Смесь выпаривали до сухого состояния и остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 140 мг указанного в заголовке соединения в виде смеси диастереомеров.
ЖХ/МС (метод LC1): Rt = 1,32 мин; м/z = 353,1 [MNa+], 683,2 [2MNa+]
Стадия 2: Метиловый эфир 4-метокси-3-(3-м-толилпропил)-бензойной кислоты
Соединение стадии 1 (140 мг, 0,0424 ммоль) растворяли в этаноле (10 мл) и 12н. хлористоводородной кислоте (0,2 мл), добавляли палладий на угле (10%) и смесь гидрировали под давлением водорода 6 бар при комнатной температуре в течение ночи. После фильтрации и выпаривания остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 80 мг указанного в заголовке соединения.
1H-ЯМР: δ = 7,83 (дд, 1H); 7,72 (д, 1H); 7,16 (дд, 1H); 7,06 (д, 1H); 7,03-6,96 (м, 3H); 3,85 (с, 3H); 3,80 (с, 3H); 2,65-2,53 (м, 4H); 2,27 (с, 3H); 1,82 (м, 2H)
Стадия 3: 2-[4-метокси-3-(3-м-толилпропил)-бензоиламино]-индан-2-карбоновая кислота
Из соединения стадии 2 получали указанное в заголовке соединение в результате гидролиза эфирной группы по аналогии с примером 2, реакции полученной карбоновой кислоты с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 1 примера 15 и гидролиза эфирной группы по аналогии с примером 2.
1H-ЯМР: δ = 12,3 (уш.с, 1H); 8,61 (с, 1H); 7,73 (дд, 1H); 7,66 (д, 1H); 7,25-7,20 (м, 2H); 7,19-7,12 (м, 3H); 3,81 (с, 3H); 3,57 (д, 2H); 3,38 (д, 2H); 2,61-2,52 (м, 4H); 2,26 (с, 3H); 1,86-1,78 (м, 2H)
Пример 117
2-(4-метокси-3-фенилацетиламинобензоиламино)-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 2-(4-метокси-3-нитробензоиламино)-индан-2-карбоновой кислоты
К гидрохлориду метилового эфира 2-аминоиндан-2-карбоновой кислоты (0,40 г, 1,77 ммоль) и 4-метокси-3-нитробензойной кислоты (0,35 г, 1,77 ммоль) в 4 мл ДМФ добавляли NMM (0,59 мл, 5,32 ммоль), HOBT (0,31 г, 2,31 ммоль) и EDC (0,44 г, 2,31 ммоль). Смесь перемешивали при 60°С до тех пор, пока анализ ЖХ/МС показал полную конверсию. Неочищенный продукт очищали хроматографией на силикагеле (градиент HEP/EA) с получением 0,43 г указанного в заголовке соединения.
Стадия 2: Метиловый эфир 2-(3-амино-4-метоксибензоиламино)-индан-2-карбоновой кислоты
Соединение стадии 1 (0,43 г, 1,16 ммоль) растворяли в метаноле (30 мл), добавляли 10% палладия на угле (200 мг) и колбу продували аргоном. Подсоединили надувной шар, наполненный водородом, и смесь перемешивали при комнатной температуре в течение ночи. Надувной шар удаляли, колбу продували аргоном, катализатор отфильтровывали через целит и фильтрат выпаривали под вакуумом с получением 0,38 г указанного в заголовке соединения.
Стадия 3: Метиловый эфир 2-(4-метокси-3-фенилацетиламинобензоиламино)-индан-2-карбоновой кислоты
Соединение стадии 2 (0,042 г, 0,12 ммоль) и фенилуксусную кислоту (0,013 г, 0,092 ммоль) растворяли в ДХМ (3 мл) и ДМФ (1 мл), добавляли NMM (0,031 мл, 0,28 ммоль), HOBT (0,016 г, 0,12 ммоль) и EDC (0,021 г, 0,12 ммоль) и смесь перемешивали в течение ночи. Анализ ЖХ/МС показал полную конверсию. Смесь фильтровали, фильтрат подвергали препаративной ОФ ВЭЖХ (градиент вода/ACN), и фракции, содержащие указанное в заголовке соединение, лиофилизировали. Выход: 0,042 г.
Стадия 4: 2-(4-метокси-3-фенилацетиламинобензоиламино)-индан-2-карбоновая кислота
Соединение стадии 3 (42 мг, 0,091 ммоль) растворяли в метаноле (3 мл) и воде 1 мл), добавляли гидрат гидроксида лития (5,3 мл, 0,12 ммоль) и оставляли реакцию при комнатной температуре в течение ночи. Анализ ЖХ/МС показал полную конверсию. Смесь фильтровали, фильтрат подвергали препаративной ОФ ВЭЖХ (градиент вода/ACN), и фракции, содержащие указанное в заголовке соединение, лиофилизировали. Выход: 27 мг.
ЖХ/МС (метод LC5): Rt = 1,95 мин; м/z = 445,48 [MH+]
1H-ЯМР: δ = 12,4 (уш.с, 1H); 9,39 (с, 1H); 8,67 (с, 1H); 8,31 (с, 1H); 7,62 (д, 1H); 7,38-7,30 (м, 4H); 7,28-7,20 (м, 3H); 7,18-7,12 (м, 2H); 7,08 (д, 1H); 3,87 (с, 3H); 3,72 (с, 2H); 3,55 (д, 2H); 3,35 (д, 2H)
По аналогии с примером 117 подготовили образцы соединений формулы Ip, перечисленные в таблице 3. Соединения могут быть названы как 2-[3-(R92-карбониламино)-4-метоксибензоиламино]-индан-2-карбоновая кислота, например, как 2-[3-[3-фторбензоиламино]-4-метоксибензоиламино)-индан-2-карбоновая кислота в случае примера 120.

Пример 121
2-[3-(4-Фторбензоиламино)-4-метоксибензоиламино]-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 2-[3-(4-фторбензоиламино)-4-метоксибензоиламино]-индан-2-карбоновой кислоты
Соединение стадии 2 примера 117 (0,042 г, 0,12 ммоль) и 4-фторбензальдигида (0,0115 г, 0,092 ммоль) растворяли в ТГФ (3 мл) и уксусной кислоте (0,5 мл). Добавляли полимерно-связанный цианоборгидрид натрия (0,2 ммоль) и смесь перемешивали при комнатной температуре до тех пор, пока анализ ЖХ/МС показал полную конверсию. Смолу отфильтровывали, фильтрат подвергали препаративной ОФ ВЭЖХ (градиент вода/ACN) и фракции, содержащие указанное в заголовке соединение, лиофилизировали с получением 33 мг указанного в заголовке соединения.
Стадия 2: 2-[3-(4-Фторбензоиламино)-4-метоксибензоиламино]-индан-2-карбоновая кислота
Соединение стадии 1 (30 мг, 0,053 ммоль) растворяли в метаноле (3 мл) и воде (1 мл). Добавляли гидрат гидроксида лития (3,8 мг, 0,09 ммоль) и смесь перемешивали при комнатной температуре до тех пор, пока анализ ЖХ/МС показал полную конверсию. Смесь отфильтровывали, фильтрат подвергали препаративной ОФ ВЭЖХ (градиент вода/ACN) и фракции, содержащие указанное в заголовке соединение, лиофилизировали с получением 19 мг указанного в заголовке соединения.
ЖХ/МС (метод LC6): Rt = 1,66 мин; м/z = 435,19 [MH+]
1H-ЯМР: δ = 12,2 (уш.с, 1H); 8,45 (с, 1H); 7,38-7,32 (м, 2H); 7,22-7,20 (м, 2H); 7,20-7,14 (м, 3H); 7,14-7,09 (м, 2H); 6,92 (с, 1H); 6,81 (д, 1H); 4,32 (с, 2H); 3,80 (с, 3H); 3,72 (с, 2H); 3,53 (д, 2H); 3,32 (д, 2H)
По аналогии с примером 121 подготовили образцы соединений формулы Ir, перечисленные в таблице 4. Соединения могут быть названы как 2-[3-(R93-амино)-4-метоксибензоиламино]-индан-2-карбоновая кислота, например, как 2-[3-[2-фенилэтиламино)-4-метоксибензоиламино]-индан-2-карбоновая кислота в случае примера 122.

Пример 125
Стандартная методология твердофазного синтеза
0,5 г смолы AM RAM на основе полистирола с FMOC-защищенным линкером (0,5 ммоль/г или 0,75 ммоль/г, соответственно; Rapp Polymere GmbH, Tübingen, Germany) обрабатывали 50-процентным раствором пиперидина в ДМФ в течение 20 мин и тщательно промывали ДМФ. Соответствующую FMOC-защищенную 2-аминоиндан-2-карбоновую кислоту (5 эквивалентов), HOBT (5 эквивалентов) и ДИК (5 эквивалентов) растворяли в ДМФ (5 мл) и добавляли к смоле. Смесь встряхивали в течение ночи при комнатной температуре. Смолу повторно промывали ДМФ и FMOC-защищенную группу удаляли путем обработки смолы 50-процентным раствором пиперидина в DMF в течение 20 мин. Смолу повторно промывали ДМФ.
Для ацилирования аминогруппы раствор соответствующей гидрокси-замещенной бензойной кислоты (5 эквивалентов), HOBT (5 эквивалентов) и ДИК (5 эквивалентов) в ДМФ (5 мл) добавляли к смоле и смесь встряхивали в течение ночи при комнатной температуре. Смолу промывали DMF и обрабатывали раствором 2н. диметиламина в ТГФ в течение ночи, или в некоторых случаях - 50-процентным раствором пиперидина в ДМФ в течение 2 ч, для гидролиза эфира, образованного в результате ацилирования гидроксильной группы. Смолу активно промывали ДМФ, ДХМ и ТГФ.
Для реакции Мицунобу в гидроксильной группе трифенилфосфин (10 эквивалентов) и соответствующий спирт (10 эквивалентов) растворяли в 5 мл сухого ТГФ и добавляли к смоле. Суспензию охлаждали до 0°С и ДИАД (10 эквивалентов) добавляли в охлажденную смесь и оставляли для прохождения реакции в течение ночи при комнатной температуре. Смолу повторно промывали ДХМ.
Для расщепления полученного соединения смолу обрабатывали чистой ТФУ в течение 2 ч. ТФК удаляли под вакуумом и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). В большинстве случаев карбоновую кислоту выделяли после расщепления ТФУ. В некоторых случаях выделяли амид карбоновой кислоты, который преобразовывали в карбоновую кислоту путем гидролиза в 50-процентном водном растворе ТФУ при 60°С в течение ночи, частичного удаления ТФК под вакуумом и лиофилизации в водном растворе.
В соответствии со стандартной методологией, описанной в примере 125, соединения формулы Is, перечисленные в таблице 5, синтезировали. В формулах групп R95 в таблице 5 линия, перечеркнутая знаком






























Пример 156
5-Бром-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали в соответствии со стандартной методологией, описанной в примере 125.
ЖХ/МС (метод LC7): Rt = 5,06 мин; м/z = 524,1/526,1 [MH+]
Пример 157
5-Фтор-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали в соответствии со стандартной методологией, описанной в примере 125.
ЖХ/МС (метод LC7): Rt = 4,74 мин; м/z = 464,2 [MH+]
Пример 158
2-[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-5,6-диметилиндан-2-карбоновая кислота

Указанное в заголовке соединение получали в соответствии со стандартной методологией, описанной в примере 125.
ЖХ/МС (метод LC7): Rt = 5,03 мин; м/z = 474,2 [MH+]
Пример 159
5-Метокси-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота
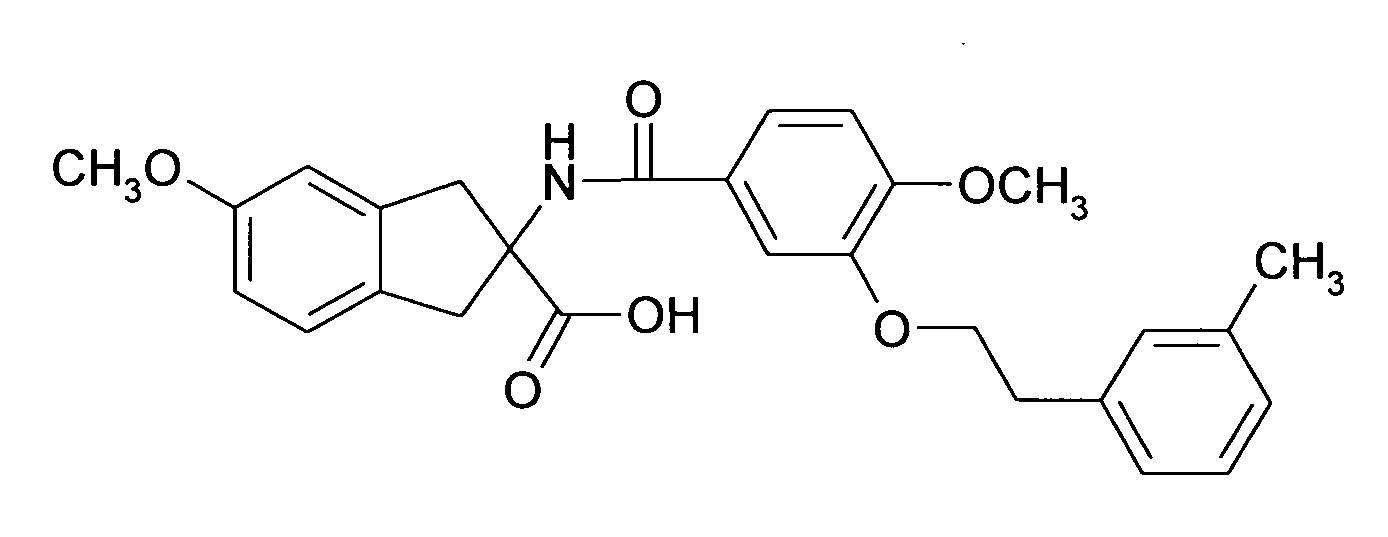
Указанное в заголовке соединение получали в соответствии со стандартной методологией, описанной в примере 125.
ЖХ/МС (метод LC7): Rt = 4,60 мин; м/z = 476,2 [MH+]
Пример 160
Амид 2-[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты

Соединение примера 14 (100 мг, 0,224 ммоль) добавляли в тионилхлорид (0,5 мл) и перемешивали в течение 30 мин при 60°C. Летучие фракции выпаривали, добавляли диоксан (1 мл) и смесь повторно выпаривали до сухого состояния. Полученный сырой хлорангидрид растворяли в ДХМ и добавляли к перемешанной смеси EA, насыщенному раствору гидрокарбоната натрия и аммиаку (30-процентный водный раствор, 0,015 мл). После перемешивания при комнатной температуре в течение 90 мин слои разделяли и водную фазу экстрагировали EA. Объединенные органические экстракты сушили над сульфатом натрия и выпаривали до сухого состояния. Остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN).
ЖХ/МС (метод LC1): Rt = 1,55 мин; м/z = 445,1 [MH+]
Пример 161
2-{4-[2-(3-Хлорфенил)-этокси]-3-метоксибензоиламино}-индан-2-карбоновая кислота

Указанное в заголовке соединение получали в соответствии со стандартной методологией, описанной в примере 125.
ЖХ/МС (метод LC7): Rt = 4,70 мин; м/z = 466,1/468,1 [MH+]
Пример 162
2-{4-[2-(2-Хлорфенил)-этокси]-3-метоксибензоиламино}-индан-2-карбоновая кислота

Указанное в заголовке соединение получали в соответствии со стандартной методологией, описанной в примере 125.
ЖХ/МС (метод LC7): Rt = 4,69 мин; м/z = 466,1/468,1 [MH+]
Пример 163
2-[4-Амино-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали в соответствии со стандартной методологией, описанной в примере 125, с использованием 0,1 г смолы (0,5 ммоль/г). На стадии ацилирования использовали 3-гидрокси-4-нитробензойную кислоту. На последней стадии нитрогруппу восстанавливали 1 М раствором дигидрата хлорида олова(II) в ДМФ в течение ночи при комнатной температуре. Смолу тщательно промывали ДМФ, метанолом и ДХМ и продукт отщепляли от смолы путем обработки ТФУ в течение 2 ч. ТФУ удаляли под вакуумом и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Выход: 12,4 мг.
ЖХ/МС (метод LC9): Rt = 4,33 мин; м/z = 431,2 [MH+]
1H-ЯМР (300 МГц, D6-ДМСО + 2 % ТФУ): δ = 2,29 (с, 3H); 3,06 (т, J=6,97 Hz, 2H); 3,30-3,43 (м, 2H); 3,54-3,65 (м, 2H); 4,28 (т, J=7,06 Hz, 2H); 7,04 (д, J=7,35 Hz, 1H); 7,09-7,26 (м, 8H); 7,43-7,54 (м, 2H); 8,71 (с, 1H)
Пример 164
2-[4-Метиламино-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 163, с использованием 0,12 г смолы (0,5 ммоль/г). После восстановления нитрогруппы аминогруппу метилировали, используя 37-процентный водный раствор формальдегида (10 эквивалентов) и цианоборгидрид натрия (8 эквивалентов, 1 M раствора в ТГФ) в смеси ДХМ и ACN (3:1), содержащей 2% уксусной кислоты. Смесь встряхивали в течение ночи, затем смолу промывали, и процедура повторялась со свежими реагентами. Для расщепления смолу обрабатывали ТФУ в течение 2 ч, удаляли ТФУ под вакуумом и остаток растворяли в 50-процентном водном растворе ТФУ. Раствор нагревали до 50°С в течение 48 ч, TFA частично удаляли под вакуумом и водный раствор лиофилизировали. Остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Выход: 9,8 мг.
ЖХ/МС (метод LC7): Rt = 4,41 мин; м/z = 445,2 [MH+]
1H-ЯМР (300 МГц, D6-ДМСО + 2 % ТФУ): δ = 2,28 (с, 3H); 2,75 (с, 3H); 3,04 (т, J=6,78 Гц, 2H); 3,30-3,41 (м, 2H); 3,52-3,63 (м, 2H); 4,21 (т, J=6,88 Гц, 2H); 6,61-6,71 (м, 1H); 7,03 (д, J=7,16 Гц, 1H); 7,10-7,25 (м, 7H); 7,37 (д, J=1,70 Гц, 1H); 7,47 (дд, J=8,19/1,79 Гц, 1H); 8,49 (уш.с, 1H)
Пример 165
2-[4-Диметиламино-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 164, с использованием 0,12 г смолы (0,5 ммоль/г) и с повторением процесса метилирования еще три раза со свежими реагентами до полной конверсии промежуточного метиламинового соединения в диметиламиновое соединение. Выход: 6,6 мг.
ЖХ/МС (метод LC7): Rt = 3,37 мин; м/z = 459,2 [MH+]
1H-ЯМР (300 МГц, D6-ДМСО + 2 % ТФУ): δ = 2,28 (с, 3H); 3,04 (с, 6H); 3,12 (т, J=6,50 Гц, 2H); 3,33-3,44 (м, 2H); 3,62 (д, J=16,77 Гц, 2H); 4,43 (т, J=6,69 Гц, 2H); 7,04 (д, J=6,97 Гц, 1H); 7,11-7,27 (м, 7H); 7,55-7,71 (м, 3H); 8,93 (с, 1H)
Пример 166
2-[4-Изопропиламино-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 163, с использованием 0,25 г смолы (0,5 ммоль/г). После восстановления нитрогруппы аминогруппу алкилировали, используя 2-метоксипропен (10 эквивалентов) в 2 мл смеси ДХМ и ACN (3:1), содержащей 2% уксусной кислоты и 1 мл 1 M раствора цианоборгидрида натрия в ТГФ. Алкилирование повторяли три раза со свежими реагентами. Расщепление и обработку проводили по аналогии с примером 164. Выход: 13,4 мг.
ЖХ/МС (метод LC7): Rt = 4,19 мин; м/z = 473,2 [MH+]
Пример 167
2-{3-[2-(2-Фторфенил)-2-гидроксиэтокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Указанное в заголовке соединение получали в соответствии со стандартной методологией, описанной в примере 125, с использованием 0,25 г смолы (0,5 ммоль/г). После связывания 2-аминоиндан-2-карбоновой кислоты со смолой происходило ацилирование с 3-гидрокси-4-метоксибензойной кислотой и обработка 50% пиперидина в ДМФ в течение 2 ч. После активного промывания ДМФ и ДХМ смола вступала в реакцию с 2-бром-1-(2-фторфенил)-этаноном (3 эквивалента) в присутствии 1,8-диазабицикло[5.4.0]ундек-7-ена (DBU; 3 эквивалента) в 3 мл ДХМ в течение ночи при комнатной температуре. Соединение отщепляли от смолы при помощи чистой ТФУ в течение 2 ч. и ТФУ выпаривали под вакуумом. Неочищенный промежуточный продукт растворяли в 4 мл ТГФ, добавляли 10 мг боргидрида лития и реакционную смесь встряхивали в течение 3 ч. Затем реакционную смесь гасили уксусной кислотой, выпаривали до сухого состояния и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Выход: 3,7 мг.
ЖХ/МС (метод LC7): Rt = 4,01 мин; м/z = 466,2 [MH+]
Пример 168
2-[4-Циано-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали в соответствии со стандартной методологией, описанной в примере 125, с использованием 0,3 г смолы (0,75 ммоль/г). На стадии ацилирования использовали 3-гидрокси-4-йодбензойную кислоту. Наконец, смолу с йодсодержащим соединением обрабатывали цианидом цинка и тетракис(трифенилфосфин)палладием(0) в 5 мл ДМФ/EDIA (2:1) в микроволновом реакторе (90 Вт) при 150°С в течение 10 мин. Смолу сливали с ДХМ, затем тщательно промывали ДМФ и ДХМ и продукт расщепляли чистой ТФУ в течение 2 ч. ТФУ удаляли в вакууме и остаток растворяли в 50% водном растворе ТФУ и нагревали при темп. 50°С в течение ночи. ТФУ частично выпаривали и водный раствор лиофилизировали. Соединение очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Выход: 11,4 мг.
ЖХ/МС (метод LC7): Rt = 4,78 мин; м/z = 441,2 [MH+]
1H-ЯМР (300 МГц, D6-ДМСО + 2 % ТФУ): δ = 2,28 (с, 3H); 3,05 (т, J=6,69 Гц, 2H); 3,33-3,43 (м, 2H); 3,55-3,65 (м, 2H); 4,36 (т, J=6,59 Гц, 2H); 7,03 (д, J=7,16 Гц, 1H); 7,09-7,26 (м, 7H); 7,49-7,58 (м, 2H); 7,80 (д, J=7,91 Гц, 1H); 9,04 (с, 1H)
Пример 169
2-[4-Метокси-3-(3-фенилпропил)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали в соответствии со стандартной методологией, описанной в примере 125, с использованием 0,3 г смолы (0,75 ммоль/г). В качестве ацилирующего агента использовали 3-йод-4-метоксибензойную кислоту вместо гидрокси-замещенной бензойной кислоты. Наконец, смола с йодосодержащим соединением вступала в реакцию в условиях реакции Соногаширы с 3-фенил-1-пропином (10 эквивалентов), растворенным в 4 мл ДМФ вместе с триэтиламином (20 эквивалентов), йодидом меди(I) (0,1 эквивалента) и хлоридом бис(трифенилфосфин)палладия (II) (0,1 эквивалента). Реакционную смесь встряхивали при комнатной температуре в течение 48 ч. Смолу промывали ДМФ, ДХМ и промежуточный продукт расщепляли чистой ТФУ в течение 2 ч. ТФУ удаляли под вакуумом и остаток растворяли в воде/ACN (3:2) и лиофилизировали. Выделенный промежуточный продукт растворяли в 6 мл метанола, добавляли 100 мг 10% палладия на угле и смесь гидрировали в реакторе Парра под давлением около 3,5 бар в течение 2 ч. После фильтрации метанол выпаривали и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Выход: 13,7 мг.
ЖХ/МС (метод LC7): Rt = 4,97 мин; м/z = 430,2 [MH+]
1H-ЯМР (300 МГц, D6-ДМСО + 2 % ТФУ): δ = 1,83 (дкв, J=7,91, 7,66 Гц, 2H); 2,59 (кв., J=7,72 Гц, 4H); 3,31-3,45 (м, 2H); 3,52-3,63 (м, 2H); 3,81 (с, 3H); 6,98 (д, J=8,67 Гц, 1H); 7,08-7,32 (м, 10H); 7,66 (д, J=2,26 Гц, 1H); 7,74 (дд, J=8,48/2,26 Гц, 1H); 8,62 (с, 1H)
Пример 170
2-[4-Ацетил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали в соответствии со стандартной методологией, описанной в примере 125, с использованием 0,3 г смолы (0,75 ммоль/г). На стадии ацилирования использовали 3-гидрокси-4-йодбензойную кислоту. Наконец, смола с йодсодержащим соединением вступала в реакцию с триметилсилилацетиленом (10 эквивалентов), растворенным в 4 мл ДМФ вместе с триэтиламином (20 эквивалентов), йодидом меди(I) (0,1 эквивалента) и хлоридом бис(трифенилфосфин)палладия(II) (0,1 эквивалента). Смолу промывали ДМФ и ТГФ и обрабатывали 1 М раствором тетрабутиламмонийфторида в ТГФ в течение 30 мин. После тщательного промывания ДХМ, 10-процентным раствором уксусной кислоты в ДХМ и ДМФ соединение отщепляли от смолы чистой ТФУ в течение 2 ч. Амид карбоновой кислоты преобразовывали в карбоновую кислоту в соответствии со стандартной методологией, описанной в примере 125, и указанное в заголовке соединение очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Выход: 5,8 мг.
ЖХ/МС (метод LC7): Rt = 4,77 мин; м/z = 458,2 [MH+]
Пример 171
2-[4-Метокси-3-(2-м-толилэтиламино)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали в соответствии со стандартной методологией, описанной в примере 125, с использованием 0,5 г смолы (0,75 ммоль/г). На стадии ацилирования вместо гидрокси-замещенной бензойной кислоты использовали 4-метокси-3-нитробензойную кислоту и впоследствии восстанавливали нитрогруппу 1 М раствора дигидрата хлорида олова(II) в ДМФ в течение ночи. Смолу промывали ДМФ, ДХМ и проводили реакцию с 2,4-динитробензолсульфонилхлоридом (5 эквивалентов) и 2,6-лутидином (10 эквивалентов), растворенными в 5 мл ДХМ в течение 5 ч. После промывания ДХМ и ТГФ раствор трифенилфосфина (10 эквивалентов) и 2-(3-метилфенил)-этанола (10 эквивалентов) в ТГФ добавляли в смолу и суспензию охлаждали до 0°C. ДИАД добавляли в охлажденную смесь и реакционную смесь встряхивали в течение ночи при комнатной температуре. Смолу промывали ДХМ и обрабатывали меркаптоуксусной кислотой (5 эквивалентов) и триэтиламином (10 эквивалентов) в ДХМ в течение 10 мин. Данную стадию повторяли со свежеприготовленным раствором. Смолу промывали ДМФ и ДХМ. Соединение отщепляли от смолы при помощи чистой ТФУ в течение 2 ч, амид карбоновой кислоты преобразовывали в карбоновую кислоту в соответствии со стандартной методологией, описанной в примере 125, и указанное в заголовке соединение очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Выход: 36,4 мг.
ЖХ/МС (метод LC7): Rt = 3,80 мин; м/z = 445,2 [MH+]
1H-ЯМР (300 МГц, D6-ДМСО + 2 % ТФУ): δ = 2,27 (с, 3H); 2,83-2,92 (м, 2H); 3,33-3,48 (м, 4H); 3,53-3,64 (м, 2H); 3,90 (с, 3H); 6,99-7,25 (м, 10H); 7,56 (с, 1H); 7,63 (с, 1H); 8,72 (с, 1H)
Пример 172
2-{4-Метокси-3-[метил-(2-м-толилэтил)-амино]-бензоиламино}-индан-2-карбоновая кислота

Сначала синтез проводили согласно методологии, описанной в примере 171, с использованием 0,25 г смолы (0,75 ммоль/г). Впоследствии для N-метилирования смолу обрабатывали 37-процентным водным раствором формальдегида (10 эквивалентов) в смеси ДХМ/ACN (3:1), содержащей 2% уксусной кислоты и 1,5 мл 1 М раствора цианоборгидрида натрия в ТГФ в течение ночи. Реакцию метилирования повторяли три раза со свежими реагентами. Расщепление, выделение и очистку соединения проводили согласно методологии, описанной в примере 171. Выход: 21,4 мг.
ЖХ/МС (метод LC7): Rt = 3,19 мин; м/z = 459,2 [MH+]
1H-ЯМР (300 МГц, D6-ДМСО + 2 % ТФУ): δ = 2,22 (с, 3H); 2,72 (т, J=7,82 Гц, 2H); 3,23 (с, 3H); 3,34-3,45 (м, 2H); 3,63 (д, J=16,95 Гц, 2H); 3,78 (т, J=8,67 Гц, 2H); 3,99 (с, 3H); 6,89-6,96 (м, 2H); 7,00 (д, J=7,91 Гц, 1H); 7,10-7,29 (м, 6H); 7,33 (д, J=8,85 Гц, 1H); 8,04 (дд, J=8,67/1,70 Гц, 1H); 8,16 (с, 1H); 8,84 (с, 1H)
Пример 173
2-[4-Циано-3-(2-м-толилэтиламино)-бензоиламино]-индан-2-карбоновая кислота

0,1 г смолы PL Wang (Polymer Laboratories, Amherst, MA, USA; 1,7 ммоль/г) ацилировали FMOC-защищенной 2-аминоиндан-2-карбоновой кислотой (3 эквивалента) в присутствии ДИК (3 эквивалента), HOBT (3 эквивалента) и 1-метилимидазола в ДМФ в течение ночи. FMOC-защищенную группу удаляли путем обработки 50% пиперидином в ДМФ и полученную аминокислоту ацилировали 4-циано-3-фторбензойной кислотой (3 эквивалента) в присутствии ДИК (3 эквивалента) и HOBT (3 эквивалента) в ДМФ. Смолу обрабатывали 1 М раствором 2-(3-метилфенил)-этиламина в ДМФ в течение ночи при комнатной температуре. Реакцию повторяли со свежим раствором амина. Смолу промывали ДМФ и ДХМ и соединение отщепляли от смолы чистой ТФУ в течение 1,5 ч. ТФК удаляли под вакуумом и соединение очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Выход: 9,7 мг.
ЖХ/МС (метод LC7): Rt = 4,66 мин; м/z = 440,2 [MH+]
1H-ЯМР (300 МГц, D6-ДМСО + 2 % ТФУ): δ = 2,27 (с, 3H); 2,83 (т, J=7,44 Гц, 2H); 3,33-3,47 (м, 4H); 3,54-3,65 (м, 2H); 6,98-7,11 (м, 4H); 7,11-7,26 (м, 7H); 7,54 (д, J=8,10 Гц, 1H); 8,95 (с, 1H)
Пример 174
2-[4-Циано-3-[3-фенилпирролидин-1-ил)-бензоиламино]-индан-2-карбоновая кислота

Синтез проводили согласно методологии, описанной в примере 174, с использованием 0,15 г смолы PL Wang (1,7 ммоль/г). Вместо 2-(3-метилфенил)-этиламина на последней стадии смола вступала в реакцию с 3-фенилпирролидином (8 эквивалентов) в диметилацетамиде при 90°С в течение ночи. Расщепление, выделение и очистку соединения проводили согласно методологии, описанной в примере 173.
Выход: 11,3 мг.
ЖХ/МС (метод LC7): Rt = 4,82 мин; м/z = 452,2 [MH+]
Пример 175
2-{4-Циано-3-(2-фторфенил)-этиламино]-бензоиламино}-индан-2-карбоновая кислота
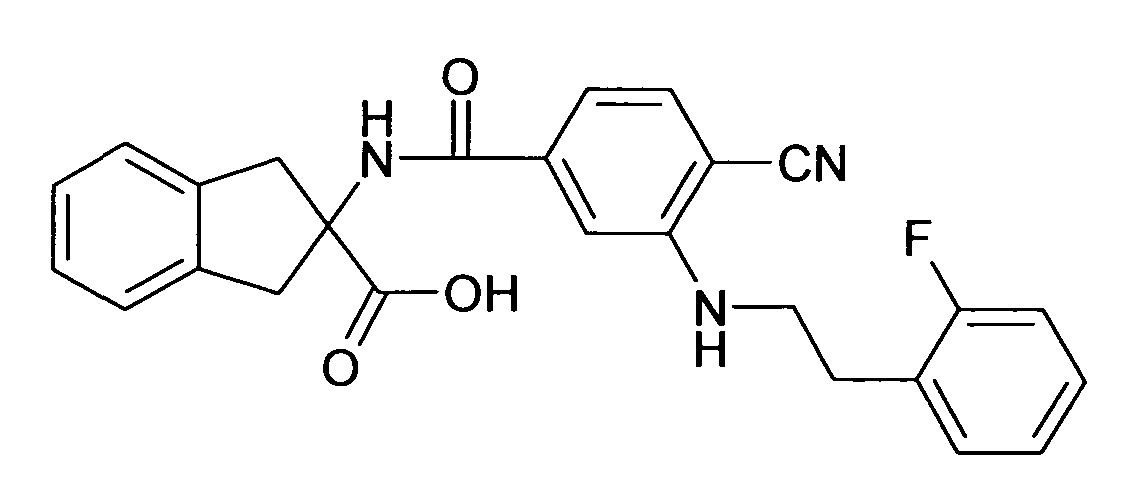
Синтез проводили согласно методологии, описанной в примере 173, с использованием 0,1 г смолы PL Wang (1,7 ммоль/г). Вместо 2-(3-метилфенил)-этиламина на последней стадии смола вступала в реакцию с 2-(2-фторфенил)-этиламином (10 эквивалентов) в диметилацетамиде в микроволновом реакторе при 150°С в течение 1 ч. Расщепление, выделение и очистку соединения проводили согласно методологии, описанной в примере 173.
Выход: 1,7 мг.
ЖХ/МС (метод LC7): Rt = 4,51 мин; м/z = 444,2 [MH+]
Пример 176
2-{3-[2-(3-Хлорфенил)-этокси]-4-метилбензоиламино}-индан-2-карбоновая кислота

Синтез проводили с использованием 0,15 г смолы PL Wang (1,7 ммоль/г). Связывание 2-аминоиндан-2-карбоновой кислоты и ацилирование 3-гидрокси-4-метилбензойной кислотой выполняли согласно методологии, описанной в примере 173. После стадии ацилирования смолу промывали ТГФ и раствором трифенилфосфина (10 эквивалентов) и добавляли в смолу 2-(3-хлорфенил)-этанол (10 эквивалентов). Суспензию охлаждали до 0°С, добавляли в охлажденную смесь ДИАД (10 эквивалентов) и реакционную смесь встряхивали в течение ночи при комнатной температуре. Смолу промывали ДХМ. Расщепление, выделение и очистку соединения проводили согласно методологии, описанной в примере 173. Выход: 2,3 мг.
ЖХ/МС (метод LC7): Rt = 5,11 мин; м/z = 450,2 [MH+]
Пример 177
2-{3-[2-(2-Фторфенил)-этокси]-4-метилбензоиламино}-индан-2-карбоновая кислота

Указанное в заголовке соединение получали в соответствии с методами, описанными в примере 176. Выход: 3,8 мг.
ЖХ/МС (метод LC7): Rt = 4,87 мин; м/z = 434,2 [MH+]
Пример 178
2-[4-Этокси-3-(2-м-толилэтиламино)-бензоиламино]-индан-2-карбоновая кислота

Синтез проводили с использованием 0,15 г смолы PL Wang (1,7 ммоль/г). Связывание 2-аминоиндан-2-карбоновой кислоты и ацилирование с использованием 4-фтор-3-нитробензойной кислоты выполняли согласно методологии, описанной в примере 173. После стадии ацилирования смолу встряхивали с этанолом (5 эквивалентов) в присутствии бис(триметилсилил)-амида натрия (5 эквивалентов) в 3 мл диметилацетамида. Смолу промывали ДМФ, 10-процентной уксусной кислотой/ДМФ, ДМФ и в завершение ДХМ. Восстановление нитрогруппы хлоридом олова(II), сульфонилирование 2,4-динитробензолсульфонилхлоридом, алкилирование 2-(3-метилфенил)-этанолом и удаление сульфонильной группы выполняли согласно методологии, описанной в примере 171, и соединение очищали препаративной ОФ ВЭЖХ (градиента вода / ACN). Выход: 2,4 мг.
ЖХ/МС (метод LC7): Rt = 3,73 мин; м/z = 459,2 [MH+]
Пример 179
2-[4-Гидрокси-3-(2-м-толилэтиламино)-бензоиламино]-индан-2-карбоновая кислота

10 мг соединения примера 171 растворяли в ДХМ и обрабатывали 200 мкл 1 М раствора трибромида бора в ДХМ в течение 5 ч. Добавляли 2 М раствора карбоната натрия и смесь выпаривали под вакуумом. Продукт очищали препаративной ОФ ВЭЖХ (градиент вода/ACN).
ЖХ/МС (метод LC7): Rt = 3,34; м/z = 431,2 [MH+]
1H-ЯМР (300 МГц, D6-ДМСО + 2 % ТФУ): δ = 2,21 (с, 3H); 2,81-2,92 (м, 2H); 3,24-3,37 (м, 2H); 3,37-3,48 (м, 2H); 3,48-3,60 (м, 2H); 6,88-7,04 (м, 5H); 7,04-7,21 (м, 6H); 7,66-7,79 (м, 2H); 8,66 (с, 1H)
Пример 180
2-[4-Метокси-3-(2-м-толилэтилсульфанил)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: 3-(5-Карбокси-2-метокси-фенилдисульфанил)-4-метоксибензойная кислота
45 г (179,5 ммоль) 3-хлорсульфонил-4-метоксибензойной кислоты суспендировали в 200 мл уксусной кислоты и нагревали до 40°C. Затем раствор 85,1 г (448,8 ммоль) хлорида олова(II) в 100 мл хлористоводородной кислоты добавляли в течение 15 мин и смесь перемешивали в течение 2 ч. с обратным холодильником. Горячий раствор добавляли по каплям до 2000 мл воды со льдом. Образовавшийся осадок собирали отсасыванием, промывали водой и сушили. Получили 32,8 г указанного в заголовке соединения.
Стадия 2: 4-Метокси-3-(2-м-толилэтилсульфанил)-бензойная кислота
732,8 мг (2 ммоль) соединения стадии 1 растворяли в 30 мл абсолютного метанола и порциями постепенно добавляли 151,3 мг (4 ммоль) боргидрида натрия. После перемешивания в течение ночи добавляли раствор 796,4 мг (4 ммоль) 1-(2-бромэтил)-3-метилбензола в 10 мл ДХМ и смесь перемешивали в течение ночи. Затем добавляли 202,4 мг (4 ммоль) триэтиламина и продолжали перемешивание в течение 2 ч при комнатной температуре и в течение 2 ч при темп. 40°C. После охлаждения смесь экстрагировали раствором гидрокарбоната натрия и органическую фазу сушили и выпаривали. Остаток использовали на следующей стадии без дополнительной очистки.
Стадия 3: 2-[4-Метокси-3-(2-м-толилэтилсульфанил)-бензоиламино]-индан-2-карбоновая кислота
700 мг неочищенного соединения стадии 2 растворяли в 5 мл ДМФ и добавляли 598 мг (4,63 ммоль) EDIA и 968 мг (2,55 ммоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата. Затем добавляли раствор 527 мг (2,32 ммоль) гидрохлорида метилового эфира 2-аминоиндан-2-карбоновой кислоты в 5 мл ДМФ. После перемешивания в течение ночи добавляли EA и водный раствор хлорида лития (4%), органическую фазу отделяли, промывали один раз раствором хлорида лития и два раза раствором гидрокарбоната натрия, сушили и выпаривали. Твердый остаток растворяли в 10 мл 9:1 смеси ТГФ и воды и добавляли 131 мг (5,47 ммоль) гидроксида лития. После перемешивания в течение ночи смесь выпаривали до сухого состояния. Остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением 222 мг указанного в заголовке соединения.
ЖХ/МС (метод LC3): Rt = 1,97 мин; м/z = 462,23 [MH+]
1H-ЯМР: δ = 12,45 (уш.с, 1H); 8,87 (с, 1H); 7,78 (с, 1H); 7,74 (д, 1H); 7,20-7,26 (м, 2H); 7,13-7,20 (м, 3H); 7,09 (с, 1H); 6,98-7,08 (м, 3H); 3,87 (с, 3H); 3,60 (д, 2H); 3,18 (д, 2H); 2,82 (т, 2H); 2,29 (с, 3H)
Пример 181
2-{3-[2-(3-Хлорфенил)-этилсульфанил]-4-метилбензоиламино}-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 180, с использованием 1-(2-бромэтил)-3-хлорбензола вместо 1-(2-бромэтил)-3-метил-бензола на стадии 2.
ЖХ/МС (метод LC3): Rt = 1,97 мин; м/z = 482,19 [MH+]
Пример 182
2-(3-Бензилсульфанил-4-метоксибензоиламино)-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 180, с использованием бензилбромида вместо 1-(2-бромэтил)-3-метилбензола на стадии 2.
ЖХ/МС (метод LC3): Rt = 1,80 мин; м/z = 434,26 [MH+]
Пример 183
2-[4-Метокси-3-(2-м-толилэтансульфонил)-бензоиламино]-индан-2-карбоновая кислота

55 мг (119,2 ммоль) соединения примера 180 растворяли в 5 мл ДХМ и обрабатывали раствором 88,2 мг (357,6 ммоль) 3-хлорпербензойной кислоты в 5 мл ДХМ. После перемешивания при комнатной температуре в течение ночи растворитель выпаривали и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением 28 мг указанного в заголовке соединения.
ЖХ/МС (метод LC3): Rt = 1,68 мин; м/z = 494,25 [MH+]
1H-ЯМР: δ = 12,45 (уш.с, 1H); 9,00 (с, 1H); 8,30 (с, 1H); 7,21-7,28 (м, 3H); 7,13-7,20 (м, 2H); 7,08 (т, 1H); 6,90-6,95 (м, 2H); 6,88 (с, 1H); 3,94 (с, 3H); 3,70 (т, 2H); 3,60 (д, 2H); 3,42 (д, 2H); 2,82 (т, 2H); 2,18 (с, 3H)
Пример 184
2-{3-[2-(3-Хлорфенил)-этансульфонил]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 183, начиная с соединения примера 181.
ЖХ/МС (метод LC3): Rt = 1,69 мин; м/z = 514,20 [MH+]
Пример 185
2-[3-(2-м-Толилэтокси)-4-трифторметилбензоиламино]-индан-2-карбоновая кислота

Стадия 1: 3-Ацетокси-4-трифторметилбензойная кислота
3,5 г (17 ммоль) 3-гидрокси-4-трифторметилбензойной кислоты (полученной согласно описанию в WO 2006/128184) растворяли в 35 мл ангидрида уксусной кислоты и нагревали с обратным холодильником в течение 3 ч. Добавляли 60 мл воды и продолжали нагревание с обратным холодильником в течение 10 мин. После охлаждения и перемешивания в течение ночи образовавшийся осадок собирали отсасыванием и сушили с получением 3,0 г указанного в заголовке соединения.
Стадия 2: Метиловый эфир 2-(3-ацетокси-4-трифторметилбензоиламино)-индан-2-карбоновой кислоты
К раствору 2,7 г (10,9 ммоль) соединения стадии 1 в 16,3 мл 2 М раствора оксалилхлорида в ДХМ (32,6 ммоль) добавляли 80 мг ДМФ и смесь перемешивали в течение 30 мин при комнатной температуре. Растворитель выпаривали, добавляли 20 мл ДХМ и смесь повторно выпаривали. Остаток растворяли в 20 мл ДХМ и раствор добавляли через 5 мин при 0°С к раствору 2,48 г (10,9 ммоль) гидрохлорида метилового эфира 2-аминоиндан-2-карбоновой кислоты в 50 мл ДХМ и 30 мл насыщенного раствора гидрокарбоната натрия. После перемешивания в течение ночи фазы разделяли, органическую фазу сушили над сульфатом натрия, выпаривали, остаток очищали хроматографией на силикагеле (градиент HEP/EA). Получали 1,1 г указанного в заголовке соединения.
Стадия 3: Метиловый эфир 2-[3-(2-м-толилэтокси)-4-трифторметилбензоиламино]-индан-2-карбоновой кислоты
200 мг (0,48 ммоль) соединения стадии 2 растворяли в 5 мл метанола, добавляли 13,1 мг (0,1 ммоль) карбоната калия и смесь перемешивали в течение 30 мин. Смесь подкисляли 1н. хлористоводородной кислотой и экстрагировали три раза порциями 20 мл EA. Объединенные органические фазы сушили и выпаривали. Остаток растворяли в 5 мл ТГФ, добавляли 96,9 мг (0,71 ммоль) 2-(3-метилфенил)-этанола и 186,7 мг (0,71 ммоль) трифенилфосфина, смесь охлаждали в ледяной бане и добавляли 191,9 мг (0,95 ммоль) ДИАД. После перемешивания в течение ночи смесь выпаривали до сухого состояния и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Получали 104 мг указанного в заголовке соединения.
Стадия 4: 2-[3-(2-м-толилэтокси)-4-трифторметилбензоиламино]-индан-2-карбоновая кислота
Указанное в заголовке соединение получали из соединения стадии 3 путем гидролиза с гидроксидом лития по аналогии со стадией 3 примера 180.
ЖХ/МС (метод LC3): Rt = 2,15 мин; м/z = 484,19 [MH+]
1H-ЯМР: δ = 12,50 (уш.с, 1H); 9,00 (с, 1H); 7,69 (д, 1H); 7,59 (с, 1H); 7,55 (д, 1H); 7,20-7,26 (м, 2H); 7,13-7,20 (м, 4H); 7,11 (д, 1H); 7,03 (д, 1H); 4,35 (т, 2H); 3,60 (д, 2H); 3,39 (д, 2H); 3,02 (т, 2H); 2,29 (с, 3H)
По аналогии с примером 185 образцы соединений формулы It, перечисленные в таблице 6, получали с помощью соответствующего 2-(замещенный фенил)-этанола вместо 2-(3-метилфенил)-этанола на стадии 3. Соединения могут быть названы как 2-{3-[2-(R96)-этокси)-4-трифторметилбензоиламино]-индан-2-карбоновая кислота, например, как 2-{3-[2-(3-хлорфенил)-этокси]-4-трифторметилбензоиламино}-индан-2-карбоновая кислота в случае примера 186.

Пример 193
2-{3-[2-(5-Хлор-2-фторфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Стадия 1: 2-(5-Хлор-2-фторфенил)-этанол
Раствор 5 г (26,51 ммоль) 5-хлор-2-фторфенилуксусной кислоты в 60 мл ТГФ добавляли по каплям к суспензии 2,012 г (53,02 ммоль) алюмогидрида лития в 26,5 мл ТГФ. Добавляли 30 мл ТГФ и смесь нагревали с обратным холодильником в течение 3 ч. После охлаждения до 0°C осторожно добавляли раствор 929,7 мг (16,57 ммоль) гидроксида калия в 4 мл воды и смесь перемешивали в течение ночи при комнатной температуре. Образовавшийся осадок фильтровали отсасыванием и промывали ТГФ. Объединенные фильтраты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (ДХМ/метанол 98:2) с получением 3,8 г указанного в заголовке соединения.
Стадия 2: 2-{3-[2-(5-Хлор-2-фторфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота
Указанное в заголовке соединение получали по аналогии с примером 185, с использованием 2-(5-хлор-2-фторфенил)-этанола вместо 2-(3-метилфенил)-этанола на стадии 3.
ЖХ/МС (метод LC4): Rt = 2,35 мин; м/z = 484,13 [MH+]
1H-ЯМР: δ = 12,45 (уш.с, 1H); 8,61 (с, 1H); 7,55 (м, 1H) 7,50 (д, 1H); 7,45 (с, 1H); 7,30-7,37 (м, 1H); 7,18-7,26 (м, 3H); 7,11-7,19 (м, 2H); 7,00 (д, 1H); 4,21 (т, 2H); 3,80 (с, 3H); 3,59 (д, 2H); 3,35 (д, 2H); 3,06 (т, 2H)
Пример 194
2-[4-Метокси-3-(4-трифторметилфенилэтинил)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 2-(3-бром-4-метоксибензоиламино)-индан-2-карбоновой кислоты
3-Бром-4-метоксибензойной кислоты (22,8 г, 98,8 ммоль) растворяли в тионилхлориде (42 мл) и перемешивали при 60°С в течение 30 мин. Летучие фракции выпаривали под вакуумом и остаток десорбировали диоксаном. Полученный хлорангидрид растворяли в ДХМ (50 мл). Гидрохлорид метилового эфира 2-аминоиндан-2-карбоновой кислоты (15,0 г, 65,9 ммоль) суспендировали в ДХМ (100 мл), добавляли EDIA (10,2 г, 79,1 ммоль), смесь охлаждали в ледяной бане и постепенно добавляли раствор хлорангидрида. Смесь перемешивали в течение ночи при комнатной температуре и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (градиент ДХМ/метанол) с последующей кристаллизацией от EA с получением 21,8 г указанного в заголовке соединения.
ЖХ/МС (метод LC3): Rt = 1,495 мин; м/z = 404,0/406,0 [MH+]
Стадия 2: Метиловый эфир 2-[4-метокси-3-(4-трифторметилфенилэтинил)-бензоиламино]-индан-2-карбоновой кислоты
300 мг (0,74 ммоль) соединения стадии 1 и 90,1 мг (0,89 ммоль) триэтиламина растворяли в 10 мл сухого толуола. Добавляли 171,5 мг (148 мкмоль) тетракис(трифенилфосфин)палладия(0) и 14,1 мг (74 мкмоль) йодида меди(I) и смесь перемешивали в течение 30 мин. Впоследствии добавляли 126,2 мг (0,74 ммоль) 1-этинил-4-трифторметилбензола и смесь нагревали до 100°С в течение 10 ч. Смесь фильтровали, растворитель выпаривали и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением 40 мг указанного в заголовке соединения.
Стадия 3: 2-[4-Метокси-3-(4-трифторметилфенилэтинил)-бензоиламино]-индан-2-карбоновая кислота
Из соединения стадии 2 указанное в заголовке соединение получали путем гидролиза с гидроксидом лития по аналогии со стадией 3 примера 180 и очистки хроматографией на силикагеле (ДХМ/метанол 98:2). Выход: 32 мг.
ЖХ/МС (метод LC4): Rt = 2,57 мин.; м/z = 480,17 [MH+]
1H-ЯМР: δ = 12,45 (уш.с, 1H); 8,80 (с, 1H); 8,09 (с, 1H); 7,94 (д, 1H); 7,80 (д, 2H); 7,74 (д, 2H); 7,60-7,65 (м, 1H); 7,13-7,25 (м, 4H); 3,91 (с, 3H); 3,57 (д, 2H); 3,40 (д, 2H)
Пример 195
2-[3-(4-Трет-бутилфенилэтинил)-4-метоксибензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 194, с использованием 1-трет-бутил-4-этинилбензола вместо 1-этинил-4-трифторметилбензола.
ЖХ/МС (метод LC4): Rt = 2,79 мин.; м/z = 486,24 [MH+]
Пример 196
2-[(3'-Изопропил-6-метоксибифенил-3-карбонил)-амино]-индан-2-карбоновая кислота

250 мг (0,62 ммоль) соединения стадии 1 примера 194 и 152,1 мг (0,93 ммоль) 3-изопропилфенилбороновой кислоты растворяли в 5 мл ДМФ и 5 мл толуола в атмосфере аргона. Добавляли 187,9 мг (1,24 ммоль) фторида цезия и 35,73 мг (0,05 ммоль) тетракис(трифенилфосфин)палладия(0) и смесь перемешивали в течение ночи при температуре 100°C. После охлаждения смесь фильтровали и растворитель выпаривали. Полученный метиловый эфир 2-[(3'-изопропил-6-метоксибифенил-3-карбонил)-амино]-индан-2-карбоновой кислоты растворяли в 10 мл смеси ТГФ и воды (9:1), добавляли 42,2 мг (1,80 ммоль) гидроксида лития и смесь перемешивали в течение ночи. Растворитель выпаривали и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Получали 71 мг указанного в заголовке соединения.
ЖХ/МС (метод LC4): Rt = 2,50 мин; м/z = 430,30 [MH+]
1H-ЯМР: δ = 12,5 (уш.с, 1H); 8,72 (с, 1H); 7,89 (д, 1H); 7,80 (с, 1H); 7,13-7,37 (м, 9H); 3,81 (с, 3H); 3,60 (д, 2H); 2,92 (м, 1H); 1,24 (д, 6H)
По аналогии с примером 196 образцы соединений формулы Iu, перечисленные в таблице 7, получали с помощью соответствующей замещенной фенилбороновой кислоты вместо 3-изопропилфенилбороновой кислоты. В случае примеров 198 и 199 промежуточный метиловый эфир 2-[(замещенный бифенил-3-карбонил)-амино]-индан-2-карбоновой кислоты очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) до проведения гидролиза. Соединения могут быть названы как 2-[(замещенный бифенил-3-карбонил)-амино]-индан-2-карбоновая кислота, например, как 2-[(3'-цианометил-6-метоксибифенил-3-карбонил)-амино]-индан-2-карбоновая кислота в случае примера 199, в котором группа R97 представляет собой 3-цианометилфенил и, с учетом правил номенклатуры, группа 3-(R97)-4-метоксифенил-C(O), представленная в формуле Iu, таким образом, называется 3'-цианометил-6-метоксибифенил-3-карбонильной.

Пример 204
2-{3-[2-(2,5-дифторфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 2-{3-[2-(2,5-дифторфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновой кислоты
300,1 мг (0,88 ммоль) метилового эфира 2-(3-гидрокси-4-метоксибензоиламино)-индан-2-карбоновой кислоты, 208,6 мг (1,32 ммоль) 2-(2,5-дифторфенил)-этанола и 346 мг (1,32 ммоль) трифенилфосфина растворяли в 10 мл ТГФ. Смесь охлаждали в ледяной бане и добавляли 355,5 мг (1,76 ммоль) ДИАД. После перемешивания в течение ночи смесь выпаривали до сухого состояния и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Получили 340 мг указанного в заголовке соединения.
Стадия 2: 2-{3-[2-(2,5-дифторфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота
Из соединения стадии 1 указанное в заголовке соединение получали путем гидролиза с гидроксидом лития по аналогии со стадией 3 примера 180 и очистки препаративной ОФ ВЭЖХ (градиент вода/ACN). Выход: 260 мг.
ЖХ/МС (метод LC3): Rt = 1,81 мин; м/z = 468,11 [MH+]
1H-ЯМР: δ = 12,5 (уш.с, 1H); 8,63 (с, 1H); 7,51 (д, 1H); 7,44 (с, 1H); 7,30-7,10 (м, 6H); 7,00 (д, 1H); 4,22 (т, 2H); 3,79 (с, 3H); 3,59 (д, 2H); 3,38 (д, 2H); 3,08 (т, 2H)
Пример 205
2-{3-[2-(5-Этилпиридин-2-ил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 204, с использованием 2-(5-этилпиридин-2-ил)-этанола вместо 2-(2,5-дифторфенил)-этанола на стадии 1.
ЖХ/МС (метод LC3): Rt = 1,22 мин; м/z = 461,34 [MH+]
Пример 206
2-{4-Метокси-3-[2-(4-метилтиазол-5-ил)-этокси]-бензоиламино}-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 204, с использованием 2-(4-метилтиазол-5-ил)-этанола вместо 2-(2,5-дифторфенил)-этанола на стадии 1.
ЖХ/МС (метод LC4): Rt = 2,70 мин; м/z = 453,11 [MH+]
Пример 207
6-[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-6,7-дигидро-5H-циклопентапиразин-6-карбоновая кислота

Указанное в заголовке соединение получали из 2,3-бис-хлорметилпиразина (K. Yoshiizumi et al., Bioorg. Med. Chem. 11 (2003), 433-450) по аналогии с примерами 97 и 98, с использованием N-метилпирролидона вместо ДМФ как растворителя на начальной стадии циклизации. Промежуточный эфир аминокислоты не очищали, но использовали в качестве сырья.
ЖХ/МС (метод LC1): Rt = 1,32 мин; м/z = 448,0 [MH+]
Пример 208
2-{[6-Метокси-5-(2-м-толилэтокси)-пиридин-3-карбонил]-амино}-индан-2-карбоновая кислота
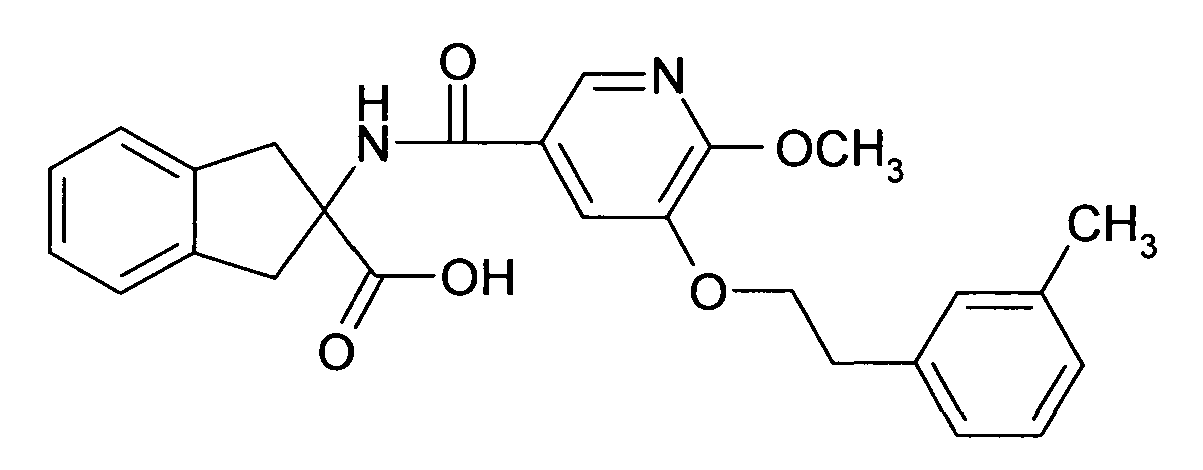
Метиловый эфир 6-хлор-5-нитроникотиновой кислоты получали в соответствии с методами, описанными в WO 2005/021544, и преобразовывали в метиловый эфир 5-гидрокси-6-метоксиникотиновой кислоты в соответствии с методами, описанными в WO 95/04045, с последующим преобразованием в указанное в заголовке соединение путем этерификации с 2-м-толилэтанолом по аналогии со стадией 1 примера 1, гидролиза эфирной группы по аналогии с примером 2, проведения реакции полученной карбоновой кислоты с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 1 примера 15 и гидролиза эфирной группы по аналогии с примером 2.
1H-ЯМР: δ = 12,5 (с, 1H); 8,75 (с, 1H); 8,21 (д, 1H); 7,63 (д, 1H); 7,75-7,13 (м, 6H); 7,11 (д, 1H); 7,02 (д, 1H); 4,21 (т, 2H); 3,91 (с, 3H); 3,61 (д, 2H); 3,37 (д, 2H); 3,01 (т, 2H); 2,28 (с, 3H)
Пример 209
2-[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-1-метилиндан-2-карбоновая кислота
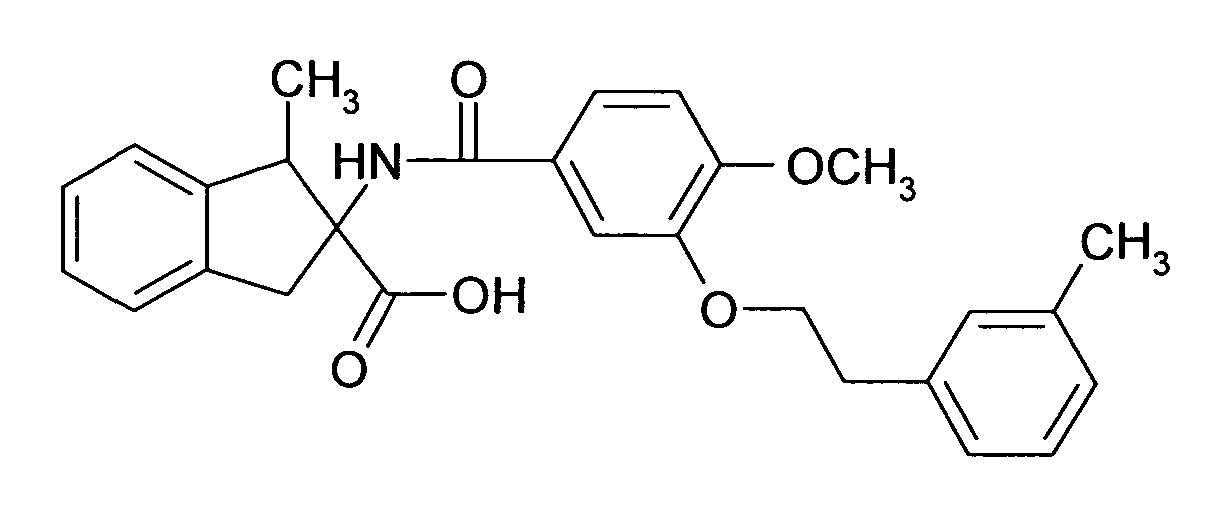
Стадия 1: 1-(1-Хлорэтил)-2-хлорметилбензол
1-(2-Гидроксиметилфенил)-этанол (полученный в соответствии с методологией, описанной в P. Canonne et al., Tetrahedron 44 (1988), 2903-2912) (0,376 г, 2,47 ммоль) растворяли в ДХМ. Добавляли тионилхлорид (2,94 г, 24 ммоль) и оставляли для прохождения реакции в течение 1 ч. Смесь разделяли в EA, воде и избытке водного раствора гидрокарбоната натрия. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния под вакуумом. Остаток очищали хроматографией на силикагеле с HEP с получением 0,228 г указанного в заголовке соединения.
1H-ЯМР: δ = 7,68 (дд, 1H); 7,46-7,41 (м, 2H); 7,35 (дд, 1H); 5,66 (кв., 1H); 4,95 (д, 1H); 4,90 (д, 1H); 1,82 (д, 3H)
Стадия 2: 2-[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-1-метилиндан-2-карбоновая кислота
Указанное в заголовке соединение получали из соединения стадии 1 по аналогии с примерами 97 и 98, с использованием N-метилпирролидона вместо ДМФ как растворителя на начальной стадии циклизации. Промежуточный эфир аминокислоты не очищали, но использовали в качестве сырья.
ЖХ/МС (метод LC1): Rt = 1,66 мин; м/z = 460,2 [MH+]
Пример 210
2-(3-{2-[3-(2-Гидрокси-1-гидроксиметил-1-метилэтил)-фенил]-этокси}-4-метоксибензоиламино}-индан-2-карбоновая кислота

Стадия 1: Диметиловый эфир 2-(3-метоксикарбонилметилфенил)-малоновой кислоты
Трис(дибензилиденацетон)дипалладий(0) (0,104 г, 0,113 ммоль), тетрафторборат три-(трет-бутил)фосфония (65,8 мг, 0,227 ммоль) и гидрид натрия (295 мг, 60% дисперсия в минеральном масле) поместили в колбу в атмосфере аргона. Метиловый эфир (3-бромфенил)уксусной кислоты (1,30 г, 5,67 ммоль) растворяли в ТГФ (10 мл) и добавляли в смесь. Затем добавляли диметилмалонат (0,995 г, 7,37 ммоль) и смесь перемешивали с обратным холодильником в течение ночи. Смесь фильтровали через тонкий слой силикагеля, выпаривали до сухого состояния и остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 0,704 г указанного в заголовке соединения.
ЖХ/МС (метод LC1): Rt = 1,28 мин; м/z = 281,1 [MH+]
Стадия 2: Диметиловый эфир 2-(3-метоксикарбонилметилфенил)-2-метилмалоновой кислоты
Соединение стадии 1 (0,353 г, 1,26 ммоль) растворяли в ДМФ (1,5 мл), добавляли трет-бутоксид калия (151 мг, 1,32 ммоль), смесь перемешивали при комнатной температуре в течение 10 мин и затем добавляли йодометан (0,542 г, 3,78 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч и разделяли в EA и 2н. хлористоводородной кислоте. Объединенные экстракты промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и выпаривали до сухого состояния. Остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением 0,128 г указанного в заголовке соединения.
ЖХ/МС (метод LC1): Rt = 1,36 мин; м/z = 295,0 [MH+]
Стадия 3: 2-[3-(2-Гидроксиэтил)-фенил]-2-метилпропан-1,3-диол
Соединение стадии 2 (0,128 г, 0,435 ммоль) растворяли в 2 мл ТГФ и осторожно добавляли в ледяную суспензию алюмогидрида лития (174 мг, 4,35 ммоль) в ТГФ. Через несколько минут добавляли диэтиловый эфир (12 мл) и затем 200 мкл EA. Впоследствии постепенно и осторожно добавляли воду - до образования на дне колбы светлосерой массы солей окиси алюминия. Супернатант сливали и осадок промывали EA. Объединенные экстракты сушили над сульфатом натрия и выпаривали до сухого состояния. Остаток использовали на следующей стадии без дополнительной очистки.
ЖХ/МС (метод LC1): Rt = 0,69 мин; м/z = 228,1 [MNH4+]
Стадия 4: 2-(3-{2-[3-(2-Гидрокси-1-гидроксиметил-1-метилэтил)-фенил]-этокси}-4-метоксибензоиламино}-индан-2-карбоновая кислота
Соединение стадии 3 вступало в реакцию с метил-3-гидрокси-4-метоксибензоатом по аналогии со стадией 1 примера 94. Из полученного промежуточного продукта получали указанное в заголовке соединение по аналогии со стадией 1 примера 15.
ЖХ/МС (метод LC1): Rt = 1,28 мин; м/z = 520,1 [MH+]
Пример 211
2-[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-октагидроинден-2-карбоновая кислота

Ацетилхлорид (22 мг, 0,282 ммоль) осторожно растворяли в этаноле (2 мл). Добавляли 2-аминоиндан-2-карбоновую кислоту (50 мг, 0,282 ммоль) и двуокись платины (25 мг) и смесь гидрировали при комнатной температуре под давлением водорода 5 бар в течение 5 ч. Раствор фильтровали через тонкий слой целита и выпаривали до сухого состояния. Для преобразования в метиловый эфир 2-аминооктагидроинден-2-карбоновой кислоты остаток суспендировали в метаноле (2 мл), добавляли тионилхлорид (0,5 мл) и смесь перемешивали в течение ночи при комнатной температуре. Смесь выпаривали до сухого состояния с получением 99 мг сырья, использованного на следующей стадии без дополнительной очистки. Из полученного промежуточного продукта указанного в заголовке соединение получали по аналогии со стадией 1 примера 15 и в результате гидролиза эфирной группы по аналогии с примером 2.
1H-ЯМР: δ = 12,0 (с, 1H); 8,40 (с, 1H); 7,48 (дд, 1H); 7,41 (дд, 1H); 7,22-7,16 (м, 2H); 7,11 (д, 1H); 7,04 (д, 1H); 7,00 (д, 1H); 4,18 (т, 2H); 3,80 (с, 3H); 3,01 (т, 2H); 2,29 (с, 3H); 2,20-2,00 (м, 6H); 1,53-1,40 (м, 6H); 1,32-1,20 (м, 2H)
Пример 212
2-{3-[2-(3-Хлорфенил)-2,2-дифторэтокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Стадия 1: 2-(3-Хлорфенил)-2,2-дифторэтанол
1,00 г (4,26 ммоль) этилового эфира (3-хлорфенил)-дифторуксусной кислоты (полученного в соответствии с методологией, описанной в WO 2006/122788) растворяли в 100 мл метанола и обрабатывали 120 мг (0,75 ммоль) боргидрида натрия на ледяной бане. После перемешивания в течение ночи смесь выпаривали и остаток очищали хроматографией на силикагеле (ДХМ/метанол 98:2) с получением 700 мг указанного в заголовке соединения.
Стадия 2: 2-(3-Хлорфенил)-2,2-дифторэтиловый эфир трифторметансульфоновой кислоты
700 мг (3,64 ммоль) соединения стадии 1 растворяли в 10 мл дихлорметана и обрабатывали при температуре 0°C 78 мкл (4,36 ммоль) EDIA и 1,23 г (4,36 ммоль) ангидридом трифторметансульфоновой кислоты. После завершения реакции (контроль с помощью тонкослойной хроматографии) (ДХМ/метанол 98:2) смесь выливали на воду и фазы разделяли. Органическую фазу промывали один раз насыщенным раствором хлорида натрия, сушили над сульфатом натрия и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (ДХМ) с получением 450 мг указанного в заголовке соединения.
Стадия 3: Метиловый эфир 2-{3-[2-(3-хлорфенил)-2,2-дифтор-этокси]-4-метоксибензоиламино}-индан-2-карбоновой кислоты
К смеси метилового эфира 230 мг (0,67 ммоль) 2-(3-гидрокси-4-метоксибензоиламино)-индан-2-карбоновой кислоты и 227 мг (1,62 ммоль) карбоната калия в 6 мл ацетона и 1,7 мл ДМФ постепенно добавляли раствор 437 мг (1,65 ммоль) соединения стадии 2. Реакционную смесь перемешивали в течение 3 дней при комнатной температуре, а затем выпаривали. Остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением 20 мг указанного в заголовке соединения.
Стадия 4: 2-{3-[2-(3-Хлорфенил)-2,2-дифторэтокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота
20 мг соединения стадии 3 растворяли в 5 мл смеси ТГФ и воды (9:1) и добавляли 1,9 мг (77,5 мкмоль) гидроксида лития. После перемешивания при комнатной температуре в течение 3 дней смесь подкисляли 1 М хлористоводородной кислотой и выпаривали. Остаток очищали хроматографией на силикагеле (ДХМ/метанол 95:5) и ОФ ВЭЖХ (градиент вода/ACN) с получением 8 мг указанного в заголовке соединения.
ЖХ/МС (метод LC1): Rt = 1,64 мин; м/z = 502,10 [MH+]
1H-ЯМР: δ = 12,45 (уш.с, 1H); 8,62 (с, 1H); 7,75 (с, 1H); 7,50-7,65 (м, 4H); 7,48 (с, 1H); 7,23 (м, 2H) 7,17 (м, 2H); 7,04 (д, 1H); 4,65 (т, 2H); 3,80 (с, 3H); 3,59 (д, 2H); 3,38 (д, 2H)
Пример 213
2-[3-(2,2-Дифтор-2-фенилэтокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 212, начиная с 2,2-дифтор-2-фенилэтанола.
ЖХ/МС (метод LC1): Rt = 1,53 мин; м/z = 468,15 [MH+]
1H-ЯМР: δ = 12,45 (уш.с, 1H); 8,62 (с, 1H); 7,64 (д, 2H); 7,46-7,59 (м, 5H); 7,22 (м, 2H) 7,17 (м, 2H); 7,04 (д, 1H); 4,60 (т, 2H); 3,80 (с, 3H); 3,58 (д, 2H); 3,38 (д, 2H)
Пример 214
4,7-Дифтор-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: (3,6-Дифтор-2-гидроксиметилфенил)-метанол
Алюмогидрид лития (792 мг, 19,8 ммоль) суспендировали в ТГФ (6 мл) и охлаждали в ледяной бане. Раствор 4,7-дифторизобензофуран-1,3-диона (730 мг, 3,97 ммоль) в ТГФ (6 мл) добавляли в течение 5 мин. После завершения реакции (5 мин) добавляли диэтиловый эфир (30 мл). Затем добвляли 2 мл EA для того, чтобы разложить избыток алюмогидрида лития, после чего постепенно добавляли воду до осаждения окиси алюминия. Супернатант сливали и осадок дважды промывали EA. Объединенные экстракты сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния с получением 360 мг указанного в заголовке соединения.
1H-ЯМР: δ = 7,16 (т, 2H); 5,13 (т, 2H); 4,60 (д, 4H)
Стадия 2: 2,3-бис-хлорметил-1,4-дифторбензол
Соединение стадии 1 (360 мг, 2,07 ммоль) растворяли в ацетилхлориде (2,3 мл) в ампуле. Через 10 мин добавляли хлорид цинка (843 мг, 6,21 ммоль) и смесь нагревали до 130°С в микроволновом реакторе в течение 30 мин. После охлаждения смесь разделяли в диэтиловом эфире и насыщенном растворе гидрокарбоната натрия и водную фазу экстрагировали диэтиловым эфиром. Объединенные органические фазы сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния с получением 200 мг указанного в заголовке соединения.
1H-ЯМР: δ = 7,40 (т, 2H); 4,90 (с, 4H)
Стадия 3: 4,7-Дифтор-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота
Соединение стадии 2 преобразовали в указанное в заголовке соединение по аналогии с примерами 97 и 98.
ЖХ/МС (метод LC13): Rt = 2,60 мин; м/z = 482,2 [MH+]
Пример 215
4-фтор-2-[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 214, начиная с (3-фтор-2-гидроксиметилфенил)-метанола.
ЖХ/МС (метод LC11): Rt = 1,91 мин; м/z = 464,2 [MH+]
Пример 216
2-[4-Метокси-3-(2-м-толилэтокси)-бензоиламино]-4-метилиндан-2-карбоновая кислота
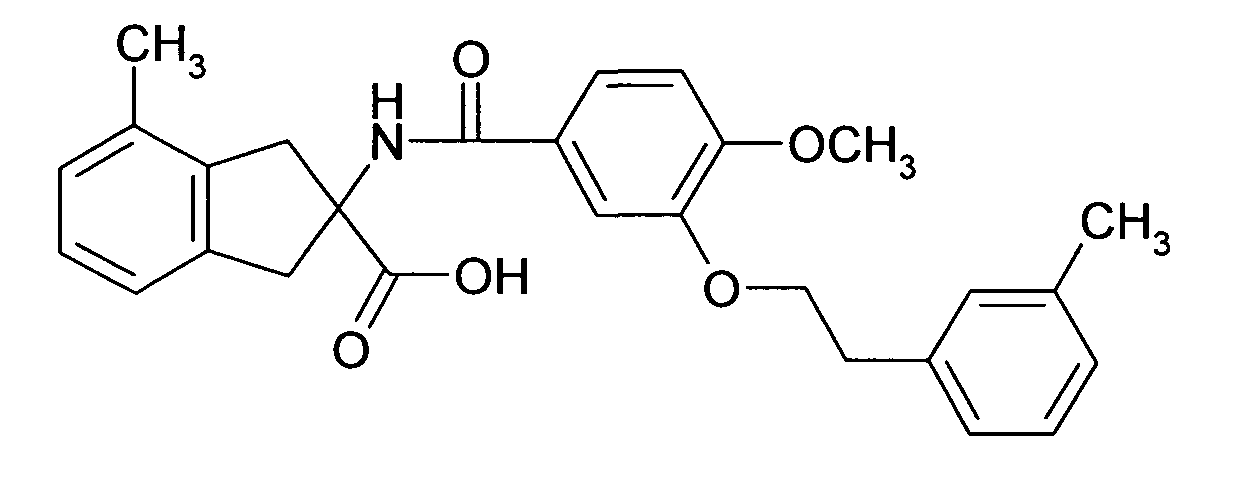
Указанное в заголовке соединение получали по аналогии с примером 214, начиная с 4-метилизобензофуран-1,3-диона.
ЖХ/МС (метод LC12): Rt = 3,74 мин; м/z = 460,2 [MH+]
Пример 217
4-Хлор-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 214, начиная с 4-хлоризобензофуран-1,3-диона.
ЖХ/МС (метод LC13): Rt = 2,67 мин; м/z = 480,2 [MH+]
Пример 218
5-Циано-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: Этиловый эфир 5-бром-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты
Начиная с 5-бром-3H-изобензофуран-1-она, промежуточный 4-бром-1,2-бис-хлорметилбензол получали по аналогии со стадиями 1 и 2 примера 214. Данный промежуточный продукт преобразовывали в указанное в заголовке соединение по аналогии с примером 97.
ЖХ/МС (метод LC13): Rt = 3,03 мин; м/z = 552,2 [MH+]
Стадия 2: Этиловый эфир 5-циано-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты
Соединение стадии 1 (50 мг, 0,091 ммоль) добавляли к смеси цианида цинка (10,6 мг, 0,091 ммоль) и тетракис(трифенилфосфин)палладия(0) (5,2 мг, 0,004 ммоль) в ДМФ (0,16 мл) при 150°С и перемешивали в течение 2 ч. После охлаждения трет-бутиловый эфир добавляли и смесь фильтровали через целит. Фильтрат промывали водой, органическую фазу сушили над сульфатом магния, фильтровали и выпаривали до сухого состояния с получением указанного в заголовке соединения, которое использовали без дополнительной очистки.
Стадия 3: 5-Циано-2-[4-метил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота
Соединение стадии 2 преобразовывали в указанное в заголовке соединение по аналогии со стадией 3 примера 94.
ЖХ/МС (метод LC13): Rt = 2,54 мин; м/z = 471,3 [MH+]
Пример 219
5-Карбамоил-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Соединение стадии 2 примера 218 преобразовывали в указанное в заголовке соединение путем гидролиза по аналогии с примером 98 (время гидролиза 3 ч).
ЖХ/МС (метод LC14): Rt = 3,68 мин; м/z = 489,3 [MH+]
Пример 220
1-Гидрокси-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: Этиловый эфир 2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-1-оксоиндан-2-карбоновой кислоты
Этиловый эфир 2-амино-1-оксоиндан-2-карбоновой кислоты (L. Benati et al., J. Org. Chem. 64 (1999), 7836-7841) (460 мг, 2,10 ммоль) вступил в реакцию с 4-метокси-3-(2-м-толилэтокси)-бензойной кислотой по аналогии со стадией 2 примера 13 с получением 0,331 г указанного в заголовке соединения.
ЖХ/МС (метод LC12): Rt = 4,09 мин; м/z = 488,2 [MH+]
Стадия 2: Этиловый эфир 1-гидрокси-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты
Соединение стадии 1 (0,439 г, 0,900 ммоль) растворяли в ТГФ (4 мл). Смесь охлаждали до -30°С и добавляли боргидрид натрия (35 мг, 0,90 ммоль) с последующим добавлением по каплям метанола. Через 30 мин смесь разделяли в диэтиловом эфире и 2н. хлористоводородной кислоте, водную фазу экстрагировали диэтиловым эфиром и объединенные органические фазы сушили над сульфатом натрия и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением указанного в заголовке соединения в виде смеси стереоизомеров.
ЖХ/МС (метод LC12): Rt = 3,77 мин; м/z = 490,3 [MH+]
Стадия 3: 1-Гидрокси-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота
Соединение стадии 2 гидролизировали по аналогии с примером 2. В результате очистки ОФ ВЭЖХ (градиент вода/ACN) получали один из диастереомеров (диастереомер А) указанного в заголовке соединения в чистом виде (как рацемат) и смесь другого диастереомера с диастереомером А (относительная стереохимия диастереомеров неизвестна).
Диастереомер А:
ЖХ/МС (метод LC12): Rt = 3,43 мин; м/z = 462,2 [MH+]
1H-ЯМР: δ = 12,1 (уш.с, 1H); 8,43 (с, 1H); 7,48 (дд, 1H); 7,41 (д, 1H); 7,32 (дд, 1H); 7,28-7,13 (м, 5H); 7,10 (д, 1H); 7,02 (д, 1H); 7,00 (д, 1H); 5,70 (уш.с, 1H); 5,40 (с, 1H); 4,16 (т, 2H); 3,90 (д, 1H); 3,09 (д, 1H); 3,00 (т, 2H); 2,28 (с, 3H)
Пример 221
2-{[5-(3-Изопропилфенил)-6-метоксипиридин-3-карбонил]-амино}-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 5-(3-изопропилфенил)-6-метоксиникотиновой кислоты
В атмосфере аргона смесь метилового эфира 5-бром-6-метоксиникотиновой кислоты (W. J. Thompson и J. Gaudino, J. Org. Chem. 49 (1984), 5237-5243) (100 мг, 0,406 ммоль), 3-изопропилфенилбороновой кислоты (73 мг, 0,447 ммоль), тетрафторбората три-(трет-бутил)фосфония (7 мг, 0,024 ммоль), трис(дибензилиденацетон)дипалладия(0) (11 мг, 0,012 ммоль) и фторида калия (78 мг, 1,34 ммоль) в колбе суспендировали в диоксане (1,5 мл) и нагревали до 45°С в течение 3 ч. После охлаждения его фильтровали через тонкий слой силикагеля и выпаривали до сухого состояния. Очистка остатка хроматографией на силикагеле (градиент HEP/EA) и последующей ОФ ВЭЖХ (градиент вода/ACN) дали 63 мг указанного в заголовке соединения.
ЖХ/МС (метод LC13): Rt = 2,95 мин; м/z = 286,1 [MH+]
Стадия 2: 5-(3-Изопропилфенил)-6-метоксиникотиновая кислота
Соединение стадии 1 (60 мг, 0,63 ммоль) гидролизировали по аналогии со стадией 3 примера 94 с получением 57 мг указанного в заголовке соединения.
ЖХ/МС (метод LC13): Rt = 2,48 мин; м/z = 272,1 [MH+]
Стадия 3: 2-{[5-(3-Изопропилфенил)-6-метоксипиридин-3-карбонил]-амино}-индан-2-карбоновая кислота
Соединение стадии 2 вступало в реакцию с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 2 примера 13, и полученный эфир гидролизировали по аналогии с примером 2.
ЖХ/МС (метод LC14): Rt = 3,52 мин; м/z = 431,2 [MH+]
Пример 222
2-{[6-Метокси-5-(3-метилсульфанилфенил)-пиридин-3-карбонил]-амино}-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 2-[(5-бром-6-метоксипиридин-3-карбонил)-амино]-индан-2-карбоновой кислоты
Метиловый эфир 5-бром-6-метоксиникотиновой кислоты (W. J. Thompson и J. Gaudino, J. Org. Chem. 49 (1984), 5237-5243) (2,00 г, 8,13 ммоль) гидролизировали по аналогии с примером 2. Полученная кислота вступала в реакцию с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 2 примера 13.
ЖХ/МС (метод LC14): Rt = 3,28 мин; м/z = 405,0 [MH+]
Стадия 2: 2-{[6-Метокси-5-(3-метилсульфанилфенил)-пиридин-3-карбонил]-амино}-индан-2-карбоновая кислота
Соединение стадии 1 вступало в реакцию с 3-метилсульфанилфенилбороновой кислотой по аналогии со стадией 1 примера 221. Промежуточный эфир гидролизировали по аналогии с примером 2.
ЖХ/МС (метод LC14): Rt = 3,30 мин; м/z = 435,1 [MH+]
По аналогии с примером 221 и примером 222, соответственно, образцы соединений формулы Iv, перечисленные в таблице 8, получали с помощью соответствующей замещенной фенилбороновой кислоты. Если первоначальная реакция сочетания палладия в процессе, аналогичном примеру 221, протекала неудовлетворительно, ее повторяли еще раз. Соединения могут быть названы как 2-{[5-(замещенный фенил)-6-метоксипиридин-3-карбонил]-амино}-индан-2-карбоновая кислота, например, как 2-{[6-метокси-5-(3-метилфенил)-пиридин-3-карбонил]-амино}-индан-2-карбоновая кислота в случае примера 224.

(a) получение по аналогии с примером 221
(b) получение по аналогии с примером 222
(c) [(2M-H)‾] вместо [MH+]
Пример 237
2-{[5-(3-Этансульфонилфенил)-6-метоксипиридин-3-карбонил]-амино}-индан-2-карбоновая кислота

Соединение примера 233 (0,50 мг, 0,11 ммоль) растворяли в уксусной кислоте (3,8 мл). Добавляли перекись водорода (30-процентный раствор в воде, 0,034 мл, 0,33 ммоль) и проводили реакцию смеси при комнатной температуре в течение 72 ч. Смесь разделяли в EA и водном растворе сульфита натрия (с концентрацией около 1%). Органическую фазу сушили над сульфатом магния, фильтровали и выпаривали до сухого состояния. Остаток очищали ОФ ВЭЖХ (градиент вода/ACN). После выпаривания фракции продукта остаток перемешивали со смесью диэтилового эфира / HEP, фильтровали и сушили под вакуумом.
ЖХ/МС (метод LC14): Rt = 3,88 мин; м/z = 481,2 [MH+]
Пример 238
2-(4-Метокси-3-о-толилоксибензоиламино)-индан-2-карбоновая кислота

Стадия 1: 4-Метокси-3-о-толилоксибензойная кислота
Карбонат калия (1,20 г, 8,66 ммоль), о-крезол (468 мг, 4,33 ммоль), медный порошок (28 мг, 0,43 ммоль) и 3-бром-4-метоксибензойную кислоту (1,00 г, 4,33 ммоль) суспендировали в ДМФ (5 мл) и нагревали до 165°С в течение ночи. Повторно добавляли карбонат калия (1,20 г, 8,66 ммоль) и о-крезол (468 мг, 4,33 ммоль) и продолжали нагревать еще 2 ч. Неочищенную смесь разделяли в EA и 2н. хлористоводородной кислоте, водную фазу экстрагировали EA и объединенные органические фазы сушили над сульфатом магния, фильтровали и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 600 мг указанного в заголовке соединения.
ЖХ/МС (метод LC14): Rt = 3,00 мин; м/z = 300,1 [(M+CH3CN+H)+]
Стадия 2: 2-(4-Метокси-3-о-толилоксибензоиламино)-индан-2-карбоновая кислота
Соединение стадии 1 вступало в реакцию с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 2 примера 13. Полученный эфир гидролизировали по аналогии с примером 2.
ЖХ/МС (метод LC12): Rt = 3,57 мин; м/z = 418,1 [MH+]
Пример 239
2-(4-Метокси-3-м-толилоксибензоиламино)-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 238, с использованием м-крезола вместо о-крезола.
ЖХ/МС (метод LC12): Rt = 3,54 мин; м/z = 418,1 [MH+]
Пример 240
2-[4-Метокси-3-(2-метилбензоил)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 4-метокси-3-(2-метилбензоил)-бензойной кислоты
Метиловый эфир 4-метоксибензойной кислоты (5,00 г, 30,1 ммоль) и 2-метилбензоилхлорид (4,88 г, 31,6 ммоль) растворяли в хлорбензоле (10 мл), осторожно добавляли хлорид олова(IV) (9,41 г, 36,1 ммоль) и смесь нагревали до 140°С в течение 3 ч. Добавление хлорангидрида и тетрахлорида олова повторяли два раза и затем смесь нагревали до 140°С в течение 3 ч каждый раз. Смесь выливали в 300 мл воды со льдом и экстрагировали ДХМ. Объединенные экстракты сушили над сульфатом магния, фильтровали и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (градиент HEP/EA) с последующей ОФ ВЭЖХ (градиент вода/ACN) с получением 200 мг указанного в заголовке соединения.
1H-ЯМР: δ = 8,12 (дд, 1H); 7,91 (д, 1H); 7,71 (д, 1H); 7,50-7,41 (м, 1H); 7,31-7,22 (м, 2H); 7,06 (д, 1H); 3,81 (с, 3H); 3,72 (с, 3H); 2,42 (с, 3H)
Стадия 2: 2-[4-Метокси-3-(2-метилбензоил)-бензоиламино]-индан-2-карбоновая кислота
Соединение стадии 1 гидролизировали по аналогии с примером 2, и полученная кислота вступала в реакцию с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 2 примера 13. Полученный эфир гидролизировали по аналогии с примером 2.
ЖХ/МС (метод LC14): Rt = 3,16 мин; м/z = 430,1 [MH+]
Пример 241
2-[3-(Гидрокси-о-толилметил)-4-метоксибензоиламино]-индан-2-карбоновая кислота

Соединение примера 240 (70 мг, 0,163 ммоль) растворяли в смеси метанола (1,5 мл) и этанола (1,5 мл) и охлаждали в ледяной бане. Добавляли двумя порциями боргидрид натрия (18,9 мг, 0,49 ммоль), и реакция смеси протекала при охлаждении льдом до завершения (3 ч.). Летучие фракции выпаривали и остаток разделяли в диэтиловом эфире и 1н. хлористоводородной кислоте. Водную фазу экстрагировали диэтиловым эфиром и объединенные органические фазы сушили и выпаривали. Остаток очищали ОФ ВЭЖХ (градиент вода/ACN) с получением 13 мг указанного в заголовке соединения.
ЖХ/МС (метод LC12): Rt = 3,22 мин; м/z = 432,2 [MH+]
Пример 242
2-[4-Метокси-3-(2-метилбензил)-бензоиламино]-индан-2-карбоновая кислота

Соединение примера 241 (32 мг, 0,074 ммоль) растворяли в этаноле (10 мл), добавляли палладий (10%) на угле (10 мг) и смесь гидрировали при комнатной температуре в течение 1 ч под давлением водорода 5 бар. После завершения реакции смесь фильтровали на силикагеле и выпаривали до сухого состояния. Остаток обрабатывали диэтиловым эфиром, фильтровали и сушили под вакуумом с получением 25 мг указанного в заголовке соединения.
ЖХ/МС (метод LC14): Rt = 3,45 мин; м/z = 416,3 [MH+]
Пример 243
2-[4-Метокси-3-бензилбензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примерами 240, 241 и 242.
ЖХ/МС (метод LC14): Rt = 3,33 мин; м/z = 402,2 [MH+]
Пример 244
2-[4-Метокси-3-(3-метилбензил)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примерами 240, 241 и 242.
ЖХ/МС (метод LC12): Rt = 3,74 мин; м/z = 416,1 [MH+]
Пример 245
2-[4-Метокси-3-(4-метилбензил)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примерами 240, 241 и 242.
ЖХ/МС (метод LC12): Rt = 3,68 мин; м/z = 416,2 [MH+]
Пример 246
2-(3-{2-[3-(2-Аминоэтил)-фенил]-этокси}-4-метоксибензоиламино)-индан-2-карбоновая кислота
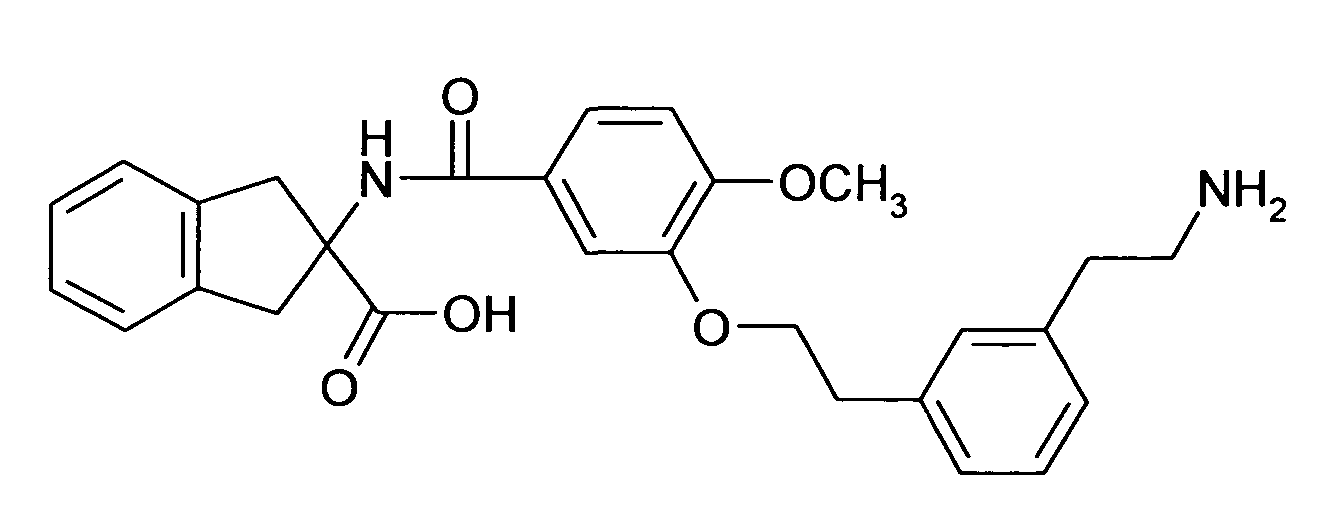
Стадия 1: Метиловый эфир 2-(3-{2-[3-(2-азидоэтил)-фенил]-этокси}-4-метоксибензоиламино)-индан-2-карбоновой кислоты
300 мг (0,613 ммоль) метилового эфира 2-(3-{2-[3-(2-гидроксиэтил)-фенил]-этокси}-4-метоксибензоиламино)-индан-2-карбоновой кислоты (промежуточный метиловый эфир примера 27) и трифенилфосфин (0,241 г, 0,920 ммоль) растворяли в ТГФ (5 мл) и охлаждали в ледяной бане. Последовательно добавляли дифенилфосфорилазид (0,258 г, 0,920 ммоль) и DIAD (0,198 г, 0,920 ммоль), после удаления ледяной бани смесь перемешивали в течение 2 ч при комнатной температуре. Смесь выпаривали до сухого состояния и очищали хроматографией на силикагеле (градиент HEP/EA) с получением 0,188 мг указанного в заголовке соединения.
ЖХ/МС (метод LC14): Rt = 3,68 мин; м/z = 515,3 [MH+]
Стадия 2: Метиловый эфир 2-(3-{2-[3-(2-аминоэтил)-фенил]-этокси}-4-метоксибензоиламино)-индан-2-карбоновой кислоты
Соединение стадии 1 (0,185 г, 0,360 ммоль) и трифенилфосфин (0,149 г, 0,539 ммоль) растворяли в смеси 3 мл ТГФ и 3 мл воды и раствор перемешивали в течение ночи при комнатной температуре. Смесь выпаривали до сухого состояния и очищали хроматографией на силикагеле (градиент ДХМ/метанол/28% аммиака, 70:30:0 до 0:100:0 до 0:90:10) с получением 0,17 г указанного в заголовке соединения.
ЖХ/МС (метод LC14): Rt = 2,68 мин; m/z = 489,2 [MH+]
Стадия 3: 2-(3-{2-[3-(2-Аминоэтил)-фенил]-этокси}-4-метоксибензоиламино)-индан-2-карбоновая кислота
Соединение стадии 2 (85 мг, 0,174 ммоль) гидролизировали по аналогии с примером 2 с получением 31 мг указанного в заголовке соединения.
ЖХ/МС (метод LC12): Rt = 2,68 мин; м/z = 475,2 [MH+]
Пример 247
2-(3-{2-[3-(2-Ацтиламиноэтил)-фенил]-этокси}-4-метоксибензоиламино)-индан-2-карбоновая кислота

Соединение стадии 2 примера 246 (85 мг, 0,174 ммоль) растворяли в ангидриде уксусной кислоты и перемешивали с обратным холодильником в течение 30 мин. Добавляли избыточное количество воды и смесь нагревали с обратным холодильником в течение 10 мин. После охлаждения смесь экстрагировали EA, объединенные экстракты сушили сульфатом натрия и выпаривали до сухого состояния. Остаток очищали ОФ ВЭЖХ (градиент вода/ACN) с получением метилового эфира указанного в заголовке соединения. Гидролиз этого эфира по аналогии с примером 2 дал 17 мг указанного в заголовке соединения.
ЖХ/МС (метод LC16): Rt = 3,95 мин; м/z = 1031,2 [(2M-H)‾]
Пример 248
2-{3-[2-(3-Карбамоилметилфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Стадия 1: (3-Метоксикарбонилметилфенил)-уксусная кислота
(3-Карбоксиметилфенил)-уксусную кислоту (7,4 г, 38,1 ммоль) суспендировали в метаноле (20 мл). Добавляли тионилхлорид (4,5 г, 38 ммоль) при темп. около -30°С (бурная реакция) и затем смесь перемешивали при комнатной температуре в течение 90 мин. После завершения реакции смесь выпаривали до сухого состояния с получением диэфира в виде маслянистого вещества желтого цвета. Данный материал растворяли в метаноле (20 мл), добавляли твердый гидроксид лития (0,948 г, 1 эквивалент) и смесь перемешивали при комнатной температуре в течение 1 ч. После выпаривания метанола остаток разделяли в 2н. хлористоводородной кислое и EA и водную фазу экстрагировали EA. Объединенные органические фазы сушили над сульфатом натрия и выпаривали до сухого состояния. Неочищенную смесь деэфира, моноэфира и дикарбоновой кислоты очищали ОФ ВЭЖХ (градиент вода/ACN) с получением 3,1 г указанного в заголовке соединения.
ЖХ/МС (метод LC16): Rt = 3,18 мин; м/z = 415,3 [(2M-H)‾]
Стадия 2: Метиловый эфир (3-карбамоилметилфенил)-уксусной кислоты
Соединение стадии 1 (0,4 г, 1,92 ммоль) растворяли в тионилхлориде (2,7 мл) и перемешивали при 60°C в течение 1 ч. Летучие фракции выпаривали, остаток растворяли в ДХМ и добавляли к перемешанной смеси EA и 28-процентному водному раствору аммиака. После завершения реакции смесь разделяли в воде и EA и водную фазу экстрагировали EA. Объединенные органические фазы сушили над сульфатом натрия и выпаривали до сухого состояния с получением 0,278 г указанного в заголовке соединения.
ЖХ/МС (метод LC16): Rt = 2,56 мин; м/z = 252,0 [(M+HCOOH-H)‾]
Стадия 3: 2-[3-(2-Гидроксиэтил)-фенил]-ацетамид
Соединение стадии 2 (0,151 г, 0,729 ммоль) растворяли в 0,5 мл ТГФ и добавляли в суспензию алюмогидрида лития (58 мг, 1,46 ммоль) в ТГФ (1,5 мл) при -78°C. Через 2 мин добавляли диэтиловый эфир (6 мл) и затем EA (0,2 мл). После доведения до комнатной температуры постепенно добавляли воду - до образования густой суспензии окиси алюминия, из которой можно было легко слить супернатант. Суспензию повторно промывали EA. Объединенные экстракты сушили сульфатом натрия и выпаривали до сухого состояния с получением 0,101 г указанного в заголовке соединения.
ЖХ/МС (метод LC15): Rt = 2,40 мин; м/z = 180,2 [MH+]
Стадия 4: 2-{3-[2-(3-Карбамоилметилфенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота
Соединение стадии 3 преобразовывали в указанное в заголовке соединение по аналогии со стадией 3 примера 15 с последующим гидролизом по аналогии с примером 2.
ЖХ/МС (метод LC12): Rt = 2,93 мин; м/z = 489,3 [MH+]
Пример 249
2-(3-{2-[3-(2-Гидрокси-2-метилпропил)-фенил]-этокси}-4-метоксибензоиламино)-индан-2-карбоновая кислота

Стадия 1: [3-(2-Гидрокси-2-метилпропил)-фенил]-уксусная кислота
(3-Метоксикарбонилметилфенил)-уксусную кислоту (500 мг, 2,40 ммоль) растворяли в ТГФ (3,5 мл) и постепенно добавляли хлорид метилмагния (2,8 мл, 3 М раствора в ТГФ) при комнатной температуре. После перемешивания в течение 30 мин реакция была завершена. Осторожно добавляли воду и смесь разделяли в EA и 2н. хлористоводородной кислоте. Водную фазу экстрагировали EA и объединенные органические фазы сушили над сульфатом натрия и выпаривали до сухого состояния. Остаток очищали ОФ ВЭЖХ (градиент вода/ACN) с получением 0,34 г указанного в заголовке соединения.
ЖХ/МС (метод LC16): Rt = 2,92 мин; м/z = 415,2 [(2M-H]‾]
Стадия 2: 1-[3-(2-Гидроксиэтил)-фенил]-2-метилпропан-2-ол
Соединение стадии 1 (0,132 г, 0,634 ммоль) растворяли в ТГФ (0,5 мл) и добавляли в нагреваемую с обратным холодильником суспензию алюмогидрида лития (122 мг, 3,1 ммоль) в ТГФ (1 мл). Смесь перемешивали в течение 1 ч с обратным холодильником и охлаждали до комнатной температуры. Добавляли диэтиловый эфир (6 мл) и затем EA (0,4 мл). Затем постепенно добавляли воду - до образования густой суспензии окиси алюминия, из которой можно было легко слить супернатант. Суспензию повторно промывали EA, объединенные экстракты сушили сульфатом натрия и выпаривали до сухого состояния. Остаток очищали ОФ ВЭЖХ (градиент вода/ACN) с получением 49 мг указанного в заголовке соединения.
1H-ЯМР: δ = 7,15 (т, 1H); 7,06-7,00 (м, 3H); 4,59 (т, 1H); 4,23 (с, 1H); 3,58 (дт, 2H); 2,69 (т, 2H); 2,60 (с, 2H); 1,04 (с, 6H)
Стадия 3: 2-(3-{2-[3-(2-Гидрокси-2-метилпропил)-фенил]-этокси}-4-метоксибензоиламино}-индан-2-карбоновая кислота
Соединение стадии 2 преобразовывали в указанное в заголовке соединение по аналогии со стадией 3 примера 15 с последующим гидролизом по аналогии с примером 2.
ЖХ/МС (метод LC14): Rt = 3,05 мин; м/z = 504,2 [MH+]
Пример 250
2-[4-Метокси-3-(3-фенилоксетан-3-илметокси)-бензоиламино]-индан-2-карбоновая кислота

Соединение стадии 2 примера 15 и (3-фенилоксетан-3-ил)-метанол (S. Kanoh et al., Tetrahedron 58 (2002), 7065-7074) вступали в реакцию по аналогии со стадией 3 примера 15 и полученный метиловый эфир гидролизировали по аналогии с примером 16.
ЖХ/МС (метод LC14): Rt = 3,10 мин; m/z = 474,4 [MH+]
Пример 251
2-{3-[2-(3-Гидроксифенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота

Стадия 1: 3-(2-Гидроксиэтил)-фениловый эфир уксусной кислоты
2-(3-гидроксифенил)этанол (400 мг, 2,90 ммоль) растворяли в смеси 4 мл диоксана и 4 мл воды и добавляли гидрокарбонат натрия (2,43 г, 29 ммоль) с последующим добавлением ангидрида уксусной кислоты (2,96 г, 29 ммоль) при охлаждении льдом. Смесь перемешивали в течение ночи при комнатной температуре и затем разделяли в 2н. хлористоводородной кислоте и EA. Водную фазу экстрагировали EA, объединенные органические фазы сушили над сульфатом натрия и выпаривали до сухого состояния с получением неочищенного указанного в заголовке соединения, которое использовали без дополнительной очистки.
Стадия 2: 2-{3-[2-(3-Гидроксифенил)-этокси]-4-метоксибензоиламино}-индан-2-карбоновая кислота
Соединение стадии 1 и соединение стадии 2 примера 15 вступали в реакцию по аналогии со стадией 3 примера 15 и полученный эфир гидролизировали по аналогии с примером 16.
ЖХ/МС (метод LC12): Rt = 3,17 мин; м/z = 448,2 [MH+]
Пример 252
2-[3-Метокси-4-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: Этиловый эфир 3-ацетокси-4-гидроксибензойной кислоты
Этиловый эфир 3,4-дигидроксибензойной кислоты (550 мг, 3,02 ммоль) растворяли в ДМФ (5 мл), добавляли трет-бутоксид калия (210 мг, 2,87 ммоль) и смесь перемешивали в течение 10 мин. Добавляли ангидрид уксусной кислоты (339 мг, 3,32 ммоль) и продолжали перемешивать в течение 10 мин. Смесь разделяли в EA и 2н хлористоводородной кислоте и водную фазу экстрагировали EA. Объединенные органические фазы сушили над хлоридом натрия, сливали и выпаривали до сухого состояния. Остаток очищали ОФ ВЭЖХ (градиент вода/ACN).
ЖХ/МС (метод LC15): Rt = 3,97 мин; м/z = 225,2 [MH+]
Стадия 2: Этиловый эфир 3-ацетокси-4-(2-м-толилэтокси)-бензойной кислоты
Соединение стадии 1 (350 мг, 1,56 ммоль) вступало в реакцию с 2-м-толилэтанолом по аналогии со стадией 3 примера 15 с получением 450 мг указанного в заголовке соединения.
ЖХ/МС (метод LC14): Rt = 3,86 мин; м/z = 343,2 [MH+]
Стадия 3: Метиловый эфир 3-метокси-4-(2-м-толилэтокси)-бензойной кислоты
Соединение стадии 2 (150 мг, 0,438 ммоль) растворяли в метаноле (3 мл), добавляли трет-бутоксид калия (73 мг, 0,657 ммоль) и смесь перемешивали с обратным холодильником в течение ночи. Затем повторно добавляли карбонат калия (60 мг, 0,44 ммоль) и йодометан (124 мг, 0,876 ммоль) с интервалами в 1 ч, при перемешивании с обратным холодильником до завершения реакции. Летучие фракции выпаривали под вакуумом, остаток разделяли в EA и 2н. хлористоводородной кислоте и водную фазу экстрагировали EA. Объединенные органические фазы сушили над хлоридом натрия, сливали и выпаривали до сухого состояния с получением указанного в заголовке соединения.
ЖХ/МС (метод LC15): Rt = 5,27 мин; м/z = 301,2 [MH+]
Стадия 4: 2-[3-Метокси-4-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота
Соединение стадии 3 гидролизировали по аналогии с примером 2, полученная карбоновая кислота вступала в реакцию с метиловым эфиром 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 1 примера 15 и полученный эфир гидролизировали по аналогии с примером 2.
ЖХ/МС (метод LC14): Rt = 3,41 мин; м/z = 446,1 [MH+]
Пример 253
2-[4-Бензилокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: Этиловый эфир 3-ацетокси-4-бензилоксибензойной кислоты
Соединение стадии 1 примера 252 (20 г, 89,2 ммоль) растворяли в ДМФ (100 мл) и охлаждали в ледяной бане. Добавляли карбонат калия (18,4 г, 134 ммоль) и, непосредственно после него, бензилбромид (15,2 г, 89,2 ммоль). Смесь перемешивали в течение 30 мин при комнатной температуре, фильтровали в смесь 2н. хлористоводородной кислоты и диэтилового эфира. Твердый продукт повторно промывали диэтиловым эфиром. Объединенные эфирные фазы промывали водой, сушили над хлоридом натрия, сливали и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 21 г указанного в заголовке соединения.
ЖХ/МС (метод LC14): Rt = 3,68 мин; м/z = 315,1 [MH+]
Стадия 2: Этиловый эфир 4-бензилокси-3-гидроксибензойной кислоты
Соединение стадии 1 (10 г, 31,8 ммоль) растворяли в метаноле, добавляли карбонат калия (88 мг, 0,636 ммоль) и смесь перемешивали в течение 2 ч с обратным холодильником. После выпаривания до сухого состояния остаток использовали на следующей стадии без дополнительной очистки.
ЖХ/МС (метод LC14): Rt = 3,32 мин; м/z = 273,1 [MH+]
Стадия 3: Метиловый эфир 2-[4-бензилокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты
Соединение стадии 2 вступало в реакцию с 2-м-толилэтанолом по аналогии со стадией 1 примера 1, и полученный эфир гидролизировали по аналогии с примером 2. Полученная карбоновая кислота вступала в реакцию с метиловым эфиром 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 2 примера 13.
ЖХ/МС (метод LC14): Rt = 4,05 мин; м/z = 536,3 [MH+]
Стадия 4: 2-[4-Бензилокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота
Соединение стадии 3 гидролизировали по аналогии с примером 2.
ЖХ/МС (метод LC14): Rt = 3,79 мин; м/z = 522,2 [MH+]
Пример 254
2-[4-Гидрокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 2-[4-гидрокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты
Соединение стадии 3 примера 253 (800 мг, 1,49 ммоль) растворяли в EA (15 мл) и гидрировали в присутствии палладия (10%) на угле (200 мг) под давлением водорода 5 бар и при комнатной температуре в течение 6 ч. Смесь фильтровали на силикагеле и выпаривали до сухого состояния. Остаток очищали ОФ ВЭЖХ (градиент вода/ACN) с получением 300 мг указанного в заголовке соединения.
ЖХ/МС (метод LC14): Rt = 3,47 мин; м/z = 446,2 [MH+]
Стадия 2: 2-[4-Гидрокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота
Соединение стадии 1 гидролизировали по аналогии с примером 2.
ЖХ/МС (метод LC14): Rt = 3,22 мин; м/z = 432,2 [MH+]
Пример 255
2-[4-Изопропокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Соединение стадии 1 примера 254 вступало в реакцию с 2-пропанолом по аналогии со стадией 1 примера 1 и полученный эфир гидролизировали по аналогии с примером 2.
ЖХ/МС (метод LC14): Rt = 3,71 мин; м/z = 474,2 [MH+]
По аналогии с примером 255 следующие образцы соединений формулы Iw, перечисленные в таблице 9, получали с помощью соответствующего спирта вместо 2-пропанола. В формулах групп R99 в таблице 9 линия, перечеркнутая знаком
















Пример 270
2-[4-(2-Гидроксиэтокси)-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: 2-[4-(2-Ацетоксиэтокси)-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота
Соединение стадии 1 примера 254 (70 мг, 0,157 ммоль) растворяли в ДМФ (1 мл). Добавляли карбонат калия (108 мг, 0,786 ммоль), а затем 2-бромэтилацетат (39 мг, 0,235 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч и затем разделяли в EA и воде. Водную фазу экстрагировали EA и объединенные органические фазы сушили над сульфатом натрия и выпаривали до сухого состояния. Остаток очищали ОФ ВЭЖХ (градиент вода/ACN).
ЖХ/МС (метод LC18): Rt = 2,59 мин; м/z = 532,2 [MH+]
Стадия 2: 2-[4-(2-Гидроксиэтокси)-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота
Соединение стадии 1 гидролизировали по аналогии с примером 2, используя 6 эквивалентов гидроксида лития.
ЖХ/МС (метод LC18): Rt = 2,21 мин; м/z = 476,2 [MH+]
Пример 271
2-[4-Карбометокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 270 из соединения стадии 1 примера 254 и 2-бромацетамида. На заключительной стадии гидролиза использовали 6 эквивалентов гидроксида лития, проводя в результате гидролиз эфирной группы и ацетамидной группы.
ЖХ/МС (метод LC18): Rt = 2,23 мин; м/z = 490,1 [MH+]
Пример 272
2-[4-Циклопропокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: 2-[4-(1-Фенилсульфанилциклопропокси)-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота
Соединение стадии 1 примера 254 (50 мг, 0,116 ммоль), (1-йодоциклопропилсульфанил)-бензол (G. J. Hollingworth et al., Tetrahedron Lett. 40 (1999), 2633-2636) (64 мг, 0,232 ммоль) и карбонат серебра (64 мг, 0,232 ммоль) в толуоле (1 мл) перемешивали в течение ночи при 50°C. Смесь фильтровали, фильтрат выпаривали до сухого состояния и остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 44 мг указанного в заголовке соединения.
ЖХ/МС (метод LC14): Rt = 4,23 мин; м/z = 594,2 [MH+]
Стадия 2: 2-[4-Циклопропокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота
3-Хлорпербензойную кислоту (43 мг, 0,177 ммоль) добавляли к соединению стадии 1 (35 мг, 0,059 ммоль) в ДХМ (2 мл) и насыщенном водном растворе гидрокарбоната натрия (2 мл). Смесь перемешивали в течение 30 мин при комнатной температуре, а затем разделяли в EA и растворе карбоната натрия. Водную фазу экстрагировали EA и объединенные органические фазы сушили над сульфатом натрия и выпаривали до сухого состояния. Остаток (44 мг) растворяли в смеси 0,5 мл ТГФ и 1 мл метанола. Добавляли гидрофосфат динатрия (40 мг, 0,28 ммоль) и амальгаму натрия и ртути (5% натрия) (250 мг), смесь перемешивали в течение 30 мин. при комнатной температуре и хранили в течение 6 дней при температуре 5°C. Затем смесь разделяли в 2н. хлористоводородной кислоте и EA, водную фазу экстрагировали EA и объединенные органические экстракты сушили над хлоридом натрия, сливали и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (градиент ДХМ/метанол/28% аммиака, 90:10:1 до 85:15:1,5). Фракции продукта выпаривали до сухого состояния и остаток разделяли в 2н. хлористоводородной кислоте и EA. Водную фазу экстрагировали EA и объединенные органические экстракты сушили над хлоридом натрия, сливали и выпаривали до сухого состояния.
ЖХ/МС (метод LC14): Rt = 3,60 мин; м/z = 472,2 [MH+]
Пример 273
2-{[5-Этил-4-(2-м-толилэтокси)-тиазол-2-карбонил]-амино}-индан-2-карбоновая кислота

Этиловый эфир 5-этил-4-гидрокситиазол-2-карбоновой кислоты (F. A. J. Kerdesky et al., J. Med. Chem. 34 (1991), 2158-2165) (100 мг, 0,497 ммоль) вступал в реакцию с 2-м-толилэтанолом по аналогии со стадией 1 примера 1, а затем эфирную группу гидролизировали по аналогии с примером 2. Полученная карбоновая кислота вступала в реакцию с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 2 примера 13 и полученный эфир гидролизировали по аналогии с примером 2.
ЖХ/МС (метод LC12): Rt = 4,17 мин; м/z = 451,2 [MH+]
Пример 274
2-({5-[2-(2-Фтор-5-метилфенил)-этокси]-6-метоксипиридин-3-карбонил]-амино}-индан-2-карбоновая кислота

Указанное в заголовке соединение получали по аналогии с примером 208, с использованием 2-(2-фтор-5-метилфенил)-этанола вместо 2-м-толилэтанола.
ЖХ/МС (метод LC12): Rt = 3,64 мин; м/z = 465,2 [MH+]
Пример 275
2-[(5-{2-[3-(2-Гидроксиэтил)-фенил]-этокси}-6-метоксипиридин-3-карбонил)-амино}-индан-2-карбоновая кислота

Метиловый эфир 6-хлор-5-нитроникотиновой кислоты получали в соответствии с методами, описанными в WO 2005/021544, и преобразовывали в метиловый эфир 5-гидрокси-6-метоксиникотиновой кислоты в соответствии с методами, описанными в WO 95/04045. Последнее названное соединение преобразовали в указанное в заголовке соединение путем этерификации с 2-[3-(2-гидроксиэтил)-фенил]-этанолом по аналогии со стадией 1 примера 1, гидролиза эфирной группы по аналогии с примером 2, проведения реакции полученной карбоновой кислоты с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 3 примера 1 и гидролиза эфирной группы по аналогии с примером 2.
ЖХ/МС (метод LC14): Rt = 2,90 мин; м/z = 477,2 [MH+]
Пример 276
2-(3-Фтор-5-{2-[3-(2-гидроксиэтил)-фенил]-этокси}-4-метоксибензоиламино}-индан-2-карбоновая кислота

Стадия 1: 2-[3-(2-Гидроксиэтил)-фенил]-этиловый эфир уксусной кислоты
2-[3-(2-Гидроксиэтил)-фенил]-этанол (2,49 г, 15,0 ммоль) растворяли в ACN (5 мл) и добавляли ангидрид уксусной кислоты (3,06 г, 30 ммоль). Смесь перемешивали с обратным холодильником в течение 1 ч, а затем выпаривали до сухого состояния. Обработка остатка хроматографией на силикагеле (градиент HEP/EA) дала 1,30 г указанного в заголовке соединения (моно-ацетилированный продукт).
1H-ЯМР: δ = 7,20 (т, 1H); 7,10-7,05 (м, 3H); 4,61 (т, 1H); 4,19 (т, 2H); 3,59 (дт, 2H); 2,82 (т, 2H); 2,69 (т, 2H); 1,98 (с, 3H)
Стадия 2: Метиловый эфир 2-фтор-3-гидрокси-4-метоксибензойной кислоты и метиловый эфир 3-фтор-5-гидрокси-4-метоксибензойной кислоты
Метиловый эфир 3-ацетокси-4-метоксибензойной кислоты (WO 2005/009389) (3,58 г, 16,0 ммоль) и 1-хлорметил-4-фтор-1,4-диазониабицикло[2.2.2]октан бис(тетрафторборат) (Selectfluor ®) (14,1 г, 39,9 ммоль) в ACN нагревали партиями (7 партий) до 170°С в течение 7 мин в микроволновом реакторе. Объединенные партии разделяли в 2н. хлористоводородной кислоте и диэтиловом эфире. Водную фазу экстрагировали диэтиловым эфиром и объединенные органические фазы сушили над сульфатом натрия, фильтровали и выпаривали до сухого состояния. Остаток очищали хроматографией на силикагеле (градиент HEP/EA) с получением 0,7 г смеси изомерных фторсодержащих соединений с ацетильной группой и без нее. Данную смесь растворяли в метаноле (5 мл) и после добавления карбоната калия (80 мг) нагревали с обратным холодильником в течение 3 ч. После выпаривания до сухого состояния остаток разделяли в 2н. хлористоводородной кислоте и EA, водную фазу экстрагировали EA и объединенные экстракты сушили над сульфатом натрия и выпаривали до сухого состояния. Остаток разделяли ОФ ВЭЖХ (градиент вода/ACN) с получением 0,14 г метилового эфира 2-фтор-3-гидрокси-4-метоксибензойной кислоты и 0,27 г метилового эфира 3-фтор-5-гидрокси-4-метоксибензойной кислоты.
Метиловый эфир 2-фтор-3-гидрокси-4-метоксибензойной кислоты:
1H-ЯМР: δ = 10,55 (с, 1H); 7,61 (дд, 1H); 6,80 (дд, 1H); 3,90 (с, 3H); 3,88 (с, 3H)
Метиловый эфир 3-фтор-5-гидрокси-4-метоксибензойной кислоты:
1H-ЯМР: δ = 10,25 (с, 1H); 7,30 (уш.с, 1H); 7,21 (дд, 1H); 3,87 (с, 3H); 3,81 (с, 3H)
Стадия 3: 2-(3-Фтор-5-{2-[3-(2-гидроксиэтил)-фенил]-этокси}-4-метоксибензоиламино}-индан-2-карбоновая кислота
Указанное в заголовке соединение получали из метилового эфира 3-фтор-5-гидрокси-4-метоксибензойной кислоты путем этерификации с соединением стадии 1 по аналогии со стадией 1 примера 1, гидролиза обеих эфирных групп полученного соединения с 6 эквивалентами гидроксида лития по аналогии с примером 2, проведения реакции полученной карбоновой кислоты с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 3 примера 1 и гидролиза метилового эфира по аналогии с примером 2.
ЖХ/МС (метод LC14): Rt = 3,11 мин; м/z = 494,2 [MH+]
Пример 277
2-[2-Фтор-4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновая кислота

Метиловый эфир 2-фтор-3-гидрокси-4-метоксибензойной кислоты этерифицировали с 2-м-толилэтанолом по аналогии со стадией 1 примера 1, полученный эфир гидролизировали по аналогии с примером 2, полученная карбоновая кислота вступала в реакцию с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 3 примера 1 и метиловый эфир гидролизировали по аналогии с примером 2.
ЖХ/МС (метод LC14): Rt = 3,92 мин; м/z = 464,2 [MH+]
Пример 278
2-{4-Метокси-3-[2-(3-метилциклогексил)-этокси]-бензоиламино}-индан-2-карбоновая кислота

Стадия 1: 4-Метокси-3-[2-(3-метилциклогексил)-этокси)-бензойная кислота
Соединение стадии 1 примера 13 (100 мг) растворяли в этаноле (5 мл). Добавляли оксид платины(IV) (12 мг), смесь гидрировали в течение 1 ч при комнатной температуре под давлением водорода 1 бар, фильтровали через целит и выпаривали до сухого состояния с получением 99 мг указанного в заголовке соединения.
ЖХ/МС (метод LC12): Rt = 3,87 мин; м/z = 334,2 [(M+CH3CN+H)+]
Стадия 2: 2-{4-Метокси-3-[2-(3-метилциклогексил)-этокси]-бензоиламино}-индан-2-карбоновая кислота
Соединение стадии 1 вступало в реакцию с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 4 примера 3, и полученный эфир гидролизировали по аналогии с примером 2.
ЖХ/МС (метод LC18): Rt = 2,72 мин; м/z = 452,2 [MH+]
Пример 279
2-[4-Метокси-3-(3-метилбензилоксиметил)-бензоиламино]-индан-2-карбоновая кислота

Стадия 1: Метиловый эфир 2-(3-формил-4-метоксибензоиламино)-индан-2-карбоновой кислоты
3-формил-4-метоксибензойная кислота (F. D. Chattaway и F. Calvet, J. Chem. Soc. (1928), 2913-2918) (1,017 г, 5,65 ммоль) вступала в реакцию с гидрохлоридом метилового эфира 2-аминоиндан-2-карбоновой кислоты по аналогии со стадией 4 примера 3. Полученный продукт (1,895 г) растворяли в уксусной кислоте (20 мл), добавляли ацетат натрия (0,57 г, 6,96 ммоль) и смесь перемешивали с обратным холодильником в течение 24 ч. Летучие фракции выпаривали под вакуумом, остаток разделяли в насыщенном водном растворе гидрокарбоната натрия и EA и водную фазу экстрагировали EA. Объединенные органические фазы сушили над сульфатом натрия и выпаривали до сухого состояния с получением 1,52 г указанного в заголовке соединения.
ЖХ/МС (метод LC14): Rt = 3,00 мин; м/z = 354,1 [MH+]
Стадия 2: Метиловый эфир 2-(3-гидроксиметил-4-метокси-бензоиламино)-индан-2-карбоновой кислоты
Соединение стадии 1 (0,500 г, 1,42 ммоль) растворяли в ТГФ (5 мл) и охлаждали в ледяной бане. Добавляли боргидрид натрия (0,164 г, 4,25 ммоль). Затем добавляли по каплям метанол (2 мл). Через 1 ч выпаривали летучие фракции, остаток разделяли в диэтиловом эфире и насыщенном растворе гидрокарбоната натрия и водную фазу экстрагировали диэтиловым эфиром. Объединенные органические фазы сушили над сульфатом натрия и выпаривали до сухого состояния.
ЖХ/МС (метод LC14): Rt = 2,74 мин; м/z = 356,1 [MH+]
Стадия 3: 2-[4-Метокси-3-(3-метилбензилоксиметил)-бензоиламино]-индан-2-карбоновая кислота
Соединение стадии 2 (50 мг, 0,14 ммоль) растворяли в DMF (3 мл) и добавляли гидрид натрия (60% дисперсию в минеральном масле, 6,2 мг, 0,15 ммоль), а затем 1-бромметил-3-метилбензол (27 мг, 0,14 ммоль). Смесь перемешивали в течение ночи. Затем добавляли гидроксид лития (1 М раствор в воде, 0,42 мл) и диоксан (1 мл) и смесь нагревали до 60°С в течение 1 ч. Смесь разделяли в 2н хлористоводородной кислоте и EA и водную фазу экстрагировали EA. Объединенные органические фазы сушили над сульфатом натрия и выпаривали до сухого состояния. Остаток очищали ОФ ВЭЖХ (градиент вода/ACN) с получением 5 мг указанного в заголовке соединения.
ЖХ/МС (метод LC14): Rt = 3,44 мин; м/z = 446,2 [MH+]
По аналогии с примером 196 образцы соединений формулы Iu, перечисленные в таблице 10, получали с помощью соответствующей замещенной фенилбороновой кислоты вместо 3-изопропилфенилбороновой кислоты. В случае примеров 282, 283 и 284 промежуточный метиловый эфир 2-[3-(R97)-4-метоксибензоиламино]-индан-2-карбоновой кислоты очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) перед проведением гидролиза. Соединения могут быть названы как 2-[3-(R97)-4-метоксибензоиламино]-индан-2-карбоновая кислота, например, как 2-[3-[5-хлорпиридин-3-ил)-4-метоксибензоиламино]-индан-2-карбоновая кислота в случае примера 282.

Таблица 10. Образцы соединений формулы Iu
Пример 285
Стандартная методология получения 2-(3-арил-4-метоксибензоиламино)-индан-2-карбоновых кислот
0,3 ммоль соответствующей борной кислоты поместили в реакционную ампулу для микроволновой обработки. Добавляли 0,2 ммоль метилового эфира 2-(3-бром-4-метоксибензоиламино)-индан-2-карбоновой кислоты в 2 мл 1,2-диметоксиэтана и 0,4 ммоль фторида цезия в 1 мл метанола, затем добавляли 0,01 ммоль тетракис(трифенилфосфин)палладия(0) в 0,5 мл метанола. Ампулу закрывали обжимной крышкой и облучали в микровом реакторе при 130°С в течение 5 мин. Охлажденный раствор обрабатывали 0,25 мл 4н. водного раствора гидроксида натрия и облучали еще 5 минут при 130°С в микровом реакторе. Охлажденный раствор нейтрализовали 0,25 мл 4н. водного раствора хлористоводородной кислоты и выпаривали. Остаток растворяли в 2 мл ДМФ, фильтровали и подвергали препаративной ОФ ВЭЖХ (градиент вода/ACN).
В соответствии со стандартной методологией, описанной в примере 285, получали соединения формулы Iu, перечисленные в таблице 11. Они могут быть названы как -[3-(R97)-4-метоксибензоиламино]-индан-2-карбоновая кислота, например, как 2-[(3'-этансульфонил-6-метоксибифенил-3-карбонил)-амино]-индан-2-карбоновая кислота в случае примера 312, в котором группа R97 представляет 3-этансульфонилфенил и, с учетом правил номенклатуры, группа 3-(R97)-4-метоксифенил-C(O), представленная в формуле Iu, таким образом, называется 3'-этансульфонил-6-метоксибифенил-3-карбонильной.

По аналогии с примерами, перечисленными в таблице 1, получали образцы соединений формулы Im, перечисленные в таблице 12. В формулах групп R90 в таблице 12 линия, перечеркнутая знаком






Пример 356 (исходное соединение)
2-(2-Фтор-5-трифторметоксифенил)-этанол
3,00 г (12,6 ммоль) 2-(2-фтор-5-трифторметоксифенил)-уксусной кислоты растворяли в 50 мл сухого тетрагидрофурана и добавляли по каплям при темп. 0°C к суспензии 956 мг (25,2 ммоль) алюмогидрида лития в 11 мл ТГФ. После перемешивания в течение ночи добавляли 150 мл ТГФ с последующим добавлением 3 мл EA. Добавляли по каплям 15 мл воды и из полученной суспензии сливали супернатант. Суспензию три раза экстрагировали 20 мл EA. Объединенные органические фазы сушили над сульфатом натрия и выпаривали. Маслянистый остаток (2,5 г) использовали на следующей стадии без дополнительной очистки.
По аналогии с примером 356 получали исходные соединения 2-(бензо[D]изоксазол-3-ил)-этанол и 2-(4-метилфуразан-3-ил)-этанол из 2-(бензо[D]изоксазол-3-ил)-уксусной кислоты и 2-(4-метилфуразан-3-ил)-уксусной кислоты, соответственно.
Пример 357
2-[(3'-Этансульфонил-5-фтор-6-метоксибифенил-3-карбонил)-амино]-индан-2-карбоновая кислота
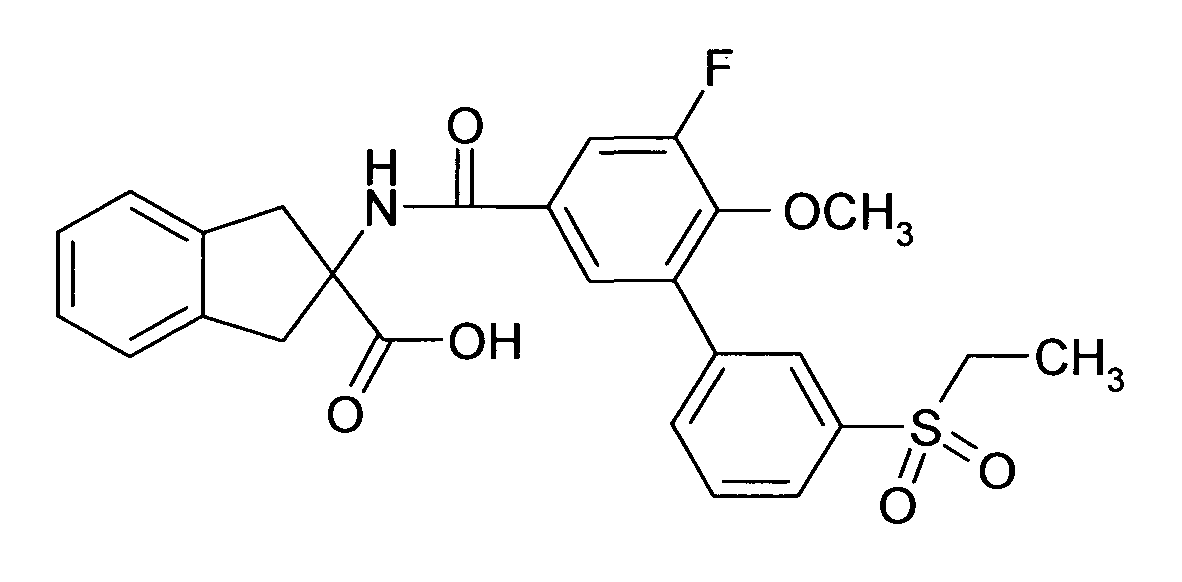
Стадия 1: Метиловый эфир 3-бром-5-фтор-4-гидроксибензойной кислоты
В течение 30 мин. добавляли 5,64 г (35,27 ммоль) брома к раствору 5,00 г (29,39 ммоль) метилового эфира 3-фтор-4-гидроксибензойной кислоты в 30 мл дихлорметана и 30 мл уксусной кислоты при 0°C. После перемешивания в течение ночи добавляли 200 мл метилацетата. Полученный раствор экстрагировали раствором 7,56 г (60 ммоль) сульфита натрия в 50 мл воды, насыщенного раствора хлорида натрия и воды. Органическую фазу сушили над сульфатом натрия, фильтровали и выпаривали. Полученное твердое вещество белого цвета (7,2 г) использовали на следующей стадии без дополнительной очистки.
Стадия 2: Метиловый эфир 3-бром-5-фтор-4-метоксибензойной кислоты
6 г (24,09 ммоль) продукта, полученного на стадии 1, растворяли в 60 мл ацетона, добавляли 10,13 г (2,270 ммоль) карбоната калия и 6,84 г (48,18 ммоль) йодометана и смесь перемешивали в течение 4 дней. Затем ее фильтровали и выпаривали. Полученный продукт (5,8 г) использовали на следующей стадии без дополнительной очистки.
Стадия 3: 3-Бром-5-фтор-4-метоксибензойная кислота
5,8 г продукта стадии 2 растворяли в 100 мл смеси ТГФ и воды (9:1), добавляли 1,06 г (44,1 мкмоль) гидроксида лития и смесь перемешивали в течение 3 дней. Растворитель выпаривали и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Получали 2,9 г указанного в заголовке соединения.
Стадия 4: Метиловый эфир 2-(3-бром-5-фтор-4-метоксибензоиламино)-индан-2-карбоновой кислоты
2,4 г (9,64 ммоль) соединения стадии 3 растворяли в 40 мл ДМФ и добавляли 2,49 г (19,27 ммоль) EDIA и 4,03 г (10,60 ммоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'--тетраметилуроний гексафторфосфата. Затем добавляли раствор 2,19 г (9,64 ммоль) гидрохлорида метилового эфира 2-аминоиндан-2-карбоновой кислоты в 10 мл ДМФ. После перемешивания в течение ночи смесь выпаривали до сухого состояния и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Получали 3,6 г указанного в заголовке соединения.
Стадия 5: 2-[(3'-Этансульфонил-5-фтор-6-метоксибифенил-3-карбонил)-амино]-индан-2-карбоновая кислота
300 мг (0,71 ммоль) соединения стадии 4 и 175 мг (1,07 ммоль) 3-этансульфонилфенилбороновой кислоты растворяли в 4 мл ДМФ и 4 мл 1,2-диметоксиэтана в атмосфере аргона. Добавляли 216 мг (1,42 ммоль) фторида цезия и 41,08 мг (0,04 ммоль) тетракис(трифенилфосфин)палладия(0) и смесь нагревали до 130°С в микроволновом реакторе в течение 15 мин. После охлаждения растворитель выпаривали и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN) с получением метилового эфира указанного в заголовке соединения. 135,1 мг (0,26 ммоль) метилового эфира растворяли в 5 мл смеси ТГФ и воды (9:1), добавляли 12,65 мг (0,53 ммоль) гидроксида лития и смесь перемешивали в течение 3 дней. Растворитель выпаривали и остаток очищали препаративной ОФ ВЭЖХ (градиент вода/ACN). Получали 123 мг указанного в заголовке соединения.
ЖХ/МС (метод LC14): Rt = 3,07 мин; м/z = 498,19 [MH+]
1H-ЯМР: δ = 12,5 (уш.с, 1H); 8,90 (с, 1H); 7,98 (с, 1H); 7,75-7,95 (м, 6H); 7,28 (д, 4H); 3,30-3,64 (м, 6H); 1,13 (т, 3H)
По аналогии с примером 357 образцы соединений формулы Ix, перечисленные в таблице 13, получили с помощью соответствующей замещенной фенилбороновой кислоты вместо 3-этансульфонилфенилбороновой кислоты. Они могут быть названы как 2-[3-(R100)-5-фтор-4-метоксибензоиламино]-индан-2-карбоновая кислота, например, как 2-[(5'-фтор-3'-изопропил-6-метоксидифенил-3-карбонил)-амино]-индан-2-карбоновая кислота в случае примера 360, в котором группа R100 представляет 3-изопропилфенил и, с учетом правил номенклатуры, группа 3-(R100)-5-фтор-4-метоксифенил-C(O), представленная в формуле Ix, таким образом, называется 5-фтор-3'-изопропил-6-метоксидифенил-3-карбонильной.

Пример 362
2-{3-[2-(2,5-Дихлорфенил)-этокси]-4-трифторметилбензоиламино}-индан-2-карбоновая кислота

Указанное в заголовке соединение синтезировали по аналогии с примером 185, с использованием 2,5-дихлорфенилэтанола вместо 2-(3-метилфенил)-этанола на стадии 3.
ЖХ/МС (метод LC13): Rt = 538,01 мин; м/z = 3,06 мин [MH+]
Фармакологические испытания
А) Определение ингибирования рецептора Edg-2 по измерениям с помощью флуориметрического планшетного считывателя (FLIPR)
Ингибирование рецептора Edg-2 (рецептора LPA1) соединениями настоящего изобретения количественно оценивали по ингибирующему эффекту на опосредованное LPA высвобождение кальция в клеточном флуориметрическом анализа кальция на клетках яичников китайского хомяка (CHO), в которых проводили стабильную сверхэкспрессию рецептора Edg-2 человека (система Flp-In, Invitrogen). С тем чтобы стимулировать связывание G-белка и направить передачу сигнала на высвобождение Ca2+, сверхэкспрессированный рецептор, кроме того, имел C-терминальную последовательность модифицированного G-белка (Gαi4qi4) (WO 02/04665). Изменения внутриклеточного кальция определяли по флуориметрическим измерениям с использованием чувствительного к кальцию красителя fluo-4 (Invitrogen) на флуориметрическом планшетном считывателе (FLIPR, Molecular Dynamics).
Клетки CHO, стабильно сверхэкспрессирующие рецептор Edg-2 человека, высевали (40 000 на лунку) в черные, с прозрачным дном, обработанные поли-D-лизином 96-луночные планшенты (Becton Dickinson, Biocoat cellware) примерно за 18-24 ч до экспериментов. Клетки выращивали в инкубаторе при 37°C, 5% двуокиси углерода и влажности 95% в клеточной культуральной среде на основе среды F-12 glutamax (Gibco, #31765) с добавлением 1% (об./об.) пенициллин/стрептомицин (PAN, #P06-07100), 10% (об./об.) фетальной телячей сыворотки (FCS, PAA, #A15-151) и гигромицина B (Invitrogen, #10687-010) 300 мг/л (конечные концентрации).
Перед экспериментом FLIPR клетки нагружали фтор-4-ацетоксиметиловым эфиром (fluo-4 AM, Invitrogen, #F14202) в течение 60 мин в инкубаторе при 37°C, 5% двуокиси углерода и 95% влажности в буфере загрузки красителя, содержащем сбалансированный солевой раствор Хэнкса (HBSS, Invitrogen, #14065049) с добавкой fluo-4 AM в концентрации 2 мкМ (все данные приводятся для конечной концентрации), Pluronic® F-127 0,05% (об/об) (Invitrogen, #P-3000MP), HEPES 20 мМ (Gibco, #15630), пробенецид 2,5 мМ (Sigma, #P-8761) и бычий сывороточный альбумин 0,05% (BSA, Sigma, #A-6003), скорректированный до pH 7,5 добавлением гидроксида натрия. В процессе загрузки клеток fluo-4 AM расщепляется внутриклеточной эстеразой, что приводит к захвату красителя fluo-4 внутри клеток. Загрузку останавливали отмывкой клеток в устройстве для отмывки клеток (Tecan Power washer), которую проводили трижды в указанном выше буфере, но без добавления fluo-4 AM и BSA. Последний буфер также использовали в качестве буфера в дальнейших измерениях флуоресценции клеток.
Загруженные красителем и отмытые клетки предварительно инкубировали в течение примерно 5 мин с различными концентрациями испытываемого соединения, которое добавляли в растворе в DMSO (0,3% об./об. максимальная конечная концентрация DMSO), или же только с DMSO в соответствующих концентрациях (положительный контроль). Последующее добавление LPA (18:1, 1-олеил-sn-глицерин-3-фосфат; 100 нМ конечная концентрация) приводит к высвобождению внутриклеточного кальция из внутренних депо, что вызывает заметное временное увеличение сигнала флуоресценции fluo-4, который отслеживается примерно в течение 3 мин. Процент ингибирования, вызываемого испытываемым соединением определяли по максимальному отклику флуоресценции после добавления LPA к клеткам, преинкубированным с соединением, по сравнению с максимальным откликом флуоресценции после добавления LPA к клеткам, преинкубированным только с DMSO. Все величины флуоресценции скорректированы с учетом фоновых значений флуоресценции, регистрируемых от клеток, преинкубированных только с DMSO и не проходивших обработку LPA (контроль по исходному фону). Все измерения проводили по три раза. По проценту ингибирования определяли ингибирующую концентрацию IC50.
Ингибирующие концентрации IC50 различных образцов соединений приведены в таблице 14, где "a" обозначает IC50 менее 0,1 мкм, "b" обозначает IC50 от 0,1 мкм до 1 мкм, и "c" обозначает IC50 от 1 мкм до 30 мкм.
Таблица 14
Ингибирующие концентрации IC50 для ингибирования рецептора Edg-2
B) In vivo антигипертрофическая и ренопротекторная активность
Можно провести исследование фармакологической активности in vivo для соединений настоящего изобретения, например на модели DOCA-солечувствительных крыс с односторонней нефрэктомией. Коротко говоря, в данной модели проводится односторонняя нефрэктомия левой почки (UNX) на крысах Sprague Dawley весом от 150 г до 200 г. После операции, а также в начале каждой последующей недели, крысам вводили 30 мг/кг веса тела DOCA (дезоксикортикостерона ацетат) посредством подкожной инъекции. Получавшие DOCA крысы после нефрэктомии получали питьевую воду, содержащую 1% хлорида натрия (крысы UNX/DOCA). У крыс UNX/DOCA развивалось высокое артериальное давление, эндотелиальная дисфункция, миокардическая гипертрофия и фиброз, а также дисфункция почек. В тестовой группе (UNX/DOCA тест) и в группе плацебо (UNX/DOCA плацебо), в которую входили рандомизированные крысы UNX/DOCA, крысам через ротовой зонд дважды в день в 06.00 ч и в 18.00 ч вводили дневную дозу испытываемого соединения (например 10 мг/кг веса тела, растворенные в носителе) или только носитель, соответственно. В контрольной группе (контроль), в которую входят животные, не подвергшиеся UNX и не получавшие DOCA, животные получали обычную питьевую воду и принимали чистый носитель. Через пять недель лечения с помощью метода хвостовой манжеты неинвазивным образом измеряли систолическое кровяное давление (SBP) и частоту сердечных сокращений (HR). Для определения альбуминурии и креатинина в метаболических клетках собирали суточную мочу. Эндотелиальную функцию оценивали по иссеченным кольцам торакальной аорты, как это описывалось ранее (W. Linz et al., JRAAS (Journal of the renin-angiotensin-aldosterone system) 7 (2006), 155-161). В качестве показателя миокардиальной гипертрофии и фиброза на препарированном сердце определяли вес сердца, вес левого желудочка и соотношение гидроксипролина и пролина.
Реферат
Изобретение относится к соединению формулы I, где кольцо A является циклоалкановым кольцом с числом членов от 3-х до 7-и, бензольным кольцом или моноциклическим 5-членным или 6-членным ароматическим гетероциклическим кольцом, содержащим 1 гетерочлен кольца, выбранный из группы, содержащей N и S, причем бензольное и гетероциклическое кольца могут необязательно иметь один или два одинаковых или разных заместителей, выбранных из группы, содержащей галоген, HO-, R-O-, HN-C(O)- и NC-; Y выбирают из группы, содержащей S, C(R)=C(R) и C(R)=N; Z выбирают из группы, содержащей C(R); R, R, R, R, Rи Rнезависимо от каждой другой группы R, R, R, R, Rи Rвыбирают из группы, содержащей (C-C)-алкил, (C-C)-алкенил, (C-C)-циклоалкил и (C-C)-циклоалкил-(C-C)-алкил-, причем все они могут необязательно иметь один или более одинаковых или разных заместителей R; Rи Rпредставляют собой водород; Rи Rвыбирают, независимо друг от друга, из группы, содержащей водород и (C-C)-алкил; R, R, Rи Rвыбирают, независимо друг от друга, из группы, содержащей водород, галоген и ON-; Rвыбирают из группы, содержащей водород и (C-C)-алкил; одна из групп Rи Rявляется группой формулы II: R-R-, а другую из групп Rи Rвыбирают из группы, содержащей водород, галоген, R, HO-, R-O-, R-S(O)-, HN-, R-NH-, R-N(R)-, R-C(O)- и NC-; Rявляется цепью, содержащей от 1 до 5 членов цепи, из которых 0 или 1 член цепи является гетерочленом цепи, выбранным из группы, содержащей N(R), O, S, тогда как другие члены цепи являются одинаковыми или разными группами C(R)(R), где две соседние группы C(R)(R) могут соединяться друг с другом двойной связью; Rвыбирают из группы, содержащей водород, R, R-O-, R-NH-, R-N(R)-, R-C(O)-NH-, HO-C(O)- и моноциклическое, бициклическое или трициклическое кольцо с числом членов от 5-и до 10-и,
Формула

где
кольцо A является циклоалкановым кольцом с числом членов от 3-х до 7-и, бензольным кольцом или моноциклическим 5-членным или 6-членным ароматическим гетероциклическим кольцом, содержащим 1 гетерочлен кольца, выбранный из группы, содержащей N и S, причем бензольное и гетероциклическое кольца могут необязательно иметь один или два одинаковых или разных заместителей, выбранных из группы, содержащей галоген, HO-, R1-O-, H2N-C(O)- и NC-;
Y выбирают из группы, содержащей S, C(R12)=C(R13) и C(R15)=N;
Z выбирают из группы, содержащей C(R16);
R1, R30, R33, R35, R54 и R55 независимо от каждой другой группы R1, R30, R33, R35, R54 и R55 выбирают из группы, содержащей (C1-C6)-алкил, (C2-C6)-алкенил, (C3-C7)-циклоалкил и (C3-C7)-циклоалкил-(C1-C4)-алкил-, причем все они могут необязательно иметь один или более одинаковых или разных заместителей R70;
R3 и R5 представляют собой водород;
R4 и R6 выбирают, независимо друг от друга, из группы, содержащей водород и (C1-C4)-алкил;
R12, R13, R15 и R16 выбирают, независимо друг от друга, из группы, содержащей водород, галоген и O2N-;
R20 выбирают из группы, содержащей водород и (C1-C4)-алкил;
одна из групп R21 и R22 является группой формулы II

а другую из групп R21 и R22 выбирают из группы, содержащей водород, галоген, R30, HO-, R30-O-, R30-S(O)m-, H2N-, R30-NH-, R30-N(R30)-, R30-C(O)- и NC-;
R23 является цепью, содержащей от 1 до 5 членов цепи, из которых 0 или 1 член цепи является гетерочленом цепи, выбранным из группы, содержащей N(R25), O, S, тогда как другие члены цепи являются одинаковыми или разными группами C(R26)(R26), где две соседние группы C(R26)(R26) могут соединяться друг с другом двойной связью;
R24 выбирают из группы, содержащей водород, R31, R31-O-, R31-NH-, R31-N(R31)-, R31-C(O)-NH-, HO-C(O)- и моноциклическое, бициклическое или трициклическое кольцо с числом членов от 5-и до 10-и, которое является насыщенным или ненасыщенным и содержит 0, 1, 2 или 3 одинаковых или разных гетерочлена кольца, выбранных из группы, содержащей N, N(R32), O, S, причем кольцо может необязательно иметь на атомах углерода кольца один или 2-3 одинаковых или разных заместителей, выбранных из группы, содержащей галоген, R33, R33-O-, R33-S(O)m-, R33-C(O)-NH-, R33-S(O)2-NH-, R33-C(O)-, HO-C(O)-, H2N-C(O)-, R33-NH-C(O)-, R33-N(R33)-S(O)2-, NC-, оксо, фенил и Het;
при условии, что общее число атомов C, N, O и S, присутствующих в двух группах R23 и R24, составляет не менее 5;
R25 выбирают из группы, содержащей водород и (C1-C4)-алкил;
R26, независимо от каждой другой группы R26, выбирают из группы, содержащей водород, фтор, (C1-C4)-алкил и HO-, или две группы R26, вместе с входящими в них членами цепи, образуют моноциклическое кольцо с числом членов 4-е, которое является насыщенным и содержит 1 гетерочлен кольца, выбранный из группы, содержащей O;
R31 выбирают из группы, содержащей (C1-C6)-алкил, который необязательно может иметь один заместитель R70;
R32 выбирают, независимо друг от друга, из группы, содержащей водород, R35 и фенил;
R50 выбирают из группы, содержащей R51-O- и R52-N(R53)-;
R51 выбирают из группы, содержащей водород и R54;
R52 выбирают из группы, содержащей водород;
R53 выбирают из группы, содержащей водород;
R70 выбирают из группы, содержащей HO-, R71-O-, H2N-, R71-NH-, R71-N(R71)-, R71-C(O)-NH-, HO-C(O)-, H2N-C(O)- и фенил;
R71, независимо от каждой другой группы R71, выбирают из группы, содержащей (C1-C4)-алкил;
Het, независимо от каждой другой группы Het, является моноциклическим гетероциклическим кольцом с числом членов 5, которое содержит 1 или 2 одинаковых или разных гетерочлена кольца, выбранных из группы, содержащей N и S, причем кольцо является насыщенным или ненасыщенным и необязательно замещенным одним или более одинаковыми или разными заместителями, выбранными из группы, содержащей (C1-C4)-алкил;
m, независимо от каждого другого числа m, является целым числом, выбранным из группы, содержащей 0, 1 и 2;
фенил, независимо от каждой другой группы фенила, может необязательно иметь один или более одинаковых или разных заместителей, выбранных из группы, содержащей галоген и (C1-C4)-алкил.
кольцо A является циклогексановым кольцом, бензольным кольцом, пиридиновым кольцом или тиофеновым кольцом, причем бензольное, пиридиновое и тиофеновое кольца могут необязательно иметь один или два одинаковых или разных заместителей, выбранных из группы, содержащей галоген и NC-;
Y выбирают из группы, содержащей S, C(R12)=C(R13) и C(R15)=N;
Z является C(R16).
кольцо A является бензольным кольцом, пиридиновым кольцом или тиофеновым кольцом, которые могут необязательно иметь один или два одинаковых или разных заместителей, выбранных из группы, содержащей галоген;
Y выбирают из группы, содержащей S, C(R12)=C(R13) и C(R15)=N;
Z является C(R16);
R3 и R5 выбирают, независимо друг от друга, из группы, содержащей водород;
R4 и R6 представляют собой водород;
R12, R13, R15 и R16 выбирают, независимо друг от друга, из группы, содержащей водород и галоген;
R20 - водород.
R21 выбирают из группы, содержащей галоген, (C1-C4)-алкил, HO-(C1-C4)-алкил-, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, H2N-, (C1-C4)-алкил-NH-, ди((C1-C4)-алкил)N-, (C1-C4)-алкил-C(O)- и NC-;
R22 является группой формулы II;

R23 представляет собой цепь, содержащую 2, 3 или 4 члена цепи, из которых 0 или 1 член цепи является гетерочленом цепи, выбранным из группы, содержащей N(R25), O и S, и другие члены цепи являются одинаковыми или разными группами C(R26)(R26), где две соседние группы C(R26)(R26) могут соединяться друг с другом двойной связью.
R24 - моноциклическое кольцо с числом членов от 5 до 7 или бициклическое кольцо с числом членов от 7 до 10, которое является насыщенным или ненасыщенным и содержит 0, 1 или 2 одинаковых или разных гетерочлена кольца, выбранных из группы, содержащей N, N(R32), O и S, причем эти кольца могут необязательно иметь на атомах углерода кольца один или 2-3 одинаковых или разных заместителей, выбранных из группы, содержащей галоген, R33, R33-O-, R33-S(O)m-, R33-C(O)-NH-, R33-S(O)2-NH-, HO-C(O)-, H2N-C(O)-, R33-NH-C(O)-, NC-, оксо, фенил и Het;
R32 выбирают из группы, содержащей водород и фенил.
кольцо А является бензольным кольцом, пиридиновым кольцом или тиофеновым кольцом, которые могут необязательно иметь один или два одинаковых или разных заместителей, выбранных из группы, содержащей галоген;
Y выбирают из группы, содержащей S, C(R12)=C(R13) и C(R15)=N;
Z является C(R16);
R3 и R5 выбирают, независимо друг от друга, из группы, содержащей водород;
R4 и R6 представляют собой водород;
R12, R13, R15 и R16 выбирают, независимо друг от друга, из группы, содержащей водород и галоген;
R20 - водород;
R21 выбирают из группы, содержащей галоген, (C1-C4)-алкил, HO-(C1-C4)-алкил-, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, H2N-, (C1-C4)-алкил-NH-, ди((C1-C4)-алкил)N-, (C1-C4)-алкил-C(O)- и NC-;
R22 является группой формулы II;

R23 представляет собой цепь, содержащую 2, 3 или 4 члена цепи, из которых 0 или 1 член цепи являются гетерочленами цепи, выбранными из группы, содержащей N(R25), O и S, и другие члены цепи являются одинаковыми или разными группами C(R26)(R26), где две соседние группы C(R26)(R26) могут соединяться друг с другом двойной связью;
R24 - моноциклическое кольцо с числом членов от 5 до 7 или бициклическое кольцо с числом членов от 7 до 10, которое является насыщенным или ненасыщенным и содержит 0, 1 или 2 одинаковых или разных гетерочлена кольца, выбранных из группы, содержащей N, N(R32), O и S, причем кольца могут необязательно иметь на атомах углерода кольца один или 2-3 одинаковых или разных заместителей, выбранных из группы, содержащей галоген, R33, R33-O-, R33-S(O)m-, R33-C(O)-NH-, R33-S(O)2-NH-, HO-C(O)-, H2N-C(O)-, R33-NH-C(O)-, NC-, оксо, фенил и Het;
R32 выбирают из группы, содержащей водород, R35 и фенил.
кольцо A является бензольным кольцом, которое может необязательно иметь необязательно один или два одинаковых или разных заместителей, выбранных из группы, содержащей галоген;
Y представляет собой C(R12)=C(R13);
Z является C(R16);
R3, R4, R5 и R6 представляет собой водород;
R12, R13 и R16 выбирают независимо друг от друга из группы, содержащей водород и галоген;
R20 является водородом;
R21 выбирают из группы, содержащей галоген, (C1-C4)-алкил, HO-(C1-C4)-алкил-, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, (C1-C4)-алкил-C(O)- и NC-;
R22 является группой формулы II;

R23 представляет собой цепь, содержащую 2, 3 или 4 члена цепи, из которых 0 или 1 член цепи являются гетерочленами цепи, выбранными из группы, содержащей N(R25), O и S, и другие члены цепи являются одинаковыми или разными группами C(R26)(R26);
R24 является бензольным кольцом, которое может необязательно иметь один или 2-3 одинаковых или разных заместителей, выбранных из группы, содержащей галоген, R33, R33-O-, R33-S(O)m-, R33-C(O)-NH-, R33-S(O)2-NH-, HO-C(O)-, H2N-C(O)-, R33-NH-C(O)- и NC-;
при условии, что общее число атомов C, N, O и S, присутствующих в двух группах R23 и R24, составляет не менее 5;
R25 выбирают из группы, содержащей водород и (C1-C4)-алкил;
R26, независимо от каждой другой группы R26, выбирают из группы, содержащей водород, фтор, (C1-C4)-алкил и HO-;
R33 выбирают, независимо от каждой другой группы R33, из группы, содержащей (C1-C4)-алкил, (C3-C7)-циклоалкил и (C3-C7)-циклоалкил-(C1-C2)-алкил-, причем все они могут необязательно иметь один или более одинаковых или разных заместителей R70;
R50 выбирают из группы, содержащей R51-O- и R52-N(R53)-;
R51 выбирают из группы, содержащей водород и (C1-C4)-алкил;
R52 представляет собой водород;
R53 представляет собой водород;
R70 выбирают из группы, содержащей HO- и R71-O-;
R71 представляет собой (C1-C4)-алкил;
m, независимо от каждого другого числа m, является целым числом, выбранным из группы, содержащей 0 и 2.
2-[4-метилсульфанил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-ацетил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-этил-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-этокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-метокси-3-(1-м-толилциклопропилметокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-{3-[2-(3-цианофенил)-этокси]-4-метокс-бензоиламино}-индан-2-карбоновой кислоты,
5-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-5,6-дигидро-4Н-циклопента[с]тиофен-5-карбоновой кислоты,
5-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-5,6-дигидро-4Н-циклопента[b]тиофен-5-карбоновой кислоты,
2-{[5-ацетил-4-(2-м-толилэтокси)-тиофен-2-карбонил]-амино}-индан-2-карбоновой кислоты,
2-[3-фтор-4-метокси-5-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-метокси-3-(2-м-толилоксиэтил)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-метокси-3-(3-м-толилпропил)-бензоиламино]-индан-2-карбоновой кислоты,
5-фтор-2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-метокси-3-(2-м-толилэтокси)-бензоиламино]-5,6-диметилиндан-2-карбоновой кислоты,
2-[4-циано-3-(2-м-толилэтокси)-бензоиламино]-индан-2-карбоновой кислоты,
2-[4-метокси-3-(2-м-толилэтиламино)-бензоиламино]-индан-2-карбоновой кислоты,
2-{3-[2-(3-хлорфенил)-этокси]-4-метилбензоиламино}-индан-2-карбоновой кислоты,
2-[4-метокси-3-(2-м-толилэтилсульфанил)-бензоиламино]-индан-2-карбоновой кислоты,
2-{[6-метокси-5-(2-м-толилэтокси)-пиридин-3-карбонил]-амино}-индан-2-карбоновой кислоты, и
2-[3-(2,2-дифтор-2-фенилэтокси]-4-метоксибензоиламино}-индан-2-карбоновая кислоты.

где кольцо A и группы Y, Z, R3 по R6, R20 по R22 и R50 в соединениях формул III и IV определены как в соединениях формулы I, и группа G в соединении формулы IV представляет HO-, (C1-C4)-алкил-O- или галоген.
Комментарии