Соединение для применения в лечении нейрогенной ортостатической гипотензии - RU2723095C1
Код документа: RU2723095C1
Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
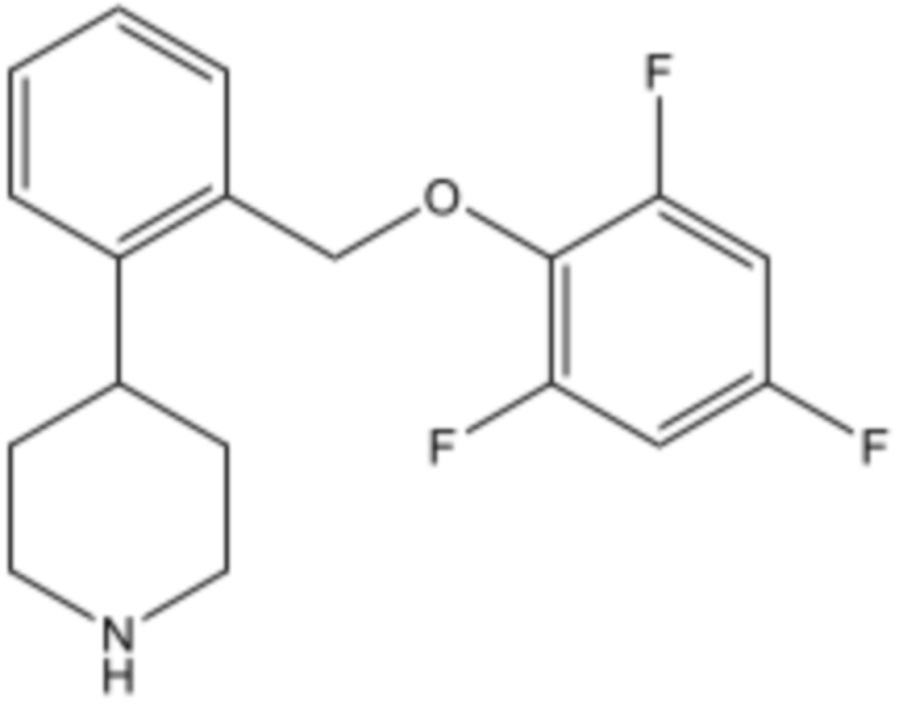
Настоящее изобретение относится к применению 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина или его фармацевтически приемлемой соли для лечения нейрогенной ортостатической гипотензии и ее симптомов.
УРОВЕНЬ ТЕХНИКИ
Ортостатическая гипотензия (ОГ), также известная как постуральная гипотензия, является одной из форм низкого кровяного давления, которое возникает, когда человек встает. Говоря в терминах медицины, ОГ определяют как падение систолического артериального давления на по меньшей мере 20 мм рт. ст. или диастолического артериального давления на по меньшей мере 10 мм рт. ст. в пределах трех минут при изменении положения тела из положения лежа на спине (супинированного положения) в вертикальное положение (Neurology 1996; 46:1470). ОГ может вызывать широкий спектр симптомов, включая головокружение, предобморочное состояние и обморок (потерю сознания). Эти симптомы ОГ часто приводят к ограничению или даже являются препятствием для повседневной деятельности, требующей стояния на ногах или ходьбы. Кроме того, ОГ связана с повышенной заболеваемостью и смертностью. См., например, Jones et al, Expert Review of Cardiovascular Therapy, 2015; 13:11, 1263-1276; Kuritzky et al., Postgrad. Med. 2015; 127(7):702-715; и Low et al, J. Clin. Neurol., 2015; 11(3):220-226.
Причины, лежащие в основе развития ОГ, в широком смысле можно разделить на две категории: нейрогенного и ненейрогенного характера. Нейрогенная ортостатическая гипотензия (нОГ) является формой ОГ, в которой задействована нервная система, например, ОГ, вызванная неврологическим заболеванием периферической или центральной нервной системы, таким как первичная вегетативная недостаточность (включая истинную вегетативную недостаточность, множественную системную атрофию и болезнь Паркинсона), и вегетативную нейропатию (дизавтономию) (включая диабетическую и недиабетическую вегетативную нейропатию) (Arbique et al., JAMDA 15 (2014) 234-239). Такие заболевания могут вызывать дефицит или дисрегуляцию выработки норэпинефрина, который является основным нейротрансмиттером, регулирующим артериальное давление в ответ на изменения положения тела (Loavenbruck et al, Curr. Med. Res. Opin., 2015; 31:2095-2104). В результате, вегетативная (автономная) нервная система становится неспособной регулировать должным образом артериальное давление во время изменения положения тела, и у пациента происходит сильное падение артериального давления, приводящее, например, к головокружению, предобморочному состоянию или обмороку.
Соответственно, одной из задач лечения нОГ является увеличение у пациентов уровня норэпинефрина. Одним из способов увеличения уровня норэпинефрина является введение агента, генерирующего норэпинефрин. Например, дроксидопа (L-трео-3-4-дигидроксифенилсерин) является аминокислотой, которая в результате декарбоксилирования превращается в норэпинефрин как в центральной, так и периферической нервной системе, таким образом увеличивая уровень норэпинефрина (Kaufmann et al., Circulation, 2003; 108:724-728; Kaufmann, Clin. Auton. Res. (2008) 18[Suppl 1]:19-24); and Isaacson et al., Vascular Health and Risk Management, 2014, 10:169-176). Препарат дроксидопа одобрен в США для лечения ортостатического головокружения, предобморочного состояния или ʺощущения, будто вы вот-вот потеряете сознаниеʺ у взрослых пациентов с клиническими проявлениями нОГ, вызванной первичной вегетативной недостаточностью (болезнью Паркинсона, множественной системной атрофией и истинной вегетативной недостаточностью), недостаточностью дофамин-бета-гидролазы и недиабетической вегетативной нейропатией. Основным побочным эффектом дроксидопа является гипертензия в супинированном положении, и относительно этого серьезного побочного эффекта в инструкции по применению этого лекарственного препарата также имеется особое предостережение.
Альтернативно, уровень норэпинефрина у пациентов может увеличиваться путем подавления транспортера норэпинефрина, ответственного за обратный захват норэпинефрина. Например, атомоксетин, одобренный США для лечения синдрома дефицита внимания и гиперактивности (ADHD), является селективным ингибитором обратного захвата норэпинефрина. Было показано, что атомоксетин увеличивает артериальное давление у пациентов с центральной вегетативной недостаточностью (Ramirez et al., Hypertension, 2014; 64:1235-40; и Shibao et al., Hypertension, 2007; 50:47-53). Однако метаболизм атомоксетина происходит в основном через ферментативный путь CYP2D6 и, следовательно, его фармакокинетические свойства меняются в зависимости от того, является ли активность CYP2D6 у субъекта пониженной (слабый метаболизатор) или нормальной (быстрый метаболизатор) (Ring et al., Drug Metabolism and Distribution, 2002, 30:319-323). Инструкция по применению атомоксетина также включает несколько предупреждений относительно возможных взаимодействий с другими лекарственными веществами. Кроме того, атомоксетин, когда применяется для лечения ADHD, вызывает несколько нежелательных явлений со стороны ЖКТ, включая сухость во рту и тошноту. Атомоксетин не одобрен для лечения нОГ.
Другие агенты, используемые для лечения нОГ включают агониста α1-адренорецептора, мидодрин (и его активный метаболит, дезглимидодрин); синтетический минералокортикоид, флудрокортизон; и ингибитор холинэстеразы, пиридостигмин. Побочные эффекты, вызываемые этими агентами, могут включать, в случае мидодрина, гипертензию в супинированном положении, парестезию (включая покалывание кожи головы), пилоэрекцию (гусиную кожу) и неотложный позыв к мочеиспусканию или задержку мочи; в случае флудрокортизона, гипокалемию, головные боли, периферический отек, сердечную недостаточность и гипертензию в супинированном положении; и в случае пиридостигмина, неприятные ощущения в животе и неотложный позыв к мочеиспусканию.
Соответственно, было бы желательно наличие других вариантов лечения нОГ. В частности, было бы желательно предоставить безопасный и хорошо переносимый ингибитор транспортера норэпинефрина с предсказуемыми фармакокинетическими свойствами для применения в лечении нОГ. Более того, поскольку у пациентов с нОГ часто наблюдается гипертензия в супинированном положении, и многие из существующих лекарственных средств могут вызывать или усугублять гипертензию в супинированном положении, крайне желательно предоставить соединения для лечения нОГ, которые не вызывают или не усугубляют гипертензию в супинированном положении.
В патентах США № 8,304,432 B2 и 8,604,058 B2 раскрыты соединения 4-[2-(2-фторфеноксиметил)фенил]пиперидина, которые представляют собой ингибиторы обратного захвата серотонина и норэпинефрина. Конкретное соединение, раскрытое в этих патентах представляет собой 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин, имеющий формулу:

Это соединение также известно как TD-9855. Дополнительно, в патентах США № 8,304,433 B2 и 9,073,859 B2 раскрыта кристаллическая гидрохлоридная соль 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина. В этих патентах раскрыты различные варианты применения 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина, включая лечение болевых расстройств, депрессивных расстройств, когнитивных расстройств, стрессовое недержание мочи, синдром хронической усталости, ожирение, вазомоторные симптомы, связанные с менопаузой, хроническую боль в нижней части спины, остеоартрит и другие нарушения. Эти патенты не описывают применение 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина для лечения нОГ; однако на вебсайте ClinicalTrials.gov 9 марта 2016 г. была опубликована информация, относящаяся к фазе 2 клинических исследований.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к применению 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина или его фармацевтически приемлемой соли для лечения нейрогенной ортостатической гипотензии и ее симптомов.
Соответственно, в одном из аспектов настоящее изобретение относится к соединению формулы I:
или его фармацевтически приемлемой соли для применения для лечения нейрогенной ортостатической гипотензии и ее симптомов у пациента-человека; где соединение вводят пациенту в количестве от примерно 1 мг/сутки до примерно 20 мг/сутки, причем применение характеризуется (a) увеличением систолического артериального давления у сидящего пациента; (b) увеличением времени нахождения пациента в положении стоя; или (c) уменьшением головокружения или ощущения предобморочного состояния, испытываемого пациентом. В одном из вариантов осуществления применение характеризуется увеличением у пациента систолического артериального давления в сидячем положении. В другом варианте осуществления применение характеризуется увеличением времени нахождения пациента в положении стоя. В другом варианте осуществления применение характеризуется уменьшением головокружения или ощущения предобморочного состояния, испытываемого пациентом.
В другом аспекте настоящее изобретение относится к соединению формулы I или его фармацевтически приемлемой соли для применения для увеличения систолического артериального давления у пациента, страдающего от симптоматической нейрогенной ортостатической гипотензии, в сидячем положении.
В другом аспекте настоящее изобретение относится к соединению формулы I или его фармацевтически приемлемой соли для применения для увеличения времени нахождения пациента, страдающего от симптоматической нейрогенной ортостатической гипотензии, в положении стоя.
В другом аспекте настоящее изобретение относится к соединению формулы I или его фармацевтически приемлемой соли для применения для лечения у пациента ортостатического головокружения или предобморочного состояния, вызванного симптоматической нейрогенной ортостатической гипотензией.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение формулы I или его фармацевтически приемлемую соль, для применения для лечения нейрогенной ортостатической гипотензии и ее симптомов у пациента-человека; где композицию вводят пациенту для обеспечения количества соединения от примерно 1 мг/сутки до примерно 20 мг/сутки, и где применение характеризуется (a) увеличением систолического артериального давления у сидящего пациента; (b) увеличением времени нахождения пациента в положении стоя; или (c) уменьшением головокружения или ощущения предобморочного состояния, испытываемого пациентом.
В другом аспекте настоящее изобретение относится к применению соединения формулы I или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения нейрогенной ортостатической гипотензии и ее симптомов у человека-пациента; где соединение вводят пациенту в количестве от примерно 1 мг/сутки до примерно 20 мг/сутки, и где применение характеризуется (a) увеличением систолического артериального давления у сидящего пациента; (b) увеличением времени нахождения пациента в положении стоя; или (c) уменьшением головокружения или ощущения предобморочного состояния, испытываемого пациентом.
В другом аспекте настоящее изобретение относится к способу лечения нейрогенной ортостатической гипотензии и ее симптомов у человека-пациента, содержащему введение пациенту соединения формулы I или его фармацевтически приемлемой соли.
В другом аспекте настоящее изобретение относится к способу лечения нейрогенной ортостатической гипотензии и ее симптомов у человека-пациента, содержащему введение пациенту соединения формулы I или его фармацевтически приемлемой соли; где соединение вводят пациенту в количестве от примерно 1 мг/сутки до примерно 20 мг/сутки, и где способ характеризуется (a) увеличением систолического артериального давления у сидящего пациента; (b) увеличением времени нахождения пациента в положении стоя; или (c) уменьшением головокружения или ощущения предобморочного состояния, испытываемого пациентом.
Если не указано иное, для каждого описанного в настоящей заявке аспекта изобретения применимы приведенные ниже различные варианты осуществления.
В одном из вариантов осуществления соединение представляет собой гидрохлорид 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина.
В одном из вариантов осуществления соединение представляет собой кристаллическую гидрохлоридную соль 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина, отличающуюся порошковой рентгенограммой, имеющей дифракционные пики при значениях 2θ 4,44±0,20, 10,22±0,20, 17,16±0,20 и 21,78±0,20. В другом варианте осуществления кристаллическая гидрохлоридная соль 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина дополнительно отличается наличием одного или более дополнительных дифракционных пиков при значениях 2θ, выбранных из 8,11±0,20, 13,18±0,20, 16,06±0,20, 18,38±0,20, 23,76±0,20, 26,32±0,20, 27,24±0,20, 29,60±0,20 и 31,94±0,20.
В одном из вариантов осуществления соединение представляет собой кристаллическую гидрохлоридную соль 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина, отличающуюся графиком дифференциальной сканирующей калориметрии, имеющим температуру плавления примерно 197±2°С.
В одном из вариантов осуществления пациент имеет неврологическое нарушение, выбранное из первичной вегетативной недостаточности (включая истинную вегетативную недостаточность, множественную системную атрофию и болезнь Паркинсона); вегетативной нейропатии (включая диабетическую и недиабетическую вегетативную нейропатию). В одном из вариантов осуществления пациент имеет первичную вегетативную недостаточность. В другом варианте осуществления пациент имеет вегетативную нейропатию. В другом варианте осуществления пациент имеет болезнь Паркинсона. В конкретном варианте осуществления пациент имеет множественную системную атрофию.
В одном из вариантов осуществления соединение вводят в количестве от примерно 0,5 мг/сутки до примерно 20 мг/сутки. В другом варианте осуществления соединение вводят в количестве от примерно 1 мг/сутки до примерно 20 мг/сутки. В другом варианте осуществления соединение вводят в количестве от примерно 1 мг/сутки до примерно 10 мг/сутки. В другом варианте осуществления соединение вводят в количестве, выбранном из примерно 1, 2, 3, 4, 5, 6, 7, 8, 9 и 10 мг/сутки.
В одном из вариантов осуществления соединение вводят один раз в сутки.
В одном из вариантов осуществления соединение вводят в виде фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение формулы I или его фармацевтически приемлемую соль.
В одном из вариантов осуществления соединение вводят с агентом, выбранным из агониста α1-адренорецептора, антагониста α2-адренергического рецептора, кортикостероида, предшественника норэпинефрина и ингибитора холинэстеразы; или их комбинации. В конкретном варианте осуществления соединение или его фармацевтически приемлемую соль вводят с мидодрином, флудрокартизона ацетатом, дроксидопой или пиридостигмином, или, в каждом случае, фармацевтически приемлемой солью; такой как мидодрина гидрохлорид или пиридостигмина бромид.
В настоящей заявке описаны и другие аспекты и варианты осуществления настоящего изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В различных аспектах и вариантах осуществления настоящее изобретение относится к применению 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина или его фармацевтически приемлемой соли для лечения нейрогенной ортостатической гипотензии и ее симптомов.
Определения
Используемые в настоящей заявке термины имеют приведенные ниже значения, если не указано иное.
Термины в единственном числе также включают соответствующие термины во множественном числе, если из контекста очевидным образом не следует иное.
Термин ʺпримерноʺ означает ±5 процентов от определенного значения.
Термин "температура плавления" означает температуру, при которой наблюдается максимальный эндометрический тепловой поток в дифференциальной сканирующей калориметрии для выявления теплого перехода, соответствующего фазовому переходу от твердого тела к жидкости.
Термин "фармацевтически приемлемый" означает допустимый для введения пациенту (например, с приемлемой безопасностью для указанного применения).
Термин ʺфармацевтически приемлемая сольʺ означает соль, полученную из кислоты или основания (включая цвиттер-ионы), которая допустима для введения пациенту (например, приемлемо безопасная соль для заданного режима дозирования).
Термин ʺтерапевтически эффективное количествоʺ означает количество, достаточное для эффективного лечения при введении нуждающемуся в лечении пациенту, например, количество, необходимое для получения требуемого терапевтического эффекта.
Термин ʺлечениеʺ или ʺтерапияʺ означает облегчение или купирование медицинского состояния или нарушения, подлежащего лечению; или облегчение симптомов медицинского состояния или нарушения.
Термин "стандартная лекарственная форма" или ʺединичные дозыʺ означает физически дискретную единицу, подходящую для дозирования пациенту, т.е., каждую единицу, содержащую заданное количество терапевтического агента, вычисленное для генерации терапевтического эффекта либо при введении одной единицы, либо в комбинации с одной или более дополнительными единицами. Примеры включают капсулы, таблетки и т.п.
Все другие используемые в настоящей заявке термины имеют общепринятое значение, понятное любому специалисту в области, к которой они относятся.
Соединение формулы I
В настоящем изобретении используется 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин, имеющий формулу I:
или его фармацевтически приемлемая соль.
4-[2-(2,4,6-Трифторфеноксиметил)фенил]пиперидин может быть получен согласно тому, как описано в настоящей заявке в примерах или методами и процедурами, описанными в патентах США №№ 8,304,432 B2; 8,304,433 B2; и 8,247,433 B2; и родственных патентах.
В одном из вариантов осуществления 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин используется в форме фармацевтически приемлемой соли. Примеры фармацевтически приемлемых солей включают соли следующих кислот (с соответствующим анионом, указанным в скобках): уксусную (ацетат), аскорбиновую (аскорбат), бензолсульфоновую (бензолсульфонат или бесилат), бензойную (бензоат), камфорсульфоновую (камфорсульфонат), хлортеофиллиновую (хлортеофиллинат), лимонную (цитрат), этансульфоновую (этансульфонат), этандисульфоновую (этандисульфонат), фумаровую (фумарат), гентизиновую (гентизинат), глюконовую (глюконат), глюкуроновую (глюконат), глюкогептоновую (глюкогептонат), глутаминовую (глутамат), гиппуровую (гиппурат), бромистоводородную (бромид), соляную (хлорид), иодистоводородную (йодид), изетионовую (изетионат), молочную (лактат), лактобионовую (лактобионат), лаурилсульфоновую (лаурилсульфонат), малеиновую (малеат), яблочную (малат), миндальную (манделат), метансульфоновую (метансульфонат или мезилат), метилсульфоновую (метилсульфонат), муциновую (мукат), нафталинсульфоновую (нафталинсульфонат или напсилат), нафталин-1,5-дисульфоновую (нафталин-1,5-дисульфонат), нафталин-2,6-дисульфоновую (нафталин-2,6-дисульфонат), нафтойную (нафтоат), никотиновую (никотинат), азотную (нитрат), октадеканоновую (октадеканоат), олеиновую (олеат), оротовую (оротат), щавелевую (оксалат), памовую (памоат), пантотеновую (пантотеат), фосфорную (фосфат), полигалактуроновая (полигалактуронатная), полигалактуроновую (полигалактуронат), янтарную (сукцинат), сульфосалициловую (сульфосалицилат), серную (сульфат), винную (тартрат), п-толуолсульфоновую (п-толуолсульфонат или тозилат) и ксинафоевую (ксинафоат) кислоту и т.п. Такие соли иногда называют аддуктами кислот.
Соли могут быть получены путем приведения в контакт одного молярного эквивалента 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина с примерно 0,95-1,05 молярным эквивалентом кислотных протонов в фармацевтически приемлемой кислоте. Например, один молярный эквивалент 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина может взаимодействовать с примерно одним молярным эквивалентом соляной кислоты с образованием гидрохлорида 4-[2-(2,4,6-трифторфеноксиметил))фенил]пиперидина; или один молярный эквивалент 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина может взаимодействовать с примерно 0,5 молярного эквивалента серной кислоты с образованием 4-[2-(2,4,6-трифторфеноксиметил)фенила]пиперидиновой соли серной кислоты.
Такие реакции обычно проводят в разбавителе, таком как дихлорметан, этанол, этилацетат, изопропилацетат и т.п., при температуре от примерно -20°С до примерно 50°С в течение от примерно 0,5 до примерно 12 часов или до тех пор, пока реакция по существу не завершится. По завершении реакции продукт обычно выделяют, используя обычные процедуры, такие как фильтрация, хроматография, перекристаллизация и т.п. Продукт таких реакций может быть или не быть кристаллическим.
В одном из вариантов осуществления соединение, используемое в настоящем изобретении, представляет собой гидрохлорид 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина. В другом варианте осуществления соединение представляет собой кристаллический гидрохлорид 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина, отличающийся порошковой рентгенограммой с характерными дифракционными пиками при значениях 2θ, равных 4,44±0,20, 10,22±0,20, 17,16±0,20 и 21,78±0,20. В другом варианте осуществления кристаллическая гидрохлоридная соль дополнительно характеризуется наличием одного или более дополнительных дифракционных пиков при значениях 2θ, выбранных из 8,11±0,20, 13,18±0,20, 16,06±0,20, 18,38±0,20, 23,76±0,20, 26,32±0,20, 27,24±0,20, 29,60±0,20 и 31,94±0,20. В другом варианте осуществления соединение представляет собой кристаллический гидрохлорид 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина, отличающийся графиком дифференциальной сканирующей калориметрии, имеющим температуру плавления примерно 197±2°С.
Используемые в настоящем изобретении кристаллические гидрохлоридные соли 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина могут быть получены согласно тому, как описано в настоящей заявке в примерах или методами и процедурами, описанными в патентах США №№ 8,304,432 B2; 8,304,433 B2; и 8,247,433 B2; и родственных патентах.
Фармацевтические композиции, составы и лекарственные формы
Используемый в настоящем изобретении 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемую соль обычно вводят пациенту в форме фармацевтической композиции или состава. При обсуждении в настоящей заявке фармацевтических композиций или составов 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемая соль могут упоминаться как "активный агент", что позволяет отличить его от других компонентов состава, таких как носитель или наполнитель. Таким образом, термин "активный агент" включает 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемую соль. К тому же, термины ʺносительʺ и ʺнаполнительʺ, используемые в настоящей заявке, являются взаимозаменяемыми и имеют одинаковое значение, если не указано иное.
Фармацевтические композиции по настоящему изобретению обычно содержат терапевтически эффективное количество активного агента. Однако специалистам в данной области техники будет понятно, что фармацевтическая композиция может содержать количество, больше терапевтически эффективного количества, т.е. многодозовая композиция, или меньше терапевтически эффективного количества, т.е. отдельные стандартные дозы, предназначенные для многократного введения с целью достижения терапевтически эффективного количества.
Как правило, фармацевтическая композиция может содержать от примерно 0,01 до примерно 95 мас.% активного агента, в том числе от примерно 0,01 до примерно 30 мас.%, например от примерно 0,01 до примерно 10 мас.%, причем фактическое количество зависит от состава, пути введения, частоты введения и т.д. Например, фармацевтическая композиция, подходящая в качестве пероральной лекарственной формы, может содержать от примерно 0,1 до примерно 10 мас.%, в том числе от примерно 0,5 до примерно 5 мас.% активного агента.
В одном из приведенных в качестве примера вариантов осуществления фармацевтическая композиция содержит от примерно 0,5 до примерно 20 мг активного агента на единичную дозу, в том числе от примерно 1 до примерно 10 мг активного агента на единичную дозу. Например, активный агент может быть находиться в лекарственной форме в виде 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 мг единичных доз, например, 1 мг, 3 мг, 5 мг и 10 мг единичных доз.
В фармацевтических композициях по настоящему изобретению может быть использован любой обычный или подходящий фармацевтически приемлемый носитель. Выбор конкретного носителя или комбинации носителей зависит от различных факторов, таких как способ введения, количество дозы, частота дозирования, время высвобождения активного агента и т.п. В этом отношении приготовление подходящей фармацевтической композиции для конкретного способа введения находится в пределах компетенции специалистов в области фармацевтики, и носители, используемые в таких композициях, являются коммерчески доступными. В качестве дополнительной иллюстрации, традиционные составы и методы приготовления составов описаны, например, в Remington: The Science and Practice of Pharmacy, 20th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); и H. C. Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
Типичные примеры фармацевтически приемлемых носителей включают, без ограничения: сахара, такие как лактоза, глюкоза и сахароза; крахмал, такой как кукурузный крахмал и картофельный крахмал; целлюлоза, такая как микрокристаллическая целлюлоза, и ее производные, такие как натрий карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин; тальк; наполнители, такие как масло какао и воски для суппозиториев; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический солевой раствор; раствор Рингера; этиловый спирт; фосфатные буферные растворы; сжатые газы-вытеснители, такие как хлорфторуглероды и гидрофторуглероды; и другие нетоксичные совместимые вещества, используемые в фармацевтических композициях.
Фармацевтические композиции обычно готовят путем основательного и тщательного перемешивания или смешивания активного агента с фармацевтически приемлемым носителем и любыми необязательными ингредиентами. Полученную однородно перемешанную смесь затем можно формовать или загружать в таблетки, капсулы, пилюли, канистры, картриджи, флаконы, бутыли, дозаторы и т.п., используя обычные процедуры и оборудование.
В одном из вариантов осуществления фармацевтическая композиция подходит для перорального введения. Фармацевтические композиции для перорального введения могут быть в форме, например, капсул, таблеток, пилюль, пастилок, облаток, драже, порошков, гранул, растворов, суспензий, эмульсий, эликсиров, сиропов и т.п., где каждая из указанных форм содержит заранее определенное количество активного агента.
Фармацевтическая композиция в твердой лекарственной форме (такой как капсулы, таблетки и т.п.), предназначенная для перорального введения, обычно содержит активный агент и один или более фармацевтически приемлемых твердых носителей, таких как цитрат натрия или дикальция фосфат. Твердые лекарственные формы также могут содержать: наполнители или сухие разбавители, такие как крахмалы, микрокристаллическая целлюлоза, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; связующие агенты, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или акация; увлажнители, такие как глицерин; разрыхлители, такие как кроскармеллоза натрия, агар-агар, карбонат кальция, крахмал картофельный или тапиоки, альгиновая кислота, некоторые силикаты и/или карбонат натрия; замедляющие растворение агенты, такие как парафин; ускорители абсорбции, такие как четвертичные аммониевые соединения; смачивающие агенты, такие как цетиловый спирт и/или моностеарат глицерина; абсорбенты, такие как каолиновая и/или бентонитовая глина; лубриканты, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и/или их смеси; красители; буферные агенты; регуляторы скорости высвобождения; покрывающие агенты; подсластители, ароматизаторы и отдушки; и консерванты и антиоксиданты.
Типичные покрывающие агенты для таблеток, капсул, пилюль и т.п. включают вещества, которые используются для энтеросолюбильных покрытий, такие как ацетатфталат целлюлозы, поливинилацетатфталат, фталат гидроксипропилметилцеллюлозы, сополимеры сложных эфиров метакриловой кислоты и метакриловой кислоты, тримеллитат ацетата целлюлозы, карбоксиметилэтилцеллюлозу, гидроксипропил метилцеллюлозы ацетат сукцинат, поливиниловый спирт и т.п.
Типичные антиоксиданты включают: водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфат натрия, сульфит натрия и т.п.; маслорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол, бутилированный гидрокситолуол, лецитин, пропилгаллат, альфа-токоферол и т.п.; и агенты, образующие комплексные соединения с металлом, такие как лимонная кислота, этилендиаминтетрауксусная кислота, сорбит, винная кислота, фосфорная кислота и т.п.
Фармацевтические композиции также могут быть составлены таким образом, чтобы обеспечивалось медленное или контролируемое высвобождение активного агента с использованием, например, гидроксипропилметилцеллюлозы в различных пропорциях или других полимерных матриц, липосом и/или микросфер. Кроме того, фармацевтическая композиция может содержать опалесцирующие агенты и может находиться в лекарственной форме составленной таким образом, чтобы в определенной части желудочно-кишечного тракта высвобождался только или предпочтительно активный агент, необязательно, с задержкой. Примеры заливочных композиций, которые можно использовать, включают полимерные вещества и воски. Активный агент также может находиться в микрокапсулированной форме, если необходимо, с одним или более из вышеописанных наполнителей.
Подходящие жидкие дозированные формы для перорального введения включают, например, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Жидкие лекарственные формы обычно содержат активный агент и инертный разбавитель, такой как, например, вода, сок или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (например, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и эфиры жирных кислот с сорбитаном и их смеси. Суспензии могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, сложные эфиры полиоксиэтиленсорбитола и сорбитана, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант и их смеси.
В другом варианте осуществления фармацевтическая композиция подходит для местного введения, такого как трансдермальное введение. Для такого введения могут быть использованы известные системы трансдермальной доставки и наполнители. Например, активный агент может быть смешан с усилителями проницаемости, такими как пропиленгликоль, монолаурат полиэтиленгликоля, азациклоалкан-2-оны и т.п., и включен в пластырь или подобную систему доставки. При необходимости в таких трансдермальных композициях могут использоваться дополнительные наполнители, включая гелеобразующие агенты, эмульгаторы и буферы.
В другом варианте осуществления фармацевтическая композиция подходит для парентерального введения (например, путем подкожной, внутривенной, внутримышечной или внутрибрюшинной инъекции). Для такого введения активный агент предоставляется в стерильном растворе, суспензии или эмульсии. Типичные носители для приготовления таких составов включают воду, физиологический раствор, низкомолекулярные спирты, такие как пропиленгликоль, полиэтиленгликоль, масла, желатин, сложные эфиры жирных кислот, такие как этилолеат, и т.п. Парентеральные составы также могут содержать один или более солюбилизаторов, стабилизаторов, консервантов, смачивающих агентов, эмульгаторов и диспергирующих агентов. Эти составы можно сделать стерильными путем использования стерильной инъекционной среды, стерилизующего агента, фильтрации, облучения или нагревания.
Типичным составом для внутривенного введения является стерильный водный раствор с рН 4-7, содержащий активный агент и физиологически приемлемый водный носитель. Типичные физиологически приемлемые водные носители включают, например, стерильную воду для инъекций, USP; Инъекцию декстрозы, USP (например, 2,5, 5,0, 10, 20% декстрозу, включая инъекцию 5% декстрозы (D5/W)); Инъекцию декстрозы и хлорида натрия, USP (например, от 2,5 до 10% декстрозы и от 0,12 (19 мг-экв натрия) до 0,9% (154 мг-экв натрия) хлорида натрия); Инъекцию маннита, USP (например, 5, 10, 15, 20 и 25% маннита); Инъекцию раствора Рингера, USP (например, 147 мг-экв натрия, 4 мг-экв калия, 4,5 мг-экв кальция и 156 мг-экв хлорида на литр); Инъекцию лактата Рингера, USP (например, 2,7 мг-экв кальция, 4 мг-экв калия, 130 мг-экв натрия и 28 мг-экв лактата на литр); Инъекцию хлорида натрия, USP (например, 0,9% хлорида натрия) и т.п. При введении пациенту активный агент обычно разводят в количестве от примерно 0,1 мл до примерно 10 мл водного носителя на мг активного агента, например, от примерно 0,5 до примерно 5 мл на мг. Затем дозируемый раствор обычно вводят пациенту путем внутривенной инфузии.
В качестве иллюстрации, типичные фармацевтические композиции могут быть получены согласно тому, как описано в приведенных ниже примерах.
А. Твердые желатиновые капсулы
Активный агент (5 г), высушенную распылением лактозу (485 г) и стеарат магния (10 г) тщательно смешивают. Затем полученную композицию загружают в твердые желатиновые капсулы (500 мг композиции на капсулу). Каждая капсула содержит 5 мг активного агента на единичную дозу, подходящую для перорального введения.
В. Твердые желатиновые капсулы
Активный агент (2 г) тщательно смешивают с крахмалом (98 г), микрокристаллической целлюлозой (98 г) и стеаратом магния (2 г). Затем смесь пропускают через сито N 45 меш США и загружают в твердые желатиновые капсулы (200 мг композиции на капсулу). Каждая капсула содержит 2 мг активного агента на единичную дозу, подходящую для перорального введения.
C. Мягкие желатиновые капсулы
Активный агент (5 г) тщательно смешивают с полиоксиэтиленсорбитан моноолеатом (65 г) и крахмальным порошком (335 г). Затем смесь загружают в мягкие желатиновые капсулы (400 мг композиции на капсулу). Каждая капсула содержит 5 мг активного агента на единичную дозу, подходящую для перорального введения.
D. Мягкие желатиновые капсулы
Активный агент (1 г) тщательно смешивают с микрокристаллической целлюлозой (290 г) и стеаратом магния (9 г). Затем смесь загружают в мягкие желатиновые капсулы (300 мг композиции на капсулу). Каждая капсула содержит 1 мг активного агента на единичную дозу, подходящую для перорального введения.
E. Таблетки
Активный агент (10 г), крахмал (45 г) и микрокристаллическую целлюлозу (35 г) пропускают через сито № 20 меш США и тщательно перемешивают. Полученные гранулы сушат при 50-60°С и пропускают через сито N 16 меш США. Раствор поливинилпирролидона (4 г в виде 10% раствора в стерильной воде) смешивают отдельно с карбоксиметилкрахмалом натрия (4,5 г), стеаратом магния (0,5 г) и тальком (1 г), и эту смесь пропускают через сито № 16 меш США. Затем полученную смесь добавляют к гранулам. После тщательного перемешивания смесь прессуют на таблеточном прессе с образованием таблеток массой 100 мг каждая. Каждая таблетка обеспечивает 10 мг активного агента на единичную дозу, подходящую для перорального введения.
F. Таблетки
Активный агент (40 г) тщательно смешивают с микрокристаллической целлюлозой (445 г), парами диоксида кремния (10 г) и стеариновой кислотой (5 г). Затем смесь прессуют на таблеточном прессе с получением таблеток весом 100 мг каждая. Каждая таблетка содержит 8 мг активного агента на единичную дозу, подходящую для перорального введения.
G. Таблетки
Активный агент (10 г) тщательно смешивают с кукурузным крахмалом (50 г), кроскармеллозой натрия (25 г), лактозой (110 мг) и стеаратом магния (5 мг). Затем смесь прессуют на таблеточном прессе с получением таблеток весом 200 мг каждая. Каждая таблетка содержит 10 мг активного агента на единичную дозу, подходящую для перорального введения.
H. Таблетки
Активный агент (10 г) тщательно перемешивают с кукурузным крахмалом (230 г) и водным раствором желатина (50 г). Смесь сушат и измельчают до мелкого порошка. Затем микрокристаллическую целлюлозу (100 г) и стеарат магния (10 г) смешивают с желатиновой композицией, гранулируют, и полученную смесь прессуют на таблеточном прессе с получением таблеток весом 200 мг каждая. Каждая таблетка содержит 5 мг активного агента на единичную дозу, подходящую для перорального введения.
I. Сироп
Приведенные ниже ингредиенты тщательно перемешивают до растворения всех твердых ингредиентов:
Полученный сироп содержит 5 мг активного агента на 10 мл сиропа, подходящего для перорального введения.
J. Стерильный раствор для внутривенного введения
Активный агент (5 мг) смешивают с 0,4 М буферным раствором ацетата натрия (2,0 мл). Значение pH полученного раствора, при необходимости, доводят до pH=4, используя 0,5 н водную соляную кислоту или 0,5 н водный гидроксид натрия, и затем добавляют достаточное количество воды для инъекций для получения общего объема, равного 20 мл. Затем смесь фильтруют через стерильный фильтр (0,22 микрона) для получения стерильного раствора, подходящего для введения путем внутривенной инфузии.
Совместное введение и комбинации
При желании 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемую соль можно вводить в комбинации с одним или более другими терапевтическими агентами (ʺвторичными агентамиʺ) для лечения нОГ и ее симптомов.
Типичные классы терапевтических средств, которые можно вводить в комбинации с 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидином или его фармацевтически приемлемой солью, включают, например, агонисты α1-адренергических рецепторов (α1-адренорецепторов), антагонисты α2-адренергических рецепторов (α2-адренорецепторов), кортикостероиды, предшественники норэпинефрина, ингибиторы холинэстеразы; или их комбинации. Специалистам в данной области понятно, что термины ʺагонист α1-адренергического рецептораʺ, ʺантагонист α2-адренергического рецептораʺ, ʺкортикостероидʺ, ʺпредшественник норэпинефринаʺ и ʺингибитор холинэстеразыʺ включают все формы соединений, обладающие указанной активностью после введение пациенту, такие как фармацевтически приемлемые соли, сольваты, кристаллические формы, полиморфы, пролекарства и т.п. Аналогично, термин ʺвторичный агентʺ включает все формы вторичного агента, такие как фармацевтически приемлемые соли, сольваты, кристаллические формы, полиморфы, пролекарства и т.п.
Типичные примеры агонистов α1-адренергического рецептора включают дезглимидодрин, этилэфрин, метараминол, мидодрин и т.п. или, в каждом случае, фармацевтически приемлемую соль. Мидодрин является пролекарством дезглимидодрина, который является агонистом α1-адренергических рецепторов. В одном из вариантов осуществления вторичный агент представляет собой мидодрин или его фармацевтически приемлемую соль, такую как мидодрина гидрохлорид.
Типичные примеры антагонистов 2-адренергических рецепторов включают йохимбин и т.п. или его фармацевтически приемлемые соли.
Типичные примеры кортикостероидов включают флудрокортизон, ацетат флудрокортизона и т.п. или, в каждом случае, их фармацевтически приемлемые соли. Флудрокортизона ацетат является пролекарством флудрокортизона. В одном из вариантов осуществления вторичный агент представляет собой ацетат флудрокортизона.
Типичные примеры предшественников норэпинефрина включают дроксидопу или ее фармацевтически приемлемые соли. В одном из вариантов осуществления вторичный агент представляет собой дроксидопу.
Типичные примеры ингибиторов холинэстеразы включают пиридостигмин или его фармацевтически приемлемую соль. В одном из вариантов осуществления вторичный агент представляет собой пиридостигмин или его фармацевтически приемлемую соль, такую как пиридостигмина бромид.
4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемая соль и вторичный агент могут быть либо физически смешаны с образованием композиции, содержащей оба агента; или каждый агент можно вводить пациенту по отдельности, либо одновременно, либо последовательно. Например, 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемая соль могут быть объединены со вторичным агентом при помощи традиционных процедур и оборудования для образования комбинации агентов, включающих 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемую соль и вторичный агент. Кроме того, агенты могут быть объединены с фармацевтически приемлемым носителем для образования фармацевтической композиции, содержащей 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемую соль, вторичный агент и фармацевтически приемлемый носитель. В этом варианте осуществления компоненты композиции обычно перемешивают или смешивают для создания физической смеси. Затем физическую смесь вводят пациенту любым подходящим способом введения, таким как пероральный, местный или парентеральный способы введения.
Альтернативно, перед введением пациенту агенты могут храниться раздельно. В этом варианте осуществления агенты физически не смешивают друг с другом перед введением, а вводят одновременно или в разные моменты времени в виде отдельных композиций. Такие композиции могут быть упакованы по отдельности или могут быть упакованы вместе в наборе. При введении в разные моменты времени вторичный агент обычно вводят менее чем через 24 часа после введения 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина или его фармацевтически приемлемой соли, например, в любой момент времени, начиная с параллельного введения до примерно через 24 часа после введения дозы активного агента. Это также называется последовательным введением. Так, например, 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемую соль можно вводить перорально одновременно или последовательно со вторичным агентом, используя две таблетки (например, по одной таблетке на каждый активный агент), где последовательное введение включает введение непосредственно перед или после введения 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина или его фармацевтически приемлемой соли или в другое время (например, за один час до или после; или за три часа до или после и т.д.). Альтернативно, комбинация может быть введена различными способами введения, например, одна перорально, а другая местно или парентерально.
Используемый в настоящем изобретении вторичный агент применяют в терапевтически эффективном количестве, т.е., в количестве, которое обеспечивает терапевтически полезный эффект при совместном введении с 4-[2-(2,4,6-трифторфеноксиметил)фенилом]пиперидином или его фармацевтически приемлемой солью. Например, такие агенты обычно используют в утвержденных для них дозировках. Например, гидрохлорид мидодрина обычно вводят перорально в количестве от примерно 2,5 до примерно 10 мг до трех раз в сутки; и дроксидопу обычно вводят перорально в количестве от примерно 100 до примерно 600 мг до трех раз в сутки.
Применимость
Предполагается, что 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин можно будет применять для лечения нейрогенной ортостатической гипотензии (нОГ) и симптомов, связанных с нОГ, таких как головокружение, предобморочное состояние или ощущение у пациента того, что он или она вот-вот потеряет сознание.
4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин обладает различными свойствами, благодаря которым он может оказаться особенно полезным для лечения нОГ. Например, в отличие от атомоксетина, метаболизм 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина катализируется несколькими ферментами CYP450, и, следовательно, не предполагается, что его воздействие на людей, фенотипически являющихся слабыми метаболизаторами по CYP2D6 (PM), может оказать значимое улучшение (Baldwin et al., ʺTD-9855, a Novel Norepinephrine and Serotonin Reuptake Inhibitor (NSRI), Demonstrates Significantly Reduced Dependence on CYP2D6 Metabolism Relative to Atomoxetineʺ Poster Presented at American Association of Pharmaceutical Scientists Annual Meeting and Exposition, Poster No. W4278, San Antonio, TX (2013); Abstract No. AAPS2013-000666). Более конкретно, химическое ингибирование CYP2D6 оказало минимальный эффект на метаболизм 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина (снижение на 7%) и, в основном, опосредовано через CYP3A4 и CYP1A2 (снижение на 34% и 25%, соответственно). При этом, на метаболизм атомоксетина химическое ингибирование CYP2D6, напротив, оказало существенное влияние (снижение на 94%), причем ингибирование других ферментов CYP450 не оказало существенного влияния. Таким образом, предполагается, что по сравнению с атомоксетином 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин обладает значительно более низким потенциалом в отношении взаимодействия с лекарственными средствами и пониженной восприимчивостью к негативным эффектам, связанным с полиморфизмом CYP2D6.
В одном из вариантов осуществления 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемая соль используется для лечения симптоматической нОГ, вызванной первичной вегетативной недостаточностью (включая истинную вегетативную недостаточность, множественную системную атрофию и болезнь Паркинсона), или вегетативной нейропатии (включая диабетическую и недиабетическую вегетативную нейропатию).
В конкретном варианте осуществления 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемая соль используется для лечения симптоматической нОГ, вызванной первичной вегетативной недостаточностью. В этом варианте осуществления пациент может быть диагностирован как имеющий истинную вегетативную недостаточность, множественную системную атрофию и/или болезнь Паркинсона. В одном из вариантов осуществления пациент имеет истинную вегетативную недостаточность. В другом варианте осуществления пациент имеет множественную системную атрофию. В другом варианте осуществления у пациента болезнь Паркинсона.
Первичная вегетативная недостаточность (также называемая первичной дизавтономией) является категорией дизавтономии, т.е., состоянием, при котором вегетативная нервная система не функционирует должным образом. При первичной вегетативной недостаточности вегетативная дисфункция возникает как первичное состояние, а не как вторичное состояние, возникающее в результате другого заболевания, такого как диабет. Например, вегетативная недостаточность обычно классифицируется как "первичная", когда она возникает в результате хронического состояния, характеризующегося дегенерацией вегетативной нервной системы, или когда вегетативная недостаточность является преобладающим симптомом, и ее причина неизвестна. Состояния, классифицируемые как первичная вегетативная недостаточность, включают истинную вегетативную недостаточность, множественную системную атрофию и болезнь Паркинсона.
Истинная вегетативная недостаточность (PAF), также известная как синдром Брэдбери-Эгглстона или идиопатическая ортостатическая гипотензия, является дегенеративным заболеванием вегетативной нервной системы. Основным симптомом PAF является ортостатическая гипотензия. Другие симптомы могут включать снижение потоотделения, непереносимость тепла, задержку мочи, спазмы мочевого пузыря (возможно, вызывающие недержание мочи), эректильную дисфункцию, недержание кала или запоры, а также зрачковые нарушения. Причина PAF до конца не выяснена, но было показано, что у пациентов с PAF наблюдается потеря клеток в латеральном промежуточном столбе спинного мозга. Кроме того, PAF может быть связан с ненормальным накоплением альфа-синуклеина.
Болезнь Паркинсона (БП) является хроническим и прогрессирующим двигательным расстройством. Причина неизвестна, но БП включает дисфункцию и гибель нейронов в области среднего мозга, называемой черной субстанцией. Эти нейроны продуцируют дофамин, который играет ключевую роль в движении и координации. По мере прогрессирования БП количество дофамина, вырабатываемого в мозге, уменьшается, приводя к проблемам, связанным с контролем и координацией движений. Симптомы включают тремор, ригидность, замедленность движений и постуральную нестабильность. Однако у некоторых пациентов с БП также имеются не связанные с моторикой симптомы, включая ортостатическую гипотензию, вызванную изменениями в автономной нервной системе, т.е., БП плюс симптомы нОГ. Кроме того, у некоторых пациентов с симптомами БП наблюдается состояние, известное как синдром Паркинсонизма-плюс (или расстройство множественной системной дегенерации). Синдромы Паркинсонизма-плюс - это группа нейродегенеративных заболеваний, которые вызывают классические симптомы БП (тремор, ригидность, акинезию/брадикинезию и постуральную нестабильность) с дополнительными признаками, которые отличают их от простой идиопатической БП. Клинические признаки, отличающие синдром Паркинсонизма-плюс от идиопатической БП, включают симметричное начало, отсутствие или нерегулярный тремор в покое и сниженный ответ на дофаминергические препараты (включая леводопу). Дополнительные признаки включают брадикинезию, раннюю постуральную нестабильность, повышенную ригидность в аксеальных мышцах, дизавтономию, синдром чужеродных конечностей, надъядерный парез взора, апраксию, вовлечение мозжечка, включая пирамидальные клетки, и в некоторых случаях значительное нарушение когнитивных функций.
Множественная системная атрофия (MSA), также известная как синдром Шай-Дрейджера, является прогрессирующим нейродегенеративным расстройством, характеризующимся комбинацией симптомов, которые влияют как на вегетативную нервную систему, так и на движение. Начальные симптомы MSA часто трудно отличить от начальных симптомов болезни Паркинсона, которые включают медлительность, тремор или ригидность (скованность); неловкость или несогласованность; нарушение речи, хриплый, дрожащий голос; обмороки или предобморочное состояние из-за ортостатической гипотензии; проблемы с контролем мочевого пузыря, такие как внезапное желание помочиться или затруднение опорожнения мочевого пузыря. MSA подразделяется на два различных типа в зависимости от наиболее выраженных симптомов, наблюдаемых при оценке индивидуума: паркинсонический тип (MSA-P) с основными характеристиками, схожими с таковыми при болезни Паркинсона (такими как медленное движение, скованность и тремор), наряду с проблемами баланса, координации и дисфункции вегетативной нервной системы; и церебеллярный тип (MSA-C) с основными симптомами, характеризующимися атаксией (проблемой с балансом и координацией), затруднением глотания, нарушениями речи или дрожанием голоса и аномальными движениями глаз. Причина MSA неизвестна. Отличительной особенностью MSA является накопление белка альфа-синуклеина в глие, клетках, которые поддерживают нервные клетки головного мозга. Эти отложения альфа-синуклеина особенно характерны для олигодендроглии, типа клеток, которые производят миелин (покрытие нервных клеток, позволяющее им быстро проводить электрические сигналы). Недавнее исследование показало, что причиной заболевания может быть прионная форма белка альфа-синуклеина (Prusiner et al, PNAS, (2015) 112: E5308-17).
4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемая соль особенно полезны для лечения MSA, что связано с фармакологией MSA. Более конкретно, MSA характеризуется дегенерацией центрального вегетативного пути; однако периферические постганглионарные норадренергические волокна и механизмы обратного захвата катехоламинов, по-видимому, интактны у пациентов с MSA, таким образом, поддерживая симпатический тонус (Biaggioni, Pharmacolgical Reviews (2017) 69 (1):53-62). Обычно повышение концентрации норадреналина в периферической крови в положении стоя противодействует симпатолитической активности ЦНС, опосредованной активированными норэпинефрином центральными α2-адренорецепторами, таким образом, смягчая эффект периферического прессора и поддерживая постуральную нормотензию. Однако при MSA неповрежденные периферические симпатические постганглионарные адренергические волокна по существу ʺотключеныʺ от модуляции ЦНС, что позволяет выявить полный эффект норадреналина. Хотя из-за этого ʺотключенияʺ остаточный симпатический тонус у этих пациентов не может быть модулирован через пути барорефлекса или входные сигналы ЦНС, он может быть мишенью для лекарственных средств. Используя преимущества этой уникальной патофизиологии, увеличение периферического симпатического синаптического норэпинефрина благодаря использованию 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина или его фармацевтически приемлемой соли должно вызывать прессорный ответ у пациентов с MSA с симптомом нОГ. Соответственно, в конкретном варианте осуществления 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемая соль используется для лечения симптоматического нОГ, вызванного MSA.
В другом конкретном варианте осуществления 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемая соль используется для лечения симптоматической нОГ, вызванной вегетативной нейропатией. Автономная нейропатия, или дизавтономия, относится к различным состояниям, при которых вегетативная нервная система (ANS) не работает должным образом. Автономная невропатия - это тип нейропатии, поражающей нервы, которые передают информацию от головного и спинного мозга к сердцу, мочевому пузырю, кишечнику, потовым железам, зрачкам и кровеносным сосудам. Основные симптомы вегетативной нейропатии, которые могут меняться у разных людей, включают: ортостатическую гипотензию; сухость во рту; учащенное сердцебиение; туннельное зрение; трудность глотания; недержание кишечника; нечеткое зрение; недержание мочи; запор; ангидроз; и сексуальную дисфункцию. Вегетативная нейропатия может быть вызвана наследственными или дегенеративными неврологическими заболеваниями (первичная дизавтономия) или может возникнуть из-за повреждения вегетативной нервной системы от приобретенного расстройства (вторичная дизавтономия).
4-[2-(2,4,6-Трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемую соль, используемую для лечения нОГ или симптомов нОГ, обычно вводят пациенту в количестве от примерно 0,5 мг до примерно 20 мг в сутки; или по мере необходимости. В одном из вариантов осуществления количество, вводимое пациенту, составляет от примерно 1 до примерно 10 мг в сутки. В другом варианте осуществления количество, вводимое пациенту, составляет от примерно 3 до примерно 10 мг в сутки. В различных вариантах осуществления количество, вводимое пациенту, составляет 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 мг в сутки, включая 1 мг, 3 мг, 5 мг или 10 мг в сутки. Количество, вводимое пациенту, путь введения и частота введения, как правило, определяются лечащим врачом.
4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемую соль можно вводить пациенту любым приемлемым способом введения, включая, например, пероральный, местный (включая трансдермальный) и парентеральный (в том числе внутривенный) способы введения.
В одном из вариантов осуществления 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемую соль вводят пациенту перорально в твердой или жидкой лекарственной форме. В конкретном варианте осуществления форма, вводимая пациенту, представляет собой твердую лекарственную форму, включающую таблетку или капсулу. В другом конкретном варианте осуществления форма, вводимая пациенту, представляет собой жидкую лекарственную форму, включающую раствор, сироп, суспензию или эмульсию.
В другом варианте осуществления используется местный способ введения. В конкретном варианте осуществления способ введения является трансдермальным при помощи трансдермального пластыря.
В другом варианте осуществления способ введения является парентеральным. В конкретном варианте осуществления способ введения представляет собой внутривенное введение.
4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемую соль можно вводить пациенту в виде однократной суточной дозы (например, один раз в сутки); в виде многократных доз в сутки (например, два раза, три раза или четыре раза в сутки); или в виде нескольких доз в неделю (например, два раза, три раза, четыре раза, пять раз или шесть раз в неделю). Альтернативно, фармацевтическую композицию можно вводить непрерывно при помощи, например, трансдермального пластыря. В конкретном варианте осуществления 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин или его фармацевтически приемлемую соль вводят пациенту один раз в сутки.
ПРИМЕРЫ
Приведенные ниже примеры являются иллюстрацией различных репрезентативных вариантов осуществления и аспектов настоящего изобретения и не предназначены для ограничения объема изобретения, если не указано иное.
Все реагенты, исходные материалы и растворители, использованные в приведенных ниже примерах, приобретали у коммерческих поставщиков (таких как Sigma-Aldrich, St. Louis, MO и его филиалах) и использовали без дополнительной очистки, если не указано иное.
Приведенные ниже сокращения имеют следующие значения, если не указано иное:
Другие сокращения, используемые в настоящей заявке, которые специально не определены, используются в общепринятом значении, понятном специалистам в области техники, к которой они относятся.
Спектры1Н ЯМР регистрировали на 400 МГц спектрометре Varian AS400, если не указано иное. Химические сдвиги представлены в виде значений δ в миллионных долях относительно тетраметилсилана (TMS) в качестве внутреннего стандарта. Константы спин-спинового взаимодействия (значения J) приведены в герцах (Гц), а мультиплетность представлена в виде следующих сокращений: s=синглет, d=дублет, t=триплет, q=квартет, m=мультиплет, br=уширенный, nd=не определено.
Пример 1
Получение сложного трет-бутилового эфира 4-(2-гидроксиметилфенил)пиперидин-1-карбоновой кислоты
Сложный трет-бутиловый эфир 4-(2-карбоксифенил)пиперидин-1-карбоновой кислоты (5,0 г, 16 ммоль, 1,0 экв.) и THF (130 мл, 1,7 мол) объединяли при комнатной температуре в атмосфере азота. Боран-диметилсульфидный комплекс (2,9 мл, 33 ммол, 2,0 экв.) добавляли по каплям, и смесь перемешивали в течение 5 минут, затем нагревали с обратным холодильником в течение 1 часа. Смесь охлаждали до комнатной температуры, и реакцию останавливали добавлением по каплям MeOH (40 мл). Затем смесь концентрировали путем ротационного выпаривания, и полученный материал подвергали азеотропной перегонке с MeOH (2×40 мл). После этого смесь разбавляли EtOAc (100 мл) и промывали водным раствором соляной кислоты (1 M; 2×50 мл), затем водным насыщенным раствором бикарбоната натрия (2×50 мл), затем водным насыщенным раствором хлорида натрия (1×50 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением сложного трет-бутилового эфира 4-(2-гидроксиметилфенил)пиперидин-1-карбоновой кислоты (4,8 г) в виде прозрачного, светло-желтого масла, которое отверждали путем отстаивания.
1H ЯМР (CDCl3) δ (ppm) 7,34-7,22 (м, 3H); 7,19 (дт, J=1,6 Гц, 7,2, 1H); 4,73 (с, 2H); 4,32-4,14 (м, 2H); 3,00 (тт, J=4,0 Гц, 12,0, 1H); 2,80 (т, J=11,6 Гц, 2H); 1,78-1,56 (м, 4H); 1,47 (м, 9H).
Пример 2
Получение сложного трет-бутилового эфира 4-[2-(толуол-4-сульфонилоксиметил)фенил]пиперидин-1-карбоновой кислоты
Сложный трет-бутиловый эфир 4-(2-гидроксиметилфенил)пиперидин-1-карбоновой кислоты (0,4 г, 1,0 ммол, 1,0 экв.) и триэтилендиамин (220 мг, 2,0 ммол, 1,4 экв.) растворяли в DCM (11 мл, 170 ммол). Смесь охлаждали при 0°C в атмосфере азота, и добавлялип-толуолсульфонил хлорид (290 мг, 1,5 ммол, 1,1 экв.). Полученную смесь перемешивали при 0°C в течение 60 минут. Смесь разбавляли EtOAc (50 мл) и промывали водой (2×25 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали путем ротационного выпаривания с получением указанного в заголовке соединения (500 мг), которое использовали без дополнительной очистки.
1H ЯМР (CDCl3) δ (ppm) 7,81 (т, J=2,0 Гц, 1H); 7,79 (т, J=2,0 Гц, 1H); 7,37-7,32 (м, 4H); 7,25-7,21 (м, 1H); 7,21-7,13 (м, 1H), 5,12 (с, 2H); 4,34-4,12 (м, 2H); 2,81-2,61 (м, 3H); 2,45 (с, 3H); 1,70-1,52 (м, 4H); 1,48 (с, 9H).
Пример 3
Получение 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидиновой соли трифторуксусной кислоты
Сложный трет-бутиловый эфир 4-[2-(толуол-4-сульфонилоксиметил)фенил]пиперидин-1-карбоновой кислоты (2,1 г, 4,7 ммол, 1,0 экв.) растворяли в MeCN (46 мл, 890 ммол) и добавляли к карбонату калия (1,9 г, 14 ммол, 3,0 экв.) и 2,4,6-трифторфенолу (1,0 г, 7,0 ммол, 1,5 экв.). Смесь встряхивали при 50°C в течение ночи, затем охлаждали до комнатной температуры. Супернатант отделяли от карбоната калия и других твердых веществ. К супернатанту добавляли TFA (7 мл, 90 ммол, 20,0 экв.), и смесь встряхивали в течение ночи при комнатной температуре. Затем раствор концентрировали, и остаток растворяли в 1:1 смеси уксусная кислота/вода (5,0 мл). Дополнительно добавляли уксусную кислоту (2,0 мл), и смесь фильтровали и очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (1,3 г, 97,5% чистоты). МС m/z: [M+H]+ вычисл. для C18H18F3NO, 322,13; полученное 322,2.
1H ЯМР (CDCl3) δ (ppm) 9,83 (уш.с, 1H); 9,32 (уш.с, 1H); 7,46-7,39 (м, 2H); 7,32 (д, J=6,8 Гц, 1H); 7,26-7,21 (м, 1H); 6,76-6,66 (м, 2H); 5,07 (с, 2H); 3,69-3,50 (м, 2H); 3,38 (т, J=11,6 Гц, 1H); 3,20-3,02 (м, 2H); 2,19 (к, J=12,8 Гц, 2H); 2,12-2,01 (м, 2H).
Пример 4
Получение сложного трет-бутилового эфира 4-(2-гидроксиметилфенил)пиперидин-1-карбоновой кислоты
Сложный трет-бутиловый эфир 4-(2-карбоксифенил)пиперидин-1-карбоновой кислоты (5,0 г, 160 ммол, 1,0 экв.) и THF (100 мл, 1,0 мол) объединяли при комнатной температуре в атмосфере азота. Боран-THF комплекс в THF (1,0 M, 32,7 мл, 32,7 ммол, 2,0 экв.) добавляли по каплям в течение 10 минут (5°C экзотерм., выделение газа). Реакционную смесь перемешивали при комнатной температуре в течение 5 минут, затем нагревали при 50°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, и реакцию останавливали медленным добавлением MeOH (30 мл) (слабый экзотермический эффект, сильное выделение газа). Затем смесь концентрировали путем ротационного выпаривания. Полученный материал подвергали азеотропной перегонке с MeOH (2×50 мл). Неочищенный продукт растворяли в EtOAc (100 мл) и промывали насыщенным водным раствором бикрбоната натрия (50 мл) и затем насыщенным водным раствором хлорида натрия (50 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением сложного трет-бутилового эфира 4-(2-гидроксиметилфенил)пиперидин-1-карбоновой кислоты (4,4 г) в виде прозрачного, светло-желтого масла, которое отверждали путем отстаивания.
Пример 5
Получение сложного трет-бутилового эфира 4-(2-метансульфонилоксиметилфенил)пиперидин-1-карбоновой кислоты
Сложный трет-бутиловый эфир 4-(2-гидроксиметилфенил)пиперидин-1-карбоновой кислоты (50,0 г, 172 ммол, 1,0 экв.) растворяли в DCM (500 мл, 8000 ммол). Смесь охлаждали при 0°C в атмосфере азота, и одной порцией добавляли метансульфоновый ангидрид (44,8 г, 257 ммол, 1,5 экв.). Диизопропилэтиламин (47,8 мл, 274 ммол, 1,6 экв.) добавляли по каплям в течение 5 минут, и смесь перемешивали при 0°C в течение 90 минут. Добавляли воду (400 мл, 20 мол), и смесь перемешивали в течение 5 минут. Фазы разделяли, и органический слой промывали водой (300 мл), сушили над безводным сульфатом натрия, и растворитель удаляли с получением указанного в заголовке соединения (70 г) в виде густого масла, которое использовали без дополнительной очистки.
1H ЯМР (400 MГц, DMSO-d6) δ (ppm) 7,37-7,43 (м, 3H), 7,31 (д, 1H), 7,22 (м, 2H), 5,38 (с, 2H), 4,28 (м, 2H), 2,92-3,10 (м, 1H), 2,92 (с, 3H), 2,80-2,92 (м, 2H), 1,63-1,81 (м, 4H), 1,51 (с, 9H).
Пример 6
Получение сложного трет-бутилового эфира 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин-1-карбоновой кислоты
Сложный трет-бутиловый эфир 4-(2-метансульфонилоксиметилфенил)пиперидин-1-карбоновой кислоты (27,0 г, 60,6 ммол, 1,0 экв.) растворяли в MeCN (540 мл) и добавляли к карбонату калия (25 г, 180 ммол, 3,0 экв.) и 2,4,6-трифторфенолу (13,5 г, 90,9 ммол, 1,5 экв.). Смесь интенсивно перемешивали при 50°C в течение 6 часов, нагревание прекращали и продолжали перемешивать в течение ночи. Смесь охлаждали при комнатной температуре и разбавляли EtOAc (700 мл) и водой (700 мл). Фазы разделяли, и органический слой промывали водным раствором гидроксида натрия (1,0 M; 2×400 мл) и насыщенным водным раствором хлорида натрия (1×400 мл), а затем сушили над безводным сульфатом натрия. Затем растворитель удаляли с получением неочищенного сложного трет-бутилового эфира 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин-1-карбоновой кислоты (25,0 г). Неочищенный продукт объединяли с более мелкими порциями с получением общего количества, равного 30 г, и очищали хроматографией (0-10% EtOAc в гексанах) с получением сложного трет-бутилового эфира 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин-1-карбоновой кислоты (22,0 г).
Пример 7
Получение гидрохлорида 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина
Сложный трет-бутиловый эфир 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин-1-карбоновой кислоты (22,0 г, 31,3 ммол, 1,0 экв.) объединяли с 1,25 M HCl в EtOH (250 мл, 310 ммол, 10,0 экв.). Смесь перемешивали при комнатной температуре в течение 8 часов, затем хранили при -10°C в течение примерно 48 часов. Большую часть растворителя удаляли путем ротационного выпаривания. К полученной густой суспензии добавляли EtOAc (80 мл) и перемешивали при комнатной температуре в течение 2 часов. Первый сбор кристаллов осуществляли фильтрованием, и фильтровальную лепешку промывали EtOAc (20 мл) и сушили с получением указанного в заголовке соединения (8,5 г, >99% чистоты) в виде белого твердого вещества. ВЭЖХ фильтрата показала ~25% площадь продукта. Во время второго сбора растворитель удаляли путем ротационного выпаривания, и полученное твердое вещество (~10 г) суспендировали EtOAc (40 мл) сначала при комнатной температуре, а затем при 60°C и снова при комнатной температуре с получением указанного в заголовке соединения в виде гидрохлоридной соли (1,7 г, >99% чистоты).
Две партии гидрохлоридной соли (18,5 г, 51,7 ммол) объединяли с EtOAc (75 мл, 770 ммол). Полученную густую, но свободно текучую суспензию нагревали при 65°C в течение 30 минут, охлаждали до комнатной температуры и фильтровали. Колбу и фильтровальную лепешку промывали EtOAc (20 мл), и твердые вещества сушили под глубоким вакуумом при комнатной температуре в течение ночи с получением кристаллической гидрохлоридной соли (18,2 г, 99,3% чистоты).
Пример 8
Получение гидрохлорида 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина
Ацетилхлорид (83,5 мл, 1170 ммол) медленно добавляли к EtOH (140 мл, 2,4 мол). Добавляли сложный трет-бутиловый эфир 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин-1-карбоновой кислоты (55,0 г, 117 ммол), растворенный в EtOH (100 мл, 2,0 мол), и полученную смесь перемешивали при комнатной температуре в течение 6 часов. Большую часть растворителя удаляли путем ротационного выпаривания. К полученной густой суспензии добавляли EtOAc (300 мл) с последующим частичным удалением растворителя до ~100 мл. Добавляли EtOAc (200 мл), и полученную суспензию перемешивали в течение 1 часа, фильтровали и сушили с получением указанного в заголовке соединения (28,0 г, ~99% чистоты). Фильтрат концентрировали с получением густой пасты, и добавляли IPAc (100 мл), перемешивали в течение 1 часа, фильтровали и сушили с получением 5,0 г гидрохлоридной соли (~99% чистоты).
Две партии гидрохлоридной соли (83,0 г, 230 ммол, ~99% чистоты) объединяли с EtOAc (250 мл, 2,6 мол). Полученную суспензию нагревали при 70°C и затем медленно охлаждали до комнатной температуры с последующим перемешиванием в течение ночи. Полученную свободнотекучую суспензию фильтровали, и фильтровальную лепешку промывали EtOAc (50 мл), затем сушили под глубоким вакуумом в течение примерно 48 часов с получением кристаллической гидрохлоридной соли (81,0 г, >99% чистоты).
Кристаллическую гидрохлоридную соль (50,0 г, 1,40 мол, >99% чистоты) растворяли в IPA (250 мл, 3,3 мол), и полученную суспензию нагревали до 75°C. Добавляли воду (25 мл, 1,4 мол). Через 5 минут наблюдали полное растворение, а внутренняя температура раствора составила 65°C. Раствор медленно охлаждали до комнатной температуры и затем перемешивали при комнатной температуре в течение ночи. Полученные твердые вещества фильтровали и сушили на воздухе в течение 2 часов с получением полусухого продукта. Затем твердые вещества сушили под глубоким вакуумом при комнатной температуре в течение примерно 48 часов с получением указанной в заголовке кристаллической гидрохлоридной соли (44,1 г, 99,5% чистоты). Этот материал использовали в анализах PXRD и DSC, описанных в примерах 9 и 10.
Пример 9
Порошковая рентгеновская дифракция
Порошковые рентгенограммы получали на Rigaku Miniflex PXRD дифрактометре с источником излучения CuKα (30,0 кВ, 15,0 мА). Анализ выполняли на гониометре, работающем в режиме непрерывного сканирования с углом 2° (2θ) в мин с размером шага 0,03° в диапазоне изменения угла два-тэта от 2 до 40°. Образцы получали на кварцевом держателе образцов в виде тонкого слоя порошкообразного материала. Инструмент калибровали с помощью стандарта, металлического кремния, в пределах ±0,02° угла два-тэта. Перед тестированием образцы вручную размельчали для уменьшения влияния размера частиц на относительное значение интенсивности. Типичная рентгенограмма PXRD для кристаллической гидрохлоридной соли примера 8 показана на фиг. 1 патента США № 8,304,433 B2. Пики PXRD в порядке убывания относительной интенсивности показаны в таблице 1. Все интенсивности пиков PXRD корректировали путем вычитания соответствующей фоновой интенсивности для каждого пика.
Таблица 1
Данные порошковой рентгенограммы
В одном из вариантов осуществления кристаллическая гидрохлоридная соль, используемая в настоящем изобретении, характеризуется порошковой рентгенограммой, характеризующейся наличием дифракционных пиков при значениях 2θ, равных 4,44±0,20, 10,22±0,20, 17,16±0,20 и 21,78±0,20. В другом варианте осуществления кристаллическая гидрохлоридная соль дополнительно характеризуется наличием одного или более дополнительных дифракционных пиков при значениях 2θ, выбранных из 8,11±0,20, 13,18±0,20, 16,06±0,20, 18,38±0,20, 23,76±0,20, 26,32±0,20, 27,24±0,20, 29,60±0,20 и 31,94±0,20.
Пример 10
Дифференциальная сканирующая калориметрия
Дифференциальную сканирующую калориметрию (DSC) выполняли, используя модуль TA Instruments Model Q-100 с контроллером Thermal Analyst. Данные собирали и анализировали при помощи программного обеспечения TA Instruments Thermal Solutions. Точное взвешивание 2,8 мг образца кристаллической гидрохлоридной соли примера 8 выполняли в закрытой алюминиевой чашке. После 5-минутного периода изотермического уравновешивания при 22°С образец нагревали, постепенно линейно повышая температуру со скоростью 10°С/мин от 22 до 250°С. Типичная термограмма DSC показана на фиг. 2 патента США № 8,304,433 B2. Как следует из термограммы DSC, кристаллическая гидрохлоридная соль обладает превосходной термостойкостью с температурой плавления примерно 196,9°С.
В одном из вариантов осуществления кристаллическая гидрохлоридная соль, используемая в настоящем изобретении, характеризуется графиком дифференциальной сканирующей калориметрии, имеющим температуру плавления примерно 197±2°С.
Пример 11
Анализ связывания радиолиганда и захвата нейротрансмиттера
Получали in vitro фармакологические характеристики 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина по отношению к транспортерам человеческого рекомбинантного и крысиного нативного моноаминов получали согласно тому, как описано у Smith et al., Inter. J. Neuropsychopharmcol. (2015) 1-11; и Tsuruda et al., J. Pharmacol. Toxicol. Meth. (2010) 61:192-204. См. также, например, патенты США №№ 8,304,432 B2 и 8,304,433 B2. Радиолиганды получали на коммерческой основе (Perkin Elmer LifeSciences или GE Healthcare Life Sciences).
Вкратце, мембраны, полученные из клеток HEK293 (эмбриональной почки человека) или CHO-K1 (яичника китайского хомячка), стабильно трансфицированных рекомбинантным SERT человека (HEK293-hSERT), NET (HEK293-hNET) или DAT (CHO-K1- hDAT) инкубировали в течение 1 часа при 22°C в отсутствие или в присутствии 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина и [3H]-циталопрама (1,0 нМ) в случае SERT, [3H]-низоксетина (2,0 нМ) в случае NET и [3H]-WIN35428 (3,0 нМ) в случае DAT в 50 мМ Трис-HCl, 120 мМ NaCl, 5 мМ KCl, 0,025% BSA, 100 мкМ аскорбиновой кислоты, рН 7,4. Препараты кортикальной мембраны крысы инкубировали с [3H]-циталопрамом (2,0 нМ) в случае SERT или [3H]-низоксетином (4,0 нМ) в случае NET в течение 1 часа при 22°C. В анализах захвата нейротрансмиттеров, перед 10 минутной инкубацией с [3H]-5-HT (20 нМ), [3H]-NE (40 нМ) или [3H]-DA (100 нМ), клетки HEK293-hSERT, hNET или hDAT, соответственно, предварительно инкубировали в течение 30 мин при 37°C в отсутствие или в присутствии 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина в 7,5 мМ HEPES, 12,5 мМ Трис-HCl, 2,2 мМ Na-фосфата, 120 мМ NaCl, 5 мМ KCl, 0,4 мМ MgCl2, 7,5 мМ глюкозы, 1,7 мМ CaCl2, 250 мкМ аскорбиновой кислоты, 150 мкМ паргилина, 0,025% BSA, pH 7,4. Кортикальные синаптосомы крыс инкубировали с [3H]-5-HT или [3H]-NE в течение 6 минут, а стриарные синаптосомы инкубировали с [3H]-DA в течение 6 минут. Анализы связывания и захвата завершали быстрой фильтрацией, и определяли радиоактивность методом жидкостной сцинтилляционной спектроскопии. Конечные концентрации [3H]-нейротрансмиттера были значительно ниже соответствующего Km настолько, что pIC50 приближался к функциональному pKi. Селективность по NET (округленная до одной значимой цифры) определяли следующим образом:
Селективность=10(pKi или pIC50 при NET минус pKi или pIC50 при SERT или DAT).
Фармакологический профиль in vitro 4-[2-(2,4,6-трифторфеноксиметил)-фенил]пиперидина у транспортеров моноаминов человека и грызунов был аналогичным, как показано в таблице 2.
Таблица 2
In vitro фармакологические характеристики транспортеров человеческих рекомбинантных и крысиных нативных SERT, NET и DAT
Данные выражены в виде средних значений pIC50 (отрицательный десятичный логарифм IC50) и значений pKi (отрицательный десятичный логарифм Ki). Данные представляют собой среднее значение (с 95% доверительными интервалами, в скобках) 3-9 отдельных экспериментов. N.D.=не определено.
Из приведенных в таблице 2 данных видно, что 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин является мощным ингибитором NET и SERT, но не DAT, с эффективностью ингибирования NET, которая в 4 раза превышает таковую для SERT. Аналогично, 4-[2- (2,4,6-трифторфеноксиметил)фенил]пиперидин является мощным ингибитором захвата [3H]-NE и [3H]-5-HT в кортикальных синаптосомах крыс с очевидной функциональной селективностью (в 10 раз) для NET по сравнению с SERT, аналогичные результаты получены и для человеческих транспортеров. Согласно результатам функциональных исследований ингибирования 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин проявляет высокое сродство связывания с человеческими NET и SERT, но не с DAT (таблица 2). Кажущиеся значения сродства связывания с крысиными нативными NET и SERT в мембранах, полученных из крысиной коры, аналогичны (перекрывающиеся доверительные интервалы) соответствующим значениям, полученным для человеческих транспортеров, что согласуется с отсутствием видовой зависимости (таблица 2).
Пример 12
Ex vivo исследования заполнения транспортеров
Взрослых самцов крыс Sprague Dawley (Charles River) содержали в контролируемых лабораторных условиях (температура 21±1°C) при 12:12 часовом цикле свет-темнота. Когда животных доставили в учреждение, им был предоставлен свободный доступ к пище и воде, животных оставляли в помещении для содержания в течение не менее 48 часов для акклиматизации. Перед введением дозы, животным не давали пищу в течение 15-18 часов, но они имели свободный доступ к воде.
Крысам (n=6/временная точка/уровень дозы) вводили однократную пероральную дозу 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина (0,3, 1, 5, 10, 30 и 60 мг/кг), затем их умерщвляли путем обезглавливания в определенные моменты времени (через 0,5, 2, 4, 6 и 8 часов для уровней дозы 5 мг/кг; через 2 часа для уровней дозы 0,3, 1, 10, 30 и 60 мг/кг) после введения. Спинной мозг иссекали для оценки заполнения транспортера ex vivo и оценки ФК у одних тех же животных. Спинной мозг собирали путем гидравлической экструзии, используя забуференный фосфатом солевой раствор, а поясничный сегмент иссекали и замораживали, используя сухой лед. Оставшиеся сегменты спинного мозга собирали и гомогенизировали в воде (25% мас./мас.) для ФК анализа. До анализа все образцы хранили при -80°С.
Кинетический анализ связывания радиолиганда использовали для определения уровня заполнения NET и SERT в спинном мозге крысы, как описано ранее у Bourdet et al., J. Pharm. Exp. Ther. (2012) 341:137-145. Параметры ФК/ФД оценивали, используя подход построения компартментной модели (WinNonlin Version 5.0.1, Pharsight Corporation). Оценивали одно- и двухкомпартментные ФК модели с абсорбцией и элиминацией первого порядка. Была выбрана однокомпартментная модель. Фармакодинамическая модель представляла собой компартментную Emax-модель эффекта, непосредственно связанную с центральным ФК компартментом (WinNonlin PK Model 3, PD Model 101). Выбор моделей был основан на наилучшем соответствии согласно визуальной оценке, информационным критериям Акаике и взвешенной остаточной сумме квадратов, используя метод минимизации Гаусса-Ньютона. Оценивали следующие параметры:
Оценки параметров ФК и ФД, полученные на основе эффект-компартментного ФК/ФД анализа для уровня заполнения NET и SERT, показаны в таблице 3.
Таблица 3
Оценка фармакокинетических и фармакодинамических параметров заполнения транспортеров норэпинефрина и серотонина 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидином в спинном мозге крысы
Окончательные значения оценки параметров указаны с коэффициентом вариации (% CV), в скобках, для каждой оценки параметров.
Как показано в Таблице 3, оценочная величина EC50 для заполнения в спинном мозге крысы составила 11,7 нг/мл для NET и 50,8 нг/мл для SERT. С учетом видовых различий в связывании белков плазмы (90,2% и 79,1% у крыс и человека, соответственно) прогнозируемые значения EC50 в плазме крови человека будут составлять 5,5 нг/мл для NET и 23,9 нг/мл для SERT.
Пример 13
Модель сердечно-сосудистой системы у крыс под наркозом
Эти исследования выполняли для оценки влияния однократного введения 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина на частоту сердечных сокращений (ЧСС) и среднее артериальное давление (СрАД) у анестезированных крыс. Используя эту сердечно-сосудистую модель, истинное воздействие на ЧСС и ингибирование прессорного эффекта тирамина оценивали как суррогатные меры, отражающие способность 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина ингибировать транспортеры норадреналина в периферической крови.
А. Структура эксперимента
Самцов крыс Sprague-Dawley весом от 250 до 350 г анестезировали внутрибрюшинной инъекцией (IP) тиобутабарбитала (инактина). Во время операции и на протяжении всего исследования всех животных содержали под полной анестезией (т.е., отсутствием реакции на тест сдавливания лапы). Обнажали правую общую сонную артерию и яремную вену, и вводили катетер. В трахею вводили трубку для обеспечения проходимости дыхательных путей во время исследования. После завершения операции артериальный катетер подключали к датчику давления, и записывали исходное значение артериального давления [систолическое (САД), среднее артериальное (СрАД) и диастолическое (ДАД)] и частоту сердечных сокращений (ЧСС), сбор данных производили, используя систему Notocord-HEM. Через по меньшей мере 60 мин от исходной точки (т.е., последние 10 мин были стабильными), вводили носитель (10% Tween 20, 2 мл/кг, IP), и отслеживали любую потенциальную реакцию в течение по меньшей мере 10 мин. После этого периода крысам вводили либо носитель, либо 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин (0,01-30 мг/мл, 2 мл/кг, IP), и отслеживали изменения в СрАД и ЧСС в течение 25 мин. Затем через яремную вену крысам внутривенно вводили некумулятивные болюсные дозы тирамина (0,03, 0,1, 0,3 и 1 мг/кг, 1 мл/кг, IV) с интервалами 5 минут. После последней дозы тирамина сбор данных продолжали еще в течение 10 минут, прежде чем эксперимент был прекращен. У отдельной группы животных собирали кровь для оценки концентрации 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина в плазме через 15 минут и 60 минут после введения дозы. Концентрации в свободной плазме, полученные через 15 мин, использовали для построения кривой зависимости концентрация-ответ (CRC) для СрАД и ЧСС, в то время как концентрации через 60 мин использовали для построения кривой CRC для тирамина. Животных умерщвляли асфиксией углекислым газом с последующей торакотомией.
В. Анализ данных
Внутренние гемодинамические эффекты описывали как максимальное изменение СрАД или ЧСС, вызванное 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидином до введения тирамина. Ингибирование эффекта тирамина нормализовали относительно ответа на 1 мг/кг дозу тирамина в контрольной группе носителя. Кривые зависимости концентрация-ответ (CRC) для СрАД, ЧСС и ингибирования эффекта тирамина анализировали путем итеративной подгонки кривой к логистическому уравнению с помощью программы Prism 5,00 TM (GraphPad, Inc.). Используемое уравнение является следующим:
Y=(Bottom+Top-Bottom)/(1+10^((LogEC50-X)*HillSlope)),
где X представляет собой логарифм дозы, Y представляет собой ответ, Y начинается снизу (Bottom) (общее ограничение - значение 0) и растет сигмоидально до уровня Top. При уменьшении изменений СрАД или ЧСС при более высокой(их) дозе(ах) более точную оценку Top (т.е., максимальной эффективности) получали путем переноса максимального эффекта, наблюдаемого при более низкой дозе, на все последующие более высокие дозы. Для CRC тирамина TOP ограничен значением 100. Величину эффекта прессора выражали как СрАД PC10, представляющее собой концентрацию свободной плазмы, которая приводит к изменению СрАД на 10 мм рт.ст., а величину эффекта ЧСС выражали как ЧСС PC25, представляющее собой концентрацию свободной плазмы, которая приводит к изменению ЧСС на 25 ударов в минуту. Наконец, способность ингибировать эффект тирамина выражали как Tyr EC50, представляющее собой концентрацию, вызывающую 50% ингибирование индуцированного тирамином (1 мг/кг, IV) увеличения САД.
Ингибирующие активности (значения pIC50), полученные при измерении ингибирования захвата меченого 5-НТ транспортерами серотонина у крыс (rSERT) в кортикальных синаптосомах крыс, были преобразованы в значения IC50 при помощи следующей формулы:
Y=(10^(-x))x(10^9)
С. Результаты
4-[2-(2,4,6-Трифторфеноксиметил)фенил]пиперидин (0,01-30 мг/кг, IP) приводил к дозозависимому увеличению СрАД и ЧСС у анестезированных крыс. При построении графика зависимости соответствующей концентрации свободной плазмы для каждой дозы, оцененные величины для СрАД и ЧСС составили 101,4 нМ (СрАД EC10) и 8,6 нМ (ЧСС EC25). Максимальные изменения СрАД и ЧСС составили 13,2 (4,4-22,1) мм рт.ст. и 28,1 (22,4-33,7) ударов в минуту, соответственно. 4-[2-(2,4,6-Трифторфеноксиметил)фенил]пиперидин также ингибировал вызванное тирамином увеличение САД. Предполагаемая активность 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина, выраженная в % ингибирования ответа на 1 мг/кг внутривенно введенного тирамина, составляла 0,86 нМ.
D.Заключение
В модели сердечно-сосудистой системы у анестезированных крыс было показано, что 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидин является сильным ингибитором прессорного эффекта тирамина и тахикардии, что согласуется с ингибированием NET на периферической крови.
Пример 14
Получение растворов для перорального дозирования
Растворы для перорального дозирования гидрохлорида 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина готовили в два этапа. Сначала готовили водный раствор с концентрацией 3 мг/мл, а затем пероральные растворы в отфильтрованном яблочном соке, имеющие разную дозировку, перед введением дозы.
А. Получение маточного раствора
Гидрохлорид 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина (500 мг, чистота 89,9%) добавляли в 250 мл прозрачную стеклянную бутылку. Добавляли стерильную воду для инъекций (150 мл), и колбу закрывали. Бутылку осторожно вращали круговыми движениями до образования твердого вещества (примерно 20 минут). При необходимости бутылку также можно обработать ультразвуком. Бутылку метили биркой, и маточный раствор (3 мг/мл) использовали в течение 2 часов или хранили в холодильнике при 2-8°С до использования. Любой маточный раствор, не использованный в течение 6 дней после первоначального приготовления, удаляли.
В. Получение растворов для перорального дозирования
Растворы для получения семи разных дозировок для перорального введения готовили для использования либо в Части А, либо в Части B клинических исследований. Полученные дозы и количества, использованные для приготовления каждой дозы, были такими, как показано в таблицах 4 и 5:
Таблица 4
Растворы для получения доз для перорального введения (Часть А)
Таблица 5
Растворы для получения доз для перорального введения (Часть B)
Для приготовления растворов для перорального введения из холодильника вынимали маточный раствор (3 мг/мл), который визуально оценивали на наличие осадков. Если осадок присутствовал, маточный раствор готовили повторно.
Яблочный сок (не менее 40 мл, 100% оригинальный яблочный сок Mott) набирали в шприц объемом 50 мл, и к шприцу прикрепляли шприцевой фильтр (25-миллиметровый PVDF-шприцевой фильтр, 0,2 мкм, Pall Life Sciences). Яблочный сок фильтровали через шприц-фильтр, отбрасывая первые 3 мл, а оставшийся яблочный сок собирали в 125 мл флакон янтарного цвета.
Затем в новую 125-мл бутылку янтарного цвета добавляли количество маточного раствора (3 мг/мл), как показано в таблицах 4 или 5 (исходный объем), и в эту же бутылку добавляли соответствующее количество отфильтрованного яблочного сока, как показано в таблице 4 или 5 (объем яблочного сока). Бутылку закрывали крышкой и содержимое перемешивали круговыми движениями в течение более 2 минут. Полученный раствор хранили при комнатной температуре до 18 часов до введения дозы пациенту. Перед введением дозы пациенту аликвоту 10 мл раствора для перорального введения переносили в новую 125 мл бутылку янтарного цвета.
Пример 15
Получение таблеток для перорального введения
Микрокристаллическую целлюлозу (4,476 кг; AVICEL Microcrystalline Cellulose, NF, Ph. Eur Type PH-112) и безводную лактозу (2,450 кг; безводная 60M NF, EP) загружали в смесительный бункер и перемешивали в течение 5 минут со скоростью 20 об/мин. Полученную смесь переносили в контейнер с двойным полиэтиленовым покрытием (ʺПремикс 1ʺ) и разделяли на две части, одна часть весила примерно 1 кг (ʺПремикс 1Аʺ), а другая часть составляла остаток (ʺПремикс 1Вʺ). Примерно половину Премикса 1А добавляли в бункер, затем добавляли 4-[2-(2,4,6-трифторфеноксиметил) фенил]пиперидина гидрохлорид (38,93 г; измельченный), и после этого добавляли вторую половину Премикса 1А. Полученную смесь перемешивали в течение 10 минут со скоростью 20 об/мин, а затем переносили в контейнер с двойным полиэтиленовым покрытием (ʺАктивный Премикс 2ʺ). Примерно половину Премикса 1В пропускали через мельницу, снабженную 1,0 мм ситом, при 1000 об/мин (800-1200 об/мин), с направленным ударным воздействием, затем добавляли Активный Премикс 2 и вторую половину Премикса 1В. Размолотые материалы собирали в контейнер с двойным полиэтиленовым покрытием (ʺИзмельченный Премикс 3ʺ). Измельченный Премикс 3 пропускали через ручное сито размером 40 меш, и просеянный материал переносили в смесительный бункер и перемешивали в течение 10 минут со скоростью 20 об/мин (ʺАктивная смесьʺ). Брали одну полную ложку активной смеси и смешивали вручную со стеаратом магния (35,0 г, NF) в полиэтиленовом пакете. Затем эту смесь пропускали через ручное сито размером 40 меш. После этого просеянную смесь добавляли обратно в смесительный бункер, и всю смесь перемешивали в течение 5 минут при 20 об/мин (ʺКонечная смесьʺ). Конечную смесь прессовали в таблетки по 200 мг, используя пресс для таблеток, и таблетки пропускали через пылеуловитель и металлоискатель. Приемлемые таблетки собирали в контейнеры с двойным полиэтиленовым покрытием.
В сосуд из нержавеющей стали для приготовления раствора добавляли очищенную воду (1,587 кг, USP), и скорость смесителя регулировали таким образом, чтобы образовался вихревой поток. В вихревой поток в смесительном резервуаре добавляли пленочное покрытие на основе поливинилового спирта (280,0 г; OPADRY II Pink 85G64744, Colorcon, Inc., West Point, PA), и скорость перемешивания уменьшали до исчезновения вихревого потока, и продолжали перемешивать в течение по меньшей мере 45 минут или до равномерного распределения цвета, согласно визуальной оценке. До и во время процесса нанесения покрытия продолжали осуществлять осторожное перемешивание. Приготовленные выше таблетки загружали в поддон для нанесения покрытия, и скорость вращения поддона устанавливали, равной на 10 об/мин. Раствор для покрытия закачивали в поддон для нанесения покрытия. При закачке в поддон раствора для покрытия контролировали изменение его массы, и раствор для покрытия прекращали добавлять, когда масса высушенной таблетки увеличивалась на 4% за счет использования суспензии для покрытия. Полученные розовые таблетки содержали примерно 1 мг 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина (эквивалент свободного основания).
Желтые таблетки, содержащие примерно 5 мг 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина (эквивалент свободного основания), также готовили, используя аналогичную процедуру и следующие количеств материалов: микрокристаллическая целлюлоза (4320 кг); безводная лактоза (2450 кг); 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина гидрохлорида (194,7 г); стеарат магния (35,0 г); очищенная вода (1587 кг); и пленочное покрытие на основе поливинилового спирта (280,0 г; OPADRY II Yellow 85G620027).
Пример 16
Клиническое исследование субъектов с нОГ
Фазу 2 клинических исследований проводят, используя 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина гидрохлорид (ʺИсследуемое соединениеʺ). Исследование представляет собой многоцентровое, рандомизированное, состоящее из двух частей, одностороннее (Часть A) и двойное слепое (Часть B) изучение исследуемого соединения относительно плацебо у субъектов с нОГ. Также проводят открытое расширенное исследование для оценки эффективности, безопасности и переносимости исследуемого соединения (часть C). Часть А соответствует схеме приема суточной разовой возрастающей дозы, начиная с плацебо в день 1, затем 1 мг исследуемого соединения в день 2, с последующим ежедневным увеличением дозы исследуемого соединения до максимальной дозы, равной 10 мг, исходя из безопасности, переносимости, для определения прессорного эффекта. Часть B соответствует рандомизированному, плацебо-контролируемому исследованию в параллельных группах для оценки дозы, которая, как было определено, в целом является хорошо переносимой и имеет прессорный эффект у заданного субъекта из Части A. Часть C - это открытое расширенное исследование для оценки эффективности (хронический прессорный ответ, симптомы нОГ), безопасности и переносимости исследуемого соединения, вводимого один раз в сутки в течение до 20 недель субъектам, у которых в части А наблюдали эффект на лечение.
Субъектами, включенными в исследование, являются мужчины или женщины в возрасте от 40 до 80 лет (включительно) с диагностированной симптоматической ортостатической гипотензией, вызванной истинной вегетативной недостаточностью, множественной системной атрофией или болезнью Паркинсона (т.е., БП плюс симптомы). При скрининге проводят тестирование вегетативной функции для подтверждения диагноза вегетативной дисфункции, включая синусовую аритмию и маневр Вальсальвы. У субъектов должно(-ы): (1) наблюдаться падение САД до 30 мм рт.ст. в течение 5 минутного стояния на ногах; (2) наблюдаться нарушение вегетативных рефлексов, определяемое отсутствием овершута АД во время фазы IV маневра Вальсальвы; (3) возникать головокружение, предобморочное состояние или обморок во время стояния на ногах; и (4) отсутствовать другие идентифицируемые причины вегетативной невропатии.
А. Изучение эскалации дозы (часть А)
В части A все дозы (исследуемое соединение и плацебо) вводят слепым методом (т.е. субъект остается маскированным) с использованием раствора для перорального введения. В части А принимают участие до 30 субъектов. За два дня до начала части А субъектов помещают в центр клинических исследований (CRC), где они остаются в течение всего периода эскалации дозы. В первый день субъекты получают одну дозу плацебо. На второй день субъекты получают одну 1 мг дозу исследуемого соединения. В каждый последующий день субъекты получают одну увеличивающуюся дозу исследуемого соединения: 2,5 мг в день 3; 5 мг в день 4; 10 мг в день 5, если отсутствуют следующие заданные критерии остановки лечения:
(a) определение исследователем (в сотрудничестве со спонсором) того, что введение следующей дозы может быть сопряжено с проблемой соблюдения безопасности;
(b) САД ≥ 180 мм или ДАД ≥ 110 мм в сидячем положении повторяются более 2 раз в течение часа;
(с) недопустимые побочные эффекты, определенные исследователем; или же
(d) субъект получает максимальную дозу исследуемого препарата, указанную в протоколе.
В случае возникновения у субъекта того или иного события, удовлетворяющего любому из вышеперечисленных критериев, дальнейшая эскалация дозы не проводится. По окончании наблюдения в клинике после введения доз субъекта выписывают из CRC.
Субъекты, у которых не наблюдалось клинически значимое увеличение САД по результатам определения исследователя (в сотрудничестве со спонсором), или участие которых в исследовании лекарственного средства было остановлено до достижения критериев остановки лечения, не участвуют в части B исследований. Для участия в части B исследований не требуется, чтобы субъекты получили все четыре дозы исследуемого соединения. Все субъекты, которые получили по меньшей мере одну дозу исследуемого соединения, которая обеспечила эффективное увеличение АД в сидячем положении, и которая считается хорошо переносимой, имеют право на участие в части B и/или части C исследований.
В. Период выведения препарата из организма
После завершения части А субъекты проходят период выведения лекарственного препарата из организма (период вымывания) (минимум 8 дней, но не более 36 дней). Ежедневно в течение первых 72 часов после выписки, а затем по меньшей мере еженедельно в течение оставшейся части периода вымывания, исследователь или назначенное им лицо связывается с субъектом по телефону для проверки состояния здоровья субъекта. Любые нежелательные явления, сообщаемые по телефону, регистрируются и отслеживаются в соответствии с медицинскими показаниями, установленными исследователем. Под руководством исследователя субъекты отслеживают свои симптомы нОГ во время периода вымывания, используя другие прописанные лекарственные средства.
С. Рандомизированное, двойное слепое исследование в параллельных группах (часть B)
После периода вымывания субъекты могут вернуться в CRC за два дня до начала части B, где их распределяют по группам для получения одной дозы либо плацебо, либо исследуемого соединения. Назначение лечения в Части B является двойным слепым с использованием исследуемого соединения в виде раствора для перорального введения, приготовленного фармацевтом в CRC без маскирования. В части B, при сотрудничестве исследователя и спонсора, субъекты получают дозу исследуемого соединения, которая достигает пикового воздействия сопоставимого с дозой, определенной в части A, для обеспечения эффективного увеличения АД в сидячем положении, и которая считается хорошо переносимой. Индивидуализированную дозу, выбранную для части B, определяют путем фармакокинетического моделирования для достижения сопоставимого воздействия исследуемого соединения, соответствующего конечной дозе, вводимой в части A, как показано в таблице 6.
Таблица 6
Определение дозы для части В
D. Открытое клиническое исследование (часть С)
В части C исследуемое соединение находится в виде 1 мг или 5 мг таблеток, упакованных в 40 бутылок из полиэтилена высокой плотности с этикетками. В 1-й день пациент получает дозу, равную самой высокой дозе, которая оказалась хорошо переносимой в части А. Во 2-й день и далее пациент получает дозу, равную 50% от дозы, вводимой в 1-й день части С (с округлением до ближайшего 1 мг). Например, если самая высокая доза в Части A составила 10 мг/сутки, то в День 1 части C пациент получает 10 мг, а в День 2 и далее пациент получает 5 мг/сутки. Аналогично, если самая высокая доза в части А составила 5 мг/сутки, то в День 1 части С пациент получает 5 мг, а в День 2 и далее пациент получает 3 мг/сутки. Если у пациента развивается или происходит ухудшение ранее наблюдаемой гипертензии в супинированном положении (САД ≥ 180 мм рт.ст. или ДАД ≥ 110 мм рт.ст.) или других нежелательных явлений, предполагающих непереносимость, введение дозы прекращают в течение 3 дней и возобновляют на уровне дозы, равной 50% (округляется до ближайшего 1 мг) предыдущей дозы. Если после уменьшения дозы гипертензия в супинированном положении или другие нежелательные явления сохраняются, такому пациенту прекращают введение дозы и назначают соответствующую альтернативную терапию и уход, определяемый главным исследователем. Кроме того, если снижение дозы происходит, когда пациент находится дома, такой пациент доставляется в клинику в пределах 2 недель от момента снижения дозы.
Е. Оценка исследований
1. Оценка эффективности
Оценку эффективности проводят на основании анализа систолического артериального давления в сидячем положении и стоя, а также заполненного опросника по оценке симптомов ортостатической гипотензии (OHSA). В части C используется амбулаторный мониторинг артериального давления, по возможности, путем измерения артериального давления каждые 2 часа в течение 24-часового периода перед введением дозы в первый день; и за 72-48 часов до посещения клиники в день 15, день 29, день 85 и день 169. Кроме того, артериальное давление измеряют за 20-30 минут до еды каждое утро при нахождении в супинированном положении (под углом 30 градусов) не менее 10 минут.
2. Оценка безопасности
Оценку безопасности проводят по наличию неблагоприятных явлений, результатам клинических лабораторных анализов (включая гематологию, химию сыворотки и анализ мочи), показателям жизнедеятельности, ЭКГ в 12 отведениях, использованию сопутствующих лекарств и результатам физического обследования.
3. Оценка фармакокинетических характеристик (РК)
В части A для оценки концентрации исследуемого соединения в плазме крови образцы крови берут перед введением дозы (в течение 30 минут до введения дозы и сразу после оценки АД в положении стоя и ортостатической оценки) и снова через 6-8 часов после введения дозы сразу после оценки АД в положении стоя и ортостатической оценки дозы в дни 3 (2,5 мг) и 4 (5 мг). Один дополнительный образец для оценки ФК берут через 24 часа после введения последней дозы после достижения критериев остановки лечения или в конце последовательности эскалации дозы.
В части B образцы крови для ФК берут перед введением дозы (в течение 30 минут до введения дозы), между 6 и 8 часами после введения дозы (один раз) и через 24 часа после введения дозы. Все образцы для ФК собирают после оценки АД и ортостатической оценки в зависимости от ситуации. Для каждого сбора крови регистрируют фактическое время сбора.
В части C образцы крови собирают за 30 минут до введения дозы в дни 1 и 2 и снова через 6-9 часов после введения дозы в день 1. Образцы крови также собирают при посещении пациентом клиники.
4. Оценка фармакодинамачисеких характеристик (ФД)
В части A образцы крови для оценки ФД и дигидроксифенилгликоля (DHPG) собирают в следующие дни введения дозы: день 1 (плацебо), 3 (2,5 мг) и 4 (5 мг) два раза перед введением дозы (в пределах 30 минут до введения дозы и сразу после каждой оценки АД в положении лежа и стоя и ортостатической оценки) и через 6-8 часов после введения дозы (сразу после оценки АД в положении стоя и ортостатической оценки). Два дополнительных образца для оценки ФД берут через 24 часа после введения последней дозы (в положении лежа и стоя) после достижения критериев остановки лечения или в конце последовательности эскалации дозы.
В части B образцы крови для анализа NE и DHPG собирают в день 1 введения дозы два раза перед дозированием (за 30 минут до введения дозы и сразу после каждой оценки АД в положении лежа и стоя и ортостатической оценки) и через 6-8 часов после введения дозы (сразу после оценки АД в положении стоя и ортостатической оценки).
В части C образцы крови собирают за 30 минут до введения дозы в дни 1 и 2 и снова через 6-9 часов после введения дозы в день 1. Образцы крови также собирают при посещении пациентом клиники.
F. Конечные точки исследования
Для обеих частей A и B первичной конечной точкой исследования является отличие от плацебо систолического артериального давления в сидячем положении через 6-8 часов после введения препарата. В части А плацебо относится к визиту в день 1, а отличие от плацебо относится к согласованной по времени разнице от каждого дня введения дозы исследуемого соединения (дни со 2 по 5) относительно введения плацебо (день 1). В Части C основной конечной точкой исследования является улучшение на 4-й неделе согласно шкале Лайкерта оценки ʺголовокружения, предобморочного состояния, слабости или ощущения того, что вы можете потерять сознаниеʺ (вопрос 1 OHSA).
Вторичные конечные точки включают: изменение от плацебо в САД в положении стоя через 6-8 часов после введения дозы; улучшение по шкале Лайкерта оценки ʺголовокружения, предобморочного состояния, слабости или ощущения того, что вы можете потерять сознаниеʺ (оценка симптомов у пациента в среднем за неделю до вопроса); результаты по другим анкетам; увеличение САД в положении стоя (площадь под кривой измеряют через 1, 3, 5 и 10 мин в положении стоя); увеличение САД в сидячем положении (площадь под кривой в течение 10 часов после введения препарата); и продолжительность стояния на ногах во время ортостатического теста в положении стоя.
Предварительные результаты в части А показаны в таблице 7. В таблице 7 галочка (√) указывает на то, что связанное с дозой улучшение наблюдалось при введении пациенту 4-[2-(2,4,6-трифторфеноксиметил)фенил]пиперидина гидрохлорида относительно плацебо. Все пациенты в таблице 7 получали в Части А максимальную дозу, равную 10 мг/сутки, за исключением Пациента № 5, который Части А получал максимальную дозу, равную 5 мг/сутки.
Таблица 7
Предварительные результаты исследования - Часть А
1 MSA=множественная системная атрофия
PAF=истинная вегетативная недостаточность
БП+=болезнь Паркинсона плюс симптомы нОГ
2√=наблюдаемое связанное с дозой улучшение по сравнению с плацебо.
Данные в таблице 7 демонстрируют, что у 10 из 12 пациентов (83%), завершивших часть A, наблюдалось улучшение по сравнению с плацебо, по меньшей мере, в одном из следующих параметров: (а) систолическое артериальное давление в сидячем положении; (b) время нахождения в положении стоя; или (с) ощущение головокружения или предобморочного состояния. Четыре пациента (не показаны в таблице 7) прекратили прием лекарства в части А или не прошли полную оценку из-за ранее существовавшей гипертензии в супинированном положении (3 пациента) или мерцательной аритмии (1 пациент). Из MSA пациентов, завершающих часть A, 6 из 6 пациентов (100%) показали улучшение по сравнению с плацебо по меньшей мере по одному параметру, приведенному в таблице 7.
Реферат
Группа изобретений относится к области медицины, а именно к внутренним болезням, и предназначена для лечения нейрогенной ортостатической гипотензии и ее симптомов у пациента-человека. Способ лечения указанной гипотензии и ее симптомов у пациента-человека включает введение пациенту соединения формулы I или фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение формулы I:или его фармацевтически приемлемую соль один раз в сутки в количестве от 1 мг/сут до 20 мг/сут. При этом пациент имеет множественную системную атрофию, истинную вегетативную недостаточность или болезнь Паркинсона. Введение соединения или фармацевтической композиции пациенту приводит к одному или более из: (а) увеличение систолического артериального давления у пациента в положении сидя, (b) увеличение времени нахождения пациента в положении стоя и (c) уменьшение головокружения или ощущения предобморочного состояния, испытываемого пациентом. 2 н. и 18 з.п. ф-лы, 7 табл., 16 пр.
Формула
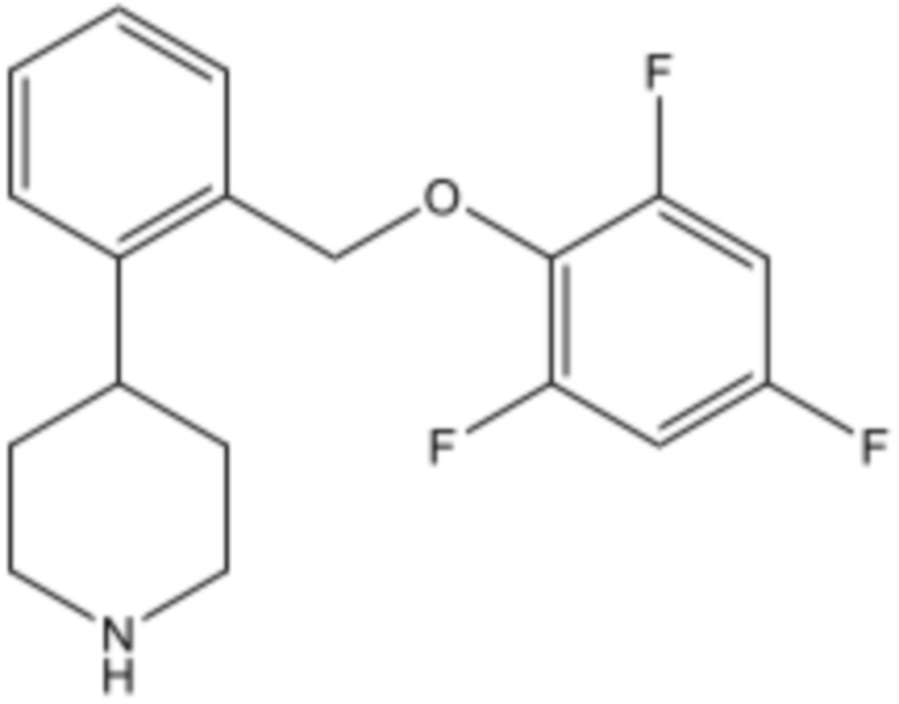
Комментарии