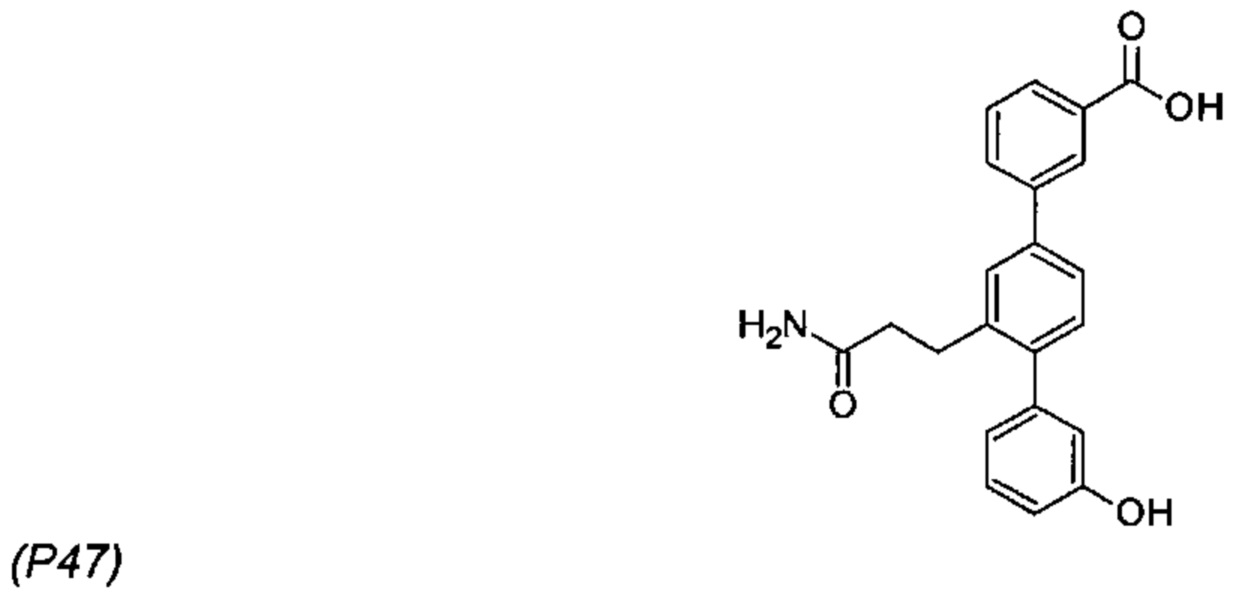
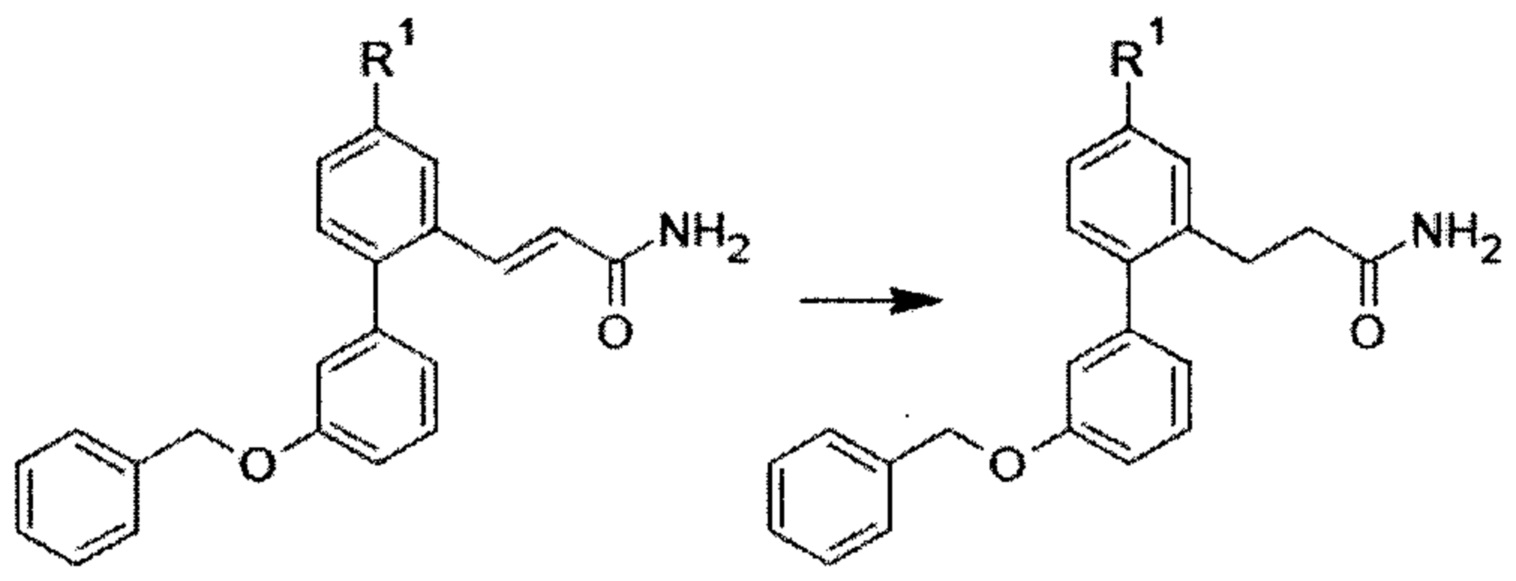
Композиции для лечения заболевания почек и/или печени - RU2712624C2
Код документа: RU2712624C2
Чертежи











Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] Настоящее изобретение относится к новым соединениям и их применению при профилактическом и/или терапевтическом лечении заболевания почек и/или печени.
[0002] Настоящее изобретение было разработано в первую очередь для профилактического и/или терапевтического лечения заболевания почек и/или печени и будет описано ниже в данном документе со ссылкой на настоящую заявку. Однако следует принимать во внимание, что настоящее изобретение не ограничено данной конкретной областью применения.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[0003] Любое обсуждение уровня техники по всему настоящему описанию никоим образом не должно рассматриваться как допущение того, что такой уровень техники широко известен или образует часть общеизвестных знаний в данной области.
[0004] Заболевание почек включает в себя различный диапазон этиологии, в том числе среди прочих иммунологические, механические, метаболические и токсические поражения (Hewitson, Fibrogenesis & Tissue Repair 2012, 5 (Suppl 1): S14). Независимо от этиологии все пациенты с хроническим заболеванием почек демонстрируют ухудшение почечной функции с течением времени, что неизбежно приводит к почечной недостаточности последней стадии - состоянию, при котором необходим пожизненный диализ или пересадка почки (Hakim & Lazarus, Am J Kidney Dis 1989, 14: 396-401). Прогрессирующая потеря почечной функции связана не только с развитием гломерулосклероза, но также с развитием интерстициального фиброза. Интерстициальный фиброз характеризуется разрушением почечных канальцев и интерстициальных капилляров, а также накоплением белков внеклеточного матрикса (Fukagawa et al., Nephrol Dial Transplant 1999, 14: 2793-2795). Фиброз почек может приводить к гипертензии вследствие увеличенного системного сосудистого сопротивления, в этом случае наблюдается гипертензия у 85-95% пациентов с хроническим заболеванием почек. (Rao et al., Am J Kidney Dis. 2008, 51 (suppl 2): S30-S37).
[0005] При лечении ингибиторами ангиотензинпревращающего фермента (АСЕ) отдельно или в комбинации с блокаторами рецепторов ангиотензина (ARB) наблюдалось замедление скорости прогрессирования почечной недостаточности, при этом они не обеспечивают лечение заболевания почек, т.е. они не обеспечивают обращение существующего фиброза и не восстанавливают нормальную структуру ткани. Кроме того, ингибиторы АСЕ и ARB могут вызывать побочные эффекты, такие как низкое артериальное давление, ангионевротический отек, гиперкалиемия и хронический сухой кашель.
[0006] Заболевание печени может быть наследственным или может быть вызвано различными факторами, которые приводят к поражению печени, такими как ожирение, диабет, инфекции или злоупотребление алкоголем. Примеры заболевания печени включают гепатит, жировую болезнь печени и цирроз.
[0007] При жировой болезни печени большие вакуоли триглицеридного жира могут накапливаться в клетках печени из-за стеатоза (т.е. аномального удерживания липидов внутри клетки). Такое накопление жира может вызывать воспаление, отмирание клеток и рубцевание.
[0008] Повреждение, вызванное жировой болезнью печени и другими заболеваниями печени, если его не лечить, приводит к накоплению фиброзной ткани, что приводит к циррозу, печеночной недостаточности и портальной гипертензии; при этом зачастую необходима пересадка печени.
[0009] Пока что не существует стандартного лечения фиброза печени. Хотя экспериментальные исследования показали мишени для предотвращения развития фиброза у грызунов, эффективность большинства способов лечения не была доказана по отношению к людям (Bataller & Brenner, J Clin Invest. 2005, 115 (2): 209-18). В настоящее время способ лечения обычно нацелен на лечение причины фиброза печени и ожидание того, что печень восстановится. Способы лечения, нацеленные на обращение фиброза, обычно являются слишком токсичными для длительного применения (например, кортикостероиды, пеницилламин) или не обладают доказанной эффективностью (например, колхицин).
[0010] Сейчас не существует фармакологической терапии накопления жира в печени.
[0011] Существует необходимость в средствах, которые предотвращают или лечат заболевание почек и/или заболевание печени. В частности, существует необходимость в средствах, которые предотвращают, ослабляют или замедляют прогрессирование фиброза почек и/или печени, уменьшают развившийся фиброз почек и/или печени, предотвращают, снижают степень или замедляют отмирание клеток почечных канальцев, восстанавливают нормальную структуру ткани в почках и/или печени и/или предотвращают, снижают степень или замедляют накопление жира в печени.
[0012] Целью настоящего изобретения является преодоление или устранение по меньшей мере одного из недостатков уровня техники или предоставление полезной альтернативы.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0013] Согласно одному аспекту настоящее изобретение относится к соединению формулы:

где
А представляет собой:


каждый из R1-R9 независимо представляет собой С, N, О или S;
Q независимо выбран из С1-6алкила, галогена, С0-6алкил-карбоновой кислоты, амино, гидрокси и С1-6алкокси;
n равняется 0, 1, 2, 3 или 4; и
X представляет собой -ОН или

или его фармакологически приемлемая соль, стереоизомер, диастереомер, энантиомер, рацемат, гидрат и/или сольват,
где если X представляет собой -ОН, А не может представлять собой незамещенный фенил.
[0014] В одном варианте осуществления Q независимо выбран из -СН3, -С(O)ОН, -F, -NH2, -ОН и -ОСН3.
[0015] В одном варианте осуществления каждый из R5-R9 независимо представляет собой С или N.
[0016] В одном варианте осуществления n равняется 0, 1 или 2.
[0017] В одном варианте осуществления С0-6алкил-карбоновая кислота представляет собой карбоновую кислоту.
[0018] В одном варианте осуществления X представляет собой -ОН.
[0019] В одном варианте осуществления X представляет собой

[0020] В одном варианте осуществления соединение выбрано из









или его фармакологически приемлемой соли, глюкуронида, стереоизомера, диастереомера, энантиомера, рацемата, гидрата и/или сольвата.
[0021] В одном варианте осуществления соединение представляет собой

или его фармакологически приемлемая соль, стереоизомер, диастереомер, энантиомер, рацемат, гидрат и/или сольват.
[0022] Согласно другому аспекту настоящее изобретение относится к фармацевтической композиции, содержащей соединение по настоящему изобретению и фармацевтически приемлемое вспомогательное вещество.
[0023] Согласно другому аспекту настоящее изобретение относится к способу терапевтического лечения заболевания почек и/или печени у субъекта, включающему введение субъекту соединения или фармацевтической композиции согласно настоящему изобретению.
[0024] Согласно другому аспекту настоящее изобретение относится к способу профилактического лечения заболевания почек и/или печени у субъекта, включающему введение субъекту соединения или фармацевтической композиции согласно настоящему изобретению.
[0025] Согласно другому аспекту настоящее изобретение относится к соединению или фармацевтической композиции по настоящему изобретению для применения в способе терапевтического лечения заболевания почек и/или печени.
[0026] Согласно другому аспекту настоящее изобретение относится к соединению или фармацевтической композиции по настоящему изобретению для применения в способе профилактического лечения заболевания почек и/или печени.
[0027] Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного препарата для терапевтического лечения заболевания почек и/или печени.
[0028] Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного препарата для профилактического лечения заболевания почек и/или печени.
[0029] В одном варианте осуществления соединение, фармацевтическая композиция или лекарственный препарат по настоящему изобретению предотвращают, ослабляют или замедляют прогрессирование фиброза почек и/или печени.
[0030] В одном варианте осуществления соединение, фармацевтическая композиция или лекарственный препарат по настоящему изобретению уменьшают развившийся фиброз почек и/или печени.
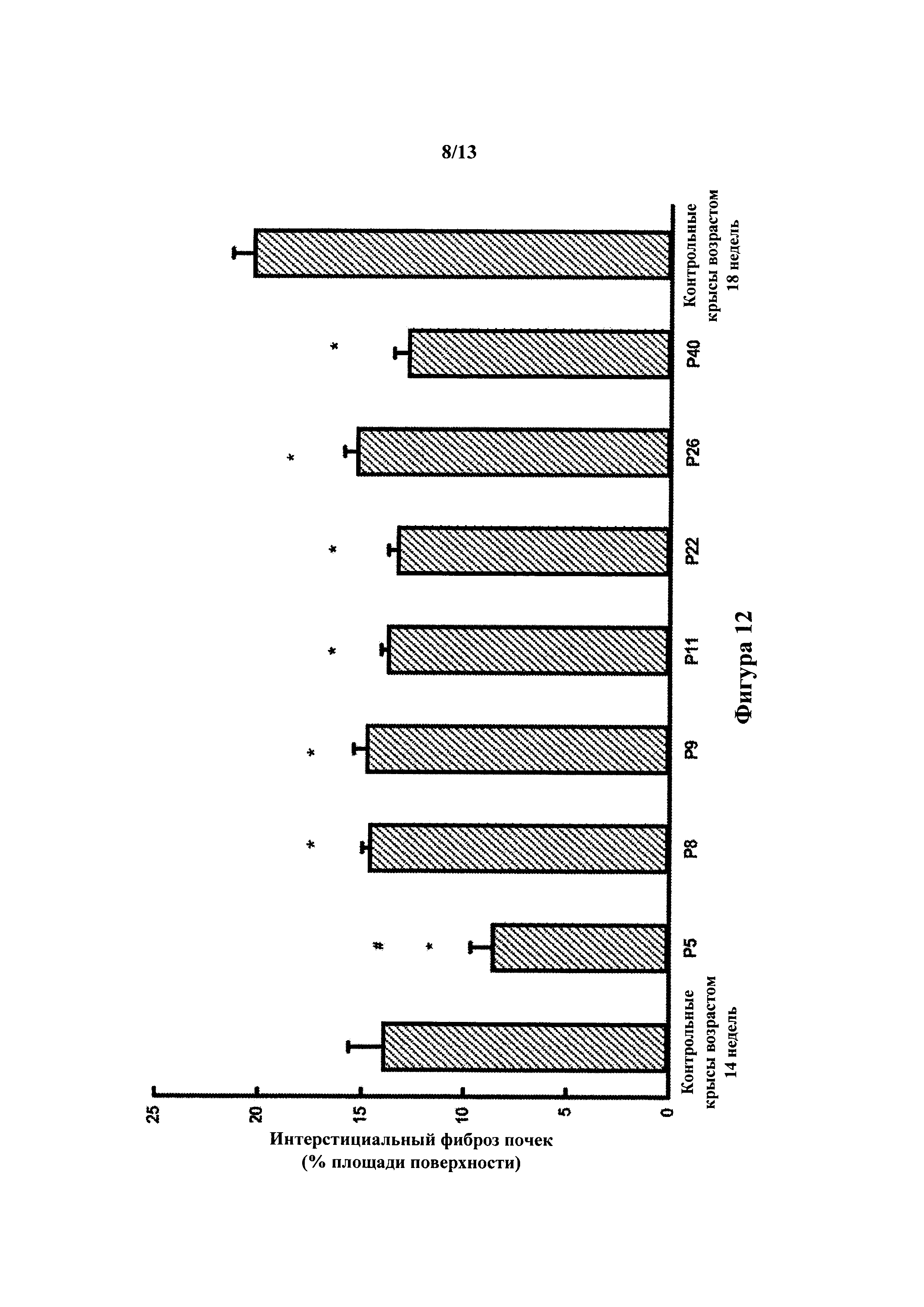
[0031] В одном варианте осуществления соединение, фармацевтическая композиция или лекарственный препарат по настоящему изобретению предотвращают, снижают степень или замедляют отмирание клеток почечного канальца.
[0032] В одном варианте осуществления соединение, фармацевтическая композиция или лекарственный препарат по настоящему изобретению предотвращают, снижают степень или замедляют накопление жира в печени.
[0033] В одном варианте осуществления соединение, фармацевтическая композиция или лекарственный препарат по настоящему изобретению восстанавливают нормальную структуру ткани в почках и/или печени.
[0034] Согласно другому аспекту настоящее изобретение относится к соединению формулы


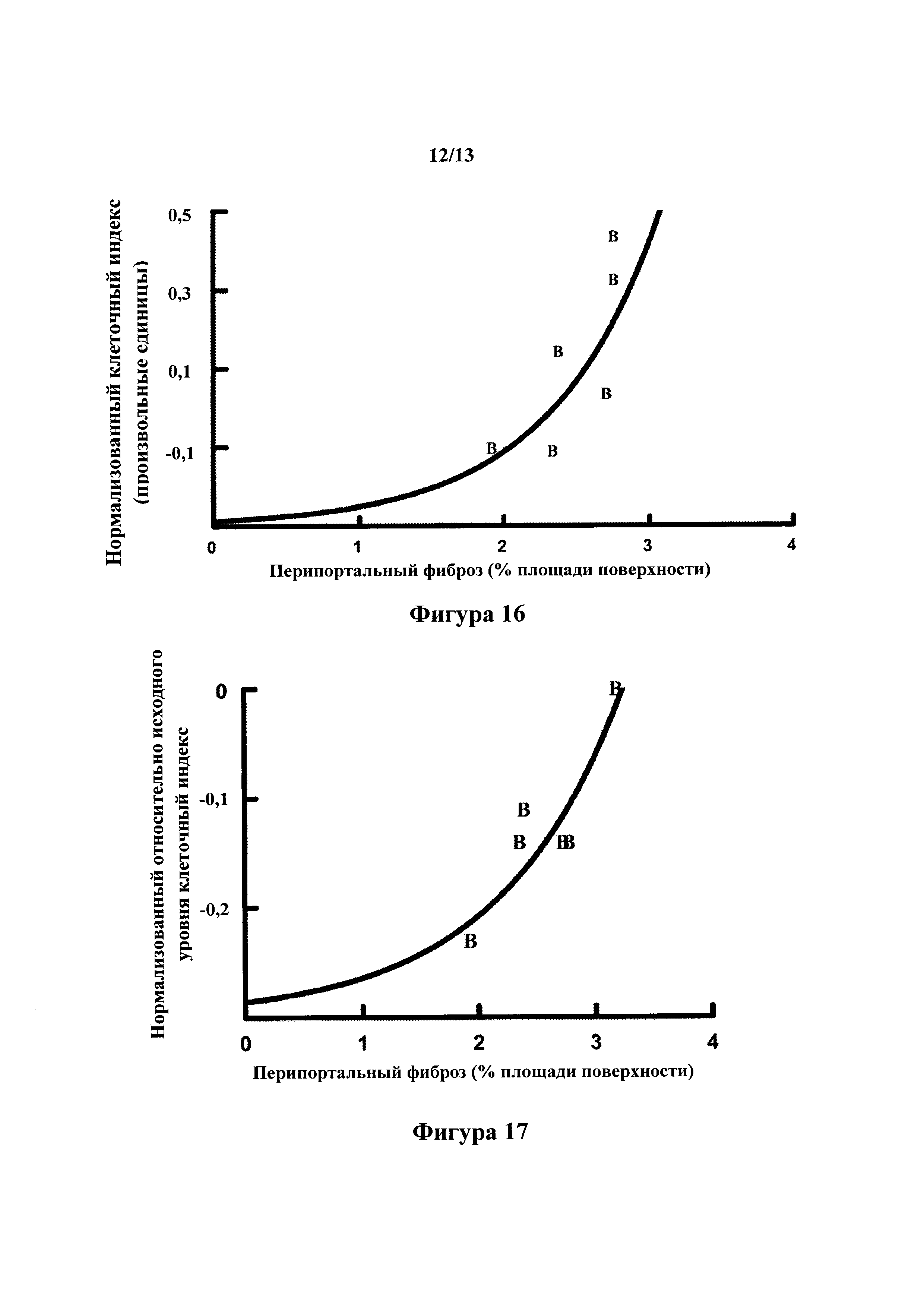
или его фармакологически приемлемая соль, глюкуронид, стереоизомер, диастереомер, энантиомер, рацемат, гидрат и/или сольват.
[0035] Если контекст явно не требует иного, по всему описанию и формуле изобретения слова "содержит", "содержащий" и тому подобное должны быть истолкованы во включающем смысле в противоположность исключающему или исчерпывающему смыслу; то есть в смысле "включая без ограничения".
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0036] Фигура 1. Схема синтеза Р5, Р8, Р11, Р22, Р26, Р40 и Р41.
[0037] Фигура 2. Схема синтеза тиазолпинаколового эфира бороновой кислоты.
[0038] Фигура 3. Схема синтеза Р9.
[0039] Фигура 4. Схема синтеза (2Е)-3-[3'-(бензилокси)-4-трифторметансульфонат-бифенил-2-ил]проп-2-енамида.
[0040] Фигура 5. Схема синтеза промежуточных соединений Р3, Р46, Р47, Р48, Р49 и Р50.
[0041] Фигура 6. Схема синтеза Р3, Р46, Р47, Р48, Р49 и Р50.
[0042] Фигура 7. Схема синтеза Р104.
[0043] Фигура 8. Клеточный импеданс гладкомышечных клеток сосудов А10, обработанных тестовыми соединениями в концентрациях, составляющих 62,5 мкМ (белые столбцы), 125 мкМ (серые столбцы) или 250 мкМ (черные столбцы).
[0044] Фигура 9. Клеточный импеданс бычьих аортальных эндотелиальных клеток, обработанных тестовыми соединениями в концентрациях, составляющих 62,5 мкМ (белые столбцы), 125 мкМ (серые столбцы) или 250 мкМ (черные столбцы).
[0045] Фигура 10. Способность тестовых соединений (30 мкМ) уберечь клетки почечных канальцев от цитотоксичности в результате лечения цисплатином (5 мкг/мл).
[0046] Фигура 11. Эффект тестовых соединений на систолическое артериальное давление.
[0047] Фигура 12. Эффект тестовых соединений на фиброз в почке у SHR на рационе с 2,2% соли после 4 недель обработки тестовым соединением в питьевом растворе с 5% этанола или только питьевым раствором.
[0048] Фигура 13. Фиброз печени у 18-недельных SHR после 4 недель обработки тестовыми соединениями (500 пмоль/кг/мин.) в питьевом растворе с 5% этанола или только питьевым раствором (контроль возрастом 18 недель).
[0049] Фигура 14. Срезы ткани, окрашенные с помощью трехцветного окрашивания по Массону, на которых видно портальные тракты контрольных крыс (А), а также крыс, обработанных Р8 (В), Р9 (С) и Р26 (D).
[0050] Фигура 15. Эффект тестовых соединений на накопление жира в печени у SHR на рационе с 2,2% соли после 4 недель обработки тестовым соединением в питьевом растворе или только питьевым раствором.
[0051] Фигура 16. Сравнение клеточного импеданса гладкомышечных клеток сосудов А10 и уровня фиброза печени у SHR, обработанных тестовыми соединениями.
[0052] Фигура 17. Сравнение клеточного импеданса бычьих аортальных эндотелиальных клеток и уровня фиброза печени у SHR, обработанных тестовыми соединениями.
[0053] Фигура 18. Сравнение степени сохранности клеток почечных проксимальных канальцев от цитотоксичности, вызванной цисплатином, и уровня фиброза почек у SHR, обработанных тестовыми соединениями.
[0054] Фигура 19. Сравнение клеточного импеданса бычьих аортальных эндотелиальных клеток и уровня содержания жира в печени у SHR, обработанных тестовыми соединениями.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0055] Настоящее изобретение относится к соединениям, эффективным при лечении заболеваний почек и/или печени. Настоящее изобретение также относится к соединениям, эффективным в предотвращении, ослаблении или замедлении прогрессирования фиброза почек и/или печени, в уменьшении развившегося фиброза почек и/или печени, в предотвращении, снижении степени или замедлении отмирания клеток почечных канальцев, в восстановлении нормальной структуры ткани в почках и/или печени и/или в предотвращении, снижении степени или замедлении накопления жира в печени.
[0056] Соединения по настоящему изобретению представлены формулой:

где
А представляет собой:


каждый из R1-R9 независимо представляет собой С, N, О или S;
Q независимо выбран из С1-6алкила, галогена, С0-6алкил-карбоновой кислоты, амино, гидрокси и С1-6алкокси;
n равняется 0, 1, 2, 3 или 4; и
X представляет собой -ОН или

или его фармакологически приемлемая соль, стереоизомер, диастереомер, энантиомер, рацемат, гидрат и/или сольват,
где если X представляет собой -ОН, А не может представлять собой незамещенный фенил.
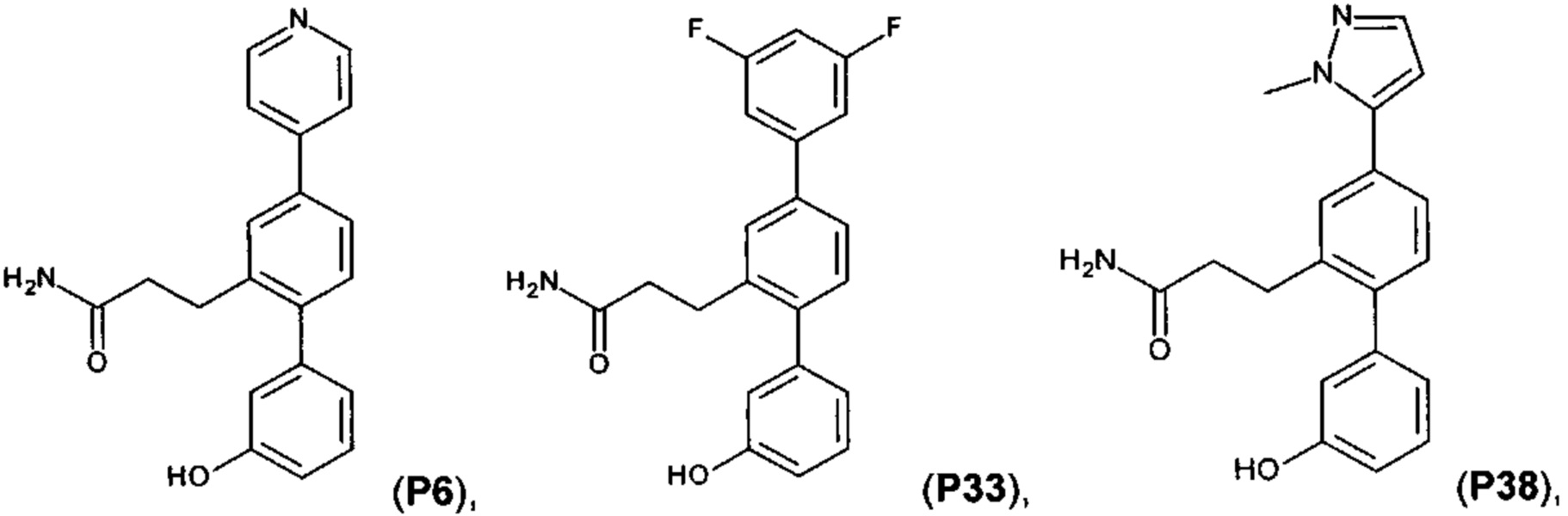
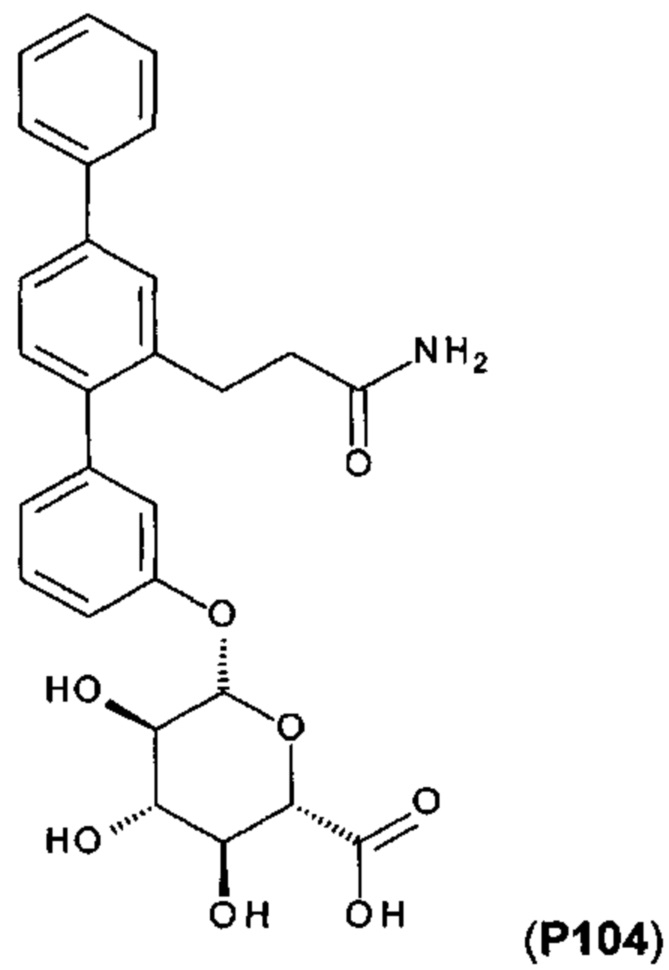
[0057] Следующие соединения являются конкретными, но не ограничивающими примерами соединений по настоящему изобретению:







[0058] Применяемый в данном документе термин "алкил", отдельно или в комбинации, означает алкильный радикал с прямой цепью или с разветвленной цепью формулы -CnH(2n+1). Примеры алкилов включают метил, этил, н-пропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изоамил, гексил, октил и т.п.
[0059] Применяемый в данном документе термин "алкокси", отдельно или в комбинации, означает алкил, связанный с кислородом, где термин "алкил" определен выше. Примеры алкокси включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси и т.п.
[0060] Применяемый в данном документе термин "галоген" означает -F, -Cl, -Br или -I.
[0061] Применяемый в данном документе термин "гидрокси" означает -ОН.
[0062] Применяемый в данном документе термин "амино" или "амин" означает -NH2.
[0063] Применяемый в данном документе термин "карбоновая кислота" означает -С(O)ОН.
[0064] Применяемый в данном документе термин "глюкуронид" включает соединения, где глюкуроновая кислота связана с соединением посредством гликозидной связи.
[0065] Применяемые в данном документе аббревиатуры Me, Et, Ph, Ms представляют собой метил, этил, фенил и метансульфонил соответственно. Более полный перечень аббревиатур, используемых специалистами в области органической химии, представлен в первом выпуске каждого тома Journal of Organic Chemistry; этот перечень обычно представлен в таблице под названием "Стандартный перечень аббревиатур". Аббревиатуры, содержащиеся в упомянутом перечне, и все аббревиатуры, используемые специалистами в области органической химии, включены в данный документ с помощью ссылки.
[0066] Соединения по настоящему изобретению могут существовать в определенных геометрических или стереоизомерных формах. Настоящее изобретение предполагает все подобные соединения, включая цис- и трансизомеры, (R) - и (S)-энантиомеры, диастереомеры, (d)-изомеры, (I)-изомеры, их рацемические смеси и другие их смеси, как попадающие в объем настоящего изобретения. Все подобные изомеры, а также их смеси предназначены для включения в настоящее изобретение.
[0067] Если, например, необходим определенный энантиомер соединения по настоящему изобретению, его можно получить путем асимметричного синтеза или путем получения производного с хиральным вспомогательным веществом, где разделяют полученную диастереомерную смесь и отщепляют вспомогательную группу с получением необходимых чистых энантиомеров. В качестве альтернативы, диастереомерные соли могут быть образованы с помощью подходящей оптически активной кислоты или основания с последующим разделением образованных таким образом диастереомеров фракционной кристаллизацией или хроматографическими средствами, хорошо известными из уровня техники, и последующим восстановлением чистых энантиомеров.
[0068] В целом, соединения по настоящему изобретению могут быть получены с помощью способов, проиллюстрированных на общих схемах реакций, как, например, описано ниже, или путем их модификаций с использованием легко доступных исходных веществ, реагентов и общепринятых методик синтеза. В данных реакциях также можно воспользоваться вариантами, которые сами по себе известны, но не упомянуты в данном документе.
[0069] Если не указано другое, способы синтеза соединения основаны на хорошо разработанных способах, описанных, например, в March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (2013) Micheal B. Smith; Advanced Organic Chemistry, Part A: Structure and Mechanisms (2008) и Advanced Organic Chemistry: Part B: Reaction and Synthesis (2010) Francis A. Carey and Richard J. Sunberg; и Greene's Protective Groups in Organic Synthesis (2014) Peter G. M. Wuts.
[0070] Настоящее изобретение также предусматривает фармацевтически приемлемые соли соединений. Термин "фармацевтически приемлемая соль" включает соли присоединения как кислоты, так и основания и относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований или кислот и которые не являются биологически или иным образом нежелательными. Фармацевтически приемлемые соли образуются с неорганическими или органическими кислотами или основаниям и могут быть получены in situ во время окончательного выделения и очистки соединений или путем отдельной реакции очищенного соединения в форме его свободного основания или кислоты с соответствующей органической или неорганической кислотой или основанием и выделения образованной таким образом соли.
[0071] В дополнение к лечению развившегося заболевания почек и/или печени соединения по настоящему изобретению можно применять профилактически для субъектов с риском развития заболевания почек и/или печени. Примеры субъектов в категории риска развития фиброза почек включают субъектов имеющих повреждение почек или хроническое заболевание почек, субъектов, имеющих диабет или получающих лекарственные средства, применяемые при химиотерапии рака (такие как даунорубицин, цисплатин), субъектов, имеющих злокачественные опухоли (такие как миелома и лимфома), генетическую предрасположенность (синдром Альпорта, поликистозное заболевание почек, рефлюкс-нефропатию), инфекции (гепатит В, гепатит С), субъектов, получающих лекарственные средства для лечения гипомании (литий), отторжения трансплантата (циклоспорин, такролимус), артритов (NSAID, пеницилламин, золото), и субъектов подверженных воздействию тяжелых металлов, таких как свинец и кадмий. Примеры субъектов в категории риска развития фиброза печени включают субъектов, имеющих гепатит А, гепатит В, гепатит С, хронический алкоголизм, аутоиммунный гепатит, первичный билиарный цирроз, первичный склерозирующий холангит, гемохроматоз, жировую болезнь печени, гепатическую энцефалопатию, накопление жира в печени, камни в желчном пузыре, рак или острое повреждение печени.
[0072] Термин "профилактический", применяемый в контексте настоящего изобретения, предназначен inter alia для охвата способов лечения, применяемых для предотвращения или замедления развития заболевания почек и/или печени у находящихся в группе риска. Субъекты, которым может быть предоставлено профилактическое лечение, уже могут иметь признаки ранней стадии почечной и/или печеночной недостаточности.
[0073] Применяемый в данном документе термин "фиброз" относится к образованию избыточной волокнистой соединительной ткани в органе или ткани.
[0074] Все органы характеризуются специфическим, но различным, расположением тканей (структурой) для нормального функционирования. Заболевание и/или отложения фиброзной ткани могут приводить к дисфункции или недостаточному функционированию органа. Таким образом, восстановление нормальной структуры ткани позволяет органам вернуть нормальную функцию.
[0075] Настоящее изобретение также предусматривает фармацевтические композиции, которые включают соединения по настоящему изобретению в сочетании с приемлемыми фармацевтическими вспомогательными веществами. Термин "фармацевтически приемлемое вспомогательное вещество", применяемый в контексте настоящего изобретения, означает любой фармацевтически приемлемый неактивный компонент композиции. Как хорошо известно из уровня техники, вспомогательные вещества включают разбавители, буферы, связующие вещества, смазывающие вещества, разрыхлители, красящие вещества, антиоксиданты/консерванты, регуляторы рН и т.д. Вспомогательные вещества выбирают, исходя из желаемых физических аспектов конечной формы: например, получение таблетки с необходимой твердостью и ломкостью, являющейся быстро диспергируемой и легко проглатываемой и т.д. Требуемая скорость высвобождения активного вещества из композиции после ее приема также играет определенную роль в выборе вспомогательных веществ. Фармацевтические композиции могут включать в себя любой тип лекарственной формы, такой как таблетки, капсулы, порошки, жидкие составы, замедленного или длительного высвобождения, пластыри, средства для вдыхания через нос, назальные спреи и тому подобное. Физическая форма и содержание предусмотренных фармацевтических композиций представляют собой традиционные препараты, которые могут быть составлены специалистом в области фармацевтических составов и основаны на хорошо установленных принципах и композициях, описанных, например, в Remington: The Science and Practice of Pharmacy, 19th Edition, 1995; British Pharmacopoeia 2000, а также аналогичных текстах и руководствах по составам.
[0076] Например, если соединения или композиции подлежат введению перорально, их можно составить в виде таблеток, капсул, гранул, порошков или сиропов; или для парентерального введения их можно составить в виде инъекций (внутривенных, внутримышечных или подкожных), препаратов для капельного вливания или суппозиториев. Для применения через слизистую оболочку глаза их можно составить в виде глазных капель или глазных мазей. Эти составы можно получить с помощью обычных средств, и, если необходимо, активный ингредиент можно смешивать с любой традиционной добавкой, такой как вспомогательное вещество, связующее вещество, разрыхляющее средство, смазывающее вещество, модификатор лекарственных средств, солюбилизирующее средство, суспендирующее вспомогательное средство, эмульгирующее средство или покрывающее средство.
[0077] Когда соединение(-ия) по настоящему изобретению вводят в виде фармацевтических препаратов человеку и животным, их можно принимать per se или в виде фармацевтической композиции, содержащей, например, от 0,1 до 99,5% (более предпочтительно от 0,5 до 90%) активного ингредиента в комбинации с фармацевтически приемлемым носителем.
[0078] Дозу соединения и частоту введения, которые следует использовать, также может легко определить практикующий врач для получения требуемой реакции.
[0079] Несмотря на то, что доза будет варьироваться в зависимости от симптомов, возраста и массы тела пациента, природы и тяжести расстройства, подлежащего лечению или предупреждению, пути введения и формы лекарственного средства, в целом суточная доза от 0,0001 мг до 200 мг соединения по настоящему изобретению может быть подходящим эффективным количеством для взрослого пациента-человека, и ее можно вводить в виде одной дозы или в виде раздельных доз.
[0080] "Пациент" или "субъект", подлежащий лечению заявленным способом, может означать человека или субъекта, не относящегося к человеку.
[0081] "Эффективное количество" заявленного соединения относительно способа лечения относится к количеству терапевтического средства в препарате, которое при применении в виде части требуемого режима дозирования обеспечивает пользу согласно клинически приемлемым стандартам лечения или профилактики определенного нарушения.
[0082] Настоящее изобретение далее будет описано более подробно со ссылкой на конкретные, но не ограничивающие примеры, в которых описаны конкретные композиции и способы применения. Однако следует понимать, что подробное описание конкретных процедур, композиций и способов включено исключительно для целей иллюстрации настоящего изобретения. В любом случае его не следует понимать в качестве ограничения широкого описания концепции изобретения, как изложено выше.
ПРИМЕРЫ
Пример 1. Синтез Р5, Р8, Р11, Р22, Р26, Р40 и Р41
[0083] Синтетический путь, применяемый для получения Р5, Р8, Р11, Р22, Р26, Р40 и Р41, показан на фигуре 1. Во-первых, 3-гидроксикоричную кислоту эстерифицировали с получением сложного эфира (1), который затем гидрировали с получением этилпропионата (2) и обрабатывали бромом с получением арилбромида (3). При реакции перекрестного сочетания Судзуки между арилбромидом (3) и 3-бензилоксифенилбороновой кислотой получали бифенил (4), который затем вводили в реакцию аминолиза с аммиаком с получением амида (5). При реакции соединения 5 с N-фенилтрифламидом получали арилтрифлат (6), который затем обрабатывали смесью тиоанизол/TFA с получением арилтрифлата (7).
[0084] После серии реакций перекрестного сочетания Судзуки между арилтрифлатом (7) и подходящими арилбороновыми кислотами/сложными эфирами получали Р5, Р8, Р11, Р22, Р26, Р40 и Р41. Результаты реакций перекрестного сочетания Судзуки между арилтрифлатом (7) и подходящими бороновыми кислотами/сложными эфирами изложены в таблице 1.

[0085] Для синтеза Р40 требовалось получить тиазолпинаколовый эфир (9) бороновой кислоты. Таким образом, 2,4-диметилтиазол бромировали с получением 5-бром-2,4-диметилтиазола (8), который в свою очередь металлировали и обрабатывали пинакол-изопропокси-эфиром бороновой кислоты с образованием тиазолпинаколового эфира 9 бороновой кислоты (фигура 2).
Получение (Е)-этил-3-(3-гидроксифенил)акрилата (1)
[0086] К перемешанному раствору (Е)-3-(3-гидроксифенил)акриловой кислоты (60,70 г, 370,0 ммоль) в этаноле (600 мл) добавляли концентрированную серную кислоту (6 мл) и реакционную смесь нагревали с применением обратного холодильника в течение 3 часов и затем при температуре окружающей среды в течение 18 часов. Этанол удаляли посредством ротационного выпаривания и остаток разделяли между водой и этилацетатом. Слои разделяли и органическую фазу промывали насыщенным раствором бикарбоната натрия и солевым раствором и концентрировали до сухого состояния. Затем горячее масло растирали с дихлорметаном и гептаном. Полученное твердое вещество собирали посредством фильтрации с получением (Е)-этил-3-(3-гидроксифенил)акрилата (1) (62,77 г, 88%) в виде бежевого твердого вещества. Т. пл. 63,8-65,2°С;1Н ЯМР (400 МГц, CDCl3) δ 7,74 (d, 1Н,3Jтpaнс 16 Гц), 7,35 (m, 1Н), 7,19 (d, 1Н, J 7,6 Гц), 7,14 (m, 1Н), 6,99 (m, 1Н), 6,51 (d,1H,3Jтpaнс 16 Гц), 5,97 (br s, 1Н), 4,38 (q, 2Н, J 7,1 Гц), 1,44 (t, 3Н, J 7,1 Гц).
Получение этил-3-(3-гидроксифенил)пропаноата (2)
[0087] (Е)-этил-3-(3-гидроксифенил)акрилат (1) (62,62 г, 326,0 ммоль) и 10% палладий на угле (50 вес.% воды) в этаноле (260 мл) перемешивали в автоклаве в атмосфере водорода при 140 фунтах на кв. дюйм в течение 1 часа в 3 партии. Объединяли 3 партии, фильтровали через целит и тщательно промывали этанолом. Фильтрат концентрировали с получением этил-3-(3-гидроксифенил)пропаноата (2) в виде бледного желто-коричневого масла (63,23 г, 100%).1Н ЯМР (400 МГц, CDCl3) δ 7,23 (m, 1Н), 6,92 (br s, 1Н), 6,85-6,80 (m, 3Н), 4,24 (q, 2Н, J 7,1 Гц), 3,00 (t, 2Н, J 7,5 Гц), 2,72 (t, 2Н, J 7,5 Гц), 1,34 (t, 3H, J 7,1 Гц).
Получение этил-3-(2-бром-5-гидроксифенил)пропаноата (3)
[0088] К тщательно перемешанной смеси 3-(3-гидроксифенил)пропаноата (2) (50,0 г, 0,258 моль) и карбоната кальция (33,5 г, 0,335 моль) в сухом DCM (500 мл) медленно добавляли бром (13,25 мл, 0,258 моль) в течение периода, составляющего 2 часа. Затем добавляли метабисульфит натрия (12,5 г, 65,79 ммоль) в воде (60 мл). Затем реакционную смесь высушивали, фильтровали и концентрировали с получением этил-3-(2-бром-5-гидроксифенил)пропаноата (3) в виде бледного желто-коричневого масла (69,27 г, 98%).1Н ЯМР (400 МГц, CDCl3) δ 7,32 (d, 1Н, J 8,6 Гц), 6,75 (d, 1Н, J 3,0 Гц), 6,58 (dd, 1Н, J 8,6, 3,0 Гц), 6,28 (s, 1Н), 4,12 (q, 2Н, J 7,2 Гц), 2,96 (t, 2Н, J 7,5 Гц), 2,62 (t, 3H, J 7,5 Гц), 1,22 (q, 3H, J 7,2 Гц).13С ЯМР(100 МГц, CDCl3) δ 174,2, 155,6, 140,6, 133,6, 117,5, 115,6, 114,3, 61,3, 34,3, 31,5, 14,2. EIMS: масса/заряд обнаруженное: М+• 272,0028, для C11H13BrO3 необходимо 272,0043. EIMS: масса/заряд 272 (М+• 5%), 193 (86), 165 (100).
Получение этил-3-(3'-бензилокси-4-гидрокси-[1,1'-бифенил]-2-ил)пропаноата (4)
[0089] Раствор этил-3-(2-бром-5-гидроксифенил)пропаноата (3) (35,0 г, 128,0 ммоль) в диметоксиэтане (650 мл) дегазировали азотом в течение 10 минут. Добавляли тетракис(трифенилфосфин)палладий(0) (3,50 г, 3,03 ммоль) и реакционную смесь перемешивали еще 15 минут. Добавляли 2 М водный раствор карбоната калия (200 мл, 0,40 ммоль) с последующим добавлением 3-бензилоксифенилбороновой кислоты (35,0 г, 154,0 ммоль). Реакционную смесь нагревали с применением обратного холодильника в течение 2 часов, затем охлаждали до температуры окружающей среды и разделяли между 2 М хлористоводородной кислотой и этилацетатом. Слои разделяли и водный слой еще раз экстрагировали этилацетатом. Объединенные органические экстракты промывали водой и солевым раствором и концентрировали с получением неочищенного продукта в виде желто-коричневого масла. Неочищенный материал предварительно абсорбировали на целит, затем хроматографировали (DCVC) с элюированием с применением градиента DCM в гептане (50-100% DCM) и затем с применением градиента этилацетата в DCM (2-6% этилацетата) с получением после концентрирования вещества в виде желтого масла (47,6 г, 99%). Его перекристаллизовывали из DCM и гептана с получением этил-3-(3'-(бензилокси)-4-гидрокси-[1,1'-бифенил]-2-ил)пропаноата (4) в виде бледно-желтого твердого вещества (38,47 г, 80%) в 3 порциях; т. пл. 85,7-87,2°С.1Н ЯМР (400 МГц, CDCl3) δ 7,43 (m, 2Н), 7,37 (m, 2Н), 7,33-7,24 (m, 2Н), 7,06 (d, 1Н, J 8,2 Гц), 6,94 (m, 1Н), 6,87 (m, 2Н), 6,75 (d, 1Н, 2,6 Гц), 6,70 (dd, 1Н, 8,2, 2,6 Гц), 5,43 (br s, 1Н), 5,07 (s, 2Н), 4,06 (q, 2Н, J 7,1 Гц), 2,86 (t, 2Н, J 8,1 Гц), 2,39 (t, 2Н, J 8,1 Гц), 1,18 (t, 3H, J 7,1 Гц).13С ЯМР (100 МГц, CDCl3) δ 173,7, 158,7, 155,4, 142,9, 139,5, 137,2, 134,4, 131,5, 129,4, 128,8, 128,1, 127,7, 122,4, 116,1, 115,9, 113,6, 113,5, 70,2, 60,8, 35,5, 28,6, 14,3. EIMS: масса/заряд обнаруженное: М+• 376,1658, для С24Н24О4 необходимо 376,1669. EIMS: масса/заряд 376 (М+•, 24%), 91 (100).
Получение 3-(3'-бензилокси-4-гидрокси-[1,1'-бифенил]-2-ил)пропанамида (5)
[0090] Этил-3-(3'-(бензилокси)-4-гидрокси-[1,1'-бифенил]-2-ил)пропаноат (4) (30,0 г, 79,80 ммоль), метанол (150 мл) и 30% водный раствор аммиака (450 мл) перемешивали при температуре окружающей среды в течение 1 недели. Полученное твердое вещество собирали посредством фильтрации. Неочищенный материал перекристаллизовывали из DCM и гептана с получением 3-(3'-(бензилокси)-4-гидрокси-[1,1'-бифенил-2-ил)пропанамида (5) в виде бесцветных квадратных пластинок (12,8 г, 46%); т. пл. 119,5-120,5°С.1Н ЯМР (400 МГц, DMSO- d6) δ 9,39 (br s, 1H), 7,46 (m, 2H), 7,39 (t, 2H, J 7,1 Гц), 7,31 (q, 2H, J 7,6 Гц), 7,23 (br s, 1H), 6,96 (m, 2H), 6,87 (m, 1H), 6,83 (d, 1H, J 7,6 Гц), 6,73 (br s, 1H), 6,71 (d, 1H, J 2,4 Гц), 6,64 (dd, 1H, J 8,2, 2,5 Гц), 5,12 (s, 2H), 2,67 (t, 2H, J 7,7 Гц), 2,21 (t, 2H, J 7,7 Гц).13C ЯМР (100 МГц, DMSO-d6) δ 175,4, 158,7, 155,9, 143,0, 139,3, 137,2, 133,9, 131,6, 129,5, 128,8, 128,2, 127,7, 122,3, 116,3, 116,2, 113,8, 113,6, 70,2, 36,8, 29,2. EIMS: масса/заряд обнаруженное: М+• 347,1515, для C22H21NO3 необходимо 347,1516. EIMS: масса/заряд 347 (М+•, 19%), 91 (100).
Получение 2-(3-амино-3-оксопропил)-3'-(бензилокси)-[1,1'-бифенил]-4-ил-трифторметансульфоната (6)
[0091] К смеси 3-(3'-(бензилокси)-4-гидрокси-[1,1'-бифенил-2-ил)пропанамида (5) (8,0 г, 21,0 ммоль) в DCM (100 мл) добавляли N-фенилтрифламид (8,21 г, 23,0 ммоль) с последующим добавлением триэтиламина (3,2 мл, 23,0 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 20 часов, затем переносили в делительную воронку, промывали водой (2x) и солевым раствором, затем концентрировали с получением желто-коричневого масла. Неочищенное масло предварительно абсорбировали на целит, затем хроматографировали (DCVC) с элюированием с применением градиента этилацетата в DCM (0-25% этилацетата). Подобные фракции объединяли и перекристаллизовывали из DCM и гептана с получением 2-(3-амино-3-оксопропил)-3'-(бензилокси)-[1,1'-бифенил]-4-ил-трифторметансульфоната (6) в виде бесцветных игл (10,73 г, 65%); т. пл. 104,0-106,0°С.1Н ЯМР (400 МГц, CDCl3) δ 7,41-7,27 (m, 6Н), 7,23 (d, 1Н, J8,2 Гц), 7,17 (d, 1Н, J2,6 Гц), 7,11 (dd, 1Н, J8,4, 2,6 Гц), 6,98 (m, 1Н), 6,82 (m, 2Н), 5,57 (br s, 1Н), 5,16 (br s, 1Н), 5,06 (s, 2Н), 2,89 (t, 2Н, J 7,9 Гц), 2,21 (t, 2Н, J 7,9 Гц).13С ЯМР (100 МГц, CDCl3) δ 173,9, 158,9, 148,9, 142,2, 141,2 (два совпадающих сигнала), 136,9, 132,0, 129,8, 128,8, 128,3, 127,7, 122,0, 121,8, 119,2, 118,9 (d, J 320.6 Гц) 115,8, 114,4, 70,2, 36,3, 28,8. EIMS: масса/заряд обнаруженное: М+• 479,1004, для

Получение 2-(3-амино-3-оксопропил)-3'-гидрокси-[1,1'-бифенил]-4-ил-трифторметансульфоната (7)
[0092] 2-(3-Амино-3-оксопропил)-3'-(бензилокси)-[1,1'-бифенил]-4-ил-трифторметансульфонат (6) (10,29 г, 22,0 ммоль) и тиоанизол (5,05 мл, 43,0 ммоль) в трифторуксусной кислоте (10 мл) перемешивали при температуре окружающей среды в колбе с притертой пробкой в течение 2 дней. Реакционную смесь охлаждали в ледяной бане, затем выливали в ледяную воду и переносили в делительную воронку. Продукт экстрагировали этилацетатом. Органическую фазу промывали водой и солевым раствором и концентрировали до сухого состояния. Неочищенный материал предварительно абсорбировали на целит, затем хроматографировали (DCVC) с элюированием с применением градиента DCM в гептане (50, 75 и 100% DCM) с последующим применением градиента метанола в DCM (1-5% метанола). Фракции, содержащие чистое вещество, объединяли и концентрировали, затем перекристаллизовывали из метанола и 1,2-дихлорэтана с получением 2-(3-амино-3-оксопропил)-3'-гидрокси-[1,1'-бифенил]-4-ил-трифторметансульфоната (7) в виде бесцветных игл (6,19 г, 74%); т. пл. 126,2-127,3°С.1Н ЯМР (400 МГц, DMSO-d6) δ 9,60 (s, 1Н), 7,42 (s, 1Н), 7,38-7,20 (m, 4Н), 6,86-6,66 (m, 4Н), 2,80 (t, 2Н, J 8,1 Гц,), 2,28 (t, 2Н, J 8,1 Гц,).13С ЯМР (100 МГц, DMSO-d6) δ 173,0, 157,3, 148,3, 142,2, 141,9, 140,7, 131,7, 129,5, 121,4, 119,6, 118,8, 116,7, 115,8, 114,6, 35,5, 28,0. EIMS: масса/заряд обнаруженное: М+• 389,0533, для

Получение 3-(3'-гидрокси-4-(пиридин-3-ил)-[1,1'-бифенил]-2-ил)пропанамида (Р5)
[0093] Смесь 2-(3-амино-3-оксопропил)-3'-гидрокси-[1,1'-бифенил]-4-ил-трифторметансульфоната (7) (0,50 г, 1,29 ммоль), пиридин-3-бороновой кислоты (0,20 г, 1,60 ммоль) и водного раствора карбоната натрия (1 М) (3,0 мл, 3,0 ммоль) в толуоле (10 мл) и этаноле (2 мл) дегазировали азотом в течение 10 минут. Добавляли тетракис(трифенилфосфин)палладий(0) (0,10 г, 0,09 ммоль) и реакционную смесь нагревали в герметизированном сосуде при 85°С до расходования исходного материала в виде трифлата. Реакционную смесь охлаждали до температуры окружающей среды, затем разделяли между 2 М хлористоводородной кислотой и этилацетатом. Слои разделяли. Органический слой проверяли посредством TLC и обнаружили очень низкое содержание требуемого продукта и его отбраковывали. Повышали основность водного слоя и повторно экстрагировали этилацетатом (2x). Объединенные органические слои промывали водой и солевым раствором и концентрировали до сухого состояния с получением твердого вещества кремового цвета (260 мг). Неочищенный материал предварительно абсорбировали на целит, затем хроматографировали (DCVC) с элюированием с применением градиента метанола в DCM (0-10% метанола). Фракции, содержащие чистый материал, объединяли и перекристаллизовывали из DCM и метанола с получением 3-(3'-гидрокси-4-(пиридин-3-ил)-[1,1'-бифенил]-2- ил)пропанамида (Р5) в виде бесцветного твердого вещества (0,09 г, 21%); т. пл. 196-198°С.1Н ЯМР (400 МГц, DMSO-d6) δ 9,55 (s, 1Н), 8,92 (m, 1Н), 8,58 (m, 1Н), 8,10 (m, 1Н), 7,67 (m, 1Н), 7,59 (dd, 1Н, J 2,0, 7,9 Гц), 7,50 (m, 1Н), 7,25 (m, 3Н), 6,78 (m, 4Н), 2,84 (m, 2Н), 2,33 (m, 2Н).13С ЯМР (100 МГц, DMSO-d6) δ 173,4, 157,1, 148,4, 147,6, 141,9, 141,3, 139,4, 136,0, 135,4, 134,0, 130,4, 129,3, 127,4, 124,3, 123,8, 119,6, 115,8, 114,1, 36,1, 28,1. EIMS: масса/заряд обнаруженное: М+• 318,1358, для C20H18N2O2 необходимо 318,1363. EIMS: масса/заряд 318 (М+•, 92%), 273 (38), 259 (100). HPLC чистота (40% ACN/H2O, 264 нм): 98,90%.
Получение 3-(3'-гидрокси-4-(тиофен-3-ил)-[1,1'-бифенил]-2-ил)пропанамида (Р8)
[0094] Получали в соответствии со способом Р5 из 2-(3-амино-3-оксопропил)-3'-гидрокси-[1,1'-бифенил]-4-ил-трифторметансульфоната (7) (0,32 г, 0,82 ммоль), тиофен-3-бороновой кислоты (0,132 г, 1,03 ммоль), тетракис(трифенилфосфин)палладия(0) (0,056 г, 0,05 ммоль) и водного раствора карбоната натрия (1 М) (2,0 мл, 2,0 ммоль) в толуоле (10 мл) и этаноле (2 мл). Неочищенный материал очищали посредством хроматографии (DCVC) с элюированием с применением градиента метанола в DCM (0-5% метанола). Фракции, содержащие чистый материал, объединяли и перекристаллизовывали из DCM и метанола с получением 3-(3'-гидрокси-4-(тиофен-3-ил)-[1,1'-бифенил]-2-ил)пропанамида (Р8) в виде бежевого твердого вещества (0,13 г, 50%); т. пл. 211-212°С.1Н ЯМР (400 МГц, DMSO-d6) δ 9,50 (s, 1Н), 7,88-7,84 (m, 1Н), 7,68-7,63 (m, 2Н), 7,59-7,54 (m, 2Н), 7,27-7,19 (m, 2Н), 7,16 (d, 1Н, J 7,9 Гц), 6,80-6,68 (m, 4Н), 2,80 (t, 2Н, J 7,9 Гц), 2,30 (t, 2Н, J 7,9 Гц).13С ЯМР (100 МГц, DMSO-d6) δ 173,4, 157,1, 142,2, 141,3, 140,2, 139,0, 134,2, 130,1, 129,3, 127,0, 126,6, 126,2, 123,7, 120,8, 119,7, 115,8, 113,9, 36,2, 28,2. EIMS: масса/заряд обнаруженное: М+• 323,0964, для

Получение 3-(4-(фуран-3-ил)-3'-гидрокси-[1,1'-бифенил]-2-ил)пропанамида (Р11)
[0095] Получали в соответствии со способом Р5 из 2-(3-амино-3-оксопропил)-3'-гидрокси-[1,1'-бифенил]-4-ил-трифторметансульфоната (7) (0,50 г, 1,29 ммоль), фуран-3-бороновой кислоты (0,18 г, 1,60 ммоль), тетракис(трифенилфосфин)палладия(0) (0,10 г, 0,09 ммоль) и водного раствора карбоната натрия (1 М) (2,5 мл, 2,50 ммоль) в толуоле (10 мл) и этаноле (2 мл). Неочищенный материал очищали посредством хроматографии (DCVC) с элюированием с применением градиента метанола в DCM (0-10% метанола) и затем перекристаллизовывали из DCM и метанола с получением 3-(4-(фуран-3-ил)-3'-гидрокси-[1,1'-бифенил]-2-ил)пропанамида (Р11) в виде бежевых стержней (0,23 г, 58%); т. пл. 191-192°С.1Н ЯМР (400 МГц, DMSO-d6) δ 9,49 (s, 1Н),8,20-8,16 (m, 1Н), 7,78-7,73 (m, 1Н), 7,58-7,54 (m, 1Н), 7,49-7,44 (m, 1Н), 7,26-7,18 (m, 2Н), 7,14 (d, 1Н, J 7,9 Гц), 6,99-6,94 (m, 1Н), 6,80-6,67 (m, 4Н), 2,79 (t, 2Н, J 8,3 Гц), 2,29 (t, 2Н, J 8,3 Гц).13С ЯМР (100 МГц, DMSO-d6) δ 173,4, 157,1, 144,3, 142,2, 140,1, 139,2, 139,0, 130,9, 130,0, 129,2, 126,0, 125,6, 123,1, 119,6, 115,8, 113,9, 108,7, 36,1, 28,1. EIMS: масса/заряд обнаруженное: М+• 307,1204, для C19H17NO3 необходимо 307,1203. EIMS: масса/заряд 307 (М+•, 100%), 248 (50). HPLC чистота (40% ACN/H2O, 265 нм): 99,33%.
Получение 3-(3'-гидрокси-4-(1Н-пиразол-4-ил)-[1,1'-бифенил]-2-ил)пропанамида (Р22)
[0096] Получали в соответствии со способом Р5 из 2-(3-амино-3-оксопропил)-3'-гидрокси-[1,1'-бифенил]-4-ил-трифторметансульфоната (7) (0,50 г, 1,29 ммоль), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-карбоксилата (0,47 г, 1,61 ммоль), тетракис(трифенилфосфин)палладия(0) (0,10 г, 0,09 ммоль) и водного раствора карбоната натрия (1 М) (3,0 мл, 3,00 ммоль) в толуоле (10 мл) и этаноле (2 мл). Неочищенный материал очищали посредством хроматографии (DCVC) с элюированием с применением градиента метанола в DCM (0-20% метанола). Материал дополнительно очищали посредством радиальной хроматографии с элюированием градиентом этилацетата в DCM (50-100% этилацетата) и затем градиентом метанола в этилацетате (1-5% метанола) с получением 3-(3'-гидрокси-4-(1H-пиразол-4-ил)-[1,1'-бифенил]-2-ил)пропанамида (Р22) в виде бежевых кристаллов (0,10 г, 26%); т. пл. 161,5-163,2°С.1Н ЯМР (400 МГц, DMSO-d6) δ 12,95 (s, 1Н), 9,44 (s, 1Н), 8,13 (br s, 1Н), 7,99 (br s, 1Н), 7,58-7,54 (m, 1Н), 7,49-7,42 (m, 1Н), 7,28-7,19 (m, 2Н), 7,10 (d, 1Н, J7.9 Гц), 6,79-6,67 (m, 4Н), 2,78 (t, 2Н, J 7,9 Гц), 2,29 (t, 2Н, J 7,9 Гц).13С ЯМР (100 МГц, DMSO-d6) δ 173,6, 157,1, 142,4, 139,0, 138,9, 136,3, 131,9, 130,1, 129,2, 125,5 (два совпадающих сигнала), 122,8, 121,0, 119,7, 115,9, 113,8, 36,2, 28,2. EIMS: масса/заряд обнаруженное: М+• 307,1314, для C18H17N3O2 необходимо 307,1315. EIMS: масса/заряд 307 (М+•, 100%), 248 (57). HPLC чистота (35% ACN/0,1% TFA, 270 нм): 99,08%.
Получение 3-(3'-гидрокси-4-(пиримидин-5-ил)-[1,1-бифенил]-2-ил)пропанамида (Р26)
[0097] Получали в соответствии со способом Р5 из 2-(3-амино-3-оксопропил)-3'-гидрокси-[1,1'-бифенил]-4-ил-трифторметансульфоната (7) (1,00 г, 2,58 ммоль), пиримидин-5-бороновой кислоты (0,40 г, 3,20 ммоль), тетракис(трифенилфосфин)палладия(0) (0,20 г, 0,18 ммоль) и водного раствора карбоната натрия (1 М) (6,0 мл, 6,00 ммоль) в толуоле (20 мл) и этаноле (4 мл). Неочищенный материал очищали посредством хроматографии (DCVC) (×2) с элюированием с применением градиента метанола в DCM (0-7,5% метанола) и затем перекристаллизовывали из метанола с получением 3-(3'-гидрокси-4-(пиримидин-5-ил)-[1,1-бифенил]-2-ил)пропанамида (Р26) в виде твердого вещества бледно-лимонного цвета (0,17 г, 20%); т. пл. 191,9-193,5°С.1Н ЯМР (400 МГц, DMSO-d6) δ 9,56 (s, 1Н), 9,20 (s, 1Н), 9,17 (s, 2Н), 7,77-7,75 (m, 1Н), 7,69-7,66 (m, 1Н), 7,32-7,22 (m, 2Н), 7,25 (br s, 1Н), 6,81-6,71 (m, 4Н), 2,87-2,80 (m, 2Н), 2,37-2,13 (m, 2Н).13С ЯМР (100 МГц, DMSO-d6) δ 173,4, 157,24, 157,18, 154.7 (два совпадающих сигнала), 142,1, 141,7, 139,6, 133,1, 132,7, 130,5, 129,3, 127,4, 124,4, 119,6, 115,8, 114,2, 36,0, 28,1. EIMS: масса/заряд обнаруженное: М+• 319,1310, для C19H17N3O2 необходимо 319,1315. EIMS: масса/заряд 319 (М+•, 70%), 274 (48), 260 (100). HPLC чистота (40% ACN/H2O, 265 нм): 99,87%.
Получение 5-бром-2,4-диметилтиазола (8)
[0098] К тщательно перемешанной смеси 2,4-диметилтиазола (23,37 г, 0,207 моль) и карбоната кальция (26,90 г, 270 ммоль) в DCM (200 мл) медленно добавляли раствор брома (11,10 мл, 217 ммоль) в DCM (100 мл). Реакцию проверяли через 3 часа посредством TLC (DCM), и она не завершилась. Для протекания реакции до завершения требовалось добавление двух порций брома (2×3,00 мл, 117,10 ммоль) в DCM (2×20 мл). К реакционной смеси медленно добавляли метабисульфит натрия (16,0 г, 84,17 ммоль) в воде (60 мл). Добавляли больше воды и реакционную смесь переносили в делительную воронку. Слои разделяли и водный слой еще раз экстрагировали с помощью DCM. Объединенные органические слои промывали 1 М раствором карбоната натрия (2х) и водой и концентрировали с получением 5-бром-2,4-диметилтиазола (8) в виде бледного желто-коричневого масла (38,40 г, 97%).1Н ЯМР (400 МГц, CDCl3) δ 2,51 (s, 3Н), 2,24 (s, 3Н).
Получение 2,4-диметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазола (9)
[0099] По каплям в течение периода, составляющего один час, добавляли раствор 5-бром-2,4-диметилтиазола (8) (5,00 г, 26,0 ммоль) и 1,2-дибромэтана (0,24 г, 1,3 ммоль) в THF (20 мл) в колбу, содержащую магниевую стружку (0,65 г, 26,8 ммоль). Реакционную смесь нагревали до 75°С в течение 4 часов, охлаждали до температуры окружающей среды, затем переносили в капельную воронку на второй реакционной колбе посредством канюли. Затем к раствору изопропилпинаколбората (5,30 мл, 26,00 ммоль) в THF (10 мл) при 0°С по каплям добавляли реактив Гриньяра. После завершения добавления реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение 20 часов. Реакционную смесь охлаждали до ~10°С и затем медленно добавляли уксусную кислоту (1,03 мл, 25,50 ммоль) с обеспечением рН 7 реакционной смеси. Растворитель удаляли посредством ротационного выпаривания, затем добавляли этилацетат и также удаляли посредством ротационного выпаривания. Неочищенное масло предварительно абсорбировали на целит, затем хроматографировали (DCVC) с элюированием с применением градиента этилацетата в гептане (0-30% этилацетата). Фракции, содержащие требуемый материал, объединяли и концентрировали с получением 2,4-диметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазола (9) в виде бледного желто-коричневого масла, которое затвердевало (1,65 г, 26%).1Н ЯМР (400 МГц, DMSO-d6) δ 2,63 (s, 3Н), 2,53 (s, 3Н), 1,26 (s, 12Н).13С ЯМР (50 МГц, DMSO-d6) δ 170,4, 163,2, 84,1, 24,9, 19,1, 17,6 (один сигнал не наблюдался). EIMS: масса/заряд обнаруженное: М+• 239,1143, для

Получение 3-(4-(2,4-диметилтиазол-5-ил)-3'-гидрокси-[1,1'-бифенил]-2-ил)пропанамида (Р40)
[0100] Получали в соответствии со способом Р5 из 2-(3-амино-3-оксопропил)-3'-гидрокси-[1,1'-бифенил]-4-ил-трифторметансульфоната (7) (1,00 г, 2,58 ммоль), 2,4-диметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазола (0,76 г, 3,20 ммоль), тетракис(трифенилфосфин)палладия(0) (0,24 г, 0,21 ммоль) и водного раствора карбоната натрия (1 М) (6,0 мл, 6,00 ммоль) в толуоле (20 мл) и этаноле (4 мл). Неочищенный материал очищали посредством хроматографии (DCVC) с элюированием с применением градиента метанола в дихлорметане (0-5% метанола) и затем перекристаллизовывали из метанола с получением 3-(4-(2,4- диметилтиазол-5-ил)-3'-гидрокси-[1,1'-бифенил]-2-ил)пропанамида (Р40) в виде желтых кристаллов (0,23 г, 26%); т. пл. 196,6-199,4°С.1Н ЯМР (400 МГц, DMSO-d6) δ 9,54 (s, 1Н), 7,38 (s, 1Н), 7,32-7,19 (m, 4Н), 6,81-6,69 (m, 4Н), 2,80 (t, 2Н, J 7,8 Гц), 2,63 (s, 3Н), 2,42 (s, 3Н), 2,27 (t, 2Н, J 7,8 Гц).13С ЯМР (100 МГц, DMSO-d6) δ 173,3, 162,5, 157,2, 146,7, 141,8, 140,8, 139,2, 130,7, 130,4, 130,1, 129,3, 129,1, 126.2, 119,6, 115,8, 114,1, 35,9, 27,9, 18,7, 16,0. EIMS: масса/заряд обнаруженное: М+• 352,1230, для

Получение 3-(4-(3,5-диметилизоксазол-4-ил)-3'-гидрокси-[1,1'-бифенил]-2-ил)пропанамида (Р41)
[0101] Получали в соответствии со способом Р5 из 2-(3-амино-3-оксопропил)-3'-гидрокси-[1,1'-бифенил]-4-ил-трифторметансульфоната (7) (0,50 г, 1,29 ммоль), 3,5-диметилизоксазол-4-бороновой кислоты (0,23 г, 1,60 ммоль), тетракис(трифенилфосфин)палладия(0) (0,10 г, 0,09 ммоль) и водного раствора карбоната натрия (1 М) (2,5 мл, 2,50 ммоль) в толуоле (10 мл) и этаноле (2 мл). Неочищенный материал очищали посредством хроматографии (DCVC) с элюированием с применением градиента метанола в DCM (0-5% метанола) и затем перекристаллизовывали из DCM и метанола с получением 3-(4-(3,5-диметилизоксазол-4-ил)-3'-гидрокси-[1,1'-бифенил]-2-ил)пропанамида (Р41) в виде твердого вещества бледно-лимонного цвета (0,15 г, 33%); т. пл. 203-204°С.1Н ЯМР (400 МГц, DMSO-d6) δ 9,52 (s, 1Н), 7,32-7,30 (m, 1Н), 7,28-7,20 (m, 4Н), 6,81-6,70 (m, 4Н), 2,52-2,49 (m, 2Н), 2,44 (s, 3Н), 2,32-2,25 (m, 2Н), 2,27 (s, 3Н).13С ЯМР (50 МГц, DMSO-d6) δ 173,3, 165,1, 158,2, 157,1, 142,0, 140,5, 139,0, 130,0, 129.3, 129,2, 128,8, 126,2, 119,6, 115,8, 115,7, 114,0, 35,9, 27,9, 11,4, 10,6. EIMS: масса/заряд обнаруженное: М+• 336,1459, для C19H17N3O2 необходимо 336,1468. EIMS: масса/заряд обнаруженное: М+• 336,1459, для C20H20N2O3 необходимо 336,1468. EIMS: масса/заряд 336 (М+• 86%), 292 (100). HPLC чистота (40% ACN/H2O, 275 нм): 97,36%.
Пример 2. Синтез Р9
[0102] Синтетический путь, применяемый для получения Р5, Р8, Р11, Р22, Р26, Р40 и Р41, показан на фигуре 3. Вкратце, арилтрифлатный сложный эфир (11) получали из бифенилового сложного эфира (4) посредством осуществления реакции с N-фенилтрифламидом с получением защищенного арилтрифлата (10) с последующей обработкой смесью тиоанизол/TFA. Затем в результате катализируемой палладием реакции перекрестного сочетания между арилтрифлатным сложным эфиром (11) и пирролом получали требуемое терарильное соединение (12), которое подвергали аминолизу с получением Р9.
Получение этил-3-(3'-(бензилокси)-4-(((трифторметил)сульфонил)окси)-[1,1'-бифенил]-2-ил)пропаноата (10)
[0103] Получали в соответствии со способом, применяемым для получения соединения 6, из этил-3-(3'-(бензилокси)-4-гидрокси-[1,1'-бифенил]-2-ил)пропаноата (4) (8,0 г, 21,00 ммоль), N-фенилтрифламида (8,21 г, 23,00 ммоль) и триэтиламина (3,2 мл, 23,00 ммоль) в (100 мл). Неочищенный материал очищали посредством его пропускания через пробку из силикагеля с элюированием с получением этил-3-(3'-(бензилокси)-4-(((трифторметил)сульфонил)окси)-[1,1'-бифенил]-2-ил)пропаноата (10) в виде желтого масла (количественный выход) с достаточной чистотой для применения на следующей стадии.1Н ЯМР (400 МГц, DMSO-d6) δ 7,46-7,23 (m, 7Н), 7,20-7,11 (m, 2Н), 7,02-6,97 (m, 1Н), 6,88-6,83 (m, 2Н), 5,08 (s, 2Н), 4,06 (q, 2Н, J 7,2 Гц), 2,90 (t, 2Н, J 7,9 Гц), 2,39 (t, 2Н, J 7,9 Гц), 1,18 (t, 3H, J 7,2 Гц).13С ЯМР (100 МГц, DMSO-d6) δ 172,6, 158,9, 148,9, 142,4, 141,2, 141,1, 137,0, 132,0, 129,8, 128,8, 128,3, 127,7, 123,7, 121,9, 121,8, 119,1, 115,8, 114,4, 70,3, 60,8, 34,9, 28,4, 14,3. EIMS: масса/заряд обнаруженное: М+• 508,1160, для C25H23F3O632S необходимо 508,1162. EIMS: масса/заряд 508 (М+•, 10%), 91 (100).
Получение этил-3-(3'-гидрокси-4-(((трифторметил)сульфонил)окси)-[1,1'-бифенил]-2-ил)пропаноата (11)
[0104] Получали в соответствии со способом, применяемым для получения соединения 7, из этил-3-(3'-(бензилокси)-4-(((трифторметил)сульфонил)окси)-[1,1'-бифенил]-2-ил)пропаноата (10) (10,67 г, 21,0 ммоль) и тиоанизола (5 мл, 42,62 ммоль) в TFA (10 мл). Неочищенный материал предварительно абсорбировали на целит, затем хроматографировали (DCVC) с элюированием с применением градиента DCM в гептане (50-100% DCM) с последующей рекристаллизацией из DCM и гептана с получением этил-3-(3'-гидрокси-4-(((трифторметил)сульфонил)окси)-[1,1'-бифенил]-2-ил)пропаноата (11) в виде бесцветных призм (4,84 г, 55%); т. пл. 90,8-91,9°С.1Н ЯМР (400 МГц, DMSO-d6) δ 7,30-7,23 (m, 2Н), 7,19-7,11 (m, 2Н), 6,87-6,78 (m, 2Н), 6,76-6,73 (m, 1Н), 5,73 (s, 1Н), 4,07 (q, 2Н, J 7,2 Гц), 2,95 (t, 2Н, J 7.9 Гц), 2,44 (t, 2Н, J 7.9 Гц), 1,19 (t, 3H, J 7,2 Гц).13С ЯМР (100 МГц, DMSO-d6) δ 173,0, 155,9, 148,9, 142,2, 141,3, 140,9, 132,0, 130,0, 121,8, 121,6, 119,0 (q, J=321,2 Гц) 119,2, 116,2, 115,0, 61,1, 35,1, 28,5, 14,3. EIMS: масса/заряд обнаруженное: М+• 418,0690, для

Получение этил-3-(3'-гидрокси-4-(1Н-пиррол-1-ил)-[1,1'-бифенил]-2-ил)пропаноата (12)
[0105] Высушенный в сушильной камере сосуд для нагревания под воздействием микроволнового облучения (2-5 мл), содержащий 1,4-диоксан (4,5 мл), дегазировали в течение 10 мин., после чего в него добавляли Pd2(dba)3 (0,07 ммоль, 66 мг), 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил (DavePhos) (0,07 ммоль, 28 мг) и K3PO4 (1,1 ммоль, 228 мг) и обеспечивали перемешивание в течение 20 минут. Затем добавляли этил-3-(3'-гидрокси-4-(((трифторметил)сульфонил)окси-[1,1'-бифенил]-2-ил)пропаноат (11) (300 мг, 0,72 ммоль) и пиррол (4,30 ммоль, 298 мкл), сосуд герметизировали и реакционную смесь нагревали при 100°С в течение 16 ч. Растворитель выпаривали и остаток фильтровали через небольшую колонку из силикагеля с элюированием смесью 3:7 этилацетат: РЕ. После выпаривания растворителя остаток очищали посредством DCVC с элюированием смесью 5:95 этилацетат: РЕ→1:4 этилацетат: РЕ с получением этил-3-(3'-гидрокси-4-(1H-пиррол-1-ил)-[1,1'-бифенил]-2-ил)пропаноата (12) в виде желтого масла (159 мг, 72%).1Н ЯМР (400 МГц, MeOH-d4) δ 7,39 (m, 1Н), 7,36 (dd, 1Н, J 8,4, 2,4 Гц), 7,30-7,18 (m, 4Н), 6,84-6,72 (m, 3Н), 6,28 (m, 2Н), 4,02 (q, 2Н, J7,1 Гц), 2,97 (m, 2Н), 2,45 (m, 2Н), 1,14 (t, 3H, J7,1 Гц).13С ЯМР(100 МГц, MeOH-d4) δ 174,7, 158,6, 143,7, 141,4, 140,9, 140,8, 132,4, 130,6, 121,8, 121,6, 120,1, 118,9, 117,3, 117,1, 111,5, 61,7, 36,2, 29,8, 14,6. EIMS: масса/заряд обнаруженное: М+• 335,1510, для C21H21NO3 необходимо 335,1516. EIMS: масса/заряд 335 (М+•, 100%).
Получение 3-(3'-гидрокси-4-(1Н-пиррол-1-ил)-[1,1'-бифенил]-2-ил)пропанамида (Р9)
[0106] Этил-3-(3'-гидрокси-4-(1Н-пиррол-1-ил)-[1,1'-бифенил]-2-ил)пропаноат (12) (145 мг, 0,43 ммоль) растворяли в метаноле (4 мл), к которому добавляли 30% водный раствор аммиака (2,5 мл) и обеспечивали перемешивание реакционной смеси при комнатной температуре в течение 16 ч. Затем добавляли дополнительное количество аммиака (1 мл) с последующим добавлением (1 мл) через 24 ч. После непрерывного перемешивания в течение дополнительных 16 ч. добавляли этилацетат (20 мл) и воду (20 мл). Смесь разделяли, высушивали органическую фазу и выпаривали растворитель. Остаток растворяли в горячем метаноле, добавляли обесцвечивающий уголь и реакционную смесь фильтровали через теплую фильтровальную бумагу с получением прозрачного раствора. Через некоторое время образовывалось твердое вещество, которое собирали и промывали холодным метанолом с получением 3-(3'-гидрокси-4-(1H-пиррол-1-ил)-[1,1'-бифенил]-2-ил)пропанамида (Р9) в виде белых кристаллов (81 мг, 61%); т. пл. 221-224°С.1Н ЯМР (400 МГц, DMSO-d6) δ 9,52 (br s, 1Н); 7,50 (m, 1Н), 7,42 (dd, 1Н, J 2,4, 8,4 Гц), 7,36 (m, 2Н), 7,25-7,19 (m, 3Н), 6,79-6,69 (m, 4Н), 6,28 (m, 2Н), 2,80 (m, 2Н), 2,31 (m, 2Н).13С ЯМР (100 МГц, DMSO-d6) δ 173,3, 157,1, 141,7, 140,1, 139,0, 138,5, 130,7, 129,3, 119,7 (два совпадающих сигнала), 118,9, 116,9, 115,9, 114,0, 110,4, 35,9, 28,2. EIMS: масса/заряд обнаруженное: М+• 306,1355, для C19H18N2O2 необходимо 306,1363. EIMS: масса/заряд 306 (М+•, 28%), 288 (100). HPLC чистота (35% ACN/0,1% TFA, 270 нм): 99,33%.
Пример 3. Синтез промежуточного соединения (2Е)-3-[3'-(бензилокси)-4-трифторметансульфонат-бифенил-2-ил]проп-2-енамида
[0107] Синтетический путь, применяемый для получения промежуточного соединения (2Е)-3-[3'-(бензилокси)-4-трифторметансульфонат-бифенил-2-ил]проп-2-енамида, показан на фигуре 4.
Получение (2Е)-3-(2-бром-5-гидроксифенил)проп-2-еновой кислоты
[0108] К смеси 4-бром-3-формилфенола (25,0 г, 0,124 моль) и малоновой кислоты (15,53 г, 0,149 моль) в пиридине (150 мл) добавляли пиперидин (1,47 мл) и нагревали до температуры флегмы в течение 4 ч. Реакционную смесь быстро охлаждали перед добавлением хлористоводородной кислоты (2 М, 500 мл) и подкисляли до рН 1-2 концентрированной хлористоводородной кислотой (33%, прибл. 50-100 мл). Суспензию охлаждали до приблизительно 10°С и твердое вещество собирали вакуумной фильтрацией, промывая хлористоводородной кислотой (2 М, 60 мл), и высушивали под вакуумом в течение 18 ч. Данный неочищенный материал содержал воду и пиридина гидрохлорид, как показано с помощью1Н ЯМР , и его переносили в этилацетат (1,3 л) и промывали хлористоводородной кислотой (2 М, 2×750 мл), высушивали над сульфатом магния и фильтровали. Фильтрат концентрировали до сухого состояния с получением указанного в заголовке соединения в виде серого порошка (23,63 г, 0,0972 моль, 78%).1Н ЯМР (400 МГц, DMSO-d6) d ppm 12,62 (br. s., 1H) 9,91 (s, 1H) 7,76 (d, J=16,0 Гц, 1H) 7,48 (d, J=8,6 Гц, 1H) 7,19 (d, J=2,3 Гц, 1H) 6,80 (dd, J=8,8, 2,5 Гц, 1H) 6,41 (d, J=16,0 Гц, 1H); HPLC (градиент вода/ACN+0,1% TFA) 98,9% при 220 нм; LCMS [М+Н]+=242,9, [М-Н]-=242,0. прибл. 2-5 мол. % неизвестных примесей, как показано с помощью1Н ЯМР-анализа.
Получение (2Е)-3-(2-бром-5-гидроксифенил)проп-2-енамида
[0109] В течение 10 мин. добавляли оксалилхлорид (16 мл, 0,19 моль) к суспензии (2Е)-3-(2-бром-5-гидроксифенил)проп-2-еновой кислоты (23,50 г, 0,0967 моль) в дихлорметане (200 мл) и диметилформамиде (0,5 мл) при 0°С. Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 1 ч. Добавляли дополнительное количество оксалилхлорида (16 мл, 0,19 моль) и нагревали до температуры флегмы в течение 5 ч. и затем перемешивали в течение 16 ч. при комнатной температуре. Реакционную смесь концентрировали до сухого состояния с получением неочищенного промежуточного соединения в виде хлорангидрида.
[0110] Неочищенный хлорангидрид растворяли в 1,4-диоксане (100 мл) и выливали в водный раствор аммиака (28%, 68 мл, 1,12 моль) в 1,4-диоксане (200 мл). Данную смесь перемешивали в течение 30 мин. перед разбавлением реакционной смеси водой (500 мл). Реакционную смесь концентрировали до сухого состояния с получением твердого вещества серого цвета. Твердое вещество серого цвета суспендировали в хлористоводородной кислоте (1 М, 200 мл) и собирали вакуумной фильтрацией, промывали хлористоводородной кислотой (1 М, 60 мл) и водой (60 мл), затем высушивали на роторном испарителе (70°С) в течение 45 мин. и затем под высоким вакуумом в течение 4 ч. с получением неочищенного указанного в заголовке соединения (30,51 г) в виде серого порошка, содержащего неизвестную примесь, как показано с помощью1Н ЯМР-анализа. Порцию данного материала (29,6 г) перемешивали в этилацетате (500 мл) и фильтровали, промывая осадок на фильтре этилацетатом (200 мл). Фильтраты концентрировали до сухого состояния с получением указанного в заголовке соединения в виде светло-коричневого порошка (21,73 г, 98%).1Н ЯМР (400 МГц, DMSO-d6) d 9,89 (s, 1Н) 7,63 (br. s., 1H) 7,59 (d, J=15,7 Гц, 1H) 7,45 (d, J=9,0 Гц, 1H) 7,23 (br. s., 1H) 7,06 (d, J=2,7 Гц, 1H) 6,76 (dd, J=8,6, 2,7 Гц, 1H) 6,53 (d, J=15,7 Гц, 1H); HPLC (градиент вода/ACN+0,1% TFA) 95,3% при 220 нм; LCMS [M+H]+=244,1, [M+Na]+=264,0.
Получение (2Е)-3-[3'-(бензилокси)-4-гидроксибифенил-2-ил]проп-2-енамида
[0111] Через смесь (2E)-3-(2-бром-5-гидроксифенил)проп-2-енамида (10,00 г, 41,31 ммоль), 3-бензилоксифенилбороновой кислоты (12,22 г, 53,58 ммоль) и карбоната калия (17,34 г, 0,125 моль) в смеси воды (60 мл), толуола (160 мл) и этанола (100 мл) в течение 10 мин. барботировали азот перед добавлением тетракис(трифенилфосфин)палладия(0) (1,21 г, 10,5 ммоль) и нагреванием смеси до температуры флегмы в течение 2,5 ч. Смесь быстро охлаждали, разбавляли водой (200 мл) и подкисляли посредством добавления хлористоводородной кислоты (2 М, прибл. 400 мл, рН: 0-1) и экстрагировали этилацетатом (3×300 мл). Объединенные органические экстракты высушивали над сульфатом магния и фильтровали. Фильтрат концентрировали до сухого состояния и остаток очищали посредством флэш-хроматографии (силикагель, градиент 10-100% этилацетат/гексаны) с получением указанного в заголовке соединения в виде коричневой твердой пены (14,37 г, 101%).1Н ЯМР (400 МГц, DMSO-d6) d 9,71 (s, 1Н) 7,39-7,48 (m, 4Н) 7,29-7,38 (m, 4Н) 7,17 (d, J=8,2 Гц, 1H) 7,09 (s, 1Н) 7,06 (d, J=2,4 Гц, 1H) 7,01 (dd, J=8,2, 2,4 Гц, 1H) 6,83-6,90 (m, 2Н) 6,81 (d, J=7,4 Гц, 1H) 6,49 (d, J=15,6 Гц, 1H) 5,12 (s, 2Н); HPLC (градиент вода/ACN+0,1% TFA) 88,2% при 220 нм; LCMS [М+Н]+=346,2, [М-Н]-=344,1. прибл. 11 вес.% этилацетата и 14 мол. % неизвестной примеси, как показано с помощью1Н ЯМР-анализа.
Получение (2Е)-3-[3'-(бензилокси)-4-трифторметансульфонат-бифенил-2-ил]проп-2-енамида
[0112] N-фенил-бис(трифторметансульфонамид) (16,35 г, 45,77 ммоль) добавляли порциями в течение 1 мин. к раствору (2E)-3-[3'-(бензилокси)-4-гидроксибифенил-2-ил]проп-2-енамида (14,27 г, 41,33 ммоль) и карбоната калия (11,63 г, 84,15 ммоль) в ацетонитриле (200 мл), который охлаждали в ледяной бане. Реакционную смесь нагревали до комнатной температуры и тщательно перемешивали в течение 1 ч. Добавляли силикагель и смесь концентрировали и очищали посредством флэш-хроматографии (диоксид кремния, градиент 10-100% этилацетат/гексаны) с получением указанного в заголовке соединения в виде светло-коричневой твердой пены (15,95 г, 81%).1Н ЯМР (400 МГц, DMSO-d6) d 7,78 (d, J=2,4 Гц, 1H) 7,52-7,61 (m, 3Н) 7,43-7,50 (m, 2Н) 7,37-7,43 (m, 3 3Н) 7,29-7,37 (m, 2Н) 7,21 (br. s., 1H) 7,11 (dd, J=8,2, 2,4 Гц, 1H) 6,96-7,03 (m, 1H) 6,90 (d, J=7,4 Гц, 1H) 6,69 (d, J=15,6 Гц, 1H) 5,15 (s, 2H); HPLC (градиент вода/ACN+0,1% TFA) 95,0% при 220 нм; LCMS [М+Н]+=478,1. незначительные примеси, как показано с помощью1Н ЯМР-анализа.
Пример 4. Синтез промежуточных соединений Р3, Р46, Р47, Р48, Р49 и Р50
[0113] Синтетический путь, применяемый для получения промежуточных соединений Р3, Р46, Р47, Р48, Р49 и Р50, показан на фигуре 5.
[0114] Азот барботировали через смесь (2Е)-3-[3'-(бензилокси)-4-трифторметансульфонат-бифенил-2-ил]проп-2-енамида (1 экв.), фенилбороновой кислоты (1,3 эквив.) и карбоната калия (3 экв.) в смеси воды (3 мл), толуола (8 мл) и этанола (5 мл) в течение 5 мин. перед добавлением тетракис(трифенилфосфин)палладия(0) (0,1 экв.) и нагреванием при 80°С-90°С в герметизированном флаконе или с применением обратного холодильника с конденсатором в атмосфере азота до тех пор, пока сохранялся (2E)-3-[3'-(бензилокси)-4-трифторметансульфонат-бифенил-2-ил]проп-2-енамид, определяемый с помощью TLC, LCMS и/или HPLC. Реакционные смеси охлаждали и адсорбировали на силикагель перед очисткой посредством флэш-хроматографии (диоксид кремния, градиент 10-100% этилацетат/гексаны) с получением неочищенных требуемых соединений. Для некоторых соединений требовалась дополнительная очистка, описанная ниже.
[0115] С помощью данной процедуры получали следующие соединения.
3'-[(1Е)-3-Амино-3-оксопроп-1-ен-1-ил]-3''-бензилокси-1,1':4',1''-терфенил-3-карбоновая кислота

[0116] Неочищенное указанное в заголовке соединение (243 мг) содержало трифенилфосфиноксид, обнаруженный с помощью HPLC- и LCMS-анализов. Материал дополнительно очищали посредством флэш-хроматографии (диоксид кремния, градиент 50-100% этилацетат/дихлорметан с последующим градиентом 0-20% метанол/дихлорметан) с получением указанного в заголовке соединения в виде белого порошка (112 мг, 16%).1Н ЯМР (400 МГц, DMSO-d6) d 13,18 (br. s, 1H) 8,26 (t, J=1,56 Гц, 1H) 7,95-8,05 (m, 3H) 7,79 (dd, J=8,22, 1,96 Гц, 1H) 7,65 (t, J=7,83 Гц, 1H) 7,53 (br. s., 1H) 7,38-7,51 (m, 7 H) 7,29-7,37 (m, 1H) 7,14 (br. s., 1H) 7,06-7,12 (m, 1Н) 6,97-7,03 (m, 1Н) 6,90-6,96 (m, 1Н) 6,77 (d, J=15,65 Гц, 1H) 5,16 (s, 2Н); HPLC (градиент вода/ACN+0,1% TFA) 94,5% при 220 нм; LCMS [М+Н]+=450,1, [M+Na]+=472,1. прибл. 2 вес.% этилацетата и других незначительных примесей, как показано с помощью1Н ЯМР-анализа.
(2Е)-3-(3-Бензилокси-4''-фтор-1,1':4',1''-терфенил-2'-ил)проп-2-енамид

[0117] Грязно-белая твердая пена (215 мг, 32%).1Н ЯМР (400 МГц, DMSO-d6) d 7,93 (d, J=1,2 Гц, 1H) 7,80 (dd, J=8,6, 5,5 Гц, 2H) 7,72 (dd, J=7,8, 657 Гц, 1H) 7,28-7,53 (m, 11Н) 7,13 (br. s., 1H) 7,09 (dd, J=8,2, 2,0 Гц, 1H) 6,98 (s, 1H) 6,92 (d, J=7,4 Гц, 1H) 6,75 (d, J=16,0 Гц, 1H) 5,12-5,20 (m, 2H); HPLC (градиент вода/ACN+0,1% TFA) 83,7% при 220 нм; LCMS [М+Н]+=424,2, [M+Na]+=446,2. прибл. 3% этилацетата и 10 мол. % других неизвестных примесей, как показано с помощью1Н ЯМР-анализа.
(2Е)-3-(3-Бензилокси-4''-нитро-1,1':4'1''-терфенил-2'-ил)проп-2-енамид

[0118] Неочищенное указанное в заголовке соединение содержало трифенилфосфиноксид и (2E)-3-[3'-(бензилокси)бифенил-2-ил]проп-2-енамид, обнаруженные с помощью HPLC- и LCMS-анализов. Посредством дополнительной очистки посредством двух разделений с помощью флэш-хроматографии (диоксид кремния, градиент 50-100% этилацетат/дихлорметан) получали указанное в заголовке соединение в виде желтого порошка (158 мг, 22%).1Н ЯМР (400 МГц, DMSO-d6) d 8,36 (d, J=8,6 Гц, 2H) 8,07 (m, J=8,6 Гц, 3H) 7,87 (dd, J=8,0, 1,4 Гц, 1H) 7,53 (d, J=8,2 Гц, 1H) 7,44-7,51 (m, 4Н) 7,41 (s, 3Н) 7,34 (s, 1Н) 7,15 (br. s., 1H) 7,11 (dd, J=8,2, 1,96 Гц, 1H) 7,00 (s, 1H) 6,94 (d, J=7,4 Гц, 1H) 6,77 (d, J=15,6 Гц, 1H) 5,18 (s, 2H); HPLC (градиент вода/ACN+0,1% TFA) 95,9% при 220 нм; LCMS [М+Н]+=452,3, [M+Na]+=473,2.
(2Е)-3-(3-Бензилокси-3''-метил-1,1':4',1''-терфенил-2'-ил)проп-2-енамид

[0119] Белая твердая пена (272 мг, 62%).1Н ЯМР (400 МГц, DMSO-d6) d 7,94 (d, J=1,6 Гц, 1H) 7,73 (dd, J=8,2, 1,6 Гц, 1H) 7,58 (s, 1Н) 7,54 (d, J=7,8 Гц, 1H) 7,45-7,51 (m, 4Н) 7,37-7,44 (m, 5Н) 7,33 (m, J=7,0 Гц, 1H) 7,23 (d, J=7,4 Гц, 1H) 7,13 (br. s., 1H) 7,09 (dd, J=8,4, 2,2 Гц, 1H) 6,99 (s, 1H) 6,92 (d, J=7,4 Гц, 1H) 6,75 (d, J=15,7 Гц, 1H) 5,16 (s, 2H) 2,41 (s, 3H); HPLC (градиент вода/ACN+0,1% TFA) 97,7% при 220 нм; LCMS [М+Н]+=420,3, [M+Na]+=442,3. прибл. 4 вес.% этилацетата и незначительные примеси, как показано с помощью1Н ЯМР-анализа.
(2Е)-3-(3-Бензилокси-3''-гидрокси-1,1':4',1''-терфенил-2'-ил)проп-2-енамид

[0120] Грязно-белая твердая пена (485 мг, 66%).1Н ЯМР (400 МГц, DMSO-d6) d 9,58 (br. s., 1H) 7,91 (s, 1H) 7,69 (d, J=8,2 Гц, 1H) 7,46-7,54 (m, 4H) 7,39-7,46 (m, 4H) 7,26-7,39 (m, 2H) 7,06-7,22 (m, 4H) 6,91-7,04 (m, 2H) 6,84 (d, J=7,8 Гц, 1H) 6,76 (d, J=15,7 Гц, 1H) 5,17 (s, 2H); HPLC (градиент вода/ACN+0,1% TFA) 89,3% при 220 нм; LCMS 444,2=[M+Na]+. прибл. 7 вес.% этилацетата и 16 мол. % неизвестной примеси, как показано с помощью1Н ЯМР-анализа.
(2Е)-3-(3-Бензилокси-3''-метокси-1,1':4',1''-терфенил-2'-ил)проп-2-енамид

[0121] Неочищенное указанное в заголовке соединение (456 мг, 68%) содержало (2E)-3-[3'-(бензилокси)бифенил-2-ил]проп-2-енамид и трифенилфосфиноксид. Осуществляли дополнительную очистку посредством флэш-хроматографии (диоксид кремния, градиент 20-100% этилацетат/дихлорметан) с получением неочищенного указанного в заголовке соединения (361 мг), содержащего (2E)-3-[3'-(бензилокси)бифенил-2-ил]проп-2-енамид. Осуществляли дополнительную очистку посредством препаративной HPLC (С18, 30-90% ацетонитрил в воде (+0,1% TFA)) с получением указанного в заголовке соединения в виде бесцветного стеклообразного твердого вещества (218 мг, 32%).1Н ЯМР (400 МГц, DMSO-d6) d 7,94 (s, 1Н) 7,74 (dd, J=7,8, 1,2 Гц, 1H) 7,44-7,54 (m, 4Н) 7,37-7,44 (m, 5Н) 7,29-7,37 (m, 2Н) 7,27 (s, 1Н) 7,12 (br. s., 1H) 7,09 (dd, J=8,4, 1,8 Гц, 1H) 6,95-7,03 (m, 2H) 6,92 (d, J=7,4 Гц, 1H) 6,75 (d, J=15,7 Гц, 1H) 5,16 (s, 2H) 3,85 (s, 3H); HPLC (градиент вода/ACN+0,1% TFA) 98,4% при 220 нм; LCMS [М+Н]+=436,3, [M+Na]+=458,3.
Пример 5. Синтез Р3, Р46, Р47, Р48, Р49 и Р50
[0122] Синтетический путь, применяемый для получения Р3, Р46, Р47, Р48, Р49 и Р50, показан на фигуре 6.
[0123] Добавляли палладий на активированном угле (10% вес/вес, 10 мг на 100 мг производного бензилокси-терфенила) к раствору производного бензилокси-терфенила (1 эквив.) в этилацетате или метаноле (5-15 мл) и триэтиламине (100 мкл на 1 мл этилацетата или метанола) и помещали в атмосферу водорода и нагревали до температуры флегмы до завершения реакции, что определяли с помощью TLC, HPLC и/или LCMS. Процедуры обработки и очистки для каждого соединения отличались и описаны ниже.
[0124] Следующие соединения получали данным способом. 3'-(3-Амино-3-оксопропил)-3''-гидрокси-1,1':4',1''-терфенил-3-карбоновая кислота
[0125] Реакционную смесь охлаждали, разбавляли хлористоводородной кислотой (2 М, 10 мл) и этилацетатом (20 мл) и фильтровали через целит, промывая подушку из целита этилацетатом (2×20 мл). К фильтратам добавляли хлористоводородную кислоту (2 М, 20 мл) и собирали органический слой. Водный слой экстрагировали этилацетатом (2×30 мл). Объединенные органические экстракты концентрировали с получением указанного в заголовке соединения в неочищенной форме. Данный материал промывали смесью метанол: дихлорметан (1:3, 3×0,5 мл) и высушивали под вакуумом с получением указанного в заголовке соединения в виде грязно-белого порошка после высушивания под вакуумом при 40°С (41 мг, 47%).1Н ЯМР (400 МГц, DMSO-d6) δ 9,52 (s, 1Н) 8,22 (s, 1Н) 7,95 (d, J=7,8 Гц, 2Н) 7,65 (br. s., 1Н) 7,62 (t, J=7,8 Гц, 1H) 7,56 (dd, J=7,8, 1,2 Гц, 1H) 7,19-7,29 (m, 3Н) 6,77 (t, J=8,6 Гц, 2H) 6,72 (br. s., 2H) 2,84 (t, J=8,0 Гц, 2H) 2,31 (t, J=8,6 Гц, 2H); HPLC (градиент вода/ACN+0,1% TFA) 97,4% при 220 нм; LCMS [М+Н]+=362,2 [M+Na]+=384,2.
3-(4''-Фтор-3-гидрокси-1,1':4',1''-терфенил-2'-ил)пропанамид (Р3)

[0126] Реакционную смесь охлаждали до комнатной температуры и полученную смесь разбавляли хлористоводородной кислотой (2 М, 15 мл) и этилацетатом (15 мл) и фильтровали через целит, промывая подушку из целита этилацетатом (2×20 мл). К фильтратам добавляли дополнительное количество хлористоводородной кислоты (2 М, 20 мл) и собирали органический слой. Водный слой экстрагировали этилацетатом (2×20 мл). Объединенные органические экстракты концентрировали и остаток кристаллизовали посредством добавления дихлорметана (2 мл). Смесь концентрировали с получением неочищенного указанного в заголовке соединения (75 мг) в виде грязно-белого порошка, который промывали этанолом (3×0,5 мл) и высушивали под вакуумом с получением указанного в заголовке соединения в виде грязно-белого порошка (30 мг, 37%) после высушивания под вакуумом.1Н ЯМР (400 МГц, DMSO-d6) δ 9,51 (s, 1Н) 7,73 (dd, J=8,4, 5,7 Гц, 2Н) 7,58 (s, 1Н) 7,50 (dd, J=8,2, 1,6 Гц, 1Н) 7,31 (t, J=8,8 Гц, 2Н) 7,15-7,27 (m, 3Н) 6,65-6,83 (m, 4Н) 2,82 (t, J=7,8 Гц, 2Н) 2,30 (t, J=8,0 Гц, 2Н); HPLC (градиент вода/ACN+0,1% TFA) 97,6% при 220 нм; LCMS [М+Н]+=336,2, [M+Na]+=358,1.
3-(4''-Амино-3-гидрокси-1,1':4',1''-терфенил-2'-ил)пропанамид (Р49)

[0127] Реакционную смесь охлаждали до комнатной температуры и полученную смесь разбавляли хлоридом аммония (насыщ., 30 мл) и этилацетатом (30 мл) и фильтровали через целит, промывая подушку из целита этилацетатом (2×20 мл). Органический слой фильтрата отделяли и водный слой экстрагировали этилацетатом (2×30 мл). Объединенные органические экстракты высушивали над сульфатом магния и концентрировали с получением грязно-белого порошка (67 мг). Неочищенный продукт очищали посредством флэш-хроматографии (диоксид кремния, градиент 30-100% этилацетат/гексаны) с получением указанного в заголовке соединения в виде белого порошка после высушивания под вакуумом (41 мг, 37%).1Н ЯМР (400 МГц, DMSO-d6) δ 9,48 (s, 1Н) 7,47 (s, 1Н) 7,38 (d, J=8,6 Гц, 3Н) 7,18-7,27 (m, 2Н) 7,12 (d, J=7,8 Гц, 1Н) 6,68-6,79 (m, 4Н) 6,65 (d, J=8,2 Гц, 2Н) 5,22 (s, 2Н) 2,79 (t, J=7,8 Гц, 2Н) 2,28 (t, J=7,8 Гц, 2Н); HPLC (градиент вода/ACN+0,1% TFA) 100,0% при 220 нм; LCMS [М+Н]+=333,2, [M+Na]+=355,2.
3-(3-Гидрокси-3''-метил-1,1':4',1''-терфенил-2'-ил)пропанамид (Р46)

[0128] Реакционную смесь охлаждали, подкисляли смесью хлористоводородная кислота-диэтиловый эфир (до рН 4-6), добавляли диоксид кремния и смесь концентрировали. Посредством очистки посредством флэш-хроматографии (диоксид кремния, градиент 10-100% этилацетат/гексаны) получали требуемое соединение. Данный материал измельчали до мелкодисперсного порошка и высушивали под вакуумом в течение 2 дней с получением указанного в заголовке соединения в виде белой твердой пены (109 мг, 55%).
[0129]1Н ЯМР (400 МГц, DMSO-d6) δ 9,51 (s, 1Н) 7,59 (s, 1Н) 7,43-7,55 (m, 3Н) 7,36 (t, J=7,6 Гц, 1Н) 7,14-7,29 (m, 4Н) 6,64-6,84 (m, 4Н) 2,83 (t, J=7,8 Гц, 2Н) 2,39 (s, 3Н) 2,30 (t, J=7,8 Гц, 2Н); HPLC (градиент вода/ACN+0,1% TFA) 98,9% при 220 нм; LCMS [М+Н]+=332,3 [М+Н]+, [M+Na]+=354,2
3-(3,3''-Дигидрокси-1,1':4',1''-терфенил-2'-ил)пропанамид (Р48)

[0130] Реакционную смесь разбавляли хлористоводородной кислотой (2 М, 5 мл) и этилацетатом (30 мл) и фильтровали через целит, промывая подушку из целита этилацетатом (2×20 мл). Фильтрат разбавляли хлористоводородной кислотой (2 М, 20 мл), отделяли органический слой и экстрагировали водный слой этилацетатом (2×30 мл). Объединенные органические экстракты высушивали над сульфатом магния, концентрировали и очищали посредством препаративной HPLC (С18, 20-70% ацетонитрил в воде (+0,1% TFA)) с получением указанного в заголовке соединения в виде белого порошка (24 мг, 15%) после высушивания под вакуумом.1Н ЯМР (400 МГц, DMSO-d6) δ 9,52 (br. s., 2Н) 7,53 (s, 1Н) 7,44 (d, J=7,8 Гц, 1Н) 7,14-7,30 (m, 4Н) 7,09 (d, J=7,8 Гц, 1Н) 7,05 (s, 1Н) 6,65-6,81 (m, 5Н) 2,81 (t, J=7,8 Гц, 2Н) 2,29 (t, J=7,8 Гц, 2Н); HPLC (градиент вода/ACN+0,1% TFA) 96,8% при 220 нм; LCMS [М+Н]+=334,2, [M+Na]+=356,1.
3-(3-Гидрокси-3''-метокси-1,1':4',1''-терфенил-2'-ил)пропанамид (Р50)

[0131] Реакционную смесь охлаждали до комнатной температуры и разбавляли хлористоводородной кислотой (2 М, 10 мл) и этилацетатом (20 мл) и фильтровали через целит, промывая подушку из целита этилацетатом (2×30 мл). Фильтрат разбавляли хлористоводородной кислотой (2 М, 25 мл), отделяли органический слой и экстрагировали водный слой этилацетатом (2×30 мл). Объединенные органические экстракты высушивали над сульфатом магния, концентрировали с получением белого порошка, который дополнительно измельчали и высушивали под вакуумом с получением указанного в заголовке соединения в виде белого порошка (90 мг, 54%).1Н ЯМР (400 МГц, DMSO-d6) δ 9,51 (br. s., 1Н) 7,60 (s, 1Н) 7,52 (dd, J=7,8, 1,2 Гц, 1Н) 7,39 (t, J=7,8 Гц, 1Н) 7,16-7,30 (m, 5Н) 6,95 (dd, J=8,0, 1,8 Гц, 1Н) 6,67-6,84 (m, 4Н) 3,84 (s, 3Н) 2,83 (t, J=7,8 Гц, 2Н) 2,31 (t, J=8,0 Гц, 2Н); HPLC (градиент вода/ACN+0,1% TFA) 98,4% при 220 нм; LCMS [М+Н]+=348,2, [M+Na]+=370,2.
Пример 6. Синтез Р1, Р6 и Р33
[0132] Синтетический путь, применяемый для получения Р1, Р6 и Р33, описан ниже.
Процедура сочетания А

[0133] Сосуд для нагревания под воздействием микроволнового облучения объемом 20 мл наполняли смесью 2-[(1E)-3-амино-3-оксопроп-1-ен-1-ил]-3'-бензилоксибифенил-4-ил-трифторметансульфоната (1,0 экв.), бороновой кислоты (1,3 экв.) и карбоната калия (3,0 экв.) в растворе вода/этанол/толуол (1:2:3 0,05 М). Азот барботировали через раствор в течение 5 мин. перед тем, как добавляли тетракис(трифенилфосфин)палладий(0) (10 мол. %) и реакционную смесь герметизировали и помещали в микроволновой реактор на 3 ч. при 110°С. После расходования исходного материала, как показано с помощью TLC и/или LCMS, смесь охлаждали, абсорбировали на силикагеле и очищали посредством флэш-хроматографии (этилацетат/гексаны) с получением следующих соединений.
(2Е)-3-(3''-Фтор-3-бензилокси-1,1':4',1''-терфенил-2'-ил)проп-2-енамид

[0134] (2Е)-3-(3''-Фтор-3-бензилокси-1,1':4',1''-терфенил-2'-ил)проп-2-енамид получали в виде грязно-белого твердого вещества (0,300 г, 68%).1Н ЯМР (400 МГц, CDCl3) δ 7,83 (d, J=1,9 Гц, 1Н), 7,70 (d, J=15,8 Гц, 1Н), 7,62 (dd, J=1,9, 8,0 Гц, 1Н), 7,47.7,44 (m, 3Н), 7,44-7,40 (m, 3Н), 7,40-7,37 (m, 2Н), 7,36-7,32 (m, 3Н), 7,04-7,00 (m, 1Н), 6,99-6,97 (m, 1Н), 6,96-6,93 (m, 1Н), 6,45 (d, J=15,7 Гц, 1Н), 5,42 (br. s., 2Н), 5,10 (s, 2Н); LCMS [M+H]+=424,2, [M+Na]+=446,1. Незначительные примеси, обнаруживали с помощью1Н ЯМР.
(2Е)-3-[3'-Бензилокси-4-(пиридин-4-ил)бифенил-2-ил]проп-2-енамид

[0135] (2Е)-3-[3'-Бензилокси-4-(пиридин-4-ил)бифенил-2-ил]проп-2-енамид получали в виде грязно-белого твердого вещества (0,150 г, 70%).1Н ЯМР (400 МГц, CDCl3) δ 8,90 (d, J=1,8 Гц, 1Н), 8,64 (dd, J=1,5, 4,8 Гц, 1Н), 7,95-7,91 (m, 1Н), 7,83 (d, J=1,8 Гц, 1Н), 7,70 (d, J=15,8 Гц, 1Н), 7,63 (dd, J=1,9, 8,0 Гц, 1Н), 7,50-7,43 (m, 3Н), 7,43-7,30 (m, 5Н), 7,04-7,00 (m, 1Н), 6,99-6,96 (m, 1Н), 6,96-6,92(m, 1Н),6,45 (d, J=15,7 Гц, 1Н), 5,50 (br. s., 2Н), 5,10 (s, 2H); LCMS [M+H]+=407,15, [M+Na]+=429,2. Незначительные примеси, обнаруживали с помощью1Н ЯМР.
(2Е)-3-(3'',5''-Дифтор-3-бензилокси-1,1':4',1''-терфенил-2'-ил)проп-2-енамид

[0136] (2Е)-3-(3'',5''-Дифтор-3-бензилокси-1,1':4,,1''-терфенил-2'-ил)проп-2-енамид получали в виде грязно-белого твердого вещества (0,090, 39%).1Н ЯМР (400 МГц, CDCl3) δ 7,80 (d, J=1,9 Гц, 1Н), 7,69 (d, J=15,8 Гц, 1Н), 7,59 (dd, J=2,0, 8,0 Гц, 1Н), 7,48-7,43 (m, 3Н), 7,42-7,32 (m, 4Н), 7,18-7,13 (m, 2Н), 7,02 (ddd, J=0,9, 2,6, 8,3 Гц, 1Н), 6,98-6,95 (m, 1Н), 6,95-6,90 (m, 1Н), 6,83 (d, J=8,8 Гц, 1Н), 6,45 (d, J=15,7 Гц, 1Н), 5,45 (br. s., 2Н), 5,10 (s, 2H); LCMS [M+H]+=442,1, [M+Na]+=464,2.
Незначительные примеси, обнаруживали с помощью1Н ЯМР .
Процедура гидрирования А
[0137] Добавляли палладий на активированном угле (10% вес/вес, 10 мг на 100 мг производного (бензилокси)-1,1':4',1''-терфенил-2'-ил)проп-2-енамида) к раствору производного (бензилокси)-1,1':4',1''-терфенил-2'-ил)проп-2-енамида (1 экв.) в этилацетате или метаноле (5-15 мл) и триэтиламине (100 мкл на 1 мл этилацетата или метанола) и помещали в атмосферу водорода. Смесь нагревали до температуры флегмы до завершения реакции (24-48 ч.), как показано с помощью TLC, HPLC и/или LCMS. После расходования исходного материала реакционную смесь фильтровали через HPLC нейлоновый шприцевой фильтр, концентрировали и очищали посредством флэш-хроматографии (метанол/дихлорметан) с получением следующих соединений.
3-(3''-Фтор-3-гидрокси-1,1':4',1''-терфенил-2'-ил)пропанамид (Р1)

[0138] Р1 получали в виде белого порошка (0,105 г, 58%).1Н ЯМР (400 МГц, CDCl3) δ 9,51 (s, 1Н), 7,64 (d, J=1,8 Гц, 1Н), 7,58-7,50 (m, 4Н), 7,27-7,17 (m, 4Н), 6,80-6,70 (m, 4Н), 2,86-2,79 (m, 2Н), 2,33-2,29 (m, 2Н); HPLC (градиент вода/ACN+0,1% TFA) 98,28% при 220 нм; LCMS [М+Н]+=336,1, [M+Na]+=358,1.
3-[3'-Гидрокси-4-(пиридин-4-ил)бифенил-2-ил]пропанамид (Р6)

[0139] Р6 получали в виде бледно-желтого порошка (0,080 г, 64%).1Н ЯМР (400 МГц, DMSO) δ 9,52 (s, 1Н), 8,92 (d, J=1,8 Гц, 1Н), 8,58 (dd, J=1,6, 4,8 Гц, 1Н), 8,12-8,06 (m, 1Н), 7,67 (d, J=1,9 Гц, 1Н), 7,59 (dd, J=2,0, 7,9 Гц, 1Н), 7,50 (ddd, J=0,8, 4,8, 7,9 Гц, 1Н), 7,28-7,21 (m, 3Н), 6,79 (ddd, J=1,0, 2,4, 8,1 Гц, 1Н), 6,77-6,71 (m, 3Н), 2,84 (dd, J=6,9, 8,9 Гц, 2Н), 2,32 (dd, J=7,0, 8,9 Гц, 2Н); HPLC (градиент вода/ACN+0,1% TFA)>99% при 220 нм; LCMS [М+Н]+=319,2.
3-(3'',5''-Дифтор-3-гидрокси-1,1':4', 1''-терфенил-2'-ил)пропанамид (Р33)

[0140] Р33 получали в виде белого порошка (0,090 г, 75%).1Н ЯМР (400 МГц, DMSO) δ 9,52 (s, 1Н), 7,69 (d, J=1,9 Гц, 1Н), 7,60 (dd, J=2,0, 8,0 Гц, 1Н), 7,51-7,44 (m, 2Н), 7,27-7,19 (m, 4Н), 6,79 (ddd, J=1,0, 2,4, 8,1 Гц, 1Н), 6,77-6,72 (m, 2Н), 6,72-6,69 (m, 1Н), 2,82 (dd, J=7,0, 8,9 Гц, 2Н), 2,33 (dd, J=7,1, 8,9 Гц, 2Н); HPLC (градиент вода/ACN+0,1% TFA) 98,83% при 220 нм; LCMS [M+H]+=354,1, [M+Na]+=376,1.
Пример 7. Синтез Р38, Р42, Р43, Р44 и Р45
[0141] Синтетический путь, применяемый для получения Р38, Р42, Р43, Р44 и Р45, описан ниже.
2-(3-Амино-3-оксопропил)-3'-гидроксибифенил-4-ил-трифторметансульфонат

[0142] Добавляли оксид платины (10 вес/вес %, 10 мг на 100 мг субстрата) к раствору 2-[(1E)-3-амино-3-оксопроп-1-ен-1-ил]-3'-бензилоксибифенил-4-ил-трифторметансульфоната (5,0 г, 10,4 ммоль) в этаноле (250 мл), помещали в атмосферу водорода и нагревали до температуры флегмы до завершения реакции (24 ч.), как показано с помощью LCMS. При охлаждении реакционную смесь фильтровали через HPLC нейлоновый шприцевой фильтр, концентрировали и неочищенный материал очищали посредством флэш-хроматографии (этилацетат/гексаны) с получением 2-(3-амино-3-оксопропил)-3'-гидроксибифенил-4-ил-трифторметансульфоната (2,6 г, 64%) в виде бледно-розового порошка.1Н ЯМР (400 МГц, DMSO-d6) δ 9,57 (s, 1Н), 7,42 (d, J=2,5 Гц, 1Н), 7,38-7,33 (m, 1Н), 7,33-7,30 (m, 1Н), 7,27-7,22 (m, 2Н), 6,80 (ddd, J=0,8, 2,4, 8,1 Гц, 1Н), 6,76 (br. s., 1Н), 6,74-6,71 (m, 1Н), 6,69-6,67 (m, 1Н), 2,82-2,75 (m, 2Н), 2,30-2,23 (m, 2Н).
3-[3'-Гидрокси-4-(1-метил- 1Н-пиразол-5-ил)6ифенил-2-ил]пропанамид (Р38)

[0143] Сосуд для нагревания под воздействием микроволнового облучения объемом 20 мл наполняли 2-(3-амино-3-оксопропил)-3'-гидроксибифенил-4-ил-трифторметансульфонатом (0,35 г, 0,899 ммоль), 1-метил-1H-пиразол-5-ил-5-бороновой кислотой (0,17 г, 1,35 ммоль) и карбонатом калия (0,37 г, 2,69 ммоль) в растворе толуола (8 мл), этанола (5 мл) и воды (1 мл). Азот барботировали через смесь в течение 5 мин. перед тем, как добавляли тетракис(трифенилфосфин)палладий(0) (10 мол. %, 0,103 г, 0,089 ммоль), реакционный сосуд герметизировали и помещали в микроволновой реактор на 4 ч. при 120°С. При охлаждении добавляли воду (10 мл), 2 М хлористоводородную кислоту (10 мл) и этилацетат (10 мл), отделяли органическую фазу и водную фазу обратно экстрагировали этилацетатом (2×10 мл). Объединенные органические фазы высушивали над безводным сульфатом магния, фильтровали, концентрировали и неочищенный материал очищали посредством флэш-хроматографии (этилацетат/дихлорметан/метанол) с получением Р38 (0,045 г, 16%) в виде белого твердого вещества.1Н ЯМР (400 МГц, DMSO-d6) δ 9,53 (s, 1Н), 7,48 (d, J=2,0 Гц, 1Н), 7,46 (d, J=1,6 Гц, 1Н), 7,39 (dd, J=2,0, 7,8 Гц, 1Н), 7,27-7,21 (m, 3Н), 6,81-6,76 (m, 2Н), 6,76-6,71 (m, 2Н), 6,41 (d, J=1,8 Гц, 1Н), 3,89 (s, 3Н), 2,82 (t, J=7,8 Гц, 2Н), 2,33-2,25 (m, 2Н); HPLC (градиент вода/ACN+0,1% TFA) 96,07% при 220 нм; LCMS [M+H]+=322,20.
3-[4-(3,5-Диметил-1Н-пиразол-4-ил)-3'-гидроксибифенил-2-ил]пропанамид (Р42)

[0144] Сосуд для нагревания под воздействием микроволнового облучения объемом 20 мл наполняли 2-(3-амино-3-оксопропил)-3'-гидроксибифенил-4-ил-трифторметансульфонатом (0,50 г, 1,28 ммоль), третбутил-4-(пинаколовый-сложный-эфир-бороновой-кислоты)-3,5-диметил-1H-пиразол-1-карбоксилатом (0,50 г, 1,54 ммоль) и карбонатом цезия (0,83 г, 2,56 ммоль) в растворе из воды (10 мл) и 1,4-диоксана (1 мл). Азот барботировали через смесь в течение 5 мин. перед тем, как добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном(0) (10 мол. %, 0,105 г, 0,128 ммоль), реакционный сосуд герметизировали и помещали в микроволновой реактор на 96 ч. при 80°С. При охлаждении добавляли воду (50 мл) и смесь промывали этилацетатом (50 мл). Органическую фазу отделяли и промывали 2 М хлористоводородной кислотой (20 мл). Водные фазы объединяли, дополнительно подкисляли до рН 1 и промывали этилацетатом (3×50 мл). Объединенные органические фазы высушивали над безводным сульфатом магния, фильтровали, концентрировали и неочищенный остаток очищали посредством флэш-хроматографии (метанол/дихлорметан) с получением грязно-белого твердого вещества. Данный материал дополнительно очищали посредством растирания из смеси дихлорметан/хлороформ с получением Р42 (0,030 г, 7%) в виде грязно-белого твердого вещества.1Н ЯМР (400 МГц, DMSO-d6) δ 9,47 (s, 1Н), 8,19 (s, 1Н), 7,25-7,19 (m, 3Н), 7,15 (d, J=1,2 Гц, 2Н), 6,78-6,73 (m, 2Н), 6,73-6,70 (m, 2Н), 2,78 (t, J=7,8 Гц, 2Н), 2,30-2,25 (m, 2Н), 2,23 (s, 6Н); HPLC (градиент вода/ACN+0,1% TFA) 96,21% при 220 нм; LCMS [M+H]+=336,20.
3-[3'-Гидрокси-4-(3-метилтиофен-2-ил)бифенил-2-ил]пропанамид (Р43)

[0145] Сосуд для нагревания под воздействием микроволнового облучения объемом 20 мл наполняли 2-(3-амино-3-оксопропил)-3'-гидроксибифенил-4-ил-трифторметансульфонатом (0,50 г, 1,28 ммоль), 4,4,5,5-тетраметил-2-[3-(метил)тиофен-2-ил]-1,3,2-диоксабороланом (0,35 г, 1,54 ммоль) и карбонатом цезия (1,25 г, 3,84 ммоль) в растворе воды (1 мл) и 1,4-диоксана (10 мл). Азот барботировали через смесь в течение 5 мин. перед тем, как добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном(0) (10 мол. %, 0,105 г, 0,128 ммоль) и реакционный сосуд герметизировали и помещали в микроволновой реактор на 9 ч. при 130°С. При охлаждении смесь абсорбировали на силикагель и очищали посредством флэш-хроматографии (этилацетат/дихлорметан/метанол) с получением желтого твердого вещества (0,260 мг), представляющего собой смесь требуемого продукта и исходного материала в соотношении 85:15. Данный материал (0,2 г) растворяли в тетрагидрофуране (2 мл) и раствор охлаждали до 0°С на ледяной бане. Добавляли тетра-н-бутиламмония фторид (5,93 мл, 1 М раствор в THF, 5,93 ммоль), обеспечивали достижение реакционной смесью комнатной температуры и ее перемешивали в течение 60 ч. Добавляли этилацетат (20 мл), органическую фазу промывали последовательно 1 М хлористоводородной кислотой (10 мл), водой (10 мл) и солевым раствором (10 мл), высушивали над безводным сульфатом магния, фильтровали, концентрировали и неочищенный остаток очищали посредством флэш-хроматографии (дихлорметан/этилацетат/метанол) с получением бледно-желтого твердого вещества. Данный материал дополнительно кристаллизовали из смеси дихлорметан/метанол с получением Р43 (0,055 г, 28%) в виде бледно-желтого твердого вещества.1Н ЯМР (400 МГц, DMSO-d6) δ 9,51 (s, 1Н), 7,46 (d, J=5,1 Гц, 1Н), 7,40 (d, J=1,8 Гц, 1Н), 7,32 (dd, J=1,9, 7,9 Гц, 1Н), 7,27-7,18 (m, 3Н), 7,01 (d, J=5,1 Гц, 1Н), 6,80-6,69 (m, 4Н), 2,83-2,76 (m, 2Н), 2,33 (s, 3Н), 2,27 (dd, J=7,0, 8,8 Гц, 2H); HPLC (градиент вода/ACN+0,1% TFA) 97,08% при 220 нм; LCMS [М+Н]+=338,2, [M+Na]+=360,1.
3-[3'-Гидрокси-4-(4-метилтиофен-3-ил)бифенил-2-ил]пропанамид (Р44)

[0146] Сосуд для нагревания под воздействием микроволнового облучения объемом 20 мл наполняли 2-(3-амино-3-оксопропил)-3'-гидроксибифенил-4-ил-трифторметансульфонатом (0,50 г, 1,28 ммоль), 4,4,5,5-тетраметил-2-[4-(метил)тиофен-3-ил][1,3,2]диоксабороланом (0,35 г, 1,54 ммоль) и карбонатом цезия (1,25 г, 3,84 ммоль) в растворе из воды (1 мл) и 1,4-диоксана (10 мл). Азот барботировали через смесь в течение 5 мин. перед тем, как добавляли комплекс [1,1'-бис(дифенилфосфино) ферроцен]дихлорпалладия(II) с дихлорметаном(0) (10 мол. %, 0,105 г, 0,128 ммоль) и реакционный сосуд герметизировали и помещали в микроволновой реактор на 7,5 ч. при 130°С. При охлаждении смесь абсорбировали на силикагель и очищали посредством флэш-хроматографии (метанол/дихлорметан) с получением желтого твердого вещества. Данный материал дополнительно очищали посредством растирания из дихлорметана с получением Р44 (0,110 г, 25%) в виде белого твердого вещества.1Н ЯМР (400 МГц, DMSO-d6) δ 9,49 (s, 1Н), 7,48 (d, J=3,1 Гц, 1Н), 7,37 (d, J=1,8 Гц, 1Н), 7,31-7,27 (m, 2Н), 7,26-7,20 (m, 2Н), 7,17 (d, J=7,8 Гц, 1Н), 6,80-6,70 (m, 4Н), 2,83-2,76 (m, 2Н), 2,31-2,24 (m, 5Н); HPLC (градиент вода/ACN+0,1% TFA) 98,31% при 220 нм; LCMS [М+Н]+=338,15, [M+Na]+=360,10.
3-[3'-Гидрокси-4-(3-метил-1Н-пиразол-4-ил)бифенил-2-ил]пропанамид (Р45)

[0147] Сосуд для нагревания под воздействием микроволнового облучения объемом 20 мл наполняли 2-(3-амино-3-оксопропил)-3'-гидроксибифенил-4-ил-трифторметансульфонатом (0,25 г, 0,642 ммоль), третбутил-3-метил-4-(пинаколовый-сложный-эфир-бороновой-кислоты)-1H-пиразол-1-карбоксилатом (0,30 г, 0,96 ммоль) и карбонатом цезия (0,42 г, 1,28 ммоль) в растворе из воды (1 мл) и 1,4-диоксана (10 мл). Азот барботировали через смесь в течение 5 мин. перед тем, как добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном(0) (10 мол. %, 0,052 г, 0,064 ммоль) и реакционный сосуд герметизировали и помещали в микроволновой реактор на 12 ч. при 80°С. При охлаждении добавляли воду (10 мл) и смесь экстрагировали этилацетатом (3×10 мл). Объединенные органические фазы высушивали над безводным сульфатом магния, фильтровали и концентрировали. Неочищенный остаток переносили в этилацетат и промывали 2 М хлористоводородной кислотой (20 мл). Водный кислотный слой отстаивали и образовывался осадок, который выделяли посредством фильтрации с получением указанного в заголовке соединения (0,060 г, 29%) в виде мелкодисперсных бледно-желтых кристаллов.1Н ЯМР (400 МГц, DMSO-d6) δ 7,86 (s, 1Н), 7,39 (d, J=1,7 Гц, 1Н), 7,31 (dd, J=1,9, 7,9 Гц, 1Н), 7,28-7,19 (m, 2Н), 7,14 (d, J=7,9 Гц, 1Н), 6,79-6,69 (m, 4Н), 2,82-2,75 (m, 2Н), 2,41 (s, 3Н), 2,28 (dd, J=7,1, 8,9 Гц, 2Н), NH и ОН не обнаружены; HPLC (градиент вода/ACN+0,1% TFA) 97,39% при 220 нм; LCMS [М+Н]+=322,20.
Пример 8. Синтез Р4
[0148] Синтетический путь, применяемый для получения Р4, описан ниже.
(2Е)-3-[4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-1-ил)-3'-бензилоксибифен-2-ил]проп-2-енамид

[0149] Сосуд для нагревания под воздействием микроволнового облучения объемом 20 мл наполняли 2-(3-амино-3-оксопропил)-3'-бензилоксибифенил-4-ил-трифторметансульфонатом (1,00 г, 2,10 ммоль), бис(пинаколато)дибором (0,69 г, 2,73 ммоль) и ацетатом калия (0,62 г, 6,30 ммоль) в 1,4-диоксане (15 мл). Азот барботировали через смесь в течение 5 мин. перед тем, как добавляли комплекс [1,1'-бис(дифенил фосфино)ферроцен]дихлорпалладия(II) с дихлорметаном(0) (20 мол. %, 0,343 г, 0,42 ммоль) и реакционный сосуд герметизировали и помещали в микроволновой реактор на 5 ч. при 130°С. При охлаждении смесь абсорбировали на силикагель и очищали посредством флэш-хроматографии (этилацетат/дихлорметан) с получением желтой смолы. Данный материал дополнительно очищали посредством растирания из дихлорметана с получением (2Е)-3-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-1-ил)-3'-бензилоксибифен-2-ил]проп-2-енамида (0,650 г, 68%) в виде желтого твердого вещества.1Н ЯМР (400 МГц, CDCl3) δ=8,13-8,08 (m, 1Н), 7,84 (dd, J=1,2, 7,6 Гц, 1Н), 7,66 (d, J=15,8 Гц, 1Н), 7,46-7,44 (m, 1Н), 7,44-7,42 (m, 1Н), 7,40-7,30 (m, 5Н), 6,99 (ddd, J=0,9, 2,6, 8,3 Гц, 1Н), 6,94-6,92 (m, 1Н), 6,92-6,88 (m, 1Н), 6,48 (d, J=15,7 Гц, 1Н), 5,61 (br. s., 2Н), 5,08 (s, 2Н), 1,37 (s, 12Н); LCMS [М+Н]+=456,2, [M+Na]+=478,2.
3-[4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-1-ил)-3'-гидроксибифен-2-ил]пропанамид

[0150] Добавляли оксид платины (10 вес/вес %, 10 мг на 100 мг субстрата) к раствору (2Е)-3-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-1-ил)-3'-бензилоксибифен-2-ил]проп-2-енамида (0,30 г, 0,66 ммоль) в этаноле (15 мл), реакционную смесь помещали в атмосферу водорода и нагревали до температуры флегмы в течение 24 ч. При охлаждении смесь фильтровали через HPLC нейлоновый шприцевой фильтр, концентрировали и неочищенный остаток очищали посредством флэш-хроматографии (гексаны/этилацетат/метанол) с получением (0,160 г) белого порошка в виде смеси 3-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-1-ил)-3'-гидроксибифен-2-ил]пропанамида и соответствующей бороновой кислоты в соотношении 65:35, как показано с помощью HPLC. LCMS [М+Н]+=368,2 (3-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-1-ил)-3'-гидроксибифен-2-ил]пропанамид) и [М+Н]+=286,1 (бороновая кислота). 3-[4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-1-ил)-3'-гидроксибифен-2-ил]пропанамид1Н ЯМР (400 МГц, MeOD) δ 7,71 (s, 1Н), 7,59 (dd, J=1,2, 7,6 Гц, 1Н), 7,23 (t, J=7,8 Гц, 1Н), 7,16 (d, J=7,6 Гц, 1Н), 6,80-6,76 (m, 2Н), 6,76-6,71 (m, 1Н), 2,94-2,88 (m, 2Н), 2,37-2,32 (m, 2Н), 1,36 (s, 12Н), NHH и ОН не обнаружено.
3-[3'-Гидрокси-4-(пиридин-2-ил)бифенил-2-ил]пропанамид (Р4)

Сосуд для нагревания под воздействием микроволнового облучения объемом 20 мл наполняли неочищенным 3-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-1-ил)-3'-гидроксибифен-2-ил]пропанамидом (0,15 г), 2-бромпиридином (0,14 г, 0,86 ммоль) и карбонатом калия (0,11 г, 0,82 ммоль) в растворе из воды (0,5 мл) и диметоксиэтана (5,0 мл). Азот барботировали через смесь в течение 5 мин. перед тем, как добавляли тетракис(трифенилфосфин)палладий(0) (10 мол. %, 0,047 г, 0,041 ммоль), реакционный сосуд герметизировали и помещали в микроволновой реактор на 4 ч. при 110°С. При охлаждении смесь абсорбировали на силикагеле и очищали посредством флэш-хроматографии (дихлорметан/этилацетат/метанол) с получением (0,090 г) бледно-желтого твердого вещества. Данный материал кристаллизовали из ацетона с получением Р4 (0,07 г, 54%) в виде мелкодисперсного грязно-белого порошка.1Н ЯМР (400 МГц, DMSO-d6) δ 9,51 (s, 1Н), 8,70 -8,65 (m, 1Н), 8,04 (d, J=1,8 Гц, 1Н), 7,99-7,95 (m, 1Н), 7,95-7,86 (m, 2Н), 7,36 (ddd, J=1,1, 4,8, 7,4 Гц, 1Н), 7,25 (td, J=3,9, 7,9 Гц, 3Н), 6,81-6,70 (m, 4Н), 2,88-2,81 (m, 2Н), 2,34-2,27 (m, 2Н); HPLC (градиент вода/ACN+0,1% TFA) >99% при 220 нм; LCMS [М+Н]+=319,15.
Пример 9. Синтез Р104
[0151] Синтетический путь, применяемый для получения Р104, показан на фигуре 7.
[0152] Стадия 1 - a) pTSA, ZnCl2 или SnCl4 для X=ОАс; b) TMSOTf или DEAD/PPh3 для X=ОН; или с) BF3.OEt2 для X=OC(NH)CCl3.
[0153] Стадии 2 и 3 LiOH (водн.), EtOH.
[0154] Стадии 1-3 описаны в a) Atzrodt et al, ARKIVOC 2012 (iii) 257-278; b) Jacquinet, J-C, Carbohydrate Research, 199 (1990) 153-181; c) Stachulski and Jenkins, Natural Product Reports, 1998, p 173-186 и d) Engstrom et al, J. Org. Chem., 2006, 8378-8383.
Пример 10. Скрининг соединений in vitro
[0155] Систему xCELLigence SP (Roche) использовали для измерения изменений клеточного импеданса (клеточного индекса) после обработки эмбриональных гладкомышечных клеток сосудов А10 (АТСС, CRL-1476) тестовым соединением. В этой in vitro клеточной экспериментальной системе отрицательный профиль импеданса коррелирует со снижением артериального давления у крыс - снижение импеданса связано с вазодилатацией, и увеличение импеданса связано с вазоконстрикцией (Stallaert W., Dorn J.F., van der Westhuizen E., Audet M. & Bouvier M. Impedance responses reveal β-adrenergic signaling pluridensitometry and allow classification of ligands with distinct signalling profiles, PLoS ONE 2012; 7 (1): e29420, doi:10.1371/journal.pone.0029420).
[0156] Вкратце, 50 мкл среды для культивирования клеток (DMEM с низким уровнем глюкозы, дополненная 10% фетальной бычьей сыворотки при 37°С) добавляли в каждую лунку E-Plate 96 (Roche) и измеряли фоновый импеданс в каждой лунке. Затем 50 мкл суспензии клеток А-10 (10000 клеток/лунка) добавляли в соответствующие лунки E-Plate 96. Отслеживали клеточный индекс каждой лунки E-Plate 96 при помощи установки RTCA SP внутри инкубатора для культивирования клеток. После инкубирования в течение ночи на протяжении 16-20 часов при 5% CO2 и влажности 95% 100 мкл тестового соединения (тестовые соединения готовили в DMSO и разбавляли средой для культивирования клеток до конечной концентрации DMSO 0,25%) добавляли в соответствующие лунки E-Plate 96 и значения клеточного индекса измеряли сразу же после обработки соединением через каждые 20 секунд в течение 3 часов. Значение клеточного индекса корректировали относительно исходного уровня путем вычитания клеточного индекса обработанных носителем клеток и нормализовали путем деления на клеточный индекс в момент времени непосредственно перед добавлением соединения. Нормализованный относительно исходного уровня клеточный индекс в зависимости от времени наносили на график с помощью программного обеспечения Roche RTCA.
[0157] С помощью соединений может достигаться снижение артериального давления при взаимодействии с гладкомышечными клетками сосудов, вызывая расслабление этих клеток, приводящее к вазодилатации и снижению артериального давления. Они называются прямыми вазодилататорами. Реакция с отрицательным импедансом для гладкомышечных клеток сосудов А10 указывает на то, что тестовое соединение является прямым вазодилататором.
[0158] Систему xCELLigence SP также использовали для измерения изменений клеточного импеданса после обработки бычьих аортальных эндотелиальных клеток (Европейская коллекция клеточных культур) тестовым соединением. Используемый способ является тем же, что и для эмбриональных гладкомышечных клеток сосудов А10, описанных выше, но со средой для культивирования клеток, дополненной 15% фетальной бычьей сывороткой вместо 10%.
[0159] Соединения могут взаимодействовать с эндотелиальными клетками сосудов, вызывая высвобождение веществ, таких как оксид азота и эндотелиальный фактор гиперполяризации, которые, в свою очередь, действуют на гладкомышечные клетки сосудов, вызывая вазодилатацию и снижение артериального давления. Такие соединения называются непрямыми вазодилататорами. Реакция с отрицательным импедансом для аортальных эндотелиальных клеток указывает на то, что исследуемое соединение является непрямым вазодилататором.
[0160] Реакции с отрицательным импедансом для гладкомышечных клеток сосудов А10 наблюдали для Р3, Р5, Р8, Р9, Р11, Р33 и Р46 (фигура 8), что указывает на то, что данные соединения являются прямыми вазодилататорами.
[0161] Реакции с отрицательным импедансом для бычьих аортальных эндотелиальных клеток наблюдали для Р1, Р3, Р4, Р6, Р8, Р9, Р11, Р22, Р26, Р33, Р38, Р41, Р42, Р43, Р44, Р46, Р47, Р48, Р49 и Р50 (фигура 9), что указывает на то, что эти соединения являются непрямыми вазодилататорами.
[0162] Клетки проксимальных почечных канальцев крысы (NRK-52E), выращенные в DMEM + 10% FBS + 1% NEAA + 2 мМ глутамина, помещали в 96-луночные планшеты в концентрации, составляющей 10000 клеток/лунка, и инкубировали при 37°С с 5% CO2 в течение ночи. Тестовые соединения в концентрации, составляющей 30 мкМ, инкубировали с клетками почечных проксимальных канальцев человека или крысы в течение 2 часов при 37°С и 5% CO2. Затем добавляли цис-диамминдихлорплатину(III) (цисплатин) в концентрации 5 мк/мл и затем каждую клеточную популяцию инкубировали в течение 24 или 48 часов при 37°С с 5% CO2. Придерживались изначальных концентраций тестовых соединений. Для оценки цитотоксических эффектов цисплатина на клетки проксимальных почечных канальцев крысы использовали имеющую высокую растворимость в воде тетразолиевую соль, WST-8, которая восстанавливается при помощи дегидрогеназ в клетках с образованием формазана, водорастворимого, имеющего желтый цвет индикаторного красителя, следуя инструкциям изготовителя (в частности, анализ с набором 8 для определения количества клеток (ССК-8) от Sigma). Степень абсорбции в планшетах реагента WST-8 (ССК-8) затем измеряли при 450 нм при помощи считывающего устройства для планшетов Thermo Scientific Multiskan EX.
[0163] Показатель отмирания клеток, индуцированного цисплатином, снижался в культурах клеток проксимальных почечных канальцев, обработанных Р1, Р3, Р4, Р5, Р6, Р8, Р9, Р11, Р22, Р26, Р33, Р38, Р40, Р41, Р42, Р43, Р44, Р45, Р46, Р49 и Р50 (фигура 10), что указывало на то, что данные соединения снижают степень отмирания клеток почечных проксимальных канальцев.
Пример 11. Скрининг соединений in vivo
[0164] Четырнадцатинедельных SHR (на рационе с 2,2% соли; Glen Forrest Stockfeeders) случайным образом распределяли в группы: контрольную в начальный момент времени (крысы возрастом 14 недель), обработки тестовым соединением в питьевом растворе (500 пмоль/кг/мин. в деионизированной дистиллированной воде) или контрольным питьевым раствором (5% этанол в деионизированной дистиллированной воде). Крыс, распределенных в контрольную группу в начальный момент времени (крысы возрастом 14 недель), анестезировали и собирали их почки и печень, в то время как крыс, распределенных в группы для обработки контролем и тестовым соединением, взвешивали дважды в неделю и отслеживали их потребление питьевого раствора, чтобы обеспечить регулирование концентрации тестового соединения в питьевом растворе для поддержания постоянной дозы в течение 4-недельного периода исследования. Значение артериального давления измеряли дважды в неделю при помощи плетизмографии с использованием хвостовой манжеты (PowerLab, ADInstruments, Касл Хилл, Новый Южный Уэльс, Австралия). Через 4 недели крыс анестезировали (крысы возрастом 18 недель) и собирали их почки и печень.
[0165] Для количественной оценки фиброза тканей и/или содержания жира, срезы тканей толщиной ≤3 мм фиксировали в 10% забуференном формалине в течение 24 часов, обрабатывали и заливали в парафин. Поперечные срезы толщиной три микрометра окрашивали с использованием трехцветного окрашивания по Массону. Минимум 20 случайных полей при ×20 увеличении с поперечных срезов (5 на каждом из 2 уровней) оцифровывали и определяли степень фиброза и содержания жира как процент от площади поля каждого оцифрованного изображения с использованием Image-Pro Plus V.7 (Media Cybernetics, Бетесда, Мэриленд, США), затем усредняли для определения уровня фиброза и/или содержания жира для каждой крысы.
[0166] Артериальное давление снижалось у крыс, обработанных Р5, Р8, Р22, Р28 и Р40 (фигура 11).
[0167] Степень проявления фиброза в почке через 4 недели лечения с помощью 500 пмоль/кг/мин. Р5, Р8, Р9, Р11, Р22, Р26, Р40 снижалась по сравнению с контрольными крысами возрастом 18 недель (фигура 12, * р<0,05), что указывало на то, что данные соединения снижают развитие фиброза почек. Степень проявления фиброза в почке через 4 недели лечения с помощью 500 пмоль/кг/мин. Р5 также снижалась по сравнению с контрольными крысами возрастом 14 недель (фигура 12, # р<0,05), что указывало на то, что данное соединение способствует устранению развившегося фиброза почек.
[0168] Степень проявления фиброза в печени через 4 недели лечения с помощью 500 пмоль/кг/мин. Р8, Р9, Р11, Р22, Р26, Р40 снижалась по сравнению с контрольными крысами возрастом 18 недель (фигура 13, * р<0,05, ** р<0,025 и *** р<0,01), что указывало на то, что данные соединения снижают развитие фиброза печени.
[0169] На срезах, окрашенных с применением трехцветного окрашивания по Массону, на которых видны портальные тракты, отчетливо видно, что фиброз окружает портальный тракт и начинает распространяться на синусоидальное пространство (стрелки) контроля (фигура 14А). На срезах, полученных от крыс, обработанных с помощью Р8 (фигура 14В), Р9 (фигура 14С) и Р26 (фигура 14D), волокнистая ткань ограничена базальной мембраной стенок сосудов и восстановлена нормальная структура ткани.
[0170] Количество жира в печени через 4 недели лечения с помощью 500 пмоль/кг/мин. Р22, Р26 и Р40 снижалось по сравнению с контрольными крысами возрастом 18 недель (фигура 15, * р<0,05, ** р<0,025 и *** р<0,01) что указывало на то, что данные соединения снижают степень накопления жира в печени.
Пример 12. Сравнение скрининга соединений in vitro и in vivo
[0171] Сравнение клеточного импеданса гладкомышечных клеток сосудов А10 и уровня фиброза печени у SHR, обработанных различными тестовыми соединениями, показало, что анализ in vitro указывает на способность тестовых соединений уменьшать фиброз в печени (фигура 16, R2=0,618).
[0172] Сравнение клеточного импеданса бычьих аортальных эндотелиальных клеток и уровня фиброза печени у SHR, обработанных различными тестовыми соединениями показало, что анализ in vitro указывает на способность тестовых соединений уменьшать фиброз в печени (фигура 17, R2=0,759).
[0173] Сравнение степени сохранности клеток почечных проксимальных канальцев от цитотоксичности, вызванной цисплатином, и уровня фиброза почек у SHR, обработанных различными тестовыми соединениями показало, что анализ in vitro указывает на способность соединения уменьшать фиброз в почках (фигура 18, R2=0,914).
[0174] Сравнение клеточного импеданса бычьих аортальных эндотелиальных клеток и уровня содержания жира в печени у SHR, обработанных различными тестовыми соединениями показало, что анализ in vitro указывает на способность тестовых соединений уменьшать содержание жира в печени (фигура 19, R2=0,996).
Реферат
Изобретение относится к соединениям указанной ниже формулы или их фармакологически приемлемым солям, которые могут найти применение для профилактического или терапевтического лечения фиброза почек и/или печени, для предотвращения, снижения степени или замедления отмирания клеток почечных канальцев, предотвращения, снижения степени или замедления накопления жира в печени или восстановления нормальной структуры ткани у субъекта. В указанной формуле А представляет собойкаждый из R-Rнезависимо представляет собой С, N, О или S; Q независимо выбран из Салкила, галогена, Салкилкарбоновой кислоты, амино, гидрокси и Cалкокси; n равняется 0, 1, 2, 3 или 4 и X представляет собой -ОН илипричем, если X представляет собой -ОН, А не может представлять собой незамещенный фенил. Изобретение относится также к фармацевтической композиции, способу профилактического или терапевтического лечения указанных выше заболеваний и состояний, применению предлагаемых соединений для изготовления лекарственного препарата для профилактического или терапевтического лечения указанных выше заболеваний и состояний, а также к промежуточным соединениям, используемым в их синтезе. 6 н. и 18 з.п. ф-лы, 19 ил., 1 табл., 12 пр.
Формула





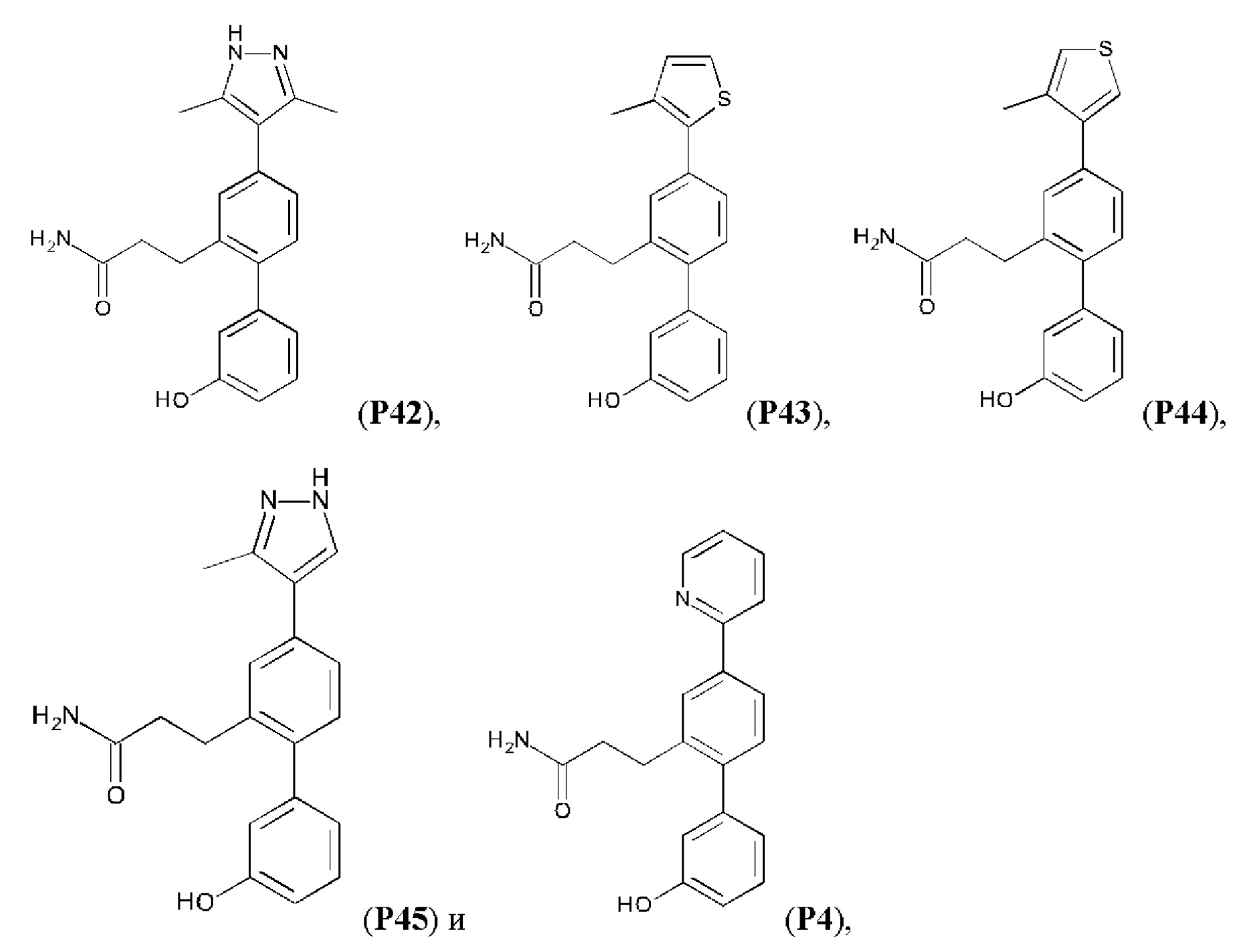



Документы, цитированные в отчёте о поиске
Композиции и способы лечения расстройств почки
Комментарии