Комбинация, содержащая новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты и другой активный ингредиент, для активирования фермента рецептора g-белка 40 - RU2680248C1
Код документа: RU2680248C1
Чертежи










Описание
Область техники, к которой относится изобретение
Настоящее изобретение испрашивает приоритет на основании заявки на патент Кореи №10-2014-0141216, поданной в патентное ведомство Кореи 17 октября 2014, раскрытие которой включено сюда посредством ссылки.
Настоящее изобретение относится к фармацевтической композиции для предотвращения или лечения метаболических нарушений, в которой новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты и по меньшей мере одно соединение, выбранное из группы, состоящей из лекарственных средств на основе ингибиторов дипептидилпептидазы IV (DPPIV), на основе сульфонилмочевины, на основе тиазолидиндиона (TZD), на основе бигуанида и на основе ингибитора натрий/глюкоза котранспортера 2 (SGLT2), могут быть введены в качестве различных активных ингредиентов, в комбинации или в виде композиционного препарата.
Уровень техники
Диабет является тяжелым заболеванием, которое постоянно угрожает нашему здоровью и по меньшей мере сто миллионов человек страдают им во всем мире. Диабет можно классифицировать на две категории по клиническим симптомам, которые являются диабетом типа I и диабетом типа II. Диабет типа I, также известный как инсулинозависимый сахарный диабет (ИЗСД), вызывается аутоиммунным разрушением бета-клеток поджелудочной железы, которые продуцируют инсулин, так что он требует регулярного введения экзогенного инсулина. Диабет типа II, также известный как инсулиннезависимый сахарный диабет (ИНСД), является результатом нарушения регуляции уровня сахара в крови. Таким образом, у тех людей, у которых есть диабет типа II, характерно проявляется дефект секреции инсулина или резистентности к инсулину, что указывает на то, что они практически не имеют эффективного инсулина, секретируемого in vivo, или не могут эффективно использовать инсулин.
Диабет характеризуется высокой концентрацией глюкозы в крови и моче, из-за чего это заболевание вызывает полиурию, жажду, голод и другие проблемы, связанные с метаболизмом липидов и белков. Диабет может вызвать осложнения, угрожающие жизни, такие как потеря зрения, почечная недостаточность и сердечные заболевания. Диабет также является причиной повреждения сетчатки и увеличивает риск возникновения катаракты и глаукомы. Диабет также снижает реакцию на боль, связанную с повреждением нервов в ногах и стопах, и может быть причиной серьезной инфекции.
Современные препараты для лечения диабета включают инсулины, секретогены инсулина, эффекторы снижения уровня глюкозы, активаторы рецепторов, активируемых пролифераторами пероксисом и т.д. Однако последние методы лечения имеют проблемы индуцирования низкого уровня сахара в крови, увеличения массы тела, потери восприимчивости к лекарственному средству с течением времени, вызывают проблемы желудочно-кишечного тракта и отеки и т.д. Поэтому в настоящее время ведутся исследования по внедрению более мощного и эффективного способа лечения. Одна из таких попыток заключается в использовании рецептора, связанного с G-белком (GPCR).
Недавно GPR40 был идентифицирован как один из рецепторов, связанных с G-белком (GPCR). Он известен как рецептор свободной жирной кислоты I, который сверхэкспрессируется в β-клетках поджелудочной железы. Концентрация внутриклеточного кальция повышается соединениями, которые активируют GPR40 (FFAR1) и, соответственно, стимулируется секреция инсулина, индуцированная глюкозой (GSIS) (непатентный документ 1). Когда активатор GPR40 вводили нормальной мыши или трансгенной мыши, предрасположенной к диабету, и затем проводили тест толерантности к глюкозе, он показал повышенную толерантность к глюкозе. Обработанные мыши продемонстрировали кратковременное увеличение инсулина в плазме крови. Из исследования функций GPR40 было подтверждено, что свободная жирная кислота, которая является лигандом GPR40, действует в панкреатических β-клетках, и в результате β-клетки, секретирующие инсулин, зависимы от концентрации глюкозы. В анализе мышей с нокаутированным GPR было подтверждено, что GPR40 вовлечен в ожирение и диабет (непатентный документ 2). Таким образом, GPR40 рассматривается как новая цель для исследования диабета.
Таки образом, заявители настоящего изобретения установили, что совместная обработка новым производным 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты и по меньшей мере одним соединением, выбранным из группы, состоящей из лекарственных средств на основе ингибиторов дипептидилпептидазы IV (dipeptidyl peptidase-4, DPPIV), на основе сульфонилмочевины, на основе тиазолидиндиона (TZD), на основе бигуанида и на основе ингибитора натрий/глюкоза котранспортера 2 (SGLT2), в качестве различных активных ингредиентов, показали улучшенный эффект снижения уровня глюкозы в крови и затем завершили настоящее изобретение.
На протяжении всего изобретения ссылаются на многие документы и патентные документы и представлены ссылки на них. Раскрытие цитируемых работ и патентных документов полностью включено в настоящее описание посредством ссылки, а уровень технической области, к которой относится настоящее изобретение, и подробности настоящего изобретения поясняются более ясно.
Документы предшествующего уровня техники:
(Непатентный документ 0001) Current Drug Targets, 2008, 9, 899-910
(Непатентный документ 0002) Can J Diabetes 2012, 36, 275-280.
Подробное описание изобретения
Техническая проблема
Одним аспектом настоящего изобретения является получение фармацевтической композиции для предотвращения или лечения метаболических нарушений, в которой новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты, или его оптический изомер, гидрат или сольват, или его фармацевтически приемлемая соль, и по меньшей мере одно соединение, выбранное из группы, состоящей из лекарственных средств на основе ингибиторов дипептидилпептидазы IV (DPPIV), на основе сульфонилмочевины, на основе тиазолидиндиона (TZD), на основе бигуанида и на основе ингибитора натрий/глюкоза котранспортера 2 (SGLT2), могут быть введены в качестве различных активных ингредиентов, в комбинации или в виде составного (композиционного) препарата.
Другие цели и преимущества настоящего изобретения будут более понятны, исходя из следующего подробного описания изобретения, формулы изобретения и фигур.
Техническое решение
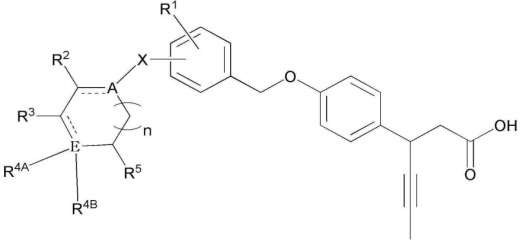
В соответствии с воплощением настоящего изобретения, предложена композиция для предотвращения или лечения метаболических нарушений, композиция, содержащая: (a), в качестве первого активного ингредиента, соединение формулы 1, или его оптический изомер, гидрат или сольват, или его фармацевтически приемлемую соль; и (b), в качестве второго активного ингредиента, по меньшей мере одно соединение, выбранное из группы, состоящей из соединений на основе ингибиторов дипептидилпептидазы IV (DPPIV), на основе сульфонилмочевины, на основе тиазолидиндиона (TZD), на основе бигуанида и на основе ингибитора натрий/глюкоза котранспортера 2 (SGLT2):
[Формула 1]

(В формуле 1

A и E каждый независимо представляет собой C, N или O;
n представляет собой целое число 0-5;
X представляет собой одинарную связь, или C1-10 линейный или разветвленный алкилен;
R1 представляет собой -H, -OH, галоген, C1-10 линейный или разветвленный алкил, C1-10 линейный или разветвленный алкокси, C5-10 циклоалкил, или C5-10 циклоалкенил;
R2, R3, и R5 независимо представляют собой -H, -OH, галоген, C1-10 линейный или разветвленный алкил, или C1-10 линейный или разветвленный алкокси,
где R2 и R3, вместе с атомами, к которым они присоединены, могут образовывать C5-10 циклоалкил, C6-10 арил, 5-10 членный гетероциклоалкил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S, или 5-10 членный гетероарил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S;
R4A представляет собой -H, -OH, =O, незамещенный или замещенный C6-10 арил, или незамещенный или замещенный C5-10 гетероарил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S,
где замещенный C6-10 арил и замещенный C5-10 гетероарил могут быть независимо замещены по меньшей мере одним заместителем, выбранным из группы, состоящей из -OH, галогена, нитрила, C1-5 линейного или разветвленного алкила незамещенного или замещенного по меньшей мере одним атомом галогена, C1-5 линейного или разветвленного алкокси незамещенного или замещенного по меньшей мере одним атомом галогена, C1-10 линейного или разветвленного алкилсульфонила,
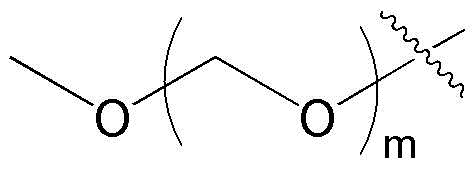
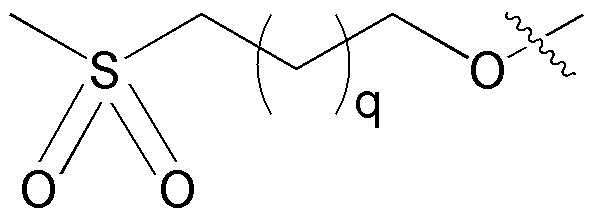
также фенил может быть конденсированным с незамещенным или замещенным C5-10 гетероарилом,
где, R3 и R4A, вместе с атомами, к которым они присоединены, могут образовывать C5-10 циклоалкил, C6-10 арил, 5-10 членный гетероциклоалкил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S, или 5-10 членный гетероарил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S,
также, C5-10 циклоалкил, C6-10 арил, 5-10 членный гетероциклоалкил, и 5-10 членный гетероарил могут быть независимо замещены C1-5 линейным или разветвленным алкокси; и
R4B отсутствует, или R4B, вместе с атомами, к которым R4B присоединен, и R4A, могут образовывать C5-10 гетерокольцо, содержащее по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S.)
В настоящем изобретении соединение на основе ингибитора дипептидилпептидазы IV может включать ситаглиптин, вилдаглиптин, саксаглиптин, линаглиптин, тенелиглиптин, алоглиптин, гемиглиптин, дутоглиптин, берберин, лупеол, красную ольху и одуванчиковый кофе, а соединение на основе сульфонилмочевины является любым, выбранным из группы, состоящей из цербутамида, ацетогексамида, хлорпропамида, толбутамида, глипизида, гликлазида, глибенкламида, глиборнурида, глихидона, глизоксепида, гликлопирамида и глимепирида.
Соединение на основе тиазолидиндионов может быть любым, выбранным из группы, состоящей из розиглитазона, пиоглитазона, троглитазона, нетоглитазона, ривоглитазона, циглитазона и роданина, и соединение на основе бигуанида может быть любым, выбранным из группы, состоящей из метформина, фенформина, буформина, прогуанила, хлорпрогуанила, хлоргексидина, полиаминопропила бигуанид (PAPB), полигексанида и алексидина.
Соединением на основе ингибитора натрий/глюкоза котранспортера 2 (SGLT2) является любое, выбранное из группы, состоящей из эмпаглифлозина, канаглифлозина и дапаглифлозина.
В соответствии с другим аспектом настоящего изобретения, предложен способ предотвращения или лечения метаболических нарушений, способ, включающий введение субъекту фармацевтически эффективного количества композиции, содержащей: (a), в качестве первого активного ингредиента, соединение формулы 1, или его оптический изомер, гидрат или сольват, или его фармацевтически приемлемую соль; и (b), в качестве второго активного ингредиента, по меньшей мере одно соединение, выбранное из группы, состоящей из соединений на основе ингибиторов дипептидилпептидазы-IV (DPP-IV), на основе сульфонилмочевины, на основе тиазолидиндиона (TZD), на основе бигуанида и на основе ингибитора натрий/глюкоза котранспортера 2 (SGLT2), в качестве второго активного ингредиента:
[Формула 1]
В настоящем изобретении Формула 1 является такой, как описано в подробном описании композиции для предотвращения или лечения метаболических нарушений.
Смешанная композиция первого активного ингредиента и второго активного ингредиента в частности не ограничивается массовым соотношением смешивания, так как никаких побочных эффектов или снижения эффективности не вызываются массовым соотношением смешивания, и, принимая во внимание патологические состояния пациентов, известные характеристики второго активного ингредиента и т.д., первый активный ингредиент и второй активный ингредиент могут быть смешаны в подходящих количествах и введены в комбинации. В одном воплощении массовое соотношение смешивания составляет от 0,03:1 до 100:1. В другом воплощении массовое соотношение при смешивании составляет от 0,03:1 до 30:1, а еще в одном варианте массовое соотношение смешивания составляет от 0,03:1 до 10:1.
Положительные эффекты
Комбинированное лечение новым производным 3-(4-(бензилокси)фенил)гекс-4-ановой кислоты, или его оптического изомера, гидрата или сольвата, или его фармацевтически приемлемой соли и по меньшей мере одним соединением, выбранным из группы, состоящей из лекарственных средств на основе ингибиторов дипептидилпептидазы IV (DPPIV), на основе сульфонилмочевины, на основе тиазолидиндиона (TZD), на основе бигуанида и на основе ингибитора натрий/глюкоза котранспортера 2 (SGLT2), в качестве различных активных ингредиентов, показало значительно улучшенный эффект снижения уровня глюкозы в крови при различных диабетических заболеваниях у животных, и таким образом, композиция настоящего изобретения, в которой производное, оптический изомер, гидрат или сольват, или его фармацевтически приемлемая соль и по меньшей мере одно соединение, выбранное из группы, состоящей из лекарственных средств на основе ингибиторов дипептидилпептидазы IV (DPPIV), на основе сульфонилмочевины, на основе тиазолидиндиона (TZD), на основе бигуанида и на основе ингибитора натрий/глюкоза котранспортера 2 (SGLT2) могут быть введены, в качестве различных активных ингредиентов, в комбинации или в виде составного препарата, может выгодно применяться в качестве фармацевтической композиции для предотвращения или лечения метаболических нарушений, таких как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
Краткое описание Фигур
Фиг. 1 представляет собой график, иллюстрирующий паттерн активации белка GPR40, измеренный в соответствии с концентрациями соединений примера 9, Сравнительного Примера 1, и Сравнительного Примера 3.
Фиг. 2 представляет собой график, иллюстрирующий концентрацию GLP-1 в крови при пероральном введении крысам Спрег-Доули (SD) соединений примера 9 и Сравнительного Примера 1.
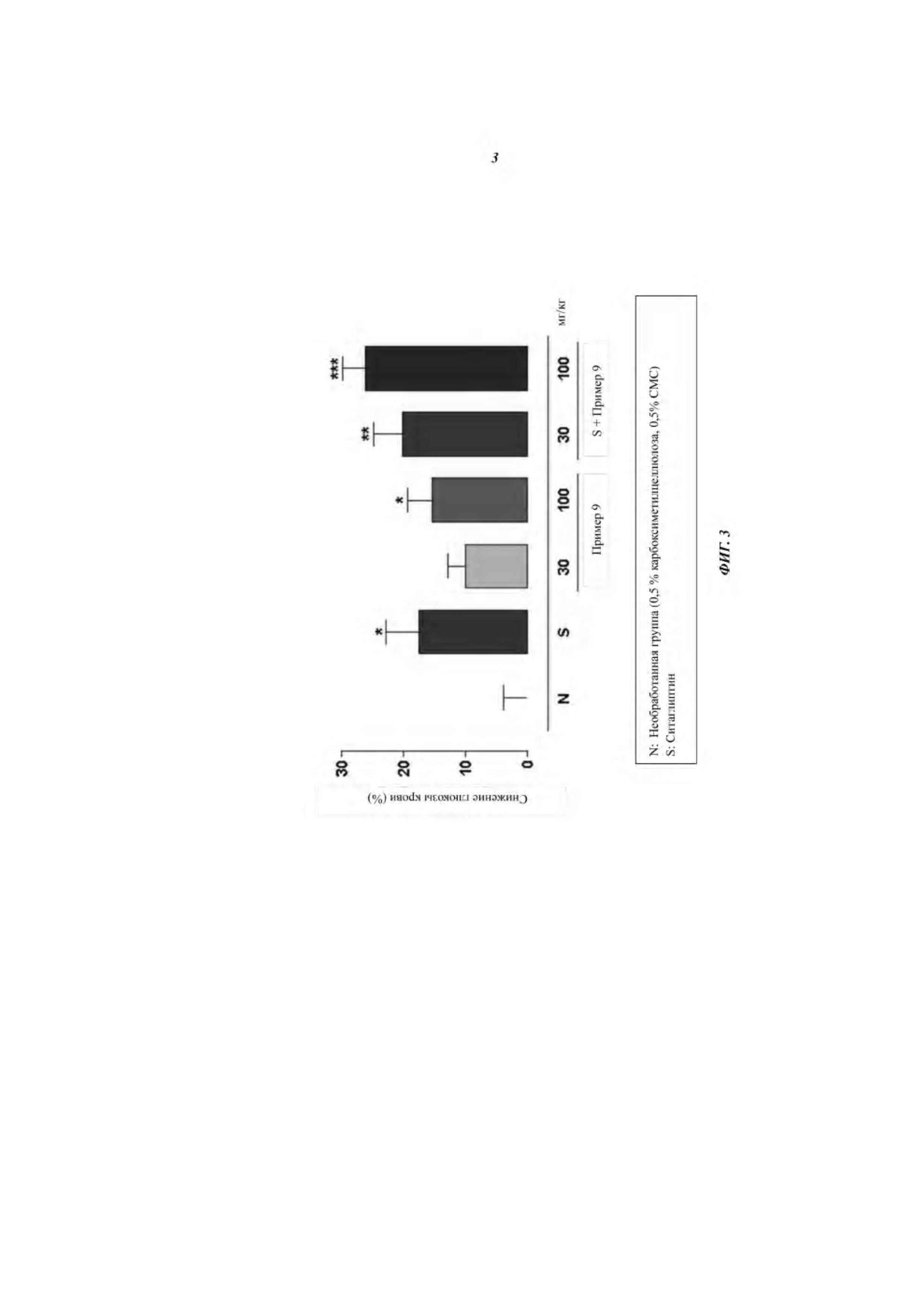
Фиг. 3 представляет собой график, иллюстрирующий снижение глюкозы крови (%), показанное при введении мышам с алиментарным ожирением (DIO) соединения примера 9 или ситаглиптина самостоятельно или при совместном введении соединения примера 9 и ситаглиптина.
Фиг. 4 представляет собой график, иллюстрирующий снижение глюкозы крови (%), показанное при введении мышам с алиментарным ожирением (DIO) соединения примера 9 или глимепирида самостоятельно или при совместном введении соединения примера 9 и глимепирида.
Фиг. 5 представляет собой график, иллюстрирующий снижение глюкозы крови (%), показанное при введении мышам с алиментарным ожирением (DIO) соединения примера 9, росиглитазона, или пиоглитазона самостоятельно или при совместном введении соединения примера 9 и росиглитазона или соединения примера 9 и пиоглитазона.
Фиг. 6 представляет собой график, иллюстрирующий снижение глюкозы крови (%) при введении тучным диабетическим крысам Цукера (ZDF) соединения примера 9 или метформина самостоятельно или совместном введении соединения примера 9 и метформина.
Фиг. 7 представляет собой график, иллюстрирующий снижение глюкозы крови (%), когда крысам Спрег-Доули (SD) вводили соединение примера 9 или линаглиптин самостоятельно или совместно вводили соединение примера 9 и линаглиптин.
Фиг. 8 представляет собой график, иллюстрирующий снижение глюкозы крови (%) при введении крысам Спрег-Доули (SD) соединения примера 9 или эмпаглифлозина самостоятельно или совместном введении соединения примера 9 и эмпаглифлозина.
Фиг. 9 представляет собой график, иллюстрирующий снижение глюкозы крови (%) при введении крысам Спрег-Доули (SD) соединения примера 9 или метформина самостоятельно или совместном введении соединения примера 9 и метформина.
Фиг. 10 показывает результаты in vitro эксперимента анализа секреции GLP-1 с использованием клеток NCI-H716.
Фиг. 11 показывает результаты in vitro эксперимента секреции инсулина с использованием клеток INS-1 (клеточная линия инсулиномы крыс).
Варианты осуществления изобретения
Далее настоящее изобретение будет описано более детально.
В соответствии с воплощением настоящего изобретения, предложена композиция для предотвращения или лечения метаболических нарушений, композиция, содержащая: (a), в качестве первого активного ингредиента, соединение формулы 1, или его оптический изомер, гидрат или сольват, или его фармацевтически приемлемую соль, в качестве первого активного ингредиента; и (b), в качестве второго активного ингредиента, по меньшей мере одно соединение, выбранное из группы, состоящей из соединений на основе ингибиторов дипептидилпептидазы IV (DPPIV), на основе сульфонилмочевины, на основе тиазолидиндиона (TZD), на основе бигуанида и на основе ингибитора натрий/глюкоза котранспортера 2 (SGLT2):
[Формула 1]

(В формуле 1
A и E каждый независимо представляет собой C, N или O;
n представляет собой целое число 0-5;
X представляет собой одинарную связь, или C1-10 линейный или разветвленный алкилен;
R1 представляет собой -H, -OH, галоген, C1-10 линейный или разветвленный алкил, C1-10 линейный или разветвленный алкокси, C5-10 циклоалкил, или C5-10 циклоалкенил;
R2, R3, и R5 независимо представляют собой -H, -OH, галоген, C1-10 линейный или разветвленный алкил, или C1-10 линейный или разветвленный алкокси,
где R2 и R3, вместе с атомами, к которым они присоединены, могут образовывать C5-10 циклоалкил, C6-10 арил, 5-10 членный гетероциклоалкил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S, или 5-10 членный гетероарил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S;
R4A представляет собой -H, -OH, =O, незамещенный или замещенный C6-10 арил, или незамещенный или замещенный C5-10 гетероарил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S,
где замещенный C6-10 арил и замещенный C5-10 гетероарил могут быть независимо замещены по меньшей мере одним заместителем, выбранным из группы, состоящей из -OH, галогена, нитрила, C1-5 линейного или разветвленного алкила незамещенного или замещенного по меньшей мере одним атомом галогена, C1-5 линейного или разветвленного алкокси незамещенного или замещенного по меньшей мере одним атомом галогена, C1-10 линейного или разветвленного алкилсульфонила,
также фенил может быть конденсированным с незамещенным или замещенным C5-10 гетероарилом,
где R3 и R4A, вместе с атомами, к которым они присоединены, могут образовывать C5-10 циклоалкил, C6-10 арил, 5-10 членный гетероциклоалкил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S, или 5-10 членный гетероарил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S,
также C5-10 циклоалкил, C6-10 арил, 5-10 членный гетероциклоалкил, и 5-10 членный гетероарил могут быть независимо замещены C1-5 линейным или разветвленным алкокси; и
R4B отсутствует, или R4B, вместе с атомами, к которым R4B присоединен, и R4A могут образовывать C5-10 гетерокольцо, содержащее по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S).
В одном из воплощений настоящего изобретения,
A и E независимо представляют собой C, N, или O;
n представляет собой целое число 0-3;
X представляет собой одинарную связь, или C1-5 линейный или разветвленный алкилен;
R1 представляет собой -H, -OH, галоген, C1-5 линейный или разветвленный алкил, C1-5 линейный или разветвленный алкокси, C5-8 циклоалкил, или C5-8 циклоалкенил;
R2, R3, и R5 независимо представляют собой -H, -OH, галоген, C1-5 линейный или разветвленный алкил, или C1-5 линейный или разветвленный алкокси,
где, R2 и R3, вместе с атомами, к которым они присоединены, могут образовывать C5-8 циклоалкил, C6-8 арил, 5-8 членный гетероциклоалкил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S, или 5-8 членный гетероарил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S;
R4A представляет собой -H, -OH, =O, незамещенный или замещенный C6-8 арил, или незамещенный или замещенный C5-8 гетероарил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S,
где замещенный C6-8 арил и замещенный C5-8 гетероарил могут быть независимо замещены по меньшей мере одним заместителем, выбранным из группы, состоящей из -OH, галогена, нитрила, C1-5 линейного или разветвленного алкила незамещенного или замещенного по меньшей мере одним атомом галогена, C1-5 линейного или разветвленного алкокси незамещенного или замещенного по меньшей мере одним атомом галогена, C1-8 линейного или разветвленного алкилсульфонила,
также фенил может быть конденсированным с незамещенным или замещенным C5-8 гетероарилом,
где R3 и R4A, вместе с атомами, к которым они присоединены, могут образовывать C5-8 циклоалкил, C6-8 арил, 5-8 членный гетероциклоалкил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S, или 5-8 членный гетероарил, содержащий по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S;
также C5-8 циклоалкил, C6-8 арил, 5-8 членный гетероциклоалкил, и 5-8 членный гетероарил могут быть независимо замещены C1-5 линейным или разветвленным алкокси; и
R4B отсутствует, или R4B, вместе с атомами, к которым R4B присоединен, и R4A могут образовывать C5-8 гетерокольцо, содержащее по меньшей мере один гетероатом, выбранный из группы, состоящей из N, O, и S.)
В одном из воплощений настоящего изобретения,
A и E независимо представляют собой C или N;
n представляет собой целое число 0-1;
X представляет собой одинарную связь, или C1-3 линейный или разветвленный алкилен;
R1 представляет собой -H или

R2, R3, и R5 независимо представляют собой -H,
где R2 и R3, вместе с атомами, к которым они присоединены, могут образовывать фенил;
R4A представляет собой -H, -OH, =O,




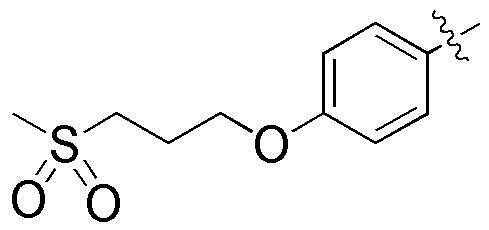


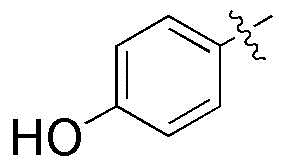
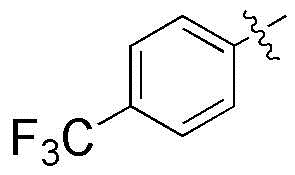



где R3 и R4A, вместе с атомами, к которым они присоединены, могут образовывать фенил, и фенил может быть замещен метокси группой; и
R4B отсутствует, или R4B, вместе с атомами, к которым R4B присоединен, и R4A могут образовывать

В одном из воплощений настоящего изобретения, соединение, представленное формулой 1, является любым соединением, выбранным из следующей группы:
(1) 3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновая кислота
(2) L-лизина 3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат
(3) 4-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновая кислота
(4) 3-(4-(3-(4-оксоциклогекс-1-енил)бензилокси)фенил)гекс-4-иновая кислота
(5) 3-(4-(3-(4-гидроксициклогекс-1-енил)бензилокси)фенил)гекс-4-иновая кислота
(6) L-лизина 3-(4-(3-(4-гидроксициклогекс-1-енил)бензилокси)фенил)гекс-4-иноат
(7) (3S)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновая кислота
(8) (3R)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновая кислота
(9) L-лизина (3S)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат
(10) L-лизина (3R)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат
(11) натрия (3S)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат
(12) 3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(13) 3-(4-(3-циклогексенил-4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(14) 3-(4-(4-((4-фенил-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(15) 3-(4-(4-((4-фенилпиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(16) 3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(17) 3-(4-(4-((4-фенилпиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(18) 3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(19) 3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(20) 3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(21) (S)-3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(22) (S)-3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(23) (S)-3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(24) калия (S)-3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иноат
(25) (S)-3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(26) (S)-3-(4-(4-((4-фенилпиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(27) (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иновая кислота
(28) (S)-3-(4-(4-((4-фенил-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(29) (S)-3-(4-(4-((4-(4-(метоксиметокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(30) (S)-3-(4-(4-((4-(5-изопропил-1,2,4-оксадиазол-3-ил)пиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(31) (S)-3-(4-(4-((4-(5-изопропил-1,2,4-оксадиазол-3-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(32) (S)-3-(4-(4-((4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(33) (S)-3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(34) (3S)-3-(4-(4-(1-(3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иновая кислота
(35) (S)-3-(4-(4-((4-(4-гидроксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(36) (S)-3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(37) натрия (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иноат
(38) L-лизина (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иноат
(39) (S)-3-(4-(4-((4-(4-фторфенил)-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(40) (S)-3-(4-(4-((4-(4-метоксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(41) натрия (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иноат
(42) калия (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иноат
(43) (S)-3-(4-(4-((4-(бензо[d]тиазол-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(44) (S)-3-(4-(4-((4-(5-пропилпиримидин-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(45) (S)-3-(4-(4-((4-(5-цианопиридин-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(46) (3S)-3-(4-(4-((3-фенилпирролидин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота
(47) натрия (S)-3-(4-(3-((4-(4-метоксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иноат;
(48) (S)-3-(4-(4-(2-(6-метокси-3,4-дигидроизохинолин-2(1Н)-ил)этил)бензилокси)фенил)гекс-4-иновая кислота
(49) (S)-3-(4-(4-(2-(изоиндолин-2-ил)этил)бензилокси)фенил)гекс-4-иновая кислота
(50) (S)-3-(4-(4-(2-(3,4-дигидроизохинолин-2(1Н)-ил)этил)бензилокси)фенил)гекс-4-иновая кислота и
(51) натрия (S)-3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1Н)-ил)метил)бензилокси)фенил)гекс-4-иноат.
Соединение, представленное формулой 1 настоящего изобретения, может быть использовано в форме фармацевтически приемлемой соли, в которой соль представляет собой кислотно-аддитивную соль, образованную фармацевтически приемлемой свободной кислотой. Кислотно-аддитивная соль получена из: неорганических кислот, таких как соляная кислота, азотная кислота, фосфорная кислота, серная кислота, бромистоводородная кислота, иодистоводородная кислота, азотистая кислота и фосфористая кислота; нетоксичных органических кислот, таких как алифатический моно и диакарбоксилат, фенилзамещенный алканоат, гидроксиалканоат и алкандиоат, ароматические кислоты и алифатические и ароматические сульфокислоты; или органических кислот, таких как уксусная кислота, бензойная кислота, лимонная кислота, молочная кислота, малеиновая кислота, глюконовая кислота, метансульфоновая кислота, 4-толуолсульфоновая кислота, винная кислота и фумарова кислота. Примеры фармацевтической нетоксичной соли включают сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, иодид, фторид, ацетат, пропионат, деканоат, каприлат, акрилат, формиат, изобутилат, капрат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, кабакат, фумарат, малеат, бутин-1,4-диоат, гексан-1,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат Метоксибензоат, фталат, терефталат, бензолсульфонат, толуолсульфонат, хлорбензолсульфонат, ксиленсульфонат, фенилацетат, фенилпропионат, фенилбутилат, цитрат, лактат, β-гидроксибутилат, гликолят, малат, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, и манделат.
Кислотно-аддитивную соль настоящего изобретения можно получить общепринятым способом, и, например, кислотно-аддитивная соль может быть получена растворением производного формулы 1 в органическом растворителе, таком как метанол, этанол, ацетон, метиленхлорид или ацетонитрил, добавлением органической кислоты или неорганической кислоты для образования осадка, а затем фильтрованием и высушиванием осадка, или могут быть получены путем отгонки растворителя и избытка кислоты при пониженном давлении с последующей сушкой и кристаллизацией в органическом растворителе.
В дополнение, фармацевтически приемлемая соль металла может быть получена с использованием основания. Например, соль щелочного металла или щелочноземельного металла, получают растворением соединения в избытке гидроксида щелочного металла или раствора гидроксида щелочноземельного металла, фильтрованием нерастворимой соли соединения и затем выпариванием и сушкой фильтрата. В данном случае, в качестве соли металла, натриевую, калиевую или кальциевую соль предпочтительно получают, исходя из фармацевтических предпочтений. В дополнение, соответствующая соль получалась путем взаимодействия соли щелочного или щелочноземельного металла с подходящей солью (например, нитратом серебра).
Кроме того, фармацевтически приемлемая соль может быть получена с использованием аминокислоты, в которой аминогруппа присоединена к органической кислоте, и в качестве соли аминокислоты может быть использована природная аминокислота, такая как глицин, аланин, фенилаланин, валин, лизин или глутаминовая кислота, предпочтительно получают из фармацевтического аспекта, и L-лизин наиболее предпочтительно получают из фармацевтического аспекта.
В дополнение, настоящее изобретение включает не только соединение, представленное формулой 1, и его фармацевтически приемлемую соль, но также его сольват, оптический изомер, гидрат и т.д., которые могут быть получены из него.
Фармацевтическая композиция настоящего изобретения может содержать фармацевтически приемлемый носитель в дополнение к активным ингредиентам. В препарате обычно используется фармацевтически приемлемый носитель, содержащийся в фармацевтической композиции настоящего изобретения, и его примеры могут включать, без ограничения, лактозу, декстрозу, сахарозу, сорбит, маннит, крахмал, камедь акации, фосфат кальция, альгинат, желатин, кальция силикат, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, патоку, метилцеллюлозу, метилгидроксибензоат, пропилгидроксибензоат, тальк, стеарат магния и минеральное масло. Фармацевтическая композиция настоящего изобретения может дополнительно содержать, в дополнение к вышеуказанным ингредиентам, смазывающее вещество, смачивающий агент, подслащивающий агент, вкусовой агент, эмульгатор, суспендирующий агент, консервант и т.д. Подходящие фармацевтически приемлемые носители и агенты подробно описаны в Remington's Pharmaceutical Sciences (19th ed., 1995).
В дополнение, эффективная доза соединения настоящего изобретения на организм человека может варьироваться в зависимости от возраста, массы тела, пола, формы введения, состояния здоровья и тяжести заболевания пациента и обычно составляет около 0,001-100 мг/кг/день, и предпочтительно может быть использовано 0,01-35 мг/кг/день. На основании взрослого пациента массой 70 кг доза обычно составляет 0,07-7000 мг/день, и предпочтительно может быть использовано 0,7-2500 мг/день, и доза может вводиться один или несколько раз в день в заранее определенный интервал времени согласно решению врача или фармацевта.
Фармацевтическая композиция настоящего изобретения может вводиться перорально или парентерально, и примеры парентерального введения могут включать местное нанесение на кожу, внутривенную инъекцию, подкожную инъекцию, мышечную инъекцию, внутрибрюшинную инъекцию и трансдермальное введение. Фармацевтическая композиция настоящего изобретения предпочтительно может вводиться перорально. Примеры твердого препарата для перорального введения включают таблетку, пилюлю, порошок, гранулу, капсулу, пастилку и т.д., и такие твердые препараты готовят путем смешивания одного или нескольких соединений настоящего изобретения с по меньшей мере, одним эксципиентом, таким как крахмал, карбонат кальция, сахароза или лактоза, или желатин. В дополнение к простым эксципиентам также используются любриканты, такие как стеарат магния и тальк. Примеры жидкого препарата для перорального введения могут включать суспензию, жидкость для внутреннего применения, эмульсию, сироп, и т.д. В дополнение к обычно применяемым простым разбавителям, таким как вода и жидкий парафин, различные эксципиенты, такие как увлажняющие агенты, подсластители, ароматизаторы и консерванты могут быть включены в жидкий препарат.
Примеры препаратов для перорального введения включают стерилизованный водный раствор, неводный растворитель, суспензионный растворитель, эмульсию, лиофилизатор и суппозиторий. В качестве неводного растворителя и суспензионного растворителя можно использовать пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, инъецируемый сложный эфир, такой как этилолат, или т.п. В качестве субстрата для суппозиториев может быть использован витепсол, макрогол, твин 61, какао-бумагу, лаурин, глицерин, желатин и т.д.
Фармацевтическая композиция настоящего изобретения может быть составлена с использованием фармацевтически приемлемого носителя и/или эксципиента в соответствии со способом, который может быть легко выполнен специалистом в данной области, к которой относится настоящее изобретение, и может быть получен в виде единичной дозы или может быть приготовлен путем упаковки в контейнер с несколькими дозами. Здесь дозированная форма может быть раствором в масляной или водной среде, суспензией, эмульсией, экстрактом, порошком, гранулой, таблеткой или капсулой и может дополнительно содержать диспергатор или стабилизатор.
В одном воплощении настоящего изобретения, соединение на основе ингибитора дипептидилпептидазы IV является любым соединением, выбранным из группы, состоящей из ситаглиптин вилдаглиптин, саксаглиптин, линаглиптин, тенелиглиптин, алоглиптин, гемиглиптин, дутоглиптин, берберин, лупеола, красная ольха и одуванчиковый кофе.
В одном воплощении настоящего изобретения, соединение на основе сульфонилмочевины является любым соединением, выбранным из группы, состоящей из цербутамида, ацетогексамида, хлорпропамида, толбутамида, глипизида, гликлазида, глибенкламида, глиборнурида, глихидона, глизоксепида, гликлопирамида и глимепирида.
В одном воплощении настоящего изобретения, соединение на основе тиазолидиндиона является любым соединением, выбранным из группы, состоящей из розиглитазона, пиоглитазона, троглитазона, нетоглитазона, ривоглитазона, циглитазона и роданина.
В одном воплощении настоящего изобретения, соединение на основе бигуанида является любым соединением, выбранным из группы, состоящей из метформина, фенформина, буформина, прогванила, хлорпрогуанила, хлоргексидина, полиаминопропила бигуанид (PAPB), полигексанида и алексидина.
В одном воплощении настоящего изобретения, соединение на основе ингибиторов SGLT2 является любым соединением, выбранным из группы, состоящей из эмпаглифлозина, канаглифлозина и дапаглифлозина.
В одном воплощении настоящего изобретения, массовое соотношение смеси первого активного ингредиента и второго активного ингредиента настоящего изобретения составляет 0.03:1 до 100:1. В другом воплощении, массовое соотношение смеси составляет 0.03:1 до 30:1, и в еще одном воплощении, массовое соотношение смеси составляет 0.03:1 до 10:1. Однако композиция настоящего изобретения не ограничивается массовым соотношением смешивания, так как никаких побочных эффектов или снижения эффективности не вызываются массовым соотношением смешивания, и, принимая во внимание патологические состояния пациентов, известные характеристики второго активного ингредиента и т.д., первый активный ингредиент и второй активный ингредиент могут быть смешаны в подходящих количествах и введены в комбинации.
В одном из воплощений настоящего изобретения композиция настоящего изобретения активирует фермент рецептора G-белка 40 (GPR40). GPR40 представляет собой рецептор, связанный с G-белком (GPCR), который в основном экспрессируется в клетках поджелудочной железы, секретирующих инсулин. Профиль экспрессии GPR40 обладает потенциальной практичностью для лечения различных метаболических нарушений, включая ожирение и диабет.
В настоящем изобретении, в качестве оценки активности рецептора GPR40, когда соединение, представленное формулой 1, его оптический изомер или его фармацевтически приемлемую соль, в качестве первого активного ингредиента использовали отдельно, можно увидеть, что соединения всех примеров настоящего изобретения активировали рецептор GPR40 на 50% (EC50) при низких концентрациях, и, таким образом, эффекты активации соединениями настоящего изобретения были превосходящими (см. Экспериментальные Примеры 1 и 2, и Фиг. 1).
В дополнение, в настоящем изобретении в результате оценки ингибиторной активности CYP-фермента лекарственными метаболитами соединения, представленного формулой 1, его оптическим изомером или его фармацевтически приемлемой солью, в качестве первого активного ингредиента, было установлено, что соединения примеров настоящего изобретения имели низкую ингибирующую активность CYP-фермента, не вызывая токсичности, обусловленной концентрацией во время совместного введения с другими лекарственными средствами, и, таким образом, соединения настоящего изобретения могут быть совместно введены с другими лекарственными средствами при осложнениях заболеваний (См. Экспериментальный пример 3).
Кроме того, в настоящем изобретении в качестве результата проведения перорального теста толерантности к глюкозе соединения, представленного формулой 1, его оптического изомера или его фармацевтически приемлемой соли в качестве первого активного ингредиента можно увидеть, что соединения всех Примеров настоящего изобретения показали аналогичные или превосходящие эффекты снижения сахара в крови по сравнению с активатором GPR40, который был известен в уровне техники, и, таким образом, соединения настоящего изобретения имели значительно лучший эффект активации GPR40 in vivo (см. Экспериментальные примеры 4, 5 и 6).
Кроме того, в настоящем изобретении в результате проведения эксперимента для оценки скорости увеличения концентрации GLP-1 в крови после перорального введения соединения, представленного формулой 1, его оптического изомера или его фармацевтически приемлемой соли, в качестве первого активного ингредиента, было установлено, что по сравнению с группой, обработанной глюкозой (Veh), соединение Сравнительного Примера 1 не показало эффекта увеличения концентрации GLP-1 в крови после введения, но соединение примера 9 увеличивало концентрацию GLP-1 в крови при введении крысам SD (см. Экспериментальный пример 7 и фиг. 2).
Кроме того, было подтверждено, что совместное введение нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения и типичного лекарственного средства, такого как ингибитор на основе дипептидилпептидазы IV (DPPIV), на основе на сульфонилмочевины, на основе тиазолидиндиона (TZD), на основе бигуанида, и препараты на основе ингибиторов SGLT2, имело превосходящий эффект снижения уровня глюкозы в крови по сравнению с введением указанных лекарств самостоятельно (см. Таблицы 8-14 в экспериментальных примерах 8-12 и Фиг. 3-12).
Вкратце, фармацевтическая композиция настоящего изобретения обладает превосходящим эффектом активации белка GPR40, что приводит к превосходящему эффекту секреции инсулина и может быть совместно введено вместе с другими лекарственными средствами, а также обладает значительно превосходящим эффектом активации белка GPR40 in vivo.
В одном варианте осуществления настоящего изобретения метаболическое заболевание является любым, выбранным из группы, состоящей из ожирения, диабета типа I, диабета типа II, нарушенной толерантности к глюкозе, синдрома резистентности к инсулину, гипергликемии, гиперлипидемии, гипертриглицеридемии, гиперхолестеринемии, дислипидемии и синдрома Х. Композицию настоящего изобретения можно с преимуществом применять для профилактики или лечения вышеупомянутых метаболических нарушений через эффект снижения сахара в крови.
Способ получения соединения, представленного формулой 1 настоящего изобретения описан далее:
Способ получения 1
Соединение, представленное формулой 1 настоящего изобретения, может быть получено, как показано на Реакционной Схеме 1 ниже, включающей стадии:
проведения реакции конденсации соединения, представленного формулой 2 и соединения, представленного формулой 3 с получением соединения, представленного формулой 4 (Стадия 1); и
проведения реакции восстановления соединения, представленного формулой 4, полученного на стадии 1 с получением соединения, представленного формулой 1 (Стадия 2).
[Реакционная Схема 1]

(где в Реакционной Схеме 1
R1, R2, R3, R4A, R4B, R5, A, E, n, и X являются такими, как определено в формуле 1; и
Y представляет собой C1-10 линейный или разветвленный алкил).
Далее способ получения соединения, представленного формулой 1 настоящего изобретения, будет описан подробно по стадиям.
В способе получения соединения, представленного формулой 1 настоящего изобретения, стадия 1) представляет собой получение соединения, представленного формулой 4, посредством проведения реакции связывания между соединением, представленным формулой 2, и соединением, представленным формулой 3. Более конкретно, соединение, представленное формулой 2, азокарбоксилатный реагент, медленно добавляют по каплям в раствор, в котором смешаны соединение, представленное формулой 3, и трифенилфосфин, при температуре от -5° до 10° для проведения реакции Мицунобу с получением соединения, представленного формулой 4.
В данном случае, в качестве азокарбоксилатного реагента могут быть использованы диэтилазодикарбоксилат (DEAD) или диизопропилазодикарбоксилат (DIAD), и предпочтительно может быть использован диизопропилазодикарбоксилат (DIAD).
В дополнение, в качестве реакционного растворителя может быть использован, тетрагидрофуран (ТГФ), дихлорметан (ДХМ), толуол, или ацетонитрил, и предпочтительно может быть использован тетрагидрофуран (ТГФ).
Кроме того, реакционная температура предпочтительно находится между 0° до температуры кипения растворителя, и реакционное время специфично не ограничено, но взаимодействие может предпочтительно проводиться в течение 0.5-10 часов.
В способе получения соединения, представленного формулой 1 настоящего изобретения, стадия 2) представляет собой получение соединения, представленного формулой 1 посредством проведения реакции восстановления соединения, представленного формулой 4, полученного на стадии 1), в присутствии основания. Более конкретно, соединение, представленное формулой 4 полученное на стадии 1) взаимодействует с основанием при комнатной температуре с получением соединения, представленного формулой 1, где эфирная группа, содержащаяся в соединении, представленном формулой 4, восстанавливается в карбоксильную группу.
В данном случае, в качестве основания могут использоваться гидроксид калия (KOH), гидроксид натрия (NaOH), или гидроксид лития (LiOH), и предпочтительно может быть использован гидроксид калия (KOH).
В дополнение, в качестве реакционного растворителя может быть использован, тетрагидрофуран (ТГФ), дихлорметан (ДХМ), толуол, или ацетонитрил, и предпочтительно может быть использован тетрагидрофуран (ТГФ).
Кроме того, реакционная температура предпочтительно находится между 0° до температуры кипения растворителя, и реакционное время специфично не ограничено, но взаимодействие может предпочтительно проводиться в течение 0.5-10 часов.
Способ получения исходного материала (соединение, представленное формулой 2)
В Реакционной Схеме 1 в настоящем изобретении соединение, представленное формулой 2 и используемое в качестве исходного материала, может быть получено способом, включающим следующие стадии, как показано на Реакционной Схеме 2 ниже:
Взаимодействие соединения, представленного формулой 8 с соединением, представленным формулой 9 с получением соединения, представленного формулой 10 (Стадия 1);
Взаимодействие соединения, представленного формулой 10, полученного на стадии 1) с соединением, представленным формулой 11 с получением соединения, представленного формулой 12 (Стадия 2); и
проведение реакции восстановления соединения, представленного формулой 12, полученного на стадии 2) с получением соединения, представленного формулой 2 (Стадия 3).
[Реакционная Схема 2]

(где в Реакционной Схеме 2
R1, R2, R3, R4A, R4B, R5, A, E, n, и X являются такими, как определено в формуле 1; и
-OTf представляет собой трифторметансульфонатную группу.
Далее способ получения соединения настоящего изобретения, представленного формулой 2, будет описан подробно по стадиям.
В способе получения соединения, представленного формулой 2 настоящего изобретения, стадия 1) представляет собой получение соединения, представленного формулой 10, посредством взаимодействия соединения, представленного формулой 8 с соединением, представленным формулой 9. Более конкретно, соединение, представленное формулой 8 и соединение, представленное формулой 9 растворяют в органическом растворителе при -80° до -70°, и затем к ним медленно добавляют бис(триметилсилил)амида металлический комплекс по каплям, и смесь перемешали, при этом температура повысилась до комнатной температуры, с получением, таким образом, соединения, представленного формулой 10.
В данном случае, в качестве бис(триметилсилил)амида металлического комплекса может использоваться калия бис(триметилсилил)амид, лития бис(триметилсилил)амид, или натрия бис(триметилсилил)амид, и предпочтительно может быть использован калия бис(триметилсилил)амид.
В дополнение, в качестве органического растворителя может быть использован тетрагидрофуран (ТГФ), диэтиловый эфир, дифениловый эфир, диизопропиловый эфир (DIPE), диметилформамид (DMF), диметилацетамид (DMA), диметилсульфоксид (DMSO), дихлорметан (ДХМ), хлорбензол, толуол, и бензол.
Кроме того, реакционная температура предпочтительно находится между -80° до температуры кипения растворителя, и реакционное время специфично не ограничено, но взаимодействие может предпочтительно проводиться в течение 0.5-10 часов.
В способе получения соединения, представленного формулой 2 настоящего изобретения, стадия 2) представляет собой получение соединения, представленного формулой 12 посредством взаимодействия соединения, представленного формулой 10, полученного на стадии 1) с соединением, представленным формулой 11. Более конкретно, реакция сочетания Сузуки соединения, представленного формулой 10, полученного на стадии 1) и соединения, представленного формулой 11, проводится в присутствии палладиевого катализатора с получением соединения, представленного формулой 12.
В данном случае, в качестве палладиевого катализатора может быть использован тетракис(трифенилфосфин) (Pd(PPh3)4), бис(трифенилфосфин)палладий(II) дихлорид (PdCl2(PPh3)2), дихлорид палладия (PdCl2), или ацетат палладия (Pd(OCOCH3)2), и предпочтительно может быть использован, тетракис(трифенилфосфин) (Pd(PPh3)4).
В дополнение, в качестве органического растворителя может быть использован тетрагидрофуран (ТГФ), диэтиловый эфир, дифениловый эфир, диизопропиловый эфир (DIPE), диметилформамид (DMF), диметилацетамид (DMA), диметилсульфоксид DMSO), дихлорметан (ДХМ), хлорбензол, толуол, или бензол, и предпочтительно может быть использован толуол.
Кроме того, реакционная температура предпочтительно находится между 0° до температуры кипения растворителя, и реакционное время специфично не ограничено, но взаимодействие может предпочтительно проводиться в течение 0.5-10 часов.
В способе получения соединения, представленного формулой 2 настоящего изобретения, стадия 3) представляет собой получение соединения, представленного формулой 2 посредством проведения реакции восстановления соединения, представленного формулой 12, полученного на стадии 2), в присутствии основания. Более конкретно, соединение, представленное формулой 12, полученное на стадии 2) растворяют в органическом растворителе, и добавляют основание, получив таким образом соединение, представленное формулой 2, где альдегидная группа, содержащаяся в соединении, представленном формулой 12 восстанавливается в карбоксильную группу.
В данном случае, в качестве органического растворителя может быть использован, метанол, этанол, этилацетат, тетрагидрофуран, диэтиловый эфир, или смесь двух или более из вышеуказанных, и предпочтительно может быть использован мешанный раствор тетрагидрофуран : метанол (4:1).
В дополнение, в качестве основания могут использоваться, борогидрид натрия (NaBH3) или алюмогидрид лития (LiAlH4), и предпочтительно может быть использован борогидрид натрия (NaBH3).
Кроме того, реакционная температура предпочтительно находится между 0° до температуры кипения растворителя, и реакционное время специфично не ограничено, но взаимодействие может предпочтительно проводиться в течение 0.5-10 часов.
Способ получения 2
Соединение, представленное формулой 1, настоящего изобретения может быть получено, как показано на Реакционной Схеме 3 ниже, посредством включения стадий: проведение реакции связывания соединения, представленного формулой 5, и соединения, представленного формулой 3, с получением соединения, представленного формулой 6 (Стадия 1);
Проведение реакции мезилата соединения, представленного формулой 6, полученного на стадии 1) с получением соединения, представленного формулой 7 (Стадия 2);
Замена мезилатного участка соединения, представленного формулой 7, полученного на стадии 2) соединением, представленным формулой 13, с получением соединения, представленного формулой 4 (Стадия 3); и
Проведение реакции восстановления соединения, представленного формулой 4, полученного на стадии 3), с получением соединения, представленного формулой 1 (Стадия 4).
[Реакционная Схема 3]

(где в Реакционной Схеме 3
R1, R2, R3, R4A, R4B, R5, A, E, n, и X являются такими, как определено в формуле 1; и
Y представляет собой C1-10 линейный или разветвленный алкил).
Далее способ получения соединения, представленного формулой 1 настоящего изобретения, будет описан подробно по стадиям.
В способе получения соединения, представленного формулой 1 настоящего изобретения, стадия 1) представляет собой получение соединения, представленного формулой 6, посредством проведения реакции связывания соединения, представленного формулой 5, и соединения, представленного формулой 3.
В данном случае, в качестве органического растворителя может быть использован, тетрагидрофуран (ТГФ), диэтиловый эфир, дифениловый эфир, диизопропиловый эфир (DIPE), диметилформамид (DMF), диметилацетамид (DMA), диметилсульфоксид DMSO), дихлорметан (ДХМ), хлорбензол, толуол, или бензол, и предпочтительно может быть использован, диметилформамид (DMF).
В дополнение, в качестве основания могут использоваться, карбонат цезия (Cs2CO3), борогидрид натрия (NaBH3), или алюмогидрид лития (LiAlH4), и предпочтительно может быть использован, карбонат цезия (Cs2CO3).
Кроме того, реакционная температура предпочтительно находится между 0° до температуры кипения растворителя, и реакционное время специфично не ограничено, но взаимодействие может предпочтительно проводиться в течение 0.5-10 часов.
В способе получения соединения, представленного формулой 1 настоящего изобретения, стадия 2) представляет собой получение соединения, представленного формулой 7, посредством проведения реакции мезилата соединения, представленного формулой 6, полученного на стадии 1) в растворителе.
В данном случае, в качестве реагента, используемого в реакции мезилата, может выступать метансульфонилхлорид (MsCl).
В дополнение, в качестве органического растворителя может быть использован, триэтиламин (TEA), тетрагидрофуран (ТГФ), диэтиловый эфир, дифениловый эфир, диизопропиловый эфир (DIPE), диметилформамид (DMF), диметилацетамид (DMA), диметилсульфоксид (DMSO), дихлорметан (ДХМ), хлорбензол, толуол, или бензол, и предпочтительно может быть использован, триэтиламин (TEA).
Кроме того, реакционная температура предпочтительно находится между 0° до температуры кипения растворителя, и реакционное время специфично не ограничено, но взаимодействие может предпочтительно проводиться в течение 0.5-10 часов.
В способе получения соединения, представленного формулой 1 настоящего изобретения, стадия 3) представляет собой получение соединения, представленного формулой 4 посредством замены участка мезилата соединения, представленного формулой 7, полученного на стадии 2) соединением, представленным формулой 13.
В данном случае, в качестве органического растворителя может быть использован тетрагидрофуран (ТГФ), диэтиловый эфир, дифениловый эфир, диизопропиловый эфир (DIPE), диметилформамид (DMF), диметилацетамид (DMA), диметилсульфоксид (DMSO), дихлорметан (ДХМ), хлорбензол, толуол, или бензол, и предпочтительно может быть использован, дихлорметан (ДХМ).
В дополнение, в качестве основания могут использоваться карбонат цезия (Cs2CO3), борогидрид натрия (NaBH3), или алюмогидрид лития (LiAlH4), и предпочтительно может быть использован карбонат цезия (Cs2CO3).
Кроме того, реакционная температура предпочтительно находится между 0° до температуры кипения растворителя, и реакционное время специфично не ограничено, но взаимодействие может предпочтительно проводиться в течение 0.5-10 часов.
В способе получения соединения, представленного формулой 1 настоящего изобретения, стадия 4) представляет собой получение соединения, представленного формулой 1, посредством проведения реакции восстановления соединения, представленного формулой 4, полученного на стадии 3), в присутствии основания. Более конкретно, соединение, представленное формулой 4 полученное на стадии 3) взаимодействует с основанием при комнатной температуре с получением соединения, представленного формулой 1, где эфирная группа, содержащаяся в соединении, представленном формулой 4, восстанавливается до карбоксильной группы.
В данном случае, в качестве основания могут использоваться гидроксид калия (KOH), гидроксид натрия (NaOH), и гидроксид лития (LiOH), и предпочтительно может быть использован гидроксид калия (KOH).
В дополнение, в качестве реакционного растворителя может быть использован, тетрагидрофуран (ТГФ), дихлорметан (ДХМ), толуол, и ацетонитрил, и предпочтительно может быть использован тетрагидрофуран (ТГФ).
Кроме того, реакционная температура предпочтительно находится между 0° до температуры кипения растворителя, и реакционное время специфично не ограничено, но взаимодействие может предпочтительно проводиться в течение 0.5-10 часов.
Способ Получения 3
Соединение, представленное формулой 1 настоящего изобретения, может быть получено, как показано на Реакционной Схеме 4 ниже, посредством включения стадий для проведения реакции открытия кольца соединения, представленного формулой 1a с получением соединения, представленного формулой 1b (Стадия 1).
[Реакционная Схема 4]

(где в Реакционной Схеме 4
R1 является таким, как определено в формуле 1; и
Соединения, представленные формулами 1a и 1b, входят в состав соединения, представленного формулой 1).
Далее способ получения соединения, представленного формулой 1 настоящего изобретения, будет описан подробно по стадиям.
В способе получения соединения, представленного формулой 1 настоящего изобретения, стадия 1) представляет собой получение соединения, представленного формулой 1b посредством проведения реакции открытия цикла соединения, представленного формулой 1a в присутствии кислоты. Более конкретно, соединение, представленное формулой 1a, входящее в соединение представленное формулой 1 подвергается реакции открытия цикла в присутствии кислоты, получив, таким образом, соединение, представленное формулой 1b, которое содержит карбонил открытого кольца гетерокольца соединения, представленного формулой 1a.
В данном случае, в качестве кислоты можно использовать неорганическую кислоту, такую как соляная кислота, серная кислота или фосфорная кислота, и предпочтительно может быть использована соляная кислота.
В дополнение, в качестве реакционного растворителя может быть использован, тетрагидрофуран (ТГФ), дихлорметан (ДХМ), толуол, и ацетонитрил, и предпочтительно может быть использован тетрагидрофуран (ТГФ).
Кроме того, реакционная температура предпочтительно находится между 0° до температуры кипения растворителя, и реакционное время специфично не ограничено, но взаимодействие может предпочтительно проводиться в течение 0.5-10 часов.
Способ получения 4
Соединение, представленное формулой 1 настоящего изобретения, может быть получено, как показано на Реакционной Схеме 5 ниже, посредством включения стадии для проведения реакции восстановления соединения, представленного формулой 1b, с получением соединения, представленного формулой 1c (Стадия 1).
[Реакционная Схема 5]

(где в Реакционной Схеме 5
R1 является таким, как определено в формуле 1; и
Соединения, представленные формулами 1b и 1c, входят в состав соединения, представленного формулой 1).
Далее способ получения соединения, представленного формулой 1 настоящего изобретения, будет описан подробно по стадиям.
В способе получения соединения, представленного формулой 1 настоящего изобретения, стадия 1) представляет собой получение соединения, представленного формулой 1c посредством проведения реакции восстановления соединения, представленного формулой 1b в присутствии основания. Более конкретно, соединение, представленное формулой 1b, которое является одним из соединений, представленных формулой 1, подвергается реакции восстановления в присутствии основания, получив таким образом соединение, представленное формулой 1c, в котором карбонильная группа соединения, представленного формулой 1b восстанавливается до гидроксильной группы.
В дополнение, в качестве основания могут использоваться борогидрид натрия (NaBH3) или алюмогидрид лития (LiAlH4), и предпочтительно может быть использован борогидрид натрия (NaBH3).
В дополнение, в качестве реакционного растворителя может быть использован, тетрагидрофуран (ТГФ), дихлорметан (ДХМ), толуол, и ацетонитрил, и предпочтительно может быть использован тетрагидрофуран (ТГФ).
Кроме того, реакционная температура предпочтительно находится между 0° до температуры кипения растворителя, и реакционное время специфично не ограничено, но взаимодействие может предпочтительно проводиться в течение 0.5-10 часов.
Фармацевтическая композиция настоящего изобретения характеризуется активацией фермента GPR40.
GPR40 представляет собой рецептор, связанный с G-белком (GPCR), который в основном экспрессируется в клетках поджелудочной железы, секретирующих инсулин. Профиль экспрессии GPR40 обладает потенциальной практичностью для лечения различных метаболических нарушений, включая ожирение и диабет.
В связи с этим, в качестве результата оценки активности рецептора GPR40 соединения, представленного формулой 1, его оптического изомера или его фармацевтически приемлемой соли, было обнаружено, что соединения всех примеров настоящего изобретения активировали рецептор GPR40 на 50% (EC50) при низких концентрациях, и, таким образом, эффекты активации соединениями настоящего изобретения были превосходящими (см. Экспериментальные Примеры 1 и 2, и Фиг. 1).
В дополнение, в качестве результата оценки ингибиторной активности CYP-фермента лекарственными метаболитами соединения, представленного формулой 1, его оптическим изомером или его фармацевтически приемлемой солью, было установлено, что соединения всех примеров настоящего изобретения имели низкую ингибирующую активность CYP-фермента, не вызывая токсичности, обусловленной концентрацией, во время совместного введения с другими лекарственными средствами, и, таким образом, соединения настоящего изобретения могут быть совместно введены с другими лекарственными средствами при осложнениях заболеваний (См. Экспериментальный пример 3).
Кроме того, в качестве результата проведения перорального теста толерантности к глюкозе соединения, представленного формулой 1, его оптического изомера или его фармацевтически приемлемой соли можно увидеть, что соединения всех Примеров настоящего изобретения показали аналогичные или превосходящие эффекты снижения сахара в крови по сравнению с активатором GPR40, который был известен в уровне техники, и, таким образом, соединения настоящего изобретения имели значительно лучший эффект активации GPR40 in vivo (см. Экспериментальные примеры 4, 5 и 6).
В дополнение в качестве результата проведения эксперимента для оценки скорости увеличения концентрации GLP-1 в крови после перорального введения соединения, представленного формулой 1, его оптического изомера или его фармацевтически приемлемой соли, было установлено, что по сравнению с группой, обработанной глюкозой (Veh), соединение Сравнительного Примера 1 не показало эффекта увеличения концентрации GLP-1 в крови после введения, но соединение примера 9 увеличивало концентрацию GLP-1 в крови при введении крысам SD (см. Экспериментальный пример 7 и фиг. 2).
Кроме того, было подтверждено, что совместное введение нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения и типичного лекарственного средства, такого как ингибитор на основе дипептидилпептидазы IV (DPPIV), на основе на сульфонилмочевины, на основе тиазолидиндиона (TZD), на основе бигуанида, имело превосходящий эффект снижения уровня глюкозы в крови по сравнению с введением указанных лекарств самостоятельно (см. Таблицы 8, 9, 10 и 11 в экспериментальных примерах 8, 9, 10 и 11 и Фиг. 3, 4, 5 и 6).
Таким образом, соединения, представленные формулой 1, обладают превосходящим эффектом активации белка GPR40, что приводит к превосходящему эффекту секреции инсулина и могут быть совместно введены вместе с другими лекарственными средствами, а также обладают значительно превосходящим эффектом активации белка GPR40 in vivo, и таким образом композиция, содержащая соединение, представленное формулой 1, в качестве активного ингредиента может выгодно применяться в качестве фармацевтической композиции для предотвращения или лечения метаболических нарушений, таких как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
Соединение, представленное формулой 1, настоящего изобретения может быть введено в нескольких лекарственных формах для перорального или парентерального введения во время клинического введения и может быть составлено с использованием разбавителя или эксципиента, такого как наполнитель, экстендер, связующее вещество, смачивающий агент, разрыхлитель или поверхностно-активное вещество, которые обычно используют.
Твердые препараты для перорального введения включают таблетку, пилюлю, порошок, гранулу, капсулу, пастилку и т.д. Эти твердые препараты могут быть приготовлены путем смешивания по меньшей мере одного соединения настоящего изобретения с по меньшей мере, одним эксципиентом, таким как крахмал, карбонат кальция, сахароза или лактоза, или желатин, или аналогичные. В дополнение к простым эксципиентам также используются любриканты, такие как стеарат магния и тальк. Примеры жидкого препарата для перорального введения могут включать суспензию, жидкость для внутреннего применения, эмульсию, сироп, и т.д. В дополнение к обычно применяемым простым разбавителям, таким как вода и жидкий парафин, различные эксципиенты, такие как увлажняющие агенты, подсластители, ароматизаторы и консерванты могут быть включены в жидкий препарат.
Препараты для перорального введения включают стерилизованный водный раствор, неводный растворитель, суспензионный растворитель, эмульсию, лиофилизатор и суппозиторий. В качестве неводного растворителя и суспензионного растворителя можно использовать пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, инъецируемый сложный эфир, такой как этилолат, или т.п. В качестве субстрата для суппозиториев может быть использован витепсол, макрогол, твин 61, какао-бумагу, лаурин, глицерин, желатин и т.д.
В дополнение, эффективная доза соединения настоящего изобретения на организм человека может варьироваться в зависимости от возраста, массы тела, пола, формы введения, состояния здоровья и тяжести заболевания пациента и обычно составляет около 0,001-100 мг/кг/день, и предпочтительно может быть использовано 0,01-35 мг/кг/день. На основании взрослого пациента массой 70 кг доза обычно составляет 0,07-7000 мг/день, и предпочтительно может быть использовано 0,7-2500 мг/день, и доза может вводиться один или несколько раз в день в заранее определенный интервал времени согласно решению врача или фармацевта.
Далее здесь настоящее изобретение будет описано детально со ссылкой на примеры и экспериментальные примеры.
Однако следующие примеры не предназначены для ограничения объема настоящего изобретения, а скорее предназначены для иллюстрации настоящего изобретения.
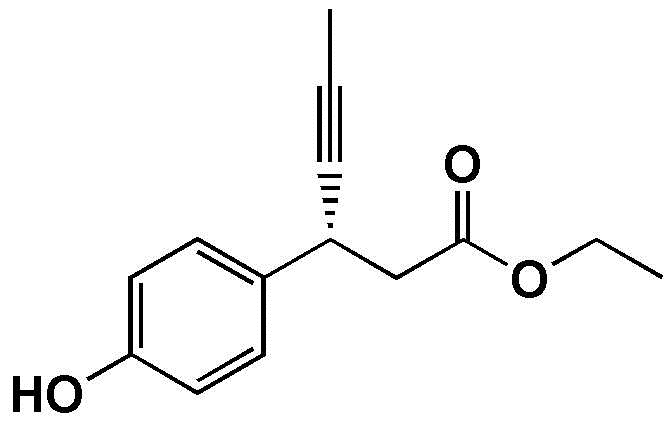
Пример Получения 1. Получение этил 3-(4-гидроксифенил)гекс-4-иноата
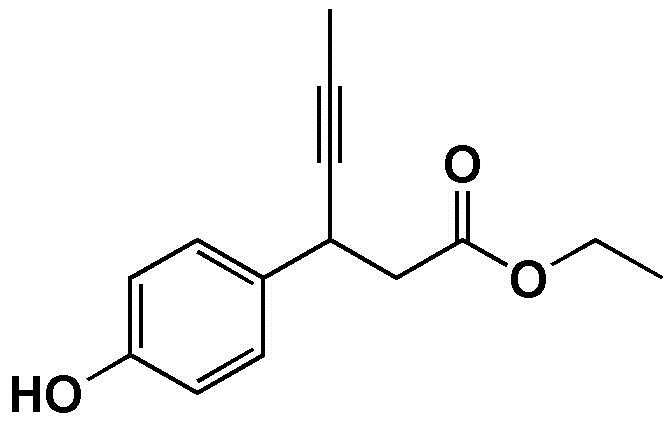
В атмосфере азота 3-(4-гидроксифенил)-гекс-4-иновую кислоту (20.0 г) и этанол (200 мл) загрузили в 250-мл колбу и перемешивали для растворения, и затем, серную кислоту (9.6 мл) медленно добавили по каплям при комнатной температуре. После этого реакционную смесь перемешивали при кипении с обратным холодильником в течение 6 часов или дольше. После завершения реакции, дистиллированную воду (150 мл) медленно добавили по каплям, с последующей экстракцией с использованием этилацетата (200 мл). Экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке (19.5 г, 85.7%).
1H NMR (400MHz, CDCl3): δ 7.25(2H, d), 6.78(2H, d), 4.95(1H, s), 4.14(2H, m), 4.04(1H, m), 2.68(2H, m), 1.84(3H, d), 1.29(3H, t).
Пример Получения 2. Получение (S)-этил 3-(4-гидроксифенил)гекс-4-иноата

В атмосфере азота (S)-3-(4-гидроксифенил)-гекс-4-иновую кислоту (20.0 г) и этанол (200 мл) загрузили в 250-мл колбу и перемешивали для растворения, и затем серную кислоту (9.6 мл) медленно добавили по каплям при комнатной температуре. После этого реакционную смесь перемешивали при кипении с обратным холодильником в течение 6 часов или дольше. После завершения реакции, дистиллированную воду (150 мл) медленно добавили по каплям, с последующей экстракцией с использованием этилацетата (200 мл). Экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке (21.2 г, 93.2%).
1H NMR (400MHz, CDCl3): δ 7.25(2H, d), 6.78(2H, d), 4.95(1H, s), 4.14(2H, m), 4.04(1H, m), 2.68(2H, m), 1.84(3H, d), 1.29(3H, t).
Пример Получения 3. Получение (S)-этил 3-(4-гидроксифенил)гекс-4-иноата
В атмосфере азота (R)-3-(4-гидроксифенил)-гекс-4-иновую кислоту (20.0 г) и этанол (200 мл) загрузили в 250-мл колбу и перемешивали для растворения, и затем серную кислоту (9.6 мл) медленно добавили по каплям при комнатной температуре. После этого реакционную смесь перемешивали при кипении с обратным холодильником в течение 6 часов или дольше. После завершения реакции, дистиллированную воду (150 мл) медленно добавили по каплям, с последующей экстракцией с использованием этилацетата (200 мл). Экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке (20.6 г, 90.6%).
1H NMR (400MHz, CDCl3): δ 7.25 (2H, d), 6.78 (2H, d), 4.95 (1H, s), 4.14 (2H, m), 4.04 (1H, m), 2.68 (2H, m), 1.84 (3H, d), 1.29 (3H, t).
Пример Получения 4. Получение (3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)фенил)метанола

Стадия 1: Получение 1,4-диоксаспиро[4.5]дец-7-ен-8-ил трифторметансульфоната
В атмосфере азота 1.4-диоксаспиро[4.5]декан-8-он (30.0 г) и толуол (300 мл) загрузили в 1000-мл колбу и перемешивали для растворения, и затем добавили N-фенил бис(трифторметансульфонимид) (64.3 г). После этого 0.7 M раствор калия бис(триметилсилил)амида (257 мл) медленно добавили по каплям туда с использованием капельной воронки при -78°, и затем смесь перемешивали в течение 4 часов или дольше при этом температура медленно выросла до комнатной температуры. После завершения реакции, дистиллированную воду (200 мл) медленно добавили по каплям, с последующей экстракцией с использованием этилацетата (300 мл), и затем экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке (54.7 г, 98.8%).
1H NMR (400MHz, CDCl3): δ 5.68 (1H, t), 4.01 (4H, s), 2.55 (2H, t), 2.42 (2H, d), 1.92 (2H, t).
Стадия 2: Получение 3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензальдегида
В атмосфере азота 1.4-диоксаспиро[4.5]дец-7-ен-8-ил трифторметансульфонат (54.70 г) и толуол (300 мл) загрузили в 1000-мл колбу и перемешивали для растворения, и затем добавили 3-формилфенилбороновую кислоту (28.7 г) и карбонат цезия (156 г). Реакционную смесь охладили до 0°, и тетракис(трифенилфосфин)палладий (11.09 г) медленно добавили, и затем смесь перемешивали в течение 3 часов или дольше, при этом температура снова повысилась до комнатной температуры. После завершения реакции дистиллированную воду (200 мл) медленно добавили по каплям, с последующей экстракцией с использованием этилацетата (300 мл), и затем экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке (45.9 г, 99%).
1H NMR (400MHz, CDCl3): δ 10.03 (1H, s), 7.92 (1H, s), 7.76 (1H, d), 7.67 (1H, d), 7.47 (1H, t), 6.11 (1H, s), 4.05 (4H, s), 2.71 (2H, t), 2.51 (2H, s), 1.97 (2H, t).
Стадия 3: Получение (3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)фенил)метанола
В атмосфере азота 3-(1.4-диоксаспиро[4.5]дец-7-ен-8-ил)бензальдегид (46.9 г), тетрагидрофуран (160 мл), и метанол (40 мл) добавили в 500-мл колбу и перемешивали для растворения, и затем смесь охладили до 0°. После этого борогидрид натрия (10.9 г) медленно добавили, и смесь перемешивали в течение 3 часов или дольше, при этом температура повысилась до комнатной температуры. После завершения реакции, дистиллированную воду (150 мл) медленно добавили по каплям, с последующей экстракцией с использованием этилацетата (150 мл), и затем экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке (37.8 г, 81.7%).
1H NMR (400MHz, CDCl3): δ 7.34 (1H, s), 7.25 (3H, m), 6.01 (1H, m), 4.69 (2H, d), 4.04 (4H, s), 2.68 (2H, m), 2.48 (2H, s), 1.94 (2H, t), 1.80 (1H, t).
Пример Получения 5. Получение (4-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)фенил)метанола

Стадия 1: Получение 4-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензальдегида
В атмосфере азота 1,4-диоксаспиро[4.5]дец-7-ен-8-ил трифторметансульфонат (3.0 г) и толуол (50 мл) загрузили в 250-мл колбу и перемешивали для растворения, и затем добавили 3-формилфенилбороновую кислоту (1.8 г) и карбонат цезия (8.47 г), с последующим охлаждением до 0°. После этого тетракис(трифенилфосфин)палладий (601 мг) медленно добавили, и затем смесь перемешивали в течение 3 часов или дольше, при этом температура повысилась. После завершения реакции дистиллированную воду (500 мл) медленно добавили по каплям, с последующей экстракцией с использованием этилацетата (100 мл), и затем экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке (2.0 г, 78.7%).
1H NMR (400MHz, CDCl3): δ 10.00 (1H, s), 7.84 (2H, d), 7.57 (2H, d), 6.19 (1H, s), 4.06 (4H, s), 2.71 (2H, t), 2.53 (2H, s), 1.97 (2H, t).
Стадия 2: Получение (4-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)фенил)метанола
В атмосфере азота 4-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензальдегид (2.0 г), тетрагидрофуран (40 мл), и метанол (10 мл) добавили в 250-мл колбу и перемешивали для растворения, и затем смесь охладили до 0°. После этого борогидрид натрия (619 мг) медленно добавили, и смесь перемешивали в течение 3 часов или дольше, при этом температура повысилась до комнатной температуры. После завершения реакции, дистиллированную воду (50 мл) медленно добавили по каплям, с последующей экстракцией с использованием этилацетата (100 мл), и затем экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке (1.6 г, 52.9%).
1H NMR (400MHz, CDCl3): δ 7.40 (2H, d), 7.32 (2H, d), 6.01 (1H, m), 4.70 (2H, d), 4.13 (4H, s), 2.68 (2H, t), 2.49 (2H, s), 1.93 (2H, t), 1.60 (1H, t).
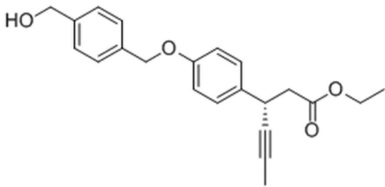
Пример Получения 6. Этил 3-(4-(4-((метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноат
Стадия 1: Получение (4-(бромметил)фенил)метанола

В атмосфере азота 4-(бромметил)бензоат (5.0 г) и MC (20 мл) добавили в 1-л колбу и перемешивали для растворения, и затем DIBAL-H (70 мл) медленно добавили по каплям. После перемешивания в течение 5 часов, после завершения реакции температуру снизили до 0°, и дистиллированную воду медленно добавили по каплям, с последующей экстракцией с использованием MC. Экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.42(2H, d), 7.38(2H, d), 4.73(2H, s), 4.52(2H, m).
Стадия 2: Получение этил 3-(4-(4-(гидроксиметил)бензилокси)фенил)гекс-4-иноата

В атмосфере азота этил 3-(4-гидроксифенил)гекс-4-иноат (4.0 г), полученный в Примере Получения 1 и (4-(бромметил)фенил)метанол (5.0 г), полученный на стадии 1, добавили в ДМФ (50 мл) в 500-мл колбу и перемешивали для растворения, с последующим добавлением по каплям Cs2CO3 (9.0 г), и затем смесь перемешивали при комнатной температуре в течение 12 часов. После завершения реакции, дистиллированную воду медленно добавили по каплям, экстрагировали этилацетатом, промыли солевым раствором, высушили над безводным сульфатом магния, и затем сконцентрировали. После этого реакционный продукт разделили с помощью колоночной хроматографии на силикагеле с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.42(2H, d), 7.38(2H, d), 7.29(2H, d), 6.93(2H, d), 5.06(2H, s), 4.73(2H, d), 4.15(2H, m), 4.06(1H, m), 2.68(2H, m), 1.84(3H, s), 1.69(1H, m), 1.24(3H, m).
Стадия 3: Получение этил 3-(4-(4-(метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноата

В атмосфере азота 3-(4-(4-(гидроксиметил)бензилокси)фенил)гекс-4-иноат (3.0 г), полученный на стадии 2, добавили к MC (30 мл) в 500-мл колбу и перемешивали для растворения, и затем TEA (4.0 мл) добавили по каплям при 0°. Через 30 минут MsCl (2.1 мл) медленно добавили по каплям. После завершения реакции через один час, дистиллированную воду медленно добавили по каплям, с последующей экстракцией с использованием MC. Экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.49(4H, m), 7.29(2H, d), 6.93(2H, d), 5.27(2H, s), 5.08(2H, s), 4.15(2H, m), 4.06(1H, m), 2.95(3H, s), 2.68(2H, m), 1.84(3H, s), 1.69(1H, m), 1.24(3H, m).
Пример Получения 7. Получение (S)-этил 3-(4-(4-((метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноата
Стадия 1: Получение (S)-этил 3-(4-(4-(гидроксиметил)бензилокси)фенил)гекс-4-иноата
Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 2 в Примере Получения 6, за исключением того, что (S)-этил 3-(4-гидроксифенил)гекс-4-иноат использовали вместо этил 3-(4-гидроксифенил)гекс-4-иноата.
1H NMR (400MHz, CDCl3): δ 7.42(2H, d), 7.38(2H, d), 7.29(2H, d), 6.93(2H, d), 5.06(2H, s), 4.73(2H, d), 4.15(2H, m), 4.06(1H, m), 2.68(2H, m), 1.84(3H, s), 1.69(1H, m), 1.24(3H, m).
Стадия 2: Получение (S)-этил 3-(4-(4-((метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноата

Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 3 в Примере Получения 6, за исключением того, что (S)-этил 3-(4-(4-(гидроксиметил)бензил)фенил)гекс-4-иноат, полученный на стадии 1, использовали вместо этил 3-(4-(4-(гидроксиметил)бензилокси)фенил)гекс-4-иноата.
1H NMR (400MHz, CDCl3): δ 7.49(4H, m), 7.29(2H, d), 6.93(2H, d), 5.27(2H, s), 5.08(2H, s), 4.15(2H, m), 4.06(1H, m), 2.95(3H, s), 2.68(2H, m), 1.84(3H, s), 1.69(1H, m), 1.24(3H, m).
Пример Получения 8. Получение 6-метокси-1,2,3,4-тетрагидроизохинолина
Стадия 1: Получение этил 3-метоксифенэтилкарбамата

В атмосфере азота 2-(3-метоксифенил)этиламин (25 г) добавили к MC (300 мл) и перемешивали для растворения, и затем TEA (24.2 мл) добавили по каплям при 0°. Через 30 минут, этилхлорформиат (16.6 мл) медленно добавили по каплям. После завершения реакции через один час, дистиллированную воду медленно добавили по каплям, с последующей экстракцией с использованием MC. Экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.25(1H, m), 6.79(3H, m), 4.70(1H, s), 4.13(2H, m), 3.81(3H, s), 3.46(2H, m), 2.80(2H, m), 1.25(3H, m).
Стадия 2: Получение 6-метокси-3,4-дигидроизохинолинe-1(2H)-она

В атмосфере азота 36 г этил 3-метоксифенэтилкарбамата, полученного на стадии 1, перемешивали для растворения в 120 г полифисфорной кислоты в 500-мл колбе, и смесь нагревали при кипении с обратным холодильником в течение 3 часов или дольше. После того, как температура снизилась до комнатной температуры, этилацетат и дистиллированную воду медленно добавили по каплям, с последующей экстракцией три раза или более. Экстрагированный органический слой промыли солевым раствором, и высушили над безводным сульфатом магния, и затем сконцентрировали. После этого реакционный продукт разделили с помощью колоночной хроматографии на силикагеле с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 8.03(1H, d), 6.87(1H, d), 6.72(1H, s), 6.44(1H, s), 3.86(3H, s), 3.57(2H, m), 2.98(2H, m).
Стадия 3: Получение 6-метокси-1,2,3,4-тетрагидроизохинолина

В атмосфере азота 10 г 6-метокси-3,4-дигидроизохинолин-1(2H)-она, полученного на стадии 2, перемешивали для растворения в 150 мл ТГФ, и 4.3 г LAH медленно добавили по каплям при 0°. После нагревания при кипении с обратным холодильником в течение 5 часов или дольше, после завершения реакции, дистиллированную воду медленно добавили по каплям, экстрагировали этилацетатом, промыли солевым раствором, высушили над безводным сульфатом магния, и затем сконцентрировали. После этого соединение, указанное в заголовке, получили затвердением.
1H NMR (400MHz, CDCl3): δ 6.94(1H, d), 6.73(1H, d), 6.65(1H, s), 4.14(2H, s), 3.80(3H, s), 3.13(2H, m), 2.79(2H, m).
Пример Получения 9. Получение 4-(4-(метилсульфонил)фенил)-1,2,3,6-тетрагидропиридина гидрохлорида
Стадия 1: Получение трет-бутил 4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата

В атмосфере азота 3.31 г трет-бутил 4-(трифторметилсульфонилокси)-5,6-дигидропиридин-1(2H)-карбоксилата и 50 мл толуола загрузили в 1000-мл колбу и перемешивали для растворения, и затем добавили 2.0 г 4-(метилсульфонил)фенилбороновой кислоты и 6.6 г карбоната цезия. После этого смесь охладили до 0°, с последующим медленным добавлением 1.16 г тетракис(трифенилфосфин)палладия, и затем перемешивали в течение 3 часов или дольше, при этом температура снова повысилась до комнатной температуры. После завершения реакции, дистиллированную воду медленно добавили по каплям, и затем экстрагировали этилацетатом. Экстрагированный органический слой высушили при пониженном давлении, и затем разделили с помощью колоночной хроматографии на силикагеле с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.92(2H. d), 7.56(2H, d), 6.21(1H, s), 4.14(2H, d), 3.68(2H, m), 3.07(3H, s), 2.56(2H, s), 1.49(9H, s).
Стадия 2: Получение 4-(4-(метилсульфонил)фенил)-1,2,3,6-тетрагидропиридина гидрохлорида

1.4 г трет-бутил 4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилат (1.4 г), полученный на стадии 1, растворили в 20 мл MC, 10.4 мл 4 н HCl добавили по каплям. После завершения реакции через пять часов диэтиловый эфир добавили по каплям и произошло отвердевание с получением соединения, указанного в заголовке.
1H NMR (400MHz, D2O): δ 7.92(2H. d), 7.56(2H, d), 6.21(1H, s), 4.14(2H, d), 3.68(2H, m), 3.07(3H, s), 2.56(2H, s).
Пример Получения 10. Получение 4-(1,2,3,6-тетрагидропиридин-4-ил)фенола гидрохлорида
Стадия 1: Получение трет-бутил 4-(4-гидроксифенил)-5,6-дигидропиридин-1(2H)-карбоксилата

Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 1 в Примере Получения 9, за исключением того, что 4-гидроксифенилбороновую кислоту использовали вместо 4-(метилсульфонил)фенилбороновой кислоты.
1H NMR (400MHz, CDCl3): δ 7.26(2H, d), 6.83(2H, d), 5.93(1H, s), 5.47(1H, s), 4.07(2H, s), 3.66(2H, m), 2.50(2H, s), 1.52(9H, s).
Стадия 2: Получение 4-(1,2,3,6-тетрагидропиридин-4-ил)фенола гидрохлорид
Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 2 в Примере Получения 9, за исключением того, что трет-бутил 4-(4-гидроксифенил)-5,6-дигидропиридин-1(2H)-карбоксилат, полученный на стадии 1, использовали вместо трет-бутил 4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата.
1H NMR (400MHz, D2O): δ 7.26(2H, d), 6.83(2H, d), 5.93(1H, s), 5.47(1H, s), 4.07(2H, s), 3.66(2H, m), 2.50(2H, s).
Пример Получения 11. Получение 4-(4-(3-(метилсульфонил)пропокси)фенил)-1,2,3,6-тетрагидропиридина гидрохлорида
Стадия 1: Получение 3-(метилтио)пропил 4-метилбензолсульфонат

В атмосфере азота 25.4 г 3-(метилтио)пропан-1-ола добавили в 500 мл MC в 500-мл колбу и перемешивали для растворения, и затем туда добавили 44 мл TEA по каплям при 0°. Через 30 минут 46 г TsCl медленно добавили по каплям, и после завершения реакции через один час, дистиллированную воду медленно добавили по каплям, с последующей экстракцией с использованием MC. Экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.81(2H, d), 7.38(2H, d), 4.16(2H, m), 2.53(2H, m), 2.47(3H, s), 2.05(3H, s), 1.94(2H, m).
Стадия 2: Получение 3-(метилсульфонил)пропил 4-метилбензолсульфонат
В атмосфере азота 62 г 3-(метилтио)пропил 4-метилбензолсульфоната, полученного на стадии 1, добавили в ТГФ/дистиллированную воду (150/100 мл) в колбе и перемешивали для растворения, и затем 310 г оксона добавили по каплям при 0°. После перемешивания при комнатной температуре в течение 12 часов или дольше, после завершения реакции, дистиллированную воду медленно добавили по каплям, экстрагировали этилацетатом, промыли солевым раствором, и высушили над безводным сульфатом магния с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.81(2H, d), 7.38(2H, d), 4.20(2H, m), 3.13(2H, m), 2.93(3H, s), 2.48(3H, s), 2.23(2H, m).
Стадия 3: Получение трет-бутил 4-(4-(3-метилсульфонил)пропокси)фенил)-5,6-дигидропиридин-1(2H)-карбоксилат

Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 2 в Примере Получения 6, за исключением того, что использовали трет-бутил 4-(4-гидроксифенил)-5,6-дигидропиридин-1(2H)-карбоксилат, полученный на стадии 1, и 3-(метилсульфонил)пропил 4-метилбензолсульфонат, полученный на стадии 2.
1H NMR (400MHz, CDCl3): δ 7.34(2H, d), 6.85(2H, d), 6.00(1H, s), 4.12(2H, s), 3.28(2H, m), 3.18(2H, s), 2.97(3H, s), 2.72(2H, m), 2.56(2H, m), 2.36(2H, m), 1.52(9H, s).
Стадия 4: Получение 4-(4-(3-(метилсульфонил)пропокси)фенил)-1,2,3,6-тетрагидропиридина гидрохлорида

Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 2 в Примере Получения 9, за исключением того, что трет-бутил 4-(4-(3-метилсульфонил)пропокси)фенил)-5,6-дигидропиридин-1(2H)-карбоксилат, полученный на стадии 3, использовали вместо трет-бутил 4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата.
1H NMR (400MHz, D2O): δ 7.34(2H, d), 6.85(2H, d), 6.00(1H, s), 4.12(2H, s), 3.28(2H, m), 3.18(2H, s), 2.97(3H, s), 2.72(2H, m), 2.56(2H, m), 2.36(2H, m).
Пример Получения 12. Получение (3S)-этил 3-(4-(4-(1-бромэтил)бензилокси)фенил)гекс-4-иноата
Стадия 1: Получение 1-(4-(бромметил)фенил)этанона

В атмосфере азота 5.0 г 1-п-толил этанона добавили в 100 мл CCl4 в колбе и перемешивали для растворения, и 14.6 г NBS и 6.7 г AIBN добавили по каплям при 0°. После нагревания при кипении с обратным холодильником в течение 5 часов или дольше, после завершения реакции, дистиллированную воду медленно добавили по каплям, экстрагировали MC, промыли солевым раствором, высушили над безводным сульфатом магния, и затем сконцентрировали. После этого реакционный продукт разделили с помощью колоночной хроматографии на силикагеле с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.95(2H, d), 7.50(2H, d), 4.52(2H, s), 2.62(3H, s).
Стадия 2: Получение (S)-этил 3-(4-(4-(ацетилбензилокси)фенил)гекс-4-иноата

Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 2 в Примере Получения 6, за исключением того, что использовали (S)-этил 3-(4-гидроксифенил)гекс-4-иноат, полученный в Примере Получения 2, и 1-(4-(бромметил)фенил)этанон, полученный на стадии 1.
1H NMR (400MHz, CDCl3): δ 7.99(2H, d), 7.53(2H, d) 7.31(2H, d), 6.92(2H, d), 5.13(2H, s), 4.15(2H, m), 4.09(1H, m), 2.75(2H, m), 2.64(3H, s), 1.84(3H, d), 1.24(3H, m).
Стадия 3: Получение (3S)-этил 3-(4-(4-(1-гидроксиэтил)бензилокси)фенил)гекс-4-иноата

В атмосфере азота 1.0 г (S)-этил 3-(4-(4-ацетилбензилокси)фенил)гекс-4-иноата, полученного на стадии 2, добавили к 50 мл ТГФ в колбе и перемешивали для растворения, и затем 0.16 г NaBH4 добавили по каплям при 0°. После перемешивания при комнатной температуре в течение 2 часов или дольше, после завершения реакции, дистиллированную воду медленно добавили по каплям, экстрагировали EA, промыли солевым раствором, высушили над безводным сульфатом магния, и затем сконцентрировали с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 8.02(2H, d), 7.57(2H, d) 7.36(2H, d), 6.99(2H, d), 5.21(2H, s), 4.23(2H, m), 4.17(1H, m), 3.81(1H, s), 2.75(2H, m), 2.64(3H, s), 1.84(3H, d), 1.24(3H, m).
Стадия 4: Получение (3S)-этил 3-(4-(4-(1-бромэтил)бензилокси)фенил)гекс-4-иноата

В атмосфере азота 0.76 г (3S)-этил 3-(4-(4-(1-гидроксиэтил)бензилокси)фенил)гекс-4-иноата, полученного на стадии 3, добавили к 50 мл MC в колбе и перемешивали для растворения, и затем 0.6 г трифенилфосфина и 0.75 г CBr4 добавили по каплям при 0°. После перемешивания при комнатной температуре в течение 2 часов или дольше, после завершения реакции, дистиллированную воду медленно добавили по каплям, экстрагировали EA, промыли солевым раствором, высушили над безводным сульфатом магния, и затем сконцентрировали с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 8.02(2H, d), 7.57(2H, d) 7.36(2H, d), 6.99(2H, d), 5.21(2H, s), 4.23(2H, m), 4.17(1H, m), 3.92(1H, s), 2.85(2H, m), 2.44(3H, s), 1.86(3H, d), 1.27(3H, m).
Пример Получения 13. Получение 2-(пиперазин-1-ил)бензо[d]тиазола гидрохлорида
Стадия 1: Получение трет-бутил 4-(бензо[d]тиазол-2-ил)пиперазин-1-карбоксилата

В атмосфере азота 2.0 г трет-бутил пиперазин-1-карбоксилата добавили в AN/дистиллированную воду (100/50 мл) в колбе и перемешивали для растворения, и затем 2.1 мл DIPEA добавили по каплям при 0°. После этого 0.9 г 2-хлорбензо[d]тиазол добавили туда по каплям, и смесь нагревали при кипении с обратным холодильником в течение 2 часов или дольше. После завершения реакции, дистиллированную воду медленно добавили по каплям, экстрагировали EA, промыли солевым раствором, высушили над безводным сульфатом магния, и затем сконцентрировали с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.61(1H, d), 7.60(1H, d), 7.29(1H, m), 7.09(1H, m), 3.77(4H, m), 2.62(4H, m), 1.52(9H, s).
Стадия 2: Получение 2-(пиперазин-1-ил)бензо[d]тиазола гидрохлорида

Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 2 в Примере Получения 9, за исключением того, что трет-бутил 4-(бензо[d]тиазол-2-ил)пиперазин-1-карбоксилат, полученный на стадии 1, использовали вместо трет-бутил 4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата.
1H NMR (400MHz, D2O): δ 7.61(1H, d), 7.60(1H, d), 7.29(1H, m), 7.09(1H, m), 3.77(4H, m), 2.62(4H, m).
Пример Получения 14. Получение 2-(пиперазин-1-ил)-5-пропилпиримидина гидрохлорида
Стадия 1: Получение трет-бутил 4-(5-пропилпиримидин-2-ил)пиперазин-1-карбоксилата

Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 1 в Примере Получения 13, за исключением того, что 2-хлор-5-пропилпиримидин использовали вместо 2-хлорбензо[d]тиазола.
1H NMR (400MHz, CDCl3): δ 8.19(2H, s), 3.77(4H, m), 2.62(4H, m), 2.41(2H, m), 1.61(2H, m), 1.52(9H, s), 0.96(3H, m).
Стадия 2: Получение 2-(пиперазин-1-ил)-5-пропилпиримидина гидрохлорида

Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 2 в Примере Получения 9, за исключением того, что трет-бутил 4-(5-пропилпиримидин-2-ил)пиперазин-1-карбоксилат, полученный на стадии 1, использовали вместо трет-бутил 4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата.
1H NMR (400MHz, D2O): δ 8.19(2H, s), 3.77(4H, m), 2.62(4H, m), 2.41(2H, m), 1.61(2H, m), 0.96(3H, m).
Пример Получения 15. Получение 6-(пиперазин-1-ил)никотинонитрил гидрохлорида
Стадия 1: Получение трет-бутил 4-(5-цианопиридин-2-ил)пиперазин-1-карбоксилата

Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 1 в Примере Получения 13, за исключением того, что 6-хлороникотинонитрил использовали вместо 2-хлорбензо[d]тиазола.
1H NMR (400MHz, CDCl3): δ 8.41(1H, s)7.61(1H, d), 6.59(1H, d), 3.77(4H, m), 2.62(4H, m), 1.52(9H, s).
Стадия 2: Получение 6-(пиперазин-1-ил)никотинонитрил гидрохлорида

Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 2 в Примере Получения 9, за исключением того, что трет-бутил 4-(5-цианопиридин-2-ил)пиперазин-1-карбоксилат, полученный на стадии 1, использовали вместо трет-бутил 4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата.
1H NMR (400MHz, D2O): δ 8.41(1H, s)7.61(1H, d), 6.59(1H, d), 3.77(4H, m), 2.62(4H, m).
Пример Получения 16. Получение (S)-этил 3-(4-(4-(2-(метилсульфонилокси)этил)бензилокси)фенил)гекс-4-иноата
Стадия 1: Получение 2-(4-(бромметил)фенил)этанола
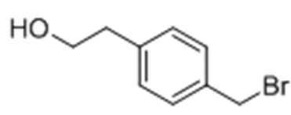
В атмосфере азота 5 г 2-(4-(бромметил)фенил)уксусной кислоты и 100 мл ТГФ загрузили в колбу и перемешивали для растворения, и затем 70 мл раствора боран-ТГФ медленно добавили по каплям при 0°. После перемешивания в течение 2 часов, после завершения реакции, температуру снизили до 0°, и дистиллированную воду медленно добавили по каплям, с последующей экстракцией с использованием EA. Экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 37(2H, d), 7.24(2H, d), 4.51(2H, s), 3.89(2H, m), 2.89(2H, m).
Стадия 2: Получение (S)-этил 3-(4-(4-(2-гидроксиэтил)бензилокси)фенил)гекс-4-иноата

Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 2 в Примере Получения 5, за исключением того, что 2-(4-(бромметил)фенил)этанол, полученный на стадии 1, использовали вместо 4-(бромметил)фенил)метанола.
1H NMR (400MHz, CDCl3): δ 7.40(2H, d), 7.30(2H, d), 7.27(2H, d), 6.95(2H, d), 5.04(2H, s), 4.18(2H, m), 4.11(1H, m), 3.89(2H, m), 2.91(2H, m), 2.71(2H, m), 1.84(3H, s), 1.38(1H, m), 1.25(3H, m).
Стадия 3: Получение (S)-этил 3-(4-(4-(2-(метилсульфонилокси)этил)бензилокси)фенил)гекс-4-иноата

Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 3 в Примере Получения 6, за исключением того, что (S)-этил 3-(4-(4-(2-гидроксиэтил)бензилокси)фенил)гекс-4-иноат, полученный на стадии 2, использовали вместо этил 3-(4-(4-(гидроксиметил)бензилокси)фенил)гекс-4-иноата.
1H NMR (400MHz, CDCl3): δ 7.40(2H, d), 7.30(2H, d), 7.27(2H, d), 6.95(2H, d), 5.04(2H, s), 4.18(2H, m), 4.11(1H, m), 3.99(2H, m), 2.95(3H, s), 2.93(2H, m), 2.71(2H, m), 1.84(3H, s), 1.25(3H, m).
Пример 1. Получение 3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты

Стадия 1: Получение этил 3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата
В атмосфере азота (3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)фенил)метанол (19.54 г), полученный в Примере Получения 4, и тетрагидрофуран (80 мл) загрузили в 500-мл колбу и перемешивали для растворения, и затем этил 3-(4-гидроксифенил)гекс-4-иноат (18.42 г), полученный в Примере Получения 1, и трифенилфосфин (31.21 г) медленно добавили. После этого диизопропилазодикарбоксилат (23.4 мл) медленно добавили по каплям с использованием капельной воронки при 0°, и затем перемешивали в течение 4 часов или дольше, при этом температура повысилась до комнатной температуры. После завершения реакции, дистиллированную воду (200 мл) медленно добавили по каплям и экстрагировали этилацетатом (300 мл), и затем экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке (32.1 г, 87.9%).
1H NMR (400MHz, CDCl3): δ 7.46(1H, s), 7.31(5H, m), 6.93(2H, d), 6.02(1H, m), 5.04(2H, s), 4.13(2H, m), 4.08(1H, m), 4.04(4H, s), 2.69(4H, m), 2.49(2H, s), 1.94(2H, t), 1.84(3H, d), 1.31(3H, t).
Стадия 2: Получение 3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты
В атмосфере азота этил 3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат (32.1 г), полученный на стадии 1, метанол (50 мл), и дистиллированную воду (50 мл) загрузили в 500-мл колбу и перемешивали для растворения, и затем гидроксид калия (19.5 г) медленно добавили при комнатной температуре, с последующим перемешиванием в течение одного часа или дольше. После завершения реакции смесь подкислили с помощью 1 М водного раствора соляной кислоты до pH 2-3, экстрагировали этилацетатом (300 мл), и высушили при пониженном давлении с получением соединения, указанного в заголовке (24.1 г, 79.9%).
1H NMR (400MHz, CDCl3): δ 7.44(1H, s), 7.34(5H, m), 6.91(2H, d), 6.00(1H, t), 5.02(2H, s), 4.08(1H, m), 4.04(4H, s), 2.73(4H, m), 2.48(2H, s), 1.92(2H, t), 1.82(3H, s).
Пример 2. Получение L-лизина 3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата

В атмосфере азота 3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновую кислоту (24.1 г), полученную в примере 1, и этанол (170 мл) загрузили в 500-мл колбу и перемешивали для растворения, и затем туда добавили L-лизин (7.33 г). После этого реакционную температуру повысили до 50°, и смесь перемешивали в течение 30 минут при 50°, и снова охладили до комнатной температуры, с последующим перемешиванием в течение 30 минут. После завершения реакции, полученный осадок отфильтровали с получением соединения, указанного в заголовке (31.5 г, 73.3%).
1H NMR (400MHz, D2O): δ 7.11(3H, m), 6.99(3H, m), 6.64(2H, d), 5.65(1H, s), 4.59(2H, s), 3.79(5H, s), 3.60(1H, t), 2.88(2H, t), 2.35(2H, d), 2.23(2H, s), 2.14(2H, s), 1.75(2H, m), 1.59(7H, m), 1.38(2H, m).
Пример 3. Получение 4-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты

Стадия 1: Получение этил 4-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата
В атмосфере азота (4-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)фенил)метанол (1.5 г), полученный в Примере Получения 5 и тетрагидрофуран (20 мл) загрузили в 100-мл колбу и перемешивали для растворения, и затем медленно добавили этил 3-(4-гидроксифенил)гекс-4-иноат (1.41 г), полученный в Примере Получения 1, и трифенилфосфин (2.39 г). После этого диизопропилазодикарбоксилат (9.38 мл) медленно добавили по каплям с использованием капельной воронки при 0°, и затем перемешивали в течение 4 часов или дольше, при этом температура повысилась до комнатной температуры. После завершения реакции, дистиллированную воду (50 мл) медленно добавили по каплям, с последующей экстракцией с использованием этилацетата (100 мл), и затем экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке (1.38 г, 49.2%).
1H NMR (400MHz, CDCl3): δ 7.42(2H, d), 7.37(2H, d), 7.30(2H, d), 6.92(2H, d), 6.01(1H, s), 5.01(2H, s), 4.14(2H, m), 4.06(5H, m), 2.70(4H, m), 2.49(2H, s), 1.94(2H, t), 1.84(3H, d), 1.24(3H, t).
Стадия 2: Получение 4-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты
В атмосфере азота этил 4-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат (1.38 г), полученный на стадии 1, метанол (10 мл), и дистиллированную воду (10 мл) загрузили в 500-мл колбу и перемешивали для растворения, и затем гидроксид калия (1.25 г) медленно добавили при комнатной температуре, с последующим перемешиванием в течение одного часа или дольше. После завершения реакции смесь подкислили с помощью 1 М водного раствора соляной кислоты до pH 2-3, экстрагировали этилацетатом (50 мл), и высушили при пониженном давлении с получением соединения, указанного в заголовке (0.98 г, 75.6%).
1H NMR (400MHz, CDCl3): δ 7.41(2H, d), 7.36(2H, d), 7.29(2H, d), 6.92(2H, d), 6.01(1H, s), 5.01(2H, s), 4.04(5H, m), 2.77(4H, m), 2.49(2H, s), 1.96(2H, t), 1.83(3H, d).
Пример 4. Получение 3-(4-(3-(4-оксоциклогекс-1-енил)бензилокси)фенил)гекс-4-иновой кислоты

В атмосфере азота 3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновую кислоту (1 г), полученную в примере 1, и тетрагидрофуран (5 мл) загрузили и перемешивали для растворения, и затем добавили 6 н водный раствор соляной кислоты (5 мл), и смесь перемешивали при комнатной температуре в течение 1 часа или дольше. После завершения реакции, дистиллированную воду (50 мл) медленно добавили по каплям, с последующей экстракцией с использованием этилацетата (50 мл), и затем экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке (0.76 г, 84.6%).
1H NMR (400MHz, CDCl3): δ 7.48(1H, s), 7.40(5H, m), 6.94(2H, d), 6.13(1H, s), 5.07(2H, s), 4.05(1H, m), 3.10(1.5H, t), 2.93(1.5H, t), 2.82(2H, m), 2.67(2H, t), 1.85(3H, s).
Пример 5. Получение 3-(4-(3-(4-гидроксициклогекс-1-енил)бензилокси)фенил)гекс-4-иновой кислоты

В атмосфере азота 3-(4-(3-(4-оксоциклогекс-1-енил)бензилокси)фенил)гекс-4-иновую кислоту (1 г), полученную в примере 4, и этанол (10 мл) загрузили в 100-мл колбу и перемешивали для растворения, и затем добавили борогидрид натрия (0.3 г), с последующим перемешиванием при комнатной температуре в течение 3 часов или дольше. После завершения реакции смесь подкислили с помощью 1 М водного раствора соляной кислоты до pH 4-5, и экстрагировали этилацетатом (100 мл) и дистиллированной водой (100 мл). Экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке (0.81 г, 80.6%).
1H NMR (400MHz, CDCl3): δ 7.44(1H, s), 7.33(5H, m), 6.93(2H, d), 6.02(1H, s), 5.03(2H, s), 4.08(2H, s), 2.78(2H, m), 2.55(2.5H, m), 2.22(1H, m), 2.04(1H, m), 1.85(3H, s).
Пример 6. Получение L-лизина 3-(4-(3-(4-гидроксициклогекс-1-енил)бензилокси)фенил)гекс-4-иноата

В атмосфере азота 3-(4-(3-(4-гидроксициклогекс-1-енил)бензилокси)фенил)гекс-4-иновую кислоту (1 г), полученную в примере 5, и этанол (170 мл) загрузили в 100-мл колбу и перемешивали для растворения, и затем туда добавили L-лизин (0.7 г). После этого реакционную температуру повысили до 50°, и смесь перемешивали в течение 30 минут при 50°, и снова охладили до комнатной температуры, с последующим перемешиванием в течение 30 минут. После завершения реакции полученный осадок отфильтровали с получением соединения, указанного в заголовке (0.95 г, 69.1%).
1H NMR (400MHz, D2O): δ 7.11(3H, m), 6.99(3H, m), 6.64(2H, d), 5.65(1H, s), 4.59(2H, s), 3.79(1H, s), 3.60(1H, t), 2.88(2H, t), 2.35(2H, d), 2.23(2H, s), 2.14(2H, s), 1.75(2H, m), 1.59(7H, m), 1.38(2H, m).
Пример 7. Получение (3S)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты

Стадия 1: Получение этил-(3S)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата
В атмосфере азота (3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)фенил)метанол (19.54 г), полученный в Примере Получения 4, и тетрагидрофуран (80 мл) загрузили в 500-мл колбу и перемешивали для растворения, и затем медленно добавили (S)-этил 3-(4-гидроксифенил)гекс-4-иноат (18.42 г), полученный в Примере Получения 2, и трифенилфосфин (31.21 г). После этого диизопропилазодикарбоксилат (23.4 мл) медленно добавили по каплям с использованием капельной воронки при 0°, и затем перемешивали в течение 4 часов или дольше, при этом температура повысилась до комнатной температуры. После завершения реакции дистиллированную воду (200 мл) медленно добавили по каплям и экстрагировали этилацетатом (300 мл), и затем экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.46(1H, s), 7.31(5H, m), 6.93(2H, d), 6.02(1H, m), 5.04(2H, s), 4.13(2H, m), 4.08(1H, m), 4.04(4H, s), 2.69(4H, m), 2.49(2H, s), 1.94(2H, t), 1.84(3H, d), 1.31(3H, t).
Стадия 2: Получение (3S)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты
В атмосфере азота этил-(3S)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат (32.1 г), полученный на стадии 1, метанол (50 мл), и дистиллированную воду (50 мл) загрузили в 500-мл колбу и перемешивали для растворения, и затем гидроксид калия (19.5 г) медленно добавили при комнатной температуре, с последующим перемешиванием в течение одного часа или дольше. После завершения реакции смесь подкислили с помощью 1 М водного раствора соляной кислоты до pH 2-3, экстрагировали этилацетатом (300 мл), и высушили при пониженном давлении с получением соединения, указанного в заголовке (24.1 г, 66.2%).
1H NMR (400MHz, CDCl3): δ 7.44(1H, s), 7.34(5H, m), 6.91(2H, d), 6.00(1H, t), 5.02(2H, s), 4.08(1H, m), 4.04(4H, s), 2.73(4H, m), 2.48(2H, s), 1.92(2H, t), 1.82(3H, s).
Пример 8. Получение (3R)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты

Стадия 1: Получение этил-(3R)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата
В атмосфере азота (3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)фенил)метанол (19.54 г), полученный в Примере Получения 4, и тетрагидрофуран (80 мл) загрузили в 500-мл колбу и перемешивали для растворения, и затем медленно добавили (R)-этил 3-(4-гидроксифенил)гекс-4-иноат (18.42 г), полученный в Примере Получения 3, и трифенилфосфин (31.21 г). После этого диизопропилазодикарбоксилат (23.4 мл) медленно добавили по каплям с использованием капельной воронки при 0°, и затем перемешивали в течение 4 часов или дольше, при этом температура повысилась до комнатной температуры. После завершения реакции, дистиллированную воду (200 мл) медленно добавили по каплям и экстрагировали этилацетатом (300 мл), и затем экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.46(1H, s), 7.31(5H, m), 6.93(2H, d), 6.02(1H, m), 5.04(2H, s), 4.13(2H, m), 4.08(1H, m), 4.04(4H, s), 2.69(4H, m), 2.49(2H, s), 1.94(2H, t), 1.84(3H, d), 1.31(3H, t).
Стадия 2: Получение (3R)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты
В атмосфере азота этил-(3R)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат (32.1 г), полученный на стадии 1, метанол (50 мл), и дистиллированную воду (50 мл) загрузили в 500-мл колбу и перемешивали для растворения, и затем гидроксид калия (19.5 г) медленно добавили при комнатной температуре, с последующим перемешиванием в течение одного часа или дольше. После завершения реакции смесь подкислили с помощью 1 М водного раствора соляной кислоты до pH 2-3, экстрагировали этилацетатом (300 мл), и высушили при пониженном давлении с получением соединения, указанного в заголовке (17.3 г, 47.5%).
1H NMR (400MHz, CDCl3): δ 7.44(1H, s), 7.34(5H, m), 6.91(2H, d), 6.00(1H, t), 5.02(2H, s), 4.08(1H, m), 4.04(4H, s), 2.73(4H, m), 2.48(2H, s), 1.92(2H, t), 1.82(3H, s).
Пример 9. Получение L-лизина (3S)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата

В атмосфере азота (3S)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновую кислоту (24.1 г), полученную в примере 7, и этанол (170 мл) загрузили в 500-мл колбу и перемешивали для растворения, и затем туда добавили L-лизин (7.33 г). После этого реакционную температуру повысили до 50°, и смесь перемешивали в течение 30 минут при 50°, и снова охладили до комнатной температуры, с последующим перемешиванием в течение 30 минут. После завершения реакции полученный осадок отфильтровали с получением соединения, указанного в заголовке (22.5 г, 69.8%).
1H NMR (400MHz, D2O): δ 7.11(3H, m), 6.99(3H, m), 6.64(2H, d), 5.65(1H, s), 4.59(2H, s), 3.79(5H, s), 3.60(1H, t), 2.88(2H, t), 2.35(2H, d), 2.23(2H, s), 2.14(2H, s), 1.75(2H, m), 1.59(7H, m), 1.38(2H, m).
Пример 10. Получение L-лизина (3R)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата
В атмосфере азота (3R)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновую кислоту (24.1 г), полученную в примере 8, и этанол (170 мл) загрузили в 500-мл колбу и перемешивали для растворения, и затем добавили туда L-лизин (7.33 г). После этого реакционную температуру повысили до 50°, и смесь перемешивали в течение 30 минут при 50°, и снова охладили до комнатной температуры, с последующим перемешиванием в течение 30 минут. После завершения реакции полученный осадок отфильтровали с получением соединения, указанного в заголовке (16.2 г, 71.4%).
1H NMR (400MHz, D2O): δ 7.11(3H, m), 6.99(3H, m), 6.64(2H, d), 5.65(1H, s), 4.59(2H, s), 3.79(5H, s), 3.60(1H, t), 2.88(2H, t), 2.35(2H, d), 2.23(2H, s), 2.14(2H, s), 1.75(2H, m), 1.59(7H, m), 1.38(2H, m).
Пример 11. Получение натрия (3S)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата

В атмосфере азота (3S)-3-(4-(3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновую кислоту (1 г), полученную в примере 7, и этанол (170 мл) загрузили в 500-мл колбу и перемешивали для растворения, и затем водный раствор 3 н гидроксида натрия (0.77 мл) добавили по каплям. После этого смесь перемешивали при комнатной температуре, и после завершения реакции, реакционный раствор сконцентрировали при пониженном давлении, с последующим добавлением изопропилового спирта (10 мл), и полученный осадок отфильтровали с получением целевого соединения (0.73 г, 69.2%).
1H NMR (400, CDCl3): δ 7.44(1H, s), 7.34(5H, m), 6.91(2H, d), 6.00(1H, t), 5.02(2H, s), 4.08(1H, m), 4.04(4H, s), 2.73(4H, m), 2.48(2H, s), 1.92(2H, t), 1.82(3H, s)
Пример 12. Получение 3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
Стадия 1: Получение этил 3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата
В атмосфере азота 0.5 г 1,2,3,4-тетрагидроизохинолин добавили в 20 мл ДМФ в колбе и перемешивали для растворения, и затем 1.2 г карбоната цезия добавили при комнатной температуре. Через 30 минут, 1.0 г этил 3-(4-(4-((метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноата добавили по каплям, с последующим перемешиванием при комнатной температуре в течение 12 часов. После завершения реакции дистиллированную воду медленно добавили по каплям, экстрагировали этилацетатом, промыли солевым раствором, высушили над безводным сульфатом магния, и затем сконцентрировали. После этого реакционный продукт разделили с помощью колоночной хроматографии на силикагеле с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.38(2H,d), 7.31(2H,d), 7.22(2H,d), 7.16(3H,m), 6.97(3H,m), 4.98(2H,s), 4.14(2H,m), 4.09(1H,s), 3.91(1H,d), 3.70(3H,m), 2.92(4H,s), 2.73(2H,m), 1.83(3H,s), 1.29(3H,m).
Стадия 2: Получение 3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
В атмосфере азота 0.7 г этил 3-(4-(4-(3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата, полученного на стадии 1, добавили в ТГФ, метанол, и дистиллированную воду в колбе и перемешивали для растворения, и затем, 0.7 г гидроксид лития медленно добавили при комнатной температуре, с последующим перемешиванием в течение 1 часа или дольше. После завершения реакции смесь подкислили с помощью 1 М водного раствора соляной кислоты до pH 2-3, экстрагировали этилацетатом, и высушили при пониженном давлении с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.38(2H,d), 7.31(2H,d), 7.22(2H,d), 7.16(3H,m), 6.97(3H,m), 4.98(2H,s), 4.09(1H,s), 3.91(1H,d), 3.70(3H,m), 2.92(4H,s), 2.73(2H,m), 1.83(3H,s).
Пример 13. Получение 3-(4-(3-циклогексенил-4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Стадия 1: Получение этил 3-(4-(3-циклогексенил-4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата
В атмосфере азота 1.0 г (3-циклогексенил-4-((3,4-дигидроизохинолин-2(1H)-ил)метил)фенил)метанола и 30 мл тетрагидрофурана загрузили в колбе и перемешивали для растворения, и затем 0.8 г этил 3-(4-гидроксифенил)гекс-4-иноата, полученного в Примере Получения 1, и 0.6 г трифенилфосфина медленно добавили. После этого 0.5 мл диизопропилазодикарбоксилата медленно добавили по каплям с использованием капельной воронки при 0°, и затем перемешивали в течение 4 часов или дольше, при этом температура повысилась до комнатной температуры. После завершения реакции дистиллированную воду медленно добавили по каплям, с последующей экстракцией с использованием этилацетата, и экстрагированный органический слой высушили при пониженном давлении с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 12.56(1H,s), 8.26(1H,d), 7.43(2H,d), 7.25(6H,m), 7.21(1H,d), 7.02(1H,d), 6.89(2H,d), 5.46(1H,s), 5.03(2H,s), 4.14(2H,m), 4.05(1H,s), 3.92(1H,s), 3.70(1H,s), 3.35(1H,s), 3.27(1H, s), 3.03(1H,s), 2.83(2H,m), 2.01(4H,m), 1.84(3H,d), 1.51(4H,m), 1.29(3H,m).
Стадия 2: Получение 3-(4-(3-циклогексенил-4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
Соединение, указанное в заголовке, получили способом, аналогичным приведенному на стадии 2 в примере 12, за исключением того, что этил 3-(4-(3-циклогексенил-4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноат использовали вместо этил 3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата.
1H NMR (400MHz, CDCl3): δ 12.56(1H,s), 8.26(1H,d), 7.43(2H,d), 7.25(6H,m), 7.21(1H,d), 7.02(1H,d), 6.89(2H,d), 5.46(1H,s), 5.03(2H,s), 4.05(1H,s), 3.92(1H,s), 3.70(1H,s), 3.35(1H,s), 3.27(1H, s), 3.03(1H,s), 2.83(2H,m), 2.01(4H,m), 1.84(3H,d), 1.51(4H,m).
Пример 14. Получение 3-(4-(4-((4-фенил-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 12, за исключением того, что 4-фенил-1,2,3,6-тетрагидропиридина гидрохлорид использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.25(2H, d), 6.78(2H, d), 4.95(1H, s), 4.14(2H, m), 4.04(1H, m), 2.68(2H, m), 1.84(3H, d), 1.29(3H, t).
Пример 15. Получение 3-(4-(4-((4-фенилпиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 12, за исключением того, что 1-фенилпиперазин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.37(2H,d), 7.29(4H,m), 7.11(2H,d), 6.93(5H,m), 4.96(2H,s), 4.13(1H,s), 3.66(2H,m), 3.23(4H,s), 2.83(2H,m), 2.66(2H,s), 1.82(3H,s).
Пример 16. Получение 3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 12, за исключением того, что 6-метокси-1,2,3,4-тетрагидроизохинолин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.40(4H,q), 7.26(2H,d), 6.92(3H,q), 6.66(2H,d), 5.06(2H,s), 3.94(1H,s), 3.73(3H,s), 3.63(2H,s), 3.35(3H,s), 2.78(2H,t), 2.62(2H,t), 2.58(2H,s), 1.77(3H,s)
Пример 17. Получение 3-(4-(4-((4-фенилпиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 12, за исключением того, что 4-фенилпиперидин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.44(2H,d), 7.32(2H,d), 7.23(5H,t), 7.13(2H,d), 6.96(2H,d), 4.92(2H,s), 4.16(1H,s), 3.85(2H,q), 3.33(2H,t), 2.90(1H,d), 2.78(1H,m), 2.58(1H,t), 2.38(2H,t), 2.02(2H,m), 1.83(5H,m).
Пример 18. Получение 3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 12, за исключением того, что 1-(4-фторфенил)пиперазин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.60(2H,d), 7.46(2H,d), 7.30(3H,d), 6.97(2H,t), 6.86(4H,m), 5.01(2H,s), 4.21(2H,s), 4.04(1H,t), 3.50(4H,d), 3.25(4H,s), 2.78(2H,m), 1.80(3H,d).
Пример 19. Получение 3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 12, за исключением того, что 1-(4-(трифторметил)фенил)пиперазин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.63(2H,d), 7.51(4H,d), 7.21(2H,d), 6.93(2H,d), 6.74(2H,s), 5.03(2H,s), 4.13(2H,m), 4.01(1H,t), 3.73(4H,s), 2.96(4H,s), 2.71(2H,m), 1.78(3H,s).
Пример 20. Получение 3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 12, за исключением того, что 1-(4-(3-(метилсульфонил)пропокси)фенил)пиперазина гидрохлорид использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.65(2H,d), 7.49(2H,d), 7.30(2H,d), 6.87(6H,m), 5.07(2H,s), 4.20(2H,d), 4.08(2H,t), 4.01(1H,t), 6.63(2H,s), 3.49(4H,m), 3.26(2H,t), 3.01(2H,s), 2.97(3H,s), 2.71(2H,m), 2.34(2H,m), 1.83(2H,d).
Пример 21. Получение (S)-3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Стадия 1: Получение этил (S)-3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата
В атмосфере азота 0.5 г 1,2,3,4-тетрагидроизохинолин добавили к 20 мл ДМФ в колбе и перемешивали для растворения, и затем 1.1 г карбоната цезия добавили при комнатной температуре. Через 30 минут 1.0 г (S)-этил 3-(4-(4-((метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноата, полученного в Примере Получения 7, добавили по каплям, с последующим перемешиванием при комнатной температуре в течение 12 часов. После завершения реакции дистиллированную воду медленно добавили по каплям, экстрагировали этилацетатом, промыли солевым раствором, высушили над безводным сульфатом магния, и затем сконцентрировали. После этого реакционный продукт разделили с помощью колоночной хроматографии на силикагеле с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.38(2H,d), 7.31(2H,d), 7.22(2H,d), 7.16(3H,m), 6.97(3H,m), 4.98(2H,s), 4.14(2H,m), 4.09(1H,s), 3.91(1H,d), 3.70(3H,m), 2.92(4H,s), 2.73(2H,m), 1.83(3H,s), 1.29(3H,m).
Стадия 2: Получение (S)-3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
В атмосфере азота 0.5 г этил (S)-3-(4-(4-(3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата, полученного на стадии 1, добавили в ТГФ, метанол, и дистиллированную воду в колбе и перемешивали для растворения, и затем, 0.5 г гидроксида лития медленно добавили при комнатной температуре, с последующим перемешиванием в течение 1 часа или дольше. После завершения реакции смесь подкислили с помощью 1 М водного раствора соляной кислоты до pH 2-3, экстрагировали этилацетатом, и высушили при пониженном давлении с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.38(2H,d), 7.31(2H,d), 7.22(2H,d), 7.16(3H,m), 6.97(3H,m), 4.98(2H,s), 4.09(1H,s), 3.91(1H,d), 3.70(3H,m), 2.92(4H,s), 2.73(2H,m), 1.83(3H,s).
Пример 22. Получение (S)-3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 1-(4-(трифторметил)фенил)пиперазин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.65(2H,d), 7.51(4H,m), 7.30(2H,d), 6.61(2H,d), 6.85(2H,d), 5.05(2H,s), 4.21(2H,s), 4.03(1H,t), 3.68(4H,s), 3.49(2H,s), 2.84(2H,s), 2.70(2H,m), 1.82(3H,s).
Пример 23. Получение (S)-3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 1-(4-фторфенил)пиперазин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.39(2H,d), 7.30(2H,d), 7.19(2H,d), 6.96(4H,m), 6.87(2H,m), 4.97(2H,s), 4.10(2H,s), 3.81(1H,d), 3.51(1H,d), 3.15(4H,s), 2.80(6H,m), 1.82(3H,s).
Пример 24. Получение калия (S)-3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иноата

В атмосфере азота 0.4 г (S)-3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты, полученной в примере 23, и 10 мл этанола загрузили в колбу и перемешивали для растворения, и затем 0.3 мл 3 н водного раствора гидроксида калия добавили по каплям. После этого смесь перемешивали при комнатной температуре, и после завершения реакции, реакционный раствор сконцентрировали при пониженном давлении, с последующим добавлением изопропилового спирта, и полученный осадок отфильтровали с получением целевого соединения.
1H NMR (400MHz, D2O): δ 7.10(4H,m), 6.98(2H,d), 6.57(4H,d), 6.38(2H,s), 4.55(2H,s), 3.82(1H,s), 3.07(2H,s), 2.59(4H,s), 2.36(2H,s), 2.13(4H,s), 1.51(3H,s).
Пример 25. Получение (S)-3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
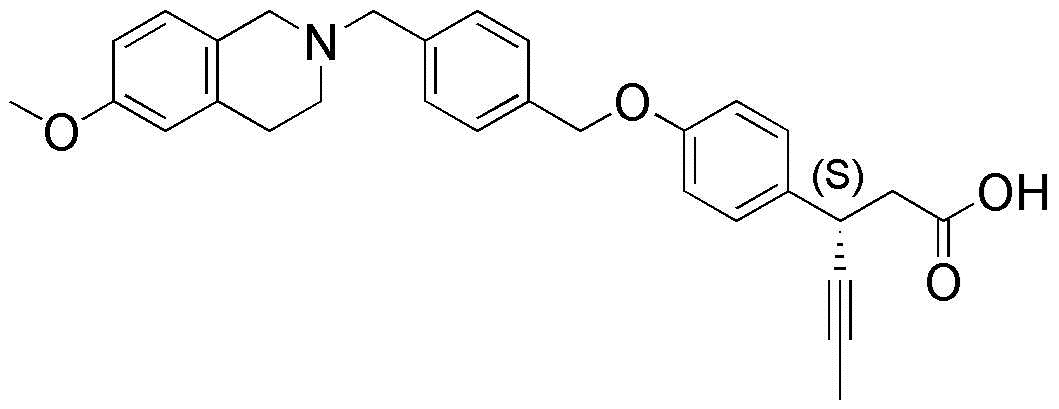
Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 6-метокси-1,2,3,4-тетрагидроизохинолин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, DMSO): δ 7.40(4H,q), 7.26(2H,d), 6.94(3H,m), 6.68(2H,m), 5.06(2H,s), 3.95(1H,t), 3.70(3H,s), 3.51(2H,s), 3.43(2H,s), 2.77(2H,t), 2.66(2H,t), 2.57(2H,d), 1.75(3H,d).
Пример 26. Получение (S)-3-(4-(4-((4-фенилпиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
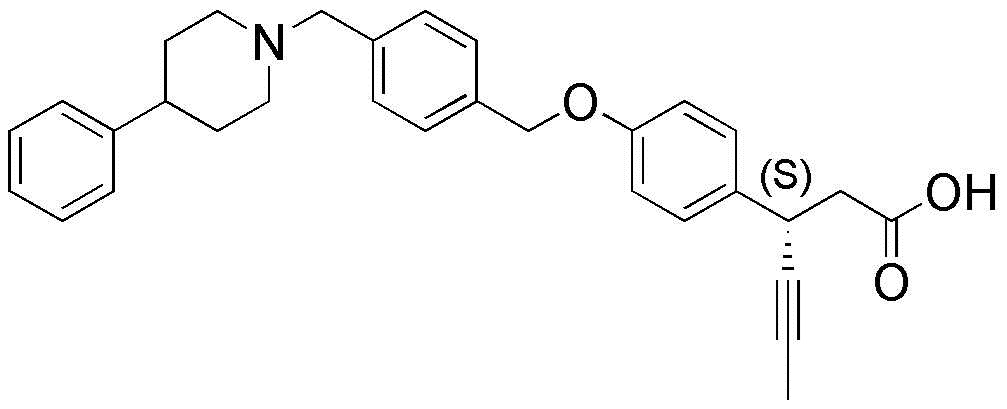
Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 4-фенилпиперидин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.66(2H,d), 7.49(2H,d), 7.30(7H,m), 6.87(2H,d), 5.04(2H,s), 4.19(2H,s), 4.06(1H,t), 3.59(2H,d), 2.73(7H,m), 2.00(2H,d), 1.82(3H,s).
Пример 27. Получение (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что изоиндолин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.68(2H,d), 7.47(2H,d), 7.38(2H,m), 7.30(4H,m), 6.87(2H,d), 5.06(2H,s), 4.90(2H,s), 4.32(4H,m), 4.05(1H,t), 2.81(2H,m), 1.83(3H,s).
Пример 28. Получение (S)-3-(4-(4-((4-фенил-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 4-фенил-1,2,3,6-тетрагидропиридина гидрохлорид использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.47(2H,d), 7.36(9H,m), 6.88(2H,d), 5.99(1H,s), 4.99(2H,s), 4.18(1H,m), 4.06(2H,m), 3.53(2H,s), 3.22(2H,s), 2.82(4H,m), 1.82(3H,s).
Пример 29. Получение (S)-3-(4-(4-((4-(4-(метоксиметокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 1-(4-(метоксиметокси)фенил)пиперазин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.57(2H,d), 7.46(2H,d), 7.26(2H,d), 6.97(2H,d), 6.87(2H,d), 6.80(2H,d), 5.13(2H,s), 5.01(2H,s), 4.13(2H,s), 4.02(1H,t), 3.51(11H,m), 2.72(2H,m), 1.79(3H,s).
Пример 30. Получение (S)-3-(4-(4-((4-(5-изопропил-1,2,4-оксадиазол-3-ил)пиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 3-изопропил-5-(пиперидин-4-ил)-1,2,4-оксадиазол использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.63(2H,d), 7.46(2H,d), 7.30(2H,d), 6.86(2H,d), 5.05(2H,d), 4.13(2H,m), 4.03(1H,t), 3.61(1H,s), 3.43(2H,s), 3.10(1H,m), 2.92(4H,m), 2.73(2H,m), 2.30(2H,m), 1.83(3H, s), 1.32(6H,d).
Пример 31. Получение (S)-3-(4-(4-((4-(5-изопропил-1,2,4-оксадиазол-3-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 3-изопропил-5-(пиперазин-1-ил)-1,2,4-оксадиазол использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.61(2H,d), 7.49(2H,d), 7.30(2H,d), 6.87(2H,d), 5.05(2H,s), 4.15(4H,m), 4.02(1H,t), 3.49(3H,m), 2.81(3H,m), 1.83(3H,s), 1.24(6H,d).
Пример 32. Получение (S)-3-(4-(4-((4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 4-(4-(метилсульфонил)фенил)-1,2,3,6-тетрагидропиридина гидрохлорид, полученный в Примере Получения 9, использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, DMSO): δ 7.95(2H,d), 7.75(2H,d), 7.63(2H,d), 7.44(2H,d), 7.30(2H,d), 6.98(2H,d), 6.37(1H,s), 5.14(2H,s), 4.45(2H,t), 6.97(1H,s), 6.82(4H,m), 3.27(4H,s), 2.84(2H,s), 2.59(2H,d), 1.77(3H,s).
Пример 32. Получение (S)-3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 4-(4-(3-(метилсульфонил)пропокси)фенил)-1,2,3,6-тетрагидропиридина гидрохлорид, полученный в Примере Получения 11 использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.66(2H,d), 7.49(2H,d), 7.32(2H,d), 7.15(2H,d), 6.90(2H,d), 6.82(2H,d), 5.06(2H,s), 4.18(2H,s), 4.09(3H,m), 3.58(2H,s), 3.26(2H,m), 2.97(3H,s), 2.81(5H,m), 2.62(3H,s), 2.32(2H,m), 1.96(2H,d), 1.83(3H,s).
Пример 34. Получение (3S)-3-(4-(4-(1-(3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что (3S)-этил 3-(4-(4-(1-бромэтил)бензилокси)фенил)гекс-4-иноат, полученный в Примере Получения 12, использовали вместо (S)-этил 3-(4-(4-((метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноата.
1H NMR (400MHz, CDCl3): δ 12.98(1H,s), 7.61(6H,m), 7.30(4H,m), 6.92(2H,t), 5.08(2H,s), 4.29(2H,s), 4.06(1H,s), 3.81(1H,s), 3.51(2H,s), 3.21(2H,m), 2.75(2H,m), 1.95(2H,d), 1.84(3H,s).
Пример 35. Получение (S)-3-(4-(4-((4-(4-гидроксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 4-(1,2,3,6-тетрагидропиридин-4-ил)фенола гидрохлорид, полученный в Примере Получения 10, использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 8.80(1H,s), 7.41(2H,d), 735(2H,d), 7.28(2H,d), 6.94(2H,d), 6.74(2H,d), 6.63(2H,d), 5.06(2H,s), 3.94(1H,t), 3.62(3H,s), 2.95(4H,s), 2.61(2H,d), 1.77(3H,s).
Пример 36. Получение (S)-3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 1-(4-(3-(метилсульфонил)пропокси)фенил)пиперазина гидрохлорид использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 12.32(1H,s), 7.42(4H,m), 7.29(2H,d), 6.96(2H,d), 6.83(4H,q), 5.06(2H,s), 4.02(2H,t), 3.92(1H,t), 3.52(2H,s), 3.25(2H,t), 3.01(7H,m), 2.61(2H,d), 2.09(2H,m), 1.77(3H,d).
Пример 37. Получение натрия (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иноат
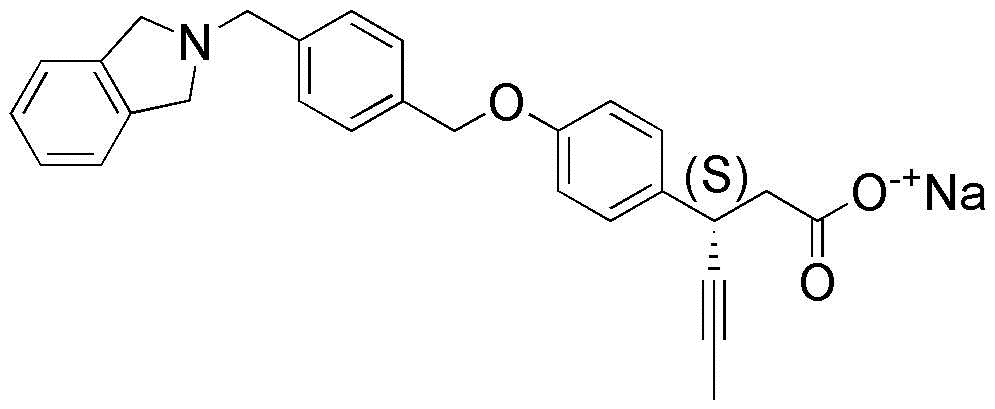
В атмосфере азота 0.4 г (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иновой кислоты, полученной в примере 27, и этанол загрузили в 500-мл колбу и перемешивали для растворения, и затем 0.3 мл 3 н водного раствора гидроксида натрия добавили по каплям. После этого смесь перемешивали при комнатной температуре, и после завершения реакции реакционный раствор сконцентрировали при пониженном давлении, с последующим добавлением изопропилового спирта, и полученный осадок отфильтровали с получением целевого соединения.
1H NMR (400MHz, CDCl3): δ 7.09(2H,d), 7.03(2H,d), 6.97(2H,d), 6.85(2H,m), 6.75(2H,m), 6.57(2H,d), 4.54(2H,s), 3.81(1H,t), 3.36(4H,s), 3.31(2H,s), 2.33(2H,d), 1.54(3H,d).
Пример 38. Получение L-лизина (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иноата
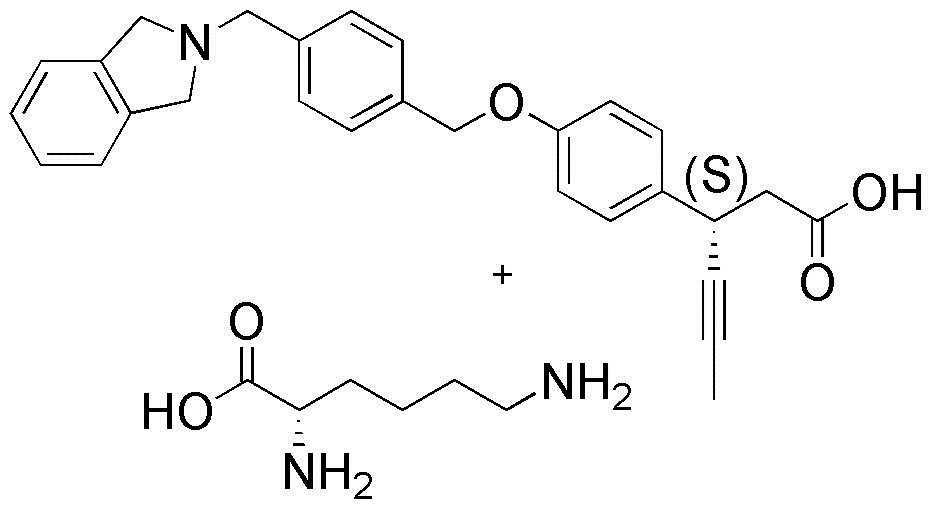
В атмосфере азота 0.4 г (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иновой кислоты, полученной в примере 27, и этанол загрузили в колбу и перемешивали для растворения, и затем добавили 0.12 г L-лизина. После этого реакционную температуру повысили до 50°, и смесь перемешивали в течение 30 минут при 50°, и снова охладили до комнатной температуры, с последующим перемешиванием в течение 30 минут. После завершения реакции полученный осадок отфильтровали с получением соединения, указанного в заголовке.
1H NMR (400MHz, D2O): δ 7.03(6H,s), 6.83(2H,s), 6.74(2H,s), 6.54(2H,s), 4.53(2H,s), 3.77(1H,s), 3.54(5H,m), 2.88(2H,t), 2.28(2H,s), 1.74(2H,m), 1.62(3H,m), 1.42(3H,s), 1.35(3H, m).
Пример 39. Получение (S)-3-(4-(4-((4-(4-фторфенил)-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 4-(4-фторфенил)-1,2,3,6-тетрагидропиридина гидрохлорид использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.69(2H,d), 7.48(2H,d), 7.32(4H,m), 7.04(2H,t), 6.86(2H,d), 5.90(1H,s), 5.03(2H,s), 4.30(2H,s), 4.02(1H,t), 3.71(2H,s), 3.54(2H,s), 3.31(2H,s), 2.73(2H,m), 1.81(3H,d).
Пример 40. Получение (S)-3-(4-(4-((4-(4 -метоксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 4-(4-метоксифенил)-1,2,3,6-тетрагидропиридин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.64(2H,d), 7.48(2H,d), 7.31(2H,d), 6.94(2H,s), 6.86(4H,t), 5.04(2H,s), 4.21(2H,s), 4.03(1H,t), 3.78(3H,s), 3.60(2H,s), 3.47(2H,s), 3.05(2H,s), 2.73(2H,m), 1.82(3H,s).
Пример 41. Получение натрия (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иноата

Стадия 1: Получение (S)-3-(4-(4-((3,4-дигидроизохинолин-1(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 1,2,3,4-тетрагидрохинолин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 7.02(2H,d), 6.76(2H,d), 6.69(2H,d), 6.43(4H,m), 6.21(1H,s), 6.02(1H,s), 4.24(2H,s), 3.84(3H,s), 2.68(2H,s), 2.37(2H,d), 2.14(2H,s), 1.47(3H,s), 1.35(2H,s).
Стадия 2: Получение натрия (S)-3-(4-(4-((3,4-дигидрохинолин-1(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата
Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 37, за исключением того, что (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновую кислоту полученную на стадии 1, использовали вместо (S)-3-(4-(4-(изоиндолин-2-ил-метил)бензилокси)фенил)гекс-4-иновой кислоты.
1H NMR (400MHz, D2O): δ 7.01(2H,d), 6.74(2H,d), 6.68(2H,d), 6.42(4H,m), 6.15(1H,s), 6.02(1H,s), 4.25(2H,s), 3.79(3H,s), 2.62(2H,s), 2.34(2H,d), 2.12(2H,s), 1.45(3H,s), 1.32(2H,s).
Пример 42. Получение калия (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иноата

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 25, за исключением того, что (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновую кислоту, полученную на стадии 1 в примере 41, использовали вместо (S)-3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
1H NMR (400MHz, D2O): δ 6.97(2H,d), 6.71(2H,d), 6.63(2H,d), 6.45(2H.s), 6.38(2H,d), 6.13(1H,s), 5.98(1H,s), 4.20(2H,s), 3.71(3H,m), 2.58(2H,s), 2.32(2H,s), 2.15(2H,s), 1.43(3H,s), 1.29(2H,s).
Пример 43. Получение (S)-3-(4-(4-((4-(бензо[d]тиазол-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
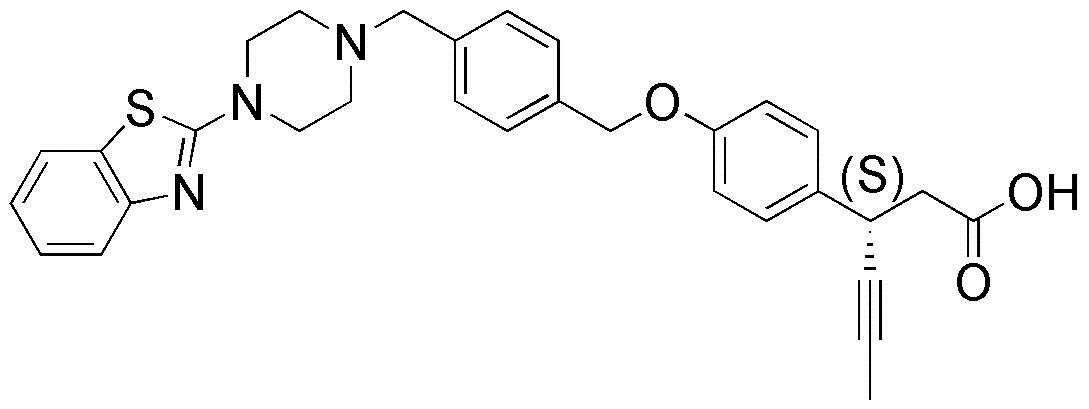
Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 2-(пиперазин-1-ил)бензо[d]тиазола гидрохлорид, полученный в Примере Получения 13, использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, DMSO): δ 10.87(1H,s), 7.85(1H,d), 7.55(5H,m), 7.31(3H,m), 7.14(2H,t), 6.96(2H,d), 5.13(2H,s), 4.40(2H,s), 4.17(2H,d), 3.95(1H,t), 3.57(3H,t), 3.22(3H,s), 2.57(2H,d), 1.78(3H,d).
Пример 44. Получение (S)-3-(4-(4-((4-(5-пропилпиримидин-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 2-(пиперазин-1-ил)-5-пропилпиримидина гидрохлорид, полученный в Примере Получения 14, использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 8.20(2H,s), 7.62(2H,d), 7.47(2H,d), 7.30(2H,d), 6.85(2H,d), 5.08(2H,s), 4.80(2H,d), 4.17(2H,s), 4.03(1H,t), 3.84(1H,t), 3.43(2H,s), 2.74(4H,m), 2.43(2H,t), 1.83(3H,d), 1.59(2H,q), 0.94(3H,t).
Пример 45. Получение (S)-3-(4-(4-((4-(5-цианопиридин-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 6-(пиперазин-1-ил)никотинонитрила гидрохлорид, полученный в Примере Получения 15, использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, DMSO): δ 11.20(1H,s), 8.56(1H,s), 7.99(1H,d), 7.63(1H,d), 7.55(1H,d), 7.27(2H,d), 7.04(1H,d), 6.95(2H,d), 5.12(2H,s), 4.57(2H,d), 4.35(2H,s), 3.95(1H,t), 3.39(5H,m), 2.90(2H,m), 2.59(2H,d), 1.77(3H,d).
Пример 46. Получение (3S)-3-(4-(4-((3-фенилпирролидин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 21, за исключением того, что 3-фенилпирролидин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1Н NMR (400 MHz, CDCl3): (12.64 (1H, s), 7.66 (2H, s), 7.46 (2H, d), 7.32 (7H, m), 6.86 (2H, d), 5.02 (2H, s), 4.28 (2H, m), 4.04 (1H, t), 3.87 (2H, s), 3.73 (1H, s), 3.18 (1H, s), 2.89 (1H, m), 2.84 (3H, m), 2.61 (1H, s), 2.41 (1H, s), 2.19 (1H, s), 1.81 (3H, d).
Пример 47. Получение натрия (S)-3-(4-(3-((4-(4-метоксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иноата
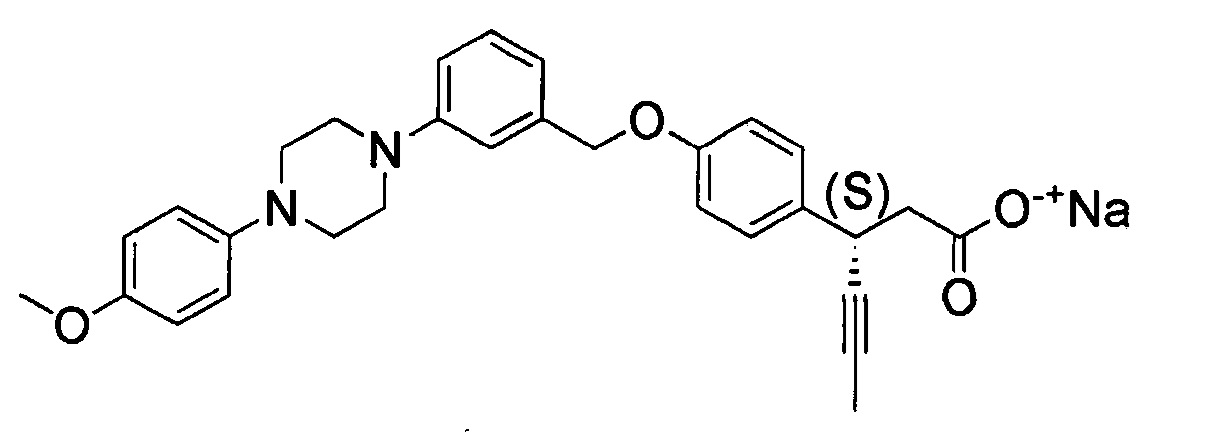
Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 37, за исключением того, что (S)-3-(4-(4-((4-(4-метоксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновую кислоту, полученную в примере 40, использовали вместо (S)-3-(4-(4-(изоиндолин-2-ил-метил)бензилокси)фенил)гекс-4-иновой кислоты.
1Н NMR (400 MHz, МЕОС): (7.33 (2H, d), 7.26(1H, d), 7.11 (1H, s), 6.96 (8H, m), 5.04 (2H, s), 4.04 (1H, t), 3.76 (3H, s), 3.32 (4H, m), 3.21 (4H, m), 2.52 (2H, m), 1.80 (3H, s).
Пример 48. Получение (S)-3-(4-(4-(2-(6-метокси-3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иновой кислоты

Стадия 1: Получение этил (S)-3-(4-(4-(2-(6-метокси-3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иноата
В атмосфере азота 0.5 г 6-метокси-1,2,3,4-тетрагидроизохинолин добавили к 20 мл ДМФ в колбе и перемешивали для растворения, и затем 1.1 г карбоната цезия добавили при комнатной температуре. Через 30 минут, 1.0 г (S)-этил 3-(4-(4-(2-(метилсульфонилокси)этил)бензилокси)фенил)гекс-4-иноата, полученного в Примере Получения 16, добавили по каплям, с последующим перемешиванием при комнатной температуре в течение 12 часов. После завершения реакции дистиллированную воду медленно добавили по каплям, экстрагировали этилацетатом, промыли солевым раствором, высушили над безводным сульфатом магния, и затем сконцентрировали. После этого реакционный продукт разделили с помощью колоночной хроматографии на силикагеле с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.35(2H,d), 7.30(2H,d), 7.23(2H,d), 7.00(1H,d), 6.85(2H,d), 6.80(1H,d), 6.70(1H,d), 5.00(2H,s), 4.30(2H,m), 4.13(2H,m) 4.03(1H,t), 3.80(3H,s), 3.58(6H,m), 3.30(2H,s), 2.78(2H,m), 1.86(3H,d), 1.28(3H,m).
Стадия 2: Получение (S)-3-(4-(4-(2-(6-метокси-3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иновой кислоты
В атмосфере азота 0.5 г этил (S)-3-(4-(4-(2-(6-метокси-3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иноат, полученный на стадии 1 добавили в ТГФ, метанол, и дистиллированную воду в колбе и перемешивали для растворения, и затем, 0.5 г гидроксида лития медленно добавили при комнатной температуре, с последующим перемешиванием в течение 1 часа или дольше. После завершения реакции смесь подкислили с помощью 1 М водного раствора соляной кислоты до pH 2-3, экстрагировали этилацетатом, и высушили при пониженном давлении с получением соединения, указанного в заголовке.
1H NMR (400MHz, CDCl3): δ 7.35(2H,d), 7.30(2H,d), 7.23(2H,d), 7.00(1H,d), 6.85(2H,d), 6.80(1H,d), 6.70(1H,d), 5.00(2H,s), 4.30(2H,m), 4.03(1H,t), 3.80(3H,s), 3.58(6H,m), 3.30(2H,s), 2.78(2H,m), 1.86(3H,d).
Пример 49. Получение (S)-3-(4-(4-(2-(изоиндолин-2-ил)этил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 48, за исключением того, что изоиндолин использовали вместо 6-метокси-1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, CDCl3): δ 13.57(1H,s), 7.38(3H,m), 7.29(7H,m), 6.90(2H,d), 5.03(4H,m), 4.28(2H,s), 4.08(1H,t), 3.48(2H,m), 3.34(2H,m), 2.80(2H,m), 1.83(3H,d).
Пример 50. Получение (S)-3-(4-(4-(2-(3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иновой кислоты

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 48, за исключением того, что 1,2,3,4-тетрагидроизохинолин использовали вместо 6-метокси-1,2,3,4-тетрагидроизохинолина.
1H NMR (400MHz, DMSO): δ 7.44(2H,d), 7.38(2H,d), 7.27(5H,m), 7.22(1H,d), 6.94(2H,d), 5.07(2H,s), 4.64(1H,d), 4.38(1H,s), 3.95(1H,t), 3.77(1H,s), 3.39(2H,s), 3.16(4H,m), 2.26(2H,d), 1.77(3H,d), 1.84(3H,d), 1.29(3H,t).
Пример 51. Получение натрия (S)-3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата

Соединение, указанное в заголовке, получили способом, аналогичным описанному в примере 37, за исключением того, что (S)-3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновую кислоту, полученную в примере 25, использовали вместо (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иновой кислоты.
1H NMR (400MHz, D32O): δ 7.10(2H,d), 7.02(2H,d), 6.95(2H,d), 6.55(2H,d), 6.40(1H,d), 6.34(2H,s), 4.53(2H,s), 3.83(1H,t), 3.39(3H,s), 3.17(2H,s), 3.05(2H,s), 2.37(4H,m), 2.20(2H,s), 1.57(3H,s).
Сравнительный Пример 1. Получение [(3S)-6-({(2',6'-диметил-4'-[3-(метансульфонил)пропокси]-[1,1'-бифенил]-3-ил)}метокси)-2,3-дигидро-1-бензофуран-3-ил]уксусной кислоты

[(3S)-6-({(2',6'-диметил-4'-[3-(метансульфонил)пропокси]-[1,1'-бифенил]-3-ил)}метокси)-2,3-дигидро-1-бензофуран-3-ил]уксусную кислоту получили способом, известным из WO 2008/001931.
Сравнительный Пример 2. Получение (3S)-3-(4-{[4-(1'H-спиро[инден-1,4'-пиперидин]-1'-илметил)бензил]окси}фенил)гекс-4-иновой кислоты

(3S)-3-(4-{[4-(1'H-спиро[инден-1,4'-пиперидин]-1'-илметил)бензил]окси}фенил)гекс-4-иновую кислоту получили способом, известным из WO 2011/046851.
Сравнительный Пример 3. Получение 4-(3-феноксибензиламино)фенилпропиновой кислоты
4-(3-феноксибензиламино)фенилпропиновую кислоту получили известным способом.
В Таблице 1 суммированы химические структуры соединений, полученных в примерах 1-51.
Таблица 1













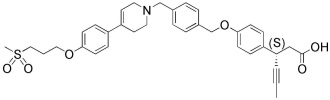



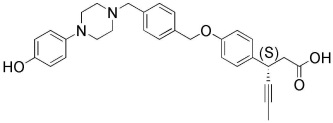


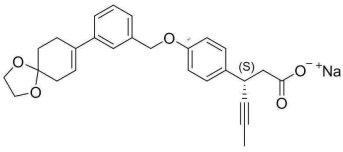









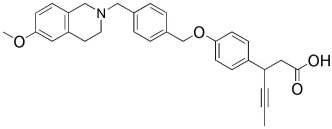




















В соответствии с другим аспектом настоящего изобретения, предложен способ предотвращения или лечения метаболических нарушений, способ, включающий введение субъекту фармацевтически эффективного количества композиции, содержащей: (a), в качестве первого активного ингредиента, соединение формулы 1, или его оптический изомер, гидрат или сольват, или его фармацевтически приемлемую соль; и (b), в качестве второго активного ингредиента, по меньшей мере одно соединение, выбранное из группы, состоящей из соединений на основе ингибиторов дипептидилпептидазы-IV (DPP-IV), на основе сульфонилмочевины, на основе тиазолидиндиона (TZD), на основе бигуанида и на основе ингибитора натрий/глюкоза котранспортера 2 (SGLT2), в качестве второго активного ингредиента:
Формула 1

В данном случае, Формула 1 является такой, как описано в подробном описании композиции для предотвращения или лечения метаболических нарушений.
Смешанная композиция первого активного ингредиента и второго активного ингредиента в частности не ограничивается массовым соотношением смешивания, так как никаких побочных эффектов или снижения эффективности не вызываются массовым соотношением смешивания, и, принимая во внимание патологические состояния пациентов, известные характеристики второго активного ингредиента и т.д., первый активный ингредиент и второй активный ингредиент могут быть смешаны в подходящих количествах и введены в комбинации. В одном воплощении массовое соотношение смешивания составляет от 0,03: 1 до 100: 1. В другом воплощении массовое соотношение при смешивании составляет от 0,03:1 до 30:1, а еще в одном варианте массовое соотношение смешивания составляет от 0,03: 1 до 10: 1.
Экспериментальный Пример 1. Оценка активации белка GPR40 производным 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты
С целью оценить активацию GPR40 новым производным 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения, проводился следующий эксперимент.
Активация белка GPR40 новым производным 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения оценивалась на основе изменения внутриклеточной концентрации кальция в результате активности белка GPR40. Сначала, клетки HEK-293 трансфицировали с помощью ДНК GPR40 человека (Origene, RC218370) посредством Fugene HD (Promega, E2311). Трансфицированные клетки HEK-293 засевали в 96-луночные черные с прозрачным дном планшеты (Costar, 3603) и культивировали. Через 24 часа клеточную питательную среду удалили, и заменили на среду Игла, модифицированную по Дульбекко (DMEM, 50 мкл) с добавлением 1% фетальной бычьей сыворотки (FBS). Для измерения концентрации кальция 50 мкл реагента Fluo-4 (Invitrogen, F10471) добавили в каждую лунку и культивировали в инкубаторе при 37° в течение 2 часов. Во время культивирования соединения примеров и соединения Сравнительных Примеров 1 и 2 разбавили 1 x солевым раствором буфера Хэнка (HBSS) содержащего 20 мМ буфера 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновой кислоты (HEPES) для получения образцов, которыми будут обрабатывать клетки. Через 2 часа после инициации культуры полученные образцы автоматически инъецировали в клетки с использованием Flexstation 3 (Molecular Devices), и затем измеряли изменение концентрации внутриклеточного кальция с использованием программного обеспечения SoftMax®Pro в течение 120 секунд. В данном случае, для необрабатываемых групп, в клетки инъецировался диметилсульфоксид (DMSO) для измерения изменений в концентрации кальция. Активность белка GPR40 рассчитывалась на основе измеренных значений концентрации кальция с использованием уравнения 1 ниже, и получили активность GPR40 (значение EC50) для полученных образцов. Результаты показаны на Фиг. 2.
Уравнение 1
Активность GPR 40 =(концентрация внутриклеточного кальция, увеличенная образцом)/( концентрация внутриклеточного кальция необработанной группы)×100
Таблица 2
В Таблице 2,
A: менее 0.20 мкМ;
B: 0.20-0.30 мкМ; и
C: более 0.30 мкМ.
Как показано в Таблице 2, соединения примеров настоящего изобретения обладают улучшенными эффектами активации белка GPR 40 при низких концентрациях. В частности, соединения примеров 7, 9, 11, 12, 14, 27, 28, 37, и 38 активируют белок GPR40 на 50% при очень низких концентрациях, таких как 0.20 мкМ или менее, указывая на то, что их способность увеличивать концентрацию внутриклеточного Ca2+ было гораздо выше по сравнению с соединением Сравнительного Примера 1 (B, 0.28 мкМ).
Таким образом, новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения индуцирует увеличенную активность белка GPR40 и, особенно, проявляет аналогичную или улучшенную активность белка GPR40 по сравнению с обычными противодиабетическими лекарствами (Сравнительный Пример 1), которые как известно активируют белок GPR40 для улучшения секреции инсулина, и таким образом, фармацевтическая композиция, содержащая соединение настоящего изобретения в качестве активного ингредиента может выгодно применяться в качестве фармацевтической композиции для предотвращения или лечения метаболических нарушений, таких как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
Экспериментальный Пример 2. Анализ потока кальция
С целью оценки потока кальция в соответствии с активацией GPR40 посредством нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения, эксперимент проводился компанией Millipore, которая специализируется на анализах GPCR.
Соединения примеров настоящего изобретения, растворенные в ДМСО (диметилсульфоксид), PBS (фосфатно-солевом буфере), и DW (дистиллированной воде) три раза разбавили специальным буфером для анализа EMD Millipore's GPCR. Аналогично, необработанную группу (носитель) и группы положительного контроля (Сравнительные Примеры 1 и 3) использовали для верификации правильности анализа. Каждую лунку подготовили с использованием специального буфера для анализа EMD Millipore's GPCR. Специальный буфер для анализа EMD Millipore's GPCR представлял собой сбалансированные солевой раствор Хэнкса (HBSS), содержащий 20 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота) и 2.5 мМ пробенецид (4-(дипропилсульфамоил)бензойная кислота), доведенный до pH 7.4.
Соединение примера дублировали для каждой концентрации. Группы положительного контроля (Сравнительные Примеры 1 и 3) для каждого рецептора, связанного с G белком (GPCR), получили таким же способом, как и необработанную группу (носитель). Группы положительного контроля (Сравнительные Примеры 1 и 3) для каждого GPCR включали в Emax в качестве концентрации, проявляющей максимальную активность. Агонистический анализ проводился с использованием FLIPRTETRA, при этом измеряли базовые линии флуоресценции и люминесценции. Соединения примеров, необработанной группы, и групп положительного контроля (Сравнительные Примеры 1 и 3) добавили к планшетам для анализа. Для измерения активности примера, анализ GPCR активности проводили в течение 180 секунд.
Значения флуоресценции за вычетом базовой линии сравнивались со значениями Emax положительных контролей (Сравнительные Примеры 1 и 3) и необработанной группы, и затем рассчитывались в виде процента (%). Полученные данные показывают ингибирование (%) в результате сравнения EC50 с необработанной группой, и качество каждого планшета было оценено с помощью статистических данных, отражающих % активности на основе значений повторов. Когда данные анализа были недостаточно согласованными, проводился дополнительный эксперимент.
Все графики зависимости от концентрации строились с использованием GraphPad Prism. График модифицировали сигмоидальным дозозависимым эффектом, и минимальное значение принимали в качестве 0, а максимальное значение принимали в качестве 100 для предсказания значений лучшего эффекта.
Результаты показаны на Фиг. 1 и Таблице 3.
Таблица 3
Фиг. 1 представляет собой график, иллюстрирующий паттерн активации белка GPR40, измеренный в соответствии с концентрациями соединений примера 9, Сравнительного Примера 1, и Сравнительного Примера 3.
Как показано на Фиг. 1, можно увидеть, что концентрация, необходимая для достижения 50% активности GPR40 была очень низкой (ниже чем измеримая концентрация в 1 нМ) для соединения примера по сравнению с соединениями Сравнительных Примеров 1 и 3. Особенно, как показано в Таблице 3, соединение примера настоящего изобретения активировало GPR40 при намного более низкой концентрации, чем соединения Сравнительного Примера 1 (14 нМ) и Сравнительного Примера 3 (27 нМ).
Таким образом, новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения индуцирует превосходящую активность белка GPR40, и в частности, показывает существенное увеличение активности белка GPR40 по сравнению с традиционными противодиабетическими лекарствами (Сравнительные Примеры 1 и 3), которые, как известно, активируют GPR40 белок для стимулирования секреции инсулина, и таким образом, фармацевтическая композиция, содержащая новое соединение по изобретению в качестве активного ингредиента может выгодно применяться в качестве фармацевтической композиции для предотвращения или лечения метаболических нарушений, таких как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
Экспериментальный Пример 3. Анализ ингибирования CYP
C целью оценки взаимодействия между новым производным 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения и лекарственного средства проводился следующий эксперимент.
Ферменты CYP участвуют в метаболизме лекарственных средств, и в зависимости от ингибирующих эффектов этих ферментов, может быть предсказана доза лекарственного средства и токсичность в результате совместного введения, концентрация для совместного введения с другим лекарственным средством. Таким образом, заявители измерили ингибирующие эффекты соединения примеров по изобретению на CYP3A4, CYP2C9, CYP1A2, CYP2D6, и CYP2C19, существующие у человека. В данном случае, набор Invitrogen (P2862) использовали в качестве набора, ингибирующего CYP2D6, и набор BD GENTEST (459100, 459300, 459400, 459500) использовали в качестве ингибирующих наборов CYP1A2, CYP2C9, CYP2C19, и CYP3A4. Для набора Invitrogen тестовый образец разбавили в дистиллированной воде при 2.5× конечной экспериментальной концентрации.
Реагент P450 BACULOSOMES® и дупликатор (100X), представленные в наборе Invitrogen, разбавили в реакционном буфере Vivid® CYP450 (2×) до концентрации, соответствующей целевому CYP450. Полученный образец 2.5× (80 мкл) и разбавленная смесь реагента P450 BACULOSOMES® (100 мкл) смешали в 96-луночном планшете с U-образным дном, с последующим предварительным культивированием в течение 20 минут. Субстрат Vivid® CYP450 и NADP+ (100×) разбавили в реакционном буфере Vivid® CYP450 (2×) до концентрации, которая соответствовала целевому CYP450 и виду субстрата. После завершения предварительного культивирования смесь субстрат-никотинамид аденин динуклеотидфосфат (NADP) (20 мкл) добавили туда, с последующим взаимодействием в течение 1 часа. После завершения реакции, взаимодействующее вещество перенесли на белый планшет, и затем измерили флуоресценцию с помощью микропланшетного ридера (CYP 2D6 волна возбуждения: 400 нм, длина волны поглощения: 502 нм).
Относительно набора BD GENTEST, тестовый образец разбавили в ацетонитриле при 50× конечной экспериментальной концентрации. Смесь NADPH-кофермента получили разбавлением коферемента, G6PDH, и регуляторного белка, присутствующих в наборе, дистиллированной водой до концентрации, указанной в наборе. Полученный образец 50× (4 мкл) и смесь NADPH-кофермента (96 мкл) смешали в 96-луночном планшете с U-образным дном, с последующим предварительным культивированием в течение 10 минут в 37° инкубаторе. Смесь фермент/субстрат получили разбавлением буфера (0.5 M фосфат калия, pH 7.4) и каждой смеси CYP450 фермент/субстрат дистиллированной водой до концентрации, указанной в инструкции в соответствии с видом CYP450. После завершения предварительного культивирования, 100 мкл смеси фермент/субстрат добавили к планшету, с последующим взаимодействием в 37° инкубаторе в течение 15 минут (CYP 1A2), 30 минут (CYP 3A4 и CYP 2C19) ил полутора часов (CYP 2C9). После завершения реакции, взаимодействующее вещество перенесли в белый планшет, и затем измеряли флуоресценцию с помощью микропланшетного ридера (длина волны возбуждения: 410 нм, длина волны поглощения: 460 нм для CYP 1A2 и CYP 2C19; и длина волны возбуждения: 409 нм, длина волны поглощения: 530 нм для CYP 2C9 и CYP 3A4). Измеренные выше значения конвертировали в % ингибирования образцом по равнению с необработанной группой. Результаты показаны в Таблице 4.
Таблица 4
Как показано в Таблице 4, соединения примеров настоящего изобретения проявили низкую активность в отношении ингибирования CYP450, предполагая, что риск побочных эффектов в результате взаимодействия лекарственных средств является низким. Более конкретно, соединения почти всех примеров настоящего изобретения показали ингибирование фермента приблизительно 50% или менее для CYP 1A2, CYP 2C9, CYP 2C19, CYP 2D6, и CYP 3A4 ферментов. В частности, соединения примеров показали относительную очень низкую ингибиторную активность для фермента CYP 2C9, по сравнению с соединением Сравнительного Примера 1 (81.2%), которое использовали в качестве обычного противодиабетического лекарственного средства, которое может стимулировать секрецию инсулина посредством активации белка GPR40. В дополнение, соединения примеров настоящего изобретения показали относительно низкую ферментативную ингибиторную активность для фермента CYP 2D6, по сравнению с соединением Сравнительного Примера 2 (63.2%).
Поскольку новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения обладает существенно низкой ингибиторной активностью фермента CYP, фармацевтическая композиция, содержащая указанное новое соединение в качестве активного ингредиента может вводиться совместно с другими лекарственными средствами, и таким образом может выгодно применяться при лечении осложнений, включая метаболические заболевания, такие как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
Экспериментальный Пример 4. Пероральный тест толеранстности к глюкозе (OGTT) 1
С целью оценки эффекта снижения глюкозы в крови in vivo для нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения, проводился следующий эксперимент.
Самцы крыс Спрег-Доули в возрасте 8-10 недель акклиматизировали в течение по меньшей мере 7 дней, и затем только здоровых животных использовали для перорального теста толерантности к глюкозе (тест OGTT). После голодания в течение 16-18 часов, пять крыс из группы случайным образом группировали и им перорально вводили соединения, полученный в примерах 2, 3, 4, 6, 9, 12, 14, 16, 25, 29, 36, 37, 41, 43, и 44 в дозировке 10 мг/кг каждое. В данном случае в качестве необработанной группы (носитель) перорально вводили раствор (PEG 400/Tween 80/0.25% CMC, 5%/5%/90%, об./об./об.) содержащий 5% полиэтиленгликоль/5% твин 80/0.25% карбоксиметилцеллюлозу (CMC) в такой же дозировке. Глюкозу (4 г/кг) перорально вводили в дозировке 5 мл/кг через 30 минут после введения каждого образца. Затем измеряли уровень глюкозы крови с использованием полосок Accu-chek active strip (Roche diagnostic Co.). Время для измерения задали как 30 минут перед введением глюкозы (-30), 0 минут, 20 минут, 40 минут, 60 минут, и 120 минут после введения глюкозы, и уровень глюкозы крови измеряли прокалыванием хвостовой вены. Снижение (%) AUC глюкозы крови измеряли и результаты показаны в Таблице 5 ниже.
Таблица 5
Как показано в Таблице 5, соединения примеров настоящего изобретения обладают эффектом снижения глюкозы крови, в среднем, на 21.9% по сравнению с необработанной группой, предполагая, что соединения примеров обладают превосходящими благоприятными эффектами in vivo. Более конкретно, соединение Сравнительного Примера 1, известное как обычный активатор белка GPR40, было установлено как обладающее эффектом снижения глюкозы крови на 16.2%, при этом соединения примеров настоящего изобретения показали более превосходящие эффекты снижения глюкозы крови по сравнению с соединением Сравнительного Примера 1. Особенно, соединения примеров 9, 12, 14, 29, и 37 показали эффекты снижения глюкозы крови на 24.7%, 31.0%, 27.7%, 27.1%, и 28.5%, соответственно, и таким образом проявили более превосходящую эффективность по сравнению с соединением Сравнительного Примера 1.
Таким образом, новые производные 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения обладают превосходящим эффектом активации белка GPR40, и таким образом обладают превосходящим эффектом активации секреции инсулина, приводящим к существенно превосходящему эффекту снижения глюкозы крови, и так, фармацевтическая композиция, содержащая указанное новое соединение в качестве активного ингредиента может выгодно применяться в качестве фармацевтической композиции для лечения метаболических нарушений, таких как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
Экспериментальный Пример 5. Пероральный тест толерантности к глюкозе (OGTT) 2
С целью оценки in vivo эффект снижения глюкозы крови нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения, проводился следующий эксперимент.
Самцы крыс Гото-Какизаки (GK) в возрасте 22-23 недель, в качестве модели сахарного диабета II типа без ожирения, акклиматизировали в течение по меньшей мере 7 дней, и затем только здоровых животных использовали для проведения перорального теста толерантности к глюкозе (тест OGTT). После голодания в течение 16-18 часов, пять крыс на группу были случайно сгруппированы и им перорально вводили соединения, полученные в примерах 5, 9, 14, 28, 37, и 47 в дозировке 0.3-10 мг/кг каждого. В данном случае, в качестве необработанной группы (носитель), перорально вводили раствор (PEG 400/Tween 80/0.25% CMC, 5%/5%/90%, об./об./об.) содержащий 5% полиэтиленгликоль/5% твин 80/0.25% карбоксиметилцеллюлозу (CMC) в такой же дозировке. Глюкозу (4 г/кг) перорально вводили в дозировке 5 мл/кг через 60 минут после введения каждого образца. Затем уровень глюказы крови измеряли с использованием полосок Accu-chek active strip (Roche diagnostic Co.). Время для измерения было задано как 30 минут перед введением глюкозы (-30), 0 минут, 20 минут, 40 минут, 60 минут, и 120 минут после введения глюкозы, и уровень глюкозы крови измеряли прокалыванием хвостовой вены. Снижение (%) AUC глюкозы крови измеряли, и результаты показаны в Таблице 6 ниже.
Таблица 6

В таблице 6
A: более 35.0%;
B: 25.0-35.0%; и
C: менее 25.0%.
Как показано в Таблице 6, соединения примеров настоящего изобретения показали эффект снижения глюкозы крови на, в среднем, по меньшей мере 30.0% по сравнению с необработанной группой в такой же дозировке соединения Сравнительного Примера 1 (10 мг/кг). Более конкретно, соединение Сравнительного Примера 1 показало эффект снижения глюкозы крови на 25.3% (B) в дозировке 10 мг/кг, при этом соединения примеров 5, 9, 14, 28, 37, и 47 показали аналогичный эффект снижения глюкозы крови в дозировке 3 мг/кг по сравнению с соединением Сравнительного Примера 1. В частности, соединения примеров 9 и 37 показали эффект снижения глюкозы крови на 35.0% или более в дозировке 10 мг/кг, указывая на более превосходящую эффективность по сравнению с соединением Сравнительного Примера 1.
Таким образом, новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения обладает превосходящим эффектом активации белка GPR40, и таким образом обладает превосходящим эффектом активации секреции инсулина, приводящим к существенно превосходящему эффекту снижения глюкозы крови, и так, фармацевтическая композиция, содержащая указанное новое соединение в качестве активного ингредиента, может выгодно применяться в качестве фармацевтической композиции для лечения метаболических нарушений, таких как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
Экспериментальный Пример 6. Пероральный Тест Толерантности к Глюкозе (OGTT) 3
С целью оценки in vivo эффекта снижения глюкозы крови нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения, проводился следующий эксперимент.
Самцов крыс со спонтанным ожирением Otsuka Long-Evans Tokushima (OLETF) возрастом 29-30 недель, в качестве модели диабета II типа с ожирением, акклиматизировали в течение по меньшей мере 7 дней, и затем только здоровых животных использовали для проведения перорального теста толерантности к глюкозе (OGTT тест). После голодания в течение 16-18 часов, пять крыс на группу были случайно сгруппированы, и им перорально вводили соединения, полученные в примерах 5, 9, 14, 28, 37, и 47 в дозировке 1-10 мг/кг каждого. В данном случае, в качестве необработанной группы (носитель), раствор (PEG 400/Tween 80/0.25% CMC, 5%/5%/90%, об./об./об.), содержащий 5% полиэтиленгликоль/5% твин 80/0.25% карбоксиметилцеллюлозу (CMC), перорально вводили в такой же дозировке. Глюкозу (4 г/кг) перорально вводили в дозировке 5 мл/кг 60 минут после введения каждого образца. Затем уровень глюкозы крови измеряли с использованием полосок Accu-chek active strip (Roche diagnostic Co.). Время для измерения было задано как 60 минут перед введением глюкозы (-60), 0 минут, 20 минут, 40 минут, 60 минут, и 120 минут после введения глюкозы, и уровень глюкозы крови измеряли прокалыванием хвостовой вены. Снижение (%) AUC глюкозы крови измеряли, и результаты показаны в Таблице 7 ниже.

В Таблице 7
A: более 35.0%;
B: 25.0-35.0%; и
C: менее 25.0.
Как показано в Таблице 7, соединения примеров настоящего изобретения показали эффект снижения глюкозы крови на, в среднем 35.0% или более, по сравнению с необработанной группой в такой же дозировке соединения Сравнительного Примера 1 (10 мг/кг). Более конкретно, соединение Сравнительного Примера 1 показало эффект снижения глюкозы крови на 31.6% (B) в дозировке 10 мг/кг, при этом соединения примеров 9 и 37 показали более превосходящие эффекты снижения глюкозы крови в дозировке 1 мг/кг по сравнению с соединением Сравнительного Примера 1. В частности, соединения примеров 9 и 37 показали эффект снижения глюкозы крови на 35.0% или более в дозировке 10 мг/кг, указывая на более превосходящую эффективность по сравнению с соединением Сравнительного Примера 1.
Таким образом, новые производные 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения обладают превосходящим эффектом активации белка GPR40, и таким образом обладают превосходящим эффектом активации секреции инсулина, приводящим к существенно превосходящему эффекту снижения глюкозы крови, и так, фармацевтическая композиция, содержащая указанное новое соединение в качестве активного ингредиента, может выгодно применяться в качестве фармацевтической композиции для лечения метаболических нарушений, таких как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
Экспериментальный Пример 7. Измерение увеличения концентрации в крови глюкагонподобного пептида-1 (GLP-1) после перорального введения
С целью оценки скорости увеличения концентрации GLP-1 в крови после введения нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения, проводился следующий эксперимент.
Самцов крыс Спрег-Доули (SD) в возрасте 10-12 недель акклиматизировали в течение по меньшей мере 7 дней, и затем только здоровые животные использовались для следующего эксперимента. После голодания в течение 16-18 часов, пять крыс на группу были случайно сгруппированы и им перорально вводили соединение, полученное в примере 9, в дозировке 10-100 мг/кг каждого (объем растворителя для введения: 5 мл/кг). В данном случае, в качестве необработанной группы (носитель), раствор (PEG 400/Tween 80/0.25% CMC, 5%/5%/90%, об./об./об.), содержащий 5% полиэтиленгликоль/5% твин 80/0.25% карбоксиметилцеллюлозу (CMC), перорально вводили в такой же дозировке. Через 20 минут, около 0.5 мл цельной крови собрали прямым сбором крови через сердечную инъекцию, и собранную кровь немедленно поместили в пробирку для образцов, обработанную ингибитором дипептидилпептидазы IV (DPPIV) и этилендиаминтетрауксусной кислотой (EDTA), и поместили в контейнер, содержащий лед. Собранную кровь центрифугировали при 3600 об/мин в течение 10 минут для отделения плазмы, и отделенную плазму анализировали на концентрацию в плазме GLP-1 с помощью набора GLP-1 ELISA (Millipore, USA). Результаты показаны на Фиг. 2.
Фиг. 2 представляет собой график, иллюстрирующий концентрацию GLP-1 в крови, когда крысам Спрег-Доули (SD) перорально вводили соединения примера 9 и Сравнительного Примера 1.
Как показано на Фиг. 2, по сравнению с обработанной глюкозой группой (Veh.), соединение Сравнительного Примера 1 не показало эффект увеличения концентрации гормона GLP-1, который активирует секрецию инсулина, после введения, но соединение примера 9 увеличивало концентрацию GLP-1 в крови при дозе, вводимой крысам SD.
Таким образом, новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения обладает превосходящим эффектом активации секреции гормона GLP-1, по сравнению с соединением Сравнительного Примера 1, и особенно, показало сильно превосходящую эффективность на животной модели диабета. В дополнение, можно ожидать, что новые соединения настоящего изобретения предотвращают дисфункцию бета клеток и набор веса посредством активации секреции GLP-1, и может выгодно применяться в качестве фармацевтической композиции для лечения метаболических нарушений, таких как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
При этом соединения, представленные формулой 1 настоящего изобретения могут быть составлены в различные формы в соответствии с назначением применения. Далее приводятся примеры некоторых препаратов, содержащих соединение формулы 1 настоящего изобретения в качестве активного ингредиента, однако настоящее изобретение не ограничено ими.
Экспериментальный Пример 8. Пероральный Тест Толерантности к Глюкозе (OGTT) при совместном введении с ингибитором дипептидилпептидазы IV (DPPIV)
С целью оценки in vivo эффекта снижения глюкозы крови при совместном введении с новым производным 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения и ингибитором дипептидилпептидазы IV (DPPIV), проводился следующий эксперимент.
8-1. Эксперимент на мышиной модели
После голодания в течение 16-18 часов, самцов мышей с алиментарным ожирением (DIO) в возрасте 29 - 30 недель случайно группировали по пять животных на каждую группу, и затем перорально вводили соединение, полученное в примере 9 в дозировке 30-100 мг/кг (объем растворителя для введения: 5 мл/кг). В данном случае, в качестве необработанной группы, 5% карбоксиметилцеллюлозу (CMC) перорально вводили в такой же дозировке. В дополнение, 10 мг/кг ситаглиптина, который является хорошо известным как лекарство-ингибитор дипептидилпептидазы IV (DPPIV) вводили самостоятельно. Кроме того, 10 мг/кг ситаглиптина и 30-100 мг/кг соединения, полученного в примере 9, вводили совместно. Каждый тестовый образец и 0.5% карбоксиметилцеллюлозы (CMC) перорально вводили при 5 мл/кг.
Через 60 минут глюкозу (4 г/кг) перорально вводили в дозировке 5 мл/кг. Глюкозу крови измеряли с ипользованием полосок Accu-chek active strip (Roche diagnostic Co.). Время для измерения было задано как 60 минут перед введением глюкозы (-60), 0 минут, 20 минут, 40 минут, 60 минут, и 120 минут после введения глюкозы, и уровень глюкозы крови измеряли прокалыванием хвостовой вены. Результаты показаны на Фиг. 3 и в Таблице 3 ниже в виде снижения (%) AUC глюкозы крови.
Таблица 8
Как показано на Фиг. 3 и в Таблице 8 выше, эффект снижения глюкозы крови был более сильным, когда ситаглиптин (10 мг/кг) и соединение (30 мг/кг или 100 мг/кг), полученное в примере 9, вводили совместно по сравнению с тем, когда ситаглиптин (10 мг/кг) использовали самостоятельно.
8-2. Эксперимент на модели крыс Спрег-Доули (SD)
Самцов крыс Спрег-Доули (SD) возрастом 8-10 недель акклиматизировали в течение по меньшей мере 7 дней, и затем только здоровых животных использовали для эксперимента OGTT. После голодания в течение 16-18 часов, шесть крыс на группу были случайно сгруппированы и им вводили носитель (0.5%, карбоксиметилцеллюлоза (CMC)) или соединение примера 9 (3 мг/кг) или линаглиптин (1, 3, или 10 мг/кг), или совместно вводили соединение примера 9 (3 мг/кг) плюс линаглиптин (1, 3, или 10 мг/кг). Носитель и соединение примера 9 перорально вводили при 10 мл/кг. Через 30 минут после введения носителя или соединение примера 9, глюкозу (4 г/кг) перорально вводили в дозировке 5 мл/кг. Глюкозу крови измеряли с ипользованием полосок Accu-chek active strip (Roche diagnostic Co.). Время для измерения было задано как 30 минут перед введением глюкозы (-30), 0 минут, 20 минут, 40 минут, 60 минут, и 120 минут после введения глюкозы, и уровень глюкозы крови измеряли прокалыванием хвостовой вены. Результаты показаны в виде снижения (%) AUC глюкозы крови. Снижение (%) AUC показано в Таблице 9 ниже и на Фиг. 7.
Результаты эксперимента были выражены в виде среднего и стандартного отклонения (Среднее ± SE), и различия между контрольной и экспериментальной группами тестировались с помощью одномерного анализа ANOVA (способ Даннета) программного обеспечения “GraphPad Prism 4” (Graphpad co., La Jolla, CA, USA). В данном случае, p<0.05 рассматривалось как статистически значимое значение.
При совместном введении различных доз линаглиптина и соединения примера 9, снижение (%) AUC наблюдалось до достижения значений, близких к пороговому значению, которое можно наблюдать в экспериментальном окружении, и каждая экспериментальная группа показала эффект уменьшения площади под кривой (AUC) на приблизительно 5.8-24.4% по сравнению с носителем. Эти результаты показывают, что совместное введение соединения примера 9 и лекарственного средства на основе дипептидилпептидазы IV может максимизировать эффективность лекарственного средства на основе дипептидилпептидазы IV при снижении дозы лекарственного средства на основе дипептидилпептидазы IV.
Таблица 9
Таким образом, совместное введение нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения и лекарственных средств на основе дипептидилпептидазы IV (DPPIV) показывает превосходящий эффект снижения глюкозы крови по сравнению с введением лекарственного средства самостоятельно, и так, фармацевтическая композиция, содержащая производное настоящего изобретения и другой активный ингредиент может с преимуществом применяться для предотвращения или лечения метаболических нарушений, таких как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
Экспериментальный Пример 9. Пероральный Тест Толерантности к Глюкозе (OGTT) при совместном введении с лекарственным средством на основе сульфонилмочевины
С целью оценки in vivo эффекта снижения глюкозы крови посредством совместного введения нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения и лекарственного средства на основе сульфонилмочевины, проводился следующий эксперимент.
После голодания в течение 16-18 часов, самцов мыши с алиментарным ожирением (DIO) возраста 29-30 недель случайно группировали по пять животных на каждую группу, и затем им перорально вводили соединение, полученное в примере 9, в дозировке 10-100 мг/кг (объем растворителя для введения: 10 мл/кг). В данном случае, в качестве необработанной группы, 5% карбоксиметилцеллюлозу (CMC) перорально вводили в такой же дозировке. В дополнение, 10 мг/кг глимепирида, который хорошо известен в качестве лекарственного средства на основе сульфонилмочевины, вводили самостоятельно. Кроме того, 10 мг/кг глимепирида и 10-100 мг/кг соединения, полученного в примере 9 вводили совместно. Физраствор и тестируемые соединения перорально вводили при 5 мл/кг.
Через 60 минут глюкозу (4 г/кг) перорально вводили в дозировке 5 мл/кг. Глюкозу крови измеряли с использованием полосок Accu-chek active strip (Roche diagnostic Co.). Время для измерения было задано как 60 минут перед введением глюкозы (-60), 0 минут, 20 минут, 40 минут, 60 минут, и 120 минут после введения глюкозы, и уровень глюкозы крови измеряли прокалыванием хвостовой вены. Результаты показаны на Фиг. 4 и в Таблице 10 ниже в виде снижения (%) AUC глюкозы крови.
[Таблица 10]
Как показано на Фиг. 4 и в Таблице 10 выше, эффект снижения глюкозы крови был более превосходящим, когда глимепирид (10 мг/кг) и соединение (30 мг/кг или 100 мг/кг), полученное в примере 9, вводили совместно, по сравнению с тем, когда ситаглптин (10 мг/кг) использовали самостоятельно.
Таким образом, совместное введение нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения и лекарственного средства на основе сульфонилмочевины показывает превосходящий эффект снижения глюкозы крови по сравнению с введением лекарственного средства самостоятельно, и так, фармацевтическая композиция в соответствии с настоящим изобретением может с преимуществом применяться для предотвращения или лечения метаболических нарушений, таких как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
Экспериментальный Пример 10. Пероральный Тест Толерантности к Глюкозе (OGTT) при совместном введении с лекарственным средством на основе тиазолидиндиона (TZD)
С целью оценки in vivo эффекта снижения глюкозы крови посредством совместного введения нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения и лекарственного средства на основе тиазолидиндиона (TZD), проводился следующий эксперимент.
После голодания в течение 16-18 часов, самцов мышей с алиментарным ожирением (DIO) возраста 29 - 30 недель случайно группировали по пять животных на каждую группу, и затем им перорально вводили соединение, полученное в примере 9, в дозировке 10-30 мг/кг (объем растворителя для введения: 10 мл/кг). В данном случае, в качестве необработанной группы, перорально вводили 5% карбоксиметилцеллюлозу (CMC) в такой же дозировке. В дополнение, 10 мг/кг росиглитазона и пиоглитазона, которые хорошо известны как лекарственные средства на основе тиазолидиндиона (TZD), вводились самостоятельно. Кроме того, 10 мг/кг росиглитазона и пиоглитазона каждого и 10-30 мг/кг соединения, полученного в примере 9, вводили совместно. Физраствор и тестируемые соединения перорально вводили при 5 мл/кг.
Через 60 минут, глюкозу (4 г/кг) перорально вводили в дозировке 5 мл/кг. Глюкозу крови измеряли с использованием полосок Accu-chek active strip (Roche diagnostic Co.). Время для измерения было задано как 60 минут перед введением глюкозы (-60), 0 минут, 20 минут, 40 минут, 60 минут, и 120 минут после введения глюкозы, и уровень глюкозы крови измеряли прокалыванием хвостовой вены. Результаты показаны на Фиг. 5 и в Таблице 11 ниже в виде снижения (%) AUC глюкозы крови.
Таблица 11
Как показано на Фиг. 5 и в Таблице 11 выше, эффект снижения глюкозы крови был более превосходящим, когда росиглитазон (5 мг/кг) и соединение (10 мг/кг или 30 мг/кг), полученное в примере 9 вводили совместно, по сравнению с тем, когда росиглитазон (5 мг/кг) использовали самостоятельно. Эффект снижения глюкозы крови также был более превосходящим, когда пиоглитазон (10 мг/кг) и соединение (10 мг/кг), полученное в примере 9, вводили совместно по сравнению с тем, когда пиоглитазон (10 мг/кг) использовали самостоятельно
Таким образом, совместное введение нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения и лекарственного средства на основе тиазолидиндиона (TZD) показывает превосходящий эффект снижения глюкозы крови по сравнению с введением лекарственного средства самостоятельно, и так, фармацевтическая композиция, содержащая производное настоящего изобретения и другой активный ингредиент, может с преимуществом применяться для предотвращения или лечения метаболических нарушений, таких как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
Экспериментальный Пример 11. Пероральный Тест Толерантности к Глюкозе (OGTT) при совместном введении с лекарственным средством на основе бигуанида
С целью оценки in vivo эффекта снижения глюкозы крови посредством совместного введения нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения и лекарственного средства на основе бигуанида, проводился следующий эксперимент.
11-1. Эксперимент на крысиной модели сахарного диабета II типа
После голодания в течение 16-18 часов, самцов тучных диабетических крыс Zucker (ZDF) возрастом 8 недель случайно группировали по пять животных на каждую группу, и затем им перорально вводили соединение, полученное в примере 9, в дозировке 1-10 мг/кг (объем растворителя для введения: 5 мл/кг). В данном случае, в качестве необработанной группы, перорально вводили носитель (5% карбоксиметилцеллюлоза (CMC)) в такой же дозировке. В дополнение, 50 мг/кг метформина, который хорошо известен в качестве лекарственного средства на основе бигуанида, вводили самостоятельно. Кроме того, 50 мг/кг метформина и 1-10 мг/кг соединения, полученного в примере 9, вводили совместно.
Через 60 минут, глюкозу (4 г/кг) перорально вводили в дозировке 5 мл/кг. Глюкозу крови измеряли с использованием полосок Accu-chek active strip (Roche diagnostic Co.). Время для измерения было задано как 60 минут перед введением глюкозы (-60), 0 минут, 20 минут, 40 минут, 60 минут, и 120 минут после введения глюкозы, и уровень глюкозы крови измеряли прокалыванием хвостовой вены. Результаты показаны на Фиг. 6 и в Таблице 12 ниже в виде снижения (%) AUC глюкозы крови.
Таблица 12
Как показано на Фиг. 6 и в Таблице 12 выше, эффект снижения глюкозы крови был более превосходящим, когда метформин (50 мг/кг) и соединение (1 мг/кг, 3 мг/кг, или 10 мг/кг), полученное в примере 9, вводили совместно, по сравнению с тем, когда метформин (50 мг/кг) использовали самостоятельно.
11-2. Эксперимент на модели крыс SD
Самцов крыс Спрег-Доули (SD) возрастом 8-10 недель акклиматизировали в течение по меньшей мере 7 дней, и затем только здоровых животных использовали для эксперимента OGTT. После голодания в течение 16-18 часов, шесть крыс на группу были случайно сгруппированы и им вводили носитель (0.5%, карбоксиметилцеллюлоза (CMC)) или соединение примера 9 (3 мг/кг) или метформин (10, 50, или 100 мг/кг), или совместно вводили соединение примера 9 (3 мг/кг) плюс метформин (10, 50, или 100 мг/кг). Носитель и соединение примера 9 перорально вводили при 10 мл/кг. Через 30 минут после введения носителя или соединения примера 9, глюкозу (4 г/кг) перорально вводили в дозировке 5 мл/кг. Глюкозу крови измеряли с использованием полосок Accu-chek active strip (Roche diagnostic Co.). Время для измерения было задано как 30 минут перед введением глюкозы (-30), 0 минут, 20 минут, 40 минут, 60 минут, и 120 минут после введения глюкозы, и уровень глюкозы крови измеряли прокалыванием хвостовой вены. Результаты показаны на Фиг. 8 и в Таблице 13 ниже в виде снижения (%) AUC глюкозы крови.
Результаты эксперимента были выражены в виде среднего и стандартного отклонения (Среднее ± SE), и различия между контрольной и экспериментальной группами тестировались с помощью одномерного анализа ANOVA (способ Даннета) программного обеспечения “GraphPad Prism 4” (Graphpad co., La Jolla, CA, USA). В данном случае, p<0.05 рассматривалось как статистически значимое значение.
При совместном введении различных доз метформина и соединения примера 9, снижение (%) AUC наблюдалось до достижения значений, близких к пороговому значению, которое можно наблюдать в экспериментальном окружении, и каждая экспериментальная группа показала эффект уменьшения площади под кривой (AUC) на приблизительно 3.9-20.2% по сравнению с носителем. Эти результаты показывают, что совместное введение соединения примера 9 и лекарственного средства на основе бигуанида может максимизировать эффективность лекарственного средства на основе бигуанида при снижении дозы лекарственного средства на основе бигуанида.
Таблица 13
Таким образом, совместное введение новых производных 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения и лекарственного средства на основе бигуанида показывает превосходящий эффект снижения глюкозы крови по сравнению с введением лекарственного средства самостоятельно, и так, фармацевтическая композиция в соответствии с настоящим изобретением может с преимуществом применяться для предотвращения или лечения метаболических нарушений, таких как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
Экспериментальный Пример 12. Пероральный Тест Толерантности к Глюкозе (OGTT) при совместном введении с лекарственным средством на основе ингибитора котранспортера натрий/глюкозы 2 (SGLT2)
С целью оценки in vivo эффекта снижения глюкозы крови посредством совместного введения нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения и лекарственного средства на основе ингибитора котранспортера натрий/глюкозы 2 (SGLT2) проводился следующий эксперимент.
Самцов крыс Спрег-Доули (SD) возрастом 8-10 недель акклиматизировали в течение по меньшей мере 7 дней, и затем только здоровых животных использовали для эксперимента OGTT. После голодания в течение 16-18 часов, шесть крыс на группу были случайно сгруппированы и им вводили носитель (0.5%, карбоксиметилцеллюлоза (CMC)) или соединение примера 9 (3 мг/кг) или эмпаглифлозин (1, 3, или 10 мг/кг), или совместно вводили соединение примера 9 (3 мг/кг) плюс эмпаглифлозин (1, 3, или 10 мг/кг). Носитель и соединение примера 9 перорально вводили при 10 мл/кг. Через 30 минут после введения носителя или соединение примера 9, глюкозу (4 г/кг) перорально вводили в дозировке 5 мл/кг. Глюкозу крови измеряли с использованием полосок Accu-chek active strip (Roche diagnostic Co.). Время для измерения было задано как 30 минут перед введением глюкозы (-30), 0 минут, 20 минут, 40 минут, 60 минут, и 120 минут после введения глюкозы, и уровень глюкозы крови измеряли прокалыванием хвостовой вены. Результаты показаны на Фиг. 9 и в Таблице 14 ниже в виде снижения (%) AUC глюкозы крови.
Результаты эксперимента были выражены в виде среднего и стандартного отклонения (Mean ± SE), и различия между контрольной и экспериментальной группами тестировались с помощью одномерного анализа ANOVA (способ Даннета) программного обеспечения “GraphPad Prism 4” (Graphpad co., La Jolla, CA, USA). В данном случае, p<0.05 рассматривалось как статистически значимое значение.
При совместном введении различных доз эмпаглифлозина и соединения примера 9, снижение (%)AUC наблюдалось до достижения значений, близких к пороговому значению, которое можно наблюдать в экспериментальном окружении, и каждая экспериментальная группа показала эффект уменьшения площади под кривой (AUC) на приблизительно 6.5-36.6 % по сравнению с носителем. Эти результаты показывают, что совместное введение соединения примера 9 и лекарственного средства на основе ингибитора SGLT2 может максимизировать эффективность лекарственного средства на основе ингибитора SGLT2 при снижении дозы лекарственного средства на основе ингибитора SGLT2.
Таблица 14
Таким образом, совместное введение нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты настоящего изобретения и лекарственного средства на основе ингибитора котранспортера натрий/глюкозы 2 (SGLT2) показывает превосходящий эффект снижения глюкозы крови по сравнению с введением лекарственного средства самостоятельно, и так, фармацевтическая композиция настоящего изобретения может с преимуществом применяться для предотвращения или лечения метаболических нарушений, таких как ожирение, диабет I типа, диабет II типа, нарушение толерантности к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х.
Экспериментальный Пример 13. Эксперимент анализа секреции GLP-1
NCI-H716 клетки засеяли в 12-луночный планшет, покрытый Матригелем (BD), при плотности 1×106 клеток/лунка, и культивировали в инкубаторе (37°) в течение 48 часов. После удаления супернатанта, клетки промыли средой DMEM с низким содержанием глюкозы (5.5 мМ; содержащей 2% ФБС, 2 мМ L-глутамина, 100 МЕ/мл пенициллина, и 100 мкг/мл стрептомицина), и выращивали клетки в аналогичной среде в течение 4 часов. После удаления супернатанта, среду заменили на среду DMEM с высоким содержанием глюкозы (25 мМ), содержащую разведенный ситаглиптин (0.1, 1, или 10 мкМ), и затем предварительно обработали в течение 30 минут. Через 30 минут, среду обработали соединением примера 9 каждой дозой (1, 10, или 30 мкМ), и затем клетки культивировали при 37° в течение 2 часов. В качестве контрольной группы использовали 0.1% ДМСО. Количество GLP-1, секретируемого клетками NCI-H716 измеряли с помощью набора для глюкагонподобного пептида 1 (GLP-1) (Millipore) с использованием супернатанта клеток после завершения эксперимента (см. Таблицу 15 и Фиг. 10).
Таблица 15
В качестве результата наблюдали, что секреция GLP-1 существенно увеличилась в группах, совместно обработанных ситаглиптином и соединением примера 9, по сравнению с группами, обработанными только ситаглиптином.
Экспериментальный Пример 14. Эксперимент секреции инсулина
INS-1 клетки (клеточная линия инсулиномы крыс) засеяли в 24-луночный планшет при плотности 5×105 клеток/лунка, и культивировали в течение 48 часов. Затем клетки промыли 3 мМ буфером глюкоза-KRB (118 мМ NaCl, 4.7 мМ KCl, 1.2 мМ KH2PO4, 1.16 мМ MgCl2, 10 мМ HEPES, 2.5 мМ CaCl2, 25.5 мМ NaHCO3, 0.2% БСА, pH 7.4), и культивировали в аналогичном буфере в течение 2 часов, таким образом, что концентрация внутриклеточной глюкозы могла быть на низком уровне. Тестируемое соединение (см. Таблицу 16) разбавили до конечной концентрации 0.1-10 M в 25 мМ буфере глюкоза-KRB, и затем использовали для обработки клеток после завершения культивирования в среде с 3 мМ глюкозой в течение 1 часа, активируя таким образом секрецию инсулина. Количество секретированного инсулина измеряли с помощью набора для инсулина ELISA (Morinaga) с использованием супернатанта клеток после завершения эксперимента (см. Таблицу 16 и Фиг. 11).
[Таблица 16]
В качестве результата, было установлено, что секреция инсулина увеличивалась сильнее в группах совместного введения глибенкламида и соединения примера 9, по сравнению с группами введения только соединения примера 9.
Препаративный Пример 1. Получение фармацевтического препарата
1-1. Получение порошков
Порошки получили смешиванием всех вышеуказанных ингредиентов и последующей упаковки смеси в герметический пакетик.
1-2. Получение таблеток
Таблетки получили смешиванием вышеуказанных ингредиентов и последующего таблетирования смеси в соответствии с обычным способом для получения препаратов таблеток.
1-3. Получение капсул
Капсулы получили смешиванием вышеуказанных ингредиентов и последующего заполнения смесью желатиновых капсул в соответствии с обычным способом для получения препаратов капсул.
1-4. Получение инъекций
Инъекции получили заполнением вышеуказанных ингредиентов в ампулы (2 мл) в соответствии с обычным способом для получения инъекций.
1-5. Получение жидких препаратов
В соответствии с обычным способом получения жидкого препарата, каждый ингредиент растворили в очищенной воде, в которую добавили лимонный ароматизатор, и затем вышеуказанные ингредиенты смешали, к чему добавили очищенную воду для доведения общего объема до 100 мл. Смесью заполнили коричневую бутылку и стерилизовали для получения жидких препаратов.
Хотя настоящее изобретение было подробно описано со ссылкой на конкретные признаки, специалистам в данной области техники будет очевидно, что это описание относится только к предпочтительному варианту осуществления и не ограничивает объем настоящего изобретения. Таким образом, существенный объем настоящего изобретения будет определяться прилагаемой формулой изобретения и ее эквивалентами.
Реферат
Настоящее изобретение относится к фармацевтической комбинации для активирования фермента рецептора G-белка 40 (GPR40), которая содержит новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты и второй активный ингредиент, который выбран из группы, состоящей из лекарственных средств на основе ингибиторов дипептидилпептидазы-4 (DPPIV), на основе сульфонилмочевины, на основе тиазолидиндиона (TZD), на основе бигуанида и на основе ингибитора натрий/глюкоза котранспортера 2 (SGLT2). Новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты, предпочтительно, представляет собой натрия (S)-3-(4-(3-((4-(4-метоксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иноат. Применение комбинации настоящего изобретения может обеспечить значительно превосходящий эффект снижения сахара крови в различных животных моделях диабетических заболеваний, и комбинация настоящего изобретения может благоприятно применяться для предотвращения или лечения метаболических нарушений, таких как ожирение, диабет типа I, диабет типа II, нарушенная толерантность к глюкозе, синдром резистентности к инсулину, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром Х. 2 н. и 3 з.п. ф-лы, 11 ил., 16 табл.
Формула











Комментарии