Композиции для лечения фиброза и связанных с фиброзом состояний - RU2712140C2
Код документа: RU2712140C2
Чертежи














Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] Настоящее изобретение относится к новым соединениям и их применению при профилактическом и/или терапевтическом лечении фиброза и связанных с фиброзом состояний.
[0002] Настоящее изобретение было разработано в первую очередь для лечения фиброза и будет описано ниже в данном документе со ссылкой на настоящую заявку. Однако следует принимать во внимание, что настоящее изобретение не ограничено данной конкретной областью применения.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[0003] Любое обсуждение уровня техники по всему настоящему описанию никоим образом не должно рассматриваться как допущение того, что такой уровень техники широко известен или образует часть общеизвестных знаний в данной области.
[0004] Репарация поврежденных тканей является основным биологическим процессом. Процесс репарации включает две различные стадии: регенеративную фазу, при которой пораженные клетки замещаются нормальными клетками того же типа; и фазу, известную как фиброз, при которой соединительная ткань замещает нормальную паренхиматозную ткань. В большинстве случаев обе стадии необходимы для замедления или устранения повреждения, вызванного повреждающим средством. Однако даже первоначально благоприятный процесс заживления может стать патогенным, если он продолжается бесконтрольно, вызывая существенное ремоделирование ткани и образование постоянной рубцовой ткани. Фиброзное рубцевание зачастую определяют как заживляющую раны реакцию, которая пошла не так.
[0005] Фиброзные изменения могут происходить во всех основных тканях и системах органов, в том числе в сердце, почке и печени, причем правительство США установило, что 45% смертей в США можно объяснить фиброзными нарушениями (Wynn, Nat Rev Immunol, 2004, 4 (8): 583-594). Например:
- фиброзные изменения в сердце приводят в результате к утолщению сердечных клапанов и потере эластичности сердечной мышцы, что может вызвать сердечную недостаточность;
- фиброзные изменения в почке могут привести в результате к разрушению почечных канальцев и внутренних капилляров, вызывая прогрессирующую потерю почечной функции; и
- жировая болезнь печени (при которой большие вакуоли триглицерида накапливаются в клетках печени) приводит в результате к накоплению фиброзной ткани в печени, вызывая цирроз, печеночную недостаточность и портальную гипертензию.
[0006] Существует необходимость в средствах, которые предотвращают или лечат фиброз и связанные с фиброзом состояния. В частности, существует необходимость в средствах, которые предотвращают, ослабляют или замедляют прогрессирование фиброза, уменьшают развившийся фиброз, предотвращают, снижают степень или замедляют отмирание клеток почечных канальцев, предотвращают, снижают степень или замедляют накопление жира в печени и/или восстанавливают нормальную структуру ткани.
[0007] Целью настоящего изобретения является преодоление или устранение по меньшей мере одного из недостатков уровня техники или предоставление полезной альтернативы.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0008] Согласно одному аспекту настоящее изобретение предусматривает соединение формулы:



где
А выбран из необязательно замещенного насыщенного, частично насыщенного или ненасыщенного 5- или 6-членного гетероциклила; необязательно замещенного С1-6алкоксиламина; необязательно замещенного С1-6алкиламина; необязательно замещенной С0-6алкил-карбоновой кислоты; необязательно замещенного С1-6алкилгидроксила; необязательно замещенного насыщенного или ненасыщенного С0-6алкил-бициклического-гетероциклила и необязательно замещенного насыщенного или ненасыщенного С1-6алкоксил-бициклического-гетероциклила, или их фармакологически приемлемая соль, стереоизомер, диастереомер, энантиомер, рацемат, гидрат и/или сольват.
[0009] В одном варианте осуществления насыщенный, частично насыщенный или ненасыщенный 5- или 6-членный гетероциклил содержит один или несколько атомов N, S или О, необязательно замещенных одним или несколькими заместителями, представляющими собой оксо, С1-6алкил, амино, гидроксил или галоген.
[0010] В одном варианте осуществления насыщенный, частично насыщенный или ненасыщенный 5- или 6-членный гетероциклил выбран из пирролила, пиразолила, имидазолила, триазолила, имидазолидинила, пирролидинила, пирролидинилидена, дигидропирролила, изоксазолил-дигидрооксазолила, изоксазолидинила, оксазолидинила и оксазолила, необязательно замещенных одним или несколькими заместителями, представляющими собой оксо, С1-6алкил, амино, гидроксил или галоген.
[0011] В одном варианте осуществления С1-6алкоксиламин представляет собой аминооксиметил.
[0012] В одном варианте осуществления С1-6алкиламин необязательно замещен одним или несколькими из С1-6алкила, С1-6галогеналкила, гидроксила или галогена, предпочтительно моно-, ди- или тризамещенного галогеналкила, наиболее предпочтительно трифторметана.
[0013] В одном варианте осуществления С0-6алкил-карбоновая кислота представляет собой карбоновую кислоту.
[0014] В одном варианте осуществления С1-6алкилгидроксил представляет собой метилгидроксил.
[0015] В одном варианте осуществления С0-6алкил-бициклический-гетероциклил выбран из индолила, изоиндолила, индолинила и изоиндолинила, необязательно замещенных одним или несколькими оксо, предпочтительно диоксо.
[0016] В одном варианте осуществления С1-6алкоксил-бициклический-гетероциклил выбран из индолила, изоиндолила, индолинила и изоиндолинила, необязательно замещенных одним или несколькими оксо, и где С1-6алкоксил представляет собой метокси или этокси.
[0017] В одном варианте осуществления А выбран из:




[0018] В одном варианте осуществления соединение выбрано из группы, состоящей из:






или его фармакологически приемлемой соли, стереоизомера, диастереомера, энантиомера, рацемата, гидрата и/или сольвата.
[0019] Согласно другому аспекту настоящее изобретение относится к фармацевтической композиции, содержащей соединение по настоящему изобретению и фармацевтически приемлемое вспомогательное вещество.
[0020] Согласно другому аспекту настоящее изобретение относится к способу терапевтического лечения фиброза у субъекта, включающему введение субъекту соединения или фармацевтической композиции согласно настоящему изобретению.
[0021] Согласно другому аспекту настоящее изобретение относится к способу профилактического лечения фиброза у субъекта, включающему введение субъекту соединения или фармацевтической композиции согласно настоящему изобретению.
[0022] Согласно другому аспекту настоящее изобретение относится к соединению или фармацевтической композиции по настоящему изобретению для применения в способе терапевтического лечения фиброза.
[0023] Согласно другому аспекту настоящее изобретение относится к соединению или фармацевтической композиции по настоящему изобретению для применения в способе профилактического лечения фиброза.
[0024] Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного препарата для терапевтического лечения фиброза.
[0025] Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного препарата для профилактического лечения фиброза.
[0026] В одном варианте осуществления соединение, фармацевтическая композиция или лекарственный препарат по настоящему изобретению предотвращает, ослабляет или замедляет прогрессирование фиброза.
[0027] В одном варианте осуществления соединение, фармацевтическая композиция или лекарственный препарат по настоящему изобретению уменьшает развившийся фиброз.
[0028] В одном варианте осуществления соединение, фармацевтическая композиция или лекарственный препарат по настоящему изобретению восстанавливает нормальную структуру ткани.
[0029] В одном варианте осуществления фиброз представляет собой фиброз миокарда.
[0030] В одном варианте осуществления фиброз представляет собой фиброз почки.
[0031] В одном варианте осуществления фиброз представляет собой фиброз печени.
[0032] Согласно другому аспекту настоящее изобретение относится к способу предотвращения, снижения степени или замедления накопления жира в печени субъекта, включающему введение субъекту соединения или фармацевтической композиции согласно настоящему изобретению.
[0033] Согласно другому аспекту настоящее изобретение относится к способу предотвращения, снижения степени или замедления отмирания клеток почечных канальцев у субъекта, включающему введение субъекту соединения или фармацевтической композиции согласно настоящему изобретению.
[0034] Согласно другому аспекту настоящее изобретение относится к способу восстановления нормальной структуры ткани у субъекта, включающему введение субъекту соединения или фармацевтической композиции согласно настоящему изобретению.
[0035] Согласно другому аспекту настоящее изобретение относится к соединению или фармацевтической композиции по настоящему изобретению для применения в способе предотвращения, снижения степени или замедления накопления жира в печени.
[0036] Согласно другому аспекту настоящее изобретение относится к соединению или фармацевтической композиции по настоящему изобретению для применения в способе предотвращения, снижения степени или замедления отмирания клеток почечных канальцев.
[0037] Согласно другому аспекту настоящее изобретение относится к соединению или фармацевтической композиции по настоящему изобретению для применения в способе восстановления нормальной структуры ткани.
[0038] Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного препарата для предотвращения, снижения степени или замедления накопления жира в печени.
[0039] Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного препарата для предотвращения, снижения степени или замедления отмирания клеток почечных канальцев.
[0040] Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного препарата для восстановления нормальной структуры ткани.
[0041] Согласно другому аспекту настоящее изобретение относится к соединению формулы:

[0042] Если контекст явно не требует иного, по всему описанию и формуле изобретения слова "содержит", "содержащий" и тому подобные должны быть истолкованы во включающем смысле в противоположность исключающему или исчерпывающему смыслу; то есть в смысле "включая без ограничения".
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0043] Фигура 1: схема синтеза А32.
[0044] Фигура 2: схема синтеза А6.
[0045] Фигура 3: схема синтеза А30.
[0046] Фигура 4: схема синтеза A56f, A56g и А56.
[0047] Фигура 5: схема синтеза A56k.
[0048] Фигура 6: схема синтеза промежуточного соединения А31-4.
[0049] Фигура 7: схема синтеза А26 и А27.
[0050] Фигура 8: схема синтеза А31.
[0051] Фигура 9: схема синтеза А35.
[0052] Фигура 10: схема синтеза А45.
[0053] Фигура 11: схема синтеза А79.
[0054] Фигура 12: схема синтеза А81.
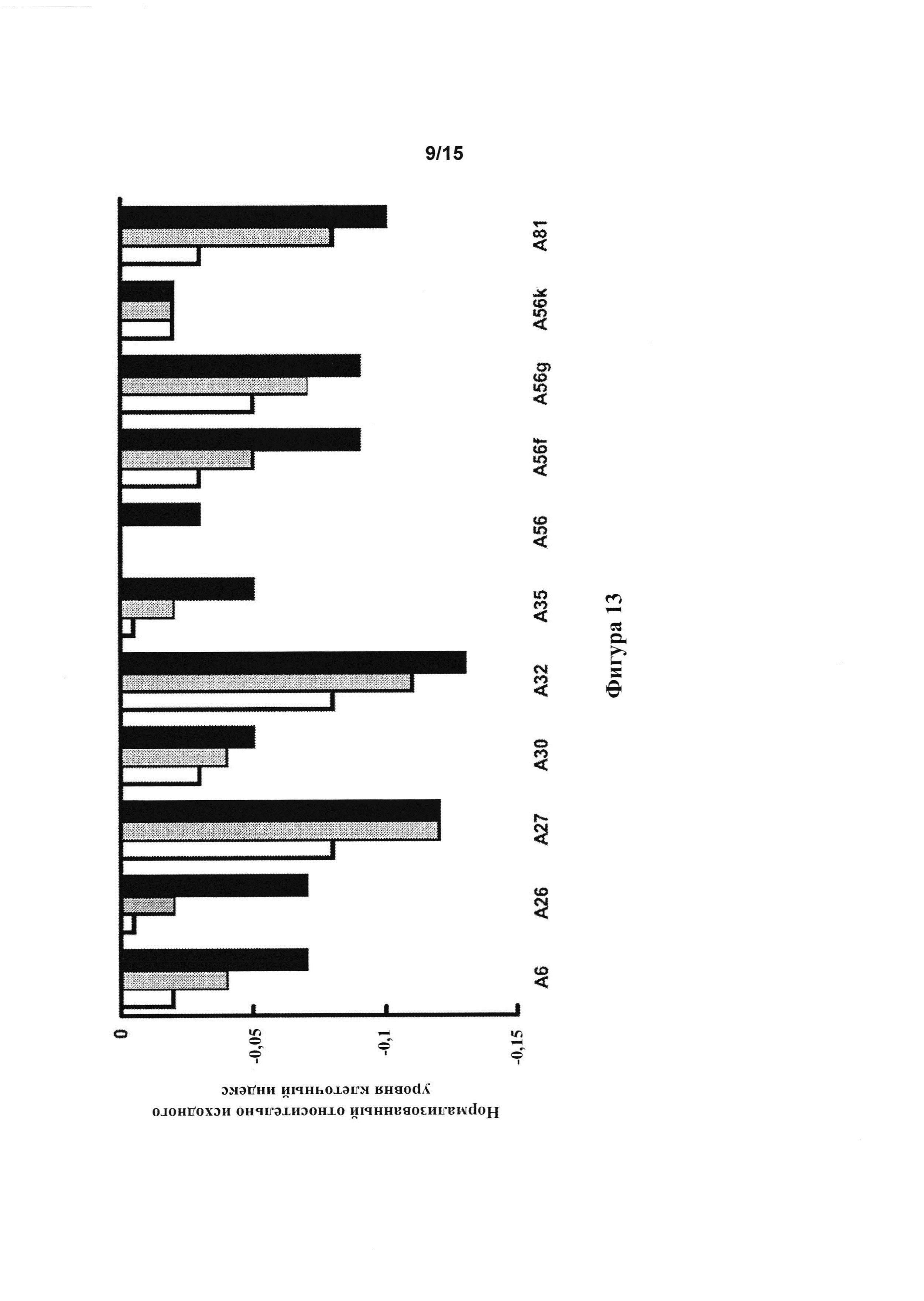
[0055] Фигура 13: клеточный импеданс бычьих аортальных эндотелиальных клеток, обработанных тестовыми соединениями в 3 концентрациях, составляющих 62,5 мкМ (белые столбцы), 125 мкМ (серые столбцы) и 250 мкМ (черные столбцы).
[0056] Фигура 14: отмирание клеток для клеток проксимальных почечных канальцев человека, инкубированных с исключительно 5 мкг/мл цис-диамминдихлорплатины (III) (цисплатина) (сплошные столбцы), 5 мкг/мл цисплатина и 32 мкМ А32 (заштрихованные столбцы), 5 мкг/мл цисплатина и 63 мкМ А32 (белые столбцы), и отмирание клеток для клеток проксимальных почечных канальцев крысы, инкубированных с исключительно 12,5 мкг/мл цисплатина (сплошные столбцы), 12,5 мкг/мл цисплатина и 32 мкМ А32 (заштрихованные столбцы), 12,5 мкг/мл цисплатина и 63 мкМ А32 (белые столбцы). Все инкубации длились 24 ч.
[0057] Фигура 15: эффект 500 пмоль/кг/мин. А32 в течение 4 недель на интерстициальный фиброз в почке у SHR на рационе с 2,2% соли и питьевом растворе с 5% этанола.
[0058] Фигура 16: эффект 500 пмоль/кг/мин. А32 в течение 4 недель на фиброз миокарда у SHR на рационе с 2,2% соли и питьевом растворе с 5% этанола.
[0059] Фигура 17: эффект 500 пмоль/кг/мин. А32 в течение 6 недель на фиброз печени у SHR на рационе с высоким содержанием жира и питьевом растворе с 10% этанола.
[0060] Фигура 18: срезы, окрашенные с применением трехцветного окрашивания по Массону, на которых видны портальные тракты контрольных крыс (А), а также крыс, обработанных А32 (В), А6 (С), А27 (D), А56 (Е) и A56f (F).
[0061] Фигура 19: срезы, окрашенные с применением трехцветного окрашивания по Массону, на которых видна ткань сердца контрольных крыс (А) и крыс, обработанных А32 (В).
[0062] Фигура 20: эффект тестовых соединений в отношении накопления жира в печени у SHR на солевом рационе с высоким содержанием жира после 6 недель обработки соединением в питьевом растворе (10% этанола) или только питьевым раствором.
[0063] Фигура 21: эффект обработки тестовым соединением в течение 6 недель на уровни аминотрансферазы (AST) в плазме крови у SHR на рационе с высоким содержанием жира и питьевом растворе с 10% этанола.
[0064] Фигура 22: Сравнение клеточного импеданса бычьих аортальных эндотелиальных клеток и уровня фиброза печени у SHR на рационе с высоким содержанием жира, обработанных тестовыми соединениями.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0065] Настоящее изобретение относится к соединениям, которые проявляют противофиброзные и родственные эффекты. Настоящее изобретение также относится к соединениям, которые эффективны в предотвращении, ослаблении или замедлении прогрессированния фиброза, уменьшении развившегося фиброза, предотвращении, снижении степени или замедлении отмирания клеток почечных канальцев, предотвращении, снижении степени или замедлении накопления жира в печени и/или восстановлении нормальной структуры ткани.
[0066] Соединения по настоящему изобретению представлены формулами:



где
А выбран из необязательно замещенного насыщенного, частично насыщенного или ненасыщенного 5- или 6-членного гетероциклила; необязательно замещенного С1-6алкоксиламина; необязательно замещенного С1-6алкиламина; необязательно замещенной С0-6алкил-карбоновой кислоты; необязательно замещенного С1-6алкилгидроксила; необязательно замещенного насыщенного или ненасыщенного С0-6алкил-бициклического-гетероциклила и необязательно замещенного насыщенного или ненасыщенного С1-6алкоксил-бициклического-гетероциклила, или их фармакологически приемлемая соль, стереоизомер, диастереомер, энантиомер, рацемат, гидрат и/или сольват.
[0067] Следующие соединения являются конкретными, но не ограничивающими примерами соединений по настоящему изобретению:






[0068] Применяемый в данном документе термин "алкил", отдельно или в комбинации, означает алкильный радикал с прямой цепью или с разветвленной цепью формулы -CnH(2n+1). Примеры алкилов включают метил, этил, н-пропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изоамил, гексил, октил и т.п.
[0069] Применяемый в данном документе термин "алкокси", отдельно или в комбинации, означает алкил, связанный с кислородом, где термин "алкил" определен выше. Примеры алкокси включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изо-бутокси, втор-бутокси, трет-бутокси и т.п.
[0070] Применяемый в данном документе термин "галоген" означает -F, -Cl, -Br или -I.
[0071] Применяемый в данном документе термин "гидрокси" означает -ОН.
[0072] Применяемые в данном документе термины "амино" или "амин" означают -NH2.
[0073] Применяемый в данном документе термин "карбоновая кислота" означает -С(O)ОН.
[0074] Применяемый в данном документе термин "окси" означает -О-.
[0075] Применяемый в данном документе термин "оксо" означает =O.
[0076] Применяемые в данном документе аббревиатуры Me, Et, Ph, Ms представляют собой метил, этил, фенил и метансульфонил соответственно. Более полный перечень аббревиатур, используемых специалистами в области органической химии, представлен в первом выпуске каждого тома Journal of Organic Chemistry; этот перечень обычно представлен в таблице под названием "Стандартный перечень аббревиатур". Аббревиатуры, содержащиеся в упомянутом перечне, и все аббревиатуры, используемые специалистами в области органической химии, включены в данный документ с помощью ссылки.
[0077] Соединения по настоящему изобретению могут существовать в определенных геометрических или стереоизомерных формах. Настоящее изобретение предполагает все подобные соединения, включая цис- и трансизомеры, (R)- и (S)-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры, их рацемические смеси и другие их смеси, как попадающие в объем настоящего изобретения. Все подобные изомеры, а также их смеси предназначены для включения в настоящее изобретение.
[0078] Если, например, необходим определенный энантиомер соединения по настоящему изобретению, его можно получить путем асимметричного синтеза или путем получения производного с хиральным вспомогательным средством, где разделяют полученную диастереомерную смесь и отщепляют вспомогательную группу с получением необходимых чистых энантиомеров. В качестве альтернативы, диастереомерные соли могут быть образованы с помощью подходящих оптически активных кислоты или основания с последующим разделением полученных таким образом диастереомеров фракционной кристаллизацией или хроматографическими средствами, хорошо известными из уровня техники, и последующим восстановлением чистых энантиомеров.
[0079] В целом, соединения по настоящему изобретению могут быть получены с помощью способов, проиллюстрированных на общих схемах реакций, как, например, описано ниже, или путем их модификаций с использованием легко доступных исходных веществ, реагентов и общепринятых методик синтеза. В данных реакциях также можно воспользоваться вариантами, которые сами по себе известны, но не упомянуты в данном документе.
[0080] Если не указано другое, способы синтеза соединения основаны на хорошо разработанных способах, описанных, например, в March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (2013) Micheal B. Smith; Advanced Organic Chemistry, Part A: Structure and Mechanisms (2008) и Advanced Organic Chemistry: Part B: Reaction and Synthesis (2010) Francis A. Carey and Richard J. Sunberg; и Greene's Protective Groups in Organic Synthesis (2014) Peter G.M. Wuts.
[0081] Настоящее изобретение также предусматривает фармацевтически приемлемые соли соединений. Термин "фармацевтически приемлемая соль" включает соли присоединения как кислоты, так и основания и относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований или кислот и которые не являются биологически или иным образом нежелательными. Фармацевтически приемлемые соли образуются с неорганическими или органическими кислотами или основаниям и могут быть получены in situ во время окончательного выделения и очистки соединений или путем отдельной реакции очищенного соединения в форме его свободных основания или кислоты с соответствующими органическими или неорганическими кислотой или основанием и выделения образованной таким образом соли.
[0082] Термин "фиброз", применяемый в контексте настоящего изобретения, относится к образованию избыточной волокнистой соединительной ткани в органе или ткани и включает фиброз миокарда, фиброз почек и/или фиброз печени.
[0083] Все органы характеризуются специфическим, но различным, расположением тканей (структурой) для нормального функционирования. Заболевание и/или отложения фиброзной ткани могут приводить к дисфункции или недостаточному функционированию органа. Таким образом, восстановление нормальной структуры ткани позволяет органам вернуть нормальную функцию.
[0084] В дополнение к лечению развившегося фиброза соединения по настоящему изобретению можно применять профилактически для субъектов с риском развития фиброза. В качестве примера субъектов из категории риска развития фиброза являются те, у которых имеются гипертензия, диабет, миокардит, ишемическая болезнь сердца, синдром Конна, феохромоцитома, злокачественные опухоли (как например, миелома и лимфома), наследственная предрасположенность (синдром Альпорта, болезнь Вильсона, дефицит ингибитора трипсина α1, гемохроматоз), инфекции (гепатит В, гепатит С), которые находятся на рационе с высоким содержанием соли и/или принимают лекарственные средства, используемые в химиотерапии рака (как например, даунорубицин, цисплатин, блеомицин), для лечения гипомании (литий), отторжения трансплантата (циклоспорин, такролимус), связанных с артритом состояний (NSAID, пеницилламин, золото), и те, которые подвержены воздействию тяжелых металлов, таких как свинец и кадмий. Термин "профилактический", применяемый в контексте настоящего изобретения, предназначен inter alia для охвата способов лечения, используемых для предотвращения или замедления развития фиброза у находящихся в группе риска.
[0085] Настоящее изобретение также предусматривает фармацевтические композиции, которые включают соединения по настоящему изобретению в сочетании с приемлемыми фармацевтическими вспомогательными веществами. Термин "фармацевтически приемлемое вспомогательное вещество", применяемый в контексте настоящего изобретения, означает любой фармацевтически приемлемый неактивный компонент композиции. Как хорошо известно из уровня техники, вспомогательные вещества включают разбавители, буферы, связующие вещества, смазывающие вещества, разрыхлители, красящие вещества, антиоксиданты/консерванты, регуляторы pH и т.д. Вспомогательные вещества выбирают, исходя из необходимых физических аспектов конечной формы: например, получение таблетки с необходимой твердостью и ломкостью, являющейся быстро диспергируемой и легко проглатываемой и т.д. Требуемая скорость высвобождения активного вещества из композиции после ее приема также играет определенную роль в выборе вспомогательных веществ. Фармацевтические композиции могут включать в себя любой тип лекарственной формы, такой как таблетки, капсулы, порошки, жидкие составы, замедленного или длительного высвобождения, пластыри, средства для вдыхания через нос, назальные спреи и тому подобное. Физическая форма и содержимое предусмотренных фармацевтических композиций представляют собой традиционные препараты, которые могут быть составлены специалистом в области фармацевтических составов и основаны на хорошо установленных принципах и композициях, описанных, например, в Remington: The Science and Practice of Pharmacy, 19th Edition, 1995; British Pharmacopoeia 2000, а также аналогичных текстах и руководствах по составам.
[0086] Например, если соединения или композиции подлежат введению перорально, их можно составить в виде таблеток, капсул, гранул, порошков или сиропов; а если для парентерального введения, их можно составить в виде препаратов для инъекций (внутривенных, внутримышечных или подкожных), препаратов для капельного вливания или суппозиториев. Для применения через слизистую оболочку глаза их можно составить в виде глазных капель или глазных мазей. Эти составы можно получить с помощью обычных средств, и, если необходимо, активный ингредиент можно смешивать с любой традиционной добавкой, такой как вспомогательное вещество, связующее вещество, разрыхляющее средство, смазывающее вещество, модификатор лекарственных средств, солюбилизирующее средство, суспендирующая добавка, эмульгирующее средство или покрывающее средство.
[0087] Когда соединение(-ия) по настоящему изобретению вводят в виде фармацевтических препаратов людям и животным, их можно принимать per se или в виде фармацевтической композиции, содержащей, например, от 0,1 до 99,5% (более предпочтительно от 0,5 до 90%) активного ингредиента в комбинации с фармацевтически приемлемым носителем.
[0088] Дозу соединения и частоту введения, которые следует применять, также может легко определить практикующий врач для получения требуемой реакции.
[0089] Несмотря на то, что доза будет варьировать в зависимости от симптомов, возраста и массы тела пациента, природы и тяжести нарушения, подлежащего лечению или предотвращению, пути введения и формы лекарственного средства, в целом суточная доза от 0,0001 мг до 200 мг соединения по настоящему изобретению может быть подходящим эффективным количеством для взрослого пациента-человека, и ее можно вводить в виде одной дозы или в виде раздельных доз.
[0090] "Пациент" или "субъект", подлежащий лечению заявленным способом, может означать либо человека, либо субъекта, не относящегося к человеку.
[0091] "Эффективное количество" заявленного соединения, применительно к способу лечения, относится к количеству терапевтического средства в препарате, которое в случае применения в рамках требуемого режима дозирования обеспечивает пользу согласно клинически приемлемым стандартам лечения или профилактики определенного нарушения.
[0092] Настоящее изобретение далее будет описано более подробно со ссылкой на конкретные, но не ограничивающие примеры, в которых описаны конкретные композиции и способы применения. Однако следует понимать, что подробное описание конкретных процедур, композиций и способов включено исключительно для целей иллюстрации настоящего изобретения. В любом случае его не следует понимать в качестве ограничения обширного описания концепции изобретения, как изложено выше.
ПРИМЕРЫ
Пример 1. Синтез А32
[0093] Синтетический путь, применяемый для получения А32, показан на фигуре 1. Вкратце, 2-формиларилтрифлат 14 получали посредством реакции перекрестного сочетания Судзуки между 5-бром-2-гидроксибензальдегидом и фенилбороновой кислотой с получением 2-гидрокси-5-фенилбензальдегида 13, затем осуществляли реакцию которого с N-фенилтрифламидом. В другой реакции Судзуки между 2-формиларилтрифлатом 14 и 3-бензилоксифенилбороновой кислотой получали терфенилальдегид 15, который подвергали реакции Хорнера-Уодсворта-Эммонса (HWE) с диэтил-5-гидантоилфосфонатом с образованием ненасыщенного гидантоина 16. В присутствии водорода и Pd/C соединение 16 подвергали одновременному восстановлению олефина и снятию защитной группы с фенола с получением А32.
Получение 2-гидрокси-5-фенилбензальдегида (13)
[0094] 5-Бромсалицилальдегид (2,49 г, 12,4 ммоль), фенилбороновую кислоту (1,51 г, 12,4 ммоль), ацетат палладия (II) (14 мг, 0,5 мол. %) и карбонат калия (5,14 г, 37,2 ммоль) перемешивали в дегазированной воде (75 мл) при температуре окружающей среды в течение 2 ч. в атмосфере аргона. Реакцию отслеживали при помощи TLC (1:1 дихлорметан/пентан). Добавляли воду (75 мл) и подкисляли реакционную смесь (pH 6) с помощью 10% HCl, затем экстрагировали этилацетатом

Получение 3-формилбифенил-4-ил-трифторметансульфоната (14)
[0095] 2-Гидрокси-5-фенилбензальдегид (100 мг, 0,50 ммоль), N-фенилтрифлимид (180,0 мг, 0,51 ммоль) и карбонат калия (209 мг, 1,51 ммоль) перемешивали в сухом THF в герметично закрытой пробирке и нагревали при 120°C в течение 6 мин. с помощью микроволнового излучения. Удаляли растворитель при пониженном давлении; добавляли воду и дихлорметан и разделяли слои. Водный слой дополнительно экстрагировали дихлорметаном


Получение 2'-[3-бензилокси-(1,1':4',1''-терфенил)]карбальдегида (15)
[0096] 3-Формилбифенил-4-ил-трифторметансульфонат (153 мг, 0,463 ммоль), 3-бензилоксифенилбороновую кислоту (116 мг, 0,51 ммоль), тетракис(трифенилфосфин)палладий (0) (13 мг, 2,5 мол. %) и безводный фосфат калия (147 мг, 0,695 ммоль) помещали в колбу Шленка в атмосфере аргона. Добавляли дегазированный 1,4-диоксан (2 мл) и смесь продували аргоном. Реакционную смесь нагревали при 85°C до тех пор, пока не наблюдали полное превращение (отслеживаемое с помощью GCMS), обычно требующее времени реакции в течение ночи. Реакционную смесь разбавляли бензолом (4 мл) и обрабатывали 30% водным раствором перекиси водорода (10 мл). Продукт экстрагировали диэтиловым эфиром

Получение (Е/Z)-5-((3-(бензилокси)-[1,1':4',1''-терфенил]-2'-ил)метилен)имидазолидин-2,4-диона (16)
[0097] 2,-[3-Бензилокси-(1,1':4',1''-терфенил)]карбальдегид (15) (978 мг, 2,7 ммоль), диэтил-5-гидантоилфосфонат (949 мг, 4,0 ммоль), порошкообразный гидроксид калия (301 мг, 5,4 ммоль), этанол (20 мл) и воду (0,5 мл) объединяли в 20 мл реакционной пробирке и нагревали при 150°C в течение 1 ч. с помощью микроволнового излучения (300 ватт). Смесь выливали в воду и твердое вещество собирали посредством фильтрации с использованием жесткой беззольной фильтровальной бумаги Whatman 542, тщательно промывая водой. Твердое вещество переносили в горячий этанол и повторно медленно выливали в воду при перемешивании с получением мелкодисперсного осадка. Твердое вещество собирали посредством фильтрации (жесткая беззольная фильтровальная бумага Whatman 542), промывали тщательно водой, затем сушили in vacuo при 40°C с получением (Е/Z)-5-((3-(бензилокси)-[1,1':4',1''-терфенил]-2'-ил)метилен)имидазолидин-2,4-диона (16) (1,04 г, 87%) в виде бледно-желтого твердого вещества. Дополнительная очистка не требовалась.1Н ЯМР (200 МГц, DMSO-d6) δ 10,99 (br s, 2Н); 7,90-7,21 (m, 14Н), 7,17-6,89 (m, 3Н), 6,21 (s, 1Н), 5,14 (s, 2Н).13С ЯМР (50 МГц, DMSO-d6) δ 165,5, 158,3, 155,9, 141,0, 140,5, 139,8, 139,6, 137,0, 131,2, 130,5, 129,5, 129,2, 128,8, 128,4, 127,8, 127,6 (два совпадающих сигнала), 127,1, 126,9, 122,1, 115,9, 114,2, 106,8, 69,4 (один сигнал не наблюдался). EIMS: масса/заряд Найденное значение:
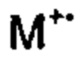
Получение 5-((3-гидрокси-[1,1':4',1''-терфенил]-2'-ил)метил)имидазолидин-2,4-диона (А32)
[0098] (Е/Z)-5-((3-(Бензилокси)-[1,1':4',1''-терфенил]-2'-ил)метилен)имидазолидин-2,4-дион (16) (1,02 г, 2,3 ммоль) и 10% палладий на угле (50 вес.% в H2O, 200 мг) в метаноле (50 мл) перемешивали при комнатной температуре в атмосфере водорода при 50 фунтах на кв. дюйм в течение 1 ч. Метанол удалял, остаток переносили в DCM и подвергали гравитационной фильтрации через бумагу GF. Очищали с помощью радиальной хроматографии (3:97 метанол:DCM → 5:95 метанол:DCM) и препаративной HPLC (соединение предварительно абсорбировали на диоксиде кремния Chromatorex С18, 45% ACN/H2O, 80 мл/мин., 240 нм, 300×40 мм колонка Deltaprep С18) с получением 5-((3-гидрокси-[1,1':4',1''-терфенил]-2'-ил)метил)имидазолидин-2,4-диона (А32) (285 мг, 35%) в виде мелкодисперсного белого порошка; т.пл. 214-216°C.1Н ЯМР (200 МГц, DMSO-d6) δ 10,59 (br s, 1Н), 9,54 (br s, 1Н), 7,90 (m, 1Н), 7,80-7,31 (m, 7Н), 7,30-7,15 (m, 2Н), 6,84-6,67 (m, 3Н), 4,22 (m, 1Н), 3,12 (dd, 1Н, J 4,5, 14,6 Гц), 2,81 (dd, 1Н, J 9,0, 14,6 Гц).13С ЯМР (50 МГц, DMSO-d6) δ 175,4, 157,3, 157,1, 141,8, 141,3, 139,9, 139,1, 134,4. 130,3, 129,2, 128,8, 128,0, 127,4, 126,8, 124,8, 119,8, 116,0, 114,1, 57,9, 34,8. EIMS: масса/заряд Найденное значение:
Пример 2. Синтез А6
[0099] Синтетический путь, применяемый для получения А6, показан на фигуре 2.
Получение диэтил[2-амино-3,3,3-трифторпроп-1-ен-1-ил]фосфоната
[0100] Раствор диэтилметилфосфоната (1,000 г, 6,57 ммоль) в безводн. тетрагидрофуране (33 мл) получали в атмосфере азота и охлаждали в охлаждающей ванне при -80°C. Добавляли по каплям 1,21 М раствор метиллития в диэтиловом эфире (5,5 мл, 6,6 ммоль). Смесь перемешивали при -80°C в атмосфере азота в течение 1 ч.
[00100] Добавляли по каплям трифторуксусную кислоту (0,71 мл, 9,6 ммоль) к безводн. пиридину (11,7 мл, 145 ммоль) в атмосфере азота. Мутные пары становились прозрачными в потоке азота. Круглодонную колбу на 50 мл наполняли трифторацетамидом (3,177 г, 33,4 ммоль) и растворяли в смеси пиридина/трифторуксусной кислоты в атмосфере азота. В свободное пространство над данным раствором помещали канюлю, другой конец канюли помещали в фосфонатный раствор. Триметилацетилхлорид (7,3 мл, 59,3 ммоль) добавляли по каплям к раствору трифторацетамида в течение периода 80 мин. Во время добавления фосфонатный раствор перемешивали при -80°C, затем еще в течение 4 ч., после чего давали возможность нагреться до комнатной температуры в течение ночи.
[0101] Реакционную смесь разделяли между дихлорметаном (20 мл) и водой (60 мл). Фазы разделяли. Водный слой экстрагировали дихлорметаном (10 мл). Объединенные слои дихлорметана промывали солевым раствором (20 мл), сушили над безводн. сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (этилацетат/гексаны) с получением указанного в заголовке соединения в виде бледно-желтого порошка (638 мг, 39%).1Н ЯМР (400 МГц, CDCl3) 5,71 (br. s, 2Н), 4,46 (d, J=8,6 Гц, 1H), 3,98-4,15 (m, 4Н), 1,34 (t, J=7,0 Гц, 6H). [Ссылка: F. Palacios et al., J. Org. Chem. 2004, 69, 8767-8774].
Получение 1,1,1-трифтор-4-[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]бут-3-ен-2-амина
[0102] Растворы диэтил[2-амино-3,3,3-трифторпроп-1-ен-1-ил]фосфоната (638 мг, 2,58 ммоль) в безводн. тетрагидрофуране (7,7 мл) и 3-(бензилокси)-1,1':4',1''-терфенил-2'-карбальдегида (944 мг, 2,59 ммоль) в безводн. тетрагидрофуране (7,7 мл) получали в атмосфере азота. Фосфонатный раствор охлаждали до -5°C. Добавляли по каплям 1,47 М раствор бутиллития в гексанах (1,8 мл, 2,65 ммоль). Смесь перемешивали при -5°C в атмосфере азота в течение 1 ч. При помощи шприца добавляли по каплям раствор альдегида. Смесь перемешивали в атмосфере азота при -5°C в течение 15 мин., затем при комнатной температуре в течение 70 мин.
[0103] Реакционную смесь охлаждали до -78°C. Добавляли боргидрид натрия (196 мг, 5,18 ммоль) с последующим добавлением по каплям метанола (15 мл). Смесь перемешивали при -78°C в течение 80 мин., затем давали возможность нагреться до комнатной температуры в течение ночи.
[0104] Осторожно добавляли хлористоводородную кислоту (1 М, 5 мл). Смесь перемешивали при комнатной температуре в течение 45 мин., регулировали pH до 11 (универсальный индикатор) путем добавления гидроксида натрия (253 мг, 6,33 ммоль) и экстрагировали этилацетатом (30 мл). Водный слой разделяли между этилацетатом (10 мл) и водой (10 мл) и разделяли фазы. Объединенные слои этилацетата промывали солевым раствором (2×20 мл), сушили над безводн. сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (дихлорметан) с получением смеси указанного в заголовке соединения (72 мол. %) и бензилового спирта в результате восстановления исходного альдегида (28 мол. %) (467 мг, 39%).1Н ЯМР (400 МГц, CDCl3); выбранные резонансы); указанное в заголовке соединение С(3)Н: 6,14 (dd, J=15,8, 6,8 Гц, 1Н), бензиловый спирт ArCH2OH: 4,65 (d, J=5,7 Гц, 2Н).
Получение 2'-(3-амино-4,4,4-трифторбутил)-1,1':4',1''-терфенил-3-ола (А6)
[0105] Раствор неочищенного 1,1,1-трифтор-4-[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]бут-3-ен-2-амина (576 мг, 1,25 ммоль) в уксусной кислоте (20 мл) добавляли к 10% палладию на угле (131 мг, 0,12 ммоль относительно Pd). Смесь гидрировали при 2,1 бар в течение 18 ч. Смесь фильтровали через целит. Осадок на фильтре промывали уксусной кислотой (2×20 мл). Объединенные фильтраты выпаривали до сухого состояния. Остаток разделяли между этилацетатом (20 мл) и насыщ. раствором гидрокарбоната натрия (20 мл). Слой этилацетата промывали насыщ. раствором гидрокарбоната натрия (20 мл) и солевым раствором (20 мл), сушили над безводн. сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (этилацетат/гексаны) с получением указанного в заголовке соединения в виде бледного оранжево-коричневого масла, которое затвердевало при отстаивании (323 мг, 69%). Продукт суспендировали в смеси 7,5% дихлорметана/гексанов и выделяли посредством фильтрации с получением грязно-белого порошка.1Н ЯМР (400 МГц, CDCl3) 7,58-7,67 (m, 2Н), 7,42-7,55 (m, 4Н), 7,33-7,40 (m, 1Н), 7,27-7,33 (m, 2Н), 6,88-6,95 (m, 1Н), 6,79-6,87 (m, 2Н), 5,26 (br. s, 1Н), 2,93-3,08 (m, 2Н), 2,69-2,82 (m, 1 H), 1,85-1,98 (m, 1 H), 1,48-1,61 (m, 1 H), 1,17 (br. s, 2H); HPLC (градиент вода/ACN+0,1% TFA) 99,40% при 220 нм; LCMS [M+H]+=372,2.
Пример 3. Синтез A30
[0106] Синтетический путь, применяемый для получения А30, показан на фигуре 3.
Получение 3-(трифенил-I5-фосфанилидин)пирролидин-2,5-диона
[0107] Суспензию малеимида (3,17 г, 32,7 ммоль) и трифенилфосфина (8,56 г, 32,6 ммоль) в ацетоне (165 мл) нагревали при температуре возврата флегмы в атмосфере азота в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и фильтровали. Осадок на фильтре промывали ацетоном (3×20 мл) и сушили под вакуумом с получением указанного в заголовке соединение в виде белого порошка (7,21 г, 61%).1Н ЯМР (400 МГц, DMSO-d6) 9,73 (br. s, 1Н), 7,66-7,75 (m, 3Н), 7,53-7,65 (m, 12Н), 2,89 (s, 2H).
[0108] Фильтраты вышеуказанного объединяли и концентрировали с удалением прибл. 120 мл растворителя. Оставшийся материал нагревали при температуре возврата флегмы в атмосфере азота в течение 2 ч., давали возможность охладиться до комнатной температуры и фильтровали. Осадок на фильтре промывали ацетоном (3×10 мл) и сушили под вакуумом с получением дополнительной порции указанного в заголовке соединения в виде белого порошка (2,63 г, 22%). [Ссылка: G. Brackman et al., Bioorg. Med. Chem. 2013, 21, 660-667].
Получение 3-{[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]метилиден}пирролидин-2,5-диона
[0109] Смесь 3-{[3-(бензилокси)-1,1':4',1''-терфенил-2'-карбальдегида (1,71 г, 4,69 ммоль) и 3-(трифенил-I5-фосфанилидин)пирролидин-2,5-диона (1,69 г, 4,69 ммоль) в метаноле (15 мл) нагревали при температуре возврата флегмы в атмосфере азота в течение 1,5 ч. Реакционную смесь фильтровали теплой. Осадок на фильтре промывали метанолом (2×25 мл) и сушили на воздухе с получением указанного в заголовке соединения в виде желтого порошка (1,05 г, 50%).1Н ЯМР (400 МГц, CDCl3) 8,18 (s, 1Н), 7,67-7,69 (m, 3Н), 7,61 (d, J=8,0 Гц, 2Н), 7,33-7,51 (m, 10Н), 7,02 (dd, J=8,0, 2,0 Гц, 1Н), 6,95 (s, 1Н), 6,92 (d, J=7,6 Гц, 1Н), 5,10 (s, 2Н), 3,56 (s, 2Н); LCMS [М+Н]+=446,3, [M+Na]+=468,2, [М-Н]-=444,2.
Получение 3-[(3-гидрокси-1,1':4',1''-терфенил-2'-ил)метил]пирролидин-2,5-диона (А30)
[00101] Смесь 3-{[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]метилиден}пирролидин-2,5-диона (2,25 г, 5,05 ммоль), этилацетата (200 мл) и триэтиламина (40 капель) дегазировали путем барботирования азота (2 л) через смесь в течение периода, составляющего 5-10 мин. Добавляли 10% палладий на угле (0,23 г) в атмосфере азота. Смесь гидрировали при атмосферном давлении при температуре возврата флегмы в течение ночи. Горячую реакционную смесь фильтровали через целит и осадок на фильтре промывали этилацетатом (3×50 мл). Объединенные фильтраты выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (этилацетат/гексаны). Продукт концентрировали из этанола (100 мл) и сушили под высоким вакуумом в течение 3 дней с получением указанного в заголовке соединения в виде бесцветного стеклообразного вещества (1,61 г, 89%).1Н ЯМР (400 МГц, CDCl3) 7,89 (br. s, 1Н), 7,61 (d, J=6,8 Гц, 2Н), 7,53 (dd, J=8,0, 2,0 Гц, 1Н), 7,44-7,48 (m, 3Н), 7,37 (t, J=7,2 Гц, 1Н), 7,31 (t, J=7,2 Гц, 2Н), 6,81-6,90 (m, 3Н), 5,40 (br. s, 1Н), 3,51 (dd, J=14,0, 4,8 Гц, 1Н), 3,01 (m, 1Н), 2,87 (dd, J=14,0, 10,4 Гц, 1Н), 2,56 (dd, J=18,4, 9,2 Гц, 1Н), 2,25 (dd, J=18,4, 5,6 Гц, 1Н); HPLC 99,01% при 220 нм; LCMS [М+Н]+=358,2, [М+Na]+=380,1, [М-Н]-=356,2.
Пример 4. Синтез A56f, A56g и А56
[0110] Синтетический путь, используемый для получения A56f, A56g и А56, показан на фигуре 4.
Получение 2-[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]-1-(метилсульфанил)этенилметилсульфоксида
[0111] 3-(Бензилокси)-1,1':4',1''-терфенил-2'-карбальдегид (5,330 г, 14,6 ммоль) растворяли в тетрагидрофуране (65 мл). Добавляли метил(метилсульфинил)метилсульфид (2,745 г, 22,1 ммоль) и гидроксид натрия (654 мг, 16,4 ммоль). Смесь нагревали при температуре возврата флегмы в атмосфере азота в течение ночи. Реакционную смесь разделяли между этилацетатом (400 мл) и водой (200 мл). Водный слой экстрагировали этилацетатом (2×200 мл). Объединенные слои этилацетата промывали водой (2×200 мл) и солевым раствором (200 мл), сушили над безводн. сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (этилацетат/дихлорметан) с получением указанного в заголовке соединения в виде бледно-оранжевого масла (3,733 г, 54%).1Н ЯМР (400 МГц, CDCl3) 8,14 (d, J=1,4 Гц, 1Н), 7,62-7,72 (m, 4Н), 7,42-7,53 (m, 5H), 7,39 (t, J=7,3 Гц, 3Н), 7,29-7,36 (m, 2H), 6,90-7,01 (m, 3Н), 5,10 (s, 2H), 2,70 (s, 3H), 2,28 (s, 3H).
Получение этил[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]ацетата
[0112] 2-[3-(Бензилокси)-1,1':4',1''-терфенил-2'-ил]-1-(метилсульфанил)этенилметилсульфоксид (3,733 г, 7,93 ммоль) растворяли в этаноле (70 мл). Добавляли конц. хлористоводородную кислоту (6,6 мл) и смесь нагревали при температуре возврата флегмы в течение 5 дней. Реакционную смесь разделяли между этилацетатом (500 мл) и водой (250 мл). Слой этилацетата промывали водой (200 мл) и солевым раствором (200 мл), сушили над безводн. сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (дихлорметан) с получением указанного в заголовке соединения в виде желто-оранжевого масла (2,129 г, 64%).1Н ЯМР (400 МГц, CDCl3) 7,60-7,68 (m, 2Н), 7,59 (br. s, 1Н), 7,55 (dd, J=8,0, 1,6 Гц, 1Н), 7,42-7,50 (m, 4Н), 7,29-7,42 (m, 6Н), 6,92-7,04 (m, 3Н), 5,09 (s, 2Н), 4,10 (q, J=7,0 Гц, 2Н), 3,65 (s, 2Н), 1,21 (t, J=7,1 Гц, 3Н); LCMS [М+Н]+=423,1.
Получение 2-[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]этанола
[0113] Этил[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]ацетат (2,129 г, 5,04 ммоль) растворяли в безводн. тетрагидрофуране (20 мл) в атмосфере азота. Получали суспензию алюмогидрида лития (306 мг, 8,06 ммоль) в безводн. тетрагидрофуране (10 мл) в атмосфере азота и охлаждали в водяной ванне со льдом. В суспензию алюмогидрида лития добавляли по каплям раствор сложного эфира. Смесь перемешивали при комнатной температуре в атмосфере азота в течение ночи. Реакционную смесь охлаждали в водяной ванне со льдом. Избыточный алюмогидрид лития гасили путем добавления по каплям воды (0,37 мл), 15% раствора гидроксида натрия (0,37 мл) и воды (1,5 мл). Смесь перемешивали в течение 30 мин. Добавляли этилацетат (60 мл) и смесь фильтровали через целит. Осадок на фильтре промывали этилацетатом (2×30 мл). Объединенные фильтраты выпаривали до сухого состояния с получением указанного в заголовке соединения в виде бледно-оранжевого масла (2,046 г, 107%).1Н ЯМР (400 МГц, CDCl3) 7,59-7,67 (m, 2Н), 7,55 (d, J=1,6 Гц, 1Н), 7,50 (dd, J=7,9, 1,9 Гц, 1Н), 7,43-7,48 (m, 4Н), 7,28-7,42 (m, 6Н), 6,92-7,03 (m, 3Н), 5,11 (s, 2Н), 3,66-3,74 (m, 2Н), 2,92 (t, J=6,8 Гц, 2Н), 1,21 (t, J=5,9 Гц, 1Н); LCMS [М+Н-H2O]+=363,3, [2М+Н]+=761,6.
Получение 2-{2-[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]этокси}-1Н-изоиндол-1,3(2Н)-диона
[0114] Смесь 2-[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]этанола (2,046 г, 5,38 ммоль), трифенилфосфина (1,707 г, 6,51 ммоль) и N-гидроксифталимида (1,053 г, 6,45 ммоль) суспендировали в безводном тетрагидрофуране (30 мл) в атмосфере азота. Смесь охлаждали в водяной ванне со льдом. Добавляли по каплям диэтилазодикарбоксилат (1,130 г, 6,49 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (дихлорметан/гексаны) с получением указанного в заголовке соединения в виде бледно-желтого воскообразного твердого вещества (2,509 г, 89%).1Н ЯМР (400 МГц, CDCl3) 7,75-7,82 (m, 2Н), 7,69-7,74 (m, 2Н), 7,61-7,68 (m, 3Н), 7,43-7,53 (m, 5Н), 7,32-7,43 (m, 4Н), 7,28 (d, J=8,0 Гц, 1Н), 7,21 (t, J=7,9 Гц, 1Н), 6,86-6,96 (m, 2Н), 6,79 (dd, J=8,3, 1,9 Гц, 1Н), 5,06 (s, 2Н), 4,26 (t, J=7,6 Гц, 2Н), 3,19 (t, J=7,5 Гц, 2Н).
Получение 0-{2-[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]этил}гидроксиламина
[0115] 2-{2-[3-(Бензилокси)-1,1':4',1''-терфенил-2'-ил]этокси}-1H-изоиндол-1,3(2Н)-дион (1,769 г, 3,37 ммоль) суспендировали в абс. этаноле (65 мл). Добавляли гидразин-гидрат (230 мкл, 3,69 ммоль) и смесь нагревали при 65°C в атмосфере азота в течение 8 ч., затем давали возможность отстаиваться при комнатной температуре в течение ночи. Реакционную смесь фильтровали. Осадок на фильтре промывали этанолом (2×30 мл). Объединенные фильтраты выпаривали до сухого состояния. Остаток суспендировали в дихлорметане (60 мл) и смесь фильтровали. Осадок на фильтре промывали дихлорметаном (2×30 мл). Объединенные фильтраты выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (этилацетат/дихлорметан) с получением указанного в заголовке соединения в виде прозрачного масла (1,317 г, 99%).1Н ЯМР (400 МГц, CDCl3) 7,59-7,68 (m, 2Н), 7,55 (d, J=1,8 Гц, 1Н), 7,42-7,52 (m, 5Н), 7,27-7,42 (m, 6Н), 6,93-7,03 (m, 3Н), 5,22 (br. s, 2Н), 5,11 (s, 2Н), 3,77 (t, J=6,9 Гц, 2Н), 2,96 (t, J=6,9 Гц, 2Н).
Получение 2'-(2-гидроксиэтил)-1,1':4',1''-терфенил-3-ола (A56f)
[0116] Раствор O-{2-[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]этил}гидроксиламина (1,317 г, 3,33 ммоль) в этилацетате (40 мл) добавляли к 10% палладию на угле (346 мг, 0,33 ммоль относительно Pd). Смесь гидрировали при 2,1 бар в течение 65 ч. Смесь фильтровали через целит. Осадок на фильтре промывали этилацетатом (2×40 мл). Объединенные фильтраты выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (метанол/дихлорметан) с получением указанного в заголовке соединения в виде белого порошка (864 мг, 89%).1Н ЯМР (400 МГц, CDCl3) 7,59-7,68 (m, 2Н), 7,53-7,58 (m, 1Н), 7,41-7,53 (m, 3Н), 7,33-7,40 (m, 1Н), 7,26-7,33 (m, 2Н), 6,88-6,96 (m, 1Н), 6,79-6,87 (m, 2Н), 5,14 (br. s, 1Н), 3,71-3,84 (m, 2Н), 2,97 (t, J=6,8 Гц, 2H), 1,35-1,48 (m, 1Н); HPLC (градиент вода/ACN + 0,1% TFA) 99,19% при 220 нм; LCMS [М+Н-H2O]+=273,2, [M+Na]+=313,2.
Получение 2-[2-(3-гидрокси-1,1':4',1''-терфенил-2'-ил)этокси]-1Н-изоиндол-1,3(2Н)-диона (A56g)
[0117] Смесь 2'-(2-гидроксиэтил)-1,1':4',1''-терфенил-3-ола (864 мг, 2,98 ммоль), трифенилфосфина (946 мг, 3,61 ммоль) и N-гидроксифталимида (587 мг, 3,60 ммоль) суспендировали в безводном тетрагидрофуране (30 мл) в атмосфере азота. Смесь охлаждали в водяной ванне со льдом. Добавляли по каплям диэтилазодикарбоксилат (626 мг, 3,59 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (этилацетат/дихлорметан) с получением указанного в заголовке соединения в виде белого порошка (1,265 г, 98%).1Н ЯМР (400 МГц, CDCl3) 7,79-7,88 (m, 2Н), 7,71-7,79 (m, 2Н), 7,56-7,68 (m, 3Н), 7,41-7,54 (m, 3Н), 7,31-7,40 (m, 2Н), 7,23 (t, J=7,9 Гц, 1Н), 6,93-7,00 (m, 1Н), 6,88 (d, J=7,6 Гц, 1Н), 6,75 (dd, J=8,1, 2,1 Гц, 1Н), 5,72 (s, 1Н), 4,40 (t, J=7J Гц, 2Н), 3,21 (t, J=7,7 Гц, 2H); HPLC (градиент вода/ACN + 0,1% TFA) 98,52% при 220 нм; LCMS [M+Na]+=458,1.
Получение 2'-[2-(аминоокси)этил]-1,1':4',1''-терфенил-3-ола (А56)
[0118] 2-[2-(3-Гидрокси-1,1':4',1''-терфенил-2'-ил)этокси]-1H-изоиндол-1,3(2Н)-дион (999 мг, 2,29 ммоль) суспендировали в абс. этаноле (45 мл). Добавляли гидразин-гидрат (150 мкл, 2,40 ммоль). Смесь нагревали при 65°C в течение 4 ч. в атмосфере азота, затем давали возможность отстаиваться при комнатной температуре в течение ночи. Реакционную смесь фильтровали. Осадок на фильтре промывали этанолом (2×20 мл). Объединенные фильтраты выпаривали до сухого состояния. Остаток суспендировали в дихлорметане (40 мл) и смесь фильтровали. Осадок на фильтре промывали дихлорметаном (2×20 мл). Объединенные фильтраты выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (метанол/дихлорметан) с получением указанного в заголовке соединения в виде белого порошка (648 мг, 92%).1Н ЯМР (400 МГц, CDCl3) 7,60-7,66 (m, 2Н), 7,54 (d, J=1,6 Гц, 1Н), 7,41-7,51 (m, 3Н), 7,33-7,39 (m, 1Н), 7,26-7,32 (m, 2Н), 6,89-6,96 (m, 1Н), 6,79-6,87 (m, 2Н), 5,29 (br. s, 3Н), 3,82 (t, J=6,9 Гц, 2Н), 2,98 (t, J=7,0 Гц, 2Н); HPLC (градиент вода/ACN + 0,1% ТРА) 95,36% при 220 нм; LCMS [М+Н]+=306,2, [М+Na]+=328,1.
Пример 5. Синтез A56k
[0119] Синтетический путь, применяемый для получения A56k, показан на фигуре 5.
Получение 2-[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]-1-(метилсульфанил)этенилметилсульфоксида
[0120] 3-(Бензилоксин)-1,1':4'1''-терфенил-2'-карбальдегид (5,330 г, 14,6 ммоль) растворяли в тетрагидрофуране (65 мл). Добавляли метил(метилсульфинил)метилсульфид (2,745 г, 22,1 ммоль) и гидроксид натрия (654 мг, 16,4 ммоль). Смесь нагревали при температуре возврата флегмы в атмосфере азота в течение ночи. Реакционную смесь разделяли между этилацетатом (400 мл) и водой (200 мл). Водный слой экстрагировали этилацетатом (2×200 мл). Объединенные слои этилацетата промывали водой (2×200 мл) и солевым раствором (200 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (этилацетат/дихлорметан) с получением указанного в заголовке соединения в виде бледно-оранжевого масла (3,733 г, 54%).1Н ЯМР (400 МГц, CDCl3) 8,14 (d, J=1,4 Гц, 1Н), 7,62-7,72 (m, 4Н), 7,42-7,53 (m, 5Н), 7,39 (t, J=7,3 Гц, 3Н), 7,29-7,36 (m, 2Н), 6,90-7,01 (m, 3Н), 5,10 (s, 2Н), 2,70 (s, 3Н), 2,28 (s, 3Н).
Получение этил[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]ацетата
[0121] 2-[3-(Бензилокси)-1,1':4',1''-терфенил-2'-ил]-1-(метилсульфанил)этенилметилсульфоксид (3,733 г, 7,93 ммоль) растворяли в этаноле (70 мл). Добавляли конц. хлористоводородную кислоту (6,6 мл) и смесь нагревали при температуре возврата флегмы в течение 5 дней. Реакционную смесь разделяли между этилацетатом (500 мл) и водой (250 мл). Слой этилацетата промывали водой (200 мл) и солевым раствором (200 мл), сушили над безводн. сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (дихлорметан) с получением указанного в заголовке соединения в виде желто-оранжевого масла (2,129 г, 64%).1Н ЯМР (400 МГц, CDCl3) 7,60-7,68 (m, 2Н), 7,59 (br. s, 1Н), 7,55 (dd, J=8,0, 1,6 Гц, 1Н), 7,42-7,50 (m, 4Н), 7,29-7,42 (m, 6Н), 6,92-7,04 (m, 3Н), 5,09 (s, 2Н), 4,10 (q, J=7,0 Гц, 2H), 3,65 (s, 2Н), 1,21 (t, J=7,1 Гц, 3Н); LCMS [M+H]+=423,1.
Получение [3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]уксусной кислоты
[0122] Этил[3-(бензилокси)-1,1':4',1''-терфенил-2'-ил]ацетат (414 мг, 0,98 ммоль) растворяли в этаноле (15 мл). Добавляли раствор гидроксида натрия (1 М, 3 мл, 3 ммоль) и смесь нагревали при 70°C в течение 1 ч. Реакционную смесь разделяли между этилацетатом (45 мл) и хлористоводородной кислотой (1 М, 15 мл). Слой этилацетата промывали водой (15 мл) и солевым раствором (15 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния с получением указанного в заголовке соединения в виде бледно-коричневого порошка (350 мг, 91%).1Н ЯМР (400 МГц, CDCl3) 7,59-7,65 (m, 2Н), 7,53-7,59 (m, 2Н), 7,40-7,48 (m, 4Н), 7,27-7,39 (m, 6Н), 6,96-7,02 (m, 2Н), 6,91-6,96 (m, 1Н), 5,08 (s, 2Н), 3,68 (s, 2Н).
Получение (3-гидрокси-1,1':4',1''-терфенил-2'-ил)уксусной кислоты (A56k)
[0123] [3-(Бензилокси)-1,1':4',1''-терфенил-2'-ил]уксусную кислоту (350 мг, 0,89 ммоль) суспендировали в уксусной кислоте (9 мл) и конц. хлористоводородной кислоте (2,2 мл). Смесь нагревали при 100°C в течение 2,25 ч. Реакционную смесь выливали в воду (60 мл) и смесь экстрагировали этилацетатом (60 мл). Слой этилацетата промывали водой (2×30 мл) и солевым раствором (30 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния. Остаток суспендировали в толуоле (20 мл) и выпаривали до сухого состояния. Данный процесс повторяли. Остаток очищали посредством флэш-хроматографии (этилацетат/дихлорметан) с получением указанного в заголовке соединения в виде воскообразного коричневого твердого вещества (189 мг, 70%). Продукт суспендировали в смеси 7,5% дихлорметана/гексанов и выделяли посредством фильтрации с получением бледно-бежевого порошка.1Н ЯМР (400 МГц, CDCl3) 7,58-7,65 (m, 2Н), 7,51-7,58 (m, 2Н), 7,40-7,48 (m, 2Н), 7,31-7,39 (m, 2Н), 7,26-7,30 (m, 1Н), 6,87-6,92 (m, 1Н), 6,79-6,86 (m, 2Н), 3,69 (s, 2Н); HPLC (градиент вода/ACN + 0,1% TFA) 95,58% при 220 нм; LCMS [М-Н]-=303,1, [2M-H]-=607,3.
Пример 6. Синтез промежуточного соединения А31-4
[0124] Синтетический путь, применяемый для получения А31-4, показан на фигуре 6.
Получение метил-2-бром-5-иодбензоата
[0125] Смесь 2-бром-5-иодбензойной кислоты (20,070 г, 61,4 ммоль) и карбоната калия (12,698 г, 91,9 ммоль) суспендировали в ОМР (45 мл). Добавляли подметан (11,373 г, 80,1 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разделяли между диэтиловым эфиром (400 мл) и водой (250 мл). Слой простого эфира промывали водой (2×120 мл) и солевым раствором (120 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния с получением указанного в заголовке соединения в виде оранжевого масла (20,451 г, 98%).1Н ЯМР (400 МГц, CDCl3) 8,10 (d, J=2,0 Гц, 1Н), 7,62 (d, J=8,4, 2,1 Гц, 1Н), 7,38 (d, J=8,2 Гц, 1Н), 3,93 (s, 3Н). [Ссылка: WO 2004/048314].
Получение метил-4-бромбифенил-3-карбоксилата
[0126] Метил-2-бром-5-иодбензоат (10,019 г, 29,4 ммоль), фенилбороновую кислоту (3,571 г, 29,3 ммоль) и карбонат калия (8,112 г, 58,7 ммоль) растворяли в смеси толуола (200 мл), абс. этанола (50 мл) и воды (25 мл). Реакционную колбу продували азотом и азот барботировали через смесь в течение 30 мин. Добавляли тетракис(трифенилфосфин)палладий (3,401 г, 2,94 ммоль) в потоке азота. Азот барботировали через реакционную смесь в течение 15 мин. Смесь нагревали при температуре возврата флегмы в течение 12 ч., затем давали возможность отстаиваться при комнатной температуре. Реакционную смесь разделяли между толуолом (100 мл) и водой (300 мл). Водный слой экстрагировали толуолом (100 мл). Объединенные слои толуола промывали солевым раствором (150 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния. Остаток очищали посредством флэш-хроматографии (дихлорметан/гексаны) с получением указанного в заголовке соединения в виде оранжевого масла (8,092 г, 84%).1Н ЯМР (400 МГц, CDCl3) 8,01 (d, J=2,3 Гц, 1Н), 7,72 (d, J=8,4 Гц, 1Н), 7,52-7,60 (m, 3Н), 7,43-7,49 (m, 2Н), 7,36-7,42 (m, 1Н), 3,96 (s, 3Н).
Получение (4-бромбифенил-3-ил)метанола
[0127] Получали раствор метил-4-бромбифенил-3-карбоксилата (6,992 г, 24,0 ммоль) в безводном тетрагидрофуране (80 мл) в атмосфере азота. Получали суспензию алюмогидрида лития (692 мг, 18,2 ммоль) в безводн. тетрагидрофуране (60 мл) в атмосфере азота и охлаждали в водяной ванне со льдом. Раствор сложного эфира переносили в суспензию алюмогидрида лития с помощью канюли. Смесь перемешивали в водяной ванне со льдом в течение 40 мин. Избыточный алюмогидрид лития гасили путем добавления по каплям воды (1,75 мл), 15% раствора гидроксида натрия (1,75 мл) и воды (7 мл). Смесь перемешивали при комнатной температуре в течение 40 мин. Добавляли этилацетат (290 мл) и смесь фильтровали через целит. Осадок на фильтре промывали этилацетатом (2×140 мл). Объединенные фильтраты выпаривали до сухого состояния. Остаток объединяли с неочищенными продуктами подобных реакций с метил-4-бромбифенил-3-карбоксилатом (1,024 г, 3,51 ммоль) и смесь очищали с помощью флэш-хроматографии (дихлорметан/гексаны) с получением указанного в заголовке соединения в виде бледно-оранжевого масла, которое затвердевало при отстаивании (6,729 г, 93%).1Н ЯМР (400 МГц, CDCl3) 7,71 (d, J=2,1 Гц, 1Н), 7,53-7,64 (m, 3Н), 7,41-7,48 (m, 2Н), 7,32-7,41 (m, 2Н), 4,82 (d, J=6,4 Гц, 2Н), 2,01 (t, J=6,4 Гц, 1Н).
Получение 4-бромбифенил-3-карбальдегида (А31-4)
[0128] Активированный оксид марганца (I\/) (19,279 г, 222 ммоль) добавляли в раствор (4-бромбифенил-3-ил)метанола (5,844 г, 22,2 ммоль) в толуоле (90 мл). Смесь перемешивали при 60°C в атмосфере азота в течение 16 ч. Реакционную смесь фильтровали через целит. Осадок на фильтре промывали толуолом (2×20 мл). Объединенные фильтраты выпаривали до сухого состояния с получением указанного в заголовке соединения в виде бледно-желтого масла, которое затвердевало при отстаивании (4,782 г, 82%).1H ЯМР (400 МГц, CDCl3) 10,41 (s, 1Н), 8,14 (d, J=2,1 Гц, 1Н), 7,70-7,74 (m, 1Н), 7,65-7,70 (m, 1Н), 7,56-7,63 (m, 2Н), 7,43-7,51 (m, 2Н), 7,36-7,43 (m, 1Н).
Пример 7. Синтез А26 и А27
[0129] Синтетический путь, применяемый для получения А26 и А27, показан на фигуре 7.
Стадия 1 - i) тетрафторборат 1-карбоэтоксициклопропилтрифенилфосфония, DMF, 2 ч., 80°C, ii) А31-4, 18 ч., 80°C (по материалам Chung et al Org. Letts, 2011 Vol. 13, No. 19, 5338-5341).
Стадия 2 - TFA, DCM (по материалам WO2009/89359).
Стадия 3 - 3-гидроксибензолбороновая кислота, K2CO3, H2O, 1,4-диоксан, Pd(PPh3)4, 18 ч., 75°C.
Стадия 4 - Н2 (атмосфера), 5 мол. % Pd/C в МеОН, 18 ч., 50°C (по материалам WO 2005/90300).
Пример 8. Синтез А31
[0130] Синтетический путь, применяемый для получения А31, показан на фигуре 8.
Стадия 1 - N-ацилглицин, Ac2O, AcONa, нагрев, 6 ч.
Стадия 2 - 3 M HCl, нагрев.
Стадия 3 -

Стадия 5 - метоксикарбонилметилтрифенилфосфоран, толуол, получение смеси Е- и Z-изомеров.
Стадия 5 - NaOH, нагрев
(стадии 1-5 по материалам Wong et al, Synthesis, 1992, 793-797 и

Стадия 6 - мочевина, толуол, нагрев (Е-изомер оставался непрореагировавшим - как описано в WO 2008/15139).
Стадия 7 - 3-гидроксибензолбороновая кислота, K2CO3, H2O, 1,4-диоксан, Pd(PPh3)4, 18 ч., 75°С.
Пример 9. Синтез А35
[0131] Синтетический путь, применяемый для получения А35, показан на фигуре 9.
Стадия 1 - NaBH4, МеОН, к.т.
Стадия 2 - PBr3, THF (как описано в Yu et al, Org. Letts, 2009, vol. 11(2), 469-472).
Стадия 3 - (4-формил-5-метилизоксазол-3-ил)-карбаминовой кислоты третбутиловый сложный эфир, нBuLi, THF (как описано в Konoike et al Tet. Letts, Vol. 37, No. 19, 3339-3342, 1996).
Стадия 4 - TFA, DCM, к. т.
Стадия 5 - 3-гидроксибензолбороновая кислота, K2CO3, H2O, 1,4-диоксан, Pd(PPh3)4, 18 ч., 75°C.
Пример 10. Синтез А45
[0132] Синтетический путь, применяемый для получения А45, показан на фигуре 10.
Стадия 1 - HNMeOMe.HCl, CDI, DIPEA, DCM (по материалам WO 2011/119518).
Стадия 2 - бензолбороновая кислота, Pd(PPh3)4, K2CO3, толуол : этанол : вода, нагрев, 12 ч.
Стадия 3 - MeMgBr, THF, 0°C (как описано в ЕР 2455380).
Стадия 4 - Br2, EtOH, к. т.(как описано в WO 2008/157726).
Стадия 5 - 1,3-тиазолидин-2,4-дион, K2CO3, TBAI, DMF, к.т., 2 ч. (по материалам Nagarapu et al, Euro. J. Med. Chem. 71, (2014), 91-97).
Стадия 6 - NaOH в МеОН или NEt3 в EtOH) (по материалам Shvaika et al, J. Org. Chem. USSR, 1983, vol. 19, #8, 1533-1543).
Стадия 7 - 3-гидроксибензолбороновая кислота, K2CO3, H2O, 1,4-диоксан, Pd(PPh3)4, 18 ч., 75°C.
Пример 11. Синтез А79
[0133] Синтетический путь, применяемый для получения А79, показан на фигуре 11.
Стадия 1 - 4-оксазолидинон-2-тион, NaOAc, НОАс.
Стадия 2 -

Стадия 3 - HCl, EtOH, H2O
(стадии 1-3 по материалам Unangst et al, J. Med. Chem. 1994, 37, 322-328).
Стадия 4 - 3-гидроксибензолбороновая кислота, K2CO3, H2O, 1,4-диоксан, Pd(PPh3)4, 18 ч., 75°C.
Стадия 5 - Н2 (атмосфера), 5 мол. % Pd/C в МеОН, 18 ч., 50°C.
Пример 12. Синтез А81
[0134] Синтетический путь, применяемый для получения А81, показан на фигуре 12.
Стадия 1.
i) Из 5,02 г А31-3 получали требуемый продут А31-4 (5,027 г, выход 96%).
ii) Из 20,1 г, мол. А31-3 получали требуемый продут А31-4 (20,451 г, выход 98%).
Стадия 2.
i) 0,984 г А31-4 (1,0 экв. PhB(OH)2, 0,05 экв. Pd(dppf)Cl2, 2 экв. K2CO3, диоксан/этанол/вода, 85°C, 16 ч.). При помощи TLC наблюдали полное расходование А31-4. Неочищенный продукт разделяли на фракции колоночной хроматографией. Чистых образцов не получили, но благодаря1Н ЯМР анализу выявляли по меньшей мере 4 продукта, в том числе бифенил, соответствующие А31-5.
ii) Из 10 г А31-4 получали требуемый продут А31-5 (8,1 г, 84%).
Стадия 3.
i) Из 0,809 г А31-5 (1,5 экв. LiAlH4, к.т., 16 ч.) получали требуемый продут А31-6 (0,340 г, 47%) и нежелательное des-бромосоединение (бифенил-3-илметанол): 0,208 г, 41%.
ii) Из 0,510 г A31-5 (1,5 экв. LiAlH4, 0°C, 50 мин.) получали требуемый продут (неочищенный) А31-6 (0,435 г, 95%). Согласно 1Н ЯМР в нем содержалось 10 мол. % des-бромосоединения. Очистка в составе предстоящей более крупной партии.
iii) Из 0,514 г А31-5 (0,75 экв. LiAlH4, 0°C, 50 мин.) получали требуемый продут (неочищенный) А31-6 (0,453 г, 98%). Согласно 1Н ЯМР в нем содержалось 5,5 мол. % des-бромосоединения.
iv) Из 6,992 г А31-5 (0,75 экв. LiAlH4, 0°C, 40 мин.) получали неочищенный продукт А31-6 (6,183 г), который согласно 1Н ЯМР содержал 5,5 мол. % des-бромосоединения. Объединяли его с продуктами двух экспериментальных реакций и очищали при помощи колоночной хроматографии с получением А31-6 (6,729 г, 93%).
Стадия 4.
Из 0,102 г А31-6 (1,2 экв. 3-(НО)С6Н4В(ОН)2, 0,1 экв. Pd(PPh3)4, 3 экв. K2CO3, смеси толуола/этанола/воды, Δ, 17 ч.) получали требуемый продут А81 (0,054 г, выход 50%).
Пример 13. Скрининг соединений in vitro
[0135] Систему xCELLigence SP (Roche) использовали для измерения изменений клеточного импеданса (клеточного индекса) после обработки бычьих аортальных эндотелиальных клеток (Европейская коллекция клеточных культур) тестовым соединением. В этой in vitro клеточной экспериментальной системе отрицательный профиль импеданса коррелирует со снижением артериального давления у крыс -снижение импеданса связано с вазодилатацией, и увеличение импеданса связано с вазоконстрикцией (Stallaert W., Dorn J.F., van der Westhuizen E., Audet M. & Bouvier M. Impedance responses reveal β-adrenergic signaling pluridensitometry and allow classification of ligands with distinct signalling profiles, PLoS ONE 2012; 7 (1): e29420, doi: 10.1371 /journal.pone.0029420).
[0136] Вкратце, 50 мкл среды для культивирования клеток (DMEM с низким уровнем глюкозы, дополненная 15% фетальной бычьей сыворотки при 37°C) добавляли в каждую лунку E-Plate 96 (Roche) и измеряли фоновый импеданс в каждой лунке. Затем 50 мкл суспензии бычьих аортальных эндотелиальных клеток (10000 клеток/лунка) добавляли в соответствующие лунки E-Plate 96. Отслеживали клеточный индекс каждой лунки E-Plate 96 при помощи установки RTCA SP внутри инкубатора для культивирования клеток. После инкубации в течение ночи на протяжении 16-20 часов при 5% CO2 и влажности 95% 100 мкл раствора тестового соединения (тестовые соединения готовили в DMSO и разбавляли средой для культивирования клеток до концентрации, составляющей 62,5 мкМ, 125 мкМ или 250 мкМ тестового соединения, с конечной концентрацией DMSO 0,25%) добавляли в соответствующие лунки E-Plate 96 и значения клеточного индекса измеряли сразу же после обработки соединением через каждые 20 секунд в течение 3 часов. Значение клеточного индекса корректировали относительно исходного уровня путем вычитания клеточного индекса обработанных носителем клеток и нормализовали путем деления на клеточный индекс в момент времени непосредственно перед добавлением соединения. Нормализованный относительно исходного уровня клеточный индекс в зависимости от времени наносили на график с помощью программного обеспечения Roche RTCA.
[0137] Реакции с отрицательным импендансом для бычьих аортальных эндотелиальных клеток, которые наблюдали с А6, А26, А27, А30, А32, А35, А56, A56f, A56g, A56k и А81 (фигура 13), указывали на то, что эти соединения являются вазодилататорами.
[0138] Клетки проксимальных почечных канальцев человека (HPCT-wt-05) и крысы (NRK-52E) (выращенные в среде II для кератиноцитов + добавка для роста кератиноцитов + 5 нг/мл рекомбинантного эпидерамального фактора роста человека + 5% FBS + 2 мМ глутамина или DMEM + 10% FBS + 1% NEAA + 2 мМ глутамина соответственно) помещали в 96-луночные планшеты в концентрации, составляющей 10000 клеток/лунка, и инкубировали при 37°C с 5% CO2 в течение ночи. Тестовые соединения в концентрациях, составляющих 32 мкМ или 63 мкМ, инкубировали с клетками проксимальных почечных канальцев человека или крысы в течение 2 часов при 37°C и 5% CO2. Затем добавляли цис-диамминдихлорплатину (III) (цисплатин) в концентрации, составляющей 5 мкг/мл для клеток человека и 12,5 мкг/мл для клеток крысы. Далее каждую клеточную популяцию инкубировали в течение 24 часов при 37°C с 5% CO2. Придерживались изначальных концентраций тестового соединения А32. Для оценки цитотоксических эффектов цисплатина на клетки проксимальных почечных канальцев человека и крысы использовали имеющую высокую растворимость в воде тетразолиевую соль, WST-8, которая восстанавливается при помощи дегидрогеназ в клетках с образованием формазана, водорастворимого, имеющего желтый цвет индикаторного красителя, следуя инструкциям изготовителя (в частности, анализ с набором 8 для определения количества клеток (CCK-8) от Sigma). Степень абсорбции в планшетах реагента WST-8 (CCK-8) затем измеряли при 450 нм с помощью считывающего устройства для планшетов Thermo Scientific Multiskan EX.
[0139] Показатель клеточной смерти, индуцированной цисплатином, который снижался в культурах клеток проксимальных почечных канальцев человека и крысы, обработанных 32 мкМ или 63 мкМ А32 в течение 24 часов (фигура 14), указывал на то, что это соединение снижает степень отмирания клеток проксимальных почечных канальцев.
Пример 14. Скрининг соединений in vivo
[0140] Четырнадцатинедельных SHR на рационе с 2,2% соли (Glen Forrest Stockfeeders) случайным образом распределяли в контрольную группу в начальный момент времени, группу обработки тестовым соединением (500 пкмоль/кг/мин.) в питьевом растворе или контрольным питьевым раствором (5% этанола в деионизированной дистиллированной воде (в каждой группе n=5). Крыс, распределенных в контрольную группу в начальный момент времени (крысы возрастом 14 недель), анестезировали и собирали их почки и сердца, в то время как крыс, распределенных в группы для обработки контролем и тестовым соединением, взвешивали дважды в неделю и отслеживали их потребление питьевого раствора, чтобы обеспечить регулирование концентрации тестового соединения в питьевом растворе для поддержания постоянной дозы в течение 4-недельного периода исследования (крысы возрастом 18 недель). По завершению периода исследования крыс анестезировали и собирали их почки и сердца.
[0141] Четырнадцатинедельных SHR на рационе с высоким содержанием жира (Glen Forrest Stockfeeders) случайным образом распределяли в группы питьевого раствора с тестовым соединением (500 пмоль/кг/мин. тестового соединения в 10% этаноле в деионизированной дистиллированной воде) или контрольного питьевого раствора (10% этанола в деионизированной дистиллированной воде). Через 4 недели крыс анестезировали, брали образцы крови для анализа уровней аминотрансферазы (AST) в плазме крови и собирали печенки.
[0142] Для количественной оценки фиброза тканей и/или содержания жира срезы тканей толщиной ≤3 мм фиксировали в 10% забуференном формалине в течение 24 часов, обрабатывали и заливали в парафин. Поперечные срезы толщиной три микрометра окрашивали с использованием трехцветного окрашивания по Массону. Минимум 20 случайных полей при

[0143] Уровни AST в плазме крови измеряли при помощи аппарата RefloVET Plus (Roche) с использованием расходных пластинок с магнитными идентификаторами анализа, узнаваемыми аппаратом. Калибровочный стандарт использовали в аппарате перед каждым применением и устройство эксплуатировали согласно инструкциям изготовителя. Результаты представлены в виде международных единиц на литр (IU/L).
[0144] Степень проявления фиброза в почке через 4 недели лечения с помощью 500 пмоль/кг/мин. А32, которая снижалась по сравнению с контрольными крысами возрастом 18 недель (фигура 15), указывала на то, что это соединение предотвращает развитие фиброза почек.
[0145] Степень проявления фиброза миокарда через 4 недели лечения с помощью 500 пмоль/кг/мин. А32, которая снижалась по сравнению с контрольными крысами возрастом 14 и 18 недель (фигура 16), указывала на то, что это соединение предотвращает развитие фиброза миокарда и способствует устранению развившегося фиброза миокарда.
[0146] Степень проявления фиброза печени через 6 недель лечения с помощью 500 пмоль/кг/мин. А6, А27, А32 и A56f, которая снижалась по сравнению с контрольными крысами (фигура 17, * р<0,025, ** р<0,01, *** р<0,005), указывала на то, что эти соединения предотвращают развитие фиброза печени.
[0147] На срезах, окрашенных с применением трехцветного окрашивания по Массону, где видны портальные тракты, можно видеть, что фиброзные тяжи тянуться от портальных трактов (стрелки) и нарушают структуру ткани контрольной крысы (фигура 18А). На срезах, полученных от крыс, обработанных с помощью А32 (фигура 18В), А6 (фигура 18С), А27 (фигура 18D), А56 (фигура 18Е) и A56f (фигура 18F), была восстановлена нормальная структура ткани.
[0148] На срезах, окрашенных с применением трехцветного окрашивания по Массону, на которых видна ткань сердца, полученная от контрольных крыс (фигура 19А), фиброзная ткань присутствует на всем срезе, при этом находится между мышечными волокнами, а в некоторых случаях окружает и заменяет мышечные волокна (стрелки). На секциях, на которых видна ткань сердца, полученная от крыс, обработанных с помощью А32 (фигура 19В), присутствует минимальное количество фиброзной ткани, и была восстановлена нормальная структура ткани.
[0149] Количество жира в печени через 4 недели лечения с помощью 500 пмоль/кг/мин. А27, А32 и А56, которое снижалось по сравнению с контрольными крысами возрастом 18 недель (фигура 20, * р<0,05), указывало на то, что эти соединения снижают степень накопления жира в печени.
[0150] Уровни AST в плазме крови, которые снижались у крыс, обработанных с помощью А32 и A56f, по сравнению с контрольными крысами (фигура 21, * р<0,025), указывали на то, что эти соединения предотвращают повреждение печени.
Пример 15. Сравнение скрининга соединений in vitro и in vivo
[0151] Сравнение клеточного импеданса бычьих аортальных эндотелиальных клеток и уровня фиброза печени у SHR, обработанных различными тестовыми соединениями, показало, что анализ in vitro указывает на способность тестовых соединений уменьшать фиброз в печени (фигура 22, R2=0,925).
Реферат
Настоящее изобретение относится к новому соединению формулы:или к его фармакологически приемлемой соли, применяемым для профилактического или терапевтического лечения фиброза. В указанных структурных формулах А выбирают из: частично насыщенного или ненасыщенного 5- или 6-членного гетероциклила, выбранного из пирролила, пиразолила, имидазолила, триазолила, имидазолидинила, пирролидинила, пирролидинилидена, дигидропирролила, изоксазолил-дигидрооксазолила, изоксазолидинила, оксазолидинила и оксазолила, где указанный частично насыщенный или ненасыщенный 5- или 6-членный гетероциклил необязательно замещен одним или несколькими заместителями, представляющими собой оксо, Cалкил, амино, гидроксил или галоген; Cалкоксиламина; Cалкиламина, необязательно замещенного одним или несколькими заместителями, представляющими собой Cалкил, Cгалогеналкил, гидроксил или галоген; Салкил-карбоновой кислоты; Cалкилгидроксила; насыщенного или ненасыщенного Cалкил-бициклического-гетероциклила, выбранного из индолила, изоиндолила, индолинила и изоиндолинила, где указанный насыщенный или ненасыщенный Cалкил-бициклический-гетероциклил необязательно замещен одним или несколькими оксо; и насыщенного или ненасыщенного Cалкоксил-бициклического-гетероциклила, выбранного из индолила, изоиндолила, индолинила и изоиндолинила, где указанный Cалкоксил-бициклический-гетероциклил необязательно замещен одним или несколькими оксо. 11 н. и 16 з.п. ф-лы, 22 ил., 15 пр.
Формула











Комментарии