Алкилзамещенные циклические амины в качестве селективных лигандов d3-допамина - RU2185372C2
Код документа: RU2185372C2
Чертежи






Описание
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Изобретение относится к
арилзамещенным циклическим аминам для лечения болезней ЦНС, таких как шизофрения, болезнь Паркинсона, поздняя дискинезия, тучное компульсивное нарушение, депрессия и тревожное состояние, которые
преимущественно связаны с рецептором D3-допамина. Рецептор D3-допамина недавно был клонирован Sokoloff et al. (Nature, 347, 146 (1990)). Было высказано предположение, что этот подтип рецептора играет
роль при действии антипсихотических средств. Интересно, что данный рецептор имеет относительно высокое содержание в областях мозга, связанных с эмоциональными и познавательными функциями.
Соединения с таким профилем действия можно использовать для лечения нарушений ЦНС, например шизофрении, мании, депрессии, гериатрических нарушений, злоупотреблении лекарственными средствами и аддикции, болезни Паркинсона, тревожных нарушений, нарушений сна, нарушений циркадного ритма и слабоумия.
Описание уровня техники.
В патентной публикации РСТ W090/07490 описаны 2-аминотетралины и 2-аминоинданы с замещением в ароматическом кольце OCH3 или ОН вместе с заместителем Br.
В патентной публикации РСТ W095/04713 описаны 2-аминоинданы, которые связываются с рецептором D3-допамина.
В патентной заявке РСТ PCT/US96/00020 описаны 2-аминоинданы, имеющие сульфонамидное замещение в кольце бензола и пригодные для лечения шизофрении.
В патенте США 4.968 679 описаны 2-аминотетралины, имеющие замещение в положении 8, которые являются агонистами/антагонистами серотонина.
В PJMurry, Novel 6-substituted 2-Aminotetralins, Bioorg. And Med. Chem. Lett. 1996, 403, описаны соединения, имеющие селективность относительно рецептора D3-допамина, которые имеют кольцо бензола, замещенное 5-метокси и 6-арилэтилом и другими группами с 6-CH2SO2 (4-метоксифенил или 4-I-фенил).
В J. Med. Chem. 1987, 30, 494 и Eur. J. Med. Chem. Chim. Ther. 1984, 19, 451, описаны циклические амины, аналогичные соединениям общей формуле III, если "n" равно 2 и "X" представляет NH2.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В одном аспекте
изобретение относится к соединениям и фармацевтически приемлемым солям формулы I:
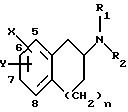
где X и Y находятся в положении 5, 6 или 7 вместо водорода (т.е. замещают водород группы СН), так что
i) когда n равно 1, Х может быть (СН2)mCONR4R5 (где m равно 0 или 1), (CH2)mSO2NR4R5, (СН2)mNR4CONHR5, (СН2)mNHSO2R3, (CH2)mNHCOR3,
C(O)R4 или (CH2)mSO2R3 (где для (СН2)mSO2R3, Y не может быть водородом или галогеном) и
Y представляет R4, (СН2)pCONR4R5 (где р равно 0 или 1), (CH2)p CN, (CH2)pSO2NR4R5, OR6, (CH2)pSO2R3, (СН2)pNHSO2R3, галоген или (СН2)pNHCOR3; или
ii) когда n равно 0 или 1, то Х и Y могут быть в 0-положении относительно друг друга и вместе представляют:
N-R10-замещенный имид, такой как -С(O)NR10C(O)-, C(O)NR4(CH2)xNR10C(O) - (где х равно 0 или 1)
лактам, такой как -CH2NR10C(O)-,
- (CH2)2NR10C(O) -,
- CH2C(O)NR10-,
- N(R3)-C(O)-N(R3)-,
- N(R3)-С(O)-O-,
- N=C(R7)-N(R3) - или
циклический амин, такой как -CH2N(R8)CH2-;
(iii) когда n равно 0 и Y представляет OR9, X может быть (CH2)mCONR4R5 (где m равно 0 или 1), (СН2)mSO2NR4R5, (CH2)mNR4СONHR5, (CH2)mSO2R3,
(CH2)mNHSO2R3, (CH2 )mNHCOR3 или C(O)R4;
R1 и R2 представляют, независимо, Н, С1-С8-алкил, включая его изомерные формы, или С1-С8-алкиларил;
R3 представляет С1-С8-алкил, C1-C6-алкиларил или арил;
R4 и R5 представляют, независимо, Н, C1-C8-алкил, С1-С6-алкиларил или арил;
R6 представляет Н, SO2CF3, SO2 -С1-С8-алкил, SO2-C1-C6-алкиларил, SO2-арил, C1-C8-алкил, С1-С6-алкиларил или арил;
R7 представляет водород, CON(R4)2, SO2N(R4)2 или SO2R4;
R8 представляет С1 -С8-алкил, С1-С6-алкиларил, арил, CON(R4)2, COR4, SO2N(R4)2 или SO2R4 (при условии, что для CON(R4)2, COR4, SO2N(R4)2 или SO2R4, R4 не может быть водородом);
R9 представляет С2-С8-алкил (необязательно замещенный 1-3 галогенами), С1-С6-алкиларил, арил и
R10 представляет Н, С1 -С8-алкил, C1-C6-алкиларил, арил или (СН2)0-6SO2-арил.
В другом аспекте изобретение относится к соединениям и
фармацевтически приемлемым солям формулы II:
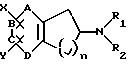
где один из А, В, С и D представляют азот и остальные положения представляют СН и n равно 1 или 2;
R1 и R2 представляют, независимо, Н, С1-С8-алкил и его изомерные формы, С1-С8-алкиларил;
Х и Y могут быть замещены в положениях А, В, С или D вместо водорода (т. е. замещают водород группы СН), где
i) Х представляет (CH2)mCONR4R5, (CH2)mCN, (CH2)mSO2NR4R5, (CH2)mNR4CONHR5, (CH2 )mSO2R3, (СН2)mNHSO2R3, (СН2)mNHCOR3 или С(О)R4 (где m равно 0 или 1, за исключением того, что когда m равно 0, то Y не может быть водородом или галогеном) и
Y представляет R4, (СН2)pCONR4R5, (CH2 )pCN, (СН2)pSO2NR4R5, OR6, OSO2R3, (СН2)pSO2R3, (СН2)pNHSO2R3, галоген или (СН2)pNHCOR3 (где р равно 0 или 1); или
(ii) Х и Y, когда они находятся в 0-положении относительно друг друга, совместно представляют:
N-R4 - замещенный имид, такой как -С(O)NR4C(O)-, лактам, такой как -CH2NR4C(O)- или -СН2 С(O)NR4-, или циклический амин, такой как -CH2NR4CH2-;
R3 представляет С1-С8-алкил, C1-C6 -алкиларил или арил;
R4 и R5 представляют, независимо, Н, С1-С8-алкил, С1-С6-алкиларил или арил; и
R6 представляет Н, SO2CF3, SO2CH3, SO2-арил, С1-С8-алкил, C1-C6-алкиларил или арил.
В
другом аспекте изобретение относится к соединениям и фармацевтически приемлемым солям формулы III:
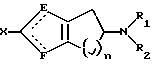
где один из Е и F представляет N и другой представляет S и n равно 1 или 2;
R1 и R2 представляют, независимо, Н, С1-С8-алкил и его изомерные формы или С1-С8-алкиларил;
где Х представляет (СН2)mCONR4R5, (CH2)mCN, (CH2)m SO2NR4R5, CH2NR4CONHR5, (СН2)mSO2R3, (СН2)mNHSO2R3, CH2NHCOR3 или C(O)R4; R3 представляет С1-С8-алкил, С1-С6-алкиларил или арил;
R4 и R5 представляют, независимо, Н, C1-С8-алкил, С1-С6-алкиларил или арил; и где m равно 0 или 1.
В другом аспекте изобретение относится к соединениям и фармацевтически приемлемым солям указанной выше формулы I, II или III, включая рацемические смеси и оба энантиомера.
Еще в одном аспекте изобретение относится к способу лечения шизофрении введением терапевтически эффективного количества соединения формулы I, II или III пациенту, страдающему шизофренией. Соединения формулы I, II или III можно вводить пациенту, страдающему шизофренией, манией, депрессией, гериатрическими нарушениями, злоупотребляющему лекарственными средствами и страдающему аддикцией, болезнью Паркинсона, нарушениями сна, нарушениями циркадного ритма, нарушениями, связанными с тревогой или слабоумием. Соединения можно вводить в количестве от около 0,25 мг до около 100 мг/человека.
Еще в одном аспекте изобретение относится к способу лечения нарушений центральной нервной системы, связанных с активностью рецептора D3-допамина, у пациента, нуждающегося в таком лечении, включающему введение субъекту терапевтически эффективного количества соединения формулы I, II или III для облегчения такого нарушения. Обычно соединение формулы I, II или III вводят в форме фармацевтической композиции, содержащей фармацевтически приемлемый носитель или разбавитель.
Еще в одном аспекте изобретение относится к фармацевтической композиции для лечения нарушений центральной нервной системы, связанных с активностью рецептора D3-допамина, содержащей эффективное количество соединения формулы I, II или III вместе с фармацевтически приемлемым носителем или разбавителем.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение относится к соединениям или фармацевтически приемлемым солям формулы I, II или III, как описано выше, либо в рацемической форме, либо в форме чистых
энантиомеров.
"Алкил" представляет алкил, содержащий до восьми или шести атомов углерода, такой как метил, этил, пропил, бутил, пентил, гексил, гептил, октил и их изомерные формы.
"Галоген" представляет атом фтора, хлора, брома или иода.
"Арил" включает фенил, пиридинил, имидазолил, тиофенил, оксазолил, оксадиазол, бензотриазол, бензоксадиазол, тиазол и изоксазолил. Арил может быть замещен одним или несколькими заместителями, выбранными из фтора, хлора, брома, амино, CN, карбоксамидо, ацетамидо, метила, нитро, сульфонила, сульфонамидо, трифторметила, трифторметокси, O-алкокси, трифлата или ацетила.
Фармацевтически приемлемые соли включают соли как неорганических, так и органических кислот. Предпочтительные фармацевтически приемлемые соли включают соли следующих кислот: матансульфоновой, хлористоводородной, бромистоводородной, серной, фосфорной, азотной, бензойной, лимонной, винной, фумаровой или малеиновой.
Соединения формулы I, II и III активны перорально или парентерально. Перорально соединения формулы I, II и III можно вводить в виде твердых лекарственных форм, таких как таблетки или капсулы, или в виде жидких лекарственных форм, таких как эликсиры, сиропы или суспензии, которые известны специалистам в данной области. Предпочтительно, чтобы соединения формулы I, II или III вводились в виде твердой лекарственной формы и чтобы она была таблеткой.
Обычно соединения формулы I, II или III можно вводить в количестве от около 0,5 мг до около 250 мг/человека от одного до трех раз в день. Предпочтительно от около 5 до около 50 мг/день в виде разделенных доз.
Точная доза и частота введения зависит от конкретного используемого соединения формулы I, II или III, конкретного состояния, которое лечат, тяжести состояния, которое лечат, возраста, массы, общего физического состояния конкретного пациента, другой лекарственной терапии индивидуума, которая может проводиться, что хорошо известно специалистам данной области, и может быть более точно определена путем измерения уровня или концентрации активного соединения в крови пациента и/или реакции пациента на конкретное проводимое лечение.
Таким образом, соединения-объекты данного изобретения вместе с фармацевтически приемлемым носителем, разбавителем или буфером можно вводить в терапевтическом или фармакологическом количестве, эффективном для облегчения нарушения центральной нервной системы относительно диагностированного физиологического состояния. Соединения можно вводить внутривенно, внутримышечно, местно, чрескожно, например, с помощью накожных пластырей, трансбуккально или перорально человеку или другим позвоночным.
Композиции настоящего изобретения можно формулировать для введения людям и другим позвоночным в единичных дозированных формах, таких как таблетки, капсулы, пилюли, порошки, гранулы, стерильные парентеральные растворы или суспензии, пероральные растворы или суспензии, эмульсии масло-в-воде и вода-в-масле, содержащие подходящие количества соединения, суппозитории и жидкие суспензии или растворы.
Для перорального введения можно готовить либо твердые, либо жидкие единичные дозированные формы. Для получения твердых композиций, таких как таблетки, соединение можно смешивать с обычными ингредиентами, такими как тальк, стеарат магния, дикальцийфосфат, силикат магния и алюминия, сульфат кальция, крахмал, лактоза, аравийская камедь, метилцеллюлоэа и функционально подобные фармацевтические материалы-разбавители или -носители. Капсулы получают смешиванием соединения с инертным фармацевтическим разбавителем и наполнением смеси в твердую желатиновую капсулу подходящего размера. Мягкие желатиновые капсулы получают машинным капсулированием суспензии соединения с приемлемым растительным маслом, светлым вазелиновым маслом или другим инертным маслом.
Можно также получать жидкие единичные дозированные формы для перорального введения, такие как сиропы, эликсиры и суспензии. Эти формы можно растворять в водном наполнителе вместе с сахаром, ароматизирующими агентами и консервантами для образования сиропа. Суспензии можно получить с водным наполнителем с помощью суспендирующего агента, такого как аравийская камедь, трагакант, метилцеллюлоза и подобные.
Для парентерального введения жидкие единичные дозированные формы можно получать с использованием соединения и стерильного наполнителя. Для получения растворов соединение можно растворить в воде для инъекции и стерилизовать через фильтр до заполнения в подходящие пузырек или ампулу и закупоривания. Вспомогательные агенты, такие как местный анестетик, консервант и буферы, можно растворить в наполнителе. Композицию можно заморозить после наполнения в ампулу и воду удалить в вакууме. Лиофилизованный порошок можно затем запаять в ампуле и повторно приготовить перед использованием.
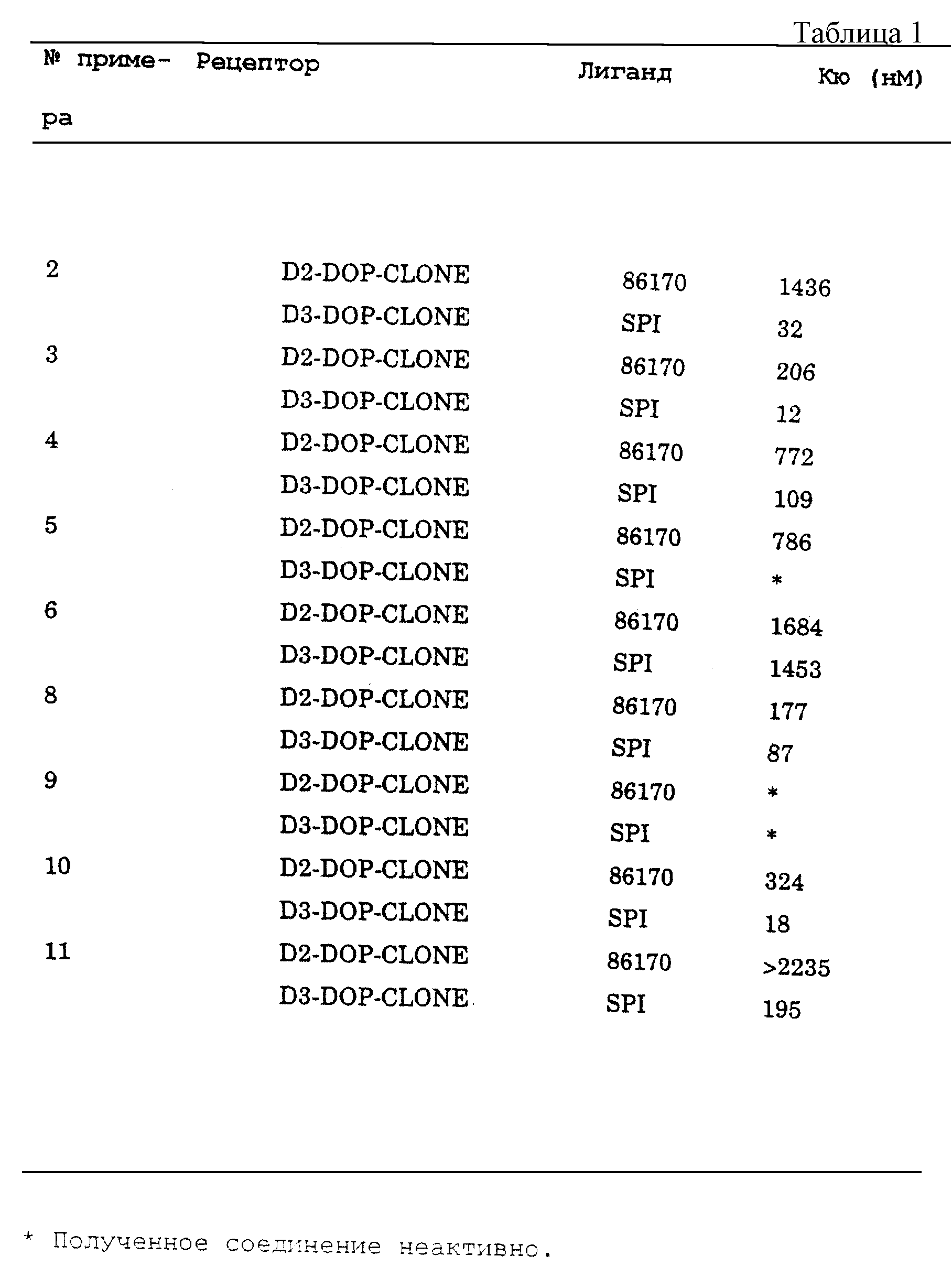
ДАННЫЕ СВЯЗЫВАНИЯ ДЛЯ СОЕДИНЕНИЙ ПРИМЕРОВ
В экспериментах по конкуретному связыванию использовали одиннадцать разбавлений
тестируемых соединений формулы I, конкурирующих с [3Н] -5-(дипропиламино)(-5,6-дигидро-4Н-имидазо(4,5,1-ij)хинолин-2(1H)-оном (R-энантиомер) ("86170") (62 Кю/ммоль, 2 нМ) и [3Н]
-спипероном ("SPI") (107 Кю/ммоль, 0,5 нМ) в сайтах связывания с D2 и D3, соответственно (Lahti, R.A. , Eur. J. Pharmacol. , 202, 289 (1991)). В каждом эксперименте использовали клонированные
рецепторы крысы, экспрессированные в клетках СНО-К1 (Chio, C. L., Nature, 343, 266 (1990); and Huff, R.M., Pharmacol. 45, 51-60 (1993)).
Результаты показаны в табл.1.
Легенда примеров: формула I, где n равно 1, R1 и R2 представляют н-пропил: Y представляет Н и Х замещен в положении 7 следующим образом:
Пример 2: Х = CH2
NHSO4-4-CN-фенил;
Пример 3: Х = CH2NHSO2-4-Cl-фенил;
Пример 4: Х = CH2NHSO2-4-NO2-фенил;
Пример 5: Х =
CH2NHSO2-3-CN-фенил;
Пример 6: Х = CH2NHSO2-4-метил-имидазол;
Пример 8: Х = CH2NHC(O)-4-Cl-фенил;
Пример 9: Х = CH2NHC(O)-4-CN-фенил;
Пример 10: Х =NHSO2-4-Cl-фенил;
Пример 11: Х =NHSO2-4-CN-фенил.
Как показано в табл.1, в экспериментах по конкурентному связыванию использовали одиннадцать разбавлений испытуемых соединений формулы I, конкурирующих с [3Н]-5-(дипропиламино)-5,6-дигидро-4Н-имидазо(4,5-1-ij)хинолин-2(1Н)-оном (R-энантиомер) ("86170") (62 Кю/ммоль, 2 нМ) и [3H]-7-OH-DPAT ("7-OH-DPAT") (107 Кю/ммоль, 0,5 нМ) за сайты связывания с D2 и D3 соответственно. (Lahti, R. A., Eur.J.Pharinacol., 202, 289 (1991)). В каждом эксперименте использовали клонированные крысиные рецепторы, экспрессированные в клетках СНО-К1 (Chio, C.L., Nature, 343, 266 (1990); and Huff, R.M. Pharmacol. 45, 51-60 (1993)). Величины Кю вычисляли по уравнению Chen-Prushoff. Результаты приводятся в табл.2 для соединений, полученных по схеме 5.
Химические синтезы:
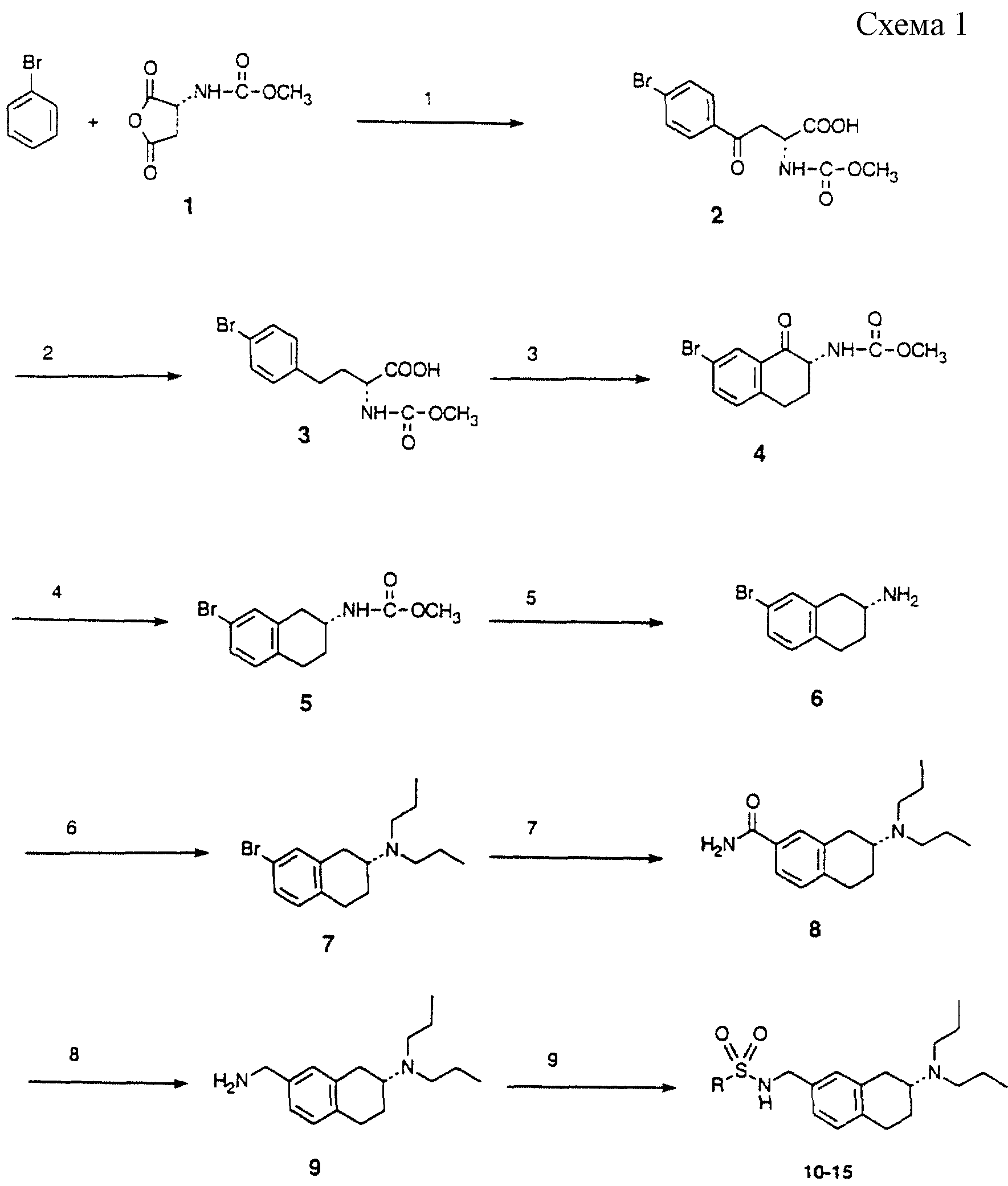
Схема 1 и схема 2: (Аналоги формулы
I)
N-Карбометоксиангидрид, полученный из D-аспарагиновой кислоты (1, схема 1), подвергали медиированной хлоридом алюминия реакции Фриделя-Крафтса с бромбензолом с получением кетона 2. Этот
кетон восстанавливали по кетонной группе три-этилсиланом, при катализе тетрахлоридом титана, получая 3. Эту карбоновую кислоту превращали в хлорангидрид, который затем подвергали катализируемой
хлоридом алюминия циклизации Фриделя-Крафтса с получением тетразона 4. Этот кетон восстанавливали триэтилсиланом, при катализе тетрахлоридом титана, получая тетралин 5. Карбаматную группу омыляли
гидроксидом, получая 7-бром-2-аминотетралин 6, который затем алкилировали, получая 7. Обработка этого арилбромида трет-бутиллитием и затем триметилсилилизоцианатом (Tetrahed. Lett. 1975, 981) с
последующим водным гидролизом давала карбоксамид 8. Кипячение с обратным холодильником этого карбоксамида в ТГФ с бораном приводит к восстановлению в первичный амин 9. Этот амин (9) обрабатывали
различными сульфонилхлоридами (методика 9), получая сульфонамиды 10-15.
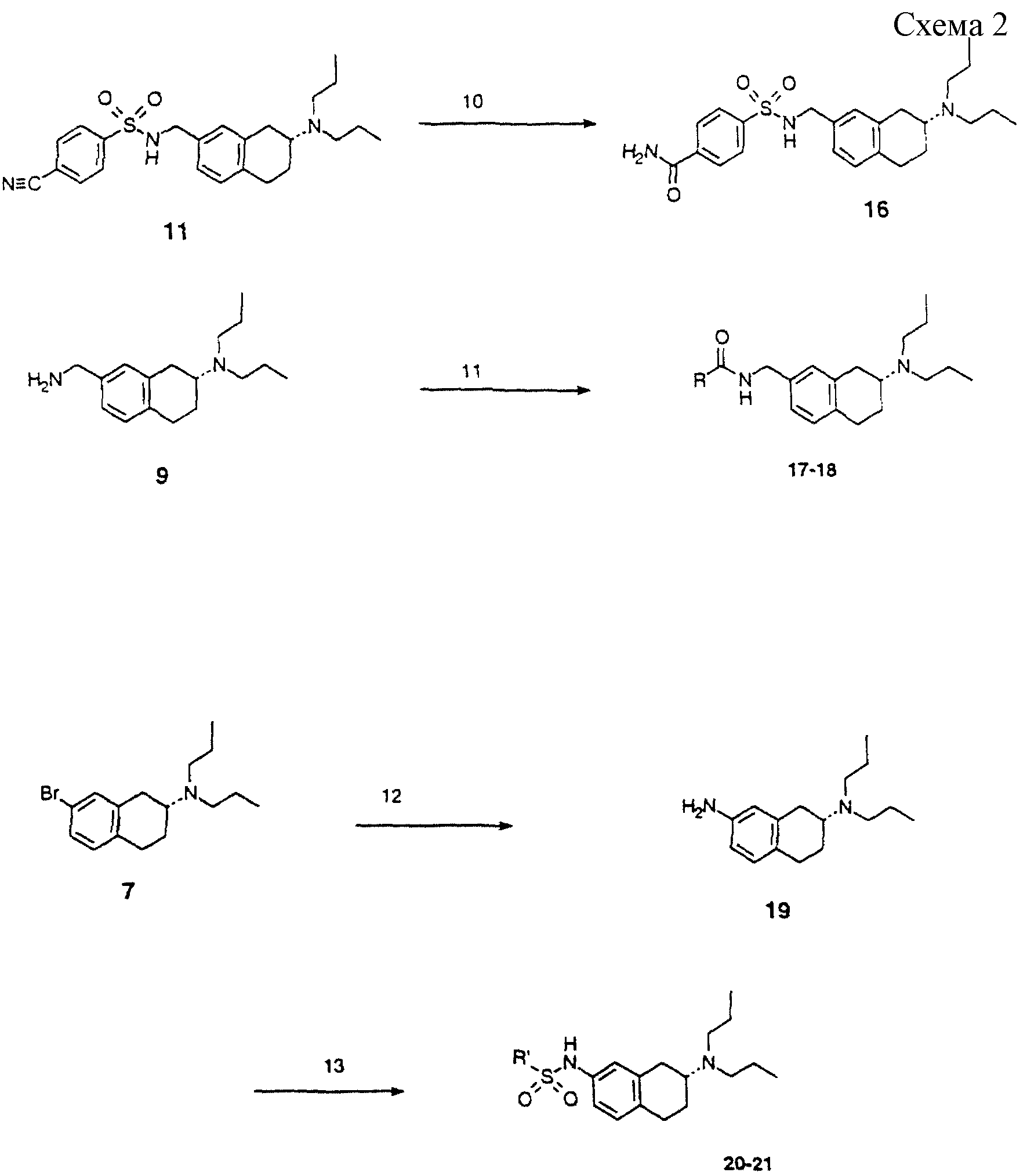
Сульфонамид 11 далее превращали в 16 (схема 2) гидролизом нитрила пероксидом водорода, получая карбоксамид 16 (Tetrahed, Lett. 1989, 949).
Первичный амин 9 также превращали в карбоксамиды 17 и 18 с использованием подходящего хлорангидрида карбоновой кислоты (схема 2).
Арилбромид 7 обрабатывали н-бутиллитием, затем дифенилфосфорилазидом и затем бис(2-метоксиэтокси)алюминийгидридом натрия (схема 2), получая амин 19 (Tetrahed, Lett. 1984, 429). Этот амин обрабатывали сульфонилхлоридами, получая сульфониламиды 20 и 21.
Схема 3: (Аналоги формулы I)
Аминотетралин 22 (J.Org.Chem. 1995, 4324) защищали ВОС-группой и затем подвергали обмену
металл-галоген с трет-бутиллитием с последующим гашением ДМФ, получая альдегид. Этот альдегид восстанавливали борогидридом натрия с получением бензилового спирта. Его обрабатывали тионилхлоридом,
получая бензилхлорид, который, в свою очередь, превращали в бензилазид реакцией с азидом натрия. Катализируемое Pd/C гидрирование азида давало бензиламин 24. Этот амин конденсировали с различными
арилсульфонилхлоридами с получением сульфонамидов. Их обрабатывали трифторуксусной кислотой для удаления ВОС-защитных групп (методика 16), получая сульфонамидные соединения 25-26. Их алкилировали
бромпропаном (методика 17), получая третичные аминсульфонамиды, представленные 27.
Амин 24 конденсировали также с хлорангидридами арилкарбоновых кислот с получением амидов, которые затем освобождали от защитной группы трифторуксусной кислотой, получая амиды 28-29. Эти амиды (28-29) нагревали с бромпропаном (методика 17) для образования аналогов третичных аминов, представленных 30.
Амин 24 конденсировали также с арилизоцианатами, получая мочевины. Их освобождали от защитной группы трифторуксусной кислотой с получением мочевин 31-33.
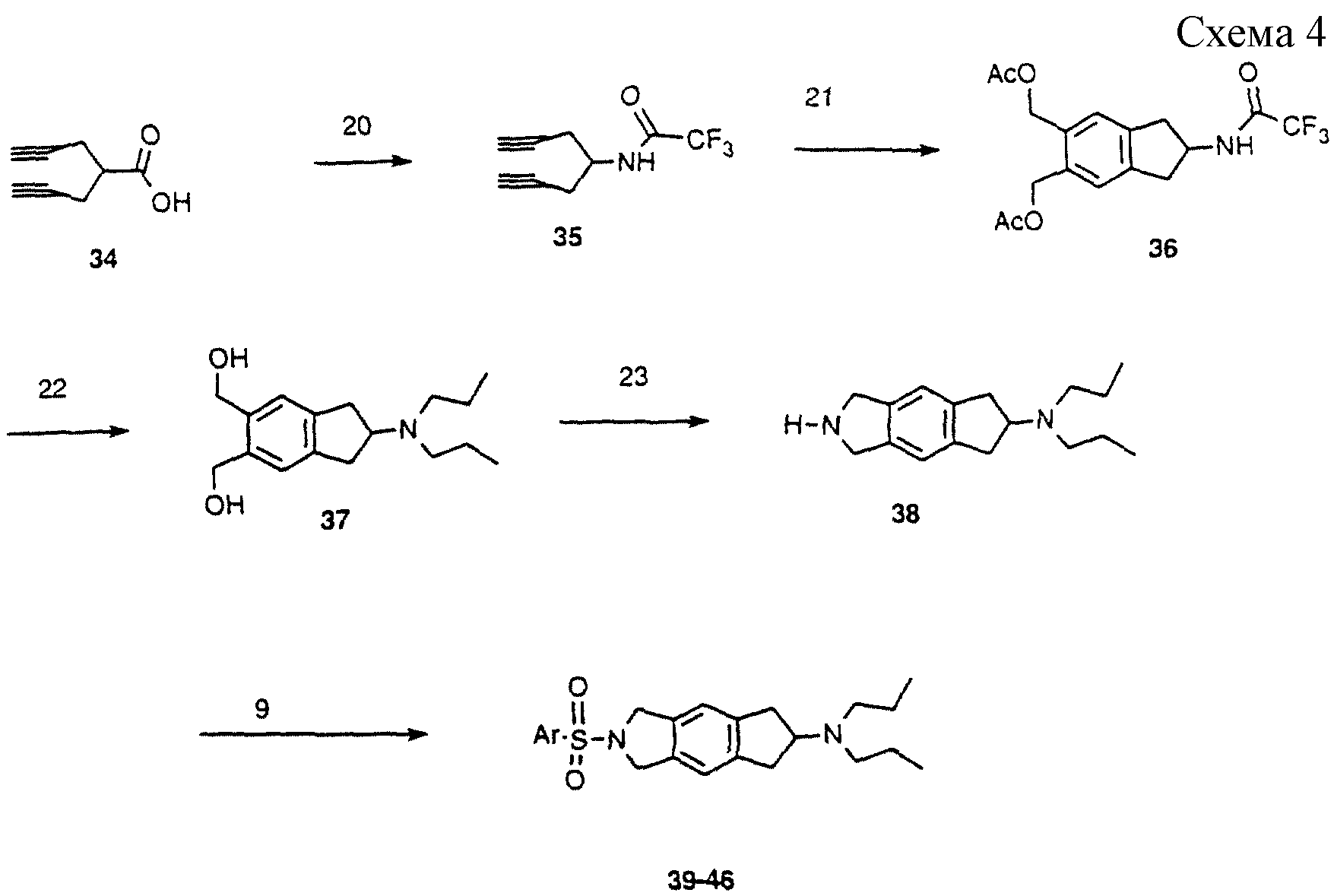
Схема 4:
(аналоги формулы I)
Получали диинкислоту 34 (J.Chem.Soc., Perkin Trans. 1, 1215-1224 (1986) и подвергали перегруппировке Курциуса под действием дифенилфосфорилазида, улавливая изоцианатный
промежуточный продукт трет-бутанолом с получением ВОС-защищенного продукта. ВОС-группу отщепляли трифторуксусной кислотой, получая первичный амин, который затем обрабатывали трифторуксусным ангидридом
с получением 35. Этот диин (35) обрабатывали катализатором Вилькинсона и 1,4-диацетокси-2-бутином для получения 36 (Tetrahed. Lett., 34, 23-26 (1993)). Ацетаты и трифторацетильную
группу отщепляли
основанием и продукт алкилировали н-бромпропаном, получая 37. Этот диол обрабатывали аллиламином, получая циклический 5-членный амин; затем аллильную группу удаляли катализом палладием, получая 38.
Амин 38 конденсировали с различными арилсульфонилхлоридами (методика 9), получая сульфонамиды 39-46.
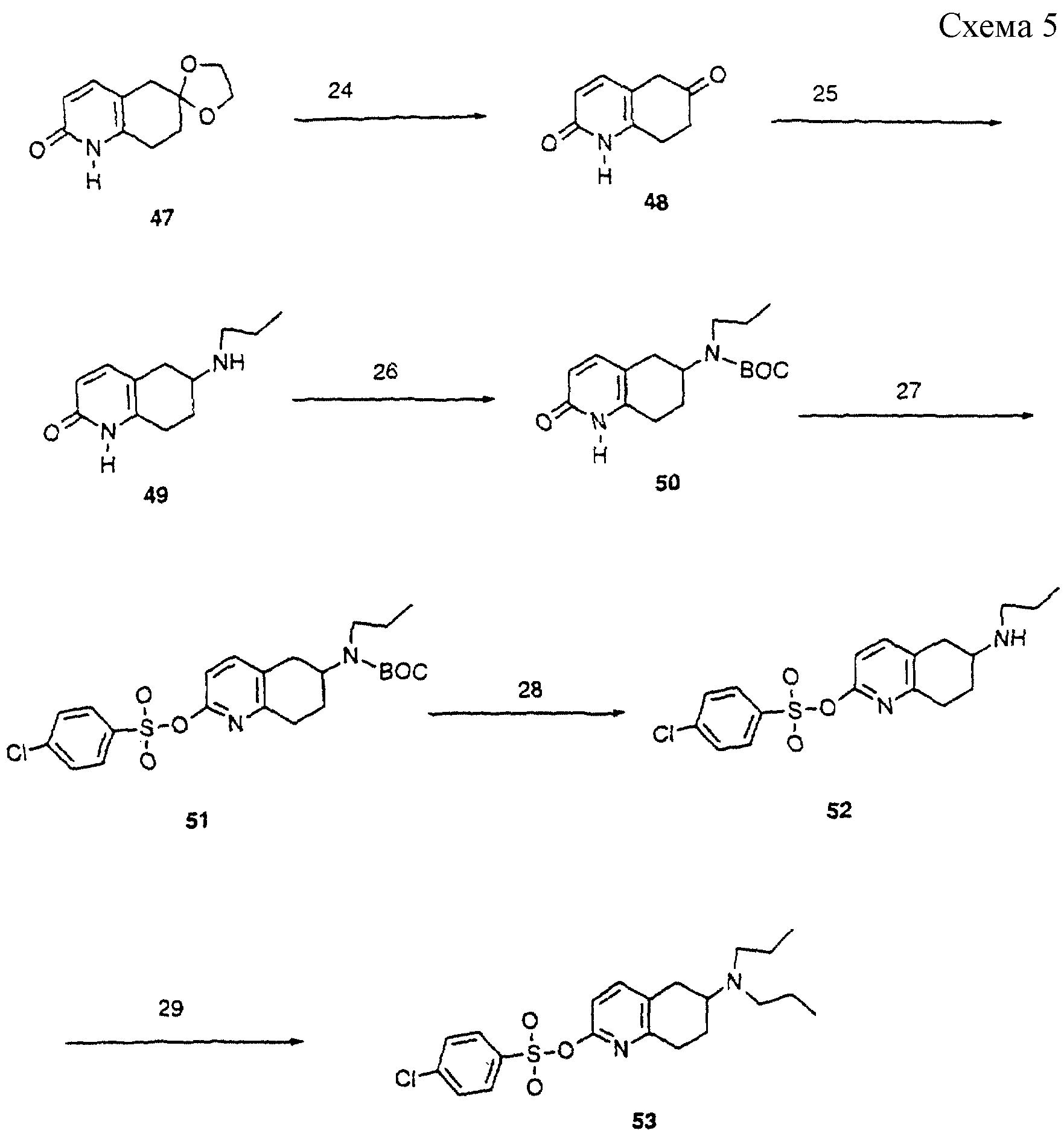
Схема 5: (аналоги формулы II)
Пиридон 47 (J.Chem.Soc.Perkin Trans, 1990,
195) гидролизовали (схема 5) водной перхлорной кислотой при 95oС с получением кетона 48. Этот кетон подвергали восстановительному аминированию н-пропиламином, используя газообразный водород
при 2,109 ат (50 psi), уксусную кислоту и оксид платины в этаноле. Пропилзамещенный амин (49) получали с хорошим выходом. Обработка ди-трет-бутилкарбонатом в ТГФ давала ВОС-защищенное соединение (50).
Его конденсировали с 4-хлорбензолсульфонилхлоридом в присутствии DMAP и триэтиламина, получая сульфонилоксизамещенный пиридин 51. Удаление ВОС-группы трифторуксусной кислотой при 25oС дает
после обработки амин 52. Его можно превращать в аналог дипропиламина (53) нагреванием с н-бромпропаном в ацетонитриле в присутствии карбоната калия.
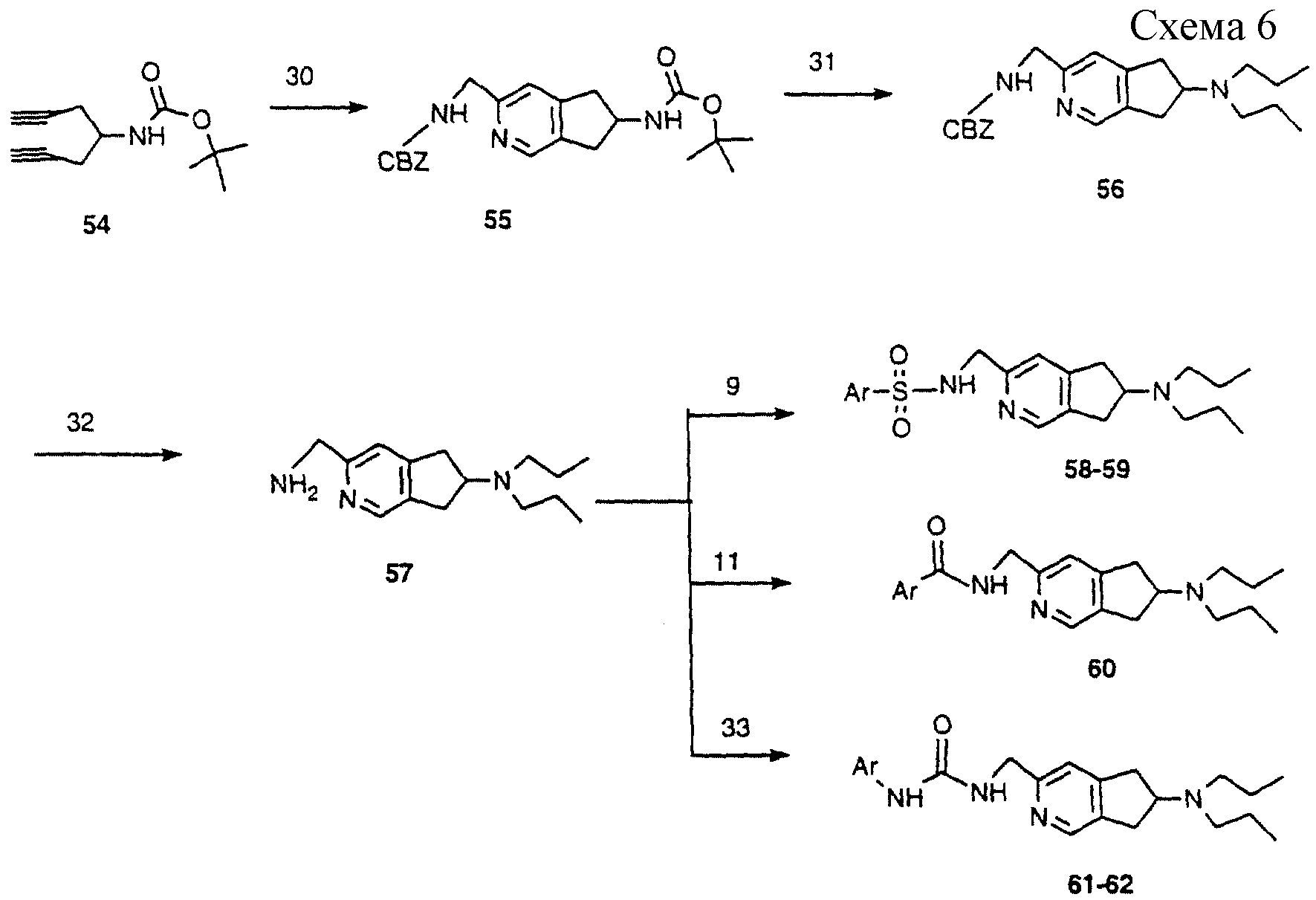
Схема 6: (аналоги формулы II)
Диин 54, получение которого уже описано, обрабатывали N-бензилокси-2-аминоацетонитрилом при воздействии кобальтовым катализатором (J.Chem.Soc., Chem. Comm. , 133-134 (1982) с получением
гетероцикла 55. ВОС-группу удаляли трифторуксусной кислотой и амин алкилировали н-бромпропаном, получая 56. CBZ-группу удаляли катализируемым палладием гидрированием с получением амина 57. Этот амин
обрабатывали арилсульфонилхлоридами (методика 9), получая сульфонамиды, представленные 58-59.
Амин 57 также обрабатывали хлорангидридами арилкарбоновых кислот (методика 11), чтобы получить амиды, представленные 60.
Амин 57 также обрабатывали арилизоцианатами (методика 33), получая мочевины, представленные 61-62.
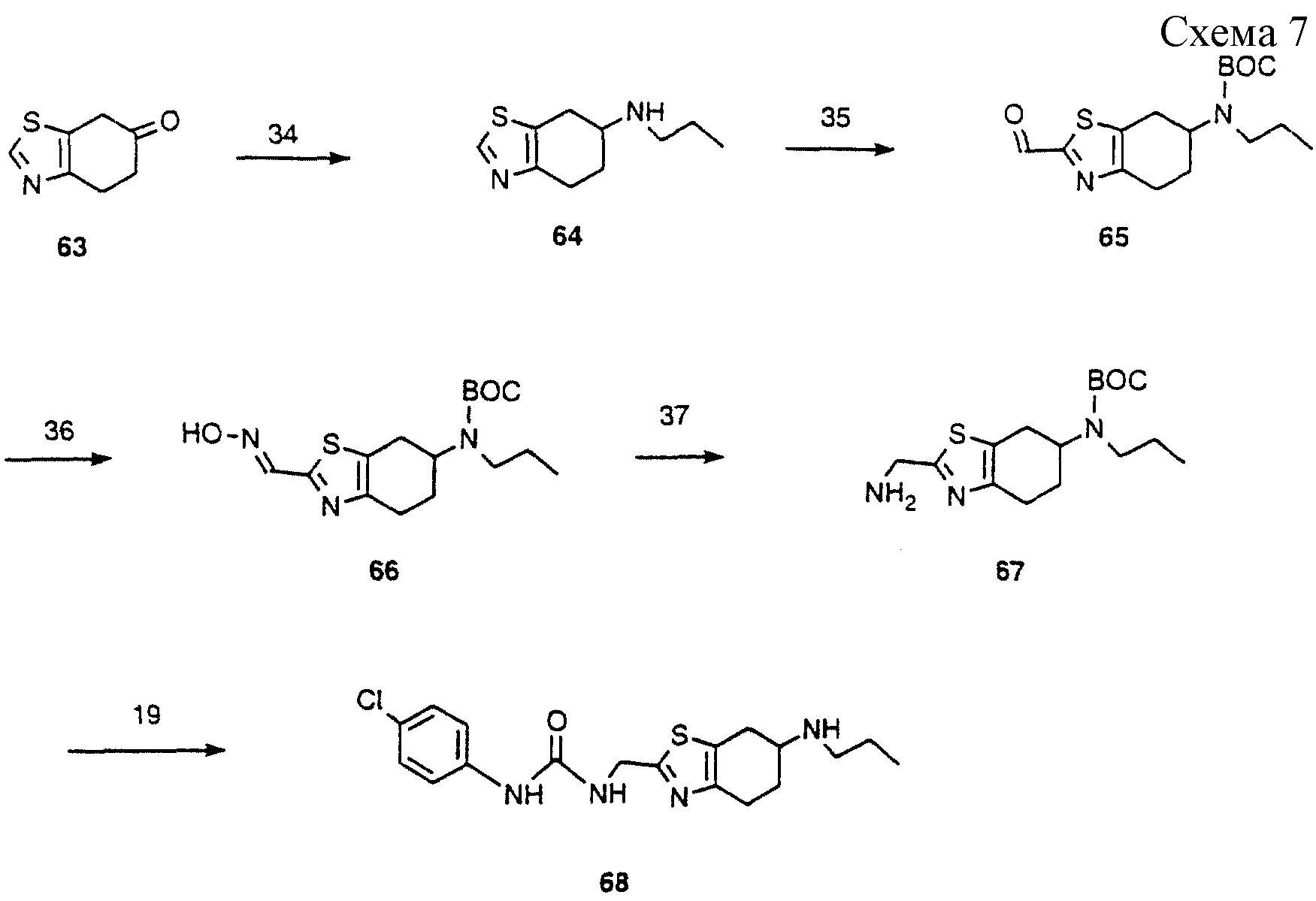
Схема 7: (Аналоги формулы III)
Кетон 63 (Helv. Chim. Acta 1994, 1256) подвергали восстановительному аминированию цианоборогидридом натрия в присутствии уксусной кислоты и пропанола с получением 64. Этот амин защищали
ВОС-группой и соединение обрабатывали н-бутиллитием при низкой температуре с последующим гашением диметилформамидом, получая альдегид 65. Этот альдегид конденсировали с гидрохлоридом гидроксиламина,
получая 66. Оксим 66 восстанавливали сплавом Davarda (сплав содержит 50% меди, 45% алюминия, 5% цинка) с получением амина 67. Этот амин обрабатывали 4-хлорфенилизоцианатом с получением мочевины 68.
Схема 8: (Аналоги формулы I)
Гидрокситрифлат 69 (патент 4714) алкилировали различными алкилгалогенидами с использованием гидрида натрия в ДМФ с получением промежуточных продуктов
70-73 (методика 38). Эти трифлатные промежуточные продукты превращали карбонилированием с использованием ацетата палладия и 1,3-бис(дифенилфосфинопропана) в атмосфере монооксида углерода (методика 39),
получая метиловые эфиры как промежуточные продукты 74-77. Эти метиловые эфиры как промежуточные продукты превращали в любой из первичных карбоксамидных продуктов 78-81 с использованием формамида и
метоксида натрия (методика 40) или превращали в алкилкарбоксамидные продукты 82-84 с использованием подходящих замещенных формамидов и метоксида натрия (методика 41). Первичный карбоксамид 81 далее
превращали с использованием гидрида натрия и алкилгалогенида (методика 42) в алкилкарбоксамидный продукт 85.
Сложный гидроксиэфир 86 (патент 4714) превращали в карбоксамид 87 с использованием метоксида натрия и формамида (методика 43) с последующим алкилированием с использованием карбоната калия и алкилгалогенидов (методика 44) с получением 88-90.
Схема 9:
(Аналоги формулы I)
Сложный диметиловый эфир 91 (патент 4714) гидролизовали в дикарбоновую кислоту 92 с использованием водной смеси NaOH/-MeOH (методика 45). Эту дикислоту затем
конденсировали и циклизовали ацетатом аммония и НСl в уксусной кислоте, получая 93 (методика 46), или, альтернативно, конденсировали с различными аминами в уксусной кислоте, получая 95-124 (методика
49). Продукты 93, 95-124 восстанавливали в соответствующие производные лактама 94, 125-133 системой цинк/уксусная кислота (методика 47). Промежуточные продукты 93 и 94 также алкилировали различными
замещенными галогенидами с получением 95-124 и 125-133 (методика 48 и 50).
Схема 10: (Аналоги формулы I)
Дикарбоновую кислоту 92 конденсировали с различными гидразинами в
уксусной кислоте с получением 134-135 (методика 51).
Следующие методики 1-9 из схемы 1 можно использовать для получения соединений примеров 1-6 данного изобретения (за исключением соединения примера 5, которое было неактивно в скрининге на допамин).
Методика 1: (R)-4-Бром-α-[(метоксикарбонил)амино]-γ-оксобензолбутановая кислота, 2
Смесь
бромбензола (373 г) и (R)-2-карбометоксиамино-янтарного ангидрида (1) (90,51 г) в дихлорметане (260 мл) охлаждали во льду и в течение 1 минуты добавляли хлорид алюминия (174,34 г) (реакция
экзотермическая!). Темно-красную смесь перемешивали при 0oС в течение 30 минут и при комнатной температуре в течение 1 часа. Смесь выливали в измельченный лед и медленно, при перемешивании
добавляли концентрированную хлористоводородную кислоту. Добавляли диэтиловый эфир и смесь перемешивали до тех пор, пока не исчезал весь темно-красный материал. Слои разделяли и водный слой
экстрагировали более двух раз диэтиловым эфиром. Объединенные эфирные экстракты промывали водой и экстрагировали водным карбонатом натрия. Объединенные экстракты промывали диэтиловым эфиром, охлаждали
во льду и подкисляли концентрированной хлористоводородной кислотой. Кислоту экстрагировали 3 раза диэтиловым эфиром. Объединенные экстракты промывали солевым раствором и сушили (MgSO4).
Растворитель удаляли в вакууме, получая указанное в заголовке соединение в виде пены. 149,6 г. [α]D=-41o(25oС, СН3ОН, с=1,0352).
Методика 2: (R)-4-Бром-α-[(метоксикарбонил)амино] бензолбутановая кислота, 3
Раствор (R)-4-Бром-α-[(метоксикарбонил)амино]-γ-оксобензолбутановой кислоты (2) (74,35 г) в
дихлорметане и три-этилсилане (182 мл) охлаждали до 0oС и в течение периода 15 минут при перемешивании по каплям добавляли тетрахлорид титана (99,0 мл). Через 5,5 часов добавляли
триэтилсилан (72 мл) и смесь перемешивали при комнатной температуре в течение 17 часов и при кипячении с обратным холодильником на паровой бане в течение 3 часов. Смесь охлаждали и выливали на лед.
Смесь экстрагировали два раза диэтиловым эфиром. Объединенные эфирные экстракты промывали водой и экстрагировали 3 раза порциями по 250 мл 10% раствора карбоната натрия. Объединенные экстракты
промывали диэтиловым эфиром, охлаждали во льду и подкисляли концентрированной хлористоводородной кислотой. Осадок фильтровали, промывали водой и сушили в вакууме, получая белый твердый продукт.
Кристаллизация из смеси этилацетат/гексан давала указанное в заголовке соединение в виде белых кристаллов (52,7 г, 74%, т.пл. 136-137oС).
[α]D=-12o(25oС, СН3ОН, с=0,8709).
Методика 3: Метил-(R)-(7-бром-1,2,3,4-тетрагидро-1-оксо-2-нафталинил)карбамат, 4
Суспензию (R)-4-бром-α
-[(метоксикарбонил)амино]бензол-бутановой кислоты (3) (97,0 г) в дихлорметане охлаждали во льду и добавляли диметилформамид (1,2 мл) и оксалилхлорид (28,1 мл). Смесь перемешивали при 0oС в
течение 5 минут и при комнатной температуре в течение 1,5 часов. Раствор охлаждали до -25oС и в течение 12 минут по частям добавляли хлорид алюминия (86,4 г). Смесь перемешивали при -20oС в течение 40 минут и выливали при перемешивании на смесь льда, 10% хлористоводородной кислоты (300 мл) и хлороформа (100 мл). Смесь экстрагировали два раза диэтиловым простым эфиром и
экстракты промывали водой, насыщенным раствором бикарбоната натрия и солевым раствором. Раствор сушили (MgSO4) и растворитель удаляли в вакууме, оставляя светло-желтый твердый продукт (91,
76 г). Кристаллизация из метанола давала бесцветные кристаллы, которые фильтровали, промывали гексаном и сушили в вакууме (69,9 г, 76%, т.пл. 116-117oС). Фильтрат выпаривали и остаток
кристаллизовали из метанола, получая дополнительное количество указанного в заголовке соединения в виде не совсем белого твердого продукта (6,81 г, т.пл. 111-112oС). [α]D
=+43o(25oС, СН3ОН, с=0,8143).
Методика 4: Метил-(R)-(7-бром-1,2,3,4-тетрагидро-2-нафталинил)-карбамат, 5
В трехгорлую колбу на 2 л, снабженную
механической мешалкой, капельной воронкой на 125 мл и вводным отверстием для N2, помещали раствор метил-(R)-(7-бром-1,2,3,4-тетрагидро-1-оксо-2-нафталинил) карбамата (4) (68,74 г) в
дихлорметане. Добавляли триэтилсилан (147 мл) и смесь охлаждали во льду. Через капельную воронку в течение 10 минут добавляли тетрахлорид титана (76,2 мл) и смесь перемешивали при комнатной
температуре в течение 24 часов. Добавляли триэтилсилан (18,5 мл) и смесь перемешивали в течение дополнительных 2 часов. Смесь выливали на лед, слои разделяли и водный слой экстрагировали два раза
дихлорметаном. Объединенные экстракты промывали два раза 5% хлористоводородной кислотой, один раз водой и один раз 5% раствором гидроксида натрия. Раствор сушили (MgSO4) и растворитель
удаляли в вакууме, получая масло, которое частично кристаллизовалось (122,9 г). Смесь разбавляли гексаном, охлаждали во льду и фильтровали, получая белый твердый продукт (60,88 г). Кристаллизация из
смеси этилацетат/гексан давала указанное в заголовке соединение в виде бесцветных кристаллов (52,69 г, 0,185 моль, 80,3%, т. пл. 99-100,5oС). Получали вторую порцию продукта (5,29 г).
Анализ хиральной ВЭЖХ; (колонка с хирагелем OD, Daicel Chem.Ind., LTD; 10% изопропанол в гексане; скорость потока 1 мл/мин; λ=215, колонка 25 см • 4,6 мм внутренний диаметр) показывает 3,
26 мин (1,5%), 8,57 мин (1,7%), 10,66 мин (96.9%), 13,97 мин (1,6%). Рацемат показывает 3,27 мин (1,2%), 8,66 мин (0,3%), 10,75 мин (48,9%), 14,30 мин (49,4%), 25,79 мин (0,2%).
[α]D=+74o(25oС, СН3ОН, с=0,88884).
Методика 5: (Z)-2-бутендиоат(R)-7-бром-1,2,3,4-тетрагидро-2-нафталинамина (1:1), 6
Метил-(R)-(7-бром-1,2,3,4-тетрагидро-2-нафталинил)карбамат (5) (51,62 г), гидроксид калия (61,2 г), воду (150 мл) и этанол (350 мл) нагревали при кипячении с обратным холодильником в течение двух
дней. Этанол удаляли в вакууме и остаток распределяли между водой и смесью 2:1 диэтиловый простой эфир/тетрагидрофуран. Водный слой экстрагировали снова тем же самым растворителем и объединенные
экстракты промывали солевым раствором и сушили (MgSO4). Растворитель удаляли в вакууме, оставляя масло (37,9 г). Образец соединения (15,28 г) смешивали с малеиновой кислотой (7,85 г) и
смесь кристаллизовали из смеси метанол/диэтиловый эфир, получая указанное в заголовке соединение в виде бесцветных кристаллов (19,24 г, т.пл. 184-184,5oС).
[α]D=+40o(25oС, СН3OH, с=0,7756). Получали вторую порцию продукта (2,05 г).
Методика 6: 4-Метилбензолсульфонат (R)-7-бром-1,2,3,4-тетрагидро-N,
N-дипропил-2-нафталинамина 7
Смесь (R)-2-амино-3-бром-1,2,3,4-тетрагидронафталина (6) (22,62 г) 1-бромпропана (36,4 мл) и карбоната калия (41,5 г) в ацетонитриле перемешивали при кипячении с
обратным холодильником в течение 16 часов. Добавляли 1-бромпропан (20 мл) и кипячение с обратным холодильником продолжали в течение 8 часов. Смесь распределяли между водой и диэтиловым простым эфиром.
Водный слой снова экстрагировали диэтиловым эфиром и объединенные эфирные экстракты промывали солевым раствором и сушили (MgSO4). Растворитель удаляли в вакууме, получая масло (29,18 г).
Очистка флэш-хроматографией (силикагель 230-400 меш. этилацетат/гексан) давала масло (26,4 г, 85%). Образец (0,997 г) смешивали с гидратом п-толуолсульфоновой кислоты (0,62 г) и кристаллизовали из
смеси метанол/диэтиловый эфир, получая указанное в заголовке соединение в виде бесцветных кристаллов (1,39 г, т.пл. 182-183,5oС). [α]D=+48o(25oС,
СН3ОН, с=0,9834).
Методика 7: (R)-7-(Дипропиламино)-5,6,7,8-тетрагидро-2-нафталинкарбоксамид, 8
Раствор (R)-2-(дипропиламино)-7-бром-1,2,3,4-тетрагидронафталина (7)
(13,02 г) в сухом тетрагидрофуране охлаждали до -78oС. Через шприц в течение 6 минут добавляли трет-бутиллитий (1,7 М в пентане, 50,6 мл) и смесь перемешивали в течение дополнительных 8
минут. В виде одной дозы добавляли триметилсилилизоцианат (13,4 мл, 85% чистота, 84,1 ммоль) и смесь перемешивали при -78oС в течение 10 минут и при комнатной температуре в течение 1,5
часа. Реакционную смесь гасили 10% хлористоводородной кислотой и после перемешивания в течение 30 минут подщелачивали 15% гидроксидом натрия. Свободное основание экстрагировали диэтиловым эфиром и
смесью 1:1 тетрагидрофуран/диэтиловый эфир. Объединенные экстракты промывали солевым раствором и сушили (MgSO4). Растворитель удаляли в вакууме, получая масло (15,25 г). Кристаллизация из
смеси этилацетат/гексан давала не совсем белые кристаллы (6,30 г), т.пл. 132oС.
[α]D =+67o(25oС, СН3OH, с=0,8139).
Методика 8: (R)-7-(Дипропиламино)-5,6,7,8-тетрагидро-2-нафталинметанамин, 9
Раствор (R)-7-(дипропиламино)-5,6,7,8-тетрагидронафталинкарбоксамида (8) (8,68 г) в сухом
тетрагидрофуране перемешивали при комнатной температуре и медленно добавляли комплекс борана и диметилсульфида (10,0 М, 11,1 мл). Когда интенсивность начальной реакции ослабевала, смесь кипятили с
обратным холодильником в течение 2 дней. Смесь охлаждали во льду и по каплям добавляли воду. Когда выделение газа прекращалось, добавляли 10% хлористоводородную кислоту (75 мл) и смесь кипятили с
обратным холодильником в течение 2 часов. Смесь охлаждали во льду и подщелачивали твердым гидроксидом натрия. Смесь экстрагировали диэтиловым эфиром и затем смесью 2:1 диэтиловый эфир/тетрагидрофуран.
Объединенные экстракты промывали солевым раствором и сушили (MgSO4). Растворитель удаляли в вакууме, получая указанное в заголовке соединение в виде желтого масла (8,15 г), которое
использовали без дальнейшей очистки.
ПРИМЕР 1: Методика 9: (R)-N-[[7-(Дипропиламино)-5,6,7,8-тетрагидро-2-нафталинил]метил]метансульфоиамид, 10
Раствор сырого
(R)-7-(дипропиламино)-5,6,7,8-тетрагидро-2-нафталинметанамина (9) (0,521 г) и триэтиламина (0,30 мл) в тетрагидрофуране охлаждали до 0oС. По каплям при перемешивании добавляли
метансульфонилхлорид (0,16 мл). Смесь перемешивали при комнатной температуре в течение 1,5 часа и гасили 10% раствором карбоната натрия. Смесь экстрагировали два раза диэтиловым эфиром и объединенные
экстракты промывали солевым раствором и сушили (MgSO4). Растворитель удаляли в вакууме, оставляя масло (0,63 г). Очистка флэш-хроматографией (этилацетат/гексан) давала указанное в заголовке
соединение в виде бесцветного масла (0,47 г).
[α]D=+58o(25oC, СН3ОН, с=0,5221).
ПРИМЕР 2:
(R)-4-Циано-N-[[7-(дипропиламино)-5,6,7,8-тетрагидро-2-нафталинил)метил]бензолсульфонамид, 11
Используя методику 9, раствор сырого (R)-7-(дипропиламино)-5,6,7,
6-тетрагидро-2-нафталинметанамина (9) обрабатывали 4-цианофенилсульфонилхлоридом. Очистка хроматографией давала вещество, которое кристаллизовали из смеси этилацетат/гексан, получая 11 в виде
бесцветных кристаллов (т.пл. 94-95,5o). [α]D=+46o(25oС, CH3OH, с=0,7967).
ПРИМЕР 3: (R)-4-Хлор-N-[[7-(дипропиламино)-5,6,7,
8-тетрагидро-2-нафталинил]метил]бензолсульфонамид, 12
Используя методику 9, (R)-7-(дипропиламино)-5,6,7,8-тетрагидро-2-нафталинметанамин (9) обрабатывали 4-хлорбензолсульфонилхлоридом.
Очистка хроматографией давала твердый продукт, который кристаллизовали из гексана, получая указанное в заголовке соединение в виде бесцветных кристаллов (т.пл. 72o). [α]D
=+47o(25oС, СН3ОН, с =0,6095).
ПРИМЕР 4: (R)4-Нитро-N-[[7-(Дипропиламино)-5,6,7,8-тетрагидро-2-нафталинил]метил]бензолсульфонамид, 13
Используя методику 9, (R)-7-(дипропиламино)-5,6,7,8-тетрагидро-2-нафталинметанамин (9) и триэтиламин в сухом тетрагидрофуране охлаждали до 0oС и добавляли 4-нитробензолсульфонилхлорид.
После экстракции, очистки флэш-хроматографией (этилацетат/гексан) получали твердый продукт, который кристаллизовали из гексана, содержащего небольшое количество этилацетата, получая 13 в виде желтых
кристаллов (т. пл. 105o). [α]D =+49o(25oС, СН3ОН, с= 0,9425).
ПРИМЕР 5: (R)-3-Циано-N-[[7-(дипропиламино)-5,6,7,
8-тетрагидро-2-нафталинил]метил]бензолсульфонамид, 14 (неактивен при скрининге на допамин)
Используя методику 9, (R)-7-(дипропиламино)-5,6,7,8-тетрагидро-2-нафталинметанамин (9) и триэтиламин
в сухом тетрагидрофуране охлаждали до 0oС и добавляли 3-цианбензолсульфонилхлорид. После экстракции, очистки флэш-хроматографией (этилацетат/гексан) получали указанное в заголовке
соединение в виде масла.
[α]D =+41o(25oС, СН3ОН, с=1,0394).
ПРИМЕР 6: (R)-N-[[7-(Дипропиламино)-5,6,7,
8-тетрагидро-2-л]-1-метил-1-метил-1Н-имидазол-4-сульфонамид, 15
Используя методику 9, (R)-7-(дипропиламино)-5,6,7,8-тетрагидро-2-нафталинметанамин (9) и триэтиламин в сухом тетрагидрофуране
охлаждали до 15oС и добавляли 1-метилимидазол-4-сульфонилхлорид. После экстракции, очистки флэш-хроматографией (тетрагидрофуран/этилацетат) получали указанное в заголовке соединение в виде
масла.
[α]D =+46o(25oС, СН3OH, с=0,7458).
ПРИМЕР 7: Методика 10. (R)-Карбоксамидо-N-1[7-(дипропиламино)-5,6,7,
8-тетрагидро-2-нафталинил]метил]бензолсульфонамид, 16
(R)-4-Циано-N-[[7-(дипропиламино)-5,6,7,8-тетрагидро-2-нафталинил]метил] бензолсульфонамид (11) обрабатывали пероксидом водорода и
гидроксидом натрия в водном ТГФ. После завершения гидролиза раствор экстрагировали смесью простой эфир/вода. Эфирный слой сушили над сульфатом натрия и растворитель удаляли. Остаток хроматографировали
смесью этил-ацетат/гексан, получая 16 в виде твердого продукта.
ПРИМЕР 8: Методика 11. (R)-4-Хлор-N-[[7-(дипропиламино)-5,6,7,8-тетрагидро-2-нафталинил]метил]бензамид, 17
(R)-7-(Дипропиламино)-5,6,7,8-тетрагидро-2-нафталинметанамин 9 (0,521 г) и триэтиламин (0,30 мл) в тетрагидрофуране (6 мл) охлаждали до 0oС и добавляли 4-хлорбензоилхлорид (0,254 мл). Смеси
давали нагреться до комнатной температуры и перемешивали в течение 3 часов. Реакцию гасили 10% раствором карбоната натрия и смесь перемешивали в течение 30 минут. Смесь экстрагировали два раза смесью
1:1 тетрагидрофуран/диэтиловый эфир и объединенные органические слои промывали солевым раствором и сушили (MgSO4). Растворитель удаляли в вакууме, получая твердый продукт, который
кристаллизовали из этилацетата, получая указанное в заголовке соединение в виде бесцветных кристаллов (0,52 г, т.пл. 209,5oС). [α]D=+42o(25oC,
СНСl3, с=0,9118).
ПРИМЕР 9: (R)-4-Циано-N-[[7-(дипропиламино)-5,6,7,8-тетрагидро-2-нафталинил]метил]бензамид, 18 (Сравнительный пример для этого изобретения)
Используя методику 11, (R)-2-(дипропиламино)-7-амино-1,2,3,4-тетрагидронафталин 9 и триэтиламин в тетрагидрофуране охлаждали до 0oС и добавляли 4-цианобензоилхлорид. После экстракции и так
далее твердый продукт кристаллизовали из этилацетата, получая указанное в заголовке соединение в виде не совсем белых кристаллов (т.пл. 183oС).
[α]D =+50o(25oС, СН3ОН, с=0,9773).
Методика 12: (R)-2-(Дипропиламино)-7-амино-1,2,3,4-тетрагидронафталин, 19
(R)-2-(Дипропиламино)-7-бром-1,2,3,
4-тетрагидронафталин 7 (5,91 г) растворяли в сухом тетрагидрофуране (50 мл) в атмосфере азота и охлаждали до -78oС. В течение 5 минут добавляли трет-бутиллитий (1,7 М в пентане, 23,0 мл), и
смесь
перемешивали при -78oС в течение дополнительных 10 минут. Этот раствор добавляли через капилляр к раствору дифенилфосфорилазида (5,24 г) в тетрагидрофуране (30 мл) в течение
10 минут. Смесь перемешивали при -78oС в течение 2 часов и нагревали до -20oС в течение 45 минут. Смесь снова охлаждали до -78oС и в течение 5 минут добавляли
бис-(2-метоксиэтокси)алюминийгидрид (3,4 М в толуоле, 22,4 мл). После перемешивания при -78oС в течение дополнительных 10 минут смесь нагревали до 0oС и перемешивали в течение 45
минут и при комнатной температуре в течение 30 минут. Смесь осторожно гасили водой и насыщали хлоридом натрия. Амин экстрагировали два раза диэтиловым простым эфиром и объединенные экстракты сушили
(MgSO4). Растворитель удаляли в вакууме, получая масло (6,3 г). Очистка флэш-хроматографией давала 19 в виде светло-янтарного масла (2,56 г).
ПРИМЕР 10: Методика 13.
(R)-4-Хлор-N-[7-(дипропиламино)-5,6,7,8-тетрагидро-2-нафталинил]бензолсульфонамид, 20
п-Хлорбензолсульфонилхлорид (0,32 г) добавляли к раствору (R)-2-(дипропиламино)-7-амино-1,2,3,
4-тетрагидронафталина 19 (0,370 г) и триэтиламина (0,30 мл) в сухом тетрагидрофуране (4 мл). Смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили 10% раствором карбоната натрия
(5 мл) и смесь перемешивали в течение 10 минут. Смесь экстрагировали два раза диэтиловым эфиром и объединенные экстракты промывали солевым раствором и сушили (MgSO4). Растворитель удаляли в
вакууме, оставляя масло (0,63 г). Очистка флэш-хроматографией (этилацетат в гексане) давала 20 в виде янтарного масла, [α]D =+52o(25oС, СН3ОН, с=1,
0535).
ПРИМЕР 11: (R)-4-Циано-N-[7-(дипропиламино)-5,6,7,8-тетрагидро-2-нафталинил]бензолсульфонамид, 21
Используя методику 13 п-цианобензолсульфонилхлорид добавляли к
раствору сырого (R)-2-(дипропиламино)-7-амино-1,2,3,4-тетрагидронафталина 19 и триэтиламина в сухом тетрагидрофуране. После экстракции и так далее очистка флэш-хроматографией (этилацетат в гексане)
давала 21 в виде янтарного масла, которое можно было кристаллизовать в виде его фумаратной соли из смеси метанол/простой эфир (т. пл. 123oС, разлож. ). [α]D =+48o(25oС, СН3ОН, с= 1,0113).
Методика 14: трет-Бутил-(R)-(6-бром-1,2,3,4-тетрагидро-N-пропил-2-нафталинамин)карбамат, 23
Пропионилхлорид (18,5 мл)
добавляли к (R)-2-амино-7-бром-1,2,3,4-тетрагидронафталину (22) (J.Org.Chem. 1995, 4324) (43 г), триэтиламину (31 мл) и дихлорметану. Через 2 часа смесь концентрировали в вакууме, добавляли ТГФ и
снова концентрировали в вакууме. Добавляли воду, охлаждали на ледяной бане и твердый продукт фильтровали. Промывание водой и сушка в вакууме давала 51 г твердого продукта, т.пл. 169-171oС.
Комплекс борана и диметилсульфоксида (27 мл, 10 М) с этим амидом (51 г) в ТГФ кипятили с обратным холодильником в течение 24 часов. Добавляли воду, затем 2 н. хлористоводородную кислоту. Смесь
кипятили с обратным холодильником в течение часа и затем подщелачивали 15% водным гидроксидом натрия и экстрагировали метил-трет-бутиловым эфиром. Эфирный слой промывали водой и солевым раствором и
сушили сульфатом натрия. Растворитель удаляли в вакууме, получая 48 г темного масла. Этот амин и ди-трет-бутилдикарбонат (44 г) смешивали в ТГФ. Через 60 минут добавляли воду (150 мл) и каталитическое
количество 4-диметиламинопиридина. Через 15 часов смесь распределяли между водой и метил-трет-бутиловым эфиром. Эфирный слой промывали 2 н. хлористоводородной кислотой, водой, насыщенным водным
бикарбонатом натрия и солевым раствором и сушили сульфатом натрия. Растворитель удаляли в вакууме, и кристаллизация остатка из гексана давала 59 г 23 в виде твердого продукта, т.пл. 67-69o
С.
Методика 15: трет-Бутил-(R)-(6-аминометил-1,2,3,4-тетрагидро-N-пропил-2-нафталинамин)карбамат, 24
Трет-бутиллитий (1,7 М в пентане) (64 мл) добавляли к
трет-бутил-(R)-(6-бром-1,2,3,4-тетрагидро-N-пропил-2-нафталинамин)-карбамату (23) (20 г) в сухом ТГФ (125 мл) при -78oС. Через 10 минут добавляли сухой N,N-диметилформамид (8 мл) и холодную
баню убирали. Через 90 минут раствор распределяли между водой и простым эфиром. Эфирный слой промывали водой и солевым раствором и сушили над сульфатом натрия. Растворитель удаляли в вакууме, получая
17 г твердого продукта, т.пл. 88-91oС. К этому альдегиду (14 г) в метаноле по частям добавляли борогидрид натрия (1,7 г) при охлаждении на ледяной бане. Через два часа добавляли воду и
раствор концентрировали в вакууме. Остаток распределяли между водой и простым эфиром. Эфирный слой промывали водой и солевым раствором и сушили сульфатом натрия. Растворитель удаляли в вакууме,
получая 14 г бензилового спирта в виде твердого продукта, т.пл. 113-114oС.
[α]D=+53o(25oС, СН3ОН, с=0,96). К бензиловому спирту (13,6 г) в ТГФ при 0oС медленно добавляли тионилхлорид (3,3 мл) и ледяную баню убирали. Через час добавляли водный бикарбонат натрия и смесь экстрагировали простым эфиром. Эфирный слой промывали водой и солевым раствором и сушили сульфатом натрия. Растворитель удаляли в вакууме и остаток хроматографировали на силикагеле с использованием смеси дихлорметан/гексан, получая 9,6 г бензилхлорида в виде твердого продукта, т.пл. 90-93oС. [α]D=+50o(25oС, СН3OH, с= 0,94). Азид натрия (8,7 г) и бензилхлорид (9,0 г) нагревали при 45oС в ДМФ в течение 18 часов и затем распределяли между смесью простой эфир/ТГФ и водой. Эфирный слой промывали водой и солевым раствором и сушили над сульфатом натрия. Растворитель удаляли в вакууме, получая 9,2 г азида в виде твердого продукта, т.пл. 73,5-75,0oС. [α]D=+50o(25oC, СН3ОН, с=1,00). Азид (9,1 г) и палладий на угле (0,5 г) встряхивали в ТГФ при давлении водорода 3,164 ат (45 psi) в течение 3 часов. Смесь фильтровали через диатомовую землю, и растворитель удаляли в вакууме, получая 7,9 г 24 в виде твердого продукта. Аналитический образец кристаллизовали из гексана, т. пл. 88-89oС. [α]D= +53o(25oС, СН3ОН, с=1,00).
ПРИМЕР 12: Методика 16; (R)-4-Хлор-N-[[5,6,7,8-тетрагидро-6-(пропиламино)-2-нафталинил]метил]бензолсульфонамид, 25
Трет-бутил-(R)-(6-аминометил-1,2,3,
4-тетрагидро-N-пропил-2-нафталинамин)карбамат (24) (0,50 г), триэтиламин (0,44 мл), 4-хлорбензолсульфонилхлорид (0,36 г) и ТГФ перемешивали в течение 3 часов. Смесь распределяли между водным
бикарбонатом натрия и смесью простой эфир/ТГФ. Эфирный слой промывали солевым раствором и сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на силикагеле
(дихлорметан/этилацетат/гексан), получая 0,77 г твердого продукта, т.пл. 117-121oС. [α]D=+32o(25oС, СН3ОН, с= 0,78). Для удаления
ВОС-защитной группы этот твердый продукт перемешивали с трифторуксусной кислотой (5 мл) в течение 90 минут и растворитель удаляли в вакууме. Смесь распределяли между водным бикарбонатом натрия и
смесью простой эфир/ТГФ. Эфирный слой промывали солевым раствором и сушили сульфатом натрия. Растворитель удаляли и получали 0,53 г кристаллического 25, т. пл. 145-147oС.
ПРИМЕР 13: (R)-4-[3,5-Диметил-N-[[5,6,7,8-тетрагидро-6-(пропиламино)-2-нафталинил]метил]изоксазолсульфонамид, 26
С использованием методики 16 трет-бутил-(R)-(6-аминометил-1,2,3,
4-тетрагидро-N-пропил-2-нафталинамин)карбамат (24) обрабатывали 3,5-диметилизоксазолсульфонилхлоридом, получая кристаллический 26 в виде его соли трифторуксусной кислоты после удаления защитной
ВОС-группы.
ПРИМЕР 14: Методика 17. (R)-4-Хлор-N-[[5,6,7,8-тетрагидро-6-(дипропиламино)-2-нафталинил]метил]бензолсульфонамид, 27
Триацетоксиборогидрид натрия (0,20 г)
добавляли к (R)-4-хлор-N-[[5,6,7,8-тетрагидро-6-(пропиламино)-2-нафталинил] метил]-бензолсульфонамиду (25) (0,29 г), пропиональдегиду (0,07 мл), ледяной уксусной кислоте (0,05 мл) и дихлорметану.
Через 3,5 часа смесь распределяли между водным бикарбонатом натрия и смесью простой эфир/ТГФ. Эфирный слой промывали водой и солевым раствором и сушили сульфатом натрия. Растворитель удаляли в вакууме,
получая 0,31 г соединения 27 в виде твердого продукта, т.пл. 101-103oС.
[α]D=+42o(25oС, СН3ОН, с=0,93).
ПРИМЕР 15: Методика 18. (R)-4-Хлор-N-[[5,6,7,8-тетрагидро-6-(пропиламино)-2-нафталинил)метил]бензамид, 28
Трет-бутил-(R)-(6-аминометил)-1,2,3,4-тетрагидро-N-пропил-2-нафталинамин)карбамат
(24) (0,50 г), триэтиламин (0,44 мл), 4-хлорбензоилхлорид (0,21 мл) и ТГФ перемешивали в течение 2,5 часов. Смесь распределяли между водным бикарбонатом натрия и смесью простой эфир/ТГФ. Эфирный слой
промывали солевым раствором и сушили сульфатом натрия. Растворитель удаляли и остаток хроматографировали на силикагеле (дихлорметан/этилацетат/гексан), получая 0,56 г твердого продукта, т.пл.
154-155oС. [α]D=+38o(25oС, СН3ОН, с=0,86). Для удаления ВОС-защитной группы этот твердый продукт перемешивали с трифторуксусной кислотой
(5 мл) в течение 60 минут и растворитель удаляли в вакууме. Остаток распределяли между водным бикарбонатом натрия и смесью простой эфир/ТГФ. Эфирный слой промывали водой и затем сушили сульфатом
натрия. Растворитель удаляли в вакууме, получая 0,34 г соединения 28 в виде твердого продукта, т.пл. 147-148oС.
[α]D=+49o(25oС,
СН3OH, с=0,87)
ПРИМЕР 16: (R)-2-Ацетил-N-[[5,6,7,8-тетрагидро-6-(дипропиламино)-2-нафталинил)метил]бензамид, 29
Используя методику 18 трет-бутил-(R)-(6-аминометил)-1,2,3,
4-тетрагидро-N-пропил-2-нафталинамин) карбамат (24) (0,3 г) можно обработать подходящим количеством 2-ацетилбензоилхлорида, получая белый твердый продукт, т.пл. 99oС. [α]D
= +35o(25oС, СН3ОН, с=0,95). Его перемешивали с трифторуксусной кислотой (5 мл) в течение 90 минут и растворитель удаляли в вакууме. Растирание с сухим диэтиловым
эфиром давало 0,28 г кристаллического 29 в виде его соли трифторуксусной кислоты, т.пл. 153-156oС. [α]D =+45o(25oC, СН3ОН, с= 0,86).
ПРИМЕР 17: (R)-4-Хлор-N-[[5,6,7,8-тетрагидро-6-(дипропиламино)-2-нафталинил]метил]бензамид, 30
Используя методику 17 (R)-4-хлор-N-[[5,6,7,
8-тетрагидро-6-(пропиламино)-2-нафталинил] метил] бензамид (28) превращали в 30 в виде твердого продукта, т.пл. 141-142oС. [α]D=+43o(25oС, СН3ОН, с=0,90).
ПРИМЕР 18: Методика 19. (R)-N-(4-Ацетил фенил)-N'-[[5,6, 7,8-тетрагидро-6-(пропиламино)-2-нафталинил]метил]мочевина, 31
Трет-бутил-(R)-(6-аминометил-1,2,3,
4-тетрагидро-N-пропил-2-нафталинамин)карбамат (24) (0,30 г), 4-ацетилфенилизоцианат (0,16 г) и ТГФ перемешивали в течение 6 часов. Смесь распределяли между водным бикарбонатом натрия и смесью простой
эфир/ТГФ. Эфирный слой промывали солевым раствором и сушили сульфатом натрия. Растворитель удаляли и остаток хроматографировали на силикагеле (дихлорметан/этилацетат/гексан), получая 0,35 г твердого
продукта, т. пл. 77-88oС. [α]D=+34o(25oС, СН3ОН, с=0,83). Его перемешивали с трифторуксусной кислотой (5 мл) в течение 90 минут и
растворитель удаляли в вакууме. Растирание с сухим диэтиловым эфиром давало 0,30 г кристаллического 31 в виде его соли трифторуксусной кислоты, т.пл. 200oС (разложение).
ПРИМЕР 19: (R)-N-(4-Хлорфенил)-N'-[[5,6,7,8-тетрагидро-6-(пропиламино)-2-нафталинил]метил]мочевина, 32
С использованием методики 19 трет-бутил-(R)-(6-аминометил-1,2,3,
4-тетрагидро-N-пропил-2-нафталинамин)карбамат (24) (0,70 г) обрабатывали 4-хлорфенилизоцианатом (0,36 г), затем трифторуксусной кислотой, получая 0,59 г кристаллического 32 в виде его соли
трифторуксусной кислоты, т.пл. 192oС (разложение).
ПРИМЕР 20: (R-N-(4-Нитрофенил)-N'-[[5,6,7,8-тетрагидро-6-(пропиламино)-2-нафталинил]метил)мочевина, 33
С
использованием методики 19 трет-бутил-(R)-(6-амино метил-1,2,3,4-тетрагидро-N-пропил-2-нафталинамин)карбамат (24) (0,30 г) обрабатывали 4-нитрофенилизоцианатом (0,17 г), затем трифторуксусной кислотой
с последующим подщелачиванием и экстракцией, получая 33 в виде твердого продукта.
Методика 20: 4-(Трифторацетиламино)гептан-1,6-диин, 35
Триэтиламин (19,5 г, 0,193 моль)
добавляли к раствору 2-(пропин-2-ил)-4-пентиновой кислоты (34, 25,0 г, 0,184 моль; J.Chem.Soc.Perkin Trans, I, 1215-1224 (1986)) в толуоле (200 мл) при охлаждении. Добавляли дифенилфосфорилазид (50,3
г, 0,184 моль) и смесь перемешивали при комнатной температуре в течение 15 минут и нагревали на паровой бане до прекращения выделения газа. Добавляли сухой трет-бутанол (150 мл) и смесь нагревали на
паровой бане в течение 24 часов. Растворитель удаляли в вакууме и смесь разбавляли водой и экстрагировали два раза диэтиловым эфиром. Объединенные экстракты промывали два раза 10% раствором карбоната
натрия и один раз солевым раствором. Раствор сушили (MgSO4), и растворитель удаляли в вакууме, оставляя твердый продукт. Очистка флэш-хроматографией на силикагеле с элюированием смесью
этилацетат/гексан давала белый твердый продукт, который кристаллизовали из гексана, получая 4- (трет-бутилоксикарбониламино)гептан-1,6-диин в виде бесцветных кристаллов (т.пл. 64-67oС).
4-(трет-Бутилоксикарбониламино)гептан-1,6-диин (30,0 г, 0,145 моль) охлаждали во льду и добавляли трифторуксусную кислоту (80 мл). Происходило энергичное выделение газа. Смесь перемешивали при комнатной температуре в течение 30 минут и избыток трифторуксусной кислоты удаляли в вакууме. Остаток распределяли между диэтиловым эфиром и водой и водный слой экстрагировали 10% хлористоводородной кислотой. Объединенные водные экстракты охлаждали на льду и подщелачивали твердым гидроксидом натрия и насыщали хлоридом натрия. Свободное основание экстрагировали три раза диэтиловым эфиром. Объединенные экстракты промывали солевым раствором и сушили (MgSO4). Растворитель удаляли в вакууме, оставляя 4-аминогептан-1,6-диин в виде бледно-розового масла.
Раствор 4-аминогептан-1,6-диина (14,63 г, 0,137 моль) и триэтиламина (20,8 г, 0,206 моль) в сухом тетрагидрофуране (100 мл) охлаждали на льду и при перемешивании в течение 30 минут добавляли трифторуксусный ангидрид (37,5 г, 0,178 моль). Смесь перемешивали при 0oС в течение 1 часа и затем оставляли при -15oС в течение ночи. Смесь перемешивали на ледяной бане и по каплям добавляли воду (100 мл). Слои разделяли и водный слой экстрагировали диэтиловым эфиром. Объединенные органические экстракты промывали 10% хлористоводородной кислотой, насыщенным раствором бикарбоната натрия (2Х) и солевым раствором. Раствор сушили (MgSO4) и растворитель удаляли в вакууме, оставляя твердый продукт. Кристаллизация из смеси гексан/этилацетат давала 35 в виде бледно-желтых кристаллов (т.пл. 55-57oС).
Методика 21: N-[5,6-Бис(ацетилокси)метил]-2,3-дигидро-1Н-инден-2-ил]-2,2,2-трифторацетамид, 36
По методике Magnus et al.
(Tetrahed. Lett., 34, 23-26 (1993), раствор диацетата 2-бутин-1,4-диола (34,03 г, 0,200 моль; Syn. Comm., 9, 789-797 (1979)) и трис(трифенилфосфин)-родийхлорида (2,78 г, 3,00 ммоль) в насыщенном
аргоном абсолютном этаноле (100 мл) кипятили с обратным холодильником и через шприц с насосом в течение 2,5 часов добавляли раствор 4-(трифторацетиламино)гептан-1,6-диина (35, 20,32 г, 0,100 моль) в
насыщенном аргоном абсолютном этаноле (70 мл). Смесь перемешивали в атмосфере аргона при 75-80oС в течение 8 часов и затем при комнатной температуре в течение 10 часов, и растворитель
удаляли в вакууме, получая темное масло. Очистка флэш-хроматографией на силикагеле при элюировании смесью этилацетат/гексан давала светло-янтарный твердый продукт. Кристаллизация из смеси
этилацетат/гексан давала 36 в виде рыжевато-коричневых кристаллов (т.пл. 98-100oC).
Методика 22:2-(Дипропиламино)-5,6-бис(гидроксиметил)индан, 37
Гидроксид калия
(10,10 г, 0,180 моль) в воде (35 мл) добавляли к раствору N-[5,6-бис(ацетилокси)метил] -2,3-дигидро-1Н-инден-2-ил]-2,2,2-трифторацетамида (36, 20,1 г, 53,8 ммоль) в метаноле (200 мл) и кипятили с
обратным холодильником в течение 2,5 часа. Растворитель удаляли в вакууме, получая полутвердый продукт. Добавляли 1-бромпропан (27,1 г, 0,220 моль), карбонат калия (22,32 г, 0.162 моль) и ацетонитрил
(100 мл) и смесь перемешивали при кипячении с обратным холодильником на паровой бане в течение 17 часов. Снова добавляли 1-бромпропан (6,8 г, 0,055 моль) и кипячение с обратным холодильником
продолжали в течение 4 часов. Смесь разбавляли этилацетатом и промывали водой и солевым раствором и раствор сушили (MgSO4). Растворитель удаляют в вакууме, оставляя коричневое масло.
Очистка флэш-хроматографией на силикагеле с элюированием смесью тетрагидрофуран/этилацетат давала твердый продукт. Кристаллизация из смеси этилацетат/гексан давала 37 в виде белых кристаллов (т.пл.
111-113oС).
Методика 23: 1,2,3,5,6,7-Гексагидро-N,N-дипропилциклопент[f] изоиндол-6-амин, 38
Тионил (20 мл) добавляли к 2-(дипропиламино)-5,
6-бис(гидроксиметил)индану (37, 5,55 г, 20,0 ммоль) при перемешивании и смесь кипятили с обратным холодильником на паровой бане в течение 1,75 часа. Избыток тионилхлорида удаляли в вакууме. Остаток
растворяли в хлороформе и растворитель удаляли в вакууме. Эту операцию повторяли, получая янтарный твердый продукт. Кристаллизация из смеси метанол/диэтиловый простой эфир давала гидрохлорид
2-(дипропиламино)-5,6-бис(хлорметил)индана в виде не совсем белых кристаллов (т.пл. 208-210oC).
Аллиламин (9,3 г, 0,16 моль) добавляли к 2-(дипропиламино)-5, 6-бис(хлорметил)индану (3,41 г, 10,9 ммоль) при перемешивании. В результате проходила экзотермическая реакция, которую регулировали при помощи бани с холодной водой. Смесь перемешивали при комнатной температуре в течение 18 часов и затем кипятили с обратным холодильником на паровой бане в течение 4 часов. Избыток аллиламина удаляли в вакууме и остаток разбавляли 10% раствором карбоната натрия и экстрагировали два раза диэтиловым эфиром. Объединенные экстракты промывали солевым раствором и сушили (MgSO4). Растворитель удаляли в вакууме, оставляя янтарное масло. Очистка флэш-хроматографией на силикагеле с элюированием смесью тетрагидрофуран/этилацет давала 2-(пропен-2-ил)-1,2,3,5,6,7-гексагидро-N,N-дипропилциклопент[f]изоиндол-6-амин в виде янтарного масла.
Смесь 2-(пропен-2-ил)-1,2,3,5,6,7-гексагидро-N, N-дипропилциклопент[f] изоиндол-6-амина (7,9 г, 26,5 ммоль), N, N'-диметилбарбитуровой кислоты (12.41 г, 79,5 ммоль), ацетата палладия (0,297 г, 1,32 ммоль) и трифенилфосфина (0,695 г, 2,65 ммоль) в дихлорметане (75 мл) дегазировали аргоном и нагревали до 40oС в течение 5 часов. Растворитель удаляли в вакууме, остаток разбавляли 10% раствором карбоната натрия и экстрагировали два раза диэтиловым простым эфиром. Объединенные экстракты промывали 10% карбонатом натрия и экстрагировали два раза 10% раствором хлористоводородной кислоты. Образовывалась эмульсия, которую осветляли разбавлением водой и фильтрованием через диатомовую землю. Объединенные экстракты промывали диэтиловым эфиром и подщелачивали твердым гидроксидом натрия. Свободное основание экстрагировали три раза диэтиловым эфиром. Объединенные экстракты промывали солевым раствором и сушили (Na2SO4). Растворитель удаляли в вакууме, оставляя 38 в виде коричневого твердого продукта, который можно было кристаллизовать реакцией с гидратом п-толуолсульфоновой кислоты и получить серокоричневую соль (т.пл. 190-193oС).
Методика 21: 2-[(4-Хлорфенил)сульфонил] -1,2,3,5,6,7-гексагидро-N,N-дипропилциклопент[f]изоиндол-6-амин, 39
С использованием методики 9 сырой 1,2,3,5,6,7-гексагидро-N,
N-дипропилциклопент[f]изоиндол-6-амин (38, 0,42 г, 1,6 ммоль) обрабатывали 4-хлорбензолсульфонилхлоридом (0,343 г, 1,63 ммоль). Очистка флэш-хроматографией на силикагеле с использованием смеси
этилацетат/гексан и последующая кристаллизация из метанола давала 39 в виде белых кристаллов (т.пл. 152-153oС).
ПРИМЕР 22: 2-[(2-Хлорфенил)сульфонил]-1,2,3,5,6,
7-гексагидро-N,N-дипропилциклопент[f]изоиндол-6-амин, 40
Используя методику 9 сырой 1,2,3,5,6,7-гексагидро-N,N-дипропилциклопент[f]изоиндол-6-амин (38, 0,40 г, 1,5 ммоль) обрабатывали
2-хлорбензолсульфонилхлоридом (0,36 г, 1,7 ммоль). Очистка флэш-хроматографией на силикагеле с использованием смеси этилацетат/гексан и кристаллизация из метанола давала 40 в виде рыжевато-коричневых
кристаллов (т.пл. 89-91oС).
ПРИМЕР 23: 2-[(3-Хлорфенил)сульфонил] -1,2,3,5,6,7-гексагидро-N,N-дипpoпилциклoпeнт[f]изoиндoл-6-aмин, 41
Используя методику 9 сырой 1,
2,3,5,6,7-гексагидро-N,N-дипропилциклопент[f]изоиндол-6-амин (38, 0,60 г, 2,3 ммоль) обрабатывали 4-хлорбензолсульфонилхлоридом (0,54 г, 2,6 ммоль). Очистка флэш-хроматографией на силикагеле с
использованием этилацетата в гексане и кристаллизация из метанола давала 41 в виде рыжевато-коричневых кристаллов (т.пл. 113-114oС).
ПРИМЕР 24:
2-[(3-Цианофенил)сульфонил]-1,2,3,5,6,7-гексагидро-N,N-дипропилциклопент[f]изоиндол-6-амин, 42
Используя методику 9 сырой 1,2,3,5,6,7-гексагидро-N,N-дипропилциклопент[f]изоиндол-6-амин (38, 0,
40 г, 1,5 ммоль) обрабатывали 3-цианобензолсульфонилхлоридом (0,35 г, 1,7 ммоль). Очистка флэш-хроматографией на силикагеле с использованием этилацетата в гексане и кристаллизация из метанола давала
42 в виде рыжевато-коричневых кристаллов (т.пл. 134-135oС).
ПРИМЕР 25: 2-[3,5-Диметилизоксазолил)-4-сульфонил]-1,2, 3,5,6,7-гексагидро-N,
N-дипропилциклопент[f]изоиндол-6-амин, 43
Используя методику 9, 1,2,3,5,6,7-гексагидро-N,N-дипропилциклопент[f] изоиндол-6-амин (38) обрабатывали 3,5-диметилизоксазол-4-сульфонилхлоридом.
Кристаллизация из метанола давала 43 в виде серых кристаллов (т.пл. 113-114oС).
ПРИМЕР 26: 2-[(Бензофуразан)-4-сульфонил]-1,2,3,5,6,7, гексагидро-N,
N-дипропилциклопент[f]изоиндол-6-амин, 44
Используя методику 9, 1,2,3,5,6,7-гексагидро-N,N-дипропилциклопент[f] изоиндол-6-амин (38) обрабатывали бензофуразан-4-сульфонилхлоридом.
Кристаллизация из метанола давала 44 в виде серо-коричневых кристаллов (т.пл. 115-118oС).
ПРИМЕР 27: 2-{ [(Бензоиламинометил)тиофен]-5-сульфонил}-1,2,3,5,6,7-гексагидро-N,
N-дипропилциклопент[f]изоиндол-6-амин, 45
Используя методику 9, 1,2,3,5,6,7-гексагидро-N,N-дипропилциклoпeнт[f] изoиндoл-6-aмин (38) обрабатывали
2-(бензоиламинометил)тиофен-5-сульфонилхлоридом. Кристаллизация из метанола давала 45 в виде рыжевато-коричневых кристаллов (т.пл. 160-161, 186-187oС).
ПРИМЕР 28: 2-[(2,
3-Дихлортиофен)-5-сульфонил]-1,2,3,5,6,7-гексагидро-N, N-дипpoпилциклoпeнт[f]изoиндoл-6-aмин, 46
Используя методику 9, 1,2,3,5,6,7-гексагидро-N,N-дипропилциклопент[f] изоиндол-6-амин (38)
обрабатывали 2,3-дихлортиофен-5-сульфонилхлоридом. Кристаллизация из метанола давала 46 в виде рыжевато-коричневых кристаллов (т. пл. 150-151oС).
Методика 30: трет-Бутил-[6,
7-дигидро-3-[[бензилоксикарбонил]-аминометил] -5Н-циклопента[с]пиридин-6-ил]карбамат, 55
Раствор бензилхлорформиата (17,1 г, 0,100 моль) в хлороформе (50 мл) добавляли по каплям при комнатной
температуре в течение 10 минут к смеси гидрохлорида аминоацетонитрила (13,89 г, 0,150 моль) и карбоната натрия (21,2 г, 0,200 моль) в воде (50 мл) и хлороформе (20 мл) в колбе, снабженной механической
мешалкой. Смесь перемешивали в течение 2 часов, разбавляли водой и экстрагировали два раза диэтиловым простым эфиром. Объединенные экстракты промывали солевым раствором и сушили (MgSO4).
Растворитель удаляли в вакууме, оставляя масло. Кристаллизация из смеси этилацетат/гексан давала N-бензилоксикарбонил-2-аминоацетонитрил в виде бесцветных кристаллов (т.пл. 61-62).
С использованием методики Vollhardt (J.Chem. Soc., Chem. Comm., 133-134 (1982)) раствор N-бензилокси-2-аминоацетонитрила (1,91 г, 10,0 ммоль) в п-ксилоле (50 мл) нагревали в атмосфере аргона при 145oС. В атмосфере аргона к нагретому раствору ксилола со скоростью 1,5 мл/ч через шприц с насосом добавляли раствор 4-(трет-бутилоксикарбониламино)гептан-1,6-диина (4,15 г, 20,0 ммоль), N-бензилокси-2-аминоацетонитрила (3,81 г, 20,0 ммоль) и циклопентадиенилкобальтдикарбонила (0,50 мл, ~2,8 ммоль) в п-ксилоле (45 мл). После того как добавление было завершено, растворитель удаляли в вакууме, получая темное масло. Очистка флэш-хроматографией на силикагеле с элюированием смесью этилацетат/гексан давала рыжевато-коричневый твердый продукт. Кристаллизация из смеси этилацетат/гексан давала 55 в виде рыжевато-коричневых кристаллов (т.пл. 113-115oС).
Методика 31: Фенилметил[[6-(дипропиламино)-6,7-дигидро-5Н-циклопента[с] пиридин-3-ил]метил]карбамат,
56
Трифторуксусную кислоту (25 мл) добавляли к трет-бутил-[6,7-дигидро-3-[[бензилоксикарбонил]аминометил]-5Н-циклопента[с]пиридин-6-ил]карбамату (55, 4,45 г, 11,2 ммоль) при комнатной
температуре и смесь перемешивали в течение 20 минут. Избыток трифторуксусной кислоты удаляли в вакууме и остаток распределяли между смесью 1: 1 тетрагидрофуран/диэтиловый эфир и 5% раствором
гидроксида натрия. Водный раствор экстрагировали более двух раз смесью 1:1 тетрагидрофуран/диэтиловый эфир и объединенные экстракты промывали солевым раствором и сушили (Na2SO4).
Растворитель удаляли в вакууме, оставляя фенилметил-[[6-амино-6,7-дигидро-5Н-циклопента[с] пиридин-3-ил] метил] карбамат в виде янтарного масла.
Смесь фенилметил-[[6-амино-6, 7-дигидро-5Н-циклопента[с]пиридин-3-ил]метил] карбамата (2,96 г, 10,4 ммоль), 1-бромпропана (5,2 г, 4,2 ммоль) и карбоната калия (3,60 г, 26,0 ммоль) в ацетонитриле (30 мл) перемешивали при кипячении с обратным холодильником в течение 17 часов. Смесь разбавляли водой и экстрагировали два раза диэтиловым эфиром. Объединенные экстракты промывали солевым раствором и сушили (MgSO4). Растворитель удаляли в вакууме, получая темное масло. Очистка флэш-хроматографией на силикагеле с элюированием смесью тетрагидрофуран/этилацетат давала темное масло. Соединение обрабатывали активированным углем в этилацетате и фильтровали через диатомовую землю, получая 56 в виде янтарного масла.
Методика 32: 3-Аминометил-6-(дипропиламино)-6,7-дигидро-5Н-циклопента[с]
пиридин, 57
Смесь фенилметил-[[6-(дипропиламино)-6,7-дигидро-5Н-циклопента[с] пиридин-3-ил] метил] карбамата (56, 1,35 г, 3,54 ммоль) и 10% палладия на угле в абсолютном этаноле гидрировали в
течение 3 часов при давлении водорода 3,515 ат (50 psi). Смесь фильтровали через диатомовую землю и фильтрат выпаривали в вакууме, получая 57 в виде желтого масла.
ПРИМЕР 29:
4-Хлор-N-[[6-(дипропиламино)-6,7-дигидро-5Н-циклопента[с]пиридин-3-ил]метил]бензолсульфонамид, 58
Используя методику 9,3-(аминометил)-6-(дипропиламино)-6,7-дигидро-5Н-циклопента [с] пиридин
(57, 0,29 г, 1,2 ммоль) обрабатывали 4-хлорбензолсульфонилхлоридом (0,25 г, 1,2 ммоль). После экстракции и так далее, очистки флэш-хроматографией на силикагеле с элюированием смесью
тетрагидрофуран/этилацетат получали не совсем белый твердый продукт. Кристаллизация из смеси этилацетат/гексан давала 58 в виде не совсем белых кристаллов (т.пл. 125-126,5oС).
ПРИМЕР 30: 2-Циано-N-[[6-(дипропиламино)-6, 7-дигидро-5Н-циклопента[с] пиридин-3-ил]метил]бензолсульфонамид, 59
Используя методику 9, 3-(аминометил)-6-(дипропиламино)-6,
7-дигидро-5Н-циклопента[с] пиридин (57, 0,42 г, 1,7 ммоль) обрабатывали 2-цианобензолсульфонилхлоридом (0,35 г, 1,7 ммоль). Очистка флэш-хроматографией на силикагеле с элюированием тетрагидрофураном в
этилацетате давала масло. Кристаллизация из смеси этилацетат/гексан давала 59 в виде не совсем белых кристаллов (т.пл. 112-113oС).
ПРИМЕР 31: 4-Хлор-N-[[6-(дипропиламино)-6,
7-дигидро-5Н-циклопента[с] пиридин-3-ил]метил]бензамид, 60
Используя методику 11, 3-(аминометил)-6-(дипропиламино)-6,7-дигидро-5Н-циклопента[с] пиридин (57, 0,43 г, 1,7 ммоль) обрабатывали
4-хлорбензоилхлоридом (0,32 г, 1,8 ммоль). Очистка флэш-хроматографией на силикагеле с элюированием смесью тетрагидрофуран/этилацетат давала рыжевато-коричневый твердый продукт. Кристаллизация из
смеси этилацетат/гексан давала 60 в виде серых кристаллов (т.пл. 101,5-103oС).
ПРИМЕР 32: Методика 33. N-(4-Хлорфенил)-N'-[[6-(дипропиламино)-6,
7-дигидро-5Н-циклопента[с]пиридин-3-ил]метил]мочевина, 61
3-(Аминометил)-6-(дипропиламино)-6,7-дигидро-5Н-циклопента[с] пиридин (57, 0,42 г, 1,7 ммоль) добавляли к раствору
4-хлорфенилизоцианата (0,28 г, 1,8 ммоль) в толуоле (5 мл). Смесь перемешивали при комнатной температуре в течение 18 часов и растворитель удаляли в вакууме, получая масло. Соединение перемешивали в
течение 10 минут с 5% раствором хлористоводородной кислоты, подщелачивали 10% раствором карбоната натрия и экстрагировали два раза этилацетатом. Объединенные экстракты промывали солевым раствором и
сушили (MgSO4). Растворитель удаляли в вакууме, оставляя масло. Очистка флэш-хроматографией на силикагеле с элюированием тетрагидрофураном в этилацетате давала светло-янтарный твердый
продукт. Кристаллизация из смеси этилацетат/гексан давала 61 в виде серых кристаллов (т.пл. 120-121oС).
ПРИМЕР 33: N-(4-Цианофенил)-N'-[[6-(дипропиламино)-6,
7-дигидро-5Н-циклопента[с]пиридин-3-ил[метил]мочевина, 62
С использованием методики 33, 3-(аминометил)-6-(дипропиламино)-6,7-дигидро-5Н-циклопента[с]пиридин (57, 0,495 г, 2,00 ммоль)
обрабатывали 4-цианофенилизоцианатом (0,29 г, 2,0 ммоль). Очистка флэш-хроматографией на силикагеле с элюированием смесью тетрагидрофуран/этилацетат давала светло-янтарное масло. Кристаллизация из
смеси этилацетат/гексан давала 62 в виде рыжевато-коричневых кристаллов (т.пл. 135,5-136,5oС).
Методика 34. 6-Пропиламино-4,5,6,7-тетрагидробензотиазол, 64
Цианоборогидрид натрия (25 г) добавляли к 4,5,6,7-тетрагидробензотиазол-6-ону (Helv. Chim. Acta 1994, 1256) (14,5 г), пропиламину (40 мл), ледяной уксусной кислоте (54 мл), метанолу (500 мл) и ТГФ
(200 мл) на ледяной бане. Через 3,5 часа реакции при комнатной температуре растворитель удаляли в вакууме и остаток распределяли между водным карбонатом натрия и смесью простой эфир/ТГФ. Эфирный слой
промывали водой и затем 2 н. хлористоводородной кислотой. Этот кислотный слой подщелачивали и затем экстрагировали смесью диэтиловый эфир/ТГФ. Этот эфирный слой промывали водой и солевым раствором и
сушили сульфатом натрия. Растворитель удаляли в вакууме, получая 4,8 г 64 в виде жидкости.
Методика 35: трет-Бутил-(2-формил-4,5,6,7-тетрагидро-N-пропил-6-бензотиазоламин)карбамат,
65
6-Пропиламино-4,5,6,7-тетрагидробензотиазол (64) (4,8 г) и ди-трет-бутилдикарбонат (5,8 г) смешивали в ТГФ при 45oС. Через 4,5 часа растворитель удаляли и остаток очищали
флэш-хроматографией на силикагеле с элюированием смесью этилацетат/гексан, получая 5,8 г жидкости после удаления растворителя. К этому карбамату (3,0 г) в сухом ТГФ (25 мл) при -78oС
добавляли н-бутиллитий (1,6 М в гексане, 7,4 мл). Через 20 минут добавляли сухой N,N-диметилформамид (2,4 мл) и холодную баню убирали. Через 90 минут раствор распределяли между водой и простым эфиром.
Эфирный слой промывали водой и солевым раствором и сушили над сульфатом натрия. Растворитель удаляли в вакууме и остаток хроматографировали на силикагеле (этилацетат/гексан), получая 4,5 г 65 в виде
жидкости.
Методика 36: трет-Бутил-(2-гидроксиимино)метил-4,5,6,7-тетрагидро-N-пропил-6-бензотиазоламин)карбамат, 66
Гидрохлорид гидроксиламина (1,1 г) добавляли к
трет-бутил-(2-формил-4,5,6,7-тетрагидро-N-пропил-6-бензотиазоламин)карбамату (65) (4,3 г), гидроксиду натрия (1,3 г) и воде. Через 5 минут через раствор барботировали диоксид углерода. Смесь
распределяли между водой и смесью простой эфир/ТГФ. Эфирный слой промывали водой и солевым раствором и сушили сульфатом натрия. Растворитель удаляли в вакууме и остаток хроматографировали на
силикагеле (дихлорметан/этил-ацетат/гексан), получая 3,9 г 66 в виде твердого продукта.
Методика 37: трет-Бутил-(2-аминометил-4,5,6,7-тетрагидро-N-пропил-6-бензотиазоламин)карбамат,
67
К трет-бутил-(2-гидроксиимино)метил-4,5,6,7-тетрагидро-N-пропил-6-бензотиазоламин)карбамату (66) (4,0 г), гидроксиду натрия (2 н. водный, 200 мл) и метанолу (50 мл) добавляли сплав Devaria
(сплав 50% меди, 45% аюминия, 5% цинка) (75 г) и смесь нагревали при 42oС в течение 30 минут. Смесь экстрагировали смесью диэтиловый эфир/ТГФ и эфирный слой промывали водой и солевым
раствором и затем сушили сульфатом натрия. Растворитель удаляли в вакууме, получая 2,9 г 67 в виде жидкости.
ПРИМЕР 34: N-(4-Хлорфенил)-N'-[[4,5,6,
7-тетрагидро-6-(пропиламино)-2-бензотиазолил]метил]мочевина, 68
Используя методику 19, трет-бутил-(R)-(2-аминометил-4,5,6,7-тетрагидро-N-пропил-6-бензотиазоламин)карбамат (67) (0,90 г)
обрабатывали 4-хлорфенилизоцианатом (0,46 г), затем трифторуксусной кислотой, получая 0,32 г 68 в виде твердого продукта, т.пл. 125-130oС.
Методика 38: 2-(Дипропиламино)-2,
3-дигидро-6-(фенилметокси)-1Н-инден-5-илтрифторметансульфонат, 70
Гидрид натрия (0,96 г, 24 ммоль) промывали гексаном в атмосфере азота и суспендировали в ДМФ (10 мл). По каплям при 0-5oС в течение 30 мин добавляли 2-(дипропиламино)-2,3-дигидро-6-гидрокси-1Н-инден-5-илтрифторметансульфонат (69, 7,6 г, 20 ммоль) в ДМФ (30 мл) и смесь перемешивали 30 мин. Добавляли бензилбромид
(5,13 г, 30 ммоль) в ДМФ (5 мл) и полученную смесь перемешивали 1 ч. Реакцию останавливали насыщенным NaHCO3 и экстрагировали EtOAc. Органический слой промывали водой, солевым раствором,
сушили (MgSO4), фильтровали и концентрировали, получая сырой продукт. Хроматографическая очистка давала чистый продукт 70 в виде масла, которое превращали в соль НСl и кристаллизовали из
смеси EtOAc/MeOH/гексан, получая белый твердый продукт (т.пл. 162-163oС).
6-Бутокси-2-(дипропиламино)-2,3-дигидро-1Н-инден-5-ил-трифторметансульфонат, 71
Используя
методику 38, 2-(дипропиламино)-2,3-дигидро-6-гидрокси-1Н-инден-5-илтрифторметансульфонат (69, 0,38 г, 1 ммоль) обрабатывали бромбутаном (0,27 г, 2 ммоль). Хроматографическая очистка давала чистый
продукт 71 в виде масла, которое превращали в соль НСl и кристаллизовали из смеси EtOAc/гексан, получая белый твердый продукт (т.пл. 148-149oС).
2-(Дипропиламино)-2,
3-дигидро-6-пропокси-1Н-инден-5-илтрифторметансульфонат, 72
С использованием методики 38, 2-(дипропиламино)-2,3-дигидро-6-гидрокси-1Н-инден-5-илтрифторметансульфонат (69, 1,9 г, 5 ммоль)
обрабатывали пропилбромидом (2,5 г, 20 ммоль). Хроматографическая очистка давала чистый продукт 72 в виде масла, которое превращали в соль НСl и кристаллизовали из смеси EtOAc/гексан/, получая белый
твердый продукт (т.пл. 169-170oС).
2-(Дипропиламино)-6-этокси-2,3-дигидро-1Н-инден-5-ил-трифторметансульфонат, 73
Используя методику 38, 2-(дипропиламино)-2,
3-дигидро-6-гидрокси-1Н-инден-5-илтрифторметансульфонат (69, 1,9 г, 5 ммоль) обрабатывали бромэтаном (2,2 г, 20 ммоль). Хроматографическая очистка давала чистый продукт 73 в виде масла, которое
превращали в соль НСl и кристаллизовали из смеси EtOAc/гексан, получая белый твердый продукт (т.пл. 181-182oС).
Методика 39: Метил-2-(дипропиламино)-2,
3-дигидро-6-(фенилметокси)-1Н-инден-5-карбоксилат, 74
Смесь 2-(дипропиламино)-2,3-дигидро-6-(фенилметокси)-1Н-инден-5-илтрифторметансульфоната (70, 2,6 г, 5,6 ммоль), ацетата палладия (0,13 г,
0,56 ммоль), 1,3-бис(дифенилфосфинопропана) (0,3 г, 0,73 ммоль), триэтиламина (0,7 мл, 6,3 ммоль), метанола (6 мл) и ДМФ (18 мл) нагревали при 60-70oС при введении газообразного СО. Через
24 ч реакционную смесь гасили насыщенным NaHCO4, экстрагировали EtOAc, промывали водой, солевым раствором, сушили (MgSO4), фильтровали и концентрировали. Хроматографическая
очистка давала продукт 74, который превращали в соль НСl и кристаллизовали из смеси EtOAc/MeOH, получая белый твердый продукт (т.пл. 181-182oС).
Метил-6-бутокси-2-(дипропиламино)-2,3-дигидро-1Н-инден-5-карбоксилат, 75
Используя методику 39, 6-бутокси-2-(дипропиламино)-2,3-дигидро-1Н-инден-5-илтрифторметансульфонат (71, 2,2 г, 5 ммоль)
превращали в метил-6-бутокси-2-(дипропиламино)-2,3-дигидро-1Н-инден-5-карбоксилат 75 и кристаллизовали из смеси EtOAc/гексан в виде соли НСl (т.пл. 140-141oС).
Метил-2-(дипропиламино)-2,3-дигидро-6-пропокси-1Н-инден-5-карбоксилат, 76
Используя методику 39, 2-(дипропиламино)-2,3-дигидро-6-пропокси-1Н-инден-5-илтрифторметансульфонат (72, 2,96 г, 7
ммоль) превращали в метил-2-(дипропиламино)-2,3-дигидро-6-пропокси-1Н-инден-5-карбоксилат 76 и кристаллизовали из смеси EtOAc/гексан в виде соли НСl (т.пл. 175-176oС).
Метил-2-(дипропиламино)-6-этокси-2,3-дигидро-1Н-инден-5-карбоксилат, 77
Используя методику 39, 2-(дипропиламино)-6-этокси-2,3-дигидро-1Н-инден-5-илтрифторметансульфонат (73, 2,3 г, 5,7 ммоль)
превращали в метил-2-(дипропиламино)-6-этокси-2,3-дигидро-1Н-инден-5-карбоксилат 77 и кристаллизовали из смеси EtOAc/гексан в виде соли НС1 (т.пл. 168-169oС).
ПРИМЕР 35:
Методика 40: 2-(Дипропиламино)-2,3-дигидро-6-(фенилметокси)-1Н-инден-5-карбоксамид, 78
К раствору метил-2-(дипропиламино)-2,3-дигидро-6-(фенилметокси)-1Н-инден-5-карбоксилата (74, 095 г, 2,5
ммоль) и формамида (1,1 г, 25 ммоль) в ДМФ (10 мл) при 100'С по каплям добавляли 25% метоксид натрия в метаноле (2,5 ммоль, 0,57 мл). Смесь перемешивали в течение 2 ч. Реакционную смесь охлаждали до
комнатной температуры и гасили водой (100 мл). Полученный осадок перемешивали в течение 0,5 ч и растворитель удаляли фильтрованием. Этот твердый продукт кристаллизовали из смеси EtOAc/MeOH, получая
белый твердый продукт, который превращали в соль НСl и кристаллизовали из EtOAc/MeOH, получая указанное в заголовке соединение 78 в виде белого твердого продукта (т.пл. 247-248oС).
ПРИМЕР 36: 6-Бутокси-2-(дипропиламино)-2,3-дигидро-1Н-инден-5-карбоксамид, 79
Используя методику 40, метил-6-бутокси-2-(дипропиламино)-2,3-дигидро-1Н-инден-5-карбоксилат (75, 1 г,
2,9 ммоль) превращали в 79 и кристаллизовали из EtOAc/MeOH в виде соли НСl (т.пл. 215-216oС).
ПРИМЕР 37: 2-(Дипропиламино)-2,3-дигидро-6-пропокси-1Н-инден-5-карбоксамид,
80
Используя методику 40, метил-2-(дипропиламино)-2,3-дигидро-6-пропокси-1Н-инден-5-карбоксилат (76, 1,1 г, 3,5 ммоль) превращали в 80 и кристаллизовали из EtOAc/MeOH в виде соли НСl (т.пл.
238-239oС).
ПРИМЕР 38: 2-(Дипропиламино)-6-этокси-2,3-дигидро-1Н-инден-5-карбоксамид, 81
Используя методику 40, метил-2-(дипропиламино)-6-этокси-2,
3-дигидро-1Н-инден-5-карбоксилат (77, 1,1 г, 3,5 ммоль) превращали в 81 и кристаллизовали из EtOAc/MeOH в виде соли НСl (т.пл. 236-237oС).
ПРИМЕР 39: Методика 41:
6-Бутокси-2-(дипропиламино)-2,3-дигидро-N-метил-1Н-инден-5-карбоксамид, 82
К смеси метил-6-бутокси-2-(дипропиламино)-2,3-дигидро-1Н-инден-5-карбоксилата (75, 0,35 г, 1 ммоль) и N-метил
формамида (0,59 г, 10 ммоль) в ДМФ (10 мл) при 100oС по каплям в течение 10 мин добавляли 25% метоксид натрия в метаноле (0,22 мл, 1 ммоль). Полученную смесь охлаждали до комнатной
температуры и гасили водой (100 мл). Полученный твердый продукт очищали хроматографией, получая масло, которое превращали в соль НСl и кристаллизовали из EtOAc/MeOH, получая указанное в заголовке
соединение 82 в виде белого твердого продукта (т.пл. 129-130oС).
ПРИМЕР 40: 6-Этокси-2-(дипропиламино)-2,3-дигидро-N-метил-1Н-инден-5-карбоксамид, 83
Используя
методику 41, метил-2-(дипропиламино)-6-этокси-2,3-дигидро-1Н-инден-5-карбоксилат (77, 0,32 г, 1 ммоль) обрабатывали N-метилформамидом (0,58 мл, 10 ммоль). Хроматографическая очистка давала чистый
продукт 83 в виде масла, которое превращали в соль НСl и кристаллизовали из EtOAc/MeOH, получая белый твердый продукт (т.пл. 156-159oC).
ПРИМЕР 41:
2-(Дипропиламино)-6-этокси-2,3-дигидро-N-(фенилметил)-1Н-инден-5-карбоксамид, 84
Используя методику 41, метил-2-(дипропиламино)-6-этокси-2,3-дигидро-1Н-инден-5-карбоксилат (77, 0,32 г, 1
ммоль) обрабатывали N-бензилформамидом (1,35 мл, 10 ммоль). Хроматографическая очистка давала чистый продукт 84 в виде масла, которое превращали в соль НСl и кристаллизовали из EtOAc/MeOH, получая
белый твердый продукт (т.пл. 219-221oС).
ПРИМЕР 42: Методика 42: 2-(Дипропиламино)-6-этокси-N-(2-фторэтил)-2,3-дигидро-1Н-инден-5-карбоксамид, 85
Гидрид натрия (0,
05 г, 1,2 ммоль) промывали гексаном в атмосфере азота и суспендировали в ДМФ (5 мл). По каплям при 0-5oС в течение 30 мин добавляли 2-(дипропиламино)-6-этокси-2,
3-дигидро-1Н-инден-5-карбоксамид (81, 0,15 г, 0,5 ммоль) в ДМФ (5 мл) и смесь перемешивали в течение 30 мин. Добавляли 1-бром-2-фторэтан (0,26 г, 2 ммоль) в ДМФ (5 мл) и полученную смесь перемешивали
3 ч. Реакционную смесь гасили насыщенным NaHCO3 и экстрагировали EtOAc. Органический слой промывали водой, солевым раствором, сушили (MgSO4), фильтровали и концентрировали,
получая сырой продукт. Хроматографическая очистка давала чистый продукт 85 в виде масла, которое превращали в соль НСl и кристаллизовали из смеси EtOAc/MeOH/гексан, получая белый твердый продукт
(т.пл. 158-159oС).
Методика 43: 2-(Дипропиламино)-2,3-дигидро-6-гидрокси-1Н-инден-5-карбоксамид, 87
К раствору метил-2-(дипропиламино)-2,
3-дигидро-6-гидрокси-1Н-инден-5-карбоксилата (86, 0,93 г, 2,9 ммоль) и формамида (1,2 мл, 29 ммоль) в ДМФ
(20 мл) при 100oС по каплям добавляли 25% метоксид натрия в метаноле (1,3 мл, 5,
8
ммоль). Смесь перемешивали в течение 4 ч. Реакционную смесь гасили насыщенным NaHCO3 и экстрагировали EtOAc. Органический слой промывали водой, солевым раствором, сушили
(MgSO4), фильтровали и концентрировали, получая сырой продукт.
Хроматографическая очистка давала чистый продукт в виде масла, которое превращали в соль НСl и кристаллизовали из EtOAc/MeOH, получая указанное в заголовке соединение 87 в виде белого твердого продукта (т.пл. 266-267oС).
ПРИМЕР 43: Методика 44:
2-(Дипропиламино)-6-(2-фторэтокси)-2,3-дигидро-1Н-инден-5-карбоксамид, 88
Смесь 2-(дипропиламино)-2,3-дигидро-6-гидрокси-1H-инден-5-карбоксамида (87, 0,14 г, 0,5 ммоль), карбоната калия (0,2
г, 1,5 ммоль), 1-бром-2-фторэтана (0,19 г, 1,5 ммоль) в ДМФ (10 мл) перемешивали при комнатной температуре в течение 5 ч. Смесь гасили водой и экстрагировали EtOAc. Водный слой промывали солевым
раствором, сушили (MgSO4), фильтровали и концентрировали. Хроматографическая очистка давала бледно-желтый твердый продукт, который превращали в соль НСl и кристаллизовывали из EtOAc/MeOH,
получая указанное в заголовке соединение 88 в виде не совсем белого твердого продукта (т.пл. 232-233oС).
ПРИМЕР 44: 2-(Дипропиламино)-6-(3-фторпропокси)-2,
3-дигидро-1Н-инден-5-карбоксамид, 89
Используя методику 44, 2-(дипропиламино)-2,3-дигидро-6-гидрокси-1Н-инден-5-карбоксамид (87, 0,19 г, 0,7 ммоль) обрабатывали 1-бром-3-фторпропаном (0,296
мл, 2,1 ммоль). Хроматографическая очистка давала чистый продукт 89 в виде масла, которое превращали в соль НСl и кристаллизовали из EtOAc/MeOH, получая белый твердый продукт (т.пл. 219-220o
С).
ПРИМЕР 45: 2-(Дипропиламино)-6-этокси-N-этил-2,3-дигидро-1Н-инден-5-кароксамид, 90
Используя методику 44, 2-(дипропиламино)-2,3-дигидро-6-гидрокси-1Н-инден-5-карбоксамид
(87, 0,36 г, 1,15 ммоль) обрабатывали бромэтаном (0,38 мл, 3,45 ммоль). Хроматографическая очистка давала чистый продукт 90 в виде масла, который превращали в соль НС1 и кристаллизовали из EtOAc/MeOH,
получая белый твердый продукт (т.пл. 145-146oС).
Методика 45: 2-(Дипропиламино)-2,3-дигидро-1Н-инден-5,6-дикарбоксилат, 92
Диметил-2-(дипропиламино)-2,
3-дигидро-1Н-инден-5,6-дикарбоксилат (91, 10,0 г, 30,0 ммоль) кипятили с обратным холодильником при 70oС с NaOH (2,4 г, 60,0 ммоль) в смеси 1:3 H2O/МеОН (100 мл) в течение 3 ч.
Реакционную смесь концентрировали и лиофилизовали. Получаемый твердый продукт нагревали в ТГФ/МеОН (1: 1) и оставляли для охлаждения на ночь. Полученный твердый продукт 92 получали фильтрованием и
сушили (т.пл. >300oС).
ПРИМЕР 46: Методика 46: 6-(Дипропиламино)-6,7-дигидроциклопент[f]изоиндол-1,3(2H,5Н)-дион, 93
Раствор 2-(дипропиламино)-2,
3-дигидро-1Н-инден-5,6-дикарбоксилата (92, 3,00 г, 8,6 ммоль), (NH4)OAc (6,63 г, 86,0 ммоль) и концентрированной НСl (1,7 мл) в ледяной НОАс (200 мл) кипятили с обратным холодильником при
119oС в течение 20 ч. Реакционную смесь охлаждали, концентрировали и подвергали азеотропной перегонке с толуолом, получая белый твердый продукт. Остаток разбавляли H2О,
подщелачивали насыщенным NaHCO3 до рН>9 и фильтровали через сплавленный стеклянный фильтр. Собранный твердый продукт разбавляли МеОН и несколько раз подвергали азеотропной перегонке
с толуолом до получения сухого продукта. Твердый продукт растворяли в горячем МеОН, фильтровали через фильтровальную бумагу и перекристаллизовали из дополнительного горячего МеОН, получая 93 в виде
рыжевато-коричневого твердого продукта. Твердый продукт превращали в соль НСl и перекристаллизовали из MeOH/EtOAc, получая белый твердый продукт (т.пл. 290-291oС).
ПРИМЕР
47: Методика 47: 6-(Дипропиламино)-3,5,6,7-тетрагидроциклопент[f] изоиндол-1(2Н)-он, 94
(Ref. Brewster, J. H. ; Fusco A.M., J.Org.Chem., 28, 501-503 (1963)). Раствор 6-(дипропиламино-6,
7-дигидроциклопент[f]изоиндол-1,3(2Н,5Н)-дион 93 (0,45 г, 1,6 ммоль) в ледяной НОАс (50 мл) обрабатывали цинковой пылью (1,05 г, 16,0 ммоль), затем кипятили с обратным холодильником при 113o
С в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли МеОН и фильтровали через подушку целита. Фильтрат концентрировали и остаток разбавляли Н2O, подщелачивали
насыщенным Na2CO3 и экстрагировали СН2Сl2. Органические слои промывали солевым раствором, сушили (MgSO4) и концентрировали. Полученный твердый
продукт очищали с использованием силикагеля с элюированием смесью 2: 1 гептан/ацетон, получая 94 в виде белого твердого продукта (т.пл. 206-208oС).
ПРИМЕР 48: Методика 48:
6-(Дипропиламино)-2-этил-6,7-дигидроциклопент[f] изоиндол-1,3(2Н, 5Н)-дион, 95
К раствору 6-(дипропиламино-6,7-дигидроциклопент[f] изоиндол-1,3(2Н, 5Н)-диона (93, 0,15 г, 0,52 ммоль) в ДМФ
(10 мл) при 0oС добавляли безводный K2СО3 (0,14 г, 1,04 ммоль), затем этилбромид (0,06 мл, 0,79 ммоль). Реакционную смесь перемешивали при 0oС в течение 2 ч,
затем давали нагреться до комнатной температуры. Перемешивание продолжали в течение ночи, затем гасили Н2O. Раствор экстрагировали CH2Cl2 и органические слои
промывали солевым раствором, сушили (MgSO4) и концентрировали. Остаток далее концентрировали в высоком вакууме для удаления любого избытка ДМФ и полученный зеленоватый твердый продукт
очищали с использованием силикагеля и элюированием смесью 3: 1 гексан/ацетон, получая 95 в виде не совсем белого твердого продукта. Твердый продукт превращали в соль НСl и перекристаллизовали из
горячей смеси Ме-OH/EtOAc, получая 95 в виде белого твердого продукта (т.пл. 225-230oС).
ПРИМЕР 49: 6-(Дипропиламино)-6,7-дигидро-2-пропилциклопент[f] изоиндол-1,3(2Н,
5Н)-дион, 96
Используя методику 48, 6-(дипропиламино-6,7-дигидроциклопент[f]изоиндол-1,3 (2Н, 5Н)-дион, (93, 0/15 г, 0,52 ммоль) обрабатывали 1-бромпропаном (0,07 мл, 0,78 ммоль). Очистка на
силикагеле с элюированием смесью 3:1 гексан/ацетон давала твердый продукт, который превращали в соль НСl и перекристаллизовали из горячей смеси MeOH/EtOAc, получая 96 в виде белого твердого продукта
(т.пл. 217-218oС).
ПРИМЕР 50: 4-[[6-(Дипропиламино)-3,5,6,7-тетрагидро-1,3-диоксоциклопент[f]изоиндол-2(1Н)-ил]метил]бензонитрил, 97
Используя методику 48,
6-(дипропиламино)-6,7-дигидроциклопент [f]изоиндол-1,3(2Н, 5Н)-дион (93, 0,12 г, 0,4 ммоль) обрабатывали альфа-бром-п-толунитрилом (0,08 мл, 0,4 ммоль). Очистка с использованием силикагеля и
элюированием смесью 3: 1 гексан/ацетон давала твердый продукт, который превращали в соль НСl и кристаллизовали из горячей смеси MeOH/EtOAc, получая 97 в виде белого твердого продукта (т.пл.
263-264oС).
ПРИМЕР 51: 2-(1Н-Бензотриазол-1-илметил)-6-(дипропиламино)-6,7-дигидроциклопент[f]изоиндол-1,3(2Н, 5Н)-дион, 98
Используя методику 48,
6-(дипропиламино-6,7-дигидроциклопент[f]изоиндол-1,3(2Н, 5Н)-дион (93, 014 г, 0,5 ммоль) обрабатывали 1-(хлорметил)-1H-бензотриазолом (0,08 мл, 0,5 ммоль). Очистка с использованием силикагеля и
элюированием смесью 3:1 гексан/ацетон давала твердый продукт, который превращали в соль НСl и кристаллизовали из горячей смеси Ме-OH/EtOAc, получая 98 в виде белого твердого продукта (т.пл.
254-256oС).
ПРИМЕР 52: 6-(Дипропиламино)-6,7-дигидро-2-[(2-метил-5-тиазолил)метил] циклопент[f]изоиндол-1,3(2Н, 5Н)-дион, 99
Используя методику 48,
6-(дипропиламино)-6,7-дигидроциклопент [f]изоиндол-1,3(2Н, 5Н)-дион (93, 0,14 г, 0,5 ммоль) обрабатывали гидрохлоридом 4-хлорметил-2-метилтиазола (0,18 мл, 1,0 ммоль). Очистка с использованием
силикагеля и элюированием смесью 4: 1 гексан/ацетон + 0,1% МеОН давала твердый продукт, который превращали в соль НСl и кристаллизовали из горячей смеси MeOH/EtOAc, получая 99 в виде белого твердого
продукта (т.пл. 235-236oС).
ПРИМЕР 53: 6-(Дипропиламино)-6,7-дигидро-2-(1,2,4-оксадиазол-3-илметил)циклопент[f]изоиндол-1,3(2Н, 5Н)-дион, 100
Используя методику 48,
6-(дипропиламино-6,7-дигидроциклопент[f]изоиндол-1,3 (2Н, 5Н)-дион (93, 0,14 г, 0,5 ммоль) обрабатывали 3-(хлорметил)-1,2,4-оксадиазол (0,09 мл, 0,75 ммоль). Очистка с использованием силикагеля и
элюированием смесью 7: 1 СН2Сl2/ацетон давала твердый продукт, который превращали в соль НСl и кристаллизовали из CH2Cl2/Et2O, получая 100 в виде
белого твердого продукта (т.пл. 190-191oС).
ПРИМЕР 54: Методика 49. 6-(Дипропиламино)-2-[(4-фтор-фенил)метил]-6,7-дигидроциклопент[f]изоиндол-1,3(2Н,5Н)-дион, 101
(O'Reilly, N. J. , Derwin, W. S. , Fertel, L.B., Lin, H.C., Synlett., (1990), 609-610). Раствор 2-(дипропиламино)-2,3-дигидро-1Н-инден-5,6-дикарбоксилата (92, 0.35 г, 1,0 ммоль) и 4-фторбензиламина (0,
15 мл, 1,3 ммоль) в ледяной НОАс (30 мл) кипятили с обратным холодильником при 113oС. Через 4 ч реакционную смесь охлаждали до комнатной температуры, концентрировали, затем подвергали
азеотропной перегонке с толуолом. Остаток разбавляли водой и подщелачивали насыщенным Na2CO3 (или насыщенным NaHCO3) до рН >12. Раствор экстрагировали СН2Сl2 и органические слои промывали солевым раствором, сушили (MgSO4) и концентрировали. Остаток очищали с использованием силикагеля и элюированием смесью 4:1 СН2
Сl2/ацетон+0,1% МеОН, получая 101 в виде светло-желтого масла, которое превращали в соль НСl и кристаллизовали из MeOH/EtOAc, получая белый твердый продукт (т.пл. 251-253oС).
ПРИМЕР 55: 2-[(4-Хлорфенил)метил]-6-(дипропиламино)-6,7-дигидроциклопент[f]изоиндол-1,3(2Н, 5Н)-дион, 102
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1Н-инден-5,
6-дикарбоксилат (92, 0,35 г, 1,0 ммоль) обрабатывали 4-хлорбензиламином (0,17 мл, 1,4 ммоль). Очистка с использованием силикагеля и элюированием смесью 5: 1 гексан/ацетон давала масло, которое
превращали в соль НСl и перекристаллизовали из горячей смеси MeOH/EtOAc, получая 102 в виде белого твердого продукта (т.пл. 260-261oC).
ПРИМЕР 56: 6-(Дипропиламино)-6,
7-дигидро-2-[(2-метоксифенил) метил] циклопент[f]изоиндол-1,3 (2Н, 5Н)-дион, 103
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,35 г, 1,0 ммоль)
обрабатывали о-метоксибензиламином (0,18 мл, 1,4 моль). Очистка с использованием силикагеля и элюированием смесью 5:1 СН2Сl2/ацетон давала масло, которое превращали в соль НСl и
перекристаллизовали из горячей смеси MeOH/EtOAc, получая 103 в виде белого твердого продукта (т.пл. 205oС).
ПРИМЕР 57: 6-(Дипропиламино)-6,
7-дигидро-2-[(3-метоксифенил)метил]циклопент[f]изоиндол-1,3(2Н, 5Н)-дион, 104
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1Н-инден-5,6-дикарбоксилат (92, 0,35 г, 1,0 ммоль)
обрабатывали м-метоксибензиламином (0,17 мл, 1,3 ммоль). Очистка с использованием силикагеля и элюированием смесью 3: 1 гексан/ацетон давала масло, которое превращали в соль НСl и перекристаллизовали
из горячей смеси MeOH/EtOAc, получая 104 в виде белого твердого продукта (т.пл. 247-248oС).
ПРИМЕР 58: 6-(Дипропиламино)-6,7-дигидро-2-[
(4-метокси-фенил)метил]циклопент[f]изоиндол-1,3(2Н, 5Н)-дион, 105
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,10 г, 0,28 ммоль) обрабатывали
п-метоксибензиламином (0,05 мл, 0,4 ммоль). Очистка с использованием силикагеля и элюированием смесью 5:1 СН2Сl2/ацетон давала масло, которое превращали в соль НСl и
перекристаллизовали из EtOAc, получая 105 в виде белого твердого продукта (т.пл. 255-256oС).
ПРИМЕР 59: 2-[(3,4-Диметоксифенил)метил]-6-(дипропиламино)-6,
7-дигидроциклопент[f]изоиндол-1,3(2Н, 5Н)-дион, 106
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,35 г, 1,0 ммоль) обрабатывали вератриламином (0,20
мл, 1,3 ммоль). Очистка с использованием силикагеля и элюированием смесью 3:1 гексан/ацетон давала масло, которое превращали в соль НСl и перекристаллизовали из горячей смеси MeOH/EtOAc, получая 106 в
виде белого твердого продукта (т.пл. 233-235oC).
ПРИМЕР 60: 6-(Дипропиламино)-6,7-дигидро-2-[[4-(трифторметокси) фенил)метил]циклопент[f]изоиндол-1,3(2Н,5Н)-дион, 107
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,35 г, 1,00 ммоль) обрабатывали 4-трифторметоксибензиламином (0,25 мл, 1,3 ммоль). Очистка с использованием
силикагеля и элюированием смесью 3:1 СН2Сl2/ацетон давала масло, которое превращали в соль НСl и перекристаллизовали из горячей смеси MeOH/EtOAc, получая 107 в виде белого
твердого продукта (т.пл. 233-235oС).
ПРИМЕР 61: 6-(Дипропиламино)-6,7-дигидро-2-[(2-фенилэтил)-циклопент[f] изоиндол-1,3(2Н, 5Н)-дион, 108
Используя методику 49,
2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,35 г, 1,00 ммоль) обрабатывали 4-трифторметилбензиламином (0,2 мл, 1,4 ммоль). Очистка с использованием силикагеля и элюированием смесью
4:1 СН2Сl2/ацетон давала масло, которое превращали в соль НСl и кристаллизовали из горячей смеси MeOH/EtOAc, получая 108 в виде белого твердого продукта (т.пл. 229-230o
С).
ПРИМЕР 62: 4-[[6-Дипропиламино)-3,5,6,7-тетрагидро-1,3-диоксоциклопент[f]изоиндол-2(1Н)-ил]метил]бензолсульфонамид, 109
Используя методику 49, 2-(дипропиламино)-2,
3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,35 г, 1,00 ммоль) обрабатывали п-аминометилбензолсульфонамидом (0,31 мл, 1,4 ммоль). Очистка с использованием силикагеля и элюированием смесью 4:1 СН2Сl2/ацетон давала твердый продукт, который перекристаллизовали из СН2Сl2 /Et2O/гексан, получая 109 в виде белого твердого продукта (т.пл. 190-194oС).
ПРИМЕР 63: Метил-4-[[6-(дипропиламино)-3,5,6,7-тетрагидро-1,3-диоксоциклопент[f]изоиндол-2(1Н)-ил)метил]бензоат, 110
Метиловый эфир 4-(аминометил)бензойной кислоты
получали этерификацией 4-(аминометил)бензойной кислоты с использованием MeOH/H2SO4. Эфир превращали в соль НСl и перекристаллизовали из EtOAc (т.пл. 238-240oС).
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,35 г, 1,00 ммоль) обрабатывали метиловым эфиром 4-(аминометил)бензойной кислоты (0,28 мл, 1,4 ммоль). Очистка с использованием силикагеля и элюированием смесью 4:1 CH2Cl2/ацетон давала масло, которое превращали в соль НСl и перекристаллизовали из MeOH/EtOAc, получая 110 в виде белого твердого продукта (т.пл. 244-246oС).
ПРИМЕР 64: 4-[[6-(Дипропиламино)-3,5,6,7-тетрадигидро-1,3-диоксоциклопент[f]изоиндол-2(1Н)-ил]метил]бензамид, 111
4-(Аминометил)бензамид получали превращением метилового эфира 4-(аминометил) бензойной кислоты реакцией с NH4OH с использованием методики Cliffton, J. E. , et al., J.Med.Chem., 1982, 25,
670-679. Амид превращали в соль НСl и перекристаллизовали из MeOH/EtOAc (т.пл. 244-249oС).
С использованием методики 49, 2-(дипропиламино)-2,3-дигидро-1Н-инден-5, 6-дикарбоксилат (92, 0,35 г, 1,00 ммоль) обрабатывали 4-(аминометил)бензамидом (0,26 мл, 1,4 ммоль). Очистка с использованием силикагеля и элюированием смесью 19:1 СН2Сl2/МеОН давала масло, которое превращали в соль НСl и перекристаллизовали из горячей смеси МеОН, получая 111 в виде белого твердого продукта (т.пл. 294oС).
ПРИМЕР 65:
6-(Дипропиламино)-6,7-дигидро-2-(2-фенил-этил)циклопент[f] изоиндол-1,3(2Н, 5Н)-дион, 112
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,35 г, 1,00
ммоль) обрабатывали фенилэтиламином (0,16 мл, 1,3 ммоль). Очистка с использованием силикагеля и элюированием смесью 3:1 СН2Сl2/ацетон давала масло, которое превращали в соль НСl
и перекристаллизовали из горячей смеси MeOH/EtOAc, получая 112 в виде белого твердого продукта (т.пл. 235-240oС).
ПРИМЕР 66: 6-(Дипропиламино)-6,
7-дигидро-2-[2-(4-метоксифенил)этил] циклопент[f]изоиндол-1,3(2Н, 5Н)-дион, 113
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,35 г, 1,0 ммоль)
обрабатывали метоксифенилэтиламином (0,19 мл, 1,3 ммоль). Очистка с использованием силикагеля и элюированием смесью 3:1 гексан/ацетон давала масло, которое превращали в соль НСl и перекристаллизовали
из горячей смеси MeOH/EtOAc, получая 113 в виде белого твердого продукта (т.пл. 211-215oС).
ПРИМЕР 67: 6-(Дипропиламино)-6,7-дигидро-2-(3-фенилпропил)-циклопент[f]
изоиндол-1,3(2Н, 5Н)-дион, 114
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,35 г, 1,0 ммоль) обрабатывали 3-фенилпропиламином (0,19 мл, 1,4 ммоль).
Очистка с использованием силикагеля и элюированием смесью 9: 1 СН2Сl2/ацетон давала масло, которое превращали в соль НСl и перекристаллизовали из горячей смеси MeOH/EtOAc,
получая 114 в виде белого твердого продукта (т.пл. 222-223oС).
ПРИМЕР 68: 6-(Дипропиламино)-6,7-дигидро-2-(2-пиридинилметил] циклопент[f]изоиндол-1,3(2Н, 5Н)-дион, 115
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,35 г, 1,00 ммоль) обрабатывали 2-аминометилпиридином (0,4 мл, 4,00 ммоль). Очистка с использованием
силикагеля и элюированием смесью 2: 1 СН2Сl2/ацетон давала масло, которое превращали в соль НСl и перекристаллизовали из горячей смеси MeOH/EtOAc, получая 115 в виде белого
твердого продукта (т.пл. 130-135oC).
ПРИМЕР 69: 6-(Дипропиламино)-6,7-дигидро-2-[(3-пиридинилметил) циклопент[f]изоиндол-1,3(2Н, 5Н)-дион, 116
Используя методику
49, 2-(дипропиламино)-2,3-дигидро-1Н-инден-5,6-дикарбоксилат (92, 0,35 г, 1,00 ммоль) обрабатывали 3-аминометилпиридином (0,14 мл, 1,4 ммоль). Очистка с использованием силикагеля и элюированием смесью
19:1 СН2Сl2/МеОН, насыщенной NH4, давала масло, которое превращали в соль НСl и перекристаллизовали из горячей смеси MeOH/EtOAc, получая 116 в виде белого твердого
продукта (т.пл. 141-146oС).
ПРИМЕР 70: 6-(Дипропиламино)-6,7-дигидро-2-(4-пиридинилметил) циклопент[f]изоиндол-1,3(2Н, 5Н)-дион, 117
Используя методику 49,
2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,35 г, 1,00 ммоль) обрабатывали 4-аминометилпиридином (0,14 мл, 1,4 ммоль). Очистка с использованием силикагеля и элюированием смесью 3:1
СН2Сl2/ацетон давала масло, которое превращали в соль НСl и перекристаллизовали из горячей смеси MeOH/EtOAc, получая 117 в виде белого твердого продукта (т.пл. 283-284o
С).
ПРИМЕР 71: 6-(Дипропиламино)-6,7-дигидро-2-[2-(1Н-имидазол-4-ил)этил] циклопент[f]изоиндол-1,3(2Н, 5Н)-дион, 118
Используя методику 49, 2-(дипропиламино)-2,
3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,35 г, 1,00 ммоль) обрабатывали гистамином (0,16 мл, 1,4 ммоль). Очистка с использованием силикагеля и элюированием смесью 9:1 СН2Сl2
/МеОН давала твердый продукт, который превращали в соль НСl и перекристаллизовали из горячей смеси MeOH/EtOAc, получая 118 в виде белого твердого продукта (т.пл. 190-191oC).
ПРИМЕР 72: 6-(Дипропиламино)-6,7-дигидро-2-(2-тиенилметил) циклопент[f] изоиндол-1,3(2Н, 5Н)-дион, 119
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,
35 г, 1,00 ммоль) обрабатывали 2-тиофенметиламином (0,13 мл, 1,3 ммоль). Очистка с использованием силикагеля и элюированием смесью 3: 1 СН2Сl2/ацетон давала масло, которое
превращали в соль НСl и перекристаллизовали из горячей смеси MeOH/EtOAc, получая 119 в виде белого твердого продукта (т.пл. 220-225oС).
ПРИМЕР 73: 6-(Дипропиламино)-6,
7-дигидро-2-метилциклопент-[f]изоиндол-1,3(2Н, 5Н)-дион, 120
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,3 г, 1,0 ммоль) обрабатывали гидрохлоридом
метиламина) (1,35 г, 20,0 ммоль). Очистка на силикагеле с элюированием смесью 2:1 гексан/ацетон давала масло, которое превращали в соль НСl и перекристаллизовали из смеси EtOAc/2-пропанол, получая 120
в виде белого твердого продукта (т. пл. 245-246oС).
ПРИМЕР 74: 6-(Дипропиламино)-6,7-дигидро-2-фенилциклопент[f] изоиндол-1,3(2Н, 5Н)-дион, 121
Используя методику
49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,35 г, 1,0 ммоль) обрабатывали анилином (0,11 мл, 1,2 ммоль). Очистка на силикагеле с элюированием смесью 2:1 гексан/ацетон давала
твердый продукт, который превращали в соль НСl и перекристаллизовали из смеси EtOAc/2 пропанол, получая 121 в виде белого твердого продукта (т.пл. 241-242oС).
ПРИМЕР 75:
4-[6-(Дипропиламино)-3,5,6,7-тетрагидро-1,3-диоксоциклопент[f]изоиндол-2(1Н)-ил]бензамид, 122
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,3 г, 1,0
ммоль) обрабатывали 4-аминобензамидом (1,4 мл, 10,0 ммоль). Очистка на силикагеле с элюированием смесью 9:1 СН2Сl2/МеОН, насыщенной NH3, давала твердый продукт,
который превращали в соль НСl и перекристаллизовали из EtOAc/MeOH, получая 122 в виде белого твердого продукта (т.пл. 275-276oС).
ПРИМЕР 76: 4-[6-(Дипропиламино)-3,5,6,
7-тетрадигидро-1,3-диоксоциклопент[f]изоиндол-2(1Н)-ил]бензонитрил, 123
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,3 г, 1,0 ммоль) обрабатывали
4-аминобензонитрилом (0,47 мл, 4,0 ммоль). Очистка на силикагеле с элюированием смесью 3:1 гексан/ацетон давала твердый продукт, который превращали в соль НСl и перекристаллизовали из смеси
EtOAc/этанол, получая 123 в виде белого твердого продукта (т.пл. 250-251oC).
ПРИМЕР 77: 6-(Дипропиламино)-6,7-дигидро-2-(фенилметил] циклопент[f]изоиндол-1,3(2Н, 5Н)-дион,
124
Используя методику 49, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,3 г, 1,0 ммоль) обрабатывали бензиламином (0,44 мл, 4,0 ммоль). Очистка на силикагеле с элюированием
смесью 3:1 гексан/ацетон давала твердый продукт, который превращали в соль НСl и перекристаллизовали из смеси EtOAc/2-пропанол, получая 124 в виде белого твердого продукта (т.пл. 247-248o
С).
ПРИМЕР 78: 6-(Дипропиламино)-3,5,6,7-тетрагидро-2-(фенилметил) циклопент[f]изоиндол-1,(2Н)-он, 125
Используя методику 47, 6-(дипропиламино)-6,
7-дигидро-2-(фенилметил)циклопент[f] изоиндол-1,3 (2Н, 5Н)-дион (124, 0,41 г, 1,0 ммоль) восстанавливали цинковой пылью (0,65 г, 10,0 ммоль) в ледяной НОАс (20 мл). Очистка на силикагале с
элюированием смесью 3:1 гексан/ацетон давала масло, которое превращали в соль НСl и перекристаллизовали из EtOAc/MeOH, получая 125 в виде белого твердого продукта (т.пл. 235-236oС).
ПРИМЕР 79: 6-(Дипропиламино)-3,5,6,7-тетрагидро-2-метилциклопент[f] изоиндол-1(2Н)-он, 126
Используя методику 47, 6-(дипропиламино)-6,7-дигидро-2-метилциклопент[f] изоиндол-1,3(2Н,
5Н)-дион (120, 0,13 г, 0,4 ммоль) восстанавливали цинковой пылью (0,26 г, 4,0 ммоль) в ледяной НОАс (10 мл). Очистка кристаллизацией из EtOaC/MEOH давала не совсем белый твердый продукт, который
превращали в соль НСl и перекристаллизовали из EtOAc/MeOH, получая 126 в виде белого твердого продукта (т.пл. 242-243oС).
ПРИМЕР 80: 6-(Дипропиламино)-3,5,6,
7-тетрагидро-2-фенилциклопент[f] изоиндол-1(2Н)-он, 127
Используя методику 47, 6-(дипропиламино)-6,7-дигидро-2-фенилциклопент[f] изоиндол-1,3 (2Н, 5Н)-дион (121, 0,09 г, 0,23 ммоль)
восстанавливали цинковой пылью (0,15 г, 2,3 ммоль) в ледяной НОАс (5 мл). Очистка на силикагеле с элюированием смесью 3:1 гексан/ацетон давала масло, которое превращали в соль НСl и
перекристаллизовали из EtOAc/MeOH, получая 127 в виде белого твердого продукта (т.пл. 248-249oC).
ПРИМЕР 81: 6-(Дипропиламино)-3,5,6,
7-тетрагидро-3-[[4-(трифторметокси)фенил)метил]циклопент[f]изоиндол-1(2Н)-он, 128
Используя методику 47, 6-(дипропиламино)-6,7-дигидро-2-[[4-трифторметокси)фенил] метил] циклопент[f]
изоиндол-1,3(2Н,5Н)-дион (107, 0,17 г, 0,37 ммоль) восстанавливали цинковой пылью (0,24 г, 3,7 ммоль) в ледяной НОАс (10 мл), полученный твердый продукт превращали в соль НСl и перекристаллизовывали
из EtOAc/MeOH, получая 128 в виде белого твердого продукта (т.пл. 210-211oС).
ПРИМЕР 82: 2-[ (4-Хлорфенил)метил]-6-(дипропиламино)-3,5, 6,7-тетрагидроциклопент
[f]изоиндол-1(2Н)-он, 129
Используя методику 47, 2-[(4-хлорфенил)метил]-6-дипропиламино)-6,7-дигидpoциклoпeнт[f] изoиндoл-1,3(2Н,5Н)-дион (102, 0,59 г, 1,43 ммоль) восстанавливали цинковой
пылью (0,94 г, 14,3 ммоль) в ледяной НОАс (100 мл). Очистка с использованием силикагеля и элюированием смесью 4:1 гексан/ацетон давала масло, которое превращали в соль НСl и перекристаллизовали из
горячей смеси EtOAc/MeOH, получая 129 в виде белого твердого продукта (т.пл. 233-234oС).
ПРИМЕР 83: 6-(Дипропиламино)-3,5,6,7-тетрагидро-2-[(4-метоксифенилметил]
циклопент[f]изоиндол-1(2Н)-он, 130
Используя методику 47, 6-(дипропиламино)-6,7-дигидро-2-[(4-метоксифенил)метил] циклопент[f] изоиндол-1,3(2Н, 5Н)-дион (105, 0,27 г, 0,67 ммоль)
восстанавливали цинковой пылью (0,44 г, 6,7 ммоль) в ледяной НОАс (20 мл). Очистка с использованием силикагеля и элюирования смесью 8:1 CH2Cl2/ацетон давала масло, которое
превращали в соль НСl и перекристаллизовали из горячей смеси EtOAc/MeOH, получая 130 в виде белого твердого продукта (т.пл. 227-228oС).
ПРИМЕР 84: 6-(Дипропиламино)-3,5,6,
7-тетрагидро-2-[[4-(трифторметил)фенил]метил]циклопент[f]изоиндол-1(2Н)-он, 131
Используя методику 47, 6-(дипропиламино)-6,7-дигидро-2-[[4-(трифторметил)фенил] метил] циклопент[f]изоиндол-1,
3(2Н, 5Н)-дион (108, 0,44 г, 1 ммоль) восстанавливали цинковой пылью (1,2 г, 18,4 ммоль) в ледяной НОАс (20 мл). Очистка с использованием силикагеля и элюирования смесью 9:1 СН2Сl2/МеОН давала масло, которое превращали в соль НСl и перекристаллизовали из смеси метиленхлорид/ацетон, получая 132 в виде белого твердого продукта (т.пл. 190-192oС).
ПРИМЕР 85: 6-(Дипропиламино)-2-[(4-фторфенил)метил]-3,5, 6,7-тетрагидроциклопент[f]изоиндол-1(2Н)-он, 132
Используя методику 47, 6-(дипропиламино)-2-[(4-фторфенил)метил]-6,
7-дигидроциклопент[f]изоиндол-1,3(2Н,5Н)-дион (101, 0,56 г, 1,43 ммоль) восстанавливали цинковой пылью (0,94 г, 14,3 ммоль) в ледяной НОАс (20 мл). Очистка с использованием силикагеля и элюирования
смесью 9:1 CH2Cl2/MeOH давала масло, которое превращали в соль НСl и перекристаллизовали из EtOAc/MeOH, получая 132 в виде белого твердого продукта (т.пл. 227-228o
С).
ПРИМЕР 86: Методика 50. 4-[[6-(Дипропиламино)-3,5,6,7-тетрагидро-1-оксоциклопент[f]изоиндол-2(1Н)-ил]метил]бензонитрил, 133
К суспензии промытого NaH (0,05 г, 1,32 ммоль)
в ДМФ (5 мл) добавляли суспензию 6-(дипропиламино)-3,5,6,7-тетрагидроциклопент [f]изоиндол-1(2Н)-она (94, 0,30 г, 1,10 ммоль) в ДМФ (10 мл) суспензию нагревали до 84oС и через 40 мин
обрабатывали раствором альфа-бром-п-толунитрила (0,43 г, 2,20 ммоль) в ДМФ (10 мл). Через 24 ч реакционную смесь гасили Н2O и экстрагировали CH2Cl2. Органические слои
промывали солевым раствором, сушили (MgSO4) и концентрировали в высоком вакууме. Остаток очищали на силикагеле с элюированием СН2Сl2/МеОН, получая масло, которое
превращали в соль НСl и перекристаллизовали, получая 133 в виде белого твердого продукта (т.пл. 141-145oС).
ПРИМЕР 87: Методика 51: 7-(Дипропиламино)-2,3,7,
8-тетрагидро-1Н-циклопента[g]фталазин-1,4(6Н)-дион, 134
Смесь 2-(дипропиламино)-2,3-дигидро-1Н-инден-5,6-дикарбоксилата (92, 0,17 г, 0,5 ммоль) и гидрохлорида гидразина (0,05 г, 0,7 ммоль) в
ледяной НОАс (10 мл) кипятили с обратным холодильником при 125oС в течение 4 ч. Реакционную смесь охлаждали, концентрировали и превращали в соль НСl. Полученный твердый продукт
перекристаллизовали из EtOAc/MeOH, получая белый твердый продукт. Этот белый твердый продукт далее очищали хроматографией с обращенной фазой, элюируя Н2О/МеОН (95:5). Полученный продукт
превращали в соль НСl и перекристаллизовали из горячего МеОН, получая 134 в виде белого твердого продукта (т.пл. >300oС).
ПРИМЕР 88: 7-(Дипропиламино)-2,3,7,
8-тетрагидро-2-(фенилметил)-1Н-циклопента[g]фталазин-1,4(6Н)-дион, 135
Используя методику 51, 2-(дипропиламино)-2,3-дигидро-1H-инден-5,6-дикарбоксилат (92, 0,349 г, 1 ммоль) обрабатывали
дигидрохлоридом бензилгидразина (0,27 мл, 1,4 ммоль) в НОАс (20 мл). Очистка с использованием силикагеля и элюированием смесью 19:1 СН2Сl2/МеОН давала твердый продукт, который
превращали в соль НСl и перекристаллизовали из EtOAc/MeOH, получая 135 в виде белого твердого продукта (т.пл. 219-220oС).
В конце текста ниже приводятся схемы 1-3,
описанные выше. На схеме 1 соединения изобретения структурно представлены следующим образом:
R - Соединение
CH3 - 10
4-CN-Ph - 11
4-Cl-Ph - 12
3-NO2-Ph - 13
3-CN-Ph - 14
4-(1-метил-1Н-имидазол) - 15
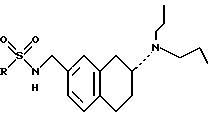
На схеме 2 соединения изобретения структурно представлены следующим образом:
R' - Соединение
4-Cl-Ph - 17
4-CN-Ph - 18
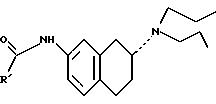
и
R' - Соединение
4-Cl-Ph - 20
4-CN-Ph - 21
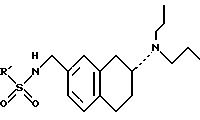
I. Тестируемые Соединения
A. Соединение по Аналогу
ЧТО наиболее близким аналогом к соединению по пункту 15 является 2-(дипропиламино)-6-метокси-2,3-дигидро-1Н-инден-5-карбоксамид, который является соединением из ПРИМЕРА 62 Международной Публикации WO 95/04713 (ХААДСМА-СВЕНССОН);
ЧТО соединение по ПРИМЕРУ 62 из ХААДСМА-СВЕНССОН (ХС-62) было получено химиком Фармация и Апджон д-ром Чу-Хонг Лин и представлено мне для биологических испытаний;
Б. Соединение по ПРИМЕРУ 38 Настоящей Патентной Заявки
ЧТО соединением по Пункту 15 настоящей патентной заявки является 2-(дипропиламино)-6-этокси-2,3-дигидро-1Н-инден-5-карбоксамид, который также является соединением по ПРИМЕРУ 38 настоящей патентной заявки;
ЧТО соединение по ПРИМЕРУ 38 настоящей патентной заявки (ПР-38) было получено д-ром Чу-Хонг Лин и направлено мне для биологических испытаний;
II. Биологические Испытания
ЧТО я сравнил ХС-62 и ПР-38 в стандартном определении сродства связывания рецептора.
А. Определение Сродства Связывания Рецептора
(1)
Теория
ЧТО определение сродства связывания применяет потому, что соединения, которые лечат шизофрению, связываются с различными рецепторами. Связывание с D2 рецептором приводит к
нежелательным побочным эффектам, поэтому желательно иметь очень низкое связывание с D2 рецептором. Связывание с D3 рецептором приводит к нужным терапевтическим эффектам. Связывание измеряют по
сродству к подходящему рецептору и представляют в виде величины Кi. Вследствие способа расчета величины константы сродства связывания (Ki) она обратно пропорциональна связыванию.
Следовательно, высокая или большая величина означает слабое связывание, и наоборот - низкая или небольшая величина означает хорошее сродство связывания. Для рецептора D2 желательно высокое значение, а
для рецептора D3 желательно низкое значение. Чем меньше величина соотношения D2/D3, тем более слабым является агент в лечении шизофрении, и чем ниже величина соотношения D2/D3, тем лучше действует
агент, причем без нежелательных побочных эффектов.
(2) Определения Связывания
ЧТО я проводил параллельные эксперименты с ХС-62 и ПР-38 в стандартном определении связывания
для рецепторов D2 и D3, используя известные методики, которые опубликованы, см. Sonesson и др., J.Med.Chem., 37, 2735-2753 (1994), копии прилагаются, а также цитированные там ссылки.
(3) Результаты Определения Связывания
ЧТО результаты определения связывания представлены ниже. Если какой-либо эксперимент проводился более одного раза, представленная в табл.3 величина
является средней.
(4) Интерпретация Данных по Связыванию
(а) D2
ЧТО вследствие большого химического структурного сходства ХС-62 и ПР-38 я ожидал, что величины D2
Ki для обоих соединений будут примерно одинаковы.
ЧТО различие между величинами D2 Кi для ХС-62 и для ПР-38 является удивительным и неожиданным.
(б) D3
ЧТО вследствие высокого химического структурного сходства ХС-62 и ПР-38 я ожидал, что величина D3 Ki для обоих соединений будет примерно одинаковой.
ЧТО различие между величинами D3 Кi для ХС-62 и для ПР-38 является удивительным и неожиданным.
(в) Соотношение D2/D3
ЧТО вследствие высокого химического структурного
сходства ХС-62 и ПР-38 я ожидал, что величина соотношения D2/D3 для обоих соединений будет примерно одинаковой.
ЧТО тот факт, что D2 ki для ХС-62 меньше, чем для ПР-38, в то время как D3 Ki для ХС-62 больше, чем для ПР-38, является весьма удивительным и неожиданным.
ЧТО большое различие между величинами соотношения D2/D3 для ХС-62 и ПР-38 является весьма удивительным и неожиданным.
5. Заключение
ЧТО свойства сродства связывания для ПР-38 являются удивительными и неожиданными с точки зрения таковых величин для
ХС-62, в особенности потому, что единственное химическое структурное различие между двумя соединениями состоит в введении этокси группы (С2Н5O-) взамен метокси группы (СН3О-).
Реферат
Изобретение относится к алкилзамещенным циклическим аминам формулы I, где Х и Y находятся в положении 5, 6 или 7, где i) n = 1, Х представляет (СН2)mCONR4R5, (CH2)mNR4CONR5, (CH2)m NHSO2R3, (где m = 0 или 1, за исключением того, что когда m = 0, то Y не может быть водородом и Y представляет водород или OR6; ii) n = 0 или 1, тогда Х и Y находятся в 0-положении относительно друг друга и вместе образуют: а) -С(О)NR10C(O)-, b) -C(O)NR4(CH2)xNR10C(O)- (где х = 0 или 1); с) -СН2 NR10C(O)-; d) -(CH2)NR10C(O)-, e) -CH2C(O)NR10, g) -N(R3)-C(O)-O-, j) -CH2N(R8)CH2-; (iii) n = 0 и Y представляет OR9, тогда Х представляет (CH2)mCONR4R5, (CH2)mNHCOR3 (где m = 0 или 1); R1 и R2 представляют, независимо, С1-С8-алкил, R3 представляет С1-С8-алкил, арил; R4 и R5 представляют, независимо, Н, С1-С8-алкил, С1-С6-алкиларил или арил; R6 представляет С1-С8-алкил, R8 представляет С1-С8 -алкил, SO2R4 (при условии, что R4 не является водородом); R10 представляет Н, С1-С8-алкил, С1-С6-алкиларил, арил, арил во всех перечисленных значениях представляет фенил, возможно замещенный метилом, нитро, хлором, фтором, бромом, амино, CN, карбоксамидо, ацетилом; или представляет тиофенил, изаксазолил, имидазолил. Соединения I являются селективными лигандами D3-допамина, что позволяет их использовать при лечении нарушений ЦНС. 15 з. п. ф-лы, 3 табл.
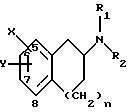
Формула
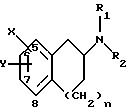
где Х и Y находятся в положении 5,6 или 7, где i) n = 1, Х представляет (СН2)m СONR4R5, (CH2)mNR4CONHR5, (CH2)mNHSO2R3 (где m = 0 или 1, за исключением того, что если m = 0, то Y не может быть водородом) и Y представляет водород, OR6; ii) n = 0 или 1, тогда Х и Y в о-положении относительно друг друга вместе образуют: а) -C(O)NR10C(O)-; b) -C(O)NR4(CH2)xNR10C(O)-, где х = 0 или 1; с) -CH2NR10C(O)-; d) -(CH2)2NR10C(O); e) -CH2 C(O)NR10-; g) -N(R3)-C(O)-O-; j) -CH2N(R8)CH2-; iii) n = 0 и Y представляет OR9, тогда Х представляет (СН2)m СONR4R5, (CH2)mNHСOR3 (где m = 0 или 1);
R1 и R2 независимо представляют С1-С8 алкил;
R3 представляет С1-С8 алкил, арил;
R4 и R5 представляют независимо Н, С1-С8 алкил, С1-С6 алкиларил или арил;
R6 представляет С1-С8 алкил;
R8 представляет С1-С8 алкил или SO2R4 (при условии, что R4 не является водородом); R9 представляет С2-С8 алкил (необязательно замещенный от 1 до 3 галогенами), С1-С6 алкиларил, арил;
R10 представляет Н, С1-С8 алкил, С1-С6 алкиларил, арил;
арил во всех перечисленных значениях представляет собой фенил, возможно замещенный заместителями, выбранными из метила, нитро, хлора, фтора, брома, амино, Сn, карбоксамидо, ацетила; тиофенил, изоксазолил, имидазолил.
Комментарии