Производные сукциноиламиногидроксиэтиламиносульфонамида, фармацевтическая композиция, способ ингибирования ретровирусной протеазы, способ лечения ретровирусной инфекции, способ лечения вич-инфекции - RU2130016C1
Код документа: RU2130016C1
Чертежи

Описание
Настоящая заявка является частичным продолжением заявки США рег. N 07/935 490, поданной 25 авг. 1992 г.
Предшествующий уровень техники
1. Область техники, к которой относится изобретение
Настоящее изобретение относится к ингибиторам
ретровирусной протеазы, а в частности к новым соединениям, композициям, и способам, предназначенным для ингибирования ретровирусных протеаз. Более конкретно, настоящее изобретение относится к
сульфонамид-содержащим гидроксиэтиламиновым соединениям, используемым в качестве ингибиторов протеазы, а также к способу ингибирования ретровирусных протеаз, таких как протеазы вируса иммунодефицита
человека (ВИЧ), и к способу лечения ретровирусных инфекций, например, ВИЧ - инфекций. Кроме того, настоящее изобретение относится к способам получения вышеуказанных соединений и их промежуточных
соединений, используемых в этих способах.
2. Известный уровень техники
В процессе репликативного цикла ретровирусов продукты генов gag и gag - poI транслируются в виде белков.
Затем эти белки подвергаются процессингу под действием протеаз (или протеиназ), кодируемых вирусным геномом, в результате чего синтезируются вирусные ферменты и структурные белки сердцевины вируса. В
основном, белки - предшественники гена gag процессируются с образованием белка сердцевины, а белки - предшественники гена poI процессируются с образованием вирусных ферментов, например, обратной
транскриптазы и ретровирусной протеазы. Было установлено, что для сборки инфекционного вириона необходим правильный процессинг белков - прешественников под действием ретровирусных протеаз. Например,
мутации со сдвигом рамки в протеазной области гена poI вируса ВИЧ могут воспрепятствовать процессингу белка-предшественника гена gag. Было также показано, что процессинг белка-предшественника гена gag
может быть предупрежден посредством сайт-направленного мутагенеза остатка аспарагиновой кислоты в ВИЧ-протеазе. Таким образом, были предприняты попытки ингибировать репликацию вируса путем
ингибирования действия ретровирусных протеаз.
Ингибирование ретровирусных протеаз может быть осуществлено посредством имитации переходного состояния путем воздействия на ретровирусную протеазу соединением - имитатором, которое, конкурируя с белками гена gag и gag-poI, связывается с ферментом, и тем самым способствует ингибированию репликации структурных белков, и, что более важно, самой ретровирусной протеазы. Таким образом может быть эффективно ингибирована репликация ретровирусных протеаз.
Для ингибирования ретровирусных протеаз, таких как протеаза ВИЧ, было предложено несколько классов соединений. Такими соединениями являются гидроксиэтиламиновые изостеры и изостеры восстановленных амидов. См., например, EP 0 346847, EP 0 342541; Roberts et al., "Rational Desing of Peptide-Based Proteinase Inhibitors", Science, 248, 358 (1990); и Erickson et al., "Design Activity, and 2,8
Известно несколько классов соединений, используемых в качестве ингибиторов протеолитического фермента ренина. См., например, патент США 4 599 198; патенты Великобритании 2184 730, 2 209 752 и 2 200 115, EP 0 264 795; и США SIR H 725. Из них в GB 2 200 115, GB 2 209 752, EP 0 264 795, США SIR H 725 и США 4 599 198 раскрываются мочевино-содержащие гидроксиэтиламиновые ингибиторы ренина. В пат. GB 2 200 115 также раскрываются некоторые сульфамоил-содержащие гидроксиэтиламиновые ингибиторы ренина. В EP 0 264 795, кроме того, раскрываются некоторые сульфонамид-содержащие соединения, ингибирующие ренин. Однако, известно, что хотя ренин и ВИЧ-протеаза, обе классифицируются как аспартил-протеазы, нельзя с уверенностью сказать, что соединения, являющиеся эффективными ингибиторами ренина, будут такими же эффективными ингибиторами ВИЧ-протеазы.
Краткое описание изобретения
Настоящее изобретение относится к вирус-ингибирующим соединениям и композициям. Более конкретно, настоящее изобретение относится к
соединениям и к композициям, ингибирующим ретровирусную протеазу, к способу ингибирования ретровирусных протеаз; к способам получения вышеуказанных соединений, а также к промежуточным соединениям,
используемым в этих способах. Соединениями настоящего изобретения являются сукциноиламиногидроксиэтиламиносульфонамидными ингибирующими соединениями.
Подробное описание изобретения
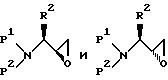
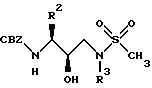
Настоящее изобретение относится к соединению, ингибирующему ретровирусные протеазы, и имеющему формулу:
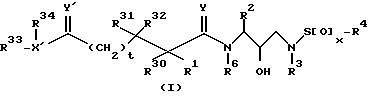
или к его фармацевтически приемлемой соли, сложному эфиру или предшественнику, где
x = 0, 1 или 2;
t = либо 0, либо 1;
R1 представляет собой водород, - CH2SO2NH2, - CO2CH3, - CONHCH3, - CON(CH3)2, -CH2C(O)NHCH3, -CH2 (O)N(CH3)2, -CONH2 -C(CH3)2(SH), -C(CH3)2 (SCH3), -C(CH3)2(S[O] CH3); -C(CH3)2(S[O]2CH3), алкил, галогеноалкил, алкенил, алкинил и циклоалкил; боковые цепи аминокислот, выбранных из аспарагина, S-метилцистеина и их соответствующих сульфоксидных и сульфоновых производных, а также боковые цепи глицина, лейцина, изолейцина, алло-изолейцина, трет-лейцина, фенилаланина, орнитина, аланина, гистидина, норлейцина, глутамина, валина, треонина, серина, о-алкилсерина, аспарагиновой кислоты, бета-цианоаланина и аллотреонина;
R2 представляет собой алкильный, арильный, циклоалкильный, циклоалкилалкильный и аралкильный радикалы, которые являются необязательно замещенными группой, выбранной из алкила и галогена; и - NO2, -CN, CF3 - OR9, - SR9, где R9 является водородом или алкилом;
R3 представляет собой водород, галогеноалкил, алкенил, алкинил, гидроксиалкил, алкоксиалкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, гетероарил, гетероциклоалкилалкил, арил, аралкил, гетероаралкил, аминоалкил и моно- и дизамещенный аминоалкил, где заместителей выбирают из алкила, арила, аралкила, циклоалкила, циклоалкилалкила, гетероарила, гетероаралкила, гетероциклоалкила и гетероциклоалкилалкила, либо, в случае дизамещенного амино алкильного радикала, указанные заместители вместе с атомом азота, с которыми они связаны, образуют гетероциклоалкильный или гетероарильный радикал;
X' представляет собой N, O и C(R17), где R17 является водородом или алкилом;
Y и Y' независимо представляют собой O, S и NR15, где R15 является водородом или радикалами, определенными выше для R3;
R4 представляет собой радикалы, определенные для R3 за исключением водорода;
R6 представляет собой водород и алкильные радикалы, такие как они были определены для R3;
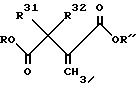
R30, R31 и R32 представляют собой радикалы, определенные выше для R1; либо один из R1 и R30, взятый вместе с одним из R31 и R32 и с атомами углерода, с которыми они связаны, образуют циклоалкильный радикал; и
R33 и R34 независимо представляют собой водород, и радикалы, определенные выше для R3; либо R33 и R34 вместе с X' представляют собой циклоалкильный, арильный, гетероциклильный и гетероарильный радикалы, при условии, что если X' представляет собой O, то R34 отсутствует.
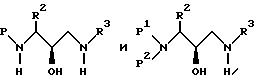
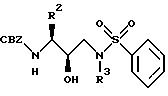
Предпочтительным классом соединений - ингибиторов ретровирусов по настоящему изобретению являются соединения формулы:
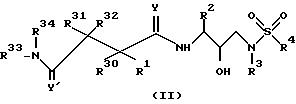
или их фармацевтически приемлемые соли, сложные эфиры или лекарства, а предпочтительно соединения, где абсолютная стереохимия у гидроксигруппы определена как (R);
где R1 представляет собой водород, -CH2SO2NH2, -CO2CH3, -CONHCH3, -CON(CH3)2, -CH2C(O)NHCH3, -CH2(O)N(CH3)2, -C(CH3)2 (SCH3), -C(CH3)2(SO[O] CH3), -C(CH3)2(S[O]2CH3), алкил, галогеноалкил, алкенил, алкинил и циклоалкил, а также боковые цепи аминокислот, выбранных из аспарагина, S-метилцистеина, и их соответствующих сульфоксидных и сульфоновых производных, и боковые цепи глицина, лейцина, изолейцина, алло-изолейцина, трет-лейцина, фенилаланина, орнитина, аланина, гистидина, норлейцина, глутамина, валина, треонина, серина, аспарагиновой кислоты, бета-цианоаланина и аллотреонина;
R2 представляет собой алкильный, арильный, циклоалкильный, циклоалкилалкильный и аралкильный радикалы, которые являются необязательно замещенными группой, выбранной из алкила и галогена, NO2, -C ≡ N, CF3, OR9 и SR9, где R9 является водородом и алкильными радикалами;
R3 представляет собой водород, галогеноалкил, алкенил, алкинил, гидроксиалкил, алкоксиалкил, циклоалкил, циклоалкилалкил, гетероциклоалкил, гетероарил, гетероциклоалкилалкил, арил, аралкил, гетероаралкил, аминоалкил и моно- и дизамещенные аминоалкилы, где заместителей выбирают из алкила, арила, аралкила, циклоалкила, циклоалкилалкила, гетероарила, гетероаралкила, гетероциклоалкила, и гетероциклоалкилалкила, либо, в случае дизамещенного аминоалкильного радикала, указанные заместители вместе с атомом азота, с которым они связаны, образуют гетероциклоалкильный или гетероарильный радикал;
R4 представляет собой радикалы, определенные для R3, за исключением водорода;
R30, R31 и R32 представляют собой радикалы, определенные выше для R1; либо один из R1 и R30, взятые вместе с одним из R31 и R32 и с атомами углерода, с которыми они связаны, образуют циклоалкильный радикал; и
R33 и R34 независимо представляют собой водород и радикалы, определенные выше для R3; либо R33 и R34, взятые вместе с атомом азота, с которыми они связаны, представляют собой гетероциклоалкильный и гетероарильный радикалы;
Y и Y' независимо представляют собой O, S и NR15, где R15 является водородом и радикалами, определенными выше для R3. Предпочтительно, если Y и Y' представляют собой O.
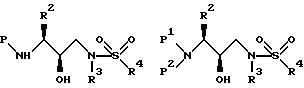
Другим предпочтительным классом соединений настоящего изобретения являются соединения формулы:
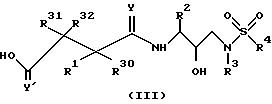
или их фармацевтически приемлемые соли, пролекарства, или сложные эфиры; а предпочтительно соединения, где стереохимия у гидроксигруппы обозначена как R; a, Y, Y', R1, R2, R3, R4, R30, R31 и R32 являются такими, как они были определены выше для формулы (II). При этом предпочтительно, если Y и Y' представляют собой O.
Используемый в настоящем описании термин "алкил" как отдельно, так и в комбинации, означает прямой или разветвленный алкильный радикал, содержащий от 1 до 10, а предпочтительно от 1 до около 8 атомов углерода. Примерами таких радикалов могут служить метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изоамил, гексил, октил и т.п. Термин "алкенил", используемый как отдельно, так и в комбинации, означает прямой или разветвленный углеводородный радикал, имеющий одну или несколько двойных связей, и содержащий от 2 до около 18 атомов углерода, а предпочтительно от 2 до около 8 атомов углерода. Примерами подходящих алкенильных радикалов являются этенил, пропенил, 1,4-бутадиенил и т.п. Термин "алкинил", используемый как отдельно, так и в комбинации, означает прямой углеводородный радикал, имеющий одну или несколько тройных связей и содержащий от 2 до около 10 атомов углерода. Примерами алкинильных радикалов являются этинил, пропинил, пропаргил и т.п. Термин "алкокси", используемый отдельно или в комбинации с другими терминами, означает алкилэфирный радикал, где термин "алкил" определен выше. Примерами подходящих алкилэфирных радикалов являются метокси, этокси, - н-пропокси, изопропокси, н-бутокси, изо-бутокси, втор-бутокси, трет-бутокси и т.п. Термин "циклоалкил", используемый отдельно или в комбинации с другими терминами, означает насыщенный или частично насыщенный моноциклический, бициклический или трициклический алкильный радикал, где каждая циклическая часть содержит от около 3 до около 8 атомов углерода. Термин "циклоалкилалкил" означает алкильный радикал, определенный выше и замещенный циклоалкильным радикалом, содержащим от около 3 до около 8, а предпочтительно от около 3 до около 6 атомов углерода. Примерами таких циклоалкильных радикалов являются циклопропил, циклобутил, циклопентил, циклогексил и т.п. Термин "арил", используемый отдельно или в комбинации с другими терминами, означает фенильный или нафтильный радикал, который необязательно имеет один или несколько заместителей, выбранных из алкила, алкокси, галогена, гидрокси, амино, нитро, циано, галогеноалкила и т.п., и примерами которого могут служить фенил, п-толуол, 4-метоксифенил, 4-(трет-бутокси)фенил, 4-фторфенил, 4-хлорофенил, 4-гидроксифенил, 1-нафтил, 2-нафтил и т. п. Термин "аралкил", используемый отдельно или в комбинации с другими терминами, означает алкильный радикал, определенный выше, в котором один атом водорода замещен арильным радикалом, определенным выше, и примерами которого могут служить бензил, 2-фенилэтил и т.п. Термин "аралкоксикарбонил", используемый отдельно или в сочетании с другими терминами, означает радикал формулы -(O)-O-аралкил, где термин "аралкил" имеет вышеуказанные значения. Примером аралкоксикарбонильного радикала является бензилоксикарбонил. Термин "арилокси" означает радикал формулы арил-O-, где термин "арил" имеет вышеуказанные значения. Термин "алканоил", используемый отдельно или в сочетании с другими терминами, означает ацильный радикал, который происходит от алканкарбоновой кислоты и примерами которого могут служить ацетил, пропионил, бутурил, валерил, 4-метилвалерил и т.п. Термин "циклоалкилкарбонил" означает ацильную группу, которая происходит от моноциклической или мостиковой циклоалканкарбоновой кислоты, такой как циклопропанкарбонил, циклогексанкарбонил, адамантанкарбонил и т.п., либо от бензконденсированной моноциклической циклоалканкарбоновой кислоты, необязательно замещенной, например, алканоиламино, такой как 1,2,3,4-тетрагидро-2-нафтоил-1,2-ацетамидо-1,2,3,4-тетрагидро-2- нафтоил. Термин "аралканоил" означает ацильный радикал, происходящий от арил-замещенной алканкарбоновой кислоты, такой как фенилацетил, 3-фенилпропионил (гидроциннамоил), 4-фенилбутурил, (2-нафтил)ацетил, 4-хлорогидроциннамоил, 4-аминогидроциннамоил, 4-метоксигидроциннамоил и т.п. Термин "ароил" означает ацильный радикал, происходящий от ароматической карбоновой кислоты. Примерами таких радикалов являются ароматические карбоновые кислоты, необязательно замещенные бензойные или нафтойные кислоты, такие как бензоил, 4-хлоробензоил, 4-карбоксибензоил, 4-(бензилоксикарбонил)бензоил, 1-нафтоил, 2-нафтоил, 6-карбокси-2-нафтоил, 6-(бензилоксикарбонил)-2-нафтоил, 3-бензилокси-2-нафтоил, 3-гидрокси-2-нафтоил, 3-(бензилоксиформамидо)-2-нафтоил и т.п. Гетероциклильной или гетероциклоалкильной частью гетероциклилкарбонильной, гетероциклилоксикарбонильной, гетероциклоалкоксикарбонильной или гетероциклиалкильной группы или т.п. является насыщенный или частично ненасыщенный моноциклический, бициклический или трициклический гетероцикл, который содержит один или несколько гетероатомов, выбранных из азота, кислорода и серы, который является необязательно замещенным на одном или нескольких атомах углерода галогеном, алкилом, алкокси, оксо и т.п., и/или на втором атоме азота (т.е., - NH) алкилом, аралкоксикарбонилом, алканоилом, фенилом или фенилалкилом, или на третьем атоме азота (т.е. =N-) оксидо-группой; и который присоединен посредством атома углерода. Гетероарильная часть гетероароильной, гетероарилоксикарбонильной, или гетероаралкоксикарбонильной группы, или т.п. представляет собой ароматический моноциклический, бициклический или трициклический гетероцикл, который содержит гетероатомы и является необязательно замещенным заместителями, определенными выше для гетероциклила. Примерами указанных гетероциклильных и гетероарильных групп являются пирролидинил, пиперидинил, пиперазинил, морфолинил, тиаморфолинил, пирролил, имидазолил (например, имидазол 4-ил, 1-бензилоксикарбонилимидазол-4-ил и т.п.), пиразолил, пиридил, пиразинил, пиримидинил, фурил, тиенил, триазолил, оксазолил, тиазолил, индолил (например, 2-индолил и т.п.), хинолинил (например, 2-хинолинил, 3-хинолинил, 1-оксидо-2-хинолинил и т. п. ), изохинолинил (например, 1-изохинолинил, 3- изохинолинил и т.п.), тетрагидрохинолинил (например, 1,2,3,4 - тетрагидро-2-хинолил и т. п.), 1,2,3,4-тетрагидроизохинолинил (например, 1,2,3,4-тетрагидро-1-оксоизохинолинил и т. п.), хиноксалинил, β- краболинил, 2-бензофуранкарбонил, 1-, 2-, 4 - или 5-бензимидазолил и т.п. Термин "циклоалкилалкоксикарбонил" означает ацильную группу, происходящую от циклоалкилалкоксикарбоновой кислоты формулы циклоалкилалкил-O-COOH, где циклоалкилалкил имеет значения, определенные выше. Термин "арилоксиалканоил" означает ацильный радикал формулы арил-O-алканоил, где арил и алканоил имеют значения, определенные выше. Термин "гетероциклилоксикарбонил" означает ацильную группу, происходящую от гетероциклил-O-COOH, где термин "гетероциклил" определен выше, Термин "гетероциклиалканоил" означает ацильный радикал, происходящий от гетероциклил-замещенной алканкарбоновой кислоты, где гетероциклил имеет значения, определенные выше. Термин "гетероциклиалкоксикабонил" означает ацильный радикал, происходящий от гетероциклил-замещенной алкан-O-COOH, где гетероциклил имеет значения, определенные выше. Термин "гетероарилоксикарбонил" означает ацильный радикал, происходящий от карбоновой кислоты формулы гетероарил-O-COOH, где гетероарил имеет значения, определенные выше. Термин "аминокарбонил", используемый отдельно или в сочетании с другими терминами, означает амино-замещенную карбонильную (карбамоильную) группу, происходящую от амино-замещенной карбоновой кислоты, где аминогруппой может быть первичная, вторичная или третичная аминогруппа, имеющая заместителей, выбранных из водорода, алкила, арила, аралкила, циклоалкила, циклоалкилалкила и т.п. Термин "аминоалканоил" означает ацильную группу, происходящую от аминозамещенной алканкарбоновой кислоты, где аминогруппой может быть первичная, вторичная или третичная аминогруппа, имеющая заместителей, выбранных из водорода, алкила, арила, аралкила, циклоалкила, циклоалкилалкила и т.п. Термин "галоген" означает фтор, хлор, бром или иод. Термин "уходящая группа" относится, в основном, к группам, которые могут быть легко заменены нуклеофилом, таким как амин, тиол или спиртовой нуклеофил. Такие уходящие группы хорошо известны специалистам. Примерами уходящих групп являются N-гидроксисукцинимид, N-гидроксибензотриазол, галиды, трифторацетаты, тозиалаты и т.п. Предпочтительные уходящие группы указаны в настоящем описании, там, где это необходимо.
Ниже описаны методики получения соединений формулы I. При этом следует отметить, что описанные общие методики относятся к получению соединений, имеющих определенную стереохимию, например соединений, где абсолютная стереохимия у гидроксильной группы обозначается (R). Однако, вообще говоря, эти методики могут быть применены к соединениям с противоположной конфигурацией, например к соединениям, где стереохимия у гидроксильной группы является (S). Кроме того, соединения, имеющие стереохимию (R), могут быть использованы для получения соединений, имеющих стереохимию (S). Например, соединение, имеющее стереохимию (R), может быть превращено в соединение, имеющее стереохимию (S), хорошо известными методами.
Получение
соединений формулы I
Соединения настоящего изобретения формулы I могут быть получены с помощью нижеследующей общей процедуры. Защищенное хлорокетоновое производное аминокислоты формулы:
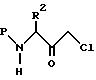
где P представляет собой аминозащитную группу, а R2 определен выше, восстанавливают от соответствующего спирта с использованием подходящего восстановителя. Подходящие аминозащитные группы хорошо известны специалистам, и примерами таких групп могут служить карбобензокси, бутирил, т-бутоксикарбонил, ацетил, бензоил и т.п. Предпочтительной аминозащитной группой является карбобензокси. Предпочтительным N-защищенным хлоркетоном является N-бензилоксикарбонил L-фенилаланинхлорметилкетон. Предпочтительным восстановителем является борогидрид натрия. Реакцию восстановления осуществляют при температуре от -10oC до около 25oC, а предпочтительно при около 0oC, в соответствующей системе растворителей, например в тетрагидрофуране, и т.п. N-защищенные хлорокетоны являются коммерчески доступными соединениями (например, Bachem Inc., Torrance California). Альтернативно, хлорокетоны могут быть получены с помощью методики, описанной S.J.Fittkau (J. Prakt. Chem. , 315, 1037 (1973), после чего осуществляют N-защиту с использованием методик, хорошо известных специалистам.
Галогеноспирт может быть использован непосредственно, как описано ниже, либо предпочтительно его подвергают реакции
предпочтительно при комнатной температуре с соответствующим основанием в подходящей системе растворителей, в результате чего получают L-защищенный аминоэпоксид формулы:
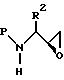
где P и R2 определены выше. Подходящей системой растворителей, используемой для получения аминоэпоксида, является этанол, метанол, изопропанол, тетрагидрофуран, диоксан и т.п., а также их смеси. Подходящими основаниями для получения эпоксида из восстановленного хлорокетона являются гидроксид калия, гидроксид натрия, н-бутоксид калия, DBU и т.п. Предпочтительным основанием является гидроксид калия.
Альтернативно, защищенный аминоэпоксид может быть получен исходя
из L-аминокислоты, которую подвергают реакции с соответствующей амино-защитной группой в соответствующем растворителе, в результате чего получают сложный эфир аминозащищенной L-аминокислоты
формулы:
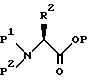
где P1 и P2 независимо представляет собой водород, бензил и аминозащитные группы, определенные выше для P, при условии, что R1 и R2 оба не являются водородом, P3 является карбоксизащитной группой, такой как метил, этил, трет-бутил, бензил и т.п. а R2 является таким, как он был определен выше.
Затем сложный эфир амино-защищенной L-аминокислоты подвергают реакции восстановления с получением
соответствующего спирта. Например, сложный эфир амино-защищенной L-амикнокислоты может быть восстановлен с использованием гидрида диизобутилалюминия при - 78oC в подходящем растворителе,
таком как толуол. После этого полученный спирт превращают, например, путем окисления Сверна в соответствующий альдегид формулы:
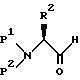
где P1, P2 и R2 определены выше. Так, например, дихлорметановый раствор спирта добавляют к охлажденному (-75o) - (-68oC) раствору оксалилхлорида в дихлорметане и ДМСО в дихлорметане, а затем размешивают в течение 35 минут.
Альдегид, полученный в результате окисления Сверна,
подвергают реакции с галогенометиллитиевым реагентом, который продуцируют in situ путем реакции алкиллития или ариллития с дихлорметаном формулы X1 CH2 X2, где X1 и X2 независимо представляют собой I, Br или Cl. Например, раствор альдегида и хлороидометана в ТГФ охлаждают до - 78oC, после чего добавляют раствор н-бутиллития в
гексане. Полученный продукт представляет собой смесь диастереомеров соответствующих аминозащищенных эпоксидов формул:
Эти диастереомеры могут быть разделены, например, путем хроматографии; либо альтернативно, они могут быть разделены после реакций этих продуктов в последующих стадиях. Для соединений, имеющих стереохимия (S), вместо D-аминокислоты может быть использована L-аминокислота.
Затем аминоэпоксид подвергают реакции в соответствующей системе
растворителей с равным количеством или предпочтительно с избыточным количеством нужного амина формулы: R3 NH2,
где R3 является водородом или имеет значения,
определенные выше. Эта реакция может быть осуществлена в широком диапазоне температур, например при температурах от около 10oC до около 100oC, а предпочтительно, хотя и
необязательно, чтобы эта реакция протекала при температуре кипения растворителя. Подходящими растворителями являются протонные, непротонные и диполярные апротонные растворители, например спирты, такие
как метанол, этанол, изопропанол, и т.п., эфиры, такие как тетрагидрофуран, диоксан, и т. п.; а также толуол, N,N-диметилформамид, диметилсульфоксид и их смеси. Предпочтительным растворителем является
изопропанол. Примерами аминов, имеющих формулу R3NH2, являются амин, изобутиламин, н-бутиламин, изопентиламин, изоамиламин, циклогексанметиламин, нафтиленметиламин и т.п.
Полученный продукт, который представляет собой производное 3-(N-зищещенный амино)-3-(R2)-1-(NHR3)-пропан-2-ола (обозначаемое далее аминоспиртом), может быть представлен
формулами:
где P, P1, P2, R2 и R3 определены выше. Альтернативно, вместо аминоэпоксида может быть использован галогеноспирт.
Затем аминоспирт, определенный выше, подвергают реакции в соответствующем растворителе с
сульфонилхлоридом (R4SO2Cl) или сульфонилангидридом в присутствии акцептора кислоты. Растворителями, подходящими для этой реакции, являются метиленхлорид, тетрагидрофуран и т.п.
Подходящим акцепторами кислоты являются триэтиламин, пиридин и т.п. Предпочтительными сульфонилхлоридами являются метансульфонилхлорид и бензолсульфонилхлорид. В зависимости от используемого эпоксида,
полученное сульфонамидное производное может быть представлено формулами:
где P, P1, P2, R2, R3 и R4 определены выше.
Сульфонилгалиды формулы R4SO2X могут быть получены с помощью реакции подходящего реагента Гриньяра или алкиллитиевого реагента с сульфонилхлоридом или с диоксидом серы с последующим окислением галогеном, предпочтительно хлором. Кроме того, тиолы могут быть окислены до сульфонилхлоридов с помощью хлора в присутствии воды и при тщательно контролируемых условиях. Помимо этого, сульфоновые кислоты могут быть превращены в сульфонилгалиды с использованием таких реагентов, как PCl5, а также в ангидриды с использованием подходящих дегитратирующих реагентов. Сульфоновые кислоты могут быть, в свою очередь, получены с использованием хорошо известных методик. Сульфоновые кислоты также являются коммерчески доступными продуктами.
Для получения соединений, где -SO2- часть заменяется частью -SO- или -S-, вместо сульфонилгалидов могут быть использованы сульфинилгалиды (R4SOX) или сульфонилгалиды (R4SX) соответственно.
После получения сульфонамидного производного аминозащитные группы P или P1 и P2 удаляют в условиях, не оказывающих неблагоприятного воздействия на оставшуюся часть молекулы. Эти методы хорошо известны специалистам, например, в этих целях может быть использован кислотный гидролиз, гидрогенолиз и т. п. Предпочтительным способом удаления защитной группы, например карбобензокси- группы, является гидрогенолиз в присутствии палладированного угля и в подходящем растворителе, таком как спирт, уксусная кислота и т.п. или их смеси. Если защитной группой является T- бутоксикарбонильная группа, то она сможет быть удалена с использованием неорганической или органической кислоты, например HCl или трифторуксусной кислоты, в подходящем растворителе, например в диоксане или метиленхлориде. Полученный продукт представляет собой производное аминовой соли. Если защитной группой является бензильный радикал, то он может быть удален путем гидрогенолиза. Затем, после нейтрализации соли, амин подвергают реакции с янтарной кислотой, как описано ниже.
Для получения янтарнокислотной части соединений формулы 1 в качестве исходного материала используют лактат формулы:
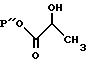
где P'' представляет собой алкильный и аралкильный радикалы, такие как этил, метил, бензил и т.п. Гидроксильную группу лактата защищают путем образования кеталя с помощью реакции в подходящем растворителе с метилизопропениловым эфиром (1,2-метоксипропен) в присутствии подходящей кислоты. Подходящими растворителями являются метиленхлорид, тетрагидрофуран и т.п., а также их смеси. Подходящей кислотой является POCl3 и т.п., при этом следует отметить, что для образования кеталя вместо метилизопропенилового эфира могут быть использованы и другие хорошо известные группы. Затем указанный кеталь подвергают реакции восстановления с использованием гидрида диизобутилалюминия (DIBL) при -78oC, в результате чего получают соответствующий альдегид, который затем обрабатывают этилидентрифенилфосфораном (реакция Виттига), и получают соединение формулы:
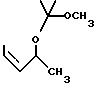
Затем кетальную защитную группу удаляют с помощью известных методик, таких как мягкий гидролиз в присутствии слабой кислоты. Полученное соединение подвергают реакции этерификации с изобутилхлоридом, в результате чего получают соединение формулы:
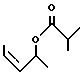
Затем это соединения обрабатывают диизопропиламидом лития при - 78oC и после нагревания реакционной смеси до комнатной температуры осуществляют перегруппировку Кляйзена ([3, 3]), в результате чего получают соответствующую кислоту формулы:
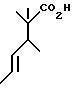
При этом каждому специалисту понятно, что описанная схема допускает различные варианты, которые предусматривают использование различных защитных групп или реагентов для осуществления аналогичных преобразований. Например, для получения соответствующих аналогов, вместо изобутилхлорида могут быть также использованы другие хлорангидриды.
В результате обработки кислоты бензилбромидом в присутствии третичного аминового
основания, например DBU, получают соответствующий сложный эфир, который затем подвергают окислительному расщеплению с получением тризамещенной янтарной кислоты:
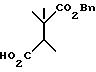
Затем тризамещенную янтарную кислоту подвергают взаимодействию с сульфонамидным изостером с помощью известных методик. Для получения соответствующей свободной кислоты бензильный сложный эфир удаляют путем гидрогенолиза. Затем полученную кислоту превращают в первичный амид стандартными способами, хорошо известными специалистам. Полученный продукт представляет собой соединение формулы I.
Альтернативный метод получения тризамещенных янтарных кислот заключается в том, что сложный эфир ацетоуксусной
кислоты формулы:
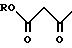
где R представляет собой подходящую защитную группу, такую как метил, этил, бензил или т-бутил, подвергают реакции с гидридом натрия и гидрокарбилгалидом (R31 X или R32 X) в подходящем растворителе, например ТГФ, в результате чего получают соответствующее дизамещенное производное формулы:
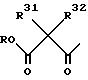
Это дизамещенное производное ацетоуксусной кислоты обрабатывают диизопропиламидом лития при температуре около -10oC и в присутствии PhN (трифторацетата)2, в результате чего получают винилтрифтолат формулы:
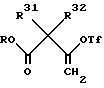
Затем винилтрифлат подвергают реакции карбонилирования с использованием палладиевого катализатора, например Pd (OAc)2 и Ph3P, в присутствии спирта (R''OH) или воды (R''=H) и основания, например триэтиламина, и в подходящем растворителе, таком как ДМФ, в результате чего получают олефиновый сложный эфир или кислоту формулы:
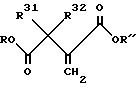
Затем олефин подвергают асимметрической гидрогенизации, как описано выше, и получают тризамещенное производное янтарной кислоты формулы:
В случае, если R'' не является H, то R'' может быть удален путем гидролиза, ацидолиза либо гидрогенолиза, с получением соответствующей кислоты, которую затем подвергают реакции взаимодействия с сульфонамидным изостером, как описано выше, после чего R - группа может быть удалена (но необязательно) с получением соответствующей кислоты, которая может быть превращена (но необязательно) в амид.
Альтернативно, сульфонамидный изостер может быть подвергнут реакции либо с соответствующим образом замещенной янтарной кислотой, либо с глутаровой кислотой формулы:
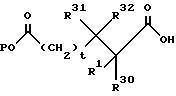
с последующим удалением защитной группы и превращением полученной кислоты в амид. Может быть также осуществлена реакция ангидрида формулы:
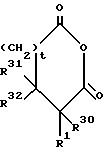
с сульфонамидным изостером с последующим разделением изомеров или превращением полученной кислоты в амид и выделением любого из изомеров.
Очевидно, что соединение Формул, имеющих R6, могут быть получены в соответствии с методикой, описанной выше и осуществляемой перед стадией реакции взаимодействия сульфонамидного производного с янтарнокислотной частью молекулы посредством восстановительного аминирования. Так, например, цианоборогидрид натрия и соответствующий альдегид или кетон могут быть подвергнуты реакции с сульфонамидным производным или соответствующим аналогом при комнатной температуре для осуществления восстановительного аминирования любого из соединений формул I-III. Очевидно также, что если R3 аминоспиртового промежуточного соединения является водородом, то ингибирующие соединения могут быть получены посредством восстановительного аминирования конечного продукта реакции между аминоспиртом и амином, либо на любой другой стадии синтеза ингибирующих соединений.
Рассмотренные эквиваленты общих формул, представленных выше для антивирусных соединений, их производных, и промежуточных соединений, во всех остальных отношениях соответствуют указанным соединениям и производным и имеют те же самые общие свойства, а одна или несколько различных групп R в указанных эквивалентах являются лишь вариантами заместителей, определенных выше, например, R является более высшей алкильной группой, чем алкильные группы, указанные выше. Кроме того, в том, случае, если в качестве заместителя указан водород, или этот заместитель может быть водородом, то конкретная химическая природа заместителя, не являющегося водородом в этом положении (например, являющегося гидрокарбильным радикалом, галогеном, гидрокси, амино или другой функциональной группой), не имеет решающего значения, если только она не оказывает неблагоприятного воздействия на общую активность и/или методику синтеза.
Вышеописанные химические реакции раскрываются исходя из наиболее широкого их применения в целях получения соединений настоящего изобретения. Однако может случиться, что данные реакции не могут быть непосредственно использованы для получения конкретного соединения, входящего в объем настоящего изобретения. Эти соединения могут быть легко определены любым специалистом. Во всех таких случаях, могут быть осуществлены либо те же самые реакции, подвергнутые лишь стандартным модификациям, известным специалистам (например, таким как соответствующая защита промежуточных групп, замена на альтернативные стандартные реагенты, обычная модификация реакционных условий и т.п. ), либо другие реакции, описанные в настоящей заявке или в других работах и подходящие для получения соответствующих соединений настоящего изобретения. В описанных препаративных методах все исходные материалы являются известными, либо они могут быть легко получены из известных исходных материалов.
Используя вышеприведенное описание изобретения, каждый специалист может применять это изобретение во всей его полноте, не прибегая при этом к дополнительному и трудоемкому экспериментированию. Вышеописанные предпочтительные варианты настоящего изобретения приводятся лишь в иллюстративных целях и не должны рассматриваться как некое ограничение его объема.
Все варианты использовали без очистки. Все протонные и углеродные ЯМР - спектры были получены либо на ЯМР - спектрометре Varian VXP-300, либо на VXP-400.
Пример 1
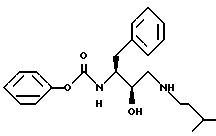
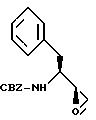
Получение N[3(S)-бензилоксикарбониламино-2 (R)-гидрокси-4-фенилбутил]-N-изомиламина
Часть A:
К раствору 75,0 г (0,226 М) N-бензилоксикарбонил-L фенилаланинхлорометилкетона в смеси 807 мл метанола и 807 мл тетрагидрофурана при - 2oC и в течение 1,4 часа добавляли 13, 17 г (0,348 М, 1,54 экв.) твердого борогидрида натрия. Растворители удаляли при пониженном давлении при 40oC, и образовавшийся остаток растворяли в этилацетате (прибл. 1 л). Затем раствор промывали 1 М - бисульфатом калия, насыщенным бикарбонатом натрия и насыщенными растворами хлорида натрия. После осушки безводным сульфатом магния и фильтрования раствор удаляли при пониженном давлении. К полученному маслообразному веществу добавляли гексан (приблизительно 1 л), и смесь нагревали до 60oC, перемешивая при этом. После охлаждения до комнатной температуры твердые вещества собирали и промывали 2 л гексана. Полученное твердое вещество перекристаллизовывали из горячего этилацетата и гексана, в результате чего получали 32,3 г (выход 0,43%) N-бензилоксикарбонил -3(S)-амино-1-хлоро-4-фенил-2(S)-бутанола,
Т.пл. 150-151oC и M + Li+=340.
Часть B:
К раствору 6,52 г (0,116 М, 1,2 экв.) гидроксида калия в 968 мл абсолютного этанола при комнатной
температуре добавляли 32,3 г (0,097 М) N-CBZ-3-(S)-амино-1-хлоро-4-фенил-2(S)-бутанола. После перемешивания в течение 50 минут растворитель удаляли при пониженном давлении, и образовавшиеся твердые
остатки растворяли в метиленхлориде. После промывания водой, осушки сульфатом магния, фильтрования и выпаривания получали 27,9 г белого твердого вещества. Это вещество перекристаллизовывали из
горячего этилацетата и гексана, в результате чего получали 22,3 г (выход = 77%) N-бензилоксикарбонил-3-(S)-амино-1,2-(S)-эпокси-4-фенилбутана,
Т.пл. 102-103oC, MH+ =
298.
Часть C:
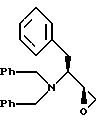
Раствор N-бензилоксикарбонил-3(S)-амино1,2(S)эпокси-4- фенилбутана (11,54 г, 38,81 ммоль) и изоамиламина (66,90 г, 767 ммоль, 19,9 экв.) в 90 мл изопропилового
спирта нагревали с обратным холодильником в течение 3,1 часа. Полученный раствор охлаждали до комнатной температуры и частично концентрировали в вакууме, после чего оставшийся раствор выливали в 200
мл гексана, перемешивая при этом, а затем образовавшийся продукт кристаллизовали из раствора. Этот продукт выделяли путем фильтрации и осушали воздухом, в результате чего получали 11,76 г (выход =
79%) N[[3-(S)-фенилметилкарбамоил)амино-2-(R)-гидрокси-4-фенилбутил] N[[(3-метилбутил)]амина, Т. пл. 118-122oC.
FAB - MC : MH+= 385.
Пример 2
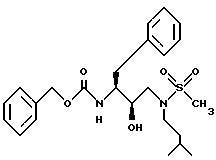
Получение фенилметил [2R-гидрокси-3-[(3-метилбутил) (метилсульфонил)амино]-1S-(фенилметил)пропил]карбамата
К раствору N[3(S)-бензилоксикарбониламино-2(R)-гидроки-4-фенилбутил] N-изоамиламина из Примера 1 (часть C, 2,0 г, 5,2 мМ) и триэтиламина (723 мкл, 5,5 мМ) в дихлорметане (20 мл) по капле добавляли метансульфонилхлорид (400 мкл, 5,2 мМ). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, после чего дихлорметановый раствор концентрировали приблизительно до объема 5 мл и переносили на колонку с силикагелем (100 г). Эту колонку элюировали хлороформом, содержащим 1% этанол и 1% метанол. Таким образом получали фенилметил [2R-гидрокси-3-[(3-метилбутил) (метилсульфонил)амино]-1S-(фенилметил)пропил]карбамат в виде белого твердого вещества.
Анализ для C24H34
H2O2S:
Вычислено: C 62,31; H 7,41; N 6,06.
Найдено: C 62,17; H 7,55; N 5,97.
Пример 3
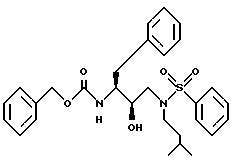
Получение фенилметил [2R-гидрокси-3-[(3-метилбутил) (фенилсульфонил)амино-1S-(фенилметил)пропил]карбамата
В результате реакции N[3(S)-бензилоксикарбониламино-2(R)-гидрокси-4-фенилбутил]N- изоамиламина (Пример 1, часть C, 1,47 г, 3,8 мМ) с триэтиламином (528 мкл, 3,8 мМ) и бензолсульфонилхлоридом (483 мкл, 3,8 мМ) получали фенилметил[2R-гидрокси-3-[(3-метилбутил) фенилсульфонил)амино] -1S-(фенилметил)пропил] карбамат. Это вещество подвергали колоночной хроматографии на силикагеле, элюируя хлороформом, содержащим 1%-ный этанол, в результате чего получали чистый продукт.
Анализ для C29H36N2O5S:
Вычислено: C 66,39; H 6,92 N 5,34;
Найдено: C
66,37; H 6,93 N 5,26.
Пример 4
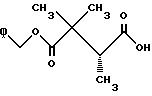
Получение бензил 2,2,3 (R)-триметилсукцината
Часть A :
Получение метил (S)-лактата, 2-метокси-2-пропилового эфира
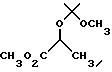
К смеси метил (S) - (-) - лактата (13,2 г, 100 мМ) и 2-метоксипропена (21,6 г, 300 мМ) в метиленхлориде (150 мл) добавляли POCl3 (7 капель). После этого полученную смесь перемешивали при комнатной температуре в течение 16 часов. После добавления Et3N (10 капель), растворители удаляли в вакууме и получали 20,0 г (98%) нужного продукта.
Часть
B :
Получение 2 (S)-гидроксипропаналя, 2-метокси-2-пропилового эфира
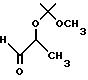
К раствору соединения Части A (20,0 г) в метиленхлориде (100 мл) по капле в течение 45 минут и при -78oC добавляли DIBAL (65 мл 1,5 М - раствор в толуоле, 97,5 мМ), а затем полученную смесь перемешивали при той же температуре в течение еще 45 минут. К этому холодному раствору добавляли MeOH (20 мл), а затем добавляли насыщенный раствор NaCl (10 мл) и полученную реакционную смесь нагревали до комнатной температуры. После разведения эфиром (200 мл), смесь перемешивали еще 2 часа, а затем добавляли 150 г сульфата магния. Полученную смесь фильтровали, а образовавшиеся твердые остатки дважды промывали эфиром. Объединенные фильтраты выпаривали на роторном испарителе и получали 11,2 г (выход = 78%) целевого альдегида.
Часть C:
Получение
2(S)-гидрокси-цис-3-бутена, 2-метокси-2-пропилового эфира
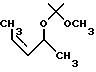
К суспензии бромида этилтрифенилфосфония (28 г, 75,5 мМ) в ТГФ (125 мл) порциями и при 0oC добавляли KN (TMS)2 (15,7 г, 95%, 75 мМ), а затем полученную смесь перемешивали в течение часа при той же температуре. Красную реакционную смесь охлаждали до -78oC и добавляли раствор альдегида Части B (11 г, 75 мМ) в ТГФ (25 мл). После завершения добавления полученную реакционную смесь оставляли для нагревания до комнатной температуры, а затем перемешивали в течение 16 часов. К этой смеси добавляли насыщенный NH4Cl (7,52 мл) и фильтровали через слой целита с тонким слоем силикагеля в верхней части. Остаточное твердое вещество дважды промывали эфиром. Объединенные фильтраты концентрировали в вакууме и получали 11,5 г неочищенного продукта. Этот неочищенный продукт очищали с помощью флеш-хроматографии (силикагель, гексан /KtOAc, 10:1) и получали 8,2 г (Выход = 69%) чистого алкена.
Часть D:
Получение 2(S)-гидрокси-цис-3-бутена
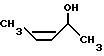
Смесь алкила Части C (8,2 г) и 30% водного раствора уксусной кислоты (25 мл) перемешивали при комнатной температуре в течение часа. К полученной смеси медленно добавляли NаHCO3 до тех пор, пока pH не становился приблизительно равным 7, после чего смесь экстрагировали эфиром (10 мл • 5). Объединенные эфирные растворы осушали сульфатом натрия и фильтровали. Фильтрат подвергали дистилляции для удаления эфира, в результате чего получали 2,85 г (64%) чистого спирта, m/e = 87 (M + H).
Часть E:
Получение 2,2,3-триметил-гекс-(транс)-4-енойной кислоты
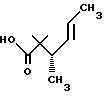
К смеси спирта Части D (2,5 г, 29 мМ) и пиридина (2,5 мл) в метиленхлориде (60 мл) медленно при 0oC добавляли изобутирилхлорида (3,1 г, 29 мМ). Полученную смесь перемешивали при комнатной температуре в течение 2 часов, а затем промывали водой (30 мл • 2) и насыщенным хлоридом натрия (25 мл). Объединенные органические фазы осушали сульфатом натрия и концентрировали с получением 4,2 г (93%) изобутирата, 2(S)-гидрокси-цис-3-бутенилизобутирата сложного эфира. Затем это вещество растворяли в тетрагидрофуране (10 мл) и добавляли (при -78oC) 1,0 М -LDA (13,5 мл 2,0 М -LDA в ТГФ и 13,5 мл ТГФ). Полученную смесь оставляли для нагревания до комнатной температуры, а затем перемешивали 2 часа и разводили 40 мл 5% NaOH. После этого органическую фазу отделяли, а водную фазу промывали 10 мл Et2O.
Водный раствор собирали и подкисляли 6 н. соляной кислотой до pH X ≈ 3. Затем смесь три раза экстрагировали эфиром (30 мл). Объединенные эфирные слои промывали насыщенным хлоридом натрия (25 мл), осушали сульфатом натрия и концентрировали с получением 2,5 г (60%) нужной кислоты, m/e = 157 (M+H).
Часть F:
Получение бензил 2,2,3(S)-триметил-транс-4-гексеноата
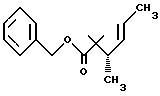
Смесь кислоты Части E (2,5 г, 16 мМ) BnBr (2,7 г, 15,8 мМ), K2CO3 (2,2 г, 16 мМ), и NaI (2,4 г) в ацетоне (20 мл) нагревали в масляной бане в течение 16 часов при 75oC. После выпаривания ацетона остаток растворяли в воде (25 мл) и эфире (35 мл). Эфирный слой отделяли, осушали сульфатом натрия и концентрировали, в результате чего получали 3,7 г (выход = 95%) бензилового сложного эфира, m/e = 247 (M + H).
Часть G:
Получение бензил 2,2,3(R)-триметилсукцината
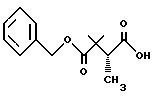
При 0oC, к тщательно перемешанной смеси KMnO4 (5,4 г, 34,2 мМ), H2O (34 мл), CH2Cl2 (6 мл) и хлорида бензилтриэтиламмония (200 мг) медленно добавляли раствор сложного эфира Части F (2,1 г, 8,54 мМ) и уксусной кислоты (6 мл) в метиленхлориде (28 мл). Полученную реакционную смесь перемешивали при той же температуре в течение 2 часов, а затем при комнатной температуре в течение 16 часов. После этого смесь охлаждали в бане из льда и воды, а затем добавляли 6 н. соляную кислоту (3 мл) и твердый NaHSO3 порциями до тех пор, пока не исчезала красная окраска. Прозрачный раствор экстрагировали метиленхлоридом (30 мл • 3). Объединенные экстракты промывали насыщенным раствором хлорида натрия, осушали сульфатом натрия и концентрировали с получением маслообразного вещества. Это вещество растворяли в Et2O (50 мл) и добавляли насыщенный гидрокарбонат натрия (50 мл). Водный слой отделяли и подкисляли путем добавления 6 н. соляной кислоты до pH ≈ 3, а затем экстрагировали 30 миллилитрами Et2O (3 раза). Объединенные экстракты промывали насыщенным раствором хлорида натрия (15 мл), осушали сульфатом натрия и концентрировали с получением 725 мг (34%) нужной кислоты, а именно бензил 2,2,3(R)-триметилсукцината, m/e = 251 (M + H).
Пример 5
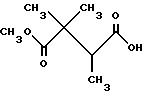
Получение (R) и (S) изомеров метил2,2-диметил-3-метилсукцината
Часть A:
Получение метил 2, 2-диметил-3-оксобутаноата
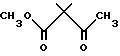
250-миллилитровую RB-колбу, снабженную магнитной мешалкой, стержнем для перемешивания и отверстием для впуска азота, загружали 100 мл безводного тетрагидрофурана и 4,57 г (180 мМ) 95% NaH. Полученную суспензию охлаждали до -20oC и по капле добавляли 10 г (87 мМ) метилацетоацетата, а затем 11,3 мл (181 мМ) CH3I. Реакционную смесь перемешивали при 0oC в течение 2 часов, после чего оставляли для охлаждения до комнатной температуры на ночь. Полученную реакционную смесь фильтровали для удаления NaI и разводили 125 мл Et2O. Органическую фазу промывали 5%-ным солевым раствором (1 • 100 л), осушали и концентрировали в вакууме, в результате чего получали темно-золотистое маслообразное вещество, которое фильтровали через слой силикагеля (30 г), элюируя гексаном. После концентрирования в вакууме получали 10,05 г целевого метилового сложного эфира в виде бледно-желтого маслообразного вещества, которое использовали без дополнительной очистки.
Часть B:
Получение метил 2,
2-диметил-3-O-(трифторометансульфонат)-бут-3- еноата
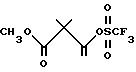
В 250-миллилитровую круглодонную колбу, снабженную магнитной стержневой мешалкой и отверстием для впуска азота загружали 80 мл тетрагидрофурана и 5,25 г (37,5 мМ) диизопропиламина. Полученный раствор охлаждали до -25oC (сухой лед/этиленгликоль) и добавляли 15 мл (37,5 мМ) 2,5 М - н-бутиллития в гексане. Через 10 минут добавляли раствор соединения Части A (5 г, 35 мМ) в 8 мл безводного тетрагидрофурана. Образовавшийся темно-желтый раствор перемешивали при -20oC в течение 10 минут, а затем добавляли N-фенил(бис)трифторометансульфонамид (35 мМ, 12,4 г). Реакционную смесь перемешивали при -10oC в течение 2 часов, концентрировали в вакууме и распределяли между этилацетатом и насыщенным гидрокарбонатом натрия. Объединенную органическую фазу промывали гидрокарбонатом натрия, а затем солевым раствором, после чего концентрировали и получали маслообразное вещество янтарного цвета, которое фильтровали через пробку с силикагелем (60 г) (элюент: 5% этилацетат (300 мл/гексан). После концентрирования в вакууме получали 9,0 г светло-желтого маслообразного вещества, которое разводили 65 мл этилацетата, промывали 5% водным раствором K2CO3 (2 • 50 мл) и солевым раствором (1 • 10 мл), а затем осушали сульфатом натрия и концентрировали в вакууме с получением 7,5 г (87%) винилового трифтороацетата (m/e = 277 (M + H), который использовали без дополнительной очистки.
Часть C:
Получение метил 2,2-диметил-3-карбоксил-бут-3-еноата
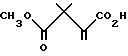
250-Миллилитровую колбу Фишера-Портера загружали соединением Части B (7,5 г, 27 мМ), безводным диметилформамидом (50 мл), трифенилфосфином (360 мг, 1,37 мМ) и 155 мг (0,69 мМ) Pd (11) (OAc)2. Реакционную смесь дважды продували азотом, а затем загружали CO под давлением 30 фунт/кв. дюйм (1,105 кг/см2). После этого раствор 20 мл безводного ДМФ и 7,56 мл (54 мМ) NEt3 охлаждали до 0oC, а затем добавляли 2,0 г (43 мМ) 99% муравьиной кислоты. Смесь тщательно встряхивали и загружали в колбу Фишера-Портера, снабженную вентиляционным отверстием. Реакционный сосуд снова загружали CO под давлением 40 фунт/кв. дюйм (2,812 кг/см2) и перемешивали в течение 6 часов при комнатной температуре. Полученную реакционную смесь концентрировали в вакууме и распределяли между этилацетатом (100 мл) и 5% водным раствором K2CO3 (75 мл). Водную фазу промывали дополнительным количеством этилацетата (1 • 40 мл), а затем подкисляли концентрированной соляной кислотой/льдом. После этого водную фазу экстрагировали этилацетатом (2 • 70 мл), а органические фазы осушали и концентрировали, в результате чего получали 3,5 г (75%) целевого соединения в виде белых кристаллов, т.пл. 72 - 75oC, m/e = 173 (M + H).
Часть D:
Получение метил 2,2-диметил-3-метилсукцината (изомер # 1)
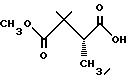
Стальной сосуд для гидрирования загружали 510 мг (3,0 мМ) акриловой кислоты Части C и 6 мг Ru (асас)2 (R - BINAP) в 10 мл дегазированного MeOH. Реакционную смесь гидрировали при комнатной температуре в течение 12 часов под давлением 50 фунт/кв.дюйм (3,515 кг/см2). Полученную реакционную смесь фильтровали через целит и концентрировали с получением 500 мг прозрачного маслообразного вещества, представленного в виде смеси (93:7) изомера N1 и N2, которую определяли с помощью ГХ-анализа с использованием колонки с 50 М β -циклодекстрином и при вращении (150oC - 15 мин, 2oC - 1 мин, изомер N1 - 17,85 мин; изомер N2 - 28 - 20 мин).
Часть E:
Получение метил 2,2-диметил-3-метилсукцината, изомера N2)
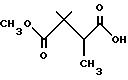
Стальной сосуд для гидрирования загружали 500 мг (2,9 мМ) акриловой кислоты из Части C и 6 мг Ru (OAc) (асас) (S - BINAP) в 10 мл дегазированного MeOH. Затем реакционную смесь гидрировали при комнатной температуре в течение 10 часов под давлением 50 фунт/кв.дюйм (3,515 кг/см2).
Эту реакционную смесь фильтровали через целит и концентрировали в вакууме с получением 490 мг продукта в виде смеси изомеров N1 и N2 (1:99), определенной с помощью хирального ГХ-анализ, описанного выше.
Аналогичным образом, бензил 2,2-диметил-3-оксо-бутаноат может быть использован для получения бензил 2,2,3-триметилсукцината (R-S-изомеров). Другие способы получения янтарных кислот, сукцинатов и сукцинамидов хорошо известны специалистам и могут быть использованы в настоящем изобретении.
Пример 6
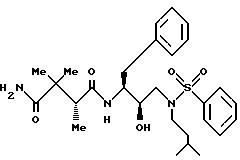
Получение [IS[IR*(S*),2S*]-N4
-[2- гидрокси-3-[(3-метилбутил)(фенолсульфонил)амино]-1-(фенилметил) пропил]-2,2,3-триметилбутандиамида
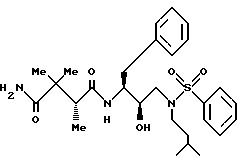
Часть A:
Получение [1S-[1R*(S*), 2S*] ] -4-[[2- гидрокси-3-[(3-метилбутил)-(фенилсульфонил)амино] -1-(фенилметил) пропил]амино]-2,2, 3-триметил-4-оксофенилметилового сложного эфира бутановой кислоты
Раствор 10,1 г (19,3 мМ) фенилметил (2R-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино] -1S-(фенилметил) пропил]-карбамата Примера 3 в 40 мл метанола гидрогенизировали с использованием 2 г 10% палладированного угля в течение 6 часов под давлением водорода в 50 фунт/кв.дюйм (3,515 кг/см2). После продувки азотом катализатор удаляли путем фильтрации через целит, а фильтрат концентрировали и получали 7,41 г (99%) 2(R)-гидрокси-3-[(3-метилбутил)(фенилсульфониламино]-1(S)- (фенилметил)пропиламина.
К раствору 2,5 г (10,0 мМ) бензил 2,2,3(R)-триметилсукцината и 2,1 г (15,0 мМ) N-гидроксибензотриазола в 10 мл безводного N,N-диметилформамида при 0oC добавляли 2,1 г (11,0 мМ) гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC). Через 2 часа добавляли раствор 3,9 г (10,0 мМ) 2(R)-гидрокси-3-[(3-метилбутил)(фенилсульфонил)-амино]-1(S)- (фенилметил)пропиламина в 3 мл диметилформамида.
После 16-часового перемешивания при комнатной температуре растворитель удаляли при пониженном давлении, и образовавшийся остаток растворяли в этилацетате, а затем промывали 0,2 н. лимонной кислотой, 5% водным раствором бикарбоната натрия и насыщенным солевым раствором. После осушки безводным сульфатом магния, фильтрования и концентрирования получали 5,74 г неочищенного продукта. Этот продукт хроматографировали на силикагеле (элюент: 1% метанол/метиленхлорид; Rf = 0,08) и получали 3,87 г. (выход = 62%) целевого продукта. M% e = 533 (M + H+).
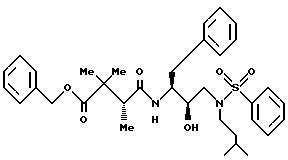
Часть B:
Получение [1S-[1R*(S*),
2S*]]-4-[[2- гидрокси-3-[3-метилбутил)(фенилсульфонил)амино] -1-(фенилметил) пропил]амино2,2,3-триметил-4-оксобутановой кислоты
Раствор 3,87 г (6,21 мМ) бензилового сложного эфира Части A в 40 мл этанола гидрогенизировали в присутствии 1,5 г 10%-ного палладированного угля под давлением водорода 50 фунт/кв.дюйм (5,313 кг/см2) в течение 4 часов. После продувки азотом, катализатор удаляли путем фильтрования через целит, а фильтрат концентрировали при пониженном давлении, в результате чего получали 3,24 г (98%) целевого продукта.
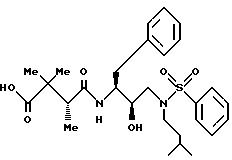
Часть C:
Получение [1S-[1R*(S*),2S*]]-N4
- [2-гидрокси-3-(3-метилбутил)(фенилсульфонил)амин]-1-(фенилметил) пропил]-2,2,3-триметилбутанамида
К раствору 3,24 г (6,1 мМ) кислоты Части B и 1,86 г (12,2 мМ) N-гидроксибензотриазола в 6 мл безводного диметилформамида при 0oC добавляли 1,75 г (9,1 мМ) гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида.
После 2-часового перемешивания при той же температуре добавляли 3,44 г (60,8 мМ) 30% водного раствора аммиака. Затем перемешивание
продолжали еще 20 часов при комнатной температуре, и растворители удаляли при пониженном давлении. Образовавшийся остаток растворяли в этилацетате, а затем промывали 0,2 г лимонной кислотой, 5% водным
раствором бикарбоната натрия и насыщенным солевым раствором, после чего осушали безводным сульфатом магния, фильтровали и концентрировали с получением 3,75 г неочищенного материала. Этот материал
пропускали через колонку со 150 г окиси основного алюминия (элюент: метиленхлорид/метанол, 20:1, об/об) для удаления непрореагировавшей кислоты. Полученный продукт осаждали из метиленхлорида с
использованием гексана, в результате чего получали 2,1 г (65%) нужного продукта. Т.пл. 110 - 112oC, m/e = 538 (M + Li)
Пример 7
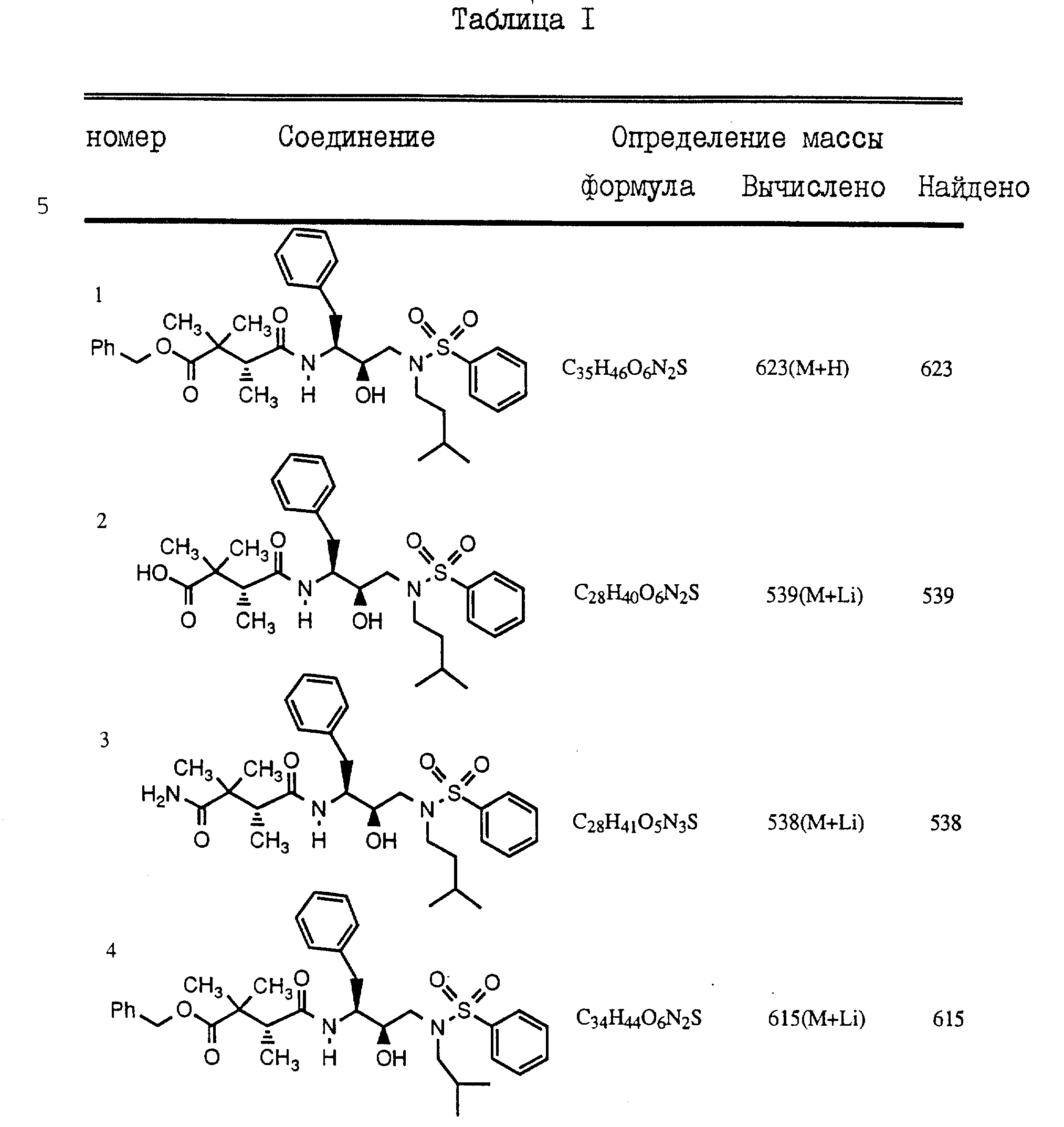
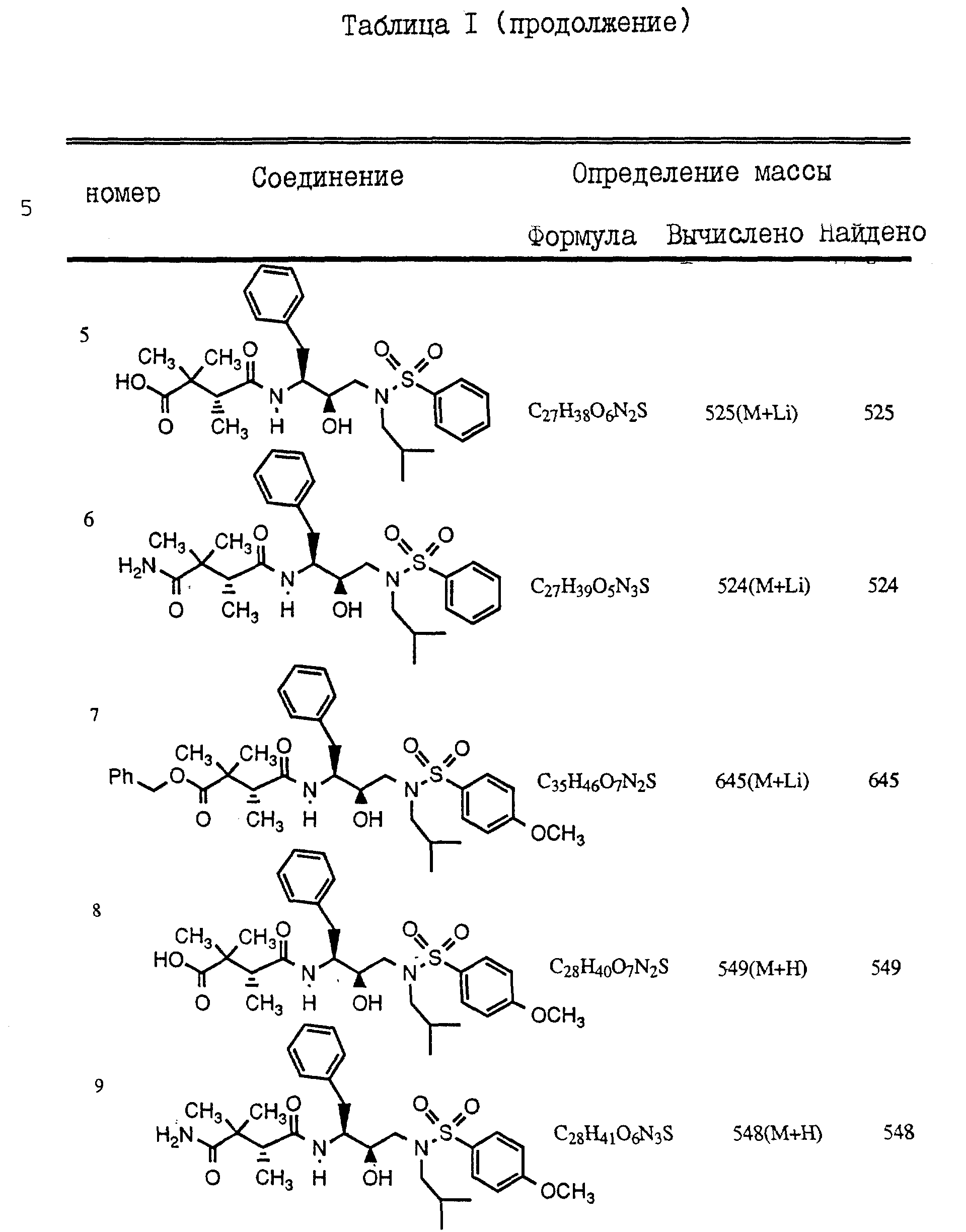
Полученные аналогичным способом соединения
проиллюстрированы в нижеследующей Таблице 1.
Пример 8
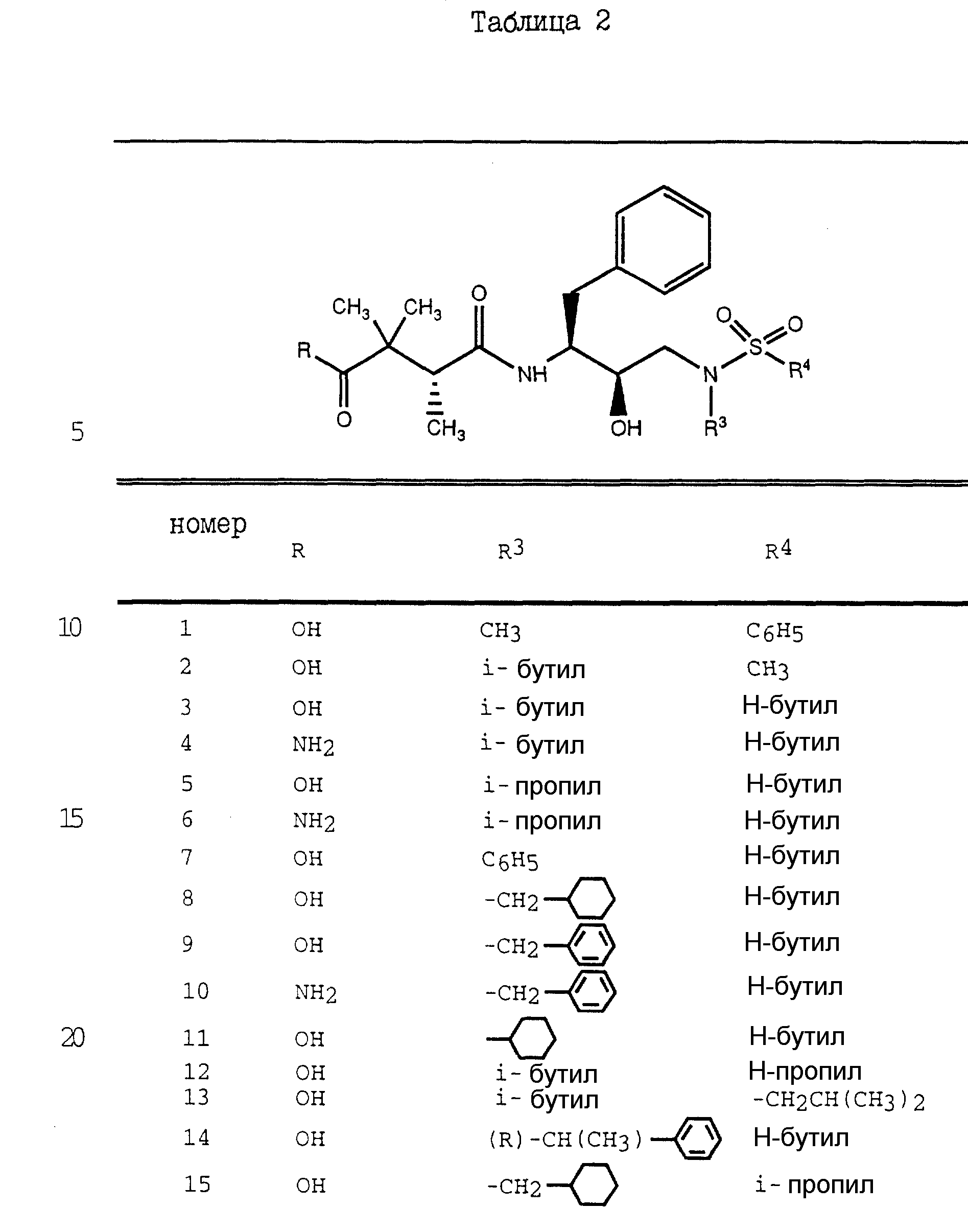
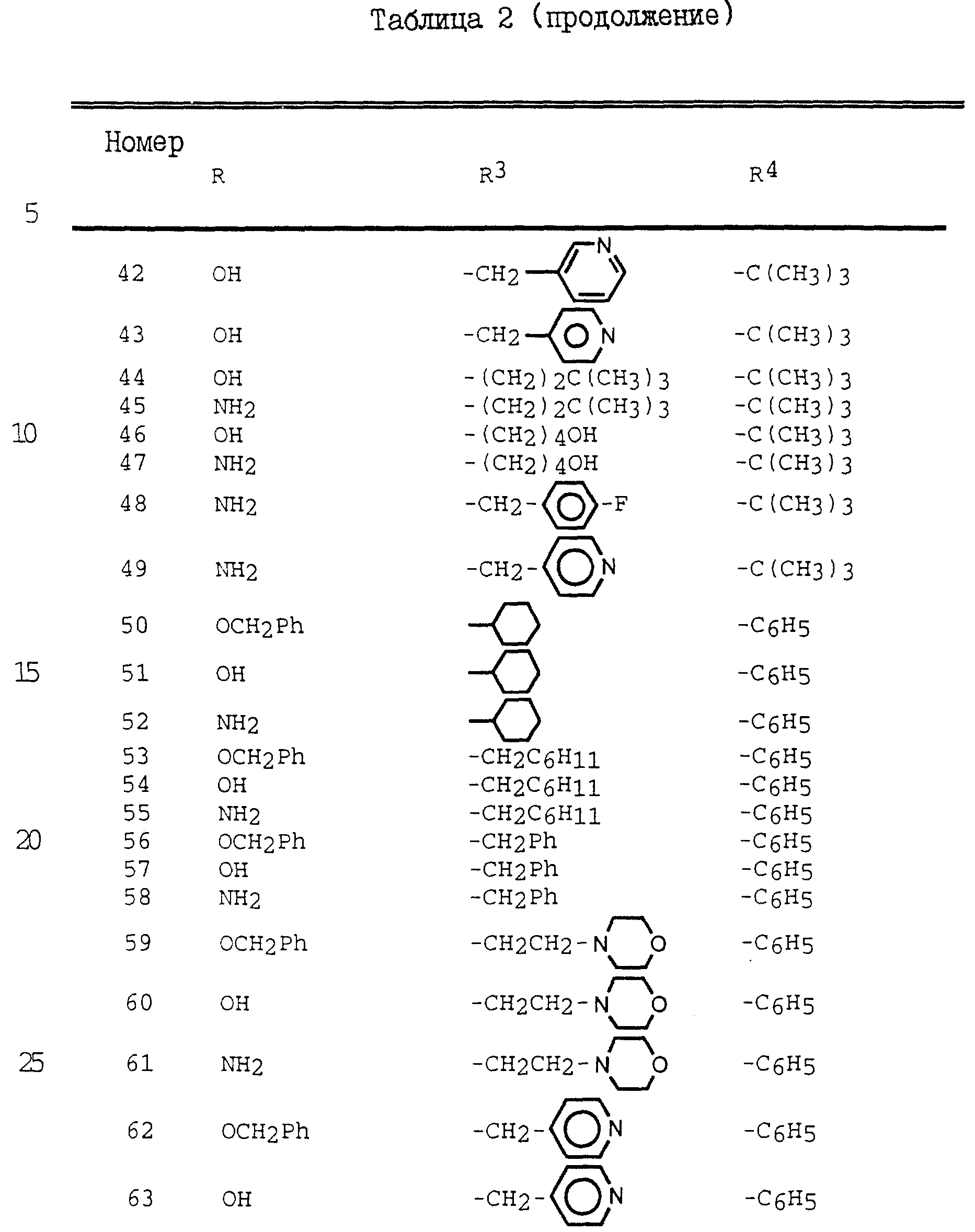
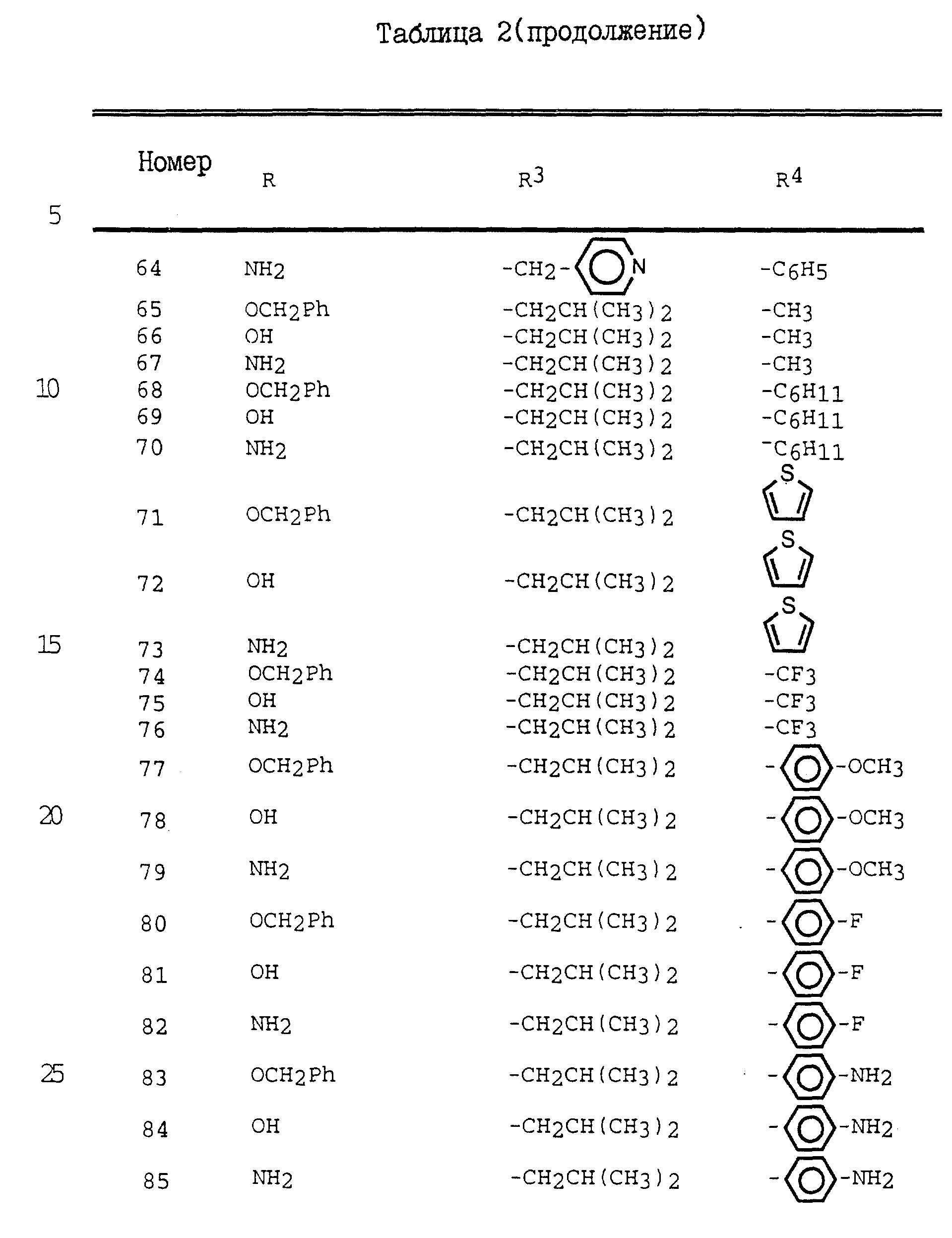
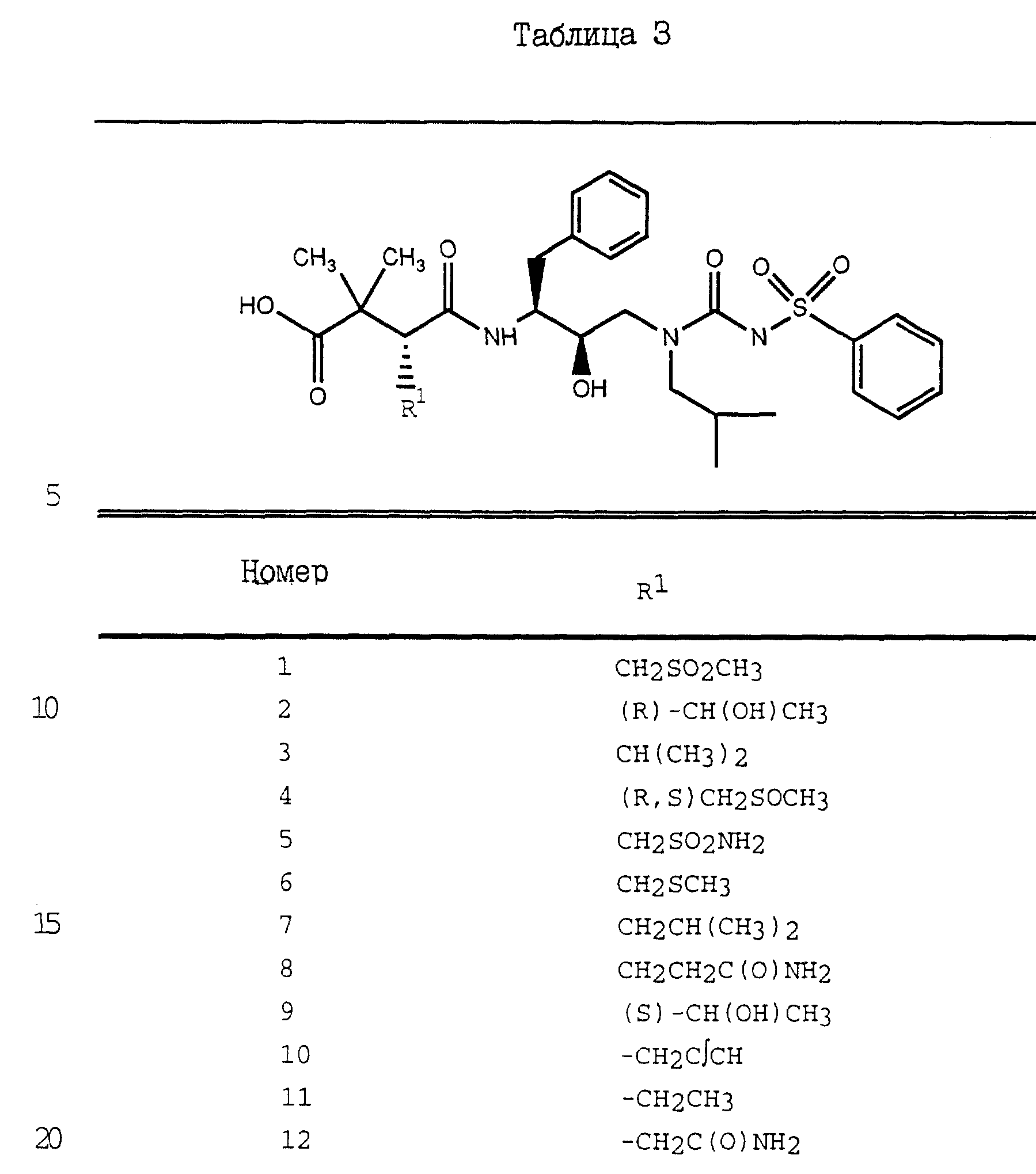
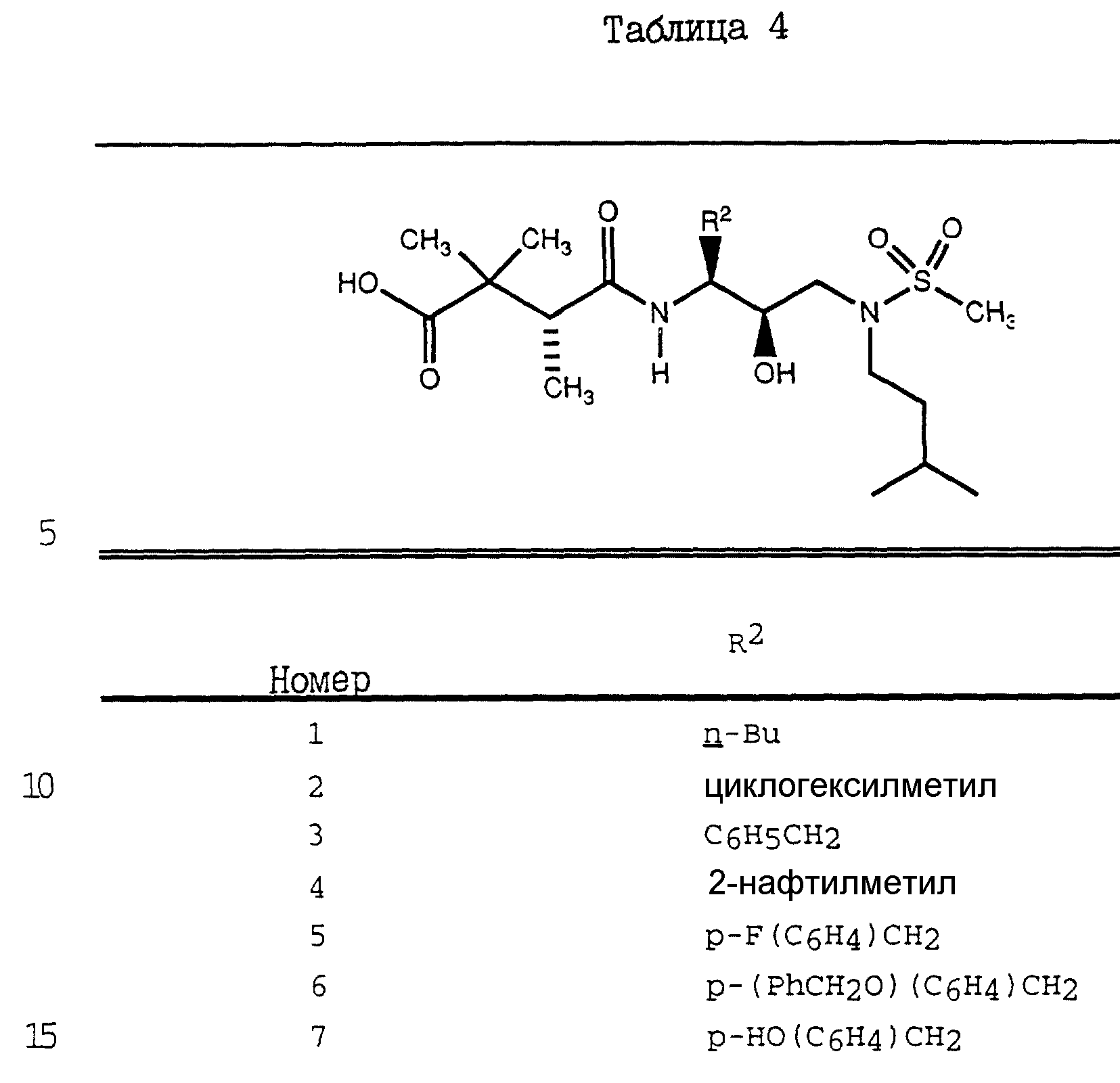
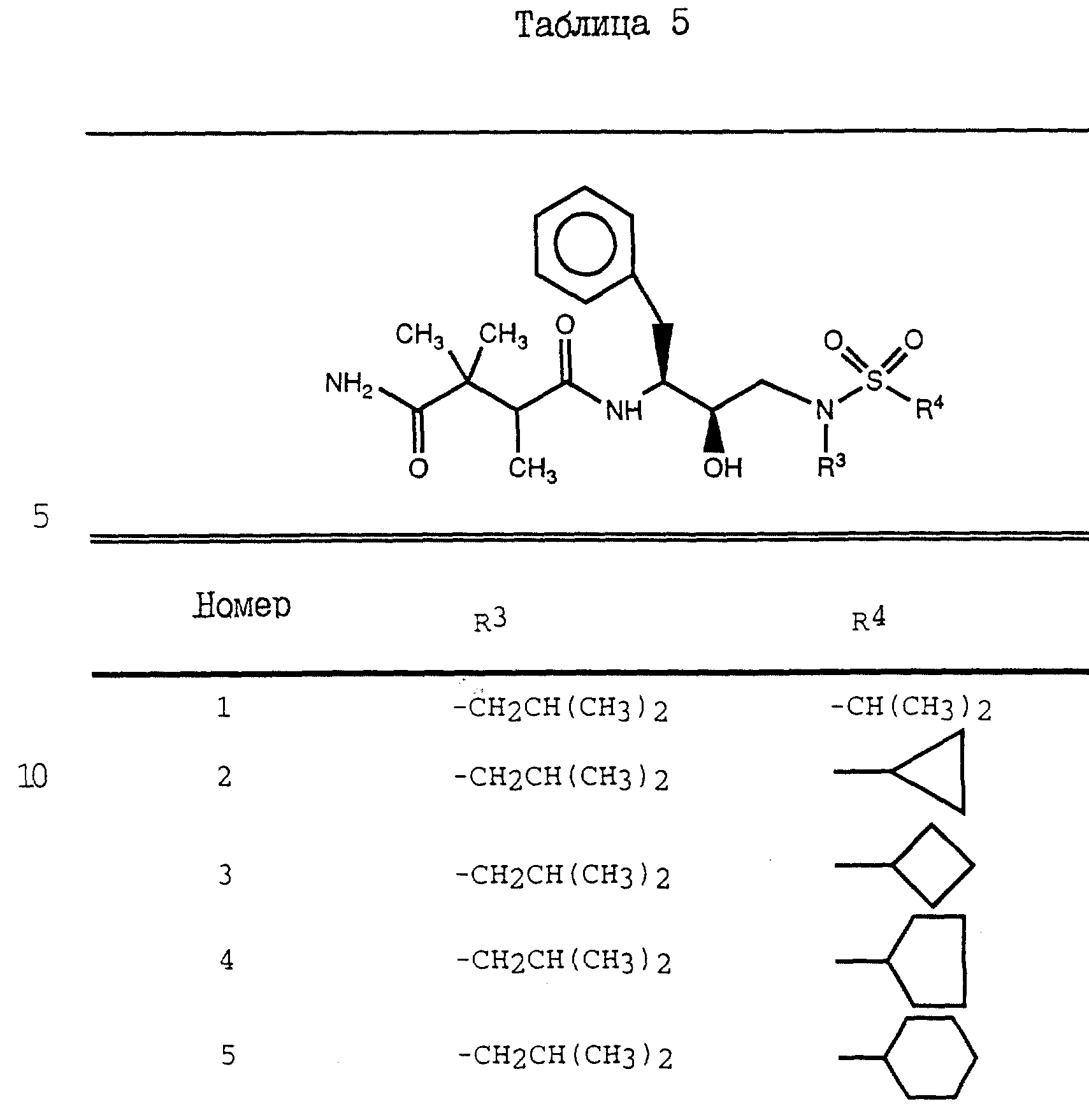
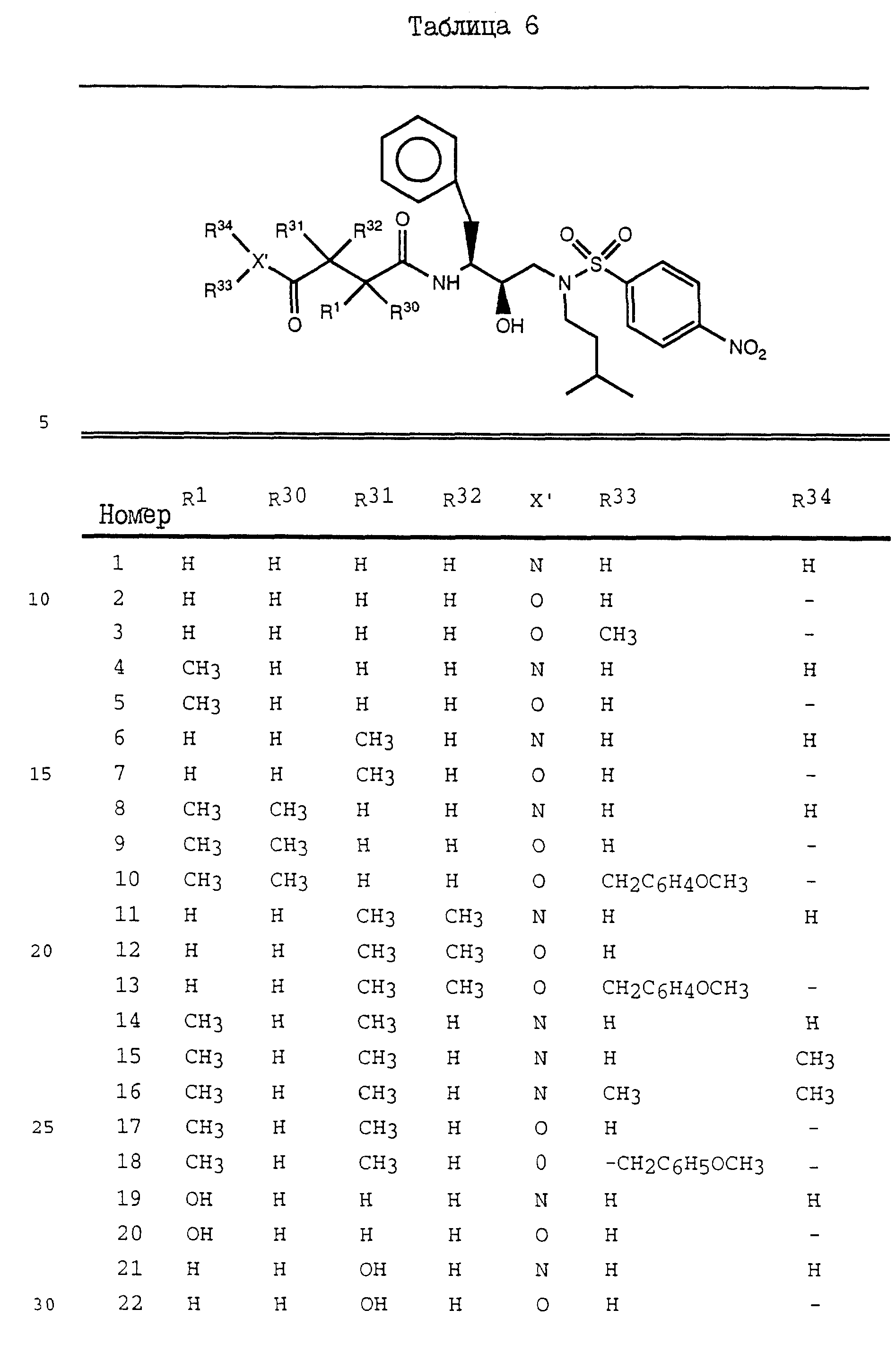
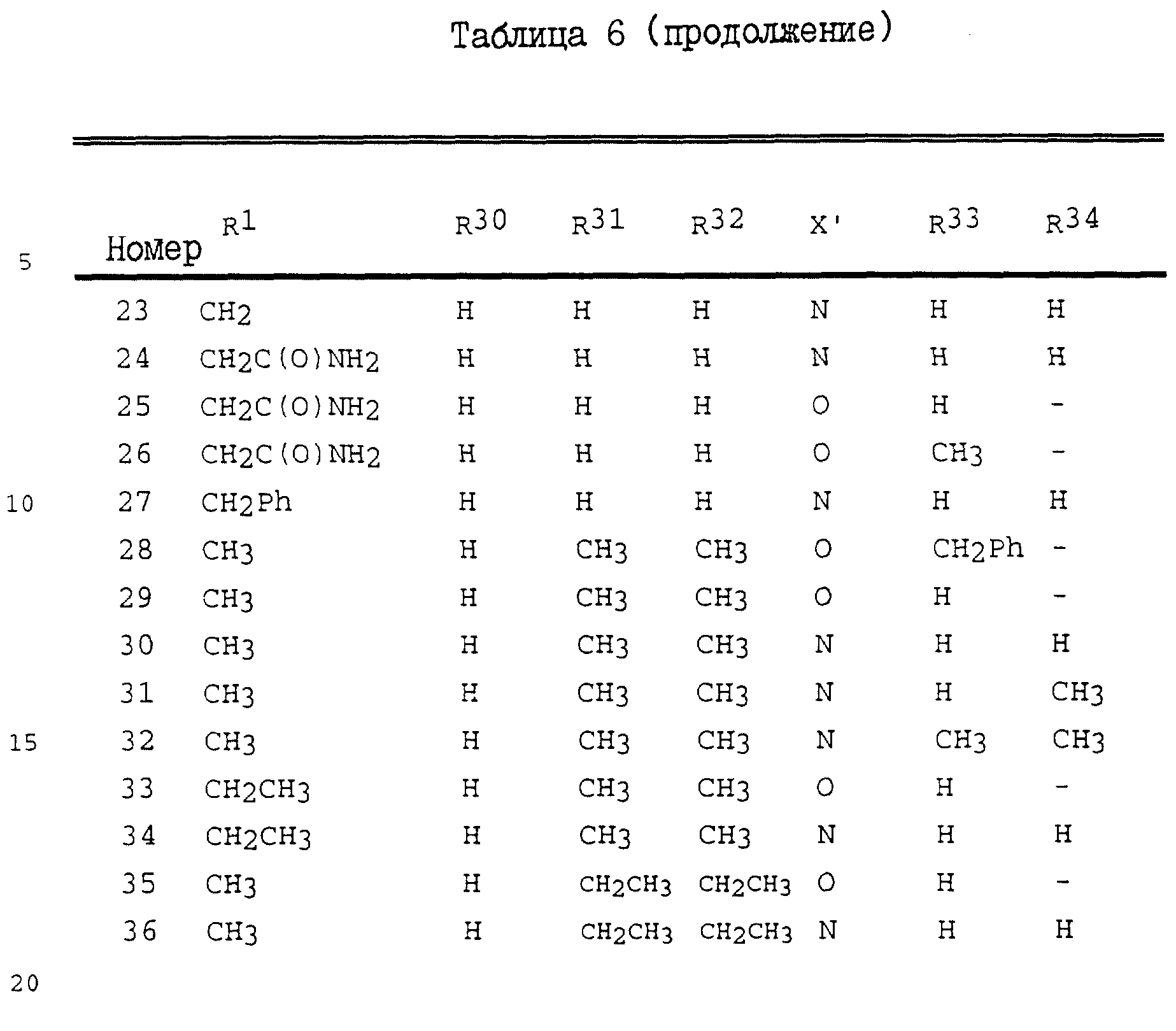
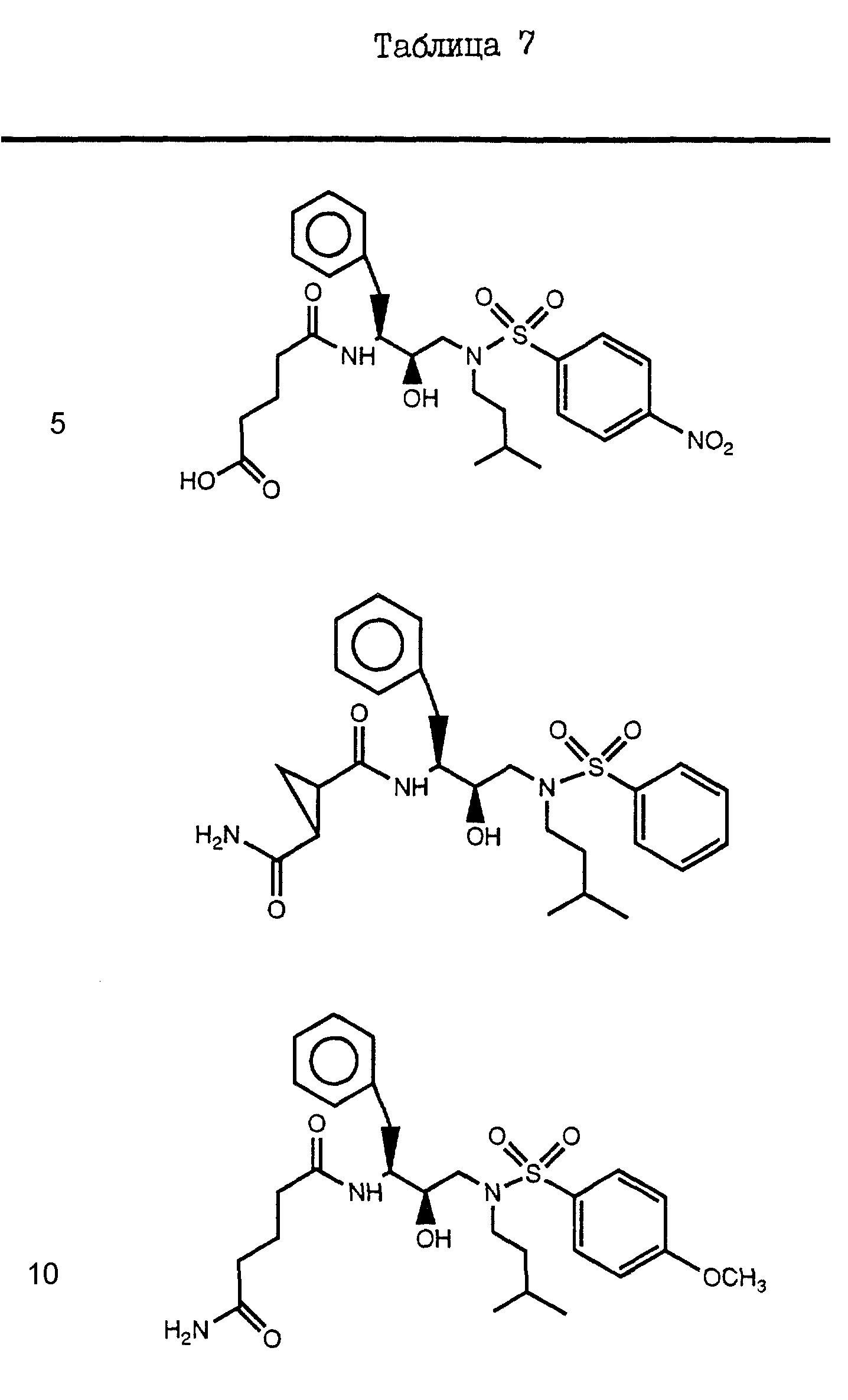
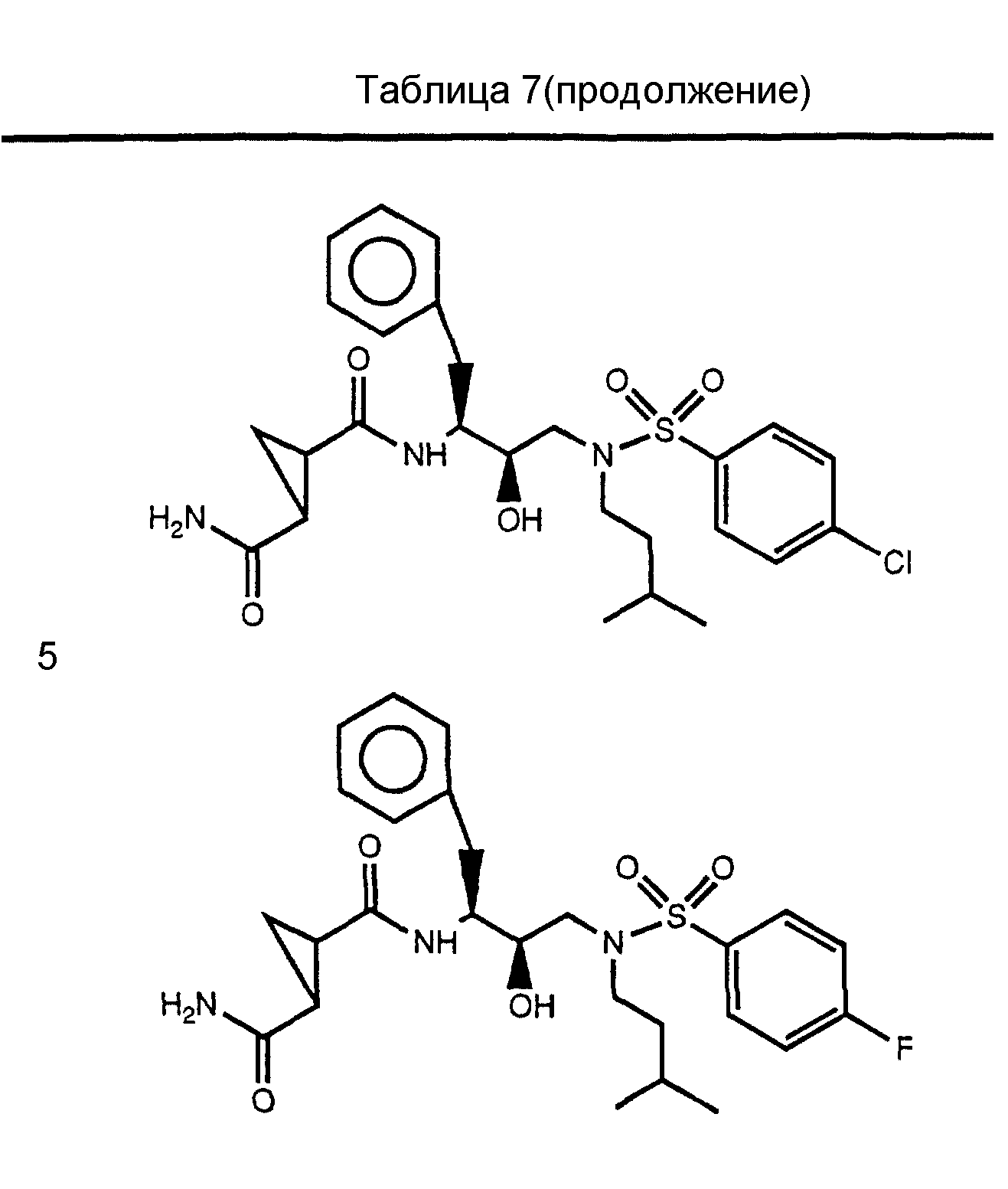
Дополнительные примеры соединений настоящего изобретения проиллюстрированы в Таблицах 2-7. Эти соединения могут быть
получены способом, аналогичным способу, описанному выше, а также в соответствии с нижеследующими общими методиками.
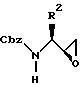
Общая методика синтеза аминоэпоксида
При -2oC, к раствору 0,226 М N-бензилоксикарбонил-L-фенилаланинхлорометилкетона в смеси 807 мл метанола и 807 мл тетрагидрофурана в течение 100 минут добавляли 1,54 эквивалента твердого борогидрида натрия. Затем растворители удаляли при пониженном давлении при 40oC, и образовавшийся остаток растворяли приблизительно в 1 л этилацетата. Полученный раствор промывали 1 М - бисульфатом калия, насыщенным бикарбонатом натрия, а затем насыщенными растворами хлорида натрия. После осушки безводным сульфатом магния и фильтрования, раствор удаляли при пониженном давлении. К полученному маслообразному веществу добавляли гексан (приблизительно 1 л), и смесь нагревали до комнатной температуры, твердые вещества собирали и промывали 2 л гексана. Полученное твердое вещество перекристаллизовывали из горячего этилацетата и гексана, в результате чего получали 32,3 г (выход = 43%) N-бензилоксикарбонил-3(S)-амино-1-хлоро-4-фенил-2-(S)-бутанола, Т.пл. 150-151oC, M + Li+ = 340.
Часть B:
К раствору гидроксида калия (1,2 экв.) в 968 мл
абсолютного этанола при комнатной температуре добавляли 0,097 M-N-CBz-3 (S)-амино-1-хлоро-4-фенил-2 (S)-бутанол. После перемешивания в течение 15 минут растворитель удаляли при пониженном давлении, а
твердые остатки растворяли в метиленхлориде. После промывания водой, осушки сульфатом магния, фильтрования и выпаривания получали белое твердое вещество. Это вещество перекристаллизовывали из горючего
этилацетата и гексана, в результате чего получали N-бензилоксикарбонил 3(S)-амино-1,2(S)-эпокси-4-фенилбутан.
Альтернативная общая методика синтеза аминоэпоксидов
Стадия A:
Раствор N-фенилаланина (50,0 г, 0,302 М), гидроксида натрия (24,2 г, 0,605 М) и карбоната калия (83,6 г, 0,605 М) в воде (500 мл) нагревали до 97oC. После этого, в течение приблизительно 25 минут, медленно добавляли бензилбромид (108,5 мл, 0,912 М). Затем эту смесь перемешивали в течение 30 минут при 97oC. Полученный раствор охлаждали до комнатной температуры и экстрагировали толуолом (2 • 250 мл). Объединенные органические слои промывали водой и солевым раствором, а затем осушали сульфатом магния, фильтровали и концентрировали с получением маслообразного продукта. Этот неочищенный продукт использовали в последующей стадии без очистки.
Стадия B:
Неочищенный бензилированный продукт, полученный в вышеописанной стадии, растворяли в толуоле (750 мл) и охлаждали до -55oC. После этого приблизительно в течение часа при
температуре от -55oC до -50oC добавляли 1,5 М - раствор DIB AL-H в толуоле (443,9 мл, 0,666 М). Полученную смесь перемешивали в течение 20 минут при -55oC. Реакцию
гасили при -55oC путем медленного добавления метанола (37 мл). Холодный раствор выливали в охлажденную при 5oC 1,5 н. соляную кислоту (1,8 л). Приблизительно 138 г осажденного
твердого вещества отфильтровывали и промывали толуолом. Образовавшийся твердый материал суспендировали в смеси толуола (400 мл) и воды (100 мл). После этого смесь охлаждали до 5oC,
обрабатывали 186 мл 2,5 н. NaOH, а затем перемешивали при комнатной температуре до растворения твердого остатка. Толуоловый слой выделяли из водной фазы и промывали водой и солевым раствором. После
осушки сульфатом магния, фильтрования и концентрирования до объема 75 мл получали 89 г остатка. К этому остатку добавляли этилацетат (25 мл) и гексан (25 мл), после чего спиртовой продукт оставляли
для кристаллизации. Через 30 минут, для дальнейшего стимулирования кристаллизации, добавляли еще 50 мл гексана. Твердое вещество отфильтровывали и промывали 50 мл гексана, в результате чего получали
приблизительно 35 г материала. Второй сбор этого материала может быть выделен путем повторного фильтрования маточного раствора. Образовавшиеся твердые вещества объединяли и перекристаллизовывали из
этилацетата (20 мл) и гексана (30 мл). Таким образом получали 2 сбора продукта, приблизительно 40 г (40% по отношению к L-фенилаланину) аналитически чистого спиртового продукта. Маточные растворы
объединяли и концентрировали с получением 34 г остатка. Остаток, происходящий из этилацетата и гексана, давал приблизительно 7 г. (выход ≈ 7%) твердого продукта с некоторым количеством
примесей. Возможно также проведение дальнейшей оптимизации выделения из маточного раствора.
Стадия C:
Раствор оксалилхлорида (8,4 мл, 0,096 М) в дихлорметане (240 мл)
охлаждали до -74oC. После этого, приблизительно в течение 1,25 часа, медленно и при температуре -74oC добавляли раствор ДМСО (12,0 мл, 0,155 М) в дихлорметане (50 мл). Полученную
смесь перемешивали в течение 5 минут, а затем в течение 20 минут при температуре от -75oC до -68oC добавляли раствор спирта (0,074 М) в 100 мл дихлорметана. Полученный раствор
перемешивали в течение 35 минут при -78oC. Затем в течение 10 минут (при температуре от -78oC до -68oC) добавляли триэтиламин (41,2 мл, 0,295 М), после чего соль
аммония осаждалась. Холодную смесь перемешивали 30 минут и добавляли 225 мл воды. Дихлорметановый слой отделяли от водной фазы и промывали водой, а затем солевым раствором. После осушки сульфатом
магния, фильтрования и концентрирования получали остаток. Этот остаток разводили этилацетатом и гексаном, а также затем фильтровали для удаления соли аммония. Фильтрат концентрировали и получали
нужный альдегидный продукт. Этот продукт использовали в последующей стадии без очистки.
Для окисления Сверна (Swern) в литературе указаны температуры выше -70oC. Возможны также другие модификации и альтернативы для окисления Сверна.
Раствор неочищенного альдегида (0,074 М) и хлороиодометана (7,0 мл, 0,096 М) в тетрагидрофуране (285 м) охлаждали до -78oC. После этого в течение 15 минут и при температуре -75oC добавляли 1,6 М - раствор н-бутиллития в гексане (25 мл, 0,040 М). После первого добавления к смеси добавляли снова 1,6 мл (0,022 М) хлороиодометана, а затем 23 мл (0,037 М) н-бутиллития, поддерживали 15 минут. Затем в течение 45 минут (при -75oC) к смеси еще 4 раза добавляли каждый из реагентов, а именно хлороиодометан (0,70 м, 0,010 М) и н-бутиллитий (5 мл, 0,008 М). Затем охлаждающую баню удаляли и раствор нагревали до 22oC в течение 1,5 часа. Полученную смесь выливали в 300 мл насыщенного водного раствора хлорида аммония. Тетрагидрофурановый слой отделяли. Водную фазу экстрагировали этилацетатом (1 • 30 мл). Объединенные органические слои промывали солевым раствором, осушали сульфатом магния и фильтровали. После концентрирования получали 27,4 г коричневого маслообразного вещества. Это вещество может быть использовано в последующей стадии без очистки. Нужный диастереомер может быть очищен путем перекристаллизации в последующей стадии получения сульфонамида.
Альтернативно, этот продукт может быть очищен с помощью хроматографии.
Общая методика синтеза производных 1,3-диамино-4-фенил-бутан- 2-Ол'а.
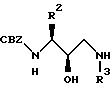
Смесь амина R3NH2 (20 экв.) в безводном изопропиловом спирте (20 мл/мМ превращаемого эпоксида) нагревали с обратным холодильником, а затем обрабатывали в течение 10 - 15 минут -NCbz-аминоэпоксидом формулы:
с использованием капельной воронки для твердых веществ. После завершения добавления раствор нагревали с обратным холодильником еще 15 минут, и ход реакции прослеживали с помощью тонкослойной хроматографии (ТСХ). Полученную реакционную смесь концентрировали в вакууме с получением маслообразного вещества, которое затем обрабатывали н-гексаном при быстром перемешивании, после чего материал с разомкнутым кольцом осаждался из раствора. Через 1 час осаждение полностью завершалось, и продукт выделяли путем фильтрации с использованием воронки Бюхнера, а затем осушали воздухом. После этого полученный продукт осушали в вакууме. Этот способ позволяет получать, в большинстве случаев, аминоспирты с чистотой, достаточной для дальнейшего применения.
Общая методика для реакции аминоспиртов с сульфонилгалидами или сульфонилангидридами
Получение сульфонамидов
К раствору N[3(S)-бензилоксикарбониламино-2(R)-гидрокси-4- фенилбутил] N-изоамиламина (2,0 г, 5,2 мМ) и триэтиламина (723 мкл, 5,5 ММ) в дихлорметане (20 мл) по капле добавляли метансульфонилхлорид (400 мкл, 5,2 мМ). Полученную реакционную смесь перемешивали в течение 2 часов при комнатной температуре, после чего дихлорметановый раствор концентрировали приблизительно до объема 5 мл и загружали на колонку с силикагелем (100 г). Эту колонку элюировали хлороформом, содержащим 1%-ный этанол и 1%-ный метанол.
Альтернативно, в результате реакции N[3 (S)-бензилоксикарбониламино-2-(R)-гидрокси-4-фенилбутил] -N- изоамиламина (1,47 г, 3,8 мМ) с триэтиламином (528 мкл, 3,8 мМ) и бензолсульфонихлоридом (483 мкл, 3,8 мМ) может быть получено соответствующее (фенилсульфонил) аминовое производное.
Общая методика удаления защитных групп путем гидрогенолиза в присутствии палладия на углероде.
А. Спиртовой
растворитель
Cbz - защищенное пептидное производное растворяли в метаноле (прибл. 20 мл/мМ), в атмосфере азота, добавляли 10%-ный палладий на углероде. Реакционный сосуд герметично закрывали
и 5 раз продували азотом, а затем 5 раз водородом. Давление поддерживали при 60 фунт/кв. дюйм (3,515 кг/см2) в течение 1-16 часов, после чего водород заменяли на азот, и полученный раствор
фильтровали через слой целита для удаления катализатора. Растворитель удаляли в вакууме и получали свободное аминовое производное, которое являлось достаточно чистым для того, чтобы использовать его в
последующей стадии.
B. Уксуснокислотный растворитель
Cbz - защищенное пептидное производное растворяли в ледяной уксусной кислоте (20 мл/мМ), а затем, в атмосфере азота,
добавляли 10%-ный палладированный уголь. Реакционный сосуд 5 раз продували азотом и 5 раз водородом, после чего в течение 2 часов поддерживали давление при 40 фунт/кв. дюйм (2,812 кг/см2).
Затем водород заменяли на азот, и реакционную смесь фильтровали через слой целита для удаления катализатора. Фильтрат концентрировали, и полученный продукт растворяли в безводном эфире, а затем
выпаривали досуха три раза. Кроме того, полученный продукт осушали в вакууме. Этот полученный продукт (ацетатная соль) являлся достаточно чистым для непосредственного использования его в последующих
реакциях.
Общая методика удаления Boc-защитных групп с использованием 4 н. соляной кислоты в диоксане
Boc-защищенную аминокислоту или пептид обрабатывали раствором 4 н. HCl в
диоксане, размешивая при комнатной температуре. Реакция разблокирования обычно завершалась через 15 минут, при этом за ходом реакции следили с помощью тонкослойной хроматографии (ТСХ). После
завершения реакции избыток диоксана и HCl удаляли путем выпаривания в вакууме. Остаточное следование количества диоксана и HCl полностью удаляли путем повторного выпаривания из безводного эфира или
ацетона. Полученную таким образом гидрохлоридную соль тщательно осушали в вакууме, после чего она может быть использована для последующей реакции.
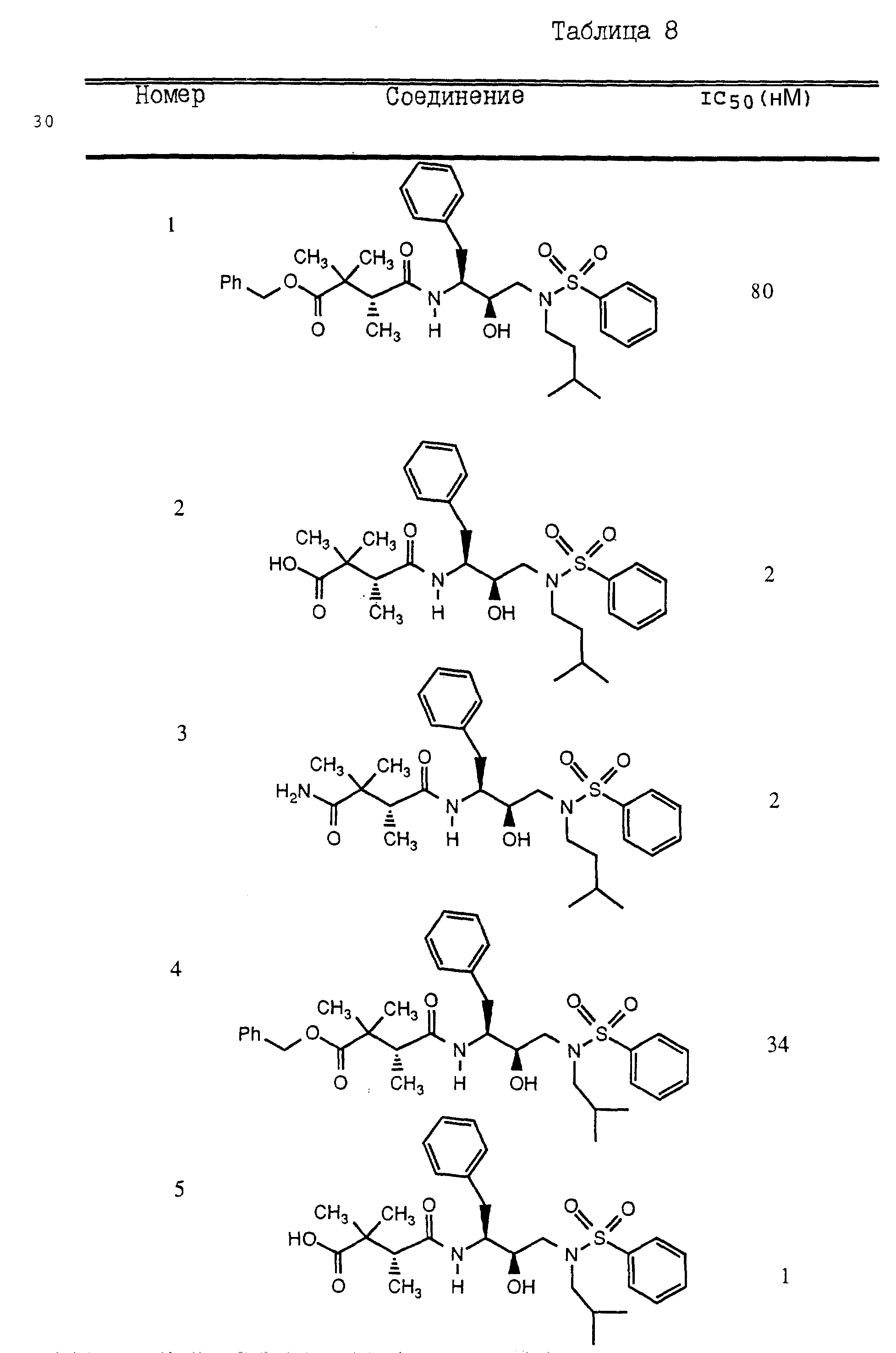
Пример 9
Соединения
настоящего изобретения являются эффективными ингибиторами ВИЧ-протеазы. Ингибирование ВИЧ-фермента соединениями настоящего изобретения проиллюстрировано с помощью ферментного анализа, описанного в
нижеприведенных примерах. Предпочтительные соединения настоящего изобретения и их вычисления IC50 - величины (50%-ная ингибирующая концентрация, то есть концентрация, при которой соединения
- ингибиторы снижают ферментную активность на 50%) показаны в Таблице 8. Указанный ферментный метод описан ниже. В качестве субстрата использовали 2-аминобензоил-Ile-Nle-Phe(p-NO2
)-Gln-ArgNH2),
а в качестве позитивного контроля использовали MVT-101 (Miller M. et. al., Science, 246, 1149 (1089)). При этом были использованы следующие условия анализа: Буфер
для анализа: 20 мМ фосфата натрия, pH 6,4
20% глицерин
1 мМ ЭДТК
1 мМ ДТТ
0,1% CHAP
Вышеуказанный субстрат растворяли в ДМСО, а затем 10-кратно разводили в
буфере для анализа. Конечная концентрация субстрата в анализе составляла 80 мКм.
ВИЧ-протеазу разводили в аналитическом буфере до конечной концентрации фермента 12,3 нМ (исходя из молекулярной массы 10780).
Конечная концентрация ДМСО составляла 14%, а конечная концентрация глицерина составляла 18%. Исследуемое соединение растворяли, а затем разводили в ДМСО до 10-х испытуемой концентрации, после чего добавляли 10 мкл ферментного препарата, полученный материал размешивали и инкубировали 15 минут при комнатной температуре. Ферментную реакцию инициировали путем добавления 40 мкл субстрата. Увеличение флуоресценции прослеживали при комнатной температуре за 4 периода времени (0, 8, 16 и 24 мин). Каждый анализ проводили в дубликатных лунках.
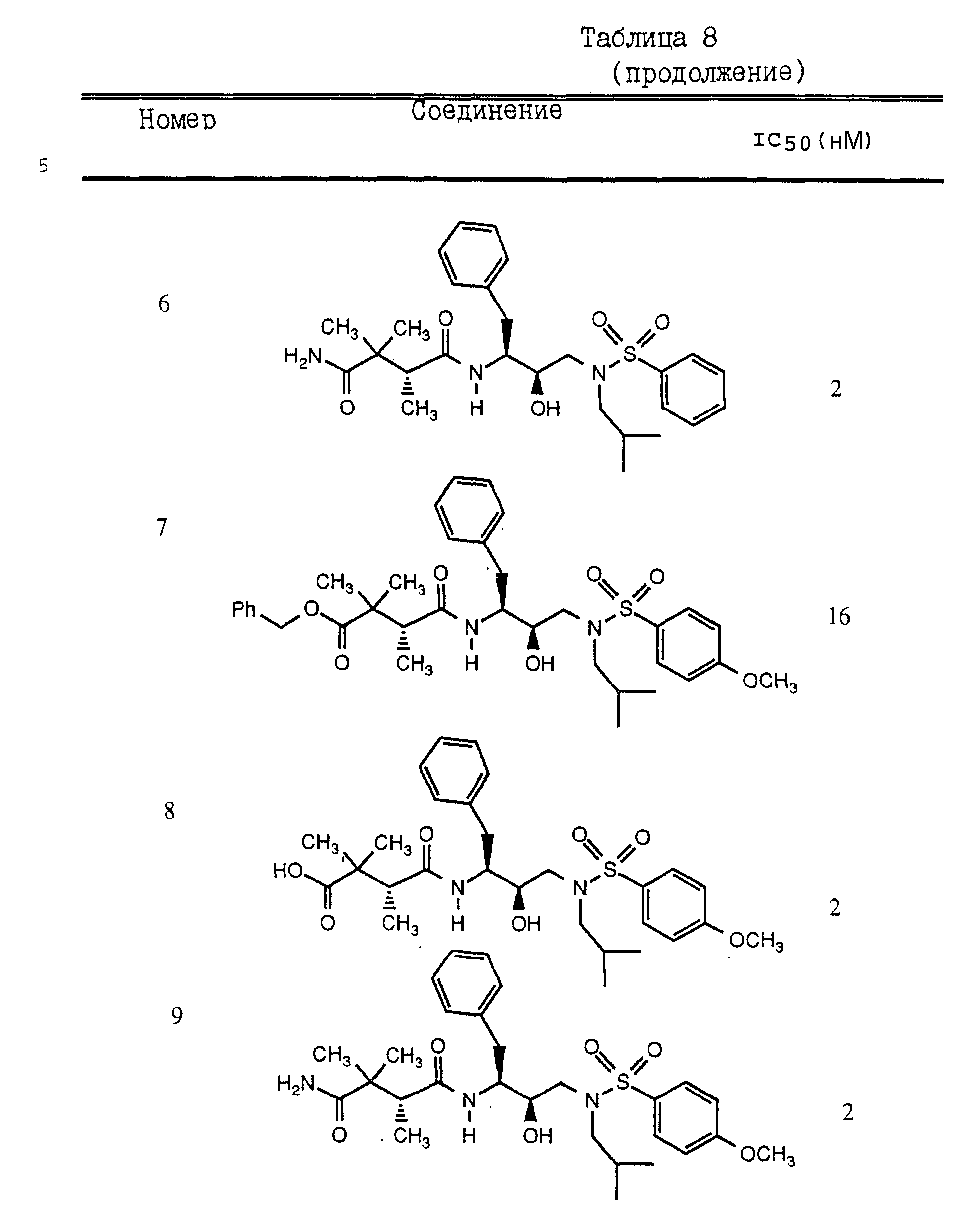
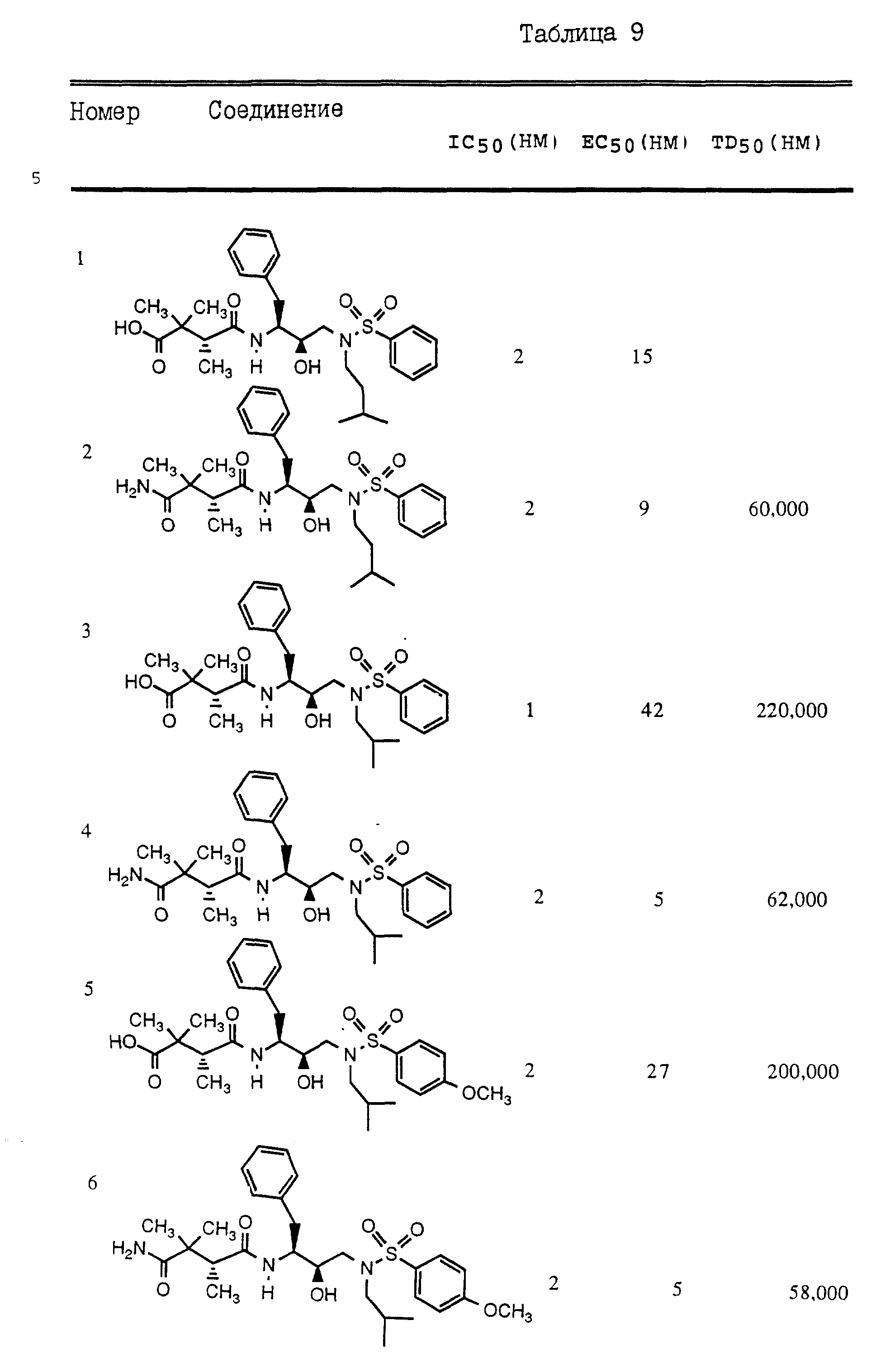
Пример 10
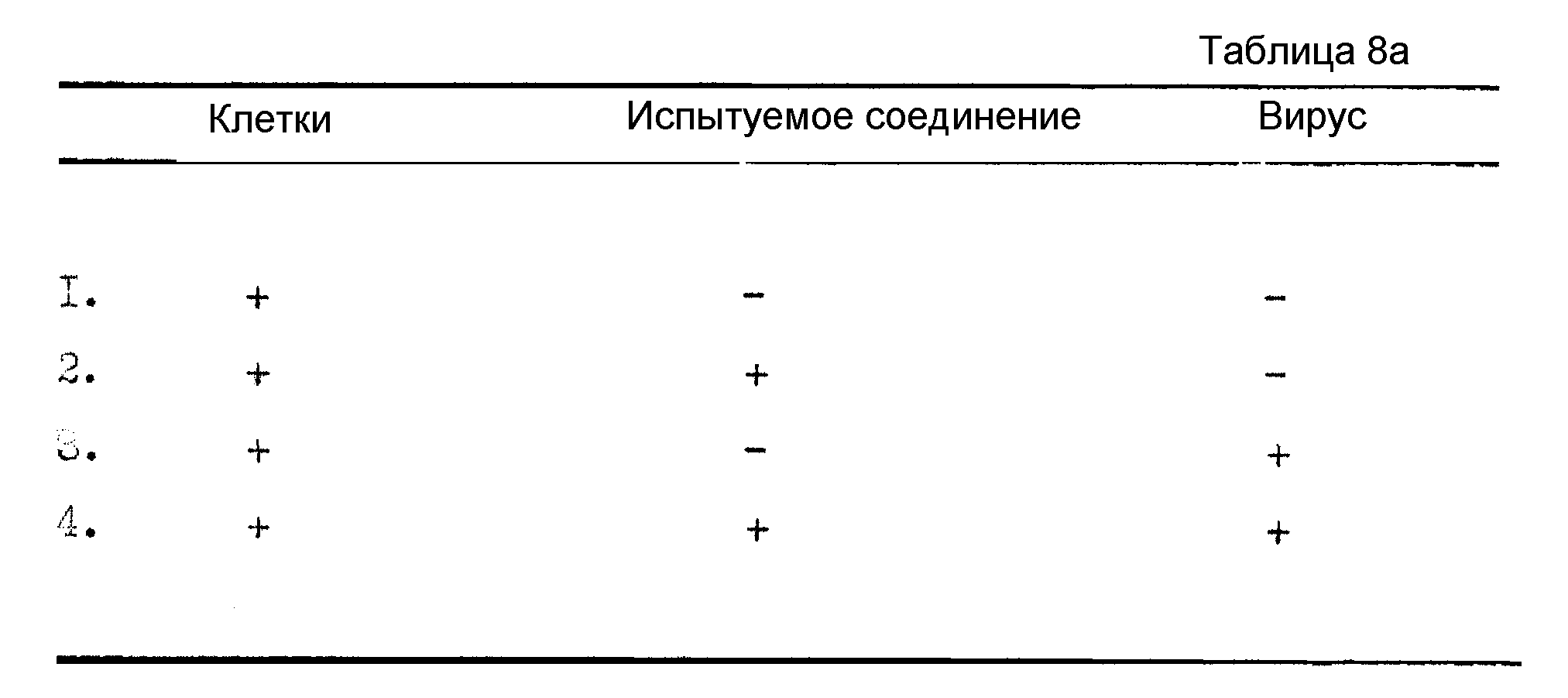
Эффективность соединений, указанных в Таблице 8, определяли с помощью вышеописанного ферментного анализа и СЕМ-клеточного анализа.
Анализ на ВИЧ-ингибирование, проводимый с использованием клеток с высоким уровнем заражения, представляет собой автоматический колометрический (на основе тетразолия) анализ, описанный, в основном, Pauwles и др., J. Virol. Methods 20, 309 - 321) (1988). Анализ осуществляли в 96 - луночных планшетах для культивирования ткани. СЕМ - клетки (клеточная линия CD4+) культивировали в среде RPM1 - 1640 (Gibco), в которую была добавлена 10% околоплодная телячья сыворотка, а затем обрабатывали полибреном (2 мкг/мл). После этого в каждую лунку планшета для культивирования тканей добавляли среду (в объеме 80 мкл), содержащую 1•104 клеток. Затем в каждую лунку добавляли испытуемое соединение (в объеме 10 мкл), растворенное в культуральной среде, (или добавляли среду, не содержащую соединения и используемую в качестве контроля) для достижения нужной конечной концентрации, после чего клетки инкубировали 1 час при 37oC. Замороженную культуру ВИЧ-1 разводили в культуральной среде до концентрации 5•104 TCID50/мл (TCID50 = доза вируса, инфицирующая 50% клеток тканевой культуры), и 20 мкл - объем вирусного препарата (содержащего TCID50 = 1000 вируса) добавляли к лункам, содержащим испытуемое соединение, и к лункам, содержащим лишь среду (инфицированные контрольные клетки). В несколько лунок добавляли культуральную среду, не содержащую вируса (неинфицированные контрольные клетки). Токсичность испытуемого соединения определяли путем добавления среды, в которой отсутствовали вирус, к нескольким лункам, содержащим испытуемое соединение. В итоге, были проведены эксперименты с использованием следующих культуральных планшетов (см. Таблицу 8a).
В экспериментах 2 и 4 конечные концентрации испытуемых соединений составляли 1, 10, 100 и 500 мкг/мл. В качестве позитивного лекарственного контроля использовали либо азидотимидин (AZT), либо дидезоксиинозин (ddI). Испытуемые соединения растворяли в ДМСО и разводили в культуральной среде так, чтобы конечная концентрация ДМСО, в любом случае, не превышала 1,5%. ДМСО добавляли ко всем контрольным лункам при соответствующей концентрации.
После добавления вируса клетки инкубировали при 37oC в течение 7 дней, в условиях влажности и в атмосфере 5% CO2. По желанию, испытуемые соединения могут быть добавлены на 0,2 и 5 день. Через 7 дней после инфицирования клетки в каждой лунке ресуспендировали, и 100 мкл образца каждой клеточной суспензии удаляли для анализа. Затем к каждой 100 мкл - клеточной суспензии добавляли 20 мкл - объем 5 мг/мл раствора 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолибромида (МТТ), и клетки инкубировали при 27oC в течение 4 часов в атмосфере 5% CO2. Во время этого инкубирования МТТ метаболически восстанавливался живыми клетками, в результате чего в клетках наблюдалось продуцирование окрашенного формазанового продукта. К каждому образцу добавляли 100 мкл 10% додецилсульфата натрия в 0,01 н. HCl (для лизиса клеток), после чего образцы инкубировали в течение ночи. Затем для каждого образца определяли оптическую плотность при 590 нм с использованием устройства для прочтения планшетов (Molecular Devices microplate reader).
Величины оптические плотности для каждой серии лунок сравнивали с величинами, полученными для вирус-инфицированного контроля, неинфицированного клеточного контроля, и испытуемых соединений, и определяли цитотоксичность и антивирусную активность.
Очевидно, что с использованием методик, описанных в вышеприведенных примерах и в общем описании заявки, могут быть получены соединения, перечисленные ниже; и эти соединения обладают ВИЧ-протеазо-ингибирующей активностью, в основном, аналогичной активности соединений, описанных в примерах.
Как показано выше, соединения настоящего изобретения являются эффективными противовирусными соединениями и, в частности, эффективными ингибиторами ретровирусов. Так, например, соединения настоящего изобретения являются эффективными ингибиторами ВИЧ-протеазы. Кроме того, соединения настоящего изобретения способны ингибировать и другие штаммы ВИЧ, такие как ВИЧ-2, а также другие вирусы, такие как вирус Т-клеточного лейкоза, вирус обезьяньего иммунодефицита, вирус лейкоза кошек, гепаднавирус, цитомегаловирус и пикорнавирус. Таким образом, соединения настоящего изобретения являются эффективными для лечения и/или профилактики ретровирусных инфекций.
Соединения настоящего изобретения могут обладать одним или несколькими асимметрическими атомами углерода, а поэтому они могут существовать в форме оптических изомеров, а также в форме их рацемических или нерацемических смесей. Оптические изомеры могут быть получены путем разделения рацемических смесей стандартными методами, например, посредством образования диастереомерных солей путем обработки оптически активной кислотой или основанием. Подходящими для этой цели кислотами являются винная, диацетилвинная, дибензоилвинная, дитолуолвинная и камфорсульфоновая кислота. Разделение смеси диастереометров осуществляют путем кристаллизации с последующим выделением оптически активных оснований из этих солей. Различные способы разделения оптических изомеров предусматривают использование хиральной хроматографической колонки, оптимально выбранной для максимизации разделения энантиомеров. Существует еще один метод разделения оптических изомеров, который предусматривает синтез ковалентных диастереоизомерных молекул посредством реакции соединений формулы I с оптически чистой кислотой в активированной форме или с оптически чистым изоцианатом. Синтезированные диастереомеры могут быть разделены стандартными способами, такими как хроматография, дистилляция, кристаллизация или сублимация, а затем гидролизованы с получением чистого энантиомерного соединения. Аналогичным образом, с использованием оптически активных исходных материалов могут быть получены активные соединения формулы I. Эти изомеры могут присутствовать в форме свободной кислоты, свободного основания, сложного эфира или соли.
Соединения настоящего изобретения могут быть использованы в виде солей, происходящих от неорганических или органических кислот. Такими солями являются (но не ограничиваются ими) ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, циклопентанпропионат, додецилсульфат, этансульфонат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидроиодид, 2-гидрокси-этансульфонат, лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, пальмоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, тозилат, мезилат и индеканоат. Кроме того, основные азот-содержащие группы могут быть кватернизированы с использованием таких агентов, как низшие алкилгалиды, например метил-, этил-, пропил- и бутил-хлориды, -бромиды, и иодиды; диалкилсульфаты, например, диметил-, диэтил-, дибутил и диамилсульфаты; галиды с длинной цепью, например децил-, лаурил-, миристил- и стеарилхлориды, -бромиды и -иодиды; аралкилгалиды, например бензил- и фенитилбромиды, и т.п. При этом могут быть получены водо- или маслорастворимые или диспергируемые продукты.
Примерами кислот, которые могут быть использованы для получения фармацевтически приемлемых кислотно-аддитивных солей, являются неорганические кислоты, такие как соляная кислота, серная кислота и фосфорная кислота, и органические кислоты, такие как щавелевая кислота, малеиновая кислота, янтарная кислота и лимонная кислота. Кроме того, соли могут быть получены с использованием оснований, например, таких как щелочные или щелочноземельные металлы, а именно натрий, калий, кальций или магний, либо органических оснований.
Общая суточная доза, вводимая индивидууму в виде разовой или дробной дозы, составляет, например, от 0,001 до 10 мг/кг веса тела в день, а в основном от 0,01 до 1 мг. Композиции, полученные в виде стандартных унифицированных форм, могут содержать вышеуказанные количества доз, поделенные на кратное число раз и составляющие, в целом, суточную дозу.
Для получения разовой стандартной формы количество активного ингредиента может быть смешано с материалом-носителем и зависит от конкретного индивидуума и от конкретного способа введения.
Выбор схемы приема лекарственного средства, а в частности соединений и/или композиции настоящего изобретения, для лечения конкретного заболевания зависит от целого ряда факторов, например, таких как тип пациента, его возраст, вес, пол, режим питания и состояние здоровья, а также от тяжести заболевания, способа введения, фармакологических свойств лекарственного средства (таких, как активность, эффективность, фармакокинетический и токсикологический профили конкретно используемого соединения) и от формы вводимого лекарственного средства (т.е. в зависимости от того, водится ли система доставки лекарственного средства к нужному участку организма, либо данное соединение вводится как часть лекарственной комбинации). Таким образом, схема приема лекарственного средства может широко варьироваться, а поэтому она может отклоняться от вышеуказанной предпочтительной схемы приема.
Соединения настоящего изобретения могут быть введены перорально, парентерально, ректально, путем ингаляции или путем местного применения, в виде унифицированных стандартных препаратов, содержащих традиционные нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители. Местное применение также предусматривает чрескожное введение лекарственного средства, например, путем использования накожных пластырей или устройств для ионофореза. Используемый в настоящем описании термин "парентеральное введение" означает подкожные, внутривенные, внутримышечные, внутригрудинные инъекции или вливания.
Препараты для инъекций,например стериальные инъецируемые водные или масляные суспензии, могут быть изготовлены стандартными способами с использованием соответствующих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильные инъецируемые препараты могут быть также изготовлены в виде стериальных инъецируемых растворов или суспензий в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. В качестве приемлемых наполнителей и растворителей могут служить вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды могут быть использованы стериальные жирные масла. Для этих целей может быть использовано любое мягкое жирное масло, включая синтетические моно- или диглицериды. Для получения инъецируемых препаратов могут быть также использованы жирные кислоты, такие как олеиновая кислота.
Суппозитории для ректального введения лекарственного средства могут быть получены путем смешивания активного соединения с подходящим нераздражающим наполнителей, таким как какао-масло и полиэтиленгликоли, которые являются твердыми при нормальных температурах, и жидкими при температуре прямой кишки, а поэтому после их введения в прямую кишку они расплавляются, высвобождая тем самым лекарственное средство.
Твердые лекарственные формы для перорального введения могут быть изготовлены в виде капсул, таблеток, пилюль, порошков и гранул. При получении указанных твердых препаратов активное соединение может быть смешано по крайней мере с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Кроме инертных разбавителей, указанные лекарственные формы могут также содержать и другие вспомогательные вещества, как это обычно принято в фармацевтической практике, например замасливающие агенты, такие как стеарат магния. Такие лекарственные формы, как капсулы, таблетки и пилюли могут также содержать забуферивающие агенты. Кроме того, таблетки и пилюли могут быть покрыты энтеросолюбильным покрытием.
Жидкие лекарственные формы для перорального введения могут быть изготовлены в виде фармацевтически приемлемых эмульсий, растворов, суспензий, сиропов и эликсиров, содержащих инертные разбавители, обычно используемые в этих целях, например, такие как вода. Указанные композиции могут также содержат другие добавки, такие как смачивающие, эмульгирующие и суспендирующие агенты, подслащивающие вещества, ароматизаторы и отдушки.
Хотя соединения настоящего изобретения могут быть введены как единственное активное фармацевтическое средство, однако они могут также быть использованы в комбинации с одним или несколькими иммуномодуляторами, противовирусными агентами или другими противоинфекционными средствами. Например, соединения настоящего изобретения могут быть введены в комбинации с AZT или с N-бутил-1-дезоксинотримицином для профилактики и/или лечения СПИДА. При введении соединений в виде комбинации терапевтические агенты могут быть изготовлены как отдельные композиции, которые вводятся одновременно или в разное время, либо эти терапевтические агенты могут быть введены как единая композиция.
Вышеприведенное описание изобретения носит лишь иллюстративный характер и не должно рассматриваться как некое ограничение его объема. Очевидно, что возможны различные варианты и изменения, не выходящие, однако, за рамки нижеследующей формулы изобретения.
Исходя из приведенного выше описания, каждый специалист может легко уяснить себе основные отличительные особенности настоящего изобретения и, не выходя за пределы его существа и объема, внести различные изменения и модификации в целях адаптации настоящего изобретения к данным конкретным условиям и применениям.
Реферат
Производные
сукциноиламиногидроксиэтиламиносульфонамида формулы I
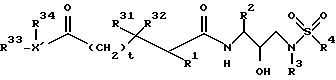
где R1-Н, C1 -C10-алкил;
R2-C1-C10-алкил, арил-C1-C10-алкил, возможно замещенный галогеном, или C3-C8-циклоалкил;
R3-C1-C10-алкил, C1-C10-галогеналкил, C3-C8-циклоалкил, C3-C8-циклоалкил-C1-C10-алкил;
R4-C1-C10-алкил, C1-C10-галогеналкил, C3-C8-циклоалкил, фенил, возможно замещенный одним или несколькими заместителями, выбранными из амино, галогена, C1-C10-алкила, C1-C10-алкокси; C1-C10-галогеналкила или гидрокси группы;
X1 - кислород, азот;
R31, R32-H, C1-C10алкил;
R33, R34 независимо H, фенил-C1-C10-алкил, при условии, что если X1 представляет кислород, то R34 отсутствует;
t-0, 1.
Используют в качестве фармацевтической композиции с антивирусной активностью, которую применяют при лечении ретровирусной инфекции. 5 с. и 31 з.п.ф-лы, 10 табл.
Формула
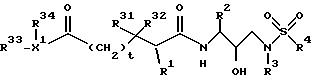
или их фармацевтически приемлемая соль,
где t - представляет собой 0 или 1;
R1 представляет собой водород или C1-C10-алкил;
R2 представляет собой C1-C10-алкил, арил C1-C10-алкил, необязательно замещенный галогеном, или C3-C8-циклоалкил;
R3 представляет собой C1-C10-алкил, арил C1-C10-галогеналкил, C3-C8-циклоалкил, C3 -C8-циклоалкил - C1-C10-алкил;
R4 представляет собой C1-C10-алкил, C1-C10-галогеналкил, C3 -C8-циклоалкил, фенил, необязательно замещенный одним или несколькими заместителями, выбранными из амино, галогена, C1-C10-алкила, C1-C10-алкокси, C1-C10-галогеналкила или гидроксигруппы;
X1 представляет собой кислород или азот;
R31 и R32 представляет собой водород или C1-C10-алкил;
R33 и R34 независимо представляют собой водород или фенил C1-C10-алкил, при условии, что если X1 представляет собой кислород, то R34 отсутствует.
[1S - [1R*(S*),2S*]]-N4 -[2-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино]-1-(фенилметил)пропил]-2,2,3-триметилбутандиамид;
[1S - [1R*(S*),2S*]]-4-[[2-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино] -1-(фенилметил)пропил] амино] -2,2,3-триметил-4-оксобутановой кислоты;
фенилметиловый эфир [1S - [1R*(S)*,2S*]]-4-[[2-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино] -1-(фенилметил)пропил] амино] -2,2,3-триметил-4-оксобутановая кислота;
[1S - [1R*(S*),2S*]]-N4-[2-гидрокси-3-[(2-метилпропил)-(фенилсульфонил)амино]-1-(фенилметил)пропил]-2,2,3-триметилбутандиамид;
фенилметиловый эфир [1S - [1R*(S*),2S*]]-4-[[2-гидрокси-3-[(2-метилпропил)(фенилсульфонил)амино] -1-(фенилметил)пропил]амино]-2,2,3-триметил-4-оксобутановой кислоты;
[1S - [1R*(S)*, 2S*]]-4-[[2-гидрокси-3-[(2-метилпропил)-(фенилсульфонил)амино] -1-(фенилметил)пропил] амино] -2,2,3-триметил-4-оксобутановая кислота;
[1S - [1R*(S*), 2S*]]-N4 -[2-гидрокси-3-[(3-метилбутил)(4-метоксифенилсульфонил)амино]-1-(фенилметил)пропил]-2,2,3-триметилбутандиамид;
фенилметиловый эфир [1S - /1R*(S*), 2S*]]-4-[[2-гидрокси-3-[(3-метилбутил)(4-метоксифенилсульфонил)амино] -1-(фенилметил)пропил] амино]-2,2,3-триметил-4-оксобутановой кислоты;
[1S - [1R*(S*), 2S*]]-4-[[2-гидрокси-3-[(3-метилбутил)-(4-метоксифенилсульфонил)амино] -1-(фенилметил)пропил]амино]-2,2,3-триметил-4-оксобутановая кислота;
[1S - [1R*(S*), 2S*] ]-4-[[2-гидрокси-3-[(2-метилпропил)-(4-метоксифенилсульфонил)амино] -1-(фенилметил)пропил] амино]-2,2,3-триметил-4-оксобутановая кислота;
фенилметиловый эфир [1S - [1R*(S*), 2S*]]-4-[[2-гидрокси-3-[(2-метилпропил)-(4-метоксифенилсульфонил)амино] -1-(фенилметил)пропил] амино]-2,2,3-триметил-4-оксобутановой кислоты;
[1S - [1R*(S*),2S*]]-N4 -[2-гидрокси-3-[(2-метилпропил)(4-метоксифенилсульфонил)амино]-1-(фенилметил)пропил]-2,2,3-триметилбутандиамид;
[1S - [1R*(S*), 2S*]]-N4 -[2-гидрокси-3-[(3-метилбутил)(4-фторофенилсульфонил)амино]-1-(фенилметил)пропил]-2,2,3-триметилбутандиамид;
фенилметиловый эфир [1S - [1R*(S*), 2S*]]-4-[[2-гидрокси-3-[(3-метилбутил)(4-фторофенилсульфонил)амино] -1-(фенилметил)пропил] амино]-2,2,3-триметил-4-оксобутановой кислоты;
[1S - [1R*(S*), 2S*] ]-4-[[2-гидрокси-3-[(3-метилбутил)-(4-фторофенилсульфонил)амино] -1-(фенилметил)пропил]амино]-2,2,3-триметил-4-оксобутановая кислота.
Комментарии