Ингибиторы катепсин-цистеинпротеазы - RU2312861C2
Код документа: RU2312861C2
Описание
ПРЕДПОСЫЛКИ К СОЗДАНИЮ ИЗОБРЕТЕНИЯ
Целый ряд нарушений у людей и других млекопитающих заключается в аномальной резорбции кости или ассоциирован с ней. Такие нарушения включают в себя, но не только, остеопороз, остеопороз, индукцированный глюкокортикоидами, болезнь Педжета (деформирующая остеодистрофия), аномально повышенный метаболизм кости, периодонтальное заболевание, выпадение зубов, переломы костей, ревматоидный артрит, остеоартпит, околопротезный остеолиз, дефект остеогенеза, метастатическое заболевание костей, гиперкальцемию злокачественной опухоли и множественную миелому. Одним из наиболее распространенных нарушений является остеопороз, наибольшая частота проявления которого имеет место у женщин в период после менопаузы. Остеопороз представляет собой системное заболевание скелета, характеризуемое низкой костной массой и разрушением микроархитектоники костной ткани с последующим увеличением ломкости кости и подверженности перелому. Остеопорозные переломы являются главной причиной заболеваемости и смертности пожилого населения. У не менее 50% женщин и трети мужчин бывает остеопорозный перелом. У большой части пожилого населения уже имеется низкая плотность кости и высокий риск переломов. Существует высокая потребность как в профилактике, так и в лечении остеопороза и других состояний, ассоциированных с резорбцией кости. Поскольку остеопороз, а также и другие нарушения, связанные с разрушением кости, в основном являются хроническими состояниями, считается, что для соответствующей терапии потребуется постоянное лечение.
Остеопороз характеризуется прогрессивным разрушением архитектуры кости и минерализации, что приводит к потере прочности кости и повышению процента переломов. Скелет постоянно реконструируется путем установления равновесия между остеобластами, которые закладывают новую кость, и остеокластами, которые разрушают, или резорбируют, кость. При некоторых болезненных состояниях и в пожилом возрасте баланс между образованием кости и резорбцией нарушен; кость ликвидируется с большей скоростью. Такой длительный дисбаланс преобладания резорбции над образованием приводит к более слабой структуре кости и к повышенному риску переломов.
Резорбция кости первоначально осуществляется остеокластами, которые представляют собой многоядерные гигантские клетки. Остеокласты резорбируют кость, образуя первоначальное клеточное прикрепление к костной ткани, за которым следует образование внеклеточного отсека или лакуны. Эти лакуны удерживаются при низком рН с помощью протонного АТФ насоса. Кислотная окружающая среда в лакуне инициирует деминерализацию кости с последующей деградацией костных белков или коллагена под действием протеаз, таких как цистеинпротеазы. См. Delaisse, J. M. et al., 1980, Biochem J 192:365-368; Delaisse, J. et al., 1984, Biochem Biophys Res Commun:441-447; Delaisse, J. M. et al.,1987, Bone 8:305-313, которые включены здесь в качестве ссылки в своем полном объеме. Коллаген составляет 95% органического матрикса кости. Следовательно, протеазы, принимающие участие в разрушении коллагена, являются важнейшими компонентами костного метаболизма и, как следствие, развития и прогрессирования остеопороза.
Катепсины принадлежат к папаиновому надсемейству цистеинпротеаз. Эти протеазы функционируют при нормальной физиологической, а также и при патологической деградации соединительной ткани. Катепсины играют большую роль при разрушении внутриклеточных белков и их обновлении и ремоделировании. В настоящее время был идентифицирован и секвенирован ряд катепсинов из различных источников. Эти катепсины обычно обнаруживают в разнообразных тканях. Например, был клонирован катепсин B, F, H, L, K, S, W и Z. Катепсин K (который также известен под аббревиатурой кат К) также известен как катепсин О и катепсин О2. См. Заявку PCT WO 96/13523, Khepri Pharmaceuticals, Inc., опубликованную 9 мая 1996, которая включена здесь в качестве ссылки в своем полном объеме. Катепсин L вовлечен в нормальный лизосомальный протеолиз, а также и в отдельные болезненные состояния, включающие в себя, но не только, метастазирование меланом. Катепсин S причастен к болезни Альцгеймера и некоторым аутоиммунным нарушениям, в том числе, но не только, юношескому диабету, рассеяному склерозу, обыкновенной пузырчатке, болезни Грависа, миастении гравис, системной красной волчанке, ревматоидному артриту и тироидиту Хошимото; аллергическим нарушениям, в том числе, но не только, астме, и аллогенным иммунным реакциям, в том числе, но не только, к отторжению органотрансплантатов или тканевых трансплантатов. В опухолях обнаружены повышенные уровени Катепсина В и перераспределение данного фермента, что подтверждает роль в опухолевой инвазии и метастазировании. Кроме того, отклоняющаяся от нормы активность Катепсина В причастна к таким болезненным состояниям, как ревматоидный артрит, остеоартрит, pneumocystiis carinii, острый панкреатит, воспалительные заболевания дыхательных путей и костные и суставные нарушения.
Известно, что ингибиторы цистеинпротеазы, такие как E-64 (транс-эпоксисукцинил-L-лейциламид-(4-гуанидино)бутан) эффективны при ингибировании резобрции кости. См. Delaisse, J. M. et al., 1987, Bone 8:305-313, включено здесь в качестве ссылки в своем полном объеме. Недавно был клонирован катепсин К, и обнаружена специфическая экспрессия в остеокластах. См. Tezuka, K. et al., 1994, J Biol Chem 269:1106-1109; Shi, G. P. et al., 1995, FEBS Lett 357:129-134; Bromme, D. и Okamoto, K., 1995, Biol Chem Hoppe Seyler 376:379-384; Bromme, D. et al., 1996. J Biol Chem 271:2126-2132; Drake, F. H. et al., 1996, J Biol Chem 271:12511-12516, включены здесь в качестве ссылок в полном объеме. Одновременно с клонированием аутосомно-рецессивное нарушение, пикнодизостоз, характеризующееся фенотипом врожденного системного остеопетроза с пониженной резорбцией кости, было картировано на мутацию, присутствующую в гене катепсина К. В настоящее время стало известно, что все мутации, выявленные в гене катепсина К, в результате приводят к инактивации белка. См. Gelb, B. D. et al., 1996, Science 273:1236-1238; Johnson, M. R. et al, 1996, Genome Res 6:1050-1055, включенные здесь в качестве ссылок в их полном объеме. Таким образом, можно предположить, что катепсин К вовлечен в опосредованную остеокластами резорбцию кости.
Катепсин К синтезируют в виде 37 КДа предшественника профермента, локализованного в лизосомальном отделе, где он предположительно самоактивируется в зрелый фермент 27 кДа при низком рН. См. McQueney, M. S. et al., 1997, J Biol Chem 272:13955-13960; Littlewood-Evans, A. et al., 1997, Bone 20:81-86, включены здесь в качестве ссылок в их полном объеме. Катепсин К наиболее близок катепсину S, имеющему 56% последовательности, идентичной на уровне аминокислот. S2P2 субстратная специфичность катепсина К сходна с таковой катепсина S с предпочтением в положениях Р1 и Р2 положительно заряженных остатков, таких как аргинин, и гидрофобных остатков, таких как фенилаланин или лейцин соответственно. См., Bromme, D. et al., 1996, J Biol Chem 271: 2126-2132; Bossard, M. J. et al., 1996, J Biol Chem 271:12517-12524, включенные здесь в качестве ссылок в их полном объеме. Катепсин К активен в широком диапазоне рН и значительно активен при рН 4-8, что, таким образом, позволяет проявляться хорошей каталитической активности в резорбционных лакунах остеокластов, где рН составляет около 4-5.
Коллаген человека I типа, основной коллаген в кости, является хорошим субстратом для катепсина К. См. Kafienah, W., et al., 1998, Biochem J 331:727-732, включено здесь в качестве ссылки в его полном объеме. Эксперименты In vitro с использованием антисмысловых олигонуклеотидов для катепсина К показали уменьшенную резорбцию кости in vitro, что возможно благодаря снижению трансляции мРНК катепсина К. См. Inui, T. et al., 1997, J Biol Chem 272:8109-8112, включено здесь в качестве ссылки в его полном объеме. Была установлена кристаллическая структура катепсина К. См. McGrath, M. E. et al., 1997, Nat Struct Biol 4:105-109; Zhao, B. et al., 1997, Nat Struct Biol 4: 109-11, которые включены здесь в качестве ссылок в их полном объеме. Также были разработаны селективные ингибиторы катепсина К, основанные на пептидах. См. Bromme, D. et al., 1996, Biochem J 315:85-89; Thompson, S. K. et al., 1997, Proc Natl Acad Sci USA 94:14249-14254, которые включены здесь в качестве ссылок в их полном объеме. Таким образом, ингибиторы катепсина К могут уменьшать резорбцию кости.
Такие ингибиторы были бы полезны при лечении нарушений, включающих резорбцию кости, таких как остеопороз.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, которые могут использоваться для лечения и/или профилактики катепсин-зависимых состояний или болезней млекопитающих, нуждающихся в этом. Один вариант осуществления настоящего изобретения иллюстрируется соединением Формулы I и его фармацевтически приемлемыми солями, стереоизомерами и N-оксидными производными

ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям следующей химической формулы:

где R1 предствляет собой водород, C1-6 алкил или C2-6 алкенил, где указанные алкильные и алкенильные группы необязательно замещены от одного до шести атомами галогена, С3-6 циклоалкил, -SR9, -SR12 -SOR9, -SOR12, -SO2R9, -SO2R12, -SO2CH(R12)(R11), -OR12, -OR9, -N(R12)2, арил, гетероарил или гетероциклил, где указанные арильная, гетероарильная и гетероциклическая группы необязательно замещены одним или двумя заместителями, независимо выбранными из C1-6 алкила, галогена, гидроксиалкила, гидрокси, алкокси или кето;
R2 предствляет собой водород, C1-6 алкил или C2-6 алкенил, где указанные алкильные и алкенильные группы необязательно замещены от одного до шести атомами галогена, С3-6 циклоалкил, -SR9, -SR12 -SOR9, -SOR12, -SO2R9, -SO2R12, -SO2CH(R12)(R11), -OR12, -OR9, -N(R12)2, арил, гетероарил или гетероциклил, где указанные арильная, гетероарильная и гетероциклическая группы необязательно замещены одним или двумя заместителями, независимо выбранными из C1-6 алкила, галогена, гидроксиалкила, гидрокси, алкокси или кето
или R1 и R2 могут быть взяты вместе с атомом углерода, к которому они присоединены, с образованием С3-8 циклоалкильного или гетероциклического кольца, где указанная кольцевая система необязательно замещена одним или двумя заместителями, независимо выбранными из С1-6 алкила, гидроксиалкила, галогеналкила или галогена;
R3 представляет собой водород, C1-6 алкил или C2-6 алкенил, где указанные алкильная и алкенильная группы необязательно замещены С3-6 циклоалкилом или от одного до шести атомами галогена;
R4 представляет собой водород, C1-6 алкил или C2-6 алкенил, где указанные алкильная и алкенильная группы необязательно замещены С3-6 циклоалкилом или от одного до шести атомами галогена
или R3 и R4 могут быть взяты вместе с атомом углерода, к которому они присоединены с образованием C3-8 циклоалкильного кольца, С5-8 циклоалкенильного кольца или пяти-семи членного гетероциклического радикала, где указанные циклоалкильная, циклоалкенильная и гетероциклическая группы необязательно замещены одним или двумя заместителями, независимо выбранными из C1-6 алкила, галогена, гидроксиалкила, гидрокси, алкокси или кето;
R5 выбран из водорода или С1-6 алкила, замещенного 1-6 атомами галогена;
R6 представляет собой арил, гетероарил, C1-6 галогеналкил, арилалкил или гетероарилалкил, где указанные арильная, гетероарильная, арилалкильная и гетероарилалкильная группы необязательно замещены одним, двумя или тремя заместителями, независимо выбранными из галогена, C1-6 алкила, C1-6 галогеналкила, С3-6 циклоалкила, галогеналкокси, -SR9, -SR12 -SOR9, -SOR12, -SO2R9, -SO2R12, -SO2CH(R12)(R11), -OR12, -N(R10)(R11), циано, или арил, необязательно замещенный -SO2R12;
каждый D независимо представляет собой C1-3 алкил, C2-3 алкенил; C2-3 алкинил, арил, гетероарил, С3-8 циклоалкил или гетероциклил, где каждая указанная арильная, гетероарильная, циклоалкильная и гетероциклильная группы, которые могут быть моноциклическими или бициклическими, необязательно замещена либо по атому углерода или по гетероатому одним - пятью заместителями, независимо выбранными из C1-6 алкила, галогеналкила, галогена, кето, алкокси, -SR9, -SR12, -OR9, -OR12, N(R12)2, -SO2R9, или -SO2R10;
R7 представляет собой водород, C1-6 алкил, C2-6 алкенил, C2-6 алкинил, C1-6 алкилокси, галоген, нитро, циано, арил, гетероарил, C3-8 циклоалкил, гетероциклил, -C(О)OR10, -C(О)OSi[CH(CH3)2]3, -OR9, -OR10, -C(О)R10, -R10C(О)R9, -C(О)R9, -C(О)N(Ra )(Rb), -C(О)N(R12)(R12), -C(О)N(R10)(R11), -C(R10)(R11)OH, -SR12, -SR9, -R10SR9, -R9, -C(R9)3, -C(R10)(R11)N(R9)2, -NR10С(О)NR10S(О)2R9, -SO2R12, -SO(R12), -SO2R9, -SOmN(Rc)(Rd), -SOmCH(R10)(R11), -SO2N(R10 )C(О)(R12), -SO2(R10)C(О)N(R12)2, -OSO2R10, -N(R10)(R11), -N(R10)C(О)N(R10)(R9), -N(R10)C(О)R9, -N(R10)C(О)R10, -N(R10)C(О)OR10, -N(R10)SO2(R10), -C(R10)(R11 )NR10C(R10)(R11)R9, -C(R10)(R11)N(R10)R9, -C(R10)(R11)N(R10)(R11), -C(R10)(R11)SC(R10)(R11)(R9), R10S-, -C(Ra)(Rb)NRaC(Ra)(Rb)(R9), -C(Ra)(Rb)N(Ra)(Rb), -C(Ra)(Rb)C(Ra)(Rb)N(Ra)(Rb), -C(О)C(Ra)(Rb )N(Ra)(Rb), -C(Ra)(Rb)N(Ra)C(О)R9, -C(О)C(Ra)(Rb)S(Ra), C(Ra)(Rb)C(О)N(Ra)(Rb), -B(OH)2, -OCH2О- или 4,4,5,5-тетраметил-1,3,2-дизоксоборолан-2-ил, где указанные группы необязательно замещены либо по атому углерода, либо по гетероатому одним - пятью заместителями, независимо выбранными из C1-6 алкила, галогена, кето, циано, галогеналкила, гидроксиалкила, -OR9, -NO2, -NH2, -NHS(О)2R8, -R9SO2R12, -SO2R12, -SO(R12), -SR12, -SR9, -SOmN(Rс )(Rd), -SOmN(R10)C(O)(R12), -C(R10)(R11)N(R10)(R11), -C(R10)(R11)OH, -COOH, -C(Ra)(Rb)C(О)N(Ra)(Rb), -C(О)(Ra)(Rb), -N(R10)C(R10)(R11)(R9), -N(R10)CO(R9), -NH(CH2)2OH, -NHC(О)OR10, -Si(CH3)3, гетероциклила, арила или гетероарила;
R8 представляет собой водород или C1-6 алкил
или R4 и R8 или могут быть взяты вместе с любым из атомов, к которому они могут быть присоединены, или находятся между ними, с образованием 4-10-членной гетероциклической кольцевой системы, где указанная кольцевая система, которая может быть моноциклической или бициклической, необязательно замещена одним или двумя заместителями, независимо выбранными из C1-6 алкила, галогена, гидроксиалкила, гидрокси, кето, -OR10, -SR10 или -N(R10)2;
R9 выбран из группы, состоящей из водорода, арила, арил(C1-4)алкила, гетероарила, гетероарил(C1-4)алкила, C3-8циклоалкила, C3-8 циклоалкил(C1-4)алкила и гетероциклил(C1-4)алкила, где указанные группы могут быть необязательно замещены одним, двумя или тремя заместителями, независимо выбранными из галогена, алкокси или -SO2R12;
R10 представляет собой водород или C1-6 алкил;
R11 представляет собой водород или C1-6 алкил;
R12 представляет собой водород, C1-6 алкил, который необязательно замещен одним, двумя или тремя заместителями, независимо выбранными из галогена, алкокси, циано, -NR10 или -SR10;
Ra представляет собой водород, C1-6 алкил, (C1-6 алкил)арил, (C1-6 алкил)гидроксил, -О(C1-6 алкил), гидроксил, галоген, арил, гетероарил, С3-8 циклоалкил, гетероциклил, где указанный алкил, арил, гетероарил, С3-8 циклоалкил и гетероциклил могут быть необязательно замещены либо по атому углерода, либо по гетероатому одним, двумя или тремя заместителями, независимо выбранными из C1-6 алкила или галогена;
Rb представляет собой водород, C1-6 алкил, (C1-6 алкил)арил, (C1-6 алкил)гидроксил, алкоксил, гидроксил, галоген, арил, гетероарил, С3-8 циклоалкил, гетероциклил, где указанный алкил, арил, гетероарил, С3-8 циклоалкил и гетероциклил могут быть необязательно замещены либо по атому углерода, либо по гетероатому одним, двумя или тремя заместителями, независимо выбранными из C1-6 алкила или галогена
или Ra и Rb могут быть взяты вместе с атомом углерда, к которому они присоединены, или находятся между ними, с образованием С3-8 циклоалкильного кольца или С3-8 гетероциклического кольца, где указанная 3-8-членная кольцевая система может быть необязательно замещена одним или двумя заместителями, независимо выбранными из C1-6 алкила и галогена;
Rc представляет собой водород или C1-6 алкил, который необязательно замещен одним, двумя или тремя заместителями, независмо выбранными из галогена или -OR9;
Rd представляет собой водород или C1-6 алкил, который необязательно замещен одним, двумя или тремя заместителями, независмо выбранными из галогена или -OR9
или Rc и Rd могут быть взяты вместе с атомом азота, к которому они присоединены, или находятся между ними с образованием C3-8 гетероциклического кольца, которое необязательно замещено одним или двумя заместителями, независимо выбранными из C1-6 алкила, галогенгидроксиалкила, гидрокси, алкокси или кето;
n независимо выбран из целого числа от нуля до трех;
каждый m независимо выбран из целого числа от нуля до двух;
и их фармацевтически приемлемым солям, стереоизомерам и N-оксидным производным.
Предпочтительно настоящее изобретение относится к соединениям следующей химической формулы:

где R1 предствляет собой водород, C1-6 алкил или C2-6 алкенил, где указанные алкильная и алкенильная группы необязательно замещены от одного до шести атомами галогена, С3-6 циклоалкил, -SR9, -SR12 -SOR9, -SOR12, -SO2R9, -SO2R12, -SO2CH(R12)(R11), -OR12, -OR9, -N(R12)2, арил, гетероарил или гетероциклил, где указанные арильная, гетероарильная и гетероциклильная группы необязательно замещены одним или двумя заместителями, независимо выбранными из C1-6 алкила, галогена, гидроксиалкила, гидрокси, алкокси или кето;
R2 предствляет собой водород, C1-6 алкил или C2-6 алкенил, где указанные алкильная и алкенильная группы необязательно замещены от одного до шести атомами галогена, С3-6 циклоалкил, -SR9, -SR12, -SOR9, -SOR12, -SO2R9, -SO2R12, -SO2CH(R12)(R11), -OR12, -OR9, -N(R12)2, арил, гетероарил или гетероциклил, где указанные арильная, гетероарильная и гетероциклильная группы необязательно замещены одним или двумя заместителями, независимо выбранными из C1-6 алкила, галогена, гидроксиалкила, гидрокси, алкокси или кето
или R1 и R2 могут быть взяты вместе с атомом углерода, к которому они присоединены, с образованием С3-8 циклоалкильного или гетероциклического кольца, где указанная кольцевая система необязательно замещена одним или двумя заместителями, независимо выбранными из С1-6 алкила, гидроксиалкила, галогеналкила или галогена;
R3 представляет собой водород, C1-6 алкил или C2-6 алкенил, где указанные алкильная и алкенильная группы необязательно замещены С3-6 циклоалкилом или от одного до шести атомами галогена;
R4 представляет собой водород, C1-6 алкил или C2-6 алкенил, где указанные алкильная и алкенильная группы необязательно замещены С3-6 циклоалкилом или от одного до шести атомами галогена
или R3 и R4 могут быть взяты вместе с атомом углерода, к которому они присоединены, с образованием C3-8 циклоалкильного кольца, С5-8 циклоалкенильного кольца или пяти-семичленного гетероциклического радикала, где указанные циклоалкильная, циклоалкенильная и гетероциклильная группы необязательно замещены одним или двумя заместителями, независимо выбранными из C1-6 алкила, галогена, гидроксиалкила, гидрокси, алкокси или кето;
R5 выбран из водорода или С1-6 алкила, замещенного 1-6 атомами галогена;
R6 представляет собой арил, гетероарил, C1-6 галогеналкил, арилалкил или гетероарилалкил, где указанные арильная, гетероарильная, арилалкильная и гетероарилалкильная группы необязательно замещены одним, двумя или тремя заместителями, независимо выбранными из галогена, C1-6 алкила, C1-6 галогеналкила, С3-6 циклоалкила, галогеналкокси, -SR9, -SR12 -SOR9, -SOR12, -SO2R9, -SO2R12, -SO2CH(R12)(R11), -OR12, -N(R10)(R11), циано или арил, необязательно замещенный -SO2R12;
каждый D независимо представляет собой C1-3 алкил, C2-3 алкенил, C2-3 алкинил, арил, гетероарил, С3-8 циклоалкил или гетероциклил, где каждая указанная арильная, гетероарильная, циклоалкильная и гетероциклильная группы, которые могут быть моноциклическими или бициклическими, необязательно замещены либо по атому углерода, либо по гетероатому одним - пятью заместителями, независимо выбранными из C1-6 алкила, галогеналкила, галогена, кето, алкокси, -SR9 , -SR12, -OR9, -OR12, N(R12)2, -SO2R9 или -SO2R10;
R7 представляет собой водород, C1-6 алкил, C2-6 алкенил, C2-6 алкинил, C1-6 алкилокси, галоген, нитро, циано, арил, гетероарил, C3-8 циклоалкил, гетероциклил, -C(О)OR10, -C(О)OSi[CH(CH3)2]3, -OR9, -OR10, -C(О)R10, -R10C(О)R9, -C(О)R9, -C(О)N(Ra)(Rb), -C(О)N(R12)(R12), -C(О)N(R10)(R11), -C(R10)(R11)OH, -SR12, -SR9, -R10SR9, -R9, -C(R9)3, -C(R10)(R11)N(R9)2, -NR10С(О)NR10S(О)2R9, -SO2 R12, -SO(R12), -SO2R9, -SOmN(Rc)(Rd), -SOmCH(R10)(R11), -SO2N(R10 )C(О)(R12), -SO2(R10)C(О)N(R12)2, -OSO2R10, -N(R10)(R11), -N(R10)C(О)N(R10)(R9), -N(R10)C(О)R9, -N(R10)C(О)R10, -N(R10)C(О)OR10, -N(R10)SO2(R10), -C(R10)(R11 )NR10C(R10)(R11)R9, -C(R10)(R11)N(R10)R9, -C(R10)(R11)N(R10)(R11), -C(R10)(R11)SC(R10)(R11)(R9), R10S-, -C(Ra)(Rb)NRaC(Ra)(Rb)(R9), -C(Ra)(Rb)N(Ra)(Rb), -C(Ra)(Rb)C(Ra)(Rb)N(Ra)(Rb), -C(О)C(Ra)(Rb )N(Ra)(Rb), -C(Ra)(Rb)N(Ra)C(О)R9, -C(О)C(Ra)(Rb)S(Ra), C(Ra)(Rb)C(О)N(Ra)(Rb), -B(OH)2, -OCH2О- или 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил, где указанные группы необязательно замещены либо по атому углерода, либо по гетероатому одним - пятью заместителями, независимо выбранными из C1-6 алкила, галогена, кето, циано, галогеналкила, гидроксиалкила, -OR9, -NO2, -NH2, -NHS(О)2R8, -R9SO2R12, -SO2R12, -SO(R12), -SR12, -SR9, -SOmN(Rс)(Rd), -SOmN(R10)C(O)(R12), -C(R10)(R11)N(R10)(R11), -C(R10)(R11)OH, -COOH, -C(Ra)(Rb )C(О)N(Ra)(Rb), -C(О)(Ra)(Rb), -N(R10)C(R10)(R11)(R9), -N(R10)CO(R9), -NH(CH2 )2OH, -NHC(О)OR10, -Si(CH3)3, гетероциклила, арила или гетероарила;
R8 представляет собой водород или C1-6 алкил;
или R4 и R8 или могут быть взяты вместе с любым из атомов, к которому они могут быть присоединены, или находятся между ними с образованием 4-10-членной гетероциклической кольцевой системы, где указанная кольцевая система, которая может быть моноциклической или бициклической, необязательно замещена одним или двумя заместителями, независимо выбранными из C1-6 алкила, галогена, гидроксиалкила, гидрокси, кето, -OR10, -SR10 или -N(R10)2;
R9 выбран из группы, состоящей из водорода, арила, арил(C1-4) алкила, гетероарила, гетероарил(C1-4)алкила, C3-8 циклоалкила, C3-8 циклоалкил(C1-4)алкила и гетероциклил(C1-4)алкила, где указанные группы могут быть необязательно замещены одним, двумя или тремя заместителями, независимо выбранными из галогена, алкокси или -SO2R12;
R10 представляет собой водород или C1-6 алкил;
R11 представляет собой водород или C1-6 алкил;
R12 представляет собой водород или C1-6 алкил, который необязательно замещен одним, двумя или тремя заместителями, независимо выбранными из галогена, алкокси, циано, -NR10 или -SR10;
Ra представляет собой водород, C1-6 алкил, (C1-6 алкил)арил, (C1-6 алкил)гидроксил, -О(C1-6 алкил), гидроксил, галоген, арил, гетероарил, С3-8 циклоалкил, гетероциклил, где указанный алкил, арил, гетероарил, С3-8 циклоалкил и гетероциклил может быть замещен либо по атому углерода, либо по гетероатому одним, двумя или тремя заместителями, независимо выбранными из C1-6 алкила или галогена;
Rb представляет собой водород, C1-6 алкил, (C1-6 алкил)арил, (C1-6 алкил)гидроксил, алкоксил, гидроксил, галоген, арил, гетероарил, С3-8 циклоалкил, гетероциклил, где указанный алкил, арил, гетероарил, С3-8 циклоалкил и гетероциклил могут быть необязательно замещены либо по атому углерода, либо по гетероатому одним, двумя или тремя заместителями, независимо выбранными из C1-6 алкила или галогена;
или Ra и Rb могут быть взяты вместе с атомом углерода, к которому они присоединены, или находятся между ними с образованием С3-8 циклоалкильного кольца или С3 -8 гетероциклического кольца, где указанная 3-8-членная кольцевая система может быть необязательно замещена одним или двумя заместителями, независимо выбранными из C1-6 алкила и галогена;
Rc представляет собой водород или C1-6 алкил, который необязательно замещен одним, двумя или тремя заместителями, независмо выбранными из галогена или -OR9;
Rd представляет собой водород или C1-6 алкил, который необязательно замещен одним, двумя или тремя заместителями, независмо выбранными из галогена или -OR9;
или Rc и Rd могут быть взяты вместе с атомом азота, к которому они присоединены, или находятся между ними с образованием C3-8 гетероциклического кольца, которое необязательно замещено одним или двумя заместителями, независимо выбранными из C1-6 алкила, галоген гидроксиалкила, гидрокси, алкокси или кето;
n независимо выбран из целого числа от нуля до трех;
каждый m независимо выбран из целого числа от нуля до двух,
и их фармацевтически приемлемым солям, стереоизомерам и N-оксидным производным.
В одном варианте осуществления данного изобретения R1 и R2 каждый представляют собой водород. В другом варианте осуществления данного изобретения R1 и R2, в случае нахождения у одного и того же атома углерода, могут быть взяты вместе с атомом углерода, к которому они присоединены, с образованием 3-8-членной циклоалкильной кольцевой системы, где указанная кольцевая система необязательно замещена C1-6 алкилом, гидроксиалкилом и галогеном. Примеры кольцевых систем, которые могут быть образованы, включают в себя циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. В предпочтительном варианте осуществления образуется циклопропил.
В другом варианте осуществления данного изобретения R1 и R2 взяты вместе с атомом углерода, к которому они присоединены, с образованием 3-8-членной гетероциклической кольцевой системы, где указаная кольцевая система необязательно замещена C1-6 алкилом, гидроксиалкилом или галогеналкилом. Примеры таких кольцевых систем, которые могут быть образованы, включают в себя пиперидинил, пирролидинил или тетрагидропиранил.
В одном варианте осуществления данного изобретения R3 и R4 каждый, независимо, представляют собой C1-4 алкил или H. В следующем варианте осуществления данного изобретения R3 представляет собой изобутил или н-пропил и R4 представляет собой H, более предпочтительно R3 представляет собой н-пропил.
В следующем варианте осуществления данного изобретения R3 представляет собой C1-6 алкил, где указанный алкил замещен С3-6 циклоалкилом или галогеном. Предпочтительно R3 представляет собой 2-фтор-2-метилпропил, 2-трифторметилпропил, 3-фтор-2-(2-фторметил)пропил, 2,2-дифторэтил, 2,2-дифторпропил, 3,3, 3-трифторпропил или 2,2-дихлорэтил и R4 представляет собой водород. Более предпочтительно R3 представляет собой 2-фтор-2-метилпропил.
В другом варианте осуществления данного изобретения R3 и R4, взятые вместе с атомом углерода, к которому они присоединены, образуют C3-8 циклоалкильное кольцо, C5-8 циклоалкенильное кольцо или пяти - семичленный гетероциклический радикал, где указанная циклоалкильная, циклоалкенильная и гетероциклическая группы необязательно замещены C1-6 алкилом, галогеном, гидроксиалкилом, гидрокси, алкокси или кето. Примеры кольцевых систем, которые могут быть образованы, включают в себя, но не только, следующие, учитывая, что гетероцикл необязательно замещен одним или более заместителями, как описано выше: циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. В одной категории осуществления изобретения образуется циклогексил.
В одном варианте осуществления данного изобретения R5 представляет собой C1-6 алкил, замещенный 1-6 атомами галогена, и R6 представляет собой C1-6 алкил, замещенный 1-6 атомами галогена. В другом варианте осуществления изобретения R5 представляет собой водород и R6 представляет собой C1-5 алкил, замещенный 1-6 атомами галогена. В следующем варианте осуществления R5 представляет собой водород и R6 представляет собой C1-6 алкил, замещенный 1-6 атомами фтора. В следующем варианте осуществления R5 представляет собой водород и R6 представляет собой C1-3 алкил, замещенный 3 атомами фтора. В другом варианте осуществления R5 представляет собой водород и R6 представляет собой трифторметил или 3,3,3,2,2-пентафторэтил, более предпочтительно R6 представляет собой трифторметил.
В другом варианте осуществления данного изобретения R5 представляет собой водород и R6 представляет собой арил или гетероарил, где указанные арил и гетероарил необязательно замещены галогеном или -SO2R12.
В другом варианте осуществления данного изобретения R1 и R2, взятые вместе с атомом углерода, к которому они присоединены, образуют циклопропил, циклобутил, циклопентил, циклогексил и циклогептил, более предпочтительно циклопропил; R3 представляет собой н-пропил, изобутил, 2-фтор-2-метилпропил, 2-трифторметилпропил, 3-фтор-2-(2-фторметил)пропил, 2,2-дифторэтил, 2,2-дифторпропил, 3,3,3-трифторпропил или 2,2-дихлорэтил; R4 и R5 представляют собой водород и R6 представляет собой C1-6 алкил, замещенный 1-6 атомами галогена; предпочтительно R6 представляет собой C1-3 алкил, замещенный 3 атомами фтора, более предпочтительно R6 представляет собой трифторметил или 3,3,3,2,2-пентафторэтил, наиболее предпочтительно трифторметил. В пределах этого варианта осуществления особенно предпочтительным вариантом осуществления является вариант, где n равно 1. Другим особенно предпочтительным вариантом осуществления является вариант, где n равно 2. Еще одним особенно предпочтительным вариантом осуществления является вариант, где n равно 3. Предпочтительно n равно 1 в тех случаях, когда D представляет собой гетероарил, который необязательно замещен галогеном или фенилом, замещенным гидроксиалкилом, -COR10 (где R10 представляет собой C1-6 алкил) или -SO2R12, или D представляет собой фенил, который необязательно замещен галогеном, -CONRаRb (где Rа представляет собой водород или C1-6 алкил и Rb представляет собой водород, C1-6 алкил, циклоалкил или C1-6 алкокси или Ra и Rb вместе с атомом азота, к которому они присоединены, образуют гетероциклил, где указанный гетероциклил необязательно замещен алкилом, гидроксиалкилом, или галогеналкилом), -SO2R12 (где R12 представляет собой C1-6 алкил), -COOR10, алкинилом, необязательно замещенным гидрокси или циклоалкилом, алкенилом, замещенным гидрокси, алкилом, необязательно замещенным гидрокси, -OR9(где R9 представляет собой арил), -OR10, -CR10R11SC10 R11R9 (где R9 представляет собой арил), -CH2S(арил), циано, -COR9 или гетероарилом.
Предпочтительно n равно 2 и каждый D представляет собой фенил, где второй фенил присоединен в положении 4 первого фенильного кольца (фенил присоединен к атому углерода, несущему группы R5 и R6) и дополнительно каждый фенил необязательно замещен одним или двумя заместителями независимо выбранными из C1-6 алкила, галогена, гидрокси, алкокси, галогеналкила, галогеналкокси или -SO2R12 (где R12 представляет собой C1-6 алкил), а второй фенил дополнительно замещен радикалом R7. Более предпочтительно каждый D представляет собой фенил, где второй фенил присоединен в положении 4 первого фенильного кольца и второй фенил необязательно замещен радикалом R7, который расположен в положении 4 фенильного кольца и представляет собой -SO2R12 (где R12 представляет собой C1-6 алкил, необязательно замещенный гидроксильной группой или галогеном), -SO2NRcRd (где Rс и Rd независимо представляют собой водород или C1-6алкил или Rc и Rd вместе с атомом азота, к которому они присоединены, образуют гетероциклическое кольцо), -SR12 (где R12 представляет собой C1-6алкил), -SOR12 (где R12 представляет собой C1-6алкил), -NHCOR10 (где R10 представляет собой C1-6алкил), -NR10R11 (где R10 и R11 представляют собой C1-6 алкил), -SO2 NHCOR10, гетероарил, галоген, -COOR10 (где R10 представляет собой водород или C1-6алкил), -OR9 (где R9 представляет собой водород или арил), -OR10 (где R10 представляет собой C1-6алкил), арил, замещенный -SO2R12 (где R12 представляет собой C1-6алкил), циано, галогеналкил, -C(R10)(R11)OH, C1-6алкил, необязательно замещенный -OR10 и галогеном, COR9 (где R9 представляет собой арил), -COR10, или -NHSO2R10 (где R10 представляет собой C1-6 алкил) и дополнительно второе фенильное кольцо необязательно замещено другим заместителем, выбранным из C1-6 алкила, -CHO, -COOR10, -COR10, -NHCOR10, галогена, галогеналкила, -OR10 (где R10представляет собой водород или C1-6 алкил, где указанный алкил необязательно замещен галогеном), -SO2NH2, -NHCOR10 (где R10 представляет собой C1-6 алкил), или -SO2R12 (где R12 представляет собой C1-6 алкил). Более предпочтительно второй фенил замещен радикалом R7 в положении 4, где R7 представляет собой -SOR12, -SO2R12 где R12 представляет собой C1-6 алкил (предпочтительно метил) или -SOmNRсRd, где Rс и Rd независимо представляют собой водород или алкил или Rс и Rd вместе образуют гетероциклил, и m равно целому числу от нуля до двух. Предпочтительно R7 представляет собой метилсульфонил, N-метиламиносульфонил, аминосульфонил или морфолин-4-илсульфонил.
Предпочтительно n равно 2, в том случае, когда первый D (D присоединен к атому углерода, несущему группы R5 и R6) представляет собой фенил, а второй D представляет собой гетероциклил (предпочтительно морфолин-4-ил, пиперазин-1-ил или пиперидин-4-ил), необязательно замещенный циклоалкилом, гетероарилом, C1-6 алкилом или гидроксиалкилом, более предпочтительно циклопропилом, метилом, этилом или гидроксиэтилом, и указанное гетероциклическое кольцо присоединено в положении 4 фенильного кольца.
Предпочтительно n равно 2, в том случае, когда первый D (D присоединен к атому углерода, несущему группы R5 и R6) представляет собой фенил, а второй D представляет собой гетероарил, замещенный одним или двумя заместителями, независимо выбранными из гидроксиалкила, -SO2 R12 (где R12 представляет собой C1-6 алкил), C1-6 алкила, галогена, галогеналкила, амино или -OR10.
Предпочтительно n равно 3, где первый и второй D представляют собой фенил, а третий D представляет собой гетероциклил и необязательно замещены, как определено выше. Более предпочтительно первый и второй D представляют собой фенил, где второй фенил присоединен в положении 4 первого фенила, а гетероциклил представляет собой морфолин-4-ил, пирролидин-1-ил, пиперидин-1-ил, пиперидин-4-ил или пиперазин-1-ил, замещенные радикалом R7. Предпочтительно R7 представляет собой водород, алкил, гидроксиалкил, галогеналкил, циклоалкил, COOR10 или -SO2R12 (где R12 представляет собой C1-6 алкил).
В другом варианте осуществления данного изобретения R1 и R2 взяты вместе с атомом углерода, к которому они присоединены, с образованием циклопропила, циклобутила, циклопентила, циклогексила и циклогептила, более предпочтительно циклопропила; R3 и R4 взяты вместе с атомом углерода, к которому они присоединены, с образованием С3-8циклоалкильного кольца, C5-8 циклоалкенильного кольца или пяти - семичленного гетероциклического радикала, где указанные циклоалкильная, циклоалкенильная и гетероциклильная группы необязательно замещены C1-6 алкилом, галогеном, гидроксиалкилом, гидрокси, алкокси или кето. Примеры кольцевых систем, которые могут быть образованы, включают в себя, но не только, следующие, учитывая, что гетероцикл необязательно замещен одним или более заместителями, как описано выше: циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, тетрагидрофуран-4-ил, пиперидин-4-ил. Предпочтительно R3 и R4, взятые вместе с атомом углерода, к которому они присоединены, образуют циклопропил, циклобутил, циклопентил, циклогексил или циклогептил. Более предпочтительным вариантом осуществления изобретения является вариант, когда R3 и R4, взятые вместе с атомом углерода, к которому они присоединены, образуют циклогексил. В этом варианте осуществления особенно предпочтительным вариантом является вариант, где радикалы R5 представляют собой водород, а R6 представляет собой C1-6 алкил, замещенный 1-6 атомами галогена, предпочтительно R6 представляет собой C1-3 алкил, замещенный 3 атомами фтора, более предпочтительно R6 представляет собой трифторметил или 3,3,3,2,2-пентафторэтил, наиболее предпочтительно трифторметил. В этом варианте осуществления особенно предпочтительным вариантом является вариант, где n равно 1. Другим особенно предпочтительным вариантом являетя вариант, где n равно 2. Еще одним особенно предпочтительным вариантом осуществления является вариант, где n равно 3. Предпочтительно n равно 1 в том случае, когда D представляет собой гетероарил, необязательно замещенный галогеном или фенилом, замещенным гидроксиалкилом, -COR10 (где R10 представляет собой C1-6 алкил) или -SO2R12, или D представляет собой фенил, необязательно замещенный галогеном, -CONRаRb (где Ra представляет собой водород или C1-6 алкил, и R5 представляет собой водород, C1-6 алкил, циклоалкил или C1-6 алкокси, или Rа и Rb вместе с атомом азота, к которому они присоединены, образуют гетероциклический радикал, где указанный гетероциклил необязательно замещен алкилом или галогеналкилом), -SO2R12 (где R12 представляет собой C1-6 алкил), -COOR10, алкинил, замещенный гидрокси, алкенил, замещенный гидрокси, алкил, необязательно замещенный гидрокси, -OR10, -CR10R11SC10R11R9 (где R9 представляет собой арил), -CH2S(арил)-COR9 или гетероарил.
Предпочтительно n равно 2 и каждый D представляет собой фенил, где второй фенил присоединен в положении 4 первого фенильного кольца (фенил присоединен к атому углерода, несущему группы R5 и R6) и, дополнительно, где каждый фенил необязательно замещен одним или двумя заместителями, независимо выбранными из C1-6 алкила, галогена, гидрокси, алкокси, галогеналкила, галогеналкокси, а второй фенил дополнительно замещен радикалом R7. Более предпочтительно каждый D представляет собой фенил, где второй фенил присоединен в положении 4 первого фенильного кольца, второй фенил необязательно замещен радикалом R7, который расположен в положении 4 фенильного кольца и представляет собой -SO2R12 (где R12 представляет собой C1-6 алкил, необязательно замещенный галогеном), -SO2NRсRd (где Rс и Rdнезависимо представляют собой водород или C1-6 алкил или Rс и Rd, вместе с атомом азота, к которому они присоединены, образуют гетероциклическое кольцо), -SR12 (где R12 представляет собой С1-6алкил), -NHCOR9 (где R9 представляет собой С1-6 алкил), -NR10R11 (где R10 и R11 представляют собой С1-6алкил), гетероарил, галоген, -COOR10 (где R10 представляет собой водород или С1-6алкил), -OR9(где R9 представляет собой водород или арил), арил, замещенный -SO2R12 (где R12 представляет собой С1-6алкил), циано, галогеналкил, -CHO, -C(R10)(R11)OH, С1-6алкил, необязательно замещенный радикалом -OR10 и галогеном, -COR10, или -NHSO2R10 (где R10 представляет собой С1-6алкил) и, дополнительно, второе фенильное кольцо необязательно замещено другим заместителем, выбранным из галогена, галогеналкила, -OR10 (где R10 представляет собой водород или С1-6алкил, где указанный алкил необязательно замещен галогеном) или -SO2R12 (где R12 представляет собой С1-6алкил). Более предпочтительно второй фенил замещен радикалом R7 в положении 4, где R7 представляет собой -SO2R12, где R12 представляет собой С1-6алкил (предпочтительно метил) или -SOmNRсRd, где Rс и Rd независимо представляют собой водород или алкил или Rс и Rd вместе образуют гетероциклил, и m представляет собой целое число от нуля до двух. Предпочтительно R7 представляет собой метилсульфонил, N-метиламиносульфонил, аминосульфонил, или морфолин-4-илсульфонил.
Предпочтительно n равно 2 в том случае, когда первый D (D, присоединенный к атому углерода, несущему группы R5 и R6) представляет собой фенил, а второй D представляет собой гетероциклический радикал (предпочтительно пиперазин-1-ил или пиперидин-4-ил), замещенный циклоалкилом, C1-6 алкилом или гидроксиалкилом, более предпочтительно циклопропилом, метилом, этилом или гидроксиэтилом, и указанное гетероциклическое кольцо присоединено в положении 4 фенильного кольца.
Предпочтительно n равно 2 в тех случаях, когда первый D (D, присоединенный к атому углерода, несущему группы R5 и R6) представляет собой фенил, а второй D представляет собой гетероарил, замещенный одним или двумя заместителями, независимо выбранными из гидроксиалкила, -SO2R12 (где R12 представляет собой C1-6 алкил), C1-6 алкил, галоген, или -OR10.
Предпочтительно n равно 3, где первый и второй D представляют собой фенил, а третий D представляет собой гетероциклический радикал и необязательно замещены, как определено выше. Более предпочтительно первый и второй D представляют собой фенил, где второй фенил присоединен в положении 4 первого фенила и гетероциклический радикал представляет собой морфолин-4-ил, пирролидин-1-ил, пиперидин-1-ил, пиперидин-4-ил или пиперазин-1-ил, замещенные радикалом R7. Предпочтительно R7 представляет собой водород, алкил, гидроксиалкил, галогеналкил или циклоалкил.
В другом варианте осуществления данного изобретения R5 представляет собой водород и R6 представляет собой арил, необязательно замещенный одним, двумя или тремя заместителями, независимо выбранными из галогена, C1-6 алкила, C1-6 галогеналкила, C3-6 циклоалкила, галогеналкокси, -SR9, -SR12, -SOR9, -SOR12, -SO2R9, -SO2R12, -SO2CH(R12)(R11), -OR12, -N(R10)(R11), циано, или арилом, который необязательно замещен -SO2R12. Более предпочтительно фенил замещен C1-6 алкилом, галогеном, галогеналкилом или галогеналкокси. В этом варианте осуществления другим предпочтительным вариантом является, когда R1 и R2 каждый представляет собой водород. В этом варианте осуществления другим предпочтительным вариантом является, когда R1 и R2 взяты вместе с атомом углерода, к которому они присоединены, для образования 3-8-членной циклоалкильной или гетероциклической кольцевой системы, где указанная кольцевая система необязательно замещена C1-6 алкилом, гидроксиалкилом и галогеном. Предпочтительные кольцевые системы, которые могут быть образованы, включают в себя циклопропил, циклобутил, циклопентил, циклогексил и циклогептил, более предпочтительно циклопропил. В этих предпочтительных и более предпочтительных вариантах осуществления еще более предпочтительным вариантом осуществления является вариант, где R3 представляет собой C1-4 алкил и R4 представляет собой H. Предпочтительно R3 представляет собой н-пропил или изобутил и R4 представляет собой H. В этих предпочтительных и более предпочтительных вариантах осуществления другим, еще более предпочтительным вариантом осуществления, является вариант, где R3 представляет собой 2-фтор-2-метилпропил, 2-трифторметилпропил, 3-фтор-2-(2-фторметил)пропил, 2,2-дифторэтил, 2,2-дифторпропил, 3,3,3-трифторпропил или 2,2-дихлорэтил и R4 представляет собой водород. В этих предпочтительных и более предпочтительных вариантах осуществления другим, еще более предпочтительным вариантом осуществления, является вариант, где R3 и R4 могут быть взяты вместе с атомом углерода, к которому они присоединены, с образованием С3-8 циклоалкильного кольца, С5-8 циклоалкенильного кольца или пяти - семичленного гетероциклического радикала, где указанные циклоалкильная, циклоалкенильная и гетероциклильная группы необязательно замещены C1-6 алкилом, галогеном, гидроксиалкилом, гидрокси, алкокси или кетоном. Примеры кольцевых систем, которые могут быть образованы, включают в себя, но не только, следующие, учитывая, что данный гетероцикл необязательно замещен одним или более заместителями, как указано выше: циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Предпочтителен вариант осуществления, когда образуется циклогексил.
В этом варианте осуществления изобретения особенно предпочтителен вариант, где n равно 1. Другой предпочтительный вариант, когда n равно 2. Еще один предпочтительный вариант, когда n равно 3. Предпочтительно n равно 1 в том случае, когда D представляет собой гетероарил, который необязательно замещен галогеном, или фенил, замещенный гидроксиалкилом, -COR10 (где R10 представляет собой C1-6 алкил) или SO2R12, или D представляет собой фенил, необязательно замещенный галогеном, -CONRаRb (где Ra представляет собой водород или C1-6 алкил и Rb представляет собой водород, C1-6 алкил, циклоалкил или C1-6 алкокси или Ra и Rb вместе с атомом азота, к которому они присоединены, образуют гетероциклический радикал, где указанный гетероциклил необязательно замещен алкилом или галогеналкилом), -SO2R12 (где R12представляет собой C1-6 алкил), -COOR10, алкинил, замещенный гидрокси, алкенил, замещенный гидрокси, алкил, необязательно замещенный гидрокси, -OR10, -CR10R11SC10R11R9 (где R9 представляет собой арил), -CH2S(арил) -COR9 или гетероарил.
Предпочтительно n равно 2, а каждый D представляет собой фенил, где второй фенил присоединен в положении 4 первого фенильного кольца (фенил присоединен к атому углерода, несущему группы R5 и R6) и, дополнительно, где каждый фенил необязательно замещен одним или двумя заместителями, независимо выбранными из C1-6 алкила, галогена, гидрокси, алкокси, галогеналкила, галогеналкокси, а второй фенил дополнительно замещен радикалом R7. Более предпочтительно каждый D представляет собой фенил, где второй фенил присоединен в положении 4 первого фенильного кольца, а второй фенил необязательно замещен радикалом R7, который расположен в положении 4 данного фенильного кольца и представляет собой -SO2R12 (где R12 представляет собой C1-6 алкил, необязательно замещенный галогеном), SO2NRcRd (где Rcи Rdнезависимо представляют собой водород или C1-6алкил или Rc и Rd вместе с атомом азота, к которому они присоединены, образуют гетероциклическое кольцо), -SR12 (где R12 представляет собой C1-6 алкил), -NHCOR9 (где R9 представляет собой C1-6 алкил), -NR10R11 (где R10и R11 представляют собой C1-6 алкил), гетероарил, галоген, -COOR10 (где R10 представляет собой водород или C1-6 алкил), -OR9(где R9 представляет собой водород или арил), арил, замещенный радикалом -SO2R12 (где R12 представляет собой C1-6 алкил), циано, галогеналкил, -CHO, -C(R10)(R11)OH, C1-6 алкил, необязательно замещенный группой -OR10 и галогеном, -COR10, или -NHSO2R10 (где R10 представляет собой C1-6 алкил) и дополнительно второе фенильное кольцо необязательно замещено другим заместителем, выбранным из галогена, галогеналкила, -OR10 (где R10 представляет собой водород или C1-6 алкил, где указанный алкил необязательно замещен галогеном) или -SO2R12 (где R12 представляет собой C1-6 алкил). Более предпочтительно второй фенил замещен радикалом R7 в положении 4, где R7 представляет собой -SO2R12, где R12 представляет собой C1-6 алкил (предпочтительно метил) или -SOmNRcRd, где Rс и Rd независимо представляют собой водород или алкил или Rс и Rd вместе образуют гетероциклил и m представляет собой целое число от нуля до двух. Предпочтительно R7 представляет собой метилсульфонил, N-метиламиносульфонил, аминосульфонил или морфолин-4-илсульфонил.
Предпочтительно n равно 2, в том случае, когда первый D (D присоединен к атому углерода, несущему группы R5и R6) представляет собой фенил, а второй D представляет собой гетероциклический радикал (предпочтительно пиперазин-1-ил или пиперидин-4-ил), замещенный циклоалкилом, C1-6 алкилом или гидроксиалкилом, более предпочтительно циклопропилом, метилом, этилом или гидроксиэтилом, и указанное гетероциклическое кольцо присоединено в положении 4 фенильного кольца.
Предпочтительно n равно 2, в том случае, когда первый D (D присоединен к атому углерода, несущему группы R5 и R6) представляет собой фенил, а второй D представляет собой гетероарил, замещенный одним или двумя заместителями, независимо выбранными из гидроксиалкила, -SO2R12 (где R12 представляет собой C1-6 алкил), C1-6 алкила, галогена или -OR10.
Предпочтительно n равно 3, когда первый и второй D представляют собой фенил, а третий D представляют собой гетероциклический радикал и необязательно замещены, как определено выше. Более предпочтительно первый и второй D представляют собой фенил, где второй фенил присоединен в положении 4 первого фенила, а гетероциклический радикал представляет собой морфолин-4-ил, пирролидин-1-ил, пиперидин-1-ил, пиперидин-4-ил или пиперазин-1-ил, необязательно замещенные группой R7. Предпочтительно R7 представляет собой водород, алкил, гидроксиалкил, галогеналкил или циклоалкил.
В одном варианте осуществления данного изобретения R5 представляет собой водород, а R6 представляет собой гетероарил, необязательно замещенный C1-6 алкилом, галогеном, галогеналкилом или галогеналкокси. Предпочтительно R6 представляет собой тиазолил, пиридинил, тетразолил, тиенил или фуранил, необязательно замещенные C1-4 алкилом или галогеном. В этом варианте осуществления предпочтительным вариантом осуществления является вариант, где R1 и R2 каждый представляет собой водород. В этом варианте осуществления другим предпочтительным вариантом является вариант, где R1 и R2 взяты вместе с атомом углерода, к которому они присоединены, образуют 3-8-членную циклоалкильную или гетероциклическую кольцевую систему, где указанная кольцевая система необязательно замещена С1-6 алкилом, гидроксиалкилом и галогеном. Предпочтительные кольцевые системы, которые могут быть образованы, включают в себя циклопропил, циклобутил, циклопентил, циклогексил и циклогептил, более предпочтительно циклопропил. В этих предпочтительных и более предпочтительных вариантах осуществления еще более предпочтительным вариантом является вариант, где R3 представляет собой C1-4 алкил и R4 представляют собой H. Предпочтительно R3 представляет собой н-пропил или изобутил и R4 представляет собой H. В этих предпочтительных и более предпочтительных вариантах осуществления другим еще более предпочтительным вариантом является вариант, где R3 представляет собой 2-фтор-2-метилпропил, 2-трифторметилпропил, 3-фтор-2-(2-фторметил)пропил, 2,2-дифторэтил, 2,2-дифторпропил, 3,3,3-трифторпропил или 2,2-дихлорэтил и R4 представляет собой водород. В этих предпочтительных и более предпочтительных вариантах осуществления другим еще более предпочтительным вариантом является вариант, где R3 и R4могут быть взяты вместе с атомом углерода, к которому они присоединены, с образованием С3-8 циклоалкильного кольца, С5-8 циклоалкенильного кольца или пяти - семичленного гетероциклического радикала, где указанные циклоалкильная, циклоалкенильная и гетероциклическая группы необязательно замещены C1-6 алкилом, галогеном, гидроксиалкилом, гидрокси, алкокси или кетоном. Примеры кольцевых систем, которые могут быть образованы, включают в себя, но не только, следующее, учитывая, что гетероцикл необязательно замещен одним или более заместителями, как описано выше: циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Предпочтителен вариант, когда образуется циклогексил.
В этом варианте осуществления особенно предпочтительным вариантом является вариант, где n равно 1. Другим, особенно предпочтительным вариантом осуществления, является вариант, где n равно 2. Еще одним особенно предпочтительным вариантом осуществления является вариант, где n равно 3. Предпочтительно n равно 1 в том случае, когда D представляет собой гетероарил, необязательно замещенный галогеном или фенилом, замещенным гидроксиалкилом, -COR10 (где R10 представляет собой C1-6 алкил) или -SO2R12, или D представляет собой фенил, необязательно замещенный галогеном, -CONRаRb (где Rа представляет собой водород или C1-6 алкил и Rb представляет собой водород, C1-6 алкил, циклоалкил или C1-6 алкокси или Rа и Rbвместе с атомом азота, к которому они присоединены, образуют гетероциклический радикал, где указанный гетероциклил необязательно замещен алкилом или галогеналкилом), -SO2R12 (где R12представляет собой C1-6 алкил), -COOR10, алкинилом, замещенным гидрокси, алкенилом, замещенным гидрокси, алкилом, необязательно замещенным гидрокси, -OR10, -CR10R11 SC10R11R9 (где R9 представляет собой арил), -CH2S(арил)-COR9 или гетероарилом.
Предпочтительно n равно 2 и каждый D представляет собой фенил, где второй фенил присоединен в положении 4 первого фенильного кольца (фенил присоединен к атому углерода, несущему группы R5 и R6) и, дополнительно, где каждый фенил необязательно замещен одним или двумя заместителями, независимо выбранными из C1-6 алкила, галогена, гидрокси, алкокси, галогеналкила, галогеналкокси, а второй фенил дополнительно замещен группой R7. Более предпочтительно каждый D представляет собой фенил, где второй фенил присоединен в положении 4 первого фенильного кольца и второй фенил необязательно замещен группой R7, которая расположена в положении 4 фенильного кольца и представляет собой -SO2R12 (где R12 представляет собой C1-6 алкил, необязательно замещенный галогеном), -SO2NRсRd (где Rc и Rd независимо представляют собой водород или C1-6 алкил или Rc и Rd вместе с атомом азота, к которому они присоединены, образуют гетероциклическое кольцо), -SR12 (где R12 представляет собой C1-6 алкил), -NHCOR9 (где R9 представляет собой C1-6 алкил), -NR10R11 (где R10 и R11 представляют собой C1-6 алкил), гетероарил, галоген, -COOR10 (где R10 представляет собой водород или C1-6 алкил), -OR9(где R9 представляет собой водород или арил), арил, замещенный группой -SO2R12 (где R12 представляет собой C1-6 алкил), циано, галогеналкил, -CHO, -C(R10)(R11)OH, C1-6 алкил, необязательно замещенный группой -OR10 и галогеном, -COR10, или -NHSO2R10 (где R10 представляет собой C1-6 алкил), и, дополнительно, второе фенильное кольцо необязательно замещено другим заместителем, выбранным из галогена, галогеналкила, -OR10 (где R10 представляет собой водород или C1-6 алкил, где указанный алкил необязательно замещен галогеном) или -SO2R12 (где R12 представляет собой C1-6 алкил). Более предпочтительно, второй фенил замещен группой R7 в положении 4, где R7 представляет собой -SO2R12, где R12 представляет собой C1-6 алкил (предпочтительно метил) или -SOmNRс Rd, где Rс и Rd независимо представляют собой водород или алкил, или Rc и Rd вместе образуют гетероциклический радикал, и m представляет собой целое число от нуля до двух. Предпочтительно R7 представляет собой метилсульфонил, N-метиламиносульфонил, аминосульфонил или морфолин-4-илсульфонил.
Предпочтительно n равно 2 в том случае, когда первый D (D присоединен к атому углерода, несущему группы R5 и R6) представляет собой фенил, а второй D представляет собой гетероциклический радикал (предпочтительно пиперазин-1-ил или пиперидин-4-ил), замещенный циклоалкилом, C1-6 алкилом или гидроксиалкилом, более предпочтительно циклопропилом, метилом, этилом или гидроксиэтилом, и указанное гетероциклическое кольцо присоединено в положении 4 фенильного кольца.
Предпочтительно n равно 2 в том случае, когда первый D (D присоединен к атому углерода, несущему группы R5 и R6) представляет собой фенил, а второй D представляет собой гетероарил, необязательно замещенный одним или двумя заместителями, независимо выбранными из гидроксиалкила, -SO2R12 (где R12 представляет собой С1-6 алкил), С1-6 алкила, галогена или -OR10.
Предпочтительно n равно 3, где первый и второй D представляют собой фенил, а третий D представляет собой гетероциклический радикал, и необязательно замещены, как описано выше. Более предпочтительно первый и второй D представляют собой фенил, где второй фенил присоединен в положении 4 первого фенила, а гетероциклический радикал представляет собой морфолин-4-ил, пирролидин-1-ил, пиперидин-1-ил, пиперидин-4-ил или пиперазин-1-ил, замещенные группой R7. Предпочтительно R7 представляет собой водород, алкил, гидроксиалкил, галогеналкил или циклоалкил.
В одном варианте осуществления данного изобретения R4 и R8 или могут быть взяты вместе с любым из атомов, к которым они могут быть присоединены, или находятся между ними с образованием 4-10-членной гетероциклической кольцевой системы, где указанная кольцевая система, которая может быть моноциклической или бициклической, необязательно замещена С1-6 алкилом, галогеном, гидроксиалкилом, гидрокси, кетогруппой, -OR10, -SR10 или -N(R10)2. В следующем варианте осуществления данного изобретения R4 и R8 определены таким образом, что они могут быть взяты вместе с атомом азота, к которому они присоединены, для образования моноциклического или бициклического гетероцикла с 5-7-членами в каждом кольце и необязательно содержащие, в дополнение к атому азота, 1 или 2 дополнительных гетероатома, выбранных из N, О и S, указанный гетероцикл необязательно замещен одним или более заместителями, выбранными из С1-6 алкила, галогена, гидроксиалкила, гидрокси, кетогруппы, -OR10, -SR10 или -N(R10)2. В следующем примере R4 и R8 определены таким образом, что они могут быть взяты вместе с атомом азота, к которому они присоединены, для образования 5- или 6-членной гетероциклической кольцевой системы. Примеры таких гетероциклов, которые, таким образом, могут быть образованы, включают в себя, но не только, пяти- или шестичленные кольца, содержащие по меньшей мере один атом азота, необязательно замещенный одним или более заместителями, как описано выше. Предпочтителен вариант осуществления, когда образуется необязательно замещенный пиролидинил.
В одном варианте осуществления данного изобретения Ra и Rb определены таким образом, что они могут быть взяты с атомом углерода или азота, к которым они присоединены, для образования моноциклического или бициклического карбоцикла или гетероцикла с 5-7 членами в каждом кольце. Данный гетероцикл может необязательно содержать, в дополнение к атому азота, 1 или 2 дополнительных гетероатома, выбранных из N, О и S. Указанный карбоцикл и гетероцикл может быть необязательно замещен одним или более заместителями, выбранными из С1-6 алкила и галогена.
Ссылка на вышеизложенные предпочтительные варианты осуществления означает включение всех комбинаций конкретных и предпочтительных групп, если не указано другого.
Другой вариант осуществления настоящего изобретения охватывает способ получения соединений по настоящему изобретению, включающий в себя:
(i) взаимодействие соединения формулы (a):

(a)
где R1-R6 и D определены выше, n1 представляет собой целое число из 1-3 и X представляет собой галоген,
с соединением формулы (b):
R7-(D)n2-Y
(b)
где R7 определен выше, n2 представляет собой целое число от 0 до 2 при условии, что n1 и n2 вместе являются целым числом от 1 до 3 и Y представляет собой бороновую кислоту или 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил; или
(ii) взаимодейстие соединения формулы (c):

где R3-R7, n и D определены выше, а Z представляет собой гидрокси, галоген или сложный эфир сукцинимида,
с соединением формулы (d):

(d)
или его солью, где R1 и R2 определены выше;
(iii) необязательно, модифицирование любой из групп R1-R7 и D;
(iv) необязательно, обработку соединения формулы (I), полученного на вышеуказанных стадиях (i)-(iii), кислотой для получения соответствующей кислотно-аддитивной соли;
(v) необязательно, обработку соединения формулы (I), полученного на вышеуказанных стадиях (i)-(iii), основанием для получения соответствующего свободного основания и
(vi) необязательно разделение смеси стереоизомеров соединения формулы (I), полученного вышеуказанных на стадиях (i), (ii), (iii), (iv) или (v), с получением отдельного стереоизомера.
Настоящее изобретение относится к способам лечения нарушений, связанных с аномальной резорбцией кости. Такие нарушения включают в себя, но не только, остеопороз, остеопороз, индуцированный глюкокортикоидами, болезнь Педжета (деформирующая остеодистрофия), аномально повышенный метаболизм кости, периодонтальное заболевание, выпадение зубов, переломы костей, ревматоидный артрит, остеоартрит, околопротезный остеолизис, недостаточность остеогенеза, заболевание костей, связанное с метастазированием, гиперкальцемию злокачественных опухолей и множественную миелому. Предпочтительный вариант осуществления включает в себя способы лечения остеопороза и заболевания костей, связанного с метастазированием. Более предпочтительный вариант осуществления включает в себя способы лечения остеопороза.
Характерные соединения настоящего изобретения представлены в Таблицах I-IV, приведенных ниже.
Соединение формулы I, где R1 R2, R4, R5 и R8 представляют собой водород, показаны в Таблице I, приведенной ниже.

Таблица I
Соединения формулы I, где R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют циклопропил, R4, R5 и R8 представляют собой водород, показаны в таблице II, приведенной ниже.

Таблица II
Соединения формулы I, где R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют циклогексил, R1, R5 и R8 представляют собой водород, показаны в таблице III, приведенной ниже.

Таблица III
Соединения формулы I, где R1, R4, R5 и R8 представляют собой водород, показаны в таблице IV, приведенной ниже.

Таблица IV
Конкретные варианты осуществления настоящего изобретения включают в себя, но не только, следующие соединения:
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2, 2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-(2,2,2-трифтор-1-фенилэтил)-L-лейцинамид;
N1 -(цианометил)-N2-[2,2,2-трифтор-1-(4-фтор-3-метилфенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1R)-2,2, 2-трифтор-1-(4-пиридин-3-илфенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(4-пиридин-3-илфенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1R)-2,2,2-трифтор-1-(4-пиридин-4-илфенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2, 2-трифтор-1-(4-пиридин-4-илфенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(lR)-2,2,2-трифтор-1-(4-{[4-(2-фторэтил)пиперазин-1-ил]карбонил}фенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[2,2,2-трифтор-1-(4-{[4-(2-фторэтил)пиперазин-1-ил]карбонил}фенил)этил]-L-лейцинамид;
N2-[1-(1,1'-дифенил-4-ил)-2,2, 2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{2,2,2-трифтор-1-[4-(3-гидрокси-3-метилбут-1-инил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(3-гидрокси-3-метилбутил)фенил]этил}-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2,3,3, 3-пентафторпропил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,3,3,3-пентафтор-1-(4-пиридин-4-илфенил)пропил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(4'-фтор-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N1-(цианометил)-N2-((1S)-2,2, 2-трифтор-1-{4-[(1E)-3-гидрокси-3-метилбут-1-енил]фенил}этил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,3,3,3-пентафтор-1-[4'-(метилтио)-1, 1'-дифенил-4-ил]пропил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,3,3,3-пентафтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]пропил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(1-оксидпиридин-3-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(морфолин-4-илкарбонил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2, 2-трифтор-1-(4-{[метокси(метил)амино]карбонил}фенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(4-тиен-3-илфенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(4-метилпиридин-2-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(5-метилпиридин-2-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(2'-фтор-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-1-[4-(3,5-диметилизоксазол-4-ил)фенил]-2,2,2-трифторэтил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[4'-(гидроксиметил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-[(1S)-1-(4'-циано-1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-1-(3',4'-дифтор-1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-L-лейцинамид;
метиловый эфир 4'-{1-[1-(цианометилкарбамоил)-3-метилбутиламино]-2,2,2-трифторэтил)дифенил-2-карбоновой кислоты;
метиловый эфир 4'-{1-[1-(цианометилкарбамоил)-3-метилбутиламино]-2,2, 2-трифторэтил}дифенил-3-карбоновой кислоты;
N1-(цианометил)-N2-[(1S)-1-(3',4'-диметокси-1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[2'-(трифторметил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-1-(3',4'-дихлор-1, 1'-дифенил-4-ил)-2,2,2-трифторэтил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(3'-формил-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(2-оксо-2,3-дигидро-1,3-бензотиазол-6-ил)фенил]этил}-L-лейцинамид;
N2-{(1S)-1-[4-(5-бромпиридин-3-ил)фенил]-2,2, 2-трифторэтил}-Nl-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(трифторметокси)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(1H-индол-4-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2, 2-трифтор-1-(4-пиримидин-5-илфенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(4-хинолин-3-илфенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(1,3-тиазол-2-ил)фенил]этил}-L-лейцинамид;
метиловый эфир 4'-{1-[1-(цианометилкарбамоил)-3-метилбутиламино]-2,2, 2-трифторэтил}дифенил-4-карбоновой кислоты;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(4-пиримидин-2-илфенил)этил]-L-лейцинамид;
N1 -(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(3-метилпиридин-2-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(3-фурил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-((1S)-2,2,2-трифтор-1-{4-[3-(трифторметил)пиридин-2-ил]фенил}этил)-L-лейцинамид;
N1-(цианометил)-N2-((1S)-2,2,2-трифтор-1-{4-[4-(трифторметил)пиридин-2-ил]фенил}этил)-L-лейцинамид;
N1-(цианометил)-N2-((1S)-2,2, 2-трифтор-1-{4-[5-(трифторметил)пиридин-2-ил]фенил}этил)-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(3'-метокси-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(3'-метокси-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N2-{(1S)-1-[4'-(ацетиламино)-3'-фтор-1, 1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(3-метилтиен-2-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(3'-фтор-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N2-{(1S)-1-[4-(5-ацетилтиен-2-ил)фенил]-2,2,2-трифторэтил}-N1-(цианометил)-L-лейцинамид;
N2-[(1S)-1-(3'-ацетил-1,1'-дифенил-4-ил)-2,2, 2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[3'-(трифторметил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(5'-фтор-2'-метокси-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-1-(3', 5'-дифтор-1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(2',3',5'-трифтор-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
3-(4'-{1-[1-(цианометилкарбамоил)-3-метилбутиламино]-2,2,2-трифторэтил}дифенил-3-ил)акриловая кислота;
N2-{(1S)-1-[4-(9-антрил)фенил]-2,2,2-трифторэтил}-N1-(цианометил)-L-лейцинамид;
N2-[(1S)-1-(4'-бензоил-1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
N2 -[(1S)-1-(3'-ацетил-4'-гидрокси-1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-1-[2'-(цианометил)-1, 1'-дифенил-4-ил]-2,2,2-трифторэтил}-L-лейцинамид;
N1-(цианометил)-N2-{2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{2,2,2-трифтор-1-[4'-(метилсульфинил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[2,2, 2-трифтор-1-(4-морфолин-4-илфенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-{(1R)-2,2,2-трифтор-1-[4-(6-метилпиридин-3-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(6-метилпиридин-3-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[2,2, 2-трифтор-1-(5-фенилтиен-2-ил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[2,2,2-трифтор-1-(4-хинолин-8-илфенил)этил]-L-лейцинамид;
N1 -(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(4-пиридин-2-илфенил)этил]-L-лейцинамид;
N2-{1-[4'-(аминосульфонил)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1 -(цианометил)-L-лейцинамид;
N2-{(1S)-1-[4'-(аминосульфонил)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1-(цианометил)-L-лейцинамид;
N1 -(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(1R)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[4'-(морфолин-4-илсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(изопропилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-{(1S)-1-[4'-(аминосульфонил)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1-(цианометил)-L-лейцинамид;
N2-((1S)-1-{4'-[(ацетиламино)сульфонил]-1,1'-дифенил-4-ил}-2,2,2-трифторэтил)-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[2'-метил-4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-[1-(5-бромтиен-2-ил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
трет-бутиловый эфир 4-(4'-{1-[1-(цианометилкарбамоил)-3-метилбутиламино]-2,2, 2-трифторэтил}дифенил-4-ил)пиперазин-1-карбоновой кислоты;
N1-(цианометил)-N2-[2,2,2-трифтор-1-(4'-пиперазин-1-ил-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N1-(цианометил)-N2-(2,2,2-трифтор-1-{4'-[4-(2-гидроксиэтил)пиперазин-1-ил]-1,1'-дифенил-4-ил}этил)-L-лейцинамид;
N1-(цианометил)-N2 -(2,2,2-трифтор-1-{4'-[4-(2-гидрокси-2-метилпропил)пиперазин-1-ил]-1,1'-дифенил-4-ил}этил)-L-лейцинамид;
N1-(цианометил)-N2-(1-{4-[(диметиламино)карбонил]фенил}-2, 2,2-трифторэтил)-L-лейцинамид;
N1-(цианометил)-N2-[2,2,2-трифтор-1-(4'-пиперазин-1-ил-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N1 -(цианометил)-N2-(1-{4-[(циклопропиламино)карбонил]фенил}-2,2,2-трифторэтил)-L-лейцинамид;
4-{1-[1-(цианометилкарбамоил)-3-метилбутиламино]-2,2,2-трифторэтил}бензойную кислоту;
N1-(цианометил)-N2-(2,2,2-трифтор-1-{4'-[4-(2-фторэтил)пиперазин-1-ил]-1,1'-дифенил-4-ил}этил)-L-лейцинамид;
N1 -(цианометил)-N2-(2,2,2-трифтор-1-{4-[(4-метилпиперазин-1-ил)карбонил]фенил}этил)-L-лейцинамид;
N1-(цианометил)-N2-[2,2, 2-трифтор-1-(4-{[4-(2-гидрокси-2-метилпропил)пиперазин-1-ил]карбонил}фенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(2-метил-1, 3-тиазол-4-ил)фенил]этил}-L-лейцинамид;
N2-{1-[4-(3-трет-бутил-1,2,4-триазин-5-ил)фенил]-2,2,2-трифторэтил}-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-[2,2,2-трифтор-1-(4-{2-[3-(метилсульфонил)фенил]-1,3-тиазол-4-ил}фенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-((1S)-2,2, 2-трифтор-1-{4-[2-(1H-пиразол-4-ил)-1,3-тиазол-4-ил]фенил}этил)-L-лейцинамид;
N1-(цианометил)-N2-(2,2,2-трифтор-1-{4'-[4-(метилсульфонил)пиперазин-1-ил]-1, 1'-дифенил-4-ил}этил)-L-лейцинамид;
N2-[1-(3-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2 -{2,2,2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-3-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[2,2,2-трифтор-1-(3-пиридин-4-илфенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[2,2,2-трифтор-1-(4'-пиперазин-1-ил-1,1'-дифенил-3-ил)этил]-L-лейцинамид;
N1-(цианометил)-N2-{2,2, 2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-3-ил]этил}-L-лейцинамид;
N-(цианометил)-1-[(2,2,2-трифтор-1-фенилэтил)амино]циклогексанкарбоксамид;
1-{[1-(4-бромфенил)-2, 2,2-трифторэтил]амино}-N-(цианометил)циклогексанкарбоксамид;
N-(цианометил)-1-{[2,2,2-трифтор-1-(4'-пиперазин-1-ил-1,1'-дифенил-4-ил)этил]амино}циклогексанкарбоксамид;
N1-(цианометил)-N2-[2,2,2-трифтор-1-(4-пиперидин-4-илфенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-{2,2, 2-трифтор-1-[4-(4-пиридин-2-илпиперазин-1-ил)фенил]этил}-L-лейцинамид;
N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-3-циклопропилаланинамид;
N1-(цианометил)-3-циклопропил-N2-[2,2,2-трифтор-1-(4-пиридин-4-илфенил)этил]аланинамид;
N1-(цианометил)-N2-[2,2, 2-трифтор-1-(4'-пиридин-4-ил-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1R)-2,2,2-трифтор-1-(1,3-тиазол-2-ил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(4'-метокси-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2, 2-трифтор-1-(4-метоксифенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(4'-пиридин-4-ил-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(4-феноксифенил)этил]-L-лейцинамид;
N2-[(1S)-1-(4'-бром-1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1 -(цианометил)-L-лейцинамид;
N2-{(1S)-1-[4-(4-хлорпиридин-3-ил)фенил]-2,2,2-трифторэтил}-N1-(цианометил)-L-лейцинамид;
N2 -{(1S)-1-[4'-(ацетиламино)-2'-метил]-1,1'-дифенил-4-ил}-2,2,2-трифторэтил}-N1-(цианометил)-L-лейцинамид;
N2-[(1S)-1-(1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(6-метоксипиридин-3-ил)фенил]этил}-L-лейцинамид;
N1 -(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(6-метоксипиридин-2-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[4"-(метилсульфонил)-1,1':4',1"-терфенил-4-ил]этил}-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1 -(цианометил)-3-(1-метилциклопропил)-L-аланинамид;
N1-(цианометил)-3-(1-метилциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-аланинамид;
N1-(цианометил)-3-(1-метилциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-аланинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(4'-метил-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N2-[(1S)-1-(4'-ацетил-1,1'-дифенил-4-ил)-2,2, 2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(гидроксиметил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-D-лейцинамид;
N1-(цианометил)-N2-{2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-D-лейцинамид;
N1-(цианометил)-N2-{2,2,2-трифтор-1-[4'-(морфолин-4-илсульфонил)-1,1'-дифенил-4-ил]этил}-D-лейцинамид;
N1-(цианометил)-N2-(2,2,2-трифтор-1-{4'-[(метиламино)сульфонил]-1,1'-дифенил-4-ил}этил)-D-лейцинамид;
N1-(цианометил)-N2-{(1R)-2,2, 2-трифтор-1-[4-(1-оксидпиридин-4-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{2,2,2-трифтор-1-[4-(1-оксидпиридин-4-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-(2,2,2-трифтор-1-{4-[6-(1-гидрокси-1-метилэтил)-1-оксидпиридин-3-ил]фенил}этил)-L-лейцинамид;
N1-(цианометил)-N2 -(2,2,2-трифтор-1-{4-[6-(метилсульфонил)пиридин-3-ил]фенил}этил)-L-лейцинамид;
N1-(цианометил)-N2-(2,2,2-трифтор-1-{4-[2-(4-метилпиперазин-1-ил)-1, 3-тиазол-4-ил]фенил}этил)-L-лейцинамид;
N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N1 -(цианометил)-N2-[2,2,2-трифтор-1-(4-пиперазин-1-илфенил)этил]-L-лейцинамид;
N2-{1-[3'-(ацетиламино)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1 -(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{2,2,2-трифтор-1-[4-(4-пропилпиперазин-1-ил)фенил]этил}-L-лейцинамид;
N1 -(цианометил)-N2-{2,2,2-трифтор-1-[4-(пиперазин-1-илкарбонил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[2,2, 2-трифтор-1-(4-{[4-(2-гидроксиэтил)пиперазин-1-ил]карбонил}фенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(4-{3-[3-(трифторметил)фенил]-1,2, 4-оксадиазол-5-ил}фенил)этил]-L-лейцинамид;
метиловый эфир 4-{1-[1-(цианометилкарбамоил)-3-метилбутиламино]-2,2,2-трифторэтил}бензойной кислоты;
N1 -(цианометил)-N2-((1S)-2,2,2-трифтор-1-{4-[(E)-2-хинолин-2-илэтенил]фенил}этил)-L-лейцинамид;
N1 -(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(3-метил-1,2, 4-оксадиазол-5-ил)фенил]этил}-L-лейцинамид;
N2-((1S)-1-{4-[3-(5-бромпиридин-3-ил)-1,2,4-оксадиазол-5-ил]фенил}-2,2,2-трифторэтил)-N1-цианометил)-L-лейцинамид;
N2-[(1S)-1-(4-бензоилфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(тиен-2-илкарбонил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(1,3-тиазол-2-илкарбонил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(4-{(Z)-2-[4-(метилсульфонил)фенил]этенил}фенил)этил]-L-лейцинамид;
N1-(цианометил)-N2 -[(1S)-2,2,2-трифтор-1-(4-{(E)-2-[4-(метилсульфонил)фенил]этенил}фенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2, 2-трифтор-1-(4-изобутирилфенил)этил]-L-лейцинамид;
N2-{(1S)-1-[4-(4-бром-1,3-тиазол-2-ил)фенил]-2,2,2-трифторэтил}-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-1-(4-цианофенил)-2,2,2-трифторэтил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-1-(4-этинилфенил)-2,2, 2-трифторэтил]-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(2'-фтор-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N1 -(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(1,3-тиазол-2-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{2,2,2-трифтор-1-[4'-(метилтио)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{2,2,2-трифтор-1-[4-(2-метилхинолин-7-ил)фенил]этил}-L-лейцинамид;
N1 -(цианометил)-N2-{2,2,2-трифтор-1-[4-(1H-индол-5-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{1-[4'-(диметиламино)-1,1'-дифенил-4-ил]-2,2, 2-трифторэтил}-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-1-(4-{[(цианометил)амино]карбонил}фенил)-2,2,2-трифторэтил]-L-лейцинамид;
N1 -(цианометил)-N2-[(1R)-1-(4-{[(цианометил)амино]карбонил}фенил)-2,2,2-трифторэтил]-L-лейцинамид;
N1-(цианометил)-N2-{2,2, 2-трифтор-1-[3'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
4'-{1-[1-(цианометилкарбамоил)-3-метилбутиламин]-2,2,2-трифторэтил}дифенил-4-карбоновую кислоту;
метоксиметиламид 4'-{1-[1-(цианометилкарбамоил)-3-метилбутиламин]-2,2,2-трифторэтил}дифенил-4-карбоновой кислоты;
N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[4-({[4-(метилсульфонил)бензил]тио}метил)фенил]этил}-L-лейцинамид;
N2-{(1S)-1-[4-(5-хлорпиридин-2-ил)фенил]-2,2,2-трифторэтил}-N1 -(цианометил)-L-лейцинамид;
N2-{(1S)-1-[3'-(аминосульфонил)-4'-бром-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1-(цианометил)-L-лейцинамид;
N2-{(1S)-1-[4'-бром-3'-(метилсульфонил)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-((1S)-2,2, 2-трифтор-1-{4-[5-метил-6-(метилсульфонил)пиридин-3-ил]фенил}этил)-L-лейцинамид;
N2-[(1S)-1-(4-{5-хлор-3-[4-(метилсульфонил)фенил]пиридин-2-ил}фенил)-2,2, 2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-((1S)-2,2,2-трифтор-1-{4-[(фенилтио)метил]фенил}этил)-L-лейцинамид;
N1-(цианометил)-N2-((1S)-2,2,2-трифтор-1-{4'-[(трифторметил)сульфонил]-1,1'-дифенил-4-ил}этил)-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2, 2-трифтор-1-(4-{[(4-фторбензоил)амино]метил}фенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(метилсульфонил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2 -{(1S)-1-[4'-(этилсульфонил)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-L-лейцинамид;
N2-((1S)-1-{4-[({[3-(2-хлор-6-фторфенил)-5-метилизоксазол-4-ил]карбонил}амино)метил]фенил}-2, 2,2-трифторэтил)-Nl-(цианометил)-L-лейцинамид;
N2-((1S)-1-{4-[(9-хлор-3-метил-4-оксоизоксазол[4,3-c]хинолин-5(4H)-ил)метил]фенил}-2,2,2-трифторэтил)-Nl -(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-метокси-3'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-{(1S)-1-[4"-хлор-4'-(метилсульфонил)-1,1':2',1"-терфенил-4-ил]-2,2,2-трифторэтил}-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[2'-метокси-4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-{(1S)-1-[2'-хлор-4'-(метилсульфонил)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1 -(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-((1S)-2,2,2-трифтор-1-{4'-[(2-гидроксиэтил)тио]-1,1'-дифенил-4-ил}этил)-L-лейцинамид;
N1 -(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[3'-фтор-4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-((1S)-2,2, 2-трифтор-1-{4'-[(2-гидроксиэтил)сульфонил]-1,1'-дифенил-4-ил}этил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[3'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-({2-[метокси(метил)амино]-2-оксоэтил}сульфонил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-((1S)-2,2,2-трифтор-1-{4'-[(2-гидрокси-2-метилпропил)сульфонил]-1,1'-дифенил-4-ил}этил)-L-лейцинамид;
N2-{(1S)-1-[4'-(аминосульфонил)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1-(1-цианоциклопропил)-L-лейцинамид;
N2-[(4-бромфенил)(2,4, 6-трифторфенил)метил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-[(4-пиридин-4-илфенил)(2,4,6-трифторфенил)метил]-L-лейцинамид;
N1-(цианометил)-N2-[[4-(4-фторбензил)фенил](фенил)метил]-L-лейцинамид;
N1-(цианометил)-N2 -{фенил[4-(пиридин-3-илметил)фенил]метил}-L-лейцинамид;
N2-{(4-бромфенил)[4-(метилсульфонил)фенил]метил}-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{[4-(метилсульфонил)фенил][4'-(метилтио)-1,1'-дифенил-4-ил]метил}-L-лейцинамид;
N1-(цианометил)-N2-{[4'-(метилсульфонил)-1, 1'-дифенил-4-ил][4-(метилсульфонил)фенил]метил}-L-лейцинамид;
N1-(цианометил)-N2-[2,2,2-трихлор-1-(4-гликолоилфенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-[2-фтор-1-(фторметил)-1-фенилэтил]-L-лейцинамид;
N1-(цианометил)-N2-{2,2, 2-трифтор-1-[4-(пирролидин-1-илацетил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{2,2,2-трифтор-1-[4-(пиперазин-1-илкарбонил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-(2,2,2-трифтор-1-{4-[2-(4-метилпиперазин-1-ил)-1,3-тиазол-4-ил]фенил}этил)-L-лейцинамид;
N1 -(1-цианоциклопропил)-3-(1-метилциклопропил)-N2-(2,2,2-трифтор-1-{4-[1-(2-гидроксиэтил)пролил]фенил}этил)-L-аланинамид;
N2 -[[4-(4-трет-бутилпиперазин-1-ил)фенил](пентафторфенил)метил]-N1-(цианометил)-L-лейцинамид;
N-(цианометил)-1-{1-[4-(4-метилпиперазин-1-ил)фенил]этил}пиперидин-2-карбоксамид;
N2-[[4-(4-трет-бутилпиперазин-1-ил)фенил](пиридин-2-ил)метил]-N1-(цианометил)-L-лейцинамид;
N2 -{[4-(4-трет-бутилпиперазин-1-ил)фенил][5-(трифторметил)пиридин-2-ил]метил}-N1-(цианометил)-L-лейцинамид;
(4S)-N-(цианометил)-4-метил-1-[(1S)-1-(4-пиперазин-1-илфенил)этил]-L-пролинамид;
(4S)-N-(цианометил)-4-метил-1-[(1R)-1-(4-пиперазин-1-илфенил)этил]-L-пролинамид;
N-(цианометил)-1-[(1S)-1-(4-пиперазин-1-илфенил)этил]-L-пролинамид;
N-(цианометил)-1-[(1R)-1-(4-пиперазин-1-илфенил)этил]-L-пролинамид;
N-(цианометил)-4, 4-дифтор-1-[(1S)-1-(4-пиперазин-1-илфенил)этил]-L-пролинамид;
Nl-(1-цианоциклопропил)-N2-[(1S)-2,2,2-трифтор-1-(4-метилфенил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)2,2,2-трифтор-1-[4-(1H-пиразол-3-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(2-метил-1,3-оксазол-4-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(4-пиразин-2-илфенил)этил]-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(2-метилпиридин-4-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(4-метилпиридин-3-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4-(1H-пиразол-4-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-[(1S)-2,2,2-трифтор-1-(4-пиридин-4-илфенил)этил]-L-лейцинамид;
N2-[(1S)-1-(3'-ацетил-1,1'-дифенил-4-ил)-2,2, 2-трифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-[(1S)-2,2,2-трифтор-1-(3'-фтор-4'-метил-1, 1'-дифенил-4-ил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-2,2,2-трифтор-1-{5-[4-(1-гидрокси-1-метилэтил)фенил]пиридин-2-ил}этил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,3,3,3-пентафтор-1-[4'-(1-гидрокси-1-метилэтил)-1,1'-дифенил-4-ил]пропил}-L-лейцинамид;
N1 -(1-цианоциклопропил)-N2-[(1S)-2,2,3,3,3-пентафтор-1-(4'-метил-1,1'-дифенил-4-ил)пропил]-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,3,3, 3-пентафтор-1-[4-(6-метоксипиридин-3-ил)фенил]пропил}-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,3,3,3-пентафтор-1-(2'-фтор-1,1'-дифенил-4-ил)пропил]-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-метокси-3'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2 -{(1S)-1-[3'-(аминосульфонил)-4'-метокси-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1-(1-цианоциклопропил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2, 2,2-трифтор-1-[4-(6-метоксипиридин-3-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,3,3, 3-пентафтор-1-[4-(5-метилпиридин-2-ил)фенил]пропил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-2,2, 2-трифтор-1-{4-[5-(метилсульфонил)пиридин-2-ил]фенил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(5-метилпиридин-2-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[2'-метил-4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1 -(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[5-(1H-пиразол-3-ил)пиридин-2-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-[(1S)-2,2, 2-трифтор-1-(5-хинолин-5-илпиридин-2-ил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-[(1S)-2,2,2-трифтор-1-(5-хинолин-6-илпиридин-2-ил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2-дифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-[(1S)-1-(4'-ацетил-1, 1'-дифенил-4-ил)-2,2,3,3,3-пентафторпропил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N2-[(1S)-1-(1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1 -(1-цианоциклопропил)-L-лейцинамид;
N2-{(1S)-1-[4'-(аминосульфонил)-1,1'-дифенил-4-ил]-2,2,3,3,3-пентафторпропил}-N1-(1-цианоциклопропил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(1-этоксивинил)фенил]-2,2,2-трифторэтил}-L-лейцинамид;
N2-[(1S)-1-(4-ацетилфенил)-2,2, 2-трифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-[(1S)-2,2,2-трифтор-1-(4-изопропилфенил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-[(1S)-2,2,2-трифтор-1-фенилэтил]-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(1-гидрокси-1-метилэтил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4-(1-метилциклопропил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-[(1S)-2,2,2-трифтор-1-(2',4',6'-триметил-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N2 -[(1S)-1-(6-хлорпиридин-3-ил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N2-{(1S)-1-[5-(4-ацетилфенил)пиридин-2-ил]-2,2,2-трифторэтил}-N1 -(1-цианоциклопропил)-L-лейцинамид;
N2-{(1S)-1-[6-(4-ацетилфенил)пиридин-3-ил]-2,2,2-трифторэтил}-N1-(1-цианоциклопропил)-L-лейцинамид;
N2-{(1S)-1-[5-(3-ацетилфенил)пиридин-2-ил]-2,2,2-трифторэтил}-N1-(1-цианоциклопропил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-2,2, 2-трифтор-1-{5-[4-(1-гидроксиэтил)фенил]пиридин-2-ил}этил)-L-лейцинамид;
N2-[(1S)-1-(1,1'-дифенил-4-ил)-2,2,3,3,3-пентафторпропил]-N'-(цианометил)-L-лейцинамид;
N2-[(1S)-1-(4'-ацетил-1,1'-дифенил-4-ил)-2,2,3,3,3-пентафторпропил]-N1-(цианометил)-L-лейцинамид;
N2-[(1S)-1-(1,1'-дифенил-4-ил)-2,2,3,3, 3-пентафторпропил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N2-(1-бензил-2,2,2-трифторэтил)-N1-(1-цианоциклопропил)-L-лейцинамид;
N2-[(1S)-1-(4-трет-бутилфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1 -(цианометил)-4-метил-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-метил-L-лейцинамид;
N1 -(1-цианоциклопропил)-N2-((1S)-2,2,2-трифтор-1-{4-[2-(1H-пиразол-4-ил)-1,3-тиазол-4-ил]фенил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(2-метил-1,3-тиазол-4-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(2-метилпиридин-4-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4-(2-метилпиридин-3-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4-(6-метилпиридин-2-ил)фенил]этил}-L-лейцинамид;
N2-[(1S)-1-(3'-ацетил-1, 1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2, 2-трифтор-1-[4-(1H-пиразол-3-ил)фенил]этил}-L-лейцинамид;
N1-[(1S)-1-цианоэтил]-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-[(1S)-1-циано-3-(метилтио)пропил]-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1 -[(1S)-1-циано-3-(метилсульфонил)пропил]-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2,3,3, 3-пентафторпропил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,3,3, 3-пентафтор-1-[4-(6-метоксипиридин-2-ил)фенил]пропил}-L-лейцинамид;
N2-[(1S)-1-(5-бромпиридин-2-ил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-((1S)-2,2,2-трифтор-1-{5-[4-(метилсульфонил)фенил]пиридин-2-ил}этил)-L-лейцинамид;
N1-(-цианоциклопропил)-N2 -{(1S)-2,2,2-трифтор-1-[4'-(1-гидрокси-1-метилэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,2-трифтор-1-(6'-метил-3, 3'-дипиридин-6-ил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4-(6-метоксипиридин-2-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(6-оксо-1,6-дигидропиридин-2-ил)фенил]этил}-L-лейцинамид;
(4S)-N1-(цианометил)-5,5,5-трифтор-N2 -{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
(4S)-N1-(1-цианоциклопропил)-5,5,5-трифтор-N2-{(1S)-2,2, 2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
(4S)-N1-(цианометил)-5,5,5-трифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилтио)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
(4S)-N1-(1-цианоциклопропил)-5,5,5-трифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
(4S)-N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-5,5,5-трифтор-L-лейцинамид;
(4S)-N2-[(1S)-1-(4-бромфенил)-2,2, 2-трифторэтил]-N1-(1-цианоциклопропил)-5,5,5-трифтор-L-лейцинамид;
N2-{(1S)-1-[4-(6-аминопиридин-3-ил)фенил]-2,2,2-трифторэтил}-N1 -(цианометил)-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид;
N1 -(цианометил)-N2-{(1S)-2,2,3,3,3-пентафтор-1-[4-(6-метилпиридин-3-ил)фенил]пропил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,3,3, 3-пентафтор-1-[4-(6-метилпиридин-3-ил)фенил]пропил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(6-метилпиридин-3-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(1-гидроксиэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(2,2,2-трифтор-1-гидроксиэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1 -(1-цианоциклопропил)-N2-{(1S)-2,2,3,3,3-пентафтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]пропил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,3,3, 3-пентафтор-1-[4'-(1-гидрокси-1-метилэтил)-1,1'-дифенил-4-ил]пропил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,3,3,3-пентафтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]пропил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,3,3,3-пентафтор-1-[4-(6-метоксипиридин-2-ил)фенил]пропил}-L-лейцинамид;
(4R)-N1-(цианометил)-5,5,5-трифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
(4R)-N1-(1-цианоциклопропил)-5,5, 5-трифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(цианометил)-N2-[(1S)-2,2,3,3, 3-пентафтор-1-(4'-метил-1,1'-дифенил-4-ил)пропил]-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,3,3,3-пентафтор-1-[4-(1,3-тиазол-2-ил)фенил]пропил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-[(1S)-1-(4-этинилфенил)-2,2,3,3,3-пентафторпропил]-L-лейцинамид;
N1-(цианометил)-N2 -{(1S)-1-[4-(циклопропилэтинил)фенил]-2,2,2-трифторэтил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(циклопропилэтинил)фенил]-2,2, 2-трифторэтил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(5-метил-1,3-тиазол-2-ил)фенил]этил}-L-лейцинамид;
N1 -(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4-(1,3-тиазол-2-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(5-метил-1,3-тиазол-2-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(циклопропилэтинил)фенил]-2,2,3,3, 3-пентафторпропил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4-(4-метил-1,3-тиазол-2-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(4-метил-1,3-тиазол-2-ил)фенил]этил}-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-1-[4-(4,5-диметил-1, 3-тиазол-2-ил)фенил]-2,2,2-трифторэтил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4'-(этилсульфонил)-1,1'-дифенил-4-ил]-2,2, 2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2,2-трифтор-1-(4-пиридин-3-илфенил)этил]-L-лейцинамид;
N1 -(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-метокси-3'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2, 2-трифторэтил]-N1-(цианометил)-L-аланинамид;
N2-{(1S)-1-[4'-(аминосульфонил)-1,1'-дифенил-4-ил]-2,2,3,3,3-пентафторпропил}-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-((1S)-2,2,3,3,3-пентафтор-1-{4'-[(2-гидрокси-2-метилпропил)сульфонил]-1,1'-дифенил-4-ил}пропил)-L-лейцинамид;
N1 -(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-аланинамид;
N1-(цианометил)-N2-{(1S)-2,2,3,3, 3-пентафтор-1-[4'-(изопропилсульфонил)-1,1'-дифенил-4-ил]пропил}-L-лейцинамид;
N1-(1-циано-1-метилэтил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-((1S)-2,2,2-трифтор-1-{4'-[(2-гидрокси-2-метилпропил)сульфонил]-1, 1'-дифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[2'-метил-4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4'-(этилсульфонил)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-L-лейцинамид;
N2-{(1S)-1-[4'-(аминосульфонил)-1, 1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид;
N1-(цианометил)-N2-{(S)-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил][4-(трифторметокси)фенил]метил}-L-лейцинамид;
N1-(цианометил)-N2-[(S)-[4'-(метилсульфонил)-1,1'-дифенил-4-ил](тиен-2-ил)метил]-L-лейцинамид;
N1-(цианометил)-N2-[(S)-(4'-пиперазин-4-иум-1-ил-1,1'-дифенил-4-ил)(тиен-2-ил)метил]-L-лейцинамид матансульфонат;
N1-(цианометил)-N2-{(S)-(4-фторфенил)[4'-(метилсульфонил)-1,1'-дифенил-4-ил]метил}-L-лейцинамид;
N1-(цианометил)-N2-{(S)-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил][4-(трифторметил)фенил]метил}-L-лейцинамид;
N2-{(S)-(4-хлорфенил)[4'-(метилсульфонил)-1,1'-дифенил-4-ил]метил}-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N2 -[(S)-(4-бромфенил)(тиен-2-ил)метил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-[(S)-[4-(4,4,5,5-тетраметил-1,3, 2-диоксаборолан-2-ил)фенил](тиен-2-ил)метил]-L-лейцинамид;
N2-{(R)-(4-бромфенил)[4-(трифторметокси)фенил]метил}-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(S)-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил][4-(трифторметокси)фенил]метил}-L-лейцинамид;
N2 -[(S)-(4-бромфенил)(2-фурил)метил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(S)-2-фурил[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]метил}-L-лейцинамид;
N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-норвалинамид;
N2 -{(R)-(4-бромфенил)[4-(трифторметил)фенил]метил}-N1-(цианометил)-L-лейцинамид,
N1-(цианометил)-N2-{1-[4'-(4-циклопропилпиперазин-1-ил)-1, 1'-дифенил-4-ил]-2,2,2-трифторэтил}-L-норвалинамид;
N2-[(R)-(4-бромфенил)(4-хлорфенил)метил]-N1-(цианометил)-L-лейцинамид;
N1 -(цианометил)-N2-{2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N2-[(S)-(4-бромфенил)(3-метилтиен-2-ил)метил]-N1 -(цианометил)-L-лейцинамид;
N2-[(S)-(4-бромфенил)(тиен-3-ил)метил]-N1-(цианометил)-L-лейцинамид;
N1-(цианометил)-N2-{(S)-(2, 4-дифторфенил)[4'-(метилсульфонил)-1,1'-дифенил-4-ил]метил}-L-лейцинамид;
N1-(цианометил)-N2-[(S)-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил](тиен-3-ил)метил]-L-лейцинамид;
N1-(цианометил)-N2-[(S)-[4'-(метилсульфонил)-1,1'-дифенил-4-ил](3-метилтиен-2-ил)метил]-L-лейцинамид;
N1-(цианометил)-N2-[(S)-[4'-(4-циклопропилпиперазин-1-ил)-1,1'-дифенил-4-ил](3-метилтиен-2-ил)метил]-L-лейцинамид;
N1-(цианометил)-N2-[(S)-[4'-(4-циклопропилпиперазин-1-ил)-1,1'-дифенил-4-ил](тиен-3-ил)метил]-L-лейцинамид;
N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-5,5, 5-трифтор-L-норвалинамид;
N1-(цианометил)-5,5,5-трифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N2-[(S)-(4-бромфенил)(3-метилтиен-2-ил)метил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-[(S)-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил](3-метилтиен-2-ил)метил]-L-лейцинамид;
N1-(цианометил)-N2-{(S)-3-фурил[4'-(метилсульфонил)-1,1'-дифенил-4-ил]метил}-L-лейцинамид;
Nl-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N2-[(1S)-1-(4-бромфенил)-2,2, 2-трифторэтил]-N1-(1-цианоциклопропил)-L-норвалинамид;
N2-[(S)-(4-бромфенил)(4-бромтиен-2-ил)метил]-N1-(цианометил)-L-лейцинамид;
N2-[(S)-(4-бромфенил)(тиен-3-ил)метил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N1-(цианометил)-N2-((S)-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]{4-[4-(метилсульфонил)фенил]тиен-2-ил}метил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-[(S)-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил](тиен-3-ил)метил]-L-лейцинамид;
N2-{(1S)-1-[4'-(аминосульфонил)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1-(1-цианоциклопропил)-L-норвалинамид;
N2-[(S)-(4-бромфенил)(4-бромтиен-2-ил)метил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N2-[(S)-[4'-(аминосульфонил)-1, 1'-дифенил-4-ил](тиен-3-ил)метил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N2-[(S)-[4'-(аминосульфонил)-1,1'-дифенил-4-ил](тиен-3-ил)метил]-N1 -(цианометил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-метокси-3'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4-(2-метилпиридин-4-ил)фенил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-5,5, 5-трифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[4-(1H-пиразол-3-ил)фенил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(1-гидрокси-1-метилэтил)-1, 1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4-(5-метилпиридин-2-ил)фенил]этил}-L-норвалинамид;
2-{[(4-бромфенил)пиридин-4-илметил]амин}пентановой кислоты цианометиламид;
2-{[(4-бромфенил)тиазол-2-илметил]амино}пентановой кислоты цианометиламид;
(1-цианоциклопропил)амид (2S)-2-[(S)-1-(4'-ацетилдифенил-4-ил)-2,2,2-трифторэтиламин]пентановой кислоты;
(1-цианоциклопропил)амид (2S)-2-[(S)-1-(2',4'-дифтордифенил-4-ил)-2,2, 2-трифторэтиламино]пентановой кислоты;
(1-цианоциклопропил)амид (2S)-2-[(S)-1-(3',4'-дифтордифенил-4-ил)-2,2,2-трифторэтиламино]пентановой кислоты;
(1-цианоциклопропил)амид (2S)-2-[(S)-1-(3'-хлор-4'-фтордифенил-4-ил)-2,2,2-трифторэтиламино]пентановой кислоты;
(1-цианоциклопропил)амид (2S)-2-[(S)-2,2, 2-трифтор-1-(4'-метансульфониламинодифенил-4-ил)этиламин]пентановой кислоты;
цианометиламид (2S)-2-{(S)-[(4-бромфенил)тиазол-2-илметил]амино}-4-метилпентановой кислоты;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-хлор-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[4'-хлор-3'-метил-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-хлор-2'-метил-1, 1'-дифенил-4-ил]этил}-L-норвалинамид;
(1-цианоциклопропил)амид (2S)-2-{(S)-2,2,2-трифтор-1-[4-(1H-индол-5-ил)фенил]этиламин}пентановой кислоты;
(1-цианоциклопропил)амид (2S)-2-[(S)-2,2,2-трифтор-1-(3'-метансульфониламиндифенил-4-ил)этиламин]пентановой кислоты;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-фтор-1, 1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-фтор-3'-метил-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[3'-фтор-4'-метил-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-N2 -{(1S)-2,2,2-трифтор-1-[4'-трифторметокси-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
(1-цианоциклопропил)амид (2S)-2-[(S)-2,2,2-трифтор-1-(4'-метилдифенил-4-ил)этиламин]пентановой кислоты;
(1-цианоциклопропил)амид (2S)-2-[(S)-1-(4'-цианодифенил-4-ил)-2,2,2-трифторэтиламин]пентановой кислоты;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[4'-метокси-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4-(бензо[1, 3]диоксол-5-ил)фенил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метоксикарбонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
(1-цианоциклопропил)амид (2S)-2-{(S)-[(4-бромфенил)тиазол-2-илметил]амино}-4-метилпентановой кислоты;
цианометиламид (2S)-2-{(S)-[(4'-метансульфонилдифенил-4-ил)тиазол-2-илметил]амин}-4-метилпентановой кислоты;
(1-цианоциклопропил)амид (2S)-2-[(S)-2,2, 2-трифтор-1-(4'-трифторметилдифенил-4-ил)этиламино]пентановой кислоты;
(цианоциклопропил)амид (2S)-2-[(S)-2,2,2-трифтор-1-(2'-трифторметилдифенил-4-ил)этиламин]пентановой кислоты;
(1-цианоциклопропил)амид (2S)-2-{(S)-[(2',4'-дифтордифенил-4-ил)тиазол-2-илметил]амино}-4-метилпентановой кислоты;
(1-цианоциклопропил)амид (2S)-2-{(S)-[(4'-метансульфонилдифенил-4-ил)тиазол-2-илметил]амино}-4-метилпентановой кислоты;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2, 2-трифтор-1-[4-(1-оксидо-2,3-дигидро-1-бензотиен-5-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(1,1-диоксидо-2, 3-дигидро-1-бензотиен-5-ил)фенил]-2,2,2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(1,1-диоксидо-2,3-дигидро-1, 2-бензизотиазол-5-ил)фенил]-2,2,2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфинил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(1,1-диоксидо-3-оксо-2,3-дигидро-1,2-бензизотиазол-5-ил)фенил]-2,2, 2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[2'-(1-гидрокси-1-метилэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4,4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(цианометил)-4, 4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(цианометил)-4-фтор-N2-{(1S)-2,2, 2-трихлор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трихлор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4,4-дифтор-N2-{(1S)-2,2,2-трихлор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(цианометил)-4,4-дифтор-N2-{(1S)-2,2,2-трихлор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
(2S)-N-(цианометил)-4, 4-дифтор-2-({(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}амин)бутанамид;
(2S)-N-(1-цианоциклопропил)-4,4-дифтор-2-({(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}амино)бутанамид;
(2S)-N-(1-цианоциклопропил)-4,4-дифтор-2-({(1S)-2,2,2-трихлор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}амин)бутанамид;
(2S)-N-(цианометил)-4,4-дифтор-2-({(1S)-2,2,2-трихлор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}амино)бутанамид;
(2S)-4,4-дихлор-N-(цианометил)-2-({(1S)-2,2, 2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}амин)бутанамид;
(2S)-4,4-дихлор-N-(1-цианоциклопропил)-2-({(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}амино)бутанамид;
(2S)-4,4-дихлор-N-(1-цианоциклопропил)-2-({(1S)-2,2,2-трихлор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}амино)бутанамид;
(2S)-4,4-дихлор-N-(цианометил)-2-({(1S)-2,2,2-трихлор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}амино)бутанамид;
N2-[(1S)-1-(1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[3'-(1-гидроксиэтил)-1, 1'-дифенил-4-ил]этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2,2-трифтор-1-(4'-метил-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[3'-(1-гидрокси-1-метилэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-[(1S)-1-(4'-ацетил-1, 1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид;
N2-[(1S)-1-(2'-ацетил-1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1 -(1-цианоциклопропил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(1-гидроксиэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[2'-(1-гидроксиэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1 -(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(1-гидрокси-1-метилэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[2'-(1-гидрокси-1-метилэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2, 2-трифтор-1-{5-[4-(метилсульфонил)фенил]пиридин-2-ил}этил)-L-лейцинамид;
Nl-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2, 2-трифтор-1-{6-[4-(метилсульфонил)фенил]пиридин-3-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2, 2-трифтор-1-(4-{6-[(метилсульфонил)амино]пиридин-3-ил}фенил)этил]-L-лейцинамид;
N1-(цианометил)-4-фтор-N2-[(1S)-2,2, 2-трифтор-1-(4-{6-[(метилсульфонил)амино]пиридин-3-ил}фенил)этил]-L-лейцинамид;
Nl-(1-цианобутил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-циано-2-циклопропилэтил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-циано-2-пиридин-3-илэтил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1 -(1-циано-3-гидрокси-3-метилбутил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(1-оксо-1,3-дигидро-2-бензофуран-5-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(3,3-диметил-1-оксо-1, 3-дигидро-2-бензофуран-5-ил)фенил]-2,2,2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(3,3-диэтил-1-оксо-1, 3-дигидро-2-бензофуран-5-ил)фенил]-2,2,2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(3-оксо-1, 3-дигидро-2-бензофуран-5-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(1,1-диметил-3-оксо-1,3-дигидро-2-бензофуран-5-ил)фенил]-2,2, 2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(1,1-диэтил-3-оксо-1,3-дигидро-2-бензофуран-5-ил)фенил]-2,2, 2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(3-оксо-1,3-дигидро-2-бензофуран-5-ил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4'-(1,1-диметил-3-оксо-1,3-дигидро-2-бензофуран-5-ил)-1,1'-дифенил-4-ил]-2,2, 2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4'-(1,1-диэтил-3-оксо-1,3-дигидро-2-бензофуран-5-ил)-1,1'-дифенил-4-ил]-2,2, 2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(1-оксо-1,3-дигидро-2-бензофуран-5-ил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4'-(3,3-диметил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)-1,1'-дифенил-4-ил]-2,2, 2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4'-(3,3-диэтил-1-оксо-1,3-дигидро-2-бензофуран-5-ил)-1,1'-дифенил-4-ил]-2,2, 2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(5-оксо-2,5-дигидрофуран-3-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(2,2-диметил-5-оксо-2,5-дигидрофуран-3-ил)фенил]-2,2,2-трифторэтил}-4-фтор-L-лейцинамид;
N1 -(1-цианоциклопропил)-N2-{(1S)-1-[4-(2,2-диэтил-5-оксо-2,5-дигидрофуран-3-ил)фенил]-2,2,2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(2-оксо-2,5-дигидрофуран-3-ил)фенил]этил}-L-лейцинамид;
Nl-(1-цианоциклопропил)-N2-{(1S)-1-[4-(5,5-диметил-2-оксо-2, 5-дигидрофуран-3-ил)фенил]-2,2,2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(5,5-диэтил-2-оксо-2,5-дигидрофуран-3-ил)фенил]-2,2, 2-трифторэтил}-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(5-оксо-4-оксаспиро[2.4]гепт-6-ен-7-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(6-оксо-5-оксаспиро[3.4]окт-7-ен-8-ил)фенил]этил}-L-лейцинамид;
N1 -(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(5-оксо-4-оксаспиро[2.4]гепт-6-ен-6-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(6-оксо-5-оксаспиро[3.4]окт-7-ен-7-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2, 2-трифтор-1-(4-хинолин-6-илфенил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-[(1S)-1-(4'-ацетил-1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N1 -(1-цианоциклопропил)-N2-[(1S)-2,2,2-трифтор-1-(4-хинолин-6-илфенил)этил]-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1 -(1-цианоциклопропил)-4,4-дифтор-L-норвалинамид;
N1-(1-цианоциклопропил)-4,4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-4,4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1 -[(1S)-1-циано-3-(метилсульфонил)пропил]-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2,2-трифтор-1-(4-хинолин-6-илфенил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфинил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(1-гидроксиэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-[(1S)-1-(4'-ацетил-1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-[(1S)-2,2, 2-трифтор-1-(4-хинолин-6-илфенил)этил]-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4,4-дифтор-L-норвалинамид;
N1-(1-цианоциклопропил)-4,4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-4, 4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2, 2-трифтор-1-{4-[5-метил-6-(метилсульфонил)пиридин-3-ил]фенил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2, 2-трифтор-1-{4-[6-(1-гидрокси-1-метилэтил)-5-метилпиридин-3-ил]фенил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2,2-трифтор-1-(4'-фтор-1, 1'-дифенил-4-ил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[3'-метил-4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(6-метилпиридин-3-ил)фенил]этил}-L-лейцинамид;
N1 -(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2, 2-трифтор-1-[4'-(1-гидрокси-1-метилэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2, 2-трифтор-1-[4-(2-метилхинолин-7-ил)фенил]этил}-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-4,4-дифтор-L-норвалинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[(1S)-1-гидроксиэтил]-1,1'-дифенил-4-ил}этил)-L-лейцинамид;
N1 -(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[(1R)-1-гидроксиэтил]-1,1'-дифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2 -{(1S)-2,2,2-трифтор-1-[4'-трифторацетил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[5-(2-нафтил)пиридин-2-ил]этил}-L-лейцинамид;
N1-(цианометил)-4,4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилтио)-1, 1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(цианометил)-4,4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(5-метил-1,3-тиазол-2-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(1,1-диоксидо-3-оксо-2,3-дигидро-1,2-бензизотиазол-5-ил)фенил]-2,2,2-трифторэтил}-4-фтор-L-лейцинамид;
N2-[(4-бромфенил)(фенил)метил]-N1 -(1-цианоциклопропил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2,2-трифтор-1-[4'-метил-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N2-[(1S)-1-(1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид;
N2-{(1S)-1-[4-(5-хлорпиридин-2-ил)фенил]-2,2, 2-трифторэтил}-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2, 2-трифтор-1-(4-пиридин-4-илфенил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[(метилсульфонил)амино]-1, 1'-дифенил-4-ил}этил)-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2-дифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N1 -(1-цианоциклопропил)-N2-{(1S)-2,2-дифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2, 2-трифтор-1-(4-пиримидин-5-илфенил)этил]-L-лейцинамид;
N2-[(1S)-1-(4'-ацетил-1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[3'-(1-гидроксиэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-[(1S)-1-(4'-ацетил-1, 1'-дифенил-4-ил)-2,2-дифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(3,5-диметилизоксазол-4-ил)фенил]-2,2, 2-трифторэтил}-4-фтор-L-лейцинамид;
N1-[(1S)-1-циано-3-(метилсульфонил)пропил]-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2,2-трифтор-1-(4-хинолин-6-илфенил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфинил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(1-гидроксиэтил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-[(1S)-1-(4'-ацетил-1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N1 -(1-цианоциклопропил)-N2-[(1S)-2,2,2-трифтор-1-(4-хинолин-6-илфенил)этил]-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1 -(1-цианоциклопропил)-4,4-дифтор-L-норвалинамид;
N1-(1-цианоциклопропил)-4,4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-4,4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1 -(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4-[5-метил-6-(метилсульфонил)пиридин-3-ил]фенил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4-[6-(1-гидрокси-1-метилэтил)-5-метилпиридин-3-ил]фенил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2, 2-трифтор-1-(4'-фтор-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[3'-метил-4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(6-метилпиридин-3-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(1-гидрокси-1-метилэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2, 2-трифтор-1-[4-(2-метилхинолин-7-ил)фенил]этил}-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-4,4-дифтор-L-норвалинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[(1S)-1-гидроксиэтил]-1,1'-дифенил-4-ил}этил)-L-лейцинамид;
N1 -(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[(1R)-1-гидроксиэтил]-1,1'-дифенил-4-ил}этил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2 -{(1S)-2,2,2-трифтор-1-[4'-(трифторацетил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[5-(2-нафтил)пиридин-2-ил]этил}-L-лейцинамид;
N1-(цианометил)-4,4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилтио)-1, 1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(цианометил)-4,4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4-(5-метил-1,3-тиазол-2-ил)фенил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(1,1-диоксидо-3-оксо-2,3-дигидро-1,2-бензизотиазол-5-ил)фенил]-2,2,2-трифторэтил}-4-фтор-L-лейцинамид;
N2-[(4-бромфенил)(фенил)метил]-N1 -(1-цианоциклопропил)-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2,2-трифтор-1-(4'-метил-1,1'-дифенил-4-ил)этил]-L-лейцинамид;
N2-[(1S)-1-(1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид;
N2-{(1S)-1-[4-(5-хлорпиридин-2-ил)фенил]-2,2, 2-трифторэтил}-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2, 2-трифтор-1-(4-пиридин-4-илфенил)этил]-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-((1S)-2,2,2-трифтор-1-{4'-[(метилсульфонил)амино]-1, 1'-дифенил-4-ил}этил)-L-лейцинамид;
N2-[(1S)-1-(4-бромфенил)-2,2-дифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N1 -(1-цианоциклопропил)-N2-{(1S)-2,2-дифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-[(1S)-2,2, 2-трифтор-1-(4-пиримидин-5-илфенил)этил]-L-лейцинамид;
N2-[(1S)-1-(4'-ацетил-1,1'-дифенил-4-ил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамид;
N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[3'-(1-гидроксиэтил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид;
N2-[(1S)-1-(4'-ацетил-1, 1'-дифенил-4-ил)-2,2-дифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамид;
N1-(1-цианоциклопропил)-N2-{(1S)-1-[4-(3,5-диметилизоксазол-4-ил)фенил]-2,2, 2-трифторэтил}-4-фтор-L-лейцинамид
и их фармацевтически приемлемые соли, стереоизомеры и N-оксидные производные.
В объем настоящего изобретения также включена фармацевтическая композиция, содержащая соединение формулы I, как описано выше, и фармацевтически приемлемый носитель. Данное изобретение также включает получение фармацевтической композиции, включающей в себя фармацевтически приемлемый носитель и любое из соединений, конкретно описанных в настоящем изобретении. Эти и другие аспекты данного изобретения будут очевидны из содержащихся здесь сведений.
Полезность
Соединения по настоящему изобретению являются ингибиторами катепсинов и, следовательно, являются полезными для лечения или профилактики катепсин-зависимых заболеваний или состояний млекопитающих, предпочтительно людей. В частности, соединения по настоящему изобретению являются ингибиторами Катепсина K, и, следовательно, полезны для лечения или профилактики заболеваний или состояний, зависимых от Катепсина K, у млекопитающих, предпочтительно, у людей.
«Катепсин-зависимые заболевания или состояния» относятся к патологическим состояниям, которые зависят от активности одного или более катепсинов. «Заболевания или состояния, зависимые от Катепсина К» относятся к патологическим состояниям, которые зависят от активности Катепсина К. Заболевания, ассоциированные с действиями Катепсина К, включают в себя остеопороз, остеопороз, индуцированный глюкокортикоидами, болезнь Педжета, аномально повышенный метаболизм кости, заболевание периодонта, выпадение зубов, переломы костей, ревматоидный артрит, остеоартрит, околопротезный остеолизис, нарушение остеогенеза, метастазирующее заболевание костей, гиперкальциемию злокачественных опухолей и множественную миелому. При лечении таких состояний заявленными в настоящем соединениями требуемое терапевтическое количество будет варьировать в соответствии с конкретным заболеванием, и оно легко может быть определено специалистом в данной области. Хотя и лечение, и профилактика рассматриваются в объеме данного изобретения, предпочтительным применением является лечение этих состояний.
Вариантом осуществления данного изобретения является способ ингибирования активности катепсина у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше.
Одной группой осуществлений является способ, в котором активность катепсина представляет собой активность катепсина К.
Другим вариантом осуществления данного изобретения является способ лечения или профилактики катепсин-зависимых состояний у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше.
Группой данного осуществления является способ, в котором активность катепсина представляет собой активность катепсина К.
Другим вариантом осуществления данного изобретения является способ ингибирования разрушения кости у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Другим вариантом осуществления данного изобретения является способ уменьшения разрушения кости у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества любого из соединений или любой из фарамацевтических композиций, описанных выше. Полезность ингибиторов катепсина К при подавлении резорбции кости, известна из литературы, см. Stroup, G.B., Lark, M.W., Veber, DF., Bhattacharrya, A., Blake, S., Dare, L.C., Erhard, K-F., Hoffman, S.J., James, I.E., Marquis, R.W., Ru, Y., Vasko-Moser, J.A., Smith, B.R., Tomaszek, T. и Gowen, M. Сильное и селективное ингибирование катепсина К человека ведет к подавлению резорбции кости in vivo у нечеловекообразных приматов. J. Bone Miner. Res., 16:1739-1746; 2001; Votta, B.J., Levy, M.A., Badger, A., Dodds, R.A., James, I.E., Thompson, S., Bossard, M.J., Carr, T., Connor, J.R., Tomaszek, T.A., Szewczuk, L., Drake, F.H., Veber, D. и Gowen, M. Пептид-альдегидные ингибиторы катепсина K подавляют резорбцию кости и in vivo, и in vitro. J. Bone Miner. Res. 12:1396-1406; 1997.
Другим вариантом данного изобретения является способ лечения или профилактики остеопороза у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества любого из соединиений или любой из вышеуказанных фармацевтических композиций, описанных выше. Полезность ингибиторов катепсина К при лечении или профилактике остеопороза известна из литературы, см. Saftig, P., Hunziker, E., Wehmeyer, О., Jones, S., Boyde, A., Rommerskirch, W., Moritz, J.D., Schu, P. и Vonfigura, K. Ослабленная резорбция кости остеокластами ведет к остеопорозу у мышей, дефицитных по катепсину К. Proc. Natl. Acad. Sci. USA 95:13453-13458; 1998.
Другим вариантом осуществления данного изобретения является способ лечения или профилактики ревматоидного артрита у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Из литературы известно, что прогрессивное разрушение отдельной кости является главной причиной дисфункции сустава и нетрудоспособности пациентов с ревматоидным артритом (РА), см. Goldring SR, «Pathogenesis of bone erosions in rheumatoid arthritis». Curr. Opin. Rheumatol. 2002; 14: 406-10. Анализ тканей сустава пациентов с РА представил доказательства, что катепсин К позитивные остеокласты представляют собой типы клеток, которые опосредуют фокальную резорбцию кости, ассоциированную с ревматоидным синовиальным повреждением, см. Hou, W-S, Li, W, Keyszer, G, Weber, E, Levy, R, Klein, MJ, Gravallese, EM, Goldring, SR, Bromme, D, «Comparision of Cathepsin K and S expression within the Rheumatoid and Osteoarthritic Synovium», Arthritis Rheumatism 2002; 46: 663-74. Кроме того, генерализованное разрушение кости является основной причиной заболеваемости, ассоциированной с тяжелым РА. Частота переломов бедра и позвоночника в значительной степени повышена у пациентов с хроническим РА, см. Gould A, Sambrook, P, Devlin J и др., «Osteoclastic activation is the principal mechanism leading to secondary osteoporosis in rheumatoid arthritis». J. Rheumatol 1998; 25: 1282-9. Полезность ингибиторов катепсина К при лечении или профилактике резорбции в субартикулярной кости и генерализованного разрушения кости представляет рациональный подход к фармакологическому вмешательству в развитие ревматоидного артрита.
Другим вариантом осуществления данного изобретения является способ лечения или профилактики развития остеоартрита у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Из литературы известно, что остеоартрит (ОА) сопровождается вполне определенными изменениями в суставах, в том числе эррозией поверхности суставного хряща, периатрикулярным внутрихрящевым окостенением/остеофитозом и субхондриальным костным склерозом и образованием кист, см. Oettmeier R, Abendroth, K, «Osteoarthritis and bone: osteologic types of osteoarthritis of the hip». Skeletal Radiol. 1989; 18: 165-74. В последнее время высказывается мнение о возможном вкладе субхондриального склероза кости в возникновение и развитие ОА. Затвердевшая субхондриальная кость, как реакция сустава на повторяющуюся импульсную нагрузку, менее способна смягчать и распределять усилия по суставу, подвергая его более значительному механическому давлению на поверхность суставного хряща. Это, в свою очередь, ускоряет изнашивание хряща и фибрилляцию, см. Radin, EL и Rose RM, «Role of subchondral bone in the initiation and progression of cartilage damage», Clin. Orthop. 1986; 213: 34-40. Подавление чрезмерной субартикулярной резорбции кости действием антирезобционного агента, такого как ингибитор катепсина К, приведет к подавлению субхондриального костного метаболизма, следовательно, может оказывать благоприятное действие при прогрессировании ОА. В дополнение к вышеуказанной гипотезе, экспрессия катепсина К была недавно выявлена в синовиальных фибробластах, макрофагподобных клетках и хондроцитах синовиального слоя и в обазцах суставного хряща, полученных от пациентов с ОА, см. Hou, W-S, Li, W, Keyszer, G, Weber, E, Levy, R, Klein, MJ, Gravallese, EM, Goldring, SR, Bromme, D, «Comparison of Cathepsin K and S expression within the Rheumatoid and Osteoarthritic Synovium», Arthritis Rheumatism 2002; 46: 663-74; Dodd, RA, Connor, JR, Drake, FH, Gowen, M, «Expression of Cathepsin K messenger RNA in giant cells and their precursors in human osteoarthritic synovial tissues». Arthritis Rheumatism 1999; 42: 1588-93; Konttinen, YT, Mandelin, J, Li, T-F, Salo, J, Lassus, J и др., «Acidic cysteine endoproteinase cathepsin K in the degeneration of the superficial articular hyaline cartilage in osteoarthritis», Arthritis Rheumatism 2002; 46: 953-60. Эти недавние исследования, таким образом, подразумевали роль катепсина K в деструкции коллагена II типа в суставном хряще, ассоциированном с развитием остеоартрита. Полезность ингибиторов катепсина К при лечении или профилактике остеоартрита, как описано в данном изобретении, следовательно, включает в себя два различных механизма: первый представляет собой ингибирование управляемого остеокластами субхондриального костного метаболизма и второй - прямое подавление дегенерации коллагена II типа в синовиальном слое и хряще у пациентов с OA.
Другим вариантом осуществления данного изобретения является способ лечения рака у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Из литературы известно, что катепсин К экспрессируется в карциноме молочной железы человека, см. Littlewood-Evans AJ, Bilbe G, Bowler WB, Farley D, Wlodarski B, Kokubo T, Inaoka T, Sloane J, Evans DB, Gallagher JA, «The osteoclast-associated protease cathepsin K is expressed in human breast carcinoma». Cancer Res 1997 Dec 1;57(23):5386-90.
Подтверждением данного изобретения на примере является применение любого из соединений, описанных выше, в приготовлении лекарственного средства для лечения и/или профилактики остеопороза у млекопитающего, нуждающегося в этом. Еще одним подтверждением данного изобретения на примере является применение любого из соединений, описанных выше, в приготовлении лекарственного средства для лечения и/или профилактики разрушения кости, резорбции кости, переломов кости, метастазирующего заболевания кости и/или нарушений, связанных с функционированием катепсина.
Соединения по данному изобретению можно вводить млекопитающим, предпочтительно людям, либо отдельно, либо предпочтительно в комбинации с фармацевтически приемлемыми носителями или разбавителями, необязательно с известными адъювантами, например алюминиевыми, в фармацевтической композиции, согласно стандартной фармацевтической практике. Данные соединения можно вводить перорально или парентерально, в том числе внутривенным, внутримышечным, внутрибрюшинным, подкожным, ректальным и местным путями введения.
В случае таблеток для перорального применения обычно используемые носители включают в себя лактозу и кукурузный крахмал, и обычно добавляют смазывающие агенты, например стеарат магния. Для перорального введения в форме капсулы применяемые разбавители включают в себя лактозу и высушенный кукурузный крахмал. Для перорального использования терапевтическое соединение согласно данному изобретению, выбранное соединение может быть введено, например, в форме таблеток или капсул, или в виде водного раствора или суспензии. Для перорального приема в форме таблетки или капсулы активный лекарственный компонент может быть объединен с пероральным, нетоксическим, фармацевтически приемлемым, инертным носителем, таким как лактоза, крахмал, сахароза, глюкоза, метилцеллюлоза, стеарат магния, дифосфат кальция, сульфат кальция, маннит, сорбит и подобным; для перорального применения в жидкой форме пероральные лекарственные компоненты могут быть объединены с любым пероральным, нетоксическим, фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и подобное. Более того, в том случае, когда это желательно или необходимо, в смесь также могут быть включены подходящие связующие, смазывающие, дезинтегрирующие агенты и красящие агенты. Подходящие связующие вещества включают в себя крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, природные и синтетические клейкие вещества, такие как аравийская камедь, трагакантовая камедь или альгинат натрия, карбоксиметилцеллюлоза, полиэтиленгликоль, воски и подобное. Смазывающие вещества, используемые в этих дозированных формах, включают в себя олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и подобное. Дезинтеграторы включают в себя, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и подобное. В тех случаях, когда для перорального применения необходимы водные суспензии, активный компонент объединяют с эмульгирующим и суспендирующим агентами. При желании могут быть добавлены определенные подсластители и/или вкусовые агенты. Для внутримышечного, внутрибрюшинного, подкожного и внутривенного применения обычно готовят стерильные растворы активного компонента, и рН растворов должен быть стабильно установлен и забуферен. Для внутривенного использования следует контролировать суммарную концентрацию растворенных веществ для предоставления препарату изотоничности.
Соединения по настоящему изобретению также можно вводить в форме липосомальных систем доставки, таких как малые однослойные везикулы, большие однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины.
Соединения по настоящему изобретению также могут быть доставлены использованием моноклональных антител в качестве индивидуальных носителей, с которыми связывают молекулы соединения. Соединения по настоящему изобретению также могут быть связаны с растворимыми полимерами в качестве наводимых носителей лекарственных средств. Такие полимеры могут включать в себя поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный остатками пальмитоила. Кроме того, соединения по настоящему изобретению могут быть соединены с классом биоразлагаемых полимеров, используемых для достижения контролируемого высвобождения лекарственного средста, например, полимолочной кислотой, полигликолевой кислотой, сополимерами полиакриловой и полигликолевой кислоты, поли-эпсилон-капролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианоакрилатами и поперечно сшитыми или амфипатически блокированными сополимерами гидрогелей.
Настоящие соединения также применимы в комбинации с известными агентами, используемыми для лечения или профилактики остеопороза, остеопороза, индуцированного глюкокортикоидами, болезни Педжета, аномального повышенного метаболизма кости, периодонтального заболевания, выпадения зубов, переломов костей, ревматоидного артрита, остеоартрита, околопротезного остеолизиса, дефекта остеогенеза, метастазирующего заболевания кости, гиперкальциемии злокачественной опухоли и множественной миеломы. Комбинации соединений, раскрытых в настоящий момент с другими агентами, используемыми при лечении или профилактике остеопороза или других костных нарушений, находятся в объеме данного изобретения. Обычный специалист в данной области может понять, какие комбинации агентов были бы полезны, на основании конкретных характеристик лекарственных средств и вызванного заболевания. Такие агенты включают в себя следующее: органический бифосфонат; модулятор рецепторов эстрогена; модулятор рецепторов андрогена; ингибитор протонной АТФазы остеокластов; ингибитор HMG-CoA редуктазы; антагонист интегринового рецептора; анаболический агент остеобластов, такой как ПТГ (РТН), и их фармацевтически приемлемые соли и смеси. Предпочтительная комбинация представляет собой соединение по настоящему изобретению и органический бифосфонат. Другая предпочтительная комбинация представляет собой соединение по настоящему изобретению и модулятор рецепторов эстрогена. Другая предпочтительная комбинация представляет собой соединение по настоящему изобретению и модулятор рецепторов андрогена. Другая предпочтительная комбинация представляет собой соединение по настоящему изобретению и анаболический агент остеобластов.
«Органический бифосфонат» включает в себя, но не только, соединения химической формулы

где n равно целому числу от 0 до 7 и где А и Х независимо выбраны из группы, состоящей из H, OH, галогена, NH2, SH, фенила, C1-C30 алкила, C3 -C30 разветвленного или циклического алкила, бициклической кольцевой структуры, содержащей два или три N, C1-C30 замещенного алкила, C1-C10 алкила, замещенного NH2, C3-C10 разветвленного или циклического алкила, замещенного NH2, C1-C10 диалкила, замещенного NH2, C1-C10 алкокси, C1-C10 алкила, замещенного тио, тиофенила, галогенфенилтио, C1-C10 алкила, замещенного фенилом, пиридилом, фуранилом, пирролидинилом, имидазолилом, имидазопиридинилом и бензилом, так, чтобы и A, и X не были выбраны из H или OH, когда n равно 0, или A и X, взятые вместе с атомом углерода, к которому они присоединены, образовывали C3-C10 кольцо.
В вышеприведенной химической формуле алкильные группы могут быть прямыми, разветвленными или циклическим при условии, что для данной химической формулы выбирают достаточные атомы. C1-C30 замещенный алкил может включать в себя широкий ряд заместителей, неограничивающие примеры которых включают в себя заместители, выбранные из группы, состоящей из фенила, пиридила, фуранила, пирролидинила, имидазонила, NH2, C1-C10 алкила или диалкила, замещенного NH2, OH, SH, и C1-C10 алкокси.
Вышеприведенная химическая формула также охватывает сложные карбоциклические, ароматические и гетороатомные структуры для А и/или Х заместителей, неограничивающие примеры которых включают в себя нафтил, хинолил, изохинолил, адамантил и хлорфенилтио.
Фармацевтически приемлемые соли и производные бифосфонатов также используются здесь. Неограничивающие примеры солей включают в себя соли, выбранные из группы, содержащей щелочной металл, аммоний и моно-, ди-, три- или тетра-C1-C30 -алкил-замещенный аммоний. Предпочтительными солями являются соли, выбранные из группы, состоящей из солей натрия, калия, кальция, магния и аммония. Более предпочтительными являются соли натрия. Неограниченные примеры производных включают в себя производные, выбранные из группы, состоящей из сложных эфиров, гидратов и амидов.
Следует отметить, что термины «бифосфонат» и «бифосфонаты», используемые здесь, в отношении терапевтических агентов настоящего изобретения, также предназначены для охвата дифосфонатов, бифосфоновых кислот и дифосфоновых кислот, а также и солей, и производных этих веществ. Использование специфической номенклатуры в отношении бифосфоната или бифосфонатов не предназначено для ограничения объема настоящего изобретения, если не указано особо. Вследствие использования в настоящее время обычными специалистами в данной области смешанной номенклатуры ссылка на удельный вес или процентный состав бифосфонатного соединения в настоящем изобретении - на основании массы активной кислоты, если здесь не оговорено особо. Например фраза «около 5 мг бифосфоната, ингибирующего резорбцию кости, выбранного из группы, состоящей из алендроната, его фармацевтически приемлемой соли, на основании массы активной алендроновой кислоты» означает, что количество выбранного бифосфонатного соединения подсчитывают на основании 5 мг алендроновой кислоты.
Неограничивающие примеры бифосфонатов, используемых здесь, включают в себя следующие:
Алендроновую кислоту, 4-амино-1-гидроксибутилиден-1,1-бифосфоновую кислоту.
Алендронат (также известный как алендронат натрия или алендронат мононатриевый тригидрат), 4-амино-1-гидроксибутилиден-1,1-бифосфоновой кислоты мононатрия тригидрат.
Алендроновая кислота и алендронат описаны в патентах США 4922007, Kieczykowski и др., опубликован 1 мая 1990; 5019651, Kieczykowski и др., опубликован 28 мая 1991; 5510517, Dauer и др., опубликован 23 апреля 1996; 5648491, Dauer и др., опубликован 15 июля 1997, каждый из которых включен здесь в качестве ссылки в своем полном объеме.
Циклогептиламинометилен-1,1-бифосфоновая кислота, YM 175, Yamanouchi (инкадронат, прежде известный как цимадронат), описанный в патенте США 4970335, Isomura и др., опубликованом 13 ноября 1990, включенном здесь в качестве ссылки в своем полном объеме.
1,1-дихлорметилен-1,1-дифосфоновая кислота (клодроновая кислота), динатриевая соль (клодронат, Procter and Gamble) описаны в патенте Бельгии 672205 (1966) и J. Org. Chem 32, 4111 (1967), включеных здесь в качестве ссылки в их полном объеме.
1-гидрокси-3-(1-пирролидинил)пропилиден-1,1-бифосфоновая кислота (EB-1053).
1-гидроксиэтан-1, 1-дифосфоновая кислота (этидроновая кислота).
1-гидрокси-3-(N-метил-N-пентиламин)пропилиден-1,1-бифосфоновая кислота, также известная как BM-210955, Boehringer-Mannheim (ибандронат), описана в патенте США 4927814, опубликованном 22 мая 1990, включенном здесь в качестве ссылки в своем полном объеме.
1-гидрокси-2-имидазо-(1,2-a)пиридин-3-илэтилиден (минодронат).
6-амино-1-гидроксигексилиден-1,1-бифосфоновая кислота (неридронат).
3-(диметиламино)-1-гидроксипропилиден-1,1-бифосфоновая кислота (олпадронат).
3-амино-1-гидроксипропилиден-1,1-бифосфоновая кислота (памидронат).
[2-(2-пиридинил)этилиден]-1,1-бифосфоновая кислота (пиридронат) описана в патенте США 4761406, включенном в качестве ссылки в своем полном объеме.
1-гидрокси-2-(3-пиридинил)этилиден-1,1-бифосфоновая кислота (ризедронат).
(4-хлорфенил)тиометан-1,1-дисфосфиновая кислота (тилудронат), описана в патенте США 4876248, Breliere и др., 24 октября 1989, который включен здесь в качестве ссылки в своем полном объеме.
1-гидрокси-2-(1H-имидазол-1-ил)этилиден-1,1-бифосфоновая кислота (золедронат).
Неограничивающие примеры бифосфонатов включают в себя аледронат, цимадронат, клодронат, этидронат, ибандронат, инкадронат, минодронат, неридронат, олпадронат, памидронат, пиридронат, ризедронат, тилудронат и золендронат и их фармацевтически приемлемые соли и сложные эфиры. Особенно предпочтительным бифосфонатом является алендронат, особенно натриевая, калиевая, кальциевая, магниевая или аммонийная соль алендроновой кислоты. Иллюстрирующим примером предпочтительного бифосфоната является натриевая соль алендроновой кислоты, особенно гидратная натриевая соль алендроновой кислоты. Данная соль может быть гидратирована целым числом молей воды или не целыми числами молей воды. Дополнительным иллюстрирующим примером предпочтительного бифосфоната является гидратная натриевая соль алендроновой кислоты, главным образом, когда данная гидратная соль представляет собой алендронат мононатрийтригидрат.
Очевидно, что может быть использована смесь двух или более бифосфонатных активных веществ.
Точная дозировка органического бифосфоната будет менятся в зависимости от расписания дозировок, конкретно выбранного бифосфоната, возраста, размера, пола и состояния млекопитающего или человека, природы и тяжести заболевания, подвергаемого лечению, и других важных медицинских и физических факторов. Следовательно, точное фармацевтически эффективное количество не может быть определено заблаговременно и может быть легко определено попечителем или клиницистом. Соответствующие количества могут быть определены в обычном эксперименте в модели на животных и клинических изучениях на людях. В общем, соответствующее количество бифосфоната выбирают для получения ингибирующего действия на резорбцию кости, т.е. вводят количество бифосфоната, ингибирующее резорбцию кости. Для людей эффективная доза бифосфоната для перорального применения обычно составляет примерно от 1,5 примерно до 6000 мкг/кг массы тела и предпочтительно примерно от 10 до 2000 мкг/кг массы тела. Для алендроната мононатрийтригидрата общепринятые вводимые дозы для человека обычно находятся в пределах примерно от 2 мг/день до 40 мг/день, предпочтительно примерно от 5 мг/день до 40 мг/день. В США апробированные в настоящее время дозы для алендроната мононатрийтригидрата составляют 5 мг/день для профилактики остеопороза, 10 мг/день для лечения остеопороза и 40 мг/день для лечения болезни Педжета.
В альтернативных режимах дозировки бифосфоната можно вводить в интервалах иных, чем ежедневные, например дозировки раз в неделю, дозировки дважды в неделю, двухнедельные дозировки и дозировки дважды в месяц. При режиме дозирования раз в неделю алендронат мононатрийтригидрат можно вводить в дозе 35 мг/неделю или 70 мг/неделю.
«Селективные модуляторы рецептора эстрогена» относятся к соединениям, которые препятствуют или подавляют связывание эстрогена с рецептором, независимо от механизма. Примеры модуляторов рецепторов эстрогена включают в себя, но не только, эстроген, прогестоген, эстрадиол, дролоксифен, ралоксифен, лазофоксифен, TSE-424, тамоксифен, идоксифен, LY353381, LY117081, торемифен, фулвестрант, 4-[7-(2,2-диметил-1-оксопропокси-4-метил-2-[4-[2-(1-пиперидинил)этокси]фенил]-2H-1-бензопиран-3-ил]фенил-2,2-диметилпропаноат, 4,4'-дигидроксибензофенон-2,4-динитрофенил-гидразон и SH646.
«Модулятором бета-рецептора эстрогена» является соединение, которое селективно действует как агонист или антагонист бета-рецептора эстрогена (ЭРβ). Агонизирующий ЭРβ повышает транскрипцию гена триптофан гидроксилазы (TPH, ключевой фермент в синтезе серотонина) через ЭРβ-опосредованное событие. Примеры агонистов бета-рецептора эстрогена могут быть найдены в публикации международной заявки PCT WO 01/82923, опубликованной 08 ноября 2001, и WO 02/41835, опубликованной 20 мая 2002, обе включены здесь в качестве ссылки в их полном объеме.
«Модулятор рецептора андрогена» относится к соединениям, которые препятствуют или ингибируют связывание андрогенов с рецептором, независимо от механизма. Примеры модуляторов рецепторов эстрогена включают в себя финастерид и другие ингибиторы 5α-редуктазы, нилутамид, флутамид, бикалутамид, лиарозол и абиратерон ацетат.
«Ингибитор протонной АТФазы остеокластов» относится к ингибитору протонной АТФазы, которая находится на апикальной мембране остеокласта и сообщалось, что играет важную роль в процессе резорбции кости. Этот протонный насос является привлекательной мишенью для разработки ингибиторов резорбции кости, которые являются потенциально полезными для лечения и профилактики остеопороза и родственных метаболических заболеваний. См. C. Farina и др., «Selective inhibitors of the osteoclast vacuolar proton ATPase as novel bone antiresorptive agents,» DDT, 4: 163-172 (1999)), включенную здесь в качестве ссылки в ее полном объеме.
«Ингибиторы HMG-CoA редуктазы» относятся к ингибиторам 3-гидрокси-3-метилглутарил-CoA редуктазы. Соединения, обладающие ингибиторующим действием в отношении HMG-CoA редуктазы, могут быть легко идентифицированы с использованием анализа, хорошо известного в данной области. Например, см. анализы, описанные или цитированные в патенте США 4231938 в кол. 6, и WO 84/02131 на стр. 30-33. Термины «HMG-CoA редуктазный ингибитор» и «ингибитор HMG-CoA редуктазы» при использовании здесь имеют одинаковое значение.
Примеры ингибиторов HMG-CoA редуктазы, которые могут быть использованы, включают в себя, но не только, ловастатин (MEVACOR®; см. патенты США 4231938, 4294926 и 4319039), симвастатин (ZOCOR®, см. патенты США 4444784, 4820850 и 4916239), правастатин (PRAVACHOL®; см. патенты США 4346227, 4537859, 4410629, 5030447 и 5180589), флувастатин (LESCOL®; см. патенты США 5354772, 4911165, 4929437, 5189164, 5118853, 5290946 и 5356896), аторвастатин (LIPITOR®; см. патенты США 5273995, 4681893, 5489691 и 5342952) и церивастатин (также известный как ривастатин и BAYCHOL®; см. патент США 5177080). Структурные формулы этих и дополнительных ингибиторов HMG-CoA редуктазы, которые могут быть использованы в настоящих способах, описаны на странице 87 M. Yalpani, «Cholesterol Lowering Drugs», Chemistry & Industry, pp. 85-89 (5 февраля 1996), и в патентах США 4782084 и 4885314. Используемое здесь понятие «ингибитор HMG-CoA редуктазы» включает в себя все фармацевтически приемлемые формы лактонов и открытых кислот (т.е., где кольцо лактона открыто в форму свободной кислоты), также как и формы солей и сложных эфиров соединений, обладающих ингибирующим действием в отношении HMG-CoA редуктазы, и, следовательно, применение таких солей, сложных эфиров, форм лактонов и открытых кислот включено в объем данного изобретения. Изображение лактонной части и соответствующей ей формы открытой кислоты приведено ниже в виде структур I и II

В ингибиторах HMG-CoA редуктазы, где может существовать форма открытой кислоты, формы солей и сложных эфиров предпочтительно могут быть образованы из открытой кислоты, и все такие формы включены в значение используемого здесь термина «ингибитор HMG-CoA редуктазы». Предпочтительно ингибитор HMG-CoA редуктазы выбирают из ловастатина и симвастатина, наиболее предпочтительно симвастатина. Здесь термин «фармацевтически приемлемые соли» в отношении ингибитора HMG-CoA редуктазы должен означать нетоксические соли соединений, используемых в данном изобретении, которые главным образом получают взаимодействием свободной кислоты с органическим или неорганическим основанием, в частности те, которые образованы из катионов, например натрия, калия, алюминия, кальция, лития, магния, цинка и тетраметиламмония, также как и те соли, которые образованы из аминов, например аммиака, этилендиамина, N-метилглюкамина, лизина, аргинина, орнитина, холина, N,N'-дибензилэтилендиамина, хлорпрокаина, диэтаноламина, прокаина, N-бензилфенэтиламина, 1-п-хлорбензил-2-пирролидин-1'-илметилбензимидазола, диэтиламина, пиперазина и трис(гидроксиметил)аминометана. Следующие примеры солевых форм ингибиторов HMG-CoA могут включать в себя, но не только, ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, эдетат кальция, камсилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эзилат, фумарат, глюцептат, глюконат, глутамат, гликолиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилсульфат, мукат, напсилат, нитрат, олеат, оксалат, памаот, пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтйодид и валерат.
Сложноэфирные производные описанных соединений ингибиторов HMG-CoA редуктазы могут действовать как предшественники лекарственных средств, которые, при абсорбции в кровоток теплокровного животного, могут расщепляться таким образом, чтобы высвобождать лекарственную форму, и дают лекарственному средству возможность предоставить улучшенную терапевтическую эффективность.
Как использовалось выше, «антагонисты интегринового рецептора» относятся к соединениям, которые селективно оказывают антагонистическое действие, ингибируют или противодействуют связыванию физиологического лиганда с αvβ3 интегрином, к соединениям, которые оказывают антагонистическое действие, ингибируют или противодействуют связыванию физиологического лиганда с αvβ5 интегрином, к соединениям, которые оказывают антагонистическое действие, ингибируют или противодействуют связыванию физиологического лиганда и с αvβ3 интегрином и с αvβ5 интегрином и к соединениям, которые оказывают антагонистическое действие, ингибируют или противодействуют активности отдельного интегрина(ов), экспрессируемых на эндотелиальных клетках капилляров. Данный термин также относится к антагонистам αv β6, αvβ8, α1β1,α2β1, α5β1, α6β1 и α6β4, интегринов. Данный термин также относится к антогонистам любой комбинации αvβ3,αvβ5,αvβ6,αvβ8,α1β1,α2β1, α5 β1, α6β1 и α6β4 интегринов. H.N. Lode с соавторами в PNAS USA 96: 1591-1596 (1999) наблюдали синергические эффекты между антагонистом антиангиогенного αv интегрина и опухолеспецифичного антитела-цитокина (интерлейкина-2) слитым белком в ликвидации спонтанных опухолевых метастазов. Их результаты говорят о том, что данная комбинация обладает потенциальными возможностями для лечения рака и метастазирующего опухолевого роста. Антагонисты рецептора αvβ3 интегрина ингибируют резорбцию кости через новый механизм, отличный от механизма всех доступных в настоящее время лекарственных средств. Интегрины представляют собой гетеродимерные трансмембранные рецепторы адгезии, которые опосредуют взаимодействия клетка-клетка и клетка-матрикс. α и β субъединицы интегринов взаимодействуют нековалентно и связывают лиганды внутриклеточного матрикса зависимым от двухвалентных катионов образом. Наиболее обильным интегрином на остеокластах является αvβ3 (>107 /остеокласт), который, по-видимому, играет скорость-лимитирующую роль в организации цитоскелета, важную для клеточной миграции и поляризации. Антагонистическое действие αvβ3выбирают из ингибирования резорбции кости, подавления рестеноза, задержки дегенерации пятна, торможения артрита и подавления роста раковой опухоли и метастазов.
«Анаболический агент остеобластов» относится к агентам, которые строят кость, такие как ПТГ. Было показано, что скачкообразное введение паратиреоидного гормона (ПТГ) или его аминоконцевых фрагментов и аналогов предупреждает, задерживает, частично обращает разрушение кости и стимулирует образование кости у животных и людей. Для обсуждения ссылаемся на D.W. Dempster и др., «Anabolic actions of parathyroid hormone on bone», Endocr Rev 14: 690-709 (1993). Изучения продемонстрировали клиническую пользу паратиреоидного гормона в стимуляции образования кости и, таким образом, увеличении массы кости и прочности. Результаты были сообщены RM Neer и др., в New Eng J Med 344 1434-1441 (2001).
Кроме того, фрагменты белка, родственного паратиреоидному гормону, или аналоги, такие как PTHrP-(1-36) продемонстрировали сильные антикальцийурические эффекты [см. M.A. Syed и др., «Parathyroid hormone-related protein-(l-36) stimulates renal tubular calcium reabsorption in normal human volunteers: implications for the pathogenesis of humoral hypercalcemia of malignancy», JCEM 86: 1525-1531 (2001)] и могут также иметь потенциальную возможность как анаболические агенты для лечения остеопороза.
В составе фиксированной дозы такие комбинации продуктов используют соединения по данному изобретению в диапазоне доз, описанном ниже, и другой фармацевтически активный агент(ы) в его установленном диапазоне доз. Соединения по настоящему изобретению могут быть использованы альтернативно с известными фармацевтически приемлемым агентом (агентами), в том случае, когда составление комбинации является неуместным.
Термин «введение» и его варианты (например, «назначение» соединения) в отношении соединения по данному изобретению означает введение данного соединения или предшественника лекарственного средства данного соединения в систему животного, нуждающегося в лечении. В том случае, когда соединение по данному изобретению или его пролекарство предоставлено в комбинации с одним или более других активных агентов (например, цитотоксическим агентом и т.д.), под «введением» и его вариантами под каждым подразумевают параллельное и последовательное введение данного соединения или его пролекарства и других агентов. Настоящее изобретение включает в своем объеме пролекарства соединений по данному изобретению. Обычно такие пролекарства будут функциональными производными соединений по данному изобретению, которые легко превращаются in vivo в желаемое соединение. Следовательно, в способах лечения по настоящему изобретению термин «назначение» будет охватывать лечение различных состояний, описанных с соединением, раскрытым отдельно или с соединением, которое может не быть раскрыто отдельно, но которое превращается в определенное соединение in vivo после введения пациенту. Обычные методики отбора и приготовления подходящих производных пролекарств описаны, например, в «Design of Prodrugs», под ред. H. Bundgaard, Elsevier, 1985, включенном здесь в качестве ссылки в его полном объеме. Метаболиты этих соединений включают в себя активные виды, продуцируемые при введении соединений по данному изобретению в биологическую среду.
Используемый здесь термин «композиция» предназначен для охвата продукта, содержащего определенные компоненты в определенных количествах, а также и любого продукта, который получается в результате, прямо или опосредовано, из комбинации определенных компонентов в определенных количествах.
Термин «терапевтически эффективное количество», используемый здесь, означает количество активного соединения или фармацевтического агента, который вызывает биологический или медицинский ответ в ткани, системе, у животного или человека, к которому стремится исследователь, ветеринар, врач или другой клиницист.
Термины «лечение» или «терапия» заболевания, используемые здесь, включают в себя профилактику заболевания, т.е. не позволить развиваться клиническим симптомам заболевания у млекопитающего, которое может подвергаться этому заболеванию или быть предрасположенным к нему, но еще не испытывающему или не проявляющему симптомов данного заболевания; задержку заболевания, т.е. остановку или уменьшение развития заболевания или его клинических симптомов, или ослабление заболевание, т.е. вызов регрессии заболевания или его клинических симптомов.
Используемый термин «резорбция кости» относится к процессу, посредством которого остеокласты разрушают кость.
Настоящее изобретение также охватывает фармацевтическую композицию, применимую для лечения остеопороза или других костных нарушений, включающего введение терапевтически эффективного количества соединений по данному изобретению, с фармацевтически приемлемыми носителями или разбавителями, или без них. Подходящие композиции по данному изобретению включают в себя водные растворы, содержащие соединения по данному изобретению и фармацевтически приемлемые носители, например физиологический раствор с уровенм рН, например, 7,4. Данные растворы могут быть введены в кровоток пациента посредством локальной болюсной инъекции.
В том случае, когда соединение по данному изобретению вводят человеку, ежедневная дозировка обычно будет определена предписанием лечащего врача с изменениями, главным образом, в соответствии с возрастом, массой и реакцией конкретного пациента, также как и тяжестью симптомов пациента.
В одном характерном применении соответствующее количество соединения вводят млекопитающему, у которого проводят лечение катепсин-зависимого состояния. Пероральные дозировки по настоящему изобретению, при использовании для получения указанных эффектов, будут находиться в диапазоне примерно между 0,01 мг на кг массы тела в день (мг/кг/день) примерно до 100 мг/кг/день, предпочтительно от 0,01 до 10 мн/кг/день и наиболее предпочтительно от 0,1 до 5,0 мг/кг/день. Для перорального введения композиции предпочтительно поставляют в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 100 и 500 миллиграмм активного компонента для симптоматической коррекции дозировки пациента, получающего лечение. Лекарственное средство типично содержит примерно от 0,01 мг примерно до 500 мг активного компонента, предпочтительно примерно от 1 мг примерно до 100 мг активного компонента. Внутривенно, наиболее предпочтительные дозы будут находиться в интервале примерно от 0,1 примерно до 10 мг/кг/минуту при постоянной скорости инфузии. Преимущественно соединения по настоящему изобретению могут быть введены в единичной дневной дозе или общую дневную дозу можно вводить разделенной дозой два, три или четыре раза в день. Более того, предпочтительные соединения для настоящего изобретения можно вводить в интраназальной форме посредством местного использования походящих интраназальных носителей или трансдермальными путями, используя те формы трансдермальных кожных кусков пластыря, хорошо известных обычным специалистам в данной области. При введении в форме трансдермальной системы доставки вводимая дозировка будет, несомненно, непрерывной, а не скачкообразной в течение режима дозирования.
Соединения по настоящему изобретению могут быть использованы в комбинации с другими агентами, используемыми для лечения катепсин-опосредованных состояний. Индивидуальные компоненты таких комбинаций могут быть введены отдельно в разное время в течение курса лечения или параллельно в разделенной или единичной комбинации форм. Понятно, что настоящее изобретение, таким образом, включает в себя все такие режимы одновременного или альтернативного лечения и термин «назначение» интерпритируют соответственно. Будет понятно, что объем комбинаций соединений данного изобретения с другими агентами, используемыми для лечения катепсин-опосредованных состояний включает в себя в принципе любую комбинацию с любой фармацевтической композицией, используемой для лечения нарушений, связанных с действием эстрогена.
Объем данного изобретения, следовательно, охватывает применение заявленных в настоящий момент соединений в комбинации с другим агентом, выбранным из органического бифосфоната; модулятора рецептора эстрогена; модулятора рецептора андрогена; ингибитора протонной АТФазы остеокластов и ингибитора HMG-CoA редуктазы; антагониста рецептора интегрина; анаболического агента остеобласта, такого как ПТГ (PTH), и их фармацевтически приемлемых солей и смесей.
Эти и другие аспекты данного изобретения будут видны из содержащихся здесь сведений.
Определения
Соединения по настоящему изобретению могут иметь асимметричные центры, хиральные оси и хиральные плоскости (описанные в E.L. Eliel и S.H. Wilen, Stereochemistry of Carbon Compounds, John Wiley & Sons, New York, 1994, страницы 1119-1190), и встречаются в виде рацематов, смесей рацематов и в виде индивидуальных диастереоизомеров, со всеми возможными изомерами и их смесями, в том числе оптические изомеры, включенные в настоящее изобретение. Кроме того, соединения, раскрытые здесь, могут существовать в виде таутомеров и обе таутомерные формы включены в объем данного изобретения, даже если изображена только одна таутомерная структура. Например, понятно, что любое притязание на соединение А, приведенное ниже, включает в себя таутомерную структуру В и наоборот, а также и их смеси.

В том случае, когда любая переменная (например R1, R2, Rа и т.д.) встречается больше одного раза в любой составляющей, его определение в каждом случае является независимым в каждом другом случае. Также комбинации заместителей и переменных допустимы, только если такие комбинации дают в результате стабильные соединения. Линии, проведенные внутрь кольцевых систем от заместителей, указывают на то, что связь может быть присоединена к любым заменяемым атомам углерода кольца. Если кольцевая система является полициклической, это означает, что связь присоединяется к любым заменяемым атомам углерода только на проксимальном кольце.
Понятно, что заместители и замещающие структуры в соединениях по настоящему изобретению могут быть выбраны обычным специалистом в данной области для получения соединений, которые являются химически стабильными и которые могут быть легко синтезированы с помощью методик, известных в данной области, а также теми способами, которые изложены ниже, из легко доступных исходных веществ. Если заместитель сам является замещенным более чем одной группой, понятно, что эти различные группы могут находиться на том же атоме углерода или на различных атомах углерода, пока в результате получается стабильная структура. Фразу «необязательно замещенный одним или более заместителями» следует принять равнозначной фразе «необязательно замещенный по меньшей мере одним заместителем», и в таких случаях предпочтительный вариант осуществления будет иметь от нуля до трех заместителей.
Используемый здесь «алкил» включает в себя как разветвленные, так и с прямой цепью насыщенные алифатические углеводородные группы, имеющие от одного до десяти атомов углерода, если не оговорено особо. Например, С1-С10, как в «С1-С10 алкил» определен для включения групп, имеющих 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода в линейном, разветвленном или циклическом расположении. Например, «С1-С10 алкил», в частности, включает в себя метил, этил, пропил, бутил, пентил, гексил, гептил, октил, нонил, децил и т.д.
«Алкокси» или «алкилокси» представляет алкильную группу, как определено выше, если не оговорено особо, где указанная алкильная группа присоединена через кислородный мостик.
Термин «циклоалкил» или «карбоцикл» будет означать циклические кольца алканов с общим числом атомов углерода от трех до восьми, если не оговорено особо, или любым числом в этом интервале (т.е. циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил).
Если число атомов углерода не определено, термин «алкенил» относится к неароматическому углеводородному радикалу, прямому или разветвленному, содержащему от 2 до 10 атомов углерода и по меньшей мере с 1 углерод-углеродной двойной связью. Предпочтительно присутствует 1 углерод-углеродная двойная связь и может присутствовать вплоть до 4 неароматических углерод-углеродных двойных связей. Таким образом, «C2-C6 алкенил» означает алкенильный радикал, имеющий от 2 до 6 атомов углерода. Алкенильные группы включают в себя этенил, пропенил, бутенил и циклогексенил. Как описано выше в отношении алкила, прямые, разветвленные или циклические части алкенильной группы могут содержать двойные связи и могут быть замещенными, если указана замещенная алкенильная группа.
Термин «циклоалкенил» будет означать циклические кольца, содержание от 3 до 10 атомов углерода, если не оговорено особо, содержащие по меньшей мере 1 углерод-углеродную двойную связь (т.е. циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил или циклооктенил).
Термин «алкинил» относится к углеводородному радикалу, прямому или разветвленному, содержащему от 2 до 10 атомов углерода, если не оговорено особо, содержащему по меньшей мере 1 углерод-углеродную тройную связь. Может присутствовать до 3 углерод-углеродных тройных связей. Следовательно, «C2-C6 алкинил» означает алкинильный радикал, имеющий от 2 до 6 атомов углерода. Алкинильные группы включают в себя этинил, пропинил и бутинил. Как описано выше в отношении алкила, прямая, разветвленная или циклическая часть алкинильной группы может содержать тройные связи и может быть замещенной, если указана замещенная алкинильная группа.
В отдельных случаях заместители могут быть определены углеродным рядом, который включает в себя ноль, например (C0-C6)алкиленарил. Если взятый арил представляет собой фенил, это определение включало бы в себя сам фенил, а также и -CH2Ph, -CH2CH2Ph, CH(CH3)CH2CH(CH3)Ph, и т.д.
Используемый здесь «арил» означает любое стабильное моноциклическое или бициклическое углеродное кольцо, содержащее до 12 атомов в каждом кольце, где по меньшей мере одно кольцо является ароматическим. Примеры таких арильных элементов включают в себя фенил, нафтил, тетрагидронафтил, инданил, бифенил, фенантрил, антрил или аценафтил. В случаях, когда арильный заместитель является бициклическим и одно кольцо является неароматическим, понятно, что присоединение происходит через ароматическое кольцо.
Термин «гетероарил», используемый здесь, представляет стабильное моноциклическое, бициклическое или трициклическое кольцо, содержащее до 10 атомов в каждом кольце, где по меньшей мере одно кольцо является ароматическим и содержит от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, N и S. Гетероарильные группы в объеме данного определения включают в себя, но не только, бензоимидазолил, бензофуранил, бензофуразанил, бензопиразолил, бензотриазолил, бензотиофенил, бензоксазолил, карбазолил, карболинил, циннолинил, фуранил, индолинил, индолил, индолазинил, индазолил, изобензофуранил, изоиндолил, изохинолил, изотиазолил, изоксазолил, нафтпиридинил, оксадиазолил, оксазолил, оксазолин, изоксазолин, пиранил, пиразинил, пиразолил, пиридазинил, пиридопиридинил, пиридил, пиримидинил, пирролил, хиназолинил, хинолил, хиноксалинил, тетразолил, тетразолпиридил, тиадиазолил, тиазолил, тиенил, триазолил, дигидробензоимидазолил, дигидробензофуранил, дигидробензотиофенил, дигидробензоксазолил, дигидроиндолил, дигидрохинолинил, метилендиоксибензен, бензотиазолил, бензотиенил, хинолинил, изохинолинил, оксазолил и тетрагидрохинолин. В случае, когда гетероарильный заместитель является бициклическим и одно кольцо является неароматическим или не содержит гетероатомов, понятно, что присоединение происходит через ароматическое кольцо или через кольцо, содежащее гетероатом, соответственно. Если гетероарил содержит атомы азота, понятно, что их соответствующие N-оксиды также охвачены этим определением.
Специалистам в данной области понятно, что «атом галогена» или «галоген», используемые здесь, включают в себя хлор, фтор, бром и йод. Термин «кето» означает карбонил (C=О). Термин «алкокси», используемый здесь, означает алкильную часть, где алкил определен выше, соединенную с остатком молекулы через атом кислорода. Примеры алкокси включают в себя метокси, этокси и подобное.
Термин «галогеналкил» означает алкильный радикал, как определено выше, если не оговорено особо, который замещен одним - пятью, предпочтительно одним - тремя атомами галогена. Характерные примеры включают в себя, но не только, трифторметил, дихлорэтил и подобное.
Термин «галогеналкокси» представляет радикал -OR, где R представляет собой алкил, как определено выше, который замещен одним - пятью, предпочтительно одним - тремя атомами галогена. Характерные примеры включают в себя, но не только, трифторметилокси, дихлорэтилокси и подобное.
Термин «арилалкил» включает в себя алкильную часть, где алкил определен выше и включает в себя арильную часть, где арил определен выше. Примеры арилалкила включают в себя, но не только, бензил, фторбензил, хлорбензил, фенилэтил, фенилпропил, фторфенилэтил и хлорфенилэтил. Примеры алкиларила включают в себя, но не только, толуил, этилфенил и пропилфенил.
Используемый здесь термин «гетероарилалкил» будет относиться к системе, которая включает в себя гетероарильную часть, где гетероарил определен выше и содержит алкильную часть. Примеры гетероарилалкила включают в себя, но не только, тиенилметил, тиенилэтил, тиенилпропил, пиридилметил, пиридилэтил и имидазоилметил.
Термин «циклоалкилалкил» включает в себя алкильную часть, где алкил определен выше, и также включает в себя циклоалкильную часть, где циклоалкил определен выше. Примеры циклоалкилалкила включают в себя, но не только, циклопропилметил, циклопентилметил, циклогексилметил, циклопропилэтил, и подобное.
Термин «гидроксиалкил» означает линейный моновалентный углеводородный радикал, содержащий от одного до шести атомов углерода, или разветвленный одновалентный углеводородный радикал, содержащий от трех до шести атомов углерода, замещенный одной или двумя гидроксигруппами, при условии, что в случае присутствия двух гидроксигрупп, они обе не находятся на одном атоме углерода. Характерные примеры включают в себя, но не только, гидроксиметил, 2-гидроксиэтил, 2-гидроксипропил, 3-гидроксипропил и подобное.
Термин «гетероцикл» или «гетероциклил», используемый здесь, означает 5-10-членное неароматическое кольцо, если не оговорено особо, содержащее от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, N, S, SO, или SO2, и включает в себя бициклические группы. «Гетероциклил», таким образом, включает в себя, но не только, следующее: пиперазинил, пиперидинил, пирролидинил, морфолинил, тиоморфолинил, тетрагидропиранил, дигидропиперидинил, тетрагидротиофенил и подобное. Если гетероцикл содержит азот, понятно, что его соответствующие N-оксиды также охватываются этим определением.
Настоящее изобретение также включает в себя N-оксидные производные и защищенные производные соединений Формулы I. Например, в том случае, когда соединения формулы I содержат окисляемый атом азота, атом азота может быть преобразован в N-оксид способами, хорошо известными в данной области. Также, в том случае, когда соединения формулы I содержат группы, такие как гидрокси, карбокси, тиол или любую группу, содержащую атом(ы) азота, эти группы могут быть защищены подходящими защищающими группами. Полный список подходящих защищающих групп можно найти в T.W. Greene, «Protective Groups in Organic Synthesis», John Wiley & Sons, Inc. 1981, раскрытие которого включено здесь в качестве ссылки в его полном объеме. Защищенные производные соединений формулы I могут быть получены способами, хорошо известными в данной области.
Когда же термин «алкил» или «арил» или любой из их стоящих впереди корней появляются в названии заместителя (например, арил C0-8 алкил) его следует интерпретировать включающим в себя те ограничения, которые данны выше для «алкила» и «арила». Обозначенное число атомов углерода (например, С1-10) будет относиться независимо к числу атомов углерода в алкильной или циклической алкильной части молекулы или к алкильной части большего заместителя, в котором алкил предстает как его стоящий впереди корень.
Фармацевтически приемлемые соли соединений по данному изобретению включают в себя общепринятые нетоксические соли соединений по данному изобретению, образованные неорганическими или органическими кислотами. Например, общепринятые нетоксические соли включают в себя соли, которые получены из неорганических кислот, таких как соляная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная и подобные, а также и соли, полученные от органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфокислота, метансульфоновая, этандисульфоновая, щавелевая, изэтионовая, трифторуксусная и подобные. Получение фармацевтически приемлемых солей описано выше и другие типичные фармацевтически приемлемые соли более полно описаны Berg et al., «Pharmaceutical Salts», J. Pharm. Sci., 1977:66:1-19, включенный здесь в качестве ссылки. Фармацевтически приемлемые соли соединений по данному изобретению могут быть синтезироаны из соединений по данному изобретению, которые содержат основную и кислотную части молекулы, традиционными химическими способами. Как правило, соли основных соединений готовят либо ионобменной хроматографией, либо взаимодействием свободного основания со стехиометрическими количествами, или с избытком желаемой сольобразующей неорганической или органической кислоты в подходящем растворителе или в различных комбинациях растворителей. Подобным образом, соли кислотных соединений образуются с помощью реакций с подходящим неорганическим или органическим основанием.
Для целей данного описания указаны значения следующих сокращений:
АсОН = уксусная кислота
BF3 = трифторид бора
Вос = трет-бутилоксикарбонил
Вос2О = ди-трет-бутилдикарбонат
BuLi = бутиллитий
CCl4 = четыреххлористый углерод
СН2Cl2 = метиленхлорид
CH3CN = ацетонитрил
CHCl3 = хлороформ
Cs2CO3 = карбонат цезия
CuI = йодид меди
DAST = трифторид диэтиламиносеры
DIPEA = диизопропилэтиламин
DMA = N,N-диметилацетамид
DMAP = 4-(диметиламино)пиридин
ДМФ = N,N-диметилформамид
ДМСО = диметилсульфоксид
DPPA = дифенилфосфорилазид
EDCI = 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид
Et2O = диэтиловый эфир
Et3N = триэтиламин
EtOAc = этилацетат
EtOH = этанол
HATU = о-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфат
HOAc = уксусная кислота
K2CO3 = карбонат калия
KHMDS = гексаметилдисилазан калия
KOBut = трет-бутоксид калия
LDA = диизопропиламид лития
LiOH = гидроксид лития
mCPBA = мета-хлорпербензойная кислота
MeOH = метанол
MeSO3H = метансульфокислота
MgSO4 = сульфат магния
Ms = метансульфонил = мезил
MsCl = метансульфонилхлорид
NaBH4 = борогидрид натрия
NaH = гидрид натрия
Nal = йодид натрия
NaCNBH3 = цианоборогидрид натрия
Na2CO3 = карбонат натрия
NaHCO3 = гидрогенкарбонат натрия
NaOH = гидроксид натрия
Na2SO4 = сульфат натрия
NBS = N-бромсукцинимид
NH3 = аммиак
NH4Cl = хлорид аммония
Pd/C = палладий на угле
PdCl2 = дихлорпалладий(II)
PdCl2(dppf) = [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)
Pd2(dba)3 = трис(дибензилиденацетон)дипаладий(0)
PG = защищающая группа
PPh3 = трифенилфосфин
(PhO)3PMeI = йодид метилтрифеноксифосфония
PPTS = пиридиний-п-толуолсульфонат
iPr2Nli = литий диизопропиламид
PyBOP = бензотриазол-1-илокситрис(пирролидин)фосфоний-гексафторфосфат
К.т. = комнатная температура
Насыщ. = насыщенный водный раствор
TFA = трифторуксусная кислота
ТГФ = тетрагидрофуран
TiCl4 = хлорид титана(IV)
tlc = тонкослойная хроматография
TMSCl = хлортриметилсилан
Me = метил
Et = этил
n-Pr = нормальный пропил
i-Pr = изопропил
n-Bu = нормальный бутил
i-Bu = изобутил
s-Bu = вторичный бутил
t-Bu = третичный бутил
Новые соединения настоящего изобретения могут быть получены в соответствии со следующими общепринятыми методами с использованием соответствующих веществ, и дополнительно иллюстрируются отдельными примерами. Соединения, проиллюстрированные в примерах, однако, не истолковывают как образующие только род, который рассматривают как данное изобретение. Следующие примеры дополнительно иллюстрируют подробности получения данных соединений по настоящему изобретению. Специалист в данной области легко поймет, что известные вариации условий и процессов следующих препаративных процессов могут быть использованы для получения этих соединений. Вся температуры даны в градусах Цельсия, если не указано отдельно.
СХЕМЫ
Соединения по настоящему изобретению могут быть получены в соответствии со Схемой 1, указанной ниже. Таким образом, к галогеналкилкетону может быть добавлен сложный α-аминоэфир для получения аминаля, который может быть дегидратирован в имин в присутствии дегидрирующего агента, такого как TiCl4, MgSO4 или изопропил трифторацетата. Восстановление имина восстанавливающим агентом, таким как цианоборогидрид натрия или борогидрид натрия, дает амин. Гидролиз сложного эфира и образование амида соответственно замещенным аминоацетонитрилом дает соединения по настоящему изобретению. Если заместителем на D системе является галоген, катализируемое палладием связывание по методу Сузуки (Suzuki) с соответствующей бороновой кислотой дает дополнительные соединения по настоящему изобретению.
СХЕМА 1
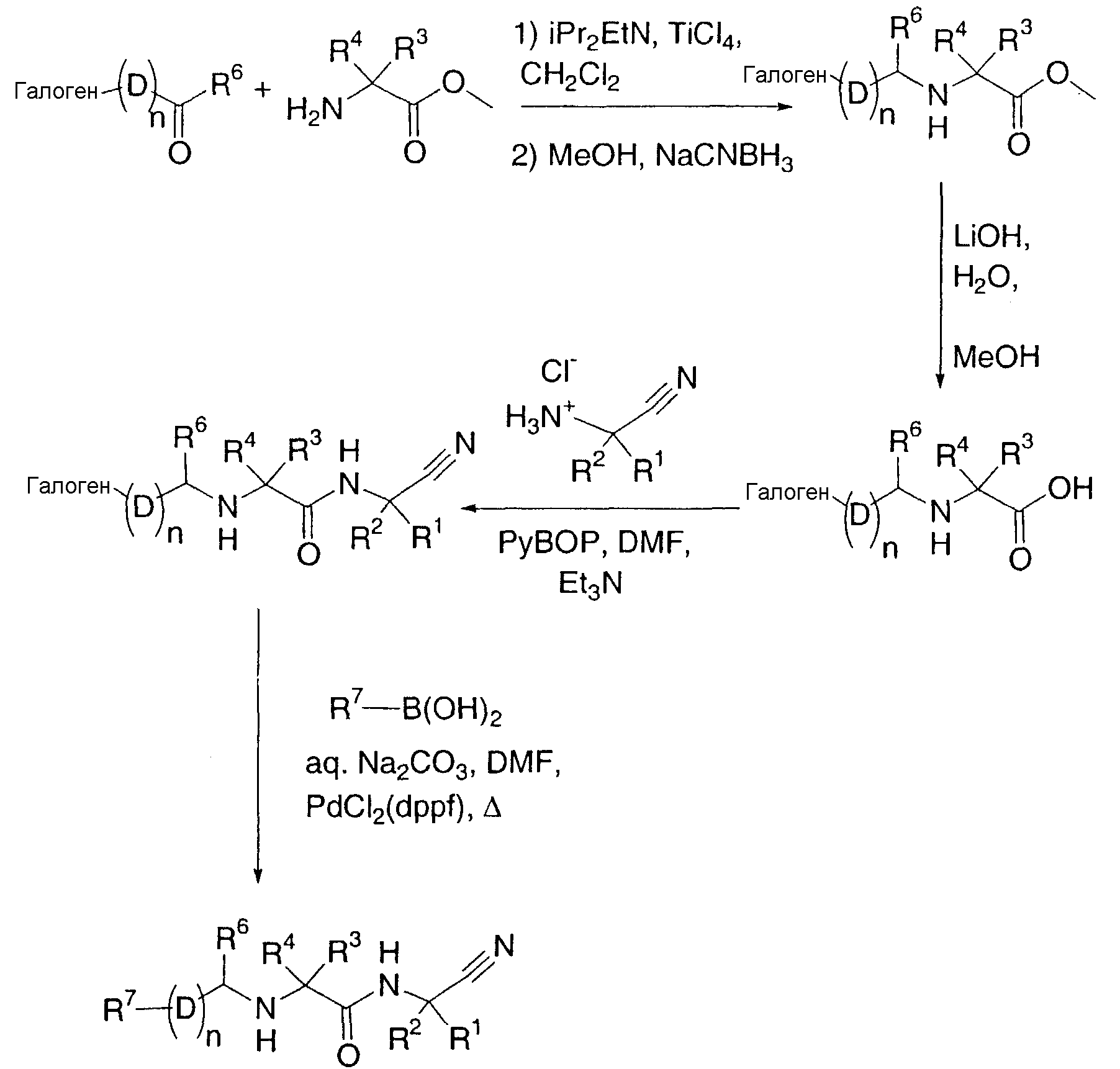
Соединения по настоящему изобретению также могут быть получены в соответствии со Схемой 2, приведенной ниже. Кетон или альдегид могут быть конденсированы с аминоспиртом с получением циклического аминаля. Обработка 3 эквивалентами реактива Гриньяра или органолитиевым реагентом дает соответствующий алкилированный аминоспирт. Окисление спирта хромовой системой, например окисление по Джонсу (Jones) или с помощью H5IO6/CrO3, или альтернативно путем двухстадийного окисления (например, оксалилхлорид/ДМСО/Et3N, затем NaClO) даст соответствующую карбоновую кислоту. Пептидное взаимодействие и реакция Сузуки, описанные в Схеме 1, дадут соединения по настоящему изобретению.
СХЕМА 2
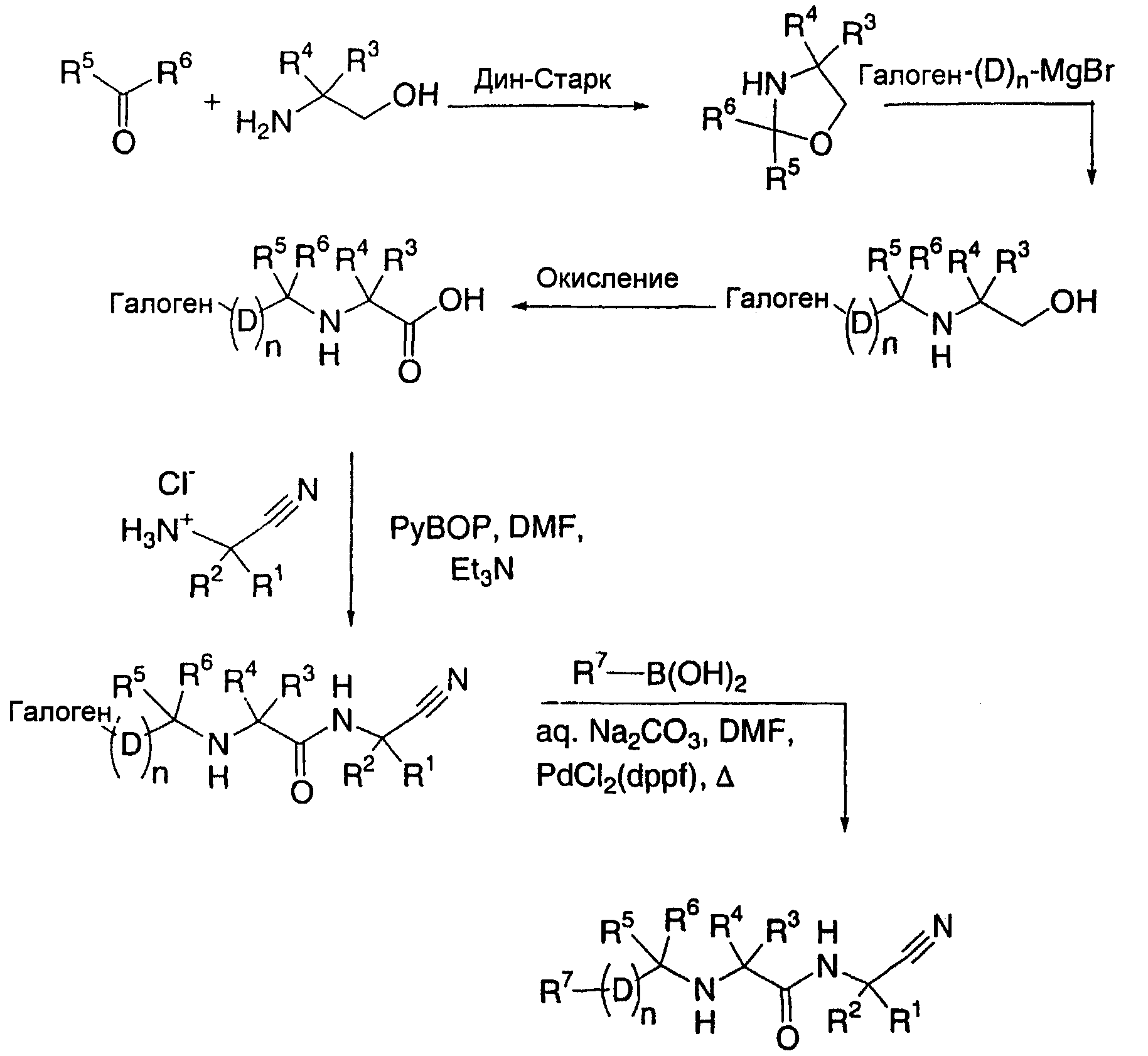
Соединения по настоящему изобретению также могут быть получены в соответствии со Схемой 3, приведенной ниже. Кетон или альдегид могут быть конденсированы с аминоспиртом с получением циклического аминаля. Обработка большим количеством эквивалентов реактива Гриньяра или органолитиевого реагента дает соответствующий алкилированный аминоспирт. Этот спирт может быть преобразован в соединение по настоящему изобретению способом, описанным в Схеме 2.
СХЕМА 3
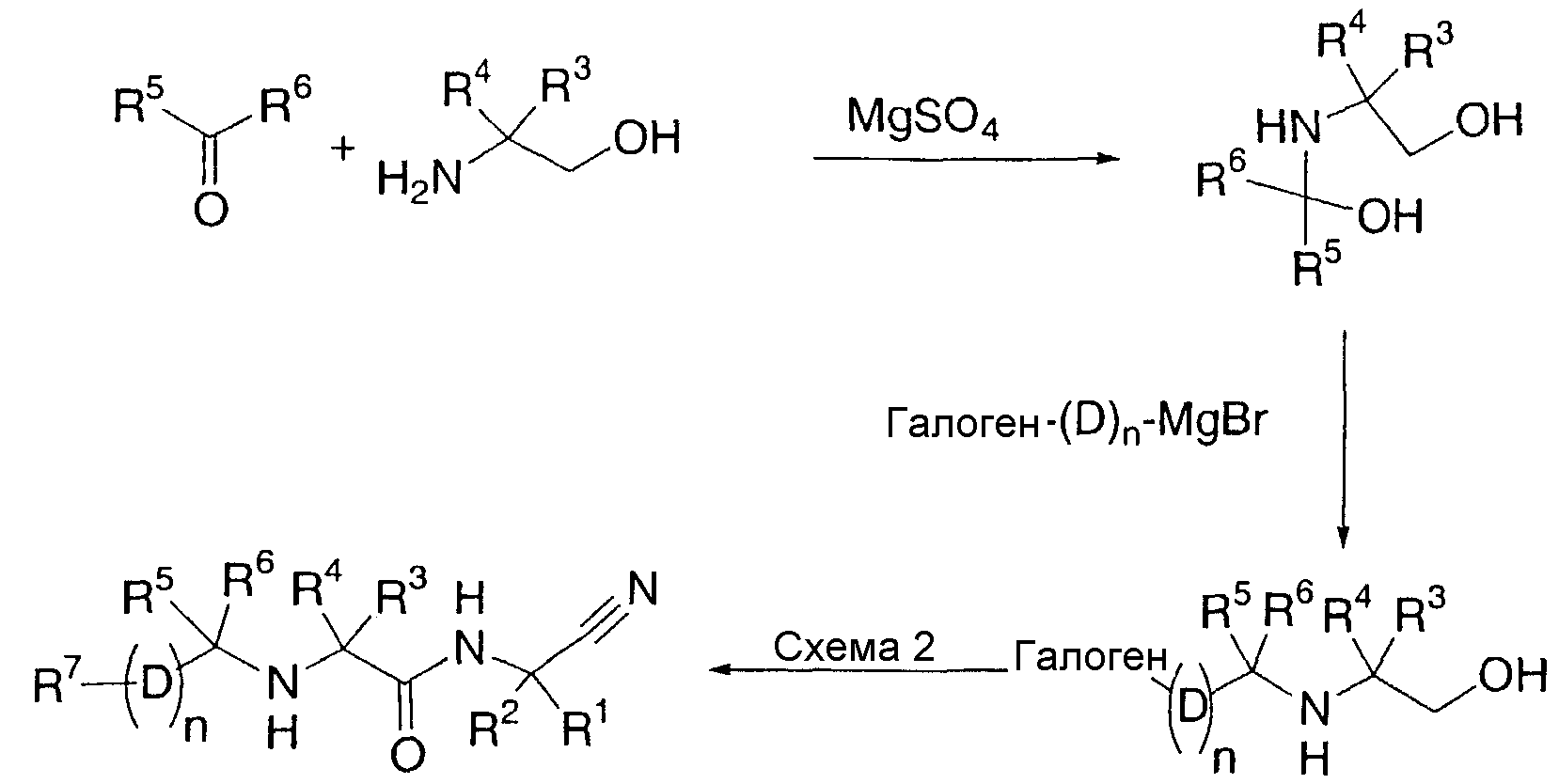
Соединения по настоящему изобретению также могут быть получены в соответствии со Схемой 4. Ацетат, замещенный соответствующим образом, может быть енолизирован подходящим основанием (в том числе, но не только, LDA, KHMDS, NaH или n-BuLi) и обработан параформальдегидом с образованием диола. Данный диол может быть преобразован в дифторид с использованием фторирующего реагента, такого как DAST. Гидролиз сложного эфира с последующией затем перегруппировкой Куртиса (Curtius) затем дает амин. Этот амин может замещать соответственно замещенный сложный альфа-бромэфир, предоставляя сложный альфа-аминоэфир. Он может быть преобразован в соединения по настоящему изобретению способом, описанным в Схеме 1.
СХЕМА 4
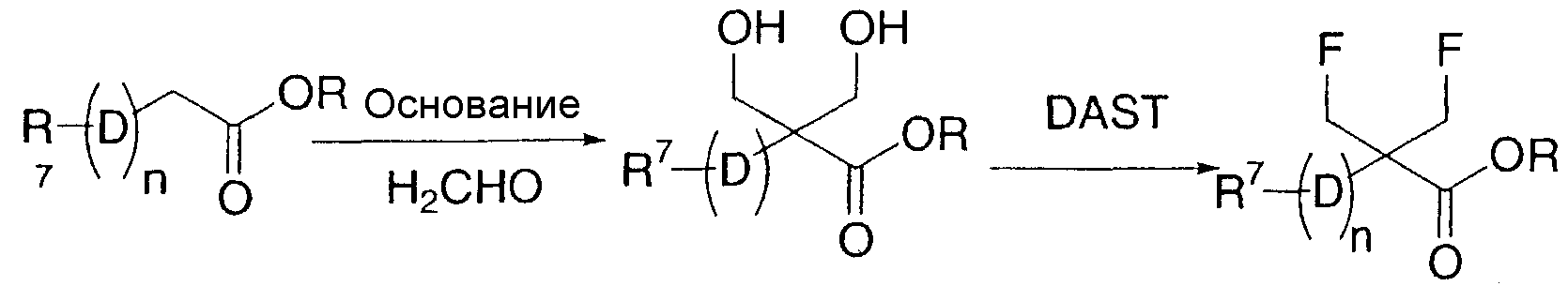

Соединения по настоящему изобретению также могут быть получены в соответствии со схемой 5, приведенной ниже. Гемиацеталь может быть конденсирован с аминоспиртом, в котором спиртовая часть молекулы защищена подходящей защищающей группой. Обработка результирующего имина реактивом Гриньяра или органолитиевым реагентом даст соответствующий аликилированный аминоспирт. Спиртовая защитная группа затем может быть удалена и спирт может быть преобразован в соединения по настоящему изобретению либо способом, описанным с Схеме 2, либо первым проведением реакции Сузуки с последующим окислением спирта воздействием H5IO6/CrO3, а затем пептидным связыванием.
СХЕМА 5

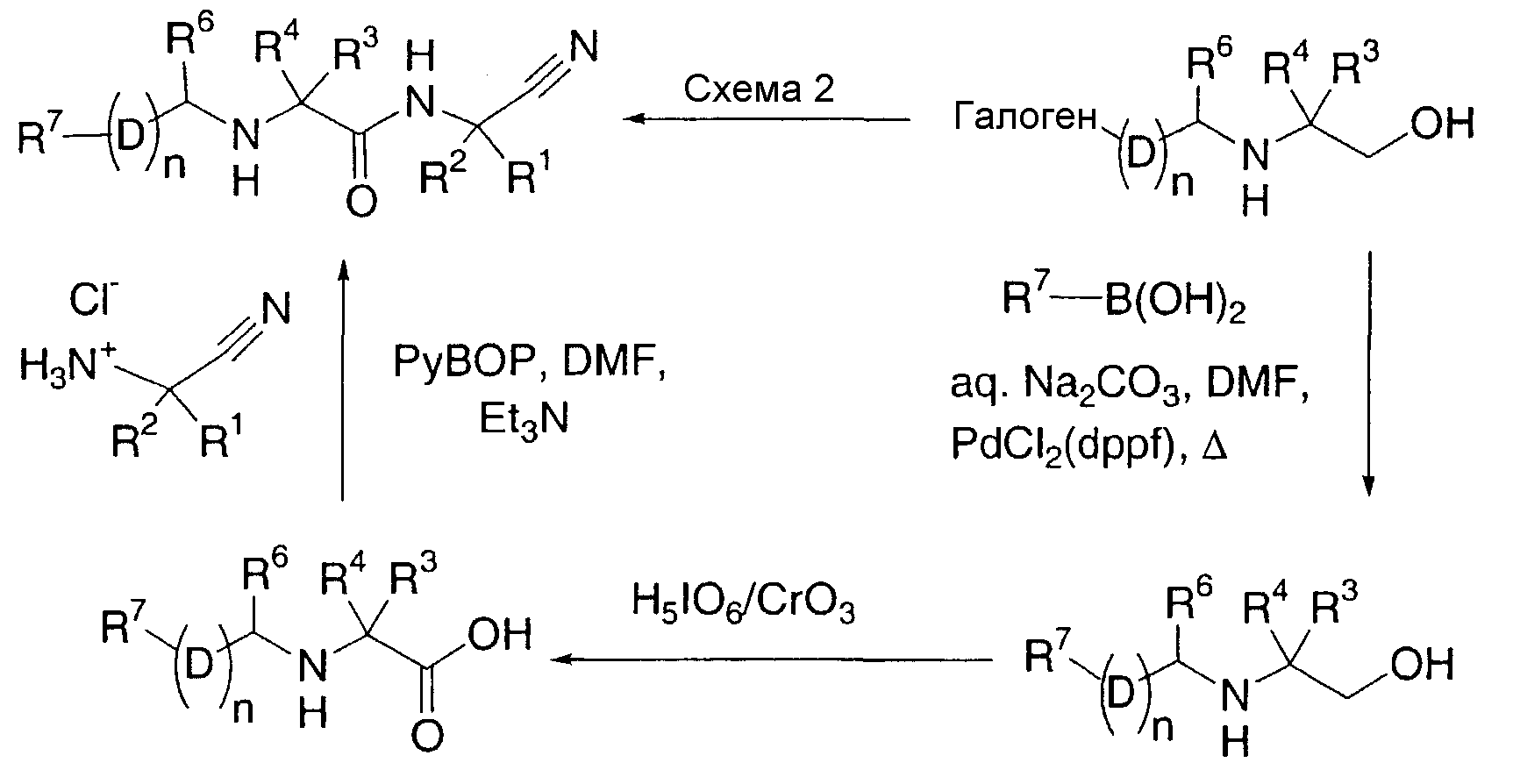
Соединения по настоящему изобретению также могут быть получены в соответствии со схемой 6, приведенной ниже. Пептидное соединение альфа-аминокислоты описано в схемах 1, 2 или 5, с альфа-аминоамидом с последующим дегидрированием полученного в результате первичного амида (Voegel, J. J.; Benner, S. A. Helv. Chem. Acta 1996, 79, 1863) дает соединения по настоящему изобретению.

Синтез некоторых аминоспиртов, используемых в качестве исходных продуктов по схемам 2, 3 и 5, представлен на схемах 7-11. Например, синтез (2S)-2-амино-4-фтор-4-метилпентан-1-ола, где R=Me, представлен на Схеме 7 ниже. Начиная с подходящей дизащищенной аспарагиновой кислоты, карбоксильная группа может быть восстановлена в спирт с использованием стандартных литературных методов (например, образование смешанного ангидрида с последующим восстановлением NaBH4). Защищенный вариант 2-амино-4-метилпентан-1,4-диола (R=Me) затем может быть образован с помощью соответствующего воздействия реактива Гриньяра или органолития. Наконец, гидроксильная часть молекулы может быть преобразована в желаемый фторозаместитель с использованием фторирующего агента, такого как DAST. Защищенный или незащищенный вариант данного аминоспирта затем может быть преобразован в соединения по настоящему изобретению согласно схемам 1, 2, 3 и 5.
СХЕМА 7

4-Фторлейцинол также может быть синтезирован с соответствии со схемой 8. 4,5-Дегидролейцин преобразован в (4S)-4-(2-метилпроп-2-енил)-1,3-оксазолидин-2-он, как представлено на схеме ниже. Это промежуточное соединение затем обрабатывают гидрофторирующим реагентом, таким как HF-пиридин, с получением (4S)-4-(2-фтор-2-метилпропил)-1,3-оксазолидин-2-она. Гидролиз основанием (т.е. Ba(OH)2 или NaOH) затем дает (2S)-2-амино-4-фтор-4-метилпентан-1-ол.

Синтез 4,4-дифтор-L-норвалина, где R=Me, представлен на схеме 9 ниже. Исходя из соответственно дизащищенного серина йодирование может быть проведено с использованием реагента, такого как (PhO)3P+MeI-. Введение цинка в полученный в результате йодид может осуществляться с использованием Zn-Cu пары и TMSCl. Полученный в результате цинкат затем подвергают реации сочетания, катализируемой палладием, с алканоилхлоридом для получения кетона. В итоге, данная кетонная часть молекулы может быть преобразована в желаемое дифторпроизводное с использованием фторирующего агента, такого как DAST. Защищенный или незащищенный вариант этой аминокислоты или аминоспирта затем может быть преобразован в соединения по настоящему изобретению согласно схемам 1, 2, 3 и 5.
СХЕМА 9

Аминоспирты, используемые в настоящем изобретении, также могут быть синтезированы в соответствии со схемой 10. Защитную аминокислоту восстанавливают восстанавливающим агентом, таким как NaBH4,в присутствии или в отсутствие LiCl, в растворителе, таком как EtOH, или в системе смешанных растворителей, таких как EtOH и ТГФ. Аминозащитную группу затем удаляют соответствующим способом в соответствии с характером защищающей группы.
СХЕМА 10

Синтез (2S,4S)-2-амино-5,5,5-трифтор-4-метилпентан-1-ола, используемого в настоящем изобретении, предствален на Схеме 11. N-Бензоил-5,5,5-трифторлейцин (Ojiima et. al. J. Org. Chem., 1989, 54, 4511 - 4522) может быть гидролизован водным раствором кислоты, такой как 6M HCl, при нагревании с обратным холодильником. Промежуточное соединение, HCl соль аминокислоты, затем преобразовывают в N-ацетил-5,5,5-трифторлейцин и аминогруппу хирального центра разлагают ферментативным способом (Synthetic Communications, 1996, 26, 1109 - 1115). Выделенный 5,5,5-трифтор-L-лейцин затем защищают с помощью защитной группы, такой как бензилкарбамат, и этерифицируют группу карбоновой кислоты. Два диастереоизомера в положении 4 затем разделяют тонкослойной колоночной хроматографией. Один из энантиомеров, (2S,4S) защитную аминокислоту, затем преобразовывают в аминоспирт, как представлено на схеме 10.
СХЕМА 11


Соединения по настоящему изобретению, где R5 представляет собой водород и R6 представляет собой арил или гетероарил, также могут быть получены в соответствии со схемой 12, как показано ниже. Конденсация арил или гетероарил альдегида с аминоспиртом, в котором спиртовая часть молекулы защищена подходящей защитной группой, с последующей обработкой полученного в результате имина реактивом Гриньяра или органолитиевым реагентом формулы галоген-(D)n-Li или галоген-(D)n-MgX (где D определен в разделе «Краткое изложение сущности изобретения»), с последующим удалением кислородзащитной группы, дает алкилированный аминоспирт. Данный алкилированный аминоспирт затем преобразовывают в соединения по настоящему изобретению либо с помощью способа, представленного на схеме 2, либо сначала проведением реакции Сузуки с бороновым сложным эфиром формулы R7-B(OH)2, затем окислением спирта подходящим окисляющим агентом, таким как H5IO6/CrO3, с получением кислоты и, в конце, обработкой данной кислоты аминоацетонитрилом в условиях пептидного связывания, описанного ранее.

Соединения по настоящему изобретению также могут быть получены в соответствии со схемой 13, показанной ниже. Взаимодействие соответветствующим образом N-защищенного аминокислотного производного с оксетантозилатом в присутствии йодида натрия в подходящем органическом растворителе, таком как диметилформамид, дает соответствующий сложный эфир оксатана, который при обработке дибораном дает сложный ортоэфир. Удаление аминозащитной группы дает амин, который при конденсации с альдегидом формулы R6CHO (где R6 представляет собой арил или гетероарил) или гемиацеталем формулы R6С(OH)(OR) (где R представляет собой алкильную группу) в реакционных условиях, описанных выше, дает имин. Обработка данного имина реактивом Гриньяра или органолитиевым реагентом в условиях реакции, описанных выше, дает N-алкилированное производное. Удаление ортоэфира дает соответствующую карбоновую кислоту, которую затем преобразовывают в соединения по настоящему изобретению конденсацией с аминоацетонитрилом в условиях пептидного связывания с последующей реакцией Сузуки, как описано выше.

Следующие примеры описывают синтез избранных соединений настоящего изобретения.
ПРИМЕР 1
Синтез N1-(цианометил)-N2-(2,2, 2-трифтор-1-фенилэтил)-L-лейцинамида

К раствору гидрохлорида метилового эфира L-лейцина (975 мг, 5,37 ммоль) в дихлорметане (30 мл) добавляли 2,2,2-трифторацетофенон (0,75 мл, 5,34 ммоль), добавляли по каплям диизопропилэтиламин (3,5 мл, 20 ммоль), TiCl4 (0,55 мл, 5, 0 ммоль) в 0,45 мл дихлорметана и данную смесь перемешивали в течение ночи. Затем еще добавляли TiCl4 (0,4 мл, 3,6 ммоль) и смесь перемешивали 3 ч. Добавляли раствор NaCNBH3 (1050 мг, 16,7 ммоль) в MeOH (20 мл) и смесь перемешивали 2 ч. Выливали в 1н NaOH и экстрагировали этилацетатом (2x). Органическую фазу промывали 1н NaOH и насыщенным солевым раствором, затем сушили над MgSO4 и выпаривали. Очистка с помощью ISCO колоночной хроматографии (градиент от 30% до 90% этилацетат/гексан) давала метил-N-(2,2,2-трифтор-1-фенилэтил)-L-лейцинат.
При комнатной температуре к раствору метил-N-(2,2,2-трифтор-1-фенилэтил)-L-лейцината (150 мг, 0,50 ммоль) в 2:1 ТГФ/MeOH добавляли 1M LiOH. Смесь перемешивали в течение ночи и концентрировали. Остаток распределяли между этилацетатом и фосфатным буфером с рН 3,5. Органическую фазу промывали насыщенным солевым раствором, сушили над MgSO4 и концентрировали с получением N-(2,2, 2-трифтор-1-фенилэтил)-L-лейцина.
Смесь N-(2,2,2-трифтор-1-фенилэтил)-L-лейцина (149 мг, 0,50 ммоль), аминоацетонитрила гидрохлорида (102 мг, 1,1 ммоль) и PyBOP (260 мг, 0,50 ммоль) растворяли в ДМФ (5 мл). Добавляли триэтиламин (0,3 мл, 2,1 ммоль) и смесь перемешивали в течение ночи, затем выливали в фосфатный буфер с pH 3 и экстрагировали 3:1 эфиром/этилацетатом. Органическую фазу затем промывали насыщенным водным NaHCO3 и насыщенным солевым раствором, сушили над MgSO4 и выпаривали. Очистка с помощью ISCO колоночной хроматографии (градиент от 20% до 50% этилацетат/гексан) давала N1-(цианометил)-N2-(2,2,2-трифтор-1-фенилэтил)-L-лейцинамид в виде 1:1 смеси диастереомеров.
MS (+APCI): 313,9 [M+1].
ПРИМЕР 2
Синтез N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамида

Используя способ Примера 1 получали N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид.
MS (-ESI): 403,9, 405,9 [M-1]-.
ПРИМЕР 3
Синтез N1-(цианометил)-N2-[2,2,2-трифтор-1-(4'-пиперазин-1-ил-1, 1'-дифенил-4-ил)этил]-L-лейцинамида

К N2-[1-(4-бромфенил)-2,2, 2-трифторэтил]-N1-(цианометил)-L-лейцинамиду (242 мг, 0,60 ммоль) и 4-[4-(трет-бутоксикарбонил)-1-пиперазинил]фенилбороновой кислоте (220 мг, 0,72 ммоль) в DME (3 мл) в токе сухого азота добавляли 2M водный карбонат натрия (0,9 мл, 1,8 ммоль) а затем катализатор PdCl2(dppf) (63 мг, 0,077 ммоль). Реационную смесь нагревали до 85°C в течение 18 часов. Добавляли воду и продукт экстрагировали EtOAc, органический слой сушили над Na2SO4 и концентрировали в вакууме. Сырой продукт очищали с помощью хроматографии с использованием EtOAc в гексане с получением трет-бутил(4'-{1-[((1S)-1-{[(цианометил)амино]карбонил}-3-метилбутил)амино]-2,2,2-трифторэтил}-1,1'-дифенил-4-ил)-1-пиперазинкарбоксилата.
К трет-бутил(4'-{1-[((1S)-1-{[(цианометил)амино]карбонил}-3-метилбутил)амин]-2,2,2-трифторэтил}-1,1'-дифенил-4-ил)-1-пиперазинкарбоксилату (275 мг, 0,47 ммоль) в сухом ТГФ (1 мл) в сухом азоте добавляли по каплям MeSO3H (125 мкл, 1,9 ммоль) в течение 15 мин и реакции давали протекать в течение 18 часов. Реакционную смесь распределяли между EtOAc и водой + насыщенный NaHCO3 для установления pH 7,5. Органический слой сушили над Na2SO4 и концентрировали в вакууме. Сырой продукт очищали с помощью хроматографии с использованием силикагеля, элюировали NH4OH конц./MeOH/CH2Cl2, (1/10/89) с получением N1-(цианометил)-N2-[2,2,2-трифтор-1-(4'-пиперазин-1-ил-1,1'-дифенил-4-ил)этил]-L-лейцинамида в виде светло-желтой пены.
MS (+ESI): 488,3 [M+1]+.
ПРИМЕР 4
Синтез N1-(цианометил)-N2-{[4'-(метилсульфонил)-1, 1'-дифенил-4-ил][4-(метилсульфонил)фенил]метил}-L-лейцинамида

Стадия 1: метил-N-{(4-бромфенил)[4-(метилсульфонил)фенил]метилен}-L-лейцинат
Раствор (4-бромфенил)[4-(метилсульфонил)фенил]метанона (202 мг, 0,59 ммоль), гидрохлорида метилового эфира L-лейцина (328 мг, 2,0 ммоль) и камфорсульфоновой кислоты (52 мг, 0,22 ммоль) в толуоле нагревали с обратным холодильником в течение 18 часов и с использованием насадки Дина-Старка. Растворитель удаляли в вакууме, полученный в результате остаток очищали с помощью хроматографии, используя EtOAc и гексан в качестве элюента, с получением 1:1 смеси указанного в заголовке соединения и исходного (4-бромфенил)[4-(метилсульфонил)фенил]метанона.
Стадия 2: метил-N-{(4-бромфенил)[4-(метилсульфонил)фенил]метил}-L-лейцинат
К раствору 1:1 смеси метил-N-{(бромфенил)[4-(метилсульфонил)фенил]метилен} лейцината и (4-бромфенил)[4-(метилсульфонил)фенил]метанона со стадии 1 (185 мг, ˜0,2 ммоль) в уксусной кислоте/метаноле (1:3, 4 мл) добавляли борогидрид натрия (˜400 мг) по частям каждые 30 мин в течение 2 дней (ночью добавление прекращали) используя воронку для прибавления твердых частиц. Реакционную смесь распределяли между EtOAc и водой, органический слой сушили над Na2SO4 и концентрировали. Полученную в результате смесь очищали с помощью хроматографии с использованием EtOAc и гексана в качестве элюента. Метил-N-{(4-бромфенил)[4-(метилсульфонил)фенил]метил]}-L-лейцинат получали в виде бесцветной смолы и (4-бромфенил)[4-(метилсульфонил)фенил]метанол получали в виде белого твердого вещества.
Стадия 3: N-{(4-бромфенил)[4-(метилсульфонил)фенил]метил}-L-лейцин
К раствору метил-N-{(4-бромфенил)[4-(метилсульфонил)фенил]метил}-L-лейцината со стадии 2 (81 мг, 0,17 ммоль) в ТГФ (1 мл) и MeOH (0,5 мл) добавляли 1н LiOH (0,3 мл, 0,3 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение 18 часов, а затем распределяли между EtOAc и водой + 1н HCl (0,5 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветной смолы.
Стадия 4: N2 -{(4-бромфенил)[4-(метилсульфонил)фенил]метил}-N1-(цианометил)-L-лейцинамид
К раствору N-{(4-бромфенил)[4-(метилсульфонил)фенил]метил}-L-лейцина со стадии 3 (76 мг, 0,17 ммоль), HATU (146 мг, 0,38 ммоль), аминоацетонитрилгидрохлорида (52 мг, 0,56 ммоль) в ДМФ (1,1 мл), охлажденному до -10°C, добавляли N,N-диизопропилэтиламин (0,13 мл, 0,75 ммоль). Реакцию оставляли протекать при комнатной температуре в течение 18 ч и распределяли между EtOAc и водой. Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью хроматографии с использованием EtOAc и гексана в качестве элюента с получением указанного в заголовке соединения в виде бесцветной смолы.
Стадия 5: N1-(цианометил)-N2-{[4-(метилсульфонил)фенил][4'-(метилтио)-1,1-дифенил-4-ил]метил}-L-лейцинамид
Гетерогенную смесь N2 -{(4-бромфенил)[4-(метилсульфонил)фенил]метил}-N1-(цианометил)-L-лейцинамида со стадии 4 (72 мг, 0,15 ммоль), 4-(метилтио)фенилбороновой кислоты (37 мг, 0,22 ммоль) в диметиловом эфире этиленгликоля (1 мл) и 2M водного карбоната натрия дегазировали в вакууме и продували азотом. К этой смеси добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II), дихлорметановый комплекс (19 мг, 0,023 ммоль) с последующей дегазацией и продуванием азотом. Реационную смесь нагревали при 85°C в течение 16 часов при эффективном перемешивании. Реакционную смесь распределяли между EtOAc и водным NH4OAc (25% мас./об.). Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью хроматографии с использованием EtOAc и гексана в качестве элюента с получением указанного в заголовке соединения в виде бесцветной смолы.
Стадия 6: N1 -(цианометил)-N2-{[4'-(метилсульфонил)-1,1'-дифенил-4-ил][4-(метилсульфонил)фенил]метил}-L-лейцинамид
К раствору N1-(цианометил)-N2 -{[4-(метилсульфонил)фенил][4'-(метилтио)-1,1'-дифенил-4-ил]метил}-L-лейцинамида (63 мг, 0,12 ммоль), натрий вольфрамат дигидрата (2 мг, 0,006 ммоль), тетрабутиламмония гидросульфата (4 мг, 0,01 ммоль) добавляли раствор 30% (мас./об.) водной перекиси водорода (100 мкл, 0,9 ммоль) и полученную в результате смесь перемешивали при комнатной температуре в течение 10 мин. Реакционную смесь распределяли между EtOAc и водой + 1M NaHSO3 (˜3:1). Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью хроматографии с использованием EtOAc и гексана в качестве элюента с получением указанного в заголовке соединения в виде бесцветной смолы.
MS (+ESI): 568,2 [M+1]+.
ПРИМЕР 5
Синтез N1-(цианометил)-N2-{2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-3-ил]этил}-L-лейцинамида

Используя методику, описанную для примера 8, где N2-[1-(4-бромфенил)-2,2, 2-трифторэтил]-N1-(цианометил)-L-лейцинамид был замещен на N2-[1-(3-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид, указанное в заголовке соединение получали в виде бесцветной смолы.
MS (+ESI): 482,2 [M+1]+.
ПРИМЕР 6
Синтез N1-(цианометил)-N2-[2,2, 2-трифтор-1-(3-пиридин-4-илфенил)этил]-L-лейцинамида

Используя методику, описанную для примера 8, где N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид был заменен на N2-[1-(3-бромфенил)-2,2,2-трифторэтил]-N1 -(цианометил)-L-лейцинамид, указанное в заголовке соединение получали в виде бесцветной смолы.
MS (+ESI): 405,1 [M+1]+.
ПРИМЕР 7
Синтез N1-(цианометил)-N2-[2,2,2-трифтор-1-(4'-пиперазин-1-ил-1,1'-дифенил-3-ил)этил]-L-лейцинамида

Используя методику, описанную для примера 3, где N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид был заменен на N2-[1-(3-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамид, получали указанное в заголовке соединение в виде бесцветной смолы.
MS (+ESI): 488,3 [M+1]+.
ПРИМЕР 8
Синтез N1-(цианометил)-N2{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамида

Стадия 1: получение (2S)-1-{[трет-бутил(диметил)силил]окси}-4-метилпентан-2-амина
При комнатной температуре к раствору L-лейцинола (6,0 г) в дихлорметане (100 мл) добавляли триэтиламин (11 мл), DMAP (0,1 г) и трет-бутилдиметилсилилхлорид (8,5 г). Данную смесь перемешивали при комнатной температуре в течение 2 часов, а затем добавляли воду. Органический слой отделяли, а водный дополнительно экстрагировали дихлорметаном. Объединенные органические слои промывали насыщенным солевым раствором, сушили сульфатом магния и растворитель удаляли в вакууме с получением осадка указанного в заголовке соединения, которое использовали как таковое в следующей реакции.
1H ЯМР (CD3COCD3) δ 3,48 (м, 2Н), 3,32 (м, 1Н), 2,76 (м, 1Н), 1,78 (м, 1Н), 1, 22-1,02 (м, 2Н), 0,88 (м, 15Н), 0,06 (с, 6Н).
Стадия 2: получение (2S)-1-{[трет-бутил(диметил)силил]окси}-4-метил-N-[(1E)-2,2,2-трифторэтилиден]пентан-2-амина
Раствор (2S)-1-{[трет-бутил(диметил)силил]окси}-4-метилпентан-2-амина со стадии 1 (50 г) в толуоле (300 мл) и трифторацетальдегид метил гемиацеталь (35 мл) нагревали до кипения с обратным холодильником в течение 16 часов, в течение этого времени воду собирали с помощью насадки Дина-Старка. Растворитель выпаривали в вакууме и остаток очищали на SiO2, используя гексан и этилацетат (9:1) в качестве элюента с получением указанного в заголовке соединения.
1H ЯМР (CD3COCD3) δ 7,88 (м, 1H), 3,76-3,45 (м, 3H), 1,60-1, 25 (м, 3H), 0,88 (м, 15H), 0,06 (с, 3H), 0,04 (с, 3H).
Стадия 3: получение (2S)-2-{[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]амино}метилпентан-1-ола.
н-BuLi (2,5 M в гексане, 42 мл) добавляли при -70°C к ТГФ-ному (400 мл) раствору 1,4-дибромбензола (25,8 г) и смесь перемешивали в течение 25 минут. Затем добавляли по каплям ТГФ-ный (30 мл) раствор (2S)-1-{[трет-бутил(диметил)силил]окси}-4-метил-N-[(1Е)-2,2,2-трифторэтилиден]пентан-2-амина (31 г) и смесь перемешивали в течение 1,5 часов. Затем медленно выливали в смесь этилацетата (500 мл), воды (2 л), льда (300 г) и хлорида аммония (100 г) при интенсивном перемешивании. Органический слой отделяли и водный дополнительно экстрагировали этилацетатом (2х500 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили сульфатом магния и растворитель удаляли в вакууме с получением остатка, который использовали как таковой. Вышеуказанный остаток растворяли в ТГФ (250 мл) и раствор охлаждали до 0°C. Добавляли по каплям 1M ТГФ-ный раствор фторида трет-бутиламмония (110 мл), смесь взаимодействовала в течение 4 часов. Смесь выливали в этилацетат (300 мл), воду (2 л) и хлорид аммония (100 г) при интенсивном перемешивании. Органический слой отделяли и водный дополнительно экстрагировали этилацетатом (2х100 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили сульфатом магния и растворитель удаляли в вакууме с получением остатка, который очищали на SiO2, используя градиент этилацетата и гексана (от 1:5 до 1:4) в качестве элюента с получением указанного в заголовке соединения.
1H ЯМР (CD3COCD3) δ 7,6 (2H, д), 7,45 (2H, д), 4,55 (1H, м), 3,65-3,7 (1H, м), 3,5-3,55 (1H, м), 3,25-3,35 (1H, м), 2,6-2,7 (1H, м), 2,25-2,35 (1Н, м), 1,65-1,75 (1H, м), 1,3-1,4 (1H, м), 1,2-1,3 (1Н, м), 0,75-0,9 (6H, дд).
Стадия 4: получение (2S)-4-метил-2-({(1S)-2,2,2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}амино)пентан-1-ола
Сквозь суспензию, полученную из бромида со стадии 3 (27,7 г), 4-(метилтио)фенилбороновой кислоты (15,7 г), 2M Na2CO3 (100 мл) и н-пропанола (500 мл), пропускали ток азота в течение 15 минут. Затем добавляли 1:3 смесь (3,5 г) Pd(OAc)2 и PPh3, реакционную смесь нагревали до 70 °C и перемешивали в атмосфере азота в течение 8 часов. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом (500 мл) и выливали на воду (2 л) и лед (500 г). Слой этилацетата отделяли и водный слой дополнительно экстрагировали этилацетатом (200 мл). Объединеннные этилацетатные экстракты промывали 0,5н NaOH (2х200 мл), водным NH4Cl, насыщенным солевым раствором и сушили сульфатом магния. После удаления растворителя оставался остаток, который очищали хроматографией на SiO2, используя градиент этилацетата и гексана (от 1:4 до 1:3), и снова ацетоном и толуолом (1:10). Остаток растворяли в горячем гексане (200 мл) и раствору давали остыть до 0°C при перемешивании. Полученное твердое вещество фильтровали и сушили с получением указанного в заголовке соединения.
1H ЯМР (CD3COCD3) δ 7,7 (2H, д), 7,65 (2H, д), 7,6 (2H, д), 7,35 (2H, д), 4,5-4,6 (1Н, м), 3,7 (1H(OH), м), 3,5-3,6 (1H, м), 3,3-3,4 (1H, м), 2,7 (1Н, м), 2,5 (3H, с), 2,3-2,4 (1H(NH), м), 1,65-1,75 (1Н, м), 1,2-1,4 (3H, м), 0, 8-0,9 (6H, дд).
Стадия 5: получение (2S)-4-метил-2-({(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}амино)пентан-1-ола
При 0°C к раствору сульфида (19 г) со стадии 4 в толуоле (400 мл) добавляли Na2WO4·2H2O (0,16 г) и Bu4NHSO4 (0,81 г). Затем медленно добавляли 30%-ную перекись водорода (12,2 мл), смесь перемешивали при комнатной температуре в течение 4,5 часов. Смесь медленно выливали на смесь льда, разбавленного водного тиосульфата натрия и этилацетата. Органический слой отделяли и водный дополнительно экстрагировали этилацетатом (2х100 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили сульфатом магния и растворитель удаляли в вакууме с получением остатка, который очищали на SiO2 с использованием этилацетата и гексана (1:1) в качесте элюента с получением данного продукта.
1H ЯМР (CD3COCD3) δ 8,05 (2H, д), 8,0 (2H, д), 7,85 (2H, д), 7,7 (2H, д), 4,6-4,7 (1Н, м), 3,75 (1H, м), 3,6 (1H, м), 3,35-3,45 (1Н, м), 3,2 (3H, с), 2,7-2,8 (1H, м), 2,35-2,45 (1H, м), 1,7-1,8 (1H, м), 1,2-1,5 (2H, м), 0,8-0,95 (6H, дд).
Стадия 6: получение N-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-лейцина
Суспензию Н5IO6/CrO3 (529 мл 0,44 M в CH3CN; см. примечание ниже) охлаждали до 0°C и добавляли по каплям раствор спирта со стадии 5 (20 г) в CH3CN (230 мл). Смесь перемешивали при 0-5°C в течение 3,5 часов. Смесь выливали в Na2HPO4приpH 4 (1,5 л) при интенсивном перемешивании и смесь экстрагировали диэтиловым эфиром (3х250 мл). Объединеные эфирные экстракты промывали водой и насыщенным солевым раствором (1:1), разбавленным водным NaHSO3 и насыщенным солевым раствором. Органический экстракт сушили сульфатом натрия, фильтровали и растворители выпаривали досуха с получением остатка, который разделяли на две части для последующей очистки.
Вышеуказанную неочищенную кислоту (10 г) растворяли в изопропилацетате (250 мл) и экстрагировали с холодным 0,1н NaOH (3х250 мл). Объединенные экстракты промывали диэтиловым эфиром (250 мл), а затем медленно подкисляли 6н HCl до pH 4. Карбоновую кислоту экстрагировали изопропилацетатом (2х250 мл), слой изопропилацетата сушили и концентрировали с получением продукта высокой степени очистки, использовали как таковой на следующей стадии.
Примечание: окисляющий реагент (H5IO6/CrO3) готовили, как описано в Tetrahedron Letters 39 (1998), 5323-5326, но с использованием CH3CN чистоты ВЭЖХ (содержит 0,5% воды); воду не добавляли.
1H ЯМР (CD3COCD3) δ 8, 05 (2H, д), 7,95 (2H, д), 7,8 (2H, д), 7,65 (2H, д), 4,45-4,55 (1H, м), 3,55-3,6 (1Н, м), 3,2 (3H, с), 2,8-3,0 (шир.м, NH/OH), 1,95-2,05 (1H, м), 1,55-1,6 (2H, м), 0,9-1,0 (6H, м).
Стадия 7: получение N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамида
К ДМФ-ному (200 мл) раствору кислоты со стадии 7 (9 г) добавляли бензотриазол-1-илокситрис(диметиламин)фосфонийгексафторфосфат (11,6 г), гидрохлорид аминацетонитрила (3,94 г) и смесь охлаждали до 0°C. Триэтиламин (9,9 мл) добавляли по каплям, смесь нагревали до комнатной температуры и перемешивали в течение 16 часов. Смесь выливали на лед и насыщенный водный бикарбонат натрия и экстрагировали диэтиловым эфиром (3х100 мл). Объединенные экстракты промывали насыщенным солевым раствором, сушили сульфатом магния и растворитель удаляли в вакууме. Остаток очищали хроматографией на SiO2, используя этилацетат и гексан (1:1). Указанное в заголовке соединение затем перемешивали в диэтиловом эфире в течение 16 часов, фильтровали и сушили (т.пл. 140,5 °C).
1H ЯМР (CD3COCD3) δ 8,0 (2H, д), 7,95 (2H, д), 7,8 (2H, д), 7,65 (2H, д), 4,35-5 4,45 (1H, м), 4,1-4,2 (2H, м), 3,45-3,55 (1H, м), 3,15 (3H, с), 2,65-2,7 (1H, м), 1,85-1,95 (1H, м), 1,4-1,6 (2H, м), 0,85-0,95 (6H, м).
ПРИМЕР 9
Синтез N2-{(1S)-1-[4'-(аминосульфонил)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1 (цианометил)-L-лейцинамида

Стадия 1: получение N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамида
Суспензию Н5IO6/CrO3 (1925 мл 0,44M в СН3CN; см. примечание на стадии 6, примера 8) охлаждали до 0°C и добавляли по каплям раствор (2S)-2-{[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]амино}-4-метилпентан-1-ола со стадии 4 примера 8 (60 г) в CH3CN (1500 мл). Смесь перемешивали при 0-5°C в течении 3,5 часов. Смесь выливали в Na2HPO4, pH 4 (2,5 л) при интенсивном перемешивании и экстрагировали диэтиловым эфиром (3х500 мл). Объединенные эфирные экстракты промывали водой и насыщенным солевым раствором (1:1), разбавленным водным NaHSO3 и насыщенным солевым раствором. Органический экстракт сушили сульфатом натрия, фильтровали и концентрировали в вакууме с получением N-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-L-лейцина, используемого как таковой в последующем соединении с аминоацетонитрилом.
К ДМФ-ному (1500 мл) раствору неочищенной кислоты (46 г) добавляли бензотриазол-1-илокситрис(диметиламин)фосфоний гексафторфосфат (71,5 г), гидрохлорид аминоацетонитрила (25,4 г) и смесь охлаждали до 0°C. Триэтиламин (60,8 мл) добавляли по каплям и смесь нагревали до комнатной температуры, и перемешивали в течение 16 часов. Смесь выливали на лед и насыщенный водный бикарбонат натрия и экстрагировали диэтиловым эфиром (3х300 мл). Объединенные экстракты промывали насыщенным солевым раствором, сушили сульфатом магния и растворитель удаляли в вакууме. Остаток очищали хроматографией на SiO2, используя градиент этилацетата и гексана (1:3 до 1:2) с получением указанного в заголовке соединения, достаточно чистого для использования на следующей стадии.
1H ЯМР (CD3COCD3) δ 7,95-8,05 (шс, NH), 7,6 (2H, д), 7,45 (2H, д), 4,4 (1H, м), 4,1 4,2 (2H, м), 3,4-3,5 (1H, м), 2,6-2,7 (1H, м), 1,8-1,95 (1H, м), 1,4-1,6 (2H, м), 0,85-0,95 (6H, м).
Стадия 2: получение N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4-(4,4,5, 5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]этил}-L-лейцинамида
Ток азота пропускали через ДМФ-ную (700 мл) суспензию N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-лейцинамида со стадии 1 (28,5 г), бис(пинаколато)диборона (23 г) и ацетата калия (24 г) в течение 15 минут с последующим добавлением [1, 1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), комплекса (1:1) с дихлорметаном (2,9 г). Смесь нагревали до 65°C и перемешивали в атмосфере азота в течение 2,5 часов. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом и гексаном (1:1, 300 мл) и выливали на воду (2 л) и лед (500 г). Органический слой отделяли и водный слой дополнительно экстрагировали этилацетатом и гексаном (1:1, 3х200 мл). Объединенные экстракты промывали насыщенным солевым раствором и сушили сульфатом магния. После удаления растворителя оставался остаток, который очищали хроматографией на SiO2, используя этилацетат и гексан (1:2) с получением бороната.
1H ЯМР (CD3COCD3) δ 7,95-8,05 (шс, NH), 7,7-7,8 (2H, д), 7,45-7,55 (2H, д), 4,3-4,4 (1H, м), 4,05-4,15 (2H, м), 3,4-3,5 (1H, м), 2,55-2,65 (1H, м), 1,85-1,95 (1Н, м), 1,45-1,55 (2H, м), 1,15-1,4 (12H, м; в качестве примеси также присутствует некоторое колличество пинакола), 0,85-0,95 (6H, м).
Стадия 3: получение N2-{(1S)-1-[4'-(аминосульфонил)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1 (цианометил)-L-лейцинамида
Ток азота пропускали через суспензию, полученную из бороната со стадии 2 (4 г), 4-бромбензолсульфонамида (3,3 г), 2M Na2CO3 (20 мл) и н-пропанола (100 мл) в течение 15 минут. Затем добавляли 1:3 смесь (0,25 г) Pd(OAc)2 и PPh3, реакционную смесь нагревали до 85°C и перемешивали в атмосфере азота в течение 3 часов. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом (100 мл) и выливали на воду (500 мл) и лед (100 г). Отделяли слой этилацетата и водный слой дополнительно экстрагировали этилацетатом (100 мл). Объединенные этилацетатные экстракты промывали разбавленным водным NaHCO3, насыщенным солевым раствором и сушили сульфатом магния. После удаления растворителя оставался остаток, который очищали хроматографией на SiO2, используя градиент этилацетата, гексана и дихлорметана (2:3:0,1 до 1:1:0,1). Продукт затем перемешивали в диэтиловом эфире в течение 16 часов, фильтровали и сушили с получением указанного в заголовке соединения.
1H ЯМР (CD3COCD3) δ 8-8,1 (3Н, м), 7,9 (2H, д), 7,8 (2H, д), 7,65 (2H, д), 6,6-6,7 (2H, м), 4,4 (1H, м), 4,1-4,2 (2H, м), 3,5 (1Н, м), 2,6-2,7 (1H, м), 1,9 (1H, м), 1,45-1,6 (2H, м), 1,4-1,6 (4H, м), 0,9-1,0 (6H, м).
ПРИМЕР 10
Синтез N1-(1-цианоциклопропил)-N2-{(1S)2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамида

К смеси N-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцина из Примера 8 (0,83 г), О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (0,78 г), циклопропиламина гидрохлорида (0,466 г) в ДМФ (18 мл) при 0°C добавляли триэтиламин (0,9 мл). Смесь выдерживали при комнатной температуре в течение 48 часов, а затем выливали в разбавленный водный хлорид аммония и диэтиловый эфир. Отделяли эфирный слой, а водный дополнительно экстрагировали диэтиловым эфиром. Объединенные эфирные экстракты промывали насыщенным солевым раствором, сушили сульфатом магния и растворитель удаляли в вакууме. Остаток очищали в SiO2, используя этилацетат и гексаны (1:1) в качестве элюента, с последующим разбавлением в диэтиловоым эфире с получением указанного в заголовке соединения.
1H ЯМР (CD3COCD3) δ 8,15 (1H, шс), 8,05 (2H, д), 8,0 (2H, д), 7,8 (2H, д), 7,65 (2H, д), 4,35-4,45 (1Н, м), 3,35-3,45 (1H, м), 3,2 (3H, с), 2,65-2,7 (1H, м), 1,85-1,95 (1H, м), 1,3-1,6 (5H, м), 1,05-1,15 (1H, м), 0,85-0,95 (6H, м).
ПРИМЕР 11
Синтез N1-(цианометил)-N2-[(1S)-2,2,3,3,3-пентафтор-1-(4-пиридин-4-илфенил)пропил]-L-лейцинамида

Стадия 1: получение(4S)-4-изобутил-2-(пентафторэтил)-1,3-оксазолидина
Пентафторпропаналь метил гемиацеталь (14,9 г, 82,8 ммоль) и L-лейцинол (9,7 г, 82,8 ммоль) растворяли в 100 мл бензола и нагревали с обратным холодильником в течение ночи в колбе, снабженной насадкой Дина-Старка. Полученный раствор охлаждали и концентрировали с получением указанного в заголовке соединения в виде масла, которое использовали непосредственно на следующей стадии.
Стадия 2: получение (2S)-2-{[(1S)-1-(4-бромфенил)-2,2,3,3,3-пентафторпропил]амин}-4-метилпентан-1-ола
К -78°C раствору дибромбензола (9,85 г, 41, 8 ммоль) в 100 мл ТГФ добавляли нBuLi (16,5 мл 2,5M раствора гексана, 41,2 ммоль), получая густую суспензию. После перемешивания в течение 10 мин добавляли раствор (4S)-4-изобутил-2-(пентафторэтил)-1, 3-оксазолидина (3,3 г, 13 ммоль) в 3 мл ТГФ, получая темно-коричневый раствор. Раствору давали нагреться до комнатной температуры, затем выливали в насыщенный водный хлорид аммония и экстрагировали эфиром. Очистка с помощью хроматографии на силикагеле (10% до 40% этилацетат/гексана градиента) дала названное соединение в виде одного диастереомера.
Стадия 3: получение N2-[(1S)-1-(4-бромфенил)-2,2,3,3,3-пентафторпропил]-N1-(цианометил)-L-лейцинамида
Суммарный образец (2S)-2-{[(1S)-1-(4-бромфенил)-2,2,3,3, 3-пентафторпропил]амин}-4-метилпентан-1-ола (1,6 г, 4,0 ммоль) преобразовывали в указанное в заголовке соединение, используя способ примера 8, стадии 6 и 7. Очистка с помощью хроматографии на силикагеле (15% - 80% этилацетат/гексана градиент) давала названное соединение в виде твердого вещества.
1H ЯМР (CD3COCD3, 500 МГц) δ 7,8 (1H, шир.), 7,55 (2H, м), 7,40 (2H, м), 4,4-4,5 (1H, м), 3,95 (2H, м), 3,33 (1H, м), 2,75 (1H, м), 1,82 (1H, м), 1,5 (1H, м), 1,38 20 (1H, м), 0,88 (6H, дд).
Стадия 4: получение N1-(цианометил)-N2-[(1S)-2,2,3,3,3-пентафтор-1-(4-пиридин-4-илфенил)пропил]-L-лейцинамида
К раствору N2-[(1S)-1-(4-бромфенил)-2,2,3,3, 3-пентафторпропил]-N1-(цианометил)-L-лейцинамида (82 мг, 0,18 ммоль), 4-пиридилбороновой кислоты (30 мг, 0,24 ммоль) и PdCl2(dppf) (14 мг, 0,02 ммоль) в 2,5 мл ДМФ добавляли 2M Na2CO3 (0,25 мл). Смесь нагревали до 95°C в течение 2,5 ч, затем охлаждали и распределяли между водным Na2CO3 и эфиром. Водную фазу промывали насыщенным солевым раствором и сушили над MgSO4. Очистка хроматографией на силикагеле (65% - 95% этилацетат/гексана градиента) дала названное соединение.
1H ЯМР (CD3COCD3, 500 МГц) δ 8,66 (2H, м), 7,85 (1H, шир.), 7,81 (2H, м), 7,70 (2H, м), 7,62 (2H, м), 4,5-4,6 (1H, м), 3,95 (2H, м), 3,4 (1H, м), 2,81 (1H, м), 1,88 (1Н, м), 1, 55 (1Н, м), 1,42 (1H, м), 0,92 (6H, дд).
ПРИМЕР 12
Синтез N1-(1-цианоциклопропил)-N2-{(1S)-2,2-дифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамида

Стадия 1: получение (2S)-1-{[трет-бутил(диметил)силил]окси}-4-метилпентан-2-амина
Получен, как на стадии 1 Примера 8.
Стадия 2: получение (2S)-1-{[трет-бутил(диметил)силил]окси}-N-[(1E)-2,2-дифторэтилиден]-4-метилпентан-2-амина
Смесь (2S)-1-{[трет-бутил(диметил)силил]окси}-4-метилпентан-2-амина (8,5 г, 36,8 ммоль) и дифторацетальдегида этилгемиацеталя (5,0 г, 39,7 ммоль) в бензоле нагревали с обратным холодильником с насадкой Дина-Старка в течение ночи. Растворитель удаляли в вакууме. Остаток пропускали через короткую колонку с силикагелем и элюировали гексан:EtOAc (10:1) с получением указанного в заголовке соединения в виде бледно-желтого масла.
1H ЯМР (CD3COCD3) δ 7,72 (м, 1H), 6,12 (дт, 1H), 3,70 (дд, 1H), 3,54 (дд, 1H), 3,36 (м, 1H), 1,48 (м, 2H), 1,32 (м, 1H), 0,95 - 0,78 (м, 15H), 0,06 (с, 3H), 0,02 (с, 3H).
Стадия 3: получение (2S)-2-{[(1S)-1-(4-бромфенил)-2,2-дифторэтил]амин}-4-метилпентан-1-ола
н-BuLi (2,5 M в гексане, 1,43 мл) добавляли к -70°C ТГФ (8,5 мл) раствору 1,4-дибромбензола (884 мг) и смесь перемешивали в течение 15 минут. Затем добавляли по капллям ТГФ (8,5 мл) раствор (2S)-1-{[трет-бутил(диметил)силил]окси}-4-метил-N-[(1Е)-2, 2-дифторэтилиден]пентан-2-амина (1,0 г) и смесь перемешивали в течение 1,5 часов. Данную смесь затем медленно выливали в ледяной насыщенный водный раствор хлорида аммония при интенсивном перемешивании и экстрагировали 3 частями этилацетата. Объединенные органические слои промывали насыщенным солевым раствором, сушили сульфатом магния и растворитель удаляли в вакууме с получением остатка, который очищали на SiO2, используя градиент гексана и этилацетата (90:10 - 75:25) в качестве элюента с получением (2S)-N-[(1S)-1-(4-бромфенил)-2, 2-дифторэтил]-1-{[трет-бутил(диметил)силил]окси}-4-метилпентан-2-амина. (2S)-N-[(1S)-1-(4-Бромфенил)-2,2-дифторэтил]-1-{[трет-бутил(диметил)силил]окси}-4-метилпентан-2-амин (200 мг) растворяли в CH3CN (4 мл) и раствор охлаждали до 0°C. Добавляли по каплям HF-пиридин (40 мкл) и смесь взаимодействовала в течение 16 часов. Смесь выливали в насыщенный раствор бикарбоната натрия, добавляли этилацетат и полученную в результате смесь интенсивно встряхивали. Органический слой отделяли, а водный дополнительно экстрагировали этилацетатом (2х50 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили сульфатом магния, растворитель удаляли в вакууме с получением остатка, который очищали на SiO2, используя градиент гексана и этилацетата (80:20 - 60:40) в качестве элюента, с получением указанного в заголовке соединения.
1H ЯМР (CD3COCD3) δ 7,6 (2H, д), 7,45 (2H, д), 6,0 (1H, дт), 4,25 (1H, м), 3,65 (1H, т), 3,5-3,55 (1H, м), 3,3-3,35 (1H, м), 2,55-2,65 (1H, м), 2,15-2,25 (1H, м), 1,6-1,7 (1H, м), 1,3-1,4 (1H, м), 1,2-1,3 (1H, м), 0,9 (3H, д), 0,8 (3H, д).
Стадия 4: получение N-[(1S)-1-(4-бромфенил)-2,2-дифторэтил]-L-лейцина
Суспензию Н5IO6/CrO3 (5,5 мл 0,40M в CH3CN; см. Примечание ниже) охлаждали до 0°C и добавляли по каплям раствор спирта со стадии 3 (250 мг) в CH3CN (3,7 мл). Смесь перемешивали при 0-5°C в течение 3,5 часов. После этого периода добавляли 2,0 мл оксиданта. После 1,5 часов смесь выливали в Na2HPO4 буфер (0,4 г в 10 мл) при интенсивном перемешивании и смесь экстрагировали диэтиловым эфиром (3х20 мл). Объединенные эфирные слои промывали водой и насыщенным солевым раствором (1:1), разбавленным водным NaHSO3 и насыщенным солевым раствором. Органический экстракт сушили сульфатом магния, фильтровали и растворитель выпаривали досуха с получением указанного в заголовке соединения, который использовали без дополнительной очистки.
Примечание: окисляющий реагент (Н5IO6/CrO3) готовили, как описано в Tetrahedron Letters 39 (1998), 5323-5326, но используя CH3CN чистоты ВЭЖХ (содержит 0,5% воды); воду не добавляли.
1H ЯМР (CD3COCD3) δ 7,55 (2H, д), 7,4 (2H, д), 6,05 (1H, дт), 3,95-4,05 (1H, м), 3,45 (1H, т), 2,7-3,0 (шм, NH/OH), 1,85-1,95 (1H, м), 1,5 (2H, т), 0, 95 (3H, д), 0,9 10 (3H, д).
Стадия 5: получение N2-[(1S)-1-(4-бромфенил)-2,2-дифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамида
К ДМФ (2 мл) раствору кислоты со стадии 4 (258 мг) добавляли О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (337 мг) и 1-аминоциклопропанкарбонитрила гидрохлорид (175 мг). После перемешивания в течение 1 мин добавляли по каплям диизопропилэтиламин (0,45 мл) и смесь перемешивали в течение 16 часов. Полученную в результате смесь выливали в насыщенный водный бикарбонат натрия и экстрагировали этилацетатом (3х15 мл). Объединенные экстракты промывали насыщенным солевым раствором, сушили сульфатом магния и растворитель удаляли в вакууме. Остаток очищали хроматографией на SiO2, используя гексан и этилацетат (80:20 - 50:50), получая названное соединение.
1H ЯМР (CD3COCD3) δ 8,05 (1H, м), 7,55 (2H, д), 7,4 (2H, д), 6,05 (1H, дт), 3,95-4,05 (1H, м), 3,25-3,3 (1H, м), 2,4-2,45 (1H, м), 1,8-1,9 (1H, м), 1,4-1,55 (2H, м), 0,95-1,1 (2H, м), 0,95 (6H, т).
Стадия 6: получение N1-(1-цианоциклопропил)-N2-{(1S)-2,2-дифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-лейцинамида
Поток азота пропускали через суспензию арилбромида со стадии 5 (65 мг), 4-(метилтио)фенилбороновой кислоты (40 мг), 2M Na2CO3 (0,22 мл) и ДМФ (1,0 мл) в течение 5 минут. Затем добавляли PdCl2 (dppf), реакционную смесь нагревали до 80°C и перемешивали в атмосфере азота в течение 4 часов. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом (20 мл) и выливали в насыщенный раствор бикарбоната натрия. Этилацетатный слой отделяли и водный дополнительно экстрагировали этилацетатом (2х15 мл). Объединенные этилацетатные экстракты промывали насыщенным солевым раствором и сушили сульфатом магния. Удаление растворителя оставляло остаток, который очищали хроматографией на SiO2 используя градиент гексана и этилацетата (90:10 - 50:50), получая названное соединение.
1H ЯМР (CD3COCD3) δ 8,1 (1H, м), 7,6-7,65 (4H, м), 7,45 (2H, д), 7,35 (2H, д), 6,05 (1H, дт), 3,9-4,0 (1H, м), 3,2-3,3 (1H, м), 2,5 (3H, с), 2,35-2,4 (1H, м), 1,8-1,9 (1H, м), 1,3-1,5 (4H, м), 0,85-1,0 (8H, м).
Стадия 7: получение N1-(1-цианоциклопропил)-N2-{(1S)-2, 2-дифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамида
К раствору сульфида (50 мг) со стадии 6 в толуоле (1,0 мл) и этилацетату (0,1 мл) добавляли Na2WO4·2H2О (1 мг) и Bu4NHSO4 (2 мг). Затем медленно добавляли 30% перекись водорода (30 мкл) и смесь перемешивали при комнатной температуре в течение 1,5 часов. Смесь выливали в разбавленный водный раствор тиосульфата натрия и этилацетат. Органический слой отделяли, а водный дополнительно экстрагировали этилацетатом (2х10 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили сульфатом магния и растворитель удаляли в вакууме с получением остатка, который очищали на SiO2, используя гексан и этилацетат (50:50 - 0:100), затем дихлорметаном и диэтиловым эфиром (90:10) в качестве элюента с получением указанного в заголовке соединения.
1H ЯМР (CD3COCD3) δ 8,15 (1H, м), 8,0 (2H, д), 7,95 (2H, д), 7,75 (2H, д), 7,55 (2H, д), 6,1 (1H, дт), 4,0-4,1 (1H, м), 3,25-3,35 (1H, м), 3,15 (3H, с), 2,4-2,5 (1H, м), 1,8-1,9 (1H, м), 1,4-1,55 (4H, м), 0,85-1,05 (8H, м).
ПРИМЕР 13
Синтез N2-[(1S)-1-(6-хлорпиридин-3-ил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамида

Стадия 1: получение (2S)-2-{[(1S)-1-(6-хлорпиридин-3-ил)-2,2, 2-трифторэтил]амин}-4-метилпентан-1-ола
К раствору 5-бром-2-хлорпиридина (2,5 г, 13 ммоль) в эфире (30 мл) при -78°C добавляли н-бутиллитий (13 ммоль, 2,5M в гексане). Смесь перемешивали при -78°C в течение 1 ч. Добавляли (2S)-1-{[трет-бутил(диметил)силил]окси}-4-метил-M-[(1E)-2,2,2-трифторэтилиден]пентан-2-амин (3,64 г, 11,7 ммоль, см. Стадия 2 Примера 8). Смесь перемешивали при -78°C в течение 2 ч. К реакционной смеси добавляли насыщенный водный NH4Cl и смесь дважды экстрагировали EtOAc. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным MgSO4 и концентрировали до масла (5,3 г). Неочищенное масло (2,0 г) затем обрабатывали (Bu)4NF (6 мл, 1M в ТГФ). Смесь перемешивали при комнатной температуре в течение 1 ч, добавляли насыщенный водный NH4Cl и смесь дважды экстрагировали EtOAc. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным MgSO4 и концентрировали до масла. Хроматография (20% EtOAc/гексан) давала указанное в заголовке соединение.
Стадия 2: получение N-[(1S)-1-(6-хлорпиридин-3-ил)-2,2,2-трифторэтил]-L-лейцина.
Готовили матричный раствор Н5IO6/CrO3 растворением Н5IO6 (68,4 г, 0,3 моль) и CrO3 (138 мг, 1,2 моль%) в CH3CN (684 мл) с получением 0,44M раствора. К раствору Н5IO6/CrO3 (16 ммоль, 36 мл, 0,44M в ТГФ) при -5°C (ледяная и солевая баня) добавляли раствор (2S)-2-{[(1S)-1-(6-хлорпиридин-3-ил)-2,2,2-трифтоэтил]амин}-4-метилпентан-1-ола (1 г) в 3 мл ТГФ по каплям. Следили за внутренней температурой и температуре реакции не давали подниматься выше 0°C. Реакцию контролировали с помощью тонкослойной хроматографии до завершения реакции (3-4 ч). К реакционной смеси добавляли Na2HPO4 (80 мл), который затем экстрагировали EtOAc. Органический экстракт промывали насыщенным солевым раствором, NaHSO3 (120 мл) и снова насыщенным солевым раствором, сушили над безводным MgSO4 и концентрировали до масла. Масло повторно растворяли в EtOAc и фильтровали через тонкий слой силикагеля, элюировали EtOAc и фильтрат концентрировали с получением указанного в заголовке соединения.
Стадия 3: получение N2-[(1S)-1-(6-хлорпиридин-3-ил)-2,2,2-трифторэтил]-N1 -(1-цианоциклопропил)-L-лейцинамида
К раствору неочищенной кислоты (0,73 г, 2,25 ммоль) со стадии 2 в ДМФ (12 мл) добавляли 1-аминоциклопропанкарбонитрила гидрохлорид (0,4 г, 3,37 ммоль) и HATU (0,86 г, 2,25 ммоль). Добавляли диизопропилэтиламин (1,96 мл, 11,24 ммоль), смесь перемешивали при комнатной температуре в течение 20 ч. Добавляли EtOAc и воду. Смесь отделяли после размешивания, органический экстракт промывали водой и насыщенным солевым раствором и сушили над безводным MgSO4. Концентрирование органического экстракта с последующей хроматографией (30-50% EtOAc/гексан) давало указанное в заголовке соединение.
MS (+ESI): 389,3 [M+1]+.
1H ЯМР (CD3COCD3, 500 МГц): δ 0,92 (д, 3H, J = 6,6 Гц), 0,93 (д, 3H, J = 6,6 Гц), 1,00 ( м, 1H), 1,09 (м, 1H), 1,45 (м, 4H), 1,90 (м, 1H), 2,81 (м, 1H), 3,43 (м, 1H), 4,47 (м, 1H), 7,54 (д, 1H, J = 8,3 Гц), 7,98 (д, 1H J = 6,2 Гц), 8,1 (с, 1H), 8,5 (с, 1Н).
ПРИМЕР 14
Синтез N2-{(1S)-1-[6-(4-ацетилфенил)пиридин-3-ил]-2,2,2-трифторэтил}-N1 -(1-цианоциклопропил)-L-лейцинамида

К раствору N2 -[(1S)-1-(6-хлорпиридин-3-ил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-лейцинамида (100 мг, 0,26 ммоль) в толуоле (1,5 мл) и н-пропанолу (0,4 мл) добавляли в потоке азота 4-(ацетил)фенилбороновую кислоту (55 мг, 0,33 ммоль), Pd(PPh3)4 (15 мг, 0,013 ммоль) и Na2CO3 (2M, 0,5 мл). Смесь дегазировали быстрым потоком азота, барботируя через смесь, и смесь нагревали до 150°C в микроволновом реакторе Smith Creator (Personal Chemistry AB, Uppsala, Sweden) в течение 800 сек. Смесь охлаждали, разбавляли EtOAc и промывали водой. Хроматография (50% EtOAc/гексан) давала указанное в заголовке соединение.
MS (+ESI): 473,2 [M+1]+.
1H ЯМР (500 МГц, CD3COCD3): δ 0,93 (д, 3H, J = 6,6 Гц), 0,94 (д, 3H, J = 6,6 Гц), 1,07 ( м, 1H), 1,40 (м, 2H), 1,49 (м, 1H), 1,55 (м, 1H), 1,92 (м, 1H), 2,66 (с, 3H), 2,83 (м, 2H), 3,45 (м, 1H), 4,50 (м, 1Н), 8,15 (м, 5H), 8,30 (д, 2H, J = 8,5 Гц), 8,79 (с, 1H).
ПРИМЕР 15
Синтез N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2, 2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамида
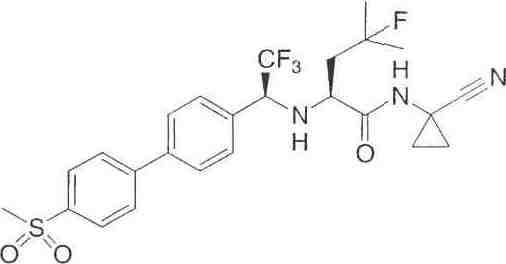
Стадия 1: получение бензил(3S)-3-[(трет-бутоксикарбонил)амино]-4-гидроксибутаноата
N-(трет-Бутоксикарбонил)-L-аспарагировой кислоты 4-бензиловый эфир (30 г) растворяли в диметоксиэтане (90 мл) и раствор охлаждали до -5°C. Добавляли N-метилморфолин (10,32 мл) с последующим медленным добавлением изобутилхлорформиата (12,66 мл) так, чтобы температура реакции удерживалась ниже -10°C. Смесь выдерживали в течение 0,5 часов. Твердые вещества быстро фильтровали и промывали диметоксиэтаном (90 мл). Фильтрат охлаждали до -50°C, медленно добавляли раствор борогидрида натрия (4,4 г) в воде (45 мл) так, чтобы температура реакции удерживалась между -30 и -15°C. Затем добавляли воду (500 мл) так, чтобы температура реакционной смеси удерживалась ниже -15°C. Суспензию фильтровали, твердое вещество промывали водой (400 мл) и сушили с получением бензил(3S)-3-[(трет-бутоксикарбонил)амин]-4-гидроксибутаноата.
1H ЯМР (CD3COCD3) δ 7,3-7,45 (5H, м), 5,85-5,95 (1H, NH), 5,15 (2H, с), 3,95-4,1 (2H, м), 3,5-3,7 (2H, м), 2,55-2,75 (2H, м), 1,4 (9H, с).
Стадия 2: получение бензил[(4S)-2-оксо-1,3-оксазолидин-4-ил]ацетата
К раствору спирта (95,7 г) со стадии 1 в дихлорэтане (925 мл) добавляли пиридин (625 мл) и смесь охлаждали до 0-5°C. Добавляли безводный п-толуолсульфоновый ангидрид (105,7 г) и смесь нагревали до комнатной температуры и перемешивали в течение 1 часа, а затем нагревали до 90°C в течение 2 часов. Смесь охлаждали, разбавляли дихлорметаном (1000 мл) и промывали 1н HCl (3х600 мл). Органический слой промывали насыщенным солевым раствором, сушили сульфатом натрия и растворители удаляли в вакууме. Остаток очищали хроматографией на SiO2 используя этилацетат и гексан в соотношении 1:1, затем этилацетатом с получением бензил[(4S)-2-оксо-1, 3-оксазолидин-4-ил]ацетата.
1H ЯМР (CD3SOCD3) δ 7,8 (1H, NH), 7,3-7,45 (5H, м), 5,05-5,15 (2H, м), 4,4-4,5 (1H, м), 4,1-4,2 (1H, м), 4,0-4,05 (1H, м), 3,6-3,8 (2H, м).
Стадия 3: получение (4S)-4-(2-гидрокси-2-метилпропил)-1,3-оксазолидин-2-она
Бромид метилмагния (227 мл 3M раствора в диэтиловом эфире) добавляли к смеси толуола (340 мл) и ТГФ (340 мл) при -20°C. Затем добавляли по каплям теплый ТГФ-ный раствор (170 мл) эфира со стадии 2 (40 г), удерживая температуру ниже -10°C. Смесь выдерживали в течение 2 часов, а затем медленно добавляли к смеси воды (1000 мл) и уксусной кислоты (200 мл) и полученную в результате смесь перемешивали в течение 2 часов при комнатной температуре. Отделяли водный слой, а органический слой экстрагировали водой (2х200 мл). Продукт экстрагировали из объединенных водных слоев, используя дихлорметан и экстракционный аппарат непрерывного действия. Дихлорметановый экстракт выпаривали досуха, используя гептан в качестве со-растворителя для азеотропной отгонки уксусной кислоты. Остаток очищали хроматографией на SiO2, используя этанол и дихлорметан (1:30), с получением (4S)-4-(2-гидрокси-2-метилпропил)-11,3-оксазолидин-2-она.
1H ЯМР (CD3COCD3) δ 6, 1-6,4 (1H, NH), 4,45-4,55 (1H, м), 4,1-4,2 (1H, м), 3,95-4,05 (1H, м), 3,7 (1H, с), 1,65-1,85 (2H, м), 1,25 (6H, м).
Стадия 4: получение (4S)-4-(2-фтор-2-метилпропил)-1,3-оксазолидин-2-она
Дихлорметановый раствор (100 мл) спирта (47,8 г) со стадии 3 добавляли к раствору трифторид(диэтиламин)серы (48,8 г) в дихлорметане (500 мл) при -70°C. Смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Затем смесь осторожно добавляли к смеси при 0°C насыщенного водного NaHCO3 (800 мл). Органический слой отделяли и промывали насыщенным водным NaHCO3. Водный слой дополнительно экстрагировали дихлорметаном (100 мл) и объединенные дихлорметановые слои сушили и концентрировали. Остаток очищали хроматографией на SiO2 используя этилацетат и гексан (1:5), затем этилацетатом с получением (4S)-4-(2-фтор-2-метилпропил)-1,3-оксазолидин-2-она.
1H ЯМР (CD3SOCD3) δ 7,6 (1H, NH), 4,4-4,5 (1H, м), 3,95-4,05 (1H, м), 3,9-3,95 (1H, м), 1,8-1,95 (2H, м), 1,25-1,4 (6H, 2с).
Стадия 5: получение (2S)-2-амино-4-фтор-4-метилпентан-1-ола
К раствору фторпроизводного (21,0 г) со стадии 4 в 90% водном этиловом спирте (216 мл) добавляли гидроксид калия (21,9 г). Смесь нагревали с обратным холодильником в течение 4 часов и охлаждали до комнатной температуры. Затем смесь концентрировали и совместно выпаривали с толуолом (3х300 мл). Остаток растворяли в дихлорметане (500 мл) и перемешивали в течение 0,5 часа. Суспензию фильтровали через целит и целит промывали дихлорметаном (3х100 мл). Фильтрат концентрировали досуха с получением (2S)-2-амино-4-фтор-4-метилпентан-1-ола.
1H ЯМР (CD3OD) δ 3,4-3,5 (1H, м), 3,2-3,3 (1H, м), 3,0-3,1 (1H, м), 1,5-1,7 (2H, м), 1,35 (3H, с), 1,3 (3H, с).
Стадия 6: получение (2S)-1-{[трет-бутил(диметил)силил]окси}-4-фтор-4-метилпентан-2-амина
Аминоспирт (21,0 г) со стадии 5 растворяли в дихлорметане (300 мл) и раствор охлаждали до 0°C. Добавляли 4-(диметиламин)пиридин (0,051 г) и хлорид трет-бутилдиметилсилила (21 г), затем триэтиламин (25 мл). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь медленно выливали в 0°C насыщенный водный хлорид аммония и экстрагировали дихлорметаном (3х300 мл). Органический слой промывали насыщенным солевым раствором, сушили сульфатом натрия, а растворители удаляли в вакууме с получением (2S)-1-{[трет-бутил(диметил)силил]окси}-4-фтор-4-метилпентан-2-амина.
1H ЯМР (CD3OD) δ 3,6-3,65 (1H, м), 3,4-3,5 (1H, м), 3,1-3,2 (1H, м), 1,6-1,8 (2H, м), 1,35-1,45 (6H, м), 0,93 (9H, с), 0,1 (6H, с).
Стадия 7: получение (2S)-1-{[трет-бутил(диметил)силил]окси}-4-фтор-4-метил-N-[(1E)-2,2,2-трифторэтилиден]пентан-2-амина
К раствору амина (31,5 г) со стадии 6 в бензоле (126 мл) добавляли трифторацетальдегид метилгемиацеталя (21,6 мл.). Раствор нагревали с обратным холодильником в течение ночи, используя насадку Дина-Старка для сбора воды. Реакционную смесь охлаждали до комнатной температуры и концентрировали досуха. Остаток очищали на SiO2, используя 4% этилацетата в гексане с получением (2S)-1-{[трет-бутил(диметил)силил]окси}-4-фтор-4-метилпентан-2-амина.
1H ЯМР (CD3COCD3) δ 7,9-7,95 (1H, м), 3,75-3,85 (1H, м), 3,7-3,75 (1H, м), 3,53-3,6 (1H, м), 1,9-2,0 (2H, м), 1,3-1,4 (6H, м), 0,9 (9H, с), 0,1 (3H, с), 0,05 (3H, с).
Стадия 8: получение (2S)-2-{[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]амин}-4-фтор-4-метилпентан-1-ола
К -75°C раствору 1, 4-дибромбензола (0,26 г) в ТГФ (4 мл) добавляли н-BuLi (0,42 мл 2,5M раствора гексана) и смесь выдерживали в течение 20 минут. Добавляли имин (0,329 г) со стадии 7 в ТГФ (2 мл) и смесь выдерживали 2 часа. Затем смесь добавляли к смеси воды (50 мл), NH4Cl (1 г) и дробленного льда. Смесь экстрагировали этилацетатом (2х25 мл) и объединенные этилацетатные слои сушили и выпаривали досуха.
Ту же процедуру повторяли в большем масштабе, используя 1,4-дибромбензол (1,2 г), н-BuLi (1,84 мл) и имин (1,38 г), и реакционную смесь обрабатывали, как указано выше. Объединенные остатки двух приготовлений растворяли в ТГФ (10 мл) и охлаждали до 0°C. Добавляли фторид н-тетрабутиламмония (6 мл из 1M раствора ТГФ), смесь перемешивали при +5°C в течение 16 ч. Смесь выливали в смесь воды (50 мл), хлорида аммония (1 г) и дробленного льда и отделяли органический слой. Водный слой дополнительно экстрагировали этилацетатом (2х15 мл), объединенные органические слои сушили и концентрировали. Остаток очищали на SiO2, используя этилацетат и гексан (1:5), с получением (2S)-2-{[(1S)-1-(4-бромфенил)-2,2, 2-трифторэтил]амин}-4-фтор-4-метилпентан-1-ола.
1H ЯМР (CD3COCD3) δ 7,65 (2H, м), 7,5 (2H, м), 4,5-4,6 (1H, м), 3,8 (1H, м), 3,6 (1H, м), 3,3-3, 4 (1H, м), 2,85-2,0 (1H, м), 2,55 (1H, м), 1,7-1,9 (2H, с), 1,3-1,4 (6H, м).
Стадия 9: получение N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамида
Суспензию Н5IO6/CrO3 (66 мл 0,44M в CH3CN; см. примечание) охлаждали до 0°C и добавляли по каплям раствор спирта со стадии 8 (1,55 г) в CH3CN (5 мл). Смесь перемешивали при 0-5°C в течение 3,5 часов. Смесь выливали в Na2HPO4, pH 4 (200 мл) при интенсивном перемешивании и смесь экстрагировали диэтиловым эфиром (3х50 мл). Объединенные эфирные экстракты промывали водой и насыщенным солевым раствором (1:1), затем разбавленным водным NaHSO3 и насыщенным солевым раствором. Смесь сушили сульфатом натрия, фильтровали и растворители выпаривали досуха с получением N-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-4-фтор-L-лейцина, который использовали как таковой на следующей стадии.
Примечание: окисляющий реагент (Н5IO6/CrO3) готовили, как описано в Tetrahedron Letters 39 (1998), 5323-5326, но используя CH3CN чистоты ВЭЖХ (содержит 0,5% воды); воду не добавляли.
Диизопропилэтиламин (4,2 мл) добавляли к 0°C суспензии кислоты (1,5 г), указанной выше, 1-амино-1-циклопропанкарбонитрила гидрохлорида (1,18 г), О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (1,94 г) и диметилформамида (5 мл), смесь взаимодействовала при комнатной температуре в течение 48 ч. Затем смесь выливали на лед и разбавленный водный хлорид аммония. Смесь экстрагировали этилацетатом и эфиром (1:1) и объединенные органические слои промывали разбавленным Na2HPO4, pH 3, и насыщенным солевым раствором. Растворители выпаривали досуха и остаток очищали хроматографией на SiO2, используя этилацетат и гексан (1:2), с получением N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4-фтор-L-лейцинамида достаточной степени чистоты для следующей стадии.
1H ЯМР (CD3COCD3) δ 8,15 (1H, NH), 7,6 (2H, м), 7,45 (2H, м), 4,35-4,45 (1H, м), 3,45-3,55 (1H, м), 1,9-2,1 (2H, м), 1,75-1, 85 (1H, NH),1,35-1,55 (8H, м), 1,1-1,15 (1H,м), 0,95-1,05 (1H, м).
Стадия 10: получение N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2, 2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-лейцинамида
Поток азота пропускали через суспензию бромида со стадии 9 (0,338 г), 4-(метилтио)фенилбороновой кислоты (0,252 г), 2M водного Na2CO3 (0,8 мл) и ДМФ (4 мл) в течение 15 минут. Затем добавляли PdCl2 dppf (0,3 г) и реакционную смесь нагревали до 85°C и перемешивали в атмосфере азота в течение 5 часов. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом (10 мл) и выливали в воду (50 мл) и лед. Отделяли слой этилацетата, а водный дополнительно экстрагировали этилацетатом. Объединенные этилацетатные экстракты сушили и растворители удаляли в вакууме. Остаток очищали хроматографией на SiO2, используя этилацетат и гексан (1:2), с получением N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-лейцинамида.
1H ЯМР (CD3COCD3) δ 8,15 (1H, NH), 7,1-7,2 (4H, м),7,5-7,55 (2H, м), 7,35-7,4 (2H, м), 4,3-4,4 (1Н, м), 3,45-3,55 (1H, м), 2,75-2,8 (1H, NH), 2,5 (3H, с), 1,9-2,05 (2H, м), 1,3-1,5 (8H, м), 1,0-1,1 (1H, м), 0, 85-0,95 (1H, м).
Стадия 11: получение N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамида
К 0°С раствору сульфида (0,265 г) со стадии 10 в толуоле (5 мл) и дихлорметана (5 мл) добавляли Na2WO4·2H2O (0,002 г) и н-Bu4NHSO4 (0,01 г). Затем медленно добавляли 30% перекись водорода (0,137 мл) и смесь перемешивали при комнатной температуре в течение 3 часов. Смесь медленно выливали на смесь льда, разбавленного водного тиосульфата натрия и этилацетата. Органический слой отделяли, а водный дополнительно экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным солевым раствором, сушили сульфатом магния и растворитель удаляли в вакууме с получением остатка, который очищали на SiO2, используя этилацетат, гексан и дихлорметан (1:1:0,1) в качестве элюента. Остаток растирали в порошок в диэтиловом эфире с получением N1-(1-цианоциклопропил)-4-фтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-лейцинамида.
1H ЯМР (CD3COCD3) δ 8,2 (1H, NH), 8,05-8,1 (2H, м), 7,95-8,0 (2H, м), 7,8 (2H, м), 7,65 (2H, м), 4,35-4,45 (1H, м), 3,5-3,6 (1H, м), 3,2 (3H, с), 2,8-2,9 (1H, NH), 1,9-2,1 (2H, м), 1,3-1,5 (8H, м), 1,05-1,15 (1H, м), 0,9-1,0 (1H, м).
ПРИМЕР 16
Синтез (4S)-N1 -(1-цианоциклопропил)-5,5,5-трифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамида

Стадия 1: получение соли 5,5,5-трифторлейцина гидрохлорида
Рацемическую диастереомерную смесь этил-N-бензоил-5,5,5-трифторлейцината (10,0 г, 31,5 ммоль), полученную в соответствии с методикой Ojima et al. (J. Org. Chem., 1989, 54, 4511-4522), нагревали с обратным холодильником в 6M водной HCl (100 мл) в течение 16 ч. После охлаждения смесь промывали Et2O и концентрировали в вакууме с получением рацемической диастереомерной смеси 5,5,5-трифторлейцин HCl соли.
1H ЯМР (метанол-d4) δ 4,10 (м, 1H), 2,65 (м, 1H), 2,35 - 1,80 (м, 2H), 1,25 (м, 3H).
Стадия 2: метил(4S)-N-[(бензилокси)карбонил]-5, 5,5-трифтор-L-лейцинат и метил(4R)-N-[(бензилокси)карбонил]-5,5,5-трифтор-L-лейцинат
К холодному (0°C) раствору соли 5,5,5-трифторлейцин·HCl (со стадии 1 выше) в H2O (30 мл) добавляли 1M водный NaOH (60 мл, 60 ммоль), затем уксусный ангидрид (3,5 мл, 36,7 ммоль). Смесь перемешивали при комнатной температуре в течение 30 мин - 1 ч. После подкисления 6M водной HCl (6 мл) смесь экстрагировали EtOAc (6x). Объединенные EtOAc экстракты промывали насыщенным солевым раствором, сушили (MgSO4) и концентрировали. Остаток разводили гексаном:Et2O (1:1) с получением N-ацетил-5,5,5-трифторлейцина в виде белого твердого вещества.
К суспензии N-ацетил-5,5,5-трифторлейцина (4,2 г, 18,5 ммоль) в H2О (35 мл) добавляли 1M водный NaOH (18,5 мл, 18,5 ммоль), смесь перемешивали в течение 15 - 30 мин с получением гомогенного раствора. Добавляли ацилазу I (EC 3.5.1.14, от Sigma, Cat. # A 3010; 55 мг), смесь перемешивали при комнатной температуре в течение ночи. ЯМР небольшой аликвоты (выпаренной в вакууме) неочищенного вещества показал соотношение 53:47 для исходного вещества и продукта. Затем смесь подкисляли 6M водной HCl (˜3,5 мл) и экстрагировали EtOAc (4x, каждую EtOAc экстракцию промывали небольшим количеством H2О). Объединенные EtOAc экстракты промывали насыщенным солевым раствором, сушили (MgSO4) и концентрировали с получением неочищенного N-ацетил-5,5,5-трифтор-D-лейцина в виде бледно-желтого твердого вещества, [α]D = + 27,6° (c 1, 5, EtOH). Водный слой концентрировали в вакууме и сушили в вакууме в течение ночи с получением 5,5,5-трифтор-L-лейцина, возможно загрязненного NaCl и HCl солями, [α]D = -4,1° (c 0,77, H2O).
К перемешанному раствору 5,5,5-трифтор-L-лейцина (12 г) в H2О (150 мл) при 0°C добавляли бензилхлорформиат (4,8 мл, 34 моль) с последующим добавлением по каплям 1M водного NaOH (120 мл, 120 ммоль). Дополнительно добавляли бензилхлорформиат (4,8 мл, 34 ммоль). Смесь дополнительно перемешивали при 0°C и pH смеси становилась ˜ 7. Смесь промывали Et2O (2x) и подкисляли водной HCl. Водный слой экстрагировали EtOAc (3x), сушили (Na2SO4) и концентрировали в вакууме с получением N-[(бензилокси)карбонил]-5,5,5-трифтор-L-лейцина. Неочищенную кислоту растворяли в Et2O, обрабатывали раствором диазометана в Et2O. Хроматография через силикагель и элюция гексаном:Et2O (7:3) давала метил(4S)-N-[(бензилокси)карбонил]-5,5,5-трифтор-L-лейцинат в виде менее полярной фракции.
1H ЯМР (ацетон-d6) δ 7,45 - 7,25 (м, 5H), 6,86 (д, 1H), 5,10 (м, 2H), 4,38 (м, 1H), 3,70 (с, 3H), 2,45 (м, 1H), 2,05 (м, 1H), 1,85 (м, 1H), 1,16 (д, 3H).
Дополнительная элюция давала (4R)-N-[(бензилокси)карбонил]-5,5,5-трифтор-L-лейцинат в виде более полярной фракции, загрязненной небольшим количеством бензилового спирта.
1H ЯМР (ацетон-d6) δ 7,40 - 7,25 (м, 5H), 6,86 (д, 1H), 5,08 (с, 2H), 4,35 (м, 1H), 3,70 (с, 3H), 2,54 (м, 1Н), 2,20 (м, 1H), 1,75 (м, 1H), 1,16 (д, 3H).
Стадия 3: (2S, 4S)-2-амино-5,5,5-трифторо-4-метилпентан-1-ол
К раствору метил(4S)-N-[(бензилокси)карбонил]-5,5,5-трифтор-L-лейцината (5,4 г, 16,2 ммоль) в EtOH (150 мл) при комнатной температуре добавляли LiCl (2,8 г, 66 моль) и смесь перемешивали в течение 10 - 15 мин с последующим добавлением NaBH4 (2,5 г, 66 ммоль). Смесь перемешивали при комнатной температуре в течение 6 ч. После разбавления H2O (60 мл) смесь гасили 6M водной HCl (18 мл). Добавляли дополнительное количество H2O и смесь экстрагировали EtOAc (2x). Объединенные EtOAc экстракты промывали насыщенным солевым раствором, сушили (Na2SO4) и концентрировали с получением неочищенного бензил(1S,3S)-4,4,4-трифтор-1-(гидроксиметил)-3-метилбутилкарбамата.
Вышеуказанный спирт растворяли в EtOH (150 мл) и добавляли 10% Pd/C (˜500 мг). Смесь перемешивали в атмосфере Н2 (баллон) в течение ночи. Катализатор отфильтровывали через целит и фильтрат концентрировали с получением указанного в заголовке соединения в виде бесцветного масла.
1H ЯМР (метанол-d4) δ 3,48 (дд, 1H), 3,38 (дд, 1H), 2,85 (м, 1H), 2,50 (м, 1H), 25 1,62 - 1,40 (м, 2H), 1,12 (д, 3H).
Стадия 4: (2S,4S)-1-{[трет-бутил(диметил)силил]окси}-5,5,5-трифтор-4-метил-N-[(1E)-2,2, 2-трифторэтилиден]пентан-2-амин
(2S,4S)-2-Амин-5,5,5-трифтор-4-метилпентан-1-ол (2,6 г, 15,2 ммоль) преобразовывали в указанное в заголовке соединение, как описано в стадиях 1 и 2 примера 8.
1H ЯМР (ацетон-d6) δ 7,98 (м, 1H), 3,80 (м, 1H), 3,60 (м, 2H), 2,18 (м, 1H), 1,98 (м, 1H), 1,65 (м, 1H), 1,12 (д, 3H), 0,88 (с, 9H), 0,06 (с, 3H), 0,02 (с, 3H).
Стадия 5: (2S,4S)-2-{[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]амино}-5,5,5-трифтор-4-метилпентан-1-ол
К холодному (-78° C) раствору 1,4-дибромбензола (7,0 г, 29,7 ммоль) в Et2О (75 мл) добавляли по каплям раствор 2,5 M н-BuLi в гексане (7,0 мл, 17,5 ммоль) и смесь перемешивали в течение 2 ч при -78°C. Добавляли раствор (2S,4S)-1-{[трет-бутил(диметил)силил]окси}-5,5,5-трифтор-4-метил-N-[(1Е)-2,2,2-трифторэтилиден]пентан-2-амина (3,7 г, 10,1 ммоль) в небольшом количестве Et2O и смесь дополнительно перемешивали при -78°C в течение дополнительного 1 ч. Затем смесь гасили H2O, экстрагировали EtOAc, сушили (MgSO4) и концентрировали.
Вышеуказанный сырой продукт растворяли в ТГФ (20 мл) и добавляли HOAc (0,3 мл). После добавления раствора 1M тетрабутиламмония фторида в ТГФ (20 мл, 20 ммоль), смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли в вакууме, остаток разводили H2O и экстрагировали EtOAc. EtOAc экстракт промывали насыщенным солевым раствором, сушили (MgSO4) и концентрировали. Хроматография через силикагель и элюция гексан:EtOAc (4:1) давали указанное в заголовке соединение в виде бледно-желтого масла.
1H ЯМР (ацетон-d6 ) δ 7,60 (д, 2H), 7,48 (д, 2H), 4,58 (м, 1H), 3,80 (т, 1H), 3,45 20 (м, 2H), 2,90 (м, 1H), 2,70 (м, 1H), 2,25 (м, 1H), 1,75 (м, 1H), 1,45 (м, 1H), 1,14 (д, 3H).
Стадия 6: (4S)-N-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-5,5,5-трифтор-L-лейцин
Указанное в заголовке соединение получали, как описано на стадии 9 Примера 15, из (2S,4S)-2-{[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]амин}-5,5,5-трифтор-4-метилпентан-1-ола.
1H ЯМР (ацетон-d6) δ 7,58 (д, 2H), 7,46 (д, 2H), 4,46 (м, 1H), 3,58 (дд, 1H), 2,80 (м, 1H), 1,92 (м, 1H), 1,72 (м, 1H), 1,20 (д, 3H).
Стадия 7: (4S)-N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1 -(1-цианоциклопропил)-5,5,5-трифтор-L-лейцинамид
Указанное в заголовке соединение получали, как описано на стадии 9 Примера 15, из (4S)-N-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-5,5, 5-трифтор-L-лейцина.
1H ЯМР (ацетон-d6) δ 8,20 (шир.с, 1H), 7,59 (д, 2H), 7,43 (д, 2H), 4,34 (м, 1H), 3,48 (м, 1H), 2,78 (м, 1H), 1,85 (м, 1H), 1,55 (м, 1H), 1,39 (м, 2H), 1,14 (д, 3H), 1,15 - 0,90 (м, 2H).
MS (+ESI): 486,488 [M+1]+.
Стадия 8: (4S)-N1-(1-цианоциклопропил)-5, 5,5-трифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-лейцинамид
Названное соединение получали, как описано на Стадиях 10 и 11 Примера 15, из (4S)-N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-5,5,5-трифтор-L-лейцинамида.
[α]D = +66° (c 0,5, ацетон).
1H ЯМР (ацетон-d6) δ 8,20 (шир.с, 1H), 8,03 (д, 2H), 7,94 (д, 2H), 7,78 (д, 2H), 7,62 (д, 2H), 4,41 (м, 1H), 3,52 (м, 1H), 3,17 (с, 3H), 2,88 (м, 1H), 1,98 (м, 1H), 1,58 (м, 1H), 1,35 (м, 2H), 1,16 (д, 3H), 1,15 - 0,85 (м, 2H).
MS (+ESI): 562 [M+1]+.
ПРИМЕР 17
Синтез N-бензил-1-(бензилокси)-4-фтор-4-метилпентан-2-амина
Стадия 1: получение N-(трет-бутоксикарбонил)-4-метиленнорвалина
К раствору дегидро-L-лейцина (2,00 г, 15,48 ммоль) и ди-трет-бутилдикарбоната (10,14 г, 46,4 ммоль) в ТГФ (ок. 100 мл) и воде (ок. 50 мл) добавляли триэтиламин (12,94 мл, 92,8 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Добавляли лимонную кислоту (ок. 100 мл 1M водного раствора) и продукт экстрагировали примерно в 400 мл метиленхлорида. Органическую фазу сушили сульфатом натрия, фильтровали и концентрировали роторным выпариванием. Очистка хроматографией на силикагеле с использованием 0-10% метанола в дихлорметане в качестве элюента давала указанное в заголовке соединение.
Стадия 2: получение трет-бутил-1-(гидроксиметил)-3-метилбул-3-енилкарбамата
К раствору Boc-дегидро-L-лейцина (2,84 г, 12,4 ммоль) и 4-метилморфолина (1,36 мл, 12,4 ммоль) в сухом ТГФ (40 мл), уравновешенному к -10°C, добавляли по каплям изобутилхлорформиат (1,61 мл, 12,4 ммоль). Реакционной смеси давали перемешаться в течение 30 минут и осадок удаляли фильтрацией. Фильтрат уравновешивали при 0°C и при перемешивании добавляли по каплям раствор борогидрида натрия (0,938 г, 24,8 ммоль) примерно в 10 мл воды. Реакционной смеси давали вернуться к температуре окружающей среды и перемешивали в течение дополнительного часа. Реакцию гасили насыщенным водным бикарбонатом натрия и продукт экстрагировали в этилацетат. Органический слой сушили сульфатом магния, фильтровали и концентрировали с помощью роторного испарителя с получением указанного в заголовке соединения.
MS (+ESI): 216,1 [M-boc-+1]+ .
Стадия 3: получение трет-бутилбензил{1-[(бензилокси)метил]-3-метилбут-3-енил}карбамата
К раствору Boc-дегидро-L-лейцинола (2,56 г, 11,91 ммоль) и бензилбромида (3,54 мл, 29,8 ммоль) в ДМФ (50 мл) добавляли гидрид натрия (1,19 г 60% дисперсии в минеральном масле, 29,8 ммоль) и реакцию оставляли перемешиваться в течение 3 часов. Добавляли дополнительное количество бензилбромида (3,54 мл) и дисперсии гидрида натрия (1,19 г) и реакционную смесь оставляли перемешиваться в течение дополнительных 16 часов. Добавляли воду (ок. 100 мл) и продукт экстрагировали дважды в дихлорметане (100 мл). Органические фазы объединяли и промывали дважды примерно 100 мл воды. Органические фазы сушили сульфатом магния, фильтровали и концентрировали с помощью роторного испарителя. Очистка с помощью хроматографии на силикагеле с использованием градиента 0-5% этилацетата в гексане давала указанное в заголовке соединение.
Стадия 4: получение N-бензил-1-(бензилокси)-4-фтор-4-метилпентан-2-амина
Раствор 70% гидрофторида в пиридине (3,75 мл) уравновешивали к 0°C в полипропиленовом резервуаре. Добавляли раствор бензилового эфира N-бензил-boc-(L)-лейцинола (1,52 г, 3,85 ммоль) и реакционную смесь оставляли перемешиваться в течение 6 часов. Реакционный сосуд затем уравновешивали на ледяной бане и оставляли для протекания реакции на 5 дней. Реакцию гасили добавлением ледяной воды и продукт экстрагировали в метиленхлорид. Очистка хроматографией на силикагеле с использованием градиента 0-10% метанола в дихлорметане давала указанное в заголовке соединение.
MS (+ESI): 316,0 [M+1]+.
ПРИМЕР 18
Синтез цианометиламида (2S)-5,5,5-трифтор-2-({(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}амино)пентановой кислоты

Стадия 1: получение метилового эфира 2-бензилоксикарбониламино-5,5, 5-трифтор-пент-2-еноевой кислоты
Триметиловый эфир N-(бензилоксикарбонил)-α-фосфонглицина и DBU растворяли в сухом метиленхлориде (50 мл) и реакционную смесь охлаждали до -30°C. 3,3,3-Трифторпропаналь (1 экв.) добавляли по каплям к охлажденному и перемешанному раствору и реакционную смесь оставляли перемешиваться в течение дополнительного часа при -30°C, а затем в течение ночи при температуре окружающей среды. Добавляли метиленхлорид (ок. 100 мл) и органическую фазу промывали 1н HCl (ок. 100 мл), затем насыщенным солевым раствором (ок. 100 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали с помощью роторного испарителя. Этот сырой продукт очищали с помощью хроматографии на силикагеле с градиентом 10-30% этилацетата в гексане с получением метилового эфира 2-бензилоксикарбониламин-5,5,5-трифторпент-2-еноевой кислоты в виде белого кристаллического твердого вещества.
MS (+ESI): 318,0 [M+1]+.
Стадия 2: получение метилового эфира (S)-2-бензилоксикарбониламин-5,5,5-трифторпентановой кислоты
Метиловый эфир 2-бензилоксикарбониламино-5,5,5-трифторпент-2-еноевой кислоты (15,37 г, 48,5 ммоль) растворяли в абсолютном этаноле (100 мл) в гидрогенизационном сосуде Парра. Раствор барботировали потоком азота, а затем добавляли (+)Duphos (350 мг). Реакционную смесь помещали в гидрогенизационный аппарат Парра и выкачивали воздух из свободного пространства над смесью, а затем создавали давление 50 фунтов/кв. дюйм водорода. Этот процесс повторяли семь раз, а затем сосуд подвергали давлению 50 фунтов/кв. дюйм водорода и перемешивали на аппарате Парра в течение ночи. Реакционную смесь затем концентрировали с помощью роторного испарения, растворяли в 1:1 этилацетат:гексане и фильтровали через слой диоксида кремния для удаления катализатора. Фильтрат концентрировали с помощью роторного испарителя с получением неочищенного метилового эфира (S)-2-бензилоксикарбониламино-5,5,5-трифторпентановой кислоты, который переносили без дополнительной очистки.
Стадия 3: получение бензилового эфира (S)-(4,4,4-трифтор-1-гидроксиметилбутил)карбаминовой кислоты
Метиловый эфир (2S)-2-бензилоксикарбониламино-5,5,5-трифторпентановой кислоты со стадии 2 растворяли в сухом ТГФ (400 мл). Раствор LiBH4 (2,11 г в 100 мл сухого ТГФ) добавляли по каплям при перемешивании и раствор оставляли перемешиваться при температуре окружающей среды в течение ночи. Реакционную смесь концентрировали с помощью роторного испарителя и добавляли 400 мл воды. Затем доводили pH до 2 добавлением концентрированной HCl, а затем продукт экстрагировали в этилацетат. Органическую фазу промывали дважды водой, сушили с помощью MgSO4, фильтровали и концентрировали с помощью роторного испарителя с получением неочищенного бензилового эфира (S)-(4, 4,4-трифтор-1-гидроксиметилбутил)карбаминовой кислоты.
MS (-ESI): 290,2 [M-1]+.
Стадия 4: получение бензилового эфира [1-(трет-бутилдиметилсиланилоксиметил)-4,4,4-трифторбутил]карбаминовой кислоты
К раствору бензилового эфира (S)-(4,4,4-трифтор-1-гидроксиметилбутил)карбаминовой кислоты (13,89 г, 47,7 ммоль) и триэтиламина (7,32 мл, 52,5 ммоль) в ДМФ (60 мл) добавляли по каплям, при перемешивании, раствор трет-бутилдиметилсилилхлорида (7,91 г в 40 мл ДМФ) при температуре окружающей среды. Раствор оставляли перемешиваться в течение ночи при температуре окружающей среды. Реакционную смесь затем концентрировали с помощью роторного испарения и добавляли этилацетат. Органическую фазу дважды промывали водой, сушили с использованием MgSO4, фильтровали и концентрировали с помощью роторного испарения. Сырой продукт очищали с помощью хроматографии на силикагеле, используя 10% этилацетат в гексане в качестве элюента, с получением бензилового эфира [1-(трет-бутилдиметилсиланилоксиметил)-4,4,4-трифтор-бутил]карбаминовой кислоты в виде белого кристаллического твердого вещества.
MS (+ESI): 406,2 [M+1]+.
Стадия 5: получение (2S)-1-{[трет-бутил(диметил)силил]окси}-5,5,5-трифторпентан-2-амина
Бензиловый эфир [1-(трет-бутилдиметилсиланилоксиметил)-4,4,4-трифторбутил]карбаминовой кислоты растворяли в абсолютном этаноле (100 мл) в гидрогенизационном сосуде Парра и раствор барботировали азотом. Добавляли 10% Pd на угле (1,8 г) и сосуд помещали в гидрогенизационный аппарат Парра. Выкачивали воздух из свободного пространства сосуда, а затем создавали давление 50 фунтов/кв. дюйм водорода. Этот процесс повторяли семь раз, а затем сосуд подвергили давлению 50 фунтов/кв. дюйм водорода и перемешивали на аппарате Парра в течение ночи. Катализатор удаляли фильтрацией через Celite®, а затем реакционную смесь концентрировали с помощью роторного испарения с получением (2S)-1-{[трет-бутил(диметил)силил]окси}-5,5,5-трифторпентан-2-амина.
MS (+ESI): 272,1 [M+1]+.
Стадия 6: получение (2S)-1-{[трет-бутил(диметил)силил]окси}-5,5,5-трифтор-N-[(1E)-2,2, 2-трифторэтилиден]пентан-2-амина
Раствор (2S)-1-{[трет-бутил(диметил)силил]окси}-5,5,5-трифторпентан-2-амина (3,00 г, 11,06 ммоль) и трифторацетальдегида этилгемиацеталя (1,6 г, 11,1 ммоль) в бензоле (20 мл) нагревали с обратным холодильником в течение 2 часов, в течение этого времени воду собирали в насадку Дина-Старка. Растворитель удаляли в вакууме с получением неочищенного (2S)-1-{[трет-бутил(диметил)силил]окси}-5,5,5-трифтор-N-[(1E)-2,2,2-трифторэтилиден]пентан-2-амина.
MS (+ESI): 352,2 [M+1]+.
Стадия 7: получение (2S)-2-{[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]амино}-5,5,5-трифторпентан-1-ола
н-BuLi (2,5 M в гексане, 21,4 мл) добавляли по каплям к перемешанному раствору 1, 4-дибромбензола (12,6 г) в сухом диэтиловом эфире (80 мл) при -30°C и реакционную смесь перемешивали в течение 30 минут. Затем добавляли по каплям раствор (2S)-1-{[трет-бутил(диметил)силил]окси}-5,5,5-трифтор-N-[(1E)-2,2,2-трифторэтилиден]пентан-2-амина (3,75 г, 10,7 ммоль) в сухом диэтиловом эфире (30 мл) и реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 16 часов. Реакционную смесь затем тушили с использованием 100 мл воды. Органическую фазу промывали насыщенным солевым раствором, сушили сульфатом магния и фильтровали. Фильтрат концентрировали с помощью роторного испарения с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле, используя 1% этилацетат в гексане в качестве элюента с получением (S)-[1-(4-бромфенил)-2,2,2-трифторэтил]-[1-(трет-бутилдиметилсиланилоксиметил)-4,4,4-трифторбутил]амина (1,8 г). (S)-[1-(4-Бромфенил)-2,2, 2-трифторэтил]-[1-(трет-бутилдиметилсиланилоксиметил)-4,4,4-трифторбутил]амин растворяли в ТГФ (50 мл) и охлаждали до 0°C и добавляли по каплям фторид трет-бутиламмония (10,6 мл, 1M ТГФ-ный раствор). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 4 часов. Реакционную смесь тушили насыщенным водным раствором хлорида аммония, а затем добавляли этилацетат и смесь интенсивно встряхивали. Органический слой отделяли и дважды промывали насыщенным солевым раствором, сушили сульфатом магния и фильтровали. Растворитель удаляли с помощью роторного испарения с получением остатка, который очищали на SiO2, используя градиент 20-25% этилацетата в гексане в качестве элюента с получением (2S)-2-{[(1S)-1-(4-бромфенил)-2,2, 2-трифторэтил]амино}-5,5,5-трифторпентан-1-ола.
MS (+ESI): 393,9, 395,8 [M+1]+.
Стадия 8: получение (2S)-5,5,5-трифтор-2-({(1S)-2,2, 2-трифтор-1-[4'-(метилсульфонид)-1,1'-дифенил-4-ил]этил}амино)пентан-1-ола
Поток азота пропускали через суспензию (2S)-2-{[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]амино}-5,5, 5-трифторпентан-1-ола (0,55 г), 2-(4-метансульфонилфенил)-4,4,5,5-тетраметил[1,3,2]диоксаборолана (0,59 г), 2,5M K2CO3 (2,75 мл) и ДМФ (0,91 мл) в течение 20 минут. Затем добавляли хлорид [1,1'-бис(дифенилфосфино)ферроцен]палладия (II) (1:1 комплекс с дихлорметаном, 34 мг), емкость герметизировали и реакционную смесь нагревали до 85°C и перемешивали в атмосфере азота в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (50 мл) и водой (50 мл) и интенсивно встряхивали. Отделяли слой этилацетата и промывали 3 раза водой, сушили сульфатом магния и фильтровали. Удаление растворителя оставляло остаток, который очищали с помощью хроматографии на SiO2, используя градиент 10-30% этилацетата в гексане. Роторное выпаривание соответствующих фракций давало (2S)-5,5,5-трифтор-2-({(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}амино)пентан-1-ол.
Стадия 9: получение (2S)-5,5,5-трифтор-2-({(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1-дифенил-4-ил]этил}амино)пентановой кислоты
Матричную суспензию окислителя получали перемешиванием периодной кислоты (2,28 г) и триоксида хрома (4,6 мг) во влажном ацетонитриле (0,75% воды) для образования суспензии общим объемом 22,8 мл. Суспензию окислителя (4,22 мл) затем добавляли по каплям к перемешанному раствору (2S)-5,5,5-трифтор-2-({(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}амино)пентан-1-ола (0,35 г, 0,74 ммоль) во влажном ацетонитриле (3,7 мл), сохраняя температуру 0-5°C. Реакционную смесь оставляли нагреваться до температуры окружающей среды через час, а затем перемешивали в течение дополнительных 4 часов. Реакцию тушили водным гидрофосфатом натрия (6 г/100 мл), добавляли толуол и смесь интенсивно встряхивали. Органическую фазу промывали 1:1 соляной раствор:вода, затем водным бисульфитом натрия (2,2 г/50 мл), затем насыщенным солевым раствором. Органический слой затем сушили сульфатом натрия, фильтровали и концентрировали с помощью роторного испарения. Сырой продукт очищали с помощью хроматографии на силикагеле, используя 5-10% метанол в метиленхлориде в качестве элюента, с получением (2S)-5,5,5-трифтор-2-({(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}амино)пентановой кислоты.
MS (-ESI): 482,0 [M-1]-.
Стадия 10: получение цианометиламида (2S)-5,5,5-трифтор-2-({(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}амино)пентановой кислоты
(2S)-5,5,5-Трифтор-2-({(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}амино)пентановую кислоту (0,55 г, 0,114 ммоль), аминоацетонитрила гидрохлорид (21 мг, 0,228 ммоль) и HATU (43 мг, 0,114 ммоль) растворяли в ДМФ (2 мл) и реакционную смесь охлаждали до -20°C. Добавляли диизопропилэтиламин (0,10 мл, 0,57 ммоль) и реакционную смесь перемешивали при -20 °C в течение трех часов, а затем при температуре окружающей среды в течение 16 часов. Добавляли этилацетат (ок.30 мл) и воду (ок.30 мл) и смесь интенсивно встряхивали. Органический слой промывали водой, сушили сульфатом магния и фильтровали. Фильтрат концентрировали с помощью роторного испарения и остаток очищали с помощью хроматографии на силикагеле, используя 30% этилацетат в гексане в качестве элюента, с получением цианометиламида (2S)-5,5,5-трифтор-2-({(1S)-2,2,2-трифтор-1-[4'-(метилсульфонид)-1,1'-дифенил-4-ил]этил}амино)пентановой кислоты в виде белого твердого вещества.
MS (+ESI): 522,3 [M+1]+.
1H ЯМР (CDCl3) δ 8,0-8,053 (2H, д), 7,74-7,78 (2H, д), 7, 63-7,66 (2H, д), 7,48-7,51 (2H, д), 6,94-7,00 (1H, bt, NH), 4,06-4,22 (3H, м), 3,36-3,43 (1H, м), 2,21-2,37 (3H, м), 1,88-2,03 (2H, м).19F ЯМР (CDCl3) δ -66,74- -66,83 (3F, т), -74,14- -74,17 (3F, д).
ПРИМЕР 19
Синтез N1-(цианометил)-N2-{(S)-(4-фторфенил)[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]метил}-L-лейцинамида

Стадия 1: получение (S)-2-[(4-фторбензилиден)амино]-4-метилпентан-1-ола
Смесь (L)-лейцинола (1,13 г, 9,67 ммоль) и 4-фторбензальдегида (1,20 г, 9,67 ммоль) растворяли в бензоле (30 мл) и нагревали с обратным холодильником в течение 1 часа, используя аппарат Дина-Старка для удаления воды. (S)-2-[(4-Фторбензилиден)амино]-4-метилпентан-1-ол выделяли концентрацией в вакууме и сразу использовали без дополнительной очистки.
Стадия 2: получение (2S)-2-{(R)-[(4-бромфенил)-(4-фторфенил)метил]амино}-4-метилпентан-1-ола
К раствору 1,4-дибромбензола (11,4 г, 48,35 ммоль) в эфире (120 мл) при -30°C добавляли н-бутиллитий (24,2 мл, 2,0M раствора циклогексана) в течение 10 минут. Реакционную смесь перемешивали в течение 45 минут, после чего добавляли по каплям раствор (S)2-[(4-фторбензилиден)амино]-4-метилпентан-1-ола (максимум 2,16 г, 9,67 ммоль с предыдущей стадии) в эфире (30 мл). Через 2 часа, во время которых реакционную смесь оставляли нагреваться до 0°C, добавляли воду (200 мл). Продукт экстрагировали этилацетатом (150 мл), промывали насыщенным солевым раствором (100 мл), сушили над MgSO4, фильтровали, концентрировали в вакууме и очищали на тонком слое силикагеля (20% этилацетат/гексан элюция) с получением (2S)-2-{(R)-[(4-бромфенил)-(4-фторфенил)метил]амино}-4-метилпентан-1-ола.
MS (+APCI): 380, 382 [M+1}+.
1H ЯМР (CDCl3) δ 0,91 (д, 6H), 1,22 (м, 1H), 1,43 (м, 1H), 1,61 (м, 1H), 2,62 (м, 1H), 3,28 (м, 1H), 3,63 (м, 1H), 4,95 (с, 1H); 7,00 (м, 2H), 7,22 (д, 2H), 7,2 (м, 2H), 7,44 (д, 2H).
Стадия 3: получение (2S)-2-{(R)-[(4-бромфенил)-(4-фторфенил)метил]амино}-4-метилпентановой кислоты
К раствору (2S)-2-{(R)-[(4-бромфенил)-(4-фторфенил)метил]амино}-4-метилпентан-1-ола (3,28 г, 8,65 ммоль) в ацетонитриле (50 мл), содержащем воду (0,375 мл), при 0-5°C добавляли, за 20 минут, раствор периодной кислоты и оксида хрома (VI) в ацетонитриле (50 мл, приготовленного растворением 11,4 г H5IO6 и 23 мг CrO3 в 100 мл CH3CN и перемешивая в течение 2 часов при комнатной температуре согласно методике, описанной в Tetrahedron Letters, 1998, vol. 39, p. 5323-5326). Реакционную смесь перемешивали в течение ночи, нагревая до комнатной температуры. Добавляли двунатрия фосфат (1,8 г/100 мл воды). Реакционную смесь экстрагировали толуолом (150 мл), промывали 1:1 насыщенным солевым раствором/водой (50 мл), свежеприготовленным раствором бисульфита натрия (2 г/50 мл воды), насыщенным солевым раствором (50 мл), сушили над MgSO4, фильтровали, концентрировали в вакууме и очищали на тонком слое силикагеля (30% этилацетат/гексан элюция для удаления неполярных примесей, затем 50% этилацетат/дихлорметан элюция) с получением (2S)-2-{(R)-[(4-бромфенил)-(4-фторфенил)метил]амино}-4-метилпентановой кислоты.
Стадия 4: получение цианометиламида (2S)-2-{(R)-[(4-бромфенил)-(4-фторфенил)метил]амино}-4-метилпентановой кислоты
К раствору (2S)-2-{(R)-[(4-бромфенил)-(4-фторфенил)метил]амино}-4-метилпентановой кислоты (1,17 г, 2,86 ммоль) в ТГФ (20 мл) при -10°C добавляли 4-метилморфолин (0,315 мл, 2,86 ммоль) и изобутилхлорформиат (0,371 мл, 2,86 ммоль). Реакционную смесь перемешивали в течение 10 минут, после чего добавляли аминоацетонитрила гидрохлорид (0,318 г, 3,43 ммоль), после этого 4-метилморфолин (0, 315 мл, 2,86 ммоль). Раствор перемешивали в течение 90 минут. Добавляли этилацетат (30 мл) и водный двунатрийфосфат (30 мл). Органическую фазу отделяли, промывали насыщенным солевым раствором, сушили над MgSO4 и выпаривали досуха. Продукт очищали на тонком слое силикагеля (10-50% этилацетат/гексан градиент элюции) с получением цианометиламида (2S)-2-{(R)-[(4-бромфенил)-(4-фторфенил)метил]амино}-4-метилпентановой кислоты.
Стадия 5: получение N1-(цианометил)-N2 -{(S)-(4-фторфенил)[4'-(метилсульфонил)-1,1'-дифенил-4-ил]метил}-L-лейцинамида
Смесь цианометиламида (2S)-2-{(R)-[(4-бромфенил)-(4-фторфенил)метил]амино}-4-метилпентановой кислоты (0, 27 г, 0,636 ммоль), 2-(4-метансульфонилфенил)-4,4,5,5-тетраметил[1,3,2]диоксаборолана (0,177 г, 0,626 ммоль) и карбоната калия (0,703 мл 2,0 M раствора) в ДМФ (5 мл) дегазировали. Добавляли [1, 1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II), комплекс дихлорметана (27 мг, 0,038 ммоль). Реакционную смесь нагревали в герметичной трубке при 80-85 °C в течение 3 часов и охлаждали до комнатной температуры. Добавляли этилацетат (15 мл). Реакционную смесь промывали насыщенным солевым раствором (10 мл), насыщенным водным NaHCO3 (10 мл), насыщенным солевым раствором (10 мл), фильтровали через слой MgSO4/DARCO активированный уголь/силикагель, концентрировали в вакууме и очищали препаративной тонкослойной хроматографией (TLC) (Chromatotron®, используя 5% этилацетат/дихлорметан для элюции, с получением N1-(цианометил)-N2-{(S)-(4-фторфенил)[4'-(метилсульфонил)-1,1'-дифенил-4-ил]метил}-L-лейцинамида.
1H ЯМР (CDCl3): δ 0,77 (д, 3H), 0,9 (д, 3H); 1,38 (м, 1H), 1,57 (м, 1H), 1,71 (м, 1H), 2,01 (шир.с, 1Н), 3,09 (с, 3H), 3,09 (м, 1H), 4,07 (м, 2H), 4,89 (1H, с), 7,03 (м, 2H), 7,21 (м, 1H), 7,33 (м, 2H), 7,42 (д, 2H), 7,53 (м, 2H), 7,68 (д, 2H), 7,97 (д, 2H).
ПРИМЕР 20
Синтез цианометиламида (2S)-2-{(S)-[(2, 4-дифторфенил)-(4'-метансульфонилдифенил-4-ил)метил]амино}-4-метилпентановой кислоты

Стадия 1: получение (2S)-2-[(2,4-дифторбензилиден)амино]-4-метилпентан-1-ола
Раствор (S)-(+)-лейцинола (2,47 г, 21 ммоль) и 2,4-дифторбензальдегида (3 г, 21 ммоль) в бензоле (50 мл) нагревали с обратным холодильником в течение 4 часов, в течение этого времени воду собирали в насадку Дина-Старка. Растворитель выпаривали в вакууме с получением (2S)-2-[(2,4-дифторбензилиден)амино]-4-метилпентан-1-ола.
1H ЯМР (CD3SOCD3) δ 8,18 (с, 1H), 7,51 (д, 1H), 6,81 (д, 2H), 4,6 (шир. с, 1H), 3,8-3, 2 (м, 3H), 1,6-1,2 (м, 3H), 0,9-0,8 (м, 6H).
Стадия 2: получение (2S)-2-{(S)-[(4-бромфенил)-(2,4-дифторфенил)метил]амино}-4-метилпентан-1-ола
К раствору 1,4-дибромбензола (24,5 г, 100 ммоль, 5 экв.) в сухом эфире (200 мл) в атмосфере азота при -30°C добавляли н-BuLi (64,75 мл, 1,6 M раствора в гексане, 5 экв.) и реакционную смесь перемешивали в течение 1 ч. Медленно добавляли раствор 2-[(2,4-дифторбензилиден)амино]-4-метилпентан-1-ола (5 г, 20 ммоль) в сухом эфире при -30°C. После перемешивания в течение 4 ч реакцию тушили водой. Эфирный слой промывали насыщенным раствором NaCl и сушили над MgSO4. Растворитель удаляли под пониженным давлением и сырой продукт подвергали флэш-хроматографии на 500 см3 силикагеля с использованием 8:2 гексан:EtOAc в качестве элюента с получением (2S)-2-{(S)-[(4-бромфенил)-(2,4-дифторфенил)метил]амино}-4-метилпентан-1-ола.
1H ЯМР (CD3SOCD3) δ 7,31 (дд, 2H), 7,05 (д, 1H), 6,94, (дд, 2H), 6,63 (д, 1H), 6,54 (д, 1H), 5,02 (с,1H), 4,45 (т, 1H), 3,5-3,3 (м, 2H), 2,3 (с, 1H), 2,15 (с, 1H), 1,85 (м, 1H), 1,18-1,4 (м, 2H), 0,9-0,8 (м, 6H).
Стадия 3: получение (2S)-2-{(S)-[(4-бромфенил)-(2,4-дифторфенил)метил]амино}-4-метилпентановой кислоты
Используя методику, приведенную на стадии 3 Примера 19, окисляли 2-{[(4-бромфенил)-(2,4-дифторфенил)метил]амино}-4-метилпентан-1-ол с получением (2S)-2-{(S)-[(4-бромфенил)-(2, 4-дифторфенил)метил]амино}-4-метилпентановой кислоты в виде твердого вещества кремового цвета.
Стадия 4: получение цианометиламида (2S)-2-{(S)-[(4-бромфенил)-(2, 4-дифторфенил)метил]амино}-4-метилпентановойкислоты
Используя методику, приведенную на стадии 4 Примера 19, (2S)-2-{(S)-[(4-бромфенил)-(2, 4-дифторфенил)метил]амино}-4-метилпентановую кислоту соединяли с аминоацетонитрилом с получением цианометиламида (2S)-2-{(S)-[(4-бромфенил)-(2,4-дифторфенил)метил]амино}-4-метилпентановой кислоты в виде белого твердого вещества.
1H ЯМР (CD3SOCD3) δ 7,31 (дд, 2H), 7,05 (д, 1H), 6,94 (дд, 2H), 6,63 (д, 1H), 6,54 (д, 1H), 4,92 (с, 1H), 4,14 (т, 2H), 3,32 (м, 1H), 3,19 (м, 1H), 2,98 ( м, 1H), 1,85 (м, 1Н), 1,46 (м, 1H), 0,9-0,8 (м, 6H).
Стадия 5: получение цианометиламида (2S)-2-{(S)-[(2, 4-дифторфенил)-(4'-метансульфонилдифенил-4-ил)метил]амино}-4-метилпентановой кислоты
Используя методику, приведенную на стадии 5 Примера 19, проводили взаимодействие Сузуки (Suzuki) с цианометиламидом (2S)-2-{(S)-[(4-бромфенил)-(2,4-дифторфенил)метил]амино}-4-метилпентановой кислоты с получением цианометиламида ((2S)-2-{(S)-[(2, 4-дифторфенил)-(4'-метансульфонилдифенил-4-ил)метил]амино}-4-метилпентановой кислоты в виде белого твердого вещества.
1H ЯМР (CD3SOCD3) δ 8,01 (дд, 2H), 7,8 (дд, 2H), 7,35 (дд, 2H), 7,11 (дд, 2H), 7,03 (д, 1H), 6,93 (д, 2H), 6,62 (д, 1H), 6,55 (д, 1H), 5,0 (с, 1H), 4,13 (т, 2H), 3,35 (м, 1H), 3,0 (м, 1H), 2,99 ( м, 1H), 2,85 (с, 3H), 1,84 (м, 1H), 1,45 (м, 1H), 0,9-0,8 (м, 6H).
ПРИМЕР 21
Синтез N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил-L-норвалинамида

Стадия 1: получение (S)-[1-(трет-бутилдиметилсиланилоксиметил)бутил]-(2,2,2-трифторэтилиден)амина
Смесь (S)-1-(трет-бутилдиметилсиланилоксиметил)бутиламина (27 г, 126 ммоль) и трифторацетальдегида метилгемиацеталя (17,2 г, 132 ммоль) в бензоле (250 мл) нагревали с обратным холодильником, используя насадку Дина-Старка для удаления воды. Через 2 часа больше воды не собирали. Реакционную смесь охлаждали и концентрировали с получением (S)-[1-(трет-бутилдиметилсиланилоксиметил)бутил]-(2,2,2-трифторэтилиден)амина, который использовали на следующей стадии без дополнительной очистки или продолжали высушивание из-за его летучести.
Стадия 2: получение (2S)-(S)-[1-(4-бромфенил)-2,2,2-трифторэтил]-[1-(трет-бутилдиметилсиланилоксиметил)бутил]амина
К раствору дибромбензола (63,7 г, 270 ммоль) в эфире (600 мл) при -30°C добавляли н-BuLi (108 мл 2,5 M гексанового раствора) за 15 минут через капельную воронку. Раствору давали нагреться до -10°C за 35 минут и снова охлаждали до -30°C. Добавляли (S)-[1-(трет-бутилдиметилсиланилоксиметил)бутил]-(2,2,2-трифторэтилиден)амин (35,12 г, 108 ммоль) в эфире (200 мл) за 20 минут. Реакционную смесь перемешивали в течение 1 часа, нагревая до 0°C. Добавляли воду (200 мл). Органическую фазу отделяли, промывали насыщенным солевым раствором (200 мл), сушили над MgSO4, фильтровали и выпаривали досуха с получением (2S)-(S)-[1-(4-бромфенил)-2,2,2-трифторэтил]-[1-(трет-бутилдиметилсиланилоксиметил)бутил]амина, который использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 3: получение (2S)-2-[(S)-1-(4-бромфенил)-2,2,2-трифторэтиламино]пентан-1-ола
К раствору (2S)-(S)-[1-(4-бромфенил)-2,2,2-трифторэтил]-[1-(трет-бутилдиметилсиланилоксиметил)бутил]амина (49,58 г, 109 ммоль) в ТГФ (200 мл) добавляли фторид тетрабутиламмония (110 мл 1,0 M раствора). Реакционную смесь перемешивали в течение ночи при комнатной температуре. После концентрации реакционной смеси добавляли эфир (300 мл). Реакционную смесь промывали водой (2x100 мл), насыщенным солевым раствором (100 мл), выпаривали досуха, а затем очищали с помощью флэш-хроматографии (3% этилацетат/гексан элюция сильно неполярных веществ, затем 40% этилацетат/гексан для элюции продукта). Фракции, содержащие продукт, объединяли, концентрировали до объема 300 мл, промывали 0,25 M лимонной кислотой (200 мл), насыщенным солевым раствором (100 мл), сушили над MgSO4, фильтровали и выпаривали с получением (2S)-2-[(S)-1-(4-бромфенил)-2,2,2-трифторэтиламино]пентан-1-ола. Это вещество использовали неосредственно в следующей стадии без дополнительной очистки. TLC (30% этилацетат/гексан) Rf = 0,28.
Стадия 4: получение (2S)-2-[(S)-1-(4-бромфенил)-2,2,2-трифторэтиламино]пентановой кислоты
К раствору неочищенного (2S)-2-[(S)-1-(4-бромфенил)-2,2,2-трифторэтиламино]пентан-1-ола (37,1 г, 109 ммоль в сухом ацетонитриле (500 мл) при -78°C добавляли раствор периодной кислоты (71,7 г) и триоксида хрома (144 мг) в сухом ацетонитриле (630 мл) в течение 1 часа. Реакционную смесь оставляли нагреваться до комнатной температуры при перемешивании в течение ночи. Реакционную смесь концентрировали под пониженным давлением до общего объема приблизительно 500 мл. Добавляли раствор лимонной кислоты (22 г в 250 мл H2O) и смесь экстрагировали этилацетатом (800 мл). Раствор промывали свежеприготовленным NaHSO3 (2 x 200 мл, 25 г твердого вещества, растворенного до 200 мл водой), насыщенным солевым раствором (300 мл), сушили над MgSO4, фильтровали и выпаривали с получением бледно-оранжевого твердого вещества, которое порошковали 5% этилацетат/гексаном с получением (2S)-2-[(S)-1-(4-бромфенил)-2,2,2-трифторэтиламино]пентановой кислоты. Дополнительное количество кислоты получали концентрацией маточных растворов, распределением остатка между эфиром и водным NaHCO3, подкислением водного слоя лимонной кислотой, экстракцией этилацетатом, высушиванием над MgSO4, фильтрацией и выпариванием растворителя.
Стадия 5: получение N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1 -(1-цианоциклопропил)-L-норвалинамида
Смесь (2S)-2-[(S)-1-(4-бромфенил)-2,2,2-трифторэтиламино]пентановой кислоты (19,0 г, 53,6 ммоль), 1-аминоциклопропанкарбонитрила гидрохлорида (12, 71 г, 107 ммоль, синтезированного согласно O'Donnell, M.J. et al. Synthesis, 1984, 127-128) и HATU (22,42 г, 59,0 ммоль) растворяли в ДМФ (200 мл) и охлаждали до -78°C. Добавляли диизопропилэтиламин (37,35 мл, 214 ммоль) и после перемешивания в течение 1 часа охлаждающую баню убирали. Реакционную смесь оставляли перемешиваться в течение дополнительного 1 часа, после чего добавляли насыщенный NaHCO3 (300 мл). Продукт экстрагировали этилацетатом (500 мл), промывали насыщенным солевым раствором (100 мл), 0,25 M лимонной кислотой (200 мл), насыщенным NaHCO3 (200 мл), насыщенным солевым раствором (200 мл), сушили над MgSO4/DARCO, фильтровали через диоксид кремния и выпаривали досуха. После элюции остатка через другой тонкий слой диоксида кремния 10% этилацетат/дихлорметаном в качестве подвижной фазы продукт концентрировали и перекристаллизовывали из эфир/гексана с получением N2-[(1S)-1-(4-бромфенил)-2,2, 2-трифторэтил]-N1-(1-цианоциклопропил)-L-норвалинамида.
MS (-ESI): 416, 418 [M-1]-.
1H ЯМР (CDCl3): δ 0,97 (3H, т), 0,98 (м, 1H), 1,07 (м, 1H), 1,41 (м, 2H), 1,49 (м, 2H), 1,62 (м, 1H), 1,72 (м, 1H), 3,27 (1H, м), 1,04 (м, 1H), 7,1 (шир.с, 1H), 1,24 (д, 25 2H), 7,75 (д, 2H).
ПРИМЕР 22
Синтез N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамида

Смесь N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-норвалинамида (6,46 г, 15,44 ммоль), 4-метансульфонилфенилбороновой кислоты (3,40 г, 16,98 ммоль), и 2M K2CO3 (19,3 мл) растворяли в ДМФ (75 мл) в толстостенной колбе. Реакционную смесь дегазировали и добавляли [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), дихлорметановый комплекс (0,678 г, 0,926 ммоль). Колбу герметизировали, нагревали при 80-85°C в течение 3 часов, охлаждали и разбавляли этилацетатом (300 мл). Раствор промывали 1:1 водой/насыщенным солевым раствором (200 мл), насыщенным солевым раствором (200 мл), фильтровали через трехслойную пробку диоксида кремния (дно) DARCO и MgSO4 (верх), концентрировали в вакууме и очищали на слое силикагеля (50-70% этилацетат/гексан элюция). Продукт затем дополнительно очищали с помощью перекристаллизации из этилацетат/гексана с получением указанного в заголовке соединения. MS (+ESI): 494 [M+1]+.
1H ЯМР (CDCl3): δ 0,97 (т, 3H), 0,98 (м, 1H), 1,1 (м, 1H), 1,42 (м, 2H), 1,29 (м, 2H), 1,63 (м, 1H), 1,77 (м, 1H), 3,13 (с, 3H), 3,28 (дд, 1H), 4,17 (кв, 1H), 7,21 (шир.с, 1H), 7,47 (д, 2H), 7,63 (д, 2H), 7,78 (д, 2H), 8,02 (д, 2H).
ПРИМЕР 23
Синтез N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-норвалинамида

N2-[1-(4-Бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-норвалинамид получали сходным образом с N1 -(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамидом, используя аминоацетонитрил на стадии взаимодействия с (2S)-2-[(S)-1-(4-бромфенил)-2,2,2-трифторэтиламино]пентановой кислотой.
MS (+ESI): 392, 394 [M+1]+.
ПРИМЕР 24
Синтез N1 -(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамида

N1-(Цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамид получали сходым образом с N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамидом, используя N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-норвалинамид, описанный выше, во взаимодействии Сузуки с 4-метансульфонилфенилбороновой кислотой или пинаколборонатом. Альтернативно, данное соединение могло быть получено в результате связывания по Сузуки между (2S)-(S)-[1-(4-бромфенил)-2,2,2-трифторэтил]-[1-(трет-бутилдиметилсиланилоксиметил)бутил]амином и 4-метансульфонилфенилбороновой кислотой в присутствии [1, 1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), дихлорметанового комплекса в соответствии с методкой, описанной выше, с последующим расщеплением силилового эфира фторидом тетрабутиламмония, окислением с использованием Н5IO6/CrO3 (см. выше) с получением (2S)-2-[(S)-2,2,2-трифтор-1-(4'-метансульфонилдифенил-4-ил)этиламино]пентановой кислоты и соединением этого вещества с аминоацетонитрилгидрохлоридом в присутствии HATU/диизопропилэтиламин/ДМФ, получая названное соединение.
MS (+ESI): 468 [M+1]+.
ПРИМЕР 25
Синтез N1-(цианометил)-N2-{(1R)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамида

Стадия 1: получение (4S)-4-пропил-2-трифторметилоксазолидина
(L)-Норвалинола гидрохлорид (5,25 г, 37,60 ммоль) и трифторацетальдегид метилгемиацеталь (5 мл, 37,6 ммоль) нагревали в бензоле (100 мл) в присутствии триэтиламина (5,26 мл, 37,6 ммоль) с обратным холодильником, используя аппарат Дина-Старка для сбора воды. Через 3 часа реакционную смесь охлаждали, разбавляли эфиром (100 мл), фильтровали и выпаривали досуха, получая (4S)-4-пропил-2-трифторметилоксазолидин, который использовали без дополнительной очистки.
Стадия 2: получение (2S)-2-[1-(4-бромфенил)-2,2,2-трифторэтиламино]пентан-1-ола
Бутиллитий (41 мл 2,0 M циклогексанового раствора) добавляли при -30°C к раствору 1,4-дибромбензола (19,3 г, 81,89 ммоль) в эфире (250 мл) за 10 минут. Через 1 час раствор (4S)-4-пропил-2-трифторметилоксазолидина (3,00 г, 16,38 ммоль) добавляли в эфир (50 мл) в течение 30 минут через капельную воронку. После перемешивания 90 минут добавляли воду (100 мл). Органическую фазу промывали насыщенным солевым раствором (100 мл), сушили над MgSO4, фильтровали и выпаривали досуха. Остаток очищали на слое силикагеля (10-30% этилацетат/гексан элюция). Приблизительно 2:1 смесь диастомеров (2S)-2-[1-(4-бромфенил)-2,2,2-трифторэтиламино]пентан-1-ола при 0,98 м.д. (1Н ЯМР) по концу CF3 на основе триплета норвалинметила наблюдали приблизительно 2:1 смесь диастомеров (2S)-2-[1-(4-бромфенил)-2,2,2-трифторэтиламино]пентан-1-ола.
Стадия 3: получение N1-(цианометил)-N2-{(1S)-2,2, 2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамида и N1-(цианометил)-N2-{(1R)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-норвалинамида
Синтез (2S)-2-[1-(4-бромфенил)-2,2,2-трифторэтиламино]пентановой кислоты и преобразование в аминоацетонитрильный продукт присоединения выполняли образом, идентичным описанному выше, для N2-[1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(цианометил)-L-норвалинамида. Взаимодействие по Сузуки данного продукта образом, идентичным описанному для N1-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамида с получением 2:1 смеси Nl-(цианометил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1-дифенил-4-ил]этил}-L-норвалинамида и N1-(цианометил)-N2-{(1R)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1-дифенил-4-ил]этил}-L-норвалинамида, который выделяли способом препаративной TLC (Chromatotron®).
MS (+ESI): 468 [M+1]+.
ПРИМЕР 26
Синтез N1-(цианометил)-N2-{1-[4'-(4-циклопропилпиперазин-1-ил)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-L-норвалинамида

Указанное в заголовке соединение получали поперечным связыванием по методу Сузуки 4-циклопропилпиперазинефенилбромида с цианометиламидом (2S)-2-{2,2,2-трифтор-1-[4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)фенил]этиламино}пентановой кислоты в присутствии [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), дихлорметанового комплекса, как описано выше, с последующей очисткой с помощью препаративной TLC (Chromatotron®).
MS (+ESI): 514 [M+1]+.
ПРИМЕР 27
Синтез N2-{(1S)-1-[4'-(аминосульфонил)-1,1'-дифенил-4-ил]-2,2,2-трифторэтил}-N1-(1-цианоциклопропил)-L-норвалинамида

Указанное в заголовке соединение получали образом, сходным описанному для N1-(цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамида (Пример 24).
MS (+ESI): 495 [M+1]+.
1H ЯМР (CDCl3): δ 0,97 (3H, т), 0,98 (м*, 1H); 1,08 (м, 1H), 1,42 (м, 2H), 1,44 (м, 2H), 1,57-1,8 (м, 4H), 3,28 (м, 1H), 4,16 (q, 1H), 4,9 (шир.с*, 1H), 7,2 (с, 1H), 7,43 (д, 2H), 7,6 (д, 2H), 7,72 (д, 2H), 7,99 (д, 2H).
ПРИМЕР 28
Синтез (1-цианоциклопропил)амида (2S)-2-[(1S)-1-(4'-ацетилдифенил-4-ил)-2,2,2-трифторэтиламино]пентановой кислоты

Указанное в заголовке соединение получали образом, сходным описанному для N1 -(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамида, посредством поперечного связывания по методу Сузуки между 4-ацетилфенилбороновой кислотой и N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-норвалинамидом в присутствии [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), дихлорметанового комплекса.
MS (+ESI): 458 [M+1]+, 480 [M+1+Na]+.
1H ЯМР (CDCl3): δ 0,97 (т, 3H), 0,98 (м, 1H), 1, 07 (м, 1H), 1,42 (м, 2H), 1,57 (с, 2H), 1,63 (м, 1H), 1,78 (м, 1H), 2,33 (с, 3H), 3,33 (дд, 1H), 4,17 (кв, 1H), 7,21 (шир.с, 1Н), 7,43 (д, 2H), 7,65-7,69 (2xд, 4H), 8,03 (д, 2H).
ПРИМЕР 29
Получение (1-цианоциклопропил)амида (2S)-2-[(1S)-1-(2',4'-дифтордифенил-4-ил)-2,2,2-трифторэтиламино]пентановой кислоты

Указанное в заголовке соединение синтезировали образом, сходным с описанным для N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамида посредством поперечного связывания по методу Сузуки между 2,4-дифторфенилбороновой кислотой и N2 -[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-норвалинамидом в присутствии [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), дихлорметанового комплекса.
MS (+ESI): 452 [M+1]+, 474 [M+1+Na]+.
1H ЯМР (CDCl3): δ 0,90 (м, 1H), 0,97 (т, 3H), 1,04 (м, 1H), 1,41-1,5 (м, 4H), 1,62 (м, 1H), 1,78 (м, 1H), 2,19 (шир.с, 1H), 3,32 (дд, 1H), 4,13 (дд, 1H), 6,95 (м, 2H), 7,19 (шир.с, 1H), 7,41 (м, 3H), 7,53 (д, 2H).
ПРИМЕР 30
Синтез (1-цианоциклопропил)амида (2S)-2-[(1S)-1-(3',4'-дифтордифенил-4-ил)-2,2,2-трифторэтиламино]пентановой кислоты

Указанное в заголовке соединение получали образом, сходным с описанным для N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамида, посредством поперечного связывания по методу Сузуки между 3,4-дифторфенилбороновой кислотой и N2 -[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-норвалинамидом в присутствии [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), дихлорметанового комплекса.
MS (+ESI): 452 [M+1]+, 474 [M+1+Na]+.
1H ЯМР (CDCl3): δ 0,97 (т, 3H), 0,98 (м, 1H), 1,07 (м, 1H), 1,42 (м, 2H), 1,5 (м, 2H), 1,62 (м, 1H), 1,77 (м, 1Н), 2,18 (шир.с, 1H), 3,29 (дд, 1H), 4,17 (кв, 1H), 7,21 (шир.с, 1H), 7,13-7,3 (м, 2H), 7,38 (м, 1H), 7,42 (д, 2H), 7,57 (д, 2H).
ПРИМЕР 31
Получение (1-цианоциклопропил)амида (2S)-2-[(1S)-1-(3'-хлор-4'-фтордифенил-4-ил)-2,2,2-трифторэтиламино]пентановой кислоты

Указанное в заголовке соединение получали образом, сходным с описанным для N1-(1-цианоциклопропил)-N2-{(1S)-2,2, 2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамида, посредством поперечного связывания по методу Сузуки между 3-хлор-4-фторфенилбороновой кислотой и N2 -[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-норвалинамидом в присутствии [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), дихлорметанового комплекса.
MS (+ESI): 468 [M+1]+.
1H ЯМР (CDCl3): δ 0,99 (т, 3H), 1,00 (м, 1H), 1,08 (м, 1H), 1,42 (м, 2H), 1,51 (м, 2H), 1,62 (м, 1Н), 1,79 (м, 1H), 2,19 (шир. с, 1H), 3,33 (дд, 1H), 4,17 (кв, 1H), 7,21 (м, 2H), 7,42 (м, 3H), 7,57 (д, 2H), 7,62 (м, 1H).
ПРИМЕР 32
Синтез (1-цианоциклопропил)амида (2S)-2-[(1S)-2,2,2-трифтор-1-(4'-метилдифенил-4-ил)этиламино]пентановой кислоты

Указанное в заголовке соединение синтезировали образом, сходным с описанным для N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1, 1'-дифенил-4-ил]этил}-L-норвалинамида, посредством поперечного связывания по методу Сузуки между п-толилбороновой кислотой и N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1 -(1-цианоциклопропил)-L-норвалинамидом в присутствии [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), дихлорметанового комплекса.
MS (+ESI): 430 [M+1]+.
1H ЯМР (CDCl3): δ 0,90 (м, 1Н), 0,98 (т, 3Н), 1,02 (м, 1H) 1,4-1,5 (м, 4H), 1,62 (м, 1H), 1,69 (м, 1H), 2,19 (шир.с, 1H), 2,41 (с, 3H), 3,35 (дд, 1H), 4,1 (кв, 1H), 7,21 (с, 1H) 7,24 (д, 2H), 7,38 (д, 2H), 7,46 (д, 2H), 7,60 (д, 2H).
ПРИМЕР 33
Синтез (1-цианоциклопропил)амида (2S)-2-[(1S)-1-(4'-цианодифенил-4-ил)-2,2, 2-трифторэтиламино]пентановой кислоты

Указанное в заголовке соединение синтезировали образом, сходным с описанным для N1-(1-цианоциклопропил)-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамида, посредством поперечного связывания по методу Сузуки между 4-цианофенилбороновой кислотой и N2-[(1S)-1-(4-бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-L-норвалинамидом в присутствии [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), дихлорметанового комплекса.
MS (+ESI): 441 [M+1]+.
ПРИМЕР 34
Синтез цианометиламида (2S)-2-{(S)-[(4-бромфенил)тиазол-2-илметил]амино}-4-метилпентановой кислоты

Стадия 1: получение 3-метил-3-(толуолсульфонилоксиметил)оксетана
п-Толуолсульфонилхлорид (57,2 г, 300 ммоль) растворяли в сухом пиридине (400 мл) в атмосфере азота. Медленно добавляли 3-метил-3-(гидроксиметил)оксетан (20,4 г, 200 ммоль) и раствор перемешивали в течение 1,5 ч. Затем добавляли дробленный лед (400 г) к интенсивно перемешаной смеси, которой давали перемешиваться в течение дополнительного 0,5 ч. Затем собирали белый осадок на фильтровальную бумагу Whatman #1 и промывали холодной водой. Продукт сушили под высоким вакуумом с получением 3-метил-3-(толуолсульфонилоксиметил)оксетана (оксетантозилата) в виде белого порошка оксетантозилата.
1H ЯМР (CDCl3): δ 7,81 (д, 2H), 7,37 (д, 2H), 4,37 (м, 4H), 4,11 (с, 2H), 2,46 (с,3H), 1,31 (с, 3H).
Стадия 2: получение 3-метилоксетан-3-илметилового эфира (2S)-2-бензилоксикарбониламино-4-метилпентановой кислоты
Cbz-L-лейцин (2 г, 7,5 ммоль) и Cs2CO3 (1,46 г, 4,5 ммоль, 0,6 экв.) растворяли в воде (20 мл). Затем воду удаляли в вакууме и полученное в результате масло лиофилизировали в течение 12 час с получением белого твердого вещества. К этому твердому веществу добавляли 3-метил-3-(толуолсульфонилоксиметил)оксетан (оксетантозилат) (1,8 г, 4,5 ммоль) и NaI (224 мг, 1,5 ммоль, 0,2 экв.), взятые в ДМФ (35 мл), и оставляли перемешиваться в атмосфере азота в течение 48 ч. Затем удаляли ДМФ в вакууме, полученное в результате твердое вещество растворяли в EtOAc (60 мл) и промывали 10% NaHCO3 (20 мл) и насыщенным NaCl (10 мл) и сушили над MgSO4. Растворитель удаляли под пониженным давлением с получением желтого масла, которое подвергали колоночной флэш-хроматографии на 200 см3 силикагеля, используя 3:1 гексан:EtOAc в качестве элюента, с получением 3-метилоксетан-3-илметилового эфира (2S)-2-бензилоксикарбониламино-4-метилпентановой кислоты (сложный эфир Cbz-Leu оксетана) в виде густого желтого масла.
1H ЯМР (CD3SOCD3) 7,75 (д, 1H), 7,26-7,38 (м, 5H), 5,05 (с, 2H), 4,0-4,4 (м, 8H), 1,45-1,7 (м, 3H), 1,25 (с, 3H), 0,85-0,9 (м, 6H).
Стадия 3: получение бензилового эфира [3-метил-1-(4-метил-2,6, 7-триоксабицикло[2.2.2]окт-1-ил)бутил]карбаминовой кислоты
(2S)-2-Бензилоксикарбониламино-4-метилпентановой кислоты 3-метилоксетан-3-илметиловый эфир (сложный эфир Cbz-Leu оксетана) (2 г, 5,7 ммоль) растворяли в сухом CH2Cl2 (10 мл) в атмосфере азота. BF3.Et2О (40 мкл, 0,3 ммоль, 0,054 экв.) разбавляли в сухом CH2Cl2 (1 мл) и добавляли в реакционный сосуд. Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 12 ч. Добавляли триэтиламин (335 мкл, 3,3 ммоль, 0,58 экв.) и реакционную смесь перемешивали в течение дополнительных 30 мин перед концентрацией в густое масло. Сырой продукт повторно растворяли в EtOAc (15 мл), промывали 3% NH4Cl (10 мл), и насыщенным NaCl (10 мл), сушили (MgSO4) и выпаривали досуха. Реакция давала бесцветное густое масло, которое кристаллизовалось при стоянии, давая бензиловый эфир [3-метил-1-(4-метил-2,6, 7-триокса-бицикло[2.2.2]окт-1-ил)бутил]карбаминовой кислоты (сложный эфир Cbz-Leu-OBO).
1H ЯМР (CD3SOCD3) 7,25-7,35 (шир. м, 5H), 6,88 (д, 1H), 5,05 (с, 2H), 3,80 (с, 6H), 3,7 (м, 1H) 1,2-1,6 (м, 3H), 0,75-0,85 (м, 6H), 0,70 (с, 3H).
Стадия 4: получение 3-метил-1-(4-метил-2,6, 7-триоксабицикло[2.2.2]окт-1-ил)бутиламина
К бензиловому эфиру [3-метил-1-(4-метил-2,6,7-триоксабицикло[2.2.2]окт-1-ил)бутил]карбаминовой кислоты (3,2 г, 9,2 ммоль) в абсолютном EtOH (30 мл) добавляли 10% Pd на активированном угле (320 мг, 10 %) в атмосфере азота. Реакционную смесь гидрогенизировали при 50 psi в течение 6 ч. Реакцию контролировали с помощью TLC и реакционную смесь фильтровали через Celite. Растворитель удаляли с получением 3-метил-1-(4-метил-2,6,7-триоксабицикло[2.2.2]окт-1-ил)бутиламина в виде белого твердого вещества.
1H ЯМР (CD3SOCD3) 3,80 (с, 6H), 3,2-3,4 (шир.с, 3H), 1,75 (м, 1H), 1,0-1,4 (м, 2H), 0,75-0,90 (м, 6H), 0,75 (с, 3H).
Стадия 5: получение [3-метил-1-(4-метил-2,6,7-триоксабицикло[2.2.2]окт-1-ил)бутил]тиазол-2-илметиленамина
Раствор 3-метил-1-(4-метил-2,6,7-триоксабицикло[2.2.2]окт-1-ил)бутиламина (1,9 г, 8,8 ммоль) и тиазол-2-карбоксальдегида (998 мг, 8,8 ммоль) в бензоле нагревали с обратным холодильником в течение 3 ч, используя насадку Дина-Старка, в течение этого времени воду собирали и оставшийся остаток выпаривали под пониженным давлением с получением [3-метил-1-(4-метил-2,6,7-триоксабицикло[2.2.2]окт-1-ил)бутил]тиазол-2-илметиленамина в виде твердого вещества оранжевого цвета.
1H ЯМР (CD3SOCD3) 8,4 (с, 1H), 7,97 (д, 1H), 7,84 (д, 1H), 3,82 (с, 6H), 3,4 (м, 1H), 1,7 (м, 1H), 1,5 (м, 1H), 1,3 (м, 1Н), 0,9 (м, 6H), 0,75 (с, 3H).
Стадия 6: получение [(4-бромфенил)тиазол-2-илметил]-[3-метил-1-(4-метил-2,6,7-триоксабицикло[2.2.2]окт-1-ил)бутил]амина
К раствору дибромбензола (760 мг, 3,2 ммоль) в сухом эфире добавляли 2,5 M раствор н-BuLi в гексане (1,3 мл, 3,2 ммоль) при -30°C. После перемешивания в течение 1 ч медленно добавляли раствор [3-метил-1-(4-метил-2,6, 7-триоксабицикло[2.2.2]окт-1-ил)бутил]тиазол-2-илметиленамина (500 мг, 1,6 ммоль) в 5 мл сухого эфира. Реакционную смесь оставляли перемешиваться при той же температуре в течение 2 ч, а затем реакцию тушили водой и экстрагировали этилацетатом. Органический слой промывали насыщенным NaCl и сушили над MgSO4. Растворитель выпаривали в вакууме. Сырой продукт подвергали колоночной флэш-хроматографии, используя 2:1 гексан:EtOAc в качестве элюента с получением [(4-бромфенил)тиазол-2-илметил]-[3-метил-1-(4-метил-2,6,7-триоксабицикло[2.2.2]окт-1-ил)бутил]амина в виде бледно-желтого твердого вещества.
1H ЯМР (CD3SOCD3) 7,64 (д, 1H), 7,59 (д, 1H), 7,5 (д, 2H), 7,28 (д, 2H), 5,68 (с, 1H), 3,82 (с, 6H), 3,3 (м, 1H), 2,65 (м, 1H), 1, 95 (м, 1H), 1,4 (м, 1H), 1,2 (м, 1H), 0,9 (д, 3H), 0,75 (с, 3H), 0,7 (д, 3H).
Стадия 7: получение (2S)-2-{(S)-[(4-бромфенил)тиазол-2-илметил]амино}-4-метилпентановой кислоты
К раствору [(4-бромфенил)тиазол-2-илметил]-[3-метил-1-(4-метил-2,6, 7-триокса-бицикло[2.2.2]окт-1-ил)бутил]амина (500 мг, 1,06 ммоль) в ТГФ (20 мл) и воде (18 мл), добавляли 1н HCl (3,5 мл). Реакционную смесь перемешивали в течение 2 ч. После сопоставления TLC на исчезновение исходного вещества добавляли Li(OH)·H2O (280,2 мг, 6,3 экв.) и перемешивали при комнатной температуре в течение 2 ч. Затем добавляли 1н HCl для установления pH 4-6 и продукт экстрагировали с помощью EtOAC, органический слой сушили над MgSO4. Растворитель выпаривали с получением (2S)-2-{(S)-[(4-бромфенил)тиазол-2-илметил]амино}-4-метилпентановой кислоты в виде желтого твердого вещества.
Стадия 8: получение цианометиламида (2S)-2-{(S)-[(4-бромфенил)тиазол-2-илметил]амино}-4-метилпентановой кислоты
В присутствии HATU (1 экв.) и диизопропилэтиламина (4 экв.) соединяли (2S)-2-{(S)-[(4-бромфенил)тиазол-2-илметил]амино}-4-метилпентановую кислоту с аминоацетонитрилом (2 экв., 1 экв. избыток) с получением цианометиламида (2S)-2-{(S)-[(4-бромфенил)тиазол-2-илметил]амино}-4-метилпентановой кислоты в виде твердого вещества кремового цвета.
1H ЯМР (CD3SOCD3) 8,73 (т, 1H), 7,66 (д, 1H), 7,64 (д, 1H), 7,5 (дд, 2H), 7,25 (дд, 10 2H) 4,92 (д, 1H), 4,14 (т, 2H), 3,32 (с, 1H), 3,19 (м, 1H), 2,98 (м, 1H), 1,85 (м, 1H), 1,46 (м, 1H), 0, 9-0,85 (м, 6H).
ПРИМЕР 35
Синтез (1-цианоциклопропил)амида (2S)-2-{(S)-[(4-бромфенил)тиазол-2-илметил]амино}-4-метилпентановой кислоты

Указанное в заголовке соединение получали соединением (2S)-2-{(S)-[(4-бромфенил)-тиазол-2-илметил]амино}-4-метилпентановой кислоты с 1-аминоциклопропанкарбонитрилом в присутствии HATU и диизопропиламина образом, сходным описанному для получения Примера 34. Синтез (2S)-2-{(S)-[(4-бромфенил)тиазол-2-илметил]амино}-4-метилпентановой кислоты проводили посредством добавления бромфениллития (образованного in situ из 1,4-дибромбензола и н-бутиллития) к продукту иминовой конденсации между тиазол-2-карбоксиальдегидом и 3-метил-1-(4-метил-2,6,7-триоксабицикло[2.2.2]окт-1-ил)бутиламином (сложный эфир L-лейцин OBO, полученный в соответствии со способом, описанным для Примера 34), с последующей ортоэфирной депротекцией.
1H ЯМР (CDCl3): δ 0,82 (д, 3H), 0,95 (д, 3H), 1,04 (м, 2Н), 1,46-1,62 (м, 4H), 1,80 (м, 1Н), 3,12 (дд, 1H), 4,95 (с, 1H), 7,22 (с, 1H), 7,23-7,25 (д, 2H), 7,31 (м, 2H), 7,47 (д, 2H), 7,74 (м, 1H).
ПРИМЕР 36
Синтез (1-цианоциклопропил)амида (2S)-2-{(S)-[(2',4'-дифтордифенил-4-ил)тиазол-2-илметил]амино}-4-метилпентановой кислоты

Указанное в заголовке соединение получали посредством поперечного связывания по методу Сузуки (1-цианоциклопропил)амида (2S)-2-{(S)-[(4-бромфенил)тиазол-2-илметил]амино}-4-метилпентановой кислоты с 2,4-дифторбензолбороновой кислотой, в присутствии [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), дихлорметанового комплекса.
MS (-ESI): 479 [M-1]-.
1H ЯМР (CDCl3): δ 0,84 (д, 3H), 0,97 (д, 3H), 1,03 (м, 2H), 1,47 (м, 2H), 1,57-1,62 (м, 4H), 1,95 (м, 1H), 3,27 (дд, 1H), 5,02 (с, 1H), 6,93 (м, 2H), 7,32 (м, 1H), 7,39 (м, 1H), 7,42 (м, 3H), 7,48 (д, 2H), 7,77 (д, 1H).
ПРИМЕР 37
Синтез (1-цианоциклопропил)амида (2S)-2-{(S)-[(4'-метансульфонилдифенил-4-ил)тиазол-2-илметил]амино}-4-метилпентановой кислоты

Указанное в заголовке соединение получали образом, сходным с описанным для (1-цианоциклопропил)амида (2S)-2-{(S)-[(2', 4'-дифтордифенил-4-ил)тиазол-2-илметил]амино}-4-метилпентановой кислоты, посредством поперечного связывания по методу Сузуки (1-цианоциклопропил)амида (2S)-2-{(S)-[(4-бромфенил)тиазол-2-илметил]амино}-4-метилпентановой кислоты с 4-метансульфонилфенилбороновой кислотой в присутствии [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), дихлорметанового комплекса.
MS (-ESI): 521 [M-1]-.
ПРИМЕР 38
Синтез N1-(1-цианоциклопропил)-4,4-дифтор-N2-{(1S)-2,2, 2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамида

Стадия 1: получение метил-N-((бензилокси)карбонил)-3-йодо-L-аланината
К раствору карбобензилокси-L-серина (25 г, 104 ммоль) в этилацетате (200 мл) добавляли раствор лиазометана в эфире до сохранения бледно-желтого цвета. Растворитель выпаривали в вакууме. К остатку добавляли N,N-диметилформамид (400 мл) и йодид метилтрифеноксифосфония (50 г, 110 ммоль). Смесь перемешивали в течение 15 минут, затем добавляли метанол (15 мл), смесь затем выливали на 20% тиосульфат натрия и экстрагировали смесью 1:1 этилацетат:гексан (2 л). Органический слой промывали водой, насыщенным солевым раствором (3x), сушили над сульфатом магния, фильтровали и растворитель выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле, используя этилацетат и гексан. Полученное соединение порошковали в диэтиловом эфире/гексане, фильтровали и сушили на воздухе с получением метил-N-((бензилокси)карбонил)-3-йодо-L-аланината.
Стадия 2: получение метил-N-((бензилокси)карбонил)-4-оксо-L-норвалината
Смесь метил-N-((бензилокси)карбонил)-3-йодо-L-аланината (10 г, 27,5 ммоль) со стадии 1, цинк-медной пары (3,3 г) в бензоле (110 мл) и N,N-диметилацетамида (7,4 мл) подвергали действию ультразвука в ультразвуковой бане в течение 2 часов. В течение этого периода добавляли 3 порции 1,2-дибромэтана (0,24 мл) и хлортриметилсилана (0,17 мл). К этой смеси затем добавляли хлорид бис(трифенилфосфин)палладия (0,958 г, 1,4 ммоль) и ацетилхлорид (2,5 мл, 35,2 ммоль) и смесь нагревали при 70°C в течение 2 часов. После охлаждения до комнатной температуры смесь фильтровали на целите с этилацетатом, затем органический слой промывали насыщенным раствором хлорида аммония, насыщенным солевым раствором (2x), сушили над сульфатом магния, фильтровали и растворитель выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле, используя этилацетат и гексан, с получением метил-N-((бензилокси)карбонил)-4-оксо-L-норвалината.
Стадия 3: получение метил-N-((бензилокси)карбонил)-4,4-дифтор-L-норвалината
К раствору метил-N-((бензилокси)карбонил)-4-оксо-L-норвалината (1,3 г, 4,65 ммоль) в дихлорметане (20 мл) и метанолу (0,019 мл) при 0°C медленно добавляли DAST (2,46 мл). Ледяную баню убирали и заменяли баней с горячей водой (57°C). Баню с горячей водой меняли 3 раза, затем смесь перемешивали в течение ночи при комнатной температуре. Смесь медленно выливали на холодный насыщенный NaHCO3, экстрагировали этилацетатом, промывали насыщенным солевым раствором, сушили над сульфатом магния, фильтровали и растворитель выпаривали в вакууме. Остаток очищали с помощью хроматографии на силикагеле, используя этилацетат и гексан, с получением метил-N-((бензилокси)карбонил)-4,4-дифтор-L-норвалината.
Стадия 4: получение бензил-(1S)-3, 3-дифтор-1-(гидроксиметил)бутилкарбамата
К раствору метил-N-((бензилокси)карбонил)-4,4-дифтор-L-норвалината (1,59 г, 5,29 ммоль) в этаноле (50 мл) добавляли хлорид лития (919 мг) и смесь перемешивали в течение 10 минут. Медленно добавляли борогидрид натрия (820 мг), смесь перемешивали в течение 2 часов. Затем добавляли другую порцию борогидрида натрия (100 мг) и перемешивание продолжали в течение 30 минут. Смесь разбавляли водой (20 мл) и медленно нейтрализовали с помощью 1н HCl с последующим добавлением другой аликвоты воды. Смесь экстрагировали этилацетатом (2x), промывали насыщенным солевым раствором, сушили над сульфатом магния, фильтровали и растворитель выпаривали в вакууме с получением бензил-(1S)-3,3-дифтор-1-(гидроксиметил)бутилкарбамата.
Стадия 5: получение (2S)-1-((трет-бутил(диметил)силил)окси)-4,4-дифторпентан-2-амина
К раствору бензил-(1S)-3,3-дифтор-1-(гидроксиметил)бутилкарбамата (со стадии 4) в этаноле (25 мл) добавляли палладий на угле (10%, 150 мг) и смесь перемешивали в атмосфере H2 (баллон) в течение 2 ч. Добавляли дихлорметан и смесь фильтровали на целите. Растворитель выпаривали в вакууме. Остаток растворяли в дихлорметане (15 мл) и добавляли триэтиламин (1 мл), N,N-диметиламинопиридин (10 мг) и хлор-трет-бутилдиметилсилан (844 мг). Смесь перемешивали в течение ночи, затем добавляли воду и соляной раствор. Смесь экстрагировали этилацетатом (2x), промывали насыщенным солевым раствором, сушили над сульфатом магния, фильтровали и растворитель выпаривали в вакууме с получением (2S)-1-((трет-бутил(диметил)силил)окси)-4,4-дифторпентан-2-амина.
Стадия 6: получение (2S)-1-((трет-бутил(диметил)силил)окси)-4, 4-дифтор-N-((1E)-2,2,2-трифторэтилиден)пентан-2-амина
Раствор (2S)-1-((трет-бутил(диметил)силил)окси)-4,4-дифторпентан-2-амина со стадии 5 и трифторацетальдегид метилгемиацеталя (80 %, 0,9 мл) в бензоле (20 мл) нагревали с обратным холодильником в течение ночи, используя аппарат Дина-Старка. Растворитель выпаривали в вакууме и остаток очищали с помощью хроматографии на силикагеле, используя этилацетат и гексаны, с получением (2S)-1-((трет-бутил(диметил)силил)окси)-4,4-дифтор-N-((1E)-2,2,2-трифторэтилиден)пентан-2-амина.
Стадия 7: получение (2S)-2-(((1S)-1-(4-бромфенил)-2,2,2-трифторэтил)амино)-4,4-дифторпентан-1-ола
К -78°C раствору 1,4-дибромбензола (330 мг) в ТГФ (5,2 мл) добавляли 2,5M n-BuLi в гексане (0,52 мл) и раствор выдерживали в течение 30 минут. Затем добавляли раствор (2S)-1-((трет-бутил(диметил)силил)окси)-4,4-дифтор-N-((1E)-2,2,2-трифторэтилиден)пентан-2-амина (333 мг) в ТГФ (5,2 мл). Смесь перемешивали при -78°C в течение 45 минут, затем выливали на холодный насыщенный хлорид аммония, экстрагировали этилацетатом (2x), промывали насыщенным солевым раствором, сушили над сульфатом магния, фильтровали и растворитель выпаривали в вакууме. Остаток растворяли в ТГФ (10 мл), охлажденном в бане лед/вода, и добавляли фторид н-тетрабутиламмония (1M в ТГФ, 1,5 мл). Смесь перемешивали при 0°C в течение 1 ч, выливали на холодную воду, экстрагировали этилацетатом (2x), промывали насыщенным солевым раствором, сушили над сульфатом магния, фильтровали и растворитель выпаривали в вакууме с получением (2S)-2-(((1S)-1-(4-бромфенил)-2,2,2-трифторэтил)амино)-4,4-дифторпентан-1-ола.
Стадия 8: получение N2-[(1S)-1-(4-бромфенил)-2,2, 2-трифторэтил]-N1-(1-цианоциклопропил)-4,4-дифтор-L-норвалинамида
(2S)-2-(((1S)-1-(4-Бромфенил)-2,2,2-трифторэтил)амино)-4,4-дифторпентан-1-ол превращали в указанное в заголовке соединение, используя способ, описанный в стадии 9 Примера 15.
Стадия 9: получение N1-(1-цианоциклопропил)-4,4-дифтор-N2-{(1S)-2, 2,2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-норвалинамида
N2-[(1S)-1-(4-Бромфенил)-2,2,2-трифторэтил]-N1-(1-цианоциклопропил)-4, 4-дифтор-L-норвалинамид превращали в указанное в заголовке соединение, используя способ, описанный на стадии 10 Примера 15.
Стадия 10: получение N1 -(1-цианоциклопропил)-4,4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилсульфонил)-1,1'-дифенил-4-ил]этил}-L-норвалинамида
N1-(1-Цианоциклопропил)-4,4-дифтор-N2-{(1S)-2,2,2-трифтор-1-[4'-(метилтио)-1,1'-дифенил-4-ил]этил}-L-норвалинамид превращали в указанное в заголовке соединение, используя способ, описанный на стадии 11 Примера 15.
Используя экспериментальные методики, аналогичные перечисленным выше, были синтезированы следующие соединения.
Фармацевтическая композиция
В качестве отдельного варианта осуществления данного изобретения 100 мг N1-(цианометил)-N2-[2,2,2-трифтор-1-[4'-пиперазин-1-ил-1,1'-дифенил-4-ил)этил]-L-лейцинамида формулировали с достаточным количеством мелко измельченной лактозы для обеспечения общего количества от 580 до 590 мг для наполнения твердой желатиновой капсулы 0 размера.
Соединения, раскрытые в настоящем изобретении, проявляют активность в следующих испытаниях. Кроме того, соединения, раскрытые в настоящем изобретении обладают расширенным фармакологическим профилем относительно ранее раскрытых соединений.
Катепсин K исследование
Серийные разведения (1/3) от 500 мкМ вплоть до 0,0085 мкМ тестируемых соединений готовили в диметилсульфоксиде (ДМСО). Затем 2 мкл ДМСО из каждого разведения добавляли к 50 мкл исследуемого буфера (MES, 50 мМ (pH 5,5); EDTA, 2,5 мМ; DTT, 2,5 мМ и 10% ДМСО) и 25 мкл катепсина K человека (0,4 нМ) в растворе исследуемого буфера. Исследуемые растворы перемешивали в течение 5-10 секунд на вибрационной пластине и инкубировали в течение 15 минут при комнатной температуре. Z-Leu-Arg-AMC (8 мкМ) в 25 мкл исследуемого буфера добавляли к исследуемым растворам. Гидролиз кумаринпокидающей группы (AMC) сопровождался спектрофлуометрией (Exλ =355 нм; Emλ = 460 нм) в течение 10 минут. Процент ингибирования подсчитывали приведением экспериментальных значений к стандартной математической модели для кривой доза-ответ.
Катепсин L исследование
Серийные разведения (1/3) от 500 мкМ вплоть до 0,0085 мкМ тестируемых соединений готовили в диметилсульфоксиде (ДМСО). Затем 2 мкл ДМСО из каждого разведения добавляли к 50 мкл исследуемого буфера (MES, 50 мМ (pH 5,5); EDTA, 2,5 мМ; DTT, 2,5 мМ и 10% ДМСО) и 25 мкл катепсина L человека (0,5 нМ) в исследуемом буферном растворе. Исследуемые растворы перемешивали в течение 5-10 секунд на вибрационной пластине и инкубировали в течение 15 минут при комнатной температуре. Z-Leu-Arg-AMC (8 мкМ) в 25 мкл исследуемого буфера добавляли к исследуемым растворам. Гидролиз кумаринпокидающей группы (AMC) сопровождался спектрофлуометрией (Exλ =355 нм; Emλ = 460 нм) в течение 10 минут. Процент ингибирования подсчитывали приведением экспериментальных значений к стандартной математической модели для кривой доза-ответ.
Катепсин B исследование
Серийные разведения (1/3) 500 мкМ вплоть до 0,0085 мкМ тестируемых соединений готовили в диметилсульфоксиде (ДМСО). Затем 2 мкл ДМСО из каждого разведения добавляли к 50 мкл исследуемого буфера (MES, 50 мМ (pH 5,5); EDTA, 2,5 мМ; DTT, 2,5 мМ и 10% ДМСО) и 25 мкл катепсина B человека (4,0 нМ) в исследуемом буферном растворе. Исследуемые растворы перемешивали в течение 5-10 секунд на вибрационной пластине и инкубировали в течение 15 минут при комнатной температуре. Z-Leu-Arg-AMC (8 мкМ) в 25 мкл исследуемого буфера добавляли к исследуемым растворам. Гидролиз кумаринпокидающей группы (AMC) сопровождался спектрофлуометрией (Exλ =355 нм; Emλ = 460 нм) в течение 10 минут. Процент ингибирования подсчитывали приведением экспериментальных значений к стандартной математической модели для кривой доза-ответ.
Катепсин S исследование
Серийные разведения (1/3) 500 мкМ вплоть до 0,0085 мкМ тестируемых соединений готовили в диметилсульфоксиде (ДМСО). Затем 2 мкл ДМСО из каждого разведения добавляли к 50 мкл исследуемого буфера (MES, 50 мМ (pH 5,5); EDTA, 2,5 мМ; DTT, 2,5 мМ и 10% ДМСО) и 25 мкл катепсина S человека (20 нМ) в исследуемом буферном растворе. Исследуемые растворы перемешивали в течение 5-10 секунд на вибрационной пластине и инкубировали в течение 15 минут при комнатной температуре. Z-Leu-Arg-AMC (8 мкМ) в 25 мкл исследуемого буфера добавляли к исследуемым растворам. Гидролиз кумаринпокидающей группы (AMC) сопровождался спектрофлуометрией (Exλ =355 нм; Emλ = 460 нм) в течение 10 минут. Процент ингибирования подсчитывали приведением экспериментальных значений к стандартной математической модели для кривой доза-ответ.
Реферат
Изобретение относится к соединениям, представленным формулой

где значения заместителей указаны в описании, и их фармацевтически приемлемым солям, которые могут использоваться для лечения и/или профилактики катепсин-зависимых состояний или болезней млекопитающих, нуждающихся в этом. Эти соединения являются полезными для лечения заболеваний, при которых показано ингибирование резорбции кости, таких как остеопороз, повышение минеральной плотности кости и снижение риска переломов. Задачей настоящего изобретения является применение заявленных соединений для изготовления лекарственного средства, обладающего ингибирующей активностью в отношении катепсина. 8 н. и 16 з.п. ф-лы, 4 табл.
Формула

Документы, цитированные в отчёте о поиске
2-сахаринилметил- или 4,5,6,7-тетрагидро-2-сахаринилметилфосфаты, -фосфонаты или -фосфинаты и фармацевтическая композиция на их основе
Комментарии