Производные гидразина или их соли, фармацевтическое средство, аминоалкилгидразины или их соли - RU2092492C1
Код документа: RU2092492C1
Чертежи















Описание
Изобретение относится к новому классу негидролизующихся аналогов, расщепляемых с помощью аспартатпротеазы пептидов, а именно к производным гидразина и их фармакологически приемлемым солям, обладающим свойствами ингибировать действие фермента ВИЧ-протеазы и антивирусной активностью; фармацевтическому средству на их основе, пригодному для борьбы с вирусными заболеваниями, а также к новым аминоалкилгидразинам промежуточным продуктам для получения производных гидразина.
В случае иммунодефицита, СПИДа ("синдром приобретенного иммунодефицита"), речь идет о заболевании со смертельным исходом. Это заболевание распространяется во всем мире в возрастающей мере, прежде всего внутри известных групп риска, и через эти группы риска. Оно затронуло уже миллионы людей, и борьба с его причиной представляет собой одну из самых значительных целей для современной медицины. До сих пор смогли идентифицировать ретровирусы ВИЧ-1 и ВИЧ-2 (ВИЧ обозначает "вирус человеческого иммунодефицита") в качестве причин заболевания и охарактеризовать их с точки зрения молекулярной биологии. Для терапии среди существовавших до сих пор средств для облегчения симптомов СПИДа и известных предохранительных средств особый интерес представляют поиски препаратов, которые препятствуют размножению самого вируса, не повреждая интактные клетки и ткани пациентов.
В качестве последних особенно обещающими успех являются соединения, которые препятствуют образованию биосинтезируемых в клетках человека протеиновых структурных элементов вируса и соединению этих структурных элементов в полные, инфекционные вирионы.
ВИЧ-1 и ВИЧ-2, смотря по обстоятельствам, в своем геноме имеют область, которая кодирует "gag-протеазу". Эта "gag-протеаза" ответственна за точное протеолитическое расщепление протеинов-предшественников, которые, в свою очередь, происходят из кодирующих "группу специфических антигенов" (gag) сегментов (отрезков) генома. При этом высвобождаются структурные протеины ядра вируса (по английски "Core" ядро, сердцевина). "gag-Протеаза" сама является составной частью кодированного за счет сегмента pol-генома ВИЧ-1 и ВИЧ-2 протеина-предшественника, который также содержит сегменты (отрезки) для "обратной транскриптазы" и "интегразы" и по всей вероятности расщепляется аутопротеолитически.
"gag-Протеаза" расщепляет основной протеин ядра ("Major Core Protein") p24 ВИЧ-1 и ВИЧ-2, предпочтительно N-терминальный пролиновых остатков, например, в двухвалентных радикалах Phe-Pro, Leu-Pro или Tyr-Pro. Речь идет о протеазе с каталитически активным аспартатным остатком в активном центре, так называемой аспартатпротеазе.
Если бы имелась возможность препятствовать действию "gag-протеазы", то у вируса не было бы в распоряжении необходимых протеинов для построения вирусного ядра. Это ограничивало бы или вовсе препятствовало бы размножению вируса. Таким образом имеется потребность в ингибиторах "gag-протеазы" для использования в качестве противовирусных средств против (СПИДа и других ретровирусных заболеваний.
Уже синтезирован ряд ингибиторов "gag-протеаз", которые содержат группы, представляющие собой не протеолитически расщепляемые пептидные изостеры (isostere). До сих пор, несмотря на интенсивные исследования, однако еще не удалось найти пригодный для применения для людей ингибитор аспартатпротеазы для борьбы со СПИДом у большей части инфицированных. Для этой цели решающими прежде всего являются фармакодинамические свойства. Кроме того, большинство известных до сих пор ингибиторов "gag-протеаз" содержат более, чем два, асимметрических атомов углерода в указанном центральном структурном элементе, что вызывает необходимость в относительно дорогостоящих стереоспецифических синтезах для разделения изомеров.
Задачей настоящего изобретения является изыскание доступного нового класса соединений-ингибиторов вирусных аспартатпротеаз с новым структурным центральным элементом в молекуле. Синтез этого центрального структурного элемента, кроме того, должен быть по возможности стерически прост. Поскольку новый структурный элемент с двух концов содержит аминогруппы при подборе заместителей можно использовать, например, аналогичные ретро-инверсно-пептидные структуры.
Согласно изобретению предлагаются производные гидразина общей формулы I.

где
R1 и R9 независимо друг от друга водород, низший алканоил, фенил-низш. алканоил, фенил-низш. алканоил, в котором остаток низшего алконоила замещен карбамоилом, морфолино-низш. алканоил, тиоморфолино-низш. алканоил, пиридил-низш. алканоил, хинолил-низш. алканоил, тетразолил-низш.алканоил, амино-низш. алканоил, замещенный у аминного азота N-морфолино- или N-тиоморфолинокарбонил, галоген-низш. алканоил, содержащий до трех атомов галогена, 2-(N-морфолино-низш.алкилкарбамоил)- низш. алкоксикарбонил, низший алкилсульфонил, N-пиридил-низш. алкил-N-низш.алкилкарбамоил, или ацильный остаток аминокислоты, выбранный из глицина, аланина, валина, лейцина, изолейцина, глютаминовой кислоты и аспарагина в форме (D)-, (L)- или (D,L)-, где α аминогруппа не замещена или ацилирована одним из вышеназванных остатков R1 или R9, при условии, что по меньшей мере один из остатков R1 и R9 означает водород;
R2, R4, R6 и R8 означают водород;
R3 низший алкил, циклогексил-низш. алкил или фенил-низш. алкил, незамещенный или замещенный галогеном, низшим алкокси или циано;
R5 означает гидроксильную группу;
R7 низший алкил, циклогексил-низш. алкил, или фенил-низш. алкил, незамещенный или замещенный галогеном, низшим алкокси или циано, или же их соли, в случае наличия солеобразующих групп.
Предпочтительны соединения формулы I, в
которой
R1
- низший алкоксикарбонил, фенил-низш. алкоксикарбонил, связанный через карбоксильную группу одновалентный радикал алифатической аминокислоты, выбранной из валина,
аланина, лейцина и изолейцина, или
связанный через карбоксильную группу, N-ацилированный по аминогруппе одним из остатков - фенил-(низший алканоил), морфолино-(низший алканоил), тиоморфолино(низший
алканоил), пиридил-(низший алканоил),
низший алкоксикарбонил или фенил-(низший алкоксикарбонил) радикал вышеуказанной алифатической аминокислоты, причем все названные аминокислоты представлены в
форме (D)- (L)-, или (D,L)-;
R2 водород;
R3 фенил-(низший алкил);
R4 водород;
R5 гидроксильная группа;
R6 водород;
R7 низший
алкил, циклогексил-низш. алкил или фенил-низш. алкил;
R8 водород; и одно из значений R1 и остатки R3 и R5 у асимметричных атомов углерода находятся
в S-конфигурации, а также фармакологически приемлемые соли этих соединений.
В частности, предпочтительны соединения формулы I, где
R1 трет.-бутоксикарбонил,
бензилоксикарбонил, связанный через карбоксильную группу одновалентный радикал аминокислоты валина или связанный через карбоксильную группу радикал аланина,
N-ацилированного по аминогруппе одним из
остатков фенилацетил, 3-пиридилацетил, морфолинокарбонил, тиоморфолинокарбонил, трет.-бутоксикарбонил или бензилоксикарбонил;
R2 водород;
R3 бензил;
R4 водород;
R5 гидроксильная группа;
R6 водород;
R7 изобутил, циклогексилметил или
бензил;
R8 водород; и
R9 одно из значений R1 и
остатки R3 и R5 у асимметричных атомов углерода находятся в
S-конфигурации, а также фармакологически приемлемые соли
этих соединений.
Особенно предпочтительны соединения формулы I, где
R1, и R9 соответственно
связанный через карбоксильную группу, N-ацилированный по
аминогруппе бензилоксикарбонилом одновалентный радикал аминокислоты-(L)-валина;
R2 и R8 водород;
R3 бензил;
R4 водород;
R5 гидроксильная группа;
R6 водород;
R7 бензил; и
остатки R3 и R5 у асимметричных атомов углерода находятся в
S-конфигурации, а также фармакологически приемлемые соли этих соединений: а также соединения формулы I, где
R1 и R9
соответственно связанный через карбоксильную группу,
N-ацилированный по аминогруппе 4-тиоморфолинокарбонилом одновалентный радикал аминокислоты (L)-валина;
R2 и R8
водород;
R3 бензил;
R4 водород;
R5 гидроксильная группа;
R6 водород;
R7 изобутил;
и
остатки R3 и R5 у асимметричных атомов
углерода находятся в S-конфигурации, а также фармакологически приемлемые соли этих соединений.
В частности, к
предпочтительным соединениям формулы I относятся соединения, выбранные
из:
Boc-[PheNNPhe]-Boc;
Boc-(L)-вал-[PheNNPhe] ← (L)-вал-Boc;
Boc-[PheNNCha]-Boc;
H-(L)-вал-[PheNNPhe] J
(L)-вал-H;
N-тиоморфолинокарбонил-(L)-вал-[PheNNPhe] J (N- тиоморфолинокарбонил-(L)-вал);
N-морфолинокарбонил-(L)-вал-[PheNNPhe] J
(N- морфолинокарбонил-(L)-вал);
фенилацетил-(L)-вал-[PheNNPhe] J (N-фенилацетил-(L)-вал);
N-(3-пиридилацетил)-(L)-вал-[PheNNPhe] J
(N-(3- пиридилацетил)-(L)-вал);
Boc-(L)-вал-[PheNNCha] J -(L)-вал-Boc;
Z-(L)-вал-[PheNNCha] J -(L)-вал-Z;
Boc-[PheNNLeu]-Boc;
Z-(L)-вал-[PheNNLeu] J -(L)-вал-Z, H-(L)-вал-[PheNNCha] J (L)-вал)-H и
N-(3-пиридилацетил)-(L)-вал-[PheNN
Leu] J (N-(3- пиридилацетил)-(L)-вал) или их соли,
где
Boc означает трет.-бутоксикарбонил,
Z бензилоксикарбонил,
остаток [PheNNPhe] означает двухвалентный
радикал
3(S)-амино-4-фенил-1- (N-бензилгидразино)-бутан-2(S)-ол и имеет формулу

где остаток [PheNNCha] означает двухвалентный радикал 3(S)-амино-4-фенил-
1-(N-циклогексилметилгидразино)-бутан-2(S)-ол и имеет формулу

где остаток [PheNNLeu] означает двухвалентный радикал 3(S)-амино-4-фенил-1-(N-изобутилгидразино)-бутан-2-(S)-ол и имеет формулу

и стрелка "←" означает поворот связи в отклонении от обычных пептидных номенклатур, причем влево амино- и вправо карбокси; а также соединения формулы I, выбранные из
Z-(L)-вал-[(p-F)PheNN(p-F)Phe] J (N-(N-(2-пиридилметил)-N- метиламинокарбонил)-(L)-вал);
Z-(L) -вал- [(p-F)PheNN(p-F)Phe] J (N-(2(R,S) -карбамоил-3-фенил-пропионил)-(L)-вал);
ацетил-(L)-вал-[PheNNCha] J (N-ацетил-(L)-вал);
ацетил-Ile- [PheNNCha] J (N-ацетил-Ile);
N-(2-пиридилметил)-N-метиламинокарбонил-(L)-вал-[PheNN (p-F)Phe] J (N-(N-(2-пиридилметил)-N-метиламинокарбонил)-(L)-вал);
Z-(L)-вал-[PheNN(p-F)Phe] J ((L)-вал)-Z;
Z-(L)-вал-[PheNN(p-CN)Phe] J ((L)-вал)-Z;
Z-(L)-Ile-[PheNNLeu] J ((L)-Ile)-Z;
изобутоксикарбонил-(L)-вал-[PheNNLeu] J (N- изобутоксикарбонил-(L)-вал);
ацетил-вал-[PheNNLeu] J (N-(2(R,S)-карбамоил-3- фенилпропионил)-вал;
N-трифторацетил-[PheNNLeu] J (N-(2(R, S)-карбамоил-3- фенилпропионил)-(L)-вал);
Z-(L)-вал-[PheNNNle] J (N-(2(R,S)-(N-(2-морфолинометил)- карбамоил)-3-метил)-бутирил);
Z-(L)-вал-[PheNNNle] J (N-(2(R, S)-(N-(2-пиридилметил)- карбамоил)-3-метил)-бутирил);
метоксикарбонил-(L)-вал-[PheNNLeu] J (N-метоксикарбонил- (L)-вал);
метоксикарбонил-(L)-вал-[PheNN(p-F)Phe] J (N- метоксикарбонил)-(L)-вал); и
метоксикарбонил-(L)-вал-[PheNN(p-CH)Phe] <-- (N- метоксикарбонил-(L)-вал) или их соли, где Z бензилоксикарбонил,
остаток [PheNNPhe] означает двухвалентный радикал 3(S)- амино-4-фенил-1-(N-бензилгидразино)-бутан-2(S)-ол и имеет формулу

где остаток [PheNNCha] означает двухвалентный радикал 3(S)-амино-4-фенил-1-(N-циклогексилметилгидразино)-бутан-2(S)-ол и имеет формулу

где остаток [PheNNLeu] означает двухвалентный радикал 3(S)-амино-4-фенил-1-(N-изобутилгидразино)-бутан-2(S)-ол и имеет формулу

остаток [PheNNNle] означает радикал 3(S)-амино-4-фенил-1-(N-n-бутилгидразино)-бутан-2(S)-ол и имеет формулу

остаток [PheNN(p-F)Phe] означает двухвалентный радикал 3(s)-амино-4-фенил-1-(N-(р-фторфенилметил)-гидразино)-бутан-2(S)-ол и имеет формулу

остаток [(p-F)PheNN(p-F)Phe] означает двухвалентный радикал 3(S)-амино-4-(p-фторфенил)-1-(N-(p-фторфенилметил)-гидразино)- бутан-2-(S)-ол и имеет формулу

остаток [PheNN(p-CN)Phe] означает двухвалентный радикал 3(S)-амино-4-фенил-1-(N-(p-цианофенилметил)-гидразино)-бутан-2(S)-ол и имеет формулу

и стрелка "<--" означает поворот связи в отклонение от обычных пептидных номенклатур, причем влево амино- и вправо карбокси.
Все соединения формулы 1 обладают свойствами ингибировать действие фермента ВИЧ-протеазы.
Выражение "низший" обозначает, что таким образом определенные группы или остатки, если не указано иное, содержат вплоть до 7 и предпочтительно вплоть до 4 C-атомов.
Замещенные радикалами R3 и R4, соответственно, R5 и R6 атомы углерода в соединениях формулы I, если они асимметричны, так же как и имеющиеся в случае необходимости другие асимметрические атомы углерода, могут быть в (R)-, (S)- или (R,S)-конфигурации. Таким образом настоящие соединения могут быть в виде смесей изомеров или в виде чистых изомеров, в особенности в виде смесей диастереомеров, пар энантиомеров или чистых энантиомеров. Предпочтительны соединения формулы I, где R3 и R5 находится в (S)- конфигурации, а другие, возможно имеющиеся асимметрические атомы углерода, независимо друг от друга, находятся в (R)-, (s)- или (R,S)- конфигурации.
Используемые в описании настоящего изобретения общие выражения и обозначения предпочтительно имеют следующие значения, причем в различных областях
определения
выше- и нижеуказанных остатков могут применяться любые комбинации или индивидуальные остатки вместо общих определений:
Ацил R1 или R9 является в первую
очередь ацильной
группой карбоновой кислоты, неполного сложного эфира угольной кислоты.
Предпочтительные ацильные группы R1 и R9 карбоновой кислоты представляют собой низший алканоил, как: формил, ацетил, пропионил, бутирил или пивалоил; или замещенный низший алканоил, как: (низший алкокси)-(низший алканоил), например, (низший алкокси)-ацетил или (низший алкокси)- пропионил, как метоксиацетил, этоксиацетил или 3-метоксипропионил; аминонизший алканоил означает, например, 2-аминоацетил или 2-амино-3-пропионил.
Предпочтительные ацильные группы R1 и R9 неполного сложного эфира угольной кислоты представляют собой (низший алкокси)-карбонил, например, метокси-, этокси-, изопропокси-, изобутокси- или трет. -(низший алкокси)- карбонил, как трет. -бутоксикарбонил или изобутоксикарбонил: 2-галоген (низший алкокси)-карбонил, как 2-хлор-, 2-бром-, 2-иод- или 2, 2, 2- трихлорэтоксикарбонил.
Предпочтительными ацильными группами R1 и R9 незамещенной или замещенной карбаминовой кислоты, наряду с пригодными, уже названными в качестве предпочтительных ацильных остатков
R1 и R9 остатками, являются:
незамещенный или замещенный N-гетероциклил N-алкилкарбамоил, где гетероциклил предпочтительно пиридил, как 2-, 3- или 4-пиридил, в первую
очередь в N-гетероциклил(низший алкил)-N-(низший алкил)-карбамоиле, например, как N-пиридил(низший алкил)-N-(низший алкил)-карбамоил, как N-(2-, 3- или 4-пиридилметил)-N-метилкарбамоил, или
N-гетероциклил-(низший алкил)-карбамоил, как, например, 2- или 3-пиридил(низший алкил)- аминокарбонил, как 2- или 3-пиридилметиламинокарбоксил; или, например, 2-(N-морфолино-N-(низший
алкил)-карбамоил)-(низший алканоил), как 2-(R,S)-[N-(2-N-морфолиноэтил)-карбамоил] -3-метилбутирил, или 2-(N-[пиридил-(низший алкил)]-карбамоил)-(низший алканоил), или (2-(R,
S)-(N-(2-пиридилметил)-карбамоил)-3-метил)-бутирил; галоген низш. алканоил, который, предпочтительно, содержит вплоть до 3-х атомов галогена, представляет собой, например, α -галогенацетил,
как
a -фтор-, a -хлор-, a -бром-, a -иод-, a,α,α -трифтор- или a,α,α -трихлорацетил-, или галогенпропионил, как b -хлор- или b -бромпропионил;
алкилсульфонил, как
метил или этилсульфонил;
фенил-низший алкоксикарбонил представляет собой, предпочтительно, бензилоксикарбонил.
Предпочтительные ацильные группы R1 и R9
незамещенной или замещенной аминокислоты образуются за счет аминокислотных остатков a- или β--аминокислоты, в особенности:
природной α -аминокислоты с L-конфигурацией, которая
обычно имеется в протеинах, или эпимера такой аминокислоты, то есть с неприродной D-конфигурацией, или ее D,L-изомерной смеси.
R2, R4, R6К и R8 обозначают водород.
R3 и R7 обозначают низший алкил, как изобутил или н-бутил; обозначают циклогексил(низший алкил) или фенилнизший алкил, незамещенный или замещенный низшим алкокси, галогеном или циано.
R5 обозначает гидроксил.
R3 и R7, прежде всего, в случае циклогексил-(низший алкил), циклогексилметил, или, в случае фенил-(низший алкил), который незамещен или замещен указанными заместителями, это бензил, 2-фенилэтил, 3-фенилпропил, 4-фтор-, 4-циано-, 4-метокси, или его соль, если имеется солеобразующая группа.
Соли соединений формулы I представляют собой в особенности соли присоединения кислот, соли с основаниями или при наличии нескольких солеобразующих групп, в случае необходимости также смешанные или внутренние соли.
Солями являются в первую очередь фармацевтически применимые нетоксичные соли соединений формулы I.
Такие соли образуются, например, соединениями формулы I с кислой группой и представляют собой, например, соли с пригодными основаниями, соли с нетоксичными металлами. Периодической системы элементов соли металлов групп IА, IБ, IIА и IIБ; в первую очередь это соли щелочных металлов, например, соли лития, натрия, или калия, или соли щелочноземельных металлов, например, соли магния или кальция; далее, соли цинка или аммония, также такие соли, которые образуются с органическими аминами, в случае необходимости замещенными гидроксилом моно-, ди- или триалкиламинами, в особенности моно-, ди- или три-(низший алкил)-аминами, или с четвертичными аммониевыми соединениями, например, с N-метил-N-этиламином, диэтиламином, триэтиламином, моно-, бис- или трис-[2-окси-(низший алкил)] -аминами, как моно-, бис- или трис-(2-оксиэтил)-амин, 2-окси-трет.-бутиламин или трис-(оксиметил)-метиламин: N, N-ди-(низший алкил)-N-(окси-(низший алкил)-аминами, как N, N-диметил-N-(2-оксиэтил)- амин или три-(2-оксиэтил)-амин, N-метил-D-глюкамином, или четвертичными аммониевыми солями, как тетрабутиламмониевые соли. Соединения формулы I с основной группой, например, аминогруппой, могут образовывать соли присоединения кислот, например, с неорганическими кислотами, как, например, галогенводородная кислота, как соляная кислота, серная кислота или фосфорная кислота, или с органическими карбоновыми, сульфо-, сульфоновыми или фосфоновыми кислотами или N-замещенными сульфаминовыми кислотами, как, например, уксусная кислота, пропионовая кислота, гликолевая кислота, янтарная кислота, малеиновая кислота, оксималеиновая кислота, метилмалеиновая кислота, фумаровая кислота, яблочная кислота, винная кислота, глюконовая кислота, глюкаровая кислота, глюкуроновая кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, салициловая кислота, 4-аминосалициловая кислота, 2-феноксибензойная кислота, 2- ацетоксибензойная кислота, эмбоновая кислота, никотиновая кислота, или изоникотиновая кислота; далее, с аминокислотами, как, например, далее называемыми a -аминокислотами, в особенности с глутаминовой кислотой и аспарагиновой кислотой, а также с метансульфоновой, этансульфокислотой, 2-оксиэтансульфокислотой, этан-1,2-дисульфокислотой, бензолсульфокислотой, 4-метилбензолсульфокислотой, нафталин-2-сульфокислотой, 2- или 3- фосфоглицератом, глюкоза-6-фосфатом, N-циклогексилсульфаминовой кислотой (при образовании цикламатов) или с другими кислыми органическими соединениями, как аскорбиновая кислота. Соединения формулы I с кислотными и основными группами также могут образовывать внутренние соли.
Для выделения или очистки также могут найти применение фармацевтически непригодные соли.
Соединения настоящего изобретения обладают ингибирующим действием на вирусные аспартатпротеазы, в особенности подавляющими gag-протеазу действием. В первую очередь предлагаемые соединения при испытании в нижеописанном тесте в концентрации 10-6 10-9 моль/л подавляют действие gag-протеазы ВИЧ-1 и ВИЧ-2 и поэтому пригодны в качестве средства против вызываемых этими или применяемыми ретровирусами заболеваний, как, например, против СПИДа.
Способность соединений формулы I ингибировать протеолитическую активность, например, ВИЧ-1-протеазы, можно продемонстрировать, например, согласно способу, описанному J. Hansen et al. The EMBO Journal, 7 1785 1791 (1988). При этом ингибирование действия gag-протеазы измеряется на субстрате, который представляет собой выраженный в E. coli, слитый протеин из gag-предшественника протеина и MS-2. Субстрат и продукты его расщепления разделяют путем полиакриламид-гель-электрофореза и проявляют за счет иммуноблоттинга с моноклональными антителами против MS-2.
В еще более просто осуществляемом тесте, который делает возможным точные количественные показания, в качестве субстрата для gag-протеазы используется синтетический пептид, который соответствует месту расщепления gag-предшественника-протеина. Этот субстрат и его продукты расщепления можно измерять с помощью жидкостной хроматографии высокого давления (ЖХВД).
Например, в качестве субстрата для рекомбинантной ВИЧ-1-протеазы [получение см. Billich S et al, J. Biol Chem. 263(34), 17905 17908 (1990)] используется синтетический хромофорный пептид-(например, HKARLV (NO2 FEANLES (Bachem, Швейцария) или Icosa-пептид, как RRSNQVSQNYPQNIQGRR (получен пептидным синтезом по известному способу), который соответствует месту расщепления gag-предшественника протеина. Этот субстрат и продукты его расщепления можно измерять с помощью ЖХВД.
Для этой цели испытуемый ингибитор формулы I растворяют в диметилсульфоксиде; ферментный тест осуществляют добавлением пригодных разбавлений ингибитора в 20 мМ b -морфолиноэтансульфокислоты [(MES)- буфер, pH 6,0] к смеси для испытания из 67,2 мкМ указанного хромофорного пептида в 0,3 М ацетата натрия, 0,1 м NaCl, pH 7,4, или 122 мкМ вышеуказанного Icosa-пептида в 20 мМ MES-буфера с pH 6,0. Объем смеси составляет 100 мкл. Реакция инициируется добавлением в первом случае 2 мкл, во втором случае - 10 мкл ВИЧ-1-протеазы и прекращается в первом случае спустя 15 мин благодаря добавке 100 мкл 0,3 М HClO4, во втором случае спустя час инкубации при 37oC благодаря добавке 10 мкл 0,3 М HClO4. Продукты реакции после центрифугирования пробы в течение 5 мин при 10000 g в 100 мкл (смесь с хромофорным пептидом), соответственно, 20 мкл (смесь с Icosa-пептидом) полученной надосадочной жидкости, после нанесения на колонку размером 125x4,6 мм Nucleosil® C 18 5 мк(ЖХВД) (Macherey and Nagel, D

При этом для соединения формулы I определяются предпочтительно IC50-значения (IC50 такая концентрация, которая снижает активность ВИЧ-1 протеазы по сравнению с контролем без ингибитора на 50%) примерно 10-6 10-9 М, в особенности, примерно 10-7-примерно 10-8 М.
В следующем тесте можно показать, что соединения настоящего изобретения защищают клетки, которые обычно инфицируются ВИЧ, от такой инфекции или при меньшей мере замедляют такую инфекцию. При этом человеческую линию клеток ЯМТ-2 с T-клеточной лейкемией [Science, 229, 563 (1985)] которая крайне чувствительна к цитопатогенному эффекту ВИЧ, инкубируют с одним ВИЧ или с ВИЧ в присутствии предлагаемых в изобретении соединений и спустя несколько дней оценивают жизнеспособность таким образом обработанных клеток.
Для этой цели МТ-2-клетки выдерживают при 37oC во влажном воздухе с 5% CO2 в RPMI-1640-среде (Gibco, Швейцария: RPMI-1640 содержит смесь аминокислот без L-Gln), которая дополнена 10% активированным L-глутамином при нагревании эмбриональной телячьей сыворотки, Hepes [2-(4-(2-оксиэтил)-1-пиперазино)-этансульфокислота] и стандартным антибиотиком. 50 мкл соответствующего испытуемого соединения в питательной среде и 100 мкл ВИЧ-1 в питательной среде (800 TCID 50/мл) (TCID 50 Tissue Culture Infections 50 доза, которая инфицирует 50% МТ-2 клеток) добавляют к 4•103 экспоненциально растущих МТ-2 клеток в 50 мкл питательной среды на углубление на микротитрационном планшете с 96-тью отверстиями.
Параллельные смеси на другом микротитрационном планшете с клетками и испытуемым соединением получают 100 мкл питательной среды без вируса. После инкубации в течение 4 дней в 100 мкл надклеточной жидкости определяют активность (RT) обратной транскриптазы. RT-активность определяется в 50 mM Трис α,α,α -трис(оксиметил) -метиламин, сверхчистый, Merck, ФРГ; pH 7,8; 75 mM KCl, 2 mM дитиотреитола, 5 mM MgCl2; 0, 05% Nonidet P-40 (детергент, Сигма, Швейцария); 50 мкг/мл полиадениловой кислоты (Фармация, Швейцария); 1,6 мкг/мл dT (12 18) (Сигма, Швейцария). Смесь отфильтровывают через 0,45 Мк Acrodisc-фильтр (Gellman Science Inc. Ann Arbor) и хранят при 20oC. К аликвотной доле этого раствора добавляют 0,1% (по объему) [альфа32P]dTTP для достижения радиоактивной конечной активности 10 мкCi/мл. 10 мкл надосадочной культуры переносят на новый, с 96-тью отверстиями микротитрационный планшет и туда добавляют 30 мкл указанного RT-"коктейля". После смешения пластину (планшет) инкубируют 1,5 3 ч при 37oC. 5 мкл этой реакционной смеси переносят на бумагу Ватман DE 81 (Ватман). Высушенные фильтры промывают 3 раза по 5 мин с помощью 300 mM NaCl/25 mM тринатрийцитрата и 1 раз с помощью 95% -ного этанола и снова высушивают на воздухе. Оценку осуществляют в Matrix Packard 96 Well Counter (Паккард). Значения ЭД90 достигаются и определяются как самая низкая концентрация соответствующего испытуемого соединения, которая снижает RT-активность на 90% по сравнению с необработанным испытуемым соединением клеточными смесями. RT-активность при этом является мерой размножения ВИЧ-1.
Предлагаемые в изобретении соединения при этом показывают ЭД90 примерно 10-5 10-8 M, предпочтительно примерно 5•10-7 5•10-8.
Данные, показывающие активность заявленных соединений.
Ингибирование in vitro ВИЧ-протеазы.
По аналогии с методом, описанным F.D.Richards et al. J. Biol. Chem. 265(14), 7733 7736, определяли способность нижеуказанных соединений согласно примерам на ингибирование ВИЧ-1-протеазы (полученной по способу S.Billich et al. J. Biol. Chem. 263(34), 17905 17908 (1990) в присутствии субстанции-аналога Icosa-пептиду RRSNQVSQNYPIVQNIQGRR (субстрат ВИЧ-1-протеазы, полученный пептидным синтезом по известным способам: J.Schneider et al. Cell 54, 363 368 (1988)), который соответствует месту расщепления gag-предшественника-протеина. Этот субстрат-аналог и продукт его расщепления анализировались количественно с помощью жидкостной хроматографии высокого давления (ЖХВД).
Соответствующая испытуемая субстанция растворялась в диметилсульфоксиде. Ферментный тест проводился путем добавления пригодных растворителей испытуемой субстанции в 20 mM b-морфолиноэтанолсульфоновой кислоты (MES)-буфер, pH 6,0 к испытуемой смеси, которая содержала вышеуказанный Icosa-пептид (122 мкМ) в 20 mM MES-буфера, pH 6,0. Испытуемый объем на все компоненты составлял 100 мкл. Реакция начиналась в результате добавления 10 мкл раствора ВИЧ-1-протеазы и заканчивалась через 1 ч инкубирования при 37oC в результате добавления 10 мкл О,3 M CHlO4. После центрифугирования пробы в течение 5 мин при 10000•г. 20 мкл полученного осадка поступали на®Nucleosil C-18-5мк-ЖХВД-колонку (Machery Nagel, Duren, Германия). Нерасщекпленные Icosa-пептиды, так же как продукты расщепления, после колонны элюировались с помощью следующих градиентов: 100% элюент 1 > 50% элюент 1 + 50% элюент 2 (элюент 1: 10% ацетонитрила, 90% H2O, 0,1% трифторуксусной кислоты; элюент 2: 75% ацетонитрила, 25% H2O, 0,08% трифторуксусной кислоты) в течение 15 мин, скорость протекания 1 мл/мин. После элюирования продукты реакции количественно анализировались с помощью пика высоты продуктов расщепления при 215 нм.
Были получены следующие результаты (табл. 1).
Значение IC50 показывает такие концентрации испытуемых соединений, при которых эти соединения проявляют половину максимального ингибирования.
Ингибирование in-vitro ВИЧ-инфекции в МТ-2-клетках культур
МТ-2-клетки применялись так, как описано выше,
но с использованием ED100, то есть концентрации испытуемой
субстанции, в которой больше не обнаруживали активности и остатков резервов транскриптазы в осадке инфицированных клеток. При
этом были получены следующие данные для ингибирования ВИЧ-1-активности
(измеренная как активность виральных ферментов резерва транскриптазы), табл. 2.
Анализ крови у мышей на содержание испытуемых соединений.
Титульное соединение согласно
примеру 55 растворялось в диметилсульфоксиде в концентрации 240 мг/мл. Полученный раствор растворялся в 20% (мас.
/объем) растворе гидроксипропил-β-циклодекстрина (в воде) до получения
концентрации испытуемого соединения 12 мг/мл. Этот раствор с помощью зонда вводился женским особям мышей в дозе 120 мг/кг,
через 30 мин после введения раствора мыши убивались и их кровь собиралась.
Исследовали 3 или 4 животных. Кровь гепаринизировалась и подготавливалась для анализа по следующей методике: кровь
депротеинировалась путем смешивания 1 объемной части крови с 1 частью ацетонитрила;
после центрифугирования осадок исследовался с помощью резервной фазы ЖХВД: анализ проводился на®
Nucleosil C-18-ЖХВД-колонне(5 мк; Machery Nagel, Duren, Германия), заполненной
мобильной фазой из 47% ацетонитрила в воде/0,1% трифторуксусной кислоты. Скорость протекания 1 мл/мин, детекция
происходила при 215 нм. Стандарты для титульного соединения по примеру 55
разрабатывались аналогично и использовались для получения калибровочной кривой, которая применялась для определения
концентраций соединения in vivo. Была установлена следующая концентрация:
Концентрация (мкМ) в крови титульного соединения по примеру 55 Время после введения (мин)
2,08 30
Аналогично были определены следующие концентрации для следующих примеров (табл.
3).
Данные о токсичности соединений
Испытанные соединения показали следующее:
а) тест
на мутагенез по Амсу мутагенез не наблюдался,
б) в опытах на клетках с
МТ-2-клетками цитотоксичность не наблюдалась,
в) в опытах на мышах с испытанными соединениями токсичность не
наблюдалась.
Активность и отсутствие токсичности заявленных соединений вытекает из представленных данных. Из этих данных следует, что соединения in vitro ингибируют ВИЧ-протеазу вирусов, вызывающих СПИД, препятствуют развитию инфекции, вызванной вирусом, в клетках и у мышей, дают удовлетворительные результаты анализов крови.
Соединения формулы I и соли
соединений по меньшей мере с одной солеобразующей группой получаются известным
способом, например:
а) производное гидразина формулы (III):

где остатки имеют вышеуказанные значения, подвергают взаимодействию с эпоксидом формулы (IV)

где остатки имеют вышеуказанные значения, причем свободные функциональные группы, за исключением принимающих участие в реакции, в случае необходимости, защищены и их в случае необходимости отщепляют; или
б) для получения соединений формулы I, где R1 и R9 обозначают алканоил, сульфонил, сульфамоил, который на азоте незамещен или замещен, имеющие вышеуказанные значения.
R2 и R8 обозначают водород,
аминосоединение формулы (V)

где остатки имеют вышеуказанные значения, конденсируют с кислотой формулы (VI):
R9 OH
или реакционноспособными производными этой кислоты, где R9 имеет вышеуказанные значения, причем свободные функциональные группы, за исключением принимающих участие в реакции, в случае необходимости, находятся в защищенной форме, и в случае необходимости затем отщепляют, или
в) для получения соединений формулы I, где R1 и R9 обозначают алканоил, сульфонил, сульфамоил, который незамещен или замещен на азоте, имеющие вышеуказанные значения,
R2 обозначает водород, R3 незамещенный или замещенный алкил, а остальные остатки имеют указанные значения,
аминосоединение формулы (VII)

где остатки имеют вышеуказанные значения, конденсируются с кислотой формулы (VIII):
R1 ОН
или реакционноспособными производными этой кислоты, где R1 имеет вышеуказанные значения, причем свободные функциональные группы, за исключением принимающих участие в реакции, в случае необходимости находятся в защищенной форме, и в случае необходимости эти защитные группы отщепляют; или
г) для получения соединений формулы I, где R1 и R9 обозначают два одинаковых остатка, выбираемых из алканоила; сульфамоила, который на азоте не замещен или замещен, сульфонила, имеющих вышеуказанные значения;
R2 обозначает водород, R3 незамещенный или замещенный алкил, а остальные остатки имеют указанные значения,
диаминосоединение формулы (IX):

где остатки имеют вышеуказанные значения, конденсируют с пригодной для введения идентичных остатков R1 и R9 кислотой или реакционноспособными производными этой кислоты, где R1 и R9 имеют вышеуказанные значения, причем свободные функциональные группы, за исключением принимающих участие в реакции, в случае необходимости находятся в защищенной форме, и в случае необходимости эти защитные группы отщепляют: или
д) для получения соединения формулы I, где вместо R7 находится остаток

в соединение формулы (I'):

где



где X обозначает отщепляемую группу, а

е) в соединении формулы I, где заместители имеют вышеуказанные значения, с тем условием, что в соответствующем соединении формулы I по меньшей мере одна функциональная группа защищена защитными группами, отщепляют имеющиеся защитные группы,
и, если желательно, получаемое по одному из вышеуказанных способов а)- е) соединение формулы I по меньшей мере с одной солеобразующей группой переводят в его соль или полученную соль переводят в свободное соединение или в другую соль, и/или в случае необходимости разделяют полученную смесь изомеров и/или предлагаемое в изобретении соединение формулы I превращают в другое, предлагаемое в изобретении, соединение формулы I.
Вышеприведенные способы ниже описываются подробнее:
Способ а) Присоединение амина к эпоксиду.
Принимающая участие в реакции аминогруппа производных гидразина формулы III, в зависимости от значения R7, содержит предпочтительно по меньшей мере один свободный атом водорода; однако, также она сама может быть дериватизирована, чтобы увеличить реакционноспособность производного гидразина.
Эпоксид формулы IV имеет в особенности структуру, которая, желательно, должна способствовать протеканию концевого присоединения производного гидразина.
Функциональные группы в исходных материалах, превращения которых нужно избегать, в особенности карбокси-, амино-, окси-, могут быть защищены пригодными защитными группами, которые обычно применяются при синтезе пептидных соединений, или цефалоспоринов и пенициллинов, а также производных нуклеиновых кислот и сахаров. Эти защитные группы могут вводиться на предварительных стадиях и должны защищать соответствующие функциональные группы от нежелательных побочных реакций, как ацилирования, этерификации до простых и сложных эфиров, окислений, сольволиза и т.д. В определенных случаях защитные группы, сверх того, могут способствовать селективному, например, стереоселективному протеканию превращений. Характерным для защитных групп является то, что они легко, то есть без нежелательных побочных реакций, отщепляются, например, сольволизом, восстановлением, протеолизом или ферментативно, например, в физиологических растворах. Аналогичные защитным группам остатки, однако, также могут иметься в конечных (целевых) веществах. Соединения формулы I с защищенными функциональными группами могут обладать повышенной метаболической стабильностью или другого рода улучшенными фармакодинамическими свойствами, чем соответствующие соединения со свободными функциональными группами. О защитных группах в более узком смысле речь шла выше и пойдет ниже, когда соответствующие остатки более не присутствуют в целевых веществах.
Защита функциональных групп такими защитными группами, сами защитные группы, а также реакции их отщепления описаны, например, в стандартных работах, как J. F. W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, Лондон и Нью-Йорк, 1973; Th.W.Greene, "Protective Groups in Organic Synthesis", Wiley, Нью-Йорк, 1981; "The Peptides, том 3 (E.Gross J.Meienhofer, Herausg.), Academic Press, Лондон и Нью-Йорк, 1981; "Методы органической химии", Губен-Вейль, 4-е издание, том 15/1, Georg Thieme Verlag, Штуттгарт, 1974; H. -D. Jakubke H.Jescheit, "Aminos γ uren, Peptide, Proteine", изд. Chemie, Weinheim, Deerfield Beach und Basel 1982; и в Jochen Lehmann, "Chemie der Kohlenhydrate: Monosaccharide und Derivate", Georg Thieme Verlag, Штуттгарт, 1974.
Карбоксигруппа защищена, например, в виде сложноэфирной группы, которая селективно расщепляется в мягких условиях. Защищенная в сложноэфирной форме карбоксигруппа этерифицирована в первую очередь низшей алкильной группой, которая предпочтительно разветвлена в 1-положении низшей алкильной группы или замещена в 1- или 2-положении низшей алкильной группы пригодными заместителями.
Защищенной карбоксигруппой, которая этерифицирована низшей алкильной группой, является, например, метоксикарбонил или этоксикарбонил.
Защищенной карбоксигруппой, которая этерифицирована низшей алкильной группой, разветвленной в 1-положении низшей алкильной группы, является, например, трет.-(низший алкокси)-карбонил, например, трет. бутоксикарбонил.
Защищенная карбоксигруппа, которая этерифицирована низшей алкильной группой, которая замещена в 1- или 2-положении низшей алкильной группы пригодными заместителями, представляет собой, например, арилметоксикарбонил с одним или двумя арильными остатками, где арил не замещен или обозначает, например, фенил, моно-, ди- или тризамещенный низшим алкилом, например, трет.-низшим алкилом, как трет.-бутил, низшим алкоксилом, например, метокси, гидроксилом, галогеном, например, хлором, и/или нитрогруппой; например, бензилоксикарбонил, замещенный указанными заместителями бензилоксикарбонил, например, 4-нитробензилоксикарбонил или 4-метоксибензилоксикарбонил, дифенилметоксикарбонил или замещенный указанными заместителями дифенилметоксикарбонил, например, ди-(4-метоксифенил)-метоксикарбонил, далее, этерифицированный низшей алкильной группой карбоксил, причем низшая алкильная группа замещена в 1-или 2-положении пригодными заместителями, как 1-[(низший алкокси)-(низший алкокси)]-карбонил, например, метоксиметоксикарбонил, 1-метоксиэтоксикарбонил или 1-этоксиэтоксикарбонил, 1-(низший алкил)-тио-(низший алкокси)- карбонил, например, 1-метил-тиометоксикарбонил или 1-этилтиоэтоксикарбонил, ароилметоксикарбонил, где ароильная группа представляет собой в случае необходимости замещенный, например, галогеном, как бром, бензоил, например, фенацилоксикарбонил, 2-галоген-(низший алкокси)-карбонил, например, 2,2,2-трихлорэтоксикарбонил, 2-бромэтоксикарбонил или 2- йодэтоксикарбонил, а также 2-(тризамещенный силил)-(низший алкокси)- карбонил, где заместители, независимо друг от друга, обозначают по алифатическому, аралифатическому, циклоалифатическому или ароматическому углеводородному остатку, замещенному в случае необходимости, например, низшим алкилом, низшим алкоксилом, арилом, галогеном, и/или нитрогруппой, как, например, в случае необходимости замещенный, как указано выше, низший алкил, фенил-(низший алкил), циклоалкил или фенил, например, 2-три-(низший алкил)-силил-(низший алкокси)-карбонил, как 2-три-(низший алкил)-силил-этоксикарбонил, например, 2-триметилсилилэтоксикарбонил или 2-(ди-н-бутил-метилсилил)-этоксикарбонил, или 2-триарилсилилэтоксикарбонил, как трифенилсилилэтоксикарбонил.
Карбоксигруппа также может быть защищена в виде органической силилкарбонильной группы. Органическая силилоксикарбонильная группа представляет собой, например, три-(низший алкил)-силилоксикарбонильную группу, например, триметилсилилоксикарбонил. Атом кремния силилоксикарбонильной группы также может быть замещен двумя низшими алкильными группами, например, как метальные группы, и аминогруппой или карбоксигруппой второй молекулы формулы I. Соединения с такими защитными группами можно получать, например, с помощью диметилхлорсилана в качестве силилирующего средства.
Защищенная карбоксигруппа представляет собой предпочтительно трет.- (низший алкокси)-карбонил, например, трет.-бутоксикарбонил. бензилоксикарбонил, 4-нитробензилоксикарбонил, 9-флуоренилметоксикарбонил или дифенилметоксикарбонил.
Защищенная аминогруппа может быть защищена защитной для аминогруппы группой, например, быть в форме ациламино-, арил-метиламино-, этерифицированной до простого эфира меркаптоамино-, 2-ацил-(низший алк-1-енил)-амино- или силиламиногруппы или в виде азидогруппы.
В соответствующей ациламино-группе ацил представляет собой, например, ацильный остаток органической карбоновой кислоты, например, с количеством атомов углерода вплоть до 18, в особенности в случае необходимости замещенной, например, галогеном или арилом, низшей алканкарбоновой кислоты или в случае необходимости замещенной, например, галогеном, низшим алкоксилом или нитро-группой бензойной кислоты, или предпочтительно неполного сложного эфира угольной кислоты. Такими ацильными группами являются, например, низший алканоил, как формил, ацетил, пропионил или пивалоил; галоген-(низший алканоил), например, 2-галогенацетил, как 2-хлор-, 2-бром-, 2-йод-, 2,2, 2-трифтор- или 2,2,2-трихлорацетил; в случае необходимости замещенный, например, галогеном, низшим алкоксилом или нитрогруппой бензоил, как бензоил, 4-хлорбензоил, 4-метоксибензоил или 4- нитробензоил; низший алкоксикарбонил, предпочтительно разветвленный в 1- положении низшего алкильного остатка или пригодным образом замещенный в 1- или 2-положении (низший алкокси)-карбонил, например, трет. -(низший алкокси)-карбонил, как трет. -бутоксикарбонил, арилметоксикарбонил с одним, двумя или тремя арильными остатками, которые представляют собой одно или многократно замещенный в случае необходимости, например, низшим алкилом, в особенности трет.-низшим алкилом, как трет.-бутил, низшим алкоксилом, как метокси, гидроксилом, галогеном, как хлор, и/или нитро-группой фенил, например, бензилоксикарбонил, 4-нитробензилоксикарбонил, дифенилметоксикарбонил, 9-флуоренилметоксикарбонил или ди-(4-метоксифенил)- метоксикарбонил, ароилметоксикарбонил, где ароильная группа представляет собой предпочтительно в случае необходимости замещенный, например, галогеном, как бром, бензоил, например, фенацетилоксикарбонил; 2-галоген(низший алкокси)-карбонил, например, 2,2, 2-трихлорэтоксикарбонил, 2- бромэтоксикарбонил или 2-йодэтоксикарбонил; 2-(тризамещенный силил)- (низший алкокси)-карбонил, например, три-(низший алкил)-силил-(низший алкокси)-карбонил, как 2-триметилсилилэтоксикарбонил или 2-(ди-н-бутилметилсилил)-этоксикарбонил, или триароилсилил-(низший алкокси)-карбонил, например, 2-трифенилсилилэтоксикарбонил.
В арилметиламиногруппе, которая представляет собой, например, моно-, ди- или в особенности триарилметиламино-группу, арильные остатки представляют собой в особенности в случае необходимости замещенные фенильные остатки. Такими группами являются, например, бензил-, дифенилметил- или в особенности трифенилметиламиногруппа.
В этерифицированной до простого эфира меркаптоаминогруппе меркаптогруппа находится в первую очередь в виде замещенного арилтио или арил(низший алкил)-тио, где арил представляет собой, например, замещенный в случае необходимости, например, низшим алкилом, как метил или трет.-бутил, низшим алкокси, как метокси, галогеном, как хлор, и/или нитрогруппой фенил, например, 4-нитрофенилтио.
В используемой в качестве защитной для аминогруппы группе 2-ацил(низший алк-1-енильный) остаток представляет собой ацил, например, соответствующий остаток низшей алканкарбоновой кислоты; замещенной в случае необходимости, например, низшим алкилом, как метил или трет.-бутил, низшим алкоксилом, как метокси, галогеном, как хлор, и/или нитрогруппой бензойной кислоты, или в особенности неполного сложного эфира угольной кислоты, как неполного сложного низшего алкильного эфира угольной кислоты. Соответствующими защитными группами в первую очередь являются 1-(низший алканоил)-(низший алк-2-ен-2-ил), например, 1-(низший алканоил)-проп-1- ен-2-ил, как 1-ацетилпроп-1-ен-2-ил, или (низший алкокси)-карбонил(низший алк-1-ен-2-ил), например, (низший алкокси)-карбонил-проп-1-ен-2- ил, как 1-этоксикарбонил-проп-1-ен-2-ил.
Силиламиногруппой является, например, три-(низший алкил)-силиламино-группа, например, триметилсилиламино- или трет. -бутил-диметилсилиламино-группа. Атом кремния силиламиногруппы также может быть замещен только двумя низшими алкильными группами, например, метильными группами, и аминогруппой или карбоксигруппой второй молекулы формулы 1. Соединения с такими защитными группами можно получать, например, с помощью хлорсиланов, как диметилхлорсилан, в качестве силилирующего средства.
Аминогруппу также можно защищать путем переведения в протонированную форму; в качестве соответствующих анионов в первую очередь принимают во внимание такие сильные неорганические кислоты, как серная, фосфорная кислоты или галогенводородные кислоты, например, хлор- или броманион, или органических сульфокислот, как п-толуолсульфокислота.
Предпочтительными защитными для аминогруппы группами являются низший алкоксикарбонил, фенил-(низший алкокси)-карбонил, флуоренил(низший алкокси)-карбонил, 2-(низший алканоил)-(низший алк-1-ен-2-ил)- или (низший алкокси)-карбонил-(низший алк-1-ен-2-ил).
Гидроксильная группа может быть защищена, например, ацильной группой, например, замещенным галогеном, как хлор, низшим алканоилом, как 2,2- дихлорацетил, или в особенности указанным для защищенных аминогрупп ацильным остатком неполного сложного эфира угольной кислоты.
Предпочтительной защитной для гидроксильной группы группой является, например, 2,2,2-трихлорэтоксикарбонил, 4-нитробензилоксикарбонил, дифенилметоксикарбонил или трифенилметил. Гидроксильная группа, далее, может быть защищена три-(низший алкил)-силилом, например, как триметилсилилом, триизопропилсилилом или трет. -бутилдиметилсилилом; легко отщепляющейся, этерифицирующей до простого эфира группой, например, алкильной группой, как трет.-низший алкил, например, трет.-бутил; окса- или тиа-алифатическим или -циклоалифатическим, в особенности 2-окса- или 2- тиаалифатическим или -циклоалифатическим углеводородным остатком, например, 1-(низший алкокси)-(низший алкилом) или 1-(низший алкил)-тио(низшим алкилом), как метоксиметил, 1-метоксиэтил, 1-этоксиэтил, метилтиометил, 1-метилтиоэтил или 1-этилтиоэтил, или 2-окса- или 2-тиациклоалкилом с 5-7 атомами в кольце, как тетрагидрофурил или 2- тетрагидропиранил, или соответствующий тиааналог, а также 1-фенил-(низшим алкилом), как бензил, дифенилметил или трифенилметил, причем фенильные остатки могут быть замещены, например, галогеном, как хлор, низшим алкоксилом, как метокси, и/или нитрогруппой.
Две, присутствующие в молекуле, в особенности соседние гидроксильные группы или соседние окси- и аминогруппа, могут быть защищены, например, двухвалентными защитными группами, как предпочтительно, например, защищенная одним или двумя низшими алкильными остатками или оксогруппой метиленовая группа, например, незамещенная или замещенная алкилиденом, например, низшим алкилиденом, как изопропилиден, циклоалкилиденом, как циклогексилиден, карбонильной группой или бензилиденом.
В качестве защитной группы, например, защитной для карбоксигруппы группы, в настоящем изобретении, нужно рассматривать также определенным образом связанный и легко отщепляющийся с защищаемой функциональной группой, например, карбоксигруппой, полимерный носитель, который пригоден, например, для синтеза по Merrifield'y. Таким пригодным полимерным носителем является, например, слабо сшитая за счет сополимеризации с дивинилбензолом полистирольная смола, которая имеет соответствующий мостик для связывания.
Присоединение соединений формулы III к эпоксидам формулы IV осуществляется предпочтительно в обычных для присоединения нуклеофилов к эпоксидам условиях.
Присоединение осуществляется в особенности в водном растворе и/или в присутствии полярных растворителей, как спирты, например, метанол, этанол или этиленгликоль, простые эфиры, как диоксан, амиды, как диметилформамид, или фенолы, как фенол, также в безводных условиях в аполярных растворителях, как бензол и толуол, или в эмульсиях бензола с водой, в случае необходимости в присутствии кислых или основных катализаторов, например, щелочей, как раствор гидроксида натрия, или в присутствии обработанных гидразином твердофазных катализаторов, как оксид алюминия, в простых эфирах, например, диэтиловом эфире, в общем при температурах примерно от 0oC вплоть до температуры кипения соответствующей реакционной смеси, предпочтительно при 20-130oC, в случае необходимости при дефлегмации, при повышенном давлении, например, в тугоплавкой трубке, при этом температура может превышать температуру кипения, и/или в атмосфере инертного газа, как азот или аргон, причем каждое из обоих соединений формул III и IV может быть в избытке, например, в молярном соотношении 1: 1-1: 100, предпочтительно в молярном соотношении 1:1-1:10, особенно предпочтительно в соотношении 1:1-1:3.
Снятие защищенных групп осуществляют в случае необходимости описанными в способе е) (отщепление защитных групп) методами.
Способ б) Получение амидной связи.
В исходных соединениях формул V и VI функциональные группы, за исключением групп, которые не должны участвовать в реакции или не должны реагировать в реакционных условиях, независимо друг от друга защищены указанными в способе а) защитными группами.
Соединения формулы VI содержат свободную карбоксигруппу или ее реакционноспособные кислотные производные, например, активированные производные сложные эфиры или реакционноспособные ангидриды, далее, реакционноспособные циклические амиды. Реакционноспособные кислотные производные могут также образовываться in situ.
Активированные сложные эфиры соединений формулы VI с карбоксигруппой представляют собой в особенности ненасыщенные сложные эфиры, например, типа сложного винилового эфира, как сложные виниловые эфиры (получаются, например, путем переэтерификации соответствующего сложного эфира с помощью винилацетата; метод активированного сложного винилового эфира); сложные карбамоильные эфиры (получаются, например, путем обработок соответствующей кислоты изоксазолиевым реагентом; 1,2-оксазолиевый метод, или метод Вудварда); или 1-(низший алкокси)-виниловые сложные эфиры (получаются, например, путем обработок соответствующей кислоты с помощью низшего алкоксиацетилена; этокси-ацетиленовый метод), или сложные амидиноэфиры нового типа, как N-N-дизамещенные амидиновые сложные эфиры (получаются, например, путем обработок соответствующей кислоты с помощью пригодного N,N-дизамещенного карбодиимида. например, N, N-дициклогексилкарбодиимида (карбодиимидный метод); или N,N-дизамещенные сложные амидиновые эфиры (получаются, например, путем обработок соответствующей кислоты с помощью N,N-дизамещенного цианамида (цианамидный метод); пригодные сложные арильные эфиры в особенности соответствующим образом замещенные электронопритягивающими заместителями сложные фенольные эфиры (получаются, например, путем обработки соответствующей кислоты соответствующим образом замещенным фенолом, например, такого как 4-нитрофенол, 4-метилсульфонилфенол, 2,4,5- трихлорфенол, 2,3,4,5,6-пентахлорфенол или 4-фенилдиазофенол, в присутствии конденсирующего средства, как N, N-дициклогексилкарбодиимид; (метод активированного сложного арилового эфира); сложные цианметиловые эфиры (получаются, например, путем обработки соответствующей кислоты хлорацетонитрилом в присутствии основания (метод цианметилового сложного эфира); сложные тиоэфиры, в особенности замещенные в случае необходимости, например, нитрогруппой, сложные фенилтиоэфиры (получаются, например, путем обработок соответствующей кислоты в случае необходимости замещенным, например, нитрогруппой, тиофенолом, в частности, с помощью ангидридного или карбодиимидного метода; метод активированного сложного тиоэфира) или в особенности амино- или аминосложные эфиры (получаются, например, путем обработок соответствующей кислоты с помощью N-оксиамино, соответственно, N-оксиамидосоединения, например, N-оксисукцинимида, N-оксипиперидина, N-оксифталимида. имида N-окси-5- норборнен-2,3-дикарбоновой кислоты, 1-оксибензтриазола или З-окси-3,4- дигидро-1,2,3-бензтриазин-4-она, например, по ангидридному или карбодиимидному методу; метод активированного сложного N-оксиэфира). Также используются внутренние сложные эфиры, например, гамма-лактоны.
Ангидриды кислот могут быть симметричными или предпочтительно смешанными ангидридами этих кислот, как, например, ангидриды с неорганическими кислотами, как галоидангидриды кислот, в особенности хлорангидриды (получаются, например, путем обработки соответствующей кислоты тионилхлоридом, пентахлоридом фосфора, фосгеном или оксалилхлоридом; метод получения хлорангидридов кислот; азиды (получаются, например, из соответствующего сложного эфира кислот через соответствующий гидразид и обработкой его с помощью азотистой кислоты; азидный метод); ангидриды со сложными неполными эфирами угольной кислоты, например, неполные сложные (низший алкиловые) эфиры угольной кислоты (получаются, например, путем обработки соответствующей кислоты с помощью низших алкиловых эфиров хлормуравьиной кислоты или с помощью 1-(низший алкокси)-карбонил-2- (низший алкокси)-1,2-дигидрохинолина; метод смешанных эфиров O- алкилугольной кислоты), или ангидриды с дигалогенированной, в особенности дихлорированной фосфорной кислотой (получаются, например, путем обработки соответствующей кислоты с помощью оксихлорида фосфора; фосфороксидхлоридный метод); ангидриды с другими производными фосфорной кислоты, например, такие, которые можно получать с помощью фенил-N-фенилфосфорамидохлоридата или путем превращения амидов алкилфосфорной кислоты в присутствии ангидридов сульфокислот и/или снижающих рацемизацию добавок, как N-оксибензтриазол, или в присутствии сложного диэтилового эфира цианофосфоновой кислоты или с производными фосфористой кислоты; или ангидриды с органическими кислотами, как смешанные ангидриды с органическими карбоновыми кислотами (получаются, например, путем обработки соответствующей кислоты с помощью в случае необходимости замещенного галоидангидрида (низший алкан)- или фенил-(низший алкан)- карбоновой кислоты, например, хлорангидрид фенилуксусной кислоты, пивалиновой кислоты или трифторуксусной кислоты: метод смешанных ангидридов карбоновых кислот) или с органическими сульфокислотами (получаются, например, путем обработки соли, как соли щелочного металла соответствующей кислоты с помощью пригодного органического галоидангидрида сульфокислоты, как хлорангидрид (низший алкан)- или арил-, например, метан- или п-толуолсульфокислоты; метод смешанных ангидридов сульфокислот); а также симметричные ангидриды (получаются, например, путем конденсации соответствующей кислоты в присутствии карбодиимида или 1- диэтиламинопропина; метод получения симметричных ангидридов).
Пригодными циклическими амидами являются в особенности амиды с пятичленными диазациклами ароматического характера, как амиды с имидазолами, например, имидазолом (получается, например, путем обработок соответствующей кислоты с помощью N,N-карбонилдиимидазола: имидазольный метод), или пиразолом, например, 3,5-диметилпиразол (получается, например, через гидразид кислоты путем обработки с помощью ацетилацетона; пиразолидный метод).
Как упоминалось, производные карбоновых кислот, которые применяются в качестве ацилирующего средства, также могут образовываться in situ. Так, например, N, N-дизамещенные амидиносложные эфиры можно получать in situ, тем, что смесь исходного соединения формулы V и используемой в качестве ацилирующего средства кислоты вводят во взаимодействие в присутствии пригодного N, N-дизамещенного карбодиимида, например, N, N-циклогексилкарбодиимида. Амино- или амидо-(сложные эфиры) используемые в качестве ацилирующего средства кислот можно получать в присутствии циклизующего исходного соединения формулы V, тем, что смесь соответствующей кислоты и аминосоединения в качестве исходных веществ вводят во взаимодействие в присутствии N,N-дизамещенного карбодиимида, например, N,N-циклогексилкарбодиимида, и N-оксиамина или N-оксиамида, например, N-оксисукцинимида, в случае необходимости в присутствии пригодного основания, например, 4- диметиламинопиридина. Активирование можно вести in situ путем превращения с N,N,N', N'-тетраалкилурониевыми соединениями, как О-бензтриазол-1-ил-N,N,N',N'-тетраметилуроний-гексафторфосфат, О ( 1,2 -дигидро 2-оксо -1 пиридил)-N,N,N',N'-тетраметилуроний-тетрафторборат или O-(3, 4-дигидро-4- оксо-1,2,3-бензтриазолин-3-ил)-N, N,N',N'-тетраметилуроний-тетрафторборат. Наконец, ангидриды фосфорной кислоты карбоновых кислот формулы VI или VII можно получать in situ, тем, что амид алкилфосфорной кислоты, как гексаметилфосфортриамид, в присутствии ангидрида сульфокислоты, как ангидрид 4-толуолсульфокислоты, вводят во взаимодействие с солью, как тетрафторборат, например, тетрафторборат натрия, или с другим производным гексаметилфосфортриамида, как бензотриазол-1-илокси-трис-(диметиламино)- фосфоний-гексафторид, предпочтительно в присутствии уменьшающей рацемизацию добавки, как N-оксибензтриазол.
Аминогруппа в соединении формулы V, которая принимает участие в реакции, содержит предпочтительно по меньшей мере один реакционноснособный атом водорода, в особенности, когда реагирующая с ним карбоксигруппа находится в реакционноспособной форме; однако, также аминогруппа может быть дериватизирована, например, путем реакции с фосфитом, как диэтилхлорфосфит, 1,2-фениленхлорфосфит, этилдихлорфосфит, этиленхлорфосфит или тетраэтилпирофосфит. Производное такого соединения с аминогруппой представляет собой, например, также галоидангидрид карбаминовой кислоты или изоцианат, причем принимающая участие в реакции аминогруппа замещена галогенкарбонилом, например, хлоркарбонилом, или соответственно, изменена, является ее предшественником, таким как изоцианатная группа, причем в последнем случае получаются только соединения формулы 1, которые содержат атом водорода на атоме азота, полученной путем реакции амидной группы.
Если соединение формулы V однократно замещено на аминогруппе низшим алкилом или арил-(низшим алкилом), то также получается соответствующее мочевинное соединение в виде реакционноспособного производного. Например, соответствующие соединения формулы 1 получают при нагревании эквимолярных количеств этого мочевинного соединения с соединением формулы VI или VIII со свободной карбоксильной группой.
Конденсацию для получения амидной связи можно осуществлять само по себе известным образом, например, как описано в стандартных работах, как "Губен-Вейль, Методы органической химии", 4-е изд. том 15/11 (1974), том IX (1955), том E 11 (1985), Georg Thieme Verlag Штуттгарт: "The Peptides" (E. Cross and J.Meienhofer, Hg.), том 1 и 2, Academic Press, Лондон и Нью-Йорк, 1979/1980, или M.Bodansky, "Principles of Peptide Synthesis", Springer-Verlag, Берлин, 1984.
Конденсацию свободной карбоновой кислоты с соответствующим амином предпочтительно можно осуществлять в присутствии обычного конденсирующего средства, или при применении ангидридов или галоидангидридов, как хлорангидридов карбоновых кислот или активированных сложных эфиров карбоновых кислот, как п-нитрофениловые сложные эфиры. Обычные конденсирующие средства представляют собой, например, карбодиимиды, например, диэтил-, дипропил-, N-этил-N'-(3-диметиламинопропил)- карбодиимид или в особенности дициклогексилкарбодиимид, далее пригодные карбонильные соединения, например, карбонилимидазол. 1,2-оксазолиевые соединения, например, 2-этил-5-фенил-1,2-оксазолий-3'-сульфонат и 2-трет. - бутил-5-метилизоксазолийперхлорат, или пригодное ациламино-соединение, например, 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин, N,N,N',N'- тетраалкилурониевые соединения, как O-бензтриазол-1-ил-N,N, N',N'- тетраметилуроний-гексафторфосфат, далее активированные производные фосфорной кислоты, например, дифенилфосфорилазид, диэтилфосфорилцианид, фенил-N-фенилфосфорамидохлоридат, хлорангидрид бис-(2-оксо-3- оксазолидинил)фосфиновой кислоты или 1-бензтриазолилокси-трис(диметиламино)фосфоний-гексафторфосфата.
В желательном случае добавляют органическое основание, предпочтительно третичный амин, например, три-(низший алкил)-амин с объемистыми остатками, например, этилдиизопропиламин, или триэтиламин, и/или гетероциклическое основание, например, 4-диметиламинопиридин или предпочтительно N-метилморфолин, или пиридин.
Конденсация активированных сложных эфиров, реакционноспособных ангидридов или реакционноспособных циклических амидов с соответствующими аминами осуществляется обычно в присутствии органического основания, например, простых три-(низший алкил)-аминов, например, как триэтиламин или трибутиламин, или одного из вышеуказанных органических оснований. В желательном случае дополнительно еще применяют конденсирующее средство, как описано для свободных карбоновых кислот.
Конденсацию ангидридов кислот с аминами можно осуществлять, например, в присутствии неорганических карбонатов, например, карбонатов или бикарбонатов аммония или щелочных металлов, как карбонат или бикарбонат натрия или калия (в желательном случае вместе с сульфатом).
Хлорангидриды карбоновых кислот, например, производные хлоругольной кислоты, производимые от кислоты формулы VI, или хлорангидриды сульфокислот конденсируются с соответствующими аминами предпочтительно в присутствии органического амина, например, вышеуказанных три-(низший алкил)-аминов или гетероциклических оснований, в случае необходимости в присутствии гидросульфата.
Конденсация осуществляется предпочтительно в инертном, апротонном, предпочтительно, безводном растворителе или смеси растворителей, например, в амиде карбоновой кислоты, например, как формамид или диметилформамид; галогенированном углеводороде, например, как метиленхлорид, четыреххлористый углерод или хлорбензол; кетоне, например, как ацетон; циклическом простом эфире, как, например, тетрагидрофуран; сложном эфире, например, как этилацетат; или нитриле, например, как ацетонитрил; или в их смеси, в случае необходимости при пониженной или повышенной температуре, например, в области температур примерно от -40oC до примерно +100oC, предпочтительно примерно от -10oC до примерно +50oC, в случае применения сложных арилсульфониловых эфиров также примерно при +100oC +200oC, и в случае необходимости в атмосфере инертного газа, например, в атмосфере азота или аргона.
Также возможны водные, например, спиртовые, например, этанол, или ароматические растворители, например, бензол или толуол. При наличии гидроксидов щелочных металлов в качестве оснований также в случае необходимости можно добавлять ацетон.
Конденсацию можно осуществлять также твердофазным методом по R.Merrifield'y, описанным, например, в Angew. Chem. 97. 801-812 (1985), Naturwissenschaften 71, 252-258 (1984) или в R.A.Houghten, Proc. Natl. Acad. Aci. USA 82,5131-5135 (1985).
В зависимости от используемых исходных соединений остатки R1 и R9 в получаемых соединениях формулы 1 могут быть одинаковыми или отличаться друг от друга.
Высвобождение защищенных групп осуществляется в случае необходимости описанными ниже в способе е) (отщепление защитных групп) методами.
Способ в) Получение амидной связи.
В исходных соединениях формул VII и VIII функциональные группы, за исключением групп, которые должны участвовать в реакции или которые не должны реагировать при реакционных условиях, независимо друг от друга, защищены указанными в способе а) защитными группами.
Способ полностью аналогичен указанному в способе б) способу, если вместо соединений формулы V используют соединения формулы VII, и вместо соединений формулы VI используют соединения формулы VIII, и при ацилировании R1 связывают вместо R9 с соединениями формулы VII вместо соединений формулы V.
В зависимости от используемых исходных соединений остатки R1 и R9 в получаемых соединениях формулы 1 могут быть одинаковыми или отличаться друг от друга.
Высвобождение защищенных групп осуществляется в случае необходимости согласно описанным в способе е) (отщепление защитных групп) методами.
Способ г) Получение амидной связи.
В исходных соединениях формулы IX и в пригодной для введения идентичных остатков R1 и R9 кислоте или ее реакционноспособных производных функциональные группы, которые не должны участвовать в реакции или не должны реагировать в реакционных условиях, независимо друг от друга, защищены одной из указанных в способе а) защитных групп.
Пригодная для введения идентичных остатков R1 и R9 кислота предпочтительно соответствует одной из формул VI или VIII.
В случае необходимости в качестве защищенных защитными группами исходных соединений формулы IX предпочтительны соединения формулы II, которые описаны ниже в разделе об исходных соединениях.
Способ полностью аналогичен указанному в пункте б) способу, причем вместо соединений формулы V используют соединения формулы IX, а вместо соединений формулы VI используют соединения формулы VI или VIII.
Высвобождение защищенных групп осуществляется в случае необходимости описанными в способе е) ниже (отщепление защитных групп) методами.
Способ д) Алкилирование вторичного атома азота.
В исходных соединениях формулы I' и в пригодном для введения остатка R7" соединении формулы XII или его реакционноспособных производных функциональные группы, которые не должны участвовать в реакции или которые не должны реагировать в реакционных условиях, независимо друг от друга, защищены указанными в способе а) защитными группами.
Удаляемая группа X в особенности представляет собой нуклеофильную удаляемую группу, выбираемую из гидроксила, этерифицированного до сложного эфира с помощью сильной неорганической или органической кислоты, как, например, этерифицированный с помощью неорганической кислоты, например, галогенводородной кислоты, как соляная, бромистоводородная или йодистоводородная кислота, или с помощью сильной органической сульфокислоты, например, в случае необходимости замещенной, например, галогеном, как фтор, (низший алкан)-сульфокислоты или ароматической сульфокислоты, например, в случае необходимости замещенной низшим алкилом, как метил, галогеном, как бром, и/или нитрогруппой бензосульфокислоты, например, метансульфо-, п-бромтолуолсульфокислоты или птолуолсульфокислоты гидроксил или этерифицированный с помощью азотистоводородной кислоты гидроксил.
Замещение может протекать в условиях нуклеофильного замещения первого или второго порядка.
Например, одно из соединений формулы VIII-XIII, где X обозначает удаляемую группу с высокой поляризуемостью электронной оболочки, например, йод, в полярном апротонном растворителе, например, ацетоне, ацетонитриле, нитрометане, диметилсульфоксиде или диметилформамиде. Реакция может проводиться в воде, к которой добавлен в случае необходимости в качестве агента растворения органический растворитель, например, этанол, тетрагидрофуран или ацетон. Реакция замещения осуществляется в случае необходимости при пониженной или повышенной температуре, например, в температурном интервале примерно от -40oC до примерно +100oC, предпочтительно примерно от -10oC до примерно 50oC, и в случае необходимости в атмосфере инертного газа, например, в атмосфере азота или аргона.
Высвобождение защищенных групп осуществляется в случае необходимости описанными в способе е) (отщепление защитных групп) методами.
Способ е)- Отщепление защитных групп.
Отщепление защитных групп, которые не являются составной частью желательных целевых продуктов формулы 1, например, защитных для карбокси-, амино-, оксигрупп осуществляется известным образом, например, путем сольволиза, в особенности гидролиза, алкоголиза или ацидолиза, или путем восстановления, в особенности гидрогенолиза или химического восстановления, а также фотолиза, в случае необходимости протекающих ступенчато или одновременно, причем также могут применяться ферментативные методы. Отщепление защитных групп, например, описано в указанных в начале в разделе о "защитных группах" стандартных работах.
Так, например, защищенный карбоксил, например, трет.-(низший алкокси)-карбонил; замещенный в 2-положении тризамещенной силильной группой или в 1-положении низшим алкоксилом или низшим алкилтио низший алкоксикарбонил или в случае необходимости замещенный дифенилметоксикарбонил, можно переводить в свободный карбоксил путем обработок с помощью пригодной кислоты, как муравьиная кислота, соляная кислота или трифторуксусная кислота, в случае необходимости при добавке нуклеофильного соединения, как фенол или анизол. Из низшего алкоксикарбонила также можно высвобождать карбоксил благодаря основаниям, как гидроксилы, например, гидроксиды щелочных металлов, как NaOH или KOH. В случае необходимости замещенный бензилоксикарбонил можно освобождать, например, с помощью гидрогенолиза, то есть путем обработок с помощью водорода в присутствии металлического катализатора гидрирования, как катализатор на основе палладия. Далее, соответствующим образом замещенный бензилоксикарбонил, как 4- нитробензилоксикарбонил, можно переводить в свободный карбоксил также путем восстановления, например, путем обработок с помощью щелочного металла, как дитионит натрия, или с помощью восстанавливающего металла, например, цинка, или восстанавливающей металлической соли, как, например, соли хрома-(II), например, хлорида хрома-(II), обычно в присутствии отдающего водород средства, которое вместе с металлом может давать атомарный водород, или в присутствии кислоты, в первую очередь пригодной карбоновой кислоты, например, в случае необходимости замещенной, например, гидроксилом, (низшей алкан)-карбоновой кислоты, например, уксусной кислоты, муравьиной кислоты, гликолевой кислоты, дифенилгликолевой кислоты, молочной кислоты, миндальной кислоты, 4-хлорминдальной кислоты или винной кислоты, или в присутствии спирта или тиола, причем предпочтительно добавляют воду. Благодаря обработке с помощью восстанавливающего металла или соли металла, как описано выше, также можно превращать в свободный карбоксил 2-галоген-(низший алкокси)-карбонил (в случае необходимости после превращения 2-бром-(низший алкокси)-карбонильной группы в соответствующую 2-йод-(низший алкокси)-карбонильную группу) или ароилметоксикарбонил. Ароилметоксикарбонил также можно расщеплять путем обработок с помощью нуклеофильного, предпочтительно солеобразующего реагента, как тиофенолят натрия или йодид натрия. 2-(Тризамещенный силил) -(низший алкокси)-карбонил, как 2-три-(низший алкил)-силил-(низший алкокси)-карбонил, также можно переводить в свободный карбоксил путем обработок с помощью дающей фторид-анион соли фтористоводородной кислоты, как фторид щелочного металла, например, фторид натрия или калия, в случае необходимости в присутствии макроциклического простого полиэфира ("краунэфира") или с помощью фторида органического четвертичного основания, как тетра-(низший алкил)-аммонийфторид или три-(низший алкил)-арил-(низший алкил)-аммонийфторид, например, тетраэтиламмонийфторид или тетрабутиламмонийфторид, в присутствии апротонного, полярного растворителя, как диметилсульфоксид или N,N-диметилацетамид. Защищенный в виде органического силилоксикарбонила, как три-(низший алкил)-силилоксикарбонил, например, триметилсилилоксикарбонил, карбоксил можно высвобождать обычным образом путем сольволиза, например, путем обработки водой, спиртом или кислотой, или, кроме фторида, как описано выше. Этерифицированный карбоксил можно освобождать также ферментативно, например, благодаря эстеразам или пригодным пептидазам, например, как этерифицированный аргинин или лизин, как сложный метиловый эфир лизина, с помощью трипсина.
Защищенную аминогруппу высвобождают в зависимости от рода защитных групп различными известными путями, предпочтительно с помощью сольволиза или восстановления. (Низший алкокси)-карбониламино. как трет.- бутоксикарбониламино, можно расщеплять в присутствии кислот, например, неорганических кислот, например, галогенводорода. как хлороводород или бромоводород, или серной или фосфорной кислоты, предпочтительно хлороводорода, или сильных органических кислот, как тригалогенуксусная кислота, например, трифторуксусная кислота, или муравьиная кислота, в полярных растворителях, как вода, или простых эфирах, предпочтительно циклических простых эфирах, как диоксан; 2-галоген-(низший алкокси)- карбониламино (в случае необходимости после превращения 2-бром-(низший алкокси)-карбониламино-группы в 2-йод-(низший алкокси)-карбониламиногруппу) можно расщеплять путем непосредственного растворения в жидкой органической карбоновой кислоте, как муравьиная кислота; ароилметоксикарбониламино или 4-нитробензилоксикарбониламино можно расщеплять, например, путем обработки пригодным восстановителем, как цинк в присутствии пригодной карбоновой кислоты, как водная уксусная кислота. Ароилметоксикарбониламино также можно расщеплять путем обработки с помощью нуклеофильного, предпочтительно солеобразующего реагента, как тиофенолят натрия, а 4- нитробензилоксикарбониламино также можно расщеплять путем обработки дитионитом щелочного металла, например, натрия. Замещенный в случае необходимости дифенилметоксикарбониламино, трет. -(низший алкокси)- карбониламино или 2-(тризамещенный силил)-(низший алкокси)- карбониламино, как 2-три-(низший алкил)-силил-(низший алкокси)- карбониламино, можно освобождать путем обработки с помощью пригодной кислоты, например, муравьиной или трифторуксусной кислоты: в случае необходимости замещенный бензилоксикарбониламино можно высвобождать, например, с помощью гидрогенолиза, то есть за счет обработки водородом в присутствии пригодного катализатора гидрирования, как катализатора на основе платины или палладия; в случае необходимости замещенный триарилметиламино или формиламино можно освобождать, например, путем обработки с помощью кислоты, как неорганическая кислота, например, хлористоводородная кислота, или органической кислоты, например, муравьиной, уксусной или трифторуксусной кислоты, в случае необходимости в присутствии воды, и защищенную в виде силиламино аминогруппу можно освобождать, например, посредством гидролиза или алкоголиза. Защищенную 2-галогенацетилом, например, 2-хлорацетилом. аминогруппу можно освобождать путем обработки с помощью тиомочевины в присутствии основания, или с помощью тиолятной соли, как тиолят щелочного металла, тиомочевины, и последующего сольволиза, как алкоголиз или гидролиз, образовавшегося продукта замещения; из трифторацетиламиногруппы аминогруппа высвобождается, например, путем гидрогенолиза с помощью оснований, как гидроксиды или карбонаты щелочных металлов, как Na2CO3 или K2CO3, в полярных растворителях, как, например, спирты, как метанол, при температурах 0-100oC, в особенности при 40-80oC. Защищенную 2- (тризамещенный силил)-(низший алкокси)-карбонилом, как 2-три-(низший алкил)-силил- (низший алкокси)-карбонил, аминогруппу можно переводить в свободную аминогруппу также путем обработки с помощью дающей фториданион соли фтористоводородной кислоты, как указано выше при освобождении соответственно защищенной карбоксигруппы. Также можно отщеплять непосредственно связанный с гетероатомом, как азот, силил, как триметилсилил, с помощью фторид-ионов.
Защищенную в виде азидогруппы аминогруппу можно переводить в свободную аминогруппу, например, путем восстановления, например, путем каталитического гидрирования с помощью водорода в присутствии катализатора гидрирования, как оксид платины, палладий или никель Ренея, путем восстановления с помощью меркапто-соединений, как дитиотреитол или меркаптоэтанол, или также путем обработки с помощью цинка в присутствии кислоты, как уксусная кислота. Каталитическое гидрирование предпочтительно осуществляется в инертном растворителе, как галогенированном углеводороде, например, в метиленхлориде, или также в воде, или смеси воды с органическим растворителем, как спирт или диоксан, примерно при 20oC-25oC, или также при охлаждении или нагревании.
Защищенную пригодной ацильной группой, три-(низший алкил)- силильной группой или в случае необходимости замещенным 1-фенил-(низшим алкилом) гидроксильную или меркапто-группу высвобождают аналогично соответственно защищенной амино-группе. Защищенная 2,2-дихлорацетилом гидроксильная, соответственно меркапто-группа, высвобождается, например, путем основного гидролиза, защищенная трет.-низшим алкилом или 2-окса- или 2-тиаалифатическим или циклоалифатическим углеводородным остатком гидроксильная, соответственно меркаптогруппа, высвобождается путем ацидолиза, например, путем обработки с помощью неорганической кислоты или сильной карбоновой кислоты, например, трифторуксусной кислоты.
При наличии нескольких защищенных функциональных групп, если желательно, защитные группы можно выбирать так, чтобы одновременно можно было отщеплять более, чем одну такую группу, например, ацидолизом, например, путем обработки с помощью трифторуксусной кислоты, или с помощью водорода и катализатора гидрирования, как катализатор палладий-наугле. Наоборот, можно выбирать группы так, чтобы они могли отщепляться не все одновременно, а в желательной последовательности, причем получаются соответствующие промежуточные продукты.
Дополнительные меры способов.
В случае дополнительных мер способов, которые осуществляются в желательном случае, функциональные группы исходных соединений, которые не должны участвовать в реакции, могут быть незащищены или быть в защищенной форме, например, благодаря одной или нескольким указанным выше в способе а) защитным группам. Защитные группы могут сохраняться в целевых продуктах или полностью, или частично отщепляться согласно указанным в способе е) методам.
Соли соединений формулы 1 по меньшей мере с одной солеобразующей группой можно получать само по себе известным образом. Так, соли соединений формулы 1 с кислотными группами образуются, например, путем обработки с помощью металлических соединений, как соли щелочных металлов пригодных органических карбоновых кислот, например, натриевая соль 2-этилгексановой кислоты, с помощью органических соединений щелочных или щелочноземельных металлов, как соответствующие гидроксиды, карбонаты или бикарбонаты, как гидроксид, карбонат или бикарбонат натрия и калия, с помощью соответствующих кальциевых соединений или с помощью аммиака или пригодного органического амина, причем предпочтительно применяют стехиометрические количества или только небольшой избыток солеобразующего средства. Соли присоединения кислот соединений формулы 1 получают обычным образом, например, путем обработки с помощью кислоты или пригодного анионообменного реагента. Внутренние соли соединений формулы 1, которые содержат кислотные и основные солеобразующие группы, например, свободную карбоксильную группу и свободную аминогруппу, могут получаться, например, путем нейтрализации солей, как солей присоединения кислот, в изоэлектрической точке, например, с помощью слабых оснований, или путем обработки ионообменниками.
Соли можно переводить в свободные соединения обычным образом; соли металлов и аммония, например, путем обработки с помощью пригодных кислот, а соли присоединения кислот, например, путем обработки с помощью пригодного основного средства.
Смеси стереоизомеров, т.е. смеси диастереомеров и/или энантиомеров, как, например, рацемические смеси, известным образом могут разделяться благодаря пригодным способам разделения на соответствующие изомеры. Так, смеси диастереомеров могут разделяться на отдельные диастереомеры путем фракционной кристаллизации, хроматографии, распределения между растворителями и т.п. Рацематы можно разделять после переведения оптических антиподов в диастереомеры, например, путем введения во взаимодействие с оптически активными соединениями, например, оптически активными кислотами или основаниями, путем колоночной хроматографией на сорбентах, покрытых оптически активными соединениями, или благодаря ферментативным методам, например, путем селективного взаимодействия только одного из обоих энантиомеров. Это разделение можно осуществлять как на стадии одного из исходных продуктов, так и в случае самих соединений формулы 1.
На отдельных центрах хиральности в соединении формулы 1 можно целенаправленно обращать конфигурацию. Например, можно обращать конфигурацию асимметрических атомов углерода, которые содержат нуклеофильные заместители, как амино или гидроксил, путем нуклеофильного замещения второго порядка, в случае необходимости после переведения связанного нуклеофильного заместителя в пригодную нуклеофильную удаляемую группу и реакции с вводящим первоначальные заместители реагентом, или можно изменять конфигурацию на атомах углерода с гидроксильными группами, как например, атом углерода с группой R5 в соединении формулы 1, путем окисления и восстановления соединений формулы 1, как описано ниже.
Остатки гидроксила R5 и водород R6 в соединении формулы 1 могут окисляться до оксо-группы, причем предпочтительно применяются такие окислители, которые селективно превращают окси-группу в кето-группу, например, хромовая кислота или ее производные, как пиридинийхромат или трет.-бутилхромат, дихромат/серная кислота, триоксид серы, в присутствии гетероциклических оснований, как пиридин/SO3, далее азотная кислота, пиролюзит или диоксид селена, или диметилсульфоксид в присутствии оксалилхлорида, в воде, водных или органических растворителях, как галогенированные растворители, например, метиленхлорид, амидах карбоновых кислот, например, как диметилформамид, или ди-(низший алкил) -сульфоксидах, как диметилсульфоксид, в присутствии или в отсутствии основных аминов, например, три-(низший алкил)-аминов, как триэтиламин, при температурах от -50oC до 100oC, предпочтительно при -10oC +50oC, например, как описано в европейской патентной заявке EP-A-0 236 734.
Наоборот, в полученных таким образом соединениях формулы 1, в которых R5 и R6 вместе образуют оксо-группу, оксо-группу можно восстанавливать до окси-группы. Для восстановления оксо-группы в соединении формулы 1 пригодны восстановители, которые при реакционных условиях способа восстанавливают изолированную кето-группу селективно или быстрее, чем имеющиеся в соединениях формулы 1 амидные группы.
В первую очередь нужно назвать пригодные боргидриды, как боргидриды щелочных металлов, в особенности боргидрид натрия, боргидрид лития или цианоборгидрид натрия, далее боргидрид цинка, или пригодные гидриды алюминия, как (низший алкокси)-алюминийгидриды щелочных металлов с объемными остатками, например, три-трет.-бутоксиалюмогидрид лития.
Восстановление можно осуществлять также с помощью водорода в присутствии пригодных катализаторов на основе тяжелых металлов, например, никеля Ренея или платиновых, или палладиевых катализаторов, например, платина-или палладий-на активном угле, или согласно Meerwein-Ponndorf-Verley с помощью алканолятов алюминия, предпочтительно 2-пропанолята алюминия или 2-этанолята алюминия.
Восстановление может осуществляться предпочтительно со стехиометрическими количествами или рационально соразмеренным избытком восстановителя в инертном растворителе при температурах от -80oC до температуры кипения растворителя, например, при -20oC +100oC, если необходимо, в атмосфере инертного газа, например, азота или аргона. Избыток восстановителя необходим в особенности тогда, когда он также реагирует с растворителем, например, с протонами протонного растворителя.
При применении боргидрида натрия пригодны полярные, протонные растворителя, например, метанол, этанол или изопропанол: при применении других восстановителей полярные, апротонные растворители, например, тетрагидрофуран.
В соединении формулы 1, в которой R1 и R9 не содержат никаких или содержат не очень реакционные арильные остатки, имеющийся в R7 и R3 арильный остаток, в особенности фенильный остаток, можно гидрировать, например, путем каталитического гидрирования, в особенности в присутствии оксидов тяжелых металлов, как смешанные родий/платиновые оксиды, например, с катализатором Nishimura, предпочтительно в полярном растворителе, как спирт, например, метанол или этанол, при температурах 0-80oC, в особенности при 10-40oC, и под давлением водорода 1-10 атм, предпочтительно, например, при нормальном давлении.
В полученном соединении формулы 1 можно замещать амино- или карбоксамидогруппу, этерифицировать до сложного эфира или амидировать находящуюся в свободной или реакционноспособной форме карбоксигруппу, соответственно, переводить этерифицированную или амидированную карбоксигруппу в свободную карбоксигруппу.
Замещение карбоксамидной группы или другой, первичной или вторичной аминогруппы, например, для введения остатков, как незамещенного или замещенного алкила, алкенила или алкинила, арил-(низшего алкила), связанного через углерод гетероциклила или гетероциклил-(низшего алкила) R1, R2, R8, или R9 в соединения формулы 1, в которых один или несколько указанных остатков обозначают водород, осуществляют, например, путем алкилирования.
Пригодными средствами для алкилирования карбоксиамидной группы в соединении формулы 1 являются, например, диазосоединения, например, диазометан. Диазометан можно разлагать в инертном растворителе, причем образующийся свободный метилен реагирует с карбоксамидной группой в соединении формулы 1. Разложение диазометана происходит предпочтительно каталитически, например, в присутствии благородного металла в тонко измельченной форме, например, меди, или соли благородного металла, например, хлорида меди-(I) или сульфата меди-(II).
Алкилирующие средства также указаны к патенту ФРГ 2331133, например, алкилгалогениды, сложные эфиры сульфокислот, соли Меервейна или 1-замещенные 3-арилтриазены, которые при указанных там реакционных условиях могут взаимодействовать с соединением формулы 1 с карбоксамидной группой.
Другие алкилирующие средства выбираются из соединений формул:
R1-X
(Х)
R2-X (ХI)
R8-X (ХIII)и
R9-X (ХIV)
где Х
обозначает удаляемую группу, а остальные остатки имеют указанные значения, кроме ацила,
незамещенного или замещенного как указано выше сульфо, фосфоно или замещенного как указано выше фосфорила.
Удаляемая группа представляет собой в особенности нуклеофильную удаляемую группу, выбираемую
из этирифицированного с помощью сильной неорганической или органической кислоты гидроксила, как с помощью
неорганической кислоты, например, галогенводородной кислоты, как соляная, бромистоводородная
или йодистоводородная кислота, или с помощью сильной органической сульфокислоты, как в случае
необходимости замещенная, например, галогеном, как фтор, низшая алкансульфокислота, или ароматической
сульфокислоты, например, как в случае необходимости замещенная низшим алкилом, как метил,
галогеном, как бром, и/или нитрогруппой бензолсульфокислота; например, этерифицированный метансульфокислотой,
триметансульфокислотой или п-толуолсульфокислотой гидроксил или этерифицированный с
помощью азотистоводородной кислоты гидроксил.
Реакция может протекать в условиях нуклеофильного замещения первого или второго порядка.
Например, одно из соединения формулы VIII-XIII, где X обозначает удаляемую группу с высокой поляризуемостью электронной оболочки, например, йод, в полярном апротонном растворителе, например, ацетоне, ацетонитриле, нитрометане, диметилсульфоксиде или диметилформамиде. Реакция может осуществляться также в воде, к которой в случае необходимости добавлен для растворения органический растворитель, например, этанол, тетрагидрофуран или ацетон. Реакция замещения осуществляется в случае необходимости при пониженной или повышенной температуре, например, в температурном интервале примерно -40oC примерно 100oC, предпочтительно примерно от -10oC до примерно 50oC, и в случае необходимости в атмосфере инертного газа, например, в атмосфере азота или аргона.
Для этерификации или амидирования карбоксигруппы в соединении формулы 1 можно, если желательно, применять свободную кислоту или переводить свободную кислоту в одно из вышеуказанных реакционноспособных производных и вводить во взаимодействие со спиртом, аммиаком, первичным или вторичным амином, или для этерификации свободную кислоту или реакционноспособную соль, например, соль цезия, вводить во взаимодействие с реакционноспособным производным спирта. Например, соль цезия карбоновой кислоты можно вводить во взаимодействие с соответствующим спирту галогенидом или сложным эфиром сульфокислоты. Этерификацию карбоксигруппы также можно осуществлять с помощью других обычных алкилирующих средств, например, с помощью диазометана, алкилгалогенидов, сложных эфиров сульфокислот, солей Меервейна или 1-замещенных 3-арилтриазенов.
Для переведения этерифицированной или амидированной карбоксигруппы в свободную карбоксигруппу можно применять один из вышеописанных при отщеплении защитных для карбоксигруппы групп методов или, в желательном случае, щелочное омыление согласно указанным в Organikum, 17, е изд. VEB Deutscher Verlag der Wissenschaften, Берлин, (Ost) 1988, реакционным условиям.
В соединении формулы 1 этерифицированную карбоксигруппу можно путем аминолиза с помощью аммиака или первичного или вторичного амина переводить в замещенную в случае необходимости карбоксамидную группу. Аминолиз может протекать при обычных, как указанные в Organikum, 15-е изд. VEB Deutscher Verlag der Wissenschaften, Берлин, (Ost), 1976, для такого рода превращений, реакционных условиях.
В соединении формулы 1 можно ацилировать имеющуюся свободную амино-группу, например, для введения одного из указанных для R1 или R9 остатков: ацил. Ацилирование протекает по методам, указанным выше в способах б), в) или г) для конденсаций, или по одному из указанных для защитных групп методов или, например, по способу, указанному в Organicum, 17-е изд. VEB Deutscher Verlag der Wissenschaften, Берлин, (Ost), 1988.
В полученном соединении формулы 1, где заместители имеют указанные значения и имеется по меньшей мере одна свободная гидроксильная группа и остальные функциональные группы находятся в защищенной форме, свободную гидроксильную группу можно ацилировать или этерифицировать до простого эфира.
Ацилирование можно осуществлять с помощью ацилирующих реагентов по одному из методов, указанных в способах б)-г), по одному из указанных для защитных групп методов или по способу, указанному в Organikum, 17-е, изд. VEB Deutscher Verlag der Wissenschaften, Берлин (Ost), 1988.
Этерификация до простого эфира может осуществляться с помощью вышеуказанных алкилирующих средств и при таких же условиях реакции, например, с помощью диазометана, алкилгалогенидов, сложных эфиров сульфокислот, солей Меервейна, 1-замещенных 3-арилтриазенов и т.д.
В соединении формулы 1 имеющиеся защитные группы или пригодные остатки R1 или R9, то есть такие, которые обозначают ацил, сульфо, замещенный сульфо, фосфоно или замещенный фосфорил, отщепляют по одному из указанных в способе е) методов, в особенности путем гидролиза, например, в присутствии оснований, как гидроксиды щелочных или щелочноземельных металлов, например, гидроксид натрия, или кислот, как органические кислоты или неорганические кислоты, например, галогенводород, как хлорводород. Гидролиз осуществляют в обычных условиях, например, в водном растворе или в безводных растворителях, в особенности в простых эфирах, как диоксан, при температурах от -50oC до температуры кипения с обратным холодильником соответствующей реакционной смеси, например, при 0-50oC С, предпочтительно в присутствии защитного газа, как аргон или азот.
Все вышеуказанные стадии способа могут осуществляться при известных, предпочтительно называемых специфическими, реакционных условиях, в отсутствии или обычно в присутствии растворителей или разбавителей, предпочтительно таких, которые инертны по отношению к используемым реагентам и растворяют их, в отсутствии или в присутствии катализаторов, конденсирующих средств или нейтрализующих агентов, например, ионообменников, как катионообменники, например, в H+ -форме, в зависимости от рода реакции и/или компонентов реакции, при пониженной, нормальной или повышенной температуре, например, в температурном интервале примерно от 100oC до примерно 190oC, предпочтительно примерно от -80oC до примерно 150oC, например, при -80oC 60oC, при комнатной температуре, при -20oC-40oC, или при температуре кипения используемого растворителя, при атмосферном давлении или в закрытом сосуде, в случае необходимости под давлением, и/или в инертной атмосфере, например, в атмосфере аргона или азота.
В случае всех исходных и промежуточных соединений могут использоваться или образовываться соли, если есть солеобразующие группы. Соли могут образовываться также по время превращения такого рода соединений.
На всех реакционных стадиях образующиеся смеси изомеров могут разделяться на отдельные изомеры, например, диастереомеры или энантиомеры, или на любые смеси изомеров, например, рацематы или смеси диастереомеров, например, аналогично методам, которые описаны под названием "Дополнительные меры способа".
В некоторых случаях, например, при гидрировании, можно достигать стереоселективности реакций, что облегчает получение отдельных изомеров.
К растворителям соответствующей реакции причисляют, например, воду, сложные эфиры, как (низший алкил)-(низший алканоат), например, диэтиловый эфир уксусной кислоты; простые эфиры, как алифатические простые эфиры, например, диэтиловый эфир, или циклические простые эфиры, например, тетрагидрофуран; жидкие ароматические углеводороды, как бензол или толуол; спирты, как метанол, этанол или 1- или 2-пропанол: нитрилы, как ацетонитрил; галогенуглеводороды, как метиленхлорид; амиды кислот, как диметилформамид; основания, как гетероциклические азотные основания, например, пиридин: ангидриды кислот, как ангидриды низших алкановых кислот, например, ацетангидрид; циклические, линейные или разветвленные углеводороды, как циклогексан, гексан или изопентан, или смеси этих растворителей, например, водные растворы, если в описании способов не указано ничего другого. Такого рода смеси растворителей могут найти применение также при обработке, например, при хроматографии или диспергирования.
Возможны также такие варианты осуществления способа, при которых исходят из получаемого на любой стадии в качестве промежуточного продукта соединения и осуществляют отсутствующие стадии или способ прерывают на любой стадии, или исходное вещество получается в реакционных условиях, или применяется в форме реакционноспособного производного или соли, или получаемое по одному из предлагаемых в изобретении способов соединение получают при условиях способа и далее обработки in situ. При этом предпочтительно исходят из таких исходных веществ, которые приводят к соединениям, которые описаны выше как предпочтительные, в особенности как особенно предпочтительные, в первую очередь предпочтительные и/или, прежде всего, предпочтительные.
Фармацевтическое средство.
Предлагаемое согласно данному изобретению фармацевтическое средство содержит эффективное количество производного гидразина формулы 1 или фармацевтически приемлемую соль этого соединения с по меньшей мере одной солеобразующей группой с по меньшей мере одним фармацевтически приемлемым носителем, способно ингибировать ВИЧ-протеазы и обладает антивирусной активностью.
В качестве носителей можно использовать защитное количество неорганических или органических, твердых или жидких фармацевтически приемлемых носителей.
К изобретению относится фармацевтическое средство, которое пригодно для введения теплокровным, в особенности людям, для лечения или профилактики заболевания, которое предназначено для ингибирования ретровирусной протеазы, в особенности ретровирусной аспартатпротеазы, как ВИЧ-1 или ВИЧ-II-gag-протеазы, например, ретровирусного заболевания, как СПИД, включающее эффективное для ингибирования ретровирусной протеазы количество соединения формулы 1 или его фармацевтически приемлемой соли, вместе с по крайней мере одним фармацевтически приемлемым носителем.
Предлагаемое средство можно использовать путем введения теплокровным людям и животным энтерально, например, через нос, ректально, орально или парентерально, например, внутримышечно или внутривенно, эффективной дозы фармакологически активного вещества индивидуально или вместе со значительным количеством фармацевтически применимого носителя. Доза биологически активного вещества зависит от вида теплокровного, веса тела, возраста и индивидуального состояния, индивидуальных фармакодинамических данных, излечиваемой болезни, а также способа введения.
Для лечения вызываемых вирусами, особенно ретровирусами, заболеваний, например, СПИДа, вводят терапевтически эффективное количество предлагаемых в изобретении соединений формулы 1 теплокровному, например, людям, которым при заболевании, в особенности СПИДом, требуется такого рода лечение. Для теплокровных, например, людей весом примерно 70 кг, дозы составляют примерно от 3 мг до примерно 3 г, предпочтительно примерно от 10 мг до примерно 1,5 г, например, примерно 300-1000 мг на человека в день, разделенные предпочтительно на 1-3 разовые дозы. которые, например, могут быть одинакового размера. Дети обычно получают половинную дозу взрослых.
Фармацевтические средства содержат примерно 1-95% предпочтительно примерно 20-90% биологически активного вещества. Предлагаемые в изобретении фармацевтические средства могут быть, например, в дозированных формах, как ампулы, пузырьки, свечи, драже, таблетки или капсулы.
Фармацевтические средства настоящего изобретения получают известным образом, например, с помощью обычных способов растворения, лиофилизации, смешения, гранулирования или дражирования.
Предпочтительно применяют растворы биологически активного вещества, в особенности изотонические водные растворы или суспензии, причем, например, при применении лиофилизированных препаратов, которые содержат биологически активное вещество индивидуально или вместе с носителем, например, маннитом, их можно готовить перед употреблением. Фармацевтические средства могут быть стерилизованы и/или могут содержать вспомогательные вещества, например, консерванты, стабилизаторы, смачиватели и/или эмульгаторы, агенты растворения, соли для регулирования осмотического давления и/или буферы, и готовятся обычным образом, например, с помощью обычных способов растворения или лиофилизации. Указанные растворы или суспензии могут содержать повышающие вязкость вещества, как карбоксиметилцеллюлозу натрия, карбоксиметилцеллюлозу, декстран, поливинилпирролидон или желатину.
Суспензии в масле содержат в качестве масляной компоненты обычные для инъекционных целей растительные, синтетические или полусинтетические масла. В качестве таковых нужно назвать в особенности жидкие сложные эфиры жирных кислот, которые в качестве кислотной компоненты содержат длинноцепочечную жирную кислоту с 8-22, в особенности 12-22, атомами углерода, как, например, лауриновая кислота, тридециловая кислота, миристиновая кислота, пентадециловая кислота, пальмитиновая кислота, маргариновая кислота, стеариновая кислота, арахидиновая кислота, бегеновая кислота или соответствующие ненасыщенные кислоты, как, например, олеиновая кислота, элаидиновая кислота, эрукановая кислота, бразидиповая кислота или линолевая кислота, в случае необходимости с добавкой антиоксидантов, как, например, витамин E, b -каротин или 3,5-ди-трет. -бутил-4-окситолуол. Спиртовая компонента этих сложных эфиров жирных кислот содержит максимально 6 C-атомов и представляет собой одно- или многоатомный, например, одно-, двух- или трехатомный спирт, например, метанол, этанол, пропанол, бутанол или пентанол, или их изомеры, прежде всего гликоли и глицерин. В качестве сложных эфиров жирных кисло поэтому предпочтительно нужно указать: этилолеат, изопропилмиристат, изопропилпальмитат, "Labrafil M 2375" (полиоксиэтиленглицеринтриолеат фирмы Gattefosse, Париж), "Miglyol 812" (триглицерид насыщенных жирных кислот с длиной цепей C8-CR12 фирмы Huls AG, Германия), особенно предпочтительны растительные масла, как масло их семян хлопка, миндальное масло, оливковое масло, касторовое масло, семазол, соевое масло и прежде всего арахисовое масло.
Приготовление инъецируемых препаратов осуществляют обычным образом в стерильных условиях, таким же образом путем заполнения в ампулы или флаконы (пузырьки), а также закупоривания емкостей.
Фармацевтические препараты для орального применения можно получать комбинацией биологически активного вещества с твердыми носителями, полученную смесь гранулируют и смесь, если желательно или необходимо, после добавления пригодных вспомогательных веществ, перерабатывают в таблетки, драже или капсулы. При этом их можно вводить также в синтетические носители, которые позволяют дозировать биологически активные вещества или позволяют им дозированно диффундировать.
Пригодными носителями являются, например, наполнители, как сахар, например, лактоза, сахароза, маннит или сорбит; целлюлозные добавки, и/или фосфаты кальция, например, трикалийфосфат или гидрофосфат кальция; связующие, как крахмальный клейстер, при применении, например, кукурузного, пшеничного, рисового или картофельного крахмалов, желатина, трагант, метилцеллюлоза, оксипропилметилцеллюлоза, натрийкарбоксиметилцеллюлоза и/или поливинилпирролидон, и/или, если желательно, расширители, как вышеуказанные крахмалы, далее, карбоксиметилкрахмалы, поперечно сшитый поливинилпирролидон, агар, альгиновая кислота или ее соль, как альгинат натрия. Вспомогательные средства представляют собой в первую очередь регуляторы текучести и смазки, например, кремневая кислота, тальк, стеариновая кислота или ее соли, как стеарат магния или кальция и/или полиэтиленгликоль. Драже снабжаются пригодными, в случае необходимости резистентными к желудочному соку покрытиями, причем применяют, в частности, концентрированные растворы сахара, которые в случае необходимости содержат гуммиарабик, тальк, поливинилпирролидон, полиэтиленгликоль и/или диоксид титана, лаковые растворы в пригодных органических растворителях или для приготовления резистентных к желудочному соку покрытий, растворы пригодных целлюлозных веществ, как этилцеллюлозофталат или оксипропилметилцеллюлозофталат. Капсулы представляют собой разъемные капсулы из желатины, а также мягкие, закрытые капсулы из желатины и пластификатора, как глицерин или сорбит. Разъемные капсулы могут содержать биологически активное вещество в форме гранулята, например, с наполнителями, как лактозы, связующими, как крахмалы, и/или мягчителями (смазками), как тальк или стеарат магния, и в случае необходимости со стабилизаторами. В мягких капсулах биологически активное вещество предпочтительно растворено или суспендировано в пригодных масляных вспомогательных веществах, как жирные масла, парафиновое масло или жидкие полиэтиленгликоли, причем также могут быть добавлены стабилизаторы и/или антибактериальные средства. В таблетки или покрытия драже и оболочки капсул могут добавлять красители или пигменты, например, для идентификации или для пометки различных доз биологически активного вещества.
Промежуточные соединения.
Объектом
настоящего изобретения являются новые промежуточные соединения для получения производных гидразина общей формулы II

где R3, R7 означают низший алкил, циклогексил-низший алкил или фенил-низший алкил, незамещенный или замещенный галогеном, низшим алкокси или циано, а также соли этих соединений, если они имеют солеобразующие группы.
При получении всех исходных материалов свободные функциональные группы, которые не должны принимать участие в соответствующей реакции, не защищены или находятся в защищенной форме, например, защищены указанными выше в способе а) защитными группами. Эти защитные группы могут высвобождаться в пригодные моменты времени за счет описанных в способе е) реакций.
Исходные вещества, используемые в способе а), известны или, если они являются новыми, могут получаться известным способом, например, из гидразина или его соответствующих производных соединений формулы III, из соответствующих аминокислот или их аналогов, например, с одной или двумя указанными значениями R3 и R4, соединений формулы IV.
Соединения формулы III, например, получаются из соединений формулы XV:
H2N-NH-R11
где R11 обозначает водород или защитную для аминогруппы группу, описанную выше в способе б), в особенности (трет.-низший алкокси)-карбонил, как трет. бутоксикарбонил, арил-(низший
алкокси)-карбонил,
как бензилоксикарбонил, или 9-флуоренилметоксикарбонил, или одну из вышеуказанных ациламинозащитных групп. Способ заключается в том, что для получения соединения формулы I, где
R7 означает


R7-NH-NH-R11
причем остатки во всех указанных соединениях имеют вышеуказанные значения и функциональные группы в участвующих реагентах, которые не должны принимать участия в реакции, в случае необходимости защищены; в случае необходимости защитную группу R11, если она не соответствует одну из остатков R9 в соединениях формулы I, и/или другие защитные группы отщепляют, и вводят остатки R9, кроме водорода, путем конденсации при вышеуказанных условиях способа б) взаимодействием с кислотами формулы VI, или путем алкилирования в соединения формулы XIII или формулы XV, как определено выше.
Используемые для получения соединений формулы XVI, пригодные для введения R7 карбонильные соединения представляют собой альдегиды или кетоны, реакционноспособные карбонильные группы которых после реакции с соединениями формулы XV и последующего восстановления приводят к получению одного из указанных остатков R7; предпочтительны альдегиды, которые пригодны для введения низшего алкила, циклогексил-(низшего алкила) или фенил-(низшего алкила).
Реакция карбонильных соединений с соединениями формулы XVI с получением соответствующих гидразонов осуществляется в обычных для реакции карбонильных соединений с аминами условиях, предпочтительно в полярных органических растворителях, например, простых эфирах, как тетрагидрофуран или диэтиловый эфир, спиртах, как метанол или этанол, амидах карбоновых кислот, как диметилформамид, или сложных эфирах, как этилацетат, или в водном растворе, предпочтительно в метаноле, далее в присутствии или в отсутствии кислых катализаторов, например, карбоновых кислот, как муравьиная кислота или уксусная кислота, или сульфокислот, как n-толуолсульфокислота, при температурах от 0oC до температуры кипения с обратным холодильником реакционной смеси, предпочтительно при температурах от 20oC до температуры кипения с обратным холодильником реакционной смеси.
Восстановление полученных гидразонов осуществляется предпочтительно путем гидрирования в присутствии пригодного катализатора. В качестве пригодных для гидрирования катализаторов применяются металлы, как никель, железо, кобальт или рутений, или благородные металлы, соответственно, их оксиды, как палладий или родий, соответственно их оксиды; в случае необходимости, например, нанесенные на пригодные носители, как сульфат бария, оксид алюминия или активный уголь, или в виде скелетных катализаторов, как никель Ренея. Используемыми растворителями для каталитического гидрирования являются, например, вода, спирты, как метанол или этанол, сложные эфиры, как этилацетат, простые эфиры, как диоксан; хлорированные углеводороды, как дихлорметан, амиды карбоновых кислот, как диметилформамид, или карбоновые кислоты, как ледяная уксусная кислота, или смеси этих растворителей. Гидрирование осуществляют при температурах 10 250oC, предпочтительно от комнатной температуры до 100oC, и под давлением водорода 1 200 бар, предпочтительно 1 10 бар, при обычных аппаратурах.
Особенно предпочтительными для получения соединений формулы XV являются реакционные условия, которые аналогичны описанным в J.Chem. Soc. Perkin I, 1712 (1975).
Соединения формулы XV, например, получаются путем восстановления аминокислот формулы:

где R10 обозначает водород или одну из указанных в способе а) защитных для аминогруппы групп, в особенности трет.-(низший алкокси)-карбонил, как трет. бутоксикарбонил, арил-(низший алкокси)-карбонил, как бензилоксикарбонил или 9-флуоренилметоксикарбонил, или одну из указанных там ацил-аминозащитных групп; R3 и R4 имеют указанные для соединений формулы I значения, предпочтительно аминокислот формулы:
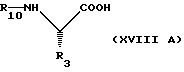
в которых остатки имеют указанные значения, с получением альдегидов формулы:

где остатки имеют указанные значения, предпочтительно альдегидов формулы:

где остатки имеют указанные значения (получаются, например, из соединений формулы XVIII А), с последующим введением во взаимодействие этих альдегидов с илид-соединением, предпочтительно с тио-илид-соединением, с получением эпоксида формулы:

где остатки имеют указанные значения, предпочтительно соединений формулы:

(получается, например, из соединений формулы XIX А), где остатки имеют указанные значения,
и, в случае необходимости, отщеплением защитной группы R11, если она не соответствует одному из остатков R8 или R9 в соединениях формулы 1, причем остатки имеют указанные значения, и алкилированием аминогруппы полученного соединения реагентами с нуклеофильными удаляемыми группами формул X или XI, где остатки имеют указанные значения, в условиях описанных для дополнительных мер способов.
Восстановление аминокислот формулы XVIII или XVIIIA до соответствующих альдегидов XIX и XIXA осуществляется, например, путем восстановления до соответствующих спиртов и последующего окисления до указанных альдегидов.
Восстановление до спиртов осуществляется, например, путем гидрирования галоидангидридов кислот или других, указанных в способе б), активированных производных карбоновых кислот при указанных для гидрирования получаемых из соединений формулы XVI гидразонов условиях, или с помощью комплексных гидридов, как боргидрид натрия. Последующее окисление полученных спиртов возможно, например, в условиях для окисления соединений формулы 1, в которых R5 обозначает гидроксил и R6 обозначает водород, до таких, в которых R5 и R6 вместе обозначают оксо-группу, как описано в дополнительных мерах способов, или благодаря окислению гидроксильной группы с помощью сульфоксида, как диметилсульфоксид, в присутствии активирующего гидроксильную группу реагента, как хлорангидрида карбоновой кислоты, например, оксалилхлорида, в инертном растворителе, например, галогенированном углеводороде, как дихлорметан, и/или ациклическом или циклическом простом эфире, как тетрагидрофуран, при -80oC-0oC, например, от 78oC до -50oC.
Также возможно прямое восстановление аминокислот в альдегиды, например, путем гидрирования в присутствии частично отравленного палладиевого катализатора или путем восстановления соответствующих сложных эфиров аминокислот, например, низших алкиловых эфиров, как этилового эфира, с помощью комплексных гидридов, например, боргидрида натрия, или предпочтительно алюминий гидридов, например, литийалюминийгидрида, литий-три-(трет.-бутокси)-алюминийгидрида или в особенности диизобутилалюминийгидрида, в аполярных растворителях, например, в углеводородах, или ароматических растворителях, как толуол, при -100oC-0oC, предпочтительно при -70oC -30oC, и последующего превращения в соответствующие семикарбазоны, например) с помощью соответствующих солей семикарбазонов, как семикарбазидгидрохлорид, в водных системах растворителей, как спирт/вода, например, этанол/вода, при температурах от 20oC до 60oC, предпочтительно при 10-30oC, и введения во взаимодействие полученных семикарбазонов с реакционноспособным альдегидом, например, формальдегидом, в инертном растворителе, например, полярном органическом растворителе, например, амиде карбоновой кислоты, как диметилформамид, при температурах от -30oC до 60oC, предпочтительно при 0-30oC, и затем вводят во взаимодействие с кислотой, например, с сильной неорганической кислотой, как галогенводород, в водном растворе, в случае необходимости в присутствии ранее используемого растворителя, при температурах от -40oC до 50oC, предпочтительно при -10oC-30oC. Соответствующие сложные эфиры получаются путем взаимодействия аминокислот с соответствующими спиртами, например, этанолом, аналогично используемым при конденсации в способе б) условиям, например, путем взаимодействия с неорганическими галоидангидридами кислот, как тионилхлорид, в смесях органических растворителей, как смеси из ароматических и спиртовых растворителей, например, толуол и этанол, при температурах от 50oC до 50oC, предпочтительно при -10oC-20oC.
Получение соединений формулы XIX и формулы XIXA осуществляется особенно предпочтительно в условиях, аналогичных указанным в J. Org. Chem. 47 3016 (1982) или J. Org. Chem. 43 3624 (1978).
Пригодным для превращения соединений формулы XIX или XIXA в эпоксиды формулы XX или XXA тио-илидом является, например, диалкилсульфонийметилид, например, диметилсульфонийметилид, алкил- или фенилдиалкиламиносульфоксонийметилид, например, метил- или фенилдиметиламиносульфоксонийметилид, или диалкилсульфоксонийметилид, например, диметил- или диэтилсульфоксонийметилид.
Соответствующее тиоилидное соединение целесообразнее получать in situ из соответствующей сульфониевой или сульфоксониевой соли и основания, например, гидрида натрия, в диполярном апротонном растворителе, например, диметилсульфоксиде, или простом эфире, например, тетрагидрофуране или 1,2- диметоксиэтане и затем вводятся во взаимодействие с соединениями формулы XIX или XIXA. Взаимодействие протекает обычно при комнатной температуре, при охлаждении, например, вплоть до -20oC, или при легком нагревании, например, вплоть до 40oC. Одновременно образующийся сульфид, сульфинамид, соответственно, сульфоксид удаляется при последующей обработке водой.
Взаимодействие с тиоилидом протекает особенно предпочтительно в условиях, аналогичных в J. Org. Chem. 50, 4615 (1985).
Соединение формулы XX (предпочтительно XXA) получается также из соединения формулы XIX (предпочтительно XIXA), как указано выше, путем введения его во взаимодействие с три- (низший алкил) -силилметильным соединением Гриньяра, например, приготовленным из соответствующего галоидметилсилана, как хлорметил-триметилсилан, в инертном растворителе, например, простом эфире, как диоксан или диэтиловый эфир, при температурах 0-50oC, например, от комнатной температуры до примерно 40oC; последующего элиминирования при удалении силильного остатка и образовании двойной связи, например, с помощью кислоты Льюиса, как BF3 причем предпочтительно также отщепляется присутствующая защитная для аминогруппы группа R10, в инертном растворителе, например, в простом эфире, как диэтиловый эфир, или галогенуглеводороде, как дихлорметан, или их смеси, при температурах от -50oC до температуры кипения с обратным холодильником, в особенности при 0-30oC, и если необходимо, путем нового ацилирования при введении защитной для аминогруппы группы, такой как R10, как указано выше, и окисления полученной двойной связи до оксирана, предпочтительно с помощью надкислоты, например, м-хлорнадбензойной кислоты, в инертном растворителе, например, галогенированном углеводороде, как дихлорметан, при температурах от -20oC до температуры кипения смеси с обратным холодильником, например, при 10-30oC.
Исходные соединения способов б), в) и г) известны или, если они являются новыми, могут получаться известными способами, например, соединение формулы V можно получать из соответствующих гидразиновых производных формулы III, в которой R9 обозначает водород, а остальные остатки имеют указанные для соединений формулы V значения, и пригодных эпоксидов формулы IV, где остатки имеют указанные для соединений формулы V значения (способ б)); соединение формулы VII можно получать из соответствующих гидразиновых производных формулы III, в которой остатки имеют указанные для соединений формулы VII значения, и соответствующих эпоксидов формулы IV, где R1 обозначает водород, а остальные остатки имеют указанные для соединений формулы VII значения (способ в)); и соединение формулы IX можно получать из соответствующих гидразиновых производных формулы III, в которой R9 обозначает водород, а остальные остатки имеют указанные для соединений формулы IX значения (способ г) и соответствующих эпоксидов формулы IV, где R1 обозначает водород, а остальные остатки имеют указанные для соединений формулы IX значения (способ г) ), по аналогии со способом а), в случае необходимости при применении и отщеплении защитных групп.
Соединения формулы I', в которой
заместители имеют
указанные выше значения, можно получать, например, из соединений формулы III':

где остатки имеют указанные для соединений формулы 1 значения, как описано в способе б), путем введения во взаимодействие с соединением формулы IV, причем имеющиеся функциональные группы, которые не должны принимать участия в реакции, как и там защищены и после реакции могут высвобождаться.
Предпочтительными для способа г) являются исходные соединения формулы:
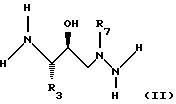
где R3 обозначает незамещенный или замещенный алкил или циклогексилалкил; фенил-низший алкил, незамещенный или замещенный; и R7 обозначает незамещенный или замещенный алкил или циклогексил-низший алкил, фенил-низший алкил, а также соли указанных соединений, если имеются солеобразующие группы, которые являются промежуточными соединениями согласно изобретению.
Эти соединения могут быть защищены в особенности по одной или по обеим аминогруппам, причем в случае наличия двух защитных для аминогруппы групп последние могут быть идентичными или отличаться друг от друга.
В качестве защитных для аминогруппы групп, например, находят применение указанные выше в способе а) защитные для аминогруппы группы. Указанные для соединений формулы II остатки R3 и R7 имеют значения, указанные выше для соединений формулы 1 при определении остатков R3 и R7.
Очень предпочтительны соединения формулы II, где R3 обозначает циклогексил-(низший алкил), фенил-(низший алкил) или п-фторфенил(низший алкил) и R7 обозначает низший алкил, циклогексил-(низший алкил), фенил-(низший алкил), п-цианофенил-(низший алкил) или п-фторфенил(низший алкил) а также соли указанных соединений, если имеются солеобразующие группы.
Очень предпочтительны в особенности соединения формулы II, где R3 обозначает фенил-(низший алкил) и R7 обозначает низший алкил, циклогексил(низший алкил) или фенил-(низший алкил), а также соли указанных соединений, если имеются солеобразующие группы.
В первую очередь предпочтительны соединения формулы II, где R3 обозначает циклогексилметил, бензил или п-фторбензил и R7 обозначает н-бутил, изобутил, циклогексилметил, бензил, п-фторбензил или п-цианобензил, а также соли указанных соединений, если имеются солеобразующие группы.
В первую очередь в особенности предпочтительны соединения формулы II, где R3 обозначает бензил и R7 обозначает изобутил, циклогексилметил или бензил, а также соли указанных соединений, если имеются солеобразующие группы.
Прежде всего предпочтительны указанные в примерах соединения формулы II.
Соединения формулы II, где заместители имеют указанные значения, или их соли, если имеются солеобразующие группы, получают, например, тем, что производное
гидразина формулы:
R7-NH-NH-R11
где R11 обозначает защитную для аминогруппы группу, присоединяют к эпоксиду формулы:

где R10 обозначает защитную для аминогруппы группу, и в случае необходимости получаемое по вышеуказанному способу А) соединение формулы II по меньшей мере с одной солеобразующей группой переводят в его соль или полученную соль переводят в свободное соединение или в другую соль и/или в случае необходимости полученную смесь изомеров разделяют и/или имеющиеся защитные группы в соединении формулы II отщепляют и/или предлагаемое согласно изобретению соединение формулы И превращают в другое, согласно изобретению, соединение формулы II.
Получение и превращение солей, разделение смесей изомеров, отщепление защитных групп и превращение соединений формулы II осуществляются аналогично описанным выше для соединений формулы I способом.
Особенно предпочтительно получение исходных продуктов формулы II, в которых заместители имеют указанные значения, путем отщепления защитных групп от соединений формулы II, в которых одна или обе аминогруппы защищены защитными для аминогрупп группами, в особенности при условиях для гидролиза соединений формулы I, как описано в дополнительных мерах способов.
Методы присоединения соединения формулы XVI к таковым формулы XXA описаны выше в способе а) при получении соединений формулы I.
Получение защитных соединений формулы I осуществляется, например, любым из до сих пор известных способов, в особенности из соединений формулы III и IV, причем функциональные группы в этих соединениях в случае необходимости защищены защитными группами, как описано в способе а).
Кислоты формул VI, VIII, XVII и XXI, а также соединения с нуклеофильными группами формул X, XI, XII, XIII, XIV и XV известны или, если они являются новыми, получаются само по себе известными способами.
Следующие примеры служат для иллюстрации изобретения, однако никоим образом не ограничивают его объема охраны.
Температуры указаны в градусах Цельсия (oC). Если нет никаких температурных данных, то реакция протекает при комнатной температуре, Rf- значения, которые указывают отношение пройденного расстояния соответствующим веществом к пройденному расстоянию фронта растворителя, определяются на пластинах с тонким слоем силикагеля путем тонкослойной хроматографии (ТСХ) в следующих системах растворителей (табл.4).
Сокращение "Rf(A)" обозначает, например, что Rf значение определено в системе растворителей A. Количественное соотношение растворителей друг к другу всегда указывается в объемных долях (табл.5).
Колонка (250х4,6 мм), заполненная с помощью материала "обратимой фазы" C18- Nucleosil® (5 мкм средний размер зерен; ковалентно связанный октадецилсиланами, дериватизированный силикагель, Macherey Nagel, Duren ФРГ). Определение через УФ-абсорбцию при 215 нм. Времена удерживания (tRet) указываются в минутах. Скорость истечения 1 мл/мин.
Для обозначения систем жидкостей (Fliessmittel) при импульсной хроматографии и хроматографии среднего давления применяют одинаковые сокращения.
Другие используемые краткие названия и сокращения имеют следующие значения (табл.6).
Масс-спектральные значения получаются либо путем обычной масс-спектрометрии, либо по FAB-MS-методу (метод "бомбардировки быстрыми атомами"). Масс-спектральные данные относятся в первом случае к непротонированным молекулам (M)+ или к протонированным молекулам (M+H)+.
Значения, получаемые с помощью спектроскопии протонного ядерного резонанса (1 Н-ЯМР), указываются в ppm (частей на миллион) в расчете на тетраметилсилан в качестве внутреннего стандарта. s синглет, d дублет, t - триплет, k квартет, m мультиплет, dd двойной дублет, br. широкий.
Величины в ИК-спектроскопии указываются в см-1, в круглых скобках указывается соответствующий растворитель. Если указано, s обозначает сильную, m обозначает среднюю и w обозначает слабую интенсивность соответствующих полос.
Остаток в значении [PheNNPhe] обозначает двухвалентный радикал
3(S)-амино-4-фенил-1-(N-бензилгидразино)-бутан-2(S)-ола и имеет формулу:

Остаток с обозначением [PheNNCha] обозначает двухвалентный радикал 3(S)-амино-4-фенил-1(N-циклогексилметилгидразино)-бутан-2(S)-ола и имеет формулу:

Остаток с обозначением [PheNNLeu] обозначает двухвалентный радикал 3(S)-амино-4-фенил-1-(N-изобутилгидразино)-бутан-2(S)-ола и имеет формулу:

Остаток со значением -[PheNNNle] обозначает радикал 3(S)-амино-4-фенил-1-(N-н-бутилгидразино)-бутан-2(S)-ола и имеет формулу:

Остаток со значением -[PheNN(p-F)Phe] обозначает двухвалентный радикал 3(S)-амино-4-фенил-1-(N-(п-фторфенилметил)-гидразино)-бутан-2-(S)-ола и имеет формулу:

Остаток со значением-[(p-F)PheNN(p-F)Phe] обозначает двухвалентный радикал 3(S)-амино-4-(п-фторфенил)-1-(N- (п-фторфенилметил)-гидразино)-бутан-2(S)-ола и имеет формулу:

Остаток со значением [PheNN(p-CN)Phe] обозначает двухвалентный радикал 3(S) -амино-4-фенил-1-(N-(п-цианофенилметил)-гидразина)-бутан-2(S)-ола и имеет формулу:

Остаток со значением-[ChaNNLeu] обозначает двухвалентный радикал 3(S)-амино-4-циклогексил-1-(N-изобутил-гидразино)-бутан-2(S)-ола и имеет формулу:

Для обозначения двухвалентных радикалов природных альфа-аминокислот применяются обычные в химии пептидов сокращения. При этом, однако, аминокислоты, которые находятся в названии соединений справа от радикалов-[PheNNPhe]-[PheNNCha]-[PheNNLeu] -[PheNNNle,-[PheNN(p-F)Phe]-[(p-F)PheNN(h-F)Phe] -[PheNN(p-CN)Phe] или-[ChaNNLeu] в отличие от обычной номенклатуры пептидов, где слева находится амино-конец, а справа карбокси-конец, связывающая карбокси-группа находится слева, что указывается стрелкой (←) которая символизирует обратное направление связи. Конфигурация у α -атома углерода, если она известна, указывается предварительными приставками (L)-или (D)-. Этерифицированные до простых эфиров на фенольной гидроксильной группе остатком R тирозиновые радикалы обозначаются с помощью Tyr(OR). Nle обозначает радикал норлейцина.
Пример 1: Boc-[PheNN
Phe]-Boc:
Раствор 300 мг (1,14 ммоль) (2R)-[1'(S)-Вос-амино-2'-фенилэ-тил]оксирана (J. Org. Chem.

Пример
2: Z-(L)-Val-[PheNN
Phe] J (L)-Val-Z):
191 мг (0,76 ммоль) Z-(L-валина, 336 мг (0,76 ммоль) ВОР и 103 мг (0,76 ммоль) HOBt растворяют в 5 мл 0,3М раствора NMM в ДМФ, спустя 10
минут добавляют 100 мг (0,25 ммоль)
H-[PheNNPhe]-H•3НСl и перемешивают в течение 2 ч при комнатной температуре в атмосфере азота. Реакционную смесь выпаривают, остаток растворяют в
метиленхлориде и промывают дважды нас,
раствором бикарбоната натрия. Органические фазы отфильтровывают через вату, выпаривают и остаток очищают с помощью хроматографии на силикагеле смесью
метиленхлорид/эфир (1: 1). После лиофилизации
содержащих продукт фракций из диоксана получают указанное в заголовке соединение в виде твердого вещества белого цвета. FAB:MS: (M+H)+ 752,
tRet (I) 27,8 мин. Rf(E) 0,
45.
Исходный материал готовят следующим образом.
а) H-[PheNNPhe]-H•3НСl
Раствор 280 мг (0,
58 ммоль) Boc-[PheNNPhe]-Boc из
примера 1 в 10 мл 4н хлороводорода в диоксане перемешивают в течение 2 ч при комнатной температуре и в атмосфере азота и затем лиофилизуют. Новая
лиофилизация из смеси диоксана с трет.-бутанолом дает
титульное соединение в виде хлопьевидного твердого вещества. FAB-MS: (M+H)+ 286; tRRet(I) 23,1 мин. Rf(C) 0,
17.
Пример 3: Boc-(L)-Val- [PheNNPhe] J ((L)-Val-Boc):
Аналогичным образом, как описано в примере 2, из 50 мг (0,13 ммоль) H-[PheNNPhe] -H•3HCI,
83 мг (0,83 ммоль) Boc-(L)-валина, 168 мг (0,38
ммоль) ВОР, 51 мг (0,38 ммоль) НОВ и 2,5 мл 0,3М NMM в ДМФ, после хроматографической очистки на силикагеле смесью хлороформа с метанолом (95:5) и
лиофилизации из диоксана, получают указанное в
заголовке соединение. FAB-MS: (M+H)+ 684; tRet(l) 27,4 мин. Rf(E) 0,38.
Пример 4: Boc-[PheNN
Cha]-Boc:
Аналогично примеру 1, из 232
мг (0,88 ммоль) (2R,3S)-1-[3-Вос-амино-2-фенилэтил] оксирана и 200 мг (0,88 ммоль) трет.-бутил-3-циклогексилметил-карбазата получают указанное в
заголовке соединение в виде осадка белого цвета из
гексана. FAB-MS: (M+H)+ 492; tRet(l) 30,4 мин; Rf(E) 0,78.
Исходный материал получают следующим
образом:
а)
трет.-бутил-3-циклогексилметил-карбазат:
10,2 г (45,1 ммоль) циклогексилкарбальдегид-трет.-бутоксикарбо- нилгидразона, растворенные в 400 мл метанола, гидрируют в
присутствии 5,1 г 5 -ной
платины-на-угле при комнатной температуре и под давлением водорода 4 атм. По окончании реакции отфильтровывают от катализатора и фильтрат выпаривают. Остаток растворяют в
метиленхлориде и промывают
водой. После выпаривания органической фазы получают указанное в заголовке соединение в виде бесцветной смолы.
1ЯМР (200 МГц, CDCI3): 6, 1 (s, br, 1Н); 3,9 (s, br, 1Н); 2,65 (d, 2H); 1,8-0,75 (m, 11H); 1,45 (s, 9H); tRet(I) 32,0 мин. Rf(E) 0,75.
б)
циклогексилкарбальдегид-трет.-бутоксикарбонилгидразон:
Раствор 10,8
г (81,2 ммоль) трет.-бутилкарбазата и 10,1 г (90 ммоль) циклогексилкарбальдегида в 400 мл этанола кипятят с обратным
холодильником в течение 2 часов. Затем отгоняют половину растворителя и путем
добавления воды осаждают указанное в заголовке соединение. Его прямо применяют далее в п. А).
Пример 5:
H-(L)-Val-[PheNNPhe] J ((L)-Val)-H•3HCI:
Раствор 40
мг (0,06 ммоль) Boc-(L)-Val-[PheNNPhe] J -((L)-Val)-Boc из примера 3 в 4 мл 4н хлороводорода в диоксане
перемешивают при комнатной температуре в течение 1 ч. Затем разбавляют диоксаном и
после лиофилизации получают указанное в заголовке соединение в виде гидрохлорида. FAB-MS: (M+H)+ 484;
tRet(II) 25,8 мин RfA) 0,45.
Пример 6:
N-тиоморфолинокарбонил-(L)-Val_[PheNNPhe] J (N-тиоморфолино-карбонил-(L) -Val):
Раствор 20 мг (0,03
ммоль) H-(L)-Val-[PheNNPhe] J ((L)-Val)-H•3НСl из примера 5 в
0,5 мл ДМФ смешивают при комнатной температуре последовательно с 35 мкл (0,25 ммоль) триэтиламина и 16 мг (0,1 ммоль)
4- тиоморфолинилкарбонилхлорида и перемешивают в течение 1 часа при комнатной
температуре. Реакционную смесь разбавляют хлороформом и промывают насыщенным раствором бикарбоната натрия. Органическую
фазу фильтруют через вату, выпаривают и остаток хроматографируют на силикагеле с
помощью градиентов хлороформ/метанол (15: 1->8: 1). Фракции продукта выпаривают и осаждают с помощью смеси
метиленхлорида с ДИПЭ. После лиофилизации из диоксана они дают указанное в заголовке
соединение в виде хлопьевидного твердого вещества. FAB-MS: (M+H)+ 742. tRet(l) 21,6 мин
Rf(D) 0,54.
Исходный материал получают следующим образом:
а) (4-тиоморфолинилкарбонил)-хлорид:
К раствору 85 мл (165 ммоль) 20-ного фосгена в толуоле при
0oC прикапывают раствор 10 г (97 ммоль) тиоморфолина в 200 мл толуола и
перемешивают суспензию белого цвета в течение 1 часа при комнатной температуре. Избыточный фосген удаляют путем
пропускания азота, суспензию отфильтровывают и фильтрат выпаривают. Получают указанное в
заголовке соединение в виде желтого масла. ИК-спектр (CH2Cl2, см-1): 1735,
1450, 1440, 1405, 1370, 1180.
Пример 7: N-морфолинокарбони-(L)-Val-[PheNNPhe] J (N-морфолоино-карбонил-(L)-Val):
Раствор 100 мг (0,25 ммоль) H-[PheNN
Phe]-H•3HCI из примера 2а, 163 мг (0,76 ммоль) N-морфолинокарбонил-(L)-валина и 288 мг (0,
76 ммоль) HBTU в 2 мл ДМФ смешивают с 210 мкл (1,52 ммоль) триэтиламина и перемешивают при комнатной
температуре в течение 16 ч в атмосфере азота. Реакционную смесь полностью выпаривают, остаток
растворяют в метиленхлориде и промывают насыщенным раствором бикарбоната натрия. Органическую фазу
фильтруют через вату, выпаривают и хроматографируют на силикагеле смесью метиленхлорид/метанола (15:
1). Указанное в заголовке соединение осаждают из метиленхлорида гексаном и после лиофилизации из
диоксана с трет.-бутанолом получают в виде хлопьевидного твердого вещества.
FAB-MS: (M+H)+ 710; tRet(I) 16,3 мин Rf(E) 0,16.
Исходный материал получают следующим образом:
а) N-морфолинокарбонил-(L)-валин:
2,7 г (8,4
ммоль) N-морфолинокарбонил-(L)-валин-бензилового сложного эфира растворяют в 75 мл
уксусного эфира и в присутствии 500 мг 10-ного палладия-на-угле под давлением водорода 1 атм и при комнатной
температуре гидрируют в течение 3 ч. Катализатор отфильтровывают и после выпаривания
растворителя получают указанное в заголовке соединение в виде бесцветного масла.1H ЯМР (300 МГц, СD3OD: 4,15 (m, 1Н); 3,65 (m, 4Н); 3,40 (m, 4Н); 2,12 (m, 1Н), 0,95 (2d,
6Н).
б) N-морфолинокарбонил-(L)-валин-бензиловый сложный эфир:
Раствор 4 г (10,5 ммоль)
4-толуолсульфоната сложного (L)-валин-бензилового эфира в 560 мл метиленхлорида
смешивают с 0,8 мл (8,1 ммоль) (морфолинокарбонил)-хлорида (получение: J. Med. Chem. 31, 2277 (1988)) и 4,1 мл (24,1
ммоль) N-этилдиизопропиламина и перемешивают в течение 24 ч при комнатной
температуре. Реакционную смесь разбавляют уксусным эфиром и промывают последовательно с помощью 1н соляной кислоты, воды,
насыщенного раствора бикарбоната натрия и рассола. Органическую фазу сушат
над сульфатом натрия и выпаривают. После хроматографии на силикагеле с помощью уксусного эфира она дает сложный
N-морфолинокарбонил-(L)-валин-бензиловый эфир в виде бесцветного масла. Сложный эфир
тотчас применяется далее в а).
Пример 8: Фенилацетил-(L)-Val-[PheNNPhe] J
(N-фенилацетил-(L)-Val:
Аналогично примеру 7, из 100 мг (0,25 ммоль) H-[PheNN
Phe]-H•3HCI из примера 2а, 143 мг (0,61 ммоль) фенилацетил-L-валина (получение: Mem. Tokyo Univ.
Agric. 20, 51 (1978)), 230 мг (0,61 ммоль) HBTU и 200 мкл (1,42 ммоль) триэтиламина, после
хроматографической очистки с помощью смеси метиленхлорид/эфир/метанол (20:20:1) и лиофилизации из смеси
диоксан/трет.-бутанол получают указанное в заголовке соединение. FAB-MS: (M+H)+ 720.
tRet(I) 23,7 мин Rf(G) 0,21.
Пример 9:
N-(3-пиридилацетил)-(L)-Val-[PheNNPhe] J (N-3-пиридилацеталил)-(L)-Val:
Аналогично примеру 7, из 100
мг (0,25 ммоль) H-[PheNNPhe]-H•3HCI из примера 2а, 576 мг (1,
52 ммоль) HBTU, 358 мг (1,52 ммоль) N-(3- пиридилацетил)-(L-валина и 316 мкл (2,3 ммоль) триэтиламина, после
хроматографической очистки с помощью смеси хлороформ/метанол (5:1) и лиофилизации из смеси
диоксана с трет.-бутанолом, получают указанное в заголовке соединение в виде твердого вещества белого цвета.
FAB-MS: (M+H)+ 722. tRet(II) 27,9 мин Rf(A) 0,71.
Исходный материал получают следующим образом:
а) N-(3-пиридилацетил)-(L)-валин:
3,
4 г N-(3-пиридилацетил)-(L)-валин-трет.-бутилового сложного эфира растворяют в 20 мл
трифторуксусной кислоты с метиленхлоридом (1:1) и перемешивают в течение 16 ч при комнатной температуре.
Реакционный раствор полностью выпаривают и остаток настаивают с ДИПЭ. Получают указанное в
заголовке соединение в виде аморфного твердого вещества белого цвета.1Н-ЯМР (200 МГц, CD3OD): 8,9-8,6 (m, br, 1Н); 8,5 (m, 1Н), 7,95 (m, 1H); 4,33 (m, 1H); 3,93 (s, 2H); 2,2 (m,
1H); 0,98 (2d, 6H).
б) N-(3-пиридилацетил)-(L)-валин-трет.-бутиловый сложный эфир:
К раствору 3,36 г (16 ммоль) трет.-бутилового эфира (L)-валина в виде гидрохлорида, 2 г (14,
5 ммоль) 3-пиридилуксусной кислоты и 2,17 мл (14,3 ммоль) диэтилового эфира цианофосфоновой кислоты и
20 мл ДМФ при 0°С прикапывают 4,2 мл триэтиламина. Реакционную смесь перемешивают в течение 48 ч
при комнатной температуре, затем разбавляют метиленхлоридом и промывают 10-ной лимонной кислотой, а
также насыщенным раствором бикарбоната натрия. Органическую фазу фильтруют через вату и после
выпаривания растворителя она дает трет.-бутиловый эфир N-(3-пиридилацетил)-(L)-валина, который тотчас
применяют далее в а).
Пример 10: Boc-(L)-Val-[PheNNCha] J
(L)-Val)-Boc:
Аналогично примеру 7, исходя из 500 мг (1,25 ммоль) H-[PheNNPhe]-H•3HCI,
1,08 г (4,98 мысль) Boc-(L-валина, 1,89 г (4,98 ммоль) HBTU и 1,39 мл (9,96 мысль)
триэтиламина после хроматографической очистки на силикагеле с помощью смеси метиленхлорида с эфиром (1:1) и
лиофилизации из диоксана, получают указанное в заголовке соединение в виде хлопьевидного
твердого вещества. FAB-MS: (M+H)+ 690; tRet(I) 29,3 мин. Rf(H) 0,48.
Исходный материал получают следующим образом:
а) H-[PheNN
Phe]-H•3НСl
1,10 г (2,2 ммоль) Boc-[PheNNCha]-Boc из примера 4 растворяют в 20 мл 4н
хлороводорода в диоксане и перемешивают при комнатной температуре в течение 3 ч. После
лиофилизации реакционного раствора получают указанное в заголовке соединение в виде гидрохлорида. FAB-MS:
(M+H)+ 292; tRet(II) 27,3 мин.
Пример 11:
Z-(L)-Val-[PheNNCha] J ((L)-Val)-Z:
Аналогично примеру 2 из 50 мг (0,12 ммоль) H-[PheNN
Cha]-H•3HCI из примера 10a, 94 мг (0,37 ммоль) Z-(L)-валина, 165 мг (0,37
ммоль) ВОР, 51 мг (0,37 ммоль) HOBt и 2,5 мл 0,3М NMM в ДМФ, после хроматографической очистки на силикагеле с помощью
смеси метиленхлорида с эфиром (1:1) и лиофилизации из диоксана, получают указанное
в заголовке соединение. FAB-MS: (M+H)+ 758; tRet(I) 29,1 мин. Rf*(H) 0,55.
Пример 12: Boc-[PheNNLeu]-Boc:
Аналогично примеру 1,
исходя из 1,0 г (3,8 ммоль) (2R)-[1'(S)-Boc-амино-2'-фенилэтил] оксирана и 715 мг (3,8 ммоль)
трет.-бутил-3-изобутилкарбазата (получение: J. Chem. Soc. Perkin I, 1712 (1975)), получают указанное в
заголовке соединение в виде осадка из гексана. FAB-MS: (M+H)+ 452; tRet(I)
27,2 мин. Rf(I) 0,55.
Пример 13: Z-(L)-Val-[PheNNLeu] J
((L)-Val)-Z:
Аналогично примеру 2, исходя из 60 мг (0,17 ммоль) H-[PheNN
Leu]-H•3НСl, 125 мг (0,50 ммоль) Z-(L)-валила, 221 мг (0,50 ммоль) ВОР, 67 мг (0,50 ммоль) HOBt и 3,3
мл 0,3М NMM в ДМФ, после хроматографической очистки на силикагеле с помощью смеси
метиленхлорида с эфиром (1:1) и лиофилизации из диоксана получают указанное в заголовке соединение. FAB- MS: (M+H)+ 718, tRet(I) 26,8 мин, Rf(H) 0,38.
Исходный материал получают следующим образом:
а) H-[PheNNLeu]-H•3НСl:
Аналогично
примеру 10a, из 1,21 г (2,48 ммоль) Boc-[PheNNLeu]-Boc из примера 12,
получают указанное в заголовке соединение в виде лиофилизата. FAB-MS: (M+H)+ 252, tRet(II) 20,9
мин, Rf(K) 0,23.
Пример 14: H-(L)-Val-[PheNNCha] J ((L)-Val)-H•3HCI:
Аналогично примеру 10a, из 632 мг (0,91 ммоль) Boc-(L)-Val- [PheNN
Cha] J ((L)-Val)-Boc из примера 10, после лиофилизации получают указанное
в заголовке соединение в виде гидрохлорида. FAB-MS: (M+H)+ 490, tRet(II) 29,4 мин, Rf(К) 0,
23.
Пример 15: N-(3-пиридилацетил)-(L)-Val-[PheNNLeu] J -(3-пиридилацетил)-(L)-Val:
Аналогично примеру 9, из 90 мг (0,25 ммоль) H-[PheNNLeu]-H•
3HCI из примера 13а, 358 мг (1,52 ммоль)
N-(3-пиридилацетил)-(L)-валила, 576 мг (1,52 ммоль) HBTU и 316 мкл (2,5 ммоль) триэтиламина, после хроматографической очистки с помощью смеси метиленхлорида с
метанолом (5:1) и лиофилизации из
диоксана с трет.-бутанолом и водой, получают указанное в заголовке соединение. FAB-MS: (M+H)+ 688, tRet(IV) 15,5 мин, Rf(D) 0,37.
Пример 16:
N-трифторацетил-[PheNN(p-F)Phe]-Boc:
Раствор 4,0 г (15,4 ммоль) 2-(R)-[1'(S)-(трифторацетиламино)-2'-фенилэтил] оксирана и 3,89 г (16,2 ммоль) трет.
-бутил-3-(п-фторфенил-метил)арбазата в 35 мл метанола нагревают в течение примерно 20 ч в тугоплавкой трубке при 80oC. Реакционную смесь выпаривают, остаток растворяют в небольшом
количестве дихлорметана, из этого раствора гексаном осаждают указанное в заголовке соединение (холодильник). Далее продукт подвергают колоночной хроматографии (SiO2, метиленхлорид/эфир,
95:7). ТСХ: Rf(J)0,57, tRet(I) 24,3 мин; FAB-MS: (M+H)+ 500.
Исходные материалы получают следующим образом: а) N-3(S)-(Boc-амино)-2 (R,S)
-гидрокси-4-aенил-1-триметилсилилбутан:
В атмосферу азота помещают предварительно 24,7 г (1,02 моль) магния в 100 мл абс. эфира и через 35 мин смешивают с небольшим количеством йода и
одновременно с 132,5 мл (0,95 моль) хлорметилтриметилсилана и 300 мл эфира, причем температура поддерживается при 38oC с помощью ледяной бани.
Полученную реакционную смесь затем перемешивают 1,5 ч при комнатной температуре. После охлаждения до-60oC смешивают с суспензией 48,6 г (0,195 моль) N-Вос-фенилаланиналя (получение: D. J. Kempf, J. Org. Chem. 51, 3921(1986)) в 1,1 л эфира в течение 40 мин. Через 90 мин реакционная смесь нагревается до комнатной температуры и следующие 90 мин ее перемешивают при этой температуре. Затем выливают в 2 л ледяной воды и 1,5 л 10-ного водного раствора лимонной кислоты. Отделенную водную фазу экстрагируют дважды с помощью 500 мл эфира. Все экстракты промывают 500 мл 10-ной лимонной кислоты и дважды рассолом. После высушивания над сульфатом натрия концентрируют в вакууме и полученное, указанное в заголовке, соединение применяют далее без всякой дополнительной очистки.
TCX: Rf (L) 0,6. FAB-MS: (М+Н)+ 338.
б) 1-Фенил-3-бутон-2 (S)-амин:
Раствор 18,8 г (0,055 моль) 3(S)-(Boc-амино)-2(R,S)-гидрокси-4-фенил- 1-триметилсилилбутана в 420 мл
метиленхлорида при 5oC в течение 10 мин смешивают с 35,6 мл (0,28 моль) примерно 48-ного раствора трифторида бора в эфире. Реакционную смесь затем перемешивают 16 ч при комнатной
температуре, охлаждают до 10oC и в течение 20 мин смешивают с 270 мл 4н раствора гидроксида натрия. Водную фазу отделяют и экстрагируют дважды по 400 мл метиленхлоридом. Объединенные
органические экстракты промывают рассолом и сушат над сульфатом натрия. Указанный в заголовке продукт применяется далее без дополнительной очистки. ТСХ: Rf(C) 0,15; ИК.-спектр
(метиленхлорид) (см-1): 3370, 3020, 2920, 1640, 1605.
в) N-трифторацетил-1-фенил-3-бутон-2 (S)-амин:
Растворенные в 210 мл метиленхлорида и 70 мл пиридина, 11,9 г
(81 ммоль) 1-фенил-3-бутен-2(S)-амина при 0oC по каплям смешивают с 17,0 мл (121 ммоль) ангидрида трифторуксусной кислоты. После перемешивания в течение получаса при 0oC
экстрагируют 2 раза разбавленной HCl, водой и рассолом. Водные фазы промывают еще 2 раза метиленхлоридом, сушат их над сульфатом натрия и выпаривают. TCX: Rf(M) 0,4.
г)
2(R)-1'(S)-(трифторацетил-амино)-2'-фенилэтил]оксиран:
Раствор 14,5 г (60 ммоль) N-трифторацетил-1-фенил-3-бутен-2(S)-амина в 600 мл хлороформа смешивают с 54,28 г (314 ммоль)
м-хлорнадбензойной кислоты и перемешивают его 24 ч при комнатной температуре. Реакционную смесь промывают 2 раза с помощью 10-ного раствора сульфита натрия, 2 раза с помощью насыщенного раствора
карбоната натрия, водой и рассолом. Водные фазы экстрагируют еще 2 раза метиленхлоридом, объединенные органические фазы сушат над сульфатом натрия и выпаривают, что дает указанное в заголовке
соединение, которое без всякой дальнейшей очистки используют в ближайшей стадии. TCX: Rf(N) 0,6.
д) п-фторфенилкарбальдегид-трет.-бутоксикарбонилгидразон:
Аналогично примеру 4б), 32 г (242 ммоль) трет.-бутилкарбазата и 30 г (242 ммоль) п-фторбензальдегида в 300 мл этанола вводят во взаимодействие в течение 3 ч при 80oC с получением
титульного
соединения, которое после охлаждения и разбавления водой выкристаллизовывается: TCX Rа(N) 0,48; tRet(I) 19,4 мин.
е)
трет.-бутил-3-(п-фторфенилметил)карбазат:
Аналогично примеру 4а), 55 г (231 ммоль) п-фторфенилкарбальдегид- трет. -бутокси-карбонилгидразона в 500 мл ТГФ гидрируют с помощью палладия
(5%)-на-угле с получением указанного в заголовке соединения.1Н-ЯМР (200 МГц, CD3OD: 7,35 (dd, 8 и 6 Гц, 2Н); 7,05 (t, 8 Гц, 2Н); 3,9 (s, 2Н); 1,45 (s, 9Н).
Пример 17: N-морфолинокарбонил-(L)-Val-[PheNN(p-F)Phe]-Boc:
Смесь 185 мг (0,80 ммоль) N-морфолинокарбонил-(L)-валина (получение: см. пример 7а), 270 мг (0,67 ммоль) H-[PheNN(p-F)Phe]-Boc, 311 мг (0,70 ммоль) ВОР и 95 мг (0,70 ммоль) HOBt при комнатной температуре растворяют в 6,8 мл NMM/ДМФ, 0,3N, и перемешивают 5 ч при комнатной температуре. Реакционную смесь
выпаривают на роторном испарителе и остаток распределяют между четырьмя порциями метиленхлорида, двумя порциями раствора карбоната натрия, водой и рассолом. Высушенные над сульфатом натрия,
объединенные органические фазы выпаривают и очищают с помощью колоночной хроматографии (SiO2, этилацетат). TCX Rf(O) 0,38; tRet(l) 21,8 мин, FAB-MS: (М+Н)+
616.
Исходный материал получают следующим образом:
а) H-[PheNN(p-F)Phe]-Boc:
При 70oC к раствору 0,3 г (0,6 ммоль) N-трифторацетил-[PheNN(p-F)Phe]-Boc (получение см. Пример 16) в 50 мл метанола в атмосфере азота прикалывают 15 мл 1М водного раствора карбоната калия и дополнительно перемешивают 25 ч при этой температуре.
Реакционную смесь выпаривают на роторном испарителе, остаток смешивают с метиленхлоридом и промывают 2 раза водой и рассолом. Водные фазы экстрагируют метиленхлоридом, органические фазы сушат над
сульфатом натрия и выпаривают. Сырой продукт используют в ближайшей стадии без дальнейшей очистки: tRetRet(l) 16,2 мин.
Пример 18: N-морфолинокарбонил-(L)-Val-[PheNN(p-F)Phe] J ((L)-Val)-Z:
Раствор 86 мг (0,34 ммоль) Z-(L)-Val и 160 мг (0,31 ммоль) N- морфолинокарбонил-(L)-Val-[PheNN(p-F)Phe]-H в 2,7 мл 0,25М NMM в CH3CN
смешивают со 129 мг (0,34 ммоль) HBTU. Спустя 4 ч при комнатной температуре выпаривают и остаток распределяют между тремя порциями метиленхлорида, двумя порциями насыщенного раствора бикарбоната
натрия и рассолом. Высушивание органической фазы над сульфатом натрия и выпаривание дает указанное в заголовке соединение, которое после настаивания получается чистым из метиленхлорида с эфиром в
соотношении 1:1. TCX Rf(P) 0,4; tRet(l) 22,4 мин; FAB-MS: (M+H)+ 749.
Исходный материал получают следующим образом:
а)
N-морфолинокарбонил-(L)-Val-[PheNN(p-F)Phe]-H:
210 мг (0,34 ммоль) N-морфонилоканбонил-(L)-Val-[PheNN(p-F)Phe]- Boc (пример 17) растворяют в 105 мл муравьиной кислоты и
перемешивают 4 ч при комнатной температуре. Затем выпаривают, остаток обрабатывают метиленхлоридом и раствор промывают насыщенным раствором бикарбоната натрия и рассолом. Экстракция водных фаз двумя
порциями метиленхлорида, высушивание органических фаз над сульфатом натрия и выпаривание дает указанное в заголовке соединение, которое без дальнейшей очистки используется в самой ближайшей стадии,
tRet(I) 12,9 мин.
Пример 19: N-морфолинокарбонил-(L)-Val-[PheNN(p-F)Phe] J ((L)-Val)-h:
При нормальном давлении 160 мг (0,21 ммоль)
N-морфолинокарбонил-L-Val-[PheNN(p-F)Phe] J ((L)-Val)-Z (пример 18) в этаноле гидрируют с помощью 40 мг палладия (10%) -на-угле. После фильтрации через цеолит цеолит (кизельгур,
вспомогательное для фильтрации средство фирмы Fluka, Швейцария), выпаривания и лиофилизации из диоксана получают указанное в заголовке соединение: tRet(гидрохлорид, I) 13,4 мин; FAB-MS:
(M+H)+ 615.
Пример 20: N-морфолинокарбонил-(L)-Val-[PheNN(p-F)Phe] <-- ((L)-Val-морфолинокарбонил-Gly):
Раствор 26,9 мг (0,143 ммоль)
N-морфолинокарбонил-глицина и 80 мг (0,130 ммоль) N-морфолинокарбонил-(L)-Val-[PheNN(p-F)Phe] <-- ((L)-Val)-H в 1,1 мл NMM/CH3CN, 0,25М, смешивают с 54 мг (0,143 ммоль)
HBTU и перемешивают 16 ч при комнатной температуре. Выпаривают и остаток распределяют между тремя порциями этилацетата, водой, двумя порциями насыщенного раствора бикарбоната натрия, водой и
рассолом.
Высушивание органических фаз над сульфатом натрия и выпаривание дает указанное в заголовке соединение, которое после растворения в небольшом количестве ДМФ и осаждения из ДИПЭ, получается
чистым:
tRet(l) 15 мин; FAB-MS: (M+H)+ 785.
Исходный материал получают следующим образом:
а) N-морфолинокарбонил-глицин-бензиловый эфир:
Аналогично
примеру 7б), 7,69 г (22,8 ммоль) 4-толуолсульфоната сложного бензилового эфира глицина и 2,8 г (19 ммоль) (морфолинокарбонил)- хлорила в 118 мл метиленхлорида и 9 мл (53 ммоль)
N-этилдиизопропиламина
вводят во взаимодействие в течение 18 ч. Указанное в заголовке соединение осаждается чистым из гексана, после экстракции метиленхлоридом и настаивания: tRet(l) 11,6
мин.
б)
N-морфолинокарбонил-глицин:
Аналогично примеру 7а, 4,8 г (18,3 мысль) сложного бензилового эфира N-морфолинокарбонилглицина в 100 мл этилацетата гидрируют с помощью
1 г палладия (10%
)-на-угле до получения указанного в заголовке соединения.1Н- ЯМР (300 МГц, CDCl3): 3,88 (s, 2Н); 3,64 (s, 4Н); 3,50 (s, 2Н), 3,35 (s, 4Н).
Пример 21:
Z-(L)-Val-[PheNN(p-F)Phe]-Boc:
Раствор 335 мг (1,33 ммоль) Z-(L)-Val и 448 мг (1,11 моль) H-[PheNN(p-F)Phe] -Boc (получение см. пример 17а) в 9,4 мл 0,25М
NMM CH3CN смешивают с 463 мг (1,22 ммоль) HBTU. После перемешивания в течение 16 ч при комнатной температуре выпаривают и остаток распределяют между тремя порциями метиленхлорида, двумя
порциями
насыщенного раствора бикарбоната натрия и рассолом. Высушивание органических фаз над сульфатом натрия и выпаривание дает указанное в заголовке соединение, которое очищается путем колоночной
хроматографии (SiO2, гексан/этилацетат 4:1 L 1:1): tRet(I) 26,6 мин; FAB-MS: (M+H)+ 637.
Пример 22: Z-(L)-Val-[PheNN(p-F)Phe]<
--((L)-Val)-Boc:
Аналогично примеру 18, 165 мг (0,76 ммоль) Boc-(L)-Val и 371 мг (0,69 ммоль) Z-(L)-Val-[PheNN(p-F)Phe] -H в 6 мл NMM/CH3CN, 0,25М, вводят во
взаимодействие с 289 мг (0,76 мысль) HBTU с получением указанного в заголовке соединения, которое может непосредственно из реакционной среды выкристаллизовываться и отфильтровываться: tRet
(I) 27,2 мин; FAB-MS: (M+H)+=736.
Исходный материал получают следующим образом:
а) Z-(L)-Val[PheNN(p-F)Phe]-H:
Аналогично примеру 18а, в 440
мг
(0,69 ммоль) Z-(L)-Val-[PheNN(p-F)Phe] -Boc удаляют защиту с помощью 212 мл муравьиной кислоты, tRet(I) 17,8 мин.
Пример 23: Z-(L)-Val-[PheNN
(p-F)Phe]<-- ((L)-Val)-H:
Аналогично примеру 18а, в 250 мг (0,34 ммоль) Z-(L)-Val-[PheNN(p-F)Phe] <--((L)-Val)-Boc (пример 22) с помощью 50 мл муравьиной кислоты
удаляют защиту для получения указанного в заголовке соединения: tRet(l) 18,0 мин; FAB-MS: (М+Н)+ 636.
Пример 24: Z-(L)-Val-[PheNN(p-F)Phe]<
-- (L)-Val)<-- (N-морфолинокарбонил-Gly):
Аналогично примеру 20, 32 мг (0,17 ммоль) N-морфолинокарбонилглицина (пример 206) и 99 мг (0,16 ммоль) Z-(L)-Val-[PheNN(p-F)Phe] L
((L)-Val)-H в 1,3 мл NMM/CH3CN, 0,25М, вводят во взаимодействие с 65 мг (0,17 ммоль) HBTU с получением указанного в заголовке соединения, которое кристаллизуется прямо из реакционного
раствора: tRet(l) 21,1 мин; FAB-MS: (M+H)+ 806.
Пример 25: Z-(L)-Asn-[PheNN(p-F)Phe]-Boc:
К раствору 2,09 г (5,2 ммоль) H-[PheNN
(p-F)Phe]-Boc (получение см. пример 17а) в 68 мл ДМФ и 2,7 мл (16 ммоль) N-этил-диизопропиламина добавляют 3,0 г (7,8 ммоль) Z-(L)-аспарагин-п-нитрофенилового сложного эфира (Bachem, Bubendorf,
Швейцария). После перемешивания в течение 16 ч при комнатной температуре выпаривают на роторном испарителе, остаток обрабатывают большим количеством метиленхлорида (плохо растворяется) и промывают
двумя порциями 5 -ного раствора карбоната калия. Водные фазы экстрагируют еще 2 раза большим количеством метиленхлорида, сушат объединенные органические фазы над сульфатом натрия и выпаривают их.
После растворения сырого продукта в небольшом количестве метанола и осаждения путем добавления толуола при -20oC получают указанное в заголовке соединение: tRet(I) 21,2 мин;
FAB-MS: (M+H)+ 652.
Пример 26: H-(L)-Asn-[PheNN(p-F)Phe]-Boc:
Аналогично примеру 19, 0,40 г (0,61 ммоль) Z-(L)-Asn-[PheNN(p-F)Phe]-Boc в 20
мл метанола гидрируют до указанного в заголовке соединения: tRet(I) 14,9 мин; FAB-MS: (M+H)+ 518.
Пример 27: хинолин-2-карбонил-(L)-Asn-[PheNN
(p-F)Phe]-Boc:
Аналогично примеру 17,134 мг (0,78 ммоль) хинолин-2-карбоновой кислоты (Fluka, Buchs, Швейцария) в 4 мл NMM/ДМФ, 0,3М, вводят во взаимодействие с 344 мг (0,78 ммоль) ВОР, 105
мг (0,78 ммоль) НОВt и 268 мг (0,52 ммоль) H-(L)-Asn-[PheNN(p-F)Phe] -Boc. Так как согласно ЖХВД после 16 часов при комнатной температуре еще имеется H-(L)-Asn-[PheNN
(p-P)Phe]-Воc, то еще раз добавляют 299 мг ВОР, 70 мг HOBt, 89 мг хинальдиновой кислоты и 113 мкл NMM. Спустя следующие 16 ч выпаривают и остаток распределяют между тремя порциями метиленхлорида,
двумя порциями насыщенного раствора бикарбоната натрия и рассолом. Объединенные органические фазы сушат над сульфатом натрия и выпаривают. Растворение сырого продукта в небольшом количестве ДМФ,
осаждение с помощью ДИПЭ и охлаждение до-20oC дает указанное в заголовке соединение: tRet(I) 22,8 мин; FAB-MS: (M+H)+ 673.
Пример 28:
Z-(L)-Asn-[PheNN(p-F)Phe] J ((L)-Val)-Z:
88 мг (0,35 ммоль) Z-(L)-Val в 3,8 мл NMM/ДМФ, 0,3N, активируют с помощью 153 мг (0,35 ммоль) ВОР и 47 мг (0,35 ммоль) HOBt и спустя 15
мин
смешивают со 144 мг (0,23 ммоль) Z-(L)-Asn-[PheNN(p-F)Phe]-H•2НСl. После перемешивания в течение 14 ч при комнатной температуре реакционную смесь выпаривают, остаток растворяют
в 2
мл метанола и распределяют между тремя порциями метиленхлорида и двумя порциями 1М раствора карбоната натрия, органические фазы высушивают над сульфатом натрия и выпаривают их. Повторное
растворение
сырого продукта в небольшом количестве ДМФ и осаждение с помощью ДИПЭ дает указанное в заголовке соединение: tRet(I) 22,2 мин; FAB-MS: (M+H)+ 785.
Исходный
материал получают следующим образом:
а) Z-(L)-Asn-PheNN(p-F)Phe]-H•2НСl:
В атмосфере азота раствор 150 мг (0,23 ммоль) Z-(L)-Asn- [PheNN
(p- F)Phe] -Вое
(пример 25) в 1 мл диоксана смешивают с 2 мл HCl/диоксан, 4N (Fluka, Buchs, Швейцария). После перемешивания в течение 1,5 часов при комнатной температуре реакционную смесь лиофилизуют
и лиофилизат
тотчас вводят во взаимодействие далее.
Пример 29: трифторацетил-[PheNN(p-F)Phe] J ((L)-Val)-Z:
Аналогично примеру 17, в течение 15 ч вводят во
взаимодействие 239
мг (0,95 ммоль) Z-(L)-Val в 10,5 мл NMM/ДМФ, 0,3N, с 421 мг (0,95 ммоль) ВОР, 129 мг (0,95 ммоль) HOBt и 0,3 г (0,63 ммоль) N-трифторацетил-[PheNN(p-F)Phe]-H. Колоночная
хроматография
(SiO2, метиленхлорид/эфир 10:1) в осаждение из раствора ДМФ с помощью ДИПЭ дает указанное в заголовке соединение: TCX Rf(O) 0,15; tRet(l) 25,9 мин;
FAB-MS: (M+H)+ 633.
Исходный материал получают следующим образом: а) N-трифторацетил- [PheNN(p-F)Phe] -H: При 0oC 0,20 г (0,40 ммоль) N-трифторацетил-[PheNN (p-F)Phe] -Boc (получение см. пример 16) в 5 мл метиленхлорида смешивают с 5 мл трифторуксусной кислоты. После перемешивания 4 ч при 0oC и 2 ч при комнатной температуре реакционную смесь выпаривают. Лиофилизация остатка из диоксана дает указанное в заголовке соединение, которое без очистки вводится во взаимодействие далее: tRet(l) 14, 7 мин.
Пример 30:
Z-(L)-Asn-[PheNNPhe]-Boc:
Аналогично примеру 25, 167 мг (0,34 ммоль) [PheNN(p-F)Phe]-Boc в 3,6 мл ДМФ и 0,18 мл (1 ммоль)
N-этил-диизопропиламина вводят во
взаимодействие с 0,20 г (0,52 ммоль) п-нитрофенилового сложного эфира Z-(L)-аспарагина с получением указанного в заголовке соединения, которое после колоночной
хроматографии (SiO2,
этилацетат) получается чистым: TCX Rf(О) 0,19; tRet(l) 20,9 мин.
Исходный материал получают следующим образом:
а)
N-трифторацетил-[PheNN
Phe]-Boc:
Аналогично примеру 16,1,82 г (7,0 ммоль) 2(R)-1'(S)-трифторацетил- амино)-2'-фенилэтил] оксирана (пример 16г) и 1,58 г (7,1 ммоль)
трет.-бутил- 3-бензил-карбазата (J. Chem. Perkin,
1,1712, (1975)) в 15 мл метанола вводят во взаимодействие в тугоплавкой трубке с получением указанного в заголовке соединения, которое выделяется с
помощью колоночной хроматографии (SiO2,
метиленхлорид/эфир 50:1): TCX Rf(J) 0,38; tRet(l) 24,5 мин.
б) H-[PheNNPhe]-Boc:
Аналогично примеру 17а, 258 мг (0,53 ммоль)
N-трифторацетил-[PheNNPhe] -Boc в 60 мл метанола вводят во взаимодействие с 10,7 мл 1М раствора карбоната калия с получением указанного в
заголовке соединения.
Пример 31:
Z-(L)-Val-[(p-F)PheNN(p-F)Phe]-Boc:
Аналогично примеру 21,18 мг (0,070 ммоль) Z-(L)-Val и 27 мг (0,064 ммоль) H-[(p-F)PheNN(p-F)Phe]-Boc в 0,6 мл NMM/CH3CN, 0,
25М, вводят во взаимодействие с 26,6 мг (0,070 ммоль) HBTU с получением указанного в заголовке соединения, которое после растворения в небольшом
количестве метиленхлорида и осаждения с помощью ДИПЭ
очищается: FAB-MS: (M+H)+ 655, Rf(CH2CI2/(C2H5)20
0,54.
Исходный материал получают следующим
образом:
а) N-Boc-(п-фторфенилаланин):
В 0,4 л смеси диоксана с водой в соотношении 1:1 вводят во взаимодействие 20 мг (109 ммоль)
п-фторфенилаланина (Fluka, Buchs, Швейцария) с 35,5
г (163 ммоль) Boc-ангидрида и 150 г (1,09 моль) карбоната калия. Спустя 4 ч реакционную смесь подкисляют раствором лимонной кислоты и экстрагируют
тремя порциями этилацетата. Органические фазы
промывают 10-ной лимонной кислотой, водой и рассолом, высушивают над сульфатом натрия и выпаривают их. Растворение остатка в небольшом количестве
метиленхлорида и кристаллизация путем добавки гексана
дает указанное в заголовке соединение: tRet(I) 16,9 мин.
б) N-Boc-(п-фторфенилаланинол):
При-5oC
-10oC раствор 17,9 г (63 ммоль)
N-Вос-(п-фторфенила-ланина) в 73 мл абс. ТГФ смешивают с 9,66 мл (69 ммоль) триэтиламина и прикапывают раствор 9,05 мл (69 ммоль) изобутилового эфира
хлормуравьиной кислоты в 44 мл абс. ТГФ. После
перемешивания в течение получаса при комнатной температуре отсасывают образовавшийся осадок. Фильтрат при охлаждении прикалывают к 4,77 г (126 ммоль)
боргидрида натрия в 28 мл воды. После
перемешивания в течение получаса при комнатной температуре подкисляют с помощью 10-ной лимонной кислоты, ТГФ выпаривают на роторном испарителе частично и остаток
распределяют между тремя порциями
этилацетата, двумя порциями 2н раствора гидроксида натрия, водой, насыщенным раствором гидрокарбоната натрия и рассолом. Высушенные над сульфатом натрия и выпаренные
органические фазы дают после
растворения в небольшом количестве метиленхлорида и кристаллизации за счет добавки гексана указанное в заголовке соединение: TCX Rf(N) 0,36; tRet(I)
16,8 мин;1Н-ЯМР
(200 МГц, CD3OD: 7,24 (dd, 8 и 5 Гц, 2Н); 6,98 (t, 8 Гц, 2Н); 3,73 (m, 1Н); 3,47 (d, 5 Гц, 2Н); 2,88 (dd, 13 и 6 Гц, 1Н); 2,62 (dd, 13 и 8 Гц, 1Н); 1,36 (s,
9Н).
в)
N-Boc-(п-фторфенилаланиналь):
В атмосфере азота к охлажденному до -60oC раствору 4,0 мл (46,8 ммоль) оксалилхлорида в 44 мл метиленхлорида прикалывают 4,
44 мл (62,4 ммоль) ДМСО,
растворенные в 76 мл метиленхлорида. После перемешивания в течение 15 мин добавляют к прозрачному реакционному раствору 8,4 г (31,2 ммоль) N-Вос-(п-фторфенилаланинола) в виде
раствора в 185 мл
метиленхлорид/ТГФ (1:1) ( J осаждение) и дополнительно перемешивают 25 мин. Затем добавляют 17,3 мл (124,8 ммоль) триэтиламина, растворенные в 38 мл метиленхлорида. После
перемешивания в течение 30
мин прикалывают 278 мл 20-ного раствора гидросульфата калия, с последующим добавлением 220 мл гексана. Оставляют нагреваться до комнатной температуры, водную фазу отделяют
и экстрагируют ее с помощью
двух порций эфира. Органические фазы после промывки насыщенным раствором бикарбоната натрия, водой и рассолом, высушивания над сульфатом натрия и выпаривания, дают
указанное в заголовке соединение,
которое без дальнейшей очистки используется в самой ближайшей стадии.1Н-ЯМР (200 МГц, CDCI3): 9,63 (s, 1Н); 6,9-7,2 (2m, 4Н); 5,04 (m, 1Н); 4,
42 (m, 1Н); 3,10 (m, 2Н); 1,43 (s,
9Н).
г)N-3(S)-(Boc-амино)-2(R, S)-гидрокси-4-(п-фторфенил)-1- триметилсилил-бутан:
Аналогично примеру 16а, 1,63 г (67 ммоль) магния в 33
мл абс. эфира вводят во
взаимодействие с 8,3 мл (60 ммоль) хлорметилтриметилсилана с получением соединения Гриньяра, которое после реакции с 13 ммолями N-Boc-(п-фторфенилаланиналя), экстракции и
колоночной хроматографии
(SiO2, гексан/этилацетат 5: 1 J 4:1) дает смесь диастереомеров указанного в заголовке соединения: TCX Rf(L) 0,32; tRet(L) 24,9 мин (22%)/25,5
мин (78%); FAB-MS:
(M+H)+ 356.
д) 1-(п-фторфенил)-3-бутен-2 (S)-амин:
Аналогично примеру 16 б, 1,1 г (3,1 ммоль) N-3(S)-(Boc-амино)-2(R,
S)- гидрокси-4-(п-фторфенил)-1-триметилсилил-бутана в 22 мл метиленхлорида вводят во взаимодействие с 1,9 мл (15,5 ммоль) примерно 48-ного раствора трифторида бора в эфире с получением указанного в
заголовке соединения:1Н-ЯМР (300 МГц, CDCI3): 7,2-7,10 и 7,05-6,9 (2m, по 2Н); 5,9-5,8 (m, 1Н); 5,2-5,0 (m, 2Н); 3,57 (m, 1Н); 2,79 (dd, 12 и 6 Гц, 1Н); 2,62 (dd, 12 и 8 Гц,
1Н);
1,7 (s, br, 2Н).
е) N-трифторацетил-1-(a-фторфенил)-3-бутон-2(S)-амин:
Аналогично примеру 16в, 364 мг (2,2 ммоль) 1-(п-фторфенил)-3-бутен-2(S)-амина в 1,8 мл
метиленхлорида
и 5,4 мл пиридина вводят во взаимодействие с 460 мкл (3,3 ммоль) ангидрида трифторуксусной кислоты с получением указанного в заголовке соединения, которое после настаивания в гексане
получается
чистым: TCX Rf(F) 0,58; MS (М)+ 261.
ж) 2 (R)-[1' (S)-(трифторацетил-амино)-2'- (п-фторфенил)этил] оксиран:
Аналогично примеру 16г, 359 мг
(1,37 ммоль)
N-трифторацетил-1-(п-фторфенил)-3-бутен-2(S)-амина в 9 мл хлороформа окисляют с помощью 1,18 г (6,87 ммоль) м-хлорнадбензойной кислоты с получением указанного в заголовке соединения: TCX
Rf
(R) 0,45.
з) N-трифторацетил-[(p-F)PheNN(p-F)Phe]-Boc:
Аналогично примеру 16, 415 мг (1,49 ммоль)
2(R)-[1'(S)-(трифторацетил-амино)-2'-(п-фторфенил)этил]оксирана
вводят во взаимодействие с 377 мг (1,57 ммоль) трет. -бутил-3-(п-фторфенил-метил)карбазата в 9 мл метанола с получением указанного в
заголовке соединения. TCX Rf(*S) 0,53; FAB-MS: (M+H)+ 518;1Н-ЯMP (300 МГц, CD3OD): 7,4-7,3 и 7,3-7,2 (2m, по 2Н); 7,05-6,9 (m, 4H);4,23 (m, 1Н); 3,90-3,65
(m, 3Н); 3,03-2,78 и 2,74-2,60 (2m, по 2Н); 1,30 (s, 9Н).
и) H-[(p-F)PheNN(p-F)Phe]-Boc:
Аналогично примеру 17а, 285 мг (0,55 ммоль) N-трифторацетил-[(р-F)PheNN(p-F)Phe] -Boc в 45 мл метанола вводят во взаимодействие
с 14 мл 1М раствора карбоната калия с получением указанного в заголовке соединения: tRet(l) 16,4 мин.
Пример 32: Z-(L)-Val-[(p-F)PheNN(p-F)Phe]-H:
Аналогично примеру 18а, в 215 мг (0,33 ммоль) Z-(L)-Val-[(p-F)PheNN(p-F)Phe] -Bос удаляют защиту с помощью 100 мл
муравьиной кислоты с получением указанного в заголовке соединения: FAB-MS:
(M+H)+ 555', tRet(I) 18 мин.
Пример 33: Z-(L)-Val-[(p-F)PheNN(p-F)Phe] J
(N-(N-(2-пиридилметилa)-N-метиламинокарбонил)- (L)-Val):
Аналогично
примеру 18, 23,6 мг (0,089 ммоль) N-(N-(2-пиридилметил)- N-метиламинокарбонил)-(L)-валила (получение см. в ЕР 402646 А1,
19 декабря 1990г. ) и 45 мг (0,081 ммоль) Z-(L)-Val-[(p-F)PheNN
(p-F)Phe]-H вводят во взаимодействие с 33,8 мг (0,089 ммоль) HBTU в 0,76 мл NMM/CH3CN (0,25М) с получением
указанного в заголовке соединения и перекристаллизовывают его из ДМФ/ДИПЭ: TCX
Rf(O) 0,39; FAB-MS: (M+H)+ 802.
Пример 34: Z-(L)-Val-[(p-F)PheNN
(p-F)Phe] J (N-2(R,S)-карбамоил-3-фенилпропионил) (L) -Val):
Аналогично
примеру 18, 26,0 мг (0,089 ммоль) N-2((R,S)-карбамоил-3- фенилпропионил)-(L)-валила (получение: Synth. struct. Fund.
Proc. Am. Pept. Symp. 7th, 85, (1981))и 45 мг (0,081 ммоль) Z-(L)-Val-[(p-F)PheNN(p- F)Phe] -H (пример 32) вводят во взаимодействие с 33,8 мг (0,089 ммоль) HBTU в 0,76 мл NMM/CH3
CN, 0,25М, до образования указанного в заголовке соединения и перекристаллизовывают
его из ДМФ/ДИПЭ: Rf(P) 0,64; FAB-MS: (M+H)+ 829.
Пример 35:
Ацетил-Val-[PheNNPhe] J (N-ацетилa-Val):
Аналогично примеру 7, из 100 мг (0,25
ммоль) H-[PheNNPhe]-H-3HCI из примера 2а, 121 мг (0,76 ммоль) N-ацетил-(L)-валина, 288 мг
(0,76 ммоль) HBTU и 0,211 мл (1,52 ммоль) триэтиламина в ДМФ получают указанное в заголовке
соединение, после лиофилизации из диоксана. FAB-MS: (M+H)+ 568, tRet(I) 15,0 мин,
Rf(В) 0,46.
Пример 36: Z-(D)-Val-[PheNNPhe] J
((D)-Val)-Z:
Аналогично примеру 2, из 50 мг (0,123 ммоль)H-[PheNNPhe] -H•3HCl из
примера 2а, 95 мг (0,38 ммоль) Z-(D)-валила, 168 мг (0,38 ммоль) ВОР, 51 мг (0,38 ммоль)
HOBt и 2,53 мл 0,3М NMM в ДМФ, получают указанное в заголовке соединение, после лиофилизации из диоксана.
FAB-MS: (M+H)+ 752; tRet(I) 26,4 мин, Rf(H) 0,21.
Пример 37: хинолин-2-карбонил-Val-[PheNNPhe] J (N-хинолин-2-карбонил)-Val):
145
мг (0,53 мысль) N-(хинолин-З-карбонил-(L)-валина, 235 мг (0,53 ммоль) ВОР и 72 мг (0,53
ммоль) HOBt растворяют в 3,5 мл 0,3М раствора NMM в ДМФ, спустя 10 мин добавляют 70 мг (0,18 ммоль) H-[PheNNPhe]-H•HCI (пример 2а) и перемешивают в течение 5 ч при комнатной
температуре в атмосфере азота. Реакционную смесь выпаривают, остаток растворяют в метиленхлориде и промывают дважды
насыщенным раствором бикарбоната натрия, один раз 10-ным раствором лимонной кислоты
и снова один раз насыщенным раствором бикарбоната натрия. Органические фазы фильтруют через вату, выпаривают и
остаток осаждают дважды из метиленхлорида с метанолом путем добавления ДИПЭ. После
лиофилизации из диоксана получают указанное в заголовке соединение в виде твердого вещества белого цвета (смесь двух,
различающихся в ЖХВД диастереомеров). FAB-MS: (M+H)+ 794; tRet(А) 29,1 и 29,3 мин; Rf(В) 0,81.
а) N-(хинолин-2-карбонил)-(L)-валин:
Раствор
2,5 г (14,5 ммоль) (L)-валил-трет.-бутилового сложного эфира и 2,5 г (14,5
ммоль) хинолин-2-карболовой кислоты в 100 мл метиленхлорида с ТГФ (10: 1) смешивают с 3,28 г (15,9 ммоль) N,
N-дициклогексилкарбодиимида и 2,0 мл (14,5 ммоль) триэтиламина и перемешивают в течение 18 ч
при комнатной температуре. Реакционную смесь охлаждают до -18oC и отфильтровывают от мочевины.
Фильтрат выпаривают, остаток растворяют в метиленхлориде и промывают по одному разу насыщенным
раствором бикарбоната натрия и водой. Органические фазы фильтруют через вату, выпаривают и после
хроматографической очистки на силикагеле с помощью смеси гексан/этилацетат (2:1) получают
N-(хинолин-2-карбонил)-(L)-валил-трет. -бутиловый эфир. 2,59 г (12,2 ммоль) его оставляют на 4,5 ч при
комнатной температуре в смеси метиленхлорид/ТФК (1:1). После выпаривания остаток очищают
хроматографией на силикагеле с помощью смеси гексан/этилацетат (2:1). Содержащие продукт фракции выпаривают,
снова растворяют в метиленхлориде и путем промывки 1н раствором гидроксида натрия и 1н
соляной кислотой титульное соединение переводят в гидрохлорид.1Н-ЯМР (200 МГц, CD3OD: 1,
05 и 1,07 (2d, J=6 Гц, 6Н); 2,40 (m, 1Н); 4,65 (m, 1H); 7,70 (m, 1H); 7,85 (m, 1H), 8,00
(dxd, 1H); 8,20 (m, 2H); 8,48 (d, 1H).
Пример 38: ацетил-(L)-Val-[PheNNCha]
<-- (N-ацетил-(L)-Val):
Аналогично примеру 37, из 160 мг (0,40 ммоль) H-[PheNNCha]-H•3HCI из примера 10а, 190 мг (1,19 ммоль) N-ацетил-(L)-валика, 525 мг (1,19
ммоль) ВОР, 160 мг (1,19 ммоль) HOBt и 7,9 мл 0,3М NMM в ДМФ получают, после осаждения из смеси
хлороформа с метанолом с помощью ДИПЭ и лиофилизации из диоксана, указанное в заголовке соединение.
FAB-MS: (M+H)+ 574, tRet(I) 18,1 мин, RfB) 0,30.
Пример 39: N-(3-пиридилацетил)-(L)-Val-[PheNNCha] <-- (N-(3 -пиридилацетил)-(L)-Val)•3HCI.
Аналогично примеру 7, из 100 мг (0,25 ммоль) H-[PheNN Cha]-H•3НСl из примера 10а, 358 мг (1,52 ммоль) N-(3-пиридилацетил)-(L)-валина из примера 9а, 576 мг (1,52 ммоль) HBTU и 0,316 мл (2,28 ммоль) триэтиламина в ДМФ, получают указанное в заголовке соединение, после хроматографической очистки на силикагеле с помощью смеси метиленхлорид/метанол (15:1) и виофилизации содержащих продукт фракций из диоксана. FAB-MS: (M+H)+ 728, tRet(I) 11,3 мин, Rf(U) 0,21.
Пример 40: ацетил-I1e-[PheNNCha] <-- (N-ацетил-I1e):
Аналогично примеру 37, из 160 мг (0,40 ммоль) H-[PheNNCha]-H•3HCI из примера 10а, 206 мг
(1,19 ммоль) N-ацетил-(L)-изолейцина, 525 мг (1,19 ммоль) ВОР, 160 мг (1,19 ммоль) HOBt и 7,9 мл 0,3М NMM в ДМФ, после осаждения из метиленхлорида с
метанолом путем добавки ДИПЭ и теофилизации из
смеси диоксан/трет. -бутанола получают указанное в заголовке соединение (смесь различающихся в HPLC двух диастереомеров); FAB-MS: (M+H)+ 602,
tRet(I) 20,4 и 20,7 мин, Rf(D) 0,33.
Пример 41: тиоморфолинокарбонил-(L)-Val-[PheNNCha] <-- (N-тиоморфолинокарбонил-(L)-Val):
Аналогично примеру 6, исходя из 70 мг (0,12
ммоль) H-(L)-Val-[PheNNCha] <-- (N-(L)-Val)-H•3HCI из примера 14, 58 мг (0,35 ммоль) (4-тиоморфолинилкарбонил)-хлорида из примера
6а и 0,127 мл триэтиламина в 2 мл ДМФ, после
хроматографической очистки на силикагеле с помощью смеси метиленхлорид/метанол (95: 5) и лиофилизации содержащих продукт фракций из диоксана, получают
указанное в заголовке соединение. FAB-MS:
(M+H)+ 748; tRet(I) 24,0 мин, Rf(В) 0,70.
Пример 42: Z-(L)-Glu-[PheNN(p-F)Phe] <
-- ((L)-Glu)-Z:
Раствор 130 мг (0,14
ммоль) Z-(L)-GIu(O-трет.-бутил)-[PheNN(p-F)Phe] <-- ((L)-Glu(O-трет.-бутил)-Z [где (Glu()-трет.-бутил) обозначает здесь
этерифицированную третичным бутильным остатком карбоксигруппа
в γ положении радикал глутаминовой кислоты] в 8 мл смеси метиленхлорид (ТФК/1:1) перемешивают в течение 3 ч при комнатной
температуре. Растворитель выпаривают при пониженном давлении и остаток
осаждают из метиленхлорида путем добавки ДИПЭ. Получают указанное в заголовке соединение после лиофилизации из смеси диоксана с
трет.-бутанолом. FAB-MS: (M+H)+ 830; tRet(I) 19,6
мин; Rf(B) 0,32.
а) Z-(L)-Glu(O-трет. -бутил)-[PheNN(p-F)Phe] ← ((L)-Glu(O-трет.
-бутил))-Z:
Аналогично примеру 37, из 100 мг (0,24 ммоль)
H-[PheNN(p-F)Phe]-H•3НСl, 245 мг (0,73 ммоль) трет.-бутилового эфира Z-(L)-глутаминовой кислоты, 321 мг (0,73
ммоль) ВОР, 98 мг (0,73 ммоль) HOBt и 4,8 мл 0,3М NMM в ДМФ, получают
указанное в заголовке соединение, после хроматографической очистки на силикагеле с помощью смеси метиленхлорида с эфиром (1:1),
tRet(I) 30,2 мин, Rf(H) 0,17.
б)
H-[PheNN(p-F)Phe]-H•3НСl:
Аналогично примеру 2а, исходя из 1,77 г (3,51 ммоль) Boc-[PheNN
(p-F)Phe] -Boc, после лиофилизации, получают указанное в заголовке
соединение, FAB-MS: (M+H)+ 304; Rf(К) 0,19.
в) Boc-[PheNN(p-F)Phe]-Boc:
Аналогично примеру 1, исходя из 2,0 г (7,60 ммоль)
(2R)-[1'(S)- Вос-амино-2'-фенилэтил] оксирана и 2,17 г (9,04 ммоль) трет.-бутил-3-(4-фторфенилметил)карбазата из примера 16е), получают указанное в
заголовке соединение, после хроматографической
очистки на силикагеле с помощью смеси гексана с этилацетатом (2:1). FAB-MS: (M+H)+ 504; tRet(I) 26,2 мин, Rf(F) 0,26.
Пример 43:
N-(2-пиридилметил)N-метил-аминокарбонил-(L)-Val-[PheNN(р-F)Phe] J (N-(N-(2-пиридилметил)-N-метиламинокарбонил)-(L)-Val):
Аналогично примеру 37, из 70 мг (0,
17 ммоль) H-[PheNN(p-F)Phe]-H•3НСl из примера 42б, 135 мг (0,51 ммоль) N-(N-(2-пиридилметил)-N-метиламинокарбонил)-(L)-валика (получение как описано в европейском патенте ЕР 0402646
А1 от 19 декабря
1990г.), 225 мг (0,51 ммоль) ВОР, 69 мг (0,51 ммоль) HOBt и 3,4 мл (0,3М) NMM в ДМФ, после хроматографии на силикагеле с помощью смеси метиленхлорида с метанолом (15:1) и лиофилизации
содержащих
продукт фракций из диоксана, получают указанное в заголовке соединение, FAB-MS: (M+H)+ 798, tRet(IV) 35 мин, Rf(U) 0,21.
Пример 44:
N-(3-тетразол-1-ил)пропионил)-(L)-Val-[PheNN (р-F)Phe] J-(N-(3-тетразол-1-ил) -пропионил)-Val:
Аналогично примеру 37, из 100 мг (0,24 ммоль) H- [PheNN(p-P)Phe]-H•
3НСl (из примера 42б), 146 мг (0,61 ммоль) (N-(3-тетразол-1-ил)-(L)-валина, 268 мг (0,061 ммоль) ВОР, 82 мг (0,61 ммоль) HOBt и 4 мл 0,3М NMM в ДМФ, после осаждения из метиленхлорида путем добавления
ДИПЭ и лиофилизации из диоксана (в HPLC различаются 4 диастереомера), получают указанное в заголовке соединение. FAB-MS: (M+H)+ 750; tRet(III) 30,8 мин, 31,4 мин, 32,4 мин и 32,
8
мин; Rf(К) 0,5.
Пример
44а: N-(3-(тетразол-1-ил)-пропионил)-(L)-валин:
Аналогично примеру 9б, исходя из 4 г (16,4 ммоль) гидрохлорида бензилового эфира
(L)-валина,
2,1 г (14,9 ммоль) 3-(тетразол-1-ил)-пропионовой кислоты (получение: патент США 4,794,109 А от 27 декабря 1988г.), 2,4 мл диэтилового эфира цианофосфоновой кислоты и 4,4 мл триэтиламина в
ДМФ, после
хроматографической очистки на силикагеле с помощью смеси метиленхлорида с метанолом (30: 1), получают бензиловый эфир N-(3-(тетразол-1-ил)-пропионил)-(L)-валина. 2,66 г (8,03 ммоль) его в
смеси
метанола с водой (9:1) гидрируют в присутствии 530 мг 10-ного палладия-на-угле под давлением водорода 1 атм и после осаждения из метанола с помощью ДИПЭ получают указанное в заголовке
соединение.1Н-ЯМР (200 МГц, CD3OD): 0,9 (d, J=7 Гц, 6Н); 2,1 (m, 1Н); 2,95 (m, 2Н); 4,29 (d, J=6 Гц, 1Н); 4,78 (m, 2Н); 9,15 (s, 1Н).
Пример 45:
Z-(L)-Val-[PheNN
N(p-F)Phe] J ((L)-Val)-Z:
Аналогично примеру 37, из 100 мг (0,24 ммоль) H-[PheNN(p-F) Phe]-H•3HCI (из примера 42б), 182 мг (0,38 ммоль)
Z-(L)-валина, 321 мг (0,73 ммоль) ВОР,
98 мг (0,73 ммоль) HOBt и 4,8 мл 0,3М NMM в ДМФ, после осаждения из метиленхлорида за счет добавления ДИПЭ и лиофилизации из диоксана, получают указанное в
заголовке соединение. FAB-MS: (M+H)+ 770; tRet(I) 26,3 мин, Rf(H) 0,25.
Пример 46: ацетил-Val-[PheNN(p-F)Phe] J (N-ацетил-Val):
Аналогично примеру 37, из 80 мг (0,19
ммоль) H-[PheNN(p-F)Phe]-H•3HCl из примера 42б, 124 мг (0,78 ммоль) N-ацетил-(L)-валина, 344 мг (0,78 ммоль) ВОР, 105 мг (0,76 ммоль) HOBt и 4,
5 мл 0,3М NMM в ДМФ, после двухкратного
переосаждения из смеси метиленхлорида с метанолом путем добавления ДИПЭ и лиофилизации из диоксана с трет.-бутанолом, получают указанное в заголовке
соединение. FAB-MS: (M+H)+ 586, tRet(I) 15,8 мин, Rf(Е) 0,32.
Пример 47: ацетил-Val-[PheNN(p-CN)Phe] J (N-ацетил-Val):
Аналогично
примеру 37, из 80 мг (0,19 ммоль) H-[PheNN(p-CN)Phe]-H•3HCl, 124 мг (0,78 ммоль) N-ацетил-(L)-валина, 344 мг (0,78 ммоль) ВОР, 105 мг (0,78 ммоль) HOBt и 4,5 мл 0,3М NMM в ДМФ,
после осаждения из смеси метиленхлорида с
метанолом за счет добавки ДИПЭ и лиофилизации из диоксана, получают указанное в заголовке соединение в виде смеси двух, различающихся по HPLC диастереомеров.
FAB-MS: (M+H)+ 593; tRet(l) 14,4 и 14,6 мин; Rf(D) 0,39.
а) H-[PheNN(p-CN)Phe]-H•3HCl:
Аналогично примеру 2а, исходя из 2,
69 г (5,27 ммоль) Boc-[PheNN
(p-CN)Phe] -Boc, после лиофилизации, получают указанное в заголовке соединение. FAB-MS: (M+H)+ 311; Rf(K) 0,16.
б)
Boc-[PheNN(p-CN)Phe]-Boc:
Аналогично примеру 1, исходя из 2,0 г (7,60 ммоль) (2R)-[1'(S)-Boc- амино-2'-фенилэтил] оксирана и 1,87 г (7,6 ммоль)
трет.-бутил-3-(4- цианофенилметил)-карбазата получают, после кристаллизации из
смеси метанола с ДИПЭ, указанное в заголовке соединение. FAB-MS: (M+H)+ 511; tRet(I) 25 мин; Rf(Y) 0,19.
в)
трет.-бутил-3-(4-цианофенил-метил)-карбазат:
Аналогично примеру 4б, 10 г (76,3 ммоль) 4-цианобензальдегида вводят во взаимодействие с 10 г (76,3 ммоль)
трет.-бутилкарбазата в этаноле с
получением 4-цианофенилкарбальдегид-трет.-бутоксикарбонилгидразона. 11,1 г этого соединения в 150 мл ТГФ гидрируют в присутствии 2 г палладия-на-угле (10-ного) под
давлением водорода 2 атм и получают
указанное в заголовке соединение.1Н-ЯМР (200 МГц, CDCI3): 7,65 (d, J=8 Гц, 2Н); 7,45 (d, J=8 Гц, 2Н); 6,08 (s, br, 1H); 4,3 (s, br, m), 4,02
(s, 2H); 1,45 (s, 9H).
Пример 48: Z-(L)-Val-[PheNN(p-CN)Phe] J ((L)-Val)-Z:
Аналогично примеру 37, из 70 мг (0,17 ммоль) H-[PheNN(p-CN)Phe]-H•
3HCl (из примера 47а), 125 мг (0,
5 ммоль) 2-(L)-валина, 221 мг (0,5 ммоль) ВОР, 68 мг (0,5 ммоль) HOBt и 3,33 мл 0,3М NMM в ДМФ, после осаждения из метиленхлорида путем добавления гексана и
лиофилизации из диоксана, получают
указанное в заголовке соединение. FAB-MS: (M+H)+ 777; tRet(l) 25,3 мин; Rf(D) 0,69.
Пример 49: Z-(L)-Ile-[PheNNLeu] J ( (L)-Ile)-Z:
Аналогично примеру 37, из 70 мг (0,19 ммоль) H-[PheNNLeu] -H•3HCI (из примера 13а), 154 мг (0,58 ммоль) Z-(L)-изолейцина, 257 мг (0,58 ммоль)
ВОР, 79 мг (0,58 ммоль) HOBt и 3,88 мл
0,3М NMM в ДМФ, после хроматографии на силикагеле с помощью смеси метиленхлорида с эфиром (3:1) и осаждения содержащих продукт фракций из метиленхлорида с
помощью ДИПЭ и лиофилизации из диоксана,
получают указанное в заголовке соединение. FAB-MS: (M+H)+ 746; tRet(I) 28,2 мин; Rf(H) 0,39.
Пример 50:
изобутоксикарбонил-(L)-Val-[PheNN
Leu] J (N-изобутоксикарбонил-(L)-Val):
Аналогично примеру 37, из 70 мг (0,19 ммоль) H-[PheNNLeu]-H•3HCI (из примера 13а), 130 мг
(0,58 ммоль)
N-(изобутоксикарбонил)-(L)-валина, 256 мг (0,58 ммоль) ВОР, 78 мг (0,58 ммоль) HOBt и 3,9 мл 0,3М NMM в ДМФ, после хроматографии на силикагеле с помощью смеси метиленхлорида с эфиром
(1:1) и
лиофилизации содержащих продукт фракций из диоксана, получают указанное в заголовке соединение. FAB-MS: (M+H)+ 650; tRet(I) 26,4 мин; Rf(H) 0,38.
а)
N-(изобутоксикарбонил)-(L)-валин:
Раствор 10 г (85,3 ммоль) (L)-валим в 100 мл 2н раствора гидроксида натрия смешивают с 11,2 мл (85,3 ммоль) изобутилхлорформиата и
перемешивают при комнатной
температуре в течение 18 часов. Реакционный раствор промывают метиленхлоридом, подкисляют 4н соляной кислотой и экстрагируют метиленхлоридом. Органические экстракты
промывают рассолом, фильтруют через
вату и после выпаривания получают указанное в заголовке соединение в виде бесцветной смолы.1Н-ЯМР (200 МГц, CD3OD): 0,95 (m, 12Н); 1,9 (m,
1Н); 2,15 (m, 1Н); 3,85 (d, J=7 Гц,
2Н); 4,05 (d, br, 1H).
Пример 51: N-(3-(тетразол-1-ил)-пропионил)-(L)-Val-[PheNNLeu] J (N-3-(тетразол-1-ил) -пропионил-L)-Val):
Аналогично примеру 37, из 150 мг (0,
42 ммоль) H-[PheNNLeu]-H•3HCI (из примера 13а), 251 мг (1,04 ммоль) N-(3-(тетразол-1-ил)-пропионил)-(L)-валина (из примера 44а), 460 мг (1,04
ммоль) ВОР, 140 мг (1,04 ммоль) HOBt и 6,
9 мл 0,3М N-метилморфолина в ДМФ, после осаждения из метиленхлорида с помощью ДИПЭ и лиофилизации из диоксана с трет.-бутанолом и водой, получают указанное в
заголовке соединение. FAB-MS: (M+H)+ 689; tRet(l) 14,7 мин; Rf(K) 0,36.
Пример 52: ацетил-Val-[PheNNLeu] J (N-ацетил-Val):
Аналогично примеру 37, из 70 мг (0,19 ммоль)
H-[PheNNLeu]-H•3HCI (из примера 13а), 184 мг (1,16 ммоль) N-ацетил-(L)-валина, 512 мг (1,16 ммоль) ВОР, 156 мг (1,16 ммоль) HOBt и 7,8
мл 0,3М NMM в ДМФ, после осаждения из смеси
метиленхлорида с метанолом за счет добавления ДИПЭ и лиофилизации из диоксана с трет.-бутанолом и водой, получают указанное в заголовке соединение
(согласно HPLC различаются 2 диастереомера). FAB-MS:
(M+H)+ 534; tRet(I) 14,7 и 15,1 мин; Rf(D) 0,35.
Пример 53: Boc-(L)-Val-[PheNNLeu] J
((L)-Val)-Boc:
Аналогично примеру 7, из 300
мг (0,83 ммоль) H-[PheNNLeu]-H•3HCI (из примера За), 722 мг (3,33 ммоль) Boc-(L)-валила, 1,262 г (3,33 ммоль) HBTU и 0,927 мл (6,
66 ммоль) триэтиламина в ДМФ, после
хроматографической очистки на силикагеле с помощью смеси метиленхлорида с эфиром (1:1), осаждения содержащих продукт фракций и лиофилизации из диоксана, получают
указанное в заголовке соединение.
FAB-MS: (M+H)+ 650; tRet(I) 26,3 мин; Rf(H) 0,64.
Пример 54: H-(L)-Val-[PheNNLeu] J ((L)-Val)-H•
3HCI:
Аналогично примеру 5,
из 396 мг (0,61 ммоль) Boc-(L)-Val-[PheNNLeu] J ((L)-Val)-Boc (из примера 53) и 10 мл хлороводорода в диоксане, после лиофилизации реакционного
раствора, получают указанное в заголовке
соединение. FAB-MS:(M+H)+ 450; tRet(II) 24,1 мин; Rf(K) 0,25.
Пример 55:
N-тиоморфолинокарбонил-(L)-Val-[PheNNLeu] J
(N-тиоморфолинокарбонил-(L) -Val):
Аналогично примеру 6, исходя из 100 мг (0,16 ммоль) H-(L)-Val- [PheNNLeu] J
(L)-Val-H•3HCI, 78,5 мг (0,47 ммоль)
(4-тиоморфоли-нилкарбонил) -хлорида (из примера 6а) и 0,172 мл триэтиламина в ДМФ, после хроматографической очистки на силикагеле с помощью смеси
метиленхлорида с метанолом (95: 5), осаждения
содержащих продукт фракций из метиленхлорида/гексана и лиофилизации из диоксана, получают указанное в заголовке соединение в виде аморфного твердого
вещества. FAB-MS: (M+H)+ 708; tRet(l) 21,4 мин; Rf(E) 0,45.
Пример 56: 2(R,S)-тетрагидрофурил-метоксикарбонил-(L)-Val-[ChaNN-Leu] J (N-2(R,
S)-тетрагидрофурил-метоксикарбонил-(L)-Val):
Аналогично примеру 37, из 80 мг (0,22 ммоль) H-[ChaNNLeu]-H•3HCl, 160 мг (0,65 ммоль) N-(2(R,
S)-тетрагидрофурил-метоксикарбонил)-(L)-валина, 289 мг (0,65 ммоль) ВОР, 88 мг (0,
65 ммоль) HOBt и 4,3 мл 0,3М NMM в ДМФ, после хроматографической очистки на силикагеле с помощью этилацетата и
лиофилизации содержащих продукт фракций из диоксана, получают указанное в заголовке
соединение. FAB-MS: (M+H)+ 712; tRet(l) 22,4 мин; Rf(Е) 0,21.
а)
H-[ChaNNLeu]-H•3HCl:
Аналогично примеру 5, из 150 мг (0,33
ммоль) Boc-[ChaNNLeu]-Boc и 10 мл 4н хлороводорода в диоксане, после лиофилизации реакционного
раствора, получают 100 мг (83%) указанного в заголовке соединения. Rf(K) 0,26.
б) Boc-[ChaNNLeu]-Boc:
Раствор 200 мг (0,24 ммоль) Boc-[PheNN
Leu]-Boc (пример 12) в 15 мл метанола гидрируют в присутствии 10 мг катализатора Нишимура
(Rh(III)-и Pt(IV)-оксид, моногидрат, Дегусса) под давлением водорода 1 атм в течение 4 часов. Катализатор
отфильтровывают, растворитель полностью выпаривают и получают указанное в заголовке соединение,
после кристаллизации из метиленхлорида с гексаном. tRet(l) 26,7 мин; Rf(V) 0,
21.
в) N-(2(R,S)- (тетрагидрофурил-метоксикарбонил)-(L)-ваплин:
Аналогично
примеру 50а, из 7 г (60 ммоль) (L)-валина и 9,8 г (60 ммоль) 2(R,
S)-тетрагидрофурилметил-хлорформиата (Heterocycles 27, 1155 (1988)) в 100 мл 2н раствора гидроксида натрия и 30 мл диоксана получают
указанное в заголовке соединение в виде смеси двух диастереомеров.
tRet(II) 23,5 и 23,8 мин.
Пример 57: Z-Val-[PheNNLeu] J
(N-(3-тетразол-1-ил)-пропионил)-Val):
Аналогично примеру 37, из 100 мг (0,21 ммоль)
Z-(L)-Val-[PheNNLeu]-H, 75 мг (0,31 ммоль) N-(3-(тетразол-1-ил)-пропионил-(L)-валина из примера
44а, 137 мг (0,31 ммоль) ВОР, 42 мг (0,31 ммоль) HOBt и 2 мл 0,3М NMM в ДМФ, после осаждения
из метиленхлорид/гексана и лиофилизации из смеси диоксана с трет.-бутанолом, получают указанное в заголовке
соединение (в HPLC различаются 2 диастереомера). FAB-MS: (M+H)+ 708; tRet(I) 21,1 и 21,1 мин; Rf(D) 0,45.
а) Z-(L)-Val-[PheNNLeu]-H:
Раствор 250 мг (0,43 моль) Z-(L)-Val-[PheNNLeu]-Boc в 5 мл муравьиной
кислоты перемешивают в течение 7,5 ч при комнатной температуре. Спустя это время, при HPLC-анализе более не
обнаруживается никакого исходного материала (tRet(I) 27,5 мин) и реакционный
раствор выпаривают. Остаток растворяют в хлороформе и промывают с помощью насыщенного раствора бикарбоната
натрия. Хлороформную фазу фильтруют через вату и получают, после испарения растворителя,
указанное в заголовке соединение красного цвета. tRet(l) 16,7 мин; Rf(K) 0,21.
б) Z-(L)-Val-[PheNNLeu]-Boc:
Аналогично примеру 37, из 230 мг
(0,653 ммоль) H-[PheNNLeu]-Boc, 247 мг (0,98 ммоль) Z-(L)-валина, 434 мг (0,98 ммоль) ВОР, 133
мг (0,98 ммоль) HOBt и 6,5 мл 0.3М NMM в ДМФ, после осаждения из метиленхлорид/метанола путем
добавления ДИПЭ, получают указанное в заголовке соединение. FAB-MS: (M+H)+ 585; tRet
(I) 27,5 мин; Rf(С) 0,71.
в) H-[PheNNLeu]-Boc:
Аналогично примеру 17а, исходя из 1,27 г (2,84 ммоль) N-трифторацетил- [PheNNLeu] -Boc и 24 мл
1н водного раствора карбоната натрия в 90 мл метанола, получают указанное в заголовке
соединение, которое осаждается из метиленхлорида за счет добавления ДИПЭ. tRet(I) 14,9 мин; Rf(K) 0,38.
г) N-трифторацетил-[PheNNLeu]-Boc:
Аналогично примеру 16, исходя из 3 г (11,57 ммоль) 2(R)-[1'(S)-(трифторацетил-амино)-2'-фенилэтил] оксирана (из
примера 16г) и 2,3 г (12,15 ммоль) трет.-бутил-3-изобутил-карбазата (получение: J.
Chem. Soc. Perkin 1, 1712 (1975)) после хроматографической очистки на силикагеле с помощью смеси метиленхлорида с
эфиром (20:1) получают указанное в заголовке соединение. tRet(I) 24,7 мин;
Rf(W) 0,36.
Пример 58: ацетил-Val-[PheNNLeu] J (N-2(R,
S)-карбамоил-3-фенилпропионилл)-Val):
Аналогично примеру 37, из 140 мг (0,3 ммоль)
ацетил-(L)-Val- [PheNNLeu] -H•2HCI, 132 мг (0,45 ммоль) N-(2(R,
S)-карбамоил-3-фенил- пропионил)-(L)-валина (получение: Synth.Struct.Funct. Ргос. Am. Pept. Symp. 7th, 85,
(1981)), 199 мг (0,45 ммоль) ВОР, 61 мг (0,45 ммоль) HOBt и 3,5 мл 0.3М NMM в ДМФ,
после осаждения из метиленхлорида с помощью ДИПЭ и лиофилизации из диоксана, получают титульное соединение (в HPLC
различаются два диастереомера). FAB-MS: (M+H)+= 667; tRet(l) 17,
9 и 18,4 мин; Rf(D) 0,33.
а) ацетил-Val-[PheNNLeu]-H•HCI:
Аналогично примеру 2а, исходя из 230 мг (0,46 ммоль) ацетил-(L)- Val-[PheNN
Leu] -Boc, после лиофилизации получают указанное в заголовке соединение, tRet(I) 10,5 мин; Rf(D) 0,38.
б) ацетил-Val-[PheNNLeu]-Boc:
Аналогично примеру 37, из 250 мг (0,71 ммоль) H-[PheNNLeu]-Boc (из примера 57в), 170 мг (1,07 ммоль)
N-ацетил-(L)-валина, 471 мг (1,07 ммоль) ВОР, 144 мг (1,07 ммоль) HOBt и 7,1 мл 0,3М NMM
в ДМФ, после осаждения из метиленхлорида за счет добавления ДИПЭ и лиофилизации из диоксана, получают
указанное в заголовке соединение. FAB-MS: (M+H)+ 493; tRet(I) 20,5 мин;
Rf(D) 0,59.
Пример 59: N-морфолинокарбонил-(L)-Val-[PheNNLeu] J
(N-(3-тетразол-1-ил)-пропионил)-Val):
Аналогично примеру 37, из 100 мг (0,19 ммоль)
N-морфолинокарбонил- (L)-Val-[PheNNLeu] -H•2HCI, 67 мг (0,38 ммоль)
N-(3-(тетразол-1-ил)- пропионил)-(L)-валина (из примера 44а), 124 мг (0,28 ммоль) ВОР, 38 мг (0,28 ммоль) HOBt и 2,
1 мл 0,3М NMM в ДМФ, после осаждения из метиленхлорида за счет добавления ДИПЭ и
лиофилизации из диоксана, получают указанное в заголовке соединение (в HPLC различаются 2 диастереомера). FAB-MS:
(M+H)+ 687; tRet(I) 15,2 и 15,4 мин; Rf(D) 0,25.
а) N-морфолинокарбонил-(L)-Val-[PheNNLeu]-H•2HCl:
Аналогично примеру 2а,
исходя из 279 мг (0,49 ммоль) N-морфолинокарбонил-(L)-Val-[PheNNLeu] -Boc,
после лиофилизации, получают указанное в заголовке соединение. FAB-MS: (M+H)+ 464; tRet(ll)
30,3 мин; Rf(D) 0,46.
б)
N-морфолинокарбонил-(L)-Val-[PheNNLeu]-Boc:
Аналогично примеру 37, из 250 мг (0,71 ммоль) H-[PheNNLeu]-Boc (из
примера 57в), 265 мг (1,07 ммоль)
N-морфолинокарбонил-(L)-валина (из примера 7а), 471 мг (1,07 ммоль) ВОР, 144 мг (1,07 ммоль) HOBt и 7,1 мл 0,3М NMM в ДМФ, после осаждения из метиленхлорида с помощью
гексана и лиофилизации из
диоксана, получают указанное в заголовке соединение. FАВ-MS: (M+H)+ 564; tRet(I) 21,5 мин; Rf(K) 0,69.
Пример 60:
N-трифторацетил-[PheNN
Leu] J (N-2(R,S)-карбамоил-3- фенилпропионил)-(L)-Val):
Аналогично примеру 37, из 136 мг (0,32 ммоль) N-трифторацетил-[PheNNLeu] -H•2HCI,
142 мг (0,49 ммоль) N-(2(R,
S)-карбамоил-3-фенил-пропионил)-(L)-валина (получение: Synth. struct, Pullet. Proc. Am. Pept. Symp. 7th, 85 (1981)), 215 мг (0,49 ммоль) ВОР, 66 мг (0,49 ммоль)
HOBt и5 мл 0,3М NMM в ДМФ,
после хроматографической очистки на силикагеле с помощью хлороформа с метанолом (15: 1), осаждения содержащих продукт фракций из метиленхлорида с помощью ДИПЭ и лиофилизации
из диоксана с
трет.-бутанолом, получают указанное в заголовке соединение (в HPLC различаются 2 диастереомера). FAB-MS: (M+H)+ 622; tRet(l) 21,6 и 22,0 Rf(K) 0,26.
а) N-трифторацетил-[PheNNLeu]-H•2HCl:
Аналогично примеру 2а, исходя из 300 мг (0,67 ммоль) N-трифторацетил-[PheNNLeu] -Boc (из примера 57г), после
лиофилизации
получают указанное в заголовке соединение. Rf(w) <0,1.
Пример 61: Z-(L)-Val-[PheNNNle] J N-(2(R,S)-(N-(2-морфолиноэтилa)-карбамоил)-3-метил)
-бутирил)
Аналогично примеру 37, из 100 мг (0,17 ммоль) Z-(L)-Val-[PheNNNle]-H•2HCl, 69 мг (0,27 ммоль) 2(R,S)-(N-(2-морфолиноэтил)-карбамоил)-3-метилмасляной кислоты
(N-(2-морфолиноэтил)-моноамидизопропилмалоновой кислоты), 119 мг (0,27 ммоль) ВОР, 36 мг (0,27 ммоль) HOBt и 2,1 мл 0,3М NMM в ДМФ, после осаждения из метиленхлорида с помощью ДИПЭ и лиофилизации из
диоксана, получают указанное в заголовке соединение (в HPLC различаются 2 диастереомера). FAB-MS: (M+H)+ 725; tRet(I) 17,2 и 17,6 мин; Rf(D) 0,56.
а)
Z-(L)-Val-[PheNNNle]-H•2HCl:
Аналогично примеру 2а, исходя из 310 мг (0,53 ммоль) Z-(L)-Val-[PheNNNle] -Boc, после лиофилизации, получают указанное в заголовке
соединение, tRet(I) 16,4 мин; Rf(U) 0,25.
б) Z-(L)-Val-[PheNNNle]-Boc:
Аналогично примеру 37, из 250 мг (0,71 ммоль) H-[PheNN
Nle]-Boc, 268 мг (1,07 ммоль) Z-(L)-валика, 472 мг (1,07 ммоль) ВОР, 144 мг (1,07 ммоль) HOBt и 7,1 мл 0,3М NMM в ДМФ, после хроматографической очистки на силикагеле с помощью смеси метиленхлорида с
метанолом (40:1) и осаждения содержащих продукт фракций из метиленхлорида с помощью ДИПЭ, получают указанное в заголовке соединение, tRet(I) 25,6 мин; Rf(X) 0,17.
в) H-[PheNNNle]-Boc:
Аналогично примеру 17а, исходя из 830 мг (1,85 ммоль) N-трифторацетил- [PheNNNle] -Boc, после осаждения из метиленхлорида с помощью ДИПЭ,
получают
указанное в заголовке соединение. tRet(l) 15,4 мин; Rf(X) 0,54.
г) N-трифторацетил-[PheNNNle]-Boc:
Аналогично примеру 16, исходя
из 1 г (3,86
ммоль) 2(R)-[1'(S)-трифторацетиламино)-2'-фенилэтил] -оксирана (из примера 16г) и 720 мг (3,86 ммоль) трет. -бутил-карбазата, после хроматографической очистки на силикагеле с помощью
смеси
метиленхлорида с эфиром (20:1), получают указанное в заголовке соединение. tRet(I) 25,3 мин; Rf(Q) 0,43.
д) трет.-бутил-3-бутил-карбазат:
Аналогично
примеру 4б, из 18,0 г (136,2 ммоль) трет.-бутилкарбазата и 12,3 мл (136,2 ммоль) н-бутанола получают соответствующий трет.-бутоксикарбонил-гидразон (25 г, 99%) в виде сырого продукта,
который, как
описано в примере 4а, гидрируют в присутствии 10 г 5-ной платины-на-угле и под давлением водорода 4 атм. Хроматографическая очистка сырого продукта на силикагеле с помощью смеси
гексан/этилацетат
(1:1) дает указанное в заголовке соединение. Rf(N) 0,44;1Н-ЯМР (200 МГц, CD3OD): 0,92 (t, J=7 Гц, 3Н); 1,43 (s, 9Н); 1,30-1,50 (m, 4Н); 2,75 (t,
J=7 Гц, 2Н).
е) 2(R,S)-(N-(2-морфолиноэтил)-карбамоил)-3-метилмасляная кислота
Аналогично примеру 9б, из 7 г (43,7 ммоль) рацемического монометилового эфира
изопропилмалоновой кислоты (Chem.
Ber. 119,1196 (1986)), 6,3 мл (48,1 ммоль) аминоэтилморфолина, 6,6 мл (43,7 ммоль) диэтилового эфира цианофосфоновой кислоты и 12,8 мл (91,8 ммоль) триэтиламина в
ДМФ получают метиловый эфир 2(R,
S)-(N-(2-морфолиноэтил)-карбамоил)-3-метилмасляной кислоты (метиловый эфир N-морфолиноэтиламина изопропилмалоновой кислоты). В течение 5 мин его перемешивают при
комнатной температуре в смеси 28 мл 2н
раствора гидроксида натрия и 28 мл диоксана, подкисляют 2н соляной кислотой и полностью выпаривают. Остаток настаивают с этанолом, отфильтровывают и получают
после выпаривания фильтрата указанное в
заголовке соединение.1Н-ЯМР (200 МГц, CD3OD): 0,95 и 1,00 (2d, J=7 Гц, 6Н); 2,25 (m, 4Н); 2,70 (m, 6Н); 2,75 (d, J=8 Гц, 1Н); 3,45 (m,
2Н); 3,75 (m, 4Н).
Пример
62: Z-(L)-Val-[PheNNNle] J (N-(3- (тетразол-1-ил)-пропионил)-Val):
Аналогично примеру 37, из 100 мг (0,18 ммоль) Z-(L)-Val-[PheNNNle]-H•2HCl (из примера 61а), 65
мг (0,27 ммоль) N-(3-(тетразол-1-ил)-пропионил)-(L)-валина (из примера 44а), 119 мг (0,27 ммоль) ВОР, 36 мг (0,27 ммоль) HOBt и 2,1 мл 0,3М
N-метилморфолина в ДМФ, после осаждения из метиленхлорида с
помощью ДИПЭ и лиофилизации из диоксана с трет.-бутанолом, получают указанное в заголовке соединение (в HPLC различимы 2 диастереомера).
FAB-MS: (M+H)+ 708; tRet 20,3 и 20,6 мин;
Rf(D) 0,43.
Пример 63: Z-(L)-Val-[PheNNNle] J (N-2(R,
S)-(N-(2-пиридилметилa)-карбамоил)-3-метил)-бутирил) (дибензолсульфонат):
Аналогично примеру 37, из
95 мг (0,17 ммоль) Z-(L)-Val-[PheNNNle]-H•2HCl (из примера 61а), 60 мг
(0,26 ммоль) N-(2-пиколил)-моноамида-(R,S)-изопропилмалоновой кислоты, 113 мг (0,26 ммоль) ВОР, 35 мг (0,26
ммоль) HOBt и 2,0 мл 0,3М NMM в ДМФ, после хроматографической очистки на силикагеле с
помощью смеси метиленхлорид/метанол (15:1) получается указанное в заголовке соединение в виде свободного амина.
Его растворяют в метиленхлориде, смешивают с двумя эквивалентами бензолсульфокислоты и
осаждают путем добавления ДИПЭ. Лиофилизация из трет.-бутанола дает дибензолсульфонатную соль (в HPLC различимы 2
диастереомера). FAB-MS: (M+H)+ 703: tRet(l) 17,7 и 18,0 мин;
Rf(D) 0,54.
а) N-(2-пиколил)-моноамид изопропилмалоновой кислоты:
Раствор 15 г
(93,6 ммоль) монометилового эфира изопропилмалоновой кислоты (получение: Chem. Ber.
119, 1196 (1986)) в 150 мл ТГФ смешивают с 10,6 мл (103 ммоль) N-метилморфолина и затем по каплям с 13,5 мл (103
ммоль) изобутилхлорформиата. Спустя 30 минут добавляют 15,3 мл (150 ммоль)
2-пиколиламина и перемешивают образующуюся суспензию в течение 2 часов. Реакционную смесь разбавляют метиленхлоридом,
промывают 1н раствором гидроксида натрия и водой, органическую фазу фильтруют
через вату и выпаривают. Кристаллизация остатка дает N-(2-пиколиламид)-метиловый эфир изопропилмалоновой кислоты, который,
как описано в примере 6е, гидролизуют в 2н растворе гидроксида натрия и
диоксане. tRet(ll) 16,0 мин.
Пример 64: Z-(L)-Val-[PheNN(p-F)Phe] J (N-(3-(тетразол-1-ил)-пропионил)-(L)-Val) (бензолсульфонат).
Аналогично примеру 37, из 100 мг (0,16 ммоль) Z-(L)-Val-[PheNN(p-F)Phe] -H (из примера 22а), 59 мг (0,25 ммоль) N-(3-тетразол-1-ил)-пропионил)-(L)-валина (из примера 44а), 109 мг (0,25 ммоль) ВОР, 33 мг (0,25 ммоль) НОВt и 1,19 мл 0,3М N-метилморфолина в ДМФ, после осаждения из метиленхлорида с помощью ДИПЭ, получают указанное в заголовке соединение в виде свободного амина. Его растворяют в смеси метиленхлорида с водой, смешивают с 1 эквивалентом бензолсульфокислоты и осаждают за счет добавления гексана. Лиофилизация из трет. -бутанола дает указанное в заголовке соединение в виде бензолсульфоната. FAB-MS: (M+H)+ 760; tRet(l) 21,6 мин; Rf(B) 0,49.
Пример 65: метилсульфонил-[PheNNPhe] J (N-фенилацетил-(L)-Val):
132 мг (0,28 ммоль) метилсульфонил-[PheNNPhe]-H•2HCI, аналогично примеру 7, вводят во
взаимодействие со 197 мг (0,84 ммоль) N-фенилацетил-(L)-валина (получение: Mem. Tokyo
Univ. Agric. 20, 51 (1978)), 317 мг (0,84 ммоль) HBTU и 0,23 мл (1,67 ммоль) триэтиламина в ДМФ, и после осаждения
из метанола за счет добавления эфира, получают указанное в заголовке соединение.
FAB-MS: (M+H)+ 581; tRet(I) 20,2 мин; Rf(B) 0,64.
а)
метилсульфонил-[PheNNPhe]-H•2HCl:
Аналогично примеру 2а, исходя из
130 мг (0,28 ммоль) метилсульфонил- [PheNNPhe]-Boc, после лиофилизации, получают указанное в
заголовке соединение. FAB-MS: (M+H)+ 364; tRet(ll) 28,5 мин, Rf
(K) 0,56.
б) метилсульфонил-[PheNNPhe]-Boc:
Аналогично примеру 16а,
исходя из 1,1 г (4,56 ммоль) 2(R)-[1'(S)- (метилсульфониламино)-2'-фенилэтил1-оксирана и 1,11
г (5,02 ммоль) трет.- бутил-3-бензилкарбазата (получение: J. Chem. Soc. Perkin 1,1712 (1975)), получают
указанное в заголовке соединение в виде смеси диастереомеров в соотношении 4:1. Путем
кристаллизации из метиленхлорида с гексаном соотношение улучшается в пользу 25-диастереомера до 10:1. FAB-MS:
(M+H)+ 464; tRet(I) 21,3 мин; Rf(N) 0,26.
в) 2(R)-[1'(S)-(метилсульфониламино) -2'-фенилэтил]оксиран:
Раствор 1 г (6,8 ммоль)
1-фенил-3-бутен-2(S)-амина (из примера 16б) в 10 мл метиленхлорида при 0oC смешивают
с 2,36 г (13,6 ммоль) ангидрида метансульфокислоты и 1,88 мл (13,6 ммоль) триэтиламина и перемешивают в
течение 1 часа. Реакционную смесь промывают водой и насыщенным раствором бикарбоната натрия,
органическую фазу фильтруют через вату и выпаривают и получают 2(S)-метилсульфониламино-1-фенил-3-бутен. 1
г (4,4 ммоль) этого сырого продукта растворяют в 30 мл метиленхлорида, смешивают при
комнатной температуре с 3,05 г (17,7 ммоль) 4-хлорнадбензойной кислоты и перемешивают в течение 18 ч. Реакционный
раствор промывают 5 раз с помощью 10-наго водного раствора сульфита натрия, фильтруют
через вату и полностью выпаривают. Сырой продукт, согласно1Н-ЯМР, содержит оба (2R)-и (2S)-эпимера в
соотношении 4: 1.1Н-ЯМР (200 МГц, CD3OD): 2,30 и 2,52 (s,
вместе 3Н); 2,6-3,2 (m, 5Н); 3,55 (m, 1Н); 7,32 (m, 5Н).
Пример 66: метоксикарбонил-(L)-Val-[PheNNLeu] J (N-метоксикарбонил-(L)-Val):
Аналогично примеру 37, из
200 мг (0,55 ммоль) H-[PheNNLeu]-H•-H•3HCl (из примера 13а), 291 мг (1,66 ммоль)
N-метоксикарбонил-(L)-валила (получение: Chem. Lett. 705, (1980)), 735 мг (1,66 ммоль) ВОР,
225 мг (1,66 ммоль) HOBt и 11 мл 0,3М NMM в ДМФ получают, после осаждения из метиленхлорида с помощью ДИПЭ и
лиофилизации из диоксана, указанное в заголовке соединение. FAB-MS: (M+H)+ 566;
tRet(I) 18,6 мин; Rf(U) 0,33.
Пример 67:
метоксикарбонил-(L)-Val-[PheNN(p-F)Phe] J (N-метокси-карбонил-(L)-Val):
Аналогично примеру 37, из
200 мг (0,48 ммоль) H-[PheNN(p-F)Phe]- H•3HCl (из примера 42б),
255 мг (1,45 ммоль) N-метоксикарбонил-(L)-валина (получение: Chem. Lett. 705, (1980)), 643 мг (1,45 ммоль) ВОР, 196
мг (1,45 ммоль) HOBt и 9,7 мл 0,3М NMM в ДМФ получают, после осаждения из
метиленхлорида с помощью ДИПЭ и лиофилизации из диоксана, указанное в заголовке соединение. FAB-MS: (M+H)+ 618;
tRet(I) 19,5 мин; Rf(U) 0,22.
Пример
68: метоксикарбонил-(L)-Val-[PheNN(p-CN)Phe] J (N-метокси-карбонил- (L)-Val):
Аналогично примеру 37, из
200 мг (0,48 ммоль) H-[PheNN(p-CN)Phe]-H•3HCl (из примера
47а), 250 мг (1,43 ммоль) N-метоксикарбонил-(L)-валина (получение: Chem. Lett. 705, (1980)), 631 мг (1,43 ммоль) ВОР, 193
мг (1,43 ммоль) HOBt и 9,5 мл 0,3М NMM в ДМФ, после хроматографической очистки
на силикагеле с помощью смеси метиленхлорид/метанол (15:1) и лиофилизации содержащих продукт фракций из диоксана,
получают указанное в заголовке соединение. FAB-MS: (M+H)+ 625; tRet(I) 18 мин; Rf(U) 0,31.
Пример 69: Z-(L)-Val-[(p-F)PheNN(p-F)Phe] J
(N-2(R,S)-(N-(2-морфолиноэтил)-карбамоил)- 3-метил)-бутирил):
Аналогично
примеру 18, 23,0 мг (0,089 ммоль) 2(R,S)-(N-(2-морфолиноэтил)-карбамоил)-3-метилмасляной кислоты (пример 61е) и 45 мг
(0,081 ммоль) Z-(L)-Val-[(p-F)PheNN(p-F)Phe]-H (пример 32) вводят во
взаимодействие с 33,8 мг (0,089 ммоль) HBTU в 0,76 мл NMM/CH3CN (0,25М) с получением указанного в заголовке
соединения и переосаждают его с помощью ДМФ/ДИПЭ. ТCX Rf(P) 0,42;
FAB-MS: (M+H)+ 795.
Пример 70: Z-(L)-Val-[(p-F)PheNN(p-F)Phe] J N-(2(R,
S)-(N-(2-пиридилметил)-карбамоил)-3-метил)-бутирил):
Аналогично примеру 18, 21,0
мг (0,089 ммоль) рацемического N-(2-пиколил)-амида изопропилмалоновой кислоты (пример 63а) и 45 мг (0,081
ммоль) Z-(L)-Val-[(p-F)PheNN(p-F)Phe] -H (пример 32) вводят во взаимодействие с 33,
8 мг (0,089 ммоль) HBTU в 0,76 мл 0,25М NMM в CH3CN для получения указанного в заголовке
соединения и переосаждают из ДМФ с помощью ДИПЭ. TCX Rf(P) 0,53; FAB-MS: (M+H)+
773.
Пример 71:
Аналогично вышеуказанному способу можно получать
следующие соединения:
а) Z-(L)-Val-[(p-F)PheNN(p-F)Phe] J ((L)-Val) J
(N-морфолинокарбонил-Gly);
б) N-морфолоинокарбонил-(L)-Val-[(p-F)PheNN(p-F)Phe] J
((L)-Val) J (N- морфолинокарбонил-Gly);
в) N-(хинолин-2-карбонил)-(L)-Asn-[PheNN(p-F)Phe] <-- ((L)-Val)-Z;
г) N-(морфолиносульфонил)-(L)-Val-[PheNNLeu]
<-- (N-(морфолиносульфонил)- (L)-Val).
Пример 72: желатиновый
раствор:
Стерильно профильтрованный водный раствор с 20% циклодекстрина в качестве агента растворения
одного из указанных в предыдущих примерах соединений формулы I в качестве биологически
активного вещества при нагревании смешивают со стерильным желатиновым раствором, который в качестве консерванта
содержит фенол, при асептических условиях, так, что 1,0 мл раствора имеет следующий
состав:
Биологически активное вещество 3 мг
Желатина 150,О мг
Фенол 4,7 мг
Дистиллированная вода с 20% циклодекстрина в качестве агента растворения
1,0
мл
Пример 73: стерильное сухое вещество для инъекции:
5 мг одного из указанных в предыдущих
примерах соединений формулы I в качестве биологически активного вещества растворяют в 1 мл
водного раствора с 20 мг маннита и 20% циклодекстрина в качестве агента растворения. Раствор стерильно
фильтруют и при асептических условиях заливают в ампулы емкостью 2 мл, подвергают глубокому
охлаждению и лиофилизации. Перед употреблением лиофилизат растворяют в 1 мл дистиллированной воды или 1 мл
физиологического раствора хлорида натрия. Раствор применяют внутримышечно или внутривенно.
Этим раствором можно также заполнять двухкамерные ампулы для инъекций.
Пример 74:
пульверизатор для носа (аэрозольная упаковка):
В смеси из 3,5 мл Myglyol 812® и 0,08 г бензилового спирта суспендируют 500 мг тонко размолотого порошка (<5,0 мкм)
одного из указанных в вышеприведенных примерах соединений формулы 1 в качестве биологически активного
вещества. Эту суспензию заливают в емкость с дозировочным вентилем. Заполняют 5,0 фреона 12® под давлением через вентиль в емкость. Путем встряхивания "фреон" растворяется в смеси
Myglyol с бензиловым спиртом. Эта емкость-пульверизатор содержит примерно 100 разовых доз,
которые могут вводиться отдельно.
Пример 75: покрытые лаком таблетки:
Для
приготовления 10000 таблеток, содержащих по 100 мг биологически активного вещества, перерабатывают
следующие составные части:
Биологически активное вещество 1000 г
Кукурузный крахмал
680 г
Коллоидальная кремневая кислота 200 мг
Стеарат магния 20 г
Стеариновая кислота 50 г
Карбоксиметилкрахмал натрия 250 г
Вода quantum satis
Смесь одного из указанных в вышеприведенных примерах соединений формулы I в качестве
биологически активного вещества, 50 г кукурузного крахмала и коллоидальной кремневой кислоты вместе с крахмальным
клейстером из 250 г кукурузного крахмала, и 2,2 кг дименирализованной воды
перерабатывают во влажную массу. Эту массу протирают через сито с размерами отверстий 3 мм и высушивают при 45oC в
течение 30 мин в сушилке с псевдоожиженным слоем. Высушенный гранулят
продавливают через сито с размером отверстий 1 мм, смешивают с заранее просеянной смесью (сито с размером отверстий 1 мм) 330 г
кукурузного крахмала, стеарата магния, стеариновой кислоты и
карбоксиметилкрахмала натрия и прессуют в слабо выпуклые таблетки.
Реферат
Использование: в медицине как обладающие свойством ингибировать действие фермента ВИЧ- протеазы и антивирусной активностью. Сущность изобретения: производные гидразина общей формулы 1: R1R2N - C(R3 R4) - C(R5R6 - N(R7)N(R8R9) , где R1 и R9 независимо друг от друга - водород, низший алканоил, фенил(низш.) алканоил, фенил(низш). алканоил, в котором остаток низшего алконоила замещен карбамоилом, морфолино(низш.) алканоил, тиоморфолино(низш.) алканоил, пиридил(низш.) алканоил, хинолил(низш.) алканоил, тетразолил(низш. ) алканоил, амино(низш. ) алканоил, замещенный у аминного азота N-морфолино- или N-тиоморфолинокарбон - ил, галоген(низш.) алканоил, содержащий до трех атомов галогена, 2-(N-морфолино(низш.) алкилкарбамоил)-низший алканоил, 2-(N-пиридил(низш. ) алкилкарбамоил)-низший алканоил, низший алкоксикарбонил, фенил(низш. ) алкоксикарбонил, тетрагидрофуранил(низш.) алкилксикарбонил, низший алкилсульфонил, N-пиридил(низш.) алкил-N-(низш.) алкилкарбамоил, или ацильный остаток аминокислоты, выбранный из глицина, аланина, валина, лейцина, изолейцина, глютаминовой кислоты и аспарагина в форме (D)-, (L)-, или (D,L)-, где α - аминогруппа незамещена или ацилирована одним из вышеназванных остатков R1 или R9, при условии, что по меньшей мере один из остатков R1 и R9 означает водород, R2, R4, R6 и R8 означают водород; R3 - низший алкил, циклогексил(низш.) алкил или фенил(низш.) алкил, незамещенный или замещенный галогеном, низшим алкокси или циано; R5 - означает гидроксильную группу; R7 - низший алкил, циклогексил(низш.) алкил, или фенил(низш. ) алкил, незамещенный или замещенный галогеном, низшим алкокси или циано, или их соли, в случае наличия солеобразующих групп, фармацевтическое средство, содержащее эффективное количество соединений формулы I, а также производное гидразина формулы II: H2N - HC(R3) - CH(OH) - N(R7)NH2 - промежуточные продукты в синтезе I. 2 с. и 8 з.п.ф-лы, 3 табл.
Формула

где R1 и R9 независимо друг от друга водород, низший алканоил, фенил-низш. алканоил, фенил-низш.алканоил, в котором остаток низшего алканоила замещен карбамоилом, морфолино-низш.алканоил, тиоморфолино-низш.алканоил, пиридил-низш. алканоил, хинолил-низш.алканоил, тетразолил-низш.алканоил, амино-низш. алканоил, замещенный у аминного азота N-морфолино- или N-тиоморфолинокарбонил, галоген-низш. алканоил, содержащий до трех атомов галогена, 2-(N-морфолино-низш. алкилкарбамоил)-низший алканоил, 2-(N -пиридил-низш.алкилкарбамоил)-низший алканоил, низший алкоксикарбонил, фенил-низш.алкоксикарбонил, тетрагидрофуранил-низш. алкоксикарбонил, низший алкилсульфонил, N -пиридил-низш.алкил- N-низш.алкилкарбамоил, или ацильный остаток аминокислоты, выбранный из глицина, аланина, валина, лейцина, изолейцина, глютаминовой кислоты и аспарагина в форме (D)-, (L)- или (D,L)-, где α -аминогруппа, не замещена или ацилирована одним из названных остатков R1 или R9 при условии, что по меньшей мере один из остатков R1 и R9 водород;
R2, R4, R6 и R8 водород;
R3 низший алкил, циклогексил-низш.алкил или фенил-низш.алкил, незамещенный или замещенный галогеном, низшим алкокси или циано;
+R5 гидроксильная группа;
R7 низший алкил, циклогексил-низш.алкил или фенил-низш.алкил, незамещенный или замещенный галогеном, низшим алкокси или циано,
или их соли в случае наличия солеобразующих групп.
а также фармакологически приемлемые соли этих соединений.
а также фармакологически приемлемые соли этих соединений.
а также фармакологически приемлемые соли этих соединений.
а также фармакологически приемлемые соли этих соединений.
Вос-[PheNN Phe] -Boc; Boc-(L)-вал- [PheNNPhe]←(L)-вал-Вос; Вос-[PheNNCha] -Boc; H-(L)-вал-[Phe NN Phe] J(L)-вал-Н; N-тиоморфолинокарбонил-(L)-вал-[PheNNPhe] J(N-тиоморфолинокар- бонил-(L)-вал); N-морфолинокарбонил-(L)-вал-[PheNNPhe] J(N-морфолинокарбонил-- (L)-вал); фенилацетил-(L)-вал-[PheNNPhe] J(N-фенилацетил-(L)-вал); N-(3-пиридилацетил)-(L)-вал-[PheNNPhe] J(N-(3-пиридилацетил)- -(L)-вал); Вос-(L)-вал-[PheNNCha] J(L)-вал-Вос; Z-(L)-вал-[PheNNCha]J(L)-вал-Z; Вос-[PheNN Leu]-Вос; Z-(L)-вал-[PheNNLeu)J(L)-вал-Z, Н-(L)-вал-[PheNNCha] J(L)- вал)- и N-(3-пиридилацетил)-(L)-вал-[PheNNLeu]J(N-(3-пиридилацетил)-(L)-вал)
или их соли,
где Вос означает трет-бутоксикарбонил,
Z бензилоксикарбонил,
остаток [PheNNPhe] означает двухвалентный радикал 3(S)-амино-4-фенил-1-(N-бензилгидразино)-бутан-2(S)-ол и имеет формулу

где остаток [PheNNCha] означает двухвалентный радикал 3(S)-амино-4-фенил-1-(N-циклогексилметилгидразино)-бутан-2(S)-ол и имеет формулу


и " ← " означает поворот связи в отклонении от обычных пептидных номенклатур, причем влево амино и вправо карбокси.
или их соли, где Z бензилоксикарбонил, остаток [PheNNPhe] означает двухвалентный радикал 3(S)-амино-4-фенил-1-(N-бензилгидразино)-бутан-2(S)-ол и имеет формулу

где остаток [PheNNChа] означает двухвалентный радикал 3(S)-амино-4-фенил1-(N-циклогексилметилгидразино)-бутан-2(S)-ол и имеет формулу

где остаток [PheNNLeu] означает двухвалентный радикал 3(S)-амино-4-фенил-1-(N-изобутилгидразино)-бутан -2(S)-ол и имеет формулу

остаток [PheNNNIe] означает радикал 3(S)-амино-4-фенил-1-(N-n-бутилгидразино)-бутан-2(S)-ол и имеет формулу

остаток [PheNN(p-F)Phe] означает двухвалентный радикал 3(S)-амино-4-фенил-1-(N-(p-фторфенил)-гидразино)-бутан-2(S)-ол и имеет формулу

остаток [(p-F)PheNN(p-F)Phe] означает двухвалентный радикал 3(S)-амино-4-фенил-(р-фторфенил)-1-(N-(p-фторфенилметил)-гидразино)-бутан- 2(S)-ол и имеет формулу

остаток [PheNN(p-СN)Phe] означает двухвалентный радикал 3(S)-амино-4-фенил-1-(N-(p- цианофенилметил)-гидразино)-бутан-2(S)-ол и имеет формулу
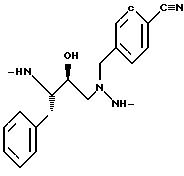
и " ← " означает поворот связи в отклонение от обычных пептидных номенклатур, причем, влево амино и вправо карбокси.

где R3 и R7 низший алкил, циклогексил-низший алкил, или фенил-низший алкил, незамещенный или замещенный галогеном, низшим алкокси или циано, или их соли, если они имеют солеобразующие группы.
Комментарии