Бензамидные производные, обладающие противораковой активностью, способ их получения и их применение - RU2565079C2
Код документа: RU2565079C2
Описание
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
[1] Настоящее изобретение относится к бензамидным производным, обладающим противораковой активностью, и их метаболитам. Настоящее изобретение также относится к способам синтеза противораковых лекарственных средств указанного типа и способам лечения раковых заболеваний с использованием указанного типа соединений.
УРОВЕНЬ ТЕХНИКИ
[2] Рак бесспорно представляет собой трудноизлечимое опасное для жизни заболевание, распространенное по всему миру. Таким образом, рак является серьезной проблемой для здравоохранения во всем мире. В Соединенных Штатах Америки рак является основной причиной смертности среди американцев моложе 85 лет и второй по распространенности причиной смертности среди американцев более пожилого возраста. По статистике в США ежедневно от рака умирают примерно 1500 человек, и ежедневно возникает примерно 3400 новых случаев раковых заболеваний, тогда как в Китае каждый год от рака умирает примерно 1,8 миллионов человек, и каждый год возникает примерно 2,6 миллионов новых случаев раковых заболеваний. Рак стал основной причиной смертности среди китайцев.
[3] Рак представляет собой заболевание, возникающее из собственных клеток организма, и представляет собой злокачественную опухоль. Такие опухолевые клетки являются крайне аномальными и претерпевают случайные и неупорядоченные деления, и, таким образом, их рост и пролиферация абсолютно неподконтрольны. Раковые клетки являются очень агрессивными; они атакуют и повреждают окружающие ткани. Они также могут покидать первоначальную опухоль и проникать в кровеносную и лимфатическую систему с образованием новых опухолей в других частях тела.
[4] Несмотря на то, что на рынке представлены различные противораковые лекарственные средства, эффективное лечение рака до сих пор является трудной задачей. Таким образом, по-прежнему существует острая потребность в продолжении исследований и разработке противораковых лекарственных средств, обладающих более высокой активностью и вызывающих менее выраженные токсические и побочные эффекты. Интересно, что соединения галогенизированного нитро- и нитрозо-эстрогена можно использовать для лечения раковых заболеваний, в частности, рака молочной железы. Еще больший интерес представляет то, что также было обнаружено, что ароматические галогенизированные нитро- и нитрозо-соединения, имеющие простую структуру, обладают очень высокой противоопухолевой активностью, в частности, в отношении рака молочной железы (Kun et al. US 5464871). Такие соединения представляют собой ингибиторы поли(АДФ-рибоза)-полимеразы (PARP), которая вовлечена в репарацию повреждений ДНК. Таким образом, репарацию ДНК можно подавлять путем ингибирования поли(АДФ-рибоза)-полимеразы для усиления терапевтического эффекта радиотерапии и химиотерапии при раковых заболеваниях (Ossovskaya et al. US 20090149397; Sherman et al. US 20090131529 and 20090123419). Фактически ароматическое нитросоединение сначала превращается в ароматическое нитрозосоединение in vivo, которое является активным соединением, подавляющим опухолевый рост. В связи с относительно малой растворимостью в воде при физиологическом значении pH и ограниченной стабильностью ароматических нитрозосоединений сложно предсказать, смогут ли они достигнуть раковые клетки. В то же время ароматические нитросоединения не имеют указанных недостатков и, таким образом, являются идеальными пролекарствами ароматических нитрозосоединений. Среди указанных ароматических нитросоединений многообещающим противораковым лекарственным средством является 4-йод-3-нитробензамид (INBA, код BSI-201). На сегодняшний день указанное соединение вошло в III фазу клинических испытаний, также исследуется его противораковое действие в комбинации с другими противораковыми лекарственными средствами, где подвергаемый лечению рак представляет собой метастатический трижды негативный рак молочной железы (МТНРМЖ).
[5] Поскольку органический йодид является очень чувствительным к воздействию света и воздуха и плохо растворяется в воде, желательна разработка не содержащих йода противораковых лекарственных средств, обладающих более высокой активностью и лучшей растворимостью в воде. Согласно настоящему изобретению, предложены новые бензамидные производные, обладающие противораковой активностью, которые представляют собой более эффективные по сравнению с INBA (BSI-201) не содержащие йода противораковые лекарственные средства.
Краткое описание изобретения
[6] Согласно одному из аспектов настоящего изобретения предложен новый тип противораковых лекарственных средств - бензамидные производные или их фармацевтически приемлемые соли.
[7] Соединение согласно настоящему изобретению может быть представлено формулой (I):

где
[8] R1 и R2 независимо выбраны из группы, состоящей из H, замещенного или незамещенного (C1-C8)алкила, замещенного или незамещенного (C3-C8)алкенила, замещенного или незамещенного (C3-C8)алкинила, замещенного или незамещенного (C3-C7)циклоалкила, -COR5 и -CO2R6; R1 и R2 также могут циклизоваться с образованием замещенного или незамещенного 4-, 5- или 6-членного кольца;
[9] R3 представляет собой нитро или нитрозо;
[10] R4 выбран из группы, состоящей из этинила, пропинила или циано;
[11] R5 выбран из группы, состоящей из замещенного или незамещенного (C1-C8)алкила, замещенного или незамещенного (C3-C8)алкенила, замещенного или незамещенного (C3-C8)алкинила, замещенного или незамещенного (C3-C7)циклоалкила и замещенного или незамещенного арила;
[12] R6 выбран из группы, состоящей из замещенного или незамещенного (C1-C8)алкила, замещенного или незамещенного (C3-C8)алкенила, замещенного или незамещенного (C3-C8)алкинила и замещенного или незамещенного (C3-C7)циклоалкила;
[13] где в случае замещенного или незамещенного (C3-C8)алкенила и замещенного или незамещенного (C3-C8)алкинила двойная связь указанного алкенила и тройная связь указанного алкинила предпочтительно не связаны напрямую с атомом азота амида, карбонилом или карбонилокси;
[14] или его фармацевтически приемлемые соли или пролекарство.
[15] При использовании в настоящей заявке, если иное специально не указано, термин «замещенный» относится к соединению, содержащему в качестве заместителя радикал, выбранный из группы, состоящей из гидрокси, (C1-C8)алкила, (C2-C8)алкенила, (C2-C8)алкинила, (C3-C7)циклоалкила, (C1-C8)алкилокси, амино, (C1-C8)алкиламино, ди(C1-C8)алкиламино, -(C1-C8)алкилтио и галогена, предпочтительно, гидрокси, метокси, амино, метиламино, диметиламино, метилтио и галогена,
[16] где R1 и R2 предпочтительно выбраны из группы, состоящей из водорода, замещенного или незамещенного (C1-C8)алкила, -COR5 или -CO2R6; R1 и R2 также могут циклизоваться с образованием замещенного или незамещенного 4-, 5- или 6-членного кольца, где R5 представляет собой замещенный или незамещенный (C1-C8)алкил, замещенный или незамещенный (C3-C8)алкенил, замещенный или незамещенный (C3-C8)алкинил, замещенный или незамещенный (C3-C7)циклоалкил, замещенный или незамещенный арил; R6 представляет собой замещенный или незамещенный (C1-C8)алкил, замещенный или незамещенный (C3-C8)алкенил, замещенный или незамещенный (C3-C8)алкинил, замещенный или незамещенный (C3-C7)циклоалкил; где в случае замещенного или незамещенного (C3-C8)алкенила и замещенного или незамещенного (C3-C8)алкинила двойная связь указанного алкенила и тройная связь указанного алкинила предпочтительно не связаны напрямую с атомом азота амида, карбонила или карбонилокси.
[17] Более предпочтительно, R1 и R2 независимо выбраны из группы, состоящей из водорода, замещенного или незамещенного (C1-C8)алкила, -COR5; или -CO2R6; R1 и R2 также могут циклизоваться с образованием замещенного или незамещенного 4-, 5- или 6-членного кольца, где R5 представляет собой замещенный или незамещенный (C1-C8)алкил, замещенный или незамещенный (C3-C7)циклоалкил, замещенный или незамещенный арил; R6 представляет собой замещенный или незамещенный (C1-C8)алкил, замещенный или незамещенный (C3-C7)циклоалкил.
[18] Наиболее предпочтительно, R1 и R2 независимо выбраны из группы, состоящей из водорода, метила, этила, пропила, -COR5 или -CO2R6; R1 и R2 также могут циклизоваться с образованием замещенного или незамещенного 5- или 6-членного кольца, и образованное кольцо может содержать один или более гетероатомов, где указанный гетероатом представляет собой атом азота, кислорода или серы, предпочтительно атом азота; R1 и R2 циклизуются с образованием замещенного или незамещенного 5- или 6-членного кольца, содержащего один или более гетероатомов, предпочтительно, пирролидинового кольца, пиперидинового кольца, морфолинового кольца и пиперазинового кольца,
[19] где R5 предпочтительно выбран из замещенного или незамещенного (C1-C8)алкила и замещенного или незамещенного арила, наиболее предпочтительно, метила и фенила;
[20] где R6 предпочтительно представляет собой замещенный или незамещенный (C1-C8)алкил, более предпочтительно, метил, этил, пропил, изопропил, н-бутил или изобутил, наиболее предпочтительно, метил или этил;
[21] где R3 предпочтительно выбран из нитро или нитрозо, более предпочтительно, нитро;
[22] где R4 предпочтительно выбран из группы, состоящей из этинила, пропинила и циано, более предпочтительно, этинила и циано.
[23] Более конкретно, где соединение формулы (I) выбрано из группы, состоящей из:
4-этинил-3-нитробензамида (I-1)
N-метоксикарбонил-4-этинил-3-нитробензамида (I-2)
N-этоксикарбонил-4-этинил-3-нитробензамида (I-3)
N-пропоксикарбонил-4-этинил-3-нитробензамида (I-4)
N-бутоксикарбонил-4-этинил-3-нитробензамида (I-5)
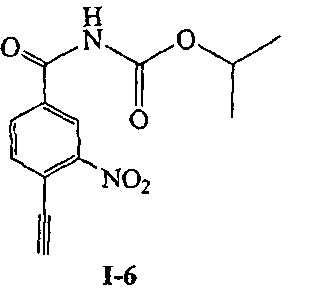
N-изопропоксикарбонил-4-этинил-3-нитробензамида (I-6)
N-изобутоксикарбонил-4-этинил-3-нитробензамида (I-7)
4-циано-3-нитробензамида (I-8)
N-метил-4-этинил-3-нитробензамида (I-9)
N,N-диметил-4-этинил-3-нитробензамида (I-10)
N-ацетил-4-этинил-3-нитробензамида (I-11)
4-(1-пропинил)-3-нитробензамида (I-12)
N-бензоил-4-этинил-3-нитробензамида (I-13)
N,N-диэтил-4-этинил-3-нитробензамида (I-14)
N,N-дипропил-4-этинил-3-нитробензамида (I-15)
N,N-дибутил-4-этинил-3-нитробензамида (I-16)
N-этил-4-этинил-3-нитробензамида (I-17)
N-пропил-4-этинил-3-нитробензамида (I-18)
N-бутил-4-этинил-3-нитробензамида (I-19)
(4-этинил-3-нитрофенил)(пирролидин-1-ил)кетона (I-20)
(4-этинил-3-нитрофенил)(пиперидин-1-ил)кетона (I-21)
(4-этинил-3-нитрофенил)(морфолин-4-ил)кетона (I-22)
(4-этинил-3-нитрофенил)(пиперазин-1-ил)кетона (I-23)
(4-этинил-3-нитрофенил)(4-метилпиперазин-1-ил)кетона (I-24)
(4-этинил-3-нитрофенил)(азетидинон-1-ил)кетона (I-25)
N-метил-4-(1-пропинил)-3-нитробензамида (I-26)
N-метил-4-циано-3-нитробензамида (I-27)
4-этинил-3-нитрозобензамида (I-28)
гидрохлорида (4-этинил-3-нитрофенил)(4-метилпиперазин-1-ил)кетона (I-29)
гидрохлорида (4-этинил-3-нитрофенил)(пиперазин-1-ил)кетона (I-30).
[24] Согласно второму аспекту настоящего изобретения также предложено применение соединения общей формулы (I) и его фармацевтической композиции для получения лекарственного средства для лечения рака. Рак включает рак толстой кишки, рак молочной железы, рак легкого, рак яичников, рак желудка, острый лейкоз, хронический лейкоз, рак предстательной железы, рак матки человека, рак поджелудочной железы, рак печени, рак мозга, рак центральной нервной системы, рак мочевого пузыря, рак почки, рак кожи, рак шеи, миосаркому, лимфому, рак кости и другие типы раковых заболеваний. При этом согласно настоящему изобретению также предложено применение соединения, представленного общей формулой (I), и его фармацевтической композиции для получения лекарственного средства для лечения состояния, вызванного, связанного или сопровождающегося нарушением в результате клеточной пролиферации и/или ангиогенеза.
[25] Согласно другому аспекту настоящего изобретения предложено применение соединения общей формулы (I) для получения лекарственного средства для лечения заболевания, связанного с ингибитором поли(АДФ-рибоза)-полимеразы (PARP). Заболевание, связанное с ингибитором поли(АДФ-рибоза)-полимеразы (PARP), включает рак, инсульт, инфаркт миокарда, нейродегенеративное заболевание и т.д. Согласно другому аспекту настоящего изобретения предложен способ лечения заболеваний, вызванных, связанных или сопровождающихся нарушением в результате клеточной пролиферации и/или ангиогенеза, с использованием эффективного количества соединения, представленного общей формулой (I), или фармацевтической композиции, содержащей указанное соединение отдельно или в комбинации с другими лекарственными средствами. Фармацевтическая композиция согласно настоящему изобретению также может содержать фармацевтически приемлемые носители, совместимые с соединением формулы (I). Соединение формулы (I) можно вводить в виде традиционных дозированных форм, таких как дозированные формы для инъекционного и перорального введения, включая капсулу, таблетку, порошок, крахмальную капсулу, суспензию или раствор, предпочтительно путем инъекции. Дозированные формы и фармацевтические композиции могут быть получены с использованием стандартных фармацевтически приемлемых вспомогательных веществ и добавок с помощью общеизвестных методов. Фармацевтически приемлемые вспомогательные вещества и добавки включают нетоксичные совместимые наполнители, связующие вещества, разрыхлители, буферы, консерванты, антиоксиданты, смазывающие вещества, ароматизаторы, загустители, красители, эмульгатор и т.д.
[26] Согласно другому аспекту настоящего изобретения предложен способ получения бензамидиновых производных.
[27] Согласно другому аспекту настоящего изобретения, предложен способ получения соединения формулы (I) (как показано в Уравнении 1). Указанный способ применим для получения соединения формулы (I), в котором R1 и R2 представляют собой водород, R3 представляет собой нитро, R4 представляет собой этинил, пропинил или циано, например, как для получения Соединения I-1, Соединения I-8 и Соединения I-12.
[28]

[29] Согласно другому аспекту настоящего изобретения, предложен способ получения соединения формулы (I) с использованием Соединения IX в качестве исходного материала (как показано в Уравнении 2). Указанный способ применим для получения соединения формулы (I), в котором R1 представляет собой H, R2 представляет собой -COR5 или -CO2R6, R3 представляет собой нитро, R4 представляет собой этинил, пропинил или циано, R5 и R6 являются такими, как определено выше, например, для получения Соединения I-2, Соединения I-3, Соединения I-4, Соединения I-5, Соединения I-6, Соединения I-7, Соединения I-11 и Соединения I-13.

[30] Согласно другому аспекту настоящего изобретения, предложен способ получения соединения формулы (I) (как показано в Уравнении 3). Указанный способ применим для получения соединения формулы (I), в котором R1 или R2 представляет собой водород, замещенный или незамещенный (C1-C8)алкил, замещенный или незамещенный (C3-C8)алкенил, замещенный или незамещенный (C3-C8)алкинил, замещенный или незамещенный (C3-C7)циклоалкил; R1 и R2 также могут циклизоваться с образованием замещенного или незамещенного 4-, 5- или 6-членного кольца, но R1 и R2 не могут одновременно оба представлять собой водород; R3 представляет собой нитро, R4 представляет собой этинил, пропинил или циано, например, как для получения Соединения I-9, Соединения I-10, Соединения I-14, Соединения I-15, Соединения I-16, Соединения I-17, Соединения I-18, Соединения I-19, Соединения I-20, Соединения I-21, Соединения I-22, Соединения I-23, Соединения I-24, Соединения I-25, Соединения I-26 и Соединения I-27.

[31] Согласно другому аспекту настоящего изобретения, предложен способ получения соединения формулы (I) (как показано в Уравнении 4). Способ применим для соединения формулы (I), в котором R1 и R2 представляют собой водород, замещенный или незамещенный (C1-C8)алкил, замещенный или незамещенный (C3-C8)алкенил, замещенный или незамещенный (C3-C8)алкинил, замещенный или незамещенный (C3-C7)циклоалкил, -COR5 и -CO2R6; R1 и R2 также могут циклизоваться с образованием замещенного или незамещенного 4-, 5- или 6-членного кольца; R3 представляет собой нитрозо; R4 представляет собой этинил, пропинил или циано; R5 представляет собой замещенный или незамещенный (C1-C8)алкил, замещенный или незамещенный (C3-C8)алкенил, замещенный или незамещенный (C3-C8)алкинил, замещенный или незамещенный (C3-C7)циклоалкил, замещенный или незамещенный арил; R6 представляет собой замещенный или незамещенный (C1-C8)алкил, замещенный или незамещенный (C3-C8)алкенил, замещенный или незамещенный (C3-C8)алкинил, замещенный или незамещенный (C3-C7)циклоалкил, например, как для получения Соединения I-28.

[32] Согласно настоящему изобретению, также предложен способ получения соединения формулы (I), включающий образование соли с кислотой: смешивание соответствующим образом соединения формулы (I) согласно настоящему изобретению с соответствующей кислотой (такой как соляная кислота, серная кислота и т.д.) для получения соответствующей соли путем последующей обработки, такой как в случае получения Соединения I-29 и I-30.
[33] Некоторые из терминов, используемых в настоящей заявке, имеют следующие далее определения:
[34] Термин «галоген» относится к фтору, хлору, брому и йоду, предпочтительно, хлору, брому и йоду.
[35] «Алкил», как группа или часть группы, относится к прямой или разветвленной алифатической углеводородной группе. (C1-C8)алкил является предпочтительным. Примеры алкильных групп включают, но не ограничиваются ими, метил, этил, н-пропил, 2-пропил, н-бутил, изобутил, трет-бутил, гексил и т.д.
[36] «Алкенил», как группа или часть группы, относится к содержащей углерод-углеродную двойную связь алифатической углеводородной группе, которая может быть прямой или разветвленной. (C3-C8)алкенил является предпочтительным. Примеры алкенильных групп включают, но не ограничиваются ими, аллил, 2-бутенил и т.д.
[37] «Алкинил», как группа или часть группы, относится к содержащей углерод-углеродную тройную связь алифатической углеводородной группе, которая может быть прямой или разветвленной. (C3-C8)алкинил является предпочтительным. Примеры алкинильной группы включают, но не ограничиваются ими, пропаргил, 2-бутинил и т.д.
[38] Термин «гетероциклическая группа» относится к ароматической или неароматической гетероциклической группе, в которой один или более атомов кольца представляют собой гетероатомы, такие как кислород, азот, сера и т.д. Ароматическая гетероциклическая группа, т.е. группа, обычно называемая «гетероарильной», предпочтительно относится к ароматическому 5-6-членному моноциклическому кольцу или 9-10-членному бициклическому кольцу, которое может содержать 1, 2 или 3 атома, выбранных из группы, состоящей из атома азота, кислорода и/или серы, например, фурилу, пиридилу, 2-оксо-1,2-дигидропиридинилу, пиридазинилу, пиримидинилу, пиразинилу, тиенилу, изоксазолилу, оксазолилу, оксадиазолилу, имидазолилу, пирролилу, пиразолилу, триазолилу, тетразолилу, тиазолилу, изотиазолилу, 1,2,3-тиадиазолилу, бензодиоксолилу, бензимидазолилу, индолилу, изоиндолилу, 1,3-диоксоизоиндолилу, хинолилу, индазолу, бензоизотиазолилу, бензоксазолилу и бензоизоксазолилу. Предпочтительный гетероарил представляет собой пиридил. Гетероарил может содержать заместители, как описано для указанного выше термина «арил». Неароматическая гетероциклическая группа относится к неароматической гетероциклической группе, предпочтительно содержащей 5-6-членное моноциклическое кольцо или 8-10-членное бициклическое или трициклическое кольцо, которое может содержать 1, 2 или 3 атома, выбранных из группы, состоящей из атома азота, кислорода и/или серы, например, морфолинил, тиоморфолинил, тетрагидропиранил, 1,1-диоксотиоморфолинил, пиперидил, 2-оксопиперидинил, пирролидинил, 2-оксопирролидинил, пиперазин-2-он, 8-окса-3-азабицикло[3,2,1]октил и пиперазинил. Гетероциклическая группа может необязательно содержать заместители, как описано для указанного выше термина «арил».
[39] Кроме того, термин «фармацевтически приемлемая соль» относится к конкретным солям указанных выше соединений, которые сохраняют исходную биоактивность и являются подходящими для медицинского применения. Фармацевтически приемлемая соль соединения, представленного формулой (I), может представлять собой соль металла, соль амина, образованную с подходящей кислотой, где указанная соль металла предпочтительно выбрана из соли щелочного металла, соли щелочно-земельного металла, и подходящая кислота включает неорганические кислоты и органические кислоты, такие как уксусная кислота, бензолсульфоновая кислота, бензойная кислота, камфорсульфоновая кислота, лимонная кислота, этансульфоновая кислота, фумаровая кислота, глюконовая кислота, глутаминовая кислота, бромоводородная кислота, соляная кислота, изэтиновая кислота, молочная кислота, яблочная кислота, малеиновая кислота, миндальная кислота, метансульфоновая кислота, азотная кислота, фосфорная кислота, сукциновая кислота, серная кислота, винная кислота, n-толуолсульфоновая кислота и т.д. Соляная кислота, бромоводородная кислота, фосфорная кислота и серная кислота являются особо предпочтительными, и гидрохлорид является наиболее предпочтительным.
[40] «Циклоалкил» относится к насыщенному или частично ненасыщенному моноциклическому, конденсированному или спиро-углеродному кольцу. Кольцо, образованное из 3-7 атомов углерода, является предпочтительным. Примеры включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил и т.д.
[41] «Алкокси» относится к группе «алкил-O-», где указанный алкил является таким, как указано в соответствующем определении в настоящей заявке. (C1-C6)алкокси является предпочтительным. Примеры указанной группы включают, но не ограничиваются ими, метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси и т.д.
[42] Термин «арил», при использовании отдельно или в комбинации, относится к карбоциклической ароматической системе, содержащей одно или два кольца, где указанные кольца могут быть соединены путем конденсации. Термин «арил» включает ароматические группы, такие как фенил, нафтил и тетрагидронафтил. Предпочтительный арил представляет собой (C6-C10)арил, более предпочтительно, арил представляет собой фенил. «Арил» может содержать один или более заместителей, таких как (C1-C6)алкил, гидрокси, галоген, галогеналкил, нитро, циано, (C1-C6)алкокси, (C1-C6)алкиламино и т.д.
[43] Ранее мы обнаружили, что соединения согласно настоящему изобретению являются высоко активными ингибиторами опухолевого роста.
[44] Настоящее изобретение также проиллюстрировано с помощью следующих примеров. Указанные примеры описывают получение типичных соединений формулы (I) и относящиеся к ним данные для структурной идентификации. Необходимо отметить, что следующие примеры приведены для иллюстрации настоящего изобретения и не ограничивают настоящее изобретение.
[45] В следующих примерах, если специально не указано иное, все температуры представляют собой температуры по Цельсию и, если специально не указано иное, различные исходные материалы и реагенты получены из коммерческих источников. Коммерчески доступные исходные материалы и реагенты непосредственно использовали без дополнительной очистки, если специально не указано иное.
[46] Стеклянную посуду сушили с использованием печи и/или путем нагревания. За реакциями следили с использованием стеклянных пластин, заполненных силикагелем 60 F254 (0,25 мм) (ТСХ). Аналитическую тонкослойную хроматографию осуществляли при подходящем отношении растворителей (по объему). Реакция завершалась, когда исходные материалы по результатам ТСХ исчезали.
[47] Спектры1H ЯМР измеряли и получали с использованием прибора Bruker (400 МГц), химические сдвиги выражали в частях на миллион (ppm). В качестве внутреннего стандарта использовали тетраметилсилан (0,00 ppm). Описание1H ЯМР: s = синглет, d = дуплет, t = триплет, m = мультиплет, br = уширенный, dd = дуплет дуплетов, dt = дуплет триплетов. Константы взаимодействия, если представлены, выражены в Герцах, Гц.
[48] Масс-спектры измерены и получены с использованием прибора для ЖХ/МС. Режим ионизации представлял собой ионизацию электрораспылением (ИЭР) или химическую ионизацию при атмосферном давлении (APCI).
[49] Все температуры плавления не скорректированы.
[50] Следующие примеры приведены исключительно для иллюстрации способов синтеза конкретных соединений согласно настоящему изобретению и никоим образом не являются лимитирующими. Соединения, которые не перечислены ниже, также могут быть получены с использованием таких же путей и способов синтеза, как описанные ниже, путем выбора подходящих исходных материалов и, при необходимости, внесения незначительных и подходящих обоснованных здравым смыслом изменений в условия реакции.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
Пример 1: Получение 4-йод-3-нитробензойной кислоты (Соединение V):

[51] В реакционную колбу добавляли 45 г (0,25 моль) 4-амино-3-нитробензойной кислоты, 400 мл воды и 100 мл концентрированной соляной кислоты. Начинали перемешивать и охлаждали смесь до температуры от 0 до 5°C, затем добавляли по каплям 50 мл водного раствора 25,9 г нитрита натрия (0,38 моль). Твердое вещество постепенно растворялось. После завершения добавления по каплям смеси позволяли реагировать при температуре от 0 до 5°C в течение 1 часа и добавляли по каплям 200 мл водного раствора 88 г (0,5 моль) йодида калия при указанной температуре. Смесь перемешивали при комнатной температуре в течение 2 часов после завершения указанного добавления по каплям и осаждали твердое вещество. Указанное твердое вещество фильтровали, промывали водой и сушили для получения 4-йод-3-нитробензойной кислоты (соединения V) в виде твердого вещества, 65 г (0,22 моль), выход 89,7%.
Пример 2: Получение метил-4-йод-3-нитробензоата (Соединение IV):

[52] В реакционную колбу добавляли 55 г (0,19 моль) 4-йод-3-нитробензойной кислоты (Соединение V), 16,5 г (0,12 моль) карбоната калия и 550 мл ацетонитрила и начинали перемешивать. Смесь охлаждали до температуры от 0 до 5°C и добавляли 52,9 мл (0,38 моль) тритиламина. Температуру поддерживали ниже 10°C и добавляли 71 мл (1,12 моль) йодметана. Смесь нагревали до температуры примерно 40°C в течение 8 часов и концентрировали при пониженном давлении после завершения реакции до выпаривания большей части ацетонитрила. Смесь затем экстрагировали 500 мл этилацетата, промывали водой три раза и органический слой сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали до получения его минимального количества, затем добавляли 300 мл петролейного эфира, перемешивали в течение 30 минут и затем фильтровали. Отфильтрованный осадок промывали петролейным эфиром и сушили для получения метил-4-йод-3-нитробензоата (Соединения IV), примерно 41,2 г (0,13 моль), выход 71,5%.1Н-ЯМР (400 МГц, ДМСО-d6): δ 3,91 (s, 3Н), 7,87 (dd, 1Н, J1=8,08 Гц, J2=1,88 Гц), 8,26 (d, 1Н, J=8,12 Гц), 8,34 (d, 1Н, J=9,12 Гц); МС (m/z): 308 [М+Н].
Пример 3: Получение метил-4-(2-триметилсилил)этинил-3-нитробензоата (соединения III):

[53] 25 г (0,08 моль) метил-4-йод-3-нитробензоата (соединение IV), 20 мл (0,14 моль) триэтиламина и 200 мл тетрагидрофурана добавляли в реакционную колбу под защитой слоя газообразного азота и перемешивали для растворения, затем добавляли 2,4 г (0,0034 моль) дихлорида бис(трифенилфосфин)палладия, 0,65 г (0,0034 моль) йодида меди и 13,7 мл (0,096 моль) триметилсилилэтина, смеси позволяли реагировать при комнатной температуре в течение 1 часа, концентрировали при пониженном давлении для выпаривания большей части тетрагидрофурана. После экстрагирования 300 мл этилацетата органический слой промывали водой три раза, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали до высыхания и отделяли с помощью колоночной хроматографии для получения метил-4-(2-(триметилсилил))этинил-3-нитробензоата (Соединения III), 15,1 г (0,055 моль), выход 66,9%.1Н-ЯМР (400 МГц, ДМСО-d6): δ 0,09 (s, 9Н), 3,74 (s, 3Н), 7,69 (d, 1Н, J=8,08 Гц), 8,00 (dd, 1Н, J1=8,08 Гц, J2=1,52 Гц), 8,30 (d, 1Н, J=l,36 Гц); МС (m/z): 278 [М+Н].
Пример 4: Получение метил-4-этинил-3-нитробензоата (Соединения II-1):

[54] 15 г (0,054 моль) метил-4-(2-триметилсилил)этинил-3-нитробензоата (Соединения III) и 225 мл тетрагидрофурана добавляли в реакционную колбу и начинали перемешивать и охлаждали до температуры от -20 до -25°C, затем добавляли по каплям 35 мл раствора 7,1 г (0,027 моль) фторида тетрабутиламмония в тетрагидрофуране и позволяли реакции продолжаться в течение 20 минут после завершения добавления по каплям. После завершения реакции, за которой наблюдали с помощью ТСХ, значение pH доводили до 4~5 путем добавления 0,5 М соляной кислоты. Смесь концентрировали при пониженном давлении для выпаривания большей части тетрагидрофурана и затем экстрагировали 300 мл этилацетата, органический слой промывали водой три раза, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали до высыхания, для получения примерно 10,5 г неочищенного продукта, который отделяли с помощью колоночной хроматографии для получения метил-4-этинил-3-нитробензоата (соединения II-1), 7,3 г (0,036 моль), выход 65,8%.1Н-ЯМР (400 МГц, ДМСО-d6): δ 3,93 (s, 3Н), 5,02 (s, 1Н), 7,93 (d, 1Н, J=8,08 Гц), 8,21 (dd, 1Н, J1=8,08 Гц, J2=1,64 Гц), 8,50 (d, 1Н, J=1,48 Гц); МС (m/z): 206 [М+Н]
Пример 5: Получение 4-этинил-3-нитробензамида (Соединение I-1):

[55] В реакционную колбу добавляли 7,3 г (0,036 моль) метил-4-этинил-3-нитробензоата (Соединения II-1), 300 мл метанола и 200 мл тетрагидрофурана, перемешивали и через полученную смесь пропускали газообразный аммоний в течение 1 часа. Затем смеси позволяли реагировать при комнатной температуре в течение 24 часов. После завершения реакции, за которой наблюдали с помощью ТСХ, смесь концентрировали при пониженном давлении для выпаривания большей части тетрагидрофурана и метанола, затем экстрагировали 300 мл этилацетата. Органический слой промывали водой три раза, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали до высыхания и отделяли с помощью колоночной хроматографии для получения 4-этинил-3-нитробензамида (соединения I-1), 3,6 г (0,0019 моль), выход 53,2%.1Н-ЯМР (400 МГц, ДМСО-d6): δ 4,93 (s, 1Н), 7,82 (s, 1Н), 7,92 (d, 1Н, J=8,08 Гц), 8,21 (dd, 1Н, J1=8,04 Гц, J2=1,68 Гц), 8,37 (s, 1Н), 8,57 (d, 1Н, J=1,56 Гц); МС (m/z): 191 [М+Н].
Пример 6: Получение N-метоксикарбонил-4-этинил-3-нитробензамида (Соединения I-2)
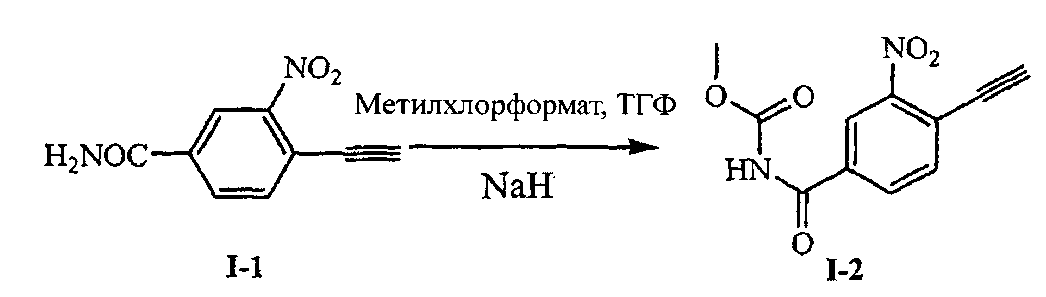
[56] В четырехгорлую реакционную колбу на 100 мл добавляли 3 г (0,016 моль) 4-этинил-3-нитробензамида (Соединения 1-1), 45 мл тетрагидрофурана и 6 мл метилхлорформата, перемешивали для растворения и охлаждали до -10°C и добавляли 7,5 г гидрида натрия и поддерживали при температуре примерно 0°C в течение 30 минут. После завершения реакции реакционную смесь выливали на колотый лед и pH доводили до кислых значений с помощью соляной кислоты, затем экстрагировали 200 мл этилацетата. Органический слой промывали водой три раза и сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали до высыхания для получения примерно 3,5 г неочищенного продукта, который отделяли с помощью колоночной хроматографии для получения N-метоксикарбонил-4-этинил-3-нитробензоатамида (Соединения I-2), 1,6 г (0,0065 моль), выход 40,8%.1Н-ЯМР (400 МГц, ДМСО-d6): δ 3,71 (s, 3Н), 4,98 (s, 1Н), 7,91 (d, 1Н, J=8,08 Гц), 8,18 (dd, 1Н, J1=8,08 Гц, J2=1,802 Гц), 8,58 (d, 1Н, J=1,72 Гц) 11,36 (s, 1Н); МС (m/z): 249 [М+Н].
[57] Следующие соединения получали согласно способу Примера 6 путем выбора подходящих реагентов, с использованием Соединения I-1 в качестве исходного материала.




Пример 12: Получение метил-4-циано-3-нитробензоата (Соединения II-8):

[58] В реакционную колбу добавляли 5 г (0,016 моль) метил-4-йод-3-нитробензоата (Соединения IV-1), 1,77 г (0,02 моль) цианида меди и 15 мл гексаметилфосфорного триамида и нагревали до 100°C и позволяли реагировать в течение 1 часа. После завершения реакции смесь охлаждали до комнатной температуры, затем экстрагировали 100 мл этилацетата и органический слой промывали водой три раза, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали до высыхания, отделяли с помощью колоночной хроматографии для получения метил-4-циано-3-нитробензоата (Соединение II-8), 2,7 г (0,013 моль), выход 80,4%.1Н-ЯМР (400 МГц, ДМСО-d6): δ 3,96 (s, 3Н), 8,34 (d, 1Н, J=7,9 Гц), 8,42 (d, 1Н, J=7,7 Гц), 8,68 (s, 1Н); МС (m/z): 229 [М+Н].
Пример 13: Получение 4-циано-3-нитробензамида (Соединение I-8):

[59] В реакционную колбу добавляли 2 г (9,71 ммоль) Метил-4-циано-3-нитробензоата (Соединение II-8) и 60 мл метанола, перемешивали и через реакционную смесь пропускали газообразный аммиак в течение 1 часа, затем позволяли реагировать в течение 8 часов при комнатной температуре. После завершения реакции смесь концентрировали при пониженном давлении для выпаривания большей части метанола. Затем смесь экстрагировали 100 мл этилацетата и органический слой промывали водой три раза, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали до высыхания для получения 1,1 г (5,76 ммоль) неочищенного продукта, который перекристаллизовывали из этилацетат-н-гексана для получения 4-циано-3-нитробензамида (соединения I-8), 0,6 г, выход 32,6%.1Н-ЯМР (400 МГц, ДМСО-d6): δ 7,97 (s, 1Н), 8,32 (d, 1Н, J=7,96 Гц), 8,39 (dd, 1Н, J1=7,96 Гц, J2=1,56 Гц), 8,53 (s, 1Н), 8,78 (d, 1Н, J=1,52 Гц); МС (m/z): 190 [М+Н].
Пример 14: Получение N-метил-4-йод-3-нитробензамида (Соединения VII-1):
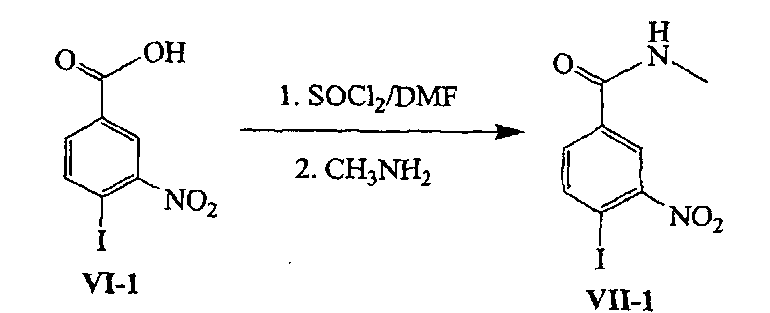
[60] В реакционную колбу добавляли 10 г (0,034 моль) 4-йод-3-нитробензойной кислоты (Соединение VI-1) и 50 мл N,N-диметилформамида, перемешивали для растворения и охлаждали до температуры ниже 10°C и добавляли 7,5 мл (0,10 моль) сульфоксидхлорида. После завершения указанного добавления по каплям смесь нагревали до комнатной температуры и позволяли реагировать в течение одного часа. Реакционную смесь выливали в 200 мл водного раствора 30% метиламина при низкой температуре, перемешивали в течение 5 минут для осаждения твердого вещества и затем смесь добавляли к 500 мл смеси лед/вода, перемешивали в течение 10 минут, фильтровали. Твердое вещество промывали водой и сушили для получения N-метил-4-йод-3-нитробензамида (Соединение VII-1), 6,5 г (0,021 моль), выход 62,3%.1Н-ЯМР (400 МГц, ДМСО-d6): δ 2,81 (d, 3Н, J=4,1 Гц), 7,84 (d, 1Н, J=7,8 Гц), 8,23 (d, 1Н, J=8,1 Гц), 8,33 (s, 1Н), 8,75 (s, 1Н); МС (m/z): 307 [М+Н].
Пример 15: Получение N-метил-4-(2-триметилсилил)этинил-3-нитробензамида (Соединения VIII-1)

[61] В четырехгорлую колбу на 100 мл добавляли 6 г (0,02 моль) 1 N-метил-4-йод-3-нитробензамида (Соединения VII-1), 60 мл тетрагидрофурана и 4,2 мл триэтиламина, перемешивали для растворения и добавляли 0,44 г (0,63 ммоль) дихлорида бис(трифенилфосфин)палладия, 0,24 г (1,26 ммоль) йодида меди и 6 мл (0,042 моль) триметилсилилацетилена и позволяли реагировать при комнатной температуре в течение 1 часа. Большую часть тетрагидрофурана выпаривали посредством концентрирования при пониженном давлении и смесь экстрагировали путем добавления 200 мл этилацетата, затем органический слой промывали водой три раза, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали до высыхания, разделяли с помощью колоночной хроматографии для получения 3,8 г (0,014 моль) N-метил-4-(2-триметилсилил)этинил-3-нитробензамида (Соединение VIII-1) в виде твердого вещества, выход 70,2%.1H-ЯМР (400 МГц, ДМСО-d6): δ 0,26 (s, 9Н), 2,81 (d, 3Н, J=4,5 Гц), 7,87 (d, 1Н, J=8,1 Гц), 8,14 (dd, 1Н, J1=8,1 Гц, J2=1,6 Гц), 8,52 (d, 1Н, J=1,5 Гц), 8,83 (d, 1 Н, J=4,4 Гц); МС (m/z): 277 [М+Н].
Пример 16: Получение N-метил-4-этинил-3-нитробензамида (Соединения 1-9)

[62] В реакционную колбу на 100 мл добавляли 3,3 г (0,012 моль) N-метил-4-(2-триметилсилил)этинил-3-нитробензамида (Соединения VIII-1) и 20 мл метанола, перемешивали для растворения и добавляли 0,53 г (0,0056 моль) дигидрата фторида калия и позволяли реагировать в течение 30 минут. После завершения реакции в смесь добавляли по каплям 60 мл воды, фильтровали и сушили для получения 2,2 г твердого вещества, которое перекристаллизовывали из этилацетат-н-гексана для получения N-метил-4-этинил-3-нитробензамида (Соединения I-9), 1,5 г (0,0074 моль), выход 61,7%.1Н-ЯМР (400 МГц, ДМСО-d6): δ 2,82 (d, 3Н, J=4,56 Гц), 4,92 (s, 1Н), 7,92 (d, 1Н, J=8,08 Гц), 8,17 (dd, 1Н, J1=8,08 Гц, J2=1,76 Гц), 8,54 (d, 1Н, J=1,64 Гц), 8,86 (d, 1Н, J=4,32 Гц); МС (m/z): 203 [М+Н].
Пример 17: Получение N,N-диметил-4-этинил-3-нитробензамида (Соединения 1-10)

[63] Соединение 1-10 готовили согласно способам Примера 14, Примера 15 и Примера 16, с использованием 4-йод-3-нитробензойной кислоты (Соединения VI-1) и диметиламина в качестве исходных материалов:1Н-ЯМР (400 МГц, CDCl3): δ 3,02 (s, 3Н), 3,14 (s, 3Н), 3,62 (s, 1Н), 7,67 (dd, 1Н, J1=7,96 Гц, J2=1,64 Гц), 7,74 (d, 1Н, J=7,92 Гц), 8,12 (d, 1Н, J=1,56 Гц); МС (m/z): 219 [М+Н].
Пример 18: Получение N-ацетил-4-этинил-3-нитробензамида (Соединения I-11)

[64] В реакционную колбу добавляли 4 г (0,021 моль) 4-этинил-3-нитробензамида (Соединения I-1), 30 мл (0,320 моль) уксусного ангидрида и 20 мл уксусной кислоты, перемешивали и нагревали до 120°C в течение 15 часов. После завершения реакции, за которой наблюдали с помощью ТСХ, смесь экстрагировали 300 мл этилацетата и органический слой промывали водой три раза, сушили над безводным сульфатом натрия, фильтровали и фильтрат концентрировали до высыхания, разделяли с помощью колоночной хроматографии для получения N-ацетил-4-этинил-3-нитробензамида (Соединения I-11), 1,1 г (0,0047 моль), выход 22,6%.1Н-ЯМР (400 МГц, ДМСО-d6): δ 2,36 (s, 3Н), 4,99 (s, 1Н), 7,94 (d, 3Н, J=8,08 Гц), 8,21 (dd, 1Н, J1=8,12 Гц, J2=1,80 Гц), 8,60 (d, 1Н, J=1,72 Гц), 11,32 (s, 1Н); МС (m/z): 231 [М+Н].
Пример 19: Получение 4-(1-пропинил)-3-нитробензамида (Соединения 1-12)

[65] Соединение I-12 готовили согласно способам Примера 3, Примера 4 и Примера 5, с использованием метил-4-йод-3-нитробензоата (Соединения IV) и триметилпропаргилсилана в качестве исходных материалов:1Н-ЯМР (400 МГц, ДМСО-d6): δ 2,16 (s, 3Н), 7,74 (s, 1Н), 7,79 (d, 3Н, J=8,16 Гц), 8,14 (dd, 1Н, J1=8,13 Гц, J2=1,77 Гц), 8,30 (s, 1Н), 8,50 (d, 1Н, J=1,71 Гц); МС (m/z): 203 [М+Н].
Пример 20: Получение N-бензоил-4-этинил-3-нитробензамида (Соединения I-13)

[66] Соединение I-13 готовили согласно способу Примера 6, с использованием Соединения I-1 и бензоилхлорида в качестве исходных материалов:1Н-ЯМР (400 МГц, ДМСО-d6): δ 4,89 (s, 1Н), 7,54 (t, 1Н, J=7,4 Гц), 7,91-7,96 (m, 3Н), 8,20 (dd, 1Н, J1=8,1 Гц, J2=1,6 Гц), 8,58 (d, 1Н, J=1,5 Гц), 11,52 (s, 1Н); МС (m/z): 293 [М+Н].
[67] Соединение I-14, Соединение I-15, Соединение I-16, Соединение I-17, Соединение I-18, Соединение I-19, Соединение I-20, Соединение I-21, Соединение I-22, Соединение I-23, Соединение I-24 и Соединение I-25 получали последовательно согласно способам Примера 14, Примера 15 и Примера 16, путем выбора соответствующих аминов с использованием триметилсилилацетилена и с использованием 4-йод-3-нитробензойной кислоты (Соединение VI-1) в качестве исходного материала; Соединение I-26 готовили последовательно согласно способам Примера 15 и Примера 16, с использованием N-метил-4-йод-3-нитробензамида (Соединение VII-1) и триметилпропаргилсилана в качестве исходных материалов:













Пример 34: Получение N-метил-4-циано-3-нитробензамида (Соединения I-27)

[68] В реакционную колбу добавляли 2 г (6,53 ммоль) N-метил-4-йод-3-нитробензамида (Соединения VII-1), 0,09 г (10,0 ммоль) цианида меди и 10 мл гексаметилфосфорного триамида. Смесь нагревали до 100°C и позволяли реагировать в течение 40 минут и после завершения реакции смесь охлаждали до комнатной температуры и затем экстрагировали этилацетатом. Органический слой промывали водой три раза, сушили над безводным сульфатом натрия, обесцвечивали с помощью активированного древесного угля, фильтровали и фильтрат концентрировали до высыхания, перекристаллизовывали из этилацетата для получения 0,74 г N-метил-4-циано-3-нитробензамида (Соединения 1-27), выход 55,2%.1Н-ЯМР (400 МГц, ДМСО-d6): δ 2,84 (d, 3Н, J=4,6 Гц), 8,27 (d, 1Н, J=8,0 Гц), 8,32 (dd, 1Н, J1=8,0 Гц, J2=1,5 Гц), 8,72 (d, 1Н, J=1,4 Гц), 8,90 (m, 1Н); MC (m/z): 204 [М+Н].
Пример 35: Получение 4-этинил-3-нитрозобензамида (Соединения I-28)

[69] Этап 1: В четырехгорлую колбу на 150 мл добавляли 5 г (26,3 ммоль) 4-этинил-3-нитробензамида (Соединение XII-1) и 75 мл тетрагидрофурана, механически перемешивали для растворения и добавляли 10 мл водного раствора 10 г (186,9 ммоль) хлорида аммония, затем порциями добавляли 10 г (178,5 ммоль) железного порошка и позволяли реагировать при комнатной температуре в течение 4 часов до завершения реакции. Смесь фильтровали и отфильтрованный осадок промывали тетрагидрофураном. Фильтрат экстрагировали этилацетатом и органическую фазу промывали водой, сушили над безводным сульфатом натрия, обесцвечивали с помощью активированного древесного угля, фильтровали и концентрировали для осаждения твердого вещества, которое фильтровали и сушили для получения 4-этинил-3-аминобензамида (Соединение XI-1, твердое вещество серого цвета), 2,4 г, выход 59,4%.1Н-ЯМР (400 МГц, ДМСО-d6): δ 4,55 (s, 1Н), 7,25 (dd, 1Н, J1=8,0 Гц, J2=1,6 Гц), 7,31 (d, 1Н, J=7,9 Гц), 7,35 (s, 1Н), 7,61 (d, 1Н, J=1,4 Гц), 7,97 (s, 1Н), 8,21 (s, 1Н), 8,65 (s, 1Н); МС (m/z): 161 [М+Н].
[70] Этап 2: В одногорлую колбу на 50 мл добавляли 0,46 г (2,87 ммоль) 4-этинил-3-аминобензамида (Соединения XI-1), 10 мл воды, затем добавляли 1,2 г 58% серной кислоты и затем 0,92 г 18,5% раствора дихромата калия. Смесь перемешивали в течение 3 минут до завершения реакции, экстрагировали этилацетатом, сушили над безводным сульфатом натрия и разделяли с помощью колоночной хроматографии для получения 0,06 г 4-этинил-3-нитрозобензамида (Соединение I-28, твердое вещество желто-зеленого цвета), выход 12%, хранили при -20°C.1Н-ЯМР (400 МГц, ДМСО-d6): δ 4,97 (s, 1Н), 6,99 (d, 1Н, J=1,3 Гц), 7,70 (s, 1Н), 8,09 (d, 1Н, J=8,0 Гц), 8,27 (s, 1Н), 8,34 (dd, 1Н, J1=8,0 Гц, J2=1,6 Гц); МС (m/z): 219 [М+Н].
Пример 36: Получение гидрохлорида (4-этинил-3-нитрофенил)(4-метилпиперазин-1-ил)кетона (Соединения I-29)

[71] В одногорлую колбу на 50 мл добавляли 0,75 г (2,74 ммоль) (4-этинил-3-нитрофенил)(4-метилпиперазин-1-ил)кетона (Соединения I-24) и 7,5 мл безводного метанола, перемешивали с помощью магнитной мешалки для растворения. pH смеси доводили до значения pH=2 путем добавления концентрированной соляной кислоты. Твердое вещество белого цвета осаждали при охлаждении с помощью ледяной воды и фильтровали. Отфильтрованный осадок промывали безводным этанолом, сушили для получения 0,46 г гидрохлорида (4-этинил-3-нитрофенил)(4-метилпиперазин-1-ил)кетона (Соединение I-29), выход: 54,1%.1Н-ЯМР (400 МГц, ДМСО-d6): δ 2,76 (s, 3Н), 3,16 (br s, 4Н), 3,66 (br s, 4Н), 4,91 (s, 1Н), 7,85 2 (dd, 1Н, J1=8,0 Гц, J2=1,4 Гц), 7,88 (d, 1Н, J=8,0 Гц), 8,21 (d, 1Н, J=1,3 Гц); МС (m/z): 274 [М+Н-HCl].
Пример 37: Получение гидрохлорида (4-этинил-3-нитрофенил)(пиперазин-1-ил)кетона (Соединения I-30)

[72] Гидрохлорид (4-этинил-3-нитрофенил)(пиперазин-1-ил)кетона (Соединение I-30) готовили согласно способу Примера 36, с использованием (4-этинил-3-нитрофенил)(пиперазин-1-ил)кетона (Соединения I-23) в качестве исходного материала.1Н-ЯМР (400 МГц, D2O): δ 3,32 (br s, 2Н), 3,45 (br s, 2H), 4,03 (br s, 2H), 4,11 (s, 1H), 7,80 (dd, 1H, J1=8,0 Гц, J2=1,6 Гц), 7,91 (d, 1H, J=8,0 Гц), 8,24 (d, 1Н, J=1,4 Гц); МС (m/z): 260 [М+Н-HCl].
[73] Фармакодинамический скрининг соединений согласно настоящему изобретению осуществляли следующим образом:
I. Фармакодинамический скрининг in vitro
1. Клеточная культура
[74] Линии клеток рака толстой кишки человека (Colo205 и НСТ-116), линии клеток рака молочной железы человека (MCF-7 и MDA-MB435), линии клеток рака легкого человека (А549, 95D и NCI460), линии клеток рака яичников человека (OVCAR-5, OVCAR-8, НО8910 и SKOV-3), линии клеток лейкоза человека (HL-60 и K562), рака предстательной железы человека (DU145 и ВХРС-3), рака матки человека (Hela), рака поджелудочной железы человека (PANC-1), гепатомы человека (HepG 2) получали из Американской коллекции типовых культур, АТСС. Клетки Colo205, HL-60, K562 культивировали в среде RPMI 1640, содержащей 2 мМ/л глютамина, 10% ЭБС, 1,0 мМ пируват натрия. Клетки НСТ-116, MCF-7, А549, 95D, НО8910, NCI460, OVCAR-5, OVCAR-8, DU145, ВХРС-3, Hela, PANC-1, HepG2 культивировали в среде DMEM, содержащей 2 мМ/л глютамина, 10% ЭБС. Клетки MDA-MB435 культивировали в среде L-15, содержащей 2 мМ/л глютамина, 10% ЭБС. Клетки SKOV-3 культивировали в среде McCoy, содержащей 2 мМ/л глютамина, 10% ЭБС. Клетки Colo205, HL-60 и K562 высевали на 96-луночные планшеты в объеме 150 мкл/лунку и концентрации 8000 клеток/лунку и указанные 96-луночные планшеты предварительно инкубировали в инкубаторе при 37°C, 5% CO2, 100% относительной влажности в течение 24 часов. Клетки НСТ-116, MCF-7, NCI460, OVCAR-5, OVCAR-8, Hela, HepG 2 и MDA-MB435 высевали в 96-луночные планшеты в концентрации 5000 клеток/лунку; клетки А549, 95D, НО8910, DU145, ВХРС-3, PANC-1 и SKOV-3 высевали в 96-луночные планшеты в концентрации 10000 клеток/лунку и указанные 96-луночные планшеты предварительно инкубировали в инкубаторе при 37°C, 5% CO2, 100% относительной влажности в течение 24 часов, для возможности прикрепления клеток.
2. Скрининг соединений
[75] 50 мкл предварительно охлажденного 50% (масса/объем) ТСА добавляли в контрольные лунки, являющиеся точкой отсчета, для каждой клеточной линии для фиксации клеток. В другие лунки добавляли по 50 мкл соединений в различных концентрациях и инкубировали в течение 48 часов, при этом каждую концентрацию лекарственного средства использовали в трех повторностях. Имелись пустые контрольные лунки (культуральная среда без клеток), контрольные лунки, не содержащие лекарственное средство (такой же объем полной питательной среды без лекарственных средств) и лунки, содержащие лекарственное средство BSI-201, являющееся положительным контролем, и планшеты инкубировали в инкубаторе при 37°C, 5% CO2, абсолютной влажности (100% относительной влажности) в течение 48 часов.
3. Выявление клеток
[76] 50 мкл предварительно охлажденного 50% (масса/объем) ТСА добавляли на поверхность культуральной среды для фиксации клеток, которые затем инкубировали при 4°C в течение 1 часа. Супернатант удаляли и каждую лунку промывали деионозированной водой 5 раз для удаления ТСА, белков сыворотки и т.д. После высушивания на воздухе в каждую лунку добавляли примерно 100 мкл состава, содержащего достаточное количество 0,4% SRB (с 1% уксусной кислотой) добавляли в каждую лунку и инкубировали при комнатной температуре в течение 20-30 мин. Жидкость из каждой лунки удаляли и указанные лунки быстро промывали 1% уксусной кислотой пять раз для удаления не связавшегося красителя до его полной отмывки. После высушивания на воздухе до исчезновения видимой жидкости в каждую лунку добавляли по 200 мкл основания Трис, для растворения полученную смесь перемешивали на шейкере для планшетов в течение 5 мин. или смешивали путем пипетирования с помощью наконечника для пипетки, затем анализировали на многофункциональном аппарате (М5 Detection System, MD Group Ltd.) при длине волны детектирования 515 нм, к нулю подводили с использованием пустой контрольной лунки при длине волны 690 нм.
[77] Кривую зависимости эффекта от дозы строили с помощью программы XL-fit для определения значения GI50.
4. Результаты скринингового анализа
[78] Результаты скрининговых анализов эффективности in vitro с использованием BSI-201 (Iniparib) в качестве лекарственного средства, являющегося положительным контролем, показали, что соединения I-1, I-2, I-3, I-4, I-5, I-6, I-7, I-8, I-9, I-10, I-11, I-12, I-13, I-14, I-15, I-16, I-17, I-18, I-19, I-20, I-21, I-22, I-23, I-24, I-25, I-26, I-27, I-28, I-29 и I-30 обладают ингибирующим пролиферацию раковых клеток действием, причем эффективность Соединения I-1 в 10 раз выше по сравнению с эффективностью лекарственного средства BSI-201, являющегося положительным контролем, для 18 исследуемых линий раковых клеток; эффективность соединений I-2, I-3, I-14, I-15, I-17, I-20, I-21, I-23, I-24, I-29 и I-30 немного ниже по сравнению с эффективностью соединений 1-1; соединения I-4, I-5, I-6, I-7, I-8, I-9, I-10, I-11, I-12, I-13, I-16, I-18, I-19, I-25, I-27 и I-28 также обладают хорошим ингибирующим пролиферацию раковых клеток действием. Результаты скринингового анализа показаны в Таблице 1.
II. Фармакодинамический скрининг in vivo
1. Установление животной модели
[79] Выращивали 5-6-недельных самок или самцов «голых» мышей линии BALB/C с массой тела примерно 18-20 г. Модель ксенотрансплантата раковой опухоли человека у «голых» мышей устанавливали с использованием следующих этапов: линии клеток рака толстой кишки человека НСТ-116 и Colo205, линию клеток рака молочной железы человека MDA-MB435, линию клеток рака предстательной железы человека ВХРС-3, линию клеток рака яичников человека SKOV-3, линию клеток рака легкого человека А549, линию клеток рака поджелудочной железы человека PANC-1 и линию клеток рака матки человека Hela получали из АТСС. Каждую клеточную линию культивировали и монослой указанных культивируемых опухолевых клеток снимали с помощью фермента со стенок культуральной посуды, собирали, ресуспендировали в бессывороточной культуральной среде и доводили до концентрации 2×106 клеток/0,2 мл, и затем приносили в виварий в холодильной камере. 0,2 мл клеточной суспензию набирали непосредственно с помощью шприца с иглой 6 калибра и трансплантировали подкожно в лопаточную часть задней области левой подмышки «голых» мышей, по 2×106 клеток/0,2 мл для каждой мыши, и объем опухоли измеряли каждые 2-3 дня. Через 2 недели опухоль удаляли в стерильных условиях из тела содержащих опухоль «голых» мышей, у которых наблюдался интенсивный опухолевый рост и отсутствие аррозии. Опухолевую ткань разрезали на кусочки размером примерно 2~3 мм и вводили подкожно в лопаточную часть задней области левой подмышки «голых» мышей. Через три пассажа, когда объем опухоли достигал 100 мм3, «голых» мышей, содержащих слишком большие или слишком маленькие опухоли, удаляли из эксперимента и оставшихся животных делили случайным образом на группы для введения.
2. Скрининг соединений
[80] Мышей делили случайным образом на 5 групп, включая группу отрицательного контроля (растворитель), группу положительного контроля (BSI-201, 80 мг/кг) и три лечебные группы, получающие высокую, среднюю и низкую дозы (40 мг/кг, 30 мг/кг и 20 мг/кг, соответственно, где высокая доза была ниже MTD). В каждой группе было по 8 «голых» мышей, за исключением того, что в группе отрицательного контроля было 16 животных. Животным вводили дозы путем внутрибрюшинной инъекции один раз в день в течение 3 следующих друг за другом недель, в течение которых определяли массу тела животных и объем опухоли указанных животных; число умерших животных фиксировали каждые 2 дня. Животных умерщвляли через 24 часа после последнего введения и измеряли объем опухоли, массу опухоли и массу тела «голых» мышей; строили кривые роста объема опухоли, кривые роста массы тела «голых» мышей и скорости подавления опухоли и смертности животных и рассчитывали относительную скорость пролиферации опухоли Т/С (%) согласно уравнению Т/С (%)=TRTV/CRTV*100%. (TRTV: RTV лечебной группы; CRTV: RTV группы отрицательного контроля, относительный объем опухоли RTV=Vt/V0, где V0 представляет собой объем опухоли на момент разделения мышей на группы для введения, Vt представляет собой объем опухоли после введения).
3. Результаты скрининга
[81] Результаты экспериментов по фармакодинамическому скринингу на предмет эффективности in vivo с использованием BSI-201 (Iniparib) в качестве лекарственного средства, являющегося положительным контролем, показали, что доза 30 мг/кг Соединения I-1 обладает эффективностью, эквивалентной эффективности дозы 80 мг/кг BSI-201, и оказывает значительное фармакодинамическое действие. Результаты скрининга показаны в Таблице 2.
[82] Все литературные источники, упоминаемые в настоящем изобретении, приведены в виде ссылок, как если бы каждый литературный источник был отдельно упомянут в виде ссылки. Также необходимо понимать, что возможно осуществление различных изменений или модификации настоящего изобретения специалистом в данной области техники после его ознакомления с вышеприведенным содержанием настоящего изобретения, и указанные эквиваленты также находятся в рамках настоящего изобретения, как определено согласно прилагаемой формуле изобретения.
Реферат
Изобретение относится к соединению, представленному формулой (I), или его фармацевтически приемлемой соли, где Rи Rнезависимо выбраны из группы, состоящей из водорода, незамещенного (C-C)алкила, -CORи -COR; Rи Rтакже могут циклизоваться с образованием замещенного или незамещенного 4-, 5- или 6-членного кольца, выбранного из морфолина, пиперидина, пирролидина, пиперазина, азетидина, 4-метилпиперазина; Rпредставляет собой нитро или нитрозо; Rвыбран из группы, состоящей из этинила, пропинила или циано; Rвыбран из группы, состоящей из незамещенного (C-C)алкила или незамещенного арила; Rпредставляет собой незамещенный (C-C)алкил. Также изобретение относится к способам получения соединения формулы (I) (вариантам). Соединения формулы (I) предназначены для изготовления фармацевтической композиции или лекарственного средства для лечения заболевания, связанного с ингибитором поли(АДФ-рибоза)-полимеразы (PARP), или для лечения рака, содержащей эффективную дозу соединения формулы (I) или его фармацевтически приемлемой соли. Технический результат - производные бензамида, предназначенные для получения лекарственного средства для лечения заболевания, связанного с ингибитором поли(АДФ-рибоза)-полимеразы (PARP), или рака. 9 н. и 11 з.п. ф-лы, 2 табл., 37 пр.
Формула

или его фармацевтически приемлемая соль,
где
R1 и R2 независимо выбраны из группы, состоящей из водорода, незамещенного (C1-C8)алкила, -COR5 и -CO2R6; R1 и R2 также могут циклизоваться с образованием замещенного или незамещенного 4-, 5- или 6-членного кольца, выбранного из морфолина, пиперидина, пирролидина, пиперазина, азетидина, 4-метилпиперазина;
R3 представляет собой нитро или нитрозо;
R4 выбран из группы, состоящей из этинила, пропинила или циано;
R5 выбран из группы, состоящей из незамещенного (C1-C8)алкила или незамещенного арила;
R6 представляет собой незамещенный (C1-C8)алкил.
4-этинил-3-нитробензамида (I-1),
N-метоксикарбонил-4-этинил-3-нитробензамида (I-2),
N-этоксикарбонил-4-этинил-3-нитробензамида (I-3),
N-пропоксикарбонил-4-этинил-3-нитробензамида (I-4),
N-бутоксикарбонил-4-этинил-3-нитробензамида (I-5),
N-изопропоксикарбонил-4-этинил-3-нитробензамида (I-6),
N-изобутоксикарбонил-4-этинил-3-нитробензамида (I-7),
4-циано-3-нитробензамида (I-8),
N-метил-4-этинил-3-нитробензамида (I-9),
N,N-диметил-4-этинил-3-нитробензамида (I-10),
N-ацетил-4-этинил-3-нитробензамида (I-11),
4-(1-пропинил)-3-нитробензамида (I-12),
N-бензоил-4-этинил-3-нитробензамида (I-13),
N,N-диэтил-4-этинил-3-нитробензамида (I-14),
N,N-дипропил-4-этинил-3-нитробензамида (I-15),
N,N-дибутил-4-этинил-3-нитробензамида (I-16),
N-этил-4-этинил-3-нитробензамида (I-17),
N-пропил-4-этинил-3-нитробензамида (I-18),
N-бутил-4-этинил-3-нитробензамида (I-19),
(4-этинил-3-нитрофенил)(пирролидин-1-ил)кетона (I-20),
(4-этинил-3-нитрофенил)(пиперидин-1-ил)кетона (I-21),
(4-этинил-3-нитрофенил)(морфолин-4-ил)кетона (I-22),
(4-этинил-3-нитрофенил)(пиперазин-1-ил)кетона (I-23),
(4-этинил-3-нитрофенил)(4-метилпиперазин-1-ил)кетона (I-24),
(4-этинил-3-нитрофенил)(азетидинон-1-ил)кетона (I-25),
N-метил-4-(1-пропинил)-3-нитробензамида (I-26),
N-метил-4-циано-3-нитробензамида (I-27),
4-этинил-3-нитрозобензамида (I-28),
гидрохлорида (4-этинил-3-нитрофенил)(4-метилпиперазин-1-ил)кетона (I-29), и
гидрохлорида (4-этинил-3-нитрофенил)(пиперазин-1-ил)кетона (I-30).

где R1 и R2 представляют собой водород, R3 представляет собой нитро, и R4представляет собой этинил, пропинил или циано.

где R1 представляет собой водород, R2 представляет собой -COR5 или -CO2R6, R3 представляет собой нитро, R4 представляет собой этинил, пропинил или циано, R5 и R6 являются такими, как определено в п.1, X представляет собой галоген.

где R1 и R2 независимо выбраны из группы, состоящей из водорода, незамещенного (С1-С8)алкила, и R1 и R2 также могут циклизоваться с образованием замещенного или незамещенного 4-, 5- или 6-членного кольца, выбранного из морфолина, пиперидина, пирролидина, пиперазина, азетидина, 4-метилпиперазина, но R1, R2 не могут оба одновременно представлять собой водород; R3 представляет собой нитро, и R4 представляет собой этинил или пропинил.

где R1 и R2 независимо выбраны из группы, состоящей из водорода, незамещенного (С1-С8)алкила, и R1 и R2 также могут циклизоваться с образованием замещенного или незамещенного 4-, 5- или 6-членного кольца, выбранного из морфолина, пиперидина, пирролидина, пиперазина, азетидина, 4-метилпиперазина, но R1, R2 не могут оба одновременно представлять собой водород; R3 представляет собой нитро, и R4представляет собой циано.

где R1 и R2 независимо выбраны из группы, состоящей из водорода, незамещенного (С1-С8)алкила, -COR5 и -CO2R6; R1 и R2 также могут циклизоваться с образованием замещенного или незамещенного 4-, 5- или 6-членного кольца, выбранного из морфолина, пиперидина, пирролидина, пиперазина, азетидина, 4-метилпиперазина; R3 представляет собой нитрозо; R4 представляет собой этинил, пропинил или циано; R5 представляет собой незамещенный (С1-С8)алкил или незамещенный арил; R6 представляет собой незамещенный (С1-С8)алкил.
Комментарии