Новые соединения как потенциальные индукторы терминальной дифференциации клеток опухоли и фармацевтическая композиция на их основе - RU2128643C1
Код документа: RU2128643C1
Чертежи







Описание
Раковая опухоль представляет собой заболевание, при котором совокупность клеток становится в различной степени невосприимчивой к механизмам контроля, которые при нормальном состоянии управляют быстрым размножением и дифференцировкой. В течение многих лет существовали две основные стратегии химотерапевтического лечения рака: а) блокировка гормонально- зависимого быстрого размножения клеток опухоли путем вмешательства с получением или периферическим действием половых гормонов; б) уничтожение раковых клеток непосредственно при воздействии на них цитотоксических веществ, которые повреждают как опухолевые, так и нормальные совокупности клеток.
Относительно недавно сделаны попытки лечения рака индуцированием терминальной дифференцировки клеток опухоли (Sporn, М.В., Roberts, А.В., and Driscoli, J.S. (1985) in Cancer; Principles and Practice of Oncology, eds. Hellman, S., Rosenberg, S.A., and DeVita, V. Т., Jr., Ed. 2, (J.B. Lippincott, Philadelphia), P. 49). В моделях культуры клеток дифференцировка объяснялась воздействием на клетки различных раздражителей, в том числе циклического АМФ (АМР) и ретеноевой кислоты (Breitman, T.R., Selonick, S.E., and Collins, S. J. (1980) Proc. Natl. Acad. Sci. USA 77: 2936-2940; Olsson, I.L. and Breitman, T. R. (1982) Cancer Res. 42: 3924-3927), акларубицина и других антрациклинов (Schwartz, E.L. and Sartorelli, A.C. (1982) Cancer Res. 42: 2651-2655).
Существует большое число доказательств того, что опухолевое перерождение необязательно разрушает способность раковых клеток к дифференцировке (Sporn, М. В. , Roberts, A. B. and Driscoll, J.S. (1985) in Cancer; Principles and Practice of Oncology, eds. Hellman, S., Rosenberg, S.A. and DeVita, V. Т., Jr. , Ed. 2, (J.B. Lippincott, Philadelphia), P. 49, Marks, P.A., Sheffery, M. and Rifkind, R. A. (1987) Cancer Res. 47: 659; Sachs, L. (1978) Nature (Lond.) 274: 535). Также существует большое число клеток опухоли, которые не реагируют на обычные регуляторы размножения и, как оказывается, блокируются при проявлении программы их дифференцировки, но все еще могут быть подвержены дифференцировке и прекращению воспроизведения. Различные агенты, в том числе ряд относительно простых полярных соединений (Marks, P.A., Sheffery, M. and Rifkind, R.A. (1987) Cancer Res. 47: 659; Friend, С., Scher, W., Holland, J. W. and Sato, T. (1971) Proc. Natl. Acad. Sci. (USA) 68: 378-382; Tanaka, M., Levy, J., Terada, M., Breslow, R., Rifkind, R.A. and Marks, P.A. (1975) Proc. Natl. Acad. Sci. (USA) 72: 1003-1006; Reuben, R.C., Wife, R.L., Breslow, R. , Rifkind, R.A. and Marks, P.A. (1976) Proc. Natl. Acad. Sci. (USA) 73: 862-866), производные витамина D и ретеноевой кислоты (Abe, Е., Miyaura, С., Sakagami, Н., Takeda, М., Коnnо, К., Yamazaki, Т., Yoshika, S. and Suda, Т. (1981) Proc. Natl. Acad. Sci. (USA) 78: 4990-4994; Schwartz, E. L. , Snoddy, J.R., Kreutter, D., Rasmussen, H. and Sartorelli, A.C. (1983) Proc. Am. Assoc. Cancer Res. 24: 18; Tanenaga, К., Hozumi, M. and Sakagami, Y. (1980) Cancer Res. 40: 914-919), стероидные гормоны (Lotem, J. and Sachs, L. (1975) Int. J. Cancer 15: 731-740), ростовые факторы (Sachs, L. (1978) Nature (Lond. ) 274: 535; Metcalf, D. (1985) Science, 229: 16-22), протеазы (Scher, W. , Scher, B.M. and Waxman, S. (1983) Exp. Hematol. 11: 490-498; Scher, W., Scher, B.M. and Waxman, S. (1982) Biochem. & Biophys. Res. Comm. 109: 348-354), опухолевые стимуляторы (Huberman, E. and Callaham, M.F. (1979) Proc. Natl. Acad. Scl. (USA) 76: 1293-1297; Lottem, J. and Sachs, L. (1979) Proc. Natl. Acad. Sci. (USA) 76: 5158-5162), и ингибиторы синтеза ДНК или РНК (Schwartz, E.L. and Sartorelli, А.С. (1982) Cancer Res. 42: 2651-2655; Terada, M., Epner, E.,Nudel, U., Salmon, J., Fibach, E., Rifkind, R.A. and Marks, P.A. (1978) Proc. Natl. Acad. Sci. (USA) 75: 2795-2799; Morin, M. J. and Sartoreili, A.C. (1984) Cancer Res. 44; 2807-2812; Schwartz, E.L., Brown, B.J., Nierenberg, M., Marsh, J.C. and Sartoreili, A.C. (1983) Cancer Res. 43; 2725-2730; Sugano, H.,Furusawa, M., Kawaguchi, T. and lkawa, Y. (1973) Bibl. Hematol. 39: 943-954; Ebert, P.S., Wars, I. and Buell, D.N. (1976) Cancer Res. 36: 1809-1813; Hayashi, M., Okabe, J. and Hozumi, M. (1979) Gann 70: 235-238), могут заставить различные линии трансформированных клеток и первичных эксплантантов опухоли человека проявлять более дифференцирующие характеристики.
Более ранние исследования заявителей выявили серию полярных соединений, которые были эффективными индукторами дифференцировки ряда линий переродившихся клеток (Tanaka, M., Levy, J., Terada, M., Breslow, R., Rifkind, R. A. and Marks, P.A. (1975) Proc. Natl. Acad. Sci. (USA) 72: 1003-1006; Reuben, R. C. , Wife, R.L., Breslow, R., Rifkind, R.A. and Marks, P.A. (1976) Proc. Natl. Acad. Scl. (USA) 73: 862-866). Среди этих соединений наиболее эффективным индуктором был гибрид полярного/неполярного соединения N,N-гексаметиленбис (ацетамид) (ГМБА)(Reuben, R.C. Wife, R.L., Breslow, R., Rifkind, R. A. and Marks, P.A. (1976) Proc. Natl, Acad. Sci. (USA) 73: 862-866). Использование этого полярного/неполярного соединения для индуцирования эритроидной дифференцировки клеток эритролейкоза мышей (ЭЛКМ) с подавлением онкогенности подтвердило полезность модели для изучения вызываемой индуктором дифференцировки переродившихся клеток (Marks, P.A., Sheffery, М. and Rifkind, R.A. (1987) Cancer Res. 47: 659; Friend, С., Scher, W., Holland, J.W. and Sato, T. (1971) Proc. Natl. Acad. Sci. (USA) 68: 378-382; Tanaka, M., Levy, J. , Terada, M. , Breslow, R., Rifkind, R.A. and Marks, P.A. (1975) Proc. Natl. Acad. Sci. (USA) 72: 1003-1006; Reuben, R.C., Wife, R.L., Breslow, R. , Rifkind, R.A. and Marks, P.A. (1976) Proc. Natl. Acad. Sci. (USA) 73: 862-866). Индуцируемая с помощью ГМБА терминальная эритроидная дифференцировка ЭЛКМ представляет собой многостадийный процесс. При добавлении ГМБА к ДКЭМ (745A-DS19) в культуре существует латентный период от 10 до 12 часов прежде чем обнаруживается коммитирование к терминальной дифференцировке. Коммитирование определяется как способность клеток к проявлению терминальной дифференцировки несмотря на удаление индуктора (Fibach, Е., Reuben, R. C. , Rifkind, R.A. and Marks, P.A. (1977) Cancer Res. 37: 440-444). При непрерывном воздействии ГМБА существует прогрессивный рекрутинг клеток для дифференцировки. Заявители сообщали, что клетки линий ЭЛКМ, резистентные к относительно низкому содержанию винкристина, становятся более чувствительными к индуцирующему действию ГМБА и их можно привести к дифференцировке при небольшом латентном периоде или без него (Melloni, Е., Pontremoli, S., Damiani, G. , Viotti, P., Weich, N., Rifkind, R.A. and Marks, P.A. (1988) Proc. Natl. Acad. Sci (USA) 85; 3835-3839).
ГМБА обладает способностью индуцировать фенотипичные изменения, согласующиеся с дифференцировкой в большом числе линий клеток (Marks, P.A., Sheffery, М. and Rifkind, R.A. (1987) Cancer Res. 47: 659). Характеристики индуцирующего эффект лекарства наиболее интенсивно изучались на эритролейкозе клеточной системы мышей (ЭЛКМ) (Marks, P.A., Sheffery, М. and Rifkind, R.A. (1987) Cancer Res. 47: 659; Fibach, E., Reuben, R.C., Rifkind, R.A. and Marks, P.A. (1977) Cancer Res. 37; 440-444; Reuben, R., Khanna, P.L., Gazitt, Y. , Breslow, R., Rifkind, R.A. and Marks, P.A. (1978) J. Biol. Chem. 253: 4214-4218; Marks, P.A. and Rifkind, R.A. (1988) International Journal of Cell Cloning 6: 230-240). Индуцирование дифференцировки ЭЛКМ зависит как от времени, так и от концентрации. Минимальная концентрация, которая необходима для того, чтобы показать эффект in vivo в большинстве линий, составляет от 2 до 3 мМ; минимальная продолжительность непрерывного воздействия, которая необходима в большинстве случаев для индуцирования дифференцировки в значительной части (более 20%) популяции без продолжения воздействия лекарства, составляет приблизительно 36 ч.
Первичная мишень действия ГМБА неизвестна. Существует доказательство того, что белок-киназа С вступает в вызываемую индуктором дифференцировку (Melloni, Е. , Pontremoli, S. , Michetti, М., Sacco, О., Cakiroglu, A.G>, Jackson, J. F., Rifkind, R.A. and Marks, P.A. (1987) Proc. Natl. Acad. Sci. (USA) 84; 5282-5286). Исследования in vivo создали основу для оценки потенциала ГМБА в качестве цитодифференцирующего агента при лечении раковых заболеваний человека (Marks, P.А. and Rifkind R.A. (1984) Cancer 54: 2766-2769). Проведен ряд клинических испытаний I фазы с использованием ГМБА (Egorin, M.J., Sigman, L.M., VanEcho, D.A., Forrest, A., Whitacre, M.Y. and Aisner, J. (1987) Cancer Res. 47: 617-623; Rowinsky, E.W., Ettinger, D.S., Grochow, L. B. , Brundrett, R.B., Cates, A.E. and Donehower, R.C. (1986) J. Clin. Oncol. 4: 1835-1844; Rowinsky, E.L., Ettinger, D.S., McGuire, W.P., Noe, D. A., Grochow, L.B. and Donehower, R.C. (1987) Cancer Res. 47: 5788-5795; Callery, P.S., Egorin, M.J., Geelhaar, L.A. and Nayer, M.S.B. (1986) Cancer Res. 46: 4900-4903; Young, C.W., Fanucchi, M.P. Walsh, T.B., Blatzer, L. , Yaldaie, S. , Stevens, Y.W., Gordon, C., Tong, W., Rifkind, R.A. and Marks, P. A. (1988) Cancer Res. 48: 7304-7309, Andreeff, М., Young, C., Clarkson, B. , Fetten, J., Rifkind, R.A. and Marks, P.A. (1988) Blood 72: 186a). Клинические испытания показали, что это соединение может вызвать терапевтический ответ у пациентов, имеющих раковую опухоль (Young, C.W., Fanucchi, М. Р. , Walsh, Т. В., Blatzer, L., Yaldaie, S., Stevens, Y.W., Gordon, C. , Tong, W., Rifkind, R.A. and Marks, P.A. (1988) Cancer Res. 48; 7304-7309; Andreeff, M., Young, C., Clarkson, B., Fetten, J., Rifkind, R.A. and Marks, P.A. (1988) Blood 72: 186a). Однако, эти клинические испытания I фазы также показали, что потенциальная эффективность ГМБА частично ограниченf зависимой от дозы токсичностью, которая не позволяет достигнуть оптимального содержания в крови, и необходимостью внутривенного введения больших количеств агента в течение длительного периода.
Недавно заявители сообщили о ряде соединений, родственных ГМБА, с полярными группами, отделенными неполярными мостиками, которые на молекулярной основе являются такими же активными (Marks, P.A., Breslow, R., Rifkind, R.A. , Ngo, L. and Singh, R. (1989) Proc. Natl. Acad. Sci. (USA) 86: 6358-6362) или в 100 раз более активными, чем ГМБА (Breslow, R., Jursic, В., Yan, Z.F., Friedman, E., Leng, L., Ngo, L., Rifkind, R.A. and Marks, P.A. (1991) Proc. Natl. Acad. Sci. (USA) 88: 5542-5546). Однако было установлено, что симметричные димеры, такие как ГМБА и родственные соединения, не являются самыми хорошими цитодифференцирующими агентами.
Неожидано было установлено, что наиболее эффективные соединения содержат две полярные концевые группы, разделенные гибкой цепочкой метиленовых групп, где одна или более полярных концевых групп представляет собой большую гидрофобную группу. Предпочтительно, чтобы полярные концевые группы отличались друг от друга и только одна из них представляла собой большую гидрофобную группу. Эти соединения являются в тысячу раз более активными, чем ГМБА и в десять раз более активными, чем родственные ГМБА соединения.
Этот новый класс соединений настоящего изобретения может быть использован для селективного индуцирования концевой дифференцировки опухолевых клеток и, следовательно, могут быть использованы для лечения рака у больных.
Краткое описание изобретения
Настоящее изобретение предлагает соединения, имеющие строение
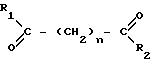
где каждый из заместителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения; если заместители R1 и R2 являются одинаковыми, каждый из них представляет собой замещенный или незамещенный ариламино, циклоалкиламино, пиридиламино, пиперидино, 9-пурин-6-амино или тиазолиламиногруппу; если заместители R1 и R2 различны, то R1 представляет собой группу R3-N-R4, где каждый из заместителей R3 и R4 независимо друг от друга принимают одинаковые или разные значения и представляют собой атом водорода, гидроксильную группу, замещенный или незамещенный, линейный или разветвленный алкил, алкенил, циклоалкил, арил, алкилокси-, арилокси-, арилалкилокси- или пиридильную группу, или заместители R3 и R4 вместе образуют пиперидиновую группу, а заместитель R2 представляет собой гидроксиламино-, гидроксильную, амино-, алкиламино-, диалкиламино- или алкоксильную группу;
n принимает целые значения от приблизительно 4 до приблизительно 8.
Настоящее изобретение предлагает также соединения, имеющие структуру
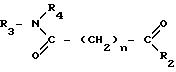
где каждый из заместителей R3 и R4 независимо друг от друга принимают одинаковые или разные значения и представляют собой атом водорода, гидроксильную группу, замещенный или незамещенный, линейный или разветвленный алкил, алкенил, циклоалкил, арил, алкилокси-, арилокси-, арилалкилокси- или пиридильную группу, или заместители R3 и R4 вместе образуют пиперидиновую группу;
заместитель R2 представляет собой гидроксиламино-, гидроксильную, амино-, алкиламино-, диалкиламино- или алкоксильную группу;
n принимает целые значения от приблизительно 4 до приблизительно 8.
Настоящее изобретение предлагает
также описанное выше соединение, имеющее структуру

где заместитель R представляет собой замещенный или незамещенный ариламино, циклоалкиламино, пиридиламино, пиперидино, 9-пурин-6-амино или тиазолиламиногруппу;
n принимает целые значения от приблизительно 4 до приблизительно 8.
Настоящее изобретение предлагает также соединения, имеющие структуру

где каждый из заместителей X и Y независимо друг от друга принимают одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламиногруппу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу;
заместитель R представляет собой атом водорода, гидроксильную группу, замещенный или незамещенный алкил, арил, алкилокси- или арилоксигруппу;
каждая из величин m и n независимо друг от друга принимает одинаковые или различные целые значения от приблизительно 0 до приблизительно 8.
Настоящее изобретение далее предлагает соединение, имеющее структуру

где каждый из заместителей X и Y независимо друг от друга принимают одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламиногруппу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу;
каждый из заместителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения и представляют собой атом водорода, гидроксильную группу, замещенный или незамещенный алкил, арил, алкилокси- или арилоксигруппу;
каждая из величин m, n и о независимо друг от друга принимает одинаковые или различные целые значения от приблизительно 0 до приблизительно 8.
Настоящее изобретение дополнительно предлагает соединение, имеющее структуру

где каждый из заместителей X и Y независимо друг от друга принимают одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламиногруппу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу;
каждый из заместителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения и представляют собой атом водорода, гидроксильную группу, замещенный или незамещенный алкил, арил, алкилокси- или арилоксигруппу;
каждая из величин m и n независимо друг от друга принимает одинаковые или различные целые значения от приблизительно 0 до приблизительно 8.
Настоящее изобретение также предлагает соединение, имеющее структуру
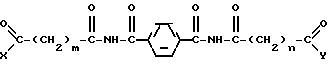
где каждый из заместителей X и Y независимо друг от друга принимают одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламиногруппу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу;
каждая из величин m и n независимо друг от друга принимает одинаковые или различные целые значения от приблизительно 0 до приблизительно 8.
Настоящее
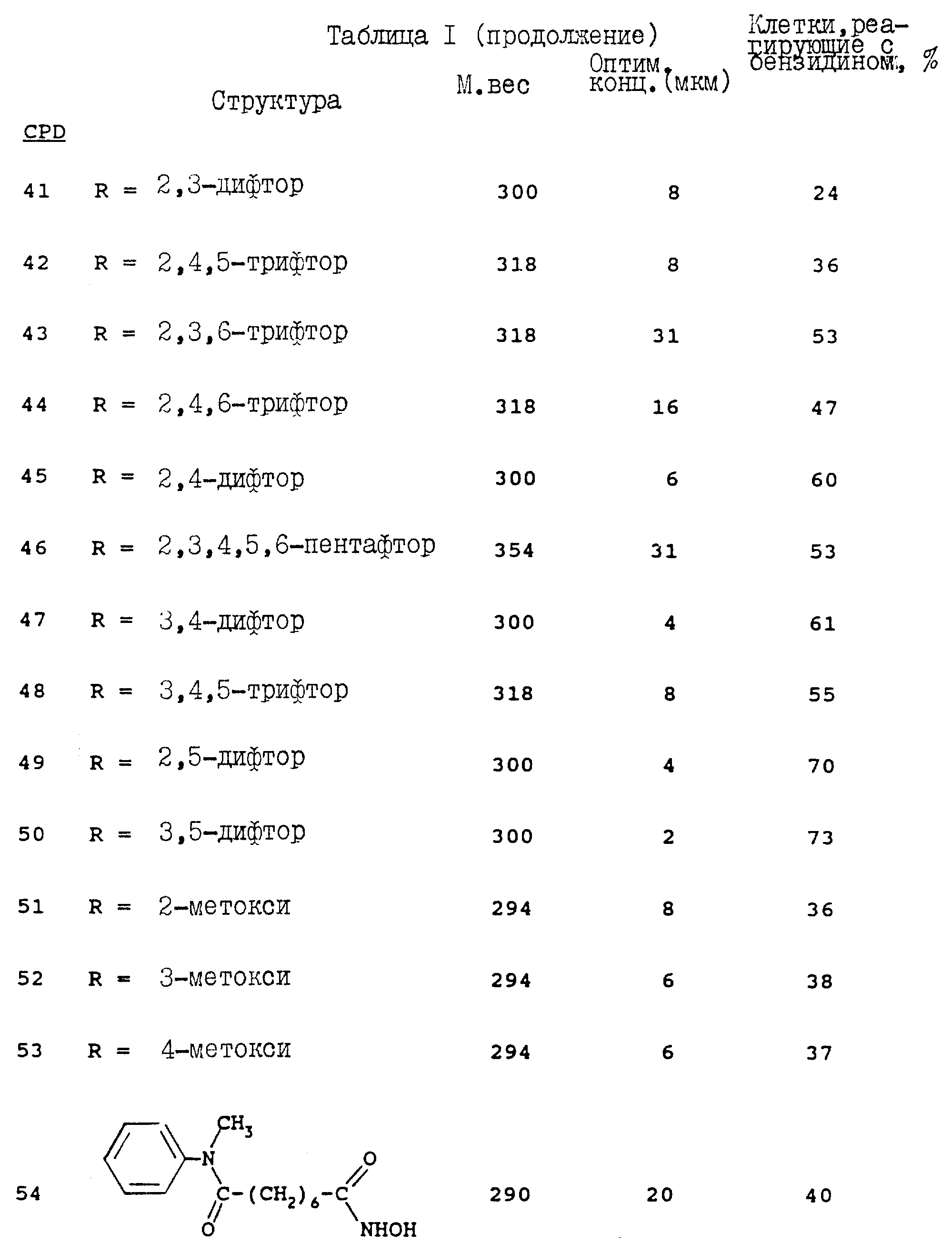
изобретение также предлагает соединение, имеющее структуру

где каждый из заместителей X и Y независимо друг от друга принимают одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламиногруппу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино-, или арилоксиалкиламиногруппу;
каждый из заместителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения и представляют собой атом водорода, гидроксильную группу, замещенный или незамещенный алкил, арил, алкилокси- или арилоксигруппу;
каждая из величин m и n независимо друг от друга принимает одинаковые или различные целые значения от приблизительно 0 до приблизительно 8.
Настоящее изобретение далее предлагает соединение, имеющее
структуру

где каждый из заместителей X и Y независимо друг от друга принимают одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламиногруппу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу;
величина n принимает целые значения от приблизительно 0 до приблизительно 8.
Настоящее изобретение
дополнительно предлагает соединение, имеющее структуру
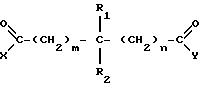
где каждый из заместителей X и Y независимо друг от друга принимают одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламиногруппу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу;
каждый из заместителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения и представляют собой атом водорода, гидроксильную группу, замещенный или незамещенный алкил, арил, алкилокси-, арилокси-, карбонилгидроксиаминогруппу или атом фтора;
каждая из величин m и n независимо друг от друга принимает одинаковые или различные целые значения от приблизительно 0 до приблизительно 8.
Настоящее изобретение далее
предлагает соединение, имеющее структуру

где каждый из заместителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения и представляют собой гидроксильную, алкокси-, амино-, гидроксиламино-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу.
Настоящее изобретение также предлагает соединение, имеющее структуру

где каждый из заместителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения и представляют собой гидроксильную, алкокси-, амино-, гидроксиламино-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу.
Настоящее изобретение дополнительно предлагает соединение, имеющее структуру

где каждый из заместителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения и представляют собой гидроксильную, алкокси-, амино-, гидроксиламино-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу.
Кроме того, настоящее изобретение также предлагает способ селективного индуцирования терминальной дифференцировки опухолевых клеток и посредством этого ингибирования быстрого размножения таких клеток, который включает контактирование клеток при приемлемых условиях с эффективным количеством описанных выше соединений, которые эффективны для селективного индуцирования терминальной дифференцировки.
Настоящее изобретение также обеспечивает способ лечения пациента, имеющего опухоль, которая характеризуется быстрым размножением опухолевых клеток, который включает назначение пациенту эффективного количества любого описанного выше соединения, которое эффективно при селективном индуцировании терминальной дифференцировки таких опухолевых клеток и посредством этого ингибируют их быстрое размножение.
Наконец, настоящее изобретение предлагает фармацевтическую композицию, содержащую фармацевтически приемлемый носитель и терапевтически приемлемое количество любого из описанных выше соединений.
Настоящее изобретение предлагает соединение, имеющее строение

где каждый из заместителей R1 и R2 независимо друг от друга принимает одинаковые или разные значения; если заместители R1 и R2 являются одинаковыми, каждый из них представляет собой замещенный или незамещенный ариламино, циклоалкиламино, пиридиламино, пиперидино, 9-пурин-6-амино или тиазолиламиногруппу; если заместители R1 и R2 различны, то R1 представляет собой группу R3-N-R4, где каждый из заместителей R3 и R4 независимо друг от друга принимают одинаковые или разные значения и представляют собой атом водорода, гидроксильную группу, замещенный или незамещенный, линейный или разветвленный алкил, алкенил, циклоалкил, арил, алкилокси-, арилокси-, арилалкилокси- или пиридильную группу, или заместители R3 и R4 вместе образуют пиперидиновую группу, а заместитель R2 представляет собой гидроксиламино-, гидроксильную, амино-, алкиламино-, диалкиламино- или алкоксильную группу;
n принимает целые значения от приблизительно 4 до приблизительно 8.
Настоящее изобретение предлагает
также соединения, имеющие структуру

где каждый из заместителей R3 и R4 независимо друг от друга принимает одинаковые или разные значения и представляют собой атом водорода, гидроксильную группу, замещенный или незамещенный, линейный или разветвленный алкил, алкенил, циклоалкил, арил, алкилокси-, арилокси-, арилалкилокси- или пиридильную группу, или заместители R3 и R4 вместе образуют пиперидиновую группу;
заместитель R2 представляет собой гидроксиламино-, гидроксильную, амино-, алкиламино-, диалкиламино- или алкоксильную группу;
n принимает целые значения от приблизительно 4 до приблизительно 8.
В предпочтительном воплощении вышеуказанного соединения заместитель R2 представляет собой гидроксиламино-, гидроксильную, амино-, метиламино, диметиламино- или метоксигруппу, n принимает значение 6. Наиболее предпочтительно заместитель R4 представляет собой атом водорода, а заместитель R3 - замещенную или незамещенную фенильную группу.
Фенильная группа может быть замещена метильной, циано-, нитро-, трифторметильной, амино-, аминокарбонильной, цианометильной группой, атомами хлора, фтора, брома, йода, 2,3-дифтор, 2,4-дифтор, 2, 5-дифтор, 3,4-дифтор, 3,5-дифтор, 2,6-дифтор, 1,2,3-трифтор, 2,3,6-трифтор, 2,4,6-трифтор, 3,4,5-трифтор, 2,3,5,6-тетрафтор, 2,3,4,5,6-пентафтор, азидо-группой, гексильной, трет.-бутильной, фенильной, карбоксильной, гидроксильной, метокси-, бензилокси-, фениламиноокси-, фенилметокси-, фениламинокарбонил-, метоксикарбонил-, метиламинокарбонил-, диметиламино-, диметиламинокарбонил- или гидроксиламинокарбонильной группой.
В других предпочтительных воплощених вышеуказанного соединения заместитель R4 представляет собой атом водорода, а заместитель R3 - циклогексильную группу; заместитель R4 представляет собой атом водорода, а заместитель R3 - метоксигруппу; заместители R3 и R4 вместе образуют пиперидиновую группу; заместитель R4 представляет собой атом водорода, а заместитель R3 - гидроксигруппу; заместитель R4 представляет собой атом водорода, а заместитель R3 - бензилоксигруппу; заместитель R4 представляет собой атом водорода, а заместитель R3 - γ-пиридильную группу; заместитель R4 представляет собой атом водорода, а заместитель R3 - α-пиридильную группу; оба заместителя R3 и R4 представляют собой метильные группы; или заместитель R4 представляет собой метильную группу, а заместитель R3 - фенильную группу.
Настоящее изобретение предлагает также соединение, имеющее структуру
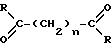
где заместитель R представляет собой замещенный или незамещенный ариламино, циклоалкиламино, пиридиламино, пиперидино, 9-пурин-6-амино или тиазолиламино-группу;
n принимает целые значения от приблизительно 4 до приблизительно 8.
В предпочтительном воплощении вышеуказанного соединения заместитель R представляет собой замещенную или незамещенную фениламиногруппу. Фениламиногруппа может быть замещена циано-, цианометильной-, нитро-, карбоксильной, аминокарбонильной, метиламинокарбонильной, диметиламинокарбонильной, трифторметильной, гидроксиламинокарбонильной, N-гидроксиламинокарбонильной, метоксикарбонильной группой, атомами хлора, фтора, метильной, метоксигруппой, группами 2,3-дифтор, 2,4-дифтор, 2,5-дифтор, 2,6-дифтор, 3,5-дифтор, 2,6-дифтор, 2,3,6-трифтор, 1,2,3-трифтор, 3,4,5-трифтор, 2,3,4,5-тетрафтор или 2,3,4,5,6-пентафтор.
В другом воплощении вышеуказанного соединения заместитель R представляет собой циклогексиламиногруппу.
Настоящее изобретение предлагает также соединение, имеющее структуру

где каждый из заместителей X и Y независимо друг от друга принимает одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламиногруппу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино, ариламино-, алкилариламино-, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу;
заместитель R представляет собой атом водорода, гидроксильную группу, замещенный или незамещенный алкил, арил, алкилокси- или арилокси-группу;
каждая из величин m и n независимо друг от друга принимает одинаковые или различные целые значения от приблизительно 0 до приблизительно 8.
В предпочтительном воплощении вышеуказанного соединения заместители X, Y и R представляют собой гидроксильные группы, а каждая из величин m и n принимает значение 5.
Настоящее изобретение также предлагает
соединение, имеющее структуру

где каждый из заместителей X и Y независимо друг от друга принимает одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламино-группу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино-, ариламино-, алкилариламино-, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу;
каждый из заместителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения и представляют собой атом водорода, гидроксильную группу, замещенный или незамещенный алкил, арил, алкилокси- или арилоксигруппу;
каждая из величин m, n и о независимо друг от друга принимает одинаковые или различные целые значения от приблизительно 0 до приблизительно 8.
В предпочтительном воплощении вышеуказанного соединения заместители X и Y представляют собой гидроксильные группы, а каждый из заместителей R1 и R2 представляет собой метильную группу. Наиболее предпочтительно, чтобы величины n и o принимали значение 6, а m принимало значение 2.
Настоящее изобретение также предлагает соединение, имеющее структуру

где каждый из заместителей X и Y независимо друг от друга принимает одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламиногруппу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу;
каждый из заместителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения и представляют собой атом водорода, гидроксильную группу, замещенный или незамещенный алкил, арил, алкилокси- или арилоксигруппу;
каждая из величин m и n независимо друг от друга принимает одинаковые или различные целые значения от приблизительно 0 до приблизительно 8.
Настоящее изобретение также предлагает соединение, имеющее структуру

где каждый из заместителей X и Y независимо друг от друга принимает одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламиногруппу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу;
каждая из величин m и n независимо друг от друга принимает одинаковые или различные целые значения от приблизительно 0 до приблизительно 8.
В предпочтительном воплощении вышеуказанного соединения каждый из заместителей X и Y представляет собой гидроксильную группу, а каждая из величин n и m принимает значение 5.
Настоящее изобретение также предлагает соединение, имеющее структуру

где каждый из заместителей X и Y независимо друг от друга принимает одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламиногруппу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу;
каждый из заместителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения и представляют собой атом водорода, гидроксильную группу, замещенный или незамещенный алкил, арил, алкилокси- или арилоксигруппу;
каждая из величин m и n независимо друг от друга принимает одинаковые или различные целые значения от приблизительно 0 до приблизительно 8.
Настоящее изобретение также предлагает соединение, имеющее структуру

где каждый из заместителей X и Y независимо друг от друга принимает одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламиногруппу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу;
величина n принимает целые значения от приблизительно 0 до приблизительно 8.
В предпочтительном воплощении вышеуказанного соединения каждый из заместителей X и Y представляет собой диметиламиногруппу, а величина n принимает значение 4 или 5.
Настоящее изобретение также предлагает соединение, имеющее структуру
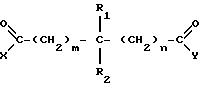
где каждый из заместителей X и Y независимо друг от друга принимает одинаковые или разные значения и представляют собой гидроксильную, амино- или гидроксиламиногруппу, замещенную или незамещенную алкокси-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу;
каждый из заместителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения и представляют собой атом водорода, гидроксильную группу, замещенный или незамещенный алкил, арил, алкилокси-, арилокси-, карбонилгидроксиламиногруппу или атом фтора;
каждая из величина m и n независимо друг от друга принимает одинаковые или различные целые значения от приблизительно 0 до приблизительно 8.
В предпочтительном воплощении вышеуказанного соединения каждый из заместителей X и Y представляет собой гидроксиламиногруппу, заместитель R1 представляет собой метил, заместитель R2 - атом водорода, а каждый из величин m и n принимает значение 2. В другом предпочтительном воплощении каждый из заместителей X и Y представляет собой гидроксиламиногруппу, заместитель R1 представляет собой карбонилгидроксиламиногруппу, заместитель R2 - атом водорода, а каждая из величин m и n принимает значение 5. В другом предпочтительном воплощении каждый из заместителей X и Y представляет собой гидроксиламиногруппу, каждый из заместителей R1 и R2 представляет собой атом фтора, а каждая из величин m и n принимает значение 2.
Настоящее изобретение также предлагает соединение, имеющее структуру

где каждый из заместителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения и представляют собой гидроксильную, алкокси-, амино-, гидроксиламино-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу.
Предпочтительно R1 представляет собой фениламиногруппу, а R2 - гидроксиламиногруппу.
Настоящее
изобретение также предлагает соединение, имеющее структуру

где каждый из заместителей R1 и R2 независимо друг от друга принимает одинаковые или разные значения и представляют собой гидроксильную, алкокси-, амино-, гидроксиламино-, алкиламино-, диалкиламино, ариламино, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу.
Предпочтительно R1 представляет собой фениламиногруппу, а R2 - гидроксиламиногруппу.
Настоящее изобретение также предлагает соединение, имеющее структуру

где каждый из заместителей R1 и R2 независимо друг от друга принимает одинаковые или разные значения и представляют собой гидроксильную, алкокси-, амино-, гидроксиламино-, алкиламино-, диалкиламино-, ариламино-, алкилариламино, алкоксиамино-, арилоксиамино-, алкоксиалкиламино- или арилоксиалкиламиногруппу.
В предпочтительном воплощении заместители R1 и R2 представляют собой гидроксиламиногруппу.
Данное изобретение также предлагает способ селективного индуцирования терминальной дифференцировки опухолевых клеток и посредством этого ингибирование быстрого размножения таких клеток, который включает контактирование клеток при приемлемых условиях с эффективным количеством описанных выше соединений, которые эффективны для селективного индуцирования терминальной дифференцировки.
Контактирование должно проводиться непрерывно в течение длительного периода, то есть в течение по меньшей мере 48 ч, предпочтительно в течение 4-5 дней или больше.
Способ может быть осуществлен на практике in vivo или in vitro. Если способ осуществляется in vitro, то контактирование проводят путем инкубирования клеток с соединением. Концентрация соединения при контакте с клетками должна составлять приблизительно от 1 мкМ до приблизительно 25 мМ, предпочтительно от 4 мкМ до приблизительно 5 мМ. Концентрация зависит от конкретного соединения и состояния опухолевых клеток.
Способ также может включать начальную обработку клеток с помощью противоопухолевого агента, так чтобы придать им устойчивость к противоопухолевому агенту, с последующим контактированием получаемых устойчивых клеток в приемлемых условиях с эффективным количеством описанных выше соединений, которые эффективны для селективного индуцирования терминальной дифференцировки таких клеток.
Противоопухолевый агент может представлять собой один из химиотерапевтических агентов, таких как алкилирующий агент, антиметаболит, гормональный агент, антибиотик, колхицин, алкалоид vinca L-аспарагиназа, прокарбазин, гидроксимочевина, митотан, нитрозомочевины или карбоксамид имидазола. Приемлемыми агентами являются такие агенты, которые стимулируют деполяризацию тубулина. Предпочтительным агентом является колхицин или алкалоид vinca; особенно предпочтительными являются винбластин и винкристин. В воплощениях, в которых противоопухолевым агентом является винкристин, клетки предпочтительно обрабатываются так, чтобы они были резистентными к винкристину при концентрации приблизительно 5 мг/мл. Обработка клеток с целью придания им резистентности к противоопухолевому агенту может быть проведена путем контактирования клеток с этим агентом в течение по меньшей мере 3-5 дней. Контактирование получаемых клеток с любым из вышеописанных соединений проводится так, как описано ранее.
Настоящее изобретение также обеспечивает способ лечения пациента, имеющего опухоль, которая характеризуется быстрым размножением опухолевых клеток, который включает назначение пациенту эффективного количества любого описанного выше соединения, которое эффективно при селективном индуцировании терминальной дифференцировки таких опухолевых клеток и посредством этого ингибируют их быстрое размножение.
Способ настоящего изобретения предназначен для лечения больных людей, имеющих опухоль. Однако также вероятно, что способ должен быть эффективен при лечении опухолей и у других млекопитающих. Понятие "опухоль" включает любую опухоль, вызванную быстрым размножением опухолевых клеток, таких как рак легких, острая лимфоидная миеломная болезнь, миеломная болезнь мочевого пузыря, рак почки, рак молочной железы или рак прямой кишки. На сегодняшний день, как доказано, внутривенное применение является эффективным. Введение соединения должно осуществляться непрерывно в течение продолжительного времени, например, в течение по меньшей мере 3 часов и, предпочтительно, более 5 дней. В наиболее предпочтительном воплощении введение проводят непрерывно в течение по меньшей мере 10 дней и повторяют время от времени, причем время каждого непрерывного применения составляет по меньшей мере 10 дней. Например, введение может быть осуществлено с промежутками от менее 5-10 дней до приблизительно 25-35 дней и непрерывно в течение по меньшей мере 10 дней для каждого из этих промежутков. Оптимальный промежуток будет изменяться в зависимости от пациента и вида опухоли. Например, в случае острого лейкоза непрерывное вливание должно быть таким продолжительным, пока пациент остается толерантным к лекарству без интоксикации при сохранении положительного ответа.
Количество введенного пациенту соединения составляет меньшее, чем количество, которое будет вызывать интоксикацию пациента. В некоторых воплощениях количество соединения, которое вводится пациенту меньше количества, которое приводит к накоплению соединения в плазме пациента в количестве, равном или превышающем токсический уровень этого соединения. Предпочтительно концентрация соединения в плазме пациента должна поддерживаться на уровне приблизительно 1,0 мМ. С помощью ГМБА установлено, что введение соединения в количестве приблизительно от 5 г/м2/сут до 30 г/м2/сут, предпочтительно около 20 г/м2/сут, является эффективным и не приводит к интоксикации пациента. Оптимальное количество соединения, которое должно быть введено пациенту, при реализации на практике настоящего изобретения будет зависеть от конкретного используемого соединения и типа ракового заболевания, которое подвергается лечению.
Настоящее изобретение помимо перечисленных выше соединений предполагает использование гомологов и аналогов таких соединений. В этой связи гомологи представляют собой молекулы, имеющие значительные структурные аналогии с описанными выше соединениями, а аналоги представляют собой молекулы, имеющие существенные аналогии в проявлении биологических свойств, независимо от структурных аналогий.
Способ также может включать предварительное введение пациенту антиопухолевого агента для придания клеткам устойчивости к противоопухолевому агенту и последующее введение пациенту эффективного количества любого из описанных выше соединений, эффективного при лесективном индуцировании терминальной дифференцировки таких опухолевых клеток и посредством этого ингибирует их быстрое размножение.
Противоопухолевый агент может представлять собой один из химиотерапевтических агентов, таких как алкилирующий агент, антиметаболит, гормональный агент, антибиотик, колхицин, алкалоид vinca L-аспарагиназа, прокарбазин, гидроксимочевина, митотан, нитрозомочевины или карбоксамид имидазола. Приемлемыми агентами являются такие агенты, которые стимулируют деполяризацию тубулина. Предпочтительным агентом является колхицин или алкалоид vinca; особенно предпочтительными являются винбластин и винкристин. В воплощениях, в которых противоопухолевым агентом является винкристин, клетки предпочтительно обрабатываются так, чтобы они были резистентными к синкристину при концентрации приблизительно 5 мг/мл. Введение агента осуществляют преимущественно так, как описано выше для введения любого из соединений. Предпочтительно введение агента осуществляют в течение по меньшей мере 3-5 дней. Введение любого из описанных выше соединений осуществляют так, как это было описано ранее.
Данное изобретение также предлагает фармацевтическую композицию, содержащую фармацевтически приемлемый носитель, такой как стерильная апирогенная вода и терапевтически приемлемое количество любого из описанных выше соединений. Предпочтительно, эффективное количество представляет собой количество, эффективное для селективного индуцирования терминальной дифференцировки опухолевых клеток, и меньше количества, которое приводит к интоксикации пациента.
И, наконец, настоящее изобретение предлагает описанную выше фармацевтическую композицию в сочетании с противоопухолевым агентом. В качестве противоопухолевого агента может быть использован любой из описанных выше агентов.
Настоящее изобретение иллюстрируется экспериментальными данными. Экспериментальные данные представлены для того, чтобы облегчить понимание изобретения, а не для того, чтобы ограничить изобретение каким-либо способом, которое определяется в приведенной ниже формуле изобретения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Клетки и материалы
Клетки ЭЛКМ 645A-DS19 и различные варианты ЭЛКМ, полученные из этой линии клеток,
обычно из резистентных к винкристину линий клеток ЭЛКМ V 3.17 и VCR. C (Breitman, T. R., Selonick, S.E., and Collins, S.J. (1980) Proc. Natl. Acad. Sci. (USA) 77: 2936-2940) 15 (Melloni, E.,
Pontremoli, S., Damiani, G. , Viotti, P., Weich, N., Rifkind, R.A. and Marks, P.A. (1988) Proc. Natl. Acad. Sci. (USA) 85: 3835-3839) и линии клеток DR10 (Ohta, Y., Tanaka, M., Terada, M., Miller,
O.J., Bank, A., Marks, P.A. and Rifkind, R.A. (1976) Proc. Natl. Acad. Sci. (USA) 73: 1232-1236), резистентных к диметилсульфоксиду, выдерживают в альфа-минимальной среде, содержащей 10%-ную фетальную
телячью сыворотку (Scher, W., Scher, B.M. and Waxman, S. (1982) Biochem. & Biophys. Res. Comm. 109: 348-354). Культуры клеток для всех опытов стимулируют клетками в фазе логарифмического роста
(клетки, выращиваемые 2 дня) при плотности 105 клеток/мл. Индуцирующие соединения добавляют в концентрациях, указанных ниже, растворенными в культуральной среде без фетальной телячьей
сыворотки, если это не оговорено особо. Плотность клеток и реакционная способность по отношению к бензидину определяют в соответствии с описанием работы (Scher, W. , Scher, B.M and Waxman, S. (1982)
Biochem. & Biophys. Res. Comm. 109: 348-354).
Комитирование к терминальной дифференцировке, которое характеризуется ограниченным делением клеток (размер колонии менее 32 клеток) и накоплением гемоглобина (колонии, реакционноспособные к бензидину), оценивают путем клонирования колонии с использованием 2%-ной метилцеллюлозы, как это описано в работе (Fibach, E., Reuben, R.C., Rifkind, R.A. and Marks, P.A. (1977) Cancer Res. 37: 440-444) (см. таблицу 1).
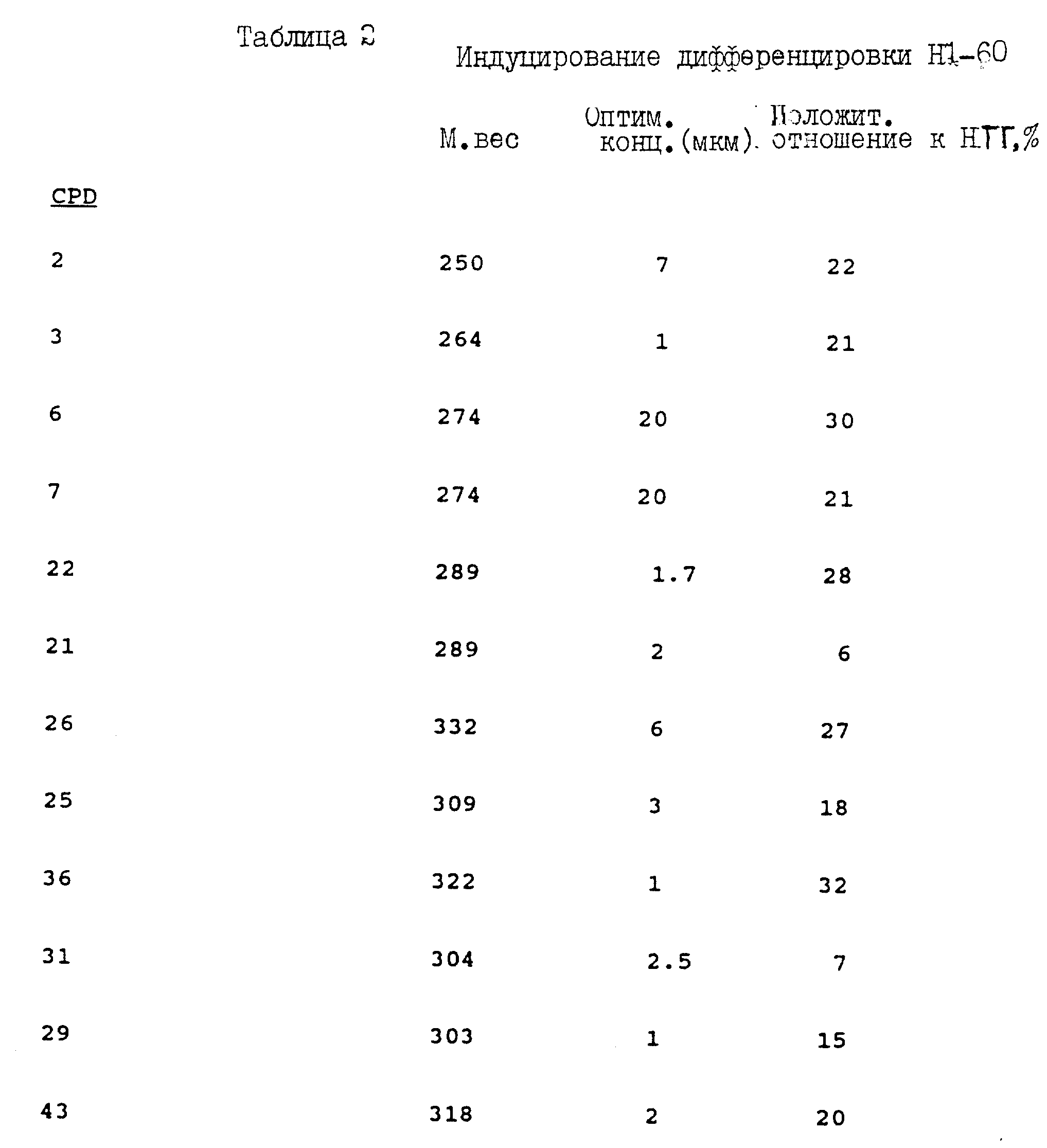
Клетки лейкоза человека HL-60 получают из периферических кровяных лейкоцитов пациента, страдающего острым промиелоцитным лейкозом (Collins, S.J., Gallo, R. C., and Gallagher, R.E. (1978) Nature (London) 270; 405-409). Индуцированную дифференцировку клеток HL-60 оценивают путем определения части клеток, которые развивают способность восстанавливать нитротетразолий голубой (НТГ) (Synder, S.W., Egorin, M.J., Geelhaar, L.A., Hamburger, A.M., and Callery, P.S. (1988) Cancer Res. 48; 3613-3616) (см. таблицу 2).
Химия
Соединение, имеющие формулу

Получение PhCH2ONHOC(CH2)6COOCH3
Раствор монометилового эфира пробковой кислоты (1.9 г, 0.01 моля), оксалилхлорида (1.75 мл, 2.54 г, 0.02 моля) и 0.1 мл ДМФА в бензоле (200 мл) перемешивают в течение ночи при комнатной температуре. Растворитель упаривают и маслянистый остаток растворяют в хлороформе (≈ 20 мл) и смешивают с раствором O-бензилгидроксиламина (2.46 г, 0.02 моля) и пиридина (1.6 мл, 1.68 г, 0.02 моля) в хлороформе (100 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, хлороформенный раствор промывают водой (50 мл), 10%-ной соляной кислотой и снова водой (2х50 мл). Органический слой сушат безводным сульфатом магния и упаривают. Твердый остаток промывают гексаном (≈100 мл) и отфильтровывают. Выход PhCH2ONHOC(CH2)6COOCH3 составляет 2.61 г (89%).

Полученный монобензилгидроксиламид монометилового эфира пробковой кислоты (1 г, 3.4 моля) растворяют в сухом метаноле (50 мл) и добавляют 5%-ный Pd/C (50 мг). Суспензию черного цвета встряхивают в атмосфере водорода при давлении ≈3.4 атм. (≈50 фунтов/кв. дюйм) в течение ночи при комнатной температуре. Катализатор отделяют фильтрацией, фильтра упаривают. Твердый остаток промывают гексаном (≈20 мл) и отфильтровывают. Выход гидроксамовой кислоты монометилового эфира пробковой кислоты составляет 900 мг (95%).
Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 10.31 (с, NHOH, 1H), 8.89 (уш. с. , NHOH, 1H), 3.57 (с, CH3, 3H), 2.27 (т, J=7.4 Гц, CH2COOCH3, 2H), 1.91 (т, J=7.4 Гц, CH2CONHOH, 2H), 1.49 (м, 4H), 1.24 (м, 4H).
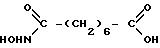
Гидроксамовую кислоту монометилового эфира пробковой кислоты (1 г, 3.4 ммоля) и гидроксид натрия (210 мг, 3.75 ммоля) растворяют в 10 мл смеси метанол-вода (4: 1). Реакционную смесь кипятят в течение двух часов и упаривают растворитель. Твердый остаток растворяют в 5 мл воды и подкисляют концентрированной соляной кислотой до pH≈5. Белый осадок отфильтровывают, сушат и перекристаллизовывают из смеси этилацетат-гексан. Выход монобензилоксиамида пробковой кислоты составляет 820 мг (86%). Продукт растворяют в метаноле (50 мл) и добавляют 5%-ный Pd/C (50 мг). Реакционную смесь встряхивают в атмосфере водорода при давлении ≈3.4 атм. (≈50 фунтов/кв. дюйм) в течение ночи. Катализатор отделяют фильтрацией, фильтрат упаривают. Твердый остаток промывают гексаном и отфильтровывают. Выход моногидроксамовой кислоты пробковой кислоты составляет 520 мг (81%).
Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 10.31 (уш. с., COOH, 1H), 10.31 (с, NHOH, 1H), 8.63 (уш. с., NHOH, 1H), 2.17 (с, J=7.4 Гц, CH2COOH, 2H), 1.91 (с, CH2CONHOH, 2H), 1.46 (м, 4H), 1.22 (м, 4H).
Соединения, имеющие формулу

Общая методика
Раствор O-бензилгидроксиламина (2.46 г, 0.02 моля), соответствующего амина и хлорангидрида пробковой кислоты в пиридине (500 мл) перемешивают при комнатной температуре в течение ночи. Растворитель упаривают и полутвердый остаток растворяют в 1000 мл смеси хлороформ-метанол (4:1); полученный раствор промывают водой (2х100мл), 10%-ной соляной кислотой (3х100мл) и снова водой (2х100 мл). Органический слой сушат безводным сульфатом магния и упаривают. Твердый остаток растворяют в метаноле (100 мл) и добавляют 5%-ный Pd/C. Суспензию черного цвета встряхивают в атмосфере водорода при давлении ≈3.4 атм. (≈50 фунтов/кв. дюйм) в течение ночи. Катализатор отделяют фильтрацией, фильтрат упаривают. Целевой продукт выделяют колоночной хроматографией на силикагеле (элюент этилацетат-тетрагидрофуран).

Выход 1,1 г (26%). Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 10.93 (с, NHOCH3, 1H), 10.32 (с, NHOH, 1H), 8.66 (с, NHOH, 1H), 3.55 (с, CH3, 3H), 1.91 (с, J=7.6 Гц, CH2CO-, 4H), 1.45 (м, 4H), 1.20 (м, 4H).

Выход 1.2 г (21%). Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 10.31 (уш. с. , NHOH, 1H), 8.60 (с, NHOH, 1H), 7.57 (д, J=7.6 Гц, NH-C6H11, 1H), 3.40 (м, CH-NH, 1H), 1.99 (т, J=7 Гц, CH2CONHC6H11, 2H), 1.91 (т, J=7.6 Гц, CH2CONHOH, 2H), 1.63 (м, 4H), 1.44 (м, 6H), 1.20 (м, 8H).

Выход 870 мг (20%). Спектр1H ЯМР (ДМСО-d6, 200 МГц) δ (м.д.): 10.31 (с, NHOH, 1H), 8.67 (уш. с., NHOH, 1H), 2.85 (д, J=30 Гц, N(CH3)2, 6H), 2.24 (т, J= 7.4 Гц, CH2CON(CH3)2, 2H), 1.91 (т, J=7.4 Гц, CH2CONHOH, 2H), 1.50 (м, 4H), 1.20 (м, 4H).

Выход 1.4 г (27%). Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 10.31 (с, NHOH, 1H), 8.67 (уш. с., NHOH, 1H), 3.40 (2т, CH2N, 4H), 2.20 (т, J=7.4 Гц, CH2 CON(CH3)2, 2H), 1.91 (т, J=7.4 Гц, CH2CONHOH, 2H), 1.10 - 160 (уш. м., 14H).
Соединение, имеющее структуру

Раствор O-бензилгидроксиламина (1.23 г, 0.01 моля), О-(триметилсилил)гидроксиламина (1.1 г, 0.01 моля), пиридина (1.6 мл, 1.7 г, 0.02 моля) и хлорангидрида пробковой кислоты (1.8 мл, 2.11 г, 0.01 моля) в хлороформе (500 мл) перемешивают при комнатной температуре в течение ночи. Полученную суспензию разбавляют метанолом (100 мл) и промывают 10%-ной соляной кислотой (3х100 мл). Органический слой сушат безводным сульфатом магния и упаривают. Твердый остаток подвергают колоночной хроматографии на силикагеле (элюент этилацетат-тетрагидрофуран, 4: 1). Выход составляет 500 мг (17%). Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 11.09 (с, NHOCH2C6H5, 1H), 10.31 (с, NHOH, 1H), 8.67 (уш.с., NHOH, 1H), 7.36 (с, C6H5, 5H), 4.76 (с, CH2C6H5, 2H), 1.92 (т, J = 7.4 Гц, CH2CO-, 4H), 1.45 (м, 4H), 1.20 (м, 4H).
Соединение, имеющее структуру

К заложенному раствору гидроксида натрия (2.24 г, 0.04 моля) и хлоргидрата O-бензилгидроксиламина (1.23 г, 0.01 моля) в 30 мл смеси тетрагидрофуран-вода (1: 1) добавляют 6-бромгексаноилхлорид (3.1 мл, 4.27 г, 0.02 моля). Реакционную смесь перемешивают при комнатной температуре в течение 1 ч. Растворитель упаривают и твердый остаток распределяют между хлороформом (200 мл) и водой (100 мл). Хлороформный слой промывают 10%-ной соляной кислотой (3х50 мл) и водой (2х50 мл). Органический слой сушат безводным сульфатом магния и упаривают. Продукт очищают кристаллизацией из смеси этилацетат-гексан. Выход N-бензилокси-6-бромгексаноиламида составляет 4.7 г (78%). Раствор N-бензилокси-6-бромгексаноиламида (4.5 г, 15 ммолей) и цианида натрия (7.35 г, 0.15 моля) в диметилсульфоксиде (250 мл) нагревают при 130oC в течение ночи. Растворитель упаривают, твердый остаток распределяют между хлороформом (300 мл) и водой (300 мл). Хлороформенный слой промывают водой (5х100 мл), сушат безводным сульфатом магния и упаривают. Маслянистый остаток очищают колоночной хроматографией на силикагеле (элюент этилацетат-третрагидрофуран, 4: 1). ВыходN-бензилокси-6-цианогексаноиламида составляет 1.62 г (43%). Продукт растворяют в метаноле (50 мл) и добавляют 5%-ный Pd/C (100 мг). Суспензию черного цвета встряхивают в атмосфере водорода при давлении ≈ 3.4 атм. (≈ 50 фунтов/кв. дюйм) в течение ночи. Катализатор отделяют фильтрацией, фильтрат упаривают. Твердый остаток промывают гексаном (≈ 20 мл) и отфильтровывают. Выход N-гидрокси-6-цианогексаноиламида составляет 900 мг (общий выход 30%).
Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 10.32 (с, NHOH, 1H), 8.65 (с, NHOH, 1H), 2.45 (т, J = 7 Гц, CH2CN, 2H), 1.93 (т, J = 7 Гц, CH2CONHOH, 2H), 1.49 (м, 4H), 1.33 (м, 4H).
Соединения, имеющие формулу

Общая методика
К охлажденному до 0oC раствору гидроксида калия (1.12 г, 0.02 моля) и соответствующего амина (0.01 моля) в 30 мл смеси тетрагидрофуран-вода (1:1) добавляют дихлорангид дикарбоновой кислоты (0.01 моля). Реакционную смесь перемешивают при комнатной температуре в течение одного часа. Растворитель упаривают и твердый остаток распределяют между хлороформом (300 мл) и водой (300 мл). При необходимости в некоторых случаях для полноты растворения твердого вещества добавляют небольшое количество метанола. Органический слой промывают 10%-ным раствором гидроксида калия (3х30 мл). Основной водный экстракт подкисляют 10%-ной соляной кислотой. Осадок отделяют фильтрацией, сушат и очищают кристаллизацией из этилацетата или колоночной хроматографией на силикагеле (элюент этилацетат-тетрагидрофуран, 4:1). Выход составляет 20-37%.

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 11.97 (с, COОH, 1H), 9.84 (с, NH, 1H), 7.57 (д, J = 7.4 Гц, орто-ароматические протоны), 7.26 (т, J = 8.4 Гц, мета-ароматические протоны, 2H), 6.99 (т, J = 7.4 Гц, пара-ароматические протоны, 1H), 2.27 (т, J = 7 Гц, CH2CONHPh, 2H), 2.18 (т, J = 7.2 Гц, 2H), 1.52 (м, 4H), 1.28 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 11.95 (с, COОH, 1H), 10.20 (с, NH, 1H), 8.10 (с, ароматический протон, 1H), 7.75 (м, ароматический протон, 1H), 7.45 (м, ароматические протоны, 2H), 2.28 (т, J = 7.4 Гц, CH2CONHAr, 2H), 2.21 (т, J = 7.2 Гц, CH2COOH, 2H), 1.46 (м, 4H), 1.20 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 11.95 (с, COОH, 1H), 10.29 (с, NH, 1H), 7.75 (м, ароматические протоны, 4H), 2.33 (т, J = 7.2 Гц, CH2CONHAr, 2H), 2.18 (т, J = 7.4 Гц, CH2COOH, 2H), 1.53 (м, 4H), 1.27 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 11.98 (с, COОH, 1H), 10.48 (с, NH, 1H), 8.21 (д, J = 9.2 Гц, ароматические протоны, 2H), 7.82 (д, J = 9.2 Гц, ароматические протоны, 2H), 2.36 (т, J = 7.4 Гц, CH2CONHAr, 2H), 2.18 (т, J = 7.2 Гц, CH2COOH, 2H), 1.55 (м, 4H), 1.29 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 12.00 (с, COОH, 1H), 10.24 (с, NH, 1H), 8.38 (д, J = 5.8 Гц, ароматические протоны, 2H), 7.55 (д, J = 5.8 Гц, ароматические протоны, 2H), 2.33 (т, J = 7.2 Гц, CH2CONHAr, 2H), 2.18 (т, J = 7.2 Гц, CH2COOH, 2H), 1.52 (м, 4H), 1.27 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 11.95 (с, COОH, 1H), 7.58 (д, J = 8 Гц, 1H), 3.50 (м, CH, 1H), 2.17 (т, J = 7.2 Гц, CH2COOH, 2H), 2.00 (т, J = 7 Гц, CH2CONH-, 2H), 1.60 (м, 4H), 1.46 (м, 6H), 1.20 (м, 8H).
Аналогично были получены и охарактеризованы
следующие соединения:

где n = 4, 5, 6, 7 и 8;
заместитель R представляет собой атом водорода, 2-, 3- и 4-циано, 2-, 3- и 4-нитро, 2-, 3- и 4-цианометил, 2-, 3- и 4-трифторметил, 2-, 3- и 4-фтор;

где n = 4, 5, 6, 7 и 8;

где n = 4, 5, 6, 7 и 8;

где n = 4, 5, 6, 7 и 8;

где n = 4, 5, 6, 7 и 8;
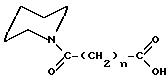
где n = 4, 5, 6, 7 и 8;

где заместитель R представляет собой 2-, 3- и 4-карбокси, 2-, 3- и 4-метиламинокарбонил, 2-, 3- и 4-диметиламинокарбонил, 2-, 3- и 4-хлор, 2-, 3- и 4-бром, 2-, 3- и 4-йод, 2-, 3- и 4-метил, 2-, 3- и 4-метокси, 2-, 3- и 4-гидрокси, 2-, 3- и 4-амино и 2-, 3- и 4-диметиламино.
Соединения, имеющие общую формулу

где n = 4, 5, 6 и 7.
Общая методика A
Суспензию хлоргидрата O-бензилгидроксиламина (3.2 г, 0.02 моля) и соответствующего дихлорангидрида дикарбоновой кислоты в пиридине (500 мл) перемешивают при комнатной температуре в течение трех
дней, добавляют воду (10 мл) и перемешивают в течение ночи. Растворитель упаривают, а твердый остаток очищают колоночной хроматографией на силикагеле (элюент тетрагидрофуран-метанол). Диацильный
продукт растворяют в метаноле (100 мл) и добавляют 5%-ный Pd/C (100 мг). Суспензию встряхивают в атмосфере водорода при давлении ≈ 3.4 атм (≈ 50 фунтов/кв. дюйм) в течение ночи.
Катализатор отделяют фильтрацией, осадок промывают горячим метанолом (5х50 мл). Объединенные метанольные фильтраты упаривают. Твердый остаток промывают ацетоном и отфильтровывают. Выход составляет
10-20%.
Общая методика Б
Раствор O-бензилгидроксиламина (2.6 г, 0.02 моля) и соответствующего хлорангидрида монобензилового эфира дикарбоновой кислоты (0.04 мл) в пиридине
(500 мл) перемешивают при комнатной температуре в течение ночи и упаривают растворитель. Полутвердый остаток растворяют в хлороформе (300 мл) и экстрагируют 5%-ной соляной кислотой (2x50 мл), 10%-ным
раствором гидроксида калия (3x100 мл) и водой (2x100 мл). Органический слой сушат безводным сульфатом магния и упаривают. Твердый остаток очищают колоночной хроматографией на силикагеле (элюент
этилацетат). Трибензильный продукт растворяют в метаноле (100 мл) и добавляют 5%-ный Pd/C (100 мг). Суспензию встряхивают в атмосфере водорода при давлении ≈ 3.4 атм (≈ 50 фунтов/кв.
дюйм) в течение ночи. Катализатор отделяют фильтрацией, осадок промывают горячим метанолом (5x50 мл). Объединенные метанольные фрагменты упаривают. Твердый остаток промывают захоложенным ацетоном и
отфильтровывают. Выход целевого продукта составляет 30 - 60%.

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 11.53 (с, COOH, 1H), 2.41 (т, J = 7.2 Гц, CH2CON(OH)COOH2, 4H), 2.18 (т, J = 7.0 Гц, CH2COOH, 4H), 1.52 (м, 8H), 1.22 (м, H). Масс-спектр [бомбардировка быстрыми атомами (ББА) глицерин] : 346 (M+1).
Соединения, имеющие структуру

Раствор хлорангидрида монометилового эфира дикарбоновой кислоты (0.02 моля) и N, N'-диметил-1, ω-диаминоалкана (0.01 моля) в пиридине (500 мл) перемешивают при комнатной температуре в течение ночи. Растворитель упаривают и маслообразный остаток растворяют в хлороформе (300 мл). Хлороформенный раствор промывают водой (3x50 мл), 10%-ным раствором гидроксида калия (3x50 мл), 10%-ной соляной кислотой (3x50 мл) и снова водой (3x50 мл). Органический слой сушат и упаривают. Маслянистый остаток растворяют в растворе гидроксида калия (1.2 г, 0,021 моля) в 80%-ном метаноле (100 мл) и реакционную смесь кипятят два часа. Растворитель упаривают, твердый остаток растворяют в воде (50 мл) и экстрагируют хлороформом (3x50 мл). Водный раствор подкисляют до pH ≈ 5 и упаривают (до объема приблизительно 10 мл). Водный раствор или суспензию охлаждают, выпавший осадок отфильтровывают. Твердый продукт очищают кристаллизацией из этилацетата. Выход составляет 40 - 60%.

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 8.15 (уш. с., COOH, 2H), 3.52 + 3.45 (2с., CH2N, 4H), 3.01 + 2.93 (2с., CH3N, 6H), 2.30 (4т, CH2CO, 8H), 1.60 (м, 8H), 1.32 (м, 8H).
Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м,д.): 3.44 + 3.336 + 3.36 (3с., CH2N, 4H), 2.94 + 2.90 + 2.79 (3с., CH3N, 6H), 2.27 + 2.23 + 2.12 (3т, CH2CO, 8H), 1.46 (м, 8H), 1.23 (м, 8H).
Соединения, имеющие структуру

Раствор 6-аминокапроновой кислоты (2.6 г, 0.02 моля) и терефталоилхлорида (2 г, 0. 01 моля) в пиридине (500 мл) перемешивают при комнатной температуре в течение ночи (≈ 12 ч) и при 90oC в течение 23 ч. Растворитель упаривают, твердый остаток перекристаллизовывают из воды (10 мл) четыре раза. Выход составляет 800 мг (19%).
Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м,д.): 12.8 (уш. с., COOH, 2H), 8.54 + 7.72 (2т, NH, 2H), 3.24 + 2.98 (2м, NHCH2, 4H), 2.20 + 2.03 (2м, CH2CO, 4H), 1.50 (м, 8H), 1.32 (м, 4H).
Соединения, имеющие структуру

К смеси анилина (2.75 г, 0.03 моля), хлоргидрата гидроксиламина (2.08 г, 0.03 моля) и гидроксида калия (5.50 г, 0.09 моля) в 50%-ном тетрагидрофуране (100 мл) медленно при комнатной температуре добавляют раствор терефталоилхлорида (6 г, 0.03 моля) в тетрагидрофуране (20 мл) и полученную суспензию перемешивают при комнатной температуре в течение 30 мин. Растворитель упаривают, твердый остаток растворяют в горячем метаноле (1000 мл) и сушат сульфатом магния. Метанольный раствор отделяют фильтрованием, фильтрат упаривают. Твердый остаток промывают 20 мл захоложенного метанола и отфильтровывают. Белый кристаллический продукт промывают эфиром (5x50 мл) и сушат. Выход составляет 4.6 г (39%).
Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 11.35 (уш. с., NHOH, 1H), 10.35 (с, NHPh, 1H), 9.19 (с, NHOH, 1H), 8.03 (д, J = 8 Гц, терефталевые протоны, 2H), 7.89 (д, J = 8 Гц, терефталевые протоны, 2H), 7.82 (д, J = 7,4 Гц, протоны орто-анилида, 2H), 7.34 (т, J = 7.4 Гц, протоны мета-анилида, 2H), 7.10 (J = 7.4 Гц, протон пара-анилида, 1H).
Соединение, имеющее структуру

Раствор 1,4-фенилендиакриловой кислоты (2.18 г, 0.01 моля) в тионилхлориде (50 мл, 81.55 г, 0.68 моля) кипятят в течение ночи, избыток тионилхлорида упаривают. Твердый остаток растворяют в тетрагидрофуране (20 мл) и добавляют к охлажденному до 0oC раствору гидроксида калия (1.12 г, 0,02 моля) и анилина в 50%-ном тетрагидрофуране. Реакционную смесь перемешивают при комнатной температуре в течение 30 мин, растворитель упаривают. Твердый остаток растворяют водой и отфильтровывают. Белый кристаллический продукт растворяют в небольшом количестве метанола и очищают колоночной хроматографией на силикагеле. Выход 3,15 мг (10%).
Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 10.80 (с, NHOH, 1H), 10.23 (с, NHPh, 1H), 9.09 (с, NHOH, 1H), 7.69 (д, J = 7.6 Гц, протоны орто-анилида, 2H), 7.64 (с, фениленовые протоны, 4H), 7.55 (д, J = 15.8 Гц, PhNHOCCH= CH-, 1H), 7.40 (д, J = 15.8 Гц, HONHOCH=CH-, 1H), 7.33 (т, J = 7.8 Гц, протоны мета-анилида, 2H), 7.06 (J = 7.2 Гц, протон пара-анилида, 1H), 6.89 (д, J = 15.8 Гц, PhNHOCCH=CH-, 1H), 6.51 (д, J = 15.8 Гц, HONHOCH=CH-, 1H).
Соединения, имеющие структуру

где n = 4, 5, 6, 7 и 8.
Раствор триэтиламина (1.4 г, 1.0 г, 0.01 моля),
соответствующего амина (0.01 моля) и дихлорангидрида дикарбоновой кислоты (0.005 моля) в хлороформе перемешивают при комнатной температуре в течение 5 ч. Если реакционная смесь прозрачна, ее промывают
водой (5x100 мл), органический слой сушат безводным сульфатом магния и упаривают до образования твердого остатка. Если в процессе реакции образовался осадок, то его отделяют фильтрацией. Белые
кристаллы после фильтрации или твердый продукт после упаривания кристаллизуют из этилацетата, тетрагидрофурана, метанола или их смеси. Выход составляет 60 - 90%,

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 10.23 (с, NH, 2H), 7.82 (д, J = 9 Гц, ароматические протоны, 4H), 7.60 (д, J = 9 Гц, ароматические протоны, 4H), 2.31 (т, J = 7,4 Гц, CH2CO, 4H), 2.61 (м, 4H), 1.32 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 10.48 (с, NH, 2H), 8.18 (д, J = 9.2 Гц, ароматические протоны, 4H), 7.81 (д, J = 9.2 Гц, ароматические протоны, 4H), 2.37 (т, J = 7.2 Гц, CH2CO, 4H), 1.60 (м, 4H), 1.33 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 9.91 (с, NH, 2H), 7.58 (д, J = 8.6 Гц, ароматические протоны, 4H), 7.26 (д, J = 8.6 Гц, ароматические протоны, 4H), 3.94 (с, CH2CN, 4H), 2.29 (т, J = 7.4 Гц, CH2-CO-, 4H), 1,60 (м, 4H), 1.31 (м, 4H).
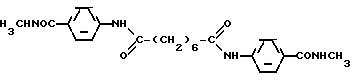
Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 10.08 (с, CONHAr, 2H), 7.79 (д, J = 8,6 Гц, ароматические протоны, 4H), 7.63 (д, J = 8 Гц, ароматические протоны, 4H), 7.22 (с, H3CHNCO, 2H), 3.32 (с, CH3, 6H), 2.31 (т, J = 7 Гц, CH2C-, 6H), 1.59 (м, 4H), 1.31 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 10.90 (уш.с., NHOH, 1H), 10.05 (с, NHAr, 2H), 8.90 (уш.с., NHOH, 2H), 7.68 (д, J = 9 Гц, ароматические протоны, 4H), 7.62 (д, J = 9 Гц, ароматические протоны, 4H), 2.31 (т, J = 7.2 Гц, CH2CO-, 4H), 1.59 (м, 4H), 1.30 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 10.06 (уш.с., NH, 2H), 8.71 (д, J = 2.6 Гц, ароматические протоны, 2H), 7.31 (д + д, ароматические протоны, 2H), 2.32 (т, J= 7,4 Гц, CH2CO-, 4H), 1.59 (м, 4H), 1.33 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д.): 12.00 (уш.с., NH, 2H), 7.43 (д. J = 3.6 Гц, ароматические протоны, 2H), 7.16 (д, J = 3.6 Гц, ароматические протоны, 2H), 2.41 (т, J = 7.2 Гц, CH2CONH-, 4H), 1.58 (м, 4H), 1.28 (м, 4H).
Аналогичным образом были получены и охарактеризованы следующие соединения:

где n = 4, 5, 6, 7 и 8.
Все соединения являются симметричными, а заместитель R принимает значения 2-, 3- и 4-циано, 2-, 3- и 4-цианометил, 2-, 3- и 4-нитро, 2-, 3- и 4-карбокси, 2-, 3- и 4-аминокарбонил,
2-, 3- и 4-метиламинокарбонил, 2-, 3- и 4-диметиламинокарбонил и 2-, 3- и 4-трифторметил;

где заместитель R представляет собой 4-гидроксиаминокарбонил, 4-метоксикарбонил, 2-, 3- и 4-хлор, 2-, 3- и 4-фтор, 2-, 3- и 4-метил, 2-, 3- и 4-метокси, 2,3-дифтор, 2,4-дифтор, 2,5-дифтор, 2,6-дифтор, 1,2,3-трифтор, 3,4,5-трифтор, 2,3,5,6-тетрафтор, 2,3,4,5,6-пентафтор.


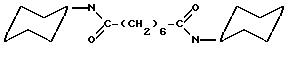

Соединения, имеющие структуру

где n = 4, 5, 6, 7 и 8.
Общая методика A
К раствору гидроксида калия (1.68 г, 0.03 моля), хлоргидрата гидроксиламина (0.7 г, 0.01
моля) и соответствующего анилина (0.01 моля) в 50%-ном тетрагидрофуране (100 мл) при перемешивании добавляют дихлорангидрид дикарбоновой кислоты (0.01 моля). Полученную реакционную смесь перемешивают
при комнатной температуре в течение 30 мин и упаривают растворитель до образования твердого остатка. Твердый остаток растворяют в метаноле (≈ 100 мл) и сушат безводным сульфатом магния.
Метанольный раствор отделяют фильтрованием, фильтрат упаривают досуха. Продукт очищают колоночной хроматографией на силикагеле [элюент этилацетат-тетрагидрофуран (в большинстве случаев 3:1)]. Выход
составляет 15-30%.
Общая методика Б
Раствор соответствующего монометилового эфира дикарбоновой кислоты (0.01 моля), оксалилхлорида (0.03 моля) и несколько капель ДМФА в
бензоле (500 мл) перемешивают в течение ночи при комнатной температуре. Растворитель упаривают и маслянистый остаток растворяют в сухом бензоле (3х50 мл) и вновь упаривают. К захоложенному раствору
соответствующего амина (0.01 моля) и пиридина (1.6 мл, 1.6 г, 0.02 моля) в тетрагидрофуране (200 мл) медленно добавляют раствор полученного монохлорангидрида моноэфира соответствующей дикарбоновой
кислоты в тетрагидрофуране (50 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Растворитель упаривают, остаток растворяют в хлороформе (300 мл) и раствор промывают
10%-ной соляной кислотой (3х50 мл), 10%-ным раствором гидроксида калия (3х50 мл). Органический слой сушат безводным сульфатом магния и упаривают, получая моноэфир-моноамид дикарбоновой кислоты.
Продукт и гидроксид калия (0.56 г, 0.01 моля) растворяют в 80%-ном метаноле. Реакционную смесь кипятят 2 часа и упаривают до образования твердого остатка. Остаток растворяют в воде (≈ 20 мл) и
подкисляют 10%-ной соляной кислотой до pH≈5. Моноамид дикарбоновой кислоты выделяют фильтрованием осадка или экстракцией водного раствора хлороформом. Выделенный моноамид дикарбоновой кислоты
смешивают с эквивалентным количеством O-бензилгидроксиламина и 1,3-дициклогексилкарбодиимида в пириде (≈ 100 мл на каждые 0.01 моля O-бензилгидроксиламина) и перемешивают при комнатной
температуре в течение ночи. Растворитель упаривают, твердый остаток распределяют между хлороформом (500 мл) и 10%-ной соляной кислотой (300 мл). Органический слой промывают водой (3х100 мл) и сушат
безводным сульфатом магния. Растворитель упаривают досуха, твердый остаток растворяют в большом количестве тетрагидрофурана и фильтруют через короткую колонку с силикагелем. Выделенный сырой продукт
растворяют в метаноле (1000 мл) и добавляют 5%-ный Pd/C (50 мг). Полученную суспензию встряхивают в атмосфере водорода при давлении ≈ 3.4 атм (≈ 50 фунтов/кв.дюйм) в течение ночи.
Катализатор отделяют фильтрацией, фильтрат упаривают досуха. Твердый остаток промывают гексаном и отфильтровывают. В большинстве случаев таким образом выделяют чистый продукт. При необходимости
дальнейшую очистку осуществляют колоночной хроматографией на силикагеле (элюент этилацетат-татрагидрофуран). Выход составляет от 35 до 65%.
Общая методика В
Раствор
O-бензилгидроксиламина (1.23 г, 0.01 моля), соответствующего амина (0.01 моля) и дихлорангидрида дикарбоновой кислоты (0.01 моля) в пиридине (500 мл) перемешивают при комнатной температуре в течение
ночи. Растворитель упаривают, получают белый твердый остаток, который по данным1H ЯМР содержит два симметричных амида и целевой несимметричный амид. Твердый остаток растворяют в метаноле и
сушат безводным сульфатом магния. Фильтрат упаривают и твердый остаток растворяют в метаноле (≈ 100 мл). К метанольному раствору добавляют 5%-ный Pd/C (100 мг) и суспензию черного цвета
встряхивают в атмосфере водорода при давлении ≈ 3.4 атм (≈ 50 фунтов/кв.дюйм) в течение ночи. Катализатор отделяют фильтрацией, фильтрат упаривают. Продукт выделяют колоночной
хроматографией на силикагеле (элюент этилацетат-тетрагидрофуран). Выход составляет от 20 до 35%.
Общая методика Г
Раствор триэтиламина (3 мл, 2.18 г, 0.0215 моля),
соответствующего амина (0.01 моля) O-(триметилсилил)гидроксиламина (1.05 г, 0.01 моля), и дихлорангидрида дикарбоновой кислоты (0.01 моля) в хлороформе перемешивают при комнатной температуре в течение
ночи. Растворитель упаривают, остаток растворяют в метаноле (≈10 мл) и к метанольному раствору добавляют 10%-ный раствор хлорида аммония (≈10 мл). Полученную суспензию перемешивают при
50oC в течение 2 часов и упаривают растворитель. Твердый продукт растворяют в метаноле и сушат безводным сульфатом магния. Метанольный раствор отделяют фильтрацией и упаривают досуха.
Продукт выделяют колоночной хроматографией на силикагеле (элюент этилацетат-тетрагидрофуран). Выход составляет от 20 до 33%.

Элементный анализ:
Вычислено C 63.62 H 7.63 N 10.60
Найдено C 63.52 H 7.59 N 10.48
Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д): 10.31 (с, NHOH, 1H), 9.83 (с, NHPh, 1H), 8.64 (с, NHOH, 1H), 7.57 (д, J = 8.2 Гц, орто-ароматические протоны, 2H), 7.26 (т, J = 8.2 Гц, мета-ароматические протоны, 2H), 6.99 (т, J = 7.4 Гц, пара-ароматические протоны, 2H), 2.27 (т, J = 7.2 Гц, CH2CONHPh, 2H), 1.93 (т, J = 7.2 Гц, CH2CONHOH, 2H), 1.52 (м, 4H), 1.26 (м, 4H).
Масс-спектр (ББА, глицерин): 172, 204, 232, 24, 265, (110%, M+1).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д): 10.31 (с, NHOH, 1H), 10.08 (с, NHPh, 1H), 8.64 (с, NHOH, 1H), 7.78 (д, J = 7.6 Гц, ароматические протоны, 1H), 7.66 (т, J = 7.4 Гц, ароматические протоны, 1H), 7.48 (д, J = 7.48 Гц, ароматические протоны, 1H), 7.29 (т, J = 7.4 Гц, ароматические протоны, 1H), 2.34 (т, J = 7 Гц, CH2CONHAr, 2H), 1.93 (т, J = 7.4 Гц, CH2CONHOH, 2H), 1.58 (м, 4H), 1.27 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д): 10.31 (с, NHOH, 1H), 10.21 (с, NHPh, 1H), 8.65 (с, NHOH, 1H), 8.09 (с, ароматический протон, 1H), 7.77 (м, ароматический протон, 1H), 7.49 (м, ароматический протон, 1H), 2.31 (т, J = 7.2 Гц, CH2CONHAr, 2H), 1.93 (т, J = 7.2 Гц, CH2CONHOH, 2H), 1.51 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д): 10.35 (с, NHPh, 1H), 10.31 (с, NHOH, 1H), 8.63 (с, NHOH + ароматический протон, 2H), 7.88 (д, J = 8 Гц, ароматические протоны, 2H), 7.57 (т, J = 8 Гц, ароматический протон, 1H), 2.33 (т, J = 7.6 Гц, CH2CONHAr, 2H), 1.93 (т, J = 7.4 Гц, CH2CONHOH, 2H), 1.52 (м, 4H), 1.27 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д): 10.33 (с, NHOH, 1H), 10.15 (с, NHAr, 1H), 10.09 (с, NHPh, 1H), 8.66 (с, NHOH, 1H), 7.91 (д, J = 8.6 Гц, ароматические протоны, 2H), 7.76 (д, J = 7.8 Гц, орто-протоны анилида, 2H), 7.71 (д, J = 8,6 Гц, ароматические протоны, 2H), 7,33 (т, J = 7.6 Гц, мета-протоны анилида, 2H), 7.07 (т, J = 7.4 Гц, пара-протоны анилида), 2.33 (т, J = 7.5 Гц, CH2CONHAr, 2H), 1.93 (т, J = 7.2 Гц, CH2CONHOH, 2H), 1.51 (м, 4H), 1.28 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д): 10.32 (с, NHOH, 1H), 10.21 (с, NHPh, 1H), 8.65 (с, NHOH, 1H), 7.31 (дд, J = 10 и 2.2 Гц, ароматические протоны, 2H), 6.84 (тт, J = 9.4 и 2.4 Гц, ароматические протоны, 1H), 2.29 (т, CH2CONHAr, 2H), 1.93 (т, J = 7.2 Гц, CH2CONHOH, 2H), 1.51 (м, 4H), 1.26 (м, 4H).
Аналогичным образом были получены и охарактеризованы следующие соединения:

где n = 4, 5, 6, 7 и 8;
заместитель R представляет собой 2-, 3- и 4-циано, 2-, 3- и 4-цианометил, 2-, 3- и 4-нитро, 2-, 3- и 4-карбокси, 2-, 3- и 4-аминокарбонил, 2-, 3- и 4-метиламинокарбонил, 2-, 3- и 4-диметиламинокарбонил и 2-, 3- и 4-трифторметил;

где заместитель R представляет собой 4-гидроксиаминокарбонил, 4-метоксикарбонил, 4-тетразолил, 2-, 3- и 4-хлор, 2-, 3- и 4-фтор, 2-, 3- и 4-метил, 2-, 3- и 4-метокси, 2,3-дифтор, 2,4-дифтор, 2,5-дифтор, 2,6-дифтор, 1,2,3-трифтор, 3,4,5-трифтор, 2,4,5-трифтор, 2,4,6-трифтор, 2,3,6-трифтор, 2,3,5, 6-тетрафтор, 2,3,4,5,6-пентафтор, 2-, 3- и 4-фенил, 2-, 3- и 4-бензилокси, 4-гексил и 4-трет.-бутил;


Соединения, имеющие структуру

где n = 4, 5, 6, 7 и 8;
заместитель R представляет собой атом водорода или метил.
К раствору гидроксида калия (1.68 г, 0.03 моля), анилина или N-метиланилина (0,01 моля) и хлоргидрата диметиламина (0.805 г, 0.01 моля) в 50%-ном тетрагидрофуране (100 мл) при перемешивании добавляют дихлорангидрид дикарбоновой кислоты (0.01 моля). Реакционную смесь перемешивают в течение 30 мин при комнатной температуре и распределяют между хлороформом (400 мл) и водой (300 мл). Органический слой промывают 10%-ной соляной кислотой (3х100 мл), 10%-ным раствором гидроксида калия (3х100 мл) и водой (2х100 мл). Органический слой сушат безводным сульфатом магния и упаривают. Твердый остаток промывают гексаном и отфильтровывают. Выход составляет 25-34%.

Спектр1H ЯМР (ДМСО-d6 , 200 МГц), δ (м.д): 9.82 (с, NHPh, 1H), 7.58 (д, J = 7.6 Гц, орто-ароматические протоны, 2H), 7.26 (т, J = 7.4 Гц, мета-ароматические протоны, 2H), 6.99 (т, J = 7.4 Гц, пара-ароматические протоны, 2H), 2.85 (д, J = 28 Гц, N(CH3)2, 6H), 2.28 (т, J = 7.2 Гц, CH2CO, 2H), 2.24 (т, J = 7.4 Гц, CH2CO, 2H), 1.51 (м, 4H), 1.29 (м, 4H).

Спектр1H ЯМР (ДМСО-d6, 200 МГц), δ (м.д): 7.30 (м, C6H5, 5H), 3.13 (с, H3CNPh, 3H), 2.83 (д, J = 26 Гц, N(CH3)2, 6H), 2.17 (т, J = 7.6 Гц, CH2CON(CH3)2, 2H), 1.98 (т, J = 7.4 Гц, CH2CON(CH3)Ph, 2H), 1.41 (м, 4H), 1.11 (м, 4H).
Пример приготовления фармацевтической композиции, включающей заявленное соединение формулы I
Раствор соединения формулы I
Соединение 2, г - 5
Стерильная апирогенная вода - Остальное до 100 мл
Далее производят необходимое разведение в зависимости от веса пациента и
вида опухоли для создания оптимальной концентрации в организме.
Реферат
Соединения формулы R1-С(О)-(СH2)2-C(О)-R2, где каждый из заменителей R1 и R2 независимо друг от друга принимают одинаковые или разные значения; если R1 и R2 являются одинаковыми, каждый из них представляет собой замещенную или незамещенную ариламино-, циклоалкиламино-, пиридиламино-, пиперидино-, 9-пурин-6-амино- или тиазолиламиногруппу; eсли R1 и R2 различны, то R1 представляет группу R3-N-R4, где R3 и R4 независимо друг от друга одинаковые или различные и каждый представляет собой -Н, ОН, замещенный или незамещенный, линейный или разветвленный алкил, алкенил, циклоалкил, арил, алкилокcи-, арилокcи-, арилалкилокси-, пиридильную группу, или R3 и R4 вместе образуют пиперидиновую группу, a R2 представляет собой гидроксиламино-, -ОН, NН2-, алкиламино-, диалкиламино-, алкоксигруппу; n = 4 - 8. Используют для приготовления фармацевтической композиции, ингибирующей пролиферацию клеток опухоли. 2 с. и 18 з.п. ф-лы, 2 табл.
Формула

где каждый из заместителей R1 и R2 независимо друг от друга принимает одинаковые или разные значения; если заместители R1 и R2 являются одинаковыми, каждый из них представляет собой замещенную или незамещенную ариламино-, циклоалкиламино-, пиридиламино-, пиперидино-, 9-пурин-6-амино или тиазолиламиногруппу; если заместители R1 и R2 различны, то R1 представляет собой группу R3-N-R4, где каждый из заместителей R3 и R4 независимо друг от друга принимает одинаковые или разные значения и представляет собой атом водорода, гидроксильную группу, замещенный или незамещенный, линейный или разветвленный алкил, алкенил, циклоалкил, арил, алкилокси-, арилокси-, арилалкилокси- или пиридиальную группу, или заместители R3 и R4 вместе образуют пиперидиновую группу, а заместитель R2 представляет собой гидроксиламино-, гидроксильную, амино-, алкиламино-, диалкиламино- или алкоксильную группу;
n принимает целые значения от 4 до 8.

где каждый из заместителей R3 и R4 независимо друг от друга принимает одинаковые или разные значения и представляет собой атом водорода, гидроксильную группу, замещенный или незамещенный, линейный или разветвленный алкил, алкенил, циклоалкил, арил, алкилокси-, арилокси-, арилалкилокси- или пиридильную группу, или заместители R3 и R4 вместе образуют пиперидиновую группу;
заместитель R2 представляет собой гидроксиламино-, гидроксильную, амино-, алкиламино-, диалкиламино- или алкоксильную группу;
n принимает целые значения от 4 до 8.

отличающееся тем, что заместитель R представляет собой замещенную или незамещенную ариламино-, циклоалкиламино-, пиридиламино-, пиперидино-, 9-пурин-6-амино или тиазолиламиногруппу, а n принимает целые значения от 4 до 8.
Комментарии