Ингибиторы неприлизина - RU2629930C2
Код документа: RU2629930C2
Описание
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Область техники, к которой относится изобретение
Настоящее изобретение относится к новым соединениям, обладающим активностью ингибирования неприлизина. Изобретение относится к фармацевтическим композициям, содержащим такие соединения, способам и промежуточным соединениям для получения таких соединений и способам применения таких соединений для лечения таких заболеваний, как гипертензия, сердечная недостаточность, легочная гипертензия и почечные заболевания.
Уровень техники
Неприлизин (нейтральная эндопептидаза, EC 3.4.24.11) (NEP) представляет собой связанную с эндотелиальной мембраной Zn2+ металлопептидазу, обнаруживаемую во многих органах и тканях, включая мозг, почки, легкие, желудочно-кишечный тракт, сердце и периферическую сосудистую сеть. NEP разрушает и инактивирует ряд эндогенных пептидов, таких как энкефалины, циркулирующий брадикинин, ангиотензиновые пептиды и натрийуретические пептиды, последние из которых оказывают несколько эффектов, включая, например, расширение сосудов и натрийурез/диурез, а также ингибирование сердечной гипертрофии и желудочкового фиброза. Таким образом, NEP играет важную роль в гомеостазе артериального давления и здоровье сердечно-сосудистой системы.
Ингибиторы NEP, такие как тиорфан, кандоксатрил и кандоксатрилат, исследовали в качестве потенциальных терапевтических средств. Известны также соединения, которые ингибируют и NEP, ангиотензин-I превращающий фермент (ACE), и они включают омапатрилат, гемпатрилат и сампатрилат. Этот последний из указанных класс соединений, именуемых ингибиторами вазопептидазы, описан в публикации Robl et al. (1999) Exp. Opin. Ther. Patents 9(12): 1665-1677.
Ksander et al. (1995) J. Med. Chem. 38: 1689-1700 описывают дипептидные ингибиторы NEP дикарбоновых кислот формулы:

Соединение 21a, которое имеет заместитель янтарной кислоты, представляет собой самое активное соединение с IC50 5 нМ. Авторы наблюдали, что «янтарная кислота в участке P2' представляется оптимальной, поскольку удлинение цепи карбоновой кислоты на одну (21e) и две (21f) метиленовые единицы снижает активность в 18 и 65 раз». Авторы, кроме того, отметили, что «уменьшение длины цепи на один метилен (21g) также проявил 18-кратное уменьшение активности» (стр. 1692, 2ая колонка).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым соединениям, которые, как было обнаружено, обладают активностью ингибирования фермента неприлизина (NEP). Соответственно, ожидается, что соединения по изобретению полезны и имеют преимущества в качестве терапевтических средств для лечения таких состояний, как гипертензия и сердечная недостаточность.
Один аспект изобретения относится к соединению формулы I:

где:
R1 выбран из H, -C1-8алкила, -C1-3алкилен-C6-10арила, -C1-3алкилен-C1-9гетероарила, -C3-7циклоалкила, -[(CH2)2O]1-3CH3, -C1-6алкилен-OC(O)R10, -C1-6алкилен-NR11R12, -C1-6алкилен-C(O)R13, -C0-6алкиленморфолинила, -C1-6алкилен-SO2-C1-6алкила,


R10 выбран из -C1-6алкила, -O-C1-6алкила, -C3-7циклоалкила, -O-C3-7циклоалкила, фенила, -O-фенила, -NR11R12, -CH(R15)-NH2, -CH(R15)-NHC(O)O-C1-6алкила и -CH(NH2)CH2COOCH3; и R11 и R12 независимо выбраны из H, -C1-6алкила и бензила; или R11 и R12 взяты вместе в виде -(CH2)3-6-, -C(O)-(CH2)3- или -(CH2)2O(CH2)2-; R13 выбран из -O-C1-6алкила, -O-бензила и -NR11R12; и R14 обозначает -C1-6алкил или -C0-6алкилен-C6-10арил; R15 обозначает H, -CH3, -CH(CH3)2, фенил или бензил;
R2 представляет собой -OR21 или -CH2OR21; и R3 представляет собой H или -CH3; где R21 представляет собой H, -C(O)-C1-6алкил, -C(O)-CH(R22)-NH2, -C(O)-CH(R22)-NHC(O)O-C1-6алкил или -P(O)(OR23)2; R22 представляет собой H, -CH3, -CH(CH3)2, фенил или бензил; R23 представляет собой H, -C1-6алкил или фенил; или
R2 взят вместе с группой R1 с образованием -OCR15R16- или -CH2O-CR15R16-, и R3 выбран из H и -CH3, где R15 и R16 независимо выбраны из H, -C1-6алкила и -O-C3-7циклоалкила, или R15 и R16 взяты вместе с образованием =O; или
R2 взят вместе с R3 с образованием -CH2-O-CH2- или -CH2-CH2-; или
и R2, и R3 представляют собой -CH3;
Z выбран из -CH- и -N-;
R4 выбран из H, -C1-8алкила, -C1-3алкилен-O-C1-8алкила, -C1-3алкилен-C6-10арила, -C1-3алкилен-O-C6-10арила, -C1-3алкилен-C1-9гетероарила, -C3-7циклоалкила, -[(CH2)2O]1-3CH3, -C1-6алкилен-OC(O)R40, -C1-6алкилен-NR41R42, -C1-6алкилен-C(O)R43, -C0-6алкиленморфолинила, -C1-6алкилен-SO2-C1-6алкила,

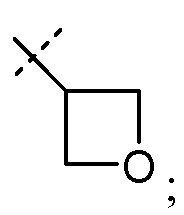
R40 выбран из -C1-6алкила, -O-C1-6алкила, -C3-7циклоалкила, -O-C3-7циклоалкила, фенила, -O-фенила, -NR41R42, -CH(R45)-NH2, -CH(R45)-NHC(O)O-C1-6алкила и -CH(NH2)CH2COOCH3; и R41 и R42 независимо выбраны из H, -C1-6алкила и бензила; или R41 и R42 взяты вместе в виде -(CH2)3-6-, -C(O)-(CH2)3- или -(CH2)2O(CH2)2-; R43 выбран из -O-C1-6алкила, -O-бензила и -NR41R42; и R44 представляет собой -C1-6алкил или -C0-6алкилен-C6-10арил; R45 представляет собой H, -CH3, -CH(CH3)2, фенил или бензил;
a равно 0 или 1; R5 выбран из галогена, -CH3, -CF3 и -CN;
b равно 0 или целому числу от 1 до 3; каждый R6 независимо выбран из галогена, -OH, -CH3, -OCH3, -CN и -CF3;
где каждая алкильная группа в R1 и R4 необязательно замещена 1-8 атомами фтора; и
где метиленовый линкер на бифениле необязательно замещен одной или двумя -C1-6алкильными группами или циклопропилом;
или его фармацевтически приемлемой соли.
Другой аспект изобретения относится к фармацевтическим композициям, содержащим фармацевтически приемлемый носитель и соединение по изобретению. Такие композиции могут необязательно содержать другие терапевтические средства. Соответственно, в еще одном аспекте изобретения, фармацевтическая композиция содержит соединение по изобретению в качестве первого терапевтического средства, одно или несколько вторичных терапевтических средств, и фармацевтически приемлемый носитель. Другой аспект изобретения относится к комбинации активных средств, содержащей соединение по изобретению и второе терапевтическое средство. Соединение по изобретению может быть составлено вместе или отдельно от дополнительного средства(-в). При включении в состав отдельно, фармацевтически приемлемый носитель может быть включен с дополнительным средством(-ами). Таким образом, еще один аспект изобретения относится к комбинации фармацевтических композиций, причем комбинация содержит: первую фармацевтическую композицию, содержащую соединение по изобретению и первый фармацевтически приемлемый носитель; и вторую фармацевтическую композицию, содержащую второе терапевтическое средство и второй фармацевтически приемлемый носитель. В другом аспекте изобретение относится к набору, содержащему такие фармацевтические композиции, например, где первая и вторая фармацевтические композиции представляют собой отдельные фармацевтические композиции.
Соединения по изобретению обладают активностью ингибирования фермента NEP, и поэтому ожидается, что они могут быть полезны в качестве терапевтических средств для лечения пациентов, страдающих заболеванием или расстройством, которое лечится ингибированием фермента NEP, или увеличением уровней его пептидных субстратов. Таким образом, один аспект изобретения относится к способу лечения пациентов, страдающих заболеванием или расстройством, которое лечится ингибированием фермента NEP, включающему введение пациенту терапевтически эффективного количества соединения по изобретению. Другой аспект изобретения относится к способу лечения гипертензии, сердечной недостаточности или почечного заболевания, включающему введение пациенту терапевтически эффективного количества соединения по изобретению. Еще один аспект изобретения относится к способу ингибирования фермента NEP у млекопитающего, включающему введение млекопитающему ингибирующее фермент NEP количество соединения по изобретению.
Поскольку соединения по изобретению обладают ингибирующей NEP активностью, то они также полезны в качестве инструментов исследований. Соответственно, один аспект изобретения относится к способу применения соединения по изобретению в качестве инструмента исследования, причем способ включает проведение биологического анализа с использованием соединения по изобретению. Соединения по изобретению можно также применять для оценки новых химических соединений. Таким образом, другой аспект изобретения относится к способу оценки испытываемого соединения в биологическом анализе, включающему: (a) проведение биологического анализа испытываемого соединения для получения первой аналитической величины; (b) проведение биологического анализа соединения по изобретению для получения второй аналитической величины; причем стадия (a) проводится перед, после или одновременно со стадией (b); и (c) сравнение первой аналитической величины со стадии (a) со второй аналитической величиной со стадии (b). Иллюстративные анализы включают анализ ингибирования фермента NEP. Еще один аспект изобретения относится к способу изучения биологической системы или образца, содержащего фермент NEP, причем способ включает: (a) приведение биологической системы или образца в контакт с соединением по изобретению; и (b) определение эффектов, вызванных соединением, на биологическую систему или образец.
Еще один аспект изобретения относится к способам и промежуточным соединениям, полезным для получения соединений по изобретению. Соответственно, другой аспект изобретения относится к способу получения соединений формулы I, включающему стадию связывания соединения формулы 1 с соединением формулы 2:
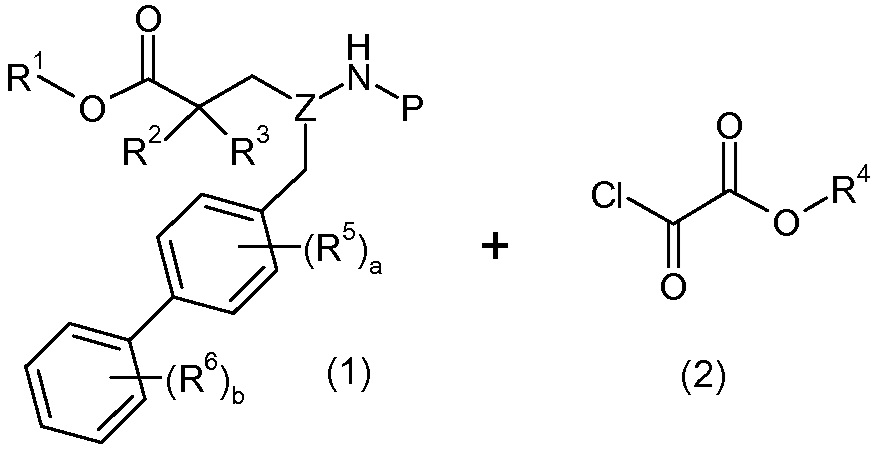
для получения соединения формулы I; где P представляет собой H или амино-защитную группу, выбранную из трет-бутоксикарбонила, тритила, бензилоксикарбонила, 9-флуоренилметоксикарбонила, формила, триметилсилила и трет-бутилдиметилсилила; и где способ дополнительно включает снятие защиты соединения формулы 1, когда P представляет собой амино-защитную группу; и где R1-R6, a, b и Z такие, как определено для формулы I. Другой аспект изобретения относится к способу получения фармацевтически приемлемой соли соединения формулы I, включающему приведение соединения формулы I в форме свободной кислоты или основания в контакт с фармацевтически приемлемым основанием или кислотой. В других аспектах изобретение относится к продуктам, полученным любым из описанных здесь способов, а также к новым промежуточным соединениям, используемым в таком способе. В одном аспекте изобретения новые промежуточные соединения или их соли имеют формулу 1, как определено в настоящем описании.
Еще один аспект изобретения относится к применению соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства, в особенности, для получения лекарственного средства, полезного для лечения гипертензии, сердечной недостаточности или почечного заболевания. Другой аспект изобретения относится к применению соединения по изобретению для ингибирования фермента NEP у млекопитающего. Еще один аспект изобретения относится к применению соединения по изобретению в качестве инструмента исследования. Другие аспекты и варианты осуществления изобретения описаны ниже.
Особая группа соединений формулы I представляет собой те соединения, которые описаны в предварительной заявке на патент США № 61/554,625, поданной 2 ноября 2011 г. Эта группа включает соединения формулы I'; где:

где: R1 выбран из H, -C1-8алкила, -C1-3алкилен-C6-10арила, -C1-3алкилен-C1-9гетероарила, -C3-7циклоалкила, -[(CH2)2O]1-3CH3, -C1-6алкилен-OC(O)R10, -C1-6алкилен-NR11R12, -C1-6алкилен-C(O)R13, -C0-6алкиленморфолинила, -C1-6алкилен-SO2-C1-6алкила,


R10 выбран из -C1-6алкила, -O-C1-6алкила, -C3-7циклоалкила, -O-C3-7циклоалкила, фенила, -O-фенила, -NR11R12, -CH[CH(CH3)2]-NH2, -CH[CH(CH3)2]-NHC(O)O-C1-6алкила и -CH(NH2)CH2COOCH3; и R11 и R12 независимо выбраны из H, -C1-6алкила и бензила; или R11и R12 взяты вместе в виде -(CH2)3-6-, -C(O)-(CH2)3- или -(CH2)2O(CH2)2-; R13 выбран из -O-C1-6алкила, -O-бензила и -NR11R12; и R14 представляет собой -C1-6алкил или -C0-6алкилен-C6-10арил; R2 выбран из -OH, -CH2OH, -OP(O)(OH)2 и -CH2OP(O)(OH)2; и R3 выбран из H и -CH3; или R2 взят вместе с R1 с образованием -OCR15R16- или -CH2O-CR15R16-, и R3 выбран из H и -CH3, где R15 и R16 независимо выбраны из H, -C1-6алкила и -O-C3-7циклоалкила, или R15 и R16 взяты вместе с образованием =O; или R2 взят вместе с группой R3 с образованием -CH2-O-CH2- или -CH2-CH2-; или оба R2 и R3 представляют собой -CH3; Z выбран из -CH- и -N-; R4 выбран из H, -C1-8алкила, -C1-3алкилен-C6-10арила, -C1-3алкилен-C1-9гетероарила, -C3-7циклоалкила, -[(CH2)2O]1-3CH3, -C1-6алкилен-OC(O)R40, -C1-6алкилен-NR41R42, -C1-6алкилен-C(O)R43, -C0-6алкиленморфолинила, -C1-6алкилен-SO2-C1-6алкила,


R40 выбран из -C1-6алкила, -O-C1-6алкила, -C3-7циклоалкила, -O-C3-7циклоалкила, фенила, -O-фенила, -NR41R42, -CH[CH(CH3)2]-NH2, -CH[CH(CH3)2]-NHC(O)O-C1-6алкила и -CH(NH2)CH2COOCH3; и R41 и R42 независимо выбраны из H, -C1-6алкила и бензила; или R41 и R42 взяты вместе в виде -(CH2)3-6-, -C(O)-(CH2)3- или -(CH2)2O(CH2)2-; R43 выбран из -O-C1-6алкила, -O-бензила и -NR41R42; и R44 представляет собой -C1-6алкил или -C0-6алкилен-C6-10арил; a равно 0 или 1; R5 выбран из галогена, -CH3, -CF3 и -CN; b равно 0 или целому числу от 1 до 3; каждый R6 независимо выбран из галогена, -OH, -CH3, -OCH3 и -CF3; и где каждая алкильная группа в R1 и R4 необязательно замещена 1-8 атомами фтора; и где метиленовый линкер на бифениле необязательно замещен одной или двумя -C1-6алкильными группами или циклопропилом; или их фармацевтически приемлемую соль.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Если не указано иное, при описании соединений, композиций, способов и процессов по изобретению следующие термины имеют следующие значения. Кроме того, пока контекст применения ясно не диктует иного, используемые в настоящем описании формы единственного числа с неопределенными и определенными артиклями включают соответствующие формы множественного числа. Термины «содержащий», «включающий» и «имеющий» предназначены для включения и означают, что могут быть дополнительные элементы, отличные от перечисленных элементов. Если не указано иное, все числа, выражающие количества ингредиентов, свойств, таких как молекулярная масса, условия реакций и т.д., используемые в настоящем описании, следует понимать как являющиеся модифицированными во всех случаях термином «примерно». Соответственно, числа, представленные в настоящем описании, представляют собой приблизительные величины, которые могут варьироваться в зависимости от требуемых свойств, к получению которых стремится настоящее изобретение. По меньшей мере, а не в качестве попытки ограничения применения доктрины эквивалентов объемом формулы изобретения, каждое число следует рассматривать по меньшей мере в свете представленных значащих цифр и применением обычных методик округления.
Термин «алкил» означает одновалентную насыщенную углеводородную группу, которая может быть линейной или разветвленной. Если не определено иное, такие алкильные группы обычно содержат от 1 до 10 атомов углерода и включают, например, -C1-4алкил, -C1-5алкил, -C2-5алкил, -C1-6алкил, -C1-8алкил и -C1-10алкил. Репрезентативные алкильные группы включают в качестве примера метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил и т.п.
Когда для конкретного термина, используемого в настоящем описании, предназначено определенное число атомов углерода, то число атомов углерода показано перед термином в виде подстрочного обозначения. Например, термин «-C1-6алкил» означает алкильную группу, имеющую от 1 до 6 атомов углерода, а термин «-C3-7циклоалкил» означает циклоалкильную группу, имеющую от 3 до 7 атомов углерода, соответственно, где атомы углерода находятся в любой приемлемой конфигурации.
Термин «алкилен» означает двухвалентную насыщенную углеводородную группу, которая может быть линейной или разветвленной. Если не определено иное, такие алкиленовые группы обычно содержат от 0 до 10 атомов углерода и включают, например, -C0-1алкилен-, -C0-6алкилен-, -C1-3алкилен- и -C1-6алкилен-. Репрезентативные алкиленовые группы включают в качестве примера метилен, этан-1,2-диил («этилен»), пропан-1,2-диил, пропан-1,3-диил, бутан-1,4-диил, пентан-1,5-диил и т.п. Понятно, что когда термин "алкилен" включает ноль атомов углерода, такой как -C0-1алкилен-, такие термины предназначены для включения отсутствия атомов углерода, то есть алкиленовая группа не присутствует, за исключением ковалентной связи, соединяющей группы, разделенные термином "алкилен".
Термин «арил» означает одновалентный ароматический углеводород, имеющий одно кольцо (т.е. фенил) или одно или несколько конденсированных колец. Конденсированные кольцевые системы включают те, которые являются полностью ненасыщенными (например, нафталин), а также те, которые являются частично ненасыщенными (например, 1,2,3,4-тетрагидронафталин). Если не определено иное, такие арильные группы обычно содержат от 6 до 10 кольцевых атомов углерода и включают, например, -C6-10арил. Репрезентативные арильные группы включают в качестве примера фенил и нафталин-1-ил, нафталин-2-ил и т.п.
Термин «циклоалкил» означает одновалентную насыщенную карбоциклическую углеводородную группу. Если не определено иное, такие циклоалкильные группы обычно содержат от 3 до 10 атомов углерода и включают, например, -C3-5циклоалкил, -C3-6циклоалкил и -C3-7циклоалкил. Репрезентативные циклоалкильные группы включают в качестве примера циклопропил, циклобутил, циклопентил, циклогексил и т.п.
Термин «галоген» означает фтор, хлор, бром и йод.
Термин «гетероарил» предназначен для обозначения одновалентного ненасыщенного (ароматического) гетероцикла, имеющего одно кольцо или два конденсированных кольца. Одновалентные ненасыщенные гетероциклы обычно называются «гетероарильными» группами. Если не определено иное, гетероарильные группы обычно содержат в общей сложности от 5 до 10 кольцевых атомов, из которых от 1 до 9 являются кольцевыми атомами углерода, и от 1 до 4 являются кольцевыми гетероатомами и включают, например, -C1-9гетероарил и -C5-9гетероарил. Репрезентативные гетероарильные группы включают в качестве примера пиррол (например, 3-пирролил и 2H-пиррол-3-ил), имидазол (например, 2-имидазолил), фуран (например, 2-фурил и 3-фурил), тиофен (например, 2-тиенил), триазол (например, 1,2,3-триазолил и 1,2,4-триазолил), пиразол (например, 1H-пиразол-3-ил), оксазол (например, 2-оксазолил), изоксазол (например, 3-изоксазолил), тиазол (например, 2-тиазолил и 4-тиазолил), и изотиазол (например, 3-изотиазолил), пиридин (например, 2-пиридил, 3-пиридил и 4-пиридил), пиридилимидазол, пиридилтриазол, пиразин, пиридазин (например, 3-пиридазинил), пиримидин (например, 2-пиримидинил), тетразол, триазин (например, 1,3,5-триазинил), индолил (например, 1H-индол-2-ил, 1H-индол-4-ил и 1H-индол-5-ил), бензофуран (например, бензофуран-5-ил), бензотиофен (например, бензо[b]тиен-2-ил и бензо[b]тиен-5-ил), бензимидазол, бензоксазол, бензотиазол, бензотриазол, хинолин (например, 2-хинолил), изохинолин, хиназолин, хиноксалин и т.п.
Термин «необязательно замещенный» означает, что рассматриваемая группа может быть незамещенной, или она может быть замещена один или несколько раз, например, от 1 до 3 раз или от 1 до 5 раз или от 1 до 8 раз. Например, алкильная группа, которая является «необязательно замещенной» атомами фтора, может быть незамещенной, или она может содержать 1, 2, 3, 4, 5, 6, 7 или 8 атомов фтора. Аналогичным образом, группа, которая является «необязательно замещенной» одной или двумя -C1-6алкильными группами, может быть незамещенной, или она может содержать одну или две -C1-6алкильные группы.
Используемая в настоящем описании фраза «имеющее формулу» или «имеющее структуру» не предназначена для ограничения и используется так же, как обычно используется термин «содержащее». Например, если изображена одна структура, то понятно, что, если не указано иное, охватываются стереоизомерные и таутомерные формы.
Термин «фармацевтически приемлемый» относится к материалу, который не является биологически или иным образом неприемлемым при использовании в изобретении. Например, термин «фармацевтически приемлемый носитель» относится к материалу, который может быть включен в композицию и вводится пациенту, не вызывая неприемлемых биологических эффектов или не взаимодействуя неприемлемым образом с другими компонентами композиции. Такие фармацевтически приемлемые материалы обычно соответствуют требуемым стандартам токсикологического и производственного тестирования и включают те материалы, которые идентифицированы как пригодные неактивные ингредиенты Управлением по контролю за пищевыми продуктами и лекарственными средствами США.
Термин «фармацевтически приемлемая соль» означает соль, полученную из основания или кислоты, которая приемлема для введения пациенту, такому как млекопитающее (например, соли, имеющие приемлемую безопасность для млекопитающих для данной схемы дозировки). Однако понятно, что не требуется, чтобы соли, охватываемые изобретением, представляли собой фармацевтически приемлемые соли, такие как соли промежуточных соединений, которые не предназначены для введения пациенту. Фармацевтически приемлемые соли могут быть получены из фармацевтически приемлемых неорганических или органических оснований и из фармацевтически приемлемых неорганических или органических кислот. Кроме того, когда соединение формулы I содержит и основную составляющую, такую как амин, пиридин или имидазол, и кислотную составляющую, такую как карбоновая кислота или тетразол, могут образовываться цвиттерионы, и они охвачены термином «соль», используемым в настоящем описании. Соли, полученные из фармацевтически приемлемых неорганических оснований, включают соли аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия и цинка и т.п. Соли, полученные из фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, включая замещенные амины, циклические амины, встречающиеся в природе амины и т.п., такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Соли, полученные из фармацевтически приемлемых неорганических кислот, включают соли борной, карбоновой, галогенводородной (бромистоводородной, хлористоводородной, фтористоводородной или йодистоводородной), азотной, фосфорной, сульфамовой и серной кислот. Соли, полученные из фармацевтически приемлемых органических кислот, включают соли алифатических гидроксикислот (например, лимонной, глюконовой, гликолевой, молочной, лактобионовой, яблочной и винной кислот), алифатических монокарбоновых кислот (например, уксусной, масляной, муравьиной, пропионовой и трифторуксусной кислот), аминокислот (например, аспарагиновой и глутаминовой кислот), ароматических карбоновых кислот (например, бензойной, п-хлорбензойной, дифенилуксусной, гентизовой, гиппуровой и трифенилуксусной кислот), ароматических гидроксикислот (например, о-гидроксибензойной, п-гидроксибензойной, 1-гидроксинафталин-2-карбоновой и 3- гидроксинафталин-2-карбоновой кислот), аскорбиновой, дикарбоновой кислот (например, фумаровой, малеиновой, щавелевой и янтарной кислот), глюкуроновой, миндальной, муциновой, никотиновой, оротовой, памоевой, пантотеновой, сульфоновой кислот (например, бензолсульфоновой, камфосульфоновой, эдисиловой, этансульфоновой, изетионовой, метансульфоновой, нафталинсульфоновой, нафталин-1,5-дисульфоновой, нафталин-2,6-дисульфоновой и п-толуолсульфоновой кислот), ксинафоевой кислоты и т.п.
Используемый в настоящем описании термин «пролекарство» в целом предназначен для обозначения неактивного предшественника лекарственного средства, который превращается в активную форму в организме в физиологических условиях, например, посредством нормальных метаболических процессов. Такие соединения могут не обладать фармакологической активностью в отношении NEP, но могут вводиться перорально или парентерально и затем метаболизироваться в организме с образованием соединений, которые являются фармакологически активными в отношении NEP. При пероральном введении такие соединения могут также обеспечить лучшую абсорбированную фракцию (т.е. лучшие pK свойства) для почечной доставки, по сравнению с пероральным введением активной формы. Иллюстративные пролекарства включают сложные эфиры, такие как C1-6алкилэфиры и арил-C1-6алкилэфиры. В одном варианте осуществления активное соединение имеет свободный карбоксил, и пролекарство представляет собой его сложноэфирное производное, т.е. пролекарство представляет собой сложный эфир, такой как -C(O)OCH2CH3. Такие сложноэфирные пролекарства затем превращаются сольволизом или в физиологических условиях в свободное карбоксильное соединение. Термин «пролекарство» также предназначен для включения менее активного предшественника лекарственного средства, который превращается в более активную форму в организме. Например, определенные пролекарства могут обладать фармакологической активностью в отношении NEP, но необязательно на требуемом уровне; такие соединения превращаются в организме в форму, имеющую требуемый уровень активности. Этот термин также предназначен для включения определенных защищенных производных соединений формулы I, которые могут быть получены перед конечной стадией снятия защиты. Таким образом, все защищенные производные и пролекарства соединений формулы I включены в объем изобретения.
Термин «терапевтически эффективное количество» означает количество, достаточное для осуществления лечения при введении нуждающемуся в этом пациенту, то есть, количество лекарственного средства, необходимое для получения требуемого терапевтического эффекта. Например, терапевтически эффективное количество для лечения гипертензии представляет собой количество соединения, необходимое, например, для уменьшения, подавления, устранения или предотвращения симптомов гипертензии, или для лечения лежащей в основе причины гипертензии. В одном варианте осуществления терапевтически эффективное количество представляет собой то количество лекарственного средства, которое требуется для снижения артериального давления, или количество лекарственного средства, которое требуется для поддержания нормального артериального давления. С другой стороны, термин «эффективное количество» означает количество, достаточное для получения требуемого результата, который может необязательно представлять собой терапевтический результат. Например, при изучении системы, содержащей фермент NEP, «эффективное количество» может представлять собой количество, требуемое для ингибирования фермента.
Используемый в настоящем описании термин «лечить» или «лечение» означает лечение заболевания или медицинского показания (такого как гипертензия) у пациента, такого как млекопитающее (в частности, человек), которое включает одно или несколько из следующих: (a) предотвращение возникновения заболевания или медицинского показания, т.е. предотвращение рецидива заболевания или медицинского показания или профилактическое лечение пациента, который предрасположен к заболеванию или медицинскому показанию; (b) облегчение течения заболевания или медицинского показания, т.е. устранение или вызов обратного развития заболевания или медицинского показания у пациента; (c) подавление заболевания или медицинского показания, т.е. замедление или остановка развития заболевания или медицинского показания у пациента; или (d) облегчение симптомов заболевания или медицинского показания у пациента. Например, термин «лечение гипертензии» может включать предотвращение возникновения гипертензии, облечение течения гипертензии, подавление гипертензии и облегчение симптомов гипертензии (например, снижение артериального давления). Термин «пациент» предназначен для включения таких млекопитающих, как люди, которые нуждаются в лечении или предотвращении заболевания, или которые в настоящее время получают лечение для предотвращения или излечения определенного заболевания или медицинского показания, а также лечения индивидов, у которых соединения по изобретению оцениваются или используются при анализе, например, на экспериментальной модели.
Предполагается, что все другие термины, используемые в настоящем описании, имеют обычное значение, понятное специалистам в данной области техники, для которых они предназначены.
В одном аспекте изобретение относится к соединениям формулы I:

или их фармацевтически приемлемым солям.
Используемый в настоящем описании термин «соединение по изобретению» включает все соединения, охватываемые формулой I, такие как виды, представленные формулами Ia и Ib, а также соединения, охватываемые формулами IIa-IIk, IIIa-IIIb и IVa-IVd. Кроме того, соединения по изобретению могут также содержать несколько основных или кислотных групп (например, амино- или карбоксильных групп), и поэтому такие соединения могут существовать в виде свободного основания, свободной кислоты или в различных солевых формах. Все такие солевые формы включены в объем изобретения. Кроме того, соединения по изобретению могут также существовать в виде пролекарств. Соответственно, специалистам в данной области техники понятно, что, если не указано иное, ссылка в настоящем описании на соединение, например, ссылка на «соединение по изобретению» или «соединение формулы I» включает соединение формулы I, а также фармацевтически приемлемые соли и пролекарства этого соединения. Кроме того, термин «или его фармацевтически приемлемая соль и/или пролекарство» предназначен для включения всех комбинаций солей и пролекарств, таких как фармацевтически приемлемая соль пролекарства. Кроме того, сольваты соединений формулы I включены в объем настоящего изобретения.
Соединения формулы I могут содержать один или несколько хиральных центров, и поэтому эти соединения могут бьыть получены и использоваться в различных стереоизомерных формах. Соответственно, если не указано иное, изобретение также относится к рацемическим смесям, чистым стереоизомерам (например, энантиомерам и диастереоизомерам), обогащенным стереоизомерами смесям и т.п. Когда в настоящем описании химическая структура изображена без какой-либо стереохимии, то следует понимать, что такой структурой охватываются все возможные стереоизомеры. Таким образом, например, термины «соединение формулы I», «соединения формулы II» и т.д. предназначены для включения всех возможных стереоизомеров соединения. Аналогичным образом, когда в настоящем описании показан или назван конкретный стереоизомер, то специалистам в данной области техники следует понимать, что, если не указано иное, в композиции по изобретению могут присутствовать небольшие количества других стереоизомеров, при условии, что возможность применения композиции в целом не исключается присутствием таких других изомеров. Отдельные стереоизомеры могут быть получены многочисленными способами, которые хорошо известны в данной области техники, включая хиральную хроматографию с использованием подходящей хиральной стационарной фазы или носителя, или химическим превращением их в диастереоизомеры, разделением диастереоизомеров обычными средствами, такими как хроматография или перекристаллизация, затем регенерацией первоначального стереоизомера.
Кроме того, где целесообразно, если конкретно не указано иное, все цис-транс или E/Z изомеры (геометрические изомеры), таутомерные формы и топоизомерные формы соединений по изобретению включены в объем изобретения.
Конкретнее, соединения формулы I могут содержать по меньшей мере два хиральных центра, когда составляющая «Z» представляет собой -CH-, и могут содержать по меньшей мере один хиральный центр, когда составляющая «Z» представляет собой -N-. Эти хиральные центры указаны символами * и ** в следующих формулах Ia и Ib:
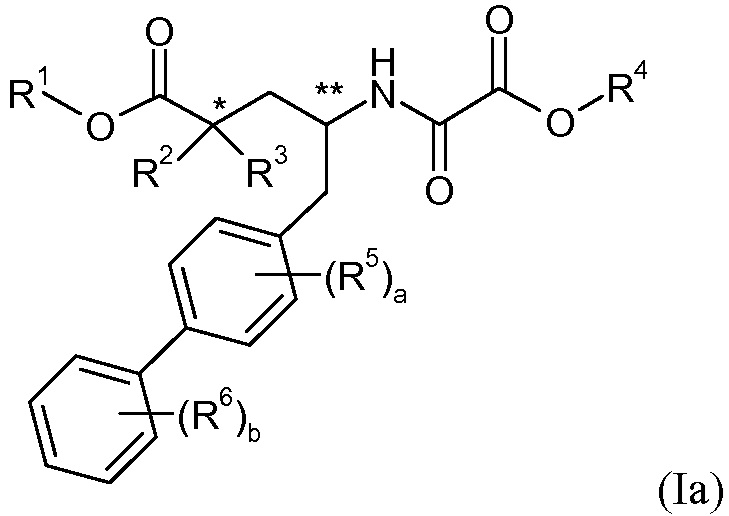

Следует, однако, отметить, что * хиральный центр отсутствует, когда R2 берется вместе с R3 с образованием - CH2-O-CH2- или -CH2-CH2-, или и R2, и R3 представляют собой -CH3.
В одном стереоизомере соединения формулы Ia оба атома углерода, идентифицированные символами * и **, имеют конфигурацию (R). В этом варианте осуществления соединения имеют конфигурацию (R,R) у атомов углерода * и ** или обогащены стереоизомерной формой, имеющей конфигурацию (R,R) у этих атомов углерода. В другом стереоизомере соединения формулы Ia, оба атома углерода, идентифицированные символами * и **, имеют конфигурацию (S). В этом варианте осуществления соединение имеют конфигурацию (S,S) у атомов углерода * и ** или обогащены стереоизомерной формой, имеющей конфигурацию (S,S) у этих атомов углерода. В еще одном стереоизомере соединения формулы Ia атом углерода, идентифицированный символом *, имеет конфигурацию (S), и атом углерода, идентифицированный символом **, имеет конфигурацию (R). В этом варианте осуществления соединения имеют конфигурацию (S,R) у атомов углерода * и ** или обогащены стереоизомерной формой, имеющей конфигурацию (S,R) у этих атомов углерода. В еще одном стереоизомере соединения формулы Ia атом углерода, идентифицированный символом *, имеет конфигурацию (R), и атом углерода, идентифицированный символом **, имеет конфигурацию (S). В этом варианте осуществления соединения имеют конфигурацию (R,S) у атомов углерода * и ** или обогащены стереоизомерной формой, имеющей конфигурацию (R,S) у этих атомов углерода.
В одном стереоизомере соединения формулы Ib атом углерода, идентифицированный символом *, имеет конфигурацию (R). В этом варианте осуществления соединения имеют конфигурацию (R) у атома углерода * или обогащены стереоизомерной формой, имеющей конфигурацию (R) у этого атома углерода. В другом стереоизомере соединения формулы Ib, атом углерода, идентифицированный символом *, имеет конфигурацию (S). В этом варианте осуществления соединения имеют конфигурацию (S) у атома углерода * или обогащены стереоизомерной формой, имеющей конфигурацию (S) у этого атома углерода.
Эти различные варианты осуществления могут быть показаны в виде формулы Ia-1:

формула Ia-2:
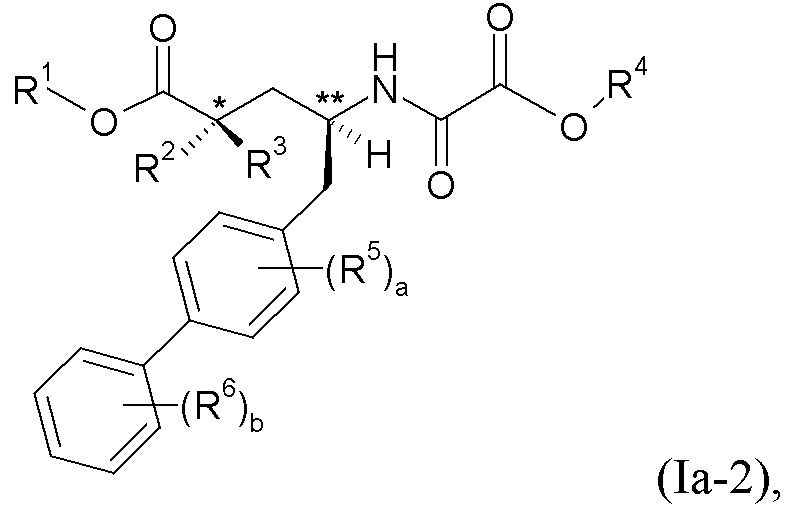
формула Ia-3:

формула Ia-4:

формула Ib-1:
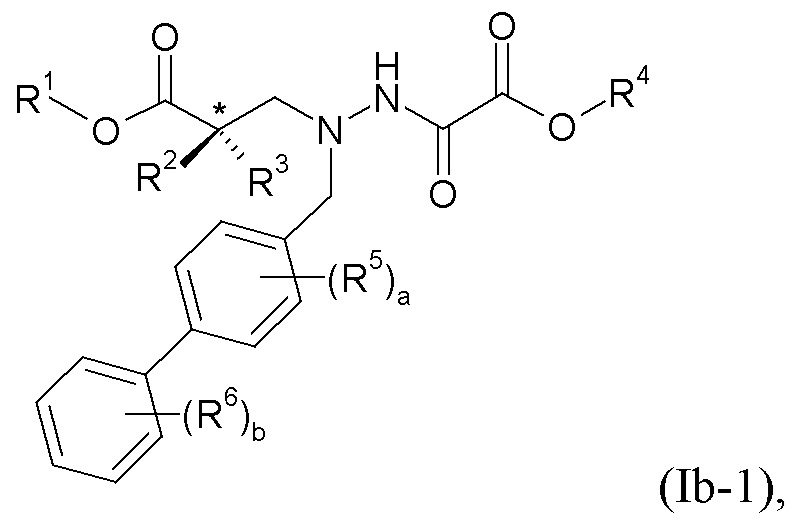
формула Ib-2:

В некоторых вариантах осуществления для оптимизации терапевтической активности соединения по изобретению, например, для лечения гипертензии, может быть желательно, чтобы атомы углерода, идентифицированные символами * и **, имели конкретную конфигурацию или были обогащены стереоизомерной формой, имеющей такую конфигурацию. Таким образом, в определенных аспектах, настоящее изобретение относится к каждому отдельному энантиомеру или обогащенной энантиомером смеси энантиомеров, содержащим преимущественно один энантиомер или другой энантиомер. В других вариантах осуществления соединения по изобретению присутствуют в виде рацемических смесей энантиомеров.
Соединения по изобретению, а также те соединения, которые используются при их синтезе, могут также включать изотопно меченные соединения, то есть, где один или несколько атомов были обогащены атомами, имеющими атомную массу, отличную от атомной массы, преимущественно обнаруживаемой в природе. Примеры изотопов, которые могут быть включены в соединения формулы I, например, включают без ограничения2H,3H,13C,14C,15N,18O,17O,35S,36Cl и18F. Особый интерес представляют соединения формулы I, обогащенные тритием или углеродом-14, которые могут использоваться, например, в исследованиях тканевого распределения; соединения формулы I, обогащенные дейтерием, в частности, в участке метаболизма, приводят, например, к получению соединений, имеющих большую метаболическую устойчивость; и соединения формулы I, обогащенные испускающими позитроны изотопами, такими как11C,18F,15O и13N, которые могут использоваться, например, при исследованиях с использованием позитронно-эмиссионной хроматографии (PET).
Номенклатура, используемая в настоящем описании для именования соединений по изобретению, иллюстрируется в разделе «Примеры» настоящего описания. Эта номенклатура была выведена с использованием имеющегося в продаже программного обеспечения AutoNom (MDL, San Leandro, California).
Репрезентативные варианты осуществления
Следующие заместители и величины предназначены для обеспечения репрезентативных примеров различных аспектов и вариантов осуществления изобретения. Эти репрезентативные величины предназначены для дополнительного определения и иллюстрации данных аспектов и вариантов осуществления и не предназначены для исключения других вариантов осуществления или ограничения объема изобретения. В этом отношении, если конкретно не указано иное, представление, что конкретная величина или заместитель является предпочтительным, ни в коей мере не предназначено для исключения других величин или заместителей из изобретения.
В одном аспекте настоящее изобретение относится к соединениям формулы I:

Группа R1 выбрана из:
H;
-C1-8алкила, например, -CH3, -CH2CH3, -(CH2)2CH3, -CH(CH3)2, -C(CH3)3, -CH2CH(CH3)2, -(CH2)3CH3, -(CH2)4CH3, -(CH2)2CH(CH3)2, -(CH2)5CH3 и -(CH2)6CH3;
-C1-3алкилен-C6-10арила, например, бензила;
-C1-3алкилен-C1-9гетероарила, например, -CH2-пиридинила и -(CH2)2-пиридинила;
-C3-7циклоалкила, например, циклопентила;
-[(CH2)2O]1-3CH3, например, -(CH2)2OCH3 и -[(CH2)2O]2CH3;
-C1-6алкилен-OC(O)R10, например, -CH2OC(O)CH3, -CH2OC(O)CH2CH3, -CH2OC(O)(CH2)2CH3, -CH2CH(CH3)OC(O)CH2CH3, -CH2OC(O)OCH3, -CH2OC(O)OCH2CH3, -CH(CH3)OC(O)OCH2CH3, -CH(CH3)OC(O)O-CH(CH3)2, -CH2CH(CH3)OC(O)-циклопентила, -CH2OC(O)O-циклопропила, -CH(CH3)-OC(O)-O-циклогексила, -CH2OC(O)O-циклопентила, -CH2CH(CH3)OC(O)-фенила, -CH2OC(O)O-фенила, -CH2OC(O)-CH[CH(CH3)2]-NH2, -CH2OC(O)-CH[CH(CH3)2]-NHC(O)OCH3 и -CH(CH3)OC(O)-CH(NH2)CH2COOCH3;
-C1-6алкилен-NR11R12, например, -(CH2)2-N(CH3)2,

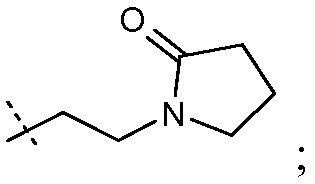
-C1-6алкилен-C(O)R13, например, -CH2C(O)OCH3, -CH2C(O)O-бензила, -CH2C(O)-N(CH3)2 и

-C0-6алкиленморфолина, например, -(CH2)2-морфолина и -(CH2)3-морфолина:
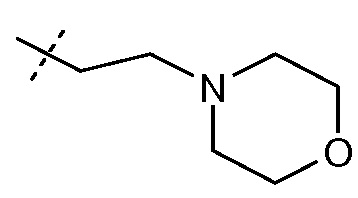

-C1-6алкилен-SO2-C1-6алкила, например, -(CH2)2SO2CH3;
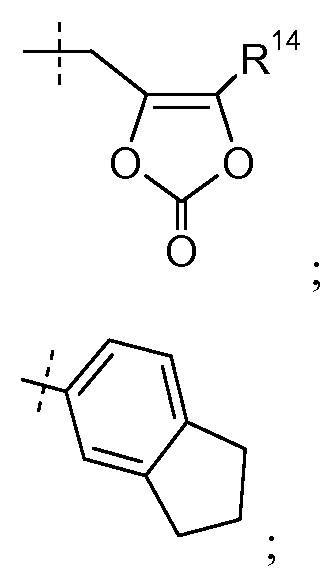

Группа R10 выбрана из:
-C1-6алкила, например, -CH3 и -CH2CH3;
-O-C1-6алкила, например, -OCH3, -O-CH2CH3 и -O-CH(CH3)2;
-C3-7циклоалкила, например, циклопентила;
-O-C3-7циклоалкила, например, -O-циклопропила, -O-циклогексила и -O-циклопентила;
фенила;
-O-фенила;
-NR11R12;
-CH(R15)-NH2, например, -CH[CH(CH3)2]-NH2;
-CH(R15)-NHC(O)O-C1-6алкила, например, -CH[CH(CH3)2]-NHC(O)OCH3; и
-CH(NH2)CH2COOCH3.
Группы R11 и R12 независимо выбраны из H, -C1-6алкила (например, CH3) и бензила. Альтернативно, группы R11 и R12 могут быть взяты вместе в виде -(CH2)3-6-, -C(O)-(CH2)3- или -(CH2)2O(CH2)2-, например, с образованием такой группы, как:


Группа R13 выбрана из -O-C1-6алкила, например, -OCH3, -O-бензила и -NR11R12, например, -N(CH3)2, и

Группа R14 представляет собой -C1-6алкил (например, -CH3 и -C(CH3)3) или -C0-6алкилен-C6-10арил. Группа R15 представляет собой H, -CH3, -CH(CH3)2, фенил или бензил.
Кроме того, каждая алкильная группа в R1 необязательно замещена 1-8 атомами фтора. Например, когда R1 представляет собой -C1-8алкил, то R1 может также обозначать такую группу, как -CH2CF3, -CH(CH3)CF3, -(CH2)2CF3, -CH2CF2CH3, -CH2CF2CF3, -CH(CF3)2, -CH(CH2F)2, -C(CF3)2CH3 и -CH(CH3)CF2CF3.
В одном варианте осуществления R1 выбран из H, -C1-8алкила, -C1-6алкилен-OC(O)R10 и

где R10представляет собой -C1-6алкил, -O-C1-6алкил или -CH[R15]-NHC(O)O-C1-6алкил; R14 представляет собой -C1-6алкил; R15 представляет собой -CH(CH3)2; и каждая алкильная группа в R1 необязательно замещена 1-8 атомами фтора. В одном определенном варианте осуществления R1 выбран из H, -CH2CH3, -CH(CH3)2, -CH2CH(CH3)2, -(CH2)3CH3, -(CH2)6CH3, -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, -CH2CF2CF3, -CH2OC(O)CH3, -CH2OC(O)CH2CH3, -CH2OC(O)(CH2)2CH3, -CH2OC(O)OCH2CH3, -CH2OC(O)-CH[CH(CH3)2]-NHC(O)O-CH3 и

где R14 представляет собой -CH3. В других вариантах осуществления эти соединения имеют формулы IIa-IId, IIi-IIk, IIIa-IIIb и IVa-IVd.
В одном варианте осуществления R1 представляет собой H. В других вариантах осуществления эти соединения имеют формулы IIa-IId, IIi-IIk, IIIa-IIIb и IVa-IVd.
В другом варианте осуществления R1 выбран из -C1-8алкила, -C1-3алкилен-C6-10арила, -C1-3алкилен-C1-9гетероарила, -C3-7циклоалкила, -[(CH2)2O]1-3CH3, -C1-6алкилен-OC(O)R10, -C1-6алкилен-NR11R12, -C1-6алкилен-C(O)R13, -C0-6алкиленморфолинила, -C1-6алкилен-SO2-C1-6алкила,


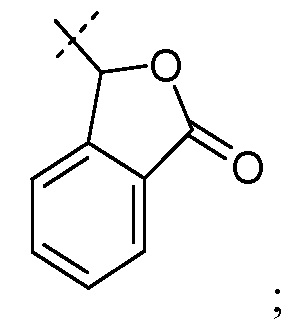
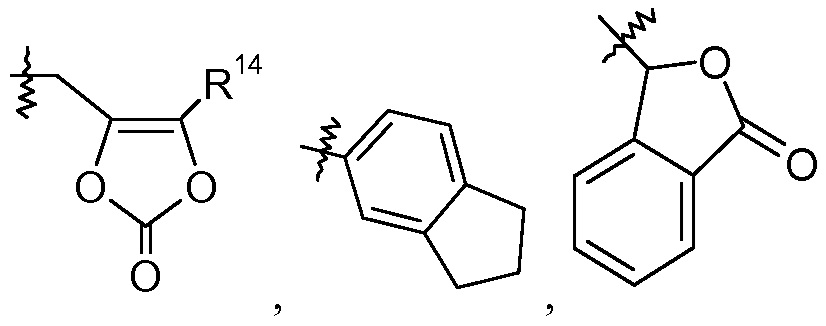
В других вариантах осуществления эти соединения имеют формулы IIa-IId, IIi-IIk, IIIa-IIIb и IVa-IVd. В одном аспекте изобретения эти соединения могут, в частности, найти применение в качестве пролекарств или в виде промежуточных соединений в описанных здесь процедурах синтеза. Определенные примеры таких пролекарственных групп включают те, где R1 представляет собой -C1-6алкилен-OC(O)R10, такие как -CH(CH3)OC(O)-O-циклогексил:

делая соединение сложным цилексетиловым эфиром; или R1 представляет собой -C0-6алкиленморфолин, такой как -(CH2)2-морфолин:

делая соединение сложным 2-морфолиноэтиловым или мофетиловым эфиром; или
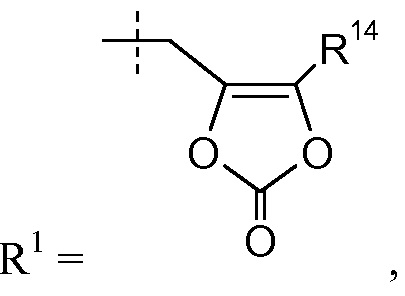
такой как -CH2-5-метил-[1,3]диоксол-2-он:
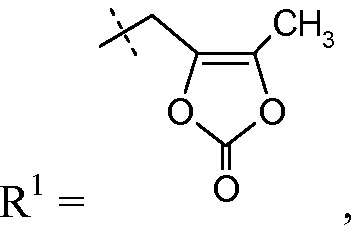
делая соединение сложным медоксомиловым эфиром.
В одном варианте осуществления R2 представляет собой -OR21 или -CH2OR21, и R3 представляет собой H или -CH3. Эти варианты осуществлений могут быть изображены в виде формул IIa-IId:

Группа R21 представляет собой H, -C(O)-C1-6алкил, -C(O)-CH(R22)-NH2, -C(O)-CH(R22)-NHC(O)O-C1-6алкил или -P(O)(OR23)2; и в одном конкретном варианте осуществления группа R21 представляет собой H. Группа R22 представляет собой H, -CH3, -CH(CH3)2, фенил или бензил. Группа R23 представляет собой H, -C1-6алкил или фенил.
В одном варианте осуществления соединения по изобретению имеют формулу IIa, и в одном иллюстративном варианте осуществления R1 выбран из H, -C1-8алкила, -C1-6алкилен-OC(O)R10 и

где R10представляет собой -C1-6алкил, -O-C1-6алкил или -CH[R15]-NHC(O)O-C1-6алкил; R14 представляет собой -C1-6алкил; R15 представляет собой -CH(CH3)2; и каждая алкильная группа в R1 необязательно замещена 1-8 атомами фтора; Z выбран из -CH- и -N-; R4 выбран из H, -C1-8алкила, -C1-3алкилен-O-C1-8алкила, -C1-3алкилен-O-C6-10арила, -[(CH2)2O]1-3CH3 и

где R44 представляет собой -C1-6алкил; и каждая алкильная группа в R4 необязательно замещена 1-8 атомами фтора; a равно 0, и b равно 0; или a равно 0, b равно 1, и R6 представляет собой галоген; или a равен 0, b равен 2, и один R6 представляет собой галоген, и другой R6 представляет собой галоген или -CH3; или a равно 1, R5 представляет собой галоген, и b равно 0; или a равно 1, R5 представляет собой галоген, b равно 1, и R6 представляет собой галоген; или a равно 1, R5 представляет собой галоген, b равен 2, и каждый R6 представляет собой галоген; и где метиленовый линкер на бифениле необязательно замещен двумя группами -CH3; и в другом иллюстративном варианте осуществления R1 выбран из H, -CH2CH3, -CH(CH3)2, -CH2CH(CH3)2, -(CH2)3CH3, -(CH2)6CH3, -CH2CF3, -(CH2)2CF3, -CH2CF2CH3, -CH2CF2CF3, -CH2OC(O)CH3, -CH2OC(O)CH2CH3, -CH2OC(O)(CH2)2CH3, -CH2OC(O)OCH2CH3, -CH2OC(O)-CH[CH(CH3)2]-NHC(O)O-CH3 и
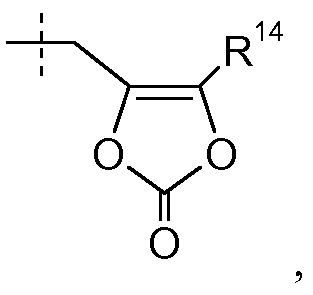
где R14 представляет собой -CH3; R4 выбран из H, -CH2CH3, -CH(CH3)2, -CH2CH(CH3)2, -(CH2)3CH3, -C(CH3)3, -(CH2)2CF3, -CH2CF2CH3, -(CH2)3-O-CH2CH3, -(CH2)2-O-фенила, -(CH2)2OCH3 и

где R44 представляет собой -CH3; и a равно 0, и b равно 0; или a равно 0, b равно 1, и R6 представляет собой 2'-фтор, 3'-фтор, 3'-хлор или 4'-фтор; или a равно 0, b равно 2, и R6 представляет собой 2'-фтор, 5'-хлор или 2'-метил, 5'-хлор или 2',5'-дихлор; или a равно 1, R5 представляет собой 3-хлор, и b равно 0; или a равно 1, R5 представляет собой 3-хлор, b равно 1, и R6 представляет собой 3'-хлор; или a равно 1, R5 представляет собой 3-хлор, b равно 2, и R6 представляет собой 2'-фтор, 5'-хлор.
В одном варианте осуществления соединения по изобретению имеют формулу IIb, и в одном иллюстративном варианте осуществления H или -C1-8алкил; Z представляет собой -N-; R4 представляет собой H или -C1-8алкил; и a и b равны 0; и в другом иллюстративном варианте осуществления R1 и R4 представляют собой H.
В одном варианте осуществления соединения по изобретению имеют формулу IIc, и в одном иллюстративном варианте осуществления R1 представляет собой H или -C1-8алкил; Z представляет собой -CH-; R4 представляет собой H или -C1-8алкил; a равно 0 или a равно 1, и R5 представляет собой галоген; b равно 0 или b равно 1 или 2, и R6 представляет собой галоген; и где метиленовый линкер на бифениле необязательно замещен двумя группами -CH3; и в другом иллюстративном варианте осуществления R1 представляет собой H, -CH2CH3 или -(CH2)3CH3; R4 представляет собой H; a равно 0 или a равно 1, и R5 представляет собой 3-хлор-; b равно 0 или b равно 1, и R6 представляет собой 2'-фтор, 3'-фтор, 3'-хлор или 4'-фтор.
В одном варианте осуществления соединения по изобретению имеют формулу IId, и в одном иллюстративном варианте осуществления R1 представляет собой H или -C1-8алкил; Z представляет собой -CH-; R4 представляет собой H или -C1-8алкил; a равно 0; и b равно 0 или b равно 1, и R6 представляет собой галоген; и в другом иллюстративном варианте осуществления R1 представляет собой H или -CH2CH3; R4 представляет собой H или -CH2CH(CH3)2; и b равно 0 или b равно 1, и R6 представляет собой 2'-фтор-, 3'-фтор-, 3'-хлор- или 4'-фтор-.
В другом варианте осуществления R2 взят вместе с R1 с образованием -OCR15R16- или -CH2O-CR15R16-, и R3 выбран из H и -CH3. Группы R15 и R16 независимо выбраны из H, -C1-6алкила и -O-C3-7циклоалкила, или R15 и R16 взяты вместе с образованием =O. Это может быть изображено в виде формул IIe-IIh:
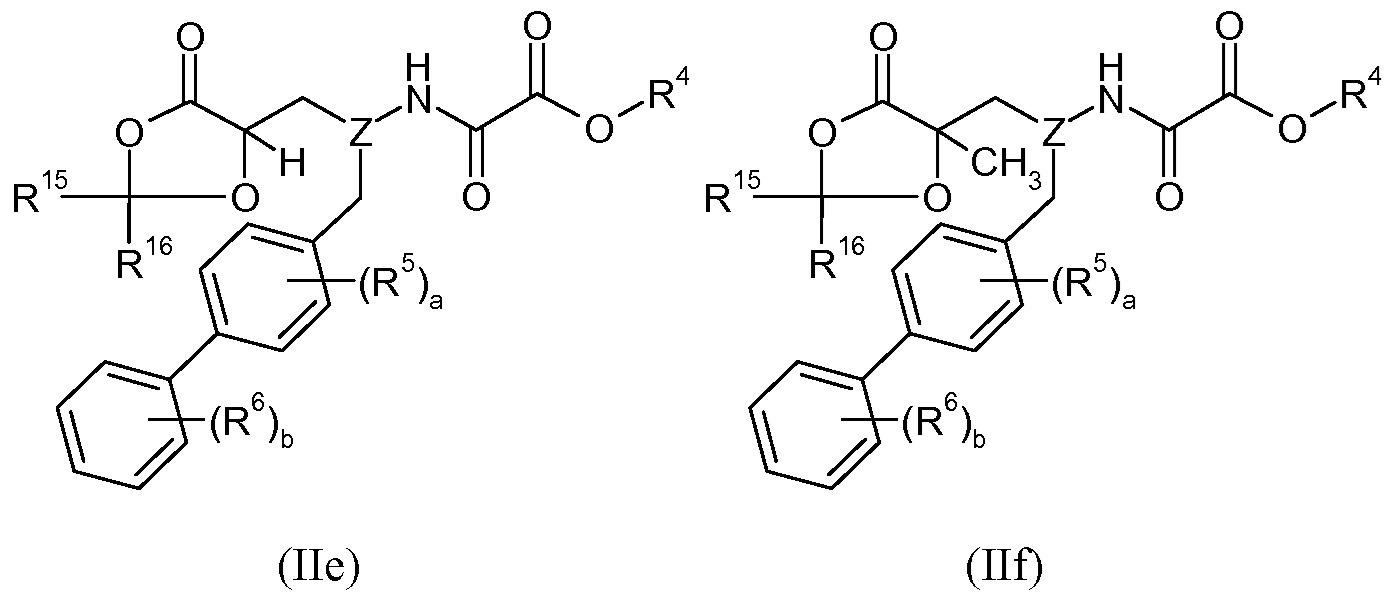

В одном аспекте изобретения эти соединения, в частности, могут найти применение в качестве пролекарств или в качестве промежуточных соединений в описанных здесь синтетических процедурах. Соединения, где R2 представляет собой -CH2OP(O)(OH)2 могут также найти применение в качестве пролекарств. В одном варианте осуществления соединений формул IIe, IIf, IIg и IIh, Z представляет собой -CH-, R4 представляет собой H, a равно 0, b равно 1, R6 представляет собой 3'-Cl, и R15 и R16 представляют собой H.
В другом варианте осуществления R2 взят вместе с R3 с образованием -CH2-O-CH2- или -CH2-, которые могут быть изображены в виде формул IIi и IIj, соответственно:
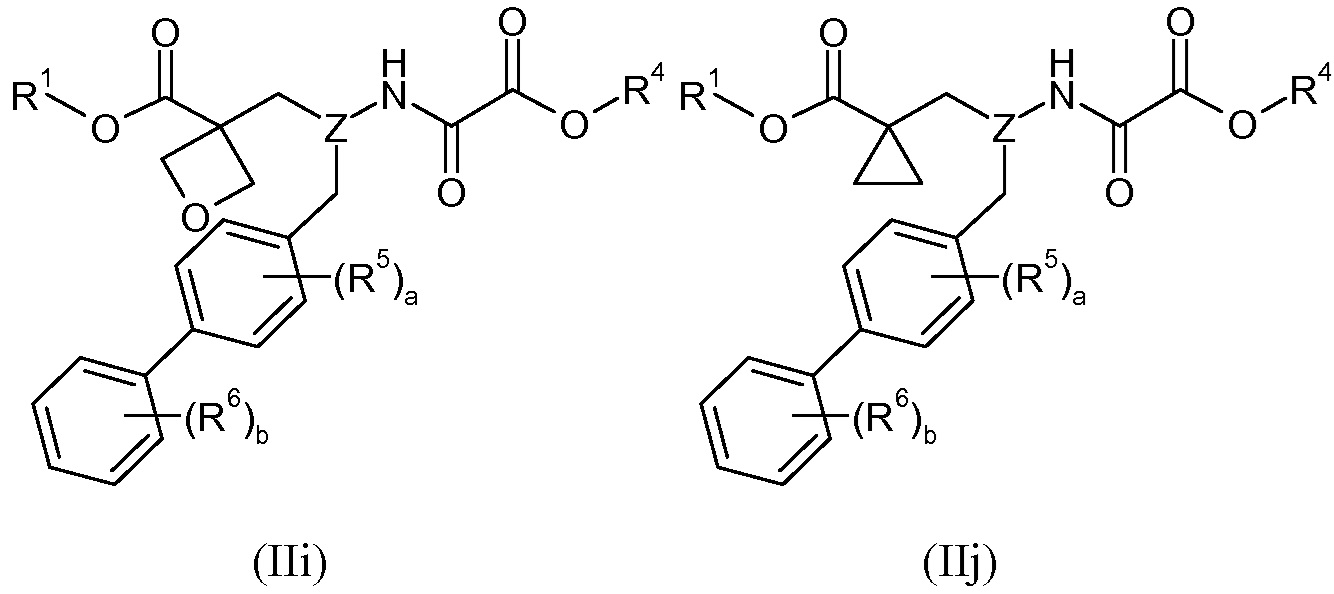
В другом варианте осуществления и R2, и R3 представляют собой -CH3, который может быть изображен в виде формулы IIk:

В одном варианте осуществления соединений формул IIi, IIj, и IIk R1 представляет собой H, Z представляет собой -CH-, R4 представляет собой -C1-8алкил (например, -CH2CH(CH3)2), a равно 0, b равно 1, и R6 представляет собой 3'-Cl.
Группа Z выбрана из -CH- и -N-. Эти варианты осуществления могут быть изображены в виде формул IIIa и IIIb:

Группа R4 выбрана из:
H;
-C1-8алкила, например, -CH3, -CH2CH3, -(CH2)2CH3, -CH(CH3)2, -C(CH3)3, -CH2CH(CH3)2, -(CH2)3CH3, -(CH2)4CH3, -(CH2)2CH(CH3)2, -(CH2)5CH3и -(CH2)6CH3;
-C1-3алкилен-O-C1-8алкила, например, -(CH2)3-O-CH2CH3;
-C1-3алкилен-C6-10арила, например, бензила;
-C1-3алкилен-O-C6-10арила, например, -(CH2)2-O-фенила;
-C1-3алкилен-C1-9гетероарила, например, -CH2-пиридинила и -(CH2)2-пиридинила;
-C3-7циклоалкила, например, циклопентила;
-[(CH2)2O]1-3CH3, например, -(CH2)2OCH3 и -[(CH2)2O]2CH3;
-C1-6алкилен-OC(O)R40, например, -CH2OC(O)CH3, -CH2OC(O)CH2CH3, -CH2OC(O)(CH2)2CH3, -CH2CH(CH3)OC(O)CH2CH3, -CH2OC(O)OCH3, -CH2OC(O)OCH2CH3, -CH(CH3)OC(O)OCH2CH3, -CH(CH3)OC(O)O-CH(CH3)2, -CH2CH(CH3)OC(O)-циклопентила, -CH2OC(O)O-циклопропила, -CH(CH3)-OC(O)-O-циклогексила, -CH2OC(O)O-циклопентила, -CH2CH(CH3)OC(O)-фенила, -CH2OC(O)O-фенила, -CH2OC(O)-CH[CH(CH3)2]-NH2, -CH2OC(O)-CH[CH(CH3)2]-NHC(O)OCH3 и -CH(CH3)OC(O)-CH(NH2)CH2COOCH3;
-С1-6алкилен-NR41R42, например, -(CH2)2-N(CH3)2,

-C1-6алкилен-C(O)R43, например, -CH2C(O)OCH3, -CH2C(O)O-бензила, -CH2C(O)-N(CH3)2 и

-C0-6алкиленморфолина, например, -(CH2)2-морфолина и -(CH2)3-морфолина:


-C1-6алкилен-SO2-C1-6алкила, например, -(CH2)2SO2CH3;


Группа R40 выбрана из:
-C1-6алкила, например, -CH3 и -CH2CH3;
-O-C1-6алкила, например, -OCH3, -O-CH2CH3 и -O-CH(CH3)2;
-C3-7циклоалкила, например, циклопентила;
-0-C3-7циклоалкила, например, -O-циклопропила, -O-циклогексила и -O-циклопентила;
фенила;
-O-фенила;
-NR41R42;
-CH(R45)-NH2, например, -CH[CH(CH3)2]-NH2;
-CH(R45)-NHC(O)O-C1-6алкила, например, -CH[CH(CH3)2]-NHC(O)OCH3 и -CH(NH2)CH2COOCH3.
Группы R41 и R42 независимо выбраны из H, -C1-6алкила (например, CH3) и бензила. Альтернативно, группы R41 и R42 могут быть взяты вместе в виде -(CH2)3-6-, -C(O)-(CH2)3- или -(CH2)2O(CH2)2-, например, с образованием такой группы, как:


Группа R43 выбрана из -O-C1-6алкила, например, -OCH3, -O-бензила и -NR41R42, например,-N(CH3)2, и

Группа R44 представляет собой -C1-6алкил (например, -CH3 и -C(CH3)3) или -C0-6алкилен-C6-10арила. Группа R45 представляет собой H, -CH3, -CH(CH3)2, фенил или бензил.
Кроме того, каждая алкильная группа в R4 необязательно замещена 1-8 атомами фтора. Например, когда R4 представляет собой -C1-8алкил, то R4 может также обозначать такую группу, как -CH2CF3, -CH(CH3)CF3, -(CH2)2CF3, -CH2CF2CH3, -CH2CF2CF3, -CH(CF3)2, -CH(CH2F)2, -C(CF3)2CH3 и -CH(CH3)CF2CF3.
В одном варианте осуществления R4 выбран из H, -C1-8алкила, -C1-3алкилен-C1-8алкила, -C1-3алкилен-O-C6-10арила, -[(CH2)2O]1-3CH3 и

где R44 представляет собой -C1-6алкил; и каждая алкильная группа в R4 необязательно замещена 1-8 атомами фтора. В одном определенном варианте осуществления R4 выбран из H, -CH2CH3, -CH(CH3)2, -CH2CH(CH3)2, -(CH2)3CH3, -C(CH3)3, -(CH2)2CF3, -CH2CF2CH3, -(CH2)3-O-CH2CH3, -(CH2)2-O-фенила, -(CH2)2OCH3 и
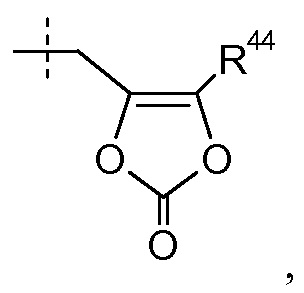
где R44 представляет собой -CH3. В других вариантах осуществления эти соединения имеют формулы IIa-IIk, IIIa-IIIb и IVa-IVd.
В одном варианте осуществления R1 представляет собой H. В других вариантах осуществления эти соединения имеют формулы IIa-IIk, IIIa-IIIb и IVa-IVd. В еще одном варианте осуществления и R1, и R4 представляют собой H. В других вариантах осуществления эти соединения имеют формулы IIa-IIh, IIm-IIo, IIIa-IIIb и IVa-IVd.
В другом варианте осуществления R4 выбран из -C1-8алкила, -C1-3алкилен-O-C1-8алкила, -C1-3алкилен-C6-10арила, -C1-3алкилен-O-C6-10арила, -C1-3алкилен-C1-9гетероарила, -C3-7циклоалкила, -[(CH2)2O]1-3CH3, -C1-6алкилен-OC(O)R40, -C1-6алкилен-NR41R42, -C1-6алкилен-C(O)R43, -C0-6алкиленморфолинила, -C1-6алкилен-SO2-C1-6алкила,


В других вариантах осуществления эти соединения имеют формулы IIa-IIk, IIIa-IIIb и IVa-IVd. В одном аспекте изобретения эти соединения могут, а частности, найти применение в качестве пролекарств или в качестве промежуточных средств в описанных здесь синтетических процедурах. В одном варианте осуществления и R1, и R4 представляют собой такие пролекарственные группы. В другом варианте осуществления один из R1 и R4 представляет собой пролекарственную группу, а другой представляет собой H. Определенные примеры таких пролекарственных групп включают те, где R4 представляет собой -C1-6алкилен-OC(O)R10, такой как -CH(CH3)OC(O)-O-циклогексил:
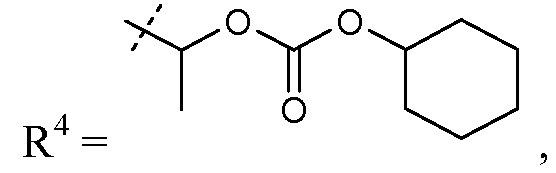
получая соединение сложный эфир цилексетил; или R4 представляет собой -C0-6алкиленморфолин, такой как -(CH2)2-морфолин:

получая соединение 2-морфолиноэтил или сложный эфир мофетила; или

такой как -CH2-5-метил-[1,3]диоксол-2-он:
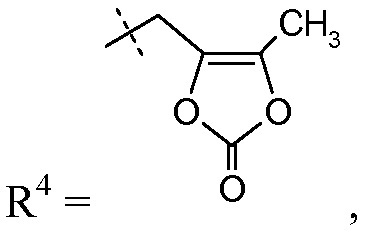
получая соединение сложный эфир медоксомила.
Нумерация для групп R5 и R6 следующая:

Целое число «a» равно 0 или 1. Группа R5, в случае ее присутствия, выбрана из галогена, -CH3, -CF3 и -CN. В одном варианте осуществления a равно 0. В другом варианте осуществления a равно 1, и R5 представляет собой галоген, такой как 3-хлор или 3-фтор. В еще одном варианте осуществления a равно 0 или a равно 1, и R5 представляет собой галоген. В других вариантах осуществления эти соединения имеют формулы IIa-IIk, IIIa-IIIb и IVa-IVd.
Целое число «b» равно 0 или целому числу от 1 до 3. Группа R6, в случае ее присутствия, независимо выбрана из галогена, -OH, -CH3, -OCH3, -CN и -CF3. В одном варианте осуществления b равно 0. В другом варианте осуществления b равно 1, и R6 выбрана из Cl, F, -OH, -CH3, -OCH3, -CN и -CF3, например, 2'-хлор, 3'-хлор, 2'-фтор, 3'-фтор, 2'-гидрокси, 3'-гидрокси, 3'-метил, 2'-метокси, 3'-циано или 3'-трифторметил. В другом варианте осуществления b равно 1, и R6 представляет собой галоген, -CH3 или -OCH3, например, 3'-хлор, 3'-метил или 2'-метокси. В другом варианте осуществления b равно 2, и R6 представляет собой 2'-фтор-5'-хлор, 2',5'-дихлор, 2',5'-дифтор, 2'-метил-5'-хлор, 3'-фтор-5'-хлор, 3'-гидрокси-5'-хлор, 3',5'-дихлор, 3',5'-дифтор, 2'-метокси-5'-хлор, 2'-метокси-5'-фтор, 2'-гидрокси-5'-фтор, 2'-фтор-3'-хлор, 2'-гидрокси-5'-хлор или 2'-гидрокси-3'-хлор. В другом варианте осуществления b равно 3, и каждая R6 представляет собой независимо галоген или -CH3, например, 2'-метил-3',5'-дихлор или 2'-фтор-3'-метил-5'-хлор. В одном конкретном варианте осуществления b равно 0 или b равно 1, и R6 представляет собой галоген, или b равно 2, и каждая R6 независимо выбрана из галогена и -CH3. В других вариантах осуществления эти соединения имеют формулы IIa-IIk, IIIa-IIIb и IVa-IVd.
В других иллюстративных вариантах осуществления a равно 0, и b равно 0; или a равно 0, b равно 1, и R6 представляет собой 2'-фтор, 3'-фтор, 3'-хлор или 4'-фтор; или a равно 0, b равно 2, и R6 представляет собой 2'-фтор, 5'-хлор или 2'-метил, 5'-хлор или 2',5'-дихлор; a равно 1, R5 представляет собой 3-хлор, и b равно 0; или a равно 1, R5 представляет собой 3-хлор-, b равно 1, и R6 представляет собой 3'-хлор; или a равно 1, R5 представляет собой 3-хлор-, b равно 2, и R6 представляет собой 2'-фтор, 5'-хлор. В других вариантах осуществления эти соединения имеют формулы IIa-IIk, IIIa-IIIb и IVa-IVd. Особый интерес представляют соединения формул:

Метиленовый линкер на бифениле необязательно замещен одной или двумя -C1-6алкильными группами или циклопропилом. Например, в одном варианте осуществления метиленовый линкер на бифениле является незамещенным; в другом варианте осуществления метиленовый линкер на бифениле замещен одной -C1-6алкильной группой (например, -CH3); и в еще одном варианте осуществления метиленовый линкер на бифениле замещен двумя -C1-6алкильными группами (например, двумя группами -CH3); в другом варианте осуществления метиленовый линкер на бифениле замещен циклопропильной группой. Эти варианты осуществления изображены соответственно в виде формул IVa-IVd:
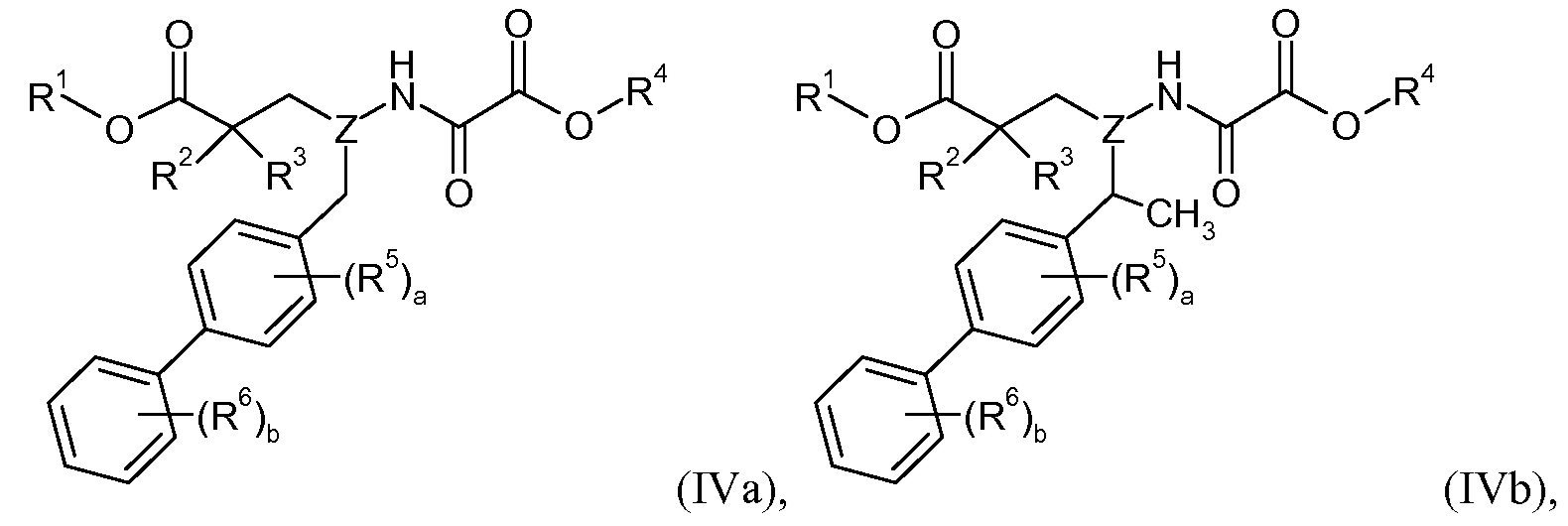

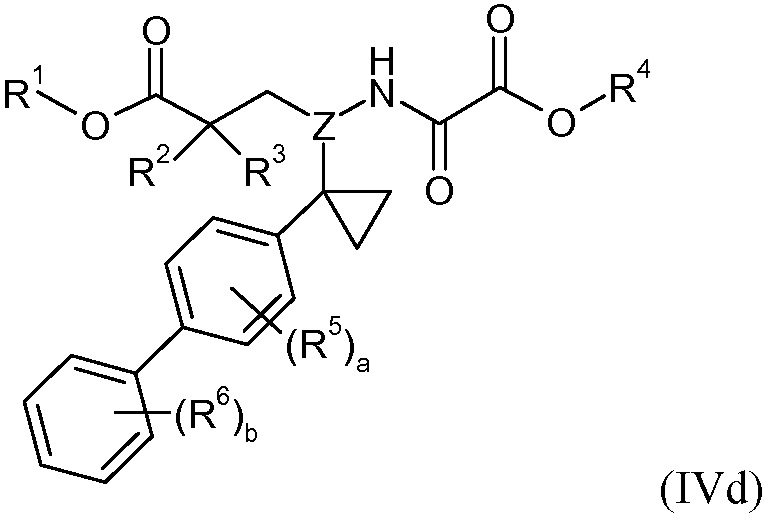
В одном варианте осуществления соединений формул IVa, IVb, IVc и IVd R1 представляет собой H, R2 представляет собой -OR21, и R21 представляет собой H, R3 представляет собой H, Z представляет собой -CH-, R4 представляет собой -C1-8алкил (например, -CH2CH(CH3)2), a равно 0, b равно 1, и R6 представляет собой 3'-Cl.
В другом варианте осуществления R1 выбран из H, -C1-8алкила, -C1-6алкилен-OC(O)R10 и

где R10представляет собой -C1-6алкил, -O-C1-6алкил или -CH[R15]-NHC(O)O-C1-6алкил; R14 представляет собой -C1-6алкил; R15 представляет собой -CH(CH3)2; и каждая алкильная группа в R1 необязательно замещена 1-8 атомами фтора;
R4 выбран из H, -C1-8алкила, -C1-3алкилен-O-C1-8алкила, -C1-3алкилен-O-C6-10арила, -[(CH2)2)O]1-3CH3 и
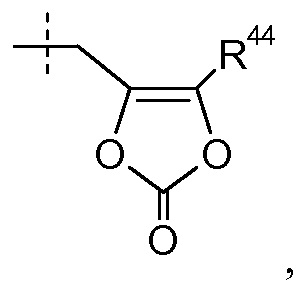
где R44 представляет собой -C1-6алкил; и каждая алкильная группа в R4 необязательно замещена 1-8 атомами фтора;
a равно 0, и b равно 0; или a равно 0, b равно 1, и R6 представляет собой 2'-фтор, 3'-фтор, 3'-хлор или 4'-фтор; или a равно 0, b равно 2, и R6 представляет собой 2'-фтор, 5'-хлор или 2'-метил, 5'-хлор или 2',5'-дихлор; или a равно 1, R5 представляет собой 3-хлор, и b равно 0; или a равно 1, R5 представляет собой 3-хлор, b равно 1, и R6 представляет собой 3'-хлор; или a равно 1, R5 представляет собой 3-хлор, b равно 2, и R6 представляет собой 2'-фтор, 5'-хлор; и где метиленовый линкер на бифениле необязательно замещен двумя группами CH3. В одном конкретном варианте осуществления этих соединений R2 представляет собой -OR21 или -CH2OR21; и R3 представляет собой H или -CH3; где R21 представляет собой H.
Кроме того, конкретные соединения формулы I, которые представляют интерес, включают такие, которые представлены в «Примерах» ниже, а также их фармацевтически приемлемые соли.
ОБЩИЕ ПРОЦЕДУРЫ СИНТЕЗА
Соединения по изобретению могут быть получены из общедоступных исходных материалов с использованием следующих общих способов, процедур, перечисленных в "Примерах", или с использованием других методов, реагентов и исходных материалов, которые известны средним специалистам в данной области техники. Хотя следующие процедуры могут иллюстрировать конкретный вариант осуществления изобретения, понятно, что аналогичным образом можно получить другие варианты осуществления изобретения, используя такие же или аналогичные способы, или используя другие способы, реагенты и исходные материалы, известные специалистам в данной области техники. Следует также понимать, что когда представляются типичные или предпочтительные условия процесса (например, температуры реакции, периоды времени, молярные отношения реагентов, растворители, величины давления и т.д.), то, если не указано иное, могут также использоваться другие условия процесса. В некоторых случаях реакции проводили при комнатной температуре, и действительную температуру не измеряли. Понятно, что комнатная температуры может быть принята как обозначающая температуру в пределах диапазона, обычно связанного с окружающей температурой в лабораторной среде, и она обычно находится в диапазоне от примерно 18°C до примерно 30°C. В других случаях, реакции проводили при комнатной температуре, и действительную температуру измеряли и регистрировали. Хотя оптимальные условия реакции обычно варьируются в зависимости от разнообразных параметров реакции, таких как конкретные используемые реагенты, растворители и количества, специалисты в данной области техники могут легко определить подходящие условия реакции, используя обычные процедуры оптимизации.
Кроме того, как очевидно для специалистов в данной области техники, обычные защитные группы могут быть необходимы или желательны для предотвращения подверженности нежелательным реакциям определенных функциональных групп. Выбор пригодной защитной группы для конкретной функциональной группы, а также подходящие условия и реагенты для защиты и снятия защиты таких функциональных групп хорошо известны в данной области техники. При желании, можно использовать защитные группы, отличные от тех, которые иллюстрируются в описанных здесь процедурах. Например, многочисленные защитные группы и их введение и удаление описаны в руководстве T.W. Greene и Г.M. Wuts, Protecting Groups in Organic Synthesis, Fourth Edition, Wiley, New York, 2006, и в приведенных в нем ссылках.
Карбокси-защитные группы пригодны для предотвращения нежелательных реакций карбоксильных групп, и примеры включают без ограничения метил, этил, трет-бутил, бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm), триметилсилил (TMS), трет-бутилдиметилсилил (TBDMS), дифенилметил (бензгидрил, DPM) и т.п. Амино-защитные группы пригодные для предотвращения нежелательных реакций у аминогрупп, и примеры включают без ограничения трет-бутоксикарбонил (BOC), тритил (Tr), бензилоксикарбонил (Cbz), 9-флуоренилметоксикарбонил (Fmoc), формил, триметилсилил (TMS), трет-бутилдиметилсилил (TBDMS) и т.п. Гидрокси-защитные группы пригодны для предотвращения нежелательных реакций у гидроксильных групп, и примеры включают без ограничения C1-6алкилы, силильные группы, включая триC1-6алкилсилильные группы, такие как триметилсилил (TMS), триэтилсилил (TES) и трет-бутилдиметилсилил (TBDMS); сложные эфиры (ацильные группы), включая C1-6алканоильные группы, такие как формил, ацетил и пивалоил и ароматические ацильные группы, такие как бензоил; арилметильные группы, такие как бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm) и дифенилметил (бензгидрил, DPM); и т.п.
Для удаления защитных групп используются стандартные технологии и реагенты снятия защиты, и они могут варьироваться в зависимости от того, какая группа используется. Например, гидроксид натрия или лития обычно используется, когда карбокси-защитная группа представляет собой метил, кислота, такая как TFA или HCl, обычно используется, когда карбокси-защитная группа представляет собой этил или трет-бутил, и H2/Pd/C может использоваться, когда карбокси-защитная группа представляет собой бензил. BOC амино-защитная группа может быть удалена с использованием кислотного реагента, такого как TFA в DCM или HC1 в 1,4-диоксане, тогда как Cbz амино-защитная группа может быть удалена с использованием условий каталитической гидрогенизации, таких как H2 (1 атм) и 10% Pd/C в спиртовом растворителе («H2/Pd/C»). H2/Pd/C обычно используется, когда гидрокси-защитная группа представляет собой бензил, тогда как NaOH обычно используется, когда гидрокси-защитная группа представляет собой ацильную группу.
Подходящие основания для использования в этих схемах включают в качестве примера, но не ограничения, карбонат калия, карбонат кальция, карбонат натрия, триэтиламин, пиридин, 1,8-диазобицикло[5.4.0]ундец-7-ен (DBU), N,N-диизопропилэтиламин (DIPEA), 4-метилморфолин, гидроксид натрия, гидроксид калия, трет-бутоксид калия и гидриды металлов.
Пригодные инертные разбавители или растворители для использования в этих схемах включают в качестве примера, но не ограничения, тетрагидрофуран (THF), ацетонитрил (MeCN), N,N-диметилформамид (DMF), N,N-диметилацетамид (DMA), диметилсульфоксид (DMSO), толуол, дихлорметан (DCM), хлороформ (CHCl3), тетрахлорид углерода (CCl4), 1,4-диоксан, метанол, этанол, воду и т.п.
Пригодные сочетающие реагенты карбоновой кислоты/амина включают гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (BOP), гексафторфосфат бензотриазол-1-илокситрипирролидинфосфония (PyBOP), гексафторфосфат N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (HATU), (гексафторфосфат 2-(6-хлор-1H-бензотриазол-1-ил)-1,1,3,3-тетраметиламмония) (HCTU), 1,3-дициклогексилкарбодиимид (DCC), N-(3-диметиламинопропил)-N'-этилкарбодиимид (EDCI), карбонилдиимидазол (CDI), 1-гидроксибензотриазол (HOBt) и т.п. Реакции сочетания проводят в инертном разбавителе в присутствии основания, такого как DIPEA, и выполняют в обычных условиях образования амидной связи.
Все реакции обычно проводят при температуре в пределах диапазона примерно от -78°C до 100°C, например, при комнатной температуре. Реакции можно контролировать, используя тонкослойную хроматографию (TLC), высокоэффективную жидкостную хроматографию (ВЭЖХ) и/или LCMS (жидкостная хроматография/масс-спектрометрия), до завершения. Реакции могут завершаться за минуты или могут занимать часы, обычно от 1-2 часов и вплоть до 48 часов. После завершения полученная смесь или продукт реакции может дополнительно обрабатываться с получением требуемого продукта. Например, полученная смесь или продукт реакции могут подвергаться одной или нескольким из следующих процедур: концентрация и разделение (например, между EtOAc и водой или между 5% THF в EtOAc и 1M фосфорной кислотой); экстракция (например, EtOAc, CHCl3, DCM, хлороформом); промывание (например, насыщенным водным NaCl, насыщенным водным NaHCO3, Na2CO3 (5%), CHCl3 или 1M NaOH); сушка (например, над MgSO4, над Na2SO4 или в вакууме); фильтрование; кристаллизация (например, из EtOAc и гексанов); концентрация (например, в вакууме); и/или очистка (например, хроматография на силикагеле, флэш-хроматография, препаративная ВЭЖХ, обращенно-фазовая ВЭЖХ или кристаллизация).
Соединения формулы I, а также их соли, могут быть получены, как показано в схеме I:
Схема I
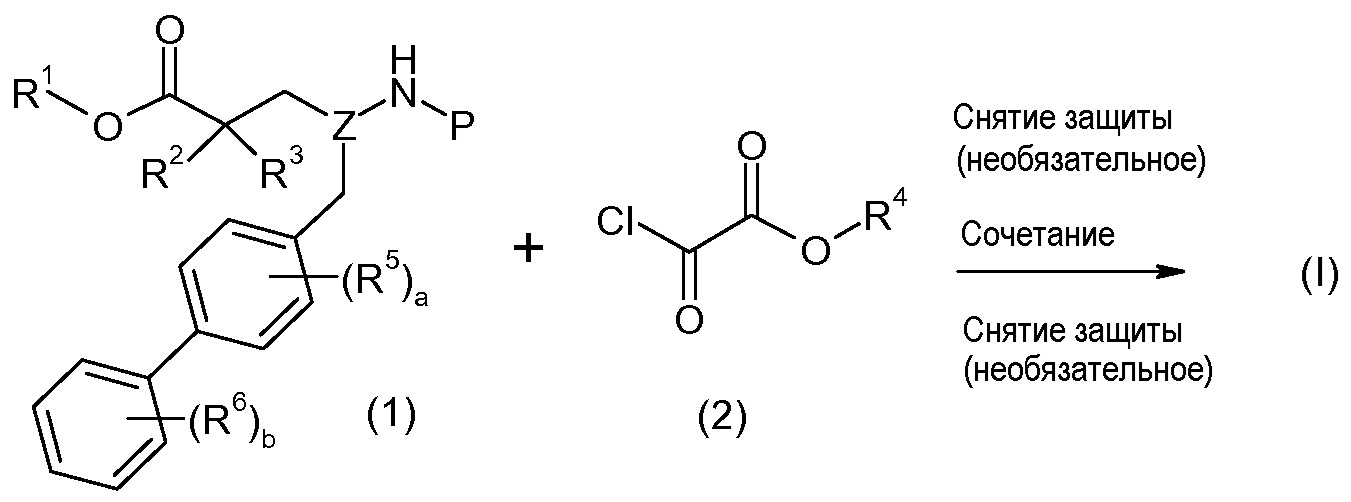
Способ включает стадию сочетания соединения 1 с соединением 2, где R1-R6, Z, a, и b такие, как определено для формулы I, и P выбран из H и подходящей амино-защитной группы, примеры которой включают трет-бутоксикарбонил, тритил, бензилоксикарбонил, 9-флуоренилметоксикарбонил, формил, триметилсилил и трет-бутилдиметилсилил. Когда P представляет собой амино-защитную группу, то способ дополнительно включает снятие защиты соединения формулы I перед стадией сочетания или одновременно с ней.
В случае, когда R1 представляет собой такую группу, как -OCH3 или -OCH2CH3, то стадия сочетания может протекать по стадии снятия защиты для получения соединения формулы I, где R1 представляет собой такую группу, как -OH. Таким образом, один способ получения соединения по изобретению включает сочетание соединений 1 и 2 с необязательной стадией снятия защиты с образованием соединения формулы I или его фармацевтически приемлемой соли.
Способы получения соединения I описаны в «Примерах». Соединение 2 обычно является коммерчески доступным или может быть получено с использованием процедур, которые известны в данной области техники.
Считается, что определенные описанные здесь промежуточные соединения являются новыми, и, соответственно, такие соединения относятся к дополнительным аспектам изобретения, включая, например, соединения формулы 1 или их соль:
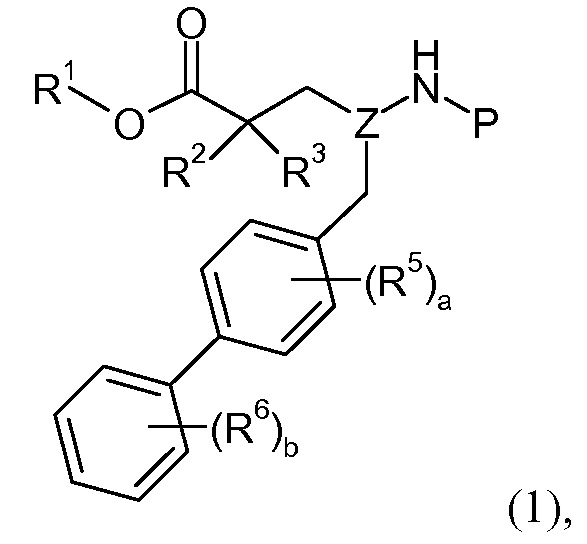
где P представляет собой H или подходящую амино-защитную группу, примеры которой включают трет-бутоксикарбонил, тритил, бензилоксикарбонил, 9-флуоренилметоксикарбонил, формил, триметилсилил и трет-бутилдиметилсилил; и R1, R2, R3, R5, R6, Z, a и b такие, как определено для формулы I.
Дополнительные детали, относящиеся к определенным условиям реакции и другим процедурам получения репрезентативных соединений по изобретению или их промежуточных соединений, описаны в изложенных ниже «Примерах».
ПОЛЕЗНОСТЬ
Соединения по изобретению обладают ингибирующей активностью в отношении неприлизина (NEP), то есть, соединения способны ингибировать ферментативно-каталитическую активность. В другом варианте осуществления соединения не проявляют значимую ингибирующую активность в отношении ангиотензин-превращающего фермента. Один показатель способности соединения ингибировать активность NEP представляет собой константу ингибирования (pKi). Величина pKi представляет собой отрицательный логарифм к числу по основанию 10 константы диссоциации (Ki), которая обычно представляется в молярных единицах. Соединениями по изобретению, представляющими особый интерес, являются те, которые имеют pKi у NEP, бьльшую или равную 6,0, в частности, те, которые имеют pKi, бьльшую или равную 7,0, и еще конкретнее, те, которые имеют pKi, бьльшую или равную 8,0. В одном варианте осуществления представляющие интерес соединения имеют pKi в диапазоне 6,0-6,9; в другом варианте осуществления представляющие интерес соединения имеют pKi в диапазоне 7,0-7,9; в еще одном варианте осуществления представляющие интерес соединения имеют pKi в диапазоне 8,0-8,9; и в еще одном варианте осуществления представляющие интерес соединения имеют pKi в диапазоне, бьльшем или равном 9,0. Такие величины можно определить методиками, которые хорошо известны в данной области техники, а также описанными здесь анализами.
Другим показателем способности соединения ингибировать активность NEP является видимая константа ингибирования (IC50), которая представляет собой молярную концентрацию соединения, которая приводит к половине максимального ингибирования превращения субстрата ферментом NEP. Величина pIC50 представляет собой отрицательный логарифм к числу по основанию 10 IC50. Соединения по изобретению, которые представляют особый интерес, включают те, которые проявляют pIC50 в отношении NEP, бьльшую или равную примерно 5,0. Представляющие интерес соединения также включают те, которые имеют pIC50 в отношении NEP ≥ примерно 6,0, или pIC50 в отношении NEP ≥ примерно 7,0. В другом варианте осуществления представляющие интерес соединения имеют pIC50 в отношении NEP в пределах диапазона примерно 7,0-11,0; и в другом варианте осуществления в пределах диапазона примерно 8,0-11,0, например, в пределах диапазона примерно 8,0-10,0.
Следует отметить, что в некоторых случаях соединения по изобретению могут обладать слабой активностью ингибирования NEP. В таких случаях, как понятно специалистам в данной области, эти соединения могут, тем не менее, быть полезны в качестве инструментов исследований.
Иллюстративные анализы для определения таких свойств соединений по изобретению, как активность ингибирования NEP, описаны в «Примерах» и включают в качестве иллюстрации, а не ограничения, анализы, которые измеряют ингибирование NEP (описаны в Анализе 1). Полезные вторичные анализы включают анализы для измерения ингибирования ACE (также описаны в Анализе 1) и аминопептидазы P (APP) (описанные в публикации Sulpizio et al. (2005) JPET 315: 1306-1313). Фармакодинамический анализ для оценки ингибирующей активности in vivo в отношении ACE и NEP у анестезированных крыс описан в Анализе 2 (см. также публикации Seymour et al. (1985) Hypertension 7(Suppl I): I-35-1-42 и Wigle et al. (1992) Can. J. Physiol. Pharmacol. 70: 1525-1528), где ингибирование ACE измеряется в виде процента ингибирования прессорной реакции на ангиотензин I, и ингибирование NEP измеряется как увеличенный выход с мочой циклического гуанозин-3',5'-монофосфата (cGMP).
Существует много анализов in vivo, которые могут использоваться для установления дополнительной применимости соединений по изобретению. Модель бодрствующих крыс со спонтанной гипертензией (SHR) представляет собой модель ренин-зависимой гипертензии, и она описана в Анализе 3. См. также публикации Intengan et al. (1999) Circulation 100(22): 2267-2275 и Badyal et al. (2003) Indian Journal of Pharmacology 35: 349-362. Модель бодрствующих крыс с гипертензией, вызванной дезоксикортикостерона ацетатом (DOCA), представляет собой модель объем-зависимой гипертензии, которая полезна для измерения активности NEP, и описана в Анализе 4. См. также Trapani et al. (1989) J. Cardiovasc. Pharmacol. 14: 419-424, Intengan et al. (1999) Hypertension 34(4): 907-913 и Badyal et al. (2003) выше). Модель DOCA, в частности, полезна для оценки способности испытуемого соединения снижать артериальное давление, а также измерения способности тестируемого соединения предотвращать или отсрочивать повышение артериального давления. Модель гипертензии у соль-чувствительных крыс Dahl (DSS) представляет собой модель гипертензии, которая чувствительна к соли рациона (NaCl), и она описана в Анализе 5. См. также Rapp (1982) Hypertension 4: 753-763. Модель вызванной монокроталином легочной артериальной гипертензии у крыс, описанная, например, в Kato et al. (2008) J. Cardiovasc. Pharmacol. 51(1): 18-23, представляет собой надежный прогностический показатель клинической эффективности лечения легочной артериальной гипертензии. Экспериментальные модели сердечной недостаточности включают крысиную модель DSS сердечной недостаточности и модель аорто-кавального свища (AV (артерио-венозный) шунт), последняя из которых описана, например, в публикации Norling et al. (1996) J. Amer. Soc. Nephrol. 7: 1038-1044. Другие экспериментальные модели, такие как горячая платина, тесты одергивания хвоста и формалиновый тест, могут использоваться для измерения анальгетических свойств соединений по изобретению, а также модель нейропатической боли с перевязкой спинномозговых нервов (SNL). См., например, Malmberg et al. (1999) Current Protocols in Neuroscience 8.9.1-8.9.15.
Ожидается, что соединения по изобретению будут ингибировать фермент NEP в любом из перечисленных выше анализов или в анализах подобной природы. Таким образом, указанные выше анализы применимы при определении терапевтической пользы соединений по изобретению, например, их пригодности в качестве антигипертензивных средств или средств против диареи. Другие свойства и применимость соединений по изобретению можно продемонстрировать с использованием других анализов in vitro и in vivo, хорошо известных специалистам в данной области техники. Соединения формулы I могут представлять собой активные лекарственные средства, а также пролекарства. Таким образом, при обсуждении активности соединений по изобретению подразумевается, что любые такие пролекарства могут не проявлять ожидаемую активность в анализе, но ожидается, что они проявят желательную активность после метаболизации.
Ожидается, что соединения по изобретению полезны для лечения и/или предотвращения медицинских показаний, реагирующих на ингибирование NEP. Таким образом, ожидается, что пациенты, страдающие заболеванием или расстройством, которое лечится ингибированием фермента NEP или увеличением уровней его пептидных субстратов, можно лечить введением терапевтически эффективного количества соединения по изобретению. Например, ожидается, что путем ингибирования NEP, соединения будут потенциировать биологические эффекты эндогенных пептидов, которые метаболизируются с помощью NEP, таких как натрийуретические пептиды, бомбезин, брадикинины, кальцитонин, эндотелины, энкефалины, нейротензин, вещество P и вазоактивный кишечный пептид. Таким образом, ожидается, что эти соединения проявляют другие виды физиологической активности, например, на почечную, центральную нервную, репродуктивную и желудочно-кишечную системы.
В одном варианте осуществления изобретения пациентов, страдающих заболеванием или расстройством, которое лечат путем ингибирования фермента NEP, лечат введением соединения по изобретению, которое представлено в его активной форме, т.е., соединения формулы I, где R1 и R4 представляют собой H, и R2, R3, R5, R6, a, b, и Z такие, как определено для формулы I.
В другом варианте осуществления пациентов лечат введением соединения, которое метаболизируется in vitro с образованием соединения формулы I, где R1 и R4 представляют собой H, и R2, R3, R5, R6, a, b, и Z такие, как определено для формулы I.
В другом варианте осуществления пациентов лечат введением соединения по изобретению, которое представлено в его пролекарственной форме у группы R1, т.е., соединение формулы I, где R1 выбрана из -C1-8алкила, -C1-3алкилен-C6-10арила, -C1-3алкилен-C1-9гетероарила, -C3-7циклоалкила, -[(CH2)2O]1-3CH3, -C1-6алкилен-OC(O)R10, -C1-6алкилен-NR11R12, -C1-6алкилен-C(O)R13, -C0-6алкиленморфолинила, -C1-6алкилен-SO2-C1-6алкила,
В еще одном варианте осуществления пациентов лечат введением соединения по изобретению, которое представлено в его пролекарственной форме у группы R4, т.е. соединения формулы I, где R4 выбрана из -C1-8алкила, -C1-3алкилен-C6-10арила, -C1-3алкилен-C1-9гетероарила, -C3-7циклоалкила, -[(CH2)2O]1-3CH3, -C1-6алкилен-OC(O)R40, -C1-6алкилен-NR41R42, -C1-6алкилен-C(O)R43, -C0-6алкиленморфолинил, -C1-6алкилен-SO2-C1-6алкила,

В еще одном варианте осуществления пациентов лечат введением соединения по изобретению, которое представлено в его пролекарственной форме у группы R1 и у группы R4.
Сердечно-сосудистые заболевания
Ожидается, что путем потенциирования эффектов вазоактивных пептидов, подобных натрийуретическим пептидам и брадикинину, соединения по изобретению найдут применение при лечении и/или профилактике медицинских показаний, таких как сердечно-сосудистые заболевания. См., например, публикации Roques et al. (1993) Pharmacol Rev. 45: 87-146 и Dempsey et al. (2009) Amer. J. of Pathology 174(3): 782-796. Представляющие особый интерес сердечно-сосудистые заболевания включают гипертензию и сердечную недостаточность. Гипертензия включает в качестве примера, а не ограничения: первичную гипертензию, которая также именуется эссенциальной гипертонией или идиопатической гипертонией; вторичную гипертензию; гипертензию с сопровождающим почечным заболеванием; тяжелую гипертензию с сопровождающим почечным заболеванием или без него; легочную гипертензию, включая легочную артериальную гипертензию; и резистентную гипертензию. Сердечная недостаточность включает в качестве примера, а не ограничения: застойную сердечную недостаточность; острую сердечную недостаточность; хроническую сердечную недостаточность, например, со сниженной фракцией изгнания левого желудочка (также именуемую систолической сердечной недостаточностью) или с сохранной фракцией изгнания левого желудочка (также именуемую диастолической сердечной недостаточностью); и острую и хроническую декомпенсированную сердечную недостаточность с сопровождающим почечным заболеванием или без него. Таким образом, один вариант осуществления изобретения относится к способу лечения гипертензии, в частности, первичной гипертензии или легочной артериальной гипертензии, включающему введение пациенту терапевтически эффективного количества соединения по изобретению.
Для лечения первичной гипертензии терапевтически эффективное количество представляет собой обычно количество, которое достаточно для снижения артериального давления пациента. Это включает и гипертензию от легкой до умеренной степени и тяжелую гипертензию. При применении для лечения гипертензии соединение может быть введено в комбинации с другими терапевтическими средствами, такими как антагонисты альдостерона, ингибиторы альдостеронсинтазы, ингибиторы ангиотензин-превращающего фермента и ингибиторы ангиотензин-превращающего фермента/неприлизина двойного действия, активаторы и стимуляторы ангиотензин-превращающего фермента 2 (ACE2), вакцины против ангиотензина-II, противодиабетические средства, противолипидные средства, антитромботические средства, антагонисты рецепторов БФ1 и антагонисты рецепторов БФ1/ингибиторы неприлизина двойного действия, антагонисты β1-адренергических рецепторов, антагонисты β-адренергических рецепторов/антагонисты α1-рецепторов двойного действия, блокаторы кальциевых каналов, диуретики, антагонисты рецепторов эндотелина, ингибиторы эндотелин-превращающего фермента, ингибиторы неприлизина, натрийуретические пептиды и их аналоги, антагонисты клиренс-рецепторов натрийуретических пептидов, доноры оксида азота, нестероидные противовоспалительные средства, ингибиторы фосфодиэстеразы (в частности, ингибиторы PDE-V), агонисты простагландиновых рецепторов, ингибиторов ренина, растворимых стимуляторов и активаторов гуанилатциклазы и их комбинации. В одном конкретном варианте осуществления изобретения соединение по изобретению комбинируется с антагонистом рецепторов БФ1, блокатором кальциевых каналов, диуретиком или их комбинацией, и применяется для лечения первичной гипертензии. В другом конкретном варианте осуществления изобретения соединение по изобретению комбинируется с антагонистом рецепторов AT1 и применяется для лечения гипертензии с сопутствующим почечным заболеванием. При применении для лечения резистентной гипертензии, соединение может быть введено в комбинации с другими терапевтическими средствами, такими как ингибиторы альдостеронсинтазы.
Для лечения легочной артериальной гипертензии терапевтически эффективное количество обычно представляет собой количество, которое достаточно для снижения легочного сосудистого сопротивления. Другими целями лечения являются улучшение способности переносить физическую нагрузку. Например, в условиях клиники терапевтически эффективное количество может представлять собой количество, которое улучшает способность пациента комфортно идти в течение 6 минут (проходя расстояние приблизительно 20-40 метров). При применении для лечения легочной артериальной гипертензии соединение можно вводить в комбинации с другими терапевтическими средствами, такими как антагонисты α-адренергических рецепторов, антагонисты β1-адренергических рецепторов, агонисты β2-адренергических рецепторов, ингибиторы ангиотензин-превращающего фермента, антикоагулянты, блокаторы кальциевых каналов, диуретики, антагонисты рецепторов эндотелина, ингибиторы PDE-V, аналоги простагландина, селективные ингибиторы обратного захвата серотонина и их комбинации. В одном конкретном варианте осуществления изобретения, соединение по изобретению комбинируется с ингибитором PDE-V или селективным ингибитором обратного захвата серотонина и применяется для лечения легочной артериальной гипертензии.
Другой вариант осуществления изобретения относится к способу лечения сердечной недостаточности, в частности, застойной сердечной недостаточности (включая и систолическую, и диастолическую застойную сердечную недостаточность), включающему введение пациенту терапевтически эффективного количества соединения по изобретению. Обычно, терапевтически эффективное количество представляет собой количество, которое достаточно для снижения артериального давления и/или улучшения почечных функций. В условиях клиники, терапевтически эффективное количество может представлять собой количество, которое достаточно для улучшения сердечной гемодинамики, как, например, снижение давления заклинивания, давления в правом предсердии, давления наполнения и сосудистого сопротивления. В одном варианте осуществления соединение вводится в виде внутривенной лекарственной формы. При применении для лечения сердечной недостаточности соединение может вводиться в комбинации с другими терапевтическими средствами, такими как антагонисты рецепторов аденозина, средства, разрушающие конечные продукты поздних стадий гликирования, антагонисты альдостерона, антагонисты рецепторов AT1, антагонисты β1-адренергических рецепторов, антагонисты β1-адренергических рецепторов/антагонисты α1-рецепторов двойного действия, ингибиторы химазы, дигоксин, диуретики, ингибиторы эндотелин-превращающего фермента (ECE), антагонисты рецепторов эндотелина, натрийуретические пептиды и их аналоги, антагонисты клиренс-рецепторов натрийуретических пептидов, доноры оксида азота, аналоги простагландина, ингибиторы PDE-V, растворимые активаторы и стимуляторы гуанилатциклазы и антагонисты вазопрессиновых рецепторов. В одном конкретном варианте осуществления изобретения соединение по изобретению комбинируется с антагонистом альдостерона, антагонистом рецепторов β1-адренергических рецепторов, антагонистом рецепторов AT1 или диуретиком и применяется для лечения застойной сердечной недостаточности.
Диарея
Ожидается, что в качестве ингибиторов NEP, соединения по изобретению будут ингибировать разрушение эндогенных энкефалинов, и, таким образом, такие соединения могут также найти применение для лечения диареи, включая инфекционную и секреторную/водянистую диарею. См., например, Baumer et al. (1992) Gut 33: 753-758; Farthing (2006) Digestive Diseases 24: 47-58; и Marcais-Collado (1987) Eur. J. Pharmacol. 144(2): 125-132. При применении для лечения диареи, соединения по изобретению могут комбинироваться с одним или несколькими дополнительными противодиарейными средствами.
Почечные заболевания
Ожидается, что путем потенциирования эффектов вазоактивных пептидов, подобных натрийуретическим пептидам и брадикинину, соединения по изобретению улучшат почечную функцию (см. Chen et al. (1999) Circulation 100: 2443-2448; Lipkin et al. (1997) Kidney Int. 52: 792-801; и Dussaule et al. (1993) Clin. Sci. 84: 31-39) и найдут применение при лечении и/или предотвращении почечных заболеваний. Представляющие особый интерес почечные заболевания включают диабетическую нефропатию, хроническое почечное заболевание, протеинурию, и в частности, острое почечное повреждение или острую почечную недостаточность (см. публикации Sharkovska et al. (2011) Clin. Lab. 57: 507-515 и Newaz et al. (2010) Renal Failure 32: 384-390). При применении для лечения почечного заболевания, соединение может вводиться в комбинации с другими терапевтическими средствами, такими как ингибиторы ангиотензин-превращающего фермента, антагонисты рецепторов AT1 и диуретики.
Профилактическая терапия
Ожидается также, что ввиду потенциирования эффектов натрийуретических пептидов, соединения по изобретению также полезны при профилактической терапии ввиду антигипертрофических и антифибротических эффектов натрийуретических пептидов (см. Potter et al. (2009) Handbook of Experimental Pharmacology 191: 341-366), например, при предотвращении прогрессирования сердечной недостаточности после инфаркта миокарда, предотвращении артериального рестеноза после ангиопластики, предотвращения утолщения стенок кровеносных сосудов после сосудистых операций, предотвращения атеросклероза и предотвращения диабетической ангиопатии.
Глаукома
Ожидается, что ввиду потенциирования эффектов натрийуретических пептидов, соединения по изобретению могут быть полезны для лечения глаукомы. См., например, публикацию Diestelhorst et al. (1989) International Ophthalmology 12: 99-101. При применении для лечения глаукомы, соединения по изобретению могут комбинироваться с одним или несколькими дополнительными средствами против глаукомы.
Облегчение боли
Ожидается, что в качестве ингибиторов NEP, соединения по изобретению будут ингибировать разрушение эндогенных энкефалинов, и, таким образом, такие соединения могут также найти применение в качестве анальгетиков. См., например, публикации Roques et al. (1980) Nature 288: 286-288 и Thanawala et al. (2008) Current Drug Targets 9: 887-894. При применении для лечения боли, соединения по изобретению могут комбинироваться с одним или несколькими дополнительными обезболивающими лекарственными средствами, такими как ингибиторы аминопептидазы N или дипептилпептидазы III, нестероидные противовоспалительные средства, ингибиторы обратного захвата моноамина, мышечные релаксанты, антагонисты рецепторов NMDA (N-метил-D-аспартата), агонисты опиоидных рецепторов, агонисты серотониновых 5-HT1D рецепторов и трициклические антидепрессанты.
Другие виды применения
Ожидается, что ввиду их свойств в качестве ингибиторов NEP, соединения по изобретению могут быть полезны в качестве противокашлевых средств, а также найти применение при лечении портальной гипертензии, связанной с циррозом печени (см. Sansoe et al. (2005) J. Hepatol. 43: 791-798), рака (см. Vesely (2005) J. Investigative Med. 53: 360-365), депрессии (см. Noble et al. (2007) Exp. Opin. Ther. Targets 11: 145-159), менструальных нарушений, преждевременных родов, преэклампсии, эндометриоза, репродуктивных расстройств (например, мужского и женского бесплодия, синдрома поликистоза яичников, несостоятельности имплантации) и сексуальной дисфункции у мужчин и женщин, включая эректильную дисфункцию у мужчин и расстройство сексуального возбуждения у женщин. Конкретнее, ожидается, что соединения по изобретению могут быть полезны при лечении сексуальной дисфункции у женщин (см. Pryde et al. (2006) J. Med. Chem. 49: 4409-4424), которая часто определяется как затруднение или неспособность пациентки найти удовлетворение в сексуальном выражении. Это включает разнообразные сексуальные расстройства у женщин, включая в качестве иллюстрации, а не ограничения, расстройство в виде гипоактивного сексуального желания, расстройства сексуального возбуждения, расстройства оргазма и расстройства в виде боли при сексуальном контакте. При применении для лечения таких расстройств, как, в частности, сексуальная дисфункция у женщин, соединения по изобретению могут комбинироваться с одним или несколькими из следующих вторичных средств: ингибиторы PDE-V, агонисты допамина, агонисты и/или антагонисты эстрогеновых рецепторов, андрогены и эстрогены. Ожидается, что ввиду их свойств в качестве ингибиторов NEP, соединения по изобретению могут обладать противовоспалительными свойствами, и ожидается, что они могут применяться как таковые, в частности, при применении в комбинации со статинами.
Недавно проведенные исследования свидетельствуют о том, что NEP играет роль в регулировании нервной функции при инсулинзависимом диабете и вызванном рационом ожирении (Coppey et al. (2011) Neuropharmacology 60: 259-266). Поэтому, ожидается, что ввиду их свойств ингибирования NEP, соединения по изобретению могут также применяться для обеспечения защиты от повреждения нервов, вызванного сахарным диабетом или вызванного рационом ожирением.
Количество соединения по изобретению, вводимое на дозу, или общее количество, вводимое в день, может быть определено заранее, или оно может быть определено на основе особенностей пациентов с учетом многочисленных факторов, включая природу и тяжесть состояния пациента, подвергаемое лечению состояние, возраст, массу тела и общее состояние здоровья пациента, переносимость пациентом активного средства, путь введения, фармакологические аспекты, такие как активность, эффективность, фармакокинетика и профили токсикологии соединения и любых вводимых вторичных средств и т.п. Лечение пациента, страдающего заболеванием или медицинским показанием (таким как гипертензия), может начинаться с заданной дозировки или дозировки, определяемой лечащим врачом, и продолжаться в течение периода времени, необходимого для предотвращения, облегчения, подавления или смягчения симптомов заболевания или медицинского показания. Обычно в соответствии с установленной практикой производится мониторинг пациентов для определения эффективности лечения. Например, при лечении гипертензии для определения эффективности можно использовать измерения артериального давления. Подобные показатели для других описанных здесь заболеваний и состояний хорошо известны и общедоступны для лечащего врача. Непрерывный мониторинг врачом гарантирует то, что в любое данное время будет введено оптимальное количество соединения по изобретению, а также обеспечится содействие определению длительности лечения. Это особенно важно, когда вводятся также вторичные средства, поскольку их выбор, дозировка и длительность лечения могут также потребовать коррекции. Таким образом, тактику лечения и схему дозировки можно корригировать в течение всего курса лечения с тем, чтобы вводилось минимальное количество активного средства, которое проявляет требуемую эффективность, и дополнительно, чтобы введение продолжалось только необходимое для успешного лечения заболевания или медицинского показания время.
Инструменты исследования
Поскольку соединения по изобретению обладают ингибирующей активностью в отношении фермента NEP, то такие соединения также полезны в качестве инструментов исследования для исследования и изучения биологических систем или образцов, содержащих фермент NEP, например, для изучения заболеваний, при которых играют роль фермент NEP или его пептидные субстраты. Любая пригодная биологическая система или образец, имеющий фермент NEP, может использоваться в таких исследованиях, которые могут проводиться или in vitro, или in vivo. Репрезентативные биологические системы или образцы, пригодные для таких исследований, включают без ограничения клетки, клеточные экстракты, плазматические мембраны, образцы ткани, изолированные органы, млекопитающих (таких как мыши, крысы, морские свинки, кролики, собаки, свиньи, люди и т.д.), и т.п., причем особый интерес представляют млекопитающие. В одном конкретном варианте осуществления изобретения активность фермента NEP у млекопитающего ингибируется введением ингибирующего NEP количества соединения по изобретению. Соединения по изобретению могут также использоваться в качестве инструментов исследования проведением биологических анализов с использованием таких соединений.
При использовании в качестве инструмента исследования, обычно обеспечивается контакт биологической системы или образца, содержащего фермент NEP, с ингибирующим фермент NEP количеством соединения по изобретению. После воздействия соединения на биологическую систему или образец, эффекты ингибирования фермента NEP определяют, используя обычные процедуры и оборудование, например, измерением связывания с рецепторами в анализе связывания или измерением лиганд-опосредованных изменений в функциональном анализе. Воздействие включает приведение клеток или ткани в контакт с соединением, введение соединения млекопитающему, например, внутрибрюшинным, пероральным, внутривенным, подкожным или ингаляционным введением и т.д. Эта стадия определения может включать измерение реакции (количественный анализ) или может включать проведение наблюдения (качественный анализ). Измерение реакции включает, например, определение эффектов соединения на биологическую систему или образец с использованием обычных процедур и оборудования, таких как анализы ферментативной активности и измерение ферментного субстрата или опосредованных продуктом изменений функциональных анализов. Результаты анализов можно использовать для определения уровня активности, а также количества соединения, необходимого для достижения требуемого результата, то есть, количества, ингибирующего фермент NEP. Обычно, стадия определения включает определение эффектов ингибирования фермента NEP.
Кроме того, соединения по изобретению могут использоваться в качестве инструментов исследования для оценки других химических соединений, и, таким образом, они также полезны при скрининговых анализах для обнаружения, например, новых соединений, обладающих NEP-ингибирующей активностью. Таким образом, соединение по изобретению применяется в качестве стандарта в анализе для обеспечения возможности сравнения результатов, полученных с тестируемым соединением, и с соединением по изобретению, для идентификации тех тестируемых соединений, которые имеют примерно равную или превосходящую активность, если она есть. Например, данные pKi для тестируемого соединения или группы тестируемых соединений сравнивают с данными pKi для соединения по изобретению для идентификации тех тестируемых соединений, которые имеют требуемые свойства, например, тестируемых соединений, имеющих величину pKi, примерно равную или превосходящую таковую соединения по изобретению, если они есть. Этот аспект изобретения включает в качестве отдельных вариантов осуществления оба поколения данных сравнения (с использованием соответствующих анализов) и анализ данных теста для идентификации тестируемых соединений, представляющих интерес. Таким образом, тестируемое соединение можно оценить в биологическом анализе способом, включающим стадии: (a) проведения биологического анализа с тестируемым соединением для получения первой величины анализа; (b) проведения биологического анализа с соединением по изобретению для получения второй величины анализа; где стадия (a) проводится либо перед, либо после, либо одновременно со стадией (b); и (c) сравнения величины первого анализа со стадии (a) с величиной второго анализа со стадии (b). Иллюстративные биологические анализы включают анализ ингибирования фермента NEP.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СОСТАВЫ
Соединения по изобретению обычно вводятся пациенту в виде фармацевтической композиции или состава. Такие фармацевтические композиции могут быть введены пациенту любым приемлемым путем введения, включая без ограничения пероральный, ректальный, вагинальный, интраназальный, ингаляционный, местный (включая трансдермальный), глазной и парентеральный пути введения. Кроме того, соединения по изобретению могут быть введены, например, перорально, множественными дозами в день (например, два, три или четыре раза в день), одной суточной дозой или одной еженедельной дозой. Следует понимать, что любая форма соединения по изобретению, (то есть свободное основание, свободная кислота, фармацевтически приемлемая соль, сольват и т.д.), которая пригодна для конкретного пути введения, может применяться в описанных здесь фармацевтических композициях.
Соответственно, в одном варианте осуществления изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение по изобретению. Композиции могут при желании содержать другие терапевтические и/или используемые при составлении средства. При обсуждении композиций «соединение по изобретению» может также называться в настоящем описании «активным средством» для отличия его от других компонентов состава, таких как носитель. Таким образом, подразумевается, что термин «активное средство» включает соединения формулы I, а также фармацевтически приемлемые соли, сольваты и пролекарства этих соединений.
Фармацевтические композиции по изобретению обычно содержат терапевтически эффективное количество соединения по изобретению. Однако специалистам в данной области техники понятно, что фармацевтическая композиция может содержать более чем терапевтически эффективное количество, например, в композиции в промышленном масштабе, или менее чем терапевтически эффективное количество, то есть, отдельные единичные дозы, предназначенные для множественного введения для достижения терапевтически эффективного количества. Обычно, композиция содержит примерно 0,01-95 масс.% активного средства, включая примерно 0,01-30 масс.%, такое количество, как примерно 0,01-10 масс.%, при действительном количестве, зависящем от самого состава, пути введения, частоты введения и т.д. В одном варианте осуществления композиция, пригодная для пероральной лекарственной формы, например, может содержать примерно 5-70 масс.% или примерно 10-60 масс.% активного средства.
Любой обычный носитель или эксципиент может использоваться в фармацевтических композициях по изобретению. Выбор конкретного носителя или эксципиента или комбинации носителей или эксципиентов зависит от пути введения, используемого для лечения конкретного пациента, или типа медицинского показания или стадии заболевания. В этом отношении, получение подходящей композиции для конкретного пути введения вполне укладывается в объем навыков специалистов в фармацевтических областях. Кроме того, носители или эксципиенты, используемые в таких композициях, являются коммерчески доступными. В качестве дополнительной иллюстрации обычные технологии получения составов описаны в Remington: The Science и Practice of Pharmacy, 20th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); и H.C. Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
Репрезентативные примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают без ограничения следующие: сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу, такую как микрокристаллическая целлюлоза и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин; тальк; эксципиенты, такие как масло какао и воски для суппозиториев; масла, такие как арахисовое масло, масло хлопковых семян, масло сафлора, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; забуферивающие агенты, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический солевой раствор; раствор Рингера; этиловый спирт; фосфатные буферные растворы; сжатые газы в качестве пропеллентов, такие как хлорфторуглероды и гидрофторуглероды; и другие нетоксичные совместимые вещества, используемые в фармацевтических композициях.
Фармацевтические композиции обычно получают тщательным смешиванием или перемешиванием активного средства с фармацевтически приемлемым носителем и одним или несколькими необязательными ингредиентами. Полученные равномерно перемешанные смеси могут быть затем профилированы или загружены в форму таблетки, капсулы, пилюли, канистры, кассеты, раздаточных устройств и т.п., с использованием обычных процедур и оборудования.
В одном варианте осуществления фармацевтические композиции пригодны для перорального введения. Подходящие композиции для перорального введения могут быть представлены в форме капсул, таблеток, пилюль, пастилок, саше, драже, порошков, гранул; растворов или суспензий в водной или неводной жидкости; эмульсий масло-в-воде или вода-в-масле; эликсиров или сиропов и т.п.; причем каждая содержит заданное количество активного средства.
Если композиция предназначена для перорального введения в твердой лекарственной форме (капсулы, таблетки, пилюли и т.п.), то она обычно содержит активное средство и один или несколько фармацевтически приемлемых носителей, таких как цитрат натрия или дикальций фосфат. Твердые лекарственные формы могут также содержать: наполнители или агенты для увеличения объема, такие как крахмалы, микрокристаллическая целлюлоза, лактоза, сахароза, глюкоза, маннит и/или кремневая кислота; связующие вещества, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или камедь; увлажнители, такие как глицерин; разрыхлители, такие как агар-агар, карбонат кальция, картофельный или полученный из тапиоки крахмал, альгиновая кислота, определенные силикаты и/или карбонат натрия; агенты, задерживающие растворение, такие как парафин; ускорители всасывания, такие как соединения четвертичного аммония; смачивающие агенты, такие как цетиловый спирт и/или моностеарат глицерина; абсорбенты, такие как каолин и/или бентонитовая глина; смазывающие вещества, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и/или их смеси; красящие агенты и забуферивающие агенты.
Высвобождающие агенты, смачивающие агенты, покрывающие агенты, подсластители, ароматизирующие агенты, консерванты и антиоксиданты могут также присутствовать в фармацевтических композициях. Иллюстративные покрывающие агенты для таблеток, капсул, пилюль и т.п. включают те, которые используются для энтеросолюбильных покрытий, такие как ацетатфталат целлюлозы, поливинилацетатфталат, фталат гидроксипропилметилцеллюлозы, сополимеры метакриловой кислоты-сложного эфира метакриловой кислоты, ацетаттримеллитат целлюлозы, карбоксиметилэтилцеллюлоза, ацетатсукцинат гидроксипропилметилцеллюлозы и т.п. Примеры фармацевтически приемлемых антиоксидантов включают: растворимые в воде антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфат натрия, сульфит натрия и т.п.; растворимые в масле антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол, бутилированный гидрокситолуол, лецитин, пропилгаллат, альфа-токоферол и т.п.; и металл-хелатирующие агенты, такие как лимонная кислота, этилендиаминтетрауксусная кислота, сорбит, винная кислота, фосфорная кислота и т.п.
Композиции также могут быть составлены для обеспечения медленного или контролируемого высвобождения активного средства с использованием в качестве примера гидроксипропилметилцеллюлозы в варьирующихся пропорциях или других полимерных матриц, липосом и/или микросфер. Кроме того, фармацевтические композиции по изобретению могут содержать придающие матовость агенты и могут быть составлены так, чтобы они высвобождали активное средство только или преимущественно в определенной части желудочно-кишечного тракта, необязательно, отсроченным образом. Примеры композиций для заливки, которые могут использоваться, включают полимерные вещества и воски. Активное средство также может быть представлено в микроинкапсулированной форме, необязательно с одним или несколькими из описанных выше эксципиентов.
Подходящие жидкие лекарственные формы для перорального введения включают в качестве иллюстрации фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Жидкие лекарственные формы обычно содержат активное средство и инертный разбавитель, такой как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (например, масла хлопковых семян, земляного ореха, кукурузы, зародышей, оливковое, касторовое и кунжутное), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоль и сложные эфиры жирных кислот сорбитана и их смеси. Суспензии могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант и их смеси.
Когда фармацевтические композиции по изобретению предназначены для перорального введения, то они могут быть упакованы в стандартную лекарственную форму. Термин «стандартная лекарственная форма» относится к физически дискретной единице, пригодной для введения пациенту, от есть, каждая единица содержит заданное количество активного средства, рассчитанное для обеспечения требуемого терапевтического эффекта или отдельно, или в комбинации с одной или несколькими дополнительными единицами. Например, такие стандартные лекарственные формы могут представлять собой капсулы, таблетки, пилюли и т.п.
В другом варианте осуществления композиции по изобретению пригодны для ингаляционного введения и обычно представлены в форме аэрозоля или порошка. Такие композиции в целом вводят, используя хорошо известные устройства доставки, такие как распылитель, ингалятор сухого порошка или ингалятор отмеренной дозы. Распылительные устройства создают высокоскоростной поток воздуха, который вызывает распыление композиции в виде тумана, который переносится в дыхательный тракт пациента. Иллюстративный распыляемый состав содержит активное средство, растворенное в носителе, с образованием раствора, или микронизируется и комбинируется с носителем с образованием суспензии микронизированных частиц вдыхаемого размера. Ингаляторы сухого порошка вводят активное средство в виде свободно текущего порошка, который диспергируется в воздушном потоке пациента во время вдоха. Иллюстративный состав в виде сухого порошка содержит активное средство, в сухом виде смешанное с эксципиентом, таким как лактоза, крахмал, маннит, декстроза, полимолочная кислота, полилактид-ко-гликолид и их комбинации. Ингаляторы отмеренной дозы выбрасывают отмеренное количество активного средства, используя сжатый газ-пропеллент. Иллюстративный состав в виде отмеренной дозы содержит раствор или суспензию активного средства в сжиженном пропелленте, таком как хлорфторуглерод или гидрофторалкан. Необязательные компоненты таких составов включают сорастворители, такие как этанол или пентан, и поверхностно-активные вещества, такие как сорбитан триолеат, олиновая кислота, лецитин, глицерин и лаурилсульфат натрия. Такие композиции обычно получают добавлением охлажденного или сжатого гидрофторалкана в пригодный контейнер, содержащий активное средство, этанол (если он присутствует) и поверхностно-активное вещество (если оно присутствует). Для получения суспензии активное средство микронизируют и затем комбинируют с пропеллентом. Альтернативно, состав в виде суспензии может быть получен распылительной сушкой покрытия поверхностно-активного вещества на микронизированные частицы активного средства. Затем состав загружают в аэрозольную канистру, которая образует часть ингалятора.
Соединения по изобретению также можно вводить парентерально (например, подкожной, внутривенной, внутримышечной или внутрибрюшинной инъекцией). Для такого введения активное средство представлено в стерильном растворе, суспензии или эмульсии. Иллюстративные растворители для получения таких составов включают воду, солевой раствор, спирты с низкой молекулярной массой, такие как пропиленгликоль, полиэтиленгликоль, масла, желатин, сложные эфиры жирных кислот, такие как этилолеат, и т.п. Парентеральные составы также могут содержать один или несколько антиоксидантов, солюбилизаторов, стабилизаторов, консервантов, смачивающих агентов, эмульгаторов и диспергирующих агентов. Поверхностно-активные вещества, дополнительные стабилизирующие агенты или регулирующие pH агенты (кислоты, основания или буферы) и антиоксиданты особенно полезны для обеспечения стабильности составов, например, для минимизации или во избежание гидролиза сложноэфирных и амидных связей, которые могут присутствовать в соединении. Эти составы можно стерилизовать, используя стерильную инъецируемую среду, стерилизующее средство, фильтрование, облучение или нагревание. В одном конкретном варианте осуществления парентеральный состав содержит водный раствор циклодекстрина в качестве фармацевтически приемлемого носителя. Пригодные циклодекстрины включают циклические молекулы, содержащие шесть или более единиц α-D-глюкопиранозы, связанных в положениях 1,4 связями, как в амилазе, β-циклодекстрин или циклогептаамилозу. Иллюстративные циклодекстрины включают производные циклодекстрина, такие как простые гидроксипропиловые и сульфобутиловые эфиры циклодекстринов, такие как гидроксипропил-β-циклодекстрин и простой сульфобутиловый эфир β-циклодекстрина. Иллюстративные буферы для таких составов включают буферы на основе карбоновых кислот, такие как цитратные, лактатные и малеатные буферные растворы.
Соединения по изобретению также могут введены трансдермально с использованием известных систем трансдермальной доставки и эксципиентов. Например, соединение может быть смешано с усилителями проникновения, такими как пропиленгликоль, монолаурат полиэтиленгликоля, азациклоалкан-2-оны и т.п., и включено в трансдермальную систему или аналогичную систему доставки. Дополнительные эксципиенты, включающие желатинирующие агенты, эмульгаторы и буферы, можно при желании использовать в таких трансдермальных композициях.
Вторичные средства
Соединения по изобретению могут применяться в виде отдельного средства для лечения заболевания или может быть комбинировано с одним или несколькими дополнительными терапевтическими средствами для получения требуемого терапевтического эффекта. Таким образом, в одном варианте осуществления фармацевтические композиции по изобретению содержат другие лекарственные средства, которые совместно вводят с соединением по изобретению. Например, композиция может дополнительно содержать одно или несколько лекарственных средств (также именуемых «вторичными лекарственными средствами»). Такие терапевтические средства хорошо известны в данной области техники и включают антагонисты аденозиновых рецепторов, антагонисты α-адренергических рецепторов, антагонисты β1-адренергических рецепторов, агонисты β2-адренергических рецепторов, антагонисты β-адренергических рецепторов/антагонисты α1-рецепторов двойного действия, разрушители конечных продуктов поздних стадий гликирования, антагонисты альдостерона, ингибиторы альдостеронсинтазы, ингибиторы аминопептидазы N, андрогены, ингибиторы ангиотензин-превращающего фермента и ингибиторы двойного действия ангиотензин-превращающего фермента/неприлизина, активаторы и стимуляторы ангиотензин-превращающего фермента 2, вакцины против ангиотензина-II, антикоагулянты, противодиабетические средства, средства против диареи, средства против глаукомы, антилипидные средства, обезболивающие средства, антитромботические средства, антагонисты AT1 рецепторов и двойного действия антагонисты AT1 рецепторов/ингибиторы неприлизина и многофункциональные блокаторы рецепторов ангиотензина, антагонисты рецепторов брадикинина, блокаторы кальциевых каналов, ингибиторы химазы, дигоксин, диуретики, агонисты допамина, ингибиторы эндотелин-превращающего фермента, антагонисты рецепторов эндотелина, ингибиторы HMG-CoA редуктазы, эстрогены, агонисты и/или антагонисты эстрогеновых рецепторов, ингибиторы обратного захвата моноаминов, мышечные релаксанты, натрийуретические пептиды и их аналоги, антагонисты клиренс-рецепторов натрийуретических пептидов, ингибиторы неприлизина, доноры оксида азота, нестероидные противовоспалительные средства, антагонисты рецепторов N-метил-d-аспартата, агонисты опиоидных рецепторов, ингибиторы фосфодиэстеразы, аналоги простагландина, агонисты рецепторов простагландина, ингибиторы ренина, селективные ингибиторы обратного захвата серотонина, блокаторы натриевых каналов, растворимые стимуляторы и активаторы гуанилатциклазы, трициклические антидепрессанты, антагонисты вазопрессиновых рецепторов и их комбинации. Определенные примеры этих средств подробно представлены в настоящем описании.
Соответственно, в еще одном аспекте изобретения, фармацевтическая композиция содержит соединение по изобретению, второе активное средство и фармацевтически приемлемый носитель. В композицию может быть также включено третье, четвертое и т.д. активные средства. При комбинированной терапии, количество соединения по изобретению, которое вводится, а также количество вторичных средств может быть меньше, чем количество, обычно вводимое при монотерапии.
Соединения по изобретению могут быть физически смешаны со вторым терапевтическим средством с образованием композиции, содержащей оба средства; или каждое средство может присутствовать в отдельных и обособленных композициях, которые вводят пациенту одновременно или в отдельные моменты времени. Например, соединение по изобретению может быть комбинировано со вторым активным средством, используя обычные процедуры и оборудование, с образованием комбинации активных средств, содержащей соединение по изобретению и второе активное средство. Кроме того, активные средства могут быть комбинированы с фармацевтически приемлемым носителем с образованием фармацевтической композиции, содержащей соединение по изобретению, второе активное средство и фармацевтически приемлемый носитель. В этом варианте осуществления компоненты композиции обычно смешивают для создания физической смеси. Затем физическую смесь вводят в терапевтически эффективном количестве с использованием любого из описанных здесь путей.
Альтернативно, активные средства могут оставаться отделенными и обособленными перед введением пациенту. В этом варианте осуществления средства не смешиваются физически вместе перед введением, но вводятся одновременно или в отдельные моменты времени в виде отдельных композиций. Такие композиции могут быть упакованы отдельно или могут быть упакованы вместе в наборе. При введении в отдельные моменты времени вторичное средство обычно вводится менее чем через 24 часа после введения соединения по изобретению, в диапазоне от примерно одновременного введения с соединением по изобретению до примерно 24 часов после его введения. Это также называется последовательным введением. Таким образом, соединение по изобретению можно вводить перорально одновременно или последовательно с другим активным средством, используя две таблетки, по одной таблетке для каждого активного средства, где последовательное введение может означать введение непосредственно после введения соединения по изобретению или позднее в некоторое заданное время (например, через один час или через три часа). Предусматривается также, что вторичное средство может вводиться через более чем 24 часа после введения соединения по изобретению. Альтернативно, комбинация может быть введена различными путями введения, то есть, одно перорально, а другое ингаляцией.
В одном варианте осуществления набор включает первую лекарственную форму, содержащую соединение по изобретению и по меньшей мере одну дополнительную лекарственную форму, содержащую одно или несколько вторичных средств, указанных в настоящем описании, в количествах, достаточных для осуществления способов по изобретению. Первая лекарственная форма и вторая (или третья и т.д.) лекарственная форма вместе содержат терапевтически эффективное количество активных средств для лечения или профилактики заболевания или медицинского показания у пациента.
Вторичное средство(-а) при его включении присутствует в терапевтически эффективном количестве, так что оно обычно вводится в количестве, которое обеспечивает терапевтически благоприятный эффект при совместном введении с соединением по изобретению. Вторичное средство может быть представлено в форме фармацевтически приемлемой соли, сольвата, оптически чистого стереоизомера и т.д. Вторичное средство может быть также представлено в форме пролекарства, например, соединения, имеющего группу карбоновой кислоты, которая была этерифицирована. Таким образом, вторичные средства, перечисленные здесь, предназначены для включения всех таких форм и являются коммерчески доступными или могут быть получены с использованием обычных процедур и реагентов.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с антагонистом аденозиновых рецепторов, репрезентативные примеры которого включают без ограничения наксифиллин, ролофиллин, SLV-320, теофиллин и тонапофиллин.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с антагонистом α-адренергических рецепторов, репрезентативные примеры которого включают без ограничения доксазозин, празозин, тамсулозин и теразозин.
Соединения по изобретению могут также вводиться в комбинации с антагонистом β1-адренергичесикх рецепторов («β1-блокаторами»). Репрезентативные β1-блокаторы включают без ограничения ацебутолол, адпренолол, амосулалол, аротинолол, атенолол, бефунолол, бетаксолол, бевантолол, бисопролол, бопиндолол, буциндолол, букумолол, буфетолол, буфуралол, бунитролол, бупранолол, бубридин, бутофилолол, каразолол, картеолол, карведилол, целипролол, цетамолол, клоранолол, дилевалол, эпанолол, эсмолол, инденолол, лабетолол, левобунолол, мепиндолол, метипранолол, метопролол, такой как метопролол сукцинат и метопролол тартрат, мопролол, надолол, надоксолол, небивалол, нипрадилол, окспренолол, пенбутолол, пербутолол, пиндолол, практолол, пронеталол, пропранолол, соталол, суфиналол, талиндол, тертатолол, тилисолол, тимолол, толипролол, ксибенолол и их комбинации. В одном конкретном варианте осуществления β1-антагонист выбран из атенолола, бисопролола, метопролола, пропранолола, соталола и их комбинаций. Обычно, β1-блокатор вводится в количестве, достаточном для обеспечения дозы примерно 2-900 мг на дозу.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с агонистом β2-адренергических рецепторов, репрезентативные примеры которого включают без ограничения албутерол, битолтерол, фенотерол, формотерол, индакатерол, изоэтарин, левалбутерол, метапротеренол, пирбутерол, сальбутамол, сальмефамол, салметерол, тербуталин, вилантерол и т.п. Обычно агонист β2-адренергических рецепторов вводится в количестве, достаточном для обеспечения примерно 0,05-500 мкг на дозу.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с разрушителем конечного продукта поздних стадий гликирования (AGE), примеры которого включают в качестве иллюстрации, а не ограничения, алагебриум (или ALT-711) и TRC4149.
В другом варианте осуществления соединения по изобретению вводятся в комбинации с антагонистом альдостерона, репрезентативные примеры которого включают без ограничения эплеренон, спиронолактон и их комбинации. Обычно, антагонист альдостерона вводится в количестве, достаточном для обеспечения примерно 5-300 мг в день.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с аминопептидазой N или ингибитором дипептидилпептидазы III, примеры которых включают в качестве иллюстрации, а не ограничения, бестатин и PC 18 (2-амино-4-метилсульфонилбутантиол, метионинтиол).
Соединения по изобретению также могут быть введены в комбинации с ингибитором ангиотензин-превращающего фермента (ACE). Репрезентативные ингибиторы ACE включают без ограничения аккуприл, алацеприл, беназеприл, беназеприлат, каптоприл, церанаприл, цилазаприл, делаприл, эналаприл, эналаприлат, фосиноприл, фосиноприлат, имидаприл, лизиноприл, моэксиприл, моноприл, мовелтиприл, пентоприл, периндоприл, хинаприл, хинаприлат, рамиприл, рамиприлат, саралазинацетат, спираприл, темокаприл, трандолаприл, зофеноприл и их комбинации. В конкретном варианте осуществления ингибитор ACE выбран из: беназеприла, каптоприла, эналаприла, лизиноприла, рамиприла и их комбинации. Обычно ингибитор ACE вводится в количестве, достаточном для обеспечения примерно 1-150 мг в день.
В другом варианте осуществления соединения по изобретению вводятся в комбинации с ингибитором ангиотензин-превращающего фермента/неприлизина (ACE/NEP) двойного действия, примеры которого включают без ограничения: AVE-0848 ((4S,7S,12bR)-7-[3-метил-2(S)-сульфанилбутирамидо]-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]-бензазепин-4-карбоновую кислоту); AVE-7688 (илепатрил) и его родительское соединение; BMS-182657 (2-[2-оксо-3(S)-[3-фенил-2(S)-сульфанилпропионамидо]-2,3,4,5-тетрагидро-1H-1-бензазепин-1-ил]уксусную кислоту); CGS-35601 (N-[1-[4-метил-2(S)-сульфанилпентанамидо]циклопентилкарбонил]-L-триптофан); фасидотрил; фасидотрилат; эналаприлат; ER-32935 ((3R,6S,9aR)-6-[3(S)-метил-2(S)-сульфанилпентанамидо]-5-оксопергидротиазоло[3,2-a]азепин-3-карбоновую кислоту); гимпатрилат; MDL-101264 ((4S,7S,12bR)-7-[2(S)-(2-морфолиноацетилтио)-3-фенилпропионамидо]-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновую кислоту); MDL-101287 ([4S-[4α,7α(R*),12β]]-7-[2-(карбоксиметил)-3-фенилпропионамидо]-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновую кислоту); омапатрилат; RB-105 (N-[2(S)-(меркаптометил)-3(R)-фенилбутил]-L-аланин); сампатрилат; SA-898 ((2R,4R)-N-[2-(2-гидроксифенил)-3-(3-меркаптопропионил)тиазолидин-4-илкарбонил]-L-фенилаланин); Sch-50690 (N-[1(S)-карбокси-2-[N2-(метансульфонил)-L-лизиламино]этил]-L-валил-L-тирозин); и также могут быть включены их комбинации. В одном конкретном варианте осуществления ингибитор ACE/NEP выбран из: AVE-7688, эналаприлата, фазидотрила, фазидотрилата, омапатрилата, сампатрилата и их комбинации.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с активатором или стимулятором ангиотензин-превращающего фермента 2 (ACE2).
В одном варианте осуществления соединения по изобретению вводятся в комбинации с вакциной против ангиотензина-II, примеры которой включают без ограничения ATR12181 и CYT006-AngQb.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с антикоагулянтом, репрезентативные примеры которого включают без ограничения: кумарины, такие как варфарин; гепарин; и прямые ингибиторы тромбина, такие как аргатробан, бивалирудин, дабигатран и лепирудин.
В еще одном варианте осуществления соединения по изобретению вводятся в комбинации с противодиабетическим средством. Репрезентативные противодиабетические средства включают инъецируемые лекарственные средства, а также перорально эффективные лекарственные средства и их комбинации. Примеры инъецируемых лекарственных средств включают без ограничения инсулин и производные инсулина. Примеры перорально эффективных лекарственных средств включают без ограничения: бигуаниды, такие как метформин; антагонисты глюкагона; ингибиторы α-глюкозидазы, такие как акарбоза и миглитол; ингибиторы дипептидилпептидазы IV (ингибиторы DPP-IV), такие как алоглиптин, денаглиптин, линаглиптин, саксаглиптин, ситаглиптин и вилдаглиптин; меглитиниды, такие как репаглинид; оксадиазолидиндионы; сульфонилмочевины, такие как хлорпропамид, глимепирид, глипизид, глибурид и толазамид; тиазолидиндионы, такие как пиоглитазон и росиглитазон; и их комбинации.
В другом варианте осуществления соединения по изобретению вводятся в комбинации с другими средствами лечения диареи. Репрезентативные средства лечения включают без ограничения пероральные регидрационные растворы (ORS), лоперамид, дифеноксилат и субсалицилат висмута.
В еще одном варианте осуществления соединение по изобретению вводят в комбинации со средством против глаукомы. Репрезентативные средства против глаукомы включают без ограничения: α-адренергические агонисты, такие как бримонидин; антагонисты β1-адренергических рецепторов; топические β1-блокаторы, такие как бетаксолол, левобунолол и тимолол; ингибиторы карбоангидразы, такие как ацетазоламид, бринзоламид или дорзоламид; холинергические агонисты, такие как цевимелин и DMXB-анабазеин; соединения эпинефрина; миотики, такие как пилокарпин; и аналоги простагландина.
В еще одном варианте осуществления соединения по изобретению вводятся в комбинации с антилипидным средством. Репрезентативные антилипидные средства включают без ограничения: ингибиторы белка-переносчика сложного холестерилового эфира (CETP), такие как анацетрапиб, далцетрапиб и торцетрапиб; статины, такие как аторвастатин, флувастатин, ловастатин, правастатин, росувастатин и симвастатин; и их комбинации.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с антитромботическим средством. Репрезентативные антитромботические средства включают без ограничения: аспирин; антитромбоцитарные средства, такие как клопидогрел, прасугрел и тиклопидин; гепарин и их комбинации.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с антагонистом AT1 рецепторов, также известным как блокаторы рецепторов типа 1 ангиотензина-II (ARB). Репрезентативные ARB включают без ограничения абитесартан, азилсартан (например, азилсартан медоксомил), бензиллосартан, кандесартан, кандесартан цилексетил, элисартан, эмбусартан, энолтасосартан, эпросартан, EXP3174, фонсартан, форасартан, глициллосартан, ирбесартан, изотеолин, лосартан, медоксимил, милфасартан, олмесартан (например, олмесартан медоксомил), опомисартан, пратосартан, риписартан, саприсартан, саралазин, сармезин, TAK-591, тазосартан, телмисартан, валсартан, золасартан и их комбинации. В конкретном варианте осуществления ARB выбран из азилсартана медоксомила, кандесартана цилексетила, эпросартана, ирбесартана, лосартана, олмесартана медоксомила, саприсартана, тазосартана, телмисартана, валсартана и их комбинации. Иллюстративные соли и/или пролекарства включают кандесартан цилексетил, эпросартан месилат, калиевую соль лосартана и олмесартан медоксомил. Обычно ARB вводится в количестве, достаточном для обеспечения примерно 4-600 мг на дозу, при иллюстративных суточных дозировках в диапазоне 20-320 мг в день.
Соединения по изобретению также можно вводить в комбинации со средством двойного действия, таким как антагонист рецепторов БФ1/ингибитор неприлизина (ARB/NEP), примеры которого включают без ограничения соединения, описанные в опубликованных заявках на патенты США №№ 2008/0269305 и 2009/0023228, обе поданные Allegretti et al. 23 апреля 2008 г, такие как соединение 4'-{2-этокси-4-этил-5-[((S)-2-меркапто-4-метилпентанoиламино)-метил]имидазол-1-илметил}-3'-фторбифенил-2-карбоновая кислота.
Соединения по изобретению можно также вводить в комбинации с многофункциональными блокаторами ангиотензиновых рецепторов, как описано в Kurtz & Klein (2009) Hypertension Research 32: 826-834.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с антагонистом рецепторов брадикинина, например, икатибантом (HOE-140). Ожидается, что это комбинированное лечение может предоставить преимущество предотвращения ангиоотека или других нежелательных последствий повышенных уровней брадикинина.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с блокатором кальциевых каналов. Репрезентативные блокаторы кальциевых каналов включают без ограничения амлодипин, анипамил, аранипин, барнидипин, бенциклан, бенидипин, бепридил, клентиазем, цилнидипин, циннаризин, дилтиазем, эфонидипин, элгодипин, этафенон, фелодипин, фендилин, флунаризин, галлопамил, исрадипин, лацидипин, лерканидипин, лидофлазин, ломеризин, манидипин, мибефрадил, никардипин, нифедипин, нигульдипин, нилудипин, нилвадипин, нимодипин, нисолдипин, нитрендипин, нивалдипин, пергексилин, прениламин, риосидин, семотиадил, теродилин, тиапамил, верапамил и их комбинации. В конкретном варианте осуществления блокатор кальциевых каналов выбран из амлодипина, бепредила, дилтиазема, фелодипина, исрадипина, лацидипина, никардипина, нифедипина, нигулдипина, нилудипина, нимодипина, нисолдипина, риосидина, верапамила и их комбинации. Обычно блокатор кальциевых каналов вводится в количестве, достаточном для обеспечения примерно 2-500 мг на дозу.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с ингибитором химазы, таким как TPC-806 и 2-(5-формиламино-6-оксо-2-фенил-1,6-дигидропиримидин-1-ил)-N-[{3,4-диоксо-1-фенил-7-(2-пиридилокси)}-2-гептил]ацетамид (NK3201).
В одном варианте осуществления соединения по изобретению вводятся в комбинации с диуретиком. Репрезентативные диуретики включают без ограничения: ингибиторы карбоангидразы, такие как ацетазоламид и дихлорфенамид; петельные диуретики, которые включают производные сульфонамида, такие как ацетазоламид, амбузид, азосемид, буметанид, бутазоламид, хлораминофенамид, клофенамид, клопамид, клорексолон, дисульфамид, этоксзоламид, фуросемид, мефрузид, метазоламид, пиретанид, торсемид, трипамид и ксипамид, а также несульфонамидные диуретики, такие как этакриновая кислота и другие соединения феноксиуксусной кислоты, такие как тиениловая кислота, индакринон и хинкарбат; осмотические диуретики, такие как маннит; калийсберегающие диуретики, которые включают антагонисты альдостерона, такие как спиронолактон, и ингибиторы Na+ каналов, такие как амилорид и триамтерен; тиазид и подобные тиазиду диуретики, такие как алтиазид, бендрофлуметиазид, бензилгидрохлортиазид, бензтиазид, бутиазид, хлорталидон, хлортиазид, циклопентиазид, циклотиазид, эпитиазид, этиазид, фенквизон, флуметиазид, гидрохлортиазид, гидрофлуметиазид, индапамид, метилклотиазид, метикран, метолазон, парафлутизид, политиазид, хинетазон, теклотиазид и трихлорметиазид; и их комбинации. В конкретном варианте осуществления диуретик выбран из амилорида, буметанида, хлортиазида, хлорталидона, дихлорфенамида, этакриновой кислоты, фуросемида, гидрохлортиазида, гидрофлуметиазида, индапамида, метилклотиазида, метолазона, торсемида, триамтерена и их комбинации. Диуретик вводится в количестве, достаточном для обеспечения примерно 5-50 мг в день, обычнее, 6-25 мг в день, причем обычные дозировки составляют 6,25 мг, 12,5 мг или 25 мг в день.
Соединения по изобретению можно также вводить в комбинации с ингибитором эндотелин-превращающего фермента (ECE), примеры которых включают без ограничения фосфорамидон, CGS 26303 и их комбинации.
В конкретном варианте осуществления соединения по изобретению вводятся в комбинации с антагонистом рецепторов эндотелина. Репрезентативные антагонисты рецепторов эндотелина включают без ограничения: селективные антагонисты рецепторов эндотелина, которые воздействуют на рецепторы эндотелина A, такие как авосентан, амбрисентан, атрасентан, BQ-123, клазосентан, дарусентан, ситаксентан и зиботентан; и двойные антагонисты рецепторов эндотелина, которые воздействуют и на A, и на B рецепторы эндотелина, такие как босентан, мацитентан, тезосентан).
В еще одном варианте осуществления соединение по изобретению вводится в комбинации с один или несколькими ингибиторами HMG-CoA редуктазы, которые также известны как статины. Репрезентативные статины включают без ограничения аторвастатин, флувастатин, ловастатин, питавастатин, правастатин, розувастатин и симвастатин.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с ингибитором обратного захвата моноамина, примеры которого включают в качестве примера, а не ограничения, ингибиторы обратного захвата норэпинефрина, такие как атомоксетин, бупроприон и метаболит бупроприона гидроксибупроприон, мапротилин, ребоксетин и вилоксазин; селективные ингибиторы обратного захвата серотонина (SSRI), такие как циталопрам и метаболит циталопрама десметилциталопрам, дапоксетин, эсциталопрам (например, эсциталопрам оксалат), флуоксетин и десметильный метаболит флуоксетина норфлуоксетин, флувоксамин (например, флувоксамин малеат), пароксетин, сертралин и метаболит сертралина деметилсертралин; двойные ингибиторы обратного захвата серотонина-норэпинефрина (SNRI), такие как бицифадин, дулоксетин, милнаципран, нефазодон и венлафаксин; и их комбинации.
В другом варианте осуществления соединения по изобретению вводятся в комбинации с мышечным релаксантом, примеры которого включают без ограничения: карисопродол, хлорзоксазон, циклобензаприн, дифлунизал, метаксалон, метокарбамол и их комбинации.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с натрийуретическим пептидом или аналогом, примеры которых включают без ограничения: карперитид, CD-NP (Nile Therapeutics), CU-NP, несиритид, PL-3994 (Palatin Technologies, Inc.), уларитид, цендеритид и соединения, описанные в публикации Ogawa et al (2004) J. Biol. Chem. 279: 28625-31. Эти соединения также называются агонистами рецепторов-A натрийуретических пептидов (NPR-A). В другом варианте осуществления соединения по изобретению вводятся в комбинации с антагонистом клиренс-рецепторов натрийуретических пептидов (NPR-C), таким как SC-46542, cANF (4-23) и AP-811 (Veale (2000) Bioorg Med Chem Lett 10: 1949-52). Например, AP-811 проявил синергизм при комбинации с ингибитором NEP тиорфаном (Wegner (1995) Clin. Exper. Hypert. 17: 861-876).
В другом варианте осуществления соединения по изобретению вводятся в комбинации с ингибитором неприлизина (NEP). Репрезентативные ингибиторы NEP включают без ограничения: AHU-377; кандоксатрил; кандоксатрилат; дексекадотрил (сложный бензиловый эфир (+)-N-[2(R)-(ацетилтиометил)-3-фенилпропионил]глицина); CGS-24128 (3-[3-(бифенил-4-ил)-2-(фосфонометиламино)пропионамидо]пропионовая кислота); CGS-24592 ((S)-3-[3-(бифенил-4-ил)-2-(фосфонометиламино)пропионамидо]пропионовая кислота); CGS-25155 (сложный бензиловый эфир N-[9(R)-(ацетилтиометил)-10-оксо-1-азациклодекан-2(S)-илкарбонил]-4(R)-гидрокси-L-пролина); производные 3-(1-карбамоилциклогексил)пропионовой кислоты, описанные в патенте WO 2006/027680, выданном Hepworth et al. (Pfizer Inc.); JMV-390-1 (2(R)-бензил-3-(N-гидроксикарбамоил)пропионил-L-изолейцил-L-лейцин); экадотрил; фосфорамидон; ретротиорфан; RU-42827 (2-(меркаптометил)-N-(4-пиридинил)бензолпропионамид); RU-44004 (N-(4-морфолинил)-3-фенил-2-(сульфанилметил)пропионамид); SCH-32615 ((S)-N-[N-(1-карбокси-2-фенилэтил)-L-фенилаланил]-β-аланин) и его пролекарство SCH-34826 ((S)-N-[N-[1-[[(2,2-диметил-1,3-диоксолан-4-ил)метокси]карбонил]-2-фенилэтил]-L-фенилаланил]-β-аланин); сиалорфин; SCH-42495 (сложный этиловый эфир N-[2(S)-(ацетилсульфанилметил)-3-(2-метилфенил)пропионил]-L-метионина); спинорфин; SQ-28132 (N-[2-(меркаптометил)-1-оксо-3-фенилпропил]лейцин); SQ-28603 (N-[2-(меркаптометил)-1-оксо-3-фенилпропил]-β-аланин); SQ-29072 (7-[[2-(меркаптометил)-1-оксо-3-фенилпропил]амино]гептановая кислота); тиорфан и его пролекарство рацекадотрил; UK-69578 (цис-4-[[[1-[2-карбокси-3-(2-метоксиэтокси)пропил]циклопентил]карбонил]амино]циклогексан-карбоновая кислота); UK-447,841 (2-{1-[3-(4-хлорфенил)пропилкарбамоил]циклопентилметил}-4-метоксимасляная кислота); UK-505,749 ((R)-2-метил-3-{1-[3-(2-метилбензотиазол-6-ил)пропилкарбамоил]циклопентил}пропионовая кислота); 5-бифенил-4-ил-4-(3-карбоксипропиониламино)-2-метилпентанoевая кислота и сложный этиловый эфир 5-бифенил-4-ил-4-(3-карбоксипропиониламино)-2-метилпентановой кислоты (WO 2007/056546); даглутрил [(3S,2'R)-3-{1-[2'-(этоксикарбонил)-4'-фенилбутил]-циклопентан-1-карбониламино}-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусная кислота], описанный в патенте WO 2007/106708, выданном Khder et al. (Novartis AG); и их комбинации. В конкретном варианте осуществления ингибитор NEP выбран из AHU-377, кандоксатрила, кандоксатрилата, CGS-24128, фосфорамидона, SCH-32615, SCH-34826, SQ-28603, тиорфана, и их комбинации. В конкретном варианте осуществления ингибитор NEP представляет собой такое соединение, как даглутрил или CGS-26303 ([N-[2-(бифенил-4-ил)-1(S)-(1H-тетразол-5-ил)этил]амино]метилфосфоновая кислота), которые обладают активностью и как ингибиторы эндотелин-превращающего фермента (ECE), и NEP. Можно также применять другие соединения ECE/NEP двойного действия. Ингибитор NEP вводится в количестве, достаточном для обеспечения примерно 20-800 мг в день, причем обычные суточные дозировки находятся в диапазоне 50-700 мг в день, обычнее 100-600 или 100-300 мг в день.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с донором оксида азота, примеры которого включают без ограничения никорандил; органические нитраты, такие как пентраэритрита тетранитрат; и сиднонимины, такие как линзидомин и молсидомин.
В еще одном варианте осуществления соединения по изобретению вводятся в комбинации с нестероидным противовоспалительным средством (NSAID). Репрезентативные NSAID включают без ограничения: ацеметацин, ацетилсалициловую кислоту, алклофенак, алминопрофен, амфенак, амиприлозу, алоксиприн, аниролак, апазон, азапропазон, бенорилат, беноксапрофен, безпиперилон, броперамол, буклоксовую кислоту, карпрофен, клиданак, диклофенак, дифлунизал, дифталон, эноликам, этодолак, эторикоксиб, фенбуфен, фенклофенак, фенклозовую кислоту, фенопрофен, фентиазак, фепразон, флуфенамовую кислоту, флуфенизал, флупрофен, флурбипрофен, фурофенак, ибуфенак, ибупрофен, индометацин, индопрофен, изоксепак, изоксикам, кетопрофен, кеторолак, лофемизол, лорноксикам, меклофенамат, меклофенамовую кислоту, мефенамовую кислоту, мелоксикам, месаламин, миропрофен, мофебутазон, набуметон, напроксен, нифлумовую кислоту, оксапрозин, окспинак, оксифенбутазон, фенилбутазон, пироксикам, пирпрофен, пранопрофен, салсалат, судоксикам, сульфасалазин, сулиндак, супрофен, теноксикам, тиопинак, тиапрофеновую кислоту, тиоксапрофен, толфенамовую кислоту, толметин, трифлумидат, зидометацин, зомепирак и их комбинации. В конкретном варианте осуществления NSAID выбран из этодолака, флурбипрофена, ибупрофена, индометацина, кетопрофена, кеторолака, мелоксикама, напроксена, оксапрозина, пироксикама и их комбинации.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с антагонистом рецепторов N-метил-d-аспартата (NMDA), примеры которого включают в качестве иллюстрации, а не ограничения, амантадин, декстрометорфан, декстропропоксифен, кетамин, кетобемидон, мемантин, метадон и т.д.
В еще одном варианте осуществления соединения по изобретению вводятся в комбинации с агонистом опиоидных рецетпоров (также называемые опиоидными анальгетиками). Репрезентативные агонисты опиоидных рецетпоров включают без ограничения: бупренорфин, буторфанол, кодеин, дигидрокодеин, фентанил, гидрокодон, гидроморфон, леваллорфан, леворфанол, меперидин, метадон, морфин, налбуфин, налмефен, налорфин, налоксон, налтрексон, налорфин, оксикодон, оксиморфон, пентазоцин, пропоксифен, трамадол и их комбинации. В определенных вариантах осуществления агонист опиоидных рецепторов выбран из кодеина, дигидрокодеина, гидрокодона, гидроморфона, морфина, оксикодона, оксиморфона, трамадола и их комбинации.
В конкретном варианте осуществления соединения по изобретению вводятся в комбинации с ингибитором фосфодиэстеразы (PDE), в частности, ингибитором PDE-V. Репрезентативные ингибиторы PDE-V включают без ограничения аванафил, лоденафил, мироденафил, силденафил (Revatio®), тадалафил (Adcirca®), варденафил (Levitra®) и уденафил.
В другом варианте осуществления соединения по изобретению вводятся в комбинации с аналогом простагландина (также именуемые простаноидами или аналогами простациклина). Репрезентативные аналоги простагландина включают без ограничения берапрост натрия, биматопрост, эпопростенол, илопрост, латанопрост, тафлупрост, травопрост и трепростинил, причем биматопрост, латанопрост и тафлупрост представляют особый интерес.
В еще одном варианте осуществления соединения по изобретению вводятся в комбинации с агонистом рецепторов простагландина, примеры которого включают без ограничения биматопрост, латанопрост, травопрост и т.д.
Соединения по изобретению также можно вводить в комбинации с ингибитором ренина, примеры которого включают без ограничения алискирен, эналкирен, ремикирен и их комбинации.
В другом варианте осуществления соединения по изобретению вводятся в комбинации с селективным ингибитором обратного захвата серотонина (SSRI). Репрезентативные SSRI включают без ограничения: циталопрам и метаболит циталопрама десметилциталопрам, дапоксетин, эсциталопрам (например, эсциталопрам оксалат), флуоксетин и десметиловый метаболит флуоксетина норфлуоксетин, флувоксамин (например, флувоксамин малеат), пароксетин, сертралин и метаболит сертралина деметилсертралин и их комбинации.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с агонистом серотониновых 5-HT1D рецепторов, примеры которого включают в качестве иллюстрации, а не ограничения, триптаны, такие как алмотриптан, авитриптан, элетриптан, фроватриптан, наратриптан, ризатриптан, суматриптан и золмитриптан.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с блокатором натриевых каналов, примеры которого включают в качестве иллюстрации, а не ограничения, карбамазепин, фосфенитоин, ламотриджин, лидокаин, мексилетин, оксарбазепин, фенитоин и их комбинации.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с растворимым стимулятором или активатором гуанилатциклазы, примеры которого включают без ограничения атацигуат, риоцигуат и их комбинации.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с трициклическим антидепрессантом (TCA), примеры которого включают в качестве иллюстрации, а не ограничения, амитриптилин, амитриптилиноксид, бутриптилин, кломипрамин, демексиптилин, дезипрамин, дибензепин, диметакрин, досулепин, доксепин, имипрамин, имипраминоксид, лофепрамин, мелитрацен, метапрамин, нитроксазепин, нортриптилин, ноксиптилин, пипофезин, пропизепин, протриптилин, хинупрамин и их комбинации.
В одном варианте осуществления соединения по изобретению вводятся в комбинации с антагонистом вазопрессиновых рецепторов, примеры которого включают в качестве иллюстрации, а не ограничения, кониваптан и толваптан.
Комбинированные вторичные терапевтические средства могут также содействовать при дополнительном комбинированном лечении соединениями по изобретению. Например, соединения по изобретению могут комбинироваться с диуретиком и ARB, или блокатором кальциевых каналов и ARB, или диуретиком и ингибитором ACE, или блокатором кальциевых каналов и статином. Конкретные примеры включают комбинацию ингибитора ACE эналаприла (в форме малеата) и диуретика гидрохлортиазида, которая продается под маркой Vaseretic®, или комбинацию блокатора кальциевых каналов амлодипина (в форме безилата) и ARB олмесартана (в форме пролекарства медоксомила), или комбинацию блокатора кальциевых каналов и статина, все могут также применяться с соединениями по изобретению. Другие терапевтические средства, такие как агонисты α2-адренергических рецепторов и антагонисты вазопрессиновых рецепторов, могут также содействовать при комбинированной терапии. Иллюстративные агонисты α2-адренергических рецепторов включают клонидин, дексмедетомидин и гуанфацин.
Следующие составы иллюстрируют репрезентативные фармацевтические композиции по изобретению.
Иллюстративные твердые желатиновые капсулы для перорального введения
Соединение по изобретению (50 г), 440 г высушенной распылением лактозы и 10 г стеарата магния тщательно перемешивают. Затем полученную композицию загружают в твердые желатиновые капсулы (500 мг композиции на капсулу). Альтернативно, соединение по изобретению (20 мг) тщательно перемешивают с крахмалом (89 мг), микрокристаллической целлюлозой (89 мг) и стеаратом магния (2 мг). Затем смесь пропускают через сито № 45 меш по стандарту США и загружают в твердую желатиновую капсулу (200 мг композиции на капсулу).
Альтернативно, соединение по изобретению (30 г), вторичное средство (20 г), 440 г высушенной распылением лактозы и 10 г стеарата магния тщательно перемешивают и обрабатывают, как описано выше.
Иллюстративный состав в виде желатиновой капсулы для перорального введения
Соединение по изобретению (100 мг) тщательно перемешивают с моноолеатом полиоксиэтиленсорбитана (50 мг) и крахмальным порошком (250 мг). Затем смесь загружают в желатиновую капсулу (400 мг композиции на капсулу). Альтернативно, соединение по изобретению (70 мг) и вторичное средство (30 мг) тщательно смешивают с моноолеатом полиоксиэтиленсорбитана (50 мг) и крахмальным порошком (250 мг), и полученную смесь загружают в желатиновую капсулу (400 мг композиции на капсулу).
Альтернативно, соединение по изобретению (40 мг) тщательно перемешивают с микрокристаллической целлюлозой (Avicel PH 103; 259,2 мг) и стеаратом магния (0,8 мг). Затем смесь загружают в желатиновую капсулу (размер #1, белая, непрозрачная) (300 мг композиции на капсулу).
Иллюстративный состав в виде таблетки для перорального введения
Соединение по изобретению (10 мг), крахмал (45 мг) и микрокристаллическую целлюлозу (35 мг) пропускают через сито № 20 меш по стандарту США и тщательно смешивают. Полученные таким образом гранулы сушат при 50-60°C и пропускают через сито № 16 меш по стандарту США. Раствор поливинилпирролидона (4 мг в виде 10% раствора в стерильной воде) смешивают с карбоксиметилкрахмалом натрия (4,5 мг), стеаратом магния (0,5 мг) и тальком (1 мг), и затем эту смесь пропускают через сито № 16 меш по стандарту США. Затем к гранулам добавляют карбоксиметилкрахмал натрия, стеарат магния и тальк. После смешивания смесь прессуют на таблетировочной машине для получения таблетки массой 100 мг.
Альтернативно, соединение по изобретению (250 мг) тщательно смешивают с микрокристаллической целлюлозой (400 мг), высокодисперсным диоксидом кремния (10 мг) и стеариновой кислотой (5 мг). Затем смесь прессуют с образованием таблеток (665 мг композиции на таблетку).
Альтернативно, соединение по изобретению (400 мг) тщательно перемешивают с кукурузным крахмалом (50 мг), кроскармеллозой натрия (25 мг), лактозой (120 мг) и стеаратом магния (5 мг). Затем смесь прессуют с образованием таблетки с одной насечкой (600 мг композиции на таблетку).
Альтернативно, соединение по изобретению (100 мг) тщательно перемешивают с кукурузным крахмалом (100 мг), с водным раствором желатина (20 мг). Смесь сушат и измельчают в мелкий порошок. Затем микрокристаллическую целлюлозу (50 мг) и стеарат магния (5 мг) смешивают с желатиновым составом, гранулируют, и полученную смесь прессуют с образованием таблеток (по 100 мг соединения по изобретению на таблетку).
Иллюстративный суспензионный состав для перорального введения
Следующие ингредиенты смешивают с образованием суспензии, содержащей 100 мг соединения по изобретению на 10 мл суспензии:
Иллюстративный жидкий состав для перорального введения
Пригодный жидкий состав представляет собой такой, который содержит буфер на основе карбоновой кислоты, такой как цитратный, лактатный и малеатный буферные растворы. Например, соединение по изобретению (которое может быть предварительно смешано с DMSO) перемешивают с 100 мМ буфера цитрата аммония, и pH доводят до pH 5, или перемешивают с 100 мМ раствора лимонной кислоты, и pH доводят до pH 2. Такие растворы могут также включать солюбилизирующий эксципиент, такой как циклодекстрин, например, раствор может включать 10 масс.% гидроксипропил-β-циклодекстрина.
Другие пригодные составы включают 5% раствор NaHCO3 с циклодекстрином или без него.
Иллюстративный инъецируемый состав для введения инъекцией
Соединение по изобретению (0,2 г) перемешивают с 0,4M буферным раствором ацетата натрия (2,0 мл). pH полученного раствора доводят до pH 4 с использованием 0,5N водного раствора хлористоводородной кислоты или 0,5N водным гидроксидом натрия, по необходимости, и затем добавляют достаточное количество воды для инъекций для обеспечения общего объема 20 мл. Затем смесь фильтруют через стерильный фильтр (0,22 мкм) для получения стерильного раствора, пригодного для введения инъекцией.
Иллюстративные композиции для введения ингаляцией
Соединение по изобретению (0,2 мг) микронизируют и затем смешивают с лактозой (25 мг). Эту смесь затем загружают в желатиновую ингаляционную кассету. Содержимое кассеты вводят, используя, например, ингалятор сухого порошка.
Альтернативно, микронизированное соединение по изобретению (10 г) диспергируют в растворе, полученном растворением лецитина (0,2 г) в деминерализованной воде (200 мл). Полученную суспензию подвергают распылительной сушке и затем микронизируют с образованием микронизированной композиции, содержащей частицы, имеющие средний диаметр менее чем примерно 1,5 мкм. Затем микронизированную композицию загружают в кассеты ингалятора отмеренных доз, содержащие находящийся под избыточным давлением 1,1,1,2-тетрафторэтан, в количестве, достаточном для обеспечения от примерно 10 мкг до примерно 500 мкг соединения по изобретению на дозу при введении ингалятором.
Альтернативно, соединение по изобретению (25 мг) растворяют в забуференном цитратом (pH 5) изотоническом солевом растворе (125 мл). Смесь перемешивают и обрабатывают ультразвуком до тех пор, пока не растворится соединение. pH раствора проверяют и при необходимости доводят до pH 5 медленным добавлением водного 1N NaOH. Раствор вводят, используя распылительное устройство, которое обеспечивает подачу от примерно 10 мкг до примерно 500 мкг соединения по изобретению на дозу.
ПРИМЕРЫ
Следующие «Способы получения» и «Примеры» представлены для иллюстрации определенных вариантов осуществления изобретения. Однако, если конкретно не указано иное, эти определенные варианты осуществления ни в коей мере не предназначены для ограничения объема изобретения.
Если не указано иное, представленные ниже аббревиатуры имеют следующие значения, и любые другие аббревиатуры, используемые здесь и не определенные, имеют свое стандартное, обычно принятое значение:
Если не указано иное, все материалы, такие как реагенты, исходные материалы и растворители закупали у коммерческих поставщиков (таких как Sigma-Aldrich, Fluka Riedel-de Haen и т.п.) и использовали без дополнительной очистки.
Если не указано иное, реакции проводили в атмосфере азота. Ход реакций контролировали тонкослойной хроматографией (TLC), аналитической высокоэффективной жидкостной хроматографией (анал. ВЭЖХ) и масс-спектрометрией, подробности которых представлены в определенных примерах. Растворители, используемые в аналитической ВЭЖХ, были следующие: растворитель A представлял собой 98% H2O/2% MeCN/1,0 мл/л TFA; растворитель B представлял собой 90% MeCN/10% H2O/1,0 мл/л TFA.
Реакции проводили, как конкретно описано в каждом случае получения, например; обычно реакционные смеси очищали экстракцией и другими способами очистки, такими как зависимая от температуры и растворителя кристаллизация и осаждение. Кроме того, реакционные смеси обычным образом очищали препаративной ВЭЖХ, обычно используя наполнители колонки Microsorb CI8 и Microsorb BDS и обычные элюенты. Ход реакций обычно измеряли жидкостной хроматографией/масс-спектрометрией (LCMS). Характеристику изомеров осуществляли спектроскопией ядерного эффекта Оверхаузера (NOE). Характеристику продуктов реакции обычным образом осуществляли масс- и1H-ЯМР спектрометрией. Для измерения ЯМР образцы растворяли в дейтерированном растворителе (CD3OD, CDCl3 или DMSO-d6), и спектры1H-ЯМР получали прибором Varian Gemini 2000 (400 МГц) в стандартных условиях наблюдения. Масс-спектрометрическую идентификацию соединений обычно проводили, используя способ электрораспылительной ионизации (ESMS) прибором Applied Biosystems (Foster City, CA) модель API 150 EX или Agilent (Palo Alto, CA) модель 1200 LC/MSD.
Получение 1
Сложный этиловый эфир ацетокси(диэтоксифосфорил)уксусной кислоты

Этил-2-оксоацетат (50%) (74 г, 724,8 ммоль) добавляли по каплям с перемешиванием при 0°C к раствору диэтилгидрофосфата (50 г, 362,1 ммоль) в толуоле (100 мл), в атмосфере азота. Et3N (110 г, 1,1 моль) добавляли по каплям с перемешиванием при 0°C. Полученный раствор перемешивали в течение 1 часа при комнатной температуре. К смеси по каплям добавляли уксусный ангидрид (37 г, 362,4 ммоль) с перемешиванием при 0°C. Полученный раствор перемешивали в течение ночи при комнатной температуре. Величину pH раствора доводили до 6 2N HCl. Полученный раствор экстрагировали DCM (3×150 мл), и органические слои объединяли, сушили над Na2SO4, и концентрировали в вакууме. Остаток загружали на колонку с силикагелем с EtOAc : гексанами (1:2-1:5) с получением указанного в заголовке соединения (52 г) в виде светло-желтой жидкости.
Получение 2
Сложный этиловый эфир (R)-4-амино-5-бифенил-4-ил-2-гидроксипентаноевой кислоты
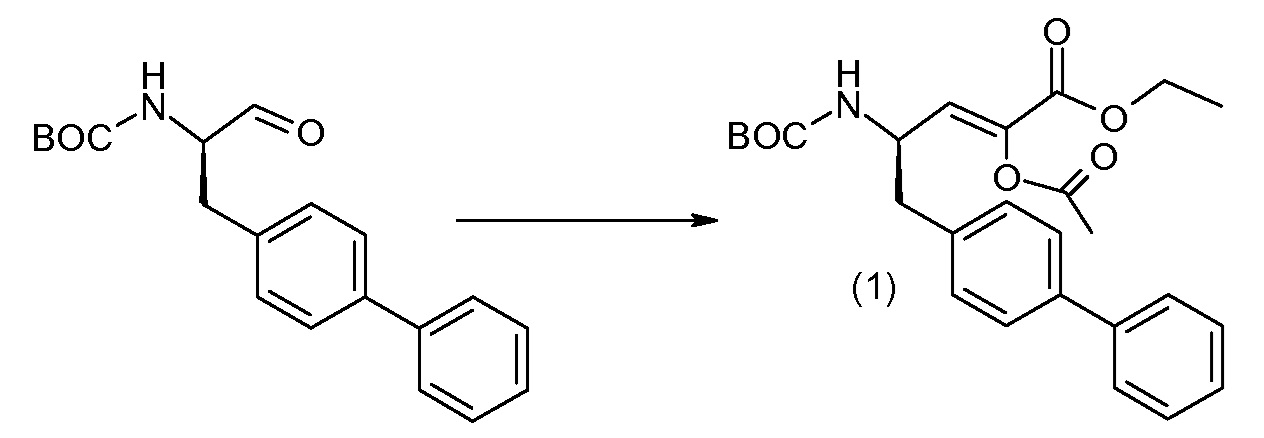
Раствор сложного этилового эфира ацетокси(диэтоксифосфорил)уксусной кислоты (15,6 г, 55,3 ммоль, 1,2 экв.) в THF (сухой) (150 мл), в атмосфере азота, охлаждали до -78°C. LiHMDS (1M в THF) (55,3 мл) добавляли по каплям с перемешиванием при -78°C. После перемешивания в течение 30 минут при этой температуре по каплям в течение 15 минут добавляли раствор неочищенного сложного трет-бутилового эфира ((R)-2-бифенил-4-ил-1-формилэтил)карбаминовой кислоты (15,0 г, 1,0 экв.) в THF (сухой) (30 мл). Перемешивание продолжали в течение 1,5 часов при -78°C перед тем, как смесь выливали в холодный раствор с водой (200 мл) и EtOAc (200 мл). Органический слой повторно отделяли, и водный слой повторно экстрагировали EtOAc (2×100 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и выпаривали, и остаток очищали флэш-хроматографией (EtOAc/гексаны=0~1:10) с получением соединения 1 (10,5 г) в виде белого твердого вещества.

Перемешанный раствор соединения 1 (10,5 г, 23,2 ммоль) в EtOH (безводный) (100 мл) объединяли с палладированным углем (1,0 г), в атмосфере азота. Смесь продували четыре раза водородом, и затем водород барботировали в течение 2 часов при комнатной температуре. Палладированный уголь отфильтровывали, и фильтрат концентрировали в вакууме с получением неочищенного соединения 2 (10,0 г) в виде бледно-желтого масла, которое использовали без дополнительной очистки.
Соединение 2 (10,0 г, 22,0 ммоль) в EtOH (безводный) (100 мл) объединяли с карбонатом калия (6,1 г, 44,1 ммоль), и полученный раствор перемешивали в течение 2 часов при комнатной температуре. Твердые вещества отфильтровывали, и фильтрат концентрировали в вакууме. Остаток загружали на колонку с силикагелем (EtOAc/гексаны=0-1:5) с получением соединения 3 (6,0 г) в виде белого твердого вещества.

Соединение 3 (6,0 г, 14,5 ммоль) растворяли в DCM (сухой) (120 мл), и HCl барботировали в смесь в течение 5~6 часов при комнатной температуре. Наблюдали выпадение твердого осадка. Смесь концентрировали до половины объема, затем фильтровали. Твердые вещества собирали и промывали холодным EtOAc и сушили при пониженном давлении с получением указанного в заголовке соединения (4,2 г) в виде не совсем белой твердой соли HCl. LC-MS (ES, m/z): 314 [M-HCl+H]+.
1H ЯМР (300 MГц, DMSO): δ (м.д.) = 8,07 (с, 1,9H), 7,96 (с, 1,2H), 7,65-7,69 (м, 4,0H), 7,45-7,50 (м, 2,0H), 7,33-7,39 (м, 3,0H), 6,05-6,07 (м, 0,63H), 5,88-5,90 (м, 0,88H), 4,32-4,38 (м, 0,80H), 4,18-4,31 (м, 0,51H), 4,05-4,11 (м, 2H), 3,50 (с, 1H), 2,75-3,05 (м, 2,8H), 1,83-1,94 (м, 1H), 1,71-1,82 (м, 1H), 1,10-1,20 (м, 3,3H).
Получение 3
Сложный трет-бутиловый эфир (S)-2-(4-бромбензил)-5-оксопирролидин-1-карбоновой кислоты
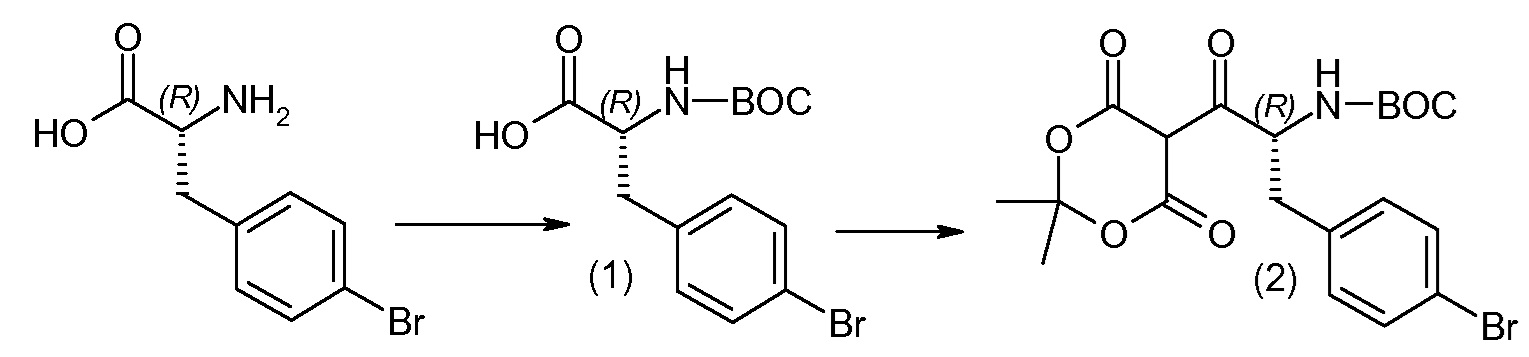
К раствору (R)-2-амино-3-(4-бромфенил)пропионовой кислоты (50 г, 0,2 моль) в MeCN (700 мл) добавляли раствор NaOH (16,4 г, 0,4 моль) в воде (700 мл) при -5°C. После перемешивания в течение 10 минут, добавляли раствор (BOC)2O (44,7 г, 0,2 моль) в MeCN (100 мл). Смесь нагревали до комнатной температуры и перемешивали в течение ночи. После выпаривания MeCN остаток разбавляли DCM (800 мл) и 1 M HCl до pH 2 при -5°C. Водный слой экстрагировали DCM (3×200 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (500 мл), сушили над Na2SO4 и концентрировали с получением соединения 1 (66,5 г) в виде белого твердого вещества. LC-MS: 366 (M+Na), 709 (2M+Na).
К раствору соединения 1 (66,5 г, 193 мкмоль), кислоты Мельдрума (33,4 г, 232 ммоль) и DMAP (37,7 г, 309 ммоль) в безводном DCM (600 мл) по каплям добавляли раствор DCC (47,9 г, 232 ммоль) в безводном DCM (200 мл) в течение 1 часа при -5°C в атмосфере азота. Смесь перемешивали при -5°C в течение 8 часов, затем выдерживали в холодильнике в течение ночи. Наблюдали выпадение кристаллов дициклогексилмочевины. Смесь фильтровали, промывали 5% KHSO4 (5×200 мл) и насыщенным водным раствором NaCl (200 мл), затем сушили над безводным MgSO4 в условиях отрицательной температуры в течение ночи. Раствор затем выпаривали с получением неочищенного соединения 2 (91 г) в виде светло-желтого твердого вещества. LC-MS: 492(M+Na), 961(2M+Na).

К раствору неочищенного соединения 2 (91 г, 193 ммоль) в безводном DCM (1 л) добавляли AcOH (127,5 г, 2,1 моль) при -5°C в атмосфере азота. Смесь перемешивали при -5°C в течение 30 минут, затем небольшими порциями добавляли NaBH4 (18,3 г, 483 ммоль) в течение 1 часа. После перемешивания в течение еще 1 часа при -5°C добавляли насыщенный водный раствор NaCl (500 мл). Органический слой промывали насыщенным водным раствором NaCl (2×300 мл) и водой (2×300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали промыванием Et2O с получением соединения 3 (68 г) в виде светло-желтого твердого вещества. LC-MS: 478 (M+Na), 933 (2M+Na).
Раствор соединения 3 (68 г, 149 ммоль) в безводном толуоле (500 мл) кипятили с обратным холодильником в атмосфере азота в течение 3 часов. После выпаривания растворителя остаток очищали хроматографией (гексаны:EtOAc=10:1) с получением указанного в заголовке соединения (38 г) в виде светло-желтого масла. LC-MS: 376 (M+Na), 729 (2M+Na).
Получение 4
Сложный этиловый эфир (2R,4R)-4-амино-5-(4-бромфенил)-2-гидроксипентановой кислоты

К раствору сложного трет-бутилового эфира (S)-2-(4-бромбензил)-5-оксопирролидин-1-карбоновой кислоты (38 г, 107 ммоль) в безводном DCM (250 мл) добавляли TFA (20 мл, 0,27 моль) при -5°C в атмосфере азота. Смесь нагревали до комнатной температуры и перемешивали в течение ночи. После выпаривания растворителя остаток разбавляли EtOAc (300 мл) и промывали насыщенным водным раствором NaHCO3 (3×200 мл), водой (200 мл), насыщенным водным раствором NaCl (250 мл), сушили над Na2SO4 и концентрировали с получением неочищенного соединения 1 (24 г) в виде светло-желтого твердого вещества. LC-MS: 254 [M+H].
К раствору NaH (8,6 г, 250 ммоль) в безводном THF (200 мл) по каплям добавляли раствор соединения 1 (24 г, 94 ммоль) в безводном THF (200 мл) в течение 30 минут при 0°C в атмосфере азота. Смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. После охлаждения до 0°C, пивалоилхлорид (18 г, 150 ммоль) добавляли по каплям в течение 30 минут. Смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакцию гасили насыщенным водным раствором NH4Cl (300 мл) и экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=25:1) с получением соединения 2 (18 г) в виде светло-желтого твердого вещества. LC-MS: 360 (M+Na).
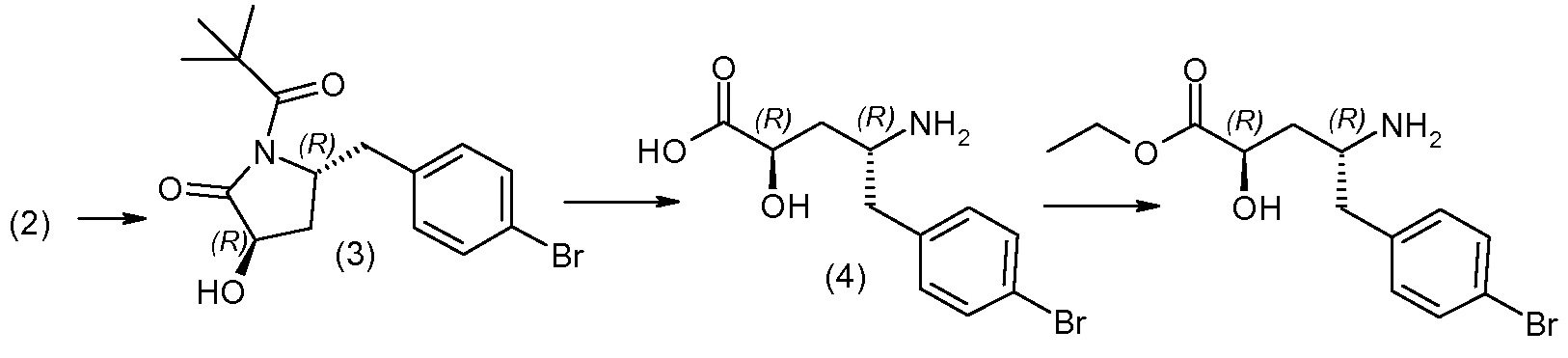
К раствору соединения 2 (18 г, 53 ммоль) в безводном THF (250 мл) добавляли по каплям NaHMDS (47,7 мл, 96 ммоль) в течение 30 минут при -78°C в атмосфере азота. После перемешивания при -78°C в течение 90 минут, раствор (+)-(8,8-дихлоркамфорилсульфонил)оксазиридина (31,6 г, 106 ммоль) добавляли по каплям в течение 30 минут. После перемешивания при -78°C в течение 2 часов, реакцию гасили насыщенным водным раствором NH4Cl (400 мл) и экстрагировали EtOAc (3×300 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=15:1) с получением соединения 3 (8,9 г) в виде светло-желтого твердого вещества. LC-MS: 376 (M+Na).
Раствор соединения 3 (8,9 г, 25 ммоль) в концентрированной HCl (81 мл, 81 ммоль) нагревали при 100°C в течение 16 часов. Смесь затем концентрировали с получением неочищенного продукта, который дополнительно очищали промыванием Et2O с получением соединения 4 (7 г) в виде светло-желтой соли HCl. LC-MS: 323 (M+ H).
Раствор соединения 4 (7 г, 22 ммоль) в EtOH (10 мл) объединяли с 8M HCl в EtOH (120 мл, 960 ммоль) при комнатной температуре. Смесь нагревали при 50°C в течение 16 часов, затем концентрировали. Неочищенный продукт дополнительно очищали промыванием Et2O с получением указанного в заголовке соединения (6 г) в виде светло-желтой твердой соли HCl. LC-MS: 352 (M+H).
Получение 5
(3R,5R)-5-(3'-Хлорбифенил-4-илметил)-1-(2,2-диметилпропионил)-3-гидроксипирролидин-2-он

К раствору сложного трет-бутилового эфира (S)-2-(4-бромбензил)-5-оксопирролидин-1-карбоновой кислоты (15 г, 43 ммоль) в 1,4-диоксане (600 мл) добавляли 3-хлорфенилбороновую кислоту (8 г, 51 ммоль) и Pd(dppf)2Cl2 (3,1 г, 4,2 ммоль) при комнатной температуре в атмосфере азота. После перемешивания в течение 10 минут добавляли раствор K2CO3 (11,7 г, 85 ммоль) в воде (60 мл). Смесь нагревали до 60°C и перемешивали в течение ночи. После выпаривания растворителя добавляли воду (200 мл) и экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (400 мл), сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который дополнительно очищали колоночной хроматографией (гексаны:EtOAc=6:1) с получением соединения 1 (15 г) в виде светло-желтого твердого вещества. LC-MS: 408 (M+Na).
К раствору соединения 1 (15 г, 0,039 моль) в безводном DCM (250 мл) добавляли TFA (20 мл, 270 ммоль) при -5°C в атмосфере азота. Смесь нагревали до комнатной температуры и перемешивали в течение ночи. После выпаривания растворителя остаток разбавляли EtOAc (300 мл), затем промывали насыщенным водным раствором NaHCO3 (3×200 мл), водой (200 мл) и насыщенным водным раствором NaCl (250 мл), затем сушили над Na2SO4 и концентрировали с получением неочищенного соединения 2 (11 г) в виде светло-желтого твердого вещества. LC-MS: 286 [M+H].

К раствору NaH (2,3 г, 98 ммоль) в безводном THF (200 мл) по каплям добавляли раствор соединения 2 (11 г, 39 ммоль) в безводном THF (100 мл) в течение 30 минут при 0°C в атмосфере азота. Смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. После охлаждения до 0°C пивалоилхлорид (6 г, 51 ммоль) добавляли по каплям в течение 30 минут. Смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакцию гасили насыщенным водным раствором NH4Cl (200 мл) и экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=25:1) с получением соединения 3 (10,5 г) в виде светло-желтого твердого вещества. LC-MS: 391 (M+Na).
К раствору соединения 3 (10,5 г, 29 ммоль) в безводном THF (120 мл) добавляли по каплям NaHMDS (29 мл, 58 ммоль) в течение 30 минут при -78°C в атмосфере азота. После перемешивания при -78°C в течение 90 минут, раствор (+)-(8,8-дихлоркамфорилсульфонил)оксазиридина (15,6 г, 52 ммоль) добавляли по каплям в течение 30 минут. После перемешивания при -78°C в течение 2 часов реакцию гасили насыщенным NH4Cl (400 мл) и экстрагировали EtOAc (3×300 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=15:1) с получением указанного в заголовке соединения (9,6 г) в виде светло-желтого твердого вещества. LC-MS: 408 (M+Na).
Получение 6
Сложный этиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты

Раствор (3R,5R)-5-(3'-хлорбифенил-4-илметил)-1-(2,2-диметилпропионил)-3-гидроксипирролидин-2-она (9,6 г, 25 ммоль) в концентрированной HCl (81 мл, 81 ммоль) нагревали при 100°C в течение 16 часов. Смесь затем концентрировали с получением неочищенного продукта, который дополнительно очищали промыванием Et2O с получением соединения 1 (5,7 г) в виде светло-желтой твердой соли HCl. LC-MS: 320 (M+H).
К раствору соединения 1 (5,7 г, 18 ммоль) в EtOH (10 мл) добавляли 8M HCl в EtOH (120 мл, 960 ммоль) при комнатной температуре. Смесь нагревали при 50°C в течение 16 часов. После концентрирования неочищенный продукт дополнительно очищали промыванием Et2O с получением указанного в заголовке соединения (2,1 г) в виде светло-желтой твердой соли HCl. LC-MS: 348 (M+H).
Получение 7
(2R,4R)-4-Амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановая кислота

1M водную HCl (2,0 ммоль) добавляли к сложному этиловому эфиру (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (150,0 мг, 431 мкмоль), и смесь перемешивали при 100°C в течение 2 часов. Смесь концентрировали в вакууме в течение 3 часов, и остаток очищали ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения (117 мг) в виде белого твердого вещества.
Получение 8
Сложный изопропиловый эфир хлороксоуксусной кислоты

Изопропанол (158 мкл, 2,1 ммоль, 1,0 экв.) добавляли по каплям в течение 5 минут к оксалилхлориду (350 мкл, 4,14 мкмоль, 2,0 экв.) при 0°C, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Избыток оксалилхлорида удаляли роторным выпариванием (40°C, 50 мм рт. ст.) и использовали без дополнительной очистки.
Получение 9
Сложный изобутиловый эфир хлороксоуксусной кислоты
Изобутанол (191 мкл, 2,1 ммоль, 1,0 экв.) добавляли по каплям в течение 5 минут к оксалилхлориду (350 мкл, 4,14 мкмоль, 2,0 экв.) при 0°C, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Избыток оксалилхлорида удаляли роторным выпариванием (40°C, 40 мм рт. ст.) и использовали без дополнительной очистки.
Получение 10
трет-Бутилоксалилхлорид

Оксалилхлорид (274 мкл, 3,2 ммоль) добавляли к раствору трет-бутилового спирта (289 мкл, 3,0 ммоль) в эфире (2,0 мл, 19,0 ммоль), и смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали в вакууме с получением прозрачной бесцветной жидкости. Приблизительно 1M раствор трет-бутилоксалилхлорида получали растворением полученной прозрачной бесцветной жидкости в DCM (~3,0 мл).
Получение 11
Сложный 2-метоксиэтиловый эфир хлороксоуксусной кислоты

Раствор 2-метоксиэтанола (295 мг, 3,9 ммоль) в DCM (общий объем: 0,5 мл) добавляли к раствору оксалилхлорида (0,5 мл, 5,8 ммоль) в DCM (общий объем 1,0 мл) при 0°C, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Смесь концентрировали в вакууме, и полученный остаток растворяли в DCM (3,9 мл) с получением 1,0M раствора в DCM.
Получение 12
Сложный 3-этоксипропиловый эфир хлороксоуксусной кислоты
Раствор 3-этоксипропан-1-ола (404 мг, 3,9 ммоль) в DCM (общий объем: 0,5 мл) добавляли к раствору оксалилхлорида (0,5 мл, 5,8 ммоль) в DCM (общий объем 1,0 мл) при 0°C, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Смесь концентрировали в вакууме, и полученный остаток растворяли в DCM (3,9 мл) с получением 1,0M раствора в DCM.
Получение 13
Сложный 2-феноксиэтиловый эфир хлороксоуксусной кислоты
Раствор 2-феноксиэтанола (536 мг, 3,9 ммоль) в DCM (общий объем: 0,5 мл) добавляли к раствору оксалилхлорида (0,5 мл, 5,8 ммоль) в DCM (общий объем 1,0 мл) при 0°C, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Смесь концентрировали в вакууме, и полученный остаток растворяли в DCM (3,9 мл) с получением 1,0M раствора в DCM.
Получение 14
(2R,4R)-4-Бутоксикарбониламино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановая кислота

Раствор (3R,5R)-5-(3'-хлорбифенил-4-илметил)-1-(2,2-диметилпропионил)-3-гидроксипирролидин-2-она (4,5 г, 11,7 ммоль) в концентрированной HCl (30 мл) перемешивали при 100°C в течение 16 часов. Смесь концентрировали в вакууме с получением соединения 1 (4 г) в виде белой твердой соли HCl. LC-MS: 321 [M+H]+.
К раствору NaOH (1,8 г, 45,2 ммоль) в воде (100 мл) по каплям добавляли соединение 1 (4 г, 11,3 ммоль) в MeCN (100 мл). Смесь перемешивали в течение 10 минут при 0°C. Добавляли ди-трет-бутилдикарбонат (7,17 г, 33,8 ммоль), и смесь перемешивали в течение 15 часов при комнатной температуре. Полученную смесь концентрировали в вакууме для удаления MeCN, затем разбавляли DCM (300 мл), и pH доводили до pH=5-6 с помощью 1N водной HCl. Затем органический слой собирали, и остаток экстрагировали DCM (3×300 мл). Объединенные органические слои коцентрировали и промывали гексанами (150 мл) с получением указанного в заголовке соединения (4 г) в виде белого твердого вещества. LC-MS: 442 [M+Na]+.
Получение 15
Сложный 2,2,3,3,3-пентафторпропиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентанoевой кислоты

К раствору (2R,4R)-4-трет-бутоксикарбониламино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (0,9 г, 6 ммоль) и 2,2,3,3,3-пентафторпропан-1-ола (450 мг, 3 ммоль) в DCM (30 мл) добавляли DCC (880 мг, 4,3 ммоль) и DMAP (260 мг, 2,1 ммоль). Полученную смесь перемешивали в течение 15 часов при комнатной температуре, затем концентрировали в вакууме. Остаток растворяли в EtOAc (100 мл) и промывали водой (30 мл) и насыщенным водным раствором NaCl (30 мл). Органический слой собирали и концентрировали и очищали колоночной хроматографией (гексаны/EtOAc=5:1) с получением соединения 3 (0,4 г) в виде белого твердого вещества. LC-MS: 574 [M+Na]+.

Раствор соединения 3 (0,4 г, 690 мкмоль) в 1,4M HCl в растворе 1,4-диоксана (15 мл) перемешивали в течение ночи, и затем концентрировали в вакууме. Остаток диспергировали в EtOAc (10 мл), и осадок собирали фильтрованием с получением указанного в заголовке соединения в виде не совсем белой твердой соли HCl (165 мг). LC-MS: 452 [M+H]+.
1H ЯМР: (DMSO-d6) 1,95-1,82 (м, 2H), 2,99-2,98 (м, 2H), 3,56 (ушир., 1H), 4,41-4,38 (м, 1H), 4,92-4,82 (м, 2H), 6,35 (с, 1H), 7,71-7,38 (м, 8H), 8,09 (с, 3H).
Получение 16
Сложный 5-метил-2-оксо[1,3]диоксол-4-илметиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты
Суспензию (2R,4R)-4-трет-бутоксикарбониламино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (740 мг, 1,8 ммоль), 4-(бромметил)-5-метил-1,3-диоксол-2-она (340 мг, 1,8 ммоль), йодида калия (58 мг, 350 мкмоль) и K2CO3 (486 мг, 3,5 ммоль) в DMF (20 мл) перемешивали в течение 4 часов при комнатной температуре. Смесь разбавляли EtOAc (150 мл) и промывали водой (30 мл). Органический слой собирали и концентрировали и очищали колоночной хроматографией (гексаны/EtOAc=1:1) с получением белого твердого вещества (490 мг). LC-MS: 554 [M+23]+. Раствор этого твердого вещества (476 мг, 890 мкмоль) в 3N HCl в 1,4-диоксане (20 мл) перемешивали в течение ночи и затем концентрировали в вакууме. Остаток диспергировали в EtOAc (10 мл), и осадок собирали фильтрованием с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (290 мг). LC-MS: 432 [M+H]+.
1H ЯМР: (DMSO-d6) 1,92-1,82 (м, 2H), 2,16 (с, 3H), 2,99 (ушир., 2H), 3,56 (ушир., 1H), 4,35-4,32 (м, 1H), 5,017 (с, 2H), 6,17 (с, 1H), 7,39-7,36 (м, 4H), 7,71-7,68 (м, 4H), 8,05 (с, 3H).
Получение 17
Сложный бутирилоксиметиловый эфир (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил-2-гидроксипентановой кислоты
Раствор (2R,4R)-4-трет-бутоксикарбониламино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (900 мг, 2,1 ммоль), хлорметилбутирата (350 мг, 2,6 ммоль), йодида натрия (481 мг, 3,21 ммоль) и DIPEA (828 мг, 6,42 ммоль) в DMF (20 мл) перемешивали в течение 16 часов при 30°C. Смесь разбавляли EtOAc (150 мл) и промывали водой (50 мл) и насыщенным водным раствором NaCl (50 мл). Органический слой собирали и концентрировали и очищали колоночной хроматографией (гексаны/EtOAc=5:1) с получением белого твердого вещества (240 мг). LC-MS: 542 [M+Na]+. Раствор этого твердого вещества (240 мг, 460 мкмоль) в 1,4M HCl в 1,4-диоксане (15 мл) перемешивали в течение ночи, и затем концентрировали в вакууме. Остаток диспергировали в EtOAc (10 мл), и осадок собирали фильтрованием с получением указанного в заголовке соединения в виде не совсем белой твердой соли HCl (140 мг). LC-MS: 420 [M+H]+.
1H ЯМР: (DMSO) 0,85 (т, J=7,5 Гц, 3H), 1,61-1,52 (м, 2H), 1,89-1,86 (м, 2H), 2,30 (т, J=7,5 Гц, 2H), 2,98 (ушир., 2H), 3,56 (ушир., 1H), 4,33-4,30 (м, 1H), 5,74-5,68 (м, 2H), 6,21 (с, 1H), 7,37-7,35 (м, 4H), 7,70-7,767 (м, 4H), 8,01 (ушир.с, 3H).
Получение 18
Сложный этиловый эфир (2R,4R)-4-амино-5-(2',5'-дихлорбифенил-4-ил)-2-гидроксипентановой кислоты

К раствору сложного трет-бутилового эфира (S)-2-(4-бромбензил)-5-оксопирролидин-1-карбоновой кислоты (33,5 г, 95 ммоль) в 1,4-диоксане (1,2 л) добавляли 2,5-дихлорфенилбороновую кислоту (21,7 г, 114 ммоль) и Pd(dppf)2Cl2 (3,5 г, 4,7 ммоль) при комнатной температуре в атмосфере азота. После перемешивания в течение 10 минут добавляли раствор K2CO3 (26,1 г, 189 ммоль) в воде (120 мл). Смесь нагревали до 60°C и перемешивали в течение ночи. После выпаривания растворителя добавляли воду (400 мл) и экстрагировали EtOAc (3×400 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (500 мл), сушили над безводным Na2SO4 и концентрировали с получением неочищенного продукта, который дополнительно очищали колоночной хроматографией (гексаны:EtOAc=6:1) с получением соединения 1 (35,8 г) в виде светло-желтого твердого вещества. LC-MS: 442 [M+Na].
К раствору соединения 1 (35,8 г, 85 ммоль) в безводном DCM (300 мл) добавляли TFA (30 мл, 405 ммоль) при -5°C в атмосфере азота. Смесь нагревали до комнатной температуры и перемешивали в течение ночи. После выпаривания растворителя остаток разбавляли EtOAc (500 мл), затем промывали насыщенным водным раствором NaHCO3 (3×300 мл), водой (200 мл) и насыщенным водным раствором NaCl (250 мл), затем сушили над Na2SO4 и концентрировали с получением неочищенного соединения 2 (26 г) в виде светло-желтого твердого вещества. LC-MS: 320 [M+H].

К раствору соединения 2 (26 г, 81 ммоль) в безводном THF (500 мл) добавляли по каплям n-BuLi в гексане (39 мл, 97 ммоль) в течение 1 часа при -78°C в атмосфере азота. После перемешивания при -78°C в течение 2 часов, реакцию гасили добавлением пивалоилхлорида (12,7 г, 105 ммоль) по каплям в течение 30 минут. После перемешивания при -78°C в течение 2 часов реакцию гасили насыщенным водным раствором NH4Cl (200 мл) и экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (300 мл), сушили над безводным MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=25:1) с получением соединения 3 (33 г) в виде светло-желтого твердого вещества. LC-MS: 426 [M+Na].
К раствору соединения 3 (10 г, 0,025 моль) в безводном THF (120 мл) добавляли по каплям NaHMDS (18,6 мл, 37 ммоль) в течение 30 минут при -78°C в атмосфере азота. После перемешивания при -78°C в течение 2 часов, раствор (+)-(8,8-дихлоркамфорилсульфонил)оксазиридина (11,1 г, 37 ммоль) в THF (80 мл) добавляли по каплям в течение 30 минут. После перемешивания при -78°C в течение 2 часов, реакцию гасили насыщенным водным раствором NH4Cl (500 мл) и экстрагировали EtOAc (3×300 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=15:1) с получением соединения 4 (4,2 г) в виде светло-желтого масла. LC-MS: 442 [M+Na].
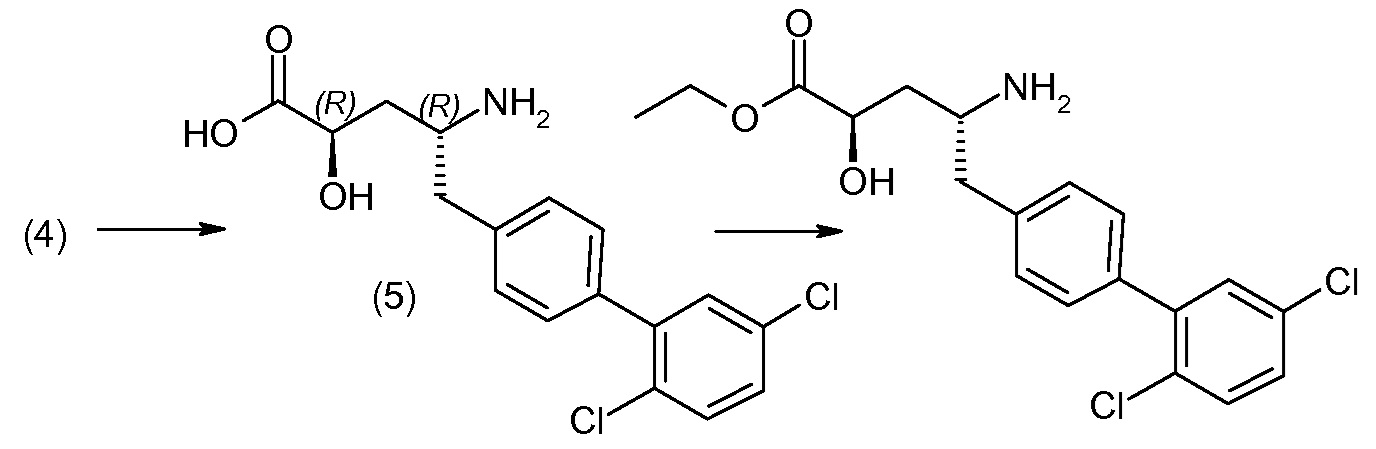
Раствор соединения 4 (4,2 г, 10 ммоль) в концентрированной HCl (80 мл, 0,96 моль) нагревали при 100°C в течение 16 часов. Смесь затем концентрировали с получением неочищенного продукта, который дополнительно очищали промыванием Et2O, с получением соединения 5 (3,8 г) в виде белого твердого вещества. LC-MS: 354 [M+H].
К раствору соединения 5 (3,8 г, 10 ммоль) в EtOH (5 мл) добавляли 4M HCl в EtOH (100 мл, 0,4 моль) при комнатной температуре. Смесь нагревали при 50°C в течение 16 часов. После концентрации неочищенный продукт дополнительно очищали промыванием Et2O с получением указанного в заголовке соединения (3,3 г) в виде белого твердого вещества. LC-MS: 382 [M+ H].
Получение 19
(3R,5R)-5-Амино-6-(4-бром-2-хлорфенил)-2-этоксигекс-1-ен-3-ол

К суспензии 4-бром-2-хлорбензальдегида (50 г, 22,8 ммоль) в MeOH (500 мл) порциями добавляли NaBH4 (17,3 г, 45,6 ммоль) при 0°C. Смесь перемешивали в течение 30 минут и затем добавляли водный NH4Cl для гашения реакции. Смесь концентрировали в вакууме. Остаток экстрагировали EtOAc (2×200 мл), и объединенные органические слои сушили над безводным Na2SO4 и концентрировали в вакууме с получением соединения 1 (48 г) в виде белого твердого вещества.
К раствору соединения 1 (46,8 г, 21,1 ммоль) в сухом DCM (500 мл) по каплям добавляли трибромид фосфора (68,6 г, 25,3 ммоль) при 0°C в атмосфере азота. Смесь перемешивали в течение 2 часов и затем промывали насыщенным водным раствором NaHCO3 (2×200 мл) и насыщенным водным раствором NaCl (200 мл), сушили над безводным Na2SO4, концентрировали в вакууме с получением соединения 2 (36 г) в виде бесцветного масла.

К перемешанному раствору (R)-пирролидин-2-карбоновой кислоты (57,7 г, 0,5 моль) и KOH (84 г, 1,5 моль) в изопропиловом спирте (330 мл) по каплям добавляли бензилхлорид (70 мл, 0,6 моль) при 0°C в течение 3 часов. Смесь затем перемешивали в течение ночи при такой же температуре. Полученную смесь нейтрализовали концентрированной HCl до pH=6 с последующим добавлением хлороформа (200 мл). Смесь перемешивали в течение 30 минут, затем фильтровали, и осадок промывали хлороформом (3×100 мл). Объединенные растворы с хлороформом сушили над безводным Na2SO4 и концентрировали в вакууме с получением соединения 3 (52 г) в виде белого твердого вещества. LC-MS: 206 [M+H]+.
К раствору соединения 3 (10 г, 48,8 ммоль) в сухом DCM (50 мл) добавляли SO2Cl2 (7,3 г, 61 ммоль) при -20°C в атмосфере азота. Смесь перемешивали при -20°C в течение 3 часов с последующим добавлением раствора (2-аминофенил)(фенил)метанона (6 г, 30,5 ммоль) в сухом DCM (25 мл), и смесь перемешивали в течение ночи при комнатной температуре. Добавляли раствор Na2CO3 (10,3 г) в воде (40 мл) при 0°C. Органический слой отделяли, и водный слой экстрагировали DCM (50 мл × 3). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток промывали MTBE (2×50 мл) с получением соединения 4 (8,5 г) в виде желтого твердого вещества. LC-MS: 385 [M+H]+.
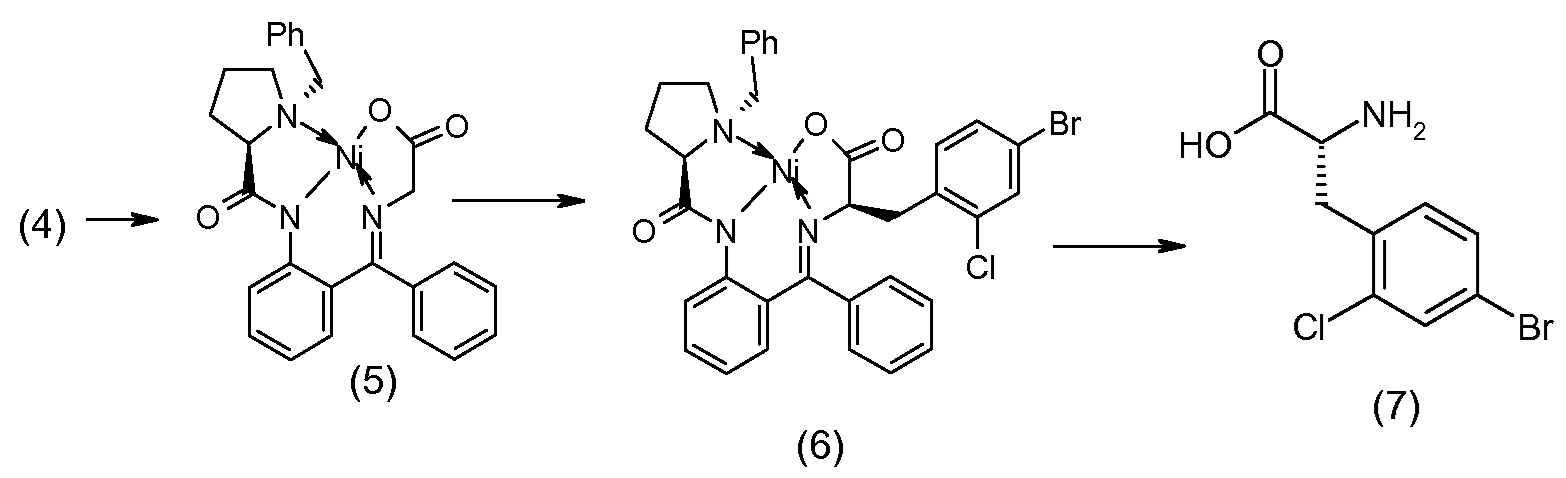
К раствору cоединения 4 (29,4 г, 76,5 ммоль), глицина (28,7 г, 382,4 ммоль) и Ni(NO3)2·6H2O (44,5 г, 152,9 ммоль) в MeOH (280 мл) добавляли раствор KOH (30 г, 535,3 ммоль) в MeOH (100 мл) при 45°C в атмосфере азота. Смесь перемешивали при 60°C в течение часа. Полученный раствор нейтрализовали AcOH (31 мл) и выливали в ледяную воду (380 мл). Полученное твердое вещество фильтровали и растворяли в DCM (450 мл), которое промывали насыщенным водным раствором NaCl (150 мл), сушили над безводным Na2SO4 и концентрировали. Остаток промывали EtOAc (2×50 мл) с получением соединения 5 (38 г) в виде красного твердого вещества. LC-MS: 498 [M+H]+.
Соединение 5 (14,3 г, 28,7 ммоль) и NaOH (3,4 г, 81,6 ммоль) добавляли в колбу, которую дважды продували азотом. Добавляли безводный DMF (100 мл), и смесь перемешивали в течение 5 минут при 0°C перед добавлением раствора соединения 2 (8,6 г, 30,1 ммоль) в DMF (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут до полного потребления соединения 4 (проверяемого TLC). Полученную смесь выливали в 5% водный раствор AcOH (120 мл), которую затем экстрагировали DCM (3×150 мл), и объединенные органические слои промывали насыщенным водным раствором NaCl (150 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток перекристаллизовывали DCM/Et2O (1:1) с получением соединения 6 (15,5 г) в виде красного твердого вещества. LC-MS: 702 [M+H]+.
К раствору соединения 6 (46 г, 65,6 ммоль) в MeOH (300 мл) добавляли 3N HCl (200 мл). Смесь кипятили с обратным холодильником до тех пор, пока красный цвет ни переходил в зеленый. Полученный раствор концентрировали в вакууме и добавляли концентрированный NH3·H2O (100 мл), и с последующей экстракцией DCM (2×200 мл). Водную фазу концентрировали в вакууме и подвергали обработке катионообменной смолой (элюировали NH3·H2O/EtOH, 1:1) с получением соединения 7 (15 г) в виде белого твердого вещества. LC-MS: 280 [M+H]+.

К суспензии соединения 7 (15 г, 53,9 ммоль) в MeCN (150 мл) добавляли раствор NaOH (4,3 г, 107,7 ммоль) в воде (150 мл) при 0°C и с последующим добавлением (BOC)2O (17,6 г, 80,8 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. Полученный раствор концентрировали в вакууме с последующей экстракцией DCM (2×150 мл). Водную фазу подкисляли 1N HCl до pH=3 и экстрагировали EtOAc (3×150 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (150 мл), сушили над безводным Na2SO4 и концентрировали в вакууме с получением соединения 8 (12,3 г, 60%) в виде белого твердого вещества. LC-MS: 402 [M+Na]+.
К суспензии соединения 8 (18,4 г, 48,5 ммоль) и кислоты Мельдрума (8,4 г, 58,2 ммоль) в DCM (400 мл) добавляли DMAP (9,5 г, 77,6 ммоль) при -5°C. После перемешивания в течение 10 минут, добавляли по каплям раствор DCC (12 г, 58,2 ммоль) в DCM (100 мл) при -5°C. Смесь перемешивали в течение ночи при комнатной температуре. Полученный раствор охлаждали до 0°C и фильтровали. Фильтрат промывали водным раствором лимонной кислоты (3×200 мл) и насыщенным водным раствором NaCl (200 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток промывали Et2O (2×50 мл) с получением соединения 9 (22 г) в виде светло-желтого твердого вещества.

К раствору соединения 9 (22 г, 43,6 ммоль) в DCM (400 мл) добавляли AcOH (28,8 г, 479,4 ммоль) при 0°C. После перемешивания в течение 10 минут, порциями добавляли NaBH4 (4,1 г, 109 ммоль). Смесь перемешивали в течение часа при 0°C. Полученный раствор промывали насыщенным водным раствором NaHCO2 (2×200 мл) и насыщенным водным раствором NaCl (200 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток промывали простым эфиром (2×100 мл) с получением соединения 10 (18,6 г) в виде не совсем белого твердого вещества. LC-MS: 514 [M+Na]+.
Раствор соединения 10 (18,6 г, 37,9 ммоль) в толуоле (350 мл) нагревали при кипячении с обратным холодильником в течение 2 часов. После охлаждения, смесь выпаривали досуха с получением соединения 11 (14 г) в виде желтого сиропа. LC-MS: 334 [M-tBu+H]+.
К раствору соединения 11 (14 г, 36,0 ммоль) в DCM (250 мл) добавляли TFA (20 мл). Смесь перемешивали в течение 4 часов при 0°C. Полученный раствор концентрировали в вакууме для удаления TFA. Остаток растворяли в DCM (400 мл) и промывали насыщенным водным раствором NaHCO3 (2×200 мл), сушили над безводным Na2SO4 и концентрировали с получением соединения 12 (10 г) в виде желтого твердого вещества. LC-MS: 290 [M+H]+.
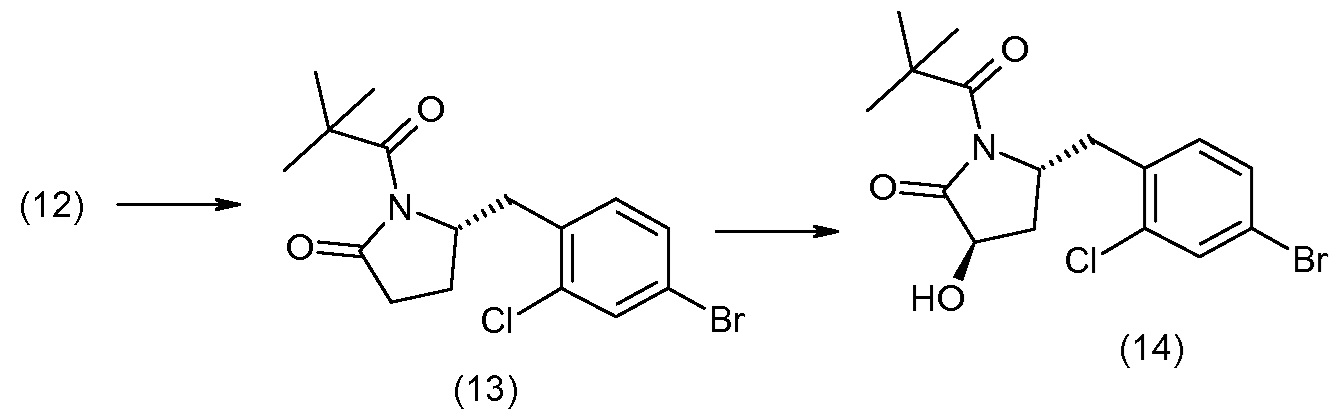
К раствору соединения 12 (10 г, 34,7 ммоль) в сухом THF (250 мл) добавляли NaH (2,4 г, 69,3 ммоль, 70%) при 0°C. Смесь перемешивали в течение одного часа при 0°C в атмосфере азота. Затем добавляли пивалоилхлорид (5 г, 41,6 ммоль). После перемешивания в течение еще 2 часов добавляли насыщенный водный раствор NaHCO3 (100 мл) для гашения реакции. Полученную смесь концентрировали и экстрагировали EtOAc (3×100 мл), и объединенные органические слои промывали насыщенным водным раствором NaCl (100 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (гексаны/EtOAc, 5:1) с получением соединения 13 (11,8 г) в виде белого твердого вещества. LC-MS: 374[M+H]+.
К раствору соединения 13 (11,8 г, 31,8 ммоль) в сухом THF (70 мл) по каплям добавляли NaHMDS (24 мл, 47,7 ммоль, 2,0M в THF) при -78°C в атмосфере азота. После перемешивания в течение 30 минут, добавляли по каплям раствор (+)-(8,8-дихлоркамфорилсульфонил)оксазиридина (15,2 г, 50,8 ммоль) в THF (70 мл) при -78°C. Смесь перемешивали в течение еще одного часа при такой же температуре перед добавлением водного раствора NH4Cl (70 мл) для гашения реакции. Полученную смесь экстрагировали EtOAc (3×150 мл), и объединенные органические слои промывали насыщенным водным раствором NaCl (150 мл), сушили над безводным Na2SO4, концентрировали в вакууме и очищали хроматографией на силикагеле (гексаны/EtOAc, 20:1-5:1) с получением неочищенного продукта (5 г), который дополнительно очищали препаративной ВЭЖХ с получением соединения 14 (4 г) в виде желтого твердого вещества. LC-MS: 390 [M+H]+.
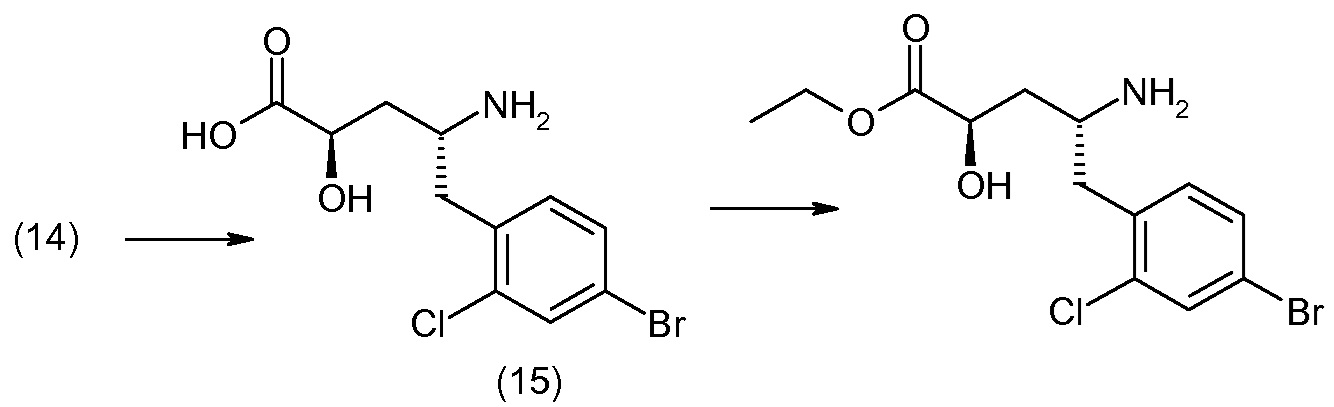
Раствор соединения 14 (4 г, 10,3 ммоль) в концентрированной HCl (50 мл) нагревали при кипячении с обратным холодильником в течение ночи. Смесь концентрировали в вакууме, и полученное твердое вещество промывали Et2O (2×50 мл) с получением соединения 15 (3,1 г) в виде белой твердой соли HCl. LC-MS: 324 [M+H]+.
Раствор соединения 15 (3,1 г, 8,6 ммоль) в HCl/EtOH (6,7M, 40 мл) перемешивали в течение ночи при 50°C. Полученную смесь концентрировали в вакууме, и остаток промывали эфиром (2×50 мл) с получением указанного в заголовке соединения (2,9 г) в виде не совсем белой твердой соли HCl. LC-MS: 352 [M+H]+.
1H ЯМР: (CD3OD) 1,268 (т, J=6,9 Гц, 3H), 1,862-1,946 (м, 1H), 2,068-2,143 (м, 1H), 3,104-3,199 (м, 2H), 3,769-3,809 (м, 1H), 4,162-4,209 (м, 2H), 4,274-4,881 (м, 1H), 7,325 (дд, J=8,1, 2,1 Гц, 1H), 7,522 (дд, J=8,3, 3,0 Гц, 1H), 7,696 (д, J=1,8 Гц, 1H).
Получение 20
Сложный трет-бутиловый эфир [(R)-1-бифенил-4-илметил-2-(2,2-диметил-4,6-диоксо-[1,3]диоксан-5-ил)-2-оксоэтил]карбаминовой кислоты

К раствору (R)-3-бифенил-4-ил-2-трет-бутоксикарбониламинопропионовой кислоты (50 г, 146 ммоль), кислоты Мельдрума (23,3 г, 161 ммоль) и DMAP (27,8 г, 227 ммоль) в безводном DCM (500 мл) добавляли раствор DCC (33,3 г, 161 ммоль) в безводном DCM (200 мл) в течение 1 часа при -5°C в атмосфере азота. Смесь перемешивали при -5°C в течение 8 часов, затем выдерживали в холодильнике в течение ночи, в течение которых осаждались мельчайшие кристаллы дициклогексилмочевины. После фильтрования, смесь промывали 5% KHSO4 (4×200 мл) и насыщенным водным раствором NaCl (1×200 мл), затем сушили в условиях отрицательной температуры с MgSO4 в течение ночи. Раствор выпаривали с получением указанного в заголовке соединения (68 г, светло-желтое твердое вещество), которое использовали без дополнительной очистки. LC-MS: 490 [M+Na], 957 [2M+Na].
Получение 21
Сложный этиловый эфир (2R,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-гидроксиметилпентановой кислоты (соединение 6) и сложный этиловый эфир (2S,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-гидроксиметилпентанoевой кислоты (соединение 7)

К раствору неочищенного сложного трет-бутилового эфира [(R)-1-бифенил-4-илметил-2-(2,2-диметил-4,6-диоксо-[1,3]диоксан-5-ил)-2-оксоэтил]карбаминовой кислоты (68 г, 147 ммоль) в безводном DCM (1 л) добавляли AcOH (96,7 г, 1,6 моль) при -5°C в атмосфере азота. Смесь перемешивали при -5°C в течение 0,5 часа, затем небольшими порциями добавляли NaBH4 (13,9 г, 366 ммоль) в течение 1 часа. После перемешивания в течение еще 1 часа при -5°C, добавляли насыщенный водный раствор NaCl (300 мл). Органический слой промывали насыщенным водным раствором NaCl (2×300 мл) и водой (2×300 мл), сушили над MgSO4, фильтровали и выпаривали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=5:1) с получением соединения 1 (46 г, светло-желтое твердое вещество). LC-MS: 476 [M+Na], 929 [2M+Na].

Раствор соединения 1 (46 г, 101 ммоль) в безводном толуоле (300 мл) кипятили с обратным холодильником в атмосфере азота в течение 3 часов. После выпаривания растворителя остаток очищали хроматографией (гексаны:EtOAc=10:1) с получением соединения 2 (27 г, светло-желтое твердое вещество). LC-MS: 374 [M+Na], 725 [2M+Na].
1H ЯМР (300 MГц, CDCl3): δ 7,64-7,62 (м, 4H), 7,51-7,46 (м, 2H), 7,42-7,39 (м, 1H), 7,39-7,30 (м, 2H), 4,50-4,43 (м, 1H), 3,27-3,89 (м, 1H), 2,88-2,80 (м, 1H), 2,48-2,42 (м, 2H), 2,09-1,88 (м,2H), 1,66 (с,9H).
Смесь соединения 2 (27 г, 77 ммоль) и трет-бутокси-N,N,N',N'-тетраметилметандиамина (40,3 г, 231 ммоль) нагревали до 80°C в атмосфере азота. После перемешивания в течение 3 часов при 80°C, смесь разбавляли EtOAc (300 мл), промывали водой (2×150 мл) и насыщенным водным раствором NaCl (2×150 мл), сушили над MgSO4, фильтровали и выпаривали с получением неочищенного соединения 3 (29,7 г, светло-желтое масло). LC-MS: 425 [M+H], 835 [2M+H].

К раствору неочищенного соединения 3 (29,7 г, 73 ммоль) в THF (200 мл) добавляли 1M HCl (81 мл) при 0°C в атмосфере азота. После перемешивания в течение 1 часа при комнатной температуре смесь разбавляли EtOAc (100 мл) и доводили насыщенным водным раствором NaHCO3 до pH 7. Водный слой экстрагировали EtOAc (2×150 мл), и объединенные органические слои промывали водой (2×150 мл) и насыщенным водным раствором NaCl (1×150 мл), сушили над MgSO4, фильтровали и выпаривали с получением неочищенного соединения 4 (29,4 г, желтое масло). LC-MS: 402 [M+Na], 781 [2M+Na].
К раствору соединения 4 (29,4 г, 77 ммоль) в безводном THF (300 мл) добавляли безводный EtOH (30 мл) и AcOH (92,5 г, 1,5 моль) при -5°C в атмосфере азота. Смесь перемешивали при -5°C в течение 0,5 часа, затем небольшими порциями добавляли NaBH3CN (19,4 г, 308 ммоль) в течение 1 часа. После перемешивания в течение одного дополнительного часа при -5°C, pH смеси доводили насыщенным водным раствором NaHCO3 до pH 7. Водные слои экстрагировали EtOAc (2×200 мл), и объединенные органические слои промывали водой (2×150 мл) и насыщенным водным раствором NaCl (1×150 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=5:1) с получением соединения 5 (11,2 г, светло-желтое твердое вещество). LC-MS: 404 [M+Na], 785 [2M+Na].

К раствору соединения 5 (11,2 г, 29 ммоль) в безводном EtOH (500 мл) добавляли безводный K2CO3 (8,0 г, 58 ммоль) при 0°C в атмосфере азота. После перемешивания в течение 1 часа при 0°C, смесь нагревали до комнатной температуры и перемешивали в течение 16 часов. После фильтрования, фильтрат концентрировали, и остаток разбавляли водой (150 мл), DCM (200 мл) и насыщенным водным раствором NaCl (50 мл). После отделения водный слой экстрагировали DCM (2×150 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (2×200 мл), сушили над MgSO4 и концентрировали с получением неочищенного продукта, который дополнительно очищали колоночной хроматографией (гексаны:EtOAc=5:1) с получением соединений 6 и 7 (8,3 г, светло-желтое твердое вещество).
Соединение 6: LC-MS: 450 [M+Na], 877 [2M+Na].
1H ЯМР (300 MГц, CDCl3): δ 7,58-7,23 (м, 9H), 4,46-4,43 (д, 1H), 4,20-4,13 (м, 2H), 3,94 (с, 1H), 3,82-3,70 (м, 2H), 2,85-2,70 (м, 3H), 2,25-2,22 (д, 1H), 2,01-1,92 (м, 1H), 1,47 (с, 9H), 1,26-1,24 (м, 3H).
Соединение 7: LC-MS: 450 [M+Na], 877 [2M+Na].
1H ЯМР (300 MГц, CDCl3): δ 7,58-7,55 (м, 4H), 7,50-7,43 (м, 2H), 7,40-7,30 (м, 1H), 7,26-7,23 (м, 1H), 4,46 (м, 1H), 4,21-4,13 (м, 2H), 3,94 (м, 1H), 3,82-3,77 (м, 2H), 2,83-2,81 (д, 2H), 2,66-2,63 (м, 1H), 2,24 (м, 1H), 1,83-1,81 (м, 2H), 1,38 (с, 9H), 1,30-1,25 (м, 3H).
Получение 22
(2S,4S)-5-Бифенил-4-ил-4-трет-бутоксикарбониламино-2-гидроксиметилпентановая кислота (R7=H; P2=BOC) и сложный этиловый эфир (2S,4S)-4-амино-5-бифенил-4-ил-2-гидроксиметилпентановой кислоты (R7= -CH2CH3: группа P2 удалена)

Сложный этиловый эфир (2S,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-гидроксиметилпентановой кислоты (210 мг) омыляли LiOH с получением BOC-защищенной кислоты (R7= H; P2=BOC) (120 мг). Сложный этиловый эфир (2S,4S)-5-бифенил-4-ил-4-трет-бутоксикарбониламино-2-гидроксиметилпентановой кислоты (~180 мг) подвергали снятию защиты HCl с получением аминоэфира (R7= -CH2CH3; группа P2 удалена) в виде соли HCl (120 мг).
Получение 23
Сложный этиловый эфир (2S,4S)-4-амино-5-(2'-фторбифенил-4-ил)-2-гидроксиметилпентановой кислоты

К раствору сложного трет-бутилового эфира (S)-2-(4-бромбензил)-5-оксопирролидин-1-карбоновой кислоты (18,4 г, 52 ммоль) в 1,4-диоксане (500 мл) добавляли 2-фторфенилбороновую кислоту (8,7 г, 63 ммоль) и Pd(dppf)2Cl2 (3,8 г, 5,2 ммоль) при комнатной температуре в атмосфере азота. После перемешивания в течение 10 минут добавляли раствор K2CO3 (14,4 г, 104 ммоль) в воде (50 мл). Смесь нагревали до 80°C и перемешивали при этой температуре в течение 5 часов. После выпаривания растворителя добавляли воду (300 мл), и смесь экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (400 мл), сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который дополнительно очищали колоночной хроматографией (гексаны:EtOAc=8:1) с получением соединения 1 (17,3 г) в виде красного масла. LC-MS: 392 [M+Na].
Смесь соединения 1 (17,3 г, 46,7 ммоль) и трет-бутокси-N,N,N',N'-тетраметилметандиамина (24,4 г, 140 ммоль) нагревали до 80°C в атмосфере азота. После перемешивания в течение 3 часов при 80°C смесь разбавляли EtOAc (300 мл) и промывали водой (2×150 мл), насыщенным водным раствором NaCl (150 мл), сушили над MgSO4, фильтровали и выпаривали с получением неочищенного соединения 2 (20,6 г) в виде красного масла. LC-MS: 425 [M+H], 849 (2M+H).

К раствору неочищенного соединения 2 (20,6 г, 48,6 ммоль) в THF (300 мл), добавляли 1M HCl (58 мл, 58 ммоль) при 0°C в атмосфере азота. После перемешивания в течение 1 часа при комнатной температуре смесь разбавляли EtOAc (100 мл) и доводили насыщенным водным раствором NaHCO3 до pH 7. Водный слой экстрагировали EtOAc (2×150 мл), и объединенные органические слои промывали водой (2×150 мл) и насыщенным водным раствором NaCl (150 мл), сушили над MgSO4, фильтровали и выпаривали с получением неочищенного соединения 3 (18,9 г) в виде красного масла. LC-MS: 420 (M+Na), 817 (2M+Na).
К раствору неочищенного соединения 3 (18,9 г, 47,6 ммоль) в безводном THF (400 мл) добавляли безводный EtOH (50 мл) и AcOH (57,2 г, 952 ммоль) при -5°C в атмосфере азота. Смесь перемешивали при -5°C в течение 30 минут, затем небольшими порциями добавляли NaBH3CN (15 г, 238 ммоль) в течение 1 часа. После перемешивания в течение дополнительного 1 часа при -5°C, pH смеси доводили насыщенным водным раствором NaHCO3 до pH 7. Водный слой экстрагировали EtOAc (3×200 мл), и объединенные органические слои промывали водой (2×150 мл) и насыщенным водным раствором NaCl (150 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=6:1) с получением соединения 4 (7,1 г) в виде светло-желтого твердого вещества. LC-MS: 422 (M+Na), 821 (2M+Na).
К раствору соединения 4 (7,1 г, 17,7 ммоль) в безводном EtOH (500 мл) добавляли безводный K2CO3 (9,8 г, 70,8 ммоль) при 0°C в атмосфере азота. После перемешивания в течение 1 часа при 0°C, смесь нагревали до комнатной температуры и перемешивали в течение 16 часов. После фильтрования фильтрат концентрировали, и остаток разбавляли водой (150 мл), DCM (200 мл) и насыщенным водным раствором NaCl (50 мл). После отделения водный слой экстрагировали DCM (2×150 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (2×200 мл), сушили над MgSO4 и концентрировали с получением неочищенного продукта, который дополнительно очищали колоночной хроматографией (гексаны:EtOAc=5:1) с получением соединения 5 (2 г) в виде светло-желтого твердого вещества. 2,1 г (R,S) изомера также получали в виде светло-желтого твердого вещества.

Соединение 5 (400 мг, 0,9 ммоль) растворяли в MeCN (3 мл) и 4M HCl в диоксане (0,5 мл). Смесь перемешивали при комнатной температуре в течение 1 часа, затем концентрировали с получением указанного в заголовке соединения в виде соли HCl (340 мг), которое образовывалось в виде масла и застывало в течение ночи.
Получение 24
Сложный трет-бутиловый эфир [(R)-2-бифенил-4-ил-1-(2,2,5-триметил-4,6-диоксо-1,3-диоксан-5-илметил)этил]карбаминовой кислоты

AcOH (8,6 мл) добавляли к раствору неочищенного сложного трет-бутилового эфира [(R)-1-бифенил-4-илметил-2-(2,2-диметил-4,6-диоксо[1,3]диоксан-5-ил)-2-оксо-этил]карбаминовой кислоты (6,4 г, 14 ммоль) в безводном MeCN (90 мл) при -5°C в атмосфере азота. Смесь перемешивали при -5°C в течение 30 минут, затем небольшими порциями добавляли боргидрид натрия (1,3 г, 34,5 ммоль) в течение 2 часов. После перемешивания в течение еще 1 часа при -5°C, добавляли насыщенный водный раствор NaCl и 1,7M NaCl в воде (30 мл). Слои разделяли, и органический слой промывали насыщенным водным раствором NaCl (2×30 мл) и водой (2×30 мл), сушили в атмосфере MgSO4, фильтровали и выпаривали. Полученный неочищенный продукт дополнительно очищали хроматографией (5:1 гептан:EtOAc) с получением сложного трет-бутилового эфира [(S)-2-бифенил-4-ил-1-(2,2-диметил-4,6-диоксо-[1,3]диоксан-5-илметил)этил]карбаминовой кислоты (1,1 г, чистота 98,4%) в виде светло-желтого твердого вещества.
Сложный трет-бутиловый эфир [(S)-2-бифенил-4-ил-1-(2,2-диметил-4,6-диоксо-[1,3]диоксан-5-илметил)этил]карбаминовой кислоты (5,0 г, 11 ммоль) и K2CO3 (1,8 г, 13,2 ммоль) растворяли в DMF (33,9 мл) и охлаждали до 0°C при перемешивании в атмосфере азота. Добавляли метилйодид (892 мкл), и полученную смесь перемешивали при 0°C в течение 1 часа. Смеси давали возможность нагреться до комнатной температуры. Добавляли насыщенный водный раствор NaCl (35 мл) и EtOAc (35 мл), и полученную смесь перемешивали в течение 2 минут. Слои разделяли, и органический слой выпаривали. Остаток перетирали с EtOAc (20 мл). Твердое вещество фильтровали и сушили в вакууме. Фильтрат концентрировали и снова перетирали с EtOAc с получением указанного в заголовке соединения (3,9 г).
Получение 25
(2S,4R)-5-Бифенил-4-ил-4-трет-бутоксикарбониламино-2-гидроксиметил-2-метилпентановая кислота (P2=BOC) и (2S,4R)-4-амино-5-бифенил-4-ил-2-гидроксиметил-2-метилпентановая кислота (группа P2 удалена)
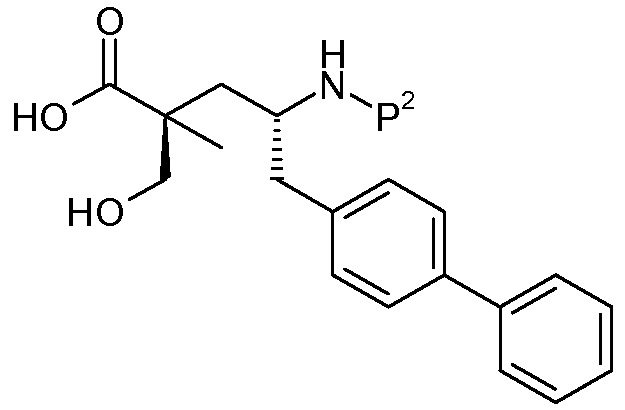
Дистиллированную воду (140 мл) продували 30 минут азотом, затем канюлировали в сосуд, содержащий 0,1M дийодидида самария в THF (800 мл), соблюдая предосторожность, чтобы не дать возможность какому-либо воздуху вступать в контакт с раствором. При поддержании атмосферы азота через канюлю добавляли дегазированный раствор сложного трет-бутилового эфира [(R)-2-бифенил-4-ил-1-(2,2,5-триметил-4,6-диоксо-1,3-диоксан-5-илметил)этил]карбаминовой кислоты (3,7 г, 8,0 ммоль) и THF (100 мл). Полученную смесь перемешивали в течение 15 минут, затем подвергали воздействию воздуха. Добавляли насыщенный водный раствор NaCl (12 мл), 10% лимонную кислоту (6 мл) и EtOAc (30 мл). Смесь перемешивали в течение 5 минут, затем оба слоя экстрагировали. Органический слой сушили над Na2SO4и концентрировали в вакууме. Неочищенный продукт очищали хроматографией (330 г, колонка gold, 50% EtOAc с градиентом 0,5% AcOH/эфир) с получением BOC-защищенной кислоты. (P2=BOC) (1,4 г). BOC-защищенную кислоту растворяли в MeCN (10 мл) с последующим добавлением 4N HCl в диоксане (10 мл). Растворитель выпаривали, и продукт азеотропировали толуолом (2×) с получением кислоты. (P2 удалена) (1,0 г).
Получение 26
Сложный этиловый эфир (2S,4R)-4-амино-5-бифенил-4-ил-2-гидроксиметил-2-метилпентановой кислоты

(2S,4R)-4-Амино-5-бифенил-4-ил-2-гидроксиметил-2-метилпентановую кислоту (0,3 г, 957 мкмоль) объединяли с EtOH (6 мл) и 4M HCl в 1,4-диоксане (718 мкл) и перемешивали в течение ночи. Растворители выпаривали, и продукт азеотропировали толуолом (2×) с получением указанного в заголовке соединения (295 мг), которое использовали без дополнительной очистки.
Получение 27
Сложный трет-бутиловый эфир [(R-1-(3'-фторбифенил-4-илметил)-2-(2,2,5-триметил-4,6-диоксо-[1,3]диоксан-5-ил)этил]карбаминовой кислоты

К раствору (R)-2-амино-3-(4-бромфенил)пропионовой кислоты (50 г, 0,2 моль) в MeCN (700 мл) добавляли раствор NaOH (16,4 г, 0,4 моль) в воде (700 мл) при -5°C. После перемешивания в течение 10 минут добавляли раствор (BOC)2O (44,7 г, 0,2 моль) в MeCN (100 мл). Смесь нагревали до комнатной температуры и перемешивали в течение ночи. После выпаривания MeCN, остаток разбавляли DCM (800 мл) и подкисляли 1M HCl до pH 2 при -5°C. Водный слой экстрагировали DCM (3×200 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (500 мл), сушили над безводным Na2SO4 и концентрировали с получением соединения 1 (64,2 г, белое твердое вещество). LC-MS: 366 [M+Na], 709 [2M+Na].
К раствору соединения 1 (64,2 г, 187 ммоль) в 1,4-диоксане (500 мл) добавляли 3-фторфенилбороновую кислоту (31,3 г, 224 ммоль) и Pd(dppf)2Cl2 (13,7 г, 19 ммоль) при комнатной температуре в атмосфере азота. После перемешивания в течение 10 мин добавляли раствор K2CO3(51,7 г, 374 ммоль) в воде (250 мл). Смесь нагревали до 100°C и перемешивали в течение ночи. После выпаривания растворителя добавляли воду (200 мл). Водный слой подкисляли 1M HCl до pH 2 и экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (400 мл), сушили над безводным Na2SO4 и концентрировали с получением неочищенного продукта, который дополнительно очищали колоночной хроматографией (гексаны:EtOAc=4:1) с получением соединения 2 (45 г, светло-желтое масло). LC- MS: 382 [M+Na], 741 [2M+Na].

К раствору соединения 2 (45 г, 125 ммоль), кислоты Мельдрума (23,5 г, 163 ммоль) и DMAP (26,0 г, 213 ммоль) в безводном DCM (500 мл) добавляли раствор DCC (33,3 г, 163 ммоль) в безводном DCM (200 мл) в течение 1 часа при -5°C в атмосфере азота. Смесь перемешивали при -5°C в течение 8 часов, затем выдерживали в холодильнике в течение ночи, и в этот период осаждались мельчайшие кристаллы дициклогексилмочевины. После фильтрования смесь промывали 5% KHSO4 (4×200 мл) и насыщенным водным раствором NaCl (1×200 мл), затем сушили в условиях отрицательной температуры с безводным MgSO4 в течение ночи. Раствор выпаривали с получением неочищенного соединения 3 (57,7 г, светло-желтое масло). LC-MS: 508 [M+Na], 993 [2M+Na].

К раствору неочищенного соединения 3 (57,7 г, 119 ммоль) в безводном DCM (1 л) добавляли AcOH (78,4 г, 1,3 моль) при -5°C в атмосфере азота. Смесь перемешивали при -5°C в течение 0,5 часа, затем небольшими порциями добавляли NaBH4 (11,3 г, 0,3 моль) в течение 1 часа. После перемешивания в течение еще одного часа при -5°C добавляли насыщенный водный раствор NaCl (300 мл). Органический слой промывали насыщенным водным раствором NaCl (2×300 мл) и водой (2×300 мл), сушили над безводным MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=6:1) с получением соединения 4 (28 г, светло-желтое масло). LC-MS: 494 [M+Na], 965 [2M+Na].
К раствору соединения 4 (28 г, 60 ммоль) в безводном DMF (250 мл) добавляли K2CO3 (9,9 г, 72 ммоль) и CH3I (25,6 г, 180 ммоль) при 0°C в атмосфере азота. После перемешивания в течение 1 часа при 0°C, смесь нагревали до комнатной температуры и перемешивали в течение ночи. Смесь разбавляли водой (3 л) и экстрагировали EtOAc (3×300 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (500 мл), сушили над безводным Na2SO4 и концентрировали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=5:1) с получением указанного в заголовке соединения (11,7 г, светло-желтое твердое вещество). LC-MS: 508 [M+Na], 993 [2M+Na].
1H ЯМР (300 MГц, CD3OD): δ 7,52-7,49 (м, 2H), 7,41-7,39 (м, 2H), 7,32-7,27 (м, 3H), 7,07-7,01 (м, 1H), 6,21-6,18 (д, 1H), 3,79 (м, 1H), 2,78-2,61 (м, 2H), 2,35-2,20 (м, 2H), 1,76 (с, 6H), 1,59 (с, 3H), 2,21 (с, 1H), 1,28 (с, 9H).
Получение 28
(2S,4R)-4-трет-Бутоксикарбониламино-5-(3'-фторбифенил-4-ил)-2-гидроксиметил-2-метилпентановая кислота (P2=BOC) и (2S,4R)-амино-5-(3'-фторбифенил-4-ил)-2-гидроксиметил-2-метилпентановая кислота (P2 удалена)

Дистиллированную воду (181 мл) продували 1 час азотом, затем через канюлю направляли в сосуд, содержащий 0,1M дийодид самария в THF (800 мл). При поддержании атмосферы азота, аналогичным образом дегазировали раствор сложного трет-бутилового эфира [(R)-1-(3'-фторбифенил-4-илметил)-2-(2,2,5-триметил-4,6-диоксо-[1,3]диоксан-5-ил)этил]карбаминовой кислоты (4,9 г, 10,0 ммоль) и THF (20 мл) добавляли через канюлю. Полученную смесь перемешивали в течение 15 минут, затем подвергали воздействию воздуха. Растворитель выпаривали и добавляли EtOAc (200 мл), насыщенным водным раствором NaCl (50 мл) и 10% лимонную кислоту (20 мл). Смесь перемешивали в течение 5 минут, затем оба слоя экстрагировали. Органический слой сушили над Na2SO4 и концентрировали в вакууме. Неочищенный продукт очищали хроматографией (330 г, колонка gold, 1:1 эфир:EtOAc с 0,5% AcOH) с получением BOC-защищенной кислоты. (P2=BOC) (1,5 г). Часть BOC-защищенной кислоты растворяли в 4M HCl в диоксане (6 мл) и MeCN (10 мл). Растворитель выпаривали в вакууме с получением кислоты (P2 удалена).
Получение 29
Сложный метиловый эфир 3-(N-бифенил-4-илметил-N'-трет-бутоксикарбонилгидразино)-2-гидрокси-2-метилпропионовой кислоты
4-(Бромметил)бифенил (2,00 г, 8,09 ммоль) и DIPEA (1,4 мл, 8,1 ммоль) растворяли в DMF (40,0 мл), затем добавляли трет-бутилкарбазат (2,1 г, 16,2 ммоль), и смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции смесь частично концентрировали, и остаток разделяли между EtOAc и насыщенным водным раствором NaHCO3. Слой EtOAc сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали флэш-хроматографией (0-60% EtOAc/гексаны с 0,5% DIPEA) с получением соединения 1 (1,3 г.)
Соединение 1 (460 мг, 1,5 ммоль) растворяли в изопропиловом спирте (10,0 мл), затем добавляли метил 2-метилглицидат (180 мкл, 1,7 ммоль), и смесь нагревали до 85°C в течение ночи. После завершения реакции, смесь разделяли между EtOAc и насыщенным водным раствором NaHCO3. Слой EtOAc затем сушили над Na2SO4 и концентрировали с получением указанного в заголовке соединения (0,5 г), которое использовали без дополнительной очистки.
Получение 30
Сложный метиловый эфир (R)-3-[N-(4-бромбензил)-N'-трет-бутоксикарбонилгидразино]-2-гидроксипропионовой кислоты

4-Бромбензилбромид (5,0 г, 20 ммоль) и DIPEA (3,48 мл, 20,0 ммоль) растворяли в DMF (20 мл). Добавляли трет-бутилкарбазат (7,9 г, 60,0 ммоль), и смесь перемешивали при комнатной температуре до завершения реакции. Смесь частично концентрировали, затем остаток разделяли между EtOAc и насыщенным водным раствором NaHCO3. Слой EtOAc затем сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали флэш-хроматографией с получением соединения 1 (3,8 г).

Соединение 1 (1,9 г, 6,3 ммоль) растворяли в изопропиловом спирте (26,4 мл). Добавляли метил-(2R)-глицидат (1,1 мл, 12,6 ммоль), и смесь нагревали при 90°C до завершения реакции (~4 дня). Смесь охлаждали до комнатной температуры и концентрировали с получением указанного в заголовке соединения (2,5 г) в виде белого твердого вещества.
Получение 31
Сложный этиловый эфир (R)-3-[N-(3'-хлорбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты
Сложный метиловый эфир (R)-3-[N-(4-бромбензил)-N'-трет-бутоксикарбонилгидразино]-2-гидроксипропионовой кислоты (600 мг, 1 ммоль), 3-хлорфенилбороновую кислоту (419 мг, 2,7 ммоль) и K2CO3 (617 мг, 4,5 ммоль) объединяли в EtOH (5 мл) и воде (1 мл) с последующим добавлением SilicaCat®Pd(0) (0,09 ммоль/г загрузки, 1160 мг, 104 мкмоль). Смесь нагревали в микроволновом реакторе при 120°C до завершения реакции (~30 минут). Смесь фильтровали и концентрировали. Остаток растворяли в MeCN/AcOH и очищали обращенно-фазовой хроматографией (колонка 55 г; градиент 30-95% MeCN в воде с 0,1% TFA). Чистые фракции собирали, концентрировали и затем растворяли в 4M HCl в диоксане (6 мл) и EtOH (6 мл). Смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали с получением указанного в заголовке соединения (250 мг), которое использовали без дополнительной очистки.
Получение 32
Сложный трет-бутиловый эфир (S)-2-(1-бифенил-4-ил-1-метилэтил)-5-оксо-пирролидин-1-карбоновой кислоты

К раствору 2-(4-бромфенил)ацетонитрила (130,0 г, 0,7 моль) и йодметана (103,9 мл, 1,7 моль) в THF (1,0 л) небольшими порциями добавляли NaH (60% дисперсию в минеральном масле, 66,7 г, 1,7 моль) при 10°C. После завершения добавления смесь перемешивали при 10°C в течение еще 2 часов. Смесь выливали в ледяную воду (2,0 л) и экстрагировали EtOAc (1,5 л). Органический слой промывали насыщенным водным раствором NaCl, сушили над безводным MgSO4 и концентрировали с получением соединения 1 (175 г, содержащего минеральное масло) в виде желтого масла, которое использовали непосредственно без дополнительной очистки.
1H ЯМР (CDCl3, 300 MГц) δ 7,52 (д, J=9,0 Гц, 2H), 7,38 (д, J=9,0 Гц, 2H), 1,72 (с, 6H).
К раствору соединения 1 (175 г, содержащему минеральное масло) в DCM (1,0 л) по каплям добавляли DIBAL (1,0M раствор в DCM, 700 мл, 0,70 моль) при -78°C. Реакционную смесь перемешивали при -78°C в течение 1,5 часов и затем осторожно гасили 3,0N HCl (1,0 л). Полученную смесь перемешивали при комнатной температуре в течение ночи, и органический слой промывали насыщенным водным раствором NaCl, сушили над безводным Na2SO4 и концентрировали с получением соединения 2 (180 г) в виде желтого масла, которое использовали непосредственно без дополнительной очистки.
1H ЯМР (CDCl3, 300 MГц) δ 9,48 (с, 1H), 7,53 (д, J=11,0 Гц, 2H), 7,17 (д, J=11,0 Гц, 2H), 1,46 (с, 6H).

К водному раствору NaCN (32,7 г в 1,0 л H2O, 0,7 моль) добавляли (NH4)2CO3 (380 г, 4,0 моль) и соединение 2 (180 г). Реакционную смесь кипятили с обратным холодильником в течение ночи и затем концентрировали при пониженном давлении при 75°C. К остатку добавляли воду (350 мл), и смесь концентрировали снова. Остаток суспендировали в простом петролейном эфире (700 мл) и воде (250 мл), и полученную смесь перемешивали при комнатной температуре в течение 15 минут. Осадок собирали фильтрованием и сушили с получением соединения 3 (150 г) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 MГц) δ 10,39 (с, 1H), 8,05 (с, 1H), 7,48 (д, J=9,0 Гц, 2H), 7,28 (д, J=9,0 Гц, 2H), 4,17 (с, 1H), 1,42 (с, 3H), 1,34 (с, 3H).
Суспензию соединения 3 (150 г, 0,51 моль) в 6,0N NaOH (400 мл) и этан-1,2-диола (300 мл) перемешивали при 120°C в течение 38 часов. Смесь охлаждали до комнатной температуры и нейтрализовали раствором HCl. Осадок собирали фильтрованием и сушили с получением соединения 4 (250 г, содержавшего соль NaCl) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 300 MГц) δ 7,35 (д, J=9,0 Гц, 2H), 7,17 (д, J=9,0 Гц, 2H), 3,22 (с, 1H), 1,16 (с, 3H), 1,15 (с, 3H).
К суспензии соединения 4 (250 г, содержащей соль HCl) в MeOH (1,0 л) по каплям добавляли тионилхлорид (72,0 мл, 1,0 моль) при 5°C. Смесь кипятили с обратным холодильником в течение ночи, и растворитель удаляли при пониженном давлении. Остаток разделяли между DCM (1,0 л) и насыщенным водным раствором NaHCO3 (1,5 л). Органический слой промывали насыщенным водным раствором NaCl, сушили над безводным Na2SO4 и концентрировали с получением соответствующего сложного метилового эфира (90,0 г). К раствору сложного эфира (90,0 г) и Et3N (56,5 мл, 0,41 моль) в DCM (1,0 л) добавляли по каплям 2-фенилацетилхлорид (48,6 г, 0,32 моль) при 0°C, и смесь перемешивали при 0°C в течение 30 минут. Смесь промывали 1,0N HCl (500 мл) и насыщенным водным раствором NaCl, соответственно. Органический слой сушили над безводным Na2SO4 и концентрировали с получением соединения 5 (120 г).
1H ЯМР (CDCl3, 300 MГц) δ 7,32 (м, 5H), 7,18 (м, 2H), 6,95 (м, 2H), 5,68 (ушир.с, 1H), 4,76 (д, J=9,0 Гц, 1H), 3,57 (с, 3H), 3,53 (д, J=5,0 Гц, 2H), 1,30 (с, 3H), 1,25 (с, 3H).
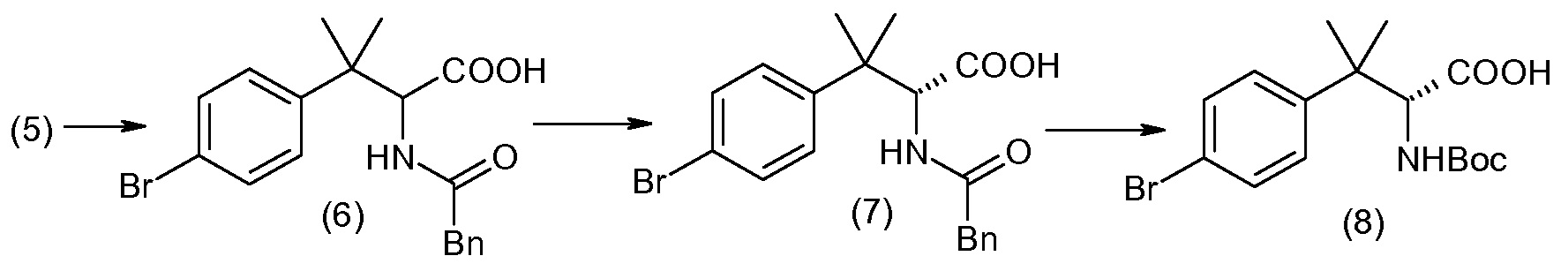
К раствору соединения 5 (120 г, 0,30 моль) в MeOH (500 мл) добавляли 4,0N NaOH (200 мл). Смесь перемешивали при комнатной температуре в течение 4 часов, и затем pH доводили до pH=1 с помощью 3,0N HCl. Полученную смесь экстрагировали EtOAc (2×300 мл). Объединенные экстракты промывали насыщенным водным раствором NaCl, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток перекристаллизовывали из EtOAc/гексанов с получением соединения 6 (82,0 г).
1H ЯМР (DMSO-d6, 300 MГц) δ 7,41 (д, J=6,0 Гц, 2H), 7,22 (м, 5H), 6,99 (д, J=6,0 Гц, 2H), 4,65 (д, J=9,0 Гц, 1H), 3,52 (д, J=14,0 Гц, 1H), 3,36 (д, J=14,0 Гц, 1H), 1,34 (с, 3H), 1,30 (с, 3H).
Суспензию соединения 6 (82,0 г, 0,21 моль) в дистиллированной воде (3,0 л) доводили до pH=8,5 3,0N LiOH, и образовывался прозрачный раствор. Добавляли иммобилизованную пенициллиназу (20,0 г), и полученную смесь перемешивали при 37°C в течение 60 часов. Смесь фильтровали, и фильтрат доводили до pH=1 с помощью 3,0N HCl и экстрагировали EtOAc. Объединенные экстракты промывали насыщенным водным раствором NaCl, сушили над безводным Na2SO4 и концентрировали с получением соединения 7 (59,0 г, 80% ee, содержащего 2-фенилуксусную кислоту).
Суспензию соединения 7 (59,0 г, содержащую 2-фенилуксусную кислоту) в 6,0 N HCl (500 мл) кипятили с обратным холодильником в течение ночи. Смесь промывали EtOAc (300 мл), и водную фазу концентрировали при пониженном давлении с получением соответствующей аминокислоты в виде ее гидрохлорида. Соль растворяли в воде (300 мл), и pH раствора доводили до pH=l1. Добавляли раствор (BOC)2O (33,0 г, 0,2 моль) в ацетоне (200 мл), и смесь перемешивали при комнатной температуре в течение 2 часов. Смесь промывали гексанами (200 мл), и pH водной фазы доводили до pH=2. Полученную смесь экстрагировали EtOAc (2×300 мл). Объединенные экстракты промывали насыщенным водным раствором NaCl, сушили над безводным Na2SO4 и концентрировали с получением соединения 8 (37,0 г) в виде белого твердого вещества.
1H ЯМР (CDCl3, 300 MГц) δ 9,48 (ушир.с, 1H), 7,46 (д, J=7,0 Гц, 2H), 7,26 (д, J=7,0 Гц, 2H), 5,02 (ушир.с, 1H), 4,56 (д, J=9.0 Гц, 1H), 1,39 (с, 9H).

Смесь соединения 8 (37,0 г, 0,1 моль) в диоксане (200 мл) и 1,0N K2CO3 (200 мл) дегазировали в течение 30 минут азотом с последующим добавлением фенилбороновой кислоты (13,4 г, 0,1 моль) и Pd(PPh3)4 (1,6 г, 1,4 ммоль). Смесь нагревали при 75°C в течение 8 часов и затем охлаждали до комнатной температуры. Смесь промывали EtOAc/гексанами (150 мл, 1:1), и pH водной фазы доводили до pH=2 и экстрагировали EtOAc (2×300 мл). Объединенные экстракты промывали насыщенным водным раствором NaCl, сушили над безводным Na2SO4 и концентрировали с получением соединения 9 (31,0 г, выход 84%) в виде белого твердого вещества.
Раствор соединения 9 (31,0 г, 84 ммоль), кислоты Мельдрума (13,3 г, 92 ммоль) и DMAP (15,4 г, 0,13 моль) в DCM (400 мл) охлаждали до -5°C и добавляли раствор DCC (19,0 г, 92 ммоль) в DCM (200 мл) в течение 1 часа. Смесь перемешивали при -5°C в течение ночи. Осадок отфильтровывали, и фильтрат промывали 1,0N HCl (2×700 мл) и насыщенным водным раствором NaCl, соответственно. После сушки органического слоя, содержащего соединение 10, над безводным MgSO4, его использовали непосредственно для следующей стадии без концентрации.

Раствор соединения 10 в DCM (600 мл) охлаждали до -5°C и добавляли AcOH (45,0 мл). Затем небольшими порциями добавляли NaBH4 (7,0 г, 0,2 моль) в течение 30 минут, и смесь перемешивали при -5°C в течение 3 часов. По каплям добавляли воду (50,0 мл) с последующим добавлением насыщенного водного раствора NaCl (450 мл). Органический слой промывали водой (2×300 мл) и насыщенным водным раствором NaHCO3 (2×300 мл), сушили над безводным MgSO4 и концентрировали с получением соединения 11 (32?0 г, 75% ee) в виде не совсем белого твердого вещества. После перекристаллизации из EtOH, получали хирально чистое соединение 11 (13,0 г).
1H ЯМР (CDCl3, 300 MГц) δ 7,61 (м, 10H), 4,46 (ушир.с, 1H), 4,26 (м, 1H), 3,72 (ушир.с, 1H), 2,23 (м, 1H), 1,79 (с, 3H), 1,76 (с, 3H), 1,48 (с, 6H), 1,39 (с, 9H).
Раствор соединения 11 (13,0 г, 27,0 ммоль) в толуоле (100,0 мл) кипятили с обратным холодильником в течение 3 часов. После выпаривания растворителя, остаток перекристаллизовывали из гексанов/EtOAc (3:1) с получением указанного в заголовке соединения (8,0 г) в виде белого твердого вещества.
Получение 33
Сложный этиловый эфир (2R,4S)-4-амино-5-бифенил-4-ил-2-гидрокси-5-метилгексановой кислоты

Смесь сложного трет-бутилового эфира (S)-2-(1-бифенил-4-ил-1-метилэтил)-5-оксопирролидин-1-карбоновой кислоты (14,0 г, 36,9 ммоль, рацемическая) в 3,0N растворе HCl-EtOAc (150 мл) перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли при пониженном давлении с получением соединения 1 (10,0 г) в виде белого твердого вещества.
К раствору соединения 1 (10,0 г, 35,8 ммоль) в THF (80,0 мл) добавляли BuLi (2,5M в гексанах, 15,0 мл) по каплям при -78°C. После перемешивания смеси в течение 30 минут по каплям добавляли пивалоилхлорид (4,8 мл, 39,4 ммоль). Смесь перемешивали при -78°C в течение 1 часа и затем гасили насыщенным водным раствором NH4Cl. Полученную смесь экстрагировали EtOAc, и объединенные экстракты промывали насыщенным водным раствором NaCl, сушили над безводным MgSO4 и концентрировали. Остаток очищали флэш-хроматографией на силикагеле с получением соединения 2 (9,0 г) в виде белого твердого вещества.

К раствору соединения 2 (9,0 г, 24,7 ммоль) в THF (50,0 мл) по каплям добавляли бис(триметилсилила)амид натрия (2,0M в THF, 18,5 мл, 37,0 ммоль) при -78°C. Смесь перемешивали в течение 20 минут и по каплям добавляли раствор производного оксазиридина (10,8 г, 37,0 ммоль) в THF (30,0 мл). Смесь перемешивали при -78°C в течение 30 минут и затем гасили насыщенным водным раствором NH4Cl. Полученную смесь экстрагировали EtOAc (1,0 л), и экстракт промывали 1,0N HCl и насыщенным водным раствором NaCl, сушили над безводным MgSO4 и выпаривали для удаления большей части растворителя. Осадок отфильтровывали, и фильтрат концентрировали. Остаток очищали флэш-хроматографией на силикагеле (DCM:гексаны=1:1 к DCM) с получением соединения 3 (4,3 г, рацемическое). Этот рацемат подвергали хиральной AD-колоночной хроматографии с получением хирально чистого соединения 3 (1,4 г).
1H ЯМР (DMSO-d6, 300 MГц) δ 7,63 (м, 4H), 7,49 (м, 4H), 4,83 (д, 1H), 3,29 (м, 1H), 2,31 (м, 2H), 1,40 (с, 3H), 1,36 (с, 3H), 1,28 (с, 9H).
LC-MS (ESI): m/z 380,1 [M+H]+.
Раствор соединения 3 (1,7 г, 160 ммоль) в EtOH (15,0 мл) и 12,0N HCl (15,0 мл) нагревали при 90-95°C в течение 20 часов. Растворитель удаляли, и остаток обрабатывали 3,0N раствором HCl-EtOH (25,0 мл) при кипячении с обратным холодильником в течение еще 3 часов. После удаления растворителя, остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (0,6 г) в виде пенистой твердой соли HCl.
1H ЯМР (DMSO-d6, 300 MГц) δ 7,88 (ушир.с, 3H), 7,68 (м, 4H), 7,49 (м, 4H), 7,35 (м, 1H), 6,11 (ушир.с, 1H), 4,11 (ушир.с, 1H), 4,05 (q, 2H), 3,61 (ушир.с, 1H), 1,67 (м, 2H), 1,40 (с, 3H), 1,36 (с, 3H), 1,09 (т, 3H).
LC-MS (ESI): m/z 342,1 [M+H]+.
Получение 34
Сложный этиловый эфир (2S,4S)-4-Амино-5-бифенил-4-ил-2-гидроксиметил-5-метилгексановой кислоты

Смесь сложного трет-бутилового эфира (S)-2-(1-бифенил-4-ил-1-метилэтил)-5-оксопирролидин-1-карбоновой кислоты (8,0 г, 21,2 ммоль) и трет-бутокси-N,N,N',N'-тетраметилметандиамина (10,0 г, 63,6 ммоль) нагревали при 80°C в течение 3 часов. Смесь охлаждали до комнатной температуры и разбавляли EtOAc (200 мл). Полученный раствор промывали водой (2×100 мл) и насыщенным водным раствором NaCl, сушили над безводным MgSO4 и концентрировали с получением соединения 1 (9,2 г, количественный) в виде масла.
1H ЯМР (CDCl3, 300 MГц) δ 7,53 (м, 9H), 6,95 (с, 1H), 4,60 (ушир.с, 1H), 2,90 (с, 1H), 2,62 (м, 2H), 1,61 (с, 9H), 1,39 (с, 3H), 1,34 (с, 3H).
К раствору соединения 1 (9,2 г, 21,2 ммоль) в THF (80,0 мл) добавляли 1,0N HCl (25,0 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 2 часов и затем разбавляли EtOAc (100 мл). Полученную смесь нейтрализовали насыщенным водным раствором NaHCO3 и экстрагировали EtOAc (2×100 мл). Объединенные экстракты промывали водой (2×100 мл) и насыщенным водным раствором NaCl, сушили над безводным MgSO4 и концентрировали с получением соединения 2 (8,6 г, количественный) в виде масла. LC-MS (ESI): m/z 430,1 [M+Na]+.

К раствору соединения 2 (8,6 г, 21,2 ммоль) в THF (150 мл) и EtOH (15,0 мл) добавляли AcOH (24,3 мл, 0,4 моль) при -5°C. После перемешивания смеси при -5°C в течение 30 минут, небольшими порциями добавляли NaBH3CN (5,3 г, 84,8 ммоль) в течение 1 часа. Смесь перемешивали при -5°C в течение 1 часа и нейтрализовали насыщенным водным раствором NaHCO3. Полученную смесь экстрагировали EtOAc (2×100 мл). Объединенные экстракты промывали водой (2×100 мл) и насыщенным водным раствором NaCl, сушили над безводным MgSO4 и концентрировали с получением соединения 3 (8,67 г, количественный) в виде пенистого твердого вещества.
К раствору соединения 3 (3,5 г, 8,6 ммоль) в EtOH (30,0 мл) добавляли K2CO3 (2,4 г, 17,1 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 1 часа и затем давали возможность нагреться до комнатной температуры и перемешивали в течение ночи. Смесь фильтровали, и фильтрат концентрировали. Остаток обрабатывали водой (20 мл), и полученную смесь экстрагировали DCM (3×25 мл). Объединенные экстракты промывали насыщенным водным раствором NaCl, сушили над безводным MgSO4 и концентрировали. Остаток очищали флэш-хроматографией на силикагеле (гексаны:EtOAc=6:1) с получением соединения 4 (2,2 г) в виде пенистого твердого вещества.
1H ЯМР (CDCl3, 300 MГц) δ 7,53 (м, 9H), 4,35 (ушир.с, 1H), 4,15 (м, 2H), 3,95 (ушир.с, 1H), 3,65 (м, 2H), 2,61 (ушир.с, 1H), 1,79 (м, 1H), 1,45 (с, 9H), 1,35 (с, 3H), 1,29 (с, 3H), 1,25 (т, 3H).
LC-MS (ESI): m/z 478,2 [M+Na]+.

Смесь соединения 4 (2,2 г, 4,8 ммоль) в 2,0N растворе HCl-EtOH (30,0 мл) перемешивали при комнатной температуре в течение 3 часов. Удаление растворителя при пониженном давлении обеспечивало выход указанного в заголовке соединения (1,6 г) в виде пенистой твердой соли HCl.
1H ЯМР (DMSO-d6, 300 MГц) δ 8,08 (ушир.с, 3H), 7,55 (м, 9H), 4,95 (ушир.с, 1H), 3,95 (м, 2H), 3,48 (м, 2H), 2,75 (ушир.с, 1H), 1,79 (м, 2H), 1,47 (с, 3H), 1,40 (с, 3H), 1,09 (т, 3H).
LC-MS (ESI): m/z 356,1 [M+H]+.
Получение 35
Сложный этиловый эфир 3-[N-(4-Бромбензил)гидразино]-2-гидроксипропионовой кислоты
Раствор сложного метилового эфира (R)-3-[N-(4-бромбензил)-N'-трет-бутоксикарбонилгидразино]-2-гидроксипропионовой кислоты (25 г, 62 ммоль) в EtOH/HCl (310 мл, 1,0M, 0,3 моль) перемешивали в течение ночи. Смесь концентрировали, и остаток промывали EtOAc (120 мл) и фильтровали с получением указанного в заголовке соединения в виде белой твердой соли HCl (15 г).
Получение 36
Сложный этиловый эфир (R)-2-[N-(4-бромбензил)-N'-этоксиоксалилгидразино]-1-этоксикарбонилэтилщавелевой кислоты
Этилоксалилхлорид (70 мкл, 630 мкмоль) добавляли по каплям к раствору сложного этилового эфира 3-[N-(4-бромбензил)гидразино]-2-гидроксипропионовой кислоты (200 мг, 630 мкмоль) и Et3N (220 мкл, 1,6 ммоль) в DCM (4,0 мл, 62,2 ммоль) при 0°C. Полученную смесь перемешивали в течение 15 минут при 0°C и в течение 15 минут при комнатной температуре. Добавляли воду (3 мл), слои разделяли, и водный слой экстрагировали DCM (2×2 мл). Слои DCM объединяли, сушили над MgSO4 и концентрировали с получением указанного в заголовке соединения (275 мг).
Получение 37
Сложный трет-бутиловый эфир N'-(4-бромбензил)гидразинкарбоновой кислоты

К перемешиваемому раствору трет-бутилкарбазата (50 г, 0,4 моль) в сухом THF (400 мл) по каплям добавляли раствор 4-бромбензальдегида (70 г, 0,4 моль) в сухом THF (200 мл). Смесь перемешивали при комнатной температуре в течение 2 часов и затем концентрировали в вакууме с получением соединения 1 в виде желтого твердого вещества (113,8 г). LC-MS: 243 [M-tBu+H]+.
К раствору соединения 1 (113,8 г, 0,4 моль) в сухом THF (1 л) порциями добавляли NaCNBH3 (36 г, 0,6 моль) при 0°C. По каплям добавляли AcOH (180 мл), и полученную смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду (2 л) и EtOAc (1,5 л), и pH водной фазы доводили до pH=7 насыщенным водным раствором Na2CO3. Органический слой отделяли, промывали насыщенным водным раствором NaCl и водой (200 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток обрабатывали MeOH (2 л) и 1N NaOH (1,5 л), и затем перемешивали при комнатной температуре в течение 2 часов. После удаления растворителя MeOH, осадок собирали фильтрованием с получением указанного в заголовке соединения в виде белого твердого вещества (112 г). LC-MS: 245 [M-tBu+H]+.
Получение 38
Сложный метиловый эфир (R)-3-[N'-трет-бутоксикарбонил-N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты

К раствору сложного трет-бутилового эфира N'-(4-бромбензил)гидразинкарбоновой кислоты (60 г, 0,2 моль) в 1,4-диоксане (1,5 мл) добавляли 5-хлор-2-фторфенилбороновую кислоту (38 г, 0,2 моль) и Pd(dppf)Cl2 (7,3 г). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 10 минут, и затем добавляли K2CO3 (55,2 г, 0,4 моль) в воде (240 мл). Полученную смесь перемешивали при 60°C в течение 3 часов и затем охлаждали до комнатной температуры и концентрировали в вакууме. Остаток экстрагировали EtOAc (3×300 мл). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали в вакууме. Продукт очищали колоночной хроматографией (PE:EtOAc=10:1-5:1) с получением соединения 1 в виде розового твердого вещества (56 г). LC-MS: 701 [2М+З]+.

К раствору соединения 1 (20 г, 57 ммоль) в изопропиловом спирте (250 мл) добавляли метил-(2R)-глицидат (8,7 г, 86 ммоль) в атмосфере азота. Смесь перемешивали при 85°C в течение 3 дней, затем охлаждали до комнатной температуры. Осажденное твердое вещество собирали фильтрованием с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (18,5 г). LC-MS: 397 [M-tBu+H]+.
Получение 39
Сложный этиловый эфир (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты

Раствор сложного метилового эфира (R)-3-[N'-трет-бутоксикарбонил-N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (20 г, 16 ммоль) в HCl/EtOH (1,1M, 200 мл) перемешивали в течение ночи и затем концентрировали в вакууме. Остаток диспергировали в EtOAc (2×40 мл), и осадок собирали фильтрованием с получением указанного в заголовке соединения в виде не совсем белой твердой соли HCl (8,8 г). LC-MS: 367 [M+H]+.
1H ЯМР (300 MГц, DMSO-d6) δ 1,05 (т, J=7,2 Гц, 3H), 3,05-3,03 (кв, J=7,2 Гц, 2H), 4,06-3,95 (м, 4H), 4,42 (ушир., 1H), 6,46 (ушир., 1H), 7,62-7,40 (м, 7H), 9,42 (с, 3H).
Получение 40
Сложный 2-оксо-2-фенилэтиловый эфир (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты
Сложный этиловый эфир 3-[N-(4-бромбензил)гидразино]-2-гидроксипропионовой кислоты (3,1 г, 9,6 ммоль) объединяли с ди-трет-бутилдикарбонатом (4,2 г, 19,2 ммоль) и DCM (92,4 мл, 1,4 моль). Добавляли DIPEA (5,0 мл, 28,8 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 24 часов. Смесь концентрировали, и остаток растворяли в DCM и очищали флэш-хроматографией (10-95% EtOAc в гексанах). Чистые фракции собирали и концентрировали с получением соединения 1 в виде белого порошка (4,0 г).

Соединение 1 (3,5 г, 8,4 ммоль) объединяли с 5-хлор-2-фторфенилбороновой кислотой (1,8 г, 10,1 ммоль) и K2CO3 (3,5 г, 25,2 ммоль) в EtOH (29,4 мл, 503 ммоль) и водой (7,6 мл, 419 ммоль). Полученную смесь помещали в атмосферу азота и затем добавляли SilicaCat DPP-Pd (0,28 ммоль/г загрузки; 3,0 г, 839 мкмоль). Смесь обрабатывали микроволновым излучением при 120°C в течение 15 минут. Смесь затем фильтровали и выпаривали при пониженном давлении. Неочищенный остаток очищали, используя флэш-хроматографию (10-90% EtOAc в гексанах) с получением соединения 2 (2,0 г).
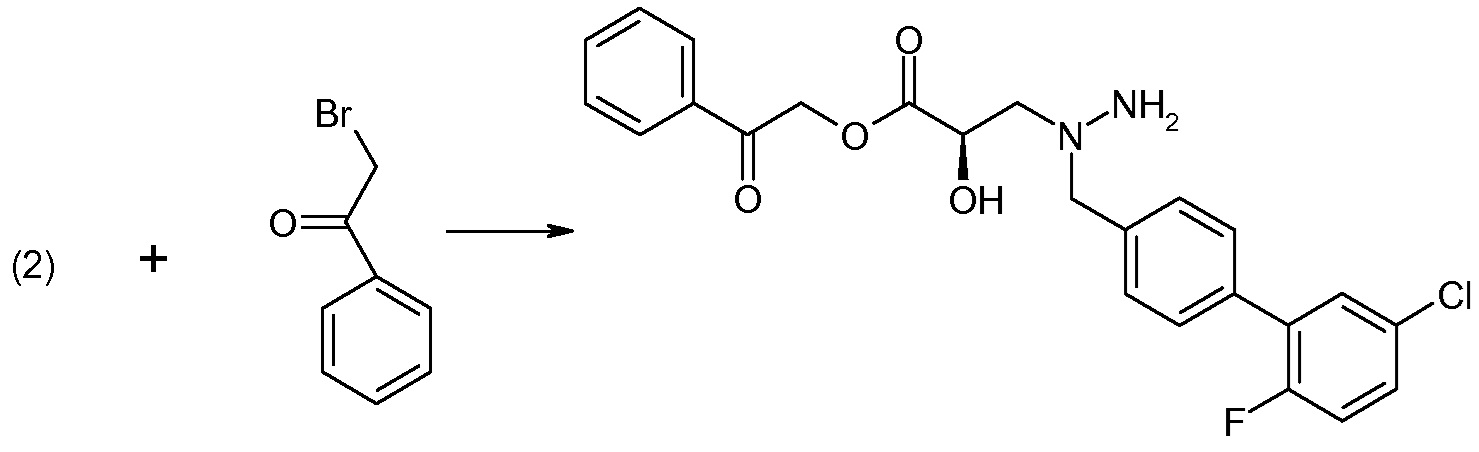
Соединение 2 (500 мг, 1 ммоль) объединяли с K2CO3 (315 мг, 2,3 ммоль) в DMF (5,3 мл, 68,4 ммоль). Затем добавляли 2-бромацетофенон (249 мг, 1,3 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 15 минут. Смесь затем очищали, используя флэш-хроматографию (50-100% EtOAc в гексанах). Этот очищенный материал (605 мг) затем растворяли в MeCN (3,6 мл, 68,4 ммоль). Затем добавляли раствор 4M HCl в 1,4-диоксане (1,4 мл, 5,7 ммоль), и полученную смесь перемешивали в течение 1 часа с получением указанного в заголовке соединения (245 мг).
Получение 41
(R)-3-[N'-трет-Бутоксиоксалил-N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино1-2-гидроксипропионовой кислоты

трет-Бутилоксалилхлорид получали добавлением оксалилхлорида (102 мкл, 1,2 ммоль) к раствору трет-бутилового спирта (35 мкл, 361 мкмоль) в эфире (632 мкл, 6,0 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 15 минут и затем концентрировали в вакууме с получением прозрачной бесцветной жидкости.
Сложный 2-оксо-2-фенилэтиловый эфир (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (55,0 мг, 120 мкмоль) растворяли в DCM (463 мкл, 7,2 ммоль). Добавляли трет-бутилоксалилхлорид, и полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме. Полученный остаток растворяли в AcOH (411 мкл, 7,2 ммоль). К смеси добавляли цинк (394 мг, 6,0 ммоль), которую затем перемешивали при комнатной температуре в течение 10 минут. Смесь фильтровали и очищали (колонка Interchim для обращенно-фазовой колонки) с получением указанного в заголовке соединения (25,0 мг).
Получение 42
Сложный 5-метил-2-оксо-[1,3]диоксол-4-илметиловый эфир (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты

Гидрат LiOH (3 г, 73 ммоль) в воде (60 мл) добавляли к сложному метиловому эфиру (R)-3-[N'-трет-бутоксикарбонил-N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (16,5 г, 36,5 ммоль) в MeOH (300 мл). Смесь перемешивали при комнатной температуре в течение 2 часов, и MeOH выпаривали в вакууме. pH смеси доводили до pH=5 1M водной HCl, и остаток экстрагировали EtOAc (2×300 мл). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали в вакууме с получением соединения 1 в виде белого твердого вещества (18 г). LC-MS: 383 [M-iBu+H].

К раствору соединения 1 (1,5 г, 3,42 ммоль), K2CO3 (0,95 г, 6,84 ммоль) и йодида калия (20 мг) в DMF (40 мл) добавляли 4-(бромметил)-5-метил-1,3-диоксол-2-он (0,8 г, 4,1 ммоль) в DMF (15 мл). Полученную смесь перемешивали в течение 4 часов при комнатной температуре. Добавляли насыщенный водный раствор NaCl (30 мл), и смесь экстрагировали EtOAc (2×50 мл). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток очищали колоночной хроматографией (гексаны/EtOAc=1:1) с получением соединения 2 в виде желтого твердого вещества (930 мг). LC-MS: 495 [M-t-Bu+H]+.

Соединение 2 (400 мг, 0,73 ммоль) растворяли в MeCN (20 мл) и охлаждали до 0°C. Добавляли по каплям N-триметилсилилаимидазол (290 мг, 1,46 ммоль), и полученную смесь перемешивали в течение 2 часов. Для гашения реакции добавляли MeOH (50 мл). Смесь промывали насыщенным водным раствором NaCl (2×50 мл) и экстрагировали DCM (2×80 мл). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали в вакууме. Продукт собирали с получением указанного в заголовке соединения в виде желтого твердого вещества (200 мг). LC-MS: 451 [M+H]+.
Получение 43
(R)-3-[N'-трет-бутоксиоксалил-N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовая кислота

К смеси сложного 2-оксо-2-фенилэтилового эфира (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (2,0 г, 4,4 ммоль) в DCM (10 мл) добавляли по каплям трет-бутил-2-хлор-2-оксоацетат (1,5 г, 8,8 ммоль) при 0°C в атмосфере азота. Затем по каплям добавляли DIPEA (1,15 г, 8,8 ммоль), и полученную смесь перемешивали в течение 5 минут при 0°C. Растворитель удаляли выпариванием, и остаток очищали колоночной хроматографией (PE:EtOAc=2:1) с получением соединения 1 в виде желтой жидкости (2,0 г). LC-MS: 585 [M+H]+.

Смесь соединения 1 (2,0 г, 3,4 ммоль) и Zn (15,5 г, 240 ммоль) в AcOH (15 мл) перемешивали в течение 1 часа при комнатной температуре, затем фильтровали. К фильтрату добавляли воду (30 мл), и смесь экстрагировали EtOAc (3×40 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (2×50 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Остаток очищали колоночной хроматографией (DCM/MeOH=10:1) с получением указанного в заголовке соединения в виде желтой жидкости (1,4 г). LC-MS: 467 [M+H]+.
ПРИМЕР 1
(R)-5-Бифенил-4-ил-2-гидрокси-4-(оксалиламино)пентановая кислота

Сложный этиловый эфир (R)-4-амино-5-бифенил-4-ил-2-гидроксипентановой кислоты (соль HCl; 47 мг, 0,2 ммоль) и этилоксалилхлорид (18,4 мкл, 1,1 экв.) объединяли с DIPEA (52,2 мкл, 0,3 ммоль) в DMF (0,3 мл)/DCM (0,3 мл). Смесь перемешивали при комнатной температуре до завершения реакции. Растворитель удаляли, и остаток растворяли в EtOH (750 мкл) и 1M водном NaOH (750 мкл) и перемешивали при комнатной температуре в течение ночи. Растворитель удаляли, и остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (28,2 мг, чистота 100%). MS m/z [M+H]+ рассчитано для C19H19NO6, 358,12; найдено 358,0.
ПРИМЕР 2
A. (2R,4R)-5-(3'-Хлорбифенил-4-ил-2-гидрокси-4-(оксалиламино)пентановая кислота

Раствор этилоксалилхлорида (70,7 мкл, 0,6 ммоль) в DIPEA (165 мкл, 0,9 ммоль) добавляли к раствору сложного этилового эфира (2R,4R)-4-амино-5-(4-бромфенил)-2-гидроксипентановой кислоты (100 мг, 0,3 ммоль) и DCM (0,7 мл), и полученную смесь перемешивали при комнатной температуре в течение 10 минут с последующим выпариванием растворителя при пониженном давлении. Добавляли 3-хлорфенилбороновую кислоту, сложный пинаколовый эфир (112 мг, 468 мкмоль), K2CO3 (97 мг, 702 мкмоль), EtOH (2 мл) и воду (0,6 мл), с последующим добавлением SilicaCat®Pd(0) (0,09 ммоль/г загрузки, 260 мг, 23,4 мкмоль). Смесь нагревали при 120°C в течение 20 минут. Реакционную смесь концентрировали и добавляли 10M водный NaOH (316 мкл) и THF (4,0 мл) с 1 каплей MeOH. Полученную смесь перемешивали при комнатной температуре в течение 1 часа. Остаток растворяли в AcOH и очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (9 мг, чистота 95%). MS m/z [M+H]+ рассчитано для C19H18ClNO6, 392,08; найдено 392,4.
B. Сложный этиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
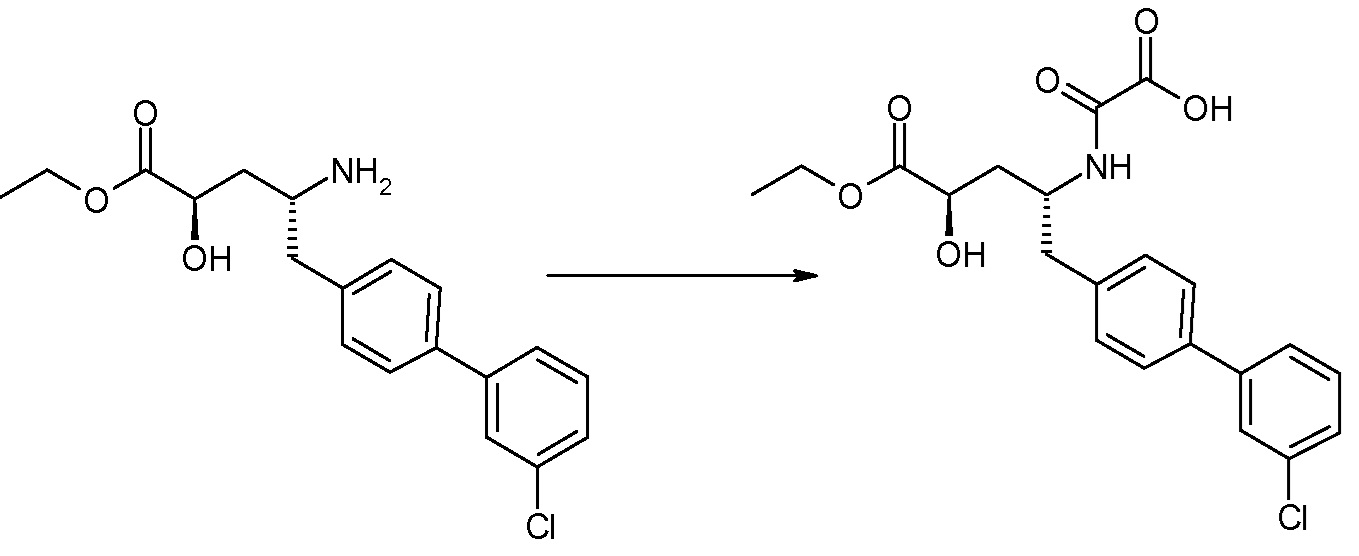
Оксалилхлорид (54,5 мкл, 0,6 ммоль) добавляли к раствору трет-бутилового спирта (56,0 мкл) в эфире (1,0 мл), и смесь перемешивали в течение 1 часа при комнатной температуре. Смесь концентрировали в вакууме, и раствор сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (67,9 мг, 0,2 ммоль) и DIPEA (102 мкл, 0,6 ммоль) в DCM (1,0 мл) добавляли к полученному прозрачному бесцветному жидкому остатку. Полученную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали в вакууме с получением прозрачной желтой жидкости. 1:1 TFA/DCM (1,6 мл) добавляли к неочищенной жидкости, и реакционную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали в вакууме с получением прозрачной желтой жидкости. Неочищенную жидкость очищали обращенно-фазовой препаративной ВЭЖХ (40-90% MeCN/H2O) с получением указанного в заголовке соединения (25,0 мг, чистота 95%) в виде белого твердого вещества. MS m/z [M+H]+ рассчитано для C21H22ClNO6, 420,11; найдено 420,1.
C. Сложный этиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксипентановой кислоты

Раствор этилоксалилхлорида (24,6 мкл, 0,2 ммоль) в DCM (0,4 мл) добавляли к раствору сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (69,6 мг, 0,2 ммоль) и Et3N (69,7 мкл, 0,5 ммоль) в DCM (1,0 мл) при 0°C в течение периода 10 минут. Полученную смесь перемешивали при 0°C в течение 30 минут, и затем в течение 15 минут при комнатной температуре. Добавляли воду (2 мл), слои разделяли, и водный слой экстрагировали DCM (2×2 мл). Слои DCM объединяли, сушили над Na2SO4 и концентрировали с получением прозрачной желтой жидкости. Неочищенную жидкость очищали флэш-хроматографией (4 г колонка, 16 мл/мин с использованием 35% EtOAc/гексанов (2 мин), 35-50% (1 мин), 50% (4 мин), 50-70% (1 мин) и 70% EtOAc/гексанов (3 мин)) с получением указанного в заголовке соединения (63,9 мг, чистота 90%) в виде прозрачной бесцветной жидкости, которая застывала при стоянии в белое твердое вещество. MS m/z [M+H]+ рассчитано для C23H26ClNO6, 448,14; найдено 448,2.
D. Сложный бутиловый эфир (2R,4R)-4-(бутоксиоксалиламино)-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты
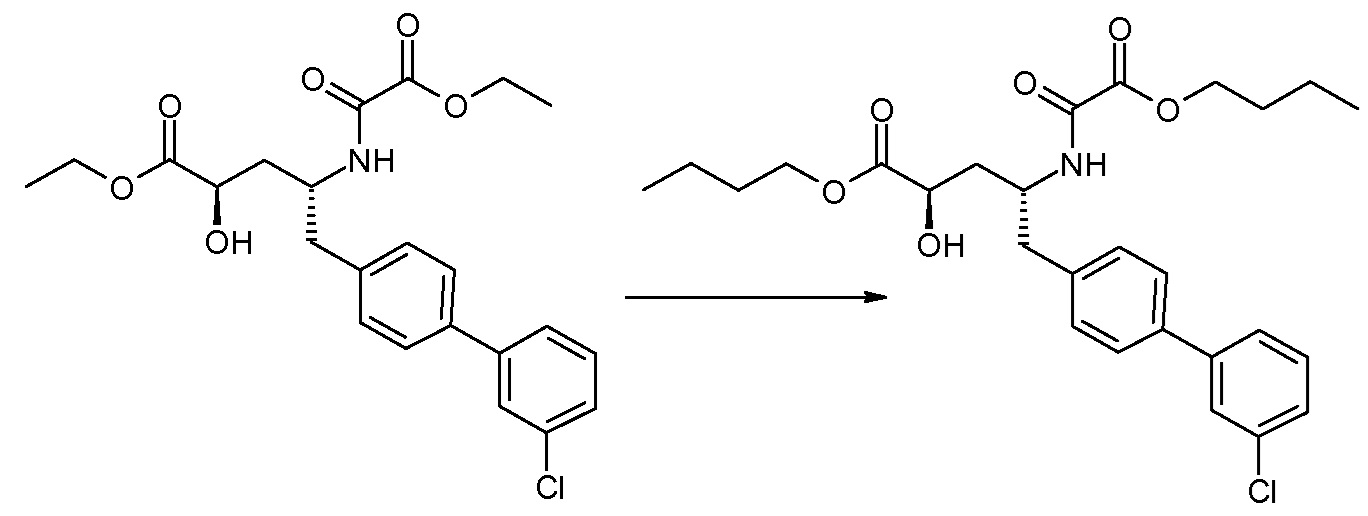
Моногидрат п-толуолсульфоновой кислоты (849 мкг, 4 мкмоль) добавляли к раствору сложного этилового эфира (2R,4R)-5-(3'-хлорбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксипентановой кислоты (20,0 мг, 45 мкмоль) в 1-бутаноле (0,5 мл). Реакционную смесь перемешивали при 80°C в течение 14 часов, при 90°C в течение 4 часов, и затем давали возможность охладиться до комнатной температуры. Добавляли насыщенный водный раствор NaHCO3 (2 мл), и водный слой экстрагировали DCM (3×2 мл). Слои DCM объединяли, сушили над Na2SO4 и концентрировали в вакууме с получением прозрачной бесцветной жидкости. Неочищенную жидкость очищали флэш-хроматографией (4 г колонка, 40% EtOAc/гексаны) с получением указанного в заголовке соединения (18,1 мг, чистота 99%) в виде прозрачной бесцветной жидкости. MS m/z [M+H]+ рассчитано для C27H34ClNO6, 504,21; найдено 504,2.
E. Сложный 5-метил-2-оксо-[1,3]диоксол-4-илметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-[(5-метил-2-оксо-[1,3]диоксол-4-илметоксиоксалил)амино]пентановой кислоты

Оксалилхлорид (22,4 мкл, 264 мкмоль) добавляли к раствору 4-гидроксиметил-5-метил-[1,3]диоксол-2-она (29,1 мг, 224 мкмоль) в эфире (1,5 мл), и смесь перемешивали при комнатной температуре в течение 2 часов. Простой эфир удаляли в вакууме, и остаток растворяли в DMF (1,5 мл). Полученный раствор добавляли к раствору (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (65,0 мг, 203 мкмоль) и NaHCO3 (51,2 мг) при 0°C. Полученную смесь перемешивали при комнатной температуре в течение 3 часов, затем концентрировали в вакууме. Затем остаток очищали обращенно-фазовой препаративной ВЭЖХ (30%-90% MeCN/H2O) с получением соединения 1 (19,1 мг) в виде белого твердого вещества.

1-Гидроксибензотриазол (7,7 мг, 56,8 мкмоль) и N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорид (10,9 мг, 56,8 мкмоль) добавляли к раствору соединения 1 (19,1 мг, 37,9 мкмоль) в DCM (1,0 мл), и смесь перемешивали при комнатной температуре в течение 10 минут. Добавляли 4-гидроксиметил-5-метил-[1,3]диоксол-2-он (14,8 мг, 114 мкмоль) и 4-метилморфолин (7,7 мг, 75,8 мкмоль), и полученную смесь перемешивали при комнатной температуре в течение 6 часов. Добавляли воду, и смесь экстрагировали DCM (3×1,5 мл). Слои DCM объединяли, сушили над Na2SO4 и концентрировали с получением желтой жидкости. Неочищенную жидкость очищали обращенно-фазовой препартивной ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (5,1 мг). MS m/z [M+H]+ рассчитано для C29H26ClNO12, 616,11; найдено 616,1.
F. (2R,4R)-5-(3'-Хлорбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксипентановая кислота
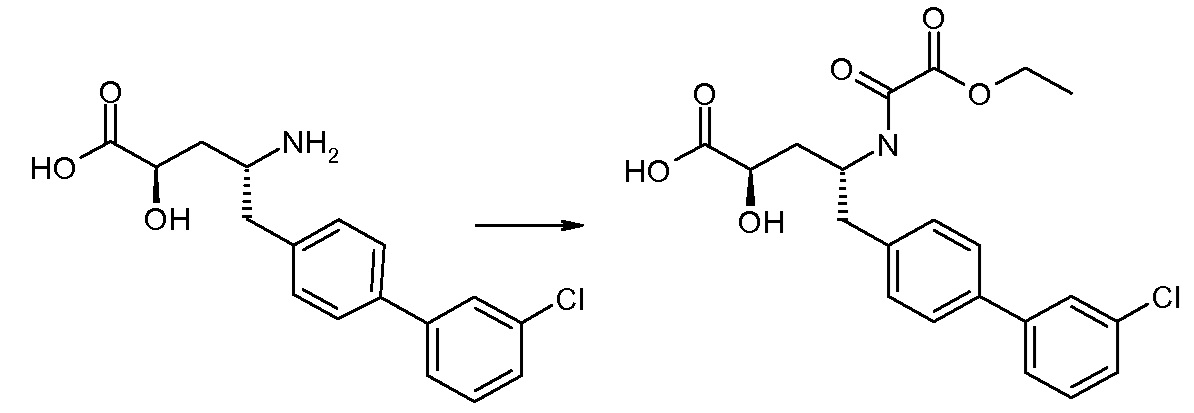
Этилоксалилхлорид (46,1 мкл, 0,4 ммоль) добавляли к раствору (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (120 мг, 0,4 ммоль) и Et3N (157 мкл, 1,1 ммоль) в DMF (2,0 мл, 25,8 ммоль) при 0°C, и полученную смесь перемешивали при комнатной температуре в течение 20 минут. Добавляли дополнительный этилоксалилхлорид (30 мкл), и смесь перемешивали еще 10 минут. Добавляли воду (2 мл), и смесь экстрагировали DCM (3×2 мл). Экстракты объединяли, сушили над Na2SO4 и концентрировали с получением желтой жидкости. Неочищенную жидкость очищали (хроматография на колонке C-18, 20 г; 40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (28,5 мг) в виде белого твердого вещества. MS m/z [M+H]+ рассчитано для C21H22ClNO6, 420,11; найдено 420,2.
G. (2R,4R)-5-(3'-Хлорбифенил-4-ил)-2-гидрокси-4-(изопропоксиоксалиламино)пентановая кислота

Сложный изопропиловый эфир хлороксоуксусной кислоты (62,1 мг, 413 мкмоль; ~53 мкл) добавляли по каплям к раствору (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (100 мг, 313 мкмоль) и Et3N (157 мкл, 1,1 ммоль) в DMF (2,0 мл, 25,8 ммоль) при 0°C, и полученную смесь перемешивали при комнатной температуре в течение 10 минут. Добавляли дополнительный этилоксалилхлорид (50 мкл), и смесь перемешивали еще 10 минут. Добавляли насыщенный водный раствор NaHCO3 (5 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь экстрагировали DCM (3×3 мл), экстракты объединяли, сушили над Na2SO4 и концентрировали с получением желтой жидкости. Неочищенную жидкость очищали (препаративная ВЭЖХ на колонке C-18, маленькая колонка, с использованием 40-95% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (53,0 мг) в виде белого твердого вещества. MS m/z [M+H]+ рассчитано для C22H24ClNO6, 434,13; найдено 434,1.
H. (2R,4R)-5-(3'-Хлорбифенил-4-ил)-2-гидрокси-4-(изобутоксиоксалиламино)пентановая кислота

1,0M водную HCl (2,5 мл, 2,5 ммоль) добавляли к сложному этиловому эфиру (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (100 мг, 287 мкмоль), и полученную смесь перемешивали при 100°C в течение 1 часа. Смесь концентрировали с получением (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты.
Сложный изобутиловый эфир хлороксоуксусной кислоты (99,4 мг, 604 мкмоль) добавляли по каплям к раствору (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты и Et3N (160 мкл, 1,2 ммоль) в DMF (2,0 мл, 25,8 ммоль) при 0°C и перемешивали при комнатной температуре в течение 10 минут. Добавляли насыщенный водный раствор NaHCO3 (5 мл), и смесь перемешивали при комнатной температуре в течение 2 часов. Смесь экстрагировали DCM (3×5 мл), экстракты DCM объединяли, промывали насыщенным водным раствором NaCl, сушили над Na2SO4 и концентрировали с получением белого твердого остатка. Неочищенное твердое вещество очищали препаративной ВЭЖХ на колонке C18 (маленькая колонка; 40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (40,0 мг) в виде белого твердого вещества. MS m/z [M+H]+ рассчитано для C23H26ClNO6, 448,14; найдено 448,1.
I. Сложный изобутиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
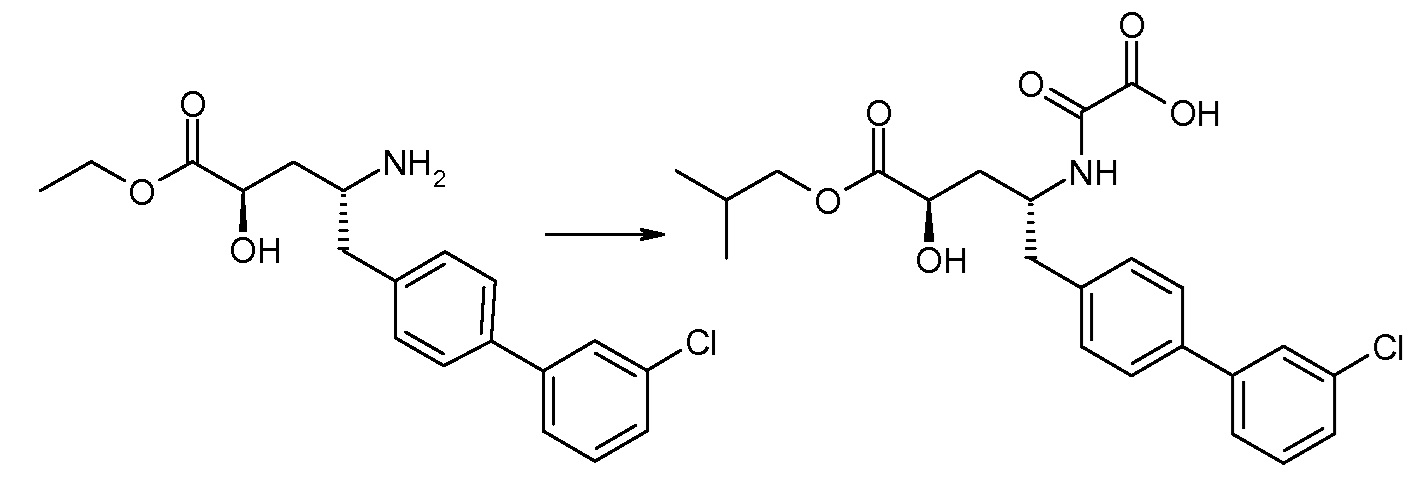
4,0M HCl в 1,4-диоксане (216 мкл, 862 мкмоль) добавляли к суспензии сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (75,0 мг, 216 мкмоль) в изобутиловом спирте (0,5 мл, 5,4 ммоль), и полученную смесь перемешивали при 60°C в течение 2 часов. Смесь затем концентрировали в вакууме с получением белого твердого вещества. Белое твердое вещество растворяли в DCM и DIPEA (113 мкл, 647 мкмоль) и затем добавляли к смеси с последующим добавлением по каплям ~0,22 мл 1M раствора трет-бутилоксалилхлорида в DCM (0,2 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением желтой жидкости. Раствор TFA/DCM (1:1, 1,3 мл, 7,7 ммоль) добавляли к желтой жидкости, и полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (хроматография ВЭЖХ препаративного масштаба на колонке C18, 40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (70,5 мг, чистота 99%) в виде белого твердого вещества. MS m/z [M+H]+ рассчитано для C23H26ClNO6, 448,14; найдено 448,1.
J. Сложный изопропиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты

4,0M HCl в 1,4-диоксане (216 мкл, 862 мкмоль) добавляли к суспензии сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (75,0 мг, 216 мкмоль) в изопропиловом спирте (0,5 мл, 6,5 ммоль), и полученную смесь перемешивали при 60°C в течение ночи. Смесь затем концентрировали в вакууме с получением белого твердого вещества. Белое твердое вещество растворяли в DCM, и затем к смеси добавляли DIPEA (113 мкл, 647 мкмоль) с последующим добавлением по каплям ~0,22 мл 1M раствора трет-бутилоксалилхлорида в DCM (0,2 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением желтой жидкости. Раствор TFA/DCM (1:1, 1,3 мл, 7,7 ммоль) добавляли к желтой жидкости, и полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (хроматография ВЭЖХ препаративного масштаба на колонке C18, 40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (62,8 мг, чистота 98%) в виде белого твердого вещества. MS m/z [M+H]+ рассчитано для C22H24ClNO6, 434,13; найдено 434,1.
K. Сложный гептиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
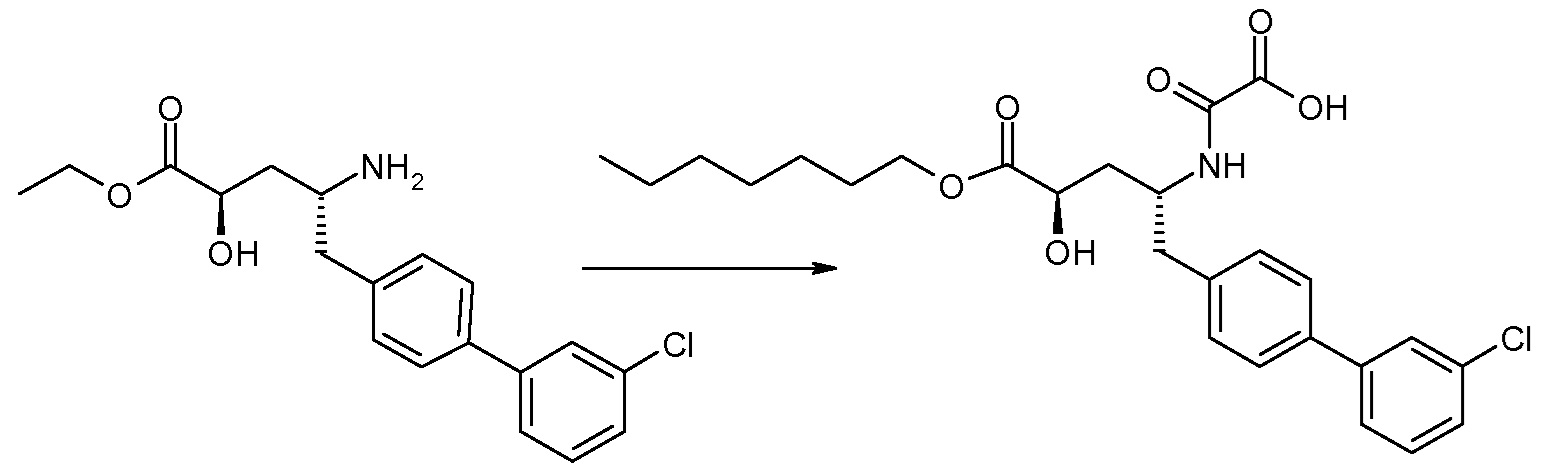
4,0M HCl в 1,4-диоксане (216 мкл, 862 мкмоль) добавляли к суспензии сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (75,0 мг, 216 мкмоль) в 1-гептаноле (250 мкл, 1,8 ммоль), и полученную смесь перемешивали при 60°C в течение 2 часов. Смесь затем концентрировали в вакууме с получением белого твердого вещества, которое очищали (колонка Interchim для обращенно-фазовой хроматографии; градиент 30-90% MeCN в воде с 0,5% TFA). Очищенное белое твердое вещество растворяли в DCM, и затем к смеси добавляли DIPEA (113 мкл, 647 мкмоль) с последующим добавлением по каплям ~0,22 мл 1M раствора трет-бутилоксалилхлорида в DCM (0,2 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением желтой жидкости. Раствор TFA/DCM (1:1, 1,3 мл, 7,7 ммоль) добавляли к желтой жидкости, и полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (ВЭЖХ в препаративном масштабе на колонке C18, 40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (43,3 мг, чистота 99%) в виде белого твердого вещества. MS m/z [M+H]+ рассчитано для C26H32ClNO6, 490,19; найдено 490,2.
L. Сложный 3,3,3-трифторпропиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты

4,0M HCl в 1,4-диоксане (287 мкл, 1,2 ммоль) добавляли к суспензии сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50,0 мг, 144 мкмоль) в 3,3,3-трифторпропан-1-оле (492 мг, 4,3 ммоль), и полученную смесь перемешивали при 80°C в течение 12 часов. Смесь затем концентрировали в вакууме с получением белого твердого вещества, которое растворяли в DCM (1,0 мл) и ~0,2 мл 1M раствора трет-бутилоксалилхлорида в DCM (0,2 ммоль). Затем по каплям добавляли DIPEA (75,1 мкл, 431 мкмоль), и полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением желтой жидкости. Раствор TFA/DCM (1:1, 1,3 мл, 7,7 ммоль) добавляли к желтой жидкости, и полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (ВЭЖХ в препаративном масштабе на колонке C18, 40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (44,9 мг, чистота 99%) в виде белого твердого вещества. MS m/z [M+H рассчитано для C22H21ClF3NO6, 488,10; найдено 488,1.
M. Сложный 2,2,2-трифторэтиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
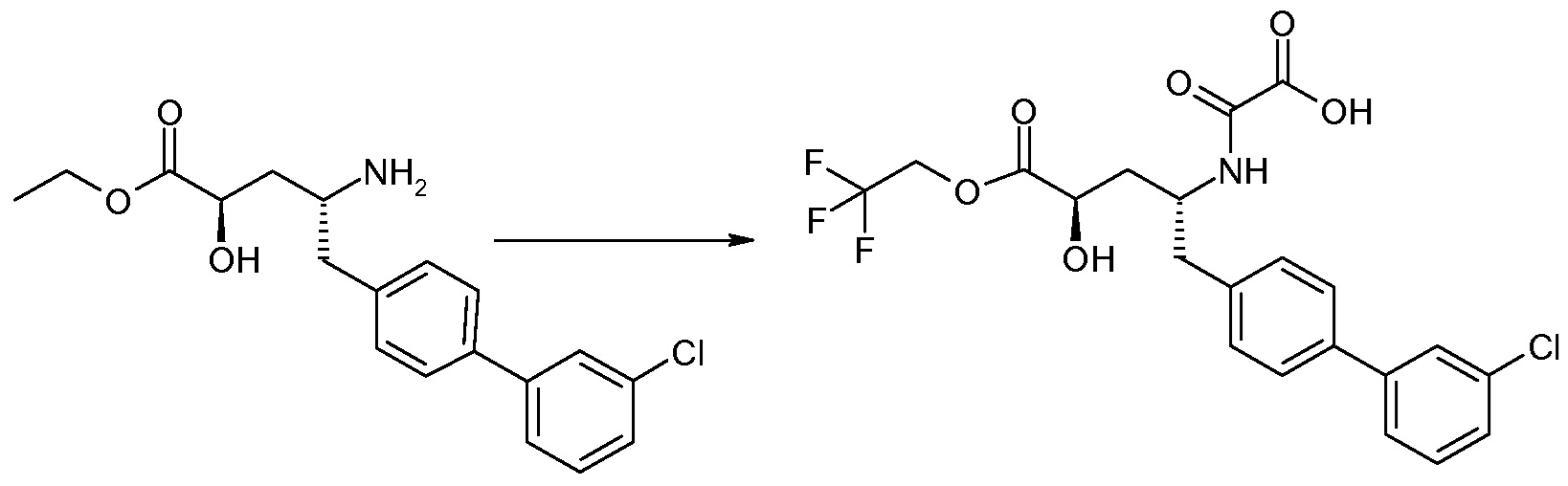
4,0M HCl в 1,4-диоксане (287 мкл, 1,2 ммоль) добавляли к суспензии сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50,0 мг, 144 мкмоль) в 2,2,2-трифторэтаноле (0,5 мл, 6,9 ммоль), и полученную смесь перемешивали при 110°C в течение 12 часов. Смесь затем концентрировали в вакууме с получением белого твердого вещества, которое растворяли в DCM (1,0 мл) и ~0,2 мл 1M раствора трет-бутилоксалилхлорида в DCM (0,2 ммоль). Затем по каплям добавляли DIPEA (75,1 мкл, 431 мкмоль), и полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением желтой жидкости. Раствор TFA/DCM (1:1, 1,3 мл, 7,7 ммоль) добавляли к желтой жидкости, и полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (ВЭЖХ в препаративном масштабе на колонке C18, 40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (22,5 мг, чистота 98%) в виде белого твердого вещества. MS m/z [M+H]+ рассчитано для C21H19ClF3NO6, 474,09; найдено 474,1.
N. (2R,4R)-5-(3'-Хлорбифенил-4-ил)-2-гидрокси-4-[(3,3,3-трифторпропоксиоксалил)амино]пентановая кислота

Бензиловый спирт (13,0 мл, 126 ммоль) добавляли к сложному этиловому эфиру (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (1,9 г, 5,3 ммоль) с последующим добавлением 4,0M HCl в 1,4-диоксане (5,3 мл, 21,3 ммоль), и смесь перемешивали при 60°C в течение 1 часа. Смесь очищали (колонка Interchim для обращенно-фазовой хроматографии; 30-90% MeCN в воде с 0,05% TFA) с получением сложного бензилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (2,2 г) в виде белого твердого вещества, (выпаривали в вакууме с водой (4×300 мл) для удаления избытка бензилового спирта).
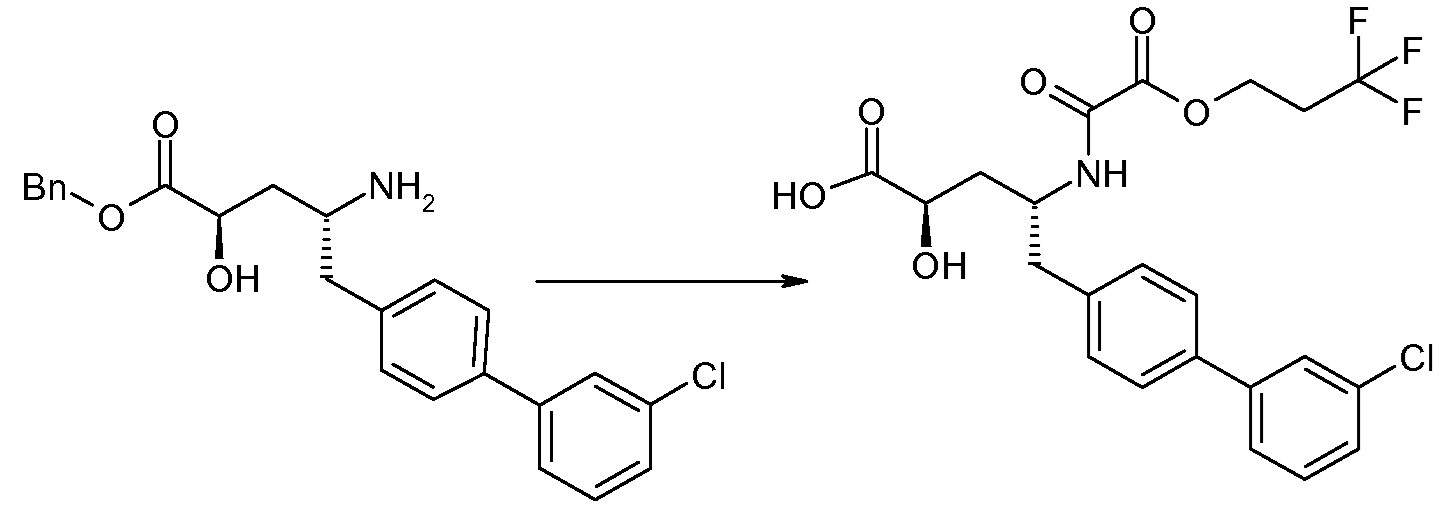
3,3,3-Трифторпропилоксалилхлорид получали добавлением оксалилхлорида (51,6 мкл, 610 мкмоль) к раствору 3,3,3-трифторпропан-1-ола (62,6 мг, 549 мкмоль) в эфире (500 мкл, 4,8 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали в вакууме с получением прозрачной бесцветной жидкости. ~1M раствор оксалилхлорида получали растворением полученной жидкости в ~0,61 мл DCM.
Раствор 3,3,3-трифторпропилоксалилхлорида (~140 мкл) добавляли к раствору сложного бензилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50,0 мг, 122 мкмоль) в DCM (1,0 мл) при 0°C, и смесь перемешивали при 0°C в течение 15 минут. Добавляли насыщенный водный раствор NaHCO3 (1 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Слои разделяли, и водный слой экстрагировали DCM (2×2 мл). Слои DCM объединяли, сушили над Na2SO4 и концентрировали с получением прозрачной желтой жидкости. К раствору желтой жидкости в DCM и THF (1,0 мл) добавляли 10% Pd/C, 50% влажности (0,45 ммоль/г загрузки; 13,6 мг, 6,1 мкмоль), и смесь перемешивали в атмосфере водорода в течение 30 минут. Смесь фильтровали, и фильтрат концентрировали с получением прозрачной желтой жидкости. Неочищенную жидкость очищали препаративной ВЭЖХ (40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (16,5 мг, чистота 99%) в виде белого твердого вещества. MS m/z [M+H]+ рассчитано для C22H21ClF3NO6, 488,10; найдено 4880.
O. Сложный 2,2,3,3,3-пентафторпропиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты

~1M раствор трет-бутилоксалилхлорида (~0,2 мл) добавляли к раствору сложного 2,2,3,3,3-пентафторпропилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50,0 мг, 111 мкмоль) в DCM (1,0 мл) при 0°C с последующим добавлением по каплям в течение 10 минут DIPEA (21,2 мкл, 122 мкмоль). Смесь перемешивали при 0°C в течение 15 минут. Добавляли насыщенный водный раствор NaHCO3 (5 мл), и смесь экстрагировали DCM (3×5 мл). Экстракты DCM объединяли, сушили над Na2SO4 и концентрировали с получением прозрачной бесцветной жидкости. Неочищенную жидкость очищали флэш-хроматографией (50% EtOAc/гексаны) с получением прозрачной бесцветной жидкости. 1:1 TFA/DCM (1,0 мл) добавляли к раствору бесцветной жидкости и перемешивали при комнатной температуре в течение 30 минут. Смесь концентрировали в вакууме с получением прозрачной желтой жидкости. Неочищенную жидкость очищали препаративной ВЭЖХ (40%-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (21,6 мг, чистота 98%) в виде белого твердого вещества. MS m/z [M+H]+ рассчитано для C22H19ClF5NO6, 524,08; найдено 524,0.
P. Сложный 5-метил-2-оксо-[1,3]диоксол-4-илметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты

~1M раствор трет-бутилоксалилхлорида (160 мкл) добавляли к раствору сложного 5-метил-2-оксо[1,3]диоксол-4-илметилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50,0 мг, 116 мкмоль) в DCM (1,00 мл, 15,6 ммоль) при 0°C с последующим добавлением по каплям в течение 10 минут N,N-диизопропиламина (17,8 мкл, 127 мкмоль). Полученную смесь перемешивали при 0°C в течение 15 минут, затем концентрировали в вакууме. 1:1 TFA/DCM (1,0 мл, 6,2 ммоль) добавляли к остатку, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Смесь концентрировали в вакууме с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (хроматография в препаративном масштабе на колонке C18, маленькая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого вещества (9,6 мг). MS m/z [M+H]+ рассчитано для C24H22ClNO9, 504,10; найдено 504,0.
Q. Сложный бутирилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты

~1M раствор трет-бутилоксалилхлорида (160 мкл) добавляли к раствору сложного бутирилоксиметилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (48,6 мг, 116 мкмоль) в DCM (1,00 мл, 15,6 ммоль) при 0°C с последующим добавлением по каплям в течение 10 минут N,N-диизопропиламина (17,8 мкл, 127 мкмоль). Полученную смесь перемешивали при 0°C в течение 15 минут, затем концентрировали в вакууме. 1:1 TFA/DCM (1,0 мл, 6,2 ммоль) добавляли к остатку, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Смесь концентрировали в вакууме с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (хроматография в препаративном масштабе на колонке C18, маленькая колонка, с использованием 30-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения в виде белого твердого вещества (10,2 мг). MS m/z [M+H]+ рассчитано для C24H26ClNO8, 492,13; найдено 492,0.
R. Сложный ацетоксиметиловый эфир (2R,4R)-5-(3'-хлор-бифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
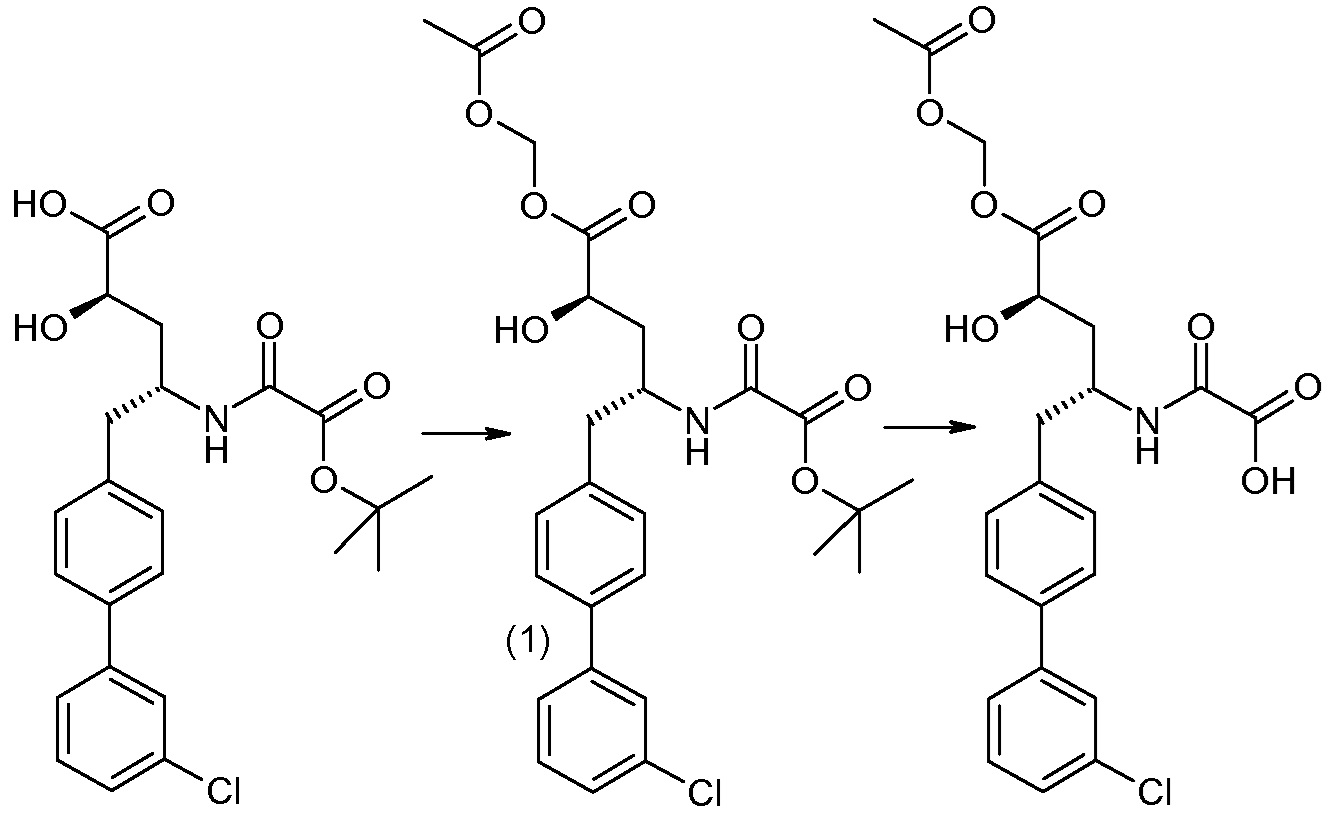
К раствору (2R,4R)-4-(трет-бутоксиоксалиламино)-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (200 мг, 450 мкмоль) и бромметилацетата (97 мг, 0,9 ммоль) в DMF (2 мл) добавляли 2,6-лутидин (144 мг, 1,3 ммоль) и NaI (67 мг, 450 мкмоль). После перемешивания при комнатной температуре в течение 24 часов смесь разбавляли водой (20 мл) и экстрагировали EtOAc (2×20 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (2×70 мл), сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали препаративной TLC (PE:EtOAc=2:1) с получением соединения 1 (100 мг) в виде желтого твердого вещества. LC-MS: 542 [M+Na]+.
К раствору соединения 1 (100 мг, 0,2 ммоль) в DCM (5 мл) добавляли TFA (2 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 2 часов, растворитель удаляли, и остаток дополнительно очищали препаративной TLC (DCM:MeOH=8:1) с получением указанного в заголовке соединения в виде белого твердого вещества (10 мг). LC-MS: 464 [M+H]+.
1H ЯМР (400 MГц, MeOD) δ 7,61 (с, 1H), 7,55 (д, J=8,0 Гц, 3H), 7,42 (т, J=7,8 Гц, 1H), 7,34 (д, J=8,1 Гц, 3H), 5,78 (с, 2H), 4,40 (с, 1H), 4,31 (т, J=5,9 Гц, 1H), 2,94 (ддд, J=22,0, 13,8, 7,2 Гц, 2H), 2,09 (м, 5H).
S. Сложный этоксикарбонилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
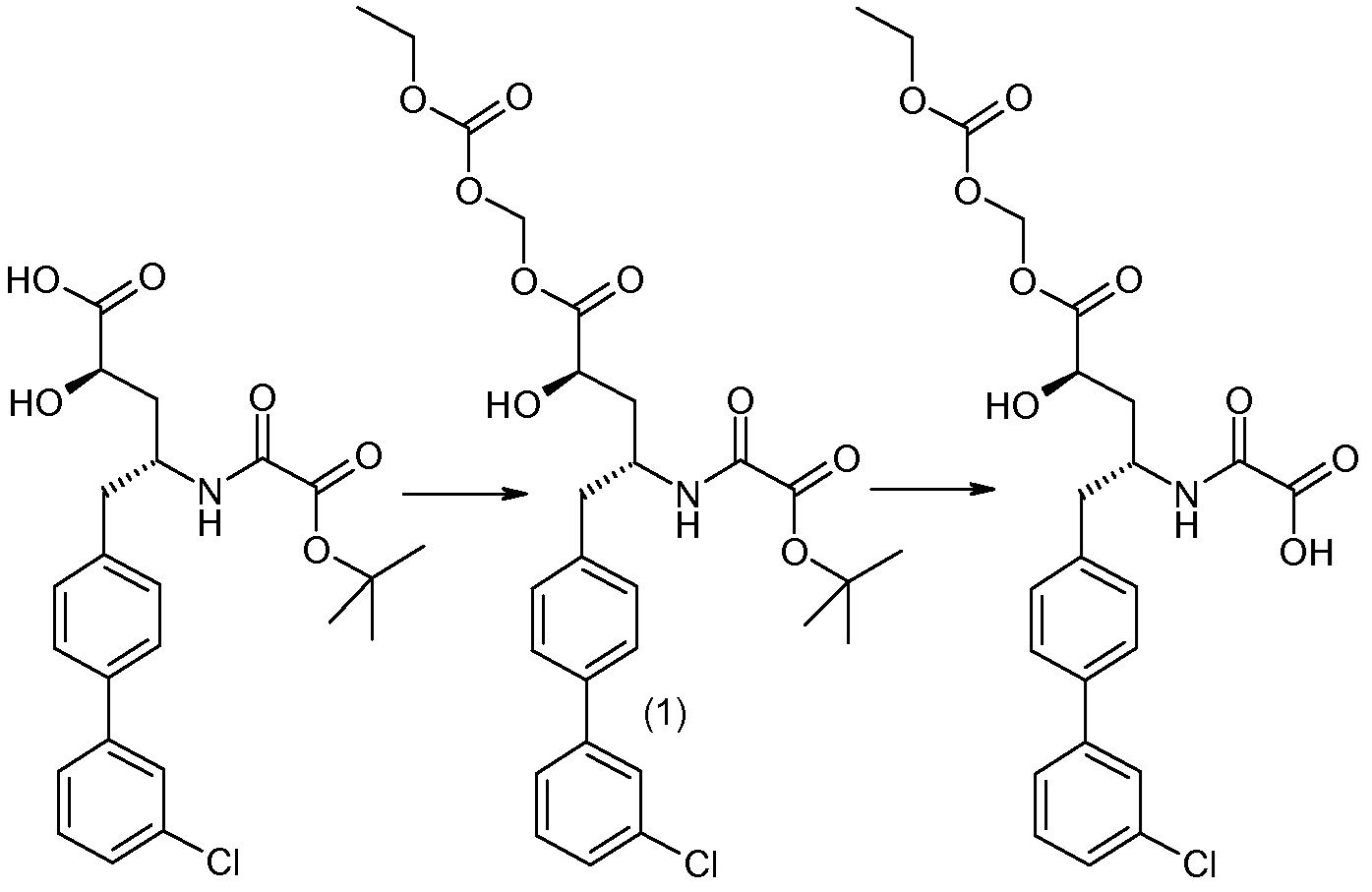
К раствору (2R,4R)-4-(трет-бутоксиоксалиламино)-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (100 мг, 220 мкмоль) и хлорметилэтилкарбоната (61 мг, 440 мкмоль) в DMF (3 мл) добавляли 2,6-лутидин (72 мг, 660 мкмоль) и NaI (33 мг, 220 мкмоль). После перемешивания при комнатной температуре в течение 24 часов смесь разбавляли водой (20 мл) и экстрагировали EtOAc (2×20 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (2×70 мл), сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали препаративной TLC (PE:EtOAc=2:1) с получением соединения 1 (40 мг) в виде желтого твердого вещества. LC-MS: 572 [M+Na]+.
К раствору соединения 1 (40 мг, 70 мкмоль) в DCM (3 мл) добавляли TFA (1 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 2 часов, растворитель удаляли, и остаток дополнительно очищали препаративной TLC (DCM:MeOH=8:1) с получением указанного в заголовке соединения в виде белого твердого вещества (18 мг). LC-MS: 494 [M+H]+.
1H ЯМР (400 MГц, MeOD) δ 7,55 (м, 4H), 7,38 (м, 4H), 5,80 (д, J=18,6 Гц, 2H), 4,34 (с, 2H), 4,21 (дд, J=14,3, 7,1 Гц, 2H), 2,95 (м, 2H), 2,07 (д, J=28,0 Гц, 2H), 1,29 (дд, J=12,6, 5,5 Гц, 3H).
MS m/z [M+H]+ рассчитано для C23H24ClNO9, 494,11; найдено 494.
T. (2R,4R)-5-(3'-Хлорбифенил-4-ил)-2-гидрокси-4-[(2-метоксиэтоксиоксалил)амино]пентановая кислота

DIPEA (64 мкл, 366 мкмоль) добавляли к раствору сложного бензилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50,0 мг, 122 мкмоль) в DCM (3 мл) с последующим добавлением по каплям 1,0M раствора сложного 2-метоксиэтилового эфира хлороксоуксусной кислоты (22 мг, 134 мкмоль) в DCM. Полученную смесь перемешивали при комнатной температуре в течение 30 минут, затем концентрировали с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (хроматографическая колонка Interchim C18, 20 г, 340-90% MeCN в воде с 0,05% TFA). THF (3 мл) добавляли к очищенному материалу, с последующим добавлением палладированного угля (10 масс.% на угле, влага 50 г, 12,9 мг, 12 мкмоль), и смесь перемешивали в атмосфере азота в течение 30 минут. Смесь фильтровали и концентрировали в вакууме, и остаток растворяли в AcOH (0,5 мл) и очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (9,8 мг). MS m/z [M+H]+ рассчитано для C22H24ClNO7, 450,12; найдено 450,2.
U. (2R,4R)-5-(3'-Хлорбифенил-4-ил)-2-гидрокси-4-[(2-феноксиэтоксиоксалил)амино]пентановая кислота

DIPEA (64 мкл, 366 мкмоль) добавляли к раствору сложного бензилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50,0 мг, 122 мкмоль) в DCM (3 мл) с последующим добавлением по каплям 1,0M раствора сложного 2- феноксиэтилового эфира хлороксоуксусной кислоты (31 мг, 134 мкмоль) в DCM. Полученную смесь перемешивали при комнатной температуре в течение 30 минут, затем концентрировали с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (хроматографическая колонка Interchim C18, 20 г, 340-90% MeCN в воде с 0,05% TFA). THF (3 мл) добавляли к очищенному материалу, с последующим добавлением палладированного угля (10 масс.% на угле, влага 50 г, 12,9 мг, 12 мкмоль), и смесь перемешивали в атмосфере азота в течение 30 минут. Смесь фильтровали и концентрировали в вакууме, и остаток растворяли в AcOH (0,5 мл) и очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (3,5 мг). MS m/z [M+H]+ рассчитано для C27H26ClNO7, 512,14; найдено 512,2.
V. (2R,4R)-5-(3'-Хлорбифенил-4-ил)-4-[(3-этоксипропоксиоксалил)амино]-2-гидроксипентановая кислота

DIPEA (64 мкл, 366 мкмоль) добавляли к раствору сложного бензилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50,0 мг, 122 мкмоль) в DCM (3 мл) с последующим добавлением по каплям 1,0M раствора сложного 3-этоксипропилового эфира хлороксоуксусной кислоты (26 г, 134 мкмоль) в DCM. Полученную смесь перемешивали при комнатной температуре в течение 30 минут, затем концентрировали с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (хроматографическая колонка Interchim C18, 20 г, 340-90% MeCN в воде с 0,05% TFA). THF (3 мл) добавляли к очищенному материалу, с последующим добавлением палладированного угля (10 масс.% на угле, влага 50 г, 12,9 мг, 12 мкмоль), и смесь перемешивали в атмосфере азота в течение 30 минут. Смесь фильтровали и концентрировали в вакууме, и остаток растворяли в AcOH (0,5 мл) и очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (10,5 мг). MS m/z [M+H]+ рассчитано для C24H28ClNO7, 478,16; найдено 478,2.
W. Сложный (S)-2-метоксикарбониламино-3-метилбутирилоксиметиловый эфир (2R,4R)-5-(3'-хлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты

Следуя описанным здесь способам, также получали указанное в заголовке соединение (12,6 мг). MS m/z [M+H]+ рассчитано для C27H31ClN2O10, 579,17; найдено 579,2.
ПРИМЕР 3
A. (2R,4R)-5-(2',5'-Дихлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановая кислота
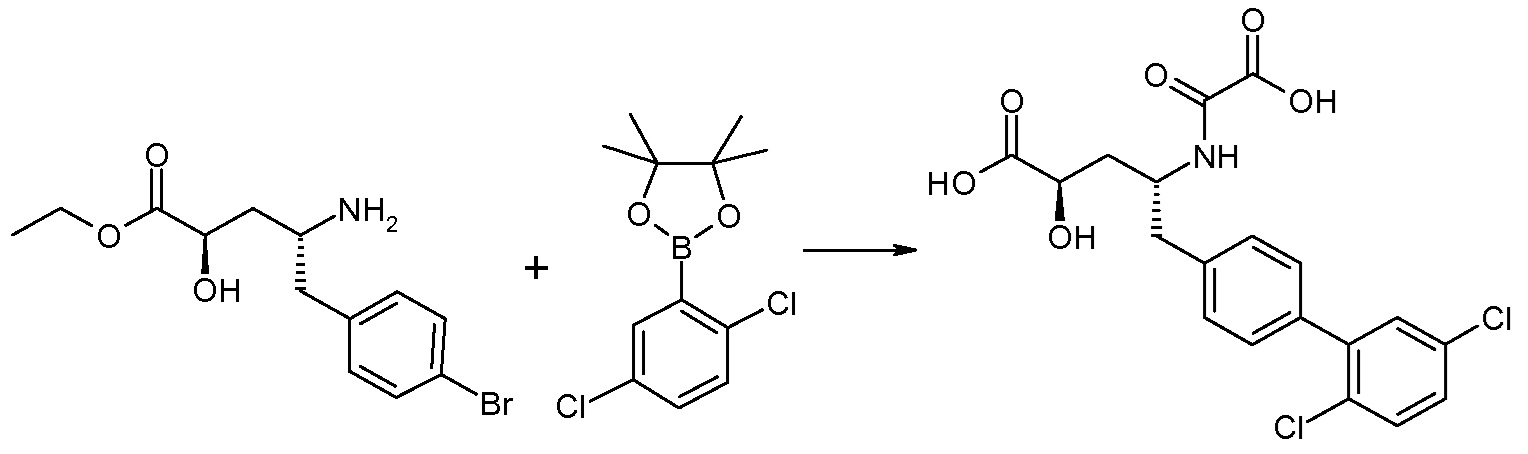
Раствор этилоксалилхлорида (42,4 мкл, 0,4 ммоль) в DCM (0,4 мл, 6 ммоль) добавляли к раствору сложного этилового эфира (2R,4R)-4-амино-5-(4-бромфенил)-2-гидроксипентановой кислоты (80 мг, 0,2 ммоль) и Et3N (0,1 мл, 0,8 ммоль) в DCM (1 мл), и полученную смесь перемешивали при комнатной температуре в течение 30 минут, затем выпаривали при пониженном давлении. Породукт затем объединяли с 2,5-дихлорфенилбороновой кислотой (72,4 мг, 0,4 ммоль), K2CO3 (104,9 мг, 759 мкмоль), EtOH (0,9 мл) и водой (0,2 мл). Смесь помещали в атмосферу азота и добавляли SilicaCat®DPP-Pd (0,28 ммоль/г загрузки, 90,4 мг, 25,3 мкмоль). Смесь обрабатывали микроволновым излучением при 120°C в течение 20 минут, затем фильтровали. Добавляли 1M водный LiOH (2,5 мл, 2,5 ммоль) с получением указанного в заголовке соединения (11,9 мг, чистота 100%). MS m/z [M+H]+ рассчитано для C19H17Cl2NO6, 426,04; найдено 426,0.
B. Сложный изобутиловый эфир (2R,4R)-5-(2',5'-дихлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
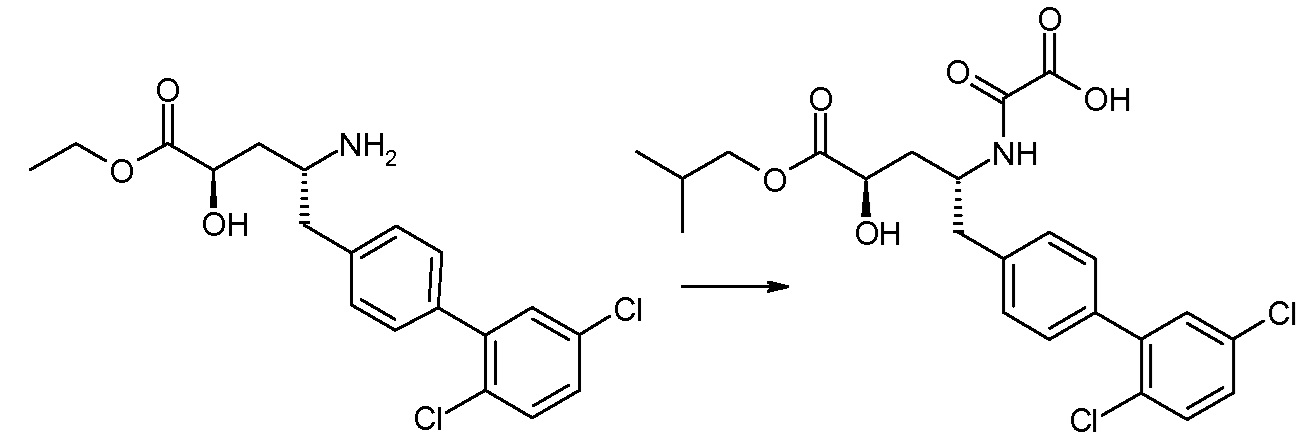
4,0M HCl в 1,4-диоксане (196 мкл, 785 мкмоль) добавляли к суспензии сложного этилового эфира (2R,4R)-4-амино-5-(2',5'-дихлорбифенил-4-ил)-2-гидроксипентановой кислоты (75,0 мг, 196 мкмоль) в изобутиловом спирте (0,5 мл, 5,4 ммоль), и полученную смесь перемешивали при 60°C в течение 2 часов. Смесь затем концентрировали в вакууме с получением белого твердого вещества. Белое твердое вещество растворяли в DCM (1 мл) и затем к смеси добавляли DIPEA (102 мкл, 588 мкмоль) с последующим добавлением по каплям ~0,2 мл 1M раствора трет-бутилоксалилхлорида в DCM (0,2 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением желтой жидкости. Раствор TFA/DCM (1:1, 1,1 мл, 7,0 ммоль) добавляли к желтой жидкости, и полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (колоночная хроматография в масштабе препаративной ВЭЖХ на колонке C18, 40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (80 мг, чистота 99%) в виде белого твердого вещества. MS m/z [M+H]+ рассчитано для C23H25Cl2O6, 482,11; найдено 482,1.
C. Сложный изопропиловый эфир (2R,4R)-5-(2',5'-дихлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты

4,0M HCl в 1,4-диоксане (196 мкл, 785 мкмоль) добавляли к суспензии сложного этилового эфира (2R,4R)-4-амино-5-(2',5'-дихлорбифенил-4-ил)-2-гидрокси-пентановой кислоты (75,0 мг, 196 мкмоль) в изопропиловом спирте (0,5 мл, 6,5 ммоль), и полученную смесь перемешивали при 60°C в течение ночи. Смесь затем концентрировали в вакууме с получением белого твердого вещества. Белое твердое вещество растворяли в DCM (1 мл) и затем к смеси добавляли DIPEA (102 мкл, 588 мкмоль) с последующим добавлением по каплям ~0,2 мл 1M раствора трет-бутилоксалилхлорида в DCM (0,2 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением желтой жидкости. Раствор TFA/DCM (1:1, 1,1 мл, 7,0 ммоль) добавляли к желтой жидкости, и полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме с получением прозрачной желтой жидкости. Неочищенную жидкость очищали (колоночная хроматография в масштабе препаративной ВЭЖХ на колонке C18, 40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (60,6 мг, чистота 98%) в виде белого твердого вещества. MS m/z [M+H]+ рассчитано для C22H23Cl2O6, 468,09; найдено 468,1.
D. (2R,4R)-5-(2',5'-Дихлорбифенил-4-ил)-2-гидрокси-4-(изобутоксиоксалиламино)пентановая кислота

1,0M водную HCl (3,5 мл, 3,5 ммоль) добавляли к сложному этиловому эфиру (2R,4R)-4-амино-5-(2',5'-дихлорбифенил-4-ил)-2-гидроксипентановой кислоты (155 мг, 405 мкмоль), и смесь перемешивали при 100°C в течение 1 часа, затем концентрировали. Продукт объединяли с Et3N (226 мкл, 1,6 ммоль) в DMF (2,5 мл, 32,3 ммоль). По каплям добавляли сложный изобутиловый эфир хлороксоуксусной кислоты (140 мг, 851 мкмоль) при 0°C, и полученную смесь перемешивали при комнатной температуре в течение 10 минут. Добавляли насыщенный водный раствор NaHCO3 (5 мл), и смесь перемешивали при комнатной температуре в течение 2 часов. Смесь экстрагировали DCM (3×5 мл), экстракты объединяли, промывали насыщенным водным раствором NaCl, сушили над Na2SO4 и концентрировали с получением белого твердого остатка. Неочищенное твердое вещество очищали посредством препаративной ВЭЖХ (колонка C18; 40-90% MeCN в воде с 0,05% TFA) с получением указанного в заголовке соединения (98,0 мг, чистота 99%) в виде белого твердого вещества. MS m/z [M+H]+ рассчитано для C23H25C12NO6, 482,11; найдено 482,1.
ПРИМЕР 4
(2R,4R)-5-(3-Хлорбифенил-4-ил-2-гидрокси-4-(оксалиламино)пентановая кислота

Раствор этилоксалилхлорида (41 мкл, 0,4 ммоль) в DCM (0,5 мл) добавляли к раствору (3R,5R)-5-амино-6-(4-бром-2-хлорфенил)-2-этоксигекс-1-ен-3-ола (96 мг, 0,3 ммоль) и Et3N (0,1192 мл, 0,8556 ммоль) в DCM (1,4 мл) и перемешивали в течение 20 минут при комнатной температуре. Смесь выпаривали при пониженном давлении и объединяли с фенилбороновой кислотой (52,2 мг, 0,4 ммоль), K2CO3 (100 мг, 0,9 ммоль), водой (0,2 мл), и EtOH (1 мл). Полученную смесь помещали в атмосферу азота и добавляли SilicaCat®DPP-Pd (0,28 ммоль/г загрузки; 100 мг, 0,03 ммоль). Смесь нагревали при 120°C в течение 20 минут до завершения реакции. Смесь фильтровали и добавляли 1M водный раствор LiOH (3 мл, 3 ммоль). Продукт затем очищали (на обращенно-фазовой хроматографической колонке Interchim) с получением указанного в заголовке соединения (12,6 мг). MS m/z [M+H]+ рассчитано для C19H18ClNO6 392,08; найдено 392,2.
ПРИМЕР 5
Следуя процедурам, описанным в разделе «Примеры» настоящего описания, и замещая соответствующие исходные материалы и реагенты, получали следующие соединения:
1. (2R,4R)-5-(5'-Хлор-2'-метилбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановая кислота
2. Сложный изопропиловый эфир (2R,4R)-5-(5'-хлор-2'-метилбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
3. Сложный этиловый эфир (2R,4R)-5-(5'-хлор-2'-метилбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
4. Сложный изобутиловый эфир (2R,4R)-5-(5'-хлор-2'-метилбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
5. (2R,4R)-5-(5'-Хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановая кислота
6. Сложный этиловый эфир (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
7. Сложный изобутиловый эфир (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
8. Сложный изопропиловый эфир (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
9. (2R,4R)-5-(3,3'-Дихлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановая кислота
10. (2R,4R)-5-(3,3'-Дихлорбифенил-4-ил)-2-гидрокси-4-(изобутоксиоксалиламино)пентановая кислота
11. Сложный изобутиловый эфир (2R,4R)-5-(3,3'-дихлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
12. Сложный изопропиловый эфир (2R,4R)-5-(3,3'-дихлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
13. (R)-3-[N-(3,5'-Дихлор-2'-фторбифенил-4-илметил)-N'-оксалилгидразино]-2-гидроксипропионовая кислота
14. (2R,4R)-5-(5'-Хлор-2'-фторбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксипентановая кислота
15. (2R,4R)-5-(5'-Хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-(изопропоксиоксалиламино)пентановая кислота
16. (2R,4R)-5-(5'-Хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-(изобутоксиоксалиламино)пентановая кислота
17. Сложный этиловый эфир (2R,4R)-5-(3,3'-дихлорбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты
ПРИМЕР 6
A. (2S,4S)-5-Бифенил-4-ил-2-гидроксиметил-4-(оксалиламино)пентановая кислота

Этилоксалилхлорид (27 мкл, 0,2 ммоль, 1,1 экв.) добавляли к раствору сложного этилового эфира (2S,4S)-4-амино-5-бифенил-4-ил-2-гидроксиметилпентановой кислоты (соль HCl; 80 мг, 0,22 ммоль) в DMF (0,5 мл)/DCM (0,5 мл) и перемешивали при комнатной температуре в течение 20 минут. Растворитель удаляли, и остаток растворяли в LiOH (моногидрат; 92,2 мг, 2,2 ммоль), воде (1,0 мл) и EtOH (2,0 мл) и перемешивали при комнатной температуре в течение 30 минут. Реакцию гасили AcOH, и растворитель удаляли. Остаток растворяли в AcOH/MeCN и очищали препаративной ВЭЖХ. Чистые фракции объединяли и лиофилизировали с получением указанного в заголовке соединения (37 мг, чистота 95%). MS m/z [M+H]+ рассчитано для C20H21NO6, 372,14; найдено 372,2.
B. Сложный этиловый эфир (2S,4S)-5-Бифенил-4-ил-2-гидроксиметил-4-(оксалиламино)пентановой кислоты
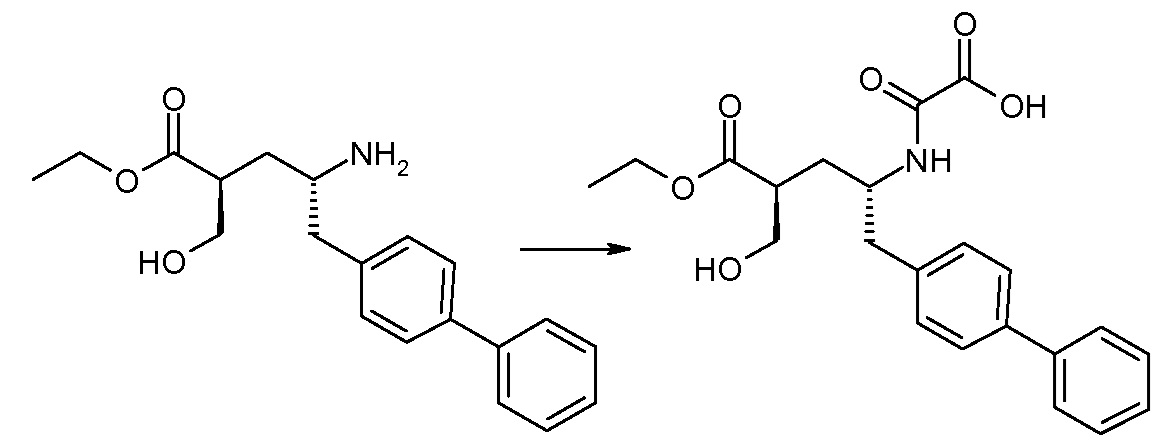
Оксалилхлорид (232 мкл, 2,8 ммоль) и трет-бутиловый спирт (228 мкл) объединяли в эфире (6,7 мл) в атмосфере азота при 0°C. Полученную смесь перемешивали в течение 30 минут при комнатной температуре. Растворитель выпаривали в вакууме с образованием сложного трет-бутилового эфира хлороксоуксусной кислоты, который затем растворяли в DCM (10 мл) и объединяли со сложным этиловым эфиром (2S,4S)-4-амино-5-бифенил-4-ил-2-гидроксиметилпентановой кислоты (соль HCl; 667 мг, 1,8 ммоль), который был растворен в DCM Et3N (2,6 мл) при 0°C. Полученную смесь перемешивали в течение 5 минут при комнатной температуре. Неочищенный продукт концентрировали, растворяли в DCM и очищали флэш-хроматографией (20-80% EtOAc/гексаны). Растворитель удаляли, и остаток растворяли в DCM (5 мл) и TFA (1 мл) и перемешивали в течение 1 часа. Продукт сушили в вакууме и очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (135 мг, чистота 95%). MS m/z [M+H]+ рассчитано для C22H25NO6, 400,17; найдено 400,2.
ПРИМЕР 7
A. Сложный бутиловый эфир (2S,4S)-5-(2'-фторбифенил-4-ил)-2-гидроксиметил-4-(оксалиламино)пентановой кислоты

Оксалилхлорид (44,1 мкл, 0,5 ммоль) и трет-бутиловый спирт (46,5 мкл) объединяли в эфире (1 мл) и перемешивали в течение 30 минут при комнатной температуре. Растворитель выпаривали в вакууме с образованием сложного трет-бутилового эфира хлороксоуксусной кислоты, который затем растворяли в DCM (2 мл). Сложный этиловый эфир (2S,4S)-4-амино-5-(2'-фторбифенил-4-ил)-2-гидроксиметилпентановой кислоты (соль HCl; 120 мг, 0,3 ммоль) объединяли с 1-бутанолом (3 мл) и 4M HCl в 1,4-диоксане (3 мл) и перемешивали при 60°C в течение 2 часов. Растворитель выпаривали и азеотропировали толуолом (2×), и продукт растворяли в Et3N (155 мкл) и DCM, затем объединяли со сложным трет-бутиловым эфиром хлороксоуксусной кислоты. Полученную смесь перемешивали в течение 20 минут при комнатной температуре. Растворитель выпаривали, и остаток повторно растворяли в 1:1 TFA:DCM и перемешивали в течение 20 минут при 40°C. Добавляли AcOH, и продукт очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (30 мг, чистота 95%). MS m/z [M+H]+рассчитано для C24H28FNO6, 446,19; найдено 446,4.
B. (2S,4S)-5-(2'-Фторбифенил-4-ил)-2-гидроксиметил-4-(оксалиламино)пентановая кислота

Этилоксалилхлорид (13,8 мкл, 0,1 ммоль) и DIPEA (39,2 мкл, 0,2 ммоль) объединяли со сложным этиловым эфиром (2S,4S)-4-амино-5-(2'-фторбифенил-4-ил)-2-гидроксиметилпентановой кислоты (соль HCl; 43 мг, 0,1 ммоль), растворенном в DCM (0,9 мл). Смесь перемешивали при комнатной температуре в течение 10 минут, затем концентрировали в вакууме. Добавляли 1M водный раствор LiOH (0,9 мл) и EtOH (0,9 мл), и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь гасили AcOH, и растворитель выпаривали. Остаток растворяли в AcOH/MeCN и очищали препаративной ВЭЖХ. Чистые фракции объединяли и лиофилизировали с получением указанного в заголовке соединения (32,7 мг, чистота 95%). MS m/z [M+H]+рассчитано для C20H20FNO6, 390,13; найдено 390,2.
ПРИМЕР 8
Следуя процедурам, описанным в разделе «Примеры» настоящего описания, и замещая соответствующие исходные материалы и реагенты, получали следующие соединения:

1. Сложный этиловый эфир (2S,4S)-5-(3'-фторбифенил-4-ил)-2-гидроксиметил-4-(оксалиламино)пентановой кислоты
2. (2S,4S)-5-(3'-Фторбифенил-4-ил)-2-гидроксиметил-4-(оксалиламино)пентановая кислота
3. (2S,4S)-5-(3'-Хлорбифенил-4-ил)-2-гидроксиметил-4-(оксалиламино)пентановая кислота

4. (2S,4S)-5-(4'-Фторбифенил-4-ил)-2-гидроксиметил-4-(оксалиламино)пентановая кислота

5. (2S,4S)-5-(3-Хлорбифенил-4-ил)-2-гидроксиметил-4-(оксалиламино)пентановая кислота
6. (2R,4S)-5-(3-Хлорбифенил-4-ил)-2-гидроксиметил-4-(оксалиламино)пентановая кислота
ПРИМЕР 9
A. (2S,4R)-5-Бифенил-4-ил-2-гидроксиметил-2-метил-4-(оксалиламино)пентановая кислота
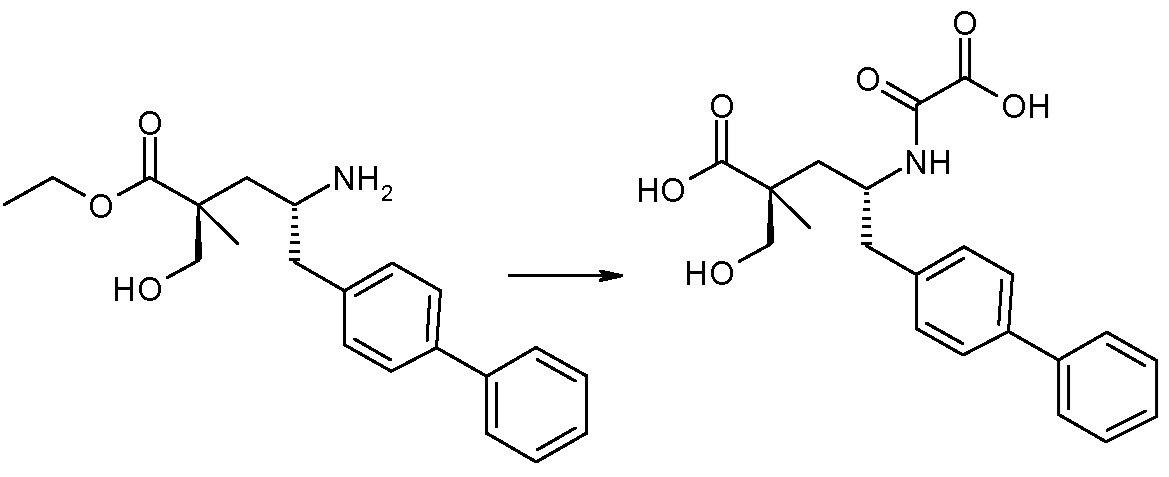
Этилоксалилхлорид (13,1 мкл, 0,1 ммоль) объединяли со сложным этиловым эфиром (2S,4R)-4-амино-5-бифенил-4-ил-2-гидроксиметил-2-метилпентановой кислоты (40 мг, 0,1 ммоль), растворяли в DCM (0,3 мл) и небольшом количестве DMF. Смесь перемешивали при комнатной температуре в течение 20 минут, затем концентрировали в вакууме. Добавляли 1M водный раствор NaOH (117 мкл) и THF (1,5 мл), и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Остаток растворяли в AcOH и очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (8 мг, чистота 95%). MS m/z [M+H]+ рассчитано для C21H23NO6, 386,15; найдено 386,0.
B. Сложный этиловый эфир (2S,4R)-5-бифенил-4-ил-2-гидроксиметил-2-метил-4-(оксалиламино)пентановой кислоты

Оксалилхлорид (12,4 мкл, 0,1 ммоль) и трет-бутиловый спирт (13,1 мкл) объединяли в эфире (0,3 мл) в атмосфере азота при 0°C. Полученную смесь перемешивали в течение 30 минут при комнатной температуре. Растворитель выпаривали в вакууме с образованием сложного трет-бутилового эфира хлороксоуксусной кислоты, который затем растворяли в DCM (0,7 мл) и объединяли со сложным этиловым эфиром (2S,4R)-4-амино-5-бифенил-4-ил-2-гидроксиметил-2-метилпентановой кислоты (33,4 мг, 98 мкмоль). Добавляли Et3N (43,6 мкл) при 0°C, и полученную смесь перемешивали в течение 30 минут при комнатной температуре. Растворитель выпаривали, и остаток растворяли в 1:1 TFA:DCM и перемешивали в течение 1 часа. Добавляли AcOH, и продукт очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (7 мг, чистота 95%). MS m/z [M+H]+ рассчитано для C23H27NO6, 414,18; найдено 414,4.
ПРИМЕР 10
(2S,4R)-5-(3'-Фторбифенил-4-ил)-2-гидроксиметил-2-метил-4-(оксалиламино)пентановая кислота
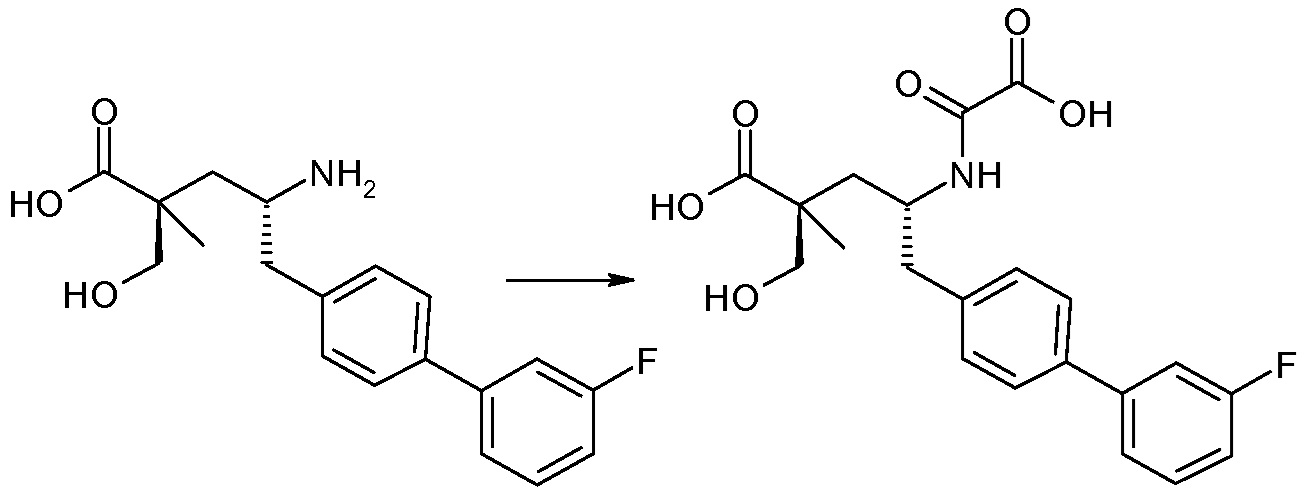
Этилоксалилхлорид (9,1 мкл, 0,1 ммоль) объединяли с (2S,4R)-4-амино-5-(3'-фторбифенил-4-ил)-2-гидроксиметил-2-метилпентановой кислотой (27 мг, 0,1 ммоль) растворяли в DCM (0,2 мл) и небольшом количестве DMF. Смесь перемешивали при комнатной температуре в течение 20 минут. Растворитель выпаривали и добавляли 10M водный раствор NaOH (81,5 мкл) и THF (1,0 мл), и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Остаток растворяли в AcOH и очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (6 мг, чистота 95%). MS m/z [M+H]+ рассчитано для C21H22FNO6, 404,14; найдено 404,4.
ПРИМЕР 11
Следуя процедурам, описанным в разделе «Примеры» настоящего описания, и замещая соответствующие исходные материалы и реагенты, получали следующие соединения:

1. (2S,4R)-5-(2'-Фторбифенил-4-ил)-2-гидроксиметил-2-метил-4-(оксалиламино)пентановая кислота
2. (2S,4R)-5-(5'-Хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метил-4-(оксалиламино)пентановая кислота

3. (2S,4R)-5-(4'-Фторбифенил-4-ил)-2-гидроксиметил-2-метил-4-(оксалиламино)пентановая кислота
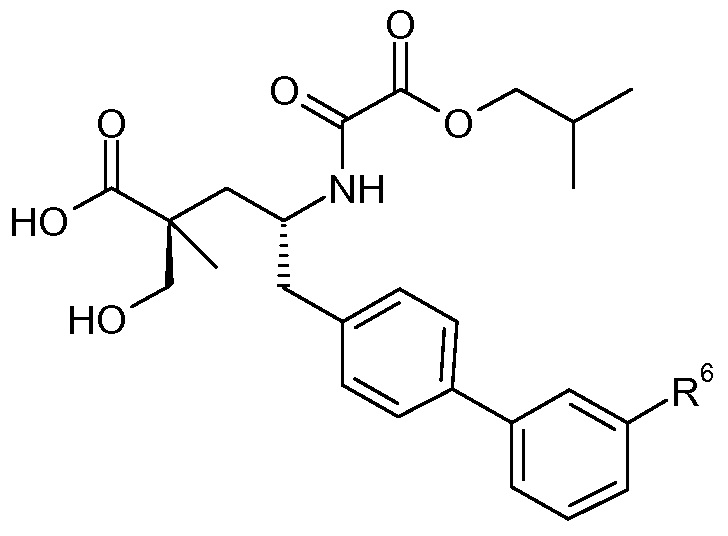
4. (2S,4R)-5-(3'-Хлорбифенил-4-ил)-2-гидроксиметил-4-(изобутоксиоксалиламино)-2-метилпентановая кислота
ПРИМЕР 12
3-(N-Бифенил-4-илметил-N'-оксалилгидразино)-2-гидрокси-2-метилпропионовая кислота
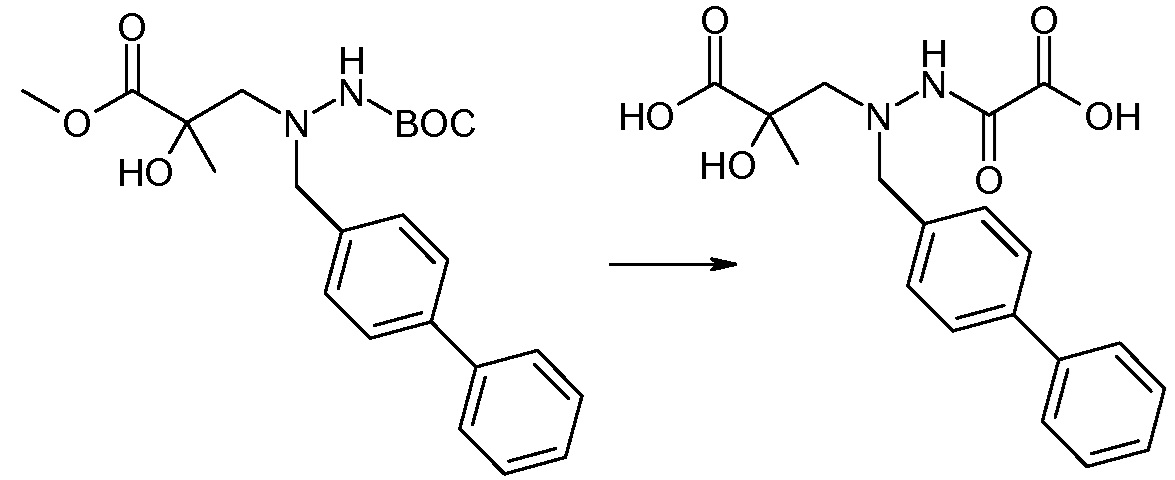
Сложный метиловый эфир 3-(N-бифенил-4-илметил-N'-трет-бутоксикарбонилгидразино)-2-гидрокси-2-метилпропионовой кислоты (0,1 г, 241 мкмоль) растворяли в DCM (1,0 мл), затем добавляли TFA (1,0 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали, и остаток растворяли в DMF (2,00 мл). Добавляли DIPEA (126 мкл, 724 мкмоль) с последующим добавлением этилоксалилхлорида (29,6 мкл, 265 мкмоль), и полученную смесь перемешивали при комнатной температуре до завершения реакции (~3 часов). Смесь концентрировали, и остаток растворяли в THF (1,5 мл), затем добавляли моногидрат гидроксида лития (101 мг, 2,4 ммоль) в воде (1,50 мл), и смесь перемешивали при комнатной температуре в течение 30 минут. Реакцию гасили AcOH, и раствор концентрировали. Неочищенный продукт очищали препаративной ВЭЖХ (10-70% MeCN/H2O) с получением указанного в заголовке соединения (10,9 мг, чистота 95%). MS m/z [M+H]+ рассчитано для C19H20N2O6, 373,13; найдено 373,2.
ПРИМЕР 13
(R)-3-[N-(3'-Хлорбифенил-4-илметил)-N'-оксалилгидразино]-2-гидроксипропионовая кислота
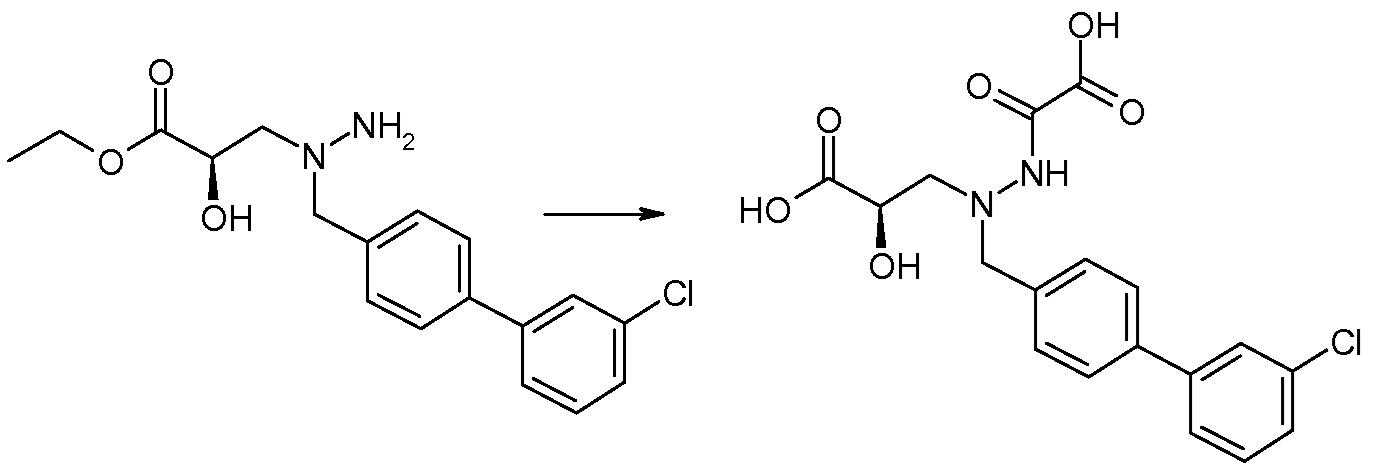
Сложный этиловый эфир (R)-3-[N-(3'-хлорбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (70 мг, 0,2 ммоль) растворяли в DCM (1,5 мл) с последующим добавлением этилоксалилхлорида (24,7 мкл, 221 мкмоль) и DIPEA (69,9 мкл, 401 мкмоль). Смесь перемешивали при комнатной температуре до завершения реакции (~10 минут). Смесь концентрировали в вакууме. Добавляли 1M водный раствор гидроксида лития (1,6 мл, 1,6 ммоль) и EtOH (1,5 мл), и смесь перемешивали при комнатной температуре до завершения реакции (~2 часа). Реакцию гасили AcOH, и растворитель выпаривали. Остаток растворяли в AcOH/MeCN и очищали препаративной ВЭЖХ. Чистые фракции объединяли и лиофилизировали с получением указанного в заголовке соединения (8,3 мг, чистота 95%). MS m/z [M+H]+ рассчитано для C18H17ClN2O6, 393,08; найдено 393,2.
ПРИМЕР 14
A. (R)-3-[N-(5'-Хлор-2'-фторбифенил-4-илметил)-N'-оксалилгидразино]-2-гидроксипропионовая кислота

Сложный (R)-2-[N-(4-бромбензил)-N'-этоксиоксалилгидразино]-1-этоксикарбонилэтиловый эфир щавелевой кислоты (675 мг, 1,3 ммоль) объединяли с 5-хлор-2-фторфенилбороновой кислотой (273 мг, 1,6 ммоль) и K2CO3 (541 мг, 3,9 ммоль) в EtOH (4,6 мл, 78,3 ммоль) и водой (1,2 мл, 65,2 ммоль). Полученную смесь помещали в атмосферу азота, и затем добавляли SilicaCat®DPP-Pd (0,28 ммоль/г загрузки; 466 мг, 130 мкмоль). Смесь обрабатывали микроволновым излучением при 120°C в течение 10 минут, затем фильтровали и выпаривали при пониженном давлении. Остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (40 мг). MS m/z [M+H]+ рассчитано для C18H16ClFN2O6, 411,07; найдено 411,0.
B. Сложный этиловый эфир (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)-N'-оксалилгидразино]-2-гидроксипропионовой кислоты

~1M раствор трет-бутилоксалилхлорида в DCM (136 мкл) добавляли к перемешиваемому раствору сложного этилового эфира (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (соль HCl; 55,0 мг, 136 мкмоль) в DCM (1,3 мл, 20 ммоль) при 0°C. После перемешивания при комнатной температуре в течение 2 часов добавляли по каплям DIPEA (11,9 мкл, 68 мкмоль) в растворе DCM (80 мкл). Через одну минуту добавляли дополнительный DIPEA (10 мкл) в DCM (80 мкл), и смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали, и полученный остаток очищали флэш-хроматографией (4 г силикагеля, 0-100% EtOAc/гексаны). Требуемые фракции объединяли и концентрировали с получением бесцветного масла (60 мг). Часть этого масла (20 мг) обрабатывали смесью 1:1 DCM:TFA (0,2 мл) при комнатной температуре в течение 20 минут. Смесь концентрировали, остаток растворяли в 50% воды/AcOH (1,5 мл), фильтровали, и очищали обращенно-фазовой препаративной ВЭЖХ с получением указанного в заголовке соединения (10 мг) в виде соли TFA. MS m/z [M+H]+ рассчитано для C20H20ClFN2O6, 439,10; найдено 439,4.
C. (R)-3-[N-(5'-Хлор-2'-фторбифенил-4-илметил)-N'-изобутоксиоксалилгидразино]-2-гидроксипропионовая кислота

Сложный изобутиловый эфир хлороксоуксусной кислоты получали добавлением оксалилхлорида (21 мкл, 252 мкмоль) к раствору изобутанола (21 мкл, 226 мкмоль) в эфире (206 мкл, 2,0 ммоль). Смесь перемешивали при комнатной температуре в течение 15 мин и затем выпаривали.
Сложный изобутиловый эфир хлороксоуксусной кислоты затем добавляли к раствору сложного 2-оксо-2-фенилэтилового эфира (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (23,0 мг, 50 мкмоль) в DCM (413 мкл, 6,4 ммоль) при 0°C. Полученную смесь перемешивали при 0°C в течение 15 минут. Затем добавляли насыщенный водный раствор NaHCO3 и слои разделяли. Водный слой экстрагировали DCM. Слои DCM объединяли, сушили над MgSO4 и концентрировали с получением прозрачной желтой жидкости. Цинк (164 мг, 2,5 ммоль) добавляли к раствору этой желтой жидкости в AcOH (172 мкл, 3,0 ммоль), и смесь перемешивали при комнатной температуре в течение 10 минут. Смесь фильтровали, используя AcOH и воду, растворители выпаривали в вакууме, и остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (9,0 мг). MS m/z [M+H]+ рассчитано для C22H24ClFN2O6, 467,13; найдено 467,1.
D. (R)-3-[N-(5'-Хлор-2'-фторбифенил-4-илметил)-N'-(2,2-дифторпропоксиоксалил)гидразино]-2-гидроксипропионовая кислота

2,2-Дифторпропилоксалилхлорид получали добавлением оксалилхлорида (21 мкл, 252 мкмоль) к раствору 2,2-дифторпропанола (21,8 мг, 226 мкмоль) в эфире (206 мкл, 2,0 ммоль). Смесь перемешивали при комнатной температуре в течение 15 минут и затем выпаривали.
2,2-Дифторпропилоксалилхлорид затем добавляли к раствору сложного 2-оксо-2-фенилэтилового эфира (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (23,0 мг, 50 мкмоль) в DCM (413 мкл, 6,4 ммоль) при 0°C. Полученную смесь перемешивали при 0°C в течение 15 минут. Затем добавляли насыщенный водный раствор NaHCO3, и слои разделяли. Водный слой экстрагировали DCM. Слои DCM объединяли, сушили над MgSO4 и концентрировали с получением прозрачной желтой жидкости. Цинк (164 мг, 2,5 ммоль) добавляли к раствору этой желтой жидкости в AcOH (172 мкл, 3,0 ммоль), и смесь перемешивали при комнатной температуре в течение 10 минут. Смесь фильтровали, используя AcOH и воду, растворители выпаривали в вакууме, и остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (1,1 мг). MS m/z [M+H] рассчитано для C21H20ClF3N2O6, 489,10; найдено 489,0.
E. Сложный 5-метил-2-оксо-[1,3]диоксол-4-илметиловый эфир (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)-N'-оксалилгидразино]-2-гидроксипропионовой кислоты

К раствору сложного 5-метил-2-оксо-[1,3]диоксол-4-илметилового эфира (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (350 мг, 780 мкмоль) в безводном DCM (15 мл) добавляли трет-бутилоксалилхлорид (193 мг, 1,2 ммоль) и DIPEA (302 мг, 2,3 ммоль) при 0°C. Полученную смесь перемешивали при комнатной температуре в течение 5 часов. Смесь затем промывали насыщенным водным раствором NaCl (2×30 мл) и экстрагировали DCM (3×50 мл). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали в вакууме с получением белого твердого вещества (300 мг). LC-MS: 523 [M-t-Bu+H]+.
Это твердое вещество (100 мг, 170 мкмоль) растворяли в TFA (5 мл) и DCM (15 мл). Полученную смесь перемешивали в течение ночи. Смесь выпаривали в вакууме, и остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (20 мг). LC-MS: 523,1 [M+H]+.
1H-ЯМР: (DMSO-d6): δ 2,14 (с, 3H), 3,17-3,16 (м, 2H), 4,11-4,08 (м, 2H), 4,26 (ушир., 1H), 4,98 (ушир., 2H), 5,50 (ушир., 1H), 7,58-7,36 (м, 7H), 9,94 (с, 1H), 13,8 (ушир., 1H).
F. (R)-3-[N-(5'-Хлор-2'-фторбифенил-4-илметил)-N'-этоксиоксалилгидразино]-2-гидроксипропионовая кислота
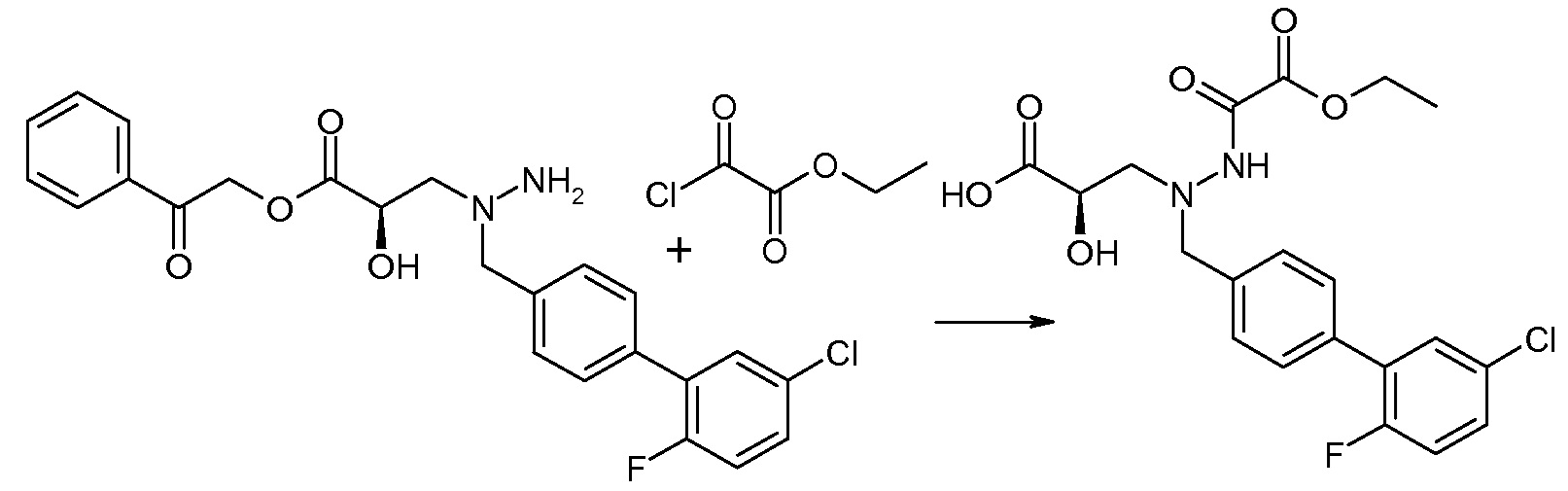
Этилоксалилхлорид (12,4 мкл, 111 мкмоль) добавляли к раствору сложного 2-оксо-2-фенилэтилового эфира (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (23,0 мг, 50 мкмоль) в DCM (413 мкл, 6,4 ммоль) при 0°C, и полученную смесь перемешивали при 0°C в течение 15 минут. Затем добавляли насыщенный водный раствор NaHCO3 (1 мл), и слои разделяли. Водный слой экстрагировали DCM (2×2 мл). Слои DCM объединяли, сушили над MgSO4 и концентрировали. Цинк (164 мг, 2,5 ммоль) добавляли к раствору этого остатка в AcOH (172 мкл, 3,0 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 10 минут. Смесь фильтровали, и остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (10 мг). MS m/z [M+H]+ рассчитано для C20H20ClFN2O6, 439,10; найдено 439,1.
G. Сложный 2,2-дифторпропиловый эфир (R)-3-[N-(5'-хлор-2'-фторфенил-4-илметил)-N'-оксалилгидразино]-2-гидроксипропионовой кислоты

(R)-3-[N'-трет-Бутоксиоксалил-N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]2-гидроксипропионовую кислоту (15,0 мг, 32 мкмоль) объединяли с HOBt (26,0 мг, 193 мкмоль) и EDC (34 мкл, 0,2 ммоль) в DCM (0,2 мл, 4 ммоль). Раствор перемешивали в течение 10 минут и добавляли 2,2-дифторпропанол (24,7 мг, 257 мкмоль). Реакционную смесь перемешивали при комнатной температуре и проводили мониторинг для выявления завершения реакции. Через 2 часа смесь концентрировали роторным выпариванием, и растворитель удаляли в вакууме. Полученный остаток растворяли в DCM (124 мкл, 1,9 ммоль). Добавляли TFA (124 мкл, 1,6 ммоль), и полученную смесь перемешивали в течение 2 часов. Растворитель удаляли в вакууме, и остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (2,2 мг). MS m/z [M+H]+ рассчитано для C21H20ClF3N2O6, 489,10; найдено 489,1.
H. Сложный изобутиловый эфир (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)-N'-оксалилгидразино]-2-гидроксипропионовой кислоты
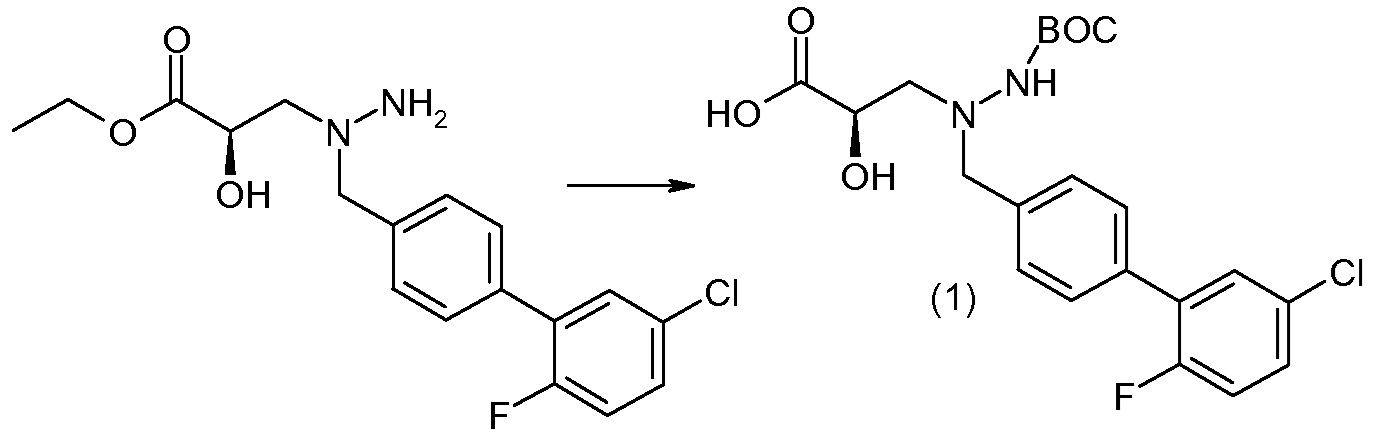
К смеси сложного этилового эфира (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (соль HCl; 500,0 мг, 1,3 ммоль) в DCM (6,0 мл, 94 ммоль) при комнатной температуре добавляли ди-трет-бутилдикарбонат (342 мкл, 1,5 ммоль) и DIPEA (216 мкл, 1,3 ммоль). После перемешивания при комнатной температуре в течение ночи смесь концентрировали, и остаток очищали флэш-хроматографией (12 г силикагеля, 0-50% EtOAc/гексаны). Требуемые фракции объединяли и концентрировали с получением светлого желтоватого масла. Только этот остаток растворяли в MeOH (6,0 мл, 150 ммоль) и воде (1,0 мл, 56 ммоль), затем обрабатывали моногидратом LiOH (104 мг, 2,5 ммоль) при комнатной температуре в течение 30 минут. Смесь концентрировали, и остаток разбавляли водой (2,0 мл) и EtOAc (10,0 мл), затем подкисляли 1N водным раствором HCl до pH~2,0 при энергичном перемешивании. Органический слой промывали насыщенным водным раствором NaCl (2×2,0 мл), сушили над Na2SO4, фильтровали и концентрировали с получением соединения 1 в виде белого твердого вещества (528,6 мг).

Соединение 1 (65,0 мг, 148 мкмоль) растворяли в изобутиловом спирте (684 мкл, 7,4 ммоль). Добавляли 4,0M раствор HCl в 1,4-диоксане (1,2 мл, 4,9 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 2 часов, затем при 60°C еще в течение пары часов до завершения реакции. Растворитель удаляли в вакууме с получением соединения 2, которое использовали без дополнительной очистки.

трет-Бутилоксалилхлорид получали добавлением оксалилхлорида (63 мкл, 741 мкмоль) к раствору трет-бутилового спирта (43 мкл, 444 мкмоль) в эфире (778 мкл, 7,4 ммоль). Смесь перемешивали при комнатной температуре в течение 15 минут, затем концентрировали в вакууме. Соединение 2 (58,5 мг, 148 мкмоль) растворяли в DCM (570 мкл, 8,9 ммоль) и добавляли трет-бутилоксалилхлорид. Полученную смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали в вакууме. Остаток растворяли в растворе 1:1 DCM:TFA и перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли в вакууме, и остаток очищали препаративной ВЭЖХ с получением указанного в заголовке соединения (8,5 мг). MS m/z [M+H]+ рассчитано для C22H24ClFN2O6, 467,13; найдено 467,0.
I. Сложный этиловый эфир (R)-3-[N'-трет-бутоксиоксалил-N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты

К раствору сложного этилового эфира (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (200 мг, 0,5 ммоль) в DCM (2,0 мл) по каплям добавляли раствор трет-бутилоксалилхлорида (165 мг, 1,0 ммоль) при 0°C в атмосфере азота. Полученную смесь перемешивали в течение 5 минут и затем по каплям добавляли DIPEA (130 мг, 1,0 ммоль). Растворитель удаляли выпариванием, и остаток очищали колоночной хроматографией (петролейный эфир/EtOAc=4:1) с получением указанного в заголовке соединения в виде желтой жидкости (144 мг). LC-MS: 495 [M+H]+.
1H ЯМР (CDCl3, 400 MГц): δ 1,30 (т, J=7,1 Гц, 3H), 1,56 (с, 9H), 3,37-3,24 (м, 2H), 4,27-4,16 (м, 4H), 4,38-4,30 (м, 1H), 7,14-7,09 (м, 1H), 7,30-7,28 (м, 1H), 7,48-7,41 (м, 3H), 7,56-7,50 (м, 2H), 8,05 (с, 1H).
J. Сложный этоксикарбонилоксиметиловый эфир (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)-N'-оксалилгидразино]-2-гидроксипропионовой кислоты
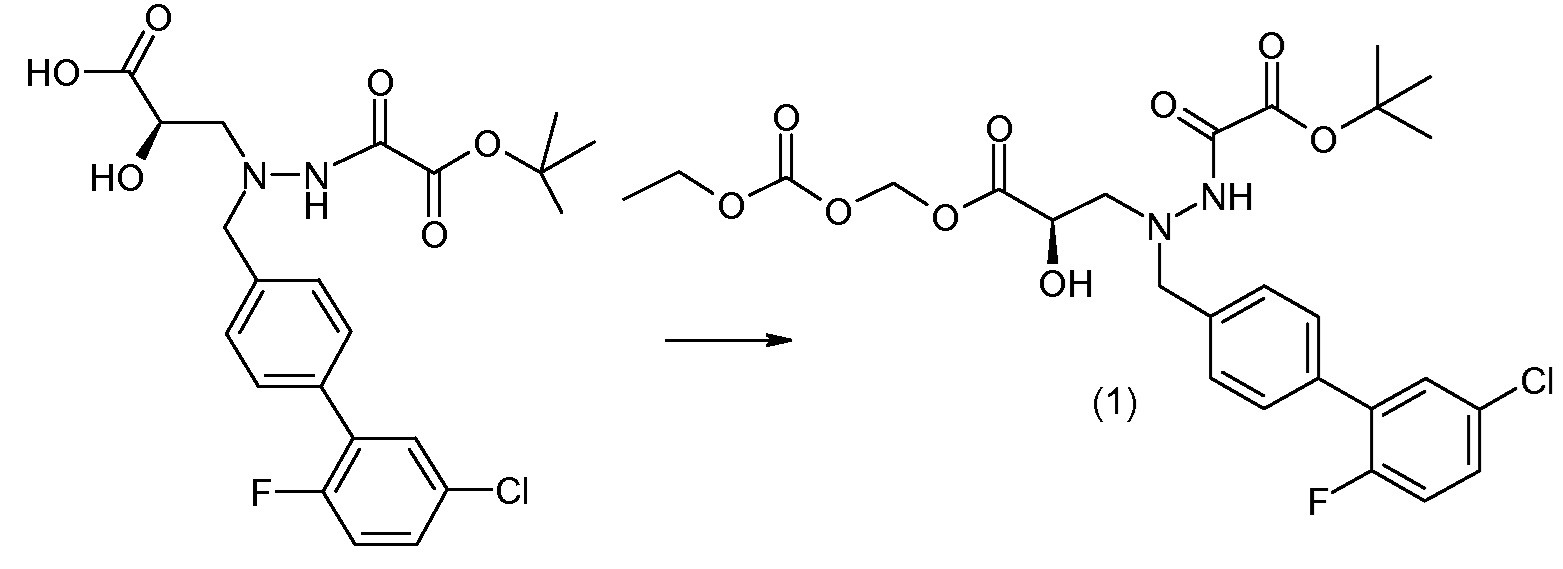
Смесь (R)-3-[N'-трет-бутоксиоксалил-N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (270 мг, 580 мкмоль), хлорметилэтилкарбоната (160 мг, 1,16 ммоль), NaI (174 мг, 1,2 ммоль) и 2,6-диметилпиридина (620 мг, 5,8 ммоль) в DMF (10 мл) перемешивали при комнатной температуре в течение ночи. Смесь выливали в воду (30 мл), и смесь затем экстрагировали EtOAc (3×30 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (2×30 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Неочищенное соединение 1 (300 мг) использовали без очистки. LC-MS: 569 [M+H]+.

TFA (1,0 мл) добавляли по каплям при комнатной температуре к раствору соединения 1 (300 мг, 530 мкмоль) в DCM (5 мл). Полученную смесь перемешивали в течение 2 часов при комнатной температуре, и растворитель затем удаляли. Остаток очищали колоночной хроматографией (DCM/MeOH, 10:1) с получением указанного в заголовке соединения в виде желтой жидкости (10 мг). LC-MS: 512,9 [M+H]+.
1H ЯМР (400 MГц, MeOD) δ 1,28 (т, J=7,3 Гц, 3H), 3,24-3,28 (м, 2H), 4,18-4,20 (м, 4H), 4,41 (ушир., 1H), 5,80 (дд, J=11,6, 5,8 Гц, 2H), 7,22 (д, J=10,1 Гц, 1H), 7,34-7,40 (м, 1H), 7,48-7,51 (м, 5H).
K. Сложный (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)-N'-оксалилгидразино]-2-гидроксипропионилоксиметиловый эфир масляной кислоты

Смесь (R)-3-[N'-трет-бутоксиоксалил-N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (300 мг, 430 мкмоль), хлорметилбутирата (175 мг, 1,3 ммоль), NaI (192 мг, 1,3 ммоль) и 2,6-диметилпиридина (680 мг, 6,4 ммоль) в DMF (10 мл) перемешивали при комнатной температуре в течение ночи. Смесь выливали в воду (30 мл), и смесь затем экстрагировали EtOAc (3×20 мл). Органический слой отделяли, промывали насыщенным водным раствором NaCl (30 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Неочищенное соединение 1 (300 мг) использовали без очистки. LC-MS: 567 [M+H]+.

TFA (1,0 мл) добавляли по каплям при комнатной температуре к раствору соединения 1 (300 мг, 464 мкмоль) в DCM (5 мл). Полученную смесь перемешивали в течение 2 часов при комнатной температуре, и растворитель затем удаляли. Остаток очищали колоночной хроматографией (DCM/MeOH, 10:1) с получением указанного в заголовке соединения в виде желтого масла (21 мг). LC-MS: 511,1 [M+H]+.
1H ЯМР (400 MГц, MeOD) δ 0,94 (т, J=7,4 Гц, 3H), 1,62 (дд, J=14,8, 7,4 Гц, 2H), 2,33 (т, J=7,3 Гц, 2H), 3,28 (д, J=6,1 Гц, 2H), 4,14 (кв, J=13,2 Гц, 2H), 4,38 (дд, J=6,0, 4,2 Гц, 1H), 5,80 (ушир., 2H), 7,16-7,26 (м, 1H), 7,33-7,40 (м, 1H), 7,47-7,52 (м, 5H).
L. Сложный ацетоксиметиловый эфир (R)-3-[N-(5'-хлор-2'-фторбифенил-4-илметил)-N'-оксалилгидразино]-2-гидроксипропионовой кислоты
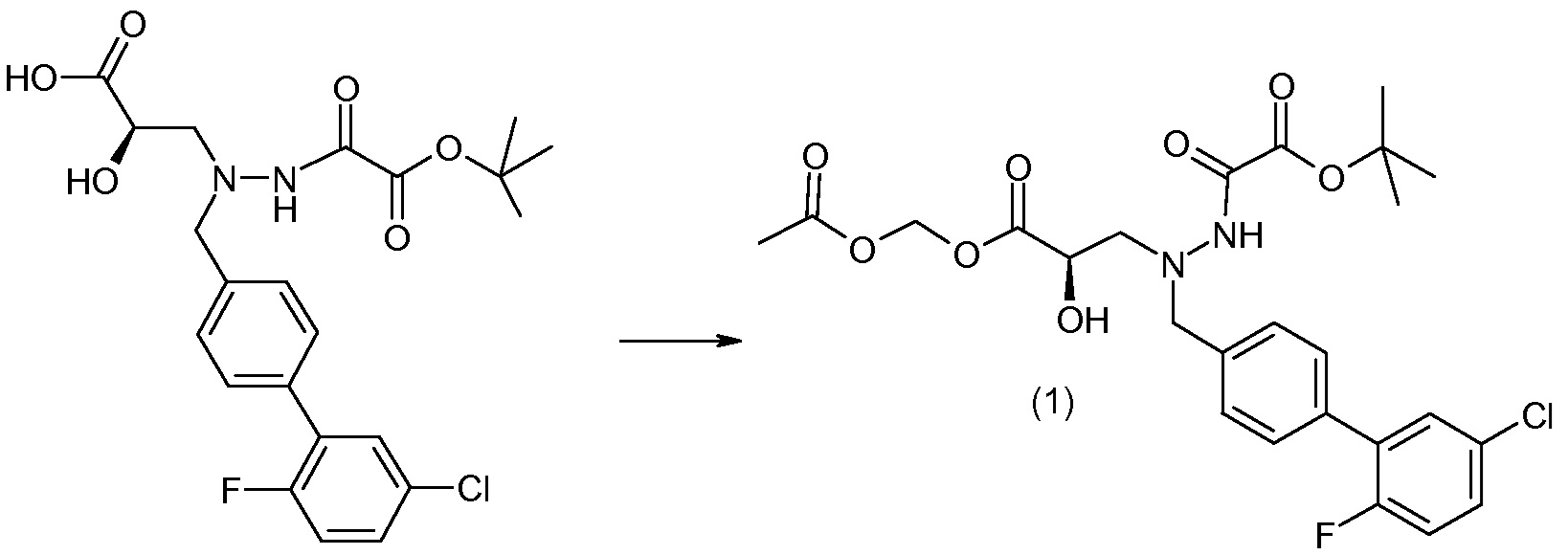
Смесь (R)-3-[N'-трет-бутоксиоксалил-N-(5'-хлор-2'-фторбифенил-4-илметил)гидразино]-2-гидроксипропионовой кислоты (300 мг, 640 мкмоль), бромметилацетата (196 мг, 1,3 ммоль), NaI (192 мг, 1,3 ммоль) и 2,6-диметилпиридина (680 мг, 6,4 ммоль) в DMF (10 мл) перемешивали при комнатной температуре в течение ночи. Смесь выливали в воду (30 мл), и смесь затем экстрагировали EtOAc (3×20 мл). Органический слой отделяли, промывали насыщенным водным раствором NaCl (30 мл), сушили над безводным Na2SO4 и концентрировали в вакууме. Неочищенное соединение 1 (300 мг) использовали без очистки. LC-MS: 539 [M+H]+.

TFA (1,0 мл) добавляли по каплям при комнатной температуре к раствору соединения 1 (300 мг, 550 мкмоль) в DCM (5 мл). Полученную смесь перемешивали в течение 2 часов при комнатной температуре, и растворитель затем удаляли. Остаток очищали колоночной хроматографией (DCM/MeOH, 10:1) с получением указанного в заголовке соединения в виде желтого масла (15 мг). LC-MS: 482,9 [M+H]+.
1H ЯМР (400 MГц, MeOD) δ 2,07 (с, 3H), 3,25-3,28 (м, 2H), 4,14 (кв, J=13,2 Гц, 2H), 4,38 (т, J=5,9 Гц, 1H), 5,88-5,71 (м, 2H), 7,25-7,17 (м, 1H), 7,41-7,31 (м, 1H), 7,70-7,46 (м, 5H).
ПРИМЕР 15
(2R,4S)-5-Бифенил-4-ил-2-гидрокси-5-метил-4-(оксалиламино)гексановая кислота

Сложный этиловый эфир (2R,4S)-4-амино-5-бифенил-4-ил-2-гидрокси-5-метилгексановой кислоты (70 мг, 0,2 ммоль) растворяли в DCM (5 мл) и перемешивали в течение 2 минут с последующим добавлением этилоксалилхлорида (23 мкл, 0,2 ммоль) и DIPEA (79 мг, 0,6 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, затем выпаривали при пониженном давлении. Смесь концентрировали в вакууме. Остаток растворяли в EtOH и добавляли достаточно эквивалентов 10N NaOH для того, чтобы сделать раствор осньвным. Мониторинг хода реакции проводили в течение 1 часа до завершения конечного снятия защиты. Раствор подкисляли равным объемом AcOH и выпаривали при пониженном давлении. Затем продукт очищали с использованием обращенно-фазовой хроматографии (градиент 10-70% MeCN с получением указанного в заголовке соединения (37 мг, чистота 95%). MS m/z [M+H]+ рассчитано для C21H23NO6, 386,15; найдено 386,4.
ПРИМЕР 16
(2S,4S)-5-Бифенил-4-ил-2-гидроксиметил-5-метил-4-(оксалиламино)гексановая кислота

Сложный этиловый эфир (2S,4S)-4-амино-5-бифенил-4-ил-2-гидроксиметил-5-метилгексановой кислоты (соль HCl; 40 мг, 0,1 ммоль) растворяли в DCM и DMF (1 мл) с последующим добавлением этилоксалилхлорида (17 мкл, 0,2 ммоль) и DIPEA (53,3 мкл, 0,3 ммоль). Смесь перемешивали при комнатной температуре до завершения реакции (~5 минут). Реакцию гасили водой. Продукт экстрагировали EtOAc, и полученный органический слой концентрировали. Добавляли 1M водный раствор гидроксида лития (1,0 мл, 1,0 ммоль) и EtOH (2,0 мл), и смесь перемешивали при комнатной температуре до завершения реакции (~2 часа). Реакцию гасили AcOH, и продукт очищали препаративной ВЭЖХ. Чистые фракции объединяли и лиофилизировали с получением указанного в заголовке соединения (19 мг, чистота 95%). MS m/z [M+H]+ рассчитано для C22H25NO6, 400,17; найдено 400,2.
Дополнительные соединения по изобретению могут быть получены с использованием следующих исходных материалов:
Сложный этиловый эфир (R)-4-амино-5-бифенил-4-ил-2-гидрокси-2-метилпентановой кислоты

К раствору (R)-3-бифенил-4-ил-2-трет-бутоксикарбониламино-пропионовой кислоты (50 г, 0,1 моль), кислоты Мельдрума (23,3 г, 0,2 моль) и DMAP (27,8 г, 0,2 моль) в безводном DCM (500 мл) добавляли раствор DCC (33,3 г, 0,2 моль) в безводном DCM (200 мл) в течение 1 часа при -5°C в атмосфере азота. Смесь перемешивали при -5°C в течение 8 часов, затем выдерживали в холодильнике в течение ночи, в течение которой осаждались мельчайшие кристаллы дициклогексилмочевины. После фильтрования смесь промывали 5% KHSO4 (4×200 мл), насыщенным водным раствором NaCl (200 мл) и сушили в условиях отрицательной температуры с MgSO4 в течение ночи. Полученный раствор выпаривали с получением неочищенного соединения 1 в виде светло-желтого твердого вещества (68 г). LC-MS: 490 [M+Na], 957 [2M+Na].
К раствору неочищенного соединения 1 (68 г, 0,1 моль) в безводном DCM (1 л) добавляли AcOH (96,8 г, 1,6 моль) при -5°C в атмосфере азота. Смесь перемешивали при -5°C в течение 0,5 часа, затем небольшими порциями в течение 1 часа добавляли NaBH4 (13,9 г, 0,4 моль). После перемешивания при -5°C в течение еще одного часа добавляли насыщенный водный раствор NaCl (300 мл). Органический слой промывали насыщенным водным раствором NaCl (2×300 мл) и водой (2×300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=5:1) с получением соединения 2 в виде светло-желтого твердого вещества (46 г). LC-MS: 476 [M+Na], 929 [2M+Na].

К раствору соединения 2 (46 г, 0,1 моль) в третичном бутиловом спирте (100 мл) добавляли диметилметилениммониййодид (46,3 г, 0,3 моль) при комнатной температуре в атмосфере азота. Смесь нагревали до 65°C и перемешивали при этой температуре в течение 16 часов. После фильтрования фильтрат концентрировали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=20:1-10:1) с получением соединения 3 в виде светло-желтого твердого вещества) (18 г). LC-MS: 460 [M+Na], 897 [2M+Na].
К раствору соединения 3 (18 г, 44 ммоль) в ацетоне (430 мл) и воде (22 мл) добавляли Судан красный в качестве индикатора. Атмосферу озона вводили в смесь при 0°C до исчезновения красного цвета Судана красного. Добавляли диметилсульфид (45 мл), и смесь перемешивали при комнатной температуре в течение ночи. Смесь затем концентрировали, и остаток очищали хроматографией (гексаны:EtOAc=15:1-7:1) с получением соединения 4 в виде светло-желтого твердого вещества (9,5 г). LC-MS: 434 [M+H], 845 [2M+H].

К раствору соединения 4 (9,5 г, 23 ммоль) в безводном THF (120 мл) добавляли раствор метилмагнийбромида в THF (9,2 мл, 28 ммоль) при -70°C в атмосфере азота. Смесь перемешивали при -60°C в течение 3 часов, и реакцию затем гасили насыщенным водным раствором NH4Cl (50 мл). Органический слой отделяли и сушили над MgSO4. Смесь затем концентрировали, и остаток очищали хроматографией (гексаны:EtOAc=l0:1-5:1) с получением соединения 5 в виде масла (7,9 г). LC-MS: 450 [M+H], 877 [2M+H].
В раствор соединения 5 (7,9 г, 18,4 ммоль) в безводном DCM (300 мл) прокачивали атмосферу HCl при 0°C в течение 6 часов. Смесь затем концентрировали, и остаток промывали безводным Et2O с получением указанного в заголовке соединения в виде белой твердой соли HCl (5,8 г). LC-MS: 364 [M+H], 727 [2M+H].
1H ЯМР (300 MГц, DMSO): δ 8,00-7,97 (д, 4H), 7,67-7,62 (м, 6H), 7,47-7,28 (м, 8H), 6,32 (с, 1H), 6,09 (с, 1H), 4,13-4,06 (м, 2H), 3,95-3,78 (м, 2H), 3,60 (с, 1H), 3,22-3,08 (м, 3H), 2,95-2,65 (м, 2H), 1,99-1,79 (м, 4H), 1,30-0,87 (м, 9H).
Сложный этиловый эфир (R)-4-амино-5-бифенил-4-ил-2,2-диметилпентановой кислоты
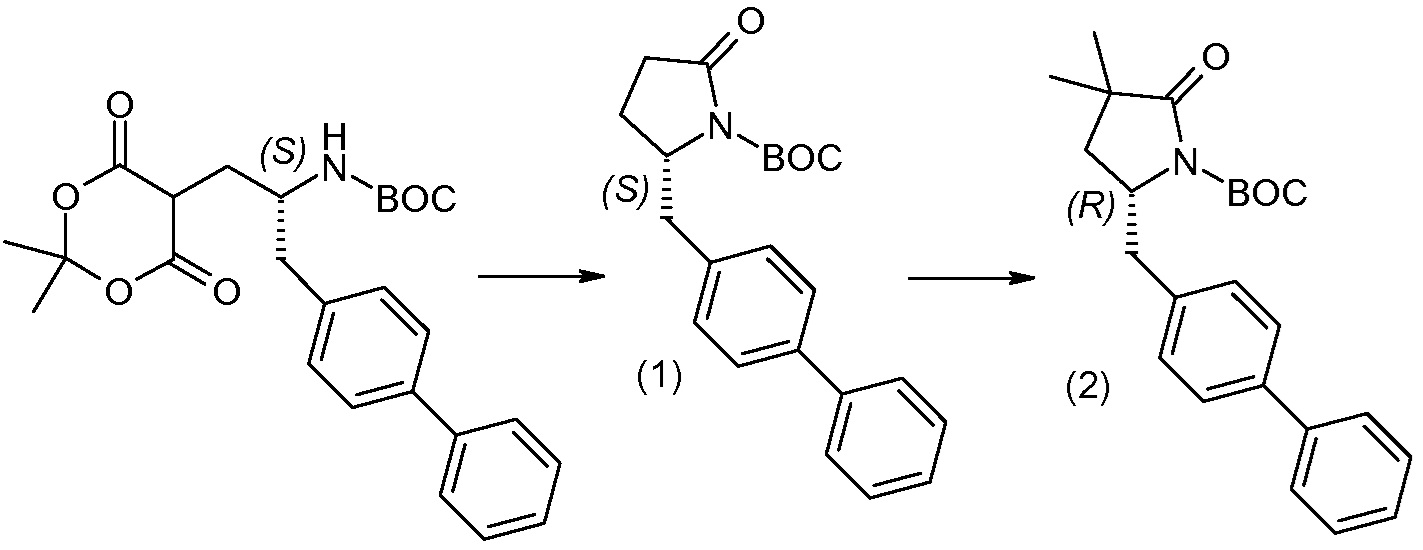
Раствор сложного трет-бутилового эфира [(S)-1-бифенил-4-илметил-2-(2,2-диметил-4,6-диоксо-[1,3]диоксан-5-ил)этил]карбаминовой кислоты (46 г, 0,1 моль) в безводном толуоле (300 мл) кипятили с обратным холодильником в течение 3 часов в атмосфере азота. После выпаривания растворителя остаток очищали хроматографией (гексаны:EtOAc=10:1) с получением соединения 1 в виде светло-желтого твердого вещества (27 г). LC-MS: 374 [M+Na], 725 [2M+Na].
К раствору соединения 1 (6,2 г, 17,6 ммоль) в безводном THF (100 мл) добавляли раствор LiHMDS в THF (39 мл, 39 ммоль) при -78°C в атмосфере азота. Смесь перемешивали при -78°C в течение 2 часов и затем добавляли метилйодид (7,5 г, 53 ммоль). После перемешивания в течение 0,5 часа при -78°C, смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 3 часов. После охлаждения смеси до -10°C, реакцию гасили насыщенным водным раствором NH4Cl (100 мл) и экстрагировали EtOAc (100 мл×4). Объединенные органические слои промывали насыщенным водным раствором NaCl (300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который дополнительно очищали хроматографией (гексаны:EtOAc=10:1) с получением соединения 2 в виде светло-желтого твердого вещества (5,7 г). LC-MS: 402 [M+Na], 781 [2M+Na].
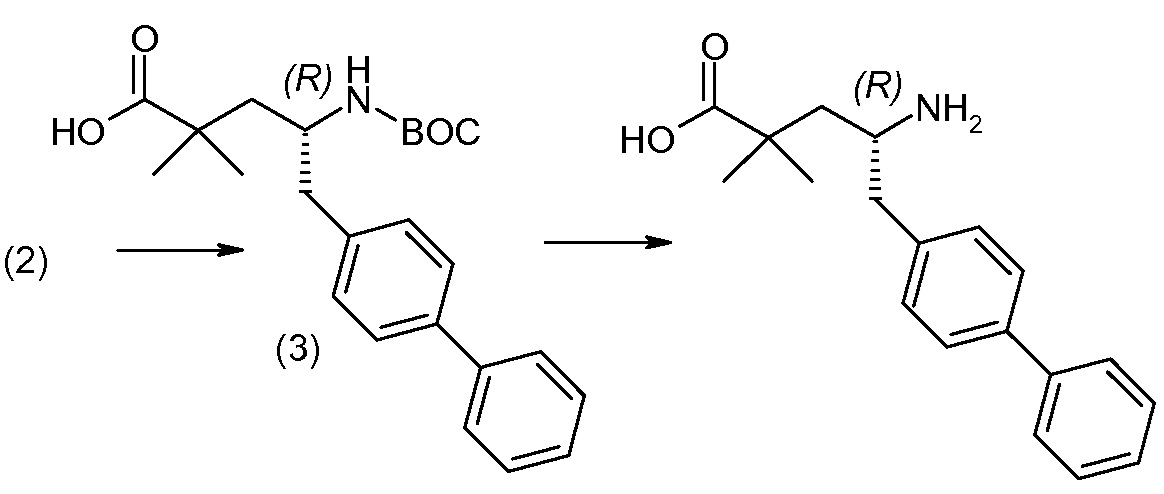
К раствору соединения 2 (5,7 г, 15 ммоль) в ацетоне (120 мл) добавляли 1M NaOH (60 мл, 60 ммоль) при -5°C в атмосфере азота. Смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 20 часов. Смесь концентрировали, и остаток разбавляли водой (250 мл) и промывали EtOAc (150 мл). pH водного слоя доводили до 2 с помощью 6M HCl при 0°C, и твердое вещество фильтровали и сушили в вакууме с получением неочищенного соединения 3 в виде белого твердого вещества (5 г). LC-MS: 420 [M+Na], 817 [2M+Na].
К раствору неочищенного соединения 3 (5 г, 12,7 ммоль) в безводном EtOH (300 мл) добавляли SOCl2 (13,4 мл, 190 ммоль) при -30°C в атмосфере азота. Смесь нагревали до комнатной температуры и перемешивали в течение 20 часов при комнатной температуре. Смесь концентрировали, и остаток промывали безводным Et2O с получением указанного в заголовке соединения в виде белой твердой соли HCl (3,7 г). LC-MS: 326 [M+H], 651 [2M+H].
1H ЯМР (300 MГц, DMSO): δ 7,86 (с, 3H), 7,67-7,64 (м, 4H), 7,49-7,33 (м, 5H), 4,09-3,97 (м, 2H), 3,42 (м, 1H), 2,90-2,80 (м, 2H), 1,88-1,84 (м, 2H), 1,17-1,12 (м, 9H).
1-((R)-2-Амино-3-бифенил-4-илпропил)циклопропанкарбоновая кислота

В колбу, содержащую BOC-D-4,4'-бифенилаланин (11,3 г, 33,1 ммоль, 1,0 экв.), 4-диметиламинопиридин (6,5 г, 53,0 ммоль, 1,6 экв.), 2,2-диметил-1,3-диоксан-4,6-дион (5,3 г, 36,4 ммоль, 1,1 экв.) в DCM (100 мл) добавляли 1M DCC в DCM (38,1 мл) при 0°C в течение 30 минут. Смесь поддерживали при 0°C в течение 6 часов, и полученный осадок отфильтровывали. Фильтрат промывали водным 10% KHSO4 (2×50 мл), затем сушили. Раствор подкисляли AcOH (20 мл) при 0°C, и 3 порциями добавляли боргидрид натрия (3,1 г, 82,7 ммоль, 2,5 экв.) в течение 30 минут. Смесь поддерживали при 0°C в течение 3 часов, промывали водой и сушили, затем концентрировали в вакууме. Неочищенный материал очищали хроматографией (градиент 0-40% EtOAc/гексаны). Добавляли соль Эшенмозера (15,9 г, 86,0 ммоль) в трет-бутиловом спирте (70 мл), и полученную смесь перемешивали при 65°C в течение ночи. Смесь концентрировали и добавляли Et2O (10 мл). Органический раствор затем промывали насыщенным водным раствором NaHCO3 (10 мл) и 10% KHSO4 (10 мл). Органический раствор сушили над Na2SO4и концентрировали. Неочищенный продукт очищали хроматографией (градиент 0-40% EtOAc/гексаны) с получением соединения 1 (3,3 г).

Триметилсульфоксоний йодид (2,0 г, 9,2 ммоль, 1,0 экв.) в диметилсульфоксиде (50 мл) объединяли с NaH (366 мг, 9,2 ммоль, 1,1 экв.) и перемешивали в течение 15 минут при комнатной температуре. К этому добавляли соединение 1 (3,6 г, 8,3 ммоль, 1,0 экв.) растворенное в диметилсульфоксиде (50 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи. Раствор смешивали с насыщенным водным раствором NaCl (50 мл) и экстрагировали EtOAc (3×10 мл), и органический слой промывали насыщенным водным раствором NaCl (2×50 мл) и сушили над безводным Na2SO4. После выпаривания растворителя неочищенный продукт реакции очищали хроматографией (градиент 0-40% EtOAc/гексаны) с получением соединения 2, сложный трет-бутиловый эфир 1-((R)-3-бифенил-4-ил-2-т-бутоксикарбониламинопропил)циклопропанкарбоновой кислоты. Добавляли TFA (200 мкл) и DCM (500 мкл), и полученную смесь перемешивали в течение 30 минут. Растворитель выпаривали в вакууме и азеотропировали толуолом (2×) с получением указанного в заголовке соединения.
АНАЛИЗ 1
Анализы in vitro для количественного определения ингибирующей активности в отношении человеческого и крысиного NEP и человеческого ACE
Ингибирующую активность соединений в отношении человеческого и крысиного неприлизина (EC 3.4.24.11; NEP) и человеческого ангиотензин-превращающего фермента (ACE) определяли, используя описанные ниже анализы in vitro.
Выявление активности NEP в почках крыс
Крысиный NEP получали из почек взрослых крыс Sprague Dawley. Целые почки промывали в холодном солевом растворе, забуференном фосфатом (PBS), и помещали в ледяной лизисный буфер (1% Triton X-l14, 150 мМ NaCl, 50 мМ трис(гидроксиметил)аминометан (Tris) pH 7,5; Bordier (1981) J. Biol. Chem. 256: 1604-1607) в соотношении 5 мл буфера на каждый грамм почечной ткани. Образцы гомогенизировали на льду, используя портативный измельчитель ткани Polytron. Гомогенаты центрифугировали при 1000×g в бакет-роторе в течение 5 минут при 3°C. Клеточный осадок после центрифугирования ресуспендировали в 20 мл ледяного лизисного буфера и инкубировали на льду в течение 30 минут. Образцы (15-20 мл) затем наслаивали на 25 мл ледяного прокладочного буфера (6% масс./об. сахарозы, 50 мМ Tris pH 7,5, 150 мМ NaCl, 0,06%, Triton X-l14), нагревали до 37°C в течение 3-5 минут и центрифугировали при 1000×g в бакет-роторе при комнатной температуре в течение 3 минут. Два верхних слоя удаляли аспирацией, оставляя вязкий маслянистый осадок, содержащий обогащенную мембранную фракцию. Глицерин добавляли до концентрации 50%, и образцы хранили при -20°C. Величины концентрации белка количественно определяли, используя систему выявления BCA с применением бычьего сывороточного альбумина (BSA) в качестве стандарта.
Анализы ингибирования ферментов
Рекомбинантный человеческий NEP и рекомбинантный человеческий ACE получали из коммерческих источников (R&D Systems, Minneapolis, MN, католожные номера 1182-ZN и 929-ZN, соответственно). Флуорогенный пептидный субстрат Mca-D-Arg-Arg-Leu-Dap-(Dnp)-OH (Medeiros et al. (1997) Braz. J. Med. Biol. Res. 30: 1157-62; Anaspec, San Jose, CA) и Abz-Phe-Arg-Lys(Dnp)-Pro-OH (Araujo et al. (2000) Biochemistry 39:8519-8525; Bachem, Torrance, CA) использовали в анализах NEP и ACE, соответственно.
Анализы выполняли в 384-луночных белых непрозрачных планшетах при 37°C с использованием флуорогенных пептидных субстратов в концентрации 10 мкМ в аналитическом буфере (NEP: 50 мМ HEPES, pH 7,5, 100 мм NaCl, 0,01% полиэтиленгликольсорбитан монолаурата (Твин-20), 10 мкМ ZnSO4; ACE: 50 мМ HEPES, pH 7,5, 100 мМ NaCl, 0,01% Твин-20, 1 мкМ ZnSO4). Соответствующие ферменты использовали в концентрациях, которые приводили к количественному протеолизу 1 мкМ субстрата после 20 минут при 37°C.
Тестируемые соединения анализировали по диапазону концентраций от 10 мкМ до 20 пМ. Тестируемые соединения добавляли к ферментам и инкубировали в течение 30 минут при 37°C перед инициацией реакции путем добавления субстрата. Реакции прекращали после 20 минут инкубации при 37°C добавлением ледяной уксусной кислоты до конечной концентрации 3,6% (об./об.).
Планшеты считывали на флуориметре при длинах волн возбуждения и эмиссии, установленных на 320 нм и 405 нм, соответственно. Константы ингибирования получали нелинейной регрессией данных с использованием уравнения (программное обеспечение GraphPad Software, Inc., San Diego, CA):

где ν представляет собой скорость реакции, ν0 представляет собой скорость неингибированной реакции, I представляет собой концентрацию ингибитора, и K' представляет собой видимую константу ингибирования.
Соединения по изобретению тестировали в данном анализе, и было обнаружено, что они имеют следующие величины pKi для NEP. В целом, либо пролекарственные соединения не ингибировали фермент в этом анализе in vitro, либо пролекарства не тестировали (n.d.), поскольку их активность не следовало ожидать.
АНАЛИЗ 2
Фармакодинамический (PD) анализ активности ACE и NEP у анестезированных крыс
Самцов крыс Sprague Dawley с нормальным артериальным давлением анестезировали 120 мг/кг (внутрибрюшинно) инактина. После анестезии вставляли катетеры в яремную вену, сонную артерию (трубка PE 50) и мочевой пузырь (трубка PE 50 с раструбом) и выполняли трахеотомию (тефлоновой иглой 14 калибра) для содействия спонтанному дыханию. Затем животным давали возможность стабилизироваться в течение периода 60 минут и поддерживали непрерывную инфузию 5 мл/кг/ч солевого раствора (0,9%) в течение всего теста для предотвращения дегидратации и обеспечения продукции мочи. Температуру тела поддерживали в течение всего эксперимента использованием греющей накладки. В конце 60-минутного периода стабилизации животным внутривенно (в/в) вводили две дозы AngI (1,0 мкг/кг, для выявления ингибирующей активности в отношении ACE) с интервалом 15 минут. Через 15 минут после введения второй дозы AngI, животных вводили носитель или тестируемое соединение. Через пять минут животных дополнительно обработали болюсной в/в инъекцией предсердного натрийуретического пептида (ANP; 30 мкг/кг). Сбор мочи (в предварительно взвешенные пробирки Эппендорфа) начинали сразу после введения ANP и продолжали в течение 60 минут. Через 30 и 60 минут после начала сбора мочи у животных проводили пробу с повторным введением AngI. Измерения артериального давления проводили, используя устройство Notocord (Kalamazoo, MI). Образцы мочи замораживали при -20°C до использования для анализа cGMP. Концентрации cGMP в моче определяли иммуноферментным анализом с использованием имеющегося в продаже набора (Assay Designs, Ann Arbor, Michigan, каталожный № 901-013). Объем мочи определяли гравиметрически. Выход cGMP с мочой рассчитывали как продукт диуреза и концентрации cGMP в моче. Ингибирование ACE оценивали количественным определением % ингибирования прессорной реакции на AngI. Ингибирование NEP оценивали количественным определением потенциирования вызванного ANP повышения выхода cGMP с мочой.
АНАЛИЗ 3
Оценка in vivo антигипертензивных эффектов на модели гипертензии у бодрствующих крыс SHR
Спонтанно гипертензивным крысам (SHR, в возрасте 14-20 недель) предоставляли возможность акклиматизироваться минимум в течение 48 часов после доставки в место тестирования при свободном доступе к пище и воде. Для регистрации артериального давления этим животным хирургически имплантировали радиопередатчики для мелких грызунов (телеметрический узел; DSI Models TA1 1PA-C40 или C50-PXT, Data Science Inc., USA). Кончик катетера, соединенный с передатчиком, вводили в нисходящую аорту над подвздошным разветвлением и фиксировали на месте тканевым клеем. Передатчик удерживали внутрибрюшинно и фиксировали к брюшной стенке при зашивании брюшного разреза нерассасываемой нитью. Наружный слой кожи зашивали нитями и скобками. Животным давали возможность восстановиться при должном послеоперационном уходе. В день эксперимента животных в их клетках помещали поверх блоков телеметрических приемников для акклиматизации к среде тестирования и регистрации исходных показателей. По меньшей мере через 2 часа после измерения исходных показателей животным затем вводили носитель или тестируемое соединение, и в последующем проводили контрольное измерение артериального давления в течение 24 часов после введения. Данные регистрировали непрерывно в течение всего исследования с использованием программного обеспечения Notocord (Kalamazoo, MI) и хранили в виде электронных цифровых сигналов. Измеряемыми параметрами были артериальное давление (систолическое, диастолическое и среднее артериальное давление) и частота сердечных сокращений.
АНАЛИЗ 4
Оценка in vivo антигипертензивных эффектов на модели гипертензии, вызванной DOCA, у бодрствующих крыс
Крысам CD (взрослым самцам с массой тела 200-300 граммов, Charles River Laboratory, USA) предоставляли возможность акклиматизироваться в течение по меньшей мере 48 часов после доставки в место тестирования, перед тем как им назначали рацион с высоким содержанием соли. Через одну неделю после начала рациона с высоким содержанием соли (8% в корме или 1% NaCl в питьевой воде), подкожно имплантировали пеллету деоксикортикостерона ацетата (DOCA) (100 мг, время высвобождения 90 дней, Innovative Research of America, Sarasota, FL) и выполняли одностороннюю нефрэктомию. В это время животным также хирургически имплантировали радиопередатчики для мелких грызунов для измерения артериального давления (детали описаны в анализе 3). Животным давали возможность восстановиться при должном послеоперационном уходе. Структура исследования, регистрация данных и измеряемые параметры аналогичны тем, которые описаны для анализа 3.
АНАЛИЗ 5
Оценка in vivo антигипертензивных эффектов на модели гипертензии у бодрствующих крыс Dahl/SS
Самцам чувствительных к соли крысам Dahl (Dahl/SS, в возрасте 6-7, Charles River Laboratory, USA) предоставляли возможность акклиматизироваться в течение по меньшей мере 48 часов после доставки в место тестирования перед тем, как им назначали рацион с высоким содержанием соли (8% NaCl) (TD.92012, Harlan, USA), затем хирургически имплантировали радиопередатчики для мелких грызунов для измерения артериального давления (детали описаны в анализе 3). Животным давали возможность восстановиться при должном послеоперационном уходе. Ожидалось, что через приблизительно 4-5 недель от начала скармливания рациона с высоким содержанием соли у этих животных разовьется гипертензия. После подтверждения уровня гипертензии этих животных использовали для исследования при продолжении скармливания рациона с высоким содержанием соли для поддержания у них уровня гипертензии. Структура исследования, регистрация данных измеряемые параметры аналогичны тем, которые описаны для анализа 3.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 1
(2R,4S)-5-Бифенил-4-ил-2-метил-4-(оксалиламино)пентановая кислота (Сравнительное соединение A; R = -C(O)-COOH)
Сложный этиловый эфир (2R,4S)-4-амино-5-бифенил-4-ил-2-метилпентановой кислоты (соль HCl; 527 мг, 0,2 ммоль) и этилоксалилхлорид (18,4 мкл, 1,1 экв.) объединяли с DIPEA (52,2 мкл, 0,3 ммоль) в DMF (0,3 мл)/DCM (0,3 мл). Смесь перемешивали при комнатной температуре до завершения реакции. Растворитель удаляли, и остаток растворяли в EtOH (750 мкл) и 1M водном NaOH (750 мкл) и перемешивали при комнатной температуре в течение ночи. Растворитель удаляли, и остаток очищали препаративной ВЭЖХ с получением сравнительного соединения A (11,2 мг, чистота 100%). MS m/z [M+H]+ рассчитано для C20H21NO5, 356,14; найдено 356,2.
(2R,4S)-5-Бифенил-4-ил-4-(3-карбоксипропиониламино)-2-метилпентановая кислота (сравнительное соединение B; R=-C(O)-(CH2)2-COOH)
Сложный этиловый эфир (2R,4S)-5-бифенил-4-ил-4-(3-карбоксипропиониламино)-2-метилпентановой кислоты (соль Na; 400 мг, 923 мкмоль) смешивали с EtOH (7 мл, 0,1 моль), затем THF (6 мл, 0,1 моль). Затем добавляли 1M водный NaOH (2,8 мл, 2,8 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 4 часов и затем концентрировали. Продукт очищали препаративной ВЭЖХ (10-60% MeCN:вода масс./0,5% TFA) с получением сравнительного соединения B (150 мг, чистота 97%). MS m/z [M+H]+ рассчитано для C22H25NO5, 384,17; найдено 384,6.
Сравнительные соединения A и B тестировали, как описано в анализе 1, и было найдено, что они имели следующие величины pKi в отношении человеческого NEP:
Эти данные показывают, что Сравнительные соединения A и B имеют одинаковые величины pKi для ингибирования NEP.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 2

(R)-5-Бифенил-4-ил-4-(2-карбоксиацетиламино)-2-гидроксипентановая кислота (Сравнительное соединение C; R=-C(O)-CH2-COOH)
Сложный этиловый эфир (R)-4-амино-5-бифенил-4-ил-2-гидроксипентановой кислоты (соль HCl; 60,3 мг, 0,2 ммоль) и метилмалонилхлорид (21 мкл, 0,2 ммоль) объединяли с DIPEA (84 мкл, 0,5 ммоль) в DMF (5 мл). Смесь перемешивали при комнатной температуре до завершения реакции (1 час). Растворитель удаляли, и остаток растворяли в MeOH (3 мл) и 10N NaOH (250 мкл) и перемешивали при 60°C до завершения реакции (1 час). Добавляли ледяную уксусную кислоту (250 мкл), и продукт выпаривали при пониженном давлении и очищали препаративной ВЭЖХ с получением сравнительного соединения C (6,3 мг, чистота 98%). MS m/z [M+H]+ рассчитано для C20H21NO6, 372,14; найдено 372,2.
(R)-5-Бифенил-4-ил-4-(3-карбоксипропиониламино)-2-гидроксипентановая кислота (сравнительное соединение D; R=-C(O)-(CH2)2-COOH)
Сложный этиловый эфир (R)-4-амино-5-бифенил-4-ил-2-гидроксипентановой кислоты (соль HCl; 60,3 мг, 0,2 ммоль) и 3-(карбометокси)пропионилхлорид (24 мкл, 0,2 ммоль) объединяли с DIPEA (84 мкл, 0,5 ммоль) в DMF (5 мл). Смесь перемешивали при комнатной температуре до завершения реакции (1 час). Растворитель удаляли, и остаток растворяли в MeOH (3 мл) и 10N NaOH (250 мкл), и перемешивали при 60°C до завершения реакции (1 час). Добавляли ледяную уксусную кислоту (250 мкл), и продукт выпаривали при пониженном давлении и очищали препаративной ВЭЖХ с получением сравнительного соединения D (8,0 мг, чистота 100%). MS m/z [M+H]+ рассчитано для C21H23NO6, 386,15; найдено 386,2.
(R)-5-Бифенил-4-ил-4-(4-карбоксибутириламино)-2-гидроксипентановая кислота (сравнительное соединение E; R=-C(O)-(CH2)3-COOH)
Сложный этиловый эфир (R)-4-амино-5-бифенил-4-ил-2-гидроксипентановой кислоты (соль HCl; 60,3 мг, 0,2 ммоль) и метил-5-хлор-5-оксовалерат (31,7 мг, 0,2 ммоль) объединяли с DIPEA (84 мкл, 0,5 ммоль) в DMF (5 мл). Смесь перемешивали при комнатной температуре до завершения реакции (1 час). Растворитель удаляли, и остаток растворяли в MeOH (3 мл) и 10N NaOH (250 мкл), и перемешивали при 60°C до завершения реакции (1 час). Добавляли ледяную уксусную кислоту (250 мкл), и продукт выпаривали при пониженном давлении и очищали препаративной ВЭЖХ с получением сравнительного соединения E (8,7 мг, чистота 100%). MS m/z [M+H]+ рассчитано для C22H25NO6, 400,17; найдено 400,2.
Соединение примера 1 и сравнительные соединения C, D и E тестировали, как описано в анализе 1, и было найдено, что они имели следующие величины pKi в отношении человеческого NEP:
Эти данные показывают, что соединение примера 1 обладало более высокой активностью в отношении NEP, чем сравнительные соединения C, D и E.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 3

(2S,4S)-5-Бифенил-4-ил-4-(2-карбоксиацетиламино)-2-гидроксиметилпентановая кислота (сравнительное соединение F; R=-C(O)-CH2-COOH)
(2S,4S)-4-Амино-5-бифенил-4-ил-2-гидроксиметилпентановую кислоту (соль HCl; (5 мг, 10 мкмоль) растворяли в 1M водном NaOH (119 мкл, 119 мкмоль) и медленно добавляли к раствору метилмалонилхлорида (1,9 мкл, 18 мкмоль) и MeCN (0,5 мл, 10 ммоль). Полученный раствор перемешивали при комнатной температуре до завершения реакции (в течение ночи), и продукт очищали препаративной ВЭЖХ с получением сравнительного соединения F (1,0 мг, чистота 95%). MS m/z [M+H]+ рассчитано для C21H23NO6, 386,15; найдено 386,1.
(2S,4S)-5-Бифенил-4-ил-4-(3-карбоксипропиониламино)-2-гидроксиметилпентановая кислота (сравнительное соединение G; R=-C(O)-(CH2)2-COOH)
(2S,4S)-4-Амино-5-бифенил-4-ил-2-гидроксиметилпентановую кислоту (соль HCl; (5 мг, 10 мкмоль) растворяли в 1M водном NaOH (119 мкл, 119 мкмоль) и медленно добавляли к раствору 3-(карбометокси)пропионилхлорида (2,2 мкл, 18 мкмоль) и MeCN (0,5 мл, 10 ммоль). Полученный раствор перемешивали при комнатной температуре до завершения реакции (в течение ночи), и продукт очищали препаративной ВЭЖХ с получением сравнительного соединения G (3,4 мг, чистота 95%). MS m/z [M+H]+ рассчитано для C22H25NO6, 400,17; найдено 400,3.
(2S,4S)-5-Бифенил-4-ил-4-(4-карбоксибутириламино)-2-гидроксиметилпентановая кислота (сравнительное соединение H; R=-C(O)-(CH2)3-COOH)
(2S,4S)-4-Амино-5-бифенил-4-ил-2-гидроксиметилпентановую кислоту (соль HCl; (5 мг, 10 мкмоль) растворяли в 1M водном NaOH (119 мкл, 119 мкмоль) и медленно добавляли к раствору метил-5-хлор-5-оксовалерата (2,5 мкл, 18 мкмоль) и MeCN (0,5 мл, 10 ммоль). Полученный раствор перемешивали при комнатной температуре до завершения реакции (в течение ночи), и продукт очищали препаративной ВЭЖХ с получением сравнительного соединения H (3,0 мг, чистота 95%). MS m/z [M+H]+ рассчитано для C23H27NO6, 414,18; найдено 414,7.
Соединение примера 5A и сравнительных соединений F, G и H тестировали, как описано в примере 1, и было найдено, что они имели следующие величины pKi в отношении человеческого NEP:
Эти данные показывают, что соединение примера 5A обладало более высокой активностью в отношении NEP, чем сравнительные соединения F, G, и H.
СРАВНИТЕЛЬНЫЙ ПРИМЕР 4

(2R,4R)-4-(2-Карбоксиацетиламино)-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановая кислота (сравнительное соединение I; R=-C(O)-CH2-COOH)
Метилмалонилхлорид (18,5 мкл, 172 мкмоль) добавляли к раствору сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50,0 мг, 144 мкмоль) и DIPEA (75,1 мкл, 431 мкмоль) в DCM (1,5 мл, 23,4 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Смесь затем концентрировали с получением желтой жидкости. 1M водный раствор LiOH (719 мкл, 719 мкмоль) добавляли по каплям к маслу, и смесь перемешивали при 60°C в течение 1 часа. Смесь концентрировали в вакууме, и полученный остаток растворяли в AcOH (1,0 мл), очищали препаративной ВЭЖХ с получением сравнительного соединения I (2,0 мг, чистота 100%). MS m/z [M+H]+ рассчитано для C20H20ClNO6, 406,10; найдено 406,1.
(2R,4R)-4-(3-Карбоксипропиониламино)-5-(3'-хлорбифенил-4-ил)-2-гидроксипентанoевая кислота (сравнительное соединение J; R=-C(O)-(CH2)9-COOH)
3-(Карбометокси)пропионилхлорид (21,2 мкл, 172 мкмоль) добавляли к раствору сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50,0 мг, 144 мкмоль) и DIPEA (75,1 мкл, 431 мкмоль) в DCM (1,5 мл, 23,4 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Смесь затем концентрировали с получением желтой жидкости. 1M водный раствор LiOH (719 мкл, 719 мкмоль) добавляли по каплям к маслу, и смесь перемешивали при 60°C в течение 1 часа. Смесь концентрировали в вакууме, и полученный остаток растворяли в AcOH (1,0 мл), очищали препаративной ВЭЖХ с получением сравнительного соединения J (31,1 мг, чистота 100%). MS m/z [M+H]+ рассчитано для C21H22ClNO6, 420,11; найдено 420,2.
(2R,4R)-4-(4-Карбоксибутириламино)-5-(3'-хлор-бифенил-4-ил)-2-гидроксипентанoевая кислота (сравнительное соединение K; R=-C(O)-(CH2)3-COOH)
Метил 5-хлор-5-оксовалерат (23,8 мкл, 172 мкмоль) добавляли к раствору сложного этилового эфира (2R,4R)-4-амино-5-(3'-хлорбифенил-4-ил)-2-гидроксипентановой кислоты (50,0 мг, 144 мкмоль) и DIPEA (75,1 мкл, 431 мкмоль) в DCM (1,5 мл, 23,4 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Смесь затем концентрировали с получением желтой жидкости. 1M водный раствор LiOH (719 мкл, 719 мкмоль) добавляли по каплям к маслу, и смесь перемешивали при 60°C в течение 1 часа. Смесь концентрировали в вакууме, и полученный остаток растворяли в AcOH (1,0 мл), очищали препаративной ВЭЖХ с получением сравнительного соединения K (29,2 мг, чистота 100%). MS m/z [M+H]+ рассчитано для C22H24ClNO6, 434,13; найдено 434,2.
Соединение примера 2A и сравнительные соединения I, J и K тестировали, как описано в анализе 1, и было найдено, что они имели следующие величины pKi в отношении человеческого NEP:
Эти данные показывают, что соединение примера 2A обладало более высокой активностью в отношении NEP, чем сравнительные соединения I, J и K.
Хотя настоящее изобретение было описано со ссылкой на его определенные аспекты или варианты осуществления, специалистам в данной области техники следует понимать, что могут быть сделаны различные изменения или эквивалентные замены без отхода от действительной сущности и объема изобретения. Кроме того, в той степени, которая допускается применимыми патентными законами и нормативными актами, все публикации, патенты и патентные заявки, приведенные в настоящем описании, полностью включены в него путем ссылки в такой же степени, как если бы каждый документ был отдельно включен в него путем ссылки.
Реферат
Изобретение относится к соединениям формулы I или их фармацевтически приемлемым солям, которые обладают способностью ингибировать активность неприлизина (NEP). В формуле I Rвыбирают из Н, -Салкила, -Cалкилен-ОС(О)R, -СН-пиридинила, -(СН)-пиридинила и; Rвыбирают из -Cалкила, -O-Cалкила и -CH(R)-NHC(О)O-Cалкила; Rпредставляет собой -Cалкил; Rпредставляет собой -СН(СН); Rпредставляет собой -ORили -CHOR; и Rпредставляет собой Н или -СН; где Rпредставляет собой Н; или Rвзят вместе с Rс образованием -СН-СН-; Z выбирают из -СН- и -N-; Rвыбирают из Н, -Cалкила, -Салкилен-O-Салкила, -Cалкилен-Сарила, -[(СН)О]СНи; Rпредставляет собой -Cалкил; а равно 0 или 1; Rвыбирают из галогена, -СН, -CFи -CN; b равно 0 или целому числу от 1 до 3; каждый Rнезависимо выбирают из галогена, -ОН, -СН, -ОСН, -CN и -CF, причем каждая алкильная группа в Rи Rнеобязательно замещена 1-8 атомами фтора и метиленовый линкер на бифениле необязательно замещен одной или двумя -Cалкильными группами. Изобретение относится также к способу получения соединений формулы I, содержащей их фармацевтической композиции и применению указанных соединений для получения лекарственного средства для лечения гипертензии, сердечной недостаточности или почечного заболевания. 4 н. и 36 з.п. ф-лы, 16 пр.
Формула
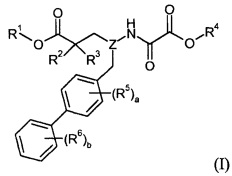
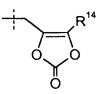
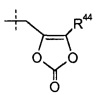



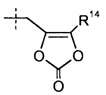





Документы, цитированные в отчёте о поиске
Способ получения 5-бифенил-4-амино-2-метилпентановой кислоты
Комментарии