Способ улучшения биоактивации лекарственных веществ - RU2550969C2
Код документа: RU2550969C2
Чертежи


Описание
Настоящее изобретение относится к способу улучшения биоактивации лекарственных веществ.
Условием терапевтического эффекта лекарственного вещества после перорального введения является его всасывание из желудочно-кишечного тракта. Важнейшим механизмом такого эффекта является пассивная диффузия. Степень резорбции на пути пассивной диффузии зависит, кроме прочего, от липофилии.
Следующей проблемой при медикаментозном лечении многих болезней является необходимость прохождения гематоэнцефалического барьера. Гематоэнцефалический барьер является эффективным барьером в отношении поглощения веществ в головном мозге. Он обеспечивает избирательное поглощение и препятствует проникновению веществ. Кроме того, гематоэнцефалический барьер действует не только как физический, но и как ферментантивный барьер. В пенетрации веществ в мозг задействованы различные процессы. В продаже имеется мало, по сравнению с другими показаниями, лекарственных средств, которые предназначены для воздействия на центральную нервную систему (ЦНС). Из них преобладающая часть попадает в ЦНС путем диффузии. Таким способом лечат такие болезни как эпилепсия, хронические боли или депрессии. Другие имеющие большое значение функциональные расстройства, как, например, опухоли мозга или боковой амиотрофический склероз, в настоящее время плохо лечатся таким способом.
Чтобы быть способным преодолеть биомембраны пассивной диффузией, вещество должно быть липофильным, иметь молекулярный вес меньше 500 Да, и оно должно быть незаряженным. Чтобы специфично поглощать маленькие сильно полярные молекулы, как аминокислоты или сахар, в биомембранах с барьерной функцией (желудочно-кишечный тракт, гематоэнцефалический барьер) экспрессируются различные транспортные системы, как, например, транспортер нуклеозида, транспортер притока и истечения для органических анионов или катионов, транспортер глюкозы, транспортер пептидов и транспортер аминокислот.
Поэтому для улучшения фармакокинетических свойств привлекаются различные системы пролекарств. Под пролекарством имеется в виду фармакологически неактивное или очень малоактивное лекарственное вещество, которое переходит в активный метаболит только после метаболизации в организме.
N-гидроксиамидины (амидоксимы) и N-гидроксигуанидины представляют собой известные концепции пролекарств для повышении я пероральной биодоступности амидинов [Clement, B. Способы лечения и профилактики пневмонии Pneumocystis carinii (PCP) и других заболеваний, а также соединения и композиции для применения в указанных способах. [DE 4321444]] и гуанидинов. Так как атомы азота амино- и иминогруппы находятся в солях амидинов и гуанидинов в мезомерном равновесии, эти идеи могут применяться к обоим атомам азота.
Преобразование в активный метаболит протекает при этом, в зависимости от принципа, лежащего в основе пролекарства, через различные ферментные системы. Ферментной системой, имеющейся практически во всех формах жизни, является цитохром-P450 (CYP450), который, наряду с прочими, катализирует следующие реакции:
N-окисление, S-окисление, N-дезалкилирование, O-дезалкилирование, S-дезалкилирование, дезаминирование, дегалогенирование, а также гидроксилирование ароматических и алифатических соединений.
Разносторонность ферментной системы CYP450 влечет за собой то, что различные субстраты и лекарственные средства конкурируют при их превращении в систему. Происходят интеракции, взаимодействия и нежелательные взаимные влияния. Поэтому при разработке пролекарств стремятся к биоактивации, не зависящей от CYP450.
Поэтому задачей изобретения является создать пролекарственную систему, использующую способ биоактивации, который не зависит от фермента цитохром-P450 (CYP450). Настоящее изобретение, охарактеризованное в формуле изобретения, позволяет решить эту задачу. Зависимые пункты указывают предпочтительные формы осуществления изобретения.
Согласно изобретению, в первом аспекте задача решена обеспечением пролекарства, содержащего фрагмент структуры, имеющий общую формулу (I) или (II)

причем R1 и R2 означают водород, алкильный или арильный остаток.
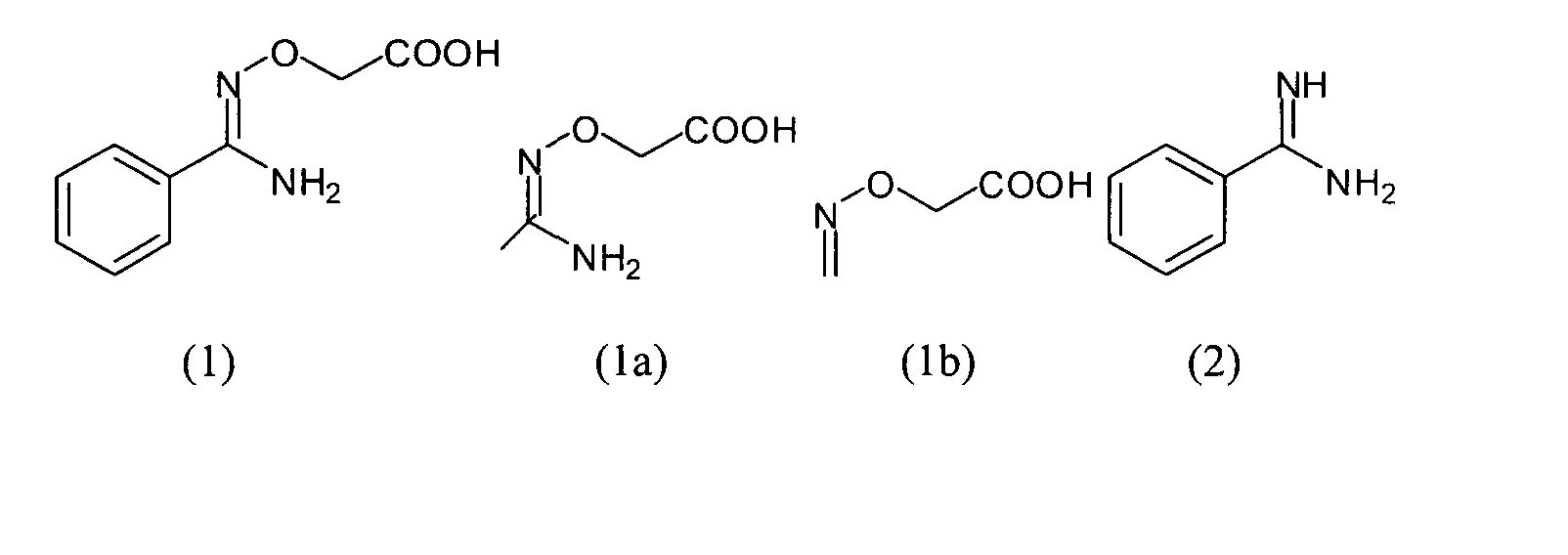
В одном предпочтительном варианте осуществления изобретения термин "фрагмент структуры", как он применяется здесь, означает, что структурный элемент, задаваемый соответствующей формулой, является частью формулы вещества, предпочтительно пролекарства. Например, соединение O-карбоксиметилбензамидоксим (1) является соответствующим пролекарством лекарственного вещества бензамидин, причем фрагмент структуры является фрагментом структуры формулы (II), и каждый из R1 и R2 является атомом водорода. Этот фрагмент структуры является заместителем у бензольного кольца и вместе с ним представляет лекарственное вещество бензамидин.
В одном предпочтительном варианте осуществления изобретения термин "пролекарство", как он используется здесь, означает вещество, которое само по себе является неактивным или лишь очень фармакологически малоактивным веществом, которое лишь в результате обмена веществ (метаболизации) в организме переходит в фармакологически активное лекарственное вещество. Пролекарство может, но не обязательно, иметь лучшую пероральную биодоступность, чем собственно активно-действующее лекарственное вещество. Альтернативно, пролекарство может применяться, если оно по сравнению с лекарственным веществом имеет лучшую растворимость, биоактивацию, проходимость гематоэнцефалического барьера, физико-химическую стабильность, меньшую токсичность и/или допустимый или приятный вкус. Так, 2'-этилсукцинат из эритромицина A назначается детям как пролекарство только из-за горького вкуса эритромицина A, а не из-за его недостаточной резорбции или растворимости (Bhadra et al. (2005), J. Med. Chem.).
В следующем предпочтительном варианте осуществления изобретения изначальное пролекарство метаболизируется из пролекарства в лекарственное вещество не в одностадийной реакции, а через несколько реакционных стадий, причем каждый метаболит, образованный на реакционной стадии, может иметь одно или несколько одинаковых и/или разных выгодных свойств по сравнению с исходным пролекарством. При этом возможно, что не все метаболиты имеют выгодные свойства по сравнению с пролекарством. Например, первый продукт метаболизма пролекарства может иметь более высокую фармакологическую активность, чем пролекарство, второй продукт метаболизма, полученный из первого продукта метаболизма, также может иметь повышенную фармакологическую активность по сравнению с пролекарством, а третий продукт метаболизма, произведенный из второго продукта метаболизма, может иметь повышенные по сравнению с пролекарством проходимость гематоэнцефалического барьера и физико-химическую стабильность.
В одном предпочтительном варианте осуществления изобретения под термином "физико-химическая стабильность", как он используется здесь, понимается способность вещества, например, пролекарства или лекарственного вещества, храниться и/или применяться в форме релевантного водного раствора, например, растворенного в воде, буфере или физиологическом растворе поваренной соли, без химического разложения, например, гидролиза. В следующем предпочтительном варианте осуществления изобретения это понятие, как оно используется здесь, означает, что вещество может быть синтезировано в стабильной и синтетической форме. В следующем предпочтительном варианте осуществления изобретения это понятие, как оно используется здесь, означает, что при синтезе вещества отдельные, релевантные стадии синтеза более стабильны по сравнению с аналогичными продуктами, предшественниками или промежуточными продуктами в случае других веществ, которые были получены по аналогичной или такой же стратегии синтеза, настолько, что продукты или промежуточные продукты последующего синтеза получены в более стабильной форме или вообще могут быть получены только так.
В одном варианте осуществления задача решена путем предоставления пролекарства, отличающегося тем, что фрагмент структуры, входящий в пролекарство, является элементом гидроксиламина, N-оксида, нитрона, диазенийдиолята (NONOат) или подобного Ν-O-содержащего донора моноксида азота; гидроксамовой кислоты, гидроксимочевины, оксима, амидоксима (N-гидроксиамидина), N-гидроксиамидиногидразона или N-гидроксигуанидина.
Например, в случае, когда карбоксиметилбензамидоксим (1) является пролекарством лекарственного вещества бензамидин, фрагмент структуры является фрагментом структуры формулы (II), и каждый из R1 и R2 означает атом водорода, а имеющийся в пролекарстве фрагмент структуры является элементом амидоксима (Ν-гидроксиамидина).
В одном варианте осуществления задача решена предоставлением пролекарства, отличающегося тем, что пролекарство метаболизируется с образованием лекарственного вещества, которое является лекарственным веществом для лечения заболевания, вызванного дефицитом моноксида азота.
В одном варианте осуществления задача решена предоставлением пролекарства, отличающегося тем, что пролекарство или соответствующее лекарственное вещество выбрано из группы, которая содержит ингибиторы протеазы, интеркалирующие ДНК и РНК соединения, ингибиторы вирусных ферментов и антагонисты рецептора N-метил-D-аспартата.
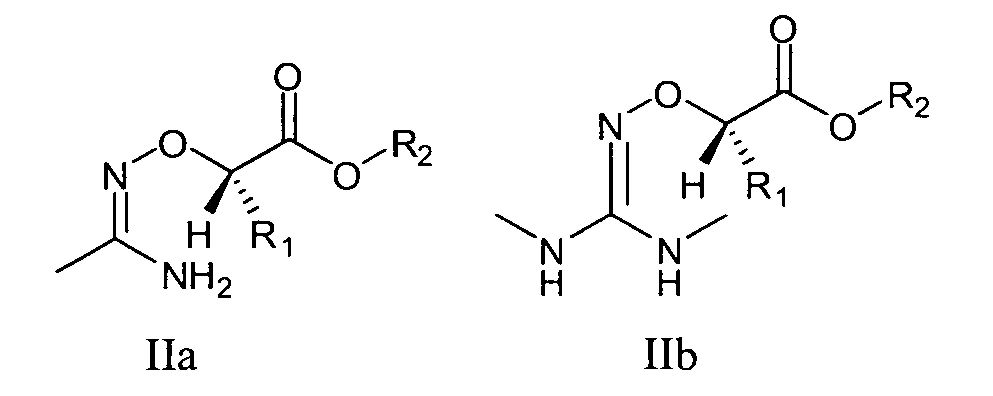
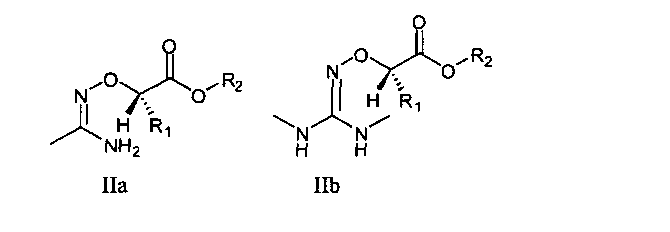
В одном предпочтительном варианте осуществления настоящего изобретения понятие "фрагмент структуры более высокого уровня", как он используется здесь, следует понимать так, что этот фрагмент структуры более высокого уровня, с одной стороны, включает в себя фрагмент структуры формулы (I) или (II), а с другой стороны является частью полной структуры соответствующего вещества. Например, в случае, когда предшественником лекарственного вещества бензамидин (2) является пролекарство карбоксиметилбензамидоксим (1), фрагмент структуры более высокого уровня, обозначенный здесь (1a), является фрагментом структуры формулы (IIa), в которой R1 и R2 означают водород, а фрагмент структуры, обозначенный здесь (1b), является фрагментом структуры формулы (II), причем R1 и R2 также означают водород.
В одном варианте осуществления задача решена предоставлением пролекарства, отличающегося тем, что фрагмент структуры имеет общую формулу IIa или IIb.

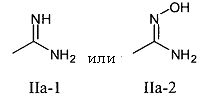
Например, в случае карбоксиметилбензамидоксима (1), являющегося пролекарством лекарственного вещества бензамидин, вышеуказанный фрагмент структуры является фрагментом структуры формулы (IIa), в которой R1 и R2 означают водород, тогда фрагмент структуры является фрагментом структуры формулы (II), причем R1 и R2 также означают водород, и лекарственное вещество вместо фрагмента структуры формулы (IIa) в пролекарстве имеет структуру (IIa-1).
В одном варианте осуществления изобретения задача решается предоставлением пролекарства, отличающегося тем, что пролекарство является предшественником лекарственного вещества, причем фрагмент структуры общей формулы IIa после метаболизации имеет структуру, отвечающую формуле
а фрагмент структуры общей формулы IIb после метаболизации имеет структуру, отвечающую формуле

В следующем аспекте изобретения задача решена применением фрагмента структуры, имеющего общую формулу (I) или (II)
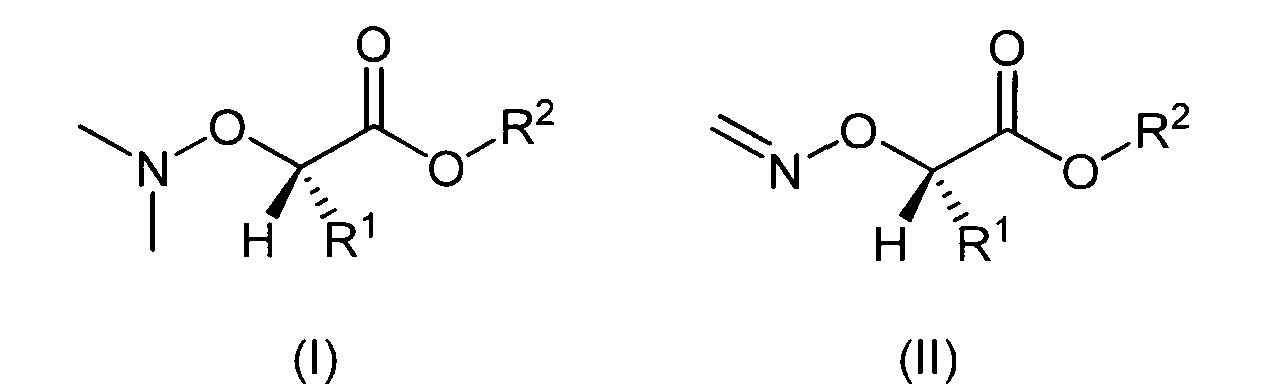
в качестве компонента полной структуры пролекарства, причем это пролекарство является предшественником лекарственного вещества, причем R1 и R2 означают водород, алкильный или арильный остаток.
В одном варианте осуществления задача решена применением пролекарства, причем фрагмент структуры имеет общую формулу (II) и является частью фрагмента структуры более высокого уровня, IIa или IIb,
в качестве замены амидиновой или гуанидиновой группы лекарственного вещества для улучшения растворимости, пероральной биодоступности, проходимости гематоэнцефалического барьера, вкуса и/или физико-химической стабильности.
В одном варианте осуществления задача решена применением пролекарства, причем пролекарство является предшественником лекарственного вещества, имеющего такую же структуру, что и пролекарство, за исключением того, что оно вместо фрагмента структуры более высокого уровня, IIa, содержит фрагмент структуры IIa-1 или IIa-2,
или вместо фрагмента структуры более высокого уровня, IIb, имеет фрагмент структуры IIb-1 или IIb-2.
В одном варианте осуществления задача решена применением пролекарства для активации лекарственного вещества посредством пептидилглицин-α-амидирующей монооксигеназы (PAM).
В одном предпочтительном варианте осуществления изобретения термин "активация пролекарства посредством пептидилглицин-α-амидирующей монооксигеназы (PAM)", "активация пролекарства способом PAM-активации", биоактивация или подобное, как это используется здесь, означает, что пролекарство распознается PAM как субстрат и метаболизируется. В одном предпочтительном варианте осуществления изобретения выражение "введение лекарственного вещества в путь PAM-активации, включающий в себя получение пролекарства лекарственного вещества", как оно используется здесь, понимается, что из вводимого в путь PAM-активации лекарственного вещества получена соответствующая пролекарственная форма, которая распознается PAM и метаболизируется. В одном предпочтительном варианте осуществления сродство пролекарства к PAM, как может определить специалист с помощью значений KM, в 1-1000 раз, в 2-100 раз, в 3-50 раз, в 4-40 раз, в 5-20 раз или в 6-15 раз выше, чем сродство к лекарственному веществу.
В одном варианте осуществления задача решена за счет применения пролекарства, отличающегося тем, что фрагмент структуры является элементом гидроксиламина, N-оксида, нитрона, диазенийдиолята (NONOат) или подобного N-O-содержащего донора моноксида азота; гидроксамовой кислоты, гидроксимочевины, оксима, амидоксима (N-гидроксиамидина), N-гидроксиамидиногидразона или N-гидроксигуанидина.
В следующем аспекте изобретения задача решена способом введения лекарственного вещества, содержащего свободную амидиновую или гуанидиновую функциональную группу, в путь PAM-активации, содержащим получение пролекарства лекарственного вещества.
В следующем аспекте изобретения задача решена способом лечения пациента, включающим введение пациенту пролекарства.
В следующем аспекте изобретения задача решена путем применения пролекарства для получения лекарственного вещества.
В одном предпочтительном варианте осуществления изобретения лекарственное вещество является лекарственным веществом, соответственно пролекарство является пролекарством для борьбы с вирусными инфекциями, как грипп, для борьбы с ВИЧ-инфекцией, для профилактики и лечения висцерального и кожного лейшманиоза, для профилактики пневмонии, вызванной Pneumocystis carinii (PcP), для лечения трипаносомоза (африканской сонной болезни), для лечения малярии, для лечения бабезиоза, для подавления свертывания крови, например, для первичной профилактики венозных тромбоэмболических осложнений, для профилактики апоплексии у пациентов с мерцанием предсердий, для снижения кровяного давления, для подавления роста злокачественных опухолей, для нейропротекции, для борьбы с вирусными инфекциями, как грипп, для (с точки зрения диуретики) вымывания воды из тела, например, при сердечной недостаточности, отеке легких, отравлениях, почечной недостаточности или циррозе печени, для лечения аллергии, для лечения астмы, для лечения воспалительных заболеваний, например ревматизма или панкреатита, для профилактики ишемии (недостаточное кровоснабжение).
В следующем аспекте изобретения задача решена путем применения пролекарства по одному из пп.7-11 и п.14 или способом по п.13 формулы изобретения, причем применение или способ являются применением, соответственно способом лечения заболеваний, вызванных дефицитом моноксида азота.
В одном варианте осуществления задача решена путем применения пролекарства, отличающегося тем, что лекарственное вещество или пролекарство выбраны из группы ингибиторов протеазы, соединений, интеркалирующих ДНК и РНК, ингибиторов вирусных ферментов и антагонистов рецептора N-метил-D-аспартата.
В одном варианте осуществления задача решена путем применения пролекарства, причем применение означает применение для профилактики и/или лечения висцерального и/или кожного лейшманиоза, трипаносомоза, трипаносомоза второй стадии или пневмонии, вызванной Pneumocystis carinii, для подавления роста злокачественных опухолей, для подавления свертывания крови, для снижения кровяного давления, для нейропротекции или для борьбы с вирусными инфекциями, включая грипп и ВИЧ-инфекции.
В следующем аспекте изобретения задача решена лекарственным веществом с фрагментом структуры, имеющим общую формулу (I) или (II)

причем R1 и R2 означают водород, алкильный или арильный остаток.
В одном варианте осуществления задача решена лекарственным веществом с фрагментом структуры, имеющим общую формулу (I) или (II), отличающимся тем, что фрагмент структуры является элементом гидроксиламина, N-оксида, нитрона, диазенийдиолята (NONOат) или подобных, N-O-содержащих доноров моноксида азота; гидроксамовой кислоты, гидроксимочевины, оксима, амидоксима (N-гидроксиамидина), N-гидроксиамидиногидразона или N-гидроксигуанидина.
В одном варианте осуществления задача решена лекарственным веществом по одному из предыдущих пунктов, отличающимся тем, что лекарственное вещество подходит для лечения заболеваний, связанных с дефицитом моноксида азота.
В одном варианте осуществления задача решена лекарственным веществом, отличающимся тем, что лекарственное вещество выбрано из группы ингибиторов протеазы, соединений, интеркалирующих ДНК и РНК, ингибиторов вирусных ферментов и антагонистов рецептора N-метил-D-аспартата.
В следующем аспекте изобретения задача решена применением O-карбоксиалкилированной функциональной группы, содержащей N-O, для получения лекарственного вещества с фрагментом структуры, имеющим общую формулу (I) или (II)

причем R1 и R2 означают водород, алкильный или арильный остаток, для улучшения растворимости, биодоступности, проходимости гематоэнцефалического барьера, биоактивации и/или физико-химической стабильности лекарственного вещества.
В одном варианте осуществления задача решена применением лекарственного вещества с O-карбоксиалкилированной N-O-содержащей функциональной группой для активации лекарственного вещества посредством пептидилглицин-α-амидирующей монооксигеназы (PAM).
В одном варианте осуществления задача решена применением лекарственного вещества, отличающимся тем, что фрагмент структуры является элементом гидроксиламина, N-оксида, нитрона, диазенийдиолята (NONOат) или подобного, N-O-содержащего донора моноксида азота; гидроксамовой кислоты, гидроксимочевины, оксима, амидоксима (N-гидроксиамидина), N-гидроксиамидиногидразона или N-гидроксигуанидина.
В одном варианте осуществления задача решена применением лекарственного вещества, отличающимся тем, что лекарственное вещество подходит для лечения заболеваний, связанных с дефицитом моноксида азота.
В одном варианте осуществления задача решена применением лекарственного вещества, отличающимся тем, что лекарственное вещество выбрано из группы ингибиторов протеазы, соединений, интеркалирующих ДНК и РНК, ингибиторов вирусных ферментов и антагонистов рецептора N-метил-D-аспартата.
В одном варианте осуществления задача решена применением лекарственного вещества, отличающимся тем, что лекарственное вещество подходит для профилактики и/или лечения висцерального и/или кожного лейшманиоза, трипаносомоза, трипаносомоза второй стадии или пневмонии, вызванной Pneumocystis carinii, для подавления роста злокачественных опухолей, для подавления свертывания крови, для снижения кровяного давления, для нейропротекции или для борьбы с вирусными инфекциями, включая грипп и ВИЧ-инфекции.
В следующем аспекте изобретения даются фармацевтические соединения, фармацевтические композиции и лекарственные средства, которые содержат соединения согласно изобретению и/или их соли. Предпочтительно, фармацевтические композиции содержат носители и/или вспомогательные вещества и идеально совместимы фармацевтически. Подобные носители и вспомогательные вещества специалисту общеизвестны. Предоставляются также соединения согласно изобретению для применения в медицине.
Достаточно, если лекарственное вещество содержит по меньшей мере одну или несколько активных амидиновых, N-гидроксиамидиновых (амидоксим), гуанидиновых или N-гидроксигуанидиновых функциональных групп в предложенной форме. Соответственно, лекарственное вещество может содержать, например, несколько амидоксимовых функциональных групп (например, две, как в случае пентоксимового эфира) или N-гидроксигуанидиновых функциональных групп, причем тогда по меньшей мере одна из этих групп модифицирована вышеописанным способом. Могут также применяться смеси лекарственных веществ, из которых по меньшей мере одно модифицировано согласно изобретению.
Прием соединения согласно изобретению может проводиться однократно как прием болюса, ежедневно, еженедельно или ежемесячно. Способ введения также легко определить. В принципе, подходят пероральное, ректальное, парентеральное, как внутривенное, внутримышечное, подкожное, чрескожное введение, внутрилегочное введение, а также прием в виде аэрозоля, внутрипузырного капельного введения, внутрибрюшинных или внутрисердечных инъекций, впитывание через слизистую оболочку или интравагинальное введение, например, посредством суппозиториев. Пероральная форма применения может быть выполнена как жидкая, полутвердая или твердая готовая форма, в частности, как таблетка, драже, гранула или микрокапсула. При этом для таких форм осуществления, в которых применяются жидкие готовые формы, активное вещество или смесь активных веществ введены в подходящий, нетоксичный растворитель, как, например, вода, одноатомные спирты, в частности этанол, многоатомные спирты, в частности глицерин и/или пропандиол, полигликоли, в частности полиэтиленгликоли и/или миглиол, глицеринформаль, диметилизосорбит, натуральные или синтетические масла. Для получения полутвердых или твердых готовых форм подходят обычные основы, как, например, бентонит, вигум, гуаровая мука и/или производные целлюлозы, в частности метилцеллюлоза и/или карбоксиметилцеллюлоза, а также полимеры из виниловых спиртов и/или винилпирролидонов, альгинаты, пектины, полиакрилаты, твердые и/или жидкие полиэтиленгликоли, парафины, жирные спирты, вазелины и/или воски, жирные кислоты и/или сложные эфиры жирных кислот.
Кроме того, в твердых готовых формах могут содержаться известные наполнители, такие, например, как коллоидная кремниевая кислота, тальк, молочный сахар, крахмальный порошок, сахар, желатин, оксиды металлов и/или соли металлов. В качестве дальнейших добавок подходят стабилизаторы, эмульгаторы, диспергаторы, а также консерванты.
Неожиданно оказалось, что O-карбоксиалкилированные, N-O-содержащие функциональные группы общей формулы (I) или (II), которые через соединение с азотом (N) связываются с молекулой лекарственного вещества,

причем (I) и (II) могут являться, например, элементом гидроксиламина, N-оксида, нитрона, диазенийдиолята (NONOат) или аналогичного, N-O-содержащего донора моноксида азота; гидроксамовой кислоты, оксима, амидоксима (N-гидроксиамидина), N-гидроксиамидиногидразона или N-гидроксигуанидина, и R1 (который должен быть в конфигурации pro-R) и R2 могут означать водород, алкильный или арильный остаток, использует путь биоактивации, который не зависит от фермента цитохром P450 (CYP450). Это является неожиданным результатом, так как известно, что фермент CYP450, вообще говоря, катализирует окислительное O-дезалкилирование, которое также потребовалось бы в предлагаемой здесь концепции пролекарства, чтобы выделить собственно лекарственное вещество.
Предложенная переэтерификация Ν-O-содержащих функциональных групп с карбоксиалкильными остатками дает то особое преимущество, что для биоактивации может использоваться другой фермент, отличный от фермента CYP450, а именно, пептидилглицин-α-амидирующая монооксигеназа (PAM). Благодаря этому можно, например, избежать побочных эффектов и обсужденных выше взаимодействий с другими, одновременно принимаемыми лекарственными веществами.
Пептидилглицин-α-амидирующая монооксигеназа (PAM) является в высших организмах (позвоночных) бифункциональным ферментом, который состоит из домена монооксигеназы (PHM, пептидилглицин-α-гидроксилирующая монооксигеназа, EC 1.14.17.3) и домена лиазы (PAL, пептидил-α-гидроксиглицин α-амидирующая лиаза, EC 4.3.2.5). В совокупности, PAM подвергается сильно тканеспецифической и зависящей от разработки регуляции через сплайсинг и экспрессию. Что касается пост-трансляционной модификации, PAM способна активировать различные пептидные гормоны физиологического происхождения, медиаторы и факторы роста (например, вещество P, нейропептид Y, окситоцин, вазопрессин, кальцитонин). При этом пептиды амидируются C-терминально тем, что концевой глицин отщепляется в результате окислительного N-дезалкилирования в реакции монооксигеназы.
Особым преимуществом предлагаемой согласно изобретению переэферификации Ν-O-содержащей функциональной группы карбоксиалкильными остатками является улучшение растворимости благодаря введению карбоновой кислоты, отрицательно заряженной в физиологических условиях (pH 6-8).
Следующее преимущество обусловлено тем, что благодаря предлагаемой согласно изобретению переэтерификации N-O-содержащих функциональных групп (при использовании (алкоксикарбонил)алкиловых эфиров или (арилоксикарбонил)алкиловых эфиров) липофилия повышается настолько, что становится возможной пассивная диффузия и тем самым улучшается биодоступность и/или проходимость гематоэнцефалического барьера.
При этом преимуществом является также возможность использовать сравнительно малый остаток, в простейшем случае карбоксиметильный остаток, как пролекарственную группу, так что размер молекулы лекарственного вещества увеличивается лишь незначительно.
Wand et al. [Metabolism 1985, 34, 11, 1044] исследовали активность PAM в различных тканях человека и обнаружили наивысшую активность в тканях ЦНС (прежде всего в гипофизе). Напротив, в классических метаболизирующих посторонние вещества органах: печени и почках не было установлено никакой активности. В плазме, сердце и легких также была обнаружена активность, которая могла использоваться для предполагаемой концепции пролекарства.
В частности, высокую активность этого фермента в ЦНС можно использовать, чтобы транспортировать O-карбоксиалкилированное пролекарство через гематоэнцефалический барьер, которое затем можно преобразовать. Но возможна также и биоактивация в сердечно-сосудистой системе после перорального введения и поглощения из желудочно-кишечного тракта.
При этом пролекарственная система согласно изобретению позволяет применение на разных лекарственных веществах, которые содержат амидиновую или гуанидиновую группу. Особенно предпочтительны при этом следующие лекарственные вещества:
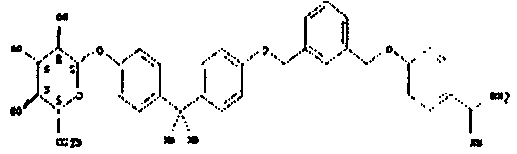
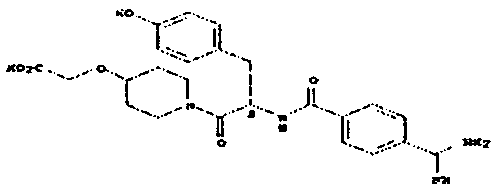
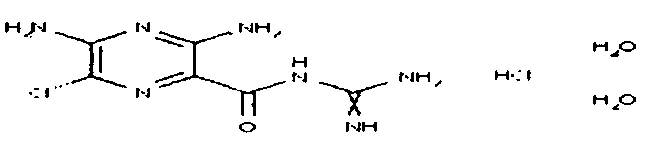
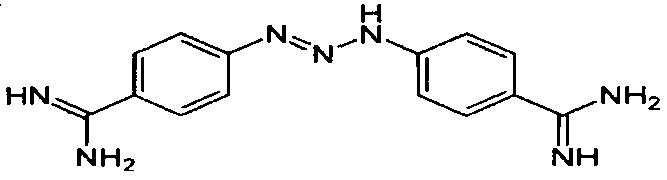
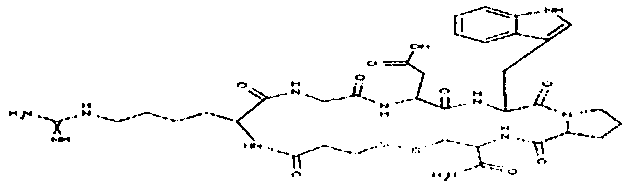
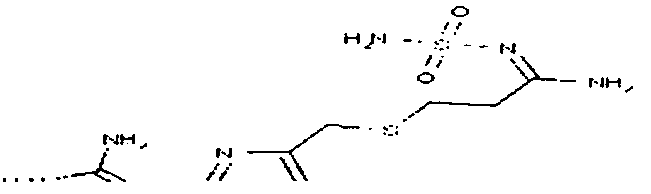
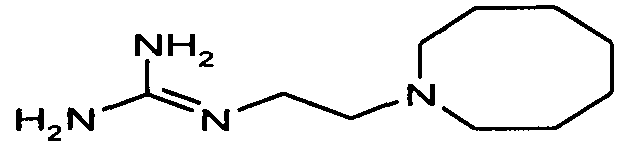
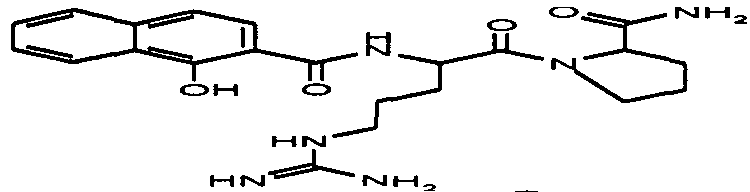
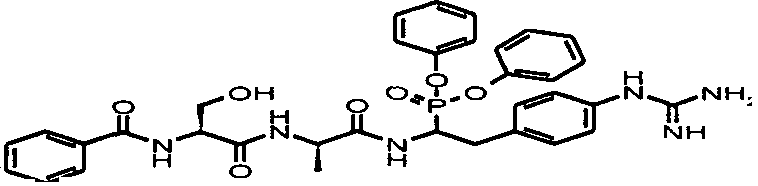
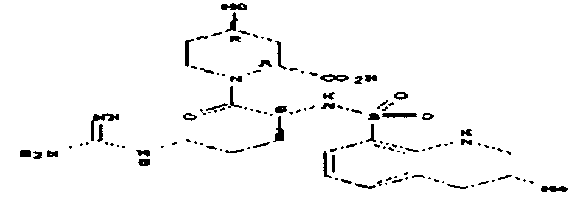
пентамидин, дабигатран, BSF 411693 (Abbott), идазоксан гидрохлорид, ирбесартан, линоглирид, лофексидин гидрохлорид, тетрагидрозолин гидрохлорид, толазолин, ксилометазолин гидрохлорид, пентамидин изетионат, тарибавирин, тиамин (витамин B1), бозентан, дибромпропамидин изетионат, гидроксистильбамидин изетионат, сибрафибан, орбофибан, ксемилофибан, аргатробан, ксимелагатран, мелагатран, 2-пиперидиновая кислота, орбофибанацетат, эпинастин (релестат), RO 43-8857, AB1 (хлорамбицил, аналоги), AMG-126737, AY-0068, B-623, BABIM, BIBT-986 (Boehringer Ingelheim), CI-1031 (фирма Biosciences)), CJ-1332 (фирма Curacyte), CJ-463 (фирма Curacyte), CJ-672 (фирма Curacyte), CT50728 (Portolla Pharmaceuticals), CVS-3983, DX-9065a, ламифибан (Roche), LB-30870 (фирма LG LifeSciences Ltd), LY-178550 (фирма Lilly), PHA-927F и аналоги, RO-44-3888 (Roche), сепимостат, FUT-187 (Torii), вирамидин (Ribapharm), WX-FX4 (Wilex), YM-60828 (Yamanouchi Pharmaceutical Co Ltd), ZK-807191 (Berlex Biosciences), NAPAP (SR 25477), BIIL 315 (Boehringer Ingelheim), BIIL 260 (Boehringer Ingelheim), BIIL 284/260 (Boehringer Ingelheim), таногитран, моксилубант, стильбамидин, панамидин, фрадафибан, диминацен, роксифибан, фурамидин, PD0313052, PHA 927F, PHA 798, фидексабан, отамиксабан, тромстоп (тромбостоп), занамивир, амилоридгидрохлорид, анагрелидгидрохлорид, прогуанил, циметидин, клонидин гидрохлорида, гуанокса, перамивир, ромифидин, тирапазамин, тизанидин, толонидин нитрат, метмофин, диминазен, дебризоквин, сульфаметазин, эптифибатид, фамотидин (Bayer-Arzneistoff), стрептомицин, нафамостат, FUT-175, иногатран, гуанетидин (тилодигон), 3DP-10017, APC-366, CVS-1123, производное дифенилфосфоната, E-64, FOY-305, MBGB, MIBG, RWJ-422521, синталин, WX-293, WX-340, BMS-189090, JTV-803, (Japan Tabacco), напсагатран, исмелин, Tan 1057A, хидикал, фенформикс (Retardo), нетропсин (синаномицин), BIIB 722 (сабипорид), гуанадрел, деоксиспергуалин, BMS 262084, сиамформет (Orabet), PPACK (Pebac), MERGETPA (ингибитор карбоксипетидазы Пламмера), перамивир, фамотидин, залтидин.
В приложении находится таблица с химическими формулами, номерами по CAS и показаниями для лекарственных веществ.
Далее в качестве примера показано 4 пролекарства согласно изобретению:

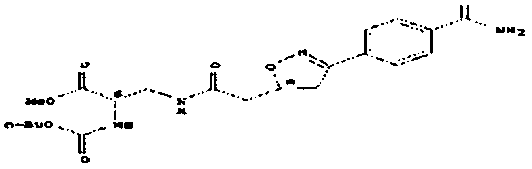
карбоксиэтокси-предшественник занамивира

карбоксиметокси-предшественник занамивира

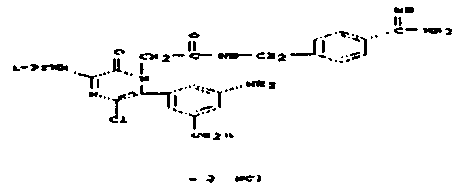
бис(карбоксиметокси)-предшественник пентамидина

Карбоксиметилбензамидоксим.
Неожиданное открытие, что в качестве субстратов для PAM допустимы также непептидные O-карбоксиалкилированные N-O-содержащие функциональные группы, демонстрируется в примерах осуществления на модельных соединениях, основанных на амидоксиме и N-гидроксигуанидине.
В качестве модельного соединения для амидоксима тестировался O-карбоксиметилбензамидоксим (1) на его свойства как субстрата для PAM. O-карбоксиметилбензамидоксим является возможным предшественником лекарственного вещества бензамидин. Катализируемая PAM биоактивация O-карбоксиметилбензамидоксима (1) с получением бензамидоксима (2) протекает с одновременным выделением глиоксалевой кислоты.

На фигуре 1 показаны результаты колориметрического определения образования глиоксилата. Определенные концентрации глиоксилата являются средними значениями ± стандартное отклонение по двум инкубациям, каждая из которых измерялась дважды. Образование глиоксилата как продукта отщепления PAM-каталазы из соединения 1 можно подтвердить концентрационно-зависимым способом. Инкубации при оптимальном pH PAM (pH 6,0) приводят к заметно более высоким конверсиям по сравнению с инкубацией при pH 7,4. В колориметрическом испытании проводилась 5-точечная калибровка глиоксилата параллельно испытанию соединения 1. Калибровка в измеренной области концентраций была линейной (r2=1,000).
Так как O-карбоксиметилбензамидоксим (1) согласно этим результатам признан как субстрат для PAM, реакцию более точно характеризовали через определение значений KM и Vmax.
Для этой цели была разработана методика ВЭЖХ-анализа. Калибровочная прямая для бензамидоксима была в исследуемой области концентраций линейной (r2=1,000), и степень повторного обнаружения составляла 130,6% (r2=0,999). Из двух независимых экспериментов (n=2) получено значение KM=307±80 мкМ и значение Vmax=393±40 нмоль/мин/мг PAM. На фигуре 2 такое определение представлено репрезентативно.
Для изучения субстрата CYP450 вышеуказанный метод ВЭЖХ-анализа был изменен таким образом, чтобы дополнительно можно было детектировать возможные метаболиты бензамидина, как продукт N-восстановления бензамидоксима (2). При pH 6,0, а также pH 7,4 ни с одним из использованных источников фермента CYP450 не удалось обнаружить ни бензамидоксима (возможный предшественник бензамидина), ни бензамидина.
На основе соединения 1, моделирующего бензамидоксим, в рамках реакции монооксигеназы O-карбоксиметильная группа удаляется только из PAM, но не из цитохрома P450.
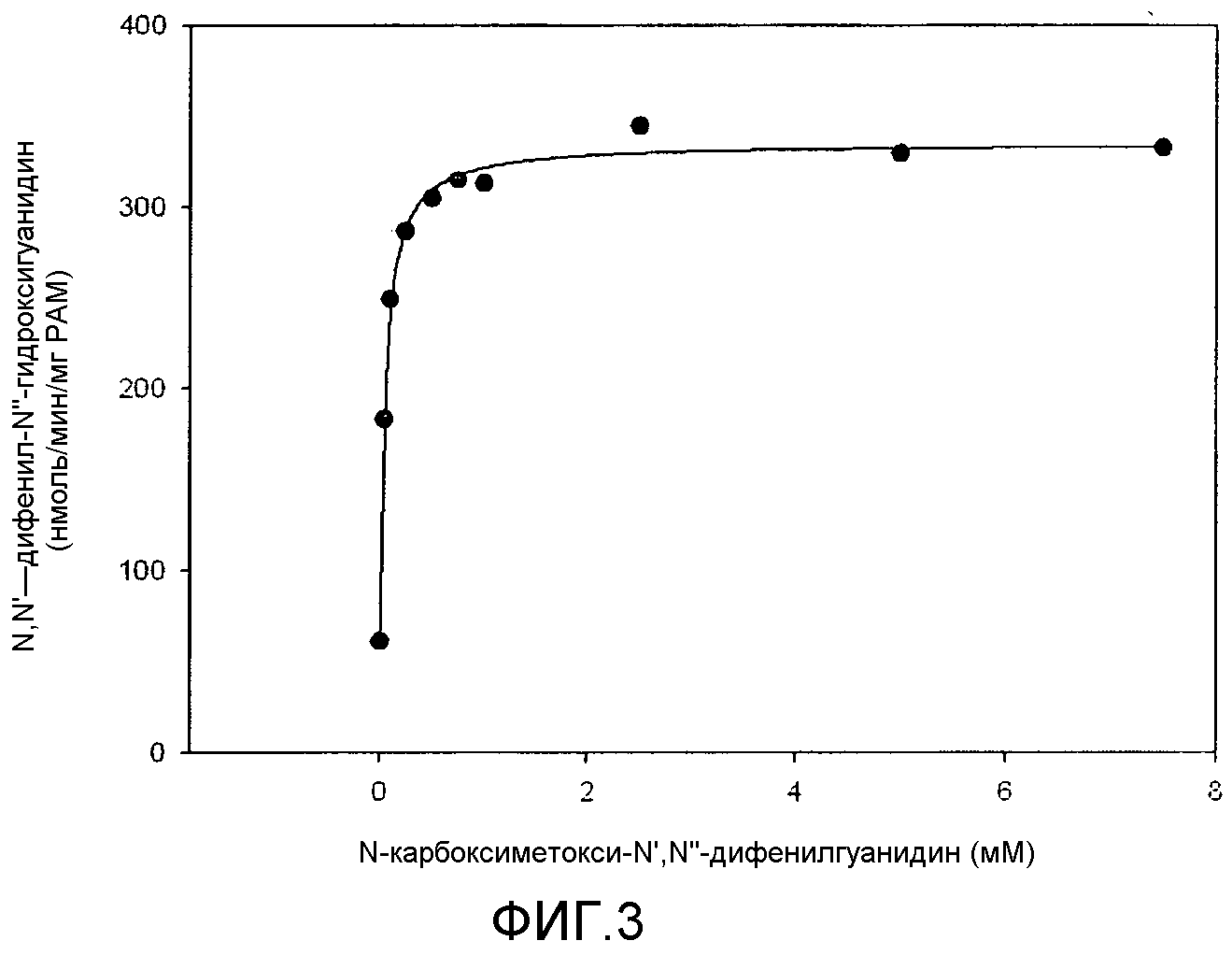
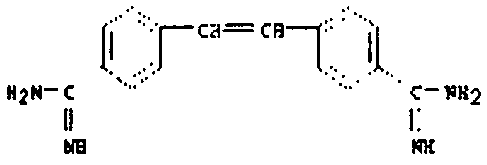
В качестве модельного соединения для гидроксигуанидина испытывали N-карбоксиметокси-N',N"-дифенилгуанидин (3) на его свойства как субстрата PAM.
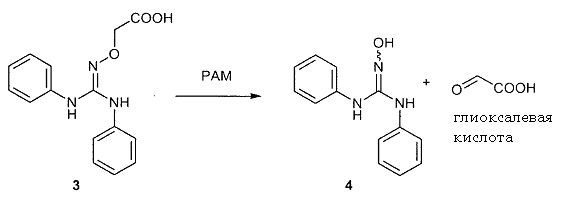
Катализируемая PAM биоактивация N-карбоксиметокси-N',N"-дифенилгуанидина (3) в N,N'-дифенил-N"-гидроксигуанидин (4) протекает с одновременным выделением глиоксалевой кислоты.
Результаты колориметрического испытания с соединением 3 были сравнимы с результатами для моделирующего амидоксим соединения 1. Таким образом, для определения значений KM и Vmax была разработана методика ВЭЖХ-анализа, которая способна разделить пролекарство 3 и гидроксигуанидин 4 на RP-колонке в пределах 15 минут. Калибровочная прямая для N,N'-дифенил-N"-гидроксигуанидина (4) была в измеренной области концентраций линейной (r2=0,999), и степень повторного обнаружения составляла 111,7% (r2=0,999). Из двух независимых экспериментов (n=2) получилось значение KM=37±5 мкМ и значение Vmax=373±53 мкмоль/мин/мг PAM. На фигуре 3 такое определение представлено репрезентативно.
Из установленного значения KM можно сделать вывод о в 8 раз более высоком сродстве к PAM по сравнению с амидоксимовым пролекарством 1, тогда как степени превращения сравнимы.
Для изучения субстрата CYP450 разработанная для изучения PAM методика ВЭЖХ-анализа была изменена таким образом, чтобы дополнительно можно было детектировать возможные метаболиты N,N'-дифенилгуанидина как продукта N-восстановления гидроксигуанидина 4. При pH 6,0, а также pH 7,4 ни с одним из использованных источников фермента CYP450 не удалось после 180 минут инкубации обнаружить ни соединения 4, ни N,N'-дифенилгуанидина.
По аналогии с O-карбоксиметилбензамидоксимом (1) на основе моделирующего гидроксигуанидин соединения 3 в рамках реакции монооксигеназы O-карбоксиметильная группа удалялась только из PAM, но не из цитохрома P450.
Материал и методы
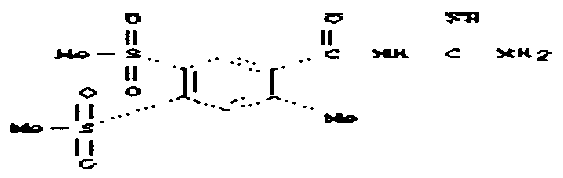
Натриевая соль O-карбоксиметилбензамидоксима, моногидрат (1)
Модифицированный предшественник согласно Koch [Ber. Dtsch. Chem. Ges. 1889, 22, 3161]:

Раствор 681 мг бензамидоксима (5,0 ммоль), 1,04 г бромуксусной кислоты (7,5 ммоль) и 500 мг таблеток едкого натра (12,5 ммоль) в 5 мл этанола кипятили 5 часов с обратным холодильником. Затем растворитель удаляли в вакууме до тех пор, пока не начинал образовываться осадок. Это осадок оставляли до полного осаждения, отфильтровывали и сушили. Продукт перекристаллизовывали из смеси этанол (96%)/вода (95:5).
Выход: 937 мг белых, мелких спутанно-волокнистых кристаллов (80%)
Т.пл.: 226°C (разл.)
1H-ЯМР (DMSO-d6): δ/м.д.=4,13 (с, 2H, O-CH2), 6,09 (шир.с, 2H, NH2), 7,37 (м, 3H, 3',4',5'-CH), 7,67 (м, 2H, 2',6'-CH).
13C-ЯМР (CDCl3): δ/м.д.=73,6 (O-CH2), 125,7, 128,0, 129,0 (ArCH), 132,8 (ArC), 151,4 (C=N), 173,2 (CO).
МС (ESI): m/z=217 [M+Na]+, 195 [M+H]+, 119 [M-C4H2-C2H2+H]+, 105 [C6H5N2]+.
C9H9N2NaO3·1,0 H2O (234,18)
Рассчитано: C 46,16 H 4,73 N 11,96
Найдено: C 46,43 H 4,44 N 11,65
N'-карбоксиметокси-N',N"-дифенилгуанидин (3)

546 мг семихлорида аминооксиуксусной кислоты (5 ммоль) и 697 мкл триэтиламина (5 ммоль) перемешивали 30 мин в 10 мл сухого ДМФ. Осадок отфильтровывали и в фильтрат добавляли 970 мг N,N'-дифенилкарбодиимида (5 ммоль). Смесь перемешивали четыре часа при комнатной температуре, извлекали этилацетатом и продукт перекристаллизовывали из этанола.
Выход: 285 мг белого твердого вещества (20%)
Т.пл.: 176°C
DC: Rf=0,29 (дихлорметан/метанол, 9:1)
1H-ЯМР (DMSO-d6): δ/м.д.=4,37 (с, 2H, O-CH2), 6,75-6,87 (м, 2H, ArH), 7,03-7,20 (м, 8H, ArH), 8,02, 8,21 (2 × шир.с, 1H, NH), 12,05 (шир.с, 1H, COOH).
13C-ЯМР (DMSO-d6): δ/м.д.=70,0 (O-CH2), 116,7, 118,7, 119,8, 121,0, 128,5 (ArCH), 140,7, 142,3 (ArC), 147,5 (C=N), 171,8 (CO).
МС (ESI): m/z=308 [M+Na]+, 286 [M+H]+, 210 [M-C2H4O3]+.
МС (EI): m/z (%)=209 (38), 208 (37), 119 (20), 118 (38), 93 (100), 91 (47), 77 (43), 66 (31), 51 (30).
C15H15N3O3·0,3H2O (290,71)
Рассчитано: C 61,97 H 5,41 N 14,45
Найдено: C 62,18 H 5,72 N 14,57
ВЭЖХ-система:
ВЭЖХ-система Waters Breeze с насосом Waters 1525, детектором поглощения Waters 2487, автоматическим пробоотборником Waters 717 Plus и программой сбора и обработки данных Breeze (версия 3.30), термостат колонного типа Gynkotek STH 585.
Колонки ВЭЖХ:
Synergi Max-RP 80 A (250 × 4,6 мм, 4 мкм) с предколонкой C-18 (4 × 3 мм) (фирма Phenomenex);
LiChroCART, LiChrospher 100, RP-8 (125 × 4 мм, 5 мкм) с предколонкой LiChrospher 60, RP-select B (4 × 4 мм, 5 мкм) (фирма Merck);
LiChroCART, LiChrospher RP-select B (250 × 4,6 мм, 5 мкм) с предколонкой LiChrospher 60, RP-select B (4 × 4 мм, 5 мкм) (фирма Merck).
Другие приборы и материалы:
Фотометр Cary 50 UV-Vis (фирма Varian); 96-луночные планшеты (фирма Greiner); водяная баня-шейкер GFL-1083 (Gesellschaft für Labortechnik, Burgwedel); микролитровая центрифуга (фирма Hettich GmbH); pH-метр InoLab pH Level 1 (Wissenschaftlich-Technische Werkstätten GmbH, Weilheim) с pH-электродом LiQ Plast (фирма Hamilton); вихревой встряхиватель VF2 (Janke & Kunkel GmbH & Co. KG, Staufen); реакционный сосуд на 1,5 мл (Sarstedt AG & Co., Nümbrecht).
Источники ферментов:
Используемая рекомбинантная пептидилглицин-α-амидирующая монооксигеназа (PAM, крысы, EC 1.14.17.3) была предоставлена фирмой Unigene Laboratories, Inc. (New Jersey, USA) (удельная активность=5,8·106 ед./мг белка); каталаза бычьей печени (EC 1.11.1.6), удельная активность=12600 ед./мг твердого вещества (фирма Aldrich).
Получение используемого источника фермента цитохрома-P450 осуществлялось рабочей группой Clement von Grünewald по следующей инструкции:
Печеночные микросомы свиней и 9000 г избыточной фракции:
Свиная печень была приобретена у местного мясника (Bordesholm), и органы сразу после забоя переносили в 20 мМ охлаждаемого льдом фосфатного буфера (1 мМ Na2-EDTA, pH 7,4). Для дальнейшей обработки доли печени сначала перфузировали и промывали в 50 мМ фосфатного буфера (1 мМ Na2-EDTA, pH 7,4). Ткань разрезали на куски и пропускали через стандартную мясорубку. Суспензию разбавляли равным объемом фосфатного буфера и гомогенизировали с помощью проточного гомогенизатора. Дальнейшее получение микросом и 9000 г избыточной фракции проводилось путем дифференциального ультрацентрифугирования. Для хранения полученные препараты аликвотировали и замораживали при -80°C.
Печеночные микросомы человека и 9000 г избыточной фракции:
Для получения микросом человека ткань человеческой печени получали в хирургическом отделении университетской клиники университета Christian-Albrechts от раковых больных, которые должны были подвергнуться резекции половины печени.
Куски печеночной ткани мгновенно замораживали в содержащем сахарозу фосфатном буфере (10 мМ K2HPO4, 10 мМ KH2PO4, 250 мМ сахарозы, 1 мМ Na2-EDTA, pH 7,4, 4°C). После того, как в наличии появилось достаточное число частей органа (>3), соответствующие куски размораживали и соединяли, чтобы компенсировать различие из-за колебаний между отдельными индивидуумами. Куски ткани при 4°C разрезали на мелкие части, несколько раз промывали буферным раствором (без EDTA) и перерабатывали гомогенизатором в суспензию. Получение микросом и 9000 г избыточной фракции проводилось из этой суспензии путем дифференциального ультрацентрифугирования. Для хранения полученные препараты аликвотировали и замораживали при -80°C.
PAM-анализ: условия инкубации
Типичная смесь для инкубации объемом 300 мкл (полный объем) содержала 25000 ед./мл пептидилглицин-α-амидирующей монооксигеназы (PAM, фирма Unigene Laboratories), 250 ед./мл каталазы, 1 мкМ меди(II) (введенной как ацетат/моногидрат), 2 мМ аскорбата натрия, 5 мМ йодида калия и соответствующий субстрат в концентрации 0,1 мМ или 1 мМ, в буфере с разными значениями pH. В качестве буферной системы использовалось 30 мМ MES для инкубации при pH 6,0 и 50 мМ HEPES для инкубации при pH 7,4. Значение pH устанавливали разбавленным раствором едкого натра. Инкубировали 60 мин при 37°C в водяной бане-шейкере, отбирали 100 мкл и реакцию останавливали 50 мкл смеси 10%-ная TFA(водн.)/ацетонитрил (2:3). Остальную исходную смесь инкубировали еще 180 мин при 37°C и реакцию останавливали с помощью 100 мкл смеси 10%-ная TFA(водн.)/ацетонитрил (2:3).
Остановленные пробы встряхивали 5 мин (вихревой встряхиватель) и замораживали при -80°C. Для анализа проб их размораживали, 5 мин встряхивали и выпавший белок центрифугировали при 10000 об/мин. Избыточную фракцию использовали для колориметрического определения глиоксилата и/или ВЭЖХ-анализа.
Для определения значений KM и Vmax получение 100 мкл исходной смеси при pH 6,0 проводили в вышеописанных условиях, но с тем отличием, что время инкубации составляло 30 мин.
Колориметрическое определение глиоксилата
200 мкл освобожденной от белка инкубационной смеси соединяли с 20 мкл раствора фенилгидразина (20 мг в 2 мл бидистиллированной воды) и встряхивали 5 мин на водяной бане-шейкере при 37°С. Затем охлаждали за 15 мин до 0°C, добавляли 100 мкл очень холодной HCl 6н. и оставляли еще на 5 мин при 0°C. Затем добавляли 20 мкл раствора калийгексацианоферрата(III) (100 мг в 2 мл бидистиллированной воды). Смесь оставляли на 15 мин при комнатной температуре и отбирали 200 мкл для измерения с помощью планшет-ридера (фотометр Cary 50 UV-Vis, 520 нм).
Калибровка
Для 5-точечной калибровки глиоксалевую кислоту измеряли, как описано выше, в концентрациях 2, 5, 10, 50 и 100 мкМ в смеси 2:1 буфера для анализа (pH 6,0) и смеси (2:3) 10%-ная TFA(водн.)/ацетонитрил. Эта калибровка проводилась параллельно при каждом проводимом анализе испытуемого соединения.
Методика ВЭЖХ для разделения O-карбоксиметилбензамидоксима (1) и бензамидоксима (2)
Калибровка и повторное обнаружение
Для калибровки вышеописанным способом ВЭЖХ проводили измерения с бензамидоксимом в восьми концентрациях в диапазоне 0,1-500 мкМ, растворенным в буфере для анализа (30 мМ MES, 1 мкМ ацетат меди(II), 2 мМ аскорбата натрия, 5 мМ йодида калия, pH 6,0).
Для определения повторного обнаружения получали одинаковые концентрации в буфере для анализа (конечный объем=100 мкл). Кроме того, добавляли O-карбоксиметилбензамидоксим (0,5 мМ) и 250 ед./мл каталазы, а затем также 50 мкл смеси 10%-ная TFA(водн.)/ацетонитрил (2:3). Пробы встряхивали с помощью вихревого встряхивателя и замораживали при -80°C. Для измерения пробы размораживали, 5 мин встряхивали на вихревом встряхивателе и центрифугировали 5 мин при 10000 об/мин.
Методика ВЭЖХ для разделения N-карбоксиметокси-N',N"-дифенилгуанидина (3) и N-гидрокси-N',N"-дифенилгуанидина (4)
Калибровка и повторное обнаружение
Для калибровки вышеописанной методикой ВЭЖХ-анализа проводили измерения с N-гидрокси-N,N"-дифенилгуанидином (4) в восьми концентрациях в диапазоне 0,1-500 мкМ, растворенным в буфере для анализа (30 мМ MES, 1 мкМ ацетат меди(II), 2 мМ сорбат натрия, 5 мМ йодид калия, pH 6,0).
Для определения повторного обнаружения получали одинаковые концентрации в буфере для анализа (конечный объем=100 мкл). Кроме того, добавляли N-карбоксиметокси-N,N"-дифенилгуанидина (3) (0,5 мМ) и 250 ед./мл каталазы, а затем также 50 мкл смеси (2:3) 10%-ная TFA(водн.)/ацетонитрил. Пробы встряхивали с помощью вихревого встряхивателя и замораживали при -80°C. Для измерения пробы размораживали, 5 мин встряхивали на вихревом встряхивателе и центрифугировали 5 мин при 10000 об/мин.
Испытание на CYP450: условия инкубации
Типичная исходная смесь для инкубации объемом 500 мкл (полный объем) содержала 0,3 мг белка (источник фермент печени свиньи или человека), 0,1 мМ (или 1 мМ) испытуемого соединения в 100 мМ фосфатного буфера (pH 6,0 или pH 7,4) и 1 мМ ΝADH (или ΝADPH). Инкубацию начинали после 5-минутной предварительной инкубации фермента и испытуемого соединения в буфере путем добавления ΝADH (или ΝADPH) и встряхивали 60 мин, соответственно 180 мин при 37°C в водяной бане-шейкере. Смесь останавливали добавлением равного объема ацетонитрила, встряхивали на вихревом встряхивателе и замораживали при -80°C.
Для анализа пробы размораживали, встряхивали 5 мин на вихревом встряхивателе и в результате 5-минутного центрифугирования при 10000 об/мин отделяли белок. Избыточная фракция применялась для ВЭЖХ-анализа.
Методика ВЭЖХ-анализа для разделения O-карбоксиметилбензамидоксима (1), бензамидоксима (2) и бензамидина
Методика ВЭЖХ-анализа для разделения N-карбоксиметокси-N',N"-дифенилгуанидина (3), N-гидрокси-N'N"-дифенилгуанидина (4) и N',N"-дифенилгуанидина
Далее следует таблица лекарственных веществ, в которых предпочтительно может применяться система пролекарств согласно изобретению:







































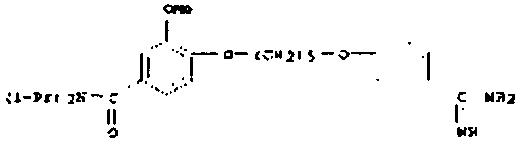
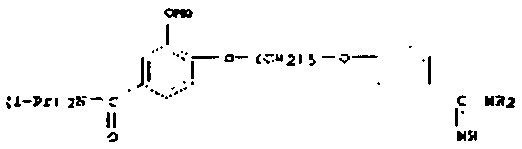





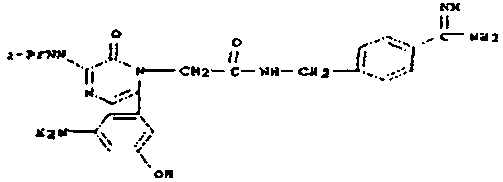















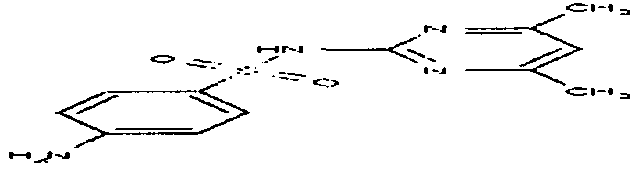

















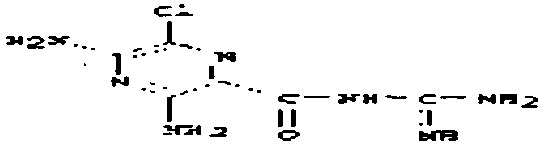







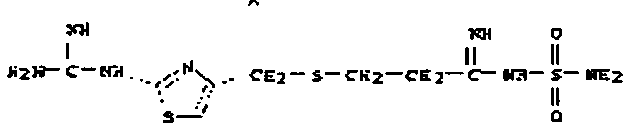


Реферат
Изобретение относится к фармацевтике. Описано пролекарство, содержащее фрагмент структуры общей формулы (IIa) или (IIb). Пролекарство метаболизируется в лекарственное вещество по пути, не зависящему от фермента цитохром-P450 (CYP450). Также описан способ лечения и способ получения N-алкоксигуанидина. Технический результат обеспечивается применением пролекарства для улучшения фармакокинетических свойств лекарственного вещества. 4 н. и 7 з.п. ф-лы, 3 ил., 1 табл.
Формула

причем R1 и R2 означают водород или алкильный остаток, где пролекарство метаболизируется в лекарственное вещество по пути, не зависящему от фермента цитохром-P450 (CYP450), и где лекарственное вещество выбрано из группы, состоящей из пентамидина, дабигатрана, BSF 411693 (Abbott), идазоксана гидрохлорида, ирбесартана, линоглирида, лофексидин гидрохлорида, тетрагидрозолин гидрохлорида, толазолина, ксилометазолин гидрохлорида, пентамидин изетионата, тарибавирина, тиамина (витамин B1), бозентана, дибромпропамидин изетионата, гидроксистильбамидин изетионата, сибрафибана, орбофибана, ксемилофибана, аргатробана, ксимелагатрана, мелагатрана, 2-пиперидиновой кислоты, орбофибанацетата, эпинастина (релестата), RO 43-8857, AB1 (хлорамбицил, аналоги), AMG-126737, AY-0068, В-623, BABIM, BIBT-986 (Boehringer Ingelheim), CI-1031 (фирма Biosciences)), CJ-1332 (фирма Curacyte), CJ-463 (фирма Curacyte), CJ-672 (фирма Curacyte), CT50728 (Portolla Pharmaceuticals), CVS-3983, DX-9065a, ламифибана (Roche), LB-30870 (фирма LG LifeSciences Ltd), LY-178550 (фирма Lilly), PHA-927F и аналогов, RO-44-3888 (Roche), сепимостата, FUT-187 (Torii), вирамидина (Ribapharm), WX-FX4 (Wilex), YM-60828 (Yamanouchi Pharmaceutical Co Ltd), ZK-807191 (Berlex Biosciences), NAPAP (SR 25477), BIIL 315 (Boehringer Ingelheim), BIIL 260 (Boehringer Ingelheim), BIIL 284/260 (Boehringer Ingelheim), таногитрана, моксилубанта, стильбамидина, панамидина, фрадафибана, диминацена, роксифибана, фурамидина, PD0313052, PHA 927F, PHA 798, фидексабана, отамиксабана, тромстопа (тромбостопа), занамивира, амилоридгидрохлорида, анагрелидгидрохлорида, прогуанила, циметидина, клонидин гидрохлорида, гуанокса, перамивира, ромифидина, тирапазамина, тизанидина, толонидин нитрата, метмофина, диминазена, дебризоквина, сульфаметазина, эптифибатида, фамотидина (Вауеr-Arzneistoff), стрептомицина, нафамостата, FUT-175, иногатрана, гуанетидина (тилодигона), 3DP-10017, АРС-366, CVS-1123, производного дифенилфосфоната, E-64, FOY-305, MBGB, MIBG, RWJ-422521, синталина, WX-293, WX-340, BMS-189090, JTV-803, (Japan Tabacco), напсагатрана, исмелина, Tan 1057А, хидикала, фенформикса (Retardo), нетропсина (синаномицина), BIIB 722 (сабипорида), гуанадрела, деоксиспергуалина, BMS 262084, сиамформета (Orabet), РРАСК (Pebac), MERGETPA (ингибитора карбоксипетидазы Пламмера), перамивира, фамотидина, залтидина.
а фрагмент структуры общей формулы IIb после метаболизирования имеет структуру, отвечающую формуле

фармакокинетических свойств лекарственного вещества, где пролекарство содержит фрагмент структуры, имеющей общую формулу (IIa) или (IIb)
причем R1 и R2 означают водород или алкильный остаток, где пролекарство метаболизируется в лекарственное вещество по пути, не зависящему от фермента цитохром-P450 (CYP450), и где лекарственное вещество выбрано из группы, состоящей из пентамидина, дабигатрана, BSF 411693 (Abbott), идазоксана гидрохлорида, ирбесартана, линоглирида, лофексидин гидрохлорида, тетрагидрозолин гидрохлорида, толазолина, ксилометазолин гидрохлорида, пентамидин изетионата, тарибавирина, тиамина (витамин B1), бозентана, дибромпропамидин изетионата, гидроксистильбамидин изетионата, сибрафибана, орбофибана, ксемилофибана, аргатробана, ксимелагатрана, мелагатрана, 2-пиперидиновой кислоты, орбофибанацетата, эпинастина (релестата), RO 43-8857, АВ1 (хлорамбицил, аналоги), AMG-126737, AY-0068, В-623, BABIM, BIBT-986 (Boehringer Ingelheim), CI-1031 (фирма Biosciences)), CJ-1332 (фирма Curacyte), CJ-463 (фирма Curacyte), CJ-672 (фирма Curacyte), CT50728 (Portolla Pharmaceuticals), CVS-3983, DX-9065a, ламифибана (Roche), LB-30870 (фирма LG LifeSciences Ltd), LY-178550 (фирма Lilly), PHA-927F и аналогов, RO-44-3888 (Roche), сепимостата, FUT-187 (Torii), вирамидина (Ribapharm), WX-FX4 (Wilex), YM-60828 (Yamanouchi Pharmaceutical Co Ltd), ZK-807191 (Berlex Biosciences), NAPAP (SR 25477), BIIL 315 (Boehringer Ingelheim), BIIL 260 (Boehringer Ingelheim), BIIL 284/260 (Boehringer Ingelheim), таногитрана, моксилубанта, стильбамидина, панамидина, фрадафибана, диминацена, роксифибана, фурамидина, PD0313052, PHA 927F, PHA 798, фидексабана, отамиксабана, тромстопа (тромбостопа), занамивира, амилоридгидрохлорида, анагрелидгидрохлорида, прогуанила, циметидина, клонидин гидрохлорида, гуанокса, перамивира, ромифидина, тирапазамина, тизанидина, толонидин нитрата, метмофина, диминазена, дебризоквина, сульфаметазина, эптифибатида, фамотидина (Bayer-Arzneistoff), стрептомицина, нафамостата, FUT-175, иногатрана, гуанетидина (тилодигона), 3DP-10017, АРС-366, CVS-1123, производного дифенилфосфоната, E-64, FOY-305, MBGB, MIBG, RWJ-422521, синталина, WX-293, WX-340, BMS-189090, JTV-803, (Japan Tabacco), напсагатрана, исмелина, Tan 1057A, хидикала, фенформикса (Retardo), нетропсина (синаномицина), BIIB 722 (сабипорида), гуанадрела, деоксиспергуалина, BMS 262084, сиамформета (Orabet), РРАСК (Pebac), MERGETPA (ингибитора карбоксипетидазы Пламмера), перамивира, фамотидина, залтидина.

или вместо вышеуказанного фрагмента структуры IIb содержит фрагмент структуры IIb-I или IIb-2


включающий взаимодействие карбодиимида формулы R1-N=C=N-R3 с аминоокси-соединением формулы H2N-O-R5 или его солью, причем в случае использования соли аминооксисоединения реакция проводится в присутствии основания, где R1 и R3, независимо друг от друга, выбраны из группы, содержащей H, необязательно замещенный алкил, необязательно замещенный арил, необязательно замещенный гетероарил, необязательно замещенный арилоксикарбонил, необязательно замещенный аминоацил, необязательно замещенный алкоксикарбонил, необязательно замещенный алкоксикарбонилалкокси, необязательно замещенный гетероарил, необязательно замещенный алкилциклоалкил, необязательно замещенный гетероалкилциклоалкил, необязательно замещенный аралкил и необязательно замещенный циклоалкил,
каждый R2 и R4 представляют собой водород, и
R5 выбран из группы, которая содержит алкоксикарбонил, (алкоксикарбонил) алкокси и карбоксиалкокси.
Комментарии