Фармацевтическая композиция для лечения связанных с ожирением заболеваний, содержащая конъюгат инсулинотропного пептида - RU2446816C2
Код документа: RU2446816C2
Чертежи






Описание
Область техники
Настоящее изобретение относится к композиции для лечения связанных с ожирением заболеваний, содержащей конъюгат инсулинотропного пептида, более конкретно к композиции для лечения связанных с ожирением заболеваний, содержащей конъюгат, полученный ковалентным связыванием инсулинотропного пептида с веществом-носителем через непептидильный линкер, и к способу лечения связанных с ожирением заболеваний с использованием этих композиций.
Предпосылки изобретения
Ожирение является хроническим заболеванием, связанным с высокой распространенностью и смертностью, вызванным накоплением жировой ткани вследствие нарушенной регуляции энергетического баланса или избыточного питания. Ожирение и связанные с ним заболевания представляют собой серьезную проблему для здоровья людей в Соединенных Штатах и по всему миру. За последние семь лет количество людей с ожирением в США увеличилось с 22,9% до 30,6%. Согласно данным проводимого в 1999-2002 годах Центром по контролю и профилактике заболеваний США исследования здоровья и питания населения 29,8% взрослых людей в возрасте 20 лет и старше имеют избыточный вес, а 30,4% страдают ожирением. Распространенность крайней степени ожирения среди взрослого населения составляла 4,9% (Hedley et al., JAMA 2004; 291:2847-50). Количество людей с ожирением сильно возросло не только в США, но также и во всех странах, которые приняли «западный» стиль питания и культуры. На сегодняшний момент во всем мире насчитывается 250 миллионов людей с ожирением, а к 2025 году, предположительно, около 300 миллионов людей будет страдать от ожирения. Ожирение само по себе представляет проблему для здоровья, а также коррелирует с рядом других осложнений, таких как гипертония, гиперлипидемия, сердечно-сосудистые заболевания и диабет. Примерно 80% пациентов с ожирением имеют одно или несколько из вышеперечисленных заболеваний (Mantzoros et al., J Clin Endocrinol Metab 2000; 85:4000-2), и приблизительно 300000 людей ежегодно умирают вследствие осложнений, вызываемых ожирением (Allison et al., JAMA 1999; 282: 1530-8). Было показано, что увеличение веса всего на 1 кг увеличивает риск сердечно-сосудистых заболеваний на 3,1%, а риск диабета - на 4,5-9%, и было показано, что потеря веса на примерно 11% снижает смертность на 25%. Поэтому так необходима срочная разработка стратегий лечения ожирения (Arbeeny et al., Obes Res 2004; 12:1191-6). В 1893 году для лечения ожирения использовали лекарственные средства на основе гормонов щитовидной железы для усиления термогенеза под действием норадреналина и адреналина. Однако эти лекарственные соединения усиливали потерю массы нежировых тканей и приводили к отрицательному азотному балансу, что вызывало кардиотоксичность в качестве побочного эффекта вместо желаемого эффекта уменьшения жировой ткани. Поэтому, в настоящий момент, их применение ограничено применением при гипертиреоидизме. В 1930-е гг. в качестве лекарственного средства для подавления аппетита использовали амфетамин, но его длительное применение было запрещено вследствие возникновения зависимости. Для лечения ожирения использовали фентермин, диэтилпропион и фенфлурамин, которые не вызывают зависимости. Однако их применение также было запрещено, поскольку многие из них вызывали сердечно-сосудистые заболевания, гипертонию, нарушение сердечного ритма, легочную гипертонию и психические расстройства, такие как потеря памяти. Современные стратегии для лечения ожирения включают средства, подавляющие аппетит, которые стимулируют центральные адренергические рецепторы или предупреждают обратный захват серотонина, термогенные бета-3-адренергические агонисты, ингибиторы пищеварительных липаз и гормональные регуляторы, такие как лептин и PYY (Dunstan et al., Nature reviews drug discovery 2006; 5:919-931). Среди них ингибиторы липаз, орлистат и сибутрамин, которые предупреждают повторный захват серотонина, подавляя аппетит, являются единственными лекарственными соединениями, одобренными Управлением по контролю за лекарственными соединениями и продуктами питания США, но они имеют побочные эффекты, включающие стеаторею, головные боли и повышенное кровяное давление. Таким образом, достаточно трудно разработать безопасное и, при этом, эффективное лекарственное соединение.
GLP-1, гормон, продуцируемый в тонком кишечнике, обычно вызывает биосинтез и секрецию инсулина и ингибирует секрецию глюкагона, регулируя концентрацию глюкозы в крови. Было опубликовано, что GLP-1 способен подавлять потребление пищи и снижать вес тела при введении мышам (Meeran et al, Endorinology 1999; 140:244-50), и эти эффекты были показаны как на нормальных мышах, так и на мышах с ожирением, что указывало на его потенциал в качестве агента против ожирения. Однако GLP-1 быстро деградирует под действием дипептидил-пептидазы IV (DPPIV), и, поэтому, его потенциал в качестве лекарственного средства крайне ограничен. С другой стороны, экзендин представляет собой пептид, найденный в яде ящерицы ядозуба, распространенной в Аризоне и Мексике. Он обладает активностью, аналогичной GLP-1, но устойчив к действию DPPIV, что делает возможным его использование в качестве терапевтического агента при диабете и ожирении. Экзендин продается в качестве средства для лечения диабета, которое вводят дважды в день. В патентах США №№ US6956026, US6989366 и US7115569 раскрыт способ подавления потребления пищи с помощью экзендина и его производных, в котором была продемонстрированная эффективность подавления потребления пищи, но эффект сохранялся в течение всего 6 часов после максимального введения. Для лечения хронических заболеваний, таких как ожирение, эти соединения необходимо вводить пациенту с помощью инъекций несколько раз в день, что опять же трудно для пациентов. Кроме того, описанные в патентах производные экзендина отличаются друг от друга по эффективности, дозо-зависимости и длительности действия, а их эффективность подавления потребления пищи не превышает значительно эффективность нативного экзендина.
Описание изобретения
Проблема, решаемая изобретением
Таким образом, в качестве способа максимального повышения эффекта увеличения времени полужизни инсулинотропного пептида и сохранения активности in vivo авторы изобретения использовали способ получения, в котором иммуноглобулиновая Fc-область, непептидильный линкер и инсулинотропный пептид ковалентно связаны друг с другом. Авторами было обнаружено, что конъюгат (в частности, экзендин-4, дезамино-гистидил-экзендин-4 с удаленной N-концевой аминогруппой, бета-гидрокси-имидазо-пропионил-экзендин-4, полученный замещением N-концевой аминогруппы гидроксильной группой, диметил-гистидил-экзендин-4, полученный модификацией N-концевой аминогруппы двумя метильными группами, и имидазо-ацетил-экзендин-4 с удаленным первым атомом углерода в первом гистидине) заметно лучше подавляют потребление пищи и обладают заметно большим временем действия in vivo, благодаря чему изобретение было выполнено.
Решение проблемы
Целью настоящего изобретения является композиция для лечения связанных с ожирением заболеваний, подавляющая потребление пищи и уменьшающая количество телесного жира, которая содержит конъюгат, полученный ковалентным связыванием инсулинотропного пептида с носителем через непептидильный линкер.
Другой целью настоящего изобретения является способ лечения связанных с ожирением заболеваний, подавляющий потребление пищи и уменьшающий количество телесного жира путем применения композиции, содержащей конъюгат.
Полезные эффекты
Эффекты подавления потребления пищи для уменьшения количества телесного жира и лечения связанных с ожирением заболеваний композиции, содержащей конъюгат инсулинотропного пептида по настоящему изобретению, превышает аналогичные эффекты нативного инсулинотропного пептида. Поэтому композиция пригодна для максимального увеличения терапевтического эффекта в отношении связанных с ожирением заболеваний.
Краткое описание чертежей
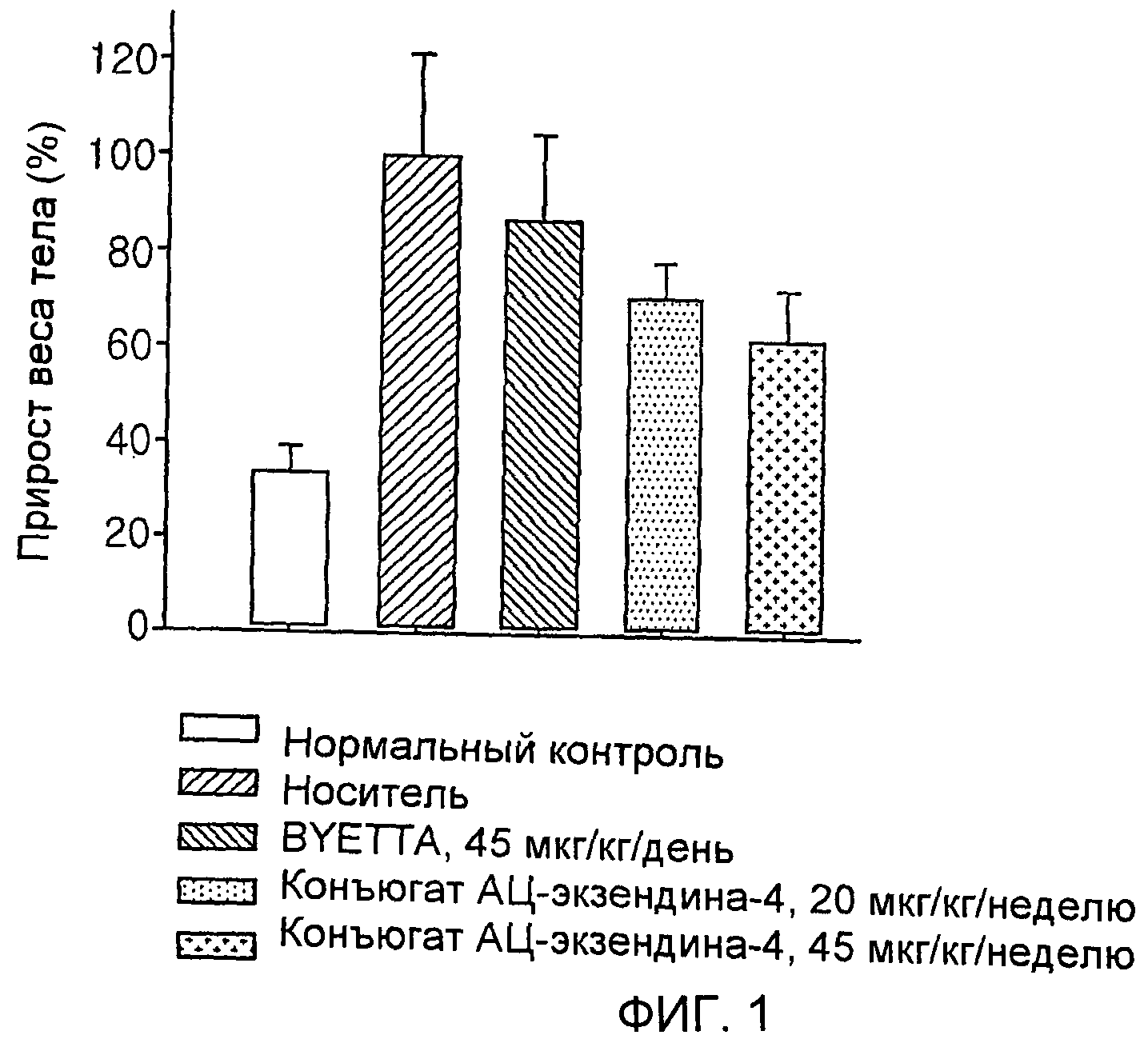
Фиг. 1 представляет собой график, на котором показана эффективность уменьшение количества телесного жира у мышей ob/ob;
Фиг. 2 представляет собой график, на котором показано изменение количества телесного жира у мышей ob/ob;
Фиг. 3 представляет собой график, на котором показана эффективность снижения веса тела на животной модели DIO;
Фиг. 4 представляет собой график, на котором показана эффективность уменьшения количества телесного жира у крыс ZDF; и
Фиг. 5 представляет собой график, на котором показана эффективность снижения потребления пищи у крыс ZDF.
Наилучший способ осуществления изобретения
Согласно одному аспекту настоящее изобретение относится к композиции для лечения связанных с ожирением заболеваний, подавляющих потребление пищи или уменьшающих количество телесного жира, которая содержит конъюгат, получаемый ковалентным связыванием инсулинотропного пептида с носителем через непептидильный линкер.
Используемый в настоящем описании термин «связанные с ожирением заболевания» может быть выбран из группы, состоящей из избыточного веса, компульсивного переедания и булимии, гипертонии, диабета, повышенной концентрации инсулина в плазме, инсулинорезистентности, гиперлипидемии, метаболического синдрома, синдрома инсулинорезистентности, связанного с ожирением гастроэзофагеальной рефлюксной болезни, атеросклероза, гиперхолестеринемии, гиперурикемии, боли в нижнем отделе спины, гипертрофии сердечной мышцы и гипертрофии левого желудочка, липодистрофии, неалкогольного стеатогепатита, сердечно-сосудистых заболеваний и синдрома поликистозных яичников, а субъекты с этими связанными с ожирением заболеваниями включают тех, кто желает снизить свой вес.
Используемый в настоящем описании термин «инсулинотропный пептид» представляет собой пептид, обладающий инсулинотропным действием, усиливающим синтез и экспрессию инсулина в бета-клетках поджелудочной железы. Эти пептиды включают пептид-предшественник, агонист, производное, фрагмент и вариант и, предпочтительно, GLP (глюкагон-подобный пептид)-1, экзендин-3 и экзендин-4 или их производные.
Производное инсулинотропного пептида по настоящему изобретению представляет собой производное, имеющее химически модифицированный N-концевой остаток гистидина, или производное, имеющее химически модифицированную аминогруппу N-концевого остатка гистидина. Кроме того, производное экзендина-4 или экзендина-3 относится к пептиду, полученному в результате замены, делеции или инсерции одной или нескольких аминокислот в нативном пептиде (или из нативного пептида) или в химически модифицированном пептиде, полученном алкилированием, ацилированием, этерификацией или амидированием одной или нескольких аминокислот в нативном пептиде, и относится к пептиду, имеющему нативную активность. Примеры производного экзендина-3 или экзендина-4 включают, но не ограничены аналогами экзендина, имеющими частично удаленный С-конец или замену неприродной аминокислотой норлейцином, которые раскрыты в WO97/46584, аналоги экзендина, имеющие замены неприродными аминокислотами, такими как пентилглицин, гомопролин и трет-бутилглицин, которые раскрыты в WO99/07404, и аналоги экзендина, имеющие более короткую аминокислотную последовательность, чем нативный экзендин, в результате частичного удаления С-концевого аминокислотного остатка, и аналоги экзендина, имеющие замены другими аминокислотами, раскрытые в US 2008/0119390, содержание которых полностью включено в настоящее описание путем ссылки. Более предпочтительно, чтобы инсулинотропным пептидом являлся экзендин-4 или его производные.
В частности, производное инсулинотропного пептида по настоящему изобретению может включать его производное с удаленной N-концевой аминогруппой (дезамино-гистидильное производное), его производное, получаемое замещением аминогруппы гидроксильной группой (бета-гидрокси-имидазопропионильное производное), его производное, получаемое модификацией аминогруппы двумя метильными остатками (диметил-гистидильное производное), его производное, получаемое замещением N-концевой аминогруппы карбоксильной группой (бета-карбоксиимидазопропионильное производное), или его производное, получаемое удалением положительного заряда с аминогруппы, при котором удаляют атом углерода в альфа-положении (альфа-углерод) N-концевого остатка гистидина, оставляя только имидазольную группу, и другие производные с модифицированной N-концевой аминогруппой.
В настоящем изобретении более предпочтительно, чтобы производным инсулинотропного пептида являлось производное экзендина-4, имеющее химически модифицированные N-концевую аминогруппу и аминокислотный остаток, еще более предпочтительно - производное экзендина-4, которое получают удалением или заменой альфа-аминогруппы, присоединенной к альфа-углероду N-концевого остатка His1 экзендина-4, или удалением или заменой альфа-углерода. Еще более предпочтительными являются производные, показанные на следующих формулах -

Носитель, который можно использовать в настоящем изобретении, представляет собой вещество, ковалентно связанное с инсулинотропным пептидом через непептидильный линкер и заметно увеличивающее время полужизни пептида в кровотоке, которое можно выбрать из группы, состоящей из иммуноглобулиновой Fc-области, сывороточного альбумина, трансферина, коллагена и их фрагментов, фибронектина и его фрагментов, и ПЭГ, а предпочтительным носителем является иммуноглобулиновая Fc-область. Используемый в настоящем изобретении термин «иммуноглобулиновая Fc-область» относится к белку, который включает в себя 2-ю константную область тяжелой цепи (СН2) и 3-ю константную область тяжелой цепи (СН3) иммуноглобулина, 1-ю константную область тяжелой цепи (СН1) и 1-ю константную область легкой цепи (CL1) иммуноглобулина за исключением вариабельных областей тяжелой и легкой цепей. Он дополнительно может включать шарнирный участок константной области тяжелой цепи. Также иммуноглобулиновая Fc-область по настоящему изобретению может содержать часть или всю Fc-область, включая 1-ю константную область тяжелой цепи (СН1) и/или 1-ю константную область легкой цепи (CL1) за исключением вариабельных областей тяжелой и легкой цепей, при условии, что эта конструкция обладает физиологическими функциями, по существу аналогичными (или лучшими) свойствам нативного белка. Также иммуноглобулиновая Fc-область может представлять собой фрагмент с удаленной относительно большой частью аминокислотной последовательности СН2 и/или СН3. А именно иммуноглобулиновая Fc-область по настоящему изобретению может содержать: 1) СН1-домен, СН2-домен, СН3-домен и СН4-домен, 2) СН1-домен и СН2-домен, 3) СН1-домен и СН3-домен и 4) СН2-домен и СН3-домен, 5) комбинацию одного или нескольких доменов и шарнирного участка иммуноглобулина (или части шарнирного участка) и 6) димер каждого домена константных областей тяжелой цепи и константной области легкой цепи. Иммуноглобулиновая Fc-область по настоящему изобретению включает нативную аминокислотную последовательность и ее производную (мутанта). Производное аминокислотной последовательности представляет собой последовательность, отличающуюся от нативной аминокислотной последовательности вследствие наличия делеции, инсерции, неконсервативной или консервативной замены (или их комбинации) одного или более аминокислотных остатков. Например, в Fc иммуноглобулина IgG аминокислотные остатки 214-238, 297-299, 318-322 или 327-331, известные как важные для связывания, можно использовать в качестве подходящей мишени для модификации. Также возможны различные другие производные, в том числе производные, в которых удалена область, способная образовывать дисульфидные связи, или удалены определенные аминокислотные остатки на N-конце нативной формы Fc, или к ней добавлен остаток метионина. Кроме того, для удаления эффекторных функций можно провести делецию на участке связывания комплемента, таком как участок связывания C1q, и ADCC-участке (антитело-зависимой клеточно-опосредованной цитотоксичности). Методики получения таких производных иммуноглобулиновой Fc-области раскрыты в WO 97/34631 и WO 96/32478. В данной области известны замены аминокислот в белках и пептидах, которые в общем не влияют на активность белков и/или пептидов (H. Neurath, R. L. Hill, The Proteins, Academic Press, New York, 1979). Наиболее часто распространенными заменами являются Ala/Ser, Val/Ile, Asp/Glu, Thr/Ser, Ala/Gly, Ala/Thr, Ser/Asn, Ala/Val, Ser/Gly, Thy/Phe, Ala/Pro, Lys/Arg, Asp/ Asn, Leu/Ile, Leu/Val, Ala/Glu и Asp/Gly, в любом направлении. В дополнение, при желании модификацию Fc-области можно провести с помощью фосфорилирования, сульфирования, акрилирования (добавления остатков акриловой кислоты), гликозилирования, метилирования, фарнезилирования, ацетилирования, амидирования и т.п. Перечисленные выше производные Fc представляют собой производные, которые имеют биологическую активность, идентичную Fc-области по настоящему изобретению, или улучшенную структурную стабильность, например, к нагреванию, рН и т.п. В дополнение, эти Fc-области могут быть получены из нативных форм, выделенных из человека и других животных, в том числе коров, коз, свиней, мышей, кроликов, хомяков, крыс и морских свинок, или могут представлять собой их рекомбинатные формы или производные, полученные из трансформированных животных клеток или микроорганизмов. При этом они могут быть получены из нативного иммуноглобулина путем выделения полноразмерных иммуноглобулинов из организма человека или животных и обработки их протеолитическим ферментом. Папаин расщепляет нативные иммуноглобулины на Fab- и Fc-области, а обработка пепсином приводит к pF'c- и F(ab)2-фрагментам. Например, для выделения Fc или pF'c эти фрагменты можно разделить с помощью гель-хроматографии. Предпочтительно, чтобы полученная от человека Fc-область представляла собой рекомбинантную Fc-область иммуноглобулина, получаемую из микроорганизмов. В дополнение, иммуноглобулиновая Fc-область по настоящему изобретению может находиться в форме, имеющей нативные сахарные цепи, увеличенное число сахарных цепей относительно нативной формы или уменьшенное число сахарных цепей по сравнению с нативной формой, или она может находится в дегликозилированной форме (т.е. без присоединенных сахаров). Увеличить/уменьшить количество сахарных цепей Fc-области иммуноглобулина или удалить их можно общеизвестными в данной области способами, такими как химический, ферментативный или генно-инженерный способ с использованием микроорганизмов. Удаление цепей сахара с Fc-области приводит к резкому снижению аффинности связывания с C1q частью первого компонента С1 комплемента и снижению или потере антитело-зависимой клеточно-опосредованной цитотоксичности или комплемент-зависимой цитотоксичности, вследствие чего не возникает ненужный иммунный ответ in vivo. В этом отношении иммуноглобулиновая Fc-область в дегликозилированной или агликозилированной форме может лучше подходит в качестве носителя лекарственного средства для цели настоящего изобретения. Используемый в настоящем описании термин «дегликозилированный» относится к ферментативно удаленным с Fc-области сахарам, а термин «агликозилированный» относится к Fc-области, которая продуцируется в негликозилированной форме прокариотами, предпочтительно, E. coli. С другой стороны, иммуноглобулиновую Fc-область можно получить из иммуноглобулинов человека или других животных, в том числе коров, коз, свиней, мышей, кроликов, хомяков, крыс и морских свинок и, предпочтительно, человека. В дополнение иммуноглобулиновая Fc-область может представлять собой Fc-область, полученную из IgG, IgA, IgD, IgE и IgM, или может быть создана из их комбинации или гибрида. Предпочтительно, чтобы она была получена из IgG или IgM, которые относятся к наиболее распространенным белкам в крови человека, и, наиболее предпочтительно, из IgG, который, как известно, увеличивает время полужизни лиганд-связывающих белков. С другой стороны, используемый в настоящем описании термин «комбинация» означает, что полипептиды, кодирующие одноцепочечные иммуноглобулиновые Fc-области из разных источников, присоединены к одноцепочечному полипептиду, происходящему из другого источника, образуя димер или мультимер. То есть димер или мультимер могут быть образованы двумя или несколькими фрагментами, выбранными из группы, состоящей из Fc-фрагментов IgG, IgA, IgM, IgD и IgE. Используемый в настоящем описании термин «гибрид» означает, что последовательности, кодирующие два или несколько иммуноглобулиновых Fc-областей различного происхождения, присутствуют в одноцепочечной иммуноглобулиновой Fc-области. В настоящем изобретении возможны различные типы гибридов. То есть в доменные гибриды могут входить от одного до четырех доменов, выбранных из группы, состоящих из СН1, СН2, СН3 и СН4 Fc-областей IgG, IgM, IgA, IgE и IgD, и могут включать шарнирный участок. С другой стороны, IgG подразделяются на подклассы IgG1, IgG2, IgG3 и IgG4, а настоящее изобретение включает их комбинации и гибриды. Предпочтительными является подклассы IgG2 и IgG4, а наиболее предпочтительной является Fc-область IgG4, редко имеющего эффекторные функции, такие как CDC (комплемент-зависимая цитотоксичность). То есть в качестве лекарственного носителя по настоящему изобретению наиболее предпочтительной иммуноглобулиновой Fc-областью является негликозилированная Fc-область, полученная из IgG4 человека. Полученная из иммуноглобулина человека Fc-область является более предпочтительной, чем Fc-область, полученная из иммуноглобулина биологического вида, кроме человека, который может действовать как антиген в организме человека и вызывать нежелательные иммунные ответы, такие как продукция нового антитела против антигена.
В конъюгате, содержащемся в композиции по настоящему изобретению, инсулинотропный пептид присоединен к носителю через непептидильный линкер. Используемый в настоящем описании термин «непептидильный линкер» относится к моносоединению или биосовместимому полимеру, включающему две или несколько повторяющихся единиц, соединенных друг с другом. Непептидильный линкер, который можно использовать в настоящем изобретении, может иметь любую химическую структуру, и преимущественно функционирует в качестве линкера, связывающего инсулинотропный пептид и носитель друг с другом ковалентной связью. Таким образом непептидильный линкер отличается тем, что является химическим соединением, имеющим реакционные группы, способный ковалентно связывать пептид/носитель на двух концах, в котором концевые реакционно-способные группы на двух концах являются одинаковыми или отличаются друг от друга. Реакционно-способные группы на двух концах непептидильного линкера могут быть одинаковыми или различными. Например, непептидильный линкер может иметь малеимидную группу на одном конце и альдегидную группу, пропиональдегидную группу или бутиральдегидную группу на другом конце. Реакционно-способные группы на двух концах непептидильного линкера предпочтительно выбирать из группы, состоящей из реакционно-способной альдегидной группы, пропиональдегидной группы, бутиральдегидной группы, малеимидной группы и сукцинимидного производного. Сукцинимидным производным может быть сукцинимидилпропионат, гидроксисукцинимидил, сукцинимидил-карбоксиметил или сукцинимидилкарбонат. В частности, когда непептидильный линкер имеет реакционно-способную альдегидную группу на двух концах, он эффективен для присоединения на двух концах физиологически активного полипептида и иммуноглобулиновой Fc-области с минимальными неспецифическими реакциями. Конечный продукт, генерируемый восстановительным алкилированием с помощью альдегидной связи, является гораздо более стабильным, чем соединенный амидной связью. Альдегидная реакционная группа селективно связывает N-конец белка при низких значениях рН и может связывать остаток лизина с образованием ковалентной связи при высоком рН, таком как рН 9,0. Когда в качестве непептидильного полимера используют полиэтиленгликоль, имеющий реакционно-способные гидроксильные группы на своих двух концах, гидроксильные группы можно превратить в различные реакционно-способные группы с помощью известных химических реакций, или можно использовать доступный в продаже полиэтиленгликоль с модифицированными реакционно-способными группами, группы для получения конъюгата инсулинотропного пептида по настоящему изобретению. Непептидильный полимер, который можно использовать в настоящем изобретении, может представлять собой (но не ограничен этими соединениями) SMCC (сукцинимидил-4-(N-малеимидо-метил)циклогексан-1-карбоксилат) или SFB (сукцинимидил-4-формилбензоат), которые могут быть ковалентно присоединены к аминным или сульфгидрильным группам пептида. Непептидильный полимер может быть выбран из группы, состоящей из полиэтиленгликоля, полипропиленгликоля, поливинилпирролидона, сополимеров этиленгликоля и пропиленгликоля, полиоксиэтилированных многоатомных спиртов, поливинилового спирта, полисахаридов, декстрана, поливинилэтилового эфира, биоразлагаемых полимеров, таких как PLA (полимолочная кислота) и PLGA (полимолочная-гликолевая кислота), липидных полимеров, хитинов, гиалуроновой кислоты и их комбинаций, а предпочтительным является полиэтиленгликоль. Также в объем настоящего изобретения включены их производные, хорошо известные в данной области, которые легко может получить специалист в данной области.
Предпочтительные примеры конъюгата, который содержится в композиции для лечения связанных с ожирением заболеваний по настоящему изобретению, полученного ковалентным связыванием инсулинотропного пептида с носителем через непептидильный линкер, раскрыты в WO08/082274 и представлены следующей формулой 1.
<Формула 1>
R1-X-R2-L-F,
в которой R1 выбран с группы, состоящей из дезамино-гистидила, диметил-гистидила, бета-гидрокси-имидазопропила, 4-имидоазоацетила и бета-карбокси-имидазо-пропионила,
R2 выбран из группы, состоящей из -NH2, -OH и -Lys,
Х выбран из группы, состоящей из Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Y-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Z-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser, Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Y-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Z-Asn-Gly-Gly и Ser-Asp-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Y-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Z-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser,
Y выбран из группы, состоящей из Lys, Ser и Arg,
Z выбран из группы, состоящей из Lys, Ser и Arg,
L представляет собой непептидильный линкер и
F является Fc-областью иммуноглобулина.
В дополнение, фармацевтическая композиция, содержащая конъюгат по настоящему изобретению, может включать в себя фармацевтически приемлемый носитель. Для перорального введения фармацевтически приемлемый носитель может включать связующее вещество, смазывающее вещество, разрыхлитель, вспомогательное вещество, растворитель, диспергирующий агент, стабилизатор, суспендирующий агент, краситель и отдушку. Для препаратов для инъекций фармацевтически приемлемый носитель может включать буферный агент, консервант, обезболивающее вещество, растворитель, изотонический агент и стабилизатор. Для местных препаратов фармацевтически приемлемый носитель может включать основу, вспомогательное вещество, смазывающее вещество и консервант. Фармацевтическая композиция по настоящему изобретению может быть введена в состав различных лекарственных форм в комбинации с вышеупомянутыми фармацевтически приемлемыми носителями. Например, для перорального введения фармацевтическая композиция может быть введена в состав таблеток, пастилок, капсул, эликсиров, суспензий, сиропов или облаток. Для препаратов для инъекций фармацевтическая композиция может быть введена в состав стандартной лекарственной формы, такой как мультидозовый контейнер, или в ампулу, в качестве однодозовой лекарственной формы. Фармацевтическая композиция может быть также введена в состав растворов, суспензий, таблеток, пилюль, капсул и препаратов длительного действия. С другой стороны, примеры носителя, вспомогательного вещества и разбавителя, подходящих для фармацевтических составов, включают лактозу, декстрозу, сахарозу, сорбит, маннит, ксилит, эритрит, мальтит, крахмал, гуммиарабик, альгинат, желатин, фосфат кальция, силикат кальция, целлюлозу, метилцеллюлозу, микрокристаллическую целлюлозу, поливинилпирролидон, воду, метилгидроксибензоат, пропилгидроксибензоат, тальк, стеарат магния и минеральные масла. В дополнение, фармацевтические составы могут, кроме того, включать наполнители, антикоагулянты, смазывающие вещества, увлажнители, отдушки и антисептики. Вводимую дозу фармацевтической композиции по настоящему изобретению можно определить с помощью нескольких связанных факторов, включая тип заболевания, против которого будет проводиться лечение, пути введения, возраст, пол, вес и тяжесть болезни пациента. Поскольку фармацевтическая композиция по настоящему изобретению обладает превосходными показателями по продолжительности действия in vivo и титру, это может заметно снизить частоту введения и дозу фармацевтических лекарственных препаратов, содержащих композицию по настоящему изобретению.
Конъюгат инсулинотропного пептида, содержащийся в композиции по настоящему изобретению, обладает продолжительным эффектом подавления потребления пищи при гораздо более меньшей концентрации, чем нативный инсулинотропный пептид, вследствие чего используется для лечения заболеваний, таких как ожирение и острый коронарный синдром. В дополнение, благодаря эффекту подавления потребления пищи (подавления аппетита) конъюгат инсулинотропного пептида можно использовать для уменьшения количества телесного жира, такого как холестерин, и жировой ткани. Для лечения ожирения и связанных с ожирением заболеваний необходимо снижение телесного жира, но нежелательна потеря нежировой ткани, то есть потеря белка. Поскольку нежировая масса тела состоит из мышц, органов жизнеобеспечения, костей, соединительной ткани и других нежировых тканей, полагают, что потеря нежировой массы ткани вредна для здоровья человека. Соответственно, потеря веса вследствие подавления аппетита с помощью композиции по настоящему изобретению приводит к потере жировой ткани, а не уменьшает массу нежировой ткани, и, таким образом, действует в качестве очень важного фактора при лечении связанных с ожирением заболеваний.
Поскольку инсулинотропные пептиды, такие как GLP-1, амилин, ССК и экзендин, сохраняют свою эффективность по подавлению аппетита в течение короткого времени от 1 до 6 часов после введения, для лечения хронических заболеваний, таких как ожирение и связанные с ожирением заболевания, их необходимо вводить повторно. Конъюгат инсулинотропного пептида, содержащийся в композиции по настоящему изобретению, сохраняет свою эффективность при низкой дозировке в течение недели, таким образом, проявляя максимальную терапевтическую эффективность.
По еще одному аспекту настоящее изобретение относится к способу лечения связанных с ожирением заболеваний, способу подавления потребления пищи и способу уменьшения количества телесного жира путем использования композиции. В частности, способ по настоящему изобретению может включать в себя стадию введения терапевтически эффективного количества композиции.
Используемый в настоящем описании термин «введение» означает введение определенного количества вещества пациенту с помощью некоторого подходящего способа. Содержащую конъюгат композицию можно вводить любыми общепринятыми путями, при условии, что он способен достичь целевой ткани. Предусмотрен ряд вариантов введения, включая интраперитонеальное, внутривенное, внутримышечное, подкожное, внутрикожное, пероральное, местное, интраназальное, внутрилегочное и интратекальное, но настоящее изобретение не ограничено этими приведенными для примера вариантами введения. Однако вследствие того что пептиды расщепляются при пероральном введении, активные ингредиенты композиции для защиты от разрушения в желудке должны быть покрыты оболочкой или введены в фармацевтический состав. Предпочтительно, чтобы настоящую композицию можно было вводить в виде инъекций. В дополнение, фармацевтическую композицию по настоящему изобретению можно вводить с использованием некоторых приборов, способных переносить активные ингредиенты в клетку-мишень. При этом терапевтически приемлемую дозу композиции можно определить исходя из различных вышеупомянутых факторов.
По еще одному аспекту настоящее изобретение относится к фармацевтической композиции для лечения связанных с ожирением заболеваний с использованием только одного конъюгата инсулинотропного пептида или в комбинации с одним или несколькими лекарственными соединениями против ожирения. Примеры веществ, входящих в фармацевтическую композицию для лечения связанных с ожирением заболеваний в комбинации с конъюгатом инсулинотропного пептида, включают вещества, имеющие подавляющую аппетит или усиливающую метаболизм активность, активность, подавляющую деградацию липидов, активность, снижающую чувство пустоты в желудке, активность, ингибирующую белковую тирозин-фосфатазу (РТР)-1b, и DPPIV-ингибирующую активность, такую как активность GLP-1 и его производных (Patricia, Trends in endocrinology and metabolism 2007; 18:240-245), амилин, PYY (пептид YY) (Lynn et al., Bioorganic & Medicinal Chemistry Letters 2007;17:1916-1919), лептин, холецитокинин (CCK), оксинтомодулин, грелиновый антагонист, антагонист NPY (Elena et al., Nutrition, Metabolism & Cardiovascular Disease 2008; 18:158-168; Sarika et al., Neuropeptides 2006;40:375-401), римонабант, сибутрамин и орлистат (Alan Dove., Nature biotechnology 2001; 19:25-28), но не ограничены ими.
Осуществление изобретения
Далее в настоящем документе для лучшего понимания изобретения следуют примеры, приведенные для иллюстрации изобретения, которые не следует истолковывать как ограничивающие настоящее изобретение.
[Пример 1] Получение конъюгата инсулинотропного пептида (АЦ-экзендина)
Поводили реакцию присоединения (пегилирования) PropionALD(2)PEG (ПЭГ с двумя пропиональдегидными группами, 3400 кД, IDB Inc., Korea) к остатку лизина имидазо-ацетил-экзендина-4 (Bachem, Swiss) при 4°С в течение ночи с молярным соотношением пептида к ПЭГ 1:15 при концентрации пептида 5 мг/мл. На данном этапе реакцию проводили в буфере при рН 7,5 и к реакции в качестве восстанавливающего агента добавляли 20 мМ SCB (NaCNBH3) для проведения реакции. Монопегилированный экзендин и изомеры выделяли на колонке SOURCE S (XK 16 мл, Amersham Biosciences) при следующих условиях.
Колонка: SOURCE S (XK 16 мл, Amersham Biosciences)
Скорость потока: 2,0 мл/мин
Градиент: A 0→100% B за 45 мин (A: 20 мМ лимонная кислота, рН 3,0, B: A+0,5 M KCl)
Выделенный монопегилированный АЦ-экзендин-4 присоединяли к Fc иммуноглобулина. Реакцию проводили в соотношении пептида к Fc иммуноглобулина - 1:4 и общей концентрации белков 50 мг/мл при 4°С в течение 16 часов. Реакцию проводили в 100 мМ растворе K-P (pH 6,0) и к реакции добавляли 20 мМ SCB в качестве восстанавливающего агента. После реакции присоединения проводили двухстадийную очистку на колонках SOURCE Phe (16 мл) и SOURCE Q (16 мл).
Колонка: SOURCE Phe (HR l6 мл, Amersham Biosciences)
Скорость потока: 2,0 мл/мин
Градиент: B 100→0% В за 30 мин (A: 20 мМ Tris pH 7,5, B: A+1,5 M NaCl)
Колонка: SOURCE Q (XK 16 мл, Amersham Biosciences)
Скорость потока: 2,0 мл/мин
Градиент: A 0→25% В за 60 мин (A: 20 мМ Tris pH 7,5, B: A+1 M NaCl)
[Пример 2] Эффект потери веса у ob/ob мышей в результате действия конъюгата инсулинотропного пептида
Служащих в качестве хорошо известной животной модели ожирения 8-9 недельных самок ob/ob мышей (C57BL/6JHamSlc-ob/bo) разделяли на 4 группы (по 5 мышей в каждой группе) и им вводили носитель и Byetta (Amylin-Lily, экзендин-4, 45 мкг/кг, ежедневно с помощью подкожной инъекции) и конъюгат инсулинотропного пептида, полученный в примере 1 (45 мкг или 100 мкг/кг, один раз в неделю с помощью подкожной инъекции). Затем в течение 28 дней проводили измерения изменения веса тела и после окончания введения измеряли содержание в крови параметров липидного метаболизма, таких как холестерин и свободные жирные кислоты. После завершения тестирования вырезали печень и жировую ткань и взвешивали. Эффект потери веса у ob/ob мышей под действием конъюгата инсулинотропного пептида показан в таблице 1.
Как показано в таблице 1 и на фиг. 1 и 2, конъюгат инсулинотропного пептида демонстрировал эффект снижения веса тела и уровня холестерина, превышающий эффект Byetta, при дозировке 1/7 от дозировки Byetta, и эффекты были дозозависимыми. В дополнение, эффективность при введении конъюгата инсулинотропного пептида раз в неделю сохранялась дольше эффективности при ежедневном введении экзендина-4.
[Пример 3] Эффект потери веса у DIO мышей в результате действия конъюгата инсулинотропного пептида
Служащих в качестве хорошо известной животной модели ожирения 25-недельных самцов DIO (вызванное диетой ожирение) мышей (C57BL/6NCrjBgi) разделяли на 5 групп (по 5 мышей в каждой группе) и затем им вводили носитель и Byetta (100 мкг/кг, ежедневно с помощью подкожной инъекции) и конъюгат инсулинотропного пептида, полученный в примере 1 (20, 50, 100 мкг/кг, один раз в неделю с помощью подкожной инъекции). Затем в течение 2 недель проводили измерения изменения веса тела. Эффект потери веса у DIO мышей под действием конъюгата инсулинотропного пептида показан в таблице 2.
Как показано в таблице 2 и на фиг. 3, конъюгат инсулинотропного пептида демонстрировал эффект снижения веса тела, превышающий эффект Byetta, в 1/17,5 дозировке, и эффекты были дозозависимыми. В дополнение, эффективность при введении конъюгата инсулинотропного пептида раз в неделю сохранялась дольше эффективности при ежедневном введении экзендина-4.
[Пример 4] Эффект потери веса у ZDF (диабетическое ожирение Зукера) крыс в результате действия конъюгата инсулинотропного пептида
7-недельных самцов ZDF-крыс, которых обычно используют в качестве модели диабета и которые имеют признаки, аналогичные ob/ob мышам (ZDF/Gmi-fa/fa), разделяли на 5 групп (по 5 крыс в каждой группе) и затем им вводили носитель и Byetta (100 мкг/кг, ежедневно с помощью подкожной инъекции) и конъюгат инсулинотропного пептида, полученный в примере 1 (20, 50, 100 мкг/кг, один раз в неделю с помощью подкожной инъекции). Затем в течение 8 недель проводили измерения изменения веса тела и потребления пищи и после окончания введения измеряли содержание в крови параметров липидного метаболизма, таких как холестерин. После завершения тестирования вырезали жировую ткань и взвешивали. Эффект потери веса у ZDF крыс под действием конъюгата инсулинотропного пептида показан в таблице 3.
Как показано в таблице 3 и на фиг. 4 и 5, конъюгат инсулинотропного пептида демонстрировал эффекты снижения веса тела, уменьшения количества телесного жира и подавления потребления пищи, превышающие эффект Byetta, в 1/35 дозировке, и эффекты были дозозависимыми. В дополнение, эффективность при введении конъюгата инсулинотропного пептида раз в неделю сохранялась дольше эффективности при ежедневном введении экзендина-4.
Реферат
Изобретение относится к медицине и касается композиции для лечения связанных с ожирением заболеваний, содержащей конъюгат инсулинотропного пептида и фармацевтически приемлемый носитель, где инсулинотропный пептид выбран из группы, состоящей из производных GLP-1, экзендина-3, экзендина-4. Изобретение обеспечивает улучшение эффективности подавления потребления пищи, большее время действия in vivo. 10 н. и 7 з.п. ф-лы, 4 пр., 5 ил., 3 табл.
Формула
R1-X-R2-L-F <Формула 1>,
в которой R1 выбран из группы, состоящей из дезамино-гистидила, диметил-гистидила, бета-гидрокси-имидазопропионила, 4-имидоазоацетила и бета-карбокси-имидазопропионила,
R2 выбран из группы, состоящей из -NH2, -ОН и -Lys,
Х выбран из группы, состоящей из Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Y-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Z-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser, Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Y-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Z-Asn-Gly-Gly и Ser-Asp-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Y-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Z-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser,
Y выбран из группы, состоящей из Lys, Ser и Arg,
Z выбран из группы, состоящей из Lys, Ser и Arg,
L представляет собой непептидильный линкер, выбранный из группы, состоящей из SMCC (сукцинимидил-4-(N-малеимидо-метил)циклогексан-1-карбоксилата), SFB (сукцинимидил-4-формилбензоата), полиэтиленгликоля, полипропиленгликоля, поливинилпирролидона, сополимеров этиленгликоля и пропиленгликоля, полиоксиэтилированных многоатомных спиртов, поливинилового спирта, полисахаридов, декстрана, поливинилэтилового эфира, биоразлагаемых полимеров, таких как PLA (полимолочная кислота) и PLGA (полимолочная-гликолевая кислота), липидных полимеров, хитинов, гиалуроновой кислоты и их комбинаций, и
F является Fc-областью иммуноглобулина.
R1-X-R2-L-F <Формула 1>,
в которой R1, X, R2, L и F являются точно такими же, как в п.3.
R1-X-R2-L-F <Формула 1>,
в которой R1, X, R2, L и F являются точно такими же, как в п.3.
Комментарии