Соединения, активные при новом сайте на оперируемых рецептором кальциевых каналах, применимые для лечения неврологических нарушений - RU2246300C2
Код документа: RU2246300C2
Описание
Данная заявка является частичным продолжением находящейся на одновременном рассмотрении заявки U.S. Serial No. 08/485038, поданной 7 июня 1995 года, которая является частичным продолжением одновременно рассматриваемой Международной патентной публикации No. PCT/US94/12293, поданной 26 октября 1994 года, которая является частичным продолжением одновременно рассматриваемой заявки U.S. Serial No. 08/288688, поданной 9 августа 1994 года, которая является частичным продолжением ожидающей одновременного рассмотрения заявки U.S. Serial No. 08/194210, поданной 8 февраля 1994 года, которая является частичным продолжением U.S. Serial No. 08/014813, поданной 8 февраля 1993 года, в настоящее время абандонированной. Все они включены здесь в качестве ссылок во всей их полноте.
Область изобретения
Данное изобретение относится к соединениям, которые могут использоваться в качестве нейропротекторов, антиконвульсивных средств, транквилизаторов, аналгетиков, миорелаксантов или адъювантов для общих анестетиков. Данное изобретение относится также к способам, которые могут использоваться для лечения неврологических нарушений и заболеваний, в том числе, но не только, общего и очагового ишемического и геморрагического удара (внезапного приступа), повреждения спинного мозга, индуцированного гипоксией повреждения нервных клеток, такого как остановка сердца или неонатальный дистресс, эпилепсии, состояния тревоги и нейродегенеративных заболеваний, таких как болезнь Альцгеймера, болезнь Хангтингтона, болезнь Паркинсона и боковой амиотрофический склероз (ALS). Данное изобретение относится также к способам скрининга на соединения, активные при новом сайте на оперируемых рецепторами кальциевых каналах и вследствие этого имеющие терапевтическую применимость в качестве нейропротекторов, антиконвульсивных средств, транквилизаторов, аналгетиков и/или имеющие потенциальную терапевтическую применимость для лечения неврологических нарушений и заболеваний, описанных выше.
Предпосылки изобретения
Далее следует описание родственной области знаний, причем ни один из приводимых примеров не является прототипом пунктов формулы изобретения.
Глутамат представляет собой основной нейротрансмиттер (нейромедиатор) возбуждения в мозгу млекопитающего. Глутамат связывается или взаимодействует с одним или несколькими рецепторами глутамата, которые можно дифференцировать фармакологически на несколько подтипов. В центральной нервной системе млекопитающего (ЦНС) имеется три основных подтипа ионотропных рецепторов глутамата, определяемых фармакологически при помощи селективных агонистов N-метил-D-аспартата (NMDA), каината (КА) и α -амино-3-гидрокси-5-метилизоксазол-4-пропионовой кислоты (АМРА). Считалось, что NMDA-рецептор принимает участие во многих неврологических патологиях, в том числе во внезапном приступе (инсульте), травме головы, повреждении спинного мозга, эпилепсии, состоянии тревоги и нейродегенеративных заболеваниях, таких как болезнь Альцгеймера (Watkins and Collingridge. The NMDA Receptor, Oxford: IRL Press, 1989). Роль рецепторов NMDA в восприятии боли и аналгезии была также хорошо постулирована (Dickenson, A cure for wind-up: NMDA receptor antagonists as potential analgesics. Trends Pharmacol. Sci. 11: 307, 1990). Более недавно широко исследовались АМРА-рецепторы на их возможное участие в таких нейрологических патологиях (Fischer and Bogousslavsky, Evolving toward effective therapy for acute ischemic stroke. J. Amer. Med. Assoc. 270: 360, 1993; Yamaguchi et al., Anticonvulsant activity of AMPA/kainate antagonists: Comparison of GYKI 52466 and NBQX in maximal electroshok and chemoconvulsant seizure models. Epilepsy Res. 15: 179, 1993).
При активации глутаматом эндогенный нейротрансмиттер, рецептор NMDA, делает возможным приток внеклеточного кальция (Са2+) и натрия (Na+) через ассоциированный ионный канал. NMDA-рецептор делает возможным значительно больший приток Са2+, чем каинатный или АМРА-рецепторы (см. ниже), и является примером оперируемого рецептором Са2+-канала. В норме этот канал является открытым только недолго, позволяя локализованное и временное увеличение концентрации внутриклеточного Са2+ ([Са2+]), которая, в свою очередь, изменяет функциональную активность этой клетки. Однако пролонгированные увеличения [Са2+]i, происходящие из-за хронической стимуляции NMDA-рецептора, являются токсическими для клетки и приводят к гибели клетки. Считают, что хроническое повышение [Ca2+]i, происходящее в результате стимуляции NMDA-рецепторов, является первичной причиной дегенерации нейронов после инсульта (удара) (Choi, Glutamate neurotoxicity and diseases of the nervous system. Neuron 1: 623, 1988). Считают также, что сверхстимуляция NMDA-рецепторов участвует в патогенезе некоторых форм эпилепсии (Dingledine et al., Excitatory amino acid receptors in epilepsy. Trends Pharmacol. Sci. 11:334, 1990), состоянии тревоги (Wiley and Dalster, Preclinical evaluation of N-methyl-D-aspartate antagonists for antianxiety effects: A review. In: Multiple Sigma and PCP Receptor Ligands: Mechanisms for Neuromodulation and Neuroprotection? NPP Books, Ann Arbor, Michigan, pp.801-815, 1992), нейродегенеративных заболеваний (Meldrum and Garthwaite, Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol. Sci. 11: 379, 1990) и гипералгезических состояниях (повышенной болевой чувствительности) (Dickenson, A cure for wind-up: NMDA receptor antagonists as potential analgesics. Trends Pharmacol. Sci. 11: 307, 1990).
Активность комплекса NMDA-рецептор-ионофор регулируется множеством модуляторных сайтов, на которые могут быть нацелены селективные антагонисты. Конкурентные антагонисты, такие как фосфонат АР5, действуют при глутаматсвязывающем сайте, тогда как неконкурентные антагонисты, такие как фенциклидин (РСР), МК-801 или магний (Mg2+), действуют внутри ассоциированного ионного канала (ионофора). Имеется также сайт связывания глицина, который может селективно блокироваться такими соединениями, как 7-хлоркинуреновая кислота. Имеется доказательство, предполагающее, что глицин действует как коагонист, так что как глутамат, так и глицин являются необходимыми для индукции полных опосредованных NMDA-рецептором ответных реакций. Другие потенциальные сайты модулирования функции NMDA-рецептора включают цинк (Zn2+)-связывающий сайт и сигма-лигандсвязывающий сайт. Кроме того, считают, что эндогенные полиамины, такие как спермин, связываются со специфическим сайтом и таким образом усиливают функцию NMDA-рецептора (Ransom and Stec, Cooperative modulation [3H]MK-801 binding to the NMDA receptorion channel complex by glutamate, glycine and polyamines. J. Neurochem. 51:830, 1988). Потенциирующий эффект полиаминов на функцию NMDA-рецептора может быть опосредован через специфический рецепторный сайт для полиаминов; были описаны полиамины, демонстрирующие агонистическую, антагонистическую и обращенную агонистическую активность (Reynolds, Arcaine is a competitive antagonist of the polyamine site on the NMDA receptor. Europ. J. Pharmacol. 177: 215, 1990; Williams et al., Characterization of polyamines having agonist, antagonist, and inverse agonist effects at the polyamine recognition site of the NMDA receptor. Neuron 5: 199, 1990). Исследования связывания радиолиганда продемонстрировали дополнительно, что более высокие концентрации полиаминов ингибируют функцию NMDA-рецептора (Reynolds and Miller, Ifenprodil is a novel type of NMDA receptor antagonist: Interaction with polyamines. Molec. Pharmacol. 36: 758, 1989; Williams et al., Effects of polyamines on the binding of [3H]MK-801 to the NMDA receptor: Pharmacological evidence for the existence of a polyamine recognition site. Molec. Pharmacol. 36: 575, 1989; Sacaan and Johnson, Characterization of the stimulatory and inhibitory effects of polyamines on [3H]TCP binding to the NMDA receptor-ionophore complex. Molec. Pharmacol. 37: 572, 1990). Это ингибиторное действие полиаминов на NMDA-рецепторы является, возможно, неспецифическим эффектом (т.е. не медиированным через рецептор полиамина), поскольку электрофизиологические исследования (пэтч-кламп, patch-clamp)) показали, что это ингибирование осуществляется соединениями, которые, как показано ранее, действуют при рецепторе полиамина либо как агонисты, либо как антагонисты (Donevan et al., Arcaine Blocks N-Methyl-D-Aspartate Receptor Responses by an Open Channel Mechanism: Whole-Cell and Single-Channel Recording Studies in Cultured Hippocampal Neurons. Molec. Pharmacol. 41: 727, 1992; Rock and Macdonald, Spermine and Related Polyamines Produce a Voltage-Dependent Reduction of NMDA Receptor Single-Channel Conductance. Volec. Pharmacol. 42: 157, 1992).
Недавние исследования показали молекулярное разнообразие рецепторов глутамата (описанных в обзоре Nakanishi, Molecular Diversity of Glutamate Receptors and Implications for Brain Function. Science 258: 597, 1992). К настоящему времени были идентифицированы по меньшей мере пять отличающихся субъединиц NMDA-рецептора (NMDAR1 и NMDAR2A - NMDAR2D), каждая из которых кодируется отдельным геном. Также в NMDAR1 альтернативный сплайсинг дает по меньшей мере шесть дополнительных изоформ. По-видимому, NMDAR1 является необходимой субъединицей и комбинирование NMDAR1 с различными членами форм NMDAR2 образует полностью функциональный комплекс NMDA-рецептор-ионофор. Комплекс NMDA-рецептор-ионофор, таким образом, может быть определен как гетероолигомерная структура, состоящая по меньшей мере из субъединиц NMDAR1 и NMDAR2; существование дополнительных, еще не идентифицированных, субъединиц не исключается этим определением. Было показано, что NMDAR1 обладает сайтами связывания для глутамата, глицина, Мg2+, МК-801 и Zn2+. Сайты связывания для сигма-лигандов и полиаминов не были еще определены на субъединицах NMDA-рецептора, хотя недавно сообщалось, что ифенпродил является более сильным при субъединице NMDAR2B, чем при субъединице NMDAR2A (Williams, Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol. Pharmacol. 44: 851, 1993).
Несколько отдельных подтипов АМРА- и каинатных рецепторов также были клонированы (обзор Nakanishi, Molecular diversity of glutamate receptors and implications for brain function. Science 258: 597, 1992). Особое отношение к данному вопросу имеют АМРА-рецепторы, названные GluR1, GluR2, GluR3 и GluR4 (также называемые GluRA-GluRD), каждый из которых существует в одной из двух форм, названных flip и flop, которые возникают в результате альтернативного сплайсинга РНК. GluR1, GluR3 и GluR4 при экспрессии в виде гомомерных или гетеромерных рецепторов являются проницаемыми для Са2+, и, следовательно, они являются примерами оперируемых рецепторами Са2+-каналов. Экспрессия только GluR2 одного или в комбинации с другими субъединицами приводит к рецептору, который является в большей мере непроницаемым для Сa2+. Поскольку наиболее нативные АМРА-рецепторы, изученные in situ, являются непроницаемыми для Са2, считают, что такие рецепторы in situ обладают по меньшей мере одной субъединицей GluR2.
Кроме того, высказывается гипотеза, что субъединица GluR2 является функционально отличающейся, благодаря тому факту, что она содержит остаток аргинина в вероятном образующем пору трансмембранном районе II; GluR1, GluR3 и Glu4 содержат (все) остаток глутамина в этом решающем районе (названный сайтом Q/R, где Q и R являются буквенными обозначениями для глутамина и аргинина соответственно). Активность АМРА-рецептора регулируется рядом модуляторных сайтов, на которые могут быть нацелены селективные антагонисты (Honore et al., Quinoxalinediones: potent competitive non-NMDA glutamate receptor antagonists. Science 241: 701, 1988; Donevan and Rogawski, GYKI 52466, a 2,3-benzodiazepine, is a highly selective, noncompetitive antagonist of AMPA/kainate receptor responses. Neuron 10: 51, 1993). Конкурентные антагонисты, такие как NBQX, действуют при сайте связывания глутамата, тогда как такие соединения, как GYKI 52466, действуют, по-видимому, неконкурентно при ассоциированном с ними аллостерическом сайте.
Сообщается, что соединения, действующие как конкурентные или неконкурентные антагонисты при NMDA-рецепторе, являются эффективными в предотвращении гибели нервных клеток в различных тестах нейротоксичности in vitro (Meldrum and Garthwaite, Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol. Sci. 11: 379, 1990) и в моделях in vivo удара (острого внезапного приступа) (Scatton, Therapeutic potential of NMDA-receptor antagonists in ischemic cerebro-vascular disease in Drug Strategies in the Prevention and Treatment of Stroke, IBC Technical Services Ltd., 1990). Такие соединения являются также эффективными противосудорожными средствами (Meldrum, Excitatory amino acid neurotransmission in epilepsy and anticonvulsant therapy in Excitatory Amino Acids. Meldrum, Moroni, Simon, and Woods (Eds.), New York: Raven Press, p.655, 1991), транквилизаторами (Wiley and Balster, Preclinical evaluation of N-methyl-D-aspartate antagonists for antianxiety effects: A review. In: Multiple Sigma and PCP Receptor Ligands: Mechanisms for Neuromodulation and Neuroprotection? NPP Books, Ann Arbor, Michigan, pp.801-815, 1992), и аналгетиками (Dickenson, A cure for wind-up: NMDA receptor antagonists as potential analgesics. Trends Pharmacol. Sci. 11: 307, 1990), и некоторые антагонисты NMDA-рецептора могут ослаблять деменцию, связанную с болезнью Альцгеймера (Hughes, Merz's novel approach to the treatment of dementia. Script No. 1666: 24, 1991).
Подобным образом, антагонисты АМРА-рецептора были подвергнуты интенсивному критическому изучению в качестве потенциальных терапевтических агентов для лечения таких неврологических нарушений и заболеваний. Было показано, что антагонисты АМРА-рецепторов обладают нейропротекторной (Fischer and Bogousslavsky, Evolving toward effective therapy for acute ischemic stroke. J. Amer. Med. Assoc. 270: 360, 1993), противосудорожной (Yamaguchi et al., Anticonvulsant activity of AMPA/kainate antagonists: comparison of GYKI 52466 and NBQX in maximal electroshock and chemoconvulsant seizure models. Epilepsy Res. 15: 179, 1993) активностью в моделях животных ишемического удара и эпилепсии соответственно.
Никотиновый холинергический рецептор, присутствующий в ЦНС млекопитающих, является другим примером оперируемого рецептором Са2+-канала (Deneris et al., Pharmacological and functional diversity of neuronal nicotinic acetylcholine receptors. Trends Pharmacol. Sci. 12: 34, 1991). Были клонированы отличающиеся субъединицы рецептора, и эти субъединицы могут экспрессироваться, например, в ооцитах Xenopus с образованием функциональных рецепторов с их катионными каналами. Высказывается гипотеза, что такие комплексы рецептор-ионофор являются гетеропентамерными структурами. Возможная роль оперируемых рецепторами Са2+-каналов в патологии неврологических нарушений и заболеваний, таких как ишемический удар, эпилепсия и нейродегенеративные заболевания, в большой степени остается невыясненной.
Ранее было показано, что некоторые яды пауков и ос содержат арилалкиламиновые токсины (также называемые полиаминовыми токсинами, ариламиновыми токсинами, ацилполиаминовыми токсинами или полиаминамидными токсинами) с активностью против рецепторов глутамата в ЦНС млекопитающих (в качестве обзоров см. Jackson and Usherwood, Spider toxines as tools for dissecting elements of excitatory amino acid transmission. Trends Neurosci. 11: 278, 1988; Jackson and Parks, Spider Toxins: Recent Applications in Neurobiology. Annu. Rev. Neurosci. 12: 405, 1989; Saccomano et al., Polyamine spider toxins: Unique pharmacological tools. Annu. Rep. Med. Chem: 24: 287, 1989; Usherwood and Bladbrough, Spider Toxines Affecting Glutamate Receptors: Polyamines in Therapeutic Neurochemistry, Pharmacol. Therap. 52:245, 1991: Kawai, Neuroactive Toxins of Spider Venoms. J. Toxicol. Toxin Rev. 10: 131, 1991). Вначале сообщалось, что арилалкиламиновые токсины являются селективными антагонистами АМРА/каинатных подтипов глутаматных рецепторов в ЦНС млекопитающих (Kawai et al., Effect of a spider toxin on glutaminergic synapses in the mammalian brain. Biomed. Res. 3: 353, 1982; Saito et al., Spider Toxin (JSTX) blocks glutamate synapse in hippocampal pyramidal neurons. Brain Res. 346: 397, 1985; Saito et al., Effects of a spider toxin (JSTX) on hippocampal CA1 neurons in vitro. Brain Res. 481: 16, 1989; Akaike et al., Spider toxin blocks excitatory amino acid responses in isolated hippocampal pyramidal neurons. Neurosci. Lett. 79: 326, 1987; Ashe et al., Argiotoxin-636 blocks excitatory synaptic transmission in rat hippocampal CA1 pyramidal neurons. Res. 480: 234, 1988; Jones et al., Philanthotoxin blocks quisqualate-induced, AMPA-induced and kainate-induced, but not NMDA-induced excitation of rat brainstem neurons in vivo. Br. J. Pharmacol. 101: 968, 1990). Последующие исследования показали, что, хотя некоторые арилалкиламиновые токсины являются не сильнодействующими и неселективными при различных глутаматных рецепторах, другие арилалкиламины являются очень сильнодействующими и селективными при антагонистических ответах, опосредованных активацией NMDA-рецепторов в ЦНС млекопитающих (Mueller et al., Effects of polyamine spider toxines on NMDA receptor-mediated transmission in rat hippocampus in vitro. Soc. Neurosci. Abstr. 15: 945, 1989; Mueller et al., Arylaminefepider toxins antagonize NMDA receptor-mediated synaptic transmission in rat hippocampal slices. Synapse 9: 244, 1991; Parke et al., Polyamine spider toxins block NMDA receptor-mediated increases in cytosolic calcium in cerebellar granule neurons. Soc. Neurosci. Abst. 15: 1169, 1989; Parks et al., Arylamine toxins from funnel-web spider (Agelenopsis aperta) venom antagonize N-methyl-D-aspartate receptor function in mammalian brain. J. Biol. Chem. 266: 21523, 1991; Priestley et al., Antagonism of responses to excitatory amino acids on rat cortical neurones by the spider toxin, argiotoxin-636. Br. J. Pharmacol. 97: 1315, 1989; Draguhn et al., Argiotoxin-636 inhibits NMDA-activated ion channels expressed in Xenopus oocytes. Neurosci. Lett. 132: 187, 1991; Kiskin et al., A highly potent and selective N-methyl-D-aspartate receptor antagonist from the venom of the Agelenopsis aperta spider, Neuroscience 51: 11, 1992: Brackley et al., Selective antagonism of native and cloned kainate and NMDA receptors by polyamine-containing toxins. J. Pharmacol. Exp. Therap. 266: 1573, 1993; Williaiya, Effects of Agelenopsis aperta toxins in the N-methyl-D-aspartate receptor: Polyamine-like and high-affinity antagonist actions. J. Pharmacol. Exp. Therap. 266: 231, 1993). Также сообщалось ингибирование никотиновых холинергических рецепторов арилалкиламиновым токсином филантотоксином (Rozental et al., Allosteric inhibition of nicotinic acetylcholine receptors of vertebrates and insects by philanthotoxin. J. Pharmacol. Exp. Therap. 249: 123, 1989). Parks et al. (Arylamine toxins from funnel-web spider (Agelenopsis aperta) venom antagonize N-methyl-D-aspartate receptor function in mammalian brain. J. Biol. Chem. 266: 21523, 1991) описывают арилалкиламиновые токсины паука (α -агатоксины), которые являются антагонистами функции NMDA-рецептора в мозгу млекопитающих. Эти авторы обсуждают механизм действия арилалкиламиновых токсинов и показывают, что оперируемый NMDA-рецепторами ионный канал является возможным сайтом действия α -агатоксинов и, наиболее вероятно, арилалкиламинов яда других пауков. Они утверждают:
"Открытие, что эндогенные полиамины в мозгу позвоночных модулируют функцию NMDA-рецепторов, предполагает, что ариламиновые токсины могут осуществлять их антагонизм через полиамин-связывающий сайт на глутаматных рецепторах. Brackly et al. исследовали действия спермина и филантотоксина 433 на ответные реакции, индуцируемые применением возбуждающих аминокислот в ооцитах Xenopus, инъецированных мРНК из мозга крысы или цыпленка. Эти авторы сообщили, что при концентрации, более низкой, чем концентрация, являющаяся антагонистической для глутамат-рецепторной функции, как спермин, так и филантотоксин потенциируют действия передающих возбуждение аминокислот и некоторых других нейротрансмиттеров. На основании этих и других данных Brackley et al. предположили, что ариламиновые токсины могут путем неспецифического связывания с мембранами возбуждаемых клеток уменьшать текучесть мембран и изменять функцию рецептора. Обоснованность этой интригующей идеи для функции NMDA-рецептора не находит хорошего подтверждения в двух недавних исследованиях по связыванию. Reynolds сообщил, что аргиотоксин 636 ингибирует связывание [3H]MK-801 с мембранами мозга крысы способом, который является нечувствительным к глутамату, глицину или спермидину. Этот автор сделал вывод, что аргиотоксин 636 проявляет новое ингибиторное действие на комплекс NMDA-рецептора путем связывания с одним из сайтов Мg2+, локализованных в ионном канале с NMDA-воротами. Данные по связыванию, сообщенные Williams et al., поддерживают вывод, что аргиотоксин 636 не действует первично в модуляторном сайте полиамина на NMDA-рецепторе, а скорее действует непосредственно путем образования зависимого от активности блока ионного канала. Уже известно, что такие соединения, как фенциклидин и кетамин, могут блокировать ионные каналы, связанные с мышечными глутаматными рецепторами членистоногих и NMDA-рецепторами млекопитающих. Таким образом, представляется возможным, что глутаматные рецепторы позвоночных и беспозвоночных имеют общие дополнительные сайты связывания для аллостерических модуляторов функции рецептора, возможно, родственные сайтам связывания двухвалентных катионов. Понятно, что необходимы значительные дополнительные исследования для определения, определяют ли ариламины такой новый регуляторный сайт".
Usherwood and Blagbrough (Spider Toxins Affecting Glutamate Receptors: Polyaminesin Therapeutic Neurochemistry. Pharmacol. Therap. 52: 245, 1991) описывают предполагаемый внутриклеточный сайт связывания для арилалкиламиновых токсинов (полиаминамидных токсинов), расположенный в поле мембранного потенциала, называемом фильтром селективности QUIS-R-канала. Эти авторы постулируют, что сайт связывания для полиаминамидных токсинов может встречаться вблизи внутреннего входа канала с воротами QUIS-R мышцы саранчи. Авторы также отмечают, что один такой токсин, аргиотоксин-636, является селективным антагонистом NMDA-рецептора в культивируемых кортикальных нейронах крысы.
Gullak et al. (CNS binding sites of the novel NMDA-antagonist Arg-636. Soc. Neurosci. Abst. 15: 1168, 1989) описывают аргиотоксин-636 (Arg-636) как полиаминовый (арилалкил-аминовый) компонент токсина яда паука. Указывается, что этот токсин ингибирует индуцируемое NMDA повышение цГМФ неконкурентным способом. Авторы утверждают, что:
"[125I]Аrg-636 связывался с мембранами переднего мозга крысы с величинами Kd и Вmах 11,25 мкМ и 28,95 пмоль/мг белка (со специфичностью 80%). Способность других известных полиаминов и недавно обнаруженных полиаминов из Agelenopsis aperta ингибировать связывание соответствовала нейроактивности функциональных антагонистов NMDA. Никакие другие испытанные соединения не были способны ингибировать специфическое связывание".
Затем эти авторы утверждают, что полиамины (арилалкил-амины) могут быть антагонистами в отношении ответов на NMDA посредством взаимодействия с мембранными ионными каналами.
Seymour and Mena (In vivo NMDA antagonist activity of the polyamine spider venom component, argiotoxm-636. Soc. Neurosci. Abst. 15: 1168, 1989) описывают исследования, которые должны были показать, что аргиотоксин-636 не влияет значительно на локомоторную активность при дозах, которые являются эффективными против аудиосенсорных эпилептических припадков у мышей DBA/2, и что он значительно противодействует NMDA-индуцируемым приступам эпилепсии с минимальной эффективной дозой 32 мг/кг, вводимой подкожно (s.c.).
Herold and Yakash (Anesthesia and muscle relaxation with intrathecal injections of AR636 and AG489, two acylpolyamine spider toxins, in rats. Anesthesiology 77: 507, 1992) описывают исследования, которые показывают, что арилалкиламиновый аргиотоксин-636 (ARG636), но не агатоксин-489 (AG489) производит миорелаксацию и анестезию после внутриоболочечного введения в крыс.
Williams (Effects of Agelenopsis aperta toxins on the N-methyl-D-aspartate receptor: Polyamine-like and high-affinity antagonist actions. J. Pharmacol. Exp. Therap. 266: 231, 1993) сообщает, что α -агатоксины (арилалкиламины) Agel-489 и Agel-505 усиливают связывание [3H]MK-801 с NMDA-рецепторами на мембранах, полученных из мозга крысы, посредством действия при стимуляторном полиаминовом рецепторе; агонисты полиаминового рецептора устраняли стимуляторные действия Agel-489 и Agel-505, а антагонисты полиаминового рецептора ингибировали стимуляторное действие Agel-505. Более высокие концентрации Agel-489 и Agel-505 и аргиотоксин-636 при всех испытанных концентрациях проявляли ингибирующие действия на связывание [3H]MK-801. В ооцитах Xenopus с зажимами с напряжением при -70 мВ Agel-505 ингибировал ответы на NMDA с IC50 13 нМ; это действие Agel-505 имело место при концентрациях, приблизительно в 10000 раз более низких, чем концентрации, влияющие на связывание [3H]MK-801. Ответные реакции на каинат ингибировались только на 11% 30 нМ Agel-505. Антагонизм Agel-505 в отношении NMDA-индуцируемых токов сильно зависел от напряжения, что согласуется с ингибирующим действием на открытые каналы этого токсина. Williams утверждает:
"Хотя α -агатоксины могут взаимодействовать при положительном аллостерическом сайте полиамина на NMDA-рецепторе, стимулирующие действия, производимые этим взаимодействием, могут маскироваться в функциональных тестах вследствие отдельного действия этих токсинов в качестве высокоаффинных, неконкурентных антагонистов этого рецептора".
Brackley et al. (Selective antagonism of native and cloned kainate and NMDA-receptors by polyamine-containing toxins, J. Pharmacol. Exp. Therap. 266: 1573, 1993) сообщили, что полиаминсодержащие токсины (арилалкиламины) филантотоксин-343 (PhTX-343) и аргиотоксин-636 (Arg-636) производят обратимое, неконкурентное, частично потенциал-зависимое антагонистическое действие на индуцируемые каинатом и NMDA токи в ооцитах Xenopus, инъецированных РНК мозга крысы. Было показано, что Arg-636 является селективным для индуцируемых NMDA ответов (IС50=0,7 мкМ), тогда как PhTX-343 был селективным для индуцируемых каинатом ответов (IC50=0,12 мкМ) по сравнению с NMDA-индуцируемыми ответами (IС50=2,5 мкМ). Аrg-636 более сильно противодействовал ответам на NMDA в ооцитах Xenopus, экспрессирующих клонированные субъединицы NMDAR1 (IC50=0,05 мкМ), чем ответам на каинат в ооцитах, экспрессирующих клонированные субъединицы GluR1 (IС50=3,4 мкМ) или субъединицы GluR1+GluR2 (IC50=300 мкМ). РhТХ-343, с другой стороны, был эквипотентным при противодействии NMDAR1 (IС50=2,19 мкМ) и GluR1 (IC50=2,8 мкМ), но гораздо менее сильным против субъединиц GluR1+GluR2 (IС50=270 мкМ).
Raditsch et al. (Subunit-specific block of cloned NMDA receptors by argiotoxin-636. FEBS Lett. 324: 63, 1993) сообщили, что Arg-636 более сильно противодействует ответам в ооцитах Xenopus, экспрессирующих субъединицы NMDAR1+NMDAR2A (IС50=9 нМ) или субъединицы NMDAR1+NMDAR2 С (IС50=460 нМ), даже хотя все эти субъединицы рецептора содержат остаток аспарагина в вероятном образующим пору трансмембранном районе II (сайте Q/R, обсужденном выше). Авторы утверждают, что большое различие в чувствительности к Arg-636 между каналами NMDAR1+NMDAR2A и NMDAR1+NMDAR2C "должно создаваться другими структурными детерминантами".
Herlitz et al. (Argiotoxin detects molecular differences in AMPA receptor channels. Neuron 10: 1131, 1993) сообщают, что Arg-636 является антагонистом подтипов АМРА-рецепторов с потенциал-зависимым и зависимым от использования действием, что согласуется с ингибированием открытых каналов. Arg-636 сильно противодействует проницаемым для Са2+ АМРА-рецепторам состоящим из субъединиц GluRAi (Ki =0,35 мкМ), GluRCi (Ki=0,23 мкМ) или GluRDi (Ki=0,43 мкМ), хотя является по существу неэффективным против непроницаемых для Са2+ субъединиц GluRBi при концентрациях до 11 мкМ. Другие данные, сообщаемые этими исследователями, убедительно подтверждают предположение, что сайт Q/R в предполагаемом образующем пору трансмембранном районе II имеет первичное значение в определении силы Аrg-636 и проникаемости Са2+.
Blaschke et al. (A single amino acid determines the subunit-specific spider toxin block of α -amino-3-hydroxy-5-methylisoxasol-4-propionate/kainate receptor channels. Proc. Natl. Acad. Sci. USA 90: 6528, 1993) сообщают, что арилалкиламин JSTX-3 сильно противодействует ответам на каинат в ооцитах Xenopus, экспрессирующих субъединицы GluR1 (IC50=0,04 мкМ) или GluR3 (IC50=0,03 мкМ), но на экспрессируемые рецепторы, в которых присутствует субъединица GluR2, по существу не действует этот токсин. Исследования с сайт-направленным мутагенезом дают убедительные указания на то, что сайт Q/R является первичным сайтом, влияющим на силу токсина.
Nakanishi et al. (Bioorganic studies of transmitter receptors with philanthotoxin analogs. Pure Appl. Chem., in press) синтезировали ряд очень сильнодействующих фотоаффинно меченных аналогов филантотоксина (PhTX). Такие аналоги исследовались на экспрессируемых никотиновых холинергических рецепторах в качестве модельной системы для оперируемых рецепторами кальциевых каналов. Эти исследователи предполагают, что эти аналоги PhTX блокируют ионный канал гидрофобным головным участком токсина, связывающимся с сайтом вблизи цитоплазматической поверхности, тогда как полиаминовый хвост простирается в ионный канал со стороны цитоплазмы.
Сущность изобретения
Заявителем были исследованы разнообразные по структуре и биологической активности арилалкиламины (иногда называемые ариламиновыми токсинами, полиаминовыми токсинами, ацилполиаминовыми токсинами или полиаминамидными токсинами) в яде пауков и ос и обнаружено, что некоторые из арилалкиламинов, присутствующих в этих ядах, действуют как сильные неконкурентные антагонисты оперируемых глутаматным рецептором Са2+-каналов в центральной системе млекопитающих. Хотя эти арилалкиламиновые соединения содержат в их структуре полиаминовую часть, они не похожи на другие известные простые полиамины в связи с тем, что они обладают чрезвычайно сильными и специфическими действиями на определенные типы оперируемых рецепторами Са2+-каналов.
С использованием нативных арилалкиламинов в качестве ведущих (основных) структур синтезировали и испытали ряд аналогов. Первоначальные открытия на арилалкиламинах, выделенных и очищенных из яда, были подтверждены с применением синтетических арилалкиламинов. Эти соединения являются небольшими молекулами (мол. масса < 800) с продемонстрированной эффективностью в моделях in vivo ишемического удара и эпилепсии. В качестве модели оперируемых рецепторами Са2+-каналов использовали комплекс NMDA-рецептор-ионофор. Было показано, что выбранные арилалкиламины ингибируют опосредованные NMDA-рецептором ответы по новому механизму. Кроме того, уникальный поведенческий фармакологический профиль этих соединений предполагает, что они вряд ли индуцируют РСР-подобную психодисплетическую (галлюциногенную) активность и нарушение познавательной способности, которые характеризуют другие ингибиторы NMDA-рецептора. Наконец, арилалкиламины являются уникальными среди антагонистов NMDA-рецептора в том смысле, что они способны противодействовать определенным подтипам клонированных и экспрессированных АМРА-рецепторов, а именно тем, которые проницаемы для Са2+. Следовательно, арилалкиламины являются единственным известным классом соединений, способных противодействовать опосредованным глутаматным рецептором увеличениям цитозольного Са2+, независимо от фармакологического определения подтипа рецептора. Кроме того, арилалкиламины ингибируют оперируемый другим рецептором Са2+-канал никотиновым холинергическим рецептором. Если избыточные и пролонгированные увеличения количества цитозольного Са2+ связывали с этиологией нескольких неврологических нарушений и заболеваний, такие арилалкиламины являются ценными, являющимися маленькими молекулами, исходными материалами для разработки новых терапевтических средств для различных неврологических нарушений и заболеваний.
Заявитель определил, что выбранные арилалкиламины связываются с высокой аффинностью при новом сайте на комплексе NMDA-рецептор-ионофор, который до настоящего времени не был идентифицирован, и что арилалкиламины не связываются с высокой аффинностью при любых известных сайтах (глутаматсвязывающем сайте, глицинсвязывающем сайте, МК-801-связывающем сайте, Мg2+-связываюшем сайте, Zn2+-связывающем сайте, полиаминсвязывающем сайте, сигма-связывающем сайте) на комплексе NMDA-рецептор-ионофор. Это определение позволило заявителю разработать способы и протоколы, при помощи которых могут быть идентифицированы соединения, которые обеспечивают как терапевтически применимые соединения, так и исходные соединения для разработки других терапевтически применимых соединений. Эти соединения могут быть идентифицированы при помощи скрининга на соединения, которые связываются при новом сайте связывания арилалкиламина, и путем определения, имеет ли такое соединение требующиеся биологические, фармакологические и физиологические свойства.
Способ включает стадию идентификации соединения, которое связывается с оперируемым рецептором Са2+-каналом при сайте, связываемом арилалкиламиновыми соединениями, названными здесь как соединение 1, соединение 2 или соединение 3 и имеющими структуру, показанную ниже.



Под "терапевтически применимым соединением" имеют в виду соединение, которое потенциально применимо в лечении нарушения или патологического состояния. Соединение, обнаруживаемое этим способом скрининга, характеризуется как имеющее потенциальную терапевтическую применимость в лечении, поскольку еще не были проведены клинические тесты для определения действительной терапевтической применимости.
Под "неврологическим нарушением или заболеванием" имеют в виду нарушение или заболевание нервной системы, в том числе (но не только) глобальный или очаговый ишемический или геморрагический удар, травму головы, повреждение спинного мозга, ишемию спинного мозга, индуцированное ишемией или гипоксией повреждение нервных клеток, вызываемое гипоксией повреждение нервных клеток, например, при остановке сердца или неонатальном дистрессе, эпилепсию, состояние тревоги, нейропсихиатрические заболевания или нарушения познавательной способности вследствие ишемии или гипоксии, такие как нарушения, которые часто встречаются как следствие хирургии сердца при экстракорпоральном (искусственном) кровообращении, и нейродегенеративное заболевание. Под "неврологическим нарушением или заболеванием" подразумевают также такие болезни и состояния, при которых могут быть показаны, полезны, рекомендованы или предписаны нейропротекторы, антиконвульсивные средства, транквилизаторы, аналгетики, миорелаксанты и/или вспомогательные средства в общей анестезии.
Под "нейродегенеративным заболеванием" подразумевают заболевания, включающие (но не только) болезнь Альцгеймера, болезнь Хантингтона, болезнь Паркинсона и боковой амиотрофический склероз (ALS).
"Под "нейропротектором" имеют в виду соединение, способное предотвращать повреждение или гибель нейронов, связанные с неврологическим нарушением или заболеванием.
Под "противосудорожным средством" подразумевают соединение, способное к уменьшению судорог, вызываемых такими состояниями, как простые парциальные эпилептические припадки, сложные парциальные эпилептические припадки, эпилептический статус и вызванные травмой эпилептические припадки, которые встречаются после повреждения головы, в том числе хирургии головы.
Под "трансквилизатором" подразумевают соединение, способное облегчать боль путем изменения восприятия болевых стимулов, без индуцирования анестезии или потери сознания.
Под " миорелаксантом" имеют в виду соединение, которое уменьшает мышечное напряжение.
Под "вспомогательным средством в общей анестезии" имеют в виду соединение, применимое в соединении с анестезирующими агентами, в индуцировании потери способности воспринимать боль, связанной с потерей сознания.
Под "сильнодействующим" или "активным" имеют в виду, что соединение имеет активность при оперируемых рецепторами кальциевых каналах, в том числе NMDA-рецепторах, Са2+-проникаемых АМРА-рецепторах и никотиновых холинергических рецепторах, с величиной IС50 менее 10 мкМ, более предпочтительно менее 100 нМ и даже более предпочтительно менее 1 нМ.
Под термином "селективный" имеют в виду, что соединение является сильнодействующим при оперируемых рецепторами кальциевых каналах, как описано выше, но является в 10 раз менее сильнодействующим, более предпочтительно в 50 раз и даже более предпочтительно в 100 раз менее сильнодействующим при других нейротрансмиттерных рецепторах, оперируемых нейротрансмиттерными рецепторами ионных каналах или потенциалзависимых ионных каналах.
Под "биохимическими и электрофизиологическими тестами функции оперируемых рецепторами кальциевых каналов" имеют в виду тесты, предназначенные для обнаружения биохимическими или электрофизиологическими средствами функциональной активности оперируемых рецепторами кальциевых каналов. Примеры таких тестов включают, но не только, fura-2-флуорометрический тест для цитозольного кальция в культивируемых зернистых клетках мозжечков крыс (см. пример 1 и пример. 2), электрофизиологические тесты (patch сlаmр="пэтч-кламп") (см. пример 3 и пример 27), тесты синаптической передачи возбуждения на срезах гиппокампа (см. пример 5), тесты связывания радиолигандов (см. пример 4, пример 24, пример 25 и пример 26) и нейропротекторные тесты in vitro (см. пример 6).
Под "эффективностью" подразумевают, что с выбранным соединением детектируется значимый уровень желаемой активности; под "значимым" имеют в виду статистическую значимость при уровне р<0,05.
Под "нейропротекторной активностью" подразумевают эффективность в лечении неврологических нарушений или заболеваний, в том числе, но не только, глобального и очагового ишемического и геморрагического удара, травмы головы, повреждения спинного мозга, ишемии спинного мозга, индуцируемого ишемией или гипоксией повреждения нервных клеток, индуцируемого гипоксией повреждения нервных клеток, например, при остановке сердца или неонатальном дистрессе, нейропсихиатрических нарушений или нарушений познавательной способности вследствие ишемии или гипоксии, таких как нарушения, которые часто встречаются как следствие хирургии сердца при экстракорпоральном кровоснабжении, и нейродегенеративные заболевания, такие как болезнь Альцгеймера, болезнь Хантингтона, болезнь Паркинсона и боковой амиотрофический склероз (ALS) (см. примеры 7 и 8 ниже).
Под "антисудорожной активностью" имеют в виду эффективность в уменьшении судорог, вызываемых такими состояниями, как простой парциальный эпилептический припадок, сложный парциальный эпилептический припадок, эпилептический статус и индуцируемый травмой эпилептический припадок, такой как встречающийся после повреждения головы, в том числе при хирургии головы (см. примеры 9 и 10 ниже).
Под "анксиолитической" активностью имеют в виду, что соединение уменьшает чувство страха, неопределенности и тревоги, которые характерны для состояния тревоги.
Под ″ аналгезирующей активностью (болеутоляющей)" имеют в виду, что соединение вызывает отсутствие боли в ответ на стимул, который в норме был бы болезненным. Такая активность могла бы использоваться в клинических состояниях, таких как острая и хроническая боль, в том числе, но не только, для преразгрузочной предоперативной аналгезии; для периферических невропатий, встречающихся при сахарном диабете и множественном склерозе; фантомной боли; каузалгии; невралгиях, таких, которые встречаются при простом герпесе; боли, такой, которая наблюдается при повреждениях спинного мозга; гипералгезии и аллодинии.
Под "каузалгией" имеют в виду болезненное нарушение, связанное с повреждением периферических нервов.
Под "невралгией" имеют в виду боль в расположении нерва или нервов.
Под "гипералгезией" подразумевают повышенный ответ на стимул, который является в норме болезненным.
Под "аллодинией" имеют в виду боль, вызываемую стимулом, который в норме не вызывает боли (см. пример 11 - пример 14 ниже).
Под "индукцией долгосрочного потенциирования в срезах гиппокампа" имеют в виду способность вызывающей судороги электрической стимуляции афферентных коллатеральных волокон Шаффера для индуцирования долгосрочных увеличений в силе синаптической трансмиссии (передаче нервного импульса) в пути коллатеральных-СА1 пирамидальных нейронов Шаффера в срезах гиппокампа крысы, сохраняемых in vitro (см. пример 19).
Под "терапевтической дозой" имеют в виду количество соединения, которое облегчает до некоторой степени один или несколько симптомов заболевания или состояния пациента. Кроме того, под "терапевтической дозой" подразумевают количество, которое возвращает к нормальным, частично или полностью, физиологические или биохимические параметры, связанные с заболеванием или состоянием или являющиеся причинами заболевания или состояния. Как правило, это количество между 1 нмоль и 3 нмоль соединения, в зависимости от его EC50 (IC50 в случае антагониста) и от возраста, размера и заболевания, связанного с пациентом.
Под "повреждением познавательной способности" имеют в виду способность ухудшения восприимчивости памяти или выполнения выученного задания (см. пример 20). Под "повреждением познавательной способности" подразумевают способность противодействовать нормальным процессам рационального мышления и обоснования.
Под "нарушением двигательной функции" имеют в виду способность значительно изменять опорно-двигательную активность (см. пример 15) или вызывать значительную атаксию, потерю установочного рефлекса, седативный эффект или мышечную релаксацию (см. пример 16).
Под "опорно-двигательной активностью" подразумевают способность выполнять нормальные амбулаторные движения.
Под "потерей установочного рефлекса" подразумевают способность животного, обычно грызуна, самостоятельно выпрямляться после помещения его в супинированное положение.
Под "вакуолизацией нейронов" подразумевают образование вакуолей в нейронах поясного или ретросплениального кортекса (см. пример 16).
Под "сердечно-сосудистой активностью" имеют в виду способность вызывать значимые изменения в параметрах, включающих в себя (но не только) среднее артериальное кровяное давление и частоту сердечных сокращений (см. примеры 21 и 22).
Под "повышенной возбудимостью" подразумевают повышенную чувствительность к возбуждающему стимулу. Повышенная возбудимость часто проявляется как значительное увеличение в опорно-двигательной активности у грызунов, которым введено лекарственное средство (см. пример 15).
Под "седативным эффектом" подразумевают успокаивающий эффект или ослабление активности и возбуждения. Седативный эффект часто проявляется как значительное уменьшение опорно-двигательной активности у грызунов, которым введено лекарственное средство (см. пример 15).
Под "РСР-подобным потенциалом злоупотребления (токсикомании)" подразумевают потенциал лекарственного средства быть неправильно употребляемым, как в случае рекреационного использования РСР (т.е. "ангельского порошка") людьми. Считают, что РСР-подобный потенциал злоупотребления может быть предсказан по способности лекарственного средства обобщаться с РСР в грызунах, приученных к отличению РСР от солевого раствора (см. пример 17).
Под "обобщением с РСР" подразумевают, что соединение воспринимается как РСР в грызунах, обученных отличать РСР от солевого раствора (см. пример 17).
Под "РСР-подобной психотомиметической активностью" подразумевают способность лекарственного средства вызывать у людей поведенческий синдром, напоминающий острый психоз, включающий в себя галлюцинации, паранойю, ажитацию и дезориентацию во времени и пространстве (спутанность сознания). Считают, что РСР-подобная психотомиметическая активность может быть предсказана у грызунов по способности лекарственного средства вызывать РСР-подобные стереотипии, в том числе атаксию, верчение головой из стороны в сторону, повышенную возбудимость и обобщение с РСР у грызунов, обученных отличать РСР от солевого раствора (см. пример 16 и пример 17).
Под "атаксией" подразумевают недостаточность в мышечной координации.
Под "верчением головой" подразумевают стереотипическое поведение, вызываемое у грызунов РСР, при котором голова периодически движется медленно и широко от одной стороны к другой.
Под "фармацевтической композицией" имеют в виду терапевтически эффективное количество соединения данного изобретения в фармацевтически приемлемом носителе, т.е. препарат, к которому может быть добавлено соединение для растворения или облегчения другим образом введения этого соединения. Примеры фармацевтически приемлемых носителей включают воду, солевой раствор и физиологически забуференный солевой раствор. Такую фармацевтическую композицию обеспечивают в подходящей дозе. Такими композициями являются в общем композиции, которые одобряются для использования в лечении описываемого нарушения FDA или соответствующей организацией в других странах.
В родственном аспекте данное изобретение относится к способу лечения неврологического заболевания или нарушения, включающему стадию введения фармацевтической композиции, содержащей соединение, которое связывается с оперируемым рецептором кальциевым каналом при сайте, связанном одним из арилалкиламинов (соединением 1, соединением 2 и соединением 3), причем это соединение является сильнодействующим и селективным неконкурентным антагонистом при таком оперируемом рецептором кальциевом канале и имеющим одно или несколько следующих фармакологических и физиологических свойств: эффективность в биохимических и электрофизиологических тестах in vitro функции оперируемого рецептором кальциевого канала, противосудорожную активность in vivo, нейропротекторную активность in vivo, транквилизирующую активность in vivo и аналгезирующую активность in vivo; указанное соединение обладает также одним или несколькими из следующих фармакологических свойств: соединение не препятствует индукции долгосрочного потенциирования в срезах гиппокампа крыс и при терапевтической дозе не ухудшает познавательную способность, не нарушает двигательную функцию, не вызывает вакуолизации нейронов, имеет минимальную сердечно-сосудистую активность, не производит седативного эффекта или повышенной возбудимости, имеет минимальный РСР-подобный потенциал злоупотребления и имеет минимальную РСР-подобную психотомиметическую активность. Под "минимальным" имеют в виду, что любое побочное действие этого лекарственного средства переносится средним индивидуумом и, следовательно, что лекарственное средство может быть использовано для терапии заболевания-мишени. Такие побочные действия хорошо известны в данной области и рутинно рассматриваются FDA как минимальные при одобрении лекарственного средства для заболевания-мишени.
Лечение включает в себя стадии идентификации сначала пациента, который страдает от неврологического заболевания или нарушения, стандартной клинической методологией и затем лечение такого пациента композицией данного изобретения.
В следующем аспекте данное изобретение относится к соединениям, которые могут использоваться для лечения пациента, имеющего неврологическое заболевание или нарушение, причем соединение является соединением типа полиамина или его аналога (т.е. полигетероатомной молекулой), имеющим формулу

где Аr обозначает подходящим образом замещенное ароматическое кольцо, циклическую систему или другую гидрофобную часть молекулы; Аг может быть ароматическим (например, карбоциклическими арильными группами, такими как фенил, или бициклическими карбоциклическими арильными циклическими системами, такими как нафтил, 1,2,3,4-тетрагидронафтил, инданил и инденил), гетероароматическими (например, индолилом, дигидроиндолилом, хинолинилом и изохинолинилом и их соответствующими 1,2,3,4-тетрагидро- и 2-оксопроизводными), алициклическими (циклоалифатическими) или гетероалициклическими кольцом или циклической структурой (моно-, би- или трициклической структурой), имеющими 5-7-членное (членные) кольцо (кольца), необязательно замещенные 1-5 заместителями, независимо выбранными из низшего алкила с 1-5 атомами углерода, низшего галогеналкила из 1-5 атомов углерода, замещенного 1-7 атомами галогена, низшего алкокси из 1-5 атомов углерода, галогена, нитро, амино, низшего алкиламино из 1-5 атомов углерода, амидо, низшего алкиламидо из 1-5 атомов углерода, циано, гидроксила, сульфгидрила, низшего ацила из 2-4 атомов углерода, сульфонамидо, низшего алкилсульфонамидо из 1-5 атомов углерода, низшего алкилсульфоксида из 1-5 атомов углерода, низшего гидроксиалкила из 1-5 атомов углерода, низшего алкилкето из 1-5 атомов углерода или низшего тиоалкила из 1-5 атомов углерода;
каждый m равен целому числу от 0 до 3 включительно,
каждый k равен целому числу от 1 до 10 включительно,
каждый j, одинаковые или различные, обозначает целое число от 1 до 12 включительно,
каждый R1 и R2 независимо выбран из группы, включающей водород, низший алкил из 1-5 атомов углерода, низший алкиламино из 1-5 атомов углерода, низший алкиламидо из 1-5 атомов углерода, низший моно-, ди- или трифторалкил из 1-5 атомов углерода, гидрокси, амидино, гуанидино или боковую цепь типичной обычной аминокислоты, или со связанным атомом углерода R1 и R2, взятые вместе, образуют карбонил, и каждый Z выбран из группы, включающей азот, кислород, серу, амидо, сульфонамидо и углерод.
Предпочтительные ароматические головные группы включают (но не ограничены ими) следующее:






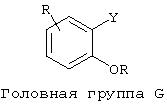
где Y =

Из данного изобретения исключены известные соединения, химические структуры которых охватываются общей формулой, представленной выше.
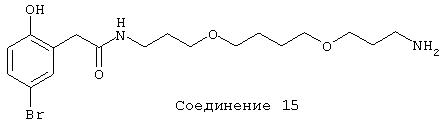
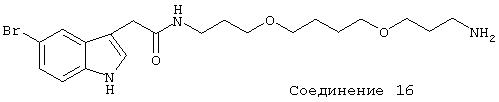
В дальнейших предпочтительных вариантах соединение выбрано из группы Соединений 4-18, где эти соединения имеют формулы:













Заявитель определил также (см. пример 23 ниже), что упрощенные арилалкиламины (см. ниже) являются сильнодействующими, неконкурентными антагонистами комплекса NMDA-рецептор-ионофор. Упрощенные арилалкиламины отличаются от арилалкил-аминов, представленных соединениями 4-18, описанными выше. Например, такие соединения связываются с сайтом, меченным [3H]MK-801, при концентрациях, приблизительно в 1-400 раз более высоких, чем концентрации, которые противодействуют опосредованной NMDA-рецептором функции. Такие упрощенные арилалкиламины обладают одним или несколькими следующими дополнительными свойствами: значительной нейропротекторной активностью, значительной противосудорожной активностью, значительной аналгезирующей активностью, отсутствием РСР-подобного стереотипического поведения у грызунов (повышенной возбудимости и верчения головой) при эффективных нейропротекторных, противосудорожных и аналгезирующих дозах, отсутствием обобщения с РСР в тесте дискриминации РСР при эффективных нейропротекторных, противосудорожных и аналгезирующих дозах, отсутствием вакуолизации нейронов при эффективных нейропротекторных, противосудорожных и аналгезирующих дозах, значительно менее сильной активностью против потенциал-зависимых кальциевых каналов и минимальной гипотензивной активностью при эффективных нейропротекторных, противосудорожных и аналгезирующих дозах. Однако такие соединения могут ингибировать индукцию LTP в срезах гиппокама крыс и могут вызывать ухудшение двигательной способности при нейропротекторных, противосудорожных и аналгезирующих дозах.
Один аспект данного изобретения относится к способу лечения пациента, имеющего неврологическое заболевание или нарушение, предусматривающему введение соединения формулы I:

где
R1 и R5 независимо выбраны из группы, включающей фенил, бензил и фенокси (каждый из которых необязательно замещен (X)m), -Н, алкил, гидроксиалкил, -ОН, -O-алкил и -O-ацил;
R2 и R6 независимо выбраны из группы, включающей -Н, алкил и гидроксиалкил; или R2 и R6 вместе представляют собой имино; или R1 и R2 вместе представляют собой -(СН2)n- или -(CH2)n-N(R3)-(CH2)n-;
R3 независимо выбран из группы, включающей –Н, алкил, 2-гидроксиэтил и алкилфенил; n равно целому числу от 0 до 6, но только 1 n может быть 0;
R4 выбран из группы, включающей тиофуран, пиридил, фенил, бензил, фенокси и фенилтио (каждый из которых необязательно замещен (Х)m), -Н, алкил и циклоалкил;
Х независимо выбран из группы, включающей фенил, бензил и фенокси (каждый из которых необязательно замещен (X)m), -Н, -Br, -Cl, -I, -F, -СF3, алкил, -ОН, -ОСF3, -O-алкил и -O-ацил;
m независимо равно целому числу от 0 до 5;
Y обозначает –NR3R3, за исключением того, что, когда R1 и R2 вместе обозначают - (СН2)n-N(R3)-(СН2)n-, тогда Y обозначает -Н;
и его фармацевтически приемлемых солей и комплексов, при этом соединение является активным при NMDA-рецепторе.
Под "пациентом" подразумевают любое животное, имеющее клетку с NMDA-рецептором. Предпочтительно это животное является млекопитающим. Наиболее предпочтительно оно является человеком.
Под "алкилом" имеют в виду разветвленную или неразветвленную углеводородную цепь, содержащую 1-6, предпочтительно 1-4, атомов углерода, например метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, 2-метилпентил, циклопропилметил, аллил и циклобутилметил.
Под "низшим алкилом" имеют в виду разветвленную или неразветвленную углеводородную цепь, содержащую 1-4 атома углерода, примеры которой перечислены здесь.
Под "гидроксиалкилом" имеют в виду алкильную группу, описанную выше, замещенную гидроксильной группой.
Под "алкилфенилом" имеют в виду алкильную группу, описанную выше, замещенную фенильной группой.
Под "ацилом" имеют в виду -C(O)R, где R является Н или алкилом, определенным выше, например формил, ацетил, пропионил или бутирил;
или R обозначает -O-алкил, такой как в алкилкарбонатах, или R обозначает N-алкил, такой как в алкилкарбаматах.
Под "циклоалкилом" имеют в виду разветвленную или неразветвленную циклическую углеводородную цепь, содержащую 3-12 атомов углерода.
В предпочтительных аспектах данного изобретения
Y выбран из группы, включающей –NH2 и -NH-метил;
R4 представляет собой тиофуран, пиридил, фенил, бензил, фенокси или фенилтио, каждый из которых необязательно замещен (Х)m;
(Х)m независимо выбран из группы, включающей мета-фтор, мета-хлор, орто-O-низший алкил, орто-метил, орто-фтор, орто-хлор, мета-O-низший алкил, мета-метил, орто-ОН и мета-ОН; и
R1, R2, R5 и R6 обозначают -Н; или R2 обозначает метил, и R1, R5 и R6 обозначают -Н; или R1 обозначает метил и R2, R5 и R6обозначают -Н.
В других предпочтительных аспектах данного изобретения R1и R5 независимо выбраны из группы, включающей -Н, низший алкил, гидроксиалкил, -ОН, -O-алкил и -O-ацил;
R2 и R6 независимо выбраны из группы, включающей –Н, низший алкил и гидроксиалкил;
или R1 и R2 вместе обозначают -(СН2)n- или - (CH2)n-N(R3)- и Y обозначает -Н;
R3 независимо выбран из группы, включающей –Н и низший алкил;
n равно целому числу от 1 до 6;
R4 выбран из группы, включающей тиофуран, пиридил, фенил, бензил, фенокси и фенилтио (каждый из которых необязательно замещен (Х)m), -Н, низший алкил и циклоалкил;
Х независимо выбран из группы, включающей –Н, -Br, -Cl, -F, -I, -СF3, низший алкил, -ОН и –ОСF3;
m независимо равно целому числу от 0 до 5;
Y обозначает -NHR3 или водород, когда R1 и R2 вместе представляют собой -(CH2)n-N(R3)-;
и их фармацевтически приемлемые соли и комплексы, при условии, что
(a) когда R1 и R2 вместе обозначают - (CH2)n-N(R3)-, тогда R5, R6 и Y обозначают водород; и
(b) когда R1 и R2 вместе не обозначают - (CH2)n-N(R3)-, тогда Y обозначает -NHR3.
В следующем предпочтительном аспекте данное изобретение относится к способу лечения пациента, имеющего неврологическое заболевание или нарушение, предусматривающий введение соединений формулы II:
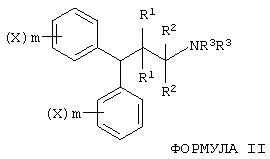
где X независимо выбран из группы, включающей -Н, -Br, -Cl, -F, -I, -СF3, алкил, -ОН, -ОСF3, -O-алкил и -O-ацил; R1 независимо выбран из группы, включающей -Н, алкил, гидроксиалкил, -ОН, -O-алкил и -O-ацил; R2 независимо выбран из группы, включающей -Н, алкил и гидроксиалкил, или оба R2 вместе обозначают имино; R3 независимо выбран из группы, включающей -Н, алкил, 2-гидроксиэтил и алкилфенил; и m независимо равно целому числу от 0 до 5; или соединений формулы III:

где Х независимо выбран из группы, включающей -Н, -Br, -C1, -F, -I, -СF3, алкил, -ОН, -ОСF3, -O-алкил и -O-ацил; R1 независимо выбран из группы, включающей -Н, алкил, гидроксиалкил, -ОН, -O-алкил и -O-ацил; R2 независимо выбран из группы, включающей –Н, алкил и гидроксиалкил, или оба R2 вместе обозначают имино; R3 независимо выбран из группы, включающей -Н, алкил, 2-гидроксиэтил и алкилфенил; R4 выбран из группы, включающей тиофуран, пиридил, фенил, бензил, фенокси и фенилтио (каждый из которых необязательно замещен (Х)m), алкил и циклоалкил; и m независимо равно целому числу от 0 до 5; или соединений формул IV и V.


где n равно целому числу от 1 до 6; Х независимо выбран из группы, включающей –Н, -Br, -Cl, -F, -I, -СF3, алкил, -ОН, -ОСF3, -O-алкил и -O-ацил; R1 независимо выбран из группы, включающей –Н, алкил, 2-гидроксиэтил и алкилфенил; и m независимо равно целому числу от 0 до 5; или соединений формул VI и VII:
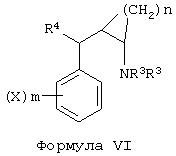

где n равно целому числу от 1 до 6; Х независимо выбран из группы, включающей -Н, -Br, -Cl, -F, -I, -СF3, алкил, -ОН, -ОСF3, -O-алкил и -O-ацил; R3 независимо выбран из группы, включающей –Н, алкил, 2-гидроксиэтил и алкилфенил; R4 выбран из группы, включающей тиофуран, пиридил, фенил, бензил, фенокси и фенилтио (каждый из которых необязательно замещен (Х)m), алкил и циклоалкил; и m независимо равно целому числу от 0 до 5.
Более предпочтительные аспекты являются вариантами, в которых (Х)m независимо выбран из группы, включающей мета-фтор, мета-хлор, орто-O-низший алкил, орто-метил, орто-фтор, орто-хлор, мета-O-низший алкил, мета-метил, орто-ОН и мета-ОН; NR3выбран из группы, включающей NH, N-метил и N-этил; NR3R3 выбран из группы, включающей NH2, NH-метил и NH-этил; каждый R1независимо выбран из группы, включающей -Н и метил; каждый R2выбран из группы, включающей -Н и метил; и R4 выбран из группы, включающей фенил, бензил и фенокси, каждый из которых необязательно замещен (Х)m.
Особенно предпочтительными аспектами являются варианты, в которых (Х)m обозначает мета-фтор; NR3 выбран из группы, включающей NH и N-метил; и NR3R3 выбран из группы, включающей NH2 и NH-метил, каждый из R1 и R2 обозначает -Н; и R4 выбран из группы, включающей фенил, бензил и фенокси, каждый из которых необязательно замещен (Х)m.
В предпочтительных вариантах способы лечения включают введение соединения, выбранного из Соединений 19-150 или их фармацевтически приемлемых солей и комплексов. Предпочтительно это соединение имеет IС50≤10 мкМ при NMDA-рецепторе, более предпочтительно ≤ 2,5 мкМ и наиболее предпочтительно ≤ 0,5 мкМ при NMDA-рецепторе.
В следующих предпочтительных вариантах способы лечения включают введение соединения с IC50≤10 мкМ при NMDA-рецепторе, выбранного из группы, включающей соединения 19, 20, 21, 22, 23, 24, 25, 27, 28, 29, 30, 31, 32, 33, 34, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 75, 76, 77, 78, 79, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 100, 101, 102, 103, 105, 106, 107, 108, 109, 111, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138 (потенциальное пролекарство), 139, 141, 142, 143, 144, 145, 146, 147, 148, 149 и 150 и их фармацевтически приемлемых солей и комплексов.
В более предпочтительных вариантах способы лечения включают введение соединения с IC50≤2,5 мкМ при NMDA-рецепторе, выбранного из группы, включающей соединения 19, 20, 21, 22, 23, 24, 25, 27, 28, 29, 30, 31, 32, 33, 34, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 70, 75, 76, 81, 82, 83, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 100, 101, 102, 103, 105, 106, 108, 109, 111, 115, 118, 119, 120, 121, 122, 125, 126, 127, 128, 129, 130, 131, 132, 133, 135, 136, 137, 138 (потенциальное пролекарство), 139, 142, 144, 145, 146, 147, 148, 149 и 150 и их фармацевтически приемлемых солей и комплексов. В других вариантах соединение выбрано из группы, включающей соединения 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 76, 82, 83, 88, 89, 90, 92, 93, 94, 95, 96, 101, 102, 103, 105, 109, 111, 115, 118, 119, 120, 121, 122, 125, 126, 127, 129, 130, 131, 135, 136, 137, 138, 139, 142, 144, 145, 148, 149 и 150 и их фармацевтически приемлемых солей и комплексов.
В особенно предпочтительных вариантах способы лечения включают введение соединения с IC50≤0,5 мкМ при NMDA-рецепторе, выбранного из группы, включающей соединения 19, 20, 21, 22, 23, 24, 25, 27, 28, 30, 31, 32, 33, 38, 39, 43, 44, 46, 47, 49, 50, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 82, 83, 89, 90, 91, 93, 94, 95, 96, 97, 103, 111, 118, 119, 120, 122, 126, 135, 136, 137, 138 (потенциальное пролекарство), 142, 144, 145, 147, 148, 149 и 150 и их фармацевтически приемлемых солей и комплексов.
В более предпочтительных вариантах способы лечения включают введение соединения, выбранного из группы, включающей соединения 20, 24, 25, 33, 50, 60, 66, 69, 103, 111, 118, 119, 120, 122, 136, 137, 138 (потенциальное пролекарство), 142, 144, 145, 148, 149 и 150 и их фармацевтически приемлемых солей и комплексов.
В особенно предпочтительных вариантах способы лечения включают введение соединения, выбранного из группы, включающей соединения 20, 33, 50, 60, 119 и 144 и их фармацевтически приемлемых солей и комплексов.
В других особенно предпочтительных вариантах способы лечения включают введение соединения, выбранного из группы, включающей соединения 33, 50, 60, 119 и 144 и их фармацевтически приемлемых солей и комплексов.
Данное изобретение обеспечивает упрощенные арилалкиламины, включающие соединения формул I-VII и все предпочтительные аспекты формул I-VII, описанные выше.
Примеры таких упрощенных арилалкиламинов включают, но не ограничены ими, соединения 19-150, структуры которых даны выше. Предпочтительно соединение имеет IC50≤10 мкМ при NMDA-рецепторе. Более предпочтительно соединение имеет IC50≤5 мкМ, более предпочтительно ≤ 2,5 мкМ и наиболее предпочтительно ≤ 0,5 мкМ при NMDA-рецепторе.
В предпочтительных вариантах соединение имеет IC50≤10 мкМ при NMDA-рецепторе и выбрано из группы, включающей соединения 21, 22, 23, 24, 25, 27, 28, 29, 33, 34, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 76, 78, 79, 82, 83, 84, 88, 89, 90, 92, 93, 94, 95, 96, 98, 101, 102, 103, 105, 107, 108, 109, 111, 115, 116, 118, 119, 120, 121, 122, 124, 125, 126, 127, 129, 130, 131, 134, 135, 136, 137, 138 (потенциальное пролекарство), 139, 141, 142, 143, 144, 145, 148, 149 и 150. В других вариантах соединение выбрано из группы, включающей соединения 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 76, 82, 83, 88, 89, 90, 92, 93, 94, 95, 96, 101, 102, 103, 105, 109, 111, 115, 118, 119, 120, 121, 122, 125, 126, 127, 129, 130, 131, 135, 136, 137, 138, 139, 142, 144, 145, 148, 149 и 150.
В более предпочтительных вариантах соединение имеет IC50≤2,5 мкМ при NMDA-рецепторе и выбрано из группы, включающей соединения 21, 22, 23, 24, 25, 27, 28, 29, 33, 34, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 76, 82, 83, 88, 89, 90, 92, 93, 94, 95, 96, 101, 102, 103, 105, 108, 109, 111, 115, 118, 119, 120, 121, 122, 125, 126, 127, 129, 130, 131, 135, 136, 137, 138 (потенциальное пролекарство), 139, 142, 144, 145, 148, 149 и 150.
В особенно предпочтительных вариантах соединение имеет IC50≤0,5 мкМ при NMDA-рецепторе и выбрано из группы, включающей соединения 21, 22, 23, 24, 25, 27, 28, 33, 38, 39, 43, 44, 46, 47, 49, 50, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 82, 83, 89, 90, 93, 94, 95, 96, 103, 111, 118, 119, 120, 122, 126, 135, 136, 137, 138 (потенциальное пролекарство), 142, 144, 145, 148, 149 и 150.
В предпочтительных вариантах соединение выбрано из группы, включающей соединения 24, 25, 33, 50, 60, 66, 69, 103, 111, 118, 119, 120, 122, 136, 137, 138, 142, 144, 145, 148, 149 и 150.
В особенно предпочтительных вариантах соединение выбрано из группы, включающей соединения 20, 33, 50, 60, 119 и 144.
В еще более предпочтительных вариантах соединение выбрано из группы, включающей соединения 33, 50, 60, 119 и 144.
Из состава данного аспекта изобретения исключены известные соединения, химические структуры которых охватываются общими формулами, представленными выше.
В аспекте данного изобретения обеспечены также фармацевтические композиции, применимые для лечения пациента, имеющего неврологическое заболевание или нарушение. Эти фармацевтические композиции обеспечены в фармацевтически приемлемом носителе и в подходящей дозе. Фармацевтические композиции могут быть в форме фармацевтически приемлемых солей и комплексов, как это известно специалистам в данной области.
Фармацевтические композиции содержат соединения формул I-VII и включают все предпочтительные аспекты формул I-VII, описанные выше.
Предпочтительные фармацевтические композиции содержат соединения 19-150. Предпочтительно соединение имеет IC50≤10 мкМ при NMDA-рецепторе. Более предпочтительно соединение имеет IC50≤5 мкМ, еще более предпочтительно ≤ 2,5 мкМ и наиболее предпочтительно ≤ 0,5 мкМ при NMDA-рецепторе.
В следующих предпочтительных вариантах фармацевтическая композиция содержит соединение с IC50≤10 мкМ при NMDA-рецепторе, выбранное из группы, включающей соединения 20, 21, 22, 23, 24, 25, 27, 28, 29, 30, 31, 32, 33, 34, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 75, 76, 77, 78, 79, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 100, 101, 102, 103, 105, 106, 107, 108, 109, 111, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138 (потенциальное пролекарство), 139, 141, 142, 143, 144, 145, 146, 147, 148, 149 и 150. Предпочтительно соединение выбрано из группы, включающей соединения 21, 22, 23, 24, 25, 26, 27, 28, 29, 33, 34, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 76, 78, 79, 82, 83, 84, 88, 89, 90, 92, 93, 94, 95, 96, 98, 101, 102, 103, 105, 107, 108, 109, 111, 115, 116, 118, 119, 120, 121, 122, 124, 125, 126, 127, 129, 130, 131, 134, 135, 136, 137, 138 (потенциальное пролекарство), 139, 141, 142, 143, 144, 145, 148, 149 и 150.
В других вариантах соединение выбрано из группы, включающей соединения 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 76, 82, 83, 88, 89, 90, 92, 93, 94, 95, 96, 101, 102, 103, 105, 109, 111, 115, 118, 119, 120, 121, 122, 125, 126, 127, 129, 130, 131, 135, 136, 137, 138, 139, 142, 144, 145, 148, 149 и 150.
В более предпочтительных вариантах фармацевтическая композиция содержит соединение с IC50≤2,5 мкМ при NMDA-рецепторе, выбранное из группы, включающей соединения 20, 21, 22, 23, 24, 25, 27, 28, 29, 30, 31, 32, 33, 34, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 70, 75, 76, 81, 82, 83, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 100, 101, 102, 103, 105, 106, 108, 109, 111, 115, 118, 119, 120, 121, 122, 125, 126, 127, 128, 129, 130, 131, 132, 133, 135, 136, 137, 138 (потенциальное пролекарство), 139, 142, 144, 145, 146, 148, 149 и 150. Предпочтительно соединение выбрано из группы, включающей соединения 21, 22, 23, 24, 25, 27, 28, 29, 33, 34, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 76, 82, 83, 88, 89, 90, 92, 93, 94, 95, 96, 101, 102, 103, 105, 108, 109, 111, 115, 118, 119, 120, 121, 122, 125, 126, 127, 129, 130, 131, 135, 136, 137, 138 (потенциальное пролекарство), 139, 142, 144, 145, 148, 149 и 150.
В особенно предпочтительных вариантах, фармацевтическая композиция содержит соединение с IС50 ≤0,5 мкМ при NMDA-рецепторе, выбранное из группы, включающей соединения 20, 21, 22, 23, 24, 25, 27, 28, 30, 31, 32, 33, 38, 39, 43, 44, 46, 47, 49, 50, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 82, 83, 89, 90, 91, 93, 94, 95, 96, 97, 103, 111, 118, 119, 120, 122, 126, 135, 136, 137, 138 (потенциальное пролекарство), 142, 144, 145, 148, 149 и 150. Предпочтительно соединение выбрано из группы, включающей соединения 21, 22, 23, 24, 25, 27, 28, 33, 38, 39, 43, 44, 46, 47, 49, 50, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 69, 82, 83, 89, 90, 93, 94, 95, 96, 103, 111, 118, 119, 120, 122, 126, 135, 136, 137, 138 (потенциальное пролекарство), 142, 144, 145, 148, 149 и 150.
В более предпочтительных вариантах фармацевтическая композиция содержит соединение, выбранное из группы, включающей соединения 20, 24, 25, 33, 50, 60, 66, 69, 103, 111, 118, 119, 120, 122, 136, 137, 138, 142, 144, 145, 148, 149 и 150. Предпочтительно соединение выбрано из группы, включающей соединения 24, 25, 33, 50, 60, 66, 69, 103, 111, 118, 119, 120, 122, 136, 137, 138, 142, 144, 145, 148, 149 и 150.
В наиболее предпочтительных вариантах фармацевтическая композиция содержит соединение, выбранное из группы, включающей соединения 20, 33, 50, 60, 119 и 144.
Предпочтительно соединение выбрано из группы, состоящей из соединения 33, 50, 60, 119 и 144.
В случае таких соединений, как 20 или 60, могут быть произведены структурные модификации, которые не изменяют существенно иллюстрируемое здесь соотношение структуры-активности (SAR). Например, могут быть выполнены успешная биоизостерическая замена или замещение необязательно замещенных фенильных групп, таких, какие встречаются в соединениях 20 или 60, другими липофильными или полуполярными ароматическими (например, нафтилом, нафтокси, бензилом, фенокси, фенилтио), алифатическими (алкилом, например изопропилом), циклоалифатическими (циклоалкилом, например циклогексилом), гетероциклическими [например, пиридилом, фуранилом, тиофуранилом (тиофенилом)] или другими функциональными группами или системами, как это хорошо известно в данной области, которые дадут клинически применимые соединения (структурные гомологи, аналоги и/или близкие по природе соединения) со сходными биофармацевтическими свойствами и активностью при NMDA-рецепторе (например, сравните соединения 37, 75, 79, 83, 89, 119-122, 125, 126, 128, 130, 132, 137, 144 и 145). Например, такие замены или замещения использовали с большим успехом в разработке SAR среди других групп очень клинически и коммерчески успешных синтетических фармацевтических средств, таких как классические H1-антигистаминные средства, антихолинергические средства (антимускариновые средства; например средства против болезни Паркинсона), антидепрессанты (в том числе трициклические соединения) и опиоидные аналгезирующие средства (См. Foye et al. (Eds.) Principles of Medicinal Chemistry, 4th ed., Lea and Febiger/Williams and Wilkins, Philadelphia, PA, 1995, pp.233, 265, 281-282, 340-341, 418-427 и 430; Prous, J.R., The Year's Drug News, Therapeutic Targets - 1995 Edition, Prous Science Publishers, Barcelona, Spain, 1995, pp.13, 55-56, 58-59, 74, 89, 144-145, 152, 296-297 и 317). Также биоизостерическая замена или замещение метиленовых или метиновых групп в пропильном каркасе таких соединений, как 20 или 60, например кислородом, серой или азотом, будет давать клинически применимые активные в отношении NMDA-рецептора соединения с применимыми подобным образом биофармацевтическими свойствами, такие как 88 (модифицированная структура "классического H1-антигистаминного типа"), которая может быть оптимизирована далее для приобретения активности при NMDA-рецепторе получением, например, соответствующего соединения (соединений), содержащих, например, (бис)-(3-фторфенил)-группу (группы), как описано в данном изобретении. Пропильная структура соединений, таких как 20 и 60, также может быть успешно модифицирована включением циклических систем (как в соединениях 102 и 111) и/или ненасыщенностью (например, двойной связью, как в соединениях 81, 106, 109 и 139) с получением дополнительных клинически применимых NMDA-рецептор-активных соединений данного изобретения (сравните соединения, цитированные выше).
В родственном аспекте данное изобретение относится к способу получения терапевтического агента, предусматривающему стадии скрининга на такой агент путем определения, является ли этот агент активным на оперируемом рецептором кальциевом канале, и синтеза этого терапевтического агента в количестве, достаточном для обеспечения его в терапевтически эффективном количестве для пациента. Такие скрининги могут быть выполнены способами, известными специалистам с обычной квалификацией в данной области, и могут быть, например, выполнены способами, изложенными здесь. Специалисты в данной области знакомы также со способами, применяемыми для синтеза терапевтических средств в количествах, достаточных для терапевтических целей.
В предпочтительном варианте оперируемый рецептором кальциевый канал представляет собой NMDA-рецептор. В более предпочтительном аспекте указанный выше способ включает стадию добавления фармацевтически приемлемого носителя к указанному агенту. В следующем предпочтительном аспекте (варианте) указанный терапевтический агент включает соединение формулы I, представленной здесь. Еще в одном предпочтительном аспекте (варианте) этот терапевтический агент включает соединение Формул II, III, IV, V, VI или VII, представленных здесь. В особенно предпочтительных аспектах терапевтический агент включает соединение, имеющее структуру, выбранную из группы, включающей Формулы I-VII, и все предпочтительные аспекты этих формул, описанные здесь. В дальнейших предпочтительных вариантах терапевтический агент выбран из группы, включающей соединения 19-150. В особенно предпочтительном аспекте, указанный терапевтический агент используют для лечения пациента, имеющего неврологическое заболевание или нарушение. В родственном аспекте указанный скрининг включает стадию идентификации соединения, которое связывается с оперируемым рецептором кальциевым каналом при сайте, связанном одним из арилалкиламинов, соединением 1, соединением 2 и соединением 3.








Другие признаки и преимущества данного изобретения будут очевидны из следующего далее описания его предпочтительных вариантов и из формулы изобретения.
Описание предпочтительных вариантов
Далее следует подробное описание способов и тестов, при помощи которых терапевтически ценные соединения могут идентифицироваться и использоваться для лечения неврологических нарушений и заболеваний. В тестах в качестве примера используют соединение 1, соединение 2 или соединение 3, но другие соединения, имеющие сходную биологическую активность в этих тестах, также могут быть использованы (как это обнаружено) для улучшения тестов. Основные соединения, такие как соединение 1, соединение 2 или соединение 3, могут быть использованы для молекулярного моделирования с применением стандартных процедур, или существование новых соединений в библиотеках природных продуктов может подвергаться скринингу согласно описанным ниже способам.
Один ключевой способ является средством, при помощи которого соединения могут быть подвергнуты быстрому скринингу стандартными способами связывания радиолиганда (тест связывания радиоактивно меченного арилалкиламина) для идентификации соединений, которые связываются при том же сайте на оперируемых рецептором Са2+-каналах, что и соединение 1, соединение 2 или соединение 3. Данные из таких исследований связывания радиолиганда также будут подтверждать, что указанные соединения не ингибируют связывание [3H]-арилалкиламина через действие при известных сайтах на оперируемых рецептором Са2+-каналах (таких как глутамат связывающий сайт, глицинсвязывающий сайт, МК-801-связывающий сайт, Zn2+-связывающий сайт, Мg2+-связывающий сайт, сигма-связывающий сайт или полиаминсвязывающий сайт на комплексе NMDA-рецептор-ионофор). Этот тест-скрининг позволит идентифицировать многочисленные количества потенциально применимых соединений и подвергнуть их скринингу на активность в других тестах. Специалистам в данной области будет понятно, что могут быть придуманы другие быстрые тесты для обнаружения связывания с сайтом арилалкиламинов на оперируемых рецептором Са2+-каналов и использованы в данном изобретении.
Дополнительное тестирование использует электрофизиологическую (patch сlamp-″ пэтч-кламп") методологию для расширения результатов, полученных при помощи вышеупомянутого теста связывания радиолиганда. Такие результаты будут подтверждать, что соединения, связывающиеся с сайтом арилалкиламинов, являются функциональными, неконкурентными антагонистами оперируемых рецепторами Са2+-каналов со следующими свойствами, общими с самими арилалкиламинами: ингибированием открытого канала, проявляющимся как зависимое от использования ингибирование, и потенциал-зависимым появлением и обращением ингибирования. Такие результаты будут также подтверждать, что эти соединения не имеют их первичной активности при описанных ранее сайтах на оперируемых рецептором Са2+-каналах (таких как глутаматсвязывающий сайт, глицинсвязывающий сайт, МК-801-связывающий сайт, Zn2+-связывающий сайт, Мg2+-связывающий сайт, сигма-связывающий сайт или полиаминсвязывающий сайт на комплексе NMDA-рецептор-ионофор).
Кроме того, может быть использована технология рекомбинантных ДНК, используемая для того, чтобы сделать такое тестирование даже еще более быстрым. Например, при помощи стандартных процедур могут быть идентифицированы и клонированы ген (гены), кодирующие новый арилалкиламинсвязывающий сайт (т.е. рецептор). Это может быть выполнено одним из нескольких путей. Например, можно приготовить аффинную арилалкиламиновую колонку и пропускать соллюбилизированные мембраны из клеток или тканей через эту колонку. Рецепторные молекулы связываются с колонкой и, следовательно, их можно выделить. Затем можно получить информацию о частичной аминокислотной последовательности, которая позволяет выделить ген, кодирующий рецептор. Альтернативно получают экспрессионные библиотеки кДНК и субфракции этой библиотеки тестируют на их способность наделять арилалкиламиновыми рецепторами клетки, которые обычно не экспрессировали такие рецепторы (например, клетки СНО, мышиные L-клетки, клетки НЕК 293 или ооциты Xenopus). Таким путем идентифицируют фракцию библиотеки, содержащую клон, кодирующий рецептор. Последовательное субфракционирование активных фракций библиотеки и тестирование в конце концов приводят к одному клону, кодирующему рецептор арилалкиламина. Подобным образом, может быть использовано hybrid-arrest (с задержкой гибрида) и hybrid-depletion (с истощением гибрида) клонирование. Ооциты Xenopus инъецировали мРНК из подходящего источника ткани или клеток (например, из ткани мозга человека). Экспрессию рецептора арилалкиламина детектировали, например, как стимулируемый NMDA или глутаматом приток кальция, который может быть блокирован соединением 1, соединением 2 или соединением 3. Клоны кДНК тестировали на их способность ингибировать экспрессию этого рецептора при гибридизации кДНК или кРНК с выбранной мРНК, перед инъекцией в ооциты Xenopus. Затем клон, ответственный за это действие, выделяли согласно описанному выше способу. После выделения гена рецептора использовали стандартные способы для идентификации полипептида или его части (частей), которая (которые) достаточны для связывания арилалкиламинов (арилалкиламин связывающего домена (доменов)). Далее при помощи стандартных процедур весь рецептор или арилалкиламинсвязывающий домен (домены) можно экспрессировать рекомбинантной технологией. Рецептор или связывающий домен (домены) могут быть выделены и использованы в качестве биохимического реагента таким образом, что скорее может быть использован простой тест прямого связывания, чем конкурентный анализ, описанный ниже. То есть скрининг проводят на соединения, которые связываются при новом рецепторе арилалкил-амина. Таким путем можно скринировать одновременно большие количества соединений, например, проведением через колонку, содержащую новый рецептор арилалкиламинов или домен связывания арилалкиламинов, и анализ выполняют на соединениях, которые связываются с этой колонкой.
Дополнительное тестирование использует сочетание молекулярно-биологических способов (экспрессию клонированных рецепторов NMDA, АМРА или никотиновых холинергических рецепторов) и электрофизиологических (patch clamps="пэтч-кламп") способов. Конкретно аналоги арилалкиламина могут быть подвергнуты быстрому скринингу на их действие при клонированных и экспрессированных субъединицах вышеупомянутых комплексов рецептор-ионофор. Для идентификации, какие аминокислотные остатки могут быть важными в определении силы действия арилалкиламина, можно использовать сайт-направленный мутагенез.
Тесты на сильнодействующие и селективные антагонисты оперируемых рецепторами кальциевых каналов в ЦНС млекопитающих
Желательные свойства лекарственного средства включают высокую аффинность и селективность в отношении оперируемых рецепторами Са2+-каналов, таких, какие присутствуют в комплексах NMDA-, АМРА- и никотиновый холинергический рецептор-ионофор (по сравнению с ответами, медиируемыми через другие нейротрансмиттерные рецепторы, оперируемые нейротрансмиттерами ионные каналы или потенциал-зависимые ионные каналы), и неконкурентный антагонизм оперируемых рецепторами Са2+-каналов.
Комплекс NMDA-рецептор-ионофор используют в качестве примера оперируемого рецептором Са2+-канала. Активация рецептора NMDA открывает катион-избирательный канал, что делает возможным приток внеклеточных Са2+ и Na+ и приводит к увеличению [Са2+]i и деполяризации клеточной мембраны. Измерения [Са2+]i использовали в качестве первичных тестов для детектирования активности арилалкиламиновых соединений на рецепторах NMDA. Очищенные арилалкиламины, синтетические арилалкиламины и синтетические аналоги арилалкиламинов испытывали на активность в тестах in vitro, способных измерять активность рецептора глутамата. Для подробного исследования были выбраны арилалкиламины, присутствующие в яде различных видов пауков. Арилалкиламины, присутствующие в этих ядах, структурно отличаются, но имеют основную структуру класса, представленного соединениями 1-3. Другие, более упрощенные синтетические аналоги в общем состоят из подходящим образом замещенных ароматических хромофорных групп, присоединенных к алкил(поли)аминовой части (см. соединения 19-69 ниже).
Был разработан первичный тест, который обеспечивает функциональный показатель активности рецептора глутамата и который делает возможным высокопроизводительный скрининг. Первичные культуры крысиных мозжечковых зернистых клеток, нагруженных флуорометрическим индикатором fura-2, использовали для измерения изменений в [Ca2+]i, вызываемых NMDA и его коагонистом глицином. Этот тест обеспечивает чрезвычайно чувствительный и точный показатель активности NMDA-рецептора. Увеличения [Са2+]i, вызываемые NMDA, зависят от присутствия глицина и ингибируются внеклеточным Mg2+ или антагонистами, действующими при сайтах связывания глутамата, глицина или МК-801. Увеличения [Ca2+]i, вызываемые NMDA/глицином, легко отличить от увеличений, являющихся результатом деполяризации, по их устойчивости к ингибированию блокаторами потенциал-зависимых Са2+-каналов. Точность, с которой измерения [Са2+]i подтверждают результаты, полученные электрофизиологическими исследованиями и исследованиями связывания лиганда, предполагает, что такие измерения близко отражают активацию комплекса NMDA-рецептор-ионофор.
Пример 1: Сильнодействующее неконкурентное ингибирование функции NMDA-рецептора
Измеряли предпочтительные ингибиторные действия арилалкиламинов на медиируемое NMDA-рецептором увеличения [Са2+]i в культивируемых крысиных мозжечковых зернистых клетках. Увеличения [Са2+]i вызывали добавлением NMDA/глицина (50 мкМ/1 мкМ) в присутствии или в отсутствие различных концентраций каждого из тестируемых соединений. Величины IC50 получали для каждого тест-соединения с использованием от 2 до 8 отдельных экспериментов на тест-соединение, и уровень стандартной ошибки был менее 10% от средней величины для каждого соединения.
Все испытанные арилалкиламины ингибировали увеличения [Са2+]i в мозжечковых зернистых клетках, вызываемые NMDA/глицином. Некоторые арилалкиламины, сходные по структуре с соединением 1 или соединением 2, были почти такими же сильнодействующими, что и МК-801 (IC50=34 нМ), который является наиболее сильнодействующим соединением, предпочтительно ингибирующим NMDA-рецепторы, согласно литературе. Соединение 3 имело IС50=2 нМ, т.е. было в 17 раз более сильным, чем конкурентные антагонисты, такие как АР5 (IС50=15 мкМ). Ингибирующие действия арилалкиламинов не снимались увеличением концентраций NMDA или глицина. То есть, не наблюдали изменения в ЕС50 ни для NMDA, ни для глицина. Таким образом, арилалкиламины являются неконкурентными антагонистами при комплексе NMDA-рецептор-ионофор и не действуют ни при сайте связывания глутамата, ни при сайте связывания глицина.
Пример 2: Активность против функции рецептора каината и АМРА
Измерения [Ca2+]i в мозжечковых зернистых клетках могут быть также использованы для наблюдения активации нативных рецепторов каината и АМРА, присутствующих в этой ткани. Хотя увеличения [Ca2+]i, вызываемые этими агонистами, имеют меньший размер, чем вызываемые NMDA/глицином, такие ответы являются сильными и могут быть использованы для точной оценки специфичности действия арилалкиламинов на фармакологически определенные подтипы рецептора глутамата. Сравнительные измерения [Ca2+]i выявили четкое различие в избирательности рецептора арилалкиламинами. Некоторые, такие как JSTX-3 (токсин паука Joro из паука Nephila clavata), были более сильными антагонистами ответов, вызываемых каинатом (100 мкМ) или АМРА (30 мкМ). С другой стороны, было обнаружено, что арилалкиламины в двух структурных классах, определяемых соединением 1 и соединением 2, ингибировали предпочтительно ответы, вызываемые NMDA (показывая приблизительно 100-кратное различие в силе действия). Таким образом, арилалкиламины, такие как соединение 1 и соединение 2, являются сильнодействующими и избирательными ингибиторами медиируемых NMDA-рецептором ответов в мозжечковых зернистых клетках.
Пример 3: Электрофиэиологические (patch clamp) исследования
Электрофизиологические исследования на выделенных нейронах коры или гиппокампа из мозга взрослых крыс обеспечили дополнительное понимание механизма действия соединения 1, соединения 2 и соединения 3. Эти исследования выявили сильное и избирательное ингибиторное действие арилалкиламинов на ответы, опосредованные NMDA-рецепторами. Так, соединения, такие как соединение 1, ингибировали ответы на NMDA при наномолярных концентрациях без влияния на ответы, вызываемые каинатом. Эти результаты, показывающие избирательные ингибирующие действия арилалкиламинов в нейронах коры головного мозга и гиппокампа, указывают на то, что арилалкиламины нацелены на NMDA-рецепторы в отличающихся районах в ЦНС млекопитающих. Кроме того, было обнаружено, что ингибирующие эффекты этих соединений были зависимыми от использования и потенциал-зависимыми. Это позволяет предположить, что эти соединения блокируют открытый канал и посредством этого действия ведут себя как неконкурентные антагонисты NMDA-рецепторов. Однако важно, чтобы арилалкиламины можно было отличать как от Мg2+, так и от МК-801, особенно в отношении потенциал-зависимости их появления действия и обратимости эффекта.
Пример 4: Тесты связывания радиолиганда
Исследования связывания радиолиганда показали, что арилалкиламины, такие как соединение 1 и соединение 2, имеют уникальный сайт действия. Хотя они действуют подобно МК-801 в некотором отношении (неконкурентное ингибирование открытого канала, обсуждаемое выше), они не могут заменять связывание [3H] МК-801 при концентрациях, которые полностью блокируют опосредованные NMDA-рецептором ответы. Тесты, подобные этим, показывают также, что арилалкиламины не связываются с высокой аффинностью с известными сайтами связывания МК-801, Мg2+ или полиамина на комплексе NMDA-рецептор-ионофор. Также арилалкиламины не связываются непосредственно с сайтами связывания глутамата, глицина или сигма-связывающим сайтом при концентрациях, которые ингибируют опосредованные NMDA-рецептором ответные реакции. В качестве радиолиганда синтезировали [3H] соединение 2 для применения в исследованиях по связыванию для дальнейшего выяснения механизма действия соединения 2 и, в частности, для применения в высокопроизводительном скрининге для оценки активности других аналогов и для обнаружения новых ведущих (основных) структур. Подобный подход был использован для [3H] соединения 5. Ясно, что соединения, подобные соединению 1 и соединению 2, нацелены на сайт на NMDA-рецептор-ионофорном комплексе, для которого в настоящее время не существует других известных соединений. Новый сайт действия арилалкиламинов на этом молекулярном уровне транслируется в заметные терапевтические преимущества на поведенческом уровне. Как описано выше, арилалкиламины обладают очень отличающимся поведенческим профилем от других неконкурентных антагонистов NMDA-рецептора.
Пример 5: Исследования синаптической передачи нервного импульса
Описанные выше открытия показывают, что некоторые арилалкиламины, в частности родственные по структуре соединению 1 и соединению 2, действуют через новый механизм и сайт действия для сильного и селективного ингибирования медиируемых NMDA-рецептором ответов на нейронах из нескольких различных участков мозга. Для дальнейшей оценки избирательных ингибиторных действий арилалкиламинов оценивали их действия на синаптическую передачу нервного импульса, медиируемую рецепторами NMDA или АМРА.
Медиируемую глутаматом передачу нервных импульсов при синапсах коллатеральных волокон Шаффера и пирамидных клеток СА1 измеряли в срезах мозга крысы, содержащих гиппокамп. Этот тест измеряет электрофизиологически, постсинаптическую деполяризацию, вызываемую пресинаптическим высвобождением глутамата, и может легко различить синаптическую трансмиссию, медиируемую рецепторами NMDA или АМРА. Опять было обнаружено, что арилалкиламины, подобные соединению 1, соединению 2 и соединению 3, проявляли предпочтительные ингибиторные действия на NMDA-рецептор-медиируемые ответы и подавляли медиируемые АМРА-рецепторами ответы при гораздо более высоких концентрациях. Например, соединение 1 имело IC50 для NMDA-рецептор-медиируемого ответа 20 мкМ, но IС50 Для АМРА-рецептор-медиируемого ответа 647 мкМ. Эти результаты показывают, что арилалкиламины могут избирательно ингибировать синаптическую трансмиссию возбуждения рецепторами NMDA. Другие природно-встречающиеся арилалкиламины, присутствующие в яде Agelenopsis aperta, также проявляют сильные и селективные ингибиторные действия на медиируемые NMDA-рецептором ответы в гиппокампе крысы.
Взятые вместе результаты этих различных исследований являются дополняющими друг друга и вместе идентифицируют новый класс соединений с сильной и селективной ингибиторной активностью в отношении NMDA-рецепторов в ЦНС млекопитающих. Кроме того, эти соединения нацелены на уникальный сайт на NMDA-рецептор-ионофорном комплексе. Соединение 1, соединение 2 и соединение 3 были выбраны для дополнительного исследования в различных тестах in vitro и in vivo, которые моделируют терапевтически важные конечные цели.
Нейропротекторная активность
Желательные свойства нейропротекторного лекарственного средства включают следующее. (1) Лекарственное средство может вводиться перорально или инъекцией (т.е. оно не разрушается значительно в желудке, кишечнике или сосудистой системе и, следовательно, достигает тканей, которые должны быть подвергнуты лечению, в терапевтически эффективном количестве). Такие лекарственные средства легко испытывать на грызунах для определения их биодоступности. (2) Лекарственное средство проявляет нейропротекторную активность (т.е. эффективность) при предоставлении после ишемического инсульта (удара, асфиксии) или травматического повреждения (травмы головы, повреждения спинного мозга). (3) Лекарственное средство не имеет побочных действий или имеет минимальные побочные действия, такие как ухудшение познавательной способности, нарушение двигательной способности, седативный эффект или повышенная возбудимость, вакуолизация нервных клеток, сердечно-сосудистая активность, РСР-подобный потенциал злоупотребления или РСР-подобную психотомиметическую активность.
Хотя глутамат представляет собой физиологический синаптический нейротрансмиттер (нейромедиатор), хроническое экспонирование с глутаматом ведет к гибели нервных клеток. Большинство нейродегенеративных состояний, вызываемых глутаматом, по-видимому, опосредовано NMDA-рецепторами и является непосредственным результатом хронически повышенных уровней Са2+ в цитозоле. В настоящее время имеется сильное экспериментальное подтверждение мнения, что рецепторы NMDA и АМРА играют основную роль в медиировании дегенерации нейронов после удара (кровоизлияния) и других ишемических/гипоксических событий (Choi, Glutamate neurotoxicity and diseases of the nervous system. Neuron 1: 623, 1988). Большая часть этих доказательств основана на способности конкурентных и неконкурентных антагонистов рецептора NMDA или АМРА эффективно ингибировать гибель нервных клеток в моделях ишемического удара как in vitro, так и in vivo. Поэтому соединение 1, соединение 2 и соединение 4 испытывали на нейропротекторные действия в стандартных тестах для обнаружения такой активности.
Пример 6: Защита кортикальных нейронов
Для оценки нейропротекторного действия арилалкиламинов in vitro кортикальные нейроны мышей, выращенные в культуре, выдерживали в течение 5 минут с NMDA и гибель клеток после 24 часов наблюдали путем измерения высвобождения лактатде-гидрогеназы (LDH), цитоплазматического фермента, который высвобождается из погибающих клеток (Choi et al., Glutamate neurotoxicity in cortical cell culture. J. Neurosci. 7: 357, 1987). Выдерживание с NMDA убивало приблизительно 80% кортикальных нейронов. Соединение 1 или соединение 2, введенные вместе с NMDA, предотвращали гибель клеток с величинами IC50 70 мкМ и 30 мкМ соответственно. Эффективные концентрации арилалкиламинов являются более высокими, чем эффективные концентрации других неконкурентных антагонистов NMDA-рецептора, но сходными с эффективными концентрациями конкурентных антагонистов. Эффективные концентрации антагонистов NMDA-рецептора варьируют в зависимости от конкретных экспериментальных условий и от типа исследованных клеток (кортикальных клеток, клеток гиппокампа, клеток полосатого тела). Нейропротекторное действие, вероятно, является результатом способности этих соединений ингибировать приток внеклеточного Са2+, запускаемый NMDA-рецептором.
Более строгое испытание для определения потенциальной терапевтической эффективности было проведено на моделях ишемического удара in vivo. В этих моделях подачу крови временно блокировали пережиманием основных артерий, идущих к мозгу. Две модели in vivo такого типа использовали для определения способности соединения 1, соединения 2 и соединения 4 предотвращать потерю нервных клеток.
Пример 7: Билатеральная окклюзия сонной артерии
Первым тестом была модель ишемии переднего мозга у песчанок, вызываемой билатеральной окклюзией общей сонной артерии (Karpiak et al., Animal models for the study of drugs in ischemic stroke. Ann. Rev. Pharmacol. Toxicol. 29: 403, 1989; Ginsberg and Busto, Rodent models of cerebral ischemia. Stroke 20: 1627, 1989). Ток крови к мозгу был прерванным в течение 7 минут зажиманием сонных артерий. Тестируемые соединения вводили в виде разовой дозы внутрибрюшинно (i.p.) через 30 минут после возобновления кровотока. Во время хода этого эксперимента животных поддерживали при 37° С для предотвращения какой-либо гипотермической реакции. Было показано, что многие антагонисты NMDA-рецептора вызывают гипотермию и этот эффект мог составлять большую часть защитного эффекта этих соединений. Мозг испытывали на гибель нервных клеток через 4 дня после этого путем окрашивания серебром срезов мозга и количественного определения гибели при помощи морфометрического анализа. Соединение 2 (20 мг/кг) значимо (р<0,05) защищало от гибели нервные клетки во всех исследованных участках мозга (районе СА1 гиппокампа, полосатом теле и неокортексе). Такие низкие дозы, как 1 мг/кг, давали полную защиту (>98%) полосатого тела. Степень защиты сравнима с достигаемой с такими же дозами неконкурентного антагониста NMDA-рецептора, МК-801.
В последующих экспериментах соединение 1 (10 мг/кг) давало 23%-ное уменьшение количества гибели нейронов в районе СА1 гиппокампа песчанки, измеренное при 7 днях пост-ишемии, тогда как соединение 4 (10 мг/кг) обеспечило 90%-ную защиту.
Пример 8: Окклюзия средней мозговой артерии
Модель ишемического удара средней мозговой артерии, выполняемая на крысах (Karpiak et al., Animal models for the study of drugs in ischemic stroke. Ann. Rev. Pharmacol. Toxicol. 29: 403, 1989; Ginsberg and Busto, Rodent models of cerebral ischemia. Stroke 20: 1627, 1989), отличается от модели песчанок, поскольку она приводит к более ограниченному церебральному инфаркту и приближается поэтому к приступам другого типа (очаговому тромботическому повреждению). В первом исследовании с применением этой модели одномозговую артерию подвергали перманентной окклюзии хирургическим лигированием. Тест-соединения вводили через 30 минут после окклюзии в виде разовой внутрибрюшинной (i.p.) инъекции. В ходе этих экспериментов температуру внутри тела животных поддерживали при 37° С для предотвращения какой-либо гипотермической реакции. Мозг оценивали гистологически на потерю нервных клеток спустя 24 часа. Объемы инфаркта рассчитывали по площади гистологической бледности из 10 слайдов и интегрировали расстояние между последовательными срезами. Было обнаружено, что одна доза (30 мг/кг) соединения 1 значимо (р<0,05) защищает против потери нервных клеток, так же как и максимальная эффективная доза (10 мг/кг), МК-801 (приблизительно 15%-ная защита). Предварительные исследования с соединением 2 (20 мг/кг) показали сходную тенденцию.
Во втором исследовании очаговой церебральной ишемии у крыс среднюю мозговую артерию подвергали перманентной окклюзии проведением небольшого куска шовной нити через сонную артерию до участка средней мозговой артерии. Температуру внутри тела поддерживали при 37° С. Соединение 4, 10 мг/кг i.р., введенное сразу же после появления ишемического события, давало статистически значимое уменьшение объема инфаркта мозга (20%), зарегистрированное спустя 24 часа.
В третьей модели очаговой церебральной ишемии у крыс ишемический инфаркт индуцировали фототромботическим способом с применением красителя Rose Bengal. Соединение 4, 10 мг/кг i.р., введенное через 30 минут после ишемического события, давало 20%-ное уменьшение объема инфаркта, сходное с уменьшением, наблюдаемым с неконкурентным антагонистом NMDA-рецептора, МК-801.
В четвертой модели очаговой церебральной ишемии у крыс среднюю мозговую артерию временно закупоривали проведением небольшого куска шовной нити через сонную артерию к району средней мозговой артерии. Шовную нить вытаскивали после ишемического периода 2 часа. Температуру внутри тела поддерживали при 37° С. Соединение 4, введенное при 10 мг/кг i.p. после появления ишемического события, давало статистически значимое уменьшение объема инфаркта мозга (37%), регистрируемое спустя 72 часа.
Из этих результатов, полученных in vivo, очевидны несколько важных свойств основных соединений. Во-первых, что наиболее важно, соединение 1, соединение 2 и соединение 4 демонстрируют нейропротекторные эффекты в нескольких различных моделях ишемии in vivo. Тест на песчанках является моделью временной общей мозговой ишемии и гипоксии, такой как остановка сердца или перинатальная гипоксия. Тесты на крысах являются моделями перманентной и временной очаговой церебральной ишемии. Обнаружение, что соединение 1 и соединение 4 являются нейропротекторными в моделях перманентного очагового инфаркта, является неожиданным, вследствие того, что доступ лекарственного средства к месту инфаркта ограничен затененным участком (penumbral region), который обычно не является большим. Тем не менее соединение 1 и соединение 4 значимо (р<0,05) ограничивали степень повреждения. Во-вторых, эти соединения эффективны при введении их после ишемического события. Это является важным, поскольку считают, что имеется "окно благоприятной возможности" после инфаркта, во время которого лекарственные средства могут эффективно останавливать некротическое повреждение. Насколько длителен этот период времени у людей, не было точно определено, и он, по-видимому, изменяется в зависимости от типа инфаркта. Однако существенным является наблюдение, что эти соединения могут предотвращать распространение гибели нервных клеток, как только начался этот дегенеративный процесс. Наконец, соединения 1, 2 и 4 являются эффективными при парентеральном введении, что свидетельствует о том, что они могут преодолевать гематоэнцефалический барьер.
Противосудорожная активность
Желательные свойства противосудорожного лекарственного средства включают в себя следующие свойства: лекарственное средство может вводиться перорально или посредством инъекции, лекарственное средство проявляет эффективную противосудорожную активность против нескольких типов эпилептических припадков, в том числе но не только, простых эпилептических парциальных припадков, сложных эпилептических парциальных припадков, эпилептического статуса и индуцированных травмой припадков, таких, какие встречаются после повреждения головы, в том числе хирургии головы; и лекарственное средство не должно иметь побочных действий или имеет минимальные побочные действия, такие как ухудшение познавательной способности, седативный эффект или повышенная возбудимость, вакуолизация нейронов, сердечно-сосудистая активность, РСР-подобный потенциал злоупотребления или РСР-подобная психотомиметическая активность.
Глутамат представляет собой основной медиатор возбуждения в мозгу и, следовательно, может играть основную роль в активности эпилептических припадков и участвовать в патогенезе эпилепсии. Много доказательств в пользу основной роли рецепторов глутамата в эпилепсии получено из фармакологических исследований, демонстрирующих, что агонисты рецептора глутамата вызывают эпилептические припадки и что антагонисты рецептора NMDA или АМРА являются эффективными противосудорожными средствами при введении их in vivo. Имеются многочисленные модели in vivo различного рода эпилептических припадков и поведенческих эффектов, которые относятся к клинически различным формам эпилепсии. Таким образом, благоразумным было бы испытание на эти эффекты в нескольких моделях, поскольку может быть сверх упрощением предполагать, что один и тот же механизм лежит в основе всех форм эпилептических припадков.
Пример 9: Блокирующая конвульсант активность
В исходных исследованиях испытывали способность арилалкил-аминов блокировать эпилептические припадки, индуцируемые каинатом, пикротоксином или бицукуллином. Каждый из этих вызывающих судороги агентов (конвульсантов) действует по отличающемуся механизму, и эпилептические припадки, индуцируемые каинатом, качественно отличаются от припадков, индуцируемых пикротоксином или бицукуллином. В этих экспериментах фракцию яда Agelenopsis aperta, содержащую некоторые арилалкиламиновые токсины, вводили внутривенно (iv) за 5 минут перед введением пикротоксина или бицукуллина и за 5 минут после введения каината. Арилалкиламины уменьшали эпилептические припадки, индуцируемые всеми тремя агентами. Эффекты пикротоксина или бицукуллина были настолько тяжелыми, что все 19 контрольных животных погибли в пределах 25 минут. В противоположность этому не было гибели среди 9 животных, предварительно получивших арилалкиламины. Фактически только половина животных, получивших арилалкиламины, обнаружила вообще судороги, и эти симптомы ослабевали в пределах часа. Эти результаты показывают отчетливые противосудорожные эффекты арилалкиламинов и побудили авторов к дальнейшим исследованиям с использованием очищенных арилалкиламинов и их аналогов.
Пример 10: Стимулы, вызывающие эпилептические припадки
Три различных индуцирующих припадки тест-примера использовали первоначально в этой второй группе исследований, и такие арилалкиламины, как соединение 1, оказались эффективными в двух таких тест-примерах. Первые две модели использовали мышей DBA/2, которые склонны к аудиогенным эпилептическим припадкам. Припадки вызывали звуком (тоном звонка при 109 дБ) или внутрибрюшинным (i.p.) введением NMDA (56 мг/кг). Тестируемые вещества вводили за 15-30 минут перед стимулом, вызывающим эпилептический припадок (конвульсантом). Число клонических судорог регистрировали в течение 1 минуты после аудиогенного стимула или в течение 15 минут после введения NMDA. Соединение 1, соединение 2 и некоторые другие арилалкиламины, такие как соединение 3 и соединение 4, подавляли судороги, вызываемые каждым стимулом. Например, соединение 2 имело ED50 0,13 мг/кг s.c. для аудиогенного стимула и 0,083 мг/кг s.c. для NMDA-стимула. Подобным образом ED50 для соединения 4 в аудиогенной модели эпилептического припадка (0,08 мг/кг) приближалось к ED50 для МК-801 (0,02 мг/кг). В противоположность этому ни соединение 1, ни соединение 2 не было эффективным при дозах до 50 мг/кг в уменьшении эпилептических припадков в мышах CF-1, вызываемых NMDA (i.р.).
Во второй независимой серии экспериментов было обнаружено, что соединение 1 и соединение 4 предотвращают эпилептические припадки, индуцированные звуком в другой модели рефлекторной эпилепсии генетически чувствительной мыши (Frings мыши), после внутрибрюшинной инъекции с величинами IC5 14,3 мг/кг и ~15 мг/кг соответственно. Эти соединения были значительно более сильнодействующими против аудиогенных эпилептических припадков в мышах Frings после интрацеребровентрикулярной (i.c.v.) инъекции, с величинами IC50 0,63 мкг (соединение 1) и 4,77 мкг (соединение 4). Было также обнаружено, что соединение 1 является эффективным против эпилептических припадков, вызываемых максимальным электрошоком в мышах CF-1 при дозе 4 мкг (i.c.v.).
В дальнейших исследованиях с использованием модели рефлекторной эпилепсии генетически чувствительной мыши (мыши Frings) соединение 9, соединение 12 и соединение 14, вводимые i.c.v. инъекцией, предотвращали индуцируемые звуком эпилептические припадки с величинами IC50 4,77 мкг, 12,2 мкг и 13,9 мкг соответственно.
Эти взятые вместе открытия демонстрируют, что арилалкиламины, такие как соединение 1, соединение 2 и соединение 4, являются эффективными в предотвращении эпилептических (аудиогенных) и не эпилептических (вызываемых химическими агентами) припадков. Этот общий характер активности предполагает, что арилалкиламины клинически применимы в контролировании эпилептических припадков. Кроме того, эффективность соединения 1, соединения 2 и особенно соединения 4 в моделях эпилептической активности in vivo показывает, что эти соединения могут иметь терапевтические эффекты при парентеральном введении в низких дозах и являются особенно эффективными при введении непосредственно в желудочки мозга.
Аналгезирующая активность
Желательные свойства болеутоляющего лекарственного средства таковы: лекарственное средство может вводиться перорально или инъекцией, лекарственное средство проявляет аналгезирующую активность, лекарственное средство не имеет побочных действий или имеет минимальные побочные действия, такие как ухудшение познавательной способности, нарушение двигательной способности, седативный эффект или повышенная возбудимость, вакуолизация нейронов, РСР-подобный потенциал злоупотребления или РСР-подобная психотомиметическая активность.
Опосредованные глутаматом и NMDA-рецептором ответные реакции могут играть роль в некоторых типах восприятия боли (Dickenson, A cure for wind up: NMDA receptor antagonists as potential analgesics. Trends Pharmacol. Scl. 11: 302, 1990). Таким образом, исследовали возможные аналгезирующие действия соединения 1, соединения 2, соединения 3 и соединения 4.
Пример 11: Тест со скорчиванием от боли
В первой серии экспериментов животным вводили неприятный стимул (2-фенил-1,4-бензохинон, PBQ), который вызывает ответ скорчивания (вытягивание живота). Обычно регистрируют число корчей, наблюдающихся за 5-минутный период наблюдения. Классические аналгезирующие лекарственные средства, такие как морфин, эффективны в уменьшении числа вызываемых PBQ корчей (100%-ное ингибирование скорчивающего ответа при 4 мг/кг i.p.). Нестероидные противовоспалительные средства также эффективны в этой модели. Соединение 1 (2 мг/кг), соединение 2 (2 мг/кг) и соединение 3 (1 мг/кг) подавляли реакцию скорчивания более чем на 95%, при введении s.c. или i.p. за 30 минут перед введением PBQ. Эти результаты показывают, что соединение 1, соединение 2 и соединение 3 ослабляют висцеральную боль.
В сходной серии исследований было обнаружено, что соединение 1 и соединение 4 подавляют вызываемые уксусной кислотой корчи у мышей после инъекции i.p. с величинами IC50 10 мг/кг и 1 мг/кг соответственно.
Пример 12: Тест горячей пластинки
Соединение 1 тестировали на аналгезирующую активность: в дополнительном тесте. В этой модели аналгезирующей активности мышам вводили тест-вещества s.c. за 30 минут перед помещением их на горячую пластинку (50° С). Время до облизывания лапок или спрыгивания с пластинки является показателем аналгезирующей активности, и эффективные аналгетики увеличивают период латентного состояния до облизывания лапок или соскакивания с пластинки. Морфин (5,6 мг/кг) увеличивал период до соскакивания на 765%. Соединение 1 было также эффективно в этом тесте и при дозах 4 и 32 мг/кг увеличивало латентный период до облизывания лапок на 136% и латентный период до спрыгивания на 360% соответственно.
Следует отметить, что аналгезирующие эффекты соединения 1 в тесте горячей пластинки не сопровождались пониженной активностью в тесте с обращенной решеткой (см. ниже). Это показывает, что увеличение латентного периода для спрыгивания с горячей пластинки просто не отражает ухудшения двигательной способности. Взятые вместе эти данные предполагают, что соединение 1 обладает значительной аналгезирующей активностью.
В последней серии экспериментов было показано, что соединение 1 и соединение 4 обладают значительной аналгезирующей активностью в крысах при внутриоболочечном введении (i.th.). В этих экспериментах в качестве болевого стимула использовали горячую (52° С) пластинку. Соединение 1 (0,3-3 нмоль) и соединение 4 (0,3-3 нмоль) производили зависимое от дозы и зависимое от времени болеутоляющее действие; эти арилалкиламины были одинаковы с морфином (0,3-3 нмоль) по силе и эффективности. С другой стороны, антагонист NMDA-рецептора, МК-801, был неэффективен в этом тесте (3-30 нмоль).
Пример 13: Тест с резкими движениями хвоста
В этом стандартном тесте термический болевой стимул представлял собой нагретую до 52° С воду с латентным периодом до резкого движения хвостом или до отдергивания, взятых за конечную точку. Соединение 1 (0,3-3 нмоль) и соединение 4 (0,3-3 нмоль) обнаруживали зависимое от дозы и зависимое от времени аналгезирующее действие после введения i.th. Эти арилалкиламины были одинаковы с морфином (0,3-3 нмоль) в отношении силы действия и эффективности. С другой стороны, антагонист NMDA-рецептора, МК-801, был неэффективен в этом тесте (3-30 нмоль).
Пример 14: Формалиновый тест
Самцов крыс Sprague-Dawley приучали к камере наблюдения в течение по меньшей мере 1 часа перед получением инъекции разбавленного формалина (5%) в объеме 50 мкл в левую заднюю лапку. Поведенческие ответные реакции наблюдали сразу же после s.с. инъекции формалина в дорсальную поверхность лапки подсчетом числа вздрагиваний животного. Поведение наблюдали в течение по меньшей мере 50 минут после инъекции формалина и регистрировали как ответы ранней фазы (0-10 минут после инъекции формалина) и ответы поздней фазы (20-50 минут после инъекции формалина). Соединения инъецировали внутриоболочечно (i.th.) за 10 минут перед введением формалина (предобработка) или через 10 минут после введения формалина (пост-обработка) в объеме 5 мкл.
Введение формалина внутрь подошвы производило типичный двухфазный ответ поведения вздрагивания, обычно описываемый как ответы ранней и поздней фазы. Внутриоболочечное введение соединения 1 (0,3-10 нмоль) или соединения 4 (0,3-10 нмоль) в виде предобработки перед введением формалина эффективно ингибировало как вздрагивания ранней фазы, так и вздрагивания поздней фазы. Это действие предобработки арилалкиламинами было сходно с действием предобработки морфином (1-10 нмоль) или МК-801 (1-30 нмоль).
Соединение 1 (0,3-10 нмоль i.th.), введенное после формалина, производило некоторое ингибирование поздней фазы вздрагиваний, хотя значимость достигалась только при дозе 10 нмоль. Соединение 4 (0,3-10 нмоль), введенное после формалина, производило значимое ингибирование поздней фазы вздрагиваний, причем значимость достигалась при дозах 3 и 10 нмоль. Этот аналгезирующий профиль активности арилалкиламинов сходен с профилем введения морфина после формалина (1-10 нмоль); постформалиновое введение МК-801 (1-30 нмоль), однако, не влияло на позднюю фазу вздрагиваний.
Взятые вместе полученные результаты тестов с горячей пластинкой, резких движений хвоста и формалиновых тестов показывают, что арилалкиламины, такие как соединение 1 и соединение 4, имеют значимую аналгезирующую активность в нескольких моделях грызунов с острой болью. Формалиновый тест дополнительно демонстрирует, что арилалкиламины эффективны в моделях хронической боли животных. Важно, что арилалкиламины обладают аналгезирующей активностью при введении после формалинового стимула. Этот профиль активности явно отличает эти арилалкиламины от стандартных антагонистов NMDA-рецептора, таких как МК-801.
Побочные эффекты арилалкиламинов
При условии, что NMDA-рецепторы играют важную роль в разнообразных функциях мозга, неудивительно обнаружить, что антагонисты этого рецептора, как правило, ассоциированы с определенными нежелательными побочными эффектами. Фактически именно это свойство создает основное препятствие для разработки терапий, нацеленных на NMDA-рецепторы. Основными побочными действиями, которые свойственны как конкурентным, так и неконкурентным антагонистам, являются РСР-подобная психотомиметическая активность, ухудшение двигательной активности, седативный эффект или повышенная возбудимость, ухудшение познавательной способности, вакуолизация нервных клеток или сердечно-сосудистые эффекты (Willets et al., The Behavioral pharmacology of NMDA receptor antagonists. Trends Pharmacol. Sci. 11: 423, 1990; Olney et al., Patological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science 244: 1360, 1989). Психотомиметический эффект, ассоциированный с ингибированием опосредованных NMDA-рецептором ответных реакций, возникает в ответ на фенциклидин (РСР) или "ангельский порошок", который действует при сайте связывания МК-801. Нарушение познавательной способности ассоциировано с важной ролью, которую NMDA-рецепторы обычно играют в обучении и памяти.
Относительно меньше известно в отношении профиля побочных эффектов антагонистов рецептора АМРА. Однако становится ясно, что такие соединения также вызывают ухудшение двигательной активности, атаксию и сильный седативный эффект.
Активность арилалкиламинов испытывали в моделях животных, которые показывают ухудшение двигательной активности, седативную и психотомиметическую активность, а также в моделях познавательной способности и памяти in vitro и in vivo.
(а) РСР-подобная психотомиметическая активность
В грызунах как конкурентные, так и неконкурентные антагонисты NMDA-рецептора вызывают РСР-подобное стереотипическое поведение, характеризующееся гиперактивностью, верчением головы из стороны в сторону и атаксией (Willets et al., The behavioral pharmacology of NMDA receptor antagonists. Trends Pharmacol. Sci. 11: 423, 1990; Snell and Johnson, In: Excitatory Ammo Acids in Health and Disease, John Wiley and Sons, p.261, 1988). Заявители исследовали, вызывают ли арилалкиламины такое поведение. Кроме того, заявители исследовали, будут ли арилалкиламины заменять РСР в крысах, обученных отличать РСР от солевого раствора (Willets et al., The behavioral pharmacology of NMDA receptor antagonists. Trends Pharmacol. Sci. 11: 423, 1990), и вызывают ли арилалкиламины РСР-подобную вакуолизацию нейронов (Olney et al., Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science 244: 1360, 1989).
Пример 15: Опорно-двигательная активность
В первом тесте просто наблюдают опорно-двигательную активность во время первых часов после периферического введения (s.c. или i.p.) тест-вещества. Мыши получали дозу соединения 1 за 15 минут перед помещением в камеры для определения активности. Активность измеряли количественно подсчетом числа прерываний в сетке фотоэлементов за период 60 минут. В этом тесте МК-801 (0,25 мг/кг р.о.) вызывает 2-3-кратное увеличение в опорно-двигательной активности. Однако соединение 1, даже при тестировании при 32 мг/кг s.c., не вызывало повышенной активности и фактически имело тенденцию ее подавления. Этот результат, полученный с применением очищенного арилалкиламина в мышах, дополняет более ранние результаты, полученные в крысах, где вся содержащая арилалкиламины фракция из Agelenopsis aperta при внутривенном введении не вызывала РСР-подобного поведенческого синдрома, а по-видимому, производила мягкое седативное действие.
Пример 16: Ухудшение двигательной функции
В первом тесте на общее ухудшение двигательной функции соединение 1 испытывали в тесте с перевернутой сеткой. В этом тесте животных помещали на сетку с проволочными отверстиями, подвешенную от вращающегося металлического стержня, которая может быть перевернута. Затем животных оценивали в баллах на их способность взбираться наверх или удерживаться на сетке. Этот тест обеспечивает показатель "поведенческого нарушения", которое может быть результатом атаксии, потери выпрямительного рефлекса, седативного эффекта или миорелаксации. В этих тестах соединение 1, введенное при 32 мг/кг s.c., не ухудшало способность мышей DBA/2 самостоятельно выпрямляться при переворачивании сетки (р>0,05). Соединение 2 также не действовало (р>0,05) на двигательную способность в мышах DBA/2 при введении при дозе 20 мг/кг s.c. Эти дозы значительно выше доз, которые требовались для предотвращения индуцируемых звуком рефлекторных эпилептических припадков у мышей DBA/2 (см. пример 10 выше).
Второй тест острого ухудшения двигательной способности был тест с вращающимся стержнем. В этом тесте мышей Frings и CF1 инъецировали тест-соединением и помещали на насеченный стержень, который вращается при скорости 6 об/мин. Определяли способность мышей сохранять равновесие в течение длительных периодов времени. Мышей, которые были неспособны сохранять равновесие на вращающемся стержне в течение 1 минуты в каждом из трех испытаний, считали имеющими нарушения. Соединение 1 производило острое двигательное нарушение в мышах Frings с TD50 (доза, которая дает моторную токсичность у 50% тестированных животных) 16,8 мг/кг i.p. Эта доза одинакова с дозой, которая предотвращает индуцируемые звуком эпилептические припадки в мышах Frings (см. пример 10 выше). Однако существует гораздо более явное разделение между эффективными и токсическими дозами соединения 1 в случае мышей Frings при введении соединения i.c.v. В этом случае не наблюдали явной моторной токсичности до тех пор, пока доза соединения 1 не превысила 1,56 мкг i.c.v. (> в два раза, чем ED50 0,63 мкг). Наконец, ухудшение двигательной способности у мышей CF1 наблюдали с соединением 1 после введения i.c.v. 4 мкг.
Соединение 4, соединение 9, соединение 12 и соединение 14 вводили мышам Frings i.с.v.-инъекцией и измеряли острое нарушение двигательной функции. Величины TD50 для Соединений 4, 8, 12 и 14 были 8-16 мкг, 14,8 мкг, 30,2 мкг и 30,8 мкг соответственно. Эти величины ТD50 были в 2-3 раза выше, чем эффективные величины IC50 для противосудорожной эффективности (см. пример 10 выше); отмечали явное разделение между эффективной и токсической дозами.
Пример 17: Различение РСР
В этом тесте крысы, которые были приучены поднимать рычагом пресс для пополнения запаса корма, должны выбрать, который из двух рычагов в их клетках является правильным. Единственным стимулом, который они имеют для выбора правильного рычага, является их способность обнаруживать, получили они инъекцию РСР или инъекцию носителя. После приблизительно двух месяцев тренировки крысы начинают очень хорошо отличать РСР от инъекций носителя и после этого их можно тестировать с другими лекарственными средствами для определения, могут ли они их отличать как РСР. При тестировании по этому способу РСР заменяют другими лекарственными средствами, которые, как известно, производят РСР-подобную интоксикацию. Эти лекарственные средства включают различные аналоги РСР, такие как кетамин, и неконкурентный антагонист NMDA-рецептора, МК-801.
Соединение 1 (1-30 мг/кг i.p.) не могло заменять РСР и, следовательно, не имело РСР-подобных эффектов различения стимула. При 30 мг/кг i.p. только одно из 7 испытанных животных вообще отвечало на тот или другой рычаг. Таким образом, очевидно, что был оценен диапазон доз соединения 1, эффективных для поведения животных. Поскольку считают, что способность тест-соединений производить РСР-подобные эффекты в крысах предсказывает их способность производить РСР-подобную психотомиметическую активность и склонность к злоупотреблению соединением у людей, эти результаты предполагают, что арилалкиламины, такие как соединение 1, не будут иметь таких неблагоприятных побочных действий у людей.
Пример 18
Введение таких соединений, как РСР и МК-801, крысам производит нейротоксическое действие, названное нейронной вакуолизацией. После одной дозы таких соединений вакуоли были найдены в конкретных центральных нейронах, особенно в нейронах коры района пояса и в ретросплениальной коре. Такой вакуолизации не было в крысах, получивших соединение 1 в единственной высокой дозе 100 мг/кг i.p.
Взятые вместе результаты по опорно-двигательной активности, ухудшению двигательной способности, различению РСР и нейронной вакуолизации предполагают, что арилалкиламины не будут иметь побочных действий у людей.
(b) Нарушение познавательной способности
Одна из главных причин постулирования роли NMDA-рецепторов в памяти и способности к познанию находится в результатах клеточных исследований по долгосрочному потенциированию (LTP) в гиппокампе крыс. LTP представляет собой длительно сохраняющееся увеличение размеров синаптических ответных реакций, продуцируемое кратким, но интенсивным синаптическим стимулированием. С момента открытия этого явления оно стало выдающейся клеточной моделью способности усвоения в мозгу позвоночных (Teyler and Discenna, Long-term potentiation, Annu. Rev. Neurosci. 10: 131, 1987). Передача возбуждения при синапсах, образуемых коллатералями Шаффера на пирамидных клетках СА1, медиируется рецепторами NMDA и АМРА. После короткого вызывающего тетаническое сокращение стимула величина потенциала действия (меры синаптической трансмиссии) сильно увеличивается и остается такой в течение часов. Было показано, что все известные конкурентные и неконкурентные антагонисты NMDA-рецепторов ингибируют LTP в гиппокампе крыс, тогда как антагонисты рецепторов не-NMDA не имеют такого действия (Collingridge and Davis, In: The NMDA Receptor, IRL Press, p.123, 1989). Это подтверждает роль NMDA-рецепторов в памяти и познавательной способности.
Пример 19: Тест LTP
Действия выбранных арилалкиламинов и взятых из литературы стандартов испытывали на срезах гиппокампа крыс. Как и ожидалось, все стандартные конкурентные (АР5 и АР7) и неконкурентные (МК-801 и ифенпродил) антагонисты NMDA-рецептора ингибировали индукцию LTP в гиппокампе. Срезы гиппокампа крысы сливали в течение 30-60 минут с тест-соединением перед подачей вызывающего тетаническое сокращение стимула, состоящего из 3 импульсов, разделяемых 500 мс, 100 Гц в течение 1 с каждый. Ответную амплитуду наблюдали еще в течение 15 минут после тетанического сокращения. Вызывающий тетаническое сокращение стимул вызывал в среднем 95%-ное увеличение в величине синаптического ответа. Индукция LTP значимо ингибировалась (р<0,05) конкурентными (АР5, АР7) или неконкурентными (МК-801, ифенпродил) антагонистами NMDA-рецептора. Совершенно неожиданно, ни один из испытанных арилалкиламинов (соединение 1, соединение 2, соединение 3 и другие) не ингибировал индукцию LTP (р>0,05), даже при высоких концентрациях (100-300 мкМ), которые вызывали некоторое ингибирование контрольного ответа.
Эти результаты выдвигают на первый план еще одну уникальную и важную особенность арилалкиламинов. Арилалкиламины представляют собой первый и в настоящее время единственный класс соединений, которые, как было показано, являются селективными и сильнодействующими антагонистами NMDA-рецептора, которые не ингибируют индукции LTP. Это, по-видимому, отражает новый механизм и сайт действия арилалкиламинов и предполагает, что лекарственные средства, которые нацелены на новый сайт на NMDA-рецепторе, не будут также оказывать влияния на LTP. Поскольку LTP представляет собой первичную клеточную модель для познавательной способности и памяти в ЦНС млекопитающих, это дополнительно предполагает, что такие лекарственные средства не будут иметь вредных побочных действий на познавательную способность.
Пример 20: Тесты на обучаемость
Предварительные эксперименты с использованием одного или нескольких сильнодействующих синтетических аналогов арилалкиламина, соединения 3, в примере обучаемости - in vivo, показывают, что эти лекарственные средства не действуют на познавательную активность. В этом тесте крыс дрессировали на чередующиеся повороты в Т-лабиринте для поощрения кормом. Для сравнения в эксперимент включали МК-801. Тест-соединения вводили i.p. за 15 минут перед тестированием. Контрольные животные делали правильный выбор приблизительно 80% времени. Увеличение доз МК-801 прогрессивно снижало правильные выборы, и это снижение в правильном поведении сопровождалось повышенной активностью. В противоположность этому соединение 3 не ухудшало способность животных делать правильный выбор (р>0,05). При наивысших испытанных дозах соединение 3 вызвало некоторое ухудшение в опорно-двигательной активности, в точности противоположный эффект наблюдали с МК-801.
Хотя МК-801 снижал познавательную способность параллельно с увеличениями опорно-двигательной активности, другие исследования с применением различных примеров на грызунах и приматах показали четкую связь между действием на обучаемость и опорно-двигательную активность. Так, как конкурентные, так и неконкурентные антагонисты NMDA-рецептора ухудшают познавательную способность при дозах, которые не вызывают какого-либо явного изменения в двигательном поведении. Это показывает, что обычные антагонисты NMDA-рецептора ухудшают обучаемость независимо от других побочных эффектов. Результаты теста с Т-лабиринтом демонстрируют, что соединение 3 и другие арилалкиламины не ухудшают познавательную способность даже при дозах, которые вызывают некоторое снижение в опорно-двигательной активности.
Из этих тестов на познавательную способность вытекает одно дополнительное наблюдение. Первый ответ животных на второй день тестирования был случайным и, следовательно, не зависел от последнего ответа тестирования в предыдущий день. Так, контрольные животные делали точно первый выбор приблизительно 50% времени. МК-801 не влияет на этот первый выбор. Однако животные с введенным соединением 3 в предыдущий день делали первый выбор правильно более часто. Затем в противоположность контрольным животным животные, получившие соединение 3, вели себя, как если бы они помнили о последнем выборе в предыдущий день.
Во второй серии экспериментов оценивали действие соединения 4 на обучаемость в водном лабиринте Морриса. В этом тесте скрытую платформу помещали в фиксированном положении в круговом стальном резервуаре и погружали ее на 2 см ниже поверхности воды. Каждой крысе давали 3 испытания в день с 10-минутным интервалом между испытаниями в течение 5 дней. Испытание начинали помещением крысы в воду носом к стенке резервуара в одном из трех заранее определенных исходных местоположений. Порядок начального местоположения варьировали ежедневно. Обучаемость измеряли как уменьшение времени, требующегося для того, чтобы крыса доплыла до платформы. Если животное не могло найти платформу в пределах 60 секунд после начала испытания, крысу направляли к платформе рукой. Животные, остающиеся на платформе в течение 10 секунд, удалялись из резервуара. Через 10 минут после последнего обучающего испытания на 5-й день животных подвергали пробному тесту. Платформу удаляли для задачи этого первого испытания и животным позволяли плавать в течение 60 секунд для оценки пространственного смещения для определения местоположения платформы. Из этой задачи регистрировали два измерения: латентный период до первого пересечения зоны, где была платформа, и общее число пересечений. Каждая крыса получала всего 5 инъекций соединения 4. В первой серии экспериментов соединение 4 вводили при 10 мг/кг i.p. ежедневно в течение 5 дней. Этот режим обработки ухудшал обучаемость; однако эти животные испытывали значительную потерю веса и необычные поведенческие признаки ("дрожание", ухудшение двигательной способности, затруднения в плавании) с повторяемым дозированием соединения 4. В следующем исследовании шесть животных получали 1 мг/кг i.p. в течение первых 4 дней обучения, тогда как два животных получали 5 мг/кг i.p. во время этого периода. В последний день обучения обе группы получили 10 мг/кг. Ни животные с 1 мг/кг, ни животные с 5 мг/кг не обнаруживали какого-либо ухудшения в обучении нахождению местоположения скрытой платформы, и конечная доза 10 мг/кг не производила какого-либо ухудшения в способности животного выполнять уже выученную задачу.
Результаты этих обучающих задач являются обнадеживающими. Они предполагают, что арилалкиламины не вызывают недостаточности познавательной способности и памяти, что является типичным для других антагонистов NMDA-рецептора. Фактически существует предположение, что арилалкиламины могут даже быть ноотропиками (усилителями памяти).
(с) Сердечно-сосудистые эффекты
Исследования in vivo с некоторыми арилалкиламинами обнаружили гипотензивное действие этих соединений, особенно при высоких дозах. На основании этих результатов проводили систематическое исследование действий арилалкиламинов на сердечно-сосудистую функцию.
Пример 21: Ингибирование Са2+-каналов
Мы обнаружили, что некоторые арилалкиламины являются сильнодействующими ингибиторами потенциал-зависимых Са2+-каналов, в частности тех, которые чувствительны к ингибированию дигидропиридинами (каналы L-типа). Можно было бы ожидать, что такие действия на гладкие мышцы сосудов расширяют кровеносные сосуды и вызывают снижения кровяного давления, создавая гипотензию.
Способность арилалкиламинов ингибировать дигидропиридин-чувствительные Са2+-каналы испытывали в мозжечковых зернистых клетках и линии клеток гладких мышц аорты крысы, A7r5. В мозжечковых зернистых клетках соединение 2 ингибировало индуцированные деполяризацией увеличения [Ca2+]i при концентрациях, превышающих в 100 раз концентрации, необходимые для ингибирования ответов на NMDA (величины IС50 24 мкМ и 161 нМ соответственно). В общем, мы наблюдали широкий диапазон активностей против потенциал-зависимых Са2+-каналов, которые не коррелируют с активностью против NMDA-рецепторов. Это позволяет предположить, что дальнейшая работа по выяснению связи структуры и функции на основе химической модификации молекулы арилалкиламина приведет к созданию соединений, которые являются очень сильнодействующими антагонистами NMDA с низкой активностью против потенциал-зависимых Са2+-каналов. Действительно, соединение 1 (с IC50 102 нМ против медиируемых NMDA-рецептором ответов в мозжечковых зернистых клетках) является относительно слабым ингибитором потенциал-чувствительных Са2+-каналов в мозжечковых зернистых клетках (IС50=257 мкМ) и фактически не действует на потенциал-чувствительный приток Са2+ в клетки А7r5 (IС50=808 мкМ).
Однако арилалкиламины не являются недискриминирующими ингибиторами потенциал-зависимых Са2+-каналов. Например, они не влияют на потенциал-зависимые Са2+-каналы в мозжечковых клетках Purkinje (каналы Р-типа) или каналы, которые считают участвующими в высвобождении нейротрансмиттеров (N-каналы). Арилалкиламины, которые действительно ингибируют потенциал-зависимые Са2+-каналы, по-видимому, нацелены специфически на Са2+-каналы L-типа. Кроме того, как упоминалось выше, существует высокая степень структурной специфичности этого действия. Например, один арилалкиламин в 57 раз более активен, чем другой арилалкиламин, в блокировании притока Са2+ через каналы L-типа, причем единственным структурным различием между этими соединениями является присутствие или отсутствие гидроксильной группы.
Пример 22: Сердечно-сосудистые исследования in vivo
Арилалкиламины соединения 1 и соединения 2 вызывают умеренные падения (20-40 мм рт. ст.) в среднем артериальном кровяном давлении (МАВР) в анестезированных крысах при дозах, которые являются эффективными в моделях инсульта in vivo (10-30 мг/кг s.c.). Гипотензивное действие соединения 4 оценивали более детально. Соединение 4 вызывало заметное снижение (40 мм рт. ст.) в среднем артериальном давлении, которое сохранялось в течение приблизительно 90-120 минут после введения при дозе 10 мг/кг i.p.; это было в той же самой группе крыс, в которой соединение 4 дало значительную нейрозащиту в модели с шовными нитями окклюзии средней мозговой артерии (см. пример 8 выше). Сходные результаты получали в исследованиях на крысах, в которых соединение 4 демонстрировало нейропротекторную активность в фототромботической модели с красителем Rose Bengal фокального ишемического удара (см. пример 8 выше). Дальнейшие исследования с использованием препаратов спинного мозга крыс сильно предполагают, что гипотензивная (понижающая артериальное давление) активность соединения 4 представляет собой опосредованный периферически эффект. Снижение артериального давления и брадикардия, вызываемые соединением 4, сохранялись у крыс, предобработанных атропином, что позволяет предположить, что эти эффекты не опосредованы холинергическим механизмом. Подобным образом, соединение 4 вызывало снижение артериального давления и брадикардию у химически десимпатизированных крыс (предобработанных ганглионарным ингибитором), что предполагает, что эти эффекты не медиируются через симпатическую нервную систему.
На основании этих открытий ожидается, что химические воздействия будут снижать сердечно-сосудистые побочные действия (1) усилением поглощения арилалкиламина в мозг, так что более низкие дозы будут необходимы для нейрозащиты, и (2) увеличением селективности (соотношения активности) арилалкиламинов в отношении оперируемых рецепторами Са2+-каналов в сравнении с потенциал-зависимыми Са2+-каналами.
Пример 23: Биологическая активность соединения 19 и аналогов
Соединения 19-139 обладают высокой активностью против NMDA-индуцируемых увеличений [Ca2+]i в крысиных мозжечковых зернистых клетках, выращенных в культуре (Таблица 1). Ингибирующее действие соединения 19 на ответы на NMDA было неконкурентным. Соединения 19-147 ингибировали связывание [3H]MK-801 в мембранах, полученных из тканей гиппокампа и коры головного мозга крысы (Таблица 1).
Соединение 19 обладало следующими дополнительными биологическими активностями: значимой (р<0,05 в сравнении с контролем) противосудорожной активностью против максимальных индуцируемых электрошоком эпилептических припадков у мышей после введения i.p. (ED50=26,4 мг/кг и TD50 (вращающийся стержень) = 43,8 мг/кг); значимой противосудорожной активностью против максимальных индуцируемых электрошоком эпилептических припадков у мышей после перорального (р.о.) введения (ED50=35 мг/кг), но ухудшением моторной активности при 30 мг/кг; значимой аналгезирующей активностью в тестах с горячей пластинкой и индуцируемого PBQ скорчивания при 16 мг/кг i.p.; отсутствием РСР-подобного стереотипического поведения (повышенной возбудимости и кручения головой из стороны в сторону) при 30 мг/кг i.p. у крыс; отсутствием обобщения с РСР в тесте различения РСР у крыс при дозах до поведенчески активной дозы 30 мг/кг i.p. Соединение 19 было значительно менее активным в противодействии увеличениям [Са2+]i, индуцируемым деполяризующими концентрациями КСl в крысиных мозжечковых зернистых клетках (IС50=10,2 мкМ), и отсутствием действия на кровяное давление при введении s.c. у крыс при дозах до 100 мг/кг. Однако соединение 19 не ингибировало индукцию LTP в срезах гиппокампа крыс при тестировании при 100 мкМ.
Соединение 20 обладало следующими дополнительными биологическими активностями; значимой противосудорожной активностью против максимальных индуцируемых электрошоком эпилептических припадков у мышей после введения i.p. (ED50=20,1 мг/кг и ТD50 (вращающийся стержень) = 20,6 мг/кг); отсутствием значимой противосудорожной активности против максимальных индуцируемых электрошоком эпилептических припадков у мышей после перорального (р.о.) введения при дозах до 30 мг/кг, но ухудшением моторной активности при 30 мг/кг; значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. (ED50=2,1 мг/кг и ТD50=19,9 мг/кг) и перорального введения (ED50=9,7 мг/кг и TD50=21,8 мг/кг); значимой противосудорожной активностью против индуцируемых максимальным электрошоком эпилептических припадков у крыс после перорального (р.о.) введения с величиной ED50 33,64 мг/кг и величиной TD50 55, 87 мг/кг; увеличением порога припадка, как показано внутривенным Metrazol-тестом на мышах при дозе 10 мг/кг i.p.; значимой нейропротекторной активностью в крысиной модели временной фокальной ишемии (51% уменьшение объема инфаркта после введения двух доз 1 мг/кг i.p., причем первую дозу вводят сразу же после окклюзии средней мозговой артерии, а вторую - спустя 6 часов); 43%-ным уменьшением объема инфаркта после введения двух доз 1 мг/кг i.p., причем первую дозу вводят через 2 часа после окклюзии средней мозговой артерии (т.е. при времени реперфузии), а вторую - спустя 6 часов; значимой нейропротекторной активностью (24%-ное уменьшение объема инфаркта) в крысиной модели перманентной фокальной ишемии после введения 1 мг/кг i.p. при 30 минутах и еще раз 4 часах после окклюзии; значимой нейропротекторной активностью (50%-уменьшение объема инфаркта) в крысиной фототромботической модели фокальной ишемии после введения 10 мг/кг i.p. при 15 мин, 3 ч и еще 6 ч после окклюзии; отсутствием значимой аналгезирующей активности при дозе 25 мг/кг i.p. в тесте горячей 52° С пластинки у крыс или в крысином тесте 48° С отдергивания хвоста; значимой аналгезирующей активностью, не ингибируемой антагонистом опиатного рецептора налоксоном, в крысином формалиновом тесте при дозе 10 мг/кг i.p.; значимой аналгезирующей активностью, не ингибируемой налоксоном, против индуцируемых уксусной кислотой брюшных корчей у мышей при дозе 10 мг/кг i.p.; отсутствием обобщения с РСР в тесте различения РСР у крыс при дозах до поведенчески активной дозы 10 мг/кг i.p.; отсутствием нейронной вакуолизации в крысах при введении при дозах 10 и 30 мг/кг i.p.; отсутствием значимой сердечно-сосудистой активности в анестезируемых крысах при дозах до 15 мкмоль/кг i.v. или 10 мг/кг i.p.; отсутствием сердечно-сосудистой активности в находящихся в сознании коротконогих гончих собаках при дозах 0,3 или 1 мг/кг i.v. (60 с болюсная инъекция); временными увеличениями среднего артериального давления и частоты сердечных сокращений у находящихся в сознании коротконогих гончих собак при дозе 3 мг/кг i.v., с большей величиной и большей продолжительностью действия при дозе 10 мг/кг i.v. (60 с болюсная инъекция); увеличенной двигательной активностью, возбуждением и тревогой, легким тремором, облизыванием рта, скулением и мочеиспусканием находящихся в сознании собак при дозе 3 мг/кг i.v. (60 с болюсная инъекция); расширенными зрачками, тремором всего тела, потерей координации, облизыванием рта, слюноотделением, учащенным дыханием, быстрым миганием глаз, скулением, тревогой, эпилептическими припадками и смертью находящихся в сознании коротконогих гончих собак при дозе 10 мг/кг i.v. (60 с болюсная инъекция); отсутствием поведенческих эффектов у находящихся в сознании самцов мышей NMRI при дозах 2 и 4 мг/кг i.p.; возбуждением и повышенной реактивностью при прикосновении к ним у находящихся в сознании самцов мышей NMRI при дозе 8 мг/кг i.p.; возбуждением, Straub-хвостом, тремором, стереотипиями, гипотермией и мидриазом у находящихся в сознании самцов мышей NMRI при дозах 16 и 32 мг/кг i.p.; судорогами и смертью находящихся в сознании самцов мышей NMRI при дозе 64 мг/кг i.p.; судорогами и смертью находящихся в сознании самцов мышей NMRI при дозах 128 и 256 мг/кг i.p.; отсутствием поведенческих эффектов у находящихся в сознании самцов крыс Wistar при дозе 2 мг/кг i.v.; возбуждением, стереотипиями, повышенной реактивностью на прикосновение, увеличенным мышечным тонусом и тремором у находящихся в сознании самцов крыс Wistar при дозах от 4 до 16 мг/кг i.v.; Straub-хвостом, конвульсиями и смертью находящихся в сознании самцов крыс Wistar при дозе 32 мг/кг i.v.
Соединение 21 обладало следующими дополнительными биологическими активностями: значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. (ЕD50=3,41 мг/кг и TD50 (ухудшение моторной активности) = 15,3 мг/кг).
Соединение 33 (энантиомер соединения 21) обладало следующими дополнительными, биологическими активностями: значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. введения (ЕD50=4,6 мг/кг и ТD50 (ухудшение моторной активности) = 27,8 мг/кг); значимой противосудорожной активностью против индуцируемых максимальным электрошоком эпилептических припадков у крыс после перорального (р.о.) введения при дозе 25 мг/кг с отсутствием моторной токсичности, наблюдаемой при этой дозе; значимой нейропротекторной активностью в крысиной модели фокального ишемического инсульта после введения i.p. 2 мг/кг за 30 минут перед окклюзией сосуда и 2 мг/кг спустя 3 часа после окклюзии; отсутствием значимой аналгезирующей активности при дозе 25 мг/кг i.p. у крыс в тесте горячей (52° С) пластинки или в тесте 48°С отдергивания хвоста у крыс; значимой аналгезирующей активностью в крысиной модели хронической невропатической боли после введения i.th. доз от 15 до 80 мкг; значимой аналгезирующей активностью в крысиной модели хронической невропатической боли после введения i.p. доз 3-10 мг/кг; отсутствием нейронной вакуолизации при введении крысам при дозе 30 мг/кг i.p.; отсутствием сердечно-сосудистой активности у анестезированных крыс при дозах до 3 мг/кг i.v.; отсутствием значимой сердечно-сосудистой активности в находящихся в сознании коротконогих гончих собаках при дозе 0,3 мг/кг i.v. (60 с болюсная инъекция); временными увеличениями среднего артериального давления у находящихся в сознании коротконогих гончих собак при дозе 1 мг/кг i.v., с большей величиной и большей продолжительностью эффектов при дозах 3-10 мг/кг i.v. (60 с болюсная инъекция); временным увеличением частоты сердечных сокращений у находящихся в сознании коротконогих гончих собак при дозе 10 мг/кг i.v. (60 с болюсная инъекция); облизыванием рта у находящихся в сознании коротконогих гончих собак при дозе 3 мг/кг i.v. (60 с болюсная инъекция); расширенными зрачками, тремором всего тела, потерей координации, облизыванием рта, слюноотделением, учащенным дыханием у находящихся в сознании коротконогих гончих собак при дозе 10 мг/кг i.v. (60 с болюсная инъекция); отсутствием значимых вызываемых лекарственным средством изменений в ЭКГ у находящихся в сознании коротконогих гончих собак при дозе 10 мг/кг i.v. (60 с болюсная инъекция); отсутствием поведенческих эффектов у находящихся в сознании самцов мышей NMRI при дозах 2 и 4 мг/кг i.p.; возбуждением и повышенной реактивностью при прикосновении к ним у находящихся в сознании самцов мышей NMRI при дозе 8 мг/кг i.p.; возбуждением, Straub-хвостом, тремором, стереотипиями, гипотермией и мидриазом у находящихся в сознании самцов мышей NMRI при дозах 16 и 32 мг/кг i.p.; судорогами находящихся в сознании самцов мышей NMRI при дозе 64 мг/кг i.p.; судорогами и смертью находящихся в сознании самцов мышей NMRI при дозах 128 и 256 мг/кг i.p.
Соединение 34 (энантиомер соединения 21) обладало следующими дополнительными биологическими активностями: значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. введения (ED50=22 мг/кг и TD50 (ухудшение моторной активности) между 10 и 15 мг/кг); гипотермией у находящихся в сознании самцов мышей NMRI при дозе 2 мг/кг i.p.; отсутствием поведенческих эффектов у находящихся в сознании самцов мышей NMRI при дозе 4 мг/кг i.p.; возбуждением и повышенной реактивностью при прикосновении к ним и гипотермией у находящихся в сознании самцов мышей NMRI при дозе 8 мг/кг i.p.; возбуждением, Straub-хвостом, тремором, вздрагиваниями, стереотипиями, гипотермией и мидриазом у находящихся в сознании самцов мышей NMRI при дозах 16 и 32 мг/кг i.p.; судорогами находящихся в сознании самцов мышей NMRI при дозе 64 мг/кг i.p.; судорогами и смертью находящихся в сознании самцов мышей NMRI при дозах 128 и 256 мг/кг i.p.
Соединение 22 обладало следующими дополнительными биологическими активностями: значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. введения (ED50=4,9 мг/кг и TD50 (перевернутая сетка) = 26,8 мг/кг) и перорального ведения (ED50 =5,1 мг/кг и LD50=18,3 мг/кг) и отсутствием значимой сердечно-сосудистой активности в анестезируемых крысах при дозах до 15 мкмоль/кг (4,47 мг/кг) i.v.
Соединение 50 (энантиомер соединения 22) обладало следующими дополнительными биологическими активностями: значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. введения (ЕD50=2,7 мг/кг и TD50 (ухудшение моторной активности) = 17,4 мг/кг); значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после р.о. введения (ED50=9,0 мг/кг и ТD50 (ухудшение моторной активности) = 18,9 мг/кг); значимой противосудорожной активностью против индуцируемых максимальным электрошоком эпилептических припадков у крыс после перорального (р.о.) введения с ED50=28 мг/кг и TD50=20 мг/кг; значимой нейропротекторной активностью в крысиной модели фокального ишемического инсульта после введения i.p. 2 мг/кг за 30 минут перед окклюзией сосуда и 2 мг/кг спустя 3 часа после окклюзии; отсутствием значимой аналгезирующей активности при дозе 25 мг/кг i.p. у крыс в тесте горячей (52° С) пластинки или в тесте 48° С отдергивания хвоста у крыс и отсутствием значимой сердечно-сосудистой активности у анестезированных крыс при дозах до 5 мг/кг i.v.
Соединение 51 (энантиомер соединения 22) обладало следующими дополнительными биологическими активностями: значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. введения (ЕD50=9,1 мг/кг и TD50 (ухудшение моторной активности) = 13,6 мг/кг).
Соединение 24 обладало следующими дополнительными биологическими активностями: значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. введения (ED50=5 мг/кг и TD50 (ухудшение моторной активности) = 16 мг/кг); значимой противосудорожной активностью против индуцируемых максимальным электрошоком эпилептических припадков у крыс после перорального введения с ED50=46 мг/кг и TD50 =51 мг/кг; отсутствием значимой нейропротекторной активности в крысиной модели фокального ишемического инсульта после введения i.p. 2 мг/кг за 30 минут перед окклюзией сосуда и 2 мг/кг спустя 3 часа после окклюзии и отсутствием значимой сердечно-сосудистой активности у анестезированных крыс при дозах до 10 мг/кг i.v.
Соединение 25 обладало следующими дополнительными биологическими активностями: значимой противосудорожной активностью против индуцируемых максимальным электрошоком эпилептических припадков у крыс после i.p. введения с ЕD50=12,47 мг/кг и TD50=32,18 мг/кг; значимой противосудорожной активностью против индуцируемых максимальным электрошоком эпилептических припадков у крыс после перорального введения с ED50=46,43 мг/кг и TD50 между 163 и 326 мг/кг.
Соединение 31 обладало следующими дополнительными биологическими активностями: значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. введения (ED50=6 мг/кг и ТD50 (ухудшение моторной активности) между 10 и 20 мг/кг).
Соединение 46 обладало следующими дополнительными биологическими активностями: значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. введения (ЕD50=25 мг/кг и TD50 (ухудшение моторной активности) между 18 и 21 мг/кг); и отсутствием значимой нейропротекторной активности в крысиной модели фокального ишемического инсульта после введения i.p. 2 мг/кг за 30 минут перед окклюзией сосуда и 2 мг/кг спустя 3 часа после окклюзии.
Соединение 57 обладало следующими дополнительными биологическими активностями: значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. введения (ED50=1 мг/кг и TD50 (ухудшение моторной активности) между 6 и 8 мг/кг).
Соединение 58 обладало следующими дополнительными биологическими активностями: значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. введения (ED50=0,9 мг/кг и TD50 (ухудшение моторной активности) = 14,5 мг/кг); отсутствием значимой нейропротекторной активности в крысиной модели фокального ишемического инсульта после введения i.p. 2 мг/кг за 30 минут перед окклюзией сосуда и 2 мг/кг спустя 3 часа после окклюзии и отсутствием значимой сердечно-сосудистой активности у анестезированных крыс при дозах до 2 мг/кг i.v.
Соединение 59 обладало следующими дополнительными биологическими активностями: значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. введения (ED50=2,7 мг/кг и ТD50 (ухудшение моторной активности) = 7,8 мг/кг); уменьшением порога припадка, как показано внутривенным Metrazol-тестом на мышах при дозе 11,7 мг/кг i.p.; отсутствием значимой нейропротекторной активности в крысиной модели фокального ишемического инсульта после введения i.p. 2 мг/кг за 30 минут перед окклюзией сосуда и 2 мг/кг спустя 3 часа после окклюзии и отсутствием значимой сердечно-сосудистой активности у анестезированных крыс при дозах до 10 мг/кг i.v.
Соединение 60 обладало следующими дополнительными биологическими активностями: значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после i.p. введения (ED50=4,4 мг/кг и TD50 (ухудшение моторной активности) = 9,2 мг/кг); значимой противосудорожной активностью против индуцируемых звуком эпилептических припадков в модели рефлекторной эпилепсии генетически восприимчивых мышей (мышей Frings) после перорального введения (ED50=10 мг/кг и TD50 (ухудшение моторной активности) = 25,6 мг/кг); значимой противосудорожной активностью против индуцируемых максимальным электрошоком эпилептических припадков у крыс после i.p. введения (ED50=8,17 мг/кг и TD50 (вращающийся стержень) = 17,30 мг/кг); отсутствием действия на порог припадка, как показано внутривенным Metrazol-тестом на мышах при дозах 8 и 17 мг/кг i.p.; значимой нейропротекторной активностью в крысиной модели временного фокального ишемического инсульта после введения i.p. 2 мг/кг за 30 минут перед окклюзией сосуда и 2 мг/кг спустя 3 часа после окклюзии; значимой нейропротекторной активностью в крысиной модели временного фокального ишемического инсульта после введения i.p. или i.v. 1 мг/кг спустя 2 часа и еще спустя 8 часов после окклюзии; значимой нейропротекторной активностью в крысиной модели временного фокального ишемического инсульта после введения i.p. или i.v. 1 мг/кг спустя 2 часа после окклюзии; отсутствием значимой нейропротекторной активности в крысиной фототромботической модели фокальной ишемии после введения 10 мг/кг i.p. при 15 мин, 3 ч и 6 ч после окклюзии; отсутствием нейронной вакуолизации при введении при дозах 20 мк/кг i.p. или 10 мг/кг i.v.; отсутствием сердечно-сосудистой активности в находящихся в сознании коротконогих гончих собаках при дозе 0,3 мг/кг i.v. (60 с болюсная инъекция); временными увеличениями среднего артериального давления у находящихся в сознании коротконогих гончих собак при дозах 1 и 3 мг/кг i.v., с большей величиной и большей продолжительностью эффектов при дозе 10 мг/кг i.v. (60 с болюсная инъекция); временными увеличениями частоты сердечных сокращений у находящихся в сознании коротконогих гончих собак при дозах 3 и 10 мг/кг i.v. (60 с болюсная инъекция); отсутствием значимых изменений в ЭКГ у находящихся в сознании коротконогих гончих собак при дозах от 0,3 до 10 мг/кг i.v. (60 с болюсная инъекция); отсутствием поведенческих эффектов у находящихся в сознании коротконогих гончих собак при дозах от 0,3 до 1 мг/кг i.v. (60 с болюсная инъекция); небольшим увеличением в частоте дыхания у находящихся в сознании коротконогих гончих собак при дозе 3 мг/кг i.v. (60 с болюсная инъекция); расширением зрачков, тремором всего тела, слюноотделением и мочеиспусканием у находящихся в сознании коротконогих гончих собак при дозе от 0,3 до 10 мг/кг i.v. (60 с болюсная инъекция); отсутствием значимых поведенческих эффектов у самцов крыс Wistar при дозах до 4 мг/кг i.v.; возбуждением, стереотипиями, повышенной реактивностью на прикосновение, увеличенным мышечным тонусом и тремором у находящихся в сознании самцов крыс Wistar при дозе 8 мг/кг i.v.; Straub-хвостом, конвульсиями и смертью находящихся в сознании самцов крыс Wistar при дозе 16 мг/кг i.v.
Взятые вместе результаты, полученные с этими упрощенными синтетическими арилалкиламинами, предполагают, что такие упрощенные молекулы не взаимодействуют специфически с сайтом связывания арилалкиламинов на оперируемом рецепторами Са2+ -каналах, как соединения 1, 2 и 3. Конкретно, соединения 19-147 связываются с сайтом, меченным [3H]MK-801, при концентрациях приблизительно в 1-400 раз более высоких, чем концентрации, которые противодействуют функции NMDA-рецептор-ионофорного комплекса. Однако факт, что соединения 19-147 при терапевтических дозах, как правило, не вызывают РСР-подобного стереотипического поведения, не заменяют РСР в тестах различения РСР и не вызывают нейронной вакуолизации, предполагает, что такие соединения могут быть применимы либо в качестве исходных соединений, либо в качестве предполагаемых лекарственных средств для неврологических нарушений и заболеваний. Сообщалось, что соединения, которые связываются с низкой активностью (по сравнению с МК-801) с сайтом, меченным [3H]МК-801, могут иметь терапевтическую применимость и обладать более благоприятным профилем побочных эффектов, чем соединения, которые обладают высокоаффинной антагонистической активностью, такие как сам МК-801 (Rogawski, Therapeutic potential of excitatory amino acid antagonists: channel blockers and 2,3-benzodiazepines. Trends Pharmacol. Sci. 14:325, 1993). Низкая аффинность некоторых соединений в рамках группы Соединений 19-147 (в сравнении с МК-801) в отношении сайта, меченного [3H]MK-801, может служить основанием для помещения этих соединений в общий класс неконкурентных антагонистов низкой аффинности.
Идентификация нового модуляторного сайта на оперируемых рецепторами кальциевых каналах
При наличии идентифицированных арилалкиламинов, имеющих полезные свойства, описанные выше, теперь могут быть идентифицированы соединения, которые действуют при критическом сайте связывания арилалкиламинов на оперируемых рецепторами Са2+-каналах, таких как присутствующие в комплексах NMDA-рецептора, АМРА-рецептора и никотинового холинергического рецептора с ионофором.
Далее приводятся примеры подходящих для этого тестов.
Пример 24: Связывание радиолиганда в коре головного мозга или мозжечке крысы
Следующий далее тест может использоваться в качестве высокопроизводительного теста для скрининга библиотек продуктов (например, библиотек природных продуктов и списков соединений в основных фармацевтических компаниях) для идентификации новых классов соединений с активностью при этом уникальном сайте арилалкиламинов. Затем эти новые классы соединений используют в качестве химических исходных (основных) структур для программы создания лекарственных средств, связывающихся с сайтом связывания арилалкиламинов на оперируемых рецепторами Са2+-каналах. Соединения, идентифицируемые этим тестом, предоставляют новый терапевтический подход для лечения неврологических нарушений или заболеваний. Примеры таких соединений включают соединения, представленные общими химическими формулами выше. Могут быть выполнены рутинные эксперименты для идентификации соединений, имеющих желательные активности.
Мембраны мозга крыс получают согласно способу Williams et al. (Effects of polyamines on the binding of [3H]MK-801 to the NMDA receptor: Pharmacological evidence for the existence of polyamine recognition site. Molec. Pharmacol. 36: 575, 1989) со следующими изменениями: самцов крыс Sprague-Dawley (Harlan Laboratories) весом 100-200 г умерщвляют обезглавливанием. Кору головного мозга или мозжечок из 20 крыс очищают и отделяют. Полученную мозговую ткань гомогенизируют при 4° С политронным гомогенизатором при самой низкой установке скорости в 300 мл 0,32 М сахарозы, содержащей 5 мМ К-ЭДТА (рН 7,0). Гомогенат центрифугируют в течение 10 минут при 1000 g и супернатант удаляют и центрифугируют при 30000 g в течение 30 минут. Полученный осадок ресуспендируют в 250 мл 5 мМ К-ЭДТА (рН 7,0), перемешивают на льду в течение 15 минут и затем центрифугируют при 30000 g в течение 30 минут. Осадок ресуспендируют в 300 мл 5 мМ К-ЭДТА (рН 7,0) и инкубируют при 32° С в течение 30 минут. Затем суспензию центрифугируют при 100000 g в течение 30 минут. Мембраны промывают ресуспендированием в 500 мл 5 мМ К-ЭДТА (рН 7,0), инкубируют при 32° С в течение 30 минут и центрифугируют при 100000 g в течение 30 минут. Процедуру промывки, в том числе 30-минутную инкубацию, повторяют. Конечный осадок ресуспендируют в 60 мл 5 мМ К-ЭДТА (рН 7,0) и хранят в виде аликвот при -80° С. Интенсивная процедура промывки, используемая в этом тесте, предусмотрена для минимизации концентрации глутамата и глицина (коагонистов при NMDA-рецептор-ионофорном комплексе), присутствующих в препарате мембран.
Для проведения теста связывания с [3 H]арилалкиламином, аликвоты SPM (синаптических плазматических мебран) оттаивают, ресуспендируют в 30 мл 30 мМ EPPS/1 мМ К-ЭДТА, рН 7,0, и центрифугируют при 100000 g в течение 30 минут. SPM ресуспендируют в буфере А (30 мМ EPPS/1 мМ К-ЭДТА, рН 7,0). К этой реакционной смеси добавляют [3H] арилалкиламин. Тесты связывания проводят в полипропиленовых тест-пробирках. Конечный инкубационный объем равен 500 мкл. Неспецифическое связывание определяют в присутствии 100 мкМ нерадиоактивного арилалкиламина. Пробы в двух повторностях инкубируют при 0° С в течение 1 часа. Тесты останавливают добавлением 3 мл охлажденного льдом буфера А с последующим фильтрованием через фильтры из стекловолокна (Schieicher and Schuel №30), которые предварительно пропитывают в 0,33% полиэтиленимине (PEI).
Фильтры промывают еще 3× 3 мл буфера А и радиоактивность определяют в сцинтилляционном счетчике при эффективности 35-40% для3H.
Для подтверждения надежности этого теста проводят следующие эксперименты:
(a) Количество неспецифического связывания [3Н]арилалкиламина с фильтрами определяют пропусканием 500 мкл буфера А, содержащего различные концентрации [3H]арилалкиламина, через предварительно пропитанные фильтры из стекловолокна. Фильтры промывают 4× 3 мл буфера А и радиоактивность, связанную с фильтрами, определяют сцинтилляционным счетом при эффективности 35-40% для3H. Было найдено, что в фильтрах, непредобработанных 0,33% PEI, 87%3 H-лиганда было связано с фильтром. Предварительное пропитывание 0,33% PEI уменьшает неспецифическое связывание до 0,5-1,0% от всего добавленного лиганда.
(b) Кривую насыщения конструируют ресуспендированием SPM в буфере А. Тест-буфер (500 мкл) содержит 60 мкг белка. Используют концентрации [3H] арилалкиламина от 1,0 нМ до 400 мкМ в полулогарифмических единицах. Кривую насыщения строят из этих данных и кажущуюся величину КD и величину Вmax определяют согласно анализу Скетчарда (Scatchard, The attractions of proteins for small molecules and ions. Ann. N. Y. Acad. Sci. 51: 660, 1949). Кооперативность связывания [3H]арилалкиламина определяют построением графика Хилла (Hill, A new mathematical treatment of changes of ionic concentration in muscle and nerve under the action of electric currents, with a theory to their mode of excitation. J. Physiol. 40: 190, 1910).
(c) Зависимость связывания от концентрации белка (рецептора) определяют ресуспендированием SPM в буфере А. Тест-буфер (500 мкл) содержит концентрацию [3H]арилалкиламина, равную его величине КD, и увеличивающиеся концентрации белка. Специфическое связывание [3H]арилалкиламина должно линейно соотноситься с количеством присутствующего белка (рецептора).
(d) Временной ход лиганд-рецепторного связывания определяют ресуспендированием SPM в буфере А. Тест-буфер (500 мкл) содержит концентрацию [3H]арилалкиламина, равную его величине КD, и 100 мкг белка. Пробы в двух повторностях инкубируют при 0° С в течение различных периодов времени; определяют время, при котором достигается равновесие, и эту временную точку рутинно используют во всех последующих анализах.
(e) Фармакология сайта связывания может быть проанализирована конкурентными экспериментами. В таких экспериментах концентрация [3H]арилалкиламина и количество белка являются постоянными, тогда как концентрацию тестируемого (конкурирующего) лекарственного средства варьируют. Этот тест позволяет определить IС50 и кажущуюся КD для конкурирующего лекарственного средства (Cheng and Prusoff, Relationship between the inhibitionconstant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. J. Biochem. Pharmacol. 22: 3099, 1973). Кооперативность связывания конкурирующего лекарственного средства определяют анализом согласно графику Хилла.
Специфическое связывание [3H] арилалкиламина представляет собой связывание с новым сайтом на оперируемых рецепторами Са2+-каналах, таких как комплексы NMDA-рецептора, АМРА-рецептора и никотинового холинергического рецептора с ионофором. Как таковые, другие арилалкиламины должны конкурировать со связыванием [3H]арилалкиламина конкурентным образом, и их активности в этом тесте должны коррелировать с их ингибиторной активностью в функциональном тесте антагонизма оперируемым рецептором Са2+-каналам (например, ингибированием индуцируемых NMDA-рецептором увеличении [Са2+]i в культурах крысиных мозжечковых зернистых клеток). Напротив, соединения, которые имеют активность при других известных сайтах оперируемых рецепторами Са2+-каналов (например, МК-801, Мg2+, полиамины), не должны вытеснять связывание [3H]арилалкиламина конкурентным образом. Скорее, можно ожидать существование сложного аллостерического модулирования связывания [3H]арилалкиламина, свидетельствующего о неконкурентных взаимодействиях. В предварительных экспериментах МК-801 не вытеснял связывания [3H]арилалкиламина при концентрациях до 100 мкМ.
(f) Исследования для оценки кинетики диссоциации проводят путем измерения связывания [3 H]арилалкиламина после установления равновесия (см. (d) выше), и большой избыток нерадиоактивного конкурирующего лекарственного средства добавляют к реакционной смеси. Затем тестируют связывание [3H]арилалкиламина при различных интервалах времени. При помощи такого анализа определяют скорости ассоциации и диссоциации связывания [3H]арилалкиламина (Titeler, Multiple Dopamine Receptors: Receptor Binding Studies In Dopamine Paharmacology. Marcel Dekker, Inc., New York, 1983). Дополнительные эксперименты включают варьирование температуры реакции (0° С-37° С) для понимания температурной зависимости этого параметра.
Пример 25: Связывание радиолиганда в мозжечковых зернистых клетках
Первичные культуры мозжечковых зернистых нейронов получают из 8-дневных крыс и высевают на квадраты пластика Aclar, покрытые поли-L-лизином. Пластиковые квадраты помещают в 24-луночные культуральные планшеты и в каждую лунку добавляют приблизительно 7,5× 105 зернистых клеток. Культуры поддерживают в среде Игла (HyClone Laboratories), содержащей 25 мМ КСl, 10% фетальную телячью сыворотку (HyClone Laboratories), 2 мМ глутамин, 100 мкг/мл гентамицина, 50 Е/мл пенициллина и 50 мкг/мл стрептомицина, при 37° С в увлажненной атмосфере 5% СO2 в воздухе в течение 24 часов перед добавлением цитозинарабинозида (10 мкМ, конечная концентрация). Никаких изменений культуральной среды не делают до тех пор, пока клетки не используют для исследования связывания рецептора через 6-8 дней после посева.
Для выполнения анализа связывания с [3H]арилалкиламином реакционная смесь состоит из 200 мкл буфера А (20 мМ K-HEPES, 1 мМ К-ЭДТА, рН 7,0) в каждой лунке 24-луночного планшета. К этой реакционной смеси добавляют [3H]арилалкиламин. Неспецифическое связывание определяют в присутствии 100 мкМ нерадиоактивного арилалкиламина. Пробы в трех повторностях инкубируют при 0° С в течение 1 часа. Тесты останавливают мануальным выскребанием клеток из квадратов Aclar и помещением их в полипропиленовые тест-пробирки. Мембраны, полученные из целых клеток таким образом, суспендируют в 10 мл охлажденного льдом буфера А и фильтруют через фильтры из стекловолокна (Schleicher and Schuell №30), которые предварительно пропитывают еще 3× 3 мл буфера А, и радиоактивность на фильтрах определяют сцинтилляционным счетом при эффективности 35-40% для3H. Тест может быть остановлен центрифугированием (вместо фильтрования) для уменьшения неспецифического связывания.
Характерные эксперименты для характеристики и подтверждения надежности этого теста проводили в основном, как описано выше, за исключением того, что вместо мембран для исходного связывания использовали клетки. Этот тест связывания позволяет определить величину IC50 и кажущуюся Ко для конкурирующего лекарственного средства, как описано, при помощи анализа Скетчарда (Scatchard, The attractions of proteins for small molecules and ions. Ann. N.Y. Acad. Sci. 51: 66.0, 1949). Кооперативность связывания конкурирующего лекарственного средства определяют Hill plot-анализом (Hill, A new mathematical treatment of changes of ionic concentration in muscle and nerve under the action of electric currents, with a theory to their mode of excitation. J. Physiol. 40: 190, 1910). Специфическое связывание [3H]арилалкиламина представляет собой связывание с новым сайтом на оперируемых рецепторами кальциевых каналах.
Пример 26: Тест связывания рекомбинантного рецептора
Здесь представлен один пример теста быстрого скрининга на ценные соединения данного изобретения. В этом тесте при помощи стандартных процедур получают клон кДНК или гена, кодирующего сайт связывания арилалкиламина (рецептор), из подходящего организма, такого как человек. Отдельные фрагменты этого клона экспрессируют в подходящем экспрессирующем векторе для получения самого маленького полипептида (полипептидов), получаемых из этого рецептора, которые сохраняют способность связывать соединение 1, соединение 2 или соединение 3. Таким путем могут быть идентифицированы полипептид или полипептиды, которые включают в себя новый рецептор арилалкиламина для этих соединений. Такие эксперименты могут быть облегчены путем использования стабильно трансфицированной линии клеток млекопитающего (например, клеток НЕК 293), экспрессирующих рецептор арилалкиламина.
Альтернативно, рецептор арилалкиламина может химически реагировать с химически модифицированным соединением 1, соединением 2 или соединением 3 таким образом, что аминокислотные остатки рецептора арилалкиламина, которые контактируют с выбранным соединением (или находятся вблизи него), модифицируются и в результате этого становятся идентифицируемыми. Фрагмент или фрагменты рецептора арилалкиламина, содержащие аминокислоты, которые, как определено, взаимодействуют с соединением 1, соединением 2 или соединением 3 и достаточны для связывания с этими молекулами, могут быть затем рекомбинантно экспрессированы, как описано выше, с использованием стандартного экспрессирующего вектора (или векторов).
Рекомбинантный полипептид или полипептиды, имеющие желаемые связывающие свойства, могут быть связаны с твердофазным носителем при помощи стандартных химических процедур. Эта твердая фаза, или аффинный матрикс, может быть затем приведена в контакт с соединением 1, соединением 2 или соединением 3 для демонстрации того, что эти соединения могут связываться с этой колонкой, и для идентификации условий, при помощи которых эти соединения могут быть удалены из твердой фазы. Затем эта процедура может быть повторена с использованием большой библиотеки соединений для определения тех соединений, которые способны связываться с этим аффинным матриксом и затем могут быть высвобождены способом, одинаковым со способом, использованным для соединения 1, соединения 2 или соединения 3. Однако могут быть использованы альтернативные условия связывания и высвобождения, отличающиеся от используемых для связывания арилалкиламина (например, условия, которые лучше имитируют физиологические условия, встречающиеся, в частности, в патологических состояниях). Таким образом, соединения, которые действительно связываются, могут быть выбраны из очень большой коллекции соединений, присутствующих в жидкой среде или экстракте.
После идентификации соединений, способных связываться с описанными здесь арилалкиламинсвязывающим полипептидом или полипептидами, эти соединения могут быть легко испытаны в различных тестах, описанных выше, для определения, являются ли они или их простые производные ценными соединениями для терапевтического лечения неврологических нарушений и заболеваний, описанных выше.
В другом способе нативный рецептор арилалкиламина может быть связан с колонкой или иным твердофазным суппортом. Затем могут быть идентифицированы соединения, которые не вытесняются конкурентно реагентами, связывающимися с другими сайтами на этом рецепторе. Такие соединения определяют новые сайты на этом рецепторе. Соединения, которые конкурентно вытесняются другими известными соединениями, связываются, следовательно, с известными сайтами или связываются с новыми сайтами, которые перекрываются с известными сайтами связывания. Тем не менее такие соединения могут структурно отличаться от известных соединений и, следовательно, могут определять новые химические классы агонистов или антагонистов, которые могут быть применимы в качестве терапевтических веществ. В заключение можно отметить, что конкурентный анализ может быть использован для идентификации ценных соединений данного изобретения.
Пример 27: Электрофизиологический "пэтч-кламп" (patch-clamp) тест
Следующий далее тест выполняют для отобранных соединений, идентифицированных в вышеупомянутых тестах связывания радио-лиганда как взаимодействующие высокоэффективно и конкурентно при новом сайте связывания арилалкиламинов на оперируемых рецепторами Са2+-каналах, таких, какие присутствуют в комплексах NMDA-рецептора, АМРА-рецептора или никотинового холинергического рецептора с ионофором. Этот "пэтч-кламп″ -тест обеспечивает дополнительные данные о сайте и механизме действия указанных предварительно выбранных соединений. Конкретно, определяют следующие фармакологические и физиологические свойства соединений, взаимодействующих при сайте связывания арилалкиламинов, с использованием комплекса NMDA-рецептор-ионофор в качестве примера оперируемых рецепторами Са2+-каналов: активность и эффективность в ингибировании опосредованных NMDA-рецептором ионных потоков, неконкурентную природу ингибирования в отношении глутамата и глицина, зависимость действия от использования, потенциал-зависимость действия, и то, и другое относительно появления ингибирования и обратимости ингибирования, кинетику блокирования и деблокирования (обратимости) и механизм блокирования открытого канала. Такие данные подтверждают, что эти соединения, взаимодействующие при сайте связывания арилалкиламинов, сохраняют уникальный биологический профиль арилалкиламинов и не имеют их первичной активности при известных сайтах на NMDA-рецептор-ионофорном комплексе (глутаматсвязывающем сайте, глицинсвязывающем сайте, МК-801-связывающем сайте, Мg2+-связывающем сайте, Zn2+-связывающем сайте, сигма-связывающем сайте, полиамин-связывающем сайте).
"Пэтч-кламп″ -регистрации нейронов млекопитающего (клеток гиппокампа, кортикальных клеток, мозжечковых зернистых клеток) проводят с использованием стандартных процедур (Donevan et al., Arraine blocks N-methyl-D-aspartate receptor responses by an open channel mechanism: whole-cell and single-channel recording studies in cultured hippocampal neurons. Molec. Pharmacol. 41: 727, 1992; Rock and Macdonald, Spermine and related polyamines produce a voltage-dependent reduction of NMDA receptor single-channel conductance. Molec. Pharmacol. 42: 157, 1992).
Альтернативно, "пэтч-кламп″ -эксперименты могут быть выполнены на ооцитах Xenopus или на стабильно трансфицированной клеточной линии млекопитающего (например, на клетках НЕК 293), экспрессирующих специфические субъединицы оперируемых рецепторами Са2+-каналов. Таким путем, например, могут быть определены активность и эффективность при различных подтипах рецептора глутамата (например, NMDAR1, NMDAR2A - NMDAR2D, GluR1 - GluR4). Дополнительная информация относительно сайта действия арилалкиламинов на этих подтипах рецепторов глутамата может быть получена с использованием сайт-направленного мутагенеза.
Пример 28: Синтез арилалкиламинов
Арилалкиламины, такие как соединение 1, соединение 2 и соединение 3, синтезируют стандартными процедурами (Jasys et al., The Total synthesis of argiotoxins 636, 659 and 673. Tetrahedron Lett. 29: 6223, 1988; Nason et al., Synthesis of neurotoxic Nephila spider venoms: NSTX-3 and JSTX-3. Tetrahedron Lett. 30: 2331, 1989). Характерные примеры синтеза аналогов арилалкиламинов 4-18 представлены в ожидающей одновременного рассмотрения Application U. S. Serial No. 08/485038, filed June 7, 1995 и ожидающей одновременного рассмотрения International Patent Application No. PCT/US94/12293, опубликованной как WO/95/21612, filed October 26, 1994, включенных здесь в качестве ссылок во всей их полноте.
Пример 29: Синтез упрощенных арилалкиламинов
Синтез соединения 20 выполняли следующим образом. Раствор гидрида натрия (1,21 г, 50 ммоль) в диметоксиэтане обрабатывали диэтилцианометилфосфонатом (8,86 г, 50 ммоль) и реакционную смесь перемешивали в течение 4 часов при комнатной температуре. К ней добавляли 3,3'-дифторбензофенон (10 г, 46 ммоль) в ДМФ. Реакционную смесь перемешивали в течение 24 часов при комнатной температуре, реакцию гасили Н2O и распределяли между диэтиловым эфиром и водой. Эфирную фракцию сушили над Na2SO4, концентрировали. ГХ/МС этого материала показали 90% продукта А и 10% исходного бензофенона.
Раствор этого материала в этаноле с каталитическим количеством Pd(OH)2 гидрировали при 55 psi в атмосфере водорода в течение 4 часов при комнатной температуре. Реакционную смесь фильтровали и катализатор промывали этанолом (3х). Фильтрат и этанольные промывки объединяли и концентрировали. ГХ/МС этого материала показали 90% продукта В и 10% исходного бензофенона.
Раствор этого материала в ТГФ обрабатывали 70 мл 1 М В2Н6(70 ммоль) в ТГФ и нагревали с обратным холодильником в течение 1 часа. После охлаждения реакционную смесь обрабатывали 6 н. НСl (50 мл) и нагревали с обратным холодильником еще в течение 1 часа. После охлаждения реакционную смесь подщелачивали до рН 14 10 н. NaOH и уравновешивали эфиром. Эфирный слой удаляли и промывали 10% НСl (3х). Кислотные промывки объединяли, подщелачивали до рН 14 10 н. NaOH и экстрагировали дихлорметаном (3х). Органические промывки объединяли, сушили над Na2SO4 с получением масла. ГХ/МС этого материла показали 100% соединение 20. GC/EI-MS (Rf=7,11 мин) m/z (относительная интенсивность) 247 (М+, 31), 230 (100), 215 (30), 201 (52), 183 (63), 134 (23), 121 (16), 101 (21), 95 (15), 77 (15). Этот материал в диэтиловом эфире фильтровали и обрабатывали 35 мл 1 М НСl в эфире. Осадок собирали, сушили и перекристаллизовывали из смеси вода-этанол с получением 1, 045 г соединения 20 в виде гидрохлоридной соли.1H-ЯМР (СDСl3) δ 8,28 (3H, ушир. с), 7,28-7,17 (2Н, м), 7,02-6,86 (6Н, м), 4,11 (1Н, т, J=8 Гц), 2,89 (2Н, ушир. т, J=8 Гц), 2,48 (2Н, ушир. т, J=7 Гц);13С-ЯМР (СDСl3) δ 164,6, 161,3, 144,8, 144,7, 130,4, 130,3, 123,3, 123,2, 114,7, 114,5, 114,1, 113,8, 47,4, 38,4, 32,7.

Синтез соединения 21, соединения 33 и соединения 34 проводили следующим образом.
Круглодонную колбу на 100 мл, оборудованную стержнем мешалки, перегородкой и источником азота, загружали соединением 1 (2,43 г, 10 ммоль) в 30 мл ТГФ. Раствор охлаждали до -78° С и обрабатывали по каплям 11 мл литий-бис(триметилсилил)амида (1 М в ТГФ) (11 ммоль). Реакционную смесь перемешивали при -78° С в течение 30 минут и обрабатывали по каплям избытком йодметана (3,1 мл, 50 ммоль). Реакционную смесь перемешивали в течение 30 минут при -58° С. GC/EI-MS-анализ аликвоты из реакционной смеси показал использование исходного нитрила 1. Реакцию гасили водой, разбавляли диэтиловым эфиром и переносили в делительную воронку. Эфирный слой промывали 10% НСl (3х), насыщенным солевым раствором (1х), сушили безводным МgSO4 и концентрировали до коричневого масла. Этот материал перегоняли (Kugelrohr, 100° C) при пониженном давлении с получением 1,5 г прозрачного масла. GC/EI-MS этого материала показал, что он содержит желаемый продукт 2. (Rf =7,35 мин) m/z (отн. инт.) 257 (М+, 3), 203 (100), 183 (59), 170 (5), 133 (4), 109 (3);1 H-ЯМР (CDCl3) δ 7,4-6,9 (8Н, м), 4,01 (1H, Д, J=10 Гц), 3,38 (1Н, дкв, J=7, 10 Гц), 1,32 (3Н, д, J=7 Гц);13С-ЯМР (CDCl3) δ 19,4, 30,5, 54,2, 114,5, 114,6, 114,7, 114,9, 115,0, 115,3, 123,3, 123,4, 123,6, 123,7, 130,5, 130,6, 131,7.
Продукт 3 синтезировали каталитическим восстановлением продукта 2 с применением никеля Ренея в смеси 95:5 EtOH:водный гидроксид натрия (2 экв.) при 60 psi (413685,42 Па) водорода. GC/EI-MS (Rt=7,25 мин) m/z (отн. инт.) 261 (М+, 20), 244 (35), 229 (16), 215 (17), 201 (80), 183 (100), 133 (42), 115 (27), 109 (47), 95 (20);1H-ЯМР (CDCl3) δ , 7,3-6,8 (8Н, м), 3,62 (1H, д, J=10 Гц), 2,70 (1H, м), 2,40 (2Н, м), 1,73 (2Н, м), 0,91 (3Н, д, J=7 Гц). Заметьте, что продукт 3 в этой реакции соответствует соединению 21.
Продукт 2 в 10% IPA-гексане (100 мг/мл) хроматографировали в виде аликвот по 500 мкл, через колонку Chiral Cel OD (2,0× 25 см) с использованием 10% IPA-гексана при 10 мл/мин с измерением оптической плотности при 254 нм. Это давало два оптически чистых энантиомера 4 и 5 (как определено аналитической хиральной ВЭЖХ; заметьте, что стереохимия этих двух соединений не была определена в это время). Эти два соединения были идентичны по их GC/EI-MS и1H-ЯМР-спектрам продукту 2 (данные, представленные выше).
Каждый из энантиомеров 4 и 5 восстанавливали отдельно с применением диметилсульфидборанового комплекса следующим образом. Раствор соединения (4 или 5) в ТГФ нагревали для кипячения с обратным холодильником, обрабатывали избытком (2 экв.) 1 М (в ТГФ) диметилсульфидборанового комплекса и реакционную смесь нагревали с обратным холодильником в течение 30 минут. После этого времени реакционную смесь охлаждали до 0° С и обрабатывали 6 н. НСl. Реакционную смесь нагревали с обратным холодильником в течение 30 минут. После этого времени реакционную смесь переносили в делительную воронку, подщелачивали до рН>12 10 н. NaOH и продукт (6 или 7) экстрагировали эфиром. Эфирный слой промывали насыщенным солевым раствором, сушили над безводным МgSO4 и концентрировали до масла. Продукт очищали препаративной ТСХ с использованием смеси 5% метанол-хлороформ. Было обнаружено, что каждый из индивидуальных энантиомеров (6 и 7) был идентичен по GC/EI-MS и1H-ЯМР-спектрам продукту 3 (данные, представленные выше). Заметьте, что продукты 6 и 7 в этой схеме соответствуют соединениям 33 и 34. Соединение 33· НСl: т.пл. = 260-270° С (разл.), [а]36526=+6,6 (с 1,0 в EtOH), [а]D26=+0,4 (с 1,0 в EtOH). Соединение 34· НСl: [а]36523=-6,1 (с 1,0 в EtOH), [a]D23=-0,1 (с 1,0 в EtOH). Соединение 33· HI: Свободное основание соединения 33 растворяли в EtOH и добавляли 47% йодистоводородную кислоту (1,1 экв.). Растворитель выпаривали в вакууме и полученный твердый гидройодид перекристаллизовывали дважды из смеси гептан/ЕtOАс медленным выпариванием: т.пл. 195-197° С. Было определено, что абсолютная конфигурация соединения 33· HI является R согласно рентгеноструктурному анализу отдельного кристалла (моноклинного игольчатого бесцветного кристалла, 0,50× 0,05× 0,03 мм) с применением дифрактометра Siemens R3m/V (3887 наблюдаемых отражений).


Синтез соединения 22 проводили, как описано ниже. Соединение 23 синтезировали аналогичным образом.
Триэтилфосфоноацетат (17,2 г, 76,8 ммоль) медленно добавляли к суспензии гидрида натрия (3,07 г, 76,8 ммоль) в N, N-диметилформамиде (350 мл). После 15 минут к раствору добавляли 3,3'-дифторбензофенон (15,2 г, 69,8 ммоль) и перемешивали еще в течение 18 часов. Реакцию гасили водой и распределяли между водой и эфиром. Объединенный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали в вакууме с получением 19,7 г этил-3, 3-бис(3-фторфенил)акрилата в виде желтого масла.
К раствору этил-3,3-бис(3-фторфенил)акрилата (19,7 г, 68,4 ммоль) в 200 мл этанола добавляли гидроксид палладия на угле (3,5 г). Смесь встряхивали при 60 psi (413685,42 Па) водорода в течение 3 часов, затем фильтровали и выпаривали в вакууме с получением 19,5 г продукта А в виде бесцветного масла.
Этиловый эфир А (19, 2 г) гидролизовали перемешиванием в течение 6 дней с 50 мл 10 н. гидроксида натрия. Затем реакционную смесь разбавляли 50 мл воды и подкисляли до рН 0 концентрированной НСl. Водную смесь экстрагировали 3 раза эфиром и эфирные экстракты сушили над сульфатом магния и выпаривали с получением 3,3-бис(3-фторфенил)пропионовой кислоты в виде белого порошка.
3, 3-бис(3-фторфенил)пропионовую кислоту (13 г, 49,6 ммоль) растворяли в 50 мл (685 ммоль) тионилхлорида и перемешивали в течение ночи при комнатной температуре. Избыток тионилхлорида удаляли в вакууме на роторном испарителе с получением 13,7 г продукта В в виде желтого масла.
К хлорангидриду В (13,7 г, 49 ммоль), растворенному в 100 мл сухого ТГФ, добавляли ацетилацетонат железа (III) (0,52 г, 1,47 ммоль). Затем добавляли метилмагнийхлорид (16,3 мл, 49 ммоль) на протяжении периода 1 ч шприцевым насосом. Реакционную смесь перемешивали еще в течение 1 часа, затем гасили выливанием ее в смесь эфир/5% НСl. Эфирный слой отделяли, промывали 5% НСl и насыщенным NaCl и сушили над сульфатом натрия. Растворитель выпаривали в вакууме с получением 4, 4-бис(3-фторфенил)-2-бутанона в виде желтого масла. Неочищенный продукт очищали на силикагеле, элюируя смесью гептан/этилацетат.
К 4,4-бис(3-фторфенил)-2-бутанону (5,7 г, 21,9 ммоль) в 25 мл этанола добавляли пиридин (1,91 г, 24,1 ммоль) и гидрохлорид метоксиламина (2,01 г, 24,1 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем выливали в эфир/5% НСl. Эфирный слой отделяли, промывали 5% НСl и насыщенным NaCl и сушили над сульфатом натрия. Растворитель выпаривали в вакууме с получением 6,26 г O-метилоксима 4,4-бис(3-фторфенил)-2-бутанона. К боргидриду натрия (4,1 г, 108,3 ммоль) в 15 мл ТГФ медленно добавляли тетрахлорид циркония (6,31 г, 27,1 ммоль). Эту смесь перемешивали в течение 15 минут, затем добавляли указанный оксим (6,26 г, 21, 7 ммоль) в 6 мл ТГФ на протяжении 5 минут. После 3 часов перемешивания при комнатной температуре реакционную смесь обрабатывали медленным добавлением 50 мМ гидроксида натрия и затем эфира. Водный слой экстрагировали 4 раза эфиром и объединенные эфирные экстракты сушили над сульфатом натрия. Растворитель выпаривали в вакууме с получением 5,3 г соединения 22.

Синтез соединения 24 проводили, как описано ниже. Соединения 25-29, 52-53, 65, 76-78, 83, 90, 96-97, 115 и 135-136 получали аналогичным образом.
Суспензию магниевых стружек (0,95 г, 39,2 ммоль) в 150 мл безводного диэтилового эфира обрабатывали 1-бром-3-фторбензолом (6,83 г, 39,2 ммоль) по каплям через шприц. После 1,5 ч раствор переносили при помощи канюли в колбу, содержащую о-анисовый альдегид (5,0 г, 36,7 ммоль) в 100 мл безводного диэтилового эфира при 0° С и перемешивали в течение 2 часов. Реакцию гасили водой и распределяли между водой и эфиром. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния с получением 7,90 г (выход 93%) продукта А.
Дихромат пиридиния (16,0 г, 42,5 ммоль) добавляли к раствору спирта А (7,90 г, 34,0 ммоль) в дихлорметане (100 мл) и реакционную смесь перемешивали в течение 12 часов. Диэтиловый эфир (300 мл) добавляли к реакционной смеси и черный раствор фильтровали через пробку из силикагеля (30 см) и промывали дополнительно 500 мл эфира. После выпаривания растворителя в вакууме твердое вещество перекристаллизовывали из ацетона с получением 7,45 г (выход 95%) продукта В.
Диэтилцианометилфосфонат (7,0 г, 39,5 ммоль) медленно добавляли к суспензии гидрида натрия (1,58 г, 39,5 ммоль) в 100 мл N,N-диметилформамида. После 30 минут к раствору добавляли кетон В и перемешивали еще в течение 2 часов. Реакцию гасили водой и распределяли между водой и эфиром. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали в вакууме с получением бледно-желтого масла.
В стеклянной бомбе масло растворяли в 100 мл этанола и 20 мл 10 н. NaOH. К раствору добавляли каталитическое количество никеля Ренея, суспендированного в воде (~15 мол.%). Реакционную смесь встряхивали при 60 psi (413685,42 Па) Н2 в течение 12 часов в гидрогенизаторе Парра. После отфильтровывания избытка никеля Ренея раствор экстрагировали хлороформом. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. После фильтрования масло пропускали через колонку с силикагелем в хлороформе и метаноле. Растворитель выпаривали в вакууме с получением бледно-желтого масла. GC/EI-MS (Rt=8,10 мин) m/z (отн. интенсивность) 259 (100), 242 (44), 213 (48), 183 (42), 136 (50), 109 (94), 91 (60), 77 (25). Затем масло подкисляли хлористым водородом в диэтиловом эфире. Выпаривание эфира давало бледно-желтое твердое вещество, которое перекристаллизовывали в горячем ацетонитриле с получением 3,45 г (выход 42,1%) белых игольчатых кристаллов соединения 24 в виде гидрохлоридной соли.

Соединения 101 и 103 синтезировали из соединений 25 и 24 соответственно расщеплением их O-метиловых эфиров трибромидом борана обычным способом.
Синтез соединения 30 выполняли, как описано ниже. Соединение 31 получали аналогичным образом.
Суспензию, содержащую магниевые стружки (0,95 г, 39,1 ммоль) в 150 мл безводного диэтилового эфира обрабатывали 1-бром-3-фторбензолом (6,85 г, 39,1 ммоль) по каплям через шприц. После 1,5 ч раствор переносили при помощи канюли в колбу, содержащую 3-хлорбензальдегид (5,0 г, 35,6 ммоль) в 100 мл безводного диэтилового эфира при 0° С и перемешивали в течение 2 часов. Реакцию гасили водой и распределяли между водой и эфиром. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния с получением 8,40 г (выход >99%) продукта А.
Хлорхромат пиридиния (15,0 г, 39,8 ммоль) добавляли к раствору спирта А (8,40 г, 35,5 ммоль) в дихлорметане (100 мл) и реакционную смесь перемешивали в течение 18 часов. Диэтиловый эфир (300 мл) добавляли к реакционной смеси и черный раствор фильтровали через пробку из силикагеля (30 см) и промывали дополнительно 500 мл эфира. После выпаривания растворителя твердое вещество перекристаллизовывали из ацетона с получением 6,31 г (выход 76%) продукта В.
Диэтилцианометилфосфонат (5,2 г, 29,6 ммоль) медленно добавляли к суспензии гидрида натрия (1,2 г, 29,6 ммоль) в 100 мл N,N-диметилформамида. После 30 минут к раствору добавляли кетон В и перемешивали еще в течение 6 часов. Реакцию гасили водой и распределяли между водой и эфиром. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали в вакууме с получением бледно-желтого масла.
В стеклянной бомбе масло растворяли в 100 мл этанола и 20 мл 10 н. NaOH. К раствору добавляли каталитическое количество родия, суспендированного на оксиде алюминия (~35 мол.%). Реакционную смесь встряхивали при 60 psi (413685,42 Па) Н2 в течение 24 часов в гидрогенизаторе Парра. После отфильтровывания избытка катализатора раствор экстрагировали хлороформом. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. После фильтрования и выпаривания растворителя в вакууме масло поглощали тетрагидрофураном (100 мл). Добавляли диборан (23,4 мл, 1,0 М) и раствор нагревали с обратным холодильником в течение 1,5 часов. Растворитель выпаривали в вакууме и осторожно добавляли 6 н. НСl (50 мл). Раствор нагревали с обратным холодильником в течение 1 часа. После охлаждения смесь подщелачивали 10 н. NaOH до рН 14 и распределяли между дихлорметаном и водой. Объединенные органические слои сушили над безводным сульфатом магния и фильтровали. После выпаривания растворителя желтое масло пропускали через колонку с силикагелем в хлороформе и метаноле. Растворитель выпаривали в вакууме с получением желтого масла. GC/EI-MS (Rt=8,15 мин) m/z (отн. интенсивность) 263 (17), 246 (21), 211 (84), 196 (33), 183 (100), 165 (19), 133 (19). Масло затем подкисляли хлористым водородом в диэтиловом эфире. Выпаривание эфира давало 0,96 г белого твердого вещества, соединения 30 в виде гидрохлоридной соли.

Синтез соединения 35 проводили, как описано ниже. Соединения 36-37 получали аналогичным образом.
Раствор 3-фторбензальдегида (3,0 г, 24,2 ммоль) при 0° С в 150 мл диэтилового эфира обрабатывали 3,0 М этилмагнийхлоридом (12,7 мл, 25,4 ммоль) в тетрагидрофуране (ТГФ) через шприц. После 4 часов реакцию гасили водой и распределяли между водой и эфиром. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния с получением 4,25 г продукта А.
Хлорхромат пиридиния (6,53 г, 30,3 ммоль) добавляли к раствору А в дихлорметане (100 мл) и перемешивали в течение 18 часов. Диэтиловый эфир (300 мл) добавляли к реакционной смеси и черный раствор фильтровали через пробку из силикагеля (30 см) и промывали дополнительно 500 мл эфира. После выпаривания растворителя твердое вещество перекристаллизовывали из ацетона с получением 3,05 г продукта В. Растворитель выпаривали в вакууме с получением бледно-желтого масла.
Диэтилцианометилфосфонат (4,7 г, 26,4 ммоль) медленно добавляли к суспензии гидрида натрия (1,1 г, 26,4 ммоль) в 100 мл N,N-диметилформамида. После 30 минут к раствору добавляли кетон В и перемешивали еще в течение 6 часов. Реакцию гасили водой и распределяли между водой и эфиром. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали в вакууме с получением бледно-желтого масла.
В стеклянной колбе масло растворяли в 100 мл этанола и 20 мл 10 н. NaOH. К раствору добавляли каталитическое количество никеля Ренея, суспендированного в воде (~15 мол.%). Реакционную смесь встряхивали при 60 psi (413685,42 Па) H2 в течение 24 часов в гидрогенизаторе Парра. После отфильтровывания избытка никеля Ренея раствор экстрагировали хлороформом. Объединенные органические слои промывали насыщенным солевым раствором и сушили над безводным сульфатом магния. После фильтрования масло пропускали через колонку с силикагелем в хлороформе и метаноле. Растворитель выпаривали в вакууме с получением бледно-желтого масла. GC/EI-MS (Rt=3,45 мин) m/z (отн. интенсивность) 167 (4), 150 (63), 135 (56), 109 (100), 96 (53), 75 (48). Затем масло подкисляли хлористым водородом в диэтиловом эфире. Выпаривание эфира давало бледно-желтое твердое вещество, которое перекристаллизовывали в горячем ацетонитриле с получением 2,2 г соединения 35 в виде гидрохлоридной соли.

Синтез соединения 38 выполняли, как описано ниже.
К раствору 3,3-бис(3-фторфенил)пропионитрила (1,5 г, 6,17 ммоль) в 250 мл ТГФ при -70° С добавляли бутиллитий (4,25 мл в гексане, 6,8 ммоль) при помощи шприца на протяжении 5 минут. Раствор перемешивали в течение 5 минут, затем добавляли метилйодид (1,75 г, 12,3 ммоль) на протяжении 1 минуты. Затем реакционной смеси давали нагреться до комнатной температуры и обрабатывали ее разбавлением эфиром и промыванием 5% НСl и водой. Эфирный слой сушили над сульфатом натрия и выпаривали с получением 1,5 г метилированного нитрила в виде желтого масла.
К 3,3-бис(3-фторфенил)-2-метил-пропионитрилу (1,46 г, 5,7 ммоль) в 50 мл дихлорметана при 0° С добавляли диизобутил-алюминийгидрид (1,02 мл, 5,7 ммоль) при помощи шприца на протяжении 10-минутного периода. Реакционную смесь перемешивали в течение 30 минут при 0° С и дополнительно 2 часа при комнатной температуре. Реакционную смесь обрабатывали добавлением 200 мл 10% НСl и перемешиванием при 40° С в течение 30 минут с последующей экстракцией продукта дихлорметаном. Органический слой сушили над сульфатом натрия и выпаривали с получением 1,36 г продукта А.
К раствору альдегида А (1,36 г, 5,23 ммоль) в 40 мл эфира при 0° С добавляли метилмагнийбромид (5,23 мл в эфире, 5,23 ммоль). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре и затем гасили разбавленной НСl. Эфирный слой отделяли, сушили над сульфатом натрия и выпаривали с получением 1,48 г 4, 4-бис (3-фторфенил)-3-метилбутан-2-ола.
К раствору этого спирта (1,4 г, 5,07 ммоль) в 300 мл дихлорметана добавляли хлорхромат пиридиния (1,2 г, 5,58 ммоль) и смесь перемешивали в течение ночи. Затем реакционную смесь разбавляли 100 мл эфира и фильтровали через пробку из силикагеля. Растворитель выпаривали с получением 1,39 продукта В.
Кетон В (1,3 г, 4,9 ммоль) добавляли к раствору гидрохлорида метоксиламина (0,45 г, 5,38 ммоль) и пиридина (0,44 мл, 5,38 ммоль) в 30 мл этанола и перемешивали в течение ночи. Затем этанол выпаривали и остаток растворяли в эфире и 10% НСl. Эфирный слой отделяли, промывали один раз 10% НСl, сушили над сульфатом натрия и выпаривали с получением 1,4 г O-метилоксима.
К суспензии боргидрида натрия (0,87 г, 23,1 ммоль) в 5 мл ТГФ добавляли тетрахлорид циркония (2,35 г, 5,8 ммоль) и раствор перемешивали в течение 15 минут с последующим добавлением еще 5 мл ТГФ. Затем добавляли O-метилоксим (1,4 г, 4,6 ммоль) в 5 мл ТГФ и смесь перемешивали в течение ночи. ТГФ удаляли выпариванием в вакууме и остаток обрабатывали 10% гидроксидом натрия. После прекращения выделения пузырьков добавляли эфир и слои разделяли. Водный слой экстрагировали 4 раза эфиром и объединенные эфирные экстракты сушили над сульфатом натрия. Эфир выпаривали с получением 1,25 г соединения 38.

Соединение 32 и соединения 39-53 синтезировали согласно стандартным процедурам, описанным выше.
Соединения 107, 116, 139 и 143 получали в качестве синтетических промежуточных продуктов, используемых в получении соединений 32, 115, 20 и 25 соответственно.
Соединение 50 также получали с применением хирального синтеза, описанного ниже.
К охлажденному на льду раствору N-бензил-(S)-α -метилбензиламина (18,0 г, 85,2 ммоль) в ТГФ (75 мл) добавляли бутиллитий (2,5 М в гексане) (37,5 мл, 93,8 ммоль) при помощи шприца на протяжении периода 10 минут при такой скорости, чтобы поддерживать температуру реакции ниже 10° С во время добавления. Затем реакционную смесь перемешивали при 0° С в течение 15 минут. Реакционную смесь охлаждали до -78° С в бане со смесью сухой лед/изопропанол и затем раствор бензилкротоната (15,0 г, 85,2 ммоль) в ТГФ (100 мл) добавляли по каплям на протяжении периода 45 минут. Реакционную смесь перемешивали при -78° С в течение 15 минут и затем добавляли насыщенный раствор NH4Cl (50 мл). Затем реакционную смесь быстро переносили в делительную воронку, содержащую насыщенный раствор NaCl (500 мл) и эфир (200 мл). Слои разделяли и водный слой экстрагировали эфиром (200 мл). Объединенные органические слои сушили, выпаривали и хроматографировали на силикагеле (50 мм × 30 см) (гексан/этил-ацетат [20:1]) с получением 21,0 г, 63,7% продукта А.1H-ЯМР показал, что диастереоселективность реакции >90%.
Смесь магния (2,58 г, 106 ммоль), ТГФ (200 мл) и 1-бром-3-фторбензола (18,60 г, 106,3 ммоль) нагревали с обратным холодильником в течение 45 минут. При продолжении нагревания с обратным холодильником добавляли продукт А (16,45 г, 42,45 ммоль) через шприц с ТГФ (25 мл) на протяжении 2-минутного периода. Нагревание продолжали в течение 1 часа и затем смеси давали остыть до комнатной температуры. Добавляли насыщенный водный раствор NH4Cl (200 мл). Затем реакционную смесь переносили в делительную воронку, содержащую насыщенный водный раствор NaCl (500 мл) и диэтиловый эфир (200 мл). Слои разделяли и водный слой экстрагировали эфиром (200 мл). Объединенные органические слои сушили над безводным сульфатом натрия и выпаривали с получением 21,41 г продукта В в виде желтой жидкости.
Продукт В (20,02 г, 42,45 ммоль, теоретически) растворяли в уксусной кислоте (120 мл) и серной кислоте (30 мл). Реакционную смесь перемешивали при 90° С в течение 1 часа. Уксусную кислоту выпаривали роторным испарением с получением коричневого шлама. Этот материал помещали в баню со льдом и добавляли холодную воду (400 мл). Продукт немедленно осаждался. К реакционной смеси медленно добавляли 10 н. NaOH (150 мл) до нейтрального рН. К этой смеси добавляли диэтиловый эфир (200 мл). Смесь встряхивали до тех пор, пока не исчезал весь нерастворенный материал. Эфирный слой отделяли, промывали водой (2 × 100 мл), сушили над сульфатом натрия и роторное испарение давало 13,14 г (выход 68,2% в расчете на эфир) густого коричневого масла. Это масло поглощали эфиром и превращали в гидрохлоридную соль хлористым водородом в диэтиловом эфире с получением продукта С в виде желто-белого твердого вещества.
Продукт С (7,17 г, 14,6 ммоль) поглощали в абсолютный этанол (200 мл). Добавляли катализатор Перлмана (Pd(OH)2/C; 2,00 г). Реакционную смесь встряхивали в атмосфере водорода (70 psi, 482632,99 Па) при 70° С в течение 20 часов и реакционную смесь фильтровали через целит. Фильтрат выпаривали на роторном испарителе с получением 3,54 г светло-желтого стеклообразного продукта. Этот материал поглощали диэтиловым эфиром (100 мл) и подщелачивали 1 н. NaOH (25 мл). Эфирный слой промывали водой (1× 25 мл), сушили над сульфатом натрия и выпаривали на роторном испарителе с получением 2,45 г светло-желтого масла. Этот материал перегоняли (Кугельрор) (90-100° С, 1 мм рт. ст.) с получением 1,17 г бесцветной жидкости. Этот материал поглощали в диэтиловый эфир и превращали в гидрохлоридную соль растворенным в эфире хлористым водородом. После роторного испарения соль перекристаллизовывали из 0,12 н. НСl (200 мг/мл). Кристаллы отфильтровывали и промывали холодной 0,12 н. НСl с получением 0,77 г (18%) соединения 50 в виде серебристо-белых кристаллов (в виде гидрохлоридной соли).
Соединение 51 синтезировали подобно синтезу соединения 50 с использованием N-бензил-(R)-α -метилбензиламина в качестве хирального исходного материала.

Синтез соединения 54 проводили, как описано ниже.
К раствору 3,3'-дифторбензофенона (5 г, 22,9 ммоль) и метилцианоацетата (3,4 г, 34,4 ммоль) в 15 мл эфира добавляли изопропоксид титана (16,9 мл, 57,25 ммоль). Этот раствор перемешивали в течение 6 дней при комнатной температуре, затем гасили 0,5 моль НСl в 300 мл воды. Смесь разбавляли 100 мл эфира и слои разделяли. Эфирный слой промывали 5% НСl и насыщенным солевым раствором, затем сушили над сульфатом натрия. Растворители выпаривали в вакууме с получением 8 г продукта А.
Соединение А растворяли в 50 мл изопропанола с последующим добавлением небольшого количества бромкрезолового зеленого. Цианоборгидрид натрия (1,52 г, 24,2 ммоль) добавляли весь сразу с последующим немедленным добавлением по каплям концентрированной НСl с такой скоростью, чтобы поддерживать желтый цвет раствора. После 2 часов реакционную смесь обрабатывали распределением между эфиром и водой. Эфирный слой промывали водой и насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали с получением продукта В.
К раствору литийалюминийхлорида (30,4 мл, 30,4 ммоль) в ТГФ добавляли продукт В (1 г, 3,04 ммоль) в 2 мл ТГФ на протяжении периода 30 секунд. Этот раствор перемешивали в течение ночи при комнатной температуре, затем гасили добавлением 20 мл этилацетата. Затем растворители удаляли в вакууме и полученное масло растворяли в водной НСl и ацетонитриле. Затем продукт очищали на колонке С-18 с градиентом 0,1% НСl - ацетонитрил с получением 82 мг соединения 54 в виде гидрохлоридной соли. EI-MS m/z (относительная интенсивность) 277 (М+, 100), 260 (2,4), 242 (8,6), 229 (28), 215 (11,7), 204 (16), 183 (12), 133 (9,5), 124 (14), 109 (6,8), 30 (22).
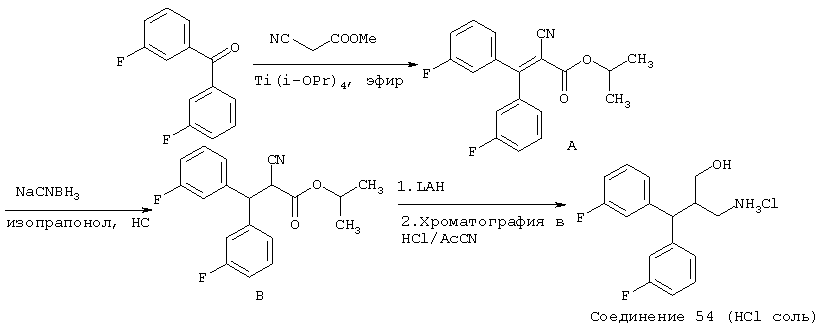
Соединение 55 синтезировали аналогично соединению 21, за исключением того, что в стадии алкилирования использовали этилйодид. GC/EI-MS (Rt=7,43 мин) m/z (относительная интенсивность) 275 (M+, 100), 258 (66), 229 (63), 204 (57), 201 (72), 183 (84), 134 (57), 124 (68), 109 (98), 72 (72).
Синтез соединения 56 выполняли следующим образом.
Спирт А синтезировали из 3-фторбромбензола и 3-фтор-2-метилбензальдегида, как описано для продукта А в синтезе соединения 24.
Спирт А (8,4 г, 36,2 ммоль) перемешивали с диоксидом марганца (12,6 г, 144,8 ммоль) в 100 мл дихлорметана в течение 4 дней. Затем реакционную смесь разбавляли эфиром и фильтровали через тефлоновый мембранный фильтр 0,2 микрон. Фильтрат концентрировали с получением 7,6 г кетона В.
Замещенный акрилонитрил С синтезировали, как описано для продукта А в синтезе соединения 20.
К нитрилу С (4 г, 15,7 ммоль) в 240 мл этанола добавляли 2 г 10% дигидроксида палладия на угле. Эту смесь гидрировали при 60-40 psi (413685,42-275790,28 Па) в течение 3 дней. Затем реакционную смесь фильтровали и концентрировали. Полученное масло растворяли в хлороформе и хроматографировали на силикагеле (30% метанол/5% изопропиламин в хлороформе) с получением амина. Этот амин растворяли в смеси водная HCl/ацетонитрил и очищали посредством ВЭЖХ на С-18 (10% ацетонитрил/0,1% НСl - 50% ацетонитрил/0,1% НСl на протяжении 60 минут), затем лиофилизировали с получением 800 мг соединения 56 в виде гидрохлоридной соли. GC/EI-MS (Rt=7,39 мин) m/z (относительная интенсивность) 261 (М+, 64), 244 (56), 229 (57), 215 (100), 203 (53), 183 (21), 133 (39), 122 (31), 109 (32).

Синтез соединения 57 выполняли следующим образом.
К раствору 5-фтор-2-метилбензонитрила (5 г, 37 ммоль) в 50 мл ТГФ добавляли 3-фторфенилмагнийбромид (46 мл, 40 ммоль) и цианид меди (1) (0,072 г, 0,8 ммоль). Этот раствор нагревали с обратным холодильником в течение 4 часов, затем выливали в смесь эфир/20% НСl и перемешивали в течение еще 2 часов. Слои разделяли и эфирный слой промывали водой и насыщенным солевым раствором. Раствор сушили над безводным сульфатом натрия и концентрировали. Неочищенное масло очищали на диоксиде кремния (гексан - 50% дихлорметан в гексане на протяжении 60 минут) с получением 6,7 г кетона А.
Кетон А превращали в соединение 57, как описано для соединения 56. GC/EI-MS (Rt=7,35 мин) m/z (относительная интенсивность) 261 (М+, 52), 244 (41), 229 (67), 215 (100), 203 (42), 201 (42), 183 (21), 133 (45), 122 (28), 109 (26).

Синтез соединения 58 выполняли следующим образом.
К раствору 5-фтор-2-метилбензоилхлорида (2,24 г, 13 ммоль) в сухом ТГФ добавляли ацетилацетонат железа (III) (0,16 г, 0,44 ммоль). Раствор охлаждали до 0° С и добавляли шприцем раствор в ТГФ 5-фтор-2-метилфенилмагнийбромида (20 мл, 15,5 ммоль) на протяжении периода 30 минут. Реакционную смесь перемешивали еще в течение 30 минут, затем медленно выливали в эфир/5% НСl. Эфирный слой отделяли, промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали с получением 3,2 г кетона А.
Сухой ТГФ (30 мл) охлаждали до -78° С с последующим добавлением бутиллития (5,85 г, 14,6 ммоль, 2,5 М раствор в гексане). Затем добавляли ацетонитрил (0,75 мл, 14,62 ммоль) на протяжении периода 2 минуты, затем оставляли перемешиваться при -78° С в течение 15 минут. К этому раствору добавляли кетон А (3 г, 12,2 ммоль) в 5 мл ТГФ. Раствор перемешивали в течение 30 минут при -78° С, затем ему давали нагреться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь распределяли между эфиром и 5% НСl. Эфирный слой отделяли, промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали с получением 2,2 г нитрила В.
Нитрил В (1 г, 3,48 ммоль) растворяли в 30 мл этанола и 3 мл 10 н. гидроксида натрия. К этому раствору добавляли 1 г 50% водной суспензии никеля Ренея и смесь гидрировали при 60 psi (413685,42 Па) в течение 20 часов. Реакционную смесь фильтровали и концентрировали до белого твердого вещества. Остаток поглощали в смесь эфир/вода и эфирный слой отделяли.
Эфирный раствор сушили над сульфатом натрия и концентрировали с получением 0,96 г гидроксиамина С.
Гидроксиамин С (0,96 г, 3,3 ммоль) растворяли в концентрированной соляной кислоте и нагревали до 70° С, что вызвало кратковременное растворение и затем осаждение алкена D. Алкен собирали фильтрованием и растворяли в 30 мл этанола и 1 мл концентрированной соляной кислоты. Дигидроксид палладия на угле (0,4 г) добавляли к раствору и смесь гидрировали при 60 psi (413685,42 Па) в течение 24 часов. Продукт выделяли отфильтровыванием катализатора и выпариванием растворителя. Остаток растворяли в 0,1% НСl и ацетонитриле и очищали на С-18 (15% ацетонитрил/0,1% НСl - ацетонитрил) с получением 0,6 г соединения 58 в виде гидрохлоридной соли. GC/EI-MS (Rt=7,82 мин) m/z (относительная интенсивность) 275 (М+, 100), 258 (20), 243 (74), 229 (38), 214 (65), 201 (31), 196 (32), 183 (20), 148 (35), 138 (42), 133 (48), 122 (69), 109 (41).

Синтез соединения 59 выполняли следующим образом.
Соединение 20 (2,0 г, 7,05 ммоль) растворяли в абсолютном EtOH (200 мл) и охлаждали до 5-10° С в бане со льдом. Добавляли ацетальдегид (0,395 мл, 7,05 ммоль, охлажденный до -4° С) с последующим добавлением никель-алюминиевого сплава (200 мг, Fluka Chemical) и реакционную смесь гидрировали на аппарате Парра при 50 psi (344737,85 Па) в течение 2 часов. GC/MS показал выход 75% продукта и 2% N,N-диэтил-побочного продукта реакции. Реакционную смесь фильтровали через диатомовую землю и фильтрат выпаривали при пониженном давлении. Неочищенный продукт растворяли в смеси изопропанол (5 мл)/эфир (60 мл)/НСl в эфире (1 М) и затем добавляли гексан (5 мл) до точки помутнения. Мутную смесь фильтровали через бумагу, затем добавляли гексан (10 мл) до точки помутнения и раствор фильтровали снова. Фильтрат закрывали пробкой и продукту давали кристаллизоваться при комнатной температуре. Кристаллы собирали и сушили с получением 0,325 г (выход 14,8%) соединения 59 в виде гидрохлоридной соли (бесцветные игольчатые кристаллы).
Синтез соединения 60 выполняли следующим образом. Соединения 66, 69, 108, 123, 142 и 145 могут быть синтезированы подобным образом с соединениями 33, 50, 32, 60, 25 и 119 в качестве исходных соединений соответственно.
Соединение 20 (в виде свободного основания) (1,0 г, 4,0 ммоль) нагревали с обратным холодильником в этилформиате (150 мл) в течение 2 часов. Растворитель затем удаляли при пониженном давлении с получением 1,2 г (выход 99%) формамида А в виде бесцветного масла. ГХ/МС показали, что продукт имеет чистоту 100%, и его использовали в следующей стадии без дополнительной очистки.
Формамид А (1,1 г, 4,0 ммоль) растворяли в сухом ТГФ (100 мл) и нагревали до кипения (без холодильника). Комплекс боран-метилсульфид (1,2 мл, 12 ммоль, 10,5 М) добавляли по каплям на протяжении периода 3 минуты к раствору. Кипение поддерживали в течение приблизительно 15 минут, открытым для доступа воздуха, пока объем реакционной смеси не уменьшался приблизительно до 30 мл. Затем смесь охлаждали на бане со льдом и лед (5 г, маленькие кусочки) осторожно добавляли с последующим добавлением H2O (25 мл) и концентрированной НСl (25 мл). Кислый раствор нагревали с обратным холодильником в течение 30 минут. Затем реакционную смесь охлаждали на бане со льдом, подщелачивали NaOH (10 н.), экстрагировали эфиром (3× 100 мл), сушили над безводным сульфатом натрия и выпаривали при пониженном давлении. Неочищенный продукт растворяли в смеси эфир (10 мл)/гексан (50 мл) и добавляли НСl в эфире (1 М) каплями для осаждения гидрохлоридной соли. Соль собирали и перекристаллизовывали из смеси изопропанол (3 мл)/эфир (40 мл) с получением 0,5 г соединения 60 в виде гидрохлоридной соли.

Альтернативно, соединение 60 синтезировали из коммерчески доступных исходных материалов в следующей включающей четыре стадии последовательности реакций. Первое промежуточное соединение в этом синтетическом пути, этил-N-бензил-N-метил-3-аминопропионат, получали добавлением конъюгата N-бензилметиламина к этилакрилату. Сложноэфирная группа первого промежуточного продукта взаимодействовала затем с двумя эквивалентами реагента Гриньяра (приготовленного из 1-бром-3-фторбензола) с образованием N-бензил-N-метил-3-гидрокси-3-(бис-3-фторфенил)пропиламина. Затем продукт реакции Гриньяра дегидрировали в смеси 6 н. HCl/уксусная кислота с получением N-бензил-N-метил-3-(бис-3-фторфенил)-2-пропенамина. Каталитическое гидрирование этого материала в виде его гидрохлоридной соли в этаноле над катализатором Перлмана [Pd(OH)2/C] давало после перекристаллизации из этилацетата бесцветные игольчатые кристаллы соединения 60 в виде гидрохлоридной соли.
В трехгорлую колбу на 500 мл, снабженную термометром, холодильником для дефлегмации и капельной воронкой на 125 мл [загруженной этилакрилатом (88,3 мл, 81,5 г, 0,815 моль)], помещали N-бензилметиламин (100 мл, 94,0 г, 0,776 моль). Этилакрилат добавляли каплями к перемешиваемой реакционной смеси на протяжении периода 80 минут. После перемешивания в течение 18 часов при комнатной температуре продукт перегоняли в вакууме и фракцию, содержащую продукт, собирали при 78-95° С (0,12-0,25 мм рт. ст.), (138 г, выход 80%): т.кип. 78-95° С (0,12-0,25 мм рт. ст.); ТСХ Rf=0,23 [гексан - EtOAc (5:1)], Rf=0,57 [МеОН-СНСl3 (100:5)]; GC, tR=6,06 мин; МС, 221 (М+), 206 (М-СН3), 192 (М-С2Н5), 176 (М-OС2H5), 144 (M-C6H5), 134 [СН3N(СН3)СН2Рh], 120 [N(CH)CHPh], 91 (СН), 77 (СН), 42 (CH2CH2N);1H-ЯМР (свободное основание, CDCl3) δ 1,25 м.д. (т, J=7,1, 3Н, CH2CH2), 2,20 (с, 3Н, NСН3), 2,51 (т, J=7,3, 2H, COCH2), 2,74 (т, J=7,2, 2H, CH2N), 3,51 (с, 2H, NCH2Ph), 4,13 (кв, J=7, 1, 2H, ОСН2СН3), 7,18-7,35 (м, 5Н, АrН);13С-ЯМР (свободное основание, СDСl3) δ 15,2 (СН2СН3), 34,0 (СОСН2), 42,9 (NСН3), 53,8 (NCH2), 61,4 (ОСH2СН3), 63,1 (CH2Ph), 128,0 (СН), 129,2 (СН), 130,0 (СН), 139,9 (кв), 173,7 (кв).
В четырехгорлую круглодонную колбу на 5 л в атмосфере азота помещали Мg [51,5 г, 2,12 моль, стружку, промытую ТГФ (2× 300 мл)] и ТГФ (2 л). Капельную воронку загружали 1-бром-3-фторбензолом (чистым, 392,8 г, 2, 24 моль). Одну двадцатую часть бромида добавляли к суспензии магния с последующим добавлением одного кристалла йода. После инициации реакции Гриньяра добавляли оставшийся 1-бром-3-фторбензол к кипящей смеси на протяжении периода 50 минут. Реакционную смесь кипятили с обратным холодильником еще в течение 45 минут. К нагреваемому раствору реагента Гриньяра добавляли раствор этил-N-бензил-N-метил-3-аминопропионата (187,5 г, 0,847 моль) в ТГФ (100 мл) на протяжении периода 20 минут. После завершения добавления этого эфира реакционную смесь нагревали с обратным холодильником в течение 1 часа. Затем реакционную смесь охлаждали в бане со льдом. Добавляли насыщенный NH4Cl (водный, 400 мл) и H2O (400 мл) и смесь переносили в делительную воронку. Органический слой отделяли и водный слой экстрагировали один раз ТГФ (400 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (2× 200 мл), сушили (Nа2SO4), фильтровали через бумагу и выпаривали на роторном испарителе с получением 281,6 г (выход 90%) неочищенного продукта в виде оранжевого вязкого масла. Этот материал (281,6 г, 0, 766 моль) растворяли в ацетонитриле (1,4 л). К перемешиваемому фильтрату добавляли концентрированную соляную кислоту (65,0 мл, 0,786 моль, 12 н.). Затем кристаллизующуюся смесь охлаждали до -20° С в течение 17 часов. Продукт собирали, промывали холодным ацетонитрилом (800 мл) и сушили с получением белого твердого вещества, 235,6 г (выход 69% в расчете на эфир). Для аналитических целей гидрохлоридную соль дополнительно очищали перекристаллизацией из ацетонитрила: т.пл. 194-197° С (некорр.); ТСХ Rf=0,23 [гексан - EtOAc (5:1)], Rf=0,85 [МеОН-СНСl3 (100:5)], Rf=0,72 [МеОН-СНСl3 (100:3)]; GC, tR=10,93 мин; МС, 367 (М+), 272 (М-С6Н4F), 258 (M-CH2Ph-H2O), 219 [(C6H4F)2CH], 148 [CH2CH2N(CH3)CH2Ph], 134 [CH2N(СН3)CH2Ph], 91 (С7Н7), 42 (CH2CH2N);1H-ЯМР (свободное основание, CDCl3) δ 2,18 (с, 3Н, NСН3), 2,41 (м, 2Н, СНСН2), 2,58 (м, 2Н, CH2N), 3,42 (с, 2Н, CH2Ph), 6,86 (дт, J1=8,5, J2=1,8, 2Н, Аr-H), 7,18-7,30 (м, 10Н, Аr-Н), 8,33 (ушир. с, 1Н, ОH);13С-ЯМР (свободное основание, CDCl3) δ 35,6 (CHCH2), 41,5 (СН3, NСН3), 54,3 (CH2, CH2N), 62,6 (CH2, CH2Ph), 113,1 (д, J=23, CH, Ar-C5,5’), 113,5 (д, J=23, CH), 121,2 (д, J=3, CH), 127,5 (CH), 128,5 (CH), 129,2 (CH), 129,5 (CH), 129,6 (CH), 137,0 (кв), 150,2 (кв), 162,8 (д, J= 243, кв, Аr-С3,3’).
В трехгорлый реакционный сосуд на 5 л, снабженный верхней механической мешалкой, дефлегмирующим холодильником и термометром, помещали гидрохлорид N-бензил-N-метил-3-гидрокси-3-бис(3-фторфенил)пропиламина (225,4 г, 0,559 моль), 6 н. НСl (1392 мл) и ледяную НОАс (464 мл). Эту суспензию нагревали в водяной бане (80-85° С) и перемешивали в течение 18 часов. После 18 часов нагревания реакционную смесь охлаждали на бане со смесью лед/метанол. К охлаждаемой реакционной смеси добавляли этилацетат (500 мл). Затем к охлаждаемой смеси добавляли NaOH (10 н., 1,7 л) на протяжении периода 25 минут при такой скорости, чтобы сохранять температуру ниже 40° С. Смесь переносили в 6-литровую делительную воронку. Органический слой отделяли и водный слой экстрагировали этилацетатом (2× 500 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (2× 100 мл), сушили Na2SO4 (250 г), выпаривали роторным испарителем и затем сушили под вакуумом с получением 185,6 г (выход 95%) свободного основания в виде жидкого коричневатого масла.
Этот материал перемешивали с гексаном (1,5 л). Полученный раствор фильтровали через бумагу. Добавляли каплями 4 М НСl в диоксане (146 мл) с перемешиванием фильтрата на протяжении периода 5 минут. Затем полупрозрачный растворитель декантировали со светло-желтого полутвердого осадка. Неочищенную гидрохлоридную соль растворяли в кипящем с обратным холодильником этилацетате (600 мл) и фильтровали. Затем фильтрат тщательно охлаждали в бане со льдом и медленно добавляли гексан (110 мл) с интенсивным перемешиванием. После охлаждения в бане со льдом в течение 2 часов вся колба наполнялась белым кристаллическим твердым веществом. Этот материал собирали на фильтрующей воронке, промывали охлажденной на льду смесью гексан/этилацетат [(1:4), 400 мл] и сушили с получением 128,7 г, 59,7% белого твердого вещества. При стоянии маточный раствор осаждал еще 14,8 г твердого вещества нестандартного белого цвета. Общий выход 128,7 г + 14,8 г = 143,5 г (67%). Т.пл. 141-142° С (некорр.); ТСХ Rf=0,20 [гексан - EtOAc (5:1)], Rf=0,75 [МеОН-СНСl3 (100:5)], Rf=0,49 [MeOH-СНСl3 (100:3)]; GC, tR=10,40 мин; МС, 349 (М+, 330, 301, 281, 258 (М-СН2Рh), 240, 229 [M-N(СН3)CH2Ph], 201, 183, 146, 133, 109, 91 (CH2C6H5), 65, 42 (С2NНСН3),1H-ЯМР (свободное основание, СDСl3) δ 2,20 м.д. (с, 3Н, NH3), 3,08 (д, J=6,8, 2Н, CH2N), 3,47 (д, J<1, 2Н, CH2Ph), 6,29 (т, J=6,8, 1H, CH), 6,85-7,04 (м, 6Н, ArH), 7,19-7,35 (м, 7Н, ArH).
Гидрохлорид N-бензил-N-метил-3-бис(3-фторфенил)аллиламина (120,0 г, 0,311 моль) растворяли в абсолютном EtOH (1250 мл).
Добавляли Pd(ОН)2/древесный уголь (10,0 г, ~20% Pd, Fluka Chemical). Реакционную смесь перемешивали при постоянном токе газообразного водорода в течение 18 часов при 25° С (при атмосферном давлении). Затем смесь фильтровали через целит*/стеклянный фильтр/ катализатор промывали EtOH (2× 50 мл) и растворитель удаляли при пониженном давлении с получением 95,4 г, 103% неочищенного продукта. Этот материал растворяли в кипящем этилацетате (300 мл) с интенсивным перемешиванием и фильтровали. Колбе давали стоять в течение 2 часов при 25° С, во время этого периода гидрохлоридная соль начинала кристаллизоваться в виде игольчатых кристаллов. Затем колбу охлаждали, продукт собирали, промывали охлажденным на льду этилацетатом (20 мл) и сушили с получением 73,7 г, 80%, соединения 60 в виде белого кристаллического твердого вещества. Т.пл. 129-130° С; UV/Vis е=2,1× 103 л· моль· см-1 (264 нм, EtOH, 25° С, линейная область: 0,05-0,20 мг/мл); ТСХ Rf=0,00 [гексан - EtOAc (5:1)], Rf=0,07 [МеОН-СНСl3 (100:5)], Rf=0,19 [МеОН-СНСl3-NH4OН (100:5:1)],; GC, tR=7,45 мин; МС, 261 (М+, 229, 215, 201, 183, 164, 150, 138, 122, 101, 83, 75, 57, 42 [CH2NHCH3],1Н-ЯМР (НСl, соль, СDСl3 + 1 gtt MeOD), δ 2,56 (м, 2Н, NCH2), 2,60 (с, 3Н, NCH), 2,85 (т, J=8,0, 2H, CHCH2), 4,11 (т, J=8,0, 1H, CH), 6,87-6,98 (м, 4Н, ArH), 7,06 (д, J=7,7, 2H, Аr2,2'Н), 7,25 (дд, J1=6, J2=8, 2H, ArH);13С-ЯМР (НСl соль, СDСl3 + 1 gt MeOD) δ 30,9 (CH2, CHCH2), 32,7 (СН3, NСН3), 47,6 (CH, CHCH2), 47,8 (СН2, CH2N), 113,9 (J=21, ArC2,2’ или АrС4,4’), 114,5 (д, J=22, ArC2,2’ или ArC4,4’), 123,2 (д, J=3, Аr-С6,6’), 130,3 (д, J=9, Ar-C5,5’), 144,7 (д, J=7, Ar-C1,1’), 162,9 (д, J=245, Аr-С3,3’); IR: KBr pellet (см-1), 3436,9, 2963,4, 2778,5, 2453,7, 1610,6, 1589,3, 1487,0, 1445,3, 1246,0, 764,5; растворимость: 2 г/мл (H2O), 1 г/мл (EtOH); аналит. рассчитано для C16H17NF2 ·HCl (Karl Fisher: 0,26% H2O: С, 64,37; Н, 6,11; N, 4,69; найдено: С, 64,14; Н, 6,13; N, 4,69;

Соединение 105 получали избирательным восстановлением его соответствующего алкена каталитическим гидрированием над Pd/C.
Соединение 61 получали из 2-бром-4-фторанизола и 3-фторбензальдегида, как описано для соединения 24. GC/EI-MS (Rt=9,22 мин) m/z (относительная интенсивность) 277 (М+, 74), 260 (46), 245 (35), 231 (44), 229 (34), 217 (24), 203 (28), 201 (31), 183 (28), 154 (24), 133 (19), 109 (100).
Соединение 62 получали из 2-броманизола и 2-метоксибензальдегида, как описано для соединения 24. GC/EI-MS (Rt=9,30 мин) m/z (относительная интенсивность) 271 (М+, 100), 254 (17), 240 (23), 225 (40), 223 (45), 207 (22), 181 (32), 165 (31), 136 (48), 121 (98), 91 (83).
Синтез соединения 63 проводили следующим образом.
Спирт А получали из 3-фторбензальдегида, как описано для продукта А синтеза соединения 24.
К спирту А (10, 275 г, 47 ммоль) в 200 мл этанола добавляли 1,6 г 10% Pd/C и 1 мл концентрированной НСl. Эту смесь гидрировали в течение 3 часов при 60 psi (413685,42 Па), затем фильтровали и концентрировали с получением дифенилметана В.
Продукт В (2,01 г, 9,86 ммоль) растворяли в 20 мл ТГФ и охлаждали до -78° С. Бутиллитий (4,4 мл, 10,8 ммоль, 2,5 М в гексане) добавляли медленно при помощи шприца и затем реакционную смесь перемешивали еще в течение 30 минут при -78° С. К этому оранжевому раствору добавляли оксид циклопентена (0,9 мл, 10,3 ммоль). Реакционную смесь оставляли перемешиваться в течение 3 часов при медленном нагревании ее до комнатной температуры. Реакционную смесь гасили 150 мл 10% НСl и экстрагировали 3 раза эфиром. Эфирный слой сушили над сульфатом натрия и концентрировали с получением 2,5 г спирта С.
К спирту С (1 г, 3,5 ммоль) в 10 мл сухого ТГФ добавляли трифенилфосфин (1,37 г, 5,2 ммоль) в 5 мл ТГФ и п-нитро-бензойную кислоту (0,87 г, 5,2 ммоль) в 5 мл ТГФ. Этот раствор охлаждали до 0° С с последующим добавлением DEAD (0,82 мл, 5,2 ммоль) и оставляли перемешиваться в течение ночи. Реакционную смесь распределяли между водой и эфиром. Эфир удаляли в вакууме и полученное масло хроматографировали на силикагеле в смеси гексан/этилацетат с получением 365 мг цис-эфира. Этот эфир гидролизовали в метаноле с карбонатом калия перемешиванием в течение ночи. После удаления метанола остаток поглощали эфиром, промывали водой, сушили над сульфатом натрия и концентрировали с получением 250 мг цис-спирта D.
К спирту D (0,25 г, 0,9 ммоль) в 5 мл сухого ТГФ добавляли трифенилфосфин (342 мг, 1,3 ммоль) в 5 мл ТГФ и фталимид (191,3 мг, 1,3 ммоль) в 5 мл ТГФ. Этот раствор охлаждали до 0° С с последующим добавлением DEAD (0,205 мл, 1,3 ммоль) и оставляли перемешиваться в течение ночи. Реакционную смесь распределяли между водой и эфиром. Эфир удаляли в вакууме и полученное масло хроматографировали на силикагеле в смеси гексан/этилацетат с получением 100 мг фталимида Е.
К раствору фталимида Е (100 мг) в 20 мл этанола добавляли 8,8 мг гидразингидрата. Раствор нагревали с обратным холодильником в течение 5 часов, затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь обрабатывали добавлением 1 мл концентрированной НСl и отфильтровыванием белого твердого вещества. Полученный раствор концентрировали досуха и твердое вещество поглощали в эфир и водный гидроксид натрия. Эфирный слой сушили над сульфатом натрия и концентрировали до белого твердого вещества. Его растворяли в небольшом количестве эфира и обрабатывали 10 каплями 1 М НСl в эфире. После перемешивания в течение ночи белое твердое вещество собирали фильтрованием и сушили с получением 50 мг соединения 63 в виде гидрохлоридной соли. GC/EI-MS (Rt=9,22 мин) m/z (относительная интенсивность) 287 (М+, 45), 270 (12), 201 (63), 183 (81), 133 (38), 109 (43), 83 (44), 56 (100), 43 (37).

Синтез соединения 64 выполняли, как описано для соединения 63, за исключением того, что пропускали стадию обратного преобразования (продукта С в продукт D) для того, чтобы получить цис-амин в качестве конечного продукта. GC/EI-MS (Rt=8,28 мин) m/z (относительная интенсивность) 287 (М+, 15), 270 (4), 201 (13), 183 (15), 133 (11), 109 (16), 84 (43), 56 (100), 43 (32).
Синтез соединения 65 выполняли следующим образом.
Кетон А синтезировали подобно кетону В в синтезе соединения 24 с использованием 2-метилфенилмагнийбромида и 2-метилбензальдегида в качестве исходных материалов. Этот кетон превращали в конечный продукт с применением процедуры, описанной для соединения 58. GC/EI-MS (Rt=7,84 мин) m/z (относительная интенсивность) 239 (М+, 88), 222 (14), 207 (100), 193 (46), 178 (71), 165 (60), 130 (39), 120 (40), 115 (51), 104 (40), 91 (38), 77 (21).

Соединение 119 синтезировали в цепи реакций из 7 стадий из коммерчески доступной транс-3-фторкоричной кислоты в качестве исходного продукта. Этот путь синтеза концептуально сходен с сообщенным в литературе [U.S. Patent 4313896 (1982)] для родственных аналогов. Однако три последние стадии выполняли с использованием значительно отличающейся цепи реакций, чем это сообщалось. Коричную кислоту восстанавливали и хлорировали в трех стадиях до соответствующего 3-(3-фторфенил)пропилхлорида.
Это соединение бромировали NBS (N-бромсукцинимидом) и полученный тригалогенид взаимодействовал затем с 3-фторфенолом. Полученный простой эфир превращали в конечный продукт с использованием синтеза Габриэля.
Транс-3-фторкоричную кислоту (25,0 г, 150,4 ммоль) растворяли в абсолютном EtOH (250 мл) и гидрировали над 10% Pd/C (2,5 г) в аппарате Парра при 60 psi (413685,42 Па), 50° С, в течение 1 часа (поглощение водорода: рассчит. 245 psig (1689214,4 Па); найдено: 260 psig (1792636,8 Па). Ракционную смесь фильтровали и выпаривали с получением кристаллического продукта (23,0 г, 89%). ГХ, tR=4,43 мин; MS, 168 (М+).
Под током сухого азота, при 0-10° С, добавляли по каплям раствор 3-фтор гидрокоричной кислоты (22,0 г, 131 ммоль) в ТГФ (100 мл) на протяжении периода 15 минут к суспензии LiAlH4 (4,23 г, 111 ммоль) в ТГФ (200 мл). Реакционную смесь нагревали с обратным холодильником в течение периода 1 час и затем обрабатывали согласно Frieser and Frieser's Reagents for Organic Synthesis (Vol. 1, 1967) с получением белого твердого вещества (20,1 г, 99%) ГХ, tR=3,74 мин; МС, 154 (М+).
Раствор 3-(3-фторфенил)-1-пропанола (15,0 г, 97,4 ммоль) и трифенилфосфина (36,0 г, 137,3 ммоль) в ССl4 (150 мл) нагревали с обратным холодильником в течение 19 часов. Дополнительное количество Р(С6Н5)3 (3× 3,0 г, 3× 11,4 ммоль) периодически добавляли на протяжении периода 24 часов. Полученный осадок удаляли фильтрованием и твердые вещества промывали гексаном. Фильтрат выпаривали под вакуумом и остаток суспендировали в гексане (200 мл) и затем фильтровали. Выпаривание фильтрата давало 16,0 г (95,1%) неочищенного продукта, который очищали флэш-хроматографией на силикагеле, элюируя гексаном с получением 14,7 г (87%) бесцветной жидкости. ГХ, tR=3,63 мин; МС, 172,174 (М+).
Раствор указанного выше хлорида (12,0 г, 69,5 ммоль), N-бромсукцинимида (17,3 г, 97,2 ммоль) и дибензоилпероксида (0,06 г) в ССl4 (75 мл) нагревали с обратным холодильником в течение 1 часа. Затем реакционную смесь охлаждали в бане со льдом, фильтровали и твердые вещества промывали гексаном. Фильтрат выпаривали с получением 17,9 г (100%) продукта. ГХ, tR=5,21 мин; МС, 251/253 (М+).
Смесь 3-бром-3-(3-фторфенил)-1-пропилхлорида (4,0 г, 15,9 ммоль), 3-фторфенола (1,98 г, 17,7 ммоль) и К2СО3 (2,65 г, 19,2 ммоль), суспендированную в ацетоне (80 мл), нагревали с обратным холодильником в течение 15 часов. Затем летучие компоненты удаляли в вакууме и полученный остаток суспендировали в смеси гексана (200 мл) и NaOH (0,1 н., 100 мл). Слои разделяли и органический слой промывали 0,1 н. NaOH и водой (100 мл), сушили (безводный Na2SO4) и выпаривали в вакууме. Полученный остаток хроматографировали на силикагеле, элюируя гексаном и затем смесью гексан/EtOAc [100:1], затем [40:1] с получением 1,64 г (37%) продукта в виде бесцветного масла. ГХ, tR=7,28 мин; МС, 282/283 (М+). ТСХ, rf=0,3, гексан/EtOAc [40:1].
Раствор 3-(3-фторфенил)-3-(3-фторфенокси)-1-пропилхлорида (1,52 г, 5,38 ммоль) и фталата калия (1,20 г, 6,48 ммоль) нагревали до 90° С в ДМФ (30 мл) в течение периода 2 часов в атмосфере азота. Затем реакционную смесь охлаждали и выливали в H2O (100 мл). Полученный раствор экстрагировали Et2O (2× 100 мл). Органический экстракт промывали насыщенным NaCl (100 мл) и Н2O (2× 100 мл), сушили (безв. Nа2SO4) и выпаривали под вакуумом с получением 2,17 г неочищенного продукта. Этот материал хроматографировали на силикагеле, элюируя смесью гексан/EtOAc [40:1] и затем [20:1] с получением после выпаривания 1,81 г (86%) продукта в виде стекловидного вещества.
Раствор N-фталоил-3-(3-фторфенил)-3-(фторфенокси)-1-пропиламина (1,74 г, 4,42 ммоль) и безводного гидразина (1,43 г, 44,6 ммоль) в абсолютном EtOH (30 мл) нагревали с обратным холодильником в течение 1 часа. Реакционную смесь охлаждали и выпаривали под вакуумом. Полученный материал суспендировали в Et2O (75 мл) и промывали 0,2 н. NaOH (2× 25 мл). Органический слой сушили (безв. Na2SO4) и выпаривали под вакуумом с получением 1,04 г (89,3%) продукта, который очищали хроматографией с обращенной фазой [Vydac Prep. C18; 264 нм; 50 мл/мин/градиентное элюирование ACN/0,1% HCl безв., 10%-50% на протяжении 20 минут; rt=17,4 мин], с получением 0,89 г (67%) соединения 119 в виде гигроскопической гидрохлоридной соли.

Соединения 118, 120-122 и 137 получали аналогично процедурам, используемым для получения соединения 119.
Соединение 113 синтезировали из коммерчески доступного 4,4-дифенилциклогексенона в трех стадиях. Сначала алкен в исходном материале восстанавливали каталитическим гидрированием.
Образование метоксиамина сопровождалось восстановлением с использованием стандартных процедур.
Соединения 67-68, 70-75, 79-82, 84-89, 91-95, 98-100, 102, 104-106, 109-114, 117, 124-134, 138 и 140-150 синтезировали стандартными процедурами, известными специалистам в данной области, как описано выше.
Газовая хроматография упрощенных арилалкиламинов
Данные газовой хроматографии и масс-спектрометрии получали на газовом хроматографе Hewlett-Packard 5890 Series II, снабженном детектором 5971 Series Mass-Selective Detector [Ultra-2 Ultra Performance Capillary Column (сшитый 5% фенилметилсиликон); длина колонки 25 м, внутренний диаметр колонки 0,20 мм; скорость тока 60 мл/мин; темп. инжектора 250° С; программа градиента температур: 20° С/мин, от 125 до 325° С в течение 10 минут, затем постоянное поддержание при 325° C в течение 6 минут].
Соединение 19. (Rt=7,40 мин), m/z (отн. инт.) 211 (M+, 13), 195 (16), 194 (100), 193 (73), 180 (8), 179 (33), 178 (19), 168 (24), 167 (50), 166 (23), 165 (72), 164 (8), 153 (10), 152 (31), 117 (13), 116 (38), 115 (26), 106 (14), 104 (14), 103 (24), 102 (8), 91 (11), 78 (14), 77 (29), 63 (9), 51 (17)
Соединение 20. (Rt=7.34 мин), m/z (отн. интен.) 247 (М+, 27), 231 (16), 230 (100), 229 (45), 215 (29), 214 (14), 204 (43), 203 (37), 202 (13), 201 (47), 184 (14), 183 (58), 181 (8), 151 (9), 135 (13), 134 (31), 133 (25), 124 (18), 122 (16), 121 (19), 109 (15), 101 (29), 96 (18), 95 (11), 83 (11), 75 (20), 57 (10), 42 (9)
Соединение 21. (Rt=7,53 мин), m/z (отн. интен.) 261 (М+, 69), 262 (13), 245 (17), 244 (100), 230 (11), 229 (42), 216 (11), 215 (15), 214 (14), 204 (45), 203 (35), 202 (16), 201 (63), 184 (12), 183 (61), 148 (11), 136 (9), 135 (27), 133 (36), 124 (21), 115 (16), 109 (43), 83 (12), 74 (8), 58 (14), 57 (11)
Соединение 22. (Rt=7.37 мин), m/z (отн. интен.) 261 (М+, 4), 244 (14), 229 (7), 204 (10), 203 (16), 201 (12), 183 (16), 138 (4), 133 (5), 109 (4), 101 (7), 75 (4), 58 (8), 57 (4), 44 (100), 42 (7)
Соединение 24. (Rt=8.21 мин), m/z (отн. инт.) 259 (М+, 122), 260 (23), 242 (44), 241 (15), 228 (15), 227 (49), 216 (15), 213 (56), 212 (16), 211 (55), 199 (32), 196 (22), 185 (34), 184 (19), 183 (67), 171 (16), 170 (38), 165 (44), 151 (20), 150 (16), 146 (13), 136 (46), 134 (17), 133 (37), 123 (15), 121 (22), 120 (13), 109 (100), 91 (34), 77 (29), 51 (15)
Соединение 25. (Rt=8.49 мин), m/z (отн. интен.) 259 (М+, 39), 243 (16), 242 (95), 241 (25), 227 (27), 217 (15), 216 (100), 215 (27), 212 (13), 211 (50), 201 (14), 200 (11), 199 (15), 196 (15), 185 (20), 184 (19), 183 (50), 171 (24), 170 (28), 165 (15), 146 (10), 136 (11), 134 (12), 133 (23), 121 (21), 77 (9)
Соединение 26. (Rt=8.69 мин), m/z (отн. интен.) 259 (М+, 11), 243 (17), 242 (100), 241 (69), 227 (10), 215 (31), 212 (11), 211 (52), 184 (14), 183 (31), 172 (13), 171 (35), 170 (23), 165 (13), 147 (21), 146 (12), 134 (19), 133 (23), 121 (13), 91 (11), 77 (10)
Соединение 27. (Rt=8.80 мин), m/z (отн. интен.) 243 (М+, 54), 226 (36), 212 (12), 211 (69), 200 (14), 199 (16), 198 (20), 197 (100), 196 (39), 185 (35), 184 (30), 183 (50), 179 (13), 178 (14), 165 (13), 134 (15), 133 (19), 120 (29), 117 (16), 115 (27), 104 (13), 101 (11), 91 (23), 77 (13)
Соединение 28. (Rt=8.77 мин), m/z (отн. интен.) 243 (М+, 25), 227 (15), 226 (85), 225 (26), 212 (19), 211 (100), 200 (22), 199 (17), 197 (18), 196 (29), 185 (46), 184 (35), 183 (64), 179 (9), 165 (11), 134 (19), 133 (23), 121 (12), 120 (18), 117 (14), 115 (24), 101 (12), 91 (25), 77 (12), 65 (11), 51 (9)
Соединение 29. (Rt=7.89 мин), m/z (отн. интен.) 243 (М+, 12), 227 (9), 226 (52), 225 (17), 212 (19), 211 (100), 199 (13), 197 (12), 196 (21), 185 (19), 184 (24), 183 (43), 179 (7), 134 (11), 133 (15), 120 (9), 117 (10), 115 (17), 91 (14)
Соединение 30. (Rt=8.36 мин), m/z (отн. интен.) 263 (М+, 21), 246 (26), 220 (13), 212 (17), 211 (100), 197 (10), 196 (25), 185 (43), 184 (30), 183 (69), 181 (9), 165 (12), 133 (18), 115 (14), 101 (15), 75 (15)
Соединение 31. (Rt=9.3l мин), m/z (отн. интен.) 279 (М+, 18), 281 (11), 262 (10), 236 (10), 229 (33), 228 (17), 227 (100), 203 (9), 201 (33), 199 (15), 192 (15), 178 (19), 166 (18), 165 (53), 164 (13), 163 (16), 140 (12), 115 (13), 103 (9)
Соединение 32. (Rt=7.30 мин), m/z (отн. интен.) 229 (М+, 21), 213 (16), 212 (100), 211 (61), 197 (33), 196 (19), 194 (14), 186 (26), 185 (30), 184 (19), 183 (69), 170 (17), 166 (16), 165 (77), 134 (25), 133 (23), 116 (17), 115 (17), 103 (18), 101 (11), 78 (13), 77 (23), 75 (13), 51 (18), 43 (13), 42 (13)
Соединение 33. (Rt=7.56 мин), m/z (отн. интен.) 261 (М+, 68), 245 (18), 244 (100), 229 (43), 215 (16), 214 (15), 204 (57), 203 (43), 202 (15), 201 (64), 184 (14), 183 (73), 148 (16), 136 (13), 135 (46), 133 (60), 124 (51), 115 (27), 111 (14), 109 (96), 107 (16), 96 (14), 83 (27), 75 (20), 58 (96), 57 (33), 56 (23), 41 (35)
Соединение 34. (Rt=7.39 мин), m/z (отн. интен.) 261 (М+, 72), 262 (14), 245 (18), 244 (100), 229 (42), 216 (9), 215 (15), 214 (14), 204 (52), 203 (38), 202 (14), 201 (54), 184 (12), 183 (62), 181 (10), 148 (13), 136 (9), 135 (31), 133 (40), 124 (30), 115 (18), 109 (57), 107 (9), 83 (13), 58 (21), 57 (11)
Соединение 35. (Rt=4.45 мин), m/z (отн. интен.) 181 (М+, 8), 165 (10), 164 (76), 138 (48), 136 (11), 135 (63), 133 (12), 123 (22), 122 (22), 121 (11), 110 (21), 109 (100), 101 (13), 96 (27), 83 (14), 75 (11), 56 (15), 45 (21), 44 (40), 42 (9), 41 (15)
Соединение 37. (Rt=4.87 мин), m/z (отн. интен.) 196 (М+, 4), 195 (17), 178 (76), 163 (20), 152 (41), 150 (22), 137 (12), 136 (29), 135 (60), 133 (19), 124 (13), 123 (20), 122 (49), 121 (17), 110 (78), 109 (100), 101 (17), 96 (29), 83 (17), 75 (12), 56 (29), 55 (12), 45 (53), 44 (45), 43 (39), 41 (30)
Соединение 38. (Rt=7.68 мин), m/z (отн. интен.) 275 (M+, l), 203 (5), 201 (6), 183 (8), 135(4), 133 (4), 109 (8), 71 (3), 45 (3), 44 (100), 42 (4)
Соединение 39. (Rt=7.67 мин), m/z (отн. интен.) 289 (М+, 6), 203 (3), 201 (5), 183 (6), 135 (2), 133 (3), 109 (7), 85 (3), 70 (3), 59 (4), 58 (100)
Соединение 40. (Rt=7.63 мин), m/z (отн. интен.) 289 (М+, 19), 203 (6), 201 (13), 183 (17), 152 (5), 135 (6), 133 (8), 109 (15), 85 (5), 70 (4), 58 (100)
Соединение 41. (Rt=7.93 мин), m/z (отн. интен.) 275 (М+, 23), 258 (20), 203 (27), 202 (14), 201 (52), 184 (9), 183 (59), 181 (10), 150 (11), 149 (18), 147 (11), 135 (24), 134 (14), 133 (40), 124 (12), 123 (19), 109 (76), 107 (9), 103 (10), 83 (15), 75 (10), 72 (100), 71 (12), 57 (18), 56 (21), 55 (41)
Соединение 43. (Rt=9.18 мин), m/z (отн. интен.) 293 (М+, 11), 276 (10), 243 (11), 241 (31), 236 (11), 235 (16), 201 (18), 199 (22), 179 (11), 178 (25), 176 (10), 166 (16), 165 (70), 164 (19), 163 (24), 103 (9), 102 (9), 75 (11), 44 (100), 43 (11), 42 (15)
Соединение 46. (Rt=9.34 мин), m/z (отн. интен.) 293 (М+, 46), 295 (28), 276 (16), 243 (24), 242 (15), 241 (75), 237 (12), 236 (18), 201 (33), 199 (31), 178 (26), 176 (13), 166 (31), 165 (100), 164 (32), 163 (43), 152 (11), 151 (13), 149 (12), 140 (30), 139 (11), 129 (12), 127 (20), 125 (31), 117 (26), 116 (26), 115 (64), 91 (12), 89 (17), 77 (13), 75 (22), 63 (14), 58 (51), 57 (15), 56 (19), 41 (19)
Соединение 50. (Rt=7.37 мин), m/z (отн. интен.) 261 (M+, 2), 244 (9), 229 (4), 204 (7), 203 (11), 201 (8), 183 (11), 101 (5), 58 (7), 44 (100), 42 (7)
Соединение 51. (Rt=7.30 мин), m/z (отн. интен.) 261 (М+, 5), 244 (20), 229 (9), 204 (14), 203 (23), 202 (6), 201 (20), 183 (27), 133 (7), 121 (6), 101 (9), 75 (6), 58 (7), 44 (100), 43 (6), 42 (11)
Соединение 52. (Rt=7.24 мин), m/z (отн. интен.) 247 (М+, 56), 231 (13), 230 (81), 229 (47), 216 (12), 215 (32), 214 (16), 204 (29), 203 (31), 202 (16), 201 (63), 196 (21), 184 (20), 183 (100), 182 (11), 181 (15), 170 (13), 151 (13), 150 (11), 135 (13), 134 (29), 133 (25), 124 (14), 122 (20), 121 (21), 109 (13), 101 (27), 96 (21), 75 (23), 43 (14), 42 (15)
Соединение 53. (Rt=7.21 мин), m/z (отн. интен.) 247 (М+, 98), 248 (17), 231 (13), 230 (84), 229 (56), 215 (38), 214 (16), 203 (33), 202 (16), 201 (68), 196 (26), 184 (16), 183 (100), 181 (15), 151 (21), 150 (15), 135 (14), 134 (35), 133 (24), 124 (19), 122 (23), 121 (25), 111 (13), 101 (31), 96 (19), 75 (19)
Соединение 55. (Rt=7.86 мин), m/z (отн. интен.) 275 (M+, 98), 276 (20), 258 (59), 229 (58), 216 (31), 215 (22), 214 (19), 204 (49), 203 (41), 202 (21), 201 (82), 184 (18), 183 (100), 181 (14), 150 (21), 135 (33), 133 (55), 124 (41), 115 (13), 109 (90), 101 (15), 83 (20), 75 (16), 72 (23), 57 (13), 56 (24)
Соединение 56. (Rt=7.79 мин), m/z (отн. интен.) 261 (М+, 67), 262 (12), 244 (54), 229 (56), 218 (27), 217 (16), 216 (19), 215 (100), 214 (45), 203 (50), 202 (32), 201 (51), 197 (16), 196 (26), 183 (24), 138 (17), 135 (20), 134 (17), 133 (39), 122 (26), 121 (13), 109 (30), 101 (17), 96 (14), 83 (16), 75 (13)
Соединение 57. (Rt=7.65 мин) m/z (отн. интен.) 261 (М+, 62), 244 (50), 229 (50), 218 (24), 217 (13), 216 (18), 215 (100), 214 (36), 203 (42), 202 (19), 201 (33), 197 (14), 196 (19), 183 (17), 138 (19), 135 (16), 134 (12), 133 (29), 122 (29), 109 (25), 101 (13)
Соединение 58. (Rt=8.15 мин), m/z (отн. интен.) 275 (М+, 134), 276 (26), 258 (23), 244 (19), 243 (100), 232 (25), 229 (53), 217 (51), 216 (23), 215 (67), 214 (97), 201 (44), 197 (21), 196 (43), 183 (23), 148 (38), 147 (21), 138 (46), 135 (46), 134 (18), 133 (64), 125 (25), 123 (28), 122 (81), 115 (27), 109 (54), 107 (17), 83 (27), 44 (19), 43 (19)
Соединение 59. (Rt=7.61 мин), m/z (отн. интен.) 275 (М+, 27), 204 (8), 203 (10), 201 (19), 183 (25), 109 (8), 101 (7), 58 (100), 57 (8), 56 (8), 44 (9)
Соединение 60. (Rt=7.34 мин), m/z (отн. интен.) 261 (М+, 55), 262 (10), 204 (16), 203 (15), 201 (31), 183 (35), 133 (11), 122 (11), 121 (10), 109 (9), 101 (16), 96 (11), 75 (10), 57 (9), 44 (100), 42 (11)
Соединение 61. (Rt=8.07 мин), m/z (отн. интен.) 277 (M+, 68), 278 (13), 260 (31), 246 (11), 245 (25), 234 (12), 231 (32), 229 (26), 217 (20), 203 (23), 201 (24), 188 (12), 183 (22), 154 (24), 151 (15), 150 (10), 133 (18), 124 (10), 109 (100), 95 (11), 44 (14)
Соединение 62. (Rt=8.93 мин), m/z (отн. интен.) 271 (М+, 155), 272 (22), 254 (16), 239 (22), 225 (36), 223 (40), 181 (33), 165 (34), 153 (13), 152 (24), 136 (39), 132 (13), 131 (16), 123 (20), 122 (13), 121 (89), 119 (13), 115 (23), 105 (17), 91 (100), 77 (22)
Соединение 63. (Rt=8.47 мин), m/z (отн. интен.) 287 (М+, 31), 241 (9), 204 (27), 203 (20), 202 (9), 201 (30), 183 (38), 150 (13), 133 (20), 109 (27), 84 (45), 83 (43), 82 (11), 57 (18), 56 (100), 43 (25)
Соединение 64. (Rt=8.57 мин) m/z (отн. интен.) 287 (М+, 63), 288 (13), 270 (14), 242 (16), 241 (17), 215 (17), 214 (18), 204 (35), 203 (27), 202 (18), 201 (70), 183 (86), 150 (18), 147 (16), 146 (17), 135 (16), 133 (45), 109 (45), 84 (31), 83 (38), 82 (13), 75 (15), 57 (21), 56 (100), 43 (44)
Соединение 65. (Rt=8.18 мин), m/z (отн. интен.) 239 (M+, 88), 240 (17), 222 (12), 208 (18), 207 (100), 195 (24), 193 (48), 192 (11), 181 (33), 180 (32), 179 (57), 178 (72), 166 (16), 165 (60), 152 (13), 130 (36), 129 (17), 120 (40), 117 (34), 116 (14), 115 (53), 107 (20), 105 (19), 104 (42), 103 (11), 91 (37), 77 (20), 65 (17)
Соединение 66. (Rt=7.46 мин), m/z (отн. интен.) 275 (М+, 7), 201 (5), 183 (6), 133 (3), 109 (6), 71 (3), 45 (3), 44 (100), 42 (3)
Соединение 67. (Rt=7.56 мин), m/z (отн. интен.) 225 (М+, 24), 194 (8), 193 (12), 179 (6), 168 (10), 167 (12), 166 (6), 165 (20), 152 (9), 120 (8), 116 (6), 115 (7), 103 (7), 77 (8), 51 (5), 44 (100)
Соединение 68. (Rt=7.85 мин), m/z (отн. интен.) 239 (М+, 22), 194 (5), 193 (10), 168 (10), 167 (12), 166 (6), 165 (19), 152 (9), 134 (6), 116 (5), 115 (7), 91 (7), 77 (6), 59 (5), 58 (100), 44 (8)
Соединение 69. (Rt=7.35 мин), m/z (отн. интен.) 275 (М+, 11), 203 (24), 202 (7), 201 (23), 183 (35), 122 (6), 121 (6), 101 (9), 58 (100), 57 (8), 56 (10)
Соединение 72. (Rt=7.90 мин), m/z (отн. интен.) 253 (М+, 25), 238 (9), 193 (7), 168 (8), 167 (14), 165 (17), 152 (9), 115 (7), 91 (11), 73 (8), 72 (100), 58 (45), 56 (7), 44 (6), 43 (9), 42 (8)
Соединение 73. (Rt=7.29 мин), m/z (отн. интен.) 239 (М+, 9), 240 (2), 167 (2), 165 (5), 152 (2), 115 (2), 77 (2), 59 (5), 58 (100), 44 (3), 42 (5)
Соединение 74. (Rt=8.01 мин), m/z (отн. интен.) 267 (М+, 7), 167 (3), 165 (6), 152 (3), 91 (4), 87 (7), 86 (100), 72 (13), 58 (10), 56 (4), 42 (4)
Соединение 79. [Rt=7.89 мин), m/z (отн. интен.) 230 (М+, 37), 214 (15), 213 (100), 212 (62), 201 (26), 200 (72), 198 (21), 195 (12), 188 (17), 187 (85), 186 (46), 185 (42), 184 (9), 157 (12), 135 (9), 133 (24), 109 (10), 107 (20), 106 (62), 80 (14), 79 (32), 78 (9), 51 (20)
Соединение 81 (Rt=7.40 мин), m/z (отн. интен.) 209 (M+, 89), 210 (14), 208 (100), 193 (17), 192 (56), 191 (42), 189 (12), 178 (20), 166 (11), 165 (45), 152 (12), 132 (86), 131 (10), 130 (53), 117 (22), 115 (48), 106 (22), 105 (1), 104 (12), 103 (16), 91 (16), 77 (22), 51 (15)
Соединение 82. (Rt=7.93 мин), m/z (отн. интен.) 275 (М+, 124), 276 (25), 232 (33), 215 (12), 214 (16), 204 (14), 203 (100), 201 (24), 196 (8), 183 (20), 150 (14), 138 (9), 136 (14), 135 (44), 133 (26), 125 (9), 124 (71), 123 (29), 121 (14), 115 (14), 111 (72), 110 (9), 109 (84), 101 (14), 83 (9), 75 (8)
Соединение 83. (Rt=7.22 мин), m/z (отн. интен.) 235 (М+, 10), 219 (17), 218 (100), 217 (62), 203 (20), 192 (10), 191 (38), 190 (7), 189 (14), 185 (17), 183 (7), 171 (9), 165 (8), 147 (10), 146 (11), 134 (12), 133 (17), 121 (8), 109 (8), 97 (8), 45 (7)
Соединение 85. (Rt=7.73 мин), m/z (отн. интен.) 239 (М+, 7), 222 (15), 179 (8), 178 (9), 168 (16), 167 (33), 166 (12), 65 (43), 161 (9), 52 (20), 146 (17), 129 (7), 120 (15),118 (7), 117 (19), 115 (25), 91 (25), 77 (7), 72 (9), 44 (100), 42 (6)
Соединение 86. (Rt=7.66 мин), m/z (отн. интен.) 239 (М+, 3), 222 (4), 168 (4), 167 (11), 166 (4), 165 (14), 152 (7), 120 (6), 117 (6), 115 (8), 91 (9), 72 (5), 44 (100), 42 (3)
Соединение 87. (Rt=7.33 мин), m/z (отн. интен.) 239 (М+, 4), 222 (9), 179 (9), 178 (11), 168 (11), 167 (27), 166 (13), 165 (48), 161 (7), 152 (22), 146 (14), 128 (7), 120 (11), 118 (8), 117 (21), 115 (31), 91 (29), 77 (9), 72 (8), 51 (7), 44 (100), 42 (9)
Соединение 88. (Rt=7.4 мин), m/z (отн. интен.) 227 (М+, 10), 183 (10), 168 (18), 167 (100), 166 (32), 165 (83), 164 (10), 163 (6), 153 (6), 152 (35), 139 (6), 115 (8), 105 (9), 77 (12), 51 (7), 45 (23)
Соединение 89. (Rt=8.74 мин), m/z (отн. интен.) 260 (М+, 220), 261 (39), 259 (89), 242 (18), 203 (17), 202 (16), 201 (61), 183 (58), 165 (100), 150 (20), 148 (25), 138 (24), 137 (61), 122 (73), 121 (31), 111 (47), 101 (23), 96 (16), 75 (16), 44 (17), 43 (29)
Соединение 90. (Rt=7.32 мин), m/z (отн. интен.) 235 (М+, 9), 219 (16), 218 (100), 217 (42), 206 (17), 205 (9), 204 (7), 203 (21), 202 (8), 193 (12), 192 (71), 191 (62), 190 (9), 189 (19), 185 (13), 171 (14), 159 (9), 147 (14), 146 (16), 134 (10), 133 (17), 121 (14), 109 (11), 101 (8), 97 (17), 45 (15)
Соединение 91. (Rt=10.67 мин), m/z (отн. интен.) 329 (М+, 6), 301 (20), 300 (81), 167 (18), 166 (6), 165 (18), 152 (10), 132 (5), 120 (45), 119 (21), 118 (11), 117 (9), 115 (11), 106 (6), 105 (5), 104 (12), 103 (5), 92 (8), 91 (100), 77 (10), 41 (6)
Соединение 92. (Rt=10.37 мин), m/z (отн. интен.) 337 (M+, 30), 336 (7), 204 (7), 203 (7), 201 (7), 183 (10), 133 (6), 121 (8), 120 (70), 106 (6), 92 (9), 91 (100)
Соединение 93. (Rt=10.25 мин), m/2 (отн. интен.) 351 (М+, 28), 352 (7), 337 (9), 336 (39), 203 (10), 201 (11), 183 (17), 135 (6), 134 (20), 133 (6), 132 (6), 120 (11), 118 (5), 109 (18), 106 (12), 105 (100), 104 (13), 103 (8), 91 (14), 79 (11), 77 (12)
Соединение 94. (Rt=10.48 мин), m/z (отн. интен.) 365 (М+, 2), 337 (25), 336 (100), 203 (8), 201 (8), 183 (14), 133 (5), 132 (6), 120 (14), 119 (13), 118 (9), 115 (5), 109 (20), 106 (5), 104 (10), 91 (52)
Соединение 95. (Rt=6.68 мин), m/z (отн. интен.) 283 (М+, 59), 284 (11), 267 (11), 266 (71), 265 (19), 251 (24), 250 (9), 241 (14), 240 (100), 239 (48), 237 (30), 232 (10), 220 (17), 219 (65), 199 (9), 152 (12), 151 (18), 142 (20), 140 (13), 139 (20), 127 (22), 119 (24), 114 (12), 101 (10), 63 (10), 44 (9)
Соединение 96. (Rt=6.93 мин), m/z (отн. интен.) 265 (М+, 46), 249 (16), 248 (100), 247 (34), 233 (27), 232 (11), 223 (9), 222 (65), 221 (39), 220 (10), 219 (36), 202 (14), 201 (54), 152 (15), 151 (14), 133 (9), 124 (12), 119 (9), 109 (9), 101 (14), 75 (9)
Соединение 97. (Rt=8.10 мин), m/z (отн. интен.) 241 (М+, 101), 242 (18), 224 (50), 223 (19), 210 (11), 209 (37), 197 (12), 196 (10), 195 (55), 194 (16), 193 (60), 181 (29), 178 (20), 167 (38), 166 (16), 165 (52), 153 (12), 152 (36), 136 (27), 133 (12), 132 (14), 116 (12), 115 (25), 103 (13), 91 (100), 77 (18)
Соединение 98. (Rt=6.69 мин), m/z (отн. интен.) 232 (М+, 3), 204 (11), 203 (37), 202 (30), 201 (100), 188 (9), 184 (14), 183 (84), 182 (10), 181 (15), 170 (9), 109 (17), 107 (10), 83 (10), 75 (8), 57 (7)
Соединение 99. (Rt=6.75 мин), m/z (отн. интен.) 233 (М+, 2), 204 (12), 203 (68), 202 (26), 201 (100), 200 (6), 188 (9), 184 (13), 183 (86), 182 (8), 181 (14), 170 (9), 133 (6), 109 (15), 107 (11), 83 (11), 81 (7), 75 (7), 57 (9)
Соединение 100. (Rt=7.66 мин), m/z (отн. интен.) 261 (М+, 150), 262 (29), 217 (11), 216 (70), 215 (28), 214 (11), 203 (30), 202 (31), 201 (100), 196 (10), 184 (15), 183 (90), 181 (11), 133 (20), 124 (12), 122 (20), 109 (39), 101 (14), 83 (10), 75 (10), 45 (43)
Соединение 101. (Rt=7.72 мин), m/z (отн. интен.) 245 (М+, 20), 229 (16), 228 (100), 227 (36), 213 (21), 211 (22), 202 (57), 201 (30), 199 (21), 183 (50), 181 (14), 171 (15), 170 (26), 165 (12), 152 (21), 134 (19), 133 (35), 122 (28), 120 (19), 120 (13), 119 (12), 109 (20), 107 (20), 106 (18), 101 (15), 94 (15), 91 (20), 77 (18), 74 (15), 65 (20), 63 (14), 55 (14), 51 (15), 44 (27), 43 (17), 42 (14)
Соединение 102. (Rt=8.33 мин), m/z (отн. интен.) 273 (М+, 19), 204 (16), 203 (16), 201 (15), 183 (18), 177 (9), 133 (8), 109 (13), 70 (41), 69 (100), 68 (20), 43 (25), 42 (5), 41 (5)
Соединение 103. (Rt=8.59 мин), m/z (отн. интен.) 245 (М+, 118), 246 (20), 229 (15), 228 (100), 227 (85), 213 (27), 211 (23), 209 (15), 207 (12), 202 (19), 201 (32), 200 (17), 199 (84), 196 (10), 183 (38), 181 (15), 171 (13), 170 (23), 152 (19), 151 (15), 150 (10), 134 (18), 133 (32), 131 (12), 122 (36), 119 (15), 109 (24), 107 (10), 106 (12), 91 (19), 77 (12)
Соединение 104. (Rt=7.72 мин), m/z (отн. интен.) 261 (М+, 94), 262 (17), 217 (15), 216 (92), 215 (18), 204 (12), 203 (86), 202 (25), 201 (100), 184 (10), 183 (69), 148 (12), 133 (13), 122 (8), 109 (26), 101 (9), 83 (8), 45 (33)
Соединение 105. (Rt=10.24 мин), m/z (отн. интен.) 351 (М+, 7), 201 (5), 183 (7), 135 (9), 134 (79), 133 (4), 109 (5), 92 (8), 91 (100), 65 (8), 42 (7)
Соединение 106. (Rt=7.52 мин), m/z (отн. интен.) 259 (М+, 77), 260 (14), 258 (31), 244 (30), 228 (13), 227 (28), 214 (14), 201 (24), 165 (12), 164 (100), 162 (29), 133 (56), 109 (44), 75 (13), 44 (80), 42 (56)
Соединение 107. (Rt=7.45 мин), m/z (отн. интен.) 227 (М+, 101), 228 (16), 226 (100), 211 (22), 210 (68), 209 (49), 207 (13), 196 (22), 184 (15), 183 (62), 150 (50), 148 (31), 133 (44), 132 (53), 130 (45), 117 (15), 115 (29), 106 (14), 77 (18), 75 (13), 51 (14)
Соединение 108. (Rt=7.46 мин), m/z (отн. интен.) 243 (М+, 34), 244 (6), 212 (6), 211 (9), 197 (6), 186 (12), 185 (10), 184 (5), 183 (19), 165 (15), 133 (6), 120 (6), 103 (5), 77 (6), 44 (100), 42 (6)
Соединение 109. (Rt=8.68 мин), m/z (отн. интен.) 285 (М+, 110), 286 (22), 284 (27), 256 (16), 228 (37), 227 (27), 225 (10), 220 (11), 207 (15), 201 (27), 191 (14), 190 (100), 163 (11), 162 (85), 161 (10), 147 (11), 146 (11), 133 (32), 109 (20), 83 (12), 82 (36)
Соединение 110. (Rt=8.66 мин), m/z (отн. интен.) 285 (М+, 91), 286 (16), 284 (100), 243 (16), 227 (26), 225 (11), 221 (10), 220 (17), 214 (12), 207 (15), 201 (23), 147 (25), 146 (16), 133 (17), 109 (20), 42 (15)
Соединение 111. (Rt=8.81 мин), m/z (отн. интен.) 287 (M+, 29), 214 (9), 204 (15), 203 (18), 202 (9), 201 (34), 183 (42), 135 (9), 133 (28), 109 (28), 84 (47), 83 (100), 82 (19), 75 (8), 70 (16), 68 (13), 57 (18), 56 (28), 44 (16), 43 (25), 42 (14)
Соединение 112. (Rt=8.85 мин), m/z (отн. интен.) 287 (М+, 141), 288 (29), 286 (22), 202 (21), 201 (62), 183 (64), 133 (23), 109 (27), 84 (100), 83 (18), 82 (31), 57 (14), 56 (58), 55 (53), 43 (14), 42 (35)
Соединение 113. (Rt=9.08 мин), m/z (отн. интен.) 251 (М+, 27), 180 (38), 179 (36), 178 (39), 174 (15), 173 (100), 166 (11), 165 (53), 158 (12), 152 (10), 132 (9), 115 (28), 91 (31), 82 (18), 77 (16), 56 (45), 51 (9), 43 (23)
Соединение 114. (Rt=8.71 мин), m/z (отн. интен.) 237 (M+, 197), 238 (37), 236 (67), 193 (15), 179 (30), 178 (40), 165 (41), 159 (43), 158 (26), 132 (24), 130 (16), 116 (17), 115 (37), 106 (21), 103 (34), 91 (50), 77 (48), 57 (68), 56 (100), 51 (32), 43 (50), 42 (34)
Соединение 115. (Rt=9.45 мин), m/z (отн. интен.) 271 (М+, 34), 255 (12), 254 (67), 253 (14), 239 (23), 229 (16), 228 (100), 227 (18), 224 (16), 223 (68), 213 (9), 212 (10), 211 (10), 197 (34), 196 (17), 195 (11), 181 (18), 169 (10), 165 (22), 153 (19), 152 (27), 146 (16), 145 (13), 141 (12), 139 (10), 136 (22), 134 (11), 133 (41), 122 (16), 121 (31), 115 (30), 91 (18), 77 (15), 65 (11), 63 (10), 44 (10)
Соединение 116. (Rt=9.50 мин), m/z (отн. интен.) 269 (M+, 41), 268 (32), 254 (8), 253 (21), 252 (100), 251 (14), 238 (23), 237 (18), 221 (10), 209 (9), 178 (8), 165 (19), 162 (22), 160 (19), 152 (18), 147 (11), 146 (8) 145, (18), 139 (9), 130 (11), 115 (10)
Соединение 117. (Rt=7.64 мин), m/z (отн. интен.) 212 (М+, 13), 183 (16), 182 (100), 180 (7), 167 (7), 152 (3), 104 (27), 91 (7), 78 (4), 77 (41), 51 (13)
Соединение 118. (Rt=7.46 мин), m/z (отн. интен.) 245 (М+, 4), 153 (8), 152 (43), 150 (9), 135 (6), 133 (10), 124 (5), 123 (36), 122 (38), 121 (17), 109 (16), 101 (14), 96 (24), 95 (16), 94 (100), 93 (7), 83 (7), 77 (21), 75 (11), 66 (15), 65 (30), 63 (10), 51 (14), 50 (6)
Соединение 119. (Rt=7.39 мин), m/z (отн. интен.) 263 (М+, 7), 171 (14), 170 (14), 152 (74), 151 (13), 150 (20), 141 (55), 135 (10), 133 (23), 123 (20), 122 (100), 121 (49), 120 (11), 113 (9), 112 (92), 111 (9), 109 (41), 107 (12), 103 (13), 102 (11), 101 (40), 97 (9), 96 (66), 95 (51), 94 (9), 84 (28), 83 (88), 82 (8), 81 (16), 77 (14), 75 (54), 74 (10), 70 (10), 69(10), 64 (10), 63 (23), 57 (62), 56 (13), 51 (15), 50 (12), 42 (8)
Соединение 120. (Rt=8.48 мин), m/z (отн. интен.) 279 (М+, 4), 159 (16), 157 (49), 153 (11), 152 (100), 150 (12), 133 (11), 130 (27), 128 (73), 123 (12), 122 (57), 121 (23), 111 (10), 109 (25), 101 (23), 99 (16), 96 (26), 95 (10), 83 (9), 75 (28), 73 (10), 65 (12), 64 (11), 63 (22), 51 (9), 50 (8)
Соединение 121. (Rt=8.30 мин), m/z (отн. интен.) 275 (M+, 2), 152 (15), 125 (8), 124 (100), 122 (14), 121 (7), 109 (35), 96 (7), 95 (10), 81 (14), 77 (9), 65 (7), 52 (11)
Соединение 122. (Rt=7.39 мин), m/z (отн. интен.) 263 (M+, 11), 170 (12), 152 (66), 151 (10), 150 (18), 141 (68), 135 (10), 133 (19), 123 (16), 122 (76), 121 (39), 112 (100), 111 (18), 109 (36), 107 (11), 103 (11), 102 (9), 101 (33), 96 (56), 95 (32), 92 (11), 83 (96), 81 (13), 77 (13), 75 (43), 64 (25), 63 (26), 57 (61), 56 (14), 51 (14), 50 (11)
Соединение 123. (Rt=5.88 мин), m/z (отн. интен.) 275 (М+, 46), 276 (9), 202 (8), 201 (30), 183 (28), 133 (8), 109 (9), 101 (9), 71 (9), 59 (12), 58 (100), 44 (8), 42 (26)
Соединение 124. (Rt=7.05 мин), m/z (отн. интен.) 229 (М+, 15), 213 (15), 212 (89), 211 (13), 198 (20), 197 (100), 196 (24), 186 (12), 185 (21), 184 (29), 183 (87), 179 (7), 178 (8), 177 (13), 176 (5), 171 (7), 170 (18), 169 (4), 166 (5), 165 (20), 152 (5), 133 (7), 75 (4), 63 (4), 57 (9), 56 (4)
Соединение 125. (Rt=7.54 мин), m/z (отн. интен.) 225 (М+, 57), 226 (13), 209 (13), 208 (75), 193 (13), 180 (14), 179 (21), 178 (20), 165 (22), 130 (34), 117 (59), 115 (28), 105 (18), 104 (94), 103 (45), 91 (100), 78 (30), 77 (38), 65 (36), 63 (13), 51 (20), 45 (17)
Соединение 126. (Rt=7.81 мин), m/z (отн. интен.) 261 (М+, 12), 244 (31), 152 (27), 151 (17), 150 (9), 136 (11), 135 (100), 133 (21), 122 (24), 115 (9), 110 (13), 109 (90), 107 (6), 96 (7), 83 (27), 56 (7)
Соединение 127. (Rt=7.93 мин), m/z (отн. интен.) 225 (М+, 23), 208 (20), 207 (6), 193 (13), 181 (7), 180 (37), 179 (100), 178 (36), 167 (9), 166 (12), 165 (36), 152 (9), 134 (30), 130 (26), 129 (9), 117 (18), 115 (22), 104 (6), 91 (38), 77 (7), 65 (7)
Соединение 128. (Rt=7.42 мин), m/z (отн. интен.) 211 (М+, 83), 212 (15), 194 (36), 193 (18), 182 (62), 181 (20), 180 (17), 179 (53), 178 (60), 176 (11), 167 (57), 166 (44), 165 (100), 152 (24), 120 (39), 116 (12), 115 (28), 104 (22), 103 (15), 91 (46), 89 (16), 78 (10), 77 (20), 65 (15), 63 (12), 51 (12)
Соединение 129. (Rt=7.39 мин), m/z (отн. интен.) 229 (М+, 104), 230 (19), 212 (28), 211 (14), 201 (13), 200 (85), 199 (22), 198 (14), 197 (50), 196 (58), 185 (73), 184 (45), 183 (100), 179 (43), 178 (55), 177 (17), 176 (17), 170 (18), 165 (33), 152 (12), 133 (22), 120 (57), 115 (17), 109 (44), 104 (23), 103 (17), 91 (32), 89 (16), 83 (20), 78 (12), 77 (22), 63 (16), 51 (13)
Соединение 130. (Rt=7.38 мин), m/z (отн. интен.) 229 (М+, 133), 230 (24), 212 (27), 211 (14), 200 (54), 199 (17), 198 (16), 197 (53), 196 (64), 185 (49), 184 (43), 183 (100), 179 (28), 178 (29), 177 (14), 170 (19), 165 (26), 133 (22), 120 (35), 115 (19), 109 (32), 104 (17), 103 (18), 91 (38), 89 (17), 83 (18), 77 (24), 63 (16)
Соединение 131. (Rt=7.40 мин), m/z (отн. интен.) 229 (М+, 146), 230 (26), 212 (48), 211 (23), 200 (51), 199 (17), 198 (16), 197 (61), 196 (70), 185 (50), 184 (43), 183 (100), 179 (28), 178 (28), 170 (20), 165 (23), 133 (21), 120 (35), 115 (20), 109 (59), 104 (25), 103 (17), 91 (27), 89 (17), 83 (22), 77 (22)
Соединение 132. (Rt=7.03 мин), m/z (отн. интен.) 0 (M+, 10), 185 (14), 164 (100), 183 (23), 181 (17), 165 (18), 155 (12), 153 (14), 152 (12), 120 (85), 119 (67), 115 (10), 106 (16), 91 (19), 89 (14), 78 (12), 77 (25), 51 (16)
Соединение 133. (Rt=7.09 мин), m/z (отн. интен.) 211 (М+, 13), 195 (16), 194 (100), 181 (27), 180 (70), 179 (31), 178 (28), 166 (25), 165 (40), 152 (9), 120 (14), 119 (14), 118 (12), 115 (10), 104 (26), 103 (53), 102 (12), 91 (62), 89 (10), 78 (13), 77 (42), 65 (17), 51 (13)
Соединение 134. (Rt=7.45 мин), m/z (отн. интен.) 211 (M+, 14), 183 (15), 162 (100), 181 (14), 179 (13), 178 (18), 167 (27), 166 (18), 165 (46), 152 (10), 115 (8), 104 (8), 103 (6), 91 (29), 89 (7), 78 (5), 77 (7), 65 (7)
Соединение 135. (Rt=8.60 мин), m/z (отн. интен.) 273 (М+, 34), 257 (14), 256 (76), 231 (16), 230 (100), 228 (18), 227 (57), 213 (14), 211 (37), 202 (30), 201 (40), 199 (26), 184 (13), 183 (50), 181 (12), 171 (17), 170 (20), 152 (15), 150 (19), 134 (15), 133 (31), 122 (14), 121 (29), 109 (16), 107 (13), 106 (17), 91 (12), 65 (12)
Соединение 136. (Rt=9.26 мин), m/z (отн. интен.) 275 (М+, 44), 277 (15), 260 (28), 259 (19), 258 (81), 257 (13), 243 (15), 234 (33), 233 (19), 232 (100), 231 (13), 229 (15), 227 (42), 224 (15), 223 (86), 208 (13), 197 (45), 196 (26), 195 (13), 182 (14), 181 (33), 179 (11), 178 (18), 166 (22), 165 (60), 164 (12), 163 (10), 153 (32), 152 (55), 151 (18), 149 (10), 139 (11), 137 (17), 136 (19), 121 (13), 115 (25), 102 (11), 91 (16), 77 (17)
Соединение 137. (Rt=7.42 мин), m/z (отн. интен.) 245 (М+, 1), 153 (8), 152 (7), 141 (64), 135 (10), 134 (100), 132 (11), 117 (6), 115 (12), 112 (56), 105 (15), 104 (55), 103 (32), 95 (8), 91 (16), 84 (8), 83 (15), 78 (24), 77 (24), 75 (9), 65 (6), 63 (8), 57 (10), 51 (9)
Соединение: 138. (Rt=9.24 мин), m/z (отн. интен.) 289 (M+, 77), 290 (16), 230 (20), 229 (21), 215 (15), 203 (22), 201 (32), 183 (36), 134 (10), 133 (13), 124 (10), 121 (9), 109 (10), 101 (10), 73 (100), 43 (23)
Соединение 139. (Rt=7.25 мин), m/z (отн. интен.) 245 (М+, 92), 246 (15), 244 (67), 229 (16), 228 (63), 227 (46), 225 (10), 224 (15), 214 (13), 201 (39), 183 (13), 151 (13), 150 (100), 149 (14), 148 (58), 135 (22), 133 (54), 124 (14), 122 (12), 109 (18), 101 (15), 75 (13)
Соединение 140a. (Rt=8.64 мин), m/z (отн. интен.) 271 (М+, 72), 272 (14), 270 (37), 255 (21), 254 (100), 242 (19), 227 (14), 226 (63), 225 (50), 199 (19), 197 (30), 196 (25), 183 (32), 176 (27), 170 (20), 150 (44), 148 (34), 146 (14), 133 (32), 131 (14), 121 (11)
Соединение 140b. (Rt=8.68 мин), m/z (отн. интен.) 271 (М+, 57), 272 (10), 270 (33), 255 (20), 254 (100), 242 (15), 227 (12), 226 (54), 225 (40), 209 (8), 199 (14), 197 (22), 196 (19), 183 (25), 176 (21), 170 (16), 150 (33), 148 (22), 146 (9), 133 (20), 131 (10)
Соединение 141. (Rt=8.44 мин), m/z (отн. интен.) 257 (М+, 48), 258 (8), 256 (36), 241 (21), 240 (100), 239 (19), 226 (22), 225 (20), 209 (11), 197 (14), 196 (18), 183 (25), 170 (16), 162 (19), 160 (10), 150 (28), 148 (26), 147 (9), 146 (8), 145 (13), 133 (20), 130 (8), 121 (10)
Соединение 142. (Rt=8.47 мин), m/z (отн. интен.) 273 (М+, 14), 217 (5), 216 (31), 215 (5), 183 (8), 170 (4), 150 (5), 121 (4), 58 (5), 45 (5), 44 (100)
Соединение 143. (Rt=9.39 мин), m/z (отн. интен.) 273 (М+, 47), 275 (16), 274 (19), 272 (36), 258 (39), 257 (26), 256 (100), 255 (17), 242 (25), 241 (15), 221 (23), 178 (25), 177 (11), 176 (14), 168 (14), 167 (11), 166 (54), 165 (34), 164 (34), 163 (16), 162 (45), 160 (19), 152 (28), 151 (22), 149 (19), 147 (18), 145 (24), 139 (11), 136 (15), 131 (15), 130 (35), 121 (15), 115 (14), 111 (11), 103 (13), 102 (19), 89 (11), 77 (16), 75 (14), 63 (16), 51 (12)
Соединение 148. (Rt=8.43 мин), m/z (отн. интен.) 261 (М+, 3), 170 (14), 169 (5), 168 (44), 153 (4), 151 (4), 140 (6), 139 (4), 138 (15), 132 (6), 125 (7), 123 (40), 115 (6), 103 (24), 102 (8), 101 (5), 95 (7), 94 (100), 89 (5), 77 (22), 75 (6), 66 (8), 65 (16), 63 (7), 51 (10), 50 (4).
Соединение 145. (Rt=9.28 мин), m/z (отн. интен.) 295 (М+, 4), 170 (32), 169 (12), 168 (100), 166 (8), 159 (22), 157 (66), 152 (11), 140 (16), 139 (11), 138 (41), 132 (11), 130 (32), 129 (8), 128 (82), 127 (10), 125 (16), 115 (12), 111 (15), 103 (55), 102 (18), 101 (15), 99 (19), 89 (10), 77 (26), 76 (8), 75 (27), 73 (11), 65 (11), 64 (10), 63 (22), 51 (11).
Соединение 150. (Rt=8.32 мин), m/z (отн. интен.) 279 (М+, 4), 171 (9), 170 (37), 169 (13), 168 (100), 166 (8), 142 (8), 141 (88), 140 (19), 139 (12), 138 (42), 132 (12), 130 (7), 125 (16), 115 (12), 113 (10), 112 (89), 111 (11), 104 (8), 103 (60), 102 (19), 101 (12), 95 (14), 89 (11), 84 (11), 83 (24), 77 (29), 76 (6), 75 (24), 63 (13), 57 (17), 51 (11).
Пример 30: Биологические свойства синтезированных арилалкиламинов
Соединения, синтезированные, как описано в примере 28 и примере 29, испытывали на различные биологические свойства, детализированные в примерах.
Таблица 1
а: Ингибирование индуцированных NMDA/глицином увеличений концентрации внутриклеточного кальция в культивируемых мозжечковых зернистых клетках крысы (RCGC) (см. пример 1). (№ в скобках указывает номер экспериментов).
b: Соль трифторуксусной кислоты (TFA).
c: Ингибирование связывания [3H]MK-801 в промытых препаратах мембран кортикальных клеток/клеток гиппокампа крыс (см. пример 4).
d: Незавершенное IС50-исследование. % ингибирования при указанной концентрации.
Сравнение величин IC50 в RCGC-тесте с величинами IC50 в тесте связывания [3H]MK-801 (Таблица 1) показывает, что арилалкиламины данного изобретения ингибируют активность NMDA-рецептора по механизму, отличающемуся от механизма связывания с сайтом связывания МК-801; концентрация соединения, которая ингибирует функцию NMDA-рецептора, на несколько порядков ниже концентрации, которая конкурирует при сайте, меченном [3H]МК-801. Однако это не так с упрощенными арилалкиламинами, представленными соединениями 19-150. Такие соединения связываются с сайтом, меченным при помощи [3H]MK-801, при концентрациях в диапазоне приблизительно в 1-400 раз более высоких концентраций, чем концентрации, которые противодействуют опосредованной NMDA-рецептором функции в тесте с крысиными мозжечковыми зернистыми клетками.
Некоторые из описанных упрощенных арилалкиламинов имеют структурные особенности, одинаковые с частями других соединений, которые используются в качестве, например, антихолинергических средств, средств против болезни Паркинсона, антигистаминных средств, антидепрессантов, ингибиторов кальциевых каналов, коронарных вазодилататоров, опиатных аналгетиков и антиаритмических средств. Однако при оценке некоторых из этих соединений на антагонистическую активность против NMDA-рецептора (Пример 1), как можно видеть в Таблице 2, ни одно из испытанных соединений, за исключением (R)- и (S)-фендилина и нисоксетина, не имело величин IC50 менее 1 мкМ. Эти данные суммированы в Таблице 2.
Таблица 2



























а: Ингибирование индуцированных NMDA/глицином увеличений концентрации внутриклеточного кальция в культивируемых крысиных мозжечковых зернистых клетках (RCGC) (см. пример 1)
b: Описано как соединение 2 в таблице 4 в Marcusson et al., Inhibition of [3 H]paroxetine binding by various serotonin uptake inhibitors: structure-activity relationships. Europ. J. Pharmacol. 215: 191-198, 1992.
с: Описано как соединение 17 в Jacobsen et al., Aryloxy-phenylpropylamines and their calcium overload blocking compositions and methods of use. U.S. Patent No. 5310756, May 10, 1994.
d: Описано как соединение 25 в Jacobsen et al., Aryloxy-phenylpropylamines and their calcium overload blocking compositions and methods of use. U.S. Patent No. 5310756, May 10, 1994.
Исследования по взаимосвязи структуры-активности были начаты с использованием соединения 19 как ведущей (основной) структуры. Исследования боковой цепи показало, что пропильная боковая цепь была оптимальной для антагонистической активности в отношении NMDA-рецептора (Таблица 3). Это открытие было подтверждено с использованием соединения 20 в качестве ведущей (исходной) структуры (Таблица 3).






а: Ингибирование индуцированных NMDA/глицином увеличений концентрации внутриклеточного кальция в культивируемых крысиных мозжечковых зернистых клетках (RCGC) (см. пример 1).
Дальнейшие исследования SAR определяли оптимальный характер замещения фенильного кольца. Начальные исследования показали, что замещение галогена (фтора или хлора) в мета-положении было оптимальным для антагонистической активности в отношении NMDA-рецептора (Таблица 4). Увеличение числа замещений фтором приводило к явному снижению активности (Таблица 4).
Таблица 4

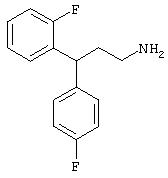










а: Ингибирование индуцированных NMDA/глицином увеличений концентрации внутриклеточного кальция в культивируемых крысиных мозжечковых зернистых клетках (RCGC) (см. пример 1).
Замена одной из групп фтора на одном фенильном кольце группами метилом, метокси или гидрокси не приводила к изменению или снижению антагонистической активности in vitro в отношении NMDA-рецептора. Орто-положение было оптимальным для групп метил, метокси или гидрокси и порядок активности для этого замещения был метил > метокси > гидрокси (Таблица 5). В Таблице 5 показаны также соединения, обладающие 3,3-бис(3-фторфенил)-частью молекулы с дополнительными заместителями метил или метокси на фенильных кольцах, часто приводящие к увеличению антагонистической активности против NMDA-рецептора. Таблица 5 иллюстрирует также соединения, имеющие 3, 3-бис(2-метилфенил)- или 3,3-бис(2-метоксифенил)-часть молекулы вместо 3,3-бис(3-фторфенил)-части; эти замещения являются приемлемыми, хотя отмечается снижение активности.
Таблица 5



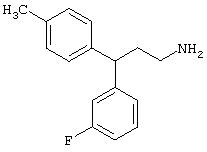

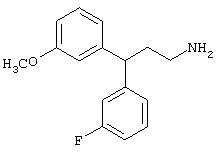











а: Ингибирование индуцированных NMDA/глицином увеличений концентрации внутриклеточного кальция в культивируемых крысиных мозжечковых зернистых клетках (RCGC) (см. пример 1).
В следующей серии экспериментов SAR исследовали действие замещений алкильной цепи (разветвленного характера) на антагонистическую активность in vitro в отношении NMDA-рецептора. Присоединение метильной группы на α - или β -углероде на пропильной стороне цепи не приводило к снижению или к изменению активности соответственно (Таблица 6).
Таблица 6














а: Ингибирование индуцированных NMDA/глицином увеличений концентрации внутриклеточного кальция в культивируемых крысиных мозжечковых зернистых клетках (RCGC) (см. пример 1).
В следующей серии экспериментов SAR исследовали влияние включения двойной связи в пропильной цепи на антагонистическую активность in vitro в отношении NMDA-рецептора (Таблица 7). Как видно из таблицы 7, включение двойной связи снижало активность последовательным образом.
Таблица 7







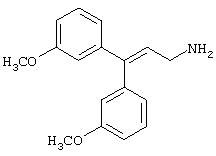
а: Ингибирование индуцированных NMDA/глицином увеличений концентрации внутриклеточного кальция в культивируемых крысиных мозжечковых зернистых клетках (RCGC) (см. пример 1).
В следующей серии экспериментов SAR исследовали влияние включения цепи пропиламина в кольцевую структуру на антагонистическую активность in vitro в отношении NMDA-рецептора (Таблица 8).
Таблица 8





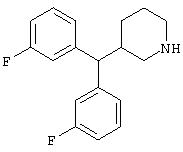

а: Ингибирование индуцированных NMDA/глицином увеличений концентрации внутриклеточного кальция в культивируемых крысиных мозжечковых зернистых клетках (RCGC) (см. пример 1).
В следующей серии экспериментов SAR исследовали влияние простого алкильного замещения на азоте на антагонистическую активность in vitro в отношении NMDA-рецептора (Таблица 9).
Таблица 9












а: Ингибирование индуцированных NMDA/глицином увеличений концентрации внутриклеточного кальция в культивируемых крысиных мозжечковых зернистых клетках (RCGC) (см. пример 1).
Некоторые упрощенные производные арилалкиламина были выбраны для оценки активности в серии тестов связывания рецептора нейротрансмиттера (нейромедиатора) и для определения активности против кальциевого канала L-типа и канала с замедленным выпрямлением калиевого тока. Эти соединения были неактивными (менее 50% ингибирования при концентрациях до 10 мкМ) в следующих тестах: неселективного α 2-адренергического рецептора (связывания [3H]RX 821002 в коре головного мозга крыс), рецептора H1 гистамина (связывания [3 H]пириламина в бычьем мозжечке), неселективного сигма-рецептора (связывания [3H]DTG в мозгу морских свинок), неселективного опиатного рецептора (связывания [3H]налоксона в переднем мозгу крыс), моноаминоксидазной активности (МАО), как МАО-А (метаболизма [14С]серотонина в митохондриях печени крысы), так и МАО-В (метаболизма [14С]фенилэтиламина в митохондриях печени крысы).
Как можно видеть в таблице 10, отмечена активность для нескольких соединений при концентрациях ниже 10 мкМ в следующих тестах: кальциевого канала L-типа, канала с замедленным выпрямлением калиевого тока, связывания центрального мускаринового холинергического рецептора и тестах связывания с поглощением моноамина (допамина, норэпинефрина и серотонина). Этот профиль активности в тестах связывания центрального мускаринового холинергического рецептора и тестах связывания с поглощением моноамина не является неожиданным, с учетом химических структур наших упрощенных арилалкиламинов (см. Таблицу 2 выше). За исключением, однако, активности соединения 19 в тесте связывания с поглощением серотонина, активности соединения 34 в тесте связывания с поглощением допамина, активности соединения 50 в тесте связывания с поглощением серотонина, активности соединений 63 и 64 в тесте связывания с поглощением допамина и активности соединения 60 в тестах связывания с поглощением допамина и серотонина, упрощенные арлиалкиламиновые соединения были наиболее активными при рецепторе NMDA.
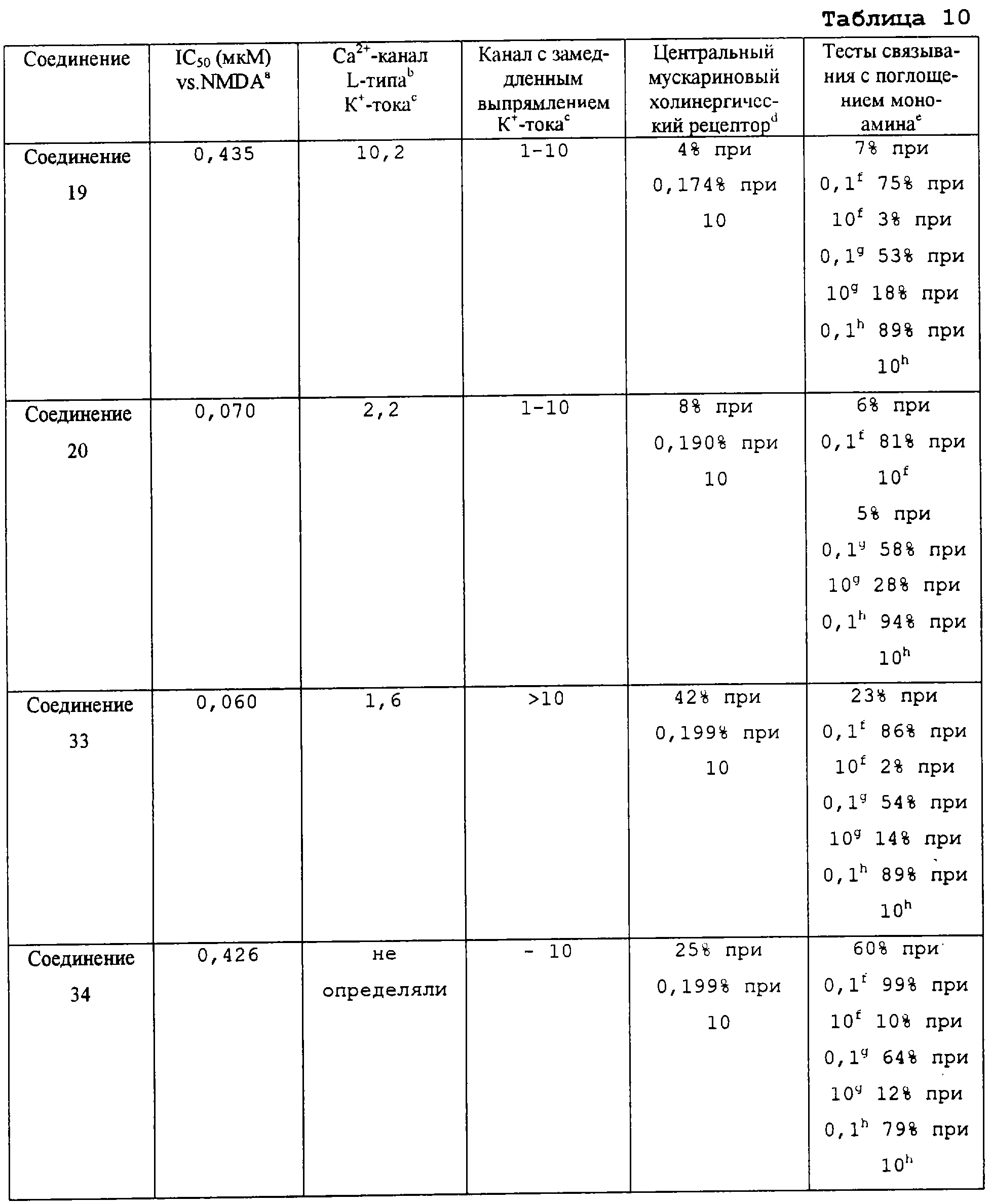


е: Ингибирование связывания [3H]WIN-35 428 с мембранами полосатого тела мозга морской свинки (тест связывания с поглощением допамина), [3H]дезипрамина с кортикальными мембранами крысы (тест связывания с поглощением норэпинефрина) или [3H]циталопрама с мембранами переднего мозга крысы (тест связывания с поглощением серотонина; процент ингибирования при указанной концентрации в мкМ или IC50, когда это доступно.
f: тест связывания с поглощением допамина.
g: тест связывания с поглощением норэпинефрина.
h: тест связывания с поглощением серотонина.
Ценные свойства арилалкиламиновых соединений данного изобретения иллюстрируются тем фактом, что концентрации, которые подавляют медиируемую NMDA-рецептором синаптическую передачу возбуждения, неспособны ингибировать LTP. Кроме того, хотя такие соединения, как соединения 9 и 11, действительно производят гипотензивное действие после системного введения крысам, гипотензивное действие, производимое этими соединениями, имеет относительно короткую продолжительность (приблизительно 30 минут). Кроме этого, соединения 12 и 14 не имеют сердечно-сосудистой активности при дозах до 37,3 мкмоль/кг i.v. и 15 мкмоль/кг i.v. соответственно.
Таблица 11
а: Концентрация, которая подавляет медиируемую NMDA-рецептором синаптическую передачу возбуждения (см. пример 5).
b: Концентрация, которая не ингибирует индукцию LTP (см. пример 19).
c: Снижение общего кровяного давления, производимое введением соединения в крыс (см. пример 22).
Приготовление лекарственных средств и введение
Как показано здесь, ценные соединения этого изобретения и их фармацевтически приемлемые соли могут быть использованы для лечения неврологических нарушения или заболеваний. Хотя эти соединения в типичном случае будут использоваться для больных людей, их можно также использовать для лечения сходных или идентичных заболеваний у других позвоночных, например других приматов, сельскохозяйственных животных, таких как свиньи, крупный рогатый скот и домашняя птица, и спортивных животных и любимых домашних животных, таких как лошади, собаки и кошки.
В терапевтических и/или диагностических приложениях соединения данного изобретения могут быть приготовлены для различных способов введения, в том числе для системного и местного, или локализованного, введения. Способы и описания приготовления в общем можно найти в Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton PA.
Фармацевтически приемлемые соли в общем хорошо известны специалистам с обычной квалификацией в данной области и могут включать в себя, например (но не в качестве ограничения), ацетат, бензолсульфонат, безилат, бензоат, бикарбонат, битайтрат, эдетат кальция, камсилат, карбонат, цитрат, эдетат, эдисилат, эстолат, эсилат, фумарат, глюцептат, глюконат, глутамат, гликоллиларсанилат, гексилресорцинат, гидробамин, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изетионат, лактат, лактобионат, малат, малеат, манделат, мезилат, мукат, напсилат, нитрат, памоат (эмбонат), пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, сульфат, таннат, тартрат или теоклат. Другие фармацевтически приемлемые соли могут быть найдены, например, в Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton PA, (18th ed, 1990).
Предпочтительные фармацевтически приемлемые соли включают, например, ацетат, бензоат, бромид, карбонат, цитрат, глюконат, гидробромид, гидрохлорид, малеат, мезилат, напсилат, памоат (эмбонат), фосфат, салицилат, сукцинат, сульфат или тартрат.
Ценные соединения данного изобретения могут также быть в форме фармацевтически приемлемых комплексов. Фармацевтически приемлемые комплексы известны специалистам с обычной квалификацией в данной области и включают, например (но не для ограничения), 8-хлортеофиллинат (теоклат).
Конкретные препарат, путь введения и доза могут быть выбраны индивидуальным врачом в связи с состоянием пациента (См., например, Fingl et al., in The Pharmacological Basis of Therapeutics, 1975, Ch. 1, p.1).
Следует отметить, что штатный врач больницы должен знать, как и когда закончить, прервать или видоизменить введение из-за токсичности или дисфункции органа. Напротив, штатный врач больницы должен также уметь корректировать лечение для использования более высоких уровней, если клинические реакции не были адекватными (предотвратить токсичность). Величина вводимой дозы в лечении представляющего интерес нарушения будет варьировать в зависимости от состояния, подлежащего лечению, и пути введения. Тяжесть заболевания может оцениваться отчасти стандартными прогностическими способами оценки. Кроме того, доза и, возможно, частота доз, также будут меняться в зависимости от возраста, веса тела и ответной реакции конкретного пациента. Программа, сравнимая с обсуждаемой выше, может быть использована в ветеринарии.
В зависимости от конкретных состояний, подлежащих лечению, такие агенты могут готовиться в виде жидких или твердых дозированных форм и вводиться системно или локально. Эти агенты могут доставляться, например, в форме с пролонгированным действием, известной специалистам в данной области. Способы приготовления и введения могут быть найдены в Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton PA. Пригодные способы введения могут включать пероральное, щечное, подъязычное, ректальное, трансдермальное, вагинальное, трансмукозное, назальное или кишечное введение; парентеральную доставку, в том числе внутримышечную, подкожную, интрамедулярную инъекции, а также внутриоболочечную, прямую интравентрикулярную, внутривенную, внутрибрюшинную, интраназальную или внутриглазную инъекции, среди прочих.
Для инъекции агенты данного изобретения могут быть приготовлены в виде водных растворов, предпочтительно в физиологически совместимых буферах, таких как раствор Хенка, раствор Рингера или физиологический солевой буфер. Для введения через слизистую оболочку в препарате используют пенетранты (смачивающие вещества), пригодные для облегчения проникновения через этот барьер. Такие пенетранты в общем известны в данной области.
Использование фармацевтически приемлемых носителей для приготовления лекарственных форм из описанных здесь соединений для практического использования изобретения в дозах, пригодных для системного введения, входит в объем данного изобретения. При правильном выборе носителя и подходящих способов приготовления композиции данного изобретения могут вводиться парентерально, например внутривенной инъекцией. Соединения могут быть легко приготовлены с использованием фармацевтически приемлемых носителей, хорошо известных в данной области, в виде лекарственных доз, пригодных для перорального введения. Такие носители позволяют готовить соединения данного изобретения в виде таблеток, пилюль, капсул, жидкостей, гелей, сиропов, суспензий и т.п., для перорального проглатывания пациентом, подлежащим лечению.
Агенты, предназначенные для внутриклеточного введения, могут вводиться способами, хорошо известными среднему специалисту в данной области. Например, такие агенты могут быть инкапсулированы в липосомы и затем введены, как описано выше. Липосомы представляют собой сферические липидные бислои с водными внутренними пространствами. Все молекулы, присутствующие в водном растворе во время образования липосом, включаются в это внутреннее пространство. Содержимое липосом защищено от наружной микросреды и вследствие слияния липосом с клеточными мембранами эффективно доставляется в цитоплазму клетки. Кроме того, вследствие гидрофобности липосом небольшие органические молекулы могут быть непосредственно введены внутрь клеток.
Фармацевтические композиции, пригодные для использования в данном изобретении, включают композиции, в которых активные ингредиенты содержатся в эффективном количестве для достижения предполагаемой цели. Определение эффективных количеств также находится в пределах квалификации специалистов в данной области, особенно в свете обеспеченного здесь подробного описания.
Кроме активных ингредиентов, фармацевтические композиции могут содержать подходящие фармацевтически приемлемые носители, включающие наполнители и вспомогательные вещества, которые облегчают обработку активных соединений для получения препаратов, которые можно использовать фармацевтически. Препараты, приготовленные для перорального введения, могут быть в форме таблеток, драже, капсул или растворов.
Фармацевтические композиции данного изобретения могут быть приготовлены способом, который сам по себе известен, например посредством обычных способов смешивания, растворения, гранулирования, изготовления драже, растирания, эмульгирования, инкапсулирования, улавливания или лиофилизации.
Фармацевтические готовые формы для парентерального введения включают водные растворы активных соединений в водорастворимой форме. Пригодные липофильные растворители или носители включают жирные масла, такие как кунжутное масло, или синтетические сложные эфиры жирных кислот, такие как этилолеат, или триглицериды, или липосомы. Водные инъекционные суспензии могут содержать вещества, которые увеличивают вязкость суспензии, такие как натрий-карбоксиметилцеллюлоза, сорбит или декстран. Необязательно, суспензия может также содержать подходящие стабилизаторы или агенты, увеличивающие растворимость этих соединений, для получения высококонцентрированных растворов.
Фармацевтические препараты для перорального использования могут быть получены объединением активных соединений с твердыми наполнителями, необязательным размалыванием полученной смеси и обработкой с получением гранул после добавления подходящих добавок, если желательно, для получения таблеток или центральных частей драже. Подходящими наполнителями являются, в частности, такие наполнители, как сахара, в том числе лактоза, сахароза, маннит или сорбит; целлюлозные препараты, например кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий-карбоксиметилцеллюлоза (CMC) и/или поливинилпирролидон (PVP: повидон). Если желательно, могут быть добавлены дезинтегрирующие агенты, такие как сшитый поливинилпирролидон, агар, или альгиновая кислота, или ее соль, такая как альгинат натрия.
Центральные участки драже обеспечивают подходящими покрытиями. Для этой цели можно использовать концентрированные растворы сахаров, которые могут необязательно содержать аравийскую камедь, тальк, поливинилпирролидон, гель карбопола, полиэтиленгликоль (PEG) и/или диоксид титана, растворы лака и подходящие органические растворители или смеси растворителей.
К таблеткам или покрытиям драже могут быть добавлены красители или пигменты для идентификации или для характеристики различных комбинаций доз активного соединения.
Фармацевтические препараты, которые могут применяться перорально, включают push-fit капсулы, приготовленные из желатина и пластификатора, такого как глицерин или сорбит. Push-fit капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связывающими веществами, такими как крахмалы, и/или смачивающими веществами, такими как тальк или стеарат магния, и необязательно стабилизаторы. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли (PEG). Кроме того, могут быть добавлены стабилизаторы.
Другие варианты находятся в рамках следующей далее формулы изобретения.
Реферат
Изобретение относится к области неврологии. Описаны арилалкиламины, специфически действующие на определенные типы оперируемых рецепторами Са2+-каналов, их применение и фармацевтические композиции для лечения неврологических расстройств или заболеваний. 6 н. и 49 з.п. ф-лы, 11 табл.
Формула



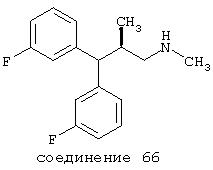




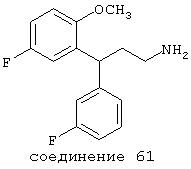
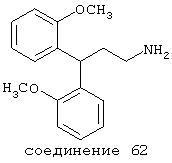










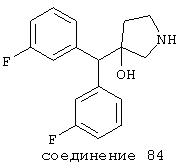
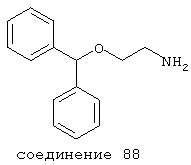





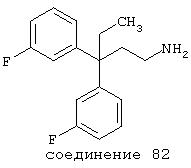



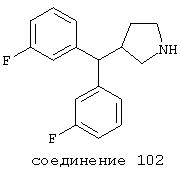







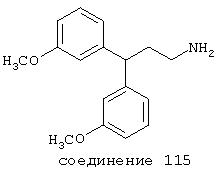
















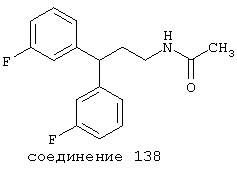







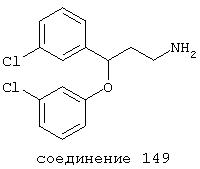
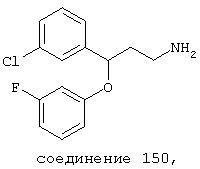







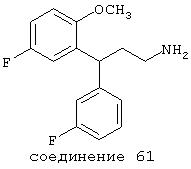







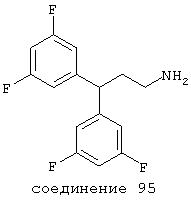








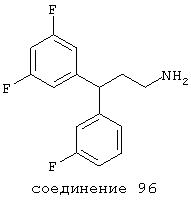
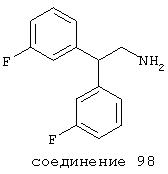




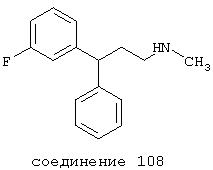












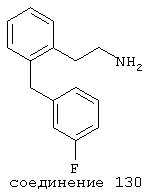




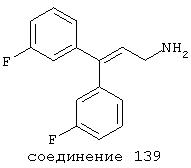



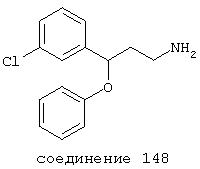








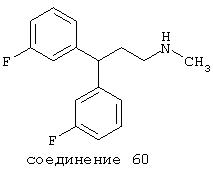








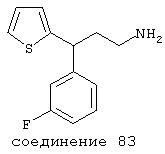

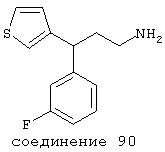

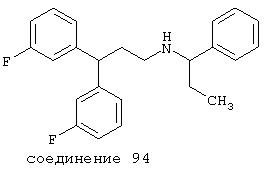




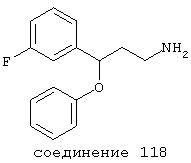

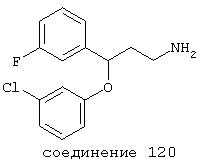


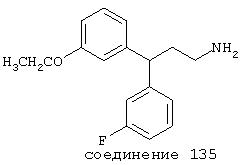



















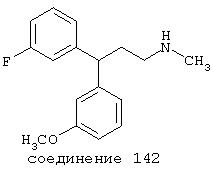


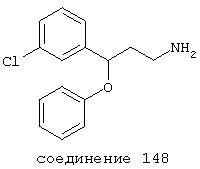





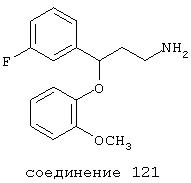









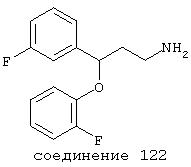




































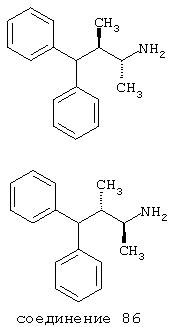



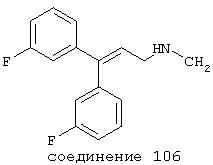





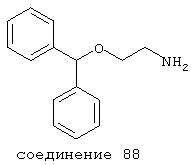












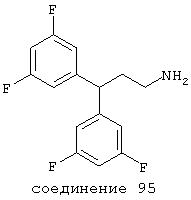



































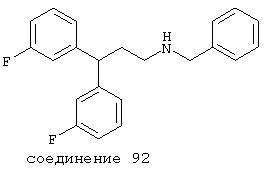







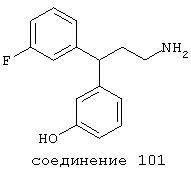










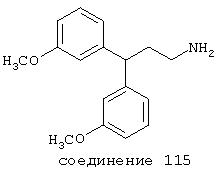














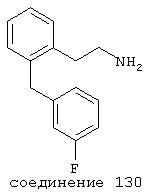










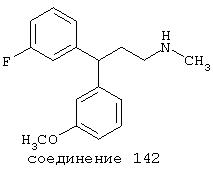




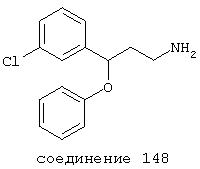























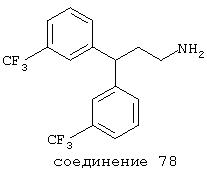

















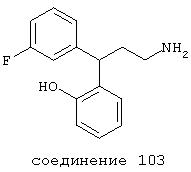







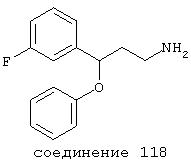










































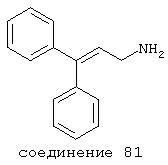










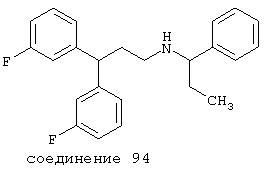


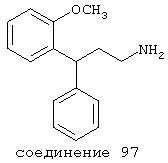




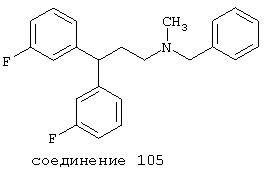






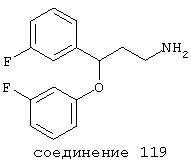









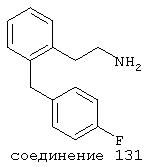






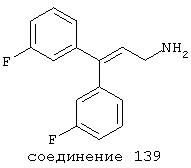

















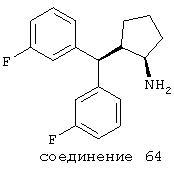









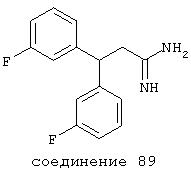
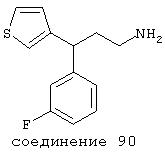








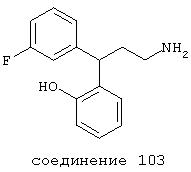












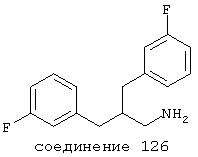






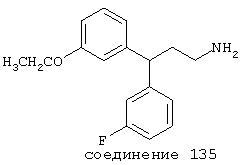
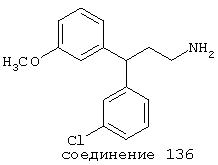


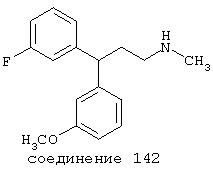
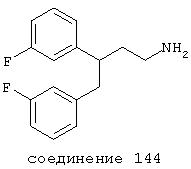













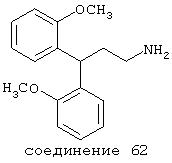


























































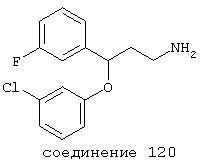
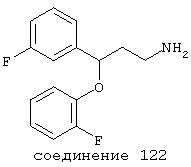















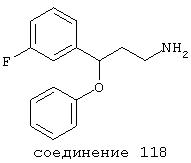









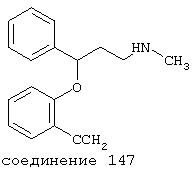









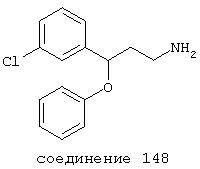















Комментарии