Замещенные производные 4-аминоциклогексана - RU2532545C2
Код документа: RU2532545C2
Описание
Настоящее изобретение относится к замещенным производным циклогексана, которые обладают сродством к µ-опиоидному рецептору и рецептору ORL1, к способу их получения, лекарственным средствам, содержащим эти соединения, и к применению этих соединений для получения лекарственных средств.
Производные циклогексана, которые обладают сродством к µ-опиоидному рецептору и к рецептору ORL1, известны из уровня техники. В связи с этим в полном объеме можно сделать ссылку, например, на WO 2002/090317, WO 2002/90330, WO 2003/008370, WO 2003/008731, WO 2003/080557, WO 2004/043899, WO 2004/043900, WO 2004/043902, WO 2004/043909, WO 2004/043949, WO 2004/043967, WO 2005/063769, WO 2005/066183, WO 2005/110970, WO 2005/110971, WO 2005/110973, WO 2005/110974, WO 2005/110975, WO 2005/110976, WO 2005/110977, WO 2006/018184, WO 2006/108565, WO 2007/079927, WO 2007/079928, WO 2007/079930, WO 2007/079931, WO 2007/124903, WO 2008/009415 и WO 2008/009416.
Однако известные соединения в некотором отношении не являются удовлетворительными, и существует потребность в других соединениях со сравнимыми или улучшенными свойствами.
Таким образом, известные соединения в пригодных анализах связывания иногда демонстрируют некоторое сродство к hERG-ионному каналу, к кальциевому ионному каналу L-типа (участки связывания фенилалкиламина, бензотиазепина, дигидропиридина) соответственно к натриевому каналу в ВТХ-анализе (батрахотоксин), что в каждом случае может быть объяснено как симптомы сердечно-сосудистых побочных действий. Далее многие из известных соединений обладают весьма незначительной растворимостью в водных средах, что, в частности, может оказывать негативное влияние на биодоступность. Кроме того, химическая стабильность известных соединений часто является недостаточной. Таким образом, иногда соединения не проявляют достаточной рН стабильности, устойчивости к УФ соответственно устойчивости к окислению, что, в частности, может отрицательно сказываться на стабильности при хранении, а также на оральной биодоступности. Далее известные соединения частично обладают неблагоприятным PK/PD (фармакокинетика/фармакодинамика) профилем, что может проявляться, например, в длительности времени действия.
Также требует улучшений метаболическая стабильность известных соединений. Улучшенная метаболическая стабильность может отражаться на повышенной биодоступности. Также слабое или отсутствующее взаимодействие с молекулами-переносчиками, которые участвуют в поступлении и выделении лекарственных веществ, следует расценивать как указание на улучшенную биодоступность и, во всяком случае, незначительные взаимодействия лекарственных средств. Далее также как можно более незначительными должны быть взаимодействия с ферментами, участвующими в расщеплении и выведении лекарственных веществ, так как такие результаты тестов равным образом указывают на то, что, во всяком случае, следует ожидать незначительные или вообще отсутствующие взаимодействия лекарственных средств.
Далее известные соединения иногда проявляют весьма незначительную избирательность в отношении каппа-опиоидного рецептора, который отвечает за побочные эффекты, в особенности дисфорию, седативный эффект, диурез. Кроме того, известные соединения иногда проявляют очень высокое сродство к µ-опиоидному рецептору, который связан с другими побочными эффектами, в особенности такими как угнетение дыхания, запор и наркотическая зависимость.
WO 01/87838 раскрывает антагонисты NK-1-рецептора.
J. Med. Chem. 1996, 9, 911-920; J. Am. Chem. Soc. 1950, 72, 2411-2417; и Tetrahedron 2006, 62, 5536-5548 в каждом случае, в частности, раскрывают геминально замещенные циклогексил-1,4-диамины, однако в которых аминогруппы всегда замещены атомами водорода.
DE 2839891 А1 в частности, раскрывает 4-(диметиламино)-1-метил-4-р-толилциклогексил ацетат.
В основе изобретения лежит задача предоставить в распоряжение соединения, которые пригодны для фармацевтических целей и имеют преимущества по сравнению с соединениями из уровня техники.
Эта задача решается с помощью объекта, представленного в формуле изобретения.
Неожиданно было обнаружено, что могут быть получены замещенные производные циклогексана, которые обладают сродством к µ-опиоидному рецептору и рецептору ORL1.
Изобретение относится к соединения общей формулы (1)

где
Y1, Y1', Y2, Y2', Y3, Y3', Y4 и Y4' в каждом случае независимо друг от друга выбраны из группы, состоящей из -Н, -F, -Cl, -Br, -I, -CN, -NO2, -CHO, -R0, -C(=O)R0, -C(=O)H, -C(=O)-OH, -C(=O)OR0, -C(=O)NH2, -C(=O)NHR0, C(=O)N(R0)2, -ОН, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, N(R0)2, -N+(R0)3, -N+(R0)2O-, -NHC(=O)R0, -NHC(=O)OR0, -NHC(=O)NH2, NHC(=O)NHR0 и -NHC(=O)N(R0)2; преимущественно в каждом случае независимо друг от друга выбраны из группы, состоящей из -Н, -F, -Cl, -CN и -C1-8-алифат; или Y1 и Y1', или Y2 и Y2', или Y3 и Y3', или Y4 и Y4' совместно означают =O;
Q означает -R0, -C(=O)-R0, -C(=O)OR0, -C(=O)NHR0, -C(=O)N(R0)2 или -C(=NH)-R0;
R0 в каждом случае независимо означает -C1-8-алифат, -С3-12-циклоалифат, -арил, -гетероарил, -С1-8-алифат-С3-12-циклоалифат, -C1-8-алифат-арил, -C1-8-алифат-гетероарил, -С3-8-циклоалифат-С1-8-алифат, -С3-8-циклоалифат-арил или -С3-8-циклоалифат-гетероарил;
R1 и R2, независимо друг от друга, означают -Н или -R0; или R1 и R2 совместно образуют кольцо и означают -CH2CH2OCH2CH2-, CH2CH2NR4CH2CH2- или -(СН2)3-6-; с указанием, что R1 и R2 преимущественно оба одновременно не означают -Н;
R3 означает -R0;
R4 в каждом случае независимо означает -Н, -R0 или -C(=O)R0;
n означает целое число от 0 до 12; преимущественно означает 0;
Х означает -O-, -S- или -NRA-; преимущественно означает -NRA-;
RA означает -Н, -R0, -S(=O)0-2R0, -C(=O)R0, -C(=O)OR0, -C(=O)NH2, -C(=O)NHR0 или -C(=O)N(R0)2;
RB означает -Н, -R0, -C(=O)H, -C(=O)R0, -C(=O)OH, -C(=O)OR0, -C(=O)NH2, C(=O)NHR0, -C(=O)N(R0)2, -S(=O)1-2-R0, -S(=O)1-2-OR0, -S(=O)1-2-NH2, S(=O)1-2-NHR0 или -S(=O)1-2-N(R0)2; или RA и RB совместно образуют кольцо и означают (СН2)2-5-, -CH2CH2OCH2CH2- или -CH2CH2NR4CH2CH2-; с указанием, что если Х означает -О- и одновременно n означает 0, то RB не означает -Н;
причем
«алифат» в каждом случае представляет собой разветвленный или неразветвленный, насыщенным или моно- или многократно ненасыщенный, незамещенный или моно- или многократно замещенный, алифатический углеводородный остаток;
«циклоалифат» в каждом случае насыщенный или моно- или многократно ненасыщенный, незамещенный или моно- или многократно замещенный, алициклический, моно- или мультициклический углеводородный остаток, число циклических атомов углерода которого преимущественно находится в указанном диапазоне (т.е. «С3-8»-циклоалифат преимущественно имеет 3, 4, 5, 6, 7 или 8 циклических атомов углерода);
причем относительно «алифат» и «циклоалифат» под «моно- или многократно замещенным» понимается однократное или многократное замещение одного или нескольких атомов водорода, например, однократное, двукратное, трехкратное или полное замещение, заместителями независимо друг от друга, выбранными из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -СНО, =O, -R0, -C(=O)R0, -C(=O)H, C(=O)OH, -C(=O)OR0, -C(=O)NH2, -C(=O)NHR0, -C(=O)N(R0)2, -ОН, -OR0, OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N+(R0)3, N+(R0)2O-, -NHC(=O)R0, -NHC(=O)OR0, -NHC(=O)NH2, -NHC(=O)-NHR0, -NH-C(=O)N(R0)2, -Si(R0)3 и -PO(OR0)2;
«арил» в каждом случае независимо означает карбоциклическую кольцевую систему с, по меньшей мере, одним ароматическим кольцом, но без гетероатомов в этом кольце, причем арильные остатки при необходимости могут быть конденсированы с другими насыщенными, (частично) ненасыщенными или ароматическими кольцевыми системами и каждый арильный остаток может быть незамещенным или моно- или многократно замещенным, причем арильные заместители могут быть одинаковыми или различными и в любом и возможном положении арила;
«гетероарил» означает 5-, 6- или 7-членный циклический ароматический остаток, который содержит 1, 2, 3, 4 или 5 гетероатомов, причем гетероатомы представляют собой одинаковый или различный азот, кислород или серу, и гетероцикл может быть незамещенным или моно- или многократно замещенным; причем в случае замещения в гетероцикле заместители могут быть одинаковыми или различными и в любом и возможном положении гетероарила; и причем гетероцикл также может быть частью бициклической или полициклической системы;
причем относительно «арил» и «гетероарил» под «моно- или многократно замещенным» понимается однократное или многократное замещение одного или нескольких атомов водорода кольцевой системы заместителями, выбранными из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -СНО, =O, -R0, -C(=O)R0, -С(=O)Н, -С(=O)ОН, -C(=O)OR0, -C(=O)NH2, -C(=O)NHR0, C(=O)-N(R0)2, -ОН, -O(CH2)1-2O-, -OR0, -OC(=O)H, -OC(=O)Ro, -OC(=O)OR0, OC(=O)NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, NH2, -NHR0, -N(R0)2, -N+(R0)3, -N+(R0)2O-, -NHC(=O)R0, -NHC(=O)OR0, -NH-C(=O)NH2, -NHC(=O)NHR0, -NHC(=O)N(R0)2, -Si(R0)3, -PO(OR0)2; причем при необходимости имеющиеся N-кольцевые атомы в каждом случае могут быть окисленными (N-оксид);
в виде отдельного стереоизомера или его смеси, свободных соединений и/или их физиологически совместимых солей и/или сольватов, причем преимущественно исключается 4-(диметиламино)-1-метил-4-р-толилциклогексил ацетат и его соли.
При обобщении различных остатков, например Y1, Y1', Y2, Y2', Y3, Y3', Y4 и Y4', a также обобщении остатков в их заместителях, таких как, например -OR0, -OC(=O)R0, -OC(=O)NHR0, один заместитель, например, R0, для двух или нескольких остатков, например -OR0, -OC(=O)R0, -OC(=O)NHR0, в пределах одного вещества может принимать различные значения.
Соединения согласно изобретению показывают хорошее связывание с рецептором ORL1 и µ-опиоидным рецептором.
В одной предпочтительной форме осуществления соединения согласно изобретению имеют соотношение ORLI/µ-сродство, по меньшей мере, в 0,1. Соотношение ORLI/µ определено как 1/[Ki(ORL1)/Ki(µ)]. Особенно предпочтительно соотношение ORLI/µ составляет, по меньшей мере, 0,2 или, по меньшей мере, 0,5, предпочтительнее, по меньшей мере, 1,0 или, по меньшей мере, 2,0, еще предпочтительнее, по меньшей мере, 3,0 или, по меньшей мере, 4,0, наиболее предпочтительно, по меньшей мере, 5,0 или, по меньшей мере, 7,5 и в особенности, по меньшей мере, 10 или, по меньшей мере, 15. В одной предпочтительной форме осуществления соотношение ORL1/µ находится в пределах от 0,1 до 30, предпочтительнее от 0,1 до 25.
В одной другой предпочтительной форме осуществления соединения согласно изобретению имеют соотношение ORLI/µ-сродство более чем в 30, предпочтительнее, по меньшей мере, 50, еще предпочтительнее, по меньшей мере, 100, наиболее предпочтительно, по меньшей мере, 200 и в особенности, по меньшей мере, 300.
Соединения согласно изобретению предпочтительно имеют Ki-значение в µ-опиоидном рецепторе самое большее в 500 нМ, предпочтительнее самое большее 100 нМ, еще предпочтительнее 50 нМ, наиболее предпочтительно самое большее 10 нМ и в особенности самое большее 1,0 нМ.
Методы определения Ki-значения в µ-опиоидном рецепторе известны специалисту в данной области техники. Предпочтительно определение происходит, как описано в связи с примерами.
Соединения согласно изобретению предпочтительно имеют Ki-значение в ORL1-рецепторе самое большее в 500 нМ, предпочтительнее самое большее 100 нМ, еще предпочтительнее 50 нМ, наиболее предпочтительно самое большее 10 нМ и в особенности самое большее 1,0 нМ.
Методы определения Ki-значения в ORL1-рецепторе известны специалисту в данной области техники. Предпочтительно определение происходит, как описано в связи с примерами.
Неожиданно оказалось, что соединения со сродством к ORL1- и µ-опиоидному рецептору, в которых определенное посредством 1/[Ki(ORL1)/Ki(µ)] соотношение ORL1 к µ находится в пределах от 0,1 до 30, преимущественно от 0,1 до 25, имеют фармакологический профиль, который по сравнению с другим лигандом опиоидного рецептора имеет явные преимущества:
1. Соединения согласно изобретению демонстрируют эффективность в моделях острой боли, которая иногда сопоставима с употребительной стадией-3 опиоидов. Но одновременно они отличаются явно улучшенной совместимостью в отношении классических µ-опиоидов.
2. В противоположность к употребительной стадии-3 опиоидов соединения согласно изобретению демонстрируют явно более высокую эффективность в моделях мононевропатической боли и полиневропатической боли, что объясняется синергизмом ORL1- и µ-опиоидного компонента.
3. В противоположность к употребительной стадии-3 опиоидов соединения согласно изобретению у невропатических животных показывают значительное, предпочтительно полное, разделение антиаллодинического соответственно антигиперальгетического действия и антиноцицептивного эффекта.
4. В противоположность к употребительной стадии-3 опиоидов соединения согласно изобретению в экспериментальных моделях на животных для хронической воспалительной боли (в частности, каррагинан-индуцированная или CFA-индуцированная гипералгезия, висцеральная воспалительная боль) показывают явное усиление действия в отношении острой боли.
5. В противоположность к употребительной стадии-3 опиоидов типичные для µ-опиоидов побочные эффекты (в частности, угнетение дыхания, вызванная опиоидами гипералгезия, физическая зависимость/ломка, психическая зависимость/мания) в соединениях согласно изобретению в терапевтически эффективном интервале дозы явно снижены, соответственно преимущественно не наблюдаются.
Исходя из сниженных µ-опиоидных побочных эффектов с одной стороны и повышенной эффективности при хронической, предпочтительно невропатической боли с другой стороны смешанные ORLI/µ-агонисты таким образом отличаются отчетливо возрастающими интервалами безопасности по сравнению с чистыми µ-опиоидами. Из этого следует явно увеличенное «терапевтическое окно» при лечении болезненных состояний, предпочтительно хронических болей, еще предпочтительнее невропатических болей.
Предпочтительны Y1, Y1', Y2, Y2', Y3, Y3', Y4 и Y4', в каждом случае независимо друг от друга выбранные из группы, состоящей из -Н, -F, -Cl, -Br, -I, -CN, -NH2, -NH-C1-6-алифат, -NH-С3-8-циклоалифат, -NH-C1-6-алифат-ОН, -N(С1-6-алифат)2, -N(С3-8-циклоалифат)2, -N(С1-6-алифат-ОН)2, -NO2, -NH-С1-6-алифат-С3-8-циклоалифат, -NH-C1-6-алифат-арил, -NH-C1-6-алифат-гетероарил, -NH-арил, -NH-гетероарил, -SH, -S-C1-6-алифат, -S-С3-8-циклоалифат, -S-С1-6-алифат-С3-8-циклоалифат, -S-C1-6-алифат-арил, -S-C1-6-алифат-гетероарил, -S-арил, -S-гетероарил, -ОН, -O-C1-6-алифат, -О-С3-8-циклоалифат, -O-C1-6-алифат-ОН, -O-С1-6-алифат-С3-8-циклоалифат, -O-C1--алифат-арил, -O-C1-6-алифат-гетероарил, -O-арил, -O-гетероарил, -O-С(=O)С1-6-алифат, -O-С(=O)С3-8-циклоалифат, -O-С(=O)С1-6-алифат-ОН, -O-С(=O)С1-6-алифат-С3-8-циклоалифат, -O-C(=O)C1-6-алифат-арил, -O-С(=O)С1-6-алифат-гетероарил, -O-С(=O)арил, -O-С(=O)гетероарил, -C1-6-алифат, -С3-8-циклоалифат, -С1-6-алифат-С3-8-циклоалифат, -C1-6-алифат-арил, -C1-6-алифат-гетероарил, -арил, -гетероарил, -С(=O)С1-6-алифат, -С(=O)С3-8-циклоалифат, -С(=O)С1-6-алифат-С3-8-циклоалифат, -С(=O)С1-6-алифат-арил, -С(=O)С1-6-алифат-гетероарил, -С(=O)арил, -С(=O)гетероарил, -CO2H, -CO2-С1-6-алифат, -CO2-С3-8-циклоалифат, -CO2-С1-6-алифат-С3-8-циклоалифат, -CO2-С1-6-алифат-арил, -CO2-С1-6-алифат-гетероарил, -CO2-арил, -CO2-гетероарил; или Y1 и Y1', или Y2 и Y2', или Y3 и Y3', или Y4 и Y4' совместно означают =O. Предпочтительны Y1, Y1', Y2, Y2', Y3, Y3', Y4 и Y4', в каждом случае независимо друг от друга выбранные из группы, состоящей из -Н, -F, -Cl, -Br, -I, -CN, -NH2 и -ОН.
В одной предпочтительной форме осуществления один из остатков Y1, Y1', Y2, Y2', Y3, Y3', Y4 и Y4' не равен -Н и остальные остатки означают -Н.
Особенно предпочтительно Y1, Y1', Y2, Y2', Y3, Y3', Y4 и Y4' в каждом случае означают -Н.
Предпочтительно Q означает -R0, -C(=O)R0 или -C(=NH)R0. Особенно предпочтительно Q означает -C1-8-алифат, -арил, -гетероарил, -С1-8-алифат-арил, -C1-8-алифат-гетероарил, -С(=O)-С1-8-алифат, -С(=O)-арил, -С(=O)-гетероарил, -С(=O)-С1-8-алифат-арил, -С(=O)-С1-8-алифат-гетероарил, -С(=NH)-C1-8-алифат, -С(=NH)-арил, -С(=NH)-гетероарил, -С(=NH)-С1-8-алифат-арил, или -С(=NH)-С1-8-алифат-гетероарил.
Особенно предпочтительно Q означает -С1-8-алифат, -арил, -гетероарил, -C1-8-алифат-арил, -С(=O)-гетероарил или -С(=NH)-гетероарил.
При этом -арил и -гетероарил в каждом случае могут быть незамещенными или моно- или многократно замещенными, преимущественно заместителями, которые независимо друг от друга выбраны из группы, состоящей из -C1-8-алифат, -ОН, -OC1-8-алифат, -С1-8-алифат-O-С1-8-алифат (например, -CH2-O-СН3), -CF3, -F, -Cl, -Br, -NO2, -CN, -гетероарил, -C1-8-алифат-арил и -С1-8-алифат-гетероарил.
В одной предпочтительной форме осуществления Q выбран из группы, состоящей из -C1-8-алкил, -фенил, -бензил, -пирролил, -фурил, -тиенил, пиридил, -индолил, -бензофурил и -бензотиенил, причем в каждом случае они могут быть незамещены или моно- или многократно замещены, преимущественно заместителями, которые независимо друг от друга выбраны из группы, состоящей из -C1-8-алифат, -ОН, -OC1-8-алифат, -С1-8-алифат-O-С1-8-алифат, -CF3, -F, -Cl, -Br, -NO2, -CN, -гетероарил, -C1-8-алифат-арил и -C1-8-алифат-гетероарил (например, -этил-4-пиридил). Особенно предпочтительно Q выбран из группы, состоящей из:





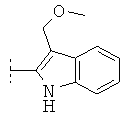








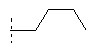









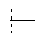
R0 предпочтительно в каждом случае независимо означает -C1-8-алифат, -С3-12-циклоалифат, -арил, -гетероарил, -С1-8-алифат-С3-12-циклоалифат, -C1-8-алифат-арил или -С1-8-алифат-гетероарил. При этом означают -С1-8-алифат-С3-12-циклоалифат, -C1-8-алифат-арил или -C1-8-алифат-гетероарил, что остатки -С3-12-циклоалифат, -арил или -гетероарил в каждом случае связаны через двухвалентный мостик -C1-8-алифат-. Предпочтительными примерами для -C1-8-алифат-арила являются -СН2-С6Н5, -CH2CH2-C6H5, и -СН=СН-С6Н5.
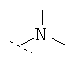
Предпочтительно R1 и R2, независимо друг от друга означают -Н; -C1-6-алифат; -С3-8-циклоалифат, -C1-6-алифат-арил, -С1-6-алифат-С3-8-циклоалифат или -C1-6-алифат-гетероарил; или остатки R1 и R2 вместе образуют кольцо и означают -CH2CH2OCH2CH2-, -CH2CH2NR4CH2CH2- или -(СН2)3-6-, с указанием, что R1 и R2 преимущественно оба не одновременно означают -Н. Предпочтительнее R1 и R2 независимо друг от друга означают -Н; -C1-5-алифат; или остатки R1 и R2 вместе образуют кольцо и означают -CH2CH2OCH2CH2-, -CH2CH2NR4-CH2CH2- или -(СН2)3-6-, причем R4 предпочтительно означает -Н или -C1-5-алифат, с указанием, что R1 и R2 преимущественно оба не одновременно означают -Н. Особенно предпочтительны соединения, где R1 и R2 независимо друг от друга означают -СН3 или -Н, причем R1 и R2 не одновременно -Н означают; или R1 и R2 образуют кольцо и означают -(СН2)3-4-. В высшей степени предпочтительны соединения, где R1 и R2 означают -СН3 или где R1 означает -Н и R2 означает -СН3.
Особенно предпочтительно R1 и R2 вместе с атомом азота, к которому они привязаны, образуют одну из следующих функциональных групп:

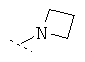
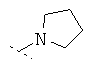
Предпочтительно R3 означает -C1-8-алифат, -С3-8-циклоалифат, -арил, -гетероарил; или в каждом случае означает через -C1-3-алифат-группу связанный -арил, -гетероарил или -С3-8-циклоалифат.
Особенно предпочтительно R3 означает -этил, -пропил, -бутил, -пентил, -гексил, -гептил, -циклопентил, -циклогексил, -фенил, -бензил, -нафтил, -антраценил, -тиофенил, -бензотиофенил, -фурил, -бензофуранил, -бензодиоксоланил, -индолил, -инданил, -бензодиоксанил, -пирролил, -пиридил, -пиримидил или -пиразинил, в каждом случае незамещенный или моно- или многократно замещенный; через насыщенную, неразветвленную -C1-3-алифат-группу связанный -С5-6-циклоалифат, -фенил, -нафтил, -антраценил, -тиофенил, -бензотиофенил, -пиридил, -фурил, -бензофуранил, -бензодиоксоланил, -индолил, -инданил, -бензодиоксанил, -пирролил, -пиримидил, -триазолил или -пиразинил, в каждом случае незамещенный или моно- или многократно замещенный.
Более предпочтительно R3 означает -пропил, -бутил, -пентил, -гексил, -фенил, -фурил, -тиофенил, -нафтил, -бензил, -бензофуранил, -индолил, -инданил, -бензодиоксанил, -бензодиоксоланил, -пиридил, -пиримидил, -пиразинил, -триазолил или -бензотиофенил, в каждом случае незамещенный или моно- или многократно замещенным; через насыщенную, неразветвленную -C1-3-алифат-группу связанный -фенил, -фурил или -тиофенил, в каждом случае незамещенный или моно- или многократно замещенный.
Еще предпочтительнее R3 означает -пропил, -бутил, -пентил, -гексил, -фенил, -фенэтил, -тиофенил, -пиридил, -триазолил, -бензотиофенил или -бензил, в каждом случае замещенный или незамещенный, особенно предпочтительно означает -пропил, -3-метоксипропил, -бутил, -пентил, -гексил, -фенил, -3-метилфенил, -3-фторфенил, -бензо[1,3]-диоксолил, -тиенил, -бензотиофенил, -4-хлорбензил, -бензил, -3-хлорбензил, -4-метилбензил, -2-хлорбензил, -4-фторбензил, -3-метилбензил, -2-метилбензил, -3-фторбензил, -2-фторбензил, -1-метил-1,2,4-триазолил или -фенэтил.
В высшей степени предпочтительно R3 означает -бутил, -этил, -3-метоксипропил, -бензотиофенил, -фенил, -3-метилфенил, -3-фторфенил, -бензо[1,3]-диоксолил, -бензил, -1-метил-1,2,4-триазолил, -тиенил или -фенэтил.
Наиболее предпочтительно R3 означает -фенил, -бензил или -фенэтил, в каждом случае незамещенный или в кольце моно- или многократно замещенный; -C1-5-алифат, -С4-6-циклоалифат, -пиридил, -тиенил, -тиазолил, -имидазолил, -1,2,4 триазолил или -бензимидазолил, незамещенный или моно- или многократно замещенный.
В частности, предпочтительно R3 означает -фенил, -бензил, -фенэтил, -тиенил, -пиридил, -тиазолил, -имидазолил, -1,2,4 триазолил, -бензимидазолил или Èбензил, незамещенный или моно- или многократно замещенный посредством -F, -Cl, -Br, -CN, -СН3, -С2Н5, -NH2, -NO2, -SH, -CF3, -ОН, -ОСН3, -OC2H5или -N(СН3)2; -этил, -n-пропил, -2-пропил, -аллил, -n-бутил, -изо-бутил, -втор.-бутил, -трет.-бутил, -n-пентил, -изо-пентил, -нео-пентил, -n-гексил, -циклопентил или -циклогексил, в каждом случае незамещенный или моно- или многократно замещенный посредством -ОН, -ОСН3 или -ОС2Н5, причем преимущественно -тиенил, -пиридил, -тиазолил, -имидазолил, -1,2,4 триазолил и -бензимидазолил являются незамещенными.
В частности, предпочтительно R3 означает -фенил, незамещенный или моно замещенный посредством -F, -Cl, -CN, -СН3; -тиенил; -этил, -n-пропил или -n-бутил, незамещенный или моно- или многократно замещенный посредством -ОСН3, -ОН или -ОС2Н5, в особенности посредством -ОСН3.
Предпочтительно R4 означает -Н, -C1-5-алифат, -С3-8-циклоалифат, -арил, -гетероарил, -C1-6-алифат-арил, -С1-6-алифат-С3-8-циклоалифат, -C1-6-алифат-гетероарил, -С(=O)арил, -С(=O)гетероарил, или -С(=O)С1-6-алифат, предпочтительнее означает -Н или -C1-5-алифат.
Предпочтительно n означает целое число от 0 до 6, предпочтительнее означает 0, 1, 2 или 3, еще предпочтительнее означает 0 или 1, особенно предпочтительно n=0.
Предпочтительно RB означает -Н, -C1-6-алифат, -С3-8-циклоалифат, -C1-6-алифат-С3-8-циклоалифат, -C1-6-алифат-арил, -C1-6-алифат-гетероарил, -арил, -гетероарил, -С(=O)Н, -С(=O)С1-6-алифат, -С(=O)С3-8-циклоалифат, -C(=O)C1-6-алифат-С3-8-циклоалифат, -С(=O)С1-6-алифат-арил, -С(=O)С1-6-алифат-гетероарил, -С(=O)-С3-8-циклоалифат-арил, -С(=O)-С3-8-циклоалифат-гетероарил, -С(=O)арил, -С(=O)гетероарил, -С(=O)ОН, -CO2-С1-6-алифат, -CO2-С3-8-циклоалифат, -CO2-С1-6-алифат-С3-8-циклоалифат, -СО2-С1-6-алифат-арил, -CO2-С1-6-алифат-гетероарил, -СО2-арил, -СО2-гетероарил, -C(=O)NH2, -С(=O)NHC1-6-алифат, -С(=O)NHC3-8-циклоалифат, -С(=O)NHC1-6-алифат-С3-8-циклоалифат, -С(=O)NHC1-6-алифат-арил, -С(=O)NHC1-6-алифат-гетероарил, -С(=O)NНарил, -С(=O)NНгетероарил, -С(=O)N(С1-6-алифат)2, -С(=O)N(С3-8-циклоалифат)2, -С(=O)N(С1-6-алифат-С3-8-циклоалифат)2, -С(=O)N(С1-6-алифат-арил)2, -С(=O)N(С1-6-алифат-гетероарил)2, -С(=O)N(арил)2, -С(=O)N(гетероарил)2, -S(=O)1-2-С1-6-алифат, -S(=O)1-2-С3-8-циклоалифат, -S(=O)1-2-С1-6-алифат-С3-8-циклоалифат, -S(=O)1-2-С1-6-алифат-арил, -S(=O)1-2-C1-6-алифат-гетероарил, -S(=O)1-2-С3-8-циклоалифат-арил, -S(=O)1-2-С3-8-циклоалифат-гетероарил -S(=O)1-2-арил, -S(=O)1-2-гетероарил, -S(=O)1-2-OC1-6-алифат, -S(=O)1-2-ОС3-8-циклоалифат, -S(=O)1-2-ОС1-6-алифат-С3-8-циклоалифат, -S(=O)1-2-ОС1-6-алифат-арил, -S(=O)1-2-ОС1-6-алифат-гетероарил, -S(=O)1-2-Оарил, -S(=O)1-2-O гетероарил, -S(=O)1-2-NH2, -S(=O)1-2-NHC1-6-алифат, -S(=O)1-2-NHC3-8-циклоалифат, -S(=O)1-2-NHC1-6-алифат-С3-8-циклоалифат, -S(=O)1-2-NHC1-6-алифат-арил, -S(=O)1-2-NHC1-6-алифат-гетероарил, -S(=O)1-2-NHарил, -S(=O)1-2-NH-гетероарил, -S(=O)1-2-N(С1-6-алифат)2, -S(=O)1-2-N(С3-8-циклоалифат)2, -S(=O)1-2-N(С1-6-алифат-С3-8-циклоалифат)2, -S(=O)1-2-N(C1-6-алифат-арил)2, -S(=O)1-2-N(С1-6-алифат-гетероарил)2, -S(=O)1-2-N(арил)2 или -S(=O)1-2-N(гетероарил)2.
Особенно предпочтительно RB означает -Н, -C1-8-алифат, -C1-8-алифат-арил, -C1-8-алифат-гетероарил, -С(=O)-С1-8-алифат, -С(=O)-С1-8-алифат-арил, -С(=O)-C1-8-алифат-гетероарил, -С(=O)-С3-8-циклоалифат-арил, -С(=O)-С3-8-циклоалифат-гетероарил, -С(=O)NH-С1-8-алифат, -S(=O)1-2-С1-8-алифат, -S(=O)1-2-арил, -S(=O)1-2-гетероарил, -S(=O)1-2-С1-8-алифат-арил, -S(=O)1-2-С1-8-алифат-гетероарил, -S(=O)1-2-С3-8-циклоалифат-арил или -S(=O)1-2-С3-8-циклоалифат-гетероарил.
Предпочтительно Х означает -О- или -NRA-, особенно предпочтительно означает -NRA-.
Если Х означает -O-, то RB предпочтительно не означает -Н. Если RB означает -Н, то n предпочтительно является 1, 2, 3 или 4. Если X означает -0-, то n предпочтительно означает 0 или 1 и RB предпочтительно означает -C1-8-алифат или -C1-8-алифат-арил.
Если Х означает -NRA-, то RA означает -Н, -R0, -S(=O)0-2R0, -C(=O)R0, -C(=O)OR0, -C(=O)NH2, -C(=O)NHR0 или -C(=O)N(R0)2; предпочтительно означает -Н или -R0 (в особенности -C1-8-алифат); особенно предпочтительно означает -Н или -СН3; или RA совместно с RB образует кольцо и означает -(СН2)3-4-.
Предпочтительно группа «RB-Х-(СН2)n-» означает








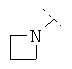


















Предпочтительными представителями группы -(СН2)n-NH-С(=O)-С1-8-алифат-арил являются представленные ниже:




В одной предпочтительной форме осуществления соединений согласно изобретению является RA=RB. В одной другой предпочтительной форме осуществления соединений согласно изобретению является RA≠RB.
В одной особенно предпочтительной форме осуществления соединений согласно изобретению является n=0 и Х означает -NRA-. Затем речь идет о геминально дизамещенных 1,4-диаминах общей формулы (1.1),

причем, по меньшей мере, одна из обеих аминогрупп, преимущественно обе аминогруппы, не одновременно соответственно могут быть замещены двумя водородными остатками.
В целях описания углеводородные остатки разделены на алифатические углеводородные остатки с одной стороны и ароматические углеводородные остатки с другой стороны.
Алифатические углеводородные остатки со своей стороны подразделены на нециклические алифатические углеводородные остатки с одной стороны (= «алифат») и циклические алифатические углеводородные остатки, т.е. алициклические углеводородные остатки, с другой стороны (= «циклоалифат»). Циклоалифаты могут быть моноциклическими или мультициклическими. Алициклические углеводородные остатки («циклоалифат») включают как чистые алифатические карбоциклы, так и алифатические гетероциклы, т.е. - если не указано отдельно - охватывает «циклоалифат» чистые алифатические карбоциклы (например, циклогексил), чистые алифатические гетероциклы (например, пиперидил или пиперазил), а также неароматические, мультициклические, при необходимости смешанные системы (например, декалинил, декагидрохинолинил).
Ароматические углеводороды со своей стороны подразделены на карбоциклические ароматические углеводороды с одной стороны (= «арил») и гетероциклические ароматические углеводороды с другой стороны (= «гетероарил»).
Упорядочение мультициклических, по меньшей мере, частично ароматических систем преимущественно зависит от того, имеет ли, по меньшей мере, одно ароматическое кольцо мультициклической системы, по меньшей мере, один гетероатом (обычно N, О или S) в кольце. Если в этом кольце имеется, по меньшей мере, один такой гетероатом, то предпочтительно речь идет о «гетероариле» (даже если при необходимости как дополнительно имеющийся цикл мультициклической системы имеется другое карбоциклическое ароматическое или неароматическое кольцо с или без гетероатома); если в одном из, при необходимости, нескольких ароматических колец мультициклической системы такой гетероатом отсутствует, то речь идет предпочтительно об «ариле» (даже если в одном при необходимости дополнительно имеющемся, неароматическом цикле мультициклической системы имеется кольцевой гетероатом).
В пределах циклических заместителей сообразно этому предпочтительно действует следующий приоритет очередности: гетероарил > арил > циклоалифат.
Для целей описания не понятийно различают одновалентные и поливалентные, например двухвалентные углеводородные остатки, т.е. охватывает «C1-3-алифат», смотря по смысловой связи, например, как -C1-3-алкил, -С1-3-алкенил и -C1-3-алкинил, так и, например, -С1-3-алкилен-, -C1-3-алкенилен- и C1-3-алкинилен-.
Предпочтительно алифат в каждом случае является разветвленным или неразветвленным, насыщенным или моно- или многократно ненасыщенным, незамещенным или моно- или многократно замещенным, алифатическим углеводородным остатком. Поскольку алифат является моно- или многократно замещенным, то заместители независимо друг от друга выбраны из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -СНО, =O, -R0, -C(=O)R0, -C(=O)-OH, -C(=O)OR0, -C(=O)NH2, -C(=O)NHR0, -C(=O)N(R0)2, -OH, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)-NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N+(R0)3, -N+(R0)2O-, -NHC(=O)R0, -NHC(=O)OR0, -NHC(=O)NH2, -NHC(=O)NHR0, -NH-C(=O)N(R0)2, -Si(R0)3, -PO(OR0)2. Таким образом «алифат» охватывает ациклические насыщенные или ненасыщенные углеводородные остатки, которые могут быть разветвленными или с прямой цепью, т.е. алканилы, алкенилы и алкинилы. При этом алкенилы имеют, по меньшей мере, одну углерод-углеродную двойную связь и алкинилы, по меньшей мере, одну углерод-углеродную тройную связь. Предпочтительные незамещенные одновалентные алифаты включают -СН3, -СН2СН3, -СН2СН2СН3, -СН(СН3)2, -СН2СН2СН2СН3, -СН(СН3)СН2СН3, -СН2СН(СН3)2, -С(СН3)3, -СН2СН2СН2-СН2СН3 и -СН2СН2-СН2СН2СН2СН3; а также -СН=СН2, -С≡СН, -СН2СН=СН2, -СН=СН-СН3, -СН2С≡СН, -С≡ССН3 и -СН=СНСН=СН2. Предпочтительные незамещенные двухвалентные алифаты включают -СН2-, -СН2СН2-, -СН2СН(СН3)-, -СН(СН3)-СН2-, -СН2СН2СН2-, -СН(СН3)СН2СН2-, -СН2СН(СН3)-СН2-, -СН2СН2СН(СН3)-, -СН-(СН2СН3)СН2- и -СН2СН2-СН2СН2-; а также -СН=СН-, -С≡С-, -СН2СН=СН-, -СН=СНСН2-, -СН2С≡С- и -С≡С-СН2-. Предпочтительные замещенные одновалентные алифаты включают -CH2F, -CHF2, -CF3, -CH2CF3, -CF2CF3, -CH2OH, -CH2CH2OH, -СН2СНОНСН3, -CH2OCH3 и CH2CH2OCH3. Предпочтительные замещенные двухвалентные алифаты включают -CF2-, -CF2CF2-, -СН2СНОН-, -СНОНСН2- и -СН2СНОНСН2-. Особенно предпочтительны метил, этил, n-пропил и n-бутил.
Предпочтительно циклоалифат в каждом случае является насыщенным или моно- или многократно ненасыщенным, незамещенным или моно- или многократно замещенным, алифатическим (т.е. неароматическим), моно- или мультициклическим углеводородным остатком. Число циклических атомов углерода преимущественно находится в указанном диапазоне (т.е. «С3-8-»циклоалифат имеет преимущественно 3, 4, 5, 6, 7 или 8 циклических атомов углерода). Для целей описания «С3-8-циклоалифат» предпочтительно является циклическим углеводородом с 3, 4, 5, 6, 7 или 8 циклическими атомами углерода, насыщенными или ненасыщенными, но неароматическими, причем при необходимости один или два атома углерода независимо друг от друга замещены одним гетероатомом S, N или О. Поскольку циклоалкил является моно- или многократно замещенным, то заместители независимо друг от друга выбраны из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -СНО,=O, -R0, -C(=O)R0, -C(=O)OH, -C(=O)OR0, -C(=O)NH2, -C(=O)NHR0, -C(=O)N(R0)2, -ОН, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)-N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N(R0)3, -N+(R0)2O-, -NHC(=O)Ro, -NHC(=O)OR0, -NHC(=O)NH2, -NHC(=O)NHR0, -NH-C(=O)N(R0)2, -Si(R0)3, -PO(OR0)2. Предпочтительно С3-8-циклоалифат выбран из группы, состоящей из циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклопентенил, циклогексенил, циклогептенил и циклооктенил, а также тетрагидропиранил, диоксанил, диоксоланил, морфолинил, пиперидинил, пиперазинил, пиразолинонил и пирролидинил.
Предпочтительно в связи с «алифатом» соответственно «циклоалифатом» под «моно- или многократно замещенным» понимают моно- или многократное замещение, например однократное, двукратное, трехкратное или четырехкратное замещение, одного или нескольких атомов водорода посредством -F, -Cl, -Br, -I, -ОН, -ОС1-6-алкил, -ОС(=O)С1-6-алкил, -SH, -NH2, -NHC1-6-алкил, -N(С1-6-алкил)2, -С(=O)ОС1-6-алкил или -С(=O)ОН. Предпочтительны соединения, где «алифат замещенный» или «циклоалифат замещенный» означает алифат или циклоалифат, замещенный посредством -F, -Cl, -Br, -I, -CN, -СН3, -С2Н5, -NH2, -NO2, -SH, -CF3, -ОН, -ОСН3, -ОС2Н5 или -N(CH3)2. Особенно предпочтительными заместителями являются -F, -Cl, -ОН, -SH, -NH2 и -С(=O)ОН.
Под многократно замещенными остатками понимают такие остатки, которые или в различных или в одинаковых атомах замещены многократно, например двукратно или трехкратно, например трехкратно в одинаковом С-атоме, как в случае -CF3 или -CH2CF3, или в различных местах, как в случае -СН(ОН)-CH=CH-CHCl2. Многократное замещение может происходить одинаковым или различным заместителем. При необходимости один заместитель со своей стороны также может быть замещенным; так -Оалифат, в частности, также охватывает -OCH2CH2O-CH2CH2-OH. Предпочтительно, если алифат или циклоалифат замещены посредством -F, -Cl, -Br, -I, -CN, -СН3, -С2Н5, -NH2, -NO2, -SH, -CF3, -ОН, -ОСН3, -ОС2Н5или -N(СН3)2. В высшей степени предпочтительно, если алифат или циклоалифат замещены посредством -ОН, -ОСН3 или -ОС2Н5.
Предпочтительно арил в каждом случае независимо означает карбоциклическую кольцевую систему с, по меньшей мере, одним ароматическим кольцом, но без гетероатомов в этом кольце, причем арильные остатки при необходимости могут быть конденсированы с другими насыщенными, (частично) ненасыщенными или ароматическими кольцевыми системами и каждый арильный остаток может быть незамещенным или моно- или многократно замещенным, причем арильные заместители могут быть одинаковыми или различными и в любом и возможном положении арила. Предпочтительными арилами являются фенил, нафтил, антраценил, Phenanthrenyl, фторантенил, фторенил, инданил и тетралинил. Особенно предпочтительны фенил и нафтил. Поскольку арил является моно- или многократно замещенным, то арильные заместители могут быть одинаковыми или различными и в любом и возможном положении арила, и независимо друг от друга выбраны из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -СНО, =O, -R0, -C(=O)R0, - C(=O)OH, -C(=O)OR0, -C(=O)NH2, -C(=O)NHR0, -C(=O)N(R0)2, -ОН, -O(CH2)1-2O, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N+(R0)3, N+(R0)2O-, -NHC(=O)R0, -NHC(=O)OR0, -NHC(=O)NH2, -NHC(=O)-NHR0, -NH-C(=O)N(R0)2, -Si(R0)3 и -PO(OR0)2. Предпочтительными замещенными арилами являются 2-фтор-фенил, 3-фтор-фенил, 4-фтор-фенил, 2,3-дифтор-фенил, 2,4-дифтор-фенил, 3,4-дифтор-фенил, 2-хлор-фенил, 3-хлор-фенил, 4-хлор-фенил, 2,3-дихлор-фенил, 2,4-дихлор-фенил, 3,4-дихлор-фенил, 2-метокси-фенил, 3-метокси-фенил, 4-метокси-фенил, 2,3-диметокси-фенил, 2,4-диметокси-фенил, 3,4-диметокси-фенил, 2-метил-фенил, 3-метил-фенил, 4-метил-фенил, 2,3-диметил-фенил, 2,4-диметил-фенил и 3,4-диметил-фенил.
Предпочтительно гетероарил означает 5-, 6- или 7-членный циклический ароматический остаток, который содержит 1, 2, 3, 4 или 5 гетероатомов, причем гетероатомы одинаково или различно представляют собой азот, кислород или серу, и гетероцикл может быть незамещенным или моно- или многократно замещенным; причем в случае замещения в гетероцикле заместители могут быть одинаковыми или различными и в любом и возможном положении гетероарила; и причем гетероцикл также может быть частью бициклической или полициклической системы. Предпочтительно «гетероарил» выбран из группы, состоящей из пирролил, индолил, фурил (фуранил), бензофуранил, тиенил (тиофенил), бензотиенил, бензотиадиазолил, бензооксадиазолил, бензотиазолил, бензооксазолил, бензотриазолил, бензодиоксоланил, бензодиоксанил, фталазинил, пиразолил, имидазолил, тиазолил, оксазолил, изоксазолил, пиридинил, пиридазинил, пиримидинил, пиразинил, пиранил, индазолил, пуринил, индолизинил, хинолинил, изохинолинил, хиназолинил, карбазолил, феназинил, фенотиазинил или оксадиазолил, причем связь может происходить через любой и возможный кольцевой член гетероарильного остатка. Поскольку гетероарил является моно- или многократно замещенным, то гетероарильные заместители могут быть одинаковыми или различными и в любом и возможном положении гетероарила, и независимо друг от друга выбраны из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -СНО, =O, -R0, -C(=O)R0, - C(=O)OH, -C(=O)OR0, -C(=O)NH2, -C(=O)NHR0, -C(=O)N(R0)2, -ОН, -O(CH2)1-2O, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N+(R0)3, N+(R0)2O-, -NHC(=O)R0, -NHC(=O)OR0, -NHC(=O)NH2, -NHC(=O)-NHR0, -NH-C(=O)N(R0)2, -Si(R0)3 и -PO(OR0)2.
Что касается «арила» или «гетероарила», то под «моно- или многократно замещенным» понимают однократное или многократное, напримердвукратное, трехкратное, четырехкратное или пятикратное замещение одного или нескольких атомов водорода кольцевой системы.
Особенно предпочтительными являются заместители арила и гетероарила в каждом случае независимо друг от друга выбранные из -F, -Cl, -Br, -I, -CN, -СНО, -CO2H, -NH2, -NO2, -NHR0, -N(R0)2, -N+(R0)3, N+(R0)2O-, -SH, -SR0, -OH, -OR0, -C(=O)R0, -CO2R0, -C(=O)NH2, -C(=O)NHR0, -C(=O)N(R0)2, -S(=O)1-2R0, -S(=O)2NH2, -SO3H, =O или -R0. Предпочтительными заместителями являются -F, -Cl, -Br, -I, -ОН, -OC1-6-алкил, -O-С(=O)-С1-6-алкил, -SH, -NH2, -NHC1-6-алкил, -N(С1-6-алкил)2, -С(=O)ОС1-6-алкил или -С(=O)ОН. Предпочтительны соединения, где «арил замещенный» или «гетероарил замещенный» означает арил или гетероарил замещенный посредством -F, -Cl, -Br, -I, -CN, -СН3, -С2Н5, -NH2, -NO2, -SH, -CF3, -ОН, -ОСН3, -ОС2Н5 или -N(СН3)2. Особенно предпочтительными заместителями являются -F, -Cl, -ОН, -SH, -NH2 и -С(=O)ОН.
Соединения согласно изобретению могут находиться в виде отдельного стереоизомера или его смеси, свободных соединений и/или их физиологически совместимых солей и/или сольватов.
Соединения согласно изобретению в зависимости от образца заместителя могут быть хиральными или ахиральными.
Соединения согласно изобретению в зависимости от замещения относительно циклогексанового кольца могут представлять собой изомеры, в которых образец заместителя в 1,4-положении (1-положении: >C(NR1R)R3; 4-положении: >CQ((СН2)nXRB) также может обозначаться посредством син/анти. «Син/анти-изомеры» являются подгруппой стереоизомеров (конфигурационные изомеры).
В одной предпочтительной форме осуществления избыток диастереомеров син-изомера составляет, по меньшей мере, 50%de, предпочтительнее, по меньшей мере, 75%de, еще предпочтительнее, по меньшей мере, 90%de, наиболее предпочтительно, по меньшей мере, 95%de и в особенности, по меньшей мере, 99%de. В одной другой предпочтительной форме осуществления составляет избыток диастереомеров анти-изомера, по меньшей мере, 50%de, предпочтительнее, по меньшей мере, 75%de, еще предпочтительнее, по меньшей мере 90%de, наиболее предпочтительно по меньшей мере 95%de и в особенности по меньшей мере 99%de.
Пригодные методы разделения изомеров (диастереомеров) известны специалисту в данной области техники. В качестве примеров могут быть названы: колоночная хроматография, препаративная ВЭЖХ, и способы кристаллизации.
Если соединения согласно изобретению являются хиральными, то преимущественно они находятся в виде рацемата или в обогащенном виде энантиомера. В одной предпочтительной форме осуществления составляет избыток энантиомеров (ее) S-энантиомера, по меньшей мере, 50%ее, предпочтительнее, по меньшей мере, 75%ее, еще предпочтительнее, по меньшей мере, 90%ее, наиболее предпочтительно, по меньшей мере, 95%ее и в особенности, по меньшей мере, 99%ее. В одной другой предпочтительной форме осуществления избыток энантиомеров (ее) R-энантиомера составляет, по меньшей мере, 50%ее, предпочтительнее, по меньшей мере, 75%ее, еще предпочтительнее, по меньшей мере, 90%ее, наиболее предпочтительно, по меньшей мере, 95%ее и в особенности, по меньшей мере, 99%ее.
Пригодные методы разделения энантиомеров известны специалисту в данной области техники. В качестве примеров могут быть названы препаративная ВЭЖХ на хиральных неподвижных фазах и переведение в диастереомерные промежуточные продукты. Переведение в диастереомерные промежуточные продукты может происходить, например, как солеобразование с помощью хиральных, энантиомерно чистых кислот. После разделения образованных таким образом диастереомеров соль затем снова может быть переведена в свободное основание или другую соль.
Если не указано определенно, то каждая ссылка на соединения согласно изобретению включает все изомеры (например, стереоизомеры, диастереомеры, энантиомеры) в любом соотношении компонентов смеси.
Если не указано определенно, то каждая ссылка на соединения согласно изобретению свободные соединения (т.е. формы, которые находятся не в виде соли) и все физиологически совместимые соли.
Для целей описания физиологически совместимые соли соединений согласно изобретению находятся в виде солей с анионами или кислот соответствующего соединения с неорганическими соответственно органическими кислотами, которые являются физиологически совместимыми - в особенности при применении у человека и/или млекопитающего.
Примерами физиологически совместимых солей определенных кислот являются соли: соляной кислоты, бромистоводородной кислоты, серной кислоты, метансульфоновой кислоты, муравьиной кислоты, уксусной кислоты, щавелевой кислоты, янтарной кислоты, яблочной кислоты, винной кислоты, миндальной кислоты, фумаровой кислоты, молочной кислоты, лимонной кислоты, глутаминовой кислоты, сахариновой кислоты, монометилсебациновой кислоты, 5-оксо-пролина, гексан-1-сульфоновой кислоты, никотиновой кислоты, 2-, 3- или 4-аминобензойной кислоты, 2,4,6-триметил-бензойной кислоты, α-липоновой кислоты, ацетилглицина, ацетилсалициловой кислоты, гиппуровой кислоты и/или аспарагиновой кислоты. Особенно предпочтительны гидрохлорид, цитрат и полуцитрат.
Физиологически совместимыми солями с катионами или основаниями являются соли соответствующего соединения - как анион с, по меньшей мере, одним, преимущественно неорганическим, катионом, которые являются физиологически совместимыми - в особенности при применении у человека и/или млекопитающего. Особенно предпочтительны соли щелочных и щелочноземельных металлов, а также соли аммония, но в особенности соли (моно-) или (ди-) натрия, (моно-) или (ди-) калия, магния или кальция.
В дальнейшем поясняются предпочтительные в каждом случае формы осуществления соединений согласно изобретению. Если не указано определенно, то имеют силу все в каждом случае поясненные ранее определения заместителей (т.е., например, от R0 до R4, от Y1 до Y4', Q и т.д.) и соответственно их соответствующие предпочтительные формы осуществления и поэтому не повторяются.
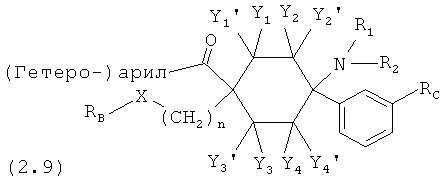
Предпочтительные формы осуществления соединений согласно изобретению общей формулы (1) имеют общую формулу (2)

причем
(гетеро-)арил означает гетероарил или арил; предпочтительно фенил; в каждом случае незамещенный или моно- или многократно замещенным, причем заместители преимущественно независимо друг от друга выбраны из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -СНО, -R0, -C(=O)R0, -C(=O)H, -C(=O)OH, C(=O)OR0, -C(=O)NH2, -С(=O)NH-R0, -C(=O)-N(R0)2, -ОН, -O(CH2)1-2O-, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N+(R0)3, N+(R0)2O-, -NHC(=O)R0, -NHC(=O)-OR0, -NH-C(=O)NH2, -NHC(=O)NHR0 и NHC(=O)N(R0)2; предпочтительнее -F, -Cl, -Br, -I, -CF3, -CN и -NO2.
Особенно предпочтительные формы осуществления соединений согласно изобретению общей формулы (2) имеют общую формулу (2.1), (2.2), (2.3), (2.4), (2.5), (2.6), (2.7), (2.8), (2.9), (2.10), (2.11), (2.12), (2.13) или (2.14)





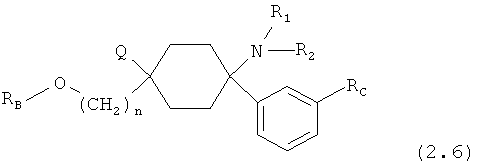







причем
RC означает -H, -F, -Cl, -Br, -I, -CN, -NO2, -CF3, -ОН или -ОСН3; и, если имеется, (гетеро-)арил означает гетероарил или арил; предпочтительно означает -фенил, -бензил или -2-индолил; в каждом случае незамещенный или моно- или многократно замещенным, причем заместители преимущественно независимо друг от друга выбраны из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -CHO, -R0, -C(=O)R0, -C(=O)H, -C(=O)OH, -C(=O)OR0, -C(=O)NH2, -C(=O)-NH-R0, -C(=O)-N(R0)2, -ОН, -O(CH2)1-2O-, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N+(R0)3, -N(R0)2O-, -NHC(=O)R0, -NHC(=O)-OR0, -NH-C(=O)NH2, -NHC(=O)NHR0 и -NHC(=O)N(R0)2; предпочтительнее -F, -Cl, -Br, -I, -CF3, -CN и -NO2; и
m означает 0, 1, 2, 3, 4, 5 или 6.
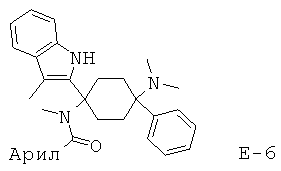
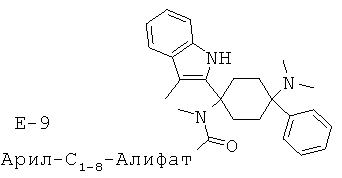
Предпочтительными представителями соединений общей формулы (2.14) являются, например, соединения от Е-1 до Е-12










Другие предпочтительные формы осуществления соединений согласно изобретению общей формулы (1) имеют общую формулу (3)

причем
Q' означает -(СН2)0-4-, -С(=O)- или -С(=NH)-; и
(гетеро-)арил означает гетероарил или арил; предпочтительно фенил; в каждом случае незамещенный или моно- или многократно замещенный, причем заместители преимущественно независимо друг от друга выбраны из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -СНО, -R0, -C(=O)R0, -C(=O)H, -C(=O)OH, -C(=O)OR0, -C(=O)NH2, -C(=O)NH-R0, -C(=O)-N(R0)2, -OH, -O(CH2)1-2O-, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N+(R0)3, -N+(R0)2O-, -NHC(=O)R0, -NHC(=O)-OR0, -NH-C(=O)NH2, -NHC(=O)NHR0 и -NHC(=O)N(R0)2; предпочтительнее -F, -Cl, -Br, -I, -CF3, -CN и -NO2.
Особенно предпочтительные формы осуществления соединений согласно изобретению общей формулы (3) имеют общую формулу (3.1), (3.2), (3.3), (3.4), (3.5), (3.6), (3.7) или (3.8):










причем, если имеется,
D означает =O или =NH;
W означает -O-, -S-, -NR11-, -CR12=CR13-, -CR12=N- или -N=CR13- означает; предпочтительно означает -O-, -S-, или -NR11-; особенно предпочтительно означает -NR11-;
R5, R6, R6', R11, R12 и R13 в каждом случае независимо друг от друга означают -Н, -F, -Cl, -Br, -I, -CN, -NO2, -СНО, -R0, -C(=O)R0, -C(=O)H, -C(=O)OH, -C(=O)OR0, -C(=O)NH2, -C(=O)NHR0, -C(=O)-N(R0)2, -OH, -O(CH2)1-2O-, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N+(R0)3, -N+(R0)2O-, -NHC(=O)R0, -NHC(=O)OR0, -NH-C(=O)NH2, -NHC(=O)NHR0, -NHC(=O)N(R0)2; или R5 и R6, или R6 и R6', или R6' и R12 вместе образуют пятичленное или шестичленное, насыщенное, частично ненасыщенное ароматическое, незамещенное или моно- или многократно замещенное кольцо, которое при необходимости включает один или два гетероциклических атома, независимо друг от друга выбранных из N, S и О;
A1 означает -N= или -CR7=,
А2 означает -N= или -CR8=,
А3 означает -N= или -CR9=,
А4 означает -N= или -CR10=;
с указанием, что самое большее два из остатков A1, А2, А3 и А4, преимущественно означают 0, 1 или 2 остатков A1, А2, А3 и А4, означают -N=;
R4, R8, R9 и R10 в каждом случае независимо друг от друга означают -Н, -F, -Cl, -Br, -I, -NO2, -CF3, -OR14, -SR14, -SO2R14, -CN, -COOR14, -CONR14, -NR15R16, =O или -R0; преимущественно означают -F, -Cl, -Br, -I, -CF3, -CN или -NO2;
R14 в каждом случае независимо означает -Н или -R0;
R15 и R16 независимо друг от друга означают -Н или -R0; или R15 и R16 вместе означают -CH2CH2OCH2CH2-, -CH2CH2NR4CH2CH2- или -(CH2)3-6-.
Если W означает, например, -CR12=CR13-, -CR12=N- или -N=CR13-, то получаются преимущественно следующие функциональные группы:



Если R6 и R6' вместе образуют, например, шестичленное ароматическое кольцо, которое не имеет гетероциклических атомов, то в каждом случае получаются следующие функциональные группы:



Образованное вместе при необходимости посредством R5 и R6, или R6 и R6', или R6' и R12 пятичленное или шестичленное, насыщенное, частично ненасыщенное или ароматическое кольцо может включать один или два гетероциклических атома, которые независимо друг от друга выбраны из N, S и О. Далее это образованное кольцо может быть незамещенным или моно- или многократно замещенным, причем заместители преимущественно независимо друг от друга выбраны из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -СНО, -Ro, -C(=O)R0, -C(=O)H, -C(=O)OH, -C(=O)OR0, -C(=O)NH2, -C(=O)NHR0, -C(=O)-N(R0)2, -ОН, -O(CH2)1-2O-, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NN2, -NHR0, -N(R0)2, -N+(R0)3, -N+(R0)2O-, -NHC(=O)R0, -NHC(=O)OR0, -NH-C(=O)NH2, -NHC(=O)NHR0 и -NHC(=O)N(R0)2; предпочтительнее -F, -Cl, -Br, -I, -CF3, -CN и -NO2.
Предпочтительно R5 означает -H, -F, -С1 или -R0; предпочтительнее означает -Н, -F, -С1-8-алифат, -C1-8-алифат-арил, -C1-8-алифат-гетероарил или -С1-8-алифат-O-С1-8-алифат (например, -СН2ОСН3).
Предпочтительно R6 и R6' вместе образуют шестичленное, насыщенное, частично ненасыщенное или ароматическое кольцо, которое при необходимости может включать один или два гетероциклических атома, которые независимо друг от друга выбраны из N, S и О. Это образованное кольцо может быть незамещенным или моно- или многократно замещенным, причем заместители преимущественно независимо друг от друга выбраны из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -СНО, -R0, -C(=O)R0, -C(=O)H, -С(=O)ОН, -C(=O)OR0, -C(=O)NH2, -C(=O)NHR0, -C(=O)-N(R0)2, -ОН, -O(CH2)1-2О-, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)-N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N+(R0)3, -N+(R0)2O-, -NHC(=O)R0, -NHC(=O)OR0, -NH-C(=O)NH2, -NHC(=O)NHR0 и -NHC(=O)N(R0)2; предпочтительнее -F, -Cl, -Br, -I, -CF3, -CN и -NO2.
R11, R12 и R13 преимущественно независимо друг от друга выбраны из группы, состоящей из -Н, -F, -Cl, -CN, -ОН, -R0 и -OR0. Особенно предпочтительны R11, R12 и R13 - если имеется - в каждом случае -Н.
Другие предпочтительные формы осуществления соединений согласно изобретению общей формулы (1) имеют общую формулу (4)

причем
(гетеро-)арил означает гетероарил или арил; предпочтительно фенил; в каждом случае незамещенный или моно- или многократно замещенным, причем заместители преимущественно независимо друг от друга выбраны из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -CHO, -R0, -C(=O)R0, -C(=O)H, -C(=O)OH, -C(=O)OR0, -C(=O)NH2, -C(=O)NH-R0, -C(=O)-N(R0)2, -OH, -O(CH2)1-2O-, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N+(R0)3, -N(R0)2O-, -NHC(=O)R0, -NHC(=O)-OR0, -NH-C(=O)NH2, -NHC(=O)NHR0 и -NHC(=O)N(R0)2; предпочтительнее -F, -Cl, -Br, -I, -CF3, -CN и -NO2.
Соединения согласно изобретению определены заместителями, например, посредством R1, R2 и R3 (заместители 1-го поколения), которые со своей стороны при необходимости замещены (заместители 2-го поколения). В зависимости от определения эти заместители заместителей со своей стороны могут быть вновь замещенными (заместители 3-го поколения). Если, например, Y1=-R0, причем R0=-C1-8-алифат (заместитель 1-го поколения), то -C1-8-алифат со своей стороны может быть замещенным, например, посредством -OR0, причем R0=-арил (заместитель 2-го поколения). Из этого получается функциональная группа -C1-8-алифат-Оарил. Затем -арил со своей стороны может быть вновь замещенным, например, посредством -Cl (заместитель 3-го поколения). Затем из этого в целом получается функциональная группа -C1-8-алифат-Оарил-Cl.
Однако в одной предпочтительной форме осуществления заместители 3-го поколения не могут быть вновь замещенными, т.е. затем нет заместителей 4-го поколения.
В одной другой предпочтительной форме осуществления заместители 2-го поколения не могут быть вновь замещенными, т.е. затем уже нет заместителей 3-го поколения. Другими словами в этой форме осуществления функциональные группы для от R0 до Y4' в каждом случае при необходимости могут быть замещенными, однако затем соответствующие заместители со своей стороны не могут быть вновь замещенными.
В одной другой предпочтительной форме осуществления уже заместители 1-го поколения не могут быть снова замещенными, т.е. затем нет ни заместителей 2-го, ни заместителей 3-го поколения. Другими словами в этой форме осуществления функциональные группы для от R0 до Y4' в каждом случае не могут быть замещенными.
Согласно изобретению особенно предпочтительными формами осуществления соединений общей формулы (1) являются соединения общей формулы (2.2)
причем
Q означает -C1-8-алифат (преимущественно -C1-8-алкил), -арил (преимущественно -фенил), -C1-8-алифат-арил (преимущественно -C1-8-алкил-фенил), -гетероарил (преимущественно -индолил), -С(=O)-гетероарил (преимущественно -С(=O)-индолил) или -С(=NH)-гетероарил (преимущественно -С(=NH)-индолил);
R1 означает -СН3;
R2 означает -Н или -СН3; или
R1 и R2 совместно образуют кольцо и означают -(СН2)3-4-;
Х означает -О- или -NRA-;
RA означает -Н или -C1-8-алифат (преимущественно -C1-8-алкил);
RB означает -Н, -C1-8-алифат (преимущественно -C1-8-алкил), - C1-8-алифат-арил (преимущественно -C1-8-алкил-фенил), -C1-8-алифат-гетероарил (преимущественно -C1-8-алкил-индолил), -С(=O)-С1-8-алифат (преимущественно -С(=O)-С1-8-алкил), -С(=O)-С1-8-алифат-арил (преимущественно -С(=O)-бензил), -С(=O)-С1-8-алифат-гетероарил (преимущественно-С(=O)-С1-8-алкил-индолил), -С(=O)-С3-8-циклоалифат-арил (преимущественно-С(=O)-циклопропил-арил), -С(=O)-С3-8-циклоалифат-гетероарил (преимущественно-С(=O)-циклопропил-гетероарил), -С(=O)NH-С1-8-алифат (преимущественно -С(=O)NH-С1-8-алкил), -S(=O)1-2-С1-8-алифат (преимущественно -S(=O)2-С1-8-алкил), -S(=O)1-2-арил (преимущественно -S(=O)2-фенил), -S(=O)1-2-гетероарил, -S(=O)1-2-С1-8-алифат-арил, -S(=O)1-2-С1-8-алифат-гетероарил, -S(=O)1-2-С3-8-циклоалифат-арил (преимущественно -S(=O)2-циклопропил-арил) или -S(=O)1-2-С3-8-циклоалифат-гетероарил (преимущественно -S(=O)2-циклопропил-гетероарил); или RA и RB совместно образуют кольцо и означают -(СН2)3-4-; с указанием, что если Х означает -О- и одновременно n означает 0, то RB не означает -Н;
RC означает -Н, -F, -Cl, -Br, -I, -CN, -NO2, -CF3, -ОН или -ОСН3; и
n означает 0, 1, 2, 3 или 4;
причем алифат, арил и гетероарил в каждом случае являются незамещенными или моно- или многократно замещенными.
В высшей степени предпочтительны соединения из группы:
- 1-(имино(1-метил-1Н-индол-2-ил)метил)-N1,N1,N4,N4-тетраметил-4-фенилциклогексан-1,4-диамин бис(2-гидроксипропан-1,2,3-трикарбоксилат)
- 4-(диметиламино)-4-фенил-1-(пирролидин-1-ил)циклогексил)(1-метил-1Н-индол-2-ил)метанон
- 1-(имино(1-метил-1Н-индол-2-ил)метил)-N1,N1,N4,N4-тетраметил-4-фенилциклогексан-1,4-диамин бис(2-гидроксипропан-1,2,3-трикарбоксилат)
- 1,4-бис(диметиламино)-4-фенилциклогексил)(1-метил-1Н-индол-2-ил)метанон
- 4-(имино(1-метил-1Н-индол-2-ил)метил)-N,N-диметил-1-фенил-4-(пирролидин-1-ил)циклогексанамин
- 4-(диметиламино)-4-фенил-1-(пирролидин-1-ил)циклогексил)(1-метил-1 Н-индол-2-ил)метанон
- N1,N1,N4-триметил-1,4-дифенилциклогексан-1,4-диамин
- N1,N1,N4,N4-тетраметил-1,4-дифенилциклогексан-1,4-диамин
- 1-бензил-N1,N1,N4,N4-тетраметил-4-фенилциклогексан-1,4-диамин
- 4-метокси-4-(3-(метоксиметил)-1Н-индол-2-ил)-N,N-диметил-1-фенилциклогексанамин 2-гидроксипропан-1,2,3-трикарбоксилат
- 4-(бензилокси)-4-(3-(метоксиметил)-1Н-индол-2-ил)-N,N-диметил-1-фенилциклогексанамин
- 4-этокси-4-(3-(метоксиметил)-1Н-индол-2-ил)-N,N-диметил-1-фенилциклогексанамин 2-гидроксипропан-1,2,3-трикарбоксилат
- N-((-4-(диметиламино)-1-метил-4-фенилциклогексил)метил)ацетамид 2-гидроксипропан-1,2,3-трикарбоксилат
- 4-хлоро-N-((-4-(диметиламино)-1-метил-4-фенилциклогексил)метил)бензолсульфонамид 2-гидроксипропан-1,2,3-трикарбоксилат
- N-((1-бутил-4-(диметиламино)-4-фенилциклогексил)метил)-4-хлоробензолсульфонамид 2-гидроксипропан-1,2,3-трикарбоксилат
- N-((-4-(диметиламино)-4-фенил-1-(4-фенилбутил)циклогексил)метанол
- N(4-((диметиламино)метил)-N,N-диметил-1,4-дифенилциклогексанамин
- 4-бензил-4-((диметиламино)метил)-N,N-диметил-1-фенилциклогексанамин
- 4-(((1Н-индол-2-ил)метиламино)метил)-N,N,4-триметил-1-фенилциклогексанамин
- N1,N1,N4,N4-тетраметил-1-(3-метил-1Н-индол-2-ил)-4-фенилциклогексан-1,4-диамин
- N-(4-(диметиламино)-1-(3-метил-1Н-индол-2-ил)-4-фенилциклогексил)-N-метилкоричный амид и
- N-(4-(диметиламино)-1-(3-метил-1Н-индол-2-ил)-4-фенилциклогексил)-N-метилацетамид
- [4-бензил-4-(диметиламинометил)-1-фенил-циклогексил]-диметил-амин
- (4-диметиламино-1,4-дифенил-циклогексил)-метил-диметил-амин (неполярный диастереомер)
- (Е)-N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-3-фенил-акриламид (неполярный диастереомер)
- N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-ацетамид (неполярный диастереомер)
- N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-метансульфоновой кислоты амид (полярный диастереомер)
- (Е)-N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-3-фенил-акриламид (полярный диастереомер)
- N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-ацетамид (полярный диастереомер)
- 3-бензил-1-(4-диметиламино-1,4-дифенил-циклогексил)-1-метил-мочевина (неполярный диастереомер)
- 3-бензил-1-(4-диметиламино-1,4-дифенил-циклогексил)-1-метил-мочевина (полярный диастереомер)
- 1-(4-диметиламино-1,4-дифенил-циклогексил)-3-этил-1-метил-мочевина (неполярный диастереомер)
- 1-(4-диметиламино-1,4-дифенил-циклогексил)-3-этил-1-метил-мочевина (полярный диастереомер)
- (4-бензил-4-((диметиламино)метил)-N-метил-1-фенилциклогексанамин (полярный диастереомер)
- (1-бензил-4-диметиламино-4-фенил-циклогексил)-метил-диметил-амин (полярный диастереомер)
- [4-(диметил-амино)-4-(3-метил-1Н-индол-2-ил)-1-фенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-диметиламино-4-(3-метил-1Н-индол-2-ил)-1-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-(диметиламинометил)-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- диметил-(4-метиламино-4-фенил-1-тиофен-2-ил-циклогексил)-амин (неполярный диастереомер)
- диметил-(4-метиламино-4-фенил-1-тиофен-2-ил-циклогексил)-амин (полярный диастереомер)
- [4-(диметил-амино)-4-фенил-1-тиофен-2-ил-циклогексил]-диметил-амин (полярный диастереомер)
- (4-диметиламино-4-фенил-1-тиофен-2-ил-циклогексил)-диметил-амин (неполярный диастереомер)
- (Е)-N-[[4-диметиламино-4-(3-фторфенил)-1-метил-циклогексил]-метил]-3-фенил-акриламид (полярный диастереомер)
- (Е)-N-[[4-диметиламино-4-(3-фторфенил)-1-метил-циклогексил]-метил]-3-фенил-акриламид (неполярный диастереомер)
- (Е)-N-[[4-диметиламино-4-(3-фторфенил)-1-метил-циклогексил]-метил]-2-фенил-этенсульфоновой кислоты амид (неполярный диастереомер)
- (Е)-N-[[4-диметиламино-4-(3-фторфенил)-1-метил-циклогексил]-метил]-2-фенил-этенсульфоновой кислоты амид (полярный диастереомер)
- (1-бутил-4-метиламино-4-фенил-циклогексил)-диметил-амин (неполярный диастереомер)
- (1-бутил-4-метиламино-4-фенил-циклогексил)-диметил-амин (полярный диастереомер)
- [4-(бутил-метил-амино)-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-(бутил-метил-амино)-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-(бензил-метил-амино)-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-(бензил-метил-амино)-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- N-[4-(диметил-амино)-1,4-дифенил-циклогексил]-N-метил-2,2-дифенил-ацетамид (полярный диастереомер)
- диметил-[4-(3-метил-1Н-индол-2-ил)-1-фенил-4-пирролидин-1-ил-циклогексил]-аминдигидрохлорид (полярный диастереомер)
- диметил-[4-(3-метил-1Н-индол-2-ил)-1-фенил-4-пирролидин-1-ил-циклогексил]-амин
- [4-(азетидин-1-ил)-4-(3-метил-1Н-индол-2-ил)-1-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
- N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-метансульфоновой кислоты амид (неполярный диастереомер)
- (4-бутил-4-диметиламино-1-фенил-циклогексил)-диметил-амин (неполярный диастереомер)
- (4-бутил-4-диметиламино-1-фенил-циклогексил)-диметил-амин (полярный диастереомер)
- [4-(циклопентил-метил)-4-диметиламино-1-фенил-циклогексил]-метил-амин (неполярный диастереомер)
- [4-(циклопентил-метил)-4-диметиламино-1-фенил-циклогексил]-метил-амин (полярный диастереомер)
- [4-(циклопентил-метил)-4-диметиламино-1-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-(циклопентил-метил)-4-диметиламино-1-фенил-циклогексил]-диметил-амин (полярный диастереомер)
- (Е)-N-[4-(циклопентил-метил)-4-диметиламино-1-фенил-циклогексил]-N-метил-3-фенил-акриламид (неполярный диастереомер)
- (Е)-N-[4-(циклопентил-метил)-4-диметиламино-1-фенил-циклогексил]-N-метил-3-фенил-акриламид (полярный диастереомер)
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-уксусная кислота (полярный диастереомер)
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-уксусная кислота (неполярный диастереомер)
- [1-(4-метоксифенил)-4-метиламино-4-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [1-(4-метоксифенил)-4-метиламино-4-фенил-циклогексил]-диметил-амин (полярный диастереомер)
- диметил-[4-метиламино-4-фенил-1-[4-(трифторметил)-фенил]-циклогексил]-амин (неполярный диастереомер)
- диметил-[4-метиламино-4-фенил-1-[4-(трифторметил)-фенил]-циклогексил]-амин (полярный диастереомер)
- [4-(диметил-амино)-4-фенил-1-[4-(трифторметил)-фенил]-циклогексил]-диметил-амин (полярный диастереомер)
- [4-диметиламино-4-фенил-1-[4-(трифторметил)-фенил]-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-(диметил-амино)-1-(4-метоксифенил)-4-фенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-диметиламино-1-(4-метоксифенил)-4-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-[(1Н-индол-3-ил-метиламино)-метил]-4-метил-1-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-[(1Н-индол-3-ил-метиламино)-метил]-4-метил-1-фенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-[(1Н-индол-3-ил-метил-метил-амино)-метил]-4-метил-1-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-[(1Н-индол-3-ил-метил-метил-амино)-метил]-4-метил-1-фенил-циклогексил]-диметил-амин (полярный диастереомер)
- [3-[[[4-(диметил-амино)-1-метил-4-фенил-циклогексил]-метил-метил-амино]-метил]-1Н-индол-1-ил]-метанол (полярный диастереомер)
- (Е)-N-[4-диметиламино-1-(3-метил-1Н-индол-2-ил)-4-фенил-циклогексил]-N-метил-3-фенил-акриламид (полярный диастереомер)
- [4-диметиламино-1-(3-метил-1Н-индол-2-ил)-4-фенил-циклогексил]-метил-амин (полярный диастереомер)
- [4-диметиламино-1-(3-метил-1Н-индол-2-ил)-4-фенил-циклогексил]-метил-амин (неполярный диастереомер)
- бензил-[4-диметиламино-1-(3-метил-1Н-индол-2-ил)-4-фенил-циклогексил]-амин 2-гидрокси-пропан-1,2,3-трикарбоновой кислоты
- диметил-[4-[метил-(пиридин-3-ил-метил)-амино]-1,4-дифенил-циклогексил]-амин (полярный диастереомер)
- [4-[[4,6-бис(метиламино)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-[[4-(4-метокси-фенокси)-6-метиламино-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-цикпогексил]-диметил-амин (неполярный диастереомер)
- N-[4-(диметил-амино)-1,4-дифенил-циклогексил]-N-метил-пиридин-3-карбоновой кислоты амид (неполярный диастереомер)
- диметил-[4-[метил-(пиридин-3-ил-метил)-амино]-1,4-дифенил-циклогексил]-амин (неполярный диастереомер)
- [4-[[4,6-бис(метиламино)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-[[4-(4-метокси-фенокси)-6-метиламино-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- N-[4-(диметил-амино)-1,4-дифенил-циклогексил]-N,1-диметил-1Н-пиразол-3-карбоновой кислоты амид (полярный диастереомер)
- N-[4-(диметил-амино)-1,4-дифенил-циклогексил]-N,1-диметил-1Н-пиразол-3-карбоновой кислоты амид (неполярный диастереомер)
- [4-(диметил-амино)-1-(3-фторфенил)-4-(3-метил-1Н-индол-2-ил)-циклогексил]-диметил-амин (полярный диастереомер)
- 4-(азетидин-1-ил)-1-(3-фторофенил)-N,N-диметил-4-(3-метил-1Н-индол-2-ил)циклогексанамин (неполярный диастереомер)
- 4-(азетидин-1-ил)-1-(3-фторофенил)-N,N-диметил-4-(3-метил-1Н-индол-2-ил)циклогексанамин (полярный диастереомер)
- [4-диметиламино-1-(3-фторфенил)-4-(3-метил-1Н-индол-2-ил)-циклогексил]-диметил-амин (неполярный диастереомер)
- N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-3-(трифторметил)-бензамид (неполярный диастереомер)
- N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-3-(трифторметил)-бензамид (полярный диастереомер)
- [4-[[4,6-бис(4-метокси-фенокси)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- (4-диметиламино-1,4-дифенил-циклогексил)-метил-[(1-метил-1Н-пиразол-3-ил)-метил]-амин (полярный диастереомер)
- (4-диметиламино-1,4-дифенил-циклогексил)-метил-[(1-метил-1Н-пиразол-3-ил)-метил]-амин (неполярный диастереомер)
- [4-[[4,6-бис(4-метокси-фенокси)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
- N-(4-диметиламино-1,4-дифенил-циклогексил)-4-метокси-N-метил-бензамид (неполярный диастереомер)
- N-(4-диметиламино-1,4-дифенил-циклогексил)-4-метокси-N-метил-бензамид (полярный диастереомер)
- (4-диметиламино-1,4-дифенил-циклогексил)-[(4-метоксифенил)-метил]-метил-амин (полярный диастереомер)
- (4-диметиламино-1,4-дифенил-циклогексил)-[(4-метоксифенил)-метил]-метил-амин (неполярный диастереомер)
- [1-(3-фторфенил)-4-(3-метил-1Н-индол-2-ил)-4-пирролидин-1-ил-циклогексил]-диметил-амин (неполярный диастереомер)
- [1-(3-фторфенил)-4-(3-метил-1Н-индол-2-ил)-4-пирролидин-1-ил-циклогексил]-диметил-амин (полярный диастереомер)
- [1-(3-фторфенил)-4-метиламино-4-(3-метил-1Н-индол-2-ил)-циклогексил]-диметил-амин
- диметил-[4-(3-метил-1Н-индол-2-ил)-1-фенил-4-пиперидин-1-ил-циклогексил]-амин (неполярный диастереомер)
- [1-(3-фторфенил)-4-(3-метил-1Н-индол-2-ил)-4-пиперидин-1-ил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-(диметил-амино)-4-(5-фтор-3-метил-1Н-индол-2-ил)-1-фенил-циклогексил]-диметил-амин (полярный диастереомер)
- (4-диметиламино-1,4-дифенил-циклогексил)-метил-[[3-(трифторметил)фенил]-метил]-амин (полярный диастереомер)
- (4-диметиламино-1,4-дифенил-циклогексил)-метил-[[3-(трифторметил)фенил]-метил]-амин (неполярный диастереомер)
- N-(4-диметиламино-1,4-дифенил-циклогексил)-3-фтор-N-метил-бензамид (неполярный диастереомер)
- N-(4-диметиламино-1,4-дифенил-циклогексил)-3-фтор-N-метил-бензамид (полярный диастереомер)
- (4-диметиламино-1,4-дифенил-циклогексил)-[(3-фторфенил)-метил]-метил-амин (неполярный диастереомер)
- (4-диметиламино-1,4-дифенил-циклогексил)-[(3-фторфенил)-метил]-метил-амин (полярный диастереомер)
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-этанол (полярный диастереомер)
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-N,N-диметил-ацетамид (полярный диастереомер)
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-N-метил-ацетамид (полярный диастереомер)
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-N,N-диметил-ацетамид (неполярный диастереомер)
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-N-метил-ацетамид (неполярный диастереомер)
- [4-диметиламино-4-(5-фтор-3-метил-1Н-индол-2-ил)-1-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-(5-фтор-3-метил-1Н-индол-2-ил)-1-фенил-4-пирролидин-1-ил-циклогексил]-диметил-амин (неполярный диастереомер)
- 2-[[4-(диметил-амино)-1,4-дифенил-циклогексил]-метил-амино]-этанол (неполярный диастереомер)
- [4-[[4,6-бис(диметиламино)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
- диметил-[4-[метил-(4-метиламино-6-пиперидин-1-ил-[1,3,5]триазин-2-ил)-амино]-1,4-дифенил-циклогексил]-амин (полярный диастереомер)
- 4-[[4-(диметил-амино)-1,4-дифенил-циклогексил]-метил-амино]-бутан-1-ол (полярный диастереомер)
- 3-[[4-(диметил-амино)-1,4-дифенил-циклогексил]-метил-карбамоил]-пропионовая кислота (полярный диастереомер)
- [4-(5-фтор-3-метил-1Н-индол-2-ил)-1-фенил-4-пирролидин-1-ил-циклогексил]-диметил-амин (полярный диастереомер)
- [1-(3-фторфенил)-4-(3-метил-1Н-индол-2-ил)-4-пиперидин-1-ил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-(азетидин-1-ил)-4-(5-фтор-3-метил-1Н-индол-2-ил)-1-(3-фторфенил)циклогексил]-диметил-амин (неполярный диастереомер)
- [4-(азетидин-1-ил)-4-(5-фтор-3-метил-1Н-индол-2-ил)-1-(3-фторфенил)-циклогексил]-диметил-амин (полярный диастереомер)
- [4-(5-фтор-3-метил-1Н-индол-2-ил)-4-морфолин-4-ил-1-фенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-(5-фтор-3-метил-1Н-индол-2-ил)-4-метиламино-1-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-(5-фтор-3-метил-1Н-индол-2-ил)-4-метиламино-1-фенил-циклогексил]-диметил-амин (полярный диастереомер)
- диметил-[4-метиламино-4-(3-метил-1Н-индол-2-ил)-1-тиофен-2-ил-циклогексил]-амин (полярный диастереомер)
- [4-(5-фтор-3-метил-1Н-индол-2-ил)-4-морфолин-4-ил-1-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-[(4-анилино-6-метиламино-[1,3,5]триазин-2-ил)-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-[[4-(изопропил-метил-амино)-6-метиламино-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-[(4-анилино-6-метиламино-[1,3,5]триазин-2-ил)-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-[[4-(бензиламино)-6-метиламино-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-[(4-бутиламино-6-метиламино-[1,3,5]триазин-2-ил)-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-[[4-(4-метокси-фенокси)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- (1,4-дифенил-4-пирролидин-1-ил-циклогексил)-метил-амин (полярный диастереомер)
- (1,4-дифенил-4-пирролидин-1-ил-циклогексил)-метил-амин (неполярный диастереомер)
- [4-[(бензил-метил-амино)-метил]-1,4-дифенил-циклогексил]-диметил-амин
- [4-диметиламино-1-(3-метил-1Н-индол-2-ил)-4-тиофен-2-ил-циклогексил]-метил-амин (неполярный диастереомер)
- (1,4-дифенил-4-пирролидин-1-ил-циклогексил)-диметил-амин (полярный диастереомер)
- (1,4-дифенил-4-пирролидин-1-ил-циклогексил)-диметил-амин (неполярный диастереомер)
- [4-[[4-(бензиламино)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- диметил-[4-[метил-(4-пиперидин-1-ил-[1,3,5]триазин-2-ил)-амино]-1,4-дифенил-циклогексил]-амин (полярный диастереомер)
- [4-[(4-бутиламино-[1,3,5]триазин-2-ил)-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-[(4-анилино-[1,3,5]триазин-2-ил)-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-[[4-(изопропил-метил-амино)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-[[4-(трет-бутиламино)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- [4-(циклогексил-метиламино)-1-(3-фторфенил)-4-(3-метил-1Н-индол-2-ил)-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-(циклопентиламино)-1-(3-фторфенил)-4-(3-метил-1Н-индол-2-ил)-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-анилино-1-(3-фторфенил)-4-(3-метил-1Н-индол-2-ил)-циклогексил]-диметил-амин
- [1-(3-фторфенил)-4-(3-метил-1Н-индол-2-ил)-4-(пиридин-4-иламино)-циклогексил]-диметил-амин
- [4-[(бутил-метил-амино)-метил]-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
- [4-[(бутил-метил-амино)-метил]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
- N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-циклогексанкарбоновой кислоты амид (полярный диастереомер)
- N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-тетрагидро-пиран-4-карбоновой кислоты амид (полярный диастереомер)
- циклогексил-метил-(4-диметиламино-1,4-дифенил-циклогексил)-метил-амин (полярный диастереомер)
- (4-диметиламино-1,4-дифенил-циклогексил)-метил-(тетрагидро-пиран-4-ил-метил)-амин (полярный диастереомер)
- N-(4-диметиламино-1,4-дифенил-циклогексил)-N,1-диметил-пиперидин-4-карбоновой кислоты амид (полярный диастереомер)
- (4-диметиламино-1,4-дифенил-циклогексил)-метил-[(1-метил-пиперидин-4-ил)-метил]-амин (полярный диастереомер)
и их физиологически совместимые соли и/или сольваты.
Соединения согласно изобретению воздействуют, например, на релевантный в связи с различными заболеваниями ORL1-рецептор, так что они пригодны в качестве фармацевтического действующего вещества в лекарственном средстве.
Поэтому другой объект изобретения относится к лекарственным средствам, которые содержат, по меньшей мере, одно соединение согласно изобретению, а также при необходимости пригодные добавки и/или вспомогательные вещества и/или при необходимости другие действующие вещества.
Соединения согласно изобретению обладают сопоставимым сродством к ORL1-рецептору, как и соединения согласно WO 03/008370. Однако по сравнению с этими соединениями они проявляют более высокую избирательность в отношении каппа-опиоидного рецептора, который отвечает за побочные эффекты, такие как, например, дисфория, седативный эффект и диурез. Кроме того, соединения согласно изобретению при благоприятном соотношении ORLI/м-сродство проявляют сбалансированное сродство к µ-опиоидному рецептору, которое не является сильным. Это представляет собой преимущество, так как µ-опиоидный рецептор устанавливает связь с побочными эффектами, в особенности такими как угнетение дыхания, запор и наркотическая зависимость. И поэтому соединения согласно изобретению особенно пригодны для разработки лекарственных средств.
Лекарственные средства согласно изобретению наряду с, по меньшей мере, одним соединением согласно изобретению при необходимости содержат пригодные добавки и/или вспомогательные вещества, а также носители, наполнители, растворители, разбавители, красители и/или связующие вещества и могут применяться как жидкие лекарственные формы в виде растворов для инъекций, капель или соков, как полутвердые лекарственные формы в виде гранул, таблеток, пеллет, пэтчей, капсул, пластырей/распылительных пластырей или аэрозолей. Выбор вспомогательных веществ и т.д., а также их используемые количества зависят от того, как должно применяться лекарственное средство: орально, перорально, парентерально, внутривенно, внутрибрюшинно, внутрикожно, внутримышечно, интраназально, буккально, ректально или местно, например, на кожу, слизистые оболочки или в глаза. Для орального применения пригодны препараты в виде таблеток, драже, капсул, гранул, капель, соков и сиропов, для парентерального, местного и ингаляционного применения пригодны растворы, суспензии, легко восстанавливаемые сухие препараты, а также спреи. Соединения согласно изобретению являются пригодными чрескожными препаратами для применения в форме с замедленным высвобождением, в растворенном виде или в пластыре, при необходимости с добавлением средств, способствующих кожному проникновению. Препаративные формы, применяемые орально или чрескожно, могут замедленно высвобождать соединения согласно изобретению. Соединения согласно изобретению могут также применяться в парентеральных формах пролонгированного действия, таких как, например, имплантаты или имплантационные насосы. В принципе, к лекарственным средствам согласно изобретению могут добавляться другие действующие вещества, известные специалисту в данной области техники.
Количества вводимого пациентам действующего вещества варьируются в зависимости от веса пациента, от вида применения, показания и степени тяжести заболевания. Обычно применяют от 0,00005 до 50 мг/кг, предпочтительно от 0,001 до 0,5 мг/кг, по меньшей мере, одного соединения согласно изобретению.
Для всех указанных выше форм лекарственных средств согласно изобретению является особенно предпочтительным, если лекарственное средство наряду с, по меньшей мере, одним соединением согласно изобретению содержит еще одно другое действующее вещество, в особенности опиоид, преимущественно сильный опиоид, в особенности морфин, или анестетик, преимущественно гексобарбитал или галотан.
В предпочтительной форме лекарственного средства содержащееся соединение согласно изобретению находится в виде чистого диастереомера и/или энантиомера.
В особенности ORL1-рецептор был идентифицирован в возникновении боли. Соответственно соединения согласно изобретению могут применяться для получения лекарственного средства для лечения боли, в особенности острой, невропатической или хронической боли.
Поэтому другой объект изобретения относится к применению соединения согласно изобретению для получения лекарственного средства для лечения боли, в особенности острой, висцеральной, невропатической или хронической боли.
Другой объект изобретения относится к применению соединения согласно изобретению для получения лекарственного средства для лечения состояний страха, стресса и связанных со стрессом синдромов, депрессий, эпилепсии, болезни Альцгеймера, старческого слабоумия, общих познавательных дисфункций, нарушений обучения и памяти (как ноотроп), синдромов отмены, злоупотребления алкоголем и/или наркотиками, и/или злоупотребления медикаментами, и/или алкогольной, наркотической, медикаментозной зависимости, сексуальных дисфункций, сердечно-сосудистых заболеваний, гипотонии, гипертензии, тиннитуса, зуда, мигрени, тугоухости, недостаточной подвижности кишечника, нарушений приема пищи, анорексии, ожирения, локомоторных расстройств, диареи, кахексии, недержания мочи соответственно как мышечный релаксант, противосудорожное средство или анестетик соответственно для совместного приема при лечении с опиоидным анальгетиком или с анестетиком, для диуреза или антинатрийуреза, анксиолизиса, для модуляции двигательной активности, для модуляции распределения нейромедиаторов и лечения связанных с этим нейродегенеративных заболеваний, для лечения синдромов отмены и/или для снижения наркотического потенциала опиоидов.
При этом в одном из указанных выше применений может быть предпочтительным, если применяемое соединение находится в виде чистого диастереомера и/или энантиомера, в виде рацемата или в виде неэквимолярной или эквимолярной смеси диастереомеров и/или энантиомеров.
Другой объект изобретения относится к способу лечения, в особенности, при одном из указанных выше показаний, нечеловеческого млекопитающего или людей, которому или которым необходимо лечение болей, в особенности хронических болей, путем введения терапевтически эффективной дозы соединения согласно изобретению или лекарственного средства согласно изобретению.
Другой объект изобретения относится к способу получения соединений согласно изобретению, как приведено в последующем описании и примерах.
а) синтез циклогексан-1,4-диаминов
Метод 1:
Структуры формулы А-2 могут быть получены путем реакции кетонов А-1 с аминами и кислыми реагентами Z-H. Пригодными реагентами Z-H являются, например, цианид водорода, 1,2,3-триазол, бензотриазол или пиразол.
Особенно предпочтительным путем для получения соединений структуры А-2 является взаимодействие кетонов с цианидами металлов и соответствующим амином в присутствии кислоты, преимущественно в спирте, при температурах от -40 до 60°С, преимущественно при комнатной температуре с цианидами щелочных металлов в метаноле.
Другим, особенно предпочтительным, путем для получения соединений структуры А-2 является взаимодействие кетонов с 1,2,3-триазолом и соответствующим амином в присутствии при водоотнимающих условиях, преимущественно при применении водоотделителя при повышенной температуре в инертном растворителе или при применении молекулярного сита или другого осушителя. Аналогично могут вводиться аналогичные А-2 структуры с бензотриазольными или пиразольными группами вместо триазольных групп.
В общем, циклогексан-1,4-диамины А-3 также могут быть получены посредством замещения пригодных уходящих групп Z в структурах формулы А-2. Пригодными уходящими группами предпочтительно являются цианогруппы; 1,2,3-триазол-1-ил-группы. Другими пригодными уходящими группами являются 1H-бензо[дн.][1,2,3]триазол-1-ил-группы и пиразол-1-ил-группы (Katritzky et al., Synthesis 1989, 66-69).
Особенно предпочтительным путем для получения соединений структуры А-3 является взаимодействие аминонитрилов А-2 с соответствующими металлорганическими соединениями, преимущественно соединениями Гриньяра, преимущественно в простых эфирах, преимущественно при КТ. Металлоорганические соединения или имеются в продаже, или могут быть получены согласно известным методам. Другим, особенно предпочтительным путем для получения соединений структуры А-3 является взаимодействие аминотриазолов А-2 с соответствующими металлорганическими соединениями, преимущественно соединениями Гриньяра, преимущественно в простых эфирах, преимущественно при КТ.
Металлоорганические соединения или имеются в продаже, или могут быть получены согласно методам, известным из литературных источников.
Метод 2:
Структуры формулы А-4 могут быть получены путем реакции кетонов А-1 с первичными аминами и кислыми реагентами Z-H. Пригодными реагентами Z-H являются, например, цианид водорода, 1,2,3-триазол, бензотриазол или пиразол.
Особенно предпочтительным путем для получения соединений структуры А-4 является взаимодействие кетонов с цианидами металлов и соответствующим амином в присутствии кислоты, преимущественно в спирте, при температурах от -40 до 60°С, преимущественно при комнатной температуре с цианидами щелочных металлов в метанол.
Другим, особенно предпочтительным, путем для получения соединений структуры А-4 является взаимодействие кетонов с 1,2,3-триазолом и соответствующим амином в присутствии при водоотнимающих условиях, преимущественно при применении водоотделителя при повышенной температуре в инертном растворителе или при применении молекулярного сита или другого осушителя. Аналогично могут быть введены аналогичные А-4 структуры с бензотриазольными или пиразольными группами вместо триазольных групп.
В общем, циклогексан-1,4-диамины А-5 также могут быть получены посредством замещения пригодных уходящих групп Z в структурах формулы А-4. Пригодными уходящими группами предпочтительно являются цианогруппы; 1,2,3-триазол-1-ил-группы. Другими пригодными уходящими группами являются 1H-бензо[дн.][1,2,3]триазол-1-ил-группы и пиразол-1-ил-группы (Katritzky et al., Synthesis 1989, 66-69).
Особенно предпочтительным путем для получения соединений структуры А-5 является взаимодействие аминонитрилов А-4 с соответствующими металлорганическими соединениями, преимущественно соединениями Гриньяра, преимущественно в простых эфирах, преимущественно при КТ. Металлоорганические соединения или имеются в продаже, или могут быть получены согласно известным методам.
Другим, особенно предпочтительным, путем для получения соединений структуры А-5 является взаимодействие аминотриазолов А-4 с соответствующими металлорганическими соединениями, преимущественно соединениями Гриньяра, преимущественно в простых эфирах, преимущественно при КТ.
Металлоорганические соединения или имеются в продаже, или могут быть получены согласно методам, известным из литературных источников.
Циклогексан-1,4-диамины типа А-3 могут быть синтезированы методами, известными специалисту в данной области техники, также из А-5. Введение (алкил-)заместителей затем может осуществляться при условиях восстановительного аминирования через альдегидный компонент. Такой, известный специалисту в данной области техники, метод может представлять собой взаимодействие с альдегидом с добавлением восстановителя, например боргидрида натрия.
b) синтез (1,4-диаминоциклогексил)(гетероарил)метанона

Замещенные (1,4-диаминоциклогексил)(гетероарил)метанона типа А-7, Х=O могут быть синтезированы известными специалисту в данной области техники методами из описанного выше исходного продукта А-2, Z=CN. Посредством взаимодействия металлированных гетероциклов типа А-6 с тройной связью А-2, Z=CN получают промежуточный продукт А-7, Х=NH. Затем при кислых условиях получается гидролиз при расщеплении имина А-7, Х=О.
с) синтез 4-алкилоксициклогексан-1-аминов

Замещенные 4-алкилоксициклогексан-1-амины типа А-10 могут быть синтезированы известными специалисту в данной области техники методами из исходного продукта А-1. Алкоголят, полученный посредством реакции металлированных алкинов с А-1, подвергают взаимодействию с соответствующими электрофилами, например, типа R0X (с Х = например, Br, I, OTos, OTf и т.д.) до получения А-8. Взаимодействие карбинолов А-9 до получения согласно изобретению замещенных 4-алкилоксициклогексан-1-аминов типа А-10 может происходить в органических растворителях, например тетрагидрофуран, диметилформамид, бензол, толуол, ксилолы, диметоксиэтан или диметиловый эфир диэтиленгликоля, в присутствии неорганического основания, например, карбоната натрия, карбоната калия или карбоната цезия или фосфата калия, в присутствии PdCl2, Pd(OAc)2, PdCl2(MeCN)2, PdCl2(PPh3)2 или хлорида [1,3-бис-(2,6-диизопропилфенил)имидазол-2-илиден]-(3-хлорпиридил)палладия(II) (PEPPSI®), при необходимости в присутствии дополнительных лигандов, например трифенил-, три-о-толил-, трициклогексил или три-t-бутилфосфин, при необходимости в присутствии катализаторов межфазного переноса, например хлорида тетра-n-бутиламмония, гидрохлорида тетра-n-бутиламмония или йодида тетра-n-бутиламмония и при температурах между 60°С и 180°С, также при воздействии микроволнами.
d) синтез 4-аминометил-циклогексил-1-аминов

Замещенные 4-аминометил-циклогексил-1-амины типа А-14 могут быть синтезированы известными специалисту в данной области техники методами из известных исходных продуктов А-1. Исходя их кетонов, таких как А-1, посредством олефинирования Виттига с фосфорилиденом получают промежуточные алкены А-11. Затем соединения формулы А-12 могут быть получены из соответствующих предшественников А-11 в присутствии комплекса кобальт-(II)-сален путем гидроцианирования (Carreira et al. Angew. Chem. Int. Ed., 46, 2006, 4519.). Взаимодействие нитрильной группы в А-12 с восстановителем, например гидридом, таким как боргидрид натрия или боргидрид лития, цианоборгидрид натрия, ацетоксиборгидрид натрия, гидрид диизобутилалюминия, три-(втор.-бутил)боргидрид лития (L-Selectride®) или гидрид литийалюминия, при необходимости в присутствии кислот Льюиса, например ZnCl2, Ni(OAc)2 или CoCl2 дает амины А-13.
Метод 1:
Амины типа А-13 известным специалисту в данной области техники способом могут быть ацилированы, сульфонилированы или карбамоилированы до получения соединений А-14. Такой, известный специалисту в данной области техники, метод может представлять собой взаимодействие с ангидридом или хлорангидридом кислоты с добавлением одного основания, например триэтиламина.
Метод 2:
Амины типа А-13 способом, известным специалисту в данной области техники, могут подвергаться восстановительному аминированию до получения соединений типа А-14. Такой, известный специалисту в данной области техники, метод может представлять собой взаимодействие с альдегидом с добавлением восстановителя, например боргидрида натрия.
d) синтез (4-аминоциклогексил)метанолов

Замещенные (4-аминоциклогексил)метанолы типа А-18 могут быть синтезированы известными специалисту в данной области техники методами из известных исходных продуктов А-15. В литературных источниках описано депротонирование сложных эфиров А-15 с основанием, например диизопропиламидом лития (LDA) и взаимодействие с соответствующими электрофилами, например, типа R0-X (с Х=, например, Br, I, OTos, OTf и т.д.) до получения А-16 (Williams et al. J. Org. Chem. 1980, 45, 5082; Shiner et al. J. Am. Chem. Soc. 1981, 103, 436; Xia et al. Org. Lett. 2005, 7, 1315.). Взаимодействие А-16 может происходить с восстановителем, например гидридом, таким как боргидрид натрия или боргидрид лития, цианоборгидрид натрия, ацетоксиборгидрид натрия, гидрид диизобутилалюминия, три-(втор.-бутил)боргидрид лития (L-Selectride®) или гидрид литийалюминия, при необходимости в присутствии кислот Льюиса, например ZnCl2, Ni(OAc)2 или CoCl2 и расщепление кеталя известными специалисту в данной области техники методами путем снятия защиты с помощью кислот. При этом Х выбран из группы алкил, алкил/алкилиден/с арил или алкил (насыщенный/ненасыщенный) замещенным алкилиденом. Защита гидроксильной группы известными специалисту в данной области техники методами, например путем взаимодействия с простыми алкилвиниловыми эфирами, приводит к получению соответствующих простых α-алкилоксиэтиловых эфиров А-17.
Структуры формулы А-18 могут быть получены путем реакции кетонов А-17 с аминами и кислыми реагентами Z-H. Пригодными реагентами Z-H являются, например, цианид водорода, 1,2,3-триазол, бензотриазол или пиразол.
Особенно предпочтительным путем до получения таких аминонитрилов является взаимодействие кетонов с цианидами металлов и соответствующим амином в присутствии кислоты, преимущественно в спирте, при температурах от -40 до 60°С, преимущественно при комнатной температуре с цианидами щелочных металлов в метанол.
Другим, особенно предпочтительным, путем до получения таких аминонитрилов является взаимодействие кетонов с 1,2,3-триазолом и соответствующим амином в присутствии при водоотнимающих условиях, преимущественно при применении водоотделителя при повышенной температуре в инертном растворителе или при применении молекулярного сита или другого осушителя. Аналогично могут быть введены подобные структуры с бензотриазольными или пиразольными группами вместо триазольных групп.
Введение остатка R3 может осуществляться посредством замещения пригодных уходящих групп Z, таких как, например, уже описаны для взаимодействия А-2 в А-3.
Соединения формулы А-18 могут быть выделены из соответствующих ацеталей, или из их солей, известными специалисту в данной области техники методами путем снятия защиты с помощью кислот. При этом PG выбран из группы известных специалисту в данной области техники ацетальных защитных групп для гидроксильных групп, например, защитной группы простого α-алкилоксиэтилового эфира.
е) синтез 4-индолилциклогексан-1,4-диаминов
Метод 1:

Стадия 1:
Кето-группа согласно методам, известным из литературных источников, в особенности при применении релевантных для синтеза А-1 отрывков литературных источников, может быть переведена в монометиламинонитрил.
Стадия 2:
Аминонитрилы А-19 с соответствующими металлорганическими соединениями, преимущественно литийорганическим соединением и соединениями Гриньяра, преимущественно в простых эфирах, преимущественно при КТ могут быть переведены в производные алкина А-20. Металлоорганические соединения или имеются в продаже, или могут быть получены согласно известным методам.
Стадия 3:
Производные алкина А-20 согласно F. Messina et al. / Tetrahedron: Asymmetry 11 (2000) 1681-1685 могут быть переведены в защищенные производные индола А-21.
Стадия 4:
Производные индола А-21 согласно методам, известным из литературных источников, (см. Protective Groups в Organic Synthesis by Peter G.M.Wuts, Theodora W.Greene, WileyBlackwell; 4-е изд.) могут подвергаться взаимодействию до получения производных индола А-22.
Стадия 5:
Производные индола А-22 посредством методов, известных специалисту в данной области техники могут быть переведены в амиды А-23. Такой, известный специалисту в данной области техники, метод может быть, например, взаимодействием с А-22 с карбоновой кислотой с добавлением реагента сочетания, например, карбонилдиимидазолом.
Производные индола А-22 посредством методов, известным специалисту в данной области техники, могут быть переведены в сульфонамиды А-23. Такой, известный специалисту в данной области техники, метод может быть, например, взаимодействием с А-22 с сульфонилхлоридом с добавлением одного основания, например триэтиламина.
Производные индола А-22 посредством методов, известным специалисту в данной области техники, могут быть переведены в амины А-23. Такой, известный специалисту в данной области техники, метод может быть, например, взаимодействием с А-22 с альдегидом с добавлением восстановителя, например боргидрида натрия.
Метод 2:

Замещенные циклогексанамины типа А-25 могут быть синтезированы из исходного продукта А-1 путем реакции с металлированными алкинами. Взаимодействие карбинолов А-8' и А-9 до получения замещенных 4-алкилоксициклогексан-1-аминов типа A-10' может осуществляться в органических растворителях, например тетрагидрофуран, диметилформамид, бензол, толуол, ксилолы, диметоксиэтан или диметиловый эфир диэтиленгликоля, в присутствии неорганического основания, например карбоната натрия, карбоната калия или карбоната цезия, или фосфата калия, в присутствии PdCl2, Pd(OAc)2, PdCl2(MeCN)2, PdCl2(PPh3)2 или хлорида [1,3-бис-(2,6-диизопропилфенил)имидазол-2-илиден]-(3-хлорпиридил)палладия(II) (PEPPSI®), при необходимости в присутствии дополнительных лигандов, например трифенил-, три-о-толил-, трициклогексил или три-t-бутилфосфин, при необходимости в присутствии катализаторов межфазного переноса, например хлорида тетра-n-бутиламмония, гидрохлорида тетра-n-бутиламмония или йодида тетра-n-бутиламмония и при температурах между 60°С и 180°С, также воздействии микроволнами. Альтернативно этому также взаимодействие металлированных гетероциклов А-9' с исходным продуктом А-1 в органических растворителях при температурах между 25°С и -100°С может приводить к получению карбинолов типа А-10'. Последующая циклизация А-10' до получения соли аммония А-24 может происходить в органических растворителях в присутствии фторирующих агентов при температурах между 25°С и -100°С. Раскрытие соли А-24 до получения согласно изобретению замещенных циклогексанаминов типа А-25 может осуществляться с пригодными нуклеофилами без или также в присутствии органических растворителей при температурах между 0°С и 180°С, также при воздействии микроволнами.
f) синтез N-гетероарил-1,4-диаминов

Замещенные циклогексанамины типа A-26 и A-27 могут быть синтезированы из исходного продукта A-5. Путем реакции гетероциклов (метод 1) или цианатов (метод 2) с пригодными уходящими группами (например, с LG=Cl или 4-ОМе-C6H4) получают промежуточный продукт А-3'. Оставшиеся уходящие группы могут быть замещены последовательно пригодными нуклеофилами (Nu) и получают соединения согласно изобретению замещенных циклогексанаминов типа A-26 и A-27.
g) предварительная стадия
Соединения общих формул А-1 и А-15 или имеются в продаже, или их получение известно из уровня техники или очевидным для специалиста в данной области техники способом вытекает из уровня техники. В частности для этого являются релевантными следующие процитированные источники: Jirkovsky et al., J. Heterocycl. Chem., 12, 1975, 937-940; Beck et al., J. Chem. Soc. Perkin 1, 1992, 813-822; Shinada et al. Tetrahedron Lett., 39, 1996, 7099-7102; Garden et al. Tetrahedron, 58, 2002, 8399-8412; Lednicer et al, J. Med. Chem, 23, 1980, 424-430; Bandini et al. J. Org. Chem. 67, 15; 2002, 5386-5389; Davis et al, J. Med. Chem. 35, 1,1992, 177-184; Yamagishi et al., J. Med. Chem. 35, 11, 1992, 2085-2094; Gleaveetal, Bioorg. Med. Chem. Lett. 8, 10, 1998, 1231-1236; Sandmeyer, Helv. Chim. Acta; 2; 1919; 239; Katzetal, J. Med. Chem. 31, 6, 1988; 1244-1250; Вас et al. Tetrahedron Lett. 1988, 29, 2819; Ma et al. J. Org. Chem. 2001, 66, 4525; Kato et al. J. dn-opine Chem. 99, 1, 1999, 5-8.
Относительно других подробностей синтеза соединений согласно изобретению может в полном объеме может быть сделана ссылка на WO 2002/090317, WO 2002/90330, WO 2003/008370, WO 2003/008731, WO 2003/080557, WO 2004/043899, WO 2004/043900, WO 2004/043902, WO 2004/043909, WO 2004/043949, WO 2004/043967, WO 2005/063769, WO 2005/066183, WO 2005/110970, WO 2005/110971, WO 2005/110973, WO 2005/110974, WO 2005/110975, WO 2005/110976, WO 2005/110977, WO 2006/018184, WO 2006/108565, WO 2007/079927, WO 2007/079928, WO 2007/079930, WO 2007/079931, WO 2007/124903, WO 2008/009415 и WO 2008/009416.
Примеры
Нижеследующие примеры служат для более подробного пояснения изобретения, однако не должны интерпретироваться как ограничивающие его.
Выходы полученных соединений не оптимизированы. Все температуры не поддаются корректировке. Указание «простой эфир» означает диэтиловый эфир, «ЕЕ» этилацетат и «ДХМ» дихлорметан. Указание «эквиваленты» означает эквиваленты количества веществ, «Тпл.» точка плавления соответственно интервал температур плавления, «Разл.» разложение, «КТ» комнатная температура, «абс.» абсолютный (безводный), «рац.» рацемический, «конц.» концентрировали, «мин» минуты, «ч» часы, «дн.» дни, «объемн.%» объемный процент, «мас.%» массовый процент и «М» является указанием концентрации в моль/л.
В качестве неподвижной фазы для колоночной хроматографии использовали силикагель 60 (0.040-0.063 мм) фирмы Е.Merck, Дармштадт. Исследования с помощью тонкослойной хроматографии проводили с помощью готовых пластин HPTLC, силикагель 60 F 254, фирмы Е.Merck, Дармштадт. Соотношение компонентов смеси растворителей для хроматографических исследований постоянно указывается в объемн./объемн.
Пример 1:
1-(имино(1-метил-1Н-индол-2-ил)метил)-N1,N1,N4,N4-тетраметил-4-фенилциклогексан-1,4-диамин бис(2-гидроксипропан-1,2,3-трикарбоксилат)
а) 1,4-бис-диметиламино-4-фенилциклогексанкарбонитрил
Смесь из метанола (50 мл) и воды (50 мл) подкисляли соляной кислотой (37%, 0,2 мл) и при охлаждении льдом и перемешивания смешивали с водным раствором диметиламино (40%, 11,5 мл, 91 ммоль). Затем к раствору добавляли 4-диметиламино-4-фенилциклогексанон (2,17 г, 10 ммоль) и KCN (1,6 г, 24,6 ммоль). Через 15 мин образовался светлый раствор. Охлаждение льдом удаляли, и состав перемешивали 2,5 ч при КТ, причем через прибл. 1 ч в осадок начало выпадать белое твердое вещество. Для дополнения осаждения состав с помощью охлаждения льдом в течение 1 ч вновь доводили до прибл. 0°С. Затем осадок отделяли с помощью фритты и высушивали в вакууме при температуре ванны в 40°С. Получали смесь диастереоизомеров титульного соединения с выходом в 1,83 г (67%) и с точкой плавления в 82-92°С.
13С ЯМР (101 МГц, CDCl3) δ част. на млн: 29.3*, 30.2, 31.2, 37.7, 38.2, 39.9, 58.4*, 60.2, 62.4*, 118.7, 119.0, 126.8, 127.4, 127.7, 128.0, 136.2*, 137.7*
* расширенные сигналы
b) 1-[имино-(1-метил-1Н-индол-2-ил)метил]-N,N,N',N'-тетраметил-4-фенил-циклогексан-1,4-диамин [неполярный диастереоизомер и полярный диастереоизомер]
При удалении влаги при 0°С к раствору n-бутиллития (2,5N в n-гексан, 4 мл, 10 ммоль) в сухом ТГФ (10 мл) добавляли N-метилиндол [1,31 г, 10 ммоль, растворенный в сухом ТГФ (10 мл)]. Состав 60 мин перемешивали, сохраняя охлаждение, причем через прибл. 10 мин в осадок начало выпадать твердое вещество. Затем происходило добавление смеси диастереоизомеров из предшествующей стадии [1,33 г, 5 ммоль, растворенной в сухом ТГФ (10 мл)] в течение 10 мин. После окончательного добавления охлаждение удаляли, и состав после достижения КТ перемешивали последующие 18 ч. Для переработки состав осторожно смешивали со смесью из ТГФ (5 мл) и воды (1 мл). Затем к смеси добавляли насыщенный раствор NaCl (30 мл). Органическую фазу отделяли, водную фазу экстрагировали этилацетатом (4×20 мл). Объединенные органические экстракты высушивали над MgSO4 и затем концентрировали. Полученный остаток (2,7 г) очищали хроматографически [силикагель 60 G (10 г); этилацетат (100 мл), этилацетат/этанол 1:1 (100 мл), EtOH 50 мл]. Неполярный диастереоизомер получали, таким образом, с выходом в 26% (526 мг), полярный диастереоизомер с выходом в 32% (650 мг).
c) 1-(имино(1-метил-1Н-индол-2-ил)метил)-N1,N1,N4,N4-тетраметил-4-фенилциклогексан-1,4-диамин бис(2-гидроксипропан-1,2,3-трикарбоксилат)
Неполярный диастереоизомер (230 мг, 0,61 ммоль) растворяли в пропан-2-оле (5 мл) при температуре кипения и смешивали с горячим раствором лимонной кислоты [382 мг, 2 ммоль, в пропан-2-оле (4 мл)]. При охлаждении раствора до КТ выпадал осадок. Состав для дополнения осаждения 20 ч выдерживали при 5°С, затем твердое вещество отделяли с помощью фритты и высушивали. Бисцитрат, таким образом, получали в виде стекловидного твердого вещества с выходом в 310 мг (64%).
13С ЯМР (101 МГц, ДМСО-d6) δ част. на млн: 25.5 (пропан-2-ол), 26.9, 27.4, 30.9, 37.5, 38.1, 43.3, 62.0 (пропан-2-ол?), 62.7, 66.1*, 71.9, 101.3*, 110.1, 119.7, 120.7, 122.1, 126.3, 128.6, 128.8, 132.1*, 137.0, 137.3, 171.2, 173.5, 175.5
* сильно расширенные сигналы
Пример 2:
(4-диметиламино-4-фенил-1-(пирролидин-1-ил)циклогексил)-(1-метил-1Н-индол-2-ил)метанон (неполярный диастереоизомер)
Во время синтеза примерного соединения 6, стадия b) также получилось аналогичное неполярное соединение в виде смеси, которую (680 мг) смешивали с 2N HCl (20 мл) и 18 ч перемешивали при КТ. В осадок выпадало твердое вещество. Для переработки реакционную смесь при комнатной температуре подщелачивали посредством 2N NaOH (30 мл). Водную фазу экстрагировали этилацетатом (3×10 мл). Объединенные органические экстракты высушивали над MgSO4 и затем концентрировали. Попытки перекристаллизации получившегося сырого продукта (из этилацетата и ДМСО) не привели к отделению примесей. Одну часть полученного сырого продукта очищали хроматографически [силикагель 60 G (10 г); циклогексан/этилацетат 2:8 (100 мл)]. Титульное соединение выделяли, таким образом, с точкой плавления в 212-218°С в количестве 59 мг.
13С ЯМР (101 МГц, CDCl3) δ част. на млн: 24.6, 26.4*, 29.9, 32.3, 37.9, 45.6, 59.9*67.6, 110.1, 110.6, 120.3, 122.7, 124.9, 125.8, 126.5, 127.3, 127.6, 134.4, 138.2*, 138.9, 198.2
* расширенные сигналы
Пример 3:
1-(имино(1-метил-1Н-индол-2-ил)метил)-N1,N1,N4,N4-тетраметил-4-фенилциклогексан-1,4-диамин бис(2-гидроксипропан-1,2,3-трикарбоксилат)(полярный диастереоизомер)
Полярный диастереоизомер из примера 1, стадия b) (248 мг, 0,66 ммоль) растворяли в пропан-2-оле (5 мл) при температуре кипения и смешивали с горячим раствором лимонной кислоты [382 мг, 2 ммоль, в горячем пропан-2-оле (4 мл)]. При охлаждении раствора до КТ выпадал осадок. Состав для дополнения осаждения выдерживали 20 ч при 5°С, затем твердое вещество отделяли с помощью фритты и высушивали. Цитрат, таким образом, получали с выходом в 380 мг (73%, точка плавления от 80°С) в виде бисцитрата.
13С ЯМР (101 МГц, ДМСО-) δ част. на млн: 24.0, 25.4 (пропан-2-ол), 28.1, 31.4, 37.4, 37.5, 43.4, 62.0, 64.4 (пропан-2-ол), 67.8, 71.8, 103.4, 110.2, 119.7, 121.0, 122.4, 126.2, 128.8, 129.2, 129.3, 130.2, 136.2, 137.8, 170.9, 171.2, 175.6
Пример 4:
(1,4-бис-диметиламино-4-фенилциклогексил)-(1-метил-1Н-индол-2-ил)метанон (неполярный диастереоизомер)
Примерное соединение 1 (250 мг, 0,62 ммоль) смешивали с 2N HCl (10 мл) и 3 ч перемешивали при КТ, а также 1 ч при 50°С (температура ванны). Во время реакции выпадал осадок. - Для переработки реакционную смесь при комнатной температуре сначала нейтрализовали с помощью К2СО3 и затем резко подщелачивали с 2N NaOH (1 мл). Водную фазу экстрагировали этилацетатом (3×10 мл). Объединенные органические экстракты высушивали над MgSO4 и затем концентрировали. Полученный остаток (240 мг) очищали хроматографически [силикагель 60 G (10 г); циклогексан/этилацетат 1:1, (100 мл)]. Таким образом, титульное соединение еще отделяли от имеющегося исходного продукта и получали с выходом в 120 мг (48%) с точкой плавления в 165-169°С (после перекристаллизации из этанола).
13С ЯМР (101 МГц, CDCl3) δ част. на млн: 24.2, 30.2, 32.3, 37.9, 38.8, 59.0, 69.6, 110.1, 111.5, 120.3, 122.9, 125.0, 125.8, 126.3, 126.8, 127.4, 134.8, 139.0, 139.3, 198.9
Пример 5:
(1,4-бис-диметиламино-4-фенилциклогексил)-(1-метил-1Н-индол-2-ил)метанон (полярный диастереоизомер)
Примерное соединение 3 (360 мг, 0,9 ммоль) смешивали с 2N HCl (10 мл) и 4 ч перемешивали при 70°С (температура ванны). - Для переработки реакционную смесь сначала нейтрализовали при комнатной температуре посредством К2СО3 и резко подщелачивали с 2N NaOH (1 мл). Водную фазу экстрагировали этилацетатом (3×10 мл). Объединенные органические экстракты высушивали над MgSO4 и затем концентрировали. Полученный остаток (240 мг) очищали хроматографически [силикагель 60 G (10 г); циклогексан/этилацетат 1:1, (150 мл), этилацетат (50 мл)]. Титульное соединение выделяли, таким образом, с выходом в 234 мг (65%) с точкой плавления в 109-111°С (после перекристаллизации из пропан-2-ола).
13С ЯМР (101 МГц, CDCl3) δ част. на млн: 25.2, 29.8, 32.5, 38.3, 38.5, 61.8, 69.7, 110.2, 111.7, 120.3, 122.9, 125.1, 125.8, 126.5, 127.8, 127.9, 133.7, 136.8, 139.2, 198.4
Пример 6:
4-(имино(1-метил-1Н-индол-2-ил)метил)-N,N-диметил-1-фенил-4-(пирролидин-1-ил)циклогексанамин (полярный диастереоизомер)
а) 4-диметиламино-4-фенил-1-(пирролидин-1-ил)циклогексанкарбонитрил
Смесь из метанола (50 мл) и воды (50 мл) подкисляли соляной кислотой (37%, 0,2 мл) и при охлаждении льдом и перемешивании смешивали с пирролидином (7,5 мл, 91 ммоль). Затем к раствору добавляли 4-диметиламино-4-фенилциклогексанон (2,17 г, 10 ммоль). Для того чтобы добиться как можно более полного растворения кетонов, состав перемешивали 10 мин. Затем осуществляли добавление KCN (1,6 г, 24,6 ммоль). Охлаждение льдом удаляли и состав 2 дн. перемешивали при КТ, причем в осадок выпадало белое твердое вещество. Для дополнения осаждения состав 1 ч с помощью охлаждения льдом вновь доводили прибл. до 0°С. Затем осадок с помощью фритты отделяли и высушивали в вакууме при температуре ванны в 40°С. Получали смесь диастереоизомеров титульного соединения с выходом в 2,7 г (90%) и с точкой плавления в 136-142°С.
AS 09460:13С ЯМР (101 МГц, CDCl3) δ част. на млн: 23.4, 23.5, 29.1*, 31.4, 32.3*, 37.7, 38.2, 48.0, 48.1, 58.8*, 60.3*, 61.8*. 62.2*, 119.7, 120.0, 126.7, 126.8, 127.4, 127.7, 127.9, 136.4*, 137.5*
* расширенные сигналы
b) 4-(имино(1-метил-1Н-индол-2-ил)метил)-N,N-диметил-1-фенил-4-(пирролидин-1-ил)циклогексанамин(полярный диастереоизомер)
При удалении влаги при 0°С к раствору n-бутиллития (2,5N в n-гексан, 4 мл, 10 ммоль) в сухом ТГФ (10 мл) добавляли М-метилиндол [1,31 г, 10 ммоль, растворенный в сухом ТГФ (10 мл)]. Состав перемешивали 60 мин, сохраняя охлаждение, причем через прибл. 10 мин. В осадок начало выпадать твердое вещество. Затем происходило добавление смеси диастереоизомеров из предшествующей стадии [1,49 г, 5 ммоль, растворяли в сухом ТГФ (20 мл)] в течение 20 мин. После окончательного добавления охлаждение удаляли, и состав после достижения КТ перемешивали последующие 18 ч. Для переработки состав осторожно смешивали со смесью из ТГФ (5 мл) и воды (1 мл). Затем к смеси добавляли насыщенный раствор NaCl (30 мл). Органическую фазу отделяли, водную фазу экстрагировали этилацетатом (4×20 мл). Объединенные органические экстракты высушивали над MgSO4 и затем концентрировали. Полученный остаток (2,78 г) очищали хроматографически [силикагель 60 G (10 г); этилацетат (200 мл), этилацетат/этанол 1:1 (50 мл)]. Таким образом, полярный диастереоизомер можно было выделить с выходом в 6% (140 мг) в виде вязкой массы, неполярный диастереомер получали в виде смеси.
13С ЯМР (101 МГц, CDCl3) δ част. на млн: 24.2, 26.0, 29.8, 31.7, 38.2, 44.9, 61.3, 64.2, 104.9, 109.8, 119.9, 121.4, 122.7, 126.5, 127.0, 127.6, 127.7, 137.0, 137.3, 138.3, 175.2
Пример 7:
(4-диметиламино-4-фенил-1-(пирролидин-1-ил)циклогексил)-(1-метил-1Н-индол-2-ил)метанон (полярный диастереоизомер)
Примерное соединение 6, стадия b) (99 мг, 0,23 ммоль) смешивали с 2N HCl (3 мл) и перемешивали 18 ч при КТ. Тотчас после добавления кислоты началось окрашивание раствора в оранжевый цвет. - Для переработки реакционную смесь при комнатной температуре подщелачивали посредством 2N NaOH (5 мл). Водную фазу экстрагировали дихлорметаном (3×10 мл). Объединенные органические экстракты высушивали над MgSO4 и затем концентрировали. Полученный остаток (84 мг) очищали хроматографически [силикагель 60 G (10 г); этилацетат (120 мл)]. Титульное соединение выделяли, таким образом, с выходом в 68 мг (68%) с точкой плавления от 134°С.
13С ЯМР (101 МГц, CDCl3) δ част. на млн: 24.1, 26.3*, 29.9, 32.4, 38.3, 45.3, 61.6*, 67.9, 110.2, 111.1, 120.3, 122.9, 125.0, 125.9, 126.5, 127.8, 127.9, 134.1, 136.9*, 139.1, 198.1
* расширенные сигналы
Пример 8:
N,N,N'-триметил-1,4-дифенил-циклогексан-1,4-диамин (неполярный диастереомер)
a) 4-диметиламино-1-метиламино-4-фенил-циклогексанкарбонитрил
К охлажденному до 0°С раствору 4N соляной кислоты (3.75 мл) и метанола (2.25 мл) добавляли 40%-ый водный раствор метиламина (8.7 мл, 69 ммоль) и 4-диметиламино-4-фенилциклогексанон (3.13 г, 14.4 ммоль), растворенный в метаноле (15 мл). Затем реакционную смесь смешивали с цианидом калия (2.25 г, 34 ммоль) и 5 дн. перемешивали при КТ. Для переработки смесь смешивали с водой (60 мл) и экстрагировали простым эфиром (3×50 мл). Объединенные органические фазы высушивали сульфатом натрия и концентрировали в вак.
Выход: 3.48 г (94%), смесь диастереомеров
1Н-ЯМР (ДМСО-d6): 1.31 (1Н, m); 1.64 (1H, m); 1.79 (2H, m); 1.93 (6H, d); 2.03 (2H, m); 2.22 и 2.34 (3H, dd); 2.77 (1H, m); 2.63 и 2.77 (1H, m); 7.33 (5H, m).
b) N,N,N'-триметил-1,4-дифенил-циклогексан-1,4-диамин (неполярный диастереомер)
Фениллитий (8.4 мл, 15 ммоль, 1.8 М раствор в дибутиловом эфире) помещали под аргоном и при КТ по каплям смешивали с раствором смеси диастереоизомеров из предшествующей стадии (1.29 г, 5 ммоль) в диэтиловом эфире (15 мл). При этом реакционный раствор нагревался до 35°С, и в осадок выпадало твердое вещество. Реакционную смесь 30 мин кипятили в колбе с обратным холодильником (ванна 50°С), затем гидролизовали в ледяной ванне (0-10°С) с 20%-ым раствором NH4Cl (10 мл) и органическую фазу отделяли. Водную фазу экстрагировали простым эфиром (2×30 мл). Объединенные органические растворы высушивали над Na2SO4 и концентрировали в вак. Остаток отделяли путем флеш-хроматографии (50 г силикагель) с хлороформ/метанолом (20:1→9:1→1:1 + 1% ТЕА).
Выход: 283 мг (18%) неполярный диастереомер, масло
1Н-ЯМР (ДМСО-d6): 1.64 (2 Н, m); 1.86 (3H, s); 1.92 (6H, s); 2.09 (6H, m); 7.25 (2H, m); 7.35 (6H, m); 7.49 (2H, m).
Пример 9:
N,N,N'-триметил-1,4-дифенил-циклогексан-1,4-диамин (полярный диастереомер)
При очистке примерного соединения 8 стадии b) также можно было выделить аналогичный полярный диастереомер.
Выход: 306 мг (20%) полярный диастереомер.
1Н-ЯМР (ДМСО-d6): 1.47 (2H, m); 1.87 (5H, m); 1.95 (6H, s); 2.13 (4H, m); 7.10 (1H, m); 7.23 (5H, m); 7.34 (4H, m).
Пример 10:
N,N,N',N'-тетраметил-1,4-дифенил-циклогексан-1,4-диамин (неполярный диастереомер)
Раствор примерного соединения 8 (242 мг, 0.78 ммоль) и формалин (1.1 мл, 37%-ый водный раствор) в ацетонитриле (10 мл) порционно смешивали с цианоборгидридом натрия (200 мг, 3.2 ммоль) и 45 мин перемешивали при КТ. Затем добавляли конц. уксусную кислоту до нейтральной реакции и 45 мин перемешивали при КТ.
Для переработки растворитель удаляли в вак., остаток ресуспендировали в 2N NaOH (10 мл) и затем экстрагировали простым эфиром (3×10 мл). Органический раствор высушивали над Na2SO4 и концентрировали в вак. Оставшийся остаток очищали путем флеш-хроматографии с CHCl3/МеОН (1:1).
Выход: 230 мг (92%)
Точка плавления: 117-118°С
1Н-ЯМР (ДМСО-d6): 1.76 (4Н, шир.); 1.96 (12Н, s); 2.28 (4Н, шир.); 7.15 (2Н, m); 7.27 (8Н, m).
Пример 11:
N,N,N',N'-тетраметил-1,4-дифенил-циклогексан-1,4-диамин (полярный диастереомер)
Раствор примерного соединения 9, стадия b) (223 мг, 0.72 ммоль) и формалин (1.0 мл, 37%-ый водный раствор) в ацетонитриле (10 мл) порционно смешивали с цианоборгидридом натрия (182 мг, 2.9 ммоль) и 45 мин при КТ перемешивали. Затем добавляли конц. уксусную кислота до нейтральной реакции и 45 мин перемешивали при КТ.
Для переработки растворитель удаляли в вак., остаток ресуспендировали в 2N NaOH (10 мл) и затем экстрагировали простым эфиром (3×10 мл). Органический раствор высушивали над Na2SO4 и концентрировали в вак. Оставшийся остаток очищали путем флеш-хроматографии с CHCl3/МеОН (9:1).
Выход: 160 мг (69%)
Точка плавления: 197-198°С
1Н-ЯМР (CD3OD): 1.47 (4Н, d); 1.91 (12Н, s); 2.75 (4Н, d); 7.32 (2Н, m); 7.46 (8H, m).
Пример 12:
1-бензил-N,N,N',N'-тетраметил-4-фенил-циклогексан-1,4-диамины (неполярный диастереомер)
a) 1,4-бис-диметиламино-4-фенил-циклогексанкарбонитрил
К смеси из 4N соляной кислоты (14 мл) и метанола (5 мл) при охлаждении льдом добавляли 40% водный раствор диметиламина (14 мл, 110.5 ммоль), 4-диметиламино-4-фенилциклогексанон (5.00 г, 23.04 ммоль) и цианид калия (3.60 г, 55.3 ммоль). Смесь 2 дн. перемешивали при комнатной температуре и затем после добавления воды (200 мл) экстрагировали простым эфиром (4×150 мл). После концентрирования раствора остаток ресуспендировали в дихлорметане (200 мл) и в течение ночи высушивали сульфатом магния, отфильтровывали и растворитель удаляли в вак. Нитрил получали в виде масла, который кристаллизовался.
Выход: 5.87 г (90%)
1Н-ЯМР (ДМСО-d6): 1.36 (1Н, m); 1.61 (1Н, m); 1.61 (2Н, m); 1.92 (8Н, m); 2.16 (4Н, m); 2.28 (3Н, s); 2.44 (1Н, m); 2.59 (1Н, m); 7.35 (5Н, m).
b) 1-бензил-N,N,N',N'-тетраметил-4-фенил-циклогексан-1,4-диамины (неполярный диастереомер)
Для титульного соединения предшествующей стадии (5.84 г, 20.5 ммоль) растворяли в ТГФ (115 мл) и при охлаждении льдом по каплям смешивали с хлоридом бензилмагния 2М (36 мл, 71.57 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь смешивали с 20%-ым раствором хлорида аммония (15 мл) и водой (10 мл) и экстрагировали диэтиловым эфиром (3×50 мл). Объединенные органические фазы промывали водой (50 мл) и нас. раствором NaCl (50 мл), высушивали над Na2SO4, фильтровали и концентрировали в вак. Остаток очищали путем флеш-хроматографии с циклогексан/этилацетатом (1:1).
Выход: 770 мг (11%) неполярный диастереомер
1Н-ЯМР (ДМСО-d6): 1.57 (4Н, m); 1.72 (2Н, m); 1.79 (6Н, s); 2.19 (6Н, s); 2.23 (2Н, m); 2.63 (2Н, s); 7.26 (10Н, m).
Пример 13:
1-бензил-N,N,N',N'-тетраметил-4-фенил-циклогексан-1,4-диамины (полярный диастереомер)
При очистке примерного соединения 12 стадия b) также можно было выделить аналогичный полярный диастереомер.
Остаток очищали путем флеш-хроматографии с циклогексан/этилацетатом (1:1). Неполярный диастереомер получали чистым. Полярный диастереомер выделяли загрязненным и еще раз очищали флеш-хроматографией с ацетонитрил/ метанол/1N NH4Cl (9:1:1).
Выход: 600 мг (9%) полярный диастереомер
1Н-ЯМР (ДМСО-d6): 0.88 (2Н, t); 1.70 (2H, m); 1.85 (6H, s); 1.90 (2H, m); 2.14 (2H, m); 2.26 (6H, s); 2.48 (2H, s); 7. 00 (6H, m); 7.18 (4H, m).
13С-ЯМР (ДМСО-d6): 27.1; 28.6; 36.3; 36.8; 37.8; 57.0; 60.5; 125.2; 125.8; 127.1; 127.2; 130.2; 136.9; 138.7.
Пример 14:
4-метокси-4-(3-(метоксиметил)-1Н-индол-2-ил)-N,N-диметил-1-фенилциклогексанамин 2-гидроксипропан-1,2,3-трикарбоксилат
а) [4-метокси-4-(3-метокси-проп-1-инил)-1-фенил-циклогексил]-диметил-амин
К 2.5 М раствору бутиллития в гексане (8.4 мл, 21.0 ммоль) при -30°С под аргоном впускали по каплям метилпропаргиловый эфир (1.47 г, 21.0 ммоль), растворенный в абс. ТГФ (15 мл). Затем при -30°С добавляли раствор 4-диметиламино-4-фенилциклогексанона (4.34 г, 20.0 ммоль) в абс. ТГФ (20 мл) и бромид лития (0.87 г, 10 ммоль), растворенный в абс. ТГФ (2.5 мл). Реакционную смесь нагревали до -5°С, по каплям смешивали с раствором метилйодида (4.25 г, 30 ммоль) в абс. ДМСО (25 мл) и 2 ч перемешивали при 50°С. Для переработки реакционной смеси при охлаждении в ледяной ванне добавляли воду (30 мл) и экстрагировали циклогексаном (4×50 мл). Органическую фазу промывали 20%-ым раствором хлорида аммония, высушивали над Na2SO4 и концентрировали в вак. Оставшийся остаток очищали путем флеш-хроматографии с CHCl3/МеОН (20:1).
Выход: 2.34 г (39%)
1Н-ЯМР (ДМСО-d6): 1.57 (2Н, m); 1.96 (10H, m); 2.25 (2H, m); 3.18 (3H, s); 3.27 (3H, m); 4.05 (2H, s); 7.37 (5H, m).
b) [4-метокси-4-(3-метокси-проп-1-инил)-1-фенил-циклогексил]-диметил-амин
2-йоданилин (328 мг, 1.5 ммоль), титульное соединение предшествующей стадии (452 мг, 1.5 ммоль) и карбонат натрия (795 мг, 7.5 ммоль) под аргоном растворяли в абс. ДМФ (10 мл). Затем добавляли катализатор (PEPPSI®, 204 мг, 0.3 ммоль) и раствор при 100°С перемешивали 24 ч. Для переработки черный реакционный раствор концентрировали в вак. До сухости, остаток растворяли в CHCl3 и промывали водой. Органическую фазу высушивали над Na2SO4 и концентрировали в вак. Оставшийся остаток очищали путем флеш-хроматографии CHCl3/МеОН (20:1→9:1).
Выход: 71 мг(12%)
1Н-ЯМР (ДМСО-d6): 1.62 (2H, m); 2.22 (10H, m); 2.63 (2H, m); 3.00 (3H, s); 3.10 (3H, m); 4.46 (2H, s); 6.95 (2H, m); 7.28 (1H, d); 7.46 (6H; m); 10.72 (1H, s).
c) 4-метокси-4-(3-(метоксиметил)-1Н-индол-2-ил)-N,N-диметил-1-фенилциклогексанамин 2-гидроксипропан-1,2,3-трикарбоксилат
Титульное соединение предшествующей стадии (217 мг, 0.55 ммоль) растворяли в горячем этаноле (4 мл) и смешивали с раствором лимонной кислоты (106 мг, 0.55 ммоль) в горячем этаноле (2 мл). После 2-часового выдерживания в холодильнике и добавления простого эфира образованное твердое вещество отсасывали и высушивали в вак.
Выход: 165 мг (51%)
точка плавления: 184-186°С
1Н-ЯМР (ДМСО-d6): 1.61 (2H, m); 2.22 (4H, m); 2.37 (6H, s); 2.52 (4 H, m); 3.01 (3H, s); 3.08 (3H, m); 4.45 (2H, s); 6.99 (2H, m); 7.25 (1H, d); 7.50 (4 H, m); 7.65 (2H, m); 10.73 (1H, s).
Пример 15:
4-(бензилокси)-4-(3-(метоксиметил)-1Н-индол-2-ил)-N,N-диметил-1-фенилциклогексанамин
а) [4-бензилокси-4-(3-метокси-проп-1-инил)-1-фенил-циклогексил]-диметил-амин
К 2.5 М раствору бутиллития в гексане (2.1 мл, 5.2 ммоль) при -30°С под аргоном впускали по каплям метилпропаргиловый эфир (0.36 г, 5.2 ммоль), растворенный в абс. ТГФ (5 мл). Затем добавляли при -30°С раствор 4-диметиламино-4-фенилциклогексанона (1.08 г, 5.0 ммоль) в абс. ТГФ (5 мл) и бромид лития (0.22 г, 2.5 ммоль), растворенный в абс. ТГФ (2.0 мл). Реакционную смесь нагревали до -5°С, по каплям смешивали с раствором бензилбромида (1.28 г, 7.5 ммоль) в абс. ДМСО (10 мл) и 2 ч перемешивали при 50°С. Для переработки реакционной смеси при охлаждении в ледяной ванне добавляли воду (10 мл) и экстрагировали циклогексаном (4×20 мл). Органическую фазу промывали 20%-ым раствором хлорида аммония, высушивали над Na2SO4 и концентрировали в вак. Оставшийся остаток очищали путем флеш-хроматографии с CHCl3/МеОН (40:1).
Выход: 541 мг (29%), неполярное соединение
1Н-ЯМР (ДМСО-d6): 1.67 (2H, m); 1.94 (6H, s); 2.04 (4H, m); 2.30 (2H, m); 3.19 (3H, s); 4.09 (2H, s); 4.60 (2H, s); 7.31 (10 H, m).
b) 4-(бензилокси)-4-(3-(метоксиметил)-1Н-индол-2-ил)-N,N-диметил-1-фенилциклогексанамин
2-ацетиламино-йоданилин (359 мг, 1.37 ммоль), титульное соединение предшествующей стадии (519 мг, 1.37 ммоль) и карбонат натрия (726 мг, 6.85 ммоль) растворяли в абс. ДМФ (10 мл) под аргоном. Затем добавляли катализатор (PEPPSI, 190 мг, 0.28 ммоль) и раствор при 100° перемешивали С 24 ч. Для переработки черный реакционный раствор концентрировали в вак. до сухости, остаток растворяли в CHCl3 и промывали водой. Органическую фазу высушивали Na2SO4 и концентрировали в вак. Оставшийся остаток очищали путем флеш-хроматографии с CHCl3/МеОН (50:1).
Выход: 210 мг (33%)
1Н-ЯМР (ДМСО-d6): 1.67 (2 Н, m); 1.61 (2H, m); 2.10 (6H, bs); 2.38 (2H, m); 2.70 (2H, m); 3.11 (3H, s); 4.13 (2H, s); 4.57 (2H, s); 7.02 (2H, m); 7.30 (12H, m); 10.78 (1H, s).
Пример 16:
4-этокси-4-(3-(метоксиметил)-1Н-индол-2-ил)-N,N-диметил-1-фенилциклогексанамин 2-гидроксипропан-1,2,3-трикарбоксилат
a) [4-этокси-4-(3-метокси-проп-1-инил)-1-фенил-циклогексил]-диметил-амин
К 2.5 М раствору бутиллития в гексане (8.4 мл, 21.0 ммоль) при -30°С под аргоном впускали по каплям метилпропаргиловый эфир (1.47 г, 21.0 ммоль), растворенный в абс. ТГФ (15 мл). Затем при -30°С добавляли раствор 4-диметиламино-4-фенилциклогексанона (4.34 г, 20.0 ммоль) в абс. ТГФ (20 мл) и бромид лития (0.87 г, 10 ммоль), растворенный в абс. ТГФ (2.5 мл). Реакционную смесь нагревали до -5°С, по каплям смешивали с раствором этилйодида (4.68 г, 30 ммоль) в абс. ДМСО (30 мл) и перемешивали 2 ч при 50°С. Для переработки реакционной смеси при охлаждении в ледяной ванне добавляли воду (30 мл) и экстрагировали циклогексаном (4×50 мл). Органическую фазу промывали 20%-ым раствором хлорида аммония, высушивали над Na2SO4 и концентрировали в вак. Оставшийся остаток путем флеш-хроматографии с CHCl3/МеОН (20:1) очищали.
Выход: 3.92 г (62%)
1Н-ЯМР (ДМСО-d6): 1.12 (3H, t); 1.58 (2H, m); 1.96 (10H, m); 2.25 (2H, m); 3.17 (3H, s); 3.51 (2H, q); 4.04 (2H, s); 7.37 (5H, m).
b) [4-этокси-4-(3-метоксиметил-1Н-индол-2-ил)-1-фенил-циклогексил]-диметил-амин
N-(2-йодфенил)-ацетамид (522 мг, 2.0 ммоль), титульное соединение предшествующей стадии (631 мг, 2.0 ммоль) и карбонат натрия (1.06 г, 10.0 ммоль) растворяли в абс. ДМФ (10 мл) под аргоном. Затем добавляли катализатор (PEPPSI, 272 мг, 0.4 ммоль) и раствор при 100°С перемешивали 24 ч. Для переработки черный реакционный раствор концентрировали в вак. до сухости, остаток растворяли в CHCl3 и промывали водой. Органическую фазу высушивали над N1Н a2SO4 и концентрировали в вак. Оставшийся остаток путем флеш-хроматографии очищали CHCl3/МеОН (50:1).
Выход: 249 мг (31%)
1Н-ЯМР (ДМСО-d6): 1.11 (3Н, t); 1.61 (2H, m); 1.99 (8H, m); 2.19 (2H, m); 2.48 (2H, m); 3.12 (5H, m); 4.53 (2H, s); 6.99 (2H, m); 7.27 (2H, d); 7.47 (5H, m); 10.61 (1H, s).
с) 4-этокси-4-(3-(метоксиметил)-1Н-индол-2-ил)-N,N-диметил-1-фенилциклогексанамин 2-гидроксипропан-1,2,3-трикарбоксилат
Титульное соединение предшествующей стадии (188 мг, 0.462 ммоль) растворяли в горячем этаноле (4 мл) и смешивали с раствором лимонной кислоты (89 мг, 0.462 ммоль) в горячем этаноле (2 мл). После 2-часового выдерживания в холодильнике и добавления простого эфира образованное твердое вещество отсасывали и высушивали в вак.
Выход: 152 мг (55%)
точка плавления: 166-167°С
1Н-ЯМР (ДМСО-d6): 1.12 (3H, t); 1.57 (2H, m); 2.17-2.35 (10H, m); 2.58 (4H, m); 2.70 (2H, m); 3.11 (3H, m); 4.51 (2H, s); 6.98 (2H, m); 7.24 (2H, d); 7.43 (4H, m); 7.62 (2H, m); 10.67 (1H, s).
Пример 17:
N-((-4-(диметиламино)-1-метил-4-фенилциклогексил)метил)ацетамид 2-гидроксипропан-1,2,3-трикарбоксилат
а) диметил-(4-метилеп-1-фенил-циклогексил)-амин
Под аргоном помещали трет-BuOK (0.550 г, 4.74 ммоль) в абс. простой эфир (10 мл) и добавляли бромид метил-трифенилфосфония (1.89 г, 4.74 ммоль). Затем нагревали до 40°С в течение 30 мин. После этого времени реакции осторожно впускали по каплям 4-диметиламино-4-фенилциклогексанон (1.00 г, 4.60 ммоль), растворенный в абс. ТГФ (10 мл), и реакционный раствор нагревали до 50°С в течение 5 ч. Реакционный состав перемешивали в течение ночи при комнатной температуре и концентрировали в вак. до сухости. Остаток ресуспендировали в диоксане (50 мл) и смешивали с HCl/диоксан (5 мл). Выпавшее в осадок твердое вещество отсасывали и промывали простым эфиром. Выделенный гидрохлорид подщелачивали с помощью 2N NaOH и экстрагировали дихлорметаном (2×80 мл). Органическую фазу высушивали над Na2SO4, отфильтровывали и концентрировали в вак.
Выход: 0.86 г (61%)
1Н-ЯМР (ДМСО-d6): 1.82 (2Н, m); 1.99 (2H, m); 2.30 (2H, m); 2.43 (6H, d); 3.01 (2H, m); 4.67 (2H, s); 7.55 (3H, m); 7.72 (2H, m).
b) 4-диметиламино-1-метил-4-фенил-циклогексанкарбонитрил
Комплекс R-R-кобальт-(II)-сален (Jacobsens Ligand, 26.0 мг, 0.04 ммоль) растворяли в дихлорметане (5 мл), смешивали с уксусной кислотой (29 µл, 0.08 ммоль, 2 экв.) и 30 мин перемешивали в открытой колбе. Затем состав концентрировали в вак. и с толуолом азеотропно удаляли избыточную уксусную кислоту. Полученный катализатор кобальт-(III) под аргоном помещали в абс. этанол (5 мл). Через 2 мин осуществляли добавление титульного соединения предшествующей стадии (0.860 г, 3.99 ммоль), растворяли в этаноле (8 мл), р-толуолсульфонилцианид (714 мг, 5.58 ммоль) вслед за фенилсиланом (0.49 мл, 3.99 ммоль). Затем еще раз добавляли этанол (5 мл) и реакционный раствор 3 дн. перемешивали при комнатной температуре. Состав до сухости концентрировали в вак. и остаток очищали флеш-хроматографией (2 × на нормальном силикагеле и 1 × на ультрамелком силикагеле) с этилацетатом. Другие исследования показали, что достаточно было однократной очистки колоночной хроматографией на ультрамелком силикагеле с хлороформ/метанолом (20:1).
Выход: 0.130 г (13%)
1Н-ЯМР (ДМСО-d6): 1.09 (2H, m); 1.16 (3H, s); 1.78 (2H, m); 1.87 (2H, m); 1.92 (6H, s); 2.56 (2H, m); 7.32 (5H, m).
13С-ЯМР (ДМСО-d6): 25.4; 29.8; 33.1; 33.7; 37.8; 60.3; 124.5; 126.5; 127.5; 127.7; 135.4.
c) (4-аминометил-4-метил-1-фенил-циклогексил)-диметил-амин
LiAlH4 (38.0 мг, 0.81 ммоль) помещали под аргоном в абс. ТГФ (5 мл), медленно смешивали с соединением, указанным в заголовке, предшествующей стадии (0.130 г, 0.54 ммоль), растворенным в абс. ТГФ (5 мл), и реакционную смесь 3 ч перемешивали в колбе с обратным холодильником. Затем при охлаждении льдом ТГФ (10 мл) и добавляли воду (4 мл) и еще перемешивали 30 мин. Осадок отфильтровывали через целит и промывали дихлорметаном (50 мл). Фильтрат концентрировали в вак. до сухости.
Выход: 0.12 г (90%)
1Н-ЯМР (ДМСО-d6): 0.72 (3H, s); 0.99 (2H, m); 1.13 (2H, t, NH2); 1.50 (2H, m); 1.84 (2H, m); 1.90 (6H, s); 2.04 (2H, m); 2.39 (2H, t); 7.24 (5H, m).
d) N-(4-диметиламино-1-метил-4-фенил-циклогексилметил)-ацетамид
Титульное соединение предшествующей стадии (0.120 г, 0.48 ммоль) растворяли в абс. ТГФ (2.5 мл) и смешивали с триэтиламином (72.0 µл, 0.53 ммоль) и ацетилхлоридом (42.0 мг, 38.0 µл, 0.53 ммоль). Реакционную смесь перемешивали 16 ч при комнатной температуре. Состав концентрировали до сухости в вак., остаток ресуспендировали в этиловом эфире уксусной кислоты (10 мл) и промывали насыщенным раствором NaHCO3 (2×10 мл) и насыщенным раствором NaCl. Органическую фазу высушивали над Na2SO4, отфильтровывали и концентрировали в вак.
Выход: 109 мг (77%)
1Н-ЯМР (ДМСО-d6): 0.71 (3H, s); 0.96 (2H, m); 1.17 (2H, m); 1.47 (2H, m); 1.84 (3H, s); 1.91 (6H, s); 2.11 (2H, m); 3.02 (2H, d) 7.30 (5H, m); 7.69 (1H, t).
е) N-((-4-(диметиламино)-1-метил-4-фенилциклогексил)метил)ацетамид 2-гидроксипропан-1,2,3-трикарбоксилат
Титульное соединение предшествующей стадии (102 мг, 0.35 ммоль) растворяли в горячем этаноле (4 мл). Лимонную кислоту (67.0 мг, 0.35 ммоль) растворяли в горячем этаноле (1.0 мл) и добавляли. Состав 2 ч перемешивали при комнатной температуре. Так как не выпадал осадок, то раствор концентрировали в вак. Остаток перемешивали с простым эфиром, вновь концентрировали в вак. и затем высушивали в вак. Желаемый цитрат получали в виде пористого твердого вещества.
Выход: 167 мг (98%)
1Н-ЯМР (ДМСО-d6): 0.62 (3Н, s); 0.92 (2H, m); 1.45 (2H, m); 1.83 (3H, s); 2.07-2.60 (14H, m); 3.07 (2H, d) 7.46 (5H, m); 7.72 (1H, t).
Пример 18:
4-хлоро-N-((-4-(диметиламино)-1-метил-4-фенилциклогексил)метил)бензолсульфонамид 2-гидроксипропан-1,2,3-трикарбоксилат
а) 4-хлор-N-(4-диметиламино-1-метил-4-фенил-циклогексилметил)-бензолсульфонамид
Титульное соединение из примера 17, стадия с) (0.160 г, 0.65 ммоль) растворяли в абс. ТГФ (3.4 мл), смешивали с триэтиламином (97 µл, 0.714 ммоль) и хлоридом 4-хлорбензолсульфоновой кислоты (151 мг, 0.71 ммоль) и 1 дн. перемешивали при комнатной температуре. Состав концентрировали в вак. до сухости и остаток очищали флеш-хроматографией: 1-я колонка с этиловым эфиром уксусной кислоты/этанол (9:1) и 2-я колонка с этиловым эфиром уксусной кислоты.
Выход: 70 мг (26%)
1Н-ЯМР (ДМСО-d6): очень плохо растворимый спектр.
b) 4-хлоро-N-((-4-(диметиламино)-1-метил-4-фенилциклогексил)метил)бензолсульфонамид 2-гидроксипропан-1,2,3-трикарбоксилат
Титульное соединение предшествующей стадии (0.070 г, 0.17 ммоль) растворяли в горячем изопропаноле (4 мл). Лимонную кислоту (32.0 мг, 0.17 ммоль) растворяли в горячем изопропанол (1.0 мл) и добавляли. Состав 2 ч перемешивали при комнатной температуре. Так как не выпадал осадок, то раствор концентрировали в вак. Остаток перемешивали с простым эфиром, вновь концентрировали в вак. и затем высушивали в вак. Желаемый цитрат получали в виде пористого твердого вещества.
Выход: 58 мг (57%)
1Н-ЯМР (ДМСО-d6): 0.64 (3Н, m); 0.90-1.04 (6H, m); 1.54 (2H, m); 1.92 (2H, m); 2.31 (6H, s); 2.73 (4H, m); 7.47-7.84 (5H, m); 10.8 (2Н, шир.).
Пример 19:
N-((1-бутил-4-(диметиламино)-4-фенилциклогексил)метил)-4-хлоробензолсульфонамид 2-гидроксипропан-1,2,3-трикарбоксилат
а) (4-бутилиден-1-фенил-циклогексил)-диметил-амин
Трет-бутилат калия (2.75 г, 23.7 ммоль) под аргоном помещали в абс. простой эфир (50 мл) и смешивали с бромидом бутил-трифенилфосфония (9.45 г, 23.7 ммоль). Состав 30 мин нагревали до 40°С. Затем осторожно впускали по каплям 4-диметиламино-4-фенилциклогексанон (5.00 г, 23.0 ммоль), растворенный в абс. ТГФ (50 мл), (экзотермическая реакция). Состав нагревали 6.5 ч до 50°С и перемешивали в течение ночи при комнатной температуре. Затем состав концентрировали до сухости в вак., ресуспендировали в диоксане (20 мл) и смешивали с HCl/диоксаном (5 мл). При этом выпадал осадок. Который отфильтровывали, промывали простым эфиром (10 мл), затем подщелачивали с помощью 2N NaOH и экстрагировали дихлорметаном (2×40 мл). Органическую фазу высушивали над Na2SO4, отфильтровывали и в вак. концентрировали до сухости.
Выход: 4.60 г (77%)
1Н-ЯМР (ДМСО-d6): 0.83 (3H, t); 1.27 (2H, m); 1.94 (9H, m); 2.09 (5H, m); 2.29 (2H, m); 5.04 (1H, t); 7.23 (1H, m); 7.36 (4H, m).
b) 1-бутил-4-диметиламино-4-фенил-циклогексанкарбонитрил
Катализатор кобальт-III (297 мг, 0.456 ммоль) помещали в абс. этанол (100 мл) под аргоном. Затем добавляли титульное соединение предшествующей стадии (11.6 г, 45.3 ммоль), растворенное в этаноле (40 мл), и затем добавляли р-толуолсульфонилцианид (13.0 г, 68.0 ммоль), фенилсилан (5.6 мл, 45.3 ммоль) и этанол (10 мл). Температура повысилась до 35°С, поэтому охлаждали ледяной водой. Состав 72 ч перемешивали при комнатной температуре и затем в вак. концентрировали до сухости. Остаток очищали путем флеш-хроматографии с хлороформ/метанолом (20:1). Предварительно очищенное вещество еще раз очищали посредством MPLC-колоночной хроматографии с хлороформ/метанол (50:1, 20:1).
Выход: 0.233 г (1.8%)
1Н-ЯМР (ДМСО-d6): 0.83 (3H, t); 1.08 (2H, m); 1.27 (6H, m); 1.75 (2H, m); 1.93 (8H, m); 2.63 (2H, m); 7.36 (5H, m).
c) (4-аминометил-4-бутил-1-фенил-циклогексил)-диметил-амин
Титульное соединение предшествующей стадии (247 мг, 0.856 ммоль) растворяли в абс. ТГФ (5 мл). Затем добавляли под аргоном LiAlH4 (64 мг, 1.71 ммоль) и состав 5.5 ч нагревали до кипения. Для переработки к составу при охлаждении льдом добавляли ТГФ (12 мл) и H2O (5 мл) и 30 мин перемешивали. Состав отфильтровывали через фритту с кизельгуром (2 см), подвергали последующей промывке дихлорметаном (50 мл) и хлороформом (50 мл) и концентрировали в вак. Остаток очищали путем флеш-хроматографии хлороформ/метанолом (20:1, 9:1, метанол).
Выход: 70 мг (28%)
1Н-ЯМР (ДМСО-d6): 0.80 (3H, t); 1.08 (9H, m); 1.45 (2H, m); 1.93 (10H, m); 2.45 (2H, m); 7.31 (5H, m).
d) N-(1-бутил-4-диметиламино-4-фенил-циклогексилметил)-4-хлор-бензолсульфонамид
Титульное соединение предшествующей стадии (65.0 мг, 0.225 ммоль) растворяли под аргоном в абс. ТГФ (5 мл) и смешивали с триэтиламином (33.5 µл, 0.247 ммоль). Затем к составу добавляли хлорид 4-хлорбензолсульфоновой кислоты (52.0 мг, 0.247 ммоль). Состав перемешивали в течение ночи при комнатной температуре. Затем его в вак. концентрировали до сухости. Остаток ресуспендировали в этилацетате (10 мл) и промывали насыщенным раствором NaHCO3 (2×10 мл) и насыщенным раствором NaCl (2×10 мл). Органическую фазу высушивали над Na2SO4, отфильтровывали и концентрировали в вак. до сухости.
Выход: 103 мг (98%)
1Н-ЯМР (ДМСО-d6): 0.76 (3Н, t); 0.97 (8H, m); 1.44 (2H, m); 1.87 (10H, m); 2.63 (2H, m); 7.35 (5H, m); 7.52 (1H, t); 7.68 (2H, m); 7.85 (2H, m).
e) N-((1-бутил-4-(диметиламино)-4-фенилциклогексил)метил)-4-хлоробензолсульфонамид 2-гидроксипропан-1,2,3-трикарбоксилат
Титульное соединение предшествующей стадии (103 мг, 0.22 ммоль) растворяли в горячем этаноле (3 мл). Лимонную кислоту (42 мг, 0.22 ммоль) растворяли в горячем этаноле (1 мл) и добавляли. Состав охлаждали до комнатной температуры и затем в вак. концентрировали до сухости.
Выход: 128 мг (88%)
Точка плавления: пористое твердое вещество
1Н-ЯМР (ДМСО-d6): 0.86 (3H, t); 0.94 (6H, m); 1.10 (2H, m); 1.50 (2H, m); 1.86 (2H, m); 2.28 (6H, s); 2.51-2.64 (6H, m); 7.52 (6H, m); 7.72 (2H, t); 7.87 (2H, m).
Пример 20:
(-4-(диметиламино)-4-фенил-1-(4-фенилбутил)циклогексил)метанол(неполярный диастереоизомер)
а) 1,4-диоксаспиро[4,5]декан-8-этиловый эфир карбоновой кислоты
Раствор этил-4-оксоциклогексанкарбоксилата (28.9 г, 169 ммоль), этиленгликоля (36.7 г, 33.0 мл, 592 ммоль) и р-толуолсульфокислоты (380 мг, 2.0 ммоль) в толуоле (90 мл) перемешивали в течение ночи при комнатной температуре. Реакционный раствор выливали в простой эфир (150 мл) и промывали водой и 5% раствором гидрокарбоната натрия (по 150 мл). Органическую фазу высушивали сульфатом натрия и концентрировали в вак. Так как сырой продукт (26.8 г) получался чистым, то его можно было использовать непосредственно далее.
Выход: 26.8 г (74%), бесцветное масло
b) 8-(4-фенилбутил)-1,4-диоксаспиро[4,5]декан-8-этиловый эфир карбоновой кислоты
2.5 М раствор n-бутиллития (2.5 г, 15.7 мл, 39.2 ммоль) при -78°С под аргоном медленно впускали по каплям к раствору диизопропиламина (3.96 г, 5.50 мл, 39.2 ммоль) в абсолютном тетрагидрофуране (50 мл). К нему впускали по каплям по очереди 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)пиримидон (DMPU, 10.0 г, 9.42 мл, 78.2 ммоль) и раствор титульного соединения предшествующей стадии (8.40 г, 39.2 ммоль) в абсолютном тетрагидрофуране (30 мл). Реакционный раствор 2 ч далее перемешивали при этой температуре, прежде чем раствор впускали по каплям 1-бром-4-фенилбутан (10.0 г, 47.0 ммоль) в абсолютном тетрагидрофуране (50 мл). Полученный раствор перемешивали в течение ночи при комнатной температуре. Затем добавляли насыщенный раствор хлорида аммония (50 мл) и экстрагировали простым эфиром (2×50 мл). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (50 мл), высушивали сульфатом натрия и концентрировали в вак. Сырой продукт (17.7 г) очищали с помощью флеш-хроматографии (400 г, 20×7.5 см) с циклогексан/этилацетатом (9:1).
Выход: 10.3 г (76%), бесцветное масло
1Н-ЯМР (ДМСО-d6): 1.12 (t, 3Н, J=7.1Hz); 1.39-1.62 (m, 12H); 1.91-2.03 (m, 2H); 2.54 (t, 2H, J=7.4 Hz); 3.82 (s, 4H); 4.05 (q, 2H, J=7.1 Hz); 7.12-7.17 (m, 3Н); 7.22-7.28 (m, 2H).
с) [8-(4-фенилбутил)-1,4-диоксаспиро[4.5]дец-8-ил]метанол
К взвеси гидрида литийалюминия (2.50 г, 67.0 ммоль) в абсолютном тетрагидрофуране (100 мл) впускали по каплям под аргоном при 65°С раствор титульного соединения предшествующей стадии (10.2 г, 33.5 ммоль) в абсолютном тетрагидрофуране (50 мл) и 3 ч перемешивали при этой температуре, после чего взаимодействие было полным. Реакционную смесь после охлаждения смешивали с водой (4.5 мл) и 4 N натровым щелоком (1.1 мл) и отфильтровывали от образовавшегося осадка. Остаток промывали тетрагидрофураном (2×60 мл) и фильтрат концентрировали в вак. Так как сырой продукт получался чистым, то его можно было использовать непосредственно далее.
Выход: 9.44 г (93%), светло-желтое масло
1Н-ЯМР (ДМСО-d6): 1.14-1.32 (m, 6H); 1.34-1.39 (m, 2H); 1.40-1.57 (m, 6H); 2.57 (t, 2H, J=7.4 Hz); 3.17 (дн., 2H, J=5.2 Hz); 3.82 (s, 4H); 4.36 (t, 1H, J=5.2 Hz); 7.13-7.19 (m, 3Н); 7.24-7.29 (m, 2H).
d) 4-гидроксиметил-4-(4-фенилбутил)циклогексанон
Раствор титульного соединения предшествующей стадии (9.40 г, 30.9 ммоль) в ацетоне (150 мл) смешивали с 1N соляной кислотой (32 мл) и перемешивали в течение ночи при комнатной температуре. Реакционный раствор с помощью 1N натрового щелока устанавливали до рН 8 и концентрировали в вак. Остаток смешивали с водой (50 мл) и затем экстрагировали дихлорметаном (3×50 мл). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (30 мл), высушивали сульфатом натрия и концентрировали в вак. Так как сырой продукт получался чистым, то его можно было использовать непосредственно далее.
Выход: 7.96 г (99%), светло-желтое масло
1Н-ЯМР (ДМСО-d6): 1.19-1.67 (m, 10Н); 2.22 (t, 4H, J=6.8 Hz); 2.59 (t, 2H, J=7.5 Hz); 3.30 (дн., 2H, J=5.2 Hz); 4.54 (t, 1H, J=5.1 Hz); 7.13-7.21 (m, 3Н); 7.24-7.29 (m, 2H).
е) 4-(1-этокси-этоксиметил)-4-(4-фенилбутил)циклогексанон
Раствор титульного соединения предшествующей стадии (7.95 г, 30.5 ммоль) в абсолютном дихлорметане (100 мл) смешивали с тозилатом пиридиния (100 мг) и простым этилвиниловым эфиром (2.64 г, 3.51 мл, 36.6 ммоль) и перемешивали в течение ночи при комнатной температуре. Реакционный раствор затем по очереди промывали 5% раствором гидрокарбоната натрия, водой (по 2×50 мл) и насыщенным раствором хлорида натрия (50 мл), высушивали сульфатом натрия и концентрировали в вакууме. Сырой продукт (8.87 г) очищали с помощью флеш-хроматографии (400 г, 20×7.5 см) с циклогексан/этилацетат (9:1).
Выход: 6.97 г (69%), бесцветное масло
1Н-ЯМР (ДМСО-d6): 1.09 (t, 3Н, J=7.0 Hz); 1.17 (дн., 3Н, J=5.3 Hz); 1.21-1.32 (m, 2H); 1.43-1.50 (m, 2H); 1.52-1.70 (m, 6H); 2.20-2.26 (m, 4H); 2.59 (t, 2H, J=7.5 Hz); 3.24 (дн., 1Н, J=9.4 Hz); 3.34-3.42 (m, 2H); 3.49-3.59 (m, 1H); 4.62 (q, 1H, J=5.3 Hz); 7.14-7.29 (m, 5H).
f) 1-диметиламино-4-(1-этоксиэтоксиметил)-4-(4-фенилбутил)циклогексанкарбонитрил
К охлажденной льдом смеси 4 N соляной кислоты (1.52 мл, 6.1 ммоль) и метанола (1.7 мл) добавляли сначала 40% раствор диметиламина (3.74 мл, 24.2 ммоль), затем титульное соединение предшествующей стадии (2.04 г, 6.1 ммоль) и цианид калия (953 мг, 14.6 ммоль). Образованную суспензию 4 ч перемешивали при комнатной температуре. Суспензию смешивали с водой (100 мл) и затем экстрагировали диэтиловым эфиром (3×100 мл). Объединенные органические фазы высушивали сульфатом натрия и концентрировали в вак.
Выход: 2.30 г (97%), светло-желтое масло
1Н-ЯМР (ДМСО-d6): 1.08 (dt, 3Н, J=2.5, 7.0 Hz, 1.075 (t, 1.5H, J=7.0 Hz); 1.085 (t, 1.5H, J=7.0 Hz); 1.14-1.18 (m, 3Н); 1.19-1.39 (m, 6H); 1.41-1.68 (m, 6H); 1.89-2.00 (m, 2H); 2.22 (s, 2.6H); 2.23 (s, 3.4H); 2.53-2.62 (m, 2H); 3.10 (дн., 0.5Н, J=9.3 Hz); 3.13 (дн., 0.5Н, J=9.2 Hz); 3.22-3.29 (m, 1H); 3.33-3.39 (m, 1H); 3.47-3.59 (m, 1H); 4.56-4.63 (m, 1H); 7.13-7.29 (m, 5H).
г) [4-(1-этоксиэтоксиметил)-1-фенил-4-(4-фенилбутил)циклогексил]диметиламин
К охлажденному льдом 2 М раствору хлорида фенилмагния (2.03 г, 7.4 мл, 14.8 ммоль) в тетрагидрофуране под аргоном медленно впускали по каплям раствор титульного соединения предшествующей стадии (смесь диастереоизомеров, 2.30 г, 5.9 ммоль) в абсолютном тетрагидрофуране (30 мл) и затем в течение ночи перемешивали при комнатной температуре. Реакционный раствор смешивали с насыщенным раствором хлорида аммония и водой (по 20 мл), фазы разделяли и водную фазу экстрагировали диэтиловым эфиром (3×30 мл). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (30 мл), высушивали сульфатом натрия и концентрировали в вак. Получалось 2.73 г сырого продукта в виде смеси диастереоизомеров, которую полностью отделяли с помощью MPLC (LiChroprep Si60 15-25 µм, 230 г, 3.6×46 см) с этилацетат/метанолом (9:1).
Неполярный диастереоизомер:
Выход: 918 мг (36%), светло-желтое масло
1Н-ЯМР (ДМСО-d6): 1.00 (t, 3Н, J=7.1 Hz); 1.06 (дн., 3Н, J=5.3 Hz); 1.10-1.20 (m, 2H); 1.21-1.29 (m, 2H); 1.32-1.48 (m, 2H); 1.51-1.62 (m, 2H); 1.90 (s, 6H); 1.92-1.99 (m, 4H); 2.59 (t, 2H, J=7.6 Hz); 2.98 (дн., 1Н, J=9.3 Hz); 3.11 (дн., 1Н, J=9.3 Hz); 3.21-3.29 (m, 2H); 3.39-3.50 (m, 2H); 4.48 (q, 1Н, J=5.3 Hz); 7.13-7.37 (m, 10Н).
h) (-4-(диметиламино)-4-фенил-1-(4-фенилбутил)циклогексил)метанол (неполярный диастереоизомер)
Раствор титульного соединения (неполярный диастереоизомер) предшествующей стадии (469 мг, 1.1 ммоль) в ацетоне (50 мл) смешивали с 2 N соляной кислотой (2 мл) и 18 ч перемешивали при комнатной температуре. Реакционный раствор с помощью 1 N натрового щелока устанавливали до рН 8, концентрировали в вак., остаток ресуспендировали в воде (50 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (30 мл), высушивали сульфатом натрия и концентрировали в вак. Сырой продукт (381 мг) очищали с помощью флеш-хроматографии (18 г, 20×2.0 см) с этилацетат/метанолом (4:1).
Выход: 325 мг (81%), белое твердое вещество
Точка плавления: 105-106°С
1Н-ЯМР (ДМСО-d6): 1.00-1.14 (m, 2H); 1.18-1.28 (m, 2H); 1.29-1.41 (m, 4H); 1.49-1.60 (m, 2H); 1.81-1.85 (m, 1H); 1.90 (s, 6H); 1.94-2.06 (m, 3H); 2.59 (t, 2H, J=7.7 Hz); 3.02 (дн., 2H, J=5.2 Hz); 4.21 (t, 1H, J=5.2 Hz); 7.13-7.37 (m, 10Н).
13С-ЯМР (ДМСО-d6): 22.4; 28.1; 28.2; 32.2; 34.1; 34.3; 35.9; 36.7; 37.9; 60.1; 66.2; 125.5; 125.7; 126.0; 127.2; 127.3; 128.1; 128.2; 137.4; 142.4.
LC-MC (метод: ASCA-7MIN-80GRAD.M): m/z: [M+1]+=366.6, Rt 2.38 мин.
Пример 21:
(-4-(диметиламино)-4-фенил-1-(4-фенилбутил)циклогексил)метанол (полярный диастереоизомер)
a) [4-(1-этоксиэтоксиметил)-1-фенил-4-(4-фенилбутил)циклогексил]диметиламин
В стадии синтеза к примеру 20, стадия g) при хроматографическом разделении также получился чистый полярный диастереоизомер.
Полярный диастереоизомер: 700 мг (27%), светло-желтое масло
1Н-ЯМР (ДМСО-d6): 0.96-1.23 (m, 14H); 1.36-1.57 (m, 4H); 1.90 (s, 6H); 1.92-2.00 (m, 4H); 2.45-2.47 (m, 1H); 3.16-3.21 (m, 1H); 3.35-3.43 (m, 1H); 3.51-3.61 (m, 1H); 4.61 (q, 1H, J=5.2Hz); 7.09-7.38 (m, 10H).
b) (-4-(диметиламино)-4-фенил-1-(4-фенилбутил)циклогексил)метанол (полярный диастереоизомер)
Раствор титульного соединения предшествующей стадии (622 мг, 1.42 ммоль) в тетрагидрофуране (5 мл) смешивали с ледяной уксусной кислотой (3.0 мл) и водой (1.5 мл) и сначала 8 ч перемешивали при кипячении в колбе с обратным холодильником. Так как реакция была еще не полной, то в течение ночи при 50°С и затем еще раз 8 ч перемешивали при кипячении в колбе с обратным холодильником. Хотя затем взаимодействие все еще было не полным, то реакционный раствор концентрировали в вак. и остаток многократно ресуспендировали в толуоле (3×10 мл) и в каждом случае снова концентрировали в вак. Остаток ресуспендировали в 5% растворе гидрокарбоната натрия (30 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (30 мл), высушивали сульфатом натрия и концентрировали в вак. Так как сырой продукт еще содержал исходное вещество, то его отделяли с помощью флеш-хроматографии (400 г, 20×7.5 см) с этилацетат/метанолом (4:1) (77 мг). Сначала получали только 245 мг очень полярного целевого соединения. Путем промывания колонки метанолом (500 мл) выделяли другой продукт (220 мг).
Выход: 465 мг (86%), белое твердое вещество
Точка плавления: 123°С
1Н-ЯМР (ДМСО-d6): 0.90-1.03 (m, 2Н); 1.11 (brs, 4Н); 1.35-1.53 (m, 4H); 1.92 (br s, 10Н); 2.47 (дн., 2Н, J=8.2 Hz); 3.25 (дн., 2Н, J=4.9 Hz); 4.35 (t, 1H, J=4.7 Hz); 7.09-7.15 (m, 3Н); 7.18-7.24 (m, 3H); 7.28-7.39 (m, 4H).
13С-ЯМР (ДМСО-d6): 22.3; 28.1; 28.2; 35.2; 35.7; 37.6; 38.9; 59.9; 65.3; 125.4; 126.1; 127.4; 128.0; 128.1; 142.3.
Пример 22:
[4-бензил-4-(диметиламинометил)-1-фенил-циклогексил]-диметил-амин
Стадия 1:
4-бензил-1-(диметиламино)-4-((диметиламино)метил)циклогексанкарбонитрил
К смеси из 4N соляной кислоты (3 мл) и метанол (1.05 мл) при охлаждении льдом добавляли 40%-ый водный раствор диметиламина (2.8 мл, 22.1 ммоль), 4-бензил-4-((диметиламино)метил)циклогексанона (1.13 г, 4.60 ммоль) и цианида калия (0.70 г, 11.0 ммоль). Смесь 2 дн. перемешивали при комнатной температуре и затем после добавления воды (200 мл) экстрагировали простым эфиром (4×150 мл). После концентрирования раствора остаток ресуспендировали в дихлорметане (200 мл) и в течение ночи высушивали сульфатом магния, отфильтровывали и растворитель в вак. удаляли. Нитрил получали в виде масла, которое кристаллизовалось.
Выход: 1.06 г (77%)
1H-ЯМР (ДМСО-d6): 1.23 (2Н, m); 1.74 (2H, m); 2.16 (6H, s); 2.24 (6H, s); 2.32 (2H, m); 2.68 (2H, s); 7.16 (5H, m).
Стадия 2:
[4-бензил-4-(диметиламинометил)-1-фенил-циклогексил]-диметил-амин
Титульное соединение стадии 1 (0.88 г, 2.94 ммоль) растворяли в ТГФ (35 мл) и при охлаждении льдом по каплям смешивали с 2М раствором фенилмагния (5.1 мл, 10.2 ммоль). Реакционный раствор 8 ч нагревали до кипения. Для переработки раствор при охлаждении льдом смешивали с 20%-ым раствором NH4Cl (0.6 мл) и вода (0.4 мл), экстрагировали простым эфиром (3×25 мл), эфирный раствор промывали водой, высушивали (Na2SO4) и концентрировали в вак. Остаток очищали колоночной хроматографией с EtOH/ЕЕ (1:20). Получали 2 фракции, причем полярная фракция (300 мг) согласно ЖХМС в частности содержала желаемое вещество.
Выход: 90 мг (9%)
1H-ЯМР (ДМСО-d6): 1.23 (4H, m); 1.39 (2H, m); 1.82 (2H, s); 1.89 (6H, s); 2.09 (6H, s); 2.10 (2H, m); 2.73 (2H, s); 7.25 (10H, m).
Пример 23:
(4-диметиламино-1,4-дифенил-циклогексил)-метил-диметил-амин
Стадия 1:
Этиловый эфир 5-циано-2-оксо-5-фенил-циклогексанкарбоновой кислоты
К раствору бензилцианида (35.6 г, 304 ммоль) и этилового эфира бромпропионовой кислоты (126 г, 694 ммоль) в сухом толуоле (1070 мл) при от 0 до 5°С порционно добавляли при помешивании в течение 3 ч NaNH2 (100 г, 2560 ммоль). Затем 6 ч нагревали до кипения (сначала реакция была экзотермической, так что временно необходимо было удалить нагревательную баню). Затем смесь охлаждали до 0°С и резко охлаждали смесью из уксусной кислоты (240 мл) и воды (120 мл). Толуольную фазу отделяли, водную фазу экстрагировали толуолом (2×200 мл) и объединенную органическую фазу промывали раствором NaHCO3 (2×200 мл) и водой (2×200 мл) и высушивали с помощью Na2SO4.
Затем растворитель удаляли в вак.
Выход: 70,8 г (86%), коричневое твердое вещество
1H-ЯМР (ДМСО-d6): 1.25 (3Н, t); 2.24-2.88 (6H, m); 4.19 (2H, q); 7.50 (5H, m).
Стадия 2:
4-оксо-1-фенил-циклогексанкарбонитрил
Титульное соединение стадии 1 (70.8 г, 261 ммоль) нагревали до кипения в смеси из уксусной кислоты (810 мл) и концентрированной соляной кислоты (354 мл) 3.5 ч под ТХ-контролем. Затем смесь охлаждали до от 0 до 5°С, разбавляли водой (1 л), экстрагировали насыщенным NaCl и холодным этиловым эфиром уксусной кислоты (3×300 мл). Фазу этилацетата промывали водой и концентрировали в вак. Твердый остаток еще раз растворяли в этилацетате, промывали раствором NaHCO3 и концентрировали до сухости.
Выход 43.3 г (83%), желтое твердое вещество
Остаток без дополнительной очистки использовали для взаимодействия с этиленгликолем.
1H-ЯМР (ДМСО-d6): 2.41 (6H, m); 2.71 (2H, m); 7.40 (3H, m); 7.60 (2H, m).
13С-ЯМР (ДМСО-d6): 35.3; 38.1; 42.3; 121.7; 125.6; 128.2; 129.0; 139.2; 206.7.
Стадия 3:
8-фенил-1,4-диокса-спиро[4.5]декан-8-карбонитрил
Титульное соединение стадии 2 (43.3 г, 217 ммоль) и этиленгликоль (27.4 г, 435 ммоль) в толуоле (430 мл) при добавлении р-толуолсульфокислоты (1.87 г, 10.9 ммоль) в водоотделителе 3 ч кипятили с обратным холодильником. После окончания реакции охлаждали, промывали раствором NaHCO3 и нас. раствором NaCl, высушивали Na2SO4 и концентрировали в вак.
Выход: 48.8 г (96%), твердое вещество
1H-ЯМР (ДМСО-d6): 1.85 (4Н, m); 2.13 (4H, m); 3.92 (4H, s); 7.44 (5H, m).
13С-ЯМР (ДМСО-d6): 32.1; 34.0; 42.5; 63.8; 106.1; 122.1; 125.5; 128.0; 128.9; 139.9.
Стадия 4:
С-(8-фенил-1,4-диокса-спиро[4.5]дец-8-ил)-метиламин
К смеси LiAlH4 (1.87 г, 49.3 ммоль) в сухом ТГФ (25 мл) под защитным газом медленно впускали по каплям раствор титульного соединения, стадии 3 (10.0 г, 41.1 ммоль) в сухом ТГФ (70 мл). Затем 3 ч перемешивали при кипячении в колбе с обратным холодильником. После охлаждения реакционной смеси при охлаждении льдом впускали по каплям раствор воды (1.87 мл, 104 ммоль), разбавленный с небольшим количеством ТГФ, и 10 мин перемешивали. После этого 15%-ый водный NaOH (1.87 мл, 8.17 ммоль), разбавленный с небольшим количеством ТГФ, впускали по каплям и затем снова добавляли воду (5.6 мл). Образовавшийся осадок отфильтровывали через кизельгур, и растворитель удаляли в вак. В качестве остатка оставался амин.
Выход: 7.96 г, (78%), желтое масло
13С-ЯМР (ДМСО-d6): 29.9; 31.0; 42.7; 53.9; 63.5; 108.3; 125.5; 126.8; 128.2; 143.8.
Стадия 5:
Диметил-(8-фенил-1,4-диокса-спиро[4.5]дец-8-илметил)-амин
Титульное соединение стадии 4 (5.40 г, 21.8 ммоль) растворяли в ацетонитриле (150 мл), причем образовался мутный раствор. Добавляли водный, 37%-ый раствор формалина (30.6 мл, 407 ммоль). Состав 20 мин перемешивали при КТ и затем смешивали с цианоборгидридом натрия (5.76 г, 91.7 ммоль). Реакцию отслеживали посредством ТХ в хлороформ/метаноле (9:1). Через 4 ч раствор устанавливали уксусной кислотой до рН 7 и концентрировали в вак. Остаток ресуспендировали в хлороформе, промывали раствором NaHCO3 и водную фазу экстрагировали простым эфиром. Объединенные органические фазы высушивали над Na2SO4 и концентрировали в вак. Сырой продукт очищали путем флеш-хроматографии хлороформ/метанолом (50:1→20:1→9:1).
Выход: 5.40 г (67%)
1H-ЯМР (ДМСО-d6): 1.32 (2Н, m); 1.56 (2H, m); 1.77 (2H, m); 1.91 (6H, s); 2.14 (2H, m); 2.28 (2H, s); 3.80 (4H, m); 7.16-7.39 (5H, m).
Стадия 6:
4-диметиламинометил-4-фенил-циклогексанон
Титульное соединение стадии 5 (5.40 г, 19.6 ммоль) растворяли в 5%-ой H2SO4 (300 мл) и 1 дн. перемешивали при КТ. Затем раствор трижды промывали простым эфиром и эфирную фазу отводили. Водную фазу при охлаждении льдом подщелачивали с помощью 5N NaOH и трижды экстрагировали дихлорметаном. Органическую фазу промывали небольшим количеством воды, высушивали над Na2SO4 и концентрировали в вак.
Выход: 4.89 г (100%)
1H-ЯМР (ДМСО-d6): 1.92 (6H, s); 1.94-2.00 (2H, m); 2.07-2.25 (4H, m); 2.39-2.46 (4H, m); 7.23 (1H, m); 7.37 (2H, m); 7.48 (2H, m).
Стадия 7:
1-диметиламино-4-диметиламинометил-4-фенил-циклогексанкарбонитрил
К смеси из 4N соляной кислоты (5 мл) и метанола (3 мл) при охлаждении льдом впускали по каплям 40%-ый, водный раствор диметиламина (12.8 мл 21.1 ммоль). Затем по очереди добавляли титульное соединение, стадии 6 (4.89 г, 21.1 ммоль) и KCN (3.30 г, 50.7 ммоль). Смесь 3 дн. перемешивали при КТ. Для переработки состав смешивали с водой (10 мл) и экстрагировали диэтиловым эфиром (3×20 мл). Эфирную фазу концентрировали в вак., остаток в CH2Cl2(20 мл) ресуспендировали, высушивали над Na2SO4 и концентрировали в вак.
Выход: 5.16 г (86%)
1H-ЯМР (ДМСО-d6): 1.28 (2Н, m); 1.69 (2H, m); 1.94 (6H, s); 2.05 (2H, m); 2.15 (6H, s); 2.26 (2H, m); 2.37 (2H, s); 7.19 (1H, m); 7.35 (4H, m).
Стадия 8:
(4-диметиламинометил-1,4-дифенил-циклогексил)-диметил-амин
К раствору титульного соединения, стадии 7 (1.00 г, 3.5 ммоль) в абс. ТГФ (10 мл) под атмосферой азота и охлаждения льдом при 10°С медленно впускали по каплям 2М раствор хлорида фенилмагния в ТГФ (3.5 мл, 7.0 ммоль). Раствор 20 ч перемешивали при КТ. Затем добавляли 20%-ый раствор NH4Cl (5 мл) и воду (2 мл) и раствор экстрагировали простым эфиром (3×5 мл). Объединенные органические фазы промывали водой (2 мл) и насыщенным раствором NaCl (2 мл), высушивали над Na2SO4 и концентрировали в вак. Посредством флеш-хроматографии остатка с хлороформ/метанолом (20:1) получали соль продукта, который выделяли посредством 1N NaOH, экстрагировали хлороформом, высушивали над Na2SO4 и в вак. освобождали от растворителя.
Выход: 336 мг (28%) неполярный диастереоизомер, пористое твердое вещество
1H-ЯМР (CDCl3): 1.39 (2H, m); 1.65-1.78 (2H, m); 1.82 (6H, s); 1.96 (6H, s); 2.20 (2H, s); 2.28-2.41 (4H, m); 7.15-7.44 (10 H, m).
Пример 24:
(Е)-N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-3-фенил-акриламид (неполярный диастереомер)
Титульное соединение из примера 8 (202 мг, 0.656 ммоль) помещали в сух. ТГФ (20 мл) и смешивали с TEA (97 µл, 0.702 ммоль). Затем добавляли хлорид коричной кислоты (116 мг, 0.702 ммоль). Состав 20 ч перемешивали при комнатной температуре. Спустя это время реакции, состав концентрировали в вак. до сухости. Остаток ресуспендировали в этилацетате (20 мл) и промывали насыщенным раствором NaHCO3 (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вак. Остаток очищали путем флеш-хроматографии хлороформом/метанол (20:1).
Выход: 95 мг (33%)
1H-ЯМР (ДМСО-d6): 1.61 (2Н, bs); 1.97 (5H, bs); 2.45 (1H, m); 2.49 (4H, m); 2.94 (3H, bs); 3.23 (1H, s); 3.35 (1H, s); 3.35 (2H, m); 7.02 (1H, m); 7.25 (2H, m); 7.34 (9H, m); 7.46 (2H, m); 7.57 (2H, m).
Пример 33:
(4-бензил-4-((диметиламино)метил)-N-метил-1-фенилциклогексанамин (полярный диастереомер)
Стадия 1:
4-бензил-4-диметиламинометил-1-метиламино-циклогексанкарбонитрил
К охлажденному до 0°С раствору 4N соляной кислоты (6.6 мл) и метанола (4.00 мл) добавляли 40%-ый водный раствор метиламина (15.3 мл, 121 ммоль) и 4-бензил-4-((диметиламино)метил)циклогексанон (6.20 г, 25.3 ммоль), растворенный в метаноле (25 мл). Затем реакционную смесь смешивали с цианидом калия (4.00 г, 60 ммоль) и 5 дн. перемешивали при комнатной температуре. Для переработки смесь разбавляли с водой (180 мл) и экстрагировали простым эфиром (3×100 мл). Объединенные органические фазы высушивали над Na2SO4, отфильтровывали и в вак. концентрировали до сухости.
Выход: 5.80 г (81%)
1H-ЯМР (ДМСО-d6): 1.35 (5H, m); 1.58 (8H, m); 2.25 (6H, m); 2.65 (4H, m); 4.35 (1H, m); 7.14 (3H, m); 7.28 (2H, m).
Стадия 2:
(4-бензил-4-((диметиламино)метил)-N-метил-1-фенилциклогексанамин (полярный диастереомер)
Фениллитий (33 мл, 60 ммоль, 1.8 М раствор в дибутиловом эфире) под аргоном и при комнатной температуре по каплям смешивали с раствором титульного соединения, из стадии 1 (5.70 г, 20 ммоль) в диэтиловом эфире (60 мл). При этом реакционный раствор нагревался до 35°С, и в осадок выпадало твердое вещество. Реакционную смесь 30 мин перемешивали при кипячении в колбе с обратным холодильником, затем гидролизовали в ледяной ванне с 20%-ым раствором NH4Cl (40 мл) и органическую фазу отделяли. Водную фазу экстрагировали простым эфиром (3×100 мл). Объединенные органические фазы высушивали над Na2SO4 и концентрировали в вак. Остаток очищали путем флеш-хроматографии с хлороформ/метанолом (20:1→9:1→1:1→1% TEA).
Полярный диастереомер получали чистым. Неполярный диастереомер выделяли неочищенным.
Выход: 1.40 г (21%), полярный диастереомер
1H-ЯМР (ДМСО-d6): 1.13 (2Н, m); 1.74 (4H, m); 1.89 (3H, m); 1.96 (4H, m); 2.23 (6H, s); 2.68 (2H, s); 7.15 (4H, m); 7.26 (2H, m); 7.33 (2H, m); 7.48 (2H, m).
Пример 34:
(1-бензил-4-диметиламино-4-фенил-циклогексил)-метил-диметил-амин (полярный диастереомер)
Раствор титульного соединения из примера 33 (1.40 г, 4.16 ммоль) и формалин (5.8 мл, 37%-ый водный раствор) в ацетонитриле (40 мл) порционно смешивали с цианоборгидридом натрия (1.03 г, 16.6 ммоль) и 45 мин перемешивали при комнатной температуре. Затем добавляли конц. уксусную кислоту до нейтральной реакции и 45 мин перемешивали при комнатной температуре. Для переработки растворитель удаляли в вак., остаток ресуспендировали в 2N NaOH (40 мл) и затем экстрагировали простым эфиром (3×40 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вак. Оставшийся остаток путем очищали флеш-хроматографии с этилацетатом (метанол → метанол + 2% TEA).
Выход: 200 мг (14%)
1H-ЯМР (ДМСО-d6): 1.10 (2Н, m); 1.56 (2H, m); 1.89 (6H, s); 2.00 (2H, m); 2.04 (2H, s); 2.11 (2H, m); 2.25 (6H, s); 2.58 (2H, m); 7.19 (10H, m).
Пример 37:
[4-(диметиламинометил)-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
К раствору титульного соединения пример 23, Стадия 7 (1.00 г, 3.5 ммоль) в абс. ТГФ (10 мл) под атмосферой азота и охлаждения льдом при 0-10°С медленно впускали по каплям 2М раствор хлорида фенилмагния в ТГФ (3.5 мл, 7.0 ммоль). Раствор 20 ч перемешивали при КТ. Затем добавляли 20%-ый раствор NH4Cl (5 мл) и вода (2 мл) и раствор экстрагировали простым эфиром (3×5 мл). Объединенные органические фазы промывали водой (2 мл) и насыщенным раствором NaCl (2 мл), высушивали над Na2SO4 и концентрировали в вак. Флеш-хроматография остатка хлороформом/метанол (20:1→9:1→4:1→1:1→1:1 + 1% NH3→ МеОН + 1% NH3) дала с 20:1 гидрохлорид неполярного диастереоизомера, который выделяли посредством 1N NaOH, экстрагировали хлороформом, высушивали над Na2SO4 и концентрировали в вак. С помощью МеОН + 1% NH3получали полярный диастереоизомер. Так как здесь первый спектр равным образом указывал на соль, то также выделяли полярный диастереоизомер посредством 1N NaOH, экстрагировали хлороформом, высушивали над Na2SO4 и концентрировали в вак.
Выход: 81 мг (7%), полярный диастереоизомер, пористое твердое вещество
1H-ЯМР (ДМСО-d6): 1.59 (2Н, шир.); 1.77-1.86 (2H, m); 1.89 (6H, s); 1.95 (6H, s); 1.97-2.05 (2H, m); 2.25 (2H, m); 2.39 (2H, s); 7.07-7.37 (10H, m).
Пример 42:
(Е)-N-[[4-диметиламино-4-(3-фторфенил)-1-метил-циклогексил]-метил]-3-фенил-акриламид (полярный диастереомер)
Стадия 1:
4-диметиламино-4-(3-фтор-фенил)-циклогексанкарбальдегид
К раствору хлорида (метоксиметил)трифенилфосфония (6.58 г, 19.2 ммоль) в абс. ТГФ (25 мл) при 0°С под аргоном впускали по каплям KOtBu (2.15 г, 19.2 ммоль), растворенный в абс. ТГФ (25 мл). Образовавшийся красный раствор через 30 мин при 0°С смешивали с раствором 4-диметил-амино-4-(3-фтор-фенил)-циклогексанон (3.0 г, 12.76 ммоль) в абс. ТГФ (25 мл) и в течение ночи перемешивали при КТ. Растворитель удаляли в вак., остаток смешивали с 1М серной кислотой (50 мл) и 2 ч перемешивали. Выпавший при этом осадок отделяли и фильтрат (рН 1) промывали простым эфиром (6×30 мл). Водный раствор устанавливали посредством 5N NaOH до рН 11 и экстрагировали этиловым эфиром уксусной кислоты (3×50 мл). Объединенные органические фазы высушивали над Na2SO4 и концентрировали в вак.
Выход: 3.20 г (100%), коричневое масло
Смесь диастереомеров 1:1
1H-ЯМР (ДМСО-d6): 1.20 (1Н, m); 1.62 (2H, m); 1.75 (3H, m); 1.93 (6H, s); 2.37 (3H, m); 7.12 (3H, m); 7.40 (1H, m); 9.50 (0.5H, s); 9.62 (0.5H, s).
Стадия 2:
4-диметиламино-4-(3-фтор-фенил)-1-метил-циклогексанкарбальдегид
Раствор титульного соединения, стадии 1 (2.73 г, 10.95 ммоль) в абс. дихлорметане (50 мл) при 0°С под аргоном смешивали с трет-BuOK (1.47 г, 13.14 ммоль) и метилйодид (747 µл, 12 ммоль). Через 30 мин состав нагревали до КТ и затем перемешивали в течение ночи (в осадок выпадало твердое вещество). Реакционную смесь смешивали с насыщенным раствором NaCl (50 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенные органические фазы высушивали над Na2SO4, концентрировали в вак. и оставшийся остаток очищали флеш-хроматографией с этиловым эфиром уксусной кислоты/МеОН 20:1.
Выход: 1.39 г (51%)
1H-ЯМР (ДМСО-d6): 0.85 (1.5Н, s); 1.00 (1.5H, s); 1.50 (1H, m); 1.54-1.77 (4H, m); 1.89-1.95 (7H, m); 2.11-2.31 (2H, m); 7.11 (3H, m); 7.38 (1H, m); 9.36 (0.5H, s); 9.44 (0.5H, s).
Стадия 3:
4-диметиламино-4-(3-фтор-фенил)-1-метил-циклогексанкарбальдегидоксим
Раствор титульного соединения, стадии 2 (1.38 г, 5.53 ммоль) и гидроксиламин-гидрохлорид (576 мг, 8.3 ммоль) в абс. этаноле (20 мл) смешивали с амберлистом А 21 (3.9 г) и 16 ч перемешивали при КТ. Ионообменник отфильтровывали, раствор концентрировали и остаток подщелачивали с помощью 1N NaOH. Водную фазу экстрагировали этиловым эфиром уксусной кислоты, высушивали над Na2SO4 и концентрировали в вак.
Выход: 1.54 г (100%)
Стадия 4:
[4-аминометил-1-(3-фтор-фенил)-4-метил-циклогексил]-диметил-амин
Гидрид литийалюминия (440 мг, 11.6 ммоль) суспендировали под аргоном в абс. ТГФ (50 мл), по каплям смешивали с раствором титульного соединения, стадии 3 (1.54 г, 5.53 ммоль) в абс. ТГФ (20 мл) и 4 ч кипятили в колбе с обратным холодильником. Затем состав при 10°С гидролизовали с водой (10 мл) и отфильтровывали через кизельгур. ТГФ удаляли в вак., остаток устанавливали посредством 1N NaOH до рН 11 и экстрагировали этиловым эфиром уксусной кислоты. Объединенные органические фазы высушивали над Na2SO4, концентрировали в вак. и оставшийся остаток отделяли флеш-хроматографией с МеОН + 2% NH3.
Выход: 435 мг (30%, неполярный диастереомер)
1H-ЯМР (ДМСО-d6): 0.85 (3H, s); 1.03 (2H, m); 1.29 (2H, m); 1.83 (2H, m); 1.91 (6H, s); 2.08 (2H, m); 2.17 (2H, s); 7.09 (3H, m); 7.38 (1H, m).
Выход: 510 мг (35%, полярный диастереомер)
1H-ЯМР (ДМСО-d6): 0.72 (3H, s); 1.00 (2H, m); 1.49 (2H, m); 1.78 (2H, m); 1.91 (6H, s); 2.07 (2H, m); 2.38 (2H, s); 7.09 (3H, m); 7.39 (1H, m).
Стадия 5:
(Е)-N-[[4-диметиламино-4-(3-фторфенил)-1-метил-циклогексил]-метил]-3-фенил-акриламид (полярный диастереомер)
Раствор титульного соединения стадии 4 (полярный диастереомер) (250 мг, 0.94 ммоль) и основание Хюнига (169 µл, 1.0 ммоль) в абс. дихлорметане (10 мл) смешивали с хлоридом коричной кислоты (166 мг 1.0 ммоль) и 24 ч перемешивали при КГ. Органический раствор промывали насыщенным раствором NaHCO3 и насыщенным раствором NaCl, высушивали над Na2SO4, концентрировали в вак. и оставшийся остаток очищали флеш-хроматографией с этиловым эфиром уксусной кислоты/МеОН 4:1.
Выход: 295 мг (80%), пористое твердое вещество
1H-ЯМР (ДМСО-d6): 0.77 (3H, s); 1.03 (2H, m); 1.53 (2H, m);1.87 (2H, m); 1.93 (6H, s); 2.12 (2H, m); 3.17 (2H, d); 6.76 (1H, d); 7.07 (3H, m); 7.37 (5 H, m); 7.56 (2H, m); 7.96 (1H, t).
Пример 48:
[4-(бутил-метил-амино)-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
Стадия 1:
N-(4-диметиламино-1,4-дифенил-циклогексил)-М-метил-бутирамид
Титульное соединение из примера 8 (308 мг, 1.0 ммоль) помещали в абс. ТГФ (15 мл) и смешивали с TEA (165 рл, 1.2 ммоль) и бутирилхлоридом (103 мг, 1.2 ммоль, V=124 µл). Состав 20 ч перемешивали при комнатной температуре и затем концентрировали в вак. до сухости. Остаток ресуспендировали в этилацетате (20 мл) и промывали насыщенным раствором NaHCO3 (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вак. Остаток очищали с помощью флеш-хроматографии хлороформом/метанол (50:1).
Выход: 206 мг (53%)
1H-ЯМР (ДМСО-d6): 0.76 (3Н, t); 1.41 (2H, q); 1.60 (2H, m); 1.95 (6H, s); 2.22 (4H, t); 2.33 (2H, m); 2.82 (3H, s); 7.20-7.39 (10H, m).
Стадия 2:
[4-(бутил-метил-амино)-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
Титульное соединение из стадии 1 (200 мг, 0.528 ммоль) растворяли в абс. ТГФ (15 мл). LiAlH4 (39 мг, 1.06 ммоль) добавляли под аргоном. Состав 7 ч кипятили в колбе с обратным холодильником. Затем состав охлаждали до комнатной температуры, при охлаждении льдом смешивали с ТГФ (12 мл) и H2O (5 мл) и 30 мин перемешивали. Состав отфильтровывали через фритту с кизельгуром и подвергали последующей промывке дихлорметаном (50 мл). Объединенные фильтраты концентрировали в вак.
Выход: 194 мг (100%), масло
1H-ЯМР (ДМСО-d6): 0.74 (3H, t); 1.12 (4H, m); 1.73 (4Н, шир.); 1.83 (6H, s); 1.90 (3H, s); 1.92 (1H, s); 1.96 (2Н, шир.); 2.25 (4Н, шир.); 7.25 (2H, m); 7.38 (8H, m).
Пример 49
[4-(бутил-метил-амино)-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
Титульное соединение из примера 9 (308 мг, 1.0 ммоль) и бутиральдегид (72 мг, 30 1.0 ммоль, V=89 µл) помещали в абс. ацетонитрил (30 мл) и смешивали с цианоборгидридом натрия (250 мг, 4.0 ммоль). Состав 45 мин перемешивали при комнатной температуре, затем смешивали с конц. уксусной кислотой (прибл. 500 µл) и 45 мин перемешивали при комнатной температуре. Для переработки состав концентрировали в вак. до сухости. Остаток смешивали с 2N NaOH и экстрагировали простым эфиром (3×20 мл). Объединенные органические фазы высушивали над Na2SO4 и концентрировали в вак. Остаток очищали с помощью флеш-хроматографии хлороформом/метанол (9:1).
Выход: 111 мг (30%), масло
1H-ЯМР (ДМСО-d6): 0.86 (3Н, t); 1.30 (6H, m); 2.05 (12H, m); 2.35 (5H, m); 7.29 (10H, m).
Пример 50:
[4-(бензил-метил-амино)-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
Стадия 1:
N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-бензамид
Титульное соединение из примера 8 (308 мг, 1.0 ммоль) помещали в абс. ТГФ (15 мл) и смешивали с TEA (165 µл, 1.2 ммоль) и бензоилхлоридом (168 мг, 1.2 ммоль, V=147 µл). Состав 16 ч перемешивали при комнатной температуре и затем концентрировали в вак. до сухости. Остаток ресуспендировали в этилацетате (20 мл), промывали насыщенным раствором NaHCO3 (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вак. Остаток очищали с помощью флеш-хроматографии хлороформом/метанол (20:1).
Выход: 169 мг (41%)
1H-ЯМР (ДМСО-d6): 1.75 (2H, m); 1.98 (6H, s); 2.38 (3H, m); 2.55 (2H, m); 2.69 (4H, s); 7.24-7.41 (13H, m); 7.54 (2H, d).
Стадия 2:
[4-(бензил-метил-амино)-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
Титульное соединение из стадии 1 (160 мг, 0.387 ммоль) растворяли в абс. ТГФ (15 мл) и смешивали с LiAlH4 (29 мг, 0.775 ммоль) под аргоном. Состав 7 ч кипятили в колбе с обратным холодильником и затем охлаждали до комнатной температуры. К составу при охлаждении льдом добавляли ТГФ (5 мл) и H2O (5 мл) и 30 мин перемешивали. Состав отфильтровывали через фритту с кизельгуром и подвергали последующей промывке дихлорметаном (50 мл). Объединенные фильтраты концентрировали в вак.
Выход: 149 мг (97%)
1H-ЯМР (ДМСО-d6): 1.78 (3Н, s); 1.85 (10H, s); 2.33 (4H, m); 3.14 (2H, bs); 7.04-7.20 (4H, m); 7.31 (2H, m); 7.40 (9H, m).
Пример 51:
[4-(бензил-метил-амино)-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
Стадия 1:
N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-бензамид
Титульное соединение из примера 9 (308 мг, 1.0 ммоль) помещали в абс. ТГФ (15 мл) и смешивали с TEA (165 µл, 1.2 ммоль) и бензоилхлоридом (168 мг, 1.2 ммоль, V=147 µл). Состав 16 ч перемешивали при комнатной температуре и затем концентрировали в вак. до сухости. Остаток ресуспендировали в этилацетате (20 мл), промывали насыщенным раствором NaHCO3 (2×20 мл) и насыщенным раствором NaCl (2×20 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вак. Остаток очищали с помощью флеш-хроматографии хлороформом/метанол (20:1).
Выход: 304 мг (74%)
1H-ЯМР (ДМСО-d6): 1.63 (2H, m); 1.92-2.00 (10H, m); 2.52 (1H, m); 2.76 (3H, s); 7.16 (1H, m); 7.28 (4H, m); 7.39-7.49 (10H, m).
Стадия 2:
[4-(бензил-метил-амино)-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
Титульное соединение из стадии 1 (290 мг, 0.702 ммоль) растворяли в абс. ТГФ (15 мл) и под аргоном смешивали с LiAlH4 (52 мг, 1.40 ммоль). Состав 7 ч кипятили в колбе с обратным холодильником и затем охлаждали до комнатной температуры. Состав отфильтровывали через фритту с кизельгуром и подвергали последующей промывке дихлорметаном (50 мл). Объединенные фильтраты концентрировали в вак.
Выход: 250 мг (89%)
1H-ЯМР (ДМСО-d6): 1.43 (1Н, m); 1.72-1.76 (1H, m); 1.89 (3H, s); 1.99 (6H, s); 2.42 (3Н, шир.); 3.25 (2H, bs); 7.16-7.39 (15H, m).
Пример 66:
2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-уксусная кислота (полярный диастереомер)
Стадия 1:
Трет-бутиловый эфир [(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-уксусной кислоты
Раствор титульного соединения из примера 9 (246 мг, 0.8 ммоль) и трет-бутиловый эфир бромуксусной кислоты (132 µл, 0.9 ммоль) в абс. ДМФ (10 мл) смешивали с карбонатом калия (124 мг, 0.9 ммоль) и 20 ч перемешивали при КТ. Затем растворитель удаляли в вак., остаток растворяли в дихлорметане (20 мл), промывали водой (2×10 мл) и насыщенным раствором NaCl (2×10 мл) и высушивали над Na2SO4. Органический раствор концентрировали в вак. и оставшийся остаток очищали флеш-хроматографией с этиловым эфиром уксусной кислоты/МеОН 20:1.
Выход: 133 мг (39%)
1H-ЯМР (CDCl3): 1.44 (9Н, s); 1.78 (2H, bs); 1.95 (2H, bs); 2.09 (6H, s); 2.21 (3H, s); 2.43 (4H, m); 2.92 (2H, s); 7.16-7.31 (10H, m).
Стадия 2:
2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-уксусная кислота (полярный диастереомер)
Титульное соединение из стадии 1 (130 мг, 0.3 ммоль) растворяли в анизоле (0.5 мл) и трифторуксусной кислоте (2.5 мл) и 20 ч перемешивали при КТ. Затем концентрировали в вак. до сухости, твердый остаток перемешивали с 1N NaOH, твердое вещество отфильтровывали, промывали водой и в вак. высушивали.
Выход: 69 мг (63%)
Точка плавления: 270-273°С
1H-ЯМР (ДМСО-d6): 1.70 (3Н, br); 1.96 (6H, s); 2.00 (3H, s); 2.25 (2H, m); 2.45 (4H, m); 3.32 (2H, s); 7.14 (2H, m); 7.25 (8H, m).
Следуя инструкции, как было описано в примере 66 за исключением того, что были применены исходные продукты, приведенные в таблице 1-1, получали следующее соединение.
Пример 68
[1-(4-метоксифенил)-4-метиламино-4-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
Стадия 1:
[8-(4-метокси-фенил)-1,4-диокса-спиро[4.5]дец-8-ил]-диметил-амин
Магний (3.65 г, 150 ммоль) и кристалл йода помещали под атмосферой азота и нагревали. Затем добавляли абс. простой эфир (10 мл) и впускали по каплям раствор 4-броманизола (18.8 мл, 150 ммоль) в абс. простой эфире (150 мл) таким образом, что простой эфир слегка кипел. Образовавшийся раствор 1 ч перемешивали при КТ и затем при КТ по каплям смешивали с раствором 8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил (10.5 г, 50.0 ммоль) в абс. ТГФ (100 мл), причем раствор во время добавления до кипения нагревался до 37-40°С. Из раствора выпадал осадок, и состав в течение ночи перемешивали при КТ. Раствор при охлаждении льдом смешивали с NH4Cl - раствор (150 мл), фазы разделяли, водную фазу трижды экстрагировали простым эфиром, объединенные органические фазы промывали насыщенным раствором NaCl и вода, высушивали над Na2SO4 и концентрировали в вак. Флеш-хроматография остатка с этилацетат/метанолом (20:1→9:1→4:1→1:4→МеОН) дала желаемый продукт.
Выход: 6.80 г (47%)
Стадия 2:
4-диметиламино-4-(4-метокси-фенил)-циклогексанон
Титульное соединение из стадии 1 (6.80 г, 23 ммоль) растворяли в простом эфире (100 мл), смешивали с 5%-ым H2SO4 (100 мл) и раствор 2 дн. сильно перемешивали при КТ. Фазы разделяли и эфирную фазу отводили. Водную фазу при охлаждении льдом подщелачивали с 5 N NaOH и трижды экстрагировали простым эфиром, затем объединенные органические фазы промывали водой, высушивали над Na2SO4 и концентрировали в вак.
Выход: 4.20 г (73%)
1H-ЯМР (ДМСО-d6): 2.00 (6Н, s); 2.01-2.14 (4Н, m); 2.42-2.48 (2H, m); 2.53-2.63 (2H, m); 3.76 (3H, s); 6.93 (2H, d); 7.34 (2H, d).
Стадия 3:
4-диметиламино-4-(4-метокси-фенил)-1-метиламино-циклогексанкарбонитрил
К смеси из 4N соляной кислоты (1.98 мл) и метанола (2.3 мл) при охлаждении льдом впускали по каплям 40%-ый водный метиламин-раствор (3.50 мл, 40.1 ммоль). Затем добавляли раствор титульного соединения, из стадии 2 (2.00 мг, 8.09 ммоль) в метаноле (30 мл) и цианид калия (1.32 г, 20.3 ммоль). Смесь 3 дн. перемешивали при КТ и затем после добавления воды (10 мл) экстрагировали простым эфиром 4х. Объединенные органические фазы высушивали над Na2SO4 и концентрировали в вак.
Выход: 2.23 г (96%), неочищенный продукт, сырым подвергали дальнейшему взаимодействию
1H-ЯМР (ДМСО-d6): 1.29 (1Н, m); 1.61 (1H, m); 1.69-1.86 (4H, m); 1.90 (6H, d); 1.93-2.04 (2H, m); 2.28 (3H, dd); 2.75 (1H, dq); 3.75 (3H, d); 6.90 (2H, d); 7.23 (2H, dd).
Стадия 4:
[1-(4-метоксифенил)-4-метиламино-4-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
Фениллитий (12.9 мл, 23.3 ммоль, 1.8 М раствор в дибутиловом эфире) помещали под аргоном и при КТ по каплям смешивали с раствором титульного соединения, стадии 3 (2.23 г, 7.76 ммоль) в абс.диэтиловом эфире (30 мл). При этом реакционный раствор нагревался до 35°С, и в осадок выпадало твердое вещество. Реакционную смесь 1 ч перемешивали при кипячении в колбе с обратным холодильником (ванна 50°С), затем гидролизовали в ледяной ванне (0-10°С) с 20%-ым раствором NH4Cl (20 мл) и органическую фазу отделяли. Водную фазу экстрагировали простым эфиром (3×50 мл). Объединенные органические растворы высушивали над Na2SO4 и концентрировали в вак. Путем флеш-хроматографии (100 г силикагель) с этиловым эфиром уксусной кислоты/метанол (20:1→9:1→МеОН→МеОН + 2% NH3) получали неполярный диастереоизомер в смешанной фракции с исходным веществом и кетоном и напоследок полярный диастереоизомер. Смешанную фракцию с неполярным диастереоизомером очищали посредством повторной флеш-хроматографии с дихлорметан/метанолом (50:1→20:1→9:1→4:1).
Выход: 232 мг (9%), неполярный диастереоизомер
1H-ЯМР (CDCl3): 1.71 (2Н, m); 1.98 (4H, m); 1.99 (6H, s); 2.11 (1H, m); 2.19-2.41 (5H, m); 3.81 (3H, s); 6.91 (2H, m); 7.27 (3H, m); 7.37 (2H, m); 7.48 (2H, m).
Пример 69
[1-(4-метоксифенил)-4-метиламино-4-фенил-циклогексил]-диметил-амин (полярный диастереомер)
Во время синтеза титульного соединения примера 68 в пределах стадии 4 также можно было выделить полярный диастереомер.
Выход: 177 мг (7%), полярный диастереоизомер
1H-ЯМР (CDCl3): 1.58-1.92 (4H, m); 2.03 (4H, m); 2.07 (6H, s); 2.10-2.18 (2H, m); 2.29 (2H, m); 3.80 (3H, s); 6.87 (2H, d); 7.14 (1H, m); 7.20-7.33 (6H, m).
Следуя инструкции, как было описано в примерах 68 и 69 за исключением того, что применяли бромиды или соответствующие реагенты Гриньяра, а также карбонитрилы, такие как приведены в таблице 1-2, получали следующие соединения.
Карбонитрилы:
8-диметиламино-1,4-диокса-спиро[4.5]декан-8-карбонитрил (CN-A)
8-(пирролидин-1-ил)-1,4-диоксаспиро[4.5]декан-8-карбонитрил (CN-B)




Пример 74:
[4-диметиламино-1-(4-метоксифенил)-4-фенил-циклогексил]-диметил-амин (полярный диастереомер)
Раствор титульного соединения, из примера 69 (111 мг, 0.33 ммоль) и формалин (0.45 мл, 37%-ый водный раствор) в ацетонитриле (3 мл) смешивали с цианоборгидридом натрия (83 мг, 1.32 ммоль) и 45 мин перемешивали при КТ. Затем добавляли конц. уксусную кислоту до нейтральной реакции и 45 мин перемешивали при КТ. Для переработки растворитель удаляли в вак., остаток ресуспендировали в 2N NaOH (5 мл) и затем экстрагировали простым эфиром (3×10 мл). Органический раствор высушивали над Na2SO4 и концентрировали в вак. Оставшийся остаток очищали путем флеш-хроматографии с этилацетат/метанолом (1:2→МеОН).
Выход: 82 мг (71%)
1H-ЯМР (CDCl3): 1.62-2.05 (4Н, m); 2.07 (12H, s); 2.37 (4H, m); 3.79 (3H, s); (6.77 (3H, s); 6.83 (2H, d); 7.20 (3H, m); 7.28 (4H, m).
Пример 75:
[4-диметиламино-1-(4-метоксифенил)-4-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
Раствор титульного соединения, из примера 68 (96 мг, 0.28 ммоль) и формалин (0.39 мл, 37%-ый водный раствор) в ацетонитриле (3 мл) смешивали с цианоборгидридом натрия (72 мг, 1.15 ммоль) и 45 мин перемешивали при КТ. Затем добавляли конц. уксусную кислоту до нейтральной реакции и 45 мин перемешивали при КТ. Для переработки растворитель удаляли в вак., остаток ресуспендировали в 2N NaOH (5 мл) и затем экстрагировали простым эфиром (3×10 мл). Органический раствор высушивали над Na2SO4 и концентрировали в вак. Оставшийся остаток очищали путем флеш-хроматографии с этилацетат/метанолом (2:1→1:1→1:1 + 2% NH3).
Выход: 62 мг (62%)
1H-ЯМР (CDCl3): 1.59 (4H, m); 1.92 (6H, s); 1.93 (6H, s); 2.48 (4H, m); 3.81 (3H, s); 6.90 (2H, m); 7.20-7.41 (7H, m).
Следуя инструкции, как было описано в примерах 74 и 75 за исключением того, что применяли приведенные в таблице 1-3 исходные продукты, получали следующие соединения.
Пример 76:
[4-[(1Н-индол-3-ил-метиламино)-метил]-4-метил-1-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
Стадия 1:
4-диметиламино-4-фенил-циклогексанкарбальдегид
К раствору хлорида (метоксиметил)трифенилфосфония (25.7 г, 75.0 ммоль) в абс. ТГФ (100 мл) при 0°С под аргоном впускали по каплям трет-BuOK (8.41 г, 75 ммоль), растворенный в абс. ТГФ (100 мл). Образовавшийся красный раствор через 30 мин при 0°С смешивали с раствором 4-диметиламино-4-фенил)-циклогексанона (10.9 г, 50.0 ммоль) в абс. ТГФ (100 мл) и в течение ночи перемешивали при КТ. Растворитель удаляли в вак., остаток смешивали с 1N серной кислотой (150 мл) и 2 ч перемешивали. Выпавший при этом осадок отделяли и фильтрат (рН 1) промывали простым эфиром (6×100 мл). Водный раствор устанавливали посредством 5N NaOH до рН 11 и экстрагировали этиловым эфиром уксусной кислоты (3×100 мл). Объединенные органические фазы высушивали над Na2SO4 и концентрировали в вак.
Выход: 11.6 г (100%), коричневое масло
Смесь диастереомеров 1:1
1H-ЯМР (ДМСО-d6): 1.18 (1Н, m); 1.59-1.91 (5H, m); 1.92 (6H, s); 2.36 (3H, m); 7.23-7.38 (5H, m); 9.48 (0.5H, s); 9.62 (0.5H, s).
Стадия 2:
4-диметиламино-1-метил-4-фенил-циклогексанкарбальдегид
Раствор титульного соединения из стадии 1 (11.6 г, 50.0 ммоль) в абс. дихлорметан (200 мл) при 0°С под аргоном смешивали с трет-BuOK (6.50 г, 58.0 ммоль) и метилйодидом (3.42 мл, 55.0 ммоль). Через 30 мин состав нагревали до КТ и затем в течение ночи перемешивали (в осадок выпадало твердое вещество). Реакционную смесь промывали водой и насыщенным раствором NaCl (50 мл), высушивали над Na2SO4, концентрировали в вак. и оставшийся остаток очищали флеш-хроматографией с этиловым эфиром уксусной кислоты/МеОН 20:1.
Выход: 5.90 г (48%)
1H-ЯМР (ДМСО-d6): 0.83 (1.5H, s); 1.00 (1.5H, s); 1.08 (1H, m); 1.55-1.82 (5H, m); 1.88 (3H, s); 1.92 (3H, s); 2.14-2.32 (2H, m); 7.27 (5H, m); 9.36 (0.5H, s); 9.45 (0.5H, s).
Стадия 3:
4-диметиламино-1-метил-4-фенил-циклогексанкарбальдегидоксим
Раствор титульного соединения из стадии 2 (5.90 г, 24.0 ммоль) и гидроксиламин-гидрохлорид (2.50 г, 36.0 ммоль) в абс.этанол (100 мл) смешивали с амберлистом А 21 (17.0 г) и 20 ч перемешивали при КТ. Ионообменник отфильтровывали, раствор концентрировали, и остаток подщелачивали посредством 1N NaOH. Водную фазу экстрагировали этиловым эфиром уксусной кислоты, высушивали над Na2SO4 и концентрировали в вак.
Выход: 6.25 г (100%)
Стадия 4:
[4-аминометил-4-метил-1-фенилциклогексил]-диметил-амин
Гидрид литийалюминия (1.82 г, 48.0 ммоль) под аргоном суспендировали в абс. ТГФ (200 мл), по каплям смешивали с раствором титульного соединения из стадии 3 (6.25 г, 24.0 ммоль) в абс. ТГФ (20 мл) и 4 ч кипятили в колбе с обратным холодильником. Затем состав при 10°С гидролизовали водой (20 мл) и отфильтровывали через кизельгур. ТГФ удаляли в вак., остаток устанавливали посредством 1N NaOH до рН 11 и экстрагировали этиловым эфиром уксусной кислоты. Объединенные органические фазы высушивали над Na2SO4, концентрировали в вак. и оставшийся остаток отделяли флеш-хроматографией с МеОН + 1% NH3.
Выход: 1.44 г (24%, неполярный диастереомер)
1H-ЯМР (ДМСО-d6): 0.86 (3H, s); 1.03 (2H, m); 1.29 (2H, m); 1.84 (2H, m); 1.91 (6H, s); 2.10 (2H, m); 2.16 (2H, s); 7.24 (1H, m); 7.32 (4H, m).
Выход: 1.53 г (26%, полярный диастереомер)
1H-ЯМР (ДМСО-d6): 0.72 (3H, s); 1.00 (2H, m); 1.49 (2H, m); 1.83 (2H, m); 1.90 (6H, s); 2.05 (2H, m); 2.39 (2H, s); 7.23 (1H, m); 7.34 (4H, m).
Стадия 5:
[4-[(1Н-индол-3-ил-метиламино)-метил]-4-метил-1-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
Индол-3-альдегид (203 мг, 1.4 ммоль) и неполярный диастереомер из стадии 4 (345 мг, 1.4 ммоль) растворяли в абс. ТГФ (10 мл), смешивали с Na2SO4 (2.0 г) и 24 ч перемешивали при КТ. Затем добавляли дихлорэтан (10 мл) и ацетоксиборгидрид натрия (423 мг, 2.0 ммоль) и последующие 24 ч перемешивали при КТ. Для переработки растворитель удаляли в вак., остаток смешивали с ЕЕ (20 мл), водой (20 мл) и 10%-ой серной кислотой (до рН 1) и фазы разделяли. Водную фазу устанавливали посредством 5N NaOH до рН 11 и трижды экстрагировали этиловым эфиром уксусной кислоты. Объединенные органические фазы высушивали над Na2SO4, концентрировали в вак. и оставшийся остаток очищали флеш-хроматографией с этиловым эфиром уксусной кислоты/МеОН 1:1 + 1% NH3.
Выход: 397 мг (76%), пористое твердое вещество
1H-ЯМР (ДМСО-d6): 0.92 (3H, s); 1.14 (2H, m); 1.32 (2H, m); 1.89 (8 H, bs); 2.05 (2H, m); 2.22 (2H, s); 3.75 (2H, s); 6.9-7.54 (10H, m); 10.75 (1H, s).
Пример 77:
[4-[(1Н-индол-3-ил-метиламино)-метил]-4-метил-1-фенил-циклогексил]-диметил-амин (полярный диастереомер)
Индол-3-альдегид (203 мг, 1.4 ммоль) и полярный диастереомер из примера 76, стадия 4 (345 мг, 1.4 ммоль), растворяли в абс. ТГФ (10 мл), смешивали с Na2SO4 (2.0 г) и 24 ч перемешивали при КТ. Затем добавляли дихлорэтан (10 мл) и ацетоксиборгидрид натрия (423 мг, 2.0 ммоль) и перемешивали последующие 24 ч при КТ. Для переработки растворитель удаляли в вак., остаток смешивали с этиловым эфиром уксусной кислоты (20 мл), водой (20 мл) и 10%-ой серной кислотой (до рН 1) и фазы разделяли. Водную фазу устанавливали посредством 5N NaOH до рН 11 и трижды экстрагировали этиловым эфиром уксусной кислоты. Объединенные органические фазы высушивали над Na2SO4, концентрировали в вак. и оставшийся остаток очищали флеш-хроматографией с этиловым эфиром уксусной кислоты/МеОН (1:1 + 1% NH3).
Выход: 370 мг (70%)
Точка плавления: 55-56°С
1H-ЯМР (ДМСО-d6): 0.78 (3Н, s); 1.02 (2H, m); 1.57 (3H, m); 1.79 (2H, m) 1.86 (6H, s); 2.02 (2H, m); 2.44 (2H, s); 3.89 (2H, s); 6.97 (1H, t); 7.06 (1H, t); 7.22-7.32 (7H, m); 7.64 (1H, d); 10.82 (1H, s).
Пример 78:
[4-[(1Н-индол-3-ил-метил-метил-амино)-метил]-4-метил-1-фенил-циклогексил]-диметил-амин (неполярный диастереомер)
Раствор титульного соединения из примера 76 (300 мг, 0.8 ммоль) и формалин (1.2 мл, 37%-ый водный раствор) в ацетонитриле (10 мл) порционно смешивали с цианоборгидридом натрия (201 мг, 3.2 ммоль) и 2 ч перемешивали при КТ. Затем добавляли конц. уксусную кислоту до нейтральной реакции и 45 мин перемешивали при КТ. Для переработки растворитель удаляли в вак., остаток ресуспендировали в 2N NaOH (10 мл) и затем экстрагировали простым эфиром (3×20 мл). Органический раствор высушивали над Na2SO4 и концентрировали в вак. Оставшийся остаток очищали путем флеш-хроматографии с этиловым эфиром уксусной кислоты/МеОН (1:1).
Выход: 189 мг (56%)
Согласно ЯМР и ЖХМС при этом речь идет о соединении гидроксиметила, которое растворяли в 1N NaOH (2 мл) и ТГФ (2 мл) и 2 ч кипятили в колбе с обратным холодильником. Затем экстрагировали простым эфиром (2×20 мл). Органический раствор высушивали над Na2SO4 и концентрировали в вак. Оставшийся остаток очищали путем флеш-хроматографии с ЕЕ/МеОН (1:1 + 1% NH3).
Выход: 119 мг (38%)
1H-ЯМР (CDCl3): 1.04 (3Н, s); 1.32 (4Н, m); 1.87 (2H, m); 2.05 (6H, s) 2.14 (2H, s); 2.15 (3H, s); 2.34 (2H, m); 3.63 (2H, s); 6.78 (1H, s); 7.08 (1H, t); 7.17 (1H, t); 7.30-7.41 (6H, m); 7.62 (1H, d); 7.99 (1H, s).
Пример 79:
[4-[(1Н-индол-3-ил-метил-метил-амино)-метил]-4-метил-1-фенил-циклогексил]-диметил-амин (полярный диастереомер)
Раствор титульного соединения из примера 77 (300 мг, 0.8 ммоль) и формалина (1.2 мл, 37%-ый водный раствор) в ацетонитриле (10 мл) порционно смешивали с цианоборгидридом натрия (201 мг, 3.2 ммоль) и 2 ч перемешивали при КТ. Затем добавляли конц. уксусную кислоту до нейтральной реакции и 45 мин перемешивали при КТ. Для переработки растворитель удаляли в вак., остаток ресуспендировали в 2N NaOH (10 мл) и затем экстрагировали простым эфиром (3×20 мл). Органический раствор высушивали над Na2SO4 и концентрировали в вак. Оставшийся остаток смешивали с ЕЕ/МеОН (1:1, 5 мл), при этом в осадок выпадало бесцветное твердое вещество, которое отделяли. Согласно ЯМР и ЖХМС при этом речь шла о соединении гидроксиметила.
Маточный раствор концентрировали (240 мг), речь также шла о соединении гидроксиметила.
Выход: 299 мг (89%)
Соединение гидроксиметила (240 мг, 0.57 ммоль) растворяли в 1N NaOH (2 мл) и ТГФ (2 мл) и 2 ч кипятили в колбе с обратным холодильником. Затем экстрагировали простым эфиром (2×20 мл). Органический раствор высушивали над Na2SO4 и концентрировали в вак. Оставшийся остаток очищали путем флеш-хроматографии с ЕЕ/МеОН (1:1 + 1% NH3).
Выход: 181 мг (82%)
1H-ЯМР (CDCl3): 0.88 (3H, s); 1.13 (2H, m); 1.74 (2H, m); 1.80 (4H, m) 2.10 (6H, s); 2.29 (3H, s); 2.44 (2H, s); 3.78 (2H, s); 7.10-7.40 (9H, m); 7.85 (1H, d); 8.24 (1H, s).
Пример 80:
[3-[[[4-(диметил-амино)-1-метил-4-фенил-циклогексил]-метил-метил-амино]-метил]-1Н-индол-1-ил]-метанол (полярный диастереомер)
В рамках синтеза примера 79 получалось соединение гидроксиметила в виде промежуточного продукта.
Выход: 299 мг (89%)
1H-ЯМР (CDCl3): 0.41 (2H, m); 0.62 (2H, m); 0.65 (3H, s); 1.27 (2H, m) 1.61 (2H, m); 1.75 (6H, s); 2.28 (2H, s); 2.47 (3H, s); 3.64 (2H, s); 5.63 (2H, s); 7.01 (2H, m); 7.14-7.40 (7H, m); 7.76 (1H, d)
Пример 86:
[4-[[4,6-бис(метиламино)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
Титульное соединение примера 103 (200 мг, 0.31 ммоль) растворяли в 33%-ом растворе метиламина в этаноле (2 мл) и перемешивали при микроволнах 30 мин при 100°С и 60 мин при 120°С. Выпавший осадок отсасывали и высушивали в вак.
Выход: 89 мг (64%)
Точка плавления: 250-252°С
1H-ЯМР (ДМСО): 1.65 (2H, m); 1.96 (6H, s); 2.41 (4H, m); 2.60 (6H, s); 3.06 (5H, m); 6.21 (2H, m); 7.16-7.43 (10H, m).
Пример 87:
[4-[[4-(4-метокси-фенокси)-6-метиламино-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
В рамках синтеза титульного соединения примера 86 маточный раствор концентрировали в вак. и оставшийся остаток очищали флеш-хроматографией с этиловым эфиром уксусной кислоты.
Выход: 25 мг (15%)
Точка плавления: 181-182°С
1H-ЯМР (ДМСО), Temp: 100°С: 1.70 (2Н, m); 1.99 (6H, s); 2.24 (2H, m); 2.38 (2H, m); 2.56 (2H, m); 2.67 (3H, d); 2.96 (3H, s); 3.76 (3H, s); 6.71 (1H, m); 6.98 (3H, m); 7.15-7.38 (10H, m).
Следуя инструкции, как было описано в примерах 86 и 87, за исключением того, что были применены исходные продукты, такие как приведены в таблице 1-4 получали, следующие соединения.
Пример 94:
[4-(диметил-амино)-1-(3-фторфенил)-4-(3-метил-1Н-индол-2-ил)-циклогексил]-диметил-амин (полярный диастереомер)
Стадия 1:
4-(диметиламино)-4-(3-фторофенил)-1-(3-метил-1Н-индол-2-ил)циклогексанол (неполярный и полярный диастереомер)
В атмосфере аргона к раствору скатола (1,00 г, 7,62 ммоль) в абсолютном тетрагидрофуране (25 мл) при -78°С медленно добавляли n-бутиллитий (8,39 ммоль, 3,35 мл, 2,5М в гексан). Образовывался бесцветный осадок. Через 10 мин раствор нагревали до комнатной температуры. Затем в реакционную смесь в течение прибл. 3 мин вводили диоксид углерода. Образовывался бесцветный раствор. Через 5 мин летучие компоненты полностью удаляли в вакууме при комнатной температуре (температура водяной ванны, ≤30°С). Бесцветный твердый остаток вновь растворяли в абсолютном тетрагидрофуране (20 мл). Светло-желтую реакционную смесь охлаждали до -78°С и впускали по каплям трет-бутиллитий (8,39 ммоль, 5,59 мл, 1,5М в пентан). Образовывался оранжевый раствор, который 1 ч перемешивали при -20°С и затем охлаждали до -78°С. Затем впускали по каплям 4-(диметиламино)-4-(3-фторофенил)циклогексанон [1,97 г, 8,39 ммоль, в абсолютном тетрагидрофуране (20 мл)] и образованный раствор 2 ч перемешивали. Затем к реакционной смеси впускали по каплям насыщенный водный раствор хлорида аммония (50 мл), 10 мин перемешивали, смесь нагревали до 0°С и 20 мин перемешивали. После этого добавляли 2N водный раствор хлороводорода (50 мл) и 10 мин перемешивали (легкое газовыделение). Затем рН-значение молочной суспензии подщелачивали насыщенным раствором гидрокарбоната натрия (50 мл) и 5N раствором гидроксида натрия (20 мл). Через 10 мин фазы разделяли. Органическая фаза содержала бесцветное твердое вещество. Фазы разделяли. Водную фазу экстрагировали дихлорметаном/метанол 20:1 (3×50 мл). Органические растворы объединяли. Летучие компоненты полностью удаляли в вакууме. Оставшийся светло-коричневый порошок экстрагировали метанолом (5×75 мл). Остаток состоял исключительно из неполярного диастереоизомера 6b/7b (450 мг (1,23 ммоль, 16%). Экстракты концентрировали в вакууме до сухости. Остаток ресуспендировали в метаноле (прибл. 30 мл). Светлое твердое вещество не растворялось. Которое отделяли с помощью фритты и затем высушивали в вакууме. Получали 980 мг (2,67 ммоль, 35%) бесцветного порошка. Который состоял из обоих диастереоизомеров. Маточный раствор отделяли хроматографически [силикагель 60 (150 г); трихлорметан/этанол 50:1 (500 мл), 19:1 (500 мл), 9:1 (300 мл), 5:1 (300 мл), 1:1 (300 мл), в каждом случае 0,5% триэтиламин, лучше начинать с трихлорметан/этанола 100:1]. Полученные фракции обоих диастереоизомеров должны были быть перекристаллизованы из метанола. Получали 93 мг (0,25 ммоль, 3%) неполярного диастереоизомера (Тпл. 197-202°С) и 146 мг (0,40 ммоль, 5%) полярного диастереоизомера (179-188°С).
13С{1Н}-ЯМР (101 МГц, ДМСО-d6, δ част. на млн, неполярный диастереоизомер): 9.5 (1 С), 28.4 (2 С), 32.5 (2 С), 37.8 (2 С), 58.2 (1 С, br), 69.4 (1 C), 102.7 (1 C), 111.0 (1 C), 113.0 (1 С, дн, J=21 Hz), 113.4 (1 С, дн., J=21 Hz), 117.3 (1 C), 117.8 (1 C) 119.9 (1 C), 122.6 (1 C, дн., J=2 Hz), 128.9 (1 C, J=8 Hz), 129.8 (1 C), 133.9 (1 C), 142.1 (1 C, br), 142.7 (1 C, дн., J=5 Hz), 161.9 (1 C, дн., J=242 Hz)
13С{1Н}-ЯМР (101 МГц, ДМСО-d6, δ част. на млн, полярный диастереоизомер): 9.0 (1 С), 26.9 (2 С, br), 33.5 (2 С), 37.6 (2 C), 55.9 (1 C, br), 68.5 (1 C), 102.3 (1 C), 110.9 (1 C), 113.5 (1 C, sbr), 115.8 (1 C, sbr), 117.2 (1 C), 117.8 (1 C) 120.0 (1 C), 125.0 (1 C, sbr), 126.6 (1 C), 130.0 (1 C, br), 133.7 (1 C), 141.1 (1 C, br), 162.4 (1 C, d, J=244 Hz), n.b. (1 С)
Стадия 2:
1-(3-фторофенил)-4-(1Н-индол-2-ил)-7,7-диметил-7-азониабицикло[2.2.1]гептанфторид
Спирт из стадии 1 (оба диастереоизомеры, 2,20 г, 6,00 ммоль) при -78°С суспендировали в абсолютном дихлорметане (50 мл). По очереди добавляли триэтиламин (3,65 г, 36,02 ммоль, 4,99 мл, 0,73 г/мл), ДМАП (16 мг, 0,12 ммоль) и DAST (2,90 г, 18,01 ммоль, 2,36 мл, 1,23 г/мл). Раствор 1 ч перемешивали при -78°С. Затем реакционную смесь в течение 10 ч (в течение ночи) нагревали до комнатной температуры. Затем добавляли насыщенный раствор гидрокарбоната натрия (50 мл) и 15 мин перемешивали (пока не окончилось газовыделение). Затем добавляли раствор гидроксида натрия - (5N, 20 мл) и 10 мин перемешивали. Фазы разделяли.
Красно-коричневую органическую фазу концентрировали в вакууме до сухости. Затем полученное коричневое твердое вещество растворяли в метаноле (50 мл).
Водную фазу равным образом концентрировали в вакууме до сухости. Светлый остаток экстрагировали с метанолом (5×75 мл).
Объединенные растворы метанола концентрировали в вакууме до сухости. Остаток сначала экстрагировали дихлорметаном (2×30 мл) и затем метанолом (5×75 мл). Оставалось светлое твердое вещество.
Экстракты метанола концентрировали в вакууме до сухости. Оставалось 1,20 г (3,26 ммоль, 54%) продукта в виде светлого твердого вещества.
Экстракты дихлорметана концентрировали в вакууме до сухости. Остаток ресуспендировали в метаноле (5 мл) и отстаивали. В осадок выпадало белое твердое вещество. Таким образом, получали другие 0,43 г (1,16 ммоль, 19%) продукта (Тпл. 175°С).
13С{1Н}-ЯМР (101 МГц, ДМСО-d6, δ част. на млн): 11.2 (1 С), 29.8 (2 С), 30.3 (2 С), 40.6 (2 С), 81.2(1 С, дн., J=2 Hz), 83.2 (1 С), 111.5 (1 С), 114.4 (1 С), 116.7 (1 С, дн., J=23 Hz), 117.6 (1 С, дн, J=21 Hz), 119.11 (1 С), 119.13 (1 С) 121.7 (1 С), 123.4 (1 С), 125.6 (1 С, J=3 Hz), 128.8 (1 С), 131.0 (1 С, дн., J=8 Hz), 132.2 (1 С, дн., J=7 Hz), 135.8 (1 С), 162.3 (1 С, дн., J=244 Hz)
Стадия 3:
[4-(диметил-амино)-1-(3-фторфенил)-4-(3-метил-1Н-индол-2-ил)-циклогексил]-диметил-амин (полярный диастереомер)
Титульное соединение из стадии 2 (500 мг, 1,36 ммоль) суспендировали в ацетонитрил/метаноле (1:1, 20 мл). Затем добавляли диметиламин (2М в тетрагидрофуране, 14 мл, 27,15 ммоль) и 2 дн. перемешивали при комнатной температуре. Раствор 6 ч перемешивали при 80°С (температура масляной бани), затем наносили на крупный силикагель и отделяли флеш-хроматографией [силикагель 60 (150 г); трихлорметан/этанол 50: 1 (1000 мл), 19:1 (500 мл), 9: 1 (1000 мл), в каждом случае 0,5% триэтиламин]. Сначала выделяли неполярный диастереоизомер. Кроме того, выделяли 250 мг смеси твердого вещества. Смесь твердого вещества растворяли в метаноле (10 мл), добавляли 50 мг гидроксида калия и 10 мин перемешивали. Летучие компоненты полностью удаляли в вакууме. Светлый остаток экстрагировали этиловым эфиром уксусной кислоты (3×20 мл). Экстракты освобождали в вакууме от летучих компонентов. Выделяли 135 мг (0,34 ммоль, 25%) полярного диастереоизомера (Тпл. 65-73°С).
13С{1Н}-ЯМР (101 МГц, ДМСО-d6, δ част. на млн, полярный диастереоизомер): 10.7 (1 С), 28.8 (2 С, br), 29.3 (2 С, br), 37.7 (2 С), 38.7 (2 С), 58.7 (1 С, br), 60.5 (1 C, br), 107.0 (1 С, br), 110.5 (1 С), 112.9 (1 С, дн., J=21 Hz), 113.7 (1 С, дн., J=21 Hz), 117.5 (1 C), 117.7 (1 C), 120.4 (1 C), 122.9 (1 C, br), 128.9 (1 С, дн., J=8 Hz), 129.0 (1 C), 132.5 (1 C, sbr), 134.5 (1 C), 141.4 (1 C, br), 161.9 (1 C, дн., J=243 Hz)
Пример 97:
[4-(диметил-амино)-1-(3-фторфенил)-4-(3-метил-1Н-индол-2-ил)-циклогексил]-диметил-амин (неполярный диастереомер)
Во время синтеза титульного соединения примера 94, стадия 3, также получался неполярный диастереомер. Выделяли 152 мг (0,39 ммоль, 29%) (Тпл. 126-132°С).
13С{1Н}-ЯМР (101 МГц, ДМСО-d6, δ част. на млн, неполярный диастереоизомер): 10.7 (1 С), 29.6 (2 С, br), 29.7 (2 C, br), 37.8 (2 С), 38.7 (2 C), 60.0 (1 C, br), 60.6 (1 C, br), 107.0 (1 C, br), 110.5 (1 C), 113.0 (1 С, дн., J=21 Hz), 114.2 (1 C, дн., J=21 Hz), 117.5 (1 C), 117.8 (1 C), 120.4 (1 C), 123.5 (1 C, br), 129.1 (1 C), 129.1 (1 C, дн., J=6 Hz), 132.2 (1 C, br), 134.6 (1 C), 140.4 (1 C, br), 162.2 (1 C, дн., J=242 Hz)
Следуя инструкции, как было описано примерах 94 и 97, за исключением того, что применяли исходные продукты, такие как приведены в таблице 1-5, получали следующие соединения.
Индолы:
скатол
5-фторо-3-метил-1Н-индол (IN-A)
Кетоны:
4-диметиламино-4-фенилциклогексанон (ВВ-А)
4-(диметиламино)-4-(3-фторофенил)циклогексанон (ВВ-В)
4-(диметиламино)-4-(тиофен-2-ил)циклогексанон (ВВ-С)
Следуя инструкции, как было описано в примере 86, за исключением того, что применяют амины и исходные продукты, как приведено в таблице 1-6, и в случае высококипящих аминов также обрабатывали без растворителя, получали следующие соединения.
Пример 100:
[4-[[4,6-бис(4-метокси-фенокси)-[1,3,5]-триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
Раствор титульного соединения примера 9 (616 мг, 2.0 ммоль) и 4-метоксифенилцианат (895 мг, 6.0 ммоль) в абс. ацетоне (20 мл) 3 дн. перемешивали при КТ. Затем растворитель удаляли в вак. и оставшийся остаток очищали флеш-хроматографией с этиловым эфиром уксусной кислоты/МеОН (20:1).
Выход: 1.16 г (92%)
1H-ЯМР (CDCl3): 1.77 (4Н, m); 1.89 (6H, s); 2.50 (4H, m); 3.07 (3H, s); 3.76 (6H, s); 6.84-7.36 (18H, m).
Пример 103:
[4-[[4,6-бис(4-метокси-фенокси)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
Раствор титульного соединения примера 8 (154 мг, 0.5 ммоль) и 4-метоксифенилцианата (224 мг, 1.5 ммоль) в абс. ацетоне (10 мл) 3 дн. перемешивали при КТ. Затем растворитель удаляли в вак. и оставшийся остаток очищали флеш-хроматографией с этилацетат/циклогексан (1:1).
Выход: 226 мг (72%)
1H-ЯМР (CDCl3): 1.80 (4H, m); 1.96 (6H, s); 2.28 (2H, m); 2.43 (2H, m); 3.04 (3H, s); 3.80 (6H, s); 6.89-7.40 (18H, m).
Следуя инструкции, как было описано в примере 24, за исключением того, что применяли реагенты ацилирования или сульфонилирования и амины, такие как приведены в таблице 1-7, получали следующие соединения.
Следуя инструкции, как было описано в примере 48, стадия 2, за исключением того, что применяли амиды, такие как приведены в таблице 1-8, получали следующие соединения.
ВВ-1:
N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-никотинамид
Раствор титульного соединения из примера 9 (308 мг, 1.0 ммоль) и триэтиламин (334 µл, 2.4 ммоль) в абс. ТГФ (15 мл) смешивали с хлоридом никотиновой кислоты гидрохлоридом (214 мг, 1.2 ммоль) и 3 дн. перемешивали при КТ. Затем растворитель удаляли в вак., оставшийся остаток растворяли в этилацетат, промывали нас. раствором NaHCO3 и нас. раствором NaCl, высушивали над Na2SO4 и очищали флеш-хроматографией с этиловым эфиром уксусной кислоты/МеОН 1:1.
Выход: 300 мг (73%), пористое твердое вещество
1H-ЯМР (ДМСО): 1.67 (2Н, m); 1.92 (2H, m); 1.98 (8H, s); 2.48 (2H, m); 2.80 (3H, s); 7.15-7.41 (10H, m); 7.52 (1H, m); 7.92 (1H, m); 8.69 (2H, m).
Пример 120:
2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-этанол (полярный диастереомер)
Стадия 1:
метиловый эфир [(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-уксусной кислоты
Титульное соединение из примера 9(463 мг, 1.50 ммоль) помещали в абс. ДМФ (10 мл) и смешивали с карбонатом калия (347 мг, 1.65 ммоль) и метиловым эфиром бромуксусной кислоты (157 µл, 1.65 ммоль). Состав 3 дн. перемешивали при комнатной температуре и затем концентрировали в вак. до сухости. Остаток ресуспендировали в дихлорметане (50 мл) и промывали водой (2×50 мл) и насыщенным раствором NaCl (50 мл), органическую фазу затем высушивали над Na2SO4 и концентрировали в вак. Остаток очищали с помощью флеш-хроматографии с этиловым эфиром уксусной кислоты/метанол (9:1).
Выход: 338 мг (59%)
1H-ЯМР (ДМСО-d6): 1.73 (4H, m); 1.96 (6H, s); 2.04 (3H, s); 2.31 (4H, m); 2.96 (2H, m); 3.58 (3H, s); 7.17 (2H, m); 7.28 (8H, m).
Стадия 2:
2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-этанол (полярный диастереомер)
Титульное соединение из стадии 1 (322 мг, 0.85 ммоль) растворяли в абс. ТГФ (15 мл), смешивали с LiAlH4 (64 мг, 1.69 ммоль) под аргоном и 3 ч кипятили в колбе с обратным холодильником. Затем состав охлаждали до комнатной температуры, при охлаждении льдом смешивали с ТГФ (10 мл) и H2O (5 мл) и 30 мин перемешивали. Состав отфильтровывали через фритту с кизельгуром, и кизельгур дополнительно промывали дихлорметаном (50 мл). Объединенные фильтраты концентрировали в вак. Сырой продукт смешивали с водой (10 мл) и экстрагировали дихлорметаном (3×20 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вак. Остаток очищали с помощью флеш-хроматографии с этиловым эфиром уксусной кислоты/метанол (1:1).
Выход: 213 мг (71%)
1H-ЯМР (ДМСО-d6): 1.72 (4H, m); 1.95 (6H, s); 2.06 (3H, s); 2.19 (2H, m); 2.29 (4H, m); 3.39 (2H, m); 4.25 (1H, m); 7.17 (2H, m); 7.27 (8H, m).
Пример 122:
2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-М-метил-ацетамид (полярный диастереомер)
Титульное соединение из примера 66 (293 мг, 0.8 ммоль) растворяли в абс. ДМФ (10 мл) и смешивали с N-гидроксибензотриазол гидрат (135 мг, 0.88 ммоль) и TEA (1.11 мл, 8.0 ммоль). Через 30 мин добавляли N-(3-диметиламинопропил)-N'-этил-карбодиимид гидрохлорид (460 мг, 2.4 ммоль) и метиламин (440 µл, 0.88 ммоль, 2М раствор в ТГФ) и в течение ночи перемешивали при КТ. Раствор фильтровали и концентрировали в вак. Посредством флеш-хроматографии остатка с этиловым эфиром уксусной кислоты/МеОН (4:1→1:1) получали соль продукта, который выделяли посредством 1N NaOH, экстрагировали с CH2Cl2, высушивали над Na2SO4 и в вак. освобождали от растворителя.
Выход: 182 мг (60%)
1H-ЯМР (ДМСО): 1.47 (2Н, m); 1.96 (7H, s); 1.99 (3H, s); 2.24 (3H, m); 2.42 (2H, m); 2.64 (6H, m); 7.24 (9H, m); 7.60 (1H, m).
Следуя инструкции, как было описано в примере 122, за исключением того, что применяли приведенные в таблице 1-9 кислоты и амины, получали следующие соединения.
Пример 127:
2-[[4-(диметил-амино)-1,4-дифенил-циклогексил]-метил-амино]-этанол (неполярный диастереомер)
Стадия 1:
метиловый эфир 4[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-уксусной кислоты
Титульное соединение из примера 8 (463 мг, 1.50 ммоль) помещали в абс. ДМФ (10 мл) и смешивали с карбонатом калия (347 мг, 1.65 ммоль) и метиловым эфиром бромуксусной кислоты (157 µл, 1.65 ммоль). Состав 3 дн. перемешивали при комнатной температуре и затем концентрировали в вак. до сухости. Остаток ресуспендировали в дихлорметане (50 мл) и промывали водой (2×50 мл) и насыщенным раствором NaCl (50 мл), затем органическую фазу высушивали над Na2SO4 и концентрировали в вак. Остаток очищали с помощью флеш-хроматографии с этиловым эфиром уксусной кислоты/метанол (9:1).
Выход: 234 мг (41%)
1H-ЯМР (ДМСО-d6): 1.72 (4Н, m); 1.84 (6H, s); 1.93 (3H, s); 2.27 (4H, m); 2.87 (2H, m); 3.48 (3H, s); 7.26 (2H, m); 7.38 (8H, m).
Стадия 2:
2-[[4-(диметил-амино)-1,4-дифенил-циклогексил]-метил-амино]-этанол (неполярный диастереомер)
Титульное соединение из стадии 1 (228 мг, 0.60 ммоль) растворяли в абс. ТГФ (10 мл), под аргоном смешивали с LiAlH4 (45 мг, 1.20 ммоль) и 3 ч кипятили в колбе с обратным холодильником. Затем состав охлаждали до комнатной температуры, при охлаждении льдом смешивали с ТГФ (10 мл) и H2O (5 мл) и 30 мин перемешивали. Состав отфильтровывали через фритту с кизельгуром, и кизельгур повторно промывали дихлорметаном (50 мл). Объединенные фильтраты концентрировали в вак. Сырой продукт смешивали с водой (10 мл) и экстрагировали дихлорметаном (3×20 мл), затем органическую фазу высушивали над Na2SO4 и концентрировали в вак. Остаток очищали с помощью флеш-хроматографии с этиловым эфиром уксусной кислоты/метанол (9:1→4:1).
Выход: 174 мг (82%)
Точка плавления: 144-149°С
1H-ЯМР (ДМСО-d6): 1.73 (4H, m); 1.84 (6H, s); 1.96 (3H, s); 2.09 (2H, m); 2.27 (4H, m); 3.23 (2H, m); 4.14 (1H, m); 7.25 (2H, m); 7.38 (8H, m).
Пример 128:
[4-[[4,6-бис(диметиламино)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
Стадия 1:
N-(4,6-дихлор-[1,3,5]триазин-2-ил)-N,N',N'-триметил-1,4-дифенил-циклогексан-1,4-диамин
Цианурхлорид (86 мг, 0.49 ммоль) помещали в абс. ТГФ (3 мл), смешивали с раствором титульного соединения из примера 8 (150 мг, 0.49 ммоль) в абс. ТГФ (6 мл) и N-этилдиизопропиламином (80 µл, 0.49 ммоль) и 16 ч перемешивали при КТ. Раствор концентрировали в вак., остаток ресуспендировали в этилацетате (20 мл) и промывали насыщенным раствором NaHCO3 (2×10 мл) и насыщенным раствором NaCl (10 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вак. Сырой продукт очищали путем флеш-хроматографии с этиловым эфиром уксусной кислоты/МеОН (20:1).
Выход: 67 мг (30%)
13С-ЯМР (CDCl3): 30.4, 31.4, 33.6, 38.0, 59.3, 66.4, 126.4, 126.7, 127.0, 127.1, 127.7, 128.2, 137.6, 143.1, 165.4, 168.0, 169.1
Стадия 2:
[4-[[4,6-бис(диметиламино)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
Титульное соединение из стадии 1 (57 мг, 0.12 ммоль) растворяли в 2М растворе диметиламина в ТГФ (2.0 мл, 4 ммоль) и при микроволнах 2 ч перемешивали при 120°С. Реакционный раствор концентрировали в вак., оставшийся остаток ресуспендировали в этилацетате (10 мл) и промывали насыщенным раствором NaHCO3 (2×5 мл) и насыщенным раствором NaCl (5 мл). Органическую фазу высушивали над Na2SO4 и концентрировали в вак. Сырой продукт очищали путем флеш-хроматографии с этиловым эфиром уксусной кислоты/МеОН (20:1).
Точка плавления: 195-197°С
Выход: 45 мг (76%)
1H-ЯМР (ДМСО-d6): 1.62 (2Н, m); 1.98 (6H, s); 2.39 (2H, m); 2.46 (2H, m); 2.91 (12H, s); 3.13 (3H, s); 7.15 (1H, m); 7.22-7.38 (9H, m).
Пример 130:
4-[[4-(диметил-амино)-1,4-дифенил-циклогексил]-метил-амино]-бутан-1-ол (полярный диастереомер)
Стадия 1:
Трет-бутиловый эфир N-(4-диметиламино-1,4-дифенил-циклогексил)-N-метил-янтарной кислоты
Титульное соединение из примера 131 (100 мг, 0.244 ммоль) помещали в абс. дихлорметан (5 мл), смешивали с ангидридом трифторуксусной кислоты (135 µл, 0.976 ммоль) и 10 мин перемешивали. К составу добавляли трет-бутанол (2 мл) и 30 мин перемешивали. Затем состав смешивали с 10%-ым NaOH и фазы разделяли. Органическую фазу промывали с помощью H2O (1×10 мл), высушивали над Na2SO4 и концентрировали в вак.
Выход: 80 мг (70%)
1H-ЯМР (ДМСО-d6): 1.38 (9H, s); 1.53 (2H, m); 1.78 (2H, m); 1.92 (6H, s); 2.37 (3H, m); 2.62 (2H, m); 2.93 (3H, s), 7.11-7.27 (6H, m); 7.36 (4H, m).
Стадия 2:
4-[[4-(диметил-амино)-1,4-дифенил-циклогексил]-метил-амино]-бутан-1-ол (полярный диастереомер)
Титульное соединение из стадии 1 (836 мг, 1.8 ммоль) растворяли в абс. ТГФ (15 мл). Под аргоном добавляли LiAlH4 (136 мг, 3.6 ммоль), 2 ч кипятили в колбе с обратным холодильником, охлаждали до комнатной температуры и перемешивали в течение ночи. При охлаждении льдом к составу добавляли ТГФ (2 мл) и H2O (2 мл) и перемешивали 30 мин. Состав фильтровали через фритту с кизельгуром, кизельгур промывали дихлорметаном (50 мл), органические фазы объединяли и концентрировали в вак. Остаток очищали посредством флеш-хроматографии хлороформом/метанол (9:1).
Выход: 405 мг (59%)
1H-ЯМР (ДМСО-d6): 1.39 (4Н, m); 1.74 (3H, m); 1.96 (6H, s); 2.01 (3H, s); 2.11 (2H, m); 2.30 (3H, m); 3.36 (2H, m); 4.41 (1H, m); 7.18 (2H, m); 7.28 (8H, m).
Пример 131:
3-[[4-(диметил-амино)-1,4-дифенил-циклогексил]-метил-карбамоил]-пропионовая кислота (полярный диастереомер)
Ангидрид янтарной кислоты (0.97 г, 9.27 ммоль) нагревали до 130°С и расплавляли. Затем добавляли титульное соединение из примера 9 (1.00 г, 3.24 ммоль) и 7 ч далее нагревали при этой температуре. Состав очищали путем флеш-хроматографии хлороформом/метанол (9:1→4:1→1:1→1:2→метанол).
Выход: 1.08 г (81%)
1H-ЯМР (ДМСО-d6): 1.55 (2H, m); 1.81 (2H, m); 1.94 (6H, s); 2.37 (4H, m); 2.62 (2H, m); 2.76 (1H, m); 2.94 (3H, s); 7.14 (3H, m); 7.17 (2H, m); 7.26 (1H, m); 7.38 (4H, m).
Пример 146:
[4-[[4-(4-метокси-фенокси)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
Стадия 1:
N-(4-хлор-[1,3,5]триазин-2-ил)-N,N',N'-триметил-1,4-дифенил-циклогексан-1,4-диамин
Раствор титульного соединения из примера 9 (462 мг, 1.5 ммоль), 2,4-дихлор-1,3,5-триазин (225 мг, 1.5 ммоль) и диизопропилэтиламин (248 µл, 1.5 ммоль) в абс. ТГФ (10 мл) перемешивали в течение ночи при КТ. Затем растворитель удаляли в вак., оставшийся остаток растворяли в этилацетате, промывали нас. раствором NaHCO3 и нас. раствором NaCl, высушивали над Na2SO4 и очищали флеш-хроматографией с этиловым эфиром уксусной кислоты/МеОН (9:1).
Выход: 166 мг (26%)
1H-ЯМР (CDCl3): 1.97 (4Н, m); 2.06 (6H, s); 2.47 (4H, bs); 3.01 (2Н, шир.); 3.34 (3H, s); 7.14-7.40 (10H, m); 8.29 (1H, s).
Стадия 2:
[4-[[4-(4-метокси-фенокси)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
Титульное соединение стадии 1 (166 мг, 0.39 ммоль), 4-метоксифенол (56 мг, 0.45 ммоль) и гидрид натрия (18 мг, 0.45 ммоль, 60%-ая дисперсия в минеральном масле) в абс. диоксане (10 мл) 4 ч перемешивали при КТ. Затем растворитель удаляли в вак., оставшийся остаток растворяли в этилацетате, промывали нас. раствором NaHCO3 и нас. раствором NaCl, высушивали над Na2SO4 и очищали флеш-хроматографией с этиловым эфиром уксусной кислоты/МеОН (4:1).
Выход: 126 мг (63%), пористое твердое вещество
1H-ЯМР (CDCl3): 1.84 (4Н, m); 2.03 (6H, s); 2.60 (4H, шир.); 3.23 (3H, s); 3.80 (3H, s); 6.87-7.38 (14H, m); 8.37 (1H, s).
Пример 149:
[4-[(бензил-метил-амино)-метил]-1,4-дифенил-циклогексил]-диметил-амин
Стадия 1:
Диметиловый эфир 4-циано-4-фенил-гептандикарбоновой кислоты
Фенилацетонитрил (11.7 г, 100 ммоль) и метилакрилат (47 мл, 500 ммоль) помещали в трет-бутанол (60 мл) и нагревали до кипения. Затем удаляли источник нагревания. Растворенный в трет.-бутаноле (23 мл) тритон В (гидроксид бензилтриметиламмония, 40%-ый в метаноле, 15.2 мл) впускали по каплям сначала медленно, затем моментально. После прикапывания состав 4 ч нагревали до кипения. В течение ночи реакционную смесь охлаждали до комнатной температуры. Для переработки состав смешивали с толуолом (100 мл) и водой (70 мл), органическую фазу отделяли и промывали водой (70 мл) и насыщенным раствором NaCl (50 мл). После высушивания с помощью Na2SO4 растворитель отгоняли. Очистка осуществлялась посредством перегонки в вакууме (Kugelrohr) при температуре от прибл. 235°С. Продукт можно было выделить в виде бесцветного, вязкотекучего вещества.
Выход: 22. 5 г (75%)
1H-ЯМР (ДМСО-d6): 2.32 (8H, m); 3.51 (6H; s); 7.40 (5H, m).
13С-ЯМР (ДМСО-d6): 22.47; 27.16; 39.28; 44.11; 113.82; 118.55; 120.83; 121.78; 129.10; 164.44.
Стадия 2:
Метиловый эфир 5-циано-2-оксо-5-фенил-циклогексанкарбоновой кислоты
Диметиловый эфир 4-циано-4-фенилгептандикарбоновой кислоты (19.8 г, 68 ммоль) растворяли в сухом тетра гидрофура не (480 мл). Затем порционно добавляли трет.-бутилат калия (13.2 г, 120 ммоль). При этом добавлении реакционная смесь окрасилась в оранжевый цвет. После этого состав 5 ч кипятили в колбе с обратным холодильником. Во время кипячения образовывался коричневый раствор. В течение ночи реакционную смесь охлаждали до комнатной температуры. При охлаждении льдом к реакционной смеси медленно впускали по каплям 2.5N уксусную кислоту (230 мл). Затем состав смешивали с толуолом (100 мл), органическую фазу отделяли и промывали насыщенным раствором NaHCO3 (3×100 мл), Н2О (3×50 мл) и раствором NaCl (1×100 мл). После высушивания с помощью Na2SO4 растворитель отгоняли в вак. Оставалось желтоватое твердое вещество.
Выход: 16.1 г (92%)
точка плавления: 75-77°С
1H-ЯМР (ДМСО-d6): 2.23-2.74 (6Н, m); 3.74 (3H; s); 7.35-7.60 (5H, m); 12.08 (1H, bs).
13С-ЯМР (ДМСО-d6): 26.95; 30.18; 34.04; 51.90; 94.79; 121.90; 125.46; 128.05; 128.85; 138.92; 169.95; 171.09.
Стадия 3:
4-оксо-1-фенил-циклогексанкарбонитрил
Метиловый эфир 5-циано-2-оксо-5-фенилциклогексанкарбоновой кислоты (16.1 г, 63 ммоль) растворяли в 10%-ой серной кислоте (218 мл) и конц. уксусной кислоте (502 мл) и 21 ч перемешивали при 100°С.
Для переработки состав при охлаждении льдом осторожно разбавляли с водой (400 мл), экстрагировали этилацетатом (3×100 мл), органическую фазу тщательно промывали водой (6×100 мл), насыщенным раствором NaHCO3 (10×100 мл) и насыщенным раствором NaCl (1×100 мл). После высушивания с помощью Na2SO4 растворитель отгоняли в вак.
Выход: 8.91 г (72%)
Точка плавления: 106-107°С
1H-ЯМР (ДМСО-d6): 2.38-2.48 (6H, m); 2.70 (2H; m); 7.36 (1H, m); 7.44 (2H, m); 7.62 (2H, m).
13С-ЯМР (ДМСО-d6): 35.31; 38.10; 42.33; 121.73; 125.65; 128.19; 129.02; 139.17; 208.79.
Стадия 4:
8-фенил-1,4-диокса-спиро[4.5]декан-8-карбонитрил
Титульное соединение из стадии 3 (8.91 г, 44.73 ммоль) ресуспендировали в толуоле (300 мл) и смешивали с этиленгликолем (6 мл, 106.8 ммоль). После добавления р-толуолсульфокислоты (0.128 г, 0.745 ммоль) состав 3.5 ч нагревали в водоотделителе до кипения. Протекание реакции наблюдали посредством ТХ.
После охлаждения реакционного состава толуольный раствор экстрагировали водой (5×60 мл) и насыщенным раствором NaCl (3×40 мл) и высушивали над Na2SO4. После удаления растворителя в вак. Получали кеталь в виде желтого твердого вещества.
Выход: 11.6 г (100%)
Точка плавления: 108-110°С
1H-ЯМР (ДМСО-d6): 1.86 (4Н, m); 2.01-2.30 (4H; m); 3.92 (4H, s); 7.38-7.53 (5H, m).
13С-ЯМР (ДМСО-d6): 32.10; 34.07; 42.49; 63.86: 106.11; 122.14; 125.51; 128.16; 129.02; 139.90.
Стадия 5:
8-фенил-1,4-диокса-спиро[4.5]декан-8-карбоновой кислоты
Титульное соединение из стадии 4 (10.9 г, 46.9 ммоль) растворяли в этиленгликоле (92 мл), смешивали с NaOH (4.00 г, 100 ммоль) и затем нагревали в колбе с обратным холодильником до кипения. Через 20 ч уже не обнаруживался нитрил. Для переработки состав смешивали со льдом (прибл. 250 г), покрывали простым эфиром (90 мл) и подкисляли посредством медленного добавления полуконц. HCl (118 мл). Водную фазу экстрагировали простым эфиром (3×70 мл), объединенные органические экстракты промывали насыщенным раствором NH4Cl (2×70 мл), высушивали над Na2SO4 и концентрировали до сухости в вак. Путем перекристаллизации оставшегося осадка из толуола выделяли желаемую карбоновую кислоту в виде кристаллического твердого вещества.
Выход: 7.42 г (59%)
Точка плавления: 134-139°С
1H-ЯМР (ДМСО-d6): 1.64 (4H, m); 1.91 (2H; m); 2.41 (2H, m); 3.86 (4H, s); 7.36 (5H, m); 12.52 (1H, bs).
13С-ЯМР (ДМСО-d6): 31.51; 32.05; 49.19; 63.65: 107.23; 125.70; 126.94; 128.39; 142.82; 175.53.
Стадия 6:
8-фенил-1,4-диокса-спиро[4.5]декан-8-карбоновой кислоты бензил-метил-амид
Титульное соединение из стадии 5 (8.00 г, 30.48 ммоль) растворяли в дихлорметане (240 мл) и при 0°С смешивали с 1,3-диизопропилкарбодиимидои (4.44 г, 5.44 мл, 35.52 ммоль) и 1-гидрокси-1H-бензотриазолом гидрат (5.44 г, 35.5 ммоль). Реакционный состав перемешивали 5 мин при охлаждении льдом и затем добавляли N-бензилметиламин (3.87 г, 4.12 мл, 32.0 ммоль). Реакционную смесь 3 дн. перемешивали при комнатной температуре. Для переработки состав концентрировали в вак. до сухости. Остаток очищали путем флеш-хроматографии с циклогексан/этилацетатом (1:1).
Выход: 7.31 г (66%)
1H-ЯМР (ДМСО-d6): 1.61 (4Н, m); 1.68 (4H, m); 2.35 (3H, m); 3.85 (6H, s); 7.28 (10H, br, m).
Стадия 7:
Бензил-метил-(8-фенил-1,4-диокса-спиро[4.5]дец-8-илметил)-амин
Титульное соединение из стадии 6 (1.20 г, 3.28 ммоль) растворяли в абс. тетрагидрофуране (160 мл), LiAlH4 (0.25 г, 6.59 ммоль) под аргоном добавляли и 5 ч перемешивали при кипячении в колбе с обратным холодильником. Затем состав охлаждали до комнатной температуры и перемешивали в течение ночи. При охлаждении льдом состав гидролизовали с ТГФ (20 мл) и H2O (20 мл) и 30 мин перемешивали. Состав отфильтровывали через фритту с кизельгуром, повторно промывали с ТГФ и дихлорметаном (50 мл) и концентрировали в вак. Остаток очищали с помощью флеш-хроматографии и циклогексан/этилацетатом (1:1).
Выход: 0.50 г (43%)
1H-ЯМР (ДМСО-d6): 1.35 (2H, m); 1.38 (2H, m); 1.72 (5 H, m); 2.20 (2H, d); 2.48 (2H, m); 3.22 (2H, s); 3.84 (4H, m); 7.25 (8H, m), 7.44 (2H d).
Стадия 8:
4-[(бензил-метил-амино)-метил]-4-фенил-циклогексанон
Титульное соединение из стадии 7 (3.40 г, 9.67 ммоль) смешивали с 5%-ой серной кислотой (300 мл) и 48 ч при комнатной температуре перемешивали. Для переработки реакционный состав смешивали с простым эфиром (100 мл), фазы разделяли и водную фазу экстрагировали простым эфиром (2×100 мл). Затем водную фазу подщелачивали посредством 5N NaOH и экстрагировали дихлорметаном (3×100 мл). Органическую фазу высушивали над Na2SO4, отфильтровывали и в вак. концентрировали до сухости.
Выход: 2.74 г (92%)
1H-ЯМР (ДМСО-d6): 1.79 (3Н, s); 2.07 (2Н, m); 2.16 (5H, m); 2.22 (1H, m); 3.26 (2H, s); 7.22 (6H, m); 7.37 (2H, t), 7.55 (2H, d).
Стадия 9:
4-[(бензил-метил-амино)-метил]-1-метиламино-4-фенил-циклогексанкарбонитрил
К охлажденному до 0°С раствору 4N соляной кислоты (2.33 мл) и метанола (1.40 мл) добавляли 40%-ый водный раствор метиламина (5.40 мл, 42.7 ммоль) и титульное соединение из стадии 8 (2.74 г, 8.91 ммоль), растворенное в метаноле (10 мл). Затем реакционную смесь смешивали с цианидом калия (1.40 г, 21.1 ммоль) и 1 дн. при комнатной температуре перемешивали. Для переработки смесь смешивали с водой (30 мл) и простым эфиром (3×50 мл) экстрагировали. Объединенные органические фазы высушивали с помощью Na2SO4, отфильтровывали и концентрировали в вак.
Выход: 2.69 г (90%)
1H-ЯМР (ДМСО-d6): 1.11 (2Н, m); 1.68 (1Н, m); 1.72 (2H, m); 1.78 (1H, m); 1.86 (2H, s); 1.92 (2H, m); 2.22 (2H, d); 2.28 (1H, m); 2.38 (2H, m); 2.67 (1H, m); 3.17 (1H, m); 3.29 (2H, m); 7.25 (10H, m).
Стадия 10:
{4-[(бензил-метил-амино)-метил]-1,4-дифенил-циклогексил}-метил-амин
Фениллитий (12.9 мл, 23.2 ммоль, 1.8 М в дибутиловом эфире) помещали под аргоном, по каплям смешивали с титульным соединением из стадии 9 (2.69 г, 7.74 ммоль) в ТГФ (15 мл) и реакционный раствор 1 ч перемешивали при кипячении в колбе с обратным холодильником. При охлаждении в ледяной ванне реакционный состав гидролизовали насыщенным раствором NH4Cl (27 мл) и фазы разделяли. Водную фазу экстрагировали простым эфиром (3×50 мл). Объединенные органические фазы высушивали над Na2SO4, отфильтровывали и концентрировали в вак. до сухости. Остаток отделяли с помощью хроматотрона и дихлорметан → дихлорметан/метанол (9:1) → метанол. Выделяли 1.20 г кетона. Целевой продукт получали в виде смеси диастереомера и как таковой подвергали дальнейшему взаимодействию.
Выход: 0.360 г (12%)
1H-ЯМР (ДМСО-d6): 1.75 (1Н, m); 1.79 (3H, s); 1.92 (1H, m); 2.02 (3H, m); 2.17 (6H, m); 2.46 (1H, m); 2.61 (2H, m); 7.25 (13H, m); 7.54 (2H, m).
Стадия 11:
[4-[(бензил-метил-амино)-метил]-1,4-дифенил-циклогексил]-диметил-амин
Раствор титульного соединения из стадии 10 (смесь диастереомеров) (0.350 г, 0.878 ммоль) и формалина (1.23 мл, 37%-ый водный раствор) в ацетонитриле (15 мл) порционно смешивали с цианоборгидридом натрия (0.250 г, 3.86 ммоль) и 45 мин перемешивали при комнатной температуре. Затем добавляли конц. уксусную кислоту до нейтральной реакции и 45 мин перемешивали при комнатной температуре. Для переработки растворитель удаляли в вак., остаток ресуспендировали в 2N NaOH (40 мл) и затем экстрагировали простым эфиром (3×40 мл). Органический раствор высушивали над Na2SO4, отфильтровывали и концентрировали в вак. Оставшийся остаток очищали с помощью хроматотрона и с циклогексан/этилацетатом 1:1. Невозможно было добиться разделения диастереомеров.
Выход: 70 мг (19%)
1H-ЯМР (ДМСО-d6): 1.60 (4H, m); 1.72 (3H, s); 1.82 (6H, s); 2.14 (2H, m); 2.49 (4H, s); 3.19 (2H, s); 6.93 (2H, m); 7.21 (5H, m); 7.40 (8H, m).
Пример 153:
[4-[[4-(бензиламино)-[1,3,5]триазин-2-ил]-метил-амино]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
Титульное соединение из примера 146, стадия 1 (100 мг, 0.236 ммоль), бензиламин (55 µл, 0.5 ммоль) и диизопропилэтиламин (50 µл, 0.3 ммоль), растворенный в абс. ТГФ (2.0 мл), при 70°С перемешивали в закрытом сосуде 5 ч. Затем растворитель удаляли в вак., оставшийся остаток растворяли в дихлорметане, промывали нас. раствором NaHCO3, высушивали над Na2SO4 и очищали флеш-хроматографией с этиловым эфиром уксусной кислоты/МеОН (4:1→1:1). Продукт еще содержал бензиламин, который удаляли в вак. при 90°С.
Выход: 82 мг (70%), масло
1H-ЯМР (CDCl3): 1.77 (4Н, m); 2.02 (6H, s); 2.37 (2H, m); 2.97 (2Н, шир.); 3.28 (3Н, s); 4.38 (2H, s); 6.01 (1H, s); 7.12-7.40 (15H, m); 8.00 (1H, s).
Следуя инструкции, как было описано в примере 153, за исключением того, что применяли приведенные в таблице 1-10 амины, получали следующие соединения.
Пример 163:
[4-[(бутил-метил-амино)-метил]-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
Стадия 1:
8-фенил-1,4-диокса-спиро[4.5]декан-8-карбоновой кислоты - бутил-метиламид
Титульное соединение из примера 149, стадия 5 (6.50 г, 24.8 ммоль) растворяли в дихлорметане (200 мл) и при 0°С смешивали с диизопропилкарбодиимидом (3.60 г, 4.41 мл, 28.8 ммоль) и 1-гидрокси-1Н-бензотриазолом гидрат (4.41 г, 28.8 ммоль). Реакционный состав 5 мин перемешивали при охлаждении льдом и затем добавляли N-метилбутиламин (2.34 г, 3.08 мл, 26.0 ммоль). Реакционную смесь 2 дн. перемешивали при комнатной температуре. Для переработки состав концентрировали в вак. до сухости. Остаток очищали путем флеш-хроматографии с циклогексан/этилацетатом (2:1). Выход: 3.50 г (43%)
1H-ЯМР (ДМСО-d6): 0.95 (6Н, m); 1.39 (2H, s); 1.80 (2H, m); 1.85 (6H, m); 2.24 (2H, m); 2.51 (1H, m); 3.10 (1H, br m).); 3.84 (4H, s); 7.23 (3H, m); 7.34 (2H, m).
Стадия 2:
Бутил-метил-(8-фенил-1,4-диокса-спиро[4.5]дец-8-илметил)-амин
Титульное соединение из стадии 1 (3.50 г, 10.6 ммоль) растворяли в абс. тетрагидрофуране (400 мл), под аргоном добавляли LiAlH4 (0.66 г, 17.5 ммоль) и 5 ч перемешивали при кипячении в колбе с обратным холодильником. Затем состав охлаждали до комнатной температуры и перемешивали в течение ночи. При охлаждении льдом состав гидролизовали с ТГФ (20 мл) и H2O (20 мл) и 30 мин перемешивали. Состав отфильтровывали через фритту с кизельгуром, повторно промывали с ТГФ и дихлорметаном (50 мл) и концентрировали в вак. Остаток очищали с помощью флеш-хроматографии и циклогексан/этилацетат (9:1→1:1).
Выход: 2.50 г (76%)
1H-ЯМР (ДМСО-d6): 0.77 (3Н, t); 1.19 (6H, m); 1.52 (2H, m); 1.77 (2H, m); 1.83 (3H, s); 2.05 (2H, m); 2.18 (2H, m); 2.31 (2H, s); 3.84 (4H, br m); 7.19 (1H, m); 7.33 (4H, m).
Стадия 3:
4-[(бутил-метил-амино)-метил]-4-фенил-циклогексанон
Титульное соединение из стадии 2 (2.50 г, 7.8 ммоль) смешивали с 5%-ой серной кислотой (300 мл) и 48 ч перемешивали при комнатной температуре. Для переработки реакционный состав смешивали с простым эфиром (100 мл), фазы разделяли и водную фазу экстрагировали простым эфиром (2×100 мл). Затем водную фазу подщелачивали посредством 5N NaOH и экстрагировали дихлорметаном (3×100 мл). Органическую фазу высушивали над Na2SO4, отфильтровывали и в вак. концентрировали до сухости.
Выход: 1.53 г (73%)
1H-ЯМР (ДМСО-d6): 0.78 (3H, t); 1.15 (4H, br, m); 1.87 (3H, s); 1.93 (2H, m); 2.13 (6H, br m); 2.45 (4H, m); 7.25 (1H, t); 7.37 (2H, t); 7.49 (2H, d).
Стадия 4:
4-[(бутил-метил-амино)-метил]-1-метиламино-4-фенил-циклогексанкарбонитрил
К охлажденному до 0°С раствору 4N соляной кислоты (1.50 мл) и метанола (0.89 мл) добавляли 40%-ый водный раствор метиламина (3.42 мл, 27 ммоль) и титульное соединение из стадии 3 (1.54 г, 5.60 ммоль), растворенное в метаноле (5 мл). Затем реакционную смесь смешивали с цианидом калия (0.901 г, 13.4 ммоль) и 3 дн. перемешивали при комнатной температуре. Для переработки смесь смешивали с водой (50 мл) и экстрагировали простым эфиром (3×100 мл). Объединенные органические фазы высушивали посредством Na2SO4, отфильтровывали и концентрировали в вак.
Выход: 1.76 г (100%)
1H-ЯМР (ДМСО-d6): 0.77 (3Н, m); 1.07 (5H, m); 1.68 (3H, m); 1.77 (1H, s); 1.84 (1H, m); 1.92 (2H, m); 2.03 (1H, m); 2.12 (2H, m); 2.21 (2H, m); 2.31 (3H, m); 2.43 (1H, m); 2.63 (1H, m); 7.19 (1H, m); 7.37 (4H, m).
Стадия 5:
{4-[(бутил-метил-амино)-метил]-1,4-дифенил-циклогексил}-метил-амин
Фениллитий (9.33 мл, 16.8 ммоль, 1.8 M в дибутиловом эфире) помещали под аргоном, по каплям смешивали с титульным соединением из стадии 4 (1.76 г, 5.61 ммоль) в простом эфире (15 мл) и реакционный раствор 1 ч перемешивали при 50°С. При охлаждении в ледяной ванне реакционный состав гидролизовали с насыщенным раствором NH4Cl (100 мл) и фазы разделяли. Водную фазу экстрагировали простым эфиром (3×50 мл). Объединенные органические фазы высушивали над Na2SO4, отфильтровывали и в вак. концентрировали до сухости. Остаток отделяли с помощью хроматотрона и дихлорметана.
Выход: 0.400 г (20%), неполярный диастереомер
1H-ЯМР (ДМСО-d6): 0.71 (3H, t); 1.05 (5H, m); 1.59 (3H, m); 1.76 (6H, s); 2.01 (6H, m); 2.40 (2H, br s); 7.19 (2H, m); 7.34 (6 H, m); 7.47 (2H, d).
Выход: 0.170 г (9%), полярный диастереомер
1H-ЯМР (ДМСО-d6): 0.76 (3H, t); 1.13 (4H, m); 1.37 (2H, m); 1.75 (4H, s); 1.86 (3H, m); 2.06 (6H, m); 2.41 (2H, s); 3.17 (1H, s); 7.13 (2H, m); 7.26 (6H, m); 7.38 (2H, m).
Стадия 6:
[4-[(бутил-метил-амино)-метил]-1,4-дифенил-циклогексил]-диметил-амин (неполярный диастереомер)
Раствор титульного соединения из стадии 6 (неполярный диастереомер) (0.400 г, 1.1 ммоль) и формалина (1.54 мл, 37%-ый водный раствор) в ацетонитриле (20 мл) порционно смешивали с цианоборгидридом натрия (0.313 г, 4.84 ммоль) и 45 мин перемешивали при комнатной температуре. Затем добавляли конц. уксусную кислоту до нейтральной реакции и 45 мин перемешивали при комнатной температуре. Для переработки растворитель удаляли в вак., остаток ресуспендировали в 2N NaOH (40 мл) и затем экстрагировали простым эфиром (3×40 мл). Органическую фазу высушивали над Na2SO4, отфильтровывали и концентрировали в вак.
Остаток очищали с помощью хроматотрона и дихлорметан → метанол.
Выход: 220 мг (53%)
1H-ЯМР (ДМСО-d6): 0.70 (3Н, t); 0.99 (4H, m); 1.42 (2H, m); 1.58 (2H, m); 1.75 (3H, s); 1.86 (6H, s); 1.95 (2H, m); 2.16 (4H, m); 2.32 (2H, m); 7.18 (1H, m); 7.38 (9H, m).
Пример 164:
[4-[(бутил-метил-амино)-метил]-1,4-дифенил-циклогексил]-диметил-амин (полярный диастереомер)
Раствор титульного соединения из примера 163, стадия 6 (полярный диастереомер) (0.170 г, 0.47 ммоль) и формалина (0.66 мл, 37%-ый водный раствор) в ацетонитриле (8.2 мл) порционно смешивали с цианоборгидридом натрия (0.134 г, 2.07 ммоль) и 45 мин перемешивали при комнатной температуре. Затем добавляли конц. уксусную кислоту до нейтральной реакции и 45 мин перемешивали при комнатной температуре. Для переработки растворитель удаляли в вак., остаток ресуспендировали в 2N NaOH (40 мл) и затем экстрагировали простым эфиром (3×40 мл). Органическую фазу высушивали над Na2SO4, отфильтровывали и концентрировали в вак. Остаток очищали с помощью хроматотрона и дихлорметан → метанол.
Выход: 75 мг (41%)
1H-ЯМР (ДМСО-d6): 0.77 (3Н, t); 1.18 (5H, m); 1.51 (2H, m); 1.76 (5H, m); 1.94 (6H, s); 2.04 (3H, m); 2.27 (2H, m); 2.40 (2H, s); 7.23 (10H, m).
Нефелометрическое исследование растворимости (фосфатный буфер рН 7.4):
Этот метод исследует растворимость вещества при установленных концентрациях (1 µM, 3 µМ, 10 µМ, 30 µМ и 100 µM) в 10 мМ растворе фосфатного буфера при рН 7.4.
Первоначально необходим 10 мМ раствор веществ в ДМСО, из которого получают 100-кратные основные растворы указанных выше уровней концентрации снова в ДМСО, конечная ДМСО концентрация в исследуемом составе составляет 1% (v/v). Эксперимент проводят с многократным определением. После добавления ДМСО основных растворов к буферу, состав 2 ч инкубируют при 37°С, прежде чем происходит определение абсорбции при 620 нм. Если абсорбция образцов повышается выше чистого раствора буфер/ДМСО, то это является индикатором образования осадка. Нижняя граница растворимости («lower bound») представляет собой концентрацию, которая предшествует той с первым образованием осадка (например, 3 µМ, если образование осадка было обнаружено при 10 µМ).
Исследования эффективности соединений согласно изобретению
Измерение ORL1-связывания
Соединения были исследованы в анализе связывания рецепторов с3H-ноцицептин/орфанин FQ с мембранами рекомбинантных CHO-ORL1 клеток. Эта тестовая система была проведена согласно методу, представленному в Ardati et al. (Mol. Pharmacol., 51, 1997, сс.816-824). Концентрация3H-ноцицептин/орфанин FQ при этом исследовании составила 0.5 нМ. Анализы связывания были проведены с помощью по 20 µг мембранного протеина по 200 µл состава в 50 мМ хепеса, рН 7,4,10 мМ MgCl2 и 1 мМ EDTA. Связывание с ORL1-рецептором определяли с применением по 1 мг WGA-SPA микрошариков (Beads) (Amersham-Pharmacia, Фрайбург), путем одночасовой инкубации состава при комнатной температуре и последующего измерения в сцинтилляционном счетчике Trilux (Wallac, Финляндия). Сродство указано в таблице 1 как наномолярное Ki-значение в или % ингибирования при с=1 µМ.
Измерение µ-связывания
Сродство рецепторов к человеческому µ-опиатному рецептору определяли в гомогенном составе в микротитрационных планшетах. Для этого в течение 90 минут при комнатной температуре инкубировали разбавленные серии в каждом случае проверяемого соединения с рецепторным мембранным препаратом (15-40 µг протеина на 250 µл инкубационного состава) СНО-К1-клеток, которые выражают человеческий µ-опиатный рецептор (RB-HOM-рецепторный мембранный препарат фирмы NEN, Zaventem, Бельгия) в присутствии 1 нмоль/л радиоактивного лиганда [3H]-налоксон (NET719, фирма NEN, Zaventem, Бельгия), а также 1 мг WGA-SPA-Beads (Wheat germ agglutinin SPA Beads фирмы Amersham/Phamnacia, Фрайбург, Германия) в общем объеме 250 µл.
В качестве инкубационного буфера применяли 50 ммоль/л трис-HCl, дополненный 0,05 мас.% азидом натрия и 0,06 мас.% альбумином сыворотки крупного рогатого скота. Для определения неспецифического связывания дополнительно добавляли 25 µмоль/л налоксона. После окончания девяностоминутного инкубационного периода микротитрационные планшеты в течение 20 минут при 1000 г отделяли центрифугированием и измеряли радиоактивность в β-счетчике (Microbeta-Trilux, фирма PerkinElmer Wallac, Фрайбург, Германия). Определяли процентное вытеснение радиоактивного лиганда из его связывания к человеческому µ-опиатному рецептору при концентрации контрольного вещества в 1 µмоль/л и указывали как процентное ингибирование (% ингибирования) специфического связывания. Исходя из процентного вытеснения, благодаря различным концентрациям контрольных соединений общей формулы I IC50, частично рассчитывали ингибирующие концентрации, которые способствовали 50-процентному вытеснению радиоактивного лиганда. Путем пересчета с помощью соотношения Чена-Прусоффа (Cheng-Prusoff) получали Ki-значения для контрольных веществ. В некоторых случаях отказывались от определения Ki-значения и определяли только ингибирование в тестовой концентрации 1 µМ.
Определение каппа-связывания
Определение происходило в гомогенном составе в микротитрационных планшетах. Для этого в течение 90 минут при комнатной температуре инкубировали разбавленные серии в каждом случае проверяемых соединений с рецепторным мембранным препаратом (7 µг протеина на 250 µл инкубационного состава) СНО-К1-клеток, которые выражают человеческий κ-опиатный рецептор в присутствии 1 нмоль/л радиоактивного лиганда [3H]-Cl-977, а также 1 мг WGA-SPA-Beads (Wheat germ agglutinin SPA Beads фирмы Amersham/Pharmacia, Фрайбург, Германия) в общем объеме 250 µл.
В качестве инкубационного буфера применяли 50 ммоль/л трис-HCl, дополненный 0,05 мас.% азидом натрия и 0,06 мас.% альбумином сыворотки крупного рогатого скота. Для определения неспецифического связывания дополнительно добавляли 100 µмоль/л налоксона. После окончания девяностоминутного инкубационного периода микротитрационные планшеты в течение 20 минут при 500 об/мин отделяли центрифугированием и измеряли радиоактивность в β-счетчике (Microbeta-Trilux 1450, фирма PerkinEImer Wallac, Фрайбург, Германия). Определяли процентное вытеснение радиоактивного лиганда из его связывания к человеческому κ-опиатному рецептору при концентрации контрольного вещества в 1 µмоль/л и указывали как процентное ингибирование (% ингибирования) специфического связывания. Исходя из процентного вытеснения, благодаря различным концентрациям контрольных соединений можно было рассчитать IC50 ингибирующие концентрации, которые способствовали 50-процентному вытеснению радиоактивного лиганда. Путем пересчета с помощью соотношения Чена-Прусоффа (Cheng-Prusoff) получали Ki-значения для контрольных веществ. Результаты приведены в нижеследующей таблице:
Исследование фармакологических свойств примерных соединений
Модель Чанга (Chung): боль при мононевропатии после спинальной лигатуры нервов
Животные: самцов крыс Sprague Dawley (140-160 г), от коммерческого животновода (Janvier, Genest St. Isle, Франция), удерживали при 12:12 ч ритме свет-темнота. По желанию животных снабжали кормом и водопроводной водой. Между доставкой животных и операцией выдержали интервал в одну неделю. После операции животных подвергали многократному тестированию в течение промежутка времени в 4-5 недель, причем было выдержано время вымывания, по меньшей мере, в одну неделю.
Описание модели: под наркозом пентобарбитала (Narcoren®, 60 мг/кг i.p., Merial GmbH, Hallbergmoos, Германия), экспонировали левые L5, L6 спинальные нервы, в то время как были удалены кусок паравертебральной мышцы и часть левого спинального процесса L5 тела поясничного позвонка. Спинальные нервы L5 и L6 были осторожно изолированы и перевязаны прочной лигатурой (NC-silk black,USP 5/0, metric 1, Braun Melsungen AG, Melsungen, Германия) (Kim and Chung 1992). После лигатуры мышцу и соседние ткани зашили и рану закрыли с помощью металлических кламмеров.
После периода отдыха в одну неделю животных поместили в клетки с проволочным дном для того, чтобы измерять механическую аллодинию. На ипсилатеральной и/или контралатеральной задней лапе каждый раз определяли порог отдергивания с помощью электронного филамента Фрея (Somedic AB, Malmö, Швеция). Срединное значение пяти стимуляций дало точку данных. Животных подвергали испытанию за 30 мин и в различные периоды времени после применения раствора испытуемого вещества или раствора лекарственной основы. Данные определяли как % максимально возможного действия (% МРЕ) из предварительных тестов отдельных животных (=0% МРЕ) и тестовых значений независимой имитационной (sham) контрольной группы (=100% МРЕ). Альтернативно были представлены пороги отдергивания в граммах.
Статистическое оценивание: ED50 значения и 95% доверительные пределы были определены посредством полулогарифмического регрессивного анализа на момент максимального действия. Данные были проанализированы дисперсионным анализом с повторными измерениями, а также с помощью вторичного анализа (post hoc) согласно Бонферрони (Bonferroni). Групповой размер обычно составлял n=10.
Ссылки: Kim, S.H. и Chung, J.M., An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation в the rat, Pain, 50 (1992) 355-363.
Результаты приведены в нижеследующей таблице (модель Chung):
Соединения согласно изобретению типа Е c W = -NHMe или -NMe2 (Прим. 9, 11 и 13) сравнивали с соответствующими соединениями типа Е с W = -ОН (V-1 и V-2):






Как подтверждают вышеуказанные данные сравнения, соединения согласно изобретению (W = -NHMe или -NMe2) по сравнению со структурально подобными веществами (W = -ОН) обладают более высокой избирательностью в отношении каппа-опиоидного рецептора (определено как 1/[Ki(ORL1)/Ki(каппа)]). Кроме того, вещества согласно изобретению при благоприятном соотношении ORLI/µ-сродство также обладают более высокой избирательностью в отношении µ-опиоидного рецептора (определено как 1/[Ki(ORL1)/Ki(µ)]).
Соединения согласно изобретению типа 1 с n=0, Х = -NMe и Q = фенил (Прим. 27, 30, 31, 32, 49, 85 и 92) сравнивали с соединениями типа F (от V-3 до V-5) с Z = NMe или NCOR, R5, R6= H, W = NH, A1-4= CH и от R1 до R3, от Y1 до Y4 и от Y1' до Y4' соответственно (1):







Как подтверждает вышеуказанное сравнение, соединения согласно изобретению из примеров 27, 30, 31, 32, 49, 85 и 92 по сравнению со структурально подобными соединениями (от V-3 до V-5) обладают лучшей растворимостью в водной среде, что в особенности должно предоставить преимущества, принимая во внимание свойства резорбции и/или биодоступность.
Реферат
Изобретение относится к новым соединениям общей формулы (1), которые обладают сродством к µ-опиоидному рецептору и ORL1-рецептору. Изобретение также относится к применению этих соединений для получения лекарственных средств, которые могут быть использованы при лечении страха, стресса и связанных со стрессом синдромов, депрессий, эпилепсии, болезни Альцгеймера, старческого слабоумия, общих познавательных дисфункций, нарушений обучения и памяти (как ноотроп), синдромов отмены, злоупотребления алкоголем и/или наркотиками и/или злоупотребления медикаментами и/или алкогольной, наркотической, медикаментозной зависимости и др. В общей формуле (1)Y, Y , Y, Y , Y, Y , Yи Y в каждом случае означают -H; Q означает -R, -C(=O)-R, -C(=O)OR, -C(=O)NHR, -C(=O)N(R)или -C(=NH)-R; Rв каждом случае независимо означает -C-алифат, -C-циклоалифат, -арил, -гетероарил, -С-алифат-С-циклоалифат, -C-алифат-арил, -C-алифат-гетероарил, -С-циклоалифат-C-алифат, -C-циклоалифат-арил или -C-циклоалифат-гетероарил; Rи Rнезависимо друг от друга означают -C--алифат; Rозначает -C-алифат, -арил, -гетероарил или -C-алифат-C-циклоалифат; n означает 0; X означает -NR-;Rозначает -C-алифат; Rозначает -C-алифат; при условии, что R, R, Rи Rодновременно не означают незамещенный -C-алифат. 4 н. и 5 з.п. ф-лы, 11 табл., 164 пр.
Формула

где
Y1, Y1', Y2, Y2', Y3, Y3', Y4 и Y4' в каждом случае означают -H;
Q означает -R0, -C(=O)-R0, -C(=O)OR0, -C(=O)NHR0, -C(=O)N(R0)2 или -C(=NH)-R0;
R0 в каждом случае независимо означает -C1-8-алифат, -C3-12-циклоалифат, -арил, -гетероарил, -С1-8-алифат-С3-12-циклоалифат, -C1-8-алифат-арил, -C1-8-алифат-гетероарил, -С3-8-циклоалифат-C1-8-алифат, -C3-8-циклоалифат-арил или -C3-8-циклоалифат-гетероарил;
R1 и R2 независимо друг от друга означают -C-1-8-алифат;
R3 означает -C1-8-алифат, -арил, -гетероарил или -C1-8-алифат-C3-12-циклоалифат;
n означает 0;
X означает -NRA-;
RA означает -C1-8-алифат;
RB означает -C1-8-алифат;
при условии что R1, R2, RA и RB одновременно не означают незамещенный -C1-8-алифат;
причем "алифат" в каждом случае представляет собой разветвленный или неразветвленный, насыщенный или моно- или многократно ненасыщенный, незамещенный или моно- или многократно замещенный, алифатический углеводородный остаток;
"циклоалифат" в каждом случае представляет собой насыщенный или моно- или многократно ненасыщенный, незамещенный или моно- или многократно замещенный, алициклический, моно- или мультициклический углеводородный остаток, число циклических атомов углерода которого преимущественно находится в указанном диапазоне (т.е. "C3-8"-циклоалифат преимущественно имеет 3, 4, 5, 6, 7 или 8 циклических атомов углерода);
причем относительно "алифат" и "циклоалифат" под "моно- или многократно замещенным" понимается моно- или многократное замещение одного или нескольких атомов водорода, например однократное, двукратное, трехкратное или полное замещение заместителями, независимо друг от друга выбранными из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -CHO, -C(=O)R0, -C(=O)H, C(=O)OH, -C(=O)OR0, -C(=O)NH2, -C(=O)NHR0, -C(=O)N(R0)2, -OH, -OR0, OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N+(R0)3, -N+(R0)2O-, -NHC(=O)R0, -NHC(=O)OR0, -NHC(=O)NH2, -NHC(=O)-NHR0, -NH-C(=O)N(R0)2, -Si(R0)3 и -PO(OR0)2;
"арил" в каждом случае независимо означает карбоциклическую кольцевую систему с по меньшей мере одним ароматическим кольцом, но без гетероатомов в этом кольце, причем арильные остатки при необходимости могут быть конденсированы с другими насыщенными, (частично) ненасыщенными или ароматическими кольцевыми системами и каждый арильный остаток может быть незамещенным или моно- или многократно замещенным, причем арильные заместители могут быть одинаковыми или различными в любом и возможном положении арила;
"гетероарил" означает 5-, 6- или 7-членный циклический ароматический остаток, который содержит 1, 2, 3, 4 или 5 гетероатомов, причем гетероатомы одинаково или различно представляют собой азот, кислород или серу, и гетероцикл может быть незамещенным или моно- или многократно замещенным; причем в случае замещения в гетероцикле заместители могут быть одинаковыми или различными в любом и возможном положении гетероарила; и причем гетероцикл также может быть частью бициклической или полициклической системы;
причем относительно "арил" и "гетероарил" под "моно- или многократно замещенным" понимается моно- или многократное замещение одного или нескольких атомов водорода кольцевой системы заместителями, выбранными из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -СНО, =O, -R0, -C(=O)R0, -C(=O)H, -C(=O)OH, -C(=O)OR0, -C(=О)NH2, -C(=O)NHR0, C(=O)-N(R0)2, -OH, -O(CH2)1-2O-, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0,-OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N+(R0)3, -N+(R0)2O-, -NHC(=O)R0, -NHC(=O)OR0, -NH-C(=O)NH2, -NHC(=O)NHR0, -NHC(=O)N(R0)2, -Si(R0)3 или -PO(OR0)2; причем при необходимости имеющиеся N-кольцевые атомы в каждом случае могут быть окисленными;
в виде отдельного стереоизомера или его смеси, свободных соединений и/или их физиологически совместимых солей.

где (гетеро-)арил означает гетероарил или арил, в каждом случае незамещенный или моно- или многократно замещенный, причем заместители независимо друг от друга выбраны из группы, состоящей из -F, -Cl, -Br, -I, -CN, -NO2, -CHO, -R0, -C(=O)R0, -С(=O)H, -С(=O)OH, C(=O)OR0, -C(=O)NH2, -C(=O)NH-R0, -C(=O)-N(R0)2, -OH, -O(CH2)1-2O-, -OR0, -OC(=O)H, -OC(=O)R0, -OC(=O)OR0, -OC(=O)NHR0, -OC(=O)N(R0)2, -SH, -SR0, -SO3H, -S(=O)1-2-R0, -S(=O)1-2NH2, -NH2, -NHR0, -N(R0)2, -N+(R0)3, -N+(R0)2O-, -NHC(=O)R0, -NHC(=O)-OR0, -NH-C(=O)NH2, -NHC(=O)NHR0 и NHC(=O)N(R0)2.

где RC означает -H, -F, -Cl, -Br, -I, -CN, -NO2, -CF3, -OH или -OCH3.
Q означает -C1-8-алифат, -арил, -C1-8-алифат-арил, -гетероарил, -C(=O)-гетероарил или -C(=NH)-гетероарил;
R1 означает -CH3;
R2 означает -CH3;
X означает -NRA-;
RA означает -C1-8-алифат;
RB означает -С1-8-алифат;
RC означает -H, -F, -Cl, -Br, -I, -CN, -NO2, -CF3, -OH или -OCH3; и
n означает 0;
причем алифат, арил и гетероарил в каждом случае являются незамещенными или моно- или многократно замещенными.
Q означает -C1-8-алкил, -фенил, -C1-8-алкил-фенил, -индолил,
-C(=O)-индолил или -C(=NH)-индолил;
R1 означает -CH3;
R2 означает -CH3;
X означает -NRA-;
RA означает -C1-8-алкил;
RB означает -C1-8-алкил;
RC означает -H, -F, -Cl, -Br, -I, -CN, -NO2, -CF3, -OH или -OCH3; и n означает 0.
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-уксусная кислота (полярный диастереомер);
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-уксусная кислота (неполярный диастереомер);
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-этанол (полярный диастереомер);
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-N,N-диметил-ацетамид (полярный диастереомер);
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-N-метил-ацетамид (полярный диастереомер);
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-N,N-диметил-ацетамид (неполярный диастереомер);
- 2-[(4-диметиламино-1,4-дифенил-циклогексил)-метил-амино]-N-метил-ацетамид (неполярный диастереомер);
- 2-[[4-(диметил-амино)-1,4-дифенил-циклогексил]-метил-амино]-этанол (неполярный диастереомер);
- 4-[[4-(диметил-амино)-1,4-дифенил-циклогексил]-метил-амино]-бутан-1-ол (полярный диастереомер);
и их физиологически совместимые соли.
Комментарии