Некоторые химические соединения, композиции и способы - RU2582676C2
Код документа: RU2582676C2
Чертежи


















Описание
Настоящая заявка испрашивает приоритет предварительной заявки на патент США №12/503776, поданной 15 июля 2009 года, которая частично является продолжением Международной заявки №PCT/US 09/00038, поданной 5 января 2009 года и Международной заявки №PCT/US 09/00042, поданной 5 января 2009 года, каждая из которых испрашивает приоритет предварительных заявок на патент США №№61/009971, поданной 4 января 2008 года, 61/194294, поданной 26 сентября 2008 года и 61/201146, поданной 5 декабря 2008 года. Все вышеупомянутые заявки на патент включены в настоящую заявку путем ссылки в полном объеме для всех целей.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Активность клеток может регулироваться внешними сигналами, стимулирующими или ингибирующими внутриклеточные события. Процесс, в результате которого стимулирующие или ингибирующие сигналы передаются в клетку и внутри нее для вызова внутриклеточной реакции, упоминается как сигнальная трансдукция. За последние десятилетия было найдено объяснение каскадам событий сигнальной трансдукции и установлено, что они играют центральную роль во множестве биологических реакций. Установлено, что дефекты различных компонентов путей сигнальной трансдукции являются причиной огромного количества заболеваний, включая многочисленные формы рака, воспалительные нарушения, метаболические расстройства, сосудистые и нейронные нарушения (Gaestel et al. Current Medicinal Chemistry (2007) 14: 2214-2234).
Киназы представляют класс важных сигнальных молекул. Киназы можно отнести, главным образом, к протеинкиназам и липидкиназам, а некоторые киназы обладают двойственной специфичностью. Протеинкиназы являются ферментами, которые фосфорилируют другие белки и/или самих себя (т.е. аутофосфорилирование). Протеинкиназы могут быть отнесены, главным образом, к трем основным группам на основании их субстратного применения: тирозинкиназы, которые преимущественно фосфорилируют субстраты на тирозиновых остатках (например, erb2, рецептор PDGF, рецептор EGF, рецептор VEGF, src, abl), серин/треонин-киназы, которые преимущественно фосфорилируют субстраты на остатках серина и/или треонина (например, mTorC1, mTorC2, ATM, ATR, DNA-PK, Akt), и киназы двойной специфичности, которые фосфорилируют субстраты на остатках тирозина, серина и/или треонина.
Липидкиназы являются ферментами, которые катализируют фосфорилирование липидов. Эти ферменты и образующиеся фосфорилированные липиды и липид-производные биологически активные органические молекулы участвуют во многих различных физиологических процессах, включая пролиферацию, миграцию, адгезию и дифференцировку. Определенные липидкиназы являются мембрансвязанными и катализируют фосфорилирование липидов, содержащихся в клеточных мембранах или связанных с ними. Примеры таких ферментов включают фосфоинозитид-киназы (такие как PI3-киназы, PI4-киназы), диацилглицерин-киназы и сфингозин-киназы.
Сигнальный путь фосфоинозитид 3-киназ (PI3K) является одной из наиболее высокомутированных систем раковых заболеваний человека. Сигнализация PI3K также является ключевым фактором во многих других заболеваниях человека. Сигнализация PI3K входит во многие болезненные состояния, включая аллергический контактный дерматит, ревматоидный артрит, остеоартроз, воспалительные заболевания кишечника, хронические обструктивные заболевания легких, псориаз, рассеянный склероз, астму, расстройства, связанные с диабетическими осложнениями и воспалительные осложнения сердечнососудистой системы, такие как острый коронарный синдром.
PI3K также являются членами уникального и консервативного семейства внутриклеточных липидкиназ, которые фосфорилируют группу 3'-ОН на фосфатидилинозитолах или фосфоинозитидах. Семейство PI3K включает 15 киназ с различной субстратной специфичностью, схемами экспрессии и способами регуляции (Katso et al., 2001). Класс I PI3K (p110α, p110β, p110δ и p110γ) обычно активируется тирозинкиназами или рецепторами, связанными с G-белком для выработки PIP3, который касается последующих эффекторов, таких как эффекторы пути Akt/PDK1, mTOR, киназы семейства Тес и ГТФазы семейства Rho. Киназы PI3 класса II и III играют ключевую роль во внутриклеточной миграции посредством PI(3)P и PI(3,4)P2. PIKK являются протеинкиназами, контролирующими клеточный рост (mTORC1) или контролирующими геномную целостность (ATM, ATR, DNA-PK и hSmg-1).
Дельта (δ) изоформа PI3K класса I участвует, в частности, в ряде заболеваний и биологических процессов. PI3K δ экспрессируется, в основном, в гематопоэтических клетках, включая лейкоциты, такие как Т-клетки, дендровидные клетки, нейтрофилы, мастоциты, В-клетки и макрофаги. PI3K δ полностью задействована в функциях иммунной системы млекопитающих, таких как функция Т-клеток, активация В-клеток, активация мастоцитов, функция дендритных клеток и активность нейтрофилов. За счет ее общей роли в функционировании иммунной системы, PI3K δ также участвует в ряде заболеваний, связанных с нежелательной иммунной реакцией, таких как аллергические реакции, воспалительные заболевания, опосредованный воспалением ангиогенез, ревматоидный артрит, аутоиммунные заболевания, такие как волчанка, бронхиальная астма, эмфизема легких и другие респираторные заболевания. Другие PI3K класса I, участвующие в функционировании иммунной системы, включают PI3K г, которые играют важную роль в лейкоцитной сигнализации и вызывают воспаление, ревматоидный артрит, аутоиммунные заболевания, такие как волчанка.
Последующие медиаторы пути сигнальной трансдукции PI3K включают Akt и мишень рапамицина в клетках млекопитающих (mTOR). Akt обладает гомологичным доменом plckstrin (РН), которые связывает PIP3, приводя к активации Akt-киназы. Akt фосфорилирует многие субстраты и является центральным последующим эффектором PI3K для разнообразных клеточных реакций. Одной важной функцией Akt является усиление активности mTOR посредством фосфорилирования TSC2 и других механизмов. mTOR является серин-треонин-киназой, связанной с липид-киназами семейства PI3K. mTOR участвует во многих биологических процессах, включая клеточный рост, клеточную пролиферацию, клеточную подвижность и выживание. Разрегуляция пути mTOR отмечена в различных типах рака. mTOR является многофункциональной киназой, которая интегрирует фактор роста и сигналы питательных веществ для регуляции трансляции белков, усвоения питательных веществ, аутофагии и митохондриальной функции.
Поэтому киназы, особенно PI3K, являются основными мишенями для разработки лекарств. Сохраняется необходимость в ингибиторах PI3K, пригодных для разработки лекарств. Настоящее изобретение направлено на удовлетворение этой необходимости и обладает относительным преимуществом, в том числе предлагая новые классы ингибиторов киназ.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
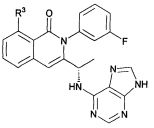
В одном аспекте настоящего изобретения предложены соединения формулы I, приведенной ниже, или их фармацевтически приемлемые соли, где

Wd является гетероциклоалкилом, арилом или гетероарилом;
В является алкилом или группировкой формулы II;

где Wc является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, a q является целым числом 0, 1, 2, 3 или 4;
X отсутствует или является -(CH(R9))z-, и z является целым числом, равным 1;
Y отсутствует или является -N(R9)-;
R1 является водородом, алкилом, алкенилом, алкинилом, алкокси, амидо, алкоксикарбонилом, сульфонамидо, галогено, циано или нитро;
R2 является алкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, гетероарилом, гетероарилалкилом, алкокси, амино, галогено, циано, гидрокси или нитро;
R3 является водородом, алкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси, нитро, арилом или гетероарилом;
R5, R6, R7 и R8 независимо являются водородом, алкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, алкокси, амидо, амино, ацилом, ацилокси, сульфонамидо, галогено, циано, гидрокси или нитро; и
каждый из R9 независимо является водородом, алкилом, циклоалкилом или гетероциклоалкилом.
В некоторых воплощениях изобретения X отсутствует или является -(CH(R9))z-, и z является целым числом, равным 1. В некоторых воплощениях изобретения X отсутствует. В некоторых воплощениях изобретения X является -(CH(R9))z-, и z является целым числом, равным 1. В некоторых воплощениях изобретения X является -CH2-, -СН(CH2CH3) или -CH(CH3)-.
В некоторых воплощениях изобретения Y отсутствует или является -N(R9)-. В некоторых воплощениях изобретения Y отсутствует. В некоторых воплощениях изобретения Y является -N(R9)-. В некоторых воплощениях изобретения Y является -NH-, -N(CH3) или -N(CH2CH3)-.
В некоторых воплощениях изобретения X отсутствует или является -(CH(R9))z-, z является целым числом, равным 1, и Y отсутствует или является -N(R9)-. В некоторых воплощениях изобретения X отсутствует, и Y является -N(R9)-. В некоторых воплощениях изобретения Y отсутствует, и X является -(CH(R9))z-, где z является целым числом, равным 1. В некоторых воплощениях изобретения присутствуют и X, и Y.
Например, в некоторых воплощениях соединений формулы I, если присутствуют X и Y, то Y является -NH- (например, X является -(CH(R9))z-, и Y является -NH-). В некоторых воплощениях изобретения X является -CH2-, -СН(CH2CH3) или -CH(CH3)-, и Y является -NH-.
В других вариантах соединений формулы I, если присутствуют X и Y, то Y является -N(CH3)- (например, X является -(CH(R9))z-, и Y является -N(CH3)-). В некоторых воплощениях изобретения X является -CH2-, -СН(CH2CH3) или -СН(CH3)-, и Y является -N(CH3)-.
В других вариантах соединений формулы I, если присутствуют X и Y, то Y является -N(CH2CH3)- (например, X является -(CH(R9))z-, и Y является -N(CH2CH3)-). В некоторых воплощениях изобретения X является -CH2-, -СН(CH2CH3) или -CH(CH3)-, и Y является -N(CH2CH3)-.
В некоторых воплощениях изобретения группа X-Y является -CH2-N(CH3), -CH2-N(CH2CH3), -CH(CH2CH3)-NH- или -CH(CH3)-NH-.
В некоторых воплощениях изобретения Wd является пиразолопиримидином формулы III(а), или пурином формулы III(b), формулы III(с) или формулы III(d), изображенных ниже:

где Ra′ в формуле III(d) является водородом, галогено, фосфатом, мочевиной, карбонатом, амино, алкилом, алкенилом, алкинилом, циклоалкилом, гетероалкилом или гетероциклоалкилом; R11 в формуле III(а) является Н, алкилом, галогено, амино, амидо, гидрокси или алкокси, и R12 в формуле III(а), формулы III(с) или формулы III(d) является Н, алкилом, алкинилом, алкенилом, галогено, арилом, гетероарилом, гетероциклоалкилом или циклоалкилом. В некоторых воплощениях изобретения Wd является пиразолопиримидином формулы III(а), где R11 является Н, алкилом, галогено, амино, амидо, гидрокси или алкокси, и R12 является циано, амино, карбоновой кислотой или амидо.
В некоторых воплощениях изобретения соединение формулы I имеет структуру формулы IV:

где R11 является H, алкилом, галогено, амино, амидо, гидрокси или алкокси, и R12 является Н, алкилом, алкинилом, алкенилом, галогено, арилом, гетероарилом, гетероциклоалкилом или циклоалкилом. В некоторых воплощениях изобретения соединение формулы I имеет структуру формулы IV, где R11 является Н, алкилом, галогено, амино, амидо, гидрокси или алкокси, и R12 является циано, амино, карбоновой кислотой или амидо.
В некоторых воплощениях соединения формулы IV, R11 является амино. В некоторых воплощениях соединения формулы IV, R12 является алкилом, алкенилом, алкинилом, гетероарилом, арилом или гетероциклоалкилом. В некоторых воплощениях соединения формулы IV, R12 является циано, амино, карбоновой кислотой, амидо, моноциклическим гетероарилом или бициклическим гетероарилом.
В некоторых воплощениях соединения формулы I, это соединение имеет структуру формулы V:

В некоторых воплощениях формулы V, NR9 является -N(CH2CH3)CH2- или N(CH3)CH2-.
В некоторых воплощениях формулы I, это соединение имеет структуру формулы VI:

В некоторых воплощениях соединения формулы VI, R3 является -Н, -CH3, -Cl или -F, и R5, R6, R7, и R8 независимо являются водородом.
В некоторых воплощениях формулы VI B является группировкой формулы II;

где Wc является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, и q является целым числом 0, 1, 2, 3 или 4.
В другом аспекте настоящего изобретения соединение и его фармацевтически приемлемые соли имеют структуру формулы I-1, где:

В является группировкой формулы II;
где Wc в B является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, и q является целым числом из 0, 1, 2, 3 или 4;
X отсутствует или является -(CH(R9))z-, и z является целым числом, равным 1;
Y отсутствует или является -N(R9)-;
если Y отсутствует, то Wd является:


R1 является водородом, алкилом, алкенилом, алкинилом, алкокси, амидо, алкоксикарбонилом, сульфонамидо, галогено, циано или нитро;
R2 является алкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, гетероарилом, гетероарилалкилом, алкокси, амино, галогено, циано, гидрокси или нитро;
R3 является водородом, алкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси, нитро, арилом или гетероарилом;
каждый из R9 независимо является водородом, C1-C10алкилом, циклоалкилом или гетероциклоалкилом; и R12 является Н, алкилом, алкинилом, алкенилом, галогено, арилом, гетероарилом, гетероциклоалкилом или циклоалкилом.
В некоторых воплощениях изобретения, соединение формулы I или формулы I-1 имеет структуру формулы IV-A:

В некоторых воплощениях соединения формулы IV-A, R12 является замещенным бензоксазолом.
В некоторых воплощениях изобретения, соединение формулы I или формулы I-1 имеет структуру формулы V-A:

В некоторых воплощениях изобретения, соединение формулы I или формулы I-1 имеет структуру формулы IV-A или формулы V-A.
В некоторых воплощениях изобретения, соединение формулы I или формулы I-1 имеет структуру формулы V-B:

В некоторых воплощениях изобретения, соединение формулы I или формулы I-1 имеет структуру формулы VI-A:

В некоторых воплощениях изобретения, соединение формулы I или формулы I-1 является соединением, где В является группировкой формулы II;

где Wc является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом; q является целым числом, равным 0 или 1; R1 является водородом, алкилом или галогено; R2 является алкилом или галогено; и R3 является водородом, алкилом или галогено. В некоторых воплощениях изобретения, если оба X и Y присутствуют, то Y является -NH-. В других воплощениях изобретения R3 является -Н, CH3, -CH2CH3, -CF3, -Cl или -F. В других воплощениях изобретения R3 является метилом или хлором. В некоторых воплощениях изобретения R3 является галогеноалкилом. Например, R3 является -CF3, -CH2F или -CHF2.
В некоторых воплощениях изобретения, соединение формулы I или формулы I-1 X является -(CH(R9))z-, где R9 является метилом, и z=1; и
Wd является

В других вариантах соединения формулы I или формулы I-1, соединение преимущественно находится в (S)-стереохимической конфигурации.
В других воплощениях соединения по настоящему изобретению, это соединение имеет структуру формулы V-A2:

или его фармацевтически приемлемой соли, где В является группировкой формулы II:

где Wc является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом; q является целым числом 0, 1, 2, 3 или 4;
R1 является водородом, алкилом, алкенилом, алкинилом, алкокси, амидо, алкоксикарбонилом, сульфонамидо, галогено, циано или нитро;
R2 является алкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, гетероарилом, гетероарилалкилом, алкокси, амино, галогено, циано, гидрокси или нитро;
R3 является водородом, алкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси, нитро, арилом или гетероарилом; и
каждый из R9 независимо является водородом, алкилом или гетероциклоалкилом.
В некоторых воплощениях соединения формулы V-A2, В является группировкой формулы II:

где Wc является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом;
q является целым числом, равным 0 или 1;
R1 является водородом, алкилом или галогено;
R2 является алкилом или галогено; и
R3 является водородом, алкилом или галогено.
Например, Wc является арилом, таким как фенил. Альтернативно, Wc является циклопропилом. В некоторых случаях Wc является замещенным по меньшей мере одним из CH3, -CH2CH3, -CF3, -Cl или -F.
В некоторых воплощениях соединения формулы V-A2, R3 является водородом, алкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, алкокси, амидо, амино, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси или нитро. Например, R3 является -Н, CH3, -CH2CH3, -CF3, -Cl или -F. В некоторых воплощениях изобретения R3 является -CH3 или -Cl. В других воплощениях изобретения R3 является галогеноалкилом. Например, R3 является -CF3, -CH2F или -CHF2.
В некоторых воплощениях соединения формулы V-A2, R9 является CH3. В других воплощениях изобретения R9 является -CH2-CH3.
В некоторых воплощениях соединения формулы V-A2 это соединение имеет формулу:

В некоторых воплощениях изобретения R2 является -Н.
В некоторых воплощениях соединения формулы V-A2 это соединение имеет формулу:

В некоторых воплощениях изобретения R1 и R2 независимо выбраны из группы, состоящей из водорода и галогено.
В некоторых воплощениях изобретения соединение имеет формулу:

В некоторых воплощениях изобретения, R1 является -Н, например, формулы:


В некоторых воплощениях изобретения, R1 является Н, и R2 является Н, например, формулы:

или его фармацевтически приемлемые соли.
В некоторых воплощениях изобретения, R1 является Н, q равен 1, и R2 является галогено (например, фторо) формулы:

или его фармацевтически приемлемые соли.
В некоторых воплощениях R1 является Н, q равен 1, и R2 является гало (например, фторо) в мета-положении, например, формулы:

или его фармацевтически приемлемые соли.
В других воплощениях изобретения, соединение формулы V-A2 имеет формулу:
В других аспектах соединение имеет формулу:


В другом аспекте настоящего изобретения предложена фармацевтическая композиция, включающая фармацевтически приемлемый носитель и одно или более соединений любой из представленных здесь формул, включая, не ограничиваясь, формулы I, I-1, IV, IV-А, V, V-A, V-A2, V-B, VI и VI-A. В некоторых воплощениях изобретения, композиция находится в жидкой, твердой, полутвердой, гелеобразной или аэрозольной форме.
В другом аспекте настоящего изобретения предложен способ ингибирования фосфатидилинозитол-3-киназы (PI3 киназы), включающий взаимодействие PI3 киназы с эффективным количеством одного или более соединений, описанных в настоящем документе. Например, стадия взаимодействия включает использование одного или более соединений любой из представленных в настоящем документе формул, включая, не ограничиваясь, формулы 1, I-1, IV, IV-А, V, V-A, V-A2, V-B, VI и VI-A. В некоторых воплощениях изобретения, стадия взаимодействия включает контактирование с клеткой, содержащей указанную PI3 киназу. В некоторых воплощениях этого способа, ингибирование происходит у субъекта, страдающего расстройством, связанным с дисфункцией одного или более типов PI3 киназ. Некоторые типичные расстройства, включающие дисфункцию одного или более типов PI3 киназ, выбраны из группы, включающей аутоиммунные заболевания, ревматоидный артрит, респираторное заболевание, аллергические реакции и различные типы рака. При необходимости, соединение, используемое в этом способе, имеет структуру формулы IV, где R11 является амино, a R12 является замещенным фенилом.
В некоторых воплощениях этого способа, ингибирование происходит у субъекта, страдающего ревматоидным артритом или респираторным заболеванием, и где соединение имеет структуру формулы IV, и где R11 является амино, a R12 является бициклическим гетероарилом.
В некоторых воплощениях изобретения способ включает введение субъекту второго терапевтического средства.
В другом аспекте настоящего изобретения предложен способ лечения заболевания, проявляющего нежелательный иммунный ответ. Способ включает стадию введения пациенту, нуждающемуся в этом, одного или более соединений, раскрытых в настоящем документе, включая соединения формулы 1, I-1, IV, IV-А, V, V-A, V-A2, V-В, VI и/или VI-A, в количестве, которое является эффективным для улучшения указанного нежелательного иммунного ответа. В некоторых воплощениях изобретения одно или более соединений ингибирует активацию В-клеток, независимую от Т-клеток, что подтверждено снижением выработки анти-TNP IgG3 по меньшей мере в пять раз при введении испытуемому животному в дозировке, меньшей, чем 30 мг/кг дважды в день.
В некоторых воплощениях изобретения заболевание, которое лечат, связано с опухолью или болью в суставах пациента. Этот способ может быть эффективным для улучшения одного или более симптомов ревматоидного артрита, что подтверждено снижением среднего диаметра сустава по меньшей мере на 10% через 17 дней и/или снижением диаметра лодыжки по меньшей мере на 5-10% или более спустя несколько дней или несколько недель лечения, включая, например, уменьшение диаметра лодыжки по меньшей мере на 5% через 7 дней лечения. В другом варианте воплощения изобретения нежелательный иммунный ответ подтверждается увеличением выработки антител против коллагена типа II, а использование одного или более соединений по изобретению снижает уровень сывороточного коллагена анти-типа II при ED50 менее, чем примерно 10 мг/кг.
ВКЛЮЧЕНИЕ ПУТЕМ ССЫЛКИ
Все публикации, патенты и заявки на патенты, упомянутые в этом описании, являются включенными в настоящую заявку путем ссылки в том же объеме, как если бы каждая отдельная публикация, патент или заявка на патент были бы указаны конкретно и отдельно для включения путем ссылки.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Далее в приложенной формуле изобретения подробно изложены новые признаки настоящего изобретения. Для лучшего понимания признаков и преимуществ настоящего изобретения ниже приведено детальное описание, представляющее дополнительные иллюстративные воплощения изобретения, в которых используются принципы настоящего изобретения, и сопровождающие графические материалы.
На Фиг. 1 отображен типичный протокол измерения независимой от Т-клеток выработки TNP-специфичных антител in vivo.
На Фиг. 2 представлено кратное снижение TNP-специфичной реакции IgG3 на антигены, полученное от соединений 7 и 53 формулы IV в сравнении с контрольным носителем, при пероральном введении.
На Фиг. 3 представлено дозозависимое влияние ежедневного двукратного перорального введения соединения 53 формулы IV на снижение роста диаметра лодыжки в зависимости от времени в модели развивающегося коллаген-индуцированного артрита на крысах. Также представлены результаты, полученные у контрольных крыс без артрита, контрольных крыс с артритом, которым вводили негативный отрицательный носитель, и контрольных крыс с артритом, которые дважды в день проходили лечение метотрексатом.
На Фиг. 4 представлено дозозависимое влияние соединений 7 и 53 формулы IV на улучшение гистопатологии лодыжки при введении в модель развивающегося коллаген-индуцированного артрита на крысах. Также представлены результаты, полученные у контрольных крыс с артритом, которым вводили отрицательный контрольный носитель или метотрексат.
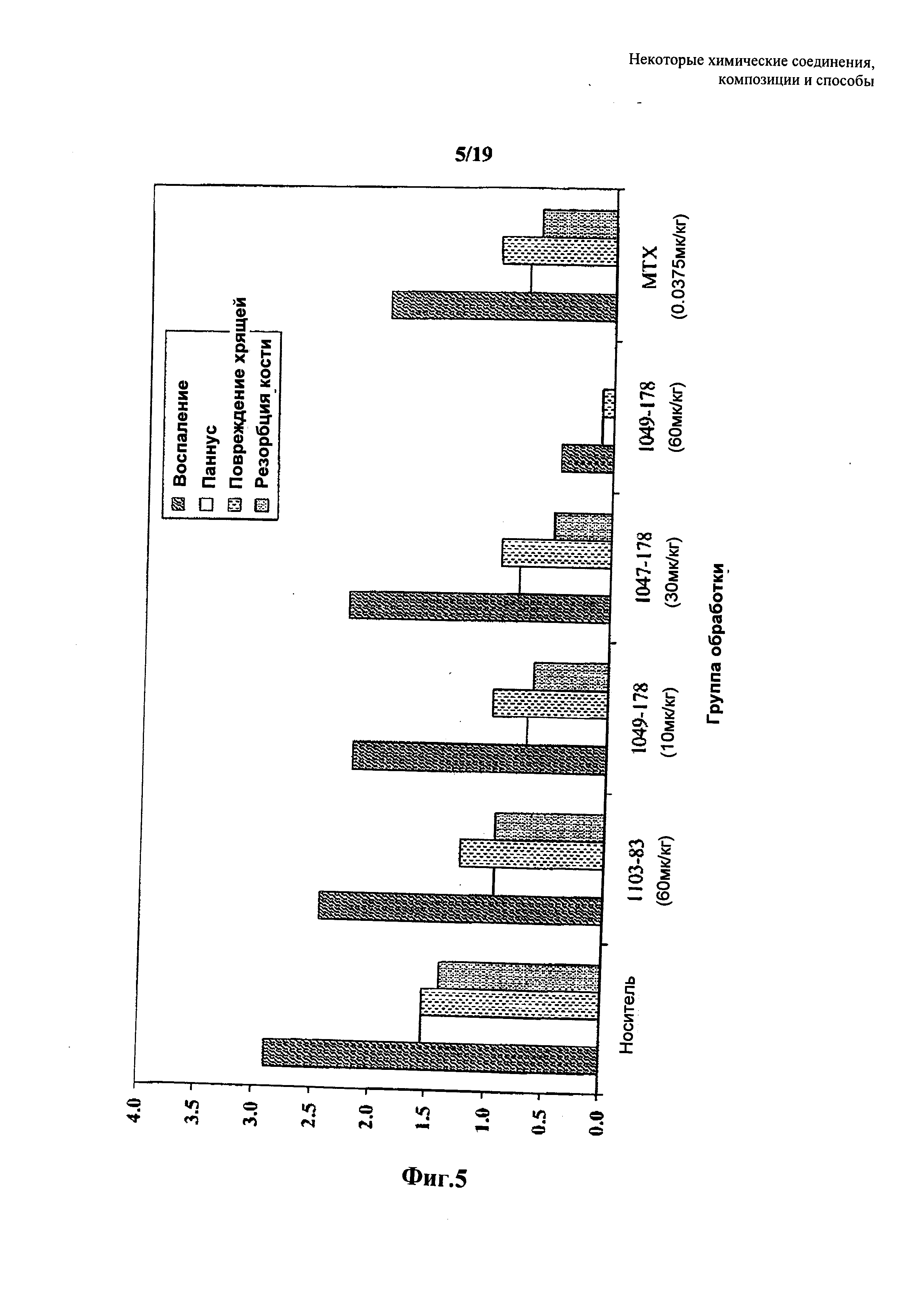
На Фиг. 5 представлено дозозависимое влияние соединений 7 и 53 формулы IV на улучшение гистопатологии колена при введении в модель развивающегося коллаген-индуцированного артрита на крысах. Также представлены результаты, полученные у контрольных крыс с артритом, которым вводили отрицательный контрольный носитель или положительный контрольный метотрексат.
На Фиг. 6 представлено дозозависимое влияние соединений 7 и 53 формулы IV на снижение уровня коллагеновых антител анти-типа II in vivo при введении в модель развивающегося коллаген-индуцированного артрита на крысах. Также представлены результаты, полученные у крыс с артритом, которым вводили отрицательный контрольный носитель или метотрексат.
На Фиг. 7 представлено дозозависимое влияние соединения 7 формулы IV на улучшение гистопатологии лодыжки при введении в модель развивающегося коллаген-индуцированного артрита на крысах. Также представлены результаты, полученные у крыс с артритом, которым вводили контрольный носитель, и у крыс с артритом, получавших метотрексат.
На Фиг. 8 представлено дозозависимое влияние ежедневного введения соединения 53 формулы IV на улучшение гистопатологии лодыжки при введении в модель устоявшегося коллаген-индуцированного артрита на крысах. Также представлены результаты, полученные у крыс с артритом, которым вводили контрольный носитель, и у крыс с артритом, получавших Энбрел.
На Фиг. 9 представлено дозозависимое влияние ежедневного двукратного введения соединения 53 формулы IV на улучшение гистопатологии лодыжки при введении в модель устоявшегося коллаген-индуцированного артрита на крысах. Также представлены результаты, полученные у крыс с артритом, которым вводили контрольный носитель, и крыс с артритом, получавших Энбрел.
На Фиг. 10 представлено дозозависимое влияние соединения 53 формулы IV на увеличение среднего объема лап в модели адъювант-индуцированного артрита.
На Фиг. 11 представлено влияние соединения 53 на средний вес крыс в зависимости от времени в моделях адъювант-индуцированного артрита крыс.
На Фиг. 12 представлено влияние соединения 292 ("Cpd-A") формулы V-A2 на снижение роста диаметра лодыжки в зависимости от времени на моделях развивающегося коллаген-индуцированного артрита на крысах.
На Фиг. 13 представлено влияние соединения 292 ("Cpd-A") формулы V-A2 на гистопатологию лодыжки на моделях устоявшегося коллаген-индуцированного артрита крыс.
На Фиг. 14 представлено влияние соединения 292 ("Cpd-A") формулы V-A2 на снижение роста диаметра лодыжки в зависимости от времени на моделях адъювант-индуцированного артрита крыс.
На Фиг. 15 представлено влияние соединения 292 ("Cpd-A") формулы V-A2 на замедление общего притока лейкоцитных нейтрофилов, вызванного липосахаридами, на моделях липосахарид-индуцированного воспаления легких крыс.
На Фиг. 16 представлено влияние соединения 292 ("Cpd-A") формулы V-A2 на замедление притока эозинофилов на моделях аллергического овальбумин-индуцированного воспаления легких у крыс.
На Фиг. 17 представлено влияние соединения 200 ("Cpd-B") формулы V-A2 на снижение роста диаметра лодыжки в зависимости от времени на моделях развивающегося коллаген-индуцированного артрита крыс.
На Фиг. 18 представлено влияние соединения 270 ("Cpd-C") формулы V-A2 на снижение роста диаметра лодыжки в зависимости от времени на моделях развивающегося коллаген-индуцированного артрита крыс.
На Фиг. 19 представлено влияние соединения 196 ("Cpd-D") формулы V-A2 на снижение роста диаметра лодыжки в зависимости от времени на моделях развивающегося коллаген-индуцированного артрита крыс.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Несмотря на то, что в настоящем документе показаны и описаны предпочтительные варианты воплощения настоящего изобретения, специалисту в данной области понятно, что эти варианты воплощения изобретения представлены лишь в качестве примеров. Специалисту в данной области понятны многочисленные варианты, изменения и отклонения в рамках настоящего изобретения. Следует понимать, что различные альтернативы вариантов воплощения настоящего изобретения, описанные в настоящем изобретении, могут использоваться в практике настоящего изобретения. Подразумевается, что приложенная формула изобретения определяет границы настоящего изобретения, и таким образом охвачены способы и структуры в этой формуле, а также их эквиваленты.
Если не определено иное, все технические и научные термины, используемые в настоящем документе, имеют то же значение, которое обычно подразумевается специалистом в области, к которой относится настоящее изобретение. Все патенты и другие публикации, перечисленные в настоящем документе, включены сюда путем ссылки.
Используемая в настоящем описании и формуле изобретения единственная форма включает множественные формы, если из контекста явно не следует обратное.
Используемый в настоящем документе термин «агент» или «биологически активный агент» относится к биологическому, фармацевтическому или химическому соединению или его части. Неограничивающие примеры включают простые или сложные органические или неорганические молекулы, пептиды, белки, олигонуклеотиды, антитела, производные антител, фрагменты антител, производные витаминов, углеводы, токсины или химиотерапевтические соединения. Могут быть синтезированы различные соединения, например, небольшие молекулы и олигомеры (например, олигопептиды и олигонуклеотиды), а также синтетические органические соединения на основе различных структур ядра. Кроме того, соединения для скрининга могут быть получены из различных природных источников, таких как растительные или животные экстракты и т.п. Специалисту в данной области понятно, что нет ограничений структурной природы агентов настоящего изобретения.
Термин «агонист», используемый в настоящем документе, относится к соединению, обладающему способностью инициировать или усиливать биологическую функцию целевого белка за счет ингибирования активности или экспрессии целевого белка. Соответственно, термин «агонист» определяется в контексте биологической роли целевого полипептида. Тогда как предпочтительные агонисты настоящего изобретения специфически взаимодействуют (например, связываются) с мишенью, соединения, инициирующие или усиливающие биологическую активность целевого полипептида за счет взаимодействия с другими членами пути сигнальной трансдукции, членом которой является целевой полипептид, также специально включены в это определение.
Термины «антагонист» и «ингибитор» используются взаимозаменяемо и относятся к соединению, обладающему способностью ингибировать биологическую функцию целевого белка за счет ингибирования активности или экспрессии целевого белка. Соответственно, термины «антагонист» и «ингибиторы» определяются в контексте биологической роли целевого белка. Тогда как предпочтительные антагонисты настоящего изобретения специфически взаимодействуют (например, связываются) с мишенью, соединения, ингибирующие биологическую активность целевого белка за счет взаимодействия с другими членами пути сигнальной трансдукции, членом которой является целевой белок, также специально включены в это определение. Предпочтительная биологическая активность, ингибируемая антагонистом, связана с развитием, ростом или распространением опухоли, или нежелательной иммунной реакции, проявляемой в аутоиммунном заболевании.
«Противораковый агент», «противоопухолевый агент» или «химиотерапевтический агент» относится к любому агенту, пригодному для лечения неопластического состояния. Один класс противораковых агентов включает химиотерапевтические средства. «Химиотерапия» означает введение раковому пациенту одного или более химиотерапевтических лекарств и/или других средств различными способами, включая внутривенное, пероральное, внутримышечное, внутрибрюшинное, внутрипузырное, подкожное, трансдермальное, буккальное или ингаляционное введение или в форме суппозиториев.
Термин «клеточная пролиферация» относится к явлению, в котором количество клеток изменяется в результате деления. Этот термин также включает рост клеток, в результате которого изменяется морфология клеток (например, увеличенный размер), в соответствии с пролиферативным сигналом.
Термины «совместное введение», «введение в комбинации с» и другие грамматические эквиваленты, используемые в настоящем документе, включают введение животному двух или более агентов, так что оба агента и/или их метаболиты присутствуют в организме животного одновременно. Совместное введение включает одновременное введение в отдельных композициях, введение в различное время в отдельных композициях или введение в композиции, в которой присутствуют оба агента.
Термин «эффективное количество» или «терапевтически эффективное количество» означает такое количество соединения, описанного в настоящем документе, которое является достаточным для действия в намеченном применении, включая, но не ограничиваясь, лечение заболевания, как определено ниже. Терапевтически эффективное количество может варьировать в зависимости от намеченного применения (in vitro или in vivo), или пациента и болезненного состояния, подлежащего лечению, например, веса и возраста пациента, серьезности болезненного состояния, способа введения и т.п., что легко может определить специалист в данной области. Этот термин также относится к дозировке, вызывающей конкретную реакцию в целевых клетках, например, снижение адгезии тромбоцитов и/или клеточной миграции. Конкретная доза варьирует в зависимости от конкретного выбранного соединения, режимов введения, возможности введения в комбинации с другими соединениями, времени введения, ткани, в которую осуществляется введение, и физической системы доставки, осуществляющей это введение.
Используемые здесь термины «лечение» или «облегчение», или «улучшение» используются взаимозаменяемо. Эти термины относятся к подходу для получения благотворных или желаемых результатов, включая, но не ограничиваясь, терапевтическую и/или профилактическую полезность. Под терапевтической полезностью понимается устранение или улучшение течения основного заболевания, подлежащего лечению. Также терапевтическая полезность достигается при устранении или улучшении одного или более физиологических симптомов, связанных с основным заболеванием, так что такое улучшение наблюдается у пациента, несмотря на то, что пациент все еще болен этим заболеванием. Для профилактической полезности композиции могут вводиться пациенту, имеющему риск развития конкретного заболевания, или пациенту, у которого наблюдается один или более физиологических симптомов заболевания, даже если не был поставлен диагноз этого заболевания.
Термин «терапевтический эффект», используемый в настоящем документе, означает терапевтическую полезность и/или профилактическую полезность, как описано выше. Профилактический эффект включает отсрочку или исключение возникновения заболевания или состояния, отсрочку или исключение возникновения симптомов заболевания или состояния, замедление, остановку или реверсирование развития заболевания или состояния или любую их комбинацию.
Термин «фармацевтически приемлемая соль» относится к солям, полученным из различных органических и неорганических противоионов, хорошо известных в данной области. Фармацевтически приемлемые соли присоединения кислоты могут быть получены из неорганических и органических кислот. Неорганические кислоты, из которых могут быть получены соли, включают, например, хлороводородную кислоту, бромоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и т.п. Органические кислоты, из которых могут быть получены соли, включают, например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, пировиноградную кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, коричную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, п-толуолсульфоновую кислоту, салициловую кислоту и т.п. Фармацевтически приемлемые соли присоединения основания могут быть образованы с неорганическими и органическими основаниями. Неорганические основания, из которых могут быть получены соли, включают, например, гидроксиды натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и т.п.
Органические основания, из которых могут быть получены соли, включают, например, первичные, вторичные и третичные амины, замещенные амины, включая встречающиеся в природе замещенные амины, циклические амины, основные ионообменные смолы и т.п., в частности, изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин и этаноламин. В некоторых воплощениях изобретения фармацевтически приемлемая соль присоединения основания выбрана из солей аммония, калия, натрия, кальция и магния.
«Фармацевтически приемлемый носитель» или «фармацевтически приемлемый наполнитель» включает любой и все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые средства, изотонические средства и агенты замедления абсорбции и т.п. Использование такой среды и агентов для фармацевтически активных веществ является известным в данной области. За исключением случаев несовместимости обычной среды или агента с активным компонентом, их использование в терапевтической композиции настоящего изобретения является предусмотренным. Дополнительные активные компоненты также можно вводить в состав композиций.
«Сигнальная трансдукция» является процессом, во время которого стимулирующие или ингибирующие сигналы передаются в клетку и внутри нее для возбуждения внутриклеточной реакции. Модулятор пути сигнальной трансдукции относится к соединению, которое модулирует активность одного или более клеточных белков, соответствующих тому же особому пути сигнальной трансдукции. Модулятор может увеличивать (агонист) или подавлять (антагонист) активность сигнальной молекулы.
Термин «селективное ингибирование» или «селективно ингибирует» в отношении биологически активного агента относится к способности этого агента селективно снижать целевую сигнальную активность по сравнению с нецелевой сигнальной активностью, за счет прямого или косвенного взаимодействия с мишенью.
Термин «В-ALL», используемый в настоящем документе, относится к В-клеткам острого лимфобластного лейкоза.
«Субъект» относится к животному, такому как млекопитающее, например, человек. Способы, описанные в настоящем документе, могут быть пригодными как для лечения людей, так и для ветеринарных целей. В некоторых воплощениях изобретения субъектом является млекопитающее, а в некоторых воплощениях изобретения субъектом является человек.
«Лучевая терапия» означает воздействие на пациента, при помощи обычных способов и композиций, известных практикующему врачу, радиационных излучателей, таких как радионуклеотиды, излучающие альфа-частицы (например, радионуклиды актиния и тория), радиационных излучателей с низкой линейной потерей энергии (LET) (например, бета-излучатели), излучателей конверсионных электронов (например, стронций-89 и самарий-153-ЭДТФА), или излучения высокой энергии, включая, но не ограничиваясь, рентгеновские лучи, гамма-лучи и нейтроны.
«Пролекарство» обозначает соединение, которое может быть превращено в физиологических условиях или путем сольволиза в биологически активное соединение, описанное в настоящем документе. Поэтому термин «пролекарство» относится к предшественникам биологически активного соединения, которое является фармацевтически приемлемым. Пролекарство может быть неактивным при введении пациенту, но превращаться in vivo в активное соединение, например, путем гидролиза. Пролекарственное соединение часто обладает преимуществом растворимости, совместимости с тканью или замедленным высвобождением в организме млекопитающего (см., например, Bundgard, Н., Design of Prodrugs (1985), pp.7-9, 21-24 (Elsevier, Amsterdam). Общий обзор пролекарств представлен в публикации Higuchi, Т., et al, Pro-drugs as Novel Delivery Systems, A.C.S. Symposium Series, T.14, а также в публикации Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, включенных сюда путем ссылки в полном объеме. Термин «пролекарство» также включает все ковалентно связанные носители, которые высвобождают активное соединение in vivo при введении такого пролекарственные средства млекопитающему. Пролекарственные средства активного соединения, описанные в настоящем документе, могут быть получены модификацией функциональных групп, присутствующих в активном соединении, таким образом, что эти модификации расщепляются обычными приемами или in vivo до родительского активного соединения. Пролекарственные средства включают соединения, в которых гидрокси, амино или меркапто-группа связана с любой группой, которая при введении пролекарственные средства активного соединения млекопитающему, расщепляется с образованием свободной гидроксильной, свободной амино или свободной меркапто-группы, соответственно. Примеры пролекарств включают, не ограничиваясь, ацетатные, формиатные и бензоатные производные спирта, или ацетамидные, формамидные или бензамидные производные аминной функциональной группы активного соединения и т.п.
Термин «in vivo» относится к событию, происходящему в организме субъекта.
Термин «in vitro» относится к событию, происходящему за пределами организма субъекта. Например, анализ in vitro включает анализы, выполняемые вне организма субъекта. Анализы in vitro включают анализы на основе клеток, в которых используются живые или мертвые клетки. Анализы in vitro включают также анализы без участия клеток, в которых не используются интактные клетки.
Если не указано иное, то структуры, представленные в настоящем документе, также включают соединения, которые отличаются только наличием одного или более изотопных атомов. Например, соединения, имеющие действительные структуры, где водород замещен дейтерием или тритием, или где атом углерода замещен углеродом13С или14С, также входят в рамки настоящего изобретения.
Соединения по настоящему изобретению могут содержать также неестественные пропорции атомных изотопов у одного или более атомов, образующих эти соединения. Например, соединения могут быть мечены радиоактивными изотопами, как, например, тритием (3Н), йодом-125 (125I) или углеродом-14 (14С). Все изотопные варианты соединений настоящего изобретения, радиоактивные или не радиоактивные, включены в рамки настоящего изобретения.
Если для физических свойств, таких как молекулярный вес, или для химических свойств, таких как химическая формула, используется диапазон, то подразумеваются включенными все комбинации и подкомбинации диапазонов и конкретных вариантов воплощения изобретения. Термин «примерно» при упоминании числа или числового диапазона означает, что число или числовой диапазон упоминается в приближении относительно экспериментальной вариабельности (или в пределах статистической экспериментальной ошибки), и поэтому число или числовой диапазон могут варьировать, например, от 1% до 15% от указанного числа или числового диапазона. Термин «включение» (и родственные термины, такие как «включают» или «включает», или «имеет», или «содержит») включает эти варианты воплощения изобретения, например, вариант любой композиции вещества, композиции, способа или процесса и т.п., который “состоит из” или “состоит, в основном, из” указанных характеристик.
Следующие аббревиатуры и термины имеют в настоящем документе указанные значения:
PI3-K = Фосфоинозитид 3-киназа; PI = фосфатидил инозитол; PDK = фосфоинозитид зависимая киназа; ДНК-РК = протеинкиназа, зависящая от дезоксирибонуклеиновой кислоты; PTEN = гомолог фосфатазы и тензина, удаленный из десятой хромосомы; PIKK = фосфоинозитид-киназа-подобная киназа; СПИД = синдром приобретенного иммунодефицита; ВИЧ = вирус иммунодефицита человека; MeI = метилйодид; POCl3 = оксихлорид фосфора; KCNS = изотиоцианат калия; ТСХ = тонкослойная хроматография; MeOH = метанол; и CHCl3 = хлороформ.
Используемые в настоящем документе аббревиатуры имеют обычное значение в рамках химической и биологической наук.
«Алкил» относится к углеводородному радикалу с прямой или разветвленной цепью, состоящему только из атомов углерода и водорода, не содержащему ненасыщенности, имеющему от одного до десяти углеродных атомов (например, C1-C10алкил). При появлении в настоящем документе числового диапазона, такого как «от 1 до 10», он относится к каждому целому числу в данном диапазоне; например, “от 1 до 10 углеродных атомов” означает, что алкильная группа может состоять из 1 углеродного атома, 2 углеродных атомов, 3 углеродных атомов и т.п., вплоть до и включая 10 углеродных атомов, при этом настоящее определение включает также наличие термина “алкил” в тех местах, где числовой диапазон не обозначен. В некоторых воплощениях изобретения он является C1-C4алкильной группой. Типичные алкильные группы включают, не ограничиваясь, метил, этил, пропил, изопропил, н-бутил, изо-бутил, втор-бутил изобутил, трет-бутил, пентил, изо-пентил, неопентил, гексил, септил, октил, нонил, децил и т.п. Алкил является присоединенным к остальной молекуле одинарной связью, например, метил (Me), этил (Et), н-пропил, 1-метилэтил (изо-пропил), н-бутил, н-пентил, 1,1-диметилэтил (трет-бутил), 3-метилгексил, 2-метилгексил и т.п. Если в настоящем описании особо не оговорено обратное, то алкильная группа является возможно замещенной одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, трифторметилом, трифторметокси, нитро, триметилсиланилом, -ORa, -SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)C(O)N(Ra)2, N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом.
«Алкиларил» относится к -(алкил)ариловому радикалу, где арил и алкил являются такими, как описано в настоящем документе, и которые являются возможно замещенными одним или более заместителями, описанными как подходящие заместители для арила и алкила, соответственно.
«Алкилгетарил» относится к -(алкил)гетариловому радикалу, где гетарил и алкил являются такими, как описано в настоящем документе, и которые являются возможно замещенными одним или более заместителями, описанными как подходящие заместители для арила и алкила, соответственно.
«Алкилгетероциклоалкил» относится к -(алкил)гетероциклиловому радикалу, где алкил и гетероциклоалкил являются такими, как описано в настоящем документе, и которые являются возможно замещенными одним или более заместителями, описанными как подходящие заместители для гетероциклоалкила и алкила, соответственно.
«Алкеновая» группа относится к группе, состоящей по меньшей мере из двух углеродных атомов и по меньшей мере одной двойной углерод-углеродной связи, и «алкиновая» группа относится к группе, состоящей по меньшей мере из двух углеродных атомов и по меньшей мере одной тройной углерод-углеродной связи. Алкильная группа, насыщенная или ненасыщенная, может быть разветвленной, прямоцепочечной или циклической.
«Алкенил» относится к группе углеводородного радикала с прямой или разветвленной цепью, состоящей только из атомов углерода и водорода, содержащей по меньшей мере одну двойную связь и имеющей от двух до десяти углеродных атомов (т.е. C2-C10алкенил). При появлении в настоящем документе числового диапазона, такого как «от 2 до 10», он относится к каждому целому числу в данном диапазоне; например, «от 2 до 10 углеродных атомов» означает, что алкениловая группа может состоять из 2 углеродных атомов, 3 углеродных атомов и т.п., вплоть до и включая 10 углеродных атомов. В некоторых воплощениях изобретения алкенил включает от двух до восьми углеродных атомов. В других воплощениях изобретения алкенил включает от двух до пяти углеродных атомов (например, C2-C5алкенил). Алкенил присоединен к остальной части молекулы одинарной связью, например, этенил (т.е. винил), проп-1-енил (т.е. аллил), бут-1-енил, пент-1-енил, пента-1,4-диенил и т.п. Если в настоящем описании особо не оговорено обратное, то алкениловая группа является возможно замещенной одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, трифторметилом, трифторметокси, нитро, триметилсиланилом, -ORa, -SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)C(O)N(Ra)2, N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом.
«Алкенил-циклоалкил» относится к -(алкенил)циклоалкильному радикалу, где алкенил и циклоалкил являются такими, как описано в настоящем документе, и которые являются возможно замещенными одним или более заместителями, описанными как подходящие заместители для алкенила и циклоалкила, соответственно.
«Алкинил» относится к группе углеводородного радикала с прямой или разветвленной цепью, состоящей только из атомов углерода и водорода, содержащей по меньшей мере одну тройную связь и имеющей от двух до десяти углеродных атомов (т.е. C2-C10алкинил). При появлении в настоящем документе числового диапазона, такого как «от 2 до 10», он относится к каждому целому числу в данном диапазоне; например, «от 2 до 10 углеродных атомов» означает, что алкиниловая группа может состоять из 2 углеродных атомов, 3 углеродных атомов и т.п., вплоть до и включая 10 углеродных атомов. В некоторых воплощениях изобретения алкинил включает от двух до восьми углеродных атомов. В других воплощениях изобретения алкинил включает от двух до пяти углеродных атомов (например, C2-C5алкинил). Алкинил присоединен к остальной части молекулы одинарной связью, например, этинил, пропинил, бутинил, пентинил, гексинил и т.п. Если в настоящем описании особо не оговорено обратное, то алкиниловая группа является возможно замещенной одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, трифторметилом, трифторметокси, нитро, триметилсиланилом, -ORa, -SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)C(O)N(Ra)2, N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом.
«Алкинил-циклоалкил» относится к -(алкинил)циклоалкильному радикалу, где алкинил и циклоалкил являются такими, как описано в настоящем документе, и которые являются возможно замещенными одним или более заместителями, описанными как подходящие заместители для алкинила и циклоалкила, соответственно.
«Карбоксальдегид» относится к радикалу -(С=O)Н.
«Карбоксил» относится к радикалу -(С=O)ОН.
«Циано» относится к радикалу -CN.
«Циклоалкил» относится к моноциклическому или полициклическому радикалу, содержащему только углерод и водород, и может быть насыщенным или частично ненасыщенным. Циклоалкильные группы включают группы, имеющие от 3 до 10 кольцевых атомов (т.е. C2-C10циклоалкил). При появлении в настоящем документе числового диапазона, такого как «от 3 до 10», он относится к каждому целому числу в данном диапазоне; например, «от 3 до 10 углеродных атомов» означает, что циклоалкильная группа может состоять из 3 углеродных атомов и т.п., вплоть до и включая 10 углеродных атомов. В некоторых воплощениях изобретения он является C3-C8циклоалкильным радикалом. В некоторых воплощениях изобретения он является C3-C5циклоалкильным радикалом. Иллюстративные примеры циклоалкильных групп включают, не ограничиваясь, следующие группы: циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклосептил, циклооктил, циклононил, циклодецил, норборнил и т.п. Если в настоящем описании особо не оговорено обратное, то циклоалкильная группа является возможно замещенной одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, трифторметилом, трифторметокси, нитро, триметилсиланилом, -ORa, -SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)C(O)N(Ra)2, N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом.
«Циклоалкил-алкенил» относится к -(циклоалкил)алкениловому радикалу, где циклоалкил и гетероциклоалкил являются такими, как описано в настоящем документе, и которые являются возможно замещенными одним или более заместителями, описанными как подходящие заместители для гетероциклоалкила и циклоалкила, соответственно.
«Циклоалкил-гетероциклоалкил» относится к -(циклоалкил)гетероциклиловому радикалу, где циклоалкил и гетероциклоалкил являются такими, как описано в настоящем документе, и которые являются возможно замещенными одним или более заместителями, описанными как подходящие заместители для гетероциклоалкила и циклоалкила, соответственно.
«Циклоалкил-гетероарил» относится к -(циклоалкил)гетероариловому радикалу, где циклоалкил и гетероциклоалкил являются такими, как описано в настоящем документе, и которые являются возможно замещенными одним или более заместителями, описанными как подходящие заместители для гетероциклоалкила и циклоалкила, соответственно.
Термин «алкокси» относится к группе -О-алкил, включающей от 1 до 8 углеродных атомов прямого, разветвленного или циклического строения и их комбинации, присоединенной к родительской структуре через кислород. Примеры включают метокси, этокси, пропокси, изопропокси, циклопропилокси, циклогексилокси и т.п. «Низший алкокси» относится к алкокси-группам, содержащим от одного до шести углеродов. В некоторых воплощениях изобретения C1-C4алкил является алкильной группой, включающей алкилы прямого и разветвленного строения, состоящие из 1-4 углеродных атомов.
Термин «замещенный алкокси» относится к алкокси, где алкильная часть является замещенной (т.е., -O-(замещенный алкил)). Если в настоящем описании особо не оговорено обратное, то алкильная часть алкокси-группы является возможно замещенной одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, трифторметилом, трифторметокси, нитро, триметилсиланилом, -ORa, -SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)C(O)N(Ra)2, N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом.
Термин «алкоксикарбонил» относится к группе формулы (алкокси)(C=О)-, присоединенной через углерод карбонила, где алкокси-группа имеет указанное количество углеродных атомов. Так, C1-C6алкоксикарбониловая группа является алкокси-группой, имеющей от 1 до 6 углеродных атомов, присоединенных через кислород к карбонильному мостику. «Низший алкоксикарбонил» относится к алкоксикарбонильной группе, где алкокси-группа является низшей алкокси-группой. В некоторых воплощениях изобретения C1-C4алкокси является алкокси-группой, включающей алкокси с прямой и разветвленной цепью, состоящие из 1-4 углеродных атомов.
Термин «замещенный алкоксикарбонил» относится к группе (замещенный алкил)-О-С(О)-, где эта группа присоединена к родительской структуре через карбонильную группу. Если в настоящем описании особо не оговорено обратное, то алкильная часть алкоксикарбонильной группы является возможно замещенной одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, трифторметилом, трифторметокси, нитро, триметилсиланилом, -ORa, SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)C(O)N(Ra)2, N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом.
"Ацил" относится к группам (алкил)-С(О)-, (арил)-С(О)-, (гетероарил)-С(О)-, (гетероалкил)-С(О)- и (гетероциклоалкил)-С(О)-, где эта группа является присоединенной к родительской структуре через карбонильную группу. В некоторых воплощениях изобретения он является C1-C10ацильным радикалом, что указывает на общее количество атомов в цепи или кольце алкильной, арильной, гетероарильной или гетероциклоалкильной части ацилокси-группы плюс карбонильный углерод ацила, т.е. три других кольцевых или цепных атома плюс карбонил. Если радикал R является гетероарилом или гетероциклоалкилом, то гетероатомы кольца или цепи вносят свой вклад в общее количество атомов цепи или кольца. Если в настоящем описании особо не оговорено обратное, то «R» в ацилокси-группе является возможно замещенной одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, трифторметилом, трифторметокси, нитро, триметилсиланилом, -ORa, SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)C(O)N(Ra)2, N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом.
«Ацилокси» относится к радикалу R(C=O)O-, где «R» является алкилом, арилом, гетероарилом, гетероалкилом или гетероциклоалкилом, которые являются такими, как описано в настоящем документе. В некоторых воплощениях изобретения он является C1-C4ацилокси-радикалом, что указывает на общее количество атомов в цепи или кольце алкильной, арильной, гетероарильной или гетероциклоалкильной части ацилокси-группы плюс карбонильный углерод ацила, т.е. три других кольцевых или цепных атома плюс карбонил. Если радикал R является гетероарилом или гетероциклоалкилом, то гетероатомы кольца или цепи вносят свой вклад в общее количество атомов цепи или кольца. Если в настоящем описании особо не оговорено обратное, то «R» в ацилокси-группе является возможно замещенным одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, трифторметилом, трифторметокси, нитро, триметилсиланилом, -ORa, -SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)C(O)N(Ra)2, N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом.
«Амино» или «амин» относится к радикальной группе -N(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом, если в настоящем описании специально не указано обратное. Если группа -N(Ra)2 имеет два Ra, отличных от водорода, они могут объединяться с атомом азота с образованием 4-, 5-, 6- или 7-членного кольца. Например, подразумевают, что -N(Ra)2 включает, не ограничиваясь, 1-пирролидинил и 4-морфолинил. Если в настоящем описании особо не оговорено обратное, то аминогруппа является возможно замещенной одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, трифторметилом, трифторметокси, нитро, триметилсиланилом, -ORa, -SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)C(O)N(Ra)2, N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом, и каждая из этих групп может быть возможно замещена так, как описано в настоящем документе.
Термин «замещенный амино» также относится к N-оксидам групп -NHRd и NRdRd, каждая из которых является такой, как описано выше. N-оксиды могут быть получены обработкой соответствующей аминогруппы, например, перекисью водорода или мета-хлорпероксибензойной кислотой. Специалисту в данной области знакомы условия реакций для выполнения N-оксидирования.
«Амид» или «амидо» относится к химической группе формулы -C(O)N(R)2 или -NHC(O)R, где R выбран из группы, состоящей из водорода, алкила, циклоалкила, арила, гетероарила (связанного через кольцевой углерод) и гетероалициклила (связанного через кольцевой углерод), и каждая из этих групп может сама быть возможно замещенной. В некоторых воплощениях изобретения он является C1-C4амидо или амидным радикалом, что включает амидный карбонил в общее количество атомов углерода в радикале. R2 в -N(R)2 амида может быть возможно взят вместе с азотом, к которому он присоединен, с образованием 4-, 5-, 6- или 7-членного кольца. Если в настоящем описании специально не указано обратное, то амидогруппа является возможно замещенной независимо одним или более заместителей, как описано в настоящем документе для алкила, циклоалкила, арила, гетероарила или гетероциклоалкила. Амид может быть молекулой аминокислоты или пептида, присоединенной к соединению формулы (I), образуя, таким образом, пролекарство. Любая аминная, гидроксильная или карбоксильная боковая цепь на соединении, описанном в настоящем документе, может быть амидирована. Способы и специальные группы для получения таких амидов известны специалистам в данной области и легко могут быть найдены в справочных источниках, таких как Greene and Wuts, Protective Groups in Organic Synthesis, 3.sup.rd Ed., John Wiley & Sons, New York, N.Y., 1999, который включен в настоящую заявку путем ссылки в полном объеме.
«Ароматический» или «арил» относится к ароматическому радикалу из шестидесяти кольцевых атомов (например, C6-C10ароматический или C6-C10арил), который имеет по меньшей мере одно кольцо, обладающее конъюгированной пи-электронной системой, и является карбоциклическим (например, фенил, флуоренил и нафтил). Двухвалентные радикалы, полученные из замещенных производных бензола и имеющие свободные валентности у кольцевых атомов, имеют названия, как у замещенных фениленовых радикалов. Двухвалентные радикалы, полученные из одновалентных полициклических углеводородных радикалов, названия которых оканчиваются на «-ил», путем удаления одного атома водорода от атома углерода со свободной валентностью, называются путем добавления «-илиден» к названию соответствующего одновалентного радикала, например, нафтильная группа с двумя точками присоединения имеет название нафтилиден. При появлении в настоящем документе числового диапазона, такого как «от 6 до 10», он относится к каждому целому числу в данном диапазоне; например, «от 6 до 10 кольцевых атомов» означает, что ариловая группа может состоять из 6 кольцевых атомов, 7 кольцевых атомов и т.п., вплоть до и включая 10 кольцевых атомов. Этот термин включает моноциклические или полициклические группы с конденсированными кольцами (т.е. кольца, которые имеют общие соседние пары кольцевых атомов). Если в настоящем описании особо не оговорено обратное, то арильная группа является возможно замещенной одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, трифторметилом, трифторметокси, нитро, триметилсиланилом, -ORa, -SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)C(O)N(Ra)2, N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом.
«Аралкил» или «арилалкил» относится к (арил)алкил- радикалу, где арил и алкил являются такими, как описано в настоящем документе, и которые являются возможно замещенными одним или более заместителями, описанными как подходящие заместители для арила и алкила, соответственно.
«Сложный эфир» относится к химическому радикалу формулы -COOR, где R выбран из группы, состоящей из алкила, циклоалкила, арила, гетероарила (связанного через кольцевой углерод) и гетероалициклила (связанного через кольцевой углерод). Любая аминная, гидроксильная или карбоксильная боковая цепь на соединениях, описанных в настоящем документе, может быть подвержена эстерификации. Способы и специальные группы для получения таких сложных эфиров известны специалистам в данной области и легко могут быть найдены в справочных источниках, таких как Greene and Wuts, Protective Groups in Organic Synthesis, 3.sup.rd Ed., John Wiley & Sons, New York, N.Y., 1999, который включен в настоящую заявку путем ссылки в полном объеме. Если в настоящем описании особо не оговорено обратное, то сложноэфирная группа является возможно замещенной одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, трифторметилом, трифторметокси, нитро, триметилсиланилом, -ORa, -SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -OC(O)N(Ra)2, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)C(O)N(Ra)2, N(Ra)C(NRa)N(Ra)2, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом.
«Фторалкил» относится к алкильному радикалу, как описано выше, замещенному одним или более фтор-радикалов, как описано выше, например, трифторметил, дифторметил, 2,2,2-трифторэтил, 1-фторметил-2-фторэтил и т.п. Алкильная часть фторалкильного радикала может быть возможно замещена, как описано выше для алкильной группы.
«Галогено», «галид» или, альтернативно, «галоген» означает фторо, хлоро, бромо или йодо. Термины «галогеноалкил», «галогеноалкенил», «галогеноалкинил» и «галогеноалкокси» включают алкильные, алкениловые, алкиниловые и алкокси-структуры, которые замещены одной или более гало-групп или их комбинацией. Например, термины «фторалкил» и «фторалкокси» включают галогеноалкильные и галоалкокси-группы, соответственно, в которых галогеном является фтор.
«Гетероалкил», «гетероалкенил» и «гетероалкинил» включают возможно замещенные алкильные, алкениловые и алкиниловые радикалы, которые имеют один или более атомов скелетной цепи, отличный от углерода, например, кислород, азот, серу, фосфор или их комбинации. Может быть задан числовой диапазон, например, C1-C4гетероалкил, что относится к длине цепи в целом, которая в данном примере имеет 4 атома в длину. Например, радикал -CH2OCH2CH3 упоминается как “С4”гетероалкил, который включает гетероатомный центр в описании длины цепи атомов. Связь с остальной частью молекулы может осуществляться через гетероатом или через углерод гетероалкильной цепи. Гетероалкильная группа может быть замещена одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, нитро, оксо, тиоксо, триметилсиланилом, -ORa, -SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом.
«Гетероалкиларил» относится к -(гетероалкил)ариловому радикалу, где гетероалкил и арил являются такими, как описано в настоящем документе, и которые являются возможно замещенными одним или более заместителями, описанными как подходящие заместители для гетероалкила и арила, соответственно.
«Гетероалкилгетероарил» относится к -(гетероалкил)гетероариловому радикалу, где гетероалкил и гетероарил являются такими, как описано в настоящем документе, и которые являются возможно замещенными одним или более заместителями, описанными как подходящие заместители для гетероалкила и гетероарила, соответственно.
«Гетероалкилгетероциклоалкил» относится к -(гетероалкил)-гетероциклоалкильному радикалу, где гетероалкил и гетероарил являются такими, как описано в настоящем документе, и которые являются возможно замещенными одним или более заместителями, описанными как подходящие заместители для гетероалкила и гетероциклоалкила, соответственно.
«Гетероалкилциклоалкил» относится к -(гетероалкил)циклоалкильному радикалу, где гетероалкил и циклоалкил являются такими, как описано в настоящем документе, и которые являются возможно замещенными одним или более заместителями, описанными как подходящие заместители для гетероалкила и циклоалкила, соответственно.
«Гетероарил» или, альтернативно, «гетероароматический» относится к 5-18-членному ароматическому радикалу (например, C5-C13гетероарилу), который включает один или более кольцевых гетероатомов, выбранных из азота, кислорода и серы, и который может быть моноциклической, бициклической, трициклической или тетрациклической кольцевой системой. При появлении в настоящем документе числового диапазона, такого как «от 5 до 18», он относится к каждому целому числу в данном диапазоне; например, «от 5 до 18 кольцевых атомов» означает, что гетероариловая группа может состоять из 5 кольцевых атомов, 6 кольцевых атомов и т.п., вплоть до и включая 18 кольцевых атомов. Двухвалентные радикалы, полученные из одновалентных гетероарильных радикалов, название которых заканчивается на «-ил», путем удаления одного атома водорода от атома со свободной валентностью, называются путем добавления «-илиден» к названию соответствующего одновалентного радикала, например, пиридильная группа с двумя точками присоединения является пиридилиденом. N-содержащие «гетероароматические» или «гетероарильные» группы относится к ароматическим группам, в которых по меньшей мере один скелетный атом кольцевой системы является атомом азота. Полициклическая гетероарильная группа может быть конденсированной или неконденсированной. Гетероатом(ы) в гетероарильном радикале является(ются) возможно окисленным(и). Один или более атомов азота, при их наличии, являются возможно кватернизованными. Гетероарил присоединен к остальной части молекулы через любой атом кольца(колец). Примеры гетероарилов включают, не ограничиваясь, азепинил, акридинил, бензимидазолил, бензиндолил, 1,3-бензодиоксолил, бензофуранил, бензооксазолил, бензо[d]тиазолил, бензотиадиазолил, бензо[b][1,4]диоксепинил, бензо[b][1,4]оксазинил, 1,4-бензодиоксанил, бензонафтофуранил, бензоксазолил, бензодиоксолил, бензодиоксинил, бензоксазолил, бензопиранил, бензопиранонил, бензофуранил, бензофуранонил, бензофуразанил, бензотиазолил, бензотиенил (бензотиофенил), бензотиено[3,2-d]пиримидинил, бензотриазолил, бензо[4,6]имидазо[1,2-а]пиридинил, карбазолил, циннолинил, циклопента[d]пиримидинил, 6,7-дигидро-5Н-циклопента[4,5]тиено[2,3-h]пиримидинил, 5,6-дигидробензо[h]хиназолинил, 5,6-дигидробензо[b]циннолинил, 6,7-дигидро-5Н-бензо[6,7]циклогепта[1,2-с]пиридазинил, дибензофуранил, дибензотиофенил, фуранил, фуразанил, фуранонил, фуро[3,2-с]пиридинил, 5,6,7,8,9,10-гексагидроциклоокта[d]пиримидинил, 5,6,7,8,9,10-гексагидроциклоокта[d]пиридазинил, 5,6,7,8,9,10-гексагидроциклоокта[d]пиридинил, изотиазолил, имидазолил, индазолил, индолил, индазолил, изоиндолил, индолинил, изоиндолинил, изохинолил, индолизинил, изоксазолил, 5,8-метано-5,6,7,8-тетрагидрохиназолинил, нафтиридинил, 1,6-нафтиридинонил, оксадиазолил, 2-оксоазепинил, оксазолил, оксиранил, 5,6,6а,7,8,9,10,10а-октагидробензо[h]хинозолинил, 1-фенил-1H-пирролил, феназинил, фенотиазинил, феноксазинил, фталазинил, птеридинил, пуринил, пиранил, пирролил, пиразолил, пиразоло[3,4-d]пиримидинил, пиридинил, пиридо[3,2-d]пиримидинил, пиридо[3,4-d]пиримидинил, пиразинил, пиримидинил, пиридазинил, пирролил, хиназолинил, хиноксалинил, хинолинил, изохинолинил, тетрагидрохинолинил, 5,6,7,8-тетрагидрохиназолинил, 5,6,7,8-тетрагидробензо[4,5]тиено[2,3-d]пиримидинил, 6,7,8,9-тетрагидро-5Н-циклогепта[4,5]тиено[2,3-d]пиримидинил, 5,6,7,8-тетрагидропиридо[4,5-с]пиридазинил, тиазолил, тиадиазолил, тиапиранил, триазолил, тетразолил, триазинил, тиено[2,3-d]пиримидинил, тиено[3,2-d]пиримидинил, тиено[2,3-с]пиридинил и тиофенил (например, тиенил). Если в настоящем описании особо не оговорено обратное, то гетероарильная группа является возможно замещенной одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, нитро, оксо, тиоксо, триметилсиланилом, -ORa, -SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2), где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом.
Замещенный гетероарил включает также кольцевые системы, замещенные одним или более оксидными (-O-) заместителями, такими как пиридинил N-оксиды.
«Гетероарилалкил» относится к группе, имеющей ариловую часть, как описано в настоящем документе, связанную с алкиленовой частью, как описано в настоящем документе, где эта связь с остальной частью молекулы осуществляется через алкиленовую группу.
«Гетероциклоалкил» относится к устойчивому 3-18-членному неароматическому кольцевому радикалу, который включает от двух до двенадцати углеродных атомов и от одного до шести гетероатомов, выбранных из азота, кислорода и серы. При появлении в настоящем документе числового диапазона, такого как «от 3 до 18», он относится к каждому целому числу в данном диапазоне; например, «от 3 до 18 кольцевых атомов» означает, что гетероциклоалкильная группа может состоять из 3 кольцевых атомов, 4 кольцевых атомов и т.п., вплоть до и включая 18 кольцевых атомов. В некоторых воплощениях он является С5-С10гетероциклоалкилом. В некоторых воплощениях изобретения он является С4-С10гетероциклоалкилом. В некоторых воплощениях изобретения он является С3-С10гетероциклоалкилом. Если в настоящем описании специально не указано обратное, то гетероциклоалкильный радикал является моноциклической, бициклической, трициклической или тетрациклической кольцевой системой и может включать конденсированные или мостиковые кольцевые системы. Гетероатомы в гетероциклоалкильном радикале могут быть возможно окислены. Один или более атомов азота, при их наличии, являются возможно кватернизованными. Гетероциклоалкильный радикал является частично или полностью насыщенным. Гетероциклоалкил может быть присоединен к остальной части молекулы через любой атом кольца(колец). Примеры таких гетероциклоалкильных радикалов включают, не ограничиваясь, диоксоланил, тиенил[1,3]дитианил, декагидроизохинолил, имидазолинил, имидазолидинил, изотиазолидинил, изоксазолидинил, морфолинил, октагидроиндолил, октагидроизоиндолил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, оксазолидинил, пиперидинил, пиперазинил, 4-пиперидонил, пирролидинил, пиразолидинил, хинуклидинил, тиазолидинил, тетрагидрофурил, тритианил, тетрагидропиранил, тиоморфолинил, тиаморфолинил, 1-оксо-тиоморфолинил и 1,1-диоксо-тиоморфолинил. Если в настоящем описании особо не оговорено обратное, то гетероциклоалкильная группа является возможно замещенной одним или более заместителями, которые независимо являются: алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, гидрокси, галогено, циано, нитро, оксо, тиоксо, триметилсиланилом, -ORa, -SRa, -OC(O)-Ra, -N(Ra)2, -C(O)Ra, -C(O)ORa, -C(O)N(Ra)2, -N(Ra)C(O)ORa, -N(Ra)C(O)Ra, -N(Ra)S(O)tRa (где t равен 1 или 2), -S(O)tORa (где t равен 1 или 2), -S(O)tN(Ra)2 (где t равен 1 или 2) или PO3(Ra)2, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероарилом или гетероарилалкилом.
«Гетероциклоалкил» также включает бициклические кольцевые системы, где одно неароматическое кольцо обычно из 3-7 кольцевых атомов содержит по меньшей мере 2 углеродных атома помимо 1-3 гетероатомов, независимо выбранных из кислорода, серы и азота, а также комбинации, включающие по меньшей мере один из вышеупомянутых гетероатомов; а также другие кольца обычно из 3-7 кольцевых атомов, возможно содержащие 1-3 гетероатома, независимо выбранные из кислорода, серы и азота и не являющиеся ароматическими.
«Изомеры» являются различными соединениями, имеющими одинаковую молекулярную формулу. «Стереоизомеры» являются изомерами, которые отличаются только способом расположения атомов в пространстве, т.е. имеют различную стереохимическую конфигурацию. «Энантиомеры» представляют собой пару стереоизомеров, которые не являются зеркальными отражениями друг друга. Смесь пары изомеров 1:1 является «рацемической» смесью. Термин «(.±.)» используется для обозначения рацемической смеси, где это уместно. «Диастереоизомеры» являются стереоизомерами, которые имеют по меньшей мере два асимметричных атома, но не являются зеркальными отражениями друг друга. Абсолютная стереохимия устанавливается в соответствии с R-S системой Кана-Ингольда-Прелога. Если соединение является чистым энантиомером, то стереохимия каждого хирального углерода может быть определена как R или S. Анализируемые соединения, абсолютная конфигурация которых является неизвестной, могут быть обозначены (+) или (-), в зависимости от направления (право- или левовращающий), в котором они вращают плоско-поляризованный свет при длине волны линии D натрия. Некоторые соединения, описанные в настоящем документе, содержат один или более асимметричных центров и поэтому могут образовывать энантиомеры, диастереомеры и другие стереоизомерные формы, которые могут быть определены, в терминах абсолютной стереохимии, как (R)-или (S)-. Химические объекты, фармацевтические композиции и способы настоящего изобретения включают все возможные изомеры, включая рацемические смеси, оптически чистые формы и промежуточные смеси. Оптически активные (R)- и (S)-изомеры могут быть получены с использованием хиральных синтонов или хиральных реагентов, или определены при помощи обычных методик. Если описанные в настоящем документе соединения содержат олефиновые двойные связи или другие центры геометрической асимметрии, и если не указано обратное, то подразумевается, что эти соединения включают Е и Z геометрические изомеры.
Термин «энантиомерная чистота», используемый в настоящем документе, относится к относительным количествам, выраженным в процентах, присутствия конкретного энантиомера по отношению к другому энантиомеру. Например, если соединение, которое потенциально может иметь (R)- или (S)-изомерную конфигурацию, присутствует в виде рацемической смеси, то энантиомерная чистота составляет примерно 50% по отношению (R)- или (S)-изомеру. Если одна изомерная форма этого соединения преобладает над другой, например, 80% (S)- и 20% (R)-, то энантиомерная чистота соединения в пересчете на (S)-изомерную форму составляет 80%. Энантиомерная чистота соединения может быть определена различными способами, известными в данной области, включая, не ограничиваясь, хроматографию с использованием хирального носителя, поляриметрическое измерение вращения поляризованного света, спектроскопию ядерного магнитного резонанса с использованием хиральных сдвиговых реагентов, которые включают, но не ограничиваясь, лантаноид-содержащие хиральные комплексы спирта Пиркла или превращение соединений с использованием хирального соединения, такого как кислота Мошера, с последующей хроматографией или спектроскопией ядерного магнитного резонанса.
«Группа» относится к специфической части или функциональной группе молекулы. Химические группы являются зачастую распознаваемыми химическими структурами, входящими в состав или присоединенными к молекуле.
«Нитро» относится к радикалу -NO2.
«Окса» относится к радикалу -О-.
«Оксо» относится к радикалу =O.
«Таутомеры» являются структурно различными изомерами, которые превращаются друг в друга в результате таутомеризации. «Таутомеризация» является формой изомеризации и включает прототропную или таутомеризацию со сдвигом протона, которая является разновидностью кислотно-основной химии. «Прототропная таутомеризация» или «таутомеризация со сдвигом протона» включает миграцию протона, сопровождающуюся изменением кратности связи, часто взаимным превращением одинарной связи и соседней двойной связи. Если таутомеризация является возможной (например, в растворе), то может быть достигнуто химическое равновесие таутомеров. Примером таутомеризации является кето-енольная таутомеризация. Конкретным примером кето-енольной таутомеризации является взаимное превращение таутомеров пентан-2,4-диона и 4-гидроксипент-3-ен-2-она. Другим примером таутомеризации является фенол-кето таутомеризация. Конкретным примером фенол-кето таутомеризации является взаимное превращение таутомеров пиридин-4-ола и пиридин-4(1Н)-она.
Термин «энантиомерно обогащенный», «энантиомерно чистый» и «нерацемический» используются в настоящем документе взаимозаменяемо и относятся к композициям, в которых весовой процент одного энантиомера больше количества этого энантиомера в контрольной смеси рацемической композиции (например, больше, чем 1:1 по весу). Например, энантиомерно обогащенная композиция (S)-энантиомера означает композицию соединения, в которой содержание (S)-энантиомера составляет более 50% по массе по сравнению с содержанием (R)-энантиомера, более предпочтительно по меньшей мере 75% по массе, и еще более предпочтительно по меньшей мере 80% по массе. В некоторых воплощениях изобретения обогащение может быть гораздо больше, чем 80% по массе, что дает "в значительной степени энантиомерно обогащенную", "в значительной степени энантиомерно чистую" или "в значительной степени нерацемическую" композицию, что относится к композициям, содержащим по меньшей мере 85% по массе одного энантиомера по сравнению с другим энантиомером, более предпочтительно по меньшей мере 90% по массе, и еще более предпочтительно по меньшей мере 95% по массе.
В предпочтительных воплощениях изобретения энантиомерно обогащенная композиция имеет более высокий потенциал в отношении терапевтической полезности на единицу массы, чем рацемическая смесь этой композиции. Энантиомеры могут быть выделены из смесей способами, известными специалистам в данной области, включая хиральную жидкостную хроматографию высокого давления (ЖХВД), и получение и кристаллизацию хиральных солей; или предпочтительные энантиомеры могут быть получены асимметричным синтезом. См., например, Jacques, et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen, S.H., et al., Тетраэдр 33: 2725 (1977); Eliel, E.L. Stereochemistry of Carbon compounds (McGraw-Hill, NY, 1962); и Wilen, S.H. Tables of Resolving Agents and Optical Resolutions p.268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN, 1972).
Соединения по настоящему изобретению могут содержать также необычные пропорции атомных изотопов у одного или более атомов, образующих эти соединения. Например, соединения могут быть мечены радиоактивными изотопами, как, например, тритием (3Н), йодом 125 (125I) или углеродом 14 (14С). Все изотопные варианты соединений настоящего изобретения, радиоактивные или нерадиоактивные, включены в рамки настоящего изобретения.
«Уходящая группа или атом» является любой группой или атомом, которые в условиях реакции отщепляются от исходного материала, активируя реакцию на заданном центре. Подходящими примерами таких групп, если не указано иное, являются атомы галогенов, мезилокси, пара-нитробензолсульфонилокси и тозилокси-группы.
«Защитная группа» имеет значение, которое обычно ассоциируется с защитной группой в органическом синтезе, т.е. группой, которая селективно блокирует один или более реактивных центров в многофункциональном соединении, так что химическая реакция может селективно протекать на другом незащищенном реактивном центре, и эта группа может быть легко удалена после завершения селективной реакции. Описаны различные защитные группы, например, в публикации Т.Н. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, Third Edition, John Wiley & Sons, New York (1999). Например, гидрокси-защищенная форма является такой, где по меньшей мере одна из гидроксильных групп, присутствующих в соединении, защищена гидрокси-защитной группой. Точно так же могут быть защищены аминные и другие реактивные группы.
«Сольват» относится к соединению (например, соединению, выбранному из формулы I или его фармацевтически приемлемой соли), находящемуся в физической связи с одной или более молекул фармацевтически приемлемого растворителя. Следует понимать, что «соединение формулы I» охватывает соединение формулы I и сольваты этого соединения, а также их смеси.
«Замещенный» означает, что указанная группа может быть замещена одной или более дополнительных групп, отдельно и независимо выбранных из ацила, алкила, алкиларила, циклоалкила, аралкила, арила, углевода, карбоната, гетероарила, гетероциклоалкила, гидрокси, алкокси, арилокси, меркапто, алкилтио, арилтио, циано, галогено, карбонила, эфира, тиокарбонила, изоцианато, тиоцианато, изотиоцианато, нитро, оксо, пергалогеноалкила, перфторалкила, фосфата, силила, сульфинила, сульфонила, сульфонамидила, сульфоксила, сульфоната, мочевины и амино, включая моно- и ди-замещенные амино группы, и их защищенные производные. Ди-замещенные аминогруппы включают такие группы, которые образуют кольцо вместе с атомом азота аминогруппы, как, например, морфолино. Сами заместители могут быть замещенными, например, циклоалкильный заместитель может иметь галоидный заместитель у одного или более кольцевых углеродов и т.п.
Защитные группы, которые могут образовывать защищенные производные перечисленных выше заместителей, известны специалистам в данной области, и могут быть найдены в справочных изданиях, таких как Greene and Wuts, указанном выше.
«Сульфанил» относится к группам: -S-(возможно замещенный алкил), -S-(возможно замещенный арил), -S-(возможно замещенный гетероарил) и -S-(возможно замещенный гетероциклоалкил).
«Сульфинил» относится к группам: -S(O)-H, -S(O)-(возможно замещенный алкил), -S(O)-(возможно замещенный амино), -S(O)-(возможно замещенный арил), -S(O)-(возможно замещенный гетероарил) и -S(O)-(возможно замещенный гетероциклоалкил).
«Сульфонил» относится к группам: -S(О2)-Н, -S(O2)-(возможно замещенный алкил), -S(O2)-(возможно замещенный амино), -S(O2)-(возможно замещенный арил), -S(O2)-(возможно замещенный гетероарил) и -S(O2)-(возможно замещенный гетероциклоалкил).
«Сульфонамидил» или «сульфонамидо» относится к радикалу -S(=O)2-NRR, где каждый R независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, арила, гетероарила (связанного через кольцевой углерод) и гетероалициклила (связанного через кольцевой углерод). Группы R в -NRR радикала -S(=O)2-NRR могут быть возможно взяты вместе с азотом, к которому они присоединены, с образованием 4-, 5-, 6- или 7-членного кольца. В некоторых воплощениях изобретения он является C1-C10сульфонамидо, где каждый R в сульфонамидо содержит 1 углерод, 2 углерода, 3 углерода или 4 углерода в целом. Сульфонамидо-группа является возможно замещенной одним или более заместителей, описанных для алкила, циклоалкила, арила, гетероарила, соответственно.
«Сульфоксил» относится к радикалу -S(=O)2OH.
«Сульфонат» относится к радикалу -S(=O)2-OR, где R выбран из группы, состоящей из алкила, циклоалкила, арила, гетероарила (связанного через кольцевой углерод) и гетероалициклила (связанного через кольцевой углерод). Сульфонатная группа является возможно замещенной у R одним или более заместителей, описанных для алкила, циклоалкила, арила, гетероарила, соответственно.
Если замещающие группы указаны в виде своих обычных химических формул, записанных слева направо, они в равной степени охватывают химически идентичные заместители, которые могут получаться при записи этой структуры справа налево, например, -CH2O- является эквивалентом -OCH2-.
Соединения по настоящему изобретению включают также кристаллические и аморфные формы этих соединений, включая, например, полиморфы, псевдополиморфы, сольваты, гидраты, несольватированные полиморфы (включая ангидриды), конформационные полиморфы и аморфные формы этих соединений, а также их смеси. «Кристаллическая форма», «полиморф» и «новая форма» могут использоваться в настоящем документе взаимозаменяемо и включают все кристаллические и аморфные формы соединения, включая, например, полиморфы, псевдополиморфы, сольваты, гидраты, несольватированные полиморфы (включая ангидриды), конформационные полиморфы и аморфные формы, а также их смеси, если не указана конкретная кристаллическая или аморфная форма.
Химические объекты включают, не ограничиваясь, соединения формул I, I-1, IV, IV-A, V, V-A, V-A2, V-B, VI или VI-A, и их фармацевтически приемлемые соли. Фармацевтически приемлемые формы соединений, упомянутых в настоящем документе, включают фармацевтически приемлемые соли, хелаты, нековалентные комплексы, пролекарственные средства и их смеси. В некоторых воплощениях изобретения соединения, описанные в настоящем документе, находятся в форме фармацевтически приемлемых солей. Поэтому термины «химический объект» и «химические объекты» включают также фармацевтически приемлемые соли, хелаты, нековалентные комплексы, пролекарственные средства и их смеси.
Кроме того, если соединение формулы I получено в виде соли присоединения кислоты, то свободное основание может быть получено подщелачиванием раствора кислой соли. И наоборот, если продукт является свободным основанием, солью присоединения, то конкретная фармацевтически приемлемая соль присоединения может быть получена растворением свободного основания в подходящем органическом растворителе и обработкой этого раствора кислотой, в соответствии со стандартными способами получения солей присоединения кислоты из основных соединений. Специалисту в данной области известны различные методики синтеза, которые могут использоваться для получения нетоксичных фармацевтически приемлемых солей.
В одном аспекте настоящего изобретения представлено соединение формулы I:

или его стереоизомеры или фармацевтически приемлемая соль, где
Wd является гетероциклоалкилом, арилом или гетероарилом;
В является алкилом, амино, гетероалкилом или группой формулы II

где Wc является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, и
q является целым числом 0, 1, 2, 3 или 4;
X отсутствует или является -(CH(R9))z-, и z является целым числом 1, 2, 3 или 4;
Y отсутствует или является -О-, -S-, -S(=O)-, -S(=O)2-, -N(R9)-, -C(=O)-(CHR9)z-, -C(=O)-, -N(R9)-C(=O)- или -N(R9)-C(=O)NH-, -N(R9)C(R9)2-, или -C(=O)-(CHR9)z-;
R1 является водородом, алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси, нитро, фосфатом, мочевиной или карбонатом;
R2 является алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси, нитро, фосфатом, мочевиной или карбонатом;
R3 является водородом, алкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси, нитро, арилом или гетероарилом;
R5, R6, R7 и R8 независимо являются водородом, C1-C4алкилом, C2-C5алкенилом, C2-C5алкинилом, C3-C5циклоалкилом, C1-C4гетероалкилом, C1-C4алкокси, C1-C4амидо, амино, ацилом, C1-C4ацилокси, C1-C4сульфонамидо, галогено, циано, гидрокси или нитро; и
каждый R9 независимо является водородом, C1-C10алкилом, С3-С7циклоалкилом, гетероциклоалкилом или C2-C10гетероалкилом.
В некоторых воплощениях изобретения, В является незамещенным или замещенным алкилом, включая, не ограничиваясь, -(CH2)2-NRaRa, где каждый Ra независимо является водородом, алкилом, фторалкилом, карбоциклилом, карбоциклилалкилом, арилом, аралкилом, гетероциклоалкилом, гетероциклоалкилалкилом, гетероарилом или гетероарилалкилом, или NRaRa объединены вместе с образованием циклической группы, которая включает, не ограничиваясь, пиперидинил, пиперазинил и морфолинил. В некоторых воплощениях изобретения, В является незамещенным или замещенным амино. В некоторых воплощениях изобретения, В является незамещенным или замещенным гетероалкилом.

В некоторых воплощениях изобретения, В является группой формулы II, где Wc является членом, выбранным из группы, состоящей из незамещенного или замещенного арила, замещенного фенила, незамещенного или замещенного гетероарила, включая, не ограничиваясь, пиридин-2-ил, пиридин-3-ил, пиридин-4-ил, пиримидин-4-ил, пиримидин-2-ил, пиримидин-5-ил или пиразин-2-ил, незамещенный или замещенный моноциклический гетероарил, незамещенный или замещенный бициклический гетероарил, гетероарил, включающий два гетероатома в качестве кольцевых атомов, незамещенный или замещенный гетероарил, включающий кольцевой атом азота, гетероарил, включающий два кольцевых атома азота, гетероарил, включающий кольцевой атом азота и атом серы, незамещенный или замещенный гетероциклоалкил, включая, но не ограничиваясь, морфолинил, тетрагидропиранил, пиперазинил и пиперидинил, незамещенный или замещенный циклоалкил, включая, но не ограничиваясь, циклопентил и циклогексил.
В некоторых воплощениях изобретения В является одной из следующих групп:
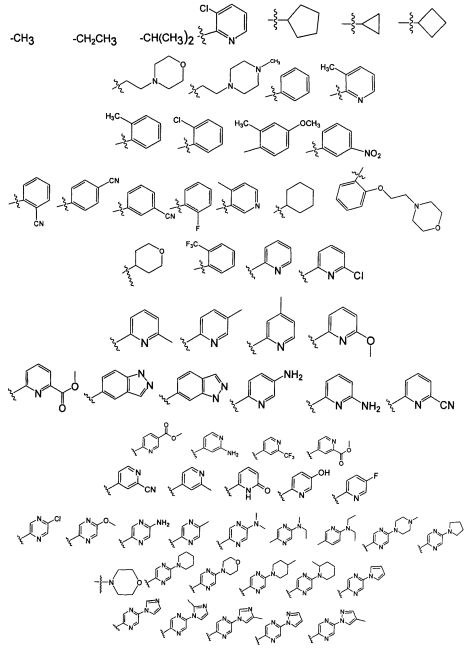

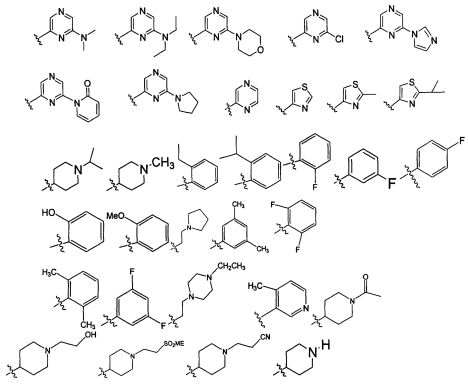
В некоторых воплощениях изобретения В замещен одним или более алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, гетероарилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси или нитро, где каждый алкил, гетероалкил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил, гетероарил, алкокси, амидо, амино, ацил, ацилокси или сульфонамидо может быть замещенным.
В некоторых воплощениях изобретения R1 является членом, выбранным из группы, состоящей из водорода, незамещенного или замещенного алкила, незамещенного или замещенного гетероалкила, незамещенного или замещенного алкенила, незамещенного или замещенного алкинила, незамещенного или замещенного циклоалкила или незамещенного или замещенного гетероциклоалкила. В некоторых воплощениях изобретения R1 является незамещенным или замещенным арилом, незамещенным или замещенным арилалкилом, незамещенным или замещенным гетероарилом, или незамещенным или замещенным гетероарилалкилом. В некоторых воплощениях изобретения R1 является незамещенным или замещенным алкокси, незамещенным или замещенным амидо, незамещенным или замещенным амино. В некоторых воплощениях изобретения R1 является незамещенным или замещенным ацилом, незамещенным или замещенным ацилокси, незамещенным или замещенным алкоксикарбонилом или незамещенным или замещенным сульфонамидо. В некоторых воплощениях изобретения R1 является галогено, который включает -Cl, -F, -I и -Br. В некоторых воплощениях изобретения R1 выбран из группы, состоящей из циано, гидрокси, нитро, незамещенного или замещенного фосфата, незамещенной или замещенной мочевины и карбоната.
В некоторых воплощениях изобретения, если R1 является алкилом, то R1 является метилом, этилом, пропилом, изопропилом, н-бутилом, трет-бутилом, втор-бутилом, пентилом, гексилом или гептилом.
В некоторых воплощениях изобретения, если R1 является алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо или гидрокси, то R1 является замещенным фосфатом, или незамещенной мочевиной, или замещенной мочевиной, или карбоновой кислотой, или карбонатом.
В некоторых воплощениях изобретения, если R1 является алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом или сульфонамидо, то R1 является замещенным одним или более алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси или нитро, где каждый алкил, гетероалкил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил, арилалкил, гетероарил, гетероарилалкил, алкокси, амидо, амино, ацил, ацилокси, алкоксикарбонил или сульфониламидо могут быть замещенными.
В некоторых воплощениях изобретения R2 является членом, выбранным из группы, состоящей из незамещенного или замещенного алкила, незамещенного или замещенного гетероалкила, незамещенного или замещенного алкенила, незамещенного или замещенного алкинила, незамещенного или замещенного циклоалкила и незамещенного или замещенного гетероциклоалкила. В некоторых воплощениях изобретения R2 является незамещенным или замещенным арилом, незамещенным или замещенным арилалкилом, незамещенным или замещенным гетероарилом или незамещенным или замещенным гетероарилалкилом. В некоторых воплощениях изобретения R2 является незамещенным или замещенным алкокси, незамещенным или замещенным амидо, незамещенным или замещенным амино. В некоторых воплощениях изобретения R2 является незамещенным или замещенным ацилом, незамещенным или замещенным ацилокси, незамещенным или замещенным алкоксикарбонилом или незамещенным или замещенным сульфонамидо. В некоторых воплощениях изобретения R2 является галогено, который является -I, -F, -Cl или -Br. В некоторых воплощениях изобретения R2 выбран из группы, состоящей из циано, гидрокси, нитро, карбоновой кислоты и карбоната. В некоторых воплощениях изобретения R2 является незамещенным или замещенным фосфатом. В некоторых воплощениях изобретения R2 является незамещенной или замещенной мочевиной. В некоторых воплощениях изобретения, если R2 является алкилом, то R2 является метилом, этилом, пропилом, изопропилом, н-бутилом, трет-бутилом, втор-бутилом, пентилом, гексилом или гептилом.
В некоторых воплощениях изобретения, если R2 является алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо или гидрокси, то он является замещенным фосфатом или замещенным мочевиной, или замещенным карбонатом.
В некоторых воплощениях изобретения, если R2 является алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, арилалкилом, гетероарилом, гетероарилалкилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом или сульфонамидо, то он является замещенным одним или более алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, гетероарилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси или нитро, где каждый алкил, гетероалкил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил, гетероарил, алкокси, амидо, амино, ацил, ацилокси, алкоксикарбонил или сульфониламидо могут быть замещенными.
В некоторых воплощениях изобретения q является целым числом, равным 0. В некоторых воплощениях изобретения q является целым числом, равным 1. В некоторых воплощениях изобретения q является целым числом, равным 2. В некоторых воплощениях изобретения q является целым числом, равным 3. В некоторых воплощениях изобретения q является целым числом, равным 4.
В некоторых воплощениях соединения формулы I, R3 является членом, выбранным из группы, состоящей из водорода, незамещенного или замещенного алкила, незамещенного или замещенного алкенила и незамещенного или замещенного алкинила. В некоторых воплощениях изобретения R3 является незамещенным или замещенным арилом, незамещенным или замещенным гетероарилом, незамещенным или замещенным циклоалкилом или незамещенным или замещенным гетероциклоалкилом. В некоторых воплощениях изобретения R3 является незамещенным или замещенным алкокси, незамещенным или замещенным амидо, незамещенным или замещенным амино. В некоторых воплощениях изобретения R3 является незамещенным или замещенным ацилом, незамещенным или замещенным ацилокси, незамещенным или замещенным алкоксикарбонилом или незамещенным или замещенным сульфонамидо. В некоторых воплощениях изобретения R3 является галогено, который является -I, -F, -Cl или -Br.
В некоторых воплощениях изобретения R3 выбран из группы, состоящей из циано, гидрокси и нитро. В некоторых воплощениях изобретения, если R3 является алкилом, то R3 является метилом, этилом, пропилом, изопропилом, н-бутилом, трет-бутилом, втор-бутилом, пентилом, гексилом или гептилом. В некоторых воплощениях изобретения R3 является -CF3, -CH2F или -CHF2.
В некоторых воплощениях изобретения, если R3 является алкилом, алкенилом, алкинилом, арилом, гетероарилом, циклоалкилом, гетероциклоалкилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом или сульфонамидо, то он является замещенным одним или более из алкила, гетероалкила, алкенила, алкинила, циклоалкила, гетероциклоалкила, арила, гетероарила, алкокси, амидо, амино, ацила, ацилокси, алкоксикарбонила, сульфонамидо, галогено, циано, гидрокси или нитро, где каждый алкил, гетероалкил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил, гетероарил, алкокси, амидо, амино, ацил, ацилокси, алкоксикарбонил или сульфониламидо могут быть замещенными.
В некоторых воплощениях соединения формулы I, R5 является водородом, незамещенным или замещенным алкилом (включая, но не ограничиваясь, незамещенный или замещенный C1-C4алкил). В некоторых воплощениях изобретения R5 является незамещенным или замещенным алкенилом, включая, но не ограничиваясь, незамещенный или замещенный C2-C5алкенил. В некоторых воплощениях изобретения R5 является незамещенным или замещенным алкинилом, включая, но не ограничиваясь, незамещенный или замещенный C2-C5алкинил. В некоторых воплощениях изобретения R5 является незамещенным или замещенным циклоалкилом, включая, но не ограничиваясь, незамещенный или замещенный С3-С5циклоалкил. В некоторых воплощениях изобретения R5 является незамещенным или замещенным гетероциклоалкилом. В некоторых воплощениях изобретения R5 является незамещенным или замещенным гетероалкилом, включая, не ограничиваясь, незамещенный или замещенный C1-C4гетероалкил. В некоторых воплощениях изобретения R5 является незамещенным или замещенным алкокси, включая, не ограничиваясь, незамещенный или замещенный C1-C4алкокси. В некоторых воплощениях изобретения R5 является незамещенным или замещенным амидо, включая, но не ограничиваясь, незамещенный или замещенный C1-C4амидо. В некоторых воплощениях изобретения R5 является незамещенным или замещенным амино. В некоторых воплощениях изобретения R5 является незамещенным или замещенным ацилом, незамещенным или замещенным ацилокси, незамещенным или замещенным C1-C4ацилокси, незамещенным или замещенным алкоксикарбонилом, незамещенным или замещенным сульфонамидо или незамещенным или замещенным C1-C4сульфонамидо. В некоторых воплощениях изобретения R5 является галогено, который является -I, -F, -Cl или -Br. В некоторых воплощениях изобретения R5 выбран из группы, состоящей из циано, гидрокси и нитро. В некоторых воплощениях изобретения R5 является -CH3, -CH2CH3, н-пропилом, изопропилом, -OCH3, -OCH2CH3 или -CF3.
В некоторых воплощениях изобретения, если R5 является алкилом, алкенилом, алкинилом, циклоалкилом, гетероалкилом, ацилом, алкокси, амидо, амино, ацилокси, алкоксикарбонилом или сульфонамидо, то R5 в некоторых случаях является замещенным одним или более алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, гетероарилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси или нитро, где каждый алкил, гетероалкил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил, гетероарил, алкокси, амидо, амино, ацил, ацилокси, алкоксикарбонил или сульфонамидо могут быть замещенными.
В некоторых воплощениях соединения формулы I, R6 является водородом, незамещенным или замещенным алкилом (включая, не ограничиваясь, незамещенный или замещенный C1-C4алкил). В некоторых воплощениях изобретения R6 является незамещенным или замещенным алкенилом, включая, но не ограничиваясь, незамещенный или замещенный C2-C5алкенил. В некоторых воплощениях изобретения R6 является незамещенным или замещенным алкинилом, включая, но не ограничиваясь, незамещенный или замещенный C2-C5алкинил. В некоторых воплощениях изобретения R6 является незамещенным или замещенным циклоалкилом, включая, не ограничиваясь, незамещенный или замещенный С3-С5циклоалкил. В некоторых воплощениях изобретения R6 является незамещенным или замещенным гетероциклоалкилом. В некоторых воплощениях изобретения R6 является незамещенным или замещенным гетероалкилом, включая, но не ограничиваясь, незамещенный или замещенный C1-C4гетероалкил. В некоторых воплощениях изобретения R6 является незамещенным или замещенным алкокси, включая, но не ограничиваясь, незамещенный или замещенный C1-C4алкокси. В некоторых воплощениях изобретения R6 является незамещенным или замещенным амидо, включая, не ограничиваясь, незамещенный или замещенный C1-C4амидо. В некоторых воплощениях изобретения R6 является незамещенным или замещенным амино. В некоторых воплощениях изобретения R6 является незамещенным или замещенным ацилом, незамещенным или замещенным ацилокси, незамещенным или замещенным C1-C4ацилокси, незамещенным или замещенным алкоксикарбонилом, незамещенным или замещенным сульфонамидо или незамещенным или замещенным C1-C4сульфонамидо. В некоторых воплощениях изобретения R6 является галогено, который является -I, -F, -Cl или -Br. В некоторых воплощениях изобретения R6 выбран из группы, состоящей из циано, гидрокси и нитро. В некоторых других воплощениях изобретения R6 является -CH3, -CH2CH3, н-пропилом, изопропилом, -OCH3, -OCH2CH3 или -CF3.
В некоторых воплощениях изобретения, если R6 является алкилом, алкенилом, алкинилом, циклоалкилом, гетероалкилом, ацилом, алкокси, амидо, амино, ацилокси, алкоксикарбонилом или сульфонамидо, то R6 в некоторых случаях является замещенным одним или более алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, гетероарилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси или нитро, где каждый алкил, гетероалкил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил, гетероарил, алкокси, амидо, амино, ацил, ацилокси, алкоксикарбонил или сульфонамидо могут быть замещенными.
В некоторых воплощениях соединения формулы I, R7 является водородом, незамещенным или замещенным алкилом (включая, но не ограничиваясь, незамещенный или замещенный C1-C4алкил). В некоторых воплощениях изобретения R7 является незамещенным или замещенным алкенилом, включая, но не ограничиваясь, незамещенный или замещенный C2-C5алкенил. В некоторых воплощениях изобретения R7 является незамещенным или замещенным алкинилом, включая, но не ограничиваясь, незамещенный или замещенный C2-C5алкинил. В некоторых воплощениях изобретения R7 является незамещенным или замещенным циклоалкилом, включая, но не ограничиваясь, незамещенный или замещенный С3-С5циклоалкил. В некоторых воплощениях изобретения R7 является незамещенным или замещенным гетероциклоалкилом. В некоторых воплощениях изобретения R7 является незамещенным или замещенным гетероалкилом, включая, не ограничиваясь, незамещенный или замещенный C1-C4гетероалкил. В некоторых воплощениях изобретения R7 является незамещенным или замещенным алкокси, включая, но не ограничиваясь, незамещенный или замещенный C1-C4алкокси. В некоторых воплощениях изобретения R7 является незамещенным или замещенным амидо, включая, не ограничиваясь, незамещенный или замещенный C1-C4амидо. В некоторых воплощениях изобретения R7 является незамещенным или замещенным амино. В некоторых воплощениях изобретения R7 является незамещенным или замещенным ацилом, незамещенным или замещенным ацилокси, незамещенным или замещенным C1-C4ацилокси, незамещенным или замещенным алкоксикарбонилом, незамещенным или замещенным сульфонамидо, или незамещенным или замещенным C1-C4сульфонамидо. В некоторых воплощениях изобретения R7 является галогено, который является -I, -F, -Cl или -Br. В некоторых воплощениях изобретения R7 выбран из группы, состоящей из циано, гидрокси и нитро. В некоторых воплощениях изобретения R7 является -CH3, -CH2CH3, н-пропилом, изопропилом, -OCH3, -OCH2CH3 или -CF3.
В некоторых воплощениях изобретения, если R7 является алкилом, алкенилом, алкинилом, циклоалкилом, гетероалкилом, ацилом, алкокси, амидо, амино, ацилокси, алкоксикарбонилом или сульфонамидо, то R7 в некоторых случаях является замещенным одним или более алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, гетероарилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси или нитро, где каждый алкил, гетероалкил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил, гетероарил, алкокси, амидо, амино, ацил, ацилокси, алкоксикарбонил или сульфонамидо могут быть замещенными.
В некоторых воплощениях соединения формулы I, R8 является водородом, незамещенным или замещенным алкилом (включая, но не ограничиваясь, незамещенный или замещенный C1-C4алкил). В некоторых воплощениях изобретения R8 является незамещенным или замещенным алкенилом, включая, но не ограничиваясь, незамещенный или замещенный C2-C5алкенил. В некоторых воплощениях изобретения R8 является незамещенным или замещенным алкинилом, включая, но не ограничиваясь, незамещенный или замещенный C2-C5алкинил. В некоторых воплощениях R8 является незамещенным или замещенным циклоалкилом, включая, но не ограничиваясь, незамещенный или замещенный С3-С5циклоалкил. В некоторых воплощениях изобретения R8 является незамещенным или замещенным гетероциклоалкилом. В некоторых воплощениях изобретения R8 является незамещенным или замещенным гетероалкилом, включая, но не ограничиваясь, незамещенный или замещенный C1-C4гетероалкил. В некоторых воплощениях изобретения R8 является незамещенным или замещенным алкокси, включая, не ограничиваясь, незамещенный или замещенный C1-C4алкокси. В некоторых воплощениях изобретения R8 является незамещенным или замещенным амидо, включая, не ограничиваясь, незамещенный или замещенный C1-C4амидо. В некоторых воплощениях изобретения R8 является незамещенным или замещенным амино. В некоторых воплощениях изобретения R8 является незамещенным или замещенным ацилом, незамещенным или замещенным ацилокси, незамещенным или замещенным C1-C4ацилокси, незамещенным или замещенным алкоксикарбонилом, незамещенным или замещенным сульфонамидо или незамещенным или замещенным C1-C4сульфонамидо. В некоторых воплощениях изобретения R8 является галогено, который является -I, -F, -Cl или -Br. В некоторых воплощениях изобретения R8 выбран из группы, состоящей из циано, гидрокси и нитро. В некоторых воплощениях изобретения R8 является -CH3, -CH2CH3, н-пропилом, изопропилом, -OCH3, -OCH2CH3 или -CF3.
В некоторых воплощениях изобретения, если R8 является алкилом, алкенилом, алкинилом, циклоалкилом, гетероалкилом, ацилом, алкокси, амидо, амино, ацилокси, алкоксикарбонилом или сульфонамидо, то R8 в некоторых случаях является замещенным одним или более алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, гетероарилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси или нитро, где каждый алкил, гетероалкил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил, гетероарил, алкокси, амидо, амино, ацил, ацилокси, алкоксикарбонил или сульфонамидо могут быть замещенными.
В некоторых воплощениях соединения формулы I, R5, R6, R7 и R8 являются Н, и соединение имеет структуру формулы I-1:

В некоторых воплощениях соединения формулы I, X отсутствует. В некоторых воплощениях X является -(CH(R9))z-, и z является целым числом из 1, 2, 3 или 4.
В некоторых воплощениях изобретения R9 является незамещенным или замещенным алкилом, включая, но не ограничиваясь, незамещенный или замещенный C1-C10алкил. В некоторых воплощениях изобретения R9 является незамещенным или замещенным циклоалкилом, включая, не ограничиваясь, незамещенный или замещенный С3-С7циклоалкил. В некоторых воплощениях изобретения R9 является этилом, метилом или водородом. В некоторых воплощениях изобретения R9 является незамещенным или замещенным гетероциклоалкилом, включая, но не ограничиваясь, незамещенный или замещенный C2-C10гетероалкил. В некоторых воплощениях изобретения R9 является незамещенным или замещенным гетероалкилом, включая, не ограничиваясь, незамещенный или замещенный C2-C10гетероалкил.
В настоящем изобретении представлены также соединения формулы I, где R9 является водородом, и X является -CH2-, -CH2CH2-, -CH2CH2CH2-,-CH(CH3)- или -СН(CH2CH3)-. В других воплощениях изобретения X является -(CH(R9))z, R9 не является водородом, и z является целым числом, равным 1. Если X является -CH(R9)-, и R9 не является водородом, то соединение может находиться в (S)- или (R)-стереохимической конфигурации относительно углерода X. В некоторых воплощениях изобретения соединение является рацемической смесью (S)- и (R)-изомеров относительно углерода X. В других вариантах настоящего изобретения представлена смесь соединений формулы I, где отдельные соединения в смеси существуют преимущественно в (S)- или (R)- изомерной конфигурации. Например, смесь соединений имеет (S)-энантиомерную чистоту более, чем примерно 55%, примерно 60%, примерно 65%, примерно 70%, примерно 75%, примерно 80%, примерно 85%, примерно 90%, примерно 95%, примерно 96%, примерно 97%, примерно 98%, примерно 99%, примерно 99,5% или более, относительно углерода X. В других воплощениях изобретения смесь соединений имеет (S)-энантиомерную чистоту примерно от 55% до примерно 99,5%, примерно от 60% до примерно 99,5%, примерно от 65% до примерно 99,5%, примерно от 70% до примерно 99,5%, примерно от 75% до примерно 99,5%, примерно от 80% до примерно 99,5%, примерно от 85% до примерно 99,5%, примерно от 90% до примерно 99,5%, примерно от 95% до примерно 99,5%, примерно от 96% до примерно 99,5%, примерно от 97% до примерно 99,5%, примерно от 98% до примерно 99,5%, примерно от 99% до примерно 99,5%, или более.
В других воплощениях изобретения смесь соединений имеет (R)-энантиомерную чистоту более, чем примерно 55%, примерно 60%, примерно 65%, примерно 70%, примерно 75%, примерно 80%, примерно 85%, примерно 90%, примерно 95%, примерно 96%, примерно 97%, примерно 98%, примерно 99%, примерно 99,5% или более, относительно углерода X. В некоторых других воплощениях изобретения смесь соединений имеет (R)-энантиомерную чистоту примерно от 55% до примерно 99,5%, примерно от 60% до примерно 99,5%, примерно от 65% до примерно 99,5%, примерно от 70% до примерно 99,5%, примерно от 75% до примерно 99,5%, примерно от 80% до примерно 99,5%, примерно от 85% до примерно 99,5%, примерно от 90% до примерно 99,5%, примерно от 95% до примерно 99,5%, примерно от 96% до примерно 99,5%, примерно от 97% до примерно 99,5%, примерно от 98% до примерно 99,5%, примерно от 99% до примерно 99,5%, или более.
В других воплощениях изобретения смесь соединений содержит идентичные химические соединения, за исключением их стереохимической ориентации, а именно (S)- или (R)-изомеры. Например, в соединениях формулы I, если X является -CH(R9)-, и R9 не является водородом, то -CH(R9)- находится в (S)- или (R)-стереохимической ориентации для каждого конкретного химического соединения. В некоторых воплощениях изобретения смесь идентичных химических соединений формулы I является рацемической смесью (S)- и (R)-изомеров по углероду, представленному X. В другом варианте воплощения изобретения смесь идентичных химических соединений (кроме их стереохимических ориентаций), содержит преимущественно (S)-изомеры или преимущественно (R)-изомеры. Например, (S)-изомеры в смеси идентичных химических соединений присутствуют в количестве примерно 55%, примерно 60%, примерно 65%, примерно 70%, примерно 75%, примерно 80%, примерно 85%, примерно 90%, примерно 95%, примерно 96%, примерно 97%, примерно 98%, примерно 99%, примерно 99,5% или более, по отношению к (R)-изомерам. В других воплощениях изобретения (S)-изомеры в смеси идентичных химических соединений, присутствует с (S)-энантиомерной чистотой более, чем примерно от 55% до примерно 99,5%, примерно от 60% до примерно 99,5%, примерно от 65% до примерно 99,5%, примерно от 70% до примерно 99,5%, примерно от 75% до примерно 99,5%, примерно от 80% до примерно 99,5%, примерно от 85% до примерно 99,5%, примерно от 90% до примерно 99,5%, примерно от 95% до примерно 99,5%, примерно от 96% до примерно 99,5%, примерно от 97% до примерно 99,5%, примерно от 98% до примерно 99,5%, примерно от 99% до примерно 99,5%, или более.
В другом воплощении изобретения (R)-изомеры в смеси идентичных химических соединений (за исключением их стереохимических ориентаций), присутствуют в количестве примерно 55%, примерно 60%, примерно 65%, примерно 70%, примерно 75%, примерно 80%, примерно 85%, примерно 90%, примерно 95%, примерно 96%, примерно 97%, примерно 98%, примерно 99%, примерно 99,5% или более, по отношению к (S)-изомерам. В некоторых воплощениях изобретения (R)-изомеры в смеси идентичных химических соединений (за исключением их стереохимических ориентаций) присутствуют с (R)-энантиомерной чистотой более, чем примерно от 55% до примерно 99,5%, примерно от 60% до примерно 99,5%, примерно от 65% до примерно 99,5%, примерно от 70% до примерно 99,5%, примерно от 75% до примерно 99,5%, примерно от 80% до примерно 99,5%, примерно от 85% до примерно 99,5%, примерно от 90% до примерно 99,5%, примерно от 95% до примерно 99,5%, примерно от 96% до примерно 99,5%, примерно от 97% до примерно 99,5%, примерно от 98% до примерно 99,5%, примерно от 99% до примерно 99,5%, или более.
В некоторых воплощениях, в соединении формулы I, X является -CH(R9)-, R9 является метилом или этилом, и соединение является (S)-изомером.
В некоторых воплощениях соединения формулы I, Y отсутствует. В некоторых воплощениях изобретения Y является -О-, -S-, -S(=O)-, -S(=O)2-, -С(=O)-, -N(R9)(C=O)-, -N(R9)(C=O)NH-, -N(R9)C(R9)2- (таким как -N(R9)CH2-, в частности, -N(CH3)CH2-, N(CH(CH3)2)CH2 - или N(CH2CH3)CH2-), -N(R9)-, -N(CH3)-, -N(CH2CH3)- или -N(CH(CH3)2)-. В некоторых воплощениях изобретения Y является -C(=O)-(CHR9)z-, и z является целым числом из 1, 2, 3 или 4.
В некоторых воплощениях изобретения присутствует по меньшей мере один из X и Y. В некоторых воплощениях соединения формулы I, -XY- является -CH2-, -CH2-N(CH3), -CH2-N(CH2CH3), -CH(CH3)-NH-, (S) -CH(CH3)-NH- или (R) -CH(CH3)-NH-. В других воплощениях изобретения X-Y является -N(CH3)-CH2-, N(CH2CH3)CH2-, -N(CH(CH3)2)CH2- или -NHCH2-. В настоящем изобретении представлены другие соединения формулы I, где если X-Y является X является -(CH(R9))zN(R9)-, z является целым числом из 1, 2, 3 или 4, и -N(R9)- не является -NH-, то -XY- не связан с пуринилом.
В некоторых воплощениях изобретения Wd в формуле, описанной в настоящем документе (включая, не ограничиваясь, I, I-1, IV, IV-А, V, V-A, V-A2, V-B, VI и VI-А), является членом, выбранным из группы, состоящей из незамещенного или замещенного гетероциклоалкила, незамещенного или замещенного арила и незамещенного или замещенного гетероарила.
В различных воплощениях изобретения Wd является незамещенным или замещенным моноциклическим гетероарилом (включая, не ограничиваясь, пиримидинил, пирролил, пиразинил, триазинил или пиридазинил) или незамещенным или замещенным бициклическим гетероарилом.
В некоторых воплощениях изобретения Wd является моноциклическим гетероарилом следующей формулы:


где Ra′ является водородом, галогено, фосфатом, мочевиной, карбонатом, незамещенным или замещенным амино, незамещенным или замещенным алкилом, незамещенным или замещенным алкенилом, незамещенным или замещенным алкинилом, незамещенным или замещенным циклоалкилом, незамещенным или замещенным гетероалкилом или незамещенным или замещенным гетероциклоалкилом; и
R12 является Н, незамещенным или замещенным алкилом, незамещенным или замещенным циано, незамещенным или замещенным алкинилом, незамещенным или замещенным алкенилом, галогено, незамещенным или замещенным арилом, незамещенным или замещенным гетероарилом, незамещенным или замещенным гетероциклоалкилом, незамещенным или замещенным циклоалкилом, незамещенным или замещенным амино, карбоновой кислотой, незамещенным или замещенным алкоксикарбонилом, незамещенным или замещенным амидо, незамещенным или замещенным ацилом или незамещенным или замещенным сульфонамидо.
В настоящем изобретении представлен моноциклический гетероарил Wd, включая, не ограничиваясь, одну из следующих формул:


В некоторых воплощениях изобретения Wd в формуле, описанной в настоящем документе (включая, не ограничиваясь, I, I-1, IV, IV-А, V, V-A, V-A2, V-B, VI и VI-А), является бициклическим гетероарилом, имеющим по меньшей мере один гетероатом, например, бициклическим гетероарилом, имеющим по меньшей мере один кольцевой атом азота. В некоторых воплощениях изобретения Wd является бициклическим гетероарилом, имеющим по меньшей мере два гетероатома, например, бициклическим гетероарилом, имеющим по меньшей мере два кольцевых атома азота. В некоторых воплощениях изобретения Wd является бициклическим гетероарилом, имеющим два гетероатома в кольце, которые связаны с XY. В некоторых воплощениях изобретения Wd является бициклическим гетероарилом, имеющим два кольцевых атома азота в кольце, к которому присоединен XY. В некоторых воплощениях изобретения Wd является бициклическим гетероарилом, имеющим четыре гетероатома, например, бициклическим гетероарилом, имеющим четыре кольцевых атома азота. В некоторых воплощениях изобретения Wd является незамещенным или замещенным 4-амино-1Н-пиразоло[3,4-d]пиримидин-1-илом, незамещенным или замещенным 7-амино-2-метил-2Н-пиразоло[4,3-d]пиримидин-3-илом, незамещенным или замещенным 6-метиленил-9Н-пурин-6-илом или незамещенным или замещенным 6-амино-9Н-пурин-9-илом.
В некоторых воплощениях изобретения Wd является одной из следующих групп:




где Ra′ является водородом, галогено, фосфатом, мочевиной, карбонатом, незамещенным или замещенным амино, незамещенным или замещенным алкилом, незамещенным или замещенным алкенилом, незамещенным или замещенным алкинилом, незамещенным или замещенным циклоалкилом, незамещенным или замещенным гетероалкилом, или незамещенным или замещенным гетероциклоалкилом;
R11 является водородом, незамещенным или замещенным алкилом, гало (который включает -I, -F, -Cl или -Br), незамещенным или замещенным амино, незамещенным или замещенным амидо, гидрокси или незамещенным или замещенным алкокси, фосфатом, незамещенной или замещенной мочевиной или карбонатом; и
R12 является Н, незамещенным или замещенным алкилом, незамещенным или замещенным циано, незамещенным или замещенным алкинилом, незамещенным или замещенным алкенилом, галогено, незамещенным или замещенным арилом, незамещенным или замещенным гетероарилом, незамещенным или замещенным гетероциклоалкилом, незамещенным или замещенным циклоалкилом, незамещенным или замещенным амино, карбоновой кислотой, незамещенным или замещенным алкоксикарбонилом, незамещенным или замещенным амидо, незамещенным или замещенным ацилом или незамещенным или замещенным сульфонамидо.
В некоторых воплощениях Wd соединений формулы I, где Ra′ является алкилом, алкинилом, циклоалкилом, гетероалкилом или гетероциклоалкилом, он является замещенным фосфатом, мочевиной или карбонатом.
В некоторых воплощениях Wd соединений формулы I, где R11 является алкилом, амино, амидо, гидрокси или алкокси, он является замещенным фосфатом, мочевиной или карбонатом.
В некоторых воплощениях соединений формулы I, -X-Y-Wd является одной из следующих групп:
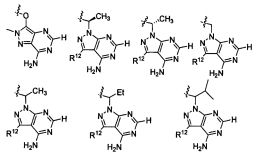











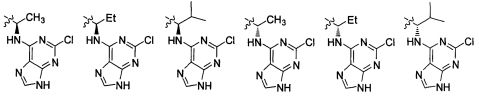









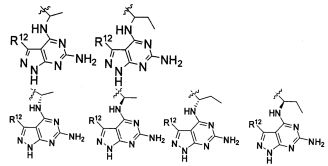
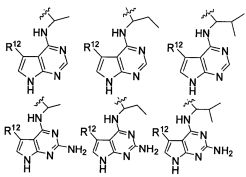

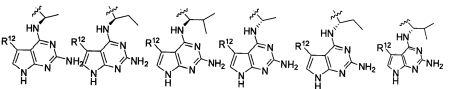





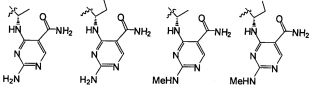


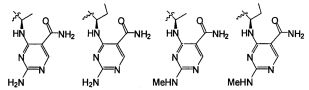



В некоторых воплощениях соединения формулы I, R12 является членом, выбранным из группы, состоящей из водорода, циано, галогено, незамещенного или замещенного алкила, незамещенного или замещенного алкинила и незамещенного или замещенного алкенила. В некоторых воплощениях изобретения R12 является незамещенным или замещенным арилом. В некоторых воплощениях изобретения R12 является незамещенным или замещенным гетероарилом, который включает, не ограничиваясь, гетероарил, имеющий 5-членное кольцо, гетероарил, имеющий шестичленное кольцо, гетероарил, имеющий по меньшей мере один кольцевой атом азота, гетероарил с двумя кольцевыми атомами азота, моноциклический гетероарил и бициклический гетероарил. В некоторых воплощениях изобретения R12 является незамещенным или замещенным гетероциклоалкилом, который включает, не ограничиваясь, гетероциклоалкил с одним кольцевым атомом азота, гетероциклоалкил с одним кольцевым атомом кислорода, R12 является гетероциклоалкилом с одним кольцевым атомом серы, 5-членным гетероциклоалкилом, 6-членным гетероциклоалкилом, насыщенным гетероциклоалкилом, ненасыщенным гетероциклоалкилом, гетероциклоалкилом, имеющим ненасыщенную группу, связанную с гетероциклоалкильным кольцом, гетероциклоалкилом, замещенным оксо-группой и гетероциклоалкилом, замещенным двумя оксо-группами. В некоторых воплощениях изобретения R12 является незамещенным или замещенным циклоалкилом, включая, не ограничиваясь, циклопропил, циклобутил, циклопентил, циклогексил, циклоалкил, замещенный одной оксо-группой, циклоалкил, имеющий ненасыщенную группу, связанную с циклоалкильным кольцом. В некоторых воплощениях изобретения R12 является незамещенным или замещенным амидо, карбоновой кислотой, незамещенным или замещенным ацилокси, незамещенным или замещенным алкоксикарбонилом, незамещенным или замещенным ацилом или незамещенным или замещенным сульфонамидо.
В некоторых воплощениях изобретения, если R12 является алкилом, алкинилом, алкенилом, арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, он является замещенным фосфатом. В некоторых воплощениях изобретения, если R12 является алкилом, алкинилом, алкенилом, арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, он является замещенным мочевиной. В некоторых воплощениях изобретения, если R12 является алкилом, алкинилом, алкенилом, арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, он является замещенным карбонатом.
В некоторых воплощениях изобретения, если R12 является алкилом, алкинилом, алкенилом, арилом, гетероарилом, гетероциклоалкилом, циклоалкилом, алкоксикарбонилом, амидо, ацилокси, ацилом или сульфонамидо, то он является замещенным одним или более алкилом, гетероалкилом, алкенилом, алкинилом, циклоалкилом, гетероциклоалкилом, арилом, гетероарилом, алкокси, амидо, амино, ацилом, ацилокси, алкоксикарбонилом, сульфонамидо, галогено, циано, гидрокси или нитро, где каждый алкил, гетероалкил, алкенил, алкинил, циклоалкил, гетероциклоалкил, арил, гетероарил, алкокси, амидо, амино, ацил, ацилокси, алкоксикарбонил или сульфонамидо могут быть замещенными.
В некоторых воплощениях соединения формулы I, R12 в Wd является одной из следующих групп:






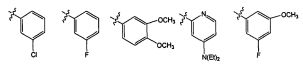




В некоторых воплощениях соединения формулы I, Wd является пиразолопиримидином формулы III:

где R11 является Н, алкилом, галогено, амино, амидо, гидрокси или алкокси, и R12 является Н, алкилом, алкинилом, алкенилом, галогено, арилом, гетероарилом, гетероциклоалкилом или циклоалкилом. В некоторых воплощениях изобретения R11 является амино, и R12 является Н, алкилом, алкинилом, алкенилом, галогено, арилом, гетероарилом, гетероциклоалкилом или циклоалкилом. В некоторых воплощениях изобретения R11 является амино, и R12 является алкилом, галогено, арилом, гетероарилом, гетероциклоалкилом или циклоалкилом. В некоторых воплощениях изобретения R11 является амино, и R12 является моноциклическим гетероарилом. В некоторых воплощениях изобретения R11 является амино, и R12 является бициклическим гетероарилом. В некоторых воплощениях изобретения R11 является амино, и R12 является циано, амино, карбоновой кислотой, ацилокси, алкоксикарбонилом или амидо.
В некоторых воплощениях настоящего изобретения соединение формулы I является соединением, имеющим структуру формулы IV:

В некоторых воплощениях соединения формулы IV, R11 является Н, алкилом, галогено, амино, амидо, гидрокси или алкокси, и R12 является Н, алкилом, алкинилом, алкенилом, галогено, арилом, гетероарилом, гетероциклоалкилом или циклоалкилом. В другом варианте воплощения изобретения R11 является амино, и R12 является алкилом, алкенилом, гетероарилом, арилом или гетероциклоалкилом. В некоторых воплощениях изобретения R11 является амино, и R12 является циано, амино, карбоновой кислотой, алкоксикарбонилом или амидо.
В некоторых воплощениях изобретения соединение формулы IV имеет структуру формулы IV-A:

В настоящем изобретении представлены также соединения формулы I, имеющие структуру формул V, V-A1, V-A2, V-B, VI, VI-A, VII-A1, VII-A2, VIII-A1, VIII-A2, IX-A1, IX-A2, X-A1, X-A2, XI-A1, XI-A2, XII-A, XII-A1, XII-A2, XIII-A, XIII-A1, XIII-A2, XIV-A, XIV-A1, XIV-A2, XV-A, XV-A1, XV-A2, XVI-A, XVI-A1, XVI-A2, XVII-A, XVII-A1, XVII-A2, XVIII-A, XVIII-A1 или XVIII-A2:


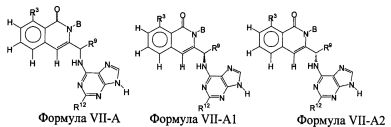
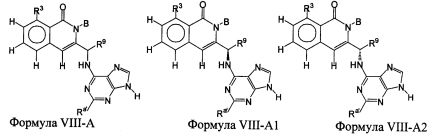


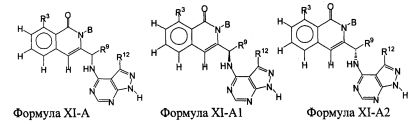




Любые из описанных элементов и их заместители в соединениях формулы I могут использоваться в любых комбинациях.
В одном аспекте, для соединений формулы I, R3 является Н, CH3, CF3, Cl или F; и В является группой формулы II:

где Wc является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом; R1 является Н, -F, -Cl, -CN, -CH3, изопропилом, -CF3, -OCH3, нитро или фосфатом; R2 является галогено, гидрокси, циано или нитро; q является целым числом из 0, 1, 2, 3 или 4; R5, R6, R7 и R8 являются Н; X отсутствует или является (CH2)z; z равен 1; Y отсутствует или является -N(R9)-; R9 является водородом, C1-C10алкилом, С3-С7циклоалкилом или C2-C10гетероалкилом; присутствует по меньшей мере один из X и Y; и Wd является пиразолопиримидином или пурином. В некоторых воплощениях изобретения, если присутствуют X и Y, и Wd является пурином, то -N(R9)- является -NH-.
В другом аспекте, для соединений формулы I, R3 является Н, CH3, CF3, Cl или F; В является группой формулы II, которая является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, R1 является Н, -F, -Cl, -CN, -CH3, изопропилом, -CF3, -OCH3, нитро или фосфатом; R2 является галогено, гидрокси, циано или нитро; q равен 0, 1 или 2; R5, R6, R7 и R8 являются Н; X отсутствует или является (CH2)z; z равен 1; Y отсутствует или является -N(R9)-; R9 является водородом, метилом или этилом; присутствует по меньшей мере один из X и Y; Wd является:


В другом аспекте для соединений формулы I, R3 является Н, CH3, CF3, Cl или F; В является группой формулы II, которая является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, R1 является Н, -F, -Cl, -CN, -CH3, изопропилом, -CF3, -OCH3, нитро или фосфатом; R2 является галогено, гидрокси, циано или нитро; q равен 0, 1 или 2; X является (CH2)z; z равен 1; R5, R6, R7 и R8 являются Н; Y отсутствует и Wd является:

В другом аспекте R3 является Н, CH3, CF3, Cl или F; В является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, R1 является Н, -F, -Cl, -CN, -CH3, изопропилом, -CF3, -OCH3, нитро или фосфатом; R2 является галогено, гидрокси, циано или нитро; q равен 0, 1 или 2; R5, R6, R7 и R8 являются Н; X является (CH2)z; z равен 1; X является (CH2)z; z равен 1; Y является -N(R9)-; R9 является водородом, метилом или этилом; и Wd является
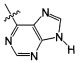
В другом аспекте для соединений формулы I, R3 является арилом, гетероарилом, Н, CH3, CF3, Cl или F; В является группой формулы II;
где Wc является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, и q является целым числом из 0, 1, 2, 3 или 4; R1 является Н, -F, -Cl, -CN, -CH3, изопропилом, -CF3, -OCH3, нитро или фосфатом; R2 является галогено, гидрокси, циано, нитро или фосфатом; q равен 0, 1 или 2; R5, R6, R7 и R8 являются Н; X отсутствует или является (CH(R9))z; z является целым числом из 1, 2, 3 или 4; Y отсутствует или является -N(R9)- или -N(R9)CH(R9)-; R9 является водородом, алкилом, циклоалкилом или гетероалкилом; присутствует по меньшей мере один из X и Y; и Wd является пиразолопиримидином или пурином. В некоторых воплощениях изобретения, если присутствует X, и Y является -N(R9)-, и Wd является пурином, то Y является -NH-.
В другом аспекте для соединений формулы I, R3 является арилом, гетероарилом, Н, CH3, CF3, Cl или F; В является алкилом или группой формулы II, которая является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, R1 является Н, -F, -Cl, -CN, -CH3, изопропилом, -CF3, -OCH3, нитро или фосфатом; R2 является галогено, гидрокси, циано, нитро или фосфатом; q равен 0, 1 или 2; R5, R6, R7 и R8 являются Н; X отсутствует или является (CH(R9))z; z является целым числом из 1, 2, 3 или 4; Y отсутствует или является -N(R9)- или -N(R9)CH(R9)-; R9 является водородом, метилом или этилом; присутствует по меньшей мере один из X и Y; Wd является:

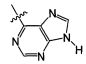
В другом аспекте для соединений формулы I, R3 является Н, CH3, CF3, Cl или F; В является алкилом или группой формулы II, которая является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, R1 является Н, -F, -Cl, -CN, -CH3, изопропилом, -CF3, -OCH3, нитро или фосфатом; R2 является галогено, гидрокси, циано, нитро или фосфатом; q равен 0, 1 или 2; R5, R6, R7 и R8 являются Н; X является (CH(R9))z; z является целым числом, равным 1; Y отсутствует; R9 является водородом, метилом или этилом; Wd является:
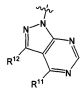
В другом аспекте для соединений формулы I, R3 является арилом, гетероарилом, Н, CH3, CF3, Cl или F; В является группой формулы II, которая является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, R1 является Н, -F, -Cl, -CN, -CH3, изопропилом, -CF3, -OCH3, нитро или фосфатом; R2 является галогено, гидрокси, циано, нитро или фосфатом; q равен 0, 1 или 2; R5, R6, R7 и R8 являются Н; X отсутствует или является (CH(R9))z; z является целым числом, равным 1; Y отсутствует или является -N(R9)- или -N(R9)CH(R9)-; R9 является водородом, метилом или этилом; присутствует по меньшей мере один из X и Y; Wd является:
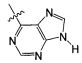
В другом аспекте, для соединений формулы I, R3 является арилом, гетероарилом, Н, CH3, CF3, Cl или F; В является группой формулы II, которая является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, R1 является Н, -F, -Cl, -CN, -CH3, изопропилом, -CF3, -OCH3, нитро или фосфатом; R2 является галогено, гидрокси, циано, нитро или фосфатом; q равен 0, 1 или 2; R5, R6, R7 и R8 являются Н; X отсутствует; Y является -N(R9)CH(R9)-; R9 является водородом, метилом или этилом; Wd является:

В другом аспекте для соединений формулы I, R3 является арилом, гетероарилом, Н, CH3, CF3, Cl или F; В является алкилом или группой формулы II, которая является арилом, гетероарилом, гетероциклоалкилом или циклоалкилом, R1 является Н, -F, -Cl, -CN, -CH3, изопропилом, -CF3, -OCH3, нитро или фосфатом; R2 является галогено, гидрокси, циано или нитро или фосфатом; q равен 0, 1 или 2; R5, R6, R7 и R8 являются Н; X отсутствует или является (CH(R9))z; z является целым числом из 1, 2, 3 или 4; Y отсутствует или является -N(R9)- или -N(R9)CH(R9)-; R9 является водородом, метилом или этилом; присутствует по меньшей мере один из X и Y; Wd является:
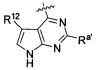
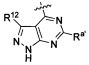

Описаны дополнительные типичные соединения по настоящему изобретению, которые имеют субструктуру формулы IV-А.

Некоторые иллюстративные соединения по настоящему изобретению, имеющие структуру формулы IV-A, включают соединения, в которых R3 является -Н, -Cl, -F или -CH3 в сочетании с любой из групп В, описанных в таблице 1, и любым из R12, описанных в таблице 2. Соединение формулы IV-A включает любые комбинации R3, В и R12. Дополнительные типичные соединения формулы IV-А представлены в таблице 4.








Другие иллюстративные соединения по настоящему изобретению имеют структуру формулы V-A, V-A1 или V-A2, где В является группой, описанной в таблице 1, в сочетании с R3, который является -Н, -Cl, -F или CH3, и R9, который является -Н, -CH3 или -CH2CH3. Соединения формулы V-A, V-A1 или V-A2 включают любые комбинации R3, В и R9
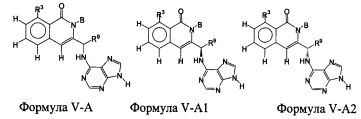
В некоторых воплощениях изобретения соединение формулы V-A2 является:

В некоторых воплощениях изобретения R1 является -Н, например, формулы:

В некоторых воплощениях изобретения q равен 1, и R2 имеет формулу:

В некоторых воплощениях изобретения R1 является Н, и R2 является Н, например, формулы:

или его фармацевтически приемлемые соли.
В некоторых воплощениях изобретения R1 является Н, q равен 1, и R2 является галогено (например, фторо) формулы:

или его фармацевтически приемлемые соли.
В некоторых воплощениях изобретения R1 является Н, q равен 1, и R2 является галогено (например, фторо) в мета-положении, например, формулы:
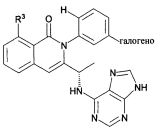
или его фармацевтически приемлемые соли.
В некоторых воплощениях формулы VA-2, R3 является галогеноалкилом. Например, R3 является -CF3, -CH2F или -CHF2.
Другие иллюстративные соединения по настоящему изобретению имеют структуру формулы V-B, где В является группой, описанной в таблице 1, в сочетании с R3, который является -Н, -Cl, -F или CH3, и R9, который является -Н, -CH3 или -CH2CH3. Соединение формулы V-B включает любые комбинации R3, В и R9
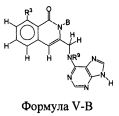
Некоторые другие иллюстративные соединения по настоящему изобретению имеют структуру формулы VI-A, где В является группой, описанной в таблице 1, в сочетании с R3, который является -Н, -Cl, -F или CH3, и R9, который является -Н, -CH3 или -CH2CH3. Соединение формулы VI-A включают любые комбинации R3, В и R9

Другие иллюстративные соединения по настоящему изобретению имеют структуру одной из формул VII-A1, VII-A2, VIII-A1, VIII-A2, IX-A1, IX-A2, X-A1, X-А2, XI-A1, XI-A2, XII-А, XII-A1, XII-A2, XIII-А, XIII-A1, XIII-A2, XIV-A, XIV-A1 или XIV-A2: где В является группой, описанной в таблице 1, любой из R12 является таким, как описано в таблице 2, в комбинации с R3, который является -Н, -Cl, -F или CH3, R9 который является -Н, -CH3 или -CH2CH3, и Ra′, который является -Н, -Cl, -F или -NH2. Соединения формулы VII-A1, VII-A2, VIII-A1, VIII-A2, IX-А1, IX-A2, X-A1, Х-А2, XI-A1, XI-A2, XII-A, XII-A1, XII-A2, XIII-А, XIII-A1, XIII-A2, XIV-A, XIV-A1 или XIV-А2: включают любые комбинации Ra, R3, В, R9 и R12.
Дополнительные типичные соединения по настоящему изобретению включают, не ограничиваясь, следующие:







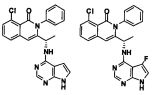
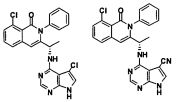





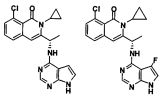



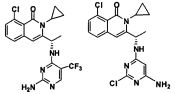

Химические соединения, описанные в настоящем документе, могут быть синтезированы по одной или более иллюстративным схемам, представленным в настоящем документе и/или с помощью способов, хорошо известных в данной области.
Если не указано иное, то описанные в настоящем документе реакции протекают при атмосферном давлении, обычно в температурном диапазоне от -10°С до 200°С. Кроме того, если не указано обратное, время и условия реакции являются приблизительными, например, протекают примерно при атмосферном давлении в температурном диапазоне от примерно -10°С до примерно 110°С, в течение периода времени от примерно 1 до примерно 24 часов; реакционная смесь, оставленная на ночь, протекает в течение среднего периода примерно 16 часов.
Термины «растворитель», «органический растворитель» и «инертный растворитель» означают растворитель, инертный в условиях реакции, описанных в связи с ней, включая, например, бензол, толуол, ацетонитрил, тетрагидрофуран ("ТГФ"), диметилформамид («ДМФ»), хлороформ, метиленхлорид (или дихлорметан), диэтиловый эфир, метанол, N-метилпирролидон ("NMP"), пиридин и т.п. Если не указано обратное, то растворители, используемые в реакциях, описанных в настоящем документе, являются инертными органическими растворителями. Если не указано обратное, то каждый грамм лимитирующего реагента, один кубический сантиметр (или мл) растворителя составляет объемный эквивалент.
Выделение и очистка химических соединений и промежуточных веществ, описанные в настоящем документе, могут быть выполнены, при необходимости, любым подходящим способом разделения или очистки, таким как, например, фильтрация, экстракция, кристаллизация, колоночная хроматография, тонкослойная хроматография или толстослойная хроматография, или комбинацией этих способов. Конкретные иллюстрации пригодных способов разделения и выделения могут быть получены по ссылке на примеры, представленные ниже. Однако могут использоваться также другие аналогичные способы разделения или выделения.
При необходимости, (R)- и (S)-изомеры соединений настоящего изобретения, при их наличии, могут быть разделены способами, известными специалистам в данной области, например, путем образования диастереоизомерных солей или комплексов, которые могут быть разделены, например, кристаллизацией; путем образования диастереоизомерных производных, которые могут быть разделены, например, кристаллизацией, газожидкостной или жидкостной хроматографией; селективной реакцией одного энантиомера с энантиомер-специфичным реагентом, например, ферментативным окислением или восстановлением с последующим разделением модифицированного и немодифицированного энантиомеров; или газожидкостной или жидкостной хроматографией в хиральной среде, например, на хиральной подложке, такой как диоксид кремния со связанным хиральным лигандом или в присутствии хирального растворителя. Альтернативно, конкретный энантиомер может быть синтезирован путем асимметричного синтеза, с использованием оптически активных реагентов, субстратов, катализаторов или растворителей, или путем превращения одного энантиомера в другой посредством асимметричного превращения.
Соединения, описанные в настоящем документе, могут в некоторых случаях взаимодействовать с фармацевтически приемлемой кислотой для получения соответствующих кислотно-аддитивных солей.
Многие возможно замещенные исходные соединения и другие реагенты являются доступными в продаже, например, производства компании Aldrich Chemical Company (Milwaukee, Висконсин), или легко могут быть получены специалистом в данной области с использованием обычных способов синтеза.
Соединения по настоящему изобретению могут быть, как правило, синтезированы путем соответствующей комбинации общеизвестных способов синтеза. Методики, пригодные для синтеза этих химических соединений являются очевидными и доступными для специалиста в данной области, на момент раскрытия настоящего изобретения.
Соединения по настоящему изобретению могут быть синтезированы соответствующей комбинацией известных в данной области способов синтеза. Описание, представленное ниже, предназначено для иллюстрации некоторых из многочисленных способов, пригодных для применения при получении соединений настоящего изобретения, и не является ограничивающим рамки реакций или реакционных последовательностей, которые могут использоваться для получения соединений настоящего изобретения.
Реакционная схема 1

Ссылаясь на схему 1, стадия 1, соединение формулы 101, где X является N или CR7, превращается в соединение формулы 103, например, через двустадийный процесс связывания Хека с соединением формулы 102 с последующей кислотно-катализируемой циклизацией в метаноле. Продукт, соединение формулы 103, выделяют. Ссылаясь на схему 1, стадия 2, соединение формулы 103 превращается в соединение формулы 404, например, по реакции с соответствующим замещенным анилином. Продукт, соединение формулы 104, выделяют. Ссылаясь на схему 1, стадия 3, соединение формулы 104 превращается в соединение формулы 105, например, путем восстановления с гидридом лития-алюминия. Продукт, соединение формулы 105, выделяют. Ссылаясь на схему 1, стадия 4, соединение формулы 105 превращается в соединение формулы 106, например, по реакции с тионилхлоридом. Продукт, соединение формулы 106, выделяют. Ссылаясь на схему 1, стадия 5, соединение формулы 106 превращается в соединение формулы 107, например, по реакции алкилирования с пиразолопиримидином с использованием основания, такого как карбонат калия. Продукт, соединение формулы 107, выделяют. Ссылаясь на схему 1, стадия 6, соединение формулы 107 превращается в соединение формулы 108, например, по реакции Сузуки. Продукт, соединение формулы 108, выделяют и в некоторых случаях очищают.
Реакционная схема 2:

Ссылаясь на схему 2, стадия 1, соединение формулы 201, где X является N или CR7, превращается в соединение формулы 202, например, с реагентом, подходящим для введения хлорида кислоты, например, оксалилхлорида. Продукт, соединение формулы 202, в некоторых случаях выделяют. Ссылаясь на схему 2, стадия 2, соединение формулы 202 превращается в соединение формулы 503, например, по реакции, например, с ариламином. Продукт, соединение формулы 203, выделяют. Ссылаясь на схему 2, стадия 3, соединение формулы 203 превращается в соединение формулы 204, например, по реакции связывания Стилле с использованием соответствующего винил-станнана. Продукт, соединение формулы 204, выделяют. Ссылаясь на схему 2, стадия 4, соединение формулы 204 превращается в третичный амид, соединение формулы 205, по реакции с хлорэтилацетатом и основанием, гидридом натрия. Продукт, соединение формулы 205, выделяют. Ссылаясь на схему 2, стадия 5, соединение формулы 205 окисляется до альдегида с использованием, например, тетраоксида осмия и периодината натрия. Продукт, соединение формулы 206, выделяют. Ссылаясь на схему 2, стадия 6, соединение формулы 206 превращается в соединение формулы 104, например, по альдольной реакции в этаноле с основанием, таким как карбонат цезия. Продукт, соединение формулы 104, выделяют. Ссылаясь на схему 2, стадия 7, соединение формулы 104 восстанавливается до первичного спирта по реакции восстановления, например, с гидридом лития-алюминия, для получения соединения формулы 105, которое выделяют. Ссылаясь на схему 2, стадия 8, соединение формулы 105 превращается в соединение формулы 207 по реакции с тетрабромидом углерода и трифенилфосфином. Продукт, соединение формулы 207, выделяют. Это соединение может быть основным промежуточным соединением в синтезе соединений настоящего изобретения.
Реакционная схема 3:

Ссылаясь на схему 3, стадия 9, соединение формулы 207, где X является N или CR7, синтезируется так, как описано в реакционной схеме 2, и превращается в соединение формулы 107 по реакции связывания с соединением формулы 208 в присутствии основания, например, трет-бутоксида калия. Продукт, соединение формулы 107, выделяют. Ссылаясь на схему 3, стадия 10, соединение формулы 107 превращается в соединение формулы 108 по реакции связывания, например, с арилбороновой кислотой в присутствии катализатора связывания и основания, например, ацетата палладия, трифенилфосфина и карбоната калия, например. Продукт, соединение формулы 108, выделяют.
Реакционная схема 4А:

Реакционная схема 4B:


Ссылаясь на реакционную схему 4А, которая иллюстрирует синтез основного класса пуринил-замещенных изохинолонов, стадия 1, сложный йодосодержащий эфир 401 реагирует с алкином формулы 400-А в присутствии палладиевого катализатора, йодида меди и триэтиламина (ТЭА) для связывания алкина в ариловое ядро соединения 401 с получением соединения формулы 402. Соединение формулы 402 в некоторых случаях выделяют. Ссылаясь на реакционную схему 4, стадия 2, соединение формулы 402 обрабатывают основанием, гидроксидом калия, для получения карбоновой кислоты, соединения формулы 403, если продукт реакции или его соль подкислены. Соединение формулы 403 в некоторых случаях выделяют. Ссылаясь на реакционную схему 4, стадия 3, соединение формулы 403 обрабатывают бис(ацетонитрил)дихлорпалладием (II) и ТЭА для замыкания внутримолекулярного кольца с получением соединения формулы 404. Соединение формулы 404 выделяют. Ссылаясь на реакционную схему 4, стадия 4, соединение формулы 404 реагирует с первичным амином для получения соединения формулы 405. Соединение формулы 405 в некоторых случаях выделяют. Ссылаясь на реакционную схему 4, стадия 5, соединение формулы 405 обрабатывают хлороводородной кислотой, снимая защитную группу на азоте, для получения соединения формулы 406. Соединение формулы 406 в некоторых случаях выделяют. Ссылаясь на реакционную схему 4, стадия 6, соединение формулы 406 реагирует с соединением формулы 407 для получения соединения формулы 408. Соединение формулы 408 выделяют.
В реакционной схеме 4В показан синтез одного подмножества пуринил-замещенных изохинолонов, где R9 является метилом и Ra является водородом, при помощи синтетических преобразований, описанных для реакционной схемы 4А.
Реакционная схема 5:
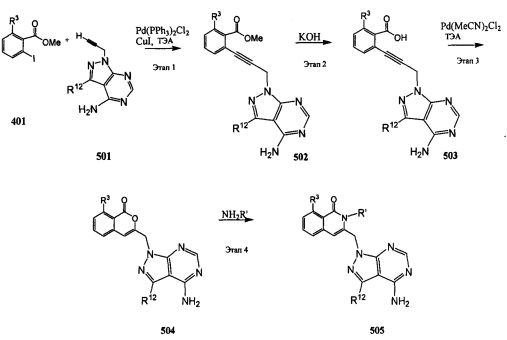
Ссылаясь на реакционную схему 5, стадия 1, сложный йодсодержащий эфир 401 реагирует с алкином 501 в присутствии палладиевого катализатора связывания, йодида меди и ТЭА, для получения соединения формулы 502. Соединение 502 в некоторых случаях выделяют. Ссылаясь на реакционную схему 5, стадия 2, соединение формулы 502 обрабатывают основанием, гидроксидом калия, для получения карбоксилата или свободной кислоты соединения формулы 503. Ссылаясь на реакционную схему 5, стадия 3, соединение формулы 503 обрабатывают бис(ацетонитрил)дихлорпалладием (II) и ТЭА для замыкания внутримолекулярного кольца с получением соединения формулы 504. Соединение формулы 504 в некоторых случаях выделяют. Ссылаясь на реакционную схему 5, стадия 4, соединение формулы 504 обрабатывают первичным амином для получения соединения формулы 505. Соединение формулы 505 выделяют.
Реакционная схема 6А:

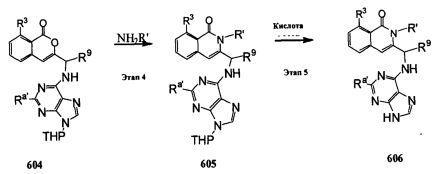
Реакционная схема 6В:

Ссылаясь на реакционную схему 6А, которая иллюстрирует синтез основного класса пуринил-замещенных изохинолонов, стадия 1, сложный йодсодержащий эфир 401 реагирует с алкином 601 в присутствии палладиевого катализатора связывания, йодида меди и ТЭА, для получения соединения формулы 602. Соединение 602 в некоторых случаях выделяют. Ссылаясь на реакционную схему 6, стадия 2, соединение формулы 602 обрабатывают основанием, гидроксидом калия, для получения карбоксилата или свободной кислоты соединения формулы 603. Ссылаясь на реакционную схему 6, стадия 3, соединение формулы 603 обрабатывают бис(ацетонитрил)дихлорпалладием (II) и ТЭА для замыкания внутримолекулярного кольца с получением соединения формулы 604. Соединение формулы 604 в некоторых случаях выделяют. Ссылаясь на реакционную схему 6, стадия 4, соединение формулы 604 обрабатывают первичным амином для получения соединения формулы 605. Соединение формулы 605 выделяют. Ссылаясь на реакционную схему 6, стадия 5, соединение формулы 605 обрабатывают кислотой для удаления ТНР защитной группы и получения соединения формулы 606. Соединение формулы 606 выделяют.
В реакционной схеме 6В показан синтез одного подмножества пуринил-замещенных изохинолонов, где R9 является метилом и Ra является водородом, при помощи синтетических преобразований, описанных для реакционной схемы 6А.
Реакционная схема 7А:
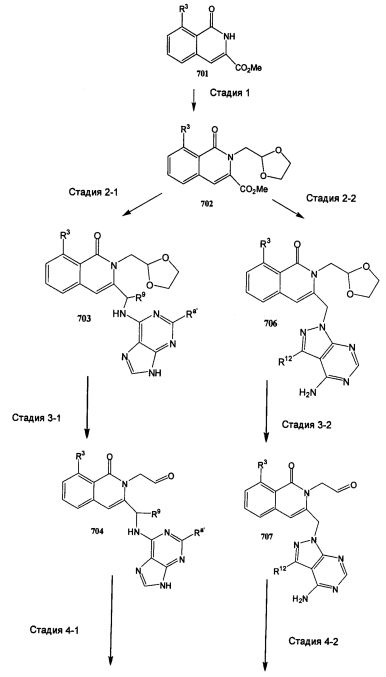

Реакционная схема 7В:

Ссылаясь на реакционную схему 7А, которая иллюстрирует синтез пуринил- или пиразолопиримидинил-замещеных изохинолонов, включая алкиламиновый заместитель в положении В формулы I, стадия 1, соединение формулы 701 синтезируется различными синтетическими путями, включая вариации схемы 1 или 2, где, например, бензиламин используется на этапе превращения соединения формулы 103 в соединение формулы 104. Бензиловая группа, являющаяся защитной группой амина, может быть удалена обычным способом снятия химической защиты для получения соединения 701. Другой пример превращения соединения формулы 103 в соединение формулы 701, представляет обработку соединения формулы 103 аммиаком для получения соединения формулы 701. Соединение формулы 701 превращается в соединение формулы 702 алкилированием амидного азота рядом 2-углеродсодержащих синтонов, у которых может быть снята защита, они могут быть окислены и повторно защищены как соответствующий кеталь, соединение формулы 702. Ссылаясь на реакционную схему 7, стадия 2-1, соединение формулы 702 превращается, например, при восстановительном аминировании сложноэфирной группы, с получением пуриниловой группы соединения 703, или альтернативно, алкилируется для введения пуриниловой группы и получения соединения формулы 703. Ссылаясь на реакционную схему 7, стадия 3-1, соединение формулы 703 обрабатывается кислотой для снятия кетальной защитной группы с получением соединения формулы 704. Соединение формулы 704 выделяют. Ссылаясь на реакционную схему 7, стадия 4-1, соединение формулы 704 подвергается восстановительному аминированию амином для получения соединения формулы 705. Соединение формулы 705 выделяют. Ссылаясь на реакционную схему 7, стадия 2-2, соединение формулы 702 превращается на этапах 7 и 8 схемы 2 и этапе 9 схемы 3 с получением пиразолопиримидиновой группы соединения формулы 706. Соединение формулы 706 выделяют. Ссылаясь на реакционную схему 7, стадия 3-2, соединение формулы 706 обрабатывают кислотой для удаления кетальной защитной группы для получения соединения формулы 707. Соединение формулы 707 выделяют.
Ссылаясь на реакционную схему 7, стадия 4-2, соединение формулы 707 подвергается восстановительному аминированию амином для получения соединения формулы 708. Соединение формулы 708 выделяют.
В реакционной схеме 7В показан синтез соединений, где R9 является метилом и Ra является водородом, с использованием этапов, описанных в схеме 7А.
Реакционная схема 8:

Ссылаясь на реакционную схему 8, стадия 1, соединение формулы 701 синтезируется так, как описано в схеме 7 или любым другим известным химическим способом. Соединение формулы 701 превращается путем алкилирования амидного азота при помощи ряда 2-углеродсодержащих синтонов, с которых может быть снята защита, и которые могут быть превращены в алкокси-защищенные частицы, как показано в соединении формулы 801, которое может быть выделено. Ссылаясь на реакционную схему 8, стадия 2, соединение формулы 801 превращается путем химических реакций, описанных на этапе 2-1 схемы 7 с получением пуриниловой группы, и это полученное соединение превращается путем снятия защиты, активации и аминирования амином в соединение формулы 802, которое выделяют.
Ссылаясь на реакционную схему 8, стадия 3, соединение формулы 801 превращается путем химических реакций, описанных на этапе 2-2 схемы 7 с получением пиразолопиримидиновой группы, и это полученное соединение превращается путем снятия защиты, активации и аминирования амином в соединение формулы 803, которое выделяют.
Реакционная схема 9:

Ссылаясь на реакционную схему 9, стадия 1, соединение формулы 901 обрабатывают амином для получения соединения формулы 902. Соединение формулы 902 выделяют. Ссылаясь на реакционную схему 9, стадия 2, соединение формулы 902 обрабатывают оксихлоридом фосфора для получения соединения формулы 903. Соединение формулы 903 выделяют. Ссылаясь на реакционную схему 9, стадия 3, соединение формулы 903 реагирует с аминопурином формулы 904 для получения соединения формулы 905. Соединение формулы 905 выделяют. Ссылаясь на реакционную схему 9, стадия 4, соединение формулы 905 обрабатывают хлороводородной кислотой для удаления защитной группы у азота в пуриновой группе для получения соединения формулы 906. Соединение формулы 906 выделяют.
Реакционная схема 10:
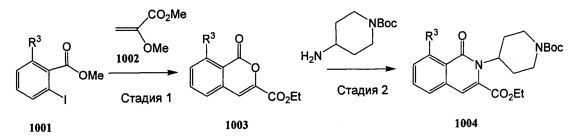
Ссылаясь на реакционную схему 10, стадия 1, соединение формулы 1001 обрабатывают винилогическим сложным эфиром 1002, используя, например, реакцию Хека с последующей циклизацией, для получения соединения формулы 1003. Соединение формулы 1003 выделяют. Ссылаясь на реакционную схему 10, стадия 2, соединение формулы 1003 реагирует с 4-амино-N-трет-бутоксикарбонил пиперидином для получения соединения формулы 1004. Соединение формулы 1004 выделяют. Соединение формулы 1004 может использоваться как промежуточный продукт в синтезе соединений настоящего изобретения.
Реакционная схема 11:

Ссылаясь на реакционную схему 11, стадия 1, соединение формулы 1101 обрабатывают алкиниловым спиртом, например, формулы 1102, в присутствии йодида меди и катализатора палладия на углероде, для получения соединения формулы 1103. Соединение формулы 1103 выделяют. Ссылаясь на реакционную схему 11, стадия 1, соединение формулы 1102 реагирует с 4-амино-N-трет-бутоксикарбонил пиперидином для получения соединения формулы 1103. Соединение формулы 1103 выделяют. Соединение формулы 1103 может использоваться как промежуточный продукт в синтезе соединений настоящего изобретения.
Реакционная схема 12:

Другой подход к синтезу соединений формулы I представлен на схеме 12. Ссылаясь на стадия 1, соединение формулы 1201 обрабатывается хлорирующим агентом, таким как оксалилхлорид, для получения хлорида кислоты формулы 1202. На этапе 2 соединение формулы 1202 реагирует с соединением формулы R′NH2 в присутствии основания, такого как триэтиламин, для получения соединения формулы 1203. В некоторых воплощениях изобретения хлорирующим агентом, используемым для превращения соединения 1201, является тионилхлорид, например, тионилхлорид в толуоле. При использовании тионилхлорида, стадия 1 и 2 могут объединяться с образованием одностадийной реакции. На этапе 3 соединение формулы 1203 обрабатывается н-бутиллитием, и затем реагирует с диалкилоксалатом, таким как диэтилоксалат, для получения соединения формулы 1204. На этапе 4, соединение формулы 1204 дефлегмируется в кислотном растворе, например, хлороводородной кислоты в метаноле, для получения соединения формулы 1205. На этапе 5, соединение формулы 1205 обрабатывается восстанавливающим агентом, таким как гидрид лития-алюминия, для получения соединения формулы 1206. На этапе 6, соединение формулы 1206 реагирует с бромирующим агентом, таким как трибромид фосфора, в присутствии диметилформамида в ацетонитриле, для получения бромсодержащего соединения формулы 1207. На этапе 7, соединение формулы 1207 реагирует с гетероариловым соединением, например, 3-йод-1Н-пиразоло[3,4-d]пиримидин-4-амином, в присутствии основания, такого как трет-бутоксид калия в диметилформамиде, для получения соединения формулы 1208.
Реакционная схема 13:
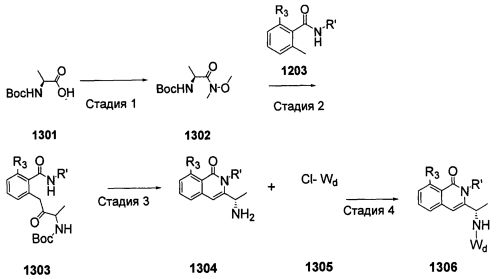
На схеме 13 представлен подход для синтеза соединений формулы I, имеющих мостик XY, где X является преимущественно или исключительно (S)-С(CH3)Н- и Y является -NH-. Wd является моноциклическим или бициклическим гетероарилом, включая, не ограничиваясь, пуринил, пиримидинил, пирролопиримидинил или пиразолопиримидинил. Ссылаясь на стадия 1 схемы 13, соединение формулы 1301 (S-изомер) связывается с N,O-диметилгидроксиламином при помощи гидроксибензотриазола (HOBt) и 1-этил-3-(3'-диметиламинопропил)карбодиимида (EDCI) в присутствии триэтиламина для получения соединения формулы 1302. На этапе 2 соединение формулы 1203, которое может быть синтезировано так, как описано в схеме 12, депротонируется н-бутиллитием в ТГФ и гексаметилфосфорамидом при -78°С в атмосфере аргона. Добавляется соединение формулы 1302, и реакционную смесь оставляют нагреваться до -50°С, погашают добавлением воды, и выделяют соединение формулы 1303. В некоторых воплощениях изобретения могут использоваться слабые нуклеофильные органомагниевые частицы, такие как изо-PrMgCl, для получения магниевого аниона соединения 1302 перед его добавлением к дианиону. На этапе 3 соединение формулы 1303 обрабатывается хлороводородной кислотой в метаноле при дефлегмации, а затем реакционная смесь подщелачивается добавлением раствора карбоната натрия до рН примерно 7-8, для получения соединения формулы 1304. Соединение формулы 1304 может быть частично эпимеризовано в результате предыдущих этапов реакции. Соединение 1304 с высокой энантиомерной чистотой может быть выделено путем получения виннокислой соли при растворении соединения формулы 1304 в метаноле с добавлением D-винной кислоты. Полученная реакционная смесь дефлегмируется в течение одного часа, затем перемешивается при комнатной температуре в течение 16 часов и оставляется для выделения соли соединения формулы 1304, где энантиомерная чистота составляет более 90% (S)-изомера. Свободный амин соединения формулы 1304 регенерируется перед его использованием на следующем этапе синтеза. Соединение формулы 1304, которое преимущественно является (S)-энантиомером, связывается с хлор-замещенным гетероарилом Wd, соединением формулы 1305, включая, не ограничиваясь, 6-хлор-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин, 2,4,5-трихлорпиримидин, 4-хлор-7Н-пирроло[2,3-d]пиримидин и 4-хлор-1Н-пиразоло[3,4-d]пиримидин в присутствии основания, такого как диизопропилэтиламин или аммиак, для получения соединения формулы 1306, и где соединение формулы 1306 является (S)-изомером.
Синтез R3-галоген-замещенных аналогов, например, хлор-замещенных аналогов изохинолона. Такая же реакционная схема 13 применима для получения соединений формулы:

Соединения, описанные в настоящем документе, могут быть синтезированы с использованием реакционных схем, описанных в настоящем документе, их разновидностей или других синтетических способов, известных в данной области.
В некоторых воплощениях изобретения соединения по настоящему изобретению обладают одной или более функциональными характеристиками, описанными в настоящем документе. Например, одно или более указанных соединений специфически связывается с PI3 киназой. В некоторых воплощениях изобретения IC50 указанного соединения для p110α, p110β, p110γ или p110δ составляет менее, чем примерно 1 мМ, менее, чем примерно 100 нМ, менее, чем примерно 50 нМ, менее, чем примерно 10 нМ, менее, чем примерно 1 нМ, менее, чем примерно 0,5 нМ, менее, чем примерно 100 пМ или менее, чем примерно 50 пМ.
В некоторых воплощениях изобретения одно или более из указанных соединений может селективно ингибировать один или более членов типа I или класса I фосфатидилинозитол 3-киназ (PI3-киназ) со значением IC50 примерно 100 нМ, 50 нМ, 10 нМ, 5 нМ, 100 пМ, 10 пМ или 1 пМ, или менее, рассчитанное по анализу киназы in vitro.
Кроме того, соединение формулы, имеющее (S)-энантиомерную конфигурацию по отношению к углероду X, может демонстрировать более высокий потенциал против одной или более целевых PI3-киназ, по сравнению с соответствующим соединением, имеющим (R)-энантиомерную конфигурацию по отношению к углероду X. Например, соединение настоящего изобретения, имеющее (S)-энантиомерную конфигурацию по отношению к углероду X, может иметь значение IC50 PI3-киназы, которое на 1, 2, 3 или 4 порядка ниже, чем значение IC50 PI3-киназы соответствующего соединения, имеющего (R)-конфигурацию. В некоторых воплощениях изобретения соединение настоящего изобретения является соединением формулы V-A2 в (S)-конфигурации относительно углерода X, имеющее значение IC50 PI3-киназы, которое на 1, 2, 3 или 4 порядка ниже, чем значение IC50 PI3-киназы соответствующего соединения, имеющего (R)-конфигурацию. Например, соединение настоящего изобретения является соединением формулы V-A2 в (S)-конфигурации относительно углерода X, имеющее значение IC50 PI3-киназы, которое на 4 порядка ниже, чем значение IC50 PI3-киназы соответствующего соединения, имеющего (R)-конфигурацию. В некоторых воплощениях изобретения соединение настоящего изобретения является соединением формулы V-A2, где R3 является С1-С3 алкилом, и В является фенилом, и где это соединение находится в (S)-конфигурации относительно углерода X и имеет значение IC50 PI3-киназы, которое по меньшей мере на 3 порядка ниже, чем значение IC50 PI3-киназы соответствующего соединения, имеющего (R)-конфигурацию. В других воплощениях изобретения соединение настоящего изобретения является соединением формулы V-A2, где R3 является галогено, и В является фенилом, и где это соединение находится в (S)-конфигурации относительно углерода X и имеет значение IC50 PI3-киназы, которое по меньшей мере на 3 порядка ниже, чем значение IC50 PI3-киназы соответствующего соединения, имеющего (R)-конфигурацию. В других воплощениях изобретения соединение настоящего изобретения является соединением формулы V-А2, где R3 является С1-С3 алкилом, и В является циклоалкилом, и где это соединение находится в (S)-конфигурации относительно углерода X и имеет значение IC50 PI3-киназы, которое на 3 порядка ниже, чем значение IC50 PI3-киназы соответствующего соединения, имеющего (R)-конфигурацию.
В некоторых воплощениях изобретения одно или более из указанных соединений могут селективно ингибировать один или два представителя типа I или класса I фосфатидилинозитол 3-киназ (PI3-киназ), состоящего из PI3-киназы α, PI3-киназы β, PI3-киназы γ и PI3-киназы δ. В некоторых аспектах указанные соединения селективно ингибируют PI3-киназу δ по сравнению со всеми остальными PI3-киназами типа I. В других аспектах некоторые из указанных соединений селективно ингибируют PI3-киназу δ и PI3-киназу γ по сравнению со всеми остальными PI3-киназами типа I. В других аспектах некоторые из указанных соединений селективно ингибируют PI3-киназу α и PI3-киназу β по сравнению со всеми остальными PI3-киназами типа I. В других аспектах некоторые из указанных соединений селективно ингибируют PI3-киназу δ и PI3-киназу α по сравнению со всеми остальными PI3-киназами типа I. В некоторых других аспектах некоторые из указанных соединений селективно ингибируют PI3-киназу δ и PI3-киназу β по сравнению со всеми остальными PI3-киназами типа I, или селективно ингибируют PI3-киназу δ и PI3-киназу α по сравнению со всеми остальными PI3-киназами типа I, или селективно ингибируют PI3-киназу α и PI3-киназу γ по сравнению со всеми остальными PI3-киназами типа I, или селективно ингибируют PI3-киназу γ и PI3-киназу β по сравнению со всеми остальными PI3-киназами типа I.
В некоторых воплощениях изобретения одно или более из указанных соединений селективно ингибируют PI3-киназу δ и PI3-киназу γ по сравнению со всеми остальными PI3-киназами типа I. В некоторых аспектах соединение настоящего изобретения демонстрирует IC50 для PI3-киназы δ, которое менее чем в 100, 90, 80, 70, 60, 50, 40, 30, 20, 10 или 5 раз ниже, чем IC50 для PI3-киназы γ. В других аспектах соединение настоящего изобретения демонстрирует IC50 для PI3-киназы δ, которое менее чем в 100, 90, 80, 70, 60, 50, 40, 30, 20, 10 или 5 раз выше, чем IC50 для PI3-киназы γ. В некоторых воплощениях изобретения соединение настоящего изобретения демонстрирует IC50 для PI3-киназы δ, которое менее чем в 10 раз ниже, чем IC50 для PI3 -киназы γ. В других воплощениях изобретения соединение настоящего изобретения демонстрирует IC50 для PI3-киназы δ, которое менее чем в 10 раз выше, чем IC50 для PI3-киназы γ. Например, соединение настоящего изобретения демонстрирует IC50 для PI3-киназы δ, которое менее чем в 5 раз выше или ниже, чем IC50 для PI3-киназы γ. В некоторых аспектах указанное соединение имеет IC50 для PI3-киназы δ, которое ниже чем IC50 для PI3-киназы γ с коэффициентом менее 20, 10, 5 или 2. Например, указанное соединение имеет IC50 для PI3-киназы δ, которое ниже, чем IC50 для PI3-киназы γ с коэффициентом менее 5.
В другом аспекте ингибитор, селективно ингибирующий один или более представителей PI3-киназ типа I, или ингибитор, селективно ингибирующий один или более сигнальных путей, опосредованных PI3-киназами типа I, альтернативно, может расцениваться как соединение, демонстрирующее 50% ингибирующей концентрации (IC50) по сравнению с данным типом PI3-киназ типа I, которое по меньшей мере в 10 раз по меньшей мере в 20 раз по меньшей мере в 50 раз по меньшей мере в 100 раз по меньшей мере в 1000 раз по меньшей мере в 10100 раз или меньше, чем IC50 ингибитора по сравнению с остальными PI3-киназами типа I.
В некоторых воплощениях изобретения одно или более из указанных соединений ингибирует p110α, р110β, DNAPK или mTor со значением IC50 больше, чем 30 нМ, и ингибирует р110δ и/или р110γ со значением IC50 менее, чем 1 мкМ. В некоторых воплощениях изобретения соединение дополнительно демонстрирует селективное ингибирование р110δ и/или p110γ по сравнению с p110α, p110β, DNAPK и/или mTor с коэффициентом по меньшей мере 3, 10, 100, 1000 или выше. Например, указанное соединение демонстрирует селективное ингибирование p110δ или p110γ по сравнению с p110α, p110β, DNAPK и/или mTor с коэффициентом по меньшей мере 3. Типичные соединения, демонстрирующие селективное ингибирование p110δ или p110γ по сравнению с p110α, p110β, DNAPK и/или mTor с коэффициентом по меньшей мере 3, включают, не ограничиваясь, соединения 328, 329, 330, 331, 332, 333, 335, 336, 337, 338, 339, 340, 341, 342, 343, 344, 345, 346, 347, 348, 349, 350, 352, 354, 357 и 361 в таблице 4.
Фармацевтические композиции
В настоящем изобретении представлены фармацевтические композиции, включающие одно или более соединений настоящего изобретения.
В некоторых воплощениях настоящего изобретения представлены фармацевтические композиции для лечения заболеваний или состояний, связанных с нежелательным, сверхактивным, вредным или разрушительным иммунным ответом у млекопитающих. Такой нежелательный иммунный ответ может быть результатом, например, астмы, эмфиземы, бронхита, псориаза, аллергии, анафилаксии, аутоиммунных нарушений, ревматоидного артрита, реакции "трансплантат против хозяина" и красной волчанки или быть связан с ними. Фармацевтические композиции настоящего изобретения могут использоваться для лечения других респираторных заболеваний, включая, но не ограничиваясь, заболевания, поражающие доли легких, плевральную полость, бронхи, трахею, верхние дыхательные пути или дыхательные нервы и мышцы.
В некоторых воплощениях, в настоящем изобретении представлены фармацевтические композиции для лечения заболеваний, таких как гиперпролиферативное заболевание, включая, но не ограничиваясь, рак, такой как острый миелоидный лейкоз, рак вилочковой железы, мозга, легкого, плоскоклеточный рак, рак кожи, глаза, ретинобластому, внутриглазную меланому, рак полости рта и ротоглотки, мочевого пузыря, желудка, поджелудочной железы, мочевого пузыря, молочной железы, шейки матки, головы, шеи, почки, печени, яичника, предстательной железы, толстой кишки, пищевода, яичка, гинекологический рак, рак щитовидной железы, ЦНС, ПНС, связанный со СПИДом (например, лимфома и саркома Капоши) или вирус-индуцированный рак. В некоторых воплощениях изобретения указанные фармацевтические композиции предназначены для лечения нераковых гиперпролиферативных заболеваний, таких как доброкачественная гиперплазия кожи (например, псориаз), простаты (например, доброкачественная гипертрофия предстательной железы (ДГПЖ) или рестеноз).
В настоящем изобретении представлены также композиции для лечения заболеваний печени (включая диабеты), заболеваний поджелудочной железы или почек (включая пролиферативный гломерулонефрит и диабет-индуцированное заболевание почек) или боли у млекопитающих.
В настоящем изобретении дополнительно представлена композиция для предотвращения имплантации бластоцитов у млекопитающих.
Настоящее изобретение относится также к композиции для лечения заболевания, связанного с васкулогенезом или ангиогенезом у млекопитающих, которое может проявляться в виде опухолевого ангиогенеза, хронических воспалительных заболеваний, таких как ревматоидный артрит, воспалительных заболеваний кишечника, атеросклероза, кожных заболеваний, таких как псориаз, экзема и склеродермия, сахарного диабета, диабетической ретинопатии, ретинопатии недоношенных, возрастной дегенерации желтого пятна, гемангиомы, глиомы, меланомы, саркома Капоши и рака яичников, молочной железы, легких, поджелудочной железы, простаты, толстой кишки и эпидермоидного рака.
Указанные фармацевтические композиции обычно изготавливаются для предоставления фармацевтически эффективного количества соединения по настоящему изобретению в качестве активного компонента, или его фармацевтически приемлемой соли, сложного эфира, пролекарственные средства, сольвата, гидрата или производного. При необходимости фармацевтические композиции содержат фармацевтически приемлемую соль и/или координационный комплекс соединения, и один или более фармацевтически приемлемых носителей, наполнителей, включая инертные твердые разбавители и наполнители, разбавители, включая стерильные водные растворы и различные органические растворители, добавки для улучшения проникновения, солюбилизаторы и адъюванты.
Указанные фармацевтические композиции могут вводиться по отдельности или в комбинации с одним или более других агентов, которые также обычно вводятся в форме фармацевтических композиций. При необходимости указанные соединения или другие агенты могут быть смешаны в композицию, или оба компонента могут быть введены в отдельные композиции для их комбинированного применения по отдельности или в разное время.
В некоторых воплощениях изобретения концентрация одного или более соединений в фармацевтических композициях настоящего изобретения составляет менее чем 100%, 90%, 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19%, 18%, 17%, 16%, 15%,14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0,5%, 0,4%, 0,3%, 0,2%, 0,1%, 0,09%, 0,08%, 0,07%, 0,06%, 0,05%, 0,04%, 0,03%, 0,02%, 0,01%, 0,009%, 0,008%, 0,007%, 0,006%, 0,005%, 0,004%, 0,003%, 0,002%, 0,001%, 0,0009%, 0,0008%, 0,0007%, 0,0006%, 0,0005%, 0,0004%, 0,0003%, 0,0002% или 0,0001% масс/масс, масс/об. или об./об.
В некоторых воплощениях изобретения концентрация одного или более соединений настоящего изобретения составляет более чем 90%», 80%, 70%, 60%, 50%, 40%, 30%, 20%, 19,75%, 19,50%, 19,25% 19%, 18,75%, 18,50%, 18,25% 18%, 17,75%, 17,50%, 17,25% 17%, 16,75%, 16,50%, 16,25% 16%, 15,75%, 15,50%, 15,25% 15%, 14,75%, 14,50%, 14,25% 14%, 13,75%, 13,50%, 13,25% 13%, 12,75%, 12,50%, 12,25% 12%, 11,75%, 11,50%, 11,25% 11%, 10,75%, 10,50%, 10,25% 10%, 9,75%, 9,50%, 9,25% 9%, 8,75%, 8,50%, 8,25% 8%, 7,75%, 7,50%, 7,25% 7%, 6,75%, 6,50%, 6,25% 6%, 5,75%, 5,50%, 5,25% 5%, 4,75%, 4,50%, 4,25%, 4%, 3,75%, 3,50%, 3,25%, 3%, 2,75%, 2,50%, 2,25%, 2%, 1,75%, 1,50%, 125%, 1%, 0,5%, 0,4%, 0,3%, 0,2%, 0,1%, 0,09%, 0,08%, 0,07%, 0,06%, 0,05%, 0,04%, 0,03%, 0,02%, 0,01%, 0,009%, 0,008%, 0,007%, 0,006%, 0,005%, 0,004%, 0,003%, 0,002%, 0,001%, 0,0009%, 0,0008%, 0,0007%, 0,0006%, 0,0005%, 0,0004%, 0,0003%, 0,0002% или 0,0001% масс/масс, масс/об. или об./об.
В некоторых воплощениях изобретения концентрация одного или более из соединений настоящего изобретения находится в диапазоне от примерно 0,0001% до примерно 50%, от примерно 0,001% до примерно 40%, от примерно 0,01% до примерно 30%, от примерно 0,02% до примерно 29%, от примерно 0,03% до примерно 28%, от примерно 0,04% до примерно 27%, от примерно 0,05% до примерно 26%, от примерно 0,06% до примерно 25%, от примерно 0,07% до примерно 24%, от примерно 0,08% до примерно 23%, от примерно 0,09% до примерно 22%, от примерно 0,1% до примерно 21%, от примерно 0,2% до примерно 20%, от примерно 0,3% до примерно 19%, от примерно 0,4% до примерно 18%, от примерно 0,5% до примерно 17%, от примерно 0,6% до примерно 16%, от примерно 0,7% до примерно 15%, от примерно 0,8% до примерно 14%, от примерно 0,9% до примерно 12%, от примерно 1% до примерно 10% масс./масс., масс./об. или об./об.
В некоторых воплощениях изобретения концентрация одного или более соединений настоящего изобретения находится в диапазоне от примерно 0,001% до примерно 10%, от примерно 0,01% до примерно 5%, от примерно 0,02% до примерно 4,5%, от примерно 0,03% до примерно 4%, от примерно 0,04% до примерно 3,5%, от примерно 0,05% до примерно 3%, от примерно 0,06% до примерно 2,5%, от примерно 0,07% до примерно 2%, от примерно 0,08% до примерно 1,5%, от примерно 0,09% до примерно 1%, от примерно 0,1% до примерно 0,09% масс/масс, масс./об. или об./об.
В некоторых воплощениях изобретения количество одного или более соединений настоящего изобретения равно или менее чем 10 г, 9,5 г, 9,0 г, 8,5 г, 8,0 г, 7,5 г, 7,0 г, 6,5 г, 6,0 г, 5,5 г, 5,0 г, 4,5 г, 4,0 г, 3,5 г, 3,0 г, 2,5 г, 2,0 г, 1,5 г, 1,0 г, 0,95 г, 0,9 г, 0,85 г, 0,8 г, 0,75 г, 0,7 г, 0,65 г, 0,6 г, 0,55 г, 0,5 г, 0,45 г, 0,4 г, 0,35 г, 0,3 г, 0,25 г, 0,2 г, 0,15 г, 0,1 г, 0,09 г, 0,08 г, 0,07 г, 0,06 г, 0,05 г, 0,04 г, 0,03 г, 0,02 г, 0,01 г, 0,009 г, 0,008 г, 0,007 г, 0,006 г, 0,005 г, 0,004 г, 0,003 г, 0,002 г, 0,001 г, 0,0009 г, 0,0008 г, 0,0007 г, 0,0006 г, 0,0005 г, 0,0004 г, 0,0003 г, 0,0002 г или 0,0001 г,
В некоторых воплощениях изобретения количество одного или более соединений настоящего изобретения составляет более, чем 0,0001 г, 0,0002 г, 0,0003 г, 0,0004 г, 0,0005 г, 0,0006 г, 0,0007 г, 0,0008 г, 0,0009 г, 0,001 г, 0,0015 г, 0,002 г, 0,0025 г, 0,003 г, 0,0035 г, 0,004 г, 0,0045 г, 0,005 г, 0,0055 г, 0,006 г, 0,0065 г, 0,007 г, 0,0075 г, 0,008 г, 0,0085 г, 0,009 г, 0,0095 г, 0,01 г, 0,015 г, 0,02 г, 0,025 г, 0,03 г, 0,035 г, 0,04 г, 0,045 г, 0,05 г, 0,055 г, 0,06 г, 0,065 г, 0,07 г, 0,075 г, 0,08 г, 0,085 г, 0,09 г, 0,095 г, 0,1 г, 0,15 г, 0,2 г, 0,25 г, 0,3 г, 0,35 г, 0,4 г, 0,45 г, 0,5 г, 0,55 г, 0,6 г, 0,65 г, 0,7 г, 0,75 г, 0,8 г, 0,85 г, 0,9 г, 0,95 г, 1 г, 1,5 г, 2 г, 2,5, 3 г, 3,5, 4 г, 4,5 г, 5 г, 5,5 г, 6 г, 6,5 г, 7 г, 7,5 г, 8 г, 8,5 г, 9 г, 9,5 г или 10 г.
В некоторых воплощениях изобретения количество одного или более соединений настоящего изобретения находится в диапазоне 0,0001-10 г, 0,0005-9 г, 0,001-8 г, 0,005-7 г, 0,01-6 г, 0,05-5 г, 0,1-4 г, 0,5-4 г или 1-3 г.
Соединения по настоящему изобретению являются эффективными в широком диапазоне дозировок. Например, для лечения взрослых людей примерами дозировок, которые могут использоваться, составляют дозировки от 0,01 до 1000 мг, от 0,5 до 100 мг, от 1 до 50 мг в день или от 5 до 40 мг в день. Примерной дозировкой является от 10 до 30 мг в день. Точная дозировка зависит от способа введения, формы, в которой вводится указанное соединение, пациента, подлежащего лечению, массы тела пациента, подлежащего лечению и предпочтений и опыта лечащего врача.
Ниже описаны не ограничивающие типичные фармацевтические композиции и способы их получения.
Фармацевтические композиции для перорального введения.
В некоторых воплощениях, в настоящем изобретении предложены фармацевтические композиции для перорального введения, содержащие соединение настоящего изобретения, и носитель, пригодный для перорального введения.
В некоторых воплощениях, в настоящем изобретении представлены твердые фармацевтические композиции для перорального введения, содержащие (1) эффективное количество соединения по настоящему изобретению; возможно (2) эффективное количество второго агента; и (3) фармацевтический носитель, пригодный для перорального введения. В некоторых воплощениях изобретения композиция дополнительно содержит (4) эффективное количество третьего агента.
В некоторых воплощениях изобретения фармацевтическая композиция может быть жидкой фармацевтической композицией, пригодной для перорального применения. Фармацевтические композиции по настоящему изобретению, пригодные для перорального введения, могут быть представлены в виде отдельных лекарственных форм, таких как капсулы, облатки или таблетки, или жидкости, или аэрозольные спреи, содержащие заданное количество активного компонента в виде порошка или в гранулах, в растворе или суспензии в водной или неводной жидкости, эмульсии масло-в-воде или жидкой эмульсии вода-в-масле. Такие лекарственные формы могут быть получены любым фармацевтическим способом, но все способы включают стадии смешивания активного компонента и носителя, который состоит из одного или более необходимых компонентов. Как правило, композиции приготавливают путем равномерного и тщательного смешивания активного компонента с жидкими носителями или тщательно измельченными твердыми носителями или обоими, а затем, при необходимости, придания продукту желаемого внешнего вида. Например, таблетки могут быть получены путем прессования или формования, возможно с одним или более вспомогательных компонентов. Спрессованные таблетки могут быть получены путем прессования в соответствующем автомате активного компонента в свободнотекучей форме, такой как порошок или гранулы, возможно смешанного с носителем, таким как, не ограничиваясь, связывающее вещество, смазывающее вещество, инертный разбавитель и/или поверхностно-активное вещество или диспергирующий агент. Формованные таблетки могут быть получены путем формования в соответствующем автомате смеси порошковых компонентов, увлажненных инертным жидким разбавителем.
В настоящем изобретении дополнительно представлены безводные фармацевтические композиции и лекарственные формы, включающие активный компонент, поскольку вода может способствовать разрушению некоторых соединений. Например, вода может быть добавлена (например, 5%) в фармацевтические изделия как средство для имитации долгосрочного хранения для определения свойств, таких как срок годности или стабильность композиций с течением времени. Безводные фармацевтические композиции и лекарственные формы по настоящему изобретению могут быть получены с использованием безводных компонентов или компонентов с низким содержанием влаги в условиях низкой влажности. Фармацевтические композиции и лекарственные формы настоящего изобретения, содержащие лактозу, могут быть получены в безводном виде, если ожидается существенный контакт с влагой при производстве, упаковке и/или хранении. Безводные фармацевтические композиции могут выпускаться и храниться так, чтобы сохранялась их безводная форма. Соответственно безводные композиции могут быть упакованы с использованием известных материалов для предотвращения воздействия воды, так что они могут быть включены в соответствующие фармакологические справочники. Примеры соответствующих упаковок включают, не ограничиваясь, герметично запаянные пленки, пластиковые или им подобные, контейнеры разовых доз, блистерные упаковки и контурные упаковки.
Активный компонент можно объединять в однородную смесь с фармацевтическим носителем в соответствии с обычными способами фармацевтического смешивания. Носитель может принимать различные формы в зависимости от заданной формы препарата для введения. При приготовлении композиций для пероральной лекарственной формы, могут использоваться любые обычные фармацевтические среды в качестве носителей, такие как, например, вода, гликоли, масла, спирты, ароматизаторы, консерванты, красители и т.п. в случае жидких пероральных препаратов (таких как суспензии, растворы или эликсиры) или аэрозоли; или могут использоваться такие носители как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазывающие агенты, связующие агенты и разрыхлители в случае твердых пероральных препаратов, в некоторых воплощениях изобретения без применения лактозы. Например, подходящие носители включают порошки, капсулы и таблетки с твердыми пероральными препаратами. При необходимости таблетки могут быть покрыты стандартными водными или неводными способами.
Связующие агенты, пригодные для применения в фармацевтических композициях и лекарственных формах, включают, не ограничиваясь, кукурузный крахмал, картофельный крахмал или другие крахмалы, желатин, природные и синтетические смолы, такие как гуммиарабик, альгинат натрия, альгиновую кислоту, другие альгинаты, порошковый трагакант, гуаровую камедь, целлюлозу и ее производные (например, этилцеллюлоза, ацетат целлюлозы, кальция карбоксиметилцеллоза, натрия карбоксиметилцеллюлоза), поливинилпирролидон, метилцеллюлозу, предварительно желатинизированный крахмал, гидроксипропил-метилцеллюлозу, микрокристаллическую целлюлозу и их смеси.
Примеры подходящих наполнителей для применения в фармацевтических композициях и лекарственных формах, описанных в настоящем документе, включают, не ограничиваясь, тальк, карбонат кальция (например, гранулы или порошок), микрокристаллическую целлюлозу, порошковую целлюлозу, декстраты, каолин, маннит, кремниевую кислоту, сорбит, крахмал, предварительно желатинизированный крахмал и их смеси.
Разрыхлители могут использоваться в композициях по настоящему изобретению для получения таблеток, которые разрушаются в водной среде. Применение слишком большого количества разрыхлителя может привести к получению таблеток, которые могут разлагаться в сосуде. Слишком маленького количества может быть недостаточно для разрушения, и это может изменить скорость и степень высвобождения активного компонента(ов) из лекарственной формы. Поэтому следует использовать достаточное количество разрыхлителя, ни слишком маленькое, ни слишком большое, во избежание вредных изменений после высвобождения активного компонента(ов) при получении лекарственных форм соединений, описанных в настоящем документе. Используемое количество разрыхлителя может изменяться в зависимости от типа препарата и режима введения, может легко быть определено специалистом в данной области. В фармацевтической композиции может использоваться от примерно 0,5 до примерно 15 массовых процентов разрыхлителя или от примерно 1 до примерно 5 массовых процентов разрыхлителя. Разрыхлители, которые могут использоваться для получения фармацевтических композиций и лекарственных форм по настоящему изобретению, включают, не ограничиваясь, агар-агар, альгиновую кислоту, карбонат кальция, микрокристаллическую целлюлозу, кроскармелозу натрия, кросповидон, полакрилин калия, натрия крахмал-гликолят, картофельный или тапиоковый крахмал, другие крахмалы, предварительно желатинизированный крахмал, другие крахмалы, глины, другие альгины, другие целлюлозы, смолы и их смеси.
Смазывающие вещества, которые могут использоваться для получения фармацевтических композиций и лекарственных форм настоящего изобретения, включают, не ограничиваясь, стеарат кальция, стеарат магния, минеральные масла, легкое минеральное масло, глицерин, сорбит, маннит, полиэтиленгликоль, другие гликоли, стеариновую кислоту, натрия лаурилсульфат, тальк, гидрогенизированное растительное масло (например, арахисовое масло, хлопковое масло, подсолнечное масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло), стеарат цинка, этилолеат, этиллаурат, агар или их смеси. Дополнительные смазывающие вещества включают, например, силоидный силикагель, коагулированный аэрозоль синтетического диоксида кремния или их смеси. Смазывающие вещества могут в некоторых случаях добавляться в количестве, составляющем менее примерно 1 весового процента от фармацевтической композиции.
Если водные суспензии и/или эликсиры разработаны для перорального применения, то основной активный компонент в них может быть объединен с различными подсластителями или ароматизаторами, красящими веществами или красителями, если это предусмотрено, эмульгирующими и/или суспендирующими агентами, вместе с такими разбавителями как вода, этанол, пропиленгликоль, глицерин и их различные комбинации.
Таблетки могут быть без покрытия или с покрытием, полученным известными способами, для замедления разложения и абсорбции в желудочно-кишечном тракте и обеспечения, таким образом, стабильного действия в течение продолжительного периода. Например, может использоваться такой материал для задержки во времени, как глицерилмоностеарат или глицерилдистеарат. Композиции для перорального применения могут применяться также в виде твердых желатиновых капсул, где активный компонент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активный компонент смешан с водой или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом.
Поверхностно-активные вещества, которые могут использоваться для образования фармацевтических композиций и лекарственных форм настоящего изобретения, включают, не ограничиваясь, гидрофильные поверхностно-активные вещества, липофильные поверхностно-активные вещества и их смеси. То есть может использоваться смесь гидрофильных поверхностно-активных веществ, смесь липофильных поверхностно-активных веществ или может использоваться смесь по меньшей мере одного гидрофильного поверхностно-активного вещества и одного липофильного поверхностно-активного вещества.
Подходящее гидрофильное поверхностно-активное вещество может иметь, в основном, значение гидрофильно-липофильного баланса (ГЛБ) по меньшей мере 10, тогда как подходящее липофильное поверхностно-активное вещество может иметь, в основном, значение ГЛБ, равное или меньше, чем примерно 10. Эмпирическим параметром, используемым для характеристики относительной гидрофильности и гидрофобности неионных амфифильных соединений, является гидрофильно-липофильный баланс (значение ГЛБ). Поверхностно-активные вещества с более низкими значениями ГЛБ являются более липофильными или гидрофобными, и имеют большую растворимость в маслах, тогда как поверхностно-активные вещества с более высокими значениями ГЛБ являются более гидрофильными и имеют большую растворимость в водных растворах. Гидрофильными поверхностно-активными веществами обычно считаются такие вещества, которые имеют значение ГЛБ более, чем примерно 10, а также анионные, катионные или цвиттер-ионные соединения, для которых обычно не используется шкала ГЛБ. Аналогично, липофильными (т.е. гидрофобными) поверхностно-активными веществами являются соединения, имеющие значение ГЛБ, равное или меньше, чем примерно 10. Однако значение ГЛБ поверхностно-активных веществ является лишь ориентиром, обычно используемым для узаконивания композиций промышленных, фармацевтических и косметических эмульсий.
Гидрофильные поверхностно-активные вещества могут быть ионными или неионогенными. Подходящие ионные поверхностно-активные вещества включают, не ограничиваясь, алкиламмониевые соли; соли фусидовой кислоты; жирно-кислотные производные аминокислот, олигопептидов и полипептидов; глицеридные производные аминокислот, олигопептидов и полипептидов; лецитины и гидрированные лецитины; лизолецитины и гидрированные лизолецитины; фосфолипиды и их производные; лизофосфолипиды и их производные; соли сложных эфиров карнитиновых жирных кислот; соли алкилсульфатов; соли жирных кислот; натрия докузат; ацилактилаты; моно- и ди-ацетилированные эфиры винной кислоты моно- и диглицеридов; сукцинилированные моно- и диглицериды; эфиры лимонной кислоты моно- и диглицеридов; и их смеси.
В рамках вышеупомянутых групп ионные поверхностно-активные вещества включают, например: лецитины, лизолецитин, фосфолипиды, лизофосфолипиды и их производные; соли сложных эфиров карнитиновых жирных кислот; соли алкилсульфатов; соли жирных кислот; натрия докузат; ацилактилаты; моно- и ди-ацетилированные сложные эфиры винной кислоты моно- и диглицеридов; сукцинилированные моно- и диглицериды; эфиры лимонной кислоты моно- и диглицеридов; и их смеси.
Ионными поверхностно-активными веществами могут быть ионизированные формы лецитина, лизолецитина, фосфатидилхолина, фосфатидилэтаноламина, фосфатидилглицерина, фосфатидиновой кислоты, фосфатидилсерина, лизофосфатидилхолина, лизофосфатидилэтаноламина, лизофосфатидилглицерина, лизофосфатидиновой кислоты, лизофосфатидилсерина, ПЭГ-фосфатидилэтаноламина, ПВП-фосфатидилэтаноламина, молочнокислые эфиры жирных кислот, стеароил-2-лактилат, стеароил лактилат, сукцинилированные моноглицериды, моно/диацетилированные сложные эфиры винной кислоты моно/диглицеридов, эфиры лимонной кислоты моно/диглицеридов, холилсаркозин, капроат, каприлат, капрат, лаурат, миристат, пальмитат, олеат, рицинолеат, линолеат, линоленат, стеарат, лаурилсульфат, терацецилсульфат, докузат, лауроил карнитины, пальмитоил карнитины, миристоил карнитины и их соли и смеси.
Гидрофильные неионогенные поверхностно-активные вещества могут включать, не ограничиваясь, алкилклюкозиды; алкилмальтозиды; алкилтиоглюкозиды; луарил макроголглицериды; полиоксиалкилен-алкильные эфиры, такие как алкильные эфиры полиэтиленгликоля; полиоксиалкилен алкилфенолы, такие как полиэтиленгликоля алкилфенолы; полиоксиалкилен алкилфеноловые эфиры жирных кислот, такие как полиэтиленгликолевые моноэфиры жирных кислоты и полиэтиленгликолевые диэфиры жирных кислот; полиэтиленгликоль глицериновые эфиры жирных кислот; полиглицериновые сложные эфиры жирных кислот; полиоксиалкилен сорбитоловые сложные эфиры жирных кислот, такие как полиэтиленгликоль сорбитола сложные эфиры жирных кислот; гидрофильные продукты трансэстерификации полиолов по меньшей мере с одним членом из группы, состоящей из глицеридов, растительных масел, гидрированных растительных масел, жирных кислот и стеринов; полиоксиэтиленстерины, их производные и аналоги; полиоксиэтилированные витамины и их производные; полиоксиэтилен-полиоксипропилен блок-сополимеры; и их смеси; полиэтиленгликоль сорбитоловые сложные эфиры жирных кислот и гидрофильные продукты трансэстерификации полиолов по меньшей мере с одним членом из группы, состоящей из триглицеридов, растительных масел и гидрированных растительных масел. Полиолом может быть глицерин, этиленгликоль, полиэтиленгликоль, сорбит, пропиленгликоль, пентаэритрит или сахарид.
Другие гидрофильные неионогенные поверхностно-активные вещества включают, не ограничиваясь, ПЭГ-10 лаурат, ПЭГ-12 лаурат, ПЭГ-20 лаурат, ПЭГ-32 лаурат, ПЭГ-32 дилаурат, ПЭГ-12 олеат, ПЭГ-15 олеат, ПЭГ-20 олеат, ПЭГ-20 диолеат, ПЭГ-32 олеат, ПЭГ-200 олеат, ПЭГ-400 олеат, ПЭГ-15 стеарат, ПЭГ-32 дистеарат, ПЭГ-40 стеарат, ПЭГ-100 стеарат, ПЭГ-20 дилаурат, ПЭГ-25 глицерилтриолеат, ПЭГ-32 диолеат, ПЭГ-20 глицериллаурат, ПЭГ-30 глицериллаурат, ПЭГ-20 глицерилстеарат, ПЭГ-20 глицерилолеат, ПЭГ-30 глицерилолеат, ПЭГ-30 глицериллаурат, ПЭГ-40 глицериллаурат, ПЭГ-40 масло пальмовых зерен, ПЭГ-50 гидрированное касторовое масло, ПЭГ-40 касторовое масло, ПЭГ-35 касторовое масло, ПЭГ-60 касторовое масло, ПЭГ-40 гидрированное касторовое масло, ПЭГ-60 гидрированное касторовое масло, ПЭГ-60 кукурузное масло, ПЭГ-6 капрат/каприлат глицериды, ПЭГ-8 капрат/каприлат глицериды, поликлицерил-10 лаурат, ПЭГ-30 холестерин, ПЭГ-25 фитостерин, ПЭГ-30 соевый стерин, ПЭГ-20 триолеат, ПЭГ-40 сорбитололеат, ПЭГ-80 сорбитоллаурат, полисорбат 20, полисорбат 80, ПОЭ-9 лауриловый эфир, ПОЭ-23 лауриловый эфир, ПОЭ-10 олеиловый эфир, ПОЭ-20 олеиловый эфир, ПОЭ-20 стеариловый эфир, токоферил ПЭГ-100 сукцинат, ПЭГ-24 холестерин, поликлицерил-10 олеат, Твин 40, Твин 60, сахарозы моностеарат, сахарозы монолаурат, сахарозы монопальмитат, серия ПЭГ 10-100 нонилфенолов, серия ПЭГ 15-100 октилфенолов и полоксамеры.
Подходящие липофильные поверхностно-активные вещества включают, только в целях примера: жирные спирты; глицериновые эфиры жирных кислот; сложные эфиры ацетилированного глицерина и жирных кислот; сложные эфиры низших спиртов и жирных кислот; пропиленгликолевые эфиры жирных кислот; сложные эфиры сорбита и жирных кислот; сложные эфиры полиэтиленгликоль сорбита и жирных кислот; производные стеринов и стерола; полиоксиэтилированные стерины и производные стерина; алкильные эфиры полиэтиленгликоля; сложные эфиры сахара; простые эфиры сахара; молочнокислые производные моно- и диглицеридов; гидрофобные продукты трансэстерификации полиолов по меньшей мере с одним членом из группы, состоящей из глицеридов, растительных масел, гидрогенированных растительных масел, жирных кислот и стеринов; маслорастворимые витамины/производные витаминов; и их смеси. В рамках этой группы предпочтительные липофильные поверхностно-активные вещества включают сложные эфиры глицерина и жирных кислот, сложные эфиры пропиленгликоля и жирных кислот и их смеси; или гидрофобные продукты трансэстерификации полиолов по меньшей мере с одним членом, из группы, состоящей из растительных масел, гидрированных растительных масел и триглицеридов.
В одном варианте воплощения изобретения композиция может включать солюбилизатор для обеспечения хорошей солюбилизации и/или растворимости соединения по настоящему изобретению и для минимизации осаждения соединения по настоящему изобретению. Это может быть особенно важным для композиций для неперорального применения, например, композиций для инъекции. Солюбилизатор может также добавляться для усиления растворимости гидрофильных лекарств и/или других компонентов, таких как поверхностно-активные вещества, или для сохранения композиции в виде устойчивого или гомогенного раствора или дисперсии.
Примеры подходящих солюбилизаторов включают, не ограничиваясь, следующие: спирты и полиолы, такие как этанол, изопропанол, бутанол, бензиновый спирт, этиленгликоль, пропиленгликоль, бутандиолы и их изомеры, глицерин, пентаэритрит, сорбит, маннит, транскутол, диметилизосорбид, полиэтиленгликоль, полипропиленгликоль, поливиниловый спирт, гидроксипропилметилцеллюлоза и другие производные целлюлозы, циклодекстрины и производные циклодекстринов; простые эфиры полиэтиленгликолей, имеющие средний молекулярный вес от примерно 200 до примерно 6000, такие как тетрагидрофурфурилового спирта ПЭГ эфир (гликофурол) или метокси ПЭГ; амиды и другие азотсодержащие соединения, такие как 2-пирролидон, 2-пиперидон, ε-капролактам, N-алкилпирролидон, N-гидроксиалкилпирролидон, N-алкилпипериддон, N-алкилкапролактам, диметилацетамид и поливинилпирролидон; сложные эфиры, такие как этилпропионат, трибутилцитрат, ацетилтриэтилцитрат, ацетилтрибутилцитрат, триэтилцитрат, этилолеат, этилкаприлат, этилбутират, триацетин, пропиленгликоля моноацетат, пропиленгликоля диацетат, ε-капролактон и его изомеры, δ-валеролактон и его изомеры, β-бутиролактон и его изомеры; и другие солюбилизаторы, известные в данной области, такие как диметилацетамид, диметилизосорбид, N-метилпирролидоны, монооктаноин, диэтиленгликоля моноэтиловый эфир и вода.
Также могут использоваться смеси солюбилизаторов. Примеры включают, не ограничиваясь, триацетин, триэтилцитрат, этилолеат, этилкаприлат, диметилацетамид, N-метилпирролидон, N-гидроксиэтилпирролидон, поливинилпирролидон, гидроксипропил метилцеллюлоза, гидроксипропил циклодекстрины, этанол, полиэтиленгликоль 200-100, гликофурол, транскутол, пропиленгликоль и диметилизосорбид. Особенно предпочтительные солюбилизаторы включают сорбит, глицерин, триацетин, этиловый спирт, ПЭГ-400, гликофурол и пропиленгликоль.
Количество солюбилизатора, подлежащее включению, специально не лимитируется. Количество данного солюбилизатора может ограничиваться до биоприемлемого количества, которое легко устанавливается специалистом в данной области. В некоторых условиях может быть полезным включать такие количества солюбилизаторов, которые намного превышают биоприемлемые количества, например, для максимизации концентрации лекарственные средства, где избыток солюбилизатора устраняется перед введением композиции пациенту при помощи стандартных способов, каких как дистилляция или упаривание. Поэтому, если солюбилизатор присутствует, его массовое содержание может составлять 10%, 25%, 50%, 100% или до 200% по массе в пересчете на общую массу лекарственные средства и других носителей. При необходимости также могут использоваться очень небольшие количества солюбилизатора: 5%, 2%, 1% или даже менее. Обычно солюбилизатор может присутствовать в количестве от примерно 1% до примерно 100%, наиболее часто от примерно 5% до примерно 25% по массе.
Композиция может дополнительно включать одну или более фармацевтически приемлемых добавок и носителей. Такие добавки и носители включают, не ограничиваясь, агенты для уменьшения вязкости, пеногасители, буферные агенты, полимеры, антиоксиданты, консерванты, хелатирующие агенты, регуляторы вязкости, регуляторы тоничности, ароматизаторы, красители, отдушки, агенты, придающие непрозрачность, суспендирующие агенты, связывающие агенты, наполнители, пластификаторы, смазывающие агенты и их смеси.
Кроме того, в композицию могут вводиться кислоты или основания для облегчения переработки, улучшения стабильности или по другим причинам. Примеры фармацевтически приемлемых оснований включают аминокислоты, сложные эфиры аминокислот, гидроксид аммония, гидроксид калия, гидроксид натрия, натрия гидрокарбонат, гидроксид алюминия, карбонат кальция, гидроксид магния, силикат магния-алюминия, синтетический алюмосиликат, синтетический гидрокальцит, гидроксид магния-алюминия, диизопропилэтиламин, этаноламин, этилендиамин, триэтаноламин, триэтиламин, триизопропаноламин, триметиламин, трис(гидроксиметил)аминометан (ТРИС) и т.п. Также пригодными являются основания, которые являются солями фармацевтически приемлемых кислот, таких как уксусная кислота, акриловая кислота, адипиновая кислота, альгиновая кислота, алкансульфоновая кислота, аминокислоты, аскорбиновая кислота, бензойная кислота, борная кислота, масляная кислота, угольная кислота, лимонная кислота, жирные кислоты, муравьиная кислота, фумаровая кислота, глюконовая кислота, гидрохиносульфоновая кислота, изоаскорбиновая кислота, молочная кислота, малеиновая кислота, щавелевая кислота, пара-бромфенилсульфоновая кислота, пропионовая кислота, п-толуолсульфоновая кислота, салициловая кислота, стеариновая кислота, янтарная кислота, дубильная кислота, винная кислота, тиогликолевая кислота, толуолсульфоновая кислота, мочевая кислота и т.п. Также могут использоваться соли многоосновных кислот, такие как фосфат натрия, динатрия гидрофосфат и натрия дигидрофосфат. Если основанием является соль, то катионом может быть любой удобный и фармацевтически приемлемый катион, такой как аммоний, щелочные металлы, щелочноземельные металлы и т.п. Примеры могут включать, не ограничиваясь, натрий, калий, литий, магний, кальций и аммоний.
Подходящими кислотами являются фармацевтически приемлемые органические или неорганические кислоты. Примеры подходящих неорганических кислот включают хлороводородную кислоту, бромоводородную кислоту, йодоводородную кислоту, серную кислоту, азотную кислоту, борную кислоту, фосфорную кислоту и т.п. Примеры подходящих органических кислот включают уксусную кислоту, акриловую кислоту, адипиновую кислоту, альгиновую кислоту, алкансульфоновую кислоту, аминокислоты, аскорбиновую кислоту, бензойную кислоту, борную кислоту, масляную кислоту, угольную кислоту, лимонную кислоту, жирные кислоты, муравьиную кислоту, фумаровую кислоту, глюконовую кислоту, гидрохиносульфоновую кислоту, изоаскорбиновую кислоту, молочную кислоту, малеиновую кислоту, метансульфоновую кислоту, щавелевую кислоту, пара-бромфенилсульфоновую кислоту, пропионовую кислоту, пара-толуолсульфоновую кислоту, салициловую кислоту, стеариновую кислоту, янтарную кислоту, дубильную кислоту, винную кислоту, тиогликолевую кислоту, толуолсульфоновую кислоту, мочевую кислоту и т.п.
Фармацевтические композиции для инъекций.
В некоторых воплощениях настоящего изобретения представлены фармацевтические композиции для инъекций, содержащие соединение настоящего изобретения и фармацевтический носитель, пригодный для инъекций. Компоненты и количества агентов в композициях описаны в настоящем документе.
Формы, в которых новые композиции по настоящему изобретению могут вводиться для применения в виде инъекций, включают водные или масляные суспензии или эмульсии, с кунжутным маслом, кукурузным маслом, хлопковым маслом или арахисовым маслом, а также эликсиры, растворы с маннитом, декстрозой или стерильные растворы в воде и аналогичных фармацевтических носителях.
Водные растворы в физрастворе также обычно используются для инъекций. Могут использоваться также этанол, глицерин, пропиленгликоль, жидкий полиэтиленгликоль и т.п. (и их соответствующие смеси), производные циклодекстрина и растительные масла. Необходимая текучесть может обеспечиваться, например, за счет использования покрытий, таких как лецитин, сохраняющих нужный размер частиц в случае дисперсий, а также при помощи поверхностно-активных веществ. Заражение микроорганизмами можно предотвратить за счет добавления различных антибактериальных и противогрибковых агентов, например, парабенов, хлорбутанола, фенола, сорбиновой кислоты, тимеросала и т.п.
Стерильные растворы для инъекций получают путем введения соединения по настоящему изобретению в заданном количестве в соответствующий растворитель с различными другими компонентами, как перечислено выше, при необходимости, с последующей фильтрацией и стерилизацией. Как правило, дисперсии получают путем введения различных стерильных активных компонентов в стерильный носитель, содержащий основную дисперсионную среду и другие необходимые компоненты из перечисленных выше. В случае стерильных порошков для приготовления стерильных растворов для инъекций, желательно использование определенных способов приготовления, таких как вакуумная сушка и лиофильная сушка, в результате которых получают порошок активного компонента, плюс дополнительный заданный компонент из его предварительно стерильно отфильтрованного раствора.
Фармацевтические композиции для местной (например, трансдермальной) доставки.
В некоторых воплощениях настоящего изобретения представлены фармацевтические композиции для трансдермальной доставки, содержащие соединение настоящего изобретения и фармацевтический носитель, пригодный для трансдермальной доставки.
Композиции настоящего изобретения могут быть приготовлены в виде препаратов в твердой, полутвердой или жидкой формах, пригодных для локального или местного введения, таких как гели, водорастворимые желе, кремы, лосьоны, суспензии, пены, порошки, взвеси, мази, растворы, масла, пасты, суппозитории, спреи, эмульсии, солевые растворы, растворы на основе диметилсульфоксида (ДМСО). В основном, носители с высокой плотностью могут обеспечивать пролонгированное воздействие активных компонентов на заданной области. Напротив, растворные композиции могут обеспечивать более быстрое воздействие активного компонента на выбранную область.
Фармацевтические композиции могут также включать подходящие твердофазные или гелеобразные носители или наполнители, которые являются соединениями, обеспечивающими усиленное проникновение, или способствующими доставке терапевтических молекул через роговой слой кожи, являющийся барьером проницаемости. Существует множество таких молекул, способствующих проникновению, которые известны опытному специалисту в области композиций для местного применения. Примеры таких носителей и наполнителей включают, не ограничиваясь, смачивающие средства (например, мочевина), гликоли (например, пропиленгликоль), спирты (например, этанол), жирные кислоты (например, олеиновая кислота), поверхностно-активные вещества (например, изопропилмиристат и натрия лаурилсульфат), пирролидоны, глицерина монолаурат, сульфоксиды, терпены (например, ментол), амины, амиды, алканы, алканолы, воду, карбонат кальция, фосфат кальция, различные сахара, крахмалы, производные целлюлозы, желатин и полимеры, такие как полиэтиленгликоли.
В другой типичной композиции для применения в способах настоящего изобретения используются устройства трансдермальной доставки («пластыри»). Такие трансдермальные пластыри могут использоваться для обеспечения непрерывной или периодической инфузии соединения настоящего соединения в контролируемых количествах, вместе с другим агентом или без него.
Конструкция и применение трансдермальных пластырей для доставки фармацевтических агентов хорошо известны в данной области. См., например, патенты США №№5023252, 4992445 и 5001139. Такие пластыри могут быть разработаны для непрерывной, пульсирующей или доставки по необходимости фармацевтических средств.
Фармацевтические композиции для ингаляции.
Композиции для ингаляции или инсуффляции включают растворы и суспензии в фармацевтически приемлемых, водных или органических растворителях, или их смесях, и порошки. Жидкие или твердые композиции могут содержать подходящие фармацевтически приемлемые носители, как описано выше. Предпочтительно эти композиции вводятся пероральным или назальным респираторным путем для достижения системного эффекта. Композиции в предпочтительных фармацевтически приемлемых растворителях могут распыляться при помощи инертных газов. Распыленные растворы можно вдыхать непосредственно из распыляющего устройства, или распылительное устройство может подключаться к маске для лица, тенту или дыхательному аппарату избыточного давления периодического действия. Композиции в растворах, суспензиях или порошках могут вводиться, предпочтительно перорально или назально через устройства, которые обеспечивают доставку композиции соответствующим образом.
Другие фармацевтические композиции.
Фармацевтические композиции могут также быть получены из композиций, описанных в настоящем документе, и одного или более фармацевтически приемлемых наполнителей, пригодных для подъязычного, буккального, ректального, внутрикостного, внутриглазного, интраназального, эпидурального или интраспинального введения. Препараты для таких фармацевтических композиций хорошо известны в данной области. См., например, Anderson, Philip О.; Knoben, James Е.; Troutman, William G, eds., Handbook of Clinical Drug Data, Tenth Edition, McGraw-Hill, 2002; Pratt and Taylor, eds., Principles of Drug Action, Third Edition, Churchill Livingston, New York, 1990; Katzung, ed., Basic and Clinical Pharmacology, Ninth Edition, McGraw Hill, 20037ybg; Goodman and Gilman, eds., The Pharmacological Basis of Therapeutics, Tenth Edition, McGraw Hill, 2001; Remingtons Pharmaceutical Sciences, 20th Ed., Lippincott Williams & Wilkins., 2000; Martindale, The Extra Pharmacopoeia, Thirty-Second Edition (The Pharmaceutical Press, London, 1999); все из которых включены в настоящую заявку путем ссылки в полном объеме.
Введение соединений или фармацевтических композиций настоящего изобретения может осуществляться любым способом, обеспечивающим доставку соединений к участку их действия. Эти способы включают пероральные пути, интрадуоденальный путь, парентеральное введение (в том числе внутривенное, внутриартериальное, подкожное, внутримышечное, внутрисосудистое, внутрибрюшинное или инфузионное), местное (например, трансдермальное применение), ректальное введение, через катетер локальней доставки или стент или путем ингаляции. Соединения могут также вводиться в жировую ткань или интратекальным способом.
Количество вводимого соединения зависит от млекопитающего, подлежащего лечению, серьезности заболевания или состояния, скорости введения, характера соединения и мнения предписывающего врача. Однако эффективная дозировка находится в диапазоне от примерно 0,001 до примерно 100 мг на кг массы тела в день, предпочтительно от примерно 1 до примерно 35 мг/кг/день, в виде единой дозы или частями. Для 70 кг человека это составляет от примерно 0,05 до 7 г/день, предпочтительно от примерно 0,05 до примерно 2,5 г/день. В некоторых случаях могут быть более правильными уровни дозировки, находящиеся ниже нижнего предела указанного диапазона, тогда как в других случаях могут использоваться гораздо большие дозировки, не вызывающие вредных побочных эффектов, например, путем разделения этих больших доз на несколько небольших доз для введения в течение дня.
В некоторых воплощениях изобретения соединение по настоящему изобретению вводится в виде разовой дозы. Как правило, такое введение осуществляется путем инъекции, например, внутривенной инъекции, для быстрого введения агента. Однако при необходимости могут использоваться другие способы. Разовая доза соединения по настоящему изобретению может использоваться для лечения острого состояния.
В некоторых воплощениях изобретения соединение по настоящему изобретению вводится в виде многократных доз. Дозирование может составлять один, два, три, четыре, пять, шесть или более шести раз в день. Дозирование может составлять примерно один раз в месяц, один раз в две недели, один раз в неделю или через день. В некоторых воплощениях изобретения соединение по настоящему изобретению и другой агент вводятся вместе, примерно от одного раза в день до примерно 6 раз в день. В другом воплощении изобретения введение соединения по настоящему изобретению и другого агента продолжается примерно в течение менее 7 дней. В другом воплощении изобретения введение продолжается в течение более 6, 10, 14, 28 дней, двух месяцев, шести месяцев или одного года. В некоторых случаях непрерывное введение достигается и сохраняется так долго, как это необходимо.
Введение агентов настоящего изобретения может продолжаться так долго, как это необходимо. В некоторых воплощениях изобретения агент настоящего изобретения вводится в течение более чем 1, 2, 3, 4, 5, 6, 7, 14 или 28 дней. В некоторых воплощениях изобретения агент настоящего изобретения вводится в течение менее чем 28, 14, 7, 6, 5, 4, 3, 2 или 1 дня. В некоторых воплощениях изобретения агент настоящего изобретения вводится постоянно в непрерывном режиме, например, для лечения хронических заболеваний.
Эффективное количество соединения по настоящему изобретению может вводиться разовой или многократной дозами с использованием одного из режимов введения агентов, имеющих аналогичное действие, включая, ректальное, буккальное, интраназальное и трансдермальное введение, введение внутри-артериальными инъекциями, внутривенно, внутрибрюшинно, парентерально, внутримышечно, подкожно, перорально, местно или путем ингаляции.
Композиции по настоящему изобретению могут также доставляться посредством пропитанных устройств или устройств с покрытием, таких как стент, например, или полимерный цилиндр, вставляемый в артерию. Такие способы введения могут, например, способствовать предотвращению или улучшению рестеноза с последующими процедурами, такими как пластика сосудов. Не ограничиваясь определенной теорией, полагают, что соединения по настоящему изобретению могут замедлять или ингибировать миграцию и пролиферацию гладких миоцитов в артериальных стенках, что вносит свой вклад в рестеноз. Соединения по настоящему изобретению могут вводиться, например, путем местно доставки через «распорки» стента, через стентовый трансплантат, из трансплантатов или из покрытия, или оболочки стента. В некоторых воплощениях изобретения соединение по настоящему изобретению вводится с матрицей. Такая матрица может быть полимерной матрицей и служить для связывания соединения со стентом. Полимерные матрицы, пригодные для такого применения, включают, например, сложные полиэфиры на основе лактона или сополиэфиры, такие как полилактид, поликапролактонгликолид, полиортоэфиры, полиангидриды, полиаминокислоты, полисахариды, полифосфазены, поли(простой эфир-сложный эфир)сополимеры (например, PEO-PLLA); полидиметилсилоксан, поли(этиленвинилацетат), акрилатные полимеры и сополимеры (например, полигидроксиэтил метилметакрилат, поливинилпирролидинон), фторированные полимеры, такие как политетрафторэтилен и эфиры целлюлозы. Подходящие матрицы могут быть неразлагаемыми или разлагаемыми с течением времени, при высвобождении соединения или соединений. Соединения по настоящему изобретению могут наноситься на поверхность стента различными способами, такими как покрытие погружением/вращением, покрытие разбрызгиванием, покрытие погружением и/или покрытие кисточкой. Соединения могут наноситься в растворителе, который затем испаряется, образуя, таким образом, слой соединения на стенте. Альтернативно, соединение может быть расположено в объеме стента или имплантата, например, в микроканалах или микропорах. При имплантации соединение диффундирует из объема стента и взаимодействует со стенкой артерии. Такие стенты могут быть получены погружением стента, имеющего такие микропоры или микроканалы, в раствор соединения по настоящему изобретению в подходящем растворителе, с последующим испарением растворителя. Избыток лекарственные средства на поверхности стента может быть устранен дополнительным быстрым промыванием в растворителе. В других воплощениях изобретения соединения по настоящему изобретению могут ковалентно линкеры, которые разлагаются in vivo, приводя к высвобождению соединения по настоящему изобретению. Для этих целей можно использовать любую биолабильную связывание, такое как сложноэфирное, амидное или ангидридное связывание. Соединения по настоящему изобретению могут дополнительно вводиться внутримышечно из надувного баллона, используемого при пластике сосудов. Для снижения рестеноза также могут использоваться экстраваскулярное введение соединения через перикард или адвенциальное введение композиций настоящего изобретения.
Разнообразные стентовые устройства, которые могут использоваться так, как описано здесь, рассмотрены, например, в следующих ссылках, все из которых включены в настоящую заявку путем ссылки: патент США №5451233; патент США №5040548; патент США №5061273; патент США №5496346; патент США №5292331; патент США №5674278; патент США №3657744; патент США №4739762; патент США №5195984; патент США №5292331; патент США №5674278; патент США №5879382; патент США №6344053.
Соединения по настоящему изобретению могут вводиться дозами. В данной области известно, что из-за вариабельности фармакокинетики соединения у различных пациентов, для оптимальной терапии необходима индивидуализация режимов дозирования. Дозирование соединения по настоящему изобретению может быть установлено путем обычных экспериментов в свете настоящего описания.
При введении соединения по настоящему изобретению в композиции, включающей один или более агентов, и если этот агент имеет более короткий период полувыведения, чем соединение настоящего изобретения, то может потребоваться соответствующая регулировка стандартных лекарственных форм этого агента и соединения по настоящему изобретению.
Указанная фармацевтическая композиция может, например, быть в форме, подходящей для перорального введения, такой как таблетки, капсулы, пилюли, порошок, композиции непрерывного высвобождения, растворы, суспензии, для парентеральной инъекции в виде стерильного раствора, суспензии или эмульсии, для местного применения, такой как мазь или крем или для ректального введения в виде суппозиториев. Фармацевтическая композиция может быть в стандартных лекарственных формах, пригодных для разового введения точной дозировки. Фармацевтические композиции включают обычный фармацевтический носитель или наполнитель и соединение согласно настоящему изобретению в качестве активного компонента. Кроме того, они могут включать другие медицинские или фармацевтические средства, носители, адъюванты и т.п.
Примеры форм для парентерального введения включают растворы или суспензии активного соединения в стерильных водных растворах, например, водных растворах пропиленгликоля или декстрозы. Такие лекарственные формы могут быть надлежащим образом буферизованы, при необходимости.
Активность соединений настоящего изобретения может быть установлена по следующей методике, а также по методике, описанной ниже в примерах. Активность киназы оценивается по измерению введения γ-33Р-фосфата из γ-33P-АТФ на N-концевой меченый субстрат His, который экспрессируется в Е. coli и очищается обычными способами в присутствии киназы. Анализ выполняется в 96-луночном полипропиленовом планшете. Инкубационная смесь (100 мкл) включает 25 мМ Hepes, рН 7,4, 10 мМ MgCl2, 5 мМ β-глицерофосфата, 100 мМ Na-ортованадата, 5 мМ DTT, 5 нМ киназы и 1 мкМ субстрата. Ингибиторы суспендируются в ДМСО, и все реакции, включая контрольные реакции, выполняются при конечной концентрации 1% ДМСО. Реакции инициируются добавлением 10 мкМ АТФ (с 0,5 мкКи γ-33Р-АТФ/лунку) и инкубируются при комнатной температуре в течение 45 минут. Добавляется равный объем 25% ТСА для прекращения реакции и осаждения белков. Осажденные белки улавливаются на фильтровальных пластинах из стекловолокна В, и избыточная меченая АТФ вымывается при помощи харвестера Tomtec MACH III. Планшеты оставляются для высушивания на воздухе, а затем добавляется 30 мкл/лунку препарата Packard Microscint 20, и планшеты подсчитываются при помощи Packard TopCount.
В настоящем изобретении также предложены наборы. Наборы включают соединение или соединения по настоящему изобретению, как описано в настоящем документе, в подходящей упаковке, и письменный материал, включающий инструкции по применению, обсуждение клинических испытаний, перечень побочных эффектов и т.п. Такие наборы могут включать также информацию, такую как ссылки на научную литературу, рекламные вкладыши, результаты клинических испытаний и/или выводы из них и т.п., которые указывают или определяют активность и/или преимущества данной композиции и/или которые описывают дозировку, введение, побочные эффекты, взаимодействие с другими лекарственные средствами и другую информацию, полезную для поставщика лекарственных средств. Такая информация может быть основана на результатах различных исследований, например, исследований с использованием экспериментальных животных, включающих модели in vivo, и исследований на основе клинических испытаний людей. Набор может дополнительно содержать другой агент. В некоторых воплощениях изобретения соединение настоящего изобретения и этот агент представлены в виде отдельных композиций в отдельных контейнерах набора. В некоторых воплощениях изобретения соединение настоящего изобретения и этот агент представлены в виде одной композиции в одном контейнере набора. Подходящая упаковка и вспомогательные изделия для применения (например, мерный стаканчик для жидких композиций, фольгированная обертка для минимизации воздействия воздуха и т.п.) являются известными в данной области и могут быть включены в набор. Наборы, описанные в настоящем документе, могут поставляться, продаваться и/или рекламироваться поставщикам лекарственных средств, включая врачей, медсестер, фармацевтов, медработников и т.п. Наборы могут также, в некоторых воплощениях изобретения, продаваться потребителю напрямую.
СПОСОБЫ
В настоящем изобретении представлены также способы применения соединений или фармацевтических композиций настоящего изобретения для лечения болезненных состояний, включая, но не ограничиваясь, заболевания, связанные с дисфункцией одного или более типов PI3 киназы. Подробное описание состояний и нарушений, опосредованных активностью киназы p110δ, представлено далее в публикации Sadu et al., WO 01/81346, которая включена в настоящую заявку путем ссылки в полном объеме для всех целей.
Способы лечения, представленные в настоящем документе, включают введение пациенту терапевтически эффективного количества соединения по настоящему изобретению. В одном варианте воплощения настоящего изобретения представлен способ лечения воспалительного расстройства, включая аутоиммунные заболевания млекопитающих. Способ включает введение указанному млекопитающему терапевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли, сложного эфира, пролекарственные средства, сольвата, гидрата или производного. Примеры аутоиммунных заболеваний включают, не ограничиваясь, острый рассеянный энцефаломиелит (ОРЭМ), болезнь Аддисона, антифосфолипидный синдром (АФС), апластическую анемию, аутоиммунный гепатит, целиакию, болезнь Крона, сахарный диабет (типа 1), синдром Гудпасчера, болезнь Грейвса, синдром Гийена-Барре (СГБ), зоб Хасимото, красную волчанку, рассеянный склероз, миастению, синдром опсоклонуса-миоклонуса, (ОМС), неврит зрительного нерва, тиреоидит Орда, пемфигус, полиартриты, первичный билиарный цирроз печени, псориаз, ревматоидный артрит, синдром Рейтера, артериит Такаясу, темопральный артериит (также известный как "гигантоклеточный артериит"), теплую аутоиммунную гемолитическую анемию, гранулематоз Вегенера, алопецию, болезнь Шагаса, синдром хронической усталости, вегетативную дистонию, эндометриоз, гнойный гидраденит, интерстициальный цистит, нейромиотонию, саркоидоз, склеродермию, неспецифический язвенный колит, витилиго и вульводинию. Другие заболевания включают резорбцию костной ткани и тромбоз.
В некоторых воплощениях изобретения способ лечения воспаления или аутоиммунных расстройств включает введение пациенту (например, млекопитающему) терапевтически эффективного количества одного или более соединений по настоящему изобретению, которые селективно ингибируют PI3K-δ и/или Р13К-γ по сравнению со всеми остальными PI3 киназами типа I. Такое селективное ингибирование PI3K-δ и/или P13K-γ может быть полезным для лечения любых заболеваний или состояний, описанных в настоящем документе. Например, селективное ингибирование PI3K-δ может замедлять воспалительные реакции, связанные с воспалительными заболеваниями, аутоиммунным заболеванием или болезнями, связанными с нежелательной иммунной реакцией, включая, не ограничиваясь, астму, эмфизему, аллергию, дерматиты, ревматоидный артрит, псориаз, красную волчанку или реакцию «трансплантат против хозяина». Селективное ингибирование PI3K-δ может дополнительно обеспечивать снижение воспаления или нежелательной иммунной реакции без сопутствующего снижения способности ослаблять бактериальную, вирусную и/или грибковую инфекцию. Селективное ингибирование обоих PI3K-δ и PI3K-γ может быть полезным для ингибирования воспалительной реакции у пациента в большей степени, по сравнению с ингибиторами, селективно ингибирующими PI3K-δ или PI3K-γ по отдельности. В одном аспекте один или более указанных способов являются эффективными для снижения антиген-специфичной выработки антител in vivo примерно в 2 раза, 3 раза, 4 раза, 5 раз, 7,5 раз, 10 раз, 25 раз, 50 раз, 100 раз, 250 раз, 500 раз, 750 раз или примерно 1000 раз или более. В другом аспекте один или более указанных способов являются эффективными для снижения антиген-специфичной выработки IgG3 и/или IgGM in vivo примерно в 2 раза, 3 раза, 4 раза, 5 раз, 7,5 раз, 10 раз, 25 раз, 50 раз, 100 раз, 250 раз, 500 раз, 750 раз или примерно 1000 раз или более.
В одном аспекте один или более указанных способов являются эффективными для улучшения симптомов, связанных с ревматоидным артритом, включая, не ограничиваясь, снижение припухлости суставов, снижение анти-коллагенового уровня в сыворотке крови и/или сокращение патологии суставов, таких как резорбция костей, повреждение хрящей, паннус и/или воспаление. В другом аспекте указанные способы являются эффективными для снижения воспаления лодыжки по меньшей мере примерно на 2%, 5%, 10%, 15%, 20%, 25%, 30%, 50%, 60% или примерно 75%-90%. В другом аспекте указанные способы являются эффективными для снижения воспаления колена по меньшей мере примерно на 2%, 5%, 10%, 15%, 20%, 25%, 30%, 50%, 60% или примерно 75%-90% или более. В другом аспекте указанные способы являются эффективными для снижения уровня коллагена анти-типа II в сыворотке крови по меньшей мере примерно на 10%, 12%, 15%, 20%, 24%, 25%, 30%, 35%, 50%, 60%, 75%, 80%, 86%, 87%, или примерно 90% или более. В другом аспекте указанные способы являются эффективными для снижения показателей гистопатологии лодыжки примерно на 5%, 10%, 15%, 20%, 25%, 30%, 40%, 50%, 60%, 75%, 80%, 90% или более. В другом аспекте указанные способы являются эффективными для снижения показателей гистопатологии колена примерно на 5%, 10%, 15%, 20%, 25%, 30%, 40%, 50%, 60%, 75%, 80%, 90% или более.
В других вариантах воплощения настоящего изобретения предложены способы применения соединений или фармацевтических композиций для лечения респираторных заболеваний, включая, не ограничиваясь, заболевания, поражающие доли легких, плевральную полость, бронхи, трахею, верхние дыхательные пути или дыхательные нервы и мышцы. Например, представлены способы лечения обструктивного заболевания легких. Хроническое обструктивное заболевание легких (ХОЗЛ) является зонтичным термином для группы заболеваний дыхательных путей, характеризующихся обструкцией или ограничением потока воздуха. Состояния, включенные в этот зонтичный термин: хронические бронхиты, эмфизема и бронхоэктазия.
В другом воплощении изобретения соединения, описанные в настоящем документе, используются для лечения астмы. Также соединения или фармацевтические композиции, описанные в настоящем документе, могут использоваться для лечения эндотоксикоза и сепсиса. В другом варианте воплощения изобретения соединения или фармацевтические композиции, описанные в настоящем документе, используются для лечения ревматоидного артрита (РА). В другом варианте воплощения изобретения соединения или фармацевтические композиции, описанные в настоящем документе, используются для лечения контактного или атопического дерматита. Контактный дерматит включает простой контактный дерматит, фототоксический дерматит, аллергический дерматит, фотоаллергический дерматит, контактную крапивницу, системные дерматиты контактного типа и т.п. Простой контактный дерматит может возникать при нанесении на кожу слишком большого количества вещества или если кожа является чувствительной к определенным веществам. Атопический дерматит, иногда называемый экземой, является видом дерматита, атопическим кожным заболеванием.
Настоящее изобретение также относится к способу лечения гиперпролиферативных заболеваний у млекопитающих, включающему введение указанному млекопитающему терапевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли, сложного эфира, пролекарственные средства, сольвата, гидрата или производного. В некоторых воплощениях изобретения указанный способ относится к лечению рака, такого как острый миелоидный лейкоз, рак вилочковой железы, мозга, легкого, плоскоклеточный рак, рак кожи, глаз, ретинобластома, внутриглазная меланома, рак полости рта и ротоглотки, мочевого пузыря, желудка, поджелудочной железы, мочевого пузыря, молочной железы, шейки матки, головы, шеи, почки, печени, яичника, предстательной железы, толстой кишки, пищевода, яичка, гинекологический рак, рак щитовидной железы, ЦНС, ПНС, связанный со СПИДом (например, лимфома и саркома Капоши) или вирусно-индуцированный рак. В некоторых воплощениях изобретения указанный способ связан с лечением нераковых гиперпролиферативных заболеваний, таких как доброкачественная гиперплазия кожи (например, псориаз) или простаты (например, доброкачественная гипертрофия предстательной железы (ДГПЖ), рестеноз.
Настоящее изобретение также относится к способу лечения заболеваний, связанных с васкулогенезом или ангиогенезом у млекопитающих, включающему введение указанному млекопитающему терапевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли, сложного эфира, пролекарственные средства, сольвата, гидрата или производного. В некоторых воплощениях изобретения указанный способ предназначен для лечения заболевания, выбранного из группы, состоящей из опухолевого ангиогенеза, хронических воспалительных заболеваний, таких как ревматоидный артрит, воспалительных заболеваний кишечника, атеросклероза, кожных заболеваний, таких как псориаз, экзема и склеродермия, сахарного диабета, диабетической ретинопатии, ретинопатии недоношенных, возрастной дегенерации желтого пятна, гемангиомы, глиомы, меланомы, саркомы Капоши и рака яичника, молочной железы, легкого, поджелудочной железы, простаты, толстой кишки и эпидермоидного рака.
В число пациентов, которые могут проходить лечение соединениями по настоящему изобретению или их фармацевтически приемлемыми солями, сложными эфирами, пролекарственные средствами, сольватами, гидратами или производными, в соответствии со способами настоящего изобретения, можно включить, например, пациентов с диагнозом псориаз; рестеноз; атеросклероз; ДГПЖ, рак молочной железы, такой как карцинома из эпителия протоков в железистой ткани молочной железы, рак мозга, слизистый рак, трубчатая карцинома и воспалительный рак молочной железы; рак яичника, в том числе эпителиальная опухоль яичника, такая как аденокарцинома в яичнике и аденокарцинома, которая мигрировала из яичника в брюшную полость, рак матки, рак шейки матки, такой как аденокарцинома шейки матки, в том числе эпителиальный плоскоклеточный рак и аденокарцинома, рак простаты, такой как рак простаты, отобранный из: аденокарциномы или аденокарциномы, которая мигрировала в кость; рак поджелудочной железы, такой как карцинома эпителия в ткани протока поджелудочной железы и аденокарцинома в протоке поджелудочной железы; рак мочевого пузыря, такой как переходно-клеточный рак мочевого пузыря, карцинома уротелия (переходная карцинома), опухоли в клетках уротелия, ограничивающих мочевой пузырь, плоскоклеточный рак, аденокарцинома, и мелкоклеточный рак; лейкемия, такая как острый миелоидный лейкоз (ОМЛ), острый лимфобластный лейкоз, хронический лимфолейкоз, хронический миелолейкоз, лейкоз ворсистых клеток, миелодисплазия, миелопролиферативные расстройства, острый миелобластный лейкоз (ОМЛ), хронический миелолейкоз (ХМЛ), мастоцитоз, хронический лимфолейкоз (ХЛЛ), множественная миелома (ММ) и миелодиспластический синдром (МДС); рак костей; рак легкого, такой как немелкоклеточный рак легкого (НМРЛ), который разделяется на плоскоклеточный рак, аденокарциному и крупноклеточную недифференцированную карциному, и мелкоклеточный рак легкого; рак кожи, такой как карцинома базальных клеток, меланома, плоскоклеточный рак и актинический кератоз, который является состоянием кожи, иногда перерастающим в плоскоклеточный рак; ретинобластома глаз; кожная или внутриглазная меланома, первичный рак печени (рак, который начинается в печени); рак почки, рак щитовидной железы, такой как папиллярный, фолликулярный, медуллярный и анапластический; лимфома, связанная со СПИДом, такая как диффузная крупноклеточная лимфома В-клеток, иммунобластная лимфома В-клеток и лимфома мелких клеток с нерасщепленным ядром, саркома Капоши, вирусно-индуцированные виды рака, в том числе вируса гепатита В (ВГВ), вируса гепатита С (ВГС) и гепатоцеллюлярная карцинома; лимфотропный вирус человека типа 1 (ЛТВЧ-1) и возрастная Т-клеточная лейкемия/лимфома, вирус папилломы человека (ВПЧ), рак шейки матки, рак центральной нервной системы (ЦНС), такой как первичная опухоль головного мозга, который включает в себя глиомы (астроцитому, анапластическую астроцитому или мультиформную глиобластому), олигодендроглиому, эпендимому, менингиому, лимфому, шванному и медуллобластому; раковые заболевания периферической нервной системы (ПНС), такие как невриномы слухового нерва и злокачественная опухоль оболочек периферических нервов (ЗООПН), включая нейрофибромы и шванномы, злокачественная фиброзная цитома, злокачественная фиброзная гистиоцитома, злокачественная менингиома, злокачественная мезотелиома, и злокачественные смешанные опухоли Мюллера, рак полости рта и ротоглотки, такой как гипофарингеальный рак, рак гортани, рак носоглотки и рак ротоглотки; рак желудка, такой как лимфома, стромальная опухоль желудка и карциноидная опухоль; рак яичек, такой как опухоль половых клеток (ОПК), которая включает семиномные, несеминомные и половые стромальные опухоли, в том числе опухоль клеток Лейдига и опухоли клеток Сертоли; рак тимуса, такой как тимома, рак вилочковой железы, болезнь Ходжкина, карциноиды неходжкинской лимфомы или карциноидные опухоли; рак прямой кишки; а также рак толстой кишки.
Пациенты, которые могут проходить лечение соединениями по настоящему изобретению или их фармацевтически приемлемыми солями, сложными эфирами, пролекарственные средствами, сольватами, гидратами или производными, в соответствии со способами настоящего изобретения, включают, например, пациентов с диагнозом состояний, включая, не ограничиваясь, невриному слухового нерва, аденокарциному, рак надпочечников, анальный рак, ангиосаркому (например, лимфангиосаркому, лимфангиоэндотелиосаркому, гемангиосаркому),
доброкачественную моноклональную гаммапатию, рак желчного пузыря (например, холангиокарциному), рак мочевого пузыря, рак молочной железы (например, аденокарциному молочной железы, папиллярный рак молочной железы, рак молочной железы, медуллярный рак молочной железы), рак мозга (например, менингиому, глиому, например, астроцитому, олигодендроглиому; медуллобластому), рак бронхов, рак шейки матки (например, аденокарциному шейки матки), хориокарциному, хордому, краниофарингиому, колоректальный рак (например, рак толстой кишки, рак прямой кишки, аденокарциному толстой кишки), эпителиальную карциному, эпендимому, эндотелиосаркому (например, саркому Капоши, множественную идиопатическую геморрагическую саркому), рак эндометрия, рак пищевода (например, аденокарциному пищевода, аденокарциному Барретта), саркому Юинга, обычную гиперэозинофилию, рак желудка (например, аденокарциному желудка), желудочно-кишечные стромальные опухоли (ЖКСО), рак головы и шеи (например, плоскоклеточную карциному головы и шеи, рак ротовой полости (например, плоскоклеточную карциному рта (ПКР)), болезнь тяжелых цепей (например, болезнь альфа-цепи, болезнь гамма-цепи, болезнь мю-цепи), гемангиобластому, воспалительные миофибробластные опухоли, иммуноцитный амилоидоз, рак почек (например, опухоль нефробластома, известную как опухоль Вильмса, клеточную карциному почек), рак печени (например, гепатоцеллюлярный рак (ГЦК), злокачественную гепатому), рак легкого (например, бронхогенную карциному, мелкоклеточный рак легкого (МРЛ), немелкоклеточный рак легкого (НМРЛ), аденокарциному легких), лейкемию (например, острый лимфобластный лейкоз (ОЛЛ), который включает ОЛЛ В-линии и ОЛЛ Т-линии, хронический лимфолейкоз (ХЛЛ), пролимфоцитную лейкемию (ПЛЛ), лейкоз ворсистых клеток (ЛВК) и макроглобулинемию Вальденстрема (MB); периферическую Т-клеточную лимфому (ПТКЛ), возрастную Т-клеточную лейкемию/лимфому (ВТЛ), кожную Т-клеточную лимфому (КТКЛ), большой гранулированный лимфолейкоз (БГЛ), болезнь Ходжкина и болезнь Рида-Стемберга; острый миелолейкоз (ОМЛ), хронический миелолейкоз (ХМЛ), хронический лимфолейкоз (ХЛЛ)), лимфомы (например, лимфому Ходжкина (ЛХ), неходжкинскую лимфому (НХЛ), фолликулярную лимфому, диффузную крупноклеточную В-клеточную лимфому (В-ККЛ), клеточная лимфому мантии (МКЛ)), лейомиосаркому (ЛМС), мастоцитоз (например, системный мастоцитоз), множественную миелому (ММ), миелодиспластический синдром (МДС), мезотелиому, миелопролиферативные расстройства (МПР) (например, полицитемию Вера (ПВ), идиопатический тромбоцитоз (ИТ), идиопатическую миелоидную метаплазию (АММ), известную под названием миелофиброз (МФ), хронический идиопатический миелофиброз, хронический миелолейкоз (ХМЛ), хронический нейтрофильный лейкоз (ХНЛ), гиперэозинофильный синдром (ГЭК)), нейробластому, нейрофиброму (например, нейрофиброматоз (НФ) типа 1 или типа 2, шванноматоз), нейроэндокринный рак (например, гастроэнтеропанкреатические нейроэндокринные опухоли (ГЭП-НЭО), карциноидные опухоли), остеосаркому, рак яичника (например, цистаденокарциному, эмбриональную карциному яичника, аденокарциному яичника), болезнь Педжета вульвы, болезнь Педжета полового члена, папиллярную аденокарциному, рак поджелудочной железы (например, панкреатическую аденокарциному, интрадуктальные папиллярные муцинозные новообразования (ИПМН)), опухоль эпифиза, примитивную нейроэктодермальную опухоль (ПНТ), рак предстательной железы (например, аденокарциному предстательной железы), рабдомиосаркому, ретинобластому, рак слюнных желез, рак кожи (например, плоскоклеточный рак (ПКР), кератоакантому (КА), меланому, базально-клеточную карциному (БКК)), мелкоклеточный рак кишечника (например, рак аппендикса), саркому мягких тканей (например, злокачественную фиброзную гистиоцитому (ЗФГ), липосаркому, злокачественную опухоль оболочек периферических нервов (ЗООПН), хондросаркому, фибросаркома, миксосаркому), рак сальной железы, рак потовых желез, синовиому, рак яичка (например, семиному, эмбриональную карциному яичка), рак щитовидной железы (например, папиллярный рак щитовидной железы, папиллярную тироидную карциному (ПТК), медуллярный рак щитовидной железы), и макроглобулинемию Вальденстрема.
Настоящее изобретение также относится к способу лечения диабета у млекопитающих, включающему введение указанному млекопитающему терапевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли, сложного эфира, пролекарственные средства, сольвата, гидрата или производного.
Кроме того, соединения, описанные в настоящем документе, могут использоваться для лечения угревой болезни.
Кроме того, соединения, описанные в настоящем документе, могут использоваться для лечения артериосклероза, включая атеросклероз. Артериосклероз является общим термином, описывающим любые уплотнения средних или крупных артерий. Атеросклероз является уплотнением артерии исключительно за счет атеросклеротических бляшек.
Далее, соединения, описанные в настоящем документе, могут использоваться для лечения гломерулонефрита. Гломерулонефрит является первичным или вторичным аутоиммунным заболеванием почек, характеризующимся воспалением клубочков. Он может быть бессимптомным или сопровождаться истинной гематурией и/или протеинурией. Существует много известных типов, которые подразделяются на острый, подострый или хронический гломерулонефрит. Причинами являются инфекции (бактериальные, вирусные или паразитические патогены), аутоиммунные или паранеопластические причины.
Кроме того, соединения, описанные в настоящем документе, могут использоваться для лечения бурсита, волчанки, острого рассеянного энцефаломиелита (ОРЭМ), болезни Аддисона, антифосфолипидного синдрома (АФС), апластической анемии, аутоиммунного гепатита, целиакии, болезни Крона, сахарного диабета (типа 1), синдрома Гудпасчера, Базедовой болезни, синдрома Гийена-Барре (СГБ), болезни Хашимото, воспалительных заболеваний кишечника, системной красной волчанки, тяжелой миастении, синдрома миоклонуса опсоклонуса (ОМС), неврита зрительного нерва, тиреоидита Орда, остеоартрита, увеоретинита, пузырчатки, полиартритов, первичного билиарного цирроза печени, синдрома Рейтера, артериита Такаясу, временного артериита, теплой аутоиммунной гемолитической анемии, гранулематоза Вегенера, алопеции, болезни Шагаса, синдрома хронической усталости, вегетативной дистонии, эндометриоза, гнойного гидраденита, интерстициального цистита, нейромиотонии, саркоидоза, склеродермии, неспецифического язвенного колита, витилиго, вульводинии, аппендицита, артериита, артрита, блефарита, бронхиолита, бронхита, цервицита, холангита, холецистита, хориоамнионита, колита, конъюнктивита, цистита, дакриоаденита, дерматомиозита, эндокардита, эндометрита, энтерита, энтероколита, эпикондилита, эпидидимита, фасциита, фиброзита, гастрита, гастроэнтерита, воспаления десен, гепатита, гидраденита, илеита, ирита, ларингита, мастита, менингита, миелита, миокардита, миозита, нефрита, омфалита, оофорита, орхита, остеомиелита, отита, панкреатита, паротита, перикардита, перитонита, фарингита, плеврита, флебита, пневмонита, проктита, простатита, пиелонефрита, ринита, сальпингита, синусита, стоматита, синовита, тендинита, тонзиллита, увеита, вагинита, васкулита или вульвита.
Дополнительно, соединения по настоящему изобретению могут использоваться для лечения круглогодичного аллергического ринита, мезентерита, перитонита, акродерматита, воспаления кожных сосудов, атопического дерматита, контактного дерматита, экземы, эритемы, опрелостей, синдрома Стивенса-Джонсона, токсического эпидермального некролиза, кожных аллергий, тяжелых аллергических реакций/анафилаксии, аллергического гранулематоза, гранулематоза Вегенера, аллергического конъюнктивита, хориоретинита, конъюнктивита, инфекционного кератоконъюнктивита, кератоконъюнктивита, офтальмии новорожденных, трахомы, увеита, глазных воспалений, блефароконъюнктивита, мастита, гингивита, перикоронита, фарингита, ринофарингита, сиаладенита, воспаления костно-мышечной системы, приобретенной болезни Стилла, болезни Бехчета, бурсита, хондрокальциноза, дактилита, синдрома Фелти, подагры, инфекционных артритов, болезни Лайма, воспалительного остеоартроза, периартрита, синдрома Рейтера, вирусной инфекции реки Росс, острых респираторных заболеваний, дистресс-синдрома, острого бронхита, острого синусита, аллергического ринита, астмы, тяжелой рефрактерной астмы, фарингита, плеврита, ринофарингита, сезонного аллергического ринита, синусита, астматического статуса, трахеобронхита, ринита, серозита, менингита, оптикомиелита, вируса полиомиелита, синдрома Алпорта, Баланита, эпидидимита, орхоэпидидимиса, фокального сегментарного нарушения, гломерулосклероза, гломерулонефрита, IgA нефропатии (болезни Берже), орхита, параметрита, воспаления тазовых органов, простатита, пиелита, пиелоцистита, пиелонефрита, гранулематоза Вегенера, гиперурикемии, аортита, артериита, хилоперикардита, синдрома Дресслера, эндартериита, эндокардита, экстракраниального височного артериита, ВИЧ-ассоциированного артериита, внутричерепного височного артериита, болезни Кавасаки, лимфангиофлебита, болезни Мондора, периартериита или перикардита.
В других аспектах соединения по настоящему изобретению применяются для лечения аутоиммунного гепатита, воспаления тощей кишки, мезентерита, мукозита, неалкогольного стеатогепатита, невирусного гепатита, аутоиммунного панкреатита, перигепатита, перитонита, поухита, проктита, псевдомембранозного колита, ректосигмоидита, сальпингоперитонита, сигмоидита, стеатогепатита, неспецифического язвенного колита, синдрома Черджа-Стросса, язвенного проктита, синдрома раздраженной кишки, желудочно-кишечных воспалений, острого энтероколита, анусита, некроза Балсера, холецистита, колита, болезни Крона, дивертикулита, энтерита, энтероколита, энтерогепатита, эозинофильного эзофагита, эзофагита, гастрита, геморрагического энтерита, гепатита, вирусной инфекции гепатита, гепатохолангита, гипертрофического гастрита, илеита, илеоцецита, саркоидоза, воспалительных заболеваний кишечника, болезни Бехтерева, ревматоидного артрита, ювенильного ревматоидного артрита, псориаза, псориатического артрита, волчанки (кожной/системной/нефрита), СПИДа, агаммаглобулинемии, комплекса, связанного со СПИДом, болезни Брутона, синдрома Чедиака Хигаси, общего вариабельного иммунодефицита, синдрома Ди Георге, дисгаммаглобулинемии, дефицита иммуноглобулина, синдрома Иова, синдрома Незелофа, бактерицидного расстройства фагоцитов, синдрома Вискотта Олдрича, асплении, слоновости, гиперспленизма, болезни Кавасаки, лимфаденопатии, лимфедемы, лимфоцеле, синдрома Нонне Милроя Межа, болезни селезенки, спленомегалии, тимомы, болезни тимуса, периваскулита, флебита, плевроперикардита, узелкового полиартериита, васкулита, артериита Такаясу, височного артериита, тромбангиита, облитерирующего тромбангиита, тромбоэндокардита, тромбофлебита или ХОБЛ.
Настоящее изобретение также связано со способом лечения сердечнососудистых заболеваний у млекопитающих, включающий введение указанному млекопитающему терапевтически эффективного количества соединения по настоящему изобретению, или его фармацевтически приемлемой соли, сложного эфира, пролекарственные средства, сольвата, гидрата или производного. Примеры сердечнососудистых состояний включают, не ограничиваясь, атеросклероз, рестеноз, окклюзию сосудов и обструктивное заболевание сонной артерии.
В другом аспекте настоящего изобретения представлены способы нарушения функции лейкоцитов или нарушения функции остеокластов. Способ включает взаимодействие лейкоцитов или остеокластов с таким количеством соединения по настоящему изобретению, которое нарушает их функцию.
В другом аспекте настоящего изобретения представлены способы лечения офтальмических заболеваний путем введения в глаз пациента одного или более из указанных соединений или фармацевтических композиций.
Дополнительно представлены способы введения соединений настоящего изобретения путем глазных капель, внутриглазной инъекции, интравитреальной инъекции, местного применения или с помощью устройства элюирования лекарств, микрокапсул, импланта или микрофлюидного устройства. В некоторых случаях соединения по настоящему изобретению вводятся с носителем или наполнителем, который увеличивает внутриглазное проникновение соединения, таким как масляная и водная эмульсия с коллоидными частицами, имеющими масляное ядро, окруженное межфазной пленкой.
В некоторых случаях коллоидные частицы включают по меньшей мере один катионный агент и по меньшей мере одно неионогенное поверхностно-активное вещество, такое как полоксамер, тилоксапол, полисорбат, производное касторового масла и полиоксиэтилена, эфир сорбита или полиоксилстеарат. В некоторых случаях катионным агентом является алкиламин, третичный алкиламин, четвертичное аммониевое соединение, катионный липид, аминоспирт, бигуанидиновая соль, катионное соединение или их смесь. В некоторых случаях катионным агентом является бигуанидиновая соль, такая как хлоргексидин, полиаминопропил бигуанидин, фенформин, алкилбигуанидин или их смесь. В некоторых случаях четвертичным аммониевым соединением является бензалкония галид, лауралкония галид, цетримид, гексадецилтриметиламмония галид, тетрадецилтриметиламмония галид, додецилтриметиламмония галид, цетримония галид, бензетония галид, бегеналкония галид, цеталкония галид, цететилдимония галид, цетилпиридиния галид, бензододециния галид, хлораллил метанамина галид, миристилалкония галид, стеаралкония галид или смесь двух или более из этих соединений. В некоторых случаях катионным агентом является бензалкония хлорид, лауралкония хлорид, бензододециния бромид, бензетения хлорид, гексадецилтриметиламмония бромид, тетрадецилтриметиламмония бромид, додецилтриметиламмония бромид или смесь двух или более из этих соединений. В некоторых случаях масляной фазой является минеральное масло и легкое минеральное масло, среднецепочечные триглицериды (СЦТ), кокосовое масло; гидрированные масла, включающие гидрированное хлопковое масло, гидрированное пальмовое масло, гидрированное касторовое масло или гидрированное соевое масло; производные полиоксиэтилена и касторового масла, включающие полиоксил-40 гидрированное касторовое масло, полиоксил-60 гидрированное касторовое масло или полиоксил-100 гидрированное касторовое масло.
В настоящем изобретении дополнительно предложены способы модулирования активности киназы путем взаимодействия киназы с таким количеством соединения по настоящему изобретению, которое является достаточным для модулирования активности этой киназы. Модулированием может быть ингибирование или активация активности киназы. В некоторых воплощениях настоящего изобретения представлены способы ингибирования активности киназы путем взаимодействия киназы с таким количеством соединения по настоящему изобретению, которое является достаточным для ингибирования активности этой киназы. В некоторых воплощениях настоящего изобретения представлены способы ингибирования активности киназы в растворе путем взаимодействия указанного раствора с таким количеством соединения по настоящему изобретению, которое является достаточным для ингибирования активности этой киназы в указанном растворе. В некоторых воплощениях настоящего изобретения представлены способы ингибирования активности киназы в клетке путем взаимодействия указанной клетки с таким количеством соединения по настоящему изобретению, которое является достаточным для ингибирования активности этой киназы в указанной клетке. В некоторых воплощениях настоящего изобретения представлены способы ингибирования активности киназы в ткани путем взаимодействия указанной ткани с таким количеством соединения по настоящему изобретению, которое является достаточным для ингибирования активности этой киназы в указанной ткани. В некоторых воплощениях настоящего изобретения представлены способы ингибирования активности киназы в организме путем взаимодействия указанного организма с таким количеством соединения по настоящему изобретению, которое является достаточным для ингибирования активности этой киназы в указанном организме. В некоторых воплощениях настоящего изобретения представлены способы ингибирования активности киназы в организме животного путем взаимодействия указанного организма животного с таким количеством соединения по настоящему изобретению, которое является достаточным для ингибирования активности этой киназы в указанном организме животного. В некоторых воплощениях настоящего изобретения представлены способы ингибирования активности киназы в организме млекопитающего путем взаимодействия указанного организма млекопитающего с таким количеством соединения по настоящему изобретению, которое является достаточным для ингибирования активности этой киназы в организме млекопитающего. В некоторых воплощениях настоящего изобретения представлены способы ингибирования активности киназы в организме человека путем взаимодействия указанного организма человека с таким количеством соединения по настоящему изобретению, которое является достаточным для ингибирования активности этой киназы в указанном организме человека. В некоторых воплощениях изобретения % активности киназы после взаимодействия киназы с соединением настоящего изобретения составляет менее чем 1, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 95 или 99% активности киназы в отсутствие указанного этапа взаимодействия.
В некоторых воплощениях изобретения киназой является липидкиназа или протеинкиназа. В некоторых воплощениях изобретения киназа выбрана из группы, состоящей из PI3 киназ, включающей различные изоформы, такие как PI3 киназа α, PI3 киназа β, PI3 киназа γ, PI3 киназа δ; ДНК-РК; mTor; Abl, VEGFR, рецептор эфрина В4 (EphB4); тирозинкиназа рецептора ТЕК (TIE2); FMS-связанная тирозинкиназа 3 (FLT-3); рецептор фактора роста тромбоцитов (PDGFR); RET; ATM; ATR; hSmg-1; Hck; Src; рецептор эпидермального фактора роста (EGFR); KIT; инсулиновый рецептор (IR) и IGFR.
Настоящее изобретение дополнительно представляет способы модулирования активности PI3 киназы путем взаимодействия PI3 киназы с таким количеством соединения по настоящему изобретению, которое является достаточным для модулирования активности этой PI3 киназы. Модулированием может быть ингибирование или активация активности PI3 киназы. В некоторых воплощениях настоящего изобретения представлены способы ингибирования активности PI3 киназы путем взаимодействия PI3 киназы с таким количеством соединения по настоящему изобретению, которое является достаточным для ингибирования активности этой PI3 киназы. В некоторых воплощениях настоящего изобретения представлены способы ингибирования активности PI3 киназы. Такое ингибирование может иметь место в растворе, в клетке, экспрессирующей одну или более PI3 киназ, в ткани, включающей клетки, которые экспрессируют одну или более PI3 киназ, или в организме, экспрессирующем одну или более PI3 киназ. В некоторых воплощениях настоящего изобретения представлены способы ингибирования активности PI3 киназы в организме животного (включая млекопитающих, таких как человек) путем взаимодействия указанного организма животного с таким количеством соединения по настоящему изобретению, которое является достаточным для ингибирования активности этой PI3 киназы в указанном организме животного.
КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ
В настоящем изобретении представлены также способы комбинированного лечения, в которых используется известное средство модулирования других путей или другие компоненты тех же путей, или даже перекрывающиеся наборы целевых ферментов, в комбинации с соединением настоящего изобретения, или его фармацевтически приемлемой солью, сложным эфиром, пролекарством, сольватом, гидратом или его производным. В одном аспекте такая терапия включает, не ограничиваясь, комбинацию указанного соединения с химиотерапевтическими средствами, терапевтическими антителами, лучевым лечением, для обеспечения синергического или аддитивного терапевтического эффекта.
В одном аспекте соединения или фармацевтические композиции настоящего изобретения могут давать синергическую или аддитивную эффективность при введении в комбинации с агентами, ингибирующими выработку или активность IgE. Такая комбинация может снижать нежелательный эффект высокого уровня IgE, связанный с использованием одного или более ингибиторов PI3Kδ, если такой эффект имеет место. Это может быть особенно полезно при лечении аутоиммунных и воспалительных нарушений (AIID), таких как ревматоидный артрит. Кроме того, введение ингибиторов PI3Kδ или Р13Кδ/γ настоящего изобретения в комбинации с ингибиторами mTOR также может демонстрировать синергию за счет усиленного ингибирования пути PI3K.
В отдельном, но родственном аспекте настоящего изобретения представлено комбинированное лечение заболевания, связанного с PI3Kδ, включающее введение ингибитора PI3Kδ и агента, ингибирующего выработку или активность IgE. Другие типичные ингибиторы PI3Kδ. является применимыми, и они описаны, например, в патенте США №6800620. Такое комбинированное лечение особенно полезно для лечения аутоиммунных и воспалительных заболеваний (AIID), включая, не ограничиваясь, ревматоидный артрит.
Агенты, ингибирующие выработку IgE, являются известными в данной области и включают, не ограничиваясь, один или более из TEI-9874, 2-(4-(6-циклогексилокси-2-нафтилокси)фенилацетамид)бензойной кислоты, рапамицина, аналогов рапамицина (т.е. рапалогов), ингибиторов TORC1, ингибиторов TORC2, и любых других соединений, ингибирующих mTORCl и mTORC2. Агенты, ингибирующие активность IgE, включают, например, анти-IgE антитела, такие как, например, омализумаб и TNX-901.
Для лечения аутоиммунных заболеваний, указанные соединения или фармацевтические композиции могут использоваться в комбинации с обычно прописываемыми лекарственные средствами, включая, не ограничиваясь, Энбрел®, Ремикад®, Хумира®, Авонекс® и Ребиф®. Для лечения респираторных заболеваний, указанные соединения или фармацевтические композиции могут использоваться в. комбинации с обычно прописываемыми лекарственные средствами, включая, не ограничиваясь, Ксолар®, Адвар®, Сингуляр® и Спирива®.
Соединения по настоящему изобретению могут входить в композиции или вводиться пациенту в сочетании с другими средствами, которые влияют на ослабления симптомов воспалительных состояний, таких как энцефаломиелит, астма и другие заболевания, описанные в настоящем документе. Эти средства включают нестероидные противовоспалительные препараты (НПВП), например, ацетилсалициловую кислоту; ибупрофен; напроксен; индометацин; набуметон; толметин; и т.п. Кортикостероиды используются для снижения воспаления и подавления активности иммунной системы. Наиболее часто прописываемым лекарством этого типа является преднизон. Хлорохин (арален) или гидроксихлорохин (плаквенил) могут быть очень полезными для некоторых пациентов с волчанкой. Их наиболее часто прописывают при кожных и суставных симптомах волчанки. Азатиоприн (имуран) и циклофосфамид (цитоксан) подавляют воспаление и склонны к подавлению иммунной системы. Другие агенты, например, метотрексат и циклоспорин, используются для регуляции симптомов волчанки. Антикоагулянты используются для предотвращения быстрого свертывания крови. Их ассортимент варьирует от аспирина в очень низких концентрациях, который предотвращает слипание тромбоцитов, до гепарина/кумадина. Другие соединения, используемые при лечении волчанки, включают белимумаб (Бенлиста®).
В другом аспекте настоящее изобретение связано также с фармацевтической композицией для ингибирования аномального клеточного роста у млекопитающих, которая включает определенное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли, сложного эфира, пролекарственные средства, сольвата, гидрата или его производного в комбинации с определенным количеством анти-ракового средства (например, биотерапевтического химиотерапевтического средства). В настоящее время в данной области известно много химиотерапевтических средств, которые могут использоваться в комбинации с соединениями настоящего изобретения. Также могут использоваться другие раковые терапии в комбинации с соединениями настоящего изобретения, которые включают, не ограничиваясь, хирургию и хирургическое лечение, а также лучевую терапию.
В некоторых воплощениях изобретения химиотерапевтические средства выбраны из группы, состоящей из митотических ингибиторов, алкилирующих агентов, анти-метаболитов, интеркалирующих антибиотиков, ингибиторов фактора роста, ингибиторов клеточного цикла, ферментов, ингибиторов топоизомеразы, модификаторов биологической реакции, анти-гормонов, ингибиторов ангиогенеза и анти-андрогенов. Неограничивающими примерами являются химиотерапевтические средства, цитотоксичные агенты, непептидные маленькие молекулы, такие как Гливек (иматиниб мезилат), Велкад (бортезомиб), Казодекс (бикалутамид), Иресса (гефитиниб) и Адриамицин, а также носители химиотерапевтических агентов. Неограничивающие примеры химиотерапевтических средств включают алкилирующие агенты, такие как тиотепа и циклофосфамид (Цитоксан™); алкилсульфонаты, такие как бусульфан, импросульфан и пипосульфан; азиридины таких как бензодопа, карбоквон, метуредопа и уредопа; этиленимины и метиламеламины, в том числе альтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметилоломеламин; азотистые иприты, такие как хлорамбуцил, хлорнафазин, холофосфамид, эстрамустин, ифосфамид, мехлоретамин, мехлоретамин оксида гидрохлорид, мелфалан, новембихин, фенестерин, преднимустин, трофосфамид, урамустин; нитрозомочевины, такие как кармустин, хлорозотоцин, фотемустин, ломустин, нимустин, ранимустин; антибиотики, такие как аклациномизин, актиномицин, аутрамицин, азасерин, блеомицин, кактиномицин, калихеамицин, карабицин, карминомицин, карзинофилин, Казодекс™, хромомицин, дактиномицин, даунорубицин, деторубицин, 6-диазо-5-оксо-L-норлейцин, доксорубицин, эпирубицин, эзорубицин, идарубицин, марцелломицин, митомицин, микофеноловая кислота, ногаламицин, оливомицин, пепломицин, потфиромицин, пуромицин, квеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, зиностатин, зорубицин; антиметаболиты, такие как метотрексат и 5-фторурацил (5-ФУ), аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметрексат; пуриновые аналоги, такие как флударабин, 6-меркаптопурин, тиамиприн, тиогуанин; пиримидиновые аналоги, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин, флоксуридин, андрогены, такие как калустерон, дромостанолон пропионат, эпитиостанол, мепитиостан, тестолактон; антинадпочечные препараты, такие как аминоглютетимид, митотан, трилостан; подкрепители фолиевой кислоты, такие как фролиновая кислота; ацеглатон; альдофосфамид гликозид; аминолевулиновая кислота; амсакрин; бестрабуцил, бисантрен; эдатраксат; дефофамин; демеколцин; диазиквон; элфомитин; эллиптиния ацетат; этоглюцид; нитрат галлия; гидроксимочевина; лентинан; лонидамин; митогуазон; митоксантрон; мопидамол; нитракрин; пентостатин; фенамет; пирарубицин; подофиллиновая кислота; 2-этилгидразид; прокарбазин; PSK.R™; разоксан; сизофиран; спирогерманий; тенуазоновая кислота; триазиквон; 2,2',2"-трихлортриэтиламин; уретан; виндезин; дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид ("Ара-С"); циклофосфамид; тиотепа; таксаны, например, паклитаксел (Таксол™, Bristol-Myers Squibb Oncology, Принстон, Нью-Джерси) и доцетаксел (Таксотер™, Rhone-Poulenc Rorer, Антоний, Франция); ретиноевая кислота; эсперамицин, капецитабин, и фармацевтически приемлемые соли, кислоты или производные любого из выше перечисленных. Также в качестве пригодных химиотерапевтических средств улучшения клеток включены антигормональные средства, которые регулируют или ингибируют действие гормонов на опухоли, такие как анти-эстрогены, включая, например, тамоксифен (Нолвадекс™), ралоксифен, ароматаза-ингибирующие 4(5)-имидазолы, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY 117018, онапристон и торемифен (Фарестон); и анти-андрогены, такие как флутамид, нилутамид, бикалутамид, лейпролид и гозерелин; хлорамбуцил; гемцитабин; 6-тиогуанин; меркаптопурин; метотрексат; платиновые аналоги, такие как цисплатин и карбоплатин; винбластин; платина; этопозид (VP-16); изофосфамид; митомицин С; митоксантрон; винкристин; винорелбин; навелбин; новантрон; тенипозид; дауномицин; аминоптерин; кселода; ибандронат; камптотецин-11 (ЦПТ-11); ингибитор топоизомеразы RFS 2000; дифторметилорнитин (ДФМО). При необходимости соединения или фармацевтические композиции настоящего изобретения могут использоваться в комбинации с обычно прописываемыми противораковыми лекарственные средствами, такими как Герцептин®, Авастин®, Эрбитукс®, Ритуксан®, Таксол®, Аримидекс®, Таксотер® и Велкейд®.
Другие химиотерапевтические средства включают, не ограничиваясь, антиэстрогены (например, тамоксифен, ралоксифен и мегестрол), агонисты ЛГРГ (например, гозерилин и лейпролид), анти-андрогены (например, флутамид и бикалутамид), фотодинамическую терапию (например, вертопорфин (BPD-MA), фталоцианин, фотосенсибилизатор РС4 и деметокси-гипокреллин (2BA-2-DMHA)), азотистые иприты (например, циклофосфамид, ифосфамид, трофосфамид, хлорамбуцил, эстрамустин и мелфалан), нитрозомочевины (например, кармустин (BCNU) и ломустин (CCNU)), алкилсульфонаты (например, бусульфан и треосульфан), триазены (например, дакарбазин, темозоломид), содержащие платину соединения (например, цисплатин, карбоплатин, оксалиплатин), алкалоиды барвинка (например, винкристин, винбластин, виндезин и винорельбином), таксоиды (например, паклитаксел или эквиваленты паклитаксела, такие как паклитаксел, связанный с наночастицами альбумина (Абраксан), паклитаксел, связанный с докозагексановой кислотой (ДГК-паклитаксел, Таксопрексин), полиглутамат-связанный паклитаксел (ПГ-паклитаксел, паклитаксела полиглумекс, КТ-2103, Ксиотакс), опухоль-активированные пролекарственные средства ANG1005 (Ангиопеп-2, связанный с тремя молекулами паклитаксела), паклитаксел-ЕС-1 (паклитаксел, связанный с еrbВ2-распознающим пептидом ЕС-1) и глюкозо-сопряженный паклитаксел, например, 2'-паклитаксел метил-2-глюкопиранозил сукцинат; доцетаксел, таксол), эпиподофиллины (например, этопозид, этопозид фосфат, тенипозид, топотекан, 9-аминокамптотецин, камптоиринотекан, иринотекан, криснатол, митомицин С), анти-метаболиты, ингибиторы DHFR (например, метотрексат, дихлорметотрексат, триметрексат, эдатрексат), ингибиторы дегидрогеназы IMP (например, микофеноловая кислота, тиазофурин, рибавирин и EICAR), ингибиторы рибонуклеотид-редуктазы (например, гидроксимочевину и дефероксамин), аналоги урацила (например, 5-фторурацил (5-ФУ), флоксуридин, доксифлуридин, ратитрексед, тегафур-урацил, капецитабин), аналоги цитозина (например, цитарабин (Ара С), цитозинарабинозид и флударабин), аналоги пурина (например, меркаптопурин и тиогуанин), аналоги витамина D3 (например, ЕВ 1089, СВ 1093 и КН 1060), ингибиторы изопрениляции (например, ловастатин), дофаминергические нейротоксины (например, ион 1-метил-4-фенилпиримидиния), ингибиторы клеточного цикла (например, стауроспорин), актиномицин (например, актиномицин D, дактиномицин), блеомицин (например, блеомицин А2, блеомицин В2, пепломицин), антрациклины (например, даунорубицин, доксорубицин, пегилированный липосомальный доксорубицин, идарубицин, эпирубицин, пирарубицин, зорубицин, митоксантрон), ингибиторы МЛУ (например, верапамил), ингибиторы Са2+АТФ-азы (например, тапсигаргин), иматиниб, талидомид, леналидомид, ингибиторы тирозинкиназы (например, акситиниб (AG013736), босутиниб (SKI-606), цедираниб (Рецентин™, AZD2171), дазатиниб (Сприцел®, BMS-354825), эрлотиниб (Тарцева®), гефитиниб (Иресса®), иматиниб (Гливек®, CGP57148B, STI-571), лапатиниб (Тикерб®, Тиверб®), лестауртиниб (СЕР-701), нератиниб (HKI-272), нилотиниб (Тасигна®), семаксаниб (семаксиниб, SU5416), сунитиниб (Сутент®, SU11248), тоцераниб (Палладиа®), вандетаниб (Зактима®, ZD6474), ваталаниб (РТК787, PTK/ZK), трастузумаб (Герцептин®), бевацизумаб (Авастин®), ритуксимаб (Ритуксан®), цетуксимаб (Эрбитукс®), панитумумаб (Вектибикс®), ранибизумаб (Люцентис®), нилотиниб (Тазигна®), сорафениб (Нексавар®), эверолимус (Афинитор®), алемтузумаб (Кэмпас®), гемтузумаба озогамицин (Милотарг®), темсиролимус (Торисел®), ENMD-2076, PCI-32765, АС220, довитиниба лактат (TKI258, CHIR-258), BIBW 2992 (Товок™), SGX523, PF-04217903, PF-02341066, PF-299804, BMS-777607, АВТ-869, МР470, BIBF 1120 (Варгатеф®), АР24534, JNJ-26483327, MGCD265, DCC-2036, BMS-690154, СЕР-11981, тивозаниб (AV-951), OSI-930, ММ-121, XL-184, XL-647 и/или XL228), ингибиторы протеасомы (например, бортезомиб (Велкейд)), ингибиторы mTOR (например, рапамицин, темсиролимус (CCI-779), эверолимус (RAD-001), ридафоролимус, АР23573 (Ариад), AZD8055 (AstraZeneca), BEZ235 (Novartis), BGT226 (Norvartis), XL765 (Sanofi Aventis), ПФ-4691502 (Pfizer), GDC0980 (Genetech), SF1126 (Semafoe) и OSI-027 (OSI)), облимерсен, гемцитабин, карминомицин, лейковорин, пеметрексед, циклофосфамид, дакарбазин, прокарбизин, преднизолон, дексаметазон, кампатецин, пликамицин, аспарагиназа, аминоптерин, метоптерин, порфиромицин, мелфалан, лейрозидин, лейрозин, хлорамбуцил, трабектедин, прокарбазин, дискодермолид, карминомицин, аминоптерин и гексаметилмеламин.
Типичные биотерапевтические средства включают, не ограничиваясь, интерфероны, цитокины (например, фактор некроза опухоли, интерферон 6, интерферон г), вакцины, гекатопоэтические факторы роста, моноклональную серотерапию, иммуностимулирующие и/или иммуномодулирующие агенты (например, IL-1, 2, 4, 6 или 12), факторы роста иммуноцитов (например, GM-CSF) и антитела (например, Герцептин (трастузумаб), T-DM1, Авастин (бевацизумаб), Эрбитукс (цетуксимаб), Вектибикс (панитумумаб), Ритуксан (ритуксимаб), Бексар (тозитумомаб)).
Настоящее изобретение дополнительно относится к способу применения соединений или фармацевтических композиций в комбинации с лучевой терапией для ингибирования аномального клеточного роста или лечения гиперпролиферативных нарушений у млекопитающих. Процедуры применения лучевой терапии известны в данной области, и эти процедуры могут использоваться в комбинированной терапии, описанной в настоящем документе. Введение соединения по настоящему изобретению в этой комбинированной терапии может быть определено так, как описано в настоящем документе.
Лучевая терапия может применяться одним из нескольких способов, или комбинацией способов, включая, без ограничения, терапию внешними лучами, внутреннюю лучевую терапию, излучение имплантируемых источников, стереотактическую радиохирургию, системную лучевую терапию, радиотерапию и постоянную или временную интерстициальную брахитерапию. Термин «брахитерапия», используемый в настоящем документе, относится к лучевой терапии, которая применяется при помощи пространственно ограниченного радиоактивного материала, внедренного в организм в опухоль или возле нее, или в другой участок пролиферативного заболевания ткани. Подразумевается, что этот термин включает, не ограничиваясь, воздействие радиоактивных изотопов (например, At-211, I-131, I-125, Y-90, Re-186, Re-188, Sm-153, Bi-212, P-32 и радиоактивных изотопов Lu). Пригодные источники радиации для использования в качестве средств, изменяющих клетку, в настоящем изобретении включают твердые вещества и жидкости. В качестве не ограничивающего примера, источником радиации может быть радионуклид, такой как I-125, I-131, Yb-169, Ir-192 в качестве твердого источника, I-125 в качестве твердого источника, или другие радионуклиды, испускающие протоны, бета-частицы, гамма-излучение или другие терапевтические лучи. Радиоактивным материалом также может быть жидкость, полученная из любого раствора радионуклида(ов), например, раствора I-125 или I-131, или радиоактивная жидкость может быть получена при помощи суспензии подходящей жидкости, содержащей небольшие частицы твердых радионуклидов, таких как Au-198, Y-90. Более того, радионуклид(ы) может быть внедрен в гель или радиоактивные микросферы.
Не ограничиваясь какой-либо теорией, полагают, что соединения по настоящему изобретению могут воздействовать на аномальные клетки, более чувствительные к обработке радиацией, в целях уничтожения и/или ингибирования роста таких клеток. Следовательно, настоящее изобретение дополнительно относится к способу сенсибилизации аномальных клеток млекопитающих для лечения радиацией, который включает введение млекопитающему такого количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, сложного эфира, пролекарственные средства, сольвата, гидрата или его производного, которое является эффективным для сенсибилизации аномальных клеток для лечения радиацией. Количество соединения, соли или сольвата в этом способе может быть определено в соответствии со средствами определения эффективных количеств таких соединений, описанных в настоящем документе.
Соединения или фармацевтические композиции по настоящему изобретению могут использоваться в комбинации с определенным количеством одного или более веществ, отобранных из агентов анти-ангиогенеза, ингибиторов сигнальной трансдукции и антипролиферативных агентов.
Агенты анти-ангиогенеза, такие как ингибиторы ММР-2 (матриксные металлопротеиназы 2), ингибиторы ММР-9 (матриксные металлопротеиназы 9) и ингибиторы СОХ-11 (циклооксигеназы 11) могут использоваться в сочетании с соединениями настоящего изобретения и фармацевтическими композициями, описанными в настоящем документе. Примеры пригодных ингибиторов СОХ-II включают Целебрекс™ (алекоксиб), валдекоксиб и рофекоксиб. Примеры пригодных ингибиторов матриксных металлопротеиназ описаны в публикациях WO 96/33172 (опубликованной 24 октября 1996 года), WO 96/27583 (опубликованной 7 марта 1996 года), заявке на Европейский патент №97304971.1 (поданной 8 июля 1997 года), заявке на Европейский патент №99308617.2 (поданной 29 октября 1999 года), WO 98/07697 (опубликованной 26 февраля 1998 года), WO 98/03516 (опубликованной 29 января 1998 года), WO 98/34918 (опубликованной 13 августа 1998 года), WO 98/34915 (опубликованной 13 августа 1998 года), WO 98/33768 (опубликованной 6 августа 1998 года), WO 98/30566 (опубликованной 16 июля 1998 года), заявке на Европейский патент №606046 (опубликованной 13 июля 1994 года), публикации Европейского патента 931788 (опубликованной 28 июля 1999 года), WO 90/05719 (опубликованной 31 мая 1990 года), WO 99/52910 (опубликованной 21 октября 1999 года), WO 99/52889 (опубликованной 21 октября 1999 года), WO 99/29667 (опубликованной 17 июня 1999 года), заявке на Международный патент РСТ № PCT/IB 98/01113 (поданной 21 июля 1998 года), заявке на Европейский патент №99302232.1 (поданной 25 марта 1999 года), заявке на патент Великобритании №9912961.1 (поданной 3 июня 1999 года), предварительной заявке на патент США №60/148464 (поданной 12 августа 1999 года), патенте США 5863949 (выданном 26 января 1999 года), патенте США 5861510 (выданном 19 января 1999 года) и публикации Европейского патента 780386 (опубликованной 25 июня 1997 года), все из которых включены в настоящую заявку путем ссылки в полном объеме. Предпочтительными ингибиторами ММР-2 и ММР-9 являются ингибиторы, которые слабо ингибируют или не ингибируют ММР-1. Более предпочтительными являются те, которые селективно ингибируют ММР-2 и/или AMP- 9 по сравнению с другими матрикс-металлопротеиназами (т.е. МАР-1, ММР-3, ММР-4, ММР-5, ММР-6, ММР-7, ММР-8, ММР-10, ММР-11, ММР-12 и ММР-13). Конкретными примерами ингибиторов ММР, используемых в настоящем изобретении, являются AG-3340, RO 32-3555 и RS 13-0830.
Настоящее изобретение также относится к способу и фармацевтической композиции для лечения сердечно-сосудистых заболеваний у млекопитающих, включающей определенное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли, сложного эфира, пролекарственные средства, сольвата, гидрата или его производного, или его производного, меченого изотопом, и определенного количества одного или более терапевтических агентов, используемых для лечения сердечно-сосудистых заболеваний.
Примерами для применения при лечении сердечно-сосудистых заболеваний являются анти-тромбозные агенты, например, простациклин и салицилаты, тромболитические агенты, например, стрептокиназа, урокиназа, активатор плазминогена ткани (TPA) и анизоилированный активатор комплекса стрептокиназы и плазминогена (APSAC), анти-тромбоцитные агенты, например, ацетилсалициловая кислота (ASA) и клопидогрель, сосудорасширяющие средства, например, нитраты, лекарственные средства, блокирующие кальциевый канал, анти-пролиферативные агенты, например, колхицин и алкилирующие агенты, интеркалирующие агенты, факторы модуляции роста, такие как интерлейкины, трансформационный фактор-бета роста и родственные тромбоцитные производные факторы роста, моноклональные антитела, направленные против факторов роста, противовоспалительные средства, как стероидные, так и нестероидные, и другие средства, которые могут модулировать тонус сосудов, функцию, артериосклероз и реакцию заживления на повреждение сосуда или органа после вмешательства. Также в комбинации могут быть включены антибиотики или покрытия, включенные в настоящее изобретение. Кроме того, покрытия могут использоваться для фокусной терапевтической доставки на стенке сосуда. Путем внедрения активного агента в разбухающий полимер, активный агент может высвобождаться при разбухании этого полимера.
Соединения, описанные в настоящем документе, могут включаться в композиции или вводиться пациенту в сочетании с жидкими или твердыми тканевыми барьерами, также известными как смазывающие вещества. Примеры тканевых барьеров включают, не ограничиваясь, полисахариды, полигликаны, сепрафильм, интерсид и гиалуроновую кислоту.
Лекарственные средства, которые могут вводиться в сочетании с соединениями, описанными в настоящем документе, включают любые пригодные лекарственные средства, эффективно доставляемые при ингаляции, например, анальгетики, например, кодеин, дигидроморфин, эрготамин, фентанил или морфин; ангинозные препараты, например, дилтиазем, противоаллергические, например, кромогликат, кетотифен или недокромил; антибактериальные средства, например, цефалоспорины, пенициллины, стрептомицин, сульфаниламиды, тетрациклин или пентамидин; антигистаминные препараты, например, метапирилен; противовоспалительные средства, например, беклометазон, флунизолид, будесонид, типредан, триамцинолона ацетонид или флутиказон; противокашлевые средства, например, носкапин; бронходилаторы, например, эфедрин, адреналин, фенотерол, формотерол, изопреналин, метапротеренол, фенилэфрин, фенилпропаноламин, пирбутерол, репротерол, римитерол, сальбутамол, сальметерол, тербуталин, изоэтарин, толубутерол, орципреналин или (-)-4-амино-3,5-дихлор-α-[[[6-[2-(2-пиридинил)этокси]гексил]-амино]метил]бензолметанол; мочегонные средства, например, амилорид; антихолинергические средства, например, ипратропий, атропин или окситропий; гормоны, например, кортизон, гидрокортизон или преднизолон; ксантины, например, эуфиллин, холина теофиллинат, лизина теофиллинат или теофиллин; и терапевтические белки и пептиды, например, инсулин или глюкагон. Специалисту в данной области понятно, что там, где уместно, эти лекарственные средства могут использоваться в форме солей (например, в виде солей щелочных металлов или аминов, или в виде солей присоединения кислоты) или сложных эфиров (например, сложных эфиров низших алкилов), или в виде сольватов (например, гидратов) для оптимизации активности и/или стабильности лекарственного средства.
Другие типичные терапевтические средства, пригодные для комбинированной терапии, включают, не ограничиваясь, агенты, описанные выше, лучевую терапию, гормональные антагонисты, гормоны и их рилизинг-факторы, тиреоидные и антитиреоидные лекарственные средства, эстрогены и прогестины, андрогены, адренокортикотропные гормоны; адренокортикоидные стероиды и их синтетические аналоги; ингибиторы синтеза и действия адренокортикоидных гормонов, инсулин, пероральные гипогликемические агенты, и фармакологию эндокринной поджелудочной железы, агенты, действующие на кальцификацию и ремоделирование кости: кальций, фосфат, паратироидный гормон, витамин D, кальцитонин, витамины, такие как водорастворимые витамины, комплекс витаминов В, аскорбиновая кислота, жирорастворимые витамины, витамины А, К и Е, факторы роста, цитокины, хемокины, агонисты и антагонисты мускариновых рецепторов; агенты антихолинэстеразы; агенты, действующие на нервно-мышечные соединения и/или вегетативные ганглии; катехоламины, симпатомиметические лекарственные средства и агонисты или антагонисты адренергических рецепторов; и агонисты и антагонисты рецепторов 5-гидрокситриптамина (5-ГТ, серотонин).
Терапевтические агенты также включают средства против боли и воспаления, такие как гистамин и антагонисты гистамина, брадикинин и антагонисты брадикинина, 5-гидрокситриптамин (серотонин), липидные вещества, полученные при биотрансформации продуктов селективного гидролиза мембранных фосфолипидов, эйкозаноиды, простагландины, тромбоксаны, лейкотриены, аспирин, нестероидные противовоспалительные средства, обезболивающие-жаропонижающие средства, агенты, ингибирующие синтез простагландинов и тромбоксанов, селективные ингибиторы индуцируемой циклооксигеназы, селективные ингибиторы индуцируемой циклооксигеназы-2, физиологически активные вещества, гормоны паракрина, соматостатин, гастрин, цитокины, опосредующие взаимодействия, входящие в гумпероральную и клеточную иммунную реакцию, липид-производные физиологически активные вещества, эйкозаноиды, β-адренергические агонисты, ипратропий, глюкокортикоиды, метилксантины, блокираторы натриевого канала, агонисты опиоидных рецепторов, блокираторы кальциевого канала, стабилизаторы мембран и ингибиторы лейкотриена.
Дополнительные терапевтические средства, рассмотренные в настоящем документе, включают диуретики, вазопрессин, агенты, действующие на удержание воды в почках, ренин, ангиотензин, агенты, пригодные для лечения ишемии миокарда, антигипертонические средства, ингибиторы ферментов, преобразующих ангиотензин, антагонисты β-адренергического рецептора, средства для лечения гиперхолестеринемии и средства для лечения дислипидемии.
Другие рассмотренные терапевтические средства включают лекарственные средства, пригодные для регуляции желудочной кислотности, агенты для лечения пептической язвы, агенты для лечения желудочно-пищеводного рефлюксов, прокинетические агенты, противорвотные средства, агенты, используемые при синдроме раздраженной толстой кишки, средства, используемые при диарее, средства, используемые при запоре, средства, используемые при воспалительной болезни кишечника, средства, используемые при желчной болезни, средства, используемые при заболевании поджелудочной железы. Терапевтические средства, используемые для лечения протозойных инфекций, препараты для лечения малярии, амебиаза, лямблиоза, трихомониаза, трипаносомоза, и/или лейшманиоза, и/или препараты, используемые в химиотерапии гельминтозов. Другие терапевтические агенты включают противомикробные препараты, сульфаниламиды, триметоприм-сульфаметоксазол хинолоны и агенты против инфекций мочевыводящих путей, пенициллины, цефалоспорины и другие, β-лактамные антибиотики, агенты, содержащие аминогликозиды, ингибиторы синтеза белка, препараты, используемые в химиотерапии туберкулеза, комплекса Mycobacterium avium и проказы, противогрибковые препараты, противовирусные средства, включая неретровирусные средства и антиретровирусные препараты.
Примеры терапевтических антител, которые можно комбинировать с соединениями настоящего изобретения, включают, не ограничиваясь, анти-рецепторные антитела тирозин-киназы (цетуксимаб, панитумумаб, трастузумаб), анти CD20 антитела (ритуксимаб, тозитумомаб) и другие антитела, такие как алемтузумаб, бевацизумаб и гемтузумаб.
Более того, в представленных в настоящем документе способах рассматриваются терапевтические агенты, используемые для иммуномодулирования, такие как иммуномодуляторы, иммуноподавляющие агенты, толерогены и иммуностимулирующие средства. Кроме того, терапевтические агенты, действующие на кровь и кроветворные органы, гематопоэтические средства, факторы роста, минералы и витамины, антикоагулянты, тромболиты и антитромбоцитарные лекарственные средства.
Дополнительные терапевтические средства, которые можно комбинировать с соединениями по настоящему изобретению, можно найти в Goodman and Gilman "The Pharmacological Basis of Therapeutics", tenth edition edited by Hardman, Limbird and Gilman, или в the Physician's Desk Reference, которые включены в настоящую заявку путем ссылки в полном объеме.
Соединения, описанные в настоящем документе, могут использоваться в комбинации с агентами, описанными в настоящем документе, или с другими пригодными агентами, в зависимости от состояния, подлежащего лечению. Поэтому в некоторых воплощениях настоящего изобретения соединения вводят совместно с другими агентами, как описано выше. При использовании в комбинационной терапии, соединения, описанные в настоящем документе, могут вводиться со вторым агентом одновременно или раздельно. Комбинированное введение может включать одновременное введение двух агентов в одной лекарственной форме, одновременное введение в отдельных лекарственных формах или раздельное введение. То есть соединение, описанное в настоящем документе и любой из агентов, описанных выше, могут быть смешаны вместе в одной лекарственной форме и вводиться одновременно. Альтернативно, соединение по настоящему изобретению и любой из агентов, описанных выше, могут вводиться одновременно, при этом оба агента присутствуют в виде отдельных композиций. В другом варианте, соединение по настоящему изобретению может вводиться непосредственно вслед за введением любого из агентов, описанных выше, или наоборот. В протоколе раздельного введения соединение по настоящему изобретению и один из агентов, описанных выше, может вводиться с интервалом в несколько минут, или в несколько часов, или в несколько дней.
Примеры и препараты, представленные ниже, дополнительно иллюстрируют и служат примером соединений настоящего изобретения и способов получения таких соединений. Следует понимать, что рамки настоящего изобретения никоим образом не ограничиваются рамками следующих примеров и препаратов. В следующих примерах молекулы с одним хиральным центром, если не указано иное, существуют в виде рацемической смеси. Молекулы с двумя или более хиральных центров, если не указано иное, существуют в виде рацемической смеси диастереомеров. Отдельные энантиомеры/диастереомеры могут быть получены способами, известными специалистам в данной области.
ПРИМЕРЫ
Пример 1: Синтез 3-((4-амино-3-(3-гидроксифенил)-1H-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-o-толилизохинолин-1(2Н)-она (соединение 1613) (способ А).
Схема 14. Синтез 3-((4-амино-3-(3-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 1613).

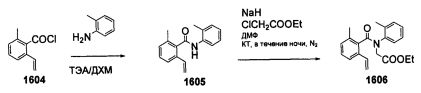



Раствор 2-амино-6-метилбензойной кислоты (1601) (106,5 г, 705 ммоль) в H2O (200 мл) охладили до 0-5°С, медленно добавили конц. НСl (250 мл). Раствор перемешивали в течение 15 минут при 0-5°С. По каплям добавили раствор нитрита натрия (58,4 г, 6,85 моль) в H2O (120 мл) при 0-5°С, и полученную смесь перемешивали в течение 30 минут. Затем полученный раствор добавили к раствору KI (351 г, 2,11 моль) в H2O (200 мл), и перемешивали полученную смесь при комнатной температуре в течение 16 часов. Раствор вылили в ледяную воду (2000 мл) и экстрагировали этилацетатом (3×1000 мл). Объединенный органический слой промыли водным раствором NaOH (15%, 3×200 мл). Водный слой подкислили до рН=1 и экстрагировали этилацетатом (3×1000 мл). Объединенный органический слой высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили желаемый продукт, 2-йод-6-метилбензойную кислоту (1602) (145 г, выход 79%) в виде твердого желтого вещества.
К перемешанной смеси 2-йод-6-метилбензойной кислоты (1602) (105 г, 400 ммоль), Pd(OAc)2 (27 г, 120 ммоль) и PPh3 (63 г, 240 моль) в ТГФ (1000 мл) при комнатной температуре добавили трибутил(винил)олово (152 г, 480 ммоль). Полученную смесь нагревали с дефлегматором в течение ночи. Смесь оставили остывать до комнатной температуры, отфильтровали через силикагель (10 г), а затем концентрировали in vacuo. Остаток вылили в ледяную воду (1000 мл) и экстрагировали этилацетатом (3×1000 мл). Объединенный органический слой промыли водным раствором NaOH (15%, 5×200 мл). Объединенный водный слой подкислили до рН=1 и экстрагировали этилацетатом (3×1000 мл). Объединенный органический слой высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили желаемый продукт 2-метил-6-винилбензойную кислоту (1603) (61 г, выход 95%) в виде твердого желтого вещества.
Смесь 2-метил-6-винилбензойной кислоты (1603) (56 г, 350 ммоль) и тионилхлорида (208 г, 1750 ммоль) в толуоле (400 мл) перемешивали с дефлегматором в течение 2 часов. Смесь концентрировали in vacuo и получили желаемый продукт, 2-метил-6-винилбензоилхлорид (1604) (63 г, выход 95%) в виде желтого маслянистого вещества. Полученный продукт использовали непосредственно на следующем этапе без очистки.
Смесь о-толуидина (45 г, 420 ммоль) и триэтиламина (71 г, 70 ммоль) в CH2Cl2 (300 мл) перемешивали в течение 10 минут при комнатной температуре. К этой смеси добавили 2-метил-6-винилбензоилхлорид (1604) (63 г, 35 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 30 минут. Раствор вылили в воду (300 мл) и экстрагировали CH2Cl2 (3×200 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили неочищенный продукт. Неочищенный продукт суспендировали в IPE (изопропиловый эфир) (300 мл), перемешивали с дефлегматором в течение 30 минут, а затем охладили до 0-5°С. Осадок собрали фильтрацией и дополнительно высушили in vacuo, и получили желаемый продукт 2-метил-N-о-толил-6-винилбензамид (1605) (81 г, выход 80%) в виде твердого желтого вещества.
К раствору 2-метил-N-о-толил-6-винилбензамида (1605) (80 г, 320 ммоль) в ДМФ (250 мл) при комнатной температуре медленно добавили NaH (60% в минеральном масле, 25,6 г, 640 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 30 минут. К этой смеси добавили этилхлорацетат (78 г, 640 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 2 ч. Раствор вылили в воду (500 мл) и экстрагировали этилацетатом (3×200 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo. Неочищенный продукт суспендировали в МеОН (160 мл), перемешивали с дефлегматором в течение 10 минут, а затем охладили до 0-5°С. Осадок собрали фильтрацией и дополнительно высушили in vacuo, и получили желаемый продукт, этил-2-(2-метил-N-о-толил-6-винилбензамидо)ацетат (1606) (67 г, выход 62%) в виде твердого белого вещества.
К перемешанной смеси этил 2-(2-метил-N-о-толил-6-винилбензамидо)ацетата (1606) (67 г, 200 ммоль) в 1,4-диоксане (300 мл) и H2O (100 мл) при комнатной температуре добавили тетроксид осмия (20 мг) и перемешивали при комнатной температуре в течение 30 минут. К этой смеси добавили периодат натрия (86 г, 400 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 16 часов. Реакционную смесь отфильтровали через силикагель (10 г), фильтрат экстрагировали этилацетатом (3×200 мл). Объединенные органические слои промыли насыщенным солевым раствором (100 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo и дополнительно высушили остаток in vacuo, получили желаемый продукт этил-2-(2-формил-6-метил-N-о-толилбензамидо)ацетат (1607) (38 г, выход 57%) в виде твердого желтого вещества.
К перемешанному раствору этил-2-(2-формил-6-метил-N-о-толилбензамидо)ацетата (1607) (38 г, 112 ммоль) в EtOH (200 мл) и этилацетате (100 мл) при комнатной температуре добавили карбонат цезия (22 г, 112 ммоль). Полученную смесь дегазировали и пропустили через нее аргон три раза, а затем перемешивали при 50°С в течение 5 ч. Смесь оставили остывать до комнатной температуры, отфильтровали через силикагель (10 г), а фильтрат концентрировали in vacuo. Остаток вылили в ледяную воду (200 мл) и экстрагировали этилацетатом (3×200 мл). Объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo. Неочищенный продукт суспендировали в IPE (120 мл), нагревали с дефлегматором в течение 10 минут, а затем охладили до 0-5°С. Осадок собрали фильтрацией и дополнительно высушили in vacuo и получили желаемый продукт этил-8-метил-1-оксо- 2-о-толил-1,2-дигидроизохинолин-3-карбоксилат (1608) (28 г, выход 77%) в виде твердого белого вещества.
К перемешанному раствору гидрида лития-алюминия (8,28 г, 218 моль) в безводном ТГФ (500 мл) при -78°С в атмосфере азота медленно, в течение 10 минут, добавили этил 8-метил-1-оксо-2-толил-1,2-дигидроизохинолин-3-карбоксилат (1608) (28 г, 87 ммоль). Полученную смесь оставили нагреваться до -30°С, перемешивали в течение 30 минут, и данные ТСХ показали завершение реакции. Реакционную смесь охладили до -78°С и медленно добавили воду (50 мл). Смесь оставили нагреваться до комнатной температуры, отфильтровали через силикагель (10 г), а затем концентрировали in vacuo. Неочищенный продукт вылили в воду (200 мл) и экстрагировали этилацетатом (3×200 мл). Объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo. Неочищенный продукт суспендировали в этилацетате (30 мл) и перемешивали в течение 10 минут. Твердое вещество собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт 3-(гидроксиметил)-8-метил-2-о-толилизохинолин-1(2Н)-он (1609) (22 г, выход 92%) в виде твердого белого вещества.
К перемешанному раствору ДМФ (11,5 г, 158 моль) в ацетонитриле (200 мл) при 0°С медленно добавили PBr3 (25,6 г, 95 ммоль), и полученную смесь перемешивали при 0°С в течение 30 минут. Медленно добавили 3-(гидроксиметил)-8-метил-2-о-толилизохинолин-1(2Н)-он (1609) (22 г, 78,8 ммоль). Затем реакционную смесь оставили нагреваться до комнатной температуры и перемешивали в течение 30 минут. Медленно добавили насыщенный водный раствор NaHCO3 (50 мл) и экстрагировали этилацетатом (3×200 мл). Объединенный органический слой промыли насыщенным солевым раствором, высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo. Неочищенный продукт суспендировали в IPE (50 мл) и затем перемешивали в течение 10 минут. Осадок собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт 3-(бромметил)-8-метил-2-о-толилизохинолин-1(2Н)-он (1610) (21 г, выход 80%) в виде твердого белого вещества.
В безводном ДМФ (150 мл) растворили 3-йод-1Н-пиразоло[3,4-d]пиримидин-4-амин (108) (10,8 г, 41,4 ммоль) и трет-бутоксид калия (4,4 г, 40 ммоль) и перемешивали при комнатной температуре в течение 30 минут. Добавили 3-(бромметил)-8-метил-2-о-толилизохинолин-1(2Н)-он (1610) (13,7 г, 40 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, вылили в ледяную воду (300 мл), а затем экстрагировали этилацетатом (3×200 мл). Объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали примерно до 100 мл in vacuo, осадок собрали фильтрацией и получили первую порцию заданного продукта 3-((4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил-8-метил-2-о-толилизохинолин-1 (2Н)-она (1611) (12 г, выход 60%) в виде твердого белого вещества. Фильтрат концентрировали in vacuo, остаток очистили колоночной флэш-хроматографией на силикагеле (2-20% МеОН/ДХМ) и получили вторую порцию заданного продукта, 3-((4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил-8-метил-2-о-толилизохинолин-1(2Н)-она (1611) (6 г, выход 30%) в виде твердого белого вещества.
3-((4-Амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил-8-метил-2-о-толилизохинолин-1(2Н)-он (1611) (13 г, 24,9 ммоль) и 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенол (1612) (6,6 г, 30 ммоль) растворили в ДМФ-EtOH-Н2О (120 мл, 40 мл, 40 мл). Последовательно добавили Pd(OAc)2 (1,684 г, 7,5 ммоль), PPh3 (3,935 г, 15 ммоль) и Na2CO3(13,25 г, 125 ммоль). Полученную смесь дегазировали и три раза пропустили через нее аргон, а затем перемешивали при 100°С в течение 1 часа. Смесь оставили остывать до комнатной температуры, отфильтровали через силикагель (10 г), а затем концентрировали in vacuo. Остаток очистили колоночной флэш-хроматографией на силикагеле (2-20% МеОН/ДХМ) и получили продукт (1613, соединение 13 в таблице 4) (9 г, выход 76%) в виде слегка желтого твердого вещества. Затем полученный продукт суспендировали в EtOH (100 мл) и нагревали с дефлегматором в течение 30 минут. Смесь оставили остывать до комнатной температуры, и собрали твердое вещество фильтрацией. Затем твердое вещество суспендировали в этилацетате (100 мл) и перемешивали в течение ночи. Осадок собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт, 3-((4-амино-3-(3-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-он (1613) (8,4 г, выход 69%) в виде твердого белого вещества.
Пример 2: Синтез 3-((4-амино-3-(3-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-o-толилизохинолин-1(2Н)-она (соединение 1613) (способ В).
Схема 15. Описан синтез 3-((4-амино-3-(3-гидроксифенил)-1Н-пиразоло [3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 1613) по способу В

В безводном ДМФ (150 мл) растворили 3-(3-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-4-амин (1701) (964 мг, 4 ммоль) и трет-бутоксид калия (0,44 г, 4 ммоль) и перемешивали при комнатной температуре в течение 30 минут. Добавили 3-(бромметил)-8-метил-2-о-толилизохинолин-1(2Н)-он (1610) (1,37 г, 4,0 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, вылили в ледяную воду (30 мл), а затем экстрагировали этилацетатом (3×50 мл). Объединенный органический слой промыли насыщенным солевым раствором (25 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, остаток очистили колоночной флэш-хроматографией на силикагеле (2-20% МеОН/ДХМ) и получили желаемый продукт, 3-((4-амино-3-(3-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил-8-метил-2-о-толилизохинолин-1(2Н)-он (1702) (1,4 г, выход 70%) в виде твердого белого вещества.
К раствору 3-((4-амино-3-(3-метоксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-она (1702) (100 мг, 0,2 ммоль) в CH2Cl2 (20 мл) при -78°С в атмосфере азота добавили BBr3 (1 мл), и перемешивали полученную смесь при -78°С в течение 3 ч. Смесь оставили нагреваться до комнатной температуры, вылили в ледяную воду (200 мл) и экстрагировали этилацетатом (3×50 мл). Объединенный органический слой промыли насыщенным солевым раствором (20 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, остаток очистили колоночной флэш-хроматографией на силикагеле (10-50% МеОН/CH2Cl2) и получили желаемый продукт, 3-((4-амино-3-(3-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил-8-метил-2-о-толилизохинолин-1(2Н)-он (1613) (87 мг, выход 91%) в виде твердого белого вещества.
Пример 3: Синтез (R)-3-((4-амино-3-(3-гидроксибут-1-инил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 1802).
Схема 16. Описан синтез (R)-3-((4-амино-3-(3-гидроксибут-1-инил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 1802).

3-((4-Амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил-8-метил-2-о-толилизохинолин-1(2Н)-он (1611) (522 мг, 1 ммоль) и (R)-бут-3-ин-2-ол (84 мг, 1,2 ммоль) растворили в безводном ТГФ (40 мл). Смесь дегазировали и через нее три раза пропустили азот. Последовательно добавили Pd(PPh3)2Cl2 (12 мг, 0,1 ммоль), CuI (47 мг, 0,25 ммоль) и (изо-Pr)2NH (505 мг, 5 ммоль). Полученную смесь дегазировали и три раза пропустили через нее аргон, а затем перемешивали с дефлегматором в течение 4 ч. Смесь оставили остывать до комнатной температуры, отфильтровали через силикагель (10 г), а затем концентрировали in vacuo. Остаток очистили колоночной флэш-хроматографией на силикагеле (2-20% МеОН/ДХМ) и получили желаемый продукт, 3 (R)-3-((4-амино-3-(3-гидроксибут-1-инил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил-8-метил-2-о-толилизохинолин-1(2Н)-он (1802, соединение 37 в таблице 4) (324 мг, выход 70%) в виде слегка желтого твердого вещества.
Пример 4: Синтез 3-((6-амино-9Н-пурин-9-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 1902).
Схема 17. Описан синтез 3-((6-амино-9Н-пурин-9-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 1902).

9Н-Пурин-6-амин (1901) (540 мг, 4,0 ммоль) растворили в безводном ДМФ (20 мл). Добавили NaH (60% в минеральном масле, 160 мг, 4,0 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Добавили 3-(бромметил)-8-метил-2-о-толилизохинолин-1(2Н)-он (1610) (1,37 г, 4,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, вылили в ледяную воду (30 мл), а затем экстрагировали этилацетатом (3×50 мл). Объединенный органический слой промыли насыщенным солевым раствором (25 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, остаток очистили колоночной флэш-хроматографией на силикагеле (2-20% МеОН/ДХМ) и получили желаемый продукт, 3-((6-амино-9Н-пурин-9-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-он (1902, соединение 5 в таблице 4) (1,1 г, выход 70%) в виде твердого белого вещества.
Пример 5: Синтез 3-((4-амино-3-(3-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-2-изопропил-8-метилизохинолин-1(2Н)-она (соединение 2009).
Схема 18. Описан синтез 3-((4-амино-3-(3-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-2-изопропил-8-метилизохинолин-1(2Н)-она (соединение 2009).
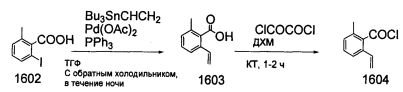


К перемешанной смеси 2-йод-6-метилбензойной кислоты (1602) (105 г, 400 ммоль), Pd(OAc)2 (27 г, 120 ммоль) и PPh3 (63 г, 240 моль) в ТГФ (1000 мл) при комнатной температуре добавили трибутил(винил)олово (152 г, 480 ммоль). Полученную смесь нагревали с дефлегматором в течение ночи. Смесь оставили остывать до комнатной температуры, отфильтровали через силикагель (10 г), а затем концентрировали in vacuo. Остаток вылили в ледяную воду (1000 мл) и экстрагировали этилацетатом (3×1000 мл). Объединенный органический слой промыли водным раствором NaOH (15%, 5×200 мл). Объединенный водный слой подкислили до рН=1 и экстрагировали этилацетатом (3×1000 мл). Объединенный органический слой высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили желаемый продукт, 2-метил-6-винилбензойную кислоту (1603) (61 г, выход 95%) в виде твердого желтого вещества.
Смесь 2-метил-6-винилбензойной кислоты (1603) (56 г, 350 ммоль) и тионилхлорида (208 г, 1750 ммоль) в толуоле (400 мл) перемешивали с дефлегматором в течение 2 ч. Смесь концентрировали in vacuo и получили желаемый продукт, 2-метил-6-винилбензоилхлорид (1604) (63 г, выход 95%) в виде желтого маслянистого вещества. Полученный продукт использовали непосредственно на следующем этапе без очистки.
Пропан-2-амин (2001) (59 г, 1,0 моль) и этилхлорацетат (122 г, 1,0 моль) растворили в толуоле (200 мл) и перемешивали смесь с дефлегматором в течение 2 ч. Реакционную смесь оставили остывать до комнатной температуры, вылили в ледяную воду (500 мл), и экстрагировали этилацетатом (3×250 мл). Объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, остаток очистили колоночной флэш-хроматографией на силикагеле (10-50% ЭА/ПЭ) и получили продукт, этил 2-(изопропиламино)ацетат (2002) (70 г, выход 51%) в виде маслянистого вещества.
Этил-2-(изопропиламино)ацетат (2002) (14,5 г, 100 ммоль) и триэтиламин (200 г, 200 ммоль) растворили в CH2Cl2 (300 мл) и перемешивали смесь в течение 10 минут при комнатной температуре. Добавили 2-метил-6-винилбензоилхлорид (1604) (18 г, 100 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 30 минут. Реакционную смесь вылили в воду (300 мл) и экстрагировали CH2Cl2 (3×200 мл). Объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили неочищенный продукт. Неочищенный продукт суспендировали в IPE (изопропиловый эфир) (300 мл), перемешивали с дефлегматором в течение 30 минут, а затем охладили до 0-5°С. Осадок собрали фильтрацией и дополнительно высушили in vacuo и получили желаемый продукт, этил 2-метил-(N-изопропил-2-метил-6-винилбензамидо)ацетат (2003) (14,5 г, выход 50%) в виде твердого желтого вещества.
К перемешанному раствору этил 2-(N-изопропил-2-метил-6-винилбензамидо)ацетата (2003) (14,0 г, 48,0 ммоль) в 1,4-диоксане (100 мл) и H2O (30 мл) добавили тетроксид осмия (20 мг), и полученную смесь перемешивали при комнатной температуре в течение 30 минут. К этой смеси добавили натрия периодат (22 г, 100 ммоль), и затем перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь отфильтровали через силикагель (10 г), фильтрат экстрагировали этилацетатом (3×200 мл). Объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo и дополнительно высушили остаток in vacuo, получили желаемый продукт, этил 2-(2-формил-N-изопропил-6-метилбензамидо)ацетат (2004) (8,33 г, выход 57%) в виде твердого желтого вещества.
К перемешанному раствору этил 2-(2-формил-N-изопропил-6-метилбензамидо)ацетата (2004) (8,3 г, 28,0 ммоль) в EtOH (100 мл) и этилацетате (50 мл) при комнатной температуре добавили карбонат цезия (5,9 г, 30 ммоль).
Полученную смесь дегазировали и пропустили через нее аргон три раза, а затем перемешивали при 50°С в течение 5 ч. Смесь оставили остывать до комнатной температуры, отфильтровали через силикагель (10 г), а фильтрат концентрировали in vacuo. Остаток вылили в ледяную воду (200 мл) и экстрагировали этилацетатом (3×200 мл). Объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo. Неочищенный продукт суспендировали в IPE (120 мл), перемешивали с дефлегматором в течение 10 минут, а затем охладили до 0-5°С. Осадок собрали фильтрацией и дополнительно высушили in vacuo и получили желаемый продукт, этил 2-изопропил-8-метил-1-оксо-1,2-дигидроизохинолин-3-карбоксилат (2005) (5,35 г, выход 70%) в виде твердого белого вещества.
К перемешанному раствору гидрида лития-алюминия (2,88 г, 76 моль) в безводном ТГФ (200 мл) при -78°С в атмосфере азота медленно, в течение 10 минут, добавили этил 2-изопропил-8-метил-1-оксо-1,2-дигидроизохинолин-3-карбоксилат (2005) (5,2 г, 19 ммоль). Полученную смесь оставили нагреваться до -30°С, перемешивали в течение 30 минут, и данные ТСХ показали завершение реакции. Реакционную смесь охладили до -78°С и медленно добавили воду (50 мл). Смесь оставили нагреваться до комнатной температуры, отфильтровали через силикагель (10 г), концентрировали фильтрат in vacuo. Неочищенный продукт вылили в воду (200 мл) и экстрагировали этилацетатом (3×200 мл). Объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo. Неочищенный продукт суспендировали в этилацетате (30 мл) и перемешивали в течение 10 минут. Твердое вещество собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт, 3-(гидроксиметил)-2-изопропил-8-метилизохинолин-1 (2Н)-он (2006) (3,51 г, выход 80%) в виде твердого белого вещества.
К раствору 3-(гидроксиметил)-2-изопропил-8-метилизохинолин-1(2Н)-она (2006) (1,61 г, 7,0 ммоль) в CH2Cl2, добавили PPh3 (3,67 г, 14,0 ммоль) и перемешивали смесь при комнатной температуре в течение 30 минут. Смесь охладили до 0°С, и частями добавили CBr4 (4,64 г, 14,0 ммоль). Полученную смесь перемешивали при температуре от 0°С до комнатной температуры в течение 30 минут, а затем концентрировали in vacuo. Неочищенный продукт очистили колоночной флэш-хроматографией на силикагеле (30-50% ЭА/ПЭ) и получили желаемый продукт, 3- (бромметил)-2-изопропил-8-метилизохинолин-1(2Н)-он (2007) (1,65 г, выход 80%) в виде твердого белого вещества.
В безводном ДМФ (20 мл) растворили 3-йод-1H-пиразоло[3,4-d]пиримидин-4-амин (108) (1,3 г, 5 ммоль) и трет-бутоксид калия (0,55 г, 5 ммоль) и перемешивали при комнатной температуре в течение 30 минут, а затем добавили 3-(бромметил)-2-изопропил-8-метилизохинолин-1(2Н)-он (2007) (1,47 г, 5 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, вылили в ледяную воду (30 мл), а затем экстрагировали этилацетатом (3×50 мл). Объединенный органический слой промыли насыщенным солевым раствором (25 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, остаток очистили колоночной флэш-хроматографией на силикагеле (2-20% МеОН/ДХМ) и получили желаемый продукт, 3-((4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил-2-изопропил-8-метилизохинолин-1(2Н)-он (2008) (1,66 г, выход 70%) в виде твердого белого вещества. К перемешанной смеси 3-((4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-2-изопропил-8-метилизохинолин-1(2Н)-она (2008) (95 мг, 0,2 ммоль) и 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенола (66 мг, 0,3 ммоль) в ДМФ-EtOH-Н2О (3:1:1, 20 мл), последовательно добавили Pd(OAc)2 (16 мг, 0,075 ммоль), PPh3 (39,3 мг, 0,15 ммоль) и Na2CO3 (132 мг, 1,25 ммоль). Полученную смесь дегазировали и пропустили через нее аргон три раза, а затем перемешивали при 100°С в течение 1 ч. Смесь оставили остывать до комнатной температуры, отфильтровали через силикагель (10 г) и концентрировали in vacuo. Остаток очистили колоночной флэш-хроматографией на силикагеле (2-20% МеОН/ДХМ) и получили желаемый продукт, 3-((4-амино-3-(3-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил-2-изопропил-8-метилизохинолин-1(2Н)-он (2009, соединение 62 в таблице 4) (53 мг, выход 61%) в виде слегка желтого твердого вещества.
Пример 6: Синтез 8-метил-3-((метил(9Н-пурин-6-ил)амино)метил)-2-o-толилизохинолин-1 (2Н)-она.
Схема 19. Описан синтез 8-метил-3-((метил(9Н-пурин-6-ил)амино)метил)-2-o-толилизохинолин-1(2Н)-она (соединение 4004).
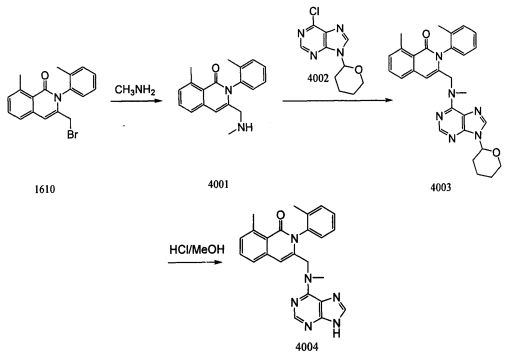
3-(Бромметил)-8-метил-2-о-толилизохинолин-1(2Н)-он (342 мг, 1,0 ммоль) 1610 растворили в растворе метиламина (100 мл) и перемешивали в течение 2 ч. Смесь вылили в ледяную воду (200 мл) и экстрагировали этилацетатом (3×50 мл). Объединенный органический слой промыли насыщенным солевым раствором (20 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили желаемый продукт, 8-метил-3-((метиламино)метил)-2-о-толилизохинолин-1(2Н)-он (4001) (250 мг, выход 86%) в виде твердого желтого вещества. Полученный продукт использовали непосредственно на следующем этапе без очистки.
8-Метил-3-((метиламино)метил)-2-о-толилизохинолин-1(2Н)-он (233 мг, 0,8 ммоль) (4001) и 6-хлор-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин (4002) (238 мг, 1,0 ммоль) растворили в EtOH (50 мл), и перемешивали полученную смесь с дефлегматором в течение 2 ч. Смесь оставили остывать до комнатной температуры и концентрировали in vacuo. Остаток очистили колоночной флэш-хроматографией на силикагеле (2-20% МеОН/ДХМ) и получили желаемый продукт, 8-метил-3-((метил(9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-ил)амино)метил-2-о-толилизохинолин-1(2Н)-он (4003) (200 мг, выход 51%) в виде слегка желтого твердого вещества.
8-Метил-3-((метил(9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-ил)амино)метил)-2-о-толилизохинолин-1(2Н)-он (4003) (180 мг, 0,36 ммоль) растворили в МеОН (НСl) (50 мл) и перемешивали смесь при комнатной температуре в течение 2 ч. К реакционной смеси добавили водный раствор NaHCO3 и довели значение рН до 9. Смесь отфильтровали и концентрировали фильтрат in vacuo, получили желаемый продукт, 8-метил-3-((метил(9Н-пурин-6-ил)амино)метил)-2-о-толилизохинолин-1(2Н)-он (4004, соединение 184 в таблице 4) (80 мг, выход 54%) в виде твердого желтого вещества.
Пример 7: Синтез 3-(1-(9Н-пурин-6-иламино)этил)-8-метил-2-о-толилизохинолин-1(2Н)-она.
Схема 20. Описан синтез 3-(1-(9Н-пурин-6-иламино)этил)-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 4106).

К перемешанному раствору 3-(гидроксиметил)-8-метил-2-о-толилизохинолин-1(2Н)-она 1609 (2,79 г, 10 ммоль) в CH2Cl2 (200 мл), добавили MnO2 (5 г), и перемешивали полученную смесь с дефлегматором в течение 3 ч. Смесь оставили остывать до комнатной температуры и концентрировали in vacuo. Остаток очистили колоночной флэш-хроматографией на силикагеле (10-50% ЭА/ПЭ) и получили желаемый продукт, 8-метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-карбальдегид 4101 (2,5 г, выход 90%) в виде твердого белого вещества.
8-Метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-карбальдегид 4101 (2,4 г, 8,6 ммоль) растворили в безводном ТГФ (280 мл) и охладили до -78°С в атмосфере азота. Медленно добавили метил MgBr (2 М, 5 мл, 10 ммоль), и полученную смесь перемешивали при -78°С в течение 2 ч. Добавили H2O (5 мл), а затем раствор вылили в ледяную воду (200 мл) и экстрагировали этилацетатом (3×50 мл). Объединенный органический слой промыли насыщенным солевым раствором, высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, и оставшийся продукт очистили колоночной флэш-хроматографией на силикагеле (10-50% ЭА/ПЭ) и получили желаемый продукт, 3-(1-гидроксиэтил)-8-метил-2-о-толилизохинолин-1(2Н)-он 4102 (1,8 г, выход 71%) в виде твердого белого вещества.
К раствору 3-(1-гидроксиэтил)-8-метил-2-о-толилизохинолин-1(2Н)-она 4102 (1,6 г, 5,5 ммоль) в CH2Cl2 добавили PPh3 (2,88 г, 11,0 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Затем к смеси частями добавили СВг4 (3,64 г, 11,0 ммоль) при 0°С. Полученную смесь оставили нагреваться до комнатной температуры, перемешивали 30 минут и концентрировали in vacuo. Неочищенный продукт очистили колоночной флэш-хроматографией на силикагеле (30-50% ЭА/ПЭ) и получили желаемый продукт, 3-(1-бромэтил)-8-метил-2-о-толилизохинолин-1(2Н)-он 4103 (1,8 г, выход 91%) в виде твердого белого вещества. К перемешанному раствору 9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-амина 4103 (436 мг, 2 ммоль) в безводном ДМФ (10 мл) добавили NaH (60% в минеральном масле, 77 мг, 2 ммоль) и перемешивали смесь в течение 30 минут. Добавили 3-(1-бромэтил)-8-метил-2-о-толилизохинолин-1(2Н)-он 4104 (700 мг, 2 ммоль). Смесь перемешивали в течение 2 ч, вылили в ледяную воду (200 мл) и экстрагировали этилацетатом (3×50 мл). Объединенный органический слой промыли насыщенным солевым раствором (20 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, остаток очистили колоночной флэш-хроматографией на силикагеле (10-50% МеОН/ДХМ) и получили желаемый продукт, 8-метил-3-(1-(9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-иламино)этил-2-о-толилизохинолин-1(2Н)-он 4105 (500 мг, выход 51%) в виде твердого белого вещества.
8-Метил-3-(1-(9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-иламино)этил)-2-о-толилизохинолин-1(2Н)-он 4105 (180 мг, 0,36 ммоль) растворили в МеОН (НСl) (50 мл) и перемешивали в течение 2 ч. К реакционной смеси добавили водный раствор NaHCO3 и довели значение рН до 9. Затем смесь отфильтровали и концентрировали фильтрат in vacuo, получили желаемый продукт, 3-(1-(9Н-пурин-6-иламино)этил)-8-метил-2-о-толилизохинолин-1(2Н)-он (4106, соединение 191 в таблице 4) (80 мг, выход 54%) в виде твердого желтого вещества.
Пример 8: Синтез 3-(4-амино-1-((8-метил-1-оксо-2-o-толил-1,2-дигидроизохинолин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенил дигидрофосфата.
Схема 21. Описан синтез 3-(4-амино-1-((8-метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенил дигидрофосфата (соединение 4303).

3-((4-Амино-3-(3-фтор-5-гидроксифенил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-он 4301 (250 мг, 0,5 ммоль) растворили в безводном ТГФ (15 мл) в круглодонной колбе в темноте (закрытой алюминиевой фольгой) и охладили до 0°С в атмосфере аргона. Добавили CBr4 (498 мг, 1,5 ммоль), а затем диэтилфосфит (129 мкл, 1,0 ммоль) и триэтиламин (417 мкл, 1,5 ммоль). Полученную смесь перемешивали в темноте при температуре от 0°С до комнатной температуры в течение 16 ч. Затем смесь разделили между этилацетатом и насыщенным солевым раствором. Органический слой высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на силикагеле, элюируя метанолом и дихлорметаном, получили желаемый продукт, 3-(4-амино-1-((8-метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенил диэтилфосфат 4302 (200 мг, выход 62%) в виде грязновато-белого твердого вещества.
3-(4-Амино-1-((8-метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенил диэтилфосфат 4302 (170 мг, 0,26 ммоль) растворили в безводном CH3CN (5 мл) и охладили до 0°С в атмосфере аргона. Медленно, через шприц, добавили TMSBr (0,34 мл, 2,64 ммоль) и перемешивали полученную смесь при температуре от 0°С до комнатной температуры в течение 16 ч. Данные ЖХ-МС показали наличие небольшого количества исходного материала, поэтому добавили дополнительное количество TMSBr (0,1 мл) и перемешивали при комнатной температуре в течение 5 ч. Данные ЖХ-МС показали полное превращение. Смесь концентрировали in vacuo, остаток растворили в Et2O (10 мл) и H2O (0,5 мл) и перемешивали в течение 30 минут. Смесь концентрировали in vacuo и получили желаемый продукт, 3-(4-амино-1-((8-метил-1оксо-2-о-толил-1,2-дигидроизохинолин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-3-ил)-5-фторфенил дигидрофосфат 4303 (140 мг, выход 91%).
Пример 9: Синтез 3-((4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 1611).
Схема 22. Описан синтез 3-((4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 1611).

Смесь 2,6-диметилбензойной кислоты (соединение 4401) (60 г, 400 ммоль) и оксалилхлорида (101 г, 800 ммоль) в CH2Cl2 (400 мл) перемешивали при комнатной температуре в течение 2 ч. Смесь концентрировали in vacuo и получили желаемый продукт, 2,6-диметилбензоилхлорид (соединение 4402) (64 г, выход 95%) в виде желтого маслянистого вещества. Полученный продукт использовали непосредственно на следующем этапе без очистки.
Смесь о-толуидина (45 г, 420 ммоль) и триэтиламина (71 г, 700 ммоль) в CH2Cl2 (300 мл) перемешивали при комнатной температуре в течение 10 минут. К этой смеси по каплям добавили 2,6-диметилбензоилхлорид (соединение 4402) (64 г, 400 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь вылили в воду (300 мл), экстрагировали CH2Cl2 (3×200 мл), высушили над безводным Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили неочищенный продукт. Неочищенный продукт суспендировали в изопропиловом эфире (300 мл), перемешивали с дефлегматором в течение 30 минут, а затем охладили до 0-5°С.Твердое вещество собрали фильтрацией и дополнительно высушили in vacuo, и получили желаемый продукт, 2,6-диметил-N-о-толилбензамид (соединение 4403) (81 г, выход 80%) в виде твердого желтого вещества.
К перемешанному раствору 2,6-диметил-N-о-толилбензамида (соединение 4403) (23,9 г, 0,1 моль, 1 экв.) и гексаметилфосфорамида (НМРА) (17.9 г, 0,1 моль, 1 экв.) в безводном ТГФ (250 мл) при -78°С в атмосфере аргона осторожно добавили н-бутиллитий (100 мл, 2,5 М, 0,25 моль, 2,5 экв.) в течение 1 часа, и поддерживали температуру реакции ниже -60°С при добавлении. Полученную смесь перемешивали при -78°С в течение 1 ч., затем быстро добавили диэтилоксалат (17,6 г, 0,12 моль, 1,2 экв.) (температура реакции увеличилась при добавлении до -20°С). Смесь перемешивали при -5°С в течение 10 минут, а затем погасили водой (100 мл). Неорганическую соль удалили фильтрацией, а фильтрат экстрагировали этилацетатом (2×100 мл). Объединенный органический слой промыли насыщенным солевым раствором (100 мл), высушили над MgSO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили неочищенный продукт в виде полутвердого маслянистого вещества. Неочищенный продукт суспендировали в изопропиловом эфире (100 мл) при комнатной температуре в течение 10 минут. Твердое вещество собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт, этил 3-(3-метил-2-(о-толилкарбамоил)фенил)-2-оксопропаноат (соединение 4404) (16,1 г, выход 47,4%) в виде твердого белого вещества.
3-(3-Метил-2-(о-толилкарбамоил)фенил)-2-оксопропаноат (соединение 4404) (11,0 г, 32,4 ммоль, 1 экв.) растворили в HCl/MeOH (10 М, 100 мл, 10 мл/1 г вещества 4404) и перемешивали с дефлегматором в течение 1 ч. Реакционную смесь концентрировали in vacuo, а остаток суспендировали в этилацетате (10 мл) при комнатной температуре в течение 30 минут. Твердое вещество собрали фильтрацией и дополнительно высушили in vacuo и получили желаемый продукт, этил 8-метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-карбоксилат (соединение 4405) (7,52 г, выход 72,5%) в виде твердого белого вещества.
К перемешанному раствору гидрида лития-алюминия (8,28 г, 218 моль) в безводном ТГФ (500 мл) при -78°С в атмосфере азота медленно, в течение 10 минут, добавили этил 8-метил-1-оксо-2-толил-1,2-дигидроизохинолин-3-карбоксилат (соединение 4405) (28 г, 87 ммоль). Полученную смесь оставили нагреваться до -30°С, перемешивали в течение 30 минут, и данные тонкослойной хроматографии показали завершение реакции. Затем смесь охладили до -78°С и медленно добавили воду (50 мл).
Смесь оставили нагреваться до комнатной температуры, отфильтровали через силикагель (10 г), концентрировали фильтрат in vacuo. Неочищенный продукт вылили в воду (200 мл) и экстрагировали этилацетатом (3×200 мл). Объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo. Неочищенный продукт суспендировали в этилацетате (30 мл) и перемешивали в течение 10 минут. Твердое вещество собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт, 3-(гидроксиметил)-8-метил-2-о-толилизохинолин-1(2Н)-он (соединение 4406) (22 г, выход 92%) в виде твердого белого вещества.
К перемешанному раствору ДМФ (11,5 г, 158 моль) в ацетонитриле (200 мл) при 0°С медленно добавили трибромид фосфора (25,6 г, 95 ммоль), и полученную смесь перемешивали при 0°С в течение 30 минут. Медленно добавили 3-(гидроксиметил)-8-метил-2-o-толилизохинолин-1(2Н)-он (соединение 4406) (22 г, 78,8 ммоль). Затем реакционную смесь оставили нагреваться до комнатной температуры и перемешивали в течение 30 минут. Медленно добавили насыщенный водный раствор NaHCO3 (50 мл) и экстрагировали этилацетатом (3×200 мл). Объединенный органический слой промыли насыщенным солевым раствором, высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo. Неочищенный продукт суспендировали в изопропиловом эфире (50 мл) и затем перемешивали в течение 10 минут. Осадок собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт, 3-(бромметил)-8-метил-2-о-толилизохинолин-1(2Н)-он (соединение 4407) (21 г, выход 80%) в виде твердого белого вещества.
В безводном ДМФ (150 мл) растворили 3-йод-1Н-пиразоло[3,4-d]пиримидин-4-амин (10,8 г, 41,4 ммоль) и трет-бутоксид калия (4,4 г, 40 ммоль) и перемешивали при комнатной температуре в течение 30 минут. Добавили 3-(бромметил)-8-метил-2-о-толилизохинолин-1(2Н)-он (соединение 4407) (13,7 г, 40 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, вылили в ледяную воду (300 мл), а затем экстрагировали этилацетатом (3×200 мл). Объединенный органический слой промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали примерно до 100 мл in vacuo, осадок собрали фильтрацией и получили первую порцию заданного продукта, 3-((4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 1611) (12 г, выход 60%) в виде твердого белого вещества. Фильтрат концентрировали in vacuo, остаток очистили колоночной флэш-хроматографией на силикагеле (2-20% МеОН/ДХМ) и получили вторую порцию заданного продукта, 3-((4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил-8-метил-2-о-толилизохинолин-1(2Н)-она (1611, соединение 6 в таблице 4) (6 г, выход 30%) в виде твердого белого вещества.
Пример 10: Синтез 3-((4-амино-3-(фторметил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 4504).
Схема 23. Описан синтез 3-((4-амино-3-(фторметил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 4504).
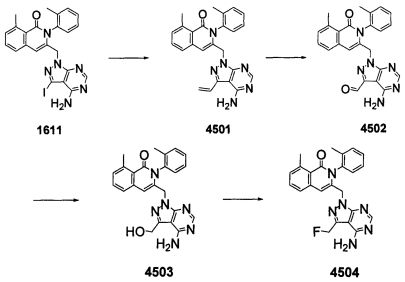
К перемешанной смеси 3-((4-амино-3-йод-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-o-толилизохинолин-1(2Н)-она (соединение 1611) (1,50 г, 2,87 ммоль) и тетракис(трифенилфосфин)палладия (166 мг, 0,14 ммоль) в безводном ДМФ (15 мл) в атмосфере аргона добавили трибутилвинилолово (1,26 мл, 4,31 ммоль), и полученную смесь перемешивали при 80°С в течение 3 ч. Смесь оставили остывать до комнатной температуры, а затем разделили между водой и этилацетатом. Органический слой промыли насыщенным солевым раствором, высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, остаток растерли с минимальным количеством безводного этилового эфира и отфильтровали, получили желаемый продукт, 3-((4-амино-3-винил-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-o-толилизохинолин-1(2Н)-он (соединение 4501) (853 мг, выход 70%) в виде твердого грязновато-белого вещества.
К перемешанному раствору 3-((4-амино-3-винил-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 4501) (853 мг, 2,0 ммоль) в 1,4-диоксане-H2O (3:1, 30 мл) в атмосфере аргона добавили тетроксид осмия (2,5 масс. % в трет-BuOH, 252 мкл, 0,020 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 30 минут. К этой смеси добавили периодат натрия (863 мг, 4,0 ммоль) и полученную смесь перемешивали в течение 3 ч. Реакционную смесь разделили между водой и этилацетатом. Органический слой промыли насыщенным солевым раствором, высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacio и получили желаемый продукт, 4-амино-1-((8-метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-3-карбальдегид в виде твердого вещества коричневатого цвета (соединение 4502) (716 мг, выход 84%).
К перемешанной смеси 4-амино-1-((8-метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-3-карбальдегида в виде твердого вещества коричневатого цвета (соединение 4502) (841 мг, 1,98 ммоль) в безводном МеОН (35 мл) при 0°С в атмосфере аргона частями добавили NaBH4 (89 мг, 2,38 ммоль). Смесь перемешивали при температуре от 0°С до комнатной температуры, а потом разделили между водой и этилацетатом. Органический слой промыли насыщенным солевым раствором, высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, получили желаемый продукт, 3-((4-амино-3-(гидроксиметил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-он (соединение 4503) (626 мг, выход 74%) в виде темно-коричневого твердого вещества.
К перемешанной суспензии 3-((4-амино-3-(гидроксиметил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-она (соединение 4503) (50 мг, 0,12 ммоль) в безводном ДХМ (2 мл) при 0°С в атмосфере аргона медленно добавили диэтиламиносеры трифторид (ДАСТ, 77 мкл, 0,59 ммоль), и полученную смесь перемешивали при температуре от 0°С до комнатной температуры в течение 5 ч. Реакцию погасили водой и экстрагировали этилацетатом. Объединенный органический слой промыли насыщенным солевым раствором, высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, остаток очистили препаративной ТСХ (7% МеОН/ДХМ) и получили желаемый продукт, 3-((4-амино-3-(фторметил)-1Н-пиразоло[3,4-d]пиримидин-1-ил)метил)-8-метил-2-о-толилизохинолин-1(2Н)-он (4504, соединение 310 в таблице 4) (10,3 мг, выход 20%) в виде твердого белого вещества.
Пример 11: Синтез 4-амино-1-((8-метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-3-карбоксамида (соединение 4602).
Схема 24. Описан синтез 4-амино-1-((8-метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-3-карбоксамида (соединение 4602).

К перемешанному раствору 4-амино-1-((8-метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-3-карбальдегида (соединение 4502) (400 мг, 0,94 ммоль) в трет-BuOH (1,8 мл) последовательно добавили раствор NaH2PO4(3,90 г, 28,27 ммоль) в воде (4,8 мл), метил-2-бутен (1,0 мл) и (по каплям) раствор NaClO2 (767 мг, 6,78 ммоль) в воде (4,8 мл). Смесь перемешивали при комнатной температуре в течение 3 часов в атмосфере аргона. Бледно-желтый раствор подкислили водным раствором HCl (2 М, 4 мл) до рН=2 и экстрагировали этилацетатом. Объединенный органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, и растерли остаток с безводным этиловым эфиром и этилацетатом. Твердое вещество собрали фильтрацией и получили желаемый продукт, 4-амино-1-((8-метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-3-карбоновую кислоту (соединение 4601) (200 мг, выход 47%) в виде твердого желтого вещества.
К перемешанному раствору 4-амино-1-((8-метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-ил)метил)-1Н-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты (соединение 4601) (150 мг, 0,34 ммоль) в безводном ДХМ (10 мл) медленно добавили оксалилхлорид (2,0 М в ДХМ, 0,22 мл), а затем каталитическое количество безводного ДМФ (1 каплю). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, а затем концентрировали in vacuo. Остаток повторно растворили в ДХМ (6 мл), и добавили избыточное количество гидроксида аммония (0,35 мл). Смесь перемешивали при комнатной температуре в течение 2 ч, а затем разделили между этилацетатом и водой. Органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, остаток очистили колоночной флэш-хроматографией на силикагеле (элюируя 5% МеОН/ДХМ) и получили желаемый продукт, 4-амино-1-((8-метил-1-оксо-2-о-толил-1,2-дигидроизохинолин-3-ил)метил)-1Н)-пиразоло[3,4-d]пиримидин-3-карбоксамид (4602, соединение 298 в таблице 4) (22 мг, выход 15%) в виде твердого белого вещества.
Пример 12: Синтез (S)-3-(1-(9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 4704) (способ А)
Схема 25. Описан синтез (S)-3-(1-(9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 4704) по способу А
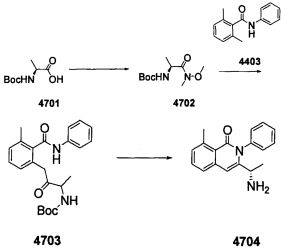
К перемешанной смеси (S)-2-(трет-бутоксикарбониламино)пропановой кислоты (соединение 4701) (189,1 г, 1 моль, 1 экв.), триэтиламина (404,8 г, 4 моль, 4 экв.) и HOBt (135 г, 1,0 моль, 1 экв.) в безводном дихлорметане (1,8 л) при 0°С частями, в течение 30 минут добавили EDCI (384,3 г, 2 моль, 2 экв.). Полученную смесь перемешивали при комнатной температуре в течение 30 минут, затем добавили N,O-диметилгидроксиламина гидрохлорид (107,3 г, 1,1 моль, 1,1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 20 ч, а затем погасили водой (1 л). Органический слой промыли водой (2×1 л) и насыщенным солевым раствором (500 мл), высушили над безводным MgSO4 и отфильтровали. Фильтрат концентрировали in vacuo. Неочищенный продукт суспендировали в петролейном эфире (1 л) и перемешивали при комнатной температуре в течение 10 минут. Твердое вещество собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт, (S)-1-трет-бутил-1-(метокси(метил)амино)-1-оксопропан-2-илкарбамат (соединение 4702) (218 г, выход 93,9%) в виде твердого белого вещества.
К перемешанной смеси 2,6-диметил-N-фенилбензамида (соединение 4403, которое может быть синтезировано так, как описано в примере 9) (30 г, 0,13 моль, 1 экв.) и гексаметилфосфорамида (НМРА) (26 г, 0,16 моль, 1,2 экв.) в безводном ТГФ (300 мл) при -78°С в атмосфере аргона осторожно добавили (по каплям) н-бутиллитий (2,5 М, 100 мл, 0,25 моль, 2,5 экв.) в течение 1 часа, и поддерживали температуру реакции ниже -60°С при добавлении. Полученную смесь перемешивали при -78°С в течение 1 ч. К этой смеси быстро добавили трет-бутил 1-(метокси(метил)амино)-1-оксопропан-2-ил карбамат (соединение 4702) (40 г, 0,173 моль, 1,3 экв.) (температура реакции поднялась при добавлении до -50°С). Смесь перемешивали при -50°С в течение 10 минут, погасили водой (300 мл) и экстрагировали этилацетатом (2×100 мл). Объединенный органический слой промыли водой (500 мл×2) и насыщенным солевым раствором (50 мл), высушили над безводным MgSO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили неочищенный продукт в виде полутвердого маслянистого вещества. Неочищенный продукт суспендировали в этилацетате и перемешивали в течение 10 минут. Твердое белое вещество удалили фильтрацией. Фильтрат концентрировали in vacuo, остаток перемешивали в смеси этилацетата (30 мл) и изопропилового спирта (200 мл) при комнатной температуре в течение 10 минут. Твердое вещество собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт, трет-бутил-4-(3-метил-2-(фенилкарбамоил)фенил)-3-оксобутан-2-ил карбамат (соединение 4703) (9,23 г, выход 17,5%) в виде твердого белого вещества.
Трет-бутил-4-(3-метил-2-(фенилкарбамоил)фенил)-3-оксобутан-2-ил карбамат (соединение 4703) (9,23 г, 23 ммоль) растворили в HCl/MeOH (100 мл) и перемешивали с дефлегматором в течение 30 минут. Смесь оставили остывать до комнатной температуры, концентрировали in vacuo, а затем добавили насыщенный раствор Na2CO3, чтобы довести рН до 7-8. Твердое вещество собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт, 3-(1-аминоэтил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 4704) (5,8 г, выход 90%, изомеры S:R=7:1) в виде твердого белого вещества.

Разделение изомеров для улучшения энантиомерной чистоты: 3-(1-Аминоэтил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 4704) (где отношение изомеров S:R=7:1) (5 г, 18 ммоль) растворили в МеОН (100 мл), добавили (D)-винную кислоту (2,7 г, 18 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут, твердое вещество выпало в осадок. Полученную смесь перемешивали с дефлегматором в течение 1 часа, а затем перемешивали при комнатной температуре 16 часов. Твердое вещество собрали фильтрацией и промыли метанолом (10 мл). Затем твердое вещество растворили в Н2О (15 мл) и добавили насыщенный раствор NaHCO3 (5 мл) для доведения рН до 8. Твердое вещество собрали фильтрацией, промыли водой (5 мл), а затем высушили in vacuo и получили энантиомерно обогащенный продукт (соединение 4704) (2,7 г, выход 58%), где отношение изомеров S:R>41:1. Это дает энантиомерную чистоту более, чем примерно 97,6% (S)-энантиомера. Отношение двух энантиомеров подтверждено связыванием с (R)-(-)-альфа-метоксифенилуксусной кислотой и обнаружением полученных диастереомеров по спектроскопии ядерного магнитного резонанса.
Пример 13: Синтез (S)-3-(1-аминоэтил)-8-метил-2-фенилизохинолин-1(2Н)-она (способ В) (соединение 4704).
Схема 26. Описан синтез (S)-3-(1-аминоэтил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 4704) по способу В

Тионилхлорид (320,8 г, 2,7 моль, 1,2 экв.) по каплям, в течение 50 минут, добавили к перемешанному безводному МеОН (2 л) при 0°С, и во время добавления поддерживали температуру реакции ниже 25°С. Смесь оставили нагреваться до комнатной температуры, а затем добавили (S)-2-аминопропановую кислоту (соединение 4801) (200 г, 2,24 моль, 1 экв.). Полученную смесь перемешивали при комнатной температуре в течение 20 ч., концентрировали in vacuo и получили желаемый продукт, (S)-метил 2-аминопропаноата гидрохлорид (соединение 4802) в виде твердого белого вещества.
К перемешанному раствору ранее полученного (S)-метил 2-аминопропаноата гидрохлорида (соединение 4802) в воде (1,6 л) при комнатной температуре последовательно добавили NaHCO3 (566,2 г, 6,741 моль, 3 экв.) и раствор ди-трет-бутилдикарбоната (490,4 г, 2,247 г, 1 экв.) в ТГФ (1,6 л). Полученную смесь перемешивали при комнатной температуре в течение 20 ч. Неорганическую соль удалили фильтрацией, а фильтрат экстрагировали этилацетатом (2×500 мл). Объединенный органический слой промыли насыщенным солевым раствором (500 мл), высушили над безводным MgSO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили желаемый продукт, (S)-метил 2-(трет-бутоксикарбониламино)пропаноат (соединение 4803) (448 г, выход 98,2%) в виде бесцветных кристаллов.
К перемешанному раствору 2,6-диметил-N-фенилбензамида (соединение 4403, которое может быть синтезировано так, как описано в примере 9) (30 г, 0,13 моль, 1 экв.) и гексаметилфосфорамида (НМРА) (26 г, 0,16 моль, 1,2 экв.) в безводном ТГФ (300 мл) при -78°С в атмосфере аргона осторожно добавили н-бутиллитий (100 мл, 2,5 М, 0,25 моль, 2,5 экв.) в течение 1 часа, и поддерживали температуру реакции ниже -60°С при добавлении. Полученную смесь перемешивали при -78°С в течение 1 ч., затем быстро добавили (S)-метил 2-(трет-бутоксикарбониламино)-пропаноат (соединение 4803) (35 г, 0,173 моль, 1,3 экв.) (температура реакции увеличилась при добавлении до -50°С). Смесь перемешивали при -50°С в течение 10 минут, погасили водой (300 мл) и экстрагировали этилацетатом (2×100 мл). Органический слой промыли водой (500 мл×2), высушили над безводным MgSO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили неочищенный продукт в виде полутвердого маслянистого вещества. Неочищенный продукт суспендировали в этилацетате (500 мл) и перемешивали в течение 10 минут. Твердое вещество удалили фильтрацией, а фильтрат концентрировали in vacuo. Маслянистый остаток перемешивали в смеси этилацетата (30 мл) и изопропилового спирта (200 мл) при комнатной температуре в течение 10 минут. Твердое вещество собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт, трет-бутил-4-(3-метил-2-(фенилкарбамоил)фенил)-3-оксобутан-2-ил карбамат (соединение 4703) (4,61 г, выход 9%) в виде твердого белого вещества.
Трет-бутил 4-(3-метил-2-(фенилкарбамоил)фенил)-3-оксобутан-2-илкарбамат (соединение 4703) (4,61 г, 0,012 моль) растворили в HCl/МеОН (50 мл) и перемешивали с дефлегматором в течение 30 минут. Смесь концентрировали in vacuo, а затем добавили насыщенный раствор Na2CO3, чтобы довести рН до 7-8. Полученное твердое вещество собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт, 3-(1-аминоэтил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 4704) (2,9 г, выход 90%, изомеры S:R=5:1) в виде твердого белого вещества.
Пример 14а: Синтез (S)-3-(1-(9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 4902)
Схема 27а. Описан синтез (S)-3-(1-(9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-она (9) (соединение 4902)

3-(1-Аминоэтил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 4704) (200 мг, 0,72 ммоль), 6-хлор-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин (344 мг, 1,44 ммоль) и диизопропилэтиламин (ДИПЭА) (279 мг, 2,16 ммоль) растворили в н-BuOH (20 мл), и полученную смесь перемешивали с дефлегматором в течение 16 ч. Реакционную смесь концентрировали in vacuo и очистили колоночной флэш-хроматографией на силикагеле (элюируя от 30% до 50% гексаны/этилацетат), получили желаемый продукт, 8-метил-2-фенил-3-((1S)-1-(9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-иламино)этил)изохинолин-1(2Н)-он (соединение 4901) (207 мг, выход 60%) в виде твердого белого вещества.
8-Метил-2-фенил-3-((1S)-1-(9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-иламино)этил)изохинолин-1(2Н)-он (соединение 4901) (200 мг, 0,42 ммоль) растворили в HCl/EtOH (3 М, 5 мл), и полученную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь погасили насыщенным водным раствором NaHCO3 и довели рН до примерно 7-8. Смесь экстрагировали CH2Cl2 (50 мл×3), высушили над безводным Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, и остаток перекристаллизовали из смеси этилацетата и гексанов (1:1). Твердое вещество собрали фильтрацией и высушили in vacuo, получили желаемый продукт, (S)-3-(1-(9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 4902) (150 мг, выход 90%) в виде твердого белого вещества.
Пример 14b: Синтез (S)-3-(1-(9Н-пурин-6-иламино)этил)-8-хлор-2-фенилизохинолин-1(2Н)-она (9) (соединение 4904)
Схема 27b. Описан синтез (S)-3-(1-(9Н-пурин-6-иламино)этил)-8-хлор-2-фенилизохинолин-1(2Н)-она (9) (соединение 4904)

Соединение формулы 4904 (соединение 292 в таблице 4) синтезировали, используя синтетические преобразования, описанные в примерах 12 и 14а, но использовали 2-хлор-6-метилбензойную кислоту (соединение 4903) вместо 2,6-диметилбензойной кислоты (соединение 4403). Таким же способом синтезировали соединение 328 в таблице 4, используя описанные синтетические преобразования, исходя из 2-хлор-6-метил-м-фторбензойной кислоты.
Пример 15а: Синтез (S)-3-(1-(9Н-пурин-6-иламино)этил)-2-циклопропил-8-метилизохинолин-1(2Н)-она (соединение 5005).
Схема 28а. Описан синтез (S)-3-(1-(9Н-пурин-6-иламино)этил)-2-циклопропил-8-метилизохинолин-1(2Н)-она.

Смесь циклопропанамина (24 г, 420 ммоль) и триэтиламина (71 г, 700 ммоль) в CH2Cl2 (300 мл) перемешивали в течение 10 минут при комнатной температуре. К этой смеси по каплям добавили 2,6-диметилбензоилхлорид (соединение 4402) (64 г, 400 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 30 минут. Реакционную смесь вылили в воду (300 мл) и экстрагировали CH2Cl2 (3×200 мл). Объединенный органический слой высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили неочищенный продукт. Неочищенный продукт суспендировали в изопропиловом эфире (IPE) (300 мл), перемешивали с дефлегматором в течение 30 минут, а затем охладили до 0-5°С. Осадок собрали фильтрацией и дополнительно высушили in vacuo, и получили желаемый продукт, N-циклопропил-2,6-диметилбензамид (соединение 5001) (61 г, выход 80%) в виде твердого желтого вещества.
К перемешанному раствору К-циклопропил-2,6-диметилбензамида (соединение 5001) (25 г, 0,13 моль, 1 экв.) и гексаметилфосфорамида (НМРА) (26 г, 0,16 моль, 1,2 экв.) в безводном ТГФ (300 мл) при -78°С в атмосфере аргона осторожно добавили н-бутиллитий (2,5 М, 100 мл, 0,25 моль, 2,5 экв.) в течение 1 часа, и поддерживали температуру реакции ниже -60°С при добавлении. Полученную смесь перемешивали при -78°С в течение 1 ч., затем быстро добавили трет-бутил 1-(метокси(метил)амино)-1-оксопропан-2-илкарбамат (40 г, 0,173 моль, 1,3 экв.) (температура реакции увеличилась при добавлении до -50°С). Смесь перемешивали при -50°С в течение 10 минут, погасили водой (300 мл) и экстрагировали этилацетатом (2×100 мл). Объединенный органический слой промыли водой (500 мл×2) и насыщенным солевым раствором (100 мл), высушили над безводным MgSO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили желаемый продукт, трет-бутил 4-(2-(циклопропилкарбамоил)-3-метилфенил)-3-оксобутан-2-илкарбамат (соединение 5002) (32 г, выход 70%) в виде желтого маслянистого вещества.
Трет-бутил-4-(2-(циклопропилкарбамоил)-3-метилфенил)-3-оксобутан-2-илкарбамат (соединение 5002) (32 г, 88 ммоль) растворили в HCl/MeOH (300 мл) и перемешивали при комнатной температуре в течение 16 ч. Смесь концентрировали in vacuo, затем добавили насыщенный водный раствор Na2CO3 для доведения рН до значения примерно 7-8. Полученное твердое вещество собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт, 3-(1-аминоэтил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 5003) (17 г, выход 80%, S:R=7:1) в виде твердого белого вещества.

К перемешанному раствору 3-(1-аминоэтил)-2-циклопропил-8-метилизохинолин-1(2Н)-она (S:R=7:1) (4,84 г, 20 ммоль) (соединение 5003) в МеОН (96,8 мл) добавили (L)-винную кислоту (3,0 г, 20 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 16 ч. Осадок собрали фильтрацией и промыли метанолом (10 мл). Твердое вещество растворили в H2O (15 мл) и добавили насыщенный раствор NaHCO3 (5 мл) для доведения рН до значения примерно 8. Полученное твердое вещество собрали фильтрацией, промыли водой (5 мл) и высушили in vacuo, получили желаемый продукт (соединение 5003) (1,94 г, выход 40%) в виде единого энантиомера (S-конфигурация). Энантиомерную чистоту подтвердили связыванием с (R)-(-)-альфа-метоксифенилуксусной кислотой и выполнением спектроскопии ядерного магнитного резонанса полученной диастереомерной смеси.
(S)-3-(1-Аминоэтил)-2-циклопропил-8-метилизохинолин-1(2Н)-он (242 мг, 1 ммоль) (соединение 5003), 6-хлор-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин (344 мг, 1,44 ммоль) и диизопропилэтиламин (ДИПЭА) (279 мг, 2,16 ммоль) растворили в н-BuOH (20 мл), и полученную смесь перемешивали с дефлегматором в течение 16 ч. Реакционную смесь концентрировали in vacuo и очистили остаток колоночной флэш-хроматографией на силикагеле (элюируя от 30% до 50% гексаны/этилацетат), получили желаемый продукт, 2-циклопропил-8-метил-3-((1S)-1-(9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-иламино)этил)изохинолин-1(2Н)-он (соединение 5004) (288 мг, выход 65%) в виде твердого белого вещества.
2-Циклопропил-8-метил-3-((1S)-1-(9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-иламино)этил)изохинолин-1(2Н)-он (соединение 5004) (222 мг, 0,5 ммоль) растворили в НСl/EtOH (3 М, 5 мл), и полученную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь нейтрализовали насыщенным раствором NaHCO3 до рН=7-8, а затем экстрагировали CH2Cl2 (50 мл×3). Объединенный органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, и остаток перекристаллизовали из смеси этилацетата и гексанов (1:1). Твердое вещество собрали фильтрацией и высушили in vacuo, получили желаемый продукт, (S)-3-(1-(9H-пурин-6-иламино)этил)-2-циклопропил-8-метилизохинолин-1(2Н)-он (5005, соединение 200 в таблице 4) (150 мг, выход 83%) в виде твердого белого вещества.
Пример 15b. Синтез (S)-3-(1-(9Н-пурин-6-иламино)этил)-2-циклопропил-8-хлор-изохинолин-1(2Н)-она (соединение 5011).
Схема 28b. Описан синтез (S)-3-(1-(9Н-пурин-6-иламино)этил)-2-циклопропил-8-хлор-изохинолин-1(2Н)-она:

Соединение формулы 5011 (соединение 270 в таблице 4) синтезировали, используя синтетические преобразования, описанные в примере 15a, но использовали 2-хлор-6-метилбензоилхлорид (соединение 5006) вместо 2,6-диметилбензоилхлорида (соединение 4402).
Пример 16: Синтез (S)-3-(1-(2-амино-5-хлорпиримидин-4-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 5102)
Схема 29. Описан синтез (S)-3-(1-(2-амино-5-хлорпиримидин-4-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 5102).

Смесь 3-(1-аминоэтил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 4704) (150 мг, 0,54 ммоль), 2,4,5-трихлорпиримидина (119 мг, 0,65 ммоль) и триэтиламина (137 мг, 1,35 ммоль) в н-BuOH (10 мл) перемешивали с дефлегматором в течение 2 ч. Смесь оставили остывать до комнатной температуры, а затем концентрировали in vacuo. Остаток очистили колоночной флэш-хроматографией на силикагеле (МеОН:CH2Cl2=1:100) и получили желаемый продукт, (S)-3-(l-(2,5-дихлорпиримидин-4-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 5101) (170 мг, выход 74%) в виде твердого белого вещества.
Смесь (S)-3-(1-(2,5-дихлорпиримидин-4-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 5101) (85 мг, 0,20 ммоль) в аммиачной воде (15 мл) в герметично закрытой пробирке перемешивали при 150°С в течение 16 ч. Раствор оставили остывать до комнатной температуры, а затем разделили между водой (30 мл) и этилацетатом (3×30 мл). Объединенный органический слой промыли насыщенным солевым раствором (2×2 мл), высушили над безводным Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили желаемый продукт, (S)-3-(1-(2-амино-5-хлорпиримидин-4-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-он (5102, соединение 249 в таблице 4) (40 мг, выход 49,6%) в виде твердого белого вещества.
Пример 17: Синтез (S)-3-(1-(2-фтор-9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 5204).
Схема 30. Описан синтез (S)-3-(1-(2-фтор-9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 5204).

К перемешанной смеси 6-хлор-2-фтор-9Н-пурина (соединение 5201) (2,07 г, 12,0 ммоль) и п-толуолсульфоновой кислоты моногидрата (34 мг, 0,18 ммоль) в этилацетате (50 мл) в атмосфере аргона добавили 3,4-дигидропиран (3,03 г, 36,0 ммоль), и полученную смесь перемешивали с дефлегматором в течение 16 ч. Реакционную смесь концентрировали in vacuo, и остаток очистили колоночной флэш-хроматографией на силикагеле (элюируя 10% смесью гексаны/этилацетат), получили желаемый продукт, 6-хлор-2-фтор-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин (соединение 5202) (1,82 г, выход 59%) в виде твердого белого вещества.
3-(1-Аминоэтил)-8-метил-2-фенилизохинолин-1(2Н)-он (200 мг, 0,72 ммоль), 6-хлор-2-фтор-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин (соединение 5202) (369 мг, 1,44 ммоль) и диизопропилэтиламин (ДИПЭА) (279 мг, 2,16 ммоль) растворили в н-BuOH (20 мл), и полученную смесь перемешивали при 120°С в течение 16 ч. Реакционную смесь концентрировали in vacuo и очистили остаток колоночной флэш-хроматографией на силикагеле (элюируя 30%-50% гексаны/этилацетатом), получили желаемый продукт, 3-(1-(2-фтор-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 5203) (167 мг, выход 47%) в виде твердого белого вещества.
3-(1-(2-Фтор-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 5203) (160 мг, 0,32 ммоль) растворили в НСl/EtOH (3 М, 5 мл), и полученную смесь перемешивали при комнатной температуре в течение 1 ч. Смесь нейтрализовали насыщенным водным раствором NaHCO3 до рН=7-8, а затем экстрагировали CH2Cl2 (50 мл×3). Объединенный органический слой промыли насыщенным солевым раствором, высушили над безводным Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, и остаток перекристаллизовали из этилацетата и гексанов. Твердое вещество собрали фильтрацией и высушили in vacuo, получили желаемый продукт, 3-(1-(2-фтор-9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-он (5204, соединение 245 в таблице 4) (125 мг, выход 94%) в виде твердого белого вещества.
Пример 18: Синтез (S)-3-(1-(2-хлор-9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 5304).
Схема 31. Описан синтез (S)-3-(1-(2-хлор-9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 5304).

К перемешанной смеси 2,6-дихлор-9Н-пурина (соединение 5301) (2,27 г, 12,0 ммоль) и пара-толуолсульфоновой кислоты моногидрата (34 мг, 0,18 ммоль) в этилацетате (50 мл) в атмосфере аргона добавили 3,4-дигидропиран (3,03 г, 36,0 ммоль), и полученную смесь перемешивали с дефлегматором в течение 16 ч.
Реакционную смесь концентрировали in vacuo, и остаток очистили колоночной флэш-хроматографией на силикагеле (элюируя 10% смесью гексаны/этилацетат), получили желаемый продукт, 2,6-дихлор-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин (соединение 5302) (2,04 г, выход 62%) в виде твердого белого вещества.
3-(1-Аминоэтил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 4704) (200 мг, 0,72 ммоль), 2,6-дихлор-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин (соединение 5302) (393 мг, 1,44 ммоль) и диизопропилэтиламин (ДИПЭА) (279 мг, 2,16 ммоль) растворили в н-BuOH (20 мл) в герметично закрытой пробирке, и полученную смесь перемешивали при 120°С в течение 16 ч. Реакционную смесь концентрировали in vacuo и очистили остаток колоночной флэш-хроматографией на силикагеле (элюируя 30%-50% гексаны/этилацетатом), получили желаемый продукт, 3-(1-(2-хлор-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 5303) (172 мг, выход 46%) в виде твердого белого вещества.
3-(1-(2-Хлор-9-(тетрагидро-2Н-пиран-2-ил)-9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 5303) (172 мг, 0,33 ммоль) растворили в HCl/EtOH (3 М, 5 мл), и полученную смесь перемешивали при комнатной температуре в течение 1 ч. Смесь нейтрализовали насыщенным водным раствором NaHCO3 до рН=7-8, а затем экстрагировали CH2Cl2 (50 мл×3). Объединенный органический слой промыли насыщенным солевым раствором, высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo, и остаток перекристаллизовали из этилацетата и гексанов. Твердое вещество собрали фильтрацией и высушили in vacuo, получили желаемый продукт, 3-(1-(2-хлор-9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-он (5304, соединение 244 в таблице 4) (128 мг, выход 90%) в виде твердого белого вещества.
Пример 19: Синтез (S)-3-(1-(2-амино-9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 5402).
Схема 32. Описан синтез (S)-3-(1-(2-амино-9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-она (соединение 5402).

(S)-3-(1-Аминоэтил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 4704) (100 мг, 0,36 ммоль), 2-амино-6-хлорпурин (соединение 5401) (60,9 мг, 0,36 ммоль) и N,N-диизопропилэтиламин (69 мкл, 0,40 ммоль) суспендировали в н-BuOH (4 мл) в герметично закрытой пробирке, и полученную смесь перемешивали при 100°С в течение 48 ч., а затем при 120°С в течение 24 ч. Смесь оставили остывать до комнатной температуры и концентрировали in vacuo для удаления н-BuOH. Остаток разделили между этилацетатом и водой. Органический слой промыли насыщенным солевым раствором, высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo. Остаток растерли с безводным этиловым эфиром и дополнительно очистили колоночной флэш-хроматографией на силикагеле (элюируя 0-8% МеОН/ДХМ), получили желаемый продукт, (S)-3-(1-(2-амино-9Н-пурин-6-иламино)этил)-8-метил-2-фенилизохинолин-1(2Н)-он в виде грязновато-белого/желтого твердого вещества (5402, соединение 323 в таблице 4) (28 мг, 20%).
Пример 20: Синтез (S)-4-(1-(8-метил-1-оксо-2-фенил-1,2-дигидроизохинолин-3-ил)этиламино)-7Н-пирроло[2,3-d]пиримидин-5-карбонитрила (соединение 5506).
Схема 33. Описан синтез (S)-4-(1-(8-метил-1-оксо-2-фенил-1,2-дигидроизохинолин-3-ил)этиламино)-7Н-пирроло[2,3-d]пиримидин-5-карбонитрила (соединение 5506).


К перемешанной смеси 4-хлор-7Н-пирроло[2,3-d]пиримидина (соединение 5501) (3,99 г, 26,0 ммоль) в сухом CH2Cl2 (150 мл) в атмосфере аргона добавили N-бромсукцинимид (6,02 г, 33,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч., разбавили МеОН (30 мл), а затем концентрировали in vacuo для получения светло-коричневого твердого вещества. Остаток растерли с H2O (150 мл), а затем перекристаллизовали из МЕОН (120 мл). Твердое вещество собрали фильтрацией и дополнительно высушили in vacuo, получили желаемый продукт, 5-бром-4-хлор-7Н-пирроло[2,3-d]пиримидин (соединение 5502) (4,0 г, выход 66%) в виде твердого белого вещества.
К перемешанному раствору 5-бром-4-хлор-7Н-пирроло[2,3-d]пиримидина (соединение 5502) (2,33 г, 10,0 ммоль) в безводном ТГФ (100 мл) при -78°С в атмосфере аргона по каплям, в течение 10 минут, добавили раствор н-BuLi (8,8 мл, 22,0 ммоль) в ТГФ (50 мл). Реакционную смесь перемешивали при -78°С в течение 1 ч., а затем по каплям, в течение 10 минут, добавили ДМФ (2,00 г, 11,0 ммоль). Реакционную смесь перемешивали при -78°С в течение 30 минут, а затем оставили медленно нагреваться до комнатной температуры и перемешивали при комнатной температуре 16 часов. Смесь разбавили H2O (50 мл), а затем концентрировали in vacuo для удаления ТГФ. Полученную суспензию обработали насыщенным водным раствором NH4Cl (50 мл), отфильтровали, промыли этилацетатом (100 мл) и высушили in vacuo, получили желаемый продукт, 4 -хлор-7Н-пирроло[2,3-d]пиримидин-5-карбальдегид (соединение 5503) (1,17 г, выход 65%) в виде твердого белого вещества.
К перемешанной смеси 4-хлор-7Н-пирроло[2,3-d]пиримидин-5-карбальдегида (соединение 5503) (1,17 г, 6,47 ммоль) в EtOH (25 мл), последовательно добавили гидроксиламина солянокислую соль (0,54 г, 7,77 ммоль) и раствор NaOH (0,311 г, 7,77 ммоль) в H2O (4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и разбавили достаточным количеством EtOH (30 мл), и продолжали перемешивание в течение 30 минут. Твердое вещество собрали фильтрацией, промыли H2O (100 мл) и высушили in vacuo, получили желаемый продукт, 4-хлор-7Н-пирроло[2,3-d]пиримидин-5-карбальдегид оксим (соединение 5504) (0,89 г, выход 70%) в виде смеси изомеров.
К перемешанной смеси 4-хлор-7Н-пирроло[2,3-d]пиримидин-5-карбальдегид оксима (соединение 5504) (865 мг, 4,40 ммоль) в CH2Cl2 (20 мл) добавили SOCl2 (3,1 мл, 43,7 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь концентрировали in vacuo. Остаток обработали этилацетатом (20 мл), H2O (20 мл), а затем концентрированным водным раствором NaHCO3 (50 мл) для доведения рН до значения примерно 3-4. Смесь перемешивали при комнатной температуре в течение 15 минут, и собрали твердое вещество фильтрацией. Фильтрат экстрагировали этилацетатом (80 мл×3), высушили над Na2SO4 и отфильтровали. Фильтрат концентрировали in vacuo и получили вторую порцию продукта. Объединенное твердое вещество перекристаллизовали из смеси этилацетата и гексанов (1:1, 20 мл). Твердое вещество собрали фильтрацией и высушили in vacuo, получили желаемый продукт, 4-хлор-7Н-пирроло[2,3-d]пиримидин-5-карбонитрил (соединение 5505) (763 мг, выход 97%).
(S)-3-(1-Аминоэтил)-8-метил-2-фенилизохинолин-1(2Н)-он (соединение 4704) (208 мг, 0,75 моль), 4-хлор-7Н-пирроло[2,3-d]пиримидин-5-карбонитрил (соединение 5505) (160 мг, 0,90 ммоль) и Et3N (228 мг, 2,25 ммоль) растворили в н-BuOH (20 мл) в герметично закрытой пробирке, и полученную смесь перемешивали при 150°С в течение 16 ч. Реакционную смесь концентрировали in vacuo, остаток очистили колоночной флэш-хроматографией на силикагеле (элюируя 50% гексаны/этилацетатом) и получили желаемый продукт, (S)-4-(1-(8-метил-1-оксо-2-фенил-1,2-дигидроизохинолин-3-ил)этиламино)-7Н-пирроло[2,3-d]пиримидин-5-карбонитрил (5506, соединение 264 в таблице 4) (90 мг, выход 28%) в виде твердого белого вещества.
Пример 21: Значения IC50 для выбранных соединений.

















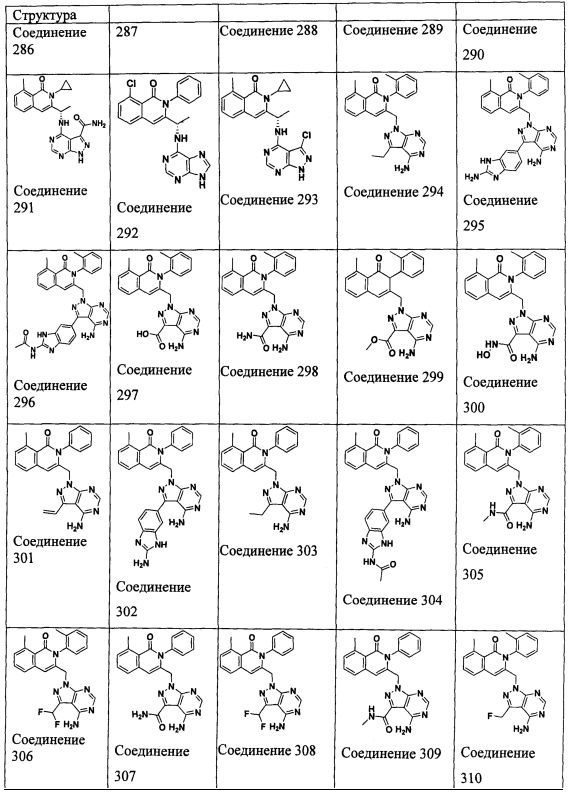


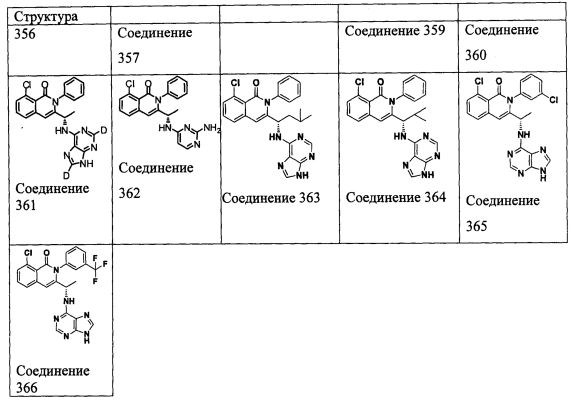
Пример 22: Анализы экспрессии и ингибирования р110α/р85α, р110β/р85α, р110δ/р85α и р110γ:
PI3-K класса I могут быть закуплены (р110α/р85α, р110β/р85α, р110δ/р85α у Upstate, и р110γ у Sigma) или экспрессированы так, как описано ранее (Knight et al., 2004). Значения IC50 измеряются с использованием стандартного ТСХ анализа для активности липидкиназы (описанного ниже) или при помощи анализа удержания на мембране. Реакции киназ выполняются путем приготовления реакционной смеси, содержащей киназу, ингибитор (конечная концентрация 2% в ДМСО), буфер (25 мМ HEPES, рН 7,4, 10 мМ MgCl2), и свежеприготовленный фосфатидилинозитол, обработанный ультразвуком (100 мкг/мл). Реакции инициируются добавлением АТФ, содержащей 10 мкКи γ-32Р-АТФ до конечной концентрации 10 или 100 мкМ, реакции выдерживаются в течение 5 минут при комнатной температуре. Для анализа ТСХ, реакции обрываются путем добавления 105 мкл 1 н. раствора HCl, а затем 160 мкл CHCl3:МеОН (1:1). Двухфазную смесь перемешивают на вортексе, быстро центрифугируют и переносят органическую фазу в новую пробирку при помощи загрузочного микродозатора, который предварительно покрыт CHCl3. Этот экстракт помещается на пластины ТСХ и выдерживается 3-4 часа в растворе н-пропанол:1М уксусная кислота 65:35. Затем пластины ТСХ высушиваются, помещаются на экран сканера Phosphorimager (Storm, Amersham), и подсчитываются. Для каждого соединения активность киназы измеряется при 10-12 концентрациях ингибитора, составляющих двукратное разбавление от максимальной испытываемой концентрации (обычно 200 мкМ). Для соединений, демонстрирующих значительную активность, определение IC50 повторяют от двух до четырех раз, и регистрируемое значение является средним из этих независимых измерений.
В продаже существуют другие наборы или системы для анализа активности PI3-К. Доступные в продаже наборы или системы могут использоваться для анализа ингибиторов и/или агонистов PI3-K, включая, не ограничиваясь, киназы PI3 α, β, δ и γ. Типичной системой является Система для анализа PI3-киназы (человека) HTRF™ производства Upstate. Анализ может выполняться по методикам, предложенным производителем. Вкратце, анализ представляет собой анализ FRET с временным разрешением, который косвенно измеряет продукт PIP3, образованный в результате активности PI3-K. Реакция киназы выполняется в микротитровальном планшете (например, 384-луночном микротитровальном планшете). Общий объем реакции составляет примерно 20 мкл на лунку. На первом этапе в каждую лунку помещают 2 мкл исследуемого соединения в 20% диметилсульфоксиде, что дает конечную концентрацию 2% в ДМСО. Затем в каждую лунку добавляют примерно 14,5 мкл смеси киназы/PIP2 (разбавленной в реакционном буфере 1X) до конечной концентрации 0,25-0,3 мкг/мл киназы и 10 мкМ PIP2. Планшет закрывают и инкубируют в течение 15 минут при комнатной температуре. Для начала реакции в каждую лунку добавляют 3,5 мкл АТФ (разбавленной в реакционном буфере 1X) до конечной концентрации 10 мкМ АТФ. Планшет закрывают и инкубируют в течение 1 часа при комнатной температуре. Реакцию прекращают добавлением в каждую лунку 5 мкл раствора для остановки реакции, а затем 5 мкл обнаруживающей смеси. Планшет закрывают, инкубируют в течение 1 часа при комнатной температуре, а затем считывают на соответствующем планшет-ридере. Данные анализируют и получают значения IC50 при помощи GraphPad Prism 5.
Пример 23: Анализ экспрессии и ингибирования Abl
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к киназе Abl может быть измерена в соответствии с любой из методик, известных в данной области, или при помощи способов, описанных ниже. Например, соединения, описанные в настоящем документе, могут быть трехкратно проанализированы по отношению к рекомбинантной Abl полной длины или Abl (T315I1) (Upstate) в образце, содержащем 25 мМ HEPES, рН 7,4, 10 мМ MgCl2, 200 мкМ АТФ (2,5 мкКи Г-32Р-АТФ) и 0,5 мг/мл BSA. Оптимизированный пептидный субстрат Abl EAIYAAPFAKKK используется в качестве фосфоакцептора (200 мМ). Реакции обрываются путем помещения на фосфоцеллюлозные листы, которые промываются 0,5% раствором фосфорной кислоты (примерно 6 раз, 5-10 минут каждый). Листы высушиваются, а перенесенная радиоактивность измеряется при помощи формирования изображения на люминесцентном фосфорном покрытии.
Пример 24: Анализ экспрессии и ингибирования Hck
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к киназе Hck может быть измерена в соответствии с любой из методик, известных в данной области, или при помощи способов, описанных ниже. Соединения, описанные в настоящем документе, могут быть трехкратно проанализированы по отношению к рекомбинантной Hck полной длины в образце, содержащем 25 мМ HEPES, рН 7,4, 10 мМ MgCl2, 200 мкМ АТФ (2,5 мкКи γ-32Р-АТФ) и 0,5 мг/мл BSA. Оптимизированный пептидный субстрат киназ семейства Src EIYGEFKKK используется в качестве фосфоакцептора (200 мкМ). Реакции обрываются путем помещения на фосфоцеллюлозные листы, которые промываются 0,5% раствором фосфорной кислоты (примерно 6 раз, 5-10 минут каждый). Листы высушиваются, а перенесенная радиоактивность измеряется при помощи формирования изображения на люминесцентном фосфорном покрытии.
Пример 25: Анализы экспрессии и ингибирования инсулинового рецептора (IR)
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к киназе рецептора IR быть измерена в соответствии с любой из методик, известных в данной области, или при помощи способов, описанных ниже. Соединения, описанные в настоящем документе, могут быть трехкратно проанализированы по отношению к домену рекомбинантной киназы инсулинового рецептора (Upstate) в образце, содержащем 25 мМ HEPES, рН 7,4, 10 мМ MnCl2, 200 мкМ АТФ (2,5 мкКи γ-32Р-АТФ) и 0,5 мг/мл BSA. В качестве субстрата используется Poly E-Y (Sigma; 2 мг/мл). Реакции обрывают путем помещения на нитроцеллюлозу, которую промывают 1 М раствором NaCl/1% фосфорной кислоты (примерно 6 раз, 5-10 минут каждый). Листы высушиваются, а перенесенная радиоактивность измеряется при помощи формирования изображения на люминесцентном фосфорном покрытии.
Пример 26: Анализы экспрессии и ингибирования Src
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к киназе Src может быть измерена в соответствии с любой из методик, известных в данной области, или при помощи способов, описанных ниже. Соединения, описанные в настоящем документе, могут быть трехкратно проанализированы по отношению к рекомбинантной Src полной длины или Src (T338I) в образце, содержащем 25 мМ HEPES, рН 7,4, 10 мМ MgCl2, 200 мкМ АТФ (2,5 мкКи γ-32Р-АТФ) и 0,5 мг/мл BSA. Оптимизированный пептидный субстрат киназ семейства Src EIYGEFKKK используется в качестве фосфоакцептора (200 мкМ). Реакции обрываются путем помещения на фосфоцеллюлозные листы, которые промываются 0,5% раствором фосфорной кислоты (примерно 6 раз, 5-10 минут каждый). Листы высушиваются, а перенесенная радиоактивность измеряется при помощи формирования изображения на люминесцентном фосфорном покрытии.
Пример 27: Анализы экспрессии и ингибирования ДНК-РК (ДНКК)
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к киназе ДНКК может быть измерена в соответствии с любой из методик, известных в данной области. ДНК-РК может быть закуплена у Promega и проанализирована при помощи аналитической системы ДНК-РК (Promega) в соответствии с инструкциями производителя.
Пример 28: Анализы экспрессии и ингибирования mTOR
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к mTOR может быть измерена в соответствии с любой из методик, известных в данной области, или при помощи способов, описанных ниже. Соединения, описанные в настоящем документе, могут быть проанализированы по отношению к рекомбинантной mTOR (Invitrogen) в образце, содержащем 50 мМ HEPES, рН 7,5, 1 мМ ЭГТК, 10 мМ MgCl2, 2,5 мМ, 0,01% Твин, 10 мкМ АТФ (2,5 мкКи µ-32Р-АТФ) и 3 мкг/мл BSA. В качестве субстрата используется рекомбинантная PHAS-1/4EBP1 крыс (Calbiochem; 2 мг/мл). Реакции обрывают путем помещения на нитроцеллюлозу, которую промывают 1 М раствором NaCl/1% фосфорной кислоты (примерно 6 раз, 5-10 минут каждый). Листы высушиваются, а перенесенная радиоактивность измеряется при помощи формирования изображения на люминесцентном фосфорном покрытии.
В продаже существуют другие наборы или системы для анализа активности mTOR. Например, можно использовать аналитический образец киназы LanthaScreen™ производства Invitrogen для тестирования ингибиторов mTOR, описанных в настоящем документе. Этот анализ представляет собой анализ на базе FRET с временным разрешением, который измеряет фосфорилирование 4ЕВР1, меченного GFP, киназой mTOR. Реакция киназы выполняется в белом 384-луночном микротитровальном планшете. Общий объем реакционной смеси составляет 20 мкл на лунку, а состав реакционного буфера включает 50 мМ HEPES рН 7,5, 0,01% Полисорбата 20, 1 мМ ЭГТК, 10 мМ MnCl2 и 2 мМ DTT. На первом этапе в каждую лунку помещают 2 мкл исследуемого соединения в 20% диметилсульфоксиде, что дает конечную концентрацию 2% в ДМСО. Затем 8 мкл mTOR, разбавленной в реакционном буфере, добавляют в каждую лунку до конечной концентрации 60 нг/мл. Для начала реакции 10 мкл смеси АТФ/GFP-4EBP1 (разбавленной в реакционном буфере) добавляют в каждую лунку до конечной концентрации 10 мкМ АТФ и 0,5 мкМ GFP-4EBP1. Планшет закрывают и инкубируют в течение 1 часа при комнатной температуре. Реакцию останавливают добавлением 10 мкл на лунку смеси антитела Tb-анти-рТ46 4ЕВР1 / ЭДТА (разбавленной в буфере TR-FRET) до конечной концентрации 1,3 нМ антитела и 6,7 мМ ЭДТА. Планшет закрывают, инкубируют в течение 1 часа при комнатной температуре, а затем считывают на планшет-ридере, настроенном на LanthaScreen™ TR-FRET. Данные анализируют, и получают значения IC50 при помощи GraphPad Prism 5.
Пример 29: Анализы экспрессии и ингибирования сосудистого эндотелиального рецептора роста (VEGF)
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к рецептору VEGF может быть измерена в соответствии с любой из методик, известных в данной области, или при помощи способов, описанных ниже. Соединения, описанные в настоящем документе, могут быть проанализированы по отношению к домену рекомбинантной киназы рецептора KDR (Invitrogen) в образце, содержащем 25 мМ HEPES, рН 7,4, 10 мМ MgCl2, 0,1% ВМЕ, 10 мкМ АТФ (2,5 мкКи м-32Р-АТФ) и 3 мкг/мл BSA. В качестве субстрата используется Poly E-Y (Sigma; 2 мг/мл). Реакции обрывают путем помещения на нитроцеллюлозу, которую промывают 1 М раствором NaCl/1% фосфорной кислоты (примерно 6 раз, 5-10 минут каждый). Листы высушиваются, а перенесенная радиоактивность измеряется при помощи формирования изображения на люминесцентном фосфорном покрытии.
Пример 30: Анализы экспрессии и ингибирования рецептора Ephrin В4 (EphB4)
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к EphB4 может быть измерена в соответствии с любой из методик, известных в данной области, или при помощи способов, описанных ниже. Соединения, описанные в настоящем документе, могут быть проанализированы по отношению к домену рекомбинантной киназы В4 рецептора Ephrin (Invitrogen) в образце, содержащем 25 мМ HEPES, рН 7,4, 10 мМ MgCl2, 0,1% ВМЕ, 10 мкМ АТФ (2,5 мкКи м-32Р-АТФ) и 3 мкг/мл BSA. В качестве субстрата используется Poly E-Y (Sigma; 2 мг/мл). Реакции обрывают путем помещения на нитроцеллюлозу, которую промывают 1 М раствором NaCl/1% фосфорной кислоты (примерно 6 раз, 5-10 минут каждый). Листы высушиваются, а перенесенная радиоактивность измеряется при помощи формирования изображения на люминесцентном фосфорном покрытии.
Пример 31: Анализы экспрессии и ингибирования рецептора эпидермального фактора роста (EGFR)
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к киназе EGFR может быть измерена в соответствии с любой из методик, известных в данной области, или при помощи способов, описанных ниже. Соединения, описанные в настоящем документе, могут быть проанализированы по отношению к домену рекомбинантной киназы рецептора EGF (Invitrogen) в образце, содержащем 25 мМ HEPES, рН 7,4, 10 мМ MgCl2, 0,1% ВМЕ, 10 мкМ АТФ (2,5 мкКи м-32Р-АТФ) и 3 мкг/мл BSA. В качестве субстрата используется Poly E-Y (Sigma; 2 мг/мл). Реакции обрывают путем помещения на нитроцеллюлозу, которую промывают 1 М раствором NaCl/1% фосфорной кислоты (примерно 6 раз, 5-10 минут каждый). Листы высушиваются, а перенесенная радиоактивность измеряется при помощи формирования изображения на люминесцентном фосфорном покрытии.
Пример 32: Анализ экспрессии и ингибирования образца KIT
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к киназе KIT может быть измерена в соответствии с любой из методик, известных в данной области, или при помощи способов, описанных ниже. Соединения, описанные в настоящем документе, могут быть проанализированы по отношению к домену рекомбинантной киназы KIT (Invitrogen) в образце, содержащем 25 мМ HEPES, рН 7,4, 1 мМ DTT, 10 мМ MgCl2, 10 мкМ АТФ (2,5 мкКи м-32Р-АТФ) и 3 мкг/мл BSA. В качестве субстрата используется Poly E-Y (Sigma; 2 мг/мл). Реакции обрывают путем помещения на нитроцеллюлозу, которую промывают 1 М раствором NaCl/1% фосфорной кислоты (примерно 6 раз, 5-10 минут каждый). Листы высушиваются, а перенесенная радиоактивность измеряется при помощи формирования изображения на люминесцентном фосфорном покрытии.
Пример 33: Анализы экспрессии и ингибирования RET
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к киназе RET может быть измерена в соответствии с любой из методик, известных в данной области, или при помощи способов, описанных ниже. Соединения, описанные в настоящем документе, могут быть проанализированы по отношению к домену рекомбинантной киназы RET (Invitrogen) в образце, содержащем 25 мМ HEPES, рН 7,4, 10 мМ MgCl2, 2,5 мМ DTT, 10 мкМ АТФ (2,5 мкКи µ-32Р-АТФ) и 3 мкг/мл BSA. Оптимизированный пептидный субстрат Abl EAIYAAPFAKKK используется в качестве фосфоакцептора (200 мкМ). Реакции обрываются путем помещения на фосфоцеллюлозные листы, которые промываются 0,5% раствором фосфорной кислоты (примерно 6 раз, 5-10 минут каждый). Листы высушиваются, а перенесенная радиоактивность измеряется при помощи формирования изображения на люминесцентном фосфорном покрытии.
Пример 34: Анализы экспрессии и ингибирования рецептора фактора роста, полученного из тромбоцитов (PDGFR)
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к киназе PDGFR может быть измерена в соответствии с любой из методик, известных в данной области, или при помощи способов, описанных ниже. Соединения, описанные в настоящем документе, могут быть проанализированы по отношению к домену рекомбинантной киназы PDG (Invitrogen) в образце, содержащем 25 мМ HEPES, рН 7,4, 10 мМ MgCl2, 2,5 мМ DTT, 10 мкМ АТФ (2,5 мкКи µ-32Р-АТФ) и 3 мкг/мл BSA. Оптимизированный пептидный субстрат Abl EAIYAAPFAKKK используется в качестве фосфоакцептора (200 мкМ). Реакции обрываются путем помещения на фосфоцеллюлозные листы, которые промываются 0,5% раствором фосфорной кислоты (примерно 6 раз, 5-10 минут каждый). Листы высушиваются, а перенесенная радиоактивность измеряется при помощи формирования изображения на люминесцентном фосфорном покрытии.
Пример 35: Анализы экспрессии и ингибирования FMS-связанной тирозинкиназы 3 (FLT-3)
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к киназе FLT-3 может быть измерена в соответствии с любой из методик, известных в данной области, или при помощи способов, описанных ниже. Соединения, описанные в настоящем документе, могут быть проанализированы по отношению к домену рекомбинантной киназы FLT-3 (Invitrogen) в образце, содержащем 25 мМ HEPES, рН 7,4, 10 мМ MgCl2, 2,5 мМ DTT, 10 мкМ АТФ (2,5 мкКи µ-32Р-АТФ) и 3 мкг/мл BSA. Оптимизированный пептидный субстрат Abl EAIYAAPFAKKK используется в качестве фосфоакцептора (200 мкМ). Реакции обрываются путем помещения на фосфоцеллюлозные листы, которые промываются 0,5% раствором фосфорной кислоты (примерно 6 раз, 5-10 минут каждый). Листы высушиваются, а перенесенная радиоактивность измеряется при помощи формирования изображения на люминесцентном фосфорном покрытии.
Пример 36: Анализы экспрессии и ингибирования тирозинкиназы рецептора ТЕК (TIE2)
Перекрестная активность или ее недостаток у одного или более соединений настоящего изобретения по отношению к киназе TIE2 может быть измерена в соответствии с любой из методик, известных в данной области, или при помощи способов, описанных ниже. Соединения, описанные в настоящем документе, могут быть проанализированы по отношению к домену рекомбинантной киназы TIE2 (Invitrogen) в образце, содержащем 25 мМ HEPES, рН 7,4, 10 мМ MgCl2, 2 мМ DTT, 10 мМ MnCl2, 10 мкМ АТФ (2,5 мкКи µ-32Р-АТФ) и 3 мкг/мл BSA. В качестве субстрата используется Poly E-Y (Sigma; 2 мг/мл). Реакции обрывают путем помещения на нитроцеллюлозу, которую промывают 1 М раствором NaCl/1% фосфорной кислоты (примерно 6 раз, 5-10 минут каждый). Листы высушиваются, а перенесенная радиоактивность измеряется при помощи формирования изображения на люминесцентном фосфорном покрытии.
Пример 37: Анализ активации и пролиферации В-клеток
Способность одного или более из указанных соединений ингибировать активацию и пролиферацию В-клеток определяется в соответствии со стандартными способами, известными в данной области. Например, существует анализ клеточной пролиферации in vitro, измеряющий метаболическую активность живых клеток. Этот анализ выполняется в 96-луночном микротитровальном планшете с использованием восстановления красителя Аламара синего. Селезеночные В-клетки Balb/c очищают при помощи градиента Ficoll-Paque™ PLUS, а затем магнитного разделения клеток с использованием набора для разделения В-клеток MACS (Miletenyi). Клетки помещают в количестве 90 мкл, 50000 клеток/лунку в среду для В-клеток (RPMI+10% FBS+Penn/Strep+50 мкМ bME+5 мМ HEPES). Соединения, описанные в настоящем документе, разбавляют в среде для В-клеток и добавляют в объеме 10 мкл. Планшеты инкубируют в течение 30 минут при 37°С и 5% CO2 (конечная концентрация ДМСО 0,2%). Затем добавляют 50 мкл стимулирующего коктейля для В-клеток, содержащего 10 мкг/мл LPS или 5 мгк/мл антимышиного IgM ослиного F(ab')2 плюс 2 нг/мл рекомбинантного мышиного IL4 в среде для В-клеток. Планшеты инкубируют в течение 72 часов при 37°С и 5% СО2. В каждую лунку добавляют реагент Аламар синий объемом 15 мкл, и инкубируют планшеты в течение 5 часов при 37°С и 5% CO2. Флуоресценцию Аламара синего считывают при 560Ех/590Em, и рассчитывают значения IC50 или ЕС50 при помощи GraphPad Prism 5.
Пример 38: Анализ пролиферации опухолевой клеточной линии
Способность одного или более из указанных соединений ингибировать пролиферацию опухолевой клеточной линии определяется в соответствии со стандартными способами, известными в данной области. Например, может быть выполнен анализ клеточной пролиферации in vitro для измерения метаболической активности живых клеток. Этот анализ выполняется в 96-луночном микротитровальном планшете с использованием восстановления красителя Аламара синего. Опухолевые клеточные линии человека закупают у АТСС (например, MCF7, U-87 MG, MDA-MB-468, РС-3), выращивают до слияния в колбах Т75, трипсинизируют с 0,25% трипсина, промывают один раз средой для опухолевых клеток (DMEM+10% FBS), и помещают на планшет в количестве 90 мкл, 5000 клеток на лунку в среде для опухолевых клеток. Соединения, описанные в настоящем документе, разбавляют в среде для опухолевых клеток и добавляют в объеме 10 мкл. Планшеты инкубируют в течение 72 часов при 37°С и 5% CO2. В каждую лунку добавляют реагент Аламар синий объемом 10 мкл, и инкубируют планшеты в течение 3 часов при 37°С и 5% CO2. Флуоресценцию Аламара синего считывают при 560Ех/590Em, и рассчитывают значения IC50 при помощи GraphPad Prism 5.
Пример 39: Противоопухолевая активность in vivo
Соединения, описанные в настоящем документе, могут быть оценены на панели моделей опухолей человека и мышей.
Паклитаксел-резистентные модели опухолей
1. Клинически полученная модель карциномы яичника.
Эта модель опухоли получена путем биопсии опухоли у пациента с раком яичника. У пациента взяли биопсию опухоли.
Соединения, описанные в настоящем документе, вводили «голым» мышам с моделью рака, используя график каждые 2 дня х 5.
2. Ксенотрансплантат карциномы яичника человека А2780Тах (мутированный тубулин)
А2780Тах является паклитаксел-резистентной моделью карциномы яичника человека. Ее получают из чувствительной родительской линии А2780 путем совместного инкубирования клеток с паклитакселем и верапамилом, MDR-реверсирующим агентом. Показано, что механизм его резистентности не связан с MDR и относится к мутации гена, кодирующего белок бета-тубулин.
Соединения, описанные в настоящем документе, могут вводиться мышам с моделью рака по графику каждые 2 дня х 5.
3. Ксенотрансплантат карциномы толстой кишки человека НСТ116/VM46 (устойчивый к различным лекарственные средствам)
HCT116/VM46 является MDR-резистентной карциномой толстой кишки, полученной из чувствительной родительской линии НСТ116. In vivo, выращенный в «голых» мышах, HCT116/VM46 устойчиво демонстрирует высокую резистентность к паклитакселу.
Соединения, описанные в настоящем документе, могут вводиться мышам с моделью рака по графику каждые 2 дня х 5.
5. Мышиная модель саркомы М5076
М5076 является мышиной фибросаркомой, которая по своей природе является резистентной к паклитекселу in vivo.
Соединения, описанные в настоящем документе, могут вводиться мышам с моделью рака по графику каждые 2 дня х 5.
Одно или более соединений настоящего изобретения могут использоваться в сочетании с другими терапевтическими средствами in vivo в ксенотрансплантатах карциномы толстой кишки человека, устойчивой ко многим лекарственные средствам, HCT/VM46, или в других моделях, известных в данной области, включая модели, описанные в настоящем документе.
Пример 40: Анализ стабильности микросом
Стабильность одного или более из указанных соединений определяется в соответствии со стандартными способами, известными в данной области. Например, стабильность одного или более соединений определяется в анализе in vitro. В частности, существует анализ стабильности микросом in vitro, который измеряет стабильность одного или более из указанных соединений при взаимодействии с микросомами мышей, крыс или человека, полученными из печени. Реакции микросом с соединениями выполняются в 1,5 мл пробирке Эппендорфа. Каждая пробирка содержит 0,1 мкл 10,0 мг/мл NADPH; 75 мкл 20,0 мг/мл микросом печени мышей, крыс или человека; 0,4 мкл 0,2 М фосфатного буфера и 425 мкл ddH2O. Пробирка негативного контроля (без NADPH) содержит 75 мкл 20,0 мг/мл микросом печени мышей, крыс или человека; 0,4 мкл 0,2 М фосфатного буфера и 525 мкл ddH2O. Реакции инициируют добавлением 1,0 мкл 10,0 мМ исследуемого соединения. Реакционные пробирки инкубируют при 37°С. Образцы объемом 100 мкл отбирают в новую пробирку Эппендорфа, содержащую 300 мкл холодного метанола, через 0, 5, 10, 15, 30 и 60 минут после начала реакции. Образцы центрифугируют при скорости 15000 об./мин для удаления белка. Надосадочную жидкость центрифугированного образца переносят в новую пробирку. Концентрацию устойчивого соединения после реакции с микросомами в надосадочной жидкости измеряют жидкостной хроматографией/масс-спектрометрией (ЖХ-МС).
Пример 41: Анализ стабильности в плазме
Стабильность одного или более из указанных соединений в плазме определяется в соответствии со стандартными способами, известными в данной области. См., например, Rapid Commun. Mass Spectrom., 10: 1019-1026. Следующая методика является ВЭЖХ-МС/МС анализом с использованием плазмы человека; могут использоваться также другие виды, включая обезьян, собак, крыс и мышей. Перед использованием замороженную, гепаринизированную плазму человека оттаивают в холодной воде и вращают в течение 10 минут при скорости 2000 об./мин при 4°С.Заданное соединение добавляют из 400 мкМ маточного раствора в аликвоту предварительно нагретой плазмы для получения конечного объема образца 400 мкл (или 800 мкл для определения периода полувыведения), содержащего 5 мкМ исследуемого соединения и 0,5% ДМСО. Реакционную смесь инкубируют при встряхивании в течение 0 минут и 60 минут при 37°С или в течение 0, 15, 30, 45 и 60 минут при 37°С для определения периода полувыведения. Реакцию прекращают перенесением 50 мкл инкубационной смеси в 200 мкл ледяного ацетонитрила, смесь перемешивают при встряхивании в течение 5 минут. Образцы центрифугируют при 6000 х г в течение 15 минут при 4°С, и 120 мкл надосадочной жидкости переносят в чистые пробирки. Затем образцы выпаривают до сухости и анализируют по ВЭЖХ-МС/МС.
При необходимости одновременно с исследуемыми соединениями анализируется одно или более контрольных или эталонных соединений (5 мкМ): одно соединение, пропоксикаин, с низкой стабильностью в плазме, и другое соединение, пропантелин, со средней стабильностью в плазме.
Образцы восстанавливают в ацетонитриле/метаноле/воде (1/1/2, об./об./об.) и анализируют (ОФ)ВЭЖХ-МС/МС с использованием выбранного мониторинга реакции (SRM). Устройство ВЭЖХ состоит из бинарного ЖХ насоса с автоматическим пробоотборником, колонки смешанного режима, С12, 2×20 мм и градиентной программы. Области пиков, соответствующие аналитам, записываются при помощи ВЭЖХ-МС/МС. Отношение родительского соединения, оставшегося через 60 минут, к количеству, оставшемуся в нулевое время, выраженное в процентах, записывается как стабильность в плазме. В случае определения времени полувыведения, время полувыведения оценивается по наклону первоначальной линейной области логарифмической кривой зависимости оставшегося соединения (%) от времени, принимая кинетику реакции за кинетику первого порядка.
Пример 42: Химическая стабильность
Химическую стабильность одного или более заданных соединений определяют в соответствии со стандартными методиками, известными в данной области. Следующие подробности представляют типичный способ определения химической стабильности заданного соединения. Буфером по умолчанию, используемым для анализа химической стабильности, является солевой фосфатный буфер (PBS) при рН 7,4; могут использоваться другие подходящие буферы. Заданное соединение добавляется из 100 мкМ маточного раствора в аликвоту PBS (в двух экземплярах) для получения конечного объема образца 400 мкл, содержащего 5 мкМ исследуемого соединения и 1% ДМСО (для определения периода полувыведения готовится образец общим объемом 700 мкл). Реакции инкубируются при встряхивании в течение 0 минут и 24 часов при 37°С; для определения периода полувыведения образцы инкубируются в течение 0, 2, 4, 6 и 24 часов. Реакции прекращают немедленным добавлением 100 мкл инкубационной смеси к 100 мкл ацетонитрила и перемешивают на вортексе в течение 5 минут. Затем образцы хранят при -20°С до проведения анализа ВЭЖХ-МС/МС. При необходимости контрольное или эталонное соединение, такое как хлорамбуцил (5 мкл) анализируется одновременно с исследуемым соединением, поскольку это соединение в значительной степени гидролизуется за 24 часа. Образцы анализируются с помощью (ОВ)ВЭЖХ-МС/МС с использованием выбранного мониторинга реакции (SRM). Устройство ВЭЖХ состоит из бинарного ЖХ насоса с автоматическим пробоотборником, колонки смешанного режима, С12, 2×20 мм и градиентной программы. Области пиков, соответствующие аналитам, записываются при помощи ВЭЖХ-МС/МС. Отношение родительского соединения, оставшегося через 24 часа, к количеству, оставшемуся в нулевое время, выраженное в процентах, записывается как химическая стабильность. В случае определения времени полувыведения, время полувыведения оценивается по наклону первоначальной линейной области логарифмической кривой зависимости оставшегося соединения (%) от времени, принимая кинетику реакции за кинетику первого порядка.
Пример 43: Анализ киназы Akt
Клетки, содержащие компоненты пути Akt/mTOR, включая, но не ограничиваясь, миобласты L6, клетки B-ALLs, В-клетки, Т-клетки, клетки лейкоза, клетки костного мозга, преобразованные клетки р190, положительные клетки филадельфийской хромосомы (Ph+) и мышиные эмбрионные фибробласты, обычно выращивают в среде для выращивания клеток, такой как DMEM, дополненной бычьей эмбриональной сывороткой и/или антибиотиками, и выращивают их до слияния.
Для сравнения действия одного или более соединений, описанных в настоящем документе, на активацию Akt, указанные клетки оставляют без сыворотки на ночь и инкубируют с одним или более соединений, описанных в настоящем документе, или примерно 0,1% ДМСО примерно в течение от 1 минуты до примерно 1 часа, а затем стимулируют инсулином (например, 100 нМ) примерно в течение от 1 минуты до примерно 1 часа. Клетки лизируют соскабливанием в ледяной лизисный буфер, содержащий детергенты, такие как натрия додецилсульфат и ингибиторы протеазы (например, PMSF). После взаимодействия клеток с лизисным буфером, раствор быстро обрабатывают ультразвуком, просветляют центрифугированием, разделяют в SDS-PAGE, переносят на нитроцеллюлозу или ПВДФ и подвергают иммуноблоттингу с использованием антител к фосфо-Akt S473, фосфо-Akt Т308, Akt и β-актину (Cell Signaling Technologies).
Результаты показали, что одно или более соединений настоящего описания ингибируют инсулин-стимулированное фосфорилирование Akt на S473. Альтернативно, некоторые соединения, описанные в настоящем документе, дополнительно ингибируют инсулин-стимулированное фосфорилирование Akt на Т308. Такой класс соединений может ингибировать Akt более эффективно, чем рапамицин и может быть индикативным для ингибиторов mTORC2 или ингибиторов предыдущих киназ, таких как PI3K или Akt.
Пример 44: Сигналинг киназ в крови
Сигналинг P13K/ Akt /mTor измеряется в клетках крови при помощи способа Phosflow (Methods Enzymol. 2007; 434:131-54). Преимуществом этого способа является то, что это природный анализ сигнальной клетки, поэтому может быть обнаружена клеточная гетерогенность, а не средняя популяция. Это позволяет выполнять конкурентное распознавание сигнальных состояний в различных популяциях, определенных другими маркерами. Phosflow также является высоко количественным анализом. Для анализа действия одного или более соединений, описанных в настоящем документе, нефракционированные спленоциты или одноядерные клетки периферийной крови стимулируют при помощи анти-CD3 для инициирования сигналинга рецепторов Т-клеток. Затем клетки фиксируют и окрашивают для поверхностных маркеров и внутриклеточных фосфопротеинов. Предполагается, что ингибиторы, описанные в настоящем документе, ингибируют анти-CD3-опосредованное фосфорилирование Akt - S473 и S6, тогда как рапамицин ингибирует фосфорилирование S6 и усиливает фосфорилирование Akt в исследуемых условиях.
Аналогично, аликвоты цельной крови инкубируют в течение 15 минут с носителем (например, 0,1% ДМСО) или ингибиторами киназы в различных концентрациях, а затем добавляют стимулятор для сшивания рецептора Т-клеток (TCR) (анти-CD3 со вторичным антителом) или рецептора В-клеток (BCR) при помощи антикаппа антитела легкой цепи (фрагменты Fab'2). Примерно через 5 и 15 минут образцы фиксируют (например, холодным 4% параформальдегидом) и используют для анализа Phosflow. Для различения Т и В клеток используют поверхностное окрашивание при помощи антител, направленных на маркеры клеточной поверхности, которые известны в данной области. Затем измеряется уровень фосфорилирования субстратов киназ, таких как Akt и S6 при помощи инкубирования фиксированных клеток мечеными антителами, специфичными к фосфорилированным изоформам этих белков. Затем популяции клеток анализируют проточной цитометрией.
Пример 45: Анализ образования колоний
Мышиные клетки костного мозга, только что преобразованные ретровирусом BCR-Abl р190 (в настоящем документе упоминаются как преобразованные клетки р190), помещают в чашки в присутствии различных комбинаций лекарств в среде метилцеллюлозы М3630 примерно на 7 дней с рекомбинантным IL-7 человека примерно в 30% сыворотке, и подсчитывают количество образовавшихся колоний при визуальном исследовании под микроскопом.
Альтернативно, одноядерные клетки периферийной крови человека получают от пациентов с положительной (Ph+) и отрицательной филадельфийской хромосомой (Ph-) при первоначальном диагнозе или рецидиве. Живые клетки выделяют и обогащают CD19+CD34+прародителями В клеток. Спустя ночь культивирования в жидкой среде, клетки помещают в чашки в Methocult GF+Н4435 (Stem Cell Tehcnologies), дополненные цитокинами (IL-3, IL-6, IL-7, G-CSF, GM-CSF, CF, лиганд Flt3 и эритропоэтин) в различных концентрациях известных химиотерапевтических средств в комбинации с соединениями настоящего описания. Колонии подсчитывают под микроскопом через 12-14 дней. Этот способ может использоваться для проверки доказательств аддитивной или синергетической активности.
Пример 46: Влияние ингибиторов киназы на лейкозные клетки in vivo
Мышь-реципиент женского пола получила летальную дозу излучения из γ-источника двумя дозами с интервалом примерно 4 ч, каждый примерно по 5 Гр. Спустя 1 час после второй дозы облучения, мыши внутривенно ввели примерно 1×106 лейкозных клеток (например, клетки Ph+человека или мыши, или преобразованные клетки костного мозга р190). Эти клетки вводятся вместе с противолучевой дозой примерно из 5×106 нормальных клеток костного мозга, полученных от донорной мыши в возрасте 3-5 недель. Реципиентам давали антибиотики в воде, и ежедневно контролировали. Мышей, заболевших примерно через 14 дней, умертвили, и собрали лимфоидные органы для анализов. Лечение ингибиторами киназы начали примерно через 10 дней после инъекции лейкозных клеток и продолжали ежедневно до момента заболевания мышей или максимально примерно 35 дней после трансплантации. Ингибиторы вводили пероральным лаважем.
Клетки периферийной крови собрали примерно на 10 день (перед лечением) и в момент эвтаназии (после лечения), обработали мечеными анти-hCD4 антителами и подсчитали проточной цитометрией. Этот способ может использоваться для демонстрации того, что синергетический эффект одного или более соединений, описанных в настоящем изобретении, в комбинации с известными химиотерапевтическими средствами значительно снижает количество лейкозных клеток крови по сравнению с лечением только известными химиотерапевтическими средствами (например, Гливек) в исследуемых условиях.
Пример 47: Лечение волчанки на мышиной модели
У мышей с недостатком ингибирующих рецепторов FcγRIIb, которые препятствуют сигналингу PI3K в В-клетках, с высокой пенетрантностью развивается волчанка. Нокаутные мыши FcrRIIb (R2KO, Jackson Labs) являются валидной моделью болезни человека, поскольку некоторые пациенты с волчанкой демонстрируют пониженную экспрессию функции FcrRIIb (S. Bolland и J.V. Ravtech 2000. Immunity 12:277-285).
У мышей R2KO развивается заболевание, подобное волчанке с анти-ядерными антителами, гломерулонефрит и протеинурея в возрасте 4-6 месяцев. Для этих экспериментов в качестве эталонного соединения используется аналог рапамицина RAD001 (выпускается LC Laboratories), который вводится перорально. Показано, что это соединение облегчает симптомы волчанки в модели B6.Sle1z.Sle3z (Т. Wu et al. J. Clin Invest 117:2186-2196).
Мышиные модели волчанки, такие как R2KO, BXSB или MLR/1pr, подвергают лечению примерно в возрасте 2 месяца, в течение примерно 2 месяцев. Мышам вводят дозы: носителя, RAD001 в дозировке примерно 10 мг/кг, или соединения, описанные в настоящем документе в дозировке от примерно 1 мг/кг до примерно 500 мг/кг. Образцы крови и мочи получают примерно на всем протяжении периода испытания, и анализируют на антиядерные антитела (в разбавленной сыворотке) или концентрацию белка (в моче). Сыворотку также анализируют на антитела анти-оцДНК и анти-дцДНК при помощи иммуноферментного твердофазного анализа ELISA. Животных умертвляют на 60 день и собирают ткани для измерения веса селезенки и заболевания почек. Гломерулонефрит определяют в сегментах почек, окрашенных Н&Е. Других животных исследуют в течение примерно двух месяцев после прекращения лечения, используя такие же конечные точки.
Эта модель, принятая в данной области техники, может использоваться для демонстрации того, что ингибиторы киназ, описанные в настоящем документе, подавляют или замедляют появление симптомов волчанки в мышиных моделях волчанки.
Пример 48: Анализ трансплантата костного мозга мышей
Мышь-реципиент женского пола получила летальную дозу излучения из γ-источника. Примерно через 1 ч. после дозы облучения мыши ввели примерно 1x106 лейкозных клеток из раннего пассажа преобразованных культур р190 (например, как описано в публикации Cancer Genet Cytogenet. 2005 Aug; 161(l):51-6). Эти клетки вводятся вместе с противолучевой дозой примерно из 5×106 нормальных клеток костного мозга, полученных от донорной мыши возрастом 3-5 недель. Реципиентам давали антибиотики в воде, и ежедневно контролировали. Мышей, заболевших примерно через 14 дней, умертвили и собрали лимфоидные органы для проточной цитометрии и/или магнитного обогащения. Лечение начали примерно на 10 день и продолжали ежедневно до наступления заболевания у мыши, или максимум в течение примерно 35 дней после трансплантации. Лекарственные средства вводили через пероральный желудочный зонд. В пилотном эксперименте определили дозу химиотерапевтического средства, которое не является лечебным, но замедляет возникновение лейкоза примерно на одну неделю или менее; контрольные образцы лечили носителем или лечили химиотерапевтическим средством, которое предварительно продемонстрировало замедление, но не излечивание лейкемогенеза в этих моделях (например, иматиниб в концентрации примерно 70 мг/кг дважды в день). Для первой фазы использовали клетки р190, экспрессирующие eGFP, и анализ после вскрытия был ограничен подсчетом процентного содержания лейкозных клеток в костном мозге, селезенке и лимфатическом узле при помощи проточной цитометрии. На второй фазе использовали клетки р190, экспрессирующие бесхвостую форму CD4 человека, и анализ после вскрытия включал магнитную сортировку клеток hCD4+из селезенки с последующим иммуноблоттинговым анализом ключевых сигнальных концевых точек: р Akt -Т308 и S473; pS6 и р4ЕВР-1. В качестве контроля для иммуноблоттингового определения отсортированные клетки инкубировали в присутствии или в отсутствие ингибиторов киназы настоящего описания перед лизисом. Возможно, использовали анализ «Phosflow» для обнаружения р Akt -S473 и pS6-S235/236 в клетках, дающих сигнал hCD4 выше порогового значения, без предварительной сортировки. Эти сигналинговые исследования являются особенно полезными, например, если у мышей, подверженных лечению лекарственные средствами, не развивается клинический лейкоз на 35 день. Строятся диаграммы выживания Каплана-Мейера и выполняется статистический анализ в соответствии со способами, известными в данной области. Результаты клеток р190 анализируются по отдельности и вместе.
Образцы периферической крови (100-200 мкл) получают раз в неделю от всех мышей, начиная с 10 дня непосредственно до начала лечения. Плазма используется для измерения концентрации лекарственные средства, а клетки анализируются на маркеры лейкоза (eGFP или hCD4) и биомаркеры сигналинга, как описано в настоящем документе.
Этот общий анализ является известным в данной области и может использоваться для демонстрации того, что эффективные терапевтические дозировки соединений, описанных в настоящем документе, могут использоваться для ингибирования пролиферации лейкозных клеток.
Пример 49: Анализ активации В-клеток, независимо от Т-клеток на TNP-фиколле
Для исследования действия соединений настоящего изобретения на подавление выработки антител, независимой от Т-клеток, использовался анализ активации В-клеток на TNP-фиколле, как описано в настоящем документе. Соединения по настоящему изобретению растворили в соответствующем носителе (например, 5% 1-метил-2-пирролидинона, 85% полиэтиленгликоля 400, 10% Солютора). Соединения вводили перорально примерно за 1 час до обработки TNP-фиколлом, мышам в возрасте 4-10 недель. Для изучения действия соединений настоящего изобретения на активацию В-клеток, одну партию мышей сгруппировали в соответствии со следующей таблицей:

Четырех животных в группе 1 и восемь животных в группах 2-7 умертвили в CO2 через 2 часа после последнего введения соединения на 7 день. Сразу собрали кровь путем сердечной пункции и хранили при 37°С в течение 1 ч. для сворачивания, а затем инкубировали в течение ночи при 4°С для сокращения сгустков. На следующий день сыворотку собрали декантацией и центрифугировали при 3000 об./мин. в течение 10 минут. Затем собранную сыворотку заморозили при -80°С для последующих анализов. Образцы сыворотки анализировали на титры антител анти-TNP при помощи иммуноферментного твердофазного анализа ELISA, как описано в настоящем документе. TNP-BSA нанесли на микротитровальный планшет Nunc Maxisorb в количестве 100 мкл на лунку в концентрации м 100 мг/мл в фосфатом буферном солевом растворе (PBS). Планшет Maxisorb инкубировали в течение 1,5 часов при комнатной температуре, а затем удалили раствор. В каждую лунку добавили 200 мкл на лунку блокирующего буфера (например, 1% BSA в PBS) и инкубировали при комнатной температуре в течение 1 часа. Планшет промыли один раз объемом 200 мкл на лунку 0,05% раствором Твин-20 в PBS (промывочный буфер). В каждую лунку первой колонки (1) микротитровального планшета добавили 1:2 разбавленную сыворотку от каждой мыши в блокирующем буфере. Затем сыворотку в каждой лунке 1 колонки 3-кратно разбавили в блокирующем буфере и поместили в колонку 2. Сыворотку в каждой лунке 2 колонки 3-кратно разбавили в блокирующем буфере и поместили в колонку 3. Процедуру повторили по всем двенадцати колонкам микротитровального планшета. Микротитровальный планшет инкубировали в течение 1 часа при комнатной температуре. Сыворотку удалили из планшета, а планшет промыли три раза промывочным буфером. В каждую лунку добавили 100 мкл на лунку козьего антимышиного IgG3-HRP, разбавленного 1:250 в блокирующем буфере, и инкубировали в течение 1 часа при комнатной температуре. Антимышиный IgG3-HRP удалили из микротитровального планшета, и промыли планшет шесть раз промывочным буфером. Субстрат HRP (200 мкл раствора ABTS+30% H2O2+10 мл цитратного буфера) поместили в каждую лунку в количестве 100 мкл на лунку, инкубировали 2-20 минут в темноте и спектрофотометрически определили количество анти-TNP IgG3 при 405 нм. Аналогично определили анти-TNP IgM и общий анти-TNP Ab, используя анти-мышиный IgM-HRP и анти-мышиный Ig-HRP, соответственно.
Результаты представлены на фиг. 2, где дополнительно показано, что при исследуемых условиях соединения №7 и №53 демонстрируют 3,4 и 6,5-кратное снижение, соответственно, уровня IgG3 по сравнению с контрольными мышами, обработанными носителем, при уровне дозировки 30 мг/кг. На фигуре 2 дополнительно показано, что соединение №53 демонстрирует 29,9-кратное снижение уровня IgG3 по сравнению с контрольными мышами, обработанными носителем, при уровне дозировки 60 мг/кг при исследуемых условиях.
Пример 50: Анализ развивающегося коллаген-индуцированного артрита типа II у крыс
Для изучения влияния соединений настоящего изобретения на аутоиммунное заболевание артритом, использовали модель развивающегося коллаген-индуцированного артрита. Крысам Lewis женского пола ввели коллагеновую инъекцию в 0 день. Бычий коллаген типа II приготовили в виде раствора концентрацией 4 мг/мл в 0,01 н. уксусной кислоте. Равные объемы коллагена и неполного адъюванта Фрейнда эмульгировали перемешиванием вручную до тех пор, пока шарики эмульгированного материала не стали удерживать свою форму в воде. Каждому грызуну ввели 300 мкл инъекции смеси при каждой инъекции, распределяя по трем подкожным точкам на спине.
Пероральное введение соединения начали в 0 день и продолжали до 16 дня с носителем (5% NMP, 85% ПЭГ 400, 10% Солютола) или с соединением настоящего изобретения в носителе или с контрольным соединением (например, метотрексатом) с 12 часовым интервалом, ежедневно. Крыс взвесили на 0, 3, 6, 9-17 дни и провели измерения толщины лодыжки на 9-17 день. Измерили конечную массу тела, а затем умертвили животных на 17 день. После эвтаназии слили кровь и удалили задние лапы и колени. Затем кровь переработали для фармакокинетических экспериментов, а также для анализа ELISA коллагенового антитела анти-типа II. Задние лапы взвесили, а затем законсервировали с коленями в 10% формалине. Затем лапы и колени обработали для микроскопии. Печень, селезенку и тимус также взвесили. Седалищные нервы подготовили для гистопатологии.
Суставы коленей и лап зафиксировали в течение 1-2 дней и декальцинировали в течение 4-5 дней. Суставы лодыжек разрезали в продольном направлении пополам, колени разрезали пополам вдоль фронтальной плоскости. Затем суставы обработали, залили, разделили и окрасили толуидиновым синим. Количественную оценку суставов выполнили в соответствии со следующими критериями:
Воспаление колен и лодыжек
0=Нормальные
1=Минимальная инфильтрация воспалительных клеток в синовиальную/вокругсуставную ткань
2=Слабая инфильтрация
3=Умеренная инфильтрация с умеренным отеком
4=Заметная инфильтрация с выраженным отеком
5=Сильная инфильтрация с сильным отеком
Паннус лодыжек
0=Нормальные
1=Минимальная инфильтрация паннуса в хрящ и субхондральную кость
2=Слабая инфильтрация (<1/4 большеберцовой кости или предплюсны в краевых зонах)
3=Умеренная инфильтрация (от 1/4 до 1/3 большеберцовой кости или небольшая часть предплюсны повреждены в краевых зонах)
4=Заметная инфильтрация (1/2-3/4 большеберцовой кости или предплюсны повреждены в краевых зонах)
5=Сильная инфильтрация (>3/4 большеберцовой кости или предплюсны повреждены в краевых зонах, сильная деформация общей конфигурации)
Паннус колен
0=Нормальные
1=Минимальная инфильтрация паннуса в хрящ и субхондральную кость
2=Слабая инфильтрация (распространяется на поверхности площадью до 1/4 поверхности субхондральной области большеберцовой кости или бедренной кости)
3=Умеренная инфильтрация (распространяется на поверхности площадью свыше 1/4, но <1/2 поверхности субхондральной области большеберцовой кости или бедренной кости)
4=Заметная инфильтрация (распространяется на поверхности площадью от 1/2 до 3/4 большеберцовой кости или бедренной кости)
5=Сильная инфильтрация (покрывает >3/4 поверхности)
Повреждение хрящей (лодыжка, выделения на небольших предплюснах)
0=Нормальные
1=Минимальное=от минимального до слабого снижение окрашивания толуидинового синего без явной потери хондроцитов или деструкции коллагена
2=Слабое=слабое снижение окрашивания толуидинового синего с очаговой слабой (поверхностной) потерей хондроцитов и/или деструкцией коллагена
3=Умеренное=умеренное снижение окрашивания толуидинового синего с многоочаговой умеренной (глубокой или средней зоны) потерей хондроцитов и/или деструкцией коллагена, более мелкие предплюсны повреждены на 1/2-3/4 глубины
4=Заметное=заметное снижение окрашивания толуидинового синего с многоочаговой заметной (глубокой и очень глубокой зоны) потерей хондроцитов и/или деструкцией коллагена, 1 или более небольших предплюсен имеют потерю хряща по всей толщине.
5=Сильное=сильное диффузное снижение окрашивания толуидинового синего с многоочаговой сильной (от очень глубокой до полной) потерей хондроцитов и/или деструкцией коллагена
Повреждение хрящей (колено, выделения на бедренных мыщелоках)
0=Нормальные
1=Минимальное=от минимального до слабого снижение окрашивания толуидинового синего без явной потери хондроцитов или деструкции коллагена
2=Слабое=слабое снижение окрашивания толуидинового синего с очаговой слабой (поверхностной) потерей хондроцитов и/или деструкцией коллагена
3=Умеренное=умеренное снижение окрашивания толуидинового синего с многоочаговой или умеренной диффузной (от глубокой до средней зоны) потерей хондроцитов и/или деструкцией коллагена
4=Заметное=заметное снижение окрашивания толуидинового синего с многоочаговой или заметной диффузной (от глубокой до очень глубокой зоны) потерей хондроцитов и/или деструкцией коллагена; или одна бедренная поверхность с полной или почти полной потерей
5=Сильное=сильное диффузное снижение окрашивания толуидинового синего с многоочаговой сильной (от очень глубокой до полной) потерей хондроцитов и/или деструкцией коллагена на обеих бедренных костях и/или большеберцовых костях
Резорбция кости (лодыжка)
0=Нормальная
1=Минимальная=неболыпие области резорбции, не сразу заметные при слабом увеличении, редкие остеокласты
2=Слабая=более многочисленные области резорбции, не сразу заметные при слабом увеличении, более частые остеокласты, резорбировано <1/4 большеберцовой кости или предплюсны в краевых зонах
3=Умеренная=заметная резорбция медуллярного трабекулярного и кортикального слоя кости без повреждения кортекса по всей толщине, потеря некоторых медуллярных трабекул, отдельные повреждения при слабом увеличении, более многочисленные остеокласты, повреждено от 1/4 до 1/3 большеберцовой кости или предплюсны в краевых зонах
4=Заметная=дефекты кортикального слоя кости по всей толщине, зачастую с искажением профиля оставшейся кортикальной поверхности, заметная потеря медуллярной кости, многочисленные остеокласты, повреждено от 1/2 до 3-4 большеберцовой кости или предплюсны в краевых зонах
5=Сильная=дефекты кортикального слоя кости по всей толщине, зачастую с искажением профиля оставшейся кортикальной поверхности, заметная потеря медуллярной кости, многочисленные остеокласты, повреждено >3/4 большеберцовой кости или предплюсны в краевых зонах, сильное искажение общей конфигурации
Резорбция кости (колено)
0=Нормальная
1=Минимальная=небольшие области резорбции, не сразу заметные при слабом увеличении, редкие остеокласты
2=Слабая=более многочисленные области резорбции, определенная потеря субхондральной кости, затрагивающая 1/4 поверхности большеберцовой кости или бедра (медиальной или поперечной)
3=Умеренная=заметная резорбция субхондральной кости, затрагивающая >1/4, но <1/2 поверхности большеберцовой кости или бедра (медиальной или поперечной)
4=Заметная=заметная резорбция субхондральной кости, затрагивающая ≥1/2, но <3/4 поверхности большеберцовой кости или бедра (медиальной или поперечной)
5=Сильная=деформация сустава в целом из-за разрушения, затрагивающего >3/4 поверхности большеберцовой кости или бедра (медиальной или поперечной)
Статистический анализ массы тела/лапы, параметры лап AUC и гистопатологические параметры были оценены с использованием t-теста Стьюдента и других соответствующих тестов (ANOVA с повторной проверкой) при заданном значении достоверности 5%. Процент ингибирования веса лап и AUC рассчитали по следующей формуле:
% ингибирования=А - В/А X 100
А=Среднее контрольного заболевания - Среднее нормальное
В=Среднее при лечении - Среднее нормальное
Результаты, представленные на фиг. 3, демонстрируют влияние соединения №53 в дозировках 10, 30, и 60 мг/кг с интервалом 12 часов на средний диаметр лодыжки с течением времени в модели развивающегося коллаген-индуцированного артрита типа II на крысах, в исследуемых условиях. По сравнению с контрольным образцом с носителем, или с контрольным образцом с метотрексатом, соединения по настоящему изобретению демонстрируют значительное снижение роста диаметра лодыжки из-за артрита с течением времени.
Результаты, представленные на фиг. 4, демонстрируют влияние соединений №7 и №53 на гистопатологию лодыжки в категориях воспаления, паннуса, повреждения хряща и костной резорбции, как описано ранее, в исследуемых условиях. Результаты демонстрируют значительное снижение в одной или более категорий в результате действия соединений настоящего изобретения (т.е. соединения №53) в исследуемых условиях. На фигуре 4 дополнительно показано, что в концентрации 60 мг/кг существует статистически значимое сокращение во всех категориях гистопатологии лодыжки для одного из соединений настоящего изобретения (т.е. соединения №53), в исследуемых условиях. Это позволяет предположить, что одно или более соединений настоящего изобретения могут быть пригодными для лечения и уменьшения симптомов заболевания артритом.
Результаты, представленные на фиг. 5, демонстрируют влияние соединений №7 и №53 на гистопатологию колена в исследуемых условиях. Результаты показывают дозозависимое снижение гистопатологии колена. Это позволяет предположить, что одно или более соединений настоящего изобретения могут быть пригодными для лечения и уменьшения симптомов заболевания артритом.
Результаты, представленные на фиг. 6, демонстрируют влияние соединений №7 и №53 на уровень сывороточного коллагена анти-типа II в исследуемых условиях. Результаты дополнительно показывают значительное снижение уровня сывороточного коллагена анти-типа II при уровнях дозировки 10, 20 и 60 мг/кг, для соединения №53, что позволяет предположить, что одно или более соединений настоящего изобретения не только могут быть пригодными для лечения и уменьшения симптомов заболевания артритом, но и могут быть полезными для ингибирования самой аутоиммунной реакции.
Результаты, представленные на фиг. 7, демонстрируют влияние соединения №7 в дозировках 10, 30 и 60 мг/кг с интервалом 12 часов на средний диаметр лодыжки с течением времени в исследуемых условиях. По сравнению с контрольным образцом с носителем, или с контрольным образцом с метотрексатом, соединения по настоящему изобретению демонстрируют снижение роста диаметра лодыжки из-за артрита в исследуемых условиях. При испытании на такой же модели по меньшей мере пять других соединений настоящего изобретения демонстрируют сравнимую или даже более высокую эффективность.
Пример 51: Анализ хронического коллаген-индуцированного артрита типа II у крыс
Для исследования эффективности соединений настоящего изобретения в зависимости от дозы при ингибировании воспалений, разрушения хрящей и резорбции кости 10-дневного хронического коллаген-индуцированного артрита типа II у крыс, соединения перорально вводили один раз или дважды в день в течение 6 дней.
Крыс Lewis женского пола анестезировали и ввели коллагеновую инъекцию, приготовленную и введенную так, как описано ранее, в 0 день. На 6 день животных анестезировали и ввели вторую инъекцию коллагена. Измерение толщины нормального (до заболевания) правого и левого голеностопных суставов выполнили на 9 день. На 10-11 день обычно появлялся артрит, и крыс случайным образом разделили на группы лечения. Рандомизацию выполнили после очевидного опухания голеностопного сустава и появления достаточных доказательств двустороннего заболевания.
После отбора животных для подготовки к исследованию, начали лечение пероральным путем. Животным давали носитель, контроль (Энбрел) или соединения по настоящему изобретению, дважды в день или один раз в день (BID или QD, соответственно). Дозы вводили на 1-6 день в объеме 2,5 мл/кг (BID) или 5 мл/кг (QD) для пероральных растворов. Крыс взвесили на 1-7 день после образования артрита, и проводили измерения толщины лодыжек ежедневно. Конечный вес тела измерили на 7 день, и умертвили животных.
Результаты, представленные на фиг. 8, демонстрируют существенное снижение роста диаметра лодыжки с течением времени для соединения №53 при однократном ежедневном приеме в исследуемых условиях. Результаты, представленные на фиг. 9, дополнительно демонстрируют существенное снижение роста диаметра лодыжки с течением времени для соединения №53 при двукратном ежедневном приеме в исследуемых условиях. Это позволяет предположить, что настоящее изобретение может быть полезным для лечения аутоиммунных заболеваний, таких как артрит. При испытании в такой же модели, по меньшей мере пять других соединений настоящего изобретения демонстрируют сравнимую или даже более высокую эффективность по сравнению с соединением №53.
Пример 52: Анализ адъювант-индуцированного артрита
Интратекальная катетеризация крыс
Крысам Lewis (200-250 г), анестезированным изофлураном, имплантировали интратекальный (IT) катетер. Спустя 6 дней периода реабилитации, всех животных, за исключением тех, у кого обнаружены сенсорные или двигательные аномалии (менее 5% от общего количества), использовали для экспериментов. Для интратекального введения использовали инъекцию 10 мкл лекарственные средства или солевого раствора с последующим введением 10 мкл изотонического солевого раствора через катетер.
Адъювантный артрит и лечение лекарственные средствами
Крыс Lewis иммунизировали у основания хвоста 0,1 мл полного адъюванта Фрейнда (CFA) на 0 день, спустя несколько дней после имплантации катетера (п=6/группа). Лечение лекарством (например, одним или более соединений настоящего изобретения или носителем) обычно начинали на 8 день и продолжали ежедневно до 20 дня. Клинические признаки артрита обычно появлялись на 10 день, и опухание лап определяли через день путем плетизмометрии вытеснения воды.
Результаты, представленные на фиг. 10, по среднему изменению объема лап при указанных режимах дозирования, демонстрируют, что при исследуемых условиях соединение №53 дает дозозависимое снижение роста среднего объема лап, что было измерено в этой системе адъювант-индуцированной модели артрита. Результаты позволяют предположить, что одно или более соединений настоящего изобретения могут быть полезными для лечения одного или более заболеваний или состояний, описанных в настоящем документе.
Результаты, представленные на фиг. 11, показывают, что соединение №53 не демонстрирует токсичности или других побочных реакций в исследуемых условиях, что было измерено по отсутствию потери веса.
Пример 53: Фармакокинетический анализ на грызунах
Для изучения фармакокинетики соединений настоящего изобретения партию мышей возрастом 4-10 недель сгруппировали в соответствии со следующей таблицей.

Соединения по настоящему изобретению растворили в соответствующем носителе (например, 5% 1-метил-2-пирролидинона, 85% полиэтиленгликоля 400, 10% Солютора) и вводили перорально с интервалом 12 часов ежедневно. Всех животных умертвили в CO2 через 2 часа после последнего введения соединения. Сразу собрали кровь и выдержали на льду для отделения плазмы. Плазму отделили центрифугированием при скорости 5000 об./мин. в течение 10 минут. Собранную плазму заморозили для фармакокинетического определения.
Результаты должны показать фармакокинетические параметры, такие как абсорбция, распределение, метаболизм, экскреция и токсичность соединений настоящего изобретения.
Пример 54: Анализ Basotest
Анализ Basotest выполняется с использованием набора реагентов Orpegen Pharma Basotest. Гепаринизированную цельную кровь предварительно инкубируют с исследуемым соединением или растворителем при 37°С в течение 20 минут. Затем кровь инкубируют со стимуляционным буфером из аналитического набора (для реакции первичных клеток), а затем с аллергеном (экстракт пылевого клеща или травяной экстракт) в течение 20 минут. Процесс дегрануляции прекращают путем инкубирования образцов крови на льду. Затем клетки помечают анти-IgE-PE для обнаружения базофильных гранулоцитов и анти-gp53-FITC для обнаружения gp53 (гликопротеин, экспрессированный на активированных базофилах). После окрашивания красные кровяные клетки лизируют добавлением лизирующего раствора. Клетки промывают и анализируют проточной цитометрией. Испытания соединений 7 и 53 в этом анализе показали, что они ингибируют аллерген-индуцированную активацию базофильных гранулоцитов в субмикромолярном диапазоне.
Пример 55: Комбинированное применение ингибиторов PI3Кд и агентов, ингибирующих выработку или активность IgE
Соединения по настоящему изобретению могут давать синергическую или аддитивную эффективность при введении в комбинации с агентами, ингибирующими выработку или активность IgE. Агенты, ингибирующие выработку IgE, включают, например, один или более из TEI-9874, 2-(4-(6-циклогексилокси-2-нафтилокси)фенилацетамид)бензойной кислоты, рапамицина, аналогов рапамицина (т.е. рапалогов), ингибиторов TORC1, ингибиторов TORC2, и любых других соединений, ингибирующих mTORC1 и mTORC2. Агенты, ингибирующие активность IgE, включают, например, анти-IgE антитела, такие как омализумаб и TNX-901.
Одно или более из исследуемых соединений, способных ингибировать PI3Kδ, являются эффективными для лечения аутоиммунных и воспалительных заболеваний (AIID), например, ревматоидного артрита. Если любое из соединений вызывает нежелательный уровень выработки IgE, то его можно выбрать для введения в комбинации с агентом, ингибирующим выработку IgE или активность IgE. Кроме того, введение ингибиторов PI3K8 или PI3Kδ/γ настоящего изобретения в комбинации с ингибиторами mTOR также может демонстрировать синергию за счет усиленного ингибирования пути PI3K. Могут использоваться различные модели in vivo и in vitro для определения влияния такого комбинированного лечения на AIID, включая, не ограничиваясь, (а) анализ выработки антител В-клеток in vitro, (b) анализ TNP in vivo, и (с) модель коллаген-индуцированного артрита у грызунов.
(a) Анализ В-клеток
Мышей умертвили, удалили селезенку, и диспергировали через нейлоновое сито для получения одноклеточной суспензии. Спленоциты промыли (с последующим удалением эритроцитов при помощи осмотического шока) и инкубировали с анти-CD43 и анти-Мас-1 антитело-конъюгированными микрогранулами (Miltenyi Biotec). Клетки, связанные с гранулами, отделили от несвязанных клеток при помощи магнитного сортировщика клеток. Намагниченная колонка удерживает нежелательные клетки, а остальные В-клетки собираются в фильтрате. Очищенные В-клетки стимулировали липополисахаридом или анти-CD40 антителом и интерлейкином 4. Стимулированные В-клетки обработали только носителем или носителем с ингибиторами PI3Kδ настоящего изобретения, такими как соединение 53 в присутствии ингибиторов mTOR, таких как рапамицин, рапалоги или ингибиторы mTORC1/C2 или без них. Результаты должны демонстрировать, что в присутствии только ингибиторов mTOR (например, рапамицина) существует лишь небольшое или несущественное действие на реакцию IgG и IgE. Однако в присутствии ингибиторов PI3Kд и mTOR, В-клетки должны демонстрировать пониженную реакцию IgG по сравнению с В-клетками, обработанными только носителем; и В-клетки должны демонстрировать пониженную реакцию IgE по сравнению с В-клетками, обработанными только ингибиторами PI3Kд.
(b) Анализ TNP
Мышей иммунизовали TNP-фиколлом или TNP-KHL и обработали: носителем, ингибитором PI3Kδ, например, соединением 53 настоящего изобретения, ингибитором mTOR, например, рапамицином или ингибитором PI3Kδ в комбинации с ингибитором mTOR, таким как рапамицин. Антиген-специфичный сывороточный IgE измерили при помощи ELISA с использованием планшетов, покрытых TNP-BSA и изотип-специфичных меченых антител. Предполагается, что мыши, обработанные только ингибитором mTOR демонстрируют небольшое или незначительное влияние на антиген-специфичную реакцию IgG3 и статистически незначимое увеличение реакции IgE по сравнению с контрольным образцом. Также предполагается, что мыши, обработанные ингибитором PI3Kδ и ингибитором mTOR, демонстрируют снижение антиген-специфичной реакции IgG3 по сравнению с мышами, обработанным только носителем. Кроме того, мыши, обработанные ингибитором PI3Кд и ингибитором mTOR, демонстрируют снижение реакции IgE по сравнению с мышами, обработанными только ингибитором PI3Кд.
(с) Модель коллаген-индуцированного артрита у крыс
Крыс Lewis женского пола анестезировали и ввели коллагеновую инъекцию, приготовленную и введенную так, как описано ранее, в 0 день. На 6 день животных анестезировали и ввели вторую инъекцию коллагена. Измерение толщины нормального (до заболевания) правого и левого голеностопных суставов выполнили на 9 день. На 10-11 день обычно появлялся артрит, и крыс случайным образом разделили на группы лечения. Рандомизацию выполнили после очевидного опухания голеностопного сустава и появления достаточных доказательств двустороннего заболевания.
После отбора животных для подготовки к исследованию, начали лечение. Животным вводили носитель, ингибитор PI3Kδ или ингибитор PI3Kδ в комбинации с рапамицином. Дозы вводили в 1-6 дни. Крыс взвесили на 1-7 день после образования артрита, и проводили измерения толщины лодыжек ежедневно. Конечный вес тела измерили на 7 день и умертвили животных.
Ожидается, что комбинированное лечение с использованием ингибитора PI3Кд и рапамицина обеспечивает более высокую эффективность, чем лечение только ингибитором PI3Кд.
Пример 56: анализ воспаления легких
Соединения по настоящему изобретению проанализировали с использованием одного или двух анализов LPS-индуцированного воспаления легких и анализа овальбумин-индуцированного воспаления легких.
Для выполнения анализа LPS-индуцированного воспаления легких, соединения вводили перорально. Группе вводили только носитель, а в другой группе использовали дексаметазон (5 мг/кг) в качестве положительного контроля. Воспаление легких определили через 6 часов после интраназального закапывания LPS (10 мкг). Оценивали следующие параметры: общее количество лейкоцитов и количество нейтрофилов в бронхоальвеолярном лаваже (BAL).
В анализе овальбумин-индуцированного воспаления легких, соединения вводили перорально. Группе вводили только носитель, а в другой группе использовали дексаметазон (5 мг/кг) в качестве положительного контроля. Воспаление легких определили через 4 дня спустя после 4 последовательных ежедневных интраназальных закапываний овальбумина. Соединения вводили через желудочный зонд за 30 минут до каждой пробы (4 пробы) в указанных дозах. Оценивали следующие параметры: общее количество лейкоцитов и количество эозинофилов в бронхоальвеолярном лаваже (BAL).
Примеры результатов представлены на фиг. 15 (LPS-индуцированный анализ) и на фиг. 16 (OVA-индуцированный анализ).
Несмотря на то, что в настоящем документе показаны и описаны предпочтительные варианты воплощения настоящего изобретения, специалисту в данной области понятно, что эти варианты воплощения изобретения представлены лишь в качестве примеров. Специалисту в данной области понятны многочисленные варианты, изменения и отклонения в рамках настоящего изобретения. Следует понимать, что различные альтернативы вариантов воплощения настоящего изобретения, описанные в настоящем изобретении, могут использоваться на практике настоящего изобретения. Подразумевается, что следующая формула изобретения определяет границы настоящего изобретения, и таким образом охвачены способы и структуры в этой формуле, а также их эквиваленты.
Реферат
Настоящее изобретение относится к новым соединениям формулы V-A или их фармацевтически приемлемым солям, обладающим свойствами ингибиторов активности PI3-киназы (фосфатидилинозитол-3-киназы). Соединения могут найти применение для лечения заболеваний и состояний, связанных с активностью РI3-киназы, в частности, в Т-клетке, В-клетке, мастоците, дендритной клетке или нейтрофиле. Такое заболевание представляет собой одно или более заболеваний, выбранных из рака, поражения костей, воспалительного заболевания, иммунного заболевания, респираторного заболевания или тромбоза. Рак представляет собой лейкоз или лимфому, или рак выбран из острого миелолейкоза, хронического лимфолейкоза и множественной миеломы, где лимфома представляет собой неходжкинскую лимфому; воспалительное или иммунное заболевание представляет собой астму, аллергию, ревматоидный артрит, воспалительное заболевание кишечника, хроническое обструктивное заболевание легких, системную красную волчанку или болезнь "трансплантат против хозяина". Соединение формулы V-A, или его фармацевтически приемлемую соль, или композицию можно применять в комбинации с рапамицином. В формуле V-A:В представляет собой группировку формулы II, где Wпредставляет собой фенил, 6-членный гетероциклоалкил с одним гетероатомом, выбранным из кислорода или азота, или С-Сциклоалкил; q равно 0 или 1; Rпредставляет собой водород, С-Салкил или галогено; Rпредставляет собой галогено или С-Салкил, возможно замещенный галогено; Rпредставляет собой галогено; Rпредставляет собой водород или С-Салкил. 13 н. и 49 з.п. ф-лы, 19 ил., 6 табл.
Формула

или его фармацевтически приемлемая соль, где В представляет собой группировку формулы II:

где
Wc представляет собой фенил, 6-членный гетероциклоалкил с одним гетероатомом, выбранным из кислорода или азота, или С3-С6циклоалкил;
q равно 0 или 1;
R1 представляет собой водород, С1-С4алкил или галогено;
R2 представляет собой галогено или С1-С4алкил, возможно замещенный галогено;
R3 представляет собой галогено;
R9 представляет собой водород или С1-С4алкил.

или его фармацевтически приемлемая соль.

или его фармацевтически приемлемая соль.

или его фармацевтически приемлемая соль.

или его фармацевтически приемлемая соль.

или его фармацевтически приемлемая соль.

или его фармацевтически приемлемая соль.





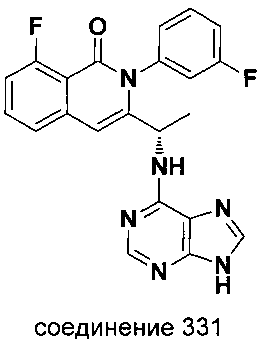





или его фармацевтически приемлемая соль.

или его фармацевтически приемлемая соль.

или его фармацевтически приемлемая соль.
Документы, цитированные в отчёте о поиске
Конденсированное 4-оксопиримидиновое производное
Комментарии