Новые производные каннабигерола - RU2684913C2
Код документа: RU2684913C2
Чертежи





Описание
Область техники
Настоящее изобретение относится к новым производным каннабигерол хинона и синтезу этих соединений. Более того, настоящее изобретение относится к их применению в качестве лекарственного средства, а также для применения в терапии, в частности, в качестве модуляторов гамма-рецептора, активируемого пролифератором пероксисом (PPARg), для лечения заболеваний и состояний, восприимчивых к модуляции PPARg. Настоящее изобретение также относится к фармацевтическим композициям, содержащим указанные соединения, и к способу лечения заболеваний с помощью указанных соединений.
Уровень техники
Ядерные рецепторы (NR) являются основной целью при поиске новых лекарств. NR представляют собой зависимые от лигандов факторы транскрипции, которые обладают способностью непосредственно взаимодействовать с ДНК, регулируя транскрипционную активность своих генов-мишеней. Эти рецепторы играют существенную роль в развитии, клеточном гомеостазе и метаболизме. Кроме того, NR вовлечены в широкий круг заболеваний и поэтому стали основной мишенью при разработке лекарственных средств для фармацевтической промышленности.
Согласно новейшей номенклатуре ядерных рецепторов, рецепторы, активируемые пролифератором пероксисом (PPAR), ядерное подсемейство 1 С (NR1C), включают три подтипа PPAR: PPARα (также называемые NR1C1), PPARβ/δ (также называемые NR1C2) и PPARγ (также называемые PPARg, рецепторы глитазона или NR1C3).
PPAR контролируют экспрессию сети генов, участвующих в адипогенезе, метаболизме липидов, воспалении и поддержании метаболического гомеостаза [Barish и др., 2006]. Эти ядерные рецепторы активируют транскрипцию путем связывания с элементами последовательностей ДНК, известными как элементы, отвечающие на пролифератор пероксисом (PPRE), в виде гетеродимера с ретиноидными X рецепторами (известными как RXR).
Подобно типичным ядерным рецепторам, PPAR состоят из различных функциональных доменов, в том числе N-концевого трансактивирующего домена (AF1), высококонсервативного ДНК-связывающего домена (DBD) и С-концевого лиганд-связывающего домена (LBD), содержащего лиганд-зависимую трансактивационную функцию (AF2) [Poulsen и др., 2012]. ДНК-связывающий домен С, состоящий из двух цинковых пальцев, связывается с элементом, отвечающим на пролифератор пероксисом, (PPRE) в регуляторной области генов-мишеней PPAR.
PPAR негативно регулируют транскрипцию генов воспалительного ответа, выступая в качестве антагонистов сигнальных путей белка-активатора 1 (АР-1), ядерного фактора-каппа В (NF-кВ), переносчика сигнала и активатора транскрипции 3 (STAT3) и ядерного фактора активированных Т- клеток (NFAT) [Vanden Berghe и др. 2003].
Гамма-рецептор, активируемый пролифератором пероксисом (PPARg), представляет особый интерес, поскольку он вовлечен в регуляцию образования адипоцитов, чувствительности к инсулину и воспаления [Fievet и др. 2006] [Stienstra и др. 2007] [Tontonoz и Spiegelman, 2008]. Этот ядерный рецептор экспрессируется в различных тканях, включая жировую ткань, клетки скелетных мышц, остеокласты, остеобласты, несколько типов клеток иммунной системы, а также в головном мозге и периферической нервной системе.
Очевидно, что PPARg является доминирующим или «главным» регулятором адипогенеза, ввиду того, что он является одновременно необходимым и достаточным для дифференциации жировых клеток. Регуляторные области большого числа генов, которые играют важную роль в липогенезе и чувствительности к инсулину, содержат сайты связывания PPARg, в том числе аР2, LPL, адипонектин и GLUT4 [Rosen и MacDougald, 2006]. Следовательно, активация PPARg в жировой ткани влияет на чувствительность к инсулину всего организма.
С другой стороны, активация PPARg оказывает противовоспалительное действие в нескольких типах клеток путем ингибирования экспрессии провоспалительных генов, тем самым снижая образование цитокинов, металлопротеиназ и белков острой фазы [Tontonoz и Spiegelman, 2008]. Он также активирует противовоспалительные цитокины и ингибирует экспрессию индуцируемиой синтазы оксида азота (iNOS) [
Было обнаружено, что PPARg играет принципиально важную роль в иммунном ответе, благодаря своей способности направлять дифференцировку клеток иммунной системы по пути противовоспалительных фенотипов [Tontonoz и Spiegelman, 2008]. Интересно отметить, что агонисты PPARg продемонстрировали противовоспалительную и нейропротекторную активность в нескольких экспериментальных моделях болезней Паркинсона, бокового амиотрофического склероза, рассеянного склероза и инсульта, а также в нескольких клинических исследованиях [Bernardo и Minghetti, 2008]. Кроме того, PPARg формально рассматривается как ген-супрессор опухолей в генетическом смысле. Он экспрессируется в различных опухолевых клетках, и активация PPARg лигандами приводит либо к ингибированию пролиферации клеток, либо к индукции апоптоза [Tachibana и др., 2008] [Tontonoz и Spiegelman, 2008].
Благоприятные эффекты активации PPARg могут быть использованы для лечения ряда заболеваний, опосредованных PPARg, как показано в Таблице 1. В настоящем описании заболевание, опосредованное PPARg, означает любой наблюдаемый патологический эффект, который может быть связан с изменением функции PPARg по сравнению с нормальными непатологическими состояниями. В данной таблице обобщены действия рецепторов PPAR при воспалительных, онкологических и других заболеваниях.
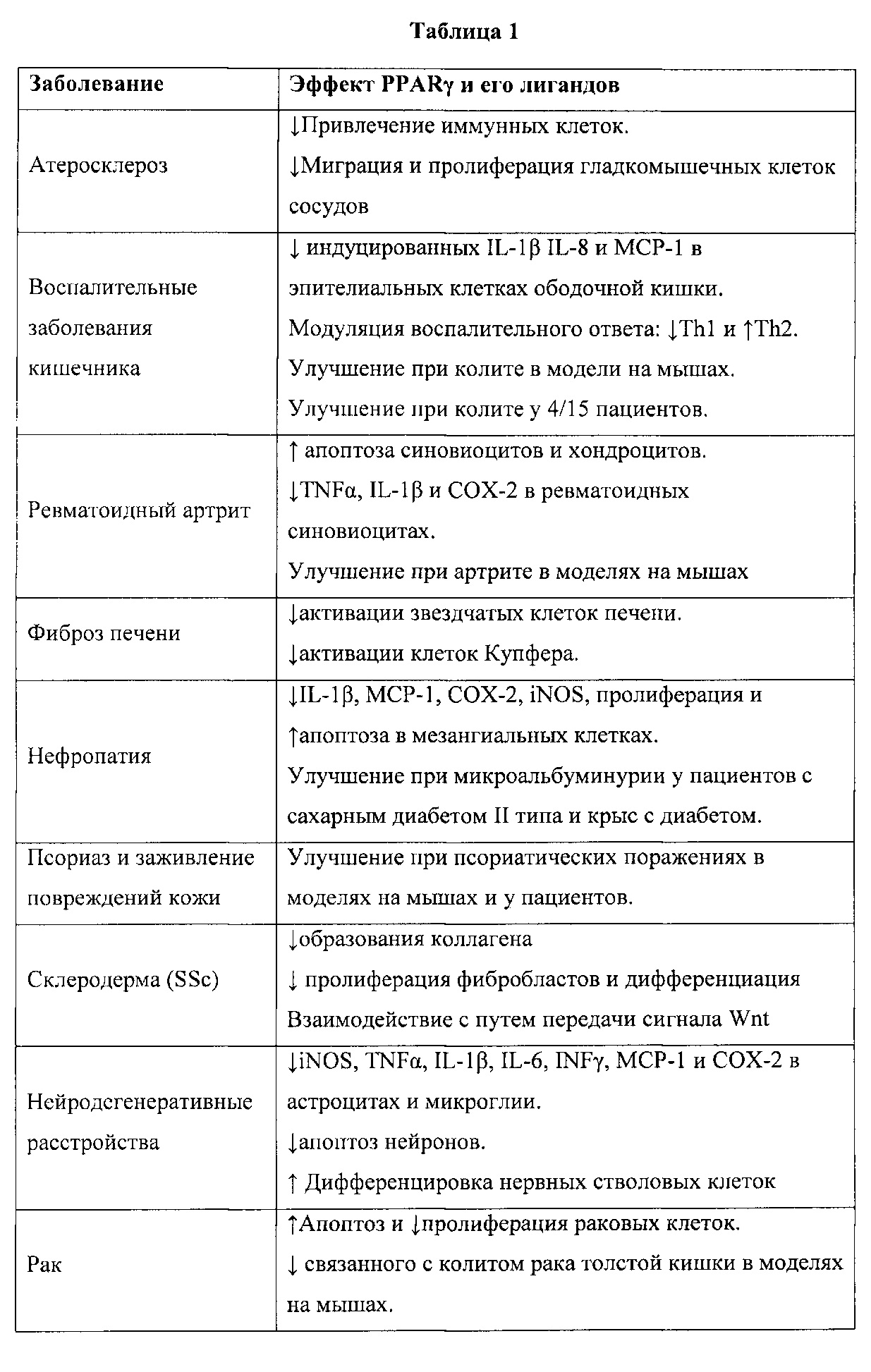
Сокращения: ↓ ингибирование, ↑ стимуляция, звездчатые клетки печени (HSC), гладкомышечные клетки сосудов (VSMC), хемоаттрактантный белокмоноцитов-1 (MCP-1), Т-хелперы (Th), фактор некроза опухоли-альфа (TNF-α), циклооксигеназа (СОХ), интерферон-гамма (INFγ), индуцируемая синтаза оксида азота (iNOS), внутриклеточные молекулы адгезии-1 (ICAM-1) [адаптировано из Kostadinova и др., 2005].
Ядерный фактор-2, подобный фактору эритроидного происхождения 2, также известный как NFE2L2 или (Nrf2), является фактором транскрипции, который в организме человека кодируется геном NFE2L2. Антиоксидантный путь ответа Nrf2 является основной клеточной защитой от цитотоксического действия окислительного стресса. Среди прочих эффектов, Nrf2 увеличивает экспрессию нескольких ферментов антиоксидантной защиты.
Сигнальный путь Keapl-Nrf2 является основным регулятором защитных ответов на эндогенные и экзогенные стрессы, вызванные активными формами кислорода (ROS) и электрофилами. Ключевым сигнальным белком пути являются транскрипционный фактор Nrf2, который вместе с малыми белками Maf, связывается с элементом, отвечающим на антиоксиданты, (ARE) в регуляторных областях генов-мишеней. В нормальном состоянии NrF2-зависимая транскрипция подавляется отрицательным регулятором Keap1 (Kelch-подобный ЕСН-ассоциированный белок 1). Когда клетки подвергаются воздействию окислительного стресса, электрофильных или хемопревентивных агентов, Nrf2 избавляется от опосредованного Keap1 подавления и активирует экспрессию генов, зависимых от элемента, отвечающего на антиоксидант (ARE), для поддержания клеточного окислительно-восстановительного гомеостаза.
Так как такой Nrf2-зависимый защитный клеточный ответ способен защитить множество органов и тканей, активация Nrf2 вовлечена в защиту от многих заболеваний человека, в том числе рака, нейродегенеративных заболеваний, сердечно-сосудистых заболеваний, острых и хронических повреждений легких, аутоиммунных заболеваний и воспаления.
Nrf2 может защитить клетки и ткани от разнообразных токсинов и канцерогенов путем повышения экспрессии ряда цитопротекторных генов. Так же, как Nrf2 защищает нормальные клетки, исследования показали, что Nrf2 может также защищать раковые клетки от химиотерапевтических агентов и способствовать опухолевой прогрессии [Na и Surh 2013].
Раковые клетки выдерживают постоянный эндогенный окислительный стресс или клеточный стресс, индуцированный активными формами кислорода (ROS), и становятся устойчивыми к некоторым противораковым агентам, которые оказывают цитотоксическое воздействие через образование ROS. В таких условиях активный Nrf2 сигнальный путь может поддерживать благоприятный окислительно-восстановительный баланс в раковых клетках, сохраняя уровень ROS в пределах диапазона, который способствует их росту и выживаемости. Предполагается, что постепенное накопление или активация Nrf2 создает для группы предраковых или раковых клеток благоприятную среду для деления, избегания апоптоза, метастазирования и устойчивости к терапевтическому вмешательству.
Известно, что ингибирование избыточной экспрессии Nrf2 изменяет фенотипические характеристики раковых клеток, что поддерживает это предположение [Sporn и Liby, 2012]. Постоянная сверхактивация Nrf2 наблюдалась в многочисленных типах злокачественных опухолей, таких как плоскоклеточный рак, рак легких, рак молочной железы, рак желчного пузыря, рак предстательной железы, рак почки, эпендимомы, эпителиальный рак яичников, рак эндометрия и рак поджелудочной железы [Na and Surh, 2013]. Больные раком с конститутивно повышенным уровнем экспрессии Nrf2 в опухоли в целом демонстрируют более низкий уровень выживаемости [Solis и др., 2010]. Поэтому Nrf2 считается прогностическим молекулярным маркером для определения состояния прогрессирования рака и способствует как внутренней, так и приобретенной устойчивости к воздействию химиотерапевтических агентов. Таким образом, этот антиоксидантный фактор транскрипции может также выступать в качестве протоонкогена, и повышенная активность Nrf2 способствует формированию и химической устойчивости солидных раков [Sporn and Liby, 2012].
CBG-Q (соединение I), предшественник химических производных CBG-Q (соединения II-XII) согласно настоящему изобретению, оказывает эффект активации в отношении PPARg. Тем не менее, CBG-Q также активирует (см. сравнительный Пример 4 и Фигуру 4) Nfr2, что вызывает нежелательный побочный эффект, и опухоли становятся устойчивыми к химиотерапевтическим агентам, а также постоянное лечение активатором Nrf2 может привести к канцерогенезу, как пояснялось выше. Таким образом, новые производные CBG согласно настоящему изобретению предлагают альтернативное более эффективное лечение рака, так как отсутствует побочный эффект индуцированной устойчивости к химиотерапии, наблюдаемый при введении CBG in vitro благодаря сверхэкспрессии Nrf2.
Среди активаторов лигандов PPARg тиазолидиндионы (TZD) имеют наибольшее клиническое значение [Lehmann и др., 1995]. По этой причине до сих пор в клинической практике преимущественно использовали розиглитазон и пиоглитазон. Они обеспечивают одинаковое воздействие на гликемический контроль, а также вызывают целый ряд сходных побочных эффектов, таких как увеличение веса, задержка жидкости, а также повышенный риск сердечной недостаточности, которые, видимо, опосредованы PPARg. Интересно отметить, что эти тиазолины отличаются по их воздействию на липидный и сердечно-сосудистый профили безопасности, что указывает на PPARg-независимый механизм. Действительно, использование розиглитазона недавно было прекращено в Европе и ограничено в США, вследствие повышенного риска сердечно-сосудистых осложнений у пациентов с сахарным диабетом 2 типа.
Несмотря на то, что тиазолидиндионы являются мощными полными агонистами PPARg (PPARg-fa), побочные эффекты, основанные на механизме их действия, ограничивают полный терапевтический потенциал этих соединений [German и др., 2007] [Ciudin и др., 2012]. Но физиологическая и терапевтическая актуальность пути PPARg способствовали новым исследованиям для разработки новых классов молекул, неблагоприятные эффекты которых слабее или отсутствуют [Ahmadian и др., 2013]. Таким образом, значительный прогресс был достигнут в открытии и развитии селективных модуляторов PPARg (PPARg-m), как более безопасных альтернатив PPARg-fa. Доклинические и клинические данные ясно показывают, что селективные PPARg-m имеют потенциал стать следующим поколением агонистов PPARg: эффективные сенсибилизаторы инсулина с превосходным профилем безопасности, как у PPARg-fa. [Doshi и др al. 2010].
В этом смысле природные и синтетические каннабиноиды считаются PPARg-m, которые ослабляют воспалительный процесс путем активации PPARg. Примерами каннабиноидов на основе PPARg-m являются аджулемовая кислота [Liu и др., 2003], [Burstein S. 2005], WIN55212-2 [Sun and Bennett, 2007],9Δ-THC и CBD [O'Sullivan 2007] и CBG и его производные [Granja и др., 2012].
Клиническая значимость ковалентной модификации поддающихся воздействию лекарственных средств белков малыми молекулами широко обсуждалась в последние несколько лет в фармацевтической промышленности, и несколько раз ковалентная модификация оказывалась в основе механизма активности успешных препаратов [Singh и др., 2011]. Тем не менее, по-прежнему существует закоренелое предубеждение против ковалентных препаратов независимо от механизма, с помощью которого они в конечном счете связываются с биомолекулами. Хиноны представляют собой класс токсических интермедиатов, которые могут проявлять различные опасные эффекты in vivo, в том числе острую цитотоксичность и иммунотоксичность [Bolton и др., 2000]. Механизмы, с помощью которых хиноны вызывают эти эффекты, могут быть достаточно сложными. Хиноны являются акцепторами в реакции Михаэля, и повреждение клеток может происходить через алкилирование важных клеточных белков и/или ДНК. В качестве альтернативы, хиноны являются высокоактивными окислительно-восстановительнами молекулами, которые могут образовывать окислительно-восстановительный цикл с их семихинонными радикалами, что приводит к образованию активных форм кислорода (ROS), которые могут вызвать сильный окислительный стресс внутри клеток через образование окисленных клеточных макромолекул, включая липиды, белки и ДНК [Monks and Jones, 2012]. Хотя существуют многочисленные примеры соединений на основе хинонов с терапевтическим применением, из-за беспокойства по поводу неспецифической токсичности и отсутствия селективности, группы акцепторов в реакции Михаэля редко вводят в качестве заместителей при дизайне лекарства.
Одним из примеров терапевтических соединений на основе хинона является вещество, опубликованное в патенте WO2011117429, который описывает синтез каннабигерол гидроксихинона (также называемого CBG-Q или VCE-003 в вышеупомянутой международной заявке на патент и, для целей настоящего описания, называемого также соединение I) и его использование при заболеваниях и состояниях, реагирующих на модуляцию PPARg. Болезни, упомянутые в WO/2011/117429 включают: атеросклероз, воспалительные заболевания кишечника, ревматоидный артрит, фиброз печени, нефропатии, псориаз, заживление повреждений кожи, регенерацию кожи, панкреатит, гастрит, нейродегенеративные расстройства, рак; артериальную гипертензию, гипертриглицеридемию, гиперхолестеринемию, ожирение и диабет типа И. Введение хиноновой группы в молекулу каннабигерола увеличивает его сродство связывания с PPARg и повышает его транскрипционную активность.
Дальнейшие исследования показывают, что каннабигерол гидроксихинон (CBG-Q или соединение I) также активирует фактор транскрипции Nrf2, клеточный сенсор окислительного/электрофильного стресса. Таким образом, введение хиноновой группы в каннабигерол приводит к двум независимым активностям, таким как агонизм к PPARg и активация Nrf2.
Чтобы улучшить только активность агониста к PPARg, не вызывая активацию Nrf2, с тем чтобы избежать индукцию резистентности к химиотерапии, в настоящем изобретении разработали библиотеку новых соединений, начиная с каннабигерол гидроксихинона в качестве основы, и удивительно, авторы настоящего изобретения обнаружили, что определенные изменения в положении 2 привели к получению новых соединений, пригодных для лечения заболеваний, связанных с PPARg из-за их высокого агонистического эффекта к PPARg без электрофильной (активация Nrf2) и цитотоксической активности.
Такие производные каннабигерол гидроксихинона согласно настоящему изобретению отличаются от соединений, описанных в WO 20011117429, так как модификации в положении 2 дают соединениям согласно настоящему изобретению способность активировать PPARg и защищать от индуцированной глутаматом цитотоксичности. Эти соединения также показали удивительную низкую цитотоксичность в клеточных линиях нейронального происхождения по сравнению с CBG-Q (соединение I), существующих на сегодняшний день. Кроме того, производные этого соединения показывают терапевтическую эффективность на животных моделях заболеваний (рассеянный склероз, болезни Паркинсона и Хантингтона), широко используемых для оценки клинической эффективности агонистов PPARg.
Краткое описание изобретения
Исходя из предшествующего уровня развития техники, задача настоящего изобретения состоит в создании производных коннабигерола гидроксихинона, которые демонстрируют активность модулятора PPARg, не вызывая активацию Nrf2.
Соединения согласно настоящему изобретению также включают их аналоги, производные, таутомеры, изомеры, стереоизомеры, полиморфы, фармацевтически приемлемые соли, фармацевтически приемлемые сольваты и композиции, содержащие те же вещества.
Для целей настоящего описания термин «аналог/и» относится к любой структурной единице производной от или гомологичной соединению формулы (I).
В контексте настоящего изобретения термин «производное/ые» соединений формулы (I) следует понимать как любой аналог CBG-Q, всегда замещенный в положении 2 и демонстрирующий фармакологические свойства, связанные с этим замещением в положении 2, как определено в настоящем документе, но также имеющий заместители в других положениях молекулы CBG-Q, отличные от групп, показанных в указанной Формуле (I).
Термин «таутомеры» означает структурные изомеры органических соединений, которые легко взаимопревращаются посредством химической реакции (таутомеризации).
Термин «изомеры» или «стереоизомеры» относится к соединениям, которые имеют одинаковый химический состав, но различаются в отношении расположения атомов или групп в пространстве.
Используемый здесь термин «полиморфная модификация» относится к кристаллическим формам одного и того же химического состава, но с разными пространственным расположением молекул, атомов и/или ионов, образующих кристалл.
Термин «фармацевтически приемлемая соль» относится к любой фармацевтически приемлемой соли, которая при введении пациенту способна обеспечить (прямо или косвенно) соединение, как описано в настоящем документе. Такие соли предпочтительно являются кислотно-аддитивными солями с физиологически приемлемыми органическими или неорганическими кислотами. Примеры кислотно-аддитивных солей включают аддитивные соли минеральных кислот, такие как, например, гидрохлорид, гидробромид, гидроиодид, сульфат, нитрат, фосфат, и органические кислотно-аддитивные соли, такие как, например, ацетат, трифторацетат, малеат, фумарат, цитрат, оксалат, сукцинат, тартрат, малат, манделат, метансульфонат и п-толуолсульфонат. Примеры основных аддитивных солей включают неорганические соли, такие как, например, натрия, калия, кальция и соли аммония, а также органические соли щелочных металлов, такие как, например, этилендиамин, этаноламин, N, N-диалкиленэтаноламин, триэтаноламин и соли основных аминокислот. Тем не менее, следует принимать во внимание, что фармацевтически неприемлемые соли также попадают в объем настоящего изобретения, так как они могут быть полезными в получении фармацевтически приемлемых солей. Процедуры получения соли являются обычными в данной области.
Термин «сольват» в соответствии с настоящим изобретением следует понимать как означающий любую форму активного соединения в соответствии с настоящим изобретением, в котором указанное соединение образует не-ковалентную связь с другой молекулой (обычно полярного растворителя), в том числе, в особенности, гидраты и алкоголяты.
Более конкретно, в настоящем изобретении соединения представляют собой производные производных каннабигерол гидроксихинона (производные CBG-Q) Формулы (I):
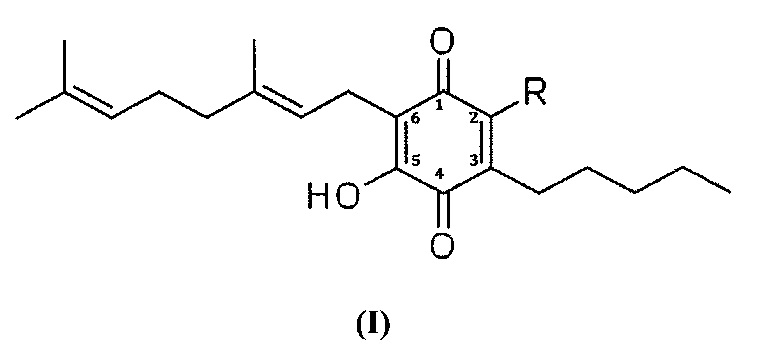
где R представляет собой атом углерода группы, представляющей собой: арильную, линейную или разветвленную алкенильную, линейную или разветвленную алкинильную или линейную или разветвленную алкоксикарбонильную группы; или где R представляет собой атом азота группы, представленной: линейной или разветвленной алкиламино, ариламино, линейной или разветвленной алкениламино или линейной или разветвленной алкиниламино группами; или, в качестве альтернативы, R представляет собой связь между 2 молекулами Формулы (I), образующими димер. В предпочтительном варианте осуществления соединения согласно настоящему изобретению являются соединениями Формулы (II), (III), (IV), (V), (VI), (VII), (VIII), (X), (XI) и (XII).
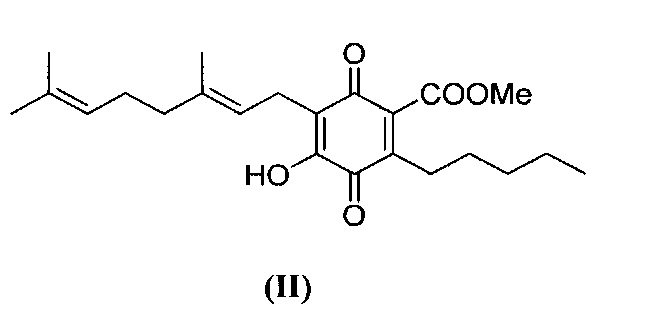
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-метоксикарбонил-[1,4]бензохинон.
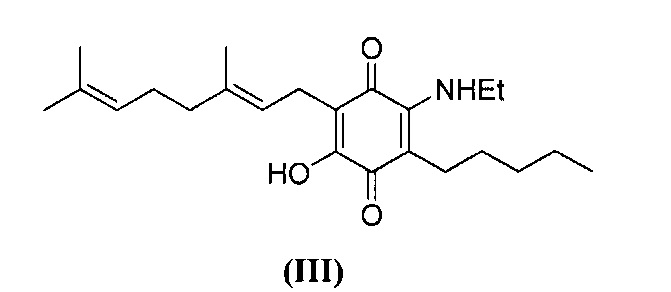
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-этиламино-[1,4]бензохинон.
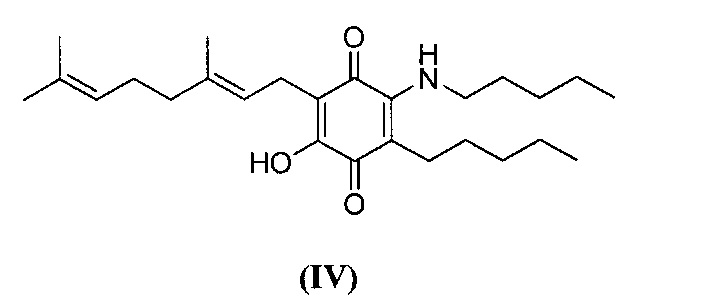
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-пентиламино[1,4]бензохинон.
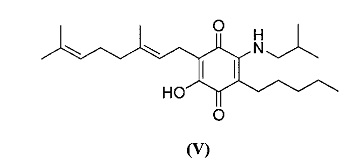
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-изобутиламино[1,4]бензохинон.
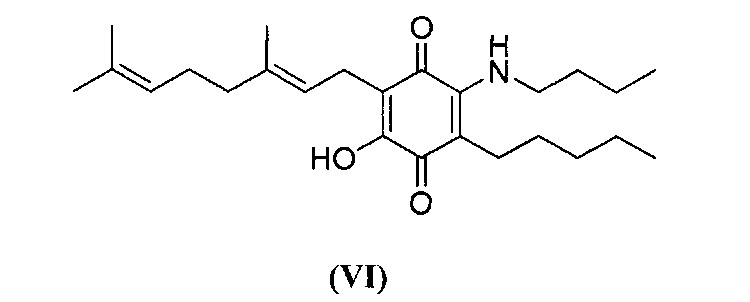
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-бутиламино[1,4]бензохинон.
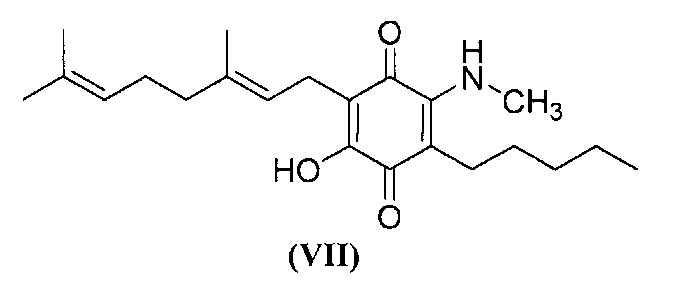
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-метиламино-[1,4]бензохинон.
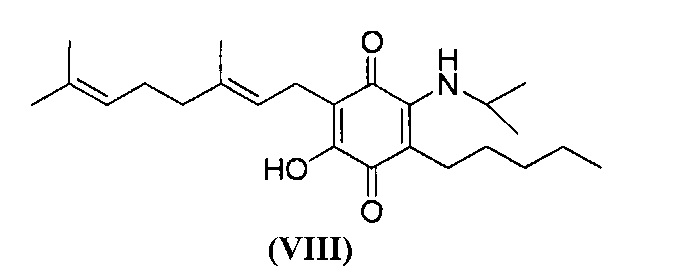
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-изопропиламино-[1,4]бензохинон.
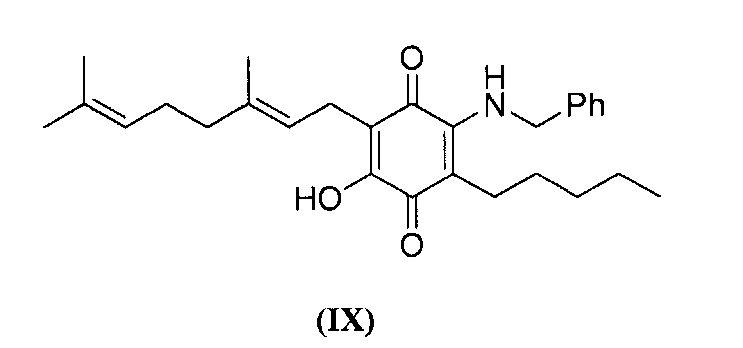
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-бензиламино[1,4]бензохинон
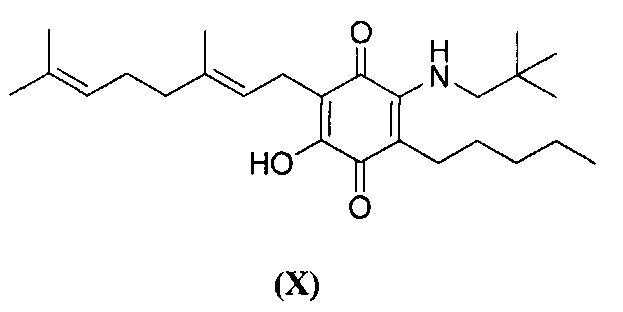
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-(2,2-диметилпропиламино)-[1,4] бензохинон.
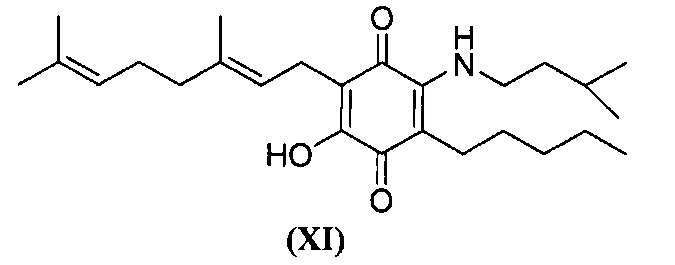
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-(3-метил-бутиламино)-[1,4]бензохинон.
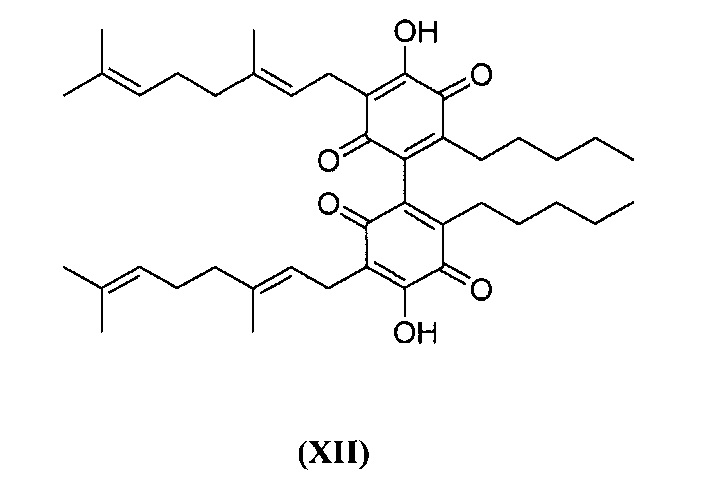
3,3'-бис((Е)-3,7-диметил-окта-2,6-диенил)-4,4'-дигидрокси-6,6'-дипентил-1,1'-би(циклогекса-3,6-диен)-2,2',5,5'-тетраон
Как следует из нижеследующих примеров и чертежей, изменения в положении 2, содержащиеся в общей Формуле I, позволяют соединениям согласно настоящему изобретению активировать PPARg и защищать от индуцированной глутаматом цитотоксичности. Эти соединения также показали удивительно низкую цитотоксичность в клеточных линиях нейронального происхождения по сравнению с CBG-Q (соединение I), существующих в настоящее время. Кроме того, соединения III и XII, в качестве представителей из этой серии, показали терапевтическую эффективность на животных моделях заболеваний (рассеянный склероз, болезни Паркинсона и Хантингтона), широко используемых для оценки клинической эффективности агонистов PPARg.
CBG-Q, соединение I, является предшественником всех производных Формулы I согласно настоящему изобретению, примеры которых представлены соединениями II-XII. Предшественник CBG-Q может быть первоначально синтезирован, исходя из природных каннабиноидов, таких как CBG (каннабигерол) и CBGA (каннабигероловая кислота) с помощью замены некоторых специфических радикалов.
Следует понимать, что ссылки на производные каннабигерол гидроксихинона также включают фармацевтически приемлемые соли таких соединений. Термин «фармацевтически приемлемые соли» относится к солям или сложным эфирам, полученным из фармацевтически приемлемых оснований или кислот, включая неорганические основания или кислоты и органические основания или кислоты, которые хорошо известны любому специалисту в данной области техники.
Еще один вариант осуществления настоящего изобретения относится к применению соединений Формулы (I) или их производных в качестве лекарственных средств, в частности, в качестве агонистов PPARg рецепторов, которые не индуцируют активацию Nfr2, в частности, при лечении таких заболеваний, как атеросклероз, воспалительные заболевания кишечника, ревматоидный артрит, фиброз печени, нефропатия, псориаз, заживлении ран кожи, регенерации кожи, панкреатит, гастрит, нейродегенеративные расстройства, нейровоспалительные расстройства, склеродермия, рак, гипертония, ожирение, сахарный диабет второго типа, а также других заболеваний, которые можно лечить с помощью агонистов PPARg.
Другой вариант осуществления настоящего изобретения относится к применению соединений Формулы (I) для приготовления композиции с более низкой цитотоксичностью для лечения связанных с PPARg заболеваний, таких как атеросклероз, воспалительные заболевания кишечника, ревматоидный артрит, фиброз печени, нефропатии, псориаз, заживление ран кожи, регенерация кожи, панкреатит, гастрит, нейродегенеративные расстройства, нейрогенные расстройства, склеродермия, рак, гипертония, ожирение, сахарный диабет II типа, а также другие заболевания, которые можно лечить с помощью агонистов PPARg.
Альтернативный вариант осуществления настоящего изобретения относится к применению указанных выше соединений Формулы (I) или их производных, отдельно или в составе композиций, в частности, фармацевтических композиций, которые содержат по меньшей мере одно из соединений согласно настоящему изобретению в сочетании с по меньшей мере еще одним активным соединением, имеющим дополнительный или синергический виды биологической активности. Альтернативно, указанные композиции могут быть получены с по меньшей мере одним инертным ингредиентом в качестве носителя или вспомогательного вещества, такого как: сорастворители, поверхностно-активные вещества, масла, увлажнители, смягчающие вещества, консерванты, стабилизаторы и антиоксиданты. Любой фармакологически приемлемый буфер может быть использован, например, TRIS или фосфатные буферы.
Для целей настоящего описания термины «активное соединение или активное начало» следует принимать в качестве синонимов, которые означают химическое соединение, которое оказывает терапевтическое действие при введении в организм человека или животного.
Типичные композиции включают в себя соединения согласно настоящему изобретению, или их производные, вместе с фармацевтически приемлемыми наполнителями, которые, например, могут являться носителем или разбавителем. Такие композиции могут быть заключены в капсулы, саше, пакет или другой контейнер. При приготовлении композиций могут быть использованы обычные методы для получения фармацевтических композиций. Например, соединение интереса, как правило, будет смешано с носителем или разбавлено носителем, или заключено в носитель, или может быть в форме ампулы, капсулы, саше, пакета или другого контейнера. Когда носитель выступает в качестве разбавителя, он может быть твердым, полутвердым или жидким материалом, который действует как носитель, наполнитель или среда для активного соединения. Соединение может быть адсорбировано на гранулированном твердом носителе, например, саше. Некоторые примеры подходящих носителей включают воду, растворы солей, спирты, полиэтиленгликоли, полигидроксиэтоксилированное касторовое масло, арахисовое масло, оливковое масло, лактозу, каолин, сахарозу, циклодекстрин, амилозу, стеарат магния, тальк, желатин, агар, пектин, аравийскую камедь, стеариновую кислоту или низшие алкиловые эфиры целлюлозы, кремниевую кислоту, жирные кислоты, амины жирных кислот, моноглицериды и диглицериды жирных кислот, сложные эфиры пентаэритрита и жирных кислот, полиоксиэтилен, гидроксиметилцеллюлозу и поливинилпирролидон. Подобным образом, носитель или разбавитель может включать любой материал для замедленного высвобождения, известный в данной области техники, такой как глицерилмоностеарат или глицерилдистеарат, отдельно или в смеси с воском. Также композиции могут включать смачивающие агенты, эмульгаторы и суспендирующие агенты, консервирующие агенты, подслащивающие агенты или отдушки. Композиции согласно настоящему изобретению могут быть приготовлены таким образом, чтобы обеспечить быстрое, длительное или замедленное высвобождение активного ингредиента после введения пациенту с использованием процедур, хорошо известных в данной области техники.
Фармацевтические композиции могут быть стерилизованы и смешаны, если это желательно, со вспомогательными агентами, эмульгаторами, солью для воздействия на осмотическое давление, буферами и/или красителями и т.п., которые не реагируют вредным образом с активными соединениями.
Композиция может быть использована для лечения таких заболеваний, как атеросклероз, воспалительные заболевания кишечника, ревматоидный артрит, фиброз печени, нефропатии, псориаз, заживление ран кожи, регенерации кожи, панкреатита, гастрита, нейродегенеративных расстройств, нейровоспалительных расстройств, склеродермии, рака, гипертонии, ожирения, сахарного диабета II типа, а также других заболеваний, которые можно лечить с помощью агонистов PPARg.
Один из предпочтительных вариантов осуществления настоящего изобретения относится к пути введения, которое может быть осуществлено любым способом, который эффективно транспортирует соединения интереса к соответствующему или требуемому месту действия, таким как пероральный, назальный, местный, легочный, чрескожное или парентеральное введения, например, ректальный, подкожный, внутривенный, внутриуретральный, внутримышечный, интраназальный, или офтальмологический раствор или мазь.
Для назального введения препарат может содержать соединение интереса в растворе или в виде суспензии в жидком носителе, в частности, водном носителе для аэрозольного применения. Носитель может содержать добавки, такие как солюбилизирующие агенты, например, пропиленгликоль, поверхностно-активные усилители абсорбции, такие как лецитин (фосфатидилхолин) или циклодекстрин, или консерванты, такие как парабены.
Для приготовления состава для местного применения соединение интереса помещают в дерматологический носитель, известный в данной области техники. Количество соединения, представляющего интерес для введения, и концентрация соединения в составе для местного применения зависят от носителя, системы доставки или выбранного устройства, клинического состояния пациента, побочных эффектов и стабильности соединения в композиции. Таким образом, врач использует соответствующий препарат, содержащий соответствующую концентрацию соединения, представляющего интерес, и выбирает количество состава, вводимого в зависимости от клинического опыта конкретного пациента или подобных пациентов.
Для офтальмологических применений, представляющее интерес соединение готовят в виде растворов, суспензий и мазей, подходящих для использования для глаз. Концентрации, как правило, используются те же, что описаны выше для местных препаратов.
Для перорального введения могут быть получены твердые или жидкие единичные готовые лекарственные формы. Для получения твердых композиций, таких как таблетки, соединение интереса смешивают в композиции с обычными ингредиентами, такими как тальк, стеарат магния, гидрофосфат кальция, алюмосиликат магния, сульфат кальция, крахмал, лактоза, аравийская камедь, метилцеллюлоза или функционально аналогичными материалами в качестве фармацевтических разбавителей или носителей.
Капсулы получают путем смешивания соединения интереса с инертным фармацевтическим разбавителем и заполнения смесью твердой желатиновой капсулы подходящего размера. Мягкие желатиновые капсулы получают путем автоматической инкапсуляции суспензии соединения интереса и приемлемого растительного масла, светлого жидкого парафина или другого инертного масла. Жидкие единичные готовые лекарственные формы для перорального введения, такие как сиропы, микстуры или суспензии, также могут быть приготовлены. Водорастворимые формы можно растворить в водном носителе вместе с сахаром, ароматизирующими агентами и консервантами с образованием сиропа. Микстуры получают с использованием водно-спиртового (например, этанол) носителя с подходящими подсластителями, такими как сахар и сахарин, вместе с ароматизирующим вкусовым агентом. Суспензии можно получить с водным наполнителем с помощью суспендирующего агента, такого как аравийская камедь, трагакант, метилцеллюлоза и тому подобное.
Подходящие лекарственные формы для парентерального применения являются очевидными для практикующего специалиста, такие как использование подходящих инъекционных растворов или суспензий. Состав, который является стерильным, подходит для различных местных или парентеральных путей введения, включая внутрикожное, внутримышечное, внутрисосудистое и подкожное.
В дополнение к соединению интереса композиции могут включать, в зависимости от состава и желаемого способа введения, фармацевтически приемлемые нетоксичные носители или разбавители, которые включают носители, обычно используемые для приготовления фармацевтических композиций для введения животным или человеку. Разбавитель выбирают таким образом, чтобы не чрезмерно влиять на биологическую активность состава.
Примерами таких разбавителей, которые особенно полезны для инъекционных лекарственных составов, являются вода, различные солевые растворы, органические или неорганические растворы солей, раствор Рингера, раствор декстрозы и раствор Хэнка. Кроме того, фармацевтическая композиция или состав может включать добавки, такие как другие носители; вспомогательные вещества; или нетоксичные, нетерапевтические, неиммуногенные стабилизаторы и тому подобное.
Кроме того, в композицию могут быть включены вспомогательные вещества. Примеры включают сорастворители, поверхностно-активные вещества, масла, увлажнители, смягчающие вещества, консерванты, стабилизаторы и антиоксиданты. Любой фармакологически приемлемый буфер может быть использован, например, трис или фосфатные буферы. Эффективными количествами разбавителей, добавок и вспомогательных веществ являются те количества, которые эффективны для получения фармацевтически приемлемого состава с точки зрения растворимости, биологической активности и т.д.
Соединение интереса может быть заключено в микросферы. Соединение может быть загружено в альбуминовые микросферы, после чего можно восстановить такие микросферы в виде сухого порошка для назального введения. Другие материалы, пригодные для получения микросфер, включают агар, альгинат, хитозан, крахмал, гидроксиэтилкрахмал, альбумин, агарозу, декстран, гиалуроновую кислоту, желатин, коллаген и казеин. Микросферы могут быть получены различными способами, известными специалистам в этой области техники, такими как сушка распылением или в процессе эмульгирования.
Например, альбуминовые микросферы могут быть получены путем добавления кроличьего сывороточного альбумина в фосфатном буфере к оливковому маслу при перемешивании с получением эмульсии воды в масле. Затем к эмульсии добавляли раствор глутарового альдегида, и эмульсию перемешивают до сшивки альбумина. Микросферы затем могут быть выделены с помощью центрифугирования, масло удаляется и сферы промывают, например, петролейным эфиром, а затем этанолом. И, наконец, микросферы могут быть просеяны, собраны и высушены с помощью фильтрации.
Крахмальные микросферы могут быть получены путем добавления теплого водного раствора крахмала, например, картофельного крахмала, к нагретому раствору полиэтиленгликоля в воде при перемешивании с образованием эмульсии. Когда образовалась двухфазная система (с раствором крахмала в качестве внутренней фазы) смесь охлаждают до комнатной температуры при перемешивании, после чего внутренняя фазу превращается в гелевые частицы. Эти частицы затем отфильтровывают при комнатной температуре и смазывают растворителем, таким как этанол, после чего частицы снова отфильтровывают и высушивают на воздухе. Микросферы можно сделать твердыми с помощью хорошо известных сшивающих процедур, таких, как термическая обработка или с использованием химических сшивающих агентов. Подходящие агенты включают диальдегиды, включая глиоксаль, малоновый альдегид, сукцинальдегид, апдипальдегид, глутаральдегид и фталевый альдегид, дикетоны, такие как бутадион, эпихлоргидрин, полифосфаты и борную кислоту. Диальдегиды используются для сшивки белков, таких как альбумин, путем взаимодействия с аминогруппами, а дикетоны образуют основания Шиффа с аминогруппами. Эпихлоргидрин активирует соединения с нуклеофилами, такими как амино или гидроксильные группы, в эпоксидные производные.
С композициями, описанными здесь, могут быть использованы медленные или замедленные системы доставки, в том числе любой из ряда биополимеров (биологические системы), системы, использующие липосомы, коллоиды, смолы и другие полимерные системы доставки или компартментализованные носители, чтобы обеспечить непрерывный или долгосрочный источник терапевтического соединения. Такие системы с медленным высвобождением применимы к композициям для доставки местным, внутриглазным, оральным и парентеральными путями.
При лечении используется эффективное количество соединения интереса. Дозировка соединений, используемых в соответствии с изобретением, изменяется в зависимости от соединения и состояния, подлежащего лечению, например, в зависимости от возраста, веса и клинического состояния пациента, получающего лечение. Другие факторы включают: способ введения, характеристики пациента, историю болезни пациента, тяжесть патологического процесса, а также активность конкретного соединения. Доза должна быть достаточной для ослабления симптомов или признаков болезни, подвергаемой лечению, не вызывая неприемлемой токсичности для пациента. В общем, эффективное количество соединения является тем, которое обеспечивает либо субъективное облегчение симптомов, либо объективно идентифицируемое улучшение, отмечаемое клиницистом или другим квалифицированным наблюдателем.
И последний вариант осуществления настоящего изобретения относится к способу лечения заболеваний, таких как атеросклероз, воспалительные заболевания кишечника, ревматоидный артрит, фиброз печени, нефропатии, псориаз, заживление ран кожи, регенерация кожи, панкреатит, гастрит, нейродегенеративные расстройства, нейровоспалительные расстройства, склеродермия, рак, гипертензия, ожирение и сахарный диабет II типа, которые можно лечить с помощью агонистов PPARg; который включает введение пациенту эффективного количества указанной выше композиции.
Сокращения:
CBG: Каннабигерол.
CBGA: Каннабигероловая кислота.
CBG-Q (соединение I): Каннабигерол гидроксихинон.
DCC: дициклогексилкарбодиимид.
Keap 1: Kelch-подобный ЕСН-ассоциированный белок 1.
NFE2L2 или (Nrf2): Ядерный фактор-2, подобный фактору эритроидного происхождения 2.
NR1C: Ядерное подсемейство 1 С.
NRs: Ядерные рецепторы.
PPAR: Рецепторы, активируемые пролифератором пероксисом.
PPARg: Гамма-рецептор, активируемый пролифератором пероксисом, также называемый PPARγ, глитазон или NR1C3.
PPARα: Альфа-рецептор, активируемый пролифератором пероксисом, также называемый NR1C1.
PPARβ/δ: Бета/дельта-рецептор, активируемый пролифератором пероксисом, также называемый NR1C2.
Описание чертежей
Фигуры изобретения кратко описаны ниже. Подробное описание каждой Фигуры включено в каждый соответствующий пример.
Сокращения, используемые в фигурах:
I: относится к CBG-Q.
II: относится к соединению Формулы (II).
III: относится к соединению Формулы (III).
IV: относится к соединению Формулы (IV).
V: относится к соединению Формулы (V).
VI: относится к соединению Формулы (VI).
VII: относится к соединению Формулы (VII).
VIII: относится к соединению Формулы (VIII).
IX: относится к соединению Формулы (IX).
X: относится к соединению Формулы (X.
XI: относится к соединению Формулы (XI).
XII: относится к соединению Формулы (XII).
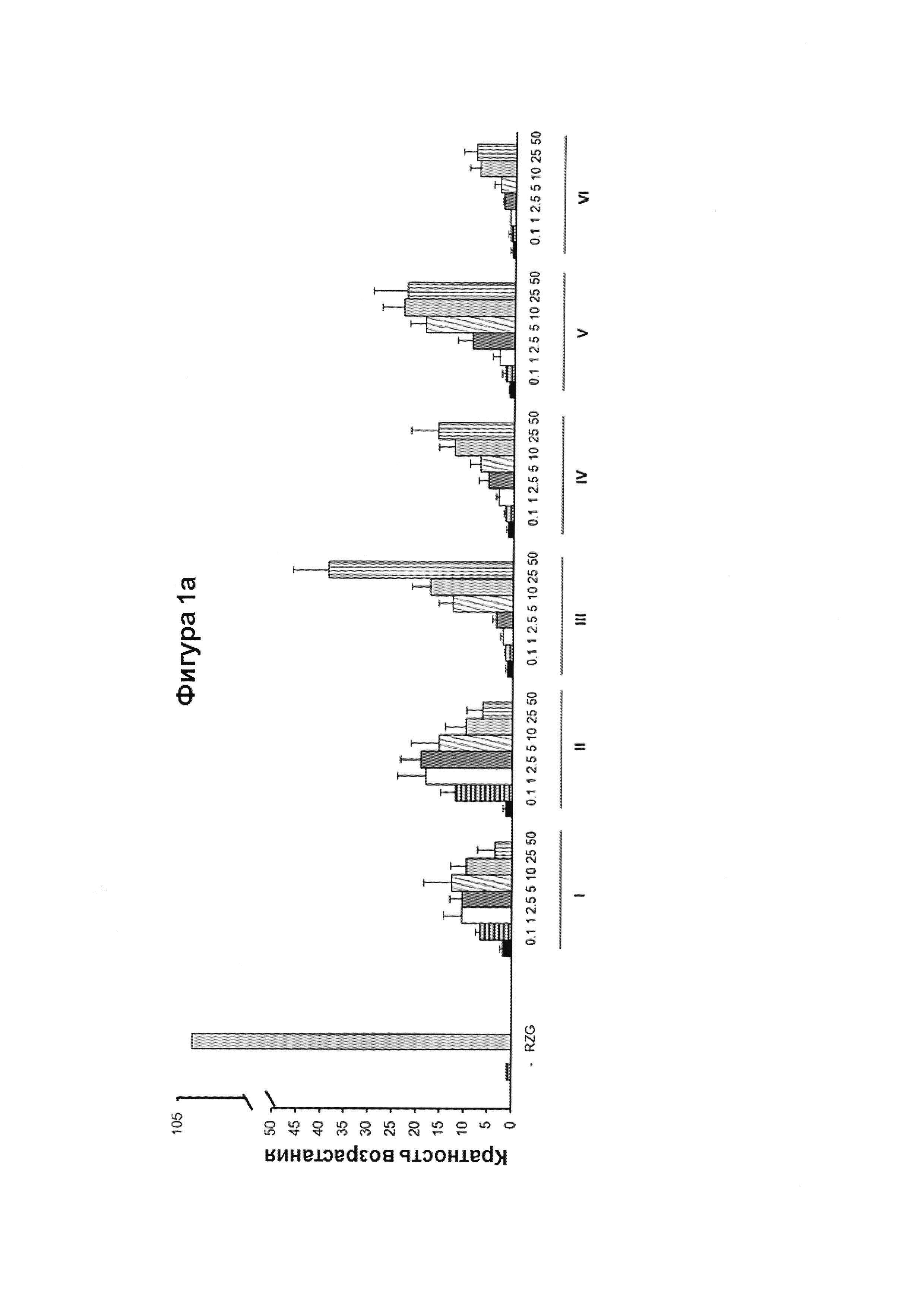
Фигура 1. Трансактивация PPAR в клетках HEK293
Концентрация тестируемого соединения (мкМ) показана на оси x, и во сколько раз активировался PPARg, показано на оси y. На этой фигуре показан эффект CBG-Q или соединения I по сравнению с действием соединений II-VI (Фигура 1а), и по сравнению с действием соединений VII-XII (Фигура 1б) на активность PPARg, который подтверждает, что производные CBG-Q (соединение I), и особенно соединения II, III, IV, V, VII, VIII и XII в настоящее время способны индуцировать активацию PPARg с более высокой эффективностью, чем CBG-Q (соединение I). Полный агонист PPARγ розиглитазон (RZG) 1 мкМ использовали в качестве контроля. Кратность возрастания активации вычисляли, принимая контрольный образец (-) без добавления любого агониста PPARg или активирующего агента в качестве базового уровня. Данные выражены в виде среднего значения ± SD по меньшей мере трех независимых экспериментов.
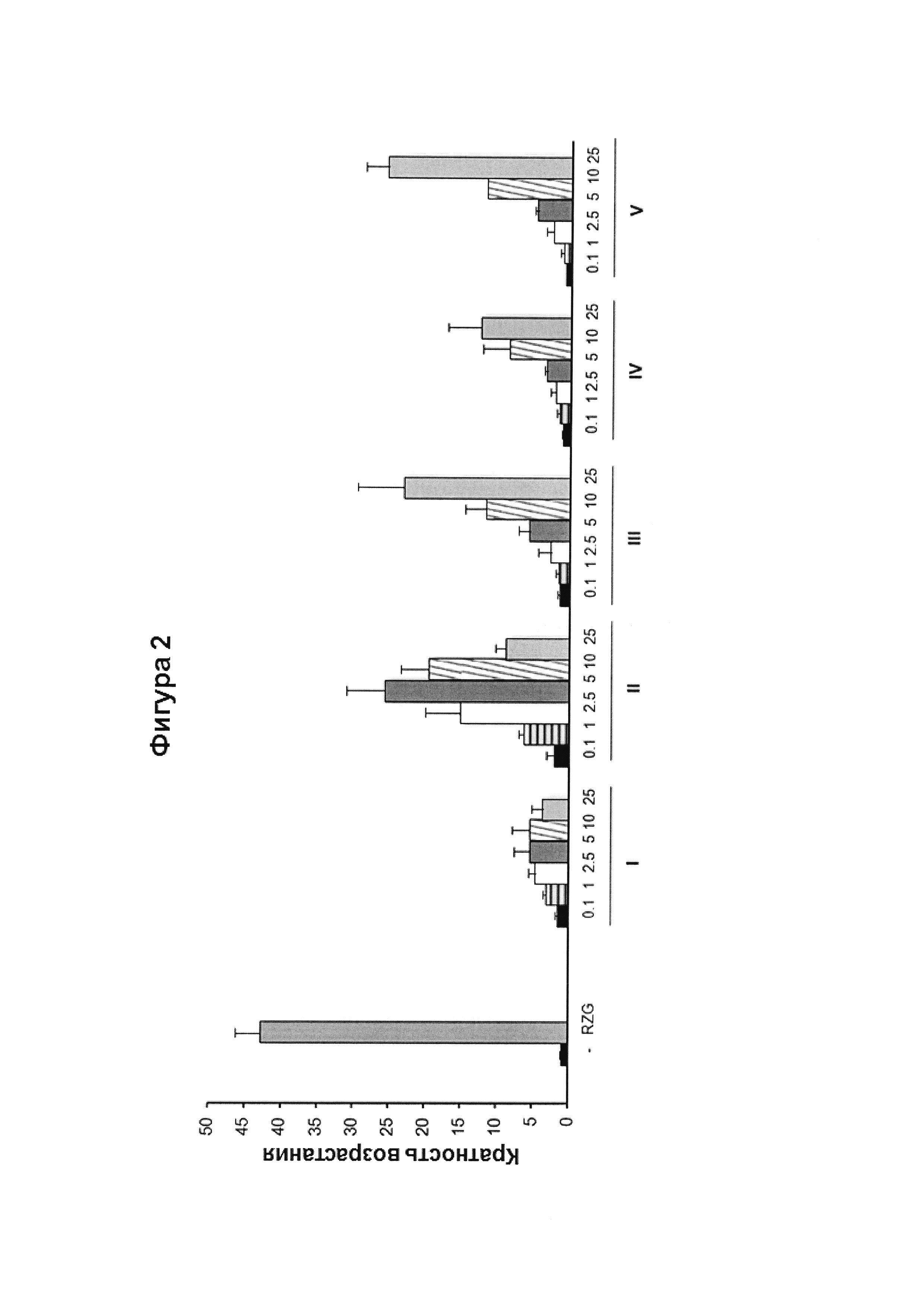
Фигура 2. Трансактивация PPAR в дермальных первичных фибробластах человека.
Концентрация тестируемого соединения (мкМ) показана на оси x, и во сколько раз активировался PPARg показано на оси y. На этой фигуре показан эффект CBG-Q или соединения I по сравнению с действием соединений II, III, IV и V на активность PPARg, который подтверждает, что соединения II, III, IV и V способны индуцировать активацию PPARg с более высокой эффективностью, чем CBG-Q (соединение I). Полный агонист PPARγ розиглитазон (RZG) 1 мкМ использовали в качестве контроля. Кратность возрастания активации вычисляли, принимая контрольный образец (-) без добавления любого агониста PPARg или активирующего агента в качестве базового уровня. Данные выражены в виде среднего значения ± SD по меньшей мере трех независимых экспериментов.
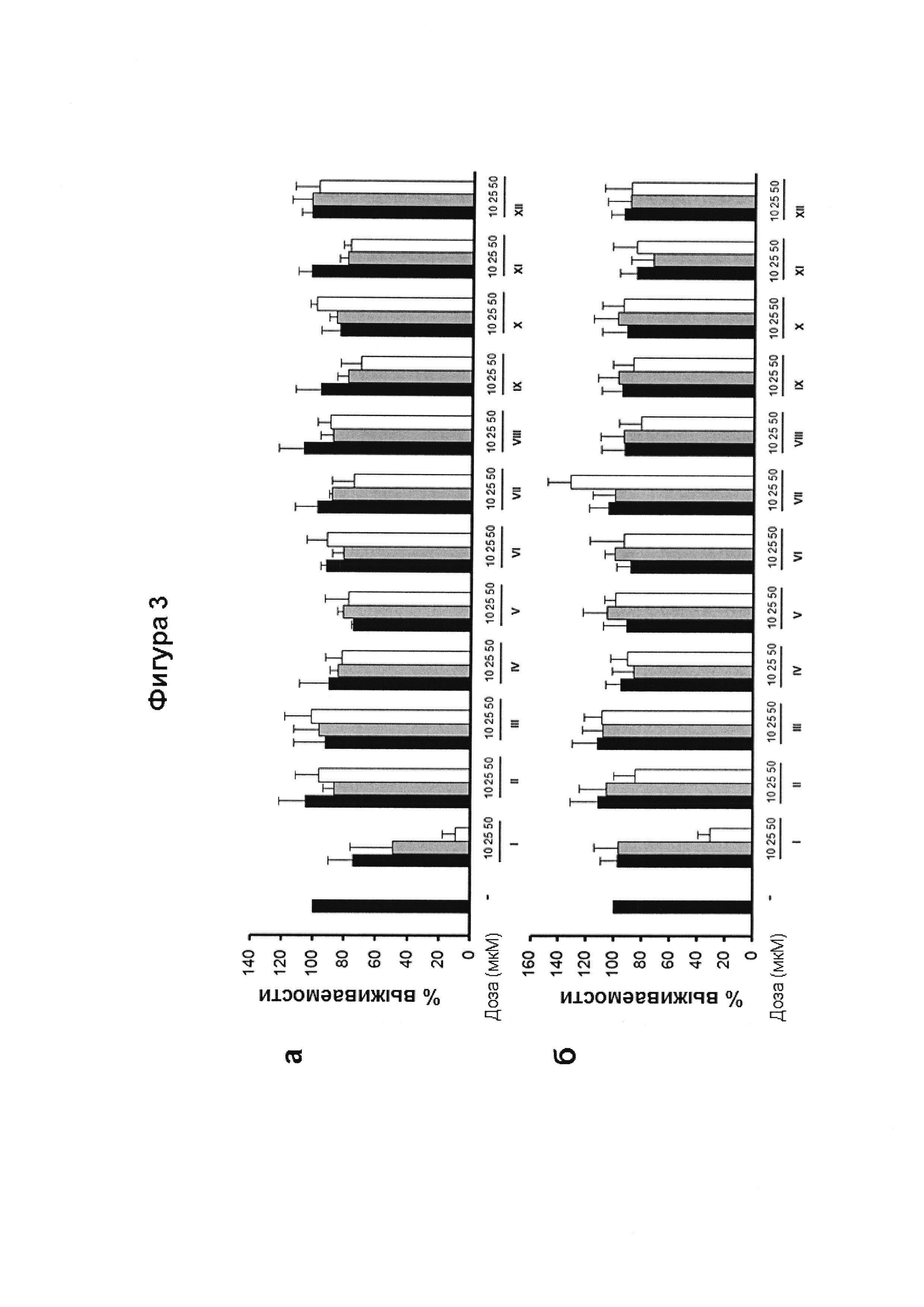
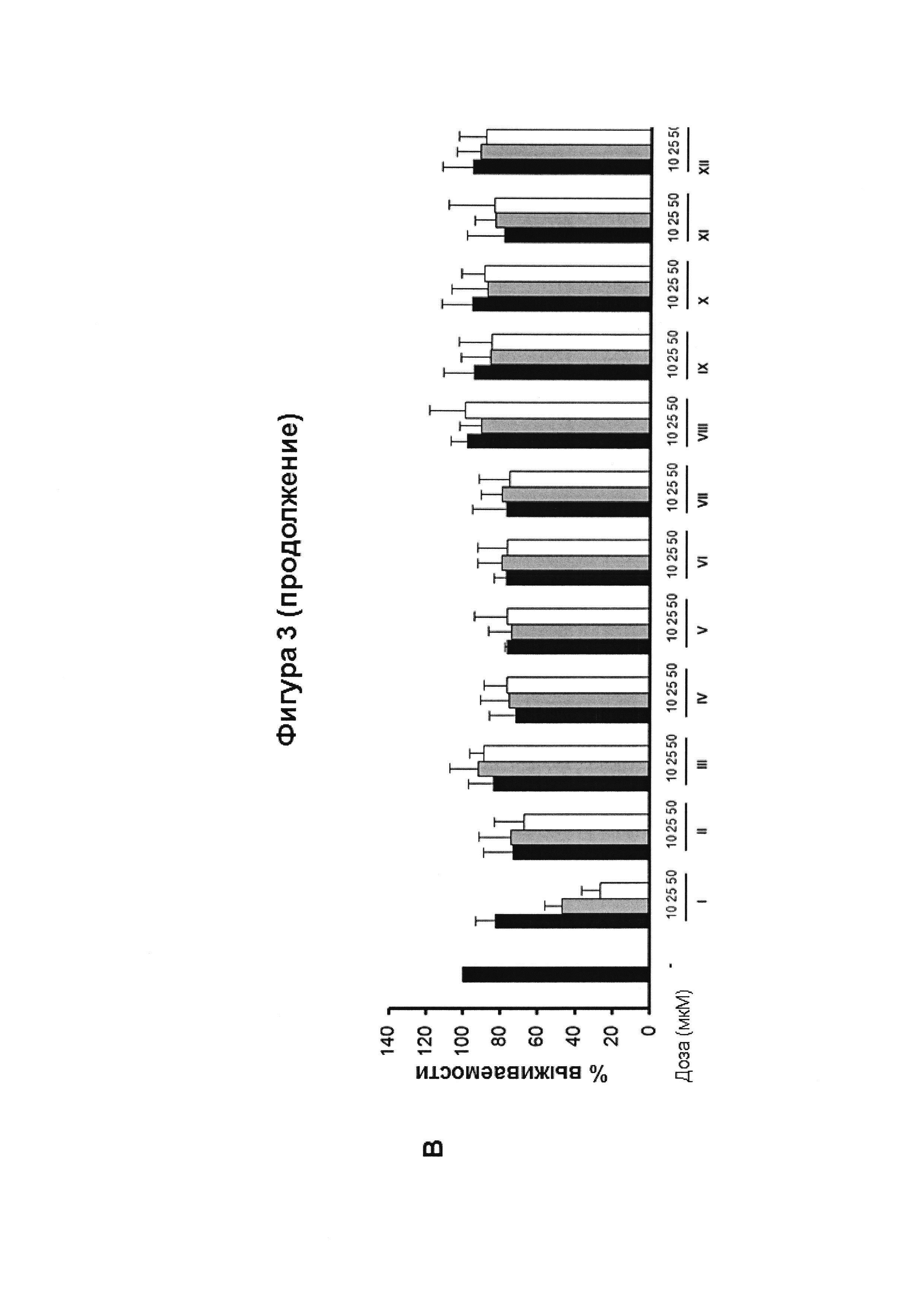
Фигура 3. Цитотоксическая активность.
Клетки линий N2a (а), НТ22 (б) и MO3.13 (в) инкубировали в течение 24 ч с указанными дозами CBG-Q (соединение I) и сравнивали с соединениями II-XII, и жизнеспособность клеток количественно определяли с помощью анализа МТТ. Результаты представлены в виде среднего значения ± SD по меньшей мере трех независимых экспериментов, и выражены в процентах жизнеспособности клеток по сравнению с контрольным образцом (-), без любого агониста PPARg или активирующего агента. Контроль определили как 100%, и данные относили к этому значению. Результаты показывают, что цитотоксическая активность, связанная с CBG-Q (соединение I) отсутствует у всех производных CBG-Q по положению 2, описанных в настоящем изобретении.
Фигура 4. Анализ транскрипции Nrf2
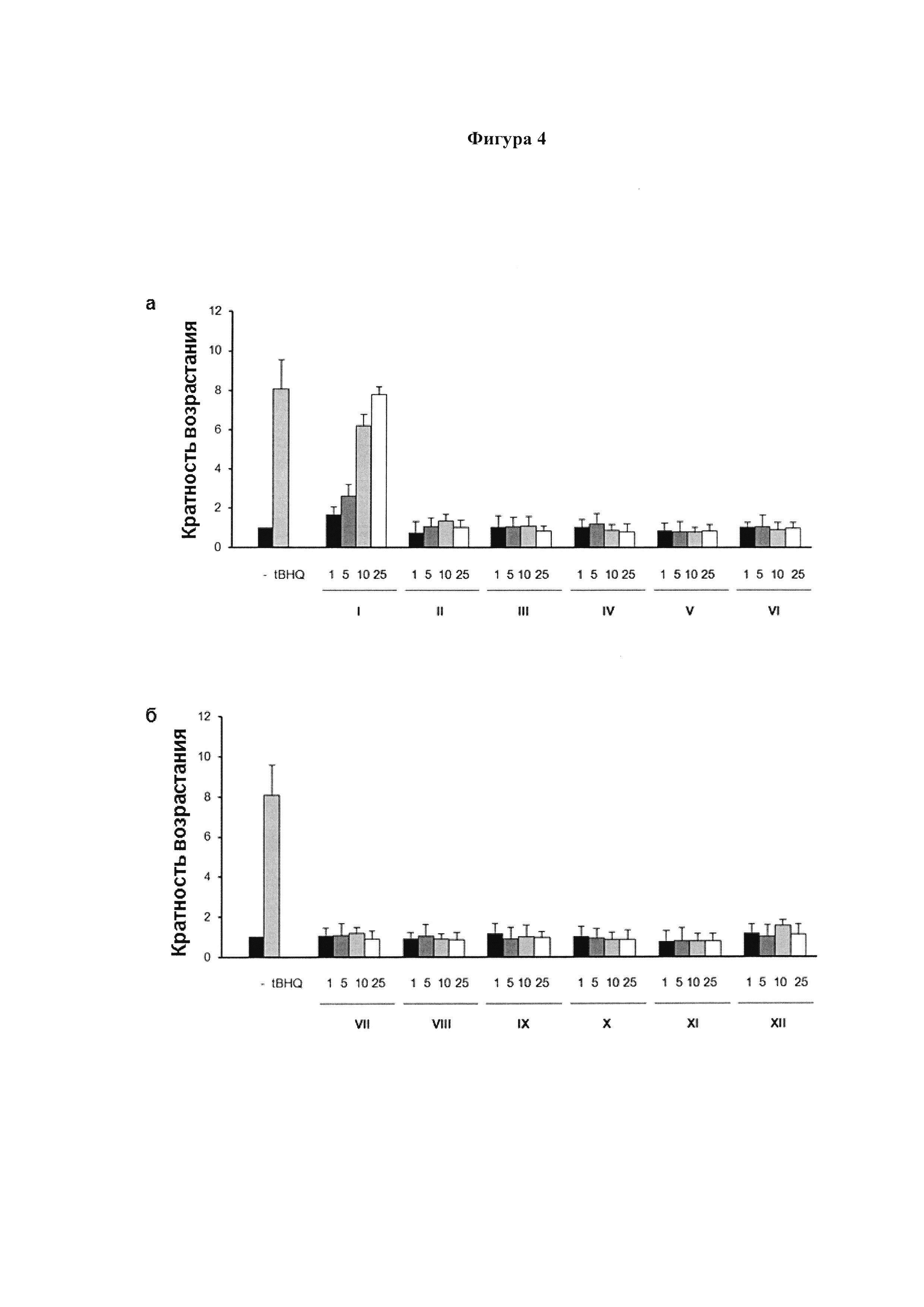
HaCaT-ARE-Luc клетки инкубировали в течение 6 ч с соединением CBG-Q (соединение I) и с соединениями I-VI (а) или с соединениями VII-XII (б) в указанных концентрациях, затем получали белковые лизаты и анализировали на активность люциферазы. Прооксидант трет-бутилгидрохинон (TBHQ) в концентрации 20 мкМ, соединение, которое вызывает клеточный окислительный стресс, использовали в качестве положительного контроля. Кратность возрастания активации вычисляли, принимая контрольный образец (-), без любого агониста PPARg или активирующего агента в качестве базового уровня. Данные выражены в виде среднего значения ± SD по меньшей мере трех независимых экспериментов. Результаты свидетельствуют, что обратная электрофильная активность, связанная с CBG-Q (соединение I) отсутствует во всех соединениях (производных по положению 2), описанных в настоящем изобретении.
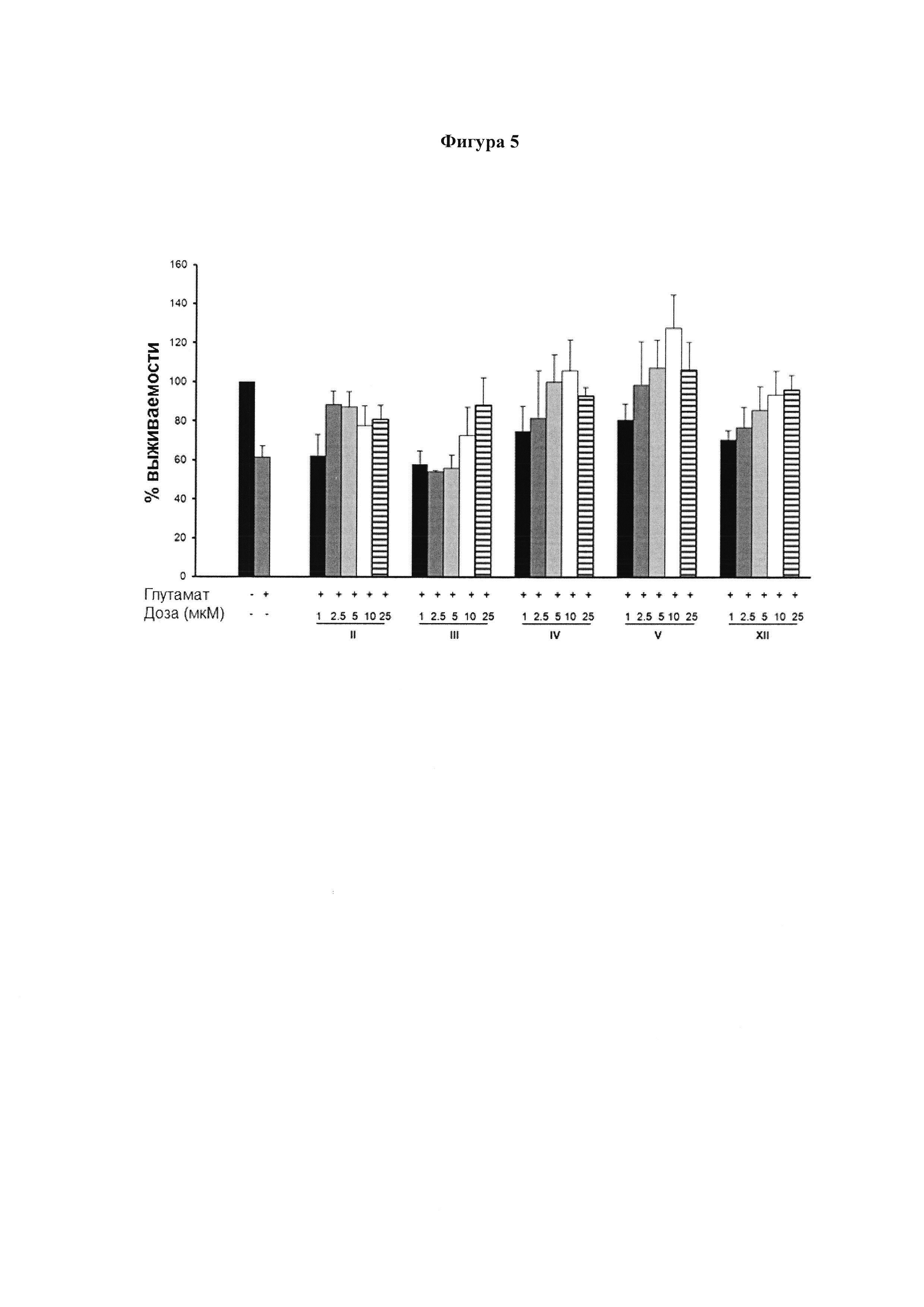
Фигура 5. Нейропротекторная активность.
N2a клетки предварительно инкубировали в течение 1 ч с соединениями (II)-(V) и (XII) в указанных концентрациях. Затем клетки обрабатывали в течение 24 ч 5 мМ глутамата, чтобы индуцировать эксайтотоксичность или цитотоксичность в нейрональных клетках, индуцированную нейротрансмиттерами. Жизнеспособность клеток определяли количественно с помощью МТТ-теста. Результаты представлены в виде среднего значения ± SD по меньшей мере трех независимых экспериментов и выражены в процентах от жизнеспособности клеток по отношению к контрольному образцу (-), без любого агониста PPARg или активирующего агента и с (-,+) или без (-, -) глутамата. Контроль приняли за 100%, и данные относили к этому значению.
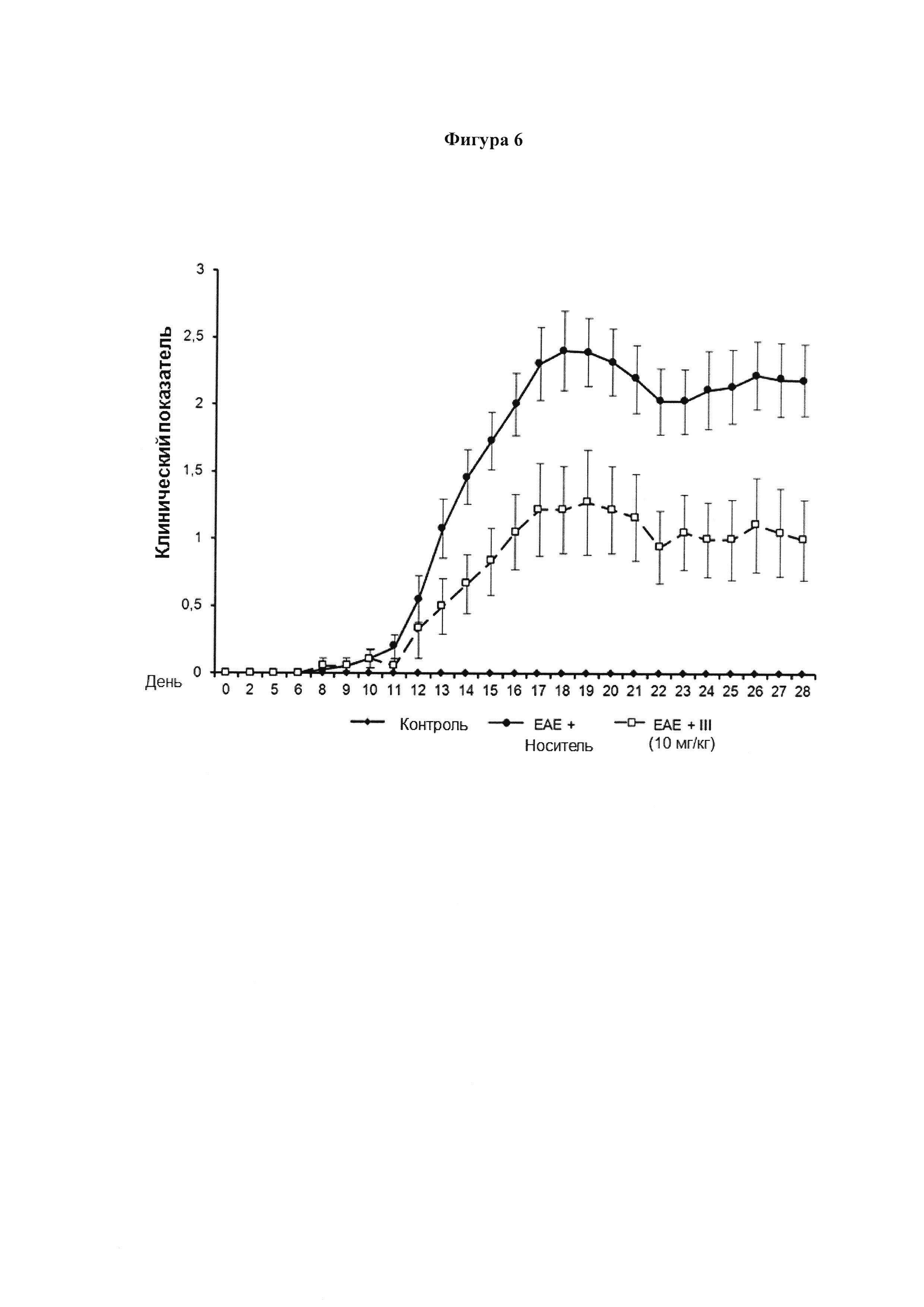
Фигура 6. Соединение (III) облегчает ЕАЕ
Мышей линии C57BL/6 иммунизировали MOG35-55 и ежедневно оценивали их клинический показатель. Мышей обрабатывали ежедневно соединением (III) (10 мг/кг) на 6-ой день после иммунизации, и 21 последующий день. График показывает среднесуточный клинический показатель (среднее ± SEM). Значения выражены как среднее ± SEM для 10 животных в каждой группе.
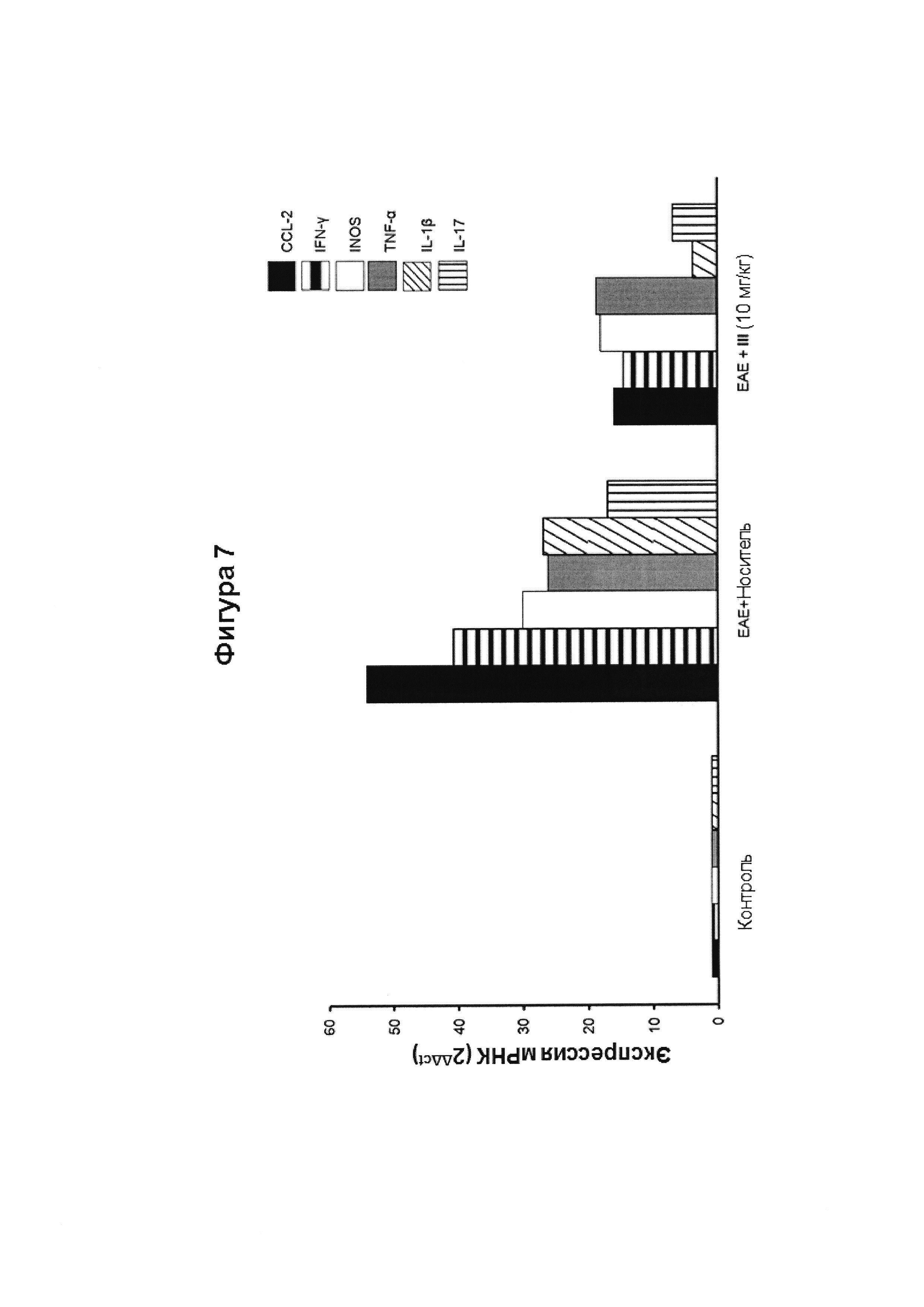
Фигура 7. Влияние соединения (III) на провоспалительные маркеры (ЕАЕ)
Экспрессия генов маркеров воспаления, включая CCL2, IFNγ, INOS, TNFα, IL-1β и IL-17 в спинном мозге была подавлена в ЕАЕ+ соединение (III) (10 мг/кг) группе мышей по сравнению с группой ЕАЕ + носитель. Уровни экспрессии рассчитали с использованием метода 2-ΔΔCt.
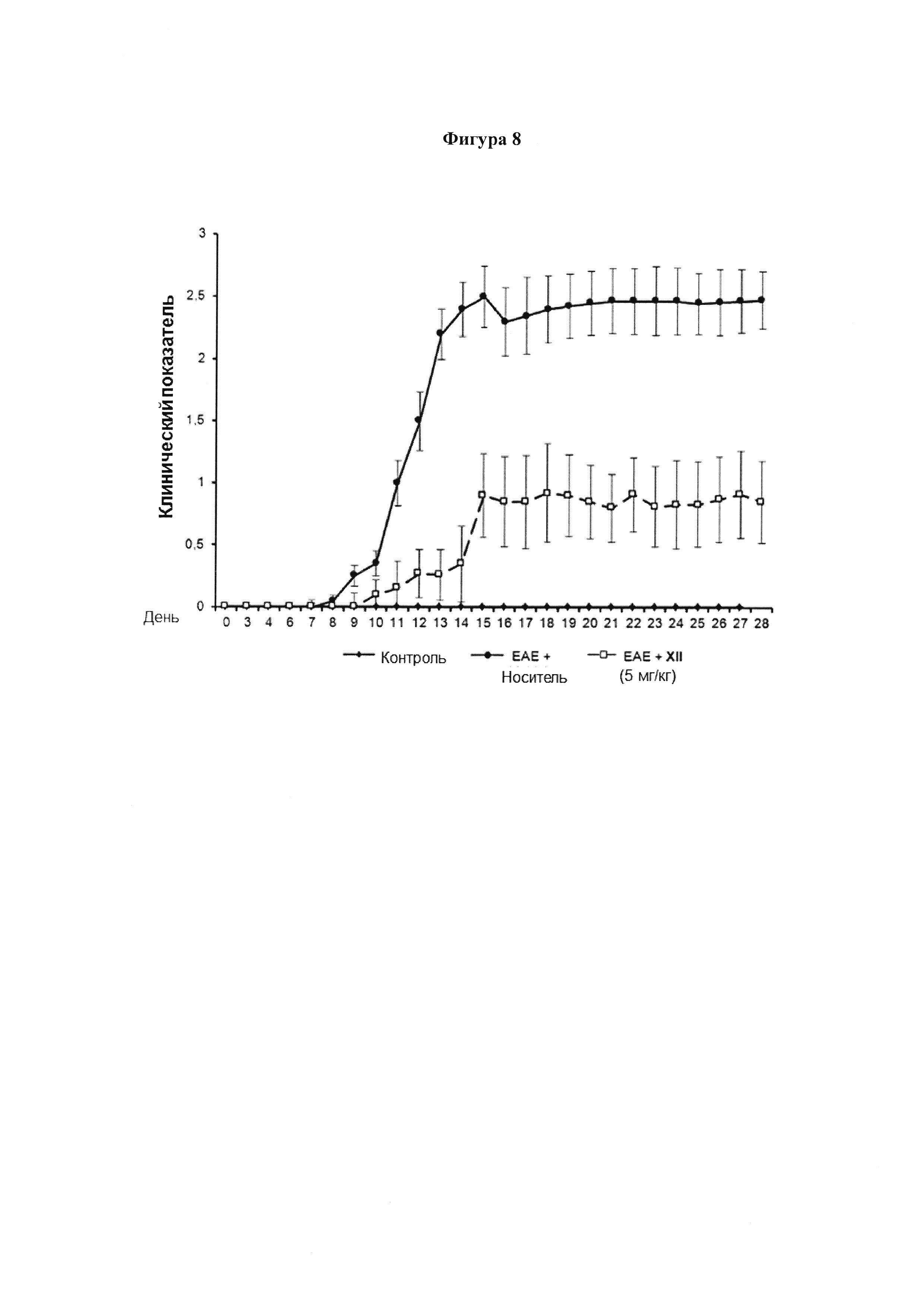
Фигура 8. Соединение (XII) ослабляет ЕАЕ
C57BL/6 мышей иммунизировали MOG35-55 и их клинический показатель оценивали ежедневно. Мышей обрабатывали ежедневно соединением (XII) (5 мг/кг) на 6-ой день после иммунизации, и 21 последующих дней. График показывает среднесуточный клинический показатель (среднее ± SEM). Значения выражены как среднее ± SEM для 6 животных в каждой группе.
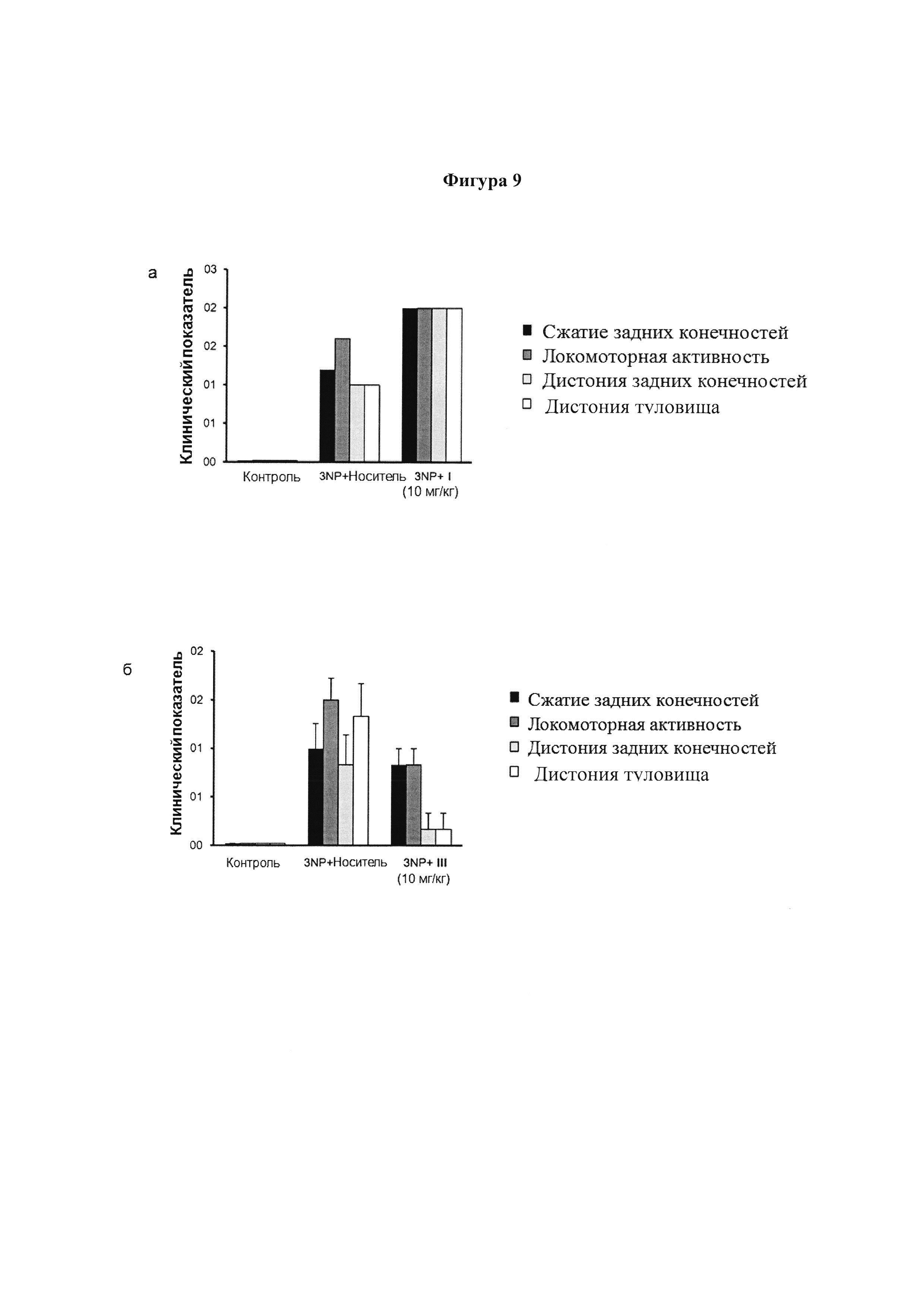
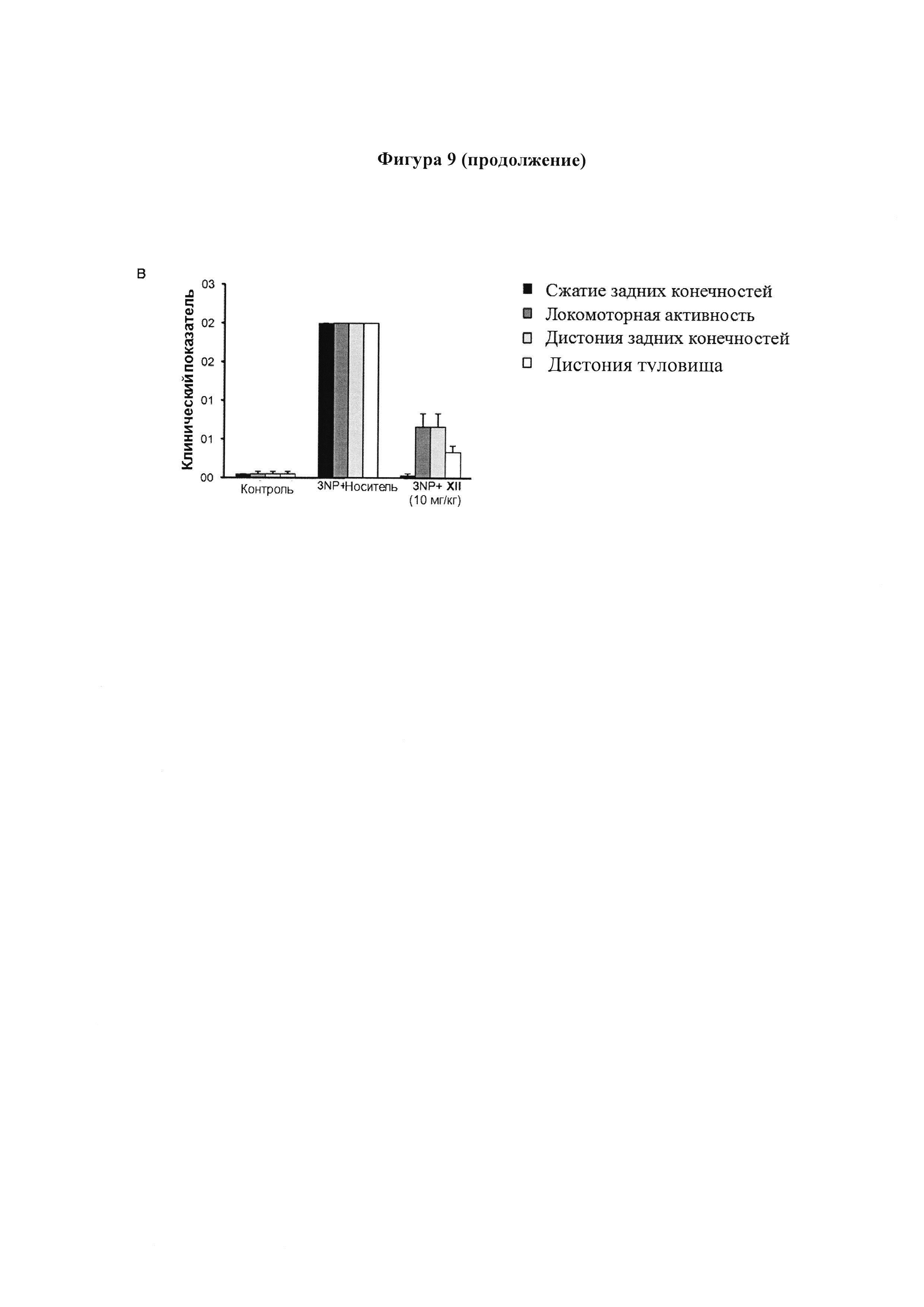
Фигура 9. Поведенческие баллы после интоксикации 3NP.
Мышей подвергли поведенческим тестам для определения их неврологического статуса после обработки соединениями I (10 мг/кг) (а), III (10 мг/кг) (б) и XII (10 мг/кг) (с). Сжатие задних конечностей, локомоторную активность, дистонию задних конечностей и дистонию туловища оценивали от 0 до 2 на основе серьезности: оценка 0 обычно указывает на нормальную функцию и 2 означает, что функция серьезно затронута. Значения выражены как среднее ± SEM для 8 животных в каждой группе.
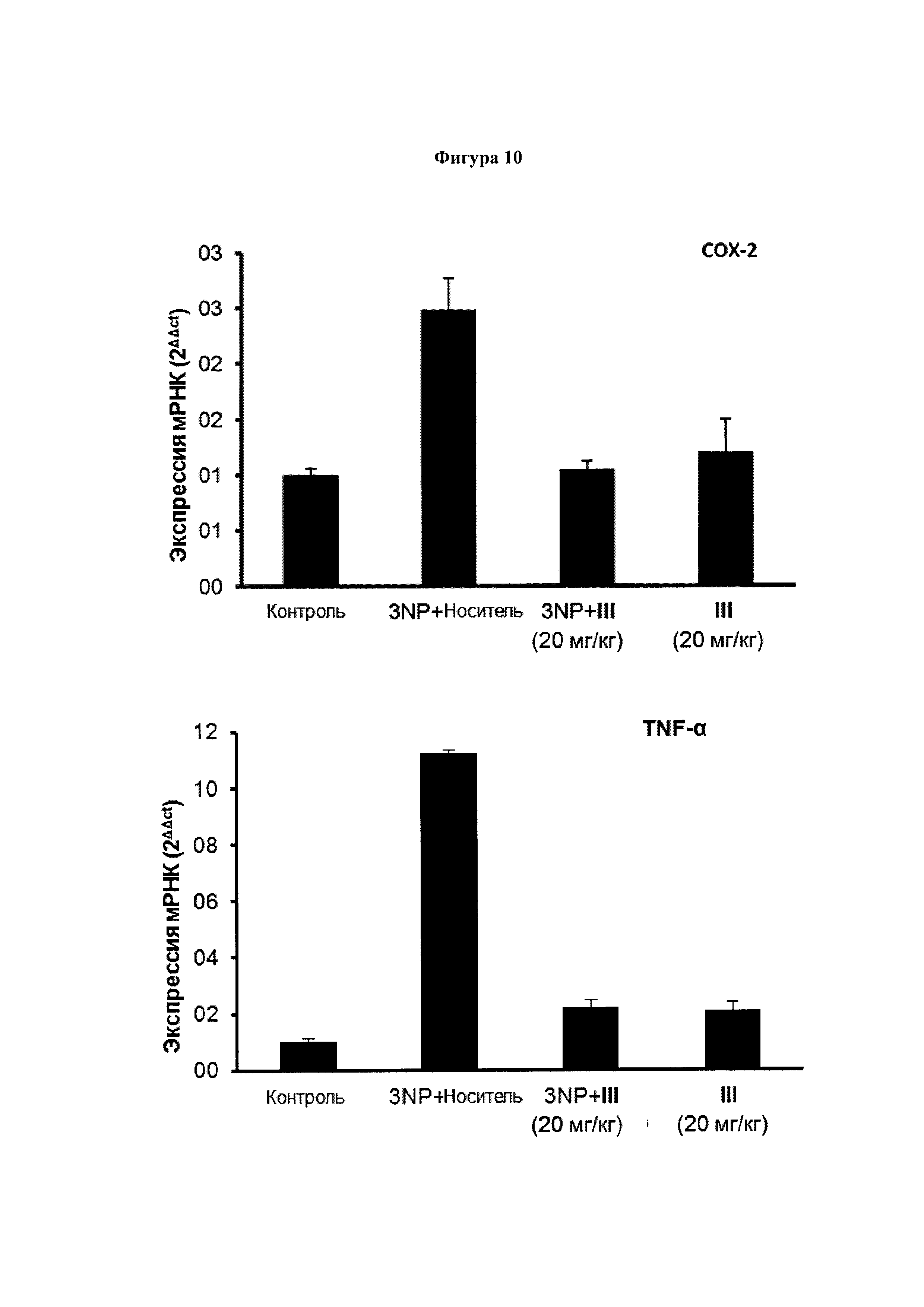
Фигура 10. Соединение III снижает экспрессию мРНК воспалительных маркеров в полосатом теле.
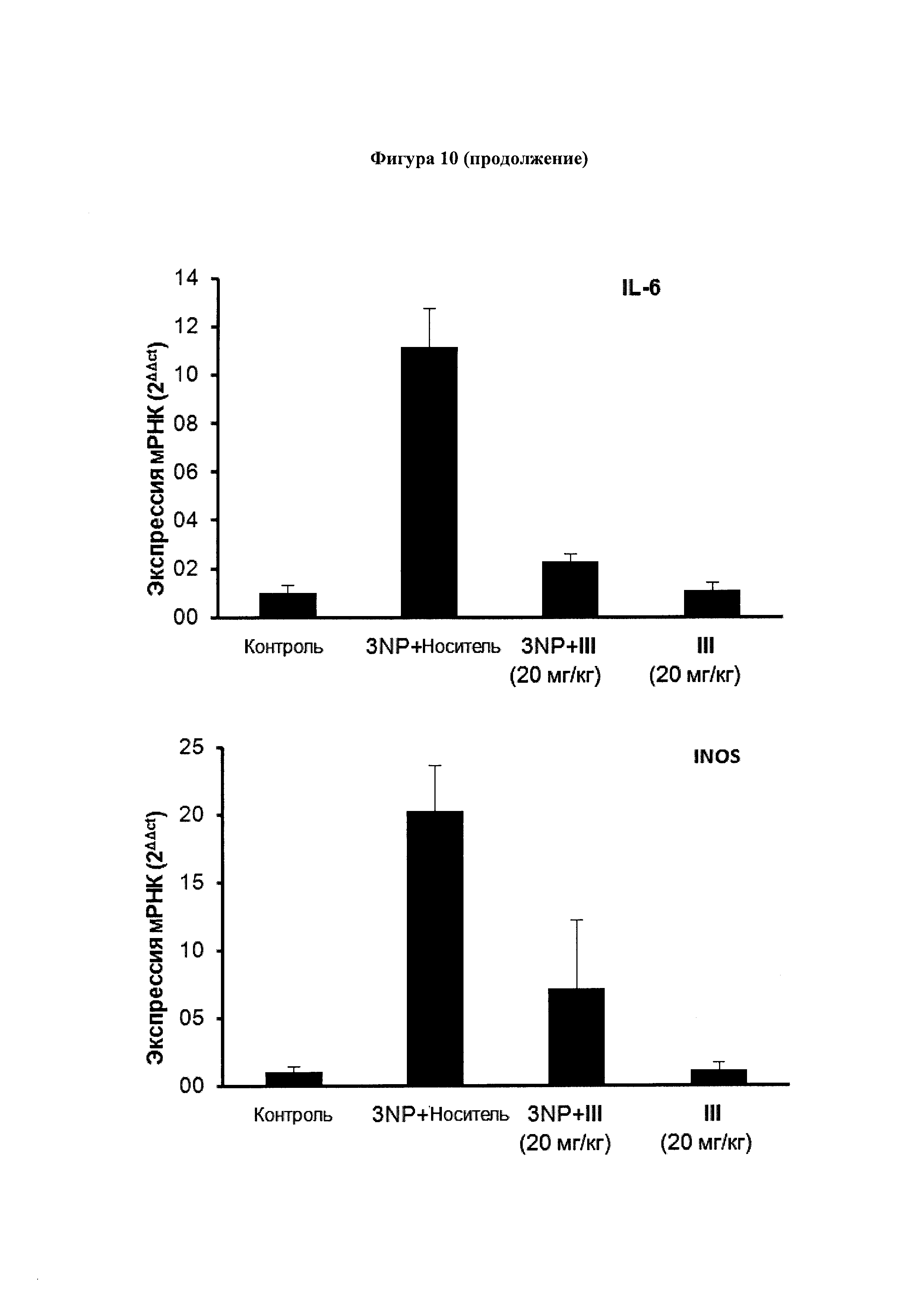
Экспрессия генов маркеров воспаления, включая СОХ-2, TNFα, IL-6 и iNOS подавлялась у обработанных 3NP + соединение III (10 мг/кг) мышей, по сравнению с мышами 3NP + носитель. Уровни экспрессии рассчитали с использованием метода 2-ΔΔCt. Значения выражены как среднее ± SEM для 6 животных в каждой группе.
Фигура 11. Соединение XII снижает экспрессию мРНК воспалительных маркеров в полосатом теле.
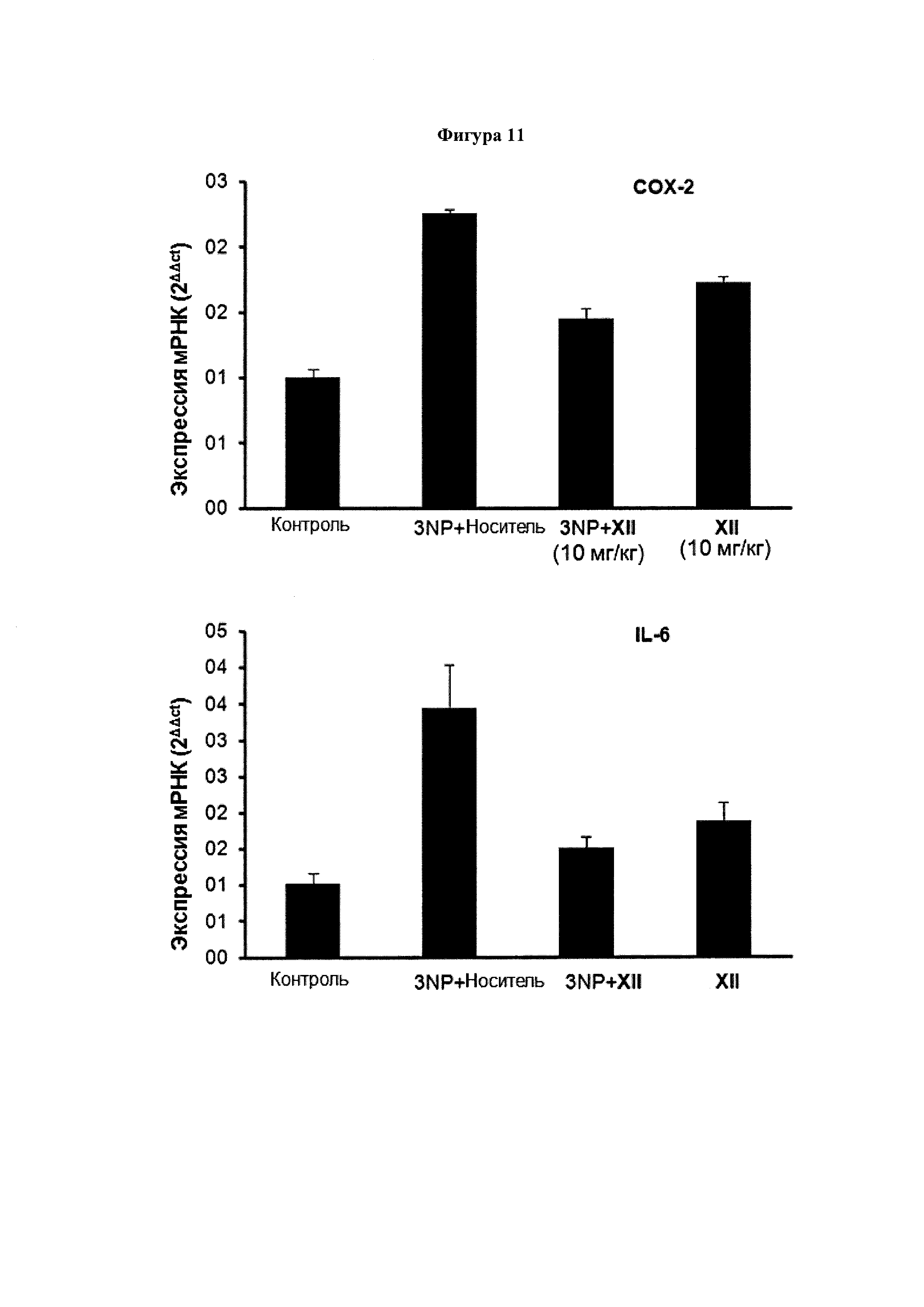
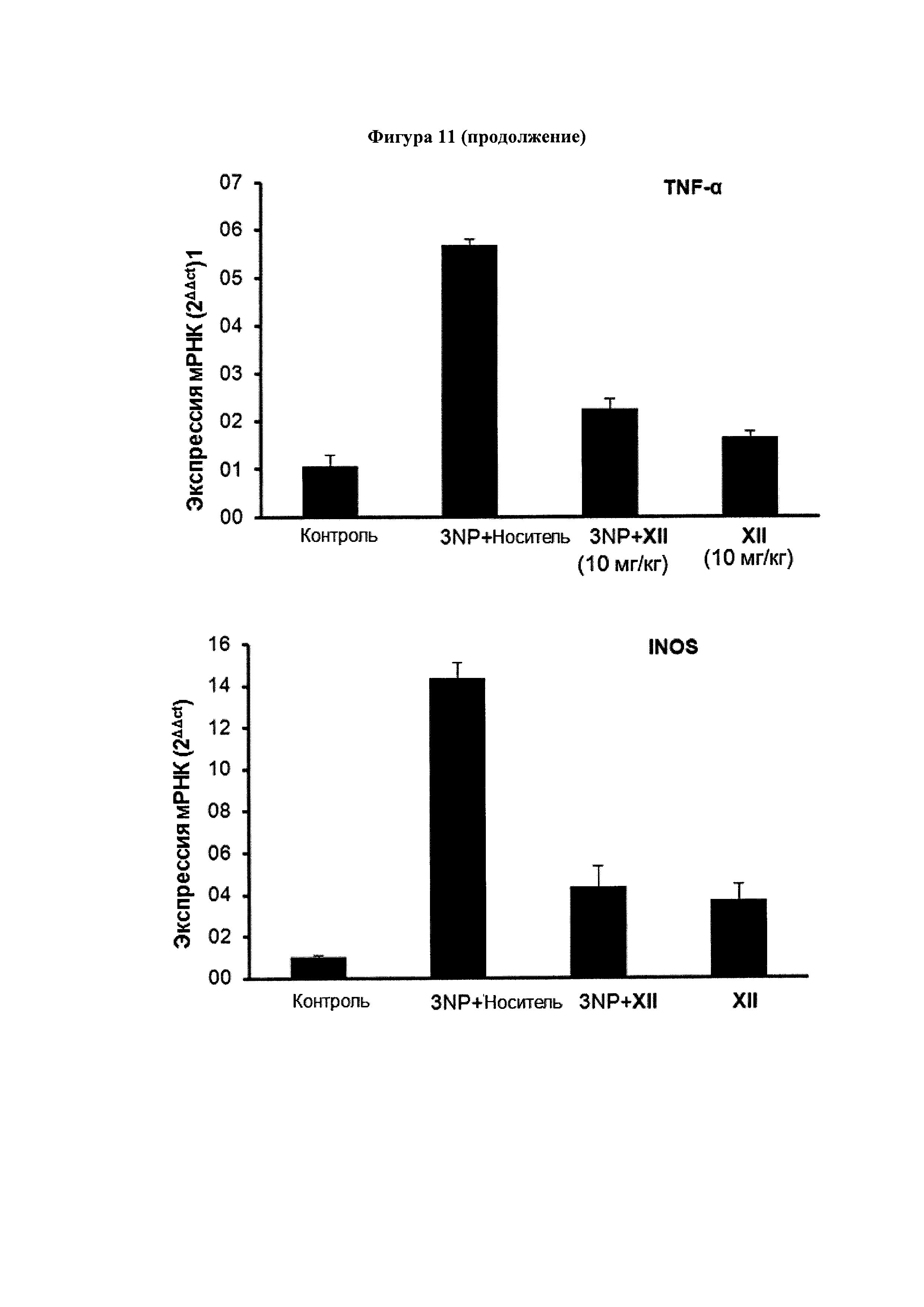
Экспрессия генов маркеров воспаления, включая СОХ-2, TNFα, IL-6 и iNOS подавлялась у обработанных 3NP + соединение XII (10 мг/кг) мышей по сравнению с мышами 3NP + носитель. Уровни экспрессии рассчитали с использованием метода 2-ΔΔCt. Значения выражены как среднее ± SEM для 6 животных в каждой группе.
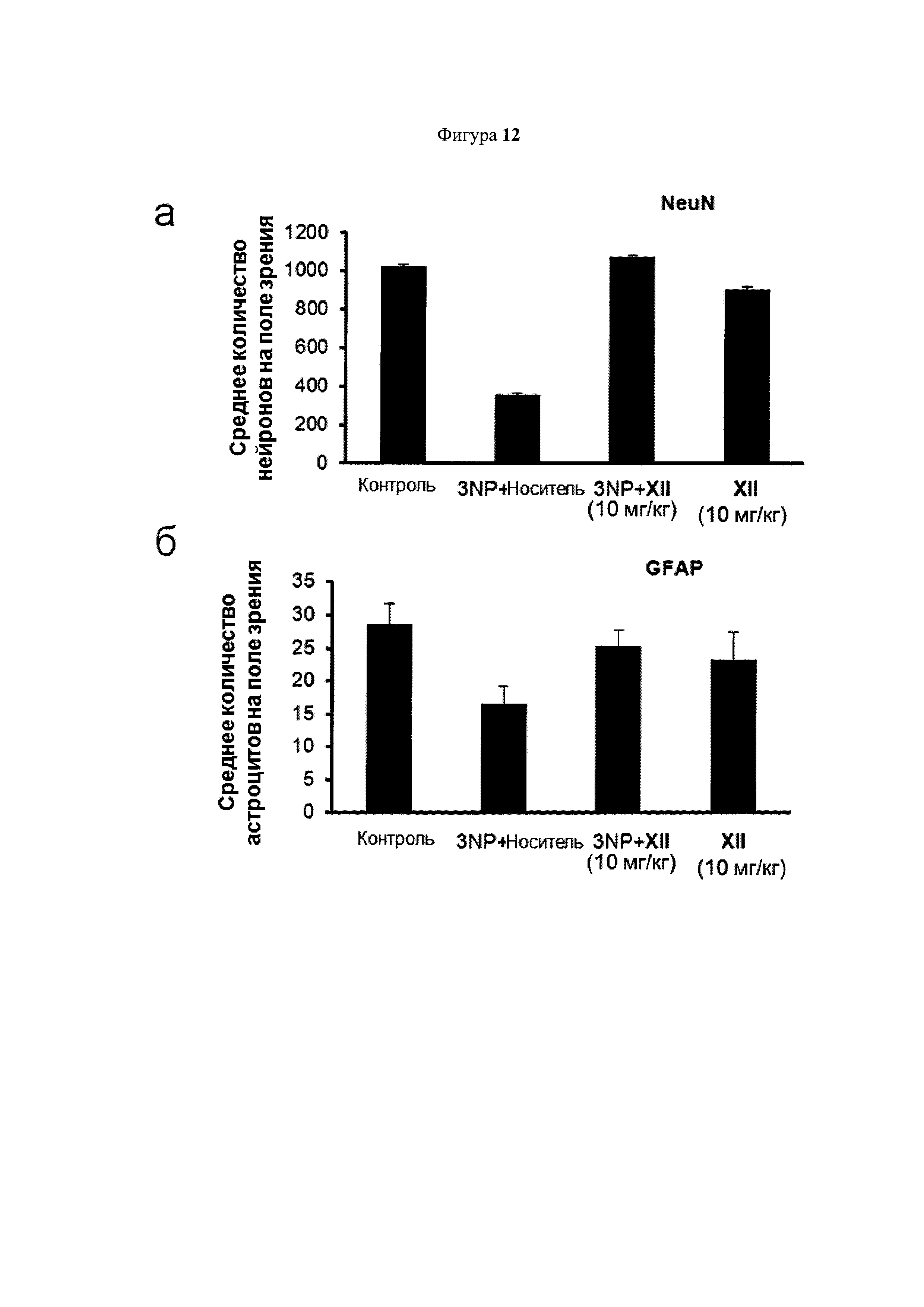
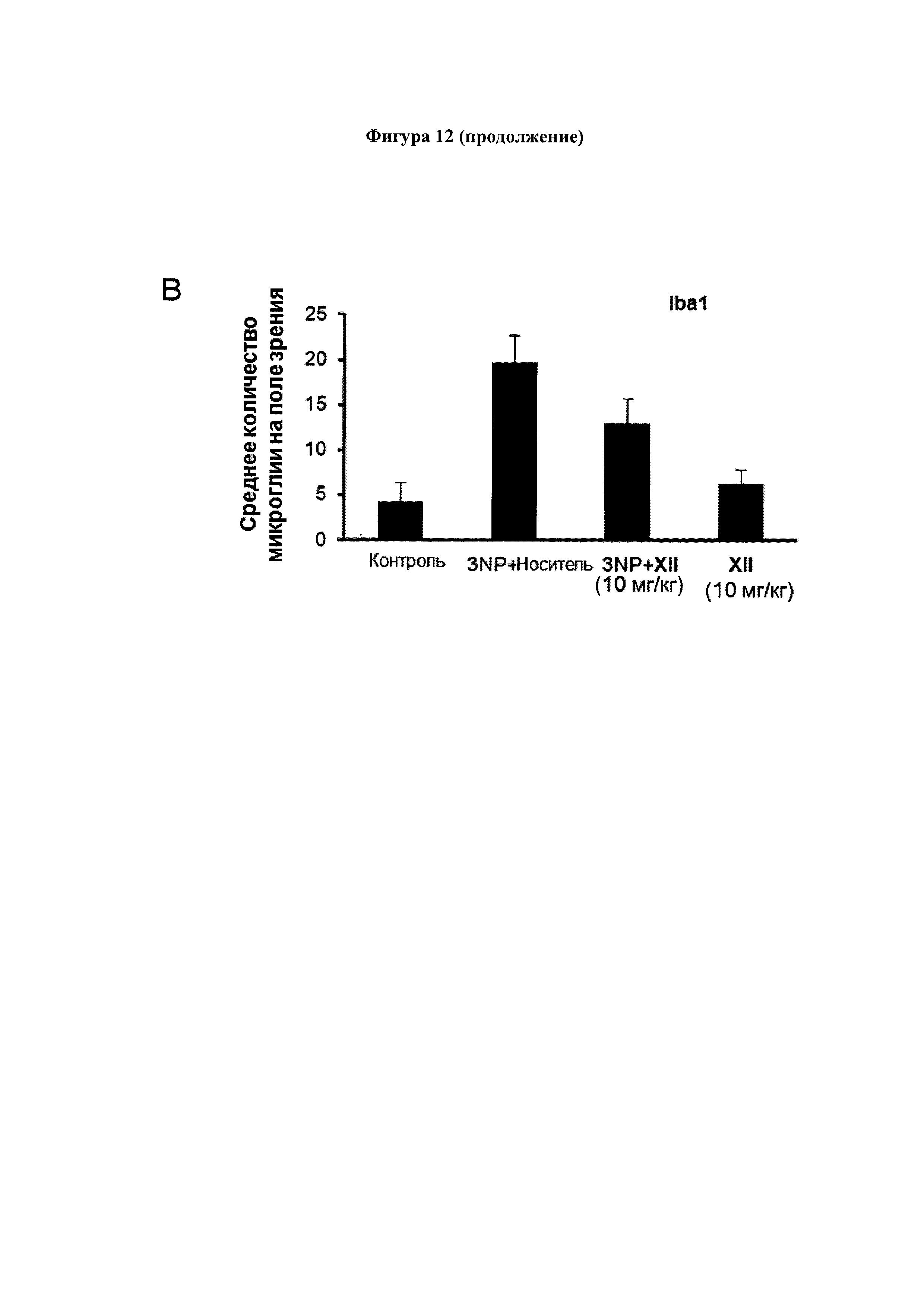
Фигура 12. Эффект соединения XII на нейродегенеративные маркеры (3NP)
NeuN (маркер нейронов), GFAP (маркер астроцитов) и Ibal (маркер микроглии) обнаруживали с помощью иммуннокрашивания в корональной секции полосатого тела мышей, обработанных носителем, 3NP + носитель, 3NP + соединение XII (10 мг/кг) и XII (10 мг/кг). Определяли количество NeuN (a), GFAP (б) и Iba1 (в) положительных клеток в полосатом теле мышей. Показано общее среднее число нейронов, астроцитов и микроглии. Значения выражены как среднее ± SEM для 6 животных в каждой группе.
Фигура 13. Эффект соединения (III) на симптоматику болезни Паркинсона, индуцированную 6-OHDA.
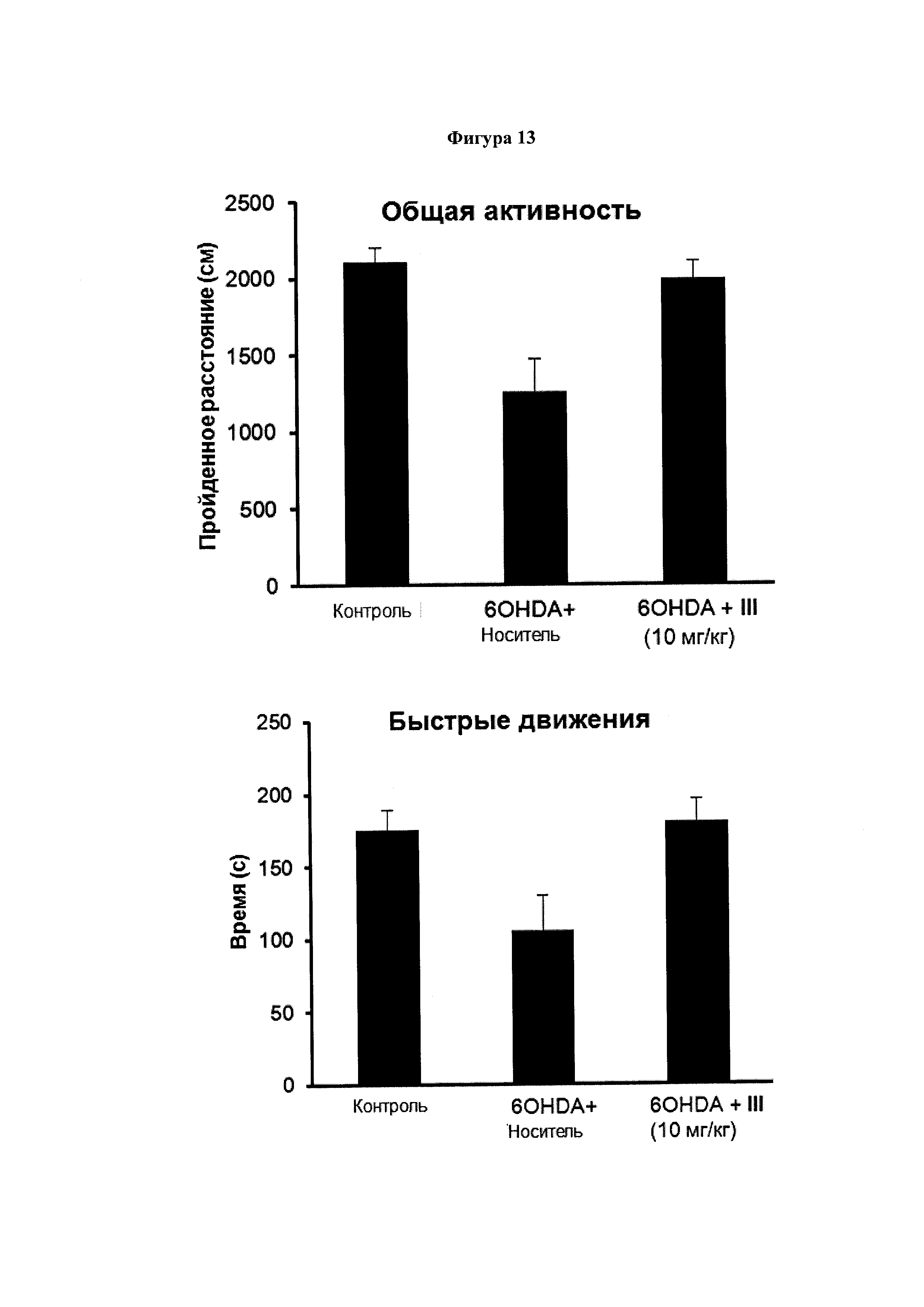
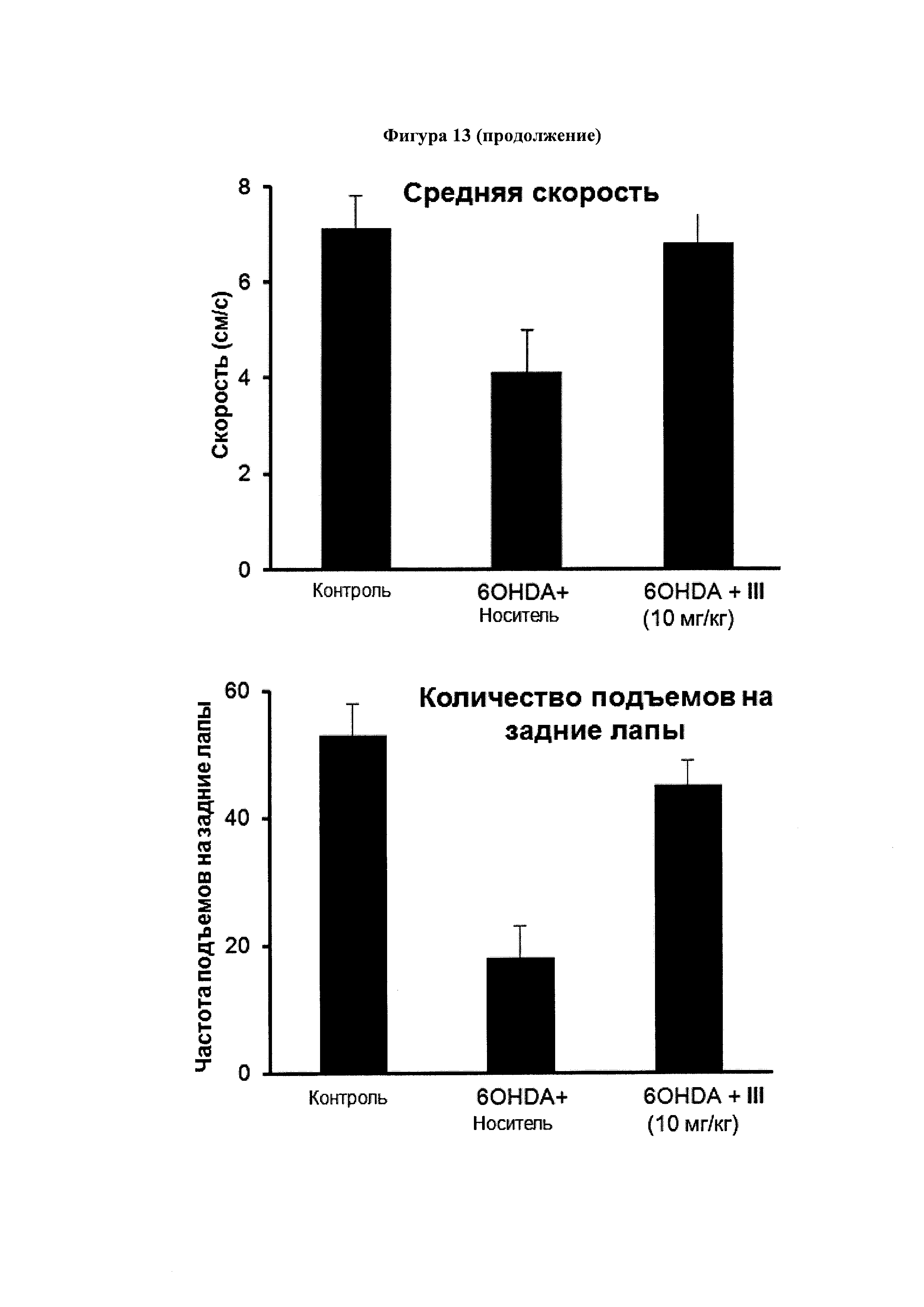
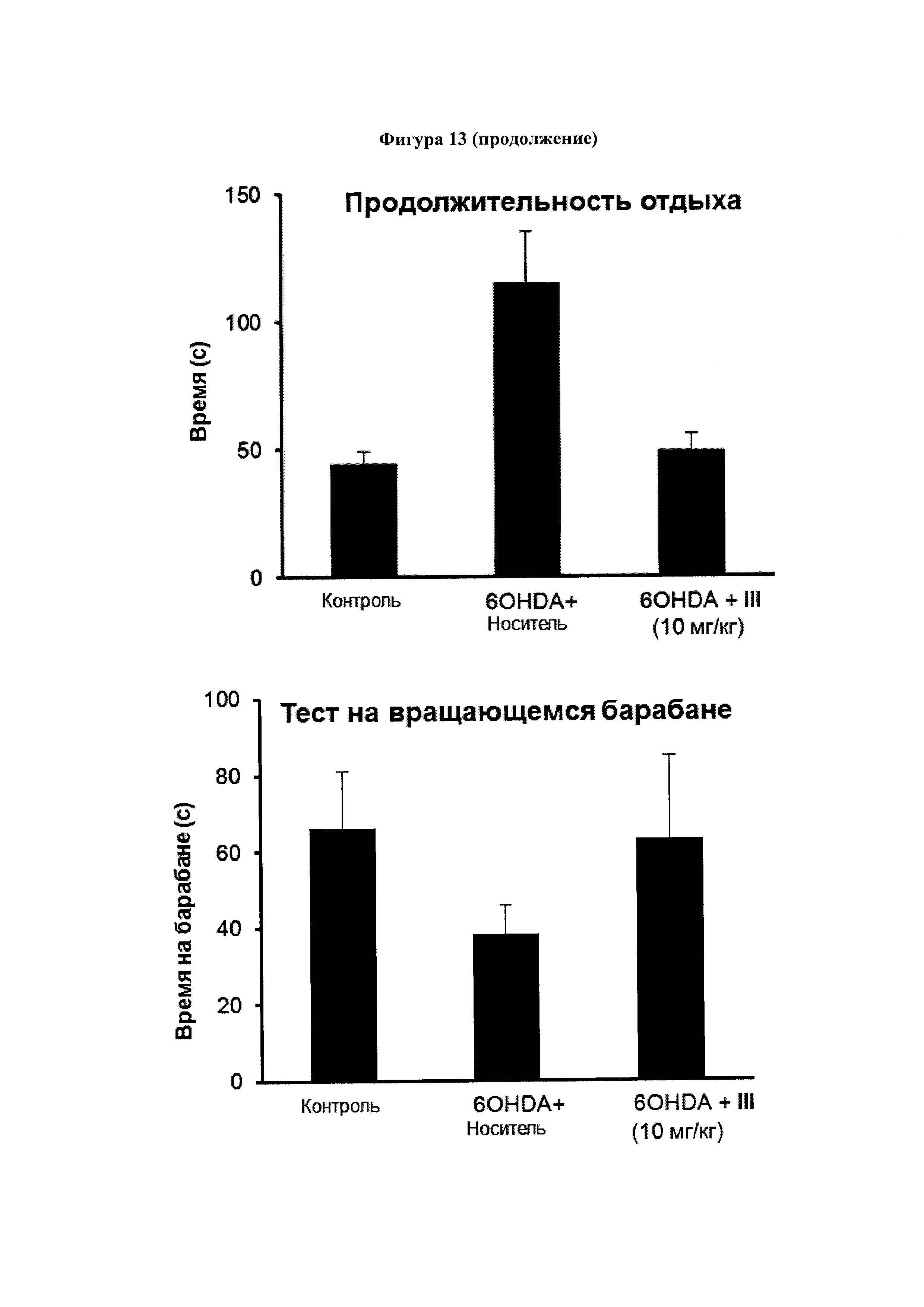
C57BL/6 мышам однократно вводили интрацеребровентрикулярно 6-гидроксидопамин (6-OHDA) или физиологический раствор (контрольные мыши) и подвергали продолжительной (14 дней) внутрибрюшинной обработке соединением III (10 мг/мл) или носителем (14 дней), начиная с 16 ч после инъекций 6-OHDA. Координацию движений оценивали по способности удерживаться на вращающемся барабане, а двигательную активность оценивали с помощью автоматизированного актиметра. Значения выражены как среднее ± SEM для 6 животных в каждой группе.
Примеры
Примеры осуществления настоящего изобретения, описанные ниже, направлены на иллюстрацию предпочтительных вариантов его осуществления, не ограничивая его объем охраны.
Пример 1. Химический синтез и ЯМР анализ
Общие процедуры для соединений, полученных из CBGA. Синтез соединений (II) и (VII))
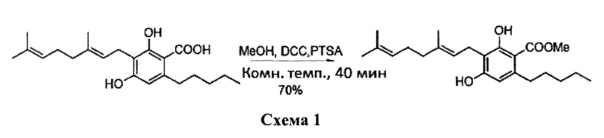
К раствору CBGA (каннабигероловой кислоты) (360 мг, 0,80 ммоль) в метаноле (10 мл) добавили дициклогексилкарбодиимид (DCC) (331 мг, 1,6 ммоль) и каталитическую п-толуолсульфоновую кислоту (около 10 мг). После перемешивания в течение 40 мин реакционную смесь обрабатывали путем выпаривания (схема 1). Остаток растворяли в толуоле (около 10 мл) и охлаждали (-18°C), чтобы осадить мочевину. Через 1 ч раствор фильтровали на фильтре из спеченного стекла, а затем остаток очищали с помощью флэш-хроматографии RP С-18 на силикагеле с получением 260 мг (Е)-метил-3-(3,7-диметилокта-2,6-диенил)-2,4-дигидрокси-6-пентилбензоата [бесцветная пена, выход: 70%].
1H NMR (CDCl3, 300 МГц) δ ppm 12.00 (bs, 1H), 6.25 (s, 1H), 5.27 (bt, J=6.5 Гц, 1H), 5.04 (bt, J=6.5 Гц, 1H), 3.90 (s, 3Н), 3.41 (d, J=6.8 Гц, 1Н), 2.05 (bm, 4Н), 1.80 (bs, 3Н), 1.66 (bs, 3Н), 0.89 (t, J=6.0 Гц, 3Н).
Получение соединения II.
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-метоксикарбонил-[1,4]бензохинон.
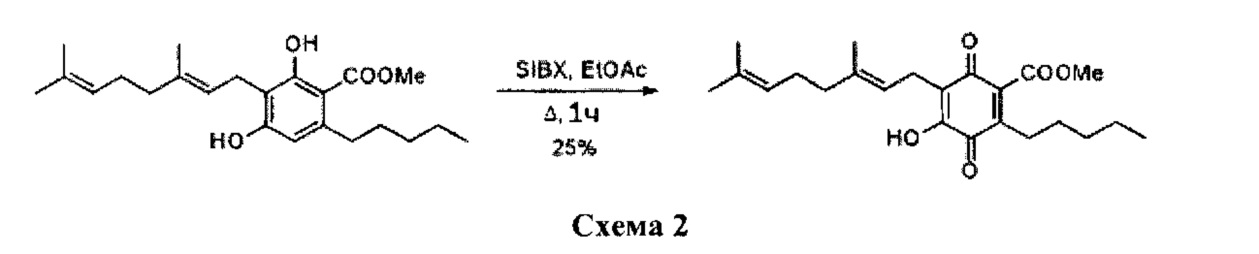
К раствору 100 мг (0,27 ммоль) (E)-метил 3-(3,7-диметилокта-2,6-диенил)-2,4-дигидрокси-6-пентилбензоата в 4 мл этилацетата добавляли SIBX (465 мг, 0,77 ммоль, 3 моль-экв.) и реакционную смесь нагревали с обратным холодильником в течение 1 ч, После охлаждения и фильтрации через целит, фильтрат последовательно промывали насыщенным NaHCO3 и насыщенным раствором соли. После сушки (Na2SO4) и выпаривания остаток очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир-CH2Cl2, 8:5 в качестве элюента) с получением 28 мг 6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-метоксикарбонил-[1,4] бензохинона. [Твердое вещество коричневого цвета, выход: 25%].
1H NMR (CDCl3, 300 МГц) δ ppm 6.95 (bs, 1H), 5.11 (bt, J=6.5 Гц, 1H), 5.04 (bt, J=6.5 Гц, 1Н), 3.89 (s, 3Н), 3.13 (d, J=6.5 Гц, 2Н), 2.38 (m, 2Н), 1.72 (bs, 3Н), 1.65 (bs, 3Н), 1.57 (bs, 3Н), 0.89 (t. J=6.5 Гц, 3Н).
Получение соединения XII
3,3'-бис ((E)-3,7-диметилокта-2,6-диенил)-4,4'-дигидрокси-6,6'-дипентил-1,1'-би(циклогекса-3,6-диен)-2,2',5,5'-тетраон
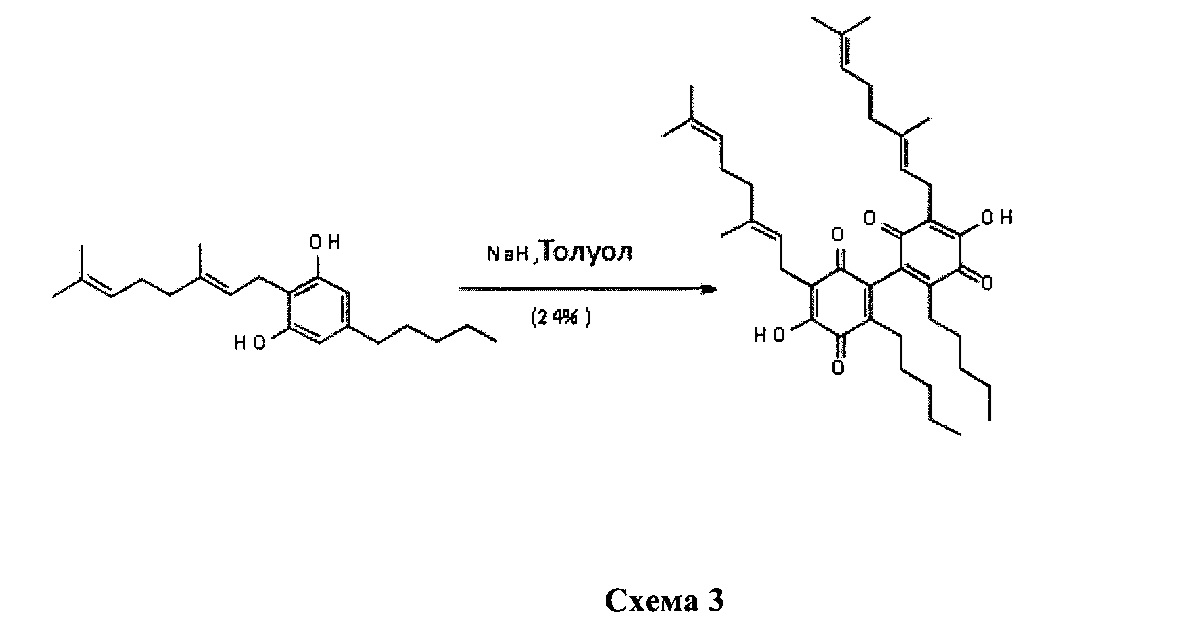
К раствору каннабигерола (CBG) (500 мг, 0,16 ммоль) в толуоле (100 мл) добавили NaH, и реакционную смесь энергично перемешивали в (95%, 0,48 ммоль, 3 мол. экв, 150 мг), оставляя колбу открытой (схема 3). Фиолетовый цвет появлялся практически мгновенно, и через 12 ч реакционную смесь обрабатывали путем подкисления с помощью 2н H2SO4 до pH 3, и разделяли солевой раствор и EtOAc. Органическую фазу сушили (Na2SO4) и выпаривали, а остаток очищали с помощью колоночной хроматографии с гравитационным элюированием на силикагеле (петролейный эфир-этилацетат 9:1 в качестве элюента) с получением 120 мг 3,3'-бис ((E)-3,7-диметилокта-2,6-диенил)-4,4'-дигидрокси-6,6'-дипентил-1,1'-би (циклогекса-3,6-диен)-2,2',5,5'-тетраон [темно-коричневая смола, выход: 24%].
1H NMR (CDCl3, 300 МГц) δ ppm: 6.99 (bs, 2Н), 5.10 (bt, J=6.5 Гц,, 2Н), 5.05 (bt, J=6.5 Гц, 2Н) 3.13 (d, J=6.5 Гц, 4Н), 1.71 (s, 6Н), 1.65 (s, 6Н), 1.57 (s, 6Н), 0.81 (t, J=7.0 Гц,
Пример 2. Химический синтез и ЯМР анализ
Общие процедуры для соединений, полученных из CBG. Синтез соединений (VIII) и (XI))
Синтез CBG-Q (соединение I), начиная с CBG (каннабигерол) проводили с использованием tBuOK в толуоле при комнатной температуре в присутствии воздуха (схема 4).
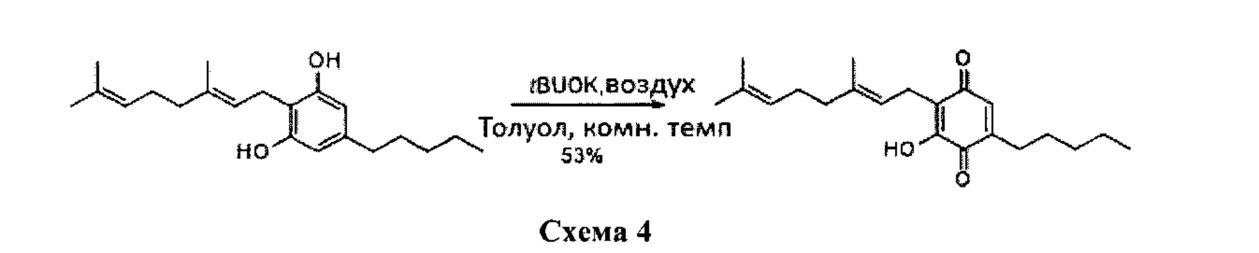
tBuOK (2,00 г, 17,824 ммоль) добавляли к раствору каннабигерола (CBG) (2,00 г, 6,319 ммоль) в толуоле (400 мл), с получением раствора фиолетового цвета. Реакционную смесь перемешивали при комнатной температуре в круглодонную колбу со свободным доступом воздуха, и реакцию контролировали с помощью анализа ТСХ (элюент: 10% EtOAc/гексан) (схема 5). Через 2 ч реакционную смесь промывали HCl (5%-ный водный раствор, 300 мл) и водный слой экстрагировали EtOAc (100 мл). Объединенные органические слои сушили над Na2SO4 (безводным), фильтровали и концентрировали. Неочищенный остаток подвергали флэш-хроматографии на SiO2 (от 2 до 4% EtOAc/гексан) с получением 1,10 г CBG-Q (соединение I) [твердое вещество оранжевого цвета, выход: 53%].
1H NMR (CDCl3, 300 МГц): δ 6.94 (s, -ОН, 1Н), 6.45 (s, 1H), 5.13 (br t, J=6.8 Гц, 1H), 5.04 (br t,J=6.8 Гц, 1H), 3.14 (s, J=6.8 Гц, 2H), 2.41 (t, J=7.8 Гц, 2H), 2.09-1.92 (m, 4H), 1.73 (br s, 3H), 1.57 (br s, 3H), ca. 1.52 (m, 2H), 1.38-1.17 (m, 4H), 0.89 (t, J=7.8 Гц, 3H).
Синтез производных, замещенных в положении 2, с алкиламино, ариламино, алкениламино или алкиниламиногруппами осуществили путем взаимодействия CBG-Q (соединение I) с большим избытком амина при комнатной температуре в реакционной системе с доступом воздуха (схема 5)
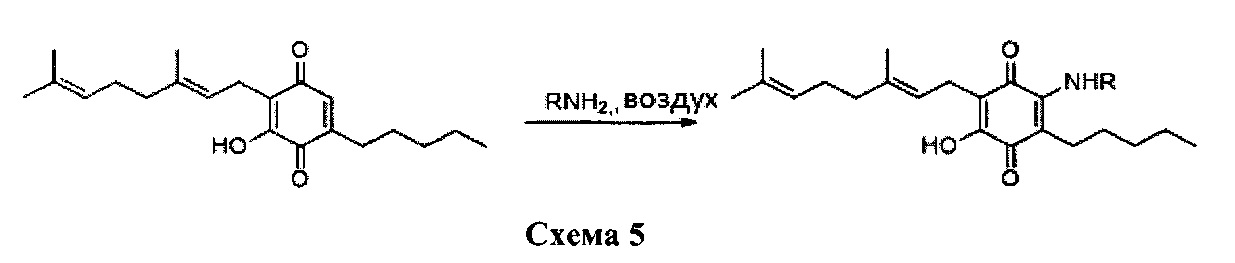
За несколько часов достигли высокой конверсии, что позволило провести реакцию до полного израсходования реагента. Растворитель концентрировали, и неочищенный остаток очищали с помощью хроматографии с обращенной фазой, с получением продуктов с чистотой около 95%.
Получение соединения III
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-этиламино-[1,4]бензохинон.
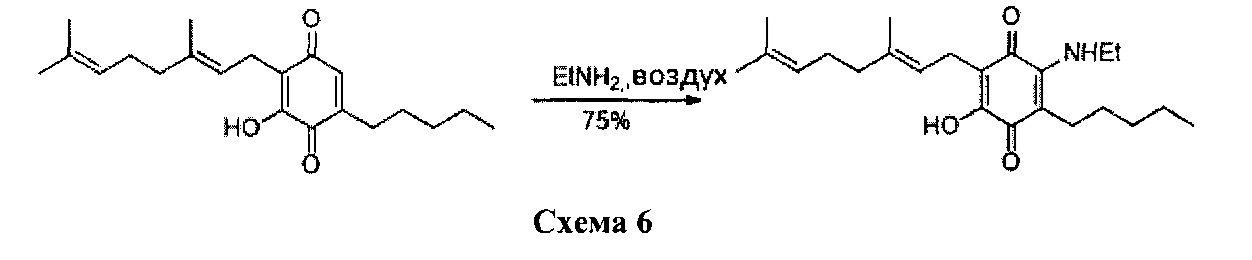
Этиламин (5,2 мл, 70% раствор в H2O, 65,403 ммоль) добавляли к раствору CBG-Q (соединение I) (510 мг, 1,543 ммоль) в этаноле (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч (схема 6). Смесь выливали в H2O (120 мл), с pH 2, доведенным с помощью HCl (10%-ный водный раствор) и экстрагировали CH2Cl2 (2×80 мл). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Сырой остаток очищали с помощью хроматографии с обращенной фазой (30 до 100% CH3CN/H2O) с получением 435 мг 2-(3,7-диметил-окта-2,6-диенил)-6-этиламино-3-гидрокси-5-пентил[1,4]бензохинона [твердое вещество фиолетового цвета, выход: 75%].
1H NMR (CDCl3 300 МГц) δ ppm: 6.39 (bs, 1H), 5.09 (m, 2Н), 3.54 (t, J=6.6 Гц, 2Н), 3.05 (d, J=6.6 Гц, 2Н), 2.49 (m, 2Н), 1.99 (m, 4Н), 1.72 (s, 3Н), 1.64 (s, 3Н), 1.57 (s, 3Н), 1.44-1.22 (m, 9Н), 0.88 (m, 3Н).
Получение соединения IV
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-пентиламино[1,4]бензохинон.
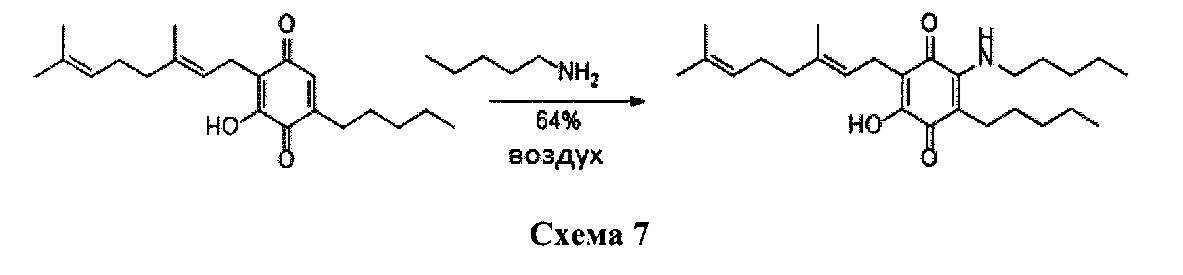
Амиламин (1,5 мл, 12,943 ммоль) добавляли к раствору соединения CBG-Q (соединение I) (109 мг, 0,330 ммоль) в этаноле (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 22 ч (схема 7). Смесь выливали в Н2О (50 мл), с pH 2, доведенным с помощью HCl (10% -ный водный раствор) и экстрагировали CH2Cl2 (30 мл). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Сырой остаток очищали с помощью хроматографии с обращенной фазой (30 до 100% CH3CN/H2O) с получением 88 мг 2-(3,7-диметил-окта-2,6-диенил)-3-гидрокси-5-пентил-6-пентиламино-[1,4]бензохинона [твердое вещество фиолетового цвета, выход: 64%].
1H NMR (CDCl3, 300 МГц) δ ppm: 6.38 (bs, 1H), 5.13 (t, J=7.1 Гц, 1H), 5.05 (t, J=6.0 Гц, 1H), 3.47 (q, J=6.6 Гц, 2Н), 3.06 (d, J=7.1 Гц, 2Н), 2.49 (m, 2Н), 2.08-1.93 (m, 4Н), 1.72 (s, 3Н), 1.65 (s, 3Н), 1.57 (s, 3Н), 1.42-1.28 (m, 12Н), 0.91 (m, 6Н).
Получение соединения V
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-изобутиламино[1,4]бензохинон.
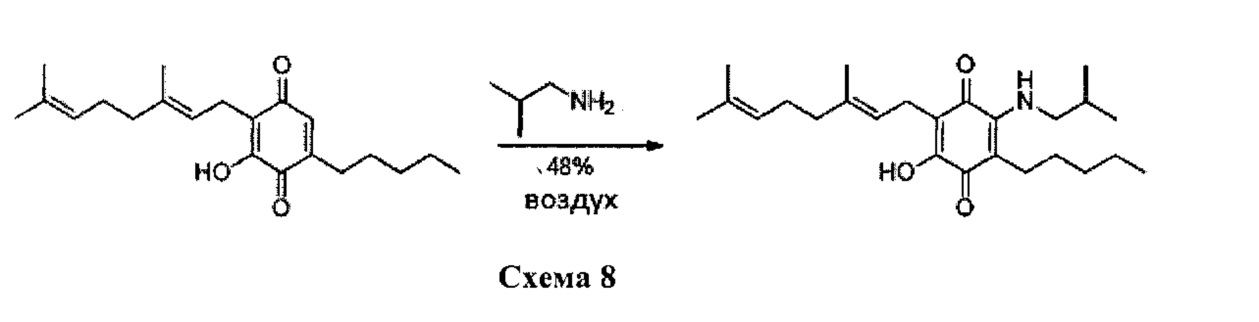
Изобутиламин (1,3 мл, 13,082 ммоль) добавляли к раствору соединения CBQ-G (соединение I) (101 мг, 0,306 ммоль) в этаноле (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 8 часов (схема 8). Смесь выливали в H2O (50 мл) с pH 2, доведенным с помощью HCl (10% -ный водный раствор) и экстрагировали CH2Cl2 (30 мл). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Сырой остаток очищали с помощью хроматографии с обращенной фазой (30 до 100% CH3CN/H2O) с получением 59 мг 2-(3,7-диметил-окта-2,6-диенил)-3-гидрокси-6-изобутиламино-5-пентил[1,4] бензохинона [твердое вещество фиолетового цвета, выход: 48%].
1H NMR (CDCl3, 250 МГц) δ ppm: 6.60 (bs, 1H), 5.11 (m, 2Н), 3.28 (t, J=6.3 Гц, 2Н), 3.06 (d, J=7.1 Гц, 2Н), 2.49 (m, 2Н), 2.07-1.84 (m, 4Н), 1.72 (s, 3Н), 1.65 (s, 3Н), 1.57 (s, 3Н), 1.41-1.27 (m, 7Н), 1.02 (s, 3Н), 0.98 (s, 3Н), 0.89 (m, 3Н).
Получение соединения VI
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-бутиламино[1,4]бензохинон.
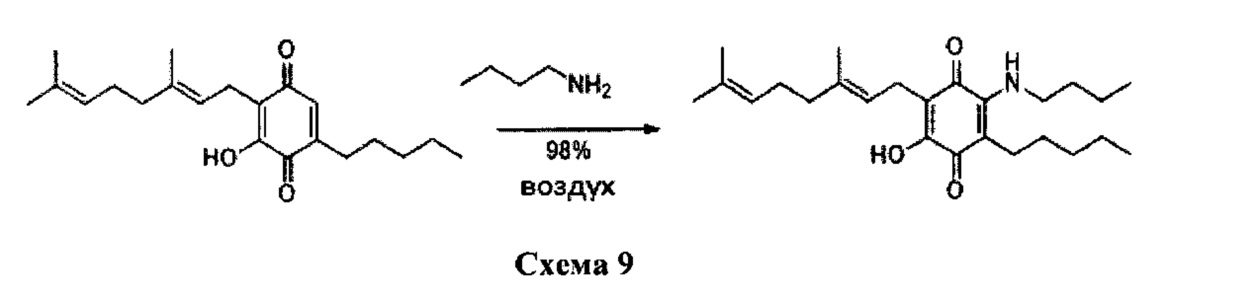
Н-бутиламин (1,2 мл, 12,143 ммоль) добавляли к раствору соединения CBG-Q (соединение I) (102 мг, 0,309 ммоль) в этаноле (12 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов (схема 9). Смесь выливали в H2O (50 мл), с pH 2, доведенным с помощью HCl (10% -ный водный раствор) и экстрагировали CH2Cl2 (30 мл). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали с получением 190 мг 2-бутиламино-6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил[1,4] бензохинона [твердое вещество фиолетового цвета, выход: 98%].
1Н NMR (CDCl3, 250 МГц) δ ppm: 6.50 (bs, 1Н), 5.09 (m, 2Н), 3.47 (q, J=7.1 Гц, 2Н), 3.05 (d, J=7.1 Гц, 2Н), 2.48 (m, 2Н), 2.08-1.90 (m, 4Н), 1.72 (s, 3Н), 1.64 (s, 3Н), 1.57 (s, 3Н), 1.50-1.22 (m, 10Н), 1.00-0.84 (m, 6Н).
Получение соединения VII
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-метиламино-[1,4]бензохинон.
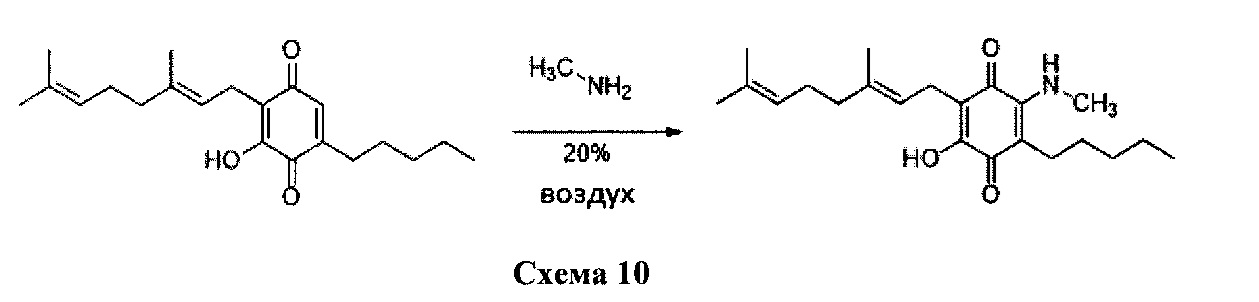
Метиламин (0,6 мл, 8М раствор в этаноле, 4,8 ммоль) добавляли к раствору соединения CBG-Q (соединение I) (102 мг, 0,309 ммоль) в этаноле (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 ч (схема 10). Смесь выливали в H2O (50 мл), с pH 2, доведенным с помощью HCl (10%-ный водный раствор) и экстрагировали CH2Cl2 (30 мл). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Сырой остаток очищали с помощью хроматографии с обращенной фазой (30 до 100%) CH3CN/H2O) с получением 23 мг 2-(3,7-диметил-окта-2,6-диенил)-3-гидрокси-6-метиламино-5-пентил[1,4]бензохинона [твердое вещество фиолетового цвета, выход: 20%].
1Н NMR (CDCl3, 300 МГц) δ ppm: 6.48 (bs, 1H), 5.12 (t, J=6.6 Гц, 1H), 5.06 (t, J=6.6 Гц, 1H), 3.20 (d, J=6.0 Гц, 3H), 3.06 (d, J=7.1 Гц, 2Н), 2.55 (t, J=7.1 Гц, 2Н), 2.07-1.92 (m, 4Н), 1.72 (s, 3H), 1.65 (s, 3H), 1.57 (s, 3H), 1.49-1.23 (m, 6Н), 0.89 (m, 3H).
Получение VIII
6-(3,7-диметилокта-2,6-диенил)-5-гидрокси-3-пентил-2-изопропиламино-[1,4]бензохинон.
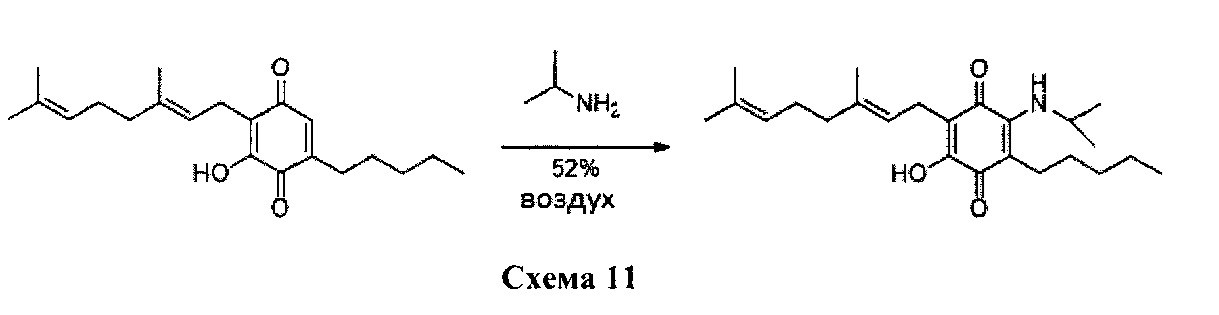
Изопропиламин (1,0 мл, 11,639 ммоль) добавляли к раствору соединения CBG-Q (соединение I) (101 мг, 0,306 ммоль) в этаноле (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов (схема 11). Смесь выливали в H2O (50 мл), с pH 2, доведенным с помощью HCl (10% -ный водный раствор) и экстрагировали CH2Cl2 (30 мл). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Сырой остаток очищали с помощью хроматографии с обращенной фазой (30 до 100% CH3CN/H2O) с получением 62 мг 2-(3,7-диметил-окта-2,6-диенил)-3-гидрокси-6-изопропиламино-5-пентил[1,4]бензохинона [твердое вещество фиолетового цвета, выход: 52%].
1Н NMR (CDCl3, 300 МГц) δ ppm: 6.37 (s, 1H), 5.13 (t, J=6.6 Гц, 1Н), 5.05 (t, J=6.6 Гц, 1Н), 3.98 (m, 1Н), 3.06 (d, J=7.1 Гц, 2Н), 2.47 (m, 2Н), 2.08-1.92 (m, 4Н), 1.72 (s, 3H), 1.65 (s, 3H), 1.57 (s, 3H), 1.42-1.29 (m, 6Н), 1.28 (s, 3H), 1.25 (s, 3H), 0.89 (m, 3H).
Получение соединения IX
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-бензиламино[1,4]бензохинон.
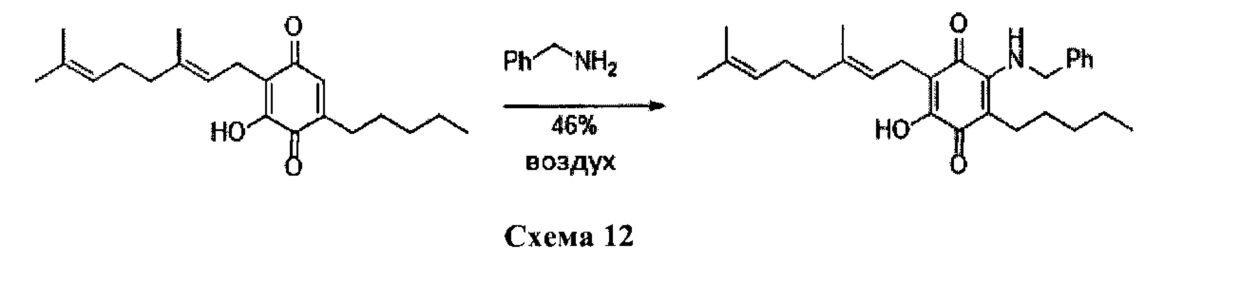
Бензиламин (1,3 мл, 11,913 ммоль) добавляли к раствору соединения CBG-Q (соединение I) (100 мг, 0,302 ммоль) в этаноле (13 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов (схема 12). Смесь выливали в Н2О (50 мл), с pH 2, доведенным с помощью HCl (10%-ный водный раствор) и экстрагировали CH2Cl2 (30 мл). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Сырой остаток очищали с помощью хроматографии с обращенной фазой (30 до 100% CH3CN/H2O) с получением указанного 61 мг 2-бензиламино-6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил[1,4] бензохинона [твердое вещество фиолетового цвета, выход: 46%].
1H NMR (CDCl3, 300 МГц) δ ppm: 7.43-7.27 (m, 5Н), 6.80 (bs, 1H), 5.18-5.02 (m, 2Н), 4.67 (d, J=5.5 Гц, 2Н), 3.07 (d, J=6.6 Гц, 2Н), 2.47 (t, J=7.7 Гц, 2Н), 2.09-1.92 (m, 4Н), 1.72 (s, 3H), 1.65 (m, 3H), 1.57 (s, 3H), 1.47-1.24 (m, 6Н), 0.88 (m, 3H).
Получение соединения X
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-(2,2-диметил-пропил амино)-[1,4]бензохинон.
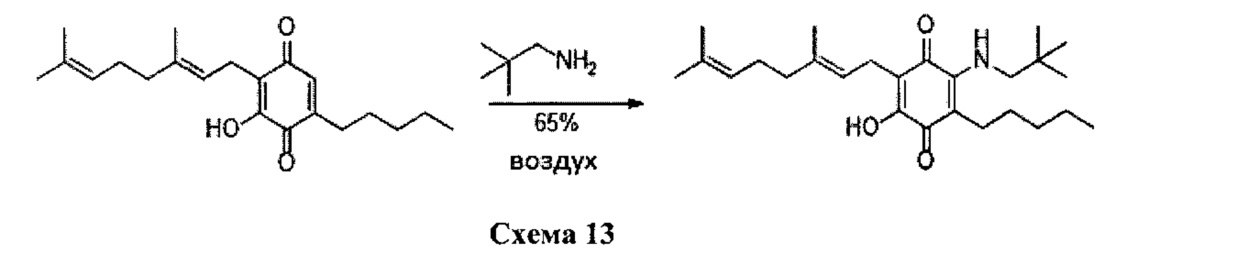
Неопентиламин (1,4 мл, 12,063 ммоль) добавляли к раствору соединения CBG-Q (соединение I) (100 мг, 0,303 ммоль) в этаноле (14 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов (схема 13). Смесь выливали в Н2О (50 мл), с pH 2, доведенным с помощью HCl (10%-ный водный раствор) и экстрагировали CH2Cl2 (30 мл). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Сырой остаток очищали с помощью хроматографии с обращенной фазой (30 до 100% CH3CN/H2O) с получением указанного 72 мг 2-(3,7-диметил-окта-2,6-диенил)-6-(2,2-диметил-пропиламино)-3-гидрокси-5-пентил[1,4]бензохинона [твердое вещество фиолетового цвета, выход: 65%].
1H NMR (CDCl3, 300 МГц) δ ppm: 6.62 (s, 1H), 5.14 (t, J=6.6 Гц, 1H), 5.05 (t, J=6.6 Гц, 1Н), 3.27 (d, J=6.0 Гц, 2Н), 3.07 (d, J=7.1 Гц, 2Н), 2.50 (t, J=7.1 Гц, 2Н), 2.09-1.92 (m, 4Н), 1.72 (s, 3H), 1.65 (s, 3H), 1.57 (s, 3H), 1.47-1.25 (m, 6Н), 1.02 (s, 9Н), 0.90 (t, J=6.6 Гц, 3Н).
Получение соединения XI
6-(3,7-диметил-окта-2,6-диенил)-5-гидрокси-3-пентил-2-(3-метил-бутиламино)-[1,4]бензохинон.
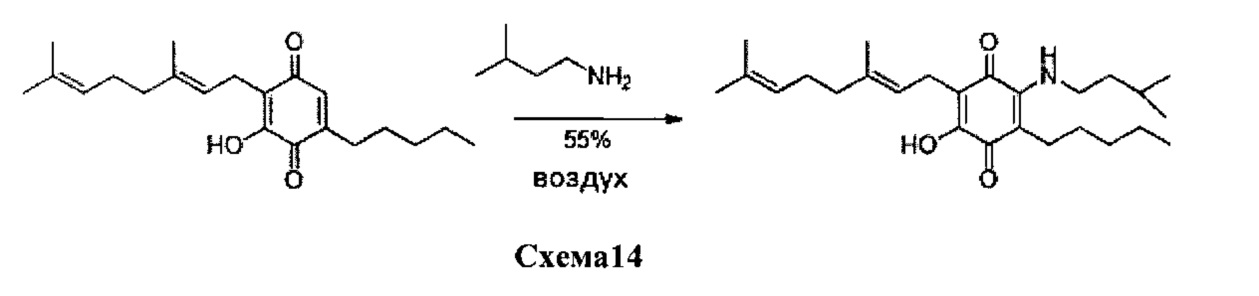
Изопентиламин (1,4 мл, 11,886 ммоль) добавляли к раствору соединения CBG-Q (соединение I) (100 мг, 0,303 ммоль) в этаноле (14 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов (схема 14). Смесь выливали в Н2О (50 мл), с pH 2, доведенным с помощью HCl (10%-ный водный раствор) и экстрагировали CH2Cl2 (30 мл). Органический слой сушили над Na2SO4 (безводным), фильтровали и концентрировали. Сырой остаток очищали с помощью хроматографии с обращенной фазой (30 до 100% CH3CN/H2O) с получением 40 мг 2-(3,7-диметил-окта-2,6-диенил)-3-гидрокси-6-(3-метилбутиламино)-5-пентил-[1,4]бензохинона [твердое вещество фиолетового цвета, выход: 55%].
1H NMR (CDCl3, 300 МГц) δ ppm: 6.38 (bs, 1H), 5.09 (m, 2Н), 3.50 (q, J=6.0 Гц, 2Н), 3.06 (d, J=7.1 Гц, 2Н), 2.51 (t, J=7.1 Гц, 2Н), 2.11-1.92 (m, 4Н), 1.72 (s, 3H), 1.65 (s, 3H), 1.57 (s, 3H), 1.48-1.24 (m, 7Н), 0.96 (s, 3H), 0.94 (s, 3H), 0.89 (m, 3H).
Анализы in vitro
Пример 2. Активность агонистов PPARg.
Для исследования биологической активности новых соединений авторы настоящего изобретения провели анализ трансактивации PPARg в клетках НЕК293 и первичных фибробластах человека.
Клетки НЕК293Т и первичные фибробласты человека инкубировали при 37°C в увлажненной атмосфере, содержащей 5% CO2 в среде DMEM с добавлением 10% эмбриональной бычьей сыворотки (FBS) и 1% (об/об) пенициллина/стрептомицина. Розиглитазон купили в Cayman Chemical Company (Ann Arbor, MI, USA). Все другие реагенты купили в Sigma Co (St Louis, MO, USA). Рассеяли HEK293T клетки (2×103/лунку) (Фигура 1) или первичные дермальные фибробласты человека (5×103/лунку) (Фигура 2) в 96-луночные плашки с прозрачным дном BD Falcon™ White Microtest™ Optilux™ и растили в течение 24 часов. Затем клетки котрансфецировали векторами, экспрессирующими GAL4-PPARγ и репортерным вектором с люциферазой GAL4-Luc с использованием Roti©-Fect (Carl Roth, Karlsruhe, Germany) в соответствии с инструкциями производителя. Через двадцать четыре часа после трансфекции клетки обрабатывали увеличивающейся дозой соединений в течение 6 часов. Затем клетки лизировали в 25 мМ трис-фосфат pH 7,8, 8 мМ MgCl2, 1 мМ DTT, 1% Triton Х-100 и 7% глицерина. Активность люциферазы измеряли в лизате клеток с использованием многорежимного микропланшет-ридера TriStar LB 941 (Berthold) и следуя инструкциям производителя Luciferase Assay Kit (Promega, Madison, WI, USA). Концентрацию белка определяли по методу Бредфорд (Bio-Rad, Richmond, СА, USA). Фоновое значение, полученное с помощью буфера для лизиса, вычитали из каждой экспериментальной величины, и рассчитывали удельную трансактивацию, выраженную в кратном возрастании индукции по сравнению необработанными клетками. Все эксперименты повторили по крайней мере три раза. Использовали плазмиду Gal4-hPPARgamma (название плазмиды: PCMV-BD-hPPARg, изготовленную в лаборатории SINAL, кафедра фармакологии, Dalhousie University) и репортерную плазмиду Gal4 luc, которая включает пять сайтов связывания ДНК Gal4, слитых с геном люциферазы. Приведенный выше анализ иллюстрируется на Фиг. 1 и Фиг. 2, которые показывают эффект CBG-Q (соединение I) и его производных на активность PPARg с помощью анализа трансактивации, выполненного на клетках, временно гиперэкспрессирющих PPARg в сочетании с геном люциферазы (PPARg-GAL4/GAL4-Luc) и обработанных соединениями в течение 6 часов. Данные приведены в виде среднего со стандартным отклонением для трех повторностей. Обнаружили значительное увеличение активности люциферазы для хиноновых производных по сравнению с необработанными клетками. Этот результат подтверждает, что соединение II является значительно более эффективным, чем соединение CBG-Q (соединение I), для активации PPARg в концентрациях от 1 до 25 мкМ. Соединения III-XII увеличивают трансактивацию PPARg зависимым от концентрации образом, соединения II, IV, V и XII наиболее активны. Кроме того, более высокие концентрации (25 и 50 мкМ) этих соединений являются особенно эффективными для активации PPARg по сравнению с CBG-Q (соединение I). Розиглитазон, полный агонист PPARg, в концентрации 1 мкМ увеличил активность PPARg более чем в 100 раз. В отличие от этого максимальная индукция активности PPARg, индуцированная концентрацией 1 мкМ соединений, описанных в настоящем изобретении, никогда не была выше, чем 12 раз (у соединения II), что означает, что эти новые соединения являются модуляторами PPARg, а не полными агонистами PPARg.
Пример 3. Анализ цитотоксичности.
Электрофильные хиноны индуцируют цитотоксичность и активируют сигнальный путь Nrf2, клеточный сенсор на образование активных форм кислорода. На Фигуре 3 анализируется индуцированная гибель клеток в трех различных типах клеток (N2a, НТ22 и MO3.13) соединением CBG-Q (соединение I) и соединениями (II)-(ХII).
Три клеточные линии, MO3.13, N2a и НТ22, инкубировали при 37°C в увлажненной атмосфере, содержащей 5% CO2 в среде DMEM, дополненной 10% фетальной бычьей сывороткой (FBS) и 1% (об/об) пенициллина/стрептомицина. Жизнеспособность клеток N2A, НТ22 и MO3.13 определяли с помощью МТТ-анализа. Коротко, клетки высевали до плотности 104 клеток/лунку в 96-луночные планшеты, 200 мкл суспензии клеток на лунку, и культивировали в течение 24 часов. Затем клетки инкубировали с несколькими концентрациями соединений в течение 24 часов. После этого 100 мкл МТТ (5 мг/мл) из смеси раствора MTT:DMEM (1:2) добавляли в каждую лунку, и клетки инкубировали в течение 4 ч при 37°C в темноте.
Затем реакцию останавливали, супернатант удаляли и добавляли 100 мкл ДМСО в каждую лунку, и инкубировали в течение 10 минут при осторожном встряхивании. Наконец, измеряли оптическую плотность при 550 нм с использованием TriStar LB 941 (Berthold Technologies, GmbH & Co. KG). Значение контрольных клеток принимали в качестве 100%, и данные относили к этому значению. Клеточные линии N2a (Фиг. 3а), НТ22 (Фигура 3б) и MO3.13 (3в) инкубировали в течение 24 ч с указанными дозами соединения CDG-Q (соединение I) и соединений (II)-(ХII), и жизнеспособность клеток количественно определяли с помощью МТТ-теста. Результаты представлены в виде среднего значения ± SD по меньшей мере трех независимых экспериментов и выражены в процентах от жизнеспособности клеток по отношению к контрольному образцу (-). Контроль приняли за 100%, и данные отнесли к этому значению. Результаты показывают, что цитотоксическая активность, связанная с CBG-Q (соединение I), коррелирует с его способностью индуцировать активацию Nrf2. В том же смысле, отсутствие цитотоксической активности для соединений II-ХII, производных по положению 2 CBG-Q, описанных в настоящем изобретении, коррелирует с их неспособностью активировать Nrf2.
Пример 4. Транскрипционная активность Nrf2.
Для изучения активности соединений на сигнальный путь Nrf2 авторы настоящего изобретения получили клеточную линию HaCaT-ARE-Luc. Плазмиды NQO1 ARE-Luc и репортерную плазмиду pPGK-Puro котрансфицировали в клетки HaCat с использованием трансфекционного реагента Lipofectamine© 2000 (Life Technologies, Carlsbad, Ca, USA). Устойчивых трансформантов отобрали и инкубировали в среде RPMI 1640, содержащей 10% FBS, 1% пенициллина-стрептомицина и 10 мкл/мл пуромицина. HaCaT-ARE-Luc клетки инкубировали в течение 6 ч с CBG-Q (соединение I) и с соединениями (II)-(VI), (а), или с соединениями (VII)-(XII) (б) в указанных концентрациях, и получали белковые лизаты, которые анализировали на активность люциферазы, как описано в примере 1. Прооксидантный трет-бутилгидрохинон (tBHQ) в концентрации 20 мкМ использовали в качестве положительного контроля. Рассчитывали кратность возрастания уровня активации, принимая контрольный образец (-) в качестве базового уровня (Фиг. 4а и 4б). Данные выражены в виде среднего значения ± SD по меньшей мере трех независимых экспериментов. Результаты подтверждают, что обратная электрофильная активность, связанная с CBG-Q (соединение I), отсутствует во всех соединениях (производных по положению 2), описанных в настоящем изобретении.
Пример 5. Анализ нейропротекторной активности.
Активация противовоспалительных ядерных рецепторов PPARg играет важную роль в нейропротекции, и известно, что агонисты PPARg предотвращают вызванную глутаматом цитотоксичность в нейрональных клетках.
Культивируемые клетки N2a предварительно инкубировали с соединениями II, III, IV, V и XII в указанных концентрациях в течение 1 ч, а затем обрабатывали 5 мМ глутамата, чтобы вызвать эксайтотоксичность, в течение 24 ч (Фигура 5). Цитотоксичность определяли с помощью метода МТТ, как описано в примере 3. Результаты представлены как средние значения ± SD по меньшей мере трех независимых экспериментов и выражены в процентах от жизнеспособности клеток по отношению к контрольному образцу (-). Контроль был установлен как 100%, и данные отнесли к этому значению.
Эти результаты показывают, что соединения II, III, IV, V и VII, которые являются модуляторами PPARg, также защищают нейрональные клетки от глутамат-индуцированного апоптоза.
Анализ in vivo
Пример 6. Индуцирование экспериментального аутоиммунного энцефаломиелита (ЕАЕ)
Модуляторы PPARg применяют для лечения нейродегенеративных и воспалительных заболеваний, и авторы настоящего изобретения исследовали эффекты двух репрезентативных соединений согласно настоящему изобретению в трех хорошо изученных животных моделях воспаления и нейродегенерации.
ЕАЕ индуцировали у самок мышей C57BL/6 в возрасте 6-8 недель путем подкожной иммунизации миелиновым олигодендроцитарным гликопротеином (MOG35-55) (300 мкг) и 200 мкг Mycobacterium tuberculosis (H37Ra, Difco, Franklin Lakes, NJ, USA) в соотношении 1:1 в смеси с неполным адъювантом Фрейнда (CFA, Sigma-Aldrich, Madrid, Spain). В тот же день и через 2 дня мышам вводили внутрибрюшинно (ip) 200 нг коклюшного токсина (Sigma-Aldrich, Madrid, Spain) в 0,1 мл PBS.
Контрольных животных (CFA) иммунизировали той же эмульсией без MOG, и они не получали коклюшный токсин. Лечение начинали на 6-й день после иммунизации (i.p.), и оно заключалось в ежедневных инъекциях соединений III (Фигура 6) и XII (Фигура 8) в указанных дозах или только носителем (ДМСО/PBS) в течение следующих 21 дней. У мышей ежедневно оценивали клинические показатели ЕАЕ и тяжесть болезни оценивали следующим образом: 0, отсутствие болезни; 1, хромота на задние конечности; 2, хромота на задние конечности и слабость задних конечностей; 3, паралич задних конечностей; 4, паралич задних конечностей и паралич передних конечностей; 5, агония и смерть. Всех животных умертвили через 28 дней (p.i.) для дальнейшего анализа. После того как животных умертвили, животных препарировали, а их спинной мозг быстро извлекали и быстро замораживали в RNAlater (Sigma-Aldric, Germany).
Как показано на Фигуре 6, соединение III явно ослабило клинические проявления экспериментального аутоиммунного энцефаломиелита (ЕАЕ), индуцированного подкожной иммунизацией (MOG35-55). У мышей, обработанных носителем, развилась тяжелая болезнь, которая достигла своего пика на 16 день после инъекции (p.i.) и достигла балла 2,5 (максимальный балл равен 3). У мышей, которые получали соединение III, болезнь достигла своего пика на 17 день после инъекции, и не достигла балла 1,3 на протяжении всего эксперимента (день 6-день 28). Клинические симптомы в ЕАЕ коррелирует с экспрессией провоспалительных генов
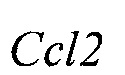
Пример 7. Индукция болезни Хантингтона (модели 3NP).
Интоксикация мышей 3-нитропропионовой кислотой (3-NP), мощным необратимым ингибитором митохондриального ферментного комплекса II, приводит к митохондриальной дисфункции и окислительному стрессу на животных моделях, что приводит к множественным неврологическим, биохимическим и гистологическим эффектам, что напоминает некоторые аспектов болезни Хантингтона. Например, обработанные 3NP мыши показывают высокие баллы в сжатии задних конечностей, дистонии, кифозе и в общей локомоторной активности по сравнению с контрольными животными.
Очаги поражения полосатого тела индуцировали с помощью 3-NP у взрослых (16-недельных; 30 г) самцов мышей C57BL/6 (Harlan
На Фигуре 9 показано, что CBG-Q (соединение I) не смогло предотвратить клинические симптомы, вызванные интоксикацией 3-NP, но соединения III и VII явно облегчают такую симптоматику.
Мы также использовали паренхиму полосатого тела 3NP-мышей для повреждения анализа некоторых гистологических и молекулярных маркеров, связанных с воспалением и нейродегенеративными заболеваниями, которые затрагиваются в этой экспериментальной модели. Экспрессия воспалительных ферментов СОХ-2 и iNOs был значительно активирована у 3NP-мышей с повреждениями параллельно с повышением экспрессии провоспалительных цитокинов TNFα и IL-6. Соединения III (Фигура 10) и XII (Фигура 11) уменьшали активацию экспрессии провоспалительных маркеров СОХ-2, iNOS, TNFα и IL-6 в полосатом теле мышей, получавших 3NP.
На Фигуре 12 показано, что паренхима полосатого тела этих пораженных 3NP животных показала высокую степень гибели нервных клеток, что было подтверждено иммуногистохимией NeuN, где обнаружили сокращение иммуномечения более чем на 50% по этому нейронному маркеру в паренхиме полосатого тела. Потеря нейронов сопровождалась заметным снижением GFAP+ клеток (астроглиоз) и повышенной экспрессии Iba-1+ клеток (реактивный микроглиоз). Соединение XII продемонстрировало защиту нейронов полосатого тела от токсичности 3NP, как показало окрашивание NeuN. Кроме того, лечение соединением XII противодействует потере GFAP+ клеток, индуцированной 3NP, и предотвращает индукцию реактивного микроглиоза (Iba-1+ клеток).
Пример 8. Индукция болезни Паркинсона (модель 6-OHDA).
Соединение III также применяли для лечения в модели болезни Паркинсона на мышах (PD).
C57BL/6 мышей, предварительно обработанных интрацеребровентрикулярно (i.c.v.), анестезировали с помощью внутрибрюшинной (ip) инъекции 200 мг/кг 2,2,2-трибромэтанола (Sigma-Aldrich) и помещали в стереотаксическую раму с адаптером для мыши (David Kopf Instruments, Tujunga, С A, USA). С помощью шприца Hamilton (Hamilton, Bonaduz, Switzerland), 4 мкл раствора 6-OHDA-HBr (5 мкг/мкл) в 0,02% аскорбиновой кислоты (Sigma-Aldrich) вводили в левое полосатое тело в два участка в следующих стереотаксических координатах (мм от темени): АР,+0,65; L, -2,0; V1, -4 и V2, -3,5, ориентированных на дорсолатеральную часть полосатого тела. После инъекции кожу зашивали, и животных удаляли из стереотаксического инструмента и помещали на грелку в течение 30 мин. Мышей подвергали длительной внутрибрюшинной обработке соединением III (10 мг/мл) или носителем (14 дней), начиная с 16 ч после инъекции 6-OHDA. Координацию движений оценивали в тесте способности удерживаться на вращающемся барабане (Ugo Basile, Рим, Италия) с увеличивающейся скоростью. Каждый день у мышей была тренировка по 1 мин на неподвижном барабане. Если мышь падала с барабана во время тренировки, ее помещали обратно. Затем мышей тестировали сеансами в течение 5 мин каждые 20 мин. Таким образом, скорость барабана составляла до 40 оборотов в минуту в течение пяти минут. Задержку падения измеряли каждый следующий день у поврежденных мышей после введения соединения III или носителя. Двигательную активность (общую активность, среднюю скорость, время отдыха, быстрые движения и количество подъемов на задние лапы) оценивали с помощью системы автоматизированного актиметра (Фигура 13).
На Фигуре 13 видно, что появление двигательных симптомов, напоминающих PD человека (изменения в общей активности, средняя скорость, время отдыха, быстрое движение, количество подъемов на задние лапы и успешность в тесте на барабане), полученных с использованием 6-гидроксидопамина (60HDA) было почти полностью подавлено лечением соединением III.
Пример 9. Гистологический анализ (пример 7).
Головной мозг мышей модели 3NP фиксировали в 4% параформальдегиде и делали срезы 5-мкм толщиной для иммуногистохимического анализа NeuN (12а), маркера нейронов, GFAP (Фиг. 12б), маркера астроцитов и Iba-1 (рис 12в), маркера клеток микроглии. Срезы для иммуногистохимического окрашивания инкубировали в течение ночи при 4°С с: (I) моноклональным антителом против NeuN мыши (Millipore, MA, USA), используемым в разведении 1/100; (II) моноклональным антителом против Iba-1 мыши (Millipore, MA, USA), которое использовали в разведении 1/50, (III), моноклональным антителом против GFAP мыши (Santa Cruz Biotechnology, СА, USA), которое использовали в разведении 1/50. После инкубации с соответствующим первичным антителом срезы промывали в 0,1 М PBS и инкубировали в течение ночи при 4° с козьим вторичным антителом против антител мыши (Millipore, MA, USA). Реакцию проявляли с помощью диаминобензидина. Отрицательные контрольные срезы получили с использованием того же протокола без первичного антитела. Все срезы для каждой иммуногистохимической процедуры были обработаны в то же время и при тех же самых условиях. Микроскоп Leica DM2500 и фотоаппарат Leica DFC 420С использовали для просмотра срезов и фотографии, и все обработку изображений выполнили с использованием программного обеспечения ImageJ, разработанного и свободно распространяемого Национальным институтом здравоохранения США (Bethesda. MD, USA).
Пример 10. Количественный ПЦР-анализ в реальном времени, используемый в изобретении (примеры 6 и 7).
Тотальную РНК выделяли из полосатых тел (модель 3NP) или спинного мозга (модель ЕАЕ) с использованием набора RNeasy Lipid Tissue Mini Kit (Qiagen, GmbH). Общее количество извлеченной РНК определяли количественно с помощью спектрофотометрии при длине волны 260 нм, а ее чистоту по соотношению между значениями оптической плотности при 260 и 280 нм. Геномную ДНК удаляли, чтобы исключить загрязнение ДНК. Однонитевую комплементарную ДНК синтезировали на матрице до 1 мкг тотальной РНК (пул из по меньшей мере 3 животных на группу) с использованием набора iScript™ cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Реакционную смесь выдерживали в замороженном виде при -20°С до ферментативной амплификации. Для количественного определения уровня мРНК использовали IQTM SYBR Green Supermix (Bio-Rad) для СОХ-2, TNF-α, IL-6, IL-17, IL-1β, IFN-γ, CCL-2 or iNOS в зависимости от болезни, которую моделировали. ПЦР в реальном времени проводили с использованием системы обнаружения CFX96 Real-Time PCR Detection System (Bio-Rad). Ген домашнего хозяйства GAPDH использовали, чтобы стандартизировать уровни экспрессии мРНК в каждом образце. Уровни экспрессии рассчитывали с использованием метода 2-ΔΔCt. Последовательности олигонуклеотидных праймеров приведены в Таблице 2.


Представленные результаты обосновывают терапевтическое применение соединений, описанных в настоящем изобретений, в частности, соединений II, III, IV, V и XII при развитии нейродегенеративных заболеваний, а также травматических расстройств головного мозга, где нейровоспаления и нейротоксичность играют существенную роль. Кроме того, соединения согласно настоящему изобретению являются особенно подходящими в качестве агонистов PPARg, особенно для лечения воспалительных заболеваний (см. Таблицу 1 уровня техники), метаболические заболевания и диабет II типа.
Список литературы:
- Ahmadian М, Sub. JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARγ signaling and metabolism: the good, the bad and the future. 2013. Nat Med. 19: 557-66
- Barish GD, Narkar VA, Evans RM. 2006. PPARδ: a dagger in the heart of the metabolic syndrome. J Clin Invest. 116: 590-597
- Bernardo, A., Minghetti, L., 2008. Regulation of Glial Cell Functions by PPAR- gamma natural and Synthetic Agonists. PPAR Res. 2008, 864140.
- Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of quinones in toxicology. 2000. Chem Res Toxicol. 3: 135-60.
- Burstein S. 2005. PPAR-gamma: a nuclear receptor with affinity for cannabinoids. Life Sci. 77:1674-84.
- Ciudin A, Hernandez C,
- Doshi LS, Brahma MK, Bahirat UA, Dixit AV, Nemmani KV. 2012. Discovery and development of selective PPAR gamma modulators as safe and effective antidiabetic agents. Expert Opin Investig Drugs. 19: 489-512.
- Fievet C, Fruchart JC, Staels B, 2006. PPAR alpha and PPAR gamma dual agonists for the treatment of type2 diabetes and the metabolicsyndrome. Curr. Opin. Pharmacol. 6: 606-614.
- Gelman, L., Feige, J.N., Desvergne, В., 2007. Molecular basis of selective PPARgamma modulation for the treatment of type 2 diabetes. Biochim. Biophys.Acta 1771: 1094-1107.
- Granja AG, Carrillo-Salinas F, Pagani A,
- Kostadinova R, Wahli W, Michalik L. 2005. PPARs in diseases: control mechanisms of inflammation. Curr Med Chem. 12: 2995-3009
- Lehmann JM, Moore LB, Smith-Oliver ТА, Wilkison WO, Willson TM, Kliewer SA. 1995. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem. 270:12953-6.
- Liu J, Li H, Burstein SH, Zurier RB, Chen JD, 2003. Activation and binding of peroxisome proliferator-activated receptor gamma by synthetic cannabinoid ajulemic acid. Mol. Pharmacol. 63: 983-992.
- Monks TJ, Jones DC. 2002. The metabolism and toxicity of quinones, quinonimines, quinone methides, and quinone-thioethers. Curr Drug Metab. 4: 425-38.
- Na HK, Surh YJ. 2013. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic Biol Med. 67: 353-365
- O'Sullivan SE. 2007. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 152: 576-82.
- Poulsen L, Siersbaek M, Mandrup S. PPARs: fatty acid sensors controlling metabolism. 2012. Semin Cell Dev Biol. 23: 631-639.
- Rosen ED, MacDougald OA. 2006. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 7: 885-96.
- Singh, J. Petter, R. C. Baillie, T. A. Whitty, A. 2011. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 10: 307-17
- Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, Corvalan AH, Biswal S, Swisher SG, Bekele BN, Minna JD, Stewart DJ, Wistuba II. 2010. Nrf2 and Keapl abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res. 16: 3743-53
- M.B. Sporn, K.T. Liby. 2012. NRF2 and cancer: the good, the bad and the importance of context. Nat. Rev. Cancer. 12: 564-57
- Stienstra R, Duval C, Muller M, Kersten S, 2007. PPARs, obesity, and inflammation. PPAR Res. 95974.
- Sun Y, Bennett A. 2007. Cannabinoids: A New Group of Agonists of PPARs. PPAR Res. 23513.
-
- Tachibana K, Yamasaki D, Ishimoto K, Doi T. 2008. The Role of PPARs in Cancer. PPAR Res. 102737.
- Tontonoz P, Spiegelman BM. 2008. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 77: 289-312.
- Vanden Berghe W, Vermeulen L, Delerive P, DeBosscher K Staels B, Haegeman G. 2003. A paradigm for gene regulation: inflammation, NF-kB and PPAR. Adv. Exp. Med. Biol. 544: 181-196.
- Vivacell Biotechnology
Реферат
Настоящее изобретение относится к новому производному каннабигерол хинона Формулы (I) или его фармацевтически приемлемой соли, применяемому для лечения связанных с PPARg заболеваний, а также к содержащей его композиции. В Формуле I R представляет собой атом углерода группы, представленной: арилом, линейным или разветвленным алкенилом, линейным или разветвленным алкинилом или линейным или разветвленным алкоксикарбонилом; или где R представляет собой атом азота линейной или разветвленной группы, представленной: линейным или разветвленным алкиламино, ариламино, линейным или разветвленным алкениламино или линейным или разветвленным алкиниламино; или, в качестве альтернативы, R представляет собой связь между 2 молекулами Формулы (I), образуя димер. 2 н. и 2 з.п. ф-лы, 13 ил., 2 табл., 10 пр.
Формула
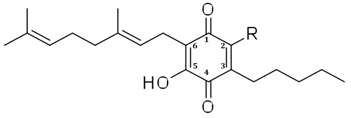
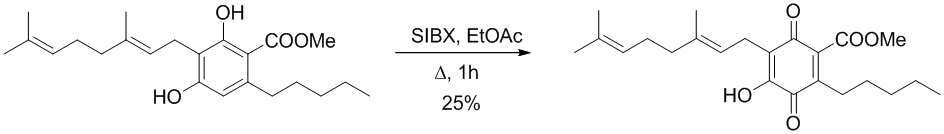
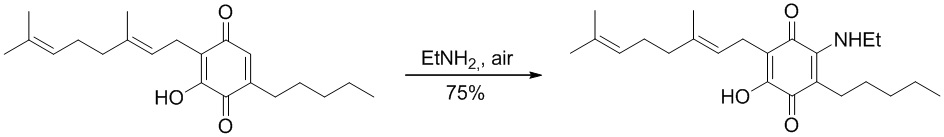
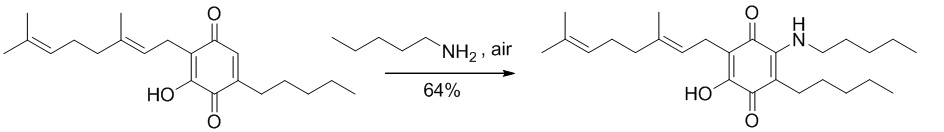
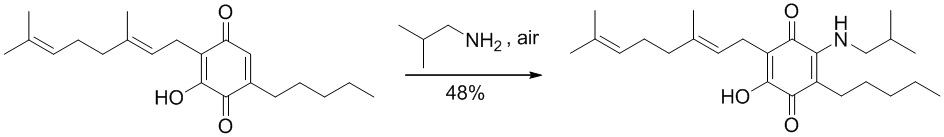
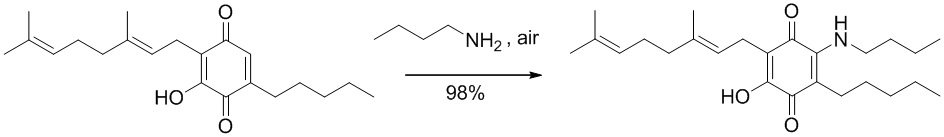
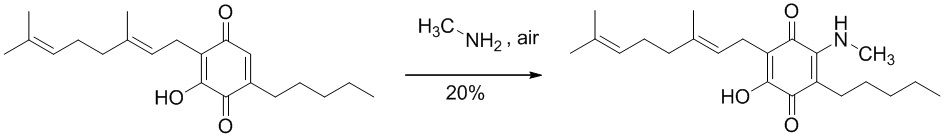
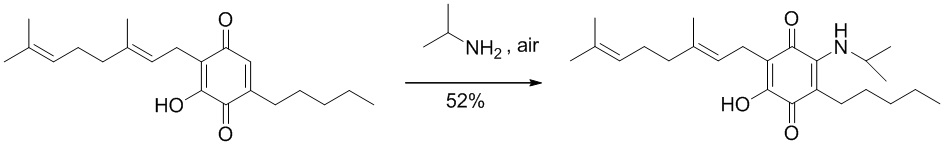
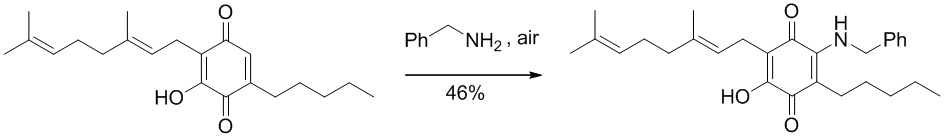
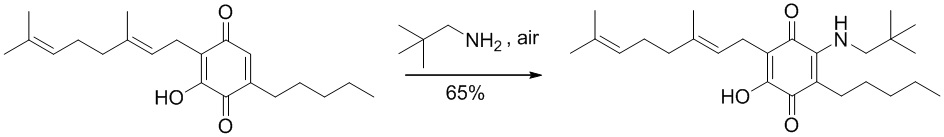
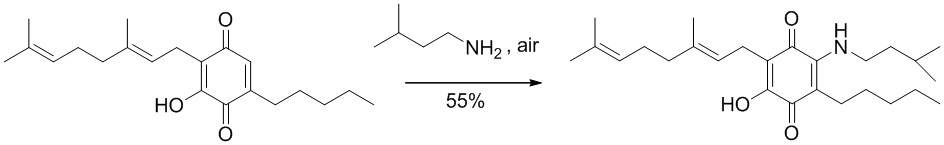
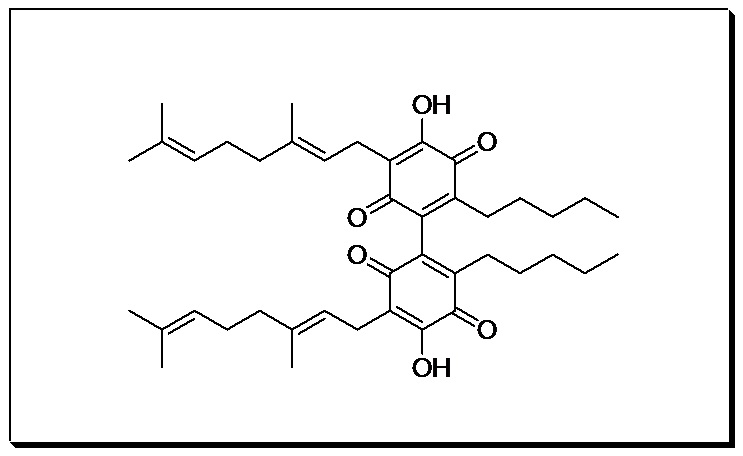
Документы, цитированные в отчёте о поиске
Хиноновые производные и их фармакологически приемлемые соли
Комментарии