Соединения, полученные из таурина, способ их получения и содержащие их фармацевтические композиции - RU2502729C2
Код документа: RU2502729C2
Чертежи








Описание
Область изобретения
Настоящее изобретение относится к новым лекарственным препаратам, полученным из таурина, преимущественно для использования в качестве нестероидных противовоспалительных (НПВП) средств, получению таких новых лекарственных препаратов и их использованию в фармацевтических композициях для лечения заболеваний, включающих воспалительные процессы, ревматоидный артрит, неспецифический язвенный колит, болезнь Крона, а также их использованию в качестве антипиретиков, анальгетиков и антиагрегантов тромбоцитов.
Предпосылки создания изобретения
Воспалительным процессам всегда уделялось большое внимание в науке как первому биологическому признаку любого нарушения состояния здоровья.
Воспаление является по своей сути защитной реакцией, вызванной физическими, химическими и биологическими раздражителями, которые могут привести к нарушениям, завершающимся некрозом тканей.
В 70-х годах, после того как Vane и др. (см. Vane, J.R. (1971), "Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs", "Nature-New Biology 231(25):232-5) продемонстрировали участие простагландинов в качестве медиаторов воспаления путем их ингибирования ацетилсалициловой кислотой, исследование было расширено с развитием большого количества групп противовоспалительных лекарственных препаратов, особенно тех, которые известны как нестероидные противовоспалительные препараты (НПВП) (см. ROBERTS, L.J.; MORROW, J.D. "Analgesic-antipyretic and antiinflamatory agents and drugs employed in the treatment of gout". HARMAN, J.G.; LIMBIRD, L.E. (Eds.). Goodman&Gilman's: the pharmacological bases of therapeutics. New York: MacGraw-Hill, 2001, стр.687-732).
НПВП являются наиболее широко применяемыми лекарственными препаратами, представляя собой важный источник лекарственных средств, несмотря на вероятность развития серьезных побочных эффектов, таких как раздражение желудка (высокая заболеваемость) и артериальная гипертензия, возникновения повреждения печени, почек, селезенки, крови и костного мозга (см. RANG, H.P.; DALE, M.M.; RITTER, J.M. Farmacologia. Fourth ed. Rio de Janeiro: Guanabara Koogan, 2001, p.692).
Механизм действия НПВП заключается в ингибировании циклооксигеназы (ЦОГ) (обозначается как ЦОГ-1 конститутивная форма и ЦОГ-2 индуцибельная форма), препятствуя синтезу простагландинов (PG) и уменьшая воспалительные реакции.
Простагландины выполняют важные физиологические функции; среди них цитопротекция в желуд очно-кишечном тракте и сосудистый гомеостаз.
ЦОГ-1 обеспечивает выработку простагландинов, отвечающих за цитопротекцию в желудочно-кишечном тракте и за синтез тромбоксана, который участвует в агрегации тромбоцитов (см. Allison, Howatson, Torrence, Lee and Russell. "Gastrointestinal Damage Associated with the Use of Nonsteroidal Antiinflammatory Grugs". N. Engl. J. Med (1992) Vol.327, pp.749-754). Известно, что ЦОГ-2 индуцируется только в очаге воспаления в ответ на провоспалительные цитокины и эндотоксины. Важно выделить тот факт, что ЦОГ-2 ингибирует синтез простагландинов, ответственных за биосинтез в воспалительных клетках (моноцитах и макрофагах), а также в центральной нервной системе (см. Masferrer, Zweifel, Manning, Hauser, Leahy, Smith, Isakson and Seibert, "Selective Inhibition of Inducible Cyclooxygenase-2 in vivo is Antiinflammatory and Nonulcerogenic", Proc. Natl. Acad. Sci. U.S.A. (1994) Vol.91, pp.3228-3232; Vane, Mitchell, Appleton, Tomlinson, Bishop-Bailey, Croxtall and Willoughby, "Inducible Isoforms of Cyclooxygenase and Nitric Oxide Synthase in Inflammation", Proc. Natl. Acad. Sci. U.S.A. (1994) Vol.91, pp.2046-2050; Harada, Hatanaka, Saito, Majima, Ogino, Kawamura, Ohno, Yang, Katori and Yamamoto, "Detection of Inducible Protaglandin H Synthase-2 in Cells in the Exudate of Rat Carrageenin-Induced Pleurisy", Biomed. Res. (1994) Vol.15, pp.127-130; Katori, Harada, Hatanaka, Kawamura, Ohno, Aizawa and Yamamoto, "Induction of Prostaglandin H Synthase-2 in Rat Carrageenin-Induced Pleurisy and Effect of a Selective COX-2 Inhibition", Advances in Prostaglandin, Thromboxana, and Leukotriene Research (1995) Vo. 23, pp.345-347; and Kennedy, Chan, Culp and Cromlish, "Cloning and Expression of Rat Prostaglandin Endoperoxide Synthase (Cyclooxigenase-2) cDNA", Biochem. Biophys. Res. Commun. (1994) Vol.197, pp.494-500).
Традиционные НПВП, такие как АСК (ацетилсалициловая кислота), диклофенак, ибупрофен и напроксен, ингибируют ЦОГ-1 и ЦОГ-2. Эта неселективность НПВП приводит также к ингибированию простагландинов, участие которых в защите желудочно-кишечного тракта является очень важным.
Для уменьшения побочных эффектов, вызванных традиционными НПВП, было изучено огромное количество селективных препаратов ЦОГ-2 (ингибиторов ЦОГ-2), некоторые из которых присутствуют на рынке.
Существуют доказательства того, что уменьшение количества побочных гастро-интестинальных эффектов, вызванных селективными ингибиторами ЦОГ-2, приводит к адаптационному ответу на повреждение желудка, который не возникает при использовании ингибиторов ЦОГ-1 (см. PESKAR, В.М.; EHRLICH, K.; PESKAR, В.А. "Interaction of cyclooxigenase-2 inhibitor and salicylate in gastric mucosal damage", European Journal of Pharmacology, v.434, n.1-2, p.65-70, 2002; YAMAMOTO, H. Те al. "Inducible types of cyclooxigenase and nitric oxide synthase in adaptive cytoprotection in rat stomachs", Journal of Physiology, v.93, p.405-12, 1999).
С другой стороны, отсутствуют исследования, демонстрирующие различие в эффективности между селективными ингибиторами ЦОГ-2, даже если существует доказательство уменьшения вызванных ими гастро-интестинальных побочных эффектов. Проблема с этими ингибиторами возникает при рассмотрении побочных эффектов со стороны сердечно-сосудистой системы, опубликованных Stacy et al. (см. STACY, Z.A.; DOBESH, P.P.; TRUJILLO, T.C. "Cardiovascular risks of cyclooxygenase inhibition", Pharmacotherapy, v.26, n.7, p.919-938, 2006), что делает предпочтительным использование неселективных противовоспалительных препаратов.
К тому же, безопасность известных ЦОГ-2 ингибиторов находится под вопросом. Самый известный случай произошел с «блокбастером» рофекоксибом, с коммерческим названием Vioxx®, производимым Merck laboratories, который удалили с рынка в 2004 году после клинических исследований, показавших, что он вызывает высокий риск инфаркта миокарда и инсульта. Другие три ингибитора ЦОГ-2, которые присутствуют на рынке Бразилии, целекоксиб (Celebra®), валдекоксиб (Bextra®) и эторикоксиб (ARCOXIA®) проходят интенсивное исследование для подтверждения безопасности их применения. К тому же, 5 апреля 2005 года, FDA (Food and Drug Administration) временно приостановили коммерческое использование Bextra в США и, кроме того, в мае 2007 года не было утверждено использование Arcoxia.
Несмотря на все эти причины, НПВП до сих пор широко применяются как важный источник лекарственных препаратов, несмотря на возможность возникновения опасных известных побочных эффектов (главным образом язвы желудка).
Кроме того, заслуживает внимания роль оксида азота в воспалительных процессах. Фактически, оксид азота (NO) привлек внимание физиологов при открытии, сделанном в 1986 году Игнарро с соавторами, которые описали его функцию как эндотелий-зависимый релаксирующий фактор (EDRF), и предположили участие оксида азота в механизмах провоспалительных действий с эффектами вазодилатации и стимулирования синтеза простагландинов, а также в механизмах противовоспалительного действия для ингибирования нейтрофилов и тромбоцитов, являясь, таким образом, зависящим от иммунорегулирующего фактора (см. MONCADA, PALMER&HIGGS, "The discovery of nitric oxide as the endogenous nitrovasodilator". Hypertension, v.l2, p.365-372, 1988).
Оксид азота представляет собой бесцветный газ, парамагнетик, растворимый в воде в пропорции 2-3 моля на дм3 и имеющий точку кипения примерно - 141.7°С. NO синтезируется in vivo посредством взаимодействия, катализируемого ферментами (синтазой оксида азота - NOS), с молекулярным кислородом и L-аргинином (в качестве субстрата). Оксид азота становится свободным радикалом, который, в отличие от многих других свободных радикалов, не димеризуется в газовой фазе при комнатной температуре и давлении, несмотря на то, что в жидком состоянии может образовывать N2O2. При потере электрона свободного радикала оксида азота происходит образование иона нитрозила (NO+).
Среди очевидных химических свойств оксида азота можно выделить возможность образования радикала и соответственно его биологическое участие как электрофила, оксиданта, соли и комплексообразующего агента. В биологической системе форма радикала оксида азота связана с другими компонентами соединений азота, такими как нитрит (NO2), нитрат (NO3) и пероксинитрит (NO4).
Конститутивные изоформы оксида азота (cNOS) подразделяются на нейрональные (nNOS) и эндотелиальные (eNOS) и, в зависимости от того, в какой ткани они обнаружены, являются кальцийзависимыми и могут быть активированы кальций-связывающим белком (калмодулин-СаМ), через агонисты, такие как ацетилхолин (Ach), адениндифосфат (ADP), брадикинин (Bk) и глутамат (см. BARRETO, R.L.; CORREIA, C.R.D.; MUSCARA, M.N., "Oxido Nitrico: propriedades e potenciais usos terapeuticos". QuimicaNova, v.28, n.6, p.1046-1054, 2005).
Дополнительно, оксид азота участвует в передаче сигналов в периферической нервной системе и мочеполовой системе, и желудочно-кишечном тракте.
Индуцибельная изоформа оксида азота (iNOS) является кальций-независимой и продуцируется в высоких концентрациях путем активации бактериальными токсинами, интерфероном и интерлейкинами.
В защитной системе NO синтезируется тучными клетками, макрофагами, клетками Купфера и нейтрофилами, вызывая окислительные поражения в клетке-мишени посредством атаки белков, которые связаны с мембраной.
Также известен способ ослабления повреждения слизистой оболочки желудка, вызванного противовоспалительными активными компонентами, который при этом гарантирует удовлетворительное поглощение таких активных компонентов путем добавления в фармацевтические композиции аргинина и аналогичных аминокислот, проявляющих протекторную активность против повреждения слизистой оболочки желудка (см. Y. Kinouchi, N. Yata, Biol. Pharm. Bull., 19(3), pp.375-378 (1996)).
Кроме того, известно, что L-аргинин (исходный субстрат для синтеза NO) защищает слизистую оболочку желудка от образования повреждения, механизм которого возможно включает увеличение кровяного потока благодаря расширению прилегающих капилляров (см. KALIA, N. Et al. "L-Arginine protects and exacerbates ethanol-induced rat gastric mucosal injury", Journal Gastroenterology and Hepatology, v.l5, n.8, p.915-24, 2000).
Исследование, которое проводили при введении L-аргинина в терапию ибупрофеном, показало уменьшение окислительного стресса и инфильтрацию нейтрофилами в слизистой оболочке желудка, уменьшая повреждение, вызванное противовоспалительным препаратом. Этот механизм повреждения, зависящий от микроциркуляции, крайне важен для явлений гастроинтестинальной токсичности, вызываемых НПВП, которые, одновременно с терапевтическим действием вызывают повреждение слизистой оболочки посредством воспалительного механизма и окислительного повреждения, контролируемых активностью миелопероксидазы, скоростью активации нейтрофилов или липидной пероксидацией, а также активацией ксантиноксидазы, глутатионпероксидазы и супероксид-дисмутазы.
Протекторную активность L-аргинина можно объяснить возникновением локального действия, которое возможно относится к ингибированию окислительного стресса в результате действия ксантиноксидазы, но не к блокированию образования свободных радикалов полиморфноядерными лейкоцитами (см. JIMENEZ, M.D. et al. "Role of L-arginine in ibuprofen-induced oxidative stress and neutrophil infiltration in gastric mucosa", Free Radical Research, v.38, n.9, p.903-11, 2004).
Известно также, что действие таурина в воспалительном процессе обусловлено его важной активностью, состоящей в ингибировании продуцирования NO и простагландина Е2, участии в супрессии индуцибельной NO-синтазы (iNOS) и в экспрессии ЦОГ-2 (см. LIU, Y. Et al. "Taurine Chloramine Inhibits Production of Nitric Oxide and Prostaglandin E2 in Actiated C6 Glioma Cells by Supressing Inducible Nitric Oxide Synthase and Cyclooxigenase-2 expression". Molecular Brain Research, v.59, p.189-195, 1998), а также в ингибировании пероксидных ионов (см. CHAEKYUN, К. et al., "The Production of Superoxide Anion and Oxide by Cultured Murine Leukocytes and the Accumulation of TNF-α in the Conditioned Media is Inhibited by Taurine Chloramine", Immunopharmacology, v.34, p.89-95, 1996).
Другое действие таурина относится к ослаблению гипераналгезирующих эффектов (см. THOMAS, G. "Oxido Nitrico" в Quimica Medicinal: Uma Introducao. Rio de Janeiro: Guanabara Koogan, p.337-61, 2003), приводя к нормальным уровням синтеза NO, угрожая таким образом активным и усиленным присутствием iNOS и ингибируя каскад арахидоновой кислоты. Кроме того, в 2001 году, Palumbo, Cioffi и D'lschia подали заявку на патент на NOS ингибирующие соединения с разным применением, включающим воспалительные процессы, с повышением требований к безопасности данной терапии (CAN 137:346227; AN 2002:894293; заявка Италии ITRM20000039 А, опубликованная 24 июля 2001 года), подтверждая результаты Moncada и Higgs (см. MONCADA, S.; HIGGS, Е.А. "Molecular mechanisms and therapeutic strategies related to nitric oxide", FASEB Journal, v.9, p.1319-1330, 1995) об использовании ингибиторов синтазы оксида азота, что является достижением в терапии воспалительных состояний.
Ингибирование окислительного стресса можно объяснить системным действием аминокислот.В этом отношении таурин обладает преимуществами, связанными с системным действием гастропротекции, возможно через подавление активности свободных радикалов кислорода, которые выполняют важную физиопатологическую роль в острой язве, вызванной НПВП и ишемией-реперфузией.
Результаты эксперимента с использованием таурина в качестве антиоксиданта при внутрижелудочном введении у крыс, предварительно обработанных 250 мг/кг или 500 мг/кг в течение от 1 (одного) до 3 (трех) дней перед индуцированном геморрагического поражения с помощью 25 мг/кг индометацина, показали уменьшение поражения с ингибированием пероксидирования липидов, помимо ингибирования активности нейтрофилов (см. SON, M. Et al "Protective effect of taurine on indometacin-induced gastric mucosal injury", Adv Exp Med Biol, v.403, p.147-55, 1996).
Известно также, что таурин обеспечивает значительное угнетение секреции кислоты и увеличение высвобождения бикарбоната из полости благодаря механизмам регулирования между продуцированием оксида азота и простагландинов, с компенсаторной реакцией, сохраняющейся в желудке (см. TAKEUCHI, K. Et al. "Nitric oxide and prostaglandins in regulation of acid secretory response in rat stomach following injury", Journal of Pharmacology Experimental and Therapeutic, v.272, n.1, p.357-63, 1995).
Кроме того, известно, что противоязвенная активность тесно связана с улучшением уменьшенного кровотока в слизистой оболочке вследствие нарушения продуцирования оксида азота, с помощью чего широко изучено влияние противоязвенных препаратов, как и в случае [2,4-диамино-6-(2,5-дихлорфенил)-8-тиазин] малеата. Согласно Takashi et al. (TAKASHI, K. et al. "Irsogladine prevents monochloramine-induced gastric mucosal lesions by improving the decrease in mucosal blood flow due to the disturbance of nitric oxide synthesis in rats", Journal of Pharmacological Sciences, v.93, p.314-20, 2003), конкретный механизм может быть продемонстрирован путем применения ингибиторов конститутивной синтазы оксида азота (cNOS) или неселективных ингибиторов, таких как NG-нитро-L-аргинин метиловый эфир (L-NAME) и селективных ингибиторов индуцибельной синтазы оксида азота (iNOS), например, аминогуанидина, где [2,4-диамино-6-(2,5-дихлорфенил)-8-тиазин] малеат блокирует ингибирующее действие cNOS без нарушения действия iNOS, ответственной за рекрутинг клеток.
Как отмечалось ранее, оксид азота выполняет важную роль в защите от язвенной болезни желудка, вызванной нестероидными противовоспалительными препаратами, с помощью механизмов, которые не ограничиваются секрецией кислоты, что приводит к новому режиму лечения язвенной болезни желудка, вызванной противовоспалительными препаратами. В доклиническом тестировании образования язвы с использованием индометацина в качестве контроля было установлено, что происходит 80% повышения кислотности желудочного сока с 22% снижением оксида азота (измерено как нитрит). С другой стороны, применение L-NAME не влияет на кислотность желудочного сока, но вызывает 50% снижение нормальных концентраций оксида азота и, следовательно, скорость поражения удваивается (см. КНАТТАВ, М.М.; GAD, M.Z.; ABDALLAH, D. "Protective role of nitric oxide in indometacin-induced gastric ulceration by a mechanism independent of gastric acid secretion", Pharmacological Research, v.43, n.5, p.463-67, 2001).
Гомеостаз тонуса периферических сосудов является очень важным для сохранения целостности функциональных прилегающих тканей, в которых изменение процесса регулирования NO вверх/вниз может привести к тромбозу и ишемическим осложнениям в случае низкого продуцирования NO.
Важно отметить, что при раздельном анализе параметров измерений NO, его подсемейства и моменты продуцирования должны быть связаны с энзиматической активностью. Это может служить ответом на тот факт, что использование простого исходного субстрата, такого как L-аргинин, не может предотвратить образование повреждения в слизистой оболочке желудка.
Следовательно, ферментный субстрат (L-аргинин) может даже повысить присутствие NO в усиленной форме в провоспалительных клетках, что, так или иначе, делает эту меру недостаточной для блокирования свободных радикалов, индуцированных в гастроинтестинальном воспалительном процессе.
В данном случае таурин играет роль медиатора микроциркуляторной реакции, помимо действия в ингибировании изоформы фермента, индуцированной в воспалительном процессе. Эта изоформа отвечает за окислительный стресс, подтверждая тем самым активность таурина как гастроинтестинального антиоксиданта и противовоспалительного лекарственного средства.
При исследовании гастропротекторных соединений наблюдалось, что таурин увеличивает клеточную резистентность на 21% с сохранением целостности повреждений мембраны, митохондрии и ядра (см. NAGY, L. Et al. "Investigation of gastroprotective compounds at subcellular level in isolated gastric mucosal cells", American Journal Physiology and Gastrointestinal Liver Physiology, v. 279, n.G1, 201-08, 2000), что подтверждает с помощью исследования слизистой оболочки на субклеточном уровне использование таурина в качестве гастропротекторного соединения.
Другое предположение о механизме действия цитопротекции основано на адаптации эндогенной реакции, вызванной простагландинами, без вовлечения эффекта защитного каскада реакций, инициируемых оксидом азота. Эта гипотеза была представлена для активности L-аргинина (NO предшественника) против повреждения желудка, вызванного у крыс, индуцированного оральным введением соляной кислоты (см. TAKEUCHI, К. et al. "Cytoprotective action of L-arginine against HCL-induced gastric injury in rats: Involvement of nitric oxide?", Japan Journal Pharmacology, v.61, p.13-21, 1993). Преимущество таурина над L-аргинином становится более очевидным из анализа результатов его использования в уменьшении повреждений слизистой оболочки желудка, так как он не проявляет активности предшественника NO.
Несмотря на то, что участие простагландинов и оксида азота в ингибировании образования повреждения, индуцированного некротическими агентами, является известным, не существует четкой корреляции со степенью важности этих медиаторов. Эксперименты по ингибированию (L-NAME) оксида азота с добавлением 16,16-диметил простагландина Е2 не выявили повреждения. С другой стороны, ингибирование простагландинов с добавлением донора оксида азота недостаточно для сохранения целостности слизистой оболочки желудка (см. UCHIDA, M et al. "Nitric oxide donating compounds inhibit HCl-induced gastric mucosal lesions mainly via prostaglandin", Japan Journal Pharmacology, v.85, p.133-38, 2001). Это исследование подтверждает самое выраженное побочное действие при терапевтическом использовании противовоспалительных препаратов, а также трудность в реверсии повреждения или гастропротекции.
Таурин действует в воспалительном процессе благодаря его важной активности в ингибировании продуцирования NO и простагландинов Е2 (PGE2) и действию в супрессии индуцибельной синтазы оксида азота (iNOS), а также в экспрессии циклооксигеназы-2 (см. LIU, Y. Et al. "Taurine inhibits production of nitric oxide and prostaglandin E2 in activated C6 glioma cells by suppressing inducible nitric oxide synthase and cyclooxigenase-2 expression". Molecular Brain Research, v.59, p.189-195, 1998), а также в ингибировании продуцирования пероксидных ионов (см. CHAEKYUN, K. et al. "The production of superoxide anion and oxide by cultured murine leukocytes and the accumulation of TNF-a in the conditioned media is inhibited by taurine chloramines", Immunopharmacology, v.34,p.89-95,1996).
Было сделано много попыток вмешаться в процесс образования повреждения желудка, вызванного НПВП. В патенте США 7,008,920 описана фармацевтическая связь между НПВП, солями желчных кислот и таурином или полиаминами на предмет снижения гастроинтестинального повреждения, индуцированного препаратами, и повышения их растворимости в воде.
Также известно, что таурин, помимо действия против повреждения желудка (см. SENER, G. et al. "Protective effect of taurine against alendronate-induced gastric damage in rats", Fundamental&Clinical Pharmacology, v.l9, p.93-100, 2004), также ослабляет почечную гипертензию (см. HAGAR, H.H.; ETTER, Е.Е.; ARAFA, M. "Taurine attenuates hypertension and renal dysfunction induced by cyclosporine A in rats", Clinical and Experimental Pharmacology and Physiology, v.33, p.189-196, 2006).
Другим важным аспектом поиска соединений, которые ослабляют побочные действия НПВП и обладают положительными эффектами этих препаратов, является разработка разных способов получения с технической и экономической точки зрения. В связи с этим были развернуты многочисленные исследования для получения новых соединений с использованием главным образом методов молекулярной модификации. Среди способов получения, очень важной является химическая модификация, целью которой является разработка пролекарств, которые заключены в неактивные формы носителя, и которые высвобождают лекарственный препарат in vivo после биотрансформации (см. WERMUTH, C.G. "The Practice of Medicinal Chemistry", London: Academic Press, 2nd ed, 2003. 768 pages; KROGSGAARD-LARSEN, P., BUNDGAARD, H. "A textbook of drug design and the development", Harwood: Academic Publish, 1991, 643 pages; SILVA, A.T.A. et al. "Advances in prodrug design", Medicinal Chemistry, v.5, n.10, p.893-914, 2005).
Самые последние терапевтические химические соединения были получены путем химической модификации оригинального лекарственного препарата, в частности с помощью этерификации и образования амидов. Проще говоря, можно сказать, что химическая модификация представляет собой процесс органического синтеза, который стремится модифицировать молекулу активного соединения или оригинального лекарственного препарата для оптимизации его фармакокинетических свойств и/или снижения его токсичности.
В последние несколько лет химическая модификация стала одним из основных инструментов для развития химиотерапевтических лекарственных препаратов, которые используются для лечения важных в настоящее время заболеваний, таких как рак и синдром иммунодефицита человека - AIDS. Поиск химически модифицированных лекарственных препаратов является оправданным, по меньшей мере, по одной из следующих причин: (i) минимизация фармакокинетических недостатков, принадлежащих оригинальному лекарственному препарату, (ii) снижение высокой токсичности оригинального препарата, (iii) улучшение слабой химической стабильности оригинального препарата, (iv) улучшение растворимости в воде оригинального препарата, (v) уменьшение недостатков, связанных с запахом и вкусом оригинального препарата и (vi) возможность сделать их пригодными для получения сложных фармацевтических составов благодаря оригинальному препарату.
Химически модифицированные лекарственные препараты, называемые пролекарствами, соответствуют оригинальным препаратам, которые являются химически трансформированными в неактивное соединение с помощью химических реакций, ферментных реакций или обоих. Пролекарство превращается в оригинальный препарат внутри организма до или после достижения точки его воздействия.
Пролекарство можно определить как любое соединение, которое подвергается биотрансформации до проявления его фармакологического действия. Пролекарство, а также аналог лекарства, представляет собой аналогичные химические структуры, однако биологические свойства этих соединений отличаются от оригинального лекарства в отношении: (i) активности, (ii) силы, (iii) биодоступности, (iv) способа синтеза, (v) спектра действия и (vi) терапевтического индекса. Пролекарство отличается от аналогичного лекарства благодаря in vivo гидролизуемой химической связи и группе переносчика.
Среди разных способов получения пролекарств, этерификация является самым используемым способом, сопровождающимся образованием амида, имида и карбамата. Одновременно, функциональные группы лекарственного препарата можно модифицировать с помощью химических реакций с получением обратимых групп, интенсивно использующихся в разработке пролекарств.
Большое количество замещений в известных молекулах, а также новых производных НПВП описано в виде методики, целью которой является нормализация не только их побочных действий, но также их противовоспалительного потенциала. Например, патент США 5905073 описывал 5-ASA и другие производные НПВП для лечения неспецифического язвенного колита.
Среди коммерчески доступных НПВП диклофенак является одним из самых используемых противовоспалительных лекарственных препаратов. Фактически, диклофенак, открытый в 1966 году и описанный в патенте США 3,558,690, является лучше всего продаваемым препаратом в мире и его эффективность, а также безопасность установлена в области противовоспалительной терапии. Разные замещения также были сделаны в 2-ариламинофенилуксусных кислотах для ослабления вредных побочных действий этого активного компонента, который описан в нескольких патентных документах, таких как, например, патент США 3,652,762; 4,173,577; 4,166,128; 4,704,468; 5,475,139; WO 9404484; WO 9709977; WO 9600716 и DE 345011.
Ацеклофенак является примером пролекарства диклофенака, описанного в патенте США 4,548,952, полученного путем этерификации карбоксильной группы маленькой алкильной цепью в попытке снижения вредных действий в желудочно-кишечном тракте при использовании в противовоспалительных терапиях. Например, патент США 6,451,858 описывает этерификации в 2-ариламинофенилуксусных кислотах как попытку повышения его селективности по отношению к ЦОГ-2.
Другие модификации в молекуле диклофенака выполняли для ослабления нежелательных побочных действий или для повышения его биодоступности с тем, чтобы, помимо орального, сделать доступными другие способы введения, указанные в: (i) патенте СШа 4,704,468, который описывает двойные пролекарства диклофенака, соединенные производными полиэтиленгликоля, для ослабления действий на желудок и (ii) патенте США 5,792,786, который описывает этерификации НПВП с жирными кислотами с длинной цепью для повышения их биодоступности при местном применении фармацевтических форм.
Кроме того, для ослабления вызванных НПВП пагубных эффектов у пациентов, проявляющих воспалительные нарушения, недавние исследования были направлены на более подробное изучение функций, выполняемых оксидом азота в биологических системах. В этой связи, патент США 5,597,847 описывает производные 2-ариламинофенилуксусной кислоты, которые были нитрированы для повышения их противовоспалительного потенциала, стремясь обеспечить оксида азота в воспалительном процессе. Аналогично, пролекарства с местным высвобождением NO описаны в патенте WO 2006125016.
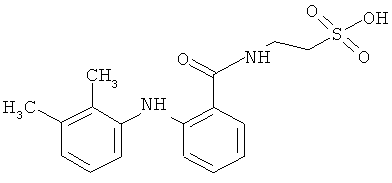
Документ WO 9109831 описывает пролекарства, полученные из НПВП, с кислыми группами, полученными путем образования ангидрида среди групп, присутствующих в НПВП или в разных НПВП, таких как АСК, СА (салициловая кислота), сулиндак, цетопрофен, индометацин, напроксен, фенопрофен, ибупрофен, дифлунизал, толметин, флурбипрофен, супрофен.
Другие примеры получения пролекарств представлены в патенте США 5,681,964, который описывает этерификации индометацина, приводящие к уменьшению повреждений желудка; в патентах США 5,607,966 и 5,811,438, которые описывают производные эфира и амиды индометацина, которые использовали в качестве антиоксидантов и ингибиторов 5-липоксигеназы, однако без проявления ЦОГ-2 селективности; в патенте США 6,399,647, который описывает сульфонамидные производные индометацина, показывающие повышение ЦОГ-2 селективности; и в патенте США 6,887,903, который описывает сульфонамидные производные, которые действуют в других путях воспалительного процесса как сигнальные молекулы полиморфно-ядерных нейтрофилов и других интерлейкинов.
Несмотря на преимущества этого разнообразия пролекарств НПВП перед оригинальными препаратами, все еще существуют разные пагубные эффекты, которые ограничивают их применение.
Таурин и другие специфические аминокислоты сами по себе являются интересными переносчиками лекарственных препаратов, способствуя улучшению не только физико-химических свойств, но также ослаблению их побочных действий. Патент США 5,059,699 представляет производные таксола (противоопухолевое средство) и таурин для увеличения его растворимости в воде, приводя к увеличению его биодоступности и стабильности в химиотерапевтических формулах. Другие примеры улучшения формулы с использованием таурина основаны на производных салицилата (СК и АСК) или сульфонамидных производных, как описано в документах JP 68003293 и JP 68004331.
Ограничения и недостатки известных пролекарств и лекарственных препаратов привели к поиску новых активных компонентов, которые минимизируют побочные эффекты НПВП. Таким образом, настоящее изобретение основано на знании механизмов действия противовоспалительных препаратов, описанных терапевтической областью, используя их потенциал в применении производных НПВП во время длительных противовоспалительных терапий.
Следовательно, целью настоящего изобретения является улучшение фармакотерапевтического лечения, включающего неотложную и длительную терапию противовоспалительными лекарственными препаратами, для ослабления или нейтрализации побочных эффектов язвы желудка, исходя из открытия того, что аминокислоты, относящиеся к противовоспалительным препаратам, вводимым орально, уменьшают распространение повреждения желудка, при этом таурин играет важную роль в этом механизме, частично благодаря его участию в процессе регуляции провоспалительных цитокинов.
Сущность изобретения
Целью настоящего изобретения является ослабление побочных и вредных эффектов нестероидных противовоспалительных (НПВП) лекарственных препаратов путем обеспечения новых соединений на основе производных таурина. В частности, изобретение основано на введении амидной связи между молекулами НПВП и таурина с образованием новых соединений по изобретению, дополнительная активность которых возникает в результате ингибирования продуцирования оксида азота, индуцированного в воспалительном процессе специфическими ферментами, присутствующими в макрофагах и нейтрофилах (индуцибельная синтаза оксида азота - iNOS), а также в результате ингибирования циклооксигеназы и вероятно медленного высвобождения активных компонентов in vivo, что позволяет контролировать токсичность НПВП с сохранением их противовоспалительной активности.
Первый вариант настоящего изобретения относится к соединениям, полученным из таурина, в которых таурин связан напрямую амидной связью или через промежуточную группу с выбранным соединением из группы нестероидного противовоспалительного соединения, называемого производным таурина и представленным Формулой (I):
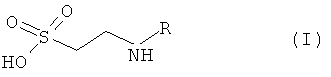
где R означает компонент с нестероидной противовоспалительной активностью.
Во втором варианте настоящее изобретение обеспечивает способ получения новых соединений Формулы (I), его солей, сольватов, гидратов, энантиомеров, диастереоизомеров и полиморфов, включающий реакцию таурина с соединением, принадлежащим к группе нестероидных противовоспалительных (НПВП) препаратов, в присутствии соответствующего катализатора для получения производного таурина посредством прямой связи таурина к НПВП или с помощью промежуточной группы.
В третьем варианте настоящее изобретение относится к фармацевтическим композициям, включающим: (а) производное таурина с противовоспалительной активностью нестероидного типа, (b) факультативно, соответствующий активный компонент для лечения заболевания, включающего воспалительные нарушения, и (с) фармацевтически приемлемый носитель.
Краткое описание Фигур
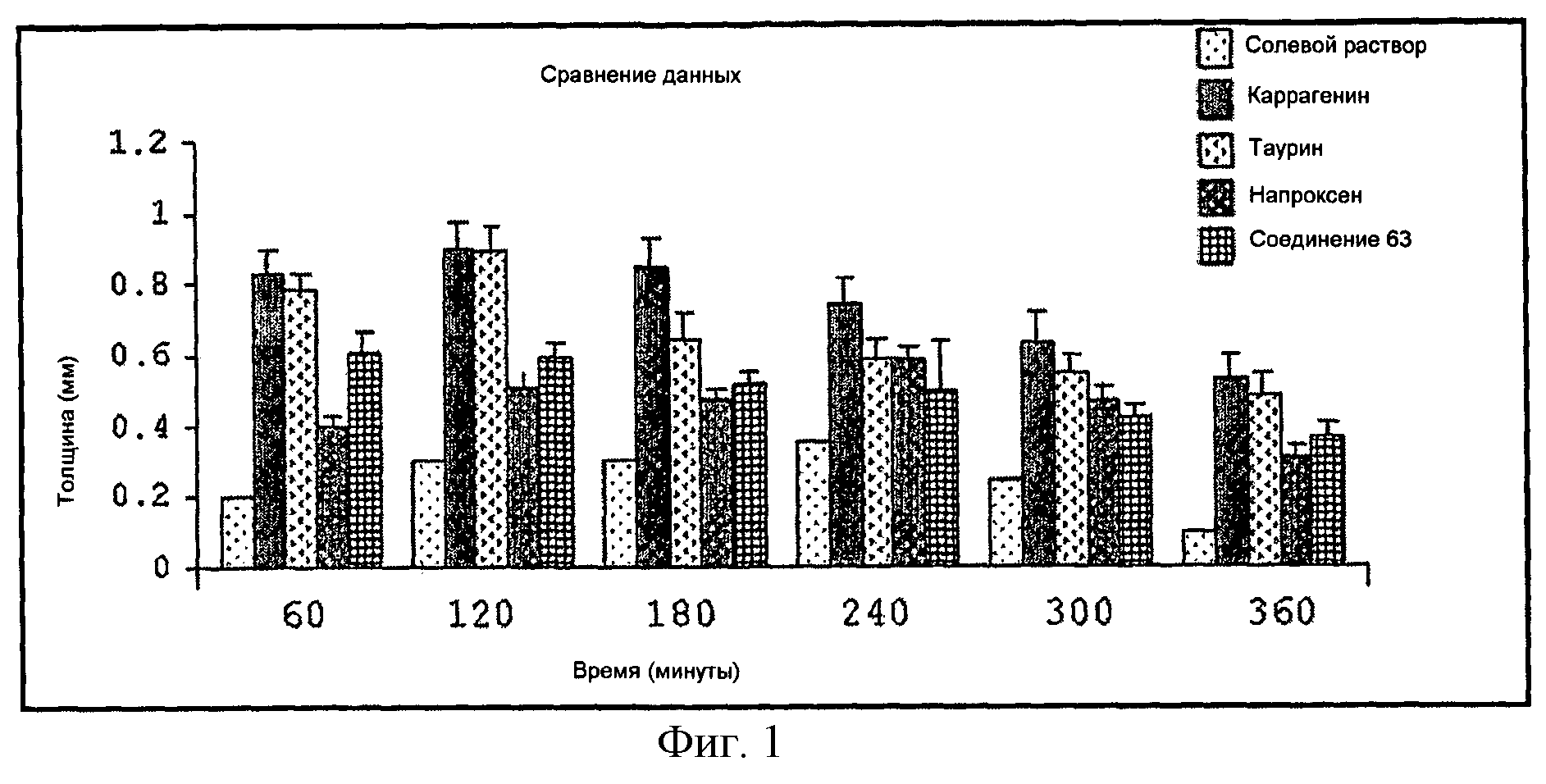
Фигура 1 показывает сравнительный анализ противовоспалительной активности на лапе крысы с использованием таурина, напроксена и его производного (Соединение 63, вариант изобретения соответствует Примеру 2).
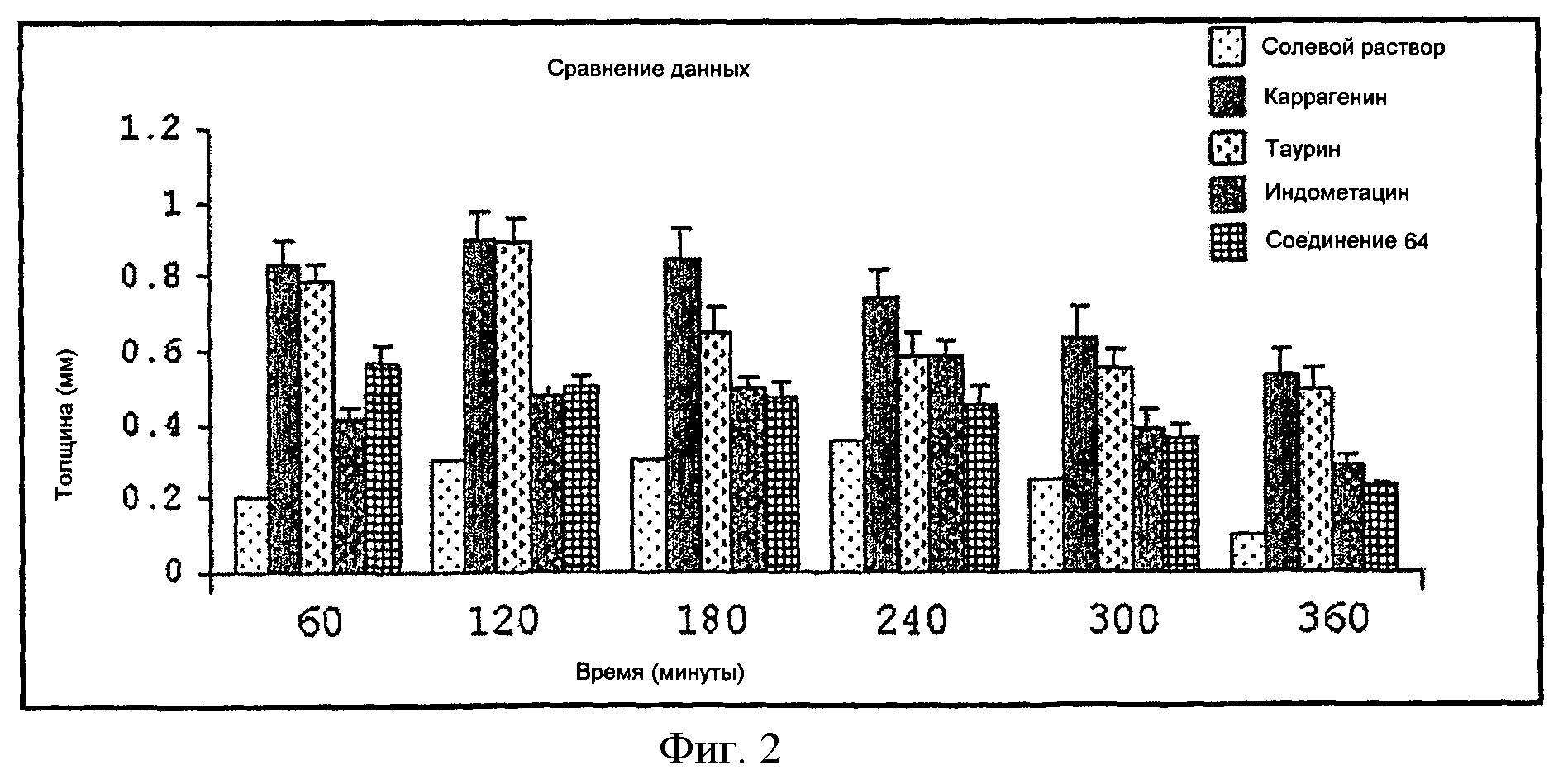
Фигура 2 показывает сравнительный анализ противовоспалительной активности на лапе крысы с использованием таурина, индометацина и его производного (Соединение 64, вариант изобретения соответствует Примеру 3).
Фигура 3 показывает сравнительный анализ противовоспалительной активности на лапе крысы с помощью таурина, ибупрофена и его производного (Соединение 27, вариант изобретения соответствует Примеру 1).
Фигура 4 графически демонстрирует профиль массы в граммах следующих органов: почки, сердце и печень относительно массы тела каждого животного, данное в процентах, при введении производного ибупрофена после анализа токсичности (Соединение 27, вариант изобретения соответствует Примеру 1).
Фигура 5 графически показывает разницы в массе органов при введении производного ибупрофена: в А=печень; и В, верхний график соответствует почкам и нижний относится к сердцу.
Фигура 6 графически представляет профиль массы в граммах органов: почки, сердце и печень относительно массы тела каждого животного, данное в процентах, при введении производного напроксена после анализа токсичности (Соединение 63, вариант изобретения соответствует Примеру 2).
Фигура 7 графически показывает разницы в массе органа при введении производного напроксена: в А - печень; и В, верхний график соответствует почкам и нижний сердцу.
Фигура 8 графически представляет профиль массы в граммах органов: почки, сердце и печень относительно массы тела каждого животного, данный в процентах, при введении производного индометацина после анализа токсичности (Соединение 64, вариант изобретения соответствует Примеру 3).
Фигура 9 графически показывает разницы в массе органа при введении производного индометацина: в А=печень; и В, верхний график соответствует почкам и нижний сердцу.
Фигура 10 графически показывает тест гастротоксичности, который проводили: (i) с НПВП - ибупрофеном, напроксеном и индометацином, (ii) с эквимолярной физической смесью таурина с этими НПВП и (in) с соединениями 63 (таурин-ибупрофен), 27 (таурин-напроксен) и 64 (таурин-индометацин) изобретения.
Подробное описание изобретения
Соединения изобретения являются производными таурина, полученными в результате деформирования амидных связей с нестероидными противовоспалительными (НПВП) препаратами напрямую, а также с помощью обеспеченного промежуточного агента.
Далее, обеспечены некоторые определения для улучшения понимания настоящего изобретения.
- Таурин - 2-аминоэтансульфоновая кислота,

- Амидная связь - химическая связь -NHCOY-типа между компонентом с нестероидной противовоспалительной активностью (НПВП) (лекарственный препарат) и таурином (переносчик).
- Нестероидные противовоспалительные (НПВП) лекарственные препараты - являются веществами с противовоспалительным, анальгетическим и антипиретическим эффектом. НПВП действуют в организме, блокируя синтез простагландинов, и включают соединения, такие как: салицилаты, пиразолоны и аналоги, производные индолилуксусной кислоты, производные арилуксусной кислоты, производные арилпропионовой кислоты, оксикамы и фенаматы.
- Промежуточный агент - промежуточная химическая группа, которая устанавливает связь между лекарственным препаратом и переносчиком. В химическом высвобождении лекарственного препарата использование промежуточного агента способствует лучшему и быстрому доступу фермента; таким образом, высвобождение активной части облегчается, что является важным фактором в реализации биологической активности.
- Производные таурина - включают изомеры, энантиомеры, аналоги и пролекарства, полученные в результате связи таурина с выбранным соединением из группы НПВП посредством прямой амидной связи или с помощью промежуточного агента.
Соединения настоящего изобретения являются соединениями, представленными общей Формулой (I):

в которой компонент таурин:

связан напрямую или через промежуточный агент с НПВП, формируя амидную связь -NHCOY-типа, чья - COY группа соответствует R заместителю общей формулы (I).
Компоненты НПВП изобретения могут быть любыми компонентами, принадлежащими к нестероидной противовоспалительной группе. Предпочтительно, чтобы компонент НПВП из числа соединений изобретения мог быть любым R заместителем, как определено в Таблице 1.

















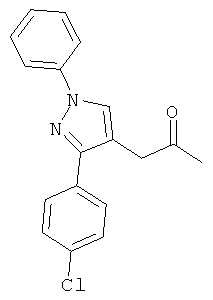
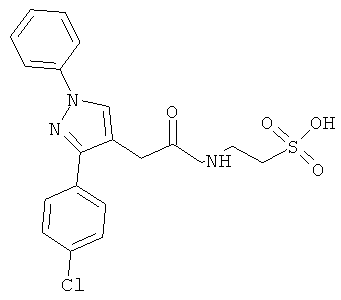



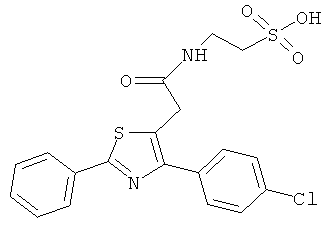

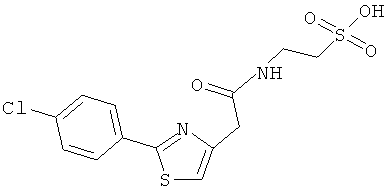




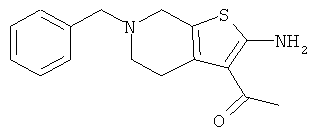


















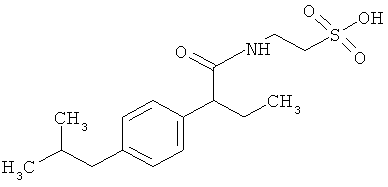








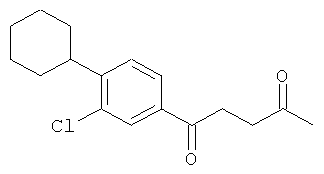






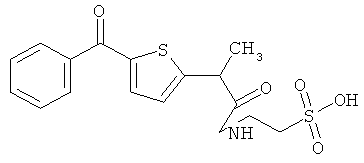





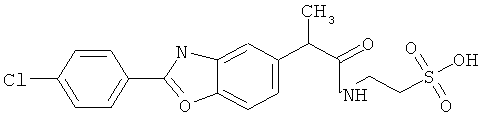

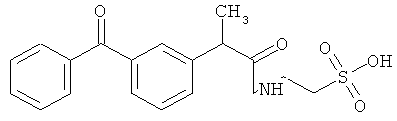




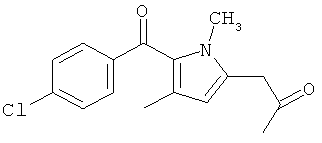



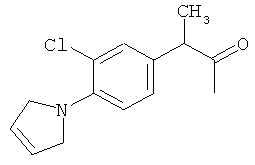












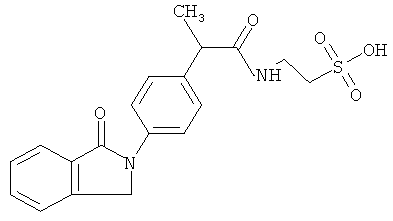







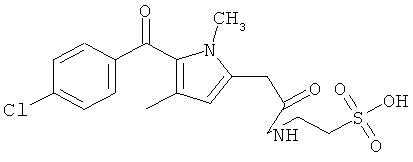

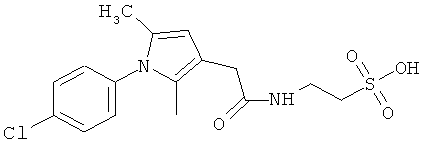


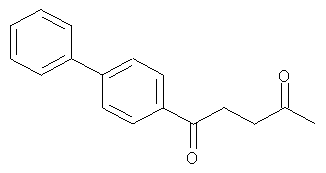
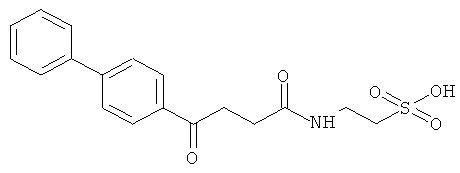



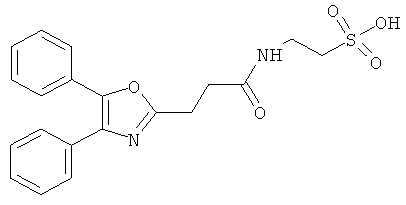

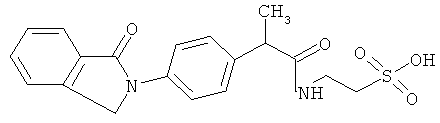



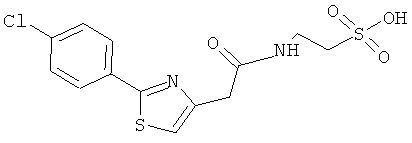
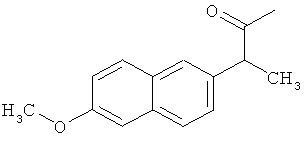



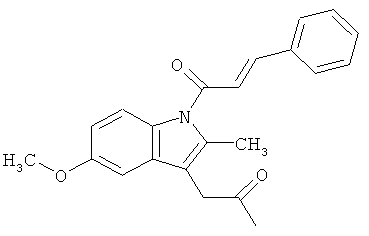
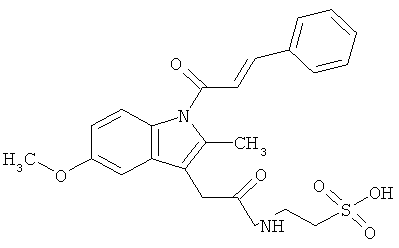

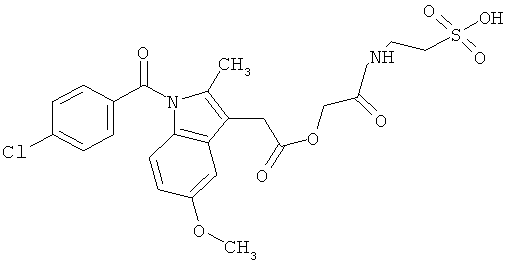




Соединения настоящего изобретения проявляют противовоспалительную активность нестероидного типа, антипиретическую, анальгетическую активность и активность против агрегации тромбоцитов, а также являются эффективными в качестве вспомогательных средств при лечении воспалительных процессов, таких как ревматоидный артрит, язвенный колит, болезнь Крона и других воспалительных заболеваний, например, нейродегенеративных заболеваний, таких как болезнь Альцгеймера, с минимальной возможностью раздражения желудка.
Особенно в отношении язвенного колита и болезни Крона, соединения настоящего изобретения обеспечивают противовоспалительную и антиоксидантную активность.
Учитывая, что таурин обнаружен в высоких концентрациях в молодом мозге и понижается с возрастом, и зная, что воспалительный процесс является одной из причин формирования амилоидных бляшек при болезни Альцгеймера, соединения настоящего изобретения могут быть полезными для предупреждения/лечения данного заболевания, так как таурин сам по себе повышает способность к обучению у взрослых животных (Е1 Idrissi, A. Taurine impoves leaning and retention in aged mice. Neuroscience Letters, 2008 DOI 10.1016/J.neulet 2008.02.070).
Соединения настоящего изобретения получены способом, который включает взаимодействие таурина с веществом, обладающим нестероидной противовоспалительной (НПВП) активностью, в присутствии соответствующего катализатора для возможности формирования прямой амидной связи или с помощью промежуточного агента между таурином и компонентом НПВП.
Вещество, обладающее нестероидной противовоспалительной активностью, может быть выбрано из группы, состоящей из следующих НПВП: салицилаты, пиразолоны и аналоги, производные индолуксусной кислоты, производные арилуксусной кислоты, производные арилпропионовой кислоты, оксикамы и фенаматы. НПВП из группы салицилатов могут быть выбраны из следующих препаратов: лизин клониксинат, бенорилат, дифлунизал, этерсалат и салсалат. НПВП из группы пиразолонов и аналогов могут быть выбраны из следующих препаратов: фенилбутазон, оксифенбутазон, аминофеназон, бумадизон, фепразон, нифеназон и суксибузон. НПВП из группы производных индолуксусной кислоты могут быть выбраны из: ацематацин, глукаметацин, индометацин, проглуметацин, оксаметацин, сулиндак и толметин. НПВП из группы производных арилуксусной кислоты могут быть выбраны из следующих препаратов: ацеклофенак, диклофенак, фентиазак и набуметон. НПВП из группы производных арилпропионовой кислоты могут быть выбраны из следующих препаратов: бутибуфен, фенбуфен, флурбипрофен, ибупрофен, ибупроксам, кетопрофен, напроксен, локсопрофен, панопрофен, оксапрозин и тиапрофен. НПВП из группы оксикамов могут быть выбраны из следующих препаратов: дроксикам, мелоксикам, пироксикам и теноксикам. НПВП из группы фенаматов могут быть выбраны из следующих препаратов: меклофенамовая кислота, мефенамовая кислота, толфенамовая кислота и нифлумовая кислота.
Предпочтительно, чтобы способ получения соединений настоящего изобретения представлял собой химическую модификацию, при этом химическую модификацию в биологически активном соединении выполняют для формирования нового соединения, которое будет высвобождать т vivo соединение или оригинальный лекарственный препарат. Химическая модификация лекарственного препарата является синонимом планирования пролекарства.
Однако следует отметить, что механизм действия соединений настоящего изобретения не объяснен полностью и, следовательно, нет возможности убедиться, что указанные соединения соответствуют пролекарствам или являются новыми химическими структурными единицами. Другими словами, соединения настоящего изобретения также могут обладать активностью без разложения in vivo пролекарства; следовательно, они могут обладать активностью per se. К тому же, при сравнении результатов испытаний гастропротекции, представленных ниже, проводимых с соединениями изобретения и их физическими смесями с известными неспецифическими и специфическими НПВП (для ингибирования ЦОГ-2), неожиданно было обнаружено, что соединения настоящего изобретения не вызывают повреждения желудка, сохраняя противовоспалительный потенциал, и проявляют отличную безопасность в соответствии со стандартами (на основе оригинальных НПВП), как показано на Фигурах от 1 до 4.
Более конкретно, способ настоящего изобретения включает реакцию выбранного НПВП, выбранного из противовоспалительных веществ, как определено здесь (оригинальные лекарственные препараты) с этансульфоновой аминокислотой (таурином) в присутствии соответствующего катализатора в среде органического растворителя.
Катализатор, который использовали в способе настоящего изобретения, может быть любым обычно используемым катализатором в процессах химической модификации. Предпочтительными являются следующие катализаторы: диэтилцианофосфонат, 1-гидроксибензотриазол, карбоимиды, триэтиламин, имидазол, пиразол, 1,2,4-триазол, 4-диметиламинопиридин, пиридин и аналогичные.
Более предпочтительным, согласно способу настоящего изобретения, является DEPC (диэтилцианофосфонат).
Способ настоящего изобретения проводят, предпочтительно в присутствии органического растворителя. Предпочтительными являются следующие растворители: ацетон, тетрагидрофуран (THF) или диметилформамид (DMF).
Реакцию можно проводить при комнатной температуре в высокоосновной среде в течение до 2 часов. В предпочтительном способе образовавшийся осадок очищают любыми известными в данной области способами для получения соединения изобретения, которое соответствует требованиям текущего законодательства по использованию в приготовлении лекарственных препаратов для применения у человека и животных, таких как C15H23NO4S (вариант согласно Примеру 1), C16H19NO5S (вариант согласно Примеру 2), C21H21C1N2O6S (вариант согласно Примеру 3).
Соединения изобретения используются в приготовлении противовоспалительных лекарственных препаратов в форме твердых, жидких, твердых-жидких или твердых-газообразных суспензий (например, аэрозолей) фармацевтических композиций, кремов, гелей, адгезивных фармацевтических форм противовоспалительных лекарственных препаратов системного применения или адекватного местного применения. Предпочтительными фармацевтическими формами являются твердые формы, аэрозоли, кремы и гели, содержащие соединения настоящего изобретения. Твердые фармацевтические формы могут быть представлены таблетками, капсулами, пилюлями и аналогичными формами. Твердые формы могут быть с быстрым высвобождением, контролируемого или пролонгированного типа. Так как производные соединения таурина настоящего изобретения являются легко растворимыми, формы для инъекций также являются предпочтительными согласно изобретению.
В случае форм для инъекций, производные соединения таурина настоящего изобретения могут вводиться парентерально, включая внутривенное введение (или внутривенозное), мышечное, подкожное и внутридермальное. Для подкожного или внутривенного введения, фармацевтическая композиция изобретения может быть в форме раствора, суспензии или эмульсии, включающей вещества, обычно используемые в таких составах, такие как солюбилизаторы, эмульгаторы или другие добавки. Соответствующими растворителями являются вода, физиологический солевой раствор или спирты, например, этанол, пропанол, глицерол и дополнительно сахарные растворы, такие как глюкоза или маннитол или смеси так называемых растворителей.
Фармацевтические композиции изобретения также могут быть в форме аэрозоля, например, растворы, суспензии или эмульсии активных инградиентов в фармацевтически приемлемом растворителе, таком как этанол или вода, или их смеси. Также могут присутствовать добавки, такие как поверхностно-активные вещества, эмульгаторы, стабилизаторы и пропелленты.
Фармацевтические композиции настоящего изобретения включают: (а) по меньшей мере, одно из соединений общей формулы (I), (b) дополнительно, по меньшей мере, один соответствующий активный компонент для лечения заболевания, включающего воспалительное нарушение и (с) фармацевтически приемлемый носитель или связующее.
Термин фармацевтически приемлемый носитель или связующее означает любое вещество или вещества, которые являются инертными, используемые в качестве носителя или разбавителей для любого из активных компонентов композиции настоящего изобретения.
В случае, если фармацевтическая форма композиции изобретения представлена в виде таблетки, она может включать один или более носителей, связующих и/или добавок, выбранных из группы, состоящей из разбавителей, дезинтеграторов, лигандов, красителей и ароматизаторов. Разбавителем может быть один или более из карбоната кальция, двухосновного фосфата кальция, сульфата кальция, микрокристаллической целлюлозы, порошкообразной целлюлозы, декстратов, декстринов, декстрозы, фруктозы, китайской глины, лактитола, лактозы, маннитола, сорбита, сахарозы, прессованного сахара и сахарной пудры, и в частности может быть лактозой. Лигандом может быть один или более из метилцеллюлозы, гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы, поливинилпирролидона, желатина, аравийской камеди, этилцеллюлозы, поливинилового спирта, пуллулана, прежелатинированного амида, агара, траганта, производного альгиновой кислоты и производного пропиленгликоля, и альгината, и в особенности может быть поливинилпирролидоном. Дезинтегратором может быть одно или более из веществ с низкой молекулярной массой, выбранных из замещенной гидроксипропилцеллюлозы, карбокиметилцеллюлозы, карбоксиметил-кальций целлюлозы, карбоксиметил-натрий целлюлозы, кроскармеллозы натрия, амида, кристаллической целлюлозы, гидроксипропиламида и частично прежелатинированного амида.
Фармацевтические композиции настоящего изобретения можно приготовить способами, известными в данной области техники.
Должно быть понятно, что примеры и варианты, описанные здесь, представлены только для иллюстрации и что разные модификации и изменения могут быть предложены специалистами в данной области и должны быть включены в объем данного описания и объем последующей формулы. Все публикации, патенты и патентные заявки, размещенные здесь, включены полностью в виде ссылки и во всех отношениях.
Соединения изобретения предпочтительно готовят химической модификацией с использованием соответствующего катализатора. Далее представлена общая методика, которую можно использовать для получения пролекарств, содержащих первый компонент, соответствующий НПВП, и второй компонент, соответствующий таурину, при этом первый и второй компоненты связаны напрямую или с помощью промежуточного агента через амидную связь. Следовательно, описанную здесь общую методику можно использовать для приготовления любого из 70 предпочтительных соединений настоящего изобретения, представленных в Таблице 1.
Общая методика получения амидов (производных таурина) соглано настоящему изобретению
Эквивалентное количество кислотной смеси (НПВП) растворяли в DMF, предварительно высушенной над молекулярным ситовым осушителем. Последовательно добавляли ледяную ванну, эквивалентное количество 1,2-диэтилцианофосфоната (DEPC), 2 эквивалента таурина и 11 эквивалентов триэтиламина, предварительно высушенного над молекулярным ситовым осушителем. Реакцию проводили в течение 2 часов, перемешивая при комнатной температуре.
В конце реакции избыток основания удаляли продуванием азота, и оставшийся растворитель удаляли выпариванием при пониженном давлении. Полученный остаток добавляли маленькими частями в насыщенный водный холодный раствор NaHCO3. Образовавшийся осадок собирали фильтрацией, отмывали с помощью небольшого количества холодной воды и высушивали над пентоксидом фосфора. Полученную сухую массу растирали с THF с образованием твердого остатка, который фильтровали высушивали.
Пример 1: Синтез [2-(4-изобутилфенил)пропаноил1амид этансульфоновой кислоты (Соединение 27) (Соединение получали из ибупрофена и таурина)
Один грамм ибупрофена растворяли в DMF, предварительно высушенном над молекулярным ситовым осушителем. Последовательно добавляли ледяную ванну, 0.9 мл диэтилцианофосфоната (DEPC), 1.212 г таурина и 7.8 мл триэтиламина, предварительно высушенного над молекулярным ситовым осушителем. Реакцию проводили в течение 2 часов при перемешивании при комнатной температуре.
В конце реакции избыток оснований удаляли пропусканием азота, и оставшийся растворитель удаляли выпариванием при пониженном давлении. Полученный остаток добавляли маленькими частями в насыщенный водный холодный раствор NaHCO3. Образовавшийся осадок собирали фильтрацией, отмывали небольшим количеством холодной воды и высушивали над пентоксидом фосфора. Полученную сухую массу растирали в THF, твердый остаток фильтровали и высушивали, и рассчитывали выход примерно 90% (анализировали высокоскоростной жидкостной хроматографией - ВЭЖХ).
Структурное подтверждение очищенного продукта, полученное в результате исследования, представлено результатами в Таблице 2.

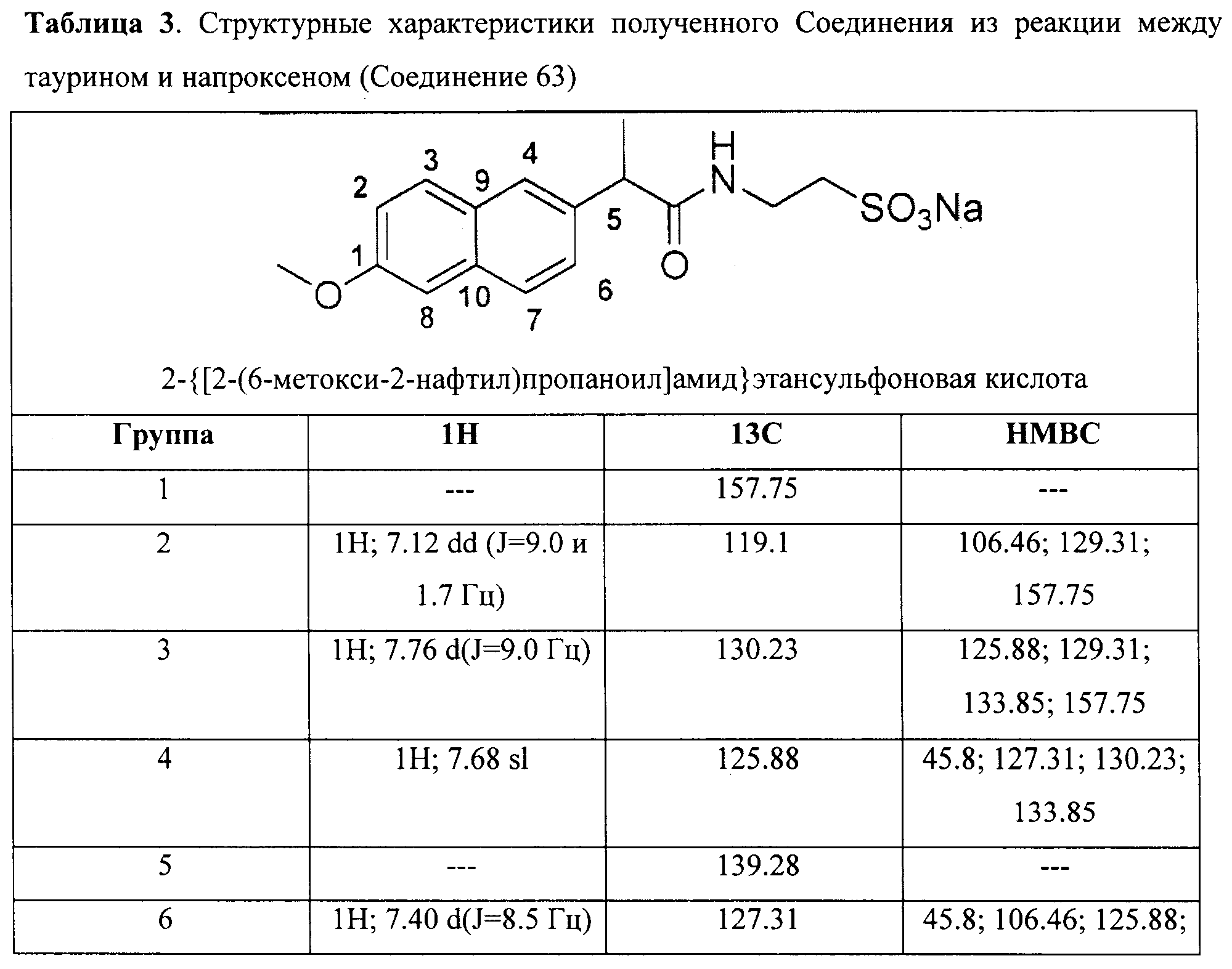
Пример 2: Синтез 2-{[2-(6-метокси-2-нафтил)пропаноил1амид}этансульфоновой кислоты (Соединение 63)
Один грамм напроксена растворяли в DMF, предварительно высушенном над молекулярным ситовым осушителем. Последовательно добавляли ледяную ванну, 0.8 мл диэтилцианофосфоната (DEPC), 1.085 г таурина и 7.0 мл предварительно высушенного над молекулярным ситовым осушителем триэтиламина. Реакцию проводили в течение 2 часов при перемешивании при комнатной температуре.
В конце реакции избыток основания удаляли пропусканием азота, и оставшийся растворитель удаляли выпариванием при пониженном давлении. Полученный остаток добавляли маленькими частями в насыщенный водный холодный раствор NaHCO3. Образовавшийся осадок собирали фильтрацией, отмывали небольшим количеством холодной воды и высушивали над пентоксидом фосфора. Полученную сухую массу растирали в THF, твердый остаток фильтровали и высушивали, и рассчитывали выход примерно 90% (анализировали ВЭЖХ).
Структурное подтверждение очищенного продукта, полученное в результате исследования, представлено результатами в Таблице 3.

Пример 3: Синтез [1-(4-хлорбензоил)-5-метокси-2-метил-1H-индол-3-ил]ацетил]амид}этансульфоновой кислоты (Соединение 64)
Один грамм индометацина растворяли в DMF, предварительно высушенном над молекулярным ситовым осушителем. Последовательно добавляли ледяную ванну, 0.5 мл диэтилцианофосфоната (DEPC), 0.700 г таурина и 4.5 мл предварительно высушенного над молекулярным ситовым осушителем триэтиламина. Реакцию проводили в течение 2 часов при перемешивании при комнатной температуре.
В конце реакции избыток основания удаляли пропусканием азота, и оставшийся растворитель удаляли выпариванием при пониженном давлении. Полученный остаток добавляли маленькими частями в насыщенный водный холодный раствор NaHCO3. Образовавшийся осадок собирали фильтрацией, отмывали небольшим количеством холодной воды и высушивали над пентоксидом фосфора. Полученную сухую массу растирали в THF, твердый остаток фильтровали и высушивали, и рассчитывали выход примерно 90% (анализировали ВЭЖХ).
Структурное подтверждение очищенного продукта, полученное в результате исследования, представлено результатами в Таблице 4.

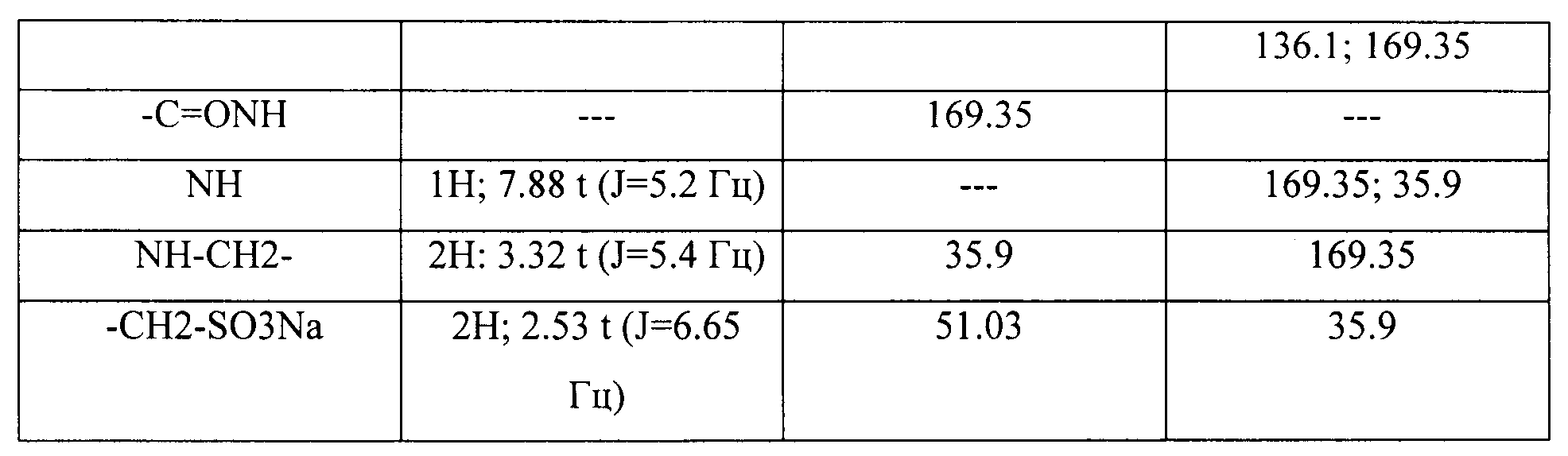
Пример 4: Биологический анализ
Поскольку таурин обладает способностью ингибировать iNOS, присутствующую в макрофагах воспалительных процессов, цель данного исследования состояла в определении, будет ли присоединение противовоспалительного компонента группы НПВП к таурину вызывать изменение этой активности, другими словами, будет ли снижена способность таурина ингибировать iNOS, тем самым аннулируя его противовоспалительную активность.
В анализе использовали максимальное продуцирование оксида азота (макрофаги, стимулированные LPS), в качестве положительного контроля использовали непрямой метод детектирования нитрита (NO2-), и в отрицательном контроле использовали аминогуанидин, искусственный ферментный субстрат, что позволило наблюдать общее ингибирование продуцирования оксида азота.
Результаты показали, что нестероидные противовоспалительные (НПВП) лекарственные препараты не проявляют NOS ингибирующей активности, когда продуцирование NO был таким же, как наблюдаемое в положительном контроле (LPS).
Результаты, полученные с использованием соединений настоящего изобретения, показали, что они были активными в продуцировании NO, аналогично таурину, что указывало на то, что присоединение НПВП к таурину не изменило эту активность.
Данные результаты предполагают, что производные таурина настоящего изобретения могут подвергаться гидролизу и таурин высвобождается на протяжении эксперимента (24 часа), а также проявляет активность per se, таким образом, не являясь пролекарствами, а аналогами (гибридами).
Для доказательства того, что снижение продуцирования NO не было вызвано гибелью клеток, выполняли тестирование клеточной жизнеспособности с использованием соединений изобретения, что являлось, таким образом, возможным доказательством достоверности предыдущего эксперимента.
Таким образом, выполняли in vivo тесты, описанные ниже, и после образования отека лапы вводили следующие соединения: C16H21NO5S (пример 2); C21H21C1N2O6S (пример 3); C15H23NO4S (пример 1), которые показали наличие противовоспалительной активности при дозах 44 мг/кг; 130 мг/кг и 182 мг/кг соответственно.
Эксперименты выполняли согласно экспериментам с эквимолярными количествами продуктов молекулярного модифицирования, описанных BANDARAGE et al. (BANDARAGE et al. "Nitrosothiol esters of diclofenac: Synthesis and pharmacological characterization as gastrointestinal-sparing prodrugs", Journal of Medicinal Chemistry, v.43, p.4005-16, 2000); BANOGLU, et al. (BANOGLU, et al. "Amide derivatives of [6-(5-Methil-3-phenylpyrarole-l-yl)-3-(2H)-pyridazinone-2-yl]acetic acids as potential analgesic and anti-inflammatory compounds", Archives of the Pharmacy and Pharmaceutical Medicinal Chemistry, v.337, p.7-14, 2004); RANATUNG, et al. (RANATUNG, et al. "Synthesis and anti-inflammatory activity of series of N-substituted naproxen glycolamides: Nitric oxide-donor naproxen prodrugs", Bioorganic and Medicinal Chemistry, v.l4, p.2589-99, 2006); OZTURK, G. et al. (OZTURK, G. et al. "New analgesic and anti-inflammatory agents 4(1H)-pyridinone derivatives", Europe Journal Medicinal Chemistry, v.37, n.10 p.829-34, 2002); LOLLI, et al. (LOLLI, et al. "A new class of ibuprofen derivatives with reduced gastrotoxicity". Journal of Medicinal Chemistry, v.44, p.3463-68, 2001). Помимо этого, тест на отек лапы проводили с использованием таурина в качестве контроля его производных соединений в дозе 10 мг/кг согласно HIRATA, Т. et al. (HIRATA, Т. et al. "Cyclo-oxygenase isozymes in mucosal ulcergenic and functional responses following barrier disruption in rat stomachs", British Journal of Pharmacology, v.122, p.447-54, 1997).
Краткое содержание эксперимента
Для подтверждения биологической активности соединений настоящего изобретения проводили тесты согласно фармакологической модели отека лапы у крыс линии Вистар с использованием группы из шести животных. Было установлено, что в указанных концентрациях в присутствии эквимолярных доз соединений, описанных в вышеупомянутых статьях, например, рекомендуемых препаратов, наблюдали уменьшение воспалительного процесса при использовании соединений изобретения.
Соединения настоящего изобретения вводили за 1 (один) час до введения раздражающего агента каррагенина в лапы животных с помощью зонда орально с использованием воды в качестве растворителя. Последующую воспалительную и противовоспалительную активность соединений изобретения оценивали измерением толщины в миллиметрах лапы крыс.
Контрольной группе вводили раздражающий агент каррагенин в нижнюю часть задних лап и орально солевой раствор. Таурин, ибупрофен, напроксен и индометацин вводили орально другим группам животных (положительный контроль) за 60 минут до каррагенина (нижняя часть лапы). Другим группам животных вводили производное таурина настоящего изобретения (варианты соответствуют Примерам 1, 2 и 3, соответственно-орально) за 60 минут до введения каррагенина (нижняя часть лапы).
Задние лапы измеряли до обработки и через каждый час в течение 6 часов после введения каррагенина с помощью измерителя толщины для измерения их объема (в мм). Результаты выражали как разность измерения лап до и после обработки.
Как показано на Фигурах 1-4, через 6 часов после введения соединения изобретения они вызывали статистически одинаковую противовоспалительную активность по сравнению с соответствующими оригинальными лекарственными препаратами.
Пример 5: Острая токсичность (Однократная доза) (LD50)
Летальная доза 50 различается в зависимости от типа используемого НПВП. Для производного ибупрофена (соединение 27) LD50 составляет 1,050 мг/кг; для производного напроксена (соединение 63) LD50 составляет 1,234 мг/кг (см. MERK INDEX, 2006-14°ed.). На основании этих данных эксперименты по определению острой токсичности соединений изобретения выполняли в дозах 1,000 мг/кг и 1,500 мг/кг.
В экспериментах по введению доз 1,000 мг/кг каждого из соединений изобретения, 27 (продукт синтеза таурина с ибупрофеном), 63 (продукт синтеза таурина с напроксеном) и 64 (продукт синтеза таурина с индометацином) (соответствуют вариантам примеров 1, 2 и 3) было установлено, что: (i) в группе, которой вводили соединение 27, смертельные случаи отсутствовали при любой из тестируемых доз, представляя величины выше величин, описанных для ибупрофена, в анализе токсичности при оральном введении (LD50=1,050 мг/кг); (ii) в группе, которой вводили соединение 63 в дозе 1,000 мг/кг, все животные выжили, и при дозе 1,5000 мг/кг было 15% смертельных случаев, что лучше данных, имеющихся в литературе для напроксена (LD50=1,234 мг/кг); и (iii) в группе, которой вводили соединение 64 в дозе 1,000 мг/кг, ни одно животное не умерло и при дозе 1,5000 мг/кг 66% популяции выжило при анализе токсичности.
Краткое содержание эксперимента:
Использовали крысиных самцов линии Вистар весом между 200 и 250 г. Контрольной группе вводили только солевой раствор. Всем изучаемым группам через зонд орально вводили дозы 1,000 или 1,500 мг/кг.
Через четырнадцать (14) дней введения и наблюдения на признаки токсичности общего вида, влияние на движение, поведение, дыхание, число смертей и форм проявления, выживших животных подвергали эйтаназии в CO2 и удаляли их органы, такие как сердце, легкие, почки, печень и желудок, и взвешивали. Для анализа результатов также учитывали массу тела.
Различие массы органов (почки, сердце и печень) тестируемых животных для трех соединений изобретения показано на Фигурах с 4 по 9, что позволяет отметить, что результаты по массе при введении соединений 27, 63 и 64 изобретения в основном близки к результатам, полученным у контрольных животных.
Пример 6: Ульцерогенез желудка
Ульцерогенез желудка устанавливали у тех же групп животных, которых использовали для модели отека лапы.
Через 6 часов после измерения лап, животных подвергали эйтаназии в CO2 и удаляли их желудок, рассекали по продольной оси и отмывали солевым раствором. Во всех экспериментах группы состояли из 6 животных, сохраняя требуемую терапевтическую активность, с отсутствием повреждений желудка, и после анализов LD50 было сделано заключение, что производные соединения таурина настоящего изобретения являются безопасными. Более того, наблюдали, что другие органы, такие как легкие и кишечник, а также макроскопическая целостность других органов были сохранены.
Краткое содержание эксперимента
При облучении слизистой оболочки наблюдали ее цвет и целостность. В случае наличия повреждений, их подсчитывали и измеряли согласно индексу ульцерогенеза желудка (G.U.I.), который соответствует численному критерию для классификации повреждений слизистой оболочки желудка: (повреждения <1 мм = 1; от 1.5 до 2.5 мм = 2; от 2.5 до 3.5 мм = 3; от 3.5 до 4.5 мм = 4 и >4.5 мм = 5). Полученные результаты представлены как средние величины ± Е.Р.М. Результаты эксперимента на повреждение в отношении введения соединений настоящего изобретения (варианты соответствуют Примерам 1, 2 и 3) не были ни макроскопическими, ни микроскопическими (640 X), представленными как индекс повреждения 0 (нулевое) значение. Кроме того, при введении этих соединений изобретения не наблюдали изменений в слизистой оболочке. Фигура 10 показывает этот удивительный результат отсутствия повреждения желудка в противовоспалительной активности соединений изобретения.
Все соединения НПВП показали максимальный индекс повреждения с установленной величиной 5 (пять), формируя повреждения с геморрагическими пятнами. Более того наблюдали, что для групп животных, которым вводили ибупрофен, их желудки показали изменение цвета слизистой оболочки в отличие от их соответствующего производного соединения (соединение 27), которое не показало никакого изменения цвета в слизистой оболочке, сохранив ее целостность.
Во всех сравнениях уменьшение области повреждения сопровождалось отсутствием изменения слизистой оболочки желудка или геморрагических пятен.
Без намерений объяснить причину отличных результатов молекулярных модификаций в противовоспалительных препаратах при введении переносчика, например, полученного с помощью соединений настоящего изобретения, преимущества, проявленные ими, могут быть связаны, что касается тестов ульцерогенеза, с дифференциальным и отличным профилем высвобождения или per su активностью, как показано на Фигуре 10.
Интересно наблюдать, что в первый час эксперимента, в тесте отека лапы, противовоспалительная активность соединений изобретения хуже по сравнению с оригинальными препаратами, но эта тенденция полностью меняется, как показано на Фигурах 1-3. Хотя смысл этих результатов не может ограничиваться теоретическим объяснением, можно сказать, что процесс химической модификации должен объяснить это поведение соединений настоящего изобретения, то есть, умеренную активность вначале (после введения), с получением плохих показателей противовоспалительной активности, и в конце отклики, аналогичные соответствующим оригинальным препаратам с преимуществом сильного уменьшения повреждений желудка. Однако, как видно для аспирина, они также могут проявлять активность как структурные аналоги и включая их метаболиты.
Все результаты были рассмотрены на изменение гомогенности (тест Левене для подтверждения гомогенности). Результаты с незначительным р (выше 0.05) были далее подвергнуты анализу отклонения (ANOVA), с последующими сравнениями (анализ post hoc) средних групповых величин (тест Newman-Keuls'); и рассматривались только величины р, если они были равны или ниже 0.05.
Все публикации и патентные заявки, упомянутые в описании, являются показателями уровня тех специалистов, к которым относится изобретение. Все публикации и патентные заявки включены здесь виде ссылок в том же масштабе, как если бы каждая отдельная публикация или каждая патентная заявка были специально и индивидуально включены в виде ссылки.
Даже если настоящее изобретение было описано в некоторых деталях с помощью иллюстрации и примеров для объяснения и понимания целей, будет очевидно, что определенные изменения и модификации могут быть выполнены в пределах объема формулы, которая прилагается к данному описанию.
Реферат
Настоящее изобретение относится к новым соединениям, полученным из таурина, представляющим Формулу (I):в которой R выбирают из 2-(6-метокси-2-нафтил)пропановой кислоты, [1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-ил] уксусной кислоты и их солей. Изобретение также относится к способу получения соединений Формулы (I) с помощью взаимодействия таурина с соединением, принадлежащим к группе нестероидных противовоспалительных препаратов (НПВП), включающей 2-(6-метокси-2-нафтил)пропановую кислоту, [1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-ил] уксусную кислоту, в присутствии катализатора в соответствующей органической среде, и к противовоспалительным фармацевтическим композициям, содержащим, по меньшей мере, одно соединение формулы 1, обладающее нестероидной противовоспалительной активностью. 3 н. и 5 з.п. ф-лы, 4 табл., 10 ил., 6 пр.
Формула

где R выбирается из 2-(6-метокси-2-нафтил)пропановой кислоты, [1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-ил]уксусной кислоты и их солей.
2-{[2-(6-метокси-2-нафтил)пропаноил]амид}этансульфоновой кислоты, [1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-ил]ацетил]амид} этансульфоновой кислоты и их солей.
(a) фармацевтически эффективное количество, по меньшей мере, одного соединения, полученного из таурина, по п.1; и
(b) фармацевтически приемлемый носитель.
Комментарии