Производные 4-замещенной феноксифенилуксусной кислоты - RU2463292C2
Код документа: RU2463292C2
Описание
Настоящее изобретение относится к новым соединениям, к содержащим данные соединения фармацевтическим композициям, к способу получения указанных соединений и к применению указанных соединений в терапии. Более конкретно, настоящая заявка относится к некоторым производным 4-замещенной феноксифенилуксусной кислоты, которые могут использоваться в лечении и профилактике аллергических заболеваний, таких как астма, аллергический ринит и атопический дерматит, и других воспалительных заболеваний, опосредованных простагландином D2 (PGD2).
Международная патентная заявка WO 2004/058164, раскрывает, среди прочего, некоторые производные 2-замещенной феноксифенилуксусной кислоты, модулирующие PGD2-селективный рецептор CRTH2 (молекула, гомологичная рецептору хемоаттрактанта, экспрессируемая клетками Th2), в настоящее время чаще называемый DP2. Утверждается, что описанные соединения применимы в лечении иммунологических заболеваний, таких как астма и аллергическое воспаление.
Теперь было обнаружено, что некоторые производные 4-замещенной феноксифенилуксусной кислоты, содержащие определенный заместитель в метаположении по отношению к остатку уксусной кислоты, представляют собой модуляторы DP2 рецептора. При использовании в данном тексте термин "модулятор" включает антагонисты.
Согласно первому аспекту, настоящее изобретение описывает соединение общей формулы (I):

или его соль, где:
R1 представляет собой Ar1-L1-W-L2-;
L2 представляет собой -(CRcRd)m-;
W представляет собой -CONR3a, или -NR3bCO-;
каждый из R3a и R3b представляет собой Н или метил;
L1 представляет собой -(CRaRb)n-, -(CH=CH)-, или -O(CRaRb), при условии, что когда W представляет собой -NR3CO-, тогда L1 не является фрагментом -(СН=СН)-;
n и m независимо равны 0,1 или 2;
каждый из Ra, Rb, Rc и Rd независимо представляет собой Н, F, ОН, метил или циклопропил, или Ra и Rb или Rc и Rd вместе с атомом углерода, к которому они присоединены, образуют циклопропильное кольцо;
Ar1 представляет собой фенил или нафтил, каждый из которых является незамещенным или замещенным одним или более заместителями, независимо выбранными из F, Cl, CN, CF3, CHF2, CH2F, SF5, метила, этила, циклопропила, т-бутила или ОМе, или Ar1 представляет собой 1,2,3,4-тетрагидронафтил, который является незамещенным или замещенным метокси-группой,
при условии, что когда Ar1 представляет собой нафтил или 1,2,3,4-тетрагидронафтил, тогда n равен 0;
R2 представляет собой Н, C1-С6алкил, остаток аминокислоты или дипептида, или CHRe(CH2)qRf;
q равен 1-6;
Re представляет собой Н, метил или этил;
Rf представляет собой NRgRh, где каждый из Rg и Rh независимо представляет собой атом водорода или C1-C4 алкильную группу, или Rg и Rh вместе с атомом азота, к которому они присоединены, образуют 5-6 членное гетероциклическое кольцо, необязательно содержащее в кольце второй гетероатом, выбранный из N и О, где указанное гетероциклическое кольцо необязательно замещено одной или более группами, независимо выбранными из C1-С6 алкила;
А представляет собой CN, CH2NH2, CH2NR4aC(=O)R5, или CH2NR4bSO2R6, Cl, ОМе, (С1-С4) алкил, циклопропил, Н, F, Br, CH2NH(C1-C4алкил), CH2N(C1-C4 алкил)2, тиенил или фенил, который является незамещенным или замещенным группой SO2Me;
каждый из R4a и R4b представляет собой Н или метил;
R5 представляет собой C1-С6 алкил, C1-С6 алкокси, С3-С6 циклоалкил, hetAr1 или Ar2;
R6 представляет собой C1-С6 алкил, NH(C1-C6 алкил), N(C1-C6 алкил)2, Ar3, или hetAr2;
hetAr1 представляет собой 6-членный гетероарил, который является незамещенным или замещенным одной или более группами, независимо выбранными из атома галогена и группы формулы -NR5aR5b, в которой каждый из R5a и R5b независимо представляет собой атом водорода или (C1-C4) алкильную группу, или вместе с атомом азота, к которому они присоединены, образуют пирролидинильную, пиперидинильную или морфолино группу;
hetAr2 представляет собой 5-6 членный гетероарил, который является незамещенным или замещенным одной или более группами, независимо выбранными из C1-C4 алкила;
Ar2 представляет собой фенил, который является незамещенным или замещенным одной или более группами, независимо выбранными из атома галогена, CN, SF5, циклопропила, C1-C4 алкильной группы, C1-C4 алкокси группы и фтор С1-С4 алкильной группы;
Ar3 такой, как описано для Ar2;
R7 и R8 независимо представляют собой Н, метил или F;
R9 представляет собой Н или метил; и
R10 представляет собой Н или F.
В некоторых вариантах осуществления, соединение формулы I имеет формулу Iа

где:
R1 представляет собой Ar1-L1-W-L2-;
L2 представляет собой -(CRcRd)m-;
W представляет собой -CONR3a- или -NR3bCO-;
каждый из R3a и R3b представляет собой Н или метил;
L1 представляет собой -(CRaRb)n, -(CH=CH)- или -O(CRaRb), при условии, что когда W представляет собой -NR3CO-, тогда L1 не является фрагментом -(СН=СН)-;
n и m независимо равны 0,1 или 2;
каждый из Ra, Rb, Rc и Rd независимо представляет собой Н, F, метил или циклопропил, или Ra и Rb или Rc и Rd вместе с атомом углерода, к которому они присоединены, образуют циклопропильное кольцо;
Ar1 представляет собой фенил или нафтил, каждый из которых является незамещенным или замещенным одним или более заместителями, выбранными независимо из F, Cl, CN, CF3, CHF2, CH2F, SF5, метила, этила, циклопропила, при условии, что когда Ar1 представляет собой нафтил, тогда b равен 0;
R2 представляет собой Н, C1-С6 алкил, остаток аминокислоты или дипептида, или CHRe(CH2)qRf;
q равен 1-6;
Re представляет собой Н, метил или этил;
Rf представляет собой NRgRh, где каждый из Rg и Rh независимо представляет собой атом водорода или C1-C4 алкильную группу, или Rg и Rh вместе с атомом азота, к которому они присоединены, образуют 5-6 членное гетероциклическое кольцо, необязательно содержащее в кольце второй гетероатом, выбранный из N и О, где указанное гетероциклическое кольцо необязательно замещено одной или более группами, независимо выбранными из C1-С6 алкила;
А представляет собой CN, CH2NH2, CH2NR4aC(=O)R5, или CH2NR4bSO2R6, Cl, OMe, (С1-С4) алкил, циклопропил;
каждый из R4a и R4b представляет собой Н или метил;
R5 представляет собой C1-С6 алкил, C1-С6 алкокси, С3-С6 циклоалкил, hetAr1 или Ar2;
R6 представляет собой C1-С6 алкил, NH(C1-C6 алкил), N(C1-C6 алкил)2, Ar3 или hetAr2;
hetAr1 представляет собой 6-членный гетероарил, который является незамещенным или замещенным одной или более группами, независимо выбранными из атома галогена и группы формулы -NR5aR5b, в которой каждый из R5a и R5b независимо представляет собой атом водорода или (C1-C4) алкильную группу, или вместе с атомом азота, к которому они присоединены, образуют пирролидинильную, пиперидинильную или морфолино группу;
hetAr2 представляет собой 5-6 членный гетероарил, который является незамещенным или замещенным одной или более группами, независимо выбранными из C1-C4 алкила;
Ar2 представляет собой фенил, который является незамещенным или замещен одной или более группами, независимо выбранными из атома галогена, CN, SF5, циклопропила, C1-C4 алкильной группы, C1-C4 алкокси группы и фторС1-С4 алкильной группы;
Ar3 такой, как описано для Ar2; и
R7 и R8 независимо представляют собой Н, метил.
Обнаружено, что соединения по настоящему изобретению являются DP2 модуляторами и пригодны для лечения иммунологических заболеваний, таких как астма и аллергическое воспаление.
Следует принять во внимание, что некоторые соединения по изобретению могут содержать один или более асимметрических центров, и поэтому они могут быть получены и выделены в виде смеси изомеров, такой как рацемическая смесь, или в энантиомерно чистой форме.
Также следует принять во внимание, что соединения формулы (I) или их соли могут быть выделены в форме сольватов, и, соответственно, что любой такой сольват включен в объем настоящего изобретения.
Соединения формулы I включают их фармацевтически приемлемые соли. Кроме того, соединения формулы I также включают другие соли таких соединений, которые необязательно представляют собой фармацевтически приемлемые соли, и которые можно использовать в качестве интермедиатов для получения и/или очистки соединений формулы I и/или для разделения энантиомеров соединений формулы I.
Термин "галоген", при использовании в данном тексте, включает F, Cl, Br и I.
Термины "С1-С4алкил" и "C1-С6алкил", при использовании в данном тексте, относятся к насыщенному одновалентному углеводородному радикалу с прямой или разветвленной цепью, содержащему от одного до четырех или от одного до шести атомов углерода, соответственно. Примеры алкильных групп включают, но не ограничены только ими, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-метил-1-пропил, 2-бутил, 2-метил-2-пропил, 2,2-диметилпропил, 1-пентил, 2-пентил, 3-пентил, 2-метил-2-бутил, 3-метил-2-бутил, 3-метил-1-бутил, 2-метил-1-бутил, 1-гексил, 2-гексил, 3-гексил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 3-метил-3-пентил, 2-метил-3-пентил, 2,3-диметил-2-бутил и 3,3-диметил-2-бутил.
Термин "гетероарил", при использовании в данном тексте, относится к одновалентному ароматическому радикалу 5-, 6-, или 7-членного кольца. Примеры гетероарильных групп включают, но не ограничены только ими, пиридинил, имидазолил, имидазопиридинил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил и пирролил.
Термин "фтор-С1-С4 алкил", при использовании в данном тексте, относится к С1-С4алкильной группе, в которой один или более атомов водорода замещен атомом фтора. Примеры включают CF3, CH2F, CHF2, CH2CH2F, CH2CHF2, CH2CF3, CH2CH2CH2F, CH2CH2CHF2, CH2CH2CF3, CF(CH3)2, CH2CF(CH3)2 и тому подобные.
Термин "С1-С6алкокси", при использовании в данном тексте, относится к C1-С6алкилокси группе. Примеры алкокси групп включают метокси, этокси, пропокси, изопропокси, бутокси, т-бутокси и изобутокси.
В одном варианте осуществления W представляет собой -CONR3a-. Примером частного значения R3a является водород. В одном варианте осуществления W представляет собой -NR3bCO-. В одном варианте осуществления R3b представляет собой водород. В другом варианте осуществления R представляет собой метил. Примерами частных значений W являются CONH, NHCO и N(СН3)СО.
В одном варианте осуществления L1 представляет собой -(CRaRb)n-. Примерами частных значений n являются 0, 1 и 2.
В одном варианте осуществления L1 представляет собой химическую связь.
В одном варианте осуществления L1 представляет собой -(CRaRb). В некоторых вариантах осуществления, Ra и Rb представляют собой водород. В некоторых вариантах осуществления, Ra представляет собой ОН. В некоторых вариантах осуществления, Ra и Rb вместе с атомом углерода, к которому они присоединены, образуют циклопропилидиновое кольцо.
В одном варианте осуществления L1 представляет собой -(CRaRb)2. В некоторых вариантах осуществления, Ra и Rb представляют собой водород. В некоторых вариантах осуществления, Ra и Rb вместе с атомом углерода, к которому они присоединены, образуют циклопропилидиновое кольцо. В некоторых вариантах осуществления, Ra и Rb присоединены к одному и тому же атому углерода. В других вариантах осуществления, Ra и Rb присоединены к разным атомам углерода.
Примерами частных значений L1 являются химическая связь, -СН2-, -CH2CH2- и циклопропилиден СН2.
Другой пример L1 включает СН(ОН)СН2.
Другие примеры L1 включают циклопропилидиновые группы, которые могут быть представлены следующими структурами:

В одном варианте осуществления L представляет собой -O(CRaR)b-. Примером варианта осуществления является -ОСН2-.
Касательно L2, примерами частных значений m являются 0 и 1. Примерами частных значений L2 являются химическая связь и -СН2-.
В некоторых вариантах осуществления сумма тип равна 0, 1 и 2. Особо следует отметить соединения, в которых сумма тип равна 0 или 2.
Примеры значений -L1-W-L2- включают -CONH-, -CH2CONH-, -CH2CH2CONH-, -CONHCH2-, -CH2CONHCH2-, -NHCO-, -CH2NHCO-, -NHCOCH2-, -CH2CH2NHCO-, -CH2NHCOCH2-, -CH2CH2NHCOCH2-, -CH2N(СН3)СОСН2-, циклопропилиден СН2NНСО и -CH2ONHCO-.
Другие примеры значений -L1-W-L2- включают -CH(OH)CH2NHCO- и -циклопропилидинNНСО-.
Частными значениями -L1-W-L2- являются -CONH-, -NHCO-, -CH2NHCO-, -NHCOCH2-, -CH2CH2NHCO-, -CH2NHCOCH2-, -CH2CH2NHCOCH2-, -CH2N(CH3)COCH2-, циклопропилиден СН2NHСО, -CH(OH)CH2NHCO- и -циклопропилидин NHCO-.
В одном варианте осуществления Ar1 представляет собой нафтильную группу или фенильную группу, которая замещена одним или двумя заместителями, независимо выбранными из F, ClСF3, ОМе, Me и т-Bu.
В одном варианте осуществления Ar1 представляет собой нафтильную группу или фенильную группу, которая является незамещенной или замещена одним или двумя заместителями, независимо выбранными из F, Cl и CF3.
В одном варианте осуществления Ar1 представляет собой нафтильную группу или фенильную группу, которая замещена одним или двумя заместителями, независимо выбранными из ОМе, Me и т-Bu.
В одном варианте осуществления Ar1 представляет собой 1,2,3,4-тетрагидронафтил, который является незамещенным или замещен ОМе. В частном варианте осуществления, А выбран из структур:

Примерами частных значений Ar1 являются нафтил, фенил, 4-фторфенил, 3,4-дифторфенил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил, 3,4-дихлорфенил, 4-хлор-3-фторфенил, 3-хлор-4-фторфенил, 4-трифторметилфенил, 3-фтор-4-трифторметилфенил, 3-трифторметилфенил, 2,6-дихлорфенил, 2,4-дихлорфенил, 3-метоксифенил, 4-метоксифенил, 4-третбутилфенил, 3-фторфенил, 4-метилфенил, 1,2,3,4-тетрагидронафт-2-ил и 6-метокси-1,2,3,4-тетрагидронафт-2-ил.
В некоторых вариантах осуществления Ar1 выбран из нафтила, фенила, 4-фторфенила, 3,4-дифторфенила, 2-хлорфенила, 3-хлорфенила, 4-хлорфенила, 3,4-дихлорфенила, 4-хлор-3-фторфенила, 3-хлор-4-фторфенила, 4-трифторметилфенила и 3-фтор-4-трифторметилфенила.
В некоторых вариантах осуществления Ar1 выбран из 3-трифторметилфенила, 2,6-дихлорфенила, 2,4-дихлорфенила, 3-метоксифенила, 4-метоксифенила, 4-третбутилфенила, 3-фторфенила и 4-метилфенила и 1,2,3,4-тетрагидронафт-2-ила и 6-метокси-1,2,3,4-тетрагидронафт-2-ила.
В одном варианте осуществления А представляет собой CN.
В одном варианте осуществления А представляет собой Н.
В одном варианте осуществления А выбран из F, Br и Cl.
В одном варианте осуществления А выбран из (С1-С4) алкила. Частные примеры включают метил и этил.
В одном варианте осуществления А представляет собой циклопропил.
В одном варианте осуществления, А выбран из CH2NH2, CH2NH(C1-C4 алкил) и CH2N(C1-C4 алкил)2. Частные примеры включают CH2NH2 и CH2NMe2.
В одном варианте осуществления А представляет собой тиенил. В частном варианте осуществления А представляет собой 2-тиенил.
В одном варианте осуществления, А представляет собой фенил, который является незамещенным или замещен группой SO2Me. Частные примеры включают фенил, 3-метилсульфонилфенил и 4-метилсульфонилфенил.
В одном варианте осуществления А представляет собой CH2NR4aC(=O)R5. Примером частного значения R4a является водород. В одном варианте осуществления R5представляет собой hetAr1. Примером частного значения для гетероарильной группы, представленной hetAr1, является пиридильная группа. Примерами необязательных заместителей в гетероарильной группе являются NH2, Cl и пирролидинил.
В другом варианте осуществления R5 представляет собой C1-С6 алкил; C1-С6 алкокси; С3-С6 циклоалкил; пиридил, который является незамещенным или замещенным атомом галогена или группой формулы -NR5aR5b, в которой каждый из R5a и R5bнезависимо представляет собой атом водорода или (C1-C4) алкильную группу, или вместе с атомом азота, к которому они присоединены, образуют пирролидинильную, пиперидинильную или морфолино группу; или фенильную группу, которая является незамещенной или замещена одним или двумя атомами галогена.
Примерами частных значений R5 являются метил, метокси, циклогексил, пирид-2-ил, пирид-3-ил, пирид-4-ил, 6-хлор-пирид-3-ил, 6-амино-пирид-3-ил, 6-пирролидин-1-илпирид-3-ил или 4-фторфенил. Дополнительным примером R5 является 6-диметиламинопирид-3-ил.
В одном варианте осуществления А представляет собой CH2-NR4bSO2R6. Примером частного значения R4b является водород. В одном варианте осуществления, R6представляет собой hetAr2. Примерами частных значений для гетероарильной группы, представленной hetAr2, являются имидазолил и пиридильная группа. Примерами необязательных заместителей в гетероарильной группе являются С1-С4 алкил, например, метил.
В другом варианте осуществления R6 представляет собой C1-С6 алкил, NH(C1-C6 алкил), N(C1-C6 алкил)2, фенильную группу, которая является незамещенной или замещена одним или двумя атомами галогена, пиридил или имидазолил, который является незамещенным или замещен C1-С3 алкильной группой.
Примерами частных значений R6 являются метил, диметиламино, 4-фторфенил, 2,4-дихлорфенил, пирид-3-ил и 1-метилимидазол-5-ил. Дополнительный пример R6представляет собой пирид-4-ил.
Примерами частных значений для А являются ацетамидометил, циклогексиламидометил, метоксикарбониламинометил, пиколинамидометил, никотинамидометил, изоникотинамидометил, 6-хлорпирид-3-иламидометил, 6-аминопирид-3-иламидометил, 6-пирролидин-1-илпирид-3-иламидометил, 4-фторбензамидометил, метилсульфонамидометил, N,N-диметилсульфамоиламино, 4-фторфенилсульфонамидометил, 2,4-дихлорфенилсульфонамидометил, 1-метилимидазол-5-илсульфонамидометил и пирид-3-илсульфонамидометил, которые могут быть представлены следующими структурами, соответственно:












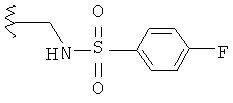



Особенно следует отметить ацетамидометил, циклогексиламидометил, метоксикарбониламинометил, пиколинамидометил, никотинамидометил, изоникотинамидометил, 4-фторбензамидометил, метилсульфонамидометил, N,N-диметилсульфамоиламино, 4-фторфенилсульфонамидометил, 2,4-дихлорфенилсульфонамидометил и пирид-3-илсульфонамидометил.
Частные значения А также включают 6-диметиламинопирид-3-иламидометил, 2-(4-фторфенилсульфонамидо)ацетамидометил, диметиламинометил и (N-метилметилсульфонамидо)метил, которые могут быть представлены следующими структурами, соответственно:




Примерами частных значений R2, когда он представляет собой C1-С6алкильную группу, являются метил, этил, пропил, изопропил и т-бутил.
В одном варианте осуществления R2 представляет собой CHRe(CH2)qRf. Примерами значений R6 являются водород и метил. В одном варианте осуществления Rfпредставляет собой ди (С1-С4) алкиламино, морфолино или пиперазинил, необязательно замещенные (С1-С4) алкилом. Примерами частных значений Rf являются диметиламино, диэтиламино, морфолино, пиперазинил и 1-метилпиперазинил. Дополнительные примеры включают NH2 и NHMe.
Примерами частных значений R2, когда он представляет собой CHRe(CH2)qRf, являются:







В одном варианте осуществления R2 представляет собой водород.
В одном варианте осуществления R7 и R8 представляют собой Н. В некоторых вариантах осуществления, R7 представляет собой Н, и R8 представляет собой метил. В других вариантах осуществления каждый из R7 и R8 представляет собой метил.
Согласно другому аспекту настоящее изобретение описывает способ получения соединения формулы (I) или его соли, как описано в данном тексте выше, который включает:
(а) для соединения формулы (I), в которой А представляет собой CN, R7 и R8 независимо представляют собой Н или Me, и R10 представляет собой Н или F, взаимодействие соответствующего соединения формулы (II):

в которой R1 представляет собой атом водорода или защитную группу для карбоксила, и Z1 представляет собой уходящий атом или группу, с соответствующим соединением формулы (III):

в которой R10a представляет собой Н или F, в присутствии основания; или
(b) для соединения формулы (I), в которой А представляет собой -CH2NH2, R10представляет собой Н, и R7 и R8 независимо представляют собой Н или Me, восстановление соответствующего соединения формулы (IV)

в которой Р2 такой, как описано для P1; или
(с) для соединения формулы (I), в которой А представляет собой -CH2NH2, R7 и R8 независимо представляют собой Н или Me, и R10 представляет собой Н, расщепление соответствующего соединения формулы (V)
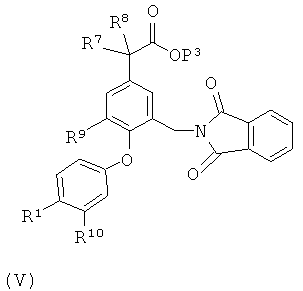
в которой Р3 такой, как описано для Р1; или
(d) для соединения формулы (I), в которой А представляет собой CH2NR4C(=O)R5или CH2NR4SO2R6, R7 и R8 независимо представляют собой Н или Me, и R10 представляет собой Н, взаимодействие соответствующего соединения формулы (VI)

в которой Р4 такой, как описано для Р1; с соединением формулы R5COZ2 или R6SO2Z3, соответственно, в которых каждый из Z2 и Z3 представляет собой уходящий атом или группу; или
(е) для соединения формулы (I), в которой R7 и R8 независимо представляют собой Н или Me, и R10 представляет собой Н, сочетание соединения формулы (VII)

в которой Р5 такой, как описано для Р1, А2 представляет собой А или его защищенную форму, и R1a представляет собой H-Xa-L2-, где Xa представляет собой HN или ОС(=O), или его реакционноспособного производного; с соединением формулы (VIII)

в которой Xb представляет собой С(=O)O или NH, или его реакционноспособным производным; или
(f) для соединения формулы (I), в которой А представляет собой Н, F или Cl, R7 и R8 независимо представляют собой Н или Me, и R10 представляет собой Н, сочетание соответствующего соединения формулы (IX)

в которой А3 представляет собой Н, F или Cl, и Р6 такой, как описано для Р1, с соответствующим соединением формулы (X)

где Е представляет собой электроноакцепторную группу, в присутствии основания; и, при необходимости, удаление указанной электроноакцепторной группы; или
(g) для соединения формулы (I), в которой А представляет собой ОМе или (С1-С4)алкил, R7 и R8 независимо представляют собой Н или Me, и R10 представляет собой Н, сочетание соответствующего соединения формулы (XI)
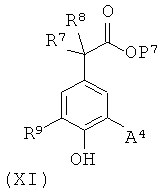
в которой А4 представляет собой ОМе или (С1-С4) алкил, соответственно, и Р7такой, как описано для Р1, с соответствующим соединением формулы (XII)

в присутствии основания, где Z4 представляет собой уходящий атом или группу, и R1x представляет собой электроноакцепторную группу, которую можно превратить в группу R1; или
(h) для соединения формулы (I), в которой А представляет собой Br или циклопропил, R7 и R8 представляют собой Н, и R10 представляет собой Н, сочетание соответствующего соединения формулы (XIV)
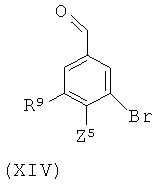
в которой Z5 представляет собой уходящую группу или атом, с соединением формулы (XV)

в присутствии основания, с последующим превращением карбонильной группы в карбоксильную группу; или
(i) для соединения формулы (I), в которой А представляет собой метил, тиенил, фенил или фенил, замещенный группой SO2Me, R9 представляет собой Н, R7 и R8независимо представляют собой Н или Me, и R10 представляет собой Н, взаимодействие соответствующего соединения формулы (XVI)

с соединением формулы A5-ZnX, в присутствии палладиевого (0) катализатора, или с соединением формулы A5B(ОН)2 в присутствии основания и палладиевого (0) катализатора, где А5 представляет собой метил, тиенил, фенил или фенил, замещенный группой SO2Me, и Х представляет собой галогенид; или
(j) для соединения формулы (I), в которой R7 представляет собой F, R8 представляет собой Н, и R10 представляет собой Н, обработка соответствующего соединения формулы (XVII)

фтороводородом;
(k) для соединения формулы (I), в которой А представляет собой
CH2NH(C1-C4 алкил) или CH2N(C1-C4 алкил)2, R7 и R8 независимо представляют собой Н или Me, и R10 представляет собой Н, взаимодействие соответствующего соединения формулы

с альдегидом формулы HC(O)(C1-C4 алкил); и удаление любой защитной группы или групп и, при необходимости, образование соли.
Что касается способа (а), уходящий атом или группа, представленная Z1, может представлять собой, например, атом галогена, такой как атом фтора. Защитная группа для карбоксила может представлять собой любую подходящую защитную группу для карбоксила, например, как описано в Greene & Wuts, eds., "Protecting Groups in Organic Synthesis", John Wiley & Sons, Inc. Примеры защитных групп для карбоксила включают (С1-С6)алкильные группы, такие как метил, этил и т-бутил. Основание может представлять собой, например, гидрид или карбонат щелочного металла, такой как гидрид натрия, карбонат натрия или карбонат калия, или третичный амин, такой как триэтиламин или N,N-диизопропилэтиламин. Подходящие растворители включают амиды, сульфоксиды и нитрилы, такие как ДМФА, ДМСО или апетонитрил. Реакцию можно осуществлять при повышенной температуре, такой как температура в диапазоне от 50 до 150°С.
Соединения формулы (II) известны или могут быть получены из соответствующего 3-галоген производного, такого как 3-бром производного, путем обработки CuCN.
Что касается способа (b), соединение формулы (IV) можно восстановить гидрированием в присутствии катализатора, представляющего собой металл VIII группы, такого как никель Ренея, со смесью метанол/аммиак. Реакцию можно осуществлять при температуре в диапазоне от 0 до 100°С.
Что касается способа (с), диоксоизоиндолинильную группу можно расщепить с использованием HBr и уксусной кислоты или гидразина.
Соединения формулы (V) можно получить взаимодействием соединения формулы (XIII)
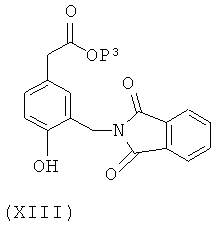
с соединением формулы (XII)

в присутствии основания, где Z4 представляет собой уходящий атом или группу, такой как атом фтора, и R1x представляет собой электроноакцепторную группу, которую можно превратить в группу R1, например нитрогруппу, которую можно восстановить до аминогруппы и затем проацилировать. Основание может представлять собой, например, гидрид или карбонат щелочного металла, такой как гидрид натрия, карбонат натрия или карбонат калия.
Соединения формулы (IX) можно получить взаимодействием 4-гидроксифенилуксусной кислоты с 2-(гидроксиметил)изоиндолин-1,3-дионом в присутствии сульфоновой кислоты, такой как метансульфоновая кислота, после чего при необходимости вводят защитную группу Р3.
Что касается способа (d), уходящий атом или группа, представленные Z2 и Z3, могут представлять собой, например, атом галогена, такой как атом фтора. Реакцию можно осуществлять в присутствии основания, например, третичного амина, такого как диизопропилэтиламин или пиридин. Подходящие растворители включают галогенированные углеводороды, такие как метиленхлорид. Реакцию можно осуществлять при температуре в диапазоне от 0 до 100°С.
Что касается способа (е), сочетание соединения формулы (VII) с соединением формулы (VIII) можно осуществлять при обычных условиях образования амидной связи, например, взаимодействием амина с реакционноспособным производным карбоновой кислоты, например галогенангидридом, таким как хлорангидрид. Примером А1, когда он представляет собой защищенную форму А, является группа формулы -CH2NR4P6, в которой Р6 представляет собой защитную группу для амина. Защитная группа для амина может представлять собой любую подходящую защитную группу для амина, например, как описано в Greene & Wuts, eds., "Protecting Groups in Organic Synthesis", John Wiley & Sons, Inc. Примеры защитных групп для амина включают ацильные и алкоксикарбонильные группы, такие как т-бутоксикарбонил (ВОС).
Что касается способа (f), примеры электроноакцепторных групп включают NO2. В вариантах осуществления, в которых электроноакцепторная группа представляет собой NO2, данную группу можно удалять, при необходимости, восстановлением нитрогруппы до аминогруппы с использованием любых подходящих условий восстановления (например, Zn и NH4Cl), с последующим отщеплением аминогруппы (например, путем обработки аминового соединения изобутилнитритом).
Что касается способа (g), основание может представлять собой, например, гидрид или карбонат щелочного металла, такой как гидрид натрия, карбонат натрия или карбонат калия. Группа Z4 представляет собой подходящий уходящий атом или группу, такой как атом фтора, и R1x представляет собой электроноакцепторную группу, которую можно превратить в группу R1, например, нитрогруппу, которую можно восстановить до аминогруппы и затем проацилировать.
Что касается способа (h), уходящий атом или группа, представленная Z5, может представлять собой, например, атом галогена, такой как атом фтора. Основание может представлять собой, например, гидрид или карбонат щелочного металла, такой как гидрид натрия, карбонат натрия или карбонат калия. Подходящие растворители включают сульфоксиды, такие как ДМСО. Реакцию можно осуществлять при повышенных температурах, например, в диапазоне 50-100°С, например, при 85°С. Карбонильную группу можно превратить в карбоксильную группу обработкой продукта сочетания метилсульфонил/метилтиометаном для получения интермедиата, имеющего формулу (XVIII)

с последующим превращением интермедиата (XVIII) в соответствующий метиловый эфир путем обработки HCl в метаноле. Полученный эфир можно превратить в соответствующую кислоту в стандартных условиях гидролиза
Что касается способа (i), галогенид, представленный X, может представлять собой F, Cl или Br. Подходящие растворители включают простые эфиры, такие как ТГФ или диоксан. Подходящие палладиевые (0) катализаторы включают Pd(PPh3)4 и бис(три-т-бутилфосфин)палладий (0). Подходящее основание, когда соединение (XVI) взаимодействует с бороновой кислотой, включает DBU. Реакцию можно осуществлять при повышенных температурах, например, в диапазоне 50-100°С, например при 60°С.
Что касается способа (j), реакцию удобно проводить в присутствии аминового основания, например пиридина.
Что касается способа (k), реакцию удобно проводить в присутствии основания, например гидрида, такого как цианоборгидрид натрия, в спиртовом растворителе, таком как метанол. Реакцию предпочтительно осуществляют в присутствии каталитического количества кислоты, например уксусной кислоты. Реакцию удобно проводить при комнатной температуре.
Способность тестовых соединений выступать в качестве модуляторов DP2 рецептора можно продемонстрировать с помощью анализа, описанного в примере А.
Соединения, являющиеся модуляторами DP2, пригодны для лечения заболеваний и нарушений, опосредованных PGD2, например заболеваний и нарушений, связанных с сверхсинтезом или нарушением регуляции PGD2.
При использовании в данном тексте, термин «лечение» включает профилактику, а также лечение существующего состояния.
Примеры нарушений и заболеваний, которые можно лечить соединениями по изобретению, включают иммунологические заболевания.
Примеры иммунологических заболеваний включают аллергические воспалительные заболевания, такие как астма, атопический дерматит, аллергический ринит, сезонные аллергии, пищевые аллергии, контактную гиперчувствительность (например, чувствительность к никелю), гиперэозинофилические синдромы и аллергический конъюнктивит.
Другие заболевания и нарушения, которые можно лечить соединениями по настоящему изобретению, включают воспалительные болезни кишечника, такие как болезнь Крона, язвенный колит, илеит и энтерит, васкулит, болезнь Бехчета, псориаз и воспалительные дерматозы, такие как дерматит, экзема, крапивница, вирусные патологии кожи, такие как патологии, вызванные вирусом папилломы человека, ВИЧ или RLV инфекциями, бактериальные, грибковые и другие паразитические патологии кожи, и кожную красную волчанку, аллергические заболевания дыхательных путей, такие как аллергические заболевания легких, хроническое обструктивное заболевание легких и тому подобные, аутоиммунные болезни, такие как артрит (включая ревматоидный и псориатический), системную красную волчанку, диабет I типа, миастению gravis, рассеянный склероз, болезнь Грейвса, гломерулонефрит и тому подобные, отторжение трансплантата (включая отторжение аллотрансплантата и болезнь отторжения трансплантата), например, отторжение кожного трансплантата, отторжение трансплантата солидного органа, отторжение трансплантата костного мозга, лихорадку, сердечно-сосудистые заболевания, такие как острая сердечная недостаточность, гипотония, гипертония, стенокардия, инфаркт миокарда, кардиомиопатия, застойная сердечная недостаточность, атеросклероз, болезнь коронарных артерий, рестеноз, тромбоз и васкулярный стеноз, церебрально-васкулярные нарушения, такие как травматическое повреждение мозга, инсульт, ишемическое реперфузионное повреждение и аневризму, рак молочной железы, кожи, предстательной железы, шейки матки, яичника, яичек, мочевого пузыря, легкого, печени, гортани, ротовой полости, толстой кишки и желудочно-кишечного тракта (например, пищевода, желудка, поджелудочной железы), мозга, щитовидной железы, крови и лимфатической системы, фиброз, болезнь соединительной ткани и саркоидоз, патологические состояния гениталий и репродуктивной системы, такие как эректильная дисфункция, желудочно-кишечные нарушения, такие как гастрит, язвы, тошнота, панкреатит и рвота; неврологические нарушения, такие как болезнь Альцгеймера, нарушения сна, такие как бессонница, нарколепсия, синдром апноэ во время сна и синдром Пиквика, боль, почечные нарушения, офтальмологические нарушения, такие как глаукома, инфекционные заболевания, вирусные инфекции, такие как ВИЧ, и бактериальные инфекции, такие как сепсис, воспаление, гиперемия, заложенность носа и отит среднего уха.
Соответственно, другой аспект настоящего изобретения описывает способ лечения заболеваний или медицинских состояний у млекопитающего, опосредованных PGD2, включающий введение указанному млекопитающему одного или более соединения формулы I или его фармацевтически приемлемой соли или пролекарства в количестве, эффективном для лечения или предотвращения указанного нарушения.
Выражение "эффективное количество" означает количество соединения, которое, при введении млекопитающему, нуждающемуся в таком лечении, достаточно для (i) лечения или предотвращения конкретного заболевания, состояния или нарушения, опосредованного PGD2, (ii) уменьшения, смягчения или устранения одного или более симптомов конкретного заболевания, состояния или нарушения, или (iii) предотвращения или замедления проявления одного или более симптомов конкретного заболевания, состояния или нарушения, описанного в данном тексте.
Количество соединения формулы I, которое соответствует такому количеству, варьирует в зависимости от таких факторов, как конкретное соединение, состояние заболевания и его тяжесть, персональные данные (например, вес) млекопитающего, нуждающегося в лечении, но, тем не менее, может определяться по стандартной методике квалифицированным специалистом в данной области техники.
При использовании в данном тексте термин "млекопитающее" относится к теплокровному животному, страдающего от или находящегося в группе риска развития заболевания, описанного в данном тексте, и включает, но не ограничивается только ими, морских свинок, собак, кошек, крыс, мышей, хомяков и приматов, включая людей.
Настоящее изобретение также описывает соединения формулы I для применения в лечении PGD2-опосредованных состояний.
Другой аспект изобретения представляет собой применение соединения формулы I в получении лекарства для терапии, такой как лечение или профилактика PGD2-опосредованных состояний.
Соединения по настоящему изобретению можно использовать в комбинации с одним или более дополнительными лекарствами, например противовоспалительным соединением, которое обладает другим механизмом действия.
Соединения по изобретению можно вводить любым подходящим способом, например, в желудочно-кишечный тракт (например, ректально или перорально), в нос, легкие, мышцы или сосудистую систему или трансдермально. Соединения можно вводить в любой подходящей форме введения, например в форме таблеток, порошков, капсул, растворов, дисперсий, суспензий, сиропов, спреев, суппозиториев, гелей, эмульсий, пластырей и т.д. Такие композиции могут содержать компоненты, традиционные для фармацевтических препаратов, например разбавители, носители, рН модификаторы, подсластители, наполнители и другие активные средства. Если желательно парентеральное введение, композиции должны быть стерильны и находиться в форме раствора или суспензии, пригодной для инъекции или инфузии. Такие композиции составляют другой аспект изобретения.
Согласно другому аспекту, настоящее изобретение описывает фармацевтическую композицию, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль, как описано в данном тексте выше. В одном варианте осуществления фармацевтическая композиция включает соединение формулы (I) вместе с фармацевтически приемлемым разбавителем или носителем.
Согласно другому аспекту настоящее изобретение описывает соединение формулы (I) или его фармацевтически приемлемую соль, для применения в терапии.
Согласно другому аспекту настоящее изобретение описывает применение соединения формулы (I) или его фармацевтически приемлемой соли, в производстве лекарства для лечения иммунологического нарушения, как описано в данном тексте выше.
Следующие далее примеры иллюстрируют настоящее изобретение. В описанных ниже примерах, если не указано иное, все температуры представлены в градусах Цельсия. Реагенты были приобретены у коммерческих поставщиков, таких как Aldrich Chemical Company, Lancaster, TCI или Maybridge и использовались без дополнительной очистки, если не указано иное. Тетрагидрофуран (ТГФ), дихлорметан (ДХМ, метиленхлорид), толуол и диоксан были приобретены у Aldrich в герметичных сосудах и использовались без дополнительной очистки.
Описанные ниже реакции обычно проводили под избыточным давлением азота или аргона или с применением осушающей трубки (если не указано иное) в безводных растворителях, и реакционные колбы в типичном случае снабжались резиновыми септами для введения исходных веществ и реагентов посредством шприца. Стеклянную посуду сушили в сушильной печи и/или прокаливали.
1Н-ЯМР спектры записывали в растворах CDCl3, CD3OD, D2O или d6-ДМСО (хим. сдвиги приведены в м.д.), используя тетраметилсилан (0,00 м.д.) или пики остаточного растворителя (CDCl3: 7,25 м.д.; CD3OD: 3,31 м.д.; D2O: 4,79 м.д.; d6-ДМСО: 2,50 м.д.) в качестве стандарта. При указании мультиплетности пиков используются следующие сокращения: с (синглет), д (дублет), т (триплет), м (мультиплет), ушир. (уширенный), дд (дублет дублетов), дт (дублет триплетов). Константы спин-спинового взаимодействия, где приведены, выражены в герцах (Гц).
Пример А
Анализ ингибирования связывания с DP-2
Кодирующую последовательность DP2 человека вводили в клеточную линию лейкемии человека K562 путем электропорации и получали стабильные клоны, экспрессирующие DP2, посредством серийных разведении с последующим окрашиванием поверхности клеток моноклональным антителом крыс, специфическим для DP2 человека. Мембраны получали из одного из данных клонов, экспрессирующих DP2, и использовали для определения способности соединений по настоящему изобретению ингибировать связывание простагландина D2(PGD2) со своим рецептором DP2, используя следующую методику. Мембраны (1,25 мкг/лунку) смешивали с3Н-меченым PGD2 и различными концентрациями анализируемых соединений в 150 мкл связывающего буфера (50 мМ Трис-HCl, рН 7,4, 40 мМ MgCl2, 0,1% бычьего сывороточного альбумина, 0,1% NaN3) в 96-луночных полипропиленовых планшетах с U-образным дном. После инкубации в течение 60 мин при комнатной температуре переносили содержимое на планшет для фильтрования (#MAFB; Millipore Corporation, Bedford, MA) и промывали три раза связывающим буфером. Радиоактивность измеряли сцинтилляционным счетчиком (TopCount; PerkinElmer Life Sciences, Boston, MA). Неспецифическое связывание определяли инкубированием в присутствии 1 мкМ немеченого PGD2 или 5 мкМ известного DP2 антагониста. Значения IC50 для ингибирования связывания определяли для каждого исследованного соединения по точке перегиба стандартной 4-параметровой логистической кривой, построенной согласно полученным значениям. Все описанные в настоящей заявке соединения имеют значения IC50 менее 1 микромоля.
Пример 1
2-(4-(4-(3,4-Дихлорбензамидо)фенокси)-3-((4-фторфенилсульфонамидо)метил)-фенил)уксусная кислота

Стадия А: 2-(4-гидроксифенил)уксусную кислоту (7,0 г, 46 ммоль) разбавляли метансульфоновой кислотой (100 мл) и охлаждали до -10°С. Порциями добавляли 2-(гидроксиметил)изоиндолин-1,3-дион (8,2 г, 46 ммоль) в течение 15 минут. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь выливали на лед и перемешивали 1 ч. Реакционную смесь фильтровали и промывали водой. Вещество высушивали на воздухе в течение ночи и затем помещали под высокий вакуум на 5 ч. Затем вещество растворяли в ДМФА при нагревании для обеспечения полного растворения вещества. Небольшие количества воды добавляли до устойчивого образования осадка. Смесь фильтровали, и выделенное твердое вещество соответствовало бисалкилированному соединению. Добавляли дополнительное количество воды для осаждения 2-(3-((1,3-диоксоизоиндолин-2-ил)метил)-4-гидроксифенил)уксусной кислоты, которую отфильтровывали и высушивали. Полученное вещество использовали в следующей стадии без дополнительной очистки.
Стадия В: Продукт стадии А (6,7 г, 22 ммоль) разбавляли HBr (21 г, 258 ммоль) и уксусной кислотой (20 мл). Реакционную смесь кипятили с обратным холодильником при перемешивании в течение ночи. Затем реакционную смесь охлаждали до 0°С и нейтрализовывали твердыми гранулами NaOH до достижения уровня рН~10, получая раствор 2-(3-(аминометил)-4-гидроксифенил)уксусной кислоты, который использовали в следующей стадии без дополнительной очистки.
Стадия С: К 2-(3-(аминометил)-4-гидроксифенил)уксусной кислоте (3,9 г, 21,5 ммоль) добавляли Boc2O (5,40 г, 24,8 ммоль) в 30 мл диоксана. После перемешивания в течение 4 ч, реакционную смесь разбавляли этилацетатом и 1 н. HCl. Органический слой высушивали над MgO4 и концентрировали. Твердую фазу растворяли в минимальном количестве метиленхлорида, фильтровали и промывали метиленхлоридом. Фильтрат концентрировали, получая 3,0 г 2-(3-((трет-бутоксикарбонил)аминометил)-4-гидроксифенил)уксусной кислоты в виде белой пены.
Стадия D: 2-(3-((трет-бутоксикарбонил)аминометил)-4-гидроксифенил)уксусную кислоту (1,6 г, 5,69 ммоль) разбавляли ТГФ (10 мл) и метанолом (5 мл), после чего по каплям добавляли TMSCHN2 (14,2 мл, 28,4 ммоль). Реакционную смесь перемешивали 30 мин и затем разбавляли этилацетатом и водой. Слои разделяли и органический слой высушивали над MgSO4, фильтровали и концентрировали. Полученное вещество очищали с использованием картриджа biotage 40S, элюируя смесью гексан:этилацетат (4:1), выход 860 мг прозрачного масла (которое позже затвердевало) метил 2-(3-((трет-бутоксикарбонил)аминометил)-4-гидроксифенил)ацетата.
Стадия Е: Метил 2-(3-((трет-бутоксикарбонил)аминометил)-4-гидроксифенил) ацетат (200 мг, 0,677 ммоль) разбавляли ацетонитрилом (2 мл), после чего добавляли K2CO3 (206 мг, 1,49 ммоль) и 1-фтор-4-нитробензол (0,103 мл, 0,948 ммоль). Реакционную смесь кипятили с обратным холодильником (при температуре около 82°С) и перемешивали в течение 5 ч. Затем реакционную смесь разбавляли этилацетатом и водой. Слои разделяли и органический слой высушивали над MgSO4, фильтровали и концентрировали. Полученное вещество очищали с использованием biotage 40S, элюируя смесью гексан:этилацетат (3:1), получая 242 мг метил 2-(3-((трет-бутоксикарбонил)аминометил)-4-(4-нитрофенокси)фенил)ацетата в виде прозрачного масла.
Стадия F: Метил 2-(3-((трет-бутоксикарбонил)аминометил)-4-(4-нитрофенокси) фенил)ацетат (242 мг, 0,581 ммоль) разбавляли ТГФ (3 мл), после чего добавляли цинковую пыль (38,0 мг, 0,581 ммоль). По каплям добавляли около 2 мл насыщенного раствора хлорида аммония. После перемешивания реакции в течение 10 мин, реакционную смесь разбавляли этилацетатом и насыщенным раствором бикарбоната натрия. Слои разделяли, и органический слой высушивали над MgSO4, фильтровали и концентрировали, получая 225 мг метил 2-(4-(4-аминофенокси)-3-((трет-бутоксикарбонил)аминометил)фенил)ацетата.
Стадия G: Метил 2-(4-(4-аминофенокси)-3-((трет-бутоксикарбонил)аминометил)фенил)ацетат (100 мг, 0,259 ммоль) разбавляли метиленхлоридом (3 мл), после чего добавляли 3,4-дихлорбензоилхлорид (81,3 мг, 0,388 ммоль) и N,N-диизопропилэтиламин (0,0451 мл, 0,259 ммоль). Реакционную смесь перемешивали 3 часа. Реакционную смесь разбавляли метиленхлоридом и водой. Слои разделяли, и органический слой высушивали над MgSO4, фильтровали и концентрировали. Полученное вещество очищали с использованием biotage 12i, элюируя смесью гексан:этилацетат (3:1), получая 130 мг метил 2-(3-((трет-бутоксикарбонил)аминометил)-4-(4-(3,4-дихлорбензамидо)фенокси) фенил)ацетата.
Стадия Н: Метил 2-(3-((трет-бутоксикарбонил)аминометил)-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)ацетат (130 мг, 0,232 ммоль) обрабатывали HCl (0,581 мл, 2,32 ммоль). После перемешивания в течение 3 ч вещество концентрировали, получая 100 мг метил 2-(3-(аминометил)-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)ацетата.
Стадия I: Продукт стадии Н (30 мг, 0,065 ммоль) разбавляли метиленхлоридом (1 мл), после чего добавляли 4-фторбензол-1-сульфонилхлорид (15 мг, 0,078 ммоль) и DIEA (0,024 мл, 0,14 ммоль). После перемешивания реакционной смеси в течение 4 ч, реакционную смесь наносили непосредственно на препаративную ТСХ пластинку с толщиной слоя 0,5 мм и элюировали смесью гексан:этилацетат (3:1), получая 30 мг метил 2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-((4-фторфенил-сульфонамидо)метил)фенил) ацетата.
Стадия J: Метил 2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-((4-фторфенил-сульфонамидо)метил)фенил)ацетат (40 мг, 0,065 ммоль) разбавляли МеОН (1 мл), после чего добавляли NaOH (0,32 мл, 1,3 ммоль). После перемешивания в течение 1 ч реакционную смесь разбавляли этилацетатом и 2 н. HCl. Слои разделяли, и органический слой высушивали над MgSO4, фильтровали и концентрировали. Вещество очищали с использованием препаративной ТСХ пластинки с толщиной слоя 0,5 мм, элюируя смесью метиленхлорид:МеОН:АсОН (90:9:1), получая 12 мг целевого соединения в виде твердого вещества.1Н ЯМР (400 МГц, CDCl3/CD3OD) 8,1 (м, 1Н), 7,85 (м, 1Н), 7,8 (м, 2Н), 7,7 (д, 1Н), 7,6 (д, 1Н), 7,25 (с, 1Н), 7,2 (м, 3Н), 6,85 (д, 2Н), 6,7 (д, 1Н), 4,15 (с, 2Н), 3,55 (с, 2Н).
Пример 2
2-(4-(4-(3,4-Дихлорбензамидо)фенокси)-3-(метилсульфонамидометил)фенил)уксусная кислота

Получено способом, описанным в примере 1, при замене 4-фторбензол-1-сульфонилхлорида на стадии Н метансульфонилхлоридом.1H ЯМР (400 МГц, d6-ДМСО) 12,4 (с, 1Н), 10,4 (с, 1Н), 8,20 (м, 1Н), 7,95 (м, 1Н), 7,85 (д, 1Н), 7,75 (д, 2Н), 7,55 (т, 1Н), 7,4 (м, 1Н), 7,19 (м, 1Н), 7,05 (д, 2Н), 6,8 (д, 1Н), 4,2 (д, 2Н), 3,55 (с, 2Н), 2,85 (с, 3Н).
Пример 3
2-(4-(4-(3,4-Дихлорбензамидо)фенокси)-3-((2,4-дихлорфенилсульфонамидо)метил)-фенил)уксусная кислота

Получено способом, описанным в примере 1, при замене 4-фторбензол-1-сульфонилхлорида на стадии Н 2,4-дихлорбензил-1-сульфонилхлоридом.1H ЯМР (400 МГц, CDCl3/CD3OD) 8,1 (с, 1Н), 7,85 (м, 2Н), 7,65 (м, 3Н), 7,50 (м, 1Н), 7,35 (д, 1Н), 7,2 (с, 1Н), 7,10 (д, 1Н), 6,90 (д, 2Н), 6,60 (д, 1Н), 4,20 (с, 2Н), 3,45 (с, 2Н).
Пример 4
2-(4-(4-(3,4-Дихлорбензамидо)фенокси)-3-(никотинамидометил)фенил)уксусная кислота
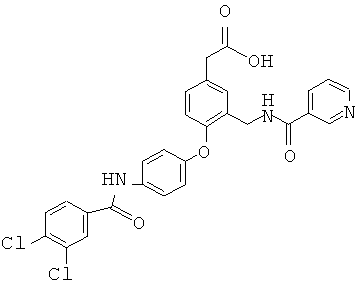
Стадия А: 4-Аминофенол (1,9 г, 17 ммоль) разбавляли ДМФА (40 мл), после чего добавляли пиридин (0,978) (1,4 мл, 17 ммоль) и 3,4-дихлорбензоилхлорид (3,0 г, 14 ммоль) в 5 мл ДМФА. После перемешивания в течение 12 ч реакционную смесь разбавляли этилацетатом и 2н. HCl. Водный слой экстрагировали этилацетатом один раз. Органические слои объединяли, промывали водой, раствором соли, высушивали над MgSO4, фильтровали и концентрировали на силикагель. Вещество очищали с использованием колонки biotage 40M, элюируя смесью гексан:этилацетат (9:1) в количестве 2 л и затем (60:40) в количестве 1 л, получая 3,4-дихлор-N-(4-гидроксифенил)-бензамид (1,5 г, 37% выход) в виде белого твердого вещества.
Стадия В: трет-бутил 2-(3-циано-4-фторфенил)ацетат (2,0 г, 8,5 ммоль) и продукт стадии А (2,9 г, 10 ммоль) разбавляли ДМСО (22 мл), после чего добавляли карбонат калия (1,4 г, 10 ммоль). Реакционную смесь нагревали до 125°С и перемешивали в течение 12 ч. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и 10%-ным водным карбонатом натрия. Слои разделяли, и органический слой промывали 10%-ным водным карбонатом натрия еще два раза, после чего промывали водой и раствором соли. Органический слой высушивали над MgSO4, фильтровали и концентрировали. Полученное вещество очищали с использованием колонки biotage 40M, элюируя смесью гексан:этилацетат (8:2), получая трет-бутил 2-(3-циано-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)ацетат (2,5 г, 59% выход) в виде белого твердого вещества.
Стадия С: Продукт стадии В (100 мг, 0,201 ммоль) разбавляли 7 н. раствором аммиак/метанол (10 мл), после чего добавляли этилацетат (10 мл). После растворения вещества добавляли никель Ренея (1,72 мг, 0,0201 ммоль) и реакционную смесь трижды продували водородом. После перемешивания в течение 12 ч реакционную смесь фильтровали через GF/F фильтр и концентрировали. Остаток очищали с использованием колонки biotage 12i, элюируя смесью метиленхлорид:МеОН:NH4OH (90:9:1), получая трет-бутил 2-(3-(аминометил)-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)ацетат (100 мг, 99,2% выход) в виде прозрачного масла.
Стадия D: Продукт стадии С (0,050 г, 0,997 ммоль) растворяли в 1 мл ДХМ в небольшой пробирке. Добавляли пиридин (0,017 г, 0,219 ммоль) и никотиноилхлорид (0,0195 г, 0,110 ммоль), и реакционную смесь перемешивали в течение 12 ч. Реакционную смесь затем разбавляли метиленхлоридом и водой. Метиленхлоридный слой отделяли и переносили непосредственно на колонку biotage 12i, элюируя смесью метиленхлорид:МеОН (95:5), получая трет-бутил 2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-(никотинамидометил)фенил)-ацетат (44 мг, 73% выход) в виде прозрачного масла.
Стадия Е: Продукт стадии D (44 мг, 0,073 ммоль) разбавляли смесью метиленхлорид:ТФУК (1:1) и перемешивали 3 ч. Реакционную смесь концентрировали, растворяли в эфире и диспергировали ультразвуком. Твердую фракцию отфильтровывали, промывали эфиром и высушивали в вакууме, получая целевое соединение (35 мг, 88% выход) в виде белого твердого вещества.1Н ЯМР (400 МГц, CDCl3/CD3OD) 8,75 (м, 1Н), 8,65 (м, 1Н), 8,15 (м, 1Н), 8,10 (м, 1Н), 8,0 (м, 1Н), 7,60 (м, 3Н), 7,45 (м, 1Н), 7,20 (м, 1Н), 6,95 (д, 2Н), 6,90 (д, 1Н), 4,65 (м, 2Н), 3,62 (с, 2Н).
Пример 5
2-(4-(4-(3,4-Дихлорбензамидо)фенокси)-3-((N,N-диметилсульфамоиламино)метил)-фенил)уксусная кислота

Получено способом, описанным в примере 4, при замене никотиноилхлорида на стадии D диметилсульфамоилхлоридом.1Н ЯМР (400 МГц, CDCl3/CD3OD) 8,05 (с, 1Н), 7,80 (м, 1Н), 7,65 (м, 2Н), 7,55 (д, 1Н), 7,35 (м, 1Н), 7,18 (м, 1Н), 7,00 (д, 2Н), 6,82 (д, 1Н), 4,25 (с, 2Н), 3,60 (с, 2Н), 2,75 (с, 6Н).
Пример 6
2-(4-(4-(3,4-Дихлорбензамидо)фенокси)-3-(метилсульфонамидометил)фенил)уксусная кислота

Получено способом, описанным в примере 4, при замене никотиноилхлорида на стадии D метансульфонилхлоридом.1H ЯМР (400 МГц, d6-ДМСО) 12,4 (с, 1Н), 10,4 (с, 1Н), 8,20 (м, 1Н), 7,95 (м, 1Н), 7,85 (д, 1Н), 7,75 (д, 2Н). 7,55 (т, 1Н), 7,4 (м, 1Н), 7,19 (м, 1Н), 7,05 (д, 2Н), 6,8 (д, 1Н), 4,2 (д, 2Н), 3,55 (с, 2Н), 2,85 (с, 3Н).
Пример 7
2-(3-(Циклогексанкарбоксамидометил)-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)-уксусная кислота

Получено способом, описанным в примере 4, при замене никотиноилхлорида на стадии D циклогексанкарбонилхлоридом.1Н ЯМР (400 МГц, CDCl3/CD3OD) 8,10 (м, 1Н), 7,80 (м, 1Н), 7,65 (д, 2Н), 7,55 (д, 1Н), 7,25 (м, 1Н). 7,15 (д, 1Н), 6,95 (д, 2Н), 6,85 (д, 1Н), 2,10 (м, 1Н), 1,6-1,8 (м, 5Н), 1,2-1,4 (м, 6Н).
Пример 8
2-(3-(Ацетамидометил)-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 4, при замене никотиноилхлорида на стадии D ацетилхлоридом.1H ЯМР (400 МГц, CDCl3/OD3OD) 8,10 (м, 1Н), 7,80 (м, 1Н), 7,60 (м, 3Н), 7,30 (м, 1Н), 7,15 (м, 1Н), 6,95 (д, 2Н), 6,85 (д, 1Н), 4,42 (с, 2Н), 3,40 (с, 2Н), 1,95 (с, 3Н).
Пример 9
2-(4-(4-(3,4-Дихлорбензамидо)фенокси)-3-(изоникотинамидометил)фенил)уксусная кислота

Получено способом, описанным в примере 4, при замене никотиноилхлорида на стадии D изоникотиноилхлоридом.1H ЯМР (400 МГц, CDCl3/CD3OD) 8,62 (м, 1Н), 8,05 (м, 1Н), 7,88 (м, 1Н), 7,80 (м, 1Н), 7,65 (м, 2Н), 7,58 (м, 3Н), 7,35 (м, 1Н), 7,20 (м, 1Н), 6,95 (д, 2Н), 6,90 (д, 1Н), 4,65 (д, 2Н), 3,62 (с, 2Н).
Пример 10
2-(4-(4-(3,4-Дихлорбензамидо)фенокси)-3-((4-фторбензамидо)метил)фенил)уксусная кислота

Получено способом, описанным в примере 4, при замене никотиноилхлорида на стадии D 4-фторбензоилхлоридом.1H ЯМР (400 МГц, CD3OD) 8,10 (с, 1Н), 7,85 (м, 1Н), 7,80 (м, 2Н), 7,70 (д, 1Н), 7,62 (д, 2Н), 7,35 (с, 1Н), 7,25 (д, 1Н), 7,35 (м, 2Н), 6,95 (д, 2Н), 6,90 (д, 1Н), 4,62 (с, 2Н), 3,60 (с, 3Н).
Пример 11
2-(4-(4-(3,4-Дихлорбензамидо)фенокси)-3-(пиколинамидометил)фенил)уксусная кислота

Получено способом, описанным в примере 4, при замене никотиноилхлорида на стадии D пиколиноилхлоридом.1Н ЯМР (400 МГц, CD3OD) 8,60 (д, 1Н), 8,10 (м, 1Н), 8,05 (м, 1Н), 7,95 (м, 1Н), 7,85 (м, 1Н), 7,70 (д, 1Н), 7,60 (д, 2Н), 7,55 (м, 1Н), 7,35 (м. 1Н), 7,20 (д, 1Н), 7,00 (д, 2Н), 6,85 (д, 1Н), 4,64 (с, 2Н), 3,60 (с, 2Н).
Пример 12
2-(4-(4-(3,4-Дихлорбензамидо)фенокси)-3-((метоксикарбониламино)метил)фенил)-уксусная кислота

Получено способом, описанным в примере 4, при замене никотиноилхлорида на стадии D метилхлорформатом.1H ЯМР (400 МГц, CDCl3) 8,10 (м, 1Н), 7,80 (м, 1Н), 7,65 (м, 2Н), 7,58 (м, 1Н), 7,30 (м, 1Н), 7,19 (м, 1Н), 6,95 (м, 2Н), 6,85 (м, 1Н), 4,40 (с, 2Н), 3,65 (с, 3Н), 3,60 (с, 2Н).
Пример 13
2-(4-(4-(3,4-Дихлорбензамидо)фенокси)-3-((пиридин-3-сульфонамидо)метил)фенил)-уксусная кислота

Получено способом, описанным в примере 4, при замене никотиноилхлорида на стадии D пиридин-3-сульфонилхлоридом.1Н ЯМР (400 МГц, CDCl3) 8,95 (с, 1Н), 8,60 (м, 1Н), 8,10 (м, 1Н), 8,05 (д, 1Н), 7,80 (д, 1Н), 7,60 (м, 3Н), 7,35 (м, 1Н), 7,22 (м, 1Н), 7,05 (д, 1Н), 6,80 (д, 2Н), 6,62 (д, 1Н), 4,25 (с, 1Н), 3,55 (с, 2Н).
Пример 14
2-(4-(4-(3,4-Дихлорфенилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)-уксусная кислота

Стадия А: трет-бутил 2-(3-циано-4-фторфенил)ацетат (2,6 г, 11,1 ммоль) и метил 4-гидроксибензоат (3,36 г, 22,1 ммоль) разбавляли ДМСО (22 мл), после чего добавляли карбонат калия (1,83 г, 13,3 ммоль). Реакционную смесь нагревали до 125°С и перемешивали 5 ч. Реакционную смесь разбавляли этилацетатом и 10%-ным водным карбонатом натрия. Слои разделяли и органический слой промывали 10%-ным водным карбонатом натрия еще два раза, после чего промывали водой и раствором соли. Органический слой высушивали над MgSO4, фильтровали и концентрировали. Полученное вещество очищали с использованием колонки biotage 40M, элюируя смесью гексан:этилацетат (9:1), получая метил 4-(4-(2-трет-бутокси-2-оксоэтил)-2-цианофенокси)бензоат (2,65 г, 65,3% выход) в виде прозрачного масла.
Стадия В: Продукт стадии А (2,5 г, 6,80 ммоль) разбавляли 7н. раствором аммиак/метанол (30 мл), после чего добавляли никеля Ренея (0,0583 г, 0,680 ммоль). Реакционную смесь трижды продували водородом и перемешивали 12 ч. Реакционную смесь затем фильтровали через GF/F фильтр и концентрировали. Остаток очищали с использованием колонки biotage 40M, элюируя смесью метиленхлорид:МеОН:NH4OH (90:9:1), получая метил 4-(2-(аминометил)-4-(2-трет-бутокси-2-оксоэтил)фенокси)бензоат (1,87 г, 74,0% выход) в виде прозрачного масла.
Стадия С: Продукт стадии В (1,87 г, 5,03 ммоль) разбавляли метиленхлоридом (2 мл), после чего добавляли пиридин (0,489 мл, 6,04 ммоль) и метансульфонилхлорид (0,779 мл, 10,1 ммоль). После перемешивания в течение 12 ч реакционную смесь разбавляли метиленхлоридом и 2н. HCl, слои разделяли и органический слой высушивали над MgSO4, фильтровали и концентрировали. Полученное вещество очищали с использованием картриджа biotage 40M, элюируя смесью гексан:этилацетат (1:1), получая метил 4-(4-(2-трет-бутокси-2-оксоэтил)-2-(метилсульфонамидометил)фенокси)бензоат (1,53 г, 67,6% выход) в виде прозрачного масла.
Стадия D: Продукт стадии С (900 мг, 2,00 ммоль) разбавляли диоксаном (10 мл), после чего добавляли LiOH-H2O (126 мг, 3,00 ммоль), растворенный в воде (2 мл). После перемешивания в течение 12 ч, реакционную смесь разбавляли этилацетатом и 2н. HCl. Слои разделяли, и органический слой высушивали над MgSO4, фильтровали и концентрировали. Полученное вещество очищали с использованием картриджа biotage 40M, элюируя смесью метиленхлорид:МеОН (95:5), получая 4-(4-(2-трет-бутокси-2-оксоэтил)-2-(метилсульфонамидо-метил)фенокси)бензойную кислоту (710 мг, 81,4% выход) в виде прозрачного масла.
Стадия Е: Продукт стадии D (62 мг, 0,14 ммоль) разбавляли метиленхлоридом (1 мл), после чего добавляли оксалилдихлорид (0,093 мл, 0,19 ммоль) и 1 каплю ДМФА. Реакционную смесь перемешивали 30 мин, после чего добавляли 3,4-дихлорбензоламин (46 мг, 0,28 ммоль). Реакционную смесь перемешивали 1 ч. Реакционную смесь переносили непосредственно на 12i sim и очищали, элюируя смесью метиленхлорид:МеОН (от 99,5:0,5 до 95:5), получая 30 мг трет-бутил 2-(4-(4-((3,4-дихлорфенил)карбамоил)фенокси)-3-(метилсульфонамидометил)фенил)ацетата в виде прозрачного масла.
Стадия F: Продукт стадии Е (22 мг, 0,038 ммоль) разбавляли метиленхлоридом (1 мл), после чего добавляли ТФУК (1 мл). После перемешивания в течение 1 ч, реакционную смесь концентрировали, разбавляли эфиром, диспергировали ультразвуком и концентрировали, получая 2-(4-(4-((3,4-дихлорфенил)карбамоил)фенокси)-3-(метилсульфонамидометил)фенил)уксусную кислоту (15 мг, 75% выход) в виде белого твердого вещества.1Н ЯМР (400 МГц, CDCl3/CD3OD) 7,92 (м, 1Н), 7,90 (д, 2Н), 7,59 (м, 1Н), 7,42 (м, 2Н), 7,25 (м, 1Н), 7,10 (д, 2Н), 6,90 (д, 1Н), 4,28 (с, 2Н), 3,62 (с, 2Н), 2,85 (с, 3Н).
Пример 15
2-(4-(4-(3,4-Дихлорфенетилкарбамоил)фенокси)-3-(метилсульфонамидометил)-фенил)уксусная кислота

Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е 2-(3,4-дихлорфенил)этанамином.1Н ЯМР (400 МГц, CDCl3/CD3OD) 7,75 (д, 2Н), 7,35 (м, 3Н), 7,22 (д, 1Н), 7,10 (д, 1Н), 6,95 (д, 2Н), 6,90 (д, 1Н), 4,25 (с, 2Н), 3,62 (м, 4Н), 3,40 (с, 2Н), 2,90 (т, 2Н), 2,85 (с, 3Н).
Пример 16
2-(3-(Метилсульфонамидометил)-4-(4-(нафталин-2-илкарбамоил)фенокси)фенил)-уксусная кислота

Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е нафталин-2-амином.1H ЯМР (400 МГц, CDCl3/CD3OD) 8,35 (с, 1Н), 7,96 (д, 2Н), 7,83 (м, 3Н), 7,70 (д, 1Н), 7,45 (м, 3Н), 7,25 (д, 1Н), 7,05 (д, 2Н), 6,95 (д, 1Н), 4,30 (д, 2Н), 3,62 (с, 2Н), 2,85 (с, 3Н).
Пример 17
2-(4-(4-(4-Фторфенетилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)-уксусная кислота

Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е 2-(4-фторфенил)этанамином.1Н ЯМР (400 МГц, CDCl3/CD3OD) 7,70 (д, 2Н), 7,40 (м, 1Н), 7,22 (м, 3Н), 7,10 (м, 2Н), 6,90 (д, 2Н), 6,70 (д, 1Н), 4,30 (с, 2Н), 3,65 (м, 4Н), 2,90 (т, 2Н), 2,85 (с, 3Н).
Пример 18
2-(3-(Метилсульфонамидометил)-4-(4-(фенетилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е фенетиламином.1H ЯМР (400 МГц, CDCl3/CD3OD) 7,70 (д, 2Н), 7,40 (м, 1Н), 7,31 (м, 1Н), 7,25 (м, 5Н), 6,95 (д, 2Н), 6,90 (д, 1Н), 4,30 (с, 2Н), 3,68 (т, 2Н), 3,61 (с, 2Н), 2,90 (т, 2Н), 2,85 (с, 3Н).
Пример 19
2-(3-(Метилсульфонамидометил)-4-(4-(фенилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е анилином.1Н ЯМР (400 МГц, CDCl3/CD2OD) 7,92 (д, 2Н), 7,65 (д, 2Н), 7,45 (с, 1Н), 7,38 (м, 2Н), 7,25 (д, 1Н), 7,18 (м, 1Н), 7,05 (д, 2Н), 6,95 (д, 1Н), 4,30 (с, 2Н), 3,65 (с, 2Н), 2,85 (с, 3Н).
Пример 20
2-(4-(4-(Бензилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)уксусная кислота

Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е бензиламином.1Н ЯМР (400 МГц, CDCl3/CD3OD) 7,80 (д, 2Н), 7,40 (м, 1Н), 7,35 (м, 4Н), 7,22 (д, 1Н), 6,95 (м, 3Н), 6,90 (д, 1Н), 4,62 (д, 2Н), 4,30 (с, 2Н), 3,62 (с, 2Н), 2,85 (с, 3Н).
Пример 21
2-(4-(4-(4-Хлорфенетилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)-уксусная кислота

Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е 2-(4-хлорфенил)этанамином.1H ЯМР (400 МГц, CDCl3/CD3OD) 7,7 (д, 2Н), 7,4 (с, 1Н), 7,25 (д, 2Н), 7,22 (д, 1Н), 7,18 (д, 2Н), 6,95 (д, 2Н), 6,90 (д, 1Н), 6,82 (ушир. т, 1Н), 4,3 (с, 2Н), 3,65 (т, 2Н), 3,4 (ушир. с, 2Н), 2,9 (т, 2Н), 2,85 (с, 3Н).
Пример 22
2-(4-(4-(2-(4-Хлорбензиламино)-2-оксоэтил)фенокси)-3-(метилсульфонамидометил)-фенил)уксусная кислота

Получено способом, описанным в примере 14, при замене метил 4-гидроксибензоата на стадии А метил 2-(4-гидроксилфенил)ацетатом и при замене 3,4-дихлорбензоламина на стадии Е 4-хлорбензиламином.1Н ЯМР (400 МГц, CDCl3/CD3OD) 7,38 (м, 1Н), 7,25 (м, 4Н), 7,20 (м, 3Н), 6,95 (д, 2Н), 6,85 (д, 1Н), 4,38 (м, 2Н), 4,32 (с, 2Н), 3,61 (с, 2Н), 3,55 (с, 2Н), 2,85 (с, 3Н).
Пример 23
2-(4-(4-(2-(Бензиламино)-2-оксоэтил)фенокси)-3-(метилсульфонамидометил)фенил)-уксусная кислота

Получено способом, описанным в примере 14, при замене метил 4-гидроксибензоата на стадии А метил 2-(4-гидроксилфенил)ацетатом и при замене 3,4-дихлорбензоламина на стадии Е бензиламином.1Н ЯМР (400 МГц, CDCl3/CD3OD) 7,38 (м, 1Н), 7,15-7,30 (м, 8Н), 6,95 (д, 2Н), 6,85 (д, 1Н), 4,40 (д, 2Н), 4,32 (с, 2Н), 3,61 (с, 2Н), 3,56 (с, 2Н), 2,82 (с, 3Н).
Пример 24
2-(3-(Метилсульфонамидометил)-4-(4-(2-оксо-2-(фенетиламино)этил)фенокси)фенил)-уксусная кислота

Получено способом, описанным в примере 14, при замене метил 4-гидроксибензоата на стадии А метил 2-(4-гидроксилфенил)ацетатом и при замене 3,4-дихлорбензоламина на стадии Е фенетиламином.1Н ЯМР (400 МГц, CDCl3/CD3OD) 7,38 (м, 1Н), 7,28 (м, 1Н), 7,25 (м, 1Н), 7,18-7,22 (м, 2Н), 7,15 (д, 2Н), 7,10 (д, 1Н), 6,92 (д, 2Н), 6,82 (д, 1Н), 4,38 (с, 2Н), 3,62 (с, 2Н), 3,48 (м, 4Н), 2,82 (с, 3Н), 2,75 (т, 2Н).
Пример 25
2-(4-(4-(2-(Бензил(метил)амино)-2-оксоэтил)фенокси)-3-(метилсульфонамидометил)-фенил)уксусная кислота

Получено способом, описанным в примере 14, при замене метил 4-гидроксибензоата на стадии А метил 2-(4-гидроксилфенил)ацетатом и при замене 3,4-дихлорбензоламина на стадии Е N-метилбензиламином.1Н ЯМР (400 МГц, CDCl3/CD3OD) 7,10-7,40 (м, 9Н), 6,94 (м, 2Н), 6,82 (м, 1Н), 4,60 (д, 2Н), 4,35 (м, 2Н), 3,75 (д, 2Н), 3,62 (м, 2Н), 2,97 (д, 3Н), 2,82 (с, 3Н).
Пример 26
2-(3-(Метилсульфонамидометил)-4-(4-(2-оксо-2-(фениламино)этил)фенокси)фенил)-уксусная кислота

Получено способом, описанным в примере 14, при замене метил 4-гидроксибензоата на стадии А метил 2-(4-гидроксилфенил)ацетатом и при замене 3,4-дихлорбензоламина на стадии Е анилином.1H ЯМР (400 МГц, CDCl3/CD3OD) 7,55 (д, 2Н), 7,30-7,35 (м, 5Н), 7,18 (м, 1Н), 7,10 (м, 1Н), 6,95 (д, 2Н), 6,85 (д, 1Н), 4,35 (с, 2Н), 3,68 (с, 2Н), 3,60 (с, 2Н), 2,85 (с, 3Н).
Пример 27
2-(4-(4-Бензамидофенокси)-3-((4-фторфенилсульфонамидо)метил)фенил)уксусная кислота

Получено способом, описанным в примере 1, при замене 3,4-дихлорбензоилхлорида на стадии G бензоилхлоридом. МС+534,9 [М+1].
Пример 28
2-(4-(4-(4-Хлорбензамидо)фенокси)-3-((4-фторфенилсульфонамидо)метил)фенил)-уксусная кислота

Получено способом, описанным в примере 1, при замене 3,4-дихлорбензоилхлорида на стадии G 4-хлорбензоилхлоридом.1Н ЯМР (400 МГц, CD3OD) 7,85 (д, 2Н), 7,70 (м, 1Н), 7,45-7,55 (м, 5Н), 7,35 (м, 1Н), 7,15 (м, 2Н), 6,60-6,70 (м, 3Н), 4,55 (с, 2Н), 3,58 (с, 2Н).
Пример 29
2-(4-(4-(3-Хлорбензамидо)фенокси)-3-((4-фторфенилсульфонамидо)метил)фенил)-уксусная кислота

Получено способом, описанным в примере 1, при замене 3,4-дихлорбензоилхлорида на стадии G 3-хлорбензоилхлоридом.1H ЯМР (400 МГц, CD3OD) 7,90 (с, 1Н), 7,80 (д, 1Н), 7,65 (м, 1Н), 7,52 (м, 4Н), 7,45 (т, 1Н), 7,35 (м, 1Н), 7,15 (м, 2Н), 6,60 (м, 4Н), 4,45 (с, 2Н), 3,50 (ушир. с, 2Н).
Пример 30
2-(4-(4-(3,4-Дифторбензамидо)фенокси)-3-(метилсульфонамидометил)фенил)уксусная кислота

Получено способом, описанным в примере 1, при замене 3,4-дихлорбензоилхлорида на стадии G 3,4-дифторбензоилхлоридом и при замене 4-фторбензол-1-сульфонилхлорида на стадии I метансульфонилхлоридом.1Н ЯМР (400 МГц, CD3OD) 7,88 (м, 1Н), 7,80 (м, 1Н), 7,65 (д, 2Н), 7,42 (м, 2Н), 7,20 (м, 2Н), 7,00 (д, 2Н). 6,82 (м, 1Н), 4,33 (с, 2Н), 3,62 (с, 2Н), 2,85 (с, 3Н).
Пример 31
2-(4-(4-(2-Хлорбензамидо)фенокси)-3-(метилсульфонамидометил)фенил)уксусная кислота

Получено способом, описанным в примере 1, при замене 3,4-дихлорбензоилхлорида на стадии G 2-хлорбензоилхлоридом и при замене 4-фторбензол-1-сульфонилхлорида на стадии I метансульфонилхлоридом.1H ЯМР (400 МГц, CD3OD) 7,68 (д, 2Н), 7,40-7,55 (м, 5Н), 7,20 (д, 1Н), 7,05 (д, 2Н), 6,85 (д, 1Н), 4,35 (с, 2Н), 3,65 (ушир. с, 2Н), 2,85 (с, 3Н).
Пример 32
2-(4-(4-(3,4-Дихлорбензамидо)фенокси)-3-метоксифенил)уксусная кислота.

Получено согласно стадиям Е, F, G и J из примера 1, при замене метил 2-(3-((трет-бутоксикарбонил)аминометил)-4-гидроксифенил)ацетата на стадии Е метил 2-(4-гидрокси-3-метоксифенил)ацетатом.1Н ЯМР (400 МГц, CDCl3) 9,64 (с, 1Н), 8,17 (д, 1Н), 7,97 (дд, 1Н), 7,72-7,75 (м, 4Н), 7,13 (д, 1Н), 6,99 (д, 1Н), 6,93 (д, 1Н), 6,87 (м, 2Н), 3,80 (с, 3Н), 3,66 (с, 2Н).
Пример 33
2-(4-(4-(бензилоксикарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)уксусная кислота
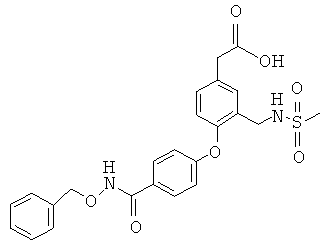
Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е гидрохлоридом O-бензилгидроксиламина.1H ЯМР (400 МГц, CD3OD) 7,76 (д, J-8,6 Гц, 2Н), 7,50 (т, J=6,3 Гц, 1Н), 7,33-7,48 (м, 5Н), 7,23 (д, J=6,3 Гц, 1Н), 6,99 (д, J=8,7 Гц, 2Н), 6,91 (д, J=8,4 Гц, 2Н), 4,91 (с, 2Н), 4,10 (д, J=6,3 Гц, 2Н), 3,60 (с, 2Н). 2,85 (с, 3Н).
Пример 34
2-(3-Хлор-4-(4-(4-хлорфенетилкарбамоил)фенокси)фенил)уксусная кислота

Стадия А: 2-(3-Хлор-4-гидроксифенил)уксусную кислоту (5,0 г, 26,8 ммоль) растворяли в 30 мл МеОН и добавляли H2SO4 (0,744 мл, 13,4 ммоль) по каплям при комнатной температуре. Реакционную смесь кипятили четыре часа и затем концентрировали в вакууме. Остаток разбавляли 150 мл EtOAc и последовательно промывали насыщенным раствором NaHCO3, водой и раствором соли, высушивали над H2SO4 и концентрировали в вакууме, получая метил 2-(3-хлор-4-гидроксифенил)ацетат (5,16 г, 96,0% выход) в виде полупрозрачного густого масла.
Стадия В: Метил 2-(3-хлор-4-гидроксифенил)ацетат (177 мг, 0,885 ммоль) разбавляли ДМСО (3 мл), после чего добавляли К2СО3 (122 мг, 0,885 ммоль) и N-(4-хлорфенетил)-4-хлор-3-нитробензамид (300 мг, 0,885 ммоль) (синтезированного на стадии С). Реакционную смесь нагревали до 85°С и перемешивали 12 ч. Реакционную смесь охлаждали, разбавляли ДХМ и промывали 10%-ным водным раствором карбоната натрия, водой и раствором соли. Слои разделяли, и органический слой высушивали над H2SO4, фильтровали и концентрировали. Полученное вещество очищали с использованием картриджа biotage 40S, элюируя смесью ДХМ:МеОН (99:1), получая метил 2-(4-(4-((4-хлорфенетил)карбамоил)-2-нитрофенокси)-3-хлорфенил)ацетат (206 мг, 46,3% выход).
Стадия С: 2-(4-Хлорфенил)этанамин (2,3 мл, 16 ммоль) разбавляли ДХМ (40 мл), после чего добавляли DIEA (2,9 мл, 16 ммоль) и 4-хлор-3-нитробензоилхлорид (3,0 г, 14 ммоль) в 10 мл ДХМ. После перемешивания в течение 30 мин реакционную смесь переносили непосредственно на картридж biotage 40M и элюировали смесью гексан:этилацетат (2:1), получая N-(4-хлорфенетил)-4-хлор-3-нитробензамид (4,0 г, 86% выход) в виде белого твердого вещества.
Стадия D: Метил 2-(4-(4-((4-хлорфенетил)карбамоил)-2-нитрофенокси)-3-хлорфенил)ацетат (110 мг, 0,219 ммоль) разбавляли ТГФ (1 мл), после чего добавляли цинковую пыль (14,3 мг, 0,219 ммоль) и насыщенный раствор NH4Cl (1 мл). После перемешивания в течение 1 ч, реакционную смесь разбавляли ДХМ и 10%-ным водным раствором карбоната натрия. Слои разделяли и органический слой высушивали над MgSO4, фильтровали и концентрировали, получая метил 2-(4-(4-((4-хлорфенетил)карбамоил)-2-аминофенокси)-3-хлорфенил)ацетат (102 мг, 98,6% выход).
Стадия Е: Изобутилнитрит (0,0751 мл, 0,634 ммоль) разбавляли ДМФА (1 мл), помещали в атмосферу азота и нагревали до 60°С. Добавляли по каплям метил 2-(4-(4-((4-хлорфенетил)карбамоил)-2-аминофенокси)-3-хлорфенил)ацетат (100 мг, 0,211 ммоль) в ДМФА (1 мл) и реакционную смесь перемешивали 1 ч. Реакционную смесь охлаждали, разбавляли этилацетатом и промывали 2н. HCl, насыщенным раствором бикарбоната натрия и раствором соли. Органический слой высушивали над MgSO4, фильтровали и концентрировали. Полученное вещество очищали с использованием колонки biotage 12i, элюируя смесью ДХМ:МеОН (99:1), получая метил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-хлорфенил)ацетат (70 мг, 72,3% выход).
Стадия F: Метил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-хлорфенил)ацетат (33 мг, 0,0720 ммоль) разбавляли ТГФ (500 мкл), после чего добавляли NaOH (0,144 мл, 0,720 ммоль) и 100 мкл воды. После перемешивания в течение 2 ч, реакционную смесь разбавляли ДХМ и 2н. HCl. Слои разделяли и органический слой высушивали над MgSO4, фильтровали и концентрировали, получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-хлорфенил)уксусную кислоту (19 мг, 59,4% выход) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3/CD3OD) δ 7,65 (д, 2Н), 7,42 (м, 1Н), 7,25 (д, 2Н), 7,20 (д, 1Н), 7,15 (д, 2Н), 7,05 (д, 1Н), 6,85 (д, 2Н), 3,65 (т, 2Н), 3,60 (с, 2Н), 2,90 (т, 2Н).
Пример 35
2-(3-циано-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)пропановая кислота

Стадия А: К перемешиваемому раствору диизопропиламина (357 мкл, 2,55 ммоль) в ТГФ (3 мл) при -78°С добавляли н-бутиллитий (2,5М раствор в гексане, 1,02 мл, 2,55 ммоль). Реакционную смесь перемешивали в течение 30 мин, по истечении этого времени добавляли по каплям раствор ацетата (500 мг, 2,13 ммоль) в ТГФ (3,5 мл) и перемешивали в течение 20 мин при -78°С, затем нагревали до 0°С. Добавляли MeI (133 мкл, 2,13 ммоль) и реакционную смесь перемешивали еще 30 мин. Реакционную смесь подкисляли 2М HCl и экстрагировали EtOAc. Объединенные органические слои затем промывали раствором соли и высушивали над MgSO4. Полученный остаток очищали на приборе Biotage Horizon (40+М, от 5% до 50% В:EtOAc, 21 мл-1008 мл), получая трет-бутил 2-(3-циано-4-фторфенил)пропаноат (0,368 г, 1,48 ммоль, 70%).1Н ЯМР (400 МГц, CDCl3) δ 7,50-7,60 (м, 2Н), 7,17 (т, J=8,7 Гц, 1Н), 3,63 (кв, J=7,l Гц, 1Н), 4,60 (д, J=7,2 Гц, 3Н), 1,40 (с, 9Н).
Стадия В: К перемешиваемому раствору 4-аминофенола (2,50 г, 22,9 ммоль) в ДМФА (55 мл) добавляли пиридин (1,85 мл, 22,9 ммоль), затем 3,4-дихлорбензоилхлорид (4,0 г, 19,1 ммоль) в ДМФА (7 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем разбавляли 2М HCl и EtOAc. Водный слой затем экстрагировали EtOAc и объединенные органические слои промывали раствором соли и высушивали над MgSO4. Полученный остаток очищали на приборе Biotage Horizon (65+M, от 5% до 75% В:EtOAc, 51 мл-2448 мл). 3,4-Дихлор-N-(4-гидроксифенил)бензамид собирали в виде белого твердого вещества (3,91 г, 13,9 ммоль, 73%).1Н ЯМР (400 МГц, CD3OD) δ 8,09 (с, 1Н), 7,84 (д, J=8,5 Гц, 1H), 7,67 (д, J=8,6 Гц, 1Н), 7,45 (д, J=8,8 Гц, 2Н), 6,78 (д, J=8,4 Гц, 2Н).
Стадия С: К перемешиваемому раствору трет-бутил 2-(3-циано-4-фторфенил)пропаноата (360 мг, 1,44 ммоль) со стадии А и 3,4-дихлор-N-(4-гидроксифенил)бензамида (488 мг, 1,73 ммоль) со стадии В в ДМСО (4 мл) добавляли K2CO3 (240 мг, 1,73 ммоль). Реакционную смесь перемешивали при 125°С в течение ночи, затем охлаждали до комнатной температуры и разбавляли EtOAc и 10%-ным водным раствором карбоната натрия. Водную фазу экстрагировали EtOAc, и объединенные органические вещества промывали 10%-ным водным раствором карбоната натрия и раствором соли, высушивали над MgSO4, концентрировали и очищали на приборе Biotage Horizon (40+M, от 5% до 50% В:EtOAc, 21 мл-1008 мл). Затем подходящие фракции объединяли и концентрировали, получая трет-бутил 2-(3-циано-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)пропаноат (0,476 г, 0,797 ммоль, 55%).1Н ЯМР (400 МГц, CDCl3) δ 7,99 (с, 1Н), 7,84 (ушир. с, N-H), 7,72 (д, J=9,3 Гц, 1Н), 7,67 (д, J=8,7 Гц, 2Н), 7,60 (м, 2Н), 7,42 (д, J=8,6 Гц, 1Н), 7,11 (д, J=7,7, 2Н), 6,83 (д, J=8,7 Гц, 1Н), 3,61 (кв, J=7,l Гц, 1Н), 1,45 (д, J-7,5 Гц, 3Н), 1,42 (с, 9Н).
Стадия D: К перемешиваемому раствору трет-бутил 2-(3-циано-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)пропаноата (25 мг, 0,049 ммоль) в ДХМ (1 мл) добавляли ТФУК (250 мкл). Реакционную смесь перемешивали при комнатной температуре 4 ч и затем концентрировали. Сырой продукт очищали препаративной ТСХ (5% МеОН/ДХМ), получая 2-(3-циано-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)пропановую кислоту (0,0174 г, 0,0382 ммоль, 78%).1Н ЯМР (400 МГц, CDCl3): 8,04 (ушир. с, N-H), 7,96 (с, 1Н), 7,64-7,71 (м, 3Н), 7,61 (с, 1Н), 7,55 (д, J=7,7 Гц, 1Н), 7,44 (дд, J=8,6, 2,2 Гц, 1Н), 7,08 (д, J-8,9 Гц, 2Н), 6,83 (д, J=8,5 Гц, 1Н), 3,74 (кв, J=7,4 Гц, 1Н), 1,53 (д, J=6,9 Гц, 3Н).
Пример 36
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-(метилсульфонамидометил)фенил)пропановая кислота

Получено способом, описанным в примере 14, при замене метил 4-(4-(2-трет-бутокси-2-оксоэтил)-2-цианофенокси)бензоата на стадии В трет-бутил 2-(3-циано-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)пропаноатом, с последующим проведением стадий С и F.1H ЯМР (400 МГц, CD3OD): 8,10 (с, 1Н), 7,85 (с, 1Н), 7,60-7,70 (м, 3Н), 7,50 (с, 1Н), 7,20 (д, 1Н), 7,1 (д, 2Н), 6,80 (д, 1Н), 4,30 (с, 2Н), 3,75 (кв, 1Н), 2,80 (2, 3Н), 1,45 (д, 3Н).
Пример 37
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-(метилсульфонамидометил)фенил)-2-метилпропановая кислота

Стадия А: К перемешиваемому раствору NaH (60% в масле, 195 мг, 4,88 ммоль) в ДМФА (10 мл) при 0°С по каплям добавляли раствор трет-бутил 2-(3-циано-4-фторфенил)ацетата (500 мг, 2,125 ммоль) и MeI (330 мкл, 5,31 ммоль) в ДМФА (1 мл). Реакционную смесь нагревали до комнатной температуры, перемешивали в течение ночи и затем разбавляли 1 н. HCl и EtOAc. Водный слой экстрагировали EtOAc и объединенные органические слои промывали раствором соли и высушивали над MgSO4. Сырой концентрированный продукт затем очищали на приборе Biotage Horizon (40+M, от 5% до 50% B:EtOAc, 18 мл-864 мл), получая трет-бутил 2-(3-циано-4-фторфенил)-2-метилпропаноат (0,423 г, 1,602 ммоль, 75%).1Н ЯМР (400 МГц, CDCl3) δ7,56-7,63 (м, 2Н), 7,17 (т, J=8,1 Гц, 1Н), 1,54 (с, 6Н), 1,38 (с, 9Н).
Стадия В: 2-(4-(4-(3,4-Дихлорбензамидо)фенокси)-3-(метилсульфонамидометил) фенил)-2-метилпропановую кислоту получали из трет-бутил 2-(3-циано-4-фторфенил)-2-метилпропаноата способом, описанным в примере 35, при замене трет-бутил 2-(3-циано-4-фторфенил)пропаноата на стадии В трет-бутил 2-(3-циано-4-фторфенил)-2-метилпропаноатом, с последующим проведением стадий В, С и F из примера 14.1Н ЯМР (400 МГц, CD3OD): 8,15 (с, 1Н), 7,85 (д, 1Н), 7,65-7,75 (м, 3Н), 7,55 (с, 1Н), 7,30 (д, 1Н), 7,05 (д, 2Н), 6,80 (д, 1Н), 4,35 (с, 2Н), 2,80 (с, 3Н), 1,55 (с, 6Н).
Пример 38
2-(3-(Метилсульфонамидометил)-4-(4-(4-(трифторметил)фенилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е 4-трифторметилбензоламином.1H ЯМР (400 МГц, CDCl3, CD3OD) 7,95 (д, 2Н), 7,82 (д, 2Н), 7,62 (д, 1Н), 7,45 (с, 1Н), 7,25 (д, 1Н), 7,05 (д, 2Н), 6,95 (д, 1Н), 4,30 (с, 2Н), 3,62 (с, 2Н), 2,82 (с, 3Н).
Пример 39
2-(3-(Метилсульфонамидометил)-4-(4-(3-(трифторметил)фенилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е 3-трифторметилбензоламином.1Н ЯМР (400 МГц, CDCl3, CD3OD) 8,05 (с, 1Н), 7,90 (м, 3Н), 7,50 (м, 1Н), 7,45 (с, 1Н), 7,40 (м, 1Н), 7,25 (д, 1Н), 7,05 (д, 2Н), 6,85 (д, 1Н), 4,30 (с, 2Н), 3,65 (с, 2Н), 2,85 (с, 3Н).
Пример 40
2-(4-(4-(4-хлор-3-фторфенилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)уксусная кислота

Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е 4-хлор-3-фторбензоламином.1Н ЯМР (400 МГц, CDCl3, CD3OD) 7,90 (д, 2Н), 7,75 (м, 1Н), 7,30-7,45 (м, 3Н), 7,25 (д, 1Н), 7,05 (д, 2Н), 6,95 (д, 1Н), 4,3 (с, 2Н), 3,65 (с, 2Н), 2,85 (с, 3Н).
Пример 41
2-(4-(4-(3-хлор-4-фторфенилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)уксусная кислота
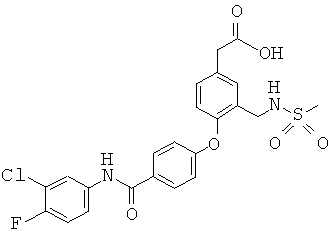
Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е 3-хлор-4-фторбензоламином.1H ЯМР (400 МГц, CDCl3, CD3OD) 7,93 (д, 2Н), 7,85 (м, 1Н), 7,55 (м, 1Н), 7,45 (с, 1Н), 7,25 (д, 1Н), 7,15 (т, 1Н), 7,05 (д, 2Н), 6,85 (д, 1Н), 4,30 (с, 2Н), 3,65 (с, 2Н), 2,85 (с, 3Н).
Пример 42
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)фенил)уксусная кислота

Стадия А: 2-(4-гидроксифенил)уксусную кислоту (1,0 г, 6,57 ммоль) разбавляли ТГФ (4 мл), после чего добавляли Реагент 2 (2,63 г, 13,1 ммоль). Реакционную смесь перемешивали 12 ч. Реакционную смесь фильтровали и переносили непосредственно на картридж biotage 40M, элюируя смесью гексан:этилацетат (3:1), получая трет-бутил 2-(4-гидроксифенил)ацетат (800 мг, 58,4% выход) в виде прозрачного масла, которое позднее затвердевало с образованием белого твердого вещества.
Стадия В: 2-(4-хлорфенил)этанамин (0,321 мл, 2,27 ммоль) разбавляли ДХМ (10 мл), после чего добавляли DIEA (d 0,742) (0,395 мл, 2,27 ммоль) и 4-фторбензоилхлорид (0,231 мл, 1,89 ммоль). После перемешивания в течение 30 мин, реакционную смесь переносили непосредственно на картридж biotage 40S и элюировали смесью гексан:этилацетат (2:1), получая N-(4-хлорфенетил)-4-фторбензамид (393 мг, 74,8% выход) в виде белого твердого вещества.
Стадия С: Трет-бутил 2-(4-гидроксифенил)ацетат (200 мг, 0,960 ммоль) разбавляли ДМСО (4 мл), после чего добавляли К2СО3 (133 мг, 0,960 ммоль) и N-(4-хлорфенетил)-4-фторбензамид (267 мг, 0,960 ммоль). Реакционную смесь перемешивали при 85°С в течение ночи и затем перемешивали 12 ч при 138°С. Реакционную смесь охлаждали до комнатной температуры, разбавляли ДХМ и 10%-ным водным раствором карбоната натрия. Слои разделяли и органический слой высушивали над MgSO4, фильтровали и концентрировали. Сырое вещество очищали хроматографией на силикагеле, элюируя смесью гексан:этилацетат (4:1), получая трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)фенил)ацетат (20 мг, 4,47% выход) в виде не совсем белого твердого вещества.
Стадия D: Трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)фенил)ацетат (20 мг, 0,043 ммоль) разбавляли ДХМ (1 мл) после чего добавляли ТФУК (1 мл). После перемешивания в течение 2 ч реакционную смесь концентрировали, разбавляли эфиром и концентрировали, получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)фенил)уксусную кислоту (17 мг, 97% выход) в виде белого твердого вещества.1Н ЯМР (400 МГц, CDCl3, CD3OD) 7,70 (д, 2Н), 7,30 (м, 4Н), 7,19 (м, 2Н), 7,00 (м, 4Н), 3,62 (м, 4Н), 2,90 (т, 2Н).
Пример 43
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-фторфенил)уксусная кислота

Стадия А: 2-(3-Фтор-4-гидроксифенил)уксусную кислоту (1,0 г, 5,88 ммоль) разбавляли смесью ТГФ:МеОН (3:1) (10 мл), после чего добавляли TMSCHN2 (5,88 мл, 11,8 ммоль). После перемешивания в течение 1 ч реакцию гасили 2н. HCl и разбавляли ДХМ. Слои разделяли и органический слой высушивали над MgSO4, фильтровали и концентрировали. Вещество очищали хроматографией на силикагеле, элюируя смесью гексан:этилацетат (2:1), получая метил 2-(3-фтор-4-гидроксифенил)ацетат (700 мг, 64,7% выход) в виде прозрачного масла.
Стадия В: 2-(4-Хлорфенил)этанамин (2,3 мл, 16 ммоль) разбавляли ДХМ (40 мл), после чего по каплям добавляли DIEA (2,9 мл, 16 ммоль) и 4-хлор-3-нитробензоилхлорид (3,0 г, 14 ммоль) в 10 мл ДХМ. После перемешивания в течение 30 мин реакционную смесь переносили непосредственно на колонку с силикагелем и элюировали смесью гексан:этилацетат (2:1), получая N-(4-хлорфенетил)-4-хлор-3-нитробензамид (4,0 г, 86% выход) в виде белого твердого вещества.
Стадия С: Метил 2-(3-фтор-4-гидроксифенил)ацетат (полученный на стадии А; 163 мг, 0,885 ммоль) разбавляли ДМСО (3 мл), после чего добавляли K2CO3 (122 мг, 0,885 ммоль) и N-(4-хлорфенетил)-4-хлор-3-нитробензамид (полученный на стадии В; 300 мг, 0,885 ммоль). Реакционную смесь перемешивали 12 ч при 80°С. Реакционную смесь охлаждали, разбавляли ДХМ и промывали 10%-ным водным раствором карбоната натрия, водой и раствором соли. Слои разделяли, и органический слой высушивали над MgSO4, фильтровали и концентрировали. Полученное вещество очищали хроматографией на силикагеле, элюируя смесью гексан:ацетон (2:1), получая метил 2-(4-(4-((4-хлорфенетил)карбамоил)-2-нитрофенокси)-3-фторфенил)ацетат (200 мг, 46,4% выход).
Стадия D: Метил 2-(4-(4-((4-хлорфенетил)карбамоил)-2-нитрофенокси)-3-фторфенил)ацетат (100 мг, 0,205 ммоль) разбавляли ТГФ (2 мл), после чего добавляли цинковую пыль (13,4 мг, 0,205 ммоль) и насыщенный водный раствор NH4Cl (1 мл). После перемешивания в течение 1 ч реакционную смесь разбавляли этилацетатом и 10%-ным водным раствором карбоната натрия. Слои разделяли и органический слой высушивали над MgSO4, фильтровали и концентрировали, получая метил 2-(4-(4-((4-хлорфенетил)карбамоил)-2-аминофенокси)-3-фторфенил)ацетат (93 мг, 99,1% выход).
Стадия Е: Изобутилнитрил (0,060 мл, 0,51 ммоль) разбавляли ДМФА (2 мл), помещали в атмосферу азота и нагревали до 60°С, после чего добавляли метил 2-(4-(4-((4-хлорфенетил)карбамоил)-2-аминофенокси)-3-фторфенил)ацетат (93 мг, 0,20 ммоль) в 500 мкл ДМФА. Реакционную смесь перемешивали 30 мин и затем охлаждали. Реакционную смесь разбавляли этилацетатом и промывали 2н. HCl и насыщенным раствором бикарбоната натрия. Слои разделяли и органический слой высушивали над MgSO4, фильтровали и концентрировали. Полученное вещество очищали хроматографией на силикагеле, элюируя смесью гексан:этилапетат (2:1), получая метил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-фторфенил)ацетат (35 мг, 39% выход).
Стадия F: Метил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-фторфенил)ацетат (32 мг, 0,072 ммоль) разбавляли диоксаном (1 мл), после чего добавляли NaOH (0,072 мл, 0,36 ммоль) и 200 мкл воды. После перемешивания в течение 3 ч, реакционную смесь разбавляли этилацетатом и 2н. HCl. Органический слой высушивали над MgSO4, фильтровали и концентрировали, получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-фторфенил)уксусную кислоту (15 мг, 48% выход) в виде белого твердого вещества.1H ЯМР (400 МГц, CDCl3, CD3OD) 7,70 (д, 2Н), 7,25 (д, 2Н), 7,19 (м, 3Н), 7,10 (м, 2Н), 6,95 (д, 2Н), 3,65 (м, 4Н), 2,90 (т, 2Н).
Пример 44
2-(3-циано-4-(4-(4-хлорфенетилкарбамоил)фенокси)фенил)уксусная кислота

Стадия А: В колбу загружали трет-бутил 2-(3-циано-4-фторфенил)ацетат (9,410 г, 40,00 ммоль), метил 4-гидроксибензоат (7,303 г, 48,00 ммоль), K2CO3 (6,634 г, 48,00 ммоль) и ДМСО (160 мл). Смесь нагревали до 90°С в атмосфере азота в течение 17 ч. Смесь охлаждали до комнатной температуры и выливали в смесь EtOAc (250 мл) и 10%-ного раствора K2CO3 (250 мл). Полученный нерастворимый остаток растворяли в воде и добавляли к EtOAc-водной смеси. Смесь перемешивали 1 ч и слои разделяли. Водный слой экстрагировали EtOAc, и объединенные экстракты промывали насыщенным раствором K2CO3, водой и раствором соли, высушивали над MgSO4, фильтровали через слой целита и концентрировали при пониженном давлении, получая 14,7 г сырого продукта в виде масла. Сырое вещество очищали хроматографией на силикагеле, получая метил 4-(4-(2-трет-бутокси-2-оксоэтил)-2-цианофенокси)бензоат (11,6 г, 79%) в виде белого твердого вещества.
Стадия В: Метил 4-(4-(2-трет-бутокси-2-оксоэтил)-2-цианофенокси)бензоат (11,60 г, 31,57 ммоль) растворяли в диоксане (160 мл) и раствор охлаждали до 10°С на ледяной бане. В раствор добавляли LiOH-H2O, 1M (37,89 мл, 37,89 ммоль) и перемешивали смесь при комнатной температуре в течение 5 ч. Полученную смесь разбавляли 2н. HCl (100 мл) и CH2Cl2 (200 мл). Слои разделяли и водный слой экстрагировали CH2Cl2. Объединенные экстракты промывали раствором соли (100 мл), высушивали над MgSO4, фильтровали через слой целита и концентрировали при пониженном давлении. Сырое вещество очищали хроматографией на силикагеле, получая 4-(4-(2-трет-бутокси-2-оксоэтил)-2-цианофенокси)бензойную кислоту (7,29 г) в виде белого твердого вещества.
Стадия С: К 4-(4-(2-трет-бутокси-2-оксоэтил)-2-цианофенокси)бензойной кислоте (0,34 г, 0,962 ммоль), 2-(4-хлорфенил)этанамину (0,165 г, 1,06 ммоль) в ДМФА (5 мл) добавляли диизопропиламин (0,200 мл, 1,15 ммоль) и HBTU (0,149 г, 1,15 ммоль). Реакционную смесь перемешивали при комнатной температуре 90 мин. Реакционную смесь разбавляли водой и продукт экстрагировали этилацетатом. Объединенные органические слои высушивали (MgSO4), фильтровали и концентрировали, получая трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-цианофенил)ацетат (0,40 г, 85%) в виде белого твердого вещества после колоночной хроматографии.
Стадия D: Трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-цианофенил)ацетату (4,40 г, 8,96 ммоль) в ДХМ (20 мл) обрабатывали ТФУК (20 мл). После перемешивания в течение 2 ч реакционную смесь концентрировали, и сырое вещество очищали хроматографией на силикагеле с градиентом от 0,5% МеОН/ДХМ, содержащего 0,5% АсОН, до 10% МеОН/ДХМ, содержащего 0,5% АсОН, получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-цианофенил)уксусную кислоту (3,30 г, 84,7% выход).1Н ЯМР (400 МГц, CD3OD) 7,85 (д, 2Н), 7,65 (с, 1Н), 7,45 (д, 1Н), 7,35 (с, 1Н), 7,25 (д, 2Н), 7,20 (д, 2Н), 7,10 (д, 2Н), 6,95 (д, 1Н), 3,62 (м, 4Н), 2,90 (т, 2Н).
Пример 45
2-(3-циано-4-(4-(2,4-дихлорфенетилкарбамоил)фенокси)фенил)уксусная кислота
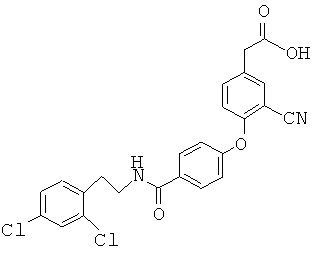
Получено способом, описанным в примере 44, при замене 2-(2,4-дихлорфенил)этанамина на 2-(4-хлорфенил)этанамин на стадии D.1H ЯМР (400 МГц, CDCl3, CD3OD) 7,79 (д, 2Н), 7,65 (м, 1Н), 7,45 (д, 1Н), 7,40 (м, 1Н), 7,21 (м, 2Н), 7,10 (д, 2Н), 6,85 (д, 1Н), 3,65 (м, 4Н), 3,05 (т, 2Н).
Пример 46
2-(3-бром-4-(4-(2,4-дихлорфенетилкарбамоил)фенокси)фенил)уксусная кислота

Стадия А: N-(4-хлорфенетил)-4-гидроксибензамид (1,5 г, 5,4 ммоль) разбавляли ДМСО (15 мл), после чего добавляли K2CO3 (0,89 г, 6,4 ммоль) и 3-бром-4-фторбензальдегид (1,0 г, 4,9 ммоль). Реакционную смесь перемешивали при 85°С в течение ночи и затем охлаждали до комнатной температуры. Реакционную смесь разбавляли этилацетатом и промывали дважды 10%-ным раствором карбоната натрия, водой и раствором соли, и затем концентрировали. Сырое вещество очищали хроматографией на силикагеле, элюируя смесью гексан:этилацетат (1:1) и затем 100%-ным этилацетатом, получая N-(4-хлорфенетил)-4-(2-бром-4-формилфенокси)бензамид (1,6 г, 71% выход).
Стадия В: N-(4-хлорфенетил)-4-(2-бром-4-формилфенокси)бензамид (1,6 г, 3,5 ммоль) разбавляли ТГФ (10 мл), после чего добавляли гидроксид N,N,N-триметил(фенил)метанаминия (0,79 мл, 1,7 ммоль) и метилсульфинил(метилтио)метан (0,87 г, 7,0 ммоль). Реакционную смесь перемешивали при 70°С в течение 2 ч. Реакционную смесь переносили непосредственно на колонку с силикагелем и элюировали смесью ДХМ:МеОН (98:2), получая (Z)-N-(4-хлорфенетил)-4-(2-бром-4-(2-(метилсульфинил)-2-(метилтио)винил)фенокси)бензамид (1,9 г, 96% выход).
Стадия С: (Z)-N-(4-хлорфенетил)-4-(2-бром-4-(2-(метилсульфинил)-2-(метилтио)винил)фенокси)бензамид (2,0 г, 3,5 ммоль) обрабатывали HCl (8,9 мл, 18 ммоль) в этаноле. Реакционную смесь перемешивали при 70°С в течение 2 ч. Реакционную смесь охлаждали и переносили непосредственно на колонку с силикагелем, элюируя смесью гексан:этилацетат (3:1), получая этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-бромфенил)ацетат.
Стадия D: Этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-бромфенил)ацетат (30 мг, 0,058 ммоль) разбавляли диоксаном (500 мкл), после чего добавляли NaOH (0,093 мл, 0,46 ммоль) и 200 мкл воды. После перемешивания в течение 3 ч, реакционную смесь разбавляли этилапетатом и 2 н. HCl. Слои разделяли и органический слой высушивали над MgSO4, фильтровали и концентрировали, получая 2-(4-(4-((4-хлорфенетил)карбамоил) фенокси)-3-бромфенил)уксусную кислоту (24 мг, 85% выход) в виде белого твердого вещества.1Н ЯМР (400 МГц, CDCl3, CD3OD) 7,70 (д, 2Н), 7,60 (с, 1Н), 7.22-7,32 (м, 3Н), 7,19 (д, 2Н), 7,05 (д, 1Н), 6,90 (д, 2Н), 3,62 (м, 4Н), 2,90 (т, 2Н).
Пример 47
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)3,5-диметилфенил)уксусная кислота

Получено способом, описанным в примере 46, при замене 4-фтор-3,5-диметилбензальдегида на 3-бром-4-фторбензальдегид на стадии А.1H ЯМР (400 МГц, CDCl3, CD3OD) 7,60 (д, 2Н), 7,25 (д, 2Н), 7,15 (д, 2Н), 7,05 (с, 2Н), 6,78 (д, 2Н), 3,65 (т, 2Н), 3,58 (с, 2Н), 2,90 (т, 2Н), 2,15 (с, 6Н).
Пример 48
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)3-метилфенил)уксусная кислота

Стадия А: Этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-бромфенил)ацетат (пример 46 стадия С; 30 мг, 0,058 ммоль) разбавляли ТГФ (1 мл), после чего добавляли бис(три-т-бутилфосфин)палладий (0) (3,0 мг, 0,0058 ммоль) и метилцинкхлорид (0,087 мл, 0,17 ммоль). После перемешивания в течение 2 ч реакционную смесь переносили непосредственно на колонку с силикагелем, элюируя смесью гексан:этилацетат (3:1), получая этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-метилфенил)ацетат (20 мг, 76% выход) в виде прозрачного масла.
Стадия В: Этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-метилфенил)ацетат (20 мг, 0,0443 ммоль) разбавляли диоксаном (500 мкл), после чего добавляли NaOH (0,0885 мл, 0,443 ммоль) и 200 мкл воды. После перемешивания в течение 3 ч реакционную смесь разбавляли этилацетатом и 2н. HCl. Слои разделяли, и органический слой высушивали над MgSO4, фильтровали и концентрировали, получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-метилфенил)уксусную кислоту (16,0 мг, 85,3% выход).1H ЯМР (400 МГц, CDCl3, CD3OD) 7,62 (д, 2Н), 7,30 (д, 2Н), 7,10-7,22 (м, 4Н), 6,92 (д, 1Н), 6,85 (д, 2Н), 3,65 (т, 2Н), 3,60 (с, 2Н), 2,90 (т, 2Н), 2,15 (с, 3Н).
Пример 49
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)3-(тиофен-2-ил)фенил)уксусная кислота

Стадия А: Этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-бромфенил)ацетат (пример 46 стадия С; 24 мг, 0,046 ммоль) разбавляли ТГФ (1 мл), после чего добавляли бис(три-т-бутилфосфин)палладий (0) (2,4 мг, 0,0046 ммоль) и 2-тиенилцинкбромид (0,23 мл, 0,12 ммоль). После перемешивания в течение 2 ч реакционную смесь очищали хроматографией на силикагеле, элюируя смесью гексан:этилацетат (3:1), получая этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-(тиофен-2-ил)фенил)ацетат (20 мг, 83% выход) в виде прозрачного масла.
Стадия В: Этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-(тиофен-2-ил)фенил)ацетат (20 мг, 0,038 ммоль) разбавляли диоксаном (500 мкл), после чего добавляли NaOH (0,052 мл, 0,26 ммоль) и 200 мкл воды. После перемешивания в течение 3 ч реакционную смесь разбавляли этилацетатом и 2 н. HCl. Слои разделяли, и органический слой высушивали над MgSO4, фильтровали и концентрировали, получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-(тиофен-2-ил)фенил)уксусную кислоту (7 мг, 37% выход) в виде белого твердого вещества.1Н ЯМР (400 МГц, CDCl3, CD3OD) 7,69 (с, 1Н), 7,64 (д, 2Н), 7,45 (д, 1Н), 7,30 (д, 2Н), 7,22 (д, 1Н), 7,15 (д, 2Н), 6,98-7,05 (м, 2Н), 6,90 (д, 2Н), 3,65 (м, 4Н), 2,90 (т, 2Н).
Пример 50
2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)бифенил-3-ил)уксусная кислота

Стадия А: Этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-бромфенил)ацетат (пример 46 стадия С; 24 мг, 0,046 ммоль) разбавляли ТГФ (1 мл), после чего добавляли бис(три-т-бутилфосфин)палладий (0) (2,4 мг, 0,0046 ммоль) и фенилцинкиодид (0,23 мл, 0,12 ммоль). После перемешивания в течение 2 ч реакционную смесь переносили непосредственно на колонку с силикагелем и элюировали смесью гексан:этилацетат (3:1), получая этил 2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)бифенил-3-ил)ацетат (15 мг, 63% выход) в виде прозрачного масла.
Стадия В: Этил 2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)бифенил-3-ил)ацетат (10 мг, 0,0195 ммоль) разбавляли диоксаном (1 мл), после чего добавляли NaOH (0,0389 мл, 0,195 ммоль) и 200 мкл воды. После перемешивания в течение 2 ч реакционную смесь разбавляли этилацетатом и 2 н. HCl. Слои разделяли, и органический слой высушивали над MgSO4, фильтровали и концентрировали, получая 2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)бифенил-3-ил)уксусную кислоту (1,6 мг, 16,9% выход) в виде белого твердого вещества. МС: ESI отриц. М-Н=485.
Пример 51
2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)-3'-(метилсульфонил)бифенил-3-ил)уксусная кислота

Стадия А: Этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-бромфенил)ацетат (пример 46 стадия С; 100 мг, 0,193 ммоль), 3-(метансульфонил)фенилбороновую кислоту (58,1 мг, 0,290 ммоль), K2CO3 (80,2 мг, 0,580 ммоль) и Pd(PPh3)4 (22,4 мг, 0,0193 ммоль) разбавляли диоксаном (2 мл) и водой (1 мл). Реакционную смесь трижды продували азотом и перемешивали при 60°С в течение ночи. Реакционную смесь охлаждали и переносили непосредственно на колонку с силикагелем, элюируя смесью гексан:этилацетат (2:1), получая этил 2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)-3'-(метилсульфонил)бифенил-3-ил)ацетат (80 мг, 69,8% выход).
Стадия В: Этил 2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)-3'-(метилсульфонил) бифенил-3-ил)ацетат (80 мг, 0,135 ммоль) разбавляли диоксаном (1 мл), после чего добавляли NaOH (0,270 мл, 1,35 ммоль) и 300 мкл воды. После перемешивания в течение 2 ч реакционную смесь разбавляли этилацетатом и 2н. HCl. Слои разделяли и органический слой высушивали над MgSO4, фильтровали и концентрировали. Вещество очищали с использованием картриджа biotage 12i, элюируя смесью 1-4% метанол/ДХМ, получая 2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)-3'-(метилсульфонил)бифенил-3-ил)уксусную кислоту (5 мг, 6,56% выход).1Н ЯМР (400 МГц, CDCl3, CD3OD) 8,05 (с, 1Н), 7,81 (д, 1Н), 7,78 (д, 1Н), 7,50-7,60 (м, 3Н), 7,42 (с, 1Н), 7,38 (д, 1Н), 7,25 (д, 2Н), 7,19 (д, 2Н), 7,05 (д, 1Н), 6,85 (д, 1Н), 3,75 (с, 2Н), 3,62 (кв, 2Н), 3,0 (с, 3Н), 2,90 (т, 2Н).
Пример 52
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-циклопропилфенил)уксусная кислота

Стадия А: В колбу загружали циклопропилмагнийбромид (29,6 мл, 14,8 ммоль) и заполняли азотом. Добавляли хлорид цинка (14,8 мл, 14,8 ммоль) и реакционную смесь перемешивали 20 мин. 3-Бром-4-фторбензальдегид (1,0 г, 4,93 ммоль) и бис(три-т-бутилфосфин)палладий (0) (0,126 г, 0,246 ммоль) разбавляли в 600 мкл ТГФ и добавляли в реакционную смесь. После перемешивания в течение 4 ч реакционную смесь нагревали до 5°С и перемешивали в течение ночи. Реакционную смесь охлаждали и гасили насыщенным раствором NH4Cl и экстрагировали ДХМ. Органический слой высушивали над MgSO4, фильтровали и концентрировали. Вещество очищали с использованием картриджа biotage 40M, элюируя смесью гексан:этилацетат (4:1), получая 3-циклопропил-4-фторбензальдегид (400 мг, 49,5% выход)
Стадия В: N-(4-Хлорфенетил)-4-гидроксибензамид (504 мг, 1,83 ммоль) разбавляли ДМСО (8 мл), и добавляли К2СО3 (379 мг, 2,74 ммоль) и 3-циклопропил-4-фторбензальдегид (300 мг, 1,83 ммоль). Реакционную смесь перемешивали при 85°С в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и дважды промывали 10%-ным раствором карбоната натрия, водой и раствором соли и концентрировали. Сырое вещество очищали хроматографией на силикагеле, элюируя смесью гексан:этилацетат (3:1), получая N-(4-хлорфенетил)-4-(2-циклопропил-4-формилфенокси)бензамид (160 мг, 20,9% выход).
Стадия С: N-(4-Хлорфенетил)-4-(2-циклопропил-4-формилфенокси)бензамид (160 мг, 0,381 ммоль) разбавляли ТГФ (3 мл), после чего добавляли гидроксид N,N,N-триметил(фенил)метанаминия (0,0866 мл, 0,191 ммоль) и метилсульфинил(метилтио) метан (94,7 мг, 0,762 ммоль). Реакционную смесь нагревали до 70°С и перемешивали 2 ч. Реакционную смесь охлаждали и переносили непосредственно на колонку с силикагелем, элюируя этилацетатом, получая (Z)-N-(4-хлорфенетил)-4-(2-циклопропил-4-(2-(метилсульфинил)-2-(метилтио)винил)фенокси)бензамид (100 мг, 49,9% выход).
Стадия D: (Z)-N-(4-хлорфенетил)-4-(2-циклопропил-4-(2-(метилсульфинил)-2-(метилтио)винил)фенокси)бензамид (100 мг, 0,190 ммоль) разбавляли HCl (0,950 мл, 1,90 ммоль), нагревали до 70°С и перемешивали в течение 4 ч. Реакционную смесь охлаждали и переносили непосредственно на картридж biotage 25 и элюировали смесью гексан:этилацетат (3:1), получая этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-циклопропилфенил)ацетат (70 мг, 77,0% выход) в виде белого твердого вещества.
Стадия Е: Этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-циклопропилфенил)ацетат (70 мг, 0,146 ммоль) разбавляли диоксаном (1 мл), после чего добавляли NaOH (0,293 мл, 1,46 ммоль) и воду (300 мкл). После перемешивания в течение 3 ч реакционную смесь разбавляли этилацетатом и 2 н. HCl. Слои разделяли и органический слой высушивали над MgSO4, фильтровали и концентрировали, получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-циклопропилфенил)уксусную кислоту (50 мг, 75,9% выход) в виде белого твердого вещества.1Н ЯМР (400 МГц, CDCl3, CD3OD) 7,65 (д, 2Н), 7,30 (д, 2Н), 7,15 (д, 2Н), 7,10 (д, 1Н), 6,90 (м, 4Н), 3,65 (т, 2Н), 3,60 (с, 2Н), 2,90 (т, 2Н), 2,05 (м, 1Н), 0,80 (м, 2Н), 0,62 (м, 2Н).
Пример 53
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-этилфенил)уксусная кислота

Стадия А: Этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-бромфенил)ацетат (пример 46 стадия С; 30 мг, 0,058 ммоль) разбавляли ТГФ (1 мл), после чего добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (II) (4,8 мг, 0,0058 ммоль) и диэтилцинк (0,13 мл, 0,15 ммоль). После перемешивания в течение 4 ч, реакционную смесь переносили непосредственно на колонку с силикагелем, элюируя смесью 5-50% этилацетат/гексан, получая этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-этилфенил)ацетат (10 мг, 37% выход).
Стадия В: Этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-этилфенил)ацетат (10 мг, 0,0215 ммоль) разбавляли диоксаном (500 мкл), после чего добавляли NaOH (0,0429 мл, 0,215 ммоль) и 5 капель воды. После перемешивания в течение 3 ч, реакционную смесь разбавляли этилацетатом и 2 н. HCl, и слои разделяли. Органический слой высушивали над MgSO4, фильтровали и концентрировали. Сырое вещество очищали хроматографией на силикагеле, элюируя смесью 10% МеОН/ДХМ, получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-этилфенил)уксусную кислоту (2 мг, 21,3% выход). (400 МГц, CDCl3, CD3OD) 7,65 (д, 2Н), 7,30 (д, 2Н), 7,10-7,25 (м, 4Н), 6,90 (м, 3Н), 3,62 (м, 2Н), 3,40 (с, 1Н), 2,90 (т, 2Н), 2,55 (кв, 2Н), 1,15 (т, 3Н).
Пример 54
2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)-4'-(метилсульфонил)бифенил-3-ил)уксусная кислота
Стадия А: Этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-бромфенил)ацетат (пример 46 стадия С; 40 мг, 0,077 ммоль), K2CO3 (32 мг, 0,23 ммоль) и 4-(метилсульфонил)фенилбороновую кислоту (23 мг, 0,12 ммоль) разбавляли смесью диоксан (1 мл)/вода (300 мкл), после чего добавляли Pd(PPh3)4 (8,9 мг, 0,0077 ммоль). Реакционную смесь продували азотом и перемешивали при 55°С в течение 12 ч. Реакционную смесь охлаждали и концентрировали. Остаток растворяли в минимальном количестве ДХМ и очищали хроматографией на силикагеле, элюируя смесью гексан:этилацетат (1:1), получая этил 2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)-4'-(метилсульфонил)бифенил-3-ил)ацетат (20 мг, 44% выход) в виде прозрачного масла.
Стадия В: Этил 2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)-4'-(метилсульфонил) бифенил-3-ил)ацетат (20 мг, 0,0338 ммоль) разбавляли диоксаном (1 мл), после чего добавляли NaOH (0,0676 мл, 0,338 ммоль) и 300 мкл воды. После перемешивания в течение 3 ч, реакционную смесь разбавляли этилацетатом и 2 н. HCl. Слои разделяли, и органический слой высушивали над MgSO4, фильтровали и концентрировали, получая 2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)-4'-(метилсульфонил)бифенил-3-ил)уксусную кислоту (10 мг, 52,5% выход). (400 МГц, CDCl3, CD3OD) 7,90 (д, 2Н), 7,70 (д, 2Н), 7,60 (д, 2Н), 7,40 (с, 1Н), 7,35 (д, 1Н), 7,25 (д, 2Н), 7,15 (д, 2Н). 7,05 (д, 1Н), 6,85 (д, 2Н), 6,55 (т, 1Н), 3,63 (м, 4Н), 3,05 (с, 3Н), 2,90 (т, 2Н).
Пример 55
2-(3-(метилсульфонамидометил)-4-(4-(3-(трифторметил)фенетилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е 2-(3-трифторметилфенил)этиламином. МС+551,1 [М+1].
Пример 56
2-(3-(метилсульфонамидометил)-4-(4-(4-трифторметил)фенетилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е 2-(4-трифторметилфенил)этиламином. МС+550,9 [М+1].
Пример 57
2-(4-(4-((1-(4-хлорфенил)циклопропил)метилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)уксусная кислота

Получено способом, описанным в примере 14, при замене 3,4-дихлорбензоламина на стадии Е (1-(4-хлорфенил)циклопропил)метанамином. МС-541,1 [М-1].
Пример 58
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-(никотинамидометил)фенил)уксусная кислота

Стадия А: Смесь трет-бутил 2-(3-циано-4-фторфенил)ацетата (1,32 г, 5,59 ммоль), М-(4-хлорфенетил)-4-гидроксибензамида (1,85 г, 671 ммоль) и карбоната калия (0,93 г, 6,70 ммоль) в ДМСО (10 мл) перемешивали при 90°С в течение 1 дня. Реакционную смесь охлаждали, разбавляли этилацетатом и промывали 10%-ным водным раствором Na2CO3. Водный слой снова экстрагировали этилацетатом. Органические слои объединяли, промывали 10%-ным раствором Na2CO3 и раствором соли, высушивали над сульфатом натрия и концентрировали. Сырое вещество очищали хроматографией на силикагеле, элюирование в градиенте от 5% этилацетат/гексан до 60% этилацетат/гексан дало в результате трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-цианофенил)ацетат (1,09 г, 40%).
Стадия В: К трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-цианофенил)ацетату (1,09 г, 2,22 ммоль) в 6 мл 7 н. раствора аммиака в метаноле при комнатной температуре добавляли никель Ренея (0,019 г) и реакционную смесь перемешивали под шариком с водородом при комнатной температуре. Реакционную смесь фильтровали и концентрировали, получая трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-(аминометил)фенил)ацетат (0,160 г, 15%).
Стадия С: Смесь трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-(аминометил)фенил)ацетата (0,015 г, 0,030 ммоль), никотиноилхлорид гидрохлорида (0,007 г, 0,039 ммоль) и триэтиламина (0,013 мл, 0,091 ммоль) в дихлорметане (1 мл) перемешивали при комнатной температуре 1 ч. Реакционную смесь переносили на силикагель, и продукт элюировали с использованием градиентной системы от 0,5% метанол/дихлорметан до 5% метанол/дихлорметан, получая трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-(никотинамидометил)фенил)ацетат (0,005 г, 275%).
Стадия D: К раствору трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-(никотинамидометил)фенил)ацетата (0,005 г, 0,008 ммоль) в дихлорметане (1,0 мл) добавляли ТФУК (1,0 мл) и реакционную смесь перемешивали при комнатной температуре. По истечении 1 ч, реакционную смесь концентрировали, получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-(никотинамидометил)фенил)уксусную кислоту (0,005 г). МС-449,1 [М-CO2H].
Пример 59
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-((пиридин-3-сульфонамидо)метил)фенил)уксусная кислота
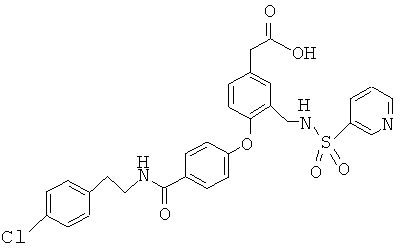
Получено способом, описанным в примере 58, при замене никотиноилхлорид гидрохлорида на стадии С пиридин-3-сульфонилхлорид гидрохлоридом. МС-578,1 [М-1].
Пример 60
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-((1-метил-1H-имидазол-5-сульфонамидо)метил)фенил)уксусная кислота

Получено способом, описанным в примере 58, при замене никотиноилхлорид гидрохлорида на стадии С 1-метил-1Н-имидазол-4-сульфонилхлоридом. МС-581,2 [М-1].
Пример 61
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-((6-диметиламино)никотинамидо)метил)фенил)уксусная кислота

Получено способом, описанным в примере 58, при замене никотиноилхлорид гидрохлорида на стадии С 6-(диметиламино)никотиновой кислотой. МС-585,2 [М-1].
Пример 62
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-((2-(4-фторфенилсульфонамидо)ацетамидо)метил)фенил)уксусная кислота

Получено способом, описанным в примере 58, при замене никотиноилхлорид гидрохлорида на стадии С 2-(4-фторфенилсульфонамидо)уксусной кислотой. МС-608,4 [М-CO2H].
Пример 63
2-(3-((6-аминоникотинамидо)метил)-4-(4-(4-хлорфенетилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 58, при замене никотиноилхлорид гидрохлорида на стадии С 6-аминоникотиновой кислотой. МС-557,1 [М-1].
Пример 64
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-((диметиламино)метил)фенил)уксусная кислота

Стадия А: К перемешиваемому раствору трет-бутил 2-(3-(аминометил)-4-(4-(фенетилкарбамоил)фенокси)фенил)ацетата (0,015 г, 0,030 ммоль) в метаноле (1 мл) добавляли уксусную кислоту (0,012 мл, 0,212 ммоль), цианборгидрид натрия (0,010 г, 0,152 ммоль) и параформальдегид (0,007 г, 0,152 ммоль). Реакционную смесь перемешивали при комнатной температуре 1 ч. Реакционную смесь непосредственно переносили на силикагель, и продукт элюировали в градиенте от 0,5% метанол/дихлорметан до 5% метанол/дихлорметан, получая трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-((диметиламино)метил)фенил)ацетат (0,005 г, 0,010 ммоль).
Стадия В: К трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-((диметиламино)метил)фенил)ацетату (0,005 г, 0,010 ммоль) в дихлорметане (1,0 мл) добавляли ТФУК (1,0 мл) и реакционную смесь перемешивали 1 ч при комнатной температуре. Реакционную смесь концентрировали, получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-((диметиламино)метил)фенил)уксусную кислоту (0,005 г). МС-465,1 [M-1].
Пример 65
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-((N-метилметилсульфонамидо)метил)фенил)уксусная кислота

Стадия А: К перемешиваемому раствору трет-бутил 2-(3-циано-4-фторфенил)ацетата (1,00 мг, 4,25 ммоль) и метил 4-гидроксибензоата (776 мг, 5,10 ммоль) в ДМСО (20 мл) добавляли K2CO3 (704 мг, 5,10 ммоль). Реакционную смесь нагревали при 90°С в течение ночи на масляной бане. Реакционную смесь разбавляли этилацетатом и 10%-ным водным раствором карбоната натрия. Водную фазу экстрагировали этилацетатом и объединенные органические слои промывали 10%-ным водным раствором карбоната натрия и раствором соли, высушивали над MgSO4, концентрировали и очищали хроматографией на силикагеле, получая метил 4-(4-(2-трет-бутокси-2-оксоэтил)-2-цианофенокси)бензоат (1,08 г).
Стадия В: К перемешиваемому раствору метил 4-(4-(2-трет-бутокси-2-оксоэтил)-2-цианофенокси)бензоата (1,086 мг, 2,96 ммоль) в 7 н. растворе аммиак/метанол (30 мл) под азотом добавляли никель Ренея (25,3 мг, 0,296 ммоль). Реакционную смесь продували водородом и перемешивали в атмосфере водорода в течение ночи. Реакционную смесь фильтровали через GF бумажный фильтр и твердый катализатор промывали метанолом и этилацетатом. Фильтрат концентрировали и сырой продукт очищали хроматографией на силикагеле, получая метил 4-(2-(аминометил)-4-(2-7трет-бутокси-2-оксоэтил)фенокси)бензоат (0,69 г).
Стадия С: К перемешиваемому раствору метил 4-(2-(аминометил)-4-(2-трет-бутокси-2-оксоэтил)фенокси)бензоата (690 мг, 1,86 ммоль) в дихлорметане (6 мл) добавляли пиридин (225 мкл, 2,78 ммоль), затем метансульфонилхлорид (287 мкл, 3,71 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч, по истечении данного времени добавляли еще пиридин (1,5 экв.) и метансульфонилхлорид (2 экв.). Реакционную смесь перемешивали еще один час и затем разбавляли этилацетатом и 1М HCl. Водный слой экстрагировали этилацетатом, и объединенные органические слои промывали раствором соли и высушивали над MgSO4. Сырой продукт затем очищали на приборе Biotage Horizon (40+М, от 5% до 75% В: этилацетат), получая метил 4-(4-(2-трет-бутокси-2-оксоэтил)-2-(метилсульфонамидометил)фенокси)бензоат (0,754 г).
Стадия D: К перемешиваемому раствору метил 4-(4-(2-трет-бутокси-2-оксоэтил)-2-(метилсульфонамидометил)фенокси)бензоата (745 мг, 1,65 ммоль) в ДМФА (10 мл) добавляли Mel (155 мкл, 2,48 ммоль), затем K2CO3 (343 мг, 2,48 ммоль). Реакционную смесь перемешивали при комнатной температуре 3 ч, затем разбавляли этилацетатом и 2М HCl. Водную фазу экстрагировали этилацетатом, и объединенные органические слои промывали раствором соли и высушивали над MgSO4. Сырую смесь очищали хроматографией на силикагеле, получая метил 4-(4-(2-трет-бутокси-2-оксоэтил)-2-((Н-метилметилсульфонамидо)метил)фенокси)бензоат (0,368 г).
Стадия Е: К перемешиваемому раствору метил 4-(4-(2-трет-бутокси-2-оксоэтил)-2-((N-метилметилсульфонамидо)метил)фенокси)бензоата (370 мг, 0,797 ммоль) в диоксане (8 мл) добавляли раствор моногидрата LiOH (43,5 мг, 1,04 ммоль) в воде (2 мл) (0,1М раствор диоксан/вода 4:1). Реакционную смесь перемешивали 5 ч, затем разбавляли дихлорметаном и 2 н. HCl. Водную фазу экстрагировали дихлорметаном, и объединенные органические слои промывали раствором соли и высушивали над MgSO4. Сырой продукт очищали хроматографией на силикагеле, получая 4-(4-(2-трет/и-бутокси-2-оксоэтил)-2-((N-метилметилсульфонамидо)метил)фенокси)бензойную кислоту (0,253 г).
Стадия F: К перемешиваемому раствору 4-(4-(2-трет-бутокси-2-оксоэтил)-2-((N-метилметилсульфонамидо)метил)фенокси)бензойной кислоты (50 мг, 0,111 ммоль) и HATU (46,5 мг, 0,122 ммоль) в ДМФА (1 мл) добавляли DIEA (23,2 мкл, 0,133 ммоль), затем 2-(4-хлорфенил)этанамин (11,2 мкл, 0,111 ммоль). Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Объединенные органические слои промывали раствором соли и высушивали над MgSO4. Сырой продукт очищали хроматографией на силикагеле, получая трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-((N-метилметилсульфонамидо)метил)фенил)ацетат (0,0115 г).
Стадия G: К перемешиваемому раствору трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-((N-метилметилсульфонамидо)метил)фенил)ацетата (11,5 мг, 0,019 ммоль) в дихлорметане (1 мл) добавляли ТФУК (500 мкл). Реакционную смесь перемешивали при комнатной температуре 4 ч и затем концентрировали. Сырой продукт затем очищали препаративной ТСХ (20% метанол/дихлорметан/0,5% АсОН), получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-((N-метилметан-2-илсульфонамидо)метил)фенил)уксусную кислоту (0,0123 г). МС+531,0 [М+1].
Пример 66
2-(3-циано-4-(4-(фенетилкарбамоил)фенокси)фенил)уксусная кислота

Стадия А: Смесь трет-бутил 2-(3-циано-4-фторфенил)ацетата (0,122 г), N-(4-хлорфенетил)-4-гидроксибензамида (0,150 г) и карбоната калия (0,086 г) растворяли в 2 мл ДМСО и перемешивали при 90°С. По истечении 1 дня реакционную смесь охлаждали, переносили на силикагель и продукт элюировали в градиентной системе растворителей от 0,5% метанол/дихлорметан до 5% метанол/дихлорметан, получая трет-бутил 2-(3-циано-4-(4-(фенетилкарбамоил)фенокси)фенил)ацетат (0,120 г).
Стадия В: К раствору трет-бутил 2-(3-циано-4-(4-(фенетилкарбамоил) фенокси)фенил)ацетата (0,120 г) в 2 мл дихлорметана добавляли 2 мл ТФУК и реакционную смесь перемешивали при комнатной температуре. По истечении 1 ч реакционную смесь концентрировали и сушили под вакуумом, получая 2-(3-циано-4-(4-(фенетилкарбамоил)фенокси)фенил)уксусную кислоту (0,100 г). МС-355,3 [М-CO2H].
Пример 67
2-(4-(4-(4-хлорфенетилкарбамоил)-3-(трифторметил)фенил)уксусная кислота

Стадия А: К перемешиваемому раствору 4-фтор-3-(трифторметил)бензальдегида (177 мкл, 1,30 ммоль) и N-(4-хлорфенетил)-4-гидроксибензамида (359 мг, 1,30 ммоль) в ДМСО (3 мл) добавляли K2CO3 (270 мг, 1,95 ммоль). Реакционную смесь перемешивали при 95°С в течение 2 ч и затем непосредственно очищали хроматографией на силикагеле, получая N-(4-хлорфенетил)-4-(4-формил-2-(трифторметил)фенокси)бензамид (0,541 г).
Стадия В: К раствору N-(4-хлорфенетил)-4-(4-формил-2-(трифторметил)фенокси) бензамида (0,225 г, 0,502 ммоль) в ТГФ (5 мл) добавляли метилсульфинил(метилтио)метан (0,125 г, 1,00 ммоль), затем гидроксид бензилтриметиламмония (0,114 мл, 0,251 ммоль) и реакционную смесь перемешивали 1 ч при 70°С. Реакционную смесь охлаждали, переносили на силикагель и продукт элюировали, используя градиент от 0,5% метанол/дихлорметан 1 до 15% метанол/дихлорметан. Выделяли (Z)-N-(4-хлорфенетил)-4-(4-(2-(метилсульфинил)-2-(метилтио)винил)-2-(трифторметил)фенокси)бензамид (0,220 г, 79,0% выход), который по данным ТСХ представлял собой смесь олефиновых изомеров в соотношении 4:1. Сырой продукт использовали непосредственно в следующей стадии.
Стадия С: К (Z)-N-(4-хлорфенетил)-4-(4-(2-(метилсульфинил)-2-(метилтио)винил)-2-(трифторметил)фенокси)бензамиду (0,220 г, 0,397 ммоль) добавляли HCl (в этаноле) (0,993 мл, 1,99 ммоль) и реакционную смесь нагревали до 70°С в течение 1 ч. Реакционную смесь концентрировали, переносили на силикагель и продукт элюировали, используя градиент от 5% этилацетат/гексан до 75% этилацетат/гексан. Выделяли этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-(трифторметил)фенил)ацетат (0,165 г, 82,1% выход) в виде белого твердого вещества.
Стадия D: К раствору этил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-(трифторметил)фенил)ацетата (0,165 г, 0,326 ммоль) в смеси метанол/ТГФ (1:1), 5 мл, добавляли LiOH-H2O (0,0684 г, 1,63 ммоль) и 3 капли воды и реакционную смесь перемешивали 1 ч. Реакционную смесь разбавляли этилацетатом, промывали 2 н. HCl и раствором соли, высушивали над сульфатом магния и концентрировали. Сырое вещество очищали хроматографией на силикагеле, элюируя в градиенте от 0,5% метанол/дихлорметан с добавлением 0,5% АсОН до 7% метанол/дихлорметан с добавлением 0,5% АсОН, получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-(трифторметил)фенил)уксусную кислоту (0,112 г). МС+478,3 [М+1].1H ЯМР (400 МГц, CD3OD) δ8,49 (дт, NH), 7,79 (д, J=8,7 Гц, 2Н), 7,68 (с, 1Н), 7,53 (д, J=4,3 Гц, 1Н), 7,28 (д, J=8,5 Гц, 2Н), 7,23 (д, J=8,6 Гц, 2Н), 7,02-7,06 (м, 3Н), 3,71 (с, 2Н), 3,56 (т, J=6,8 Гц, 2Н), 2,90 (т, J=7,3 Гц, 2Н).
Пример 68
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-цианофенил)пропановая кислота

Стадия А: К перемешиваемому раствору диизопропиламина в 2 мл ТГФ при -78°С добавляли н-бутиллитий (0,61 мл). Реакционную смесь перемешивали 30 мин, по истечении данного времени добавляли по каплям раствор трет-бутил 2-(3-циано-4-фторфенил)ацетата (0,300 г) в ТГФ (2 мл) в течение 20 мин при -78°С. Реакционную смесь нагревали до 0°С и затем переносили в колбу, содержащую 2 мл ТГФ и Mel (0,080 мл). Реакционную смесь подкисляли 2М HCl и экстрагировали этилацетатом. Объединенные органические слои затем промывали раствором соли и высушивали над MgSO4. Концентрированный продукт очищали хроматографией на силикагеле с использованием системы растворителей от 5% до 50% этилацетат/гексан, получая трет-бутил 2-(3-циано-4-фторфенил)пропаноат (0,140 г).
Стадия В: В колбу загружали трет-бутил 2-(3-циано-4-фторфенил)пропаноат (0,070 г), N-(4-хлорфенетил)-4-гидроксибензамид (0,105 г) и карбонат калия (0,047 г) в 1 мл ДМСО и перемешивали при 95°С в течение одного дня. Реакционную смесь экстрагировали дихлорметаном и промывали водой. Органический слой собирали, высушивали над MgSO4, фильтровали и концентрировали. Сырое вещество очищали хроматографией на силикагеле, элюирование системой растворителей 5%-80% этилацетат/гексан дало в результате трет-бутил 2-(4-(4-((2,4-дихлорфенетил)карбамоил)фенокси)-3-цианофенил)пропаноат (0,102 г).
Стадия С: В колбу загружали трет-бутип 2-(4-(4-((4-хлорфенетил)карбамоил) фенокси)-3-цианофенил)пропаноат (0,098 г) в 1 мл дихлорметана и 1 мл ТФУК и реакционную смесь перемешивали 1 ч. Реакционную смесь концентрировали и сушили в вакууме. Получали 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-
цианофенил)пропановую кислоту (0,080 г). МС+449,1 [М+1].
Пример 69
2-(3-циано-4-(2,4-дихлорфенетилкарбамоил)фенокси)фенил)пропановая кислота

Получено способом, описанным в примере 68, при замене N-(4-хлорфенетил)-4-гидроксибензамида на стадии В N-(2,4-дихлорфенетил)-4-гидроксибензамидом. МС+483,0 [М+1].
Пример 70
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-метоксифенил)уксусная кислота

Стадия А: Метил 2-(4-гидрокси-3-метоксифенил)ацетат (0,204 г, 1,04 ммоль), N-(4-хлорфенетил)-4-иодобензамид (0,200 г, 0,519 ммоль), 2,2,6,6-тетраметил-3,5-гептандион (0,00956 г, 0,0519 ммоль), Cu(I)Cl (0,0257 г, 0,259 ммоль) и Cs2CO3 (0,338 г, 1,04 ммоль) перемешивали в NMP (2 мл) в течение 1 ч. Реакционную смесь переносили на силикагель и продукт элюировали, используя градиент от 5% этилацетат/гексан до 100% этилацетат/гексан. Выделяли метил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-метоксифенил)ацетат (0,105 г, 44,6% выход) в виде желтого твердого вещества.
Стадия В: К раствору метил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-метоксифенил)ацетата (0,065 г, 0,143 ммоль) в смеси метанол/ТГФ (1:1, 3 мл) добавляли NaOH (0,100 мл, 0,500 ммоль). Через 5 мин реакционную смесь гасили 10 мл 2 н. HCl, и продукт экстрагировали этилацетатом. Органический слой промывали раствором соли, высушивали над сульфатом магния и концентрировали. Сырое вещество очищали хроматографией на силикагеле, элюируя в градиенте от 0,5% метанол/дихлорметан (с добавлением 0,5% АсОН) до 7,5% метанол/дихлорметан (с добавлением 0,5% АсОН), получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-метоксифенил)уксусную кислоту (0,041 г, 65,1% выход) в виде белого твердого вещества.1Н ЯМР (400 МГц, CD3OD) δ8,37-8,41 (дт, NH), 7,70 (д, J=8,7 Гц, 2Н), 7,28 (д, J=8,6 Гц, 2Н), 7,23 (д, J=8,5 Гц, 2Н), 7,08 (с, 1Н), 7,02 (д, J=8,6 Гц, 1Н), 6,91 (д, J=8,6 Гц, 2Н), 3,64 (с, 2Н), 3,56 (т, J=6,8 Гц, 2Н), 2,88 (т, J=7,4 Гц, 2Н).
Пример 71
2-(3-циано-4-(4-(2,4-дихлорфенетилкарбамоил)-3-фторфенокси)фенил)уксусная кислота

Стадия А: К перемешиваемому раствору 2-фтор-4-гидроксибензойной кислоты (200 мг, 1,28 ммоль) в ДМФА (3 мл) при комнатной температуре добавляли HATU (536 мг, 1,41 ммоль) и DIEA (268 мкл, 1,54 ммоль). Реакционную смесь перемешивали 30 мин и затем добавляли 2-(2,4-дихлорфенил)этанамин (193 мкл, 1,28 ммоль). Реакционную смесь перемешивали в течение ночи, затем концентрировали и разбавляли водой и дихлорметаном. Водный слой экстрагировали дихлорметаном и объединенные органические слои промывали раствором соли и высушивали над MgSO4. Сырой продукт очищали хроматографией на силикагеле, получая N-(2,4-дихлорфенетил)-2-фтор-4-гидроксибензамид (0,420 г).
Стадия В: К перемешиваемому раствору N-(2,4-дихлорфенетил)-2-фтор-4-гидроксибензамида (420 мг, 1,28 ммоль) и трет-бутил 2-(3-циано-4-фторфенил)ацетата (361 мг, 1,54 ммоль) в ДМСО (6 мл) добавляли K2CO3 (265 мг, 1,92 ммоль). Реакционную смесь перемешивали при нагревании при 70°С один час. Затем реакционную смесь перемешивали при 90°С в течение ночи. Реакционную смесь разбавляли водой и этилацетатом. Водную фазу экстрагировали этилацетатом, и объединенные органические слои промывали 10%-ным раствором Na2CO3 и раствором соли, высушивали над MgSO4, фильтровали и концентрировали. Сырой продукт очищали хроматографией на силикагеле, получая трет-бутил 2-(4-(4-((2,4-дихлорфенетил)карбамоил)-3-фторфенокси)-3-цианофенил)ацетат (0,0157 г).
Стадия С: К перемешиваемому раствору трет-бутил 2-(4-(4-((2,4-дихлорфенетил)карбамоил)-3-фторфенокси)-3-цианофенил)ацетата (15 мг, 0,0276 ммоль) в дихлорметане добавляли ТФУК. Реакционную смесь перемешивали 2 ч и затем концентрировали. Сырое вещество очищали препаративной ТСХ (10% метанол/0,5% АсОН/дихлорметан). Соответствующий участок собирали и затем очищали препаративной ТСХ повторно, получая 2-(4-(4-((2,4-дихлорфенетил)карбамоил)-3-фторфенокси)-3-цианофенил)уксусную кислоту (0,0042 г).1Н ЯМР (400 МГц, CD3OD) δ7,70-7,74 (м, 2Н), 7,61 (д, J=7.8 Гц, 1Н), 7,43 (с, 1Н), 7,33 (д, J=8,6 Гц, 1Н), 7,26 (д, J=7,8 Гц, 1Н), 7,13 (д, J=8,7 Гц, 1Н), 6,88-6,94 (м, 2Н), 3,52-3,65 (м, 4Н), 3,05 (т, J=7,0 Гц, 2Н).
Пример 72
2-(3-циано-4-(4-(2,6-дихлорфенетилкарбамоил)фенокси)фенил)уксусная кислота

Стадия A: TFP смолу (1,33 ммоль/г; 2,872 г, 3,820 ммоль) помещали в реактор и добавляли ДМА (5 мл) для набухания смолы. Реактор помещали на встряхиватель на 15 мин и к смеси добавляли 4-(4-(2-трет-бутокси-2-оксоэтил)-2-цианофенокси)бензойную кислоту (1,500 г, 4,245 ммоль), DIC (0,7311 мл, 4,669 ммоль) и ДМАП (0,5704 г, 4,669 ммоль) и реактор помещали на встряхиватель. Смесь фильтровали, промывали ДМА, ТГФ, дихлорметаном и эфиром, и высушивали на воздухе, получая 3,11 г связанного со смолой (4-(4-(2-трет-бутокси-2-оксоэтил)-2-цианофенокси)бензоата.
Стадия В: Связанный с TFP смолой эфир (4-(4-(2-/ирети-бутокси-2-оксоэтил)-2-цианофенокси)бензоат) (0,1968 г, 0,09840 ммоль) помещали в виалу. Добавляли ТГФ (2 мл) и оставляли смолу набухать. В виалу добавляли 2-(2,6-Дихлорфенил)этанамин (1М в ТГФ; 0,08200 мл, 0,082 ммоль), и помещали виалу на шейкер. Через 16 ч смесь декантировали и смолу промывали ТГФ (3×2 мл). Объединенный ТГФ раствор концентрировали при пониженном давлении. Остаток высушивали под высоким вакуумом, получая трет-бутил 2-(4-(4-((2,6-дихлорфенетил)карбамоил)фенокси)-3-цианофенил)ацетат (0,034 г), который использовали в следующей стадии без дополнительной очистки.
Стадия С: трет-бутил 2-(4-(4-((2,6-дихлорфенетил)карбамоил)фенокси)-3-цианофенил)ацетат (34 мг, 0,065 ммоль) растворяли в дихлорметане (1 мл) и добавляли ТФУК (1 мл). После перемешивания в течение 45 мин, смесь концентрировали при пониженном давлении, получая 2-(3-циано-4-(4-(2,6-дихлорфенетилкарбамоил)фенокси)фенил)уксусную кислоту (0,034 г).
Пример 73
2-(3-циано-4-(4-(4-фторфенетилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В 2-(4-фторфенил)этанамином.
Пример 74
2-(3-циано-4-(4-(3-метоксифенетилкарбамоил)фенокси)фенил)уксусная кислота
Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В 2-(3-метоксифенил)этанамином.
Пример 75
2-(3-циано-4-(4-(4-трет-бутилфенетилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В 2-(4-трет-бутилфенил)этанамином.
Пример 76
2-(3-циано-4-(4-(4-трифторметилфенетилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В 2-(4-трифторметилфенил)этанамином.
Пример 77
2-(3-циано-4-(4-(3-хлорфенетилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В 2-(3-хлорфенил)этанамином.
Пример 78
2-(3-циано-4-(4-(3-фторфенетилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В 2-(3-фторфенил)этанамином.
Пример 79
2-(3-циано-4-(4-(4-метоксифенетилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В 2-(4-метоксифенил)этанамином.
Пример 80
2-(3-циано-4-(4-(4-метилфенетилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В 2-(4-метилфенил)этанамином.
Пример 81
2-(3-циано-4-(4-(3,4-дихлорфенетилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В 2-(3,4-дихлорфенил)этанамином.
Пример 82
2-(4-(4-(2-(4-хлорфенил)-2-гидроксиэтилкарбамоил)фенокси)-3-цианофенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В гидрохлоридом 2-амино-1-(4-хлорфенил)этанола.
Пример 83
2-(4-(4-(1-(4-хлорфенил)циклопропилкарбамоил)фенокси)-3-цианофенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В гидрохлоридом 1-(4-хлорфенил)циклопропанамина.
Пример 84
2-(3-циано-4-(4-(2-фенилциклопропилкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,б-дихлорфенил)этанамина на стадии В 2-фенилциклопропиламином.
Пример 85
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-цианофенил)-2-фторуксусная кислота
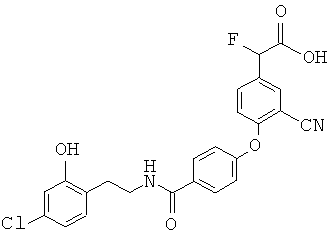
Стадия А: трет-бутил 2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-цианофенил) ацетат (0,400 г) и 4-метилбензолсульфонилазид (0,193 г) растворяли в 5 мл ацетонитрила и обрабатывали DBU (0,152 мл). Реакционную смесь перемешивали 4 ч, затем концентрировали, разбавляли этилацетатом и промывали водой. Органический слой высушивали, фильтровали, концентрировали на силикагеле и очищали хроматографией на силикагеле, получая трет-бутил 2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-цианофенил)-2-диазоацетат (0,48 г) в виде ярко-желтого твердого вещества.
Стадия В: В колбу загружали трет-бутил 2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-цианофенил)-2-диазоацетат (0,108 г), 5 мл эфира и HF пиридин (0,30 г). Реакционную смесь перемешивали при 40°С 30 мин. Реакционную смесь разбавляли этилацетатом и нейтрализовывали насыщенным водным бикарбонатным раствором. Органический слой отделяли и промывали раствором соли, высушивали, фильтровали и концентрировали на силикагеле. Сырой продукт очищали хроматографией на силикагеле, получая трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-цианофенил)-2-фторацетат (0,026 г) в виде бесцветного масла.
Стадия С: В колбу загружали трет-бутил 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-цианофенил)-2-фторацетат (0,026 г), 0,5 мл дихлорметана и 0,2 мл ТФУК. Реакционную смесь концентрировали и остаток растворяли в минимальном количестве дихлорметана и помещали в высокий вакуум, получая твердое вещество. Процедуру повторяли, получая 2-(4-(4-((4-хлорфенетил)карбамоил)фенокси)-3-цианофенил)-2-фторуксусную кислоту (0,024 г) в виде коричневатого твердого вещества.
Пример 86
2-(4-(4-((1-(4-хлорфенил)циклопропил)метилкарбамоил)фенокси)-3-цианофенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В (1-(4-хлорфенил)пиклопропил)метанамином.
Пример 87
2-(4-(4-(2-(4-хлорфенил)циклопропилкарбамоил)фенокси)-3-цианофенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В 2-(4-хлорфенил)пиклопропанамином.
Пример 88
2-(3-циано-4-(4-(1,2,3,4-тетрагидронафталин-2-илкарбамоил)фенокси)фенил)уксусная кислота

Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В гидрохлоридом 1,2,3,4-тетрагидронафталин-2-амина.
Пример 89
2-(3-циано-4-(4-(6-метокси-1,2,3,4-тетрагидронафталин-2-илкарбамоил)фенокси)фенил)уксусная кислота
Получено способом, описанным в примере 72, при замене 2-(2,6-дихлорфенил)этанамина на стадии В гидрохлоридом 6-метокси-1,2,3,4-тетрагидронафталин-2-амина.
Реферат
Предложены новые соединения формулы (I), в которой R1, R2, R7, R8, R9, R10 и А имеют значения, указанные в описании, проявляющие ингибирующую активность связывания с DP2 рецептором, пригодные для лечения иммунологических заболеваний, а также способы их получения. 7 н. и 17 з.п., 89 пр.
Формула

или его фармацевтически приемлемая соль, где:
R1 представляет собой Ar1-L1-W-L2-;
L2 представляет собой -(CRcRd)m-;
W представляет собой -CONR3a- или -NR3bCO-;
каждый из R3a и R3b представляет собой Н или метил;
L1 представляет собой -(CRaRb)n-, -(СН=СН)- или -O(CRaRb) при условии, что когда W представляет собой -NR3СО-, тогда L1 не является фрагментом -(СН=СН)-;
n и m независимо равны 0,1 или 2;
каждый из Ra, Rb, Rc и Rd независимо представляет собой Н, F, ОН, метил или циклопропил, или Ra и Rb или Rc и Rd вместе с атомом углерода, к которому они присоединены, образуют циклопропильное кольцо;
Аr1 представляет собой фенил или нафтил, каждый из которых является незамещенным или замещенным одним или более заместителями, независимо выбранными из F, Cl, CN, CF3, CHF2, CH2F, SF5, метила, этила, циклопропила, т-бутила или ОМе, или Аr1 представляет собой 1,2,3,4-тетрагидронафтил, который является незамещенным или замещенным метоксигруппой,
при условии, что когда Аr1 представляет собой нафтил или 1,2,3,4-тетрагидронафтил, тогда n равен 0;
R2 представляет собой Н, С1-С6 алкил, остаток аминокислоты или дипептида, или CHRe(CH2)qRf;
q равен 1-6;
Re представляет собой Н, метил или этил;
Rf представляет собой NRgRh, в котором каждый из Rg и Rh независимо представляет собой атом водорода или С1-С4алкильную группу, или Rg и Rh вместе с атомом азота, к которому они присоединены, образуют 5-6-членное гетероциклическое кольцо, необязательно содержащее в кольце второй гетероатом, выбранный из N и О, где указанное гетероциклическое кольцо необязательно замещено одной или более группами, независимо выбранными из C1-С6алкила;
А представляет собой CN, CH2NH2, CH2NR4aC(=O)R5 или CH2NR4bSO2R6, Сl, ОМе, (С1-С4) алкил, циклопропил, Н, F, Вr, СН2NН(С1-С4алкил), СН2N(С1-С4алкил)2, тиенил или фенил, который является незамещенным или замещенным SO2Me;
каждый из R4a и R4b представляет собой Н или метил;
R5 представляет собой С1-С6алкил, С1-С6алкокси, С3-С6циклоалкил, hetAr1или Аr2;
R6 представляет собой C1-С6алкил, NН(С1-С6алкил), N(С1-С6алкил)2, Аr3или hetAr2;
hetAr1 представляет собой 6-членный гетероарил с 1-4 атомами азота, и который является незамещенным или замещенным одной или более группами, независимо выбранными из атома галогена и группы формулы -NR5aR5b, в которой каждый из R5a и R5b независимо представляет собой атом водорода или (С1-С4) алкильную группу;
hetAr2 представляет собой 5-6-членный гетероарил с 1-4 атомами азота, и который является незамещенным или замещенным одной или более группами, независимо выбранными из С1-С4алкила;
Аr2 представляет собой фенил, который является незамещенным или замещенным одной или более группами, независимо выбранными из атома галогена, CN, SF5, циклопропила, С1-С4алкильной группы, С1-С4алкоксигруппы и фтор С1-С4алкильной группы;
Аr3 такой, как описано для Аr2;
R7 и R8 независимо представляют собой Н, метил или F;
R9 представляет собой Н или метил; и
R10 представляет собой Н или F.

в которой R1 представляет собой Аr1-L1-W-L2-;
L2 представляет собой -(CRcRd)m-;
W представляет собой -CONR3a- или -NR3bCO-;
каждый из R3a и R3b представляет собой Н или метил;
L1 представляет собой -(CRaRb)n-, -(СН=СН)- или -O(CRaRb)- при условии, что когда W представляет собой -NR3CO-, тогда L1 не является фрагментом -(СН=СН)-;
n и m независимо равны 0, 1 или 2;
каждый из Ra, Rb, Rc и Rd независимо представляет собой Н, F, метил или циклопропил, или Ra и Rb или Rc и Rd вместе с атомом углерода, к которому они присоединены, образуют циклопропильное кольцо;
Аr1 представляет собой фенил или нафтил, каждый из которых является незамещенным или замещенным одним или более заместителями, независимо выбранными из F, Cl, CN, CF3, CHF2, CH2F, SF5, метила, этила и циклопропила, при условии, что когда Аr1 представляет собой нафтил, тогда n равен 0;
R2 представляет собой Н, С1-С6алкил, остаток аминокислоты или дипептида, или CHRe(CH2)qRf;
q равен 1-6;
Re представляет собой Н, метил или этил;
Rf представляет собой NRgRh, в котором каждый из Rg и Rh независимо представляет собой атом водорода или С1-С4алкильную группу, или Rg и Rh вместе с атомом азота, к которому они присоединены, образуют 5-6-членное гетероциклическое кольцо, необязательно содержащее в кольце второй гетероатом, выбранный из N и О, где указанное гетероциклическое кольцо необязательно замещено одной или более группами, независимо выбранными из С1-С6алкила;
А представляет собой CN, CH2NH2, CH2NR4aC(=O)R5, CH2NR4bSO2R6, Cl, ОМе, (С1-С4) алкил или циклопропил;
каждый из R4a и R4b представляет собой Н или метил;
R5 представляет собой С1-С6алкил, С1-С6алкокси, С3-С6циклоалкил, hetAr1или Аr2;
R6 представляет собой С1-С6алкил, NН(С1-С6алкил), N(С1-С6алкил)2, Аr3или hetAr2;
hetAr1 представляет собой 6-членный гетероарил с 1-4 атомами азота, который является незамещенным или замещенным одной или более группами, независимо выбранными из атома галогена и группы формулы -NR5aR5b, в которой каждый из R5a и R5b независимо представляет собой атом водорода или (С1-С4) алкильную группу;
hetAr2 представляет собой 5-6-членный гетероарил с 1-4 атомами азота, который является незамещенным или замещенным одной или более группами, независимо выбранными из С1-С4алкила;
Аr2 представляет собой фенил, который является незамещенным или замещенным одной или более группами, независимо выбранными из атома галогена, CN, SF5, циклопропила, С1-С4алкильной группы, С1-С4алкоксигруппы и фтор С1-С4 алкильной группы;
Аr3 такой, как описано для Аr2; и
R7 и R8 независимо представляют собой Н или метил.
для соединения формулы (I), в которой А представляет собой CN, R7 и R8независимо представляют собой Н или Me, и R10 представляет собой Н или F, взаимодействие соответствующего соединения формулы (II)

в которой Р1 представляет собой атом водорода или защитную группу для карбоксила, и Z1 представляет собой уходящий атом или группу, с соответствующим соединением формулы (III)

в которой Rl0a представляет собой Н или F, в присутствии основания; и удаление любой защитной группы или групп и, при необходимости, образование фармацевтически приемлемой соли.
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-((4-фторфенилсульфонамидо)метил)фенил)уксусной кислоты;
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-(метилсульфонамидометил)фенил)уксусной кислоты;
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-((2,4-дихлорфенилсульфонамидо)метил)фенил)уксусной кислоты;
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-(никотинамидометил)фенил)уксусной кислоты;
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-((N,N-диметилсульфамоиламино)метил)фенил)уксусной кислоты;
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-(метилсульфонамидометил)фенил)уксусной кислоты;
2-(3-(циклогексанкарбоксамидометил)-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)-уксусной кислоты;
2-(3-(ацетамидометил)-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)уксусной кислоты;
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-(изоникотинамидометил)фенил)уксусной кислоты;
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-((4-фторбензамидо)метил)фенил)уксусной кислоты;
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-(пиколинамидометил)фенил)уксусной кислоты;
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-((метоксикарбониламино)метил)фенил)-уксусной кислоты;
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-((пиридин-3-сульфонамидо)метил)фенил)-уксусной кислоты;
2-(4-(4-(3,4-дихлорфенилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)-уксусной кислоты;
2-(4-(4-(3,4-дихлорфенетилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)уксусной кислоты;
2-(3-(метилсульфонамидометил)-4-(4-(нафталин-2-ил-карбамоил)фенокси)фенил)-уксусной кислоты;
2-(4-(4-(4-фторфенетилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)-уксусной кислоты;
2-(3-(метилсульфонамидометил)-4-(4-(фенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-(метилсульфонамидометил)-4-(4-(фенилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(4-(4-(бензилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)-уксусной кислоты;
2-(4-(4-(2-(4-хлорбензиламино)-2-оксоэтил)фенокси)-3-(метилсульфонамидометил)фенил)уксусной кислоты;
2-(4-(4-(2-(бензиламино)-2-оксоэтил)фенокси)-3-(метилсульфонамидометил)фенил)-уксусной кислоты;
2-(3-(метилсульфонамидометил)-4-(4-(2-оксо-2-(фенетиламино)этил)фенокси)фенил)-уксусной кислоты;
2-(4-(4-(2-(бензил(метил)амино)-2-оксоэтил)фенокси)-3-(метилсульфонамидометил)фенил)уксусной кислоты;
2-(3-(метилсульфонамидометил)-4-(4-(2-оксо-2-(фениламино)этил)фенокси)фенил)-уксусной кислоты;
2-(4-(4-бензамидофенокси)-3-((4-фторфенилсульфонамидо)метил)фенил)уксусной кислоты;
2-(4-(4-(4-хлорбензамидо)фенокси)-3-((4-фторфенилсульфонамидо)метил)фенил)-уксусной кислоты;
2-(4-(4-(3-хлорбензамидо)фенокси)-3-((4-фторфенилсульфонамидо)метил)фенил)-уксусной кислоты;
2-(4-(4-(3,4-дифторбензамидо)фенокси)-3-(метилсульфонамидометил)фенил)уксусной кислоты;
2-(4-(4-(2-хлорбензамидо)фенокси)-3-(метилсульфонамидометил)фенил)уксусной кислоты;
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-метоксифенил)уксусной кислоты;
2-(4-(4-(бензилоксикарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)уксусной кислоты;
2-(3-хлор-4-(4-(4-хлорфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-циано-4-(4-(3,4-дихлорбензамидо)фенокси)фенил)пропионовой кислоты;
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-(метилсульфонамидометил)фенил)пропионовой кислоты;
2-(4-(4-(3,4-дихлорбензамидо)фенокси)-3-(метилсульфонамидометил)фенил)-2-метилпропионовой кислоты;
2-(3-(метилсульфонамидометил)-4-(4-(4-(трифторметил)фенилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-(метилсульфонамидометил)-4-(4-(3-(трифторметил)фенилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(4-(4-(4-хлор-3-фторфенилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)уксусной кислоты;
2-(4-(4-(3-хлор-4-фторфенилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-фторфенил)уксусной кислоты;
2-(3-циано-4-(4-(4-хлорфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-циано-4-(4-(2,4-дихлорфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-бром-4-(4-(2,4-дихлорфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3,5-диметилфенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-метилфенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-(тиофен-2-ил)фенил уксусной кислоты;
2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)бифенил-3-ил)уксусной кислоты;
2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)-3'-(метилсульфонил)бифенил-3-ил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-циклопропилфенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-этилфенил)уксусной кислоты;
2-(6-(4-(4-хлорфенетилкарбамоил)фенокси)-4'-(метилсульфонил)бифенил-3-ил)уксусной кислоты;
2-(3-(метилсульфонамидометил)-4-(4-(3-(трифторметил)фенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-(метилсульфонамидометил)-4-(4-(4-трифторметил)фенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(4-(4-((1-(4-хлорфенил)циклопропил)метилкарбамоил)фенокси)-3-(метилсульфонамидометил)фенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-(никотинамидометил)фенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-((пиридин-3-сульфонамидо)метил)фенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-((1-метил-1H-имидазол-5-сульфонамидо)метил)фенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-((6-диметиламино)никотинамидо)метил)фенил)уксусной кислоты;
2-(3-((6-аминоникотинамидо)метил)-4-(4-(4-хлорфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-((диметиламино)метил)фенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-((N-метилметилсульфонамидо)метил)фенил)уксусной кислоты;
2-(3-циано-4-(4-(фенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-цианофенил)пропионовой кислоты;
2-(3-циано-4-(2,4-дихлорфенетилкарбамоил)фенокси)фенил)пропионовой кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-метоксифенил)уксусной кислоты;
2-(3-циано-4-(4-(2,4-дихлорфенетилкарбамоил)-3-фторфенокси)фенил)уксусной кислоты;
2-(3-циано-4-(4-(2,6-дихлорфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-циано-4-(4-(4-фторфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-циано-4-(4-(3-метоксифенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-циано-4-(4-(4-трет-бутилфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-циано-4-(4-(4-трифторметилфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-циано-4-(4-(3-хлорфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-циано-4-(4-(3-фторфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-циано-4-(4-(4-метоксифенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-циано-4-(4-(3,4-дихлорфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(3-циано-4-(4-(4-метилфенетилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(4-(4-(2-(4-хлорфенил)-2-гидроксиэтилкарбамоил)фенокси)-3-цианофенил)уксусной кислоты;
2-(4-(4-(1-(4-хлорфенил)циклопропилкарбамоил)фенокси)-3-цианофенил)уксусной кислоты;
2-(3-циано-4-(4-(2-фенилциклопропилкарбамоил)фенокси)фенил)уксусной кислоты;
2-(4-(4-(4-хлорфенетилкарбамоил)фенокси)-3-цианофенил)-2-фтор уксусной кислоты;
2-(4-(4-((1-(4-хлорфенил)циклопропил)метилкарбамоил)фенокси)-3-цианофенил)уксусной кислоты;
2-(4-(4-(2-(4-хлорфенил)циклопропилкарбамоил)фенокси)-3-цианофенил)уксусной кислоты;
2-(3-циано-4-(4-(1,2,3,4-тетрагидронафталин-2-ил-карбамоил)фенокси)фенил)уксусной кислоты; и
2-(3-циано-4-(4-(6-метокси-1,2,3,4-тетрагидронафталин-2-ил-карбамоил)фенокси)фенил)уксусной кислоты.
для соединения формулы (I), в которой R7 и R8 независимо представляют собой Н или Me, и R10 представляет собой Н, сочетание соединения формулы (VII)

в которой P5 такой, как описано для Р1, А2 представляет собой А или его защищенную форму, и Rla представляет собой H-Xa-L2-, где Хапредставляет собой HN или ОС(=O), или его реакционноспособного производного;
с соединением формулы (VIII)

в которой Хb представляет собой С(=O)O или NH, или его реакционноспособным производным; и
удаление любой защитной группы или групп и, при необходимости, образование фармацевтически приемлемой соли.
для соединения формулы (I), в которой А представляет собой Н, F или Сl, R7 и R8 независимо представляют собой Н или Me, и R представляет собой Н, сочетание соответствующего соединения формулы (IX)

в которой А3 представляет собой Н, F или Сl, и Р1 такой, как описано для Р1, с соответствующим соединением формулы (X)

где Е представляет собой электроноакцепторную группу, в присутствии основания; и, при необходимости, удаление указанной электроноакцепторной группы; и
удаление любой защитной группы или групп и, при необходимости, образование фармацевтически приемлемой соли.
Комментарии