Способ нитрования фенольных соединений - RU2318797C2
Код документа: RU2318797C2
Описание
Данное изобретение относится к способу нитрования фенольных соединений, которые являются полезными для получения ортонитрофенолов.
Нитрование ароматических соединений посредством электрофильного ароматического замещения представляет собой основную органическую реакцию, которая всесторонне описана и рассмотрена в химической литературе (Olah, G.A. et al., Nitration: Methods and Mechanisms, VCH, New York, 1989 and Taylor, R., Electrophilic Aromatic Substitution, J. Wiley & Sons, Chichester, 1990). До некоторой степени удивительно, однако, что несмотря на данное обилие информации, в большинстве важных в коммерческом отношении промышленных способах до сих пор используют "классическую" технологию, для которой необходимы смеси азотной и серной кислот. Применение таких коррозионных реагентов (обычно в избытке) создает серьезные прецеденты с защитой окружающей среды, а обработка и утилизация "использованных" кислот является дорогой. Существенными аспектами с точки зрения химии являются проблемы, связанные с избыточным нитрованием и образованием нежелательных окисленных побочных продуктов, которые часто трудно удалять из требуемого продукта. Кроме того, еще одна серьезная проблема, связанная с нитрованием ароматических соединений касается распределения продукта с точки зрения изомерного соотношения орто:мета:пара (т.е. региоселективности). Для промышленного способа нитрования желательно, чтобы он проявлял хорошую степень региоселективности в данном отношении, когда возможно образование региоизомера. Это обычно имеет место в случае, когда представляющими интерес коммерческими продуктами являются в особенности паранитроизомеры. Данная региоселективность определяется пространственными факторами и/или электронными эффектами и влиянием растворителей. Нитрование, например, ароматического кольца, содержащего, например, электронодонорный заместитель
(-алкил, -ОН, -О-алкил и т.д.) обычно приводит к увеличению содержания смеси с преобладанием орто- и паранитрованных продуктов, как правило следующей статистическому распределению. Пространственный объем нитрующего реагента и/или заместителей в ароматическом кольце имеет тенденцию оказывать благоприятное влияние на образование парапродукта. Во многих случаях, применение реагентов и катализаторов на носителях также можно использовать для оказания более благоприятного влияния на образование параизомера (Smith, K., Solid Supports and Catalysts in Organic Synthesis; Ellis Horwood: Chichester, 1992).
Следовательно, логично то, что нитрование ароматических фенольных (Ar-OH) соединений, без заместителей, отличных от атома водорода в параположении, представляет конкретные проблемы, когда возможным является образование орто- и парапродуктов вследствие сильного активирующего эффекта электронодонорной гидроксильной группы (когда присутствуют паразаместители, например алкил, параположение не является доступным для протекания электрофильного ароматического замещения и, в таких случаях, получают только ортонитрованные продукты). Применение смесей сильных кислот обычно приводит к появлению значительно окрашенных и сложных реакционных смесей вследствие окислительной деградации субстрата. Нитрования самого фенола можно легко достичь в более мягких условиях с применением разбавленной азотной кислоты в хлорированном растворителе с приемлемым объединенным выходом (61%) при соотношении орто:пара изомеров нитрофенола, равном 1:2,3 (Vollhardt, K.P.C. and Schore, N.E., Organic Chemistry, 2nd ed; W.H. Freeman, New York, 1994). Нитрование с использованием нитрата натрия в серной кислоте приводит также к объединенному выходу 61% при соотношении орто:пара изомеров, равном 1,4:1 (Vogel, A.I., Vogel's Textbook of Practical Organic Chemistry, 5th ed; J. Wiley & Sons, New York, 1989). Недавно было заявлено трехстадийное параселективное нитрование производных фенола (Kanno, H. et al., DE 19723214 A1) и сообщено о других параселективных нитрующих реагентах, включая новые комплексы нитрата металла с тетраоксидом диазота (Firouzabadi, H. et al., Synth. Commun., 27(19), 3301-3311 (1997); Iranpoor, N. et al., Synth. Commun., 28(15), 2773-2781 (1998)), нитраты металлов в неводных и апротонных условиях (Firouzabadi, H. et al., Iran. J. Chem., 16(2), 48-58 (1997)) и ионные комплексы тетраоксида диазота с 18-краун-6 (Iranpoor, N. et al., Synth. Commun., 29(19), 3295-3302 (1999)).
Неудивительно, что всего несколько способов описано для селективного ортонитрования паранезамещенных фенольных соединений. Было сообщено о нитратных солях лантанидов (III) в этилацетате при кипячении с обратным холодильником для селективного метанаправленного нитрования 3-замещенных фенолов (Gu, S. et al., Synth. Commun., 27(16), 2793-2797 (1997)), но данные лантанидные реагенты являются чрезмерно дорогими и сама реакционная смесь выделяет пары токсичного газа диоксида азота.
Селективное ортонаправленное нитрование нескольких фенольных соединений привлекло некоторое внимание вследствие возможной применимости продуктов. Была описана двухстадийная методика для селективного ортонаправленного нитрования 3-метоксифенола, включающая нитрозирование с последующим окислением для получения 2-нитро-5-метоксифенола (Maleski, R.J., Synth. Commun., 23(3), 343-348 (1993)), хотя общий выход был относительно низким и региоселективность нитрозирования была несомненно увеличена в данном конкретном случае присутствием сильно орто/пара-направляющей метоксигруппы (вследствие пространственных причин, параположение относительно метоксигруппы будет более благоприятным в данном случае). Методология одностадийного нитрования, конечно, была бы более предпочтительной, чем многостадийный подход. Так называемый эффект "сопровождения" (Strazzoloni, P. et al., Bull. Chem. Soc. Jpn., 68(4), 1155-61 (1995)), описанный для селективного ортонаправленного нитрования алкилбензолов не может быть непосредственно использован для чувствительных к окислению фенольных соединений (Strazzoloni, P. et al., J. Org. Chem., 63(4), 952-958(1998)). Было заявлено почти селективное ортонитрование фенола с использованием микроэмульсионного раствора в присутствии разбавленной азотной кислоты (Chhatre, A.S. et al., J. Colloid Interface Sci., 158(1), 183-187 (1993)), но способ имеет очевидные недостатки для общих и крупномасштабных препаративных целей. Очень высокую селективность также наблюдали для тетрафторбората нитрония и поверхностно-активного вещества в ацетонитриле (Pervez, H. et al., Tetrahedron, 44, 4555 (1988)), но данные условия и реагенты также являются неудобными для крупномасштабных нитрований.
В некоторой степени более интересным является нитрование фенола с использованием "фиксации на глине" (claycop), по существу нитрата меди на глиняном носителе, который, как сообщают обеспечивает 92% выход ортонитрофенола (Gigante, B., et al., J. Org. Chem., 60, 3445-3447 (1995)). "Фиксированный на глине" реагент, хотя и является высоко ортоселективным (13:1, орто:пара), обеспечивающим высокий выход, труднодоступен на рынке и также является очень дорогим. Получение реагента является трудоемким, нагрузка (ммоль реагента на грамм глиняного носителя) является низкой и его следует хранить в течение только очень короткого времени и при низкой температуре (˜4°С). Кроме того, предполагают, что сам нитрующий реагент фактически представляет собой образованный in situ ацетилнитрат (CH3CO-ONO2), известное и потенциально взрывчатое соединение, обычно не выделяемое. Данные реакции нитрования являются довольно экзотермическими с неопределенными индукционными периодами, и, при использовании больших количеств, иногда бурно протекающими с энергичным выделением красно-коричневого газа. Строгие меры безопасности должны применяться при использовании таких соединений, которые вследствие их опасной природы не соответствуют крупномасштабному получению. В более поздней публикации описано нитрование фенола с использованием ацилнитратов, адсорбированных на силикагеле (Rodrigues, J.A.R. et al., Tetrahedron, 55, 6733-6738 (1999)), которое было заявлено для улучшения стабильности нитрующего реагента. Хотя почти идентичные селективность и выход получают в случае вышеупомянутой методики с "фиксацией на глине", в ней не удается избежать неудобного, дорогого и опасного получения ацилнитратов, и она требует последующей адсорбции на силикагеле. Как указано авторами, вследствие угрожающей природы данных веществ, было бы опасным пытаться проводить реакцию в масштабе выше 50 ммоль. Особенно следует отметить, что, хотя реагент работает крайне эффективно для самого фенола, ортоселективность при применении к другим фенольным соединениям является значительно более низкой (например, для изованилина, 0,6:1, орто:пара), указывая на то, что способ не является универсально региоселективным. Был заявлен еще один пример нитрования изованилина с использованием 70% азотной кислоты в холодном ацетоне (Napoletano, M. et al., WO 99/32449, PCT/EP98/08292), обеспечивающий 74% объединенный выход нитрованных изомеров с только незначительно улучшенной селективностью для ортонитрованного продукта (орто:пара, 1,5:1), который далее не выделяют в чистом виде.
Применимость алкилнитратов как потенциально полезных реагентов для селективного ортонаправленного нитрования пара-не/замещенных фенольных соединений не была описана в химической литературе. Одно не относящееся к вопросу сообщение описывает сочетание н-бутилнитрата и необычного кислотного катализатора (Nafion-H) для нитрования только алкилбензолов, с очевидным предпочтением для образования наименее затрудненного пара-нитрованного продукта по отношению к ортонитрованному изомеру (Olah, G. et al., J. Org. Chem., 43(24), 4628-4630 (1978). Другие недостатки включают относительно низкую стабильность конкретного используемого алкилнитрата и применение очень дорогого катализатора. Применение метилнитрата для аналогичной реакции, описанное ранее (Olah, G. et al., Synthesis, 488 (1973)), было бы крайне нежелательным вследствие потенциально взрывчатой природы метилнитрата. Группа китайских авторов недавно заявила сходное нитрование некоторых алкилбензолов с применением различного катализатора (цеолита HZSM-5), но вновь селективность была увеличена для параположения (Peng, X. et al., Nanjing Ligong Daxue Xuebao, 23(6), 539-541 (1999)).
Патент США № 3694513 относится к способу нитрования алкилфенолов азотной кислотой в присутствии вторичного или третичного спирта, вторичного алкилнитрата, альдегида или кетона.
Следовательно, в предыдущем уровне техники отсутствует безопасная, экономичная, масштабируемая и, в целом, применимая методология нитрования, которую можно использовать для дающего высокий выход региоселективного нитрования фенольных соединений, особенно, когда возможно образование смесей изомерных нитропродуктов и/или образование окисленных побочных продуктов.
Цель изобретения состоит в предоставлении полезного, с высоким выходом и, в целом, применимого способа региоселективного ортонаправленного нитрования фенольных соединений. Дальнейшей целью изобретения является предоставление способа, в котором отсутствуют недостатки предыдущего уровня техники.
Такие соединения являются особенно полезными в качестве фармацевтически эффективных соединений или предшественников, или промежуточных соединений в их производстве. Например, такие соединения можно использовать в производстве ингибиторов катехол-О-метилтрансферазы (КОМТ), которые применяются при лечении нарушений центральной и периферической нервной системы, таких как болезнь Паркинсона.
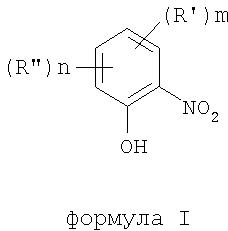
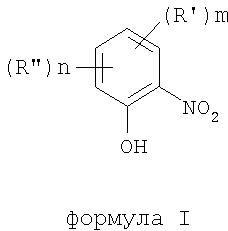
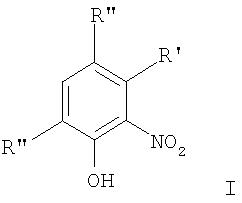
В соответствии с одним аспектом изобретения предоставлен способ получения соединений формулы I
формула I
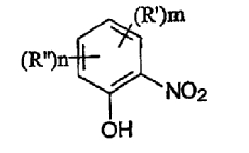
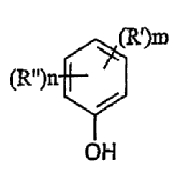
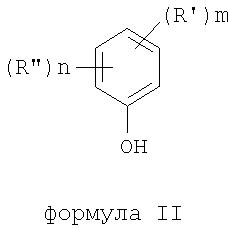
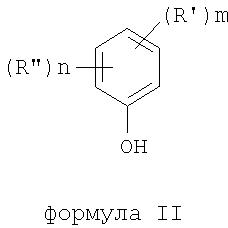
где термины R'и R" могут иметь одно и то же или различное значение и представлять: водород; низший алкил; гидрокси; низший алкокси; галоген; группу -СО-R1, где R1 означает водород, гидрокси, алкиларил, алкилгетероциклоалкил или необязательно замещенную насыщенную или частично ненасыщенную низшую алкильную или арильную группу или R1 означает группу -О-R2, где R2 означает низшую алкильную или арильную группу; группу -С=N-Ra, где Ra означает NHRa, где Ra представляет собой необязательно замещенную низшую алкильную или арильную группу или группу ORb, где Rb означает водород, низший алкил или низший алканоил; группу -С-RcRd, где Rcозначает необязательно замещенный алкилиден, где Rd представляет собой группу ORe, где Re означает необязательно замещенную низшую алканоильную или арильную группу; или R' и R", взятые вместе, означают необязательно замещенное насыщенное или частично ненасыщенное карбоциклическое кольцо; m и n независимо равны 0, 1 или 2; термин низший алкил означает углеродную цепь, прямую или разветвленную, содержащую от одного до шести атомов углерода; термин галоген означает фтор, хлор, бром или йод; термин гетероциклоалкил означает пяти- или шестичленное циклическое кольцо, включающее один или два атома кислорода, серы или азота; термин арил означает фенильную или нафтильную группу, необязательно замещенную группами алкокси, галогена или нитро; указанный способ включает взаимодействие фенольного соединения формулы II:
формула II
где термины R'и R", m и n определены выше, с алкилнитратом формулы (III)
R3-ONO2
формула III
где R3 представляет собой алкильную группу, прямую или разветвленную, содержащую от одного до шестнадцати атомов углерода или R3 представляет собой циклоалкильную группу, содержащую либо пять или шесть атомов углерода.
Реакцию предпочтительно осуществляют в присутствии кислотного катализатора в по существу инертном растворителе.
Предпочтительные алкилнитраты включают изопропилнитрат, изоамилнитрат и изооктилнитрат (2-этилгексилнитрат). Соединения формулы (III) являются известными и многие коммерчески доступны или они могут быть получены специалистами в данной области техники (напр. Olah, G. et al., Synthesis, (2), 207-208 1993). Реакцию нитрования можно осуществить перемешиванием фенольного соединения формулы II обычно с избытком предпочтительного алкилнитрата (1,2-2,5 мольных эквивалента) в инертных растворителях, таких как углеводороды, хлорированные алканы, простые эфиры или апротонные диполярные растворители или реакция может протекать в смеси вышеупомянутых растворителей. Реакция идет с применением катализаторов из минеральных или органических кислот, таких как, например, серная кислота (концентрация 20-96%), хлористоводородная кислота, фосфорная кислота, муравьиная кислота или трифторуксусная кислота, в чистом виде или, если предпочтительно, адсорбированных на инертных носителях, таких как, например, силикагель. Альтернативно, можно использовать кислоту Льюиса, такую как, например, эфират трифторида бора. Если требуется, реакция может идти с использованием сокатализатора фазового переноса, такого как галогенид тетраалкиламмония или гидросульфатной соли (1-5 мол.%). Реакцию можно осуществлять при различных температурах и давлениях, напр. между 0°С и температурой кипения реакционной смеси при применяемом давлении. Реакционные продукт(ы) могут быть простым образом выделены после промывки реакционной смеси водой и выпариванием реакционного растворителя. При необходимости, отделение основного ортонитрованного продукта от любой минорной загрязняющей примеси нитроизомера или побочных продуктов, присутствующих в неочищенном продукте, может быть быстро достигнуто перегонкой или хроматографией на подходящей неподвижной фазе, такой как силикагель или оксид алюминия, с применением соответствующей системы растворителя для элюирования. Более удобно, неочищенный продукт может быть перекристаллизован из подходящего растворителя, в котором требуемый ортонитрованный продукт имеет более ограниченную растворимость, чем любые загрязняющие нитроизомеры или побочные продукты. Очищенные продукты могут далее быть охарактеризованы аналитическим сравнением с эталонными стандартами (напр. ТСХ) и/или положение нитрования может быть быстро определено ЯМР-спектроскопией. Преимущество данного способа состоит в том, что он обеспечивает высокие выходы; общий выход данной реакции нитрования часто превышает 75%. Еще одним преимуществом данного способа является то, что он региоселективен, причем региоселективность благоприятно воздействует на преобладающее образование ортонитрованного продукта.
Чтобы избежать сомнений, авторы утверждают, что в формуле I R' и R" могут быть замещены по любому положению фенильной группы.
Дополнительно предусматривается, что соединения, представленные формулой I, могут быть использованы в качестве предшественников или промежуточных соединений при получении дальнейших фармацевтически активных/эффективных соединений.
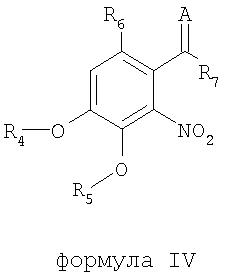
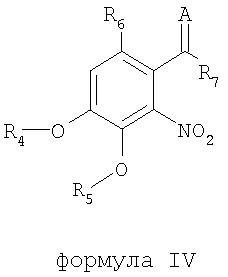
В соответствии с еще одним аспектом изобретения предоставлен способ получения соединения формулы IV:
формула IV
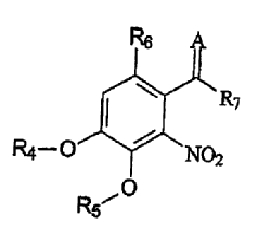
где R4 и R5 являются одинаковыми или различными и означают водород, необязательно замещенные низшие алканоил или ароил, необязательно замещенный низший алкоксикарбонил или необязательно замещенный низший алкилкарбамоил; R6 означает водород или необязательно замещенную алканоильную или ароильную группу; R7 означает необязательно замещенную насыщенную или частично ненасыщенную низшую алкильную или арильную группу или, взятая вместе с R6, означает необязательно замещенное насыщенное или частично ненасыщенное карбоциклическое кольцо; А означает атом кислорода или группу NR8, где R8 означает NHR9, где R9 означает необязательно замещенную низшую алкильную или арильную группу или группу OR10, где R10 означает водород, низший алкил или низший алканоил или А означает необязательно замещенный алкилиден, когда R7 означает группу OR11, где R11 означает необязательно замещенную низшую алканоильную или ароильную группу, и его фармацевтически приемлемых солей; указанный способ включает стадии: взятия соединения формулы I, произведенного в соответствии со способом, описанным выше, и обработки указанного соединения для получения соединения формулы IV.
В одном воплощении данных дальнейших способов обработка может включать стадию деалкилирования, которая может являться стадией деметилирования. В еще одном воплощении обработка может включать стадию ацилирования. Предпочтительно, обработка включает как стадию деалкилирования так и стадию ацилирования.
В предпочтительном воплощении данного способа m=1, n=1, где R' представляет собой COR1, где R1 представляет фенил и R" представляет метокси. Предпочтительно, соединение формулы I обрабатывают со стадией деметилирования и стадией ацилирования.
В еще одном предпочтительном воплощении R4 и R5 оба представляют бутирил; R6 представляет водород; R7 представляет фенил; и А является кислородом.
Стадия деметилирования предпочтительно включает взаимодействие соединения формулы I с акцептором метила в присутствии катализатора и кристаллизацию деметилированного продукта. Соединение формулы I может быть диспергировано в органическом растворителе, таком как этилацетат, 1,2-дихлорэтан, дихлорметан или 1,1,2,2-тетрахлорэтан. Акцептор метила может представлять пиридин, и катализатор может представлять хлорид алюминия. Альтернативно, акцептор метила и катализатор могут являться одним и тем же соединением, таким как хлорид пиридиния. Реакция может протекать в присутствии инертного газа, такого как аргон. Последовательно, в реакционную смесь может быть добавлена кислота, такая как HCl. Добавление кислоты может гасить реакцию. Осажденное твердое вещество можно удалить фильтрацией, и его предпочтительно промывают и перекристаллизуют.Стадия ацилирования предпочтительно включает в себя взаимодействие деметилированного соединения с одним или несколькими донорами ацила, таким как масляный ангидрид или этилхлорформиат, необязательно в присутствии пиридина и катализатора, такого как 4-диметиламинопиридин. Реакция может протекать предпочтительно в течение приблизительно двух часов перед тем, как продукт промывают и сушат. Стадию промывки предпочтительно проводят с применением кислоты и соляного раствора. Остаток может быть отфильтрован и упарен в вакууме и далее перекристаллизован, предпочтительно из смеси органический растворитель/петролейный эфир с получением кристаллизованного продукта.
Указано, что ряд фрагментов в формулах I, II и IV являются "необязательно замещенными" и способы изобретения применимы к широкому диапазону возможных замещений. Конкретные необязательные заместители для фрагментов включают низший алкил, алкокси, галоген, нитро, амино или циано. Таким образом, в данном описании термин "необязательно замещенные" следует читать в предпочтительном воплощении как "необязательно замещенные низшим алкилом, алкокси, галогеном, нитро, амино или циано".
Изобретение, раскрытое в данном документе, продемонстрировано следующими примерами получения, которые не следует рассматривать как ограничивающие объем раскрытия. Нужно понимать, что изобретение не ограничивается точными деталями операции или показанными структурами, так как очевидные модификации и эквиваленты будут очевидны для специалистов в данной области. Примеры 1-7 являются примерами методики нитрования. Пример 8 представляет пример методики деметилирования. Пример 9 является примером методики ацилирования. Альтернативные методики деалкилирования и ацилирования, реагенты и количества являются легко доступными для специалистов в данной области (см., например, публикации заявителя GB2344819A, EP-A-1167341 и EP-A-1167341).
Пример 1. 2-нитрофенол
К перемешиваемому раствору фенола (0,94 г, 10 ммоль) в дихлорметане (10 мл) при комнатной температуре добавляют гидросульфат тетрабутиламмония (0,17 г, 5 мол.%) с последующим добавлением изопропилнитрата (2,63 г, 25 ммоль). Далее по каплям добавляют серную кислоту (96%, 0,94 мл), и смесь становится более темной по внешнему виду по мере того, как умеренно повышается температура реакционной смеси. Через пять минут реакционную смесь выливают в воду (30 мл) и фазы разделяют. Органическую фазу промывают соляным раствором и сушат над безводным сульфатом натрия. Фильтрацией и упариванием (40°С, давление водяного аспиратора) получают темное масло, которое хроматографируют на силикагеле, используя смесь растворителей петролейный эфир/этилацетат (4:1-3:1-2:1) для градиентного элюирования. Более быстро элюируемый компонент выделяют в чистом виде с колонны в виде желто-оранжевых кристаллов (0,9 г, 65%) с т. пл. 45-46оС и идентифицируют ЯМР-спектроскопией в виде ортонитрованного продукта, указанного в заголовке, 2-нитрофенола (лит. т. пл. 44-45°С, Merck Index No 6541). Более медленно элюируемый компонент, далее извлекаемый с колонны, перекристаллизуют из смеси дихлорметан/петролейный эфир с получением бледно-красных кристаллов (0,22 г, 16%) с т. пл. 112-113°С, которые идентифицируют ЯМР-спектроскопией как паранитрованный продукт, 4-нитрофенол (лит. т. пл. 113-114°С, Merck Index No. 6542). (81% объединенный выход, орто:пара селективность, 4:1).
Пример 2. 3-гидрокси-4-метокси-2-нитробензальдегид
К перемешиваемой суспензии 3-гидрокси-4-метоксибензальдегида (изованилин, 0,76 г, 5 ммоль) в дихлорметане (10 мл) при комнатной температуре добавляют гидросульфат тетрабутиламмония (0,085 г, 5 мол.%) с последующим добавлением изопропилнитрата (1,31 г, 12,5 ммоль). Далее по каплям к смеси добавляют серную кислоту (96%, 0,76 мл) и осуществляют перемешивание при комнатной температуре в течение тридцати минут и далее выливают в воду (50 мл). Фазы разделяют и органический слой промывают соляным раствором и сушат над безводным сульфатом натрия. Фильтрацией и выпариванием растворителя (40°С, давление водяного аспиратора) получают твердый остаток, который перекристаллизуют из смеси дихлорметан/петролейный эфир с получением оранжевых кристаллов (0,74 г, 75%) с т. пл. 139-140°С, идентифицируемых ЯМР как ортонитрованный продукт, указанный в заголовке. После концентрирования маточных растворов получают небольшое количество темно-оранжевых кристаллов (0,11 г, 11%), соответствующих (ТСХ) стандартному паранитрованному продукту, 3-гидрокси-4-метокси-6-нитробензальдегиду. (86% объединенный выход, орто:пара селективность, 6,8:1).
Пример 3. 3-гидрокси-4-метокси-2-нитробензофенон
К перемешиваемому раствору 3-гидрокси-4-метоксибензофенона (10,0 г, 43,8 ммоль) в дихлорметане (100 мл) при комнатной температуре добавляют гидросульфат тетрабутиламмония (0,74 г, 5 мол.%) с последующим добавлением изопропилнитрата (11,5 г, 87,6 ммоль). Далее по каплям добавляют серную кислоту (96%, 10 мл), вызывающую умеренную экзотермическую реакцию, и, после перемешивания в течение сорока минут, реакционную смесь выливают в воду (300 мл). Фазы разделяют и водную фазу экстрагируют дихлорметаном (30 мл). Объединенные органические фазы промывают соляным раствором и сушат над безводным сульфатом натрия. Фильтрацией и выпариванием растворителя (40°С, давление водяного аспиратора) получают твердый остаток, который перекристаллизуют из небольшого объема этанола (96%, 10 мл) с получением желтых кристаллов (7,97 г, 67%) с т. пл. 137-139°С, идентифицированных ЯМР как ортонитрованный указанный в заголовке продукт. Концентрирование маточных растворов и последующая хроматография на силикагеле с применением смеси растворителей петролейный эфир:этилацетат (2:1) позволяют выделить небольшое количество минорного продукта, соответствующего (ТСХ) стандарту паранитрованного продукта, 3-гидрокси-4-метокси-6-нитробензофенона (1,43 г, 12%), т. пл. 154-156°С. (79% объединенный выход, орто:пара селективность, 5,6:1).
Пример 4. 1-(3-гидрокси-4-метокси-2-нитрофенил)-2-фенил-этанон
К перемешиваемому раствору 1-(3-гидрокси-4-метоксифенил)-2-фенил-этанонона (8,57 г, 35,4 ммоль) в дихлорметане (90 мл) при комнатной температуре добавляют сульфат тетрабутиламмония (0,6 г, 5 мол.%) с последующим добавлением изопропилнитрата (7,44 г, 70,8 ммоль). Далее по каплям добавляют серную кислоту (96%, 8,5 мл), вызывающую умеренную экзотермическую реакцию, и, после перемешивания в течение сорока минут, реакционную смесь выливают в воду (250 мл). Фазы разделяют и водную фазу экстрагируют дихлорметаном (30 мл). Объединенные органические фазы промывают соляным раствором и сушат над безводным сульфатом натрия. Фильтрацией и выпариванием растворителя (40°С, давление водяного аспиратора) получают твердый остаток, который растирают в небольшом объеме диэтилового эфира (20 мл) с получением оранжевых кристаллов (6,9 г, 68%) с т. пл. 176-177°С, идентифицированных ЯМР как ортонитрованный указанный в заголовке продукт. Концентрирование маточных растворов и последующее растирание в диэтиловом эфире (15 мл) позволяют выделить небольшое количество минорного продукта, который перекристаллизуют из смеси дихлорметан/гептан с получением желтоватых кристаллов с т. пл. 142-143°С, соответствующих (ТСХ) стандарту паранитрованного продукта, 1-(3-гидрокси-4-метокси-6-нитрофенил)-2-фенил-этанона (1,22 г, 12%). (80% объединенный выход, орто:пара селективность, 5,7:1).
Пример 5. 2-гидрокси-3-нитробензойная кислота (3-нитросалициловая кислота)
К перемешиваемой суспензии салициловой кислоты (0,69 г, 5 ммоль) в дихлорметане (10 мл) при комнатной температуре добавляют гидросульфат тетрабутиламмония (0,085 г, 5 мол.%) с последующим добавлением изопропилнитрата (1,31 г, 12,5 ммоль). Далее по каплям к смеси добавляют серную кислоту (96%, 0,69 мл) и осуществляют перемешивание при комнатной температуре в течение тридцати минут (раствор становится желтым, с последующим образованием желтого осадка) и далее выливают в воду (50 мл). Желтый осадок отфильтровывают и далее растирают в воде (10 мл). Нерастворимое вещество отфильтровывают и сушат с получением желтых кристаллов (0,42 г, 46%) с т. пл. 121-122°С, идентифицированных ЯМР как указанное в заголовке соединение (лит. т. пл. 123°С, Merck Index No. 6553). Маточные растворы концентрируют на роторном испарителе (60оС, давление водяного аспиратора) и перекристаллизуют из смеси дихлорметан/петролейный эфир с получением желто/оранжевых кристаллов (0,35 г, 39%) с т. пл. 226-228°С, идентифицированных ЯМР как 2-гидрокси-5-нитробензойная кислота (5-нитросалициловая кислота) (лит. т. пл. 228-230°С, Merck Index No.6554). (85% объединенный выход, орто:пара селективность, 1,2:1).
Пример 6. 3-гидрокси-2-нитробензальдегид и 3-гидрокси-4-нитробензальдегид
К перемешиваемой суспензии 3-гидроксибензальдегида (0,61 г, 5 ммоль) в дихлорметане (10 мл) при комнатной температуре добавляют гидросульфат тетрабутиламмония (0,085 г, 5 мол.%) с последующим добавлением изопропилнитрата (1,31 г, 12,5 ммоль). Далее по каплям к смеси добавляют серную кислоту (96%, 0,61 мл), что приводит к умеренному повышению температуры. Реакционную смесь далее перемешивают в течение пятнадцати минут (становится темно-коричневой суспензией) и далее выливают в воду (50 мл). Фазы разделяют и водную фазу экстрагируют дихлорметаном (10 мл). Объединенные органические слои промывают соляным раствором, сушат над безводным сульфатом натрия и фильтруют. Выпариванием растворителя (40°С, давление водяного аспиратора) получают коричневое твердое вещество, которое далее хроматографируют на силикагеле, используя смесь растворителей петролейный эфир/этилацетат (2:1). Более быстро элюируемый компонент получают с колонны в виде желтого твердого вещества (0,19 г, 23%), идентифицированного ЯМР как 3-гидрокси-4-нитробензальдегид. Более медленно элюируемый компонент также выделяют в виде желтого твердого вещества, идентифицированного ЯМР как 3-гидрокси-2-нитробензальдегид (0,56 г, 67%). (90% объединенный выход, оба продукта являются ортонитрованными, паранитроизомера не обнаружено).
Пример 7. 2,4-дифтор-6-нитрофенол
К перемешиваемому раствору 2,4-дифторфенола (0,65 г, 5 ммоль) в дихлорметане (7 мл) при комнатной температуре добавляют гидросульфат тетрабутиламмония (0,085 г, 5 мол.%) с последующим добавлением изопропилнитрата (1,31 г, 12,5 ммоль). Далее по каплям к смеси добавляют серную кислоту (96%, 0,65 мл), что приводит к умеренному кипячению растворителя с обратным холодильником. Реакционную смесь далее перемешивают в течение пятнадцати минут и далее выливают в воду (50 мл). Фазы разделяют и водную фазу экстрагируют дихлорметаном (10 мл). Объединенные органические слои промывают соляным раствором, сушат над безводным сульфатом и фильтруют через невысокий рыхлый слой силикагеля. Выпариванием растворителя (40°С, давление водяного аспиратора) получают желтое твердое вещество, идентифицированное ЯМР как соединение, указанное в заголовке (0,73 г, 83%). (Ортоселективность 100%, нитроизомеры не обнаружены).
Пример 8. 3,4-дигидрокси-2-нитробензофенон
К перемешиваемой суспензии 3-гидрокси-4-метокси-2-нитробензофенона (8,3 г, 30,38 ммоль) в 1,2-дихлорэтане (100 мл) при комнатной температуре в атмосфере аргона одной порцией добавляют хлорид алюминия (4,46 г, 33,45 ммоль) с последующим добавлением пиридина (9,61 г, 9,81 мл, 121,5 ммоль), что приводит к экзотермической реакции. Смесь перемешивают при кипячении с обратным холодильником в течение одного часа, позволяют охладиться до комнатной температуры и далее выливают в смесь лед-вода (300 мл). Добавляют хлористоводородную кислоту (2н., 70 мл) и смесь перемешивают в течение одного часа (первоначальный оранжевый осадок постепенно становится желтым по внешнему виду). Твердое вещество удаляют фильтрацией, промывают водой (30 мл) и сушат в вакууме с получением продукта в виде желтого твердого вещества 6,99 г, (89%) с температурой плавления 153-155°С. Органическую фазу фильтрата отделяют и водную фазу экстрагируют дихлорметаном (20 мл). Объединенные органические фазы промывают соляным раствором (30 мл), сушат над безводным сульфатом натрия и растворитель удаляют на роторном испарителе (темп.бани 40°С) с получением желтого твердого вещества (0,7 г), которое далее не очищают.
Пример 9. 3-бензоил-6-бутирилокси-2-нитрофениловый эфир масляной кислоты [3, 4-дибутирилокси-2-нитробензофенон]
К перемешиваемому раствору (3,4-дигидрокси-2-нитрофенил)фенилметанона (0,34 г, 1,29 ммоль) (5 мл) [3,4-дигидрокси-2-нитробензофенона в дихлорметане] при комнатной температуре добавляют пиридин (0,41 г, 5,19 ммоль), масляный ангидрид (0,82 г, 5,19 ммоль) и 4-диметиламинопиридин (0,01 г). Полученный раствор перемешивают в течение одного часа и далее экстрагируют холодной водой, промывают 1н. HCl и соляным раствором, далее сушат над сульфатом натрия. После фильтрации и упаривания в вакууме остаток хроматографируют на силикагеле, используя смесь этилацетат/петролейный эфир с получением не совсем белых кристаллов с т. пл. от 55 до 57°С.
Следует учитывать, что данное изобретение, описанное выше, может быть модифицировано.
Реферат
Предложен способ получения соединений формулы I, где R' и R" могут иметь одно и то же, или различные значения, указанные в п.1 формулы изобретения, m и n независимо равны 0, 1 или 2, в котором указанный способ включает взаимодействие фенольного соединения формулы II, где R' и R", m и n определены выше, с алкилнитратом формулы (III): R3-ONO2, где R3представляет собой алкильную группу с прямой или разветвленной цепью, содержащую от одного до шестнадцати атомов углерода или R3 представляет собой циклоалкильную группу, содержащую либо пять или шесть атомов углерода, причем указанную реакцию проводят в присутствии кислотного катализатора. Предложен также способ получения соединения формулы IV, где R4, R5, R6, R7 и А, имеют значения, определенные в п.11 формулы изобретения, путем деалкилирования и/или ацилирования соответствующего соединения формулы (I), полученного вышеописанным способом. Технический результат - региоселективное орто-направленное нитрование исходных фенольных соединений. 2 н. и 19 з.п. ф-лы.
Формула

Комментарии