Противомикробные пептиды - RU2392962C2
Код документа: RU2392962C2
Чертежи
Описание
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к противомикробным соединениям, которые подавляют или убивают микроорганизмы, включая грамотрицательные бактерии и грибы. Кроме того, данные соединения имеют сродство к токсинам, в частности, к токсинам грибов и бактерий, таким как липополисахариды (ЛПС) или липотейхоевая кислота (ЛТК), и они могут подавлять и нейтрализовать такие токсины. Кроме того, данное изобретение относится к терапевтическому и диагностическому использованию данных соединений и фармацевтическим композициям, содержащим данные соединения, и способам их применения.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Современная фармакотерапия была чрезвычайно успешна в борьбе с бактериальными инфекциями, которые обычно были одной из главных причин смерти в раннем возрасте до середины последнего столетия. Позже, однако, возникли растущие проблемы по причине широко распространенного использования высокоэффективных антибиотиков из-за постоянного увеличения бактериальной резистентности. Фактически за последние 25 лет устойчивость к антибиотикам, особенно множественная резистентность к широкому ряду противомикробных соединений, увеличилась фактически у каждого вида изученных бактерий. В настоящее время полагают, что антибактериальными веществами наиболее современного типа, на которые не влияют обычные механизмы резистентности, являются соединения, использование которых, по-видимому, способствует селекции мутантов со множественной резистентностью.
На основе этих разработок эксперты рекомендуют использовать антибиотики значительно более ограниченно, чем в прошлом, как в сельском хозяйстве, так и в медицине [при лечении] человека. Например, незначительные инфекции, особенно те, которые обычно вызваны даже и не бактериями, такие как обычная простуда, не нужно лечить антибиотиками, которые должны скорее резервироваться для более тяжелых патологических состояний. Кроме того, необходимо разрабатывать новые соединения для лечения бактериальных инфекций с фармакологической активностью совсем иного типа, предпочтительно, с неким действием, которое не зависит от бактериальной резистентности к обычным антибиотикам.
Одним из патологических состояний, при котором широко распространенное использование антибиотиков в результате обсуждений ставится под сомнение, является воспаление среднего уха либо в его острой форме, либо в хронической. Было показано, что число пациентов с воспалением среднего уха с выделениями (ОМЕ; ВСУВ), т.е. вид отита, характеризующийся наличием жидкости в среднем ухе, без симптомов острой инфекции, заметно повысилось после введения терапии антибиотиками на ранних стадиях острого воспаления среднего уха (АОМ; ОВСХ), что свидетельствует о том, что сами антибиотики играют роль при ВСУВ (Lim et al., Laryngoscope 92, 278-286, 1982). Полагают, что антибиотики, подобные пенициллину, мешают развитию местного иммунного ответа, такого как с продукцией местного IgМ в среднем ухе (Howie et al., Ann. Otol. Rhinol. Laringol. 85 Suppl. 25, 18-19, 1976). Другим недостатком общепринятой терапии антибиотиками является то, что бактерии погибают, но их токсины все еще остаются активными.
Предполагалось, что для лечения этих и других патологических состояний, являющихся результатом бактериальной или грибковой инфекции, преимущества может дать использование соединений, которые сами не убивают микроорганизмы или бактерии, а скорее нейтрализуют их токсины и позволяют природным механизмам защиты хозяина бороться с распространением инфекции (Nell, The Role of Endotoxin in the Pathogenesis of Otitis Media With Effusion, PhD Thesis, Leiden, 1999). В то же время эта стратегия обеспечила бы быстрое восстановление нарушенных функций слизистых оболочек.
Главную роль среди микробных токсинов, таких как токсины грибков и бактериальные токсины, участвующих в ряде патологических инфекционных состояний, таких как отит, играют эндотоксины, группа липополисахаридов (ЛПС), обнаруженных в клеточной стенке грамотрицательных бактерий, состоящих из полисахаридов, конъюгированных с высокотоксичной липидной группой, липидом А. Один из последних терапевтических подходов к лечению ВСУВ является применение соединений, которые нейтрализуют эндотоксин или ЛПС (Nell, там же).
В настоящее время известны различные соединения, способные к нейтрализации эндотоксина или ЛПС. Например, было разработано несколько антиэндотоксиновых антител, таких как НА-1А и Е5, человеческие и париетальные моноклональные IgМ антитела соответственно. Эти антитела, как было показано, улучшают степень выживаемости пациентов с некоторыми тяжелыми патологическими состояниями, такими как септический шок (Ziegler et al., New Engl. J. Med. 324, 429-436, 1991). Однако их активность и специфичность считаются неудовлетворительными.
Другая группа веществ, активных против эндотоксина, получена из человеческого эндогенного протеина, называемого бактериальным повышающим проницаемость белком (ВРI; БПП), который запасается в азурофильных гранулах нейтрофилов (Gazzano-Santoro et al., Infection and Immunity 60:11, 4754-4761, 1992). БПП, который является сильно катионным протеином, не только нейтрализует свободные эндотоксины, но также подавляет или убивает бактериальные клетки как таковые путем повышения проницаемости их наружных мембран. БПП, разумеется, является сильным природным антибиотиком, индуцируемым присутствием ЛПС и некоторых других пусковых веществ, включая фактор некроза опухолей (ФНО). Однако большая часть его активности связана с иммунными клетками, синтезирующими его, т.е. полиморфноядерными макрофагами.
Было также разработано несколько рекомбинантных белков, полученных из БПП, таких как rВРI23 (Kohn et al., 1993) и rВРI21 (Horwitz et al., 1996), которые большей частью представляют собой N-концевые части БПП с молекулярным весом в 23 и 21 кДа соответственно. Использование БПП и производных соединений БПП при лечении ВСУВ описано, например, в WО-А-00/71149.
Другим семейством природных соединений с противомикробной активностью являются кателицидины, группа пептидов, продуцируемых эпителиальными клетками дыхательных путей, альвеолярными макрофагами и другими тканями. В природной форме эти соединения являются линейными, α-спиральными, не содержащими цистеин пептидами или протеинами. Кателицидины являются катионными веществами и содержат высококонсервативную сигнальную последовательность и прообласть, кателин. Однако их С-концевой домен, кодирующий зрелый пептид, существенно гетерогенен. Данные пептиды имеют от 12 до 80 аминокислот.
Наиболее известный человеческий кателицидин является катионным противомикробным протеином САР18 в 18 кДа. 37 С-концевых аминокислот САР18, т.е. пептид LL-37, представляют собой домен, ответственный за высокое сродство и нейтрализующую способность в отношении ЛПС (Sawa et al., Antimicr. Agents Chemother. 42:12, 3269-3275, 1998). Было разработано и испытано несколько усеченных пептидов, полученных из САР18 или LL-37, таких как описанные Sawa (Sawa et al., там же), Gutsmann (Gutsmann et al., Biophys. J. 80, 2935-2945, 2001) и в патенте США № 6040291. В основном относительно небольшие пептиды предпочтительнее протеинов, таких как САР18, в качестве основных терапевтических соединений-кандидатов по нескольким соображениям. Во-первых, их можно легче оптимизировать, адаптировать и модифицировать, чтобы сохранить или усилить их желаемую активность и специфичность. Во-вторых, их легче получить или синтезировать и поэтому они более доступны. В-третьих, их легче переработать в препараты и доставить, так как протеины часто нестабильны и утрачивают свою биодоступность после непарентерального введения.
В одновременно рассматриваемой Международной патентной заявке РСТ/NL2004/00060, которая включена сюда в виде ссылки, описаны пептидные соединения, которые обладают сродством к микробным и грибковым токсинам, таким как ЛПС и ЛТК. Эти соединения состоят из аминокислотной последовательности X1KEFX2RIVX3RIKX4FLRX5LVX6 (здесь ниже называемой также коровой аминокислотной последовательностью), где Х1 собой представляет N-концевую часть последовательности, Х2 является К или Е, Х3 является Q или Е, Х4 является D или R, Х5 является N или Е, и Х6 представляет собой С-концевую часть и где одна или более из аминокислот коровой последовательности могут быть дериватизированы. Данная последовательность дополнительно характеризуется тем, что N-концевая часть ацетилирована и/или тем, что С-концевая часть амидирована, и/или тем, что аминокислотная последовательность является отличной от последовательности X1KEFKRIVQRIKDFLRNLVX6.
В указанной патентной заявке, кроме того, описаны способы получения таких соединений. Данные способы включают в себя химическое и ферментативное лигирование аминокислотных мономеров или олигомеров, со сборкой данных соединений. Они включают в себя также экспрессию последовательностей нуклеиновых кислот, кодирующих данные соединения, в клетках-хозяевах с использованием вектора для трансфицирования клеток-хозяев последовательностями нуклеиновых кислот. Способ получения соединения по любому из предшествующих пунктов, при котором аминокислотные мономеры, аминокислотные олигомеры или моно- или олигомеры из аминокислотных аналогов или миметиков собирают с помощью химического или ферментативного лигирования, которое выполняют в жидкой фазе и/или на поверхности раздела с функционализированной твердой фазой.
Эти соединения, как было обнаружено, пригодны для лечения патологических состояний, связанных с инфекциями или являющимися их результатом. Предполагают, что они могут быть терапевтически более полезны, чем обычные антибиотики, при лечении некоторых хронических инфекций, таких как воспаление среднего уха. Однако в случае тяжелых острых инфекций все еще считается обязательным эффективное подавление микробного роста, которое достигается применением антибиотиков.
ЦЕЛИ ИЗОБРЕТЕНИЯ
Несмотря на усилия, предпринятые в прототипах, существует потребность улучшения профилактического, соответственно, терапевтического лечения инфекционных заболеваний, включая системные или локальные бактериальные и грибковые инфекции. Одной из целей согласно изобретению является обеспечение новой терапии, которая безопасна, эффективна и которая приводит к эффективному подавлению инфицирующих микроорганизмов. Другая цель состоит в обеспечении терапии, которая так легко не приводит к устойчивости микроорганизмов. Кроме того, целью согласно изобретению является обеспечение терапии, которая снижает также нежелательные эффекты обычной противомикробной терапии, такие как токсические эффекты микробных токсинов, которые высвобождаются, когда микроорганизмы погибают под действием противомикробных соединений в организме.
Эти и другие цели согласно изобретению станут ясными на основе последующего описания.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В первом аспекте данное изобретение связано с применением пептидного соединения для производства лекарственного средства для профилактического или терапевтического лечения бактериальной или грибковой инфекции млекопитающего, причем данное соединение содержит аминокислотную последовательность X1KEFX2RIVX3RIKX4FLRX5LVX6, где Х1 представляет собой N-концевую часть, Х2 является К или Е, Х3 является Q или Е, Х4 является D или R, Х5 является N или Е, и Х6 представляет собой С-концевую часть. Одна или более из аминокислот коровой последовательности могут быть по желанию дериватизированы. Кроме того, N-концевая часть ацетилирована и/или С-концевая часть амидирована, и/или аминокислотная последовательность отличается от природной аминокислотной последовательности X1KEFKRIVQRIKDFLRNLVX6.
Такие соединения, как было неожиданно обнаружено, обладают не только сродством к липополисахаридам (ЛПС) или липотейхоевой кислоте (ЛТК), но также и непосредственно противомикробной активностью или микробицидной активностью. То есть эти соединения можно использовать в производстве лекарственного средства, которое убивает бактерии и грибы. Данные соединения обладают бактерицидной и фунгицидной активностью. На основании этой активности данные соединения можно использовать в терапии в качестве антибиотиков, даже при заболеваниях и патологических состояниях, которые нельзя лечить соединениями, способными только нейтрализовать микробные токсины. Примерами таких заболеваний являются острые бактериальные или грибковые инфекции, такие как септический шок, острые инфекции глаз(а), печени, почек(ки), легких, бронхов, назального или фронтального синуса, уха(ушей), влагалища, мочеиспускательного канала, кожи, центральной нервной системы, сердечной мышцы, селезенки и других органов и тканей.
В дополнительном аспекте данное изобретение относится к лекарственным средствам, которые пригодны для профилактики и/или лечения заболеваний и патологических состояний, подобных бактериальным или грибковым инфекциям, или которые связаны с бактериальными или грибковыми инфекциями.
Дополнительные аспекты согласно изобретению будут представлены ниже в подробном описании и в прилагаемой формуле изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Неожиданно было обнаружено, что пептидные соединения, описанные в одновременно рассматриваемой Международной патентной заявке РСТ/NL2004/00060, не только нейтрализуют бактериальные и грибковые токсины, такие как липополисахариды (ЛПС) и липотейхоевая кислота (ЛТК), но также обладают и значительной противомикробной активностью. Эти соединения содержат коровую аминокислотную последовательность X1KEFX2RIVX3RIKX4FLRX5LVX6, где Х1 представляет собой N-концевую часть данной последовательности, Х2 является К или Е, Х3 является Q или Е, Х4 является D или R, Х5 является N или Е, и Х6 представляет собой С-концевую часть; и где одна или более из аминокислот коровой последовательности могут быть дериватизированы. Кроме того, или N-концевая часть ацетилирована, и/или С-концевая часть амидирована, и/или аминокислотная последовательность отличается от последовательности X1KEFKRIVQRIKDFLRNLVX6. На основании сродства к микробным токсинам данные соединения можно использовать в терапии для лечения патологических состояний, связанных с наличием таких токсинов в организме. На основе их непосредственной противомикробной активности, однако, их можно использовать как антибиотики и/или вместо них. Двойная активность делает эти соединения чрезвычайно полезными при введении как отдельно, так и в комбинации с другими антибиотиками, нежелательные побочные эффекты которых, такие как порождение устойчивых микроорганизмов или токсические эффекты микробных токсинов, которые выделяются в организм, когда большое число бактерий погибает в организме, могут быть подавлены или облегчены.
В этом описании и прилагаемой формуле изобретения термины «где N-концевая часть ацетилирована» имеют следующее значение. N-концевая часть защищена путем реакции с карбоновой кислотой, с получением связанной с амидом стабилизирующей или защищающей группы. Например, возможно произвести реакцию пептида с муравьиной кислотой, с получением стабилизированного формилом пептида; с уксусной кислотой, с получением защищенного ацетилом пептида. Кроме того, можно произвести реакцию пептида с пропионовой кислотой и другими органическими кислотами, имеющими до 6 атомов углерода и даже до 10 атомов углерода в углеводородной части R. В этих органических кислотах углеводородная группа R, имеющая до 10 атомов углерода, может быть прямой или разветвленной, или циклической и/или может содержать одну или более ненасыщенных связей. Кроме того, алкильная цепь может быть замещенной, например, гидроксилом, галогеном, амино, меркапто и сульфуроксидной группами. Следовательно, на N-концевой части может присутствовать следующая группа: -С(О)-R'. Альтернативно, вместо реакции с карбоновой кислотой можно также осуществлять реакцию с сульфоновой кислотой, с получением соответствующей сульфонамидной связи. Следовательно, на N-концевой части может присутствовать группа -SО2-R. Альтернативно, указанные термины охватывают алкилирование и диалкилирование, то есть на N-концевой части может присутствовать вторичная или третичная аминогруппа -N-(R)1 или N-(R)2, где каждый R имеет указанное выше значение.
В еще одном воплощении «ацетилирование» включает в себя реакцию пептида с изоцианатом или изотиоцианатом и в этом случае соответственно получается мочевина или тиомочевина: R-N-С(О)- или R-N-С(S)-, причем R имеет значения, определяемые выше.
И, наконец, N-конец может быть защищен устойчивой к кислоте блокирующей группой, и эта группа, которую обычно вводят во время синтеза пептида, теперь не будет удалена. Хорошо известными блокирующими группами являются Fmoc и Z-группа.
В отношении значения термина «где С-концевая часть амидируется» нужно отметить следующее. Термин «амидирование» означает, что -ОН, естественно присутствующий на С-конце, заменен группой -Х, где Х является (i) -NY2-группой, причем Y независимо является Н или R, где R имеет значения, определяемые выше, или две группы Y вместе могут представлять собой циклическую группу вместе с N-группой, с которой они связаны, предпочтительно присутствует по меньшей мере одна группа R; (ii) -ОR-группой, где R дано определение выше, или (iii) -R группой. Амиды пептидов являются предпочтительными, так как они имеют наивысшую стабильность.
Пептидные соединения согласно изобретению, как было обнаружено, имеют оптимизированную стабильность по сравнению с природной аминокислотной последовательностью, исключенной из пункта 1.
Пептидные соединения являются пептидами, такими как олиго- или полипептиды, протеинами или веществами, получаемыми из пептидов. Кроме самих пептидов, пептидные соединения включают в себя также аналоги пептидов, пептидные производные, модифицированные пептиды и пептидные конъюгаты. Пептидные соединения имеют то общее свойство, что они содержат аминокислотные последовательности. Более точно, пептиды определяют как амиды, которые являются производными из двух или более аминокислот путем соединения аминогруппы одной кислоты с карбоксильной группой другой (Merriam Webster Medical Dictionary 2001). Пептидным соединением, в отличие от этого, можно также называть пептидную структуру внутри молекулы. Обычно пептиды образованы существующими в природе (L-)α-аминокислотами, в частности, аланином (Аlа или А), аргинином (Arg или R), аспарагином (Asn или N), аспарагиновой кислотой (Asp или D), цистеином (Cys или С), глутамином (Gln или Q), глутаминовой кислотой (Glu или Е), глицином (Gly или G), гистидином (His или Н), изолейцином (Ile или I), лейцином (Leu или L), лизином (Lys или К), метионином (Met или М), фенилаланином (Phe или F), пролином (Pro или P), серином (Ser или S), треонином (Thr или Т), триптофаном (Trp или W), тирозином (Tyr или Y) и валином (Val или V).
Аналоги или функциональные эквиваленты пептидов являются пептидными молекулами, обладающими одинаковой активностью и, в частности, одинаковым сродством к микробным и, в частности, бактериальным токсинам по виду, но не обязательно по количеству, и могут, например, быть модифицированными пептидами, пептоидами, пептидными аналогами или пептидомиметиками.
Модифицированные пептиды представляют собой молекулы, получаемые из пептидов путем введения заместителей или функциональных групп, которые обычно не присутствуют в существующих в природе аминокислотах. Термин включает в себя также соединения, которые получают в результате реакции пептидов с молекулами других видов химических соединений, независимо от того, встречаются ли эти молекулы в природе или нет. Например, фосфорилированные, сульфонированные и биотинилированные пептиды, гликопротеины и липопротеины часто встречаются в природе, тогда как пептиды, модифицированные полиэтиленгликолем, являются примерами химически модифицированных пептидов, которые были созданы для того, чтобы изменить некоторые, но не все из свойств пептидов.
Пептоиды, подобно пептидам, также являются пептидными соединениями. Они также обычно являются амидами из двух или более аминокислот. Однако они часто являются не непосредственными производными от встречающихся в природе аминокислот, а скорее от разных типов химически синтезированных L- и/или D-аминокислот.
Пептидомиметики, в их наиболее широком понимании, являются соединениями, которые являются по их функциональному строению более или менее похожими на пептид, но которые могут также содержать непептидные связи в их основной цепи или D-аминокислоты. В основном пептидомиметики служат в качестве заменителей для природных пептидов во взаимодействии с рецепторами и ферментами (Pharmacеutical Biotechnology, Ed. D. J. A. Crommelin and R. D. Sindelar, Harwood Academic Publishers, 1997, p. 138). Псевдопептиды, группа пептидомиметиков, являются соединениями, содержащими изостеры амидной связи вместо амидных связей (там же, рр. 137-140).
Соединения, которые используют для осуществления согласно изобретению, включают в себя также соли пептидов или функциональные эквиваленты, такие как фармацевтически приемлемые соли присоединения с кислотой или основанием и продукты присоединения. Они включают в себя также мультимеры пептидов или функциональные эквиваленты.
Кроме того, данные соединения обладают сродством по меньшей мере к одному токсину, в частности, к бактериальному токсину. При большом числе инфекционных заболеваний бактериальные токсины, такие как группа липополисахаридов (ЛПС) в случае грамотрицательных бактерий и липотейхоевая кислота (ЛТК) в случае грамположительных бактерий, участвуют в проявлении данного заболевания. Эти токсины могут вызывать существенные воспалительные реакции. Например, при инфекциях верхних дыхательных путей воспаление может приводить к повреждению слизистой оболочки эпителия среднего уха или синусов, приводя в результате к нарушению системы мукоцилиарного очищения (СМО), которая является одной из главных систем защиты верхних дыхательных путей. Сродство к грибковым или бактериальным токсинам является необходимым условием любой способности к нейтрализации и, предпочтительно, соединения согласно изобретению не только связываются с ЛПС и другими токсинами, но также обладают способностью нейтрализовать или подавлять эти токсины или иным способом снижать эффекты указанных токсинов.
Желаемый тип активности против бактерий и грибков и бактериальных и грибковых токсинов наблюдается, когда пептидные соединения удовлетворяют требованиям к структуре, которые определены в пункте 1, в соответствии с которыми данные соединения содержат аминокислотную последовательность X1KEFX2RIVX3RIKX4FLRX5LVX6, где Х1 представляет собой N-концевую часть данной последовательности, Х2 является К или Е, Х3 является Q или Е, Х4 является D или R, Х5 является N или Е, и Х6 представляет собой С-концевую часть. Этот основной мотив получен из природного противомикробного протеина САР18 или пептида LL-37, который сам получен из САР18.
В соответствии с данным описанием, N-концевая часть является группой, атомом или последовательностью, представляющей собой N-концевую группу или домен данного соединения, т.е. структуру, которая связана с концевой α-аминогруппой коровой последовательности, которая не участвует в амидной связи в данной последовательности. N-концевая часть может просто быть атомом водорода в случае свободной α-аминогруппы; или она может состоять из химической группы, связанной с атомом азота концевой α-амино, такой как ацильная группа. Она может также представлять собой большую группу, такую как последовательность из двух или более аминокислот, или химическую структуру, которая не образована аминокислотами или образована не только аминокислотами. С-концевую часть определяют аналогичным образом.
Предпочтительно соединения содержат в целом более 18 аминокислот, характеризующих коровый мотив. В одном из воплощений N-концевая часть содержит последовательность из двух или более аминокислот. Среди аминокислот, которые являются подходящими представителями данной последовательности, находятся I и G, а предпочтительной N-концевой частью является IG.
В другом воплощении С-концевая часть таже содержит аминокислотную последовательность. Данная последовательность может содержать 1 аминокислоту, 2, 3, 4 аминокислоты или более 4 аминокислот. В одном из воплощений С-концевая часть содержит 4 аминокислоты. С-концевым окончанием указанной С-концевой части 4 аминокислот может быть Е, как и в эквивалентном положении в пептиде LL-37. Однако этот С-конец также может быть определен через R. Аминокислота, которая располагается после С-концевой аминокислоты, может быть Т, как в LL-37, или может быть L. Р и R являются двумя другими предпочтительными представителями из последовательности из 4 аминокислот С-концевой части в одном из двух остальных положений. Наиболее предпочтительно, когда С-концевая часть выбрана из последовательностей РRТЕ и RРLR.
В дополнительном воплощении N-концевая часть и С-концевая часть выбраны из предпочтительных вариантов, описанных выше, с получением пептидной структуры с общим числом в 24 аминокислоты. Среди наиболее предпочтительных в настоящее время соединений можно назвать пептиды IGKEFKRIVQRIKDFLRNLVPRTE и IGKEFKRIVERIKRFLRELVRPLR, либо в виде пептидов как таковых, либо в виде модифицированных пептидов или их производных.
К предпочтительным модификациям относятся амидированные и/или ацетилированные пептиды. Одним из положений, в котором амидирование дает особенно большие преимущества, является С-конец пептида. Ацетилирование, с другой стороны, предпочтительно, выполняется по N-концевой аминокислоте. В одном из предпочтительных в настоящее время воплощений пептиды IGKEFKRIVQRIKDFLRNLVPRTE и IGKEFKRIVERIKRFLRELVRPLR являются как ацетилированными на N-конце, так и амидированными на С-конце. Предварительное испытание говорит о том, что эти модификации обладают повышенной стабильностью в присутствии экзопептидаз.
Данные соединения можно обычно получить методами, которые известны в отношении получения пептидов и подобных веществ. Соединения меньшего размера, содержащие только несколько аминокислот или подобных единиц, и предпочтительно, не более 30-50 единиц, можно получить методами химического или ферментативного связывания или используя классический подход, при котором реакции происходят в растворе или суспензии, или путем использования более современного твердофазного подхода, при котором сборка пептида происходит при его фиксации на твердой поверхности, такой как полимерная гранула. Соединения большего размера обычно синтезируют с помощью синтезаторов пептидов на твердой фазе.
Альтернативно, данные соединения могут быть получены с помощью известных методов генной инженерии. Этот подход особенно ценен, если данное соединение действительно является пептидом или слегка модифицированным пептидом. Например, ДНК-последовательность, которая кодирует данное соединение, может быть ассоциирована или соединена с вектором экспрессии, способным к трансфицированию клеток. На другой стадии данного метода клетки хозяева или клетки-мишени трансфицируют указанной ДНК путем контакта клеток с вектором и связанной с вектором ДНК в условиях, которые дают возможность трансфицирования. На дополнительной стадии клетки-хозяева или клетки-мишени культивируют в условиях, которые дают возможность экспрессии данного соединения. Затем данное соединение может быть выделено. Если само соединение не может быть закодировано или экспрессировано, но очень похоже на пептид, который кодируется или экспрессируется, данный способ может быть использован для получения пептида, которому подобно данное соединение, с последующей одной или более стадиями, когда пептид модифицируется с помощью химических или ферментативных методик, с получением данного соединения.
Для этой цели используют разные типы векторов, такие как вирусные векторы, липоплексы, полиплексы, микросферы, наносферы, дендрисферы, дендримеры, чистые ДНК, пептидные системы доставки, липиды, особенно катионные липиды или липосомы, изготовленные из них, полимерные векторы, особенно изготовленные из поликатионных полимеров. Среди предпочтительных вирусных векторов можно упомянуть ретровирусы, аденовирусы, ассоциированные аденовирусы, вирусы простого герпеса и виросомы. Предпочтительные невирусные векторы включают в себя хитозан, SРLР, полимерные системы на основе РLGА, полиэтиленимины, полилизины, полифосфоамидаты, поли(мет)акрилаты, полифосфазены, DОРЕ, DОТАР и DОТМА.
Некоторые более всеобъемлющие обобщения методов, которые можно применить при получении соединений согласно изобретению, описаны W.F. Anderson, Nature 392 Supp., 30 April 1998, p. 25-30; Pharmaceutical Biotechnology, Ed. D.J.A. Crommelin and R.D. Sindelar, Harwood Academic Publishers, 1997, p. 53-70, 167-180, 123-152, 8-20; Protein Synthesis: Methods and Protocols, Ed. R. Martin, Humana Press, 1998, p. 1-442; Solid-Phase Peptide Synthesis, Ed. G.B. Fields, Academic Press, 1997, p. 1-780; Amino Acid and Peptide Synthesis, Oxford University Press, 1997, p. 1-89.
Соли пептидов или функциональные эквиваленты получают известными методами, которые обычно включают в себя смешивание пептида или пептоида с или фармацевтически приемлемой кислотой, с образованием соли присоединения кислоты, или с фармацевтически приемлемым основанием, с образованием соли присоединения основания. Является ли кислота или основание фармацевтически приемлемым, может быть легко определено специалистом в данной области, с учетом специфического предполагаемого применения соединения. Например, не все кислоты и основания, которые приемлемы для композиций для диагностики in vitro, можно использовать в терапевтических композициях. В зависимости от предназначенного использования, фармацевтически приемлемые кислоты включают в себя органические и неорганические кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, молочная кислота, гликолевая кислота, щавелевая кислота, пировиноградная кислота, янтарная кислота, малеиновая кислота, малоновая кислота, циннамовая кислота, серная кислота, соляная кислота, гидробромистая кислота, азотная кислота, перхлорная кислота, фосфорная кислота и тиоциановая кислота, которые образуют аммониевые соли со свободными аминогруппами пептидов и функциональными эквивалентами. Фармацевтически приемлемые основания, которые образуют карбоксилатные соли со свободными карбоксильными группами пептидов и функциональными эквивалентами, включают в себя этиламин, метиламин, диметиламин, триэтиламин, изопропиламин, диизопропиламин и другие моно-, ди- и триалкиламины, а также ариламины. Кроме того, сюда включены и фармацевтически приемлемые сольваты, комплексы или соединения присоединения, такие как гидраты или этураты.
Некоторые предпочтительные модификации данных пептидов можно легко вводить во время его синтеза или в конце. Например, когда пептид синтезируют с использованием твердофазного метода, N-концевое ацетилирование можно выполнять на конце путем реакции аминокислотной последовательности, которая еще связана со смолой, с помощью уксусной кислоты вместо другой аминокислоты.
С-концевое амидирование, с другой стороны, можно выполнять путем использования специального вида смолы при твердофазном пептидном синтезе, такой как доступная для приобретения Tentagel S АМ (от Rapp, Tubingen, Germany). Эти смолы содержат химический «удерживатель», от которого освобождаются амидированные пептиды во время отщепления. Эти и другие методы модификации пептидов известны специалистам в данной области.
Как упоминалось ранее, данные соединения обладают сродством к микробным токсинам и, в частности, бактериальным токсинам, таким как липополисахариды (ЛПС) и липотейхоевая кислота (ЛТК). Поэтому данные соединения можно использовать преимущественно для профилактических, терапевтических и диагностических целей при патологических состояниях и заболеваниях, в которых участвуют такие токсины. Способность к связыванию будет обычно приводить к нейтрализации токсинов, на основании чего данные соединения можно рассматривать как антагонисты или частичные антагонисты. Кроме того, их можно использовать в качестве направляющих средств или лигандов для других соединений, которые способны к нейтрализации токсинов и которые можно специфически направить к этим токсинам либо с помощью ковалентной, либо нековалентной связи с поверхностью носителя лекарственного средства, такого как липосома, нано- или микрочастица, нано- или микрокапсула, липидный комплекс или мицелла.
При диагностике данные соединения можно использовать для детекции или количественного определения бактериальных токсинов, присутствующих в физиологических жидкостях, таких как кровь, плазма, сыворотка, слизь, покрывающая эпителий слизистой оболочки, такой как оболочка дыхательного тракта, или жидкостях, присутствие которых является результатом патологического состояния, такой как жидкость, обнаруживаемая в среднем ухе при воспалении среднего уха с выделениями (ВСУВ). С этой целью данные соединения могут быть включены в диагностические наборы для использования in vitro или в диагностические композиции, которые можно вводить пациенту. Для этой цели альтернативой является связывание данного соединения с комплексоном, который затем образует комплекс с изотопной меткой, которую можно определить с помощью соответствующей системы слежения.
При предпочтительном использовании данные соединения вводят в качестве активных лекарственных веществ для предотвращения или лечения заболеваний и патологических состояний, связанных с грибковыми и бактериальными инфекциями и наличием грибковых и бактериальных токсинов в организме. Как упоминалось выше, существуют некоторые неблагоприятные воздействия и ограничения использования у общепринятых антибиотиков при терапии острых и хронических инфекций, такие как индукция толерантности и селекция устойчивых вариантов бактерий, угнетение природных защитных систем, нарушение бактериальной флоры, в норме заселяющей слизистые, высвобождение больших количеств бактериальных токсинов, когда погибают бактерии, и т.д. Кроме того, могут быть патологические состояния и заболевания, при которых наличие токсинов и, в частности, бактериальных токсинов, а не присутствие самих микроорганизмов, является их главной причиной, такие как ВСУВ, причем локальное сохранение токсинов в среднем ухе может значительно способствовать проявлению заболевания даже в отсутствие симптомов острой инфекции.
Однако, так как данные соединения обладают также выраженной противомикробной активностью, их можно использовать при тех патологических состояниях, при которых существенно действительно снизить число микроорганизмов, инфицирующих организм, таких как тяжелые острые инфекции. В этом отношении они могут заменять или дополнять другие антибиотики даже при заболеваниях и патологических состояниях, которые нельзя вылечить только соединениями, способными к нейтрализации микробных токсинов. Примерами таких заболеваний являются острые бактериальные или грибковые инфекции, такие как септический шок, острые инфекции глаз(а), печени, почек(чки), легких, бронхов, назального и фронтального синуса, ушей(уха), влагалища, уретры, кожи, центральной нервной системы, сердечной мышцы, селезенки и других тканей и органов. Примером особенно тяжелого состояния, связанного с инфекцией, является септический шок.
Во всех этих случаях можно рекомендовать лечить заболевание не только лекарственными препаратами антибиотиков, но и веществами, которые способны к нейтрализации бактериальных токсинов. Для этой цели соединения согласно изобретению обладают особыми преимуществами, так как они проявляют высокую связывающую и нейтрализующую активность в отношении большинства значимых микробных токсинов, таких как липополисахарид (ЛПС), в случае грамотрицательных бактерий, и липотейхоевая кислота (ЛТК), в случае грамположительных бактерий. При инфекциях верхних дыхательных путей, для лечения которых соединения согласно изобретению особенно предпочтительны, эти бактериальные продукты могут вызывать воспалительную реакцию в среднем ухе или в синусах и могут вызывать повреждение слизистой оболочки эпителия верхних дыхательных путей. Нейтрализация участвующих в этом токсинов может дать возможность предотвратить, подавить или снизить повреждение слизистой, включая нарушение системы мукоцилиарного очищения (СМО), и будет, таким образом, усиливать природные защитные системы. В этих случаях, включая ВСУВ, при котором бактериальные токсины могут представлять собой главную проблему даже в отсутствие значительного числа живых бактериальных клеток, терапия, основанная на введении соединения согласно изобретению, например, непосредственно в среднее ухо, может являться главным терапевтическим подходом. Но также и при других инфекциях дыхательных путей, таких как острый или хронический синусит, или острый или хронический отит, данные соединения могут быть весьма полезны для сохранения нормальных функций слизистой оболочки и их природных систем защиты.
В более общем смысле соединения согласно изобретению являются средствами, пригодными для профилактики и терапии патологических состояний, состоящих в инфицировании и возникающих от инфицирования бактериями, включая Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis, β-гемолитических стрептококков группы А, Staphylоcoccus aureus, грамотрицательных палочковидных энтеробактерий, Streptococcus pyrogenes, Escherichia coli, грамотрицательных бацилл, Pseudomonas sp.
Инфекции, которые можно предотвращать или лечить соединениями согласно изобретению, могут быть вызваны микроорганизмами, которые являются, по существу, планктонными, то есть их внешний вид представляет собой отдельные микробы. Если планктонными микроорганизмами, например, инфицирована физиологическая жидкость млекопитающего, такая как кровь, они могут переноситься с кровотоком по организму млекопитающего.
Системные или генерализованные инфекции, которые могут быть по меньшей мере частично вызваны планктонными микроорганизмами, включают острые или хронические грибковые инфекции, такие как актиномикоз, бластомикоз, нокардиоз, криптококкоз, споротрихоз, кокцидиомикоз и аспергиллез, а также острые или хронические бактериальные инфекции, такие как сибирская язва, столбняк, гангрена, ботулизм, листериоз, тиф, болезнь легионеров, холера, желтая лихорадка и тому подобное. Острые тяжелые системные инфекции могут быть связаны с септическим шоком, который является угрожающей жизни тяжелой формой сепсиса, которая обычно возникает в результате наличия большого числа грамотрицательных бактерий и их токсинов в кровотоке, и которая дополнительно характеризуется сниженным кровотоком в органах и тканях, гипертензией, дисфункцией органов, состоянием с нарушением психики и часто с недостаточностью многих органов, и которое часто поражает людей с ослабленным иммунитетом.
В последние годы, однако, было обнаружено, что многие инфекционные заболевания и патологические состояния также по меньшей мере частично вызываются микроорганизмами, которые образуют более фиксированные сообщества, обычно называемые биопленками. Это верно как для многих системных, так и, в частности, для большого числа более локализованных инфекций. В соответствии с данным описанием, биопленка образуется фиксированным сообществом, характеризующимся клетками, которые связаны с субстратом или поверхностью раздела, или друг с другом, помещены в матрикс из экстраклеточных полимерных веществ, которые они выработали, и проявляют измененный фенотип в отношении скорости роста и транскрипции генов. Обычно экстраклеточный матрикс содержит высоко гидрированный, преимущественно анионный матриксный полимер. Биопленки могут прилипать к поверхностям и к поверхностям раздела; фактически, адгезия может запускать экспрессию генов, регулирующих продукцию экстраклеточного матрикса, и превращение ранее планктонных микроорганизмов в их фиксированные фенотипы.
Полагают, что фиксированные микроорганизмы и их биопленки играют главную роль в ряде инфекционных заболеваний, и для некоторых из них это подтверждается большим объемом данных. Среди этих заболеваний находятся, например, периодонтит, врожденный клапанный эндокардит, кистозный фиброз, хронический бактериальный простатит, бронхит, пневмония, синусит, кариес зубов, хронический тонзилит, эндокардит, некротизирующий фациит, мышечноскелетные инфекции, остеомиелит, инфекции желчевыводящих путей, инфицированные камни почек и воспаление среднего уха. Наиболее вероятно, многие другие, в частности, локальные инфекции, поражающие конкретные области или органы организма, также вызываются фиксированными микроорганизмами, такие как инфекции печени, селезенки, периодонта, глаза, почки, кожи, влагалища, уретры и сердца.
При исследовании активности пептидных соединений, как определено в п. 1, в настоящее время было установлено, что эти соединения также активны против фиксированных, образующих пленку микроорганизмов. Таким образом, они представляют соединения, пригодные для профилактики или терапии заболеваний, связанных с такими микроорганизмами или с участием микроорганизмов в фиксированном состоянии. Например, было обнаружено, что некоторые микроорганизмы способны к образованию биопленок на некоторых поверхностях, таких как ПВХ, например, Pseudomonas putida, могут быть подавлены, и образование биопленок может быть предотвращено при стандартном анализе биопленки.
Эффект подавления может быть достигнут, когда соединение согласно изобретению присутствует в концентрации, составляющей по меньшей мере примерно 0,001 мкМ, и более предпочтительно по меньшей мере 0,01 мкМ, и еще более предпочтительно по меньшей мере примерно 0,1 мкМ. В дополнительном предпочтительном воплощении концентрация данного соединения составляет примерно от 0,1 до примерно 100 мкМ. Однако в зависимости от типа и количества жидкости, которая контактирует с биопленками, и специфических микроорганизмов, образующих биопленку, эти концентрации могут быть изменены.
Кроме того, было обнаружено, что соединения согласно изобретению не только предотвращают образование биопленок микроорганизмами, которые в ином случае способны к образованию микропленки, но активны также в отношении микропленок, которые уже образовались. В зависимости от их концентрации, они способны деструктурировать или разрушать биопленки, которые можно обнаружить, например, при стандартных исследованиях биопленок. И опять, концентрация соединения должна быть выбрана с учетом типа и количества жидкости, которая контактирует с биопленками, и специфических микроорганизмов, образующих биопленку. Например, было обнаружено, что такие низкие концентрации, как примерно 0,001 мкМ, могут уже оказывать деструктурирующее действие на предварительно сформированные биопленки. Таким образом, предпочтительно, когда концентрация данного соединения выбрана так, что составляет по меньшей мере примерно 0,001 мкМ. Другие предпочтительные концентрации составляют по меньшей мере примерно 0,01 мкМ, 0,1 мкМ, 1 мкМ, 10 мкМ и 100 мкМ соответственно.
Особый риск инфекций с образованием биопленок возникает при введении или имплантации медицинских устройств в организм человека или другого млекопитающего, или в диагностических или в терапевтических целях. Например, образование биопленок было показано при инфекциях от контактных линз, сердечных клапанов, венозных катетеров, катетеров в мочевыводящем канале, внутриматочных устройств, швов, сосудистых трансплантатов и шунтов, устройств для перитонеального диализа, протезов полового члена и ортопедических протезов. Таким образом, другое предпочтительное воплощение состоит в использовании соединений согласно изобретению для профилактики или лечения инфекции, возникающей в результате присутствия таких устройств.
Особое преимущество соединений согласно изобретению над природными протеинами, из которых они получены, таких как САР18, LL-37, является их низкая степень нежелательной воспалительной активности. Эта активность связана с разными клеточными процессами, включая пролиферацию, дифференцировку и экспрессию генов, кодирующих провоспалительные медиаторы, подобные цитокинам. Цитокины являются прямыми медиаторами воспаления и влияют на развитие и направление многих иммунологических реакций. Нарушение баланса в продукции цитокинов широко признано в качестве определяющего фактора при нескольких состояниях заболевания. При таком патологическом состоянии, как воспаление среднего уха с выделениями или синусит, это равновесие уже нарушено. Пролиферация Т-клеток также неблагоприятна в этой ситуации, так как это будет дополнительно стимулировать иммунный ответ, который уже неуправляем.
Таким образом, данные соединения можно преимущественно использовать в фармацевтических композициях. В соответствии с данным изобретением такие фармацевтические композиции представляют также и сами данные соединения. В соответствии с данным описанием термин «фармацевтическая композиция» относится к терапевтическим и диагностическим композициям, а также к лекарственным средствам и диагностическим препаратам, содержащим такие композиции. Терапевтические композиции и лекарственные препараты используют для профилактики или лечения заболеваний и других патологических состояний млекопитающих, облегчение которых желательно. Диагностикумы и диагностические композиции используют для постановки диагноза таких заболеваний in vivo и in vitro.
Обычно такое лекарственное средство или композиция содержит по меньшей мере одно соединение согласно изобретению в качестве активного ингредиента и по меньшей мере один фармацевтически приемлемый носитель или наполнитель.
В одном из воплощений лекарственное средство содержит другой активный ингредиент, который может быть выбран из той же группы соединений, которая описана в пункте 1. Альтернативно, другое соединение принадлежит к другой группе. Например, оно может быть соединением, которое, как известно, обладает противомикробной и противогрибковой активностью, но с другим механизмом действия. В дополнительном воплощении другое соединение не включают в то же самое лекарственное средство, но вводят одновременно в виде отдельного препарата.
Кроме того, данную композицию перерабатывают и оформляют таким образом, что ее можно вводить человеку или животному. В соответствии с данным описанием, носитель или наполнитель представляет собой любое фармацевтически приемлемое вещество или смесь веществ, не обладающих существенной фармакологической активностью, которое можно использовать в качестве носителя или вспомогательного вещества с переработкой соединения в дозированную форму, которая стабильна и удобна для введения. Примеры фармацевтически приемлемых наполнителей известны специалистам в данной области и их можно найти в монографиях главных фармакопей.
В одном из воплощений готовят состав композиции и перерабатывают для получения препарата для парентеральной инъекции, препарата для вливания или промывания, предпочтительно для внутрисосудистых инъекций, таких как внутривенных или внутриартериальных, но также для внутримышечных, подкожных, внутрь повреждения (поражения), внутрибрюшинных, местнорегионального и других путей парентерального введения. В другом предпочтительном воплощении данную композицию применяют с воздействием непосредственно на слизистую оболочку верхних воздуховодных путей, такую как оболочка среднего уха. Те же самые принципы, которые определяют изготовление других лекарственных препаратов для этих путей введения, будут также служить указаниями специалистам в данной области, как изготовить такие композиции. Например, одним из требований для парентеральных дозированных форм является их стерильность. Другие требования описаны во всех больших фармакопеях, таких как USР 24, в монографии «General Requirements for Tests and Assays. 1. Injections», p. 1775-1777. Чтобы повысить стабильность парентерального препарата, может быть необходимо получить сухую дозированную форму, которая может быть воспроизведена перед тем, как ее можно будет ввести. Примером такой дозированной формы является высушенный из замороженного состояния или лиофилизированный препарат. Соответственно композиции согласно изобретению могут также содержать муколитический растворитель.
Может быть желательно вводить соединение согласно изобретению в виде парентеральной дозированной формы с регулируемым высвобождением, чтобы исключить частые инъекции и улучшить эффективность и удобство терапии. Разные методы получения таких депо-препаратов известны. Пролонгированное высвобождение может быть обеспечено с помощью твердых имплантатов, наночастиц, нанокапсул, микрочастиц, микрокапсул, эмульсий, суспензий, масляных растворов, липосом или подобных структур.
В случае композиций, которые нужно применять местно на пораженную слизистую, может быть полезно изготовить препарат, обладающий свойствами, которые обеспечивают увеличенный срок локального присутствия в месте применения для повышения эффективности медикаментозного лечения. Для достижения этой цели в препарат могут быть включены связывающие со слизистой вспомогательные вещества. Такие функциональные вспомогательные вещества известны специалистам в данной области; они включают такие полимеры, как полиакриловые кислоты и их производные, полиметакриловые кислоты и их производные, простые эфиры целлюлозы, включая гидроксипропилметилцеллюлозу, карбоксиметилцеллюлозу, разные виды крахмала, хитозан и т.д. Соответственно или альтернативно, композиции согласно изобретению могут также содержать муколитический растворитель. В частности, муколитические растворители используют для того, чтобы влиять на проникновение пептидного соединения согласно изобретению в слизь, например, в респираторном тракте. Подходящие растворители могут содержать известные мукорегуляторные или муколитические средства, такие как N-ацетилцистеин, S-карбоксиметилцистеин, бромгексин, амброксил, ДНКаза, эрдостеин, физиологический раствор и низостеин. Предпочтительно используют бромгексин.
Дополнительными вспомогательными веществами, которые особенно полезны для получения парентеральных препаратов в их наиболее широком определении являются растворители, сорастворители и жидкие или полужидкие носители, такие как стерильная вода, этанол, глицерин, пропиленгликоль, полиэтиленгликоль, бутандиол, жирные масла, коротко- и среднецепочечные триглицериды, лецитин, полиоксиэтиленовые производные касторового масла; вещества для регуляции осмолярности и рН, такие как сахара, особенно глюкоза, сахара-спирты, особенно маннит, хлорид натрия, карбонат натрия, лимонная кислота, ацетат, фосфат, фосфорная кислота, соляная кислота, гидроксид натрия и т.д.; стабилизаторы, антиоксиданты и консерванты, такие как аскорбиновая кислота, сульфит натрия или гидросульфит, ЭДТА, бензиловый спирт и т.д.; другие вспомогательные вещества и вспомогательные средства для лиофилизации, такие как альбумин, декстран и т.д.
Подобным же образом, может быть благоприятно вводить соединение согласно изобретению в лекарственную дозированную форму для введения через слизистую оболочку. Этот путь введения является неинвазивным и удобным для пациента; в то же самое время он обычно приводит к улучшенной биодоступности соединения согласно изобретению по сравнению с пероральным введением, особенно, если соединение нестабильно в жидкостях пищеварительной системы или если оно слишком крупное, чтобы эффективно всасываться из кишечника. Введение через слизистую оболочку возможно, например, посредством назальных, защечных, подъязычных, десневых или вагинальных дозированных форм. Эти дозированные формы могут быть получены известными методами; они могут быть изготовлены в таком составе, чтобы представлять собой назальные капли или жидкости для распыления, вкладываемые формы, пленки, пластыри, гели, мази или таблетки. Предпочтительно, вспомогательные вещества, используемые для дозированной формы для введения через слизистую, включают также одно или более из веществ, обеспечивающих адгезию на слизистой оболочке, пролонгируя, таким образом, время контакта дозированной формы с местом всасывания и тем самым потенциально повышая степень всасывания.
Альтернативно, могут быть разработаны фармацевтические композиции для перорального введения и соответственно переработаны. Соответствующие пероральные дозированные формы включают таблетки, твердые капсулы, мягкие капсулы, порошки, гранулы, пероральные дезинтегрируемые дозированные формы, сиропы, капли, суспензии, шипучие таблетки, жевательные таблетки, пероральные пленки, лиофилизированные дозированные формы, дозированные формы с длительным высвобождением, дозированные формы с регулируемым высвобождением. В одном из предпочтительных воплощений пероральная дозированная форма является твердой дозированной формой с покрытием, растворяющимся в кишечнике для обеспечения защиты соединения от кислого или протеолитического окружения в желудке.
В дополнительном воплощении данные соединения вводят через легкие (пульмонарным путем), используя дозирующий ингалятор, небулайзер, жидкость для получения аэрозоля или ингалятор для сухого порошка. Соответствующие препараты можно изготовить известными методами и по известными методикам. Трансдермальное, ректальное введение или введение в глаза также могут быть реальными в некоторых случаях.
Может быть благоприятным использование передовых способов доставки или направляющих средств для доставки соединения согласно изобретению более эффективно. Например, если выбран не парентеральный путь введения, соответствующая дозированная форма может содержать повышающее биодоступность средство, которое может быть любым веществом или смесью веществ, которое повышает биодоступность данного соединения. Это может быть достигнуто, например, путем защиты соединения от разрушения, такого как посредством ингибитора фермента или антиоксиданта. Более предпочтительно, усиливающее средство повышает биодоступность соединения путем повышения проницаемости барьера всасывания, которым обычно является слизистая оболочка. Усилители проницаемости могут действовать по разным механизмам; некоторые повышают насыщенность водой мембран слизистой, тогда как другие открывают или расширяют дизъюнкции между клетками слизистой оболочки. И еще они снижают вязкость слизи, покрывающей слой клеток слизистой оболочки. Среди предпочтительных усилителей биодоступности присутствуют амфифильные вещества, такие как производные холевой кислоты, фосфолипиды, этанол, жирные кислоты, олеиновая кислота, производные жирных кислот, ЭДТА, карбомеры, поликарбофилы и хитозан.
При использовании противомикробного и противогрибкового действия данных соединений вариантом является их использование для производства лекарственных препаратов, которые или не содержат консервантов, или содержат только в сниженном количестве. Под «сниженным количеством» подразумевается то, что содержание консерванта ниже, чем содержание консерванта, необходимое для эффективной сохранности соответствующей композиции плацебо, которая является композицией, содержащей те же самые компоненты, за исключением активного ингредиента.
Эффективно ли законсервирована композиция, можно определить соответствующими испытаниями, такими как испытание на эффективность консерванта (например, USР <51>), при котором определяли заражение пятью микроорганизмами через определенные интервалы времени, в зависимости от категории продукта. Проведенное на соответствующих сериях такое испытание можно также проводить для того, чтобы определить минимально эффективную концентрацию специфического консерванта для данной композиции, такой как не содержащая лекарственного вещества композиция, соответствующая композиции, которая описана выше.
Например, можно обнаружить, что чтобы эффективно консервировать конкретную композицию с плацебо сорбиновой кислотой, консервант должен присутствовать в концентрации, равной по меньшей мере примерно 0,1% (вес./об). В этом случае эталонная композиция, которая содержит соединение, описанное в пункте 1, могла содержать сорбиновую кислоту в существенно более низкой концентрации, такой как примерно 0,05% (в/о) или менее. В другом воплощении концентрация консерванта выбрана так, что составляет не более примерно одной пятой, а более предпочтительно, не более примерно одной десятой от концентрации, необходимой для эффективного консервирования соответствующей плацебо композиции.
Следующие примеры предназначены для дополнительной иллюстрации изобретения без ограничения объема воплощений, представленных здесь.
Пример 1: Получение соединений
Следующие пептидные соединения, каждое из них содержит 24 аминокислоты, соединения, закодированные здесь как Р60, Р60.4, Р60.Ас и Р60.4Ас, получали по твердофазным стратегиям на автоматическом синтезаторе разнообразных пептидов (SyroII, MultiSyntech, Witten, Germany). Для получения Р60 и Р60.4, Tentagel S AC (Rapp, Tubingen, Germany), привитой полимер полиэтиленгликоля и полистирола использовали в качестве смолы (загрузка 0,2 мэкв, размер частиц 90 мкм). Для получения Р60.Ас и Р60,4Ас использовали Tentagel S AМ, который дает амидированный на С-конце пептид. Повторные соединения выполняли добавлением шестикратного молярного избытка (от загрузки смолы) 0,60 М раствора соответствующей Fmoc аминокислоты в НМП, шестикратного молярного избытка 0,67 М PyBOP в НМП и двенадцатикратного молярного избытка НММ в НМП 2/1 (о/о) в реакционный сосуд. Защита боковых цепей была следующей: tВu для D, Е, S, Т; Вос для К; Тrt для N, Q и Рmс для R. Удаление защиты Fmос выполняли путем добавления 3 раза пиперидина/НМП 1/4 (о/о) в каждый реакционный сосуд. Продолжительность присоединения и удаления защиты составляла 45 мин и 3 раза по 3 минуты соответственно. Промывание после присоединения и удаления защиты Fmос выполняли 6 раз с помощью НМП. Для Р60.Ас и Р60.4Ас N-концевое ацетилирование выполняли уксусной кислотой, тогда как пептид был еще связан со смолой. После синтеза пептидил-смолы интенсивно промывали НМП, дихлорметаном, дихлорметаном/эфиром 1/1 (о/о) и эфиром соответственно и сушили на воздухе. Пептидил-смолы затем расщепляли и удаляли защиту у боковых цепей в ТФА/воде 95/5 (о/о) в течение 2,5 часов (1,5 мл на 10 мкмоль пептида), смолу удаляли фильтрованием и пептид осаждали из ТФА раствора с помощью эфира/пентана 1/1 (о/о) (10 мл на 10 мкмоль пептида). Данный раствор охлаждали в течение 1 часа при -20°С и осажденный пептид выделяли центрифугированием (-20°С, 2500g, 10 мин). После растирания и взмучивания осадка в 10 мл эфира/пентана 1/1 (о/о) и выделения с помощью такой же процедуры, пептиды сушили на воздухе при комнатной температуре в течение 1 часа. Пептиды растворяли в 2 мл воды или 2 мл 10 об.% уксусной кислоты, раствор замораживали в жидком азоте в течение примерно 5 мин и затем лиофилизировали при центрифугировании (1300 об/мин, 8-16 часов). Анализ пептидов выполняли с помощью РП-ВЭЖХ и масс-спектрометрии Малди-Тоф (Maldi-Tof).
Аминокислотные последовательности данных соединений представляют собой:
Р60 IGKEFKRIVQRIKDFLRNLVPRTE
Р60.Ас* IGKEFKRIVQRIKDFLRNLVPRTE
Р60.4 IGKEFKRIVERIKRFLRELVRPLR
Р60.4Ас* IGKEFKRIVERIKRFLRELVRPLR
*Индекс на конце Ас означает, что пептид является ацетилированным на N-конце и амидированным на С-конце.
Пример 2: Нейтрализация токсинов
Соединения, полученные в соответствии с примером 1, испытывали на их способность нейтрализовать бактериальный токсин ЛПС с помощью анализа с амебоцитным лизатом limulus (LAL) и по анализу с цельной кровью (ЦК). Нейтрализацию ЛТК также определяли с помощью анализа с цельной кровью. Пептид LL-37 использовали в качестве положительного контроля. Концентрацию пептида, при которой нейтрализовалось 50% ЛПС, использовали как меру активности пептида. Эти значения концентраций представлены в таблице 1. Различия между соединениями в каждом исследовании не были статистически значимыми. Таким образом, испытанные соединения согласно изобретению продемонстрировали примерно ту же самую степень антитоксической активности, что и у нативного противомикробного пептида LL-37.
Пример 3: Иммунологическая клеточная активация данными соединениями
Данные соединения, полученные в соответствии с примером 1, испытывали на их терапевтически нежелательную иммунологическую активность путем использования анализа Elispot, Т-клеточной пролиферации, ERK-активации и хемотаксиса нейтрофилов. Анализ Elispot применим для определения эффектов лекарственных средств, химических соединений и других соединений на секрецию цитокинов in vitro, представляя тем самым данные по их предполагаемым модуляторным эффектам на иммунную функцию in vivo. Результаты исследования представлены как часть положительных реакций на IFN-гамма. ERK-(связанные с экстраклеточным сигналом киназы)-1/2 является частью МАП-киназного метаболического пути активации, который, как было показано, задействован в разных клеточных процессах, включая пролиферацию, дифференцировку и экспрессию генов, кодирующих провоспалительные медиаторы, подобные цитокинам. Цитокины являются прямыми медиаторами воспаления и влияют на развитие и направление многих иммунологических реакций. Нарушение баланса продукции цитокинов широко признано в качестве определяющего фактора при нескольких болезненных состояниях. Этот баланс уже нарушен в случае таких патологических состояний, как воспаление среднего уха с выделениями и синусит. Т-клеточная пролиферация также неблагоприятна в этой ситуации, так как это также будет стимулировать иммунный ответ, который уже не вышел из-под контроля. Поэтому желательно, чтобы соединения согласно изобретению не стимулировали продукцию цитокинов, Т-клеточную пролиферацию, ERK-активацию или хемотаксис нейтрофилов.
Для Т-клеточной пролиферации 150000 моноцитов периферической крови (РВМС) культивировали в отсутствие или в присутствии 10 мкг/мл данных соединений в течение 5 дней в 96-луночноных круглодонных планшетах (Costar Inc. Cambridge, MA) при конечном объеме 150 мкл полной IMDM. В качестве положительного контроля РВМС культивировали в присутствии 25 Ед/мл рекомбинантного IL-2. Во время конечных 20 часов культивирования РВМС импульсно метили [3Н]тимидином (0,5 микроКи/лунку), после чего включение 3Н измеряли с помощью жидкостного сцинтилляционного счетчика. Для определения Т-клеточных цитокинов IFN и IL-10 по анализу Elispot 1,5×106 РВМС культивировали в 0,5 мл полной IMDM в отсутствие или в присутствии разных концентраций синтетического пептида. В качестве положительного контроля РВМС стимулировали 10 мкг/мл митогеном лаконоса (PWM). После 48 часов культивирования РВМС собирали путем осторожного смывания из лунок теплой IMDM, чтобы собрать несвязанные клетки, которые промывали большим объемом IMDM. Затем РВМС помещали в предварительно покрытые антителами планшеты для ELISA и культивировали в течение 5 часов в IMDM, дополненной 2% объединенной человеческой АВ сывороткой, при 37°С и 5% СО2, после чего планшеты проявляли по методике производителя (U-CyTech, Utrecht, The Netherlands). Пятна подсчитывали под микроскопом Olympus и анализировали с помощью программного обеспечения Olimpus Micro Image 4.0 (Paes Nederland, Zoeterwoude, The Netherlands). Окончательные результаты выражены как относительное количество положительных показателей стимуляции (положительно: >2).
Активацию ERK-1/2 испытывали с помощью клеток от линии мукоэпидермоидных опухолевых клеток легких NСI-Н292 (АТСС, Rockville, MD), которые культивировали в 24- и 6-луночных планшетах для культуры тканей в среде RPMI1640 (Gibco, Grand Island, NY), дополненной 2 мМ L-глутамина (Bio Wittaker, Walkersville, MD), 200 Ед/мл пенициллина (Bio Wittaker), 200 мкг/мл стрептомицина (Bio Wittaker) и 10% (о/о) инактивированной нагреванием фетальной телячьей сыворотки (Gibco). После достижения почти слияния клетки культивировали в течение ночи в бессыворотной среде. Клетки затем стимулировали в течение 15 минут указанными воздействиями. Клеточные лизаты готовили, используя буфер для лизиса (0,5% [о/о] Triton X-100, 0,1М Трис-НСl рН 7,4, 100 мМ NаСl, 1 мМ МgСl2, 1 мМ Nа3VО4, мини-полной смесью ингибитора протеазы [Boeringer Mannheim, Roche, Basel, Switzerland]). Образцы подвергали SDS-PAGE в 10% геле на основе глицина и разделенные протеины переносили на поливинилидендифторидную (ПВДФ) мембрану. Сайты неспецифического связывания блокировали (РВS) ФБР/0,05% твин-20/1% казеином. Блоты инкубировали с кроличьими поликлональными антителами к фосфорилированной ERK-1/2 (New England Biolabs, Beverly, MA) и вторыми конъюгированными с пероксидазой хрена противокроличьими IgG антителами. Для выявления иммунореактивности использовали систему регистрации повышенной хемолюминесценции (ЕСL) при Вестерн-блоттинге (Amersham Pharmacia Biotech, Upsala, Швеция).
Хемотаксис нейтрофилов определяли с помощью нейтрофилов, выделенных из периферической крови с использованием центрифугирования в среде перколл (удельная плотность: 1,082 г/мл). Клетки снова суспендировали до концентрации
2,5×106 клеток/мл в среде для хемотаксиса (20 мМ N-2-гидроксиэтилпиперазин-N'-2-этансульфоновая кислота (HEPES буфер)) HEPES, 132 мМ NаСl, 6 мМ КСl, 1,2 мМ КН2РО4, 1 мМ МgSО4, 5,5 мМ глюкозы, 0,1 мМ СаСl2 и 0,5% (в/о) человеческого сывороточного альбумина [Central Laboratory of the Netherlands Red Cross Blood Transfusion Service (CLB), Amsterdam, The Netherlands], разведенной 1:1 бессывороточной IMDM. Действие соединений на хемотаксис оценивали, используя модифицированную методику Boyden Camber. В кратком изложении, по 26 мкл действующего средства, разведенного в буфере HEPES, добавляли в лунки нижнего отделения, и 50 мкл суспензии нейтрофилов (2,5×106 клеток/мл) добавляли в верхнее отделение. Отделения были разделены двумя фильтрами: нижним фильтром с размером пор 0,45 мкм (Millipore Products, Bedford, MA) и верхним фильтром с размером пор 8 мкм (Sartorius Filter, San Francisco, CA). После инкубации в течение 90 минут при 37°С верхние фильтры удаляли, фиксировали в этаноле/бутаноле (80:20, об/об) и окрашивали раствором Weigert. Чтобы определить хемотактическую активность нейтрофилов, нейтрофилы подсчитывали в шести случайно выбранных полях при большом увеличении (×400) и процент нейтрофилов на мембране рассчитывали по сравнению с положительным контролем (10-8 М N-формилметионил-лейцил-фенилаланина (FMLP, Sigma).
Результаты представлены в таблице 2. Таким образом, испытанные соединения согласно изобретению, в частности Р60.4, индуцировали иммунный ответ на очень низком уровне, ниже чем природный пептид LL-37. При их действии показаны низкая ERK-активация и фактическое отсутствие хемотаксиса нейтрофилов.
Пример 4: Переносимость in vivo
Cоединение Р60.4Ас получали в соответствии с примером 1 и испытывали на его переносимость in vivo. Более конкретно оценивали его способность вызывать раздражение кожи и глаз на кроликах, тогда как ототоксичность изучали на модели на морских свинках. Кроме того, его ототоксичность при системном введении оценивали после внутривенного введения.
Что касается испытаний на раздражение кожи и глаз, трех кроликов подвергали воздействию 0,5 мл фосфатного буферного пептидного раствора (2 мг/мл), наносимого на выстриженную кожу на 4 часа, используя полупроницаемую повязку. Наблюдения производили через 1, 24, 48 и 72 часа после воздействия. Единственные пробы по 0,1 мл раствора пептида (2 мг/мл) в фосфатном буфере (рН 7,5) закапывали в один глаз каждого их трех кроликов, чтобы выполнить исследование на острое раздражение глаза/разъедание. Наблюдения производили через 1, 24, 48 и 72 часа после закапывания.
В результате не было обнаружено раздражения кожи. Закапывание в глаз раствора пептида приводило к покраснению конъюнктивы, которое полностью проходило через 24 часа после закапывания.
Системную токсичность Р60.4Ас оценивали при исследовании токсичности при однократном введении и при повторных введениях на крысах. Пептид вводили ежедневно внутривенно в повышающихся дозах. На этом этапе устанавливали максимальную переносимую дозу (МПД). Токсичность при повторном введении также изучали на этапе определения МПД. На этапе повышения дозы 9 крыс делили на три группы, и они получали 0,4, 2 или 8 мг/кг/сутки в течение двух дней. Клинические признаки регистрировали дважды в сутки со дня введения дозы и через один день после введения дозы, вес тела регистрировали перед введением первой дозы и через один день после введения дозы. На этапе определения МПД 5 самок и 5 самцов крыс получали 8 мг/кг/сутки на протяжении последующих 5 дней. Клинические признаки регистрировали дважды в сутки в дни введения дозы, вес тела определяли в 1 день и 6 день. Клинические лабораторные исследования выполняли перед забоем и вскрытием трупа. Макроскопию выполняли в конце этапа определения МПД.
В результате не происходило гибели при исследовании повышения дозы при системном введении. Кроме того, не было отмечено явных отклонений по клиническим признакам и весу тела. Во время этапа определения МПД также не было гибели и не отмечено явных связанных с пептидом результатов по клиническим показателям, весу тела, гематологии, показателям клинической биохимии и при макроскопическом обследовании.
Пример 5: Ототоксичность
Чтобы оценить ототоксичность пептида Р60.4-Ас этот пептид испытывали на морских свинках (HsdPoc:DH; Harlan, Horst, The Netherlands). В этом исследовании использовали семь здоровых самцов альбиносов морских свинок (500-1200 г) без внешней патологии уха. Животным проводили анестезию путем внутрибрюшинных инъекций 40 мг/кг кетамина (Eurovet Animal Health B.V., Bladel, The Netherlands) и 10 мг/кг ромпана (Bayer A.G., Leverkusen, Germany). Затем выполняли контрольное испытание слуха, слуховые полости хирургически вскрывали, чтобы поместить небольшой кусочек спонгостана в круглое окно мембраны (RWM), и на спонгостан добавляли разные растворы (примерно 10 мкл). На кожу накладывали швы и выполняли последующее испытание слуха. Нанесение на RWM выполняли на правых ушах, левые уши оставляли без воздействия растворов. Одно животное получало PBS в качестве первого плацебо раствора (плацебо I), а другое животное получало второй плацебо раствор (плацебо II), которым был 7% макрогол 10000 (Macrogol 10000) в изотоническом [NаСl] и консервированном [0,02% бензалкония хлорид и 0,1% Nа2ЭДТА] 20 мМ растворе фосфатного буфера (рН 5,5). Две морские свинки получали цисплатин (0,66 мг/мл в PBS, полученном от Sigma Chemicals, Zwijndrecht, The Netherlands), который служил при испытании в качестве положительного контроля. Пептид Р60.4-Ас (2 мг/мл) испытывали в PBS растворе (препарат I) у одного животного и в буфере, соответствующем второму плацебо (препарат II), у двух других животных.
Слуховую реакцию в стволе головного мозга (ABR) определяли перед введением и непосредственно после хирургического вмешательства, и через 3, 7, 14 и 22 дня после него, используя компьютерную систему усреднения сигнала (Tucker-Davis Technology, Alucha, FL, USA). Морским свинкам делали анестезию и головной телефон с ушным вкладышем помещали во внутренний канал уха. Подкожные электроды помещали над макушкой головы (активный) и над ипсилатеральной полостью (сравнения). Заземляющие электроды помещали над мышцами шеи. ABR регистрировали в электрически экранированной, двухстенной, экранированной от радиочастот акустической камере в ответ на 10 мс звуковые посылы при 1 кГц. Интенсивности стимулов измеряли и выражали в дБ. Порог СРСМ определяли как самую низкую интенсивность, способную вызывать воспроизводимую, визуально определяемую реакцию. Пороги ABR после воздействия препарата сравнивали с порогами ABR до воздействия.
В результате, нанесение PBS на круглое окно, который использовали в качестве контроля, не приводило к изменению порога через 22 дня после хирургического вмешательства. Буфер для препарата приводил к изменению порога в 2 дБ через 22 дня. Цисплатин, с другой стороны, индуцировал изменения порога соответственно в -49 дБ и -64 дБ, что указывало на тяжелую потерю слуха (таблица 3). Эта часть экспериментов служила в качестве положительного контроля для изучения ототоксичности. Пептид Р60.4-Ас (2 мг/мл) в PBS вызывал изменение порога в -7 дБ через 22 дня после хирургического вмешательства. У обоих животных, которые получали Р60.4-Ас в качестве препарата II, получали изменение порога в 1 дБ.
Пример 6: Противомикробная активность
Соединение Р60.4Ас получали в соответствии с примером 1 и стерилизовали путем стерилизующего фильтрования в 10 мл стеклянные флаконы с винтовыми крышками. Консервант не добавляли. Отдельно готовили соответствующий раствор плацебо, т.е. раствор с такими же компонентами, за исключением соединения Р60.4Ас. Отбирали образцы из каждого из двух препаратов для проведения испытания на противомикробную активность. В результате раствор, содержащий соединение Р60.4Ас, как было обнаружено, подавляет бактериальный рост или даже снижает число микробов, тогда как соответствующий раствор плацебо не проявил актимикробной активности.
Пример 7: Противомикробная активность
Антибактериальную и противогрибковую активность Р60.4-Ас и LL-37 in vitro определяли как минимальные подавляющие концентрации (МПК) путем определения чувствительности с помощью микроразведений в 96-луночных микротитровальных планшетах по модифицированному варианту R. Hancock's “Modified MIC method for cationic antimicrobial peptides”. Антибактериальную активность испытывали на эталонных штаммах Escherichia coli ATCC 8739, Pseudomonas aeruginosa ATCC 9027. Противогрибковую активность оценивали на Candida albicans ATCC 10231 и Aspergillus niger ATCC 14406. Исследование на противомикробную активность проводили с разными концентрациями Р60.4Ас и LL-37 для сравнения их действий на бактериальный и грибковый рост. Антибактериальную активность оценивали, используя культивируемые бактерии в логарифмической фазе роста в триптиказном соевом бульоне при 37°С. Культуры разводили 10 мМ натрийфосфатным буфером рН 7,4 с получением примерно 5,0×106 КОЕ/мл. 10 мкл разведения испытуемого штамма переносили в 96-луночный планшет и добавляли 100 мкл разных концентраций пептида в каждую лунку. Планшеты инкубировали при 37°С в течение 24 часов и затем давали оценки в баллах росту при визуальном просмотре на проекторе. Затем их возвращали на инкубацию в течение еще 24 часов, срок, после которого их снова оценивали на наличие роста. Штамм дрожжей C. albicans получали, как описано выше. Нитевидный грибок A. niger использовали в виде суспензии спор, культивируемой в чашках с глюкозным агаром Сабуро при 20-25°С в течение 6-10 дней или до тех пор, пока не происходило соответствующее спорообразование. Споры собирали разрушением роста, и если необходимо, концентрацию доводили до конечной концентрации 5×106 КОЕ/мл.
Таким образом, противомикробную активность Р60.4-Ас оценивали в отношении штаммов грамотрицательных бактерий и в отношении грибов C.albicans и A.niger и сравнивали с LL-37. Значения МПК для каждого пептида представлены в таблице 4. Р60.4-Ас продемонстрировал более высокую или равную активность в отношении штаммов грамотрицательных бактерий E.coli и P.aeruginosa в сравнении с LL-37. В некоторых случаях была определена также и бактерицидная активность для обоих пептидов. Р60.4-Ас проявил МПК при 6 мкМ в отношении С.albicans и может быть достаточно фунгицидным при 18 мкМ. Р60.4-Ас при 18 мкМ подавлял прорастание спор A.niger в течение 24 часов, тогда как LL-37 не проявлял активности против A.niger.
Пример 8: Подавление образования биопленки
Соединение Р60.4Ас получали в соответствии с примером 1 и испытывали на его эффективность как ингибитора образования биопленки Pseudomonas putida PCL1445. Использовали стандартное исследование биопланки PVC, при котором биопленки образуются на поливинилхлоридной поверхности лунок микротитровальных планшет. К суспензиям P.putida в лунках добавляли раствор Р60.4Ас (0,9 мкМ и 9 мкМ) и инкубировали в течение 10 часов. В результате было обнаружено, что Р60.4Ас при 9 мкМ подавлял образование биопленки на более, чем 90%, тогда как концентрация 0,9 мкМ давала в результате снижение примерно на 50% в сравнении с буферным раствором без пептида.
Пример 9: Подавление сформированных биопленок
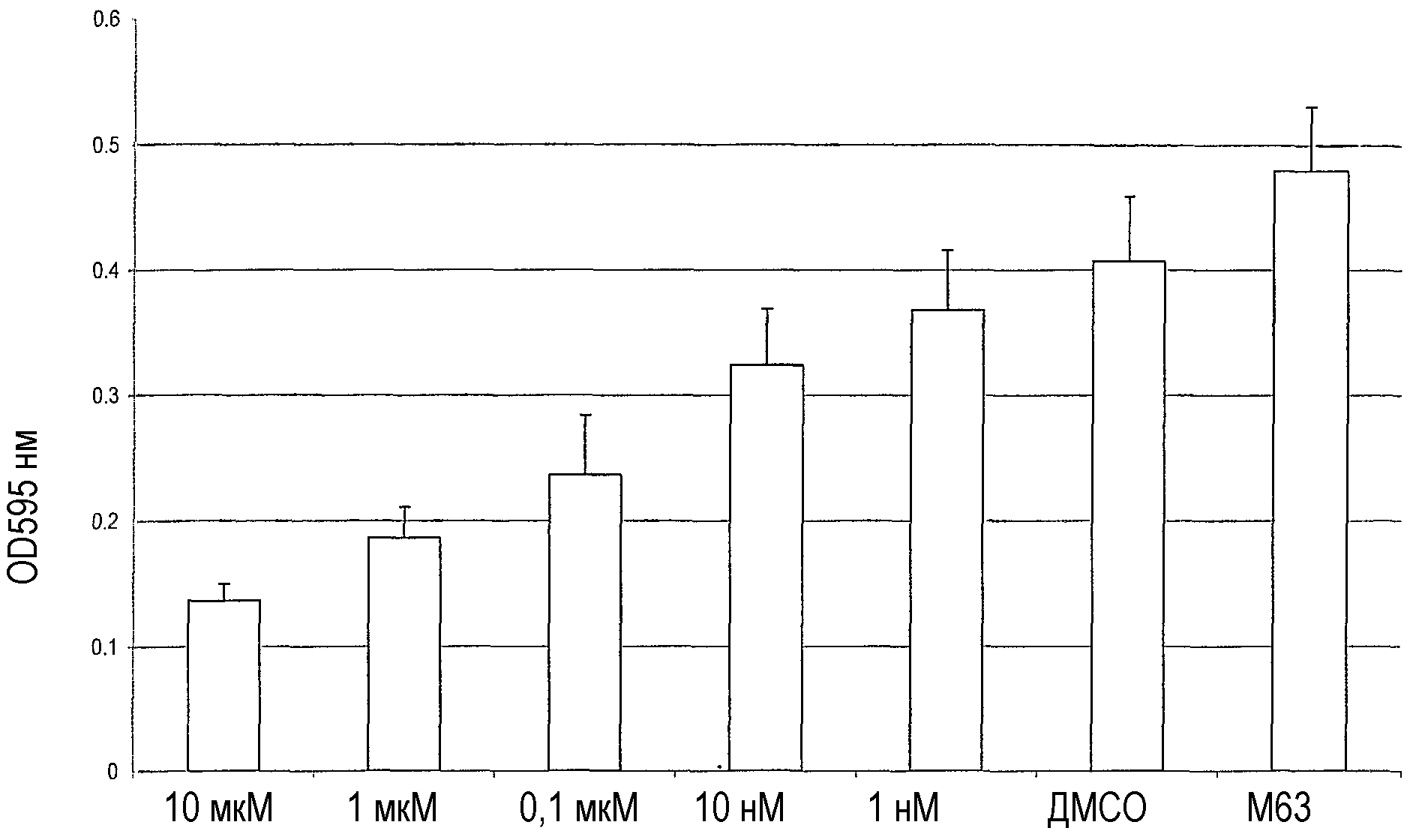
Соединение Р60.4Ас получали в соответствии с примером 1 и испытывали на его эффективность по разрушению биопленок Pseudomonas putida. Биопленки формировали, используя стандартное исследование биопланки PVC, как в примере 7. Давали образоваться биопленкам из суспензий Pseudomonas putida PCL1445 в лунках микротитровальной планшеты в течение 7 часов. После чего добавляли разные количества Р60.4Ас, а также контроли, т.е. ДМСО и раствор буферной среды, М63 соответственно. После 18 часов инкубации биопленки оценивали на их оптическую плотность при 595 нм (ОВ595). В результате ДМСО приводил только к небольшому снижению OD595 по сравнению с М63, тогда как Р60.4Ас существенно разрушал биопленки в зависимости от его концентрации.
На чертеже показаны значения OD595 для разных молярных концентраций Р60.4Ас (столбики 1-5), ДМСО и М63.
Реферат
Изобретение относится к области медицины и биотехнологии и касается пептидного соединения, которое обладает противомикробной активностью. Сущность изобретения заключается в применении аминокислотного соединения, включающего последовательность IGKEFKRIVERIKRFLRELVRPLR для производства лекарственного средства для лечения грибковой или бактериальной инфекции млекопитающего. Данное соединение обладает также сродством к токсинам и особенно к бактериальным токсинам, таким как липополисахарид или липотейхоевая кислота. Преимущество изобретения заключается в расширении области применения по сравнению с антибиотиками. 21 з.п. ф-лы, 4 табл., 1 ил.
Комментарии