Наночастицы для инкапсулирования соединений, их получение и применение - RU2718922C2
Код документа: RU2718922C2
Чертежи







Описание
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение принадлежит к отраслям фармацевтики и нанотехнологий и относится к инкапсулированию активных ингредиентов с помощью наночастиц новых полимерных конъюгатов, полученных из метилвинилового эфира-малеинового ангидрида. Настоящее изобретение также относится к способу получения указанных конъюгатов и наночастиц, композициям, содержащим указанные конъюгаты или наночастицы, и вариантам их применения.
УРОВЕНЬ ТЕХНИКИ
Парентеральное введение некоторых активных ингредиентов (лекарственных препаратов), включая многие противоопухолевые агенты, вызывает различные проблемы. Повышение качества жизни пациентов, а также снижение затрат на здравоохранение следует отдельно отметить среди основных преимуществ, связанных с введением противоопухолевых агентов пероральным путем. Пероральный способ введения должен обеспечивать постоянное воздействие противоопухолевого лекарственного препарата на раковые клетки при подходящем и устойчивом уровне концентрации, что тем самым может улучшать терапевтический индекс и уменьшать побочные эффекты.
При пероральном введении лекарственного препарата его биодоступность обычно является низкой из-за ряда факторов, таких как растворимость, стабильность в кишечнике, проницаемость и пресистемный метаболизм:
- соединения с низкой растворимостью и низкой проницаемостью вызывают сложности при изготовлении пероральных лекарственных форм.
- желудочно-кишечный тракт является проблематичной областью для введения вследствие быстрого оборота слизи и относительно постоянного времени прохождения.
- лекарственные препараты должны проходить через стенку кишечника и затем через кровоток воротной вены в печень, при этом пресистемный метаболизм протекает в обоих указанных местах.
- всасывание лекарственных препаратов также может быть ограничено механизмами оттока, особенно если соединения являются липофильными. Например, секреторный транспортер, Р-гликопротеин, расположенный на поверхности слизистой оболочки эпителиальных клеток, обуславливает низкую и изменчивую биодоступность различных лекарственных препаратов (например, паклитаксела, доцетаксела или камптотецина).
Вышеперечисленные факторы, которые можно объединить в две взаимосвязанные категории, всасывание (растворимость и проницаемость) и метаболизм, следует учитывать при оптимизации пероральной биодоступности лекарственных препаратов (Hayden Thomas et al., Expert Opin Drug Metab Toxicol 2006; 2(4):591-608).
Одной из возможных стратегий преодоления вышеупомянутых недостатков может быть применение полимерных наночастиц в качестве носителей для пероральной доставки лекарственных препаратов. Инкапсулирование лекарственных препаратов в системы на основе наночастиц защищает их от жестких условий в кишечнике, обеспечивает перенос заключенного в указанные системы лекарственного препарата до поверхности всасывающих клеток и позволяет контролировать его высвобождение. Биоадгезия дополнительно улучшает введение лекарственного препарата. Биоадгезивные системы доставки лекарственных препаратов обладают несколькими преимуществами: (i) благодаря адгезии лекарственный препарат остается дольше в месте доставки; (ii) использование специфичных биоадгезивных молекул обеспечивает нацеленное воздействие на конкретный сайт или ткань, например, в желудочно-кишечном тракте (ЖКТ); и (iii) более длительное время удержания, в комбинации с контролируемым высвобождением лекарственного препарата, может обеспечить более низкую частоту введения (Woodley, Clin Pharmacokinet 2001; 40(2):77-84). Однако для того, чтобы обладать надлежащими биоадгезивными свойствами, наночастицы должны быть способны проникать через слизистую оболочку, чтобы пересечь слизистый слой и затем начать адгезионные взаимодействия на поверхности эпителия.
Многие полимеры были описаны как материалы, пригодные для получения биоадгезивных наночастиц для пероральной доставки лекарственных препаратов, такие как хитозан, поливиниловый спирт (ПВС), гомо- и сополимеры молочной и гликолевой кислот (PLGA) или различные метилвиниловые эфиры-малеинового ангидрида (выпущен в серийное производство как гантрез (Gantrez®) компанией International Specialty Products ISP, США).
Связывание или покрытие наночастиц подходящими гидрофильными соединениями может изменить их физико-химические характеристики и тем самым изменить их распределение и взаимодействие с биологической средой, способствуя поступлению инкапсулированного лекарственного препарата в надлежащее место действия или всасывания. Одна из возможных стратегий включает использование полиэтиленгликоль (ПЭГ)-конъюгированных наночастиц, называемых пегилированными наночастицами.
В отношении использования пегилированных наночастиц при пероральном введении соединение полиэтиленгликолей со стандартными наночастицами сводит к минимуму взаимодействие таких носителей с компонентами просвета кишечника. Этот факт связан с тем, что ПЭГ способны образовывать гидрофильную корону вокруг наночастиц и обеспечивают гладкую поверхность, которая предотвращает взаимодействие с белками (Gref et al, Science 1994; 263:1600-1603) и облегчает прохождение через защитный слизистый слой слизистой оболочки кишечника.
Основным недостатком данной стратегии является стабильность связи полиэтиленгликолей с поверхностью наночастиц (Peracchia et al., Life Sci 1997; 6:749-761). Известно, что способность полиэтиленгликоля не вступать во взаимодействия с белками зависит от конфигурации, заряда, длины и гибкости полимерных цепей (Torchillin, J Microencapsul 1998;15:1-19). Модификацию поверхности наночастиц в основном осуществляют путем физической адсорбции (Stolnik et al., Adv Drug Del Rev 1995; 16:195-214) или путем ковалентного связывания в виде двойных блоков или тройных блоков (De Jaeghere et al., J Drug Target 2000; 8:143-153). Учитывая, что ковалентное связывание является предпочтительным, большинство пегилированных наночастиц были получены с использованием сополимеров полиэтиленгликоля с молочной кислотой или гликолевой кислотой. Однако способ сополимеризации требует использования нескольких катализаторов и специфичных химических условий (Beletsi et al., Int. J. Pharm 1999; 182:187-197). Помимо этого, токсичные остатки органического растворителя, используемого в органическом синтезе (метиленхлорида, толуола и т.д.), могут вызывать проблемы.
Также известны способы модификации поверхности наночастиц поли(метилвинилового эфира-малеинового ангидрида) (PVM/MA) с использованием полиэтиленгликолей. В патенте WO 2005/104648 описаны два различных способа получения пегилированных наночастиц: (i) одновременная инкубация полимера PVM/MA и полиэтиленгликоля в органическом растворителе перед десольватацией полимера, и (ii) инкубация биодеградируемых полимерных наночастиц с водным раствором полиэтиленгликоля. Оба способа подходят для получения наночастиц PVM/MA, соединенных с полиэтиленгликолем, связанным с их поверхностью, однако главным недостатком простой адсорбции является быстрая потеря покрывающего слоя в условиях in vivo (Neal et al., J Pharm Sci 1998; 87:242-1248) вследствие нестабильности указанного взаимодействия.
В WO 2005/104648 также описаны пегилированные наночастицы PVM/MA с превосходными характеристиками адгезии в кишечнике по сравнению с наночастицами PVM/MA, не содержащими полиэтиленгликоля. Указанные наночастицы продемонстрировали высокую универсальность для инкапсулирования биологически активных молекул. Например, в WO 2009/121997 раскрыто, что модификация и покрытие наночастиц PVM/MA полиэтиленгликолем позволяет получать пегилированные наночастицы, способные инкапсулировать значительные количества химически синтезированных лекарственных препаратов. Однако стабильность покрытия является низкой, и со временем покрывающий слой может быть потерян.
Несмотря на то, что биоадгезивные системы доставки лекарственных препаратов могут обеспечить улучшенное время прохождения через желудочно-кишечный тракт, благодаря адгезии на поверхности слизистой оболочки желудочно-кишечного тракта, высокая степень адгезии и более медленное прохождение не всегда приводят к улучшению биологической доступности (Davis SS. Drug Discovery Today 2005; 10(4):249-257). Если лекарственный препарат не растворяется легко или не проникает через эпителиальную мембрану из-за механизмов оттока, то время его присутствия в месте всасывания может быть недостаточным. В WO 2008/129106 описаны альтернативные наночастицы для введения лекарственного препарата, содержащие PVM/MA и циклодекстрин, которые при пероральном введении могут увеличить биодоступность лекарственных препаратов, которые являются субстратами Р-гликопротеина.
Тем не менее, по-прежнему существует потребность в улучшении биодоступности некоторых лекарственных препаратов после перорального введения. Следовательно, требуется еще много усилий для разработки новых систем введения лекарственных препаратов (например, синтез новых полимеров и сополимеров) для комбинирования гидрофильных/гидрофобных свойств нескольких лекарственных препаратов с хорошими физико-химическими свойствами (разлагаемость, стабильность и механическая прочность) и улучшенными фармацевтическими характеристиками (распределение, биоадгезия и скорость высвобождения) наночастиц, изготовленных из таких новых полимеров. Это позволило бы повысить эффективность всасывания лекарственных препаратов, обеспечивая тем самым альтернативные способы лечения, альтернативные инфузионной терапии, и снизить затраты системы здравоохранения на лечение с использованием указанного типа лекарственных препаратов и улучшить качество жизни пациента.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является разработка новых систем введения лекарственных препаратов, которые являются стабильными в ЖКТ, могут инкапсулировать значительное количество биологически активных соединений (например, лекарственных препаратов или активных ингредиентов, особенно гидрофобных лекарственных препаратов), и увеличить биодоступность перорально вводимых лекарственных препаратов, т.е. обеспечение новых полимерных конъюгатов и наночастиц, изготовленных из них, которые позволят преодолеть вышеупомянутые недостатки.
Финансирование работы, которая легла в основу настоящего изобретения, было получено в рамках Седьмой рамочной программы Европейского союза (FP 7/2007-2013) в соответствии с соглашением о гранте NMP4-LA-2011-280761.
Было обнаружено, что новые сложноэфирные полимерные конъюгаты PVM/MA с полиэтиленгликолем или производным полиэтиленгликоля, содержащим концевую реакционноспособную гидроксильную группу, могут быть синтезированы с помощью простой реакции, и продукты указанной реакции пригодны для легкого получения наночастиц с хорошими характеристиками для введения биологически активных соединений. В частности, было обнаружено, что наночастицы, образованные указанными новыми сложноэфирными полимерными конъюгатами PVM/MA с полиэтиленгликолем или его производным, содержащим концевую реакционноспособную гидроксильную группу, способны: i) инкапсулировать значительные количества биологически активных соединений (например, паклитаксела, доцетаксела и камптотецина), и ii) пересекают слизистый слой и тесно взаимодействуют с поверхностью энтероцитов (улучшая абсорбцию лекарственного препарата).
Неожиданно было обнаружено, что наночастицы, предложенные в настоящем изобретении, обеспечивают повышенную биодоступность указанных биологически активных соединений через слизистую оболочку кишечника, что приводит к достижению пролонгированного и устойчивого уровня в плазме крови в течение очень длительных периодов времени (например, по меньшей мере 72 часов в случае доцетаксела), обеспечивая способ введения, альтернативный инфузионной терапии, что позволяет снизить затраты системы здравоохранения на лечение с использованием указанного типа лекарственных препаратов и улучшить качество жизни пациента.
Таким образом, согласно первому аспекту в настоящем изобретении предложен новый сложноэфирный полимерный конъюгат PVM/MA с молекулой, содержащей концевые гидроксильные группы, причем указанная молекула с концевыми гидроксильными группами выбрана из полиэтиленгликоля и его производного, содержащего концевую реакционноспособную гидроксильную группу.
Согласно одному конкретному варианту реализации настоящего изобретения молекула с концевыми гидроксильными группами представляет собой полиэтиленгликоль, который не разветвлен и не содержит замещенных гидроксильных групп. В указанном варианте реализации настоящего изобретения применяемые полиэтиленгликоли предпочтительно имеют молекулярную массу в диапазоне от 400 до 35000 Да; согласно более конкретному варианту реализации настоящего изобретения полиэтиленгликоль выбран из группы, состоящей из полиэтиленгликоля 1000 (ПЭГ1), полиэтиленгликоля 2000 (ПЭГ2), полиэтиленгликоля 6000 (ПЭГ6) и полиэтиленгликоля 10000 (ПЭГ 10).
Согласно другому конкретному варианту реализации настоящего изобретения полиэтиленгликоль содержит одну замещенную гидроксильную группу. Согласно указанному варианту реализации настоящего изобретения молекула с концевыми гидроксильными группами является, следовательно, производным полиэтиленгликоля, содержащим концевую реакционноспособную гидроксильную группу, и предпочтительно представляет собой простой алкиловый эфир полиоксиэтилена. Согласно более конкретному варианту реализации настоящего изобретения простой алкиловый эфир полиоксиэтилена представляет собой простой метиловый эфир полиэтиленгликоля (мПЭГ) и может быть выбран из группы, состоящей из метоксиполиэтиленгликоля 1000 (мПЭГ1), метоксиполиэтиленгликоля 2000 (мПЭГ2), метоксиполиэтиленгликоля 6000 (мПЭГ6) и метоксиполиэтиленгликоля 10000 (мПЭГ10).
Согласно другому аспекту в настоящем изобретении предложен способ получения сложноэфирного полимерного конъюгата PVM/MA с молекулой, содержащей концевые гидроксильные группы, причем указанная молекула, содержащая концевые гидроксильные группы, выбрана из полиэтиленгликоля и его производного, содержащего концевую реакционноспособную гидроксильную группу, который включает следующие этапы:
a). взаимодействие PVM/MA с молекулой, содержащей концевые гидроксильные группы, согласно настоящему изобретению в органическом растворителе, и
b). удаление органического растворителя.
Указанный способ согласно настоящему изобретению является простым и может быть использован в промышленном масштабе. Согласно одному предпочтительному варианту реализации настоящего изобретения указанный способ включает дополнительный этап с) очистки сложноэфирного конъюгата.
Согласно конкретному варианту реализации настоящего изобретения массовое соотношение между PVM/MA и молекулой, содержащей концевые гидроксильные группы, в растворе согласно этапу а) составляет 1:0,01-0,25, предпочтительно 1:0,015-0,2, более предпочтительно 1:0,05-0,125.
Согласно другому аспекту в настоящем изобретении предложен сложноэфирный полимерный конъюгат, полученный указанным способом согласно настоящему изобретению.
Согласно другому аспекту в настоящем изобретении предложено применение сложноэфирного полимерного конъюгата согласно настоящему изобретению или полученного способом согласно настоящему изобретению в получении полимерных наночастиц для доставки лекарственного препарата; предпочтительно пероральной доставки лекарственного препарата.
Согласно другому аспекту в настоящем изобретении предложена композиция, содержащая i) сложноэфирный полимерный конъюгат согласно настоящему изобретению или сложноэфирный полимерный конъюгат, полученный способом согласно настоящему изобретению, и ii) носитель; а также указанная композиция согласно настоящему изобретению для применения для доставки лекарственного препарата; предпочтительно пероральной доставки лекарственного препарата.
Согласно другому аспекту в настоящем изобретении предложена наночастица, содержащая: i) матрикс из сложноэфирного полимерного конъюгата в соответствии с настоящим изобретением или сложноэфирный полимерный конъюгат, полученный способом согласно настоящему изобретению, и ii) биологически активное соединение. Согласно конкретному варианту реализации настоящего изобретения биологически активное соединение представляет собой противоопухолевый агент. Согласно более конкретному варианту реализации настоящего изобретения противоопухолевый агент выбран из группы, состоящей из доцетаксела, камптотецина и паклитаксела; предпочтительно противоопухолевый агент представляет собой доцетаксел.
Согласно другому аспекту в настоящем изобретении предложен способ получения наночастицы в соответствии с настоящим изобретением, включающий следующие этапы:
a). смешивание сложноэфирного полимерного конъюгата согласно настоящему изобретению с биологически активным соединением в органической среде, и
b). десольватация сложноэфирного полимерного конъюгата путем добавления спирта и воды в присутствии двухвалентного металла. Предпочтительно этап а) проводят посредством смешивания (i) органического раствора, содержащего сложноэфирный конъюгат согласно настоящему изобретению, с (ii) органическим раствором или дисперсией биологически активного соединения.
Предпочтительно этап b) проводят путем добавления водно-спиртовой смеси, содержащей двухвалентный металл, к смеси, полученной на этапе а).
Согласно конкретному варианту реализации настоящего изобретения двухвалентный металл выбран из группы, состоящей из кальция, магния, цинка, железа в двухвалентной форме и их комбинаций; предпочтительно двухвалентный металл представляет собой кальций.
Согласно другому конкретному варианту реализации настоящего изобретения массовое соотношение между сложноэфирным конъюгатом согласно настоящему изобретению и биологически активным соединением в смеси согласно этапу а) составляет 1:0,01-0,20, предпочтительно 1:0,02-0,15, более предпочтительно 1:0,03-0,10.
Согласно одному варианту реализации настоящего изобретения указанный способ получения наночастицы согласно настоящему изобретению включает дополнительный этап с) удаления органической среды и/или необязательно очистки; например, путем фильтрации или центрифугирования до получения осадка. Аналогичным образом, при необходимости, указанный способ получения наночастицы согласно настоящему изобретению может дополнительно включать дополнительный этап d) сушки образованных наночастиц, необязательно в присутствии защитного агента. Согласно конкретному варианту реализации настоящего изобретения указанный дополнительный этап d) выполняют путем распылительной сушки или сушки вымораживанием.
В связи с этим согласно другому аспекту в настоящем изобретении предложена наночастица, полученная указанным способом согласно настоящему изобретению.
Согласно другому аспекту в настоящем изобретении предложена наночастица в соответствии с настоящим изобретением или наночастица, полученная в соответствии со способом согласно настоящему изобретению, для применения в медицине.
Согласно другому аспекту в настоящем изобретении предложено применение наночастицы в соответствии с настоящим изобретением или применение наночастицы, полученной в соответствии со способом согласно настоящему изобретению, в изготовлении фармацевтической композиции.
Согласно другому аспекту в настоящем изобретении предложена фармацевтическая композиция, содержащая i) по меньшей мере одну наночастицу согласно настоящему изобретению или по меньшей мере одну наночастицу, полученную в соответствии со способом согласно настоящему изобретению, и ii) фармацевтически приемлемый носитель или наполнитель. Согласно конкретному варианту реализации настоящего изобретения указанный носитель или наполнитель содержит фармацевтическое вспомогательное вещество, приемлемое для введения пероральным путем.
Согласно другому аспекту в настоящем изобретении предложена указанная фармацевтическая композиция согласно настоящему изобретению для применения в медицине.
Согласно конкретному варианту реализации настоящего изобретения фармацевтическая композиция согласно настоящему изобретению содержит противоопухолевый агент. Следовательно, согласно другому аспекту в настоящем изобретении предложена указанная фармацевтическая композиция согласно настоящему изобретению, содержащая противоопухолевый агент, для применения в соответствии со способом предотвращения или лечения рака; или, в другом варианте, настоящее изобретение также относится к применению фармацевтической композиции согласно настоящему изобретению, содержащей противоопухолевый агент, в получении лекарственного средства для предотвращения или лечения рака.
Согласно более конкретному варианту реализации настоящего изобретения фармацевтическая композиция выбрана из группы, состоящей из:
фармацевтической композиции, содержащей:
a). сложноэфирный полимерный конъюгат PVM/MA с полиэтиленгликолем 2000 в количестве от 38 до 47%,
b). доцетаксел в количестве от 3% до 5%,
c). кальций в количестве от 0,1% до 0,2%, и
d). сахарид в количестве от 15 до 40%,
где все указанные содержания приведены по массе по отношению к общей массе композиции; фармацевтической композиции, содержащей:
a). сложноэфирный полимерный конъюгат PVM/MA с метоксиполиэтиленгликолем 2000 в количестве от 38% до 47%,
b). доцетаксел в количестве от 3% до 5%,
c). кальций в количестве от 0,1% до 0,2%, и
d). сахарид в количестве от 15 до 40%,
где все указанные содержания приведены по массе по отношению к общей массе композиции; и
фармацевтической композиции, содержащей:
a). сложноэфирный полимерный конъюгат PVM/MA с метоксиполиэтиленгликолем 2000 в количестве от 30 до 40%,
b). камптотецин в количестве от 0,08% до 1,5%,
c). кальций в количестве от 0,10% до 0,20%, и
d). сахарид в количестве от 15 до 40%,
где все указанные содержания приведены по массе по отношению к общей массе композиции.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
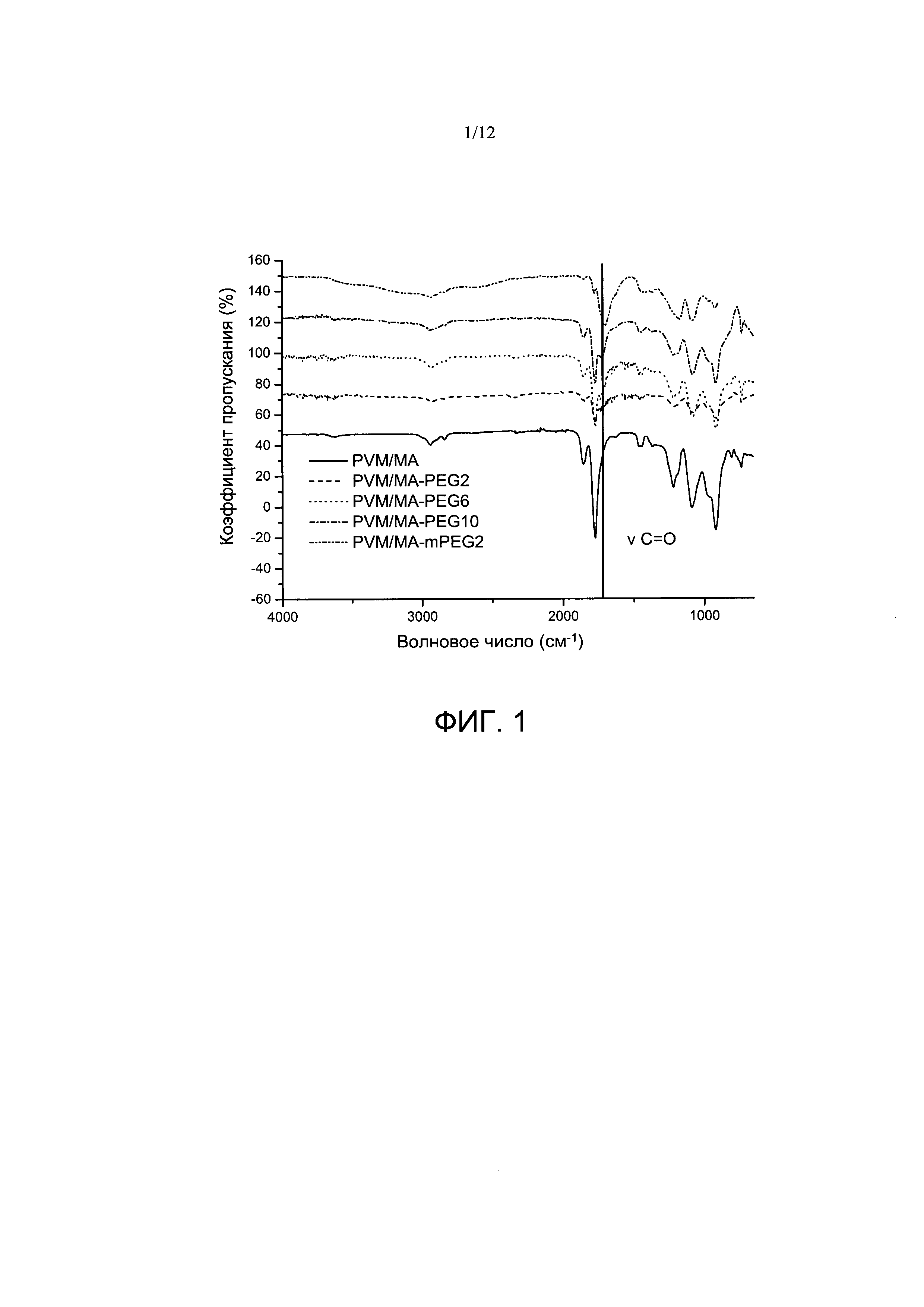
Фигура 1. Инфракрасные спектры PVM/MA и сложноэфирных полимерных конъюгатов PVM/MA с полиэтиленгликолем 2000 (РУМ/МА-ПЭГ2), полиэтиленгликолем 6000 (PVM/MA-ПЭГ6), полиэтиленгликолем 10000 (PVM/MA PEG10) и метоксиполиэтиленгликолем 2000 (РУМ/МА-мПЭГ2).
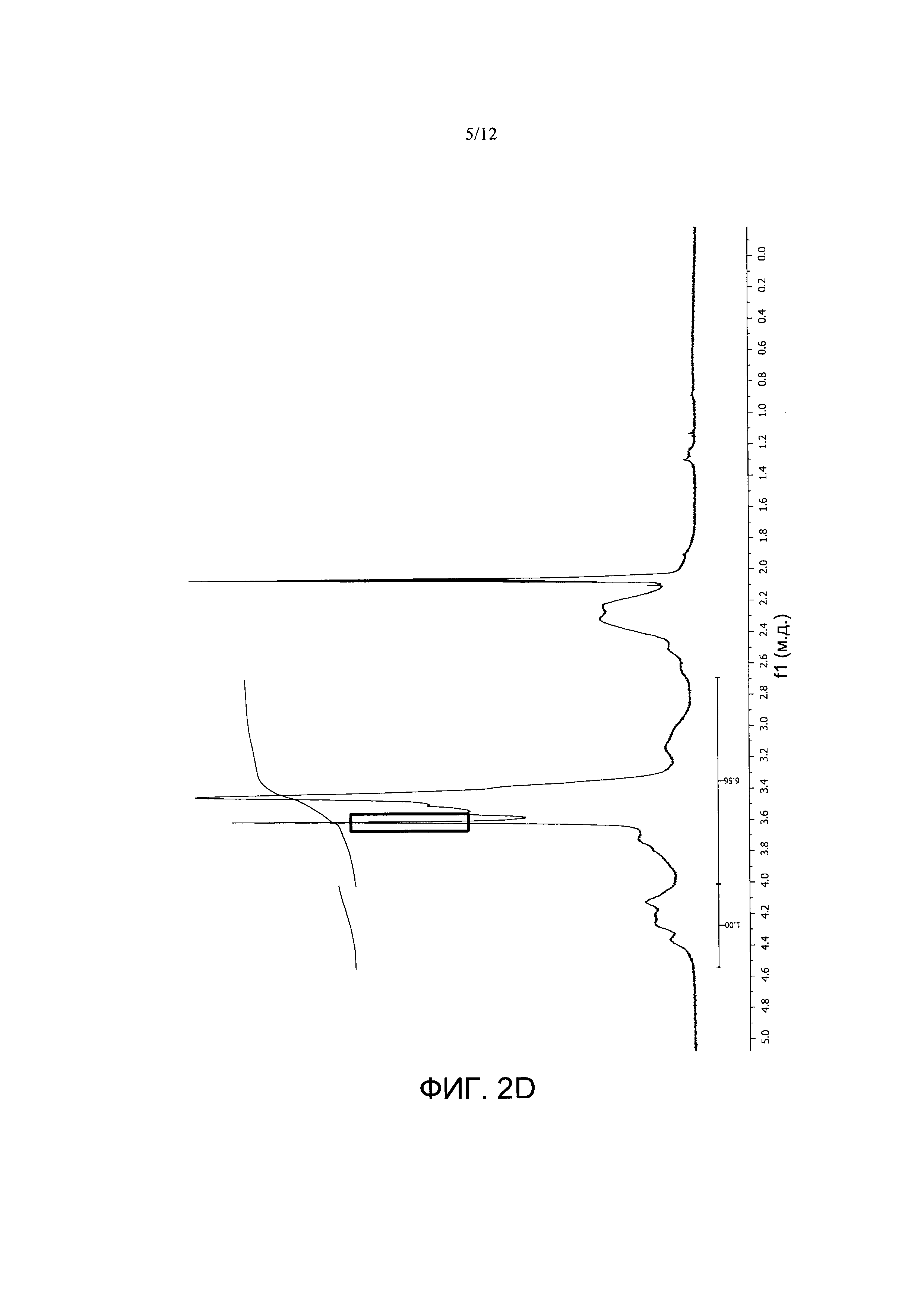
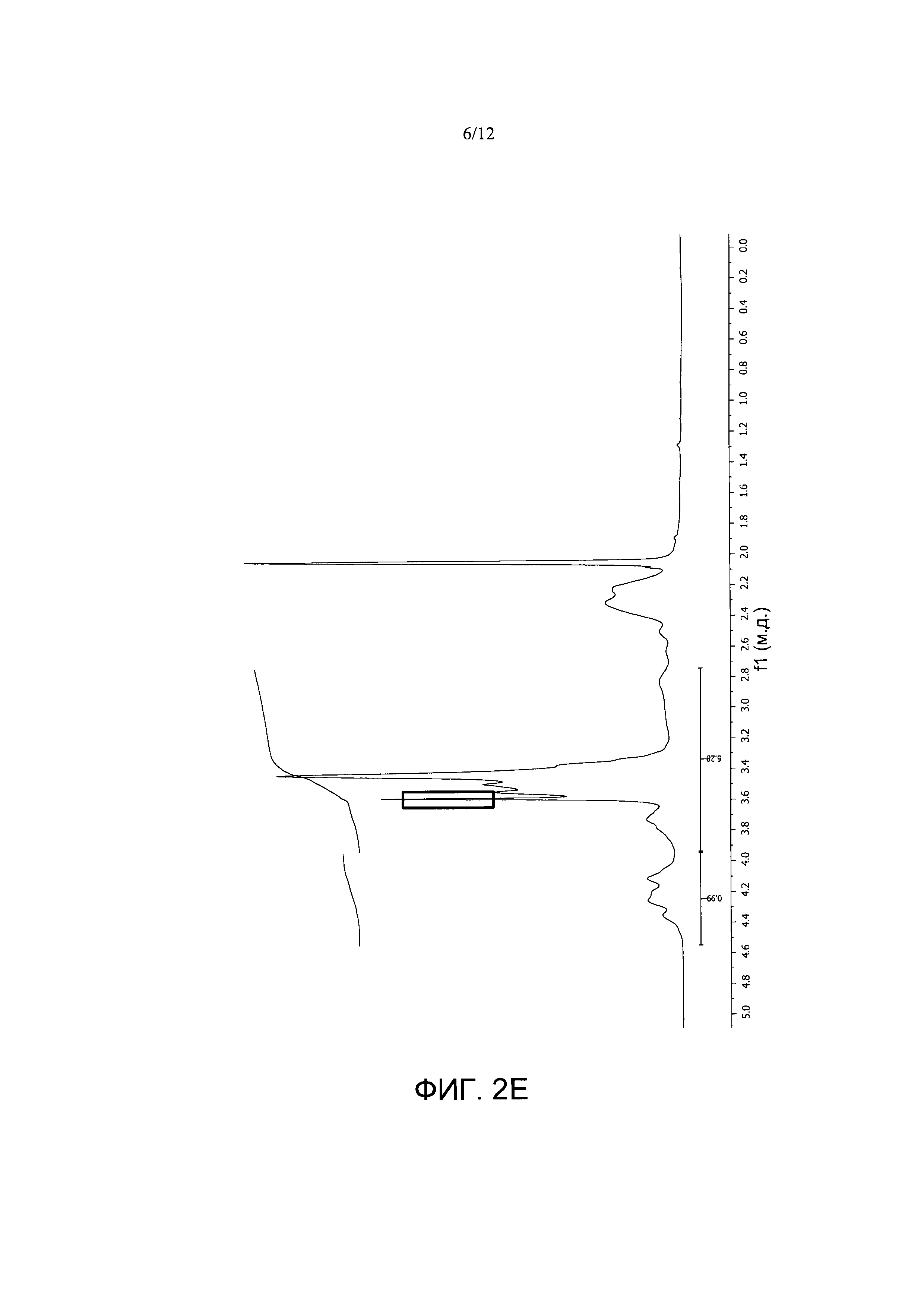
Фигура 2. Спектры1Н-ЯМР A) PVM/MA; и сложноэфирных полимерных конъюгатов PVM/MA с В) полиэтиленгликолем 2000 (РУМ/МА-ПЭГ2), С) полиэтиленгликолем 6000 (PVM/MA-ПЭГ6), D) полиэтиленгликолем 10000 (PVM/MA-ПЭГ10), Е) метоксиполиэтиленгликолем 2000 (РУМ/МА-мПЭГ2).
Фигуры 3. Результаты исследования составов наночастиц полиангидрида методом сканирующей электронной микроскопии (СЭМ). А) ПЭГ2-СРТ станд. (стандартные наночастицы PVM/MA, пегилированные с использованием ПЭГ2, инкапсулирующие камптотецин); В) ПЭГ6-СРТ станд. (стандартные наночастицы PVM/MA, пегилированные с использованием ПЭГ6, инкапсулирующие камптотецин); С) мПЭГ2-СРТ кон. (наночастицы из сложноэфирного полимерного конъюгата PVM/MA с мПЭГ2, инкапсулирующие камптотецин).
Фигура 4. Профиль высвобождения доцетаксела (DTX) из составов наночастиц DTX (стандартные наночастицы PVM/MA, инкапсулирующие доцетаксел), ПЭГ2-DТХ станд. (стандартные наночастицы PVM/MA, пегилированные с использованием ПЭГ2, инкапсулирующие доцетаксел), мПЭГ2-DТХ кон. (наночастицы из сложноэфирного полимерного конъюгата PVM/MA с мПЭГ2, инкапсулирующие доцетаксел) и ПЭГ2-DТХ (наночастицы из сложноэфирного полимерного конъюгата PVM/MA с ПЭГ2, инкапсулирующие доцетаксел) после инкубации в моделируемо желудочного сока (0-2 ч) и моделируемого кишечного сока (2-12 ч) при 37°С. Данные представлены как среднее значение ± стандартная ошибка среднего значения (n=3).
Фигура 5. Профиль зависимости концентрации доцетаксела в плазме крови от времени после внутривенного (в/в) введения 30 мг/кг дозы таксотера®. Данные представлены как среднее значение ± стандартная ошибка среднего значения, (n=3 на временную точку).
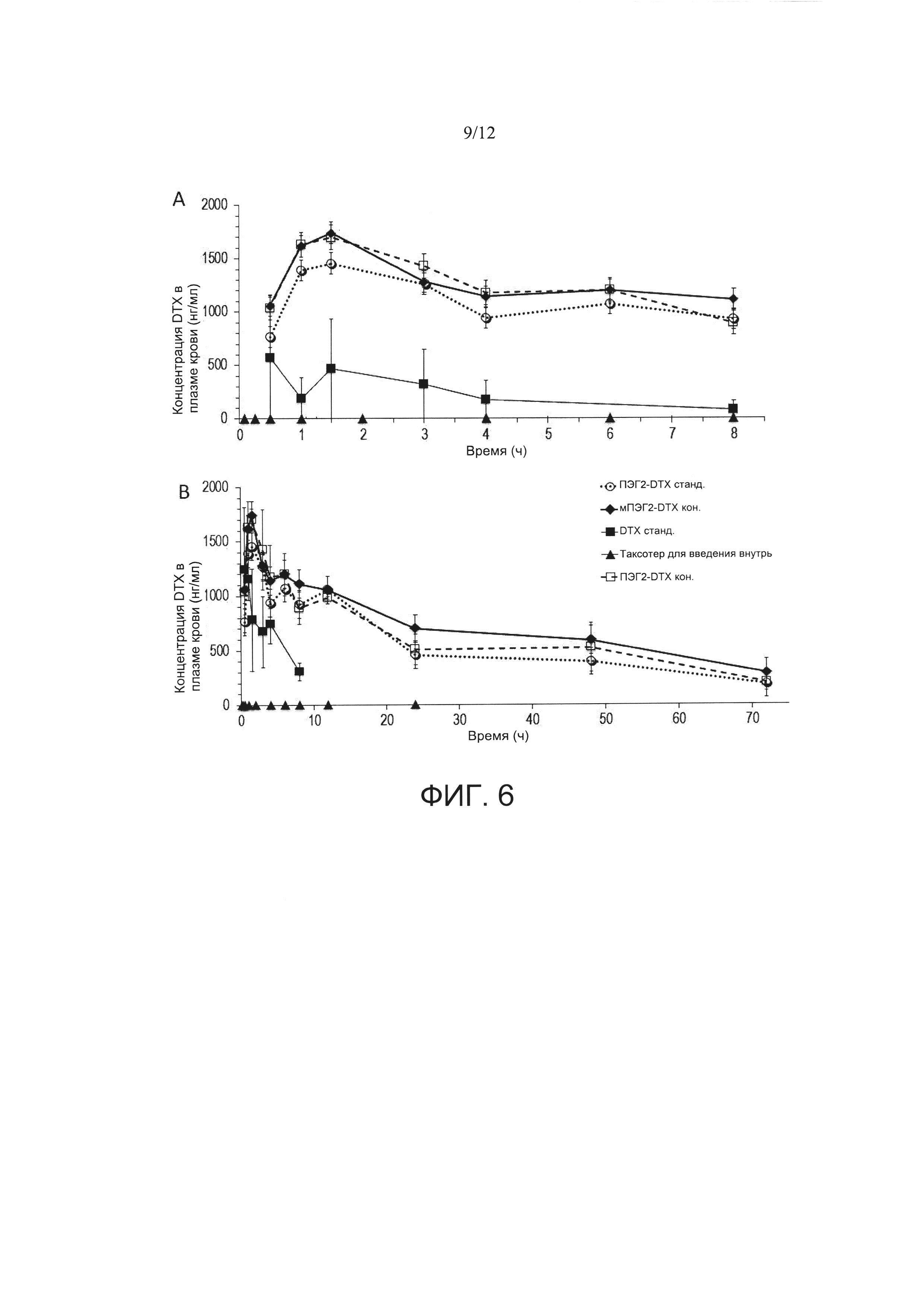
Фигура 6. Профиль зависимости концентрации доцетаксела в плазме крови от времени после введения однократной пероральной дозы 30 мг/кг. Животные получали перорально таксотер®, наночастицы DTX (стандартные наночастицы PVM/MA, инкапсулирующие доцетаксел), ПЭГ2-DТХ станд. (стандартные наночастицы PVM/MA, пегилированные с использованием ПЭГ2, инкапсулирующие доцетаксел), мПЭГ2-DТХ кон. (наночастицы из сложноэфирного полимерного конъюгата PVM/MA с мПЭГ2, инкапсулирующие доцетаксел) и ПЭГ2-DТХ кон. (наночастицы из сложноэфирного полимерного конъюгата PVM/MA с ПЭГ2, инкапсулирующие доцетаксел). А) в течение первых 8 ч, и В) в течение 72 часов. Данные представлены как среднее значение ± стандартная ошибка среднего значения (n=5).
Фигура 7. Профиль высвобождения камптотецина (СРТ) из составов ПЭГ2-СРТ станд. (стандартные наночастицы PVM/MA, пегилированные с использованием ПЭГ2, инкапсулирующие камптотецин), ПЭГ6-СРТ станд. (стандартные наночастицы PVM/MA, пегилированные с использованием ПЭГ6, инкапсулирующие камптотецин) и мПЭГ2-СРТ кон. (наночастицы из сложноэфирного полимерного конъюгата PVM/MA с мПЭГ2, инкапсулирующие камптотецин) после инкубации в моделируемом желудочном соке (0-2 ч) и моделируемой кишечной жидкости (2-14 ч) в условиях достаточного разбавления, 37°С, 60 об./мин. Данные представлены как среднее значение ± стандартная ошибка среднего значения (n=3).
Фигура 8. Профиль зависимости концентрации камптотецина в плазме крови от времени после внутривенного (в/в) введения 1 мг/кг дозы суспензии. Данные представлены как среднее значение ± стандартная ошибка среднего значения (n=3 на временную точку).
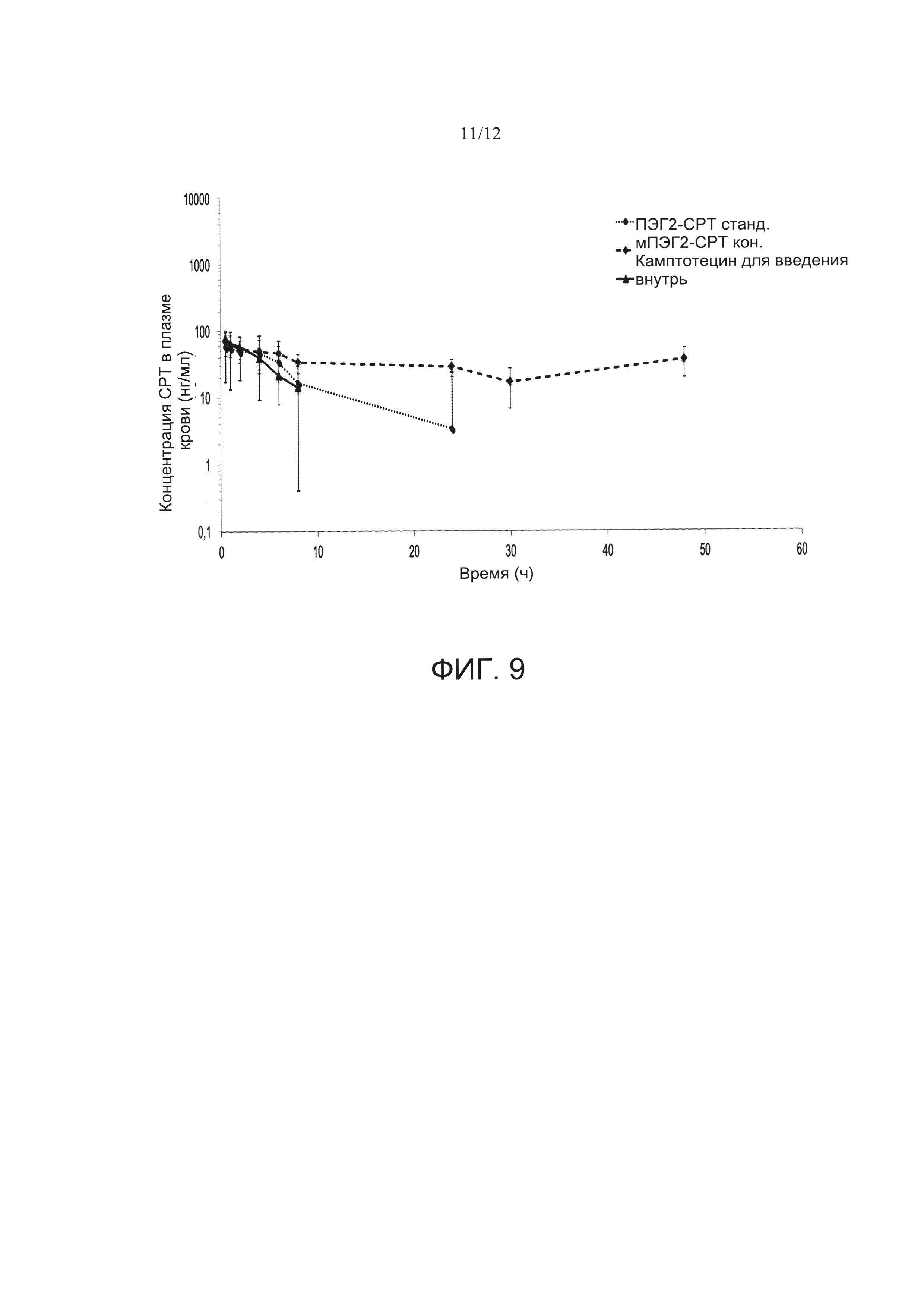
Фигура 9. Профиль зависимости концентрации камптотецина в плазме крови от времени у самцов крыс Wistar после введения однократной дозы (1 мг/кг) пероральной суспензии ПЭГ2-СТР станд. (стандартные наночастицы PVM/MA, пегилированные с использованием ПЭГ2, инкапсулирующие камптотецин) и мПЭГ2-СРТ кон. (наночастицы из сложноэфирного полимерного конъюгата PVM/MA с мПЭГ2, инкапсулирующие камптотецин). Данные представлены как среднее значение ± стандартная ошибка среднего значения (n=6).
Фигура 10. Визуализация наночастиц, флуоресцентномеченых люмогеном красным, методом флуоресцентной микроскопии в продольных срезах проксимальной подвздошной кишки крыс через два часа после перорального введения наночастиц в виде однократной дозы. (А, В) Стандартные наночастицы PVM/MA, инкапсулирующие люмоген® красный; (С, D) наночастицы, содержащие матрикс из сложноэфирного полимерного конъюгата PVM/MA с мПЭГ2, инкапсулирующего люмоген® красный (конъюгат мПЭГ2-LUM).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложены новые системы введения лекарственных препаратов (новые полимерные конъюгаты и наночастицы, изготовленные из них), которые могут инкапсулировать значительные количества биологически активных соединений, являются стабильными в ЖКТ и увеличивают биодоступность перорально вводимых лекарственных препаратов.
Неожиданно было обнаружено, что новые сложноэфирные полимерные конъюгаты PVM/MA с полиэтиленгликолем (или его производным, содержащим концевую реакционноспособную гидроксильную группу) могут быть синтезированы с помощью простой реакции, и продукты реакции пригодны для легкого получения наночастиц с улучшенными характеристиками для введения биологически активных соединений. В частности, было обнаружено, что наночастицы, образованные указанными новыми сложноэфирными полимерными конъюгатами PVM/MA с полиэтиленгликолем (или его производным, содержащим концевую реакционноспособную гидроксильную группу), способны инкапсулировать значительные количества биологически активных соединений и увеличивать их биодоступность при пероральном введении.
Определения
Для того чтобы облегчить понимание настоящего изобретения значение некоторых терминов и выражений, используемых применительно к настоящему изобретению, изложено ниже.
В настоящей заявке термин «поли(метилвиниловый эфира-малеинового ангидрида)» (PVM/MA) относится к сополимеру простого метилвинилового эфира и малеинового ангидрида (выпущенному в серийное производство International Specialty Products, ISP, под торговой маркой гантрез® AN). В этой связи выражения «PVM/МА», полиангидрид или гантрез® являются синонимами и используются в данном описании взаимозаменяемо. Сополимер гантрез® содержит чередующиеся блоки простого метилвинилового эфира и малеинового ангидрида и имеет формулу:

Фундаментальный характер этой полимеризации требует, чтобы блок малеинового ангидрида находился рядом с блоком метилвинилового эфира и наоборот, что приводит к получению истинного чередующегося сополимера. Ангидридные группы в структуре гантреза® обеспечивают химическое взаимодействие указанного полимера с гидроксил-содержащими молекулами по механизму реакции нуклеофильного замещения.
Сополимер гантрез® поставляется в виде нерастворимого в воде порошка белого цвета и широко применяется для фармацевтических и медицинских целей, в качестве клеев для зубных протезов, загустителей и суспендирующих агентов и в качестве вспомогательных веществ для получения трансдермальных пластырей. Оральная токсичность этого сополимера является довольно низкой (ЛД50 у морских свинок составляет около 8-9 г/кг), в этой связи он также был использован в качестве материала для получения наночастиц-носителей. Согласно конкретному варианту реализации настоящего изобретения сополимер PVM/MA имеет молекулярную массу в диапазоне от 80 до 2500 кДа, предпочтительно от 85 до 2000 кДа, более предпочтительно от 90 до 220 кДа.
В настоящей заявке термин «сложный эфир» относится к сложному эфиру (в том числе PVM/MA), который содержит по меньшей мере одну сложноэфирную группу (то есть по меньшей мере один ангидридный блок PVM/MA этерифицирован молекулой, содержащей концевые гидроксильные группы). В соответствии с настоящим изобретением по меньшей мере один ангидридный блок PVM/MA этерифицирован молекулой, содержащей концевые гидроксильные группы, выбранной из полиэтиленгликоля и производного полиэтиленгликоля, содержащего концевую реакционноспособную гидроксильную группу. Иллюстративные, неограничивающие примеры сложных эфиров PVM/MA включают те, в которых степень замещения составляет по меньшей мере 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95%.
В настоящей заявке термин «степень этерификации» (DE) или «степень замещения» (DS) сложноэфирного полимерного конъюгата согласно настоящему изобретению определяется как процент блоков ангидрида, которые этерифицированы молекулой, содержащей концевые гидроксильные группы, в соответствии с настоящим изобретением, т.е. процент образованных сложноэфирных связей. Согласно конкретному варианту реализации настоящего изобретения степень этерификации может быть измерена стандартными способами, известными специалисту в данной области техники, и которые описаны в качестве примера в экспериментальной части, прилагаемой к примерам, описанным ниже.
В настоящей заявке термин «молекула, содержащая концевые гидроксильные группы» представляет собой молекулу, содержащую по меньшей мере одну первичную гидроксильную группу (-ОН). В соответствии с настоящим изобретением указанная молекула, содержащая концевые гидроксильные группы, выбрана из полиэтиленгликоля и производного полиэтиленгликоля, содержащего концевую реакционноспособную гидроксильную группу, определенную ниже.
В настоящем описании под термином «полиэтиленгликоль» (ПЭГ) понимают любой гидрофильный полимер, растворимый в воде, содержащий сложноэфирные группы, связанные двумя атомами углерода, необязательно разветвленные этиленовые группы. Следовательно, указанное определение включает разветвленные или неразветвленные полиэтиленгликоли.
ПЭГ также известен как полиэтиленоксид (ПЭО) или полиоксиэтилен (ПОЭ) (три названия являются химическими синонимами и относятся к олигомеру или полимеру этиленоксида). Термин также включает производные одной из концевых гидроксильных групп, которые могут быть модифицированы (один из двух концов). Следовательно, в соответствии с настоящим изобретением могут быть использованы производные полиэтиленгликоля, содержащие одну первичную гидроксильную группу (-ОН).
Полиэтиленгликоли представляют собой водорастворимые полимеры, которые были одобрены (FDA) для перорального, парентерального и местного введения лекарственных препаратов. Полиэтиленгликоли получают путем полимеризации этиленоксида (ЭО) в присутствии воды, моноэтиленгликоля или диэтиленгликоля в качестве инициаторов реакции в щелочной среде (1,2-Epoxide Polymers: Ethylene Oxide Polymers and Copolymers" в Encyclopedia of Polymer Science and Engineering; Mark, H.F. (Ed.), John Wiley and Sons Inc., 1986, pp. 225-273). При достижении желаемой молекулярной массы (обычно контролируемой посредством измерений вязкости в ходе процесса получения), реакцию полимеризации заканчивают путем нейтрализации катализатора кислотой (молочной кислотой, уксусной кислотой или т.п.). В результате реакции образуется линейный полимер, имеющий очень простую структуру:
Н-(O-СН2-СН2)n-ОН
где (n) - количество мономеров или блоков ЭО. Термин полиэтиленгликоль обычно используется для обозначения значительного влияния концевых гидроксильных групп на физико-химические свойства указанных молекул. Термин ПЭГ обычно используется в сочетании с численным значением. В фармацевтической промышленности число указывает на среднюю молекулярную массу, тогда как в косметической промышленности число, сопровождающее буквы ПЭГ, относится к полимеризованным блокам ЭО, образующим молекулу (Handbook of Pharmaceutical Excipients, Rowev R.C., Sheskey P.J., Weller P.J. (Eds.), 4 изд., Pharmaceutical Press and American Pharmaceutical Association, London, UK, 2003). ПЭГ включены в различные фармакопеи, хотя номенклатура отличается (International Harmonisation: Polyethylene glycol (PEG): Pharmeuropa 1999, 11, 612-614). В соответствии с Handbook of Pharmaceutical Excipients (Fourth Edition), 2003 под ред. R.C. Rowe, P.J. Sheskey and P.J. Weller, опубликованном Pharmaceutical Press (London, UK), и рекомендациями Американской фармацевтической ассоциации (Вашингтон, США) полиоксиэтиленгликоли также упоминаются как полиэтиленгликоли, макроголы или ПЭГ. В Британской и Европейской фармакопеях используются термины полиэтиленгликоли и макроголы, в то время как в Фармакопее США (USP) используют термин полиэтиленгликоли.
Согласно одному варианту реализации настоящего изобретения используемые полиэтиленгликоли предпочтительно имеют молекулярную массу, составляющую от 400 до 35000 Да. Согласно одному предпочтительному варианту реализации настоящего изобретения используемый полиэтиленгликоль имеет молекулярную массу, которая равна или превышает 400, более предпочтительно равна или превышает 1000; значения, составляющие от 1000 до 20000, являются особенно предпочтительными, более предпочтительно от 2000 до 10000 Да. Иллюстративные неограничивающие примеры ПЭГ, которые могут быть использованы в настоящем изобретении, включают полиэтиленгликоль 1000, полиэтиленгликоль 2000, полиэтиленгликоль 6000 или полиэтиленгликоль 10000 (ПЭГ1, ПЭГ2, ПЭГ6 или ПЭГ10, соответственно).
ПЭГ с молекулярной массой менее 400 являются нелетучими жидкостями при комнатной температуре. ПЭГ 600 имеет температуру плавления от 17 до 22°С, тогда как ПЭГ со средними молекулярными массами от 800 до 2000 являются пастообразными материалами с низкими температурами плавления. ПЭГ с молекулярной массой выше 3000 Да являются твердыми, и ПЭГ с молекулярной массой вплоть до 35000 являются коммерчески доступными. С другой стороны, несмотря на то, что температура плавления ПЭГ увеличивается при увеличении молекулярной массы, температура кипения увеличивается до максимального значения 60°С. Аналогичным образом, при увеличении молекулярной массы растворимость ПЭГ в воде уменьшается. В любом случае, для ПЭГ 35000 в воде может быть растворено количество близкое к 50% масс/об.
С токсикологической точки зрения полиэтиленгликоли считаются достаточно нетоксичными и неиммуногенными (Hermansky S.J et al, Food Chem Toxic, 1995, 33, 139-140; Final Report on the Safety Assessment of PEGs: J.А. С.Т., 1993, 12, 429-457; Polyethylene glycol, 21 CFR 172.820, FDA). Максимальная суточная доза, определенная ВОЗ, составляет 10 мг/кг («Полиэтиленгликоли», двадцать третий доклад Объединенного экспертного комитета Организации ООН по продовольствию и сельскому хозяйству/ВОЗ по пищевым добавкам, Всемирная организация здравоохранения, Женева, Серия технических докладов 1980, 648, 17-18),
Производные полиэтиленгликоля обладают полезными признаками, которые аналогичны таковым для стандартных ПЭГ, такими как растворимость в воде, физиологическая неактивность, низкая токсичность и устойчивость при различных условиях. Подходящие производные включают очень разные продукты и характеризуются функциональной группой, замещающей первичный гидроксил. В соответствии с настоящим изобретением могут быть использованы производные полиэтиленгликоля, содержащие концевую реакционноспособную гидроксильную группу.
Согласно конкретному варианту реализации настоящего изобретения указанное производное полиэтиленгликоля, содержащее концевую реакционноспособную гидроксильную группу, представляет собой простой полиоксиэтиленалкиловый эфир. Соединения простых полиоксиэтиленалкиловых эфиров имеют общую формулу Н(2m+1)Сm(O-CH2-СН2)nОН. Они будут называться CmEn, где m указывает число атомов углерода в алкильной цепи, и n означает число этиленоксидных блоков в гидрофильном фрагменте. Иллюстративные неограничивающие примеры включают: простые метиловые эфиры полиэтиленгликоля, также известные как метоксиполиэтиленгликоли (мПЭГ); простые этиловые эфиры полиэтиленгликоля; простые пропиловые эфиры полиэтиленгликоля; и простые бутиловые эфиры полиэтиленгликоля.
Химические структуры некоторых простых алкиловых эфиров полиоксиэтилена, соответствующих вышеупомянутым группам, приведены ниже в качестве иллюстрации:
a) Н3С(O-СН2-СН2)nОН
b) Н5С2(O-СН2-СН2)nОН
c) Н7С3(O-СН2-СН2)nОН
d) Н9С4(O-СН2-СН2)nОН.
Неограничивающие примеры простых алкиловых эфиров полиоксиэтилена, которые могут быть использованы в настоящем изобретении, включают простой метиловый эфир полиэтиленгликоля 1000 или метоксиполиэтиленгликоль 1000 (мПЭГ1); простой метиловый эфир полиэтиленгликоля 2000 или метоксиполиэтиленгликоль 2000 (мПЭГ2); простой метиловый эфир полиэтиленгликоля 6000 или метоксиполиэтиленгликоль 6000 (мПЭГ6); простой метиловый эфир полиэтиленгликоля или 10000 метоксиполиэтиленгликоль 10000 (мПЭГ10).
Среди производных полиэтиленгликоля, содержащих концевую реакционноспособную гидроксильную группу, также можно выделить следующие соединения, которые могут быть использованы в настоящем изобретении:
- полиэтиленгликольметакрилат
- полиэтиленгликольакрилат
- полиэтиленгликольмонолаурат
- полиэтиленгликольмоноолеат
- полиэтиленгликоль(12)тридециловый эфир
- полиэтиленгликольтетрагидрофурфуриловый эфир.
Выбор полиэтиленгликолей и производных позволяет модулировать характеристики создаваемой системы, и использование их смесей добавляет еще один фактор изменчивости. С практической точки зрения важно адаптировать и выбрать наиболее подходящую систему для каждой активной молекулы и для каждого способа введения.
В настоящей заявке термин «молекулярная масса» определяется как средняя молекулярная масса молекулы. В отличие от малых молекул молекулярная масса полимера не является одним уникальным значением. Конкретный полимер, скорее всего, будет иметь распределение молекулярных масс в зависимости, например, от способа получения полимера. В этой связи в настоящей заявке термин молекулярная масса полимеров относится к распределению молекулярной массы или к средней молекулярной массе. Однако существует много способов расчета средней молекулярной массы. В соответствии с настоящим изобретением среднюю молекулярную массу преформированных полиэфиров согласно настоящему изобретению определяют методом1Н-ЯМР, и среднюю молекулярную массу коммерческого PVM/MA определяют с помощью эксклюзионной хроматографии с детектированием рассеивания лазерного излучения с кратными углами (SEC-MALLS), как описано, с целью иллюстрации, в экспериментальной части, прилагаемой к примерам, описанным ниже.
В настоящей заявке термин «наночастица» относится к сферическим или имеющим аналогичную форму коллоидным системам (нанокапсуле или наносфере) с размером менее 1 микрометра (мкм), предпочтительно с размером в диапазоне от 10 до 900 нанометров (нм).
В настоящей заявке термин «биологически активное соединение» (БАС) относится к любой малой молекуле (например, лекарственному препарату или активному ингредиенту лекарственного средства) или его производному, которое вводят субъекту, предпочтительно человеку, с профилактическими или терапевтическими целями; т.е. к любому веществу или химическому соединению с молекулярной массой ниже 900 Да, которое может быть использовано для лечения, излечения, предотвращения или диагностики заболевания или для улучшения физического и психического самочувствия человека и животных. В настоящей заявке термин «производное», применительно к биологически активному соединению, включает пролекарственные формы и аналоги указанного биологически активного соединения.
В настоящей заявке термины «противоопухолевый агент», «противораковый агент» или «противобластомный агент» (используемые в настоящем описании взаимозаменяемо), обычно относятся к веществам, которые ингибируют или подавляют рост и пролиферацию раковых клеток. Противоопухолевые агенты также могут включать соединения, которые разрушают раковые клетки или нарушают деление клеток, соединения, которые блокируют определенные гормоны, участвующие в патофизиологии рака, соединения, которые ингибируют или предотвращают размножение новых кровеносных сосудов (например, ингибиторы ангиогенеза), агенты, которые повреждают ДНК (например, алкилирующие агенты, такие как цисплатин, карбоплатин и оксалоплатин; антиметаболиты и ингибиторы топоизомеразы), а также соединения с противораковыми свойствами (например, таксаны, алкалоиды барвинка и растительные алкалоиды). Термин «противоопухолевый агент» также включает лучевую терапию. Противоопухолевый агент также может включать агент, специфичный в отношении нерегулируемых белков раковых клеток, такой как ингибитор рецепторных тирозинкиназ.
В настоящей заявке термин «двухвалентный металл» включает любой металл с валентностью 2, например, щелочноземельный металл, например, кальций, магний, цинк и т.д., или, если элемент имеет несколько валентностей, одна из них равна 2, например, железо и т.д., при условии, что указанный элемент является фармацевтически приемлемым.
В настоящей заявке термин «средний размер» относится к среднему диаметру популяции наночастиц, которая совместно перемещается в водной среде. Средний размер указанных систем может быть измерен стандартными способами, известными специалистам в данной области техники, и которые описаны, в качестве иллюстрации, в экспериментальной части, прилагаемой к примерам, описанным ниже. Средний размер частиц главным образом зависит от количества и молекулярной массы PVM/MA, характера и количества молекул с концевыми гидроксильными группами согласно настоящему изобретению, характера и количества биологически активной молекулы, присутствующей в наночастицах согласно настоящему изобретению (как правило, чем больше количество или молекулярная масса указанных компонентов, тем больше средний размер наночастицы), и некоторых параметров способа получения указанных наночастиц. Наночастицы согласно настоящему изобретению характеризуются тем, что средний размер частиц составляет менее 1 мкм, как правило, находится в диапазоне от 1 до 999 нм, предпочтительно от 10 до 900 нм, более предпочтительно от 100 до 500 нм, еще более предпочтительно от 150 до 400 нм. Согласно конкретному варианту реализации настоящего изобретения наночастицы согласно настоящему изобретению имеют средний размер частиц приблизительно 250 нм.
В настоящей заявке термин «фармацевтически приемлемый» означает, что соединение или комбинация соединений достаточно совместима с другими ингредиентами состава и не является вредной для субъекта вплоть до уровней, приемлемых по промышленным стандартам.
В настоящей заявке термин «носитель» относится к разбавителю, с которым вводят активный ингредиент или лекарственный препарат. Примеры фармацевтически приемлемых носителей известны в данной области техники и включают фосфатно-солевые буферные растворы, воду, эмульсии, такие как эмульсии масла/вода, различные типы смачивающих агентов, стерильные растворы и т.д. Подходящие фармацевтические носители описаны в "Remington's Pharmaceutical Sciences" by E.W. Martin, 1995. Предпочтительно носители согласно настоящему изобретению одобрены регулирующим органом федерального правительства или правительства штата или перечислены в Фармакопее США или другой общепризнанной фармакопее для применения у животных и более конкретно у человека.
В настоящей заявке термин «предотвращающий» относится к избеганию развития, подавлению или, в другом варианте, задержке начала или рецидива заболевания, расстройства или состояния, в отношении которого применяют указанный термин, или одного или нескольких симптомов, связанных с заболеванием, расстройством или состоянием. Термин «предотвращение» относится к акту предотвращения, как определено непосредственно выше для термина «предотвращающий».
В настоящей заявке термин «лечащий» относится к обращению, облегчению или ингибированию прогрессирования заболевания или состояния, в отношении которого применяют указанный термин, или одного или более симптомов указанных расстройств или состояния. Термин «лечение» относится к акту лечения, как определено непосредственно выше для термина «лечащий».
В настоящей заявке термин «субъект» означает животных, в частности млекопитающих, таких как собаки, кошки, коровы, лошади, овцы, гуси, и человека. Особенно предпочтительными субъектами являются люди обоих полов.
Сложноэфирные полимерные конъюгаты простого поли(метилвинилового эфира-малеинового ангидрида)
В настоящем изобретении предложены новые сложноэфирные полимерные конъюгаты простого поли(метилвинилового эфира-малеинового ангидрида) (PVM/MA) с молекулами, содержащими концевые гидроксильные группы. В настоящей заявке термины «сложный эфир», «поли(метилвинилового эфира-малеинового ангидрида)» и «молекула с концевыми гидроксильными группами» были определены ранее и включены в настоящее описание посредством ссылки. Следовательно, согласно настоящему изобретению по меньшей мере один ангидридный блок полимера PVM/MA этерифицирован молекулой, содержащей концевые гидроксильные группы, выбранной из: полиэтиленгликоля и производного полиэтиленгликоля, содержащего концевую реакционноспособную гидроксильную группу, в дальнейшем упоминаемой «молекула, содержащая концевые гидроксильные группы, согласно настоящему изобретению».
Следовательно, согласно первому аспекту в настоящем изобретении предложен сложноэфирный полимерный конъюгат PVM/MA с молекулой, содержащей концевые гидроксильные группы, который характеризуется тем, что указанная молекула, содержащая концевые гидроксильные группы, выбрана из полиэтиленгликоля и его производного, содержащего концевую реакционноспособную гидроксильную группу, далее «сложноэфирный полимерный конъюгат согласно настоящему изобретению».
Согласно конкретному варианту реализации настоящего изобретения по меньшей мере 5%, по меньшей мере 10%, по меньшей мере 15%, по меньшей мере 20%, по меньшей мере 25%, по меньшей мере 30%, по меньшей мере 35%, по меньшей мере 40%, по меньшей мере 45%, по меньшей мере 50%, по меньшей мере 55%, по меньшей мере 60%, по меньшей мере 65%, по меньшей мере 70%, по меньшей мере 75%, по меньшей мере 80%, по меньшей мере 85%, по меньшей мере 90% или по меньшей мере 95% ангидридных блоков полимера PVM/MA этерифицированы молекулой, содержащей концевые гидроксильные группы, согласно настоящему изобретению. Например, по меньшей мере 5% степень этерификации обеспечивает хорошие результаты.
Согласно одному варианту реализации настоящего изобретения молекула, содержащая концевые гидроксильные группы, представляет собой полиэтиленгликоль, который не является разветвленным и не содержит замещенных гидроксильных групп. Согласно указанному варианту реализации настоящего изобретения используемые полиэтиленгликоли предпочтительно имеют молекулярную массу в диапазоне от 400 до 35000 Да. Согласно одному предпочтительному варианту реализации настоящего изобретения используемый полиэтиленгликоль имеет молекулярную массу, которая равна или больше 400, более предпочтительно равна или больше 1000; значения, находящиеся в диапазоне от 1000 до 20000, являются особенно предпочтительными, более предпочтительно от 2000 до 10000 Да. Согласно более предпочтительному варианту реализации настоящего изобретения молекула, содержащая концевые гидроксильные группы, представляет собой полиэтиленгликоль, выбранный из группы, состоящей из полиэтиленгликоля 1000 (ПЭГ1), полиэтиленгликоля 2000 (ПЭГ2), полиэтиленгликоля 6000 (ПЭГ6) и полиэтиленгликоля 10000 (ПЭГ10).
В другом варианте молекула, содержащая концевые гидроксильные группы, согласно настоящему изобретению представляет собой полиэтиленгликоль с одной замещенной гидроксильной группой. Согласно указанному варианту реализации настоящего изобретения молекула, содержащая концевые гидроксильные группы, представляет собой производное полиэтиленгликоля, содержащее одну концевую реакционноспособную гидроксильную группу, предпочтительно представляет собой простой полиоксиэтиленалкиловый эфир. Согласно более конкретному варианту реализации настоящего изобретения простой полиоксиэтиленалкиловый эфир представляет собой простой метиловый эфир полиэтиленгликоля (мПЭГ). Согласно еще более предпочтительному варианту реализации настоящего изобретения простой метиловый эфир полиэтиленгликоля выбран из группы, состоящей из метоксиполиэтиленгликоля 1000 (мПЭГ1), метоксиполиэтиленгликоля 2000 (мПЭГ2), метоксиполиэтиленгликоля 6000 (мПЭГ6) и метоксиполиэтиленгликоля 10000 (мПЭГ10).
Средняя молекулярная масса сложноэфирного полимерного конъюгата согласно настоящему изобретению может изменяться в широких пределах; однако согласно конкретному варианту реализации настоящего изобретения сложноэфирный полимерный конъюгат согласно настоящему изобретению имеет среднюю молекулярную массу в диапазоне от 85 до 3000 кДа, предпочтительно от 90 до 2500 кДа, более предпочтительно от 95 до 250 кДа.
Способ получения сложноэфирного полимерного конъюгата согласно настоящему изобретению
Согласно второму аспекту настоящее изобретение относится к способу получения сложноэфирного полимерного конъюгата PVM/MA с молекулой, содержащей концевые гидроксильные группы, который характеризуется тем, что молекула, содержащая концевые гидроксильные группы, выбрана из полиэтиленгликоля и его производного, содержащего концевую реакционноспособную гидроксильную группу (т.е. сложноэфирного полимерного конъюгата согласно настоящему изобретению), именуемого в дальнейшем как «способ [1] согласно настоящему изобретению», который включает следующие этапы:
a). взаимодействие PVM/MA с молекулой, содержащей концевые гидроксильные группы, согласно настоящему изобретению в органическом растворителе, и
b). удаление органического растворителя.
Этап а) «способа [1] согласно настоящему изобретению» включает растворение и инкубацию PVM/MA и молекулы, содержащей концевые гидроксильные группы, согласно настоящему изобретению в органическом растворителе, таком как, например, ацетон, для протекания реакции. Инкубацию смеси предпочтительно осуществляют при перемешивании при температуре в диапазоне от 15°С до 80°С, более предпочтительно в диапазоне от 45°С до 65°С, наиболее предпочтительно при 55°С. Время инкубации может варьироваться от 1 до 24 ч. В конкретном предпочтительном варианте реализации инкубацию проводят в течение 2-4 ч. Например, инкубация смеси при 55°С в течение 3 ч обеспечивает хорошие результаты. В другом варианте, взаимодействие PVM/MA и молекулы, содержащей концевые гидроксильные группы, можно осуществлять при перемешивании при комнатной температуре (КТ) в течение более длительных периодов времени (24-48 ч).
Органический растворитель удаляют на этапе b) любым подходящим способом, таким как выпаривание при пониженном давлении, выпаривание при комнатной температуре или центрифугирование под вакуумом. Согласно конкретному варианту реализации настоящего изобретения этап b) включает удаление органического растворителя путем выпаривания при пониженном давлении.
Согласно одному предпочтительному варианту реализации настоящего изобретения способ [1] согласно настоящему изобретению включает дополнительный этап с) очистки сложноэфирного полимерного конъюгата согласно настоящему изобретению. Согласно одному конкретному варианту реализации настоящего изобретения дополнительный этап
c) включает несколько промывок с применением жидкости, в которой полимер нерастворим (но не молекула, содержащая концевые гидроксильные группы, не вступившая в реакцию); и фильтрацию под вакуумом до тех пор, пока следовые количества не вступившей в реакцию молекулы, содержащей концевые гидроксильные группы, не перестанут детектироваться в жидкости (например, путем измерения с помощью тонкослойной хроматографии, ТСХ). Согласно предпочтительному варианту реализации настоящего изобретения промывочная жидкость представляет собой смесь метанола/дихлорметана. Например, смесь CH2Cl2/CH3OH (9:1), используемая в качестве подвижной фазы, и йод для проявления ТСХ, обеспечивают хорошие результаты.
Характерные особенности молекулы, содержащей концевые гидроксильные группы, согласно настоящему изобретению ранее были упомянуты в разделе «Определения», а также применительно к сложноэфирному полимерному конъюгату согласно настоящему изобретению, и включены в данное описание посредством ссылки. В соответствии с вышеупомянутым в конкретном варианте реализации способом [1] согласно настоящему изобретению молекула, содержащая концевые гидроксильные группы, вступающая в реакцию с PVM/MA на этапе а), представляет собой полиэтиленгликоль; предпочтительно представляет собой полиэтиленгликоль, выбранный из группы, состоящей из полиэтиленгликоля 1000 (ПЭГ1), полиэтиленгликоля 2000 (ПЭГ2), полиэтиленгликоля 6000 (ПЭГ6) и полиэтиленгликоля 10000 (ПЭГ10). Согласно другому конкретному варианту реализации способа [1] согласно настоящему изобретению молекула, содержащая концевые гидроксильные группы, вступающая в реакцию с PVM/MA на этапе а), представляет собой производное полиэтиленгликоля, содержащее концевую реакционноспособную гидроксильную группу, предпочтительно простой алкиловый эфир полиоксиэтилена; более предпочтительно простой метиловый эфир полиэтиленгликоля (мПЭГ); наиболее предпочтительно простой метиловый эфир полиэтиленгликоля, выбранный из группы, состоящей из метоксиполиэтиленгликоля 1000 (мПЭГ1), метоксиполиэтиленгликоля 2000 (мПЭГ2), метоксиполиэтиленгликоля 6000 (мПЭГ6) и метоксиполиэтиленгликоля 10000 (мПЭГ10).
Массовое соотношение между PVM/MA и молекулой, содержащей концевые гидроксильные группы, согласно настоящему изобретению в растворе на этапе а) способа [1] согласно настоящему изобретению может изменяться в широких пределах; однако согласно конкретному варианту реализации настоящего изобретения массовое соотношение PVM/MA : мoлeкyлы, содержащей концевые гидроксильные группы, находится в диапазоне от 1:0,01 до 1:0,25; предпочтительно от 1:0,015 до 1:0,2; более предпочтительно от 1:0,05 до 1:0,125. В неограничивающем иллюстративном примере, если молекула, содержащая концевые гидроксильные группы, представляет собой полиэтиленгликоль, массовое соотношение PVM/MA : ПЭГ составляющее 1:0,05, 1:0,1, 1:0,125 или 1:0,25, обеспечивает хорошие результаты. Аналогичным образом, если молекула, содержащая концевые гидроксильные группы, представляет собой метоксиполиэтиленгликоль, массовое соотношение PVM/MA : мПЭГ, составляющее 1:0,01, 1:0,015, 1:0,025, 1:0,05,1:0,01 или 1:0,2, обеспечивает хорошие результаты.
Другими словами, массовое соотношение между молекулой, содержащей концевые гидроксильные группы, согласно настоящему изобретению и PVM/MA в растворе этапа а) способа [1] согласно настоящему изобретению находится в диапазоне от 1:4 до 1:100; предпочтительно от 1:5 до 1:66; более предпочтительно от 1:8 до 1:20. В неограничивающем иллюстративном примере, если молекула, содержащая концевые гидроксильные группы, представляет собой полиэтиленгликоль, массовое соотношение РУМ/МА : ПЭГ, составляющее 1:4, 1:8, 1:10 или 1:20, обеспечивает хорошие результаты. Аналогичным образом, если молекула, содержащая концевые гидроксильные группы, представляет собой метоксиполиэтиленгликоль, массовое соотношение PVM/MA : мПЭГ, составляющее 1:5, 1:10, 1:20, 1:40, 1:66 или 1:100, обеспечивает хорошие результаты.
Сложноэфирный полимерный конъюгат, полученный в соответствии со способом [1] согласно настоящему изобретению, т.е. сложноэфирный полимерный конъюгат PVM/MA и молекулы, содержащей концевые гидроксильные группы, который характеризуется тем, что молекула, содержащая концевые гидроксильные группы, выбрана из полиэтиленгликоля и его производного, содержащего концевую реакционноспособную гидроксильную группу, полученный с помощью способа, который включает: а) взаимодействие PVM/MA с молекулой, содержащей концевые гидроксильные группы, согласно настоящему изобретению в органическом растворителе, и b) удаление органического растворителя, представляет собой дополнительный аспект настоящего изобретения.
Варианты применения сложноэфирного полимерного конъюгата согласно настоящему изобретению
Преформированные сложноэфирные полимерные конъюгаты согласно настоящему изобретению проявили хорошие физико-химические и фармакологические свойства и могут быть использованы в качестве исходных материалов при изготовлении наночастиц для введения биологически активных соединений. Следовательно, в другом аспекте настоящее изобретение относится к применению сложноэфирного полимерного конъюгата согласно настоящему изобретению или сложноэфирного полимерного конъюгата, полученного с помощью способа [1] согласно настоящему изобретению, в получении полимерных наночастиц для доставки лекарственных препаратов; предпочтительно пероральной доставки лекарственных препаратов.
Согласно другому аспекту настоящее изобретение относится к композиции, содержащей i) сложноэфирный полимерный конъюгат согласно настоящему изобретению или сложноэфирный полимерный конъюгат, полученный с помощью способа [1] согласно настоящему изобретению, и ii) носитель, в дальнейшем именуемой «композиция [1] согласно настоящему изобретению». Согласно конкретному варианту реализации настоящего изобретения композиция [1] согласно настоящему изобретению содержит по меньшей мере 2% концентрацию (масс./об.) сложноэфирного полимерного конъюгата согласно настоящему изобретению или сложноэфирного полимерного конъюгата, полученного с помощью способа [1] согласно настоящему изобретению.
В качестве иллюстративного примера указанная композиция [1] согласно настоящему изобретению может быть использована в качестве основы для изготовления наночастиц для введения биологически активных соединений. Следовательно, согласно другому аспекту настоящее изобретение относится к указанной композиции [1] согласно настоящему изобретению для использования для доставки лекарственных препаратов; предпочтительно пероральной доставки лекарственных препаратов.
Наночастицы
Согласно другому аспекту настоящее изобретение относится к наночастице, которая содержит матрикс из сложноэфирного полимерного конъюгата согласно настоящему изобретению. Наночастицы согласно настоящему изобретению обладают способностью инкапсулировать биологически активное соединение (БАС) и могут быть использованы в качестве системы доставки лекарственных препаратов.
Следовательно, согласно конкретному варианту реализации настоящего изобретения, необязательно в комбинации с одним или более признаками различных вариантов реализации, описанных выше или ниже, наночастица дополнительно содержит биологически активное соединение и упоминается далее в настоящем описании как «наночастица согласно настоящему изобретению».
Термин «биологически активное соединение» (БАС) был определен ранее и относится к любой малой молекуле (например, лекарственному препарату или активному ингредиенту лекарственного препарата) или его производному, которое вводят субъекту, предпочтительно человеку, с профилактическими или терапевтическими целями; т.е. к любому веществу или химическому соединению с молекулярной массой ниже 900 Да, которое может быть использовано в лечении, излечении, предотвращении или диагностике заболевания или для улучшения физического и психического самочувствия человека и животных. В настоящей заявке термин «производное», применительно к малой молекуле, включает пролекарственные формы и аналоги указанной молекулы.
БАС могут быть выбраны из множества известных классов лекарственных препаратов, включая, например: противоопухолевые или противобластомные агенты, анальгетики, анестезирующие агенты, противовоспалительные агенты, противоаритмические агенты, противогипертензивные агенты, противостенокардические агенты, противоастматические агенты, антибиотики (включая пенициллины), антикоагулянты, антидепрессанты, антипсихотические агенты, противодиабетические агенты, противоэпилептические агенты, противогистаминные агенты, противокашлевые агенты, антимускариновые агенты, противомикобактериальные агенты, антиокислительные агенты, жаропонижающие, иммунодепрессанты, иммуностимуляторы, антитиреоидные агенты, глистогонные агенты, противовирусные агенты, противобактериальные агенты, противогрибковые агенты, анксиолитические седативные (снотворные и нейролептики) агенты, вяжущие агенты, бактериостатические агенты, блокаторы бета-адренорецепторов, продукты крови и заменители, бронходилататоры, буферные агенты, сердечные инотропные агенты, химиотерапевтические агенты, контрастные среды, кортикостероиды, противокашлевые (отхаркивающие и муколитические) агенты, диагностические агенты, диагностические визуализирующие агенты, диуретики, допаминэргические (противопаркинсонические) агенты и т.д. Наночастицы согласно настоящему изобретению могут включать одно или более из указанных БАС.
Примеры БАС согласно настоящему изобретению включают доцетаксел, паклитаксел, камптотецин, актиномицин D, альбендазол, альдостерон, алпразолам, амиодарон, амитриптилин, ампренавир, асимадолин, аторвастатин, бунитролол, буспирон, карбамазепин, карведилол, целипролол, циклоспорин А, циметидин, клотримазол, колхицин, кортизон, даунорубицин, дебризохин, дексаметазон, диазепам, дигитоксин, дигоксин, дилтиазем, домперидон, доксорубицин, эфавиренц, эпирубицин, эритромицин, эрготамин, эстрадиол, эстрадиола глюкуронид, эрлотиниб, этопозид, фенитоин, фентанил, фелодипин, фенотиазины, фексофенадин, фторхинолоны, фторурацил, FK-506, гентамицин, гризеофульвин, гидрокортизон, иматиниб, индинавир, итраконазол, ивермектин, кетоконазол, кемпферол, левофлоксацин, лидокаин, лоперамид, лозартан, ловастатин, мебендазол, метилпреднизолон, метотрексат, мибефрадил, мидазолам, низолдипин, морфин, нелфинавир, никардипин, нитрендипин, нифедипин, ондансетрон, пентазоцин, празиквантел, преднизолон, преднизон, кверцетин, хинидин, ранитидин, рапамицин, рифабутин, рифампицин, ритонавир, саквинавир, сиролимус, сульфаметизол, такролимус, тамоксифен, талинолол, тенипозид, терфенадин, тетрациклин, топотекан, триамцинолон, валсподар, верапамил, винбластин, винкристин, виндезин, зопиклон, их производные и их смеси.
Наночастицы согласно настоящему изобретению особенно пригодны для введения пероральным путем гидрофобного БАС (класс II и класс IV по классификации биофармацевтических систем).
Согласно одному конкретному варианту реализации настоящего изобретения БАС, присутствующее в наночастицах согласно настоящему изобретению, представляет собой противоопухолевый агент (например, доцетаксел, паклитаксел, камптотецин, доксорубицин, эпирубицин, фторурацил, циклофосфамид, метотрексат и т.д.). Согласно более конкретному варианту реализации настоящего изобретения БАС выбрано из группы, состоящей из доцетаксела, паклитаксела и камптотецина. В еще более конкретном варианте БАС представляет собой доцетаксел.
Наночастицы согласно настоящему изобретению позволяют модифицировать распределение БАС, которые они содержат, при их введении путем, обеспечивающим доступ к любой слизистой оболочке организма (например, пероральным путем и т.д.).
Указанные наночастицы согласно настоящему изобретению имеют размер частиц менее 1 мкм, как правило, размер частиц находится в диапазоне от 1 до 999 нм, предпочтительно от 10 до 900 нм, более предпочтительно от 50 до 550 нм, еще более предпочтительно от 100 до 500 нм, еще более предпочтительно от 150 до 400 нм.
Способ получения наночастиц
Согласно другому аспекту настоящее изобретение относится к способу получения наночастиц, содержащих матрикс из сложноэфирного полимерного конъюгата согласно настоящему изобретению и биологически активное соединение (т.е. наночастиц согласно настоящему изобретению), в дальнейшем именуемому «способ [2] согласно настоящему изобретению», который включает следующие этапы:
a) смешивание сложноэфирного полимерного конъюгата согласно настоящему изобретению с биологически активным соединением в органической среде, и
b) десольватация сложноэфирного полимерного конъюгата путем добавления спирта и воды в присутствии двухвалентного металла.
Согласно конкретному варианту реализации настоящего изобретения, необязательно в комбинации с одним или более признаками различных вариантов реализации, описанных выше или ниже, органическая среда на этапе а) представляет собой ацетон; и спирт, используемый на этапе b), представляет собой этанол.
Двухвалентный металл, который может быть использован для осуществления способа [2] согласно настоящему изобретению, выбран из группы, состоящей из кальция, магния, цинка, железа в двухвалентной форме, а также их комбинаций. Согласно предпочтительному варианту реализации настоящего изобретения, необязательно в комбинации с одним или более признаками различных вариантов реализации, описанных выше или ниже, двухвалентный металл представляет собой кальций; и может быть получен, например, из соли кальция. Иллюстративные неограничивающие примеры солей кальция включают хлорид кальция, ацетат кальция, глюконат кальция, лактат кальция, сорбат кальция и их смеси; предпочтительно соль кальция представляет собой хлорид кальция.
Этап а) способа [2] согласно настоящему изобретению осуществляют с помощью обычных способов, известных специалистам в данной области техники, например, путем:
- растворения сложноэфирного полимерного конъюгата согласно настоящему изобретению в органической среде и добавления биологически активного соединения; или, в другом варианте,
- смешивания (i) органического раствора, содержащего сложноэфирный полимерный конъюгат согласно настоящему изобретению, с (ii) органическим раствором или дисперсией биологически активного соединения.
Этап b) способа [2] согласно настоящему изобретению также осуществляют обычными способами, известными специалистам в данной области техники, например, путем:
- добавления (iii) спирта к смеси, полученной на этапе а), с немедленным добавлением (iv) водного раствора двухвалентного металла, или, в другом варианте,
- добавления (v) водно-спиртовой смеси, содержащей двухвалентный металл, к смеси, полученной на этапе а).
Способ [2] согласно настоящему изобретению охватывает все возможные комбинации указанных выше способов для этапов а) или b), описанных в настоящей заявке. Однако согласно предпочтительному варианту реализации настоящего изобретения, необязательно в комбинации с одним или более признаками различных вариантов реализации, описанных выше или ниже, способ [2] согласно настоящему изобретению включает следующие этапы:
a). смешивание (i) органического раствора, содержащего сложноэфирный полимерный конъюгат согласно настоящему изобретению, с (ii) органическим раствором или дисперсией биологически активного соединения, и
b). десольватация сложноэфирного полимерного конъюгата с помощью добавления водно-спиртовой смеси, содержащей двухвалентный металл, к смеси, полученной на этапе а).
Согласно конкретному варианту реализации настоящего изобретения, необязательно в комбинации с одним или более признаками различных вариантов реализации, описанных выше или ниже, водно-спиртовая смесь имеет соотношение 1:1 (об./об.) (вода/спирт). Согласно другому конкретному варианту реализации настоящего изобретения, необязательно в комбинации с одним или более признаками различных вариантов реализации, описанных выше или ниже, указанная водно-спиртовая смесь содержит 0,5-5% (об./об.) источника двухвалентного металла; предпочтительно 1-3%. Согласно другому конкретному варианту реализации настоящего изобретения указанный источник двухвалентного металла представляет собой водный раствор, содержащий 0,5-1% (масс./об.) соответствующей соли.
Согласно другому конкретному варианту реализации настоящего изобретения, необязательно в комбинации с одним или более признаками различных вариантов реализации, описанных выше или ниже, массовое соотношение между сложноэфирным полимерным конъюгатом согласно настоящему изобретению и биологически активным веществом в смеси этапа а) находится в диапазоне от 1:0,01 до 1:0,20, предпочтительно от 1:0,02 до 1:0,15, более предпочтительно от 1:0,03 до 1:0,10.
В неограничивающем иллюстративном примере, в случае если БАС представляет собой доцетаксел или паклитаксел, массовое соотношение сложноэфирный полимерный конъюгат : БАС, составляющее 1:0,05 или 1:0,10, обеспечивает хорошие результаты. Аналогичным образом, в случае если БАС представляет собой камптотецин, массовое соотношение сложноэфирный полимерный конъюгат : БАС, составляющее 1:0,03 или 1:0,06, обеспечивает хорошие результаты.
Согласно конкретному варианту реализации настоящего изобретения, необязательно в комбинации с одним или более признаками различных вариантов реализации, описанных выше или ниже, соотношение органическая/водно-спиртовая фаза (об./об.) в смеси, полученной по завершении способа [2] согласно настоящему изобретению, составляет 1:1-1:4, предпочтительно 1:2.
Согласно одному варианту реализации настоящего изобретения указанный способ получения наночастиц согласно настоящему изобретению включает дополнительный этап с) удаления органической среды (например, путем выпаривания при пониженном давлении), и/или необязательно очистки (например, с использованием методики фильтрации, центрифугирования или ультрацентрифугирования). Аналогичным образом, при необходимости, указанный способ [2] согласно настоящему изобретению может включать дополнительный этап d) сушки образованных наночастиц для получения наночастиц согласно настоящему изобретению в виде порошка. Такая форма обеспечения указанных наночастиц способствует их стабильности и дополнительно является особенно подходящей для их возможного применения в фармацевтических продуктах.
Практически любая стандартная методика или способ, пригодный для сушки наночастиц, можно применять для выполнения указанного этапа сушки; однако согласно конкретному варианту реализации настоящего изобретения сушку суспензии, содержащей наночастицы, осуществляют с помощью распылительной сушки или с помощью сушки вымораживанием (лиофилизации); предпочтительно с помощью сушки вымораживанием. Указанную обработку обычно проводят путем добавления подходящего защитного агента указанных наночастиц, такого как сахарид, например, сахароза, лактоза, трегалоза, маннит, мальтодекстрин, глюкоза, сорбит, мальтоза, и т.д., и их смеси к суспензии наночастиц. Указанный защитный агент защищает наночастицы согласно настоящему изобретению от разрушения, а также окисления во время процесса сушки.
Наночастицы, полученные с помощью способа [2] согласно настоящему изобретению, имеют характеристики наночастиц согласно настоящему изобретению. Следовательно, наночастицы, полученные с помощью способа [2] согласно настоящему изобретению, т.е. наночастицы, содержащие матрикс из сложноэфирного полимерного конъюгата согласно настоящему изобретению и биологически активное соединение, полученное с помощью способа, который включает: а) смешивание сложноэфирного полимерного конъюгата согласно настоящему изобретению с биологически активным соединением в органической среде, и b) десольватацию сложноэфирного полимерного конъюгата путем добавления спирта и воды в присутствии двухвалентного металла, представляют собой дополнительный аспект настоящего изобретения.
В частности, наночастицы, содержащие матрикс из сложноэфирного полимерного конъюгата согласно настоящему изобретению и БАС, полученные с помощью способа, который включает: а) смешение (i) органического раствора, содержащего сложноэфирный полимерный конъюгат согласно настоящему изобретению, с (b) органическим раствором или дисперсией биологически активного соединения, и b) десольватацию сложноэфирного полимерного конъюгата с помощью добавления водно-спиртовой смеси, содержащей двухвалентный металл, к смеси, полученной на этапе а), представляют собой дополнительный аспект настоящего изобретения, необязательно в комбинации с одним или более признаками различных вариантов реализации, описанных выше или ниже.
Варианты применения наночастиц
Наночастицы, предложенные в настоящем изобретении (т.е. наночастицы, содержащие матрикс из сложноэфирного полимерного конъюгата согласно настоящему изобретению, а также наночастицы, полученные непосредственно в соответствии со способом [2] согласно настоящему изобретению) обеспечивают инкапсуляцию значительных количеств биологически активных соединений и их включение в фармацевтические композиции. Указанные наночастицы могут быть обеспечены в виде суспензии, предпочтительно в водной среде, или, в другом варианте, они могут быть обеспечены в форме сухого порошка, например, в виде лиофилизата вместе с криопротекторным агентом, поддерживающим БАС в стабильном состоянии и обеспечивающим его хранение в течение длительного периода времени.
Наночастицы согласно настоящему изобретению, как представляется, способны пересекать слизистый слой и тесно взаимодействовать с поверхностью энтероцитов, улучшая всасывание лекарственного препарата через слизистую оболочку полости рта. Важно отметить, что наночастицы, обеспеченные в настоящем изобретении, повышают биодоступность БАС при пероральном введении, обеспечивая устойчивые и постоянные уровни в плазме крови. Следовательно, согласно другому аспекту настоящее изобретение относится к наночастицам согласно настоящему изобретению или наночастицам, полученным с помощью способа [2] согласно настоящему изобретению, для применения в медицине. В другом варианте, настоящее изобретение относится к способу предотвращения или лечения заболевания, который включает введение субъекту, нуждающемуся в этом, наночастицы согласно настоящему изобретению или наночастицы, полученной с помощью способа [2] согласно настоящему изобретению.
Доза загруженных наночастиц согласно настоящему изобретению, которая будет введена субъекту, нуждающемуся в лечении с использованием БАС, должна быть достаточной, чтобы вызвать положительный терапевтический ответ у пациента с течением времени. Следовательно, наночастицы вводят пациенту в количестве, достаточном для предотвращения, облегчения, уменьшения, излечения или по меньшей мере частичного купирования симптомов и/или осложнений заболевания. Количество, достаточное для реализации всего вышеперечисленного, определяют как «терапевтически эффективная доза». Доза может варьироваться в широком диапазоне и будет зависеть, помимо других признаков, от природы БАС, его активности или эффективности, количества БАС на наночастицы и т.д. В качестве иллюстративного примера доза загруженных наночастиц, которая будет введена субъекту, может составлять, например, от приблизительно 0,01 до приблизительно 10 мг на кг массы тела, предпочтительно от 0,1 до 2 мг на кг массы тела.
Подробные сведения о БАС были упомянуты в разделе «Определения», а также в связи с наночастицами согласно настоящему изобретению, и включены в настоящее описание посредством ссылки. БАС, которое входит в состав наночастиц, выбирают в зависимости от заболевания, подлежащего лечению. Согласно конкретному варианту реализации настоящего изобретения, необязательно в комбинации с одним или более признаками различных вариантов реализации, описанных выше или ниже, БАС представляет собой противоопухолевый агент; в более конкретном предпочтительном варианте БАС выбрано из группы, состоящей из доцетаксела, паклитаксела и камптотецина. Следовательно, согласно другому аспекту настоящее изобретение относится к наночастице, которая содержит матрикс из сложноэфирного полимерного конъюгата согласно настоящему изобретению, и БАС (т.е. к наночастице согласно настоящему изобретению или наночастице, полученной с помощью способа [2] согласно настоящему изобретению), причем указанное БАС представляет собой противоопухолевый агент, для применения в способе предотвращения или лечения рака; другими словами, настоящее изобретение также относится к способу предотвращения или лечения рака, который включает введение субъекту, нуждающемуся в этом, наночастицы, которая содержит матрикс из сложноэфирного полимерного конъюгата согласно настоящему изобретению, и БАС, причем указанное БАС представляет собой противоопухолевый агент.
Согласно другому аспекту настоящее изобретение относится к применению наночастицы согласно настоящему изобретению или применению наночастицы, полученной в соответствии со способом [2] согласно настоящему изобретению, в производстве фармацевтической композиции.
Согласно другому аспекту настоящее изобретение относится к фармацевтической композиции, далее «фармацевтической композиции согласно настоящему изобретению», содержащей i) по меньшей мере одну наночастицу согласно настоящему изобретению или по меньшей мере одну наночастицу, полученную с помощью способа [2] согласно настоящему изобретению, и ii) фармацевтически приемлемый носитель или наполнитель. Согласно конкретному варианту реализации настоящего изобретения указанная фармацевтическая композиция согласно настоящему изобретению содержит множество наночастиц согласно настоящему изобретению и/или множество наночастиц, полученных с помощью способа [2] согласно настоящему изобретению. Подробные сведения о наночастицах были приведены выше и включены в настоящее описание посредством ссылки.
Предпочтительно наночастицы в фармацевтической композиции согласно настоящему изобретению имеют средний размер, находящийся в диапазоне от 10 до 900 нм, предпочтительно от 50 до 550 нм, более предпочтительно от 100 до 500 нм, еще более предпочтительно от 150 до 400 нм, еще более предпочтительно приблизительно 250 нм.
Фармацевтическая композиция согласно настоящему изобретению может быть изготовлена в виде твердого вещества (например, таблеток, капсул, покрытых таблеток, гранул, суппозиториев, стерильных кристаллических или аморфных твердых веществ, которые могут быть восстановлены, чтобы обеспечить жидкие формы и т.д.), жидкости (например, суспензии или дисперсии наночастиц, и т.д.) или полутвердой (гели, помадки, кремы и т.п.) лекарственной формы. Описанные фармацевтические композиции будут содержать носители или наполнители, подходящие для каждого состава. Помимо этого, фармацевтическая композиция может содержать, в случае необходимости, стабилизаторы, суспензии, консерванты, поверхностно-активные вещества и т.п. Подходящие вспомогательные вещества будут выбраны в соответствии с выбранной лекарственной формой.
Согласно конкретному варианту реализации настоящего изобретения указанная фармацевтическая композиция изготовлена в виде лекарственной формы, подходящей для введения путем, обеспечивающим доступ к слизистой оболочке. Согласно предпочтительному варианту реализации настоящего изобретения фармацевтическую композицию, обеспеченную в настоящем изобретении, вводят перорально; следовательно, носитель или наполнитель содержит одно или более фармацевтических вспомогательных веществ, пригодных для введения пероральным путем. Пероральные составы обычно получают способами, известными специалистам в данной области техники. Обзор различных форм введения активных ингредиентов, вспомогательных веществ, которые можно применять, а также способов их получения можно найти в книге "Tratado de Farmacia


Согласно другому конкретному варианту реализации настоящего изобретения фармацевтическую композицию согласно настоящему изобретению получают в виде сухого порошка, например, в виде лиофилизата.
Фармацевтические композиции в соответствии с настоящим изобретением могут содержать БАС или активный ингредиент в количестве, которое находится в диапазоне от 0,05% до 50%, предпочтительно от 0,1% до 30%, более предпочтительно от 0,5% до 25%, еще более предпочтительно от 1% до 20%, причем указанные выше процентные доли выражены как масс/масс, по отношению к общей массе композиции или лекарственной формы. Тем не менее, подходящая доля будет зависеть от конкретного включенного БАС.
Согласно другому аспекту настоящее изобретение относится к фармацевтической композиции согласно настоящему изобретению для применения в медицине. Согласно другому аспекту настоящее изобретение относится к применению фармацевтической композиции согласно настоящему изобретению в получении лекарственного препарата для предотвращения или лечения заболевания. В другом варианте, настоящее изобретение относится к способу предотвращения или лечения заболевания, который включает введение субъекту, нуждающемуся в этом, фармацевтической композиции согласно настоящему изобретению.
Терапевтически эффективная доза фармацевтической композиции, подлежащей введению, зависит от конкретного случая и, как правило, должна быть адаптирована к условиям конкретного случая для достижения оптимального эффекта. Следовательно, указанная доза зависит, как известно, от частоты введения и от эффективности и продолжительности действия БАС, используемого в каждом случае для терапии или предотвращения, но также и от характера и степени тяжести заболевания и симптомов, а также от пола, возраста, массы, сопутствующих лекарственных средств и индивидуальной восприимчивости субъекта, подлежащего лечению, и от того, является ли лечение острым или профилактическим. Дозы могут быть адаптированы в зависимости от массы субъекта, а также для педиатрического введения.
Согласно конкретному варианту реализации настоящего изобретения, необязательно в комбинации с одним или более признаками различных вариантов реализации, описанных выше или ниже, фармацевтическая композиция согласно настоящему изобретению содержит i) по меньшей мере одну из наночастиц, содержащих матрикс из сложноэфирного полимерного конъюгата согласно настоящему изобретению, а также БАС (т.е. наночастицу согласно настоящему изобретению или наночастицу, полученную с помощью способа [2] согласно настоящему изобретению), причем указанное БАС представляет собой противоопухолевый агент, и ii) фармацевтически приемлемый носитель или наполнитель. Предпочтительно противоопухолевый агент выбран из группы, состоящей из доцетаксела, паклитаксела и камптотецина; предпочтительно представляет собой доцетаксел.
Согласно конкретному варианту реализации настоящего изобретения, в котором БАС представляет собой противоопухолевый агент, фармацевтическую композицию согласно настоящему изобретению можно применять в способе предотвращения или лечения рака.
Следовательно, в дополнительном аспекте настоящее изобретение относится к способу предотвращения и/или лечения рака, который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции согласно настоящему изобретению, содержащей противоопухолевый агент; в другом варианте, настоящее изобретение также относится к применению фармацевтической композиции согласно настоящему изобретению, содержащей противоопухолевый агент, в получении лекарственного средства для предотвращения или лечения рака.
Согласно более конкретному варианту реализации настоящего изобретения фармацевтическая композиция согласно настоящему изобретению выбрана из группы, состоящей из:
фармацевтической композиции, содержащей:
а). сложноэфирный полимерный конъюгат PVM/MA с полиэтиленгликолем 2000 в количестве от 38% до 47%,
b). доцетаксел в количестве от 3% до 5%,
c). кальций в количестве от 0,1% до 0,2%, и
d). сахарид в количестве от 15% до 40%,
где все указанные содержания приведены по массе по отношению к общей массе композиции;
фармацевтической композиции, содержащей:
a). сложноэфирный полимерный конъюгат PVM/MA с метоксиполиэтиленгликолем 2000 в количестве от 38% до 47%,
b). доцетаксел в количестве от 3% до 5%,
c). кальций в количестве от 0,1% до 0,2%, и
d). сахарид в количестве от 15% до 40%,
где все указанные содержания приведены по массе по отношению к общей массе композиции; и
фармацевтической композиции, содержащей:
a). сложноэфирный полимерный конъюгат PVM/MA с метоксиполиэтиленгликолем 2000 в количестве от 30% до 40%,
b). камптотецин в количестве от 0,08% до 1,5%,
c). кальций в количестве от 0,10% до 0,20%, и
d). сахарид в количестве от 15% до 40%,
где все указанные содержания приведены по массе по отношению к общей массе композиции.
В область настоящего изобретения включены все возможные комбинации конкретных и предпочтительных вариантов реализации, описанных в настоящем документе.
ПРИМЕРЫ
Следующие примеры описывают получение и определение характеристик сложноэфирных полимерных конъюгатов поли(винилового эфира-малеинового ангидрида) (PVM/MA или гантреза® AN) с полиэтиленгликолем или его производноым, содержащим концевую реакционноспособную гидроксильную группу. Кроме того, следующие примеры описывают получение и определение характеристик наночастиц на основе указанных сложноэфирных полимерных конъюгатов PVM/MA с различными типами полиэтиленгликолей или их производными, содержащими концевую реакционноспособную гидроксильную группу, а также биологически активных молекул. В частности, было установлено, что указанные наночастицы способны i) инкапсулировать значительное количество биологически активных соединений (например, паклитаксела, доцетаксела и камптотецина), и ii) пересекать слизистый слой и тесно взаимодействовать с поверхностью энтероцитов (повышая всасывание лекарственного препарата).
Как следует из указанных примеров, при использовании доцетаксела в качестве биологически активного соединения, его включение в указанные наночастицы позволяет поддерживать постоянные и устойчивые уровни указанного лекарственного препарата в плазме крови в течение по меньшей мере 72 ч. Аналогичным образом, при включении камптотецина в наночастицы согласно настоящему изобретению в качестве биологически активного соединения, его уровни в плазме крови поддерживаются в течение по меньшей мере 48 часов.
Общие материалы описаны в первую очередь.
МАТЕРИАЛЫ
Поли(метилвиниловый эфира-малеинового ангидрида или полиангидрида) (PVM/MA) [гантрез® AN 119] был приобретен у ISP (Барселона, Испания). Камптотецин (99,0%), паклитаксел (Фармакопея США 26, класс >99,5%) и доцетаксел (99,0%) были поставлены 21CECpharm (Лондон, Великобритания). Таксотер был получен от Aventis-Pharma (Cedex, Франция). Фосфатно-солевой буферный раствор (ФСБ) панкреатин, метоксиполиэтиленгликоль 2000 (мПЭГ2) и полиэтиленгликоль 2000, 6000 и 10000 (ПЭГ2, ПЭГ6 и ПЭГ10) были получены от Sigma-Aldrich (Испания). Пепсин, ацетон, этанол и ацетонитрил были получены от Merck (Дармштадт, Германия). Деионизированную воду (сопротивление 18,2 МОм) получали с помощью системы очистки воды (Wasserlab, Испания). Все использованные реагенты и химические вещества были аналитической степени чистоты.
Указанные примеры приведены в качестве иллюстрации и не предназначены для ограничения настоящего изобретения.
ПРИМЕР 1
Получение стандартных наночастиц PVM/MA
Получение пустых стандартных наночастиц PVM/MA (НЧ)
100 мг PVM/MA растворяли в 5 мл ацетона, и наночастицы формировали путем добавления смеси этанола и воды (1:1, об./об.). После этого удаляли органические растворители путем выпаривания при пониженном давлении, полученную в результате суспензию фильтровали через мембрану с размером пор 0,45 мкм и очищали дважды с помощью центрифугирования при 27000 g в течение 20 мин. Супернатант удаляли, и осадок повторно суспендировали в воде. В завершение, состав замораживали и лиофилизировали (Genesis 12EL, Virtis, США) с использованием сахарозы (5% масс./масс.) в качестве криопротектора. В результате были получены пустые наночастицы PVM/MA, далее составы НЧ (НЧ).
Получение стандартных наночастиц PVM/MA, инкапсулирующих паклитаксел (PTX)
Наночастицы PVM/MA, загруженные паклитакселом, получали с помощью способа замещения растворителя с некоторыми модификациями. В общих чертах, 10 мг паклитаксела диспергировали в 5 мл ацетона, содержащего 100 мг PVM/MA. Полученную смесь инкубировали при перемешивании на магнитной мешалке в течение 1 ч при комнатной температуре. По истечении этого времени наночастицы формировали путем добавления 10 мл этанола с последующим добавлением 10 мл водного раствора, содержащего 20 мг динатриевой соли этилендиаминтетрауксусной кислоты. После гомогенизации с помощью магнитной мешалки в течение 10 мин органические растворители удаляли выпариванием при пониженном давлении (

Получение стандартных наночастиц PVM/MA, инкапсулирующих доцетаксел (DTX)
Наночастицы PVM/MA, загруженные доцетакселом, получали с помощью способа замещения растворителя с некоторыми модификациями. В общих чертах, 10 мг доцетаксела диспергировали в 5 мл ацетона, содержащего 100 мг PVM/MA. Полученную смесь инкубировали при перемешивании на магнитной мешалке в течение 1 ч при комнатной температуре. По истечении этого времени, наночастицы формировали путем добавления 10 мл этанола с последующим добавлением 10 мл водного раствора, содержащего 20 мг динатриевой соли этилендиаминтетрауксусной кислоты. После гомогенизации с помощью магнитной мешалки в течение 10 мин органические растворители удаляли выпариванием при пониженном давлении (
Получение стандартных наночастиц PVM/MA, инкапсулирующих камптотецин (СРТ)
100 мг PVM/MA и 10 мг камптотецина растворяли и диспергировали в 2 и 3 мл ацетона, соответственно. Камптотецин обрабатывали ультразвуком в течение 1 мин и смешивали с раствором PVM/MA. Наночастицы немедленно формировали путем добавления смеси этанола и воды (1:1, об./об.). Затем органические растворители удаляли выпариванием при пониженном давлении, полученную суспензию фильтровали через мембрану с размером пор 0,45 мкм и дважды очищали с помощью центрифугирования при 27000 g в течение 20 мин. В завершение, состав замораживали и лиофилизировали (Genesis 12EL, Virtis, США) с использованием сахарозы (5%, масс/масс.) в качестве криопротектора. Полученные составы представляли собой СРТ-загруженные наночастицы PVM/MA, далее составы СРТ.
ПРИМЕР 2
Получение стандартных пегилированных наночастиц PVM/MA
Получение пустых стандартных пегилированных наночастиц PVM/MA (станд. ПЭГ) Пегилированные наночастицы PVM/MA получали с помощью способа замещения растворителя. В общих чертах, 12,5 мг полиэтиленгликоля (ПЭГ2, ПЭГ6 или ПЭГ10) растворяли в 3 мл ацетона и затем добавляли к раствору 100 мг PVM/MA в 2 мл аналогичного органического растворителя. Полученную смесь инкубировали при перемешивании на магнитной мешалке в течение 1 ч при комнатной температуре. Затем наночастицы формировали путем добавления 10 мл этанола с последующим добавлением 10 мл воды. Органические растворители удаляли выпариванием при пониженном давлении (
Получение стандартных пегилированных наночастиц PVM/MA, инкапсулирующих паклитаксел (ПЭГ-РТХ станд.)
Пегилированные наночастицы PVM/MA, содержащие паклитаксел, получали с помощью способа замещения растворителя с некоторыми модификациями. В общих чертах, 12,5 мг полиэтиленгликоля (ПЭГ2, ПЭГ6 или ПЭГ10) растворяли в 3 мл ацетона и затем добавляли к раствору 100 мг PVM/MA в 2 мл аналогичного органического растворителя. Полученную смесь инкубировали при перемешивании на магнитной мешалке. Одновременно с этим 10 мг паклитаксела растворяли в 0,5 мл ацетона и добавляли к смеси полимеров. Затем органическую фазу (содержащую паклитаксел, PVM/MA и ПЭГ) перемешивали с помощью магнитной мешалки в течение 1 ч при комнатной температуре. По истечении этого времени наночастицы формировали путем добавления 10 мл этанола с последующим добавлением 10 мл водного раствора, содержащего 20 мг динатриевой соли этилендиаминтетрауксусной кислоты. После гомогенизации с помощью магнитной мешалки в течение 10 мин органические растворители удаляли выпариванием при пониженном давлении (
Получение стандартных пегилированных наночастиц PVM/MA, инкапсулирующих доцетаксел (ПЭГ-DTX станд.)
Полиэтиленгликоль 2000 (12,5 мг) сначала растворяли в 3 мл ацетона и добавляли к раствору 100 мг PVM/MA также в 2 мл ацетона. Полученную смесь инкубировали при перемешивании на магнитной мешалке. Одновременно с этим доцетаксел растворяли в 0,5 мл ацетона и добавляли к раствору. Затем органическую фазу, содержащую доцетаксел, PVM/MA и ПЭГ2, инкубировали при перемешивании с помощью магнитной мешалки в течение примерно 1 ч при комнатной температуре. Затем наночастицы формировали путем добавления 10 мл этанола с последующим добавлением 10 мл водного раствора, содержащего глицин (50 мг) и динатриевую соль этилендиаминтетрауксусной кислоты (18 мг) и оставляли для гомогенизации в течение 10 мин. Органические растворители удаляли выпариванием при пониженном давлении, и конечный объем доводили до 10 мл с помощью раствора глицина. Суспензию очищали фильтрацией тангенциальным потоком в пробирках Vivaspin (300000 НОММ, Sartorius Group, Германия) при 4000 g в течение 15 мин. Осадки повторно суспендировали в воде, и этап очистки повторяли дважды. В завершение, составы замораживали и затем лиофилизировали с использованием сахарозы (5%) в качестве криопротектора. Соответственно, в результате получали DTX-загруженные стандартные пегилированные наночастицы PVM/MA, далее составы станд. ПЭГ2-DТХ.
Получение стандартных пегилированных наночастиц PVM/MA, инкапсулирующих камптотецин (станд. ПЭГ-СРТ)
Пегилированные наночастицы получали при соотношении полимер/СРТ/ПЭГ 1/0,03/0,125. ПЭГ2 или ПЭГ6 и камптотецин растворяли в ацетоне и смешивали в различных условиях. Наночастицы немедленно формировали путем добавления смеси этанола, воды, глицина и динатриевой соли этилендиаминтетрауксусной кислоты. После гомогенизации в течение 10 мин органические растворители удаляли выпариванием при пониженном давлении. Затем полученные суспензии фильтровали через мембрану с размером пор 0,45 мкм и очищали дважды с помощью пробирок Vivaspin (300000 НОММ, Sartorius group, Германия) при 3000g в течение 20 мин. В завершение, составы замораживали и затем лиофилизировали (Genesis 12EL, Virtis, USA) с использованием сахарозы (5%, масс/масс.) в качестве криопротектора. Следовательно, в результате получали СРТ-загруженные стандартные пегилированные наночастицы PVM/MA, далее составы станд. ПЭГ2-СРТ и станд. ПЭГ6-СРТ.
ПРИМЕР 3
Получение и характеристика сложноэфирных полимерных конъюгатов PVM/MA
3.1 Получение сложноэфирных полимерных конъюгатов PVM/MA
В этом случае испытывали полиэтиленгликоли различных молекулярных масс 2000, 6000 или 10000 и метоксиполиэтиленгликоль 2000.
Для получения сложноэфирных полимерных конъюгатов 5 г PVM/MA растворяли в 250 мл органического растворителя (т.е. ацетона) и инкубировали с различными количествами полиэтиленгликоля или его производного при перемешивании с помощью магнитной мешалки при 50°С в течение 3 ч. Были испытаны следующие соотношения молекула, содержащая концевые гидроксильные группы:(PVM/MA):
- мПЭГ : (PVM/MA) 1:5; для мПЭГ2
- ПЭГ : (PVM/MA) 1:20; 1:8; для ПЭГ2, ПЭГ6 и ПЭГ10
После инкубации растворитель удаляли, и полученный сложноэфирный полимерный конъюгат высушивали путем выпаривания при пониженном давлении (
3.2 Характеристика сложноэфирных полимерных конъюгатов PVM/MA
После синтеза определяли физико-химические характеристики полученных сложноэфирных полимерных конъюгатов PVM/MA и ПЭГ2, ПЭГ6 и ПЭГ10 (при соотношении ПЭГ : PVM/MA 1:8) и сложноэфирных полимерных конъюгатов PVM/MA и мПЭГ2 (при соотношении мПЭГ : PVM/МА 1:5), чтобы получить доказательства модификации остова PVM/MA, а также оценить степень замещения (DS) и молекулярную массу (ММ).
Были использованы следующие методики:
Инфракрасная спектроскопия (ИК): ИК-спектроскопию осуществляли с использованием устройства Nicolet Avatar 360FT-IR (Thermo, США) на основе технологии отражения. Эта методика позволила идентифицировать связывание функциональной -ОН группы молекулы, содержащей концевые гидроксильные группы (ПЭГ или мПЭГ), с ангидридными группами PVM/MA.
Элементный анализ (CHN): Все преформированные сложноэфирные полимерные конъюгаты исследовали на аппарате CHN-900 Leco (Leco Corporations, США). Эта методика позволяет определить состав полимеров и выявить различия в содержании С/Н, когда молекула, содержащая концевые гидроксильные группы (ПЭГ или мПЭГ), включена в структуру PVM/MA.
Титрование: PVM/MA или его сложноэфирные полимерные конъюгаты сначала гидратировали и диспергировали в воде до полного растворения. В этот момент водные растворы полимеров титровали 0,2 н раствором NaOH в присутствии фенолфталеина, используемого в качестве индикатора. Титрование использовали для измерения процентного содержания свободных карбоксильных (-СООН) групп. Уменьшение количества свободных карбоксильных групп в сложноэфирных полимерных конъюгатах в сравнении с немодифицированным полимером PVM/MA свидетельствует о связывании молекулы.
Эксклюзионная хроматография с детектированием рассеивания лазерного излучения с кратными углами (SEC-MALLS): методика SEC-MALLS является ключевой для определения молекулярной массы полимера в растворе. В указанной методике применяют калибровочную кривую времени элюирования в зависимости от молекулярной массы на колонке для эксклюзионной хроматографии, и однократное измерение одного полимера позволяет установить его молекулярную массу.
Динамическое рассеяние света (DLS): Эта методика позволяет определить гидродинамический радиус (Rh) полимеров, обеспечивая оценку наиболее вероятной конформации полимера в растворе. Измерения методом DLS проводили при угле рассеяния 90° с использованием фотоннокоррелирующего спектрометра DynaPro-MS/X, оснащенного 248-канальным мульти-тау коррелятором и термоэлектрическим модулем основанном на эффекте Пельтье (Protein Solutions Inc, США). Длина волны лазера составляла 852,2 нм при интенсивности 100%. Полимерные конъюгаты измеряли при 25°С с использованием тетрагидрофурана в качестве растворителя.
ЯМР-спектроскопия (1Н-ЯМР):1H-ЯМР-спектры регистрировали с помощью аппарата Bruker Avance 400 при частоте 400 МГц (Bruker, США) с использованием программы импульсов zg30 и временем ожидания между импульсами (D0) 1 с. Преформированные сложноэфирные полимерные конъюгаты растворяли в дейтерированном ацетоне (ацетон-d6), взятом в качестве растворителя.
3.3 Результаты
Исследование преформированных сложноэфирных полимерных конъюгатов методом инфракрасной спектроскопии выявило образование новой связи при ~1705 см-1, связанной с растяжением новой сложноэфирной карбонильной группы v (С=O), которая возникла в результате реакции гидроксильной группы ПЭГ или мПЭГ с ангидридными группами PVM/MA, см. фигуру 1.
После подтверждения факта образования связи между полимером и молекулой, содержащей концевые гидроксильные группы, методом ИК-спектроскопии, проводили дальнейшие исследования, чтобы подтвердить модификацию полимера.
В таблице 1 обобщены данные по содержанию углерода (С), водорода (Н) и кислорода (О), определенному для исходного полимера PVM/MA и его сложноэфирных полимерных конъюгатов. Элементный анализ показал уменьшение процентного содержания С и увеличение процентного содержания О, подтверждающее заметное изменение состава сложноэфирных полимерных конъюгатов PVM/MA по сравнению с немодифицированным PVM/MA, при этом соотношение С/О (соотношение С/О) ниже для сложноэфирных полимерных конъюгатов, чем для исходного сополимера.
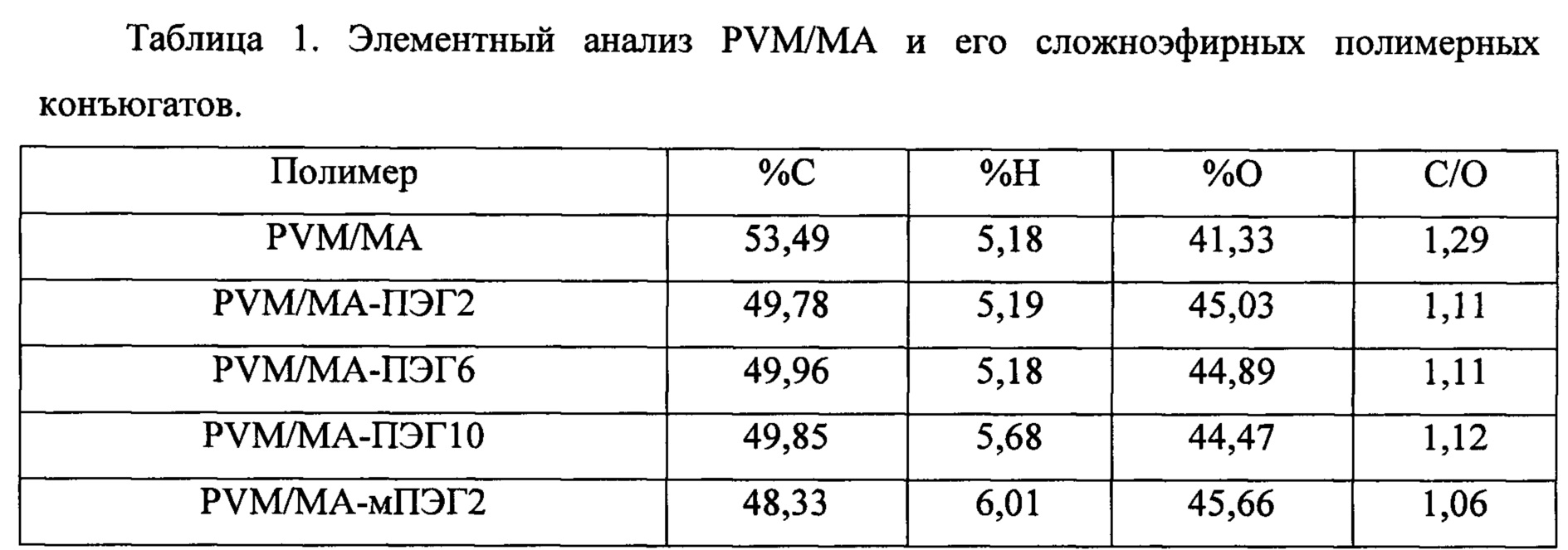
В то же время результаты титрования подтверждают уменьшение количества свободных -СООН групп, вследствие связывания кольца ангидрида с молекулой, содержащей концевые гидроксильные группы (ПЭГ или мПЭГ) (таблица 2).

Исследования методом DLS позволили определить значения гидродинамического радиуса PVM/MA и сложноэфирных полимерных конъюгатов PVM/MA с ПЭГ или мПЭГ. Во всех случаях гидродинамический радиус увеличивается относительно исходного PVM/MA, за исключением конъюгата PVM/MA-метоксиполиэтиленгликоль 2000, который имеет незначительно более низкое значение, но очень близкое, по сравнению с немодифицированным полимером PVM/MA (таблица 3). Это факт может указывать на изменение конформации молекулы, когда полимерные конъюгаты растворяют в органическом растворителе. Отсутствие атомов водорода в метоксигруппах в молекулах мПЭГ может способствовать складыванию молекулы в меньшей степени, чем гидроксильные группы, присутствующие в молекулах ПЭГ.


В завершение следует отметить, что1H-ЯМР-спектры сложноэфирных полимерных конъюгатов во всех случаях выявили наличие химического сдвига, соответствующего звеньям -О-СН2-СН2-полиэтилен в молекулах (ПЭГ или мПЭГ) (фигура 2).
Для того чтобы оценить процент молекул с концевыми гидроксильными группами в полученных сложноэфирных полимерных конъюгатах (DS), рассчитывали соотношение между площадью пика, связанного с «а» протонами в молекуле PVM/MA (см. фигуру 2А), и площадью одного характерного пика молекулы с концевыми гидроксильными группами (новый химический сдвиг в сложноэфирном полимерном конъюгате выделен на фигуре 2В-2Е).
Учитывая полученные результаты, также определяли среднюю молекулярную массу (Mw) полученных конъюгатов, принимая во внимание молекулярную массу гантреза® AN 119 равную 95,5 кДа, рассчитанную на основании измерений методом SEC-MALLS (таблица 4).
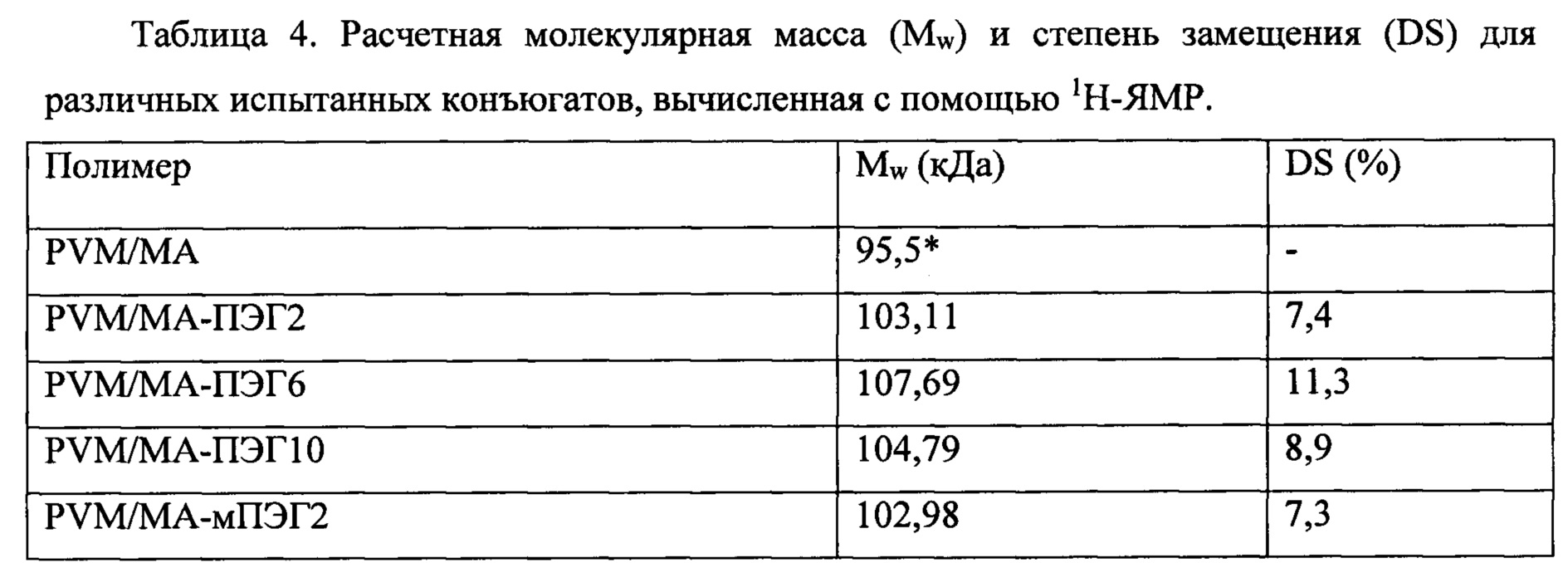
(*) Молекулярная масса, полученная с помощью SEC/MALLS
После очистки и определения характеристик сложноэфирных полимерных конъюгатов, получали наночастицы и исследовали их свойства.
ПРИМЕР 4
Получение наночастиц, содержащих матрикс из сложноэфирного полимерного конъюгата PVM/MA
Получение пустых наночастиц, содержащих матрикс из сложноэфирного полимерного конъюгата PVM/MA (ПЭГ кон, или мПЭГ кон.)
Получение наночастиц проводили следующим образом. 100 мг преформированных сложноэфирных полимеров PVM/MA, конъюгированных с полиэтиленгликолем 2000, полиэтиленгликолем 6000, полиэтиленгликолем 10000 [использованные соотношения ПЭГ2 : (PVM/МА), ПЭГ6 : (PVM/MA) и ПЭГ10 : (PVM/MA) составили 1:20] или метоксиполиэтиленгликолем 2000 [соотношение мПЭГ2 : (PVM/MA) составило 1:5] растворяли в 5 мл ацетона, и десольватировали путем добавления 10 мл водно-спиртовой смеси 1:1 (об./об.) (вода/этанол), содержащей 1% (об./об.) водного раствора CaCl2 0,8% (масс./об.) при перемешивании с помощью магнитной мешалки при комнатной температуре. Органические растворители удаляли выпариванием при пониженном давлении (
Получение наночастиц, содержащих матрикс из сложноэфирных полимерных конъюгатов PVM/MA с метоксиполиэтиленгликолем 2000, инкапсулирующих паклитаксел (мПЭГ2-РТХ кон.)
100 мг преформированного сложноэфирного полимерного конъюгата PVM/MA с метоксиполиэтиленгликолем 2000 [соотношение мПЭГ2 : (PVM/MA) 1:5] растворяли в 4 мл ацетона. Одновременно с этим 10 мг паклитаксела диспергировали в 1 мл ацетона. Раствор конъюгата и дисперсию паклитаксела смешивали при перемешивании с помощью магнитной мешалки при комнатной температуре, и затем формировали наночастицы путем добавления 10 мл водно-спиртовой смеси 1:1 (об./об.) (вода/этанол), содержащей 1% (об./об.) водного раствора CaCl2 0,8% (масс./об.). Органические растворители удаляли выпариванием при пониженном давлении (
Получение наночастиц, содержащих матрикс из сложноэфирных полимерных конъюгатов PVM/MA с полиэтиленгликолем 2000, инкапсулирующих паклитаксел (ПЭГ2-РТХ кон.)
100 мг преформированного сложноэфирного полимерного конъюгата PVM/MA с полиэтиленгликолем 2000 [соотношение ПЭГ2 : (PVM/MA) 1:20] растворяли в 4 мл ацетона. Одновременно с этим 10 мг паклитаксела диспергировали в 1 мл ацетона. Раствор конъюгата и дисперсию паклитаксела смешивали при перемешивании с помощью магнитной мешалки при комнатной температуре, и затем формировали наночастицы путем добавления 10 мл водно-спиртовой смеси 1:1 (об./об.) (вода/этанол), содержащей 1% (об./об.) водного раствора CaCl2 0,8% (масс./об.). Органические растворители удаляли выпариванием при пониженном давлении (
Получение наночастиц, содержащих матрикс из сложноэфирного полимерного конъюгата PVM/MA с метоксиполиэтиленгликолем 2000, инкапсулирующим доцетаксел (мПЭГ2-DТХ кон.)
100 мг преформированного сложноэфирного полимерного конъюгата PVM/MA с метоксиполиэтиленгликолем 2000 [соотношение мПЭГ2 : (PVM/MA) 1:5] растворяли в 4 мл ацетона. Одновременно с этим 10 мг доцетаксела диспергировали в 1 мл ацетона. Раствор конъюгата и дисперсию доцетаксела смешивали при перемешивании с помощью магнитной мешалки при комнатной температуре, и затем формировали наночастицы путем добавления 10 мл водно-спиртовой смеси 1:1 (об./об.) (вода/этанол), содержащей 1% (об./об.) водного раствора CaCl2 0,8% (масс./об.). Органические растворители удаляли выпариванием при пониженном давлении (
Получение наночастиц, содержащих матрикс из сложноэфирного полимерного конъюгата PVM/MA с полиэтиленгликолем 2000, инкапсулирующих доцетаксел (ПЭГ2-DTX кон.)
100 мг предварительно сформированного сложноэфирного полимерного конъюгата PVM/MA с полиэтиленгликолем 2000 [соотношение ПЭГ2 : (PVM/MA) 1:20] растворяли в 4 мл ацетона. Одновременно с этим 10 мг доцетаксела диспергировали в 1 мл ацетона. Раствор конъюгата и дисперсию доцетаксела затем смешивали при перемешивании с помощью магнитной мешалки при комнатной температуре, и формировали наночастицы путем добавления 10 мл водно-спиртовой смеси 1:1 (об./об.) (вода/этанол), содержащей 1% (об./об.) водного раствора CaCl2 0,8% (масс./об.). Органические растворители удаляли выпариванием при пониженном давлении (
Получение наночастиц, содержащих матрикс из сложноэфирного полимерного конъюгата PVM/MA с метоксиполиэтиленгликолем 2000, инкапсулирующих камптотецин (мПЭГ2-СРТ кон.)
100 мг преформированного сложноэфирного полимерного конъюгата PVM/MA с метоксиполиэтиленгликолем 2000 [соотношение мПЭГ2 : (PVM/MA) 1:5] растворяли в 4 мл ацетона. Одновременно с этим 3 мг камптотецина диспергировали в 1 мл ацетона и обрабатывали ультразвуком в течение 30 с. Раствор конъюгата и дисперсию камптотецина смешивали при перемешивании с помощью магнитной мешалки при комнатной температуре, и затем формировали наночастицы путем добавления 10 мл водно-спиртовой смеси в соотношении 1:1 (об./об.) (вода/этанол), содержащей 3% (об./об.) водного раствора CaCl2 0,8% (масс./об.). После удаления органических растворителей при пониженном давлении (
ПРИМЕР 5
Физико-химические характеристики наночастиц
Для выполнения данных исследований использовали составы наночастиц, описанные в примерах 1, 2 и 4.
5.1 Материалы и методы
Средний гидродинамический диаметр наночастиц и дзета-потенциал определяли с помощью фотонной корреляционной спектроскопии (ФКС) и электрофоретической лазерной доплеровской анемометрии, соответственно, с использованием анализирующей системы Zetasizer (Brookhaven Instruments Corporation, Нью-Йорк, США). Диаметр наночастиц определяли после диспергирования в сверхчистой воде (1:10) и измеряли при 25°С с помощью динамического рассеяния света под углом 90°. Дзета-потенциал определяли на этом же оборудовании следующим образом: 200 мкл тех же самых образцов разводили в 2 мл 0,1 мМ раствора KCl.
Исследование морфологии поверхности составов также проводили с помощью сканирующей электронной микроскопии (СЭМ) на цифровом сканирующем электронном микроскопе Zeiss DSM940 (Oberkochen, Германия), оснащенном цифровой системой регистрации изображений (Point Electronic GmbH, Германия). Перед проведением исследования наночастицы разбавляли деионизированной водой и центрифугировали при 17000 об./мин (Sigma 3 К30, Германия) в течение 20 мин при 4°С. Осадок высушивали и оттеняли золотым слоем толщиной 9 нм в Emitech K 550 Sputter-Coater (Ашфорд, Великобритания). И, наконец, выход составов наночастиц рассчитывали методом гравиметрии на основании разности между начальным количеством полимера, использованного для получения наночастиц, и массой лиофилизированных образцов.
Выход(%)=(НЧконеч/Полимерначальн.)*100
Содержание паклитаксела в наночастицах
Количество паклитаксела, загруженного в наночастицы, количественно определяли методом ВЭЖХ-УФ с помощью жидкостного хроматографа серии Agilent 1200 и детектора с диодной матрицей, установленной на 228 нм. Хроматографическая система была оснащена обращенно-фазовой колонкой Phenomenex Gemini C18 150 мм × 3 мм (размер частиц 5 мкм) и предварительной колонкой (Phenomenex SecurityGuard С18). Подвижная фаза, нагнетаемая со скоростью 0,5 мл/мин, представляла собой смесь фосфатного буфера (0,01 М, рН=2) и ацетонитрила (50:50, об./об.). Колонку помещали при 30°С, и впрыскиваемый объем составил 100 мкл. Доцетаксел использовали в качестве внутреннего стандарта. Калибровочные кривые были построены в диапазоне от 80 до 7000 нг/мл (r2 > 0,999). Расчетный предел количественного определения составил 40 нг/мл. Для проведения исследования наночастицы солюбилизировали в ацетонитриле (1:5 об./об.). Образцы переносили во флаконы автодозатора, закрывали и помещали в автодозатор ВЭЖХ. Каждый образец исследовали в трех повторах, и результаты выражали в виде количества паклитаксела (в мкг) на мг наночастиц.
Содержание доцетаксела в наночастицах
Количество доцетаксела, загруженного в наночастицы, количественно определяли методом ВЭЖХ-УФ с помощью жидкостного хроматографа серии Agilent 1200 и детектора с диодной матрицей, установленной на 228 нм. Хроматографическая система была оснащена обращенно-фазовой колонкой Phenomenex Gemini C18 150 мм × 3 мм (размер частиц 5 мкм) и предварительной колонкой (Phenomenex SecurityGuard С18). Подвижная фаза, нагнетаемая со скоростью 0,5 мл/мин, представляла собой смесь фосфатного буфера (0,01 М, рН=2) и ацетонитрила (50:50, об./об.). Колонку помещали при 30°С, и впрыскиваемый объем составил 100 мкл. Паклитаксел использовали в качестве внутреннего стандарта. Калибровочные кривые были построены в диапазоне от 1,25 до 320 нг/мл (r2 > 0,999). Расчетный предел количественного определения составил 60 нг/мл. Для проведения исследования наночастицы солюбилизировали в ацетонитриле (1:8 об./об.). Образцы переносили во флаконы автодозатора, закрывали и помещали в автодозатор ВЭЖХ. Каждый образец исследовали в трех повторах, и результаты выражали в виде количества доцетаксела (в мкг) на мг наночастиц.
Содержание камптотецина в наночастицах
Количество камптотецина, загруженного в наночастицы, количественно определяли методом ВЭЖХ-ДФ с помощью жидкостного хроматографа серии Agilent 1100 и флуоресцентного детектора с установленной длиной волны возбуждения и испускания 380 нм и 418 нм, соответственно. Хроматографическая система была оснащена обращенно-фазовой колонкой Phenomenex Gemini C18 150 мм × 3 мм (размер частиц 5 мкм) и предварительной колонкой (Phenomenex SecurityGuard С18). Подвижная фаза, нагнетаемая со скоростью 1 мл/мин, представляла собой смесь ацетонитрила (50:50, об./об.) и 0,01% трифторуксусной кислоты (об./об.). Колонку помещали при 30°С, и впрыскиваемый объем составил 20 мкл. Калибровочные кривые были построены в диапазоне от 0,48 до 8000 нг/мл (r2 > 0,999). Расчетный предел количественного определения составил 1,3 нг/мл.
Для проведения исследования наночастицы солюбилизировали в ацетонитриле (1:10, об./об.). Образцы переносили во флаконы авто дозатора, закрывали и помещали в автодозатор ВЭЖХ. Каждый образец исследовали в трех повторах, и результаты выражали в виде количества камптотецина (в мкг) на мг наночастиц.
5.2 Результаты
Основные физико-химические характеристики различных составов незагруженных наночастиц приведены в таблице 5.

* Содержание лиганда (молекулы, содержащей концевые гидроксильные группы) в полимерных конъюгатах рассчитывали с помощью1Н-ЯМР.
Следует отметить, что содержание молекулы с концевыми гидроксильными группами (лиганд) в наночастицах мПЭГ2-кон. значительно выше по сравнению с остальной частью наночастиц полимерных конъюгатов. Этот результат является логичным, поскольку исходное количество мПЭГ2, использованного для получения сложноэфирного полимерного конъюгата PVM/MA, значительно выше, чем ПЭГ2, ПЭГ6 или ПЭГ10.
Определение характеристик наночастиц, загруженных паклитакселом
Основные физико-химические характеристики различных составов наночастиц, загруженных паклитакселом (ПЭГ-РТХ), приведены в таблице 6. В первую очередь, наночастицы, содержащие конъюгат ПЭГ2 (сложноэфирный полимерный конъюгат PVM/MA с ПЭГ2), имеют большие размеры, чем наночастицы, содержащие конъюгат мПЭГ2 (сложноэфирный полимерный конъюгат PVM/MA с мПЭГ2). Размеры конъюгата ПЭГ2 были близки к 400 нм, в то время как для другого состава, содержащего конъюгат мПЭГ2, размеры были меньше и составляли приблизительно 300 нм.
В отношении дзета-потенциала было установлено, что загруженные наночастицы, содержащие конъюгат мПЭГ2, имели несколько более отрицательный поверхностный заряд, приблизительно -38 мВ. Наночастицы, изготовленные с использованием конъюгата ПЭГ2, имели поверхностный заряд приблизительно -33 мВ. Помимо этого, было рассчитано, что выход способа составляет 70% для обоих составов. Рассчитанное количество РТХ, загруженного в наночастицы, для конъюгатов мПЭГ2 и ПЭГ2 составило приблизительно 160 мкг/мг НЧ.
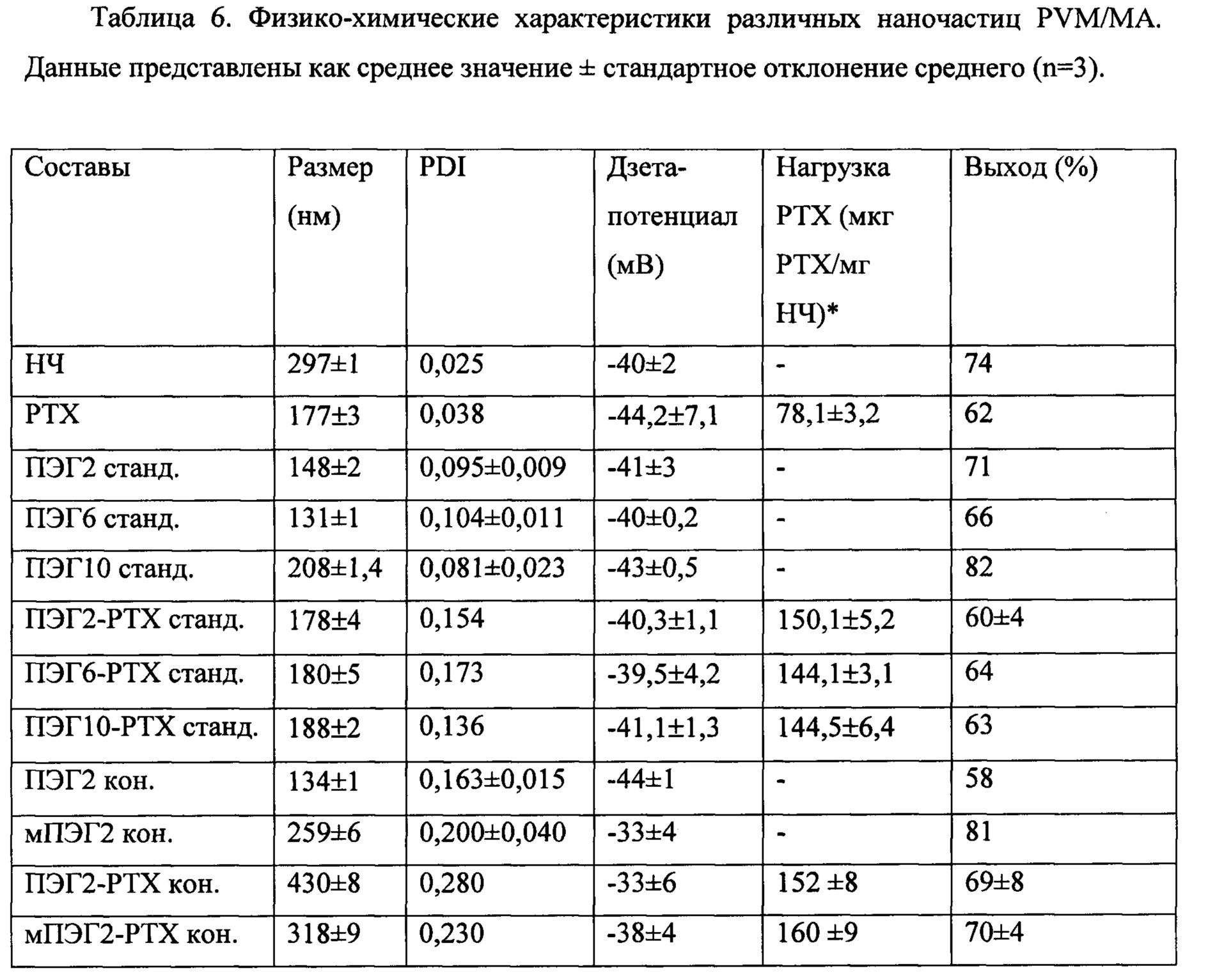
Сравнение выхода наночастиц ПЭГ2-РТХ станд. и составов ПЭГ2-РТХ кон. указывает на то, что процент образованных наночастиц выше в случае использования сложноэфирного полимерного конъюгата PVM/MA с ПЭГ2.
Определение характеристик наночастиц, загруженных доцетакселом
Наночастицы конъюгатов успешно получали методом замещения растворителя. Основные физико-химические характеристики различных составов наночастиц, загруженных доцетакселом (ПЭГ-DTX), приведены в таблице 7. В первую очередь, загруженные наночастицы, содержащие конъюгат ПЭГ2 (сложноэфирный полимерный конъюгат PVM/MA с ПЭГ2), имели большие размеры, чем загруженные наночастицы конъюгата мПЭГ2 (сложноэфирного полимерного конъюгата PVM/MA с мПЭГ2). Размеры ПЭГ2 были близки к 400 нм, в то время как для другого состава, содержащего конъюгат мПЭГ2, размеры были меньше и составляли приблизительно 300 нм.
В отношении дзета-потенциала было установлено, что загруженные наночастицы, содержащие мПЭГ2, имели несколько более отрицательный поверхностный заряд, приблизительно -39 мВ. Наночастицы, изготовленные с применением конъюгата ПЭГ2, имели поверхностный заряд приблизительно -33 мВ.
Кроме того, было рассчитано, что выход способа составляет 60% для обоих составов. Рассчитанное количество доцетаксела, загруженного в наночастицы, для конъюгатов мПЭГ2 и ПЭГ2 составило приблизительно 100 мкг/мг НЧ.

Характеристика наночастиц, загруженных камптотецином
Физико-химические характеристики полученных наночастиц, содержащих камптотецин, представлены в таблице 8. Когда наночастицы были загружены камптотецином в наночастицах мПЭГ2 кон. (сложноэфирный полимерный конъюгат PVM/MA с мПЭГ2), средний размер составил приблизительно 195 нм. Индекс полидисперсности (PDI) был ниже 0,3, что подразумевает однородные составы. В отношении дзета-потенциала наночастиц было установлено, что составы были образованы наночастицами с отрицательными поверхностными зарядами приблизительно -36 мВ. Кроме того, было рассчитано, что выход способа составляет 70%. В отношении нагрузки лекарственного препарата было установлено, что количество камптотецина, инкапсулированного в наночастицы из конъюгатов, достигло 11 мкг/мг. Количество камптотецина, загруженного в стандартные пегилированные наночастицы, было несколько выше для ПЭГ2-СРТ, чем для стандартных наночастиц ПЭГ6-СРТ и достигло 9 мкг/мг.
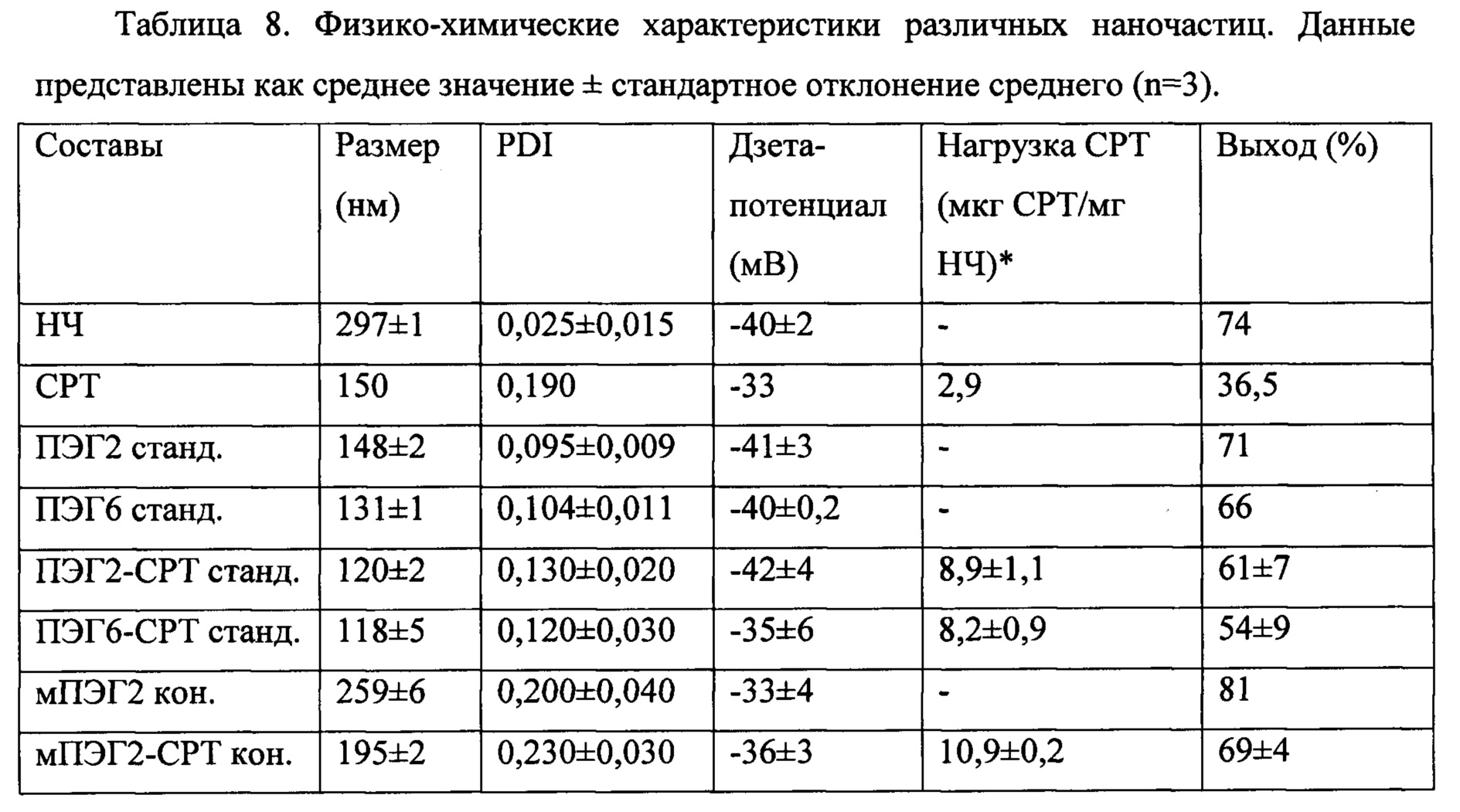
Морфологический анализ методом сканирующей электронной микроскопии (фигура 3) выявил, что все типы наночастиц полиангидрида состояли из гомогенной популяции сферических частиц, размер которых был аналогичен размеру, зарегистрированному с помощью фотонной корреляционной спектроскопии. Поверхность наночастиц оказалась гладкой, без видимых грубых участков на их поверхности.
ПРИМЕР 6
Исследования доцетаксела в условиях in vitro и in vivo
6.1 Исследования высвобождения доцетаксела в условиях in vitro
Эксперименты по высвобождению проводили в условиях достаточного разбавления при 37°С с применением модулируемого желудочного сока (SGF; рН=1,2; пепсин 0,32% масс./об.) и кишечной жидкости (SIF; рН=6,8; панкреатин 1% масс./об.), содержащей 0,5% полисорбата 80 (твин® 80) в качестве солюбилизирующего агента для доцетаксела. Исследования проводили при перемешивании в инкубаторе-встряхивателе Vortemp 56™ (Labnet International Inc., Нью-Джерси, США) после диспергирования наночастиц в соответствующей среде.
Для каждой временной точки 50 мкг доцетаксела, загруженного в наночастицы, ресуспендировали в 2 мл соответствующей модулируемой жидкости. Составы для количественных исследований включали наночастицы DTX (стандартные наночастицы PVM/MA, инкапсулирующие доцетаксел), ПЭГ2-DТХ станд. (стандартные наночастицы PVM/MA, пегилированные с применением ПЭГ2, инкапсулирующие доцетаксел), мПЭГ-2-DTX кон. (наночастицы из сложноэфирного полимерного конъюгата PVM/MA с мПЭГ2, инкапсулирующие доцетаксел) и ПЭГ2-DТХ кон. (наночастицы из сложноэфирного полимерного конъюгата PVM/MA с ПЭГ2, инкапсулирующие доцетаксел).
Концентрацию наночастиц в среде, в которой происходило высвобождение, корректировали, чтобы оценить условия достаточного разбавления для доцетаксела. Различные составы инкубировали в SGF в течение 2 часов и в SIF в течение 12 часов. В разные моменты времени пробирки с образцами собирали и центрифугировали при 27000g в течение 20 минут. В завершение, супернатанты фильтровали и количество доцетаксела, высвободившееся из составов, определяли количественно методом ВЭЖХ (калибровочные кривые свободного доцетаксела в супернатантах, полученных из SGF и SIF, r2 > 0,999). Профили высвобождения выражали в виде совокупной процентной доли высвобождения и наносили на график в зависимости от времени.
6.2 Фармакокинетические исследования наночастиц с инкапсулированным доцетакселом у мышей линии Balb/c в условиях in vivo
Введение DTX-загруженных наночастиц мышам
Фармакокинетические исследования выполняли на самках мышей линии Balb/c (20-22 г), полученных из Harlan (Барселона, Испания). Исследования проводили в соответствии с этическими принципами и требованиями к проведению исследований на лабораторных животных, утвержденными Комитетом по этике проведения экспериментов на животных исследовательского центра (протокол № Е21-12) в соответствии с европейским законодательством об экспериментах на животных (86/609/EU). Перед проведением экспериментов животные в целях адаптации получали корм в течение 1 недели при свободном доступе к пище и питьевой воде (22±2°С; 12 часовой цикл свет/темнота; 50-60% относительная влажность). Перед пероральным введением составов животные не получали корм в течение ночи, чтобы избежать нежелательного влияния на всасывание, однако имели свободный доступ к воде.
Для фармакокинетического исследования мышей случайным образом распределяли на группы в зависимости от времени отбора образцов крови. Каждая временная точка соответствовала 3 животным. Экспериментальные группы получали наночастицы DTX, ПЭГ2-DТХ станд., мПЭГ2-DТХ кон. и ПЭГ2-DТХ кон. В качестве контролей одна группа животных получала таксотер® внутривенно (в/в), и другая группа получала перорально коммерческий состав. Каждое животное получило эквивалентное количество доцетаксела в дозе 30 мг/кг массы тела перорально с помощью тупой иглы через пищевод в желудок или внутривенно через хвостовую вену.
Образцы крови собирали в установленное время после введения (0, 10 мин, 30 мин, 1 ч, 1,5 ч, 3 ч, 6 ч, 8 ч, 24 ч, 48 ч и 72 ч). ЭДТА использовали в качестве антикоагулирующего агента. Объем крови восстанавливали путем внутрибрюшинного введения равного объема физиологического раствора, предварительно нагретого до температуры тела. Образцы немедленно помещали на лед и центрифугировали при 2500 g в течение 10 минут. Плазму крови отделяли в чистые пробирки и хранили в замороженном виде при -20°С до проведения исследования методом ВЭЖХ.
Количественное определение DTX в образцах плазмы крови методом ВЭЖХ
Количество доцетаксела определяли в плазме крови методом ВЭЖХ-УФ. Калибровочные кривые использовали для преобразования площади под хроматографическим пиком DTX/PTX в концентрацию. Образцы для калибровки и контроля качества получали путем добавления соответствующих объемов стандартного доцетаксела в растворе этанола к плазме крови, не содержащей лекарственного препарата. Калибровочные кривые были построены в диапазоне от 100 до 6250 нг/мл (r2 > 0,999). Аликвоту (200 мкл) плазмы крови смешивали с 25 мкл раствора внутреннего стандарта (паклитаксел, 10 мкг/мл в этаноле, предварительно выпаренный). После перемешивания на вортексе жидкостно-жидкостную экстракцию осуществляли путем добавления 4 мл трет-бутилметилового эфира с последующим осторожным перемешиванием на вортексе (1 мин). Смесь центрифугировали в течение 10 мин при 3000 g, и затем органический слой переносили в чистую пробирку и выпаривали до сухого остатка (Savant, Барселона, Испания). И, наконец, остаток растворяли в 125 мкл раствора для восстановления (ацетонитрил-фосфатный буфер 0,01 М рН=2; 50:50, об./об.) и переносили во флаконы автоматического дозатора, закрывали и помещали в автоматический дозатор ВЭЖХ. На колонку для ВЭЖХ наносили 100 мкл аликвоты каждого образца.
Анализ фармакокинетических данных
Фармакокинетический анализ концентрации в плазме крови в зависимости от временных данных, полученных после введения различных составов DTX, анализировали с применением некомпартментной модели с помощью программного обеспечения WinNonlin 5.2 (Pharsight Corporation, США). Оценивали следующие параметры: максимальная концентрация в плазме крови (Сmax), время до достижения максимальной концентрации (Тmax), площадь под кривой концентрация-время от времени 0 до ∞ (AUC), среднее время удержания (MRT), клиренс (Cl), объем распределения (V) и период полувыведения в конечной фазе (t1/2z).
Помимо этого, относительную биодоступность доцетаксела при пероральном введении, F (%), оценивали с помощью следующего уравнения:

где AUCi.v. и AUCoral соответствовали значениям площади под кривой для внутривенного и перорального введения, соответственно.
Статистический анализ
Данные выражали как среднее значение ± стандартное отклонение среднего для по меньшей мере трех экспериментов. Непараметрический критерий Крускала-Уоллиса с последующим применением U-теста Манна-Уитни использовали для изучения статистических различий. Во всех случаях р<0,05 считали статистически значимым. Обработку всех данных проводили с применением статистического программного обеспечения GraphPad Prism 6.0 (GraphPad Software, Калифорния, США).
6.3 Результаты
Исследования высвобождения доцетаксела в условиях in vitro
Профили высвобождения доцетаксела из составов наночастиц оценивали после их инкубации в двух различных средах: моделируемого желудочного сока и кишечного сока, содержащего 0,5% твин-80 (масс./об.) в качестве солюбилизирующего агента. На фигуре 4 показаны профили высвобождения доцетаксела из наночастиц DTX, составов наночастиц ПЭГ2-DТХ станд., ПЭГ2-DТХ кон. и мПЭГ2-DТХ кон., представленные в виде совокупной процентной доли высвобожденного лекарственного препарата в зависимости от времени. Во всех случаях, когда наночастицы были диспергированы в SGF, не наблюдали высвобождения лекарственного препарата. Напротив, когда наночастицы были диспергированы в SIF, наблюдали высвобождение доцетаксела.
Профиль высвобождения для ПЭГ2-DТХ станд. в SIF характеризовался важным взрывным эффектом приблизительно 75% загруженного лекарственного препарата в первые 30 мин, с последующей более устойчивой фазой высвобождения, длящейся вплоть до 10 часов. В то же время в случае наночастиц DTX кривые высвобождения доцетаксела в SIF указывали на более медленное высвобождение доцетаксела, чем из ПЭГ2-DТХ станд., с последующим более устойчивым высвобождением до конца исследования, когда почти весь загруженный лекарственный препарат высвободился.
Оба состава наночастиц, ПЭГ2-DТХ кон. и мПЭГ2-DТХ кон., имели аналогичные профили высвобождения. В присутствии SIF высвобождение доцетаксела имело двухфазный профиль, который характеризовался быстрым первоначальным взрывным высвобождением вплоть до 90% загруженного лекарственного препарата с последующей более устойчивой фазой высвобождения. Полное высвобождение DTX для всех образцов было достигнуто через 10-11 часов после начала исследования.
Фармакокинетические исследования наночастиц. содержащих доцетаксел, у мышей линии Balb/c
Профили зависимости концентрации доцетаксела в плазме крови от времени после внутривенного введения таксотера (однократной дозы 30 мг/кг) самкам мышей линии Balb/c представлены на фигуре 5. Концентрация лекарственного препарата в плазме крови быстро снижалась с течением времени двухфазным образом, и данные корректировали с учетом некомпартментной модели. Уровни доцетаксела в плазме крови поддавались количественному определению плоть до 12 часов после введения.
На фигуре 6 представлены профили зависимости концентрации в плазме крови от времени после перорального введения доцетаксела (однократная доза 30 мг/кг) мышам линии Balb/c при введении в виде коммерческого препарата таксотера® или в инкапсулированной форме в различных составах наночастиц полиангидрида: в стандартных наночастицах PVM/MA (состав DTX), стандартных наночастицах PVM/MA, пегилированных с применением ПЭГ2 (ПЭГ2-DТХ станд.) или наночастицах из сложноэфирных полимерных конъюгатов PVM/MA с ПЭГ2 (ПЭГ2-DТХ кон.) или с мПЭГ2 (мПЭГ2-DТХ кон.). Было обнаружено, что при пероральном введении коммерческого таксотера® мышам уровни доцетаксела в плазме крови всегда были ниже предела количественного определения аналитической хроматографической методики. Напротив, если доцетаксел был загружен в наночастицы, полученные составы проявляли устойчивые уровни в плазме крови. Во всех случаях наблюдали первоначальное быстрое повышение уровней противоракового препарата в плазме крови в течение первых 1,5-2 часов до достижения Сmax, с последующим медленным снижением, которое длилось в течение по меньшей мере 8 часов для состава DTX (см. фигуру 6А) и приблизительно 70 часов для составов ПЭГ2-DТХ станд., ПЭГ2-DТХ кон. и мПЭГ2-DТХ кон. (см. фигуру 6В). При сравнении ПЭГ2-DТХ станд. с ПЭГ2-DТХ кон. и мПЭГ2-DТХ кон. было установлено, что уровни доцетаксела в плазме крови, достигнутые при использовании наночастиц из сложноэфирных полимерных конъюгатов PVM/MA с ПЭГ2000 и мПЭГ2000, были выше, чем при использовании стандартных наночастиц PVM/MA, пегилированных с применением ПЭГ2000.
В таблице 9 обобщены фармакокинетические параметры, рассчитанные с помощью некомпартментного анализа экспериментальных данных, полученных после введения различных составов доцетаксела мышам. Во-первых, среднее значение AUC для коммерческого лекарственного препарата, который вводили внутривенным путем, составило 143 мкг⋅ч/мл. Максимальная концентрация (Сmax) составила 198 мкг/мл, и Тmax было 0 ч. Значение MRT было 1,4 ч, и расчетный период полувыведения в конечной фазе (t1/2z) кривой составил 1,5 ч.
Однако значения Сmax доцетаксела в наночастицах полиангидрида находились в диапазоне от 1,3 до 2 мкг/мл, ранговый порядок этого параметра был следующим: мПЭГ2-DTX кон. = ПЭГ2-DТХ кон. > ПЭГ2-DТХ станд. > DТХ. Кроме того, Сmax была достигнута при 0 ч после внутривенного введения таксотера® и время, необходимое для достижения Сmax, было больше на 0,8 ч для состава DTX и на 1,5-2 ч для составов мПЭГ2-DТХ кон. и кон. ПЭГ2-DTX.
При сравнении составов стандартных пегилированных наночастиц и составов конъюгатов было установлено, что значения AUC для ПЭГ2-DТХ кон. и мПЭГ2-DТХ кон. были в 1,3 и 1,8 раза выше, соответственно, чем значения AUC, полученные для ПЭГ2-DTX станд., это указывает на более высокую способность таких наночастиц усиливать пероральное всасывание таксана. Помимо этого, среднее время удержания (MRT) лекарственного препарата в плазме крови и его период полувыведения в конечной фазе (t1/2z) были значительно дольше при пероральном введении в виде составов сложноэфирных полимерных конъюгатов (мПЭГ2-DТХ кон., ПЭГ2-DТХ кон.).


AUC: площадь под кривой концентрация-время от времени 0 до ∞; Сmax: максимальная концентрации в плазме крови; Тmax: время до достижения максимальной концентрации в плазме крови; t1/2z: период полувыведения в конечной фазе; Сl: клиренс; V: объем распределения; MRT: среднее время удержания; Fr: относительная биодоступность при пероральном введении. ND: не детектировали.
Аналогичным образом, объем распределения (V) противоракового лекарственного препарата, загруженного в наночастицы конъюгатов (225 мл для ПЭГ2-DТХ кон. и 240 мл для мПЭГ2-DTX кон.), был выше, чем при внутривенном введении лекарственного препарата в виде таксотера® (8 мл), и выше, чем при пероральном введении лекарственного препарата, инкапсулированного в стандартных наночастицах PVM/MA (15 мл) или стандартных пегилированных наночастицах PVM/MA (192 мл). Напротив, клиренс доцетаксела всегда был одинаков и не зависел от состава и способа введения.
И, наконец, было рассчитано, что относительная биодоступность доцетаксела при пероральной доставке в стандартных пегилированных наночастицах (ПЭГ2-DТХ станд.) составила приблизительно 32%. Было установлено, что биодоступность при пероральном введении для непегилированных наночастиц составляет лишь 5%. Значения, полученные для составов конъюгатов, были высокими и варьировались от 40 до 57% для ПЭГ2-DТХ кон. и мПЭГ2-DТХ кон. Неожиданным фактом явилось то, что составы, полученные из сложноэфирных полимерных конъюгатов, повысили относительную биодоступность доцетаксела при пероральном введении.
Подводя итоги, следует отметить, что составы ПЭГ-содержащих стандартных и конъюгированных наночастиц были пригодны для нагрузки доцетакселом и обеспечивали подходящие характеристики для их перорального введения. При пероральном введении указанные наночастицы обеспечивали длительные и устойчивые уровни доцетаксела в плазме крови в течение 3 дней. Однако для «лишенных оболочки» наночастиц концентрация доцетаксела в плазме крови была высокой в начальных условиях, но быстро снижалась, и через 12 ч после введения не удалось обнаружить поддающиеся количественному определению уровни. Кроме того, фармакокинетические исследования выявили более высокую способность конъюгированных наноносителей повышать биодоступность доцетаксела при пероральном введении, особенно для мПЭГ2-DТХ кон., при использовании которых биодоступность при пероральном введении достигала почти 57%, т.е. в 1,8 раза выше, чем для стандартных пегилированных наночастиц.
ПРИМЕР 7
Исследования камптотецина в условиях in vitro и in vivo
7.1 Исследования высвобождения камптотецина в условиях in vitro
Эксперименты по высвобождению проводили в условиях достаточного разбавления при 37°С с применением моделируемого желудочного сока (SGF; рН=1,2; пепсин 0,32% масс./об.) и кишечной жидкости (SIF; рН=6,8; панкреатин 1% масс./об.). Исследования проводили при перемешивании в инкубаторе-встряхивателе Vortemp 56™ (Labnet International Inc., Нью-Джерси, США) после диспергирования наночастиц в соответствующей среде.
Для каждой временной точки 0,8 мкг камптотецина, загруженного в стандартные пегилированные наночастицы PVM/MA с ПЭГ2 (ПЭГ2-СРТ станд.) или ПЭГ6 (ПЭГ6-СРТ станд.) и в наночастицы из сложноэфирных полимерных конъюгатов PVM/MA с мПЭГ2 (мПЭГ2-СРТ кон.), ресуспендировали в 1 мл соответствующей моделируемой жидкости. Различные составы инкубировали в SGF в течение 2 ч и в SIF в течение 14 часов. В различных временных точках образцы собирали и центрифугировали в пробирках Vivaspin (300000 НОММ, Sartorius group, Германия) при 3000 g в течение 5 мин. Количество камптотецина, высвободившегося из составов, количественно определяли с помощью ВЭЖХ (калибровочные кривые свободного камптотецина в супернатантах, полученных из SGF и SIF, r2 > 0,999).
7.2 Фармакокинетические исследования наночастиц с инкапсулированным камптотецином у крыс линии Wistar в условиях in vivo
Введение крысам наночастиц. загруженных камптотецином
Фармакокинетические исследования проводили на самцах крыс линии Wistar, полученных из Harlan (Барселона, Испания). Исследования проводили в соответствии с этическими принципами и требованиями к проведению исследований на лабораторных животных, утвержденными Комитетом по этике проведения экспериментов на животных исследовательского центра (протокол №058-12) в соответствии с европейским законодательством об экспериментах на животных (86/609/EU).
Перед проведением экспериментов животные в целях адаптации получали корм в течение 1 недели при свободном доступе к пище и питьевой воде (22±2°С; 12 часовой цикл свет/темнота; 50-60% относительная влажность). Перед пероральным введением составов животные не получали корм в течение ночи, чтобы избежать нежелательного влияния на всасывание, однако имели свободный доступ к воде.
Для фармакокинетического исследования крыс случайным образом распределяли на 2 группы (n=6). В качестве контроля одна группа животных получала перорально дозу 1 мг/кг суспензии камптотецина (с помощью тупой иглы через пищевод в желудок), и другая группа получала суспензию камптотецина внутривенно (через хвостовую вену). Экспериментальные группы получали перорально эквивалентную дозу камптотецина, изготовленного в виде наночастиц мПЭГ2-СРТ кон. и ПЭГ2-СРТ станд.
Образцы крови собирали в установленное время после введения (0, 0,5, 1, 2, 4, 6, 8, 24, 30 и 48 ч). ЭДТА использовали в качестве антикоагулирующего агента. Объем крови восстанавливали путем внутрибрюшинного введения равного объема физиологического раствора, предварительно нагретого до температуры тела. Образцы немедленно помещали на лед и центрифугировали при 2500 g в течение 10 мин. Плазму крови отделяли в чистые пробирки и хранили в замороженном виде при -20°С до проведения исследования методом ВЭЖХ.
Количественное определение камптотецина в образцах плазмы крови методом ВЭЖХ
Количество камптотецина определяли в плазме крови методом ВЭЖХ-ДФ с помощью хроматографической системы серии Agilent 1100 и детектора флуоресценции с установленной длиной волны возбуждения и испускания 380 и 418 нм, соответственно. Хроматографическая система была оснащена обращенно-фазовой колонкой Phenomenex Gemini C18 150 мм × 3 мм (размер частиц 5 мкм) и предварительной колонкой (Phenomenex Security Guard C18). Подвижная фаза, нагнетаемая со скоростью 1 мл/мин, состояла из смеси ацетонитрила и раствора трифторуксусной кислоты 0,01% (об./об.) в соотношении 50:50 (об./об.). Колонку помещали при температуре 30°С, и впрыскиваемый объем составил 100 мкл. Калибровочные кривые и кривые для контроля качества были построены в диапазоне от 0,48 до 8000 нг/мл (r2 > 0,999) путем добавления к плазме крови, не содержащей лекарственного препарата, соответствующих объемов стандартного камптотецина в смеси диметилсульфоксида/ацетонитрила/трифторуксусной кислоты в соотношении 1:8,9:0,1 (об./об./об.).
Аликвоту (100 мкл) плазмы крови смешивали с 4 объемами ацетонитрила и встряхивали в течение 2 минут, чтобы осадить белки плазмы. После центрифугирования (5000 g, 5 минут) супернатант собирали и выпаривали до сухого остатка (Savant, Барселона, Испания). Затем остаток растворяли в 120 мкл раствора для восстановления, содержащего диметилсульфоксид/ацетонитрил/трифторуксусную кислоту в соотношении 1:8,9:0,1 (об./об./об.), и переносили во флаконы автодозатора, закрывали и помещали в автодозатор ВЭЖХ. Было вычислено, что предел количественного определения составляет 2,6 нг/мл с относительным стандартным отклонением 4,6%.
Анализ фармакокинетических данных
Фармакокинетический анализ концентраций в плазме крови в зависимости от времени, полученных после введения различных составов камптотецина, анализировали с применением некомпартментной модели с помощью программного обеспечения WinNonlin 5.2 (Pharsight Corporation, США). Оценивали следующие параметры: максимальная концентрация в плазме крови (Сmax), время до достижения максимальной концентрации (Тmax), площадь под кривой концентрация-время от времени 0 до ∞ (AUC), среднее время удержания (MRT), клиренс (Cl), объем распределения (V) и период полувыведения в конечной фазе (t1/2z).
Кроме того, относительную биодоступность камптотецина при пероральном введении, Fr, выражали как соотношение площадей под кривой концентрация-время от времени 0 до ∞ (AUC) для составов в количественном исследовании и одной из введенных пероральных суспензий СРТ.
Статистический анализ
Для физико-химических и фармацевтических характеристик составов данные выражали как среднее значение ± стандартное отклонение среднего (стандартная ошибка среднего значения) по меньшей мере трех экспериментов.
Проводили статистический анализ фармакокинетических параметров в условиях in vivo. Непараметрический критерий Крускала-Уоллиса с последующим применением U-теста Манна-Уитни использовали для изучения статистических различий. Во всех случаях значения р<0,05 рассматривали как статистически значимые различия. Обработку всех данных проводили с применением статистического программного обеспечения GraphPad Prism 5.0 (GraphPad Software, США).
7.3 Результаты
Исследование высвобождения камптотецина в условиях in vitro
Кинетику высвобождения камптотецина из наночастиц оценивали в моделируемом желудочном и кишечном соках (содержащих полисорбат 80 в качестве солюбилизирующего агента для камптотецина) (фигура 7). Полученный профиль характеризуется первой стадией без высвобождения, когда наночастицы диспергировали в SGF, и стадией высвобождения (когда наночастицы диспергировали в SIF), при которой лекарственный препарат быстро высвобождался в начальный момент времени с последующей фазой более устойчивого высвобождения. Для стандартных пегилированных наночастиц (ПЭГ2-СРТ станд. и ПЭГ6-СРТ станд.) более быстрое высвобождение наблюдали в течение первых 5 часов (после инкубации в SIF), и последняя фаза устойчивого высвобождения протекала в течение 11 часов до высвобождения 100% загруженного лекарственного препарата из наночастиц. Кинетика высвобождения камптотецин из СРТ мПЭГ2 кон. выявила профиль высвобождения, который может быть разделен на три фазы. В первой из них, когда наночастицы диспергировали в SGF в течение первых 2 ч, количество высвобожденного камптотецина было очень низким (приблизительно 10%). Затем, когда наночастицы переносили в SIF, наблюдали взрывное высвобождение. Следовательно, приблизительно 90% исходного загруженного лекарственного препарата подвергалось взрывному высвобождению из состава наночастиц. И, наконец, третья фаза характеризовалась медленным и устойчивым высвобождением оставшегося лекарственного препарата, который полностью высвобождался в течение последующих 12 часов эксперимента (через 14 часов после начала исследования).
Фармакокинетические исследования наночастиц, содержащих камптотецин, у крыс линии Wistar
Профиль зависимости концентрации камптотецина в плазме крови от времени после однократного внутривенного введения в дозе 1 мг/кг представлен на фигуре 8. Камптотецин вводили в виде суспензии с размером частиц 1500±144 нм и PDI 0,44±0,05. Данные анализировали с помощью некомпартментной модели. Концентрация камптотецина в плазме крови быстро снижается после введения, и через 6 часов после введения не поддается детектированию. После введения концентрация лекарственного препарата в плазме крови достигла 430 нг/мл (Сmax), и значения AUC и t1/2z составили 0,39 мкг ч/мл и 0,69 ч, соответственно. Рассчитанные значения клиренса и объема распределения лекарственного препарата были 755 мл/ч и 683 мл, соответственно (таблица 10).
На фигуре 9 представлены профили зависимости концентрации камптотецина в плазме крови после введения однократной пероральной дозы (1 мг/кг) самцам крыс линии Wistar в виде суспензии или в виде загруженных наночастиц. При пероральном введении суспензии лекарственного препарата уровни в плазме крови быстро увеличивались, достигая Сmax через 30 мин после введения. Затем устойчивые уровни лекарственного препарата в плазме крови поддерживались в течение не менее 4 часов, после этого уровни камптотецина быстро уменьшались. Соответственно, через 10 часов после введения уровни камптотецина в плазме крови не поддавались детектированию.
Для камптотецина, загруженного в наночастицы и введенного в аналогичной дозе (1 мг/кг), основное различие с водным суспензионным составом заключалось в том, что уровни лекарственного препарата в плазме крови поддавались количественному определению в течение более длительного периода времени. Соответственно, животные, которые получали камптотецин, включенный в стандартные пегилированные наночастицы (ПЭГ2-СРТ станд.), имели первую фазу, характеризующуюся повышенными уровнями лекарственного препарата в плазме крови в течение первых 1,5 ч, с последующей фазой медленного и длительного снижения уровней, при этом уровни камптотецина поддавались количественной оценке вплоть до 24 ч. Было обнаружено, что значение AUC примерно в 2 раза выше, чем при пероральном введении лекарственного препарата в виде суспензии. Помимо этого, несмотря на то, что статистически значимые различия не были обнаружены, объем распределения лекарственного препарата при введении в виде пегилированных наночастиц был в 1,5 раз выше, чем объем распределения одного из непокрытых лекарственных препаратов при пероральном введении в виде суспензии.
Для наночастиц из сложноэфирных полимерных конъюгатов PVM/MA (мПЭГ2-СРТ кон.), введенных в аналогичной дозе (1 мг/кг), начальный профиль кривых концентраций в плазме крови был практически аналогичен; однако количество лекарственного препарата было выше, чем для обычного состава и, что более важно, уровни в плазме крови поддерживались вплоть до 48 ч после введения. В целом, уровни камптотецина, полученные в плазме крови после перорального введения мПЭГ2-СРТ кон., были выше, чем для водной суспензии и для стандартных пегилированных наночастиц (ПЭГ2-СРТ станд.), при этом значения AUC были выше в 7,6 раза и 4,2 раза, соответственно.
В таблице 10 обобщены основные фармакокинетические параметры, которые оценивали с помощью некомпартментного анализа экспериментальных данных, полученных после введения различных составов крысам. Было установлено, что значение AUC для камптотецина в мПЭГ2-СРТ кон. значительно выше (р<0,05), чем для водной суспензии лекарственного препарата. Другое различие между наночастицами и суспензией заключалось в среднем времени удержания лекарственного препарата (MRT). В этом случае значения MRT существенно различались (р<0,01) между непокрытым лекарственным препаратом и мПЭГ2-СРТ кон. (в 10 раз выше), однако не было обнаружено каких-либо существенных различий между контролем и ПЭГ2-СРТ станд. Аналогичным образом, период полувыведения в конечной фазе (t1/2z) также был значительно дольше, когда камптотецин был инкапсулирован в мПЭГ2-СРТ кон., чем при изготовлении в виде обычной суспензии, но не при изготовлении в виде наночастиц ПЭГ2-СРТ станд. В соответствии с этим результатом клиренс лекарственного препарата при введении в виде мПЭГ2-СРТ кон. был приблизительно в 11 раз ниже, чем значение, полученное при введении в виде суспензии (р<0,01). Аналогичным образом, при сравнении состава мПЭГ2-СРТ кон. с ПЭГ2-СРТ станд. было установлено, что значение AUC существенно выше (р<0,05), и статистически значимые различия также были обнаружены в отношении MRT и t1/2z (р<0,01).



*U-тест Манна-Уитни мПЭГ2-СРТ кон. по сравнению с пероральной суспензией камптотецина (р-значение <0,05).
**U-тест Манна-Уитни СРТ мПЭГ2 кон. по сравнению с пероральной суспензией камптотецина (р-значение <0,01).
AUC: площадь под кривой концентрация-время от времени 0 до ∞; Сmax: максимальная концентрации в плазме крови; Тmax: время до достижения максимальной концентрации в плазме крови; t1/2z: период полувыведения в конечной фазе; Cl: клиренс; V: объем распределения; MRT: среднее время удержания; Fr: относительная биодоступность при пероральном введении.
В заключение следует отметить, что оба типа наночастиц (ПЭГ2-СРТ станд. и мПЭГ2-СРТ кон.), инкапсулирующие камптотецин, оказались пригодными для перорального введения в количественном исследовании in vitro, принимая во внимание, что высвобождение лекарственного препарата отсутствовало до тех пор, пока их не инкубировали в условиях, моделирующих таковые в кишечнике. Однако в количественных исследованиях в условиях in vivo мПЭГ2-СРТ кон. проявили существенно более высокую биологическую доступность, которая в 4,3 раза превышала таковую для ПЭГ2-СРТ станд.
ПРИМЕР 8
Исследования биораспределения наночастиц
Для того чтобы визуализировать и оценить распределение и способность наночастиц, содержащих матрикс из сложноэфирного полимерного конъюгата PVM/MA, взаимодействовать со слизистой оболочкой кишечника, проводили исследования с применением метода флуоресцентной микроскопии.
8.1 Получение наночастиц, загруженных люмогеном
Пустые стандартные наночастицы PVM/MA и наночастицы, содержащие матрикс из сложноэфирного полимерного конъюгата PVM/MA с метоксиполиэтиленгликолем 2000 (мПЭГ2) кон., флуоресцентно метили люмогеном красным.
Наночастицы, загруженные люмогеном, получали путем добавления 0,5 мг люмогена красного® в ацетоновой фазе (5 мл), содержащей PVM/MA (гантрез® AN) или его сложноэфирный полимерный конъюгат с мПЭГ2 (мПЭГ2-LUM кон.), перед образованием наночастиц, как описано в примерах 1 и 4. После этого получали составы LUM и мПЭГ2-LUM кон., соответственно.
8.2 Исследования распределения наночастиц. загруженных люмогеном, в слизистой оболочке кишечника крыс линии Wistar в условиях in vivo
Исследования биораспределения проводили с применением флуоресцентномеченых наночастиц, инкапсулирующих люмоген красный.
Эксперименты на животных проводили в соответствии с протоколом, утвержденным Комитетом по этике и биобезопасности исследовательского центра в соответствии с европейским законодательством об экспериментах на животных (протокол №059-13). Однократную дозу 1 мл водной суспензии, содержащей 10 мг флуоресцентномеченых наночастиц, вводили перорально самцам крыс линии Wistar. Через два часа животных умерщвляли и внутренности удаляли. Части подвздошной кишки размером 1 см собирали, хранили в среде для исследования ткани Tissue-Tek® ОСТ и замораживали при -80°С. Каждую часть затем нарезали на срезы толщиной 5 мкм на криостате и прикрепляли к стеклам. В завершение, полученные образцы фиксировали формальдегидом и инкубировали с DAPI (4',6-диамидино-2-фенилиндол) в течение 15 минут до наложения покровного стекла.
Присутствие наночастиц, загруженных флуоресцентным красителем, в слизистой оболочке кишечника визуализировали на флуоресцентном микроскопе (Axioimager M1, Zeiss), оснащенном камерой (Axiocam ICc3, Zeiss) и источником света для возбуждения флуоресценции (НВО 100, Zeiss).
8.3 Результаты
Наночастицы, загруженные люмогеном, проявляли физико-химические свойства, которые были сходны со свойствами, определенными для пустых наночастиц: 215 нм, -36 мВ для LUM-наночастиц и 205 нм, -35 мВ для составов мПЭГ2-LUM кон.
Неожиданным было обнаружено, что наночастицы мПЭГ2-LUM кон. показали различное распределение в слизистой оболочке подвздошной кишки, по сравнению с наночастицами LUM. Обычные наночастицы PVM/MA, по всей видимости, застревали в слизистом слое, покрывающем эпителий кишечника (фигура 10А, 10В), тогда как наночастицы из сложноэфирных полимерных конъюгатов PVM/MA были способны пересекать слизистый слой и тесно взаимодействовать с поверхностью энтероцитов (фигуры 10С, 10D). Этот результат подтверждает, что поверхность наночастиц из сложноэфирных полимерных конъюгатов отличается от исходного PVM/MA. Можно предположить, что в процессе образования наночастиц гидрофильные участки конъюгатов будут ориентированы в водную фазу, образуя гидрофильные короны на поверхности наночастиц, которые будут придавать «скользящие» свойства полученным наночастицам, и, следовательно, возможность пересечения слизистого слой и достижения поверхности энтероцитов.
Реферат
Группа изобретений относится к области органической химии и фармацевтической промышленности, а именно: к способу получения наночастицы, к наночастице для доставки лекарственного средства, полученной указанным способом, и к противоопухолевой фармацевтической композиции, содержащей по меньшей мере одну такую наночастицу совместно с фармацевтически приемлемым носителем или наполнителем. Предложенный способ включает следующие стадии: а) сложноэфирный полимерный конъюгат поли(метилвинилового эфира-малеинового ангидрида) с полиэтиленгликолем или с полиоксиэтилен-С-С-алкиловым эфиром смешивают в органической среде с биологически активным соединением, которое является противоопухолевым агентом, выбранным из доцетаксела, паклитаксела и камптотецина; b) затем осуществляют десольватацию указанного конъюгата путем добавления спирта и воды в присутствии двухвалентного металла к смеси, полученной на стадии a). Группа изобретений обеспечивает повышение биодоступности противоопухолевого агента через слизистую оболочку кишечника, что обеспечивает пролонгированный и устойчивый уровень в плазме крови в течение длительного периода времени. 3 н. и 10 з.п. ф-лы, 8 пр., 10 табл., 14 ил.
Комментарии