Водная суспензия, содержащая микрокристаллы фармакологически активного водонерастворимого лекарственного вещества, покрытые мембранообразующим липидом (варианты), и лиофилизированный препарат водной суспензии - RU2100030C1
Код документа: RU2100030C1
Чертежи













Описание
Изобретение относится к формам подачи, обеспечивающим впрыскивание высоких концентраций водонерастворимых лекарственных препаратов млекопитающим и предоставляющим постоянное выделение вводимого лекарства. Настоящее изобретение показывает, что кристаллические водонерастворимые лекарственные препараты могут быть измельчены до субмикронных размеров и суспендированы в водных средах при высоких концентрациях в фармацевтической форме для впрыскивания посредством покрытия мембранообразующим липидом. Это покрытие обычно является фосфолипидом, но может быть получено из любого мембранообразующего липида. Микрокристалл покрывается слоем мембранообразующего липида, который стабилизирует микрокристалл путем гидрофобного, а также гидрофильного взаимодействий. Цепочки жирного ацила в фосфолипиде стабилизируют микрокристалл за счет их взаимодействия с водой. Покрытый микрокристалл может быть дополнительно стабилизирован за счет обволакивания липидом в виде двойного слоя и посредством включения избытка мембранообразующего липида в суспендирующую среду в виде пузырьков. Этот препарат совместим с тканями и обеспечивает постоянное выделение лекарства при внутримышечной, внутрикожной, подкожной инъекции или инъекций в другие ограниченные ткани или пространства (внутрисуставного, эпидурального, внутрибрюшного и др.). Препарат способен давать быстрое выделение при инъекции в кровь более или менее ограниченного отделения, в котором ожидается быстрое разбавление. Этот препарат может инъектироваться внутрь пузыря, внутрь поражения, для того чтобы образовались высокие местные дозы, без вовлечения остальной системы. Это изобретение отличается от существующих систем подачи лекарства за счет способности к инъекции, совместимости с тканями, малого размера частиц, высокой нагрузки способности к впрыскиванию и стабильности при хранении, использования фосфолипида в качестве единственного покрывающего материала и неантигенности.
Водорастворимые лекарственные препараты вводятся легко инъекцией. Нерастворимые в воде лекарства не вводятся инъекцией. Для нерастворимых в воде (или растворимых в масле) медикаментов создание форм, которые можно инъектировать, представляет собой существенную проблему. В фармацевтике имеется много примеров нерастворимых в воде лекарств, которые должны приниматься перорально, поскольку для них не существует адекватной формы для инъекции. Уровень техники ограничивается в смысле концентраций лекарственного препарата и общего объема, который может быть введен инъекцией. Использование, описанное в известном уровне техники, ограничивается проблемами местного раздражения, разрушения и др. (при внутримышечной инъекции) и тромбофлебитом, тромбоэмболизмом, блокировкой легочных капилляров и др. (при внутривенной инъекции).
В качестве введения в обсуждаемый уровень техники полезно обсудить критерии, предъявляемые к инъекционным препаратам. Для удовлетворения общим требованиям клинической практики и фармацевтической промышленности способный к инъекции продукт должен удовлетворять следующим критериям.
А. Препарат и его носитель должны быть совместимыми с тканями. Это требование одинаково важно для инъекций в ткани и в систему кровообращения. Инъекция вредных агентов внутрь мышцы может вызвать боль, раздражение, разрушение ткани, клеточные реакции, фиброз или гнойные реакции. Инъекция вредного агента в систему кровообращения может привести к тромбофлебиту, включая повреждение артерии или вены, образование сгустков в артерии или вене и блокировку кровообращения в тканях или легких. Практикуемая в настоящее время стратегия растворения, включающая использование органических растворителей, предельных значений pH и поверхностно-активных веществ, жестко ограничивается этими проблемами.
Б. Композиция не должна содержать частицы с диаметром более 10 мкм. Частицы, имеющие размеры больше, чем 10 мкм могут заблокировать кровеносные капилляры. При введении внутрь артерии они могут располагаться в капиллярах ткани, что вызывает местную ишемию. При внутривенном введении они могут располагаться в легочных капиллярах и вызывать респираторные страдания. Для соблюдения безопасности размер частиц менее 10 мкм должен выполняться и при других намечаемых способах инъекции (например, внутримышечном) из-за опасности ненамеренной внутривенной или внутриартериальной инъекции. Большинство доступных технологий получения лекарств с регулируемым выделением, направленые на пероральное дозирование, неприменимы для инъекций, поскольку они не удовлетворяют этому критерию.
В. Композиция должна обеспечивать инъекцию достаточных количеств лекарственного препарата. Композиция должна содержать высокую концентрацию лекарственного препарата. В качестве примера: если наивысшая концентрация, доступная для лекарства, составляет 2% (масса/объем) или 20 мг/мл, а наибольший практический объем для внутримышечной инъекции человеку составляет 5 мл, то за одну инъекцию вводится только 100 мг лекарства. Если это лекарство достаточно сильное, то это не создает никаких проблем. Однако имеется множество примеров, когда необходимо ввести в организм 1-2 г лекарственного препарата. Для этого потребуется или в 10-20 раз больший объем (непрактично или невозможно) или в 10-20 раз большая концентрация (недостижимая до сих пор).
Г. Композиция должна состоять из компонентов, которые не проявляют аллергических реакций. Это представляет собой специфическую проблему для инъекций в кожу и мышцы. Повторные инъекции внутрь кожи и мышц инородных белков или макромолекул может вызвать иммунную реакцию. Многие композиции с регулируемым выделением, известные из уровня техники, основаны на "пластиках", сшитых сывороточных белках или полимерах, таких как поли(D,L)-молочная кислота.
Д. Для обычного использования система подачи должна иметь высокую нагрузку. Нагрузка может быть определена как отношение массы подаваемого лекарственного препарата к массе носителя или капсулирующего вещества. Например, если в системе подачи используются 10 г воска или полимера, для того чтобы капсулировать 2 г лекарства, то его нагрузка составляет 0,2. Для системы подачи с низкой нагрузкой требуются большие количества капсулирующего вещества. Способность ткани или отделения сосуда к обмену веществ или удалению этих веществ, однако, является низкой, что ограничивает количество лекарства, которое может быть задано.
Е. Композиция должна быть стабильной, грубо гомогенной, способной к впрыскиванию и фармацевтически элегантной и должна сохранять эти свойства в течение разумного времени хранения.
Описанный в этой заявке микрокристалл, покрытый фосфолипидом, является уникальным в том, что он удовлетворяет всем критериям. Он также уникален тем, что, удовлетворяя указанным выше критериям, он обеспечивает возможность инъекции нерастворимых лекарств при столь высоких концентрациях как 40% (масса/объем).
Обзор уровня техники, который представлен как в текущей фармацевтической, так и в клинической практике, а также в патентной и научной литературе, показывает, что существующие системы не удовлетворяют всем 6 указанным выше критериям для инъекционных форм нерастворимых в воде лекарственных препаратов.
Формы, имеющиеся в продаже.
Описания форм, имеющихся в продаже, могут быть получены из обзоров, таких как "Раут Лист" (Бундесвербанд дер Фармацевтишен Индустри, (фирма) 6000 Франкфурт на Майне, Германия), в котором описаны продукты, имеющиеся в Германии, и из справочника "Физишианз Деск Референс" (PDR, фирмы Медикл Экономикс Ко. Инк. г.Ораделл, шт.Нью-Йорк), в котором описаны продукты, имеющиеся в США.
Один подход к проблеме растворения водонерастворимого медикамента состоит в ионизации его с использованием нефизиологического значения pH. В одном продукте тиопенталь подается в виде натриевой соли, которая при добавлении стерильной воды дает щелочной раствор медикамента с концентрацией 0,2-5% который может инъектироваться внутривенно. Перечисленные вредные реакции включают венозный тромбоз или флебит, распространяющийся от места инъекции (Абботт, 1988, PDR, с.556-559).
Другой подход, применяемый только для внутримышечной инъекции, состоял в инъекции раствора вещества в растительном масле. Хотя триглицериды растительного масла являются совместимыми с тканями, объемное масло в теле нелегко поглощается и подвергается метаболизму. Шарики масла "разделяются стенками" путем роста капсулирующей ткани и могут сохраняться в течение месяцев. Из-за проблем масляной гранулемы и серьезной опасности, связанной с ненамеренной внутривенной или внутриартериальной инъекцией, этот подход в значительной степени вытесняется. Остающимся примером является 7,05-ный раствор галоидперидол-деканоата в конопляном масле МакНейл, PDR, с. 1240-1241).
Другим подходом к этой проблеме является солюбилизация лекарства в органическом растворителе. В качестве примера, диазепам (зарегистрированное название "Валиум") растворяется до концентрации 5 мг/мл (0,5%) в растворе 40% пропиленгликоля, 10% этилового спирта, воды и предохранителей. Для внутривенного использования даются предостережения, для того чтобы уменьшить вероятность венозного тромбоза, флебита, местного раздражения и др. о которых сообщается как о вредных реакциях. Этот препарат не является стабильным в отношении разбавления водой (Роше, 1988, PDR, с. 1764-1766).
Из-за местных реакций органических растворителей их использование для людей является сильно ограниченным. Однако это не так при ветеринарном использовании в животноводстве, когда требуются высокие концентрации медикаментов из-за объемных ограничений шприца (20 мл) и других практических соображений. Примером является продажный 10%-ный (масса/объем) подщелоченный раствор окситетрациклина в пропиленгликоле для внутримышечной инъекции крупному рогатому скоту. Он вызывает боль при инъекции и местное повреждение ткани.
Еще одним подходом является солюбилизация или суспендирование медикамента с неионным поверхностно-активном веществом. Примером этого является суспензия ацетата кортизона, содержащая 25 мг/кг или 50 мг/мл ацетата кортизона, 4 мг/мл полисорбата 80, 5 мг/мл натрий-карбоксиметилцеллюлозы и предохранителей в изотоническом растворе соли (Мерк Шарп и Дооме, 1988 PDR, стр. 1297-1299). Указания даны только для внутримышечного использования, когда оральная терапия является невозможной. Предварительный анестезирующий успокаивающий продукт, содержащий 2 мг/мл или 4 мг/мл лоразепама и 0,18 мл/мл полиэтиленгликоля 400 (неионогенное поверхностно-активное вещество) в пропиленгликоле, является доступным для внутримышечной инъекции или внутривенной инъекции после разбавления. Сообщается, что он вызывает боль и жжение в 17% случаев внутримышечной инъекции (Вайет, 1988 PDR, стр. 2258-2259).
Многие системы в этой категории используют жирные кислоты, жиры, фосфолипиды и неионные поверхностно-активные вещества. Щелочные катионные соли жирных кислот являются "мылами", которые имеют тенденцию образовывать мицеллы диаметром не более 5 нм при смешивании с водой. Они также способны покрывать более крупные гидрофобные структуры. При этерификации жирных кислот глицерином (CH2OH-CHOH-CH2OH) образуются моноглицериды (например, моноолеат глицерина), которые являются либо маслами, либо твердыми веществами в чистом виде в зависимости от длины цепочки входящих в них жирных кислот и температуры. В некоторых обстоятельствах они способны образовывать или участвовать в образовании мембранных структур в присутствии избытка воды. При этерификации двух или трех групп-OH глицерина (диглицериды и триглицериды соответственно) это свойство исчезает. Триглицериды являются главной составной частью жира и растительного масла. Они не образуют мембран и не участвуют в мембранных структурах. Фосфолипиды представляют собой 1,2-диациловые эфиры глицерина, в которых положение-3 этерифицировано фосфатом. Они являются основным строительным блоком биологических мембран и являются весьма совместимыми с тканями. Важным и распространенным примером является лецитин (фосфатидилхолин), в котором фосфатная группа этерифицирована холином, что приводит к образованию цвиттерионной полярной головной группы. В присутствии избытка воды фосфолипиды образуют мембраны бимолекулярной толщины. Полярные головные группы ориентированы к воде, цепочки жирного ацила образуют палисадниковую структуру, причем их концы примыкают к центру мембраны. Неионные поверхностно-активные вещества, используемые в лекарственных рецептурах, являются высокомолекулярными полимерами с перемежающимися гидрофобными и гидрофильными сегментами. Часто используемым и цитируемым примером является полиэтиленгликоль, который имеет структуру H(O-CH2-CH2)nOH. Неионные поверхностно-активные вещества способны покрывать и солюбилизировать гидрофобные масла и твердые вещества в водных средах. Они не образуют мембран. Они гидролизуют биологические мембраны и таким образом являются несовместимыми с тканями.
Вретлинд с сотр. (патент США 4073943, 1978) описали применение жировых эмульсий типа используемых для внутривенной подачи, в качестве носителя для внутривенного назначения нерастворимых в воде лекарств. Эта система обладает низкой нагрузкой (г лекарства/г жира), что ограничивает количество лекарства, которое может быть введено (сравни Критерий Д выше).
Хэйнес (патент США 4725442, 1988) описал водные суспензии микрокапель нерастворимых в воде лекарств, покрытых фосфолипидом и способных инъектироваться. Эти медикаменты сами по себе являлись маслами (например, ингаляционными обезболивающими средствами) или были растворены в фармакологически приемлемом масле. Настоящее изобретение предоставляет повышенную нагрузку для кристаллических не растворимых в воде лекарств.
Липосомы, пузырьки, образующиеся из формирующих мембраны фосфолипидов, таких как лецитин, были впервые описаны Бангхэмом, Стандишем и Ваткинсом (в журнале J.Mol.Biol. 1965, т. 13, с.238). Полученные путем гомогенизации липосомы представляют собой многослойные, фосфолипидные с концентрическими двойными слоями мембраны. Липосомы, полученные обработкой звуком, представляют собой маленькие однослойные фосфолипидные пузырьки, как описано Жаунгом (в журнале Biochem. 1969, т.8, с.344). Липосомы обладают способностью удерживать полярные и сильно заряженные молекулы в своем водном окружении. Тот факт, что липосомы представляют собой неантигенные системы подачи (Критерий Г), является общепризнанным. Имеются многочисленные патенты, посвященные их свойствам удерживания и подачи растворимых в воде медикаментов. В меньшем числе патентов было показано, что липосомы способны включать растворимые в масле лекарства, однако нагрузки были низкими. Примерами являются Шранк (патент США 4411894, 1983), Мезей и Нуджент (патент США 4485054, 1984), Дингл и др. (патент США 4427649, 1984) или Абра и Сзока (патент США 4766046, 1988). Большинство медикаментов в указанных выше примерах были мембранно активными агентами, которые, как предполагалось, внедряются в структуру двойного слоя. Типичные нагрузки были меньше, чем 0,1 г/г. Верхним пределом, по-видимому, является значение 0,2 г/г, которое не может быть превзойдено без разрушения палисадниковой структуры фосфолипида в двойном слое. Одним исключением из этого предела является случай, когда само лекарство имеет липидоподобную структуру и способно подвергаться специфическому и преимущественному взаимодействию с фосфолипидами (сравни с патентом США N 4973465 Баурейна и Троуета).
Настоящее изобретение использует фосфолипид для того, чтобы суспендировать не растворимые в воде лекарства, но совершенно другим способом, чем это описано в приведенных выше патентах по липосоме. В этом изобретении скорее нет попытки растворить лекарство в липидном двойном слое, в нем лекарство удерживается в кристаллическом виде, а фосфолипид используется для покрытия кристалла. Фосфолипидный пузырек не является составной частью микрокристалла, покрытого лецитином.
В патентной литературе даны примеры лекарственных препаратов, покрытых воском или "липоидальным материалом". Однако эти примеры относятся к пероральному назначению и в некоторых случаях к местному назначению. Например, тристеарин (триглицерид глицерина и стеариновой кислоты) является обычным компонентом таблеток. Формы, включающие особо малые кристаллы, называются микрокапсулами. Триглицерид или воск, применяемые для покрытия, обычно считаются водоотталкивающими, причем восковые покрытия не придают их водным суспензиям. Покрытые воском микрокапсулы в основном предназначены для перорального использования, при котором не требуется образования стабильных суспензий в водных средах, инъектируемости и совместимости с тканями или малые размеры (Критерии А, Б и Д выше).
В патентной литературе содержится множество примеров препаратов, предназначенных для местного использования и содержащих кристаллические медикаменты, смешанные с глицеро-липидами, такими как моноолеат глицерина (Реллер, патент США N 4219548, 1988). Косметическая литература также предоставляет многочисленные примеры кремов с высоким содержанием воды (см. Ниепер и Мелсунген, патент США 3274063, 1966), в которые могут быть подмешаны кристаллические медикаменты. Эти кремы не могут считаться способными к инъекции, поскольку они представляют собой самоприклеивающуюся массу, которая не распадается с образованием частиц, достаточно мелких, чтобы пройти через кровяные капилляры (Критерий Б). Кроме того, совместимость с тканями и кровью поверхностей, представленных этими местными препаратами, не была продемонстрирована (Критерий А). Рецептуры, содержащие свободные кристаллы (см. Мезей, патент США 4761228, 1988) не были совместимы с тканями и не могли инъектироваться.
Микросферы, содержащие лекарство, внедренное в сферы диаметром 1-200 мкм из термически отвержденного сывороточного альбумина с осажденным внутрь них лекарством, были описаны Золлом (патент США 3937668, 1976). Вариации этого решения были описаны Виддером и Сенайем (патент США 4345588, 1982), Мосиером (патент США 4492720, 1985) и Моррисом (патент США 4331654, 1982). Некоторые примеры включают отвержденную смесь жирной кислоты и неионогенного поверхностно-активного вещества. Не были продемонстрированы совместимость с кровью и тканями, повторная суспензируемость и стабильность этих препаратов.
Описанные Оппенгеймом и др. наночастицы (патент США 4107288, 1978) содержат частицы диаметром 120-660 нм, сформированные желатином, отвержденным глутаровым альдегидом, включающие лекарство при низких нагрузках 0,0067-0,0979 г/г. Коувреур и др. (патент США 4329332, 1982) описал частицы размером менее 500 нм из алкилцианоакрилата, включающие лекарство с низкой нагрузкой 0,0012-0,062 и, по-видимому, стабилизированные поверхностно-активным веществом. Информация о совместимости с кровью или тканями, антигенности и стабильности суспензий не приведена.
Фиг. 1 представляет собой схематическое изображение микрокристалла, покрытого фосфолипидом. Символ 0= относится к фосфолипиду (0 это полярная головка; пара цепочек жирного ацила). Диаметр равен 0,5 мкм (интервал 0,05-10 мкм).
На фиг. 2 представлено изображение области, наблюдаемой с помощью флуоресцентного микроскопа, когда на пластинке распределяются 20% (масса/объем) окситетрациклина, 20% (масса/объем) препарата микрокристаллического яичного лецитина с добавкой красителя Найл Ред. Белый цвет указывает на высокую интенсивность флуоресценции. На верхнем рисунке показан характер флуоресценции окситетрациклина. Более мелкие частицы имеют диаметр примерно 0,2 мкм. На нижнем рисунке показана та же самая область, возбужденная облучением на длине волны красителя Найл Ред. Эта флуоресценция показывает распределение лецитина в препарате.
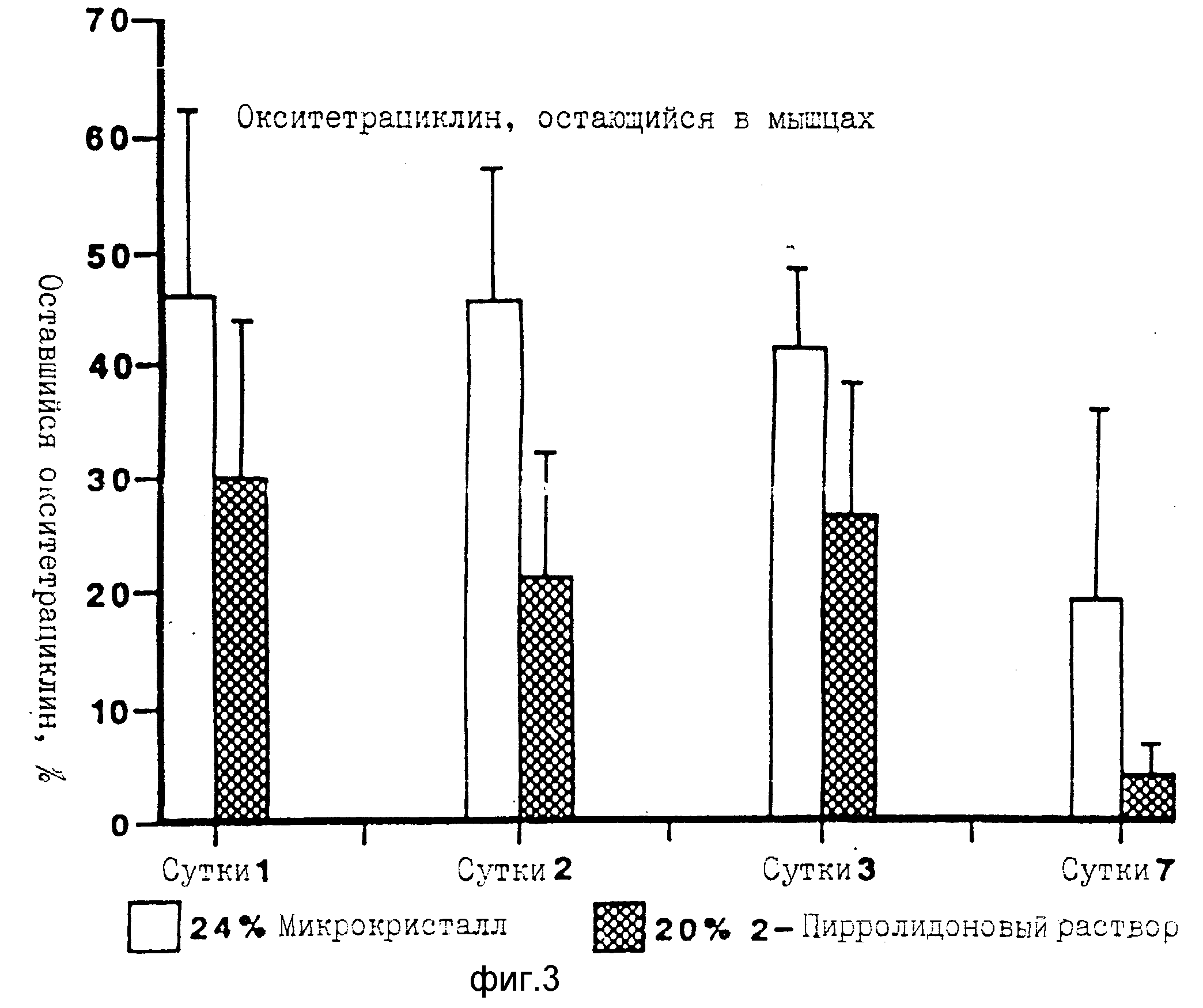
На фиг.3 показана доля окситетрациклина, остающегося в ножной мышце крыс (число их 4) после инъекции 0,1 мл 20% окситетрациклина в микрокристаллической форме, покрытой 5% яичного лецитина. Данные для 1-7 суток после инъекции сопоставлены с результатами для того же самого количества окситетрациклина, инъектированного в виде продажного раствора в 2-метилпирролидоне.
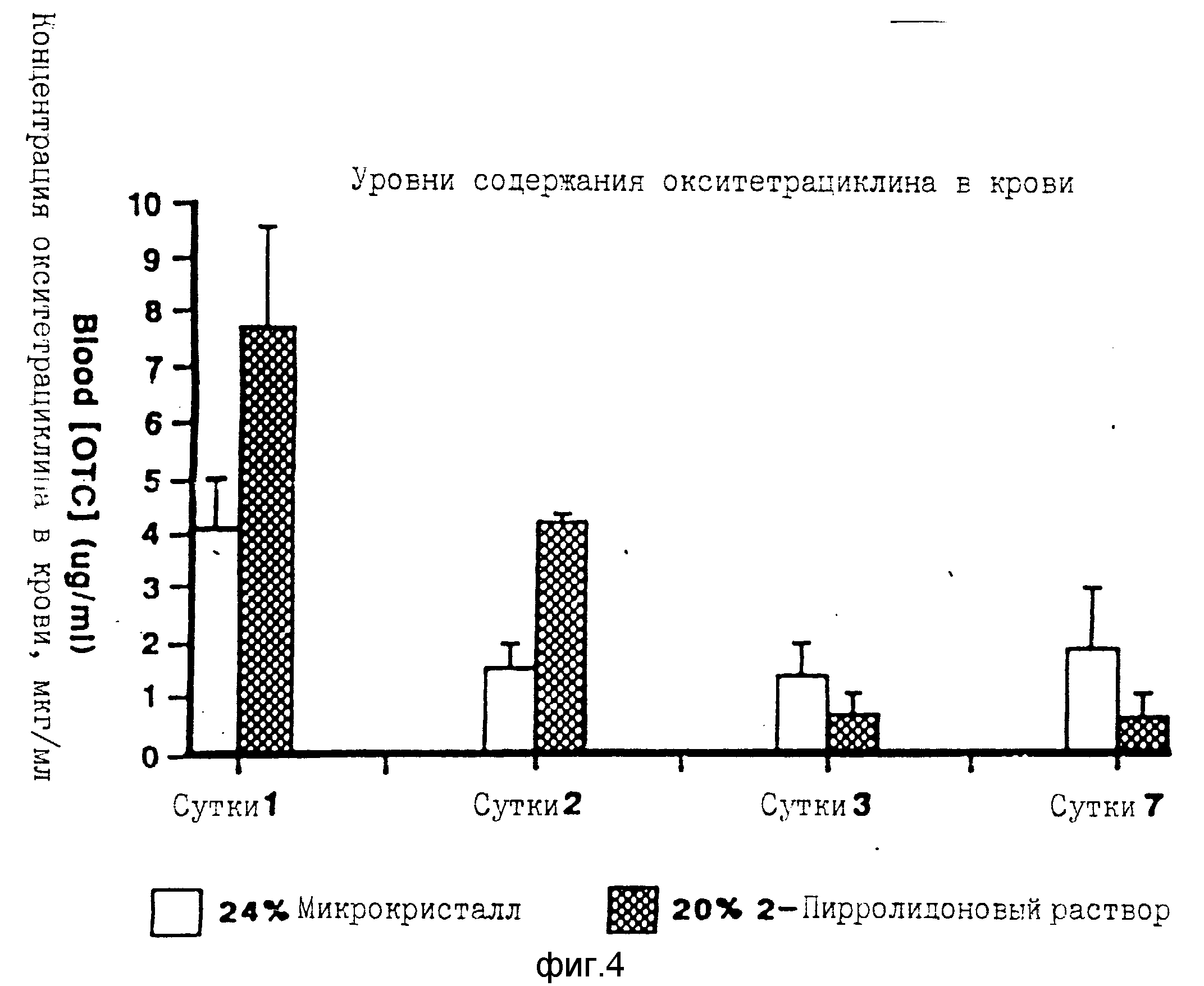
На фиг.4 показаны уровни содержания окситетрациклина в крови центральной артерии в эксперименте фиг.3.
На фиг. 5 показаны концентрации окситетрациклина в крови теленка после внутримышечной инъекции 20% (масса/объем) микрокристаллов окситетрациклина (10% масса/объем, покрыты лецитином).
На фиг.6 показано изменение защитного действия против опухоли во времени за счет 5 мг индометацина, инъектированного внутримышечно в виде микрокристаллов, покрытых лецитином, по сравнению с такой же дозой в виде щелочного раствора.
На фиг.7 показан типичный ход временной зависимости уровня анестезии для крыс, измеренного в виде порога вокализации (в миллиамперах) на внутрикожное электрическое раздражение после внутривенной инъекции микрокристаллов альфаксалона, покрытых лецитином.
На фиг.8 показано изменение во время анестезии кожи человека (булавочный укол), достигаемой с помощью микрокристаллов тетракаин-иодистоводородной кислоты, покрытых лецитином (20% масса/объем) по сравнению с растворами тетракаин HCl (фиг.9).
Это изобретение представляет средство для создания инъектируемых, совместимых с тканями суспензий не растворимых в воде лекарственных препаратов при высоких концентрациях. Это обеспечивает парентеральное (инъекция) назначение лекарственных препаратов. Вообще оно применимо для любого нерастворимого в воде лекарства, которое находится в кристаллическом состоянии при температуре 37oC. Рецептура в виде микрокристаллов, покрытых фосфолипидом, обеспечивает возможность инъекции лекарственных препаратов или иной способ парэнтерального введения. Эта композиция является уникальной, так как удовлетворяет всем шести критериям (совместимость с тканями, размер не более 10 мкм, инъектируемое количество, неантигенность, нагрузка и физическая стабильность) максимально полезной формой, способной к инъекции. Взаимная связь между микрокристаллом и покрывающим фосфолипидом изображена схематически на фиг.1.
Центральным в этом изобретении является использование амфипатического или амфифильного свойств фосфолипидов вообще и лецитина в частности. Вебстеровский словарь медицинской редакции (фирма Мерриан-Вебстер Инк. г.Спрингфилд, шт.Массачусетс, 1986) определяет термин амфипатический/амфифилический как ".состоящий из молекул, имеющих полярную водорастворимую терминальную группу, присоединенную к нерастворимой в воде углеводородной цепочке". На фиг.1 полярная головная группа фосфолипида обозначается кружочками, а гидрофобные углеводородные цепочки обозначаются "палочками". Многие вещества являются амфипатическими, включая мыла, поверхностно-активные вещества и моющие. Уникальным в этом изобретении является использование фосфолипидов с целью экранирования гидрофобной поверхности кристаллического медикамента и обеспечения дополнительных мембранных барьеров против повторного слияния кристаллов. Другие амфипатические молекулы, такие как мыла, поверхностно-активные вещества и моющие, неспособны предоставлять такие стабильные структуры, совместимые с тканями. Также уникальными в этом изобретении являются способы образования этих микрокристаллических структур, покрытых фосфолипидом. Это будет описано ниже.
Как описано в заявке, кристаллическое лекарственное вещество измельчается до размеров менее 10 мкм или субмикронных размеров в водной среде посредством обработки звуком или других обработок, включающих большой сдвиг. Лецитин (или другой образующий мембрану липид), присутствующий при озвучивании, сам разрушается на высокореакционные фрагменты, которые покрывают и обволакивают гидрофобные поверхности. Эти фрагменты покрывают и обволакивают субмикронные кристаллы, создавая первичное покрытие. Требование к этому процессу состоит в том, что лецитин и медикамент присутствуют вместе во время обработки звуком или другого высокоэнергетического процесса диспергирования. (Озвучивание кристаллов лекарства с последующим быстрым смешиванием предварительно образовавшихся фосфолипидных пузырьков не дает стабильных субмикронных водных суспензий медикамента). В подразделе, озаглавленном "Способы приготовления", оговорены альтернативные способы, включающие испарительное покрытие в полете и разбавление растворителем. Общим аспектом всех этих препаративных способов является то, что цепочки жирных ацилов фосфолипида должны иметь прямой доступ к микрокристаллу в процессе покрытия.
В этом изобретении амфипатические свойства фосфолипида удовлетворяют как гидрофильным свойствам воды, так и гидрофобным свойствам кристаллической поверхности. Кроме того, поверхность фосфолипидной мембраны служит как постоянный барьер для повторного образования макроскопических кристаллов. Вторым полезным свойством первичного покрытия является модифицирование скорости процесса растворения. Возможные структурные признаки взаимодействия фосфолипид-микрокристалл схематизированы на фиг.1.
В дополнение к использованию лецитина и других мембранообразующих липидов в качестве покрывающего и обволакивающего материала в этом изобретении описано новое использование мембранообразующих липидов в качестве механических буферов, организаторов водного объема и замедлителей рекристаллизации медикамента. Это достигается за счет избытка фосфолипида в виде однослойных и многослойных фосфолипидных пузырьков, которые образуются в качестве побочного продукта при обработке звуком в процессе первичного покрытия. Было установлено, что их удерживание в препарате улучшает длительную стабильность рецептуры. Кроме того, предварительно сформированные многослойные пузырьки (получены путем гомогенизации) или однослойные пузырьки могут быть добавлены в препарат с целью улучшения его стабильности или фармакинетики (пример 5). Вторичное покрытие может быть удалено путем повторного центрифугирования и суспендирования препарата (примеры 3 и 11).
Периферические пузырьки, образующие вторичное покрытие, стабилизируют препарат. Не желая связывать себя с какой-либо конкретной теорией или способом действия, при подробном обсуждении был предложен следующий механизм.
1. Они действуют как объемные буферы, расположенные между первично покрытыми микрокристаллами. Кристаллические и микрокристаллические медикаменты часто имеют более высокую плотность, чем фосфолипид, которые, в свою очередь, плотнее воды.
Таким образом, они будут иметь тенденцию осаждаться под действием гравитации и будут испытывать более сильные дальнодействующие взаимодействия (Ван-дер-Ваальсовское притяжение), чем два других компонента. Вторичное покрытие увеличивает расстояние наиближайшего приближения оболочек микрокристаллического лекарственного препарата, за счет чего снижается Ван-дер-Ваальсовское притяжение. Вероятно, что часть движущей силы для вторичного покрытия представляет собой Ван-дер-Ваальсовское притяжение между первично покрытым микрокристаллом и фосфолипидным пузырьком. Фосфолипиды (особенно лецитин) являются идеальными в качестве первичного и вторичного покрытий, поскольку они сильно гидратированы и принимают участие в надежно зарегистрированных репульсивных взаимодействиях ближнего порядка, которые обеспечивают их стойкость к агрегированию и слиянию.
2. Когда периферический фосфолипид присутствует в количестве 20% (масса/объем), большая часть водного объема препарата заключена внутри фосфолипидных мембран. Это служит в качестве тотологического барьера для рекристаллизации медикамента в препарате в ходе долговременного хранения. Образовавшиеся вновь кристаллы не могут быть крупнее, чем диаметр пузырьков или расстояние между ними, оба эти размера могут сохраняться малыми.
Обработка звуком наиболее удобно осуществляется при концентрации медикамента 5% (масса/объем) или менее и концентрации липида, образующего мембрану, 5% или больше. Это приводит к способной к впрыскиванию суспензии покрытых микрокристаллов с преимущественно субмикронными размерами, с частицами, для которых наблюдается броуновское движение (примеры 2, 3 и 11). В течение периода 1-2 сут микрокристаллы осаждаются, создавая отчетливую зону, в которой концентрация лекарства составляет 20-40% (масса/объем). Окончательная концентрация и объем зависят от выбора медикамента и от концентрации периферического фосфолипида. В большинстве препаратов донная зона способна повторно суспендироваться с инверсией, давая гомогенную и шприцуемую суспензию, даже после нескольких месяцев. Для препаратов, в которых это не так, способность образования повторной суспензии достигается при увеличении концентрации периферического фосфолипида.
В качестве средства концентрирования препарата может использоваться медленный процесс седиментации. Удаление объема выше зоны седиментации через 1-2 сут приводит к препаратам, в которых концентрация лекарства равна 20-40% (масса/объем). Долгосрочное хранение не приводит к дополнительному осаждению. Препараты остаются гомогенными шприцуемыми и фармацевтически приемлемыми в течение многих месяцев (см. примеры). Микроскопическое исследование этих препаратов выявило отдельные кристаллы лекарства микронного и субмикронного диаметра. Объем между этими микрокристаллами лекарства почти полностью заполнен фосфолипидными шариками, которые могут наблюдаться с помощью окрашивания красителем Найл Ред (см. примеры 2, 3 и 11). В этих концентрированных формах микрокристаллы медикамента проявляют только ограниченное броуновское движение. При обследовании в микроскопе не наблюдается изменения их положения относительно друг друга. Они колеблются или "танцуют на месте" вокруг своего центрального положения. Это частичное ограничение движения, вероятно, является важным фактором при долгосрочной стабильности препарата. При хранении концентрированные препараты разбавляются более чем тысячекратно в воде, насыщающей лекарство, причем микрокристаллы сохраняют свои микронные или субмикронные размеры.
Как отмечалось выше, основная полезность покрытых микрокристаллов состоит в их способности инъектироваться. Инъекции применимы на любых тканях или полостях тела. Они включают (но не ограничиваются) внутривенное, внутриартериальное, внутримышечное, внутрикожное, подкожное, внутрисуставное, цереброспинальное, эпидуральное, внутриреберное, внутрибрюшинное, внутриопухолевое, внутрь мочевого пузыря, внутрь поражения, субконъюнктивное и др. инъектирование. Кроме того, фосфолипидное покрытие и субмикронные размеры препарата могут оказаться выгодными для перорального использования как в качестве водной суспензии, так и в качестве лиофилизованного продукта. Аналогично водная суспензия может демонстрировать преимущества для местного наложения, вставления в глаз. Этот препарат может поставлять медикаменты посредством ингаляции в виде либо водной суспензии, либо лиофилизированной пудры. Также вероятно, что препарат может быть полезным для введения пестицидов и создания высокоценных биосовместимых продуктов, примером которого является суспензия медикаментов в питьевой воде (пример 15).
Наиболее важным фактором, определяющим скорость выделения лекарственного препарата, является выбор места инъекции. Если рецептура инъектируется внутривенно, она может выделяться из микрокристаллов весьма быстро. Если рецептура инъектируется в большом объеме внутрь ограниченного пространства, такого как мышца, общая скорость выделения может быть чрезвычайно низкой. Случай внутривенного введения будет рассмотрен первым.
Кровь представляет собой жидкую среду, которая способна разбавлять препарат в миллион раз примерно за 1 мину. Когда концентрированный микрокристаллический препарат, покрытый лецитином, разбавляется в крови, индивидуальные микрокристаллы первоначально в окружении, состоящем из других покрытых микрокристаллов, периферического липида и воды, насыщающей лекарство, переносятся в окружение, состоящее из сывороточных белков, липопротеинов сыворотки и клеточных элементов крови. Эксперименты по фракционированию вне организма, проведенные автором изобретения (примеры 3 и 11), позволяют предположить, что вторичное покрытие может быть быстро удалено. Все элементы крови способны связывать липофильные молекулы и будут так действовать со скоростью возможного растворения микрокристаллов. В случаях, когда лекарство является достаточно растворимым в воде, растворение в водной части крови является достаточным для распределения лекарства. Когда растворимость в воде является недостаточной, непрерывный процесс растворения и связывания медикаментов с элементами крови обеспечивает удаление лекарства из микрокристаллов. Скорость растворения микрокристалла будет зависеть от толщины и стабильности его первичного покрытия, растворимости лекарства в воде и других физико-химических параметров. Пример 10 показывает, что обезболивающее средство альфаксалон может удаляться из микрокристалла и проникать в мозг в пределах 10 с после внутривенной инъекции. Возможно уменьшение скорости выделения после внутривенного введения путем изменения толщины первичного покрытия или посредством включения небольших количеств нерастворимого в воде масла (такого как витамин Е) в препарат.
При инъекции в ткани, такие как мышца, препарат не претерпевает быстрого разбавления. В основном он остается в исходном элементе объема, созданного посредством инъекции. Этот элемент обычно является макроскопическим, в нем имеется незначительный поток или перемешивание. Диффузия медикамента из этого объема является медленной вследствие относительно больших расстояний потока, и она дополнительно замедляется из-за низкой растворимости медикамента в воде. Чем больше инъекционный объем и ниже растворимость в воде, тем меньше будет скорость удаления медикамента (пример 7). В пределе, на процесс выделения может потребоваться свыше 14 сут. Для больших и фиксированных объемов и концентраций медикамента скорость выделения может быть увеличена посредством внедрения гипертонической глюкозы или карбоксицеллюлозы в пузырьки вторичного покрытия. Это противодействует механическому давлению ткани, которое способствует отверждению инъектированного препарата (пример 5). Внутримышечная инъекция является полезной для создания хранилища медикамента и для получения постоянного выделения в кровь в течение периода нескольких суток. Инъекция непосредственно в целевую ткань или в пораженые ткани является полезной, так как при этом достигаются высокие и постоянные концентрации медикамента в том месте, где это необходимо, без вовлечения остальной части системы.
1. Обработка звуком. Процесс озвучивания понижает размер сверхмолекулярных структур за счет процесса кавитации. В этом процессе создаются пустые объемы, которые коллапсируют, приводя в движение материал с высокой скоростью, что приводит к раздроблению и отклонению. Это одновременно разрушает кристаллы медикамента и фосфолипидные слои на субмикронные фрагменты. Фосфолипидные мембраны раздробляются по направлениям как параллельным, так и перпендикулярным их плоскостям, с образованием поверхностей, которые могут покрывать гидрофобную поверхность микрокристалла, на который могут воссоединиться, соответственно обволакивая ее. Таким образом, концентрация фосфолипида должна соответствовать скорости покрытия и обволакивания с тем, чтобы превышать скорость воссоединения разрушенных кристаллов. Автором было обнаружено, что обычно обработка звуком удовлетворительна, когда концентрация медикамента составляет 5% (масса/объем) или ниже и когда концентрация фосфолипида равна 5% или выше. Роль вторичного покрытия из фосфолипидных пузырьков была описана выше.
Обработка звуком является неудовлетворительной, если лекарственный препарат и липид обрабатывают раздельно и добавляются друг к другу. Фактически в отсутствие фосфолипида, образующего мембрану, редко можно достигнуть измельчения медикамента до субмикронных размеров даже в течение короткого срока. Озвученный или гомогенизированный липид может быть добавлен к заранее приготовленным покрытым микрокристаллам для того, чтобы увеличить или модифицировать содержание в них периферических липидов. Как было описано выше, препарат может быть сконцентрирован до 20-40% (масса/объем), если дать ему возможность осадиться.
Этот продукт может быть переведен в сухую форму посредством лиофилизации, чтобы получить порошок, который позднее может быть вовлечен в рецептуру (пример 6). Это является целесообразным, когда долговременная химическая стабильность медикамента, подлежащего капсулированию, в водной среде является низкой.
2. Способы, включающие высокое давление и сдвиг.
Кристаллический медикамент и фосфолипид предварительно смешиваются посредством высокоскоростной гомогенизации (например, мешалкой "Варинг Блендер"). Дальнейшее измельчение и покрытие могут осуществлять по способу Микрофлюидизации (зарегистрированное название, фирма Микрофлюидикс Корп. г.Ньютон, шт. Массачузетс 02164). Этот способ основывается на высоком сдвиге, который создается за счет столкновения противоточных струй жидкости. Это устройство описано Мэйхью и сотр. в журнале Biochim. Biophys. Acta, 1984, т.775, с. 169-174. Альтернативой является гомогенизация под высоким давлением с помощью Французской ячейки давления или "Французского Пресса" (фирма Эс-Эл-Эм Инструментс, г.Урбана, шт.Иллинойс). В этом способе образец нагнетается при высоком давлении и высоком значении сдвига через узкое отверстие и подвергается быстрой декомпрессии до атмосферного давления. Другие подробности аналогичны указанным в п.1.
3. Обработка звуком или высоким сдвигом в летучих органических растворителях.
Микрокристаллы могут быть приготовлены путем суспендирования кристаллического медикамента и липида, образующего мембрану, в летучем неполярном растворителе, в котором медикамент плохо растворим, в дихлордифторметане или дихлортетрафторэтане или фреоне (например, трихлортрифторэтане, см. примеры 6 и 8). Эта суспензия измельчается посредством обработки звуком или высокоскоростным сдвигом по методам, описанным выше (п.1 или 2). Растворитель удаляется путем испарения. Образовавшийся порошок может храниться для последующего составления в рецептуре с водой или может быть составлен сразу же.
4. Измельчение в воздухе.
Кристаллы медикамента могут быть также измельчены за счет столкновения с высокой скоростью в воздухе. Они могут быть в последующем покрыты путем увлажнения раствором фосфолипида или глицерололипида в летучем растворителе, содержащем лецитин. Растворитель удаляется путем испарения. Порошкообразный продукт может быть суспендирован в воде. Альтернативно микронные кристаллы могут быть увлажнены смешивающейся с водой жидкостью, растворяющей лецитин, и быстро введены в водную среду.
5. Кристаллизация в полете.
Растворы липида и медикамента в летучем растворителе могут быть распылены, причем растворитель удаляется путем испарения в полете. Микрокристаллы собираются и сушатся на ровной поверхности. Эти микрокристаллы могут либо храниться в виде порошка для последующего составления в рецептуре с водой, либо могут составляться немедленно.
6. Разбавление растворителем.
Растворы липида и нерастворимого в воде медикамента делаются с использованием органического растворителя, смешивающегося с водой (например, этанола). Эти растворы быстро вводятся в водную среду при быстром перемешивании или озвучивании. Растворитель растворяется в воде, оставляя медикамент в микрокристаллической форме, покрытой липидом. Этот органический растворитель может быть полностью удален путем фильтрации или седиментации покрытых микрокристаллов и удаления жидкости над осадком.
Выбор лекарства, подлежащего покрытию.
Может использоваться любой медикамент, практически не растворимый в воде, который находится в кристаллическом или твердом состоянии при температуре около 37oC. Обычно это лекарство должно иметь растворимость в воде ниже 5 мг/мл при физиологических значениях pH (6,5-7,4). Использование медикаментов с более высокой растворимостью в воде не исключается, если эксперимент показывает минимальную склонность к реорганизации в макроскопические кристаллы в течение желаемого срока хранения. Обычно желательно выбирать общую концентрацию медикамента свыше 4% от растворимости его в воде с тем, чтобы по меньшей мере 80% лекарства находилось в микрокристаллическом виде. Такой выбор дает выгодные характеристики высокой нагрузки и постоянного выделения, которые связаны с покрытыми микрокристаллами. Наконец, предпочтительно, чтобы лекарство само по себе не было раздражающим. Кроме того, желательно, чтобы лекарство было химически стабильным во влажной атмосфере. В противном случае может возникнуть необходимость в получении лиофилизированных форм.
Наиболее частными применяются медикаменты, которые нерастворимы в воде, но обладают от умеренной до хорошей растворимостью в масле. Однако растворимость в масле сама по себе не является требованием для внедрения в микрокристаллы, покрытые фосфолипидом. Многие медикаменты, которые имеют плотные кристаллические структуры, высокие температуры плавления, не очень хорошо растворимы в воде или масле. Эти лекарства также могут извлекать пользу от микрокристаллической рецептуры, покрытой фосфолипидом. Аналогично, для лекарства не является необходимым быть ненаполненным, для того чтобы перевести его в микрокристаллическую форму. Необходимо только, чтобы растворимость в воде кристаллической формы медикамента была низкой.
Перевод водорастворимого лекарства в нерастворимое в воде.
Согласно настоящему изобретению использование собственно растворимого в воде медикамента при условии, что оно может быть переведено в нерастворимое в воде за счет образования комплекса. Например, используется нерастворимая соль иодистоводородной кислоты (HI) местного обезболивающего тетракаина для того, чтобы продлить срок его действия в 5 раз (пример 12). Если лекарство подается при физиологическом значении pH, оно часто может быть сделано нерастворимым путем замещения более липофильным или структурированным противоионом. Примеры перевода положительно заряженного медикамента в малорастворимое в воде состояние включают комплексование с 2-нафтиленсульфонатом (напсилат), глюконатом, 1,1-метилен-бис(2-гидрокси-3-нафталин)карбоновой кислотой (памоатом), толуолсульфонатом (тозилатом), метансульфонатом (мерзилатом), глюкогептаноатом (глюцептатом), битартратом, полиглутамовой кислотой, сукцинатом, ацетатом или бегенатом (анионная форма парафиновой жирной кислоты). При выборе анионов жирного ацила рекомендуется выбирать частицы либо с цепочкой малой длины, либо с очень длинной цепочкой, с тем чтобы свести к минимуму тенденцию к образованию мицелл. В некоторых случаях замещение бромидом, иодидом, фосфатом или нитратом является достаточным для того, чтобы перевести лекарство в менее растворимую форму. Примеры перевода отрицательно заряженного медикамента в состояние с меньшей растворимостью в воде включают комплексование с кальцием, магнием или их смесью 1:1 солей жирных кислот и с различными аминами, включающими дибензилэтилендиамин (бензатин), N,N-(дигидроабиетил)этилендиамин (гидрабамин) или полимеры, такие как полилизин. Выбор этих противоионов делается в основном на эмпирическом основании, причем первичными критериями являются стабильность произведенных кристаллов и их совместимость с водой. Поскольку выделение медикамента после разбавления или инъекции может включать удаление как заряженной, так и незаряженной форм медикаментов, а также его противоиона, эти системы проявляют сложную и многообразную кинетику. При достаточном изучении вне организма поведения микрокристаллов, покрытых фосфолипидом и полученных из ряда этих бинарных солевых систем, и при разумном выборе наиболее перспективных примеров может быть примерно оценена фармакологическая кинетика в самом организме.
Кроме того, в некоторых приложениях возможно приготовление микрокристаллов при более предельных значениях pH (4,0-6,4 или 7,5-10,0) для того, чтобы подавить ионизацию и таким образом снизить растворимость медикамента. Допустимые предельные значения pH в каждом конкретном случае будут определяться концентрацией лекарства, числом эквивалентов кислоты или основания, которое оно включает, скоростью его растворения и размером отделения, в которое производится инъекция (и в терминах времени хранения), стабильностью липида, образующего мембрану.
Первичным требованием является то, что покрывающий липид способен образовывать мембрану. Этому требованию удовлетворяют все липиды, которые в присутствии избытка воды образуют двухслойные структуры того типа, который хорошо зарегистрирован для фосфолипидных пузырьков или липосом. Этому требованию не удовлетворяют жирные кислоты, моющие, неионные поверхностно-активные вещества (например, полиэтиленгликоль) или триглицериды (растительные масла, тристеарин, жиры). Вторичным требованием является то, что липид не обладает склонностью к превращению в мицеллярные структуры. Это требование исключает фосфолипиды с малой длиной цепи (6 или менее 6 атом в углерода) или лизо-лецитин, содержащий одну цепочку жирного ацила. Высокая стабильность покрывающего материала в виде мембраны является необходимой для того, чтобы предотвратить перегруппировку материала медикамента в макроскопические кристаллы. Это составляет одну из причин, почему неионные поверхностно-активные вещества работают неудовлетворительно для решения целевой задачи.
Полезные примеры образующих мембраны липидов даны ниже.
Класс А. Первичные фосфолипиды, используемые в чистом виде, включают следующие: лецитин (фосфатидилхолин), сфингомиэлин, синтетические цвиттерионные фосфолипиды или их аналоги.
К этому классу принадлежат все фосфолипиды, которые самопроизвольно образуют мембраны при добавлении воды. Эти фосфолипиды могут быть использованы в чистом виде для того, чтобы получить покрытые микрокристаллы. Из всех этих фосфолипидов лецитин является примером наиболее применимого, поскольку он легко доступен и имеет низкую стоимость.
Класс Б. Фосфолипиды, способные к агрегации, зависящей от кальция.
Эти фосфолипиды включают следующие: фосфатидную кислоту, фосфатидилсерин, фосфатидилинозитол, кардиолипин (дифосфатидил-глицерин), фосфатидил-глицерин.
Эти липиды несут отрицательный заряд при нейтральном значении pH. Предпочтительно, эти фосфолипиды могут смешиваться с лецитином, для того чтобы получить отрицательно заряженную поверхность, которая обеспечивает отталкивание между частицами. При введении в среду, содержащую 2 ммоль/л кальция (такого как кровяной или его заместители), ожидается, что мембраны, содержащие эти фосфолипиды, обнаружат повышенную агрегируемость и реакционную способность по сравнению с клеточными мембранами. Это может быть полезным для инициирования агрегирования в тканях, что приведет к пониженным скоростям выделения. Полезность этого класса ограничивается высокой стоимостью этих фосфолипидов по сравнению с лецитином.
Класс В. Фосфатидилэтаноламин способствует агрегированию по способу, не зависящему от наличия кальция. Он может использоваться в чистом виде, чтобы покрывать микрокристаллы при pH 9. Когда значение pH доводится до 7, как при инъекции в кровь или ткани, мембраны становятся реакционноспособными, способствуя агрегированию частиц и присоединению к клеточным мембранам. Это может быть полезным свойством для снижения скорости выделения.
Класс Г. Холестерол и стероиды. Эти вещества не могут использоваться в качестве единственного покрывающего материала. В чистом состоянии они не образуют мембраны. Они могут быть добавлены к лецитину или другому покрывающему материалу для того, чтобы изменить его поверхностную активность, микровязкость или распределяемость покрытия. Стероидным гормоном (эстрогеном, андрогеном, минерало- или глюкокортикоидом) можно оказывать влияние на местную реакцию ткани в отношении микрокристалла, а также влиять на их физическое размещение.
Класс Д. Полулипоидные молекулы могут быть внедрены в фосфолипидную или глицеролипидную мембрану, и они изменяют поверхностную активность микрокапель. Включенные в этот класс молекулы перечислены ниже.
Стеариламин или другие амины с длинноцепочечным алкилом, которые могут быть первичными, вторичными, третичными или четвертично-замещенными. Они придают микрокристаллическому покрытию положительный заряд и делают его более реакционноспособным с клеточной мембраной. Хлористый бензалконий является примером из ароматического класса, который особенно полезен, поскольку он также действует в качестве предохранителя против микробиологического роста в препарате.
Жирные кислоты. Они могут быть введены в малых концентрациях (менее 0,02 г/г фосфолипида) для того, чтобы изменить упаковку и реакционную способность фосфолипида.
Класс Е. Мембраноактивные агенты, гликолипиды и гликопротеины, модифицирующие поверхностные свойства.
Примеры мембраноактивных агентов включают нистатин, амфотерицин В и грамицид, которые представляют собой поверхностно-активные антибиотики. Было показано, что они связываются с поверхностью фосфолипидных мембран и изменяют их проницаемость. Гликолипиды или гликопротеины могут быть включены как средство, изменяющее реакционную способность поверхности. Вероятно, антитела могут сочетаться с составными частями мембраны, направляя или удерживая ассоциацию микрокристаллов с целевыми клетками или тканями. (Гликолипиды, гликопротеины и антитела классифицируются как биологические реагенты. Они должны быть подвергнуты скринингу на пирогенитичность, антигенитичность и др. до их использования, причем процесс получения регламентирующего одобрения для таких рецептур может быть более сложным).
Класс Ж. Моноглицериды.
Они не являются фосфолипидами, но, как было сказано,
они способны образовывать ориентированные монослои и двойные слои в присутствии декана (Бенц и др. Biochem. Biophys. Acta, 1975, т.394, с.323-334). Таким образом было доказано, что они могут иметь
некоторое применение при покрытии микрокристаллов. Примеры таких липидов включают (но не ограничиваются) следующие:
1-монопальмитоил-(рац.)-глицерин (монопальмитин)
1-монокаприлоил-(рац.)-глицерин (монокаприлин)
1-моноолеоил-(рац.)-глицерин (С18:1, цис-9) (моноолеин)
1-моностеарил-(рац.)-глицерин (моностеарин)
Коммерчески доступные
липиды, образующие мембраны.
Имеются в виду несколько форм лецитина. В качестве примера яичный лецитин (фирма Сигма Кемикал Ко.) используется во всех представленных примерах. Он является предпочтительным из-за малой стоимости и низкой степени ненасыщенности. Лецитин также может быть получен из бычьего сердца. Более дешев лецитин соевых бобов. Он обладает повышенной степенью ненасыщенности. Доступными являются несколько синтетических разновидностей лецитина, которые различаются длиной цепи от 4 до 19 атомов углерода (фирма Супелко, Инк.). Считается, что лецитины с длинами цепочек в биологическом интервале (C10-C18) являются полезными в различных приложениях. Ненасыщенные лецитины (диолеил, дилонолеил, бета-олеоил; альфа-пальмито-бета-олеоил; альфа-пальмитоил-бета-линолеил и альфа-олеоил-бета-пальмитоил) также являются доступными. Диарахидонил-лецитин (высоконенасыщенный предшественник простогландина) также доступен.
Фосфатидиновая кислота может быть получена из яиц или как синтетические вещества (димиристоил, дипальмитоил или дистеароил, ф.Кальбиохем). Бычья фосфатидиловая сыворотка доступна из фирм Супелко или Кальбиохем.
Фосфатидил-инозитол доступен из растений (ф.Супелко) или из быков (ф.Кальбиохем). Кардиолипин может быть получен (ф.Супелко) из быков или микробного источника. Фосфатидилглицерин может быть получен из микробных (Супелко) источников или как синтетические вещества (димиристоил или дипальмитоил; ф. Кальбиохем).
Фосфатидилэтаноламин доступен в виде микробного, бычьего или плазмогенного (ф. Супелко) или в виде синтетических соединений: диоктадеканоиловый и диолеиловый аналоги и дегексадецил-, дилаурил-, димиростоил- и дипальмитоил- (Супелко и Кальбиохем).
Моноглицериды являются доступными от фирмы Сигма Кемикал Ко. 1-монопальмитоил-(рац. )-глицерин, монопальмитин; 1-монокаприлоил-(рац.)-глицерин, монокаприлин; 1-моноолеоил-(рац.)-глицерин (C18:1, цис-9), моноолеин; 1-моностеарил-(рац.)-глицерин, моностеарин.
Другие компоненты.
Возможно добавление других компонентов к микрокристаллам, для того чтобы увеличить их стабильность или изменить скорость их выделения. Например, фармакологически приемлемые масла могут быть добавлены при низкой массовой концентрации для того, чтобы способствовать контакту между микрокристаллом и фосфолипидным или глицеридолипидным покрытием. Необходимо, чтобы тип масла и его массовая концентрация выбирались таким образом, чтобы кристаллический медикамент не растворялся в масле и чтобы покрытие из мембранообразующего липида не было разрушено. Эти соотношения могут быть определены из эксперимента. Полезные масла включают (но не ограничиваются ими) витамин E, изопропиловый эфир миристиновой кислоты, бензиловый эфир бензойной кислоты, олеиловый спирт, минеральное масло, сквален и растительное масло. Пример 6 дает сведения, что внедрение витамина E в покрытый лецитином микрокристаллический препарат эритромицина снижает скорость растворения медикамента, тем самым снижается раздражение ткани этим медикаментом.
Кроме того, возможно предварительное покрытие микрокристаллов неантигенными молекулами, которые совместимы с фосфолипидом и находятся в твердом состоянии при 37oC. Примеры включают парафин, тристеарин, этилолеат, цетостеариловый спирт, цетиловый спирт, миристиловый спирт, стеариловый спирт и петролатум. Например, эти материалы могут быть внедрены в первичное покрытие посредством обработки звуком или сдвигом при температурах выше их температур плавления. Стабилизация может быть достигнута путем добавления лецитина в ходе процесса, когда температура проходит обратно к точке затвердевания этих материалов. Желательно использовать низкие массовые концентрации (менее 10%) этих компонентов, чтобы не ухудшить нагрузку и не слишком снизить скорость растворения медикамента. Кроме того, дополнительные ограничения может вызвать биологическая разлагаемость. В примере 13 описана лецитиновая совместимость парафина в форме микрочастиц.
Суспендирующая среда.
В окончательном препарате непрерывной фазой обычно является вода, доведенная буфером до физиологически приемлемого значения pH и содержащая изоосмотическую концентрацию хлористого натрия или глюкозы. В некоторых приложениях, включающих внутримышечную инъекцию больших объемов микрокристаллов при высокой концентрации, полезно увеличить осмолярность среды (т.е. концентрацию глюкозы), для того чтобы способствовать распределению материала в мышце. Как указывалось выше, это может затормозить процесс уплотнения после внутримышечной инъекции. Там, где это допустимо, могут применяться агенты, увеличивающие вязкость, такие как карбоксицеллюлоза, для того чтобы изменить фармакологическую кинетику после внутримышечной инъекции и чтобы снизить скорость осаждения микрокристаллов при хранении.
В некоторых приложениях полезно заменять воду полярным растворителем, как в примере 7, в котором албендазолсульфоксид не показал достаточно долговременной стабильности в присутствии воды (также см. пример 5). Примеры неводных полярных растворителей, которые могут применяться, включают (но не ограничиваются) следующие: глицерин (смешивающаяся с водой жидкость с диэлектрической постоянной, равной 42,5) и пропиленгликоль (смешивающаяся с водой жидкость с диэлектрической постоянной 32). Покрытые микрокристаллы могут быть получены в этих средах или могут быть подвергнуты осаждению в этих средах. Первичным требованием является то, чтобы в этом растворителе значительная часть фосфолипида или покрывающего материала находилась в виде мембраны. В такой же степени важным требованием является то, чтобы кристаллический материал не обладал достаточной растворимостью в растворителе, чтобы он мог быть перекристаллизован.
Консерванты.
Растворимые в масле консерванты могут быть добавлены в процессе формирования первичной или покрывающей фазы. Они включают бензалконий-хлорид, пропилпарабум, бутилпарабен и хлорбутанол, но не ограничиваются ими. Кроме того, существует множество водо- и маслорастворимых агентов, которые могут быть добавлены в конечный продукт в качестве предохранителей, включая бензиловый спирт, фенол, бензоат натрия, этилендиаминтетрауксусную кислоту и др.
Необязательная лиофилизация и повторное составление.
Водные микрокристаллические препараты могут быть лиофилизованы с образованием сухого продукта, который может быть вновь распущен в воде (пример 6). Это особенно полезно для медикаментов, которые не обладают долговременной стабильностью в водной среде. Также возможно проводить обработку звуком или сдвигом в летучем органическом растворителе, в котором кристаллический медикамент недостаточно растворим, и готовить сухие покрытые микрокристаллы посредством выпаривания растворителя (пример 6). Эти методики дают микрокристаллы, окруженные слоями фосфолипида в безводном состоянии. Такие формы пригодны для перорального назначения или для повторного составления с водой инъекции.
Специалист в этой области техники, следуя указаниям, которые предоставлены здесь, не
будет иметь затруднений при эмпирическом определении:
1. Наиболее удобного способа приготовления: обработка звуком, или высокое давление + сдвиг, или способы, включающие органические
растворители, или соударение в воздухе, или кристаллизация в потоке, или разбавление растворителем.
2. Наиболее выгодной формы лекарства: кристаллы нейтрального лекарственного препарата или кристаллы заряженного лекарства, или более сложные твердые формы лекарственного препарата.
3. Оптимальный липид, образующий мембрану, выбирается на основе реакционной способности и стабильности мембран, совместимости с кровью и тканями, и цены.
4. Оптимальные условия для производства, включая соотношение медикамента и фосфолипида на входе, включение небольших количеств масла или воска в качестве модифицирующих агентов, длительность обработки звуком, сдвигом и др. использование седиментации в качестве средства отбора размера, добавление более периферического липида в однослойной или многослойной форме, добавление осмотических или влияющих на вязкость агентов.
5. Оптимальный размер частиц, который может регулироваться в некоторой степени посредством подаваемого порошка, длительности обработки, концентраций медикамента и фосфолипида и который может быть отобран между 50 нм и 10 мкм с помощью скорости седиментации.
Оптимальные составы для достижения желаемого срока хранения и фармакологической кинетики.
Определяются при изучении влияния указанных выше факторов на фармакологическую кинетику после инъекции. Особенно важными являются размер частиц, содержание первичного и вторичного фосфолипида и добавок, чтобы избежать уплотнения после инъекции в ткани.
Наиболее выгодный способ введения включает инъекцию (внутривенную, внутриартериальную, внутримышечную и др.), пероральное и местное назначение, ингаляцию и др.
Все части и проценты, приведенные в описании, являются массовыми (масса/масса) или массо-объемными (масса/объем) процентами, в которых масса или объем в знаменателе представляет собой суммарную массу или объем системы. Концентрации водорастворимых компонентов в водном растворе (например, глюкозы) даны в миллимолярных единицах (ммоль/л), отнесенных к объему воды в системе. Все температуры приведены в градусах Цельсия. Диаметры и размеры даны в мм (1 мм 10-3 м), мкм (1 мкм 10-6 м), нм (1 нм 10-9 м) или единицах Ангстрема (1А 0,1 нм). Композиции изобретения могут включать или состоять из указанных материалов, причем способ или метод может включать или состоять из стадий, связанных с указанными материалами.
Пример 1. Покрытые лецитином микрокристаллы окситетрациклина (ОТЦ) были приготовлены с помощью обработки звуком следующим образом.
В стеклянный стакан емкостью 150 мл добавляют 4,4 г дигидрата окситетрациклина (фирма Сигма, 0-5740) и 16,0 г яичного лецитина (1-альфа-фосфатидилхолин из яйца, Тип XV-E, Сигма, Р-9671) и грубо перемешивают с использованием стеклянного прутка-мешалки. Затем добавляют водный раствор глюкозы 300 ммоль/л, 10 ммоль/л буфера Трис до установления pH 7,4 и доводят окончательный объем смеси до 80 мл. В жидкость погружают зонд клеточного прерывателя СенифайерR диаметром 1,0 см (фирма Хиит Систем и Ультрасоникс, г.Плэйнвью, шт.Нью-Йорк) и смесь обрабатывают звуком в течение 60 мин при значении мощности 10 (номинально 100-150 Вт). Температуру смеси регулировали с помощью водяной рубашки, прерывая время от времени озвучивание и не давая смеси нагреться выше 60oC. Значение pH 5,0 поддерживали, добавляя соляную кислоту. Обработка звуком привела к образованию мутной желтовато-бежевой суспензии, которая покрывается и осаждается в течение 24 ч. Нижняя часть из 11 мл содержала видимый осадок окситетрациклина с концентрацией 40% (масса/объем). Жидкость над осадком содержала фосфолипидные пузырьки. Придонную часть (22 мл) собирали и осадок вновь суспендировали при осторожном встряхивании. Он содержал микрокристаллы окситетрациклина, покрытые лецитином. Материал представляет собой довольно вязкую, но шприцуемую жидкость.
Методами флуориметрического анализа и жидкостной хроматографии высокого давления показано, что верхняя фаза содержит очень мало окситетрациклина. Придонная фаза, отобранная со дна (22 мл), содержала свыше 98% добавленного окситетрациклина. Образец содержал 20% (масса/объем) окситетрациклина и 20% (масса/объем) лецитина. Из обеих фаз были отобраны аликвоты, которые были разбавлены буфером, насыщенным окситетрациклином, и использованы для определения диаметра частиц (±) стандартное отклонение N 4-МД Анализатором субмикронных частиц коултера. При анализе было установлено, что верхняя фаза имеет частицы с диаметрами 30,5±8 нм (77%) и более 3000 нм, которые соответствуют монослойным и многослойным фосфолипидным пузырькам. Верхнюю фазу сливают. Фракция микрокристаллов окситетрациклина, покрытых лецитином, имела следующее распределение частиц по размерам (средне-взвешенное): 980±460 нм, 59% 2880±400 нм, 41% При анализе препарата методом электронной микроскопии с использованием отрицательного окрашивания подтверждают сделанные выше наблюдения. Несколько препаратов были сделаны, как описано выше, и ими были заполнены стеклянные ампулы, закрытые резиновой пробкой, а также стеклянные пузырьки. При хранении в течение нескольких недель наблюдалось в некоторой степени осаждение, однако препарат можно было превратить в гомогенный, перевертывая его 3 раза. Этот препарат сохраняет свои свойства, включая распределение по размеру, концентрацию окситетрациклина, химическую целостность и способность к впрыскиванию (трубка N 20 или уже) в течение периода свыше 9 мес.
Важность лецитинового покрытия была продемонстрирована следующим образом: 4,4 г окситетрациклина и 75,6 мл раствора глюкозы обрабатывали звуком в течение 60 мин, как описано выше, но в отсутствие лецитина. Была получена грубая суспензия со следующими характеристиками: (а) При немедленном отборе пробы и тысячекратном разбавлении на насыщенной окситетрациклином воде были получены частицы, видимые невооруженным глазом. Анализ субмикронных частиц анализатором Коултера показал, что частицы имели размер вне диапазона (свыше 3 мкм). При анализе не было выявлено частиц с диаметром менее 3 мкм. (б) В течение 10 мин после обработки звуком весь окситетрациклин осадился на дно. Эта донная фаза не обладала свободной текучестью и шприцуемостью. В течение 1 ч после осаждения она превратилась в твердую массу, которую не удалось вновь суспендировать при встряхивании. Аналогично было невозможно стабилизировать этот препарат путем добавления предварительно образовавшихся фосфолипидных пузырьков к обработанному звуком окситетрациклину сразу после прекращения озвучивания. Таким образом было показано, что обработка звуком с лецитином (или другим липидом, образующим мембрану) является критической стадией в этом способе приготовления.
Пример 2. Препарат примера 1 был приготовлен при следующих изменениях: объем препарата был сведен суммарно к 5,0 мл, был использован озвучивающий дуп с микронасадкой и одновременно с липидом добавляли 0,15 мг красителя Найл Ред. Этот краситель связывается с фосфолипидами и служит в качестве флуоресцирующего маркера для лецитина. Каплю препарата 20% (масса/объем) распределяют на стеклянной пластинке и наблюдают во флуоресцентном микроскопе (Лейтц Ветцлар Диалюкс 20) при высокой мощности. Фиг.2 представляет собой черно-белое изображение типичной области. Белые пятна указывают на высокую флуоресценцию. При возбуждении ультрафиолетом частицы окситетрациклина наблюдались за счет их интенсивной желто-зеленой флуоресценции. На верхней части фиг. 2 изображено флуоресцентное изображение окситетрациклина. Наблюдались отдельные частицы с диаметрами в интервале примерно от 0,2 до 3 мкм. Большая часть частиц имела диаметр менее 1 мкм (средняя величина). Эти частицы имели четкие границы, однако были окружены желто-зелеными гало. Та же область наблюдалась при возбуждении ближним ультрафиолетом, для того чтобы получить изображение Найл Ред, ассоциированного с лецитином в препарате (нижняя часть чертежа). Изображение красителя Найл Ред показывает лецитин, который окружен частицами окситетрациклина. Частицы окружены гало красной флуоресценции, простирающимся на 0,3-3 мкм за пределами микрокристалла окситетрациклина. Пустые пространства, свободные от окситетрациклина, а также от лецитина, легко могут быть распознаны среди всех частиц, расположенных вблизи края пятна. Наблюдались случайные конфигурации, которые позволяют предположить, что две частицы окситетрациклина разделяют одно фосфолипидное гало.
В этом образце наблюдалось броуновское движение. Более крупные частицы (диаметром свыше 1 мкм), которые осаждались на стеклянной пластинке, не обладали броуновским движением либо для них наблюдалось сильно ограниченное движение. Частицы менее 1 мкм (диаметр) показывали броуновское движение, двигаясь между большими частицами. Наблюдения в области низких концентраций показали, что частицы не могут передвигаться на расстояние больше примерно 1/4 их диаметра без отражения соответствующего движения гало. Более того, никогда не наблюдались прямые столкновения. Эти наблюдения объясняют замечательную стабильность суспензий микрокристаллов изобретения заявителя. Лецитин покрывает микрокристалл, предоставляя как гидрофобную поверхность для контакта с поверхностью кристалла, так и гидрофильную поверхность для контакта с водой. Поверхность покрытия обволакивается многочисленными слоями лецитина в виде мембран, что было выявлено с помощью окрашивания Найлом Ред. Стабильность оболочки показана тем фактом, что микрокристалл всегда остается внутри своего лецитинового гало, что было выявлено с помощью соответствующих им флуоресцентных сигналов. Внешние лецитиновые слои гарантируют то, что покрытые микрокристаллы не приблизятся на расстояние, достаточное для слияния.
Поведение микрокристаллов, покрытых лецитином при растворении, наблюдалось под флуоресцентным микроскопом, приводя их в контакт с большим количеством дистиллированной воды и накладывая покрывающий чехол. Меньшие частицы (примерно размером 0,2-1,0 мкм) двигались с потоком воды. Флуоресцирующее гало красителя Найл Ред двигалось вместе с флуоресцирующей частицей окситетрациклина, причем первоначально яркость этих двух флуоресцирующих сигналов остается относительно постоянной. Когда частица двигается в дистиллированной воде, сигнал окситетрациклина делается тусклым, что позволяет предположить растворение кристалла. Теряется только небольшая часть изображения красителя и интенсивность, что позволяет предположить, что лецитиновое покрытие является сохраняющейся структурой. Поведение более крупных кристаллов при растворении, которые обычно более жестко присоединяются к пластинке, было в некоторой степени другим. Течение вынуждает их сбрасывать большую часть липидов. Затем наблюдалось, что более крупные микрокристаллы расщепляются, отделяя "надкрылья", каждое из которых уносит часть гало красителя с собой.
Поведение при растворении также было изучено с использованием определителя размера субмикронных частиц Коултера N 4-МД. Как было установлено в примере 1, при растворении препарата в 1000 раз в изотоническом глюкозовом буферном растворе, насыщенном окситетрациклином, препарат является стабильным, причем наблюдаются частицы с размерами 980 ±460 и 2880±400 нм. Когда производится тысячекратное разбавление в дистиллированной воде, наблюдается быстрое изменение препарата. Ожидалось растворение, поскольку окончательная концентрация окситетрациклина стала 0,2 мг/мл, что ниже водной растворимости этого медикамента (около 1 мг/мл). Растворение наблюдалось, однако оно сопровождалось образованием некоторых частиц с диаметром более 3 мкм.
Пример 3. Был приготовлен микрокристаллический препарат окситетрациклина, покрытого лецитином и содержащего добавку Найл Ред, и был проведен следующий эксперимент по фракционированию для того, чтобы установить взаимосвязь между первичным и вторичным фосфолипидными покрытиями для покрытых микрокристаллов большего размера (1,0-1,9 мкм).
Окситетрациклин (2,0 г), 8,0 г лецитина и 1,5 мг красителя Найл Ред были добавлены к 40 мл изотонического раствора глюкозы и смесь обрабатывали звуком 30 мин. Препарату давали сконцентрироваться до 20% (масса/объем) окситетрациклина посредством седиментации в течение ночи. Объем этого препарата составлял 10 мл. Были отобраны небольшие аликвоты для флуориметрического анализа концентрации красителя, анализа фосфолипида (посредством экстракции ферроцианатом аммония) и для наблюдения под флуоресцентным микроскопом. Затем препарат был центрифугирован на клинической центрифуге со средней скоростью в течение 15 мин. Это приводило к образованию видимого осадка объемом 2,0 мл. Отделяли верхнюю фазу и анализировали аликвоты (1-ая жидкость над осадком). Донную фазу вновь суспендировали до конечного объема 10 мл путем добавления изотонического раствора глюкозы. Аликвоты этой суспензии также анализировались (1-ая промывка). Эту операцию повторяли, получая в сумме 5 промывок. По этой методике удалялись покрытые микрокристаллы небольшого диаметра (0,1-0,3 мкм) и свободно присоединенные фосфолипидные пузырьки. Это дает возможность выделить большие (диаметр 1-3 мкм) покрытые микрокристаллы и обеспечивает возможность оценки в них соотношения фосфолипид/медикамент. Результаты приведены в табл.1.
Наблюдения показали, что, когда более крупные кристаллы повторно промываются, они теряют наибольшую часть ассоциированного с ними лецитина. Количество ассоциированного липида стабилизируется между 3-ей 5-ой промывками на уровне 108±80 мкг/мл или примерно 0,4% от исходного значения (Найл Ред). Толщина этого покрытия может быть оценена из ее связи с объемом, принимая плотности окситетрациклина и лецитина равными примерно 1,4 г/см3. Из данных по Найл Ред толщина слоя составляет 15 А. Это близко к ожидаемой величине из монослоя простершихся молекул лецитина. Оценка толщины из анализа фосфолипида занижена (примерно 3 А), однако экспериментальная неопределенность здесь велика, причем эффективность экстракции для образцов 3-5 может быть значительно ниже единицы. Указанная выше методика может недооценивать толщину покрывающего слоя, если он может сбрасываться под воздействием сил центрифуги. Для малых микрокристаллов (0,1-0,3 мкм в диаметре) наблюдалось, что гало красителя тесно ассоциировано с микрокристаллом, хотя и подвергается броуновскому движению. Это позволяет предположить, что обволакивающий слой является вполне стабильным.
Пример 4. Фармакологическая кинетика микрокристаллов окситетрациклина, покрытых лецитином, была исследована на лабораторных крысах. Препарат был приготовлен практически так, как в примере 1. Он содержал 24% (масса/объем) окситетрациклина и 20% лецитина. Образцы (0,1 мл) вводились посредством глубокой внутримышечной инъекции в задние лапы лабораторных крыс. Были сделаны инъекции (дистально-проксимально) в гастрокнемус. Положительным контролем служили инъекции 0,1 мл продажного образца ОТЦ, способного к внутримышечной инъекции (ЛиквамицинR 200, фирма Пфайзер), содержащего 200 мг/мл базового окситетрациклина в виде атмотерного ОТЦ, 40% (масса/объем) 2-пирролидона, 5,0% повидона, 1,8% оксида магния, 0,2% формальдегидсульфоксилата натрия и моноэтаноламин и/или соляную кислоту, сколько требуется для установления значения pH. В обозначенные моменты времени отбиралась кровь центральной артерии, животных умерщвляли и инъецированные мышцы иссекались и исследовались внешне и под ультрафиолетовым облучением для флуоресценции окситетрациклина. Образцы крови и мышц были проэкстрагированы этанолом, и концентрация окситетрациклина была определена флуориметрически.
На фиг.3 показано, что окситетрациклин выделяется медленно из мышц, если он инъецируется в виде покрытых лецитином микрокристаллов, примерно 20% от инъецированной дозы остается в мышце спустя 7 сут. Выделение ОТЦ из микрокристаллов существенно медленнее, чем из продажного пирролидонового раствора. Из фиг.4 видно, что уровни содержания ОТЦ в крови от 4 до 1,5 мкг/мл поддерживаются непрерывно в течение 7 сут. Это можно сравнить с продажным раствором, для которого содержание ОТЦ в крови снижается до 0,5 мкг/мл или ниже в пределах 3 сут.
Пример 5. Было приготовлено большое число препаратов микрокристаллов окситетрациклина, покрытых лецитином, с концентрациями между 20 и 44% (масса/объем), как описано выше. Настоящий пример демонстрирует, как вторичное покрытие (периферические пузырьки) может быть добавлено после стадии первоначального озвучания и как гипертонический раствор глюкозы и агенты, увеличивающие вязкость, могут быть включены в вовлеченный водный объем. Яичный лецитин (20 г) и 5 г окситетрациклина-дигидрата помещали в стакан и добавляли водный 12,5-ный (масса/объем) глюкозы и 10 ммоль/л буфера Трис с pH 7,4, получая окончательный объем 100 мл. Обработку звуком и регулирование pH осуществляли, как в примере 1. Четыре отдельные загрузки были объединены и хранились в холодильнике в течение ночи в емкости с завинчивающейся крышкой. После того как препарат осадился, верхнюю его часть 87,5% (350 мл) отводят, оставляя микрокристаллы ОТЦ, покрытые лецитином, и небольшую часть верхней фазы (фосфолипид) в суспензии. Анализ показал, что нижняя фаза содержит 40% (масса/объем) ОТЦ. (Препараты ОТЦ могут быть сконцентрированы путем седиментации до 44% (масса/объем)).
Периферические фосфолипидные пузырьки были приготовлены отдельно и смешаны с концентрированными, покрытыми лецитином микрокристаллами. Добавляют 5 г яичного лецитина и 0,1 г пропилпарабена (предохранитель) к 45 мл водного раствора 12,5% глюкозы и 5% карбоксиметилцеллюлозы, и смесь обрабатывают звуком на восьмом уровне мощности в течение 15 мин. Это привело к образованию густой, но способной к впрыскиванию суспензии пузырьков лецитина с увлеченной карбоксиметилцеллюлозой и гипертонической глюкозой.
Для завершения рецептуры смешивали 33 мл микрокристаллического препарата, содержащего 40% ОТЦ, покрытого лецитином, смешивали с 33 мл указанного выше препарата периферического липида. Эту смесь хранили в запаянных ампулах. Конечные концентрации составляли в массо/объемных окситетрациклин 20, лецитин 15, пропилпарабен 0,1, причем водная фаза содержала 12,5% (масса/объем) глюкозы и 10 ммоль/л буфера Трис с pH 5,0.
Эксперименты с внутримышечной инъекцией крысам показали, что препараты, приготовленные в гипертоническом растворе глюкозы или карбоксиметилцеллюлозы, или смешанные с периферическим липидом, обработанные звуком в присутствии этих агентов, обладают большей скоростью удаления окситетрациклина из места инъекции, чем изотонический контроль.
Указанный выше препарат (обозначенный как рецептура 1) инъецировался внутримышечно трем телятам массой около 300 фунтов (135 кг) в дозе 9 мг ОТЦ на 1 кг массы тела. На фиг.5 показаны средние (+/- стандартная ошибка) концентрации окситетрациклина в крови в зависимости от времени после инъекции. Кривая временной зависимости концентрации ОТЦ в крови является монотонной, причем концентрации между 0,5 и 1,0 мкг/мл поддерживаются в интервале времен между 2 и 120 ч (5 сут). Непрерывное выделение такого типа может быть полезным, поскольку при подходящей высокой дозировке животное может получать терапевтические концентрации в течение более 5 сут без необходимости повторной инъекции.
Результаты, аналогичные приведенным на фиг.5, были получены с композициями, указанными в табл.2.
Все эти образцы показали хорошую способность к впрыскиванию и физическую стабильность. В их фармакологической кинетике имелись незначительные отличия. Они не вызывали никакой боли при инъекции или какого-либо набухания в месте инъекции. Отсутствие боли при инъекции представляет особое преимущество по сравнению с продажными растворами.
Пример 6. Эритромицин антибиотик с плохой растворимостью в воде также был составлен в рецептуре в виде покрытых лецитином микрокапель по способу, аналогичному описанному в примере 4.
Водная растворимость эритромицина выше, чем окситетрациклина при нейтральном значении pH (2 мг/мл против 1,1 мг/мл). Известно, что эритромицин раздражает ткани при высоких концентрациях. Это было подтверждено в экспериментах этой заявки, в которых суспензию кристаллов эритромицина (20 массо-объемных ), суспензированных в пропиленгликоле, инъектировали внутримышечно крысам. Это приводило к сильной боли и повреждению при инъекции, так что необходимо было немедленно умерщвлять крыс. Следующий пример иллюстрирует, как использование лецитинового покрытия снижает раздражение, присущее медикаменту (также смотри пример 6), и как это раздражение может быть дополнительно снижено посредством внедрения в покрытый лецитином микрокристалл не растворимого в воде, фармакологически приемлемого масла. В табл.3 приведены результаты для двух композиций, приготовленных обработкой звуком и испытанных на крысах при внутримышечной инъекции.
Эти данные показывают, что при добавлении витамина E снижается повреждение, вызванное лекарством, до приемлемого уровня. Дополнительный защитный эффект витамина E наиболее вероятно обусловлен его способностью внедряться между микрокристаллом эритромицина и лецитиновым покрытием, создавая дополнительный буфер и барьер в отношении растворения эритромицина. Это служит примером того, как липидное покрытие модифицируется, для того чтобы снизить доступность внедренного медикамента.
Эксперименты автора изобретения с рецептурами эритромицина также позволили определить условия, при которых лиофилизированные формы микрокристаллов, покрытых фосфолипидом, являются эффективными. Препарат с 20% эритромицина, 15% лецитина был заморожен и помещен в аппарат для лиофилизации для того, чтобы получить порошок. При смешивании и завихрении с 10-ной (масса/объем) глюкозой это дает суспензию, которая была шприцуема и физически стабильна. Оценка размера частиц дает средний диаметр 0,7 мкм идентично исходному препарату.
Дальнейшие эксперименты показали, что эквивалентный лиофилизированный продукт может быть произведен без использования воды. Обработку звуком осуществляли во фтороуглероде Фреон Ти-Эф (трихлортрифторэтан). В сосуде для озвучивания смешивают 2 г эритромицина с 0,5 г яичного лецитина, используя стеклянную магнитную мешалку. Затем к смеси добавляют 7,5 мл фреона Ти-Эф. После этого смесь обрабатывают звуком в течение 30 мин, помещают в 250 мл-колбу Эрленмейера и через образец продувают газообразный азот. Колбу вращают для того, чтобы распределить равномерно по дну высушиваемый материал. Оставшийся твердый материал был высушен в течение ночи в лиофилизаторе Лабконко Бенч Топ (модель 75034). Полученный материал представлял собой желтоватые таблетки, которые были повторно суспендированы при умеренном встряхивании в разбавителе, состоящем из предварительно озвученной водной суспензии, содержащей 20% лецитина и 5,4% глюкозы. Повторно суспендированный эритромицин был шприцуемой, физически стабильной суспензией 20% (масса/объем) эритромицина в виде частиц диаметром 0,7 мкм (определитель размера частиц Коултер N 4).
Дополнительные эксперименты показали, что для этой методики требуется обработка звуком в растворителе, таком как фреон, в котором растворимость антибиотика минимальна. Указанную выше методику повторяли, проводя обработку звуком в хлороформе или этаноле, в которых эритромицин растворим. В обоих случаях был получен порошок, который при добавлении в водную среду надлежащим образом повторно не суспендировался. Это определяет требование для медикамента оставаться почти исключительно в кристаллическом состоянии во время процессов диспергирования и выпаривания.
Пример 7. Также было показано, что покрытая лецитином микрокристаллическая форма полезна для введения противоглистных медикаментов. Для таких приложений желательно, чтобы медикамент мог поглощаться из места внутримышечной инъекции в систему в течение 2 сут. Было найдено, что препарат с 10% (масса/объем) микрокристаллов альбендазола, покрытых лецитином (8%) дает частицы диаметром 470 нм (определитель размера частиц Коултера N 4), которые способны впрыскиваться и грубо стабильны в течение 60 нед. Препараты альбендазола могут быть сконцентрированы до 20% (масса/объем) посредством седиментации. При внутримышечной инъекции 25 мкл в задние лапы лабораторных крыс были получены данные о следующем фармакинетическом поведении (табл.4).
Макроскопические наблюдения за мышцами до экстракции выявили лекарственные отложения без видимых повреждений в окружающих тканях.
Инъекции 10-20 мл этого препарата внутримышечно или подкожно овцам хорошо переносились. Вскрытие трупа на 7-ые сутки выявило отложения, представляющие основную часть медикамента, с небольшим раздражением окружающей ткани. Медленное поглощение медикамента в циркулирующую кровь, вероятно, было следствием большого инъекционного объема и малой растворимости альбендазола в воде (менее 0,1 мг/мл). Это ограничивает терапевтическую полезность покрытой лецитином микрокристаллической формы для инъекции альбендазола, однако предвещает полезность для других медикаментов с низкой растворимостью в воде, для которых желательно медленное выделение в течение нескольких недель.
Указанный выше эксперимент был повторен с альбендазолсульфоксидом, более растворимым в воде аналогом этого медикамента. Были получены повышенные скорости поглощения в кровь. Данные о растворимости альбендазолсульфоксида следующие: 0,42 мг/мл в воде; 17,7 мг/мл в пропиленгликоле и 179,0 мг/мл в этаноле. Были приготовлены препараты следующих составов: (А) 20% альбендазолсульфоксида, 20% яичного лецитина в 5,4%-ном буферном водном растворе изотонической глюкозы; (Б) 20% альбендазолсульфоксида, 5% лецитина в пропиленгликоле. Оба препарата нормально осаждались в концентрации альбендазолсульфоксида, равной 20% (масса/объем). Образцы (0,1 мл) обоих препаратов были инъектированы внутримышечно шести крысам, мышцы были исследованы при вскрытии трупа через 2 сут после инъекции. Оба препарата показали небольшие отложения медикамента без раздражения в окружающей ткани. Пропиленгликоль, который имеет две гидроксильные группы и диэлектрическую постоянную 32, является примером полярного органического вещества, которое может заменять воду. В настоящем случае он оказался выгодным для долгосрочной химической стабильности медикаментов.
Эксперименты с телятами показали, что микрокристаллический препарат с 15% (масса/объем) альбендазолсульфоксида, 3,75% лецитина в пропиленгликоле был особенно полезным. При внутримышечной инъекции примерно 6 мл этого препарата телятам (доза 6 мг/кг) они не ощущали боли и наблюдалось едва заметное набухание через 24 ч. Спустя 7 сут при вскрытии трупа не наблюдалось остатка медикамента и отмечалось только незначительное обесцвечивание мышцы. Аналогичные результаты были получены с 15% (масса/объем) микрокристаллического альбендазолсульфоксида плюс 5% яичного лецитина, суспендированных в смеси 70/30 пропиленгликоля и воды. Оба этих препарата удовлетворяли существующим критериям на полезность препарата в ветеринарной медицине. Контрольные изучения этого препарата без добавки лецитина показали, что этот медикамент является сам по себе раздражающим, что подтверждается поведением животных, микроскопическими наблюдениями и гистологией. Это показывает, что лецитиновое покрытие помогает снизить локальную реакцию на это лекарство.
Пример 8. Нитросканат является нерастворимым в воде противогельминтным веществом, для которого были бы желательны внутривенные и внутримышечные инъекционные формы. Это вещество химически не стабильно в воде или в присутствии высокой относительной влажности. Оно также химически активно по отношению к аминам. Покрытые лецитином микрокристаллы были приготовлены обработкой звуком во фреоне, как описано в примере 6, и хранились в виде порошка. Повторное составление в композиции с водным носителем дает продукт, способный к инъекции. Повторное составление этого продукта с 5,7 объемами 20% (масса/объем) обработанного звуком лецитина, содержащего 0,1% пропилпарабена в 12,5%-ном растворе глюкозы, дает 10%-ную суспензию микрокристаллов нитросканата. Эта суспензия была шприцуемой и стабильной в течение нескольких часов. Размер частиц составлял примерно 500 нм по оценке микроскопических измерений.
Пример 9. Этот пример показывает, как покрытый фосфолипидом микрокристалл может использоваться в качестве системы подачи для нестероидных противовоспалительных медикаментов при подавлении воспаления. Препарат может инъецироваться для того, чтобы создать хранилище внутри мышцы, или может инъецироваться в ткань, подлежащую защите. В качестве примера был взят индометацин. Эта молекула является карбоновой кислотой со значением константы диссоциации рКа 4,5, но ее растворимость в воде при pH 7,0 составляла только 1,376 мг/мл. Для получения 3-ного (масса/объем) раствора необходимо повысить значение pH до 9,6. Хотя растворимость этого вещества в воде мала, в разбавленном состоянии оно обладает только умеренными коэффициентами распределения масло/вода: 55/1 оливковое масло/вода и 85/1 пентанол/вода.
Микрокристаллический препарат индометацина, покрытый лецитином, был приготовлен по следующей методике. Индометацин (500 мг) смешивался с яичным лецитином (2,0 г) с помощью стеклянной магнитной мешалки и добавлялся водный раствор 300 ммоль/л глюкозы, 10 ммоль/л буфера Трис (pH 7,4) до окончательного объема 10 мл. Обработка звуком в течение 30 мин привела к гомогенной суспензии покрытых микрокристаллов. Препарату дали сконцентрироваться посредством седиментации, для того чтобы получить окончательный состав 20% (масса/объем) индометацина и 20% лецитина со средним размером частиц 100 нм (определитель размера частиц Коултер N 4). Долговременная стабильность была хорошей. Происходило дополнительное осаждение, но препарат может быть вновь суспендирован тремя встряхиваниями.
Указанный выше препарат был испытан на крысах в качестве внутримышечного хранилища для противовоспалительной активности с использованием модели опухоли лапы, индуцированной каррагенаном. Доза 5 мг индометацина задавалась внутримышечной инъекцией в заднюю лапу 0,025 мл препарата или 0,0325 мл 15,4% -ного раствора при pH 10,5. Эффективность ингибирования опухоли лапы была оценена после вызова каррагенана на 1, 24, 48 и 72-ой час после инъекции. В каждый момент времени в группе обработки было по 3 крысы. Животные умерщвлялись после испытаний, макроскопическое наблюдение мышц было проведено после вскрытия трупа. Инъекции микрокристаллического препарата были безболезненными, о чем свидетельствовало отсутствие заметной вокализации или сокращения лапы во время инъекции и нормальное поведение после инъекции. Напротив, инъекция щелочного раствора приводила к вокализации, сокращению и хромоте инъецированной лапы (12 из 12 животных). На рис.6 показан временной ход среднего процента (± стандартное отклонение) против опухоли лапы (противоположна нога) после вызова Каррагенана на 72-ом часе для двух форм лекарства. Рисунок показывает, что микрокристаллический препарат дает 89% ингибирования, тогда как щелочной раствор дает только 38% Крысы после 72 часов были умерщвлены, а инъецированные мышцы были исследованы при вскрытии трупа. Мышцы, в которые инъецировали микрокристаллический индометацин, оказались нормальными (3 из 3 животных). Напротив, мышцы, в которые инъецировали щелочной раствор, имели пораженные области размером примерно 3х6х1 мм с заметным обесцвечиванием (2 из 3 мышц).
Указанное выше демонстрирует способность микрокристаллической рецептуры вводить высокие концентрации медикамента в ткани при минимальном раздражении. Это свидетельствует о полезности ее в качестве носителя для противовоспалительных медикаментов как в виде внутримышечных избыточных инъекций, так и в виде инъекций в воспаленную ткань или пространство (например, в синовиальную жидкость).
Пример 10. Этот пример показывает, что микрокристаллы, покрытые фосфолипидом, способны быстро выделять свое содержимое при внутривенной инъекции. Не растворимый в воде анестезирующий стероид-альфаксалон, подаваемый с помощью такого способа, становится доступным мозгу в пределах 10 с после внутривенной инъекции.
Микрокристаллический препарат альфаксалона, покрытый лецитином, был приготовлен обработкой звуком двух компонентов в водном растворе 300 ммоль/л глюкозы, 10 ммоль/л буфера Трис (pH 7,4) с получением препарата с 2% (масса/объем) альфаксалона, 2% яичного лецитина. Микрокристаллы имели диаметр 548±75 нм (определитель размера частиц Коултер N 4). Этот препарат был стабильным свыше 4 месяцев. Он разделяется, давая свободно текучий осадок, который смешивается при перевертывании и полностью суспендируется вновь при встряхивании.
Следующие данные показывают, что препарат дает быстрое общее обезболивание при внутренней инъекции. Указанный выше препарат (0,11-0,25 мл) инъецировался внутривенно (в хвостовую вену) самкам крыс (Харлен Эс-Ди) массой 200-250 г, чтобы получить дозы 10, 15 или 20 мг/кг. Доза 10 мг/кг сделала 6 из 10 крыс бесчувственными. Таким образом, эта доза приближается к величине ЭК50 (концентрация, дающая 50% эффекта). Дозы 15 и 20 мг/кг сделали всех животных бесчувственными (3 из 3 и 10 из 10 соответственно). Эти животные становились бесчувственными в течение 10 с от начала инъекции. Обезболивание было столь же быстрым, сколь сама инъекция, на которую требуется 15 с. Четыре типа качественных и количественных данных записывались как функция уровня дозы: (а) характерные, самопроизвольные изменения поведения при выходе из состояния обезболивания, (б) порог вокализации от электрического стимулирования и (в) хирургическая анестезия, определяемая по надрезу брюшной полости.
(а) Первым указанием на то, что начался процесс выхода из наркоза, было начало периодических спазмов. При дозировке 20 мг/кг это происходит через 30,6±16,8 мин (± стандартное отклонение, n ± 10). За ним следует возбуждение и борьба за выпрямление, что наблюдается через 50,6±22,0 мин, выпрямление через 57,9±22,4 мин. Нормальные реакции и произвольное поведение у животных восстанавливается через 84,0±25,5 мин.
(б) Определялся порог вокализации на электрическое стимулирование от внутрикожных электродов (игольчатые электроды на коже спины на расстоянии 3 мм; блок Грасс S44 выделения стимулятора и стимула давал квадратные волны шириной 1,5 мс и частотой 50 Гц, длительность стимула 2 с; амплитуда измерялась в мА). Порог вокализации более 7,0 мА соответствовал очень сильной анестезии; порог в интервале от 2,0 до 7,0 мА соответствовал промежуточному уровню обезболивания. Для неанестезированной кожи человека (внутрикожная вставка в ноге) стимул 7 мА вызывает непереносимую боль, а стимул 2 мА - резкую боль. При всех трех дозах для всех крыс, которые были без сознания, наблюдался неизмеримо высокий порог вокализации. Не было отмечено вокализации для максимального стимула в 15 мА. На фиг.7 показаны типичные результаты для отдельных крыс.
Ниже обобщены наблюдения над группами с тремя дозами (3 крысы в группе). Неизмеримо высокая анестезия длилась в течение по меньшей мере 5 мин для всех животных без сознания при дозах 10 и 15 мг/кг и в течение по меньшей мере 15 мин для всех животных при дозе 20 мг/кг. Длительность высокого уровня анестезии возрастала в зависимости от дозы, время составляло 26,7±20,8 мин для 10 мг/кг, 35,0±20,0 мин для 15 мг/кг и 41,7±7,6 для 20 мг/кг. Аналогичное время для промежуточного уровня обезболивания составило: 35±15,0 мин, 66,7±12,6 мин и 63,3±15,3 мин. Пороги для всех инъекцрованных крыс возвращались к базовым уровням через 2 ч после инъекции.
(в) Хирургическая анестезия была определена в отдельной группе крыс при периодах времени, приближающихся к максимальному, при котором электрический порог вокализации был более 15 мА. При дозе 10 мг/кг ни одно из трех испытуемых животных не реагировало на надрезающее вскрытие брюшной полости при времени 15 мин после инъекции. При дозе 20 мг/кг ни одно из трех испытуемых животных не реагировало на вскрытие брюшной полости через 45 мин после инъекции (сразу же после этой демонстрации хирургической анестезии животные были умерщвлены удушением углекислым газом).
Приведенные выше данные показывают, что покрытые лецитином микрокристаллы альфаксалона могут растворяться, поступать в мозг и производить общую анестезию в пределах 10 с от их внутривенной инъекции. Эти данные также показывают, что этот препарат обладает полезностью в качестве индукционного агента и в качестве общего анестезирующего средства, способного к инъекции.
Пример 11. В этом примере препарат альфаксалона из примера 10 характеризуется в отношении структуры и поведения при растворении. Препарат с 2% (масса/объем) альфаксалона был получен путем обработки звуком медикамента (2%) вместе с 2% (масса/объем) яичного лецитина и 0,05% красителя Найл Ред в среде 300 ммоль/л глюкозы, 10 ммоль/л буфера Трис (pH 7,4) при высокой мощности в течение 20 мин. Как и в примере 3, Найл Ред флуоресцентный краситель, который обладает средством к липидам и фосфолипидам, был использован для визуализации и слежения за лецитином в препарате. Анализ определителем субмикронных частиц Коултер N 4 сразу же после разбавления в растворе, насыщенном альфаксалоном, дал значение среднего диаметра частиц 0,52±0,03 мкм. При визуализации препарата на пластинке с использованием флуоресцентного микроскопа Лейтц Ветцлар Диалюкс 20 были выявлены частицы этого размера. Частицы состояли из бесцветных, двоякопреломляющих кристаллов сферической или округленной формы, окруженных интенсивным гало красной флуоресценции с диаметром, примерно в 1,2-1,7 раз больше, чем диаметр кристалла. Флуоресценция была интенсивной вблизи от поверхности кристалла и была последовательно более диффузной с увеличением расстояния от поверхности кристалла.
Соотношение между первичным и вторичным лецитиновыми покрытиями было определено в эксперименте фракционирования аналогично описанному в примере 3. Препарат осаждался в клинической (для крови) центрифуге при максимальной скорости в течение 15 мин. Жидкость над осадком выливали. Аликвоты (10 мкл) исходного препарата и его надосадочной жидкости были добавлены в кювету, содержащую 2,5 мл этанола, и определялась флуоресценция (Фл) красителя, измеряемая флуорометром. Осадок был повторно суспендирован в среде глюкоза/буфер Трис для того, чтобы восстановить первоначальный объем; небольшие порции были взяты для анализа размера частиц и микроскопических наблюдений. Эта последовательность была повторена суммарно три раза. В табл.5 показаны характеристики размера частиц.
Микроскопическое наблюдение препарата 1-го суспендирования показало, что микрокристаллы сохраняют свои гало красителя, но эти гало были гораздо тоньше и менее интенсивны. Кроме того, микрокристаллы были сгруппированы в агрегаты диаметром примерно 2,5 мкм. Тонкие слои красителя были видимы между микрокристаллами в агрегате. Наблюдения препаратов 2-го и 3-го суспендирования выявили еще более крупные агрегаты (с диаметром примерно в 8 раз больше диаметра частиц исходного препарата). Эти агрегаты демонстрировали слабо розовую флуоресценцию.
Данные по флуоресценции в табл.5 показывают, что 98% лецитина в препарате представляют собой периферический фосфолипид (фиг.1), который может диссоциироваться при промывке. Как следует из данных флуоресценции красителя, 2,22% лецитина прочно ассоциированы с микрокристаллами альфаксалона. Это представляет собой первичное покрытие. Предполагая равные плотности медикамента и лецитина, легко можно рассчитать, что распределение этого количества лецитина на микрокристалле диаметром 520 нм приведет к слою толщиной 1,9 нм или 19 А. Эта величина очень близка к ожидаемой толщине монослоя лецитина. Этот эксперимент также демонстрирует роль периферического лецитина для предотвращения агрегирования микрокристаллов. Его удаление позволяет микрокристаллам приблизиться на малое расстояние, чтобы агрегироваться под действием сил дальнего порядка.
Пример 12. Следующий пример показывает, как покрытый фосфолипидом микрокристалл может использоваться в качестве средства, обеспечивающего долгосрочную анестезию кожи при отдельной инъекции. Черней (патент США 2803582, 1957) описал, каким образом водорастворимый местный обезболивающий агент тетракаин может быть переведен в нерастворимое в воде состояние путем образования соли иодистоводородной кислоты (HI). Гудман и Джиллман (Фармакологические основы терапевтики. 7-ое издание, изд-во Максиллан Паблишинг Ко. Нью-Йорк, 1985, с.312) цитируют исследование Л.С.Чернея (в журнале Anesth. Ahalg, 1963, т.42, с.477-481), демонстрирующее, что кристаллы этой соли могут разбрасываться на хирургические раны для того, чтобы обеспечить местное обезболивание длительностью 45 ч. Однако ссылка на 1988 PDR указывает, что иодистоводородная соль тетракаина не является промышленно доступной для клинического использования в США. Настоящий пример показывает, как можно увеличить полезность изобретения Чернея, используя покрытые лецитином микрокристаллы.
Нерастворимый иодистоводородный тетракаин был приготовлен путем добавления иодида калия (KI) к насыщенному водному хлористоводородному тетракаину. Осадок повторно суспендируют и промывают несколько раз водой и затем сушат. Для препарата А в пробирку добавляют 10 г иодистоводородного тетракаина и 1 г яичного лецитина, затем добавляют до окончательного объема 10 мл 5,4-ного раствора глюкозы, 10 ммол/л буфера Трис (pH 7,0). Материал обрабатывают звуком (контролируя температуру) в течение 20 мин, получая белую суспензию. Ей дают осадиться в течение ночи и верхнюю половину выливают. Препарат В получают аналогичным образом. Иодистоводородный тетракаин (0,50 г) и 1,0 г яичного лецитина совместно обрабатывают звуком в объеме 10 мл. Верхние 7,5 мл смеси выливают, а донную часть 2,5 мл повторно суспендируют, получая окончательный препарат. Оба препарата содержали 20% (масса/объем) иодистоводородного тетракаина и 10% яичного лецитина. Оба препарата обнаружили тенденцию к осаждению с образованием 30-ного (масса/объем) иодистоводородного тетракаина. Долгосрочная стабильность обоих препаратов была хорошей.
Препарат А ( 1,1 мл) был введен подкожной инъекцией в шкуру на спине крыс, поднимающих колесико диаметром примерно 1,0 см, которое было разграничено пером с фетровым наконечником. Степень анестезии в инъецированной шкуре определялась в испытании шоковой вокализации с использованием вживленных внутрикожных электродов, расположенных в центре инъецированной области. Порог вокализации был неизмеримо высок (более 15 мА) в течение первых трех часов после инъекции. Были испытаны 4 крысы в течение временного интервала 22-25 ч после инъекции. Для этой группы средний (± стандартное отклонение) порог вокализации составил 6,6±2,6 мА, что указывает на хорошую анестезию. Повторное испытание этой группы в интервале 41-44 ч после инъекции показало, что обезболивание убывает. Для трех из четырех животных состояние шкуры было нормальным. У одной крысы наблюдалось коричневатое пятно диаметром около 2 мм в середине инъецированного участка. В качестве контроля двум животным вводили инъекцию 0,1 мл раствора гидрохлорида тетракаина. Это привело к высокому уровню обезболивания (свыше 15 мА) вначале, но анестезирующий раствор вызывал сильное повреждение ткани со струпьями, которые наблюдались на вторые сутки, так что измерение анестезирующего эффекта не было ни практичным, ни разумным. Это было подтверждено результатами, полученными на двух дополнительных крысах с двумя инъекциями каждой. Все четыре инъецированные области были полностью коричневыми и покрытыми струпьями на 24-ый час, причем на 48-ой час наблюдались кратеры.
Заявитель провел эксперимент с препаратом В на самом себе. Были сделаны две внутрикожные инъекции 0,15 мл препарата В в кожу икроножной мышцы изнутри правой ноги в местах на 12 и 22 см ниже колена. Инъекция привела к полосам с диаметром примерно 1,1 см. При инъекции не ощущалось боли. Эти полосы исчезают примерно в течение 30 с. Инъецированные места были испытаны на обезболивание булавочного укола и раздражения холодом в течение следующих 24 ч.
На фиг.8 показаны результаты обезболивания булавочного укола по пятибальной шкале (4/4= полной нечувствительности, 0/4=полной чувствительности на острие иглы). На фиг.8 также приведены наблюдения для раствора 2- и 5%-ого хлористоводородного тетракаина. Микрокристаллический препарат, покрытый лецитином, продемонстрировал полное обезболивание в течение 7-9 ч после инъекции с возвращением 50%-ной чувствительности в период 12,5-14,5 ч и полной чувствительности на 16-21 ч. Инъекция не приводила к раздражению. На 11 мин или 1,5 ч наблюдалась легкая эритрема. Инъецированные области проявились и стали полностью нормальными к 24 часу. Единственным реальным средством для различения инъецированной и неинъецированной ткани была повышенная чувствительность к грубому трению в течение 1-5 сут после инъекции.
В качестве контроля в указанном выше эксперименте автор изобретения ввел себе инъекции растворов 1, 2 и 5%-ого хлористоводородного тетракаина в объеме 0,15 мл. С целью снижения степени повреждения от концентрированных растворов значения pH регулировались от 5,3 до 6,5. Физиологическая тоничность поддерживалась за счет включения глюкозы (4,3, 3,2 и 0% соответственно).
На фиг. 8 показано, что эти растворы обеспечивают полное обезболивание длительностью не более 2 ч. 1 и 2-ный растворы хлористоводородного тетракаина приводят только к мягкой эритреме с возвращением нормального цвета после убывания анестезии. Раствор 5%-ного гидрохлорида тетракаина приводил к образованию ярко-красных пятен (диаметр 7 мм) в центре и затвердению на 27 мин. Приведенные значения анестезии были взяты на их периферии. Это место было воспаленным после убывания анестезии. Красные пятна рассосались в струпья на 7-ые сутки, которые сохранялись до 21-ых суток. На 48-ые сутки на этом месте был струп диаметром 2 мм, окруженный пятном розоватой кожи (диаметр 1 см), чувствительной к прикосновению и припухлой примерно на 1 мм. Все это рассосалось в рубец, который просуществовал в течение 1 г. Такой плохой исход для 5% -ного (масса/объем) раствора тетракаина находится в полном контрасте с превосходными результатами, полученными для 20% (масса/объем) микрокристаллов гидроиодида тетракаина.
Приведенные выше данные показывают, что использование метода микрокристалла, покрытого лецитином, в сочетании с изобретением Чернея позволяет осуществлять инъекцию обезболивающего средства в 4 раза более высокой концентрации, обеспечивая безопасную, обратимую анестезию, пятикратную по длительности.
Пример 13. Этот пример показывает, что частицы воскообразных веществ могут быть покрыты и стабилизированы слоем лецитина. Эти микрочастицы, покрытые фосфолипидом, могут быть получены из твердых материалов, совместимых с фосфолипидом, которые плавятся между физиологической температурой (37oC) и 100o C. Настоящий пример иллюстрирует это использование парафинового воска. Парафин (3,35 г) расплавляют в водяной бане при 60oC. В стакан помещают 1,35 г яичного лецитина и добавляют водный раствор 300 ммоль/л глюкозы, 10 ммоль/л буфера Трис (pH 7,0) до окончательного объема 47 мл и гомогенизируют. К гомогенизированному лецитину добавляют жидкий парафин и смесь обрабатывают звуком в течение 30 мин, чтобы получить молочно-образную однородную суспензию. Стакан закрывают и дают ему охладиться до комнатной температуры. В результате получают суспензию парафиновых частиц субмикронного диаметра, покрытых лецитином, которая была стабильной свыше двух недель. Повторение этой методики, но в отсутствие фосфолипида привело к осаждению твердого парафина.
Приведенный выше пример показывает возможность получения покрытых лецитином микрочастиц из материала с температурой плавления ниже точки кипения воды и выше предполагаемой температуры использования (37oC). Фармацевтически приемлемые воски и твердые вещества биологического и синтетического происхождения включают (но не ограничиваются ими) гидрированное касторовое масло, цетостеариловый спирт, цетиловый спирт, сложный цетиловый эфир воска, миристиловый спирт, петролатум, парафин и различные воски (эмульгирующийся, микрокристаллический, белый, желтый и др.). Кроме того, возможно использование этих материалов для обеспечения воскообразного покрытия лекарственных медикаментов, которые, в свою очередь, покрыты фосфолипидом. Это воскообразное покрытие будет дополнительно снижать скорость выделения медикамента, таким образом продлевается срок его действия.
Микрокристаллы парафина, покрытые лецитином или альтернативно тристеарином в качестве биоразлагаемого воска, могут обладать очень большим временем жизни в инъецируемых тканях. Вероятно, что они могут быть полезными для фиксации и захвата в ловушку (см. пример 16) водорастворимых антигенов или мембранных фрагментов в мышце или коже, для того чтобы увеличить эффективность вакцинации (используется как вспомогательное средство).
Пример 14. Следующий пример показывает, что микрокристаллы, покрытые лецитином, могут образовываться в присутствии не смешивающихся с водой органических растворителей, в которых кристаллическое лекарство является нерастворимым. Этот пример основывается на дантролене средстве релаксации мышц. Его физическая форма представляет собой ярко-оранжевые кристаллы с температурой плавления 279-280oC и с низкой растворимостью в воде. В пробирку помещают 7,9 мг дантролена и добавляют (кумулятивно) следующие растворители, причем медикамент не растворяется: 0,3 мл минерального масла; + 0,4 мл н-дибутилового эфира; + 0,4 мл метоксифлурана; + 0,3 мл метоксифлурана; + 0,3 мл метоксифлурана; + 0,3 мл минерального масла. Указанную выше смесь обрабатывают звуком с микронаконечником в течение 15 мин. Это приводит к образованию дисперсной суспензии кристаллов дантролена, которые осаждаются примерно за 15 мин. Смесь повторно озвучивают, удаляют 0,1 мл и добавляют их в пробирку, содержащую 19,8 мг дилаурилфосфатидилхолина (лецитин). При встряхивании лецитин смачивается, но не растворяется. В пробирку добавляют 1,5 мл изотонического рассола и содержимое обрабатывают звуком. Это приводит к образованию желтоватой суспензии, напоминающей по консистенции и внешнему виду гоголь-моголь. Через 2 сут хранения содержимое пробирки разделяется на три слоя, которые удаляются из нее отдельно. Нижний слой имеет объем примерно 0,05 мл и представляет собой желто-красноватую массу, которая легко вновь суспендируется в изотоническом рассоле при осторожном встряхивании. Он содержит массу дентролена и состоит из микрокристаллов дантролена, смоченных органическими растворителями и покрытых слоем лецитина. Средний слой, который представляет собой основную массу объема, был очень мутным. Верхний слой был окрашен светлее. Средний и верхний слои представляли собой покрытые лецитином микрокапельки, которые описаны автором изобретения в патенте США N 4725442 (1988). Эти микрокапельки в среднем слое содержали больше метоксифлурана, а микрокапельки в верхнем слое содержали больше минерального масла. Этот пример показывает, что в случае выбора стабильного кристаллического лекарственного вещества, плохо растворимого в масле, покрытые лецитином микрокристаллы будут самопроизвольно образовываться при обработке звуком, даже если органический растворитель присутствует в больших количествах. Этот пример дает возможность понять суть физических взаимодействий, обуславливающих стабильность микрокристаллов, покрытых фосфолипидом.
Пример 15. Этот пример показывает, каким образом покрытые лецитином микрокристаллические препараты противогельминтного медикамента альбендазола могут быть разбавлены, чтобы получить стабильные суспензии, подходящие для назначения с питьевой водой для домашней птицы и крупного рогатого скота. Концентрированный препарат (20% (масса/объем)) альбендазола и 10% лецитина) был получен, как описано в примере 7. Аликвоту разбавляют в 400 мл водопроводной воды, получая суспензию с концентрацией 0, 25 мг/мл, которая хранится без перемешивания в закупоренной 0,5-литровой бутыли для образцов. Анализ размера частиц, осуществленный сразу после разбавления, показал, что 10% материала это частицы размером 254±200 нм, 85% имеют размеры 2,7± 0,5 мкм и 5% свыше 3 мкм. Спустя 64 ч, только 45% медикамента осадились в придонной трети бутыли. На дне бутыли находилась тонкая прозрачная пленка жидкости. Она легко вновь суспендировалась при однократном встряхивании. Анализ размера частиц показал, что 54% материала это частицы размером 17 ±11 нм, 13% частицы 3,0±0,3 нм и 32% частицы более 3 мкм. Это испытание показало, что покрытый лецитином микрокристалл в диспергированной форме может использоваться в системах автоматического разбавления (пропорционаторы), даже в случаях прекращения потока более, чем на 5 сут.
Пример 16. Этот последний пример показывает, что покрытые лецитином микрокристаллы являются полезным средством замедления выделения биологических молекул после инъекции в ткань. Эта полезность включает постоянное выделение биореагента после инъекции "про запас" и продленное удерживание вирусного или бактериального антигена в процессе вакцинации. В качестве примера водорастворимой биологической молекулы был взят альбумин бычьей сыворотки (АБС, меченный изотопом 14-С). АБС был подмешан до окончательной концентрации 27 мкг/мл к предварительно образовавшимся микрокристаллам окситетрациклина (20 массо-об. окситетрациклина, 20% лецитина), приготовленных, как в примере 1. Лабораторным крысам вводили инъекции этой смеси (а) внутрикожно или (б) внутримышечно, причем места кожной и мышечной инъекции были проанализированы на 14-С радиоактивность АБС, оставшуюся при умерщвлении спустя 2 сут. Контрольными были те же концентрации АБС в изотоническом растворе глюкозы и те же самые концентрации АБС, смешанного с лецитиновыми пузырьками (20 массо-об.), приготовленными обработкой звуком. Из табл.6 видно, что повышенные уровни радиоактивности 14-С АБС были найдены в местах на коже и в мышцах для смеси покрытых лецитином микрокристаллов окситетрациклина.
Эти данные позволяют предположить, что покрытые фосфолипидом микрокристаллы могут удерживать биореагенты и антигены в своем промежуточном водном пространстве, снижая скорости их выделения из места инъекции и таким образом продлевая их активное действие. Полезность покрытых микрокристаллов для назначения биореагентов или в качестве вакцинного вспомогательного агента (соответственно) может быть увеличена посредством включения в микрокристаллы соответствующего медикамента, подавляющего или стимулирующего иммунитет.
Реферат
Использование: в медицине для введения высоких концентраций водонерастворимого лекарственного вещества. Сущность изобретения: водная суспензия содержит микрокристаллы водонерастворимого лекарственного вещества, находящегося в твердом состоянии, диаметром от 0,05 до 10 мкм в количестве от 0,01 до 40 мас.%. Микрокристаллы покрыты капсулирующим первичным слоем мембранообразующего амфипатического липида толщиной от 0,3 нм до 3 мкм. По второму варианту микрокристаллы покрыты капсулирующим первичным слоем липида, состоящим из покрывающего и обволакивающего слоев указанного липида, и последующим вторичным слоем толщиной от 25 нм до 3 мкм, состоящим из указанного липида в форме везикул, ассоциированных с микрокристаллами лекарственного вещества, окружающих их и распределенных в суспендирующей среде. Массовое соотношение лекарственного вещества к липиду составляет от 1:1 до 1000:1. Мембранообразующий амфипатический липид представляет собой фосфолипид. 4 с. и 11 з.п. ф-лы, 9 ил., 6 табл.

Комментарии