Фармацевтические композиции, содержащие антиретровирусное лекарство и улучшитель фармакокинетики - RU2745204C2
Код документа: RU2745204C2
Чертежи




Описание
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
В данной заявке испрашивается приоритет по отношению к находящейся одновременно в процессе рассмотрения индийской предварительной заявке на патент с серийным номером 201621005051, поданной 12 февраля 2016 г., находящейся одновременно в процессе рассмотрения индийской предварительной заявке на патент с серийным номером 201621032504, поданной 23 сентября 2016 г., и находящейся одновременно в процессе рассмотрения индийской предварительной заявке на патент с серийным номером 201621040945, поданной 30 ноября 2016 г. Данные заявки включены в данный документ посредством ссылки во всей их полноте.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фармацевтическим композициям, содержащим по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики. Согласно настоящему изобретению также предложен их способ изготовления и применение указанных композиций для предупреждения, лечения или профилактики заболеваний, вызванных вирусами, конкретно вызванных ретровирусами или вирусом гепатита В.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Вирус иммунодефицита человека (ВИЧ) - вирус, который вызывает синдром приобретенного иммунодефицита (СПИД) - стал одним из самых серьезных в мире предметов озабоченности для системы здравоохранения. ВИЧ принадлежит к классу вирусов, называемых ретровирусами. Ретровирусы представляют собой РНК(рибонуклеиновая кислота)-вирусы, и для репликации (дупликации) данные вирусы должны делать копию ДНК (дезоксирибонуклеиновая кислота) их РНК. Именно гены ДНК позволяют вирусу реплицироваться. Подобно всем вирусам, ВИЧ может реплицироваться только внутри клеток, захватывая аппарат клетки для репродукции. Однако только ВИЧ и другие ретровирусы, очутившись внутри клетки, используют фермент, именуемый обратной транскриптазой, для превращения их РНК до ДНК, которая может включаться в гены клетки-хозяина.
ВИЧ разрушает CD4-позитивные (CD4) Т-клетки, которые представляют собой критически важные лейкоциты для поддержания функции человеческой иммунной системы. Разрушение данных клеток оставляет людей, инфицированных ВИЧ, уязвимыми к другим инфекциям, заболеваниям и другим осложнениям. Данные клетки, иногда именуемые «Т-хелперными клетками», играют центральную роль в иммунном ответе, сигнализируя другим клеткам в иммунной системе о выполнении их специальных функций. Поскольку ВИЧ атакует данные клетки, человек, инфицированный данным вирусом, является менее оснащенным для отражения инфекции и заболевания, что, в конечном счете, приводит к развитию СПИД.
Здоровый, неинфицированный человек обычно имеет от 800 до 1200 Т-клеток CD4 на кубический миллиметр (мм3) крови. ВИЧ, по-видимому, имеет особую аффинность в отношении человеческой лимфоцитарной клетки Т-4, которая играет жизненно важную роль в иммунной системе организма. Лейкоциты (WBC), инфицированные ВИЧ, приводят к уменьшению популяции WBC. В конечном счете, иммунная система делается неработающей и неэффективной против разных оппортунистических заболеваний. Во время не подвергающейся лечению ВИЧ-инфекции число данных клеток в крови человека прогрессивно уменьшается. При падении числа Т-клеток CD4 меньше 200/мм3 человек становится особенно уязвимым к оппортунистическим инфекциям и раковым заболеваниям, что характеризует СПИД - конечную стадию заболевания ВИЧ. Люди со СПИД часто страдают от инфекций легких, кишечного тракта, мозга, глаз и других органов, а также от ослабляющей потери массы тела, диареи, неврологических состояний и раковых заболеваний, таких как саркома Капоши и некоторые типы лимфом.
Первый случай был описан в 1981 г., в настоящее время имеется приблизительно 36,9 миллионов человек, живущих с ВИЧ, и десятки миллионов людей умерли от причин, связанных со СПИД, с начала эпидемии. В то время как новые случаи были описаны во всех регионах мира, приблизительно 70% обнаружены в Африке к югу от Сахары. Кроме того, согласно подборке данных за 2016 г. UNAIDS (Объединенная программа ООН по ВИЧ/СПИД), в 2015 г. было 36,7 миллионов человек, живущих с ВИЧ. На декабрь 2015 г., 17 миллионов человек, живущих с ВИЧ, имели доступ к антиретровирусной терапии. В 2015 г. во всем мире от причин, связанных со СПИД, умерло 1,1 миллиона человек.
ВИЧ является этиологическим фактором СПИД, который создал большую проблему для здравоохранения не только в Индии, но и в глобальном масштабе. ВИЧ вызывает постепенное разрушение иммунной системы организма, а также прогрессирующее повеждение цетральной и периферической нервной системы. С момента его исходного распознавания в начале 1980 гг. СПИД быстро распространялся и теперь достиг масштабов эпидемии в пределах относительно ограниченной доли населения. Интенсвное исследование привело к открытию ответственного агента - человеческого Т-лимфотропного ретровируса 111 (HTLV-111) - обычно именуемого вирус иммунодефицита человека или ВИЧ.
Доступные в настоящее время антиретровирусные лекарственные средства для лечения ВИЧ включают: зидовудин или AZT (Ретровир®), диданозин или DDI (Видекс®), ставудин или D4T (Зенит®), ламивудин или ЗТС (Эпивир®), зальцитабин или DDC (Хивид®), абакавира сульфат (Зиаген®), тенофовира дизопроксил фумарат (Виреад®), эмтрицитабин (Эмтрива®), Комбивир® (содержит ЗТС и AZT), Тризивир® (содержит абакавир, ЗТС и AZT), Эпзиком® (содержит абакавир и ламивудин), невирапин (Вирамун®), делавирдин (Рескриптор®), эфавиренз (Сустива®), саквинавир (Инвираза®, Фортоваза®), индинавир (Криксиван®), ритонавир (Норвир®), нелфинавир (Вирацепт®), ампренавир (Агенераза®), атазанавир (Рейатаз®), Эвотаз® (содержит атазанавир и кобицистат), фозампренавир (Лексива®), Калетра® (содержит лопинавир и ритонавир), энфувиртид (Т-20, Фузеон®), Трувада® (содержит Тенофовир и Эмтрицитабин), дарунавир (Прециста®), Презкобикс® (содержит дарунавир и кобицистат), долутегравир (Тивикай®), Триумек® (содержит долутегравир, абакавир и ламивудин), элвитегравир (Витекта®), Генвоя® (содержит элвитегравир, кобицистат, тенофовира алафенамид фумарат и эмтрицитабин), Стрибилд® (содержит элвитегравир, кобицистат, тенофовира дизопроксил фумарат и эмтрицитабин), ралтегравир (Исентресс®), Комплера® (содержит эмтрицитабин, тенофовира дизопроксил фумарат, рилпивирин) и Атрипла® (содержит тройную комбинацию фиксированной дозы тенофовира, эмтрицитабина и эфавиренза).
От 5 до 10% людей с ВИЧ также инфицированы вирусом гепатита В (часто именуемым соинфекцией). Люди с ВИЧ с меньшей вероятностью естественным образом освобождаются от гепатита В без лечения. Люди с ВИЧ и соинфекцией гепатитом могут иметь более быстрое прогрессирование заболевания печени и также могут не отвечать на лечение против гепатита В. Однако наличие гепатита В, по-видимому, не усугубляет заболевание ВИЧ. Инфекция вирусом гепатита В (HBV) является самой обычной хронической вирусной инфекцией в мире. Оценочно 2 миллиарда людей было инфицировано, и больше, чем 350 миллионов являются хроническими носителями вируса. HBV передается через контакт с инфицированной кровью или спермой.
Кроме того, вирусы СПИД (ВИЧ) и гепатита В являются замечательно сходными по наличию у них обратной транскрипции, по их происхождению от предков, общим генетическим элементам и их способам передачи. Оба являются гипермутирующими и существуют в виде квазивидов, главным образом, из-за ошибок в обратной транскрипции, хотя существует тяжелое ограничение в компетентности к репликации большинства мутантов вируса гепатита В. Они отличаются по отсутствию интегразы у вируса гепатита В и по их патогенезу у инфицированного хозяина. ВИЧ, главным образом, выживает посредством антигенной изменчивости, ускользания от иммунологического надзора и ослаблению иммунной функции посредством вирусных регуляторных контрольных элементов, ориентированных на то, чтобы ограничить смертельное повреждение хозяина. Вирус гепатита В выживает, главным образом, посредством мутации генов антигена/кора, что позволяет непосредственно уклоняться от разрушения цитотоксическими Т-клетками инфицированных клеток печени или опосредственно ограничивает разрушение инфицированных клеток посредством индукции анергии в ответе цитотоксических Т-клеток.
Кроме того, антиретровирусные лекарственные средства, такие как ламивудин, адефовир, энтекавир и тенофовир, были одобрены для лечения хронической инфекции HBV.
Активаторы или улучшители фармакокинетики используются для усиления эффективности антиретровирусных лекарственных средств. При совместном введении активатора или улучшителя фармакокинетики с антиретровирусным лекарственным средством улучшитель фармакокинетики препятствует разрушению антиретровирусного лекарственного средства, что вызывает то, что антиретровирусное лекарственное средство остается в организме в течение более длительного времени и в большей концентрации. Активаторы или улучшители фармакокинетики вызывают специфичное ингибирование ферментативной системы цитохрома Р450 3А4, приводя к увеличению плазматических концентраций совместно вводимых антиретровирусных лекарственных средств. Ингибиторы протеаз являются одним таким классом антиретровирусных лекарственных средств, которые обычно демонстрируют высокий генетический барьер в отношении резистентности к лекарственному средству и, следовательно, действительно требуют совместного введения активатора или улучшителя фармакокинетики. Из всех одобренных лекарственных средств для лечения ВИЧ, ритонавир и кобицистат называют «активаторами» или «улучшителями» фармакокинетики. Ритонавир используется из-за его способности ингибировать фермент цитохром Р450 (CYP) 3А4, метаболизирующий лекарственные средства. Ритонавир, который дают в низкой дозе, уменьшает метаболизм ингибиторов протеаз, таких как лопинавир и атазанавир, которые в значительной степени метаболизируются CYP3A4, усиливая, таким образом, экспозицию лекарственного средства. Кобицистат также является сильным ингибитором изоферментов CYP3A и увеличивает плазматические концентрации лекарственных средств, которые метаболизируются CYP3A, таких как ингибиторы протеаз, а именно атазанавир и дарунавир.
Помимо ритонавира и кобицистата имеется много встречающихся в природе веществ, которые описаны в литературе и могут исследоваться для улучшения фармакокинетической активности некоторых лекарственных средств.
Эти встречающиеся в природе вещества, которые действуют в качестве биоусилителей, представляют собой химические соединения, которые стимулируют и увеличивают биодоступность лекарственных средств, которые смешивают с ними, и не демонстрируют синергический эффект с лекарственным средством. Примеры данных биоусилителей включают пиперин, чеснок, Сагит carvi, Cuminum cyminum, лизергол, нарингин, кверцетин, ниазиридин, глицирризин, посконник крапиволистный, коровью мочу, продукт перегонки имбиря и т.д.
Данные «активаторы» или «улучшители» фармакокинетики могут уменьшать стоимость антивирусной терапии, уменьшать бремя приема таблеток для пациентов и/или уменьшать риск субтерапевтических противовирусных концентраций (например, развитие резистентности, а также увеличение приверженности к антивирусной терапии).
Однако данное фармакокинетическое усиление может быть ассоциировано с его собственными рисками. Взаимодействующее лекарственное средство, например, активатор или улучшитель, возможно должно вводиться в дозе, которая ингибирует устранение целевого лекарственного средства, а также не индуцирует его собственных побочных эффектов.
Соответственно, улучшитель, который обычно представляет собой мощный ингибитор, может ненамеренно ингибировать устранение других лекарственных средств, приводя к нежелательным вредным эффектам. Также, если доза улучшителя тщательно не корректируется или является неадекватной, или находится в избытке, она может, в конечном счете, вызывать либо уменьшение, либо увеличение концентрации целевого лекарственного средства.
Следовательно, рассматривая данные аспекты, выбор подходящей дозы усилителя играет жизненно важную роль.
Например, хотя ритонавир и имеет противовирусную активность, он вызывает нежелательные побочные эффекты, включающие желудочно-кишечные проблемы, особенно хроническую диарею и липидные нарушения. Затем был разработан кобицистат для продуцирования приблизительно такой же степени эффекта, что и ритонавир, но без противовирусной активности или любых других проблемных побочных эффектов.
Кобицистат представляет собой субстрат CYP3A4 (CYP2D6 представляет собой минорный путь метаболизма) и ингибирует его собственный метаболизм. Кроме того, кобицистат также ингибирует Р-гликопротеин (Р-gp) и CYP2D6, и, следовательно, имеется целый ряд потенциальных взаимодействий, которые могут происходить с использованием кобицистата.
Кроме того, пациенты, которых лечат против ВИЧ, всегда подвержены риску взаимодействий с другим лекарственным средством не против ВИЧ, и известно то, что кобицистат демонстрирует ключевые лекарственные взаимодействия с антацидами, бензодиазепамами, бета-блокаторами, блокаторами кальциевых каналов, лекарственными средствами против эректильной дисфункции, ингалируемыми/инъецируемыми кортикостероидами, статинами, пероральными контрацептивными прогестинами, рифампином и маравироком.
Улучшители или активаторы фармакокинетики, которые применяются в настоящее время, непреднамеренно ингибируют устранение других лекарственных средств, приводя к нежелательным вредным эффектам. Также не известно то, что применение пиперина и/или его структурных аналогов, таких как тетрагидропиперин, цис,транс-пиперин, транс,цис-пиперин, цис,цис-пиперин и транс,транс-пиперин, усиливает биодоступность таких антиретровирусных лекарственных средств.
Следовательно, сохраняется потребность в предложении комбинированной терапии активатора или улучшителя фармакокинетики с такими антиретровирусными лекарственными средствами для лечения ВИЧ, которая уменьшает дозу таких антиретровирусных лекарственных средств, побочные эффекты, демонстрируемые данными лекарственными средствами, а также поддерживает их оптимальную концентрацию. Кроме того, применение встречающегося в природе активатора или улучшителя фармакокинетики устранило бы или уменьшило бы взаимодействия с другими лекарственными средствами не против ВИЧ, которые вводились бы сопутствующе.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В некоторых воплощениях целью настоящего изобретения является предложение композиции, содержащей по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики.
В некоторых воплощениях другой целью настоящего изобретения является предложение композиции, содержащей по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики с ослабленными побочными эффектами.
В некоторых воплощениях еще одной другой целью настоящего изобретения является предложение композиции, содержащей по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики с ослабленными взаимодействиями с лекарственными средствами.
В некоторых воплощениях другой целью настоящего изобретения является предложение композиции, содержащей по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики для введения один раз или дважды в сутки.
В некоторых воплощениях другой целью настоящего изобретения является предложение композиции, содержащей по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики с пониженной дозой.
В некоторых воплощениях еще одной другой целью настоящего изобретения является предложение композиции, содержащей по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики в виде набора.
В некоторых воплощениях еще одной другой целью настоящего изобретения является предложение способа предупреждения, лечения или профилактики заболеваний, вызванных вирусами, конкретно вызванных ретровирусами, в частности синдрома приобретенного иммунодефицита или ВИЧ-инфекции, причем данный способ включает введение по меньшей мере одного антиретровирусного лекарственного средства и по меньшей мере одного активатора или улучшителя фармакокинетики.
В некоторых воплощениях еще одной другой целью настоящего изобретения является предложение способа лечения заболеваний, вызванных вирусами, конкретно вызванных вирусом гепатита В, причем данный способ включает введение по меньшей мере одного антиретровирусного лекарственного средства и по меньшей мере одного активатора или улучшителя фармакокинетики.
В некоторых воплощениях еще одной другой целью настоящего изобретения является предложение применения фармацевтической композиции, содержащей по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики для лечения или профилактики заболеваний, вызванных вирусами, конкретно вызванных ретровирусами, конкретно синдрома приобретеного иммунодефицита или ВИЧ-инфекции.
В некоторых воплощениях еще одной другой целью настоящего изобретения является предложение применения фармацевтической композиции, содержащей по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики для лечения заболеваний, вызванных вирусами, конкретно вирусом гепатита В.
Согласно одному аспекту настоящего изобретения предложена фармацевтическая композиция, содержащая по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики, и один или более чем один фармацевтически приемлемый эксципиент.
Согласно другому аспекту данного изобретения предложен способ получения фармацевтической композиции, содержащей по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики с по меньшей мере одним или более чем одним фармацевтически приемлемым эксципиентом.
Согласно другому аспекту настоящего изобретения предложен способ лечения заболеваний, вызванных вирусами, конкретно вызванных ретровирусами, особенно СПИД или ВИЧ-инфекции, причем данный способ включает введение терапевтически эффективного количества фармацевтической композиции, содержащей по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики согласно настоящему изобретению, пациенту, нуждающемуся в этом.
Согласно другому аспекту настоящего изобретения предложен способ лечения заболеваний, вызванных вирусами, конкретно вызванных вирусом гепатита В, причем данный способ включает введение терапевтически эффективного количества фармацевтической композиции, содержащей по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики согласно настоящему изобретению, пациенту, нуждающемуся в этом.
Согласно другому аспекту настоящего изобретения предложено применение фармацевтической композиции, содержащей по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики согласно настоящему изобретению, в изготовлении лекарственного средства для лечения заболеваний, вызванных вирусами, в частности, вызванных ретровирусами, особенно СПИД или ВИЧ-инфекции.
Согласно другому аспекту настоящего изобретения предложено применение фармацевтической композиции, содержащей по меньшей мере одно антиретровирусное лекарственное средство и по меньшей мере один активатор или улучшитель фармакокинетики согласно настоящему изобретению, в изготовлении лекарственного средства для лечения заболеваний, вызванных вирусами, в частности, вызванных вирусом гепатита В.
В некоторых воплощениях предложена пероральная или инъецируемая фармацевтическая композиция, содержащая терапевтически эффективное количество по меньшей мере одного антиретровирусного лекарственного средства и терапевтически эффективное количество по меньшей мере одного активатора или улучшителя фармакокинетики, или их производного.
В некоторых воплощениях предложена пероральная или инъецируемая фармацевтическая композиция, содержащая терапевтически эффективное количество по меньшей мере одного антиретровирусного лекарственного средства; терапевтически эффективное количество по меньшей мере одного активатора или улучшителя фармакокинетики, или их производного; и один или более чем один фармацевтически приемлемый эксципиент, содержащий носители, разбавители, наполнители, связующие вещества, смазки, скользящие вещества, разрыхлители, объемообразующие агенты, ароматизаторы или их любую комбинацию.
В некоторых воплощениях предложен способ лечения заболеваний, вызванных ретровирусами или вирусами геатита В, у пациента, нуждающегося в таком лечении, причем данный способ включает: введение фармацевтической композиции, содержащей (i) терапевтически эффективное количество по меньшей мере одного антиретровирусного лекарственного средства или антивирусного лекарственного средства; (ii) терапевтически эффективное количество по меньшей мере одного активатора или улучшителя фармакокинетики, или их производного; и (iii) один или более чем один фармацевтически приемлемый эксципиент, содержащий носители, разбавители, наполнители, связующие вещества, смазки, скользящие вещества, разрыхлители, объемообразующие агенты, ароматизаторы или их любую комбинацию.
В некоторых воплощениях предложен способ получения фармацевтической композиции, которая улучшает биодоступность антиретровирусного лекарственного средства, причем данный способ включает: смешивание терапевтически эффективного количества по меньшей мере одного антиретровирусного лекарственного средства и терапевтически эффективного количества по меньшей мере одного активатора или улучшителя фармакокинетики, или его производного с одним или более чем одним фармацевтически приемлемым эксципиентом с получением фармацевтической композиции.
В некоторых воплощениях предложен набор для лечения заболевания, вызванного ретровирусами или вирусами гепатита В, причем данный набор содержит терапевтически эффективное количество по меньшей мере одного антиретровирусного лекарственного средства и терапевтически эффективное количество по меньшей мере одного активатора или улучшителя фармакокинетики, или его производного, где по меньшей мере одно антиретровирусное лекарственное средство находится в отдельной композиции от по меньшей мере одного активатора или улучшителя фармакокинетики, или его производного.
В некоторых воплощениях предложен способ улучшения биодоступности перорального антиретровирусного лекарственного средства, включающий: предоставление терапевтически эффективного количества по меньшей мере одного антиретровирусного лекарственного средства и предоставление терапевтически эффективного количества по меньшей мере одного активатора или улучшителя фармакокинетики, или его производного.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
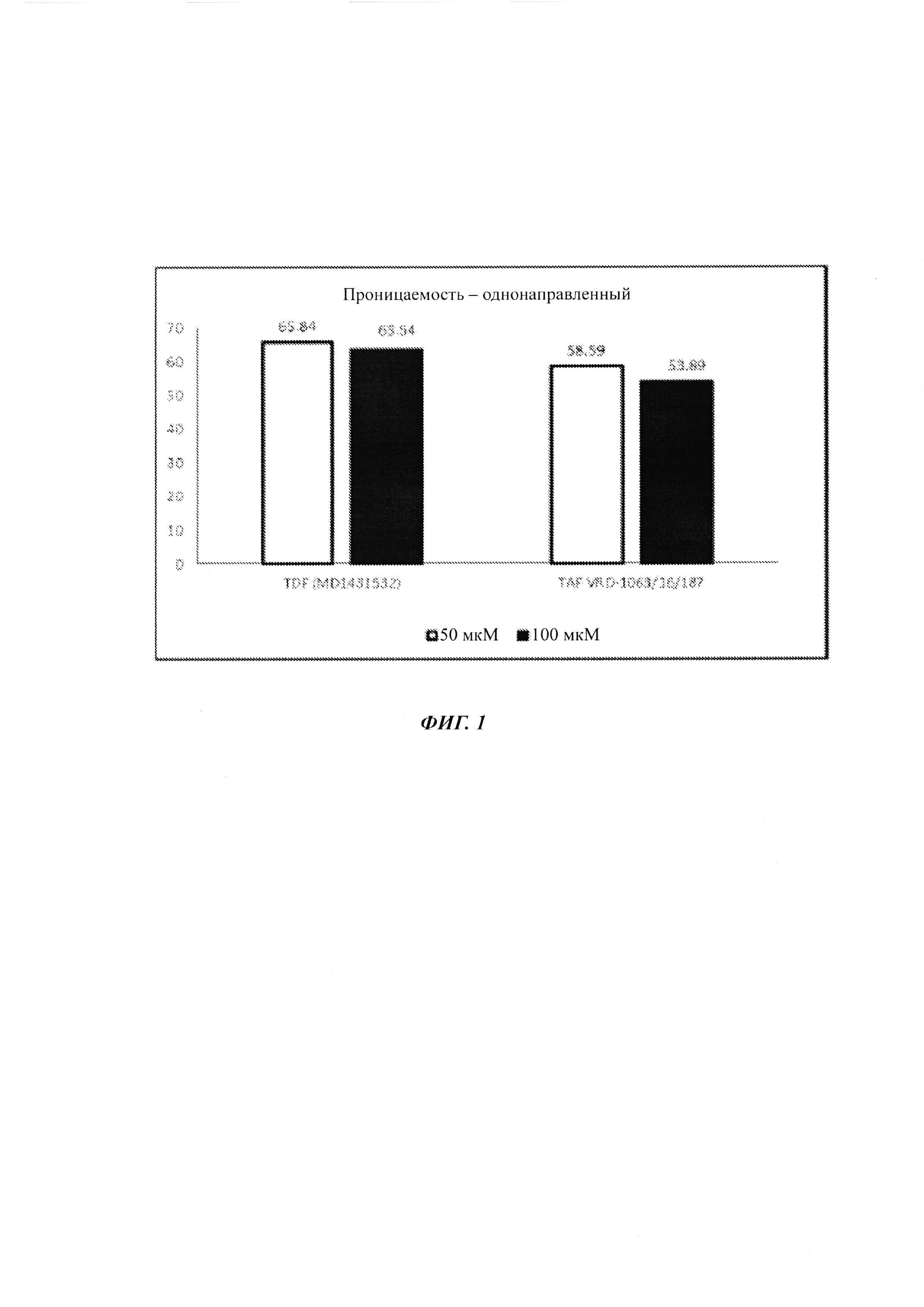
На ФИГ. 1 показана гистограмма результатов из однонаправленного анализа, показывающего проницаемость тенофовира алафенамид фумарата (TAF) и тенофовира дизопроксил фумарата (TDF). Наблюдали то, что TDF и TAF являются лекарственными средствами с проницаемостью от низкой до умеренной.
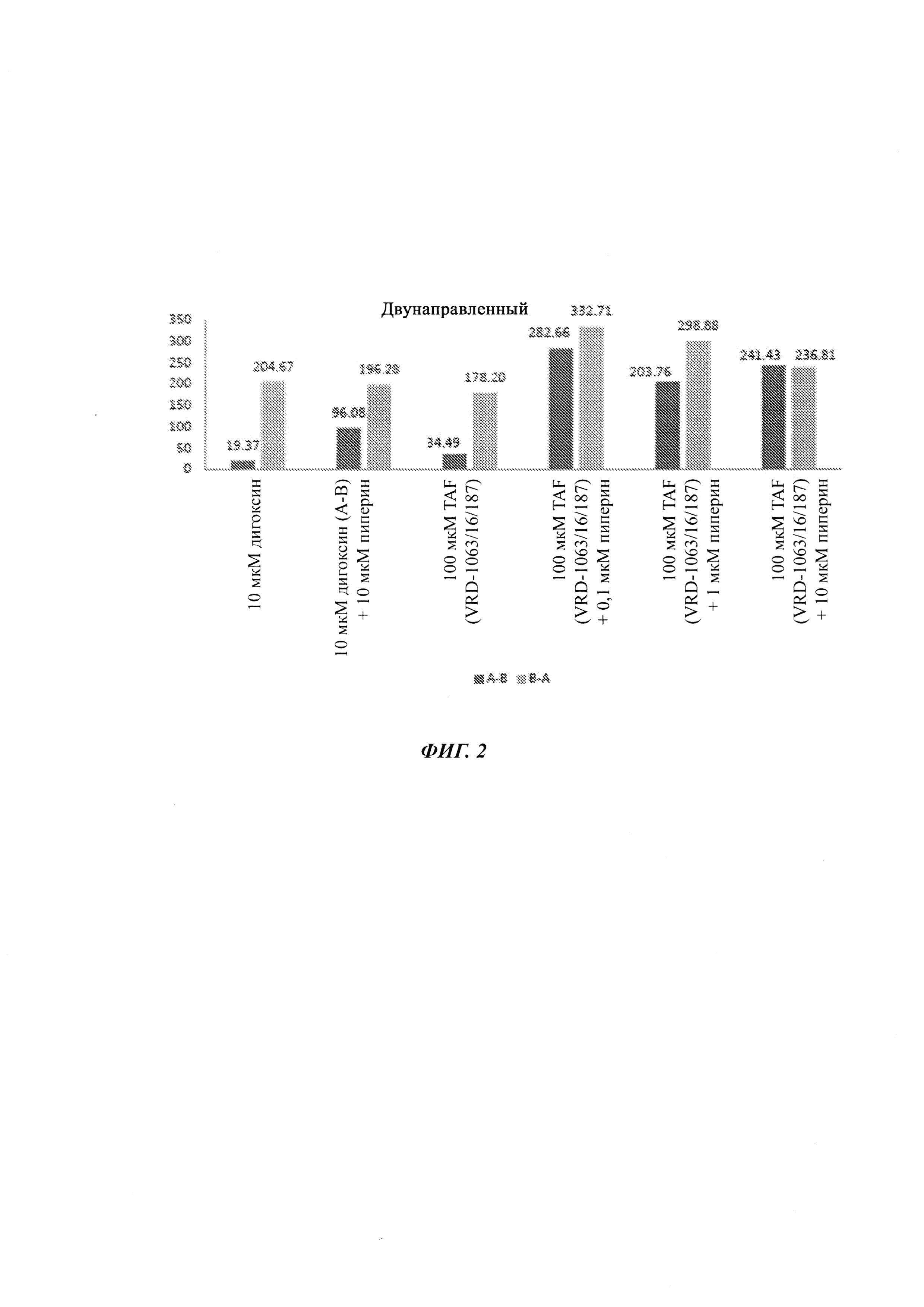
На ФИГ. 2 показана гистограмма результатов из двухнаправленного анализа 10 мкМ дигоксина, 10 мкМ дигоксина (А-Б) плюс 10 мкМ пиперина, 100 мкМ TAF
(VRD-1063/16/187), 100 мкМ TAF (VRD-1063/16/187) плюс 0,1 мкМ пиперина и 100 мкМ TAF (VRD-1063/16/187) плюс 10 мкМ пиперина. Результаты показали то, что поглощение TAF увеличивается пиперином посредством снижения отношения оттока TAF.
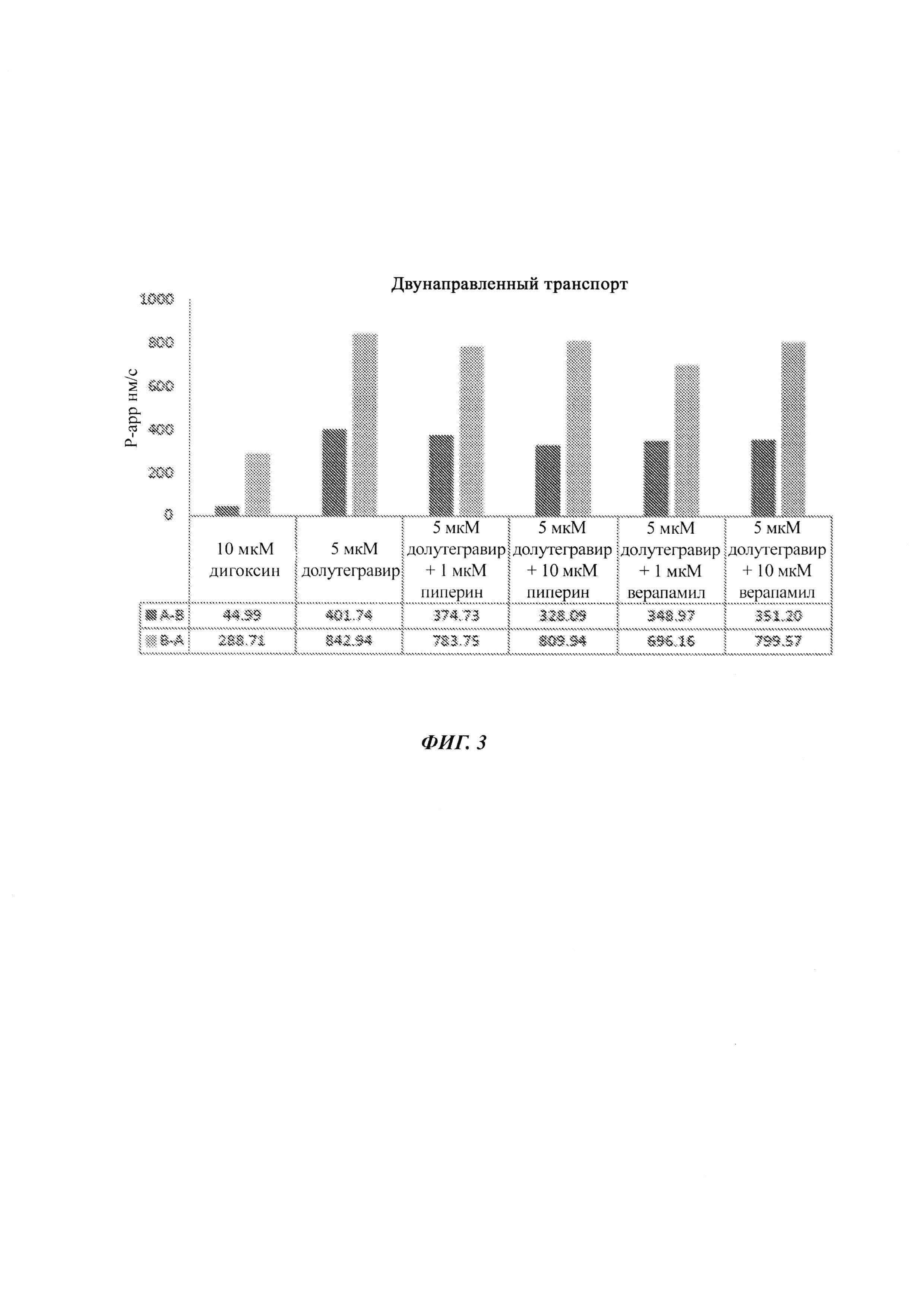
На ФИГ. 3 показана гистограмма результатов из двухнаправленного анализа 10 мкМ дигоксина, 5 мкМ долутегравира, 5 мкМ долутегравира плюс 1 мкМ пиперина, 5 мкМ долутегравира плюс 10 мкМ пиперина, 5 мкМ долутегравира плюс 1 мкМ верапамила и 5 мкМ долутегравира плюс 10 мкМ верапамила.
На ФИГ. 4 показана гистограмма результатов из двухнаправленного анализа 10 мкМ дигоксина, 40 мкМ дарунавира, 40 мкМ дарунавира плюс 1 мкМ пиперина, 40 мкМ дарунавира плюс 10 мкМ пиперина, 40 мкМ дарунавира плюс 10 мкМ кобицистата и 40 мкМ дарунавира плюс 100 мкМ кобицистата. Результаты показали то, что поглощение дарунавира увеличивается пиперином посредством снижения отношения оттока TAF.
На ФИГ. 5 показана гистограмма результатов из двухнаправленного анализа 10 мкМ дигоксина, 200 мкМ TDF, 100 мкМ TDF, 100 мкМ TDF плюс 10 мкМ пиперина и 100 мкМ TDF плюс 10 мкМ тетрагидропиперина. Результаты показали то, что поглощение TDF увеличивается пиперином посредством снижения отношения оттока, поглощение TDF увеличивается тетрагидропиперином посредством снижения отношения оттока, и сравнимое улучшение в проницаемости TDF наблюдали как посредством пиперина, так и тетрагидропиперина.
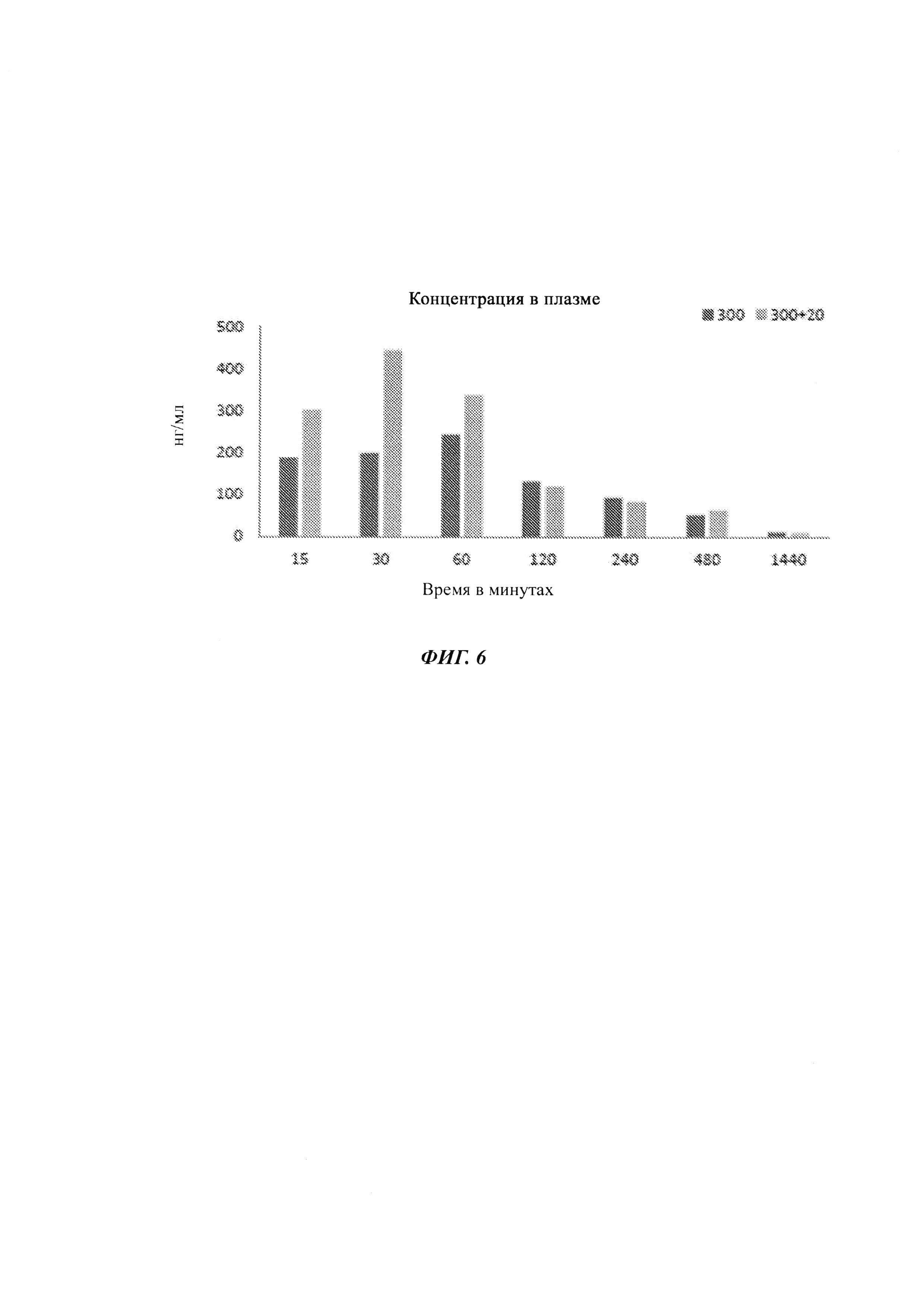
На ФИГ. 6 показана гистограмма плазматических концентраций тенофовира для 300 мг TDF и 300 мг TDF плюс 20 мг пиперина в разные моменты времени.
На ФИГ. 7 показана гистограмма плазматических концентраций тенофовира для 300 мг TDF и 300 мг TDF плюс 20 мг пиперина в разные моменты времени.
На ФИГ. 8 показаны зависящие от времени плазматические концентрации тенофовира для 300 мг TDF, 300 мг TDF плюс 20 мг пиперина и 150 мг TDF плюс 20 мг пиперина.
Следует понимать то, что графические материалы не нарисованы или сфотографированы в одном масштабе. Кроме того, соотношение между объектами на Фиг. может не приводиться в правильном масштабе и, на самом деле, может иметь обратную связь с реальным размером. Графические материалы предназначены для того, чтобы принести понимание и ясность относительно структуры каждого показанного объекта, и, таким образом, некоторые характеристики могут быть преувеличены для того, чтобы проиллюстрировать специфическую характеристику структуры.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Для лечения заболеваний, вызванных ретровирусами или вирусом гепатита В, особенно СПИД, ВИЧ-инфекции или гепатита В, важно, чтобы максимальное количество лекарственного средства достигало места действия. Большинство антиретровирусных лекарственных средств имеют или плохую растворимость и/или плохую проницаемость, что в значительной степени ухудшает биодоступность лекарственного средства.
Авторы настоящего изобретения обнаружили способы решить проблемы биодоступности таких антиретровирусных лекарственных средств. В частности, авторы изобретения обнаружили то, что свойства биодоступности данных лекарственных средств могут быть улучшены с использованием активатора или улучшителя фармакокинетики.
Повышенная биодоступность антивирусного леарственного средства раскрыта в нескольких ссылках. В статье Role of Piperine As A Bioavailability Enhancer, UMESH К PATIL et al International Journal of Recent Advances in Pharmaceutical Research October 2011; 4: 16-23 раскрыт пиперин в качестве улучшителя биодоступности.
В WO 2004067018 раскрыто применение экстрактов Carum carvi в качестве биоулучшителей либо одних, либо в комбинации с пиперином, либо с экстрактом Zinzeber officinale для улучшения биодоступности зидовудина.
Natural Bioenhancers: An overview, Deepthi V. Tatiraju et al, Journal of Pharmacognosy and Phytochemistry 2013; 2 (3): 55-60. В данной статье раскрыта комбинация пиперина с невирапином, в которой пиперин улучшал биодоступность невирапина.
Oral bioavailability enhancement of an anti-viral drug using an herbal bio-enhancer, Mohammad Asif, диссертация, представленная на рассмотрение в Ganpat University. В данной работе раскрыта комбинация пиперина с эфавирензом, в которой пиперин улучшал биодоступность эфавиренза.
Bioenhancement effect of piperine and ginger oleo resin on the bioavailability of atazanavir, Swati Prakash et al, International Journal of Pharmacy and Pharmaceutical Sciences Vol 7, Issue 10, 2015. В данной статье раскрыта комбинация пиперина с атазанвиром, в которой пиперин улучшал биодоступность атазанвира.
В WO 03084462 раскрыт способ изготовления фармацевтической композиции, содержащей антиретровирусный ингибитор протеаз, такой как индинавир, саквинавир, ампренавир, нелфинавир, лопинавир, и пиперин в одной фармацевтической композиции.
В некоторых воплощениях антиретровирусные лекарственные средства согласно настоящему изобретению включают нуклеозидные ингибиторы обратной транскриптазы (NRTI), ненуклеозидные ингибиторы обратной транскриптазы (NNRTI), ингибиторы обратной транскриптазы на основе нуклеотидного аналога, ингибиторы протеаз (PI), ингибиторы интегразы, ингибиторы слияния, ингибиторы CCR5, моноклональные антитела, ингибиторы гликопротеинов и их любые комбинации, но не ограничиваются ими.
В одном воплощении нуклеозидные ингибиторы обратной транскриптазы (NRTI) и ненуклеозидные ингибиторы обратной транскриптазы (NNRTI) включают ламивудин, абакавир, зидовудин, эмтрицитабин, диданозин, ставудин, лобукавир, энтекавир, априцитабин, ценсавудин, залцитабин, дексэльвуцитабин, аловудин, эфавиренз, амдоксовир, эльвуцитабин, фестинавир, рацивир, лерсивирин, рилпивирин, этравирин, стампидин, доравирин, дапивирин, но не ограничиваются ими.
В некоторых воплощениях нуклеозидные ингибиторы обратной транскриптазы (NRTI) и ненуклеозидные ингибиторы обратной транскриптазы (NNRTI) предпочтительно представляют собой абакавир, диданозин. Предпочительно доза абакавира варьирует от примерно 3 мг до примерно 300 мг, и доза диданозина варьирует от примерно 2 мг до примерно 200 мг для введения дважды в сутки.
В другом воплощении ингибиторы протеаз включают лопинавир, ритонавир, саквинавир, нелфинавир, ампренавир, индинавир, нелфинавир, атазанавир, ласинавир, палинавир, тирпранавир, фосампренавир, дарунавир или типранавир, но не ограничиваются ими. Предпочтительно ингибиторы протеаз представляют собой тирпранавир, дарунавир. Предпочтительно доза тирпранавира варьирует от примерно 5 мг до примерно 500 мг, и доза дарунавира варьирует от примерно 1 мг до примерно 800 мг для введения дважды в сутки. В некоторых воплощениях доза дарунавира варьирует от примерно 1 мг до примерно 500 мг, от примерно 20 мг до примерно 500 мг, от примерно 25 мг до примерно 500 мг, от примерно 30 мг до примерно 500 мг, от примерно 35 мг до примерно 500 мг, от примерно 25 мг до примерно 35 мг, от примерно 50 мг до примерно 400 мг или от примерно 100 мг до примерно 300 мг для введения дважды в сутки. В некоторых воплощениях доза дарунавира варьирует от примерно 1 мг, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, 400, 410, 420, 430, 440, 450, 460, 470, 480, 490, 500, 510, 520, 530, 540, 550, 560, 570, 580, 590, 600, 610, 620, 630, 640, 650, 660, 670, 680, 690, 700, 710, 720, 730, 740, 750, 760, 770, 780, 790 до примерно 800 мг для введения один раз в сутки или дважды в сутки. Каждая доза может находиться в одной или более чем одной стандартной лекарственной форме, как описано в данном документе.
В другом воплощении ингибиторы интегразы включают долутегравир, элвитегравир, ралтегравир, биктегравир, каботегравир, но не ограничиваются ими. Предпочтительно ингибиторы интегразы представляют собой элвитегравир, долутегравир, ралтегравир. Предпочтительно доза долутегравира варьирует от примерно 1 мг до примерно 50 мг, доза элвитегравира варьирует от примерно 1 мг до примерно 150 мг для введения один раз в сутки, и доза ралтегравира варьирует от примерно 4 мг до примерно 400 мг для введения один раз в сутки. В некоторых воплощениях доза долутегравира варьирует от примерно 5 мг до примерно 50 мг, от примерно 20 мг до примерно 50 мг, от примерно 25 мг до примерно 50 мг, от примерно 25 мг до примерно 45 мг, от примерно 30 мг до примерно 50 мг, от примерно 30 мг до примерно 40 мг или от примерно 35 мг до примерно 50 мг для введения дважды в сутки. В некоторых воплощениях доза долутегравира варьирует от примерно 1 мг, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49 до примерно 50 мг для введения один раз в сутки или дважды в сутки. Каждая доза может находиться в одной или более чем одной стандартной лекарственной форме, как описано в данном документе.
В другом воплощении ингибиторы слияния включают маравирок, энфувиртид, гриффитсин, аплавирок, викривирок, плериксафор, фостемсавир, албувиртид, но не ограничиваются ими.
В другом воплощении ингибиторы CCR5 включают аплавирок, викривирок, маравирок, ценикривирок, но не ограничиваются ими.
В другом воплощении моноклональные антитела включают ибализумаб, но не ограничиваются им.
В другом воплощении ингибиторы гликопротеинов включают сифувиртид, но не ограничиваются им.
В другом воплощении ингибиторы обратной транскриптазы на основе нуклеотидного аналога включают тенофовира алафенамид фумарат, тенофовира дизопроксил фумарат и адефовир, но не ограничиваются ими. Предпочтительно ингибиторы обратной транскриптазы на основе нуклеотидного аналога представляют собой тенофовира алафенамид фумарат и тенофовира дизопроксил фумарат. В некоторых воплощениях доза тенофовира алафенамид фумарата варьирует от примерно 1 мг до примерно 25 мг, от примерно 2,5 мг до примерно 25 мг, от примерно 5 мг до примерно 20 мг или от примерно 5 мг до примерно 15 мг для введения дважды в сутки. В некоторых воплощениях доза тенофовира алафенамида фумарата варьирует от примерно 1 мг, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 до примерно 25 мг для введения один раз в сутки или дважды в сутки. В некоторых воплощениях доза тенофовира дизопроксил фумарата варьирует от примерно 1 мг до примерно 300 мг, от примерно 1 мг до примерно 150 мг, от примерно 75 мг до примерно 250 мг, от примерно 100 мг до примерно 200 мг или от примерно 120 мг до примерно 180 мг для введения дважды в сутки. В некоторых воплощениях доза тенофовира дизопроксил фумарата варьирует от примерно 1 мг, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290 до примерно 300 мг для введения один раз в сутки или дважды в сутки. Каждая доза может находиться в одной или более чем одной стандартной лекарственной форме, как описано в данном документе.
Термин «антиретровирусное лекарственное средство» и «активатор или улучшитель фармакокинетики» используется в широком смысле для включения не только «антиретровирусного лекарственного средства» самого по себе и активатора или улучшителя фармакокинетики самого по себе, но также и его фармацевтически приемлемых производных. Подходящие фармацевтически приемлемые производные включают фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, фармацевтически приемлемые гидраты, фармацевтически приемлемые ангидраты, фармацевтически приемлемые энантиомеры, фармацевтически приемлемые сложные эфиры, фармацевтически приемлемые изомеры, фармацевтически приемлемые полиморфы, фармацевтически приемлемые пролекарства, фармацевтически приемлемые таутомеры, фармацевтически приемлемые комплексы и т.д.
Термин «активатор или улучшитель фармакокинетики» относится к алкалоиду. В некоторых воплощениях активатор или улучшитель фармакокинетики включает пиперин, тетрагидропиперин, цис-пиперин, транс-пиперин, цис-транс-пиперин, транс,цис-пиперин, цис,цис-пиперин, транс,транс-пиперин или их комбинацию. Более предпочтительно активатор или улучшитель фармакокинетики представляет собой пиперин или тетрагидропиперин и его аналоги или производные. В некоторых воплощениях активатор или улучшитель фармакокинетики увеличивает плазматические концентрации антиретровирусного лекарственного средства на 10%, 20, 30, 40, 50, 60, 70, 80, 90, 100% или больше по сравнению с ситуацией, когда активатор или улучшитель фармакокинетики не используется.
Термин «инъецируемый» относится к способу введения фармацевтической композиции. Фармацевтическую композицию можно вводить целым рядом способов. У человека фармацевтическая композиция может вводиться парентеральным путем. Например, фармацевтическая композиция может вводиться внутривенно (например, внутривенной инъекцией), подкожно, внутрикожно или посредством внутримышечной инъекции. Внутривенное введение может осуществляться смешиванием фармацевтической композиции в подходящем фармацевтическом носителе (наполнителе) или эксципиенте, как понятно практикам в данной области. Композиции, подходящие для парентерального введения, с удобством содержат стерильный водный препарат фармацевтической композиции, который может быть приготовлен так, чтобы быть изотоничным крови пациента.
Термин «терапевтически эффективное количество» или «эффективное количество» относится к такому количеству, что, при введении, фармацевтическая композиция приводит к ингибированию вируса или заболевания. Дозировка, введенная пациенту, может быть представлена в виде одной или многих доз, в зависимости от целого ряда факторов, включающих фармакокинетические свойства введенного лекарственного средства, путь введения, состояние и характеристики пациента (пол, возраст, масса тела, состояние здоровья, размер и т.д.) и степень симптомов, сопутствующие лечения, частоту лечения и желательный эффект.
Термин «лечение» или «осуществление лечения» заболевания, против вируса или состояния относится к выполнению протокола, который может включать введение одного или более чем одного лекарственного средства пациенту в попытке облегчить признаки или симптомы заболевания, воздействия вируса или состояния. Облегчение может происходить до появления признаков или симптомов заболевания, воздействия вируса или состояния, а также после их появления. Таким образом, осуществление лечения или лечение включает уменьшение, предупреждение или предотвращение заболевания, воздействия вируса или состояния. Кроме того, осуществление лечения или лечение не требует полного облегчения признаков или симптомов, не требует излечения и конкретно включает протоколы, которые имеют только минимальное влияние на пациента.
И плоды черного перца (Piper nigrum L), и длинного перца (Piper longum L.) являются важными лекарственными растениями в (традиционных индийских) аюрведической системе медицины и системе медицины унани, в которых лекарственное средство обычно состоит из смеси трав. Известен и документально подтвержден широкий спектр медицинских применений черного перца, включая его применение в лечении лейкодермы.
Пиперин может быть активатором или улучшителем фармакокинетики. Пиперин - главный алкалоид, обнаруженный в плодах черного перца (Piper nigrum L, Piperaceae), стимулирует репликацию меланоцитов и индуцирует образование меланоцитарных дендритов. Ожидается то, что пиперин вызывает повторное заселение пятен витилиго через стимулирующий эффект на меланоциты области около поражения и фолликулярные меланоциты.
Пиперин химически известен как (1-2Е, 4Е-пипериноил-пиперидин) и структурно представлен, как показано ниже:

Пиперин может повышать биодоступность лекарственного средства посредством стимулирования быстрого поглощения лекарственных средств и питательных веществ, усиливая поставку крови к желудочно-кишечному тракту, уменьшая секрецию соляной кислоты с предотвращением распада некоторых лекарственных средств, увеличивая эмульгирующее содержимое кишечника, увеличивая уровень ферментов, подобных γ-глутамилтранспептидазе, которая участвует в активном и пассивном транспорте питательных веществ в кишечные клетки.
Пиперин может увеличивать биодоступность лекарственного средства посредством ингибирования ферментов, которые участвуют в биопревращении лекарственных средств, и, таким образом, предотвращая их инактивацию и устранение. Он также ингибирует р-гликопротеин - «насосный» белок, который удаляет вещества из клеток и может уменьшать продукцию в кишечнике глюкуроновой кислоты, позволяя, посредством этого, большему числу веществ поступать в организм в активной форме.
Также было описано то, что пиперин встречается в других видах Piper, т.е. P. acutisleginum, album, argyrophylum, attenuatum, aurantiacum, betle, callosum, chaba, cubeba, guineense, hancei, khasiana, longum, macropodum, nepalense, novae hollandiae, peepuloides, retrokacturn и sylvaticum.
Тетрагидропиперин представляет собой структурный аналог пиперина. Две двойные связи в положении 2 и 4 насыщаются с образованием тетрагидроаналога. Тетрагидропиперин химически известен как 5-(1,3-бензодиоксол-5-ил)-1-пиперидин-1-илпентан-1-он и структурно представлен, как показано ниже:
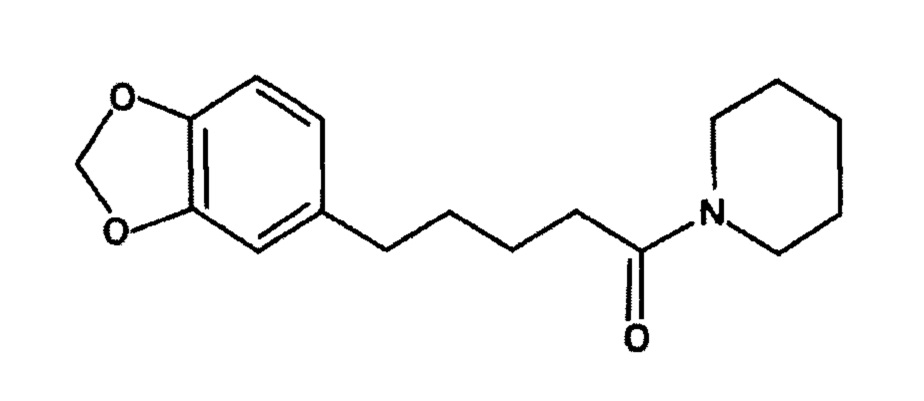
Тетрагидропиперин, подобно пиперину, встречается в природе в черном перце (примерно 0,7% в олеосмоле черного перца). Тетрагидропиперин может быть синтезирован из пиперина, который был ранее экстрагирован из олеосмолы черного перца.
Термин «аналоги или производные» тетрагидропиперина используется в широком смысле для включения алкилтетрагидропиперинов, например, метилтетрагидропиперина или этилтетрагидропиперина,
диалкилтетрагидропиперинов, например, диметилтетрагидропиперина или диэтилтетрагидропиперина, алкоксилированного тетрагидропиперина, например, метокситетрагидропиперина, гидроксилированного тетрагидропиперина, например, 1-[(5,3-бензодиоксил-5-ил)-1-гидрокси-2,4-пентадиенил]-пиперина, 1-[(5,3-бензодиоксил-5-ил)-1-метокси-2,4-пентадиенил]-пиперина, галогенированного тетрагидропиперина, например 1-[(5,3-бензодиоксил-5-ил)-1-оксо-4-галогено-2-пентенил]-пиперина и 1-[(5,3-бензодиоксил-5-ил)-1-оксо-2-галогено-4-пентенил]-пиперина, дигидропиперина, алкилдигидропиперинов, например, метилдигидропиперина или этилдигидропиперина, диалкилдигидропиперинов, например, диметилдигидропиперина или диэтилдигидропиперина, алкоксилированного дигидропиперина, например, метоксидигидропиперина и галогенированного дигидропиперина, и их фармацевтически приемлемых солей, фармацевтически приемлемых сольватов, фармацевтически приемлемых гидратов, фармацевтически приемлемых ангидратов, фармацевтически приемлемых энантиомеров, фармацевтически приемлемых сложных эфиров, фармацевтически приемлемых изомеров, фармацевтически приемлемых полиморфов, фармацевтически приемлемых пролекарств, фармацевтически приемлемых таутомеров, фармацевтически приемлемых комплексов и т.д.
В некоторых воплощениях доза пиперина предпочтительно варьирует от примерно 0,5 мг до примерно 400 мг, и доза тетрагидропиперина варьирует от примерно 0,5 мг до примерно 400 мг. В некоторых воплощениях доза пиперина и/или тетрагидропиперина варьирует от примерно 0,5 мг, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390 до примерно 400 мг. В некоторых воплощениях отношение по меньшей мере одного антиретровирусного лекарственного средства к по меньшей мере одному активатору или улучшителю фармакокинетики составляет от примерно 100:1 до примерно 1:1 по массе.
Предпочтительно фармацевтическая композиция может быть предоставлена в таких лекарственных формах, как стандартные лекарственные формы, включающие таблетки, капсулы (заполненные порошками, пеллетами, шариками, минитаблетками, пилюлями, микропеллетами, маленькими элементами в виде таблеток, таблетками из микрокапсул с активным веществом (MUPS), распадающимися таблетками, диспергируемыми таблетками, гранулами и микросферами, микрочастицами), пакетики (заполненные порошками, пеллетами, шариками, минитаблетками, пилюлями, микропеллетами, маленькими элементами в виде таблеток, MUPS, распадающимися таблетками, диспергируемыми таблетками, гранулами и микросферами, микрочастицами), порошки для растворения, чрескожные пластыри и опрыскивания, но не ограничиваясь ими, однако, можно использовать и другие лекарственные формы, такие как препараты с контролируемым высвобождением, лиофилизированные препараты, лиофилизированный порошок, препараты с модифицированным высвобождением, препараты с отложенным высвобождением, препараты с пролонгированным высвобождением, препараты с импульсным высвобождением, препараты с двойным высвобождением и тому подобное. В пределах данного изобретения также могут рассматриваться жидкие, жидкие инъецируемые или полутвердые лекарственные формы (жидкости, суспензии, растворы, дисперсии, мази, кремы, эмульсии, микроэмульсии, спреи, пластыри, растворы для капельного наесения), инъекционные препараты, парентеральные, местные, ингалируемые, буккальные, назальные и т.д. В некоторых воплощениях фармацевтическая композиция вводится посредством сиропа. Сироп может быть приготовлен добавлением активного соединения в концентрированный водный раствор сахара, например, сахарозы, в который также может быть добавлен любой вспомогательный ингредиент(ты). Такие вспомогательные ингредиенты могут включать корригенты, подходящие консерванты, агент для задержки кристаллизации сахара и агент для увеличения растворимости любого другого ингредиента, такой как многоатомный спирт, например, глицерин или сорбит.
В некоторых воплощениях стандартная лекарственная форма, такая как таблетка, может быть получена прессованием или формовкой, возможно с одним или более чем одним вспомогательным ингредиентом. Прессованные таблетки можно получать прессованием в подходящей установке с активным соединением, находящимся в свободно сыпучем виде, таком как порошок или гранулы, которое возможно смешивают со связующим веществом, разрыхлителем, смазкой, инертным разбавителем, поверхностно-активным агентом или диспергирующим агентом. Формованные таблетки, содержащие подходящий носитель, можно получать формовкой в подходящей установке.
Фармацевтические композиции по настоящему изобретению содержат по меньшей мере одно антиретровирусное лекарственное средство и пиперин или тетрагидропиперин. Данные активные ингредиенты готовят для одновременного, раздельного или последовательного введения. При последовательном введении активных ингредиентов сначала можно вводить либо по меньшей мере одно антиретровирусное лекарственное средства, либо пиперин/тетрагидропиперин. При одновременном введении активные ингредиенты можно вводить либо в той же самой, либо в разных фармацевтических композициях. Адъюнктивная терапия, например, когда один активный ингредиент используется в качестве первичного лечения, а другой(гие) активный(ные) ингредиент(ты) используется(ют)ся для содействия первичному лечению, также является воплощением настоящего изобретения.
Соответственно, предложен продукт, содержащий по меньшей мере одно антиретровирусное лекарственное средство и пиперин или тетрагидропиперин в качестве комбинированного препарата для одновременного, раздельного или последовательного применения для лечения заболеваний, вызванных ретровирусами или вирусом гепатита В, особенно СПИД или ВИЧ-инфекции, или гепатита В.
В некоторых воплощениях фармацевтические композиции по настоящему изобретению содержат тенофовира диспроксил фумарат и пиперин для лечения заболеваний, вызванных ретровирусами, особенно синдрома приобретенного иммунодефицита или ВИЧ-инфекции.
Согласно предпочтительному воплощению фармацевтические композиции по настоящему изобретению содержат тенофовира диспроксил фумарат и пиперин в соотношении от примерно 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1 до примерно 1:1 по массе.
В некоторых воплощениях фармацевтические композиции по настоящему изобретению содержат тенофовира алафенамид фумарат и пиперин для лечения заболеваний, вызванных ретровирусами, особенно СПИД или ВИЧ-инфекции.
Согласно предпочтительному воплощению фармацевтические композиции по настоящему изобретению содержат тенофовира алафенамид фумарат и пиперин в соотношении от примерно 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1 до примерно 1:1 по массе.
В некоторых воплощениях фармацевтические композиции по настоящему изобретению содержат долутегравир и пиперин для лечения заболеваний, вызванных ретровирусами, особенно синдрома приобретенного иммунодефицита или ВИЧ-инфекции.
Согласно предпочтительному воплощению фармацевтические композиции по настоящему изобретению содержат долутегравир и пиперин в соотношении от примерно 100:1, 50:1, 40:1,. 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1 до примерно 1:1 по массе.
В некоторых воплощениях фармацевтические композиции по настоящему изобретению содержат дарунавир и пиперин для лечения заболеваний, вызванных ретровирусами, особенно синдрома приобретенного иммунодефицита или ВИЧ-инфекции.
Согласно предпочтительному воплощению фармацевтические композиции по настоящему изобретению содержат дарунавир и пиперин в соотношении от примерно 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1 до примерно 1:1 по массе.
В некоторых воплощениях фармацевтические композиции по настоящему изобретению содержат тенофовира диспроксил фумарат и пиперин для лечения заболеваний, вызванных вирусом гепатита В.
Согласно предпочтительному воплощению фармацевтические композиции по настоящему изобретению содержат тенофовира диспроксил фумарат и пиперин в соотношении от примерно 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1 до примерно 1:1 по массе.
В некоторых воплощениях фармацевтические композиции по настоящему изобретению содержат тенофовира алафенамид фумарат и пиперин для лечения заболеваний, вызванных вирусом гепатита В.
Согласно предпочтительному воплощению фармацевтические композиции по настоящему изобретению содержат тенофовира алафенамид фумарат и пиперин в соотношении от примерно 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1 до примерно 1:1 по массе.
В некоторых воплощениях фармацевтические композиции по настоящему изобретению содержат тенофовира диспроксил фумарат и тетрагидропиперин для лечения заболеваний, вызванных ретровирусами, особенно синдрома приобретенного иммунодефицита или ВИЧ-инфекции.
Согласно предпочтительному воплощению фармацевтические композиции по настоящему изобретению содержат тенофовира диспроксил фумарат и тетрагидропиперин в соотношении от примерно 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1 до примерно 1:1 по массе.
В некоторых воплощениях фармацевтические композиции по настоящему изобретению содержат тенофовира алафенамид фумарат и тетрагидропиперин для лечения заболеваний, вызванных ретровирусами, особенно синдрома приобретенного иммунодефицита или ВИЧ-инфекции.
Согласно предпочтительному воплощению фармацевтические композиции по настоящему изобретению содержат тенофовира алафенамид фумарат и тетрагидропиперин в соотношении от примерно 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1 до примерно 1:1 по массе.
В некоторых воплощениях фармацевтические композиции по настоящему изобретению содержат долутегравир и тетрагидропиперин для лечения заболеваний, вызванных ретровирусами, особенно синдрома приобретенного иммунодефицита или ВИЧ-инфекции.
Согласно предпочтительному воплощению фармацевтические композиции по настоящему изобретению содержат долутегравир и тетрагидропиперин в соотношении от примерно 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1 до примерно 1:1 по массе.
В некоторых воплощениях фармацевтические композиции по настоящему изобретению содержат дарунавир и тетрагидропиперин для лечения заболеваний, вызванных ретровирусами, особенно синдрома приобретенного иммунодефицита или ВИЧ-инфекции.
Согласно предпочтительному воплощению фармацевтические композиции по настоящему изобретению содержат дарунавир и тетрагидропиперин в соотношении от примерно 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1 до примерно 1:1 по массе.
В некоторых воплощениях фармацевтические композиции по настоящему изобретению содержат тенофовира диспроксил фумарат и тетрагидропиперин для лечения заболеваний, вызванных вирусом гепатита В.
Согласно предпочтительному воплощению фармацевтические композиции по настоящему изобретению содержат тенофовира диспроксил фумарат и тетрагидропиперин в соотношении от примерно 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1 до примерно 1:1 по массе.
В некоторых воплощениях фармацевтические композиции по настоящему изобретению содержат тенофовира алафенамид фумарат и тетрагидропиперин для лечения заболеваний, вызванных вирусом гепатита В.
Согласно предпочтительному воплощению фармацевтические композиции по настоящему изобретению содержат тенофовира алафенамид фумарат и тетрагидропиперин в соотношении от примерно 100:1, 50:1, 40:1, 30:1, 20:1, 10:1, 8:1, 6:1, 5:1, 4:1, 3:1, 2:1 до примерно 1:1 по массе.
В некоторых воплощениях при введении в фармацевтической композиции активатора или улучшителя фармакокинетики, или его производного с антиретровирусным лекарственным средством частота дозирования по меньшей мере одного антиретровирусного лекарственного средства, которое вводится пациенту, уменьшается. В некоторых воплощениях по меньшей мере один активатор или улучшитель фармакокинетики, или его производное увеличивает биодоступность по меньшей мере одного антиретровирусного лекарственного средства от примерно 10% до примерно 100%, от примерно 10% до примерно 70%, от примерно 10% до примерно 50%, от примерно 10% до примерно 30% или от примерно 10% до примерно 20%. В некоторых воплощениях по меньшей мере один активатор или улучшитель фармакокинетики, или его производное увеличивает биодоступность по меньшей мере одного антиретровирусного лекарственного средства от примерно 10%, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 или 100%.
Авторы настоящего изобретения также обнаружили то, что свойства биодоступности антиретровирусных лекарственных средств также могут быть улучшены посредством доведения их до наноразмера. В некоторых воплощениях фармацевтическая композиция вводится посредством наночастиц, имеющих размер от примерно 1 нанометра (нм) до примерно 50 нм. В некоторых воплощениях наночастицы имеют размер от примерно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49 или 50 нм.
В некоторых воплощениях для приготовления лекарственных форм согласно настоящему изобретению в виде препарата можно использовать подходящие эксципиенты, такие как стабилизаторы поверхности или поверхностно-активные вещества, агенты, модифицирующие вязкость, полимеры, включающие полимеры с пролонгированным высвобождением, стабилизаторы, разрыхлители или суперразрыхлители, разбавители, пластификаторы, связующие вещества, скользящие вещества, смазки, подсластители, корригенты, средства против спекания, замутнители, противомикробные агенты, пеногасители, эмульгаторы, буферизующие агенты, крастели, носители, наполнители, антиадгезивные средства, растворители, агенты, маскирующие цвет, консерванты, антиоксиданты, улучшители консистенции, направляющие агенты, покрывающие агенты и их комбинации, но не ограничиваясь ими.
В некоторых воплощениях при предоставлении фармацевтической композиции в стандартных лекарственных формах, как обсуждалось выше, стандартная лекарственная форма может быть непокрытой или покрытой.
Эти и другие аспекты настоящей заявки будут более понятными при рассмотрении следующих Примеров, которые предназначены для иллюстрации определенных конкретных воплощений данной заявки, но не предназначены для того, чтобы ограничивать ее объем, как определено формулой изобретения.
ПРИМЕРЫ
Пример 1
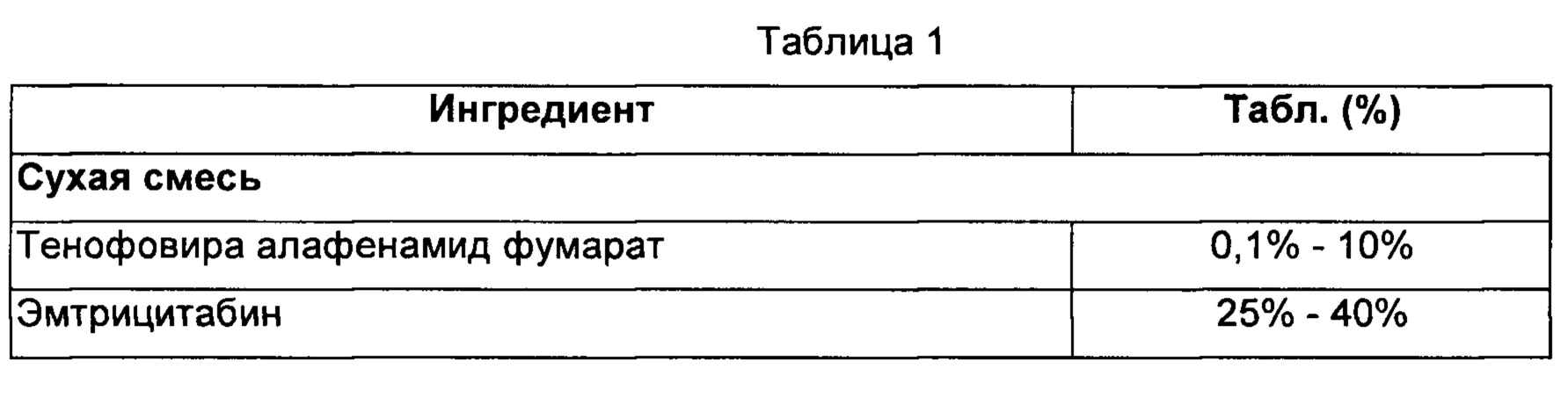
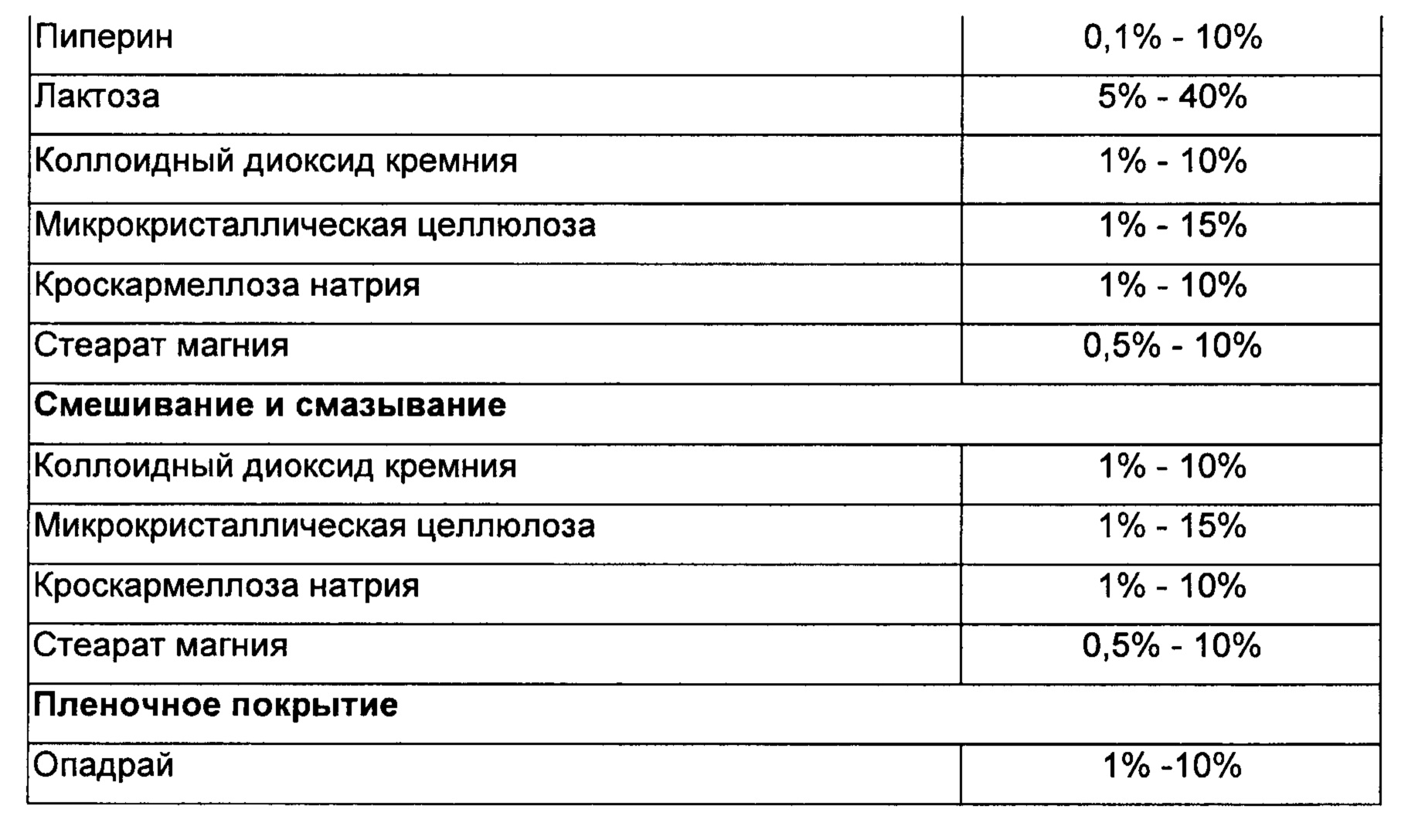
Способ:
1) Тенофовира алафенамид фумарат, эмтрицитабин, пиперин, лактозу, коллоидный диоксид кремния, микрокристаллическую целлюлозу и кроскармеллозу натрия смешивали в сухом состоянии в подходящем блендере.
2) Смесь, полученную на стадии (1), смазывали стеаратом магния, уплотняли и гранулировали в сухом состоянии.
3) Гранулы, полученные на стадии (3), коллоидный диоксид кремния, микрокристаллическую целлюлозу и кроскармеллозу натрия смешивали с образованием смеси.
4) Смесь, полученную на стадии (3), пресовали с получением таблеток и покрывали опадраем.
Пример 2


Способ:
1) Тенофовира алафенамид фумарат, пиперин, лактозу, коллоидный диоксид кремния, микрокристаллическую целлюлозу и кроскармеллозу натрия смешивали в сухом состоянии в подходящем блендере.
2) Смесь, полученную на стадии (1), смазывали стеаратом магния, прессовали с получением таблеток и покрывали опадраем.
Пример 3
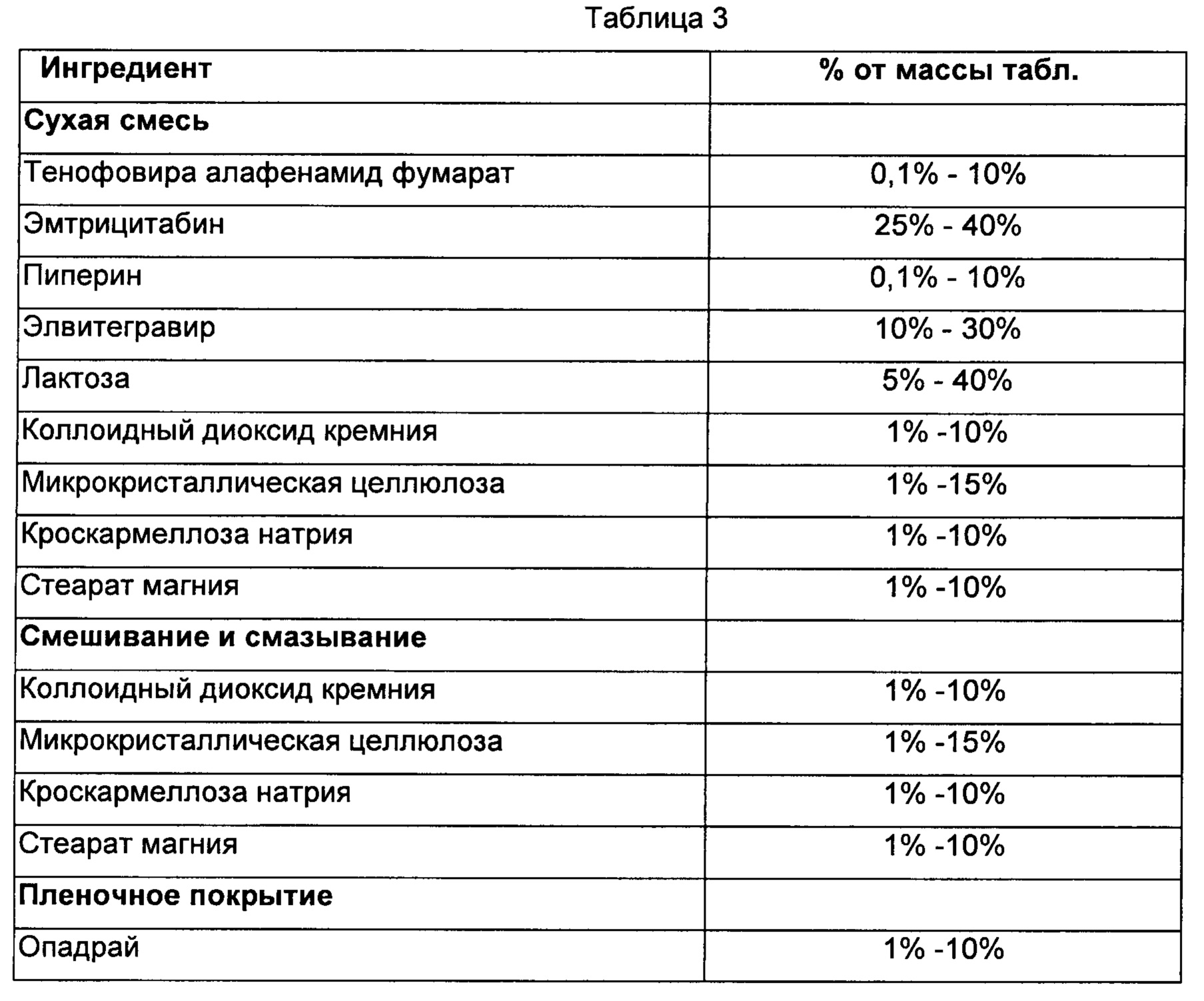
Способ:
1) Тенофовира алафенамид фумарат, эмтрицитабин, пиперин, элвитегравир, лактозу, коллоидный диоксид кремния, кроскармеллозу натрия и микрокристаллическую целлюлозу смешивали в сухом состоянии с получением смеси.
2) Смесь, полученную на стадии (1), смазывали стеаратом магния, уплотняли, прессовали до заданного размера с получением таблеток.
Пример 4

Способ:
1) Тенофовира дизопроксил фумарат, эмтрицитабин, пиперин, элвитегравир, лактозу, коллоидный диоксид кремния, кроскармеллозу натрия и микрокристаллическую целлюлозу смешивали в сухом состоянии с получением смеси.
2) Смесь, полученную на стадии (1), смазывали стеаратом магния, уплотняли, прессовали до заданного размера с получением таблеток.
Пример 5

Способ:
1) Долутегравир натрия, пиперин, маннит смешивали в сухом состоянии, а повидон растворяли в воде.
2) Сухую смесь, полученную на стадии (1), гранулировали, и полученные гранулы размалывали.
3) Гранулы, полученные на стадии (2), смешивали с маннитом, натрия крахмала гликолятом, микрокристаллической целлюлозой и коллоидным диоксидом кремния.
4) Смесь, полученную на стадии (3), смазывали стеарилфумаратом натрия, прессовали и покрывали.
Пример 6


Способ:
1) Дарунавира гидрат, пиперин, микрокристаллическую целлюлозу, кросповидон и коллоидный диоксид кремния просеивали и смешивали.
2) Сухую смесь, полученную на стадии (1), гранулировали и смазывали стеаратом магния.
3) Гранулы, полученные на стадии (2), прессовали и покрывали опадраем.
Пример 7
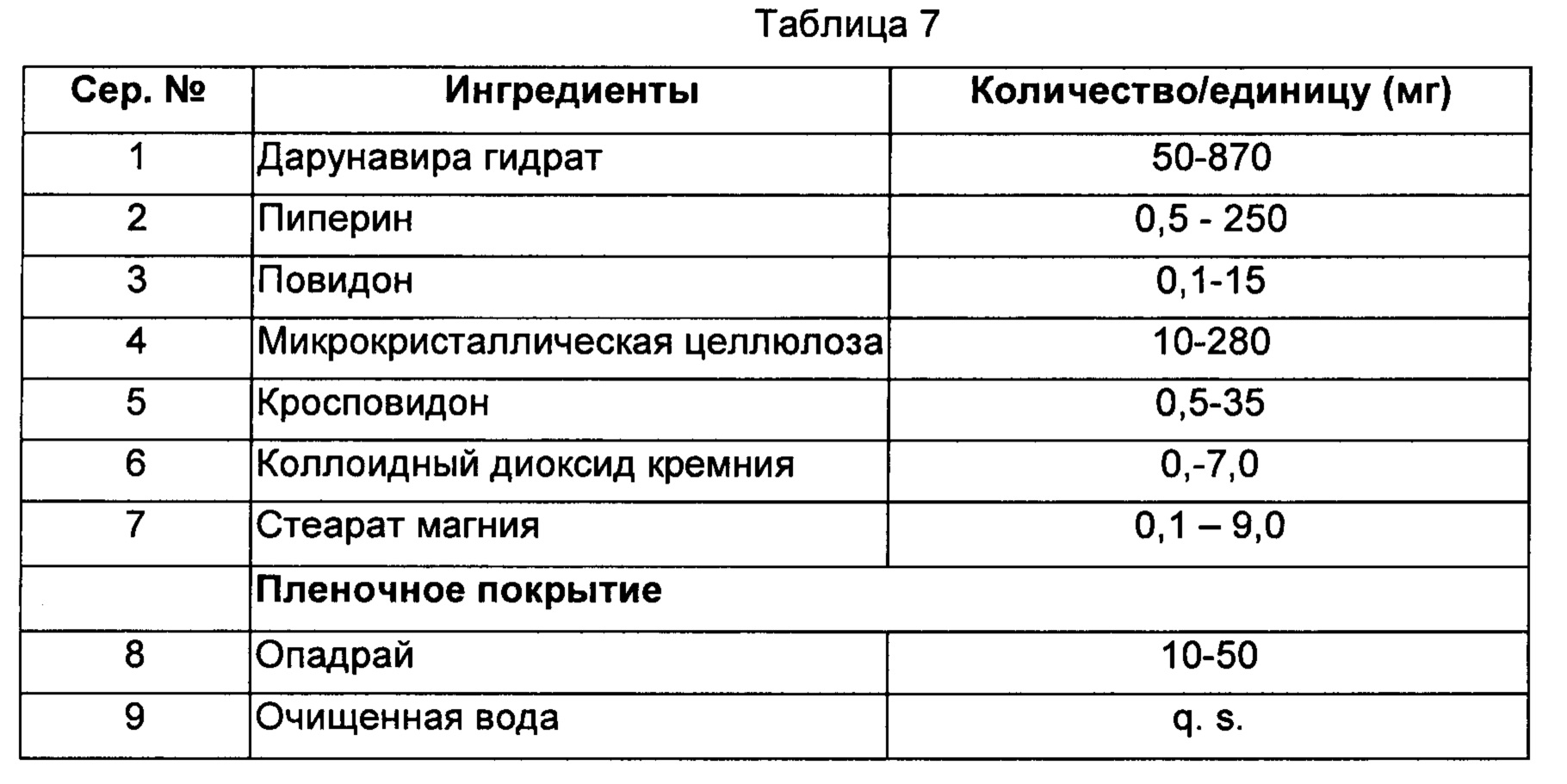
Способ:
1) Дарунавира гидрат, пиперин, повидон, микрокристаллическую целлюлозу, кросповидон и коллоидный диоксид кремния просеивали и смешивали.
2) Сухую смесь, полученную на стадии (1), гранулировали и смазывали стеаратом магния.
3) Гранулы, полученные на стадии (2), прессовали и покрывали опадраем
Пример 8

Способ:
1) Дарунавира этанолат, пиперин, повидон, силикатированную микрокристаллическую целлюлозу, кросповидон и коллоидный диоксид кремния просеивали и смешивали.
2) Сухую смесь, полученную на стадии (1), гранулировали и смазывали стеаратом магния.
3) Гранулы, полученные на стадии (2), прессовали и покрывали опадраем.
Пример 9


Способ:
1) Дарунавира этанолат, пиперин, гидроксипропилметилцеллюлозу, силикатированную микрокристаллическую целлюлозу, кросповидон и коллоидный диоксид кремния просеивали и смешивали.
2) Сухую смесь, полученную на стадии (1), гранулировали и смазывали стеаратом магния.
3) Гранулы, полученные на стадии (2), прессовали и покрывали опадраем.
Пример 10
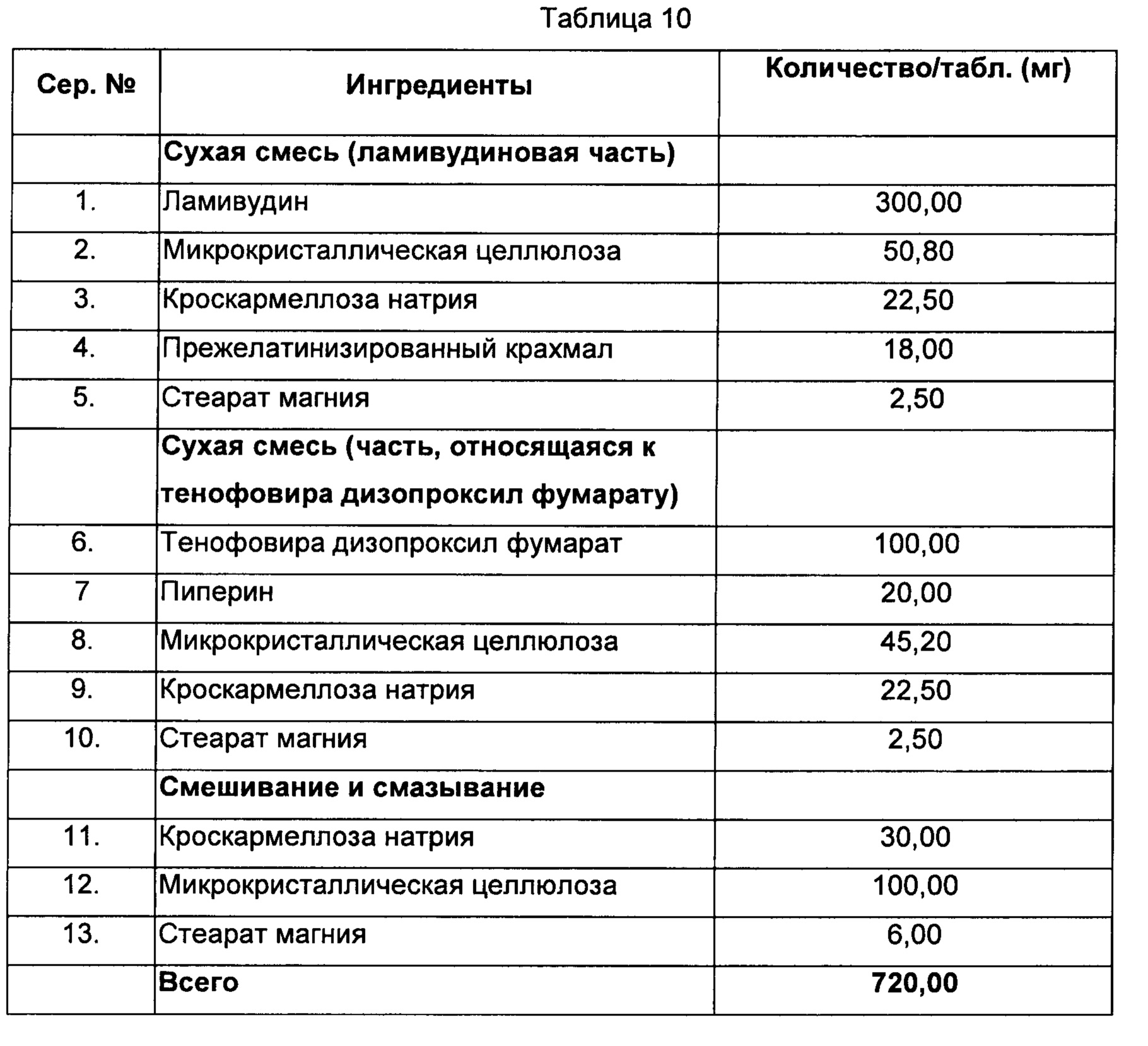
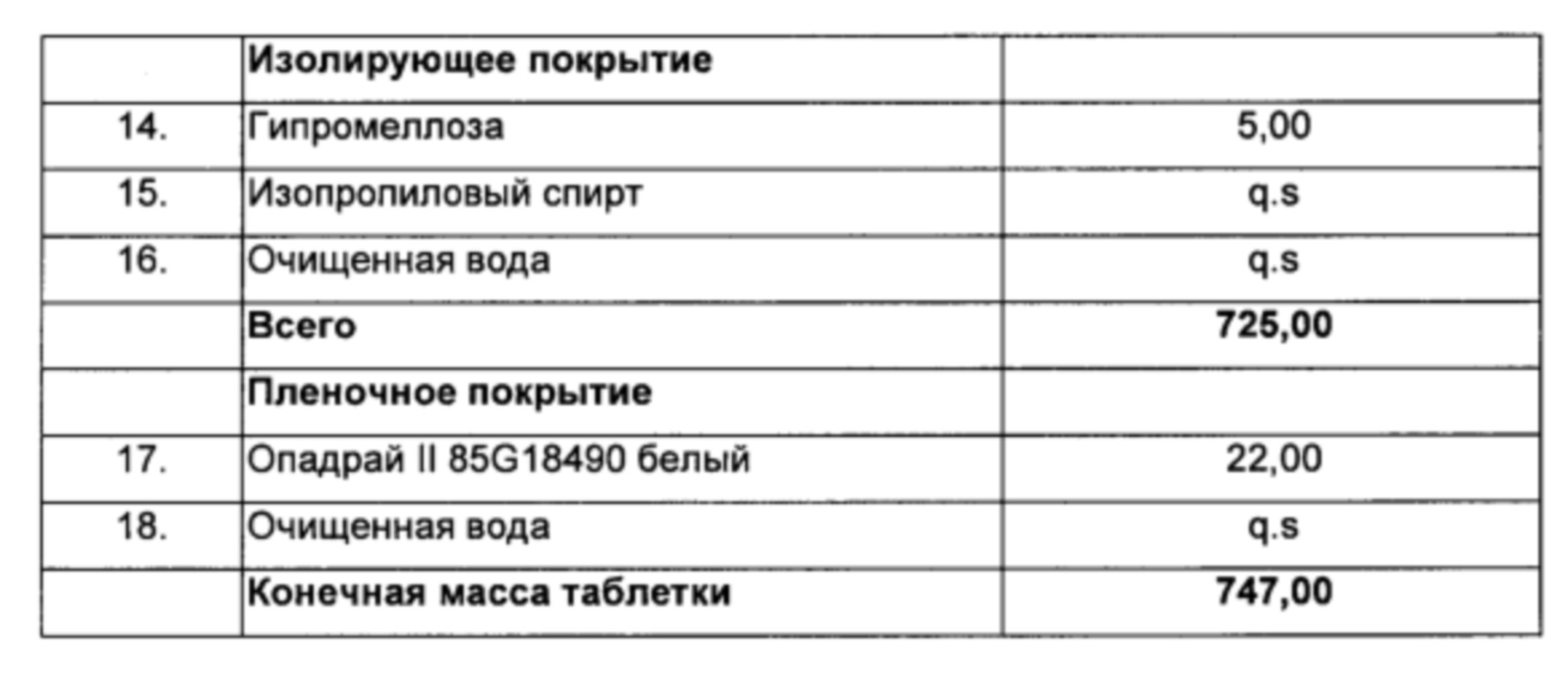
Способ:
1) Ламивудин, микрокристаллическую целлюлозу, кроскармеллозу натрия, прежелатинизированный крахмал и стеарат магния смешивали и уплотняли в гранулярную массу.
2) Тенофовира дизопроксил фумарат, пиперин, микрокристаллическую целлюлозу, кроскармеллозу натрия и стеарат магния смешивали и уплотняли в гранулярную массу.
3) Микрокристаллическую целлюлозу, кроскармеллозу натрия и стеарат магния смешивали со смесями, полученными на стадии (1) и стадии (2) и прессовали с получением таблеток с изолирующим покрытием, с последующим покрытием пленкой.
Пример 11

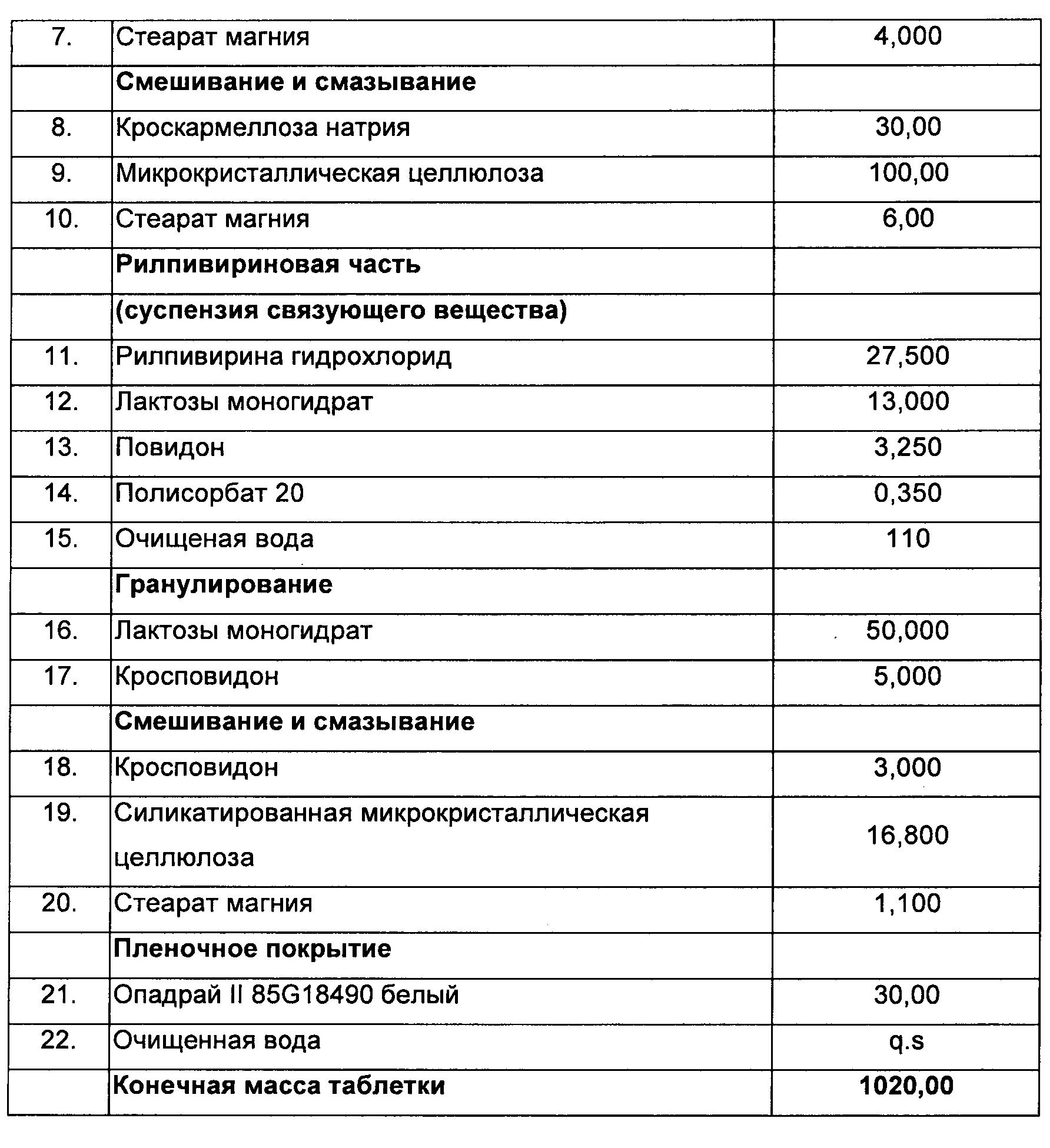
Способ:
1) Эмтрицитабин, тенофовира дизопроксил фумарат, пиперин, лактозы моногидрат, микрокристаллическую целлюлозу, кроскармеллозу натрия и стеарат магния смешивали и уплотняли до гранулярной массы.
2) Микрокристаллическую целлюлозу, кроскармеллозу натрия и стеарат магния смешивали и уплотняли до гранулярной массы.
3) Полисорбат 80, повидон и лактозу растворяли в воде.
4) Рилпивирин добавляли в раствор, полученный на стадии (3), с образованием суспензии.
5) Добавляли в суспензию, полученную на стадии (4), сухую смесь лактозы моногидрата и кросповидона.
6) Добавляли в сухую смесь, полученную на стадии (5), микрокристаллическую целлюлозу, кросповидон и стеарат магния.
7) Смесь, полученную на стадии (1), прессовали со смесью, полученной на стадии (6), с образованием двухслойной таблетки с пленочным покрытием.
Пример 12
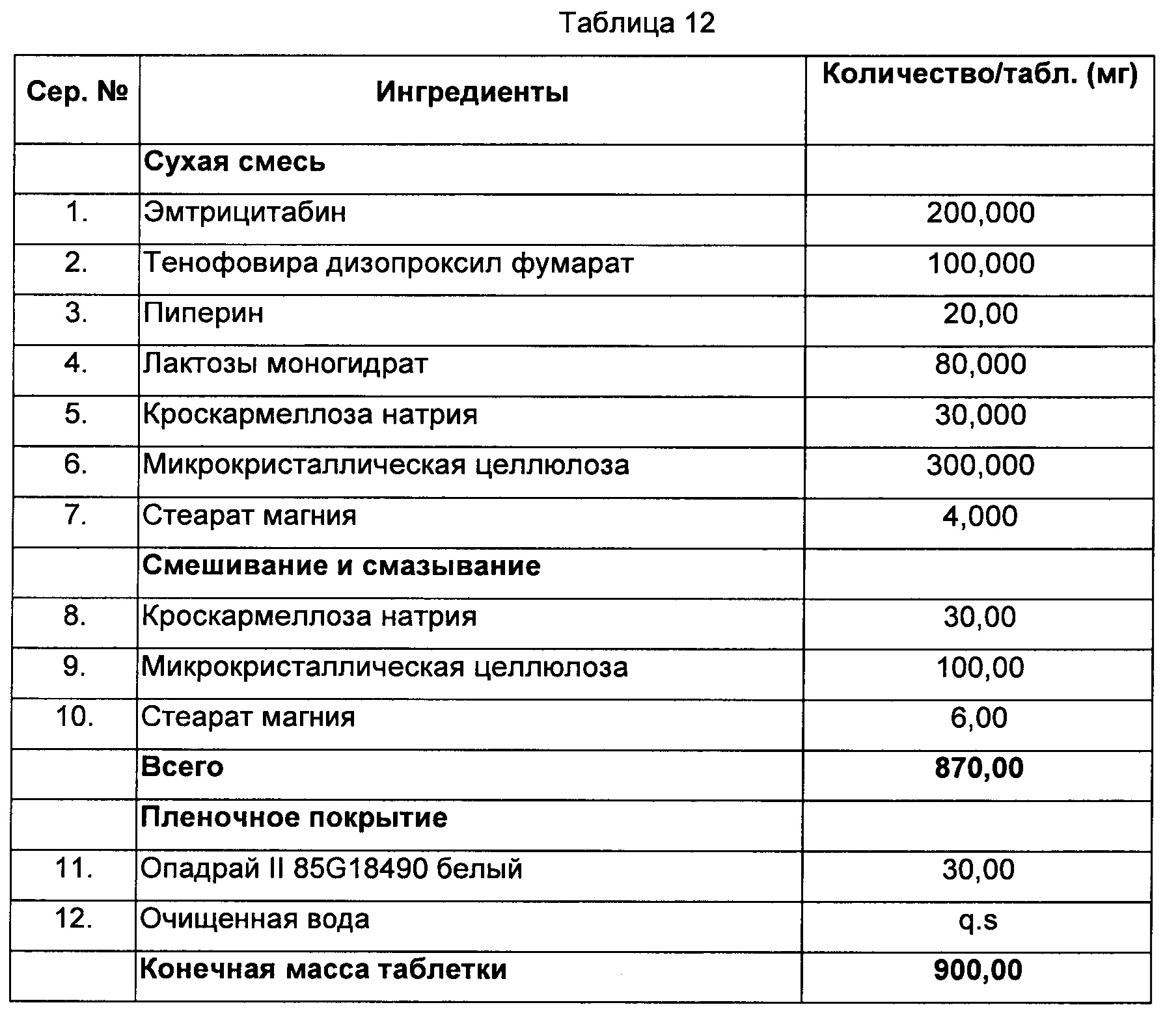
Способ:
1) Эмтрицитабин, тенофовира дизопроксил фумарат, пиперин, лактозы моногидрат, микрокристаллическую целлюлозу, кроскармеллозу натрия и стеарат магния перемешивали и смешивали с образованием гранулярной массы.
2) Микрокристаллическую целлюлозу, кроскармеллозу натрия и стеарат магния перемешивали и смешивали.
3) Смесь, полученную на стадии (1) и стадии (2), прессовали и покрывали с получением таблеток с пленочным покрытием.
Пример 13


Способ:
1) Эмтрицитабин, тенофовира дизопроксил фумарат, пиперин, лактозы моногидрат, микрокристаллическую целлюлозу, кроскармеллозу натрия и стеарат магния перемешивали и смешивали с образованием гранулярной массы.
2) Добавляли в смесь, полученную на стадии (1), микрокристаллическую целлюлозу, кроскармеллозу натрия и стеарат магния и дополнительно смешивали.
3) Эфавиренз, микрокристаллическую целлюлозу и кроскармеллозу натрия добавляли в SLS с последующим добавлением гидроксипропилцелюлозы с образованием раствора и гранулировали.
4) Лактозы моногидрат и стеарат магния смешивали и прессовали с образованием двухслойной таблетки, имеющей пленочное покрытие.
Пример 14


Способ:
1) Ламивудин, микрокристаллическую целлюлозу, кроскармеллозу натрия, прежелатинизированный крахмал и стеарат магния смешивали с образованием гранулярной массы.
2) Тенофовира дизопроксил фумарат, пиперин, микрокристаллическую целлюлозу, кроскармеллозу натрия и стеарат магния смешивали с образованием гранулярной массы.
3) Микрокристаллическую целлюлозу, кроскармеллозу натрия и стеарат магния перемешивали со смесями, полученными на стадии (1) и стадии (2).
4) Эфавиренз, микрокристаллическую целлюлозу и кроскармеллозу натрия добавляли в SLS с последующим добавлением гидроксипропилцеллюлозы с образованием раствора и гранулировали.
5) Лактозы моногидрат и стеарат магния смешивали и прессовали с образованием двухслойной таблетки, имеющей изолирующее покрытие, с последующим покрытием пленкой.
Для того чтобы данное изобретение было понятнее, изложены следующие способы получения и анализа. Данные способы служат лишь для цели иллюстрации, и их не следует истолковывать как ограничивающие объем изобретения каким-либо образом.
СПОСОБЫ ПОЛУЧЕНИЯ И АНАЛИЗА
I) ВЕЩЕСТВО
Кофеин (высокопроницаемый маркер), атенолол (низкопроницаемый маркер), дигоксин (известный субстрат P-gp), TDF (MD1431532), TAF (VRD-1063/16/187), буфер HBSS, MES гидрат, порошок HEPES, фетальная телячья сыворотка (FBS), минимальная питательная среда (MEM), люцифер желтый, пиперин (ингибитор P-gp).
СПОСОБ
1) Культура клеток сасо-2
Клетки сасо-2 культивировали в средах MEM с 10% сыворотки, высевали в плотости 75000 клеток на мл и культивировали в течение 21 суток в 24-луночном планшете с трансвелами при 37°С, 5% CO2. Целостность монослоя периодически проверяли (сутки 0-21) с использованием трансэпителиального электрического сопротивления (TEER). Клетки обрабатывали лекарственными средствами следующим образом:
2) Однонаправленный анализ (А-В)
Препараты маточных растворов: 10 мМ маточные растворы всех лекарственных средств готовили в DMSO. Тестируемые концентрации далее готовили в буфере HBSS, содержащем 10 мМ MES гидрат, рН 6,8 согласно плану планшета. Также готовили буфер HBSS с 10 мМ HEPES с рН 7,4.
План исследования
Конфигурация планшета

Протокол анализа
400 мкл образцов, приготовленных в 10 мМ MES гидрате, рН 6,8, добавляли в двойных повторностях в лунки согласно конфигурации планшета на апикальную сторону (А), а 800 мкл HBSS с 10 мМ HEPES, рН 7,4, добавляли во все базальные лунки (В). Образцы отбирали в 60, 90 и 120 минут с базальной стороны. С апикальной стороны отбирали образцы баланса массы в 0 и 120 минут. Образец анализировали на ЖХ/МС-МС (жидкостная хроматография - тандемная масс-спектрометрия).
4) Двухнаправленный анализ (А-В и В-А) для исследования эффекта пиперина (ингибитор P-gp) на проницаемость.
План планшета

Протокол анализа
400 мкл образцов добавляли в двойных повторностях в лунки согласно конфигурации планшета на апикальную сторону, с 800 мкл HBSS, рН 7,4, в базальных лунках. Образцы отбирали в 60, 90 и 120 минут с базальной стороны. С апикальной стороны отбирали образцы баланса массы в 0 и 120 минут.
Для В-А 800 мкл соответствующих разведений добавляли на базальную сторону в двойных повторностях, с 400 мкл HBSS, рН 7,4, в апикальных лунках. Образцы отбирали в 60, 90 и 120 минут с апикальной стороны. С базальной стороны отбирали образцы баланса массы в 0 и 120 минут. Образец анализировали на ЖХ/МС-МС.
Целостность монослоя проверяли в конце эксперимента с использованием люцифера желтого, и рассчитывали % отторжения люцифера желтого посредством инкубирования клеток со 100 мкг/мл люцифера.
5) Анализ данных:
Рарр рассчитывали следующим образом:
Кажущуюся проницаемость (Рарр) в единицах в секунду можно рассчитывать с использованием следующего уравнения,
Для одноточечного способа:
Рарр=(V/ (T*A))*(C0/Ct)
Для многоточечного способа:
Рарр=(dQ/dt)/ (А*С0)
% баланса массы = 100-[CR120*VR+CD120*VD/C0*VD]
Для люцифера желтого,
% прохождения люцифера желтого = [RFU (опыт) - RFU (пустой) / RFU (равновесный) - RFU (пустой)]*100
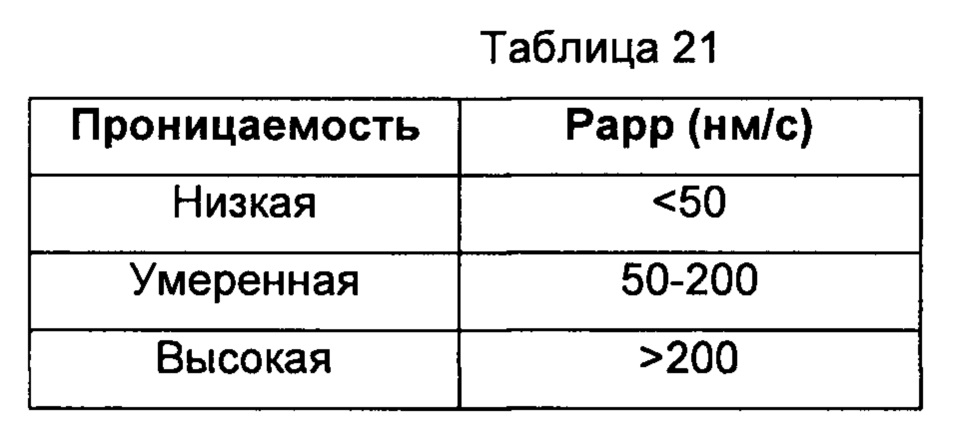
Классификация проницаемости:

Отношение оттока = Рарр В-А/Рарр А-В
Отношение оттока, большее или равное 2, показывает то, что лекарственное средство представляет собой субстрат P-gp.
Результаты
Однонаправленный анализ (ФИГ. 1)

Двунаправленный анализ (ФИГ. 2)

Заключения
Наблюдается то, что TDF и TAF представляют собой лекарственные средства с проницаемостью от низкой до умеренной. Кроме того, поглощение TAF возрастает с пиперином посредством уменьшения отношения оттока TAF. Приведенные выше данные показывают то, что TAF является субстратом выводящего переносчика, и, таким образом, его биодоступность является низкой. Как можно видеть в данных Рарр А-В составляла 34,49 нм/с, и его отношение оттока составляло 5,17. Посредством добавления пиперина, который является известным ингибитором выводящих переносчиков, Рарр А-В возрастала до более чем 282,66 нм/с, тогда как отношение оттока уменьшалось до менее, чем 1,18. Таким образом, указанное добавление пиперина улучшает проницаемость. Следовательно, можно сделать заключение о том, что применение пиперина уменьшает отношение оттока, что, в свою очередь, увеличивало бы его биодоступность.
II) Вещество
Дигоксин (известный субстрат P-gp), долутегравир (KK 1406229), буфер HBSS, MES гидрат, порошок HEPES, фетальная телячья сыворотка (FBS), минимальная питательная среда (MEM), люцифер желтый, пиперин (ингибитор Р-gp), кобицистат (ингибитор Р-gp).
Способ
1) Культура клеток сасо-2
Клетки сасо-2 культивировали в средах MEM с 10% сыворотки, высевали в плотности 75000 клеток на мл и культивировали в течение 21 суток в 24-луночном планшете с трансвелами при 37°С, 5% CO2. Целостность монослоя периодически проверяли (сутки 0-21) с использованием трансэпителиального электрического сопротивления (TEER). Клетки обрабатывали лекарственными средствами следующим образом:
2) Двунаправленный анализ (А-В и В-А) для исследования эффекта пиперина (ингибитор P-gp) на проницаемость.
План планшета

Протокол анализа
400 мкл образцов добавляли в двойных повторностях в лунки согласно конфигурации планшета на апикальную сторону, с 800 мкл HBSS, рН 7,4, в базальных лунках. Образцы отбирали в 60, 90 и 120 минут с базальной стороны. С апикальной стороны отбирали образцы баланса массы в 0 и 120 минут.
Для В-А 800 мкл соответствующих разведений добавляли на базальную сторону в двойных повторностях, с 400 мкл HBSS, рН 7,4, в апикальных лунках. Образцы отбирали в 60, 90 и 120 минут с апикальной стороны. С базальной стороны отбирали образцы баланса массы в 0 и 120 минут.
Образцы анализировали на ЖХ/МС-МС. Целостность монослоя проверяли в конце эксперимента с использованием люцифера желтого, и рассчитывали % отторжения люцифера желтого посредством инкубирования клеток со 100 мкг/мл люцифера.
3) Анализ данных:
Рарр рассчитывали следующим образом:
Кажущуюся проницаемость (Рарр) в единицах в секунду можно рассчитывать с использованием следующего уравнения,
Для одноточечного способа:
Рарр=(V/ (Т*А))*(С0/Ct)
Для многоточечного способа:
Рарр=(dQ/dt)/ (А*С0)
% баланса массы = 100-[CR120*VR+CD120*VD/C0*VD]
Для люцифера желтого,
% прохождения люцифера желтого = [RFU (опыт) - RFU (пустой) / RFU (равновесный) - RFU (пустой)]*100
Классификация проницаемости:
Отношение оттока - Рарр В-А/Рарр А-В
Отношение оттока, большее или равное 2, показывает то, что лекарственное средство представляет собой субстрат P-gp.
Результаты
Двунаправленный анализ (ФИГ. 3)

Заключения
Долутегравир представляет собой известный субстрат P-gp. Долутегравир является высокопроницаемым лекарственным средством, и пиперин не влияет на проницаемость долутегравира через монослой сасо-2. Следовательно, можно сделать вывод о том, что применение пиперина снижает отношение оттока, что, в свою очередь, увеличивало бы его биодоступность.
III) Вещество
Дигоксин (известный субстрат P-gp), дарунавир (DN0011215), буфер HBSS, MES гидрат, порошок HEPES, фетальная телячья сыворотка (FBS), минимальная питательная среда (MEM), люцифер желтый, пиперин (ингибитор P-gp), кобицистат (ингибитор P-gp).
Способ
1) Культура клеток сасо-2
Клетки сасо-2 культивировали в средах MEM с 10% сыворотки, высевали в плотности 75000 клеток на мл и культивировали в течение 21 суток в 24-луночном планшете с трансвелами при 37°С, 5% CO2. Целостность монослоя периодически проверяли (сутки 0-21) с использованием трансзпителиального электрического сопротивления (TEER). Клетки обрабатывали лекарственными средствами следующим образом:
2) Двунаправленный анализ (А-В и В-А) для исследования эффекта пиперина (ингибитор P-gp) на проницаемость.
План планшета
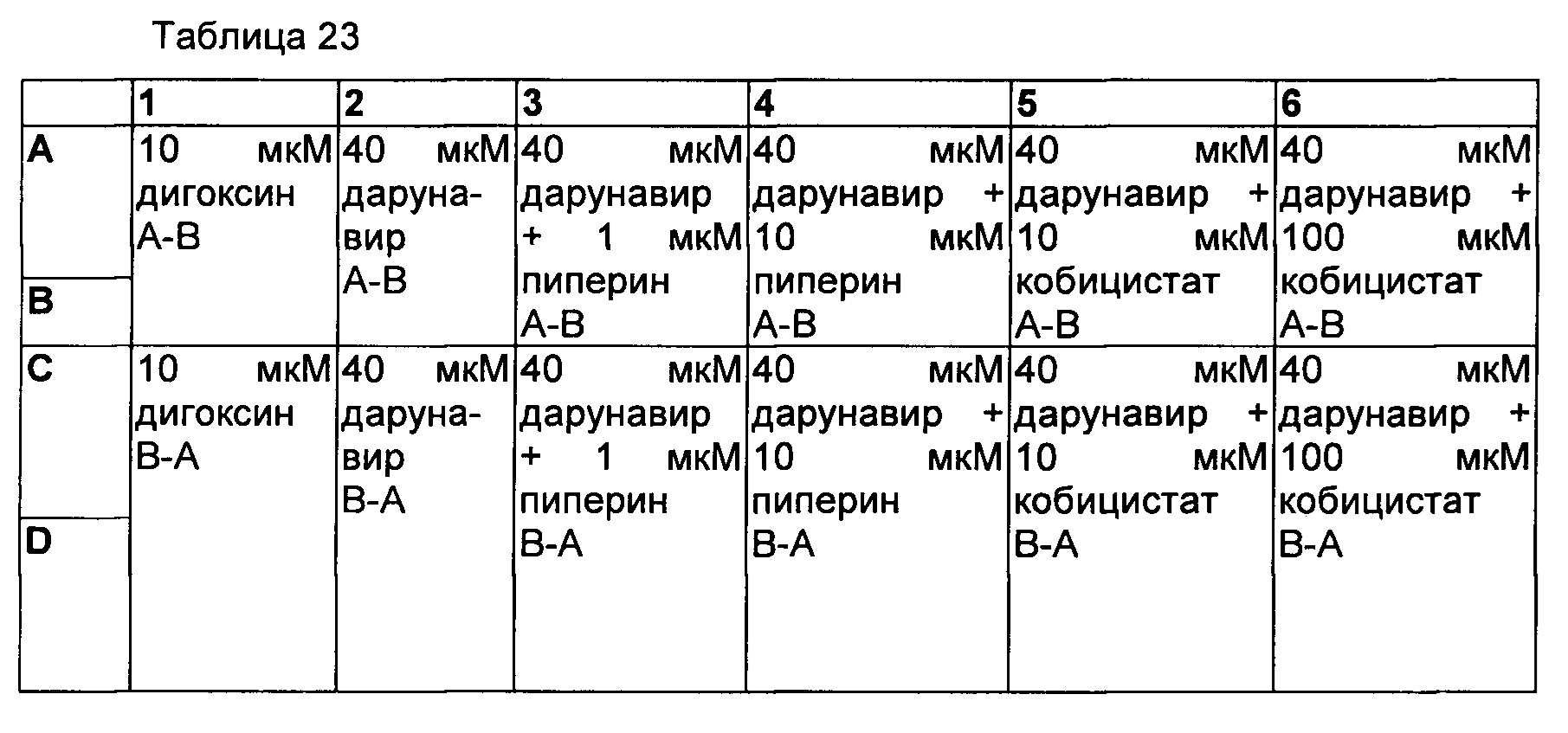
Протокол анализа
400 мкл образцов добавляли в лунки в двойных повторностях согласно конфигурации планшета на апикальную сторону, с 800 мкл HBSS, рН 7,4, в базальных лунках. Образцы отбирали в 60, 90 и 120 минут с базальной стороны. С апикальной стороны отбирали образцы баланса массы в 0 и 120 минут.
Для В-А 800 мкл соответствующих разведений добавляли на базальную сторону в двойных повторностях, с 400 мкл HBSS, рН 7,4, в апикальных лунках. Образцы отбирали в 60, 90 и 120 минут с апикальной стороны. С базальной стороны отбирали образцы баланса массы в 0 и 120 минут.
Образцы анализировали на ЖХ/МС-МС. Целостность монослоя проверяли в конце эксперимента с использованием люцифера желтого, рассчитывая % отторжения люцифера желтого посредством инкубирования клеток со 100 мкг/мл люцифера.
3) Анализ данных:
Рарр рассчитывали следующим образом:
Кажущуюся проницаемость (Рарр) в единицах в секунду можно рассчитывать с использованием следующего уравнения,
Для одноточечного способа:
Рарр=(V/(T*A))*(C0/Ct)
Для многоточечного способа:
Рарр=(dQ/dt)/(A*C0)
% баланса массы = 100-[CR120*VR+CD120*VD/C0*VD]
Для люцифера желтого,
% прохождения люцифера желтого = [RFU (опыт) - RFU (пустой) / RFU (равновесный) - RFU (пустой)]*100
Классификация проницаемости:

Отношение оттока = Рарр В-А/Рарр А-В
Отношение оттока, большее или равное 2, показывает то, что лекарственное средство представляет собой субстрат P-gp.
Результаты
Двунаправленный анализ (ФИГ. 4)
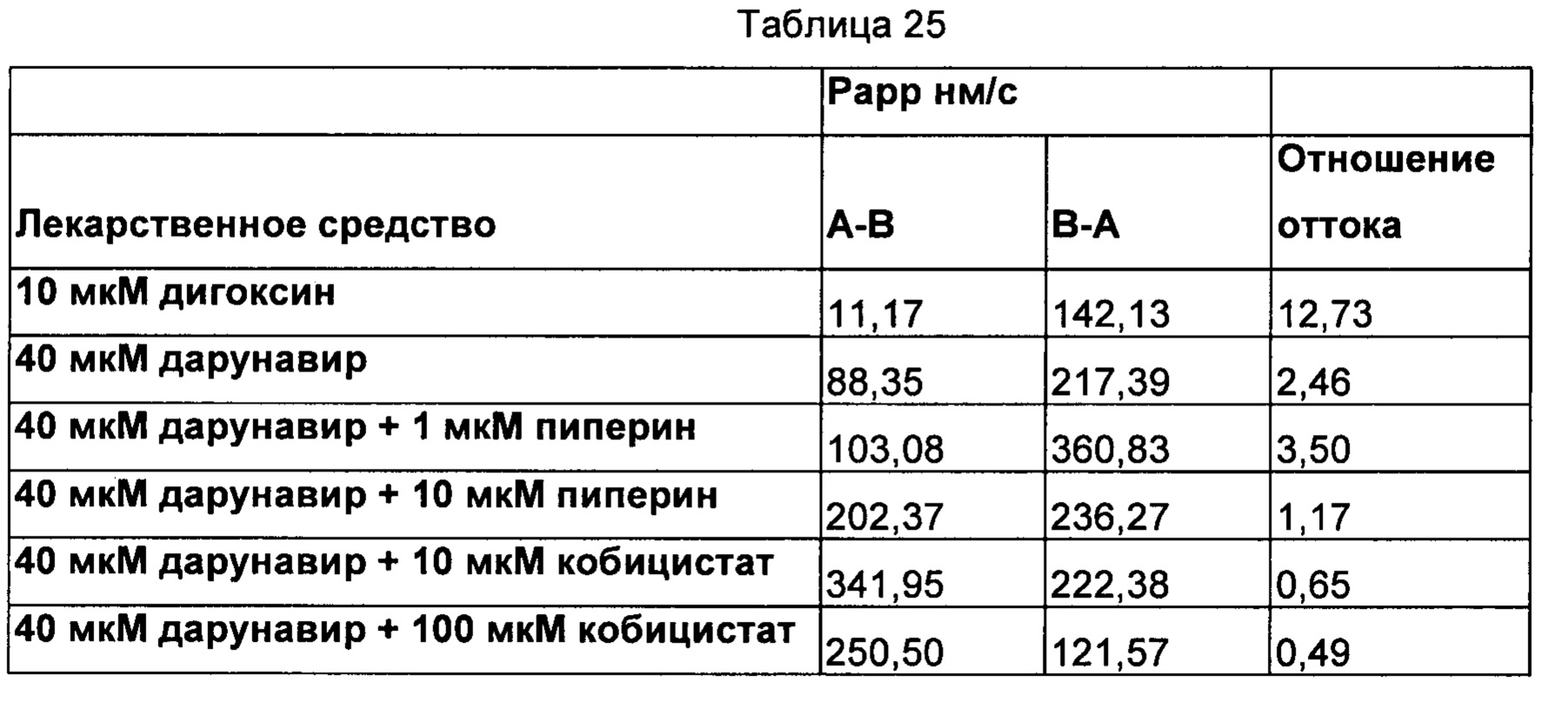
Заключения
Дарунавир представляет собой известный субстрат P-gp. Поглощение дарунавира увеличивается пиперином посредством снижения отношения оттока TAF. Следовательно, можно сделать заключение о том, что применение пиперина снижает отношение оттока, что, в свою очередь, увеличивало бы его биодоступность.
IV) Вещество
Дигоксин (известный субстрат P-gp), TDF, буфер HBSS, MES гидрат, порошок HEPES, фетальная телячья сыворотка (FBS), минимальная питательная среда (MEM), люцифер желтый, пиперин (ингибитор P-gp), кобицистат (ингибитор P-gp), тетрагидропиперин (ингибитор P-gp).
Способ
1) Культура клеток сасо-2
Клетки сасо-2 культивировали в средах MEM с 10% сыворотки, высевали в плотности 75000 клеток на мл и культивировали в течение 21 суток в 24-луночном планшете с трансвелами при 37°С, 5% CO2. Целостность монослоя периодически проверяли (сутки 0-21) с использованием трансэпителиального электрического сопротивления (TEER). Клетки обрабатывали лекарственными средствами следующим образом:
2) Двунаправленный анализ (А-В и В-А) для исследования эффекта пиперина (ингибитор P-gp) и тетрагидропиперина на проницаемость.
План планшета

Протокол анализа
400 мкл образцов добавляли в лунки в двойных повторностях согласно конфигурации планшета на апикальную сторону, с 800 мкл HBSS, рН 7,4, в базальных лунках. Образцы отбирали в 60, 90 и 120 минут с базальной стороны. С апикальной стороны отбирали образцы баланса массы в 0 и 120 минут.
Для В-А 800 мкл соответствующих разведений добавляли на базальную сторону в двойных повторностях, с 400 мкл HBSS, рН 7,4, в апикальных лунках. Образцы отбирали в 60, 90 и 120 минут с апикальной стороны. С базальной стороны отбирали образцы баланса массы в 0 и 120 минут.
Образцы анализировали на ЖХ/МС-МС. Целостность монослоя проверяли в конце эксперимента с использованием люцифера желтого, рассчитывая % отторжения люцифера желтого посредством инкубирования клеток со 100 мкг/мл люцифера.
3) Анализ данных:
Рарр рассчитывали следующим образом:
Кажущуюся проницаемость (Рарр) в единицах в секунду можно рассчитывать с использованием следующего уравнения,
Для одноточечного способа:
Рарр=(V/(T*A))*(C0/Ct)
Для многоточечного способа:
Рарр=(dQ/dt)/(A*C0)
% баланса массы = 100-[CR120*VR+CD120*VD/C0*VD]
Для люцифера желтого, % прохождения люцифера желтого = [RFU (опыт) - RFU (пустой) / RFU (равновесный) - RFU (пустой)]*100
Классификация проницаемости:

Отношение оттока = Рарр В-А/Рарр А-В
Отношение оттока, большее или равное 2, показывает то, что лекарственное средство представляет собой субстрат P-gp.
Результаты:
Двунаправленный анализ (ФИГ. 5)
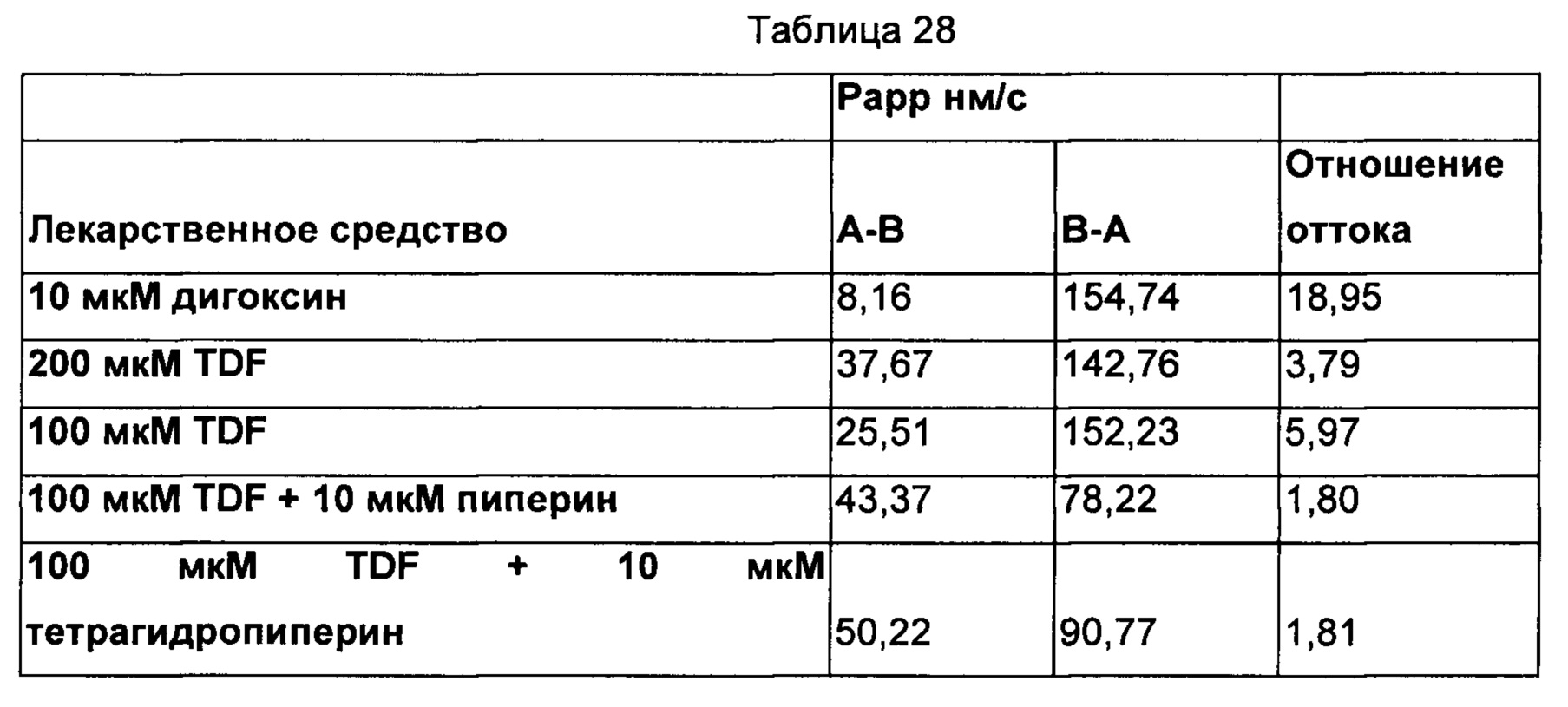
Заключения
TDF представляет собой известный субстрат P-gp. Поглощение TDF увеличивается пиперином посредством снижения отношения оттока. Кроме того, поглощение TDF увеличивается тетрагидропиперином посредством снижения отношения оттока. Посредством применения как пиперина, так и тетрагидропиперина наблюдали сравнимое улучшение проницаемости TDF. Следовательно, можно сделать заключение о том, что применение пиперина и тетрагидропиперина снижает отношение оттока, что, в свою очередь, увеличивало бы его биодоступность.
ИССЛЕДОВАНИЕ НА ЖИВОТНЫХ
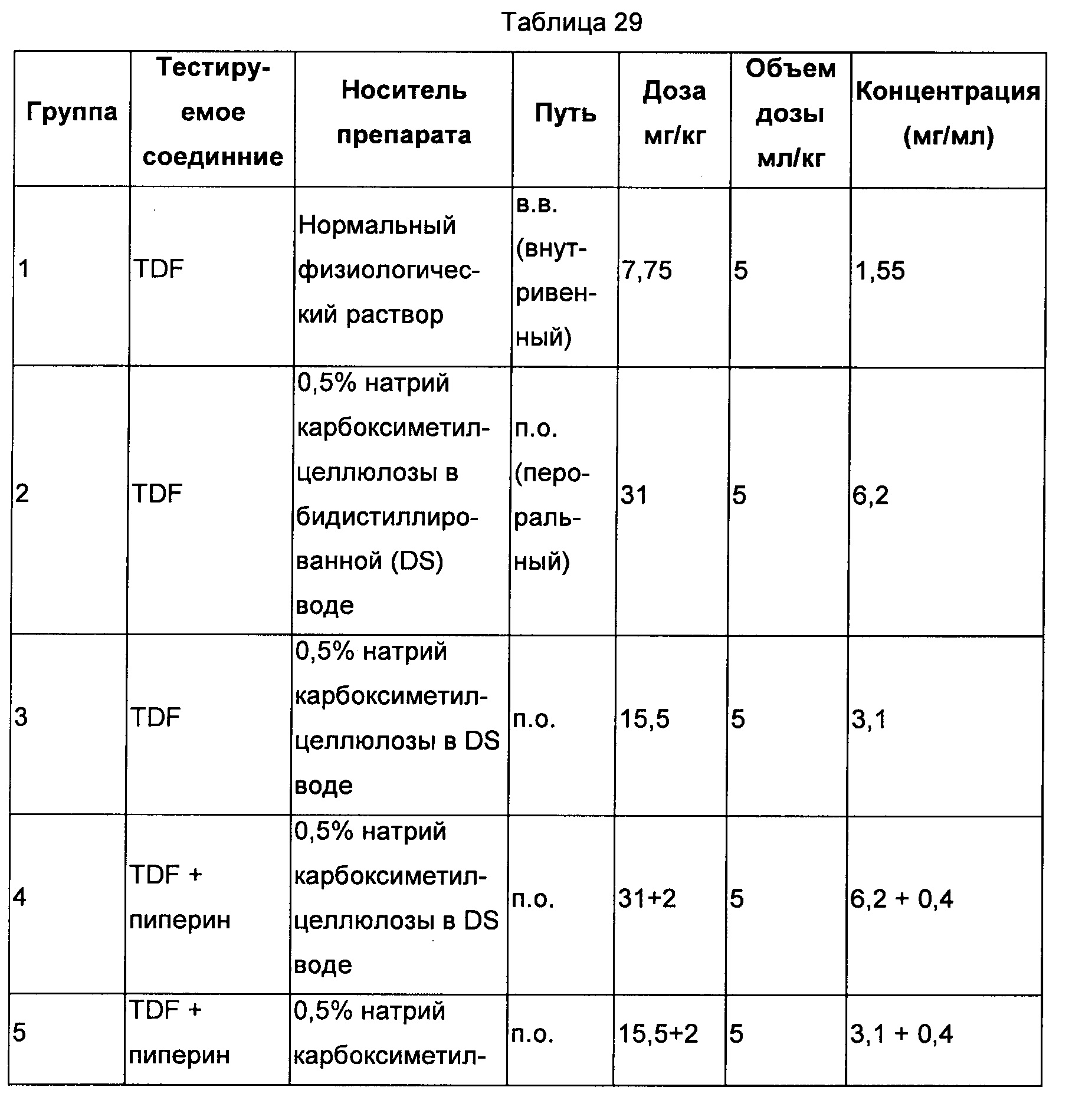
Исследование PK (фармакокинетика) у крыс in vivo
Целью данного не соответствующего GLP (надлежащая лабораторная практика) исследования было определение фармакокинетики TDF - одного и в комбинациях с пиперином в разных группах самцов крыс Wistar после внутривенного введения и перорального введения одной дозы. Данное исследвание было выполнено с одобрением Институционального комитета по этике обращения с животными (IAEC) согласно требованию Комитета для целей контроля и надзора за экспериментами на животных (CPCSEA), Индия.
Схема исследования
Данное исследование было проведено с использованием шести самцов крыс Wistar в каждой группе, как показано в Таблице 30 ниже.

Приготовление препарата
Препараты растворов готовили следующим образом: Внутривенный путь:
Взвешивали требующееся количество Соединения В (TDF) (23,03 мг), и к нему добавляли 10,94 мл носителя (нормального физиологического раствора), встряхивали на вибромешалке и обрабатывали ультразвуком в течение 2 минут с получением прозрачного препарата.
Пероральный путь:
Для группы 2: взвешивали требующееся количество соединения В (79,37 мг), и к нему добавляли 9,42 мл носителя (Na карбоксиметилцеллюлоза), встряхивали на вибромешалке и обрабатывали ультразвуком в течение 2 минут с получением препарата с концентрацией 6,2 мг/мл.
Для группы 3: взвешивали требующееся количество соединения В (46,14 мг), и к нему добавляли 10,95 мл носителя (Na карбоксиметилцеллюлоза), встряхивали на вибромешалке и обрабатывали ультразвуком в течение 2 минут с получением однородного препарата.
Для группы 4: взвешивали требующееся количество соединения В (79,56 мг), и к нему добавляли 4,72 мл носителя (Na карбоксиметилцеллюлоза), встряхивали на вибромешалке и обрабатывали ультразвуком в течение 2 минут с получением прозрачного препарата. Взвешивали требующееся количество соединения В1 (пиперин) (18,95 мг), и к нему добавляли 22,71 мл носителя (Na карбоксиметилцеллюлоза), встряхивали на вибромешалке и обрабатывали ультразвуком в течение 2 минут с получением прозрачного препарата. Равный объем (4,72 мл) каждого препарата смешивали в отдельных сосудах с получением 5 мл/кг.
Для группы 5: взвешивали требующееся количество соединения В (47,20 мг) и соединения В1 (18,95 мг), и к ним добавляли 5,61 мл носителя (Na карбоксиметилцеллюлоза), встряхивали на вибромешалке и обрабатывали ультразвуком в течение 2 минут с получением прозрачного препарата. Равный объем (5,61 мл) каждого препарата смешивали в отдельных сосудах с получением 5 мл/кг.
Биоанализ
Биоанализ проводили с использованием адаптированного для цели способа ЖХ-МС/МС для количественного измерения TDF и РМРА в образцах плазмы крысы. Калибровочная кривая (СС) для данного способа состояла из девяти ненулевых калибровочных стандартов, наряду с двойным пустым и нулевым стандартными образцами.
Фармакокинетический анализ
Фармакокинетические параметры плазмы рассчитывали с использованием инструмента для некомпартментного анализа из программы Phoenix (Версия 6.3) и определяли от индивидуальных животных в каждой группе. На основе группы с внутривенным введением рассчитывали пиковую концентрацию в плазме (Cmax), время до достижения пиковой концентрации в плазме (Tmax), площадь под кривой концентрации в плазме относительно времени (AUC0-t и AUCinf), AUC Extra (%), полупериод выведения (Т1/2), клиренс (CL), объем распределения Vd (л/кг) и среднее время удержания (MRT). На основе группы с пероральным введением рассчитывали пиковую концентрацию в плазме (Cmax), время до достижения пиковой концентрации в плазме (Tmax), AUC0-t и AUCinf, AUC Extra (%), среднее время удержания (MRT) и абсолютную пероральную биодоступность (F).
Результаты
Данные по концентрации в плазме относительно времени и фармакокинетические параметры в плазме РМРА после внутривенного и перорального введения TDF у самцов крыс Wistar представлены в Таблице 31.

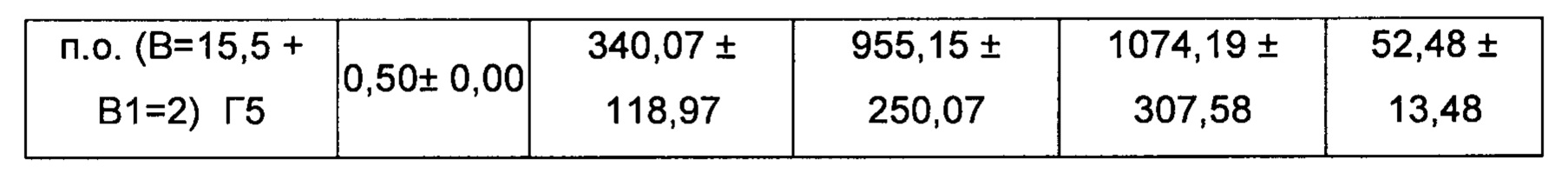
На ФИГ. 6 и 7 показаны концентрации в плазме тенофовира для 300 мг TGF и 300 мг TGF плюс 20 мг пиперина в разные моменты времени.

На ФИГ. 8 показана зависящая от времени концентрация тенофовира в плазме для 300 мг TDF, 300 мг TDF плюс 20 мг пиперина и 150 мг TDF плюс 20 мг пиперина.
Заключения
Исследование РК у крыс ясно показывают то, что пиковая концентрация тенофовира в плазме значительно возрастает при введении TDF в комбинации с пиперином. Данные результаты демонстрируют значительное увеличение биодоступности при введении 150 мг TDF с 20 мг пиперина (52,48 плюс/минус 13,48) по сравнению с 300 мг одного TDF (34,39 плюс/минус 6,07).
Композиции и способы приложенной формулы изобретения не ограничиваются по объему конкретными композициями и способами, описанными в данном документе, которые предназначены в качестве иллюстраций нескольких аспектов формулы изобретения, и подразумевается то, что любые композиции и способы, которые являются функционально эквивалентными, попадают в пределы объема формулы изобретения. Подразумевается то, что разные модификации композиций и способов, помимо композиций и способов, показанных и описанных в данном документе, попадают в пределы объема приложенной формулы изобретения. Кроме того, в то время как конкретно описываются только некоторые показательные композиции и стадии способа, раскрытого в данном документе, также подразумевается то, что другие комбинации композиций и стадий способа попадают в пределы объема приложенной формулы изобретения, даже если они конкретно не перечислены. Таким образом, комбинация стадий, элементов, компонентов или составных частей может быть прямо упомянута в данном документе, или, однако, включается меньшее число других комбинаций стадий, элементов, компонентов или составных частей, хотя они и не являются прямо изложенными. Термин «содержащий» и его вариации в том виде, в котором они используются в данном документе, используются синонимично термину «включающий» и его вариациям, и они являются открытыми, не ограничивающими терминами. Хотя термины «содержащий» и «включающий» и были использованы в данном документе для описания разных воплощений, термины «по существу состоящий из» и «состоящий из» могут использоваться вместо «содержащий» и «включающий» для предложения более конкретных воплощений изобретения, и они также раскрыты. За исключением примеров или там, где это упоминается иным образом, все числа, выражающие количества ингредиентов, условия реакции и так далее, используемые в описании и формуле изобретения, следует понимать как самые меньшие и не как попытку ограничивать применение доктрины эквивалентов к объему формулы изобретения, для истолкования в свете числа значимых цифр и обычных подходов округления.
Все патентные и непатентные публикации, процитированные в данном раскрытии, включаются в данный документ в такой степени, как если бы каждая из данных патентных и непатентных публикаций была включена в данный документ посредством ссылки во всей ее полноте. Кроме того, даже несмотря на то, что приведенное в данном документе раскрытие было описано со ссылкой на конкретные примеры и воплощения, следует понимать то, что данные примеры и воплощения просто иллюстрируют принципы и применения настоящего раскрытия. Следовательно, следует понимать то, что по отношению к иллюстративным воплощениям могут быть сделаны многочисленные модификации, и что могут быть разработаны другие механизмы без отступления от сущности и объема настоящего раскрытия, как определено следующей формулой изобретения.
Для специалистов в данной области будет очевидным то, что в отношении разных воплощений, описанных в данном документе, могут быть сделаны разные модификации и изменения без отступления от сущности или объема описанных в данном документе идей. Таким образом, подразумевается то, что разные воплощения охватывают другие модификации и изменения разных воплощений в пределах объема настоящих идей.
Реферат
Настоящее изобретение относится к пероральной или инъецируемой фармацевтической композиции, подходящей для применения в лечении вируса иммунодефицита человека (ВИЧ), содержащая терапевтически эффективное количество по меньшей мере одного антиретровирусного лекарственного средства, которое содержит (i) ингибитор обратной транскриптазы на основе нуклеотидного аналога (NRTI), выбранный из группы, включающей: тенофовира алафенамид фумарат, тенофовира дизопроксил фумарат, адефовир и их комбинацию; или (ii) ингибитор протеаз (PI), содержащий дарунавир, и терапевтически эффективное количество по меньшей мере одного активатора или улучшителя фармакокинетики, который содержит пиперин, тетрагидропиперин, цис-пиперин, транс-пиперин, цис,транс-пиперин, транс,цис- пиперин, цис,цис-пиперин, транс,транс-пиперин или их комбинацию. Также настоящее изобретение относится к способу получения пероральной или инъецируемой фармацевтической композиции, к набору для лечения вируса иммунодефицита человека (ВИЧ), к применению комбинации для увеличения биодоступности перорального антиретровирусного лекарственного средства, к пероральной или инъецируемой фармацевтической композиции, подходящей для применения в лечении вирусов гепатита В, к набору для лечения вирусов гепатита В. Техническим результатом настоящего изобретения является усиление биодоступности антиретровирусных лекарственных средств. 6 н. и 15 з.п. ф-лы, 14 пр., 31 табл., 8 ил.
Комментарии