Фармацевтический препарат с модифицированным высвобождением - RU2352323C2
Код документа: RU2352323C2
Описание
Это изобретение относится к новым фармацевтическим препаратам с модифицированным высвобождением, которые обеспечивают модифицированную доставку конкретных фармацевтических средств, к изготовлению таких препаратов и к применению такого препарата для лечения или предотвращения тромбоза.
Для поддержания желательного терапевтического уровня активного начала в плазме, жидкостях организма и/или желудочно-кишечном тракте часто бывает необходимо вводить фармацевтически активные соединения на протяжении дня с высокой частотой. Это особенно касается случаев, когда есть намерение доставлять лекарственное средство перорально и обеспечивать равномерный ответ в течение длительного периода времени.
В течение примерно последних тридцати лет лекарственные формы с модифицированным высвобождением становятся все более предпочтительным способом доставки определенных лекарственных средств пациентам, особенно пероральным путем. Такие формы могут, например, обеспечивать высвобождение лекарственного средства в течение длительного периода времени, снижая таким образом число требуемых суточных доз, причем в течение этого времени скорость высвобождения может быть по существу одинаковой и/или постоянной в пределах конкретной части желудочно-кишечного тракта или быть пульсирующей.
В данной области известны многочисленные лекарственные формы с модифицированным высвобождением, их обзор сделан, среди прочего, De Haan and Lerk в Pharmaceutisch Weekblad Scientific Edition, 6, 57 (1984); Banker в Medical Applications of Controlled Release, Vol II, eds. Langer and Wise (1984) Bocaraton, Florida, на стр.1-34; Graffner в Industrial Aspects of Pharmaceuticals, ed. Sandel, Swedish Pharmaceutical Press (1993) на стр.93-104; Proudfoot в "Dosage Regimens: Their Influence on Concentration-Time Profile of the Drug in the Body" на стр.191-211 в "Pharmaceutics: The Science of Dosage Form Design", ed. М.Е.Aulton (1988) (Churchill Livingstone).
В отличие от ближайшего аналога WO 02/19990, где в общем виде описаны препараты с модифицированным высвобождением, в настоящем изобретении предложена пероральная композиция с модифицированным высвобождением, содержащая в качестве активного ингредиента новые ингибиторы тромбина - сульфоновокислые соли соединения формулы (I).
Международная заявка на патент № PCT/SE 01/02657 (WO 02/44145, наиболее ранняя дата приоритета 1 декабря 2000, подана 30 ноября 2001, опубликована 06 июня 2002) раскрывает ряд соединений, которые представляют собой или метаболизируются в соединения, которые являются конкурентными ингибиторами трипсиноподобных протеаз, таких как тромбин. Следующие три соединения входят в число тех, что раскрыты конкретно:
(a) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe):
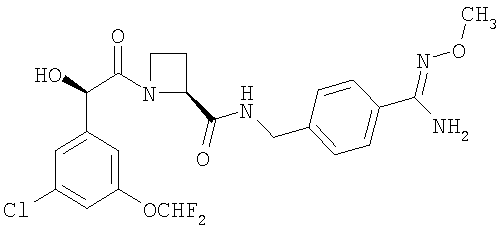
это соединение именуется здесь далее как Соединение А;
(б) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OMe):
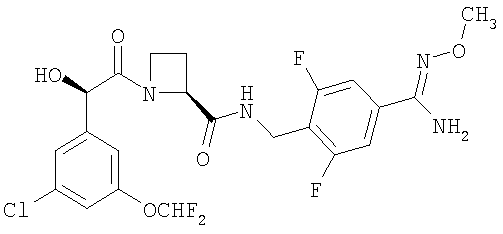
это соединение именуется здесь далее Соединение В, и
(в) Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe):
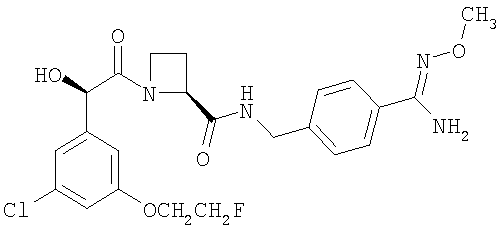
это соединение именуется здесь далее как Соединение С.
Метоксиамидиновые Соединения А, В и С метаболизируются после перорального и/или парентерального введения млекопитающему и образуют соответствующие соединения свободного амидина, причем было установлено, что именно эти соединения являются мощными ингибиторами тромбина. Таким образом:
- Соединение А метаболизируется в Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab (это соединение именуется здесь далее как Соединение D) через пролекарственное промежуточное соединение Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OH) (это соединение именуется здесь далее как Соединение G);
- Соединение В метаболизируется в Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(3)Aze-Pab(2,6-диF) (это соединение именуется здесь далее как Соединение Е) через пролекарственное промежуточное соединение Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OH) (это соединение именуется здесь далее как Соединение Н) и
- Соединение С метаболизируется в Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Ase-Pab (это соединение именуется здесь далее как Соединение F) через пролекарственное промежуточное соединение Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OH) (это соединение именуется здесь далее как Соединение J).
Способы синтеза Соединений А, В, С, D, Е, F, G и J раскрыты в Примерах 12, 40, 22, 3, 39, 21, 2 и 31 (соответственно) международной заявки на патент № PCT/SE 01/02657. Препарат с модифицированным высвобождением этих соединений или их метаболитов еще только подлежит описанию в литературе.
Авторы изобретения обнаружили, что Соединения А и С могут быть представлены в виде определенных йота-каррагинановых препаратов с модифицированным высвобождением, а также обнаружили, что соединения формулы (I) и их соли могут быть представлены в виде других фармацевтических препаратов с модифицированным высвобождением, которые легко вводить в организм, например, посредством перорального введения.
В соответствии с первым аспектом этого изобретения предложен фармацевтический препарат с модифицированным высвобождением, содержащий в качестве активного ингредиента соединение формулы (I):
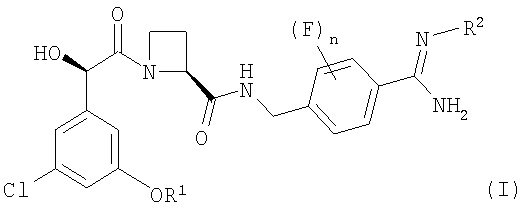
где R1 представляет собой С1-2алкил, замещенный одним или более чем одним заместителем, представляющим собой фтор;
R2 представляет собой водород, гидрокси, метокси или этокси и
n представляет собой 0, 1 или 2,
или его фармацевтически приемлемую соль, и фармацевтически приемлемый разбавитель или носитель, при условии, что когда соединение формулы (I) находится в форме соли, этот препарат может содержать только йота-каррагинан и нейтральный гелеобразующий полимер; такие препараты именуются здесь далее как "препараты по этому изобретению".
Соединения формулы (I) или их фармацевтически приемлемая соль могут быть в форме сольвата, гидрата, смешанного сольвата/гидрата или предпочтительно ансольвата, такого как ангидрат. Сольваты могут быть образованы одним или более чем одним органическим растворителем, таким как низшие (например С1-4) алкильные спирты (например, метанол, этанол или изо-пропанол), кетоны (такие как ацетон), сложные эфиры (такие как этилацетат) или их смеси.
В одном конкретном аспекте этого изобретения R1 представляет собой CHF2 или C2CH2F.
Переменная n представляет собой предпочтительно 0 или 2.
Более предпочтительные соединения формулы (I) включают соединения, у которых n представляет собой 0, или соединения, у которых n представляет собой 2, что обеспечивает два атома фтора, локализованных в положениях 2 и 6 (то есть в двух орто-положениях относительно точки присоединения бензольного кольца к группе -NH-CH2-).
Соединение формулы (I) представляет собой в особенности Соединение А, Соединение В или Соединение С.
Нейтральный гелеобразующий полимер представляет собой единственный нейтральный разлагаемый полимер или смесь более чем одного из нейтральных разлагаемых полимеров, обладающих гелеобразующими свойствами и имеющих по существу рН-независимую растворимость.
Предпочтительные соли соединений формулы (I) представляют собой соли присоединения кислот. Соли присоединения кислот включают соли присоединения неорганических кислот, такие как соли серной кислоты, азотной кислоты, фосфорной кислоты и галогеноводородных кислот, таких как бромистоводородная кислота и соляная кислота. Более предпочтительные соли присоединения кислот включают соли присоединения органических кислот, такие как соли диметилфосфорной кислоты, сахариновой кислоты, циклогексилсульфаминовой кислоты, соли присоединения карбоновых кислот (таких как малеиновая кислота, фумаровая кислота, аспарагиновая кислота, янтарная кислота, малоновая кислота, уксусная кислота, бензойная кислота, терефталевая кислота, гиппуровая кислота, 1-гидрокси-2-нафтойная кислота, памоевая кислота, гидроксибензойная кислота и тому подобные), соли присоединения гидроксикислот (таких как салициловая кислота, винная кислота, лимонная кислота, яблочная кислота (в том числе L-(-)-яблочная кислота и D,L-яблочная кислота), глюконовая кислота (в том числе D-глюконовая кислота), гликолевая кислота, аскорбиновая кислота, молочная кислота и тому подобные), соли присоединения аминокислот (таких как глутаминовая кислота (в том числе D-глутаминовая, L-глутаминовая и D,L-глутаминовая кислоты), аргинин (в том числе L-аргинин), лизин (в том числе L-лизин и L-лизина гидрохлорид), глицин и тому подобные) и особенно соли присоединения сульфоновых кислот (таких как 1,2-этандисульфоновая кислота, камфоросульфоновые кислоты (в том числе 1S-(+)-10-камфоросульфоновая кислота и (+/-)-камфоросульфоновые кислоты), этансульфоновая кислота, пропансульфоновая кислота (в том числе н-пропансульфоновая кислота), бутансульфоновая кислота, пентансульфоновая кислота, толуолсульфоновая кислота, метансульфоновая кислота, п-ксилолсульфоновая кислота, 2-мезитиленсульфоновая кислота, нафталинсульфоновые кислоты (в том числе 1,5-нафталинсульфоновая кислота и нафталинсульфоновая кислота), бензолсульфоновая кислота, гидроксибензолсульфоновые кислоты, 2-гидроксиэтансульфоновая кислота, 3-гидроксиэтансульфоновая кислота и тому подобные).
Особенно предпочтительные соли включают соли C1-6 (например C1-4) алкансульфоновых кислот, таких как этансульфоновая кислота (эзилат (esylate)) и пропансульфоновая кислота (например, н-пропансульфоновая кислота), и возможно замещенных (например, одной или более С1-2алкильными группами) арилсульфоновых кислот, таких как бензолсульфоновая кислота (безилат) и нафталиндисульфоновая кислота.
Подходящие стехиометрические соотношения кислоты и свободного основания находятся в диапазоне от 0,25:1,5 до 3,0:1, например от 0,45:1,25 до 1,25:1, в том числе от 0,50:1 до 1:1.
В соответствии с еще одним аспектом этого изобретения предложен препарат, содержащий соединение формулы (I) в по существу кристаллической форме.
Хотя авторы изобретения установили, что можно получать соединения по этому изобретению в формах, которые являются кристаллическими более чем на 80%, признак "в по существу кристаллической" включает кристаллическую более чем на 20%, предпочтительно более чем на 30% и более предпочтительно более чем на 40% (например, более чем на любое из значений, составляющих 50, 60, 70, 80 или 90%).
В соответствии с еще одним аспектом этого изобретения предложено также соединение по этому изобретению в частично кристаллической форме. Признак "частично кристаллическая" включает кристаллическую на 5% или кристаллическую на 5-20%.
Специалист может определять степень (%) кристалличности с использованием дифракции рентгеновских лучей на порошке (ДРЛП). Можно также применять другие методики, такие как ЯМР, инфракрасная спектроскопия с преобразованием Фурье (ИСФП), спектроскопия комбинационного рассеяния, дифференциальная сканирующая калориметрия (ДСК) и микрокалориметрия.
Предпочтительные соединения формулы (I), которые могут быть получены в кристаллической форме, включают в себя соли С1-6 (например С2-6, в частности С2-4) алкансульфоновых кислот, таких как этансульфоновая кислота, пропансульфоновая кислота (например, н-пропансульфоновая кислота), и возможно замещенных арилсульфоновых кислот, таких как бензолсульфоновая кислота и нафталиндисульфоновая кислота.
Термин фармацевтическая композиция "с модифицированным высвобождением" включает в себя, как понятно специалисту, любую(ой) композицию/препарат, в которой(ом) начало и/или скорость высвобождения лекарственного средства изменяется посредством галеновых манипуляций, и, таким образом, включает определение, которое дано в United States Pharmacopeia (USP XXII) на стр. xliii и xliv вводной части/преамбулы, соответствующее раскрытие в этом документе включено сюда ссылкой.
В данном случае модифицированное высвобождение может быть обеспечено соответствующим фармацевтически приемлемым носителем и/или другими средствами, причем эти носитель или средства (как целесообразно) вызывают изменение начала и/или скорости высвобождения активного ингредиента. Таким образом, этот термин должен пониматься специалистом в данной области как включающий в себя композиции, которые приготовлены с возможностью (например, как описано здесь) обеспечения "длительного", "пролонгированного" или "продленного" высвобождения лекарственного средства (где лекарственное средство высвобождается с достаточно замедленной скоростью для получения терапевтического ответа в течение требуемого периода времени, возможно включая обеспечение доступности начального количества лекарственного средства в течение предопределенного периода времени после введения для того, чтобы вызывать начальный желаемый терапевтический ответ); композиции, которые обеспечивают "отсроченное" высвобождение лекарственного средства (где высвобождение лекарственного средства отсрочено до достижения конкретного участка желудочно-кишечного тракта, после чего высвобождение лекарственного средства может быть либо пульсирующим, либо дополнительно модифицированным как указано выше); а также так называемые композиции "повторного действия" (где одна доза лекарственного средства высвобождается немедленно или через некоторое время после введения, а дальнейшие дозы высвобождаются позже).
Предпочтительно композиции по этому изобретению обеспечивают отсроченное высвобождение или более предпочтительно длительное (то есть пролонгированное или продленное) высвобождение лекарственного средства в течение некоторого периода времени. Более предпочтительные композиции по этому изобретению могут быть приготовлены с возможностью (например, как описано здесь) обеспечения достаточной дозы лекарственного средства на протяжении интервала дозирования (независимо от числа доз в единицу времени) для получения желаемого терапевтического эффекта. Высвобождение может быть равномерным и/или постоянным в течение продолжительного периода времени или иным.
Композиции по этому изобретению могут, например, быть представлены в следующих формах, хорошо известных специалистам.
(а) Пилюли, таблетки или капсулы с покрытием, которые могут быть предназначены для высвобождения по меньшей мере некоторого количества лекарственного средства, когда данный препарат достигает конкретного отдела желудочно-кишечного тракта. Такие таблетки могут быть, например, снабжены какой-либо формой устойчивого в желудке покрытия, такого как слой энтеросолюбильного покрытия, обеспечивающего высвобождение по меньшей мере части лекарственного средства, присутствующего в препарате, в конкретном отделе желудочно-кишечного тракта, таком как отделы кишечника.
(б) Системы множественных единиц или множественных частиц, которые могут быть в форме микрочастиц, микросфер или гранул, содержащих лекарственное средство (причем такие множественные единицы/множественные частицы могут обеспечивать постепенное прохождение препарата, содержащего лекарственное средство, из желудка в двенадцатиперстную кишку и далее через тонкий и толстый кишечник с высвобождением лекарственного средства с предопределенной скоростью).
(в) Препараты, содержащие дисперсии или твердые растворы активного соединения в матрице, которая может быть в форме воска, смолы или жира, особенно в форме полимера, в котором высвобождение лекарственного средства происходит посредством постепенной поверхностной эрозии таблетки и/или путем диффузии.
(г) Системы, которые содержат биоадгезивный слой, который может обеспечивать пролонгированное удержание композиции по этому изобретению в конкретном отделе желудочно-кишечного тракта (например, в желудке). Они включают в себя плавучие и погружающиеся системы (то есть системы с низкой и высокой плотностью соответственно), а также так называемые системы "увеличивающегося объема".
(д) Так называемые "висячие" устройства, в которых лекарственное средство присоединено к ионообменной смоле, которая обеспечивает постепенное высвобождение лекарственного средства под влиянием других ионов, присутствующих в желудочно-кишечном тракте, например кислой среды желудка.
(е) Устройства, в которых скорость высвобождения лекарственного средства контролируется его химическим потенциалом (например, осмотический насос).
(ж) Системы, в которых лекарственное средство высвобождается диффузией через мембраны, в том числе многослойные системы.
(з) Устройства, действующие в соответствии с внешним сигналом для высвобождения небольшого количества лекарственного средства.
(и) Активные самопрограммируемые системы, которые могут содержать чувствительный элемент, отвечающий на конкретную биологическую среду путем модуляции доставки лекарственного средства.
(к) Силастиковые депо контролируемого высвобождения, которые высвобождают лекарственное средство как функцию диффузии воды и/или желудочно-кишечных жидкостей в устройство через порт входа/выхода, что приводит к растворению и последующему высвобождению лекарственного средства.
Вышеуказанные принципы обстоятельно обсуждаются в ссылках из уровня техники, включая Pharmaceutisch Weekblad Scientific Edition, 6, 57 (1984); Medical Applications of Controlled Release, Vol II, eds. Langer and Wise (1984) Bocaraton, Florida, стр.1-34; Industrial Aspects of Pharmaceuticals, ed. Sandel, Swedish Pharmaceutical Press (1993), стр.93-104; и стр.191-211 в "Pharmaceutics: The Science of Dosage Form Design", ed. M.E.Aulton (1988) (Churchill Livingstone), а также в ссылках, которые процитированы в вышеуказанных документах, описания во всех этих документов включены сюда посредством ссылки.
В другом аспекте настоящего изобретения предложен пероральный препарат с модифицированным высвобождением, где R2 представляет собой гидрокси или метокси (например, Соединения А, В, С, G, Н или J, в частности R2представляет собой метокси, например Соединения А, В или С), или его фармацевтически приемлемая соль (в особенности его кристаллическая соль, такая как соль С1-6 (например С2-6, в частности С2-4) алкансульфоновой кислоты или соль возможно замещенной арилсульфоновой кислоты).
Это изобретение включает в себя парентеральные препараты с модифицированным высвобождением с использованием соединения формулы (I). В еще одном аспекте настоящего изобретения предложен парентеральный препарат с модифицированным высвобождением, где R2 представляет собой водород (например, Соединения D, Е или F).
В еще одном аспекте этого изобретения предложен препарат с модифицированным высвобождением, который содержит гелеобразующую матрицу. Эта матрица предпочтительно содержит гидроксипропилметилцеллюлозу (ГПМЦ), йота-каррагинан, додецилсульфат натрия (ДСН) и/или ксантановую камедь. Более предпочтительно матрица содержит гидроксипропилметилцеллюлозу (ГПМЦ), йота-каррагинан и/или поли(этиленоксид) (ПЭО). ГПМЦ может быть единственной или может представлять собой смесь двух или более ГПМЦ с разными вязкостями или молекулярными массами (как описано ниже).
Это изобретение также предлагает препарат с модифицированным высвобождением, содержащий одну или более чем одну ГПМЦ и один или более чем один дополнительный компонент, выбранный из группы, содержащей йота-каррагинан, микрокристаллическую целлюлозу, смазывающее вещество (такое как стеарилфумарат натрия) или маннит.
В еще одном аспекте данного изобретения предложет препарат с модифицированным высвобождением, содержащий ксантановую камедь или содержащий йота-каррагинан и ПЭО (как описано ниже).
Подходящие препараты с модифицированным высвобождением могут быть получены в соответствии со стандартными фармацевтическими методиками, как описано здесь или в вышеуказанных документах, и/или которые хорошо известны.
Предпочтительно в композициях по этому изобретению активный ингредиент представлен вместе с фармацевтически приемлемым носителем. В частности, предпочтительно, чтобы композиции по этому изобретению были представлены в форме активного ингредиента в полимерной матрице.
В этом отношении предпочтительно, чтобы композиции по этому изобретению были представлены для перорального введения в форме так называемой "набухающей" системы с модифицированным высвобождением или системы с модифицированным высвобождением, представляющей собой "гелеобразующую матрицу", в которой активный ингредиент представлен вместе с полимером, который набухает в водной среде (то есть "гидрофильным гелеобразующим компонентом"). Термин "водная среда" надо понимать в этом контексте как включающий в себя воду и жидкости, которые представляют собой или близки жидкостям, присутствующим в желудочно-кишечном тракте млекопитающего. Такие полимерные системы в типичных случаях содержат гидрофильные макромолекулярные структуры, которые в сухой форме могут быть в стекловидном или в по меньшей мере частично кристаллическом состоянии и которые набухают при контакте с водными средами. Модифицированное высвобождение лекарственного средства достигается, таким образом, посредством одного или более чем одного из следующих способов: транспорт растворителя в полимерную матрицу, набухание полимера, диффузия лекарственного средства через набухший полимер и/или эрозия полимера, один или более из которых могут служить для медленного высвобождения лекарственного средства из полимерной матрицы в водную среду.
Таким образом, подходящие полимерные материалы (действующие в качестве носителей), которые можно использовать в качестве гидрофильного гелеобразующего компонента гелеобразующей матрицы композиции с модифицированным высвобождением, включают в себя материалы, имеющие молекулярную массу выше 5000 г/моль, и которые
(а) являются по меньшей мере умеренно растворимыми в водных средах или
(б) набухают при контакте с водными средами (как определено здесь ранее),
тем самым делая возможным высвобождение лекарственного средства из носителя.
Таким образом, подходящие полимеры для гелеобразующей матрицы, которые могут быть синтетическими или природными, включают в себя полисахариды, такие как мальтодекстрин, ксантан, склероглюкановый декстран, крахмал, альгинаты, пуллулан, гиалуроновая кислота, хитин, хитозан и тому подобные; другие природные полимеры, такие как белки (альбумин, желатин и так далее), поли-L-лизин; полиакрилат натрия; поли(гидроксиалкилметакрилаты) (например, поли(гидроксиэтилметакрилат)); карбоксиполиметилен (например, Carbopol™); карбомер; поливинилпирролидон; камедь, такую как гуаровая камедь, гуммиарабик, камедь карайи, камедь гхатти, камедь бобов робинии, камедь тамаринда, геллановую камедь, трагакантовую камедь, агар, пектин, глютен и тому подобные; поли(виниловый спирт); этиленвиниловый спирт; поли(этиленоксид) (ПЭО); эфиры целлюлозы, такие как гидроксиметилцеллюлоза (ГМЦ), гидроксиэтилцеллюлоза (ГЭЦ), гидроксипропилцеллюлоза (ГПЦ), метилцеллюлоза (МЦ), этилцеллюлоза (ЭЦ), карбоксиэтилцеллюлоза (КЭЦ), этилгидроксиэтилцеллюлоза (ЭГЭЦ), карбоксиметилгидроксиэтилцеллюлоза (КМГЭЦ), гидроксипропилметилцеллюлоза (ГПМЦ), гидроксипропилэтилцеллюлоза (ГПЭЦ) и карбоксиметилцеллюлоза в натриевой форме (МаКМЦ), а также сополимеры и/или (простые) смеси любых вышеуказанных полимеров. Некоторые полимеры из числа вышеуказанных могут быть дополнительно подвергнуты поперечной сшивке стандартными методами.
Предпочтительно, чтобы в композициях по этому изобретению в форме систем гелеобразующей матрицы главным используемым набухающим полимером являлись ГПЦ, мальтодекстрин, склероглюкан или карбоксиполиметилен, более предпочтительно ПЭО или ксантан и особенно ГПМЦ, а также сополимеры и/или (простые) смеси любых этих полимеров. Йота-каррагинан также является предпочтительным.
Когда в гидрофильном гелеобразующем компоненте используют ПЭО, ксантан и ГПМЦ (то есть, по меньшей мере один из этих полимеров), предпочтительные молекулярные массы (то есть средневзвешенные молекулярные массы при определении стандартными методами, такими как осмометрия, ионная вытеснительная хроматография с детектором рефракции (где молекулярную массу определяют с помощью стандартных калибровочных кривых), методики светорассеяния и/или ультрацентрифугирования) этих полимеров находятся в диапазоне от 5000 г/моль до 200000000 г/моль, например до 100000000 г/моль, предпочтительно до 25000000 г/моль и более предпочтительно до 20000000 г/моль. Могут быть использованы смеси полимеров, представляющих собой ПЭО, ксантан и ГПМЦ, с разными молекулярными массами в пределах этих диапазонов.
Подходящие ГПМЦ полимеры также включают в себя полимеры, которые дают 2%-ные (мас.) растворы полимера в воде с показателями вязкости, как измерено стандартными методиками, которые в общем описаны в United States Pharmacopeia XXIV (USP XXIV/NF19) на стр.2002 et seq, а также конкретно на стр.843 и 844 (релевантные описания в этом документе включены сюда ссылкой), составляющими между 3 и 150000 сП (3-150000 мПа·с) (при 20°С), например между 10 и 120000 сП (10-120000 мПа·с), предпочтительно между 30 и 50000 сП (30-50000 мПа·с) и более предпочтительно между 50 и 15000 сП (50-15000 мПа·с). Смеси ГПМЦ полимеров с разными показателями вязкости в пределах этих диапазонов можно применять для того, чтобы, например, получать ГПМЦ смеси, которые дают растворы, как указано выше, со "средними" вязкостями (то есть вязкостями смеси) в пределах вышеуказанных предпочтительных диапазонов. Аналогичным образом можно использовать смеси ГПМЦ полимеров (с вязкостями и/или "средними" вязкостями в пределах эти диапазонов) с другими вышеупомянутыми полимерами. Подходящие ГПМЦ полимеры включают в себя полимеры, соответствующие стандартным заместительным типам 2208, 2906, 2910 и 1828 United States Pharmacopeia (см. дополнительные детали в USP XXIV/NF19). Подходящие ГПМЦ полимеры, таким образом, включают в себя полимеры, имеющиеся в продаже под товарным знаком METHOCEL™ (Dow Chemical Corporation) или товарным знаком METOLOSE™ (Shin-Etsu).
Подходящие ксантановые полимеры включают в себя полимеры, дающие 1%-ные (мас.) растворы полимера в воде с величинами вязкости, как измерено стандартными методами, описанными в United States Pharmacopeia XXIV (USP XXIV/NF19) на стр.2002 et seq, а также, конкретно, на стр.2537 и 2538 (релевантные описания в этом документе включены сюда этой ссылкой), между 60 и 2000 сП (60-2000 мПа·с) (при 24°С), например между 600 и 1800 сП (60-1800 мПа·с) и предпочтительно между 1200 и 1600 сП (1200-1600 мПа·с). Смеси ксантановых полимеров с разными величинами вязкости в пределах этих диапазонов можно использовать для того, чтобы, например, получать ксантановые смеси, которые дают растворы, как указано выше, со "средними" вязкостями (то есть вязкостями смеси) в пределах вышеуказанных предпочтительных диапазонов. Аналогичным образом можно использовать смеси ксантановых полимеров (с вязкостями и/или "средними" вязкостями в пределах этих диапазонов) с другими вышеупомянутыми полимерами. Подходящие ксантановые полимеры включают в себя полимеры, имеющиеся в продаже под товарными знаками XANTURAL™ и KELTROL™ (CPKetco) и SATIAXANE™ (Degussa, Texturant Systems).
Выбор полимера должен определяться природой активного ингредиента/лекарственного средства, использованного в композиции по этому изобретению, а также желаемой скоростью высвобождения. В частности, специалисту очевидно, например, в случае ГПМЦ, что более высокая молекулярная масса будет, в общем, обеспечивать более медленную скорость высвобождения лекарственного средства из композиции. Более того, в случае ГПМЦ разные степени замещения метоксильных групп и гидроксипропоксильных групп будут иметь результатом изменения в скорости высвобождения лекарственного средства из композиции. В этом отношении и как отмечено выше, для получения конкретного требуемого или желаемого профиля высвобождения может быть желательно представлять композиции по этому изобретению в форме систем гелеобразующей матрицы, в которых полимер-носитель получен путем смешивания двух или более полимеров, имеющих, например, разные молекулярные массы, например, как описано здесь ниже.
Авторы установили, что в случае систем в форме гелеобразующей матрицы скорость высвобождения лекарственного средства из композиции по этому изобретению можно дополнительно контролировать путем контроля соотношения лекарственное средство : полимер и соотношения площадь поверхности : объем у индивидуальных композиций (например, таблеток), содержащих лекарственное средство и систему полимера-носителя.
Композиции по этому изобретению в форме системы гелеобразующей матрицы или какой-либо иной могут содержать один или более дополнительных эксципиентов (в дополнение к системе полимера-носителя) для дальнейшего модифицирования высвобождения лекарственного средства, для улучшения физических и/или химических свойств готовой композиции, и/или для облегчения способа изготовления. Такие эксципиенты являются общепринятыми в приготовлении композиций с модифицированным высвобождением.
Например, композиции по этому изобретению могут содержать один или более следующих разбавителей: фосфат кальция (монокальцийфосфат, дикальцийфосфат и трикальцийфосфат), лактоза, микрокристаллическая целлюлоза, маннит, сорбит, диоксид титана, силикат алюминия и тому подобные. Предпочтительные разбавители включают в себя микрокристаллическую целлюлозу, а также маннит.
Композиции по этому изобретению могут содержать один или более следующих смазывающих веществ: стеарат магния, стеарилфумарат натрия и тому подобные.
Композиции по этому изобретению могут содержать скользящее вещество, такое как коллоидный диоксид кремния.
Композиции по этому изобретению могут содержать один или более следующих связующих агентов: поливинилпирролидон, лактоза, маннит, микрокристаллическая целлюлоза, полиэтиленгликоль (ПЭГ), ГПМЦ с низкой молекулярной массой, МЦ с низкой молекулярной массой, ГПЦ с низкой молекулярной массой и тому подобные. Предпочтительные связующие агенты включают в себя микрокристаллическую целлюлозу.
Композиции по этому изобретению могут содержать один или более следующих агентов контроля рН: органические кислоты (например, лимонная кислота и тому подобные) или их соли с щелочными металлами (например, натрием), фармацевтически приемлемые соли (например, соли натрия, магния или кальция) неорганических кислот (таких как угольная кислота или фосфорная кислота), оксиды магния, а также сульфаты, метабисульфаты, пропионаты и сорбаты щелочного и щелочноземельного металла (например, натрия, кальция, калия и тому подобных).
Другие дополнительные эксципиенты могут включать красители, корригенты, солюбилизаторы (такие как ДСН), покрывающие агенты, консерванты и так далее.
Можно использовать комбинации вышеуказанных дополнительных эксципиентов.
Следует понимать, что некоторые из вышеуказанных дополнительных эксципиентов, которые могут присутствовать в готовой композиции по этому изобретению, могут иметь более чем одну из вышеуказанных функций. Более того, вышеупомянутые дополнительные эксципиенты могут также функционировать как часть гидрофильного гелеобразующего компонента в системе гелеобразующей матрицы.
Общее количество дополнительных эксципиентов (за исключением, в случае систем гелеобразующей матрицы, основного(ых) полимера(ов)-носителя(ей)), которые могут присутствовать в композиции по этому изобретению, будет зависеть от природы композиции, а также от природы и количеств других составляющих этой композиции и может составлять вплоть до 85%, например между 0,1 и 75%, например от 0,2 до 65%, предпочтительно от 0,3 до 55%, более предпочтительно от 0,5 до 45% и особенно от 1 до 40%, например от 2 до 35 мас.%. В любом случае специалист может определять выбор и количество эксципиента(ов) рутинным образом (то есть без обращения к изобретательскому вкладу).
В системах гелеобразующей матрицы количество полимера в системе должно быть достаточным для того, чтобы на протяжении всего интервала дозирования обеспечивать дозу лекарственного средства, достаточную для получения желаемого терапевтического эффекта. Таким образом, для системы гелеобразующей матрицы предпочтительно, чтобы высвобождение 80% (особенно 60%) исходного количества лекарственного средства в композиции после введения пациенту в описанных здесь ниже опытных условиях занимало по меньшей мере 2 часа (предпочтительно по меньшей мере 4 часа, особенно по меньшей мере 6 часов), особенно на протяжении периода между 8 и 24 часами. Наиболее предпочтительно, чтобы по меньшей мере 80% исходного количества лекарственного средства в композиции высвобождалось за время примерно между 8 и 24 часами. Подходящие количества полимера, которые можно включить и которые должны зависеть, среди прочего, от активного ингредиента, используемого в композиции, от любых эксципиентов, которые могут присутствовать, и от природы используемого полимера, находятся в диапазоне от 5 до 99,5%, например от 10 до 95%, предпочтительно от 30 до 80 мас.%. В любом случае специалист может определить выбор и количество полимера рутинным образом.
В другом предпочтительном препарате является предпочтельным, чтобы соединения по этому изобретению присутствовали вместе в композиции гелеобразующей матрикцы, содержащей йота-каррагинан и один или более нейтральных гелеобразующих полимеров.
Йота-каррагинан предпочтительно присутствует в таком предпочтительном препарате на уровне, превышающем 15 мас.%. Предпочтительные марки йота-каррагинана включают в себя йота-каррагинан фармацевтической категории (например, доступный от FMC Biopolymer), который имеет вязкость не менее 5 сантипуаз (сП) (5 мПа·с), предпочтительно в диапазоне 5-10 сП (5-10 мПа·с) (для 1,5%-ного раствора, нагретого до 82°С, после чего вязкость измеряют при 75°С на вискозиметре Brookfield LV, снабженном шпинделем #1, действующим на скорости 30 об/мин), и йота-каррагинан технической категории (например, доступный от Fluka Biochemica), который предпочтительно имеет вязкость не менее 14 мПа·с для 0,3%-ного водного раствора, нагретого до 20°С, после чего вязкость измеряют с помощью вискозиметра с падающим шариком Haake типа, используемого совместно с термостатом Lauda C3 и Hakke Mess-System III и при использовании шариков из нержавеющей стали с золотым покрытием с плотностью 7,8 г/см3.
Нейтральный гелеобразующий полимер может быть единственным нейтральным полимером или представлять собой смесь более чем одного нейтральных полимеров, обладающих гелеобразующими свойствами и имеющих по существу рН-независимую растворимость.
Нейтральный гелеобразующий полимер предпочтительно присутствует в препарате на уровне более 10%, предпочтительно более 20 мас.%.
Подходящие нейтральные гелеобразующие полимеры включают в себя полиэтиленоксид (ПЭО), производные и члены семейства ПЭО (например, полиэтиленгликоль (ПЭГ)), предпочтительно существующие естественным образом в твердом состоянии и с подходящей молекулярной массой или вязкостью. При использовании в качестве единственного нейтрального гелеобразующего полимера ПЭО предпочтительно имеет молекулярную массу не менее 4 миллионов (4М), соответствующую диапазону вязкостей водного раствора от 1650 до 5500 мПа·с (или 1650-5500 сП, измерено для 1%-ного водного раствора при 25°С с помощью вискозиметра Brookfield RVF со шпинделем №2 при об/мин). Другие примеры подходящих ПЭО включают в себя ПЭО с молекулярной массой около 5 миллионов (5М), соответствующей диапазону вязкостей водного раствора от 5500 до 7500 мПа·с, или ПЭО с молекулярной массой около 8 миллионов (8М), соответствующей диапазону вязкостей водного раствора от 10000 до 15000 мПа·с. Этот диапазон охватывает типичные значения вязкости растворов (в сП), измеренные при 25°С, которые указаны для этого полимера в USP 24/NF 19, 2000 edition на стр.2285-2286. Если в качестве единственного нейтрального гелеобразующего полимера используют ПЭГ, он предпочтительно имеет высокую молекулярную массу, например молекулярную массу около 20000, соответствующую диапазону вязкости от 2700 до 3500 мПа·с (или 2700-3500 сП), как измерено для 50%-ного (мас.) водного раствора при 20°С с помощью капиллярного вискозиметра (Ubbelohde или его эквивалент). [Ссылка: European Pharmacopoeia 3rd Ed., 2000, Supplement, pp.908-909.]
Другие подходящие нейтральные гелеобразующие полимеры включают в себя производные целлюлозы, такие как гидроксипропилметилцеллюлоза (ГПМЦ) или гидроксиэтилцеллюлоза (ГЭЦ) с подходящим образом высокими величинами вязкости (например, ГПМЦ 50 сП, ГПМЦ 10000 сП, ГПМЦ 15000 сП, ГЭЦ типа НН или ГЭЦ типа Н). При их использовании в качестве единственного нейтрального полимера полимеры гидроксипропилметилцеллюлозы типа ГПМЦ 10000 сП и ГПМЦ 15000 сП имеют соответственно кажущиеся вязкости в диапазоне от 7500 до 14000 мПа·с (или 7500-14000 сП) и от 11250 до 21000 мПа·с (или 11250-21000 сП), измеренные при 20°С для 2%-ного (мас.) водного раствора, в рассчете со ссылкой на сухое вещество, при использовании капиллярного вискозиметра (Ubbelohde или его эквивалент). Один тип полимера гидроксиэтилцеллюлозы, например Natrosol 250 Pharma, тип НН, от Hercules Incorporated (Aqualon), показывает в типичных случаях вязкость по Брукфильду прмерно 20000 мПа·с при использовании прибора Brookfield Synchro-Lectric Model LVF, в условиях 1%-ной концентрации раствора, шпинделя №4, скорости шпинделя 30 об/мин, фактора 200 и 25°С (см. стр.21 брошюры Natrosol Physical and Chemical Properties, 33.007-E6 (1993)).
Конкретные препараты, которые могут быть упомянуты, включают в себя препараты, в которых соединение по этому изобретению приготовлено совместно с йота-каррагинаном и ГПМЦ (10000 сП (10000 мПа·с)) в соотношении 50:50 (мас.%), или совместно с йота-каррагинаном и ГПМЦ (50 сП (50 мПа·с)) & ГПМЦ (10000 сП (10000 МПа·с)) в соотношении 35:60:5 (мас.%), или совместно с йота-каррагинаном и ПЭО 4М в соотношении 50:50 (мас.%). Предпочтительные дополнительные эксципиенты в таких препаратах включают в себя смазывающие вещества, такие как стеарилфумарат натрия.
В одном аспекте этого изобретения предложен неинъецируемый препарат по этому изобретению, содержащий Соединение А, В или С или их соль, ГПМЦ и смазывающее вещество (такое как стеарилфумарат натрия). В еще одном аспекте этот препарат может содержать смесь двух или более чем двух ГПМЦ с разными показателями вязкости (например, 10000 сП (10000 мПа·с) и 50 сП (50 мПа·с)). Далее, этот препарат может дополнительно содержать солюбилизатор [такой как додецилсульфат натрия, лаурилсульфат натрия или гидрогенированное касторовое масло полиоксил 40].
Подходящие количества активного ингредиента в композиции по этому изобретению как в форме систем гелеобразующей матрицы, так и иной зависят от многих факторов, таких как природа этого ингредиента (свободное основание/соль и так далее), его требуемая доза и природа и количества других составных компонентов композиции. Однако они могут находиться в диапазоне от 0,5 до 80%, например от 1 до 75%, таком как от 3 до 70%, предпочтительно от 5 до 65%, более предпочтительно от 10 до 60% и особенно от 15 до 55 мас.%. В любом случае специалист может рутинным образом определить количество активного ингредиента для включения.
Типичная суточная доза соединения формулы (I) или его фармацевтически приемлемой соли находится в диапазоне от 0,001 до 100 мг/кг массы тела по свободному основанию (то есть в случае соли, исключая любую массу, являющуюся результатом присутствия противоиона), независимо от числа индивидуальных доз, которые вводят в течение этого дня. Предпочтительная суточная доза находится в диапазоне от 20 до 500 мг.
Композиции по этому изобретению, такие как описанные здесь ранее, можно изготавливать в соответствии с хорошо известными методиками, такими как методики, описанные в вышеупомянутых здесь ссылках. Композиции по этому изобретению, которые находятся в форме систем гелеобразующей матрицы, могут быть получены стандартными методами с помощью стандартного оборудования, известного специалисту, в том числе влажной или сухой грануляцией, прямым сжатием/прессованием, сушкой, измельчением, смешиванием, таблетированием и нанесением покрытия, а также комбинациями этих способов, например, как описано здесь далее.
Хотя композиции по этому изобретению предпочтительно адаптированы для перорального введения, их применение не ограничено этим способом введения. Парентеральные композиции с модифицированным высвобождением по этому изобретению, которые могут включать в себя системы, хорошо известные специалистам, такие как системы на основе полоксамеров, биоразлагаемых микросфер, липосом, суспензий в маслах и/или эмульсий, могут быть получены в соответствии со стандартными методиками, например, как описано Leung et al. в "Controlled Drug Delivery: Fundamentals and Applications" (Drugs and the Pharmaceutical Sciences; vol. 29), 2nd edition, eds. Robinson and Lee, Dekker (1987) в главе 10, стр.433, причем описания в этом документе включены сюда ссылкой.
Композиции по этому изобретению можно вводить один или несколько раз в сутки (предпочтительно один раз, но не более чем два раза в сутки), независимо от числа индивидуальных единиц (препаратов/композиций), которые вводят как часть одной "дозы".
Препараты по этому изобретению вводят пациентам-млекопитающим (включая людей) и, в случае соединений формулы (I), в которых R2 не представляет собой водород, они после этого метаболизируются в организме с образованием соединений формулы (I), в которых R2 представляет собой водород и которые являются фармакологически активными.
Таким образом, в соответствии с еще одним аспектом этого изобретения предложен препарат по этому изобретению для применения в качестве фармацевтического средства.
В частности, соединения формулы (I) представляют собой мощные ингибиторы тромбина или метаболизируются после введения с образованием мощных ингибиторов тромбина, например, как можно продемонстрировать в тестах, описанных среди прочего, в международной заявке на патент № PCT/SE 01/02657, а также международных заявках на патент WO 02/14270, WO 01/87879 и WO 00/42059, релевантные описания в этих документах включены сюда ссылкой.
Термин "пролекарство ингибитора тромбина" включает соединения, которые после введения метаболизируются и образуют ингибитор тромбина в количестве, экспериментально определяемом после введения.
Под "активным ингредиентом" и "активным веществом" подразумевается присутствующий в препарате фармацевтический агент (включая ингибитор тромбина и его пролекарства).
Таким образом, ожидается, что препараты по этому изобретению полезны при состояниях, когда требуется ингибирование тромбина, и/или при состояниях, когда показана антикоагулянтная терапия, включая следующее.
Лечение и/или профилактика тромбоза и гиперкоагуляции в крови и/или тканях животных, включая человека.
Известно, что гиперкоагуляция может приводить к тромбоэмболическим заболеваниям. Состояния, ассоциированные с гиперкоагуляцией и тромбоэмболическими заболеваниями, которые можно упомянуть, включают в себя наследственную или приобретенную устойчивость к активированному белку С, такую как мутация фактора V (лейденская мутация фактора V), и наследственные или приобретенные дефициты антитромбина III, белка С, белка S, кофактора II гепарина. Другие состояния, известные как ассоциированные с гиперкоагуляцией и тромбо-эмболическими заболеваниями, включают в себя циркулирующие антифосфолипидные антитела (антикоагулянт волчанки), гомоцистеинемию, вызванную гепарином тромбоцитопению и дефекты фибринолиза, а также коагуляционные синдромы (например, диссеминированное внутрисосудистое свертывание (ДВС)) и повреждения сосудов в целом (например, вследствие хирургического вмешательства).
Лечение состояний, при которых имеет место нежелательный избыток тромбина без признаков гиперкоагуляции, например при нейродегенеративных заболеваниях, таких как болезнь Альцгеймера.
Конкретные болезненные состояния, которые могут быть упомянуты, включают в себя терапевтическое и/или профилактическое лечение венозного тромбоза (например, тромбоза глубоких вен, ТГВ) и эмболии легких, артериального тромбоза (например, при инфаркте миокарда, нестабильной стенокардии, ударе вследствие тромбоза и периферическом артериальном тромбозе) и системной эмболии обычно от предсердия во время фибрилляции предсердия (например, невальвулярной фибрилляции предсердий) или от левого желудочка после трансмурального инфаркта миокарда, или вызванной застойной сердечной недостаточностью; профилактику реокклюзии (то есть тромбоза) после тромболиза, операций по чрезкожной транслюминальной ангиопластике (ЧТА) и коронарному шунтированию; предотвращение ретромбоза после микрохирургии и сосудистой хирургии вообще.
Дополнительные показания включают в себя терапевтическое и/или профилактическое лечение диссеминированного внутрисосудистого свертывания, вызванного бактериями, множественной травмой, интоксикацией или любым другим механизмом; антикоагулянтное лечение, когда кровь в организме находится в контакте с чужеродными поверхностями, такими как сосудистые трансплантаты, сосудистые стенты, сосудистые катетеры, механические и биологические протезы сосудов или любое другое медицинское устройство; и антикоагулянтное лечение, когда кровь находится в контакте с медицинскими устройствами вне организма, например во время сердечно-сосудистой хирургии с использованием аппарата сердце-легкие или при гемодиализе; терапевтическое и/или профилактическое лечение идиопатического и респираторного дистресс-синдрома у взрослых, фиброза легких после лучевой терапии или химиотерапии, септического шока, септицемии, воспалительных реакций, которые включают в себя, но без ограничения, отек, острого или хронического атеросклероза, такого как болезнь коронарных артерий и образование атеросклеротических бляшек, болезни церебральных артерий, церебрального инфаркта, церебрального тромбоза, церебральной эмболии, болезни периферических артерий, ишемии, стенокардии (включая нестабильную стенокардию), реперфузионного повреждения, рестеноза после чрезкожной транслюминальной ангиопластики (ЧТА) и шунтирования коронарных артерий.
Препарат по настоящему изобретению может также содержать любой(ые) антитромботический(е) агент(ы) с механизмом действия, отличным от соединения формулы (I), такие как один или более из числа следующего: антитромбоцитарные агенты - ацетилсалициловая кислота, тиклопидин и клопидогрел; ингибиторы рецептора и/или синтетазы тромбоксана; антагонисты рецепторов фибриногена; миметики простациклина; ингибиторы фосфодиэстеразы; антагонисты ADP-рецепторов (рецепторов аденозиндифосфата) (Р2Т) и ингибиторы карбоксипептидазы U (CPU).
Соединения формулы (I), которые ингибируют трипсин и/или тромбин, могут также быть полезными в лечении панкреатита.
Таким образом, препараты по этому изобретению показаны для терапевтического и/или профилактического лечения этих состояний.
Препараты по этому изобретению полезны в доставке соединения формулы (I) или его соли пациенту. Поскольку соединения формулы (I) и их соли полезны как в профилактике, так и в лечении тромбоза препараты по этому изобретению также полезны в лечении такого нарушения.
В соответствии с еще одним аспектом этого изобретения предложен способ лечения тромбоза, включающий введение препарата по изобретению субъекту, страдающему от такого состояния или подверженному ему.
В еще одном аспекте настоящего изобретения предложен препарат по этому изобретению для изготовления лекарственного средства для применения в лечении тромбоза.
Во избежание сомнений термин "лечение" включает терапевтическое лечение, а также профилактику состояния.
Композиции по этому изобретению обладают преимуществом, заключающимся в том, что они могут обеспечивать модифицированное высвобождение соединений формулы (I) или фармацевтически приемлемой соли любого из этих соединений для получения более равномерного и/или пролонгированного антитромботического действия и, таким образом, могут обеспечивать эффективное введение активного ингредиента предпочтительно не более чем один или два раза в сутки.
Композиции по этому изобретению могут также обладать преимуществом, заключающимся в том, что их можно изготавливать посредством установившихся способов фармацевтической обработки с использованием материалов, которые утверждены для применения в пищевых или фармацевтических продуктах или имеют аналогичный регулятивный статус.
Соединения формулы (I) могут быть получены с помощью нижеследующих методов.
Общие методы
ТСХ выполняли на силикагеле. Анализ методом хиральный ВЭЖХ (высокоэффективная жидкостная хроматография) выполняли на колонке Chiralcel OD 46 мм × 250 мм с 5-сантиметровой предохранительной колонкой. Температуру колонки поддерживали на уровне 35°С. Использовали скорость тока 1,0 мл/мин. Использовали детектор Gilson 115 UV при 228 нм. Подвижная фаза состояла из гексанов, этанола и трифторуксусной кислоты, и их надлежащие соотношения указаны для каждого соединения. В типичных случаях растворяли продукт в минимальном количестве этанола и разбавляли подвижной фазой.
В нижеуказанных «получениях A-I» ЖХ-МС/МС (жидкостная хроматография с тандемной масс-спектрометрией) выполняли с помощью прибора HP-1100, снабженного инжектором CTC-PAL и колонкой ThermoQuest, 5 Tm, 4×100 мм, Hypersil BDS-C18. Использовали масс-спектрометрический детектор API-3000 (Sciex). Скорость тока составляла 1,2 мл/мин, и подвижная фаза (градиент) состояла из 10-90%-ного ацетонитрила с 90-10% 4 мМ водн. ацетата аммония, причем оба содержали 0,2% муравьиной кислоты. В других случаях снимали масс-спектры низкого разрешения (МСНР) с помощью спектрометра Micromass ZQ в режиме переключения положительных и отрицательных ионов (posneg) с электрораспылительной ионизацией (диапазон масс m/z 100-800), а также снимали масс-спектры высокого разрешения (МСВР) с помощью спектрометра Micromass LCT в режиме электрораспылительной отрицательной ионизации (диапазон масс m/z 100-1000) с лейцин-энкефалином (С28Н37N5О7) в качестве внутреннего стандарта массы.
Спектры1H-ЯМР снимали с использованием тетраметилсилана в качестве внутреннего стандарта.
Способы синтеза соединений формулы (I) описаны в заявке на международный патент № PCT/SE 01/02657 (WO 02/44145, наиболее ранняя дата приоритета 01 декабря 2000 г., подана 30 ноября 2001 г., опубликована 06 июня 2002 г.), релевантная информация из нее включена в данное описание.
Получение А. Получение Соединения А
(1) 3-Хлор-5-метоксибензальдегид
3,5-Дихлоранизол (74,0 г, 419 ммоль) в тетрагидрофуране (ТГФ) (200 мл) добавляли по каплям к металлическому магнию (14,2 г, 585 ммоль, предварительно промыт 0,5 н. HCl) в ТГФ (100 мл) при 25°С. После этого добавления добавляли по каплям 1,2-дибромэтан (3,9 г, 20,8 ммоль). Получившуюся темно-коричневую смесь кипятили с обратным холодильником в течение 3 ч. Эту смесь охлаждали до 0°С и добавляли к ней N,N-диметилформамид (60 мл) одной порцией. Эту смесь подвергали распределению между диэтиловым эфиром (3×400 мл) и 6 н. HCl (500 мл). Объединенные органические экстракты промывали рассолом (300 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением масла. Флэш-хроматография (2×) на силикагеле с элюцией смесью Нех : EtOAc (4:1) дала указанное в подзаголовке соединение (38,9 г, 54%) в виде желтого масла.
1H-ЯМР (300 МГц, CDCl3) δ 9.90 (s, 1Н), 7.53 (s, 1Н), 7.38 (s, 1H), 7.15 (s, 1H), 3.87 (s, 3H).
(2) 3-Хлор-5-гидроксибензальдегид
Раствор 3-хлор-5-метоксибензальдегида (22,8 г, 134 ммоль; см. стадию (1) выше) в CH2Cl2 (250 мл) охлаждали до 0°С. Трибромид бора (15,8 мл, 167 ммоль) добавляли каплями на протяжении 15 мин. После перемешивания реакционной смеси в течение 2 ч медленно добавляли H2O (50 мл). Этот раствор затем экстрагировали Et2O (2×100 мл). Органические слои объединяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Флэш-хроматография на силикагеле с элюцией смесью Нех : EtOAc (4:1) дала указанное в подзаголовке соединение (5,2 г, 25%).
1H-ЯМР (300 МГц, CDCl3) δ 9.85 (s, 1H), 7.35 (s, 1H), 7.20 (s, 1H), 7.10 (s, 1H), 3.68 (s, 1H).
(3) 3-Хлор-5-дифторметоксибензальдегид
Раствор 3-хлор-5-гидроксибензальдегида (7,5 г, 48 ммоль; см. стадию (2) выше) в 2-пропаноле (250 мл) и 30%-ном КОН (100 мл) кипятили с обратным холодильником. Реакционную смесь в течение 2 ч при перемешивании барботировали CHClF2. Реакционную смесь охлаждали, подкисляли 1 н. HCl и экстрагировали EtOAc (2×100 мл). Органическую фазу промывали рассолом (100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Флэш-хроматография на силикагеле с элюцией смесью Нех : EtOAc (4:1) дала указанное в подзаголовке соединение (4,6 г, 46%).
1H-ЯМР (300 МГц, CDCl3) δ 9.95 (s, 1H), 7.72 (s, 1Н), 7.52 (s, 1H), 7.40 (s, 1H), 6.60 (t, JH-F=71.1 Гц, 1H).
(4)Ph(3-Cl)(5-OCHF2)-(R,S)CH(OTMS)CN
Раствор 3-хлор-5-дифторметоксибензальдегида (4,6 г, 22,3 ммоль; см. стадию (3) выше) в CH2Cl2 (200 мл) охлаждали до 0°С. Добавляли ZnI2 (1,8 г, 5,6 ммоль) и триметилсилилцианид (2,8 г, 27,9 ммоль) и давали реакционной смеси нагреваться до комнатной температуры при перемешивании в течение 15 ч. Эту смесь частично концентрировали в вакууме с получением указанного в подзаголовке соединения в виде жидкости, которую использовали прямо на стадии (5) ниже без дополнительной очистки или характеризации.
(5)Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(NH)OEt
Ph(3-Cl)(5-OCHF2)-(R,S)CH(OTMS)CN (6,82 г, принято за 22,3 ммоль; см. стадию (4) выше) добавляли по каплям к смеси HCl/EtOH (500 мл). Эту реакционную смесь перемешивали в течение 15 ч, затем частично концентрировали в вакууме с получением указанного в подзаголовке соединения в виде жидкости, которую использовали на стадии (6) без дополнительной очистки или характеризации.
(6)Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OEt
Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(NH)OEt (6,24 г, принято за 22,3 ммоль; см. стадию (5) выше) растворяли в ТГФ (250 мл), добавляли 0,5 М H2SO4 (400 мл) и перемешивали реакционную смесь при 40°С в течение 65 ч, охлаждали и затем частично концентрировали в вакууме для удаления большей части ТГФ. Эту реакционную смесь затем экстрагировали Et2O (3×100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в подзаголовке соединения в виде твердого вещества, которое использовали на стадии (7) без дополнительной очистки или характеризации.
(7)Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OH
Раствор Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OEt (6,25 г, принято за 22,3 ммоль; см. стадию (6) выше) в 2-пропаноле (175 мл) и 20%-ном КОН (350 мл) перемешивали при комнатной температуре в течение 15 ч. Эту реакционную смесь затем частично концентрировали в вакууме для удаления большей части 2-пропанола. Оставшуюся смесь подкисляли 1 М H2SO4, экстрагировали Et2O (3×100 мл), сушили (Na2SO4) и концентрировали в вакууме с получением твердого вещества. Флэш-хроматография на силикагеле с элюцией смесью CHCl3: МеОН : концентрированный NH4ОН (6:3:1) давала аммониевую соль указанного в подзаголовке соединения. Эту аммониевую соль затем растворяли в смеси EtOAc (75 мл) и Н2O (75 мл) и подкисляли 2 н. HCl. Органический слой отделяли и промывали рассолом (50 мл), сушили (Na2SO4) и концентрировали в вакууме с получением указанного в подзаголовке соединение (3,2 г, 57% от стадий (4)-(7)).
1H-ЯМР (300 МГц, CD3OD) δ 7.38 (s, 1H), 7.22 (s, 1H), 7.15 (s, IH), 6.89 (t, JH-F=71.1 Гц, 1H), 5.16 (s, 1H).
(8) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (а) и Ph(3-Cl)(5-OCHF2)-(S)CH(OAc)C(O)OH (б)
Смесь Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OH (3,2 г, 12,7 ммоль, см. стадию (7) выше) и липазы PS "Amano" (прибл. 2,0 г) в винилацетате (125 мл) и метил-трет-бутилового эфира (МТБЭ) (125 мл) кипятили с обратным холодильником в течение 48 ч. Эту реакционную смесь охлаждали, фильтровали через Celite® и осадок на фильтре промывали EtOAc. Фильтрат концентрировали в вакууме и подвергали флэш-хроматографии на силикагеле с элюцией смесью CHCl3: МеОН : концентрированный NH4OH (6:3:1) с получением аммониевых солей указанных в подзаголовке соединений (а) и (б). Соединение (а) в виде соли растворяли в H2O, подкисляли 2 н. HCl и экстрагировали EtOAc. Органический слой промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в подзаголовке соединения (а) (1,2 г, 37%).
Для указанного в подзаголовке соединения (а)
1H-ЯМР (300 МГц, CD3OD) δ 7.38 (s, 1H), 7.22 (s, 1H), 7.15 (s, 1H), 6.89 (t, JH-F=71.1 Гц, 1Н), 5.17 (s, 1Н).
(9) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
К раствору Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (1,1 г, 4,4 ммоль; см. стадию (8) выше) и H-Aze-Pab(Teoc) (см. международную заявку на патент WO 00/42059, 2,6 г, 5,7 ммоль) в ДМФ (50 мл) при 0°С добавляли РуВОР (2,8 г, 5,3 ммоль) и коллидин (1,3 г, 10,6 ммоль). Эту реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение дополнительных 15 ч. Эту реакционную смесь концентрировали в вакууме и подвергали флэш-хроматографии на силикагеле (3×) при элюции сначала смесью CHCl3: EtOH (9:1), затем EtOAc : EtOH (20:1) и наконец при элюции смесью СН2Cl2: СН3ОН (95:5) с получением указанного в подзаголовке соединения (1,0 г, 37%) в виде белого твердого вещества.
1H-ЯМР (300 МГц, CD3OD, смесь ротамеров) δ 7.79-7.85 (d, J=8.7 Гц, 2Н), 7.15-7.48 (m, 5H), 6.89 и 6.91 (t, JH-F=71.1 Гц, 1Н), 5.12 и 5.20 (s, 1H), 4.75-4.85 (m, 1H), 3.97-4.55 (m, 6Н), 2.10-2.75 (m, 2Н), 1.05-1.15 (m, 2Н), 0.09 (s, 9H).
МС (масс-спектрометрия) (m/z) 611 (М+1)+.
(10) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe, Teoc)
Растворяли Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,40 г, 0,65 ммоль; см. стадию (9) выше) в 20 мл ацетонитрила и добавляли 0,50 г (6,0 ммоль) гидрохлорида O-метилгидроксиламина. Эту смесь нагревали при 70°С в течение 2 ч. Растворитель выпаривали и остаток подвергали распределению между водой и этилацетатом. Водную фазу экстрагировали еще дважды этилацетатом и объединенную органическую фазу промывали водой, рассолом, сушили (Na2SO4), фильтровали и упаривали. Выход 0,41 г (91%).
1H-ЯМР (400 МГц; CDCl3): δ 7.83 (bt, 1H), 7.57 (bs, 1H), 7.47 (d, 2Н), 7.30 (d, 2Н), 7.20 (m, 1H), 7.14 (m, 1H), 7.01 (m. 1H), 6.53 (t, 1H), 4.89 (s, 1H), 4.87 (m, 1H), 4.47 (m, 2H), 4.4-4.2 (b, 1H), 4.17-4.1 (m, 3H), 3.95 (s, 3H), 3.67 (m, 1H), 2,68 (m, 1H), 2.42 (m, 1H) 0.97 (m, 2H), 0.01 (s, 9H).
(11) Соединение А
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OMe, Teoc) (0,40 г, 0,62 ммоль; см. стадию (10) выше) растворяли в 5 мл ТФУ и оставляли взаимодействовать в течение 30 мин. ТФУ выпаривали и остаток подвергали распределению между этилацетатом и водным NaHCO3. Водную фазу экстрагировали еще дважды этилацетатом и объединенную органическую фазу промывали водой, рассолом, сушили (Na2SO4), фильтровали и упаривали. Продукт сушили вымораживанием из смеси вода/ацетонитрил. Очистка была не нужна. Выход 0,28 г (85%).
1H-ЯМР (600 МГц; CDCl3): δ 7.89 (bt, 1H), 7.57 (d, 2H), 7.28 (d, 2H), 7.18 (m, 1Н), 7.13 (m, 1H), 6.99 (m, 1H), 6.51 (t, 1H), 4.88 (s, 1H), 4.87 (m, 1H), 4.80 (bs, 2H), 4.48 (dd, 1H), 4.43 (dd, 1H), 4.10 (m, 1H), 3.89 (s, 3H), 3.68 (m, 1H), 2,68 (m, 1H), 2.40 (m, 1H).
13С-ЯМР (125 МГц; CDCl3): (углероды карбонила и/или амидина, ротамеры) δ 172,9. 170,8, 152,7, 152,6.
МСВР: рассчитано для С22Н23ClF2N4O5 (М-Н)- 495,1242, найдено 495,1247.
Получение В. Получение соединения В
(1) 2,6-Дифтор-4-[(метилсульфинил)(метилтио)метил]бензонитрил
(Метилсульфинил)(метилтио)метан (7,26 г, 0,0584 моль) растворяли в 100 мл сухого ТГФ под аргоном и охлаждали до -78°С. По каплям при перемешивании добавляли бутиллитий в гексане (16 мл, 1,6 M, 0,0256 моль). Эту смесь перемешивали в течение 15 мин. Тем временем раствор 3,4,5-трифторбензонитрила (4,0 г, 0,025 ммоль) в 100 мл сухого ТГФ охлаждали до -78°С под аргоном и первый раствор добавляли через канюлю к второму раствору на протяжении периода времени 35 мин. Через 30 мин охлаждающую баню удаляли и, когда реакционная смесь достигала комнатной температуры, ее вливали в 400 мл воды. ТГФ выпаривали и оставшийся водный слой экстрагировали три раза диэтиловым эфиром. Объединенную эфирную фазу промывали водой, сушили (Na2SO4) и выпаривали. Выход 2,0 г (30%).
1H-ЯМР (500 МГц, CDCl3) δ 7.4-7.25 (m, 2H), 5.01 (s, 1H, диастереомер), 4.91 (s, 1H, диастереомер), 2.88 (s, 3H, диастереомер), 2.52 (s, 3H, диастереомер), 2.49 (s, 3H, диастереомер), 2.34 (s, 3H, диастереомер), 1.72 (широкий, 1H).
(2) 2,6-Дифтор-4-формилбензонитрил
Растворяли 2,6-дифтор-4-[(метилсульфинил)(метилтио)метил]бензонитрил (2,17 г, 8,32 ммоль; см. стадию (1) выше) в 90 мл ТГФ и добавляли 3,5 мл концентрированной серной кислоты. Эту смесь оставляли при комнатной температуре на 3 дня и затем вливали в 450 мл воды. Затем осуществляли трехкратную экстракцию EtOAc и объединенную эфирную фазу промывали дважды водным бикарбонатом натрия и рассолом, сушили (Na2SO4) и выпаривали. Выход 1,36 г (98%). Положение формильной группы определяли методом13С-ЯМР. Сигнал от фторированных углеродов при 162,7 м.д. проявлял ожидаемый паттерн взаимодействия с двумя константами взаимодействия порядка 260 Гц и 6,3 Гц соответственно, что соответствует ипсо- и мета-сопряжению от атомов фтора.
1H-ЯМР (400 МГц, CDCl3) δ 10.35 (s, 1 Н), 7.33 (m, 2H).
(3) 2,6-Дифтор-4-гидроксиметилбензонитрил
2,6-Дифтор-4-формилбензонитрил (1,36 г, 8,13 ммоль; см. стадию (2) выше) растворяли в 25 мл метанола и охлаждали на ледяной бане. Добавляли порциями при перемешивании борогидрид натрия (0,307 г, 8,12 ммоль) и оставляли реакционную смесь на 65 мин. Растворитель упаривали и остаток подвергали распределению между диэтиловым эфиром и водным бикарбонатом натрия. Эфирный слой промывали еще водным бикарбонатом натрия и рассолом, сушили (Na2SO4) и упаривали. Неочищенный продукт быстро кристаллизовался, и его можно было использовать без дополнительной очистки. Выход 1,24 г (90%).
1H-ЯМР (400 МГц, CDCl3) δ 7.24 (m, 2H), 4.81 (s, 2H), 2.10 (широкий, 1Н).
(4) 4-Циано-2,6-дифтобензилметансульфонат
К охлаждаемому льдом раствору 2,6-дифтор-4-гидроксиметилбензонтрила (1,24 г, 7,32 ммоль; см. стадию (3) выше) и метансульфонилхлорида (0,93 г, 8,1 ммоль) в 60 мл метиленхлорида добавляли триэтиламин (0,81 г, 8,1 ммоль) при перемешивании. После 3 ч при 0°С эту смесь промывали дважды 1 M HCl и один раз водой, сушили (Na2SO4) и выпаривали. Продукт можно было использовать без дополнительной очистки. Выход 1,61 г (89%).
1H-ЯМР (300 МГц, CDCl3) δ 7.29 (m, 2H), 5.33 (s, 2H), 3.07 (s, 3Н).
(5) 4-Азидометил-2,6-дифторбензонитрил
Смесь 4-циано-2,6-дифторбензилметансульфоната (1,61 г, 6,51 ммоль; см. стадию (4) выше) и азида натрия (0,72 г, 0,0111 моль) в 10 мл воды и 20 мл ДМФ перемешивали при комнатной температуре в течение ночи. Затем вливали полученную смесь в 200 мл воды и экстрагировали три раза диэтиловым эфиром. Объединенную эфирную фазу промывали пять раз водой, сушили (Na2SO4) и упаривали. Небольшую пробу упаривали для ЯМР, и продукт кристаллизовался. Остальное осторожно упаривали, но не совсем досуха. Выход (теоретически 1,26 г) считали почти количественным на основании ЯМР и аналитической ВЭЖХ.
1H-ЯМР (400 МГц, CDCl3) δ 7.29 (m, 2H), 4.46 (s, 2H).
(6) 4-Аминометил-2,6-дифторбензонитрил
Это взаимодействие проводили в соответствии с процедурой, описанной в J. Chem. Res. (M) (1992) 3128. К суспензии 520 мг 10% Pd/C (влажность 50%) в 20 мл воды добавляли раствор борогидрида натрия (0,834 г, 0,0221 моль) в 20 мл воды. Результатом было некоторое выделение газа. 4-Азидометил-2,6-дифторбензонитрил (1,26 г, 6,49 ммоль; см. стадию (5) выше) растворяли в 50 мл ТГФ и добавляли к этой водной смеси на ледяной бане на протяжении 15 мин. Эту смесь перемешивали в течение 4 ч, после чего добавляли к ней 20 мл 2 M HCI и фильтровали эту смесь через целит. Целит промывали еще водой, объединенную водную фазу промывали EtOAc и после этого подщелачивали 2 M NaOH. Затем следовала трехкратная экстракция метиленхлоридом, и объединенную органическую фазу промывали водой, сушили (Na2SO4) и выпаривали. Выход 0,87 г (80%).
1H-ЯМР (400 МГц, CDCl3) δ 7.20 (m, 2H), 3.96 (s, 2H), 1,51 (широкий, 2H).
(7) 2,6-Дифтор-4-трет-бутоксикарбониламинометилбензонитрил
Раствор 4-аминометил-2,6-дифторбензонитрила (0,876 г, 5,21 ммоль; см. стадию (6) выше) разводили в 50 мл ТГФ и добавляли ди-трет-бутилдикарбонат (1,14 г, 5,22 ммоль) в 10 мл ТГФ. Эту смесь перемешивали в течение 3,5 ч. ТГФ выпаривали и остаток подвергали распределению между водой и EtOAc. Органический слой промывали три раза 0,5 M HCl и водой, сушили (Na2SO4) и выпаривали. Продукт можно было использовать без дополнительной очистки. Выход 1,38 г (99%).
1H-ЯМР (300 МГц, CDCl3) δ 7.21 (m, 2H). 4.95 (широкий, 1Н), 4.43 (широкий, 2Н), 1,52 (s,9H).
(8) Вос-Pab(2,6-диF)(OH)
Смесь 2,6-дифтор-4-трет-бутоксикарбониламинометилбензонитрила (1,38 г, 5,16 ммоль; см. стадию (7) выше), гидрохлорида гидроксиламина (1,08 г, 0,0155 моль) и триэтиламина (1,57 г, 0,0155 моль) в 20 мл этанола перемешивали при комнатной температуре в течение 36 ч. Растворитель выпаривали и остаток подвергали распределению между водой и метиленхлоридом. Органический слой промывали водой, сушили (Na2SO4) и выпаривали. Продукт можно было использовать без дополнительной очистки. Выход 1,43 г (92%).
1H-ЯМР (500 МГц, CD3OD) δ 7.14 (m, 2H), 4.97 (широкий, 1Н), 4.84 (широкий, 2H), 4.40 (широкий, 2H), 1.43 (s, 9H).
(9) Вос-Pab(2,6-диF) × НОАс
Это взаимодействие проводили в соответствии с процедурой, описанной Judkins et al, Synth. Comm. (1998) 4351. Вос-Pab(2,6-диF)(ОН) (1,32 г, 4,37 ммоль; см. стадию (8) выше), уксусный ангидрид (0,477 г, 4,68 ммоль) и 442 мг 10% Pd/C (влажность 50%) в 100 мл уксусной кислоты гидрировали при давлении 5 атм (505 кПа) в течение 3,5 ч. Эту смесь фильтровали через целит, промывали этанолом и упаривали. Остаток подвергали сушке вымораживанием из ацетонитрила и воды и нескольких капель этанола. Указанный в подзаголовке продукт можно было использовать без дополнительной очистки. Выход 1,49 г (99%).
1H-ЯМР (400 МГц, CD3OD) δ 7.45 (m, 2H), 4.34 (s, 2H), 1.90 (s, 3H), 1.40 (s,9H).
(10) Вос-Pab(2,6-диF)(Теос)
К раствору Вос-Pab(2,6-диР) × НОАс (1,56 г, 5,49 ммоль; см. стадию (9) выше) в 100 мл ТГФ и 1 мл воды добавляли 2-(триметилсилил)этил-п-нитрофенилкарбонат (1,67 г, 5,89 ммоль). Добавляли по каплям на протяжении 5 мин раствор карбоната калия (1,57 г, 0,0114 моль) в 20 мл воды. Эту смесь перемешивали в течение ночи. ТГФ выпаривали и остаток подвергали распределению между водой и метиленхлоридом. Водный слой экстрагировали метиленхлоридом и объединенную органическую фазу промывали дважды водным бикарбонатом натрия, сушили (Na2SO4) и упаривали. Флэш-хроматография на силикагеле со смесью гептан/EtOAc = 2/1 давала 1,71 г (73%) чистого соединения.
1H-ЯМР (400 МГц, CDCl3) δ 7.43 (m, 2H), 4,97 (широкий, 1Н), 4.41 (широкий, 2H), 4.24 (m, 2H), 1.41 (s, 9H), 1.11 (m, 2H), 0.06 (s, 9H).
(11) Вос-Aze-Pab(2,6-диF)(Теос)
Вос-Pab(2,6-диF)(Теос) (1,009 г, 2,35 ммоль; см. стадию (10) выше) растворяли в 50 мл EtOAc, насыщенного HCl (газ). Эту смесь оставляли на 10 мин, выпаривали, растворяли в 18 мл ДМФ и затем охлаждали на водяной бане. Добавляли Boc-Aze-OH (0,450 г, 2,24 ммоль), РуВОР (1,24 г, 2,35 ммоль) и, наконец, диизопропилэтиламин (1,158 г, 8,96 ммоль). Эту реакционную смесь перемешивали в течение 2 ч, затем вливали в 350 мл воды и экстрагировали три раза EtOAc. Объединенную органическую фазу промывали рассолом, сушили (Na2SO4) и упаривали. Флэш-хроматография на силикагеле со смесью гептан : EtOAc (1:3) давала 1,097 г (96%) желаемого соединения.
1H-ЯМР (500 МГц, CDCl3) δ 7.46 (m, 2H), 4.65-4.5 (m, 3Н), 4.23 (m, 2H), 3.87 (m, 1H), 3.74 (m, 1H), 2.45-2.3 (m, 2H), 1.40 (s, 9H), 1.10 (m, 2H), 0.05 (s, 9H).
(12) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(Teoc)
Вос-Aze-Pab(2,6-диF)(Теос) (0,256 г, 0,500 ммоль; см. стадию (11) выше) растворяли в 20 мл EtOAc, насыщенного HCl (газ). Эту смесь оставляли на 10 мин, выпаривали и растворяли в 5 мл ДМФ. Добавляли Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)ОН (0,120 г, 0,475 ммоль; см. Получение А (8) выше), РуВОР (0,263 г, 0,498 ммоль) и, наконец, диизопропилэтиламин (0,245 г, 1,89 ммоль). Эту реакционную смесь перемешивали в течение 2 ч, затем вливали в 350 мл воды и экстрагировали три раза EtOAc. Объединенную органическую фазу промывали рассолом, сушили (Na2SO4) и выпаривали. Флэш-хроматография на силикагеле с EtOAc давала 0,184 г (60%) желаемого указанного в подзаголовке соединения.
1H-ЯМР (400 МГц, CD3OD, смесь ротамеров) δ 7.55-7.45 (m, 2H), 7.32 (m, 1H, главный ротамер), 7.27 (m, 1H, минорный ротамер), 7.2-7.1 (m, 2H). 6.90 (t, 1Н, главный ротамер), 6.86 (t, 1H, минорный ротамер), 5.15 (s, 1H, главный ротамер), 5.12 (m, 1H, минорный ротамер), 5.06 (s, 1H, минорный ротамер), 4.72 (m, 1H, главный ротамер), 4.6-4.45 (m, 2H), 4.30 (m, 1H, главный ротамер), 4.24 (m, 2H), 4.13 (m, 1H, главный ротамер), 4.04 (m, 1H, минорный ротамер), 3.95 (m, 1H, минорный ротамер), 2,62 (m, 1H, минорный ротамер), 2.48 (m, 1H, главный ротамер), 2.22 (m, 1H, главный ротамер), 2.10 (m, 1H, минорный ротамер), 1.07 (m, 2H), 0.07 (m, 9H).
(13) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(OMe, Teoc)
Смесь Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(Teoc) (64 мг, 0,099 ммоль; см. стадию (12) выше) и гидрохлорида О-метилгидроксиламина (50 мг, 0,60 ммоль) в 4 мл ацетонитрила нагревали при 70°С в течение 3 ч. Растворитель выпаривали и остаток подвергали распределению между водой и EtOAc. Водный слой экстрагировали дважды EtOAc и объединенную органическую фазу промывали водой, сушили (Na2SO4) и выпаривали. Продукт можно было использовать без дополнительной очистки. Выход 58 мг (87%).
1H-ЯМР (400 МГц, CDCl3) δ 7.90 (bt, 1H), 7.46 (m, 1H), 7.25-6.95 (m, 5H), 6.5 (t, 1H), 4.88 (s, 1H), 4.83 (m, 1H), 4.6-4.5 (m, 2H), 4.4-3.9 (m, 4H), 3.95 (s, 3H), 3.63 (m, 1H), 2,67 (m, 1H), 2.38 (m, 1H), 1.87 (широкий, 1H), 0.98 (m, 2H), 0.01 (s, 9H).
(14) Соединение В
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)(OMe, Teoc) (58 мг, 0,086 ммоль; см. стадию (13) выше) растворяли в 3 мл ТФУ, охлаждали на ледяной бане и оставляли взаимодействовать в течение 2 ч. ТФУ выпаривали и остаток растворяли в EtOAc. Органический слой промывали дважды водным карбоном натрия и водой, сушили (Na2SO4) и упаривали. Остаток подвергали сушке вымораживанием из воды и ацетонитрила с получением 42 мг (92%) соединения, указанного в заголовке.
1H-ЯМР (300 МГц, CDCl3) δ 7.95 (bt, 1H), 7.2-7.1 (m. 4H), 6.99 (m, 1H), 6.52 (t, 1H), 4.88 (s, 1H), 4.85-4.75 (m, 3H), 4.6-4.45 (m, 2H), 4.29 (широкий, 1H), 4.09 (m, 1H), 3.89 (s, 3H), 3.69 (m, 1H), 2,64 (m, 1H), 2.38 (m, 1H), 1.85 (широкий, 1H).
13С-ЯМР (100 МГц; CDCl3): (атомы углерода карбонила и/или амидина) δ 172,1, 169,8, 151,9.
APCI-MS (масс-спектрометрия с химической ионизацией при атмосферном давлении: (М+1) = 533/535 m/z.
Получение С. Получение Соединения С
(1) (2-Монофторэтил)метансульфонат
К перемешиваемому на магнитной мешалке раствору 2-фторэтанола (5,0 г, 78,0 ммоль) в CH2Cl2 (90 мл) под азотом при 0°С добавляли триэтиламин (23,7 г, 234 ммоль) и метансульфонилхлорид (10,7 г, 93,7 ммоль). Эту смесь перемешивали при 0°С в течение 1,5 ч, разбавляли CH2Cl2 (100 мл) и промывали 2 н. HCl (100 мл). Водный слой экстрагировали CH2Cl2 (50 мл) и объединенные органический экстракты промывали рассолом (75 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в подзаголовке соединения (9,7 г, 88%) в виде желтого масла, которое использовали без дополнительной очистки.
1H-ЯМР (300 МГц, CDCl3) δ 4.76 (t, J=4 Гц, 1Н), 4.64 (t, J=4 Гц, 1Н), 4.52 (t, J=4 Гц, 1 Н), 4.43 (t, J=4 Гц, 1 Н), 3.09 (s, 3Н).
2) 3-Хлор-5-монофторэтоксибензальдегид
К раствору 3-хлор-5-гидроксибензальдегида (8,2 г, 52,5 ммоль; см. Получение А (2) выше) и карбоната калия (9,4 г, 68,2 ммоль) в ДМФ (10 мл) под азотом добавляли по каплям при комнатной температуре раствор (2-монофторэтил)метансульфоната (9,7 г, 68,2 ммоль; см. стадию (1) выше) в ДМФ (120 мл). Эту смесь нагревали до 100°С в течение 5 ч и затем перемешивали в течение ночи при комнатной температуре. Реакционную смесь охлаждали до 0°С, вливали в ледяную 2 н. HCl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Коричневое масло хроматографировали на силикагеле с элюцией смесью гексан (Hex) : EtOAc (4:1) с получением указанного в подзаголовке соединения (7,6 г, 71%) в виде желтого масла.
1H-ЯМР (300 МГц, CDCl3) δ 9.92 (s, 1Н), 7.48 (s, 1Н), 7.32 (s, 1Н), 7.21 (s, 1Н), 4.87 (t, J=4 Гц, 1H), 4.71 (t, J=3 Гц, 1Н), 4.33 (t, J=3 Гц, 1Н), 4.24 (t, J=3 Гц, 1Н).
(3) Ph(3-Cl)(5-OCH2CH2F)-(R,S)CH(OTMS)CN
К раствору 3-хлор-5-монофторэтоксибензальдегида (7,6 г, 37,5 ммоль; см. стадию (2) выше) и йодида цинка (3,0 г, 9,38 ммоль) в CH2Cl2 (310 мл) добавляли по каплям при 0°С под азотом триметилсилилцианид (7,4 г, 75,0 ммоль). Эту смесь перемешивали при 0°С в течение 3 ч и при комнатной температуре в течение ночи. Реакционную смесь разбавляли Н2О (300 мл), органический слой отделяли, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в подзаголовке соединения (10,6 г, 94%) в виде коричневого масла, которое использовали без дополнительной очистки или характеризации.
(4) Ph(3-Cl)(5-OCH2CH2F)-(R,S)CH(OH)C(O)OH
Концентрированную соляную кислоту (100 мл) добавляли к Ph(3-Cl)(5-OCH2CH2F)-(R,S)CH(OTMS)CN (10,6 г, 5,8 ммоль; см. стадию (3) выше) и этот раствор перемешивали при 100°С в течение 3 ч. После охлаждения до комнатной температуры реакционную смесь дополнительно охлаждали до 0°С, медленно подщелачивали 3 н. NaOH (приблизит. 300 мл) и промывали Et2O (3×200 мл). Водный слой подкисляли 2 н. HCl (80 мл) и экстрагировали EtOAc (3×300 мл). Объединенные EtOAc экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в подзаголовке соединения (8,6 г, 98%) в виде бледно-желтого твердого вещества, которое использовали без дополнительной очистки.
Rf=0,28 (90:8:2 CHCl3: МеОН : концентрированный NH4OH).
1H-ЯМР (300 МГц, CD3OD) δ 7.09 (s, 1H), 7.02 (s, 1H), 6.93 (s, 1H), 5.11 (s, 1H), 4.77-4.81 (m, 1H), 4.62-4.65 (m, 1H), 4.25-4.28 (m, 1H), 4.15-4.18 (m, 1H).
(5) Ph(3-Cl)(5-ОСН2СН2F)-(S)СН(ОАс)С(О)OH (а) и Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)OH (б)
Раствор Ph(3-Cl)(5-OCH2CH2F)-(R,S)CH(OH)C(O)OH (8,6 г, 34,5 ммоль; см. стадию (4) выше) и липазы PS "Amano" (4,0 г) в винилацетате (250 мл) и МТБЭ (250 мл) нагревали при 70°С под азотом в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и фермент удаляли фильтрацией через Celite®. Осадок на фильтре промывали EtOAc и фильтрат концентрировали в вакууме. Хроматография на силикагеле с элюцией смесью CHCl2: МеОН : Et3N (90:8:2) давала соль триэтиламина указанного в подзаголовке соединения (а) в виде желтого масла. Дополнительно была получена соль триэтиламина указанного в подзаголовке Соединения (б) (4,0 г). Соль указанного в подзаголовке соединения (б) растворяли в H2O (250 мл), подкисляли 2 н. HCl и экстрагировали EtOAc (3×200 мл). Объединенные органические экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением указанного в подзаголовке соединения (б) (2,8 г, 32%) в виде желтого масла.
Данные для указанного в подзаголовке Соединения (б):
Rf=0,28 (90:8:2 CHCl3: МеОН : концентрированный NH4ОН).
1H-ЯМР (300 МГц, CD3OD) δ 7.09 (s, 1Н), 7.02 (s, 1Н), 6.93 (s, 1H), 5.11 (s, 1Н), 4.77-4.81 (m, 1H), 4.62-4.65 (m, 1H), 4.25-4.28 (m, 1H), 4.15-4.18 (m, 1H).
(6) Соединение С
К раствору Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)OH (818 мг, 3,29 ммоль; см. стадию (5) выше) в ДМФ (30 мл) под азотом при 0°С добавляли HAze-Pab(OMe)·2HCl (1,43 г, 4,27 ммоль, см. международную заявку на патент WO 00/42059), РуВОР (1,89 г, 3,68 ммоль) и ДИПЭА (1,06 г, 8,23 ммоль). Реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение ночи. Эту смесь концентрировали в вакууме и остаток хроматографировали два раза на силикагеле, элюируя сначала смесью CHCl3: EtOH (15:1), а затем смесью EtOAc : EtOH (20:1), с получением соединения, указанного в заголовке (880 мг, 54%).
Rf=0,60 (10:1 CHCl3: EtOH).
1H-ЯМР (300 МГц, CD3OD, сложная смесь ротамеров) δ 7.58-7.60 (d, J=8 Гц, 2Н), 7.34 (d, J=7 Гц, 2Н), 7.05-7.08 (m, 2H), 6.95-6.99 (m, 1H), 5.08-5.13 (m, 1H), 4.77-4.82 (m, 1H), 4.60-4.68 (m, 1H), 3.99-4.51 (m, 7Н), 3.82 (s, 3H), 2.10-2.75 (m, 2H).
13С-ЯМР (150 МГц; CD3OD): (атомы углерода карбонила и/или амидина) δ 173,3, 170,8, 152,5.
ХИАД-МС:(М+1)=493 m/z.
Получение Соединения D (Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab)
Соединение D
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,045 г, 0,074 ммоль; см. Получение А (9) выше) растворяли в 3 мл ТФУ и оставляли взаимодействовать в течение 1 ч. ТФУ выпаривали и остаток подвергали сушке вымораживанием из смеси вода/ацетонитрил с получением 0,043 г (100%) указанного в подзаголовке соединения в виде его ТФУ соли.
1H-ЯМР (400 МГц; CD3OD) ротамеры: δ 7.8-7.75 (m, 2H), 7.55-7.5 (m, 2H), 7.35 (m, 1H, главный ротамер), 7.31 (m, 1H, минорный ротамер), 7.19 (m, 1H, главный ротамер). 7.15 (m, 1H), 7.12 (m, 1H, минорный ротамер), 6.89 (t, 1H, главный ротамер), 6.87 (t, 1H, минорный ротамер), 5.22 (m, 1H, минорный ротамер), 5.20 (s, 1H, главный ротамер), 5.13 (s, 1H, минорный ротамер), 4.80 (m, 1H, главный ротамер), 4.6-4.4 (m, 2H), 4.37 (m, 1H, главный ротамер), 4.19 (m, 1H, главный ротамер), 4.07 (m, 1H, минорный ротамер), 3.98 (m, 1H, минорный ротамер), 2.70 (m, 1H, минорный ротамер), 2.55 (m, 1H, главный ротамер), 2.29 (m, 1H, главный ротамер), 2.15 (m, 1H, минорный ротамер).
13С-ЯМР (100 МГц; CD3OD): (атомы углерода карбонила и/или амидина, ротамеры) δ 172,6. 172,5, 172,0. 171,7, 167,0.
МС (m/z) 465 (M-1)-, 467 (M+1)+.
Получение Соединения Е (Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(2,6-диF)
Соединение Е
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-Aze-Pab(2,6-диF)(Теос) (81 мг, 0,127 ммоль; см. Получение В (12) выше) растворяли в 0,5 мл метиленхлорида и охлаждали на ледяной бане. Добавляли ТФУ (3 мл) и оставляли реакционную смесь на 75 мин. ТФУ выпаривали и остаток подвергали сушке вымораживанием из воды из ацетонитрила. Неочищенный продукт очищали препаративной жидкостной хроматографией с обращенными фазами с СН3CN : 0,1 M NH4OAc (35:65) с получением 39 мг (55%) соединения, указанного в заголовке, в виде его НОАс соли; чистота 99%.
1H-ЯМР (400 МГц, CD3DO, смесь ротамеров) δ 7.5-7.4 (m, 2H), 7.32 (m, 1H, главный ротамер), 7.28 (m, 1H, минорный ротамер), 7.2-7.1 (m, 3Н), 6.90 (t, 1H, главный ротамер), 6.86 (t, минорный ротамер), 5.15 (s, 1H, главный ротамер), 5.14 (m, 1H, минорный ротамер), 5.07 (s, 1H, минорный ротамер), 4.72 (m, 1H, главный ротамер), 4.65-4.45 (m, 2H), 4.30 (m, 1H, главный ротамер), 4.16 (m, 1H, главный ротамер), 4.03 (m, 1H, минорный ротамер), 3.95 (m, 1H, минорный ротамер), 2,63 (m, 1H, минорный ротамер), 2.48 (m, 1H, главный ротамер), 2.21 (m, 1H, главный ротамер), 2.07 (m, 1H, минорный ротамер), 1.89 (s, 3Н).
13С-ЯМР (75 МГц; CD3OD): (атомы углерода карбонила и/или амидина, смесь ротамеров) δ 171,9, 171,2, 165,0, 162,8, 160,4.
ХИАД-МС: (M+1) = 503/505 m/z.
Получение Соединения F (Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab×ТФУ)
(1) Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(Teoc)
К раствору Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)OH (940 мг, 3,78 ммоль; см. Получение С (5) выше) в ДМФ (30 мл) под азотом при 0°С добавляли HAze-Pab(Teoc)·HCl (2,21 г, 4,91 ммоль), РуВОР (2,16 г, 4,15 ммоль) и ДИПЭА (1,22 г, 9,45 ммоль). Эту реакционную смесь перемешивали при 0°С в течение 2 ч и затем при комнатной температуре в течение 4 ч. Эту смесь концентрировали в вакууме и остаток хроматографировали дважды на силикагеле, элюируя сначала смесью CHCl3: EtOH (15:1), а затем смесью EtOAc : EtOH (20:1) с получением указанного в подзаголовке соединения (450 мг, 20%) в виде ломкой белой пены.
Точка плавления 80-88°С.
Rf=0,60 (10: 1 CHCl3: EtOH).
1H-ЯМР (300 МГц, CD3DO, сложная смесь ротамеров) δ 7.79 (d, J=8 Гц, 2Н), 7.42 (d, J=8 Гц, 2Н), 7.05-7.08 (m, 1H), 6.93-6.99 (m, 2H), 5.08-5.13 (m, 1H), 4.75-4.80 (m, 2H), 4.60-4.68 (m, 1H), 3.95-4.55 (m, 8H), 2.10-2.75 (m, 2H), 1.05-1.11 (m, 2H), 0.08 (s, 9H).
ХИАД-МС: (M+1) = 607 m/z.
(2) Соединение F
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,357 г, 0,589 ммоль; см. стадию (1) выше) растворяли в 10 мл ТФУ и оставляли взаимодействовать в течение 40 мин. ТФУ выпаривали и остаток подвергали сушке вымораживанием из смеси вода/ацетонитрил с получением 0,33 г (93%) соединения, указанного в заголовке, в виде его ТФУ соли.
1H-ЯМР (600 МГц; CD3DO) ротамеры: δ 7.8-7.7 (m, 2H), 7.54 (d, 2H), 7.08 (s, 1H, главный ротамер), 7.04 (s, 1H, минорный ротамер), 6.99 (s, 1H, главный ротамер), 6.95 (s, 1H), 6.92 (s, 1H, минорный ротамер), 5.18 (m, 1H, минорный ротамер), 5.14 (s, 1H, главный ротамер), 5.08 (s, 1H, минорный ротамер), 4.80 (m, 1H, главный ротамер), 4.73 (m, 1H), 4.65 (m, 1H), 4.6-4.4 (m, 2Н), 4.35 (m, 1H, главный ротамер), 4.21 (дублет мультиплетов, 2Н), 4.12 (m, 1H, главный ротамер), 4.06 (m, 1H, минорный ротамер), 3.99 (m, 1H, минорный ротамер), 2,69 (m, 1H, минорный ротамер), 2.53 (m, 1H, главный ротамер), 2.29 (m, 1H, главный ротамер), 2.14 (m, 1H, минорный ротамер).
13С-ЯМР (150 МГц; CD3OD): (атомы углерода карбонила и/или амидина) δ 172,8,172,1,167,4.
ЭР-МС +: (M+1) = 463 (m/z).
Получение Соединения G (Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH)
(1) Ph(3-Cn(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH, Teoc)
Растворяли Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(Teoc) (0,148 г, 0,24 ммоль; см. Получение А, стадия (9) выше) в 9 мл ацетонитрила и добавляли 0,101 г (1,45 ммоль) гидрохлорида гидроксиламина. Эту смесь нагревали при 70°С в течение 2,5 ч, фильтровали через Celite® и упаривали. Неочищенный продукт (0,145 г; чистота 75%) использовали непосредственно на следующей стадии без дополнительной очистки.
(2)Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-Aze-Pab(OH, Teoc) (0,145 г, 0,23 ммоль; см. стадию (1) выше) растворяли в 0,5 мл CH2Cl2 и 9 мл ТФУ. Реакционную смесь оставляли на 60 минут. ТФУ выпаривали и остаток очищали при использовании препаративной ВЭЖХ. Фракции, представляющие интерес, объединяли и подвергали их сушке вымораживанием (2×) с получением 72 мг (выход в двух стадиях 62%) соединения, указанного в заголовке.
МС (m/z) 482 (M-1)-; 484 (M+1)+.
1H-ЯМР (400 МГц; CD3OD): δ 7.58 (d, 2H), 7.33 (m, 3Н), 7.15 (m, 2H), 6.89 (t, 1H главный ротамер), 6.86 (t, 1H минорный ротамер), 5.18 (s, 1H главный ротамер; m, 1H минорный ротамер), 5.12 (s, 1H минорный ротамер), 4.77 (m, 1H главный ротамер), 4.42 (m, 2H), 4.34 (m, 1H главный ротамер), 4.14 (m, 1H главный ротамер), 4.06 (m, 1H минорный ротамер), 3.95 (m, 1H минорный ротамер), 2,66 (m, 1H минорный ротамер), 2.50 (m, 1H главный ротамер), 2.27 (m, 1H главный ротамер), 2.14 (m, 1H минорный ротамер).
13С-ЯМР (100 МГц; CD3OD): (атомы углерода карбонила и/или амидина, ротамеры) δ 172,4, 172,3, 172,0, 171,4, 152,3, 152,1.
Получение Соединения Н Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(ОН)
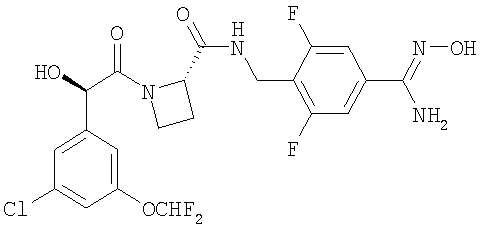
(1) Вос-(S)Aze-NHCH2-Ph(2,6-диF,4-CN)
Boc-(S)Aze-OH (1,14 г, 5,6 ммоль) растворяли в 45 мл ДМФ. Добавляли 4-аминометил-2,5-дифторбензонитрил (1,00 г, 5,95 моль, см. Пример 1(14) выше), РуВОР (3,10 г, 5,95 ммоль) и ДИПЭА (3,95 мл, 22,7 ммоль) и перемешивали этот раствор при комнатной температура в течение 2 ч. Растворитель выпаривали и остаток подвергали распределению между Н2O и EtOAc (по 75 мл каждого). Водную фазу экстрагировали 2×50 мл EtOAc и объединенную органическую фазу промывали рассолом и сушили над Na2SO4. Флэш-хроматография (SiO2, EtOAc/гептан (3/1)) давала указанное в подзаголовке соединение (1,52 г, 77%) в виде масла, которое кристаллизовалось в холодильнике.
1H-ЯМР (400 МГц; CD3OD): δ 7.19 (m, 2H), 4.65-4.5 (m, 3Н). 3.86 (m, 1H), 3.73 (m, 1H), 2.45-2.3 (m, 2H), 1.39 (s, 9H).
(2) H-(S)Aze-NHCH2-Ph(2,6-диF,4-CN) × HCl
Boc-(S)Aze-NHCH2-Ph(2,6-диF,4-CN) (0,707 г, 2,01 ммоль, см. стадию (1) выше) растворяли в 60 мл EtOAc, насыщенного HCl (газ). После перемешивания при комнатной температуре в течение 15 минут растворитель выпаривали. Остаток растворяли в смеси СН3CN/Н2O (1/1) и подвергали сушке вымораживанием с получением указанного в подзаголовке соединения (0,567 г, 98%) в виде грязно-белого аморфного порошка.
1H-ЯМР (400 МГц; CD3OD): δ 7.49 (m, 2Н), 4.99 (m, 1H), 4.58 (m, 2H), 4.12 (m, 1H), 3.94 (m, 1H), 2.80 (m, 1H), 2.47 (m, 1H).
MC (m/z) 252,0 (M+1)+.
(3) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-NHCH2-Ph(2,6-диF,4-CN)
Растворяли Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (0,40 г, 1,42 ммоль, см. Пример 1(8) выше) в 10 мл ДМФ и добавляли Н-(S)Aze-NHCH2-Ph(2,6-диF,4-CN)×HCl (0,43 г, 1,50 ммоль, см. стадию (2) выше) и РуВОР (0,779 г, 1,50 ммоль) и далее ДИПЭА (1,0 мл, 5,7 ммоль). После перемешивания при комнатной температуры в течение 2 ч растворитель выпаривали. Остаток подвергали распределению между H2O (200 мл) и EtOAc (75 мл). Водную фазу экстрагировали 2×75 мл EtOAc и объединенную органическую фазу промывали рассолом и сушили над Na2SO4. Флэш-хроматография (SiO2, EtOAc/гептан (4/1)) давала указанное в подзаголовке соединение (0,56 г, 81%) в виде масла.
1H-ЯМР (400 МГц; CD3OD) ротамеры: δ 7.43 (m, 2H), 7.31 (m, 1H, главный ротамер), 7.26 (m, 1H, минорный ротамер), 7.2-7.1 (m, 2H), 6.90 (t, 1H, главный ротамер), 6.86 (t, 1H, минорный ротамер), 5.14 (s, 1H, главный ротамер), 5.11 (m, 1H, минорный ротамер), 5.04 (s, 1H, минорный ротамер), 4.71 (m, 1H, главный ротамер), 4.6-4.45 (m, 2H), 4.30 (m, 1H, главный ротамер), 4.2-3.9 (m, 1H и 1H, минорный ротамер), 2,62 (m, 1H, минорный ротамер), 2.48 (m, 1H, главный ротамер), 2.21 (m, 1H, главный ротамер), 2.09 (m, 1H, минорный ротамер).
13С-ЯМР (100 МГц; CD3OD): (атомы углерода карбонила) δ 171,9, 171,8.
МС (m/z) 484,0, 485,9 (M-1)-, 486,0, 487,9 (M+1)+.
(4) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OH)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-NHCH2-Ph(2,6-диF,4-CN) (0,555 г, 1,14 ммоль, со стадии (3) выше) растворяли в 10 мл EtOH (95%). К этому раствору добавляли гидрохлорид гидроксиламина (0,238 г, 3,42 ммоль) и Et3N (0,48 мл, 3,44 ммоль). После перемешивания при комнатной температуре в течение 14 ч растворитель удаляли и остаток растворяли в EtOAc.
Органическую фазу промывали рассолом и Н2O и сушили над Na2SO4. Неочищенный продукт очищали препаративной ЖХ с обращенными фазами со смесью СН3CN : 0,1 М NH4OAC в качестве элюента с получением соединения, указанного в заголовке, в виде аморфного порошка (0,429 г, 72%) после сушки вымораживанием.
1H-ЯМР (400 МГц; CD3OD) ротамеры: δ 7.35-7.1 (m, 5H), 6.90 (t, 1H, главный ротамер), 6.85 (t, 1H, минорный ротамер), 5.15 (s, 1H, главный ротамер), 5.12 (m, 1H, минорный ротамер), 5.08 (s, 1H, минорный ротамер), 4.72 (m, 1H, главный ротамер), 4.6-4.4 (m, 2H), 4.30 (m, 1H, главный ротамер), 4.12 (m, 1H, главный ротамер), 4.04 (m, 1H, минорный ротамер), 3.94 (m, 1H, минорный ротамер), 2,62 (m, 1H, минорный ротамер), 2.48 (m, 1H, главный ротамер), 2.22 (m, 1H, главный ротамер), 2.10 (m, 1H, минорный ротамер).
13С-ЯМР (100 МГц; CD3OD): (атомы углерода карбонила и амидина, ротамеры) δ 172,4, 171,9, 171,0, 152,3, 151,5.
MC (m/z) 517,1, 519,0 (M-1)-, 519,1, 521,0 (M+1)+.
Получение Соединения J (Ph(3-Cl)(5-ОСН2CHF2)-(R)CH(ОН)С(O)-Aze-Pab(OH))
(1) Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(Z)
Boc-Aze-Pab(Z) (см. международную заявку на патент WO 97/02284; 92 мг, 0,197 ммоль) растворяли в 10 мл EtOAc, насыщенного HCl (газ) и оставляли взаимодействовать в течение 10 мин. Растворитель выпаривали и остаток смешивали с Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)OH (50 мг, 0,188 ммоль; см. Получение С(5) выше), РуВОР (109 мг, 0,209 ммоль) и, наконец, диизопропилэтиламином (96 мг, 0,75 ммоль) в 2 мл ДМФ. Эту смесь перемешивали в течение 2 ч и затем вливали в 50 мл воды и экстрагировали три раза EtOAc. Объединенную органическую фазу промывали водой, сушили (Na2SO4) и упаривали. Неочищенный продукт подвергали флэш-хроматографии на силикагеле со смесью EtOAc:МеОН (9:1). Выход: 100 мг (87%).
1Н-ЯМР (300 МГц, CD3OD смесь ротамеров) δ 7.85-7.75 (m, 2H), 7.45-7.25 (m, 7H), 7.11 (m, 1H, главный ротамер), 7.08 (m, 1H, минорный ротамер), 7.05-6.9 (m, 2H), 6.13 (bt, 1H), 5.25-5.05 (m, 3Н), 4.77 (m, 1H, частично скрыт сигналом от CD3ОН), 4.5-3.9 (m, 7H), 2,64 (m, 1H, минорный ротамер), 2.47 (m, 1H, главный ротамер), 2.25 (m, 1H, главный ротамер), 2.13 (m, 1H, минорный ротамер).
(2)Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(OH)
Смешивали гидрохлорид гидроксиламина (65 мг, 0,94 ммоль) и триэтиламин (0,319 г, 3,16 ммоль) в 8 мл ТГФ и обрабатывали ультразвуком в течение 1 ч при 40°С. Добавляли Ph(3-Cl)(5-OCH2CHF2)-(R)CH(OH)C(O)-Aze-Pab(Z) (96 мг, 0,156 ммоль; см. стадию (1) выше) с 8 мл ТГФ. Эту смесь перемешивали при 40°С в течение 4,5 дней. Растворитель выпаривали и неочищенный продукт очищали препаративной ЖХ с обращенными фазами СН3CN : 0,1 M NH4OAc (40:60). Выход 30 мг (38%). Чистота 99%.
1H-ЯМР (300 МГц, CD3OD, смесь ротамеров) δ 7.6-7.55 (m, 2H), 7.35-7.3 (m, 2H), 7.12 (m, 1H, главный ротамер), 7.09 (m. 1H, минорный ротамер), 7.05-6.9 (m, 2H), 6.15 (триплет мультиплетов, 1H), 5.15 (m, 1H, минорный ротамер), 5.13 (s, 1H, главный ротамер), 5.08 (s, 1H, минорный ротамер), 4.77 (m, 1H, главный ротамер), 4.5-4.2 (m, 5H), 4.08 (m, 1H, главный ротамер), 3.97 (m, 1H, минорный ротамер), 2,66 (m, 1H, минорный ротамер), 2.50 (m, 1H главный ротамер), 2.27 (m, 1H, главный ротамер), 2.14 (m, 1H, минорный ротамер).
13С-ЯМР (100 МГц; CD3OD): (атомы углерода карбонила и/или амидина, смесь ротамеров) δ 172,8, 172,2, 171,4, 159,1, 158,9, 154,2.
ХИАД-МС: (M+1) = 497/499 m/z.
Способы 1 и 2. Получение солей Соединения А
Способ 1. Общий способ получения солей
Для получения солей Соединения А использовали нижеследующий общий способ. 200 мг Соединения А (см. Получение А выше) растворяли в 5 мл МеОН. К этому раствору добавляли раствор соответствующей кислоты (1,0 молярный эквивалент), растворенной в 5 мл МеОН. После перемешивания в течение 10 минут при комнатной температуре растворитель удаляли на роторном испарителе. Оставшийся твердый материал снова растворяли в 8 мл смеси ацетонитрил:Н2O (1:1). Сушка вымораживанием давала в каждом случае бесцветный аморфный материал.
Использованные кислоты:
(1S)-(+)-10-камфорсульфоновая;
яблочная;
циклогексилсульфаминовая;
фосфорная;
диметилфосфорная;
п-толуолсульфоновая;
L-лизин;
L-лизина гидрохлорид;
сахариновая;
метансульфоновая;
хлористоводородная.
Соответствующие характерные данные показаны в Таблице 1
Все соли, образованные в этом способе, были аморфными.
Способ 2
Дополнительные аморфные соли Соединения А были получены с помощью методик, аналогичных описанным в Способе 1 выше, из следующих кислот:
бромистоводородная кислота (соль1:1);
хлористоводородная кислота (соль 1:1);
серная кислота (соль 1:0,5);
1,2-этандисульфоновая кислота (соль 1:0,5);
1S-камфорсульфоновая кислота (соль 1:1);
(+/-)-камфорсульфоновая кислота (соль 1:1);
этансульфоновая кислота (соль 1:1);
азотная кислота (соль 1:1);
толуолсульфоновая кислота (соль 1:1);
метансульфоновая кислота (соль 1:1);
п-ксилолсульфоновая кислота (соль 1:1);
2-мезитиленсульфоновая кислота (соль 1:1);
1,5-нафталинсульфоновая кислота (соль 1:0,5);
нафталинсульфоновая кислота (соль 1:1);
бензолсульфоновая кислота (соль 1:1);
сахариновая кислота (соль 1:1);
малеиновая кислота (соль 1:1);
фосфорная кислота (соль 1:1);
D-глутаминовая кислота (соль 1:1);
L-глутаминовая кислота (соль 1:1);
D,L-глутаминовая кислота (соль 1:1);
L-аргинин (соль 1:1);
L-лизин (соль 1:1);
L-лизина гидрохлорид (соль 1:1);
глицин (соль 1:1);
салициловая кислота (соль 1:1);
винная кислота (соль 1:1);
фумаровая кислота (соль 1:1);
лимонная кислота (соль 1:1);
L-(-)-яблочная кислота (соль 1:1);
D,L-яблочная кислота (соль 1:1);
D-глюконовая кислота (соль 1:1).
Способ 3. Получение аморфной соли Соединения А и этансульфоновой кислоты
Растворяли Соединение А (203 мг; см. Получение А выше) в этаноле (3 мл) и добавляли к этому раствору этансульфоновую кислоту (1 экв., 95%, 35 мкл). Эту смесь перемешивали в течение нескольких минут и затем растворитель выпаривали. Получившееся масло суспендировали в изо-октане и выпаривали досуха, пока не получали твердый материал. Наконец, это вещество снова суспендировали в изо-октане и растворитель упаривали снова с получением белого сухого аморфного твердого вещества. Это вещество подвергали вакуумной сушке при 40°С в течение ночи.
Способы 4-9. Получение кристаллической соли Соединения А и этансульфоновой кислоты
Способ 4. Кристаллизация аморфного материала
Аморфную соль Соединения А и этансульфоновой кислоты (17,8 мг; см. Способ 3 выше) суспендировали в метил-изо-бутилкетоне (600 мкл). Через 1 неделю наблюдали кристаллические иглы, которые отфильтровывали и сушили на воздухе.
Способы 5-7. Реакционные кристаллизации (без антирастворителя)
Способ 5
Соединение А (277 мг; см. Получение А выше) растворяли в метил-изо-бутилкетоне (3,1 мл). Добавляли этансульфоновую кислоту (1 экв., 95%, 48 мкл). Осаждение аморфной соли, представляющей собой этансульфонат, происходило немедленно. Добавляли еще метил-изо-бутилкетон (6 мл) и обрабатывали взвесь ультразвуком. Наконец, добавляли третью порцию метил-изо-бутилкетона (3,6 мл) и затем оставляли суспензию в течение ночи при перемешивании (магнитная мешалка). На следующий день вещество трансформировалось в кристаллические иглы. Суспензию фильтровывали, промывали метил-изо-бутилкетоном (0,5 мл) и подвергали воздушной сушке.
Способ 6
Соединение А (236 мг; см. Получение А выше) растворяли при комнатной температуре в метил-изо-бутилкетоне (7 мл). Этансульфоновую кислоту (1 экв., 41 мкл) смешивали с 2 мл метил-изо-бутилкетона во флаконе. Раствор Соединения А затравливали кристаллической солью Соединения А и этансульфоновой кислоты (см. Способы 4 и 5 выше). Затем порциями на протяжении 45 минут добавляли 250 мкл раствора этансульфоновой кислоты в метил-изо-бутилкетоне. Раствор затравливали снова и температуру увеличивали до 30°С. Затем на протяжении приблизительно 1 часа добавляли 500 мкл этого метил-изо-бутилкетонового раствора. Получившуюся суспензию оставляли на ночь и затем на протяжении 20 минут добавляли последнюю порцию раствора метил-изо-бутилкетон/кислота. Флакон ополаскивали 1,5 мл метил-изо-бутилкетона, который добавляли к суспензии. Еще через 6 часов кристаллы отфильтровывали, промывали их метил-изо-бутилкетоном (2 мл) и сушили при пониженном давлении при 40°С. В сумме получали 258 мг кристаллической соли, что соответствует выходу приблизительно 87%.
Способ 7
Соединение А (2,36 г; см. Получение А выше) растворяли в метил-изо-бутилкетоне (90 мл). К этому раствору добавляли затравочные кристаллы (10 мг) соли Соединения А и этансульфоновой кислоты (см. Способы 4-6 выше) и затем добавляли двумя порциями этансульфоновую кислоту (40 TL). Затем добавляли дополнительные затравочные кристаллы (12 мг) и две порции этансульфоновой кислоты (2×20 мкл). Эту суспензию разбавляли метил-изо-бутилкетоном (15 мл), после чего продолжали добавлять этансульфоновую кислоту. Всего на протяжении 1 часа добавили 330 мкл этансульфоновой кислоты. Добавляли небольшое количество затравочных кристаллов и, наконец, оставляли суспензию на ночь при перемешивании. На следующий день отфильтровывали кристаллы, промывали их метил-изо-бутилкетоном (2×6 мл) и сушили при пониженном давлении при 40°С. После сушки получали всего 2,57 г белого кристаллического продукта, что соответствует выходу 89%.
Способы 8 и 9. Реакционные кристаллизации (с антирастворителем)
Способ 8
Соединение А (163 мг; см. Получение А выше) растворяли в изо-пропаноле (1,2 мл). Этот раствор нагревали до 35°С. Добавляли этансульфоновую кислоту (28 мкл). Затем добавляли этилацетат (4,8 мл) и затравливали раствор кристаллической солью Соединения А и этансульфоновой кислоты (см. Способы 4-7 выше). Кристаллизация начиналась практически немедленно. Суспензию оставляли приблизительно на 80 минут при 35°С, а затем давали ей остыть до температуры окружающей среды (21°С). Через два часа отфильтровывали кристаллы, промывали их три раза этилацетатом (3×0,4 мл) и сушили при пониженном давлении при 40°С. Всего получалм 170 мг кристаллического продукта, указанного в заголовке, что соответствует выходу приблизительно 82%.
Способ 9
Растворяли Соединение А (20,0 г; см. Получение А выше) в изо-пропаноле (146,6 мл) при 40°С и к этому раствору добавляли этансульфоновую кислоту (3,46 мл, 95%, 1 экв.). К получившемуся прозрачному раствору добавляли затравочные кристаллы соли Соединения А и этансульфоновой кислоты (50 мг; см. Способы 4-8 выше). Затем в течение 10 минут добавляли этилацетат (234 мл). Получившийся мутноватый раствор затравливали еще раз (70 мг) и оставляли на один час при 40°С при перемешивании, чтобы началась кристаллизация. После этого с постоянной скоростью в течение одного часа добавляли всего 352 мл этилацетата. Когда весь этилацетат был добавлен, суспензию оставляли на 1 час, а затем охлаждали ее до 21°С на протяжении 2 часов. Кристаллизации давали осуществляться в течение 1 часа при 21°С, а затем кристаллы отфильтровывали, дважды промывали их этилацетатом (50 мл + 60 мл) и, наконец, сушили при пониженном давлении при 40°С в течение ночи. Всего получали 21,6 г белой кристаллической соли, что соответствует выходу приблизительно 90%.
Соль Соединения А и этансульфоновой кислоты характеризовали посредством ЯМР следующим образом. 23 мг соли растворяли в дейтерированном метаноле (0,7 мл). Использовали комбинации экспериментов ЯМР в режимах 1D (1Н,13С и селективный NOE (спектроскопия ЯМР с ядерным эффектом Оверхаузера)) и 2D (gCOSY, gHSQC и gHMBC). Все данные хорошо согласовывались с теоретической структурой соли, показанной ниже. В метаноле молекула существует в двух конформациях. На основании интеграла пика, обозначенного как Н5 (доминантный конформер), и пика, обозначенного как Н5' (другой конформер), было установлено, что соотношение между этими двумя конформерами составляет 70:30. Н22 не наблюдали, так как эти протоны находились в состоянии быстрого обмена с растворителем CD3OD.
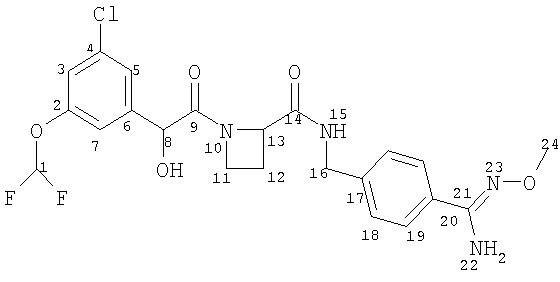
Как протонный, так и углеродный резонанс, соответствующие положению 1, расщеплены из-за спинового взаимодействия с двумя ядрами фтора в этом положении. Константы взаимодействия составляют2JHF=73 Гц и1JHF=263 Гц.
Распределение1H и13С-ЯМР химических сдвигов и протон-протонные корреляции показаны в Таблице 2.
МСВР (масс-спектрометрия высокого разрешения): рассчитано для C24H29ClF2N4O8S (M-H)- 605,1284; найдено 605,1296.
Анализировали кристаллы соли Соединения А и этансульфоновой кислоты (получена посредством одного или более чем одного из вышеприведенных Примеров 4-9) методом дифракции рентгеновских лучей на порошке (ДРЛП) и результаты представлены ниже (Таблица 3).
ДСК показала эндотерму с экстраполированной температурой начала плавления около 131°С. ТГА показал уменьшение массы примерно на 0,2% (мас./мас.) около точки плавления. Анализ методом ДСК при повторе на образце с меньшим содержанием растворителя показал температуру начала плавления около 144°С.
Способ 10
Получение аморфной соли Соединения А и бензолсульфоновой кислоты
Соединение А (199 мг; см. Получение А выше) растворяли в этаноле (2 мл). Бензолсульфоновую кислоту (1 экв. 90%, 70 мг) растворяли в этаноле (1 мл) во флаконе. Этанольный раствор кислоты добавляли к раствору Соединения А и флакон ополаскивали 1 мл этанола, который затем добавляли к смеси. Смесь перемешивали в течение нескольких минут и затем этанол выпаривали до образования масла. Добавляли этилацетат (3 мл) и снова выпаривали растворитель досуха. Образовывалось аморфное твердое вещество.
Способы 11-13. Получение кристаллической соли Соединения А и бензолсульфоновой кислоты
Способ 11. Кристаллизация аморфного материала
Аморфную соль соединения А и бензолсульфоновой кислоты (20,7 мг; см. Способ 10 выше) суспендировали в этилацетате (600 TL). Через 5 дней наблюдали в суспензии кристаллические иглы.
Способы 12 и 13. Реакционные кристаллизации
Способ 12
Соединение А (128 мг; см. Получение А выше) растворяли в этилацетате (3 мл). Раствор затравливали суспензией, полученной Способом 11 выше. Затем добавляли бензолсульфоновую кислоту (1 экв., 90%, 45 мг). Немедленно происходило осаждение соли бензолсульфоновой кислоты. К этой суспензии добавляли изопропанол (0,8 мл) и снова затравливали смесь. Через два дня вещество трансформировалось в кристаллические иглы. Эту суспезию отфильтровывали, промывали этилацетатом (3×0,2 мл) и сушили в течение короткого промежутка времени под вакуумом при 40°С. Получали в совокупности приблизительно 140 мг белого твердого вещества.
Способ 13
Соединение А (246 мг; см. Получение А выше) растворяли в изопропаноле (1,52 мл). Добавляли бензолсульфоновую кислоту (88 мг, 90%). К этому прозрачному раствору добавляли этилацетат (3 мл) и затем затравливали эту смесь для инициирования кристаллизации. Через 1 час добавляли еще этилацетат (2,77 мл). Наконец, суспензию оставляли кристаллизоваться в течение ночи, затем кристаллы отфильтровывали, промывали их этилацетатом (3×0,3 мл) и сушили при 40°С под вакуумом. В сумме получали 279 мг соли, что соответствует выходу приблизительно 86%.
Соль Соединения А и бензолсульфоновой кислоты характеризовали посредством ЯМР следующим образом. 20 мг соли растворяли в дейтерированном метаноле (0,7 мл). Использовали комбинацию экспериментов ЯМР в режиме 1D (1Н,13С и селективный NOE) и 2D (gCOSY, gHSQC и gHMBC). Все данные хорошо согласовывались с теоретической структурой соли, которая показана ниже. Молекула существует в метаноле в двух конформациях. На основании интеграла пика, обозначенного как Н12 (доминантный конформер), и пика, обозначенного как Н12' (другой конформер), установили, что соотношение между этими двумя конформерами составляло 70:30. Н22 не наблюдали, так как эти протоны находились в состоянии быстрого обмена с растворителем CD3OD.
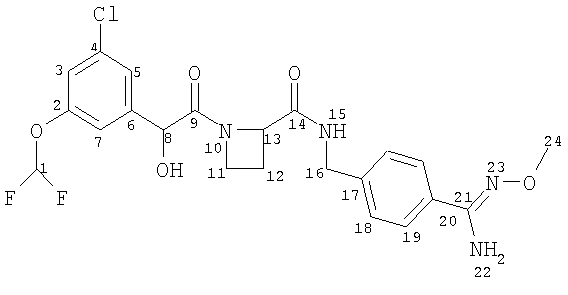
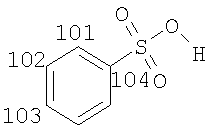
Как протонный, так и углеродный резонанс, соответствующий положению 1, были расщеплены из-за спинового взаимодействия с двумя ядрами фтора в этом положении. Константы взаимодействия составляли2JHF=74 Гц и1JHF=260 Гц.
Распределение химических сдвигов в1H-ЯМР и13С-ЯМР и протон-протонные корреляции показаны в Таблице 4.
МСВР: рассчитано для C28H29ClF2N4O8S (М-Н)- 653,1284; найдено 653,1312.
Кристаллы соли Соединения А и бензолсульфоновой кислоты (получена посредством одного или более чем одного из вышеприведенных Примеров 11-13) анализировали методом ДРЛП и результаты представлены ниже (Таблица 5).
ДСК показала эндотерму с экстраполированной температурой начала плавления около 152°С. ТГА показал снижение массы примерно на 0,1% (мас./мас.) около точки плавления.
Способ 14
Получение аморфной соли Соединения А и н-пропансульфоновой кислоты
Растворяли Соединение А (186 мг; см. Получение А выше) в изопропаноле (1,39 мл) и добавляли н-пропансульфоновую кислоту (1 экв. 95%, 39 TL). Добавляли этилацетат (5,6 мл) и выпаривали растворитель до образования сухого аморфного твердого вещества.
Способы 15 и 16. Получение кристаллической соли Соединения А и н-пропансульфоновой кислоты
Способ 15. Кристаллизация аморфного материала
Растворяли аморфную соль Соединения А и н-пропансульфоновой кислоты (20 мг; см. Способ 14 выше) в изопропаноле (60 TL) и добавляли изопропилацетат (180 TL). Через три дня наблюдали кристаллы в виде игл.
Способ 16. Реакционная кристаллизация
Растворяли соединение А (229 мг; см. Получение А выше) в изопропаноле (1,43 мл). Добавляли н-пропансульфоновую кислоту (1 экв., 95%, 48 TL). Добавляли этилацетат (2 мл) и затем затравливали раствор кристаллической солью из Способа 15 выше. Добавляли еще этилацетат (5 мл) и оставляли суспензию на ночь для кристаллизации. Отфильтровывали кристаллы, промывали их этилацетатом (3×0,3 мл) и сушили под вакуумом при 40°С.
Соль Соединения А и н-пропансульфоновой кислоты характеризовали посредством ЯМР следующим образом. 13 мг соли растворяли в дейтерированном метаноле (0,7 мл). Использовали комбинацию экспериментов ЯМЕ в режиме 1D (1Н,13С) и 2D (gCOSY). Все данные хорошо согласовывались с теоретической структурой соли, которая показана ниже. Молекула существует в метаноле в двух конформациях. На основании интеграла пика, обозначенного как Н12 (доминантный конформер), и пика, обозначенного как Н12' (другой конформер), установили, что соотношение между этими двумя конформерами составляет 65:35. Н22 не наблюдали, так как эти протоны находятся в состоянии быстрого обмена с растворителем CD3OD.
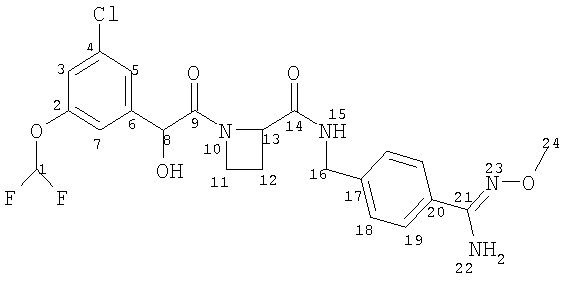
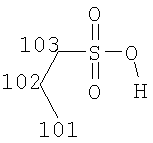
Как протонный, так и углеродный резонанс, соответствующий положению 1, был расщеплен из-за спинового взаимодействия с двумя ядрами фтора в этом положении. Константы взаимодействия составляют2JHF=74 Гц и1JHF=260 Гц.
Распределение химических сдвигов в1H-ЯМР и13С-ЯМР и протон-протонные корреляции показаны в Таблице 6.
МСВР: рассчитано для C25H31ClF2N4O8S (M-H)- 619,1441; найдено 619,1436.
Кристаллы соли Соединения А и н-пропансульфоновой кислоты (получена посредством одного или более чем одного из вышеприведенных Примеров 15 и 16) анализировали методом ДРЛП и результаты представлены ниже (Таблица 7).
ДСК показала эндотерму с экстраполированной температурой начала плавления около 135°С. ТГА не показал снижения массы около точки плавления.
Способ 17
Способ 17-А. Получение аморфной соли соединения А и н-бутансульфоновой кислоты
Растворяли аморфное Соединение А (277 мг) в IPA (1,77 мл) и добавляли бутансульфоновую кислоту (приблизительно 1 экв., 70 мкл). Добавляли этилацетат (6 мл) и выпаривали растворитель до образования сухого аморфного твердого вещества.
Способ 17-Б. Получение кристаллической соли соединения А и бутансульфоновой кислоты
Аморфную соль Соединения А и бутансульфоновой кислоты (71,5 мг; см. вышеуказанное получение) суспендировали в этилацетате (500 мкл) на протяжении ночи. Отфильтровывали кристаллы и сушили их на воздухе.
Соль Соединения А и бутансульфоновой кислоты характеризовали посредством ЯМР следующим образом.
Растворяли 21,6 мг соли в дейтерированном диметилсульфоксиде (0,7 мл) и исследовали с помощью1H-ЯМР-спектроскопии и13С-ЯМР-спектроскопии.
Спектры очень сходны со спектрами других солей того же соединения и хорошо согласуются со структурой, показанной ниже. Большинство резонансов в спектрах присутствуют в виде совокупности двух пиков из-за медленного вращения вокруг связи C9-N10, что приводит к двум атропизомерам, которые существуют в растворе одновременно. Это продемонстрировано и для других солей того же соединения.
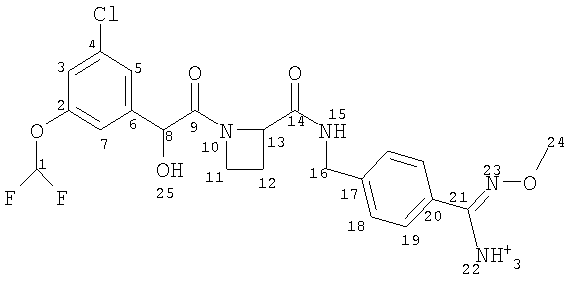
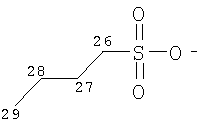
Два ядра фтора в положении 1 вызывают расщепление резонансов для протона и углерода в этом положении. Константы взаимодействия составляют2JHF=73 Гц и1JCF=258 Гц.
Химические сдвиги для протонов и углерода представлены в Таблице 8. Протоны в положении 22 и 24 не определяются из-за химического обмена. Между 8 и 9 м.д. в протонном спектре, соответствующем этим протонам, имеется очень широкий горб. Таблица 8
Распределение химических сдвигов в1H-ЯМР и13С-ЯМР у н-бутансульфоната Соединения А в дейтерированном диметилсульфоксиде при 25°С
МСВР: рассчитано для С26Н32ClF2N4O8S (М-Н)- 633,1597, найдено 633,1600.
Кристаллы соли Соединения А и н-бутансульфоновой кислоты (получена, как описано выше в Способе 17-Б) анализировали с помощью ДРЛП, и результаты представлены ниже (Таблица 9).
ДСК показала эндотерме с экстраполированной температурой начала плавления около 118°С, ТГА показал примерно 0,04%-ное снижение массы.
Способ 18. Получение солей Соединения В
Способ 18-А. Общий способ получения соли
Для получения солей Соединения В использовали нижеследующий общий способ. 200 мг соединения В (см. Получение В выше) растворяли в 5 мл МИБК (метилизобутилкетон). К этому раствору добавляли раствор соответствующей кислоты (1,0 или 0,5 молярных эквивалентов, как указано в Таблице 10), растворенной в 1,0 мл МИБК. После перемешивания в течение 10 минут при комнатной температуре растворитель удаляли на роторном испарителе. Оставшийся твердый материал снова растворяли в примерно 8 мл смеси ацетонитрил:Н2О (1:1). Сушка вымораживанием давала в каждом случае бесцветный аморфный материал.
Использованные кислоты:
Эзилат (этансульфоновая кислота)
Безилат (бензолсульфоновая кислота)
Циклогексилсульфамат
Сульфат
Бромид
п-Толуолсульфонат
2-Нафталинсульфонат
Гемисульфат
Метансульфонат
Нитрат
Гидрохлорид
Соответствующие характеристики показаны в Таблице 10.
Все соли, образованные в этом примере, были аморфными.
Способ 18-В
При использовании методик, аналогичных тем, что описаны выше в Способе 18-А, получали дополнительные аморфные соли Соединения В для следующих кислот:
1,2-Этандисульфоновая (0,5 соль)
1S-Камфорсульфоновая
(+/-)-Камфорсульфоновая
п-Ксилолсульфоновая
2-Мезитиленсульфоновая
Сахариновая
Малеиновая
Фосфорная
D-глутаминовая
L-аргинин
L-лизин
L-лизин·HCl
Способ 18-С. Получение аморфной соли соединения В и геми-1,5-нафталиндисульфоновой кислоты
Растворяли аморфное Соединение В (110,9 мг) в 2,5 мл 2-пропанола и добавляли 0,5 эквивалента тетрагидрата 1,5-нафталиндисульфоновой кислоты (растворенного в 1 мл 2-пропанола). Образец перемешивали в течение ночи. С помощью микроскопа наблюдали только мелкие (аморфные) частицы или капли масла. Образец выпаривали досуха.
Способ 18-D. Получение кристаллической соли Соединения В и геми-1,5-нафталиндисульфоновой кислоты
Эксперимент по кристаллизации проводили при температуре окружающей среды. Растворяли аморфное Соединение В (0,4 г) в этаноле (1,5 мл) и добавляли 0,5 экв. 1,5-тетрагидрата нафталиндисульфоновой кислоты (1,35 г, 10% в этаноле). Затем добавляли гептан (0,7 мл) до тех пор, пока раствор не становился мутноватым. Через примерно 15 минут раствор становился мутным. Через примерно 30 минут получали тонкую суспензию и добавляли дополнительную порцию гептана (1,3 мл). Затем оставляли суспезию на ночь для дозревания. Для разведения густой суспензии добавляли смесь этанола и гептана (1,5 мл и 1,0 мл соответственно). Через примерно 1 час суспензию фильтровали и кристаллы промывали смесью этанола и гептана (1,5:1) и в заключение чистым гептаном. Сушили кристаллы при температуре окружающей среды в течение 1 дня. Сухие кристаллы весили 0,395 г.
Способ 18-Е. Получение кристаллической соли Соединения В и геми-1,5-нафталиндисульфоновой кислоты
Растворяли аморфное Соединение В (1,009 г) в смеси 20 мл 2-пропанола + 20 мл этилацетата. Растворяли 351,7 мг тетрагидрата 1,5-нафталиндисульфоновой кислоты в 20 мл 2-пропанола, добавлявшегося капля за каплей. Выпадение осадка происходило в течение примерно 5 минут. Суспензию перемешивали в течение ночи и затем фильтровали.
Способ 18-F. Получение кристаллической соли Соединения В и геми-1,5-нафталиндисульфоновой кислоты
Растворяли 430,7 мг соли 1,5-нафталиндисульфоновой кислоты в 30 мл 1-пропанола. Этот раствор нагревали до кипения для растворения вещества. Оставляли раствор на ночь при температуре окружающей среды для кристаллизации и затем отфильтровывали кристаллы.
Способ 18-G. Получение кристаллической соли Соединения В и геми-1,5-нафталиндисульфоновой кислоты
Маточный раствор из Способа 18-F выпаривали и твердый остаток (61,2 мг) растворяли в 6 мл смеси ацетонитрил/1-пропанол, соотношение 2:1. Оставляли этот раствор на ночь при температуре окружающей среды для кристаллизации и затем отфильтровывали кристаллы.
Способ 18-Н. Получение кристаллический соли Соединения В и геми-1,5-нафталиндисульфоновой кислоты
Образец из Способа 18-С растворяли в примерно 2 мл метанола. Добавляли в качестве антирастворителя этанол (примерно 3 мл) при температуре окружающей среды и вносили затравочные кристаллы. Кристаллизация не происходила, поэтому упаривали растворители (около половины их количества) и добавляли новую порцию этанола (примерно 2 мл) и добавляли затравочные кристаллы. При перемешивании при температуре окружающей среды в течение ночи образовывались кристаллические частицы.
Способ 18-I. Получение кристаллической соли Соединения В и геми-1,5-нафталиндисульфоновой кислоты
Растворяли аморфное Соединение В (104,1 мг) в 2-пропаноле и добавляли 1 эквивалент тетрагидрата 1,5-нафталиндисульфоновой кислоты, растворенного в 2-пропаноле. В сумме количество 2-пропанола составило примерно 2,5 мл. Перемешивали этот раствор при 44°С в течение примерно 80 минут, и происходило выпадение осадка. Частицы были кристаллическими согласно данным микроскопии в поляризованном свете. Этот образец фильтровали.
Способ 18-J. Получение кристаллической соли соединения В и геми-1,5-нафталиндисульфоновой кислоты
Соль Соединения В и геми-1,5-нафталиндисульфоновой кислоты (56,4 мг) растворяли в 1,5 мл метанола. Добавляли метилэтилкетон (3 мл). В раствор вносили затравочные кристаллы, и начиналась кристаллизация. Отфильтровывали кристаллы, промывали их метилэтилкетоном и сушили на воздухе.
Способ 18-К. Получение кристаллической соли Соединения В и геми-1,5-нафталиндисульфоновой кислоты
Растворяли аморфное Соединение В (161,0 мг) в 3,5 мл 1-бутанола и нагревали раствор до 40°С. В другом химическом стакане растворяли 57,4 мг тетрагидрата нафталиндисульфоновой кислоты в 3 мл 1-бутанола. К этому раствору соединения В добавляли две капли раствора кислоты. Затем к раствору добавляли затравочные кристаллы и через 2 часа медленно добавляли остальной раствор кислоты (при 40°С). Затем медленно понижали температуру до комнатной температуры и оставляли экспериментальную смесь при перемешивании на ночь. Суспензию фильтровали, промывали 1-бутанолом и сушили под вакуумом при 44°С в течение 2 часов. Выход составлял 83%.
Снятие характеристик
Кристаллы соли Соединения В и геми-1,5-нафталиндисульфоновой кислоты, полученной вышеописанным Способом 18-D, характеризовали посредством ЯМР следующим образом.
21,3 мг соли, растворенной в дейтерированном метаноле (0,7 мл), исследовали посредством ЯМР-спектроскопии. Использовали комбинацию экспериментов ЯМР в режиме 1D (1Н,13С и селективный МОЕ) и 2D (gCOSY, gHSQC и gHMBC). Все данные хорошо согласовывались с предложенной структурой, которая показана ниже. Все углероды и протоны, присоединенные к углеродам, соотнесены. Протоны, присоединенные к гетероатомам, заменены на дейтерий из растворителя и не выявляются. Большинство резонансов в спектрах 1D1H и13С ЯМР представлены в виде совокупности двух пиков. Причиной этого является медленное вращение вокруг связи C9-N10, что приводит к двум атропоизомерам, которые существуют в растворе одновременно. Свидетельством этому является эксперимент 1D NOE. Когда резонанс одного атропоизомера иррадирован, насыщение переходит на соответствующий пик другого атропоизомера. Резонансы, соответствующие 1,5-нафталиндисульфонатному противоиону, не демонстрируют атропоизомерию
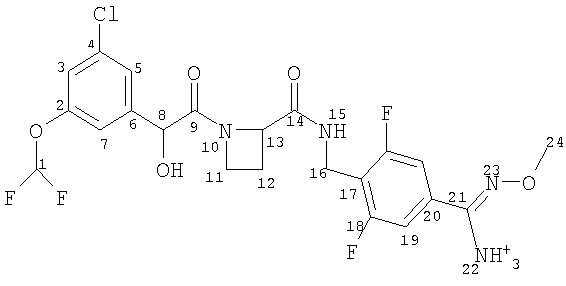
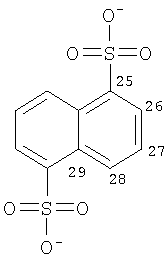
В молекуле имеется четыре атома фтора. Они вызывают расщепление резонансов для некоторых протонов и углеродов. Как протонный, так и углеродный резонанс, соответствующий положению 1, расщеплены из-за спинового взаимодействия с двумя ядрами фтора в этом положении. Константы взаимодействия составляют2JHF=73 Гц и1JCF=263 Гц. Далее, протонный резонанс, соответствующий Н19, представляет собой искаженный дублет с3JHF=6,9 Гц из-за спинового взаимодействия с ядром фтора в положении 18. Углеродные резонансы, соответствующие С17, С18, С19 и С20, также демонстрируют взаимодействие с этими ядрами фтора. Резонансы С17 и С20 представляют собой триплеты с2JCF=19 Гц и3JCF=11 Гц соответственно. Резонанс С18 представляет собой дублет дублетов с константами взаимодействия1JCF=251 Гц и3JCF=8 Гц. Резонанс С19 представляет собой мультиплет.
Сравнение величин интегралов для резонансов, соответствующих 1,5-нафталиндисульфонатному противоиону и основному соединению, дает стехиометрическое соотношение для одного 1,5-нафталиндисульфонатного противоиона, кристаллизованного с двумя молекулами основного соединения.
Распределение химических сдвигов в1H-ЯМР и13С-ЯМР и протон-протонные корреляции показаны в Таблице 11.
Кристаллы соли Соединения В и геми-1,5-нафталиндисульфоновой кислоты (получены вышеописанным Способом 18-1) анализировали методом ДРЛП, и результаты представлены ниже (Таблица 12).
ДСК показала эндотерму с экстраполированной температурой начала плавления примерно 183°С, а ТГА показал снижение массы примерно на 0,3% между 25 и 110°С.
Сокращения
Префиксы н-, втор-, изо-, трет- имеют их обычные значения: нормальный, вторичный, изо и третичный.
Это изобретение иллюстрируют, но ни коим образом не ограничивают нижеследующие примеры. Если не указано иначе, полимеры, представляющие собой ГПМЦ, были получены от Shin-Etsu (товарный знак METOLOSE™). В случае йота-каррагинана поставщиком была Fluka для всех примеров, кроме Примеров 11 и 12 (когда поставщиком была СР-Kelco). Конкретные марки и их эквиваленты по Фармакопее США (USP) представлены ниже (только один раз при первом упоминании).
Общий способ тестирования
Три индивидуальные таблетки тестировали на высвобождение лекарственного средства в 900 мл среды при использовании USP (Фармакопея США) аппарата 2 для растворения (лопасть + корзинка [сделанная по заказу четырехугольная корзинка из проволочной сетки, припаянная одной из ее верхних узких сторон к концу стального стержня. Стержень проведен через крышку емкости для растворения и зафиксирован двумя тефлоновыми гайками на расстоянии 3,2 см от центра емкости. Нижний край дна корзинки установлен на 1 см выше лопасти. Корзинку располагают вдоль потока, причем тестируемая таблетка находится на ее краю]) при 50 об/мин и 37°С. Использованными средами растворения были 0,1 М соляная кислота (рН 1) и 0,1 М натрий-фосфатный буфер (рН 6,8). Количественное определение в процессе проведения теста выполняли с помощью оптоволоконной системы С Technologies при 220 нм в качестве аналитической длины волны, когда в качестве среды растворения использовали 0,1 М HCl, и при 260 нм в качестве аналитической длины волны, когда в качестве среды растворения использовали фосфатный буфер с рН 6,8. 350 нм использовали как эталонную длину волны с обеими средами. В течение первых двух часов анализа величину высвобождения измеряли каждые 15 минут, а затем в остальной части анализа каждый час.
Пример 1
Прямое прессование Соединения А с ГПМЦ 10000 сП (10000 мПа·с) и ГПМЦ 50 сП (50 мПа·с) в соотношении 50:50.
Активное вещество и эксципиентные материалы смешивали во вспомогательном баке. Гранулят обрабатывали стеарилфумаратом натрия в качестве смазки и прессовали в таблетки с помощью эксцентрикового пресса.
Пример 2
Грануляция и прессование Соединения А с ГПМЦ, солюбилизатором и наполнителями.
Растворяли активное вещество, антиоксидант и солюбилизатор в этаноле и распределяли в эксципиентах. Эту смесь затем гранулировали с ГПЦ, растворенной в этаноле. Затем сушили гранулы в сушильной печи. Гранулят обрабатывали стеарилфумаратом натрия в качестве смазки и прессовали в таблетки с помощью эксцентрикового пресса.
Результаты не были скорректированы на значительную фоновую абсорбцию матрицы таблетки, поэтому можно видеть величины высвобождения, составляющие 120% и 132% в 0,1 М соляной кислоте и 0,1 М натрий-фосфатном буфере с рН 6,8 соответственно.
Пример 3
Грануляция и прессование Соединения А с ГПМЦ, додецилсульфатом натрия и наполнителями.
Активное вещество и эксципиентные материалы смешивали во вспомогательном баке. Гранулят обрабатывали стеарилфумаратом натрия в качестве смазки и прессовали в таблетки с помощью эксцентрикового пресса.
Пример 4. Пример препарата, содержащего эзилатную соль Соединения А, ГПМЦ и додецилсульфат натрия
Препарат приготавливали в соответствии со способом Примера 3.
Пример 5. Пример препарата с эзилатной солью Соединения А и ксантановой камедью
Препарат приготавливали в соответствии со способом Примера 3.
Пример 6. Пример препарата эзилатной соли Соединения А с ГПМЦ и йота-каррагинаном
Препарат приготавливали в соответствии со способом Примера 3.
ПРИМЕР 7. Пример препарата соли н-пропилсульфоновой кислоты и Соединения А с ГПМЦ и йота-каррагинаном
Препарат приготавливали в соответствии со способом Примера 3.
Пример 8. Пример препарата безилатной соли Соединения А с ГПМЦ и йота-каррагинаном
Препарат приготавливали в соответствии со способом Примера 3.
Пример 9
Прямое прессование эзилатной соли Соединения А с ГПМЦ 10000 сП и ГПМЦ 50 сП, соотношение 50:50.
Активное вещество и эксципиентные материалы смешивали во вспомогательном баке. Гранулят обрабатывали стеарилфумаратом натрия в качестве смазки и прессовали в таблетки с помощью эксцентрикового пресса.
Данные по высвобождению
Пример 10
Прямое прессование эзилатной соли Соединения А с ГПМЦ 10000 сП и йота-каррагинаном, соотношение 50:50
Активное вещество и эксципиентные материалы смешивали во вспомогательном баке. Гранулят обрабатывали стеарилфумаратом натрия в качестве смазки и прессовали в таблетки с помощью эксцентрикового пресса.
Пример 11
Безилатную соль Соединения А и эксципиентные материалы гранулировали в грануляторе с высоким сдвигающим усилием. Гранулят сушили, обрабатывали стеарилфумаратом натрия в качестве смазки и прессовали в таблетки с помощью эксцентрикового пресса.
Пример 12
Безилатную соль Соединения А и эксципиентные материалы гранулировали в грануляторе с высоким усилием сдвига. Гранулят сушили, обрабатывали стеарилфумаратом натрия в качестве смазки и прессовали в таблетки с помощью эксцентрикового пресса.
Пример 13
Безилатную соль Соединения А и эксципиентные материалы подвергали прямому прессованию.
Пример 14
Примеры препаратов, содержащих безилатную соль Соединения А. Примеры 14-А и 14-Б получали в соответствии с общим способом Примера 3 или 11.
Этот препарат приготавливали в соответствии со способом Примера 3.
Данные по высвобождению для Примера 14-В
Пример 15.
Примеры препаратов, содержащих соль геминафталин-1,5-дисульфоновой кислоты и Соединения В. Получают все препараты в соответствии с общим способом Примера 11.
Дополнительные примеры можно получать, как выше, причем ГПМЦ заменяют на ПЭО или смешивают с ПЭО. В случае смеси пропорция ПЭО:ГПМЦ может быть в диапазоне от 90:10 до 10:90(%). Конкретной смесью является ПЭО:ГПМЦ 80:20(%) или 75:25(%).
Пример 16
Гранулирование и прессование Соединения А с ГПМЦ, додецилсульфатом натрия и наполнителями.
Смешивали активное вещество с эксципиентными материалами во вспомогательном баке. Гранулят обрабатывали стеарилфумаратом натрия в качестве смазки и прессовали в таблетки с помощью эксцентрикового пресса.
Данные по высвобождению
Пример 17
Прямое прессование Соединения А с ксантановой камедью и йота-каррагинаном, соотношение 50:50.
Смешивали активное вещество с эксципиентными материалами во вспомогательном баке. Гранулят обрабатывали стеарилфумаратом натрия в качестве смазки и прессовали в таблетки с помощью эксцентрикового пресса.
Данные по высвобождению
Любой вышеприведенный пример, в котором используют свободное основание или соль, отличную от безилатной соли Соединения А, можно повторить при использовании безилатной соли Соединения А.
Данные по биологической активности соединений
Ингибирующая активность соединений по изобретению в отношении тромбина была подтверждена в следующих тестах.
Тест А
Определение тромбинового времени (ТТ)
Раствор ингибитора (25 мкл) инкубируют с плазмой (25 мкл) в течение 3 минут. Затем добавляют тромбин человека (Т 6769, Sigma Chem. Co или Hematologic Technologies) в буферном растворе, рН 7,4 (25 мкл, 4,0 NIH единицы/мл), и измеряют время свертывания на автоматическом приборе (КС 10, Amelung).
Тромбиновое время (ТТ) выражают в абсолютных значениях (секундах), равно как и в виде отношения ТТ без ингибитора (ТТ0) к ТТ с ингибитором (TTi). Строят кривые зависимости последних отношений (интервал 1-0) от концентрации ингибитора (в log-преобразовании) и осуществляют подгонку к сигмоидальным кривым дозового ответа согласно уравнению:
y=a/[1+(x/IC50)s],
где а = максимальный интервал, то есть 1; s = наклон кривой дозового ответа и IC50 = концентрация ингибитора, при которой происходит удвоение времени свертывания.
Вычисления выполняются на ПК с использованием программного обеспечения GraFit Version 3 с установочными параметрами уравнения, равными: старт от 0, окончание = 1 (Erithacus Software, Robin Leatherbarrow, Imperial College of Science, Лондон, Соединенное Королевство).
Тест В
Определение частичного активированного тромбопластинового времени (АРТТ)
АРТТ определяют в совокупной нормальной цитратной плазме человека с помощью реагента РТТ Automated 5, производимого Stago. Ингибиторы добавляют к плазме (10 мкл раствора ингибитора к 90 мкл плазмы), инкубируют с АРТТ-реагентом в течение 3 минут с последующим добавлением 100 мкл раствора хлорида кальция (0,025 М) и определяют АРТТ, используя анализатор коагуляции КС10 (Amelung) в соответствии с инструкциями производителя реагента.
Время свертывания выражают в абсолютных значениях (секундах), равно как и в виде отношения АРТТ без ингибитора (АРТТ0) к АРТТ с ингибитором (APTTi). Строят кривые зависимости последних отношений (интервал 1-0) от концентрации ингибитора (в log-преобразовании) и осуществляют подгонку к сигмоидальным кривым дозового ответа согласно уравнению:
y=a/[1+(x/IC50)s],
где а = максимальный интервал, то есть 1; s = наклон кривой дозового ответа и
IC50= концентрация ингибитора, при которой происходит удвоение времени свертывания. Вычисления выполняются на ПК с использованием программного обеспечения GraFit Version 3 с установочными параметрами уравнения, равными: старт от 0, окончание = 1 (Erithacus Software, Robin Leatherbarrow, Imperial College of Science, Лондон, Соединенное Королевство).
IC50APTT определяют как концентрацию ингибитора в плазме человека, увеличивающую в два раза частичное активированное тромбопластиновое время.
Тест Е
Определение тромбинового времени ex vivo
Ингибирование тромбина после перорального введения соединений по изобретению, растворенных в смеси этанол/СолютолК/вода (5:5:90) исследуют на находящихся в сознании крысах, которых за один или два дня до эксперимента снабжают катетером для отбора крови из сонной артерии. В день эксперимента в фиксированное время после введения соединения отбирают образцы крови в пластиковые пробирки из расчета 1 часть раствора цитрата натрия (0,13 моль/л) и 9 частей крови. Пробирки центрифугируют с целью получения обедненной по тромбоцитам плазмы. Образцы плазмы в объеме 50 мкл осаждают 100 мкл охлажденного ацетонитрила. Образцы центрифугируют в течение 10 минут при 40000 об/мин. 75 мкл супернатанта разбавляют 75 мкл 0,2%-ной муравьиной кислоты. Получающиеся растворы в объеме 10 мкл анализируют с помощью LC-MS/MS и, используя стандартные кривые, определяют концентрации ингибитора тромбина.
Результаты биологических тестов
Соединение Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab, соединение Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab в форме трифторуксуснокислой соли и соединение Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-(S)Aze-Pab(2,6-диF) демонстрировали значения IC50ТТ в тесте А меньше 0,02 мкМ.
Соединение Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab и соединение Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab в форме трифторуксуснокислой соли демонстрировали значения IC50АРТТ в Тесте B меньше 1 мкМ.
Анализ в соответствии с вышеприведенным Тестом Е подтвердил, что соединения являются биодоступными при пероральном введении крысам.
Реферат
Изобретение относится к области медицины и фармацевтики и касается пероральной композиции для антикоагулянтной терапии с модифицированным высвобождением, содержащей в качестве активного ингредиента соль сульфоновой кислоты и соединения формулы (I) и фармацевтически приемлемый разбавитель или носитель. Изобретение обеспечивает повышенную скорость высвобождения соединения формулы (I). 5 з.п. ф-лы, 43 табл.
Формула
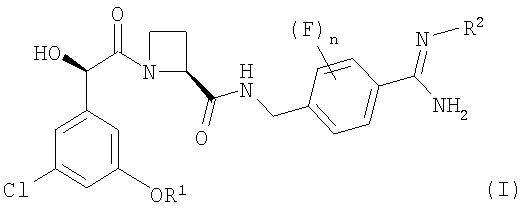
где
R1 представляет собой -CHF2 или CH2CH2F;
R2 представляет собой водород, гидрокси, метокси или этокси;
n представляет собой 0 или 2; и
гидроксипропилметилцеллюлозу (ГПМЦ) и возможно другой гелеобразующий полимер или полимеры, выбранные из:
полисахаридов (таких как мальтодекстрин, ксантан, склероглюкановый декстран, крахмал, альгинаты, гиалуроновая кислота, хитин, хитозан);
других природных полимеров (таких как белки, поли-L-лизин, поли(акриловая кислота) натрий; поли(гидроксиалкилметакрилаты);
карбоксиполиметилена;
карбомера;
поливинилпирролидона;
камедей (таких как гуаровая камедь, гуммиарабик, камедь карайи, камедь гхатти, камедь бобов робинии, камедь тамаринда, геллановая камедь, трагакантовая камедь, агар, пектин, глютен);
поли(винилового спирта);
этиленвинилового спирта;
поли(этиленоксида) (ПЭО); и
эфиров целлюлозы, такие как гидроксиметилцеллюлоза (ГМЦ), гидроксиэтилцеллюлоза (ГЭЦ), гидроксипропилцеллюлоза (ГПЦ),
метилцеллюлоза (МЦ), этилцеллюлоза (ЭЦ), карбоксиэтилцеллюлоза (КЭЦ), этилгидроксиэтилцеллюлоза (ЭГЭЦ), карбоксиметилгидроксиэтилцеллюлоза (КМГЭЦ), гидроксипропилэтилцеллюлоза (ГПЭЦ) и карбоксиметилцеллюлоза в натриевой форме (NaКМЦ);
где гидроксипропилметилцеллюлоза (ГПМЦ) и возможно другой гелеобразующий полимер или полимеры присутствуют в количестве от 5 до 99,5% мас./мас., от конечной композиции, и активный ингредиент и другие возможные эксципиенты дополняют композицию до 100% мас./мас.
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe);
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-диF)(OMe) или
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe).
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe);
Ph(3-Cl)(5-OCHF2)-(R)СН(ОН)С(O)-(S)Aze-Pab(2,6-диF)(ОМе) или
Ph(3-Cl)(5-OCH2CH2F)-(R)CH(OH)C(O)-(S)Aze-Pab(OMe).
Документы, цитированные в отчёте о поиске
Ингибиторы тромбина и промежуточные соединения для их получения
Комментарии