Циклические n, n'-диарилтиомочевины или n, n'-диарилмочевины - антагонисты андрогенных рецепторов, противораковое средство, способ получения и применения - RU2434851C1
Код документа: RU2434851C1
Чертежи
Описание
Настоящее изобретение относится к новым циклическим N,N'-диарилтиомочевинам или N,N'-диарилмочевинам, антагонистам андрогенных рецепторов, противораковому средству, фармацевтической композиции, лекарственному средству и способу лечения рака простаты.
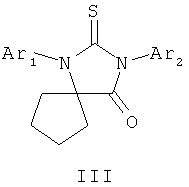
Известны антагонисты андрогенных рецепторов, представляющие собой 1,3-диарил-5,5-диметил-2-тиоксо-имидазолидин-4-оны I, 5,7-диарил-6-тиоксо-5,7-диаза-спиро[3,4]октан-8-оны II и 1,3-диарил-2-тиоксо-1,3-диаза-спиро[4,4]нонан-4-оны III, обладающие противораковой активность [WO 2006124118, WO 2007127010]. В этих рядах соединений наиболее продвинутым является 4-[5,5-диметил-4-(3-трифторметил-4-цианофенил)-4-оксо-2-тиоксо-имидазолидин-1-ил]-N-метил-2-фторбензамид MDV3100 (антагонист андрогенных рецепторов с IC50=36 nМ), который в настоящее время находится в III фазе клинических испытаний, как препарат для лечения рака простаты [Drug Data Rep., 2009, 31(6), 609].



Поиск высокоэффективных противораковых лекарственных препаратов, обладающих повышенной активностью и пониженной токсичностью, по-прежнему является одним из основных направлений создания новых фармакологических средств для лечения рака, в том числе и рака простаты. В этой связи актуальным является разработка новых противораковых субстанций, фармацевтических композиций и лекарственных препаратов, а также способов их получения и применения.
Ниже приведены определения терминов, которые использованы в описании.
«Азагетероцикл» означает ароматическую или неароматическую моноциклическую или полициклическую систему, содержащую в цикле, по крайней мере, один атом азота. Азагетероцикл может иметь один или более «заместителей циклической» системы.
«Активный компонент» (лекарственное вещество, drug-substance) означает физиологически активное вещество синтетического или иного (биотехнологического, растительного, животного, микробного и прочего) происхождения, обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного препарата (средства).
«Алкил» означает алифатическую углеводородную линейную или разветвленную группу с 1-12 атомами углерода в цепи. Разветвленная означает, что алкильная цепь имеет один или несколько «низших алкильных» заместителей. Алкил может иметь один или несколько одинаковых или различных заместителей («алкильных заместителей») включая галоген, алкенилокси, циклоалкил, арил, гетероарил, гетероциклил, ароил, циано, гидрокси, алкокси, карбокси, алкинилокси, аралкокси, арилокси, арилоксикарбнил, алкилтио, гетероарилтио, аралкилтио, арилсульфонил, алкилсульфонилгетероаралкилокси и т.п.
«Антагонисты» означают лиганды, которые связываются с рецепторами определенного типа и не вызывают активного клеточного ответа. Антагонисты препятствуют связыванию агонистов с рецепторами и тем самым блокируют передачу специфического рецепторного сигнала.
«Арил» означает ароматическую моноциклическую или полициклическую систему, включающую от 6 до 14 атомов углерода, преимуществено от 6 до 10 атомов углерода. Арил может содержать один или более «заместителей циклической системы», которые могут быть одинаковыми или разными. Представителями арильных групп являются фенил или нафтил, замещенный фенил или замещенный нафтил. Арил может быть аннелирован с неароматической циклической системой или гетероциклом.
«Гетероциклил» означает ароматическую или неароматическую насыщенную моноциклическую или полициклическую систему, включающую от 3 до 10 атомов углерода, преимущественно от 5 до 6 атомов углерода, в которой один или несколько атомов углерода заменены на гетероатом, такой как азот, кислород, сера. Приставка «аза», «окса» или «тиа» перед гетероциклилом означает наличие в циклической системе атома азота, атома кислорода или атома серы соответственно. Гетероциклил может иметь один или несколько «заместителей циклической системы», которые могут быть одинаковыми или разными. Атомы азота и серы, находящиеся в гетероциклиле, могут быть окислены до N-оксида, S-оксида или S-диоксида. Представителями гетероциклилов являются пиперидин, пирролидин, пиперазин, морфолин, тиоморфолин, тиазолидин, 1,4-диоксан, тетрагидрофуран, тетрагидротиофен и др.
«Гидрат» означает сольват, в котором вода является молекулой или молекулами растворителя.
«Заместитель» означает химический радикал, который присоединяется к скэффолду (фрагменту), например, «заместитель алкильный», «заместитель амино группы», «заместитель карбамоильный», «заместитель циклической системы», значения которых определены в данном разделе.
«Лекарственное средство (препарат)» - вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и др. готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
«Низший алкил» означает линейный или разветвленный алкил с 1-4 атомами углерода.
«Фармацевтическая композиция» обозначает композицию, включающую в себя соединение формулы 1 и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлимых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие как оливковое масло) и инъекционные органические сложные эфиры (такие как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, алгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения. Фармацевтические композиции, как правило, получают с помощью стандартных процедур, предусматривающих смешение активного соединения с жидким или тонко измельченным твердым носителем.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные. (Подробное описание свойств таких солей дано в Berge S.M., et al., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66: 1-19.) Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
Авторы обнаружили новые антагонисты андрогенных рецепторов, представляющие собой неизвестные ранее циклические N,N'-диарилтиомочевины или N,N'-диарилмочевины общей формулы 1, их оптические (R)- и (S)- изомеры и их фармацевтически приемлемые соли,

где X представляет собой атом кислорода или серы; m=0 или 1;
R1 представляет собой C1-С3алкил; R2 и R3 представляют собой атом водорода;
или R2 и R3 вместе с атомом углерода, с которым они связаны, образуют группу С=О;
или

R4 и R5 представляют собой атом водорода;
или R4 представляет собой атом водорода, а R5 представляет собой метил;
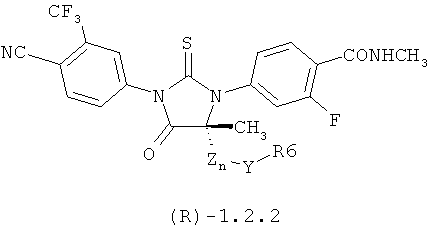
или R4 представляет собой атом водорода, метил, а R5 представляет собой группу Zn-Y- R6, в которой n=1 или 2, Z представляет собой СH2 или С=O, Y представляет собой атом кислорода или N-СН3, или Y представляет собой С=O, а Z представляет собой CH2;
R6 представляет собой атом водорода, метил, бензил, гидрокси группу или R4 и R5 вместе с атомами, с которыми они связаны, образуют пяти- или шестичленный гетероцикл, включающий по крайней мере атом кислорода или азота, последний может быть замещен метилом.
Предпочтительными являются циклические N,N'-диарилтиомочевины или N,N'-диарилмочевины общей формулы 1.2,1.3 или 1.4.
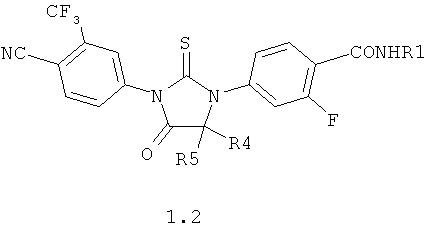
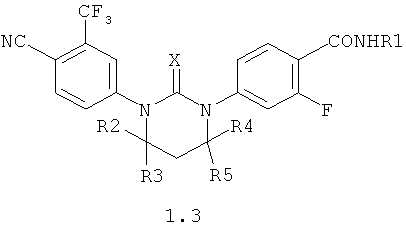

где X, R1, R2, R3, R4 и R5 имеют вышеуказанное значение.
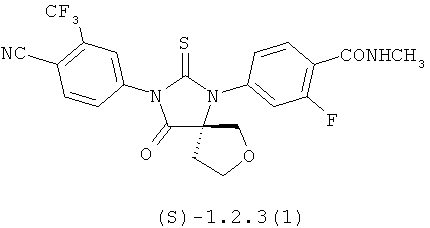
Более предпочтительными являются циклические N,N'-диарилтиомочевины формулы 1.2(1), 1.2(2), 1.2.2 и 1.2.3, и их оптические (R)-изомеры (R)-1.2(2), (R)-1.2.2 и (R)-1.2.3 и (S)-изомеры (S)-1.2(2), (S)-1.2.2 и (S)-1.2.3.
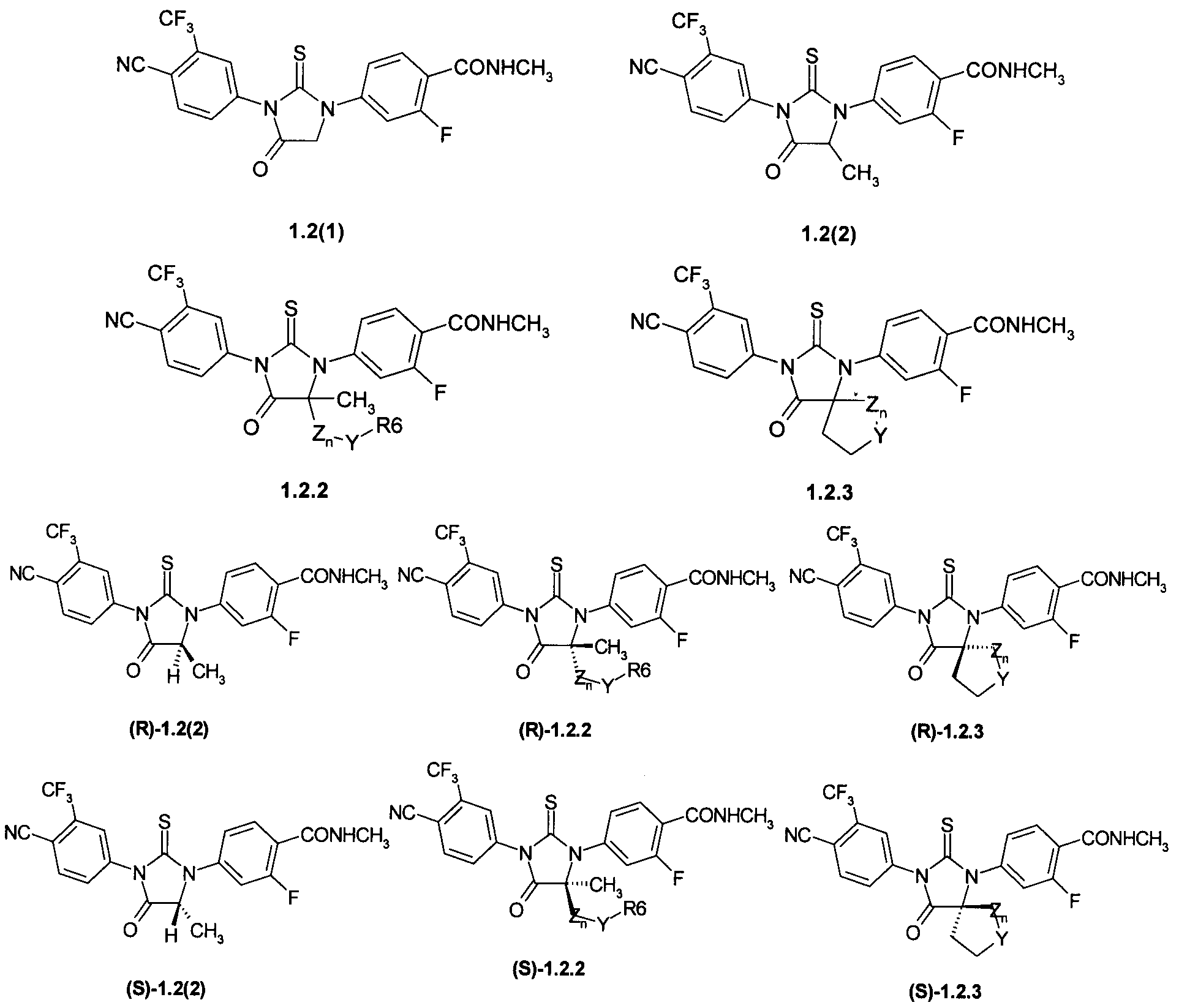
где Y, Z и R6 имеют вышеуказанное значение.
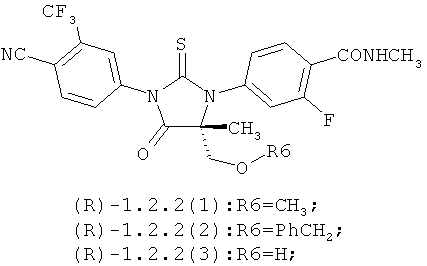
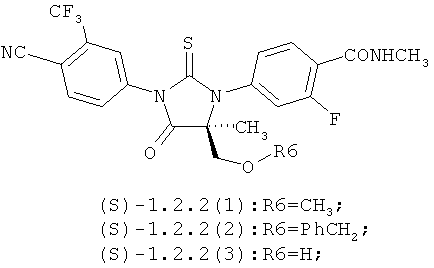
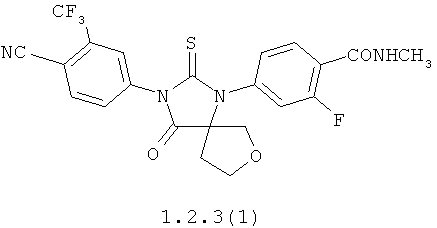
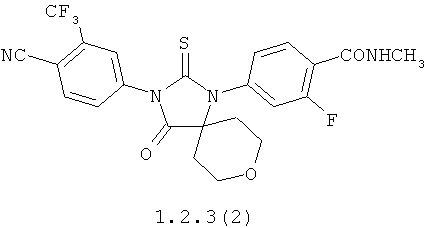
Более предпочтительными являются также соединения формулы 1.2.2(1), 1.2.2(2), 1.2.2(3), 1.2.3(1), 1.2.3(2) и 1.2.3(3), их оптические (R)-изомеры (R)-1.2.2(l), (R)-1.2.2(2), (R)-1.2.2(3), (R)-1.2.3(l), и (S)-изомеры (S)-1.2.2(l), (S)-1.2.2(2), (S)-1.2.2(3), (S)-1.2.3(l)



Предметом данного изобретения являются способы получения циклических N,N'-диарилмочевин общей формулы 1, их оптические (R)- и (S)- изомеры и их фармацевтически приемлемые соли.
1,3-Диарил-гидантоины общей формулы 1.2 получают взаимодействием соответствующих 4-(цианометил)амино-бензамидов 4.1 или (4-карбамоил-фениламино)-уксусных кислот 4.2 с изотиоцианатом 3.2 по схеме 1.
Схема 1
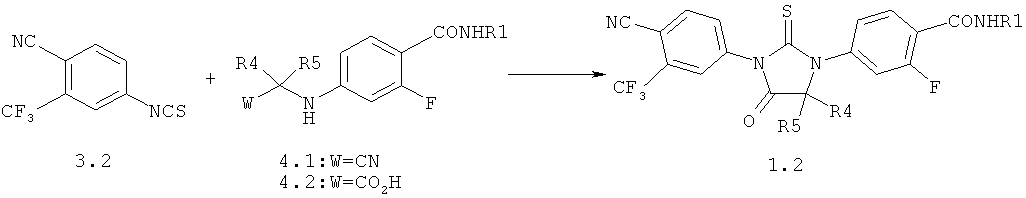
где R1, R4 и R5 имеют вышеуказанное значение.
Оптически активные изомеры (R)-1.2 и (S)-1.2 получают либо исходя из соответствующих оптических изомеров (R)-4, (S)-4, либо разделением их смесей 1.2.
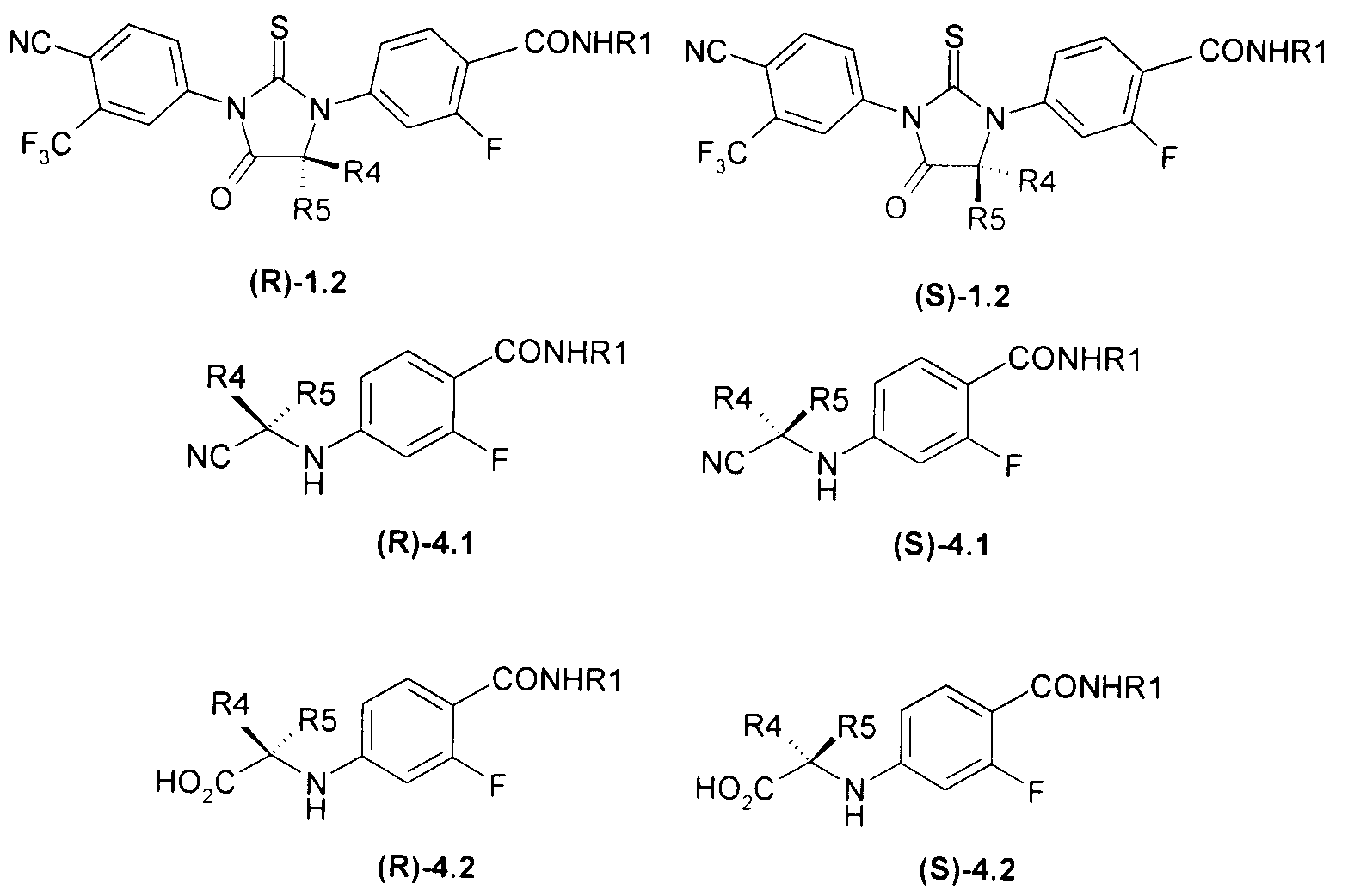
где R1, R4 и R5 имеют вышеуказанное значение.
1,3-Диарил-тетрагидро-пиримидин-2-оны общей формулы 1.3.1 получают взаимодействием соответствующих N,N'-диарилмочевин общей формулы 2 с 1,3-дибромпропаном по схеме 2.
Схема 2

где R1 имеет вышеуказанное значение.
Соединения общей формулы 1.3.2 получают взаимодействием соответствующих этил β-аланинатов общей формулы 5 с изоцианатом 3.1 или изотиоцианатом 3.2 и циклизацией образующихся мочевин общей формулы 6 по схеме 3.
Схема 3

где Х и R1 имеют вышеуказанное значение.
1,4-Диарил-[1,2,4]триазолидин-3,5-дионы общей формулы 1.4 получают взаимодействием соответствующего гидразина 7 с изоцианатом 3.1 и циклизацией образующегося семикарбозида 8 дифосгеном по схеме 4.
Схема 4
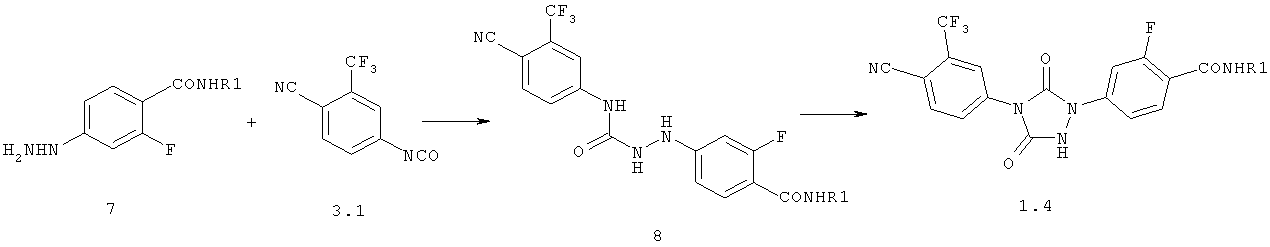
где R1 имеет вышеуказанное значение.
Новые антагонисты андрогенных рецепторов пригодны также для изучения молекулярного механизма ингибирования и активации андрогенных рецепторов.
Новые циклические N,N'-диарилтиомочевины и N,N'-диарилмочевины общей формулы 1 являются антагонистами андрогенных рецепторов, причем по активности они превосходят известные антагонисты андрогенных рецепторов, опубликованные в патентных заявках WO 2006124118, WO 2007127010, и в Drug Data Rep., 2009, 31(6), 609.
Кроме того, новый антагонист 1.2.2(1) более чем в три раза менее токсичен, чем антагонист MDV3100, так как его максимально переносимая доза (МТД), найденная в экспериментах на самцах мышей линии CD1, составляет МТД>100 мг/кг, в то время как MDV3100 имеет МТД ~ 30 мг/кг.
Предметом данного изобретения является новое противораковое средство, представляющие собой, по крайней мере, одну циклическую N,N'-диарилтиомочевину или N,N'- диарилмочевину общей формулы 1, или ее оптические (R)- и (S)-изомеры, или ее фармацевтически приемлемую соль.
Предметом данного изобретения является также фармацевтическая композиция, содержащая в качестве активного компонента, по крайней мере, одну циклическую N,N'-диарилтиомочевину или N,N'-диарилмочевину общей формулы 1, или ее оптические (R)- и (S)-изомеры, или фармацевтически приемлемую соль, обладающую противораковой активностью, в эффективном количестве.
Более предпочтительной является фармацевтическая композиция, обладающая активностью в отношении рака простаты, содержащая в качестве активного компонента, по крайней мере, одну циклическую N,N'-диарилтиомочевину или N,N'-диарилмочевину общей формулы 1, или ее оптические (R)- и (S)-изомеры, или фармацевтически приемлемую соль, обладающую противораковой активностью, в эффективном количестве.
Фармацевтические композиции могут включать фармацевтически приемлемые эксципиенты. Под фармацевтически приемлемыми экспициентами подразумеваются применяемые в сфере фармацевтики разбавители, вспомогательные агенты и/или носители. Фармацевтическая композиция, наряду с новым противораковым средством, по настоящему изобретению может включать и другие активные компоненты, в том числе обладающие противораковой активностью, при условии, что они не вызывают нежелательных эффектов.
При необходимости использования фармацевтической композиции по настоящему изобретению в клинической практике она может смешиваться с традиционными фармацевтическими носителями.
Носители, используемые в фармацевтической композиции по настоящему изобретению, представляют собой носители, которые применяются в сфере фармацевтики для получения распространенных форм, в том числе: в пероральных формах используются связующие вещества, смазывающие агенты, дезинтеграторы, растворители, разбавители, стабилизаторы, суспендирующие агенты, бесцветные агенты, корригенты вкуса; в формах для инъекций используются антисептические агенты, солюбилизаторы, стабилизаторы; в местных формах используются основы, разбавители, смазывающие агенты, антисептические агенты.
Целью настоящего изобретения также является способ получения фармацевтических композиций.
Поставленная цель достигается смешением противоракового средства с инертным наполнителем и/или растворителем, отличительная особенность которого состоит в том, что в качестве противоракового средства используют, по крайней мере, одну циклическую N,N'-диарилтиомочевину или N,N'-диарилмочевину общей формулы 1 или ее оптический (R)- и (S)-изомер или ее фармацевтически приемлемую соль.
Предметом данного изобретения является также лекарственное средство в форме таблеток, капсул, инъекций, включающее в свой состав новое противораковое средство или новую фармацевтическую композицию, предназначенную для лечения рака.
Более предпочтительным лекарственным средством, включающим в свой состав новое противораковое средство или новую фармацевтическую композицию, является средство, предназначенное для лечения рака простаты.
Предметом данного изобретения являются также терапевтические коктейли для лечения раковых заболеваний, в том числе заболеваний рака простаты, включающие в качестве одного из компонентов новое лекарственное средство или новую фармацевтическую композицию, содержащие в качестве активного компонента, по крайней мере, одну N,N'-диарилмочевину общей формулы 1 или ее оптический (R)- и (S)-изомер, или ее фармацевтически приемлемую соль.
Терапевтический коктейль для и лечения рака простаты, наряду с лекарственным средством по данному изобретению, может включать другие известные препараты, предназначенные для лечения раковых заболеваний.
В соответствии с данным изобретением способ лечения раковых заболеваний животных и людей, в том числе рака простаты, заключается во введении теплокровному животному или человеку нового лекарственного средства, новой фармацевтической композиции или нового терапевтического коктейля.
Лекарственные средства могут вводиться перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно или местно). Клиническая дозировка средства общей формулы 1 у пациентов может корректироваться в зависимости от терапевтической эффективности и биодоступности активных ингредиентов в организме, скорости их обмена и выведения из организма, а также в зависимости от возраста, пола и стадии заболевания пациента, при этом суточная доза у взрослых обычно составляет 10~500 мг, предпочтительно -50~300 мг. Поэтому во время приготовления из фармацевтической композиции лекарственного средства по настоящему изобретению в виде единиц дозировки необходимо учитывать вышеназванную эффективную дозировку, при этом каждая единица дозировки препарата должна содержать 10~500 мг средства общей формулы 1, предпочтительно -50~300 мг. В соответствии с указаниями врача или фармацевта данные препараты могут приниматься несколько раз в течение определенных промежутков времени (предпочтительно - от одного до шести раз).
Изобретение поясняется чертежами.
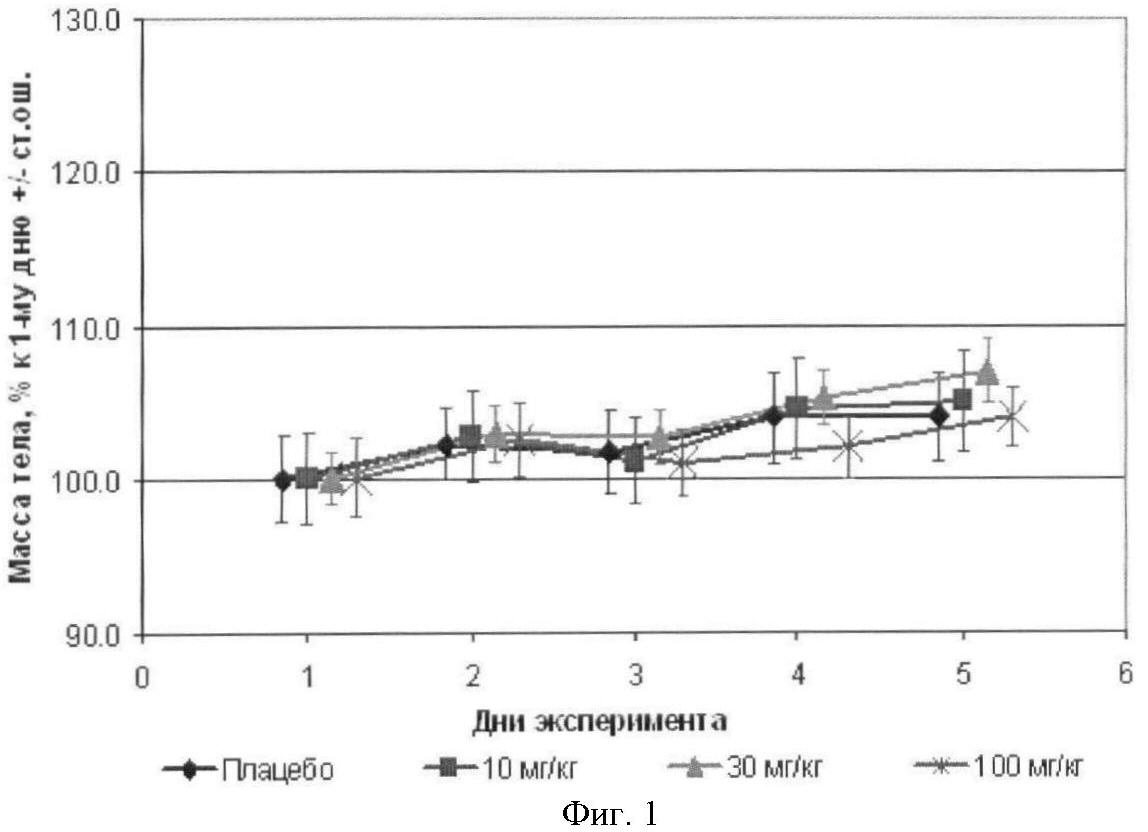
Фиг.1 - изменение веса самцов мышей при пероральном введении соединения 1.2.2(1).
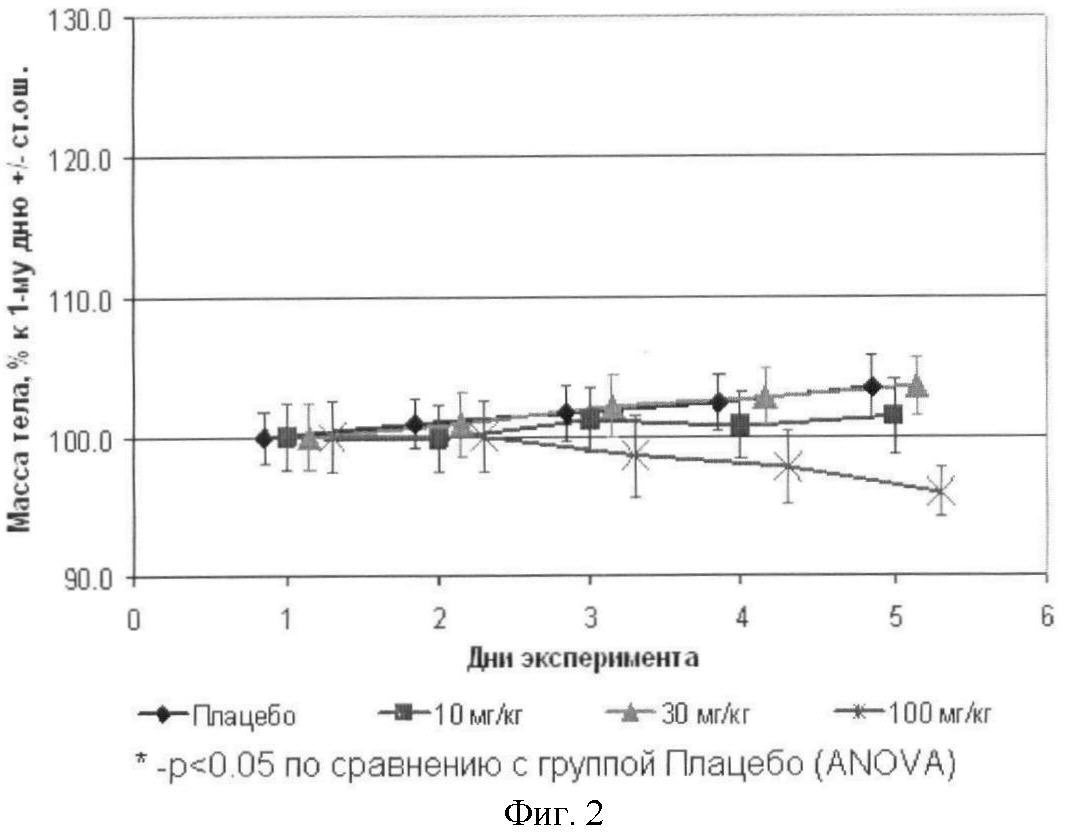
Фиг.2 - изменение веса самцов мышей при пероральном введении соединения MDV3100.
В представленных ниже примерах описан синтез N,N'-диарилтиомочевин и N,N'-диарилмочевин и их биологические испытания. Представленные ниже примеры демонстрируют, но не ограничивают, данное изобретение.
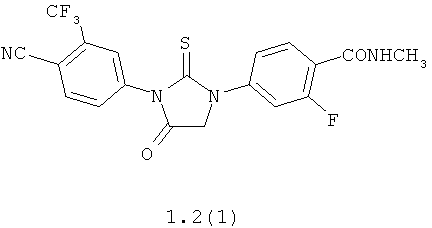
Пример 1. Синтез N-метил-4-{4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]-имидазолидин-1-ил}-2-фторбензамида 1.2(1). К раствору 279 мг (1 ммоль) 4-йод-N-метил-2-фторбензамида в 3 мл ДМФА добавляют 80 мг (1.07 ммоль) глицина и 207 мг (1.5 ммоль) К2СО3. Реакционную массу перемешивают в микроволновой печи при 140°С в течение 18 мин. Охлажденную смесь разбавляют 10 мл AcOEt и 10 мл воды, нейтрализуют НСl до рН 2-3, органический слой отделяют, а водный экстрагируют AcOEt (5×20 мл). Объединенные экстракты промывают рассолом, сушат над Na2SO4 и упаривают в вакууме. Целевой продукт выделяют колоночной хроматографией на SiO2. Получают N-(4-метилкарбамоил-2-фторфенил)глицин 4.2(1). Раствор 113 мг (0.5 ммоль) N-(4-метилкарбамоил-2-фторфенил)глицина 4.2(1) и 174 мг (1.0 ммоль) 4-изотиоцианато-2-(трифторметил)бензонитрила 3.2 в 2 мл ДМФА перемешивают при 90°С в течение 12 ч. Реакционную смесь упаривают в вакууме и выделяют методом HPLC N-метил-4-{4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]-имидазолидин-1-ил}-2-фторбензамид 1.2(1). LCMS (М+H)+ 437.
Пример 2. Общий способ синтеза N-метил-4-{5-метил-4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]имидазолидин-1-ил}-2-фторбензамида 1.2(2), N-метил-4-{(S)-5-метил-4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]имидазолидин-1-ил}-2-фторбензамида (S)-1.2(2) и N-метил-4-{(R)-5-метил-4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]имидазолидин-1-ил}-2-фторбензамида (R)-1.2(2).
К раствору 667 мг (3.9 ммоль) N-метил-2,4-дифторбензамида в 3 мл ДМСО добавляют 347 мг (7.8 ммоль) (D,L)-, (D)- или (L)-ланина и 2.54 г (7.8 ммоль) Сs2СО3. Реакционную массу перемешивают в закрытой пробирке при 90°С в течение 18 ч. Охлажденную смесь разбавляют изопропанолом, нейтрализуют 1.36 мл (15.6 ммоль) НСl, фильтруют, упаривают в вакууме и выделяют методом HPLC N-(4-метилкарбамоил-3-фторфенил)аланин 4.2(2), (S)-N-(4-метилкарбамоил-3-фторфенил)аланин (S)-4.2(2) или (R)-N-(4-метилкарбамоил-3-фторфенил)аланин (R)-4.2(2). LC-MS (M+H)+ 241.1Н ЯМР (ДМСО-d6, 400 МГц): 12.66 (уш. с, 1Н), 7.62 (м, 1Н), 7.45 (т,J=8.8 Гц, 1Н), 6.67 (уш. д, J=7.2 Гц, 1Н), 6.42 (дд, J1=8.4 Гц, J2=2.0 Гц, 1 Н), 6.29 (дд, J1=14.8 Гц, J2=2.0 Гц, 1Н), 4.03 (м, 1Н), 2.73 (д, J=4.4 Гц, 3Н), 1.37 (д, J=7.2 Гц, 3Н). Раствор 110 мг (0.46 ммоль) амина 4.2(2), (S)-4.2(2) или (R)-4.2(2) и 144 мг (0.55 ммоль) 4-изотиоцианато-2-(трифторметил)-бензонитрила 3.2 в 2 мл ДМФА перемешивают в микроволной печи при 90°С в течение 12 ч, затем добавляют еще 50 мг (0.19 ммоль) 4-изотиоцианато-2-(трифторметил)-бензонитрила 3.2 и проводят реакцию еще в течение 12 ч. Реакционную смесь упаривают в вакууме и выделяют методом HPLC N-метил-4-{5-метил-4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]имидазолидин-1-ил}-2-фторбензамид 1.2(2), N-метил-4-{(S)-5-метил-4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]имидазолидин-1-ил}-2-фторбензамид (S)-1.2(2) или N-метил-4-{(R)-5-метил-4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]имидазолидин-1-ил}-2-фторбензамид (R)-1.2(2) с высокими кажущимися константами ингибирования андрогеновых рецепторов (Кi) - высокой антагонистической активностью: (Кi1.2(2)=140.2 nМ, Кi(S)-1.2(2)=106.7 nМ и Кi(R)-1.2(2)=73.6 nМ. LC-MS (M+H)+ 451.1Н ЯМР (CDCl3, 400 МГц): 8.28 (т, J=8.6 Гц, 1Н), 8.01 (д, J=8.0 Гц, 1Н), 7.94 (д, J=1.2 Гц, 1Н), 7.81 (дд, J1=8.0 Гц, J2=1.2 Гц, 1Н), 7.48 (дд, J1=12.4 Гц, J2=1.6 Гц, 1Н), 7.36 (дд, J1=8.4 Гц, J2=1.6 Гц, 1 Н), 6.72 (уш. м, 1 Н), 4.83 (к, J=7.2 Гц, 1 Н), 3.08 (д, J=4.8 Гц, 3Н), 1.60(д, J=7.2 Гц, 3Н).
Пример 3. Синтез N-метил-4-{2-тио-3-[3-(трифторметил)-4-цианофенил]-гидантоин-1-ил}-2-фторбензамидов 1.2.2 и 1.2.3 (общая методика). Раствор 0.75 ммоль соответствующего N-мeтил-2-фтop-4-[(1-циaнoмeтил)aминo]бeнзaмидa 4.1 и 342 мг (1.5 ммоль) 4-изотиоцианато-2-(трифторметил)бензонитрила 3.2 в 3 мл ДМФА перемешивают в микроволной печи при 110°С в течение 12 ч. Реакционную смесь растворяют в 30 мл МеОН, добавляют 7.5 мл 1N HCl и кипятят в течение 1.5 ч. Раствор упаривают в вакууме, обрабатывают водой, осадок отфильтровывают, промывают водой и высушивают в вакууме. Целевой продукт выделяют методом HPLC.
Получают антагонист андрогеновых рецепторов - N-метил-4-[5-метил-5-(метоксиметил)-4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]-имидазолидин-1-ил]-2-фторбензамид 1.2.2(1) с Ki1.2.2(1)=115.9 nМ, который разделяют с помощью жидкостной хроматографии высокого давления на Chiralpak HD-H 25×1 см (Chiral Technologies Inc., USA). В качестве элюента использовали смесь 80% н-гексана, 20% 2-пропанола и 0.02% триэтиламина. Скорость потока 4 мл/мин. Получают оптически чистые изомеры (R)-l.2.2(1) и (S)-1.2.2(l), с высокой антагонистической активностью Ki(S)-1.2.2(1)=721.5 nМ и Ki(S)-1.2.2(1) - 53.3 nM. LC-MS (M+H)+ 495.1Н ЯМР (CDCl3, 400 МГц): 8.28 (т, J=8.4 Гц, 1Н), 7.99 (д,.7=8.0 Гц, 1Н), 7.92 (с, 1H), 7.80 (д, J=8.0 Гц, 1Н), 7.29 (дд, J1=8.8 Гц, J2=1.2 Гц, 1Н), 7.21 (дд, J=11.6 Гц, J=1.2 Гц. 1Н). 6.72 (к, J=4.4 Гц, 1Н),3.71 (д, J=10.0 Гц, 1Н),3.43(с, 3Н),3.35(д, J=10.0 Гц, 1Н), 3.09 (д, J=4.4 Гц, 3Н),1.52(с, 3Н).
N-Метил-4-{5-[(бензилокси)метил]-5-метил-4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]имидазолидин-1-ил}-2-фторбензамид 1.2.2(2). LC-MS (M+H)+571.1Н ЯМР (CDCl3, 400 МГц): 8.22 (т, J=8.4 Гц, 1Н), 7.96 (a,J=8.0 Гц, 1Н), 7.86 (с, 1Н), 7.70 (дд, J=8.0 Гц, J2=1.2 Гц, 1 Н), 7.39 (м. 3Н), 7.29 (м, 2Н), 7.25 (дд, J=8.4 Гц, J=1.6 Гц, 1Н),7.18(дд, J1=8.4 Гц, J2=1.6 Гц, 1Н),6.71 (к, J=4.8 Гц, 1Н), 4.59 (м, 2Н), 3.79 (д, J=10.2 Гц, 1Н), 3.45 (д, J=10.2 Гц. 1 Н). 3.08 (д, J=4.8 Гц. 31-1). 1,51 (с, 3Н).
Этил-{4-метил-3-(4-метилкарбамоил-3-фторфенил)-5-оксо-2-тиоксо-1-[3-(трифторметил)-4-цианофенил]имидазолидин-4-ил} ацетат 1.2.2(4). LC-MS (М+Н)+ 536.1Н ЯМР (CDCl3, 400 МГц): 8.26 (т, J=8.4 Гц, 1Н), 8.01 (д, J=8.0 Гц, 1Н), 8.00 (с, 1Н), 7.90 (дд, J1=8.0 Гц, J2=1.6 Гц, 1Н), 7.18 (дд, J1=8.0 Гц, J2=1.6 Гц, 1 Н), 7.10 (дд, J1=8.0 Гц, J2=1.6 Гц, 1Н), 6.78 (к, J=4.8 Гц, 1Н), 4.26(м, 1Н), 3.13 (д, J=18.0 Гц, 1Н),3.09 (д, J=4.8 Гц, 3Н), 2.64(д, J=18.0 Гц, 1Н), 1.67(с, 3Н), 1.31 (т, J=7.0 Гц, 3Н).
N-Метил-4-{4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]-7-окса-1,3-диазаспиро[4.4]нон-1-ил}-2-фторбензамид 1.2.3(1) с Ki(S)-1.2.2(1)=33.9 nM. LC-MS (M+H)+493.1Н ЯМР (CDCl3, 400 МГц): 8.30 (т, J=8.4 Гц, 1Н), 8.02 (д, J=8.4 Гц, 1Н), 7.98 (д, J=1.6 Гц, 1Н), 7.85 (дд, J,=8.4 Гц, J=1.6 Гц, 1Н), 7.34 (дд, J1=8.4 Гц, J2=1.6 Гц, 1Н), 7.25 (дд, J1=11.8 Гц, J2=1.6 Гц, 1Н), 6.78 (к, J=4.4 Гц, 1 Н), 4.43 (д, J=10.0 Гц, 1Н), 4.16 (д, 7=10.0 Гц, 1Н),3.96(м, 1Н), 3.75 (м, 1Н), 3.09 (д, J=4.4 Гц, 3Н), 2.74 (м, 1Н), 2.48 (м,1Н).
N-Метил-4-{4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]-8-окса-1,3-диазаспиро[4.5]дец-1-ил}-2-фторбензамид 1.2.3(2). LC-MS (M+H)+507.1Н ЯМР (CDCl3, 400 МГц): 8.32 (т, J=8.4 Гц, 1 Н), 8.01 (д, J=8.0 Гц, 1 Н), 7.95 (с, 1 Н), 7.83 (д, J=8.0 Гц, 1 Н), 7.20 (д, J=8.4 Гц, 1 Н), 7.10 (д, J=8.0 Гц, 1 Н), 6.73 (уш. м, 1 Н), 4.18 (м, 2Н), 3.94 (м, 2Н), 3.09 (д, J=4.4 Гц, 3Н), 2.07 (м, 4Н).
N-Метил-4-{8-метил-4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]-1,3,8-триазаспиро[4.5]дец-1-ил}-2-фторбензамид 1.2.3(3). LC-MS (M+H)+520.1Н ЯМР (ДМСО-d6, 400 МГц): 10.09 (уш. с, 1Н), 8.48 (к, J=4.4 Гц, 1Н), 8.43 (д, J=8.4 Гц, 1Н), 8.29 (с, 1 Н), 8.11 (д, J=8.4 Гц, 1 Н), 7.84 (т, J=8.0 Гц, 1 Н), 7.42 (д, J=10.4 Гц, 1 Н), 7.30 (д, J=8.0 Гц, 1Н), 3.50 (м, 4Н), 2.80 (д, J=4.4 Гц, 3Н), 2.78 (с, 3Н), 2.72(д, J=14.0 Гц, 1Н), 2.16 (м, 2Н).
Пример 4. Синтез N-метил-4-[5-(гидроксиметил)-5-метил-4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]имидазолидин-1-ил]-2-фторбензамида 1.2.2(3). К раствору 55 мг (0.11 ммоль) N-метил-4-[5-метил-5-(метоксиметил)-4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]имидазолидин-1-ил]-2-фторбензамида в 1.5 мл СНзСЬ под аргоном при -78°С по каплям прибавляют 53 мкл (0.55 ммоль) ВВr3. Реакционную массу перемешивают 3 часа при -78°С и затем еще 3 часа при комнатной температуре. По окончании реакции избыток ВВr3 нейтрализуют прибавлением 10 мл 5% раствора карбоната натрия, продукт экстрагируют AcOEt, сушат над Na2SO4, упаривают в вакууме и методом HPLC выделяют N-метил-4-[5-(гидроксиметил)-5-метил-4-оксо-2-тиоксо-3-[3-(трифторметил)-4-цианофенил]имидазолидин-1-ил]-2-фторбензамид 1.2.2(3) с Ki(S)-1.2.2(3)=46.3 nM,1Н ЯМР (ДМСО-d6, 400 МГц): 8.43 (уш. м, 1 Н), 8.39 (д, J=8.4 Гц, 1 Н), 8.13 (с, 1 Н), 7.98 (д, J=8.4 Гц, 1 Н), 7.78 (т, J=8.0 Гц, 1 Н), 7.42 (д, J=10.8 Гц, 1 Н), 7.37 (д, J=8.0 Гц, 1Н), 5.93 (т, J=4.4 Гц, 1Н), 3.81 (дд, J,=11.6 Гц, J=4.4 Гц, 1Н), 3.45 (дд, J1=11.6 Гц, J2=5.0 Гц, 1 Н), 2.79 (д, J=4.0 Гц, 3Н), 1.38 (с, 3Н).
Пример 5. Синтез {4-метил-3-(4-метилкарбамоил-3-фторфенил)-5-оксо-2-тиоксо-1-[3-(трифторметил)-4-цианофенил]имидазолидин-4-ил}уксусной кислоты 1.2.2(5). К раствору 46 мг (0.086 ммоль) эфира 1.2.2(4) 2 мл спирта добавляют раствор 7 мг (0.172 ммоль) NaOH в 0. 5 мл воды и реакционную массу перемешивают 12 ч (контроль по LC-MS). Раствор упаривают, добавляют 2 мл изопропанола и 15 мкл (0.172 ммоль) НС1, фильтруют и упаривают в вакууме. {4-Метил-3-(4-метилкарбамоил-3-фторфенил)-5-оксо-2-тиоксо-1-[3-(трифторметил)-4-цианофенил]имидазолидин-4-ил}уксусную кислоту 1.2.2(5) выделяют методом HPLC. LCMS (М+H)+ 469.1Н ЯМР (ДМСО-d6, 400 МГц): 13.31 (уш. с, 1 Н), 8.44 (м, 2Н), 8.10 (с, 1 Н), 7.95 (д, J=7.6 Гц, 1 Н), 7.81 (т, J=8.0 Гц, 1 Н), 7.25 (a, J=10.8 Гц, 1H), 7.19 (д, J=8.0 Гц. 1Н), 3.16 (д, J=17.6 Гц, 1Н), 2.79 (д, J=3.6 Гц, 3Н), 2.70 (д, J=17.6 Гц, 1 Н), 1.59 (с, 3Н).
Пример 6. Синтез 4-[3-[3-(трифторметил)-4-цианофенил]-2-оксо-тетрагидро-пиримидин-1(2H)-ил]-N-метил-2-фторбензамид 1.3(1). К раствору 100 мг (0.26 ммоль) 4-[4-циано-3-(трифторметил)-фенилкарбамоиламино]-N-метил-2-фторбензамида 1.1(1) в 2 мл ДМФА добавляют 109 мг (0.79 ммоль) К2СО3 и 32 мкл (0.32 ммоль) 1,3-дибромпропана. Смесь перемешивают при 90°С.Через 18 ч добавляют еще 109 мг К2СО3 и 32 мкл 1,3-дибромпропана и продолжают реакцию. Добавление повторяют еще 2 раза. Смесь упаривают в вакууме, растворяют в хлороформе, промывают водой, сушат над Na2SO4 и упаривают. Целевой продукт выделяют методом колоночной хроматографии на SiO2 (элюент - AcOEt). LC-MS (М+Н)+ 421.1Н ЯМР (ДМСО-d6, 400 МГц): 8.17 (уш. м, 1 Н), 8.13 (д, J=8.4 Гц, 1 Н), 8.07 (д, J=2.0 Гц, 1 Н), 7.83 (дд, J1=8.4 Гц, J2=2.0 Гц, 1 Н), 7.62 (т, J=8.4 Гц, 1 Н), 7.38 (дд, J1=12.4 Гц, J2=2.0 Гц, 1 Н), 7.29 (дд, J1=8.4 Гц, J2=2.0 Гц, 1 Н), 3.90 (т, J=5.8 Гц, 2Н), 3.81 (т, J=5.8 Гц, 2Н), 2.77 (д, J=4.8 Гц, 3Н), 2.21 (м, 2Н).
Пример 7. Синтез N-метил-4-{[3-(трифторметил)-4-цианофенил]-2,4-диоксотетрагидропиримидин-1(2H)-ил}бензамида 1.3.1(1). К раствору 9 г (53.6 ммоль) 4-амино-N-метил-2-фторбензамида в 90 мл ДМСО прибавляют 8 г (80 ммоль) этилакрилата и 0.81 г (5.4 ммоль) DBU и перемешивают 24 ч при 70°С (контроль по LC-MS). Реакционную массу подвергают лиофилизации и остаток перекристаллизовывают из водного спирта. Получают этил N-[4-(метилкарбамоил)-3-фторфенил]-β-аланинат 5. LCMS (M+H)+ 269.1Н ЯМР (ДМСО-d6, 400 МГц) δ 7.57 (уш. с.1Н). 7.48 (т, J=8.8 Гц, 1 Н), 6.47 (уш. с, 1 Н), 6.42 (д, J=8.8 Гц, 1 Н). 6.33 (д, J=14.8 Гц, 1 Н). 4.07 (к, J=7.2 Гц, 2Н), 3.32 (уш. м, 2Н), 2.73 (д, J=4.4 Гц, 3Н), 2.55 (т, J=6.4 Гц, 2Н), 1.18 (т, J=7.2 Гц, 3Н). Раствор 425 мг (1.87 ммоль) 4-изоцианато-2-(трифторметил)бензонитрила 2.1 и 500 мг (1.87 ммоль) этил-N-[4-(метилкарбамоил)-3-фторфенил]-β-аланината 5 в 10 мл СН2Сl2перемешивают 15 ч. Реакционную массу упаривают в вакууме и целевой продукт выделяют методом колоночной хроматографии на SiO2 (элюент - гексан: AcOEt: EtaN=1:1:0.03). Получают этил-N-[4-(метилкарбамоил)-3-фторфенил]-N-{[3-(трифторметил)-4-цианофенил]-карбамоил}-β-аланинат 6(1). LC-MS (M+H)+481. К раствору 500 мг (1.04 ммоль) этил-N-[4-(метилкарбамоил)-3-фторфенил]-N-{[3-(трифторметил)-4-цианофенил]-карбамоил}-β-аланината 6(1) в 5 мл АсОН добавляют 2.5 мл НС1 и перемешивают 15 ч. Реакционную массу выливают в воду, продукт экстрагируют EtOAc. Органический слой сушат над Na2SO4, упаривают в вакууме и выделяют колоночной хроматографией на SiO2 (элюент - гексан: AcOEt=1:1) N-метил-4-{[3-(трифторметил)-4-цианофенил]-2,4-диоксо-тетрагидро-пиримидин-1(2Я)-ил}бензамид 1.3.1(1). LC-MS (М+Н)+ 435.1Н ЯМР (ДМСО-d6, 400 МГц) 5 8.29 (д, J=7.6 Гц, 1Н), 8.23 (уш. м, 1Н), 8.11 (с, 1Н), 7.89 (д, J=2.0 Гц, 1 Н), 7.67 (т, J=8.4 Гц, 1 Н), 7.39 (д, J=12.4 Гц, 1 Н), 7.33 (д, J=8.4 Гц, 1 Н), 4.02 (т, J=6.4 Гц, 2Н), 3.03 (т, J=6.4 Гц, 2Н), 2.77 (д, J=4.4 Гц, 3Н).
Пример 8. Синтез N-метил-4-{[3-(трифторметил)-4-цианофенил]-4-оксо-2-тиоксотетрагидропиримидин-1(2H)-ил}бензамида 1.3.1(2). Раствор 320 мг (1.51 ммоль) 4-изотиоцианато-2-(трифторметил)бензонитрила 3.2 и 404 мг (1.51 ммоль) этил-N-[4-(метилкарбамоил)-3-фторфенил]-β-аланината 5 в 8 мл ДМФА нагревют в микроволновой печи при 60°С 8 ч. Реакционную массу упаривают в вакууме и выделяют колоночной хроматографией на SiO2 (элюент - гексан: AcOEt=1:2) этил-N-[4-(метилкарбамоил)-3-фторфенил]-N-{[3-(трифторметил)-4-цианофенил]тиокарбамоил}-β-аланинат 6(2). LC-MS (М+Н)+ 497. К раствору 200 мг (0.4 ммоль) полученного эфира 6(2) 1 мл спирта добавляют раствор 32 мг (0.8 ммоль) NaOH в 0.25 мл воды и реакционную массу перемешивают при 80°С 2 ч (контроль по LC-MS). Реакционную массу нейтрализовывают 69 мкл (0.8 ммоль) НСl, упаривают в вакууме, экстрагируют продукт реакции горячим изопропанолом и упаривают в вакууме. Получают N-[4-(метилкарбамоил)-3-фторфенил]-N-{[3-(трифторметил)-4-цианофенил]тиокарбамоил}-β-аланин 6(3). LCMS (М+Н)+ 469. К раствору 114 мг (0.24 ммоль) полученной кислоты 6.3 в 1.5 мл ДМФА добавляют 86 мг (0.36 ммоль) TBTU и 110 мг (0.84 ммоль) диизопропилэтиламина. Реакционную массу перемешивают при 45°С в течение 15 ч. По окончании реакции (контроль по LC-MS) раствор выливают в воду и экстрагируют EtOAc. Органический слой сушат над Na2S04, упаривают в вакууме и выделяют методом HPLC М-метил-4-{[3-(трифторметил)-4-цианофенил]-4-оксо-2-тиоксотетрагидропиримидин-1(2H)-ил}-2-фторбензамид 1.3.2(2) с Ki(S)-1.3.2(2)=95.2 nM. LC-MS (M+H)+ 451.1Н ЯМР (ДМСО-d6 400 МГц): 8.35 (к, J=4.4 Гц, 1Н), 8.27 (д. J=8.0 Гц, 1Н),8.06(д, J=1.6 Гц, 1 Н), 7.83 (дд, J1=8.0 Гц, J2=1.6 Гц, 1Н), 7.71 (т, J=8.2 Гц, 1Н),7.42(дд, J1=11.0 Гц, J2=1.8 Гц, 1Н), 7.33 (дд,.J1=8.2 Гц, J2=1.8 Гц, 1Н), 4.13 (т, J=6.8 Гц, 2Н), 3.17 (т, J=6.8 Гц, 2Н), 2.78 (д, J=4.4 Гц, 3Н).
Пример 9. Синтез N-Метил-2-фтор-4-[4-[3-(трифторметил)-4-цианофенил]-3,5-диоксо-1,2,4-триазолидин-1-ил]бензамида 1.4(1). К раствору 1 г (5.95 ммоль) 4-амино-N-метил-2-фторбензамида в 3.1 мл 5N НСl добавляют по каплям 2.38 мл 2.5М раствора NaNO2, поддерживая температуру смеси <5°С. Смесь перемешивают еще 30 мин при заданной температуре, после чего полученный раствор прикалывают к суспензии 4.03 г (17.9 ммоль) SnCl2*2H2O в 4.2 мл соляной кислоты при 0°С. Через 2 ч перемешивания при заданной температуре осадок отфильтровывают, растворяют в 40 мл воды и добавляют NaOH до сильнощелочной реакции. Смесь экстрагируют эфиром (3*100 мл), сушат над MgSO4 и упаривают в вакууме. Получают 4-гидразино-N-метил-2-фторбензамид 7. LC-MS (M+H)+ 184.1Н ЯМР (CDCl3, 400 МГц): 7.96 (т, J=8.4 Гц, 1Н), 6.64 (уш. м, 1 Н), 6.60 (т, J=1.6 Гц, 1 Н), 6.57 (дд, J1=7.2 Гц, J2=2.0 Гц, 1 Н), 5.60 (уш. с, 1H), 3.66 (yш. c, 2H),3.00(дд, J1=4.8 Гц, J2=1.2 Гц, 1Н). К раствору 54 мг (0.29 ммоль) 4-гидразино-N-метил-2-фторбензамида 7 в 3 мл диоксана прибавляют раствор 59 мг (0.27 ммоль) 4-изоцианато-2-(трифторметил)бензонитрила в 2 мл диоксана. Через 2 ч перемешивания диоксан отгоняют в вакууме, остаток растирают в эфире, отфильтровывают и сушат в вакууме. Получают 2-[(4-метилкарбамоил)-3-фторфенил]-N-[3-(трифторметил)-4-цианофенил]-гидразинкарбоксамид 8(1). LC-MS (М+Н)+ 405. 'Н ЯМР (ДМСО-d6, 400 МГц): 9.65 (уш. с, 1 Н), 8.72 (уш. с, 1 Н), 8.37 (с, 1 Н), 8.25 (уш. с, 1 Н), 8.03 (уш. м, 1 Н), 7.88 (д, J=8.8 Гц, 1 Н), 7.58 (м, 2Н), 6.63 (д, J=8.4 Гц. 1 Н), 6.48 (д, J=14.0 Гц, 1Н), 2.77 (д, J=4.4 Гц, 3Н). К 80 мг (0.2 ммоль) 2-[(4-метилкарбамоил)-3-фторфенил]-N-[3-(трифторметил)-4-цианофенил]-гидразинкарбоксамида 8.1 в 2 мл дихлорэтана добавляют 56 мкл (0.4 ммоль) триэтиламина и затем добавляют 27 мкл (0.22 ммоль) дифосгена. Реакционную массу перемешивают в закрытой пробирке при 80°С 15 ч. Растворитель отгоняют в вакууме и остаток хроматографируют на SiO2 (элюент -СН2Сl2: МеОН, градиент от 100:1 до 20:1). Получают N-метил-2-фтор-4-[4-[3-(трифторметил)-4-цианофенил]-3,5-диоксо-1,2,4-триазолидин-1-ил]бензамид 1.4(1). LC-MS (M+H)+422.1Н ЯМР(ДМСО-d6, 400 МГц): 11.53 (с, 1Н), 8.22 (уш. м, 1Н), 8.16 (д, J=8.8 Гц, 1 Н), 7.99 (дд, J1=8.8 Гц, J2=1.6 Гц, 1 Н). 7.95 (д, J=1.6 Гц, 1 Н), 7.81 (т, J=8.4 Гц, 1Н), 7.69 (дд, J1=8.8 Гц. J2=2.0 Гц, 1Н), 7.62 (дд, J1=12.0 Гц, J2=2.0 Гц, 1Н), 2.79 (д,,7=4.4 Гц, 3Н).
Пример 10. Определение антагонистической активности циклических N,N'-диарилмочевин общей формулы 1 и известного аналога MDV3100 по отношению к андрогенным рецепторам. Способность новых циклических N,N'-диарилмочевин общей формулы 1 и известного препарата MDV3100 блокировать андрогенные рецепторы определяли по их эффективности ингбирования стимулированной дегидротестостероном экспрессии специфического для простаты антигена (ПСА) в канцерных клетках простаты человека LNCap, полученных из Американского банка тканевых культур (АТСС, США). Эти клетки чувствительны к 5-а-дигидротестостерону (ДГТ) и продуцируют канцерный маркер (ПСА) в его присутствии. Клетки выращивались в RPMI 1640 среде (Invitrogen, США), содержащей 10% телячью сыворотку (Hyclone, США), 1% антибактериальную/антигрибковую смесь (Sigma, США) и 4,5% глюкозу. Перед экспериментами клетки отмывали и суспендировали в ту же среду, но где телячья сыворотка была заменена на сыворотку, обработанную для удаления следов гормонов активированным углем. Клетки разливали по 100 мкл (10000 клеток) в ячейки 96-луночных плашек и оставляли на 4 суток в инкубаторе при 37°С (100% влажности) в атмосфере 95% воздух/5% СO2. После 4-дневной инкубации к клеткам добавлялись циклические N,N'-диарилмочевины общей формулы 1 при различных концентрациях и затем добавлялся 20 нМ ДГТ (концентрация, соответствующая 80-90% от максимальной стимуляции). Клетки оставлялись на 5 суток для дополнительной инкубации в тех же условиях. После инкубации образцы надклеточной среды отбирались для анализа на содержание ПСА. Анализ проводили по протоколу, рекомендованному производителем набора для определения ПСА (Alpha Diagnostic International, США). После увлажнения лунок, содержащих на донышке прикрепленные антитела против ПСА, к ним добавляли по 25 мкл образцов и затем по 100 мкл антител также против ПСА, к которым конъюгирована пероксидаза хрена. После 30-минутной инкубации при комнатной температуре, содержимое лунок удаляли, лунки несколько раз промывали и в лунки заливали по 100 мкл хромогенного субстрата пероксидазы. Плашки выдерживались 15 мин при комнатной температуре, в лунки добавляли по 50 мкл стоп-раствора и развившуюся окраску измеряли по поглощению при 450 нм. Количество образовавшейся окраски пропорционально концентрации ПСА в образце. На основании полученных результатов зависимости снижения вызванной дигидротестостероном (ДГТ) наработки ПСА в присутствии разных концентраций веществ строились кривые доза-ответ, из которых определяли величины IC50, которые были использованы для определения величин кажущихся констант ингибирования (Кi) для этих испытуемых соединений общей формулы I в соответствии с уравнением Ченг-Пруссова [Cheng, Y., Prusoff, W.H. "Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (150) of an enzymatic reaction". Biochem Pharmacol. (1973) 22, 3099-3108]:
Ki=IC50/(1+L/KD),
где L это концентрация агониста (ДГТ) и КD - это константа активации рецептора, численно равная величине ЕС50, определяемой параллельно в каждом эксперименте по зависимости стимуляции синтеза ПСА от концентрации ДГТ.
Полученные результаты, представленые в соответствующих примерах, свидетельствуют о том, что новые антагонисты андрогеновых рецепторов по данному изобретению в ряде случаев более активны, чем MDV3100, испытанный в этих же условиях, как свидетель, для которого КiMDV3100 =79.5 nM.
Пример 11. Определение максимально переносимой дозы нового антагониста 1.2.2(1) и его известного аналога MDV3100. Максимально переносимые дозы (МТД) нового антагониста 1.2.2(1) и его известного аналога MDV3100 определялись в экспериментах на самцах мышей линии CD1 при пероральном введении 1 раз в сутки в течение 5 дней в дозах 10, 30 и 100 мг/кг. Вещество растворяли в стерильной воде с добавлением Твин-80. Контрольным животным (группа Плацебо) вводили стерильную воду с Твин-80. Учитывали вес тела, а также смертность животных. Статистическое сравнение групп проводили по непараметрическому критерию ANOVA, с использованием программы Statistica.
При введении соединения 1.2.2(1) в дозах до 100 мг/кг гибель мышей не наблюдалась. На 3-й -4-й дни в группе мышей, получавших изучаемое вещество в дозе 100 мг/кг, масса тела была меньше по сравнению с контрольным животным, однако статистической значимости при этом не наблюдалось (фиг.1). Эти данные свидетельствуют о том, что соединение 1.2.2(1) имеет МТД>100 мг/кг.
При введении соединения MDV3100 в дозах 10 и 30 мг/кг гибели мышей не наблюдалось. В группе мышей, которым изучаемое вещество вводили в дозе 100 мг/кг, на 3-й день масса тела начала снижаться. На 5-й день масса тела у животных из этой группы статистически значимо отличалась от массы тела животных из группы Плацебо (р=0,002, фиг.2). Погибло одно животное. Эти данные свидетельствуют о том, что соединение MDV3100 имеет МТД ~ 30 мг/кг.
Пример 12. Получение лекарственного средства в форме таблеток. Смешивают 1600 мг крахмала, 1600 мг измельченной лактозы, 400 мг талька и 1000 мг N-метил-4-[5-метил-5-метилоксиметил-3-(3-трифторметил-4-цианофенил)-4-оксо-2-тиоксо-имидазолидин-1-ил]-2-фтор-бензамид 1.2.2(1). Полученную фармацевтическую композицию спрессовывают в брусок. Полученный брусок измельчают в гранулы и просеивают через сита, собирая гранулы размером 14-16 меш. Полученные гранулы таблетируют в подходящую форму таблетки весом 560 мг каждая.
Пример 13. Получение лекарственного средства в форме капсул. Тщательно смешивают N-метил-4-[5-метил-5-метилоксиметил-3-(3-трифторметил-4-цианофенил)-4-оксо-2-тиоксо-имидазолидин-1-ил]- 2-фтор-бензамид 1.2.2(1) с порошком лактозы в соотношении 2:1. Полученную фармацевтическую композицию упаковывают по 300 мг в желатиновые капсулы подходящего размера.
Пример 16. Получение лекарственного средства в форме инъекционных композиций для внутримышечных, внутрибрюшинных или подкожных инъекций. Смешивают 500 мг N-метил-4-[5-метил-5-метилоксиметил-3-(3-трифторметил-4-цианофенил)-4-оксо-2-тиоксо-имидазолидин-1-ил]-2-фтор-бензамид 1.2.2(1) с 300 мг хлорбутанола, 2 мл пропиленгликоля и 100 мл инъекционной воды. Полученный раствор фильтруют и помещают по 1 мл в ампулы, которые запаивают.
Реферат
Настоящее изобретение относится к новым циклическим N,N'-диарилтиомочевинам или N,N'-диарилмочевинам общей формулы (1), их оптическим (R)- и (S)-изомерам и их фармацевтически приемлемым солям - антагонистам андрогенных рецепторов. Изобретение также относится к противораковому средству, фармацевтической композиции, лекарственному средству и способу лечения рака простаты с использованием соединений изобретения. В формуле (1) ! ! где Х представляет собой атом кислорода или серы; m=0 или 1; R1 представляет собой C1-С3алкил; R2 и R3 представляют собой атом водорода; или R2 и R3 вместе с атомом углерода, с которым они связаны, образуют группу С=O; или представляет собой группу NH; R4 и R5 представляют собой атом водорода; или R4 представляет собой атом водорода, а R5 представляет собой метил; или R4 представляет собой атом водорода, метил, а R5 представляет собой группу Zn-Y-R6, в которой n=1 или 2, Z представляет собой CH2, или С=O и Y-атом кислорода, или N-СН3, или Y представляет собой С=О, а Z представляет собой CH2; R6 представляет собой атом водорода, метил, бензил, гидроксигруппу или R5 и R4 вместе с атомами, с которыми они связаны, образуют пяти- или шестичленный гетероцикл, включающий по крайней мере атом кислорода или азота, который может быть замещен метилом. Изобретение также относится к способу получения соединений. 8 н. и 4 з.п. ф-лы, 2 ил.
Формула
где Х представляет собой атом кислорода или серы;
m=0 или 1;
R1 представляет собой C1-С3алкил;
R2 и R3 представляют собой атом водорода;
или R2 и R3 вместе с атомом углерода, с которым они связаны, образуют группу С=O;
или
R4 и R5 представляют собой атом водорода;
или R4 представляет собой атом водорода, а R5 представляет собой метил;
или R4 представляет собой атом водорода, метил, а R5 представляет собой группу Zn-Y-R6, в которой n=1 или 2, Z представляет собой СН2; или С=O и Y-атом кислорода или N-СН3, или Y представляет собой С=O, а Z представляет собой СН2;
R6 представляет собой атом водорода, метил, бензил, гидроксигруппу или
R5 и R4 вместе с атомами, с которыми они связаны, образуют пяти или шестичленный гетероцикл, включающий по крайней мере атом кислорода или азота, который может быть замещен метилом.
где X, R1, R2, R3, R4 и R5 имеют вышеуказанное в п.1 значение.




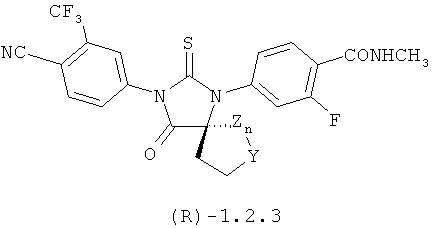


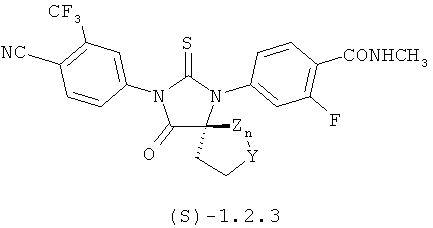
где Y, Z и R6 имеют вышеуказанное в п.1 значение.


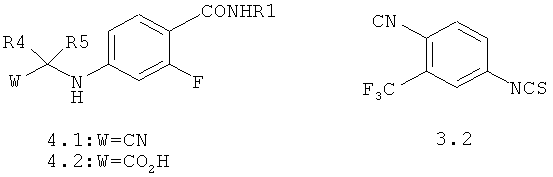
Документы, цитированные в отчёте о поиске
Замещенные фенильные производные, их получение и применение
Комментарии