Небиволол и его фармацевтически приемлемые соли, способ их получения и фармацевтические композиции небиволола - RU2378272C2
Код документа: RU2378272C2
Чертежи






Описание
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение касается улучшенного способа синтеза фармацевтически активного производного 2,2'-иминобисэтанола, то есть 2Н-1-бензопиран-2-метанол-α,α'-иминобис-(метилен)бис[6-фтор-3,4-дигидро-[2R*[R*[R*(S*)]]]], то есть небиволола, или его фармацевтически приемлемых солей, точнее гидрохлоридной соли (I)
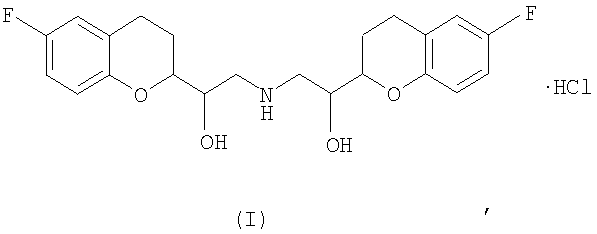
Небиволол применяют для лечения и предотвращения нарушений венечных сосудов.
Данное изобретение также касается фармацевтических композиций и способа получения твердой пероральной дозированной формы небиволола гидрохлорида формулы (I), без применения увлажняющего агента, и необязательно с применением связывающего и/или дезинтегрирующего агента.
Данное изобретение, к тому же, представляет новую полиморфную форму небиволола гидрохлорида.
ПРЕДПОСЫЛКИ ДАННОГО ИЗОБРЕТЕНИЯ
Для лечения высокого кровяного давления, борьбы с ангиной, аритмией, пост-миокардиальной инфекцией, сердечной недостаточностью, мигренью или эссенциальным дрожанием применяют бета-блокаторы.
Небиволол представляет собой высокоселективный бета-1 блокатор, который, как выяснилось, пригоден для контролирования гипертензии. Гипертензия (высокое кровяное давление) представляет собой существенный риск для здоровья, затрагивает более чем 500 миллионов человек во всем мире и требует долговременной терапии для ее контролирования.
Химически небиволол представляет собой производное 2,2'-иминобисэтанола, то есть 2Н-1-бензопиран-2-метанол-α,α'-[иминобис-(метилен)]бис[6-фтор-3,4-дигидро-[2R*[R*[R*(S*)]]]]. В европейском патенте ЕР 744946 В1 раскрывается небиволол, являющийся смесью равных количеств 2 энантиомеров, имеющих соответственно SRRR- и RSSS-конфигурацию.
Способы получения небиволола раскрыты в европейских патентах ЕР 0145067 и ЕР 0334429. Европейский патент ЕР 0145067 В1 описывает способ превращения 6-фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксальдегида (VI) в смеси изомеров 6-фтор-3,4-дигидро-2-оксиранил-2Н-1-бензопирана (VII). Эта реакция влечет за собой использование гидрида натрия в качестве основания, которое является чрезвычайно опасным. Кроме того, чистота продукта, то есть 6-фтор-3,4-дигидро-2-оксиранил-2Н-1-бензопирана (VII), полученного с применением гидрида натрия, является низкой и, как выяснили, составляет приблизительно 62-65%, что недопустимо. Смесь оксиранов, представленных формулой (VII), разделяют колоночной хроматографией, как показано на Схеме (I), для получения А-изомера (VII) (то есть VII-A) из первой фракции и В-изомера (VII) (то есть VII-B) из второй фракции.

А-изомер (VII) затем обрабатывают бензиламином для получения бензилированного А-изомера (VII) (то есть промежуточного соединения I), который реагирует с В-изомером в присутствии щавелевой кислоты для получения оксалатной соли бензилированного небиволола (VIII-a), как показано на Схеме II.
Кроме того, оксалатную соль (VIII-a), как выяснили, нужно обработать щелочью для получения свободного основания бензилированного небиволола (VIII).

Таким образом, как видно из Схемы II, реакция между промежуточным соединением I и (VII-B) включает очистку колоночной хроматографией и последующую обработку щавелевой кислотой для образования оксалатной соли. Обработка щавелевой кислотой приводит к разделению нежелательных (RSRR+SRSS) диастереомеров от желательных (RSSS+SRRR) изомеров, вследствие различия в растворимостях желательных и нежелательных изомеров. Кроме того, соль щавелевой кислоты необходимо превращать в свободное основание бензилированного небиволола обработкой щелочью. Множество этапов в описанном способе делают его громоздким, что приводит к увеличению энергозатрат и времени цикла получения активного фармацевтического ингредиента. Кроме того, чистота свободного основания бензилированного небиволола, как выяснили, является относительно низкой.
Европейский патент ЕР 334429 В1 раскрывает независимый способ получения определенного RSSS изомера небиволола. Упомянутый способ независимого получения RSSS небиволола включает применение опасных реактивов, таких как хлорид тионила, гидрид натрия и диизобутил алюминия гидрид (ДИБАГ), дорогих оптически активных реагентов, таких как (+)-1,2,3,4,4а,9,10,10а-октагидро-1,4а-диметил-7-(1-метилэтил-1-фенатренметанамин-[(+)-дегидроабиэтиламин], и применения таких условий, как колоночная хроматография и низкие температуры. Упомянутые способы также включают большое количество этапов, таким образом, повышая энергозатраты, трудовые ресурсы и время, необходимое для полного проведения цикла, что делает способ коммерчески дорогим.
Кроме того, европейский патент ЕР 744946 В1 раскрывает способ получения небиволола гидрохлорида из смеси, содержащей желательное (RSSS+SRRR) основание небиволола, загрязненное нежелательными (RSRR+SRSS) диастереомерами, с применением этанола в качестве реагента и в качестве растворителя перекристаллизации. Главный недостаток указанного способа заключается в том, что в качестве стартового материала применяют основание небиволола с примесями различных изомеров, что, таким образом, приводит к очень низкому выходу (6,6%) желательных изомеров (обладающих SRRR- и RSSS-конфигурацией) небиволола гидрохлорида. Кроме того, этанол является растворителем, который можно использовать только в контролируемых количествах из-за строгих регулирующих требований, и поэтому его применение в промышленном масштабе ограничено.
Таким образом, существует потребность развития способа синтеза смеси желательных диастереомеров более высокой чистоты с сокращением нежелательных изомерных примесей и исключения применения опасного гидрида натрия, одновременно уменьшая этапы способа синтеза небиволола.
Обнаружено, что способ синтезирования небиволола по данному изобретению не только повышает чистоту смеси желательных диастереомеров при минимальном числе этапов способа, но также исключает применение опасных химикатов в способе.
Небиволол в основном применяется для лечения и предотвращения нарушений венечных сосудов. Его принимают один или два раза в день в зависимости от потребностей пациента.
В европейском патенте ЕР 0744946 раскрыты фармацевтические композиции небиволола, где лекарство представляет собой микронизированную форму с добавлением одного или более увлажняющих агентов, в частности, полисорбатов в качестве адъювантов. В этом патенте придается особое значение необходимости микронизации и последующего увлажнения, поскольку пероральному введению небиволола гидрохлорида препятствует низкая растворимость, если он находится в нормальной кристаллической форме. Для достижения хорошей растворимости активный ингредиент должен быть достаточно увлажнен.
Микронизация небиволола гидрохлорида, необходимая в ЕР 0744946 В1 для получения фармацевтических композиций, неоправданно увеличивает время цикла производственного процесса. Также возникают чрезмерные энергозатраты, например, на размол и просеивание, что увеличивает стоимость получения конечного продукта. Кроме того, как показало сравнение растворимости таблеток, включающих кристаллический небиволол и микронизированный небиволол, скорость растворения таблетки, включающей кристаллический небиволол (Пример 6 ЕР 744946), составляет менее 50% за 45 минут.
Европейский патент ЕР 0145067 В1 представляет фармацевтическую композицию производных 2,2'-иминобисэтанола для различных дозированных форм, то есть пероральных капель, инъекционного раствора, перорального раствора, таблеток с пленочным покрытием и т.п. В европейском патенте ЕР 0145067 В1 раскрыто применение додецил сульфата натрия при получении таблетки с пленочным покрытием. Додецил сульфат натрия, то есть лаурил сульфат натрия (Wade, A. and Weller P.J., Handbook of Pharmaceutical Excipients, 2nd ed., 1994, page 448), является увлажняющим агентом в ядре композиции (таблетки с пленочным покрытием).
В международной заявке WO 2002/087508 раскрыт нитрозированный и нитрозилированный небиволол, его метаболиты и фармацевтическая композиция с применением тех же компонентов. Показано, что биодоступность композиции можно увеличить микронизацией формуляции, с помощью обычных методик, таких как размол, помол, сушка при разбрызгивании и т.п., в присутствии приемлемых наполнителей или агентов, таких как фосфолипиды или сурфактанты.
Таким образом, упомянутая литература показывает, что попытки использовать природную кристаллическую форму небиволола приводят к низкой скорости растворения и низкой биодоступности. Попытки комбинации кристаллической формы с увлажняющим агентом также в основном неудачны. Для достижения соответствующей скорости растворения или биодоступности небиволола гидрохлорида необходим микронизированный небиволол. Процесс микронизации является дорогостоящим, занимает время и требует применения увлажняющего агента.
Увлажняющий агент является сурфактантом, веществом, способным снижать поверхностное натяжение жидкости, в которой он растворен. Действие сурфактанта через оболочку кишечника является более сложным. Показано, что большинство сурфактантов взаимодействует с абсорбирующими мембранами (Bermejo, D.M. and Ruiz-Garcia, A., Business Briefing: Pharmatech 2003; pages 1-7). Усиление проницаемости и локальное повреждение - тесно связанные осложнения взаимодействия сурфактантов со стенкой кишечника (Swenson, E.S., Milisen, W.B., Curatolo, W., Pharm. Res. 1994 Aug; 11(8), pages 1132-42). Введенные сурфактанты могут облегчить проницаемость или абсорбцию потенциально токсичных или патогенных соединений, которые, в свою очередь, могут привести к неблагоприятным воздействиям на другие органы (Lieberman, Н.А., Rieger, M.M. and Banker, G.S., Eds., Pharmaceutical Dosage Forms: Disperse Systems, 2nd ed, Vol.1, page 261). Сурфактант может облегчить их проникновение и проникновение в организм другого вещества, которое таким образом входит в системную циркуляцию (Lieberman, H.A., Rieger, M.M. and Banker, G.S., Eds., Pharmaceutical Dosage Forms: Disperse Systems, 2nd ed., Vol.1, page 264).
Полисорбат 60 или 80 влияет на целостность слизистой оболочки кишечника (Lieberman, H.A., Rieger, M.M. and Banker, G.S., Eds., Pharmaceutical Dosage Forms: Disperse Systems, 2nd ed., Vol.1, page 261). Полисорбат 80 может усилить абсорбцию жирорастворимых веществ (www.lactose.co.uk/milkallergy/foodadditives400.html).
Лечение гипертензии представляет собой длительную терапию, поэтому применение увлажающих агентов в формуляции небиволола необходимо сознательно избегать. Большие дозы полисорбата 80 как увлажняющего агента в фармацевтических формуляциях вызывают абдоминальные спазмы, диарею (http://www.jtbker.com/msds/englishhtml/t7683.htm). Кроме того, установили, что полисорбат 80, содержащийся в фармацевтических композициях, вызывал аллергию у различных пациентов (http://www.hci.utah.edu/patientdocs/ hci/drugd/docetaxel.htm). Также следует избегать использования полисорбата 20 и полисорбата 40 в формуляциях, поскольку они запрещены в некоторых странах (http://www. lactose.co.uk/milkallergy/foodadditives400.html).
Таким образом, существует потребность в композициях небиволола, которые являются безопасными, эффективными и, в то же время, с эффективными для производства показателями затрат и времени.
Авторы данного изобретения обнаружили, что фармацевтические композиции по данному изобретению, приготовленные с применением небиволола в качестве активного ингредиента и без применения увлажняющего агента, показали превосходные характеристики растворимости, которые, как также было выяснено, сопоставимы с формуляцией, имеющейся в продаже.
Кроме того, неожиданно выяснили, что небиволола гидрохлорид можно разработать не только без применения увлажняющего агента, но необязательно без применения связывающего или дезинтегрирующего агента, без замедления характеристик растворения лекарства.
ЦЕЛИ ДАННОГО ИЗОБРЕТЕНИЯ
Таким образом, целью данного изобретения является обеспечение улучшенного способа синтеза фармацевтически активного производного 2,2'иминобисэтанола, то есть 2Н-1-бензопиран-2-метанол-α,α'-иминобис-(метилен)бис[6-фтор-3,4-дигидро-[2R*[R*[R*(S*)]]]], то есть основания небиволола или его гидрохлоридной соли формулы (I) выше.
Следующей целью данного изобретения является обеспечение простого и экономичного способа синтеза основания небиволола или его гидрохлоридной соли, который включает минамальное количество этапов и при котором не применяются опасные химикаты.
Еще одной целью данного изобретения является разработка фармацевтических композиций, включающих активный ингредиент небиволола гидрохлорида и один или более адъювантов, без включения увлажняющего агента для достижения необходимого профиля растворимости.
Также цель данного изобретения - разработать способ получения твердой дозированной формы, такой как таблетки или капсулы простым и экономичным способом.
Следующая цель данного изобретения заключается в получении твердой дозированной формы небиволола гидрохлорида и одного или более адъювантов, без применения увлажняющего агента, необязательно без применения связывающего агента и необязательно без применения дезинтегрирующего агента.
Другой целью данного изобретения является обеспечение новой полиморфной формы небиволола и его фармацевтически приемлемых солей, в частности гидрохлоридной соли, и способа ее получения.
Следующая цель данного изобретения состоит в обеспечении применения фармацевтической композиции небиволола гидрохлорида по данному изобретению, приготовленного без применения увлажняющего агента, для лечения гипертензии.
КРАТКОЕ ОПИСАНИЕ ДАННОГО ИЗОБРЕТЕНИЯ
Таким образом, согласно первому аспекту данного изобретения предусматривается улучшенный способ синтеза фармацевтически активного производного 2,2'иминобисэтанола, то есть 2Н-1-бензопиран-2-метанол-α,α'-иминобис-(метилен)бис[6-фтор-3,4-дигидро-[2R*[R*[R*(S*)]]]], то есть небиволола или его галогенводорода, точнее гидрохлоридной соли небиволола, как показано на Фигуре (I),
где
- 6-фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксальдегид (VI) превращается в смесь изомеров оксиранов, представленных формулой (VII), в присутствии третичного бутоксида калия в качестве основания, и
- реакцию между бензилированным А-изомером (VII) (то есть, промежуточным соединением I) и В-изомером (VII-B) и выделение продукта реакции выполняют в присутствии органического растворителя при контролированных временных и температурных условиях, что приводит к образованию свободного основания бензилированного небиволола (IX).
Согласно второму аспекту данного изобретения предусматривается небиволол, синтезированный способом по данному изобретению.
Согласно следующему аспекту данного изобретения, обеспечиваются фармацевтические композиции небиволола, включающие небиволол или его фармацевтически приемлемые соли, образованные без применения увлажняющих агентов.
Кроме того, данное изобретение также представляет твердую дозированную форму небиволола гидрохлорида и одного или нескольких адъювантов без применения увлажняющего агента, необязательно без применения связывающего агента и/или дезинтегрирующего агента и способ ее получения.
Согласно следующему аспекту данного изобретения предусматривается способ получения фармацевтической композиции по данному изобретению.
Еще один аспект данного изобретения представляет новую полиморфную форму небиволола и его фармацевтически приемлемых солей.
В следующем варианте осуществления данного изобретения предпочтительная удельная площадь поверхности небиволола гидрохлорида составляет от 0,2×103 м2/кг до 1,95×103 м2/кг.
ДЕТАЛЬНОЕ ОПИСАНИЕ ДАННОГО ИЗОБРЕТЕНИЯ
Таким образом, данное изобретение представляет улучшенный способ получения 2Н-1-бензопиран-2-метанол-α,α'-иминобис-(метилен)бис[6-фтор-3,4-дигидро-[2R*[R*[R*(S*)]]]], то есть небиволола формулы (IX), или его фармацевтически приемлемых солей, таких как гидрохлоридная соль, как показано в формуле (I)

Согласно данному изобретению небиволол получают синтезом, показанным на Схеме III:
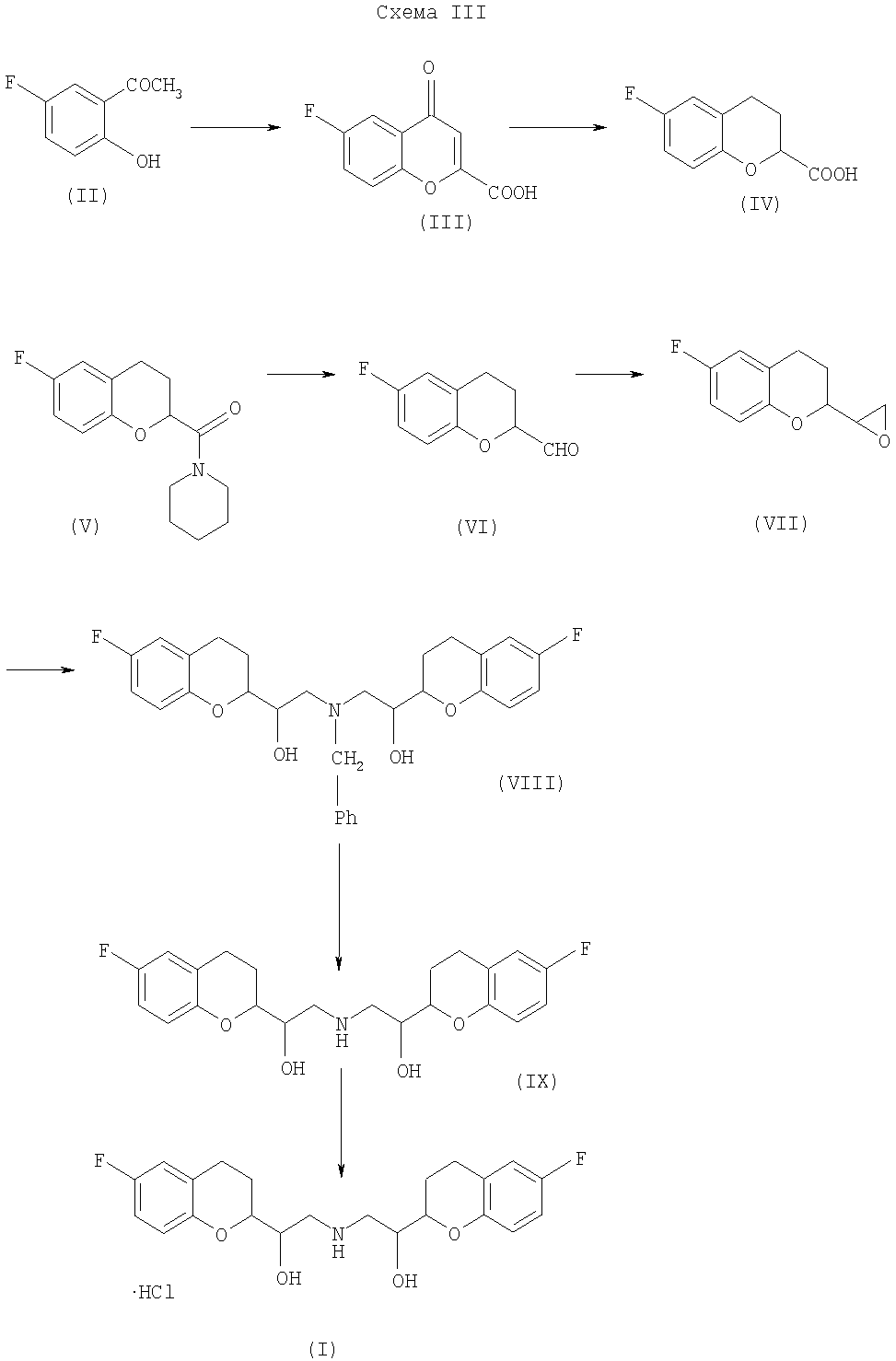
Этапы реакции для получения небиволола по данному изобретению, начиная от соединения (II), согласно вышеприведенной Схеме III, теперь описываются ниже.
5-Фтор-2-гидроксиацетофенон (II) обработали третичным бутоксидом калия и диэтил оксалатом для получения 6-фтор-4-оксо-4Н-1-бензопиран-2-карбоксильной кислоты (III) согласно Реакции 1, как приведено ниже, известным способом (патент Японии №2218675).

Проводят реакцию 6-фтор-4-оксо-4Н-1-бензопиран-2-карбоксильной кислоты (III) с 10% палладием на угле в уксусной кислоте, в качестве растворителя, в атмосфере водорода для получения 6-фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксильной кислоты (IV), как показано в Реакции 2, приведенной ниже, известным способом (патент США №4654362).

6-Фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксильную кислоту (IV) превращают в амид (V-A), используя амин RR'NH и кислотно-активирующий агент, как показано в Реакции 3(а),

где R и R' - независимо Н, алкил или фенил, необязательно связанный с гетероатомом, таким как О, N или S или не связанный с гетероатомом. Алкил представляет собой C1-С6 алкил с прямой или разветвленной цепью.
В Реакции 3(а) образования амида (V-A) из кислоты (IV) амин RR'NH может быть выбран из группы первичных или вторичных аминов. Первичный амин выбирают из алифатического или ароматического первичного амина, тогда как вторичный амин выбирают из циклических вторичных аминов или нециклических вторичных аминов, где заместитель на атоме азота является независимо алифатическим или ароматическим или их комбинацией.
Предпочтительно могут быть использованы вторичные амины. Более предпочтительно используются вторичные амины, являющиеся циклическими.
Амины могут быть выбраны из группы, включающей диметиламин, диэтиламин, N-метилфениламин, пирролидин, пиперидин, N,O-диметилгидроксиламин и морфолин. Предпочтительным амином является пиперидин.
Активирующий агент, согласно Реакции 3 (а), выбирают из группы, включающей тионил хлорид, этилхлорформиат, уксусный ангидрид, фосфора трихлорид, фосфора оксихлорид, фосфора пентахлорид, дициклогексилкарбодиимид, N,N'-карбонилдиимидазол и хлорсахарин. Предпочтительными активирующими агентами являются тионил хлорид и этилхлорформиат.
В предпочтительном варианте осуществления 6-фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксильную кислоту (IV) превращают в 6-фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксильной кислоты пиперидин амид (V) с применением тионил хлорида и пиперидина в толуоле в качестве растворителя, как показано в нижеприведенной Реакции 3.

Восстановление амида (V-A) для получения 6-фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксальдегида формулы (VI) выполняют с применением алкокси-металлогидрида, где алкокси группа является замещенным или незамещенным -O(С1-С4)алкилом, и замещение, если оно есть, - -O(C1-С4)алкилом, а металлическая часть содержит металл, выбранный из группы, включающей Li или Na.
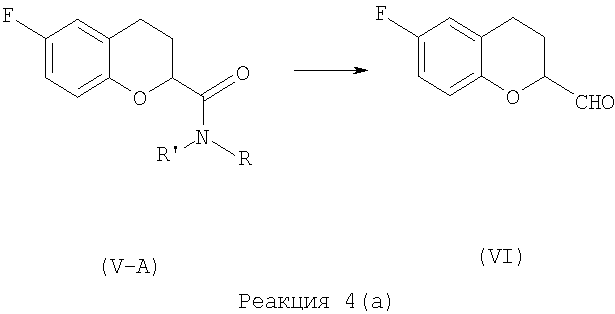
Восстановительный алкокси металлогидридный агент выбирают из группы, включающей натрия бис-(2-метоксиэтокси)алюминия гидрид, лития диэтоксиалюминия дигидрид и лития три-трет-бутокси алюминия гидрид.
Предпочтительным восстановительным агентом является натрия бис-(2-метоксиэтокси)алюминия гидрид.
6-Фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксильной кислоты пиперидин амид (V) восстанавливают с помощью Vitride®, то есть натрия бис-(2-метоксиэтокси)алюминия гидрида, в толуоле для получения 6-фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксальдегида (VI), как показано в Реакции 4.

Проводят реакцию 6-фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксальдегида (VI) с триметил сульфоксониумом йодидом в присутствии основания, третичного бутоксида калия и растворенного диметил сульфоксида для получения смеси изомеров оксиранов, представленных формулой (VII), которые могут быть необязательно дистиллированы, как показано в Реакции 5.

Таким образом, данное изобретение в приведенной выше реакции избегает применения гидрида натрия в качестве основания. Вследствие указанной замены вышеупомянутого основания, чистота продукта реакции, то есть оксирана (VII), является значительно более высокой. Чистота оксирана, полученного с применением третичного бутоксида калия в качестве основания, составляет около 75% до дистилляции продукта, для сравнения чистота продукта до дистилляции, полученного в исполнении по данному изобретению, с использованием основания гидрида натрия по методике, описанной в европейском патенте ЕР 0145067 В1, составила 60-65%. Преимущество способа по данному изобретению состоит в том, что избегается применение опасного гидрида натрия, а также повышается чистота оксирана.
Смесь оксиранов разделили колоночной хроматографией элюированием сначала чистой фракции (А-изомер, то есть VII-A), а именно (А)-6-фтор-3,4-дигидро-2-оксиранил-2Н-1-бензопирана по европейскому патенту ЕР 0145067. А затем элюированием второй фракции (В-изомер, то есть VII-В) получили (В)-6-фтор-3,4-дигидро-2-оксиранил-2Н-1-бензопиран по европейскому патенту ЕР 0145067.
Хроматографическое разделение двух изомеров А и В выполняют (ЕР 0145067) (Схема I) с применением силикагеля в качестве стационарной фазы, тогда как элюантом является смесь гексанов и этилацетата. Если имеется большое количество материала для очистки, то очистку колоночной хроматографией можно выполнять одновременно на нескольких колонках (более чем одной колонке). Колоночная хроматография сначала дает изомер А, затем изомер В. Исходя из чистоты фракций из колонки, их можно смешать вместе и использовать в дальнейшей реакции.

Проводят реакцию (А)-6-фтор-3,4-дигидро-2-оксиранил-2Н-1-бензопирана (VII-A) с бензиламином в изопропиловом спирте для получения (А)-6-фтор-3,4-дигидро-альфа-[[(фенилметил)амино]-метил]-2Н-1-бензопиран-2-метанола (промежуточное соединение I) известным способом (патент США №4654362), как показано в Реакции 6.

Проводят реакцию (А)-6-фтордигидро-α-[[(фенилметил)амино]-метил]-2Н-1-бензопиран-2-метанола (промежуточное соединение I) с (В)-6-фтор-3,4-дигидро-2-оксиранил-2Н-1-бензопираном (VII-B) в присутствии органического растворителя и выделяют при температуре от -5 до -25°С, выдерживая реакционную массу при температуре выделения более чем 2 часа, для получения бензилированного небиволола, как показано в Реакции 7.

В данном изобретение реакцию между бензилированным А-изомером (VII) (т.е. промежуточным соединением I) и В-изомером (VII-B) выполняют в органическом растворителе и выделяют при температуре от -5 до -25°С, что ведет непосредственно к образованию свободного основания бензилированного небиволола (IX), содержащего только желательные изомеры (обладающие SRRR- и RSSS-конфигурацией). В данном изобретении наблюдается, что выделение, если оно выполняется при низкой температуре, в частности при температуре от -5 до -25°С, дает в основном чистые желательные стереоизомеры (чистота ВЭЖХ (высокоэффективной жидкостной хроматографии) >90,0%), которые в дальнейшем можно необязательно очистить в спиртовом растворителе, таком как метанол, этанол, для получения желательного стереоизомера в форме с высокой степенью чистоты (чистота ВЭЖХ >98,5%), для сравнения, в способе, описанном в предыдущем уровне техники, получали желательные изомеры оксалатной соли бензилированного небиволола с гораздо меньшей чистотой (чистота ВЭЖХ <60,0%) даже после повторной очистки.
Органический растворитель, используемый в Реакции 7, выбирают из группы, включающей спирты, эфиры, кетоны и ацетонитрил.
В одном варианте осуществления данного изобретения спирт выбран из группы, включающей метанол, этанол, изопропиловый спирт, n-пропиловый спирт, n-бутанол, изобутанол. В предпочтительном варианте осуществления используют метанол. В другом варианте осуществления эфиры выбраны из группы, включающей этилацетат, n-бутилацетат. В следующем варианте осуществления кетоны выбраны из группы, включающей ацетон, метилэтилкетон, метилизобутилкетон (МИБК).
Выделение выполняют при диапазоне температур от -5 до -25°С, предпочтительно при диапазоне от -10 до -20°С, и более предпочтительно при диапазоне от -10 до -15°С.
Реакционную массу выдерживали при температуре выделения в течение 2-40 часов, предпочтительно в течение 4-30 часов, более предпочтительно в течение 8-20 часов и наиболее предпочтительно в течение 10-15 часов.
Таким образом, способ по данному изобретению обладает следующими преимуществами:
(i) избегает применения колоночной хроматографии,
(ii) избегает получения оксалатной соли и этапа ее перекристаллизации и
(iii) избегает последующего превращения оксалатной соли в свободное бензилированное основание небиволола путем обработки щелочью.
Следовательно, по сравнению со способами, известными из уровня техники, способ по данному изобретению ведет к снижению числа этапов способа, тем самым делая его более экономичным и эффективным по времени.
Далее реакцию гидрогенизации выполняют с применением 10% палладия на угле для дебензилирования бензилированного основания небиволола (VIII) до получения основания небиволола (IX) известным способом (патент США №4654362), как показано в Реакции 8.

Небиволол можно превратить в формы его фармацевтически приемлемых кислотно-аддитивных солей обработкой соответствующими кислотами. Соответствующими кислотами являются, например, неорганические кислоты, такие как галогеноводородная кислота, например, хлороводородная, бромоводородная, серная кислота, азотная кислота, фосфорная кислота; или органические кислоты, например, уксусная, пропановая, гидроксиуксусная, 2-гидроксипропановая, 2-оксопропановая, этандионовая, пропандионовая, бутандионовая, (Z)-2-бутандионовая, (Е)-2-бутандионовая, 2-гидроксибутандионовая, 2,3-дигидрокси-бутандионовая, 2-гидрокси-1,2,3-пропантрикарбоновая, метансульфоновая, этансульфоновая, бензолсульфоновая, 4-метилбензол-сульфоновая, циклогексан-сульфаминовая, 2-гидроксибензойная, 4-амино-2-гидроксибензойная кислоты. В предпочтительном варианте осуществления кислотно-аддитивной солью является гидрохлорид.
С помощью органического растворителя и хлороводородной кислоты (Реакция 9) основание небиволола (IX) превращают в небиволола гидрохлорид (I). В данном изобретении наблюдали, что получение небиволола гидрохлорида в спиртах/эфирах/кетонах дает минимальный количественный выход продукта по сравнению с применением других органических растворителей. Данный способ превращения основания небиволола в гидрохлоридную соль небиволола в присутствии спирта как растворителя дает минимальный количественный выход продукта. Таким образом, способом по данному изобретению можно получить высокий выход небиволола гидрохлорида, содержащего смесь только желательных изомеров.

Растворители для превращения в небиволола гидрохлорид или даже для дальнейшей очистки можно выбрать из группы, включающей спирты, эфиры, кетоны, галогенированные растворители, ацетонитрил и воду или их смеси. В одном варианте осуществления данного изобретения, спирт, применяемый для превращения, можно выбрать из группы, включающей метанол, этанол, n-пропанол, изопропанол, n-бутанол, изобутанол и т.п. В другом варианте осуществления эфир можно выбрать из группы, включающей этилацетат, n-бутилацетат и т.п. В еще одном варианте осуществления кетон можно выбрать из ацетона или метилизобутилкетона (МИБК). В следующем варианте осуществления галогенированные растворители, такие как метилен дихлорид, можно применять для превращения основания небиволола в небиволола гидрохлорид.
Также небиволола гидрохлорид можно получить реакцией основания небиволола и спиртового хлороводорода, такого как HCl в метаноле, HCl в этаноле, HCl в n-пропаноле, HCl в изопропаноле, HCl в n-бутаноле.
Также этого же можно достичь, пропуская газообразный НС1 сквозь раствор основания небиволола, при этом можно использовать вышеупомянутый растворитель.
Форма Т1 небиволола гидрохлорида
Данное изобретение также представляет новую аморфную форму небиволола и его фармацевтически приемлемых солей, обозначаемую формой Т1.
Полиморфизм заключается в возникновении отдельных кристаллических форм одного соединения, имеющих одну и ту же молекулярную формулу, но каждый полиморф может иметь различные физические свойства. Одно соединение может давать различные полиморфные формы, физические свойства которых могут быть индивидуальными и различными, такими как различные профили растворимости, различные точки температур плавления и различные пики дифракции рентгеновских лучей. Вследствие различия профилей растворимости полиморфных форм, идентификация фармацевтических полиморф очень важна для получения фармацевтических дозированных форм с предсказуемыми профилями растворимости.
Выражение "аморфный" в данном описании означает физическое состояние, не являющееся кристаллическим, и может быть подтверждено дифракцией рентгеновских лучей, инфракрасной спектроскопией и другими средствами, включающими изучение с помощью поляризационного микроскопа и дифференциальной сканирующей калориметрии, но не ограничивающимися ними.
Выражение "фармацевтически приемлемая соль" в данном описании относится к солям, известным как нетоксические и обычно применяемым в фармацевтической литературе. Типичные неорганические кислоты, применяемые для образования таких солей, включают хлороводородную, бромоводородную, йодоводородную, азотную, серную, фосфорную, гипофосфорную и т.п. Можно также использовать соли, производные органических кислот, таких как алифатические моно- и дикарбоксильные кислоты, фенилзамещенные алкановые кислоты, гидроксиалкановые и гидроксиалкандионовые кислоты, ароматические кислоты, алифатические и ароматические сульфоновые кислоты. Таким образом, такие фармацевтически приемлемые соли включают ацетат, фенилацетат, трифторацетат, акрилат, аскорбат, бензоат, хлорбензоат, динитробензоат, гидроксибензоат, метоксибензоат, метилбензоат, о-ацетоксибензоат, нафталин-2-бензоат, бромид, изобутират, фенилбутират, бета-гидроксибутират, хлорид, циннамат, цитрат, формиат, фумарат, гликолят, гептаноат, лактат, малеат, гидроксималеат, малонат, мезилат, нитрат, оксалат, фталат, фосфат, моногидрогенфосфат, дигидрогенфосфат, метафосфат, пирофосфат, пропионат, фенилпропионат, салицилат, сукцинат, сульфат, бисульфат, пиросульфат, сульфит, бисульфит, сульфонат, бензолсульфонат, р-бромфенилсульфонат, хлорбензолсульфонат, этансульфонат, 2-гидроксиэтансульфонат, метансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, р-толуолсульфонат, ксиленсульфонат, тартарат и т.п. Предпочтительной солью является гидрохлоридная соль.
В одном конкретном предпочтительном варианте осуществления данное изобретение представляет гидрохлоридную соль небиволола в аморфной форме.
Указанную аморфную форму можно получить методиками, такими как сушка методом разбрызгивании, сушка методом замораживания и т.п.
В предпочтительном варианте осуществления указанную форму получают обычной методикой сушки при разбрызгивании с применением сушилки с разбрызгиванием LabPlant SPD-005®. Небиволола гидрохлорид растворяют в спирте, таком как метанол, при нагревании для получения прозрачного раствора или устанавливают рН раствора основания небиволола/суспензии в спирте ниже 2,0 с помощью водного HCl/спиртового HCl/газообразного HCl, который затем сушат при разбрызгивании в течение 2-5 часов, а затем выделяют небиволола гидрохлорид формы Т1.
Выражение "выделение" включает фильтрацию, сушку или какие-либо другие методики, известные специалистам в данной области. Спирт для получения формы Т1 небиволола гидрохлорида выбирают из группы, включающей метанол, этанол, пропанол, изопропанол, n-бутанол и т.п., а также их смеси.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг.1: На данной фигуре показан график дифракции рентгеновских лучей частично аморфной формы Т1 небиволола гидрохлорида, полученной по данному изобретению.
Фиг.2: На этой фигуре показан график дифракции рентгеновских лучей кристаллической формы небиволола гидрохлорида, полученной известным способом (европейский патент ЕР 0145067).
Фиг.3: На этой фигуре показан сравнительный и суперпозиционный график дифракции рентгеновских лучей формы Т1 и кристаллической формы небиволола гидрохлорида, полученной по данному изобретению.
Фиг.4: Данная фотография является микроскопическим изображением кристаллического небиволола гидрохлорида, полученным с помощью микроскопа AXIOLAB.
Фиг.5: Данная фотография является микроскопическим изображением высушенного при разбрызгивании небиволола гидрохлорида, полученным с помощью микроскопа AXIOLAB.
Фиг.6: График зависимости среднего значения концентрации в плазме опытного небиволола от времени по сравнению с контрольной формуляцией.
Характеристика формы Т1 небиволола гидрохлорида
Форма Т1 небиволола гидрохлорида формулы (I) характеризуется следующими данными.
Форма Т1 небиволола гидрохлорида характеризуется графиком дифракции рентгеновских лучей на порошке (XRPD), как показано на Фиг.1, и может быть дифференцирована от кристаллической формы углами дифракции рентгеновских лучей на порошке (°2θ), d-значением и относительными интенсивностями, как изложено в Таблице 1.
Форма Т1 небиволола гидрохлорида отличается от кристаллической формы по следующим показателям:
отсутствие пиков при около 11,67±0,2; 16,54±0,2; 22,75±0,2; 25,41±0,2; 29,81±0,2; 31,56±0,2; 32,09±0,2 °2θ;
заметное присутствие пиков при 5,4330±0,2; 11,1544±0,2 и 19,7730±0,2 °2θ.
Фармацевтическая композиция
Данное изобретение также представляет способ получения твердой дозированной формы, предпочтительно таблетки или капсулы, небиволола гидрохлорида (I) и одного или более адъювантов без применения увлажняющего агента. Кроме того, данное изобретение также представляет твердую дозированную форму небиволола гидрохлорида и одного или более адъювантов без применения увлажняющего агента, необязательно без применения связывающего агента и необязательно без применения дезинтегрирующего агента, а также способ ее получения.
Фармацевтические композиции по данному изобретению имеет предпочтительно форму таблетки или капсулы. Активным ингредиентом, используемым в данной фармацевтической композиции, является небиволола гидрохлорид, который может быть необязательно просеянным сквозь сито со 100 отверстиями на один линейный дюйм.
Твердую дозированную форму таблетки или капсулы по данному изобретению получают с помощью активного ингредиента, то есть небиволола гидрохлорида, и фармацевтически приемлемых наполнителей, выбранных из группы, включающей разбавители, дезинтегрирующие агенты, связывающие агенты, смазывающие агенты, агенты, способствующие скольжению, и другие фармацевтически приемлемые наполнители или адъюванты, но не увлажняющий агент.
Разбавители можно выбрать из группы, включающей лактозу, крахмал, ангидрид двухосновного фосфата кальция, трехосновный фосфат кальция, каолин, сахарозу, маннитол, осажденный карбонат кальция, сорбитол, мальтодекстрин, производные целлюлозы, включая порошкообразную целлюлозу, микрокристаллическую целлюлозу, и другие материалы, известные специалисту в данной области.
Связывающие агенты можно выбрать из группы, включающей поливинилпирролидон или гидроксипропилметилцеллюлозу, гуммиарабик, альгиновую кислоту, гидроксипропилцеллюлозу, натрия карбоксиметилцеллюлозу, сжимаемый сахар, этилцеллюлозу, желатин, жидкую глюкозу, метилцеллюлозу, пептизированный крахмал и другие материалы, известные специалисту в данной области.
Дезинтегрирующие агенты можно выбрать из группы, включающей крахмал, натрия крахмал гликолят или кроскармеллозу натрия, кросповидон, альгининовую кислоту, карбоксиметилцеллюлозу натрия, гуаровую смолу и другие материалы, известные специалисту в данной области.
Смазывающие агенты можно выбрать из группы, включающей стеариновую кислоту, полиэтиленгликоль, стеарат магния, стеарат кальция, тальк, стеарат цинка, гидрогенированное касторовое масло, кремнезем, коллоидный кремнезем, кукурузный крахмал, силикат кальция, силикат магния, гидрогель кремния и другие материалы, известные специалисту в данной области.
Агенты, способствующие скольжению, можно выбрать из группы, включающей коллоидный диоксид кремния, коллоидный кремнезем, кукурузный крахмал, тальк, силикат кальция, силикат магния, коллоидный кремний, гидрогель кремния и другие материалы, известные специалисту в данной области.
Другие фармацевтические растворители выбирают из группы, включающей метанол, ацетон и очищенную воду.
Предпочтительная специфическая площадь поверхности небиволола гидрохлорида по данному изобретению составляет от 0,2×103 м2/кг до 1,95×103 м2/кг.
Композиция по данному изобретению включает ингредиенты в следующей пропорции. Количество небиволола или его фармацевтически приемлемой соли может исходить из необходимой для человека дозы.
Твердую дозированную форму по данному изобретению, содержащую небиволол или его фармацевтически приемлемую соль, получают согласно следующим этапам:
(I) а. готовят порошковую смесь, пропуская лактозу, крахмал и кроскармеллозу натрия через сито с числом отверстий на один линейный дюйм #60, а затем перемешивая ее;
b) готовят раствор небиволола или его фармацевтически приемлемой соли и гидроксипропилметилцеллюлозы или повидона в метаноле или приемлемом растворителе и воде;
с) адсорбируют раствор, полученный на вышеизложенном этапе (b) на сухой смеси разбавителей этапа 1 (а);
d) сушат продукт вышеизложенного этапа (с) и гранулируют с водой;
е) сушат влажные гранулы при 60°С и просеивают сухие гранулы через сито с числом отверстий на один линейный дюйм #30;
f) смазывают гранулы этапа (е) кроскармеллозой натрия, коллоидным диоксидом кремния, микрокристаллической целлюлозой и стеаратом магния и смешивают в смесителе;
g) прессуют гранулы, полученные на этапе (IV), в таблетки или наполняют гранулами капсулы.
ИЛИ
(I) Небиволол или его фармацевтически приемлемую соль, лактозу, крахмал и кроскармеллозу натрия пропускают через сито с числом отверстий на один линейный дюйм #60 и смешивают должным образом.
(II) Готовят раствор связывающего агента с применением гидроксипропилметилцеллюлозы или повидона в водных или безводных гранулированных растворителях.
(III) Гранулируют порошкообразную смесь этапа (I)-с или (I') с раствором связывающего агента, приготовленным на этапе II, в миксере с высокой скоростью и сушат гранулы в сушилке в кипящем слое.
(IV) Смазывают гранулы этапа (III) кроскармеллозой натрия, коллоидным диоксидом кремния, микрокристаллической целлюлозой и стеаратом магния и смешивают в контейнерном смесителе.
(V) Прессуют гранулы, полученные на этапе (IV), в таблетки или наполняют гранулами капсулы.
Таким образом, при использовании вышеупомянутого простого и менее дорогого способа растворение достигает, по меньшей мере, 75% за 45 минут без применения увлажняющего агента, как показано в релевантных примерах.
Во всем данном описании и формуле изобретения следует понимать, что слова "содержать" и "включать" и их формы, такие как "содержит", "содержащий", "включает", "включающий", должны трактоваться неисключительно, если контекст не требует обратного. То есть, использование таких слов может подразумевать также включение другого элемента или элементов, на которые прямо не указывается.
Детали данного изобретения, его цели и преимущества детально объясняются ниже в иллюстративных примерах, не являющихся ограничивающими. Примеры только поясняют, а не ограничивают идею данного изобретения, и очевидно, что различные модификации или изменения в этапах методики, а также в композициях, осуществленные специалистами в данной области без отступления от рамок данного изобретения, должны быть охвачены объемом и сущностью данного подхода и границами изобретения.
ПРИМЕРЫ
Пример 1
Получение 6-фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксильной кислоты пиперидин амида (V)
К 40 г 6-фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксильной кислоты (IV) добавляют 120 мл толуола и 0,8 мл диметил формамида при перемешивании при комнатной температуре. Добавляют тионил хлорид (32 г), реакционную смесь нагревают до 60-70°С и выдерживают в течение 30 минут. Тионил хлорид и толуол отгоняют при пониженном давлении. Затем к реакционной смеси добавляют 160 мл толуола и охлаждают. Медленно добавляют 88 мл пиперидина при комнатной температуре и взбалтывают 30 минут. Реакционную смесь подкисляют разбавленной HCl. Водный слой экстрагируют толуолом, объединенные органические слои промывают разбавленной HCl, а затем водой. Органический растворитель удаляют при пониженном давлении для получения 40 г указанного соединения в виде твердого вещества.
В качестве альтернативы также можно применять следующую методику. К 100 г 6-фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксильной кислоты (IV) добавляют 1500 мл метилен хлорида и 70 г триэтиламина при 20-40°С. Реакционную смесь охлаждают до температуры от 0 до -5°С и медленно за 30-45 минут добавляют 57 г этилхлорформиата. Реакционную смесь выдерживают при 0 - -5°С в течение 1 часа. Затем добавляют 55 г пиперидина в течение 30-45 минут, при этом поддерживают температуру 0 - -5°С. До завершения реакции реакционную смесь выдерживают при 0-5°С в течение 1,5 часа. Добавляют 1 л воды при 0-5°С и регулируют рН до значения 2,0 концентрированной хлороводородной кислотой при 15-20°С. Взбалтывают в течение 0,5 часа и слои разделяют. Слой метилен хлорида промывают 2 л воды, затем полностью отгоняют метилен хлорид под вакуумом при температуре до 45°С. Добавляют 200 мл гексана и полностью отгоняют. Снова добавляют 200 мл гексана, взбалтывают при 20-40°С в течение 0,5 часа и охлаждают до 0-5°С. Выдерживают при 0-5°С в течение 1 часа, фильтруют и промывают 100 мл охлажденного гексана при 0-5°С до получения 110 г сухого веса (сушат при 55-60°С в горячем воздухе в течение 6 часов) указанного соединения в виде твердого вещества.
Пример 2
Получение 6-фтор-3,4-дигидро-2Н-1-бензопиран-2-карбоксальдегида (VI)
К 40 г амида (V) добавляют 400 мл толуола. К реакционной смеси медленно приливают раствор Vitride® (смесь 44 г Vitride® в толуоле) при 10-15°С. К реакционной смеси добавляют метанол, затем подкисляют разбавленной HCl. Реакционную смесь экстрагируют толуолом и выпаривают при пониженном давлении до получения 26 г указанного соединения.
Пример 3
Получение 6-фтор-3,4-дигидро-2-оксиранил-2Н-1-бензопирана (VII)
К 234 мл диметил сульфоксида добавляют 32 г триметил сульфоксониум йодида. К реакционной смеси добавляют третичный бутоксид калия (16 г) и взбалтывают 1 час при 20-40°С. Реакционную смесь охлаждают и приливают раствор соединения, полученного в Примере 2, в диметил сульфоксиде (26 мл), выдерживают при температуре 20-40°С в течение 1,5 часа. Реакционную смесь гасят в холодной воде. Водный слой экстрагируют этилацетатом, промывают водой, выпаривают при пониженном давлении с последующей дистилляцией продукта до получения 19,6 г 6-фтор-3,4-дигидро-2-оксиранил-2Н-1-бензопирана (VII) в форме смеси А и В в виде масла.
Смесь изомера A (VII-A) и изомера В (VII-B) разделяют колоночной хроматографией на силикагеле в качестве стационарной фазы и смеси гексана и этилацетата в качестве элюанта. 6,75 г смеси изомеров А и В элюируют смесью гексан : этил ацетат (в соотношении 97:3) до полного отделения изомера А. Затем колонку элюируют смесью гексан: этил ацетат (в соотношении 90:10) до получения изомера В. Элюаты изомера А и изомера В выпаривают до получения 2,6 г изомера А (VII-A) в виде масла и 1,4 г изомера В (VII-B), соответственно.
Чистота полученного продукта составляет 78-82%.
Пример 4
Получение (А)-6-фтордигидро-α-[[(фенилметил)амино]-метил]-2Н-1-бензопиран-2-метанола (промежуточное соединение I)
10,65 г изомера А (VII-A) добавляют к 31,9575 мл изопропилового спирта с последующим добавлением 7,13 г бензиламина. Реакционную смесь нагревают с обратным холодильником 2 часа и охлаждают до 0-5°С. Осадок фильтруют и очищают изопропиловым спиртом (27 мл) до получения 8,52 г указанного соединения.
Пример 5
Получение бензилированного основания небиволола (VIII)
К 50 мл метанола добавляют 10 г изомера В (VII-B) и 14,70 г А амина (промежуточное соединение I) и нагревают до 65-70°С. Реакционную массу выдерживают при той же температуре 15 часов. Затем массу охлаждают до 50-55°С и добавляют 30 мл метанола. Затем массу охлаждают до -10 - -15°С и взбалтывают при той же температуре в течение 12 часов. Материал фильтруют, промывают 5 мл метанола при -10 - -15°С, фильтруют и сушат при 40-45°С в течение 8 часов до получения 11,5 г указанного соединения.
Пример 6
Получение основания небиволола (IX)
В гидрогенизатор помещают бензилированное основание небиволола (VIII) (5,85 г) вместе с 2-метокси этанолом (117 мл) и 0,351 г палладия на угле (10%). Применяют давление 160-170 фунтов на кв. дюйм с газообразным водородом и нагревают до 70-75°С. Условия температуры и давления выдерживают в течение 3 часов до полного завершения реакции. Реакционную массу охлаждают до комнатной температуры и фильтруют сквозь (18 г) слой с восходящим потоком второй фазы для отделения катализатора.
Фильтрат выпаривают до получения осадков, осадки выделяют в метаноле (38 мл) при 0-5°С. Осадок сушат до получения 4,0 г основания небиволола.
Пример 7
Получение небиволола HCl (I) с применением метанола в качестве реакционного растворителя и очистка небиволола HCl (I)
К 10 г основания небиволола (IX) добавляют 70 мл метанола и 3,5 г хлороводородной кислоты, реакционную смесь взбалтывают 4 часа при 28-32°С. Материал фильтруют и промывают охлажденным метанолом. Затем фильтруют досуха. Влажный материал дефлегмируют в 300 мл метанола 30 минут, а затем фильтруют сквозь слой с восходящим потоком второй фазы. Метанол полностью отгоняют, к остатку добавляют 100 мл изопропилового спирта, а затем взбалтывают 30 минут при 60-65°С. Охлаждают до 25-30°С. Материал снова взбалтывают при 28-32°С в течение 3 часов. Материал фильтруют, промывают 20,0 мл изопропилового спирта и сушат при 60-65°С 8 часов для получения 10,5 г небиволола гидрохлорида (I). Диапазон температуры плавления - 223-227°С. Выход (%) - 96,33%. Чистота ВЭЖХ - 99,89%.
Пример 8
Получение небиволола HCl (I) с применением изопропилового спирта в качестве реакционного растворителя и очистка небиволола HCl (I)
К 10 г основания небиволола (IX) добавляют 70 мл изопропилового спирта и 3,5 г хлороводородной кислоты, реакционную смесь взбалтывают 4 часа при 28-32°С. Материал фильтруют, промывают 10,0 мл изопропилового спирта и фильтруют досуха. Влажный материал дефлегмируют в 300 мл метанола 30 минут, а затем фильтруют сквозь слой с восходящим потоком второй фазы. Метанол полностью отгоняют, а к остатку добавляют 100 мл изопропилового спирта и затем взбалтывают 30 минут при 60-65°С. Охлаждают до 28-32°С. Материал взбалтывают при 28-32°С 3 часа, фильтруют, промывают 20,0 мл изопропилового спирта и сушат при 60-65°С 8 часов до получения 10,6 г небиволола гидрохлорида (I).
Диапазон температуры плавления - 223-227°С.
Выход (%) - 97,24%.
Чистота ВЭЖХ - 99,16%.
Пример 9
Получение небиволола HCl (I) из основания небиволола (IX) с применением этанола в качестве реакционного растворителя и очистка небиволола HCl (I)
К 10 г основания небиволола (IX) добавляют 70 мл этанола и 3,5 г хлороводородной кислоты и реакционную смесь взбалтывают 4 часа при 28-32°С. Материал фильтруют, промывают 10,0 мл этанола и фильтруют досуха. Влажный материал дефлегмируют в 300 мл метанола 30 минут, а затем фильтруют сквозь слой с восходящим потоком второй фазы. Метанол полностью отгоняют, а к остатку добавляют 100 мл изопропилового спирта и затем взбалтывают 30 минут при 60-65°С. Охлажденный до 28-32°С материал взбалтывают при 28-32°С 3 часа, фильтруют и промывают 20,0 мл изопропилового спирта и окончательно сушат при 60-65°С до получения 10,5 г небиволола гидрохлорида (I). Далее материал просеивают через сито с числом отверстий на один линейный дюйм #100.
Диапазон температуры плавления - 223-227°С.
Выход (%) - 96,33%.
Чистота ВЭЖХ - 99,45%.
Пример 10
Получение небиволола гидрохлорида (I) из бензилированного основания небиволола (VIII)
В гидрогенизатор помещают бензилированное основание небиволола (40,0 г) вместе с 2-метокси этанолом (300 мл) и 10% палладием на угле (6,0 г). Применяют давление 160-170 фунтов на кв. дюйм с газообразным водородом и нагревают до 70-75°С. Условия температуры и давления выдерживают 3,0 часа и завершают реакцию проверкой с помощью ТСХ (тонкослойной хроматографии)/ВЭЖХ. Реакционную смесь охлаждают до комнатной температуры и фильтруют сквозь слой с восходящим потоком второй фазы для отделения катализатора. Фильтрат нагревают до 65-70°С с последующим добавлением 14,0 мл хлороводородной кислоты (35%). Реакционную массу взбалтывают 2,0 часа. Материал фильтруют, затем добавляют метанол (800,0 мл) и нагревают до 60-65°С до получения прозрачного раствора. Фильтруют сквозь слой с восходящим потоком второй фазы. Фильтрат выпаривают под вакуумом, пока объем не составит 20%. Затем реакционную массу охлаждают до 0-5°С и взбалтывают при той же температуре 2,0 часа. Материал фильтруют и сушат при 55-60°С для получения 28,0 г указанного соединения.
Чистота ВЭЖХ - 99,99%.
Пример 11
Получение Формы Т1
К 2100 мл метанола добавляют 60,0 г небиволола гидрохлорида при комнатной температуре. Реакционную массу нагревают до 50-60°С до получения прозрачного раствора. Прозрачный раствор сушат при разбрызгивании 3,5 часа со скоростью подачи 10-12 мл/минута, температура на входном отверстии подачи - 65-110°С, а температура на выходном отверстии - 65-75°С. Далее продукт сушат при 60-65°С в течение 10,0 часов для получения 38,0 г формы Т1.
Пример 12
Получение формы Т1
К 2100 мл метанола добавляют 60,0 г основания небиволола при комнатной температуре. рН реакционной массы регулируют до значения ниже чем 2,0, добавлением концентрированной HCl (16,20 г) и перемешивают до получения прозрачного раствора. Затем прозрачный раствор сушат при разбрызгивании в течение 3,5 часов со скоростью подачи 10-12 мл/минута, температура на входном отверстии подачи - 65-110°С, а температура на выходном отверстии - 65-75°С. Далее продукт сушат при 60-65°С в течение 10,0 часов для получения 38,0 г формы Т1. Данные дифракции рентгеновских лучей на порошке представлены в Таблице 1.
ПОЛУЧЕНИЕ ТВЕРДОЙ ДОЗИРОВАННОЙ ФОРМЫ
Ингредиенты, применяемые при получении твердой дозированной формы, содержащей небиволола гидрохлорид по данному изобретению, приводятся ниже вместе со способом получения. Во всех последующих примерах растворение полученных таблеток определяют в лопастном аппарате. Скорость вращения составляет 50±2 оборотов в минуту, растворяющая среда представляет собой 0,1 N HCl, выдерживают при фиксированной температуре 37°С (рН 1,2, моделированная желудочная жидкость). Общий объем растворяющей жидкости составляет 500 мл.
Пример 13
Получение таблетки
Для изготовления твердой дозированной формы в рассмотренных ниже примерах используют материал, полученный в Примере 8, то есть небиволола гидрохлорид.
Методика
Небиволола гидрохлорида 5,45 мг (2,72% вес/вес) просеивают через сито с числом отверстий на один линейный дюйм #60, через такое же сито просеивают 125,05 мг лактозы (62,53% вес/вес), 5,00 мг кроскармеллозы натрия (2,5% вес/вес) и 30,00 мг крахмала (15,00% вес/вес). Просеянные материалы смешивают вместе. 3 мг (1,50% вес/вес) гидроксипропилметилцеллюлозы (6 cps) растворяют в 30 мл очищенной воды (предварительно нагретой до 70°С) и используют как раствор для грануляции для получения гранул вышеупомянутой смеси, пока не получат массу необходимой консистенции. Для этого необходимо 25 мл дополнительной очищенной воды. Влажную массу пропускают через сито с числом отверстий на один линейный дюйм #08, а влажные гранулы сушат в центробежной сушилке при 70°С, пока потеря массы при сушении не составит 1,28%. Сухие гранулы пропускают через сито с числом отверстий на один линейный дюйм #20 и смешивают с компрессионной смесью, включающей 23,0 мг (11,50% вес/вес) микрокристаллической целлюлозы (Avicel PH 102), 0,5 мг (0,25% вес/вес) коллоидного диоксида кремния, 7,0 мг (3,50% вес/вес) кроскармеллозы натрия и 1,0 мг (0,50% вес/вес) стеарата магния, предварительно просеянных через сито с числом отверстий на один линейный дюйм #60. Полученную таким образом смесь прессуют в 200 мг таблетки на таблеточном прессе с применением 7,93 мм (10/32 дюйм) круглых неглубоких пуансонов со скошенной кромкой.
Профиль растворения небиволола гидрохлорида по данному изобретению при рН 1,2 (моделированная желудочная жидкость)
Растворение таблеток, приготовленных по Примеру 13, представлено в Таблице 4.
Контроль, то есть Nebilet®, представляет собой таблетку небиволола гидрохлорида от производителя - Янссен Фармацевтика. Скорость растворения таблетки по данному изобретению, как выяснили, составляет более чем 75% за 45 минут. Таким образом, растворение таблетки по данному изобретению является приемлемым.
Пример 14
Получение таблетки
Для изготовления твердой дозированной формы в рассмотренных ниже примерах используют материал, полученный в Примере 8, то есть небиволола гидрохлорид.
Методика
Небиволола гидрохлорида 5,45 мг (2,72% вес/вес) просеивают через сито с числом отверстий на один линейный дюйм #60, через такое же сито просеивают 125,05 мг лактозы (62,53% вес/вес), 5,00 мг кроскармеллозы натрия (2,5% вес/вес) и 30,00 мг крахмала (15,00% вес/вес). Просеянные материалы смешивают вместе. 3 мг (1,50% вес/вес) поливинилпирролидона К-30 растворяют в 30 мл очищенной воды, предварительно нагретой до 70°С, и используют как раствор для грануляции при изготовлении гранул вышеупомянутой смеси, пока не получат массу необходимой консистенции. Для этого необходимо 25 мл дополнительной очищенной воды. Влажную массу пропускают через сито с числом отверстий на один линейный дюйм #08, а влажные гранулы сушат в центробежной сушилке при 70°С, пока потеря массы при сушке не составит 1,258%. Сухие гранулы пропускают через сито с числом отверстий на один линейный дюйм #20 и смешивают с компрессионной смесью, включающей 23,0 мг (11,50% вес/вес) микрокристаллической целлюлозы (Avicel PH 102), 0,5 мг (0,25% вес/вес) коллоидного диоксида кремния, 7,0 мг (3,5% вес/вес) кроскармеллозы натрия и 1,0 мг (0,50% вес/вес) стеарата магния, предварительно просеянных через сито с числом отверстий на один линейный дюйм #60. Полученную таким образом смесь прессуют в 200 мг таблетки на таблеточном прессе с применением 7,93 мм (10/32 дюйм) круглых неглубоких пуансонов со скошенной кромкой.
Профиль растворения небиволола гидрохлорида по данному изобретению при рН 1,2 (моделированная желудочная жидкость)
Растворение таблеток, приготовленных по Примеру 14, представлено в Таблице 6.
Стандарт, то есть Nebilet®, представляет собой таблетку небиволола гидрохлорида от производителя - Янссен Фармацевтика. Скорость растворения таблеток по данному изобретению, как выяснили, составляет более чем 75% за 45 минут. Таким образом, растворение таблеток по данному изобретению является приемлемым.
Пример 15
Получение таблетки
Для изготовления твердой дозированной формы в нижеприведенных примерах использовали материал, полученный в Примере 7, то есть небиволола гидрохлорид.
Получение таблеток способом влажного гранулирования (лекарство адсорбировано на наполнителях)
Ингредиенты, применяемые для получения таблеток, содержащих небиволола гидрохлорид, по данному изобретению адсорбцией лекарства на наполнителях и последующим способом влажной грануляции приводятся ниже вместе со способом получения указанных таблеток.
Этап А: Состав для адсорбции лекарства на наполнителях.
Методика
Небиволола гидрохлорида 5,45 мг (2,86% вес/вес) (2% дополнительно для компенсации потери) добавляют к 2780 мл раствора метанола при взбалтывании (1% вес/объем раствор лекарства в метаноле), пропускают 144,62 мг лактозы (75,99% вес/вес), 28,75 мг кроскармеллозы натрия (3,02% вес/вес) и 34,5 мг крахмала (18,13% вес/вес) через сито с числом отверстий на один линейный дюйм #60. Просеянные материалы смешивают вместе. Эту комбинированную смесь добавляют в чашу флюидизации, а раствор лекарства адсорбируют в ней с помощью процессора псевдоожиженного слоя (способ верхнего разбрызгивания).
Этап В: Получение таблеток способом влажного гранулирования
Ингредиенты, применяемые для получения таблеток, содержащих небиволола гидрохлорид, по данному изобретению способом влажного гранулирования приведены ниже вместе со способом получения указанных таблеток.
Методика
3,45 мг (1,50% вес/вес) гидроксипропилметилцеллюлозы (6 cps) растворяют в 35 мл очищенной воды, предварительно нагретой до 70°С, после этого охлаждают и используют как раствор для грануляции для получения гранул смешиванием смеси, адсорбирующей небиволола гидрохлорид, для получения массы, необходимой консистенции. Необходимо 25 мл дополнительно очищенной воды. Влажную массу сушат в центробежной сушилке при 70°С до высушивания на 1,48%. Высушенные гранулы просеивают через сито с числом отверстий на один линейный дюйм #20 и смешивают со смазывающими агентами, включая 26,45 мг (11,50% вес/вес) микрокристаллической целлюлозы (Avicel PH 102), 0,575 мг (0,25% вес/вес) коллоидного диоксида кремния, 8,05 мг (3,50% вес/вес) кроскармеллозы натрия и 1,15 мг (0,50% вес/вес) стеарата магния, предварительно просеянных через сито с числом отверстий на один линейный дюйм #60. Окончательную смесь, полученную таким образом, прессуют в таблетки со средней массой 230 мг на таблеточном прессе с применением 8,73 мм (11/32 дюйм) круглых неглубоких пуансонов со скошенной кромкой.
Профиль растворения небиволола гидрохлорида по данному изобретению при рН 1,2 (моделированная желудочная жидкость)
Растворение таблеток, полученных по Примеру 15, представлено в Таблице 9.
Пример 16
Получение таблетки
Для изготовления твердой дозированной формы в рассмотренных ниже примерах используют материал, полученный в Примере 8, то есть небиволола гидрохлорид.
Методика
Через сито с числом отверстий на один линейный дюйм #60 просеивают небиволола гидрохлорида 5,45 мг (2,72% вес/вес), лактозы 158,05 мг (79,03% вес/вес) и кроскармеллозы натрия 5,00 мг (2,5% вес/вес). Просеянные материалы смешивают вместе. Добавляют 35 мл очищенной воды как раствора для грануляции для изготовления гранул вышеупомянутой смеси для получения массы необходимой консистенции. Влажную массу пропускают через сито с числом отверстий на один линейный дюйм #08, влажные гранулы сушат в центробежной сушилке при 70°С до высушивания на 0,65%. Сухие гранулы пропускают через сито с числом отверстий на один линейный дюйм #20 и смешивают со смесью для прессования, включающей 23,0 мг (11,50% вес/вес) микрокристаллической целлюлозы, 0,5 мг (0,25% вес/вес) коллоидного диоксида кремня, 7,0 мг (3,5% вес/вес) кроскармеллозы натрия и 1,0 мг (0,50% вес/вес) стеарата магния, предварительно просеянных через сито с числом отверстий на один линейный дюйм #60. Окончательную смесь, полученную таким образом, прессуют в 200 мг таблетки на таблеточном прессе с применением 7,93 мм (10/32 дюйм) круглых неглубоких пуансонов со скошенной кромкой.
Профиль растворения небиволола гидрохлорида по данному изобретению при рН 1,2 (моделированная желудочная жидкость)
Растворение таблеток, полученных по Примеру 16, представлено в Таблице 11.
Контроль, то есть Nebilet®, представляет собой таблетку небиволола гидрохлорида от производителя - Янссен Фармацевтика. Выяснили, что скорость растворения таблеток, полученных по данному изобретению, составляет более 75% за 45 минут. Таким образом, растворение таблеток по данному изобретению является приемлемым.
Пример 17
Получение таблетки
Для изготовления твердой дозированной формы в рассмотренных ниже примерах используют материал, полученный в Примере 7, то есть небиволола гидрохлорид.
Методика
Небиволола гидрохлорида 5,45 мг (2,41% вес/вес) добавляют к метанолу при взбалтывании (1% вес/объем раствор лекарства в метаноле). Пропускают 179,125 мг (79,07% вес/вес) лактозы через сито с числом отверстий на один линейный дюйм #60. Эту смесь добавляют в чашу флюидизации, а раствор лекарства адсорбируют в ней с помощью аппарата псевдоожиженного слоя (способ верхнего разбрызгивания).
Получение таблеток способом влажного гранулирования
Ингредиенты, используемые при получении таблеток, содержащих небиволола гидрохлорид, по данному изобретению способом влажного гранулирования приведены ниже вместе со способом получения указанных таблеток.
Методика
Очищенную воду используют как раствор для гранулирования при приготовлении гранул смешиванием со смесью, адсорбирующей небиволола гидрохлорид, для получения массы необходимой консистенции. Влажную массу сушат в центробежной сушилке при 70°С до высушивания на 0,71%. Сухие гранулы просеивают через сито с числом отверстий на один линейный дюйм #20 и смешивают со смазывающими агентами, включающими 26,45 мг (11,67% вес/вес) микрокристаллической целлюлозы, 0,575 мг (0,25% вес/вес) коллоидного диоксида кремния, 13,8 мг (6,09% вес/вес) кроскармеллозы натрия и 1,15 мг (0,51% вес/вес) стеарата магния, предварительно просеянных через сито с числом отверстий на один линейный дюйм #60. Полученную таким образом смесь прессуют в таблетки со средней массой 226,55 мг на таблеточном прессе с применением 8,73 мм (11/32 дюйм) круглых неглубоких пуансонов со скошенной кромкой.
Профиль растворения небиволола гидрохлорида по данному изобретению при рН 1,2 (моделированная желудочная жидкость)
Растворение таблеток, полученных по Примеру 17, представлено в Таблице 13.
Пример 18
Получение таблетки
Для изготовления твердой дозированной формы в рассмотренных ниже примерах используют материал, полученный в Примере 8, то есть небиволола гидрохлорид.
Методика
5,45 мг (2,73% вес/вес) небиволола гидрохлорида и 170,05 мг (85,03% вес/вес) лактозы пропускают через сито с числом отверстий на один линейный дюйм #60. Просеянные материалы смешивают вместе. Для получения массы необходимой консистенции используют 35 мл очищенной воды как раствора для грануляции для получения гранул указанной выше смеси. Влажную массу пропускают через сито с числом отверстий на один линейный дюйм #08, влажные гранулы сушат в центробежной сушилке при 70°С до высушивания на 0,39%. Сухие гранулы пропускают через сито с числом отверстий на один линейный дюйм #20 и смешивают со смесью для прессования, включающей 23,0 мг (11,5% вес/вес) микрокристаллической целлюлозы, 0,5 мг (0,25% вес/вес) коллоидного диоксида кремния и 1,0 мг (0,50% вес/вес) стеарата магния, предварительно просеянных через сито с числом отверстий на один линейный дюйм #60. Окончательную смесь, полученную таким образом, прессуют в 200 мг таблетки на таблеточном прессе с применением 7,93 мм (10/32 дюйм) круглых неглубоких пуансонов со скошенной кромкой.
Профиль растворения небиволола гидрохлорида по данному изобретению при рН 1,2 (моделированная желудочная жидкость)
Растворение таблеток, полученных по Примеру 18, представлено в Таблице 15.
Контроль, то есть Nebilet®, представляет собой таблетку небиволола гидрохлорида от производителя - Янссен Фармацевтика. Выяснили, что скорость растворения таблеток, полученных по данному изобретению, составляет более 75% за 45 минут. Таким образом, растворение таблеток по данному изобретению является приемлемым.
Пример 19
Получение таблетки
Для изготовления твердой дозированной формы в рассмотренных ниже примерах используют материал, полученный в Примере 8, то есть небиволола гидрохлорид.
Методика
5,45 мг (2,56% вес/вес) небиволола гидрохлорида добавляют к метанолу при взбалтывании (1% вес/объем раствора лекарства в метаноле). Через сито с числом отверстий на один линейный дюйм #60179 пропускают 125 мг лактозы (84,20% вес/вес). Эту смесь добавляют в чашу флюидизации, а раствор лекарства адсорбируют в ней с помощью аппарата псевдоожиженного слоя (способ верхнего разбрызгивания).
Получение таблетки способом влажного гранулирования
Ингредиенты, применяемые для получения таблеток, содержащих небиволола гидрохлорид, по данному изобретению способом влажного гранулирования приведены ниже вместе со способом получения указанных таблеток.
Методика
Для получения массы необходимой консистенции используют 50 мл очищенной воды как раствора для гранулирования при приготовлении гранул смешиванием со смесью, адсорбирующей небиволола гидрохлорид. Влажную массу сушат в центробежной сушилке при 70°С до высушивания на 1,12%. Сухие гранулы просеивают через сито с числом отверстий на один линейный дюйм #20 и смешивают со смазывающими агентами, включающими 26,45 мг (12,43% вес/вес) микрокристаллической целлюлозы, 0,575 мг (0,27% вес/вес) коллоидного диоксида кремния и 1,15 мг (0,54% вес/вес) стеарата магния, предварительно просеянных через сито с числом отверстий на один линейный дюйм #60. Окончательную смесь, полученную таким образом, прессуют в таблетки со средней массой 212,75 мг на таблеточном прессе с применением 8,73 мм (11/32 дюйм) круглых неглубоких пуансонов со скошенной кромкой.
Профиль растворения небиволола гидрохлорида по данному изобретению при рН 1,2 (моделированная желудочная жидкость)
Растворение таблеток, полученных по Примеру 19, представлено в Таблице 17.
Контроль, то есть Nebilet®, представляет собой таблетку небиволола гидрохлорида от производителя - Янссен Фармацевтика. Выяснили, что скорость растворения таблеток, полученных по данному изобретению, составляет более 75% за 45 минут. Таким образом, растворение таблеток по данному изобретению является приемлемым.
Пример 20
Получение таблетки
Для изготовления твердой дозированной формы в рассмотренных ниже примерах используют материал, полученный в Примере 10, то есть небиволола гидрохлорид.
Методика
В 440 мг метанола и 10 мг воды готовят раствор 5,45 мг (2,37% вес/вес) небиволола гидрохлорида и 3,45 мг (1,5% вес/вес) гипромеллозы. Через сито с числом отверстий на один линейный дюйм #40 просеивают 143,475 мг (62,38% вес/вес) лактозы, 34,50 мг (15% вес/вес) кукурузного крахмала (8% добавляют дополнительно для компенсации потери) и 3,45 мг (3% вес/вес) кроскармеллозы натрия. Просеянные материалы помещают в контейнер для продукта в аппарат псевдоожиженного слоя (FBP). Раствор небиволола гидрохлорида - гипромеллозы адсорбируют в смеси, флюидизированной в контейнере аппарата псевдоожиженного слоя способом верхнего разбрызгивания. После полной адсорбции раствора лекарства и связывающего агента смесь сушат. Затем смесь гранулируют с 28 мг воды. Влажные гранулы сушат в аппарате псевдоожиженного слоя при 60°С. Сухие гранулы просеивают через сито с числом отверстий на один линейный дюйм #30 и смешивают с 26,45 мг (11,5% вес/вес) микрокристаллической целлюлозы, 3,45 мг (3% вес/вес) кроскармеллозы натрия, 0,575 мг (0,25% вес/вес) коллоидного диоксида кремния, предварительно просеянных через сито с числом отверстий на один линейный дюйм #40 и предварительно просеянным 2,30 мг (1% вес/вес) стеаратом магния. Окончательную смесь, полученную таким образом, прессуют в таблетки со средней массой 230 мг на таблеточном прессе с применением 9,0 мм круглых вогнутых пуансонов со скошенной кромкой и с бороздкой на одной стороне.
Профиль растворения небиволола гидрохлорида по данному изобретению при рН 1,2 (моделированная желудочная жидкость)
Растворение таблетки, полученной по Примеру 20, представлено в Таблице 19.
Пример 21
Сравнительные данные биодоступности таблетки небиволола по данному изобретению (опытная формуляция) по сравнению с таблеткой небилета (контрольная формуляция)
Фармакокинетика
При изучении биоэквивалентности регистрируют и рандомизируют 24 здоровых волонтера для приема опытной (небиволол 5 мг, Торрент Фармацевтикалс Лтд., Индия) или контрольной (Nebilet® 5 мг, Берлин-Хеми АГ, Германия) формуляций. Уровни небиволола в плазме измеряют с помощью проверенного метода жидкостной хроматографии - масс-спектрометрии/масс-спектрометрии (LCMS/MS).
Средние концентрации в плазме небиволола после оральной дозы 1 таблетки (5 мг небиволола) опытной формуляций или 1 таблетки (5 мг небиволола) контрольной формуляции показаны на Фигуре VI.
Оценивают фармакокинетические параметры. Для опытного небиволола и контрольных формуляций значение Сmax достигало 2,605±0,66 нг/мл и 2,574±0,78 нг/мл, соответственно. Большого различия в значениях tmax для опытной и контрольной формуляций не наблюдали. Наблюдаемое значение AUC(0-t) (величина площади под кривой "концентрация - время") для опытной и контрольной формуляций составляло 17,33±29,54 нг.час/мл и 17,71±30,77 нг.час/мл, соответственно. Наблюдаемое значение AUC(0-∞) для опытной и контрольной формуляций составляло 29,69±62,72 нг.час/мл и 21,86±41,96 нг.час/мл, соответственно.
Интервал 90%-достоверности, рассчитанный по значениям ANOVA-log (метод дисперсионного анализа) для первичного параметра, внутрииндивидуальному соотношению (Опыт/Контроль) AUC(0-t), AUC(0-∞) и Сmax небиволола, составил 0,93-1,04, 0,95-1,17 и 0,95-1,10.
Безопасность
Лекарство хорошо переносилось. Во время опыта ни один из волонтеров не сообщал о неблагоприятных явлениях. Результаты лабораторных исследований не обнаружили неблагоприятные явления или неблагоприятные реакции на лекарство.
ЗАКЛЮЧЕНИЕ
Вышеупомянутые фармакокинетические параметры небиволола опытной (небиволол 5 мг, Торрент Фармацевтикалс Лтд., Индия) и контрольной (Nebilet® 5 мг, Берлин-Хеми АГ, Германия) формуляции были биоэквивалентны по отношению к небивололу, как показано в Таблице 20.
Реферат
Изобретение относится к улучшенному способу получения 2Н-1-бензопиран-2-метанол-α,α'-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-[2R*[R*[R*(S*)]]]], то есть основания небиволола формулы (IX), или его гидрохлоридной соли ! ! а также к способу получения промежуточного соединения - бензилированного небиволола формулы (VIII), ! ! Кроме того, данное изобретение относится к фармацевтической композиции, обладающей антигепертензивным действием, без применения увлажняющего агента, и к таблетке, содержащей эту фармацевтическую композицию. 4 н. и 17 з.п. ф-лы, 20 табл., 6 ил.
Формула

при котором выполняют следующие этапы:
(а) проводят реакцию (А)-6-фтордигидро-α-[[(фенилметил)амино]-метил]-2Н-1-бензопиран-2-метанола, то есть промежуточного соединения I

промежуточное соединение I
с (В)-6-фтор-3,4-дигидро-2-оксиранил-2Н-1-бензопираном формулы (VII-В)

в присутствии приемлемого органического растворителя, такого как упомянутые здесь, выбранного из группы, включающей спирт, эфир и кетон, и выделенного при температуре от -5 до -25°С, и затем выдерживают реакционную массу при температуре выделения в течение 2 - 40 ч для получения бензилированного небиволола формулы (VIII)

(b) дебензилируют бензилированный небиволол формулы (VIII), полученный на вышеупомянутом этапе (а), до основания небиволола формулы (IX) с помощью 10% палладия на угле известным способом,
(с) превращают основание небиволола формулы (IX), полученное на вышеупомянутом этапе (b), для получения небиволола гидрохлорида формулы (I) с помощью приемлемого органического растворителя, такого как указанные здесь, выбранного из группы, включающей спирт, эфир, кетон, галогенированный растворитель, ацетонитрил, воду или их смеси, и с помощью хлороводородной кислоты

(d) выделяют продукт реакции вышеупомянутого этапа (с).

промежуточное соединение I
с (В)-6-фтор-3,4-дигидро-2-оксиранил-2Н-1-бензопираном формулы (VII-В)

в присутствии приемлемого органического растворителя, такого как упомянутые здесь, выбранного из группы, включающей спирт, эфир и кетон, и выделенного при температуре от -5 до -25°С, а затем выдерживают реакционную массу при температуре выделения в течение 2-40 ч до получения бензилированного небиволола формулы (VIII).
Документы, цитированные в отчёте о поиске
Производные дигидробензопирана, фармацевтическая композиция на их основе и способы лечения
Комментарии