Комбинированный лекарственный препарат для терапии вирусных инфекций - RU2662160C1
Код документа: RU2662160C1
Описание
Данное изобретение относится к новому комбинированному лекарственному препарату, представляющему собой твердую пероральную лекарственную форму, содержащую в качестве одного из трех активных компонентов элсульфавирин натрия, которая может быть пригодна для использования в медицине при лечении вирусных инфекций, в том числе ВИЧ и гепатита В (HBV).
Вирус иммунодефицита человека - ретровирус из рода лентивирусов, вызывающий медленно прогрессирующее заболевание - ВИЧ-инфекцию [Weiss R.A. How does HIV cause AIDS. Science 1993, 260 (5112), 1273-1279. Douek D.C., Roederer M., Koup R.A. Emerging Concepts in the Immunopathogenesis of AID». Annu. Rev. Med. 2009, 60, 471-84]. Вирус иммунодефицита человека независимо открыли в 1983 году в двух лабораториях: в Институте Пастера во Франции под руководством Люка Монтанье и в Национальном институте рака в США под руководством Роберта Галло. Результаты исследований, в которых из тканей пациентов с симптомами СПИДа впервые удалось выделить новый ретровирус, были опубликованы 20 мая 1983 года в журнале Science [

Вирус поражает клетки иммунной системы, имеющие на своей поверхности рецепторы CD4: Т-хелперы, моноциты, макрофаги, клетки Лангерганса, дендритные клетки, клетки микроглии. В результате работа иммунной системы угнетается, и развивается синдром приобретенного иммунного дефицита (СПИД), организм больного теряет возможность защищаться от инфекций и опухолей, возникают вторичные оппортунистические заболевания, которые не характерны для людей с нормальным иммунным статусом. Без врачебного вмешательства ВИЧ вызывает смерть пациента в среднем через 9-11 лет после заражения (в зависимости от подтипа вируса) [https://ru.wikipedia.org/wiki/Вирус_иммунодефецита_человека].
Согласно глобальной статистике [http://www.lenoblspid.ru/news24/postid/own_news/1166] в 2015 г во всем мире: жили с ВИЧ 36,7 миллиона человек, 2,1 миллиона человек были инфицированы ВИЧ, 1,1 миллиона человек умерли от болезней, обусловленных СПИДом, 78 миллионов человек были инфицированы ВИЧ с момента начала эпидемии, из них 35 миллионов человек умерли от болезней, обусловленных СПИДом, с момента начала эпидемии.
По состоянию на декабрь 2015 года 17 миллионов человек, живущих с ВИЧ, имели доступ к антиретровирусной терапии, в то время как в июне 2015 года это число составляло 15,8 миллиона, а в 2010 году - 7,5 миллиона человек. В 2015 году 46% всех взрослых, живущих с ВИЧ, имели доступ к лечению, в то время как в 2010 году этот показатель составлял 23%.
С 2010 года число новых случаев инфицирования ВИЧ снизилось на 6%. Во всем мире число людей, инфицированных ВИЧ в 2015 году, составило 2,1 миллиона человек, а в 2010 году это число составляло 2,2 миллиона.
По сравнению с пиковым показателем в 2005 году число смертей, связанных со СПИДом, снизилось на 45%, и в 2015 году число людей, умерших в связи со СПИДом, во всем мире составило 1,1 миллиона человек. Для сравнения в 2005 году это число составляло 2 миллиона.
ВИЧ можно ослаблять с помощью комбинированной антиретровирусной терапии (APT), состоящей из трех или более антиретровирусных препаратов. APT не излечивает эти инфекции, но контролирует репликацию вирусов в организме человека и содействует укреплению иммунной системы и восстановлению ее способностей бороться с инфекциями. При проведении APT с использованием комбинаций однокомпонентных и/или двухкомпонентных препаратов продолжительность жизни пациента может быть продлена до 70-80 лет [http://www.who.int/mediacentre/factsheets/fs360/ru/].
Гепатит В - это инфекционное воспалительное заболевание ткани печени, возникающее вследствие внедрения в организм вируса гепатита В (ВГВ или HBV). Он представляет собой серьезную глобальную проблему здравоохранения. Он может вызывать хроническую инфекцию и подвергать людей высокому риску смерти от цирроза и рака печени.
По данным всемирной организации здравоохранения (ВОЗ) около 350 млн. человек в мире страдают хроническим гепатитом В. Наиболее высокая распространенность заболевания наблюдается в Центральной и Южной Африке, большей части Азии, Амазонии, Северной части Центральной и Восточной Европы, странах Ближнего Востока, где хронически инфицированы от 5% до 10% взрослого населения. Высокая распространенность хронических инфекций также обнаружена в районе Амазонки и в южных частях Восточной и Центральной Европы. На Ближнем Востоке и в Индостане, согласно оценке, хронически инфицированы 2%-5% всего населения. Среди населения Западной Европы и Северной Америки хронически инфицированы менее 1%. Россия относится к странам со средней степенью распространенности гепатита В (≈ 7% населения). Число инфицированных в РФ по разным оценкам достигает от 3 до 6 млн. человек. Заболеваемость хроническими формами гепатита В, в целом по населению, находится на уровне 13-14 на 100 тыс. населения Российской Федерации.
Вирус гепатита В относится к гепаднавирусам - гепатотропным ДНК-содержащим вирусам. Содержание вируса гепатита В в крови во время развития болезни чрезвычайно велико и составляет до 1012 вирусных частиц в 1 мл крови. Вирус гепатита В очень устойчив и сохраняется во внешней среде в течение недели. Он в 100 раз заразнее ВИЧ (вируса иммунодефицита человека). Вирус гепатита В вызывает инфекционное заболевание - острый или хронический гепатит В, который сопровождается тяжелым воспалительным поражением печени. Одним из основных признаков данного заболевания является обнаружение в крови белка вируса HbsAg, носящего название «австралийский антиген» [http://58.rospotrebnadzor.ru/rss_all;jsessionid=1С1ВЕ5СС6СС2130С60916F6C1695C57C?p_auth=6WlU2BaT&p_p_id=101_INSTANCE_Kq6J&p_p_lifecycle=1&p_p_state=exclusive&p_p_mode=view&p_p_col_id=column1&p_p_col_count=1&_101_INSTANCE_Kq6J_struts_action=%2Fasset_publisher%2Fexport_journal_article&_101_INSTANCE_Kq6J_groupId=10156&_101_INSTANCE_Kq6J_articleId=179250&_101_INSTANCE_Kq6J_targetExtension=pdf].
Лечение ВГВ-инфекции у тex, кто в нем нуждается, снижает риск гепатоклеточной карциномы и смерти. По существующим оценкам лечение принесет пользу 20-30% лиц с ВГВ-инфекцией. Однако лекарственные препараты, активные против ВГВ, не являются широкодоступными или не используются у лиц, инфицированных ВГВ [http://www.euro.who.int/en/health-topics/communicable-diseases/hepatitis/news/news/2011/11/treatment-of-chronic-hepatitis-b-virus-infection-in-resource-constrained-settings-expert-panel-consensus/russian-version-treatment-of-chronic-hepatitis-b-virus-infection-in-resource-constrained-settings-expert-panel-consensus].
Коинфекция ВИЧ/ВГВ является обычным явлением. Хроническая инфекция ВГВ встречается у 5-10% ВИЧ-инфицированных лиц, которые подвергаются ВГВ, в 10 раз чаще, чем для населения в целом [http://hivinsite.ucsf.edu/InSite?page=kb-05-03-04#S1X].
Противовирусные препараты, рекомендованные в настоящее время для лечения инфекции, вызванной вирусом иммунодефицита человека (ВИЧ), в недостаточной степени подавляют репликацию ВГВ; это вызывает серьезную озабоченность в отношении примерно 10% ВИЧ-инфицированных в Африке, у которых диагностирована коинфекция ВГВ. Обнаружено, что у лиц с коинфекцией, которые не принимают препараты для подавления ВГВ, наблюдается прогрессирующее заболевание печени. В свете этих проблем ВОЗ считает, что хроническая ВГВ-инфекция представляет собой серьезную проблему общественного здравоохранения в развивающихся странах; все ВИЧ-инфицированные лица должны проходить скрининг на ВГВ; лица с коинфекцией ВИЧ/ВГВ должны получать APT, эффективную против обоих вирусов и снижающую вероятность развития устойчивости [http://www.euro.who.int/en/health-topics/communicable-diseases/hepatitis/news/news/2011/11/treatment-of-chronic-hepatitis-b-virus-infection-in-resource-constrained-settings-expert-panel-consensus/russian-version-treatment-of-chronic-hepatitis-b-virus-infection-in-resource-constrained-settings-expert-panel-consensus].
Примерами однокомпонентных препаратов для APT могут служить элсульфавирин формулы 1 [WO 2005/102989, RU 2389719, WO 2010/028968], проингибиторы формул 2а-2l ламивудин формулы 2а и эмтрицитабин формулы 2b и их производные формул 2с-2l
[https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/021003s015,021004s015lbl.pdf; https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/021500s019lbl.pdf; US 152221613; RU 2017106609; RU 2017106610; RU 2017106611; RU 2017106615], тенофовиры формулы 3 [https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/022577lbl.pdf] и формул 4a-4k [WO 2013025788], [US 152221613], [RU 2017106609; RU 2017106610; RU 2017106611; RU 2017106615], рилпивирина гидрохлорид формулы 5 [http://www.edurant.com/shared/prescribing-information-edurant.pdf], эфавиренц формулы 6 [https://www.accessdata.fda.gov/drugsatfda_docs/label/2005/020972s026,021360s013lbl.pdf], элвитегравир формулы 7 [https://pubchem.ncbi.nlm.nih.gov/compound/Elvitegravir#section=2D-Structure] и кобицистат формулы 8
[https://pubchem.ncbi.nlm.nih.gov/compound/Cobicistat].

где R представляет собой C2H5CON-Na+, NH2;





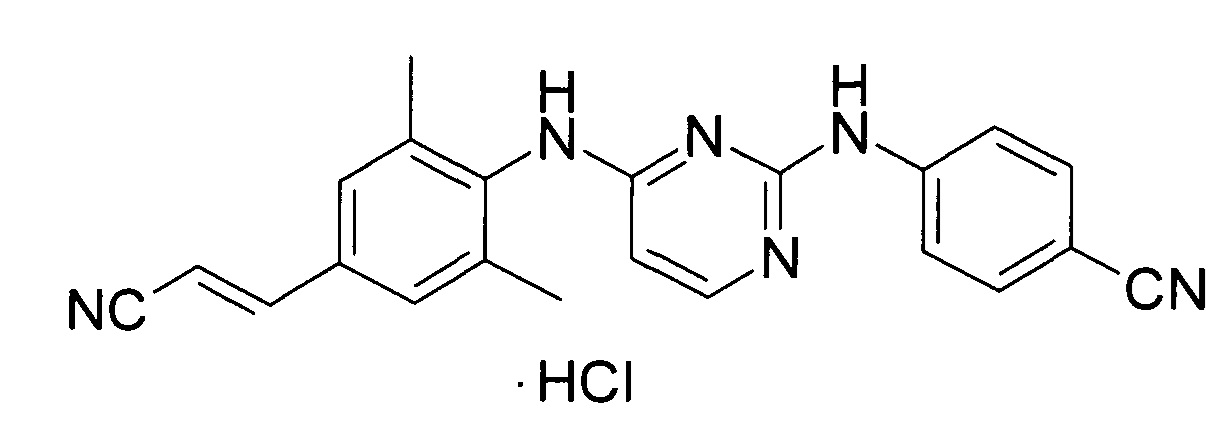


Препараты формул 1-8 имеют различный механизм действия. Так, элсульфавирин формулы 1, рилпивирин формулы 5 и эфавиренц формулы 6 являются ненуклеозидными ингибиторами обратной транскриптазы (NNRTIs), соединения формул 2а-2l являются предшественниками нуклеозидных ингибиторов обратной транскриптазы (NRTIs), а тенофовиры формул 3, 4а-4m относятся к предшественникам нуклеотидных ингибиторов обратной транскриптазы (NtRTIs). Элвитегравир формулы 5 является ингибитором интегразы (INI), а кобицистат формулы 8 - фармакокинетическим усиливающим агентом, ингибитором цитохрома Р450 3А (CYP3A). Он не проявляет противовирусную активность.
Примерами двухкомпонентных препаратов могут служить Трувада, представляющая собой таблетку, включающую в качестве активных веществ 200 мг эмтрицитабина формулы 2b и 300 мг тенофовир дизопроксил фумарата формулы 3 [WO 2004064845. WO 2006135932. http://www.gilead.com/~/media/files/pdfs/medicines/hiv/truvada/truvada_pi_old.pdf] и Дескови, представляющая собой таблетку, включающую 200 мг эмтрицитабина формулы 2b и 25 мг тенофовир алафенамид полуфумарата формулы 4f [WO 2017004244. https://www.gilead.com/~/media/files/pdfs/medicines/hiv/descovy/descovy_pi.pdf?la=en].
В настоящее время стандартное лечение вирусных заболеваний, в том числе и ВИЧ, состоит из комбинации по меньшей мере трех препаратов, обладающих различным механизмом действия (эта терапия часто называется «высокоактивной антиретровирусной терапией» или ВААРТ), которые подавляют HBV и ВИЧ.
Особенно удобными, безопасными и эффективными оказались комбинированные препараты (таблетки), включающие фиксированные дозы активных компонентов.
Наиболее продвинутыми препаратами для ВААРТ HBV и ВИЧ/СПИД являются:
Эвиплера (Комплера), включающая в качестве активных веществ 200 мг эмтрицитабина формулы 2b, 245 мг тенофовир дизопроксил фумарата формулы 3 и 27,5 мг гидрохлорида рилпивирина формулы 5, [WO 2016005327. http://www.gilead.com/~/media/files/pdfs/medicines/hiv/complera/complera_pi_old.pdf]; Атрипла, включающая в качестве активных веществ 200 мг эмтрицитабина формулы 2b, 300 мг тенофовир дизопроксил фумарата формулы 3 и 600 мг эфавиренца формулы 6 [WO 2004064845. https://aidsinfo.nih.gov/drugs/424/atripla/0/patient/. https://mini-doctor.com/pilul/atripla_tabletki_pokritie_plenochnoy_obolochkoy_CO2HCCO2H_vo_flakone-14866.html]; Дженвоя, включающая 200 мг эмтрицитабина формулы 2b, 10 мг тенофовир алафенамид полуфумарата формулы 4f, 150 мг элвитегравира формулы 7 и 150 мг кобицистата формулы 8 [https://www.gilead.com/~/media/files/pdfs/medicines/hiv/genvoya/genvoya_pi.pdf?la=en] и
Одефси, включающая 200 мг эмтрицитабина формулы 2b, 25 мг тенофовир алафенамид полуфумарата формулы 4f и 25 мг гидрохлорида рилпивирина формулы 7 [WO 2017004244. [https://www.gilead.com/~/media/files/pdfs/medicines/hiv/odefsey/odefsey_pi.pdf?la=en].
Успех эффективной и хорошо переносимой ВААРТ означает, что заболеваемость и смертность среди ВИЧ-инфицированного населения все чаще зависят от сопутствующих заболеваний, не связанных со СПИДом. В клинике все большее внимание уделяется толерантности, длительной безопасности и строгому следованию процедуры APT (Costagliola D. Demographics of HIV and aging. Curr. Opin. HIV AIDS, 2014, (4), 294). В этой связи, сохраняется существенная медицинская потребность в безопасных и эффективных новых методах ВААРТ, учитывающих возрастные популяции пациентов, не связанные с ВИЧ сопутствующие заболевания, вирусологическую резистентность и упрощение режима лечения.
Авторы изобретения обнаружили, что можно приготовить новый комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму, содержащую в качестве одного из трех активных компонентов элсульфавирин натрия формулы 1а. Эта пероральная лекарственная форма может быть пригодна для использования в медицине при лечении вирусных инфекций, в том числе ВИЧ и гепатита В (HBV).
Предметом изобретения является новый комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму, содержащую в кристаллической или поликристаллической форме терапевтически эффективное количество элсульфавирина формулы 1а, одного из предшественников NRTIs формулы 2a-2j или его фармацевтически приемлемой соли и одного из предшественников NtRTIs формулы 3 или формулы 4а-4m или его фармацевтически приемлемой соли необязательно в комбинации с эксципиентами (вспомогательными веществами).
Элсульфавирин формулы 1а является пролекарством активного соединения VM-1500А формулы 1b, которое является мощным ингибитором репликации ВИЧ-1 штамма НХВ2 в клетках линии МТ-4 и относится к классу ненуклеозидных ингибиторов обратной транскриптазы (Non-Nucleoside Reverse Transcriptase Inhibitors - NNRTI). Среднее значение IC50, полученное для VM-1500A формулы 1b на штамме НХВ2 дикого типа и для ингибирования репликации мутантных вирусов ВИЧ-1, содержащих мутации V106A, G190A, L100I/K103N и K103N/Y181C, имеют значение для: НХВ2 1,3±0,4 нМ, V106A 1,2±0,2 нМ, G190A 0,6±0,6 нМ, L100I/K103N 1,3±0,3 нМ, K103N/Y181C 1,3±0,4 нМ.
Предшественники NRTIs формул 2а-2l проявляют клиническую активность против ВИЧ и вируса гепатита В (HBV) [https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/021003s015,021004s015lbl.pdf;
https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021500s010,021896s004lbl.pdf; RU 2017106611] и представляют интерес в APT.
Тенофовир дизопроксил фумарат формулы 3 является предшественником NtRTI и обладает антивирусной активностью против ВИЧ-1 и вируса гепатита В [http://www.openaccessjoumals.com/articles/tenofovir-disoproxil-fumarate-for-the-theatment-of-hepatitis-b-virus-infection-pharmacokinetics-and-clinical-efficacy.pdf].
Тенофовиры формулы 4а-4m также являются предшественниками NtRTI и применяются для лечения инфекции ВИЧ [https://www.hepmag.com/article/fda-approves-vemlidy-tenofovir-alafenamide-taf-hepatitis-b] и хронической вирусной инфекции гепатита В.
Тенофовиры формулы 4а-4е, 4g-4m также являются предшественниками NtRTIs и, как изобретатели установили, активны против ВИЧ инфекции (Таблица 1) и гепатита В (Таблица 2).


*70% жизнеспособных клеток при 10 мкМ
Новый комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму, безопасен и упрощает режим лечения, а благодаря Элсульфавирину натрия формулы 1а эффективен при лечении вирусных заболеваний, в том числе против вируса гепатита В (HBV), дикого и мутантных вирусов ВИЧ.
Ниже приведены определения различных терминов, используемых для описания данного изобретения. Эти определения применимы к терминам, как они использованы в данном описании и формуле изобретения, если иным не ограничены в конкретных случаях либо по отдельности, либо как часть большей группы.
Термин «активный компонент» (лекарственное вещество) относится к физиологически активному веществу синтетического или иного происхождения, обладающему фармакологической активностью, которое является активным ингредиентом фармацевтической композиции.
Термин «инертный наполнитель», используемый в данном описании, относится к соединению, которое используют для получения фармацевтической композиции, и, как правило, безопасному, нетоксичному, ни биологически, ни иным образом нежелательному, и включает в себя вспомогательные вещества, которые являются приемлемыми для применения в ветеринарии, а также фармакологически приемлемыми для использования человеком. Соединения по данному изобретению могут быть введены отдельно, но обычно их будут вводить в смеси с одним или более фармацевтически приемлемыми эксципиентами, разбавителями или носителями, выбранными с учетом предполагаемого пути введения и стандартной фармацевтической практики.
Термин «комбинированный лекарственный препарат» означает твердую пероральную лекарственную форму или фармацевтическую композицию в виде таблеток, желатиновых капсул, пилюль, порошков, гранул и жевательных резинок, предназначенных для лечения и профилактики вирусных заболеваний.
Термин «кристаллическая форма» означает структуру вещества, характеризующуюся упаковкой образующих ее молекул в один из видов кристаллической решетки.
Термины «лечение» и «лечение заболевания» включают:
(1) предотвращение или снижение риска развития заболевания, то есть предотвращение развития клинических симптомов заболевания у субъекта, который может быть подвержен или предрасположен к заболеванию, но еще не испытывает или не проявляет симптомы заболевания;
(2) ингибирование заболевания, то есть остановку или уменьшение развития заболевания или его клинических симптомов; и
(3) облегчение заболевания, то есть вызывание регрессии заболевания или его клинических симптомов.
Термин «% мас. / мас.» означает массу компонента в процентах от общей массы, например слоя вещества или лекарственной формы, в которой присутствует компонент. Например, композиция, содержащая «5% мас. / мас. X», относится к композиции, в которой вес компонента X составляет 5% от общей массы композиции.
Термин «поликристаллическая форма» означает структуру вещества, имеющую поликристаллическое строение, т.е. состоящую из множества мелких монокристаллов, т.е. кристаллитов определенной кристаллической формы.
Термин «субъект» означает млекопитающее, которое включает, но не ограничивается ими, крупный рогатый скот, свиней, овец, куриц, индеек, буйволов, лам, страусов, собак, кошек и человека, предпочтительно субъектом является человек.
Термин «сегрегация», используемый в отношении определенных компонентов (например, А и В) в таблетке означает, что эти компоненты физически дискретны, так что присутствие одного компонента (например, А) существенно не влияет на стабильность при хранении другого компонента (компонентов) (например, В), от которого он отделен. Как правило, когда компоненты разделены в таблетке, они будут присутствовать в отдельных слоях в многослойной таблетке. В качестве примера компоненты А и В могут присутствовать в отдельных слоях в многослойной таблетке, где слой (а), содержащий компонент А, по существу не содержит компонента В и слой (б), содержащий компонент В, по существу не содержит компонента А. Отдельные слои могут контактировать друг с другом или могут быть разделены, например, одним или несколькими дополнительными слоями.
Термин «содержать» и его варианты, такие как «содержит» и «содержащий», должны толковаться в открытом, инклюзивном смысле, то есть «включая, но не ограничиваясь». Термин «между» по отношению к двум значениям включает в себя эти два значения, например, диапазон «между» 10 мг и 20 мг охватывает, например, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 и 20 мг.
Термин «сольват» означает молекулярный комплекс, содержащий соединение и одну или несколько фармацевтически приемлемых молекул растворителя. Примеры молекул растворителя включают воду и С1-С6-спирты, например спирт этиловый. Когда сольват представляет собой воду, можно использовать термин «гидрат».
Термин «терапевтически эффективное количество», используемый здесь, означает количество субстанции, пролекарства или лекарства, необходимое для уменьшения симптомов заболевания у субъекта. Доза субстанции, пролекарства или лекарства будет соответствовать индивидуальным требованиям в каждом конкретном случае. Эта доза может варьироваться в широких пределах в зависимости от многочисленных факторов, таких как тяжесть заболевания, подлежащего лечению, возраста и общего состояния здоровья пациента, других лекарственных средств, с помощью которых пациент проходит лечение, способа и формы введения и опыта лечащего врача. Для перорального введения суточная доза составляет приблизительно от 0,01 до 10 г, включая все значения между ними, в день в монотерапии и/или в комбинированной терапии. Предпочтительная суточная доза составляет примерно от 0,01 до 7 г в день. Как правило, лечение начинают с большой начальной «нагрузочной дозы», чтобы быстро уменьшить или устранить вирус, сопровождающей убывающую дозу до уровня, достаточного для предотвращения всплеска инфекции.
Термин «фармацевтическая композиция» обозначает композицию, включающую в себя активные компоненты и необязательно, по крайней мере, один компонент, выбранный из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых инертных наполнителей, разбавителей, носителей, вспомогательных и распределяющих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом.
Термин «фармацевтически приемлемый» по отношению к веществу относится к этому веществу, которое обычно считается безопасным и пригодным для использования без излишней токсичности, раздражения, аллергического ответа и т.п., соизмеримых с разумным соотношением выгод и риска.
Термин «фармацевтически приемлемая соль» относится к соли соединения, которое является фармацевтически приемлемым и которое обладает (или может быть преобразовано в форму, которая обладает) желаемой фармакологической активностью исходного соединения. Такие соли включают кислотно-аддитивные соли, образованные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное; или образуются с органическими кислотами, такими как уксусная кислота, бензолсульфоновая кислота, бензойная кислота, камфорсульфоновая кислота, лимонная кислота, этансульфоновая кислота, фумаровая кислота, глюкогептоновая кислота, глюконовая кислота, молочная кислота, малеиновая кислота, малоновая кислота, миндальная кислота, метансульфоновая кислота, 2-нафталинсульфоновая кислота, олеиновая кислота, пальмитиновая кислота, пропионовая кислота, стеариновая кислота, янтарная кислота, винная кислота, n-толуолсульфоновая кислота, триметилуксусная кислота и тому подобное, и соли, образующиеся, когда кислотный протон, присутствующий в исходном соединении, заменяется либо на ион металла, например ион щелочного металла, ион щелочноземельного металла или ион алюминия; или соли с органическим основанием, таким как диэтаноламин, триэтаноламин, N-метилглюкамин и тому подобное. Также в это определение включены аммониевые и замещенные или кватернизованные соли аммония. Типичные неограничивающие списки фармацевтически приемлемых солей можно найти в S.M. Berge et al., J. Pharma S Sci., 66 (1), 1-19 (1977) и Remington: Science and Practice of Pharmacy, R. Hendrickson, ed., 21-e издание, Lippincott, Williams & Wilkins, Philadelphia, PA, (2005), at п. 732, таблица 38-5, оба из которых включены здесь в качестве ссылки.
Предметом данного изобретения является новый комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму, содержащую в качестве одного из трех активных компонентов терапевтически эффективное количество элсульфавирина натрия формулы 1а в кристаллической или поликристаллической форме необязательно в комбинации с эксципиентами (вспомогательными веществами).

Более предпочтительным является комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму, содержащий в кристаллической или поликристаллической форме в терапевтически эффективном количестве элсульфавирин натрия формулы 1а, один из предшественников NRTIs формул 2a-2j или его фармацевтически приемлемую соль и один из предшественников NtRTIs формул 3, 4a-4m или его фармацевтически приемлемую соль необязательно в комбинации с эксципиентами (вспомогательными веществами).



Предпочтительным вариантом настоящего изобретения является комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки.
Предпочтительным вариантом настоящего изобретения является комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки, содержащий в качестве активных компонентов элсульфавирин натрия формулы 1а, предшественник NRTIs формул 2a-2j или его соль и тенофовир дизопроксил фумарат формулы 3 в массовом соотношении (1a):(2a-2j) или его соль: (3)≈1:10:15.
Более предпочтительным вариантом настоящего изобретения является также комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки, содержащий в качестве активных компонентов 15-25 мг элсульфавирина натрия формулы 1а, 150-300 мг предшественника NRTIs формул 2a-2j или его соли, 250-350 мг тенофовира дизопроксил фумарата формулы 3.
Более предпочтительным вариантом настоящего изобретения является также комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму в виде таблетки, содержащий в качестве активных компонентов: элсульфавирин натрия формулы 1а, предшественник NRTIs формул 2a-2j и тенофовир дизопроксил фумарат формулы 3; в качестве вспомогательных веществ: лактозу моногидрат 200, целлюлозу микрокристаллическую 102, кроскармеллозу натрия, крахмал прежелатинизированный, повидон-К30, магния стеарат, а в качестве пленочной оболочки - Vivacoat РС-8Т-181, массовое соотношение которых зависит от их природы и способа получения.
Более предпочтительным вариантом настоящего изобретения является также комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму в виде таблетки, содержащий в качестве активных компонентов 20,7 мг элсульфавирина натрия формулы 1а, 200-300 мг предшественника NRTIs формул 2a-2j, 300 мг тенофовира дизопроксил фумарата формулы 3, 386,9 мг лактозы моногидрата 200, 134,2 мг целлюлозы микрокристаллической 102, 67,1 мг кроскармеллозы натрия, 33,4 мг крахмала прежелатинизированного, 15,0 мг повидона-К30, 10,7 мг магния стеарата, а в качестве пленочной оболочки 50,0 мг Vivacoat РС-8Т-181.
Вариантом настоящего изобретения является также комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки, содержащий в качестве активных компонентов элсульфавирин натрия формулы 1а, предшественник NRTIs формул 2a-2j или его соль и тенофовир формул 4а-4m в массовом соотношении (1a):(2a-2j) или его соль: (4a-4m)≈1:10:1,25.
Более предпочтительным вариантом настоящего изобретения является комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки, содержащий в качестве активных компонентов 15-25 мг элсульфавирина натрия формулы 1а, 150-300 мг предшественника NRTIs формул 2a-2j или его соли и 10-35 мг тенофовира формул 4а-4m.
Более предпочтительным вариантом настоящего изобретения является комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки, содержащий в качестве активных компонентов 15-25 мг элсульфавирина натрия формулы 1а, 150-300 мг предшественника NRTIs формул 2a-2j или его соли и 10-35 мг тенофовира формулы 4h или 4m.
Более предпочтительным вариантом настоящего изобретения является также комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму в виде таблетки, содержащий в качестве активных компонентов: 15-25 мг элсульфавирина натрия формулы 1а, 150-300 мг предшественника NRTIs формул 2a-2j или его соли и 10-35 мг тенофовира формул 4а-4m), а в качестве эксципиентов (вспомогательных веществ): лактозы моногидрат 200, целлюлозу микрокристаллическую 102, кроскармеллозу натрия, крахмал прежелатинизированный, повидон-К30, магния стеарат, а в качестве пленочной оболочки - Vivacoat РС-8Т-181, массовое соотношение которых зависит от их природы и способа получения.
Более предпочтительным вариантом настоящего изобретения является также комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму в виде таблетки, содержащий в качестве активных компонентов 20,7 мг элсульфавирина натрия формулы 1а, 200-300 мг предшественника NRTIs формул 2a-2j, 25 мг тенофовира формулы 4f, 4h или 4m, 386,9 мг лактозы моногидрата 200, 134,2 мг целлюлозы микрокристаллической 102, 67,1 мг кроскармеллозы натрия, 33,4 мг крахмала прежелатинизированного, 15,0 мг повидона-К30, 10,7 мг магния стеарата, а в качестве пленочной оболочки 50,0 мг Vivacoat РС-8Т-181.
Более предпочтительным вариантом настоящего изобретения является также комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму в виде таблетки, содержащий в качестве активных компонентов 10-25 мг элсульфавирина натрия формулы 1а, 150-350 мг предшественника NRTIs формул 2a-2j, 5-35 мг тенофовира формулы 4f, 4h или 4m, 20-35 мг кроскармеллозы натрия, 70-120 мг микрокристаллической целлюлозы и 1-7 мг стеарата магния.
Более предпочтительным вариантом настоящего изобретения является также комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму в виде таблетки, содержащий в качестве активных компонентов 20,7 мг элсульфавирина натрия формулы 1а, 200-300 мг предшественника NRTIs формул 2a-2j, 25 мг тенофовира формулы 4f, 4h или 4m, 28 мг кроскармеллозы натрия, 105,56 мг микрокристаллов целлюлозы, 5,25 мг стеарата магния и пленочное покрытие, состоящее из Vivacoat РС-8Т-181.
Одним из вариантов данного изобретения является набор, содержащий комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму, в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки и осушитель, предпочтительно силикагель.
Одним из вариантов данного изобретения является способ получения комбинированного лекарственного препарата, представляющего собой твердую пероральную лекарственную форму, состоящую из трех активных компонентов, в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки как описано выше.
Одним из вариантов данного изобретения является способ производства комбинированного лекарственного препарата, представляющего собой таблетку, смешиванием терапевтически эффективных количеств элсульфавирина натрия формулы 1а, предшественника NRTI формул 2a-2j или его фармацевтически приемлемой соли и предшественника NtRTI формулы 3 или формул 4а-4m или его фармацевтически приемлемой соли со вспомогательными веществами с последующим прессованием.
Одним из вариантов данного изобретения является сухая гранулированная смесь элсульфавирина натрия формулы 1а, предшественника NRTIs формул 2a-2j или его фармацевтически приемлемой соли и тенофовира формулы 3, 4f, 4h или 4m.
Одним из вариантов данного изобретения является комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму, в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки для использования при терапевтическом лечении вирусной инфекции.
Одним из вариантов данного изобретения является комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму, в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки для использования при терапевтическом лечении ВИЧ-инфекции.
Одним из вариантов данного изобретения является комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму, в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки для использования при терапевтическом лечении гепатита В (HBV).
Одним из вариантов данного изобретения является способ терапевтического лечения ВИЧ-инфекции, включающий введение субъекту описанного выше нового комбинированного лекарственного препарата, представляющего собой твердую пероральную лекарственную форму, в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки.
Одним из вариантов данного изобретения является способ профилактики ВИЧ-инфекции, включающий введение субъекту комбинированного лекарственного препарата, представляющего собой твердую пероральную лекарственную форму.
Одним из вариантов данного изобретения является способ, в котором комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму, вводят с интервалом менее одного раза в день.
Одним из вариантов данного изобретения является способ, в котором комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму, вводят субъекту до и после события, которое увеличивает риск заражения ВИЧ человеком.
Изобретение относится к таблетке, содержащей элсульфавирин натрия формулы 1а, предшественник NRTIs формул 2a-2j или его соль и тенофовир формулы 3, 4f, 4h или 4m.
Таблетка покрыта оболочкой, преимущественно пленочным покрытием, таким как Vivacoat РС-8Т-181.
Предпочтительно таблетка содержит 20,7 мг элсульфавирина натрия формулы 1а, 200-300 мг предшественника NRTIs формул 2a-2j или его соли, 300 мг тенофовира дизопроксил фумарата формулы 3.
Предпочтительно таблетка содержит 20,7 мг элсульфавирина натрия формулы 1а, 200-300 мг предшественника NRTIs формул 2a-2j или его соли и 25 мг тенофовира формулы 4f, 4h или 4m.
Предпочтительно таблетка содержит в качестве вспомогательных веществ: лактозы моногидрат 200, целлюлозу микрокристаллическую 102, кроскармеллозу натрия, крахмал прежелатинизированный, повидон-К30, магния стеарат, а в качестве пленочной оболочки - Vivacoat РС-8Т-181.
Предпочтительно таблетка по изобретению имеет общую массу 1268 мг ±500 мг, или 1268±200 мг, или 1268±50 мг, или 1268 мг.
Предпочтительно таблетка содержит пленочное покрытие, предпочтительно содержащее поливиниловый спирт, полиэтиленгликоль, тальк, диоксид титана и черный оксид железа.
Предпочтительно таблетка содержит пленочное покрытие, состоящее из 50 мг Vivacoat РС-8Т-181.
В одном варианте осуществления изобретения от 29 мас. % до 68 мас. % таблетки представляет собой элсульфавирин натрия формулы 1а, предшественник NRTIs формул 2a-2j или его соль и тенофовира дизопроксил фумарат формулы 3.
В другом варианте осуществления изобретения от 19 мас. % до 50 мас. % таблетки представляет собой элсульфавирин натрия формулы 1а, предшественник NRTIs формул 2a-2j или его соль и тенофовир формулы 4f, 4h или 4m.
В одном варианте осуществления изобретения от 1,1 мас. % до 2,7 мас. % таблетки представляет собой элсульфавирин натрия формулы 1а, от 11,3 мас. % до 39,1 мас. % таблетки - предшественник NRTIs формул 2a-2j или его соль и от 16,9 мас. % до 39,1 мас. % таблетки - тенофовира дизопроксил фумарат формулы 3.
В другом варианте осуществления изобретения от 1,1 мас. % до 2,7 мас. % таблетки представляет собой элсульфавирин натрия формулы 1а, от 11,3 мас. % до 39,1 мас. % таблетки - предшественник NRTIs формул 2a-2j или его соль и от 1,4 мас. % до 3,3 мас. % таблетки - тенофовир формулы 4f, 4h или 4m.
Предметом изобретения является способ производства нового комбинированного лекарственного препарата, представляющего собой твердую пероральную лекарственную форму, в частности таблетки, который заключается в смешивании активных компонентов с эксципиентами (вспомогательными веществами) с последующим прессованием. В некоторых вариантах осуществления активные компоненты сначала смешивают и гранулируют с эксципиентами (вспомогательными веществами), например, путем сухого гранулирования. Этот этап включает, в некоторых вариантах, уплотнение валков и/или измельчение. В некоторых вариантах осуществления гранулирование смешанных активных компонентов дополнительно объединяют с экстрагранулярными эксципиентами (вспомогательными веществами), а затем прессуют.
В некоторых вариантах изобретения способ включает раздельное прессование трех активных компонентов и изготовление трехслойной таблетки.
Как правило, способы включают стадию покрытия ядер таблеток после прессования, например пленочным покрытием, как описано выше.
В общем, методы таблетирования хорошо известны в области фармации и описаны в общеизвестной книге [Remington's Pharmaceutical Sciences, 17th ed. Edited by Alfonso R. Gennaro. Mack Publishing Co., 20th and Northampton Streets, Easton, PA 18042. 1985.].
Таблетка может быть изготовлена путем прессования или формования, необязательно с одним или несколькими наполнителями. Спрессованные таблетки могут быть получены путем прессования в подходящей машине активных ингредиентов в сыпучей форме, такой как порошок или гранулы, необязательно смешанные с наполнителями.
Новый комбинированный лекарственный препарат, представляющий собой твердую пероральную лекарственную форму, в частности, таблетки, описанные выше, могут быть использованы для лечения или профилактики вирусных заболеваний, в том числе для лечения или профилактики гепатита В (HBV), ВИЧ-1 или ВИЧ-2.
Предметом данного изобретения является также способ лечения субъектов, инфицированных вирусным заболеванием (пациентов), в том числе HBV и ВИЧ, путем перорального введения пациенту нового комбинированного лекарственного препарата, представляющего собой твердую пероральную лекарственную форму, в частности, таблетку.
Предметом данного изобретения является также способ профилактики HBV и ВИЧ-инфекции у субъектов, имеющих риск заражения, путем перорального введения пациенту нового комбинированного лекарственного препарата, представляющего собой твердую пероральную лекарственную форму, в частности, таблетку.
Способы профилактики и лечения HBV и ВИЧ-инфекции у субъектов, раскрытые здесь, включают введение новой пероральной лекарственной формы, раскрытой здесь (в частности, таблетки) субъекту, обычно человеку, и обычно будут включать повторные введения, обычно один раз в день.
В некоторых вариантах осуществления раскрытые здесь способы включают повторные введения с интервалами менее одного раза в день. Например, в некоторых вариантах осуществления раскрытые здесь способы включают введение новых пероральных лекарственных форм, раскрытых здесь через день, пять раз в неделю, четыре раза в неделю, три раза в неделю, два раза в неделю или один раз в неделю.
В некоторых вариантах осуществления раскрытые здесь способы включают введение до и/или после события, которое могло бы подвергнуть человека воздействию HBV и ВИЧ или которое в противном случае увеличило бы риск заражения ВИЧ человеком, например, в качестве предварительной профилактики и/или как постконтактная профилактика. Примеры событий, которые могут повысить риск заражения ВИЧ, включают, без ограничения, использование презервативов во время анального контакта с HBV и ВИЧ-положительным партнером или партнером неизвестного HBV и ВИЧ-статуса; анальный секс с более чем 3 половыми партнерами; секс с партнером-мужчиной и диагностика инфекций, передаваемых половым путем; и отсутствие последовательного использования презервативов с сексуальным партнером, который, как известно, является HBV и ВИЧ-положительным.
В некоторых вариантах осуществления, например, при введении в виде предварительной профилактики, новые твердые пероральные лекарственные формы, раскрытые здесь, вводят от 2 до 72 часов, от 2 до 48 часов, от 2 до 24 часов или от 2 до 12 часов до события, которое увеличивает риск человека (например, до секса или другого заражения вирусом HBV и ВИЧ). В некоторых вариантах осуществления твердые пероральные лекарственные формы, раскрытые здесь, вводят в течение 72 часов, 60 часов, 48 часов, 24 часов, 12 часов, 9 часов, 6 часов, 4 часов, 3 часов, 2 часов или 1 часа до события, которое повысит риск заражения ВИЧ (например, до секса или другого заражения вирусом HBV и ВИЧ). В некоторых вариантах осуществления новые твердые пероральные лекарственные формы, раскрытые здесь, вводятся до события, которое увеличивает риск заражения HBV и ВИЧ человеком, их вводят ежедневно до события. В некоторых вариантах осуществления, когда твердые пероральные лекарственные формы, раскрытые здесь, вводят до события, которое увеличило бы риск заражения HBV и ВИЧ человеком, их вводят от одного до трех раз до события.
В некоторых вариантах осуществления, например, при введении в качестве части схемы профилактики, новые твердые пероральные лекарственные формы, раскрытые здесь, вводят от 2 до 48 часов, от 2 до 36 часов, от 2 до 24 часов или от 2 до 12 часов после события, которое повысит риск заражения ВИЧ (например, после полового контакта или другого заражения вирусом ВИЧ). В некоторых вариантах осуществления, например, при введении в виде профилактики, новые твердые пероральные лекарственные формы, раскрытые здесь, вводят в течение 7 дней, 14 дней, 21 дня, 28 дней, 30 дней или 45 дней после события, которое увеличивает индивидуальный риск приобретения ВИЧ (например, после секса или другого воздействия вируса HBV и ВИЧ). В некоторых вариантах осуществления, например, при профилактике, новые твердые пероральные лекарственные формы, раскрытые здесь, вводят в течение 30 дней после события, которое увеличивает индивидуальный риск заражения HBV и ВИЧ (например, после секса или другого воздействия вируса HBV и ВИЧ). В некоторых вариантах осуществления твердые пероральные лекарственные формы, описанные здесь, вводят менее 1 часа, 2 часа, 3 часа, 4 часа, 5 часов, 6 часов, 7 часов, 8 часов, 9 часов, 12 часов, 18 часов, 24 часов, 36 часов или 48 часов после события, которое увеличило бы риск заражения человека (например, после секса или другого воздействия вируса HBV и ВИЧ). В некоторых других вариантах осуществления твердые пероральные лекарственные формы, раскрытые здесь, вводят в течение 1 дня, 2 дней, 3 дней, 4 дней или 5 дней после события, которое увеличило бы риск заражения человека (например, после секса или другого воздействия вируса HBV и ВИЧ). В некоторых вариантах осуществления, когда новые твердые пероральные лекарственные формы, раскрытые здесь, вводятся после события, которое увеличило бы риск заражения HBV и ВИЧ человеком, они назначаются ежедневно. В некоторых вариантах осуществления, когда новые твердые пероральные лекарственные формы, раскрытые здесь, вводят после события, которое увеличивает риск заражения HBV и ВИЧ человеком, их вводят от одного до трех раз после события. В некоторых вариантах осуществления, когда твердые пероральные лекарственные формы, раскрытые здесь, вводят после события, которое увеличивает риск заражения HBV и ВИЧ человеком, их вводят один раз после события.
Настоящее изобретение далее будет описано в связи с определенными вариантами осуществления, которые не предназначены для ограничения его объема. Напротив, настоящее изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем формулы изобретения. Таким образом, следующие примеры, которые включают в себя конкретные варианты, иллюстрируют, но не ограничивают настоящее изобретение.
Пример 1. Общий способ получения твердых пероральных лекарственных форм (ТПЛФ).
Тщательно измельчают и смешивают 2,0-2,5 г элсульфавирина натрия формулы 1а, 20,0-30,0 г предшественника NRTIs формул 2a-2j и 30.0 г тенофовир дизопроксил фумарата формулы 3 или 15-35 мг тенофовира формул 4а-4m. Получают ТПЛФ (Таблица 3), которую используют для получения известным способом комбинированного лекарственного препарата в виде таблетки, желатиновой капсулы, пилюли, порошка, гранулы или жевательной резинки.

Пример 2. Определение активности против ВИЧ, цитотоксичности и индекса селективности тенофовиров формул 4f, 4h, 4k, 4m.
Определение антивирусной активности по отношению к ВИЧ активных компонентов (тестируемых веществ) проводили на линии Т-лимфоцитов, SupT1. Клетки были инфицированы ВИЧ штаммом NL4.3, несущим ген зеленого флуоресцентного белка (NL4.3-GFP). Препарат вируса получали путем трансфекции клеток 293Т провирусной ДНК. Через 48 часов после трансфекции препарат замораживали и хранили до использования. Для повышения эффективности инфекции суспензию клеток SupT1 осаждали из инфекционной смеси центрифугированием. Тестируемые вещества добавляли к клеткам непосредственно перед добавлением вируса. Через 2 часа инкубации инфекционную смесь заменяли свежей культуральной средой с тестируемыми веществами. Эффективность инфекции определяли через 45 часов путем подсчета процента флуоресцирующих клеток по сравнению с неинфицированными клеточными культурами. Цитотоксичность тестируемых веществ определяли параллельно на той же, но не инфицированной клеточной линии SupT1, используя реагент ХТТ. Использовали серийные десятикратные разведения тестируемых веществ (начиная от 10 мкМ для определения антивирусной активности, или от 100 мкМ - для цитотоксичности). В качестве негативного контроля использовали 0,1% ДМСО. Были рассчитаны значения активности ЕС50, цитотоксичности СС50 и селективного индекса СИ. Качество тестов определяли при помощи следующих контролей: отношение сигнала к фону, интегразный ингибитор ралтегравир (1 мкМ), а также воспроизводимость теста. Контролем для определения цитотоксичности служил препарат эметин (0,03, 0,09 и 0,2 мкМ). Полученные результаты представлены в Таблице 1.
Пример 3. Определение активности против ВГВ (HBV), цитотоксичности и индекса селективности тенофовиров формул 4f, 4h, 4k, 4m.
Активность против ВГВ тенофовиров формул 4f, 4h, 4k, 4m (тестируемых соединений) определяли в клеточной линии гепатомы человека АД38, несущей интегрированную ДНК вируса гепатита В (ВГВ) с терминальными повторами [Lander S, et. al, Antimicrobal Agents and Chemotherapy, 1997, pg. 1715-1720]. Данная клеточная линия была предоставлена Dr. C. Seeger, Fox Chase Cancer Center, Philadelphia, PA). Цитотоксичность соединений оценивали в параллельном режиме.
Клетки культивировали в полной среде DMEM/F12 с 2 мМ L-Глютамина (Thermo Scientific, Cat #11320033), 10% фетальной бычьей сыворотки (ThermoFisher Scientific, Cat#), 1% раствора антибиотиков-антимикотиков (ThermoFisher Scientific, Cat#15240096) и 0.3 мкг/мл тетрациклина (Sigma, Cat # T7660-5G). Клетки высевали в 96-луночные планшеты Corning Biocoat (Corning, Cat #356407) в 225 мкл полной среды без тетрациклина, 20000 клеток на лунку. Тестируемые вещества сперва растворяли в ДМСО (Sigma cat. D2650), затем в среде DMEM/F12, после чего 9 разведений с 3-кратным шагом добавляли к клеткам в объеме 225 мкл. Конечные концентрации тестируемых соединений составляли от 10 мкМ до 1 нМ. Каждое разведение препарата тестировалось на трех идентичных лунках. В качестве контроля на ингибирование использовали клетки, культивируемые в присутствии тетрациклина, т.к. тетрациклин полностью останавливает репликацию ВГВ в данной клеточной линии. Далее клетки инкубировали при 37°С в увлажненной атмосфере 5% СО2 в течение 4 дней.
Выделение секретируемой ДНК ВГВ. Через 4 дня инкубации вирусную ДНК выделяли из культуральных супернатантов при помощи набора реагентов PureLink® Pro 96 Genomic DNA Purification Kit (ThermoFisher Scientific, Cat # K183104А), используя рекомендации производителя. После элюции очищенную ДНК хранили при -20°С.
Метод количественной полимеразной цепной реакции (GWH) реального времени
(RT-qPCR) применяли с использованием прибора CFX96TM Real-Time System (Bio-Rad, Hercules, CA) и полимеразы AmpliTaq Gold® DNA Polymerase (Applied Biosystems®).
Состав реакционной смеси:
Программа циклов:
40 циклов:
Считывание флуоресцентного сигнала производили в конце каждого цикла.
Праймеры и флуоресцентные пробы были получены от компании IDT (San Diego, CA):

Значения Ct («пороговый цикл», на котором начинается заметная амплификация ДНК ВГВ), нормализованные к клеточным культурам без тестируемых соединений, определяли по формуле Е=(1/(1+100%))∧(Ct[тестируемый образец]-Ct:[K-]), где Е - нормализованный уровень ДНК ВГВ, Ct[K-] и Ct[тестируемый образец] - значения Ct для образцов без- и с тестируемым препаратом соответственно. Значения ЕС50 (Таблица 2) рассчитывали при помощи программы Graph Prizm.
Цитотоксичность тестируемых соединений определяли параллельно на той же клеточной линии АД38. Клетки культивировали в черной микроплате с прозрачным дном (96 ячеек, 104 клеток на лунку) в полной среде DMEM/F12 с 2 мМ L-Глютамина (Thermo Scientific, Cat #11320033), 10% фетальной бычьей сыворотки (ThermoFisher Scientific, Cat#), 1% раствора антибиотиков-антимикотиков (ThermoFisher Scientific, Cat#15240096). Клетки AD38 высевались в 96-луночные планшеты (7.5×103 клеток на лунку в 100 мкл питательной среды). Растворы тестируемых соединений в среде ДМЕМ готовились непосредственно перед использованием. Всего готовилось 9 серийных трехкратных разведений. Через 4 часа после высевания клеток серийные разведения препаратов добавлялись к клеткам (100 мкл на лунку). Конечная концентрация тестируемых соединений составляла от 30 мкМ до 10 нМ, а ДМСО - 0.5%. При необходимости исследовались более высокие концентрации тестируемых веществ. Далее клетки инкубировали в течение трех дней при 37°С/5% в увлажненной атмосфере СО2. Количество живых клеток определяли при помощи набора ATPLite (Perkin Elmer, Бостон, США) в соответствии с инструкциями производителя. Для каждого соединения использовали три независимых повтора. Промывали дважды каждую лунку фосфатно-солевым буфером (0,2 мл/лун) и затем лизировали клетки добавлением клеточного буфера (50 мкл/лун, все указанные реактивы входят в комплект набора ATPLite). Микроплату инкубировали в течение 5 минут на вращающейся платформе при 600 об/мин, после чего добавляли в каждую лунку 50 мкл раствора субстрата (часть набора ATPLite). Инкубировали еще 5 минут на вращающейся платформе при 600 об/мин, выдерживали 10 минут в темноте и затем измеряли люминесценцию на приборе TopCount NXT (Packard, Perkin Elmer). В качестве количественного параметра для оценки цитотоксичности использовали величину СС50, которая соответствует концентрации вещества, при которой погибает 50% клеток. Расчет параметра СС50: для расчета эффективности ингибирования (% Инг) использовали формулу: % Инг=[(Лпоз-Лэкс)/Лпоз-Лотр)]*200%, где Лпоз - положительный контроль, люминесценция в ячейках с клетками без вещества; Лотр - отрицательный контроль, люминесценция в ячейках со средой без клеток; Лэкс - люминесценция в ячейках с веществом в определенной концентрации. Значения СС50 (Таблица 2) затем расчитывали при помощи программы XLfit 4.
Пример 4. Определение острой токсичности, переносимых токсических и летальных доз ТПЛФ.
Исследование острой токсичности ТПЛФ и определение переносимых токсических и летальных доз ТПЛФ при однократном внутрижелудочном введении самцам и самкам мышей и крыс. Исследования выполнены на 24 крысах-самцах массой 235-260 г и 28 крысах-самках массой 225-250 г, а также на 24 мышах-самцах массой 21-25 г и 24 мышах-самках массой 20-24 г. Было использовано по 8 групп самцов и самок крыс и по 8 групп самцов и самок мышей. ТПЛФ вводили в максимально возможном объеме 10 мл/кг трехкратно в один день с интервалом 40 минут. Перед введением предварительно измельчали, полученную массу перетирали в ступке и перемешивали с 0,5% раствором Твина-80 для получения суспензии, пригодной для внутрижелудочного введения животным в объеме ≤10 мл/кг. Растворы для введения готовились ежедневно в день введения. Введение подготовленных суспензий ТПЛФ производили каждый день в одно и то же время (отклонение составляло не более 4-х часов). Перед введением подготовленных суспензий ТПЛФ регистрировали массу тела животных, после введения в течение 1 часа наблюдали за состоянием экспериментальных животных. Далее в течение 14 суток после введения ТПЛФ ежедневно регистрировали массу тела крыс и мышей и проводили осмотр с целью выявления случаев гибели и отклонений от нормы в состоянии животных. В результате изучения определены параметры острой токсичности ТПЛФ. Определить ЛД50 ТПЛФ технически невозможно в связи с тем, что введение максимально возможных доз ТПЛФ не вызвало гибели животных. В результате исследования установлено, что внутрижелудочное введение ТПЛФ в максимально возможной дозе 600/6000/9000 мг/кг, а также в группах, получавших препараты сравнения (соединение формулы 1а) в дозе 3000 мг/кг и Труваду (соединение формулы 2b + соединение формулы 3) в дозе 6000/9000 мг/кг не оказывало влияния на прирост массы тела - таковой не отличался в контрольных и опытных группах как мышей, так и крыс (самцов и самок). Также не было выявлено различий в потреблении корма и воды во всех опытных и контрольных группах. Внутрижелудочное введение ТПЛФ и препаратов сравнения не оказало влияния на относительную массу органов животных опытных групп, статистически значимых различий с контрольными группами не выявлено. При макроскопическом исследовании у животных, получавших препараты, каких-либо отличий от контрольных групп не выявлено.
Пример 5. Комбинированный лекарственный препарат, представляющий собой ТПЛФ в виде таблеток.
ТПЛФ в виде таблеток получали с использованием процесса обработки методом сухого гранулирования - прессования - пленочного покрытия. Сухое гранулирование ТПЛФ путем уплотнения валиком проводили для минимизирования воздействия ТПЛФ на влагу во время процесса гранулирования. Общий производственный процесс состоял из смазки ТПЛФ с помощью внутригранулярных наполнителей с последующим уплотнением и фрезерованием валков. Полученные гранулы композиций ТПЛФ затем смешивали и смазывали экстрагранулярными эксципиентами для получения конечных порошковых смесей ТПЛФ, которые прессовали в таблетки, а затем покрывали пленкой Vivacoat РС-8Т-181 или Opadry II White 85F18422.
Содержание активных компонентов в одной таблетке приведено в Таблице 4.


Реферат
Изобретение относится к новому комбинированному лекарственному препарату, для использования в виде твердой пероральной лекарственной формы, содержащему в качестве одного из трех активных компонентов элсульфавирин натрия. Лекарственный препарат может быть использован при лечении вирусных инфекций, в том числе ВИЧ и гепатита В (HBV). Комбинированный лекарственный препарат содержит терапевтически эффективное количество элсульфавирина натрия формулы 1а или его фармацевтически приемлемой соли, и в качестве двух других активных компонентов терапевтически эффективное количество нуклеозидного ингибитора обратной транскриптазы (NRTI) формул 2a-2j или его фармацевтически приемлемой соли и терапевтически эффективное количество нуклеотидного ингибитора обратной транскриптазы (NtRTI) формулы 3 или 4f, 4h, 4k, 4m или его фармацевтически приемлемой соли в комбинации со вспомогательными веществами. Структурные формулы элсульфавирина натрия формулы 1а, нуклеозидного ингибитора обратной транскриптазы (NRTI) формул 2a-2j и нуклеотидного ингибитора обратной транскриптазы (NtRTI) формулы 3 или 4f, 4h, 4k, 4m указаны в формуле изобретения. 3 н. и 17 з.п. ф-лы, 4 табл., 5 пр.
Формула





Документы, цитированные в отчёте о поиске
Комбинация анти-вич ингибиторов обратной транскриптазы и протеазы
Композиции ценикривирока и способы их получения и применения
Комментарии