Гидрогелевый препарат с длительным высвобождениемлекарства - RU2121830C1
Код документа: RU2121830C1
Чертежи






















Описание
Изобретение относится к препарату с длительным высвобождением активного ингредиента, который обладает способностью выделять лекарственное средство в течение длительного промежутка времени. Более конкретно, настоящее изобретение относится к гидрогелевому препарату с длительным высвобождением активного ингредиента, который может в достаточной степени высвобождать лекарственное средство не только в верхней части пищеварительного тракта, но и в его нижней части, в частности в толстой кишке.
До настоящего времени для реализации длительного высвобождения лекарств предлагались различные препараты гидрогелевого типа. Например, в японской заявке JP-A-62-120315 раскрывается препарат, который получают путем плавления при сжатии лекарственного средства, образующего гидрогель водорастворимого полимера и энтеросолюбильной покрывающей основы (термин "JP-A", используемый в данном описании, означает "нерассмотренная опубликованная патентная японская заявка"). В японской заявке JP-A-63-215620 раскрывается препарат гидрогелевого типа, который содержит ядро, состоящее из лекарственного средства и водорастворимого полимера, и внешний слой, который в качестве основы содержит водорастворимый полимер. В японском патенте JP-B-40-2053 раскрывается рецептура с длительным выделением, которая содержит смесь лекарства и высокомолекулярного полимера этиленоксида и в качестве необязательного компонента гидрофильное соединение (понятие "JP-B", используемое в данном описании, означает "рассмотренный японский патент").
Однако все эти препараты выполнены таким образом, чтобы непрерывно высвобождать лекарственное средство, пока введенный препарат еще удерживается в верхней части пищеварительного тракта, обычно в желудке и тонком кишечнике, и не предназначены для обеспечения высвобождения лекарства в нижней части пищеварительного тракта, обычно в толстой кишке, где присутствует небольшое количество воды. Следовательно, для любого препарата с длительным высвобождением активного соединения, предназначенного для высвобождения лекарства с целью абсорбции в процессе перемещения вниз по пищеварительному тракту, степень выделения лекарства и его абсорбция в верхней части пищеварительного тракта оказывают основное влияние на биодоступность лекарства. Однако обычно полагают, что едва ли можно ожидать высвобождения лекарства в толстой кишке вследствие незначительного содержания воды и влияния оподогенного (spodogenous) содержимого, и фактически было проведено небольшое количество исследований по высвобождению лекарств в толстой кишке (Pharm. Tech. Japan 8, (1), 41, - 1992).
Кроме того, при разработке препаратов с длительным высвобождением активного соединения большое значение также имеет биологический полупериод существования самого лекарственного средства. В общем случае считается трудным создание препарата, обеспечивающего значительное длительное выделение лекарства, которое обладает небольшим полупериодом существования (The Pharmaceuticals monthly, 25(11), 1983, 29).
В результате интенсивных исследований в области длительного высвобождения лекарственных средств авторы настоящего изобретения установили, что выделение лекарства в толстой кишке, в которой присутствует небольшое количество воды, может быть достигнуто путем создания препарата, который, находясь в верхней части пищеварительного тракта, например в желудке или тонком кишечнике, обладает способностью абсорбировать воду своим ядром и подвергаться практически полной желатинизации, а затем в форме геля перемещается в нижнюю часть пищеварительного тракта. Настоящее изобретение основано на этом открытии.
Таким образом, настоящее изобретение относится к препарату гидрогелевого
типа с
длительным выделением
лекарственного средства, который содержит:
(1) по меньшей мере одно лекарство,
(2) вспомогательное вещество, обеспечивающее проникновение воды в ядро
рецептуры
и
(3) полимер,
образующий гидрогель, и эта рецептура, находясь в верхней части пищеварительного тракта, например в желудке или тонком кишечнике, подвергается практически полной
желатинизации
и обладает способностью
выделять лекарственное средство в толстой кишке.
Понятие "практически полная желатинизация" препарата, используемое в данном описании, относится к состоянию, при котором не менее 70%, предпочтительно не менее 80% препарата желатинизировано.
Так как даже толстый кишечник может быть использован в качестве абсорбирующего участка, препарат с длительным высвобождением активного соединения настоящего изобретения в значительной степени увеличивает период абсорбции лекарственного средства и, следовательно, обеспечивает устойчивое содержание лекарства в крови. Таким образом, препарат настоящего изобретения абсорбирует воду, пока он находится в верхней части желудочно-кишечного тракта, и подвергается практически полной желатинизации, а затем перемещается в нижнюю часть желудочно-кишечного тракта, причем его поверхность постоянно подвергается эрозии и сохраняет способность высвобождать лекарство за счет дальнейшей эрозии в нижней части желудочно-кишечного тракта. В результате этого достигается длительная и достаточная абсорбция лекарственного средства даже в толстом кишечнике, где присутствует незначительное количество воды.
Ниже препарат с длительным высвобождением лекарственного средства настоящего изобретения описывается более подробно.
Лекарство или лекарственные средства, которые могут быть использованы в препарате настоящего изобретения, не ограничены по типам при условии, что они могут быть использованы в системах с длительным высвобождением.
Таким образом, конкретными примерами лекарств являются противовоспалительные, жаропонижающие, противосудорожные и/или анальгетические агенты, такие как индометацин, диклофенак, диклофенак натрия, кодеин, ибупрофен, фенилбутазон, оксифенилбутазон, мепиризол, аспирин, этензамид, ацетаминофен, аминопирин, фенацетин, бутилбромид скополамина, морфин, этомидолин, пентазоцин, фенопрофен кальция и т.д.; туберкулостатические средства, такие как изониазид, хлоргидрат этамбутола и т.д.; лекарства для сердечно-сосудистой системы, такие как изосорбитдинитрат, нитроглицерин, нифедипин, хлоргидрат барнидипина, хлоргидрат никардипина, дипиридамол, амринон, хлоргидрат инденолола, хлоргидрат гидралазина, метилдоп, фуросемид, спиронолактон, гуанетидин нитрат, резерпин, хлоргидрат амосулалола и т.д.; противопсихозные средства, такие как хлоргидрат хлорпромазина, хлоргидрат амитриптилина, немонаприд, галоперидол, хлоргидрат моперона, перфеназин, диазепам, лоразепам, хлордиазепоксид и т.д.; антигистаминные средства, такие как хлорфенираминмалеат, хлоргидрат дифенилгидрамина и т.д.; витамины, такие как тиаминнитрат, токоферолацетат, цикотиамин, пиридоксальфосфат, кобамамид, аскорбиновая кислота, никотинамид и т.д.; лекарства для лечения подагры, такие как аллопуринол, колхицин, пробенецид и т.д.; снотворные седативные средства, такие как аминобарбитал, бромвалероилмочевина, мидазолам, хлоральгидрат и т.д. ; противоопухолевые средства, такие как фторурацил, кармофур, хлоргидрат акларубицина, циклофосфамид, тиотепа и т.д.; противогиперемические средства, такие как фенилпропаноламин, эфедрин и т.д.; противодиабетические средства, такие как ацетогаксамид, инсулин, толбутамид и т.д.; диуретические средства, такие как гидрохлоротиазид, политиазид, триамтерен и т.д.; бронхолитические средства, такие как аминофиллин, формотеролфумарат, теофиллин и т.д.; противокашлевые средства, такие как фосфат кодеина, носкамин, фосфат димеморфана, декотрометофан и т.д.; противоаритмические срества, такие как хинидиннитрат, дигитоксин, хлоргидрат пропафенона, прокаинамид и т.д.; местные анестезирующие средства, такие как этиламинобензоат, лидокаин, хлоргидрат дибукаина и т.д.; противоэпилептические средства, такие как фенитоин, этосуксимид, примидон и т.д.; синтетические адренокортикальные стрероиды, такие как гидрокортизон, преднизолон, триамцинолон, бетаметазон и т.д.; лекарства пищеварительной системы, такие как фамотидин, хлоргидрат ранитидина, циметидин сукралфат, сульпирид, тепренон, плаунотол и т.д.; лекарства центральной нервной системы, такие как инделоксазин, индебенон, хлоргидрат тиаприда, хлоргидрат бифемелана, хопантенат кальция и т.д.; средства для лечения гиперлипемии, такие как правастатин натрия и т.д.; и антибиотики, такие как ампицилин фталидил гидрохлорид, цефотетан, джосамицин и т.д. Обычное лекарство среди приведенных выше лекарств представляет собой хлоргидрат никардипина. Также могут быть использованы лекарства, имеющие небольшой биологический полупериод существования. Количество лекарства представляет собой любое эффективное количество, но обычно составляет не более 85 мас.%, предпочтительно не более 80 мас.% из расчета на общую массу препарата.
Для того чтобы эти лекарства могли легко абсорбироваться в толстой кишке, в которой присутствует небольшое количество воды, предпочтительно заранее повысить их растворимость. Могут быть использованы уже известные методики повышения растворимости лекарственных средств, которые применимы к гидрогелевым препаратам. Из таких методик (солюбилизирующая обработка) может быть упомянут способ, включающий добавление поверхностно-активного вещества (например, полиоксиэтилен-гидрогенированное касторовое масло, полиоксиэтилен-сорбитановые эфиры высших жирных кислот, полиоксиэтилен-полиоксипропиленгликоли, эфиры сахарозы и жирных кислот и т.д.) и способ, включающий приготовление твердой дисперсии лекарства и солюбилизатора, такого как полимер (например, водорастворимого полимера, такого как гидроксипропилметилцеллюлозы (ГПМЦ), поливинилпирролидон (ПВП), полиэтиленгликоль (ПЭГ) и др., или энтеросолюбильного полимера, такого как карбоксиметилэтилцеллюлоза (КМЭЦ), фталат гидроксипропилметилцеллюлозы (ФГПМЦ), сополимер метилметакрилата и метакриловой кислоты (Endragit L и S; торговое название фирмы Rhom & Haas Co. и др.)). Если лекарство представляет собой основное вещество, то может быть использован способ, включающий добавление органической кислоты, такой как лимонная кислота, винная кислота и аналогичные соединения. Если необходимо, то также может быть использован способ, включающий образование клатратного соединения с использованием циклодекстрина или подобные соединения. Эти методики солюбилизации могут быть при необходимости модифицированы в зависимости от свойства конкретного лекарства ("Recent Manufacturing Pharmacy Technigue and ist Application I", Isamu Utsumi et al., Midicinal Journal, 157-159 (1983); и "Phasmacy Monogragh N 1, Bioavailability", Tsuneji Nagai et al., Softsience Co., 78 - 82 (1988)).
Из указанных способов особенно предпочтительным является способ, включающий приготовление твердой дисперсии лекарственного средства и солюбилизатора (JP-A-56-49314 и патент Франции 2460667).
Добавка, позволяющая воде проникать в ядро препарата настоящего изобретения (эта добавка, обеспечивающая проникновение воды в ядро препарата, ниже будет называться "гидрофильной основой"), представляет собой такую добавку, для которой количество воды, необходимое для растворения 1 г гидрофильной основы, составляет не более 5 мл и предпочтительно не более 4 мл при температуре 20±5oC. Чем выше растворимость гидрофильной основы в воде, тем более эффективной является основа в обеспечении проникновения воды в ядро препарата. Гидрофильная основа представляет собой, между прочим, высокогидрофильные полимеры, такие как полиэтиленгликоль (ПЭГ; например, ПЭГ400, ПЭГ1500, ПЭГ 4000, ПЭГ6000 и ПЭГ20000, производимые фирмой Nippon Oils and Fats Co.) и поливинилпирролидон (ПВП: например, ПВП К30, торговое название фирмы BASF), сахарные спирты, такие как D-сорбит, ксилит и др., сахара, такие как сахароза, безводная мальтоза, D-фруктоза, декстрин (например, декстрин 40), глюкоза и т.д., поверхностно-активные вещества, такие как полиоксиэтилен-гидрогенизированное касторовое масло (HCO, например, Cremophor RH 40, производимый фирмой BASF, HCO-40 и HCO-60, производимые фирмой Nikko Chemicals Co. ), полиоксиэтилен-полиоксипропиленовый гликоль (например, Pluronic F68, производимый фирмой Asahi Denka Kogyo K.K.), полиоксиэтиленсорбитановый эфир высокомолекулярной жирной кислоты (Tween, например, Tween 80, производимый фирмой Kanto Kagaku K. K. ) и др.; соли, такие как хлорид натрия, хлорид магния и др. , органические кислоты, такие как лимонная кислота, винная кислота и др., аминокислоты, такие как глицин, бета-аланин, хлоргидрат лизин и др., а также аминосахара, такие как Meglumin.
Предварительными гидрофильными основами являются ПЭГ6000, ПВП, D-сорбит и др.
Количество такой гидрофильной основы зависит от свойств лекарственного средства (растворимость, терапевтическая эффективность и т.д.) и содержащая лекарства, растворимости самой гидрофильной основы, характеристик используемого образующего гидрогель полимера, состояния пациента на момент приема препарата и других факторов. Однако количество гидрофильной основы предпочтительно должно быть на уровне, достаточном для обеспечения практически полной желатинизации за время нахождения препарата в верхней части пищеварительного тракта. Препарат остается в верхней части пищеварительного тракта различное время, которое зависит от вида и конкретного индивида, но составляет приблизительно 2 ч после применения в случае собак и приблизительно 4 часа после применения в случае человека (Br.J.Clien. Pharmac, 1988, 26, 435-443). При назначении человеку содержание гидрофильной основы предпочтительно должно быть достаточным для того, чтобы достигнуть практически полной желатинизации в течение от 4 до 5 часов после приема. Таким образом, содержание гидрофильной основы составляет обычно приблизительно 5-80 мас.% и предпочтительно приблизительно 5-60 мас.% из расчета на общую массу препарата.
Если содержание гидрофильной основы слишком мало, то необходимая желатинизация в ядре препарата не протекает, поэтому высвобождение лекарственного средства в прямой кишке становится недостаточным. С другой стороны, если содержание гидрофильной основы избыточно, то процесс желатинизации протекает за более короткое время, но полученный гель становится таким хрупким, что выделение лекарства осуществляется слишком быстро, что не удается достичь достаточного длительного выделения лекарства. Более того, так как количество основы слишком велико, то продукт становится объемным.
Упоминавшийся выше полимер, образующий гидрогель, должен обладать физическими характеристиками, включая вязкость в желатинизированном состоянии, которые позволяют препарату настоящего изобретения сохранять свою форму более или менее продолжительное время в процессе перемещения вниз по пищеварительному тракту, а именно в толстой кишке, и противостоять сократительным силам пищеварительного тракта, связанным с перевариванием пищи.
Полимер, образующий гидрогель, который может быть использован в препарате настоящего изобретения, предпочтительно представляет собой полимер, обладающий при желатинизации высокой вязкостью. Например, предпочтительным является полимер, имеющий вязкость 1%-ного водного раствора (при 25oC) не менее 1000 сП.
Свойства полимера зависят от его молекулярного веса. Гидрогельобразующий полимер, который может быть использован в настоящем изобретении, предпочтительно представляет собой вещество со сравнительно высоким молекулярным весом, а именно полимер, имеющий средний молекулярный вес не менее 2•106 и более предпочтительно имеющий молекулярный вес не менее 4•106.
К таким полимерам относятся полиэтиленоксид (ПЭО), имеющий молекулярный вес не менее 2•106 (например, Polyox WSR-303 со средним молекулярным весом 7• 106, вязкостью, 7500-1000 сП (1%-ный раствор в воде, 25oC); Polyox WSR Coagulant со средним молекулярным весом 5•106, вязкостью 5500-7500 сП (условия те же); Polyox WSR-301 со средним молекулярным весом 4•106, вязкостью 1650 - 5500 сП (те же условия); Polyox WSR-N-60K со средним молекулярным весом 2•106, вязкостью 2000 - 4000 сП (2%-ный раствор в воде, 25oC); - торговые названия фирмы Union Carbide Co.); гидроксипропилметилцеллюлоза (ГПМЦ) (например, Metolose 90 H100000, вязкость 4100-5600 сП 1%-ного водного раствора, 20oC, Metolose 90SH50000, вязкость 2900-3900 сП при тех же условиях, Metolose 90SH30000, вязкость 25000-35000 сП, 2%-ный раствор в воде, 20oC, все полимеры с торговой маркой Shin-Etsu Chemicals Co.), натрий-карбоксиметилцеллюлоза (КМЦ-Na) (например, Sanlose F-150MC, средний мол. вес 2•105, вязкость 1200 - 1800 сП, 1%-ный раствор в воде, 25oC, Sanlose F-1000MC, средний мол. вес 42•104, вязкость 1200-12000 сП, при условиях, аналогичных приведенным выше, Sanlose F-300MC, средний мол. вес 3•105, вязкость 2500 - 3000 сП, при условиях, аналогичных приведенным выше, все полимеры с торговой маркой фирмы Nippon Seishi Co., Ltd.); гидроксиэтилцеллюлоза (ГЭЦ) (например, HEC Daicel SE850, средний молекулярный вес 148•104, вязкостью 2400-3000 сП, 1% в воде, 25oC; HEC Daicel E900, средний молекулярный вес 156•104, вязкость 4000-5000 сП, условия те же, все торговой марки фирмы Daicel Chemical Industries); карбоксивиниловые полимеры (например, Carbopol 940, средний молекулярный вес, 25•105; фирма Goodrich Chimical Co.) и т.д.
Предпочтительным является ПЭО, имеющий средний молекулярный вес не менее 2•106. Если необходимо постоянное выделение лекарства в течение длительного периода, например в течение более 12 часов, предпочтительным является полимер, имеющий более высокий молекулярный вес, например средний молекулярный вес не менее 4•106, или более высокую вязкость, например вязкость не менее 3000 сП, при концентрации в воде 1% при 25oC.
Вышеуказанный гидрогельобразующий полимер может быть использован отдельно или в виде смеси двух или нескольких типов указанных выше полимеров. Или для получения препарата настоящего изобретения может использоваться смесь из двух или более видов других полимеров, которая имеет характеристики, удовлетворяющие изобретению.
Для того чтобы обеспечить высвобождение лекарства в толстой кишке человека, необходимо, чтобы часть препарата, подвергнутого желатинизации, оставалась в толстой кишке по меньшей мере в течение 6 - 8 часов, предпочтительно по меньшей мере в течение 12 часов после введения.
Для того чтобы получить гидрогелевый препарат, обладающий такими свойствами, хотя это и зависит от объема препарата, типа полимера и его свойств, а также от количеств лекарства и добавки, обеспечивающей проникновение воды в ядро препарата, обычно предпочтительно, чтобы препарат содержал 10-95 мас. % (предпочтительно 15-90 мас.%) гидрогельобразующего полимера из расчета на препарат весом менее 600 мг. При этом препарат содержит не менее 70 мг из расчета на препарат и предпочтительно не менее 100 мг из расчета на препарат гидрогельобразующего полимера. Если количество этого полимера меньше указанного выше уровня, препарат не будет устойчив к разрушению в желудочно-кишечном тракте в течение достаточно длительного периода и необходимая длительность высвобождения лекарственного средства может быть не достигнута.
Что касается типов и соотношений гидрофильной основы и гидрогельобразующего полимера (последний далее называется гидрогельобразующей основой), то их применимость иллюстрируются с помощью следующих экспериментов.
Экспериментальный пример. (Типы и соотношения гидрофильной основы и гидрогельобразующей основы).
(1) Временной профиль скорости желатинизации гидрогелевого препарата с длительным высвобождением лекарства в соответствии с настоящим изобретением.
Образец
Смешивают в ступке 100 вес. ч. гидрогельобразующей основы Polyox
WSR - 303 (далее называемого POLY OX303) с 150 вес.ч.
гидрофильной основы П3Г6000. Смешанную композицию
формуют под давлением с использованием масляного пресса при давлении сжатия 1 т/пуансон с
получением таблеток размером 8,0 мм и весом 200 мг
каждая.
Изучение желатинизации
Исследование желатинизации с использованием Фармакопеи Японии XII (называемой далее "JP"),
тест дезинтеграции жидкости 2 проводят в
соответствии с JP методом испытания растворения 2 (лопастной метод)
при скорости лопасти 25 об/мин. Образцы таблеток отбирают через определенные интервалы,
удаляют слой геля и измеряют диаметр
(Dнабл.) части, которая не образует геля. Используя значение
Dнабл., рассчитывают индекс желатинизации (G) (табл. 1, фиг. 1 и уравнение
1).
Используемый в данном описании "индекс желатинизации" предоставляет собой часть таблетки в процентах, которая подверглась желатинизации. Метод расчета индекса желатинизации не является строго ограниченным, и в качестве примера может быть упомянут приведенный ниже метод.
То есть исследуемая таблетка подвергается увлажнению в течение заранее определенного времени, а затем измеряется объем (или вес) части таблетки, не образовавшей гель, а полученный результат вычитается из объема (или веса) таблетки до начала испытания.
Более конкретно, гелевый слой таблетки, которая подверглась увлажнению в течение определенного промежутка времени, удаляют, а затем измеряют диаметр (или толщину) той части, которая не образовала гель. Индекс желатинизации рассчитывают по уравнению 1. Индекс желатинизации может быть также рассчитан по уравнению 2, которое приведено ниже.
В качестве варианта можно также указать способ, в котором используется различие в прочности между гелевым слоем и негелевой частью, и диаметр (или толщина) при определенном давлении, как полагают, представляет собой диаметр (или толщину) части, не образующей гель, при этом индекс желатинизации рассчитывают по уравнению 1.
Уравнение 1
Индекс желатинизации (G,%) = (1 - (Dнабл.)3/(Dнач.
)3)•100
Dнабл. - диаметр части, подвергшейся желатинизации после начала
испытания;
Dнач. - диаметр препарата до начала испытания.
Результаты
Гидрогелевая таблетка, содержащая в качестве гидрофильной основы ПЭГ6000, претерпевает
желатинизацию, при этом диаметр ее ядра постепенно уменьшается с практически
постоянной скоростью. Через два
часа после начала испытаний гидрогелевая таблетка практически прекращает
желатинизироваться (но не менее чем на 80%).
(2) Содержание гидрофильного основания.
Образцы
Одна сотая вес. часть гидрогельобразующей основы POLYOX303
смешивается в ступке с гидрофильной основой ПЭГ6000, которая используется в различных пропорциях
от 0 до 150 вес. ч.
Полученную смесь формуют под давлением с помощью масляного пресса при давлении
сжатия 1 т/пуансон и получают таблетки с диаметром 8,0 мм и весом 200 мг каждая.
Изучение
желатинизации
Исследование желатинизации с использованием JP теста дезинтеграции
жидкости 2 проводят в соответствии с JP методом испытания растворения 2 (лопастной метод) при
скорости
лопасти 25 об/мин. Образцы таблеток отбирают через определенные интервалы, снимают слой геля и
измеряют диаметр (Dнабл.) части, не образовавшей геля. Используя значение Dнабл.,
рассчитывают индекс желатинизации (G) (табл. 2, фиг. 2).
Результаты
Установлено, что введение 15 вес. ч. (13,0% от веса таблетки) гидрофильной основы ПЭГ 6000
приводит к
желатинизации не менее 80% таблетки за два часа. Также найдено, что введение 10 вес.ч. (9,1% от
веса таблетки) гидрофильной основы ПЭГ6000 приводит к не менее чем 80%-ной желатинизации за 4
ч.
(3) Выбор гидрофильной основы.
Образец
Одну сотую вес.ч.
гидрогельобразующей основы POLYOX303 смешивают в ступке со 100 вес.ч. каждой исследуемой
гидрофильной
основы и полученную смесь формуют под давлением с использованием масляного пресса при давлении
сжатия 1 т/пуансон. Получают таблетки диаметром 8,0 мм и весом 200 мг каждая.
Изучение
желатинизации
Исследование желатинизации с использованием JP теста дезинтеграции
жидкости 2 проводят в соответствии с JP методом испытания растворения 2 (лопастной метод) при
скорости
лопасти 25 об/мин. Образцы таблеток отбирают через определенные интервалы, снимают слой геля и
измеряют диаметр (Dнабл.) части, не образовавшей геля. Используя значение Dнабл.,
рассчитывают индекс желатинизации (G) (табл. 3, фиг. 3).
Результаты
При добавлении Д-маннита и лактозы, для растворения 1 г которых необходимо соответственно 6 и
8 мл воды,
системы отличаются индексами желатинизации, сравнимыми с индексом системы на основе только
одного POLYOX303. Этот факт указывает на то, что эти добавки менее эффективно способствуют
желатинизации в
ядре таблетки.
Установлено, что в качестве гидрофильной основы, обеспечивающей не менее чем 80%-ную желатинизацию в течение 2 ч, могут быть использованы высокорастворимые основы (которые требуют для растворения 1 г не более 5 мл, предпочтительно не более 4 мл воды), такие как глицин ПВП К30, ПЭГ6000 и Д-сорбит.
(4) Изучение гидрогельобразующей основы.
Используя в качестве модельных лекарств ацетаминофен и хлоргидрат инкардипина (Pd), определяют количество и молекулярный вес гидрогельобразующей основы, которая необходима для получения препарата с длительным выделением лекарства.
I. Определение
оптимального количества
Изучено соотношение между содержанием гидрогельобразующей
основы и профилем
растворения.
1. Ацетаминофен.
Компоненты, представленные в табл. 4, смешивают в указанных пропорциях в ступке, и каждую полученную композицию формуют под давлением с использованием масляного пресса при давлении сжатия 1 т/пуансон. Получают таблетки, каждая из которых содержит 50 мг ацетаминофена.
2. Хлоргидрат никардипина (Pd).
Растворяют 1 вес. ч. Pd, 0,2 вес. ч. HCO-60 и 0,4 вес. ч. гидроксипропилметилцеллюлозы (ТС-5Е, производится фирмой Shin-Etsu Chemical Co.) в смеси воды с метанолом (1:9) и полученный раствор подвергают распылительной сушке. Получают высушенный распылением продукт 1.
Приведенные в табл. 5 компоненты в указанных пропорциях смешивают в ступке и каждую полученную композицию подвергают формованию под давлением с использованием масляного пресса при давлении сжатия 1 т/пуансон. Получают таблетки весом 80 мг каждая.
Тест на растворение
Исследование
желатинизации с использованием JP теста дезинтеграции жидкости 1 или 2 проводят в соответствии с JP
методом испытания растворения 2 (лопастной метод), при этом таблетки ацетаминофена и
хлоргидрата
никардипина (Pd) служат в качестве модельных таблеток. Отбор образцов производят через заранее
установленные промежутки времени и с помощью УФ-спектроскопии определяют количество
лекарственного
средства в каждом образце (фиг. 4 и 5).
Результаты
Установлено, что
скорость растворения может быть отрегулирована путем изменения содержания
гидрогельобразующей основы
ROLYOX303. Также установлено, что при использовании 50 мг ацетаминофена в качестве основного
агента и при добавлении не более 100 мг (50% от веса таблетки) ROLYOX303
высвобождение лекарственного
средства даже при интенсивном перемешивании продолжается не менее 12 ч (скорость лопасти 200
об/мин, pH 6,8). Аналогично, использование в качестве основного компонента 80
мг Pd и включение в состав
таблетки не менее 96 мг (37,5% от веса таблетки) ROLYOX303 даже при интенсивном перемешивании
(скорость лопасти 200 об/мин, pH 1,2) обеспечивает постоянное выделение
лекарства в течение 12 ч.
Оптимальное содержание гидрогельобразующей основы наряду с другими факторами зависит от типа и количества лекарственного средства и гидрофильной основы, а также от желаемой скорости растворения, но было найдено, что чем выше содержание гидрогельобразущей основы, тем больше длительность выделения. Также установлено, что, если необходимо получить постоянное высвобождение лекарства в течение не менее 12 ч, необходимо вводить в состав препарата из расчета на его вес не менее 70 мг, предпочтительно не менее 100 мг гидрогельобразующей основы.
II. Изучение соотношения между молекулярным весом гидрогельобразующей основы и длительностью высвобождения лекарственного средства.
1. Ацетаминофен.
В качестве полиэтиленоксида (ПЭО) используются полиэтиленоксиды, имеющие средние молекулярные веса 9•105, 1•106, 2•106, 4•106, 5•106, 7•106. В каждом случае компоненты рецептуры смешиваются в ступке и подвергаются формованию под давлением с использованием масляного пресса при давлении 1 т/пуансон. Получают таблетки диаметром 9,0 мм и весом 350 мг.
2. Хлоргидрат никардипина (Pd).
В смеси воды и метанола (1: 9) растворяют 1 вес. ч. Pd, 0,4 вес. ч. HCO-40 и 0,8 вес. ч. гидроксипропилметилцеллюлозы (TC-5E, производится фирмой Shin-Etsu Chemical Co.), и полученный раствор подвергают распылительной сушке с помощью распылительной сушилки. Получают осушенный распылительной сушкой продукт 2.
В качестве полиэтиленоксида (ПЭО) используются полиэтиленоксиды, имеющие средние молекулярные веса 9•105, 1•106, 2•106, 4•106, 5•106, 7•106. В каждом случае компоненты рецептуры смешиваются в ступке и подвергаются формованию под давлением с использованием масляного пресса при давлении 1 т/пуансон. Получают таблетки диаметром 11,0 мм и весом 568 мг (содержат 80 мг Pd).
Изучение высвобождения лекарства
Препараты, содержащие ацетаминофен и никардипин, изучают в соответствии с методикой оценки растворения, описанной в
разделе 1. Определение оптимального содержания (фиг. 6 и 7).
Результаты
Скорость растворения изменяется при изменении среднего молекулярного веса гидрогельобразующей основы
- полиэтиленоксида (ПЭО). При использовании в качестве основного компонента
50
мг ацетаминофена в состав рецептуры вводят ПЭО со средним молекулярным весом не менее 4•106, что
приводит к постоянному выделению лекарственного средства в течение не менее 12 ч
при
интенсивном перемешивании (скорость лопасти 200 об/мин, pH 6,8).
Аналогично, при использовании в качестве основного компонента 80 мг Pd рекомендуется ПЭО со средним молекулярным весом не менее 2•106, что дает возможность получать постоянное высвобождение лекарства в течение не менее 12 ч.
(5) Проверка желатинизации.
Образцы
Смешивают в ступке гидрогельобразующую основу (POLYOX303) и гидрофильную основу (ПЭГ6000, ПВП К30 или
Д-сорбита) в соотношениях, указанных ниже, и каждую смесь формуют под давлением
с использованием
масляного пресса с давлением сжатия 1 т/пуансон. Получают таблетки диаметром 8,0 мм и весом 200 мг
каждая.
POLYOX303:ПЭГ6000 = 100:10, 25, 50, 100
POLYOX303:ПВП К30 = 100:10,
25, 100
POLYOX303:Д-сорбит = 100:10, 25, 100
Аутопсические испытания на
собаках
Самцам гончих собак (собаки A и B), которых не кормили в
течение 20 ч, вводят орально каждый
из исследуемых препаратов с 30 мл воды. Через 2 ч животных подвергают анестезированию с
помощью пентабарбитала натрия и после кровопускания вскрывают брюшную
полость. Таблетки извлекают из
пищеварительного тракта и определяют Dнабл.. На основе Dнабл. рассчитывают
индекс желатинизации (табл. 8).
Результаты
У
собаки A таблетки перемещаются в
толстую кишку уже через 2 ч после введения и время нахождения в пищеварительном тракте
составляет менее 2 ч. С другой стороны, все таблетки, за исключением одной,
содержащей 10 вес. ч. ПЭГ6000,
подвергаются не менее чем 80%-ной желатинизации, что в целом согласуется с данными in
vitro.
У собаки B через 2 ч после введения таблетки остаются в желудке и все таблетки подвергаются более чем 80%-ной желатинизации. Приведенные выше результаты указывают, что гидрогелевые таблетки, содержащие гидрофильную основу, обеспечивающую не менее 80% желатинизации in vitro (ПВП К30, ПЭГ6000 и Д-сорбит), при содержании в подходящих количествах, благодаря проникновению воды в ядро таблетки, легко образуют гель даже in vivo.
Если необходимо, препарат настоящего изобретения может содержать другие подходящие фармацевтически приемлемые добавки, такие как например наполнитель (например, лактозу, маннит, картофельный крахмал, пшеничный крахмал, рисовый крахмал, кукурузный крахмал и кристаллическая целлюлоза), связующие вещества (например, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, метилцеллюлоза и гуммиарабик), набухающие агенты (например, карбоксиметилцеллюлоза, кальций-карбоксиметилцеллюлоза и поперечно-сшитая натрий-карбоксиметилцеллюлоза), смазывающие вещества (например, стеариновая кислота, стеарат кальция, стеарат магния, тальк, мета-силикаталюминат магния, гидрофосфат кальция, безводный гидрофосфат кальция), разжижающие добавки (например, водная двуокись кремния, легкая безводная кремневая кислота, высушенный гель гидроксида алюминия), красители (например, желтый полуторный оксид железа и полуторный оксид железа), поверхностно-активные вещества (например лаурилстеарат натрия, эфир жирной кислоты и сахарозы), покрывающие агенты (например, зеин, гидроксипропилметилцеллюлоза и гидроксипропилцеллюлоза), ароматические добавки (например, т-ментол, перечно-мятное масло, фенхелевое масло), консерванты (например, сорбат натрия, сорбат калия, метил-п-бензоат и этил-п-бензоат), и др.
Препарат настоящего изобретения представляет собой твердый препарат, имеющий определенную форму и способность образовывать гидрогель. Препарат может быть получен обычными способами, применяемыми для приготовления гидрогелевых препаратов. Обычными способами являются таблетирование под давлением, которое включает смешение лекарства, гидрофильной основы и гидрогельобразующего полимера, а также, если необходимо, добавление других добавок и формование под давлением полученной композиции; заполнение капсул под давлением; формование экструзией, которое включает плавление смеси и отверждение расплавленной смеси; инжекторное формование и т.д. Затем может быть нанесено любое покрытие, например покрытие из сахара или пленочное покрытие, или препаратом могут быть заполнены капсулы.
Солюбилизация лекарственного средства для применения в препарате настоящего изобретения, если это необходимо, может быть проведена до начала описанного выше способа получения. Гидрофильная основа в соответствии с настоящим изобретением может заменять солюбилизатор в тех случаях, когда проводится солюбилизация. Например, препарат настоящего изобретения может быть получен с помощью способа, который включает смешение лекарства, предварительно солюбилизированного с использованием гидрофильной основы, и, если необходимо, различных добавок с гидрогельобразующим полимером, и формование под давлением полученной композиции.
Если необходимо, то препарат с длительным высвобождением лекарства настоящего изобретения может содержать некоторое количество, высвобождаемое немедленно. Например, препарат настоящего изобретения может быть снабжен такой порцией для немедленного выделения путем нанесения соответствующего покрытия.
В зависимости от предполагаемого использования продукт настоящего изобретения может быть получен в форме высушенных таблеток с покрытием. Например, если желательна высокая концентрация в крови при определенном времени после применения, ядро таблетки получают в соответствии с рецептурой, предназначенной для быстрого высвобождения лекарства (с повышенным количеством лекарства, с уменьшенным количеством гидрогельобразующей основы и/или повышенным количеством гидрофильной основы), а затем формуется внешний слой с помощью рецептуры, обеспечивающей замедленное высвобождение (с пониженным содержанием лекарства, повышенным содержанием гидрогельобразующей основы и/или уменьшенным содержанием гидрофильной основы) так, что через определенный промежуток времени скорость выделения лекарства может быть повышена.
На фиг. 1 представлены результаты оценки желатинизации гидрогелевого препарата с длительным высвобождением лекарства, содержащего ПЭГ6000.
На фиг. 2 представлены результаты оценки желатинизации препаратов с различным содержанием ПЭГ6000.
На фиг. 3 представлены индексы желатинизации различных гидрофильных основ через 2 ч.
На фиг. 4 показано соотношение между количеством POLYOX303 и профилем высвобождения лекарственного средства - ацетаминофена.
На фиг 5. показано соотношение между количеством POLYOX303 и профилем высвобождения лекарственного средства - хлоргидрата никардипина.
Фиг. 6 представляет собой соотношение между молекулярным весом ПЭО и профилем высвобождения (лекарственное средство - ацетаминофен).
Фиг. 7 представляет собой соотношением между молекулярным весом ПЭО и профилем высвобождения (лекарственное средство - хлоргидрат никардипина).
На фиг. 8 представлены результаты оценки растворения (лопастной метод) таблеток в соответствии с примером 1 и сравнительным примером 1.
На фиг. 9 представлены результаты оценки желатинизации таблеток в соответствии с примером 1 и сравнительным примером 1.
На фиг. 10 представлено временное изменение концентрации лекарства в плазме в опытах на собаках для таблеток в соответствии с примером 1 и сравнительным примером 1.
На фиг. 11 представлено сравнение данных по оценке растворения и профилем абсорбции, определенным с помощью метода раскручивания для таблеток в соответствии со сравнительным примером 1.
На фиг. 12 представлено сравнение данных по оценке растворения и профилем абсорбции, определенным с помощью метода раскручивания для таблеток в соответствии с примером 1.
На фиг. 13 представлено временное изменение концентрации лекарства в плазме в опытах на собаках для таблеток в соответствии с примером 2 и сравнительным примером 2.
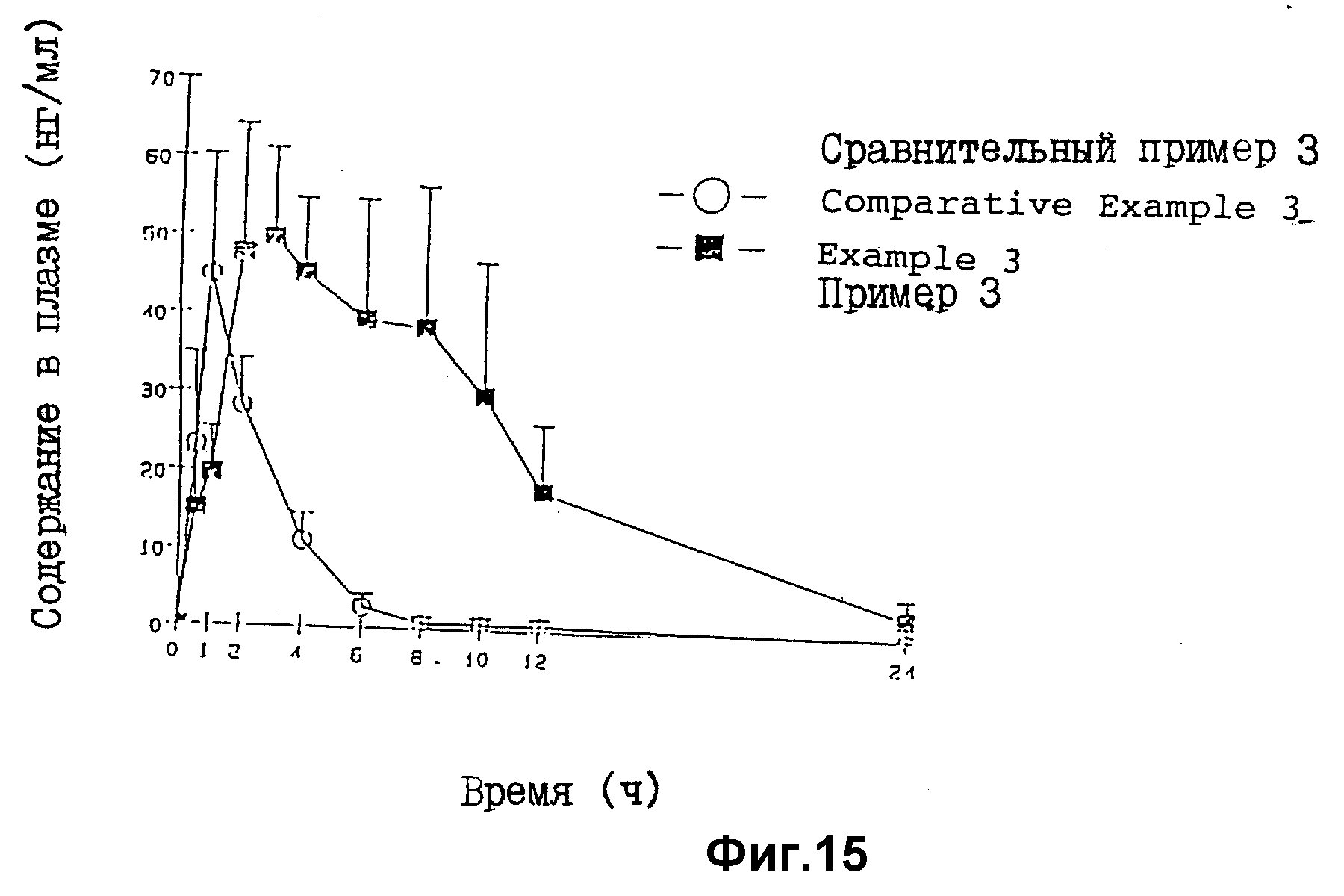
На фиг. 14 представлены результаты оценки растворения (лопастной метод) таблеток в соответствии с примером 3 (SR) и сравнительным примером 3 (SR).
На фиг. 15 представлено временное изменение концентрации лекарства в плазме в опытах на собаках для таблеток в соответствии в примером 4 и примером 5.
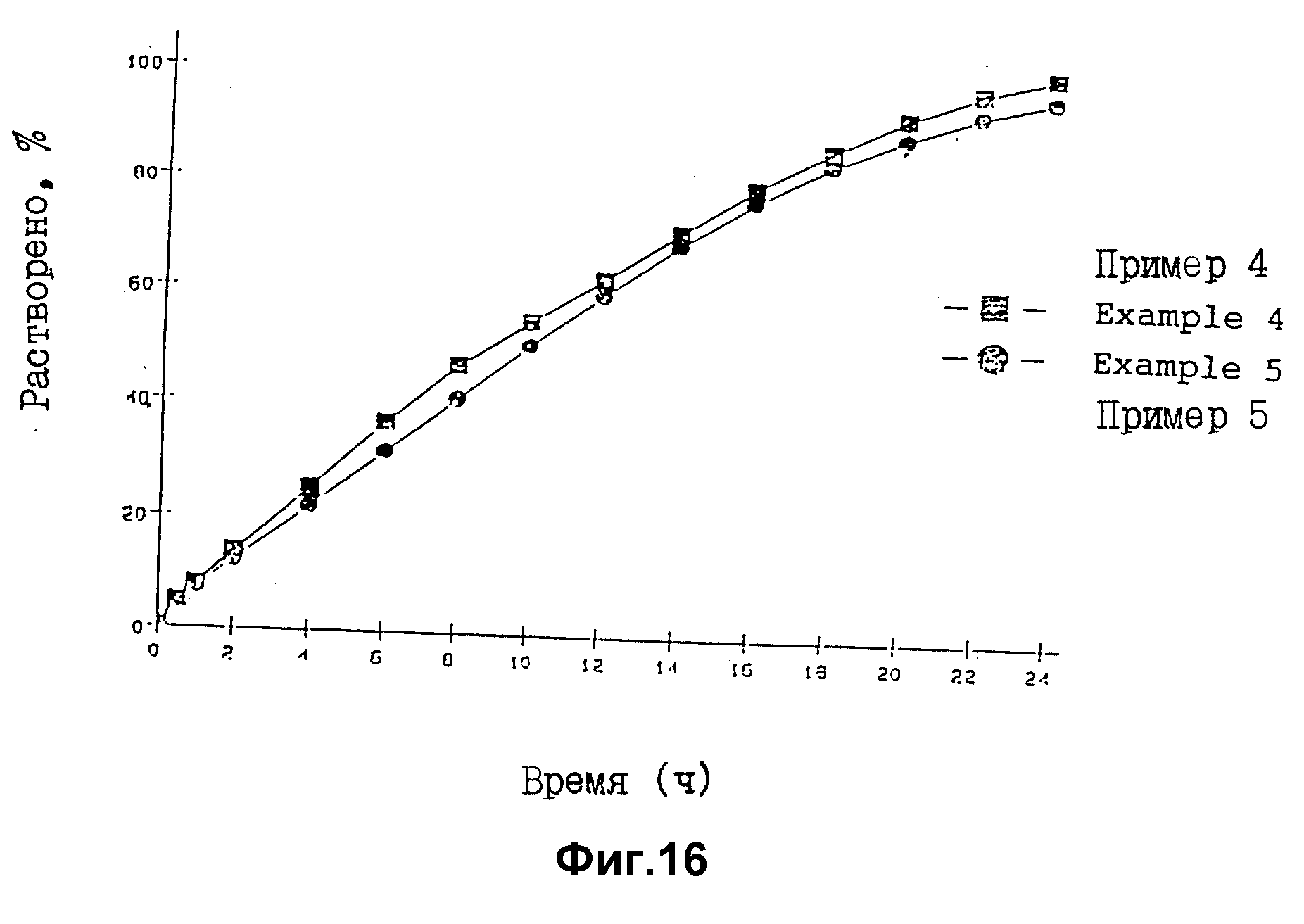
На фиг. 16 представлены результаты оценки растворения (лопастей метод) таблеток в соответствии с примером 4 и примером 5.
На фиг. 17 представлены результаты оценки растворения (лопастей метод) таблеток в соответствии с примером 6, 7 и 10.
На фиг. 18 представлены результаты оценки растворения (лопастной метод) в соответствии с примером 12 и сравнительным примером 2.
На фиг. 19 представлено временное изменение концентрации лекарства в плазме в опытах на собаках для таблеток в соответствии с примером 12 и сравнительным примером 4.
Предпочтительные воплощения изобретения.
Следующие примеры приведены для более детального описания рецептур настоящего изобретения и не могут быть интерпретированы, как ограничивающие объем притязаний изобретения.
Пример 1.
ААФ - 100 (весовых частей)
ПЭГ6000
- 400
POLYOX303 - 300
Ацетаминофен (ААФ) и ПЭГ 6000 сплавляют при 80oC, после чего охлаждают до затвердевания и распыляют. Распыленный продукт и POLYOX303
смешивают в
ступке и полученную композицию формуют под давлением с использованием масляного пресса с давлением 1 т/пуансон, получают таблетки, каждая из которых имеет диаметр 9 мм и вес 400 мг
(содержание ААФ:
50 мг).
Сравнительный пример 1.
ААФ - 100 (весовых частей)
POLYOX303 - 200
ААФ и POLYOX303 смешивают в ступке и полученную смесь
формуют под
давлением с использованием масляного пресса и давлением 1 т/пуансон получают таблетки, каждая из которых имеет диаметр 8,5 мм и вес 300 мг (содержание ААФ: 100 мг).
С использованием таблеток, полученных в соответствии с примером 1 и сравнительным примером 1, проведены следующие испытания.
(1) Оценка растворения 1.
С использованием JP теста дезинтеграции жидкости 2 оценку растворения проводят в соответствии с JP методом испытания растворения 2 (лопастной метод). Через определенные интервалы времени отбирают пробы и в каждом образце раствора определяют содержание ААФ с помощью УФ-метода (табл. 9 и фиг. 8).
(2) Оценка желатинизации.
С использованием JP теста дезинтеграции жидкости 2 оценку желатинизации проводят в соответствии с JP методом испытания растворения 2 (лопастной метод) со скоростью вращения лопасти 25 об/мин. Через определенные интервалы времени таблетки вынимают и измеряют диаметр (Dнабл.) части таблетки, не подвергнувшейся желатинизации. С использованием найденных таким образом величин Dнабл. вычисляют индекс желатинизации (G) (табл. 10 и фиг. 9).
(3) Оценка доз в опытах на собаках 1.
Самцам гончих собак (n=4), которых не кормили в течение 20 ч, с 30 мл воды вводят орально препарат примера 1 х 2 таблетки (ААФ - 100 мг) или препарата сравнительного примера 1 (ААФ - 100 мг). Через определенные промежутки времени отбирают пробы крови и с использованием метода ВЭЖХ/УФ определяют концентрацию лекарства в плазме (табл. 11 и фиг. 10). Скорость абсорбции рассчитывают с помощью метода раскручивания с использованием значений концентрации в плазме, которые получены при внутривенном введении 100 мг ААФ в воде в качестве весовой функции. Скорость абсорбции через 24 ч после применения препарата примера принимают за 100 (табл. 12).
Результаты
При оценке
растворения in vitro
получены практически одинаковые профили растворения сравнительного примера 1 и примера 1 (фиг. 8 и табл. 9), однако по скорости проникновения воды (индекс желатинизации) эти
препараты существенно
(фиг. 9, табл. 10) отличаются друг от друга. При оральном введении этих препаратов концентрация лекарства в плазме после применения препарата примера 1 поддерживается
достаточно хорошо по сравнению
со
сравнительным примером 1 (фиг. 10). Более того, как площадь под кривой зависимости "концентрация в плазме - время" (AUC), так и значение времени пребывания (MRT)
препарата сравнительного примера 1
имеют плохую сходимость, вероятно, вследствие индивидуального различия в скорости перемещения по пищеварительному тракту (табл. 11). С другой стороны, как AUC, так
и MRT препарата примера 1 не имеют
существенных различий, подтверждая тот факт, что на этот препарат скорость перемещения в пищеварительном тракте оказывает незначительное влияние. Время абсорбции
также продлевается, в результате чего,
несмотря на значительное соответствие в значениях максимальной концентрации в плазме (Cмакс) между препаратом примера 1 и препаратом сравнительного
примера 1, AUC после применения
препарата
1 имеет значение приблизительно в 1,8 раз больше.
Абсорбционный профиль, определенный с помощью метода раскручивания, сравнивается с соответствующими данными по оценке растворения. В случае препарата сравнительного примера 1 абсорбции лекарственного средства в течение первых 2 ч после применения, когда введенный препарат еще находится в верхней части пищеварительного тракта, сравнима с данными по оценке растворения in vitro. Однако абсорбция через 2 ч в значительной степени уменьшается (фиг. 11 и табл. 12). Время пребывания препарата в верхней части пищеварительного тракта у некормленных собак составляет приблизительно 2 ч и, следовательно, очевидно, что в нижней части пищеварительного тракта лекарство высвобождается и абсорбируется плохо. С другой стороны, после применения препарата примера 1 профиль абсорбции сравним с данными по оценке растворения in vitro. Поэтому очевидно, что в нижней части пищеварительного тракта лекарство высвобождается и поглощается так же эффективно, как и в верхней части пищеварительного тракта (фиг. 12 и табл. 12).
(4) Аутопсический тест на собаках.
В опыте используются три самца гончих собак, которых не кормят в течение 20 ч. За 2, 4 и 6 ч до аутопсии вместе каждый испытуемый препарат вводится орально с 30 мл воды. При аутопсии животных умерщвляют путем обескровливания при анестезии пентобарбиталом натрия, вскрывают брюшную полость и определяют местонахождение препарата в пищеварительном тракте (табл. 13). Тонкую кишку разделяют на 5 участков, которые обозначают цифрами тонк. к. 1, 2, 3, 4 и 5, начиная отсчет от самого верхнего участка.
Результаты
Очевидно, что препарат
сравнительного примера 1, который отличается
низким индексом желатинизации, и
препарат примера 1, индекс желатинизации которого увеличен путем добавления гидрофильной основы, практически идентичны с
точки зрения скорости перемещения в
пищеварительном тракте in vivo. Через 2 ч
после применения оба препарата все еще находятся в желудке одной из собак, однако у остальных собак уже находятся в
участке тонкого кишечника под номером 5 и
в толстой кишке. Этот факт, следовательно,
подтверждает то, что время пребывания в верхней части пищеварительного тракта голодных собак составляет
приблизительно 2 ч, что согласуется с ранее
установленным значением. Однако высокая концентрация
в крови через 2 ч после введения препарата примера 1 указывает на то, что этого препарата лекарство
эффективно высвобождается и абсорбируется
несмотря на тот факт, что препарат находится в нижней
части пищеварительного тракта.
Пример 2.
Pd - 160 вес.ч.
HCO-60
TC-5E - 160
ПЭГ6000 - 400
POLYOX303 - 240
Хлоргидрат никардипина (Pd), HCO-60, TC-5E и ПЭГ6000 растворяют в смеси растворителей
(метиленхлорид:метанол) и полученный раствор подвергают
распылительной сушке с помощью распылительной сушилки.
Высушенный препарат в ступке смешивают с POLYOX303 и полученную композицию подвергают
формованию под давлением с использованием масляного пресса
при давлении сжатия 1 т/пуансон. Получают таблетки с
диаметром 9,0 мм весом 346,7 мг (содержание Pd: 53,3 мг).
Сравнительный пример 2.
Pd - 130 вес. ч.
Tween 80 - 26 Длительно высвобождаемая компонента
(SR)
КМЭЦ - 130
POLYOX303 - 57,2
Pd - 30 Быстро
высвобождаемая компонента (GR)
TC-5E - 15
В
смеси растворителей (метиленхлорид:метанол) растворяют
хлоргидрат никардипина (Pd), Tween 80 и КМЭЦ и полученный раствор подвергают
распылительной сушке в распылительной сушилке. Высушенную смесь
смешивают с POLYOX303 и полученную композицию подвергают
формованию под давлением с использованием масляного пресса при давлении
сжатия 0,8 т/пуансон. Получают таблетки (SR) с диаметром 8,0 мм весом
171,6 мг (содержание Pd 65 мг). Отдельно в смеси
растворителей (метиленхлорид: метанол) растворяют Pd и TC-5E и с помощью
устройства для нанесения покрытия - Hi-Coater - полученную быстро
высвобождаемую компоненту (QR, Pd = 15 мг) наносят на
SR-компоненту (Pd=65 мг). Получают таблетки сравнительного примера 2 каждая
весом 191,4 мг (Pd=80 мг).
Используя таблетки примера 2 и сравнительного примера 2, проводят испытания, описанные ниже.
(1) Оценка растворения.
С использованием JP теста дезинтеграции жидкости 1 оценку растворения проводят в соответствии с JP методом испытания растворения 2 (лопастной метод) при скорости лопасти 200 об/мин. Через определенные интервалы времени отбирают пробы и в каждом образце раствора определяют содержание Pd с помощью УФ-метода (табл. 14).
(2) Оценка желатинизации.
С использованием JP теста дезинтеграции жидкости 1 оценку желатинизации проводят в соответствии с JP методом испытания растворения 2 (лопастной метод) при скорости лопасти 25 об/мин. Через 2 часа таблетки извлекают и определяют диаметр части, которая не образовала гель (Dнабл.). С использованием Dнабл. рассчитывают индекс желатинизации (табл. 15).
(3) Оценка доз в опытах на собаках 1.
Самцам гончих собак (n = 6), которых не кормили в течение 20 ч, с 30 мл воды вводят орально препарат примера 2х3 таблетки (Pd - 160 мг) или препарата сравнительного примера 2х2 таблетки (Pd - 160 мг). Через определенные промежутки времени отбирают пробы крови и с использованием метода ВЭЖХ/УФ определяют концентрацию лекарства в плазме (табл. 16 и фиг. 13).
Результаты
При оценке растворения
in vitro профили растворения препарата
сравнительного примера 2 (SR) и препарата примера 2 были практически одинаковы (табл. 14), однако по
скорости проникновения воды (индекс желатинизации) они
существенно (табл. 15) отличаются друг от
друга. При оральном введении собакам этих препаратов препарат примера 2 по сравнению со сравнительным
примером 2 достаточно длительно поддерживает
концентрацию лекарства в плазме.
Что касается примера 2, то концентрация лекарства в плазме значительно уменьшается через 2 часа, когда введенный препарат поступает в нижнюю часть пищеварительного тракта, указывая, что лекарство в нижней части пищеварительного тракта высвобождается и абсорбируется плохо. С другой стороны, при введении препарата 2, концентрация в плазме успешно поддерживается даже через 2 ч, когда препарат перемещается в нижнюю часть пищеварительного тракта, что указывает на то, что лекарство эффективно высвобождается и абсорбируется в нижней части пищеварительного тракта. Кроме того, хотя препарат примера 2, показывающий значение Cмакс. сравнимое со значением, получаемым после введения сравнительного примера 2, первый препарат дает значение AUC приблизительно в 3 раза больше, благодаря увеличению периода абсорбции.
Пример 3.
Pd - 65 (вес. ч.)
Tween 80 - 13 Длительно высвобождаемая
компонента
КМЭЦ - 65 (SR)
ПЭГ6000 - 65
POLYOX303 - 65
Pd - 15 Быстро выделяемая компонента (QR)
В смеси
растворителей (метиленхлорид-метанол) смешивают
хлоргидрат никардипина (Pd), Tween 80 и КМЭЦ и раствор
подвергают распылительной сушке с использованием распылительной сушилки. Высушенную смесь
смешивают с ПЭГ6000 и POLYOX303 и полученную смесь
подвергают формованию под давлением с использованием
масляного пресса при давлении сжатия 1,0 т/пуансон. Получают таблетки (SR) диаметром 8.5 мм и
весом 273 мг (SR; Pd 65 мг). Для использования в
качестве быстро выделяемой компоненты (QR) отдельно
готовят таблетки, содержащие 15 мг Pd.
Сравнительный пример 3.
Pd - 65 (вес.ч.)
Tween 80 - 13 Длительно
высвобождаемая компонента
КМЭЦ - 65 (SR)
POLYOX303 - 28,6
Pd - 15 Быстро выделяемая компонента (QP)
TC-5E - 7,
5
В смеси растворителей
(метиленхлорид-метанол) смешивают хлоргидрат никардипина (Pd) и раствор
подвергают распылительной сушке с использованием распылительной сушилки. Высушенную смесь
смешивают с POLYOX303 и полученную
смесь подвергают формованию под давлением с использованием масляного пресса
при давлении сжатия 0,8 т/пуансон. Получают таблетки (SR) диаметром 8,0 мм и весом
171.6 кг (SR, Pd 65 мг). Отдельно в
смеси растворителей (метиленхлорид-метанол) растворяют Pd и TC-5E и с помощью
устройства для нанесения покрытия - полученную быстро высвобождаемую компоненту (QR);
Pd = 15 мг) наносят на
SR-компоненту (Pd=65 мг). Получают таблетки сравнительного примера 2, каждая весом 194,1 мг
(Pd=80 мг).
(1) Оценка растворения.
С использованием JP теста дезинтеграции жидкости 2 оценку растворения проводят в соответствии с JP методом испытания растворения 2 (лопастной метод) при скорости лопасти 200 об/мин. Через определенные интервалы времени отбирают пробы и в каждом образце раствора определяют содержание Pd с помощью УФ-метода.
Результаты описанного выше опыта по оценке растворения препарата сравнительного примера 3 (JP) и примера 3 (JP) представлены на фиг. 14.
(2) Оценка желатинизации.
С использованием JP теста дезинтеграции жидкости 1, оценку желатинизации проводят в соответствии с JP методом испытания растворения 2 (лопастной метод) при скорости лопасти 25 об/мин. Через 2 часа таблетки извлекают и определяют вес части, которая не образовала гель (Bнабл.). С использованием Bнабл. по уравнению 2 рассчитывают индекс желатинизации (G) (табл. 17).
Уравнение 2
Индекс желатинизации (G,
%) = (1-(Bнабл.)/(Bнач.))•100
Bнабл. - остаточный вес после удаления гелевого слоя,
образовавшегося после начала испытания;
Bнач.
- вес таблетки
до начала испытания.
(3) Оценка доз в опытах на собаках 1.
Самцам гончих собак (n=6), которых не кормили течение 20 ч, с 30 мл воды вводят орально 2 таблетки препарата примера 1 SR и QR (Pd = 160 мг) или 2 таблетки препарата сравнительного примера 3 (Pd= 160 мг). Через определенные промежутки времени отбирают пробы крови и с использованием метода ВЭЖХ/УФ определяют концентрацию лекарства в плазме (табл. 18 и фиг.15).
(4) Аутопсический тест на собаках.
В опыте используются три самца гончих собак, которых не кормят в течение 20 ч. За 2, 4 и 6 ч до аутопсии каждый испытуемый препарат вводится орально с 30 мл воды. При аутопсии животных умерщвляют путем обескровливания при анестезии пентобарбиталом натрия, вскрывают брюшную полость и определяют местонахождение препарата в пищеварительном тракте (табл. 13). Тонкую кишку разделяют на 5 участков, которые обозначают цифрами тонк. к. 1, 2, 3, 4 и 5, начиная отсчет от самого верхнего участка.
Результаты
В опытах по оценке растворения in vitro профили
растворения препарата сравнительного примера 3 (SR)
и препарата примера 3 (SR)
были практически одинаковы (табл. 14), однако по индексам желатинизации они существенно (табл. 17) отличаются друг от
друга. При аутопсии выявляется практически одинаковая
скорость перемещения в
примере 3 и сравнительном примере 3 (табл. 19). При оральном введении этих препаратов собакам концентрации лекарства в
плазме после введения препарат примера 3 поддерживается
дольше, чем при введении
препарата сравнительного примера 3. Что касается сравнительного примера 3, то концентрация лекарства в плазме
значительно уменьшается через 2 часа, когда введенный препарат
поступает в нижнюю часть
пищеварительного тракта, указывая, что лекарство в нижней части пищеварительного тракта высвобождается и
абсорбируется плохо. С другой стороны, при введении препарата 3
концентрация в плазме успешно
поддерживается даже через 2 ч, когда препарат перемещается в нижнюю часть пищеварительного тракта, что
указывает на то, лекарство эффективно высвобождается и
абсорбируется даже в нижней части
пищеварительного тракта (фиг.15). Кроме того, хотя для препарата примера 3 значение Cмакс. сравнимо
со значением, получаемым после введения
сравнительного примера 3, первый препарат
обеспечивает значение AUC приблизительно в 4,4 раза больше, благодаря увеличению периода абсорбции.
Пример 4.
Pd - 80
мг
ПВП К30 - 32
HCO-60
- 16
POLYOX303 - 240
Смазывающее вещество - 4
В метаноле растворяют хлоргидрат никардипина (Pd),
ПВП К30 и HCO-60. С помощью
гранулятора псевдоожиженного слоя полученный
раствор распыляют на POLYOX303 и получают гранулы. К гранулам добавляют смазывающее вещество и приготовленную композицию
смешивают и формуют под
давлением. Получают таблетки с диаметром 9,5 мм весом
372 мг (Pd = 80 мг).
Пример 5.
Pd - 80 мг
TC-5E - 32
HCO-60 - 16
ПЭГ6000 - 32
POLYOX303 - 240
Смазывающее вещество - 8
Добавка, обеспечивающая текучесть - 4
В смеси вода - метанол (1:9) растворяют хлоргидрат никардипина
(Pd). TC-5E и HCO-60 и
полученный раствор подвергают распылительной сушке. К
сушенной смеси добавляют POLYOX303 и 4 мг-эквивалента смазывающего вещества с последующим гранулированием с сушкой
полученной смеси. К гранулам
добавляют 4 мг-эквивалента смазывающего вещества и
добавки, обеспечивающей текучесть, и приготовленную композицию после смешения подвергают формованию под давлением.
Получают таблетки диаметром 9,5
мм весом 412 мг (Pd = 80 мг).
Пример 6.
Pd - 80 мг
TC-5E - 32
HCO-60 - 32
ПЭГ6000 - 32
POLYOX303
- 384
Смазывающее
вещество - 11,2
Добавка, обеспечивающая
текучесть - 5,6
В смеси вода - метанол (1:9) растворяют хлоргидрат никардипина (Pd). TC-5E, HCO-60 и ПЭГ6000 и
полученный раствор подвергают
распылительной сушке. К высушенной смеси
добавляют POLYOX303 и 5,6 мг-эквивалента смазывающего вещества и смесь подвергают гранулированию с сушкой. К приготовленным таким
образом гранулам добавляют
5,6 мг-эквивалента смазывающего вещества и
добавки, обеспечивающей текучесть, и приготовленную композицию после смешения подвергают формованию под давлением. Получают
таблетки диаметром 11 мм весом
576,8 мг (Pd = 80 мг).
Пример 7.
Pd - 80 мг
TC-5E - 64
Tween 80 - 32
ПЭГ6000 - 32
POLYOX303
- 360
Смазывающее вещество
- 11,4
Добавка, обеспечивающая
текучесть - 5,7
В смеси вода - метанол (1:9) растворяют хлоргидрат никардипина (Pd), TC-5E и Tween 80 и полученный
раствор подвергают распылительной
сушке. К высушенной смеси добавляют
ПЭГ6000, POLYOX303 и 5,7 мг-эквивалента смазывающего вещества и смесь подвергают гранулированию с сушкой. К приготовленным таким
образом гранулам добавляют 5,7
мг-эквивалента смазывающего вещества и
добавки, обеспечивающей текучесть, и приготовленную композицию после смешения подвергают формованию под давлением. Получают
таблетки диаметром 11 мм весом 585,
1 мг (Pd = 80 мг).
Пример 8. В смеси метанол - вода (1:9) растворяют Pd и TC-5E и с использованием устройства для нанесения покрытия Hi-Coater на таблетки примера 7 (Pd = 80 мг) наносят компоненту с быстрым высвобождением лекарства (Pd = 20 мг). Получают таблетки весом 625,1 мг каждая (Pd = 100 мг).
Пример 9. В метаноле (1:9) растворяют Pd и HPC-SL и с использованием устройства для нанесения покрытия Hi-Coater на таблетки примера 7 (Pd = 80 мг) наносят компоненту с быстрым высвобождением лекарства (Pd = 20 мг). Получают таблетки весом 625,1 мг каждая (Pd = 100 мг).
Пример 10.
Pd - 80 мг
TC-5E - 64
HCO-40 - 32
ПЭГ6000 - 48
POLYOX303 - 344
Смазывающее вещество
- 11,4
Добавка,
обеспечивающая текучесть - 5,7
В смеси вода - метанол (1:9) растворяют хлоргидрат никардипина (Pd), TC-5E и HCO-40 и полученный раствор
подвергают распылительной
сушке. К высушенной смеси
добавляют ПЭГ6000, POLYOX303 и 5,7 мг-эквивалента смазывающего вещества и смесь подвергают гранулированию с сушкой. К приготовленным таким образом
гранулам добавляют 5,7
мг-эквивалента смазывающего
вещества и добавки, обеспечивающей текучесть, и приготовленную композицию после смешения подвергают формованию под давлением. Получают таблетки
диаметром 11 мм весом 585,
1 мг (Pd = 80 мг).
Пример 11.
Pd - 100 мг
TC-5E - 80
HCO - 40
ПЭГ6000 - 48
POKYOX303 - 300
Смазывающее вещество - 11,
4
Добавка,
обеспечивающая текучесть - 5,7
В смеси вода - метанол (1:9) растворяют хлоргидрат никардипина (Pd), TC-5E и HCO-40 и полученный раствор
подвергают распылительной сушке.
К высушенной смеси
добавляют ПЭГ6000, POLYOX303 и 5,7 мг-эквивалента смазывающего вещества и смесь повергают гранулированию с сушкой. К приготовленным таким образом
гранулам добавляют 5,7
мг-эквивалента смазывающего
вещества и добавки, обеспечивающей текучесть, и приготовленную композицию после смешения подвергают формованию под давлением. Получают таблетки
диаметром 11 мм весом 585,
1 мг (Pd = 80 мг).
(1) Оценка растворения.
С использованием JP теста дезинтеграции жидкости 1 оценку растворения проводят в соответствии с JP методом испытания растворения 2 (лопастной метод) при скорости лопасти 200 об/мин. Через определенные интервалы времени отбирают пробы и в каждом образце раствора определяют содержание Pd с помощью УФ-метода.
Результаты исследования по оценке растворения препаратов примеров 4 и 5 представлены на фиг. 16.
Результаты исследований по оценке растворения препаратов примеров 6, 7 и 10 представлены на фиг. 17.
(2) Оценка доз в опытах на собаках.
Самцам гончих собак (n = 6) один раз в день в течение 4 дней с 30 мл воды вводят орально 2 таблетки препарата примера 5 или 2 таблетки препарата примера 6. Через определенные промежутки времени отбирают пробы крови и с использованием метода ВЭЖХ/УФ определяют концентрацию лекарства в плазме.
Результаты
Оба препарата примеров 5 и 6 при назначении один раз в день обеспечивают высокое значение C24ч (концентрация в крови через 24 часа
после введения), а также
высокую биодоступность.
Пример 12.
ДФ - 37,5 мг
ПЭГ6000 - 37,5
POLYOX303 - 75,0
Смешивают в ступке дихлорфенак
(ДФ), ПЭГ6000 и Polyox 303
и с использованием
масляного пресса полученную композицию подвергают формованию под давлением при давлении сжатия 1 т/пуансон. Получают таблетки с диаметром 7 мм весом 150
мг (ДФ = 37,5 мг).
Сравнительный пример 4.
ДФ - 37,5 мг
POLYOX303 - 75,0
Смешивают в ступке дихлофенак (ДФ и Polyox 303 и с использованием масляного
пресса полученную
композицию
подвергают формованию под давлением при давлении
сжатия 1 т/пуансон. Получают таблетки с диаметром 6 мм весом 112,5 мг (ДФ=37,5 мг).
(1) Оценка растворения.
С использованием JP теста дезинтеграции жидкости 2 оценку растворения проводят в соответствии с JP методом испытания растворения 2 (лопастной метод). Через определенные интервалы времени отбирают пробы и в каждом образце раствора определяют содержание ДФ с помощью УФ-метода (фиг. 18).
(2) Оценка желатинизации.
С использованием JP теста дезинтеграции жидкости 2 оценку желатинизации проводят в соответствии с JP методом испытания растворения 2 (лопастной метод) при скорости лопасти 25 об/мин. Через 2 часы таблетки извлекают и определяют диаметр части, которая не образовала гель (Dнабл.). С использованием Dнабл. рассчитывают индекс желатинизации (G) (табл. 20).
(3) Оценка доз в опытах на собаках.
Самцам гончих собак (n = 5), которых не кормили в течение 20 ч, с 30 мл воды вводят орально препарат примера 12 (ДФ = 37,5 мг) или препарата сравнительного примера 4 (ДФ - 3,95 мг). Через определенные промежутки времени отбирают пробы крови и с использованием метода ВЭЖХ/УФ определяют концентрацию лекарства в плазме (табл. 21 и фиг. 19).
Результаты
В опытах
по
оценке растворения in vitro профили растворения препарата примера 12 и препарата сравнительного примера 4 практически одинаковы (табл. 18), однако существенно отличаются друг от
друга по скорости
проникновения воды (по индексу желатинизации) (табл. 20). При оральном введении этих препаратов собакам препарат 12 обеспечивает продление сохранения концентрации лекарства в крови
по сравнению с
препаратом сравнительного примера 4 (фиг. 19). Более того, по сравнению со сравнительным примером 4 пример 12 имеет значения AUC в 1,7 раз больше (табл. 12). Таким образом, даже в
случае дихлофенака
натрия, который представляет собой кислотное соединение, подтверждается, что применение настоящего изобретения приводит к эффективному высвобождению и абсорбции лекарственного
средства в нижней
части
пищеварительного тракта.
Пример 13.
ДФ - 75 (мг)
ПЭГ6000 - 75
POLYOX303 - 150
Дихлофенак NA (ДФ), ПЭГ6000 и
POLYOX303 смешивают в
ступке и
с использованием масляного пресса композицию формуют при давлении пресса 1 т/пуансон в таблетки диаметром 8,5 мм и массой 300 мг (содержание ДФ: 75 мг).
Пример 14.
ДФ - 75 (мг)
ПЭГ6000 - 75
POLYOX303 - 300
Дихлофенак NA (ДФ), ПЭГ6000 и POLYOX303 смешивают в ступке и с использованием масляного пресса
композицию формуют при
давлении
пресса 1 т/пуансон в таблетки диаметром 9,5 мм и массой 450 мг каждая (содержание ДФ: 75 мг).
Пример 15.
Фамотидин - 40 (мг)
ПЭГ6000 - 30
POLYOX303
- 150
Смазывающее вещество - 2
Фамотидин, ПЭГ6000, POLYOX303 и смазывающее вещество смешивают и формуют прессованием, получая таблетки диаметром 8,0
мм и массой 222 мг
каждая
(содержание фамотидина: 40 мг).
Пример 16.
Хлоргидрат барнидипина - 15 (мг)
ТС-5Е - 30
НСО-40 - 5
ПЭГ20000
- 40
POLYOX303
- 207
Смазывающее вещество - 3
Хлоргидрат барнидипина, ТС-5Е и НСО-40 растворяют в смеси воды и метанола (1 : 9). Отдельно смешивают ПЭГ20000 и POLYOX303. С
помощью гранулятора
псевдоожиженного слоя полученную смесь разбрызгивают с приготовленным раствором. Полученные таким образом гранулы сушат и после добавления смазывающего вещества композицию формуют
прессованием с
получением таблеток диаметром 9,0 мм и массой 300 мг каждая (содержание хлоргидрата барнидипина: 15 мг).
Пример 17.
Хлоргидрат амосулалола - 40 (мг)
Pluronic
F68 - 40
Polyox 303 - 196
Смазывающее вещество - 4
Хлоргидрат амосулалола, Pluronic F68, Polyox 303 и смазывающее вещество смешивают, пульверизируют и
гранулируют
сухим
способом. Затем смесь формуют прессованием с получением таблеток диаметром 8,5 мм и массой 280 мг каждая (содержание хлоридгидрата амосулалола: 40 мг).
Пример 18.
Хлоргидрат тамусулозина - 0,2 (мг)
D-сорбит - 17,8
Polyox WSR N-60K - 180
Смазывающее вещество - 2
Хлоргидрат тамусулозина, D-Сорбит и ПЭО (Polyox WSR
N-60K)
гранулируют мокрым способом с помощью этанола и затем сушат. К этим сухим гранулам добавляют смазывающее вещество, получившуюся композицию перемешивают и формуют прессованием с получением
таблеток
диаметром 8 мм и массой 200 мг каждая (содержание хлоргидрата тамусулозина: 0,2 мг).
Пример 19.
Хлоргидрат инделоксазина - 60 (мг)
Сахароза
- 37
HPMC (90SH30000) - 180
Смазывающее вещество - 3
Хлоргидрат инделоксазина, сахарозу, HPMC и смазывающее вещество смешивают и гранулируют сухим способом. Затем гранулы
формуют
прессованием с получением таблеток диаметром 9 мм и массой 280 мг каждая. (Содержание хлоргидрата инделоксазина: 60 мг).
Пример 20.
Фумарат формотерола - 0,
16
(мг)
Мальтоза безводная - 47,84
Карбопол 940 - 100
Смазывающее вещество - 2
Фумарат формотерола, безводную мальтозу, карбопол 940 и смазывающее вещество
смешивают и
полученную композицию формуют прессованием с получением таблеток диаметром 7 мм и массой 150 мг каждая (содержание фумара формотерола 0,2 мг).
Пример 21.
ААФ - 100
(мг)
ПЭГ6000 - 200
ПЭО (Polyox WSR N-60K) - 300
Ацетаминофен (ААФ), ПЭГ6000 и ПЭО (Polyox WSR N-60K, значащая молекулярная масса: 200 миллионов) смешивают в
ступке и смесь
формуют прессованием с использованием масляного пресса при давлении 1 т/пуансон с получением таблеток диаметром 11 мм и массой 600 мг каждая (содержание ААФ: 100 мг).
Сравнительный пример 5.
ААФ - 100 (мг)
ПЭО (Polyox WSR N-60K) - 300
ААФ и ПЭО (Polyox WSR N-60K) смешивают в ступке и формуют прессованием с использованием
масляного пресса при
давлении 1 т/пуансон с получением таблеток диаметром 9 мм и массой 400 мг каждая (содержание ААФ: 100 мг).
(1) Оценка растворения.
С использованием JP теста дезинтеграции жидкости 2 оценку растворения проводят в соответствии с JP методом испытания растворения 2 (лопастной метод) при скорости вращения лопасти 200 об/мин. Через определенные интервалы времени отбирают пробы и в каждом образце раствора определяют содержание ААФ с помощью УФ-метода.
(2) Оценка желатинизации.
С использованием JP теста дезинтеграции жидкости 2 оценку желатинизации проводят в соответствии с JP методом испытания растворения 2 (лопастной метод) при скорости лопасти 25 об/мин. Через 2 часа таблетки извлекают и определяют диаметр части, которая не образовала гель (Dнабл.). С использованием Dнабл. рассчитывают индекс желатинизации (G).
(3) Оценка доз в опытах на собаках.
Самцам гончих собак (n = 6), которых не кормили в течение 20 ч, с 30 мл воды вводят орально препарат сравнительного примера 5 (АФФ = 100 мг) или препарата примера 20 (ААФ - 100 мг). Через определенные промежутки времени отбирают пробы крови и с использованием метода ВЭЖХ/УФ определяют концентрацию лекарства в плазме.
Результаты
В опытах по
оценке растворения
in vitro
профили растворения препарата примера 20 и препарата сравнительного примера 5 практически одинаковы, но препарат 20, который содержит гидрофильную основу, имеет индекс
желатинизации больше,
чем
препарат сравнительного примера 5. При оральном введении этих препаратов собакам концентрация лекарства в крови поддерживается эффективнее в случае примера 21, чем в случае
препарата
сравнительного
примера 5. Максимальная концентрация (Cмакс.) лекарства после применения препарата примера 21 практически равняется концентрации лекарства после применения
препарата
сравнительного примера
5, однако первый препарат отличается превосходными значениями AUC и MRT. Более того, после применения примера 21 концентрация лекарства сохраняется на высоком уровне
в течение
12 ч.
Практическая применимость
Препарат настоящего изобретения абсорбирует воду и подвергается практически полной желатинизации за промежуток времени, в течение
которого он
находится в верхней
части пищеварительного тракта, и перемещается в нижнюю часть пищеварительного тракта, подвергаясь постоянному разрушению и непрерывному высвобождению лекарства за
счет
дополнительной эрозии. Таким
образом, этот препарат обеспечивает успешное длительное высвобождение лекарственного средства даже в толстой кишке, где присутствует незначительное количество воды,
что
обеспечивает продолжительность
выделения лекарства в течение приблизительно 6 - 18 ч (приблизительно 12 - 24 ч, если учитывать высвобождение лекарства в верхней части пищеварительного тракта) и,
следовательно, обеспечивает
стабильную концентрацию лекарства в крови.
Так как обычные препараты с длительным высвобождением лекарства выделяют последнее только в верхней части пищеварительного тракта, то длительность выделения составляет приблизительно 6 ч, а последнее сохранение концентрации лекарства определяется его полупериодом биологического существования. С другой стороны, что касается препарата настоящего изобретения, длительность высвобождения лекарства сама по себе увеличивается. Соответственно, даже когда лекарство имеет короткий полупериод существования и, следовательно, достижение его длительного выделения считается затруднительным, достаточная концентрация в крови может сохраняться в течение более 12 ч.
Таким образом, препарат настоящего изобретения обладает способностью поддерживать эффективность лекарственного средства и может быть уменьшена кратность его приема. Кроме того, побочный эффект лекарства может быть снижен вследствие подавления быстрого увеличения концентрации в крови и поддержания этой концентрации на постоянном уровне.
Как показано в примерах, описанных выше, настоящее изобретение дает возможность пролонгировать абсорбцию лекарственных средств различного типа, например таких как ацетаминофен, которое является основным лекарством, дихлофенак натрия, который представляет собой кислотное соединение. Следовательно, настоящее изобретение обеспечивает использование фармацевтической технологии, отличающейся большой гибкостью и независимостью от физических свойств лекарства.
Реферат
Изобретение может использоваться в медицине и относится к препаратам с длительным высвобождением лекарства. Препарат содержит лекарственное средство - не более 85 мас.%, добавку, которая обеспечивает проникновение воды в ядро препарата, от 5 до 80 мас.% и гидрогельобразующий полимер - от 10 до 95 мас. %. Он обладает способностью подвергаться желатинизации не менее чем на 70% в верхней части пищеварительного тракта - в желудке и в тонкой кишке. Добавка, обеспечивающая проникновение воды, имеет растворимость 1 г на не более 5 мл воды. Гидрогельобразующий полимер имеет средний молекулярный вес не менее 2000000 и может включать по меньшей мере один полиэтиленоксид. Лекарственным средством может быть хлоргидрат никардипина. Препарат позволяет выделять лекарство как в верхней, так и в нижней части пищеварительного тракта - в толстой кишке. 7 з.п.ф-лы, 19 ил., 21 табл.

Формула
18.09.92 - по пп.1,2,5,8;
08.06.93 - по пп.3 и 4;
10.09.93 - по пп.6 и 7.
Комментарии