Получение и составление композиции, содержащей ингибитор мек - RU2669391C2
Код документа: RU2669391C2
Описание
Область техники, к которой относится изобретение
В настоящем документе предложены способы получения (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты, способы получения кристаллического (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты и подходящих промежуточных соединений. В настоящем документе предложены также фармацевтические композиции, содержащие указанное кристаллическое соединение.
Уровень техники
Пролиферативные сигналы, опосредованные фактором роста, передаются из внеклеточного окружения в ядро несколькими путями, включая каскад RAS/RAF/MEK. Путь сигнальной трансдукции киназ RAS/RAF/MEK активируется за счет первоначального внеклеточного связывания и стимуляции рецепторных тирозинкиназ (RTK) соответствующими когнатными лигандами. При аутофосфорилировании определенных тирозиновых остатков в цитозольном домене RTK комплекс Grb2-Sos транслоцируется на плазматическую мембрану и превращает неактивный RAS⋅ГДФ в активный RAS⋅ГТФ. Взаимодействие между докинг-белком Grb2 и активированными киназами или фосфорилированными рецептор-связанными белками опосредовано доменом гомологии Src (SH2) сигнального белка, который распознает специфические фосфотирозиновые последовательности. RAS подвергается конформационному изменению при связывании гуанозин-5'-трифосфата (ГТФ) и приводит к рекрутированию RAF-1 к цитоплазматической мембране, где он фосфорилируется несколькими киназами и одновременно дефосфорилируется по ключевым остаткам протеинфосфатазой-2В. Активированный RAF фосфорилирует митоген-активированную протеинкиназу (MEK) по двум сериновым остаткам в активирующей петле, что приводит к активации этой протеинкиназы. Затем MEK фосфорилирует и активирует киназу, регулируемую внеклеточными сигналами (ERK), обеспечивая ее транслокацию в ядро, где она фосфорилирует факторы транскрипции, обеспечивающие экспрессию различных генов.
Примерно в 1/3 случаев рака человека путь сигнальной трансдукции RAS/RAF/MEK является дерегулированным, зачастую из-за мутаций, которые приводят к эктопической активации белка. Такое дерегулирование, в свою очередь, вызывает широкий спектр клеточных изменений, которые неразрывно связаны с этиологией и поддержанием фенотипа рака, включая, но не ограничиваясь ими, промотирование пролиферации и эвазию апоптоза (Dhillon et al., Oncogene, 2007, 26: 3279-3290). Соответственно, в фармацевтической промышленности и онкологическом сообществе интенсивно проводится разработка низкомолекулярных ингибиторов основных членов пути сигнальной трансдукции RAS/RAF/MEK.
МЕК представляет собой основной белок каскада RAS/RAF/MEK, который передает сигналы о пролиферации и выживании клетки и часто активирован в опухолях, имеющих мутации в онкогенах RAS или RAF или в тирозинкиназных рецепторах к факторам роста. МЕК играет главную роль в каскаде RAS/RAF/MEK, поскольку в каскаде он расположен после RAS и RAF. Несмотря на то, что они лишь иногда мутируют при раковых заболеваниях (Murugan et al., Cell Cycle, 2009, 8: 2122-2124; Sasaki et al., J. Thorac. Oncol., 2010, 5: 597-600), ингибиторы белков MEK1 и MEK2 также представляют собой мишени для низкомолекулярного ингибирования из-за их центрального положения в сигнальном каскаде пути сигнальной трансдукции RAS/RAF/MEK (Fremin and Meloche, J. Hematol. Oncol., 2010, 3:8). Недавно потерпели неудачу попытки продемонстрировать эффективность мощного ингибитора МЕК в клинических испытаниях на пациентах с запущенным немелкоклеточным раком легких (Haura et al., Clin. Cancer Res., 2010, 16: 2450-2457). Причина неудачи в этом испытании неясна.
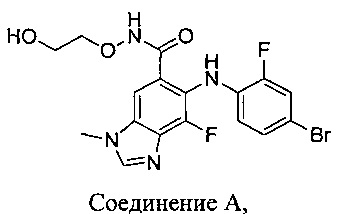
(2-Гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты (здесь и далее «соединение А») представляет собой бензимидазольное соединение, известное как мощный и селективный ингибитор белков MEK1 и MEK2, которое подходит для лечения гиперпролиферативных заболеваний, в частности, рака, у млекопитающих. Например, в опубликованном недавно испытании I фазы на 28 пациентах, страдающих неоперабельным, местнораспространенным или метастатическим раком желчных протоков, которые ранее проходили ≤1 предшествующей системной терапии, пероральное лечение соединением А (60 мг два раза в сутки) привело к 1 полной регрессии, 1 частичной регрессии и 11 диагнозам стабилизации заболевания по меньшей мере через 6 недель лечения (Finn et al., J. Clin. Oncol. 30, 2012 (дополнение 4, 2012 Gastrointestinal Cancers Symposium, реферат №220). Было также показано, что соединение А эффективно при лечении пациентов либо с BRAFV600, либо с NRAS-мутантной меланомой (Ascierto et al., J. Clin. Oncol. 30, 2012 (дополнение, 2012 ASCO Annual Meeting, реферат №8511).
Указанное соединение, а также способ его получения, описан в публикации РСТ № WO 03/077914. Производственный способ получения соединения А описан в примере 18 указанного документа. Производственный способ, описанный в нем, несмотря на свою пригодность, считается невыгодным для промышленного производства.
Благодаря высокой эффективности указанного бензимидазольного соединения, в частности, в качестве ингибитора MEK, существует необходимость в улучшенных способах производства таких соединений. В частности, существует необходимость в обеспечении способов, удовлетворяющих одному или нескольким из следующих критериев: масштабируемый, более безопасный, более простой, более производительный и более экономичный по сравнению с известными способами.
Сохраняется также необходимость в новых твердых формах для лечения рака.
Настоящее изобретение относится к улучшенному способу получения соединения А, который подходит для мелкомасштабного или крупномасштабного производства, а также подходящих промежуточных соединений. Настоящее изобретение дополнительно относится к способу получения кристаллического соединения А, а также новой фармацевтической композиции, подходящей для введения указанного кристаллического соединения. Было неожиданно обнаружено, что кристаллическое соединение А, полученное в соответствии с изобретенными способами, обладает улучшенным профилем чистоты и улучшенной физической морфологией, что является преимущественным при разработке и в производстве фармацевтических лекарств.
Раскрытие изобретения
В настоящем документе предложены способы получения (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты, способы получения кристаллического (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты и подходящих промежуточных соединений. В настоящем документе предложены также фармацевтические композиции, содержащие указанное кристаллическое соединение.
В одном аспекте в настоящем документе предложен способ получения соединения (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты (в настоящем документе упоминается как «соединение А»):

включающий стадии:
a) взаимодействия соединения формулы (I):
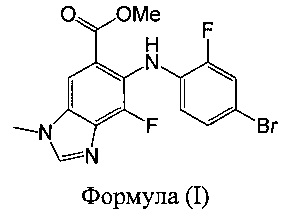
с подходящим основанием с образованием промежуточного соединения; и
b) взаимодействия указанного промежуточного соединения с соединением формулы (II):

с получением соединения формулы (III):

или его гидрата,
где P1 представляет собой защитную группу;
c) растворения указанного соединения формулы (III) или его гидрата в подходящем растворителе или системе растворителей; и
d) снятия защиты с указанного соединения формулы (III) или его гидрата при помощи подходящего реагента для снятия защиты,
где Р1 в каждом случае может быть одинаковым или различным и представляет собой подходящую защитную группу, с получением соединения А.
В другом аспекте в настоящем документе предложен способ получения соединения формулы (III):
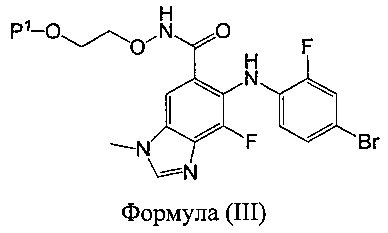
или его гидрата, включающий стадии:
a) взаимодействия соединения формулы (I):

с подходящим основанием с образованием промежуточного соединения; и
b) взаимодействия указанного промежуточного соединения с соединением формулы: (II):

где Р1 представляет собой защитную группу, с образованием соединения формулы (III) или его гидрата.
В некоторых вариантах реализации обоих способов стадии a) и b) осуществляют «однореакторным» синтезом, при этом промежуточное соединение со стадии a) взаимодействует с соединением формулы (II) без первоначального выделения из реакционной смеси со стадии a). В одном из конкретных вариантов реализации стадия a) включает взаимодействие соединения формулы (I) с подходящим основанием с образованием промежуточного соединения 1 (структура показана ниже), а стадия b) включает взаимодействие промежуточного соединения 1 с соединением формулы (II) с образованием соединения формулы (III) или его гидрата. В одном из вариантов реализации промежуточное соединение 1 не выделяют из реакционной смеси со стадии a) до стадии b). В другом варианте реализации промежуточное соединение 1 представляет собой часть раствора, содержащего растворители, выбранные из группы, состоящей из ДМФ и ТГФ.

Напротив, в других вариантах реализации промежуточное соединение, образованное на стадии a), выделяют из реакционной смеси до взаимодействия с соединением формулы (II). В одном из вариантов реализации стадия a) включает взаимодействие соединения формулы (I) с подходящим основанием с образованием промежуточного соединения формулы (V) (структура показана ниже) и выделение указанного промежуточного соединения из реакционной смеси; а стадия b) включает взаимодействие промежуточного соединения формулы (V) с соединением формулы (II) с образованием соединения формулы (III) или его гидрата. Таким образом, в указанном варианте реализации стадии a) и b) осуществляют не «однореакторным» способом, а в виде отдельных стадий получения, при этом промежуточное соединение со стадии a) выделяют до взаимодействия с соединением формулы (II) на стадии b). В одном из конкретных вариантов реализации стадия a) включает взаимодействие соединения формулы (I) с подходящим основанием с последующим взаимодействием с кислотой с образованием промежуточного соединения формулы (V) и выделение указанного промежуточного соединения из реакционной смеси; а стадия b) включает взаимодействие промежуточного соединения формулы (V) с соединением формулы (II) с образованием соединения формулы (III) или его гидрата. В одном из вариантов реализации указанного способа стадия a) включает кристаллизацию и сбор промежуточного соединения из реакционной смеси. В одном из вариантов реализации указанное промежуточное соединение кристаллизуют и собирают фильтрованием. Указанная дополнительная стадия выделения может иметь преимущество, поскольку при этом происходит удаление исходных материалов и производственных примесей до реакции связывания с соединением формулы (II). В некоторых вариантах реализации указанных способов выделение промежуточного соединения со стадии a) улучшает выход реакции синтеза.

В другом аспекте предложен способ получения кристаллической формы соединения А:

включающий стадии:
a) растворения соединения А в растворе, содержащем (i.) систему растворителей, содержащую простой эфир и необязательно спирт, и (ii.) воду, с получением раствора;
b) добавления к раствору суспензии затравочных кристаллов с получением суспензионной смеси;
d) добавления воды к суспензионной смеси с получением обработанной смеси; и
e) охлаждения обработанной смеси с получением кристаллического (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты.
В другом аспекте предложен способ получения кристаллической формы соединения А:

включающий стадии:
a) растворения соединения А в растворе, содержащем (i.) систему растворителей, содержащую простой эфир и необязательно спирт, и (ii.) воду, с получением раствора;
b) добавления к раствору суспензии затравочных кристаллов с получением суспензионной смеси;
c) охлаждения суспензионной смеси с получением охлажденной суспензионной смеси;
d) добавления воды к охлажденной суспензионной смеси с получением обработанной смеси; и
e) охлаждения обработанной смеси с получением кристаллического (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты.
В другом аспекте предложен способ получения кристаллического (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты, полученного в соответствии со способом, описанным в настоящем документе.
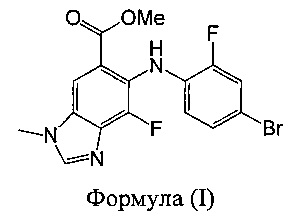
В другом аспекте предложено соединение формулы (I):

Указанное соединение формулы (I) подходит в качестве промежуточного соединения для синтеза соединения А в соответствии с настоящим изобретением.
В другом аспекте в настоящем документе предложено соединение формулы (V):

Указанное соединение формулы (V) подходит в качестве промежуточного соединения для синтеза соединения А в соответствии с настоящим изобретением.
В другом аспекте предложено соединение формулы (IV):

или его гидрат. В предпочтительном варианте реализации соединение формулы (IV) находится в форме его моногидрата. Указанное соединение формулы (IV), в том числе моногидрат, подходит в качестве промежуточного соединения для синтеза соединения А в соответствии с настоящим изобретением.
В другом аспекте предложена фармацевтическая композиция, содержащая кристаллический (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты, по меньшей мере один сахар и по меньшей мере одно целлюлозное вспомогательное вещество.
В другом аспекте предложен способ лечения пролиферативного заболевания, в частности, рака, у субъекта, включающий введение субъекту, нуждающемуся в этом, фармацевтической композиции, содержащей кристаллический (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты, по меньшей мере один сахар и по меньшей мере одно целлюлозное вспомогательное вещество.
Краткое описание чертежей
Фигура 1 иллюстрирует два микроскопических изображения агломерированной лекарственной субстанции соединения А, полученной с помощью известных ранее способов.
Фигура 2 иллюстрирует микроскопическое изображение кристаллического соединения А, полученного с помощью нового способа кристаллизации согласно настоящему изобретению: (a) без измельчения, (b) со струйным измельчением и (c) со штифтовым измельчением.
Осуществление изобретения
В настоящем документе предложены способы, подходящие для получения и синтеза эффективного и селективного ингибитора MEK 1/2, (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты («соединения А»), и подходящих промежуточных соединений. Настоящее изобретение дополнительно относится к способу синтеза кристаллического соединения А, а также фармацевтической композиции, подходящей для введения указанного кристаллического соединения.
Указанные способы имеют несколько преимуществ по сравнению с ранее известными способами (например, WO 03/077914). Например, предложенные способы получения соединения А обладают улучшенным профилем чистоты с низким содержанием палладия (менее 1 м.д.).
Общие термины, используемые в настоящем документе, определены следующими значениями, если специально не указано иное.
«Субъект» для целей настоящего изобретения включает людей и других животных, в частности, млекопитающих, и другие организмы. Следовательно, способы настоящего изобретения применимы как для лечения людей, так и для применения в ветеринарии. В предпочтительном варианте реализации пациент представляет собой млекопитающее, и в наиболее предпочтительном варианте реализации пациент представляет собой человека.
Термины «эффективное количество» или «фармацевтически эффективное количество», или «терапевтически эффективное количество» относятся к достаточному количеству агента для обеспечения заданного биологического, терапевтического и/или профилактического результата. Этим результатом может быть снижение, улучшение, облегчение, уменьшение, замедление и/или смягчение одного или более признаков, симптомов или причин заболевания, или любое другое желаемое изменение биологической системы. В отношении рака эффективное количество включает количество, достаточное для сокращения опухоли и/или снижения скорости роста опухоли (то есть, для подавления роста опухоли), или для предупреждения или отсрочки другой нежелательной клеточной пролиферации. В некоторых вариантах реализации эффективное количество представляет собой количество, достаточное для отсрочки прогрессирования. В некоторых вариантах реализации эффективное количество представляет собой количество, достаточное для предупреждения или отсрочки рецидива. Эффективное количество может быть введено за одно или более введений. Эффективное количество лекарства или композиции может: (i) снижать количество раковых клеток; (ii) уменьшать размер опухоли; (iii) подавлять, задерживать, замедлять до некоторой степени и, предпочтительно, останавливать инфильтрацию раковых клеток в периферические органы; (iv) подавлять (то есть, замедлять до некоторой степени и, предпочтительно, останавливать) метастазы опухоли; (v) подавлять рост опухоли; (vi) предупреждать или вызывать отсрочку возникновения и/или рецидива опухоли; и/или (vii) облегчать до некоторой степени один или более симптомов, связанных с раком.
Если не указано иное, «лечить» или «лечение» заболевания, расстройства или синдрома, при использовании в настоящем документе, означает подавление заболевания, расстройства или синдрома, то есть остановку его развития; и облегчение заболевания, расстройства или синдрома, то есть вызывание регрессии заболевания, расстройства или синдрома. Как известно в данной области техники, в контексте лечения может быть необходима поправка на системную доставку вместо локализованной доставки, возраст, вес тела, общее состояние здоровья, пол, питание, время введения, взаимодействие лекарств и тяжесть состояния, и она может быть установлена специалистами в данной области обычным экспериментированием.
«Предупреждение» означает предотвращение возникновения у человека заболевания, расстройства или синдрома, то есть исключение развития клинических симптомов заболевания, расстройства или синдрома у животного, которое может подвергаться или быть предрасположенным к заболеванию, расстройству или синдрому, но еще не страдает или не проявляет симптомов заболевания, расстройства или синдрома.
«Фармацевтическая композиция» означает смесь или раствор, содержащий по меньшей мере один терапевтический агент, подлежащий введению субъекту, например, млекопитающему или человеку, для предупреждения или лечения определенного заболевания или состояния, которым страдает указанное млекопитающее.
«Фармацевтически приемлемые» означает те соединения, материалы, композиции и/или лекарственные формы, которые по результатам обоснованной медицинской оценки подходят для контакта с тканями субъекта, например, млекопитающего или человека, без избыточной токсичности, раздражающей аллергической реакции и других проблемных осложнений, соразмерно с соотношением приемлемой пользы и риска.
Термины «включающий» и «содержащий» использованы в настоящем документе в неограничивающем значении, если не указано иное.
Термины в единственном числе, используемые в контексте описания настоящего изобретения (особенно в контексте следующей формулы изобретения) следует толковать как включающие единственное и множественное число, если в настоящем документе не указано иное, или очевидно не опровергается контекстом. Если для соединений, солей и т.п. использована множественная форма, то ее следует понимать и как одно соединение, соль или т.п.
При использовании в настоящем документе, термины «приблизительно» или «около», как правило, означают возможные отклонения не более чем на 10%, 5% или 1% от значения.
При использовании в настоящем документе, термин «выделенное» означает, что соединение отделено от реакционной смеси, в которой оно образовано или обнаружено. Выделенное соединение содержит менее 15%, менее 10%, менее 6% или менее 3% относительно массы органических растворителей или воды. Неограничивающие примеры способов выделения включают фильтрацию, центрифугирование, вакуумную сушку, осаждение, кристаллизацию и колоночную хроматографию.
В одном аспекте в настоящем документе предложен способ получения соединения (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты (в настоящем документе упоминается как «соединение А»)

включающий стадии:
а) взаимодействия соединения формулы (I):

с подходящим основанием с образованием промежуточного соединения; и
b) взаимодействия указанного промежуточного соединения с соединением формулы (II):

с получением соединения формулы (III):

или его гидрата,
где Р1 представляет собой защитную группу;
c) растворения указанного соединения формулы (III) или его гидрата в подходящем растворителе или системе растворителей; и
d) снятия защиты с указанного соединения формулы (III) или его гидрата при помощи подходящего реагента для снятия защиты,
где Р1 в каждом случае может быть одинаковым или различным и представляет собой подходящую защитную группу, с получением соединения А.
В способе стадии a) соединение формулы (I) взаимодействует с подходящим основанием с образованием промежуточного соединения. Примеры подходящих оснований для представленной выше реакции включают, но не ограничиваются ими, гидроксид натрия, гидроксид калия, гидроксид цезия, гидроксид лития, триметилсиланолят калия, триметилсиланолят лития и триметилсиланолят натрия. В предпочтительном варианте реализации подходящее основание представляет собой триметилсиланолят калия. В другом предпочтительном варианте реализации подходящее основание представляет собой гидроксид натрия.
Способ стадии a), в котором соединение формулы (I) взаимодействует с подходящим основанием, может быть выполнен в любом подходящем растворителе или системе растворителей. Подходящие растворители включают полярные апротонные растворители, такие как ацетон, ацетонитрил, N,N-диметилформамид, диметилсульфоксид, этилацетат, тетрагидрофуран, 2-метилтетрагидрофуран и 1,4-диоксан. Подходящие системы растворителей включают любую комбинацию подходящих растворителей. В предпочтительном варианте реализации реакцию осуществляют в смеси N,N-диметилформамида и ТГФ. Подходящие системы растворителей могут содержать также один или более подходящих растворителей в комбинации с водой. В одном конкретном варианте реализации реакцию осуществляют в смеси N,N-диметилформамида и воды.
В одном из вариантов реализации способа, описанного выше, стадии a) и b) осуществляют «однореакторным» синтезом, при этом промежуточное соединение со стадии a) взаимодействует с соединением формулы (II) без первоначального выделения из реакционной смеси стадии a) (см., например, пример 2А). В одном из вариантов реализации промежуточное соединение со стадии a) представляет собой промежуточное соединение 1:

В одном из конкретных вариантов реализации способа, включающего «однореакторный синтез», стадия a) включает взаимодействие соединения формулы (I) с подходящим основанием с образованием промежуточного соединения 1, а стадия b) включает взаимодействие промежуточного соединения 1 с соединением формулы (II) с образованием соединения формулы (III) или его гидрата. В одном из вариантов реализации промежуточное соединение 1 не выделяют из реакционной смеси стадии a) до стадии b). В другом варианте реализации промежуточное соединение 1 представляет собой часть раствора, содержащего растворители, выбранные из группы, состоящей из ДМФ и ТГФ.
В другом варианте реализации указанного способа стадии a) и b) осуществляют с выделением промежуточного соединения со стадии a) из реакционной смеси перед его взаимодействием с соединением формулы (II). В одном из вариантов реализации промежуточное соединение со стадии a) представляет собой соединение формулы (V):
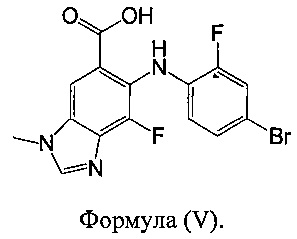
В другом варианте реализации способа, в котором промежуточное соединение со стадии a) выделяют из реакционной смеси стадии a) перед взаимодействием на стадии b), указанный способ включает выделение промежуточного соединения (например, соединения формулы (V)) из реакционной смеси со стадии a) перед стадией b). В одном из вариантов реализации указанный способ выделения включает кристаллизацию и сбор промежуточного соединения (например, соединения формулы (V)) из реакционной смеси стадии a) перед стадией b). В одном из вариантов реализации указанное промежуточное соединение кристаллизуют и собирают фильтрованием.
В другом варианте реализации стадия a) включает взаимодействие соединения формулы (I) с подходящим основанием с образованием промежуточного соединения формулы (V) и выделение указанного промежуточного соединения из реакционной смеси; а стадия b) включает взаимодействие промежуточного соединения формулы (V) с соединением формулы (II) с образованием соединения формулы (III) или его гидрата. В одном конкретном варианте реализации способа, в котором промежуточное соединение со стадии a) выделяют из реакционной смеси стадии a) перед реакцией на стадии b), стадия a) включает взаимодействие соединения формулы (I) с подходящим основанием с последующей реакцией с кислотой с образованием промежуточного соединения формулы (V) и выделение промежуточного соединения формулы (V) из реакционной смеси; а стадия b) включает взаимодействие промежуточного соединения формулы (V) с соединением формулы (II) с образованием соединения формулы (III) или его гидрата. В одном из вариантов реализации кислота представляет собой хлористоводородную кислоту. В другом варианте реализации стадия a) указанного способа включает кристаллизацию и сбор промежуточного соединения формулы (V) из реакционной смеси стадии a). В одном из вариантов реализации указанное промежуточное соединение кристаллизуют и собирают фильтрованием.
При использовании в настоящем документе, термин «защитная группа» относится к тем группам, которые используют для предотвращения участия реакционноспособных групп (таких как карбокси, амино, гидрокси и меркапто-группы) в нежелательных реакциях. В частности, подходящие защитные группы для Р1, используемого в настоящей заявке, включают кислото-неустойчивые защитные группы. Иллюстративные примеры подходящих кислото-неустойчивых защитных групп для Р1, используемого в настоящей заявке, включают, но не ограничиваются ими: алкильные группы, такие как третичные алкилы (например, третичные С4-С7 алкилы, такие как трет-бутил или третичный амил); алкенильные группы; третичные арил-алкильные группы, такие как 1-метил-1-фенилэтил (кумил) или трифенилметил (тритил); группы, которые приводят к образованию ацеталей, такие как метоксиметил, 1-этоксиэтил, 2-тетрагидропиранил или 2-тетрагидрофуранил; и силильные группы, такие как триметилсилил, триэтилсилил или трет-бутил-диметилсилил. В предпочтительном варианте реализации Р1 представляет собой трет-бутил.
Способ стадии b), в котором промежуточное соединение со стадии a) взаимодействует с соединением формулы (II), может быть осуществлен в присутствии любого связывающего агента и источника протонов. Подходящие источники протонов включают, но не ограничиваются ими, имидазола гидрохлорид, пиридиния гидрохлорид, триэтиламина гидрохлорид, N-метилморфолина гидрохлорид и сульфоновые кислоты, такие как, например, метансульфоновая кислота, и, предпочтительно, имидазола гидрохлорид. Подходящие связывающие агенты включают, но не ограничиваются ими, 1,1'-карбонилдиимидазол, изобутилхлорформиат, пивалоилхлорид, оксалилхлорид, тионилхлорид, циклический ангидрид 1-пропанфосфоновой кислоты и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (EDCI), и, предпочтительно, 1,1'-карбонилдиимидазол. В предпочтительном варианте реализации настоящего изобретения способ стадии b) осуществляют в присутствии связывающего агента 1,1'-карбонилдиимидазола и источника протонов гидрохлорида имидазола. Специалистам в данной области техники известно, как оптимизировать способ согласно настоящему изобретению для связывающих агентов, отличных от 1,1'-карбонилдиимидазола, и для источников протона, отличных от гидрохлорида имидазола.
Способ стадии c), в котором соединение формулы (III) или его гидрат растворяют в подходящем растворителе или системе растворителей и снимают с него защиту, может быть осуществлен в любом подходящем растворителе или системе растворителей. Примеры подходящих растворителей включают (a) полярные протонные растворители, такие как метанол, этанол и изопропанол, и (b) полярные апротонные растворители, такие как ацетон, ацетонитрил, N,N-диметилформамид, диметилсульфоксид, этилацетат и тетрагидрофуран. Подходящие системы растворителей включают любую комбинацию подходящих растворителей.
В одном из вариантов реализации указанную реакцию осуществляют в полярном апротонном растворителе. В предпочтительном варианте реализации указанную реакцию осуществляют в ацетонитриле.
Защитная группа для Р1 может быть снята при помощи любого подходящего агента для снятия защиты. Условия снятия защиты для гидрокси-защитных групп, безусловно, варьируются в зависимости от выбора защитной группы. Алкильные группы или алкенильные группы можно снять, например, при помощи водного раствора кислоты, такой как фосфорная кислота. Третичные арил-алкильные группы можно снять, например, при помощи водного раствора кислоты. Силильные группы можно снять, например, при помощи фторида или водного раствора кислоты. Специалистам в данной области техники понятно, что кислото-неустойчивые защитные группы можно снимать при помощи водных растворов кислот. Подходящие агенты для снятия защиты представленных выше защитных групп включают, но не ограничиваются ими, водные растворы кислот, таких как фосфорная кислота, хлористоводородная кислота или серная кислота; неводные растворы кислот, такие как хлористоводородная кислота в изопропиловом спирте или в других подходящих органических растворителях, таких как 1,4-диоксан или тетрагидрофуран, триметилсилилхлорид, трифторуксусная кислота или п-толуолсульфоновая кислота.
В одном из вариантов реализации, если защитная группа Р1 представляет собой трет-бутил, то подходящий агент для снятия защиты может быть выбран из водного раствора кислоты, такой как фосфорная кислота, хлористоводородная кислота или серная кислота; 5 М хлористоводородная кислота в изопропиловом спирте, триметилсилилхлорид или п-толуолсульфоновая кислота (гидрат). Предпочтительно, если защитная группу Р1 представляет собой трет-бутил, то подходящий агент для снятия защиты представляет собой водный раствор фосфорной кислоты.
В одном из вариантов реализации, если защитная группа Р1 представляет собой трет-бутил, то подходящий растворитель или система растворителей на стадии a) выбрана из ацетонитрила, тетрагидрофурана, метанола и этанола, а агент для снятия защиты представляет собой фосфорную кислоту.
В дополнительном варианте реализации, если защитная группа Р1 представляет собой трет-бутил, то подходящий растворитель или система растворителей на стадии a) выбрана из ацетонитрила, тетрагидрофурана, метанола и этанола, а агент для снятия защиты представляет собой хлористоводородную кислоту.
В дополнительном варианте реализации данного аспекта настоящего изобретения после стадии d) осуществляют дальнейшее превращение соединения А, полученного на стадии d), в любую его фармацевтически приемлемую соль. «Фармацевтически приемлемая соль», при использовании в настоящем документе, если не указано иное, включает соли кислотной и основной групп, которые могут присутствовать в соединениях согласно настоящему изобретению. Соединения согласно настоящему изобретению, которые являются основными по своей природе, способны образовывать широкий ряд солей с различными неорганическими и органическими кислотами. Кислоты, которые могут быть использованы для получения фармацевтически приемлемых солей присоединения кислот с такими основными соединениями согласно настоящему изобретению, представляют собой кислоты, которые образуют нетоксичные соли присоединения кислот, т.е. соли, содержащие фармацевтически приемлемые анионы, такие как ацетатные, бензоатные, бромидные, хлоридные, цитратные, фумаратные, гидробромидные, гидрохлоридные, йодидные, лактатные, малеатные, манделатные, нитратные, оксалатные, салицилатные, сукцинатные и тартратные соли. Поскольку одно соединение согласно настоящему изобретению может содержать более одного кислотного или основного фрагмента, то соединения согласно настоящему изобретению могут включать моно-, ди- или три-соли в одном соединении.
В случае наличия кислотного фрагмента в соединении согласно настоящему изобретению соль может быть образована обработкой соединения согласно настоящему изобретению основным соединением, в частности, неорганическим основанием. Предпочтительные неорганические соли представляют собой соли, образованные с щелочными и щелочноземельными металлами, такими как литий, натрий, калий, барий и кальций. Предпочтительные соли органических оснований включают, например, соли аммония, дибензиламмония, бензиламмония, 2-гидроксиэтиламмония, бис(2-гидроксиэтил)аммония, фенилэтилбензиламина, дибензил-этилендиамина и т.п. Другие соли кислотных фрагментов могут включать, например, соли, образованные с прокаином, хинином и N-метилглюкозамином, плюс соли, образованные с основными аминокислотами, такими как глицин, орнитин, гистидин, фенилглицин, лизин и аргинин. Особенно предпочтительная соль представляет собой натриевую или калиевую соль соединения согласно настоящему изобретению.
В отношении основных фрагментов соль получают при обработке соединения согласно настоящему изобретению кислотным соединением, в частности, неорганической кислотой. Предпочтительные неорганические соли данного типа могут включать, например, гидрохлоридные, гидробромидные, сульфатные, фосфатные или т.п. соли. Предпочтительные органические соли данного типа могут включать, например, соли, образованные с уксусной, янтарной, лимонной, малеиновой, фумаровой, D-глутаминовой, гликолевой, бензойной, коричной и т.п. органическими кислотами. Особенно предпочтительная соль данного типа представляет собой гидрохлоридную или сульфатную соль соединения А.
Следует понимать, что в соответствии с настоящим изобретением основание можно добавлять к кислотной реакционной смеси, образованной на стадии снятия защиты d) до достижения pH в диапазоне 5-9. Предпочтительно, основание добавляют для нейтрализации кислотной реакционной смеси, образованной на стадии снятия защиты d) до pH = приблизительно 8-8,5. Примеры подходящих оснований включают, но не ограничиваются ими, гидроксид калия, гидроксид натрия, гидроксид лития и гидроксид аммония. Предпочтительно, дополнительное основание представляет собой гидроксид калия.
В предпочтительном варианте реализации подходящее основание на стадии a) представляет собой триметилсиланолят калия, защитная группа Р1 представляет собой трет-бутил, подходящий растворитель или система растворителей на стадии c) представляет собой ацетонитрил, а подходящий агент для снятия защиты на стадии d) представляет собой водный раствор фосфорной кислоты.
В другом предпочтительном варианте реализации подходящее основание на стадии a) представляет собой гидроксид натрия, защитная группа Р1 представляет собой трет-бутил, подходящий растворитель или система растворителей на стадии c) представляет собой ацетонитрил, а подходящий агент для снятия защиты на стадии d) представляет собой водный раствор фосфорной кислоты.
В предпочтительном варианте реализации подходящее основание на стадии a) представляет собой триметилсиланолят калия, защитная группа Р1 представляет собой трет-бутил, подходящий растворитель или система растворителей на стадии c) представляет собой ацетонитрил, подходящий агент для снятия защиты на стадии d) представляет собой фосфорную кислоту, а затем добавляют основание, при этом дополнительное основание представляет собой гидроксид калия.
В другом предпочтительном варианте реализации подходящее основание на стадии a) представляет собой гидроксид натрия, защитная группа Р1 представляет собой трет-бутил, подходящий растворитель или система растворителей на стадии c) представляет собой ацетонитрил, подходящий агент для снятия защиты на стадии d) представляет собой фосфорную кислоту, а затем добавляют основание, при этом дополнительное основание представляет собой гидроксид калия.
В другом аспекте в настоящем документе предложен способ получения соединения формулы (III):

или его гидрата, включающий стадии:
a) взаимодействия соединения формулы (I):

с подходящим основанием с образованием промежуточного соединения; и
b) взаимодействия указанного промежуточного соединения с соединением формулы (II):

где Р1 представляет собой защитную группу, с образованием соединения формулы (III) или его гидрата.
Способ стадии a), в котором соединение формулы (I) взаимодействует с подходящим основанием с образованием промежуточного соединения. Примеры подходящих оснований для представленной выше реакции включают, но не ограничиваются ими, гидроксид натрия, гидроксид калия, гидроксид цезия, гидроксид лития, триметилсиланолят калия, триметилсиланолят лития и триметилсиланолят натрия. В предпочтительном варианте реализации подходящее основание представляет собой триметилсиланолят калия. В другом предпочтительном варианте реализации подходящее основание представляет собой гидроксид натрия.
Способ стадии a), в котором соединение формулы (I) взаимодействует с подходящим основанием, может быть осуществлен в любом подходящем растворителе или системе растворителей. Подходящие растворители включают полярные апротонные растворители, такие как ацетон, ацетонитрил, N,N-диметилформамид, диметилсульфоксид, этилацетат, тетрагидрофуран, 2-метилтетрагидрофуран и 1,4-диоксан. Подходящие системы растворителей включают любую комбинацию подходящих растворителей. В предпочтительном варианте реализации реакцию осуществляют в смеси N,N-диметилформамида и ТГФ. Подходящие системы растворителей также могут содержать один или более подходящих растворителей в комбинации с водой. В одном конкретном варианте реализации указанную реакцию осуществляют в смеси N,N-диметилформамида и воды.
В одном из вариантов реализации указанного способа стадии a) и b) осуществляют «однореакторным» синтезом, как описано выше. В одном из вариантов реализации промежуточное соединение со стадии a) представляет собой промежуточное соединение 1. В другом варианте реализации стадия a) включает взаимодействие соединения формулы (I) с подходящим основанием с образованием промежуточного соединения 1, а стадия b) включает взаимодействие промежуточного соединения 1 с соединением формулы (II) с образованием соединения формулы (III) или его гидрата. В одном из вариантов реализации промежуточное соединение 1 не выделяют из реакционной смеси стадии a) до стадии b). В другом варианте реализации промежуточное соединение 1 представляет собой часть раствора, содержащего растворители, выбранные из группы, состоящей из ДМФ и ТГФ.
В другом варианте реализации указанного способа стадии a) и b) осуществляют с выделением промежуточного соединения со стадии а) перед реакцией на стадии b), как описано выше. В одном из вариантов реализации промежуточное соединение со стадии а) представляет собой соединение формулы (V). В другом варианте реализации указанный способ включает выделение промежуточного соединения (например, соединения формулы (V)) из реакционной смеси стадии a) перед стадией b). В одном из вариантов реализации указанный способ выделения включает кристаллизацию и сбор промежуточного соединения (например, соединения формулы (V)) из реакционной смеси стадии a) перед стадией b). В одном из вариантов реализации указанное промежуточное соединение кристаллизуют и собирают фильтрованием.
В другом варианте реализации способа, в котором промежуточное соединение со стадии a) выделяют из реакционной смеси стадии a) перед реакцией на стадии b), стадия a) включает взаимодействие соединения формулы (I) с подходящим основанием с образованием промежуточного соединения формулы (V) и выделение указанного промежуточного соединения из реакционной смеси; а стадия b) включает взаимодействие промежуточного соединения формулы (V) с соединением формулы (II) с образованием соединения формулы (III) или его гидрата. В одном из конкретных вариантов реализации стадия a) включает взаимодействие соединения формулы (I) с подходящим основанием с последующим взаимодействием с кислотой с образованием промежуточного соединения формулы (V) и выделение промежуточного соединения формулы (V) из реакционной смеси; а стадия b) включает взаимодействие промежуточного соединения формулы (V) с соединением формулы (II) с образованием соединения формулы (III) или его гидрата. В одном из вариантов реализации кислота представляет собой хлористоводородную кислоту.
Примеры подходящих защитных групп Р1 включают группы, описанные выше, при этом текст в полном объеме включен здесь посредством ссылки.
Способ стадии b), в котором промежуточное соединение со стадии a) взаимодействует с соединением формулы (II), может быть осуществлен в присутствии любого связывающего агента и источника протонов. Подходящие источники протонов включают, но не ограничиваются ими, имидазола гидрохлорид, пиридиния гидрохлорид, триэтиламина гидрохлорид, N-метилморфолина гидрохлорид и сульфоновые кислоты, такие как, например, метансульфоновая кислота, и, предпочтительно, имидазола гидрохлорид. Подходящие связывающие агенты включают, но не ограничиваются ими, 1,1'-карбонилдиимидазол, изобутилхлорформиат, пивал оилхлорид, оксалилхлорид, тионилхлорид, циклический ангидрид 1-пропанфосфоновой кислоты и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (EDCI), и, предпочтительно, 1,1'-карбонилдиимидазол. В предпочтительном варианте реализации настоящего изобретения способ стадии b) осуществляют в присутствии связывающего агента 1,1'-карбонилдиимидазола и источника протонов гидрохлорида имидазола. Специалистам в данной области техники известно, как оптимизировать способ согласно настоящему изобретению для связывающих агентов, отличных от 1,1'-карбонилдиимидазола, и для источников протона, отличных от гидрохлорида имидазола.
В другом аспекте настоящего изобретения открыт новый способ получения новой кристаллической формы соединения А. Было показано, что известные до этого способы синтеза соединения А или его фармацевтически приемлемых солей, например, способы, описанные в WO 03/077914, имеют следующие основные недостатки для разработки фармацевтических лекарств: (a) синтезированная лекарственная субстанция, как правило, образует крупные чешуйки (агломераты) или порошок, (b) недостаточный профиль чистоты и выход, и (c) синтезированная лекарственная субстанция имеет «липкую» морфологию с плохой сыпучестью. Известные до этого способы приводят к получению очень агломерированного материала соединения А, который образует чешуйки, некоторые из которых имеют диаметр до 15 мм. Это создает значительную проблему, которая обусловливает невозможность надежного, воспроизводимого и контролируемого крупномасштабного производства фармацевтических композиций, содержащих соединение А или его фармацевтически приемлемые соли.
Был открыт новый способ получения кристаллической формы соединения А, который неожиданно приводит к получению кристаллической формы соединения А, обладающей значительно лучшим профилем чистоты и улучшенной физической морфологией (например, сниженным количеством липких кристаллов/частиц, улучшенной сыпучестью). Было обнаружено, что соединение А обладает очень слабой растворимостью в большинстве стандартных растворителей (т.е. менее 1% при комнатной температуре). Из-за такой слабой растворимости трудно выполнить кристаллизацию стандартными способами охлаждения и контролировать рост кристаллов. Однако было обнаружено, что вода, которая, в основном, представляет собой антирастворитель (растворимость <0,01% в широком диапазоне температур), также неожиданно действует в качестве растворителя соединения А при использовании в новой смеси растворителей, содержащей простой эфир и необязательно спирт и, следовательно, значительно увеличивает растворимость соединения А. Несмотря на то что добавление небольшого количества антирастворителя к растворителю обычно приводит к небольшому увеличению растворимости, добавление воды к смеси растворителей из тетрагидрофурана и метанола повышает растворимость соединения А приблизительно на 50% по сравнению со смесью растворителей из метанола и тетрагидрофурана без воды. Такое улучшение растворимости способствует улучшению профиля чистоты готовой лекарственной субстанции.
Кроме того, новый способ получения кристаллической формы соединения А приводит к получению новой кристаллической формы соединения А со сниженной характеристикой липкости и улучшенной сыпучестью. Показано такое улучшение с последующей стадией измельчения или без нее, как описано ниже, (см. фиг. 1 по сравнению с фиг. 2). Однако дополнительная стадия измельчения обеспечивает преимущество дополнительного улучшения характеристики липкости и сыпучести соединения А.
Так, в одном аспекте предложен способ получения кристаллической формы соединения А:

включающий стадии:
a) растворения соединения А в растворе, содержащем (i.) систему растворителей, содержащую простой эфир и необязательно спирт, и (ii.) воду, с получением раствора;
b) добавления к раствору суспензии затравочных кристаллов с получением суспензионной смеси;
d) добавления воды к суспензионной смеси с получением обработанной смеси; и
e) охлаждения обработанной смеси с получением кристаллического (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты.
В другом аспекте предложен способ получения кристаллической формы соединения А:
включающий стадии:
a) растворения соединения А в растворе, содержащем (i.) систему растворителей, содержащую простой эфир и необязательно спирт, и (ii.) воду, с получением раствора;
b) добавления к раствору суспензии затравочных кристаллов с получением суспензионной смеси;
c) охлаждения суспензионной смеси с получением охлажденной суспензионной смеси;
d) добавления воды к охлажденной суспензионной смеси с получением обработанной смеси; и
e) охлаждения обработанной смеси с получением кристаллического (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты.
В соответствии со стадией a) данного аспекта настоящего изобретения, соединение А растворяют в растворе, содержащем (i) систему растворителей, содержащую простой эфир и необязательно спирт, и (ii) воду. Следует понимать, что стадию a) можно осуществить любым из следующих способов: (a) добавлением соединения А к раствору, содержащему (i.) систему растворителей, содержащую простой эфир и необязательно спирт, и (ii.) воду, или (b) добавлением каждого компонента раствора к соединению А.
Подходящие простые эфиры включают ТГФ.
В соответствии с настоящим изобретением, в раствор на стадии a) не обязательно добавлять спирт. Однако предпочтителен раствор, содержащий простой эфир и спирт. Подходящие спирты включают, но не ограничиваются ими, метанол, этанол и изопропанол. Предпочтительно, спирт представляет собой метанол.
В одном из вариантов реализации соединение А растворяют в растворе, состоящем из растворителя ТГФ и воды.
В одном из вариантов реализации соединение А растворяют в растворе, содержащем или состоящем из (i) системы растворителей, содержащей простой эфир и спирт, и (ii) воды. Предпочтительно, указанный раствор содержит систему растворителей, содержащую спирт и ТГФ, и воду.
В предпочтительном варианте реализации соединение А растворяют в растворе, состоящем из (i) системы растворителей, содержащей метанол и ТГФ, и (ii) воды.
Растворение соединения А в растворе на стадии a) облегчается нагреванием смеси соединения А и раствора, содержащего (i) систему растворителей, содержащую простой эфир и спирт, и (ii) воду, до внутренней температуры около 52-56°С перед добавлением суспензии затравочных кристаллов.
В одном аспекте смесь соединения А и раствор, содержащий (i) систему растворителей, содержащую простой эфир и спирт, и (ii) воду, нагревают до внутренней температуры около 53-55°С.
К раствору на стадии b) добавляют суспензию затравочных кристаллов с получением суспензионной смеси. Указанный раствор преимущественно охлаждают (i) после нагревания смеси соединения А и раствора, содержащего (i) систему растворителей, содержащую простой эфир и спирт, и (ii) воду, до внутренней температуры около 52-56°С и (ii) перед добавлением суспензии затравочных кристаллов.
В одном аспекте к суспензионной смеси добавляют воду с получением обработанной смеси. В другом аспекте суспензионную смесь охлаждают до температуры около 30-50°С перед добавлением воды. В предпочтительном варианте реализации суспензионную смесь охлаждают до температуры около 47-48°С перед добавлением воды.
Как описано выше, в процессе кристаллизации используют охлаждающую систему антирастворителей. На стадии d) к суспензии или охлажденной суспензионной смеси добавляют воду с получением обработанной смеси. Вода, которая, как правило, действует в качестве антирастворителя (растворимость <0,01% в широком диапазоне температур), также неожиданно действует в качестве растворителя для соединения А, будучи частью системы растворителей из воды/метанола/тетрагидрофурана. Таким образом, добавление воды неожиданно вызывает значительное увеличение растворимости соединения А. При помощи указанного способа было обнаружено, что при 65°С достигается максимальная растворимость соединения А в смеси воды (24%), метанола (38%) и тетрагидрофурана (38%), при этом растворимость соединения А снижается при дальнейшем добавлении воды.
Воду можно добавлять в течение периода времени от 5 до 35 часов, так что содержание воды не превышает 70% масс./масс., предпочтительно 65% (масс./масс.) относительно системы растворителей. В одном из вариантов реализации воду добавляют в течение периода времени 10-25 часов, предпочтительно 25 часов. В другом варианте реализации воду добавляют в течение периода времени 25 часов, так что 33% добавляют в течение 15 часов, а 66% добавляют в течение 10 часов.
В другом аспекте компоненты в системе растворителей после завершения добавления воды к суспензии или охлажденной суспензионной смеси имеют окончательное соотношение спирта/простого эфира/воды в диапазоне от 40/40/20 до 15/15/70 масс./масс., при этом масс./масс. относится к массовому проценту каждого компонента относительно других компонентов в системе растворителей/антирастворителей. В предпочтительном варианте реализации окончательное соотношение спирта/простого эфира/воды составляет приблизительно 20/20/60 масс./масс. (20/20/60 масс./масс.).
На стадии e) окончательно получают кристаллизованное соединение А путем охлаждения обработанной смеси. Обработанную смесь преимущественно охлаждают в течение периода времени от 5 до 25 часов. В одном аспекте обработанную смесь охлаждают в течение периода времени от 8 до 15 часов, от 8 до 12 часов или от 9 до 11 часов. В предпочтительном варианте реализации обработанную смесь охлаждают в течение периода времени, составляющего приблизительно 10 часов. Обработанную смесь преимущественно охлаждают до внутренней температуры около 1-10°С, предпочтительно около 3-5°С. В предпочтительном варианте реализации обработанную смесь охлаждают до внутренней температуры около 3-5°С в течение приблизительно 9-11 часов.
После фильтрования кристаллизованное соединение А можно высушить, например, под вакуумом или в вакуумной печи.
В дополнительном варианте реализации кристаллизованное соединение А, полученное на стадии e) способа согласно настоящему изобретению, можно затем измельчить. Подходящие приемы измельчения известны специалистам в данной области техники и включают, но не ограничиваются ими, штифтовое измельчение или струйное измельчение.
Более мелкие исходные частицы зачастую приводят к более высокой степени агломерации. Однако после измельчения кристаллизованное соединение А неожиданно демонстрирует сниженную слипаемость даже после хранения в течение более трех месяцев.
В одном из дополнительных аспектов предложен способ получения кристаллической формы соединения А, включающий стадии:
a) взаимодействия соединения формулы (I):
с подходящим основанием с образованием промежуточного соединения; и
b) взаимодействия указанного промежуточного соединения с соединением формулы (II):

с получением соединения формулы (III):

или его гидрата, где P1 представляет собой защитную группу;
c) растворения указанного соединения формулы (III) или его гидрата в подходящем растворителе или системе растворителей;
d) снятия защиты с указанного соединения формулы (III) или его гидрата при помощи подходящего реагента для снятия защиты,
где Р1 в каждом случае может быть одинаковым или различным и представляет собой подходящую защитную группу, с получением соединения А;
e) растворения соединения А в растворе, содержащем (i) систему растворителей, содержащую простой эфир и необязательно спирт, и (ii) воду, с получением раствора;
f) добавления к раствору суспензии затравочных кристаллов с получением суспензионной смеси;
g) добавления воды к суспензионной смеси с получением обработанной смеси; и
h) охлаждения обработанной смеси с получением кристаллизованного (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты.
В другом аспекте предложен способ получения кристаллической формы соединения А, включающий стадии:
а) взаимодействия соединения формулы (I):

с подходящим основанием с образованием промежуточного соединения; и
b) взаимодействия указанного промежуточного соединения с соединением формулы (И):
с получением соединения формулы (III):

или его гидрата, где Р1 представляет собой защитную группу;
c) растворения указанного соединения формулы (III) или его гидрата в подходящем растворителе или системе растворителей;
d) снятия защиты с указанного соединения формулы (III) или его гидрата при помощи подходящего реагента для снятия защиты,
где Р1 в каждом случае может быть одинаковым или различным и представляет собой подходящую защитную группу, с получением соединения А,
e) растворения соединения А в растворе, содержащем (i) систему растворителей, содержащую простой эфир и необязательно спирт, и (ii) воду, с получением раствора;
f) добавления суспензии затравочных кристаллов к указанному раствору с получением суспензионной смеси; охлаждения суспензионной смеси с получением охлажденной суспензионной смеси;
g) добавления воды к охлажденной суспензионной смеси с получением обработанной смеси; и
h) охлаждения обработанной смеси с получением кристаллического (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты.
Способ стадии a), в котором соединение формулы (I) взаимодействует с подходящим основанием с образованием промежуточного соединения. Примеры подходящих оснований для представленной выше реакции включают, но не ограничиваются ими, гидроксид натрия, гидроксид калия, гидроксид цезия, гидроксид лития, триметилсиланолят калия, триметилсиланолят лития и триметилсиланолят натрия. В предпочтительном варианте реализации подходящее основание представляет собой триметилсиланолят калия. В другом предпочтительном варианте реализации подходящее основание представляет собой гидроксид натрия. Способ стадии a), в котором соединение формулы (I) взаимодействует с подходящим основанием, может быть осуществлен в любом подходящем растворителе или системе растворителей. Подходящие растворители включают полярные апротонные растворители, такие как ацетон, ацетонитрил, N,N-диметилформамид, диметилсульфоксид, этилацетат, тетрагидрофуран, 2-метилтетрагидрофуран и 1,4-диоксан. Подходящие системы растворителей включают любую комбинацию подходящих растворителей. В предпочтительном варианте реализации реакцию выполняют в смеси N,N-диметилформамида и ТГФ. Подходящие системы растворителей также могут содержать один или более подходящих растворителей в комбинации с водой. В одном конкретном варианте реализации указанную реакцию осуществляют в смеси N,N-диметилформамида и воды.
В одном из вариантов реализации указанного способа стадии a) и b) осуществляют «однореакторным» синтезом, как описано выше. В одном из вариантов реализации промежуточное соединение со стадии a) представляет собой промежуточное соединение 1. В другом варианте реализации стадия a) включает взаимодействие соединения формулы (I) с подходящим основанием с образованием промежуточного соединения 1, а стадия b) включает взаимодействие промежуточного соединения 1 с соединением формулы (II) с образованием соединения формулы (III) или его гидрата. В одном из вариантов реализации промежуточное соединение 1 находится в растворе, содержащем растворители, выбранные из группы, состоящей из ДМФ и ТГФ.
В другом варианте реализации указанного способа стадии a) и b) выполняют с выделением промежуточного соединения со стадии a) перед реакцией на стадии b), как описано выше. В одном из вариантов реализации промежуточное соединение со стадии a) представляет собой соединение формулы (V). В другом варианте реализации указанный способ включает выделение промежуточного соединения (например, соединения формулы (V)) из реакционной смеси стадии а) перед стадией b). В одном из вариантов реализации указанный способ включает кристаллизацию и сбор промежуточного соединения (например, соединения формулы (V)) из реакционной смеси стадии a) перед стадией b). В одном из вариантов реализации указанное промежуточное соединение кристаллизуют и собирают фильтрованием.
В другом варианте реализации способа, в котором промежуточное соединение со стадии a) выделяют из реакционной смеси стадии а) перед реакцией на стадии b), стадия a) включает взаимодействие соединения формулы (I) с подходящим основанием с образованием промежуточного соединения формулы (V) и выделение указанного промежуточного соединения из реакционной смеси; а стадия b) включает взаимодействие промежуточного соединения формулы (V) с соединением формулы (II) с образованием соединения формулы (III) или его гидрата. В одном из конкретных вариантов реализации стадия a) включает взаимодействие соединения формулы (I) с подходящим основанием с последующим взаимодействием с кислотой с образованием промежуточного соединения формулы (V) и выделение промежуточного соединения формулы (V) из реакционной смеси; а стадия b) включает взаимодействие промежуточного соединения формулы (V) с соединением формулы (II) с образованием соединения формулы (III) или его гидрата. В одном из вариантов реализации кислота представляет собой хлористоводородную кислоту.
Подходящие защитные группы и реагенты для снятия защиты на стадиях b) и d) предложены выше и включены здесь посредством ссылки в полном объеме.
Способ стадии b), в котором промежуточное соединение со стадии a) взаимодействует с соединением формулы (II), может быть осуществлен в присутствии любого связывающего агента и источника протонов. Подходящие источники протонов включают, но не ограничиваются ими, имидазола гидрохлорид, пиридиния гидрохлорид, триэтиламина гидрохлорид, N-метилморфолина гидрохлорид и сульфоновые кислоты, такие как, например, метансульфоновая кислота, и, предпочтительно, имидазола гидрохлорид. Подходящие связывающие агенты включают, но не ограничиваются ими, 1,1'-карбонилдиимидазол, изобутилхлорформиат, пивалоилхлорид, оксалилхлорид, тионилхлорид, циклический ангидрид 1-пропанфосфоновой кислоты и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (EDCI), и, предпочтительно, 1,1'-карбонилдиимидазол. В предпочтительном варианте реализации настоящего изобретения способ стадии b) осуществляют в присутствии связывающего агента 1,1'-карбонилдиимидазола и источника протонов гидрохлорида имидазола. Специалистам в данной области техники известно, как оптимизировать способ согласно настоящему изобретению для связывающих агентов, отличных от 1,1'-карбонилдиимидазола, и для источников протона, отличных от гидрохлорида имидазола.
Следует понимать, что в соответствии с настоящим изобретением основание можно добавлять к кислотной реакционной смеси, образованной на стадии снятия защиты d) до достижения pH в диапазоне 5-9. Предпочтительно, основание добавляют для нейтрализации кислотной реакционной смеси, образованной на стадии снятия защиты d) до pH = приблизительно 8-8,5. Примеры подходящих оснований включают, но не ограничиваются ими, гидроксид калия, гидроксид натрия, гидроксид лития и гидроксид аммония. Предпочтительно, дополнительное основание представляет собой гидроксид калия.
В соответствии со стадией e) данного аспекта настоящего изобретения, соединение А растворяют в растворе, содержащем (i) систему растворителей, содержащую простой эфир и необязательно спирт, и (ii) воду. Следует понимать, что стадию e) можно осуществить любым из следующих способов: (a) добавлением соединения А к предварительно смешанному раствору, содержащему (i) систему растворителей, содержащую простой эфир и необязательно спирт, и (ii) воду, или (b) добавлением каждого компонента раствора к соединению А.
Подходящие простые эфиры включают ТГФ.
В соответствии с настоящим изобретением, в раствор на стадии e) не обязательно добавлять спирт. Однако предпочтителен раствор, содержащий простой эфир и спирт. Подходящие спирты включают, но не ограничиваются ими, метанол, этанол и изопропанол. Предпочтительно, спирт представляет собой метанол.
В предпочтительном варианте реализации соединение А растворяют в растворе, состоящем из (i) системы растворителей, содержащей метанол и ТГФ, и (ii) воды.
Растворение соединения А в растворе на стадии e) облегчается нагреванием смеси соединения А и раствора, содержащего (i) систему растворителей, содержащую простой эфир и спирт, и (ii) воду, до внутренней температуры около 52-56°С перед добавлением суспензии затравочных кристаллов. В одном аспекте смесь соединения А и раствор, содержащий (i) систему растворителей, содержащую простой эфир и спирт, и (ii) воду, нагревают до внутренней температуры около 53-55°С.
К раствору на стадии f) добавляют суспензию затравочных кристаллов с получением суспензионной смеси. Указанный раствор преимущественно охлаждают (i) после нагревания смеси соединения А и раствора, содержащего (i) систему растворителей, содержащую простой эфир и спирт, и (ii) воду, до внутренней температуры около 52-56°С и (ii) перед добавлением суспензии затравочных кристаллов.
В одном аспекте к суспензионной смеси добавляют воду с получением обработанной смеси. В другом аспекте суспензионную смесь охлаждают до температуры около 30-50°С перед добавлением воды. В предпочтительном варианте реализации суспензионную смесь охлаждают до температуры около 47-48°С перед добавлением воды.
На стадии g) воду можно добавлять в течение периода времени от 5 до 35 часов, так что содержание воды не превышает 70% масс./масс., предпочтительно 65% (масс./масс.) относительно системы растворителей. В одном из вариантов реализации воду добавляют в течение периода времени 10-25 часов, предпочтительно 25 часов. В другом варианте реализации воду добавляют в течение периода времени 25 часов, так что 33% добавляют в течение 15 часов, а 66% добавляют в течение 10 часов.
В другом аспекте компоненты в системе растворителей после завершения добавления воды к суспензии или охлажденной суспензионной смеси имеют окончательное соотношение спирта/простого эфира/воды в диапазоне от 40/40/20 до 15/15/70 масс./масс., при этом масс./масс. относится к массовому проценту каждого компонента относительно других компонентов в системе растворителей/антирастворителей. В предпочтительном варианте реализации окончательное соотношение спирта/простого эфира/воды составляет приблизительно 20/20/60 масс./масс. (20/20/60 масс./масс.).
На стадии h) окончательно получают кристаллизованное соединение А путем охлаждения обработанной смеси. Обработанную смесь преимущественно охлаждают в течение периода времени от 5 до 25 часов. В одном аспекте обработанную смесь охлаждают в течение периода времени от 8 до 15 часов, от 8 до 12 часов или от 9 до 11 часов. В предпочтительном варианте реализации обработанную смесь охлаждают в течение периода времени, составляющего приблизительно 10 часов. Обработанную смесь преимущественно охлаждают до внутренней температуры около 1-10°С, предпочтительно около 3-5°С. В предпочтительном варианте реализации обработанную смесь охлаждают до внутренней температуры около 3-5°С в течение приблизительно 9-11 часов. После фильтрования кристаллизованное соединение А можно высушить, например, под вакуумом или в вакуумной печи.
Кристаллизованное соединение А, полученное с помощью способа согласно настоящему изобретению, может быть дополнительно измельчено (например, струйным измельчением или штифтовым измельчением).
В дополнительном аспекте предложен способ получения кристаллического (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты, полученного в соответствии со способом, описанным в настоящем документе, который включен здесь посредством ссылки в полном объеме. Следует понимать, что кристаллическое соединение А включает лекарственную субстанцию, полученную с дополнительным измельчением или без него.
В другом аспекте предложено соединение формулы (I):

Указанное соединение формулы (I) подходит в качестве промежуточного соединения для синтеза соединения А в соответствии с настоящим изобретением.
В другом аспекте в настоящем документе предложено соединение формулы (V):

Указанное соединение формулы (V) подходит в качестве промежуточного соединения для синтеза соединения А в соответствии с настоящим изобретением.
В другом аспекте предложено соединение формулы (IV):

или его гидрат. В предпочтительном варианте реализации соединение формулы (IV) находится в форме его моногидрата. Указанное соединение формулы (IV), в том числе его моногидрат, подходит в качестве промежуточного соединения для синтеза соединения А в соответствии с настоящим изобретением.
Фармацевтические композиции, содержащие кристаллическое соединение А
В другом аспекте предложена фармацевтическая композиция, содержащая кристаллическое соединение А и по меньшей мере один фармацевтически приемлемый носитель или вспомогательное вещество. Фармацевтическая композиция содержит кристаллическое соединение А, по меньшей мере один сахар и по меньшей мере одно целлюлозное вспомогательное вещество. Указанная композиция особенно подходит для лечения рака у субъекта, нуждающегося в этом, предпочтительно, у людей.
В фармацевтических композициях согласно настоящему изобретению кристаллическое соединение А находится в кристаллической форме, полученной с помощью способа кристаллизации, описанного выше, со стадией измельчения или без нее. В одном из вариантов реализации фармацевтической композиции, предложенной в настоящем документе, указанная фармацевтическая композиция содержит около 5-35% кристаллизованного соединения А относительно массы композиции. В дополнительном варианте реализации фармацевтическая композиция содержит около 5-11% кристаллизованного соединения А относительно массы композиции. В другом предпочтительном варианте реализации фармацевтическая композиция содержит около 6,25% кристаллизованного соединения А относительно массы композиции. В другом предпочтительном варианте реализации фармацевтическая композиция содержит около 10% кристаллизованного соединения А относительно массы композиции.
В другом варианте реализации фармацевтическая композиция содержит приблизительно 15 мг или 45 мг кристаллизованного соединения А.
Подходящие сахара для применения в фармацевтических композициях включают, но не ограничиваются ими, лактозу (например, высушенную распылением лактозу, моногидрат лактозы), мальтозу, фруктозу, галактозу, кондитерский сахар, прессуемый сахар, декстраты, декстрин, декстозы, маннит, Nu-Tab, Di-Pac, Emdex и сахарозу. В предпочтительном варианте реализации сахар, используемый в фармацевтической композиции, представляет собой лактозу, в частности, моногидрат лактозы.
В одном из вариантов реализации фармацевтической композиции, предложенной в настоящем документе, указанная фармацевтическая композиция содержит около 30-70% по меньшей мере одного сахара относительно массы композиции. В дополнительном варианте реализации фармацевтическая композиция содержит около 50-60% лактозы относительно массы композиции. В дополнительном варианте реализации фармацевтическая композиция содержит около 50-60% моногидрата лактозы относительно массы композиции. В предпочтительном варианте реализации фармацевтическая композиция содержит около 55-56% моногидрата лактозы относительно массы композиции.
Подходящие целлюлозные вспомогательные вещества включают, но не ограничиваются ими, микрокристаллическую целлюлозу, сверхтонкую целлюлозу, порошкообразную целлюлозу, метилцеллюлозу, гидроксиэтилцеллюлозу, гидроксиметилцеллюлозу, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. В предпочтительном варианте реализации целлюлозное вспомогательное вещество представляет собой микрокристаллическую целлюлозу.
В одном из вариантов реализации фармацевтической композиции, предложенной в настоящем документе, указанная фармацевтическая композиция содержит около 20-40% целлюлозного вспомогательного вещества относительно массы композиции. В дополнительном варианте реализации фармацевтическая композиция содержит около 20-40% микрокристаллической целлюлозы относительно массы композиции. В дополнительном варианте реализации фармацевтическая композиция содержит около 30-40% микрокристаллической целлюлозы относительно массы композиции. В предпочтительном варианте реализации фармацевтическая композиция содержит около 30-36% микрокристаллической целлюлозы относительно массы композиции.
Фармацевтическая композиция может быть, например, в форме, подходящей для перорального введения в виде стандартной лекарственной формы, такой как таблетка, капсула, пилюля, порошок, препараты для непрерывного высвобождения, раствор, суспензия, для парентеральной инъекции в виде стерильного раствора, суспензии или эмульсии, для местного применения, такой как мазь или крем, или для ректального введения в виде суппозиториев. Фармацевтическая композиция может быть в стандартных лекарственных формах, пригодных для однократного введения точных доз. Следует понимать, что стандартное содержание компонента комбинации, содержащегося в отдельной дозе каждой лекарственной формы, не обязательно должно составлять эффективную дозу, поскольку необходимое эффективное количество может быть достигнуто введением нескольких дозированных единиц.
Способы получения различных фармацевтических композиций с определенным количеством активного соединения являются известными или понятными специалистам в данной области техники. Примеры представлены в книге Remington, Pharmaceutical Sciences, Mack Publishing Company, Истон, штат Пенсильвания, 15-е издание (1975). Если не указано иное, то композицию согласно настоящему изобретению получают с помощью способа, известного per se, например, различными обычными способами смешивания, измельчения, прямого прессования, гранулирования, нанесения сахарного покрытия, растворения, лиофилизации, или приемами изготовления, понятными специалистам в данной области техники. Твердые композиции такого же типа также могут быть использованы в мягких и твердых наполненных желатиновых капсулах. В предпочтительном варианте реализации фармацевтическую композицию согласно настоящему изобретению получают прямым прессованием.
Как описано ниже, фармацевтическая композиция может содержать дополнительные вспомогательные вещества или носители, включая, но не ограничиваясь ими, разрыхлители, смазывающие вещества, скользящие добавки, связующие вещества, стабилизаторы и наполнители, разбавители, красители, ароматизаторы и консерванты. Специалисты в данной области техники могут выбрать один или более из вышеупомянутых носителей с учетом определенных требуемых свойств лекарственной формы при помощи обычного экспериментирования и без лишних расходов. Количество каждого используемого носителя может варьироваться в диапазонах, обычных в данной области техники. В следующих публикациях, которые включены в настоящий документ посредством ссылки, описаны приемы и вспомогательные вещества, используемые для составления пероральных лекарственных форм. См. The Handbook of Pharmaceutical Excipients, 4-ое издание, Rowe et al., ред., American Pharmaceuticals Association (2003); и Remington: the Science and Practice of Pharmacy, 20-е издание, Gennaro, ред., Lippincott Williams & Wilkins (2003). Эти необязательные, дополнительные стандартные носители могут быть включены в пероральную лекарственную форму либо путем введения одного или нескольких обычных носителей в исходную смесь, либо могут быть добавлены на стадии смешивания.
Примеры фармацевтически приемлемых разрыхлителей включают, но не ограничиваются ими, крахмалы; глины; целлюлозы; альгинаты; камеди; поперечно-сшитые полимеры, например, поперечно-сшитый поливинилпирролидон или кросповидон, например, POLYPLASDONE XL производства компании International Specialty Products (Уэйн, штат Нью-Джерси); поперечно-сшитую карбоксиметилцеллюлозу натрия или кроскармеллозу натрия (например, AC-DI-SOL производства FMC); и поперечно-сшитую карбоксиметилцеллюлозу кальция; соевые полисахариды; и гуаровую камедь. Разрыхлитель может содержаться в количестве от около 0% до около 10% относительно массы композиции. В одном из вариантов реализации разрыхлитель содержится в количестве около 0,1-5%, или около 1-3%, или около 1,5-2,5% относительно массы композиции.
В одном из вариантов реализации фармацевтическая композиция согласно настоящему изобретению содержит разрыхлитель кроскармеллозу натрия. В дополнительном варианте реализации фармацевтическая композиция согласно настоящему изобретению содержит около 2% кроскармеллозы натрия относительно массы композиции.
Примеры фармацевтически приемлемых смазывающих веществ и фармацевтически приемлемых скользящих добавок включают, но не ограничиваются ими, коллоидный диоксид кремния/коллоидный безводный диоксид кремния (например, Aerosil 200®), трисиликат магния, крахмалы, тальк, трехосновной фосфат кальция, стеарат магния, стеарат алюминия, стеарат кальция, карбонат магния, оксид магния, полиэтиленгликоль, порошкообразную целлюлозу и микрокристаллическую целлюлозу. Смазывающее вещество может содержаться в количестве от около 0% до около 10% относительно массы композиции. В одном из вариантов реализации смазывающее вещество может содержаться в количестве около 0,1-1,5%, около 0,1-1% или около 0,5-0,9% относительно массы композиции. Скользящая добавка может содержаться в количестве около 0,1-10%, около 0,1-5% или около 0,1-1% относительно массы композиции.
В одном из вариантов реализации фармацевтическая композиция согласно настоящему изобретению содержит скользящую добавку, которая представляет собой коллоидный диоксид кремния/коллоидный безводный диоксид кремния. В дополнительном варианте реализации фармацевтическая композиция согласно настоящему изобретению содержит около 0,25% (относительно массы композиции) коллоидного диоксида кремния/коллоидного безводного диоксида кремния.
В другом варианте реализации фармацевтическая композиция согласно настоящему изобретению содержит смазывающее вещество стеарат магния. В дополнительном варианте реализации фармацевтическая композиция согласно настоящему изобретению содержит около 0,75% стеарата магния относительно массы композиции.
В другом варианте реализации фармацевтическая композиция согласно настоящему изобретению содержит коллоидный диоксид кремния/коллоидный безводный диоксид кремния и стеарат магния. В дополнительном варианте реализации фармацевтическая композиция согласно настоящему изобретению содержит около 0,25% коллоидного диоксида кремния/коллоидного безводного диоксида кремния относительно массы композиции и около 0,75% стеарата магния относительно массы композиции.
Примеры фармацевтически приемлемых связующих веществ включают, но не ограничиваются ими, крахмалы; целлюлозы и их производные, например, микрокристаллическую целлюлозу, например, AVICEL РН производства FMC (Филадельфия, штат Пенсильвания), гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу и гидроксипропилметилцеллюлозу METHOCEL производства Dow Chemical Corp. (Мидланд, штат Мичиган); сахарозу; декстрозу, кукурузный сироп; полисахариды; и желатин. Связующее вещество может содержаться в количестве около 0-50% или около 2-20% относительно массы композиции.
Примеры фармацевтически приемлемых разбавителей включают, но не ограничиваются ими, кондитерский сахар, прессуемый сахар, декстраты, декстрин, декстрозу, лактозу, маннит, микрокристаллическую целлюлозу, порошкообразную целлюлозу, сорбит, сахарозу и тальк. Например, разбавитель может содержаться в количестве около 0-80%, или около 0-50%, или около 1-40%, или около 1-10% относительно массы композиции.
В конкретном варианте реализации фармацевтическая композиция дополнительно содержит одно или более из: кроскармеллозы натрия, стеарата магния и диоксида кремния.
Если водные суспензии и/или эликсиры предназначены для перорального введения, то активное соединение в них может быть смешано с различными подсластителями или ароматизаторами, красящими веществами или красителями и, при необходимости, с эмульгирующими и/или суспендирующими агентами, вместе с такими разбавителями как вода, этанол, пропиленгликоль, глицерин и их комбинации.
В конкретном варианте реализации фармацевтическая композиция содержит около 5-11% кристаллического соединения А относительно массы композиции, около 55-56% моногидрата лактозы относительно массы композиции и около 30-36% микрокристаллической целлюлозы относительно массы композиции.
В другом варианте реализации композиция содержит около 5-11% кристаллического (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты, около 55-56% моногидрата лактозы, около 30-36% микрокристаллической целлюлозы относительно массы композиции, около 1,5-2,5% кроскармеллозы натрия, около 0,5-0,9% стеарата магния и около 0,1-1% коллоидного диоксида кремния/коллоидного безводного диоксида кремния относительно массы композиции.
В другом варианте реализации композиция содержит около 5-11% кристаллического соединения А относительно массы композиции, около 55-56% моногидрата лактозы относительно массы композиции, около 30-36% микрокристаллической целлюлозы относительно массы композиции, около 2% кроскармеллозы натрия относительно массы композиции, около 0,75 процента стеарата магния относительно массы композиции и около 0,25 процента коллоидного диоксида кремния/коллоидного безводного диоксида кремния относительно массы композиции.
В другом варианте реализации фармацевтической композиции указанная фармацевтическая композиция представлена в форме таблетки. В другом варианте реализации таблетка представляет собой таблетку с покрытием.
В одном аспекте фармацевтическая композиция согласно настоящему изобретению дополнительно содержит по меньшей мере один дополнительный терапевтический агент. Примеры дополнительных терапевтических агентов включают, но не ограничиваются ими, химиотерапевтические агенты или противоопухолевые агенты, такие как митотические ингибиторы, алкилирующие агенты, антиметаболиты, интеркалирующие антибиотики, ингибиторы фактора роста, ингибиторы клеточного цикла, ферменты, ингибиторы топоизомеразы, модификаторы биологического ответа, антигормоны, ингибиторы ангиогенеза и антиандрогены.
В одном из вариантов реализации дополнительный терапевтический агент выбран из группы, состоящей из паклитаксела или эверолимуса.
Следует понимать, что кристаллическое соединение А и дополнительный агент можно вводить в одной или в различных стандартных лекарственных формах. Кроме того, такие компоненты комбинации можно вводить одновременно, отдельно или последовательно.
Способы лечения пролиферативного заболевания при помощи кристаллического соединения А
В одном аспекте настоящего изобретения предложен способ лечения пролиферативного заболевания, в частности, рака, у субъекта, включающий введение субъекту эффективного количества кристаллического (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты («соединения А»). Следует понимать, что кристаллическое соединение А представлено в кристаллической форме, полученной с помощью способа кристаллизации, описанного выше.
В дополнительном аспекте настоящего изобретения предложен способ лечения пролиферативного заболевания, в частности, рака, у субъекта, включающий введение субъекту эффективного количества фармацевтической композиции, содержащей кристаллический (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты («соединение А»), по меньшей мере один сахар и по меньшей мере одно целлюлозное вспомогательное вещество. Следует понимать, что кристаллическое соединение А представлено в кристаллической форме, полученной с помощью способа кристаллизации, описанного выше.
Дополнительно следует понимать, что настоящее изобретение включает способ лечения пролиферативного заболевания, в частности, рака, у субъекта путем введения субъекту эффективного количества фармацевтической композиции, содержащей кристаллическое соединение А, представленное в каждом из предложенных выше вариантов реализации, которые включены здесь посредством ссылки.
В соответствии со способом лечения согласно настоящему изобретению, кристаллическое соединение А вводят субъекту, нуждающемуся в этом, в терапевтически эффективном количестве.
Кристаллическое соединение А можно вводить подходящему субъекту ежедневно в виде одной или дробных доз в эффективной дозе, находящейся в диапазоне от около 0,001 до около 100 мг на кг массы тела в сутки, предпочтительно от около 1 до около 35 мг/кг/сутки, в виде одной или дробных доз. Для человека массой 70 кг это составляет от около 0,05 до 7 г/сутки, предпочтительно от около 0,05 до около 2,5 г/сутки.
Кристаллическое соединение А и/или фармацевтические композиции согласно настоящему изобретению могут быть использованы для лечения пролиферативных заболеваний, которые являются раковыми или нераковыми. Особенно предпочтительно лечение рака.
«Рак», который можно лечить при помощи фармацевтической композиции, предложенной в настоящем документе, относится к клеточно-пролиферативным болезненным состояниям, включая, но не ограничиваясь ими, солидные или жидкостные опухоли. Примеры раковых заболеваний, подходящих для лечения в соответствии с настоящим изобретением, включают, но не ограничиваются ими, рак легких, рак костей, хронические миеломоноцитарный лейкоз (CMML), рак поджелудочной железы, рак кожи, рак головы и шеи, меланому, внутриматочный рак, рак яичников, рак толстой кишки, рак прямой кишки, анальный рак, рак желудка, рак молочной железы, рак яичек, гинекологические опухоли (например, саркома матки, карцинома фаллопиевых труб, карцинома эндометрия, карцинома шейки матки, карцинома влагалища или карцинома вульвы), болезнь Ходжкина, рак пищевода, рак тонкого кишечника, рак эндокринной системы (например, рак щитовидной железы, рак паращитовидной железы или рак надпочечников), саркому мягких тканей, рак уретры, пенильный рак, рак предстательной железы, хронический или острый лейкоз, солидные опухоли в детском возрасте, лимфоцитарные лимфомы, рак мочевого пузыря, рак почек или мочеточника (например, почечноклеточная карцинома), карциному почечной лоханки, рак желчных протоков, рак головного мозга, рак мочевого пузыря, плоскоклеточный, перитонеальный рак или неоплазмы центральной нервной системы (например, первичная лимфома ЦНС, опухоли оси позвоночника, глиомы ствола головного мозга или гипофизарные аденомы). В одном из вариантов реализации рак представляет собой солидную опухоль. В конкретном варианте реализации солидная опухоль является метастатической или неоперабельной.
В одном из вариантов реализации рак представляет собой такой рак, как рак головного мозга, рак легких, плоскоклеточный рак, рак мочевого пузыря, желудка, поджелудочной железы, молочной железы, головы, шеи, ренальный рак, рак почек, яичников, предстательной железы, толстой кишки, прямой кишки, пищевода, яичек, гинекологический рак или рак щитовидной железы.
В одном из вариантов реализации рак представляет собой рак легких, плоскоклеточный рак, рак поджелудочной железы, рак молочной железы, рак головы, рак шеи, рак толстой кишки, рак прямой кишки или меланому.
В конкретном варианте реализации в настоящем документе предложен способ лечения меланомы, рака поджелудочной железы, рака яичников, карциномы фаллопиевых труб, перитонеального рака, рака желчных протоков, рака толстой кишки или рака прямой кишки у субъекта, нуждающегося в этом, включающий введение указанному субъекту кристаллического соединения А или фармацевтической композиции, предложенной в настоящем документе.
В другом варианте реализации пролиферативное заболевание представляет собой нераковое пролиферативное расстройство, такое как доброкачественная гиперплазия кожи (например, псориаз), рестеноз, заболевание предстательной железы (например, доброкачественная гипертрофия предстательной железы (ВРН)) или синдром Нунан. Примеры дополнительных терапевтических агентов включают, но не ограничиваются ими, химиотерапевтические агенты или противоопухолевые агенты, такие как митотические ингибиторы, алкилирующие агенты, антиметаболиты, интеркалирующие антибиотики, ингибиторы фактора роста, ингибиторы клеточного цикла, ферменты, ингибиторы топоизомеразы, модификаторы биологического ответа, антигормоны, ингибиторы ангиогенеза и антиандрогены.
В одном из вариантов реализации дополнительный терапевтический агент выбран из группы, состоящей из паклитаксела или эверолимуса.
Следует понимать, что кристаллическое соединение А и дополнительный агент можно вводить в одной или в различных разовых лекарственных формах. Кроме того, такие компоненты комбинации можно вводить одновременно, отдельно или последовательно.
В одном аспекте настоящего изобретения предложено применение кристаллического соединения А для получения лекарственного средства, подходящего для лечения пролиферативного заболевания, в частности, рака. Следует понимать, что кристаллическое соединение А представлено в кристаллической форме, полученной с помощью способа кристаллизации, описанного выше. В одном из вариантов реализации пролиферативное заболевание представляет собой солидную опухоль. В дополнительном варианте реализации пролиферативное заболевание представляет собой рак, как описано выше.
В другом аспекте настоящего изобретения предложено применение фармацевтической композиции согласно настоящему изобретению для получения лекарственного средства, подходящего для лечения пролиферативного заболевания, в частности, рака. В одном из вариантов реализации пролиферативное заболевание представляет собой солидную опухоль. В дополнительном варианте реализации пролиферативное заболевание представляет собой рак, как описано выше.
В конкретном варианте реализации каждого аспекта пролиферативное заболевание выбрано из меланомы, рака поджелудочной железы, рака яичников, карциномы фаллопиевых труб, перитонеального рака, рака желчных протоков, рака толстой кишки или рака прямой кишки.
В одном аспекте настоящего изобретения предложено кристаллическое соединение А для применения при лечении пролиферативного заболевания, в частности, рака. Следует понимать, что кристаллическое соединение А представлено в кристаллической форме, полученной с помощью способа кристаллизации, описанного выше. В одном из вариантов реализации пролиферативное заболевание представляет собой солидную опухоль. В дополнительном варианте реализации пролиферативное заболевание представляет собой рак, как описано выше.
В другом аспекте настоящего изобретения предложена фармацевтическая композиция согласно настоящему изобретению, как описано выше и включено здесь посредством ссылки, для применения при лечении пролиферативного заболевания, в частности, рака. В одном из вариантов реализации пролиферативное заболевание представляет собой солидную опухоль. В дополнительном варианте реализации пролиферативное заболевание представляет собой рак, как описано выше.
В конкретном варианте реализации каждого аспекта пролиферативное заболевание выбрано из меланомы, рака поджелудочной железы, рака яичников, карциномы фаллопиевых труб, перитонеального рака, рака желчных протоков, рака толстой кишки или рака прямой кишки.
Настоящее изобретение дополнительно проиллюстрировано следующими примерами, которые не следует толковать как ограничивающие настоящее изобретение в его рамках или общей идее конкретными способами, описанными в них.
Исходные материалы и различные промежуточные соединения могут быть приобретены из коммерческих источников, получены из имеющихся в продаже органических соединений или получены с помощью общеизвестных способов синтеза.
Ниже представлены иллюстративные примеры способов и композиций согласно настоящему изобретению. Однако эти примеры никоим образом не предназначены для ограничения настоящего изобретения.
ПРИМЕРЫ
Сокращения
В тексте использованы следующие сокращения:
Пример 1. Получение метилового эфира 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензимидазол-5-карбоновой кислоты

В инертизированном (N2) реакционном сосуде при внутренней температуре 20°С и при исключении попадания влаги и воздуха осуществляют реакцию соединения 1 (1,0 экв.) и соединения 2 (1,2 экв.) в присутствии карбоната цезия (2,4 экв.), трис(дибензилиденацетон)дипалладия (0) (0,035 экв.) и Xantphos (0,07 экв.) в смеси толуола и 1,4-диоксана при внутренней температуре 99°С. Через 8 часов смесь охлаждают до внутренней температуры 60°С.
Затем добавляют диметилформамид (ДМФ), вспомогательное фильтровальное вещество (CEFOK) и активированный древесный уголь (EKNS), смесь перемешивают и охлаждают до внутренней температуры 35°С. Твердые вещества отфильтровывают и промывают смесью диметилформамида и толуола. В фильтрат, содержащий продукт соединения 3, при внутренней температуре 25°С вводят газообразный хлороводород (CLC), при этом кристаллизуется HCl соль соединения 3. Остаток палладия остается, в основном, в растворе. После нагревания до 60°С и охлаждения до 0°С твердые вещества отфильтровывают при помощи центрифугирования и промывают смесью толуола и диметилформамида.
Влажную HCl соль соединения 3 загружают в реактор (оснащенный датчиком pH) вместе с диметилформамидом и нагревают до 60°С. Добавлением 4 масс. % водного раствора фосфата трикалия pH доводят до pH в диапазоне 6,8-7,6 (при заданном pH 7,2), при этом соединение 3 кристаллизуется в виде свободного основания. После охлаждения до 22°С и перемешивания твердые вещества отфильтровывают при помощи центрифуги и промывают питьевой водой. Влажное твердое вещество высушивают при 50°С под вакуумом с получением сухого, неочищенного соединения 3.
Для удаления остаточного палладия сухое, неочищенное соединение 3 растворяют в диметилформамиде при внутренней температуре 60°С и перемешивают вместе с Smopex-234 (имеется в продаже у компании Johnson Matthey) и активированным углем в течение 90 минут. Твердые вещества отфильтровывают при внутренней температуре 60°С и промывают диметилформамидом. К фильтрату добавляют питьевую воду и затравочные кристаллы соединения 3. Во время кристаллизации соединения 3 добавляют дополнительное количество питьевой воды. После охлаждения до внутренней температуры 20°С твердые вещества отфильтровывают при помощи центрифуги и промывают смесью деионизированной воды и диметилформамида, и деионизированной водой. Влажное твердое вещество высушивают при 50°С под вакуумом, получив метиловый эфир 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензимидазол-5-карбоновой кислоты (соединение 3).
Пример 2. Получение (2-трет-бутоксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензимидазол-5-карбоновой кислоты
А. «Однореакторный» синтез

В инертизированный реакционный сосуд при внутренней температуре 20-25°С под азотом к смеси ДМФ и ТГФ добавляют метиловый эфир 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты (соединение 3, 1,0 экв.). К этой суспензии добавляют раствор триметилсиланолята калия (1,05 экв.) в ТГФ при внутренней температуре 25°С в течение около 40 минут, и перемешивают полученную смесь в течение около 1 часа, получая раствор калиевой соли промежуточного соединения 1. Затем смесь ТГФ/метанола последовательно дистиллируют из указанной смеси при 85-120°С в течение около 2 часов.
Затем раствор калиевой соли добавляют к суспензии CDI (1,25 экв.) и гидрохлорида имидазола (1,40 экв.) в ТГФ при внутренней температуре 25°С в течение около 1 часа. Затем полученную смесь перемешивают приблизительно 1 час при 50°С, и образовывается следующее имидазолидное промежуточное соединение:

Имидазолидное промежуточное соединение дополнительно не выделяют.
Затем добавляют 1,2 экв. O-(2-трет-бутоксиэтил)гидроксиламина (соединение 4, CAS №1023742-13-3, имеющееся в продаже у таких поставщиков, как Huhu Technology, Inc.®) в течение около 30 минут при 50°С и перемешивают в течение 1,5 часа. Затем добавляют деминерализованную воду при 50°С, получая осадок. После охлаждения до 20°С и перемешивания в течение около 3-16 часов суспензию отфильтровывают, промывают 2 порциями ТГФ/деминерализованной воды (1:2) и тремя порциями деминерализованной воды, и высушивают при 50°С/<70 мбар в течение около 17 часов, получая (2-трет-2-бутоксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензимидазол-5-карбоновой кислоты (соединение 5) в виде моногидрата.
В. Способ синтеза с выделением промежуточного соединения со стадии a) из реакционной смеси стадии a) перед выполнением реакции на стадии b)
Альтернативно, (2-трет-бутоксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензимидазол-5-карбоновой кислоты (соединение 5) можно получить с помощью способа синтеза, представленного ниже. Соединение 3, которое представляет собой метиловый эфир, сначала превращают в карбоновую кислоту, которую затем выделяют кристаллизацией с образованием соединения 6. Затем выполняют связывание соединения 6 с соединением 4 с образованием соединения 5 в виде моногидрата. На стадии кристаллизации в указанном способе удаление исходных материалов, таких как соединение 1, технологических примесей и лиганда dba из использованного ранее катализатора, происходит до реакции связывания с соединением 4, и при этом сохраняется суммарный выход синтеза.

6-(4-Бром-2-фторфениламино)-7-фтор-3-метил-3H-бензимидазол-5-карбоновая кислота
В инертизированном (N2) реакционном сосуде при внутренней температуре 60°С соединение 3 (1,0 экв.) растворяют в ДМФ и перемешивают с волокнами, имеющимися в продаже под торговой маркой SMOPEX 234, и активированным углем для удаления палладия до его содержания не более 100 м.д. Волокна и активированный уголь удаляют фильтрованием при 60°С и промывают ДМФ.
Фильтрат (содержащий соединение 3) переносят во второй инертизированный (N2) реакционный сосуд и охлаждают до внутренней температуры 30°С. В это время может образоваться тонкодисперсная суспензия. Добавляют 30% гидроксид натрия (1,1 экв.) и воду (для промывания), и энергично перемешивают полученную реакционную смесь в течение 3 часов при внутренней температуре 30°С. Происходит омыление метилового эфира. Превращение проверяют при помощи ИПХ (ВЭЖХ). Как только соответствие критерию ИПХ достигнуто, добавляют вспомогательное фильтровальное вещество, которое имеется в продаже под торговой маркой HYFLO. Смесь перемешивают в течение 15 минут, а затем фильтруют при 30°С через листовой фильтр и фильтр тонкой очистки в третий инертизированный (N2) реакционный сосуд.
К прозрачному фильтрату в третьем сосуде добавляют 7,5% водный раствор HCl при внутренней температуре 30°С до достижения значения pH 8. Затем в раствор вносят затравочные кристаллы соединения 6 при внутренней температуре 30°С и добавляют 7,5% водный раствор HCl при энергичном перемешивании до достижения значения pH 2,8. Происходит постепенная кристаллизация продукта. Суспензию охлаждают за 60 минут до внутренней температуры 25°С и добавляют воду. Суспензию перемешивают по меньшей мере в течение 4 часов при внутренней температуре 25°С.
Полученное твердое вещество собирают центрифугированием или фильтрацией. Осадок на фильтре сначала промывают 1:1 (масс./масс.) смесью ДМФ/воды, а затем водой, выгружают и высушивают в вакууме при 50°С. Содержание воды контролируют при помощи ИПХ. Кристаллический продукт, соединение 6, выгружают по достижению соответствия критерию ИПХ.
(2-трет-бутоксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензимидазол-5-карбоновой кислоты
В инертизированный (N2) реакционный сосуд загружают соединение 6 (1,0 экв.), ДМФ и ТГФ при комнатной температуре. Суспензию нагревают до 25°С при перемешивании, пропуская поток азота. После добавления CDI (1,13 экв.) суспензия может стать более жидкой, и может наблюдаться небольшое выделение газов. После окончательного превращения суспензии в раствор, ее контролируют с помощью ИПХ (ВЭЖХ).
Как только соответствие критерию ИПХ (ВЭЖХ) достигнуто, реакционную смесь нагревают до 50°С за 20 минут и добавляют гидрохлорид имидазола (0,3 экв.), с образованием раствора промежуточного соединения 2.
К раствору промежуточного соединения 2 за 60 минут добавляют соединение 4 (1,3 экв.) при внутренней температуре 50°С, при перемешивании со скоростью 300 об./мин., пропуская поток азота. Как только соответствие критерию ИПХ (ВЭЖХ) достигнуто, смесь охлаждают до 20-25°С за 30 минут. Затем смесь выдерживают при комнатной температуре в течение ночи под азотом без перемешивания. К смеси добавляют ДМФ, затем нагревают ее до 50°С за 30 минут. Полное превращение промежуточного соединения 2 в соединение 5 подтверждают по ИПХ (ВЭЖХ).
К смеси добавляют воду при внутренней температуре 50°С за 20 минут. Затем в раствор вносят затравку соединения 5. После перемешивания при 50°С в течение 60 минут к суспензии добавляют дополнительное количество воды при 50°С за 90 минут. После энергичного перемешивания суспензию охлаждают до 20°С за 2 часа и отфильтровывают. Осадок на фильтре дважды промывают смесью ТГФ/воды (об./об.: 1:2) при 20°С, и дважды - водой при 20°С. Наконец, осадок на фильтре высушивают при 50°С под вакуумом с получением (2-трет-бутоксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензимидазол-5-карбоновой кислоты (соединения 5) в виде моногидрата.
Пример 3. Получение (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензимидазол-5-карбоновой кислоты (соединение А)

Моногидрат (2-трет-бутоксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензимидазол-5-карбоновой кислоты (соединение 5) 3 частями добавляют к предварительно смешанному раствору ацетонитрила и избытка фосфорной кислоты (85% водный раствор) при внутренней температуре 20-25°С. После перемешивания около 15 минут суспензию нагревают до внутренней температуры 50-53°С. Суспензию поддерживают при этой температуре в течение 6 часов, охлаждают до внутренней температуры 20-25°С. Затем смесь нагревают до внутренней температуры 35-37°С и разбавляют смесью этанола и воды (3:1, об./об.). Добавляют EKNS и CEFOK, реакционную смесь перемешивают приблизительно 15 минут и отфильтровывают через воронку, покрытую CEFOK. Фильтрат охлаждают приблизительно до 30°С. К охлажденному фильтрату в течение 90 минут добавляют 3 н. водный раствор гидроксида калия (KOH) до достижения значения pH около 8,1. Суспензию нагревают до внутренней температуры 60-63°С, перемешивают при этой температуре в течение периода около 2 часов, охлаждают до 20-23°С в течение около 45 минут, отфильтровывают через воронку и высушивают при 50°С и давлении <100 мбар в течение периода около 17 часов, получая (2-гидроксиэтокси)амид 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3Н-бензимидазол-5-карбоновой кислоты (соединение А) в виде белого порошка.
Пример 4. Получение кристаллического (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензимидазол-5-карбоновой кислоты (соединение А)
В сухом сосуде при комнатной температуре к предварительно смешанному раствору метанола/ТГФ/воды (35/35/30, масс./масс.) добавляют соединение А. Суспензию нагревают до внутренней температуры 53-55°С, а полученный раствор отфильтровывают в горячем состоянии при помощи глубокой и мембранной фильтрации (через бумажный фильтр и ПТФЭ мембрану) при внутренней температуре 53-56°С. Прозрачный раствор перемешивают и охлаждают до 47-48°С, и добавляют суспензию затравочных кристаллов (т.е. затравочные кристаллы кристаллизованного соединения А в воде, 10% масс./масс.) (от 0,2 до 0,5%) кристаллического соединения А относительно ожидаемого выхода массы). Примерно через 20 минут медленно добавляют воду за 25 часов (33,3% - за 15 часов, и 66,6% - за 10 часов, перемешивая по меньшей мере 10 минут после добавления воды) с получением окончательного соотношения метанола/ТГФ/воды (20/20/60, масс./масс.). После добавления воды суспензию охлаждают до внутренней температуры 3-5°С за 10 часов и перемешивают в течение 0,5 часа. Белую суспензию отфильтровывают через стеклянный вакуумный нутч-фильтр (75 мл, диаметр = 6 см, пористость 3) и один раз промывают холодной смесью метанола/ТГФ/воды (15/15/70, масс./масс. при 2-4°С) и два раза - ледяной водой (2-4°С). Высушивание выполняют в вакуумной печи для сушки при 20°С в течение 10 часов, а затем при 40°С в течение 10 часов, а затем при 60°С по меньшей мере в течение 12 часов под давлением <10 мбар, получая кристаллическое соединение А.
Пример 5. Фармацевтическая композиция
Кристаллическое соединение А составляют в композицию, как показано в таблице 1:


Реферат
Изобретение относится к области органической химии, а именно к способу получения (2-гидроксиэтокси)амида 6-(4-бром-2-фторфениламино)-7-фтор-3-метил-3H-бензимидазол-5-карбоновой кислоты (Соединение А), включающему a) взаимодействие соединения формулы (I)с подходящим основанием с последующим взаимодействием с кислотой с образованием промежуточного соединения формулы (V) и кристаллизацию промежуточного соединения формулы (V); и b) взаимодействие указанного промежуточного соединения формулы (V) с соединением формулы (II) в присутствии агента сочетания и источника протонов с получением соединения формулы (III) или его гидрата, где Pпредставляет собой трет-бутил; c) снятие защиты с указанного соединения формулы (III) или его гидрата при помощи подходящего реагента для снятия защиты в подходящем полярном апротонном растворителе, с последующим взаимодействием депротектированного соединения с подходящим основанием, с получением соединения А. Также изобретение относится к промежуточному соединению. Технический результат: разработан способ получения соединения А, обладающего улучшенным профилем чистоты с низким содержанием палладия. 2 н. и 4 з.п. ф-лы, 1 табл., 5 пр.Соединение А Формула (I) Формула (V)Формула (II), Формула (III)
Формула






Комментарии