Липосомы, содержащие проникающие в клетки пептиды и тетраэфирные липиды, для пероральной доставки макромолекул - RU2743431C2
Код документа: RU2743431C2
Чертежи






Описание
Настоящее изобретение относится к липосомным композициям, которые содержат липосомы, содержащие тетраэфирные липиды (TEL) и проникающие в клетки пептиды (CPP), где указанные CPP присоединены к соединению, являющемуся частью липидного бислоя липосомы. Настоящее изобретение, кроме того, относится к их использованию для пероральной доставки терапевтических и/или диагностических средств.
Пероральную доставку лекарственных средств считают наиболее благоприятным путем применения, в частности, для лечения хронических заболеваний, которые требуют долгосрочного и повторного введения лекарственных средств. Пероральный путь обеспечивает высокую безопасность лекарственного средства и широко принят среди пациентов из-за его удобства. Дополнительно, нестерильность пероральных форм лекарственных средств снижает стоимость производства, хранения и распространения, что может вносить вклад в усовершенствование здравоохранения стран третьего мира. По оценкам, 90% всех составов лекарственных средств на рынке предназначены для перорального использования.
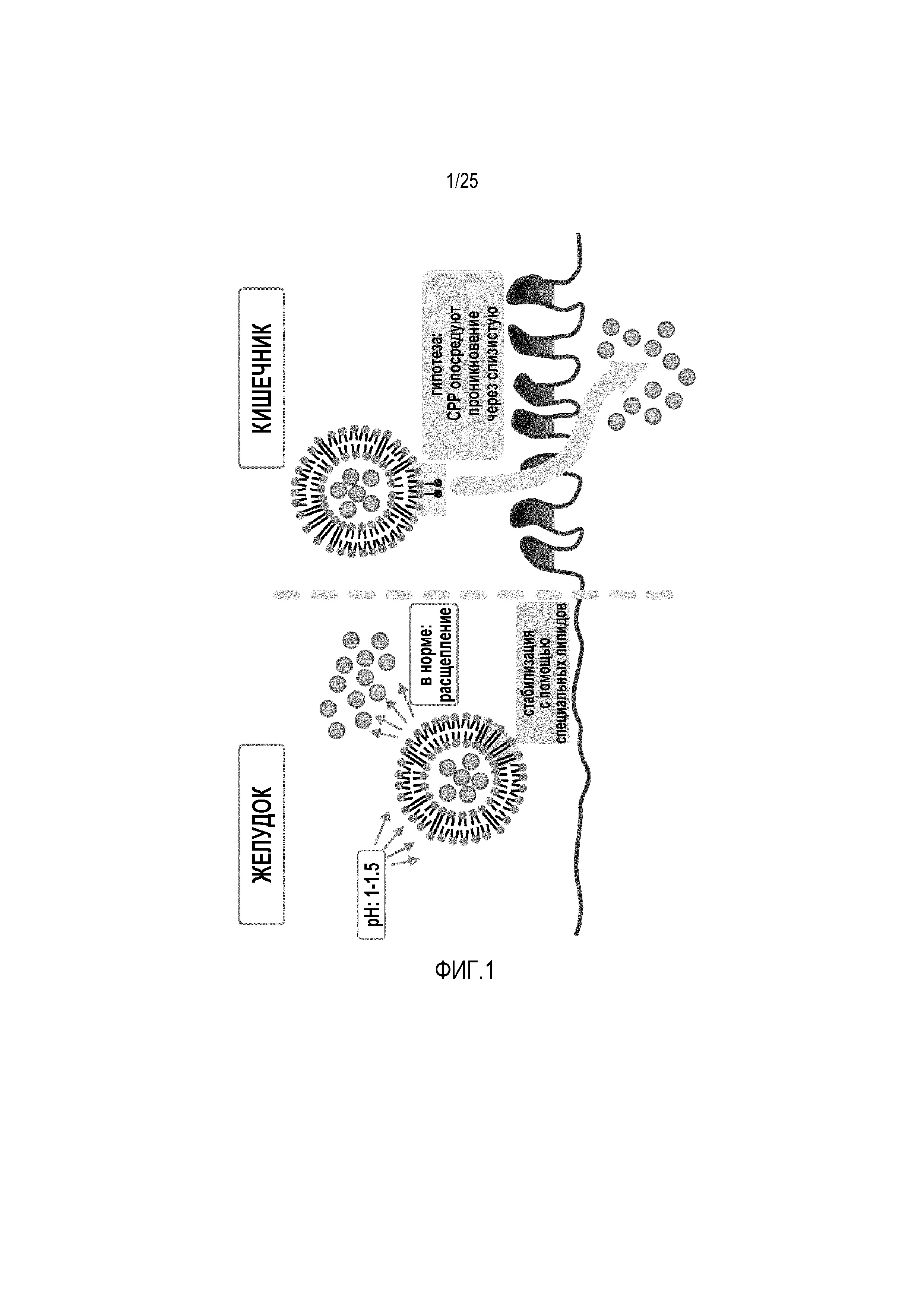
Однако многие лекарственные средства, в частности пептиды и другие макромолекулярные лекарственные средства, демонстрируют очень низкую стабильность в кислых условиях в желудке после перорального введения, а также низкую абсорбцию через желудочно-кишечный барьер (фиг. 1). Для преодоления этой проблемы в последние годы тестировали различные подходы для усовершенствования биодоступности, включая твердые липидные наночастицы, нано- или микроэмульсии или липосомы. Однако стандартные липосомные составы не были очень убедительными из-за их нестабильности в желудочно-кишечном тракте (GIT).
Значительное усовершенствование липосом можно выполнять с помощью комбинации стандартных фосфолипидов (PL) и так называемых тетраэфирных липидов, специфичных липидов, получаемых у архей, например, экстремофильного архея Sulfolobus acidocaldarius. Недавние исследования показали, что эти TEL могут усовершенствовать стабильность липосом в GIT и также опосредовать проникновение через слизистую.
S. acidocaldarius растет при температурах между 50 и 100°C, главным образом в кислых условиях, которые делают необходимой стабильную клеточную мембрану. Липиды мембран архей содержат преимущественно C20-C40 остовы из изопреноидных субъединиц, которые связаны простыми эфирными связями с мостиковой группой(ами) глицерина и/или нонитола. Мостиковая группа является незамещенной или замещенной с использованием одной из широкого спектра полярных или неполярных головных групп. Количество этих фрагментов в клеточной мембране архей меняется вместе с условиями роста и возрастает вместе с температурой окружающей среды. TEL глицерилкалдитилтетраэфир (GCTE) и диглицерилтетраэфир (DGTE) с усредненным числом от четырех до шести циклопентильных колец можно выделять из S. acidocaldarius.
Однако даже при использовании TEL, проникновение указанных липосом через слизистую остается неудовлетворительным во многих случаях. Таким образом, сохраняется необходимость улучшения проникновения через слизистую липосом.
Ввиду изложенного выше, техническая задача, лежащая в основе настоящего изобретения, состоит в получении средства доставки лекарственных средств через пероральный путь, имеющих повышенное проникновение через слизистую.
Решения вышеуказанной технической задачи достигают с помощью вариантов осуществления, охарактеризованных в формуле изобретения.
В частности, в первом аспекте настоящее изобретение относится к липосомной композиции, содержащей липосомы, причем указанные липосомы содержат
(a) тетраэфирные липиды (TEL) и
(b) проникающие в клетки пептиды (CPP),
где указанные CPP прикрепляют к соединению, которое является частью липидного бислоя липосомы.
В рамках изобретения, термин «липосомная композиция» относится к композиции, содержащей липосомы. Термин «липосома», в рамках изобретения, относится к искусственно полученным везикулам, состоящим из липидных бислоев. Липосомы можно использовать для доставки средств благодаря их уникальному свойству инкапсулировать область водного раствора внутри гидрофобной мембраны. Гидрофильные растворенные вещества не могут легко проходить через липидный бислой. Гидрофобные соединения могут растворяться в липидном бислое, и таким образом, липосомы могут нести как гидрофобные, так и гидрофильные соединения. Для того чтобы доставлять молекулы к месту приложения действия, липидный бислой может сливаться с другими бислоями, такими как клеточные мембраны, таким образом доставляя содержимое липосомы. Создавая липосомы в растворе определенного средства, его можно доставлять во внутренний просвет липосомы.
TEL, которые можно использовать для формирования липосом, конкретно не ограничены и известны в данной области. В конкретных вариантах осуществления указанные TEL получают у видов архей рода Sulfolobus, например, S. islandicus или S. acidocaldarius, где последний особенно предпочтителен. В предпочтительном варианте осуществления TEL выбирают из группы, состоящей из глицерилкалдитилтетраэфира (GCTE), диглицерилтетраэфира (DGTE) и их сочетаний.
Предпочтительно липосомы, используемые в композициях по настоящему изобретению, содержат указанные TEL в количестве от 1 до 25 моль-%, предпочтительно от 1 до 10 моль-%, более предпочтительно от 3 до 7 моль-%, более предпочтительно от 4 до 6 моль-% на основе общего количества липидов. В конкретных вариантах осуществления указанные липосомы содержат указанные TEL в количестве приблизительно 5 моль-% на основе общего количества липидов.
Помимо присутствия TEL, как описано выше, липосомы, используемые в композициях в соответствии с настоящим изобретением, конкретно не ограничены конкретными липидами. В частности, липиды, используемые для создания указанных липосом, могут представлять собой любые подходящие липиды, известные в данной области. Эти липиды включают, но без ограничения, холестерин или его производные, фосфолипиды, лизофосфолипиды или дополнительные тетраэфирные липиды. Соответственно, в предпочтительном варианте осуществления указанные липосомы содержат один или несколько липидов, выбранных из группы, состоящей из холестерина и его производных, фосфолипидов, лизофосфолипидов и тетраэфирных липидов. Предпочтительные производные холестерина в контексте настоящего изобретения представляют собой стероиды и соединения, содержащие базовую стероидную молекулярную структуру. Предпочтительно, указанные липосомы содержат фосфолипиды, где указанные фосфолипиды могут представлять собой синтетические, полусинтетические или природные фосфолипиды. В целом, подходящие липиды можно выбирать из группы, состоящей из фосфатидилхолинов, фосфатидилэтаноламинов, фосфатидилинозитов, фосфатидилсеринов, цефалинов, фосфатидилглицеринов и лизофосфолипидов. В конкретном варианте осуществления настоящего изобретения липосомы содержат яичный фосфатидилхолин (E-PC; лецитин) и холестерин, предпочтительно в количестве приблизительно 85 моль-% E-PC и приблизительно 10 моль-% холестерина. Липосомы, подлежащие использованию в соответствии с настоящим изобретением, дополнительно могут содержать какие-либо дополнительные подходящие средства, например, такие как ингибиторы ферментов, усилители проникновения или другие липофильные или гидрофильные вещества, которые можно использовать для стабилизации липосом или для изменения свойств липосом. Такие липофильные или гидрофильные вещества конкретно не ограничены и известны в данной области. Они включают, например, витамин E, жирные кислоты, воски и моно-, ди- и триглицериды. Кроме того, можно добавлять вещества, которые повышают биодоступность заключенных активных средств, такие как ингибиторы ферментов, модуляторы плотных контактов или хелатирующие средства.
CPP, которые можно использовать в связи с настоящим изобретением, конкретно не ограничены и известны в данной области. Предпочтительно, CPP представляют собой CPP, которые имеют положительный полный заряд. Соответствующие CPP известны в данной области. Более предпочтительно, CPP выбирают из группы, состоящей из линейного или циклизованного пенетратина (SEQ ID № 1; RQIKIWFQNRRMKWKK), выделенного у Drosophila melanogaster, пептида TAT (трансактиватора транскрипции) (SEQ ID № 2; CGRKKKRRQRRRPPQC), выделенного у HIV-1, MAP (модельного амфипатического пептида) (SEQ ID № 3; GALFLGFLGAAGSTMGAWSQPKSKRKV), который представляет собой искусственный пептид, R9 (SEQ ID № 4; RRRRRRRRR), который представляет собой искусственный пептид, pVEC (SEQ ID № 5; LLIILRRRIRKQAHAHSK-амид), который представляет собой CPP, полученный из кадгерина эндотелия сосудов мыши, транспортана (SEQ ID № 6; GWTLNSAGYLLGKINLKALAALAKISIL-амид), который получен из нейропептида галанина человека, и MPG (SEQ ID № 7; GALFLGFLGAAGSTMGAWSQPKSKRKV), который получен из HIV, их сочетаний и их димеров. В этом контексте все вышеуказанные пептиды могут присутствовать в линейной или циклизованной форме, где циклизованная форма является предпочтительной. Кроме того, CPP по настоящему изобретению могут состоять из L-аминокислот, D-аминокислот или их смесей, где для линейных CPP D-аминокислоты являются предпочтительными.
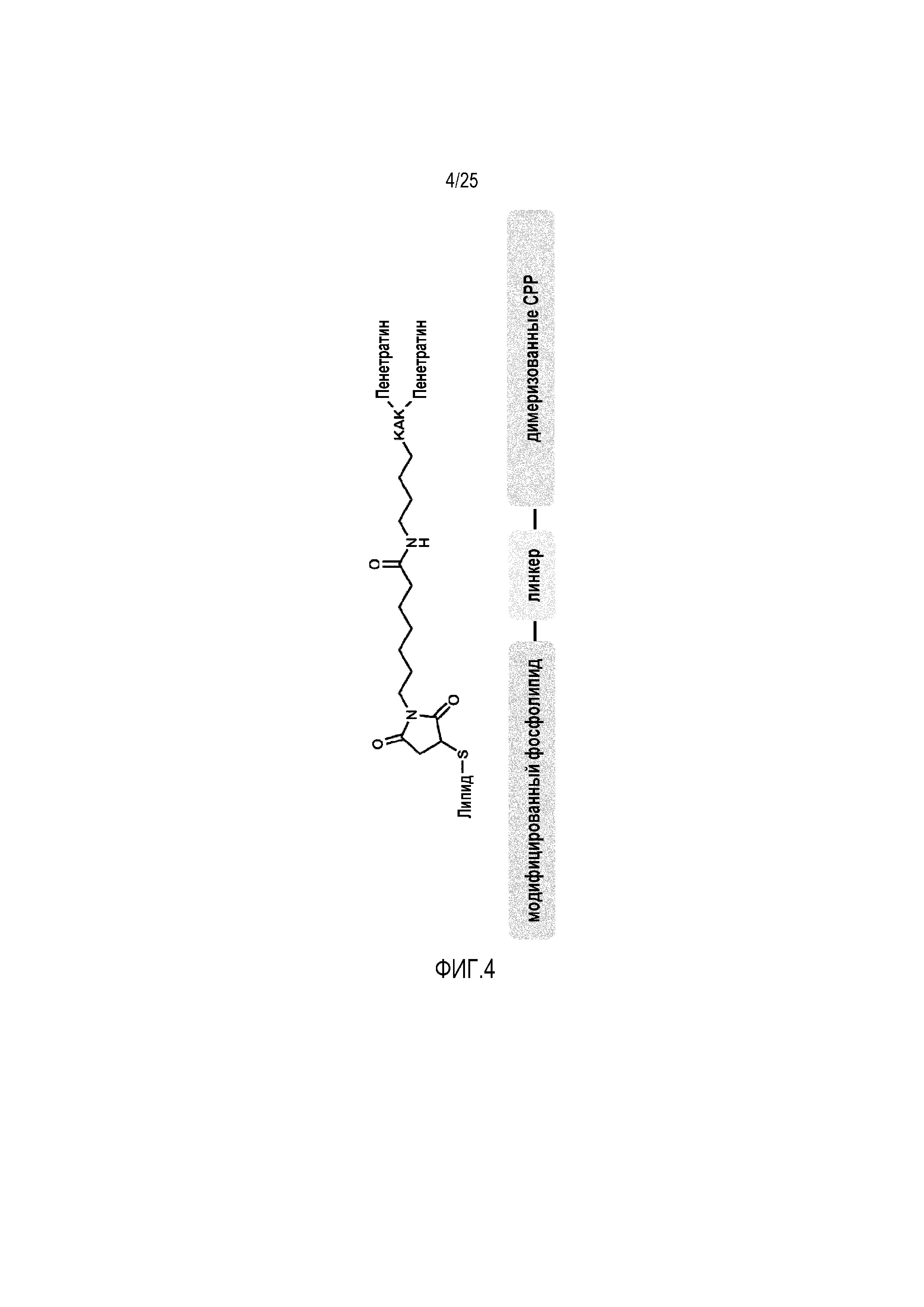
В соответствии с настоящим изобретением, CPP прикрепляют к соединению, которое является частью липидного бислоя липосомы. В этом контексте термин «является частью липидного бислоя липосомы» предназначен для того, чтобы указывать на тот факт, что указанный фосфолипид встроен в указанный липидный бислой. Предпочтительно прикрепление представляет собой ковалентное прикрепление. Соединение, к которому прикрепляют CPP и которое является частью липидного бислоя липосомы, предпочтительно представляет собой подходящий липид, как определено выше, например, липид, выбранный из группы, состоящей из холестерина и его производных, фосфолипидов, лизофосфолипидов и тетраэфирных липидов, где фосфолипиды являются предпочтительными и где указанные липиды могут представлять собой модифицированные и/или активированные липиды. Предпочтительно, CPP прикрепляют к указанному соединению через линкер. В этом контексте мономерные CPP можно ковалентно прикреплять к фосфолипиду через линкер (фиг. 3), или димеризованные CPP, среди которых возможны гомо- и гетеродимеры, ковалентно прикрепляют к фосфолипиду через линкер (фиг. 4). Димеризацию CPP можно осуществлять с помощью любого средства, известного в данной области. В конкретном варианте осуществления CPP димеризуют через трипептид KAK. Однако также возможно прикрепление без линкера, например, при использовании таких модифицированных и/или активированных липидов, например, как фосфолипиды с модифицированной малеимидом головной группой.
Предпочтительно липосомы, входящие в липосомные композиции по настоящему изобретению, содержат указанные CPP в количестве от 0,05 до 5 моль-%, предпочтительно от 0,1 до 1 моль-%, на основе общего количества липидов. В случае мономерных CPP, предпочтительно они содержатся в количестве от 0,05 до 2 моль-%, предпочтительно от 0,1 до 1 моль-%, на основе общего количества липидов. В случае димеризованных CPP, предпочтительно они содержатся в количестве от 0,05 до 0,5 моль-%, предпочтительно от 0,1 моль-%, на основе общего количества липидов.
Подходящие фосфолипиды для ковалентного прикрепления CPP конкретно не ограничены конкретными фосфолипидами. В частности, фосфолипиды, используемые для ковалентного прикрепления CPP, могут представлять собой любые подходящие фосфолипиды, известные в данной области, где указанные фосфолипиды могут представлять собой синтетические, полусинтетические или природные фосфолипиды. В целом, подходящие фосфолипиды можно выбирать из группы, состоящей из фосфатидилхолинов, фосфатидилэтаноламинов, фосфатидилинозитов, фосфатидилсеринов, цефалинов, фосфатидилглицеринов и лизофосфолипидов. Конкретным фосфолипидом в этом отношении является яичный фосфатидилхолин (E-PC; лецитин). Кроме того, подходящие фосфолипиды в этом отношении включают PEG-модифицированные версии вышеуказанных фосфолипидов, например, DSPE-PEG(2000) малеимид (1,2-дистеароил-sn-глицеро-3-фосфоэтаноламин-N-[малеимид(полиэтиленгликоль)-2000] (соль аммония)).
Подходящие линкеры для ковалентного прикрепления CPP к фосфолипидам конкретно не ограничены и известны в данной области. Они включают в целом, например, бифункциональные PEG-линкеры; например, SM(PEG)24 (пегилированный длинноцепочечный SMCC сшиватель). Конкретными образцовыми линкерами в этом отношении являются линкер SMCC (сукцинимидил-4-(N-малеимидометил)циклогексан-1-карбоксилат) и линкер из 6-малеимидогексановой кислоты. В этом контексте длина фрагмента PEG в таких линкерах влияет на эффективность инкапсулирования лекарственных средств, которые встраивают в липосомы, содержащиеся в липосомных композициях по настоящему изобретению. Соответственно, указанный фрагмент PEG предпочтительно имеет длину от 8 до 50 индивидуальных звеньев PEG. Кроме того, способы ковалентного соединения CPP с фосфолипидами через линкеры конкретно не ограничены и известны в данной области.
Липиды, используемые для получения липосом, содержащихся в композиции по настоящему изобретению, также можно прикреплять к самонаводящимся структурам, таким как пептидные последовательности, антитела, лиганды рецепторов и поверхностно-активные средства.
В предпочтительном варианте осуществления липосомы, содержащиеся в композиции по настоящему изобретению, проявляют Z-среднее, измеряемое с помощью динамического рассеяния света, после разведения в водной среде самое большее 350 нм и индекс полидисперсности (PDI) самое большее 0,3, где особенно предпочтительны Z-среднее от 100 до 200 нм и индекс полидисперсности от 0,1 до 0,3, предпочтительно приблизительно 0,2.
Способы создания липосом конкретно не ограничены и известны в данной области. Они включают, например, гомогенизацию высокого давления, экструзию и двойное асимметричное центрифугирование (DAC).
В предпочтительных вариантах осуществления липосомные композиции по настоящему изобретению дополнительно могут содержать по меньшей мере одно дополнительное терапевтическое средство и/или по меньшей мере одно диагностическое средство.
Соответствующие терапевтические средства конкретно не ограничены и включают любые средства, для которых пероральная доставка может быть интересно. Они включают, например, макромолекулы, такие как пептидные лекарственные средства (например, октреотид, глатирамер ацетат, ванкомицин, Myrcludex B, инсулин всех видов и лираглутид, а также другие аналоги GLP (глюкагоноподобного пептида), такие как эксенатид, ликсисенатид, албиглутид, дулаглутид, таспоглутид и семаглутид), и белки или антитела (например, этанерцепт; пегфилграстим; адалимумаб, инфликсимаб, ритуксимаб, эпоэтин α, тратузумаб, ранибизумаб, β-интерферон, омализумаб). Другие примеры включают фармацевтически активные средства, выбранные из группы, состоящей из гормона роста человека, рилизинг-гормона гормона роста, релизинг-пептида гормона роста, интерфероов, колониестимулирующих факторов, интерлейкинов, фактора активации макрофагов, макрофагального пептида, B-клеточного фактора, T-клеточного фактора, белка A, ингибитора аллергии, гликопротеинов некроза клеток, иммунотоксина, лимфотоксина, фактора некроза опухоли, супрессоров опухолей, фактора роста метастазов, α-1 антитрипсина, альбумина и его полипептидных фрагментов, аполипопротеина-E, эритропоэтина, фактора VII, фактора VIII, фактора IX, фактора активации плазминогена, урокиназы, стрептокиназы, белка C, C-реактивного белка, ингибитора ренина, ингибитора коллагеназы, супероксиддисмутазы, тромбоцитарного фактора роста, эпидермального фактора роста, остеогенного фактора роста, костного стимулирующего белка, кальцитонина, инсулина, атриопептина, хрящевого индуцирующего фактора, активирующего фактора соединительной ткани, фолликулостимулирующего гормона, лютеинизирующего гормона, рилизинг-гормона лютеинизирующего гормона, факторов роста нервов, паратиреоидного гормона, релаксина, секретина, соматомедина, инсулиноподобного фактора роста, гормона коры надпочечников, глюкагона, холецистокинина, панкреатического полипептида, релизинг-пептида гастрина, рилизинг-фактора кортикотропина, тиреостимулирующего гормона, моноклональных или поликлональных антител против различных вирусов, бактерий или токсинов, вакцинных антигенов вирусного происхождения, октреотида, циклоспорина, рифампицина, лопинавира, ритонавира, ванкомицина, телаванцина, оритаванцина, далбаванцина, бисфосфонатов, итраконазола, даназола, паклитаксела, циклоспорина, напроксена, капсайцина, сульфата альбутерола, сульфата тербуталина, гидрохлорида дифенгидрамина, хлорфенираминмалеата, гидрохлорида лоратидина, гидрохлорида фексофенадина, фенилбутазона, нифедипина, карбамазепина, напроксена, циклоспорина, бетаметозона, даназола, дексаметазона, преднизона, гидрокортизона, 17β-эстрадиола, кетоконазола, мефенамовой кислоты, беклометазона, алпразолама, мидазолама, миконазола, ибупрофена, кетопрофена, преднизолона, метилпреднизона, фенитоина, тестостерона, флунизолида, дифлунисала, будесонида, флутиказона, инсулина, глюкагоноподобного пептида, C-пептида, эритропоэтина, кальцитонина, лютеинизирующего гормона, пролактина, адренокортикотропного гормона, лейпролида, интерферона α-2b, интерферона β-Ia, сарграмостима, альдеслейкина, интерферона α-2a, интерферона α-n3, ингибитора α-протеиназы, этидроната, нафарелина, хорионического гонадотропина, простагландина E2, эпопростенола, акарбозы, метформина, десмопрессина, циклодекстрина, антибиотиков, противогрибковых лекарственных средств, стероидов, лекарственных средств против злокачественных опухолей, аналгетиков, противовоспалительных средств, противогельминтных средств, антиаритмических средств, пенициллинов, антикоагулянтов, антидепрессантов, противодиабетических средств, противоэпилептических средств, антигистаминных средств, антигипертензивных средств, антимускариновых средств, антимикобактериальных средств, антинеопластических средств, активных средств ЦНС, иммуносупрессантов, антитиреоидных средств, противовирусных средств, седативных анксиолитиков, снотворных средств, нейролептиков, вяжущих средств, блокаторов β-адренорецепторов, продуктов и заменителей крови, сердечных инотропных средств, контрастных веществ, кортикостероидов, средств для подавления кашля, отхаркивающих средств, муколитиков, диуретиков, дофаминэргических средств, антипаркинсонических средств, гемостатических средств, иммунологических средств, регулирующих липиды средств, мышечных релаксантов, парасимпатомиметиков, паратиреоидного кальцитонина, простагландинов, радиофармацевтических средств, половых гормонов, стероидов, противоаллергических средств, стимуляторов, анорексических средств, симпатомиметиков, тиреоидных средств, вазодилататоров, ксантинов, гепаринов, терапевтических олигонуклеотидов, соматостатинов и их аналогов, а также их фармакологически приемлемых органических и неорганических солей или их комплексных соединений с металлами.
Кроме того, соответствующие диагностические средства конкретно не ограничены и включают любые средства, для которых пероральная доставка может быть интересной.
Указанные выше средства могут присутствовать в липосомных композициях по настоящему изобретению, заключенных в липосомах, т. е. во внутреннем просвете указанных липосом, например, когда указанные средства являются гидрофильными, или встроенными в липосомную мембрану, например, когда указанные средства являются липофильными. В этом контексте инкапсулирование терапевтических и/или диагностических средств зависит от гидрофильности указанных средств и способа получения липосом.
В предпочтительном варианте осуществления содержание терапевтического и/или диагностического средства в липосомных композициях в соответствии с настоящим изобретением составляет выше 0 и самое большее 50% (масс./масс.) в отношении используемого количества средства.
В соответствии с приведенным выше аспектом, липосомные композиции по настоящему изобретению можно использовать в медицине. Предпочтительно, указанные липосомные композиции предназначены для использования при лечении сепсиса, диабета, ревматизма, акромегалии, гепатитов всех типов, злокачественных опухолей всех типов и анемии. Предпочтительно липосомные композиции для использования по настоящему изобретению предназначены для перорального введения.
Недавние исследования показывают, что предварительное лечение ингибитором протонной помпы омепразолом снижает диффузию протонов в липосомы и, как последствие, снижает денатурацию инкапсулированных средств, таких как белки, посредством повышения pH в желудке. Соответственно, в предпочтительном варианте осуществления липосомных композиций для использования в соответствии с настоящим изобретением, субъекта предварительно лечат ингибитором протонной помпы, где указанный ингибитор протонной помпы предпочтительно представляет собой омепразол (6-метокси-2-[(4-метокси-3,5-диметилпиридин-2-ил)метансульфинил]-1H-1,3-бензодиазол), пантопразол ((RS)-6-(дифторметокси)-2-[(3,4-диметоксипиридин-2-ил)метилсульфинил]-1H-бензо[d]имидазол), эзомепразол ((S)-5-метокси-2-[(4-метокси-3,5-диметилпиридин-2-ил)метилсульфинил]-3H-бензоимидазол), лансопразол ((RS)-2-([3-метил-4-(2,2,2-трифторэтокси)пиридин-2-ил]метилсульфинил)-1H-бензо[d]имидазол) и/или рабепразол ((RS)-2-([4-(3-метоксипропокси)-3-метилпиридин-2-ил]метилсульфинил)-1H-бензо[d]имидазол).
Предпочтительно, липосомные композиции по настоящему изобретению можно лиофилизировать, например, с использованием 300-500 мМ сахарозы в качестве лиопротектора, который делает возможным длительное хранение указанных композиций.
В связанном втором аспекте настоящее изобретение относится к использованию липосомной композиции по настоящему изобретению для пероральной доставки по меньшей мере одного терапевтического средства и/или по меньшей мере одного диагностического средства.
В этом контексте термин «пероральная доставка» относится к доставке одного или нескольких средств посредством перорального введения указанных средств.
В этом аспекте все релевантные ограничения и определения, предусмотренные для первого аспекта настоящего изобретения, применимы аналогичным образом. В частности, липосомные композиции, терапевтические средства и диагностические средства представляют собой то, что определено выше.
В дополнительном связанном аспекте настоящее изобретение относится к способу доставки по меньшей мере одного терапевтического средства и/или по меньшей мере одного диагностического средства субъекту, который включает стадию введения, предпочтительно перорального введения, липосомной композиции по настоящему изобретению указанному субъекту.
В этом аспекте все релевантные ограничения и определения, предусмотренные для первого аспекта настоящего изобретения, применимы аналогичным образом. В частности, липосомные композиции, терапевтические средства и диагностические средства представляют собой то, что определено выше.
В рамках изобретения, термин «приблизительно» предусмотрен в качестве модификатора±10% от точно определенного значения. В качестве примера, термин «приблизительно 5%» предназначен для того, чтобы охватывать диапазон от 4,5 до 5,5%.
Термины «содержащий/содержит», «состоящий из/состоит из» и «состоящий по существу из/состоит по существу из» используют в настоящем описании взаимозаменяемым образом, т. е. каждый из указанных терминов можно в явной форме заменять на один из двух других терминов.
Термины «композиция» и «состав» используют в настоящем описании эквивалентно, и они предназначены для того, чтобы в явной форме заменять друг друга.
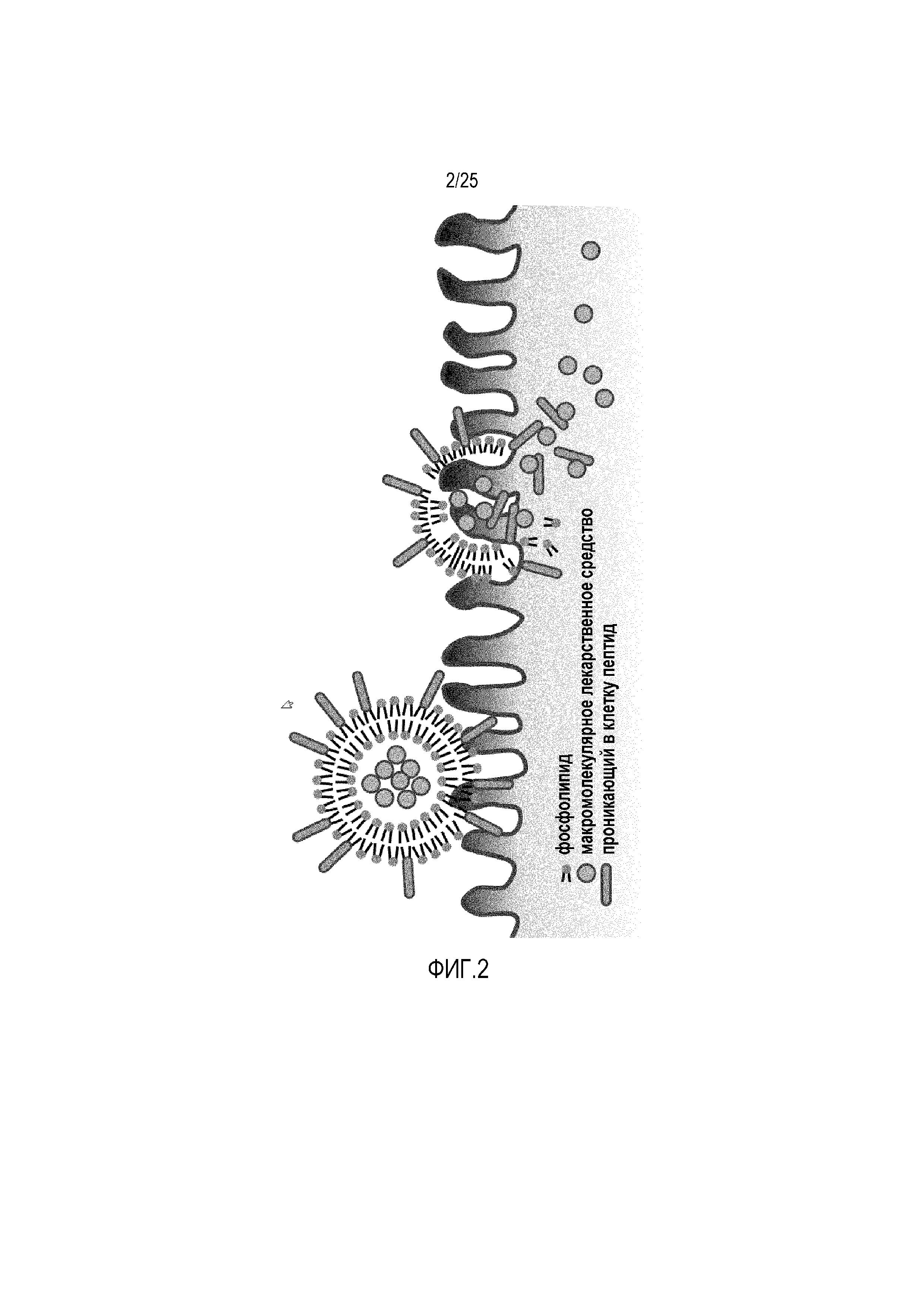
В настоящем изобретении используют CPP, которые ковалентно прикрепляют к липосомам для благоприятного увеличения проникновения через слизистую указанных липосом (фиг. 2).
На фиг. представлено:
Фиг. 1:
Разрушение в желудке и слабое проникновение через слизистую являются основными трудностями, которые препятствуют пероральной доступности биологических средств. Используя TEL-липосомы, содержащие CPP, можно преодолеть обе трудности.
Фиг. 2:
Показаны липосомы, содержащие CPP, соединенные с фосфолипидами. CPP усиливают проникновение через слизистую макромолекулярных лекарственных средств, встроенных в липосомы.
Фиг. 3:
Показана структура фосфолипида, модифицированного мономерным CPP. CPP представляет собой встречающийся в природе пептид пенетратин, линкер представляет собой SMCC-линкер.
Фиг. 4:
Показана структура фосфолипида, модифицированного димеризованными CPP. CPP представляет собой встречающийся в природе пептид пенетратин, где две молекулы пенетратина димеризуют через трипептид KAK. Линкер представляет собой линкер из 6-малеимидогексановой кислоты.
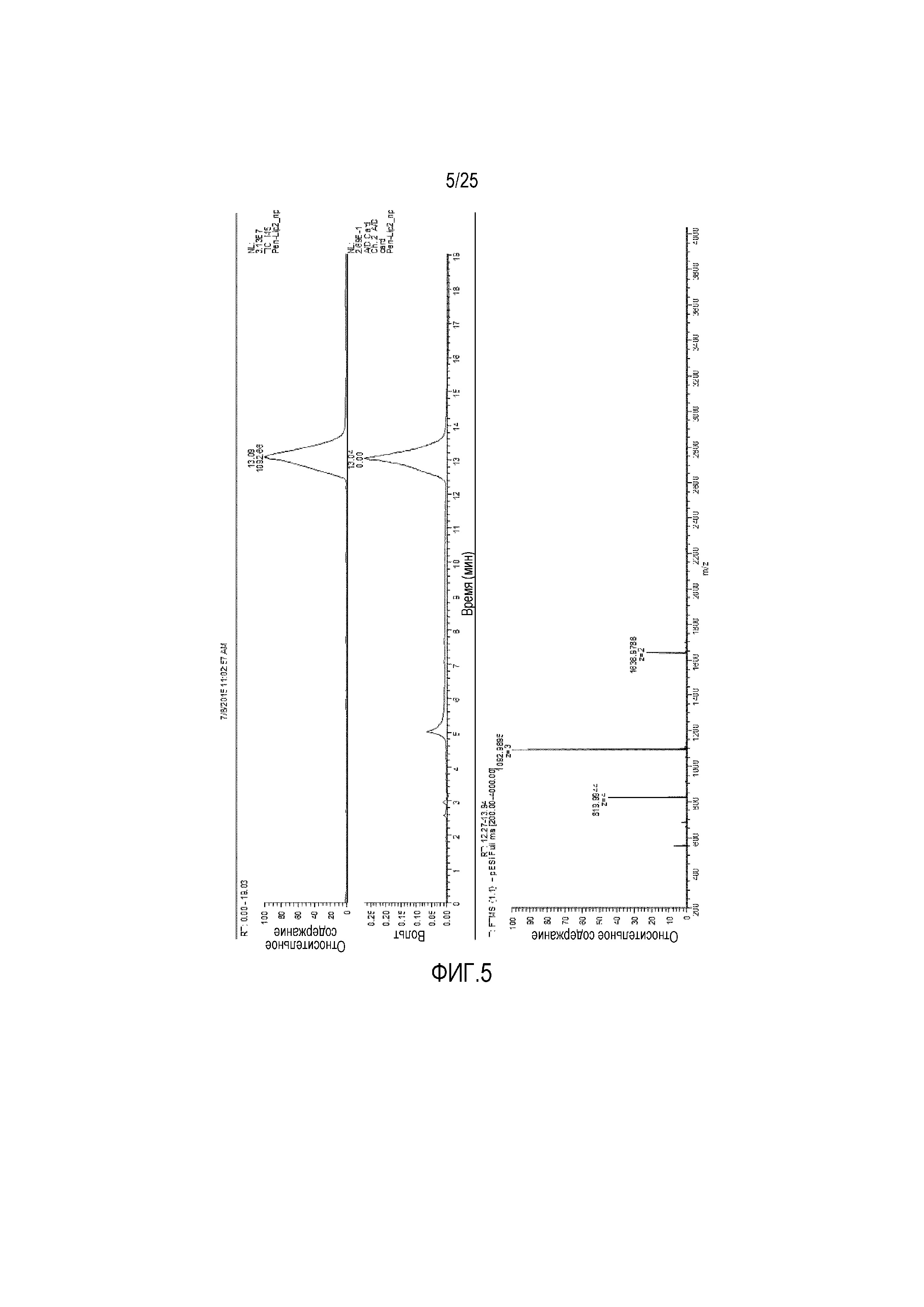
Фиг. 5:
Анализ конъюгата пенетратина-лецитина посредством масс-спектрометрии.
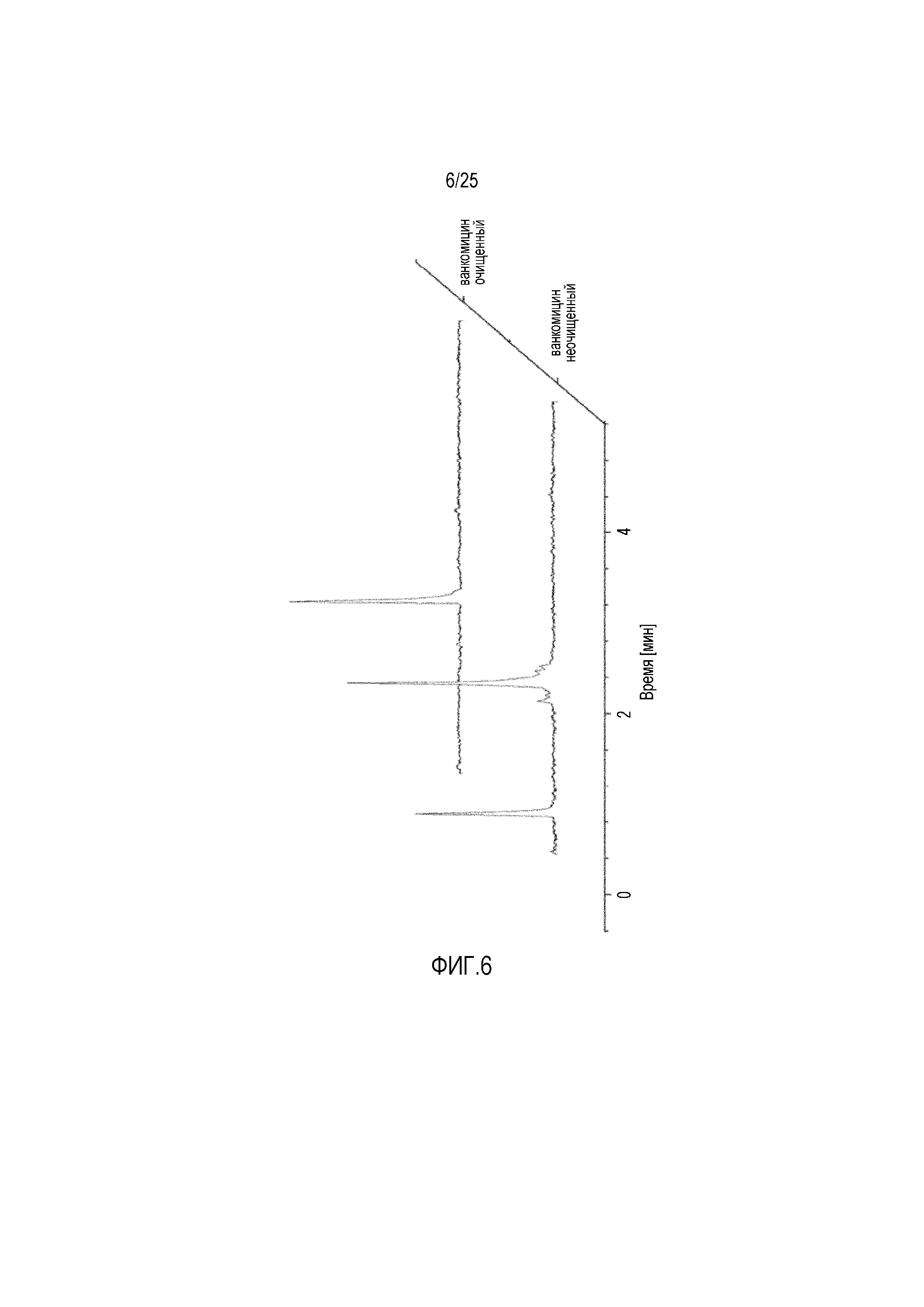
Фиг. 6:
Спектры радиоВЭЖХ для131I радиоактивно меченного ванкомицина до/после очистки посредством препаративной радиоВЭЖХ.
Фиг. 7:
Криоэлектронная микрофотография CPP-TEL-ванкомицина-липосом.
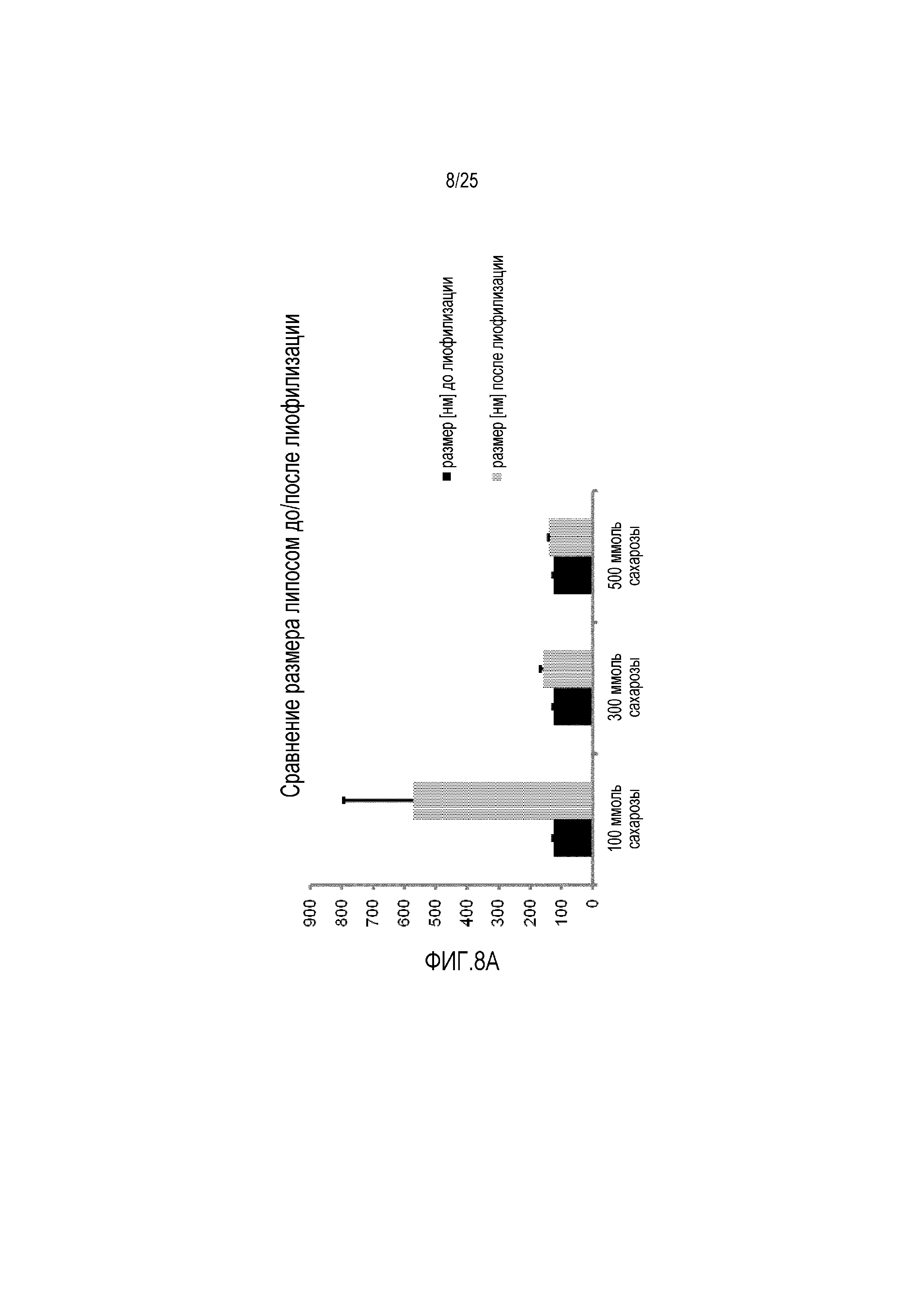
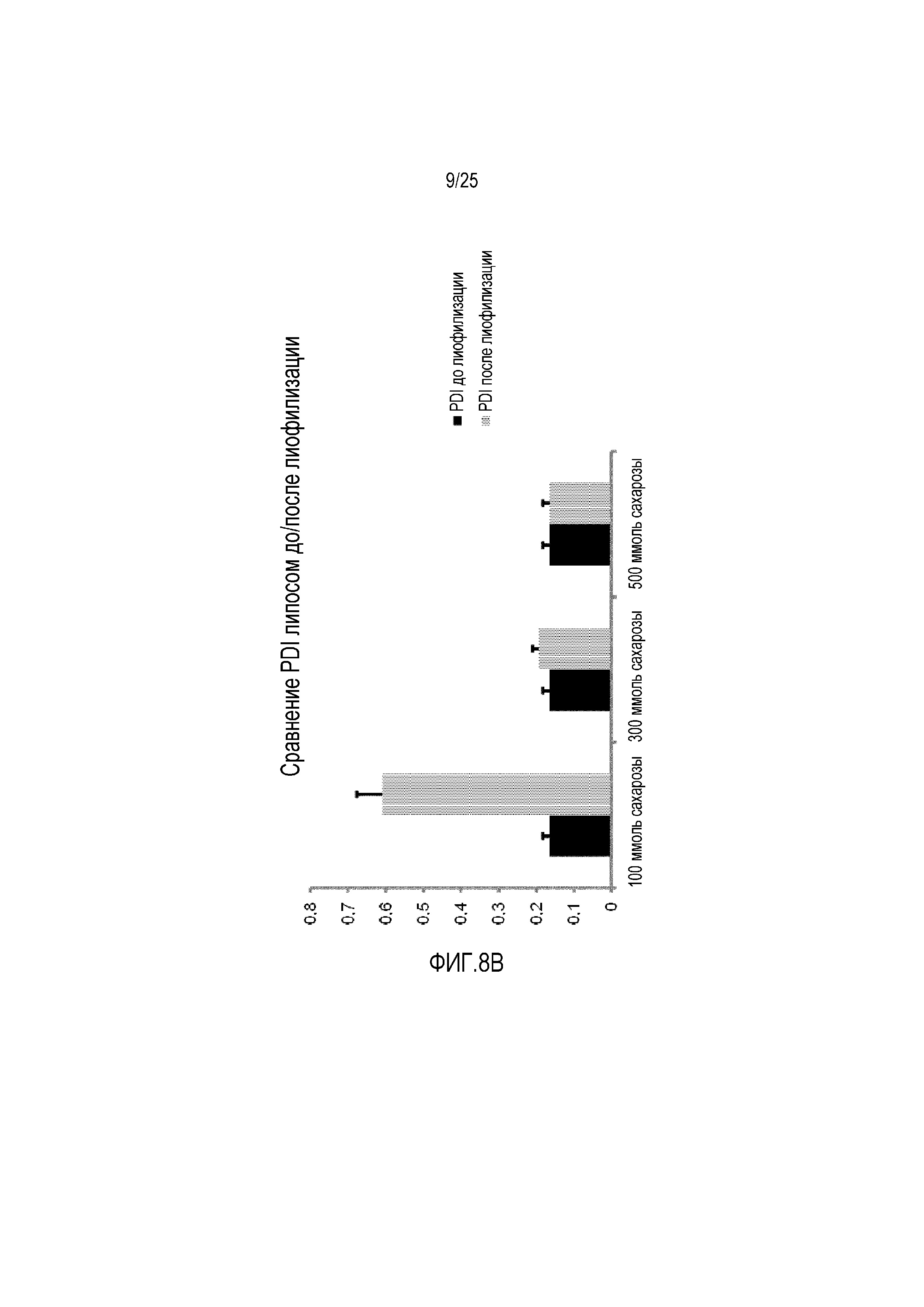
Фиг. 8:
(A) Сравнение размера липосом и (B) PDI липосом до/после процесса лиофилизации с использованием 100-500 мМ сахарозы в качестве лиопротектора.
Фиг. 9:
Сравнение уровней131I-ванкомицина в крови через 1 ч после перорального введения.
Фиг. 10:
Сравнение уровней CPP-TEL-липосом в крови.
Фиг. 11:
Новые липосомы содержат конкретные тетраэфирные липиды и конъюгаты CPP-фосфолипидов (линейные и циклические).
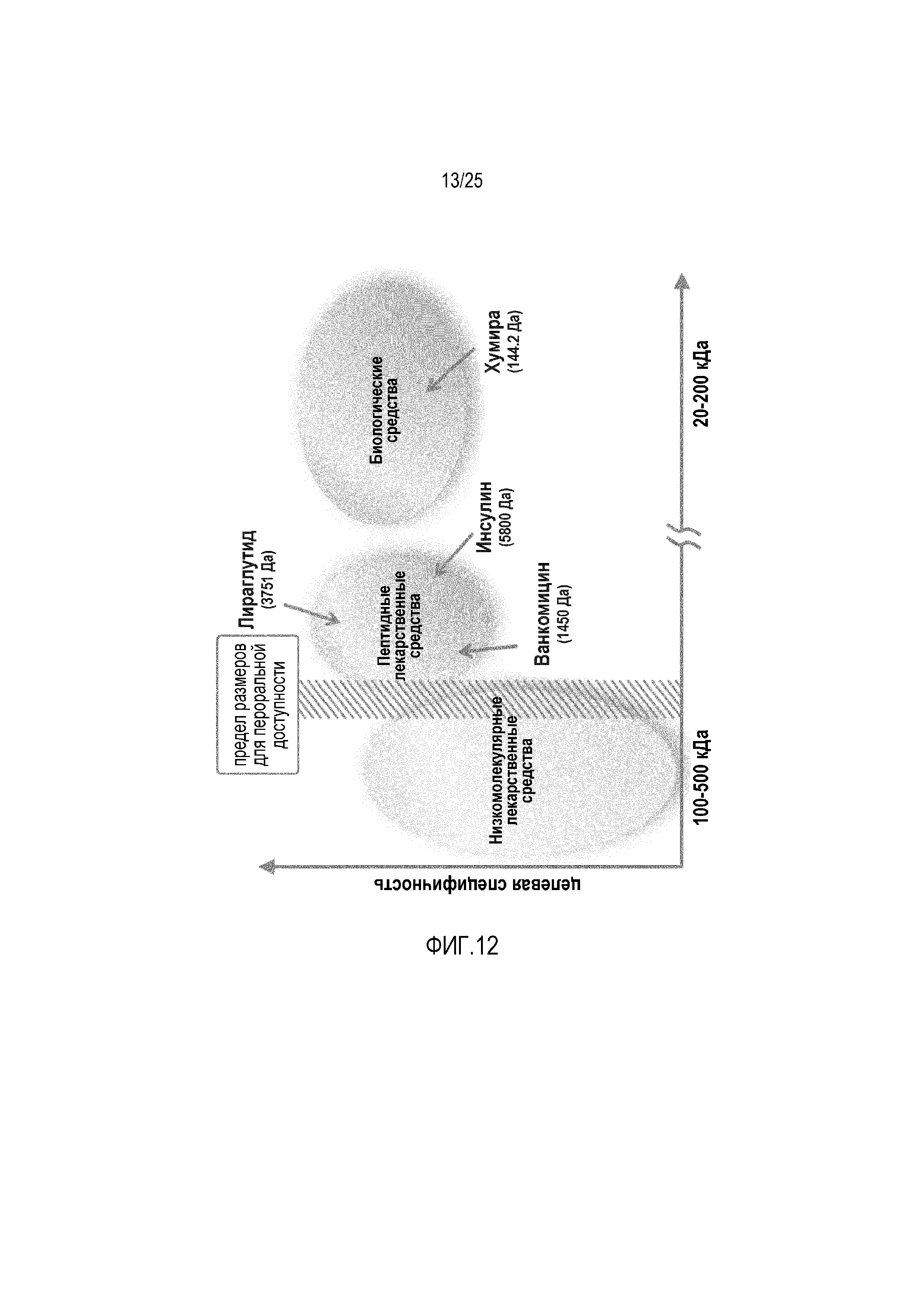
Фиг. 12:
Модельные вещества и молекулярная масса для пероральной доставки лекарственных средств.
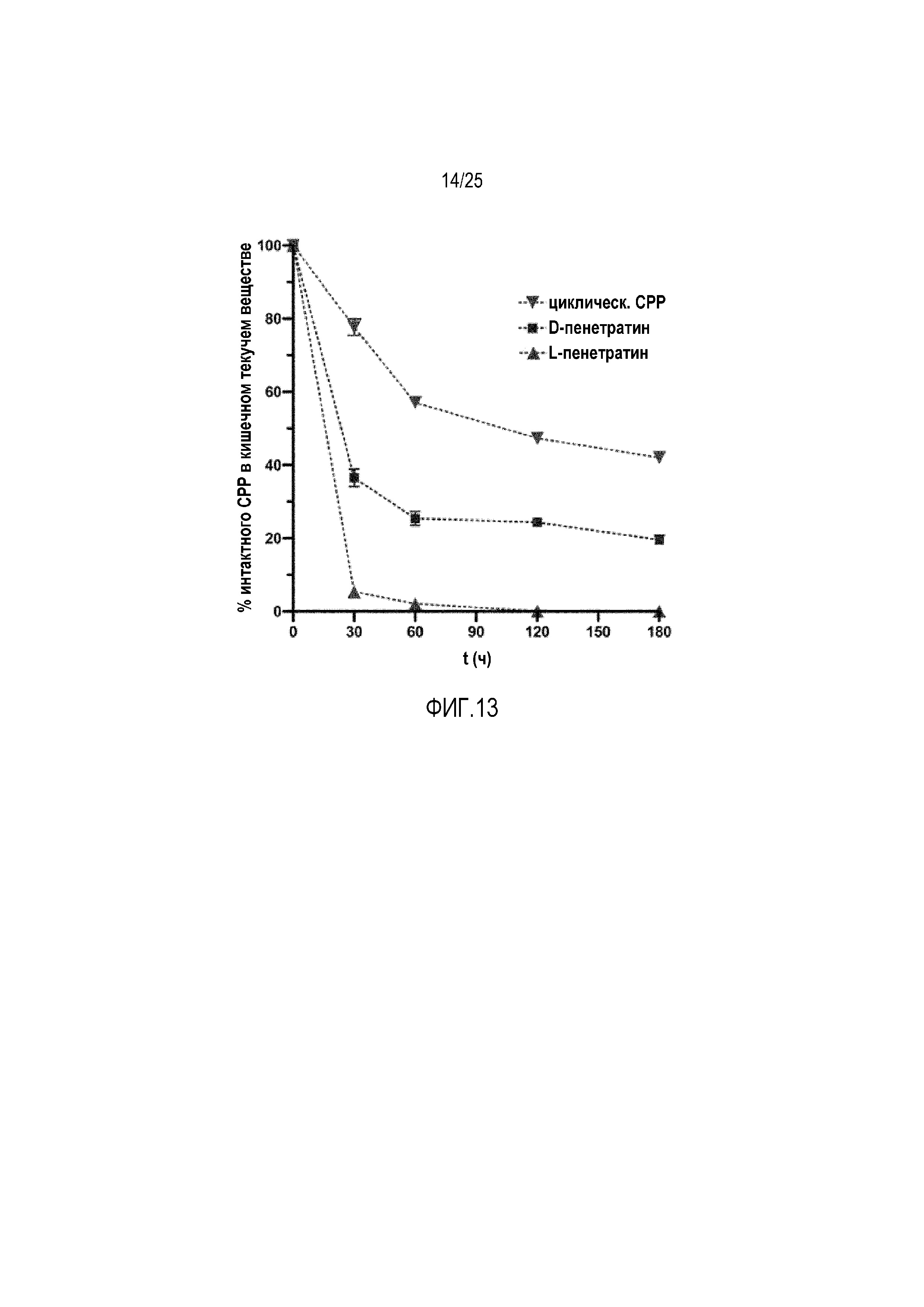
Фиг. 13:
Извлечение интактных CPP в имитированном интестинальном текучем веществе, как обнаруживали посредством ВЭЖХ/MS в несколько моментов времени.
Фиг. 14:
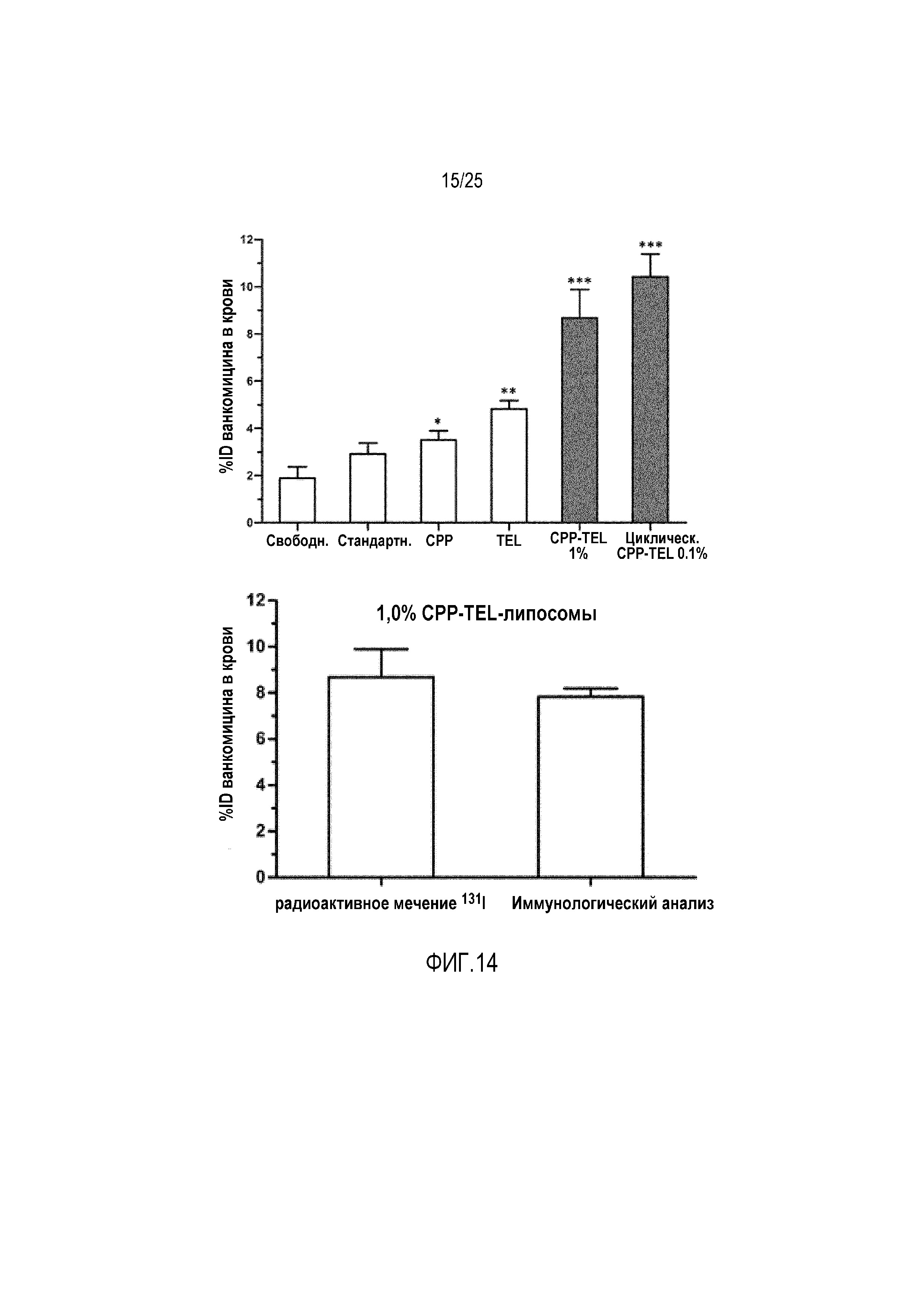
Несколько липосомных составов тестировали или с использованием радиоактивно меченного ванкомицина или посредством иммунологического анализа и определяли уровни ванкомицина в крови в несколько моментов времени.
Фиг. 15:
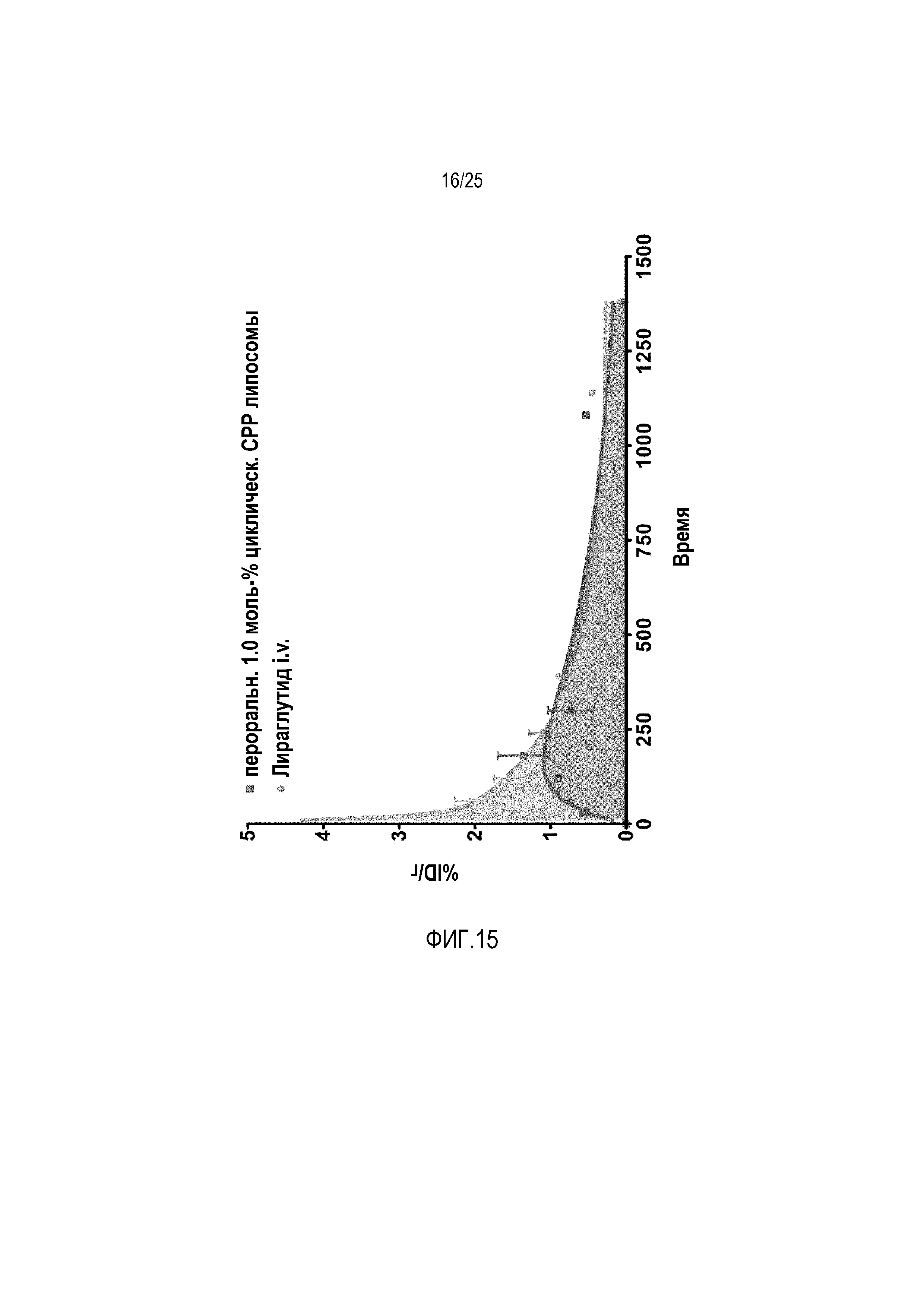
Биодоступность перорального лираглутида (1 моль-% конъюгат циклического CPP) в сравнении с i.v. применением.
Фиг. 16:
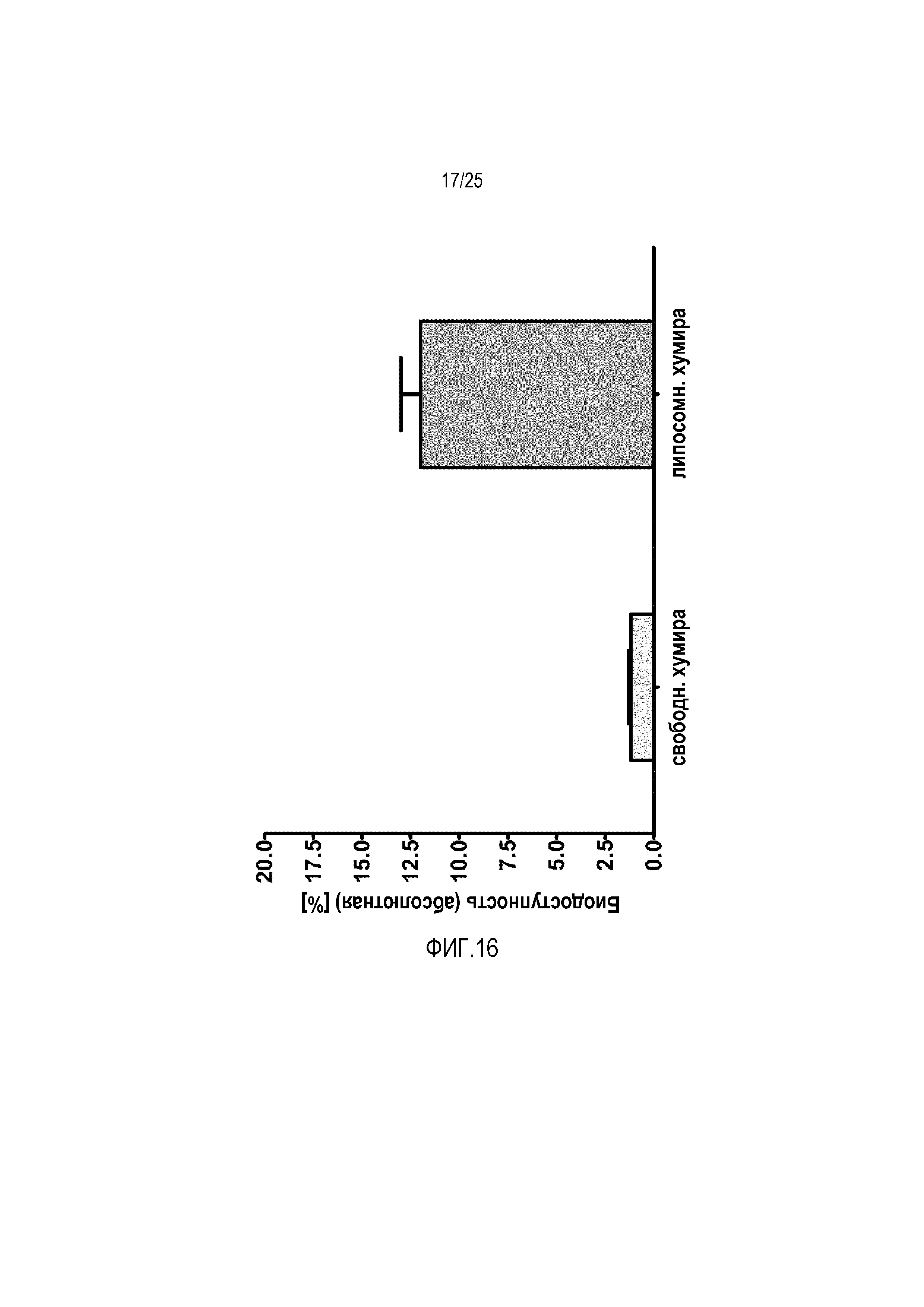
Биодоступность антитела адалимумаб (Хумира®) у самцов крыс Вистар.
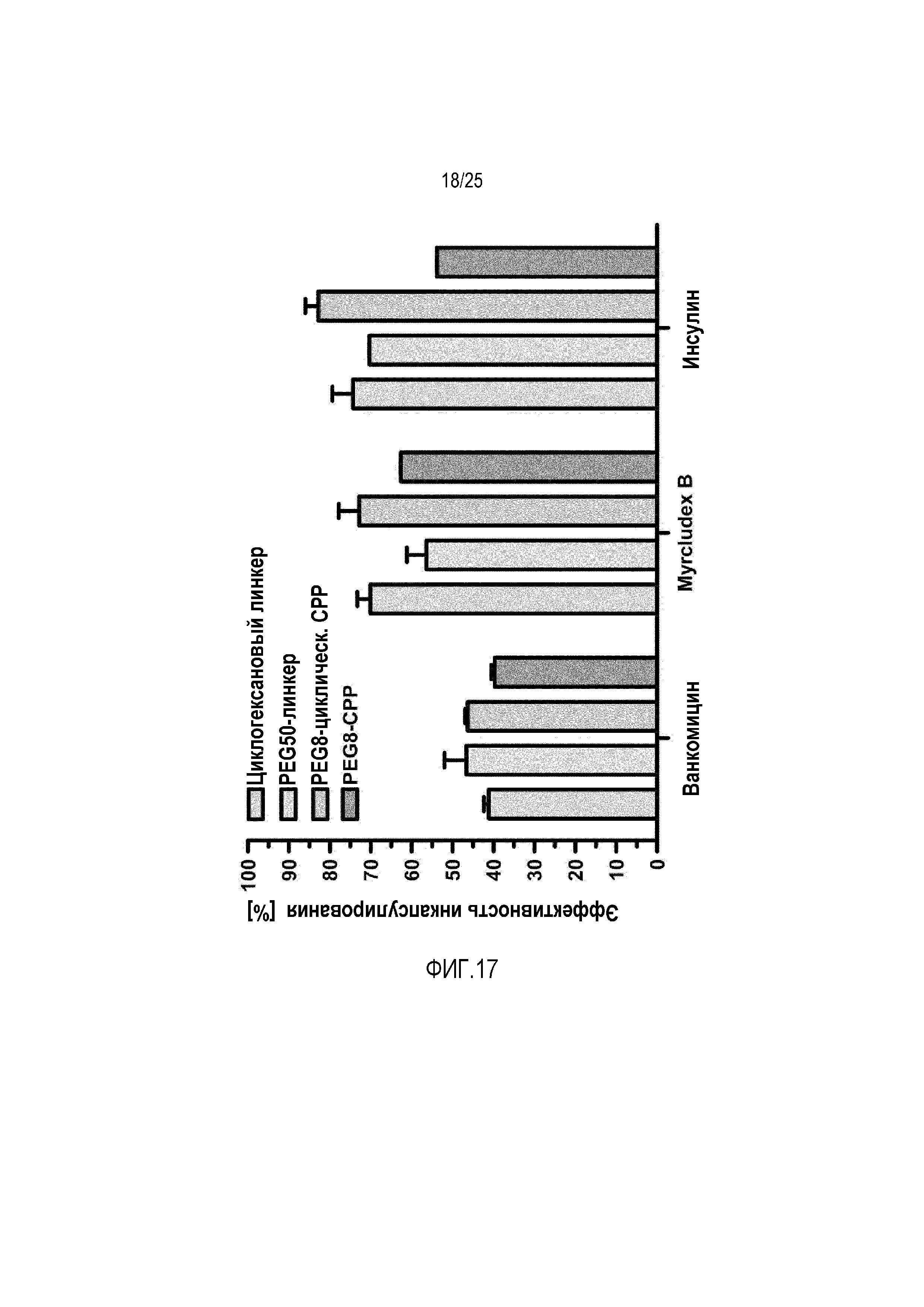
Фиг. 17:
Определение эффективности встраивания трех пептидных лекарственных средств с использованием различных PEG-линкеров (различные количества звеньев PEG).
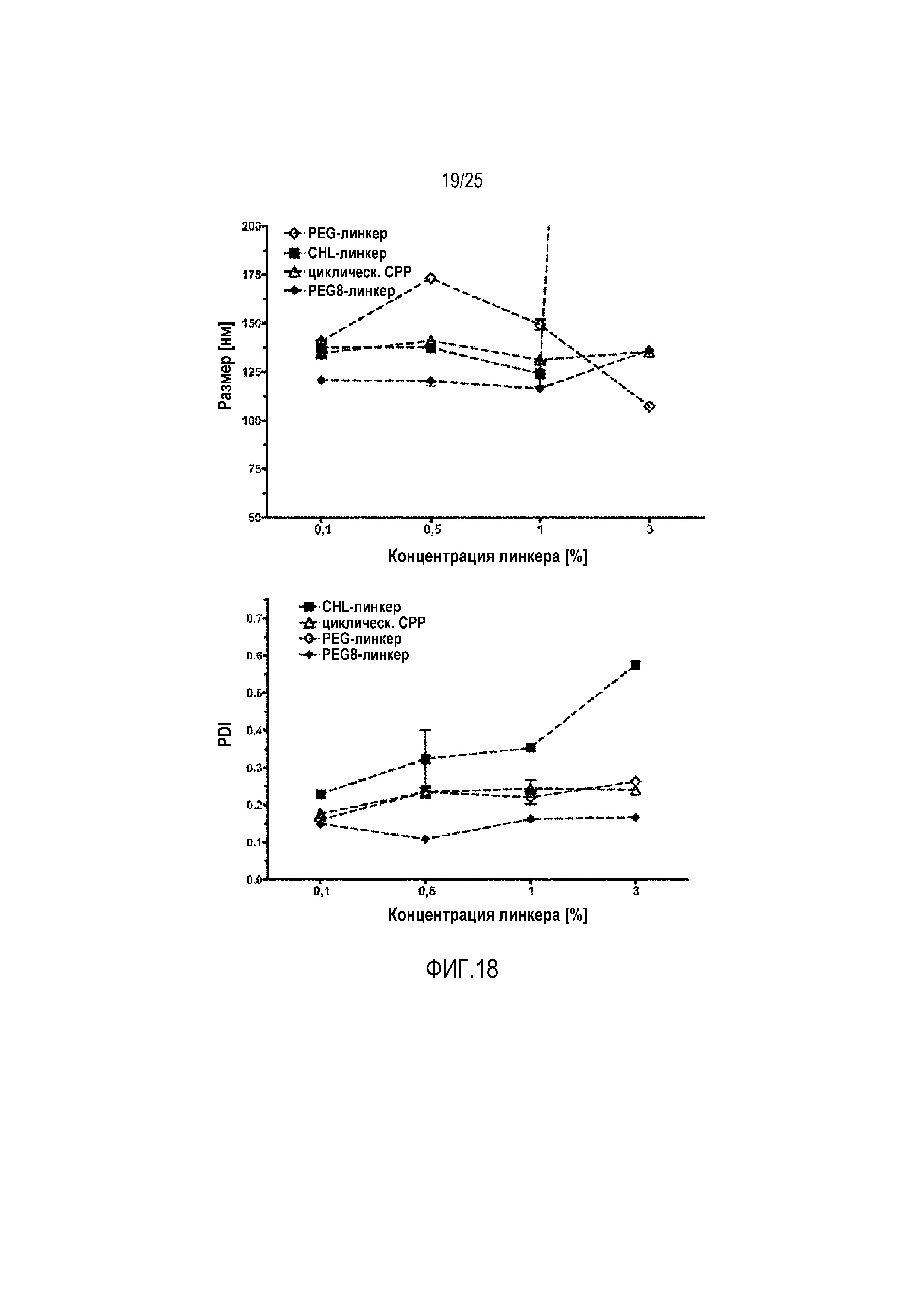
Фиг. 18:
Размер/PDI в липосомных составах, содержащих различные концентрации линкеров, которые различаются количеством звеньев PEG.
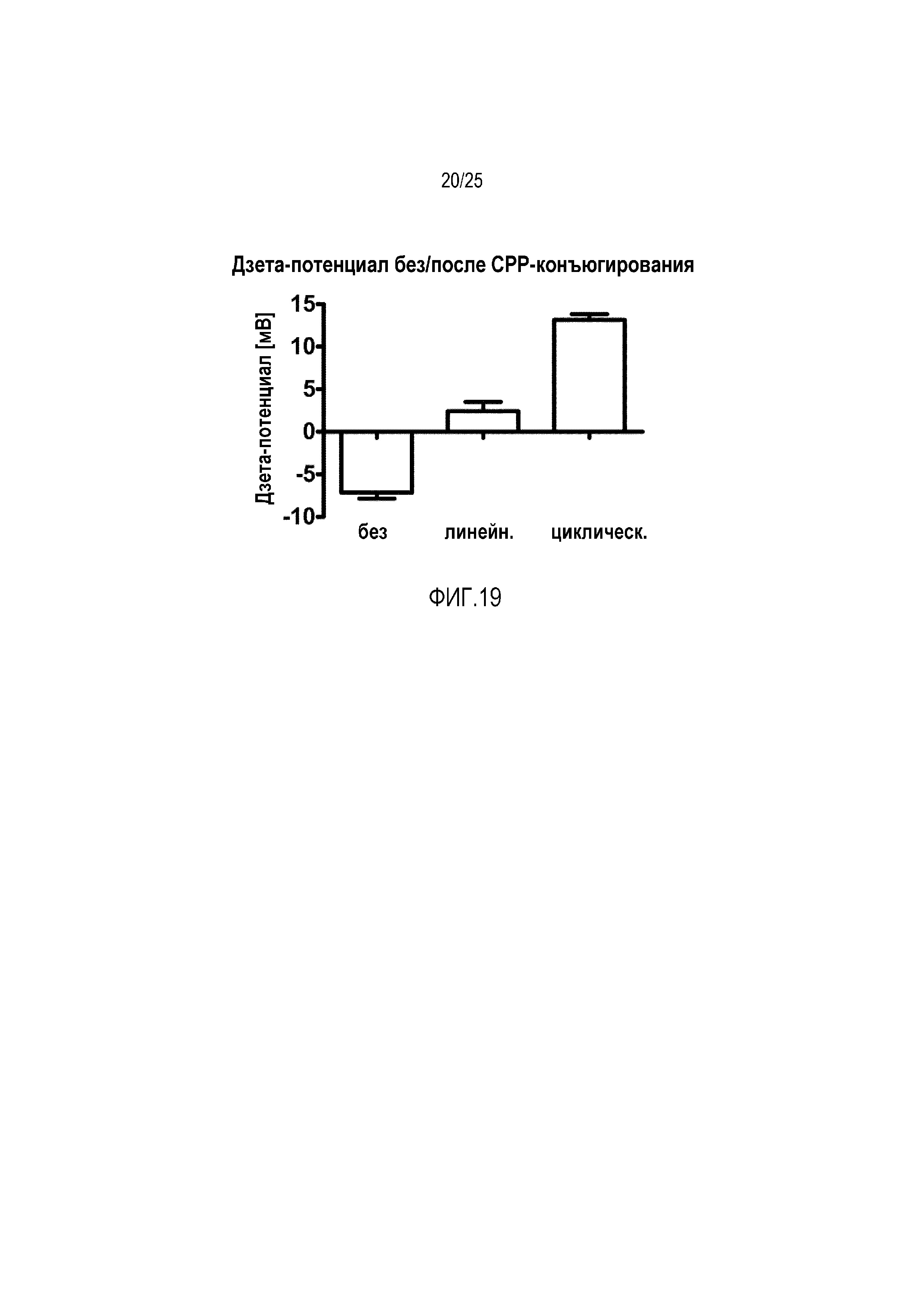
Фиг. 19:
Дзета-потенциал липосом с конъюгатами CPP-фосфолипидов (1 моль-%) в сравнении со стандартными липосомами.
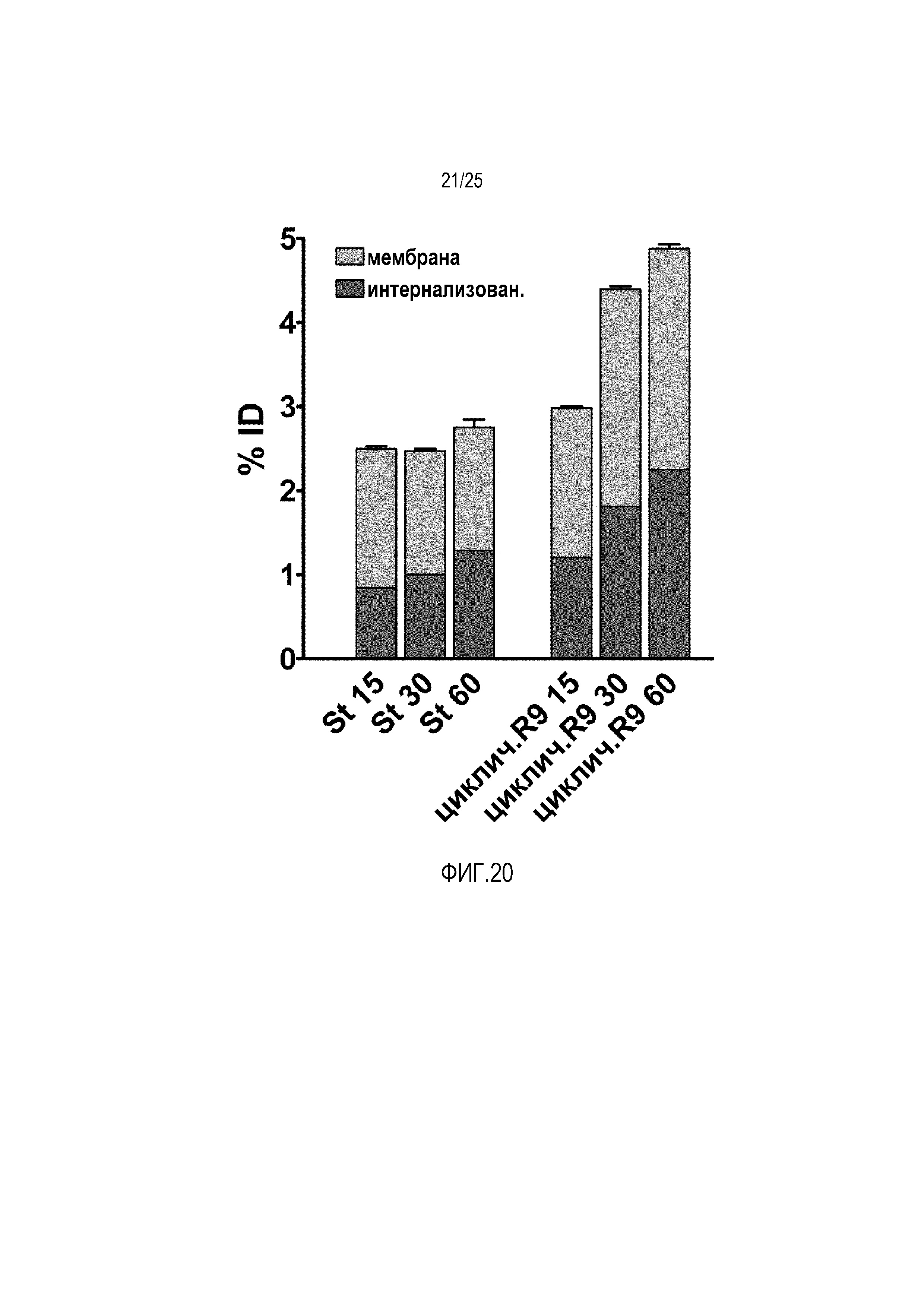
Фиг. 20:
Анализ связывания клеток для липосом, содержащих 1 моль-% конъюгатов циклических CPP-фосфолипидов, в сравнении со стандартными липосомами.
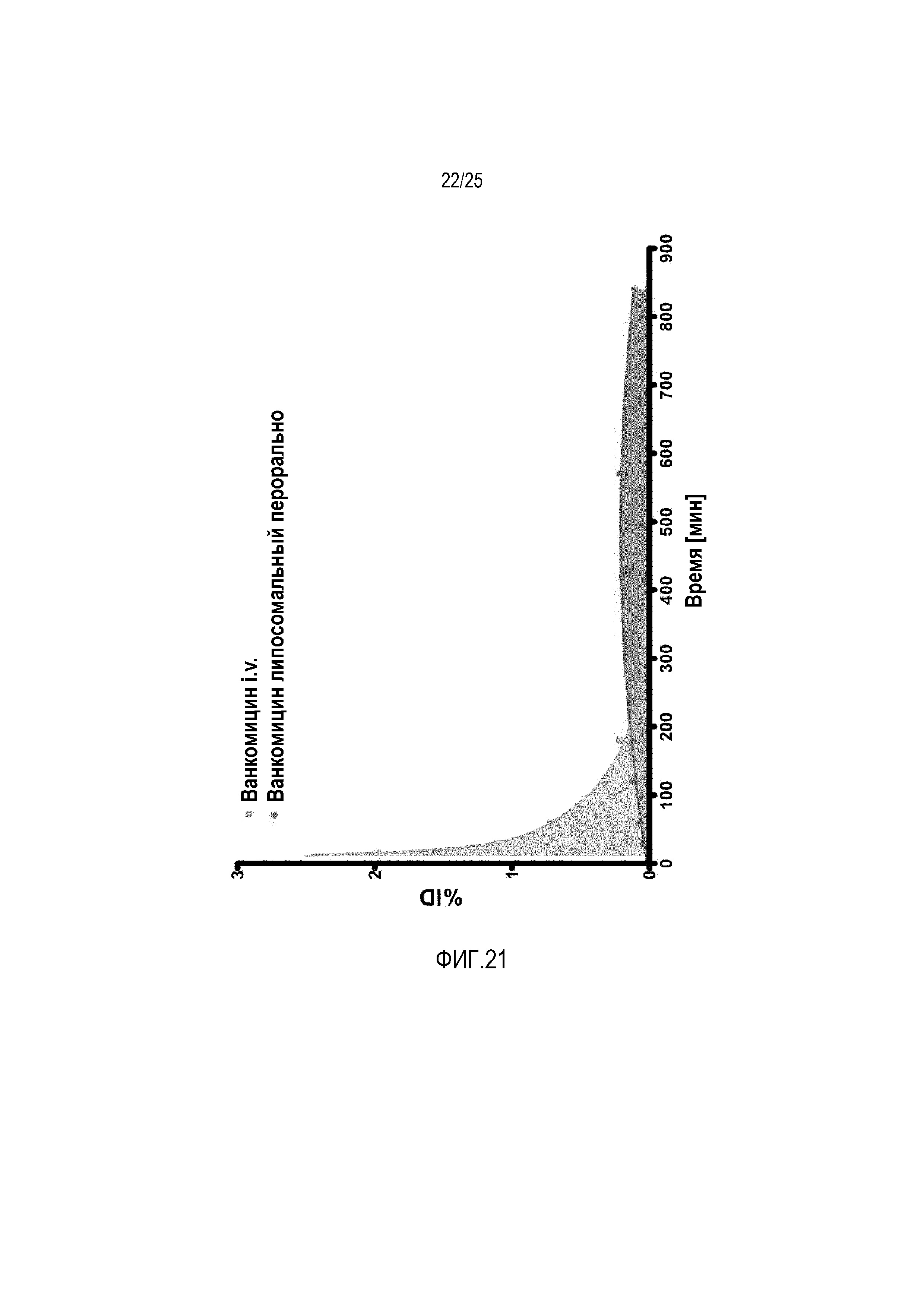
Фиг. 21:
Биодоступность перорального ванкомицина (1 моль-% конъюгат циклического CPP) в сравнении с i.v. применением.
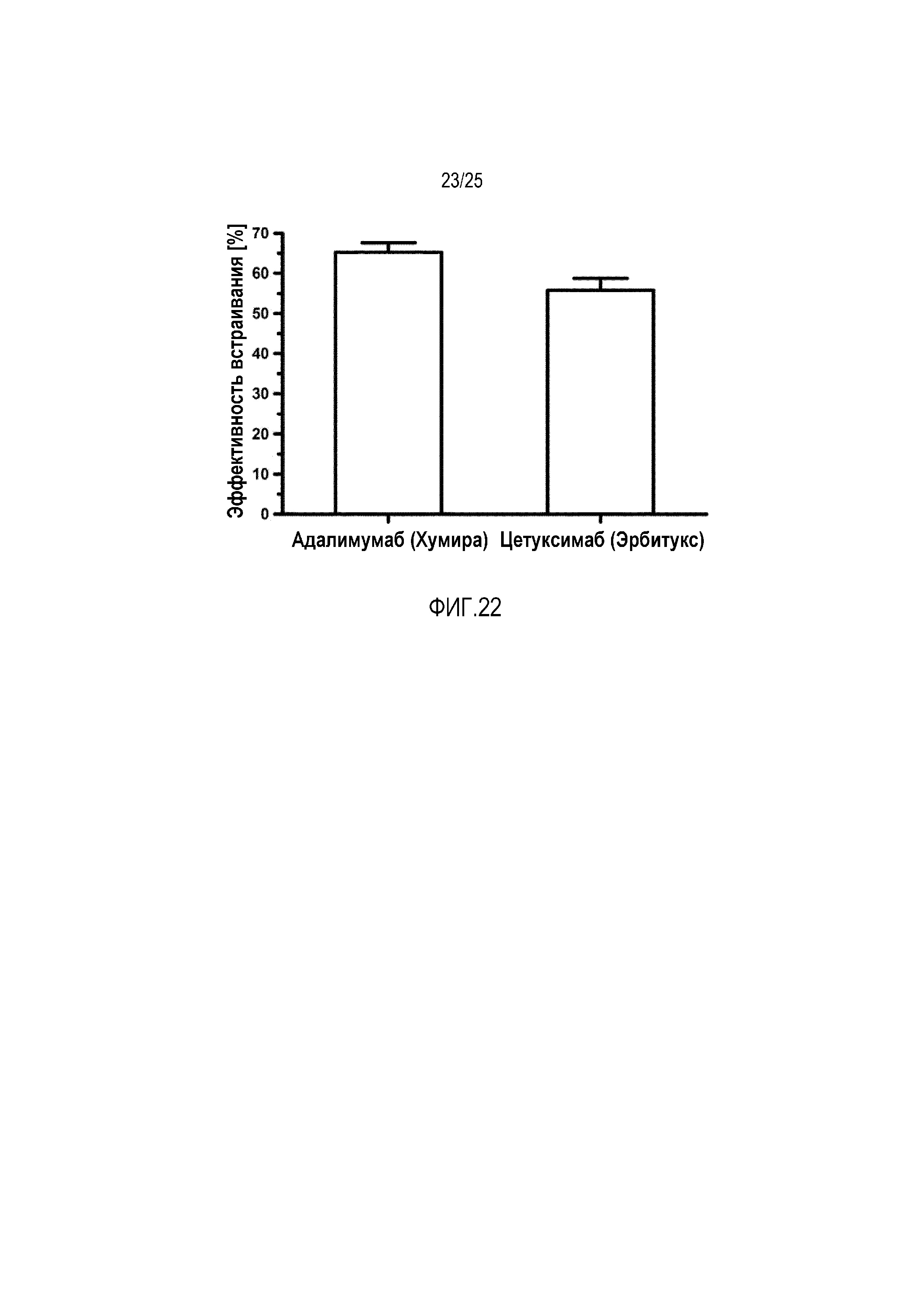
Фиг. 22:
Эффективность встраивания двух антител адалимумаб и цетуксимаб.
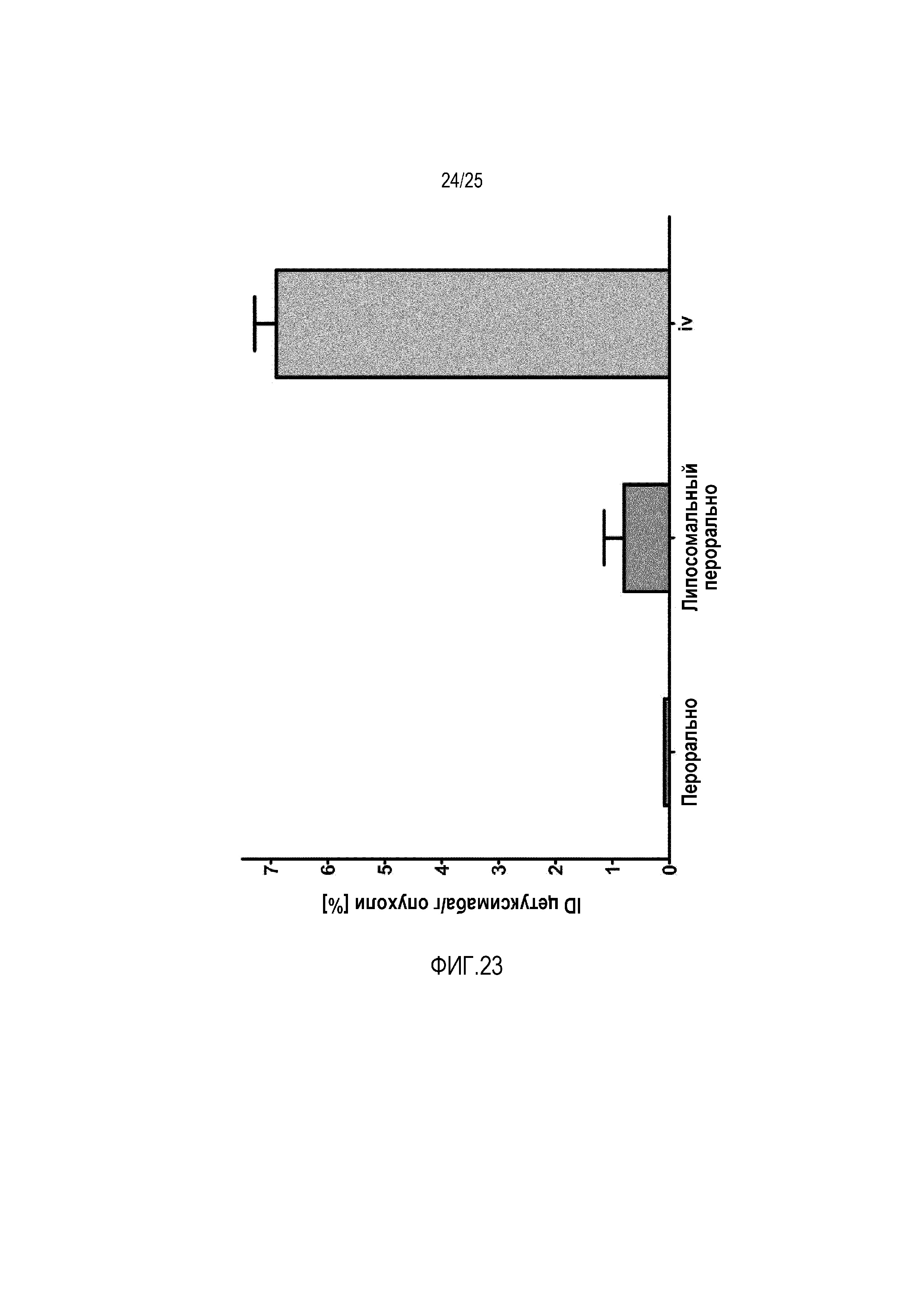
Фиг. 23:
Накопление антитела цетуксимаб в опухоли у мышей Balb/c.
Фиг. 24:
Определение характеристик частиц после встраивания конъюгата MAP-фосфолипида в липосомы.
Настоящее изобретение дополнительно проиллюстрировано следующими примерами, без ограничения ими.
Примеры
Материалы и методы
Материалы
Лецитин из Egg Bio Chemica (AppliChem GmbH, Darmstadt, Germany); холестерин (Sigma Aldrich, Taufkirchen, Germany); 1,2-дипальмитоил-sn-глицеро-3-Фосфотиоэтанол (тиол-модифицированный лецитин; полярные липиды Avanti®, Alabama, USA); тетраэфирные липиды (TEL) выделяли из S. Acidocaldarius, как раскрыто в настоящем описании далее; фосфатно-солевой буфер Дульбекко (Gibco® Life Technologies™, UK); стеклянные бусы (0,75-1,0 мм; Carl Roth GmbH+Co. KG, Karlsruhe); колонка NAP™-5 (GE Healthcare, Buckinghamshire, UK); Triton™ X-100 (Sigma Aldrich, Taufkirchen, Germany); хлороформ (Sigma Aldrich, Taufkirchen, Germany); метанол (Sigma Aldrich, Taufkirchen, Germany); Antra MUPS (омепразол, Astra Zeneca GmbH, Wedel); силикагель 60 (0,063-0,200 мм, Merck, Gernsheim, Germany); радиоактивный йод (Perkin Elmer®, Boston, USA).
Выделение TEL
Клеточный рост и экстрагирование липидов осуществляли, как известно в данной области. S. acidocaldarius выделяли из среды и лиофилизировали с использованием Delta 1-20 KD из Christ. Липиды выделяли посредством экстрагирования по Сокслету с хлороформом/метанолом (2:1), как известно в данной области. Экстрагированный растворитель удаляли посредством роторного испарения (Rotavapor-R, Büchi Labortechnik AG, Flawil, Switzerland). После этого, смесь липидов растворяли в смеси хлороформа, метанола и соляной кислоты (8:3:1). Смесь нагревали в течение 3 суток для отщепления головных групп липидов. Наконец, липиды экстрагировали хлороформом/метанолом (2:1) из водной фазы. Глицерилкалдитилтетраэфирные липиды (GCTE) разделяли посредством колоночной хроматографии на силикагеле с использованием воды/метанола (1:1) в качестве первого элюента (для предварительного промывания колонки), после чего следовали вода/метанол/хлороформ (1:2,5:1) для того, чтобы удалять нежелательные липиды, и метанол/хлороформ (1:1) для получения фракции GCTE.
Синтез конъюгата CPP (пенетратина)-фосфолипида
1. Синтез CPP пенетратина
Пенетратин получали посредством твердофазного синтеза с использованием реакции с флуоренилметоксикарбонилом/т-бутилом (Fmoc/tBu) на пептидном синтезаторе Applied Biosystems 433A. Условия соединения остатков осуществляют, как описано. Очистку осуществляли на ВЭЖХ системе LaPrep P110 (VWR International), оборудованной колонкой Reprosil™ Gold 120 C-18 (4 мкм, 150 × 20 мм). Воду и ацетонитрил, содержащие 0,1% TFA, использовали в качестве элюентов при скорости потока 20 мл/мин. Условия разделения представляли собой линейный градиент 60-90% ацетонитрила за 15 мин. В качестве элюентов использовали 0,1% трифторуксусную кислоту (TFA) в воде (элюент A) и 0,1% TFA в ацетонитриле (элюент B). Идентичность синтезированных пептидов верифицировали посредством анализа ВЭЖХ-MS (масс-спектрометрии) (Exactive, Thermo Fisher Scientific).
2. Синтез конъюгата CPP (пенетратина)-фосфолипида
В 5 мл 1 мМ раствора тиол-модифицированного фосфолипида в 2:1 смеси DCM/DMSO добавляли 20 мг пенетратин-Cys, предварительно растворенного в 200 мкл DMSO. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Растворители испаряли и неочищенный продукт растворяли в 20 мл смеси 4:1 ACN/H2O. Очистку осуществляли на ВЭЖХ системе LaPrep P110 (VWR International), оборудованной колонкой Reprosil™ Gold 120 C-18 (4 мкм, 150 × 20 мм). Воду и ацетонитрил, содержащие 0,1% TFA, использовали в качестве элюентов при скорости потока 20 мл/мин. Аналитические анализы осуществляли на ВЭЖХ системе Agilent 1100, оборудованной колонкой Chromolithe® Performance RP-C18e (100 × 4,6 мм). Воду и ацетонитрил, содержащие 0,1% TFA, использовали в качестве элюентов при скорости потока 2 мл/мин. ВЭЖХ-MS анализы выполняли на ВЭЖХ системе Agilent 1200, оборудованной колонкой Waters® Hypersil Gold aq (200 × 2,1 мм), после чего следовал масс-спектрометр Thermo Scientific Exactive. Воду и ацетонитрил, содержащие 0,05% TFA, использовали в качестве элюентов при скорости потока 200 мкл/мин.
Мечение ванкомицина радиоактивным131I
Для мечения радиоактивным131I получали 1 ммоль стоковый раствор модельного вещества ванкомицин в PBS. Необходимое количество радиоактивного131I добавляли в 25 мкл стокового раствора и осуществляли мечение с использованием способа с хлорамином T, известного в данной области. Реакционную смесь очищали посредством полупрепаративной ВЭЖХ, как известно в данной области. После этого, чистоту радиоактивного мечения определяли посредством радиоВЭЖХ (Agilent серии 1100) с использованием колонки Chromolithe® Performance RP-C18 (100-3 мм), применяя линейный градиент от 0,1% TFA в воде (элюент A) до 0,1% TFA в ацетонитриле (элюент B) в течение 5 минут; скорость потока 2 мл/мин; λ поглощения УФ=214; γ-обнаружение.
Получение липосом
Липосомы получали DAC-способом с использованием SpeedMixer™ (DAC150FVZ Hauschild Engineering GmbH & Co. KG, Hamm, Germany). Прежде всего, липиды растворяли в хлороформе/метаноле 9:1 для получения 100 ммоль стоковых растворов, тогда как CPP растворяли в хлороформе/метаноле 1:1 (1 ммоль стоковый раствор). Необходимое количество стокового раствора CPP (0,1-1,0 моль-%) добавляли в смесь липидов (85 моль-% EPC, 10 моль-% холестерина и 5 моль-% TEL) и испаряли органический растворитель с помощью потока азота. После этого получаемую липидную пленку сушили в течение 1 ч в вакуумной камере. Перед началом процесса скоростного перемешивания добавляли 20 мг 0,075-1,00 мм стеклянных бус. Липосомы получали посредством скоростного перемешивания и добавления различных количеств встраиваемого вещества (например, ванкомицина - 1 ммоль стоковый раствор, который растворяли в PBS-; раунд 1) или PBS (раунд 2/3) за 3 раунда до общего объема 250 мкл (см. таблицу 1). Для сравнения, аналогичным образом получали стандартные липосомы (85 моль-% EPC; 15 моль-% холестерина).
Таблица 1. Процесс скоростного перемешивания, который выполняли за три раунда
Эффективность инкапсулирования
Эффективность инкапсулирования ванкомицина определяли посредством ВЭЖХ с обращенной фазой (rp) (Agilent серии 1100) с использованием колонки C18 (Chromolithe® Performance RP-18e, 100-3 мм), применяя линейный градиент от 0,1% TFA в воде (элюент A) до 0,1% TFA в ацетонитриле (элюент B) в течение 5 минут; скорость потока 2 мл/мин; λ поглощения УФ=214 нм. После процесса скоростного перемешивания липосомы делили на две части по 100 мкл в каждой. Часть 1 использовали для того, чтобы вычислять 100% значение посредством разрушения липосом добавлением 50 мкл 10% Triton™ X-100 и определения AUC ванкомицина с помощью ВЭЖХ с обращенной фазой. Часть 2 очищали посредством гельфильтрационной хроматографии на Sephadex G-25 (колонка NAP™-5) и после этого осуществляли манипуляции, аналогично части 1. Эффективность инкапсулирования E(%) определяли с помощью следующего уравнения после коррекции разведения части 2:
E(%)=([AUC]части 2 ванкомицина)/([AUC] части 1 ванкомицина)*100%
Где [AUC] части 2 ванкомицина представляет собой очищенную липосомную фракцию и [AUC] части 1 ванкомицина представляет собой 100% значение после получения липосом.
Определение характеристик частиц
1. Размер частицы, индекс полидисперсности (PDI) и дзета-потенциал
Размер частицы, PDI и дзета-потенциал всех липосомных составов определяли при комнатной температуре, используя Zetasizer Nano ZS из Malvern™ (Malvern Instruments Ltd., Worcestershire, United Kingdom). Размер и PDI измеряли после разведения до концентрации 0,076 мг/мл с использованием PBS, тогда как дзета-потенциал определяли после разведения до концентрации 0,95 мг/мл 50 ммоль фосфатным буфером. Измерения проводили с использованием автоматического режима и усредненного для трех измерений. Размер определяли в нм и дзета-потенциал в мВ, тогда как PDI представляет собой безразмерное значение.
2. Криоэлектронные микрофотографии (EM)
Как известно в данной области, чтобы определять размер и слоистую структуру CPP-TEL-липосом, образцы замораживали с использованием FEI Vitrobot на сетках 2/2 Quantifoil. После этого каждый образец обрабатывали тлеющим разрядом в течение 3 с и промакивали при 4°C и 100% влажности в течение 8-10 с. Сетки наблюдали в микроскопе Krios, работающем на 200 кВ и температуре жидкого азота. Изображения CPP-липосомных образцов получали при увеличении 64000×.
Стабильность при длительном хранении
1. Лиофилизация с использованием сахарозы в качестве лиопротектора в различных молярных соотношениях
Все липосомные составы лиофилизировали в Delta 1-20 KD из Christ. Основную сушку осуществляли при -20°C в течение двух суток с последующей вторичной сушкой при 0°C в течение по меньшей мере 6 часов. Сахарозу использовали в качестве лиопротектора в диапазоне 100-500 нм, поскольку ранее обнаружено, что она является хорошим лиопротектором. В кратком изложении, липосомы получали, как описано выше, с тем отличием, что во время 3 раундов взамен буфера PBS добавляли желаемую концентрацию сахарозы в PBD. Липосомную суспензию делили по 50 мкл на сосуд Eppendorf и лиофилизировали. Лиофилизированные липосомы регидратировали в 50 мкл PBS и определяли размер и PDI, используя Zetasizer Nano ZS из Malvern™, как описано выше.
2. Остаточная влага в лиофилизатах
Остаточную влагу в CPP-TEL-липосомах определяли с помощью влагомера (Kern&Sohn, Balingen, Germany), используя 100 мг лиофилизированных липосом посредством нагрева вплоть до 120°C в течение 90 с.
Проверка концепции: исследования у животных
1. Уровни131I-меченного ванкомицина в крови для различных составов
Исследование у животных осуществляли в соответствии с местными нормами, используя самцов крыс Вистар с массой тела приблизительно 220-250 г. Для того чтобы определять уровни в крови для различных составов, ванкомицин радиоактивно метили с использованием131I и встраивали в несколько липосомных составов. Уровни ванкомицина в крови через 1 ч после перорального введения измеряли путем прямого счета радиоактивности с использованием счетчика Berthold LB 951 G в сравнении со стандартами. В кратком изложении, формировали пять групп (n=6) крыс Вистар. В сутки перед экспериментом крыс предварительно лечили суспендированным Antra MUPS™ (омепразол) посредством принудительного кормления (10 мг на крысу), поскольку недавние исследования показывают, что предварительное лечение ингибитором протонной помпы омепразолом снижает диффузию протонов в липосомы и, как следствие, снижает денатурацию инкапсулированных средств за счет повышения pH в желудке. Крыс держали без пищи 12 ч перед экспериментом, но при свободном доступе к воде. Пероральное применение происходило посредством принудительного кормления. Крыс умерщвляли через 1 ч, брали образцы крови, осуществляли взвешивание и измеряли радиоактивность в образцах крови, используя счетчик Berthold LB 951 G, в сравнении со стандартами. Ассоциированную с кровью активность связывали с полной инъецированной дозой (ID) и выражали в виде процентной доли полной инъецированной дозы на грамм ткани (%ID). После этого вычисляли общий уровень в крови (%ID).
2. Определение уровней ванкомицина в крови посредством иммунологического анализа
Для того чтобы верифицировать значения, получаемые посредством измерения радиоактивных образцов крови, осуществляли иммунологический анализ ванкомицина (ADVIA Centaur® VANC ReadyPack®, Siemens, Tarrytown, USA) в CPP-TEL-ванкомицин-липосомном составе. Подобно исследованию радиоактивного мечения, 500 мкл CPP-TEL-ванкомицин-липосом вводили 250 г самцам крыс Вистар (n=6) посредством принудительного кормления. В сутки перед экспериментом, крыс предварительно лечили суспендированным Antra MUPS™ (омепразол) посредством принудительного кормления (10 мг на крысу). Через 1 ч после перорального введения крыс умерщвляли, брали образцы крови, отделяли плазму и определяли количество ванкомицина посредством иммунологического анализа, описанного выше.
Пример 1:
Выделение TEL
В процедуре выделения получали тетраэфирные липиды в очищенной форме лишь с небольшими вариациями числа пентильных колец (от 3 до 5) в липофильных цепях, как определяют посредством масс-спектрометрии и тонкослойной хроматографии. На число колец влияет температура во время культивирования архей. Усредненно можно получать 1 г тетраэфирных липидов на 400 г влажной клеточной массы.
Пример 2:
Синтез и определение характеристик конъюгата CPP-фосфолипида
Конъюгат CPP (пенетратина)-фосфолипида можно получать с высокой чистотой, как определяют посредством масс-спектрометрии (фиг. 5). Общий выход продукта составлял приблизительно 59% после очистки посредством препаративной ВЭЖХ.
Пример 3:
Радиоактивное мечение ванкомицина с использованием131I
Радиоактивное мечение ванкомицина с использованием131I давало желаемый продукт высокой чистоты (> 95%), как определяют посредством радиоВЭЖХ (фиг. 6). Эффективность мечения с использованием способа с хлорамином T составляла приблизительно 60% от внесенной радиоактивности.
Пример 4:
Эффективность инкапсулирования
CPP-TEL-ванкомицин-липосомы демонстрировали эффективность инкапсулирования 35,38±1,65%, которая сравнима с установленным значением для стандартных липосом (38,73±1,23%).
Пример 5:
Определение характеристик частиц
1. Размер частицы, индекс полидисперсности (PDI) и дзета-потенциал
Применяемым DAC-способом получали CPP-TEL-ванкомицин-липосомы с высокой однородностью размеров, PDI и эффективности встраивания (см. таблицу 2). По сравнению со стандартными липосомами, размер и PDI были слегка увеличены, хотя CPP-TEL-ванкомицин-липосомы характеризуются более положительным дзета-потенциалом, чем стандартные липосомы.
Таблица 2. Определение характеристик частиц CPP-TEL и стандартных липосом
2. Криоэлектронные микрофотографии (EM)
На криоэлектронной микрофотографии (фиг. 7) показаны размеры и слоистая структура CPP-TEL-ванкомицин-липосом, содержащих 85 моль-% лецитина; 10 моль-% холестерина; 5 моль-% TEL и 0,1 моль-% пенетратина. Большинство изображенных липосом демонстрируют от одного соответственно вплоть до трех ламеллярных слоев.
Пример 6:
Стабильность при длительном хранении
1. Лиофилизация с использованием сахарозы в качестве лиопротектора при различных молярных соотношениях
Лиофилизация CPP-TEL-липосом, содержащих сахарозу в различных молярных соотношениях в качестве лиопротектора, приводила к сравнимым размерам и PDI для определенных молярных соотношений по сравнению с данными, которые измеряли до процесса лиофилизации (фиг. 8A, B). Что касается этого липосомного состава, наименьший предел сахарозы составляет 300 ммоль, на наилучшие результаты получали с использованием сахарозы в концентрации 500 мМ. Дальнейшее увеличение концентрации лиопротектора (больше 500 ммоль сахарозы) не обеспечивает более хороших результатов в отношении размеров и PDI липосом.
2. Остаточная влага после процесса лиофилизации
Из-за очень низкой стабильности липосомных суспензий, желательно получать лиофилизаты, которые демонстрируют очень низкую остаточную влагу для того, чтобы обеспечивать долгосрочную стабильность лиофилизированных липосом. Остаточная влага в CPP-TEL липосомном составе, содержащем 500 ммоль сахарозы в качестве лиопротектора, составляла 2,88%±0,79%.
Пример 7:
Проверка концепции: исследования у животных
1. Уровни131I-меченного ванкомицина в крови для различных составов
Исследование для проверки концепции показало 3-кратное увеличение уровней ванкомицина в крови для CPP-TEL-липосом по сравнению со стандартными липосомами. Добавление только одного дополнительного компонента (CPP или TEL) давало только небольшое увеличение уровней ванкомицина в крови. Это подчеркивает необходимость обоих компонентов в одной липосомной композиции, чтобы предоставлять инструмент для пероральной доставки макромолекул (фиг. 9).
2. Определение уровней ванкомицина в крови посредством иммунологического анализа
Определение уровней ванкомицина в крови посредством иммунологического анализа давало результаты, сравнимые с исследованием радиоактивного мечения (7,83% против 8,68%) (фиг. 10).
Пример 8:
В настоящем изобретении создан новый класс конъюгатов CPP-фосфолипидов. Использовали два различных варианта CPP (фиг. 11), с одной стороны линейные CPP (модельный CPP пенетратин, d/l-форма) и с другой стороны циклические CPP (циклическое производное R9). Циклические CPP использовали из-за более высокой ферментативной стабильности в интестинальном текучем веществе, как обнаруживали посредством масс-спектрометрии (фиг. 13). Кроме того, можно показать, что линейный CPP пенетратин более стабилен, если он состоит из D-аминокислот.
На фиг. 12 представлены четыре модельные вещества ванкомицин, инсулин, лираглутид и антитело адалимумаб (Хумира®). Эти вещества охватывают полный диапазон молекулярных масс макромолекулярных лекарственных средств и демонстрируют, что новые липосомные составы по настоящему изобретению могут служить в качестве платформы для макромолекулярных лекарственных средств в целом.
Способ анализа стабильности CPP в интестинальном текучем веществе - стабильность CPP в имитированном интестинальном текучем веществе.
Все CPP (1 мг/мл в воде) разводили 1:1 (об./об.) имитированным интестинальным текучим веществом и инкубировали при 37°C при постоянном встряхивании, как известно в данной области. После 0, 15, 30 и 60 мин образцы анализировали посредством ВЭЖХ/MS для того, чтобы обнаруживать извлечение интактного CPP, и сравнивали с начальным раствором.
Пример 9:
Тестировали различные липосомные составы с модельным веществом ванкомицин у самцов крыс Вистар (200-250 г). Измерение уровней радиоактивно меченного ванкомицина в крови показывало значительное увеличение пероральной доступности ванкомицина с использованием липосомного состава, содержащего 1 моль-% конъюгата линейного CPP-фосфолипида. Тем не менее, другой состав, содержащий только 0,1 моль-% конъюгатов циклических CPP-фосфолипидов, демонстрировал более выраженное увеличение уровней ванкомицина в крови, которому сопутствовал такой эффект, что в случае конъюгатов необходимы незначительные количества. Полученные результаты проверяли посредством иммунологического анализа ванкомицина, и можно было измерять схожие уровни в крови (фиг. 14).
Способ для исследования у животных с использованием гликопептидного лекарственного средства ванкомицин в качестве модельного вещества
Исследование у животных осуществляли в соответствии с местными нормами, используя самцов крыс Вистар с массой тела приблизительно 200-250 г. В исследовании для проверки концепции ванкомицин радиоактивно метили с использованием131I и встраивали в различные липосомные составы, как описано ранее. Через 1 ч после перорального введения уровни ванкомицина в крови измеряли посредством прямого счета в радиоактивно меченном образце. В кратком изложении, формировали группы (n ≥ 3) самцов крыс Вистар. За 12 ч до эксперимента крыс оставляли без пищи, но со свободным доступом к воде. Пероральное применение липосом и свободного пептида происходило посредством принудительного кормления. Кровь брали, взвешивали и измеряли радиоактивность с использованием счетчика Berthold LB 951 G в сравнении со стандартами. Ассоциированную с кровью активность связывали с полной инъецированной дозой (ID) и выражали в виде процентной доли от полной инъецированной дозы на грамм ткани (%ID/г). Статистические данные обрабатывали с использованием программного обеспечения Prism® (GraphPad Software, San Diego, CA, USA) и представляли в виде среднего±стандартное отклонение среднего (S.D.). Различные группы в исследовании у животных сравнивали с помощью однофакторного дисперсионного анализа с использованием программного обеспечения Prism® и при уровне значимости *p < 0,05, **p < 0,01 и ***p < 0,001 (фиг. 14).
Пример 10:
В одном дополнительном исследовании исследовали биодоступность пептидного лекарственного средства лираглутид. По этой причине лекарственное средство инъецировали i.v. и уровни в крови сравнивали с пероральным липосомным составом, содержащим 1 моль-% циклических CPP-липосом. В отношении этого пептидного лекарственного средства, биодоступность липосомного состава авторов изобретения составляет > 50% в сравнении с i.v. применением лираглутида (фиг. 15).
Способ для исследований у животных с использованием лираглутида в качестве модельного вещества для определения биодоступности.
Исследование у животных осуществляли в соответствии с местными нормами, используя самцов крыс Вистар с массой тела приблизительно 200-250 г. Для определения биодоступности антидиабетического лекарственного средства лираглутид радиоактивно метили131I и встраивали в липосомный состав, содержащий 1 моль-% конъюгата циклического CPP-фосфолипида, и сравнивали с лираглутидом i.v., как описано ранее. В несколько моментов времени уровни лираглутида в крови измеряли посредством прямого счета в радиоактивно меченном образце. В кратком изложении, формировали группы (n ≥ 3) самцов крыс Вистар. За 12 ч до эксперимента крыс оставляли без пищи, но со свободным доступом к воде. Пероральное применение липосом происходило посредством принудительного кормления. Кровь брали, взвешивали и измеряли радиоактивность с использованием счетчика Berthold LB 951 G в сравнении со стандартами. Ассоциированную с кровью активность связывали с полной инъецированной дозой (ID) и выражали в виде процентной доли от полной инъецированной дозы на грамм ткани (%ID/г). Статистические данные обрабатывали с использованием программного обеспечения Prism® (GraphPad Software, San Diego, CA, USA) и представляли в виде среднего±стандартное отклонение среднего (S.D.) (фиг. 15).
Пример 11:
В одном дополнительном пилотном исследовании исследовали пероральную доступность адалимумаба (Хумира®). По этой причине антитело радиоактивно метили131I, встраивали в липосомы, содержащие 1 моль-% конъюгата циклического CPP, перорально вводили и сравнивали с радиоактивно меченным свободным пептидом. Эффективность встраивания антитела в липосомы составляла приблизительно 60%, как обнаруживали посредством прямого измерения радиоактивно меченного антитела до и после очистки. Снова можно обнаруживать сильное увеличение пероральной доступности адалимумаба (фиг. 16). Следовательно можно заявить, что этот новый липосомный состав также подходит для макромолекулярных лекарственных средств с молекулярной массой > 100 кДа, таких как антитела.
Способ для исследования у животных с использованием антитела адалимумаб (Хумира®) в качестве модельного вещества для определения биодоступности (абсолютной).
Исследование у животных осуществляли в соответствии с местными нормами, используя самцов крыс Вистар с массой тела приблизительно 200-250 г. Для определения биодоступности (абсолютной), антитело адалимумаб (Хумира®) радиоактивно метили131I и встраивали в липосомный состав, содержащий 1 моль-% конъюгата циклического CPP-фосфолипида и сравнивали со свободным антителом, как описано ранее. После 6 ч уровни в крови измеряли посредством прямого счета в радиоактивно меченном образце. За 12 ч до эксперимента крысы оставляли без пищи, но со свободным доступом к воде. Пероральное применение липосом и радиоактивно меченного свободного антитела происходило посредством принудительного кормления. Кровь брали, взвешивали и радиоактивность измеряли с использованием счетчика Berthold LB 951 G в сравнении со стандартами. Ассоциированную с кровью активность связывали с полной инъецированной дозой (ID) и выражали в виде процентной доли от полной инъецированной дозы на грамм ткани (%ID/г) (фиг. 16).
Пример 12:
В другом исследовании исследовали влияние линкера, используемого для соединения CPP с фосфолипидом с модифицированной головной группой, и можно показать, что эффективность встраивания пептидных лекарственных средств зависит от количества звеньев PEG линкера (фиг. 17). На фиг. 18 в липосомы встраивали некоторые количества (0-3 моль-%) конъюгатов CPP-фосфолипидов с использованием различных линкеров и после этого определяли размер липосом и PDI. Можно показать, что PEG-линкеры демонстрируют наилучшие результаты, где предпочтительны PEG-линкеры, состоящие из 8-50 индивидуальных звеньев PEG.
Способ исследования различных PEG-линкеров.
Эффективность инкапсулирования пептидных лекарственных средств определяли посредством ВЭЖХ с обращенной фазой (Agilent серии 1100), используя колонку C18 (Chromolith® Performance RP-18e, 100-3 мм), с применением линейного градиента от 0,1% TFA в воде (элюент A) до 0,1% TFA в ацетонитриле (элюент B) в течение 5 минут (скорость потока 2 мл/мин; λ поглощения УФ=214 нм). После процесса скоростного перемешивания липосомы делили на две части по 100 мкл каждая. Часть 1 использовали для того, чтобы вычислять 100% значение, получаемое посредством разрушения липосом добавлением 50 мкл 1% Triton™ X-100 и определения площади под кривой (AUC) для пептидного лекарственного средства с помощью ВЭЖХ. Часть 2 очищали посредством гельфильтрационной хроматографии на Sephadex G-25 (колонки NAP™-5) и количественно определяли, как часть 1. Для того чтобы определять потенциальную потерю липидов на колонках NAP™-5 во время очистки части 2, концентрацию холестерина в липосомной суспензии измеряли непосредственно после процесса скоростного перемешивания и после очистки с использованием колонок NAP™-5. Для обоих измерений липосомы растворяли 1:10 (об./об.) в метаноле. Холестерин количественно определяли посредством ВЭЖХ, применяя изократический градиент ацетонитрила/метанола (80:20 об./об.) в течение 15 минут (скорость потока 2 мл/мин; λ поглощения УФ=208 нм) на колонке RP-18. Сравнивали концентрацию холестерина до и после стадии очистки и определяли коэффициент коррекции C для того, чтобы включать потерю липидов на колонках NAP™-5 в вычисление эффективности инкапсулирования. Эффективность инкапсулирования E (%) вычисляли с использованием следующего уравнения:
E (%)=([AUC] части 2 пептидного лекарственного средства)/([AUC] части 1 пептидного лекарственного средства) × 100% × C
В соответствии с чем [AUC] части 2 пептидного лекарственного средства представляет собой концентрацию пептидного лекарственного средства в очищенной липосомной фракции и [AUC] части 1 пептидного лекарственного средства представляет собой концентрацию пептидного лекарственного средства в липосомной суспензии. C представляет собой коэффициент коррекции для учета потери липидов на колонках NAP™-5.
Пример 13:
После очистки липосомной суспензии посредством эксклюзионной хроматографии, дзета-потенциал CPP-модифицированных липосом демонстрировал сильное увеличение по сравнению с немодифицированными липосомами из-за положительно заряженных аминокислот в CPP (фиг. 19), что подтверждает успешное встраивание конъюгатов в липосомы.
Способы измерения дзета-потенциала.
Дзета-потенциал липосом определяли при комнатной температуре с использованием автоматического режима Zetasizer Nano ZS из Malvern™ (Malvern Instruments Ltd., Worcestershire, United Kingdom). Размер частицы, PDI и дзета-потенциал всех липосомных составов определяли при комнатной температуре, используя Zetasizer Nano ZS из Malvern™ (Malvern Instruments Ltd., Worcestershire, United Kingdom). Размер и PDI измеряли после разведения до концентрации липидов 0,076 мг/мл в 10 мМ фосфатном буфере с pH 7,4, используя автоматический режим. Дзета-потенциал определяли после разведения до концентрации липидов 0,95 мг/мл 50 мМ фосфатным буфером с pH 7,4. Настройки по умолчанию для автоматического режима Zetasizer Nano ZS из Malvern™ (Malvern Instruments Ltd., Worcestershire, United Kingdom) представляют собой следующее: число измерений=3; длительность раунда=10 с; число раундов=10; время установления равновесия=60 м; показатель преломления растворителя 1,330; показатель преломления полистироловой кюветы 1,590; вязкость=0,8872 мПа×с; температура=25°C; диэлектрическая постоянная=78,5 Ф/м; режим обратного рассеяния (173°); автоматический выбор напряжения; уравнение Смолуховского, фосфатный буфер pH 7,4.
Пример 14:
Связывающую способность липосом, содержащих конъюгаты циклических CPP-фосфолипидов, в сравнении со стандартными липосомами тестировали посредством радиоактивного мечения антитела матузумаб. Липосомы, содержащие новый конъюгат, демонстрировали значительно более высокую связывающую способность, чем стандартные липосомы (фиг. 20).
Способы анализа связывания.
Клетки Caco-2 высевали в 6-луночные планшеты. После дифференцировки в течение 2 недель, 1 мл образца липосом добавляли после удаления среды для выращивания. Клетки промывали фосфатным буфером после инкубации при 37°C в течение 15, 30 и 60 минут. После этого клетки инкубировали в течение 5 мин с 1 мл 1 моль глицинового буфера с pH 2,2. Добавляли 0,3 моль NaOH и осуществляли лизис клеток с помощью 0,25% SDS. Собирали глициновый буфер и фракцию лизиса клеток и количество радиоактивности измеряли с использованием счетчика Berthold LB 951 G в сравнении со стандартами.
Пример 15:
В одном дополнительном исследовании исследовали биодоступность (абсолютную) пептидного лекарственного средства ванкомицин. По этой причине лекарственное средство инъецировали i.v. и уровни в крови сравнивали с пероральным липосомным составом, содержащим 1 моль-% циклических CPP-липосом. В отношении этого пептидного лекарственного средства, биодоступность для липосомного состава авторов изобретения составляет вплоть до 50% в сравнении с i.v. применением ванкомицина (фиг. 21).
Способ испытания у животных ванкомицина в качестве модельного вещества для определения биодоступности (абсолютной).
Исследование у животных осуществляли в соответствии с местными нормами, используя самцов крыс Вистар с массой тела приблизительно 200-250 г. Для определения биодоступности (абсолютной) гликопептидный антибиотик ванкомицин радиоактивно метили131I и встраивали в липосомный состав, содержащий 1 моль-% конъюгата циклического CPP-фосфолипида, и сравнивали с ванкомицином i.v., как описано ранее. В несколько моментов времени уровни ванкомицина в крови измеряли посредством прямого счета в радиоактивно меченном образце. В кратком изложении, формировали группы (n ≥ 3) самцов крыс Вистар. За 12 ч до эксперимента крыс оставляли без пищи, но со свободным доступом к воде. Пероральное применение липосом происходило посредством принудительного кормления. Кровь брали, взвешивали и измеряли радиоактивность с использованием счетчика Berthold LB 951 G в сравнении со стандартами. Ассоциированную с кровью активность связывали с полной инъецированной дозой (ID) и выражали в виде процентной доли от полной инъецированной дозы на грамм ткани (%ID/г).
Пример 16:
В другом исследовании определяли эффективность встраивания двух антител, а именно адалимумаб и цетуксимаб, как описано ранее (фиг. 22). Можно показать, что оба антитела также демонстрируют высокие эффективности встраивания, сравнимые с пептидными лекарственными средствами (приблизительно 50-70%).
Пример 17:
Для того чтобы определять обогащение опухоли цетуксимабом, встроенным в липосомы, содержащие 1 моль-% конъюгата циклического CPP, соответствующие липосомы вводили мышам-опухоленосителям. Для получения этих мышей клеточную линию A431 инъецировали мышам Balb/c (сверхэкспрессия EGFR). Антитело цетуксимаб радиоактивно метили, встраивали в липосомы и после этого применяли перорально у мышей. Можно показать значительное увеличение накопления в опухоли при использовании липосомного состава в сравнении со свободным пептидом (фиг. 23).
Способ определения накопления антитела цетуксимаб в опухоли.
Исследование у животных осуществляли в соответствии с местными нормами, используя самцов крыс Вистар с массой тела приблизительно 200-250 г. Для определения обогащения опухоли цетуксимабом, антитело радиоактивно метили131I и встраивали в липосомный состав, содержащий 1 моль-% конъюгата циклического CPP-фосфолипида и сравнивали со свободным цетуксимабом i.v., как описано ранее. В несколько моментов времени обогащение опухоли измеряли посредством прямого счета в радиоактивно меченном образце. В кратком изложении, формировали группы (n ≥ 3) самцов крыс Вистар. За 12 ч до эксперимента, крыс оставляли без пищи, но со свободным доступом к воде. Пероральное применение липосом происходило посредством принудительного кормления. Кровь брали, взвешивали и радиоактивность измеряли с использованием счетчика Berthold LB 951 G в сравнении со стандартами. Ассоциированную с кровью активность связывали с полной инъецированной дозой (ID) и выражали в виде процентной доли от полной инъецированной дозы на грамм ткани (%ID/г опухоли).
Пример 18:
В одном дополнительном исследовании исследовали другой CPP (MAP) для того, чтобы определять характеристики частиц. Конъюгаты синтезировали и встраивали в липосомы в диапазоне 0-1 моль-%. Размер частицы был схож со стандартными липосомами, а дзета-потенциал демонстрировал повышение, которое указывало на успешное встраивание конъюгата в липосомы (фиг. 24).
Способ определения характеристик частиц.
Размер частицы, PDI и дзета-потенциал всех липосомных составов определяли при комнатной температуре, используя Zetasizer Nano ZS из Malvern™ (Malvern Instruments Ltd., Worcestershire, United Kingdom). Размер и PDI измеряли после разведения до концентрации липидов 0,076 мг/мл 10 мМ фосфатным буфером с pH 7,4, используя автоматический режим. Дзета-потенциал определяли после разведения до концентрации липидов 0,95 мг/мл 50 мМ фосфатным буфером с pH 7,4. Настройки по умолчанию для автоматического режима Zetasizer Nano ZS из Malvern™ (Malvern Instruments Ltd., Worcestershire, United Kingdom) представляли собой следующее: число измерений=3; длительность раунда=10 с; число раундов=10; время установления равновесия=60 с; показатель преломления растворителя 1,330; показатель преломления полистироловой кюветы 1,590; вязкость=0,8872 мПа×с; температура=25°C; диэлектрическая постоянная=78,5 Ф/м; режим обратного рассеяния (173°); автоматический выбор напряжения; уравнение Смолуховского.
Заключение:
В этом изобретении можно создавать перспективную новую пероральную систему доставки для различных, например, пептидных лекарственных средств, таких как ванкомицин, лираглутид, инсулин, и антител, таких как адалимумаб или цетуксимаб, с помощью различных CPP-TEL-липосом. Эти результаты (значительно усиленный захват слизистыми) демонстрируют, что эта новая технология может служить в качестве технологической платформы для макромолекулярных лекарственных средств (пептидов, белков и антител) в целом. Все CPP-TEL-липосомы демонстрировали высокую однородность размеров, PDI и эффективности встраивания (>50%). Анализ стабильности в имитированном интестинальном текучем веществе демонстрировал, что конъюгаты циклических CPP-фосфолипидов более стабильны, чем линейные аналоги, в отношении линейных CPP, встраивание D-аминокислот является предпочтительным. В отношении нескольких линкеров, различающихся количеством звеньев PEG, можно показать, что эффективность инкапсулирования выше, если используют PEG-линкеры с 8-50 звеньями PEG. Для того чтобы демонстрировать, что характеристики липосом можно переносить на другие CPP, CPP MAP встраивали в липосомы, которые демонстрировали характеристики, схожие с другими CPP. Все липосомные составы не демонстрировали цитотоксичности при всех тестируемых концентрациях. Кроме того, длительное хранение CPP-TEL-липосом можно обеспечивать посредством лиофилизации с использованием сахарозы в качестве лиопротектора. В исследовании для проверки концепции с использованием самцов крыс Вистар (n=6), можно обнаруживать 3-кратно повышенную концентрацию радиоактивно131I меченного ванкомицина в крови по сравнению со стандартными липосомами. Для того чтобы подтверждать эти результаты, концентрацию в крови также определяли посредством иммунологического анализа, который также показывал сравнимое обогащение уровня ванкомицина в крови (7,83%ID против 8,68%ID) при использовании CPP-TEL-липосомного состава. Эти результаты (значительно повышенный захват липосомных составов слизистыми) можно подтвердить для дополнительных веществ, таких как лираглутид; инсулин; адалимумаб и цетуксимаб, посредством радиоактивного мечения веществ и определения радиоактивности в определенные моменты времени после перорального применения.
Взятые вместе, результаты демонстрируют потенциал CPP-TEL-липосомного состава в соответствии с настоящим изобретением для перорального применения всех макромолекулярных лекарственных средств в качестве технологической платформы.
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> Universit?t Heidelberg
<120> ЛИПОСОМЫ, ПРОНИКАЮЩИЕ В КЛЕТКИ, ДЛЯ ОРАЛЬНОЙ ДОСТАВКИ МАКРОМОЛЕКУЛ
<130> H 3654WO - kl
<150> EP 15 003 011.2
<151> 21.10.2015
<160> 7
<170> PatentIn version 3.5
<210> 1
<211> 16
<212> Белок
<213> Drosophila melanogaster
<400> 1
Arg Gln Ile Lys Ile Trp Phe Gln Asn Arg Arg Met Lys Trp Lys Lys
1 5 10 15
<210> 2
<211> 16
<212> Белок
<213> Вирус иммунодефицита человека 1 типа
<400> 2
Cys Gly Arg Lys Lys Lys Arg Arg Gln Arg Arg Arg Pro Pro Gln Cys
1 5 10 15
<210> 3
<211> 27
<212> Белок
<213> Искусственная последовательность
<220>
<223> искусственно разработанный пептид, проникающий в клетки
<400> 3
Gly Ala Leu Phe Leu Gly Phe Leu Gly Ala Ala Gly Ser Thr Met Gly
1 5 10 15
Ala Trp Ser Gln Pro Lys Ser Lys Arg Lys Val
20 25
<210> 4
<211> 9
<212> Белок
<213> Искусственная последовательность
<220>
<223> искусственно разработанный пептид, проникающий в клетки
<400> 4
Arg Arg Arg Arg Arg Arg Arg Arg Arg
1 5
<210> 5
<211> 18
<212> Белок
<213> Mus musculus
<220>
<221> смешанные признаки
<222> (18)..(18)
<223> K-амид
<400> 5
Leu Leu Ile Ile Leu Arg Arg Arg Ile Arg Lys Gln Ala His Ala His
1 5 10 15
Ser Lys
<210> 6
<211> 28
<212> Белок
<213> Homo sapiens
<220>
<221> смешанные признаки
<222> (28)..(28)
<223> L-амид
<400> 6
Gly Trp Thr Leu Asn Ser Ala Gly Tyr Leu Leu Gly Lys Ile Asn Leu
1 5 10 15
Lys Ala Leu Ala Ala Leu Ala Lys Ile Ser Ile Leu
20 25
<210> 7
<211> 27
<212> Белок
<213> Human immunodeficiency virus
<400> 7
Gly Ala Leu Phe Leu Gly Phe Leu Gly Ala Ala Gly Ser Thr Met Gly
1 5 10 15
Ala Trp Ser Gln Pro Lys Ser Lys Arg Lys Val
20 25
<---
Реферат
Группа изобретений относится к области медицины и фармацевтики. Первый объект представляет собой липосомную композицию, содержащую липосомы, для пероральной доставки пептидного лекарственного средства, белка или антитела, причем указанные липосомы содержат тетраэфирные липиды (TEL) и проникающие в клетки пептиды (CPP), где указанные CPP присоединены к соединению, которое является частью липидного бислоя липосомы. Второй и третий объекты представляют собой применение липосомной композиции для пероральной доставки пептидного лекарственного средства, белка или антитела. Технический результат заключается в защите пептидного лекарственного средства, белка или антитела при прохождении через желудок и улучшении абсорбции через желудочно-кишечный барьер с помощью композиции по изобретению. 3 н. и 12 з.п. ф-лы, 24 ил., 2 табл., 18 пр.
Комментарии